| • | | The Dynamic Polyconjugate® (DPC®) platform·
| ARC-LPA is an RNAi-based therapeutic designed to reduce production of apolipoprotein A (apo(a)), a small RNA delivery system that may be targeted to address multiple organ systems and cell types. It is a modular system that may be optimized on a target-by-target basis andkey component of lipoprotein(a) (Lp(a)), which has been demonstrated to promote multi-log gene knockdown in rodents and non-human primates, induce efficient endosomal escape, andgenetically linked with increased risk of cardiovascular diseases. ARC-LPA employs Arrowhead’s new hepatic delivery format being developed for subcutaneous administration. This program has wide safety margins using a variety of siRNA molecules.not yet been designated by Arrowhead as entering pre-IND development. |
RONDEL is a small RNA delivery system that has demonstrated effective systemic siRNA delivery, RNAi-mediated mRNA and protein knockdown in human melanoma patients.
Arrowhead’s Homing Peptides platform is a vast, proprietary library of short peptides that have demonstrated rapid and specific internalization into a wide variety of cell types. This library is being mined for the potential development of peptide-drug conjugates (PDCs) and companion diagnostics. Arrowhead plans to develop the targeting peptides for use with its RNA delivery platforms as well as with traditional small molecule or peptide drugs.
Primary Strategic Opportunities
RNA interference (RNAi) is a naturally occurring mechanism that effects gene expression. Short interfering RNAs (siRNAs) have been shown to trigger RNAi and are thought to be a potentially powerful and specific way of silencing expression of disease-causing gene products. However, the lack of effective and safe delivery has impeded progress of the field. Arrowhead has multiple polymer-based, non-lipid delivery systems that enable development of RNAi therapeutics. Importantly, Arrowhead’s delivery systems have been validated in multiple species and have demonstrated high levels of efficiency and specificity with wide safety margins.
Examples of guided therapeutics producing positive patient outcomes are rapidly emerging. Arrowhead’s human-derived targeting library, comprised of over 42,000 peptides, is being mined to create PDCs designed to home specifically to target cells. PDCs have the potential to produce the advantages of antibody drug conjugates while bringing new benefits such as ease of manufacturing.
1
Recent EventsArrowhead made significant progress on product and platform development during fiscal year 2015 with an expanding pipeline of RNAi therapeutics based on the Dynamic Polyconjugate (DPC™) delivery system. The following are highlights of this progress: | • | | · | Strengthened management team and R&D staff with select key hires and expanded internal capabilities in manufacturing, RNA synthesis, toxicology, QA, program management, and clinical and regulatory operations |
| · | Presented data on ARC-520 and ARC-AAT at AASLD Liver Meeting® 2014 |
ARC-520 showed statistically significant reduction in HBsAg through day 43 after a single injection (p < 0.05) Repeat dosing of ARC-AAT in primates showed reduction of approximately 90% of serum alpha 1 antitrypsin (AAT) with long duration of effect suggesting that monthly or less frequent dosing may be sufficient for sustained suppression of hepatic AAT production ARC-AAT abstract highlighted in the AASLD President's Press Conference as a promising new treatment | · | Filed for regulatory approval to begin a Phase 1 clinical trial of ARC-AAT for the treatment of liver disease associated with alpha-1 antitrypsin deficiency |
| · | Submitted an Investigational New Drug (IND) application to the U.S. Food and Drug Administration and submitted additional clinical trial authorization applications with regulatory authorities in various jurisdictions in Europe, Asia, and Australia/New Zealand for ARC-520 |
| · | Initiated dosing in a Phase 1 clinical trial of ARC-AAT |
| · | Acquired Novartis Institutes for BioMedical Research, Inc (“Novartis”) entire RNAi research and development portfolio and associated assets, including: |
Multiple patent families covering RNAi-trigger design rules and modifications that fall outside of key patents controlled by competitors, which the Company believes provides freedom to operate for any target and indication Novel intracellular targeting ligands that enhance the activity of RNAi-triggers by targeting the RNA-induced silencing complex (RISC) more effectively and improving stability once RISC is loaded An assignment of Novartis' license from Alnylam Pharmaceuticals, Inc. (“Alnylam”) granting Arrowhead access to Alnylam intellectual property, excluding delivery, for 30 gene targets chosen by Novartis A pipeline of three candidates initiated by Novartis and for which Novartis has developed varying amounts of preclinical data | · | Initiated dosing in Heparc-2004, a multiple-dose Phase 2b clinical study of ARC-520 in the U.S. |
| · | Published new data in the Journal of Controlled Release, 209 (2015) 57-66, on a subcutaneously administered formulation of its DPC™ delivery system |
| · | Completed dosing of Part A of the ARC-AAT phase 1 study in healthy volunteers, and transitioned the study into Part B in patients with PiZZ genotype alpha-1 antitrypsin deficiency |
| · | Presented data at the TIDES Conference on the development of ARC-F12, an RNAi therapeutic for factor 12 (F12) mediated hereditary angioedema and thromboembolic diseases |
| · | Received Orphan Drug Designation from the United States Food and Drug Administration |
| · | Initiated multiple-dose Heparc-2002 and Heparc-2003 Phase 2b studies of ARC-520 in Europe and Asia |
2
| · | Patient Population Enrichment StrategiesExpanded Part B of the Phase 1 study of ARC-AAT to include additional treatment sites in Europe, Australia, and New Zealand
|
Arrowhead’s targeting library can be used for companion diagnostics that identify patient populations most likely to respond to a particular treatment, thus moving toward more personalized medicine.
| • | | Improving Generics·
| Nominated ARC-HIF2 against clear cell renal cell carcinoma as Arrowhead’s first therapeutic candidate delivered using a new DPC™ designed to target tissues outside of the liver |
| · | Hosted an analyst day to discuss top-line findings from the Heparc-2001 Phase 2a clinical study of ARC-520 and findings from a study of 9 chimpanzees that have been treated monthly with ARC-520 for between 6 and 11 months. Key messages included the following: |
Arrowhead’s targeting libraryArrowhead's proprietary DPC™ platform can be used to make PDCs with generics designed to have an improved efficacyeffectively and safety profile as compared to untargeted counterparts.consistently knock down target genes in humans
Arrowhead’s internal drug pipeline is intended to drive value directly through the development of novel therapeuticsARC-520 achieves significant HBV s-Antigen (HBsAg) reductions in humans, particularly in treatment naïve, HBeAg-positive patients
Arrowhead identifies a large target HBV population for ARC-520 and to provide proof of conceptdescribes a new paradigm for the platform technologies. We actively seek collaborationHBV lifecycle ARC-520 induces deep HBsAg reduction in chronically HBV infected chimps ARC-520 has been well tolerated Arrowhead expands its HBV portfolio by nominating an additional clinical candidate that is complementary to ARC-520 | · | Presented data on ARC-LPa against cardiovascular disease, which uses a new subcutaneous delivery construct that Arrowhead has developed |
| · | Presented data at the AASLD Liver Meeting 2015 including the following: |
ARC-520 led to robust, sustained anti-viral effects in chimpanzees with chronic HBV and licensing agreementsalso described an important new discovery that HBV DNA integrated into the host genome is likely an important source of HBV surface antigen (HBsAg) production In a Phase 2a clinical study, ARC-520 effectively reduced HBV viral antigens derived from cccDNA. HBV surface antigen (HBsAg) was reduced substantially with leading biopharmaceutical companies to augment their pipelines through the applicationa maximum reduction of our technologies1.9 logs (99%) and to advance the developmenta mean maximum reduction of 1.5 logs (96.8%) in treatment naïve e-antigen (HBeAg)-positive patients | · | Presented data at Hep DART 2015 showing that ARC-520 led to immune reactivation in 7 of 9 chimps with chronic hepatitis B infection |
Acquisition of Roche and commercialization of our own technology platformsNovartis RNAi business and drug candidates. Partnerships are intended to provide access to external expertise and capital to complement our internal development and create commercialization opportunities in areas outside of our core focus.assets RECENT EVENTS
Fiscal 2012The last four years have brought substantial change to Arrowhead. We have executed on our long-term strategy of transitioning from a nanotechnology holding company in multiple industries to a focused biotech model.Arrowhead’s research and development (R&D) capabilities and strategy. We are now a unifiedan integrated RNAi therapeutics company, developing actively guidednovel drugs that interact preferentially with target tissuessilence disease causing genes based on our broad RNAi and peptide targeting technology platforms.platform.
Arrowhead made two acquisitions in fiscal 2012. These acquisitions included new technology platforms, R&D infrastructure and expertise, and operating and business development management. We believe these acquisitions provide us with a solid foundation to discover and develop drug candidates and support partnerships that we expect will drive long-term value for our shareholders. Some of the key stepsThe most significant step in this transformation were:
Acquiredtransition was our 2011 acquisition of the RNAi therapeutics business assembledof Hoffmann-La Roche, Inc. and F. Hoffmann-La Roche Ltd. (collectively, “Roche”), which included the DPC™ delivery system that we have used for all our clinical drug candidates to date. Roche built this business unit in a manner that only a large pharmaceutical company is capable of: backed by expansive capital resources, Roche whichsystematically acquired technologies, licensed expansive intellectual property rights, attracted leading scientists, developed new technologies internally, and built state-of-the-art facilities. At a time when the markets were questioning whether RNAi could become a viable therapeutic modality, we saw great promise in the technology broadly and the quality of what Roche built specifically. The acquisition provided us with three primary sources of value:
| · | Broad freedom to operate with respect to key patents directed to the primary RNAi-trigger formats: canonical, UNA, meroduplex, and dicer substrate structures; |
| · | A best-in-class RNAi delivery system, which we believe to be the targetable DPC™ platform; and |
3
| · | A state-of-the-art R&D facility in Madison, Wisconsin, including a large team of scientists experienced in RNAi and oligonucleotide delivery. |
In addition, in March 2015 we acquired the entire RNAi research and development portfolio and associated assets of Novartis. Novartis had been working in the RNAi field for over a decade and made some very important advancements in their developments of proprietary oligonucleotide formatting and modifications. Key aspects of the acquisition include the following: | · | Multiple patent families covering RNAi-trigger design rules and modifications that fall outside of key patents controlled by competitors, which we believe provides freedom to operate for any target and indication; |
| · | Novel intracellular targeting ligands that enhance the activity of RNAi-triggers by targeting the RNA-induced silencing complex (RISC) more effectively and improving stability once RISC is loaded; |
| · | An assignment of Novartis' license from Alnylam Pharmaceuticals, Inc. (“Alnylam”) granting Arrowhead access to certain Alnylam intellectual property, excluding delivery, for 30 gene targets; and |
| · | A pipeline of three candidates initiated by Novartis for which Novartis has developed varying amounts of preclinical data. |
siRNAWe see the Roche and Novartis acquisitions as a powerful combination of intellectual property, R&D infrastructure, and RNAi delivery technologies,experts. We believe we are the only company with access to all primary RNAi-trigger structures now including additional novel structures discovered by Novartis. This enables us to optimize our drug candidates on a target-by-target basis and use the structure and modifications that yield the most advanced of which is Dynamic Polyconjugates (DPCs);
Licensepotent RNAi trigger. In addition, our DPC™ delivery system enables us to multiple siRNA structuresdeliver our drug candidates efficiently to hepatocytes and chemistriesto non-hepatic tissues in key therapeutic areas;
A state-of-the-art 24,000 square foota highly specific manner. Our R&D team and facility with complete small animal facilities;
R&D staffenable rapid innovation and drive to the clinic, as evidenced by the speed at which we have advanced our pipeline to now include six proprietary drug candidates in a very short period of 40 scientists; andtime.
Multiple pre-clinical drug development programs, including an siRNA therapeutic for chronic hepatitis B infection, which is approaching an IND filing as ARC-520.
Acquired Alvos Therapeutics, Inc. providing Arrowhead with a library of peptide targeting sequences used to create PDCs as well as intellectual property that can be used to generate novel targeting antibodies;
AugmentedWe have focused our management team by hiring accomplished biopharma executives Bruce Given, M.D. as Chief Operating Officer and Head of R&D, and Brendan Rae, Ph.D., J.D., as Chief Business Officer;
Created a centralized infrastructure for the management of clinical trials; and
Integrated and consolidated R&D operations in the Madison facility, including workresources primarily on the RONDEL siRNA delivery systemDPC™ platform and CALAA-01 candidate,on advancing our suite of obesity/metabolic disease compounds including the Adipotide candidate, and the Homing Peptide discovery and development programs;
These steps have created an integrated development operation that allows Arrowhead to advance multiple programs simultaneously. Sinceproprietary DPC™-enabled RNAi therapeutic candidates through clinical trials. Additionally, with our drug development strategy is unified around actively targeted delivery, ourincreased R&D operations are synergistic across drug candidates and platforms. Additionally,headcount, Arrowhead now has the infrastructure, expertise, IP portfolio, chemistry, manufacturing and control (CMC) capabilities, and management that we believe is necessary to attract and support a broad range ofpotential partnerships and research collaborations with large biopharma companies from discovery stage through commercialization.
Pipeline Overview Our internal preclinical and clinical trials. PIPELINE OVERVIEW
Arrowhead is focused on delivering drugs preferentiallydevelopment programs are designed to their sitecreate value directly through our proprietary candidates. These programs also drive value to the technology platform as proof of action while avoiding non-specific uptake in off-target tissues. Our platform technologies are being developedconcept for the power of the programs to enable innovative new therapeutic modalities through targeted delivery and enhanced pharmacokinetics. In particular, our polymeric delivery systems, Dynamic Polyconjugates and RONDEL, have been formulated with small RNAs to develop drug candidates to address diseases such as cancer and HBV through the mechanism of RNAi. The ability to deliver the fragile siRNA molecules that induce RNAi is the key enabler of this important new field of medicine. Our Homing Peptide platform is being used in a clinical obesity therapeutic study and in preclinical studies targeting cancer.
therapies. 2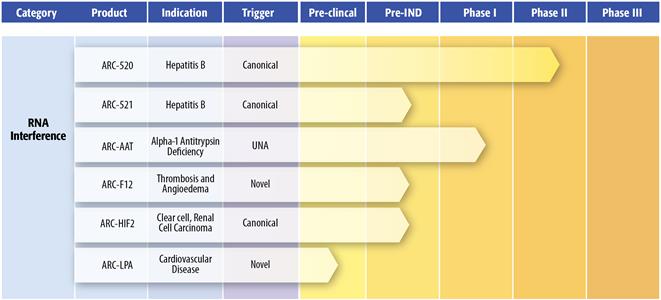
4
Clinical Trials The following table serves as a summary of our ongoing and completed clinical trials as of September 30, 2015: Drug | Study Name | Stage | Status | Description | ClinicalTrials.gov Identifier | ARC-520 | Heparc-1001 | Phase 1 | Enrollment Complete | Single dose escalation study in healthy volunteers to evaluate safety and tolerability of ARC-520 | NCT01872065 | | Heparc-1002 | Phase 1 | Enrolling | Single dose study to evaluate tolerability of increasing infusion rates of ARC-520 | NCT02535416 | | Heparc-2001 | Phase 2a | Enrollment Complete | Single dose escalation study in HBV patients to evaluate reduction in viral antigens in response to ARC-520 | NCT02065336 | | Heparc-2001 OLE | Phase 2a | Enrolling | Open label multi-dose extension study in patients completing Heparc-2001 | NCT02065336 | | Heparc-2002 | Phase 2b | Enrolling | Multi-dose study to determine the depth of hepatitis B surface antigen (HBsAg) reduction in response to ARC-520 in combination with entecavir or tenofovir in hepatitis B envelope antigen (HBeAg) negative patients | NCT02604199 | | Heparc-2003 | Phase 2b | Enrolling | Multi-dose study to determine the depth of HBsAg reduction in response to ARC-520 in combination with entecavir or tenofovir in HBeAg positive patients | NCT02604212 | | Heparc-2004 | Phase 2b | Enrolling | Multi-dose study to determine the depth of HBsAg reduction in response to ARC-520 in combination with entecavir or tenofovir in HBeAg positive patients | NCT02452528 | | Heparc-2008 (MONARCH) | Phase 2b | Not Yet Enrolling | Open label multi-dose study to evaluate ARC-520 alone and in combination with other therapeutics in patients with chronic HBV | NCT02577029 | ARC-AAT | ARCAAT-1001 | Phase 1a/1b | Enrolling | Single dose escalation study to determine the safety, tolerability and effect on circulating alpha-1 antitrypsin levels of ARC-AAT in healthy volunteers and patients | NCT02363946 |
Internal Clinical Programs – ARC-520 ARC-520 – Hepatitis B Virus Infection ARC-520 is an RNAi therapeutic candidate for the treatment of chronic hepatitis B infection with the goal of achieving a functional cure. It is the first clinical candidate to use our proprietary DPC™ technology and includes two siRNA duplexes, each conjugated to a cholesterol derivative to enhance liver delivery and cellular uptake. We have designed ARC-520 to be co-administered with an active excipient, a masked, hepatocyte targeted polymeric amine (the DPC™ delivery system). 5
| | 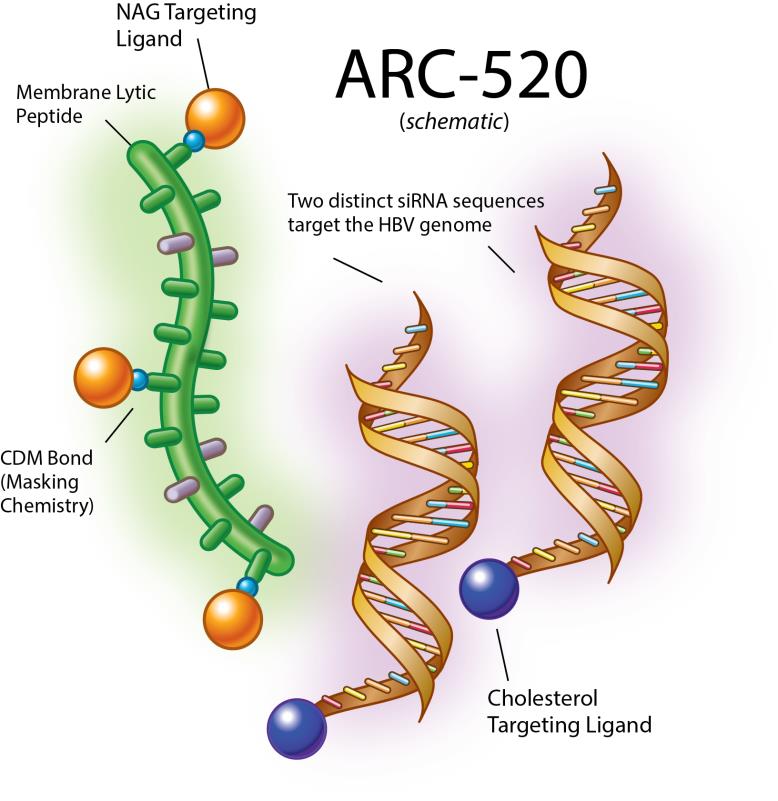
Figure 1: ARC-520 schematic |
We see the need for a next generation HBV treatment with a finite treatment period and an attractive dosing regimen, and that can be used at earlier stages of disease. We believe a novel therapeutic approach such as this that can effectively treat or provide a functional cure (seroclearance of HBsAg and with or without development of excess patient antibodies against HBsAg) has the potential to take significant market share and may expand the available market to include patients that are currently untreated. Chronic Hepatitis B Virus According to the World Health Organization, 360 million people worldwide are chronically infected with hepatitis B virus, of which 500,000 to 1,000,000 people die each year from HBV related liver disease. Chronic HBV infection is defined by the presence of hepatitis B surface antigen (HBsAg) for more than 6 months. In the immune tolerant phase of chronic infection, which can last for many years, the infected person typically produces very high levels of viral DNA and viral antigens. However, the infection is not cytotoxic and the carrier may have no symptoms of illness. Over time, the ongoing production of viral antigens causes inflammation and necrosis, leading to elevation of liver enzymes such as alanine and aspartate transaminases, hepatitis, fibrosis, and liver cancer (HCC)(hepatocellular carcinoma, or HCC). If untreated, as many as 25% to 40% of chronic HBV carriers ultimately develop cirrhosis or HCC. Antiviral therapy is generally prescribed when liver enzymes become elevated. 6
Current Treatments The current standard of care for treatment of chronic HBV infection is a daily oral dose of nucleotide/nucleoside analogs (NUCs) or a regimen of interferon injections 2 to 7 times weekly for approximately one year. NUCs are generally well tolerated, but patients may need lifetime treatment because viral replication often rebounds upon cessation of treatment. Interferon therapeutics can result in a functional cure in up to 20%10-20% of some patient types, but treatment is often associated with significant side effects, including severe flu-like symptoms, marrow suppression, and autoimmune disorders. We see the need for a next generation HBV treatment with fewer side effects, that eliminates the need for interferon based treatment, has a finite treatment period and an attractive dosing regimen, and one that can be used at earlier stagesGoal of disease. We believe a novel therapeutic approach that can effectively treat or provide a functional cure (development of patient antibodies against HBsAg) has the potential to take significant market share and may expand the available market to include patients that are currently untreated.ARC-520 Treatment
ARC-520 is an siRNA therapeutic intended for deliverydesigned to silence the active siteproduction of infection using our proprietary Dynamic Polyconjugate (DPC) technology. ARC-520 consistsall HBV gene products with the goal of two siRNA duplexes, each conjugated toachieving a cholesterol derivative to enhance liver delivery and cellular uptake. We have designed ARC-520 to be co-administered with an active excipient, a masked, hepatocyte targeted polymeric amine. Once the siRNAs and the active excipient are taken up by the hepatocytes, the polymeric amines are unmasked in the endosome and disrupt the endosomal membrane, releasing the siRNA to the cytoplasm where it can engage the RNAi machinery of the cell. 3
functional cure. The siRNAs in ARC-520 are designed to target multiple components of HBV production including the pregenomic RNA that would be reverse transcribed to generate the viral DNA. The siRNAs in ARC-520intervene at the mRNA level, upstream of where NUCs act, and target the mRNAs that produce HBsAg proteins, the viral polymerase, the core protein that forms the capsid, the pre-genomic RNA, the HBeAg, and the HBeAg. A reductionhepatitis B X antigen (HBxAg). NUCs are effective at reducing production of viral antigensparticles, but are ineffective at controlling production of HBsAg and other HBV gene products. Arrowhead believes that a reduction in the production of HBsAg and other proteins that NUCs are ineffective at controlling is considered necessary to effective HBV therapy, because the presence of viralthose proteins isare thought to be a major contributorcontributors to repression of the immune system and the persistence of liver disease secondary to HBV infection. 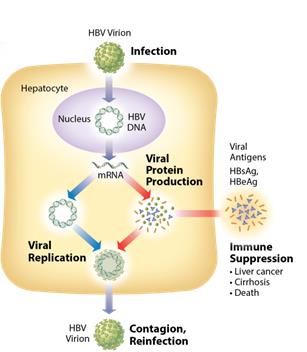
Figure 2: Chronic HBV mechanism untreated 7
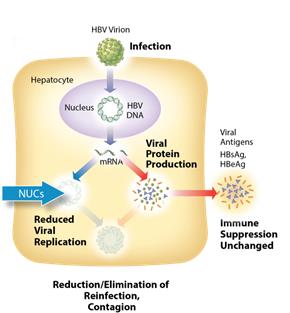
Figure 3: Mechanism of action NUCs 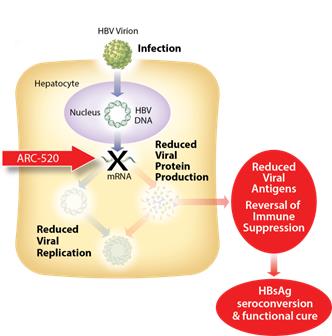 Figure 4: Mechanism of action ARC-520 8
Nonclinical Data Efficacy data in mouse models of HBV infection show that ARC-520 is capable of reducing HBsAg by greater than 3 log (99.9%), HBV DNA by approximately 3 log,logs, and HBeAg to the limit of detection. Pharmacologic effects persist for approximately one month after a single dose of ARC-520.ARC-520 in mice models. Safety data in rodents and non-human primates indicate an acceptable safety margin. WePreclinical data in a chimpanzee chronically infected with HBV demonstrate that intravenous administration of two doses (2 mg/kg on day 1, and 3 mg/kg on day 15) of ARC-520 resulted in substantial and sustained reductions in HBV DNA, HBeAg, and HBsAg, which did not return to baseline until study day 43, 43, and 71, respectively. In addition, an increase in serum alanine transaminase (ALT) occurred 4 weeks after the second dose, coincident with the nadir of circulating HBsAg. This is suggestive of a therapeutic immunological flare, which is thought to be part of a cascade that under chronic therapy may lead to HBsAg seroclearance and functional cure. Observed increases in key chemokine/cytokine mRNAs are also consistent with a T-cell mediated immunological event. Additional nonclinical data from a multiple dose study in 9 chimpanzees were reported in 2015. At the start of the study, five chimpanzees were HBeAg-positive and four chimpanzees were HBeAg-negative. Deep sequencing and phylogenetic analyses indicated the HBV sequence was a chimpanzee variant of human HBV. To reduce viral replication prior to treatment with ARC-520, chimpanzees were treated for 8-24 weeks with entecavir (ETV) or ETV+ tenofovir (TDF) in one case. Following the lead-in period, animals were administered ARC-520 intravenously at 4-week intervals. Dose levels administered were 2, 3, or 4 mg/kg ARC-520, along with maintenance doses of ETV or ETV+TDF. The study showed a robust, sustained direct anti-viral effect on HBsAg production in all HBeAg-positive and -negative chimpanzees during ARC-520 treatment. One chimpanzee achieved sustained virologic response (SVR) off-therapy and two chimpanzees exhibited signs of immune reactivation. HBeAg-positive chimpanzees displayed the highest levels of HBsAg knockdown - up to 2.7 log. In HBeAg-negative chimpanzees, HBsAg knockdown was also substantial - up to 0.9 log. ARC-520 was well tolerated after multiple doses up to 4 mg/kg ARC-520 (highest dose tested). Evidence indicates integrated HBV DNA is a significant source of total HBsAg, especially in HBeAg-negative chimpanzees. Phase 1 (Heparc-1001) Arrowhead completed nine dose cohorts in the Heparc-1001 Phase 1 trial in 2014. The study was designed to characterize the safety profile of ARC-520 across a range of doses and evaluate pharmacokinetics. It was a single-center, randomized, double-blind, placebo-controlled, single dose-escalation, first-in-human study of ARC-520 administered intravenously to healthy adult volunteers. All subjects received either placebo or ARC-520 in doses ranging from 0.01 mg/kg to 4 mg/kg. The study successfully enrolled all 54 subjects (36 received ARC-520, 18 placebo) at a single center in Melbourne, Australia. There have been no reports of serious adverse events (SAEs), no dose limiting toxicities, no discontinuations due to adverse events (AEs), and a modest overall occurrence rate of AEs without a clear dose-related increase in frequency or severity. There has been a modest occurrence rate of non-clinically significant abnormal laboratory tests. There were no reported drug related or clinically significant differences for vital signs or ECGs between subjects receiving drug versus placebo. One arrhythmia noted on telemetry occurred in a subject with similar previously undiagnosed prior cardiac atrial rhythm abnormalities and one occurrence of hypotension that appears to have been artifact from a failing automated blood pressure machine. One occurrence each of moderate flushing and urticarial rash seen at dose levels of 0.3 mg/kg and 2.0 mg/kg respectively led to the subsequent reduction, by half, in infusion rate of ARC-520 as well as the introduction of pretreatment with an oral over-the-counter antihistamine. Since the introduction of these mitigations, no additional signs of hypersensitivity or infusion reactions have been seen. There were mild creatinine elevations in 6% of subjects receiving ARC-520 and 6% receiving placebo, which were transient with rapid (<24 hours) return to baseline. There were no signs of tubular damage on laboratory evaluation and these subjects had no additional clinical symptoms. Overall, the changes in creatinine are similar between ARC-520 and placebo groups and thought to be, at least primarily, due to dietary intake of protein. There were no changes in ALT, AST or CK considered to be clinically significant by the study investigator. 9
In summary, ARC-520, when administered as a single dose up to 4 mg/kg to healthy volunteers, appears to be well tolerated. The table below shows the incidence of treatment emergent adverse events observed in greater than 5% of subjects. | | | Adverse Events >5% All Attributions | Placebo (n=18) | ARC-520 (n=36) | All AEs | 67% | 67% | Mild | 72% | 69% | Moderate | 28% | 31% | Severe | 0% | 0% | Headache | 33% | 14% | URI | 28% | 19% | Somnolence | 6% | 8% | Lethargy | 6% | 6% | Creatinine increase | 6% | 6% | Myalgia | 0% | 6% | Dizziness | 0% | 6% |
Figure 5: Phase 1 AE reporting Phase 2a (Heparc-2001) In 2015 Arrowhead reported data from the Heparc-2001 Phase 2a multicenter, randomized, double-blind, placebo-controlled, dose-escalation study designed to determine the depth and duration of hepatitis B surface antigen (HBsAg) reduction after administration of ARC-520 in combination with entecavir in patients with chronic HBV infection. Secondary objectives included the assessment of safety and tolerability of escalating single doses of ARC-520 co-administered with a fixed dose of entecavir and multiple additional secondary and exploratory endpoints. Significant details about the seven cohorts are included below. | | | | | | | Cohort | Prior entecavir (ETV) | Patient Type | ARC-520 dose | Active/Placebo | Baseline HBsAg Mean (range) Log IU/mL | Status | 1 | Yes | HBeAg-neg | 1.0 mg/kg | 6/2 | 3.4 (3.0-4.2) | Complete/Unblinded | 2 | Yes | HBeAg-neg | 2.0 mg/kg | 6/2 | 3.5 (3.2-4.3) | Complete/Unblinded | 3 | Yes | HBeAg-neg | 3.0 mg/kg | 6/2 | 3.6(3.1-4.0) | Complete/Unblinded | 4 | Yes | HBeAg-neg | 4.0 mg/kg | 6/2 | 3.4 (3.2-4.0) | Complete/Unblinded | 5 | Yes | HBeAg-pos | 4.0 mg/kg | 6/2 | 3.6 (3.1-4.2) | Complete/Unblinded | 6* | Yes | HBeAg-pos | 2 x 2.0 mg/kg (two weeks apart) | 6/0 | 3.3 (3.0-3.6) | Complete/Open label | 7 | No | HBeAg-pos HBeAg-neg | 4.0mg/kg | 6/0 6/0 | 4.0 (0.8-4.9) 2.9 (1.0-3.6) | Ongoing / Openlabel |
Figure 6: Phase 2a dose cohorts In connection with Heparc-2001, fifty-eight patients were successfully dosed with 48 receiving the drug and 10 receiving a placebo. 20 females and 38 males were enrolled, all of Chinese ethnicity, with a mean age of 41 years (range of 23 to 59). Topline results were reported at an analyst and investor day hosted by the Company and additional details were presented at the 2015 AASLD Liver Meeting. To date there have been no serious AEs, no dose limiting toxicities, no discontinuations due to medication AEs, and a modest occurrence rate of AEs. All reported AEs were deemed unrelated to study drug by the principal investigator. Safety labs continue to lack indication of end organ toxicity, with a low occurrence rate of abnormal laboratory tests with no observed relationship to timing or dose.
Adverse Events | 1 mg/kg (N=6) | 2mg/kg (N=6) | 3 mg/kg (N=6) | 4 mg/kg (N=24) | 2 mg/kg x2 (N=6) | Placebo (N=10) | All AEs | 1 | 5 | 1 | 2 | 1 | 0 | Extravasation | | 1 mild | | | | | Malaise | | 1 mod | | | | | Influenza | 1 mild | | | | | | Blood CK increase | | 1 mild | | | | | Diabetes Mellitus | | 1 mild | | | | | Pain in extremity | | | 1 mild | | | | Presyncope | | 1 mod | | | | | Headache | | | | 1 mild | | | Dizziness | | | | 1 mild | | | Fever | | | | | 1 mild | |
Figure 7: Phase 2a AE reporting Arrowhead measured the Log reduction of quantitative HBsAg, HBcrAg, and HBeAg from baseline as an assessment of ARC-520 activity. This is the first time that a reduction in HBsAg, HBcrAg, and HBeAg mediated through RNA interference has been reported in chronic HBV patients. In this study, ARC-520 effectively inhibited cccDNA-derived mRNA with reductions up to 1.9 logs (99%). ARC-520 had a direct antiviral effect that lasted up to 57 days after a single dose. In patients exhibiting a delayed response, duration of over 85 days was observed. The best qHBsAg reduction was seen in treatment naïve HBeAg-positive patients. HBeAg-negative patients showed a delayed response. HBeAg-negative, ETV experienced patients showed a dose response in HBcrAg but qHBsAg dose response was less pronounced. HBeAg positive, ETV experienced patients had a substantially higher reduction in HBeAg and HBcrAg compared to HBsAg. Variations in viral antigen reduction are consistent with lower levels of cccDNA derived mRNA transcripts in chronic ETV patients and HBeAg negative patients. These data are summarized in the figures below. | | | | | | | | | Log reduction from baseline Mean (max) | Cohort | Dose (mg/kg) | HBeAg status | Prior ETV | HBsAg | HBcrAg | HBeAg | 1 | 1 | Neg | Y | -0.2 (-0.3)* | -0.2 (-0.2) | N/A | 2 | 2 | Neg | Y | -0.2 (-0.3)* | -0.5 (-0.5) | N/A | 3 | 3 | Neg | Y | -0.3 (-0.4)* | -0.4 (-0.7) | N/A | 4 | 4 | Neg | Y | -0.4 (-0.5)* | -0.9 (-1.1) | N/A | 5 | 4 | Pos | Y | -0.3 (-0.7)* | -0.9 (-1.1) | -1.2 (-1.7) | 6‡ | 2x2 | Pos | Y | -0.5 (-0.8)+ | -0.7 (-1.2) | Pending | 7‡,† | 4 | Pos | N | -1.5 (-1.9)+ | Pending | Pending | 7‡ | 4 | Neg | N | -0.2 (-0.8)+ | Pending | N/A |
*Roche Elecsys batch analyzed data set +Abbott Architect data ‡Preliminary data, analysis is ongoing †Excluding transitional patient Figure 8: Max reduction in viral antigens 11

Figure 9: Mean reduction in HBsAg in treatment naive patients (cohort 7) Phase 2b Multiple dose and combination studies of ARC-520 aimed at producing functional cures are underway. These studies include Heparc-2001 open label extension, Heparc-2002, Heparc-2003, Heparc-2004, and the MONARCH study (Heparc-2008). There are clinical sites for these studies in the US, Europe, Asia, and Australia/New Zealand. 12
Internal Clinical Programs – ARC-AAT ARC-AAT – Liver Disease Associated with Alpha-1 Antitrypsin Deficiency Arrowhead has developed a therapeutic candidate (ARC-AAT) for the treatment of liver disease associated with Alpha-1 Antitrypsin Deficiency (AATD), a rare genetic disease that severely damages the liver and lungs of affected individuals. ARC-AAT employs a novel unlocked nucleobase analog (UNA)-containing RNAi molecule designed for systemic delivery using the DPC™ delivery system. Pre-clinical studies have demonstrated that ARC-AAT is highly effective at knocking down the Alpha-1 antitrypsin (AAT) gene transcript and reducing the hepatic production of the mutant AAT protein. | 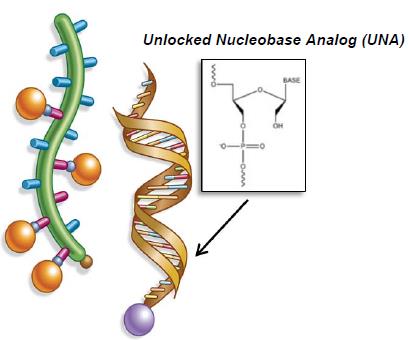
Figure 10: ARC-AAT schematic |
Alpha-1 Antitrypsin Deficiency AATD is a genetic disorder associated with liver disease in children and adults and pulmonary disease in adults. AAT is a circulating glycoprotein protease inhibitor that is primarily synthesized and secreted by liver hepatocytes. Its physiologic function is the inhibition of neutrophil proteases to protect healthy tissues during inflammation and prevent tissue damage. The most common disease variant, the Z mutant, has a single amino acid substitution that results in improper folding of the protein. The mutant protein cannot be effectively secreted and accumulates in globules in the hepatocytes. This triggers continuous hepatocyte injury, leading to fibrosis, cirrhosis, and increased risk of hepatocellular carcinoma. Current Treatments Individuals with the homozygous PiZZ genotype have severe deficiency of functional AAT leading to pulmonary disease and hepatocyte injury and liver disease. Lung disease is frequently treated with AAT augmentation therapy. However, augmentation therapy does nothing to treat liver disease, and there is no specific therapy for hepatic manifestations. There is a significant unmet need as liver transplant, with its attendant morbidity and mortality, is currently conducting IND-enablingthe only available cure. Goal of ARC-AAT Treatment The goal of treatment with ARC-AAT is prevention and potential reversal of Z-AAT accumulation-related liver injury and fibrosis. Reduction of inflammatory Z-AAT protein, which has been clearly defined as the cause of progressive liver disease in AATD patients, is important as it is expected to halt the progression of liver disease and allow fibrotic tissue repair. Preclinical Data In preclinical studies with PiZ mice, which are genetically modified to produce the mutant human AAT (Z-AAT), ARC-AAT induced a goalgreater than 95 percent reduction in circulating AAT after a single dose. The addition of chemical modifications, including UNAs, slowed the rebound in production of AAT compared to entercanonical siRNAs, and produced a substantially improved duration of effect. 13
After eight weeks of treatment in multi-dose studies in PiZ mice, soluble (monomeric) and insoluble (polymeric) forms of Z-AAT were greatly reduced in the livers of PiZ mice treated with ARC-AAT. In addition, liver globule burden was substantially reduced from baseline levels and in comparison to treatment with saline, which showed progressive globule formation (shown in the figure below). These data were submitted as an abstract to the 2014 AASLD Liver Meeting and selected for presentation at this conference. | | | Baseline
5 weeks old 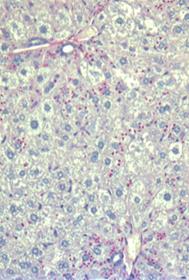
| AAT-UNA q2w 13 weeks old 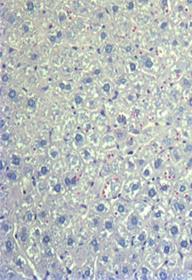
| Saline 13 weeks old 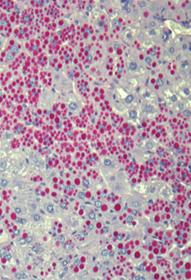
|
Figure 11: Reduction in Z-AAT globules in livers of PiZ mice In primate studies, knockdown of AAT in serum persisted for over ten weeks with greater than 80 percent knockdown observed at the six-week time point. Multi-dose studies in primates showed a sustained reduction of AAT with once every six weeks dosing, suggesting that once monthly or less frequent dosing is sufficient to maintain ~80-90% knockdown (shown in the figure below). 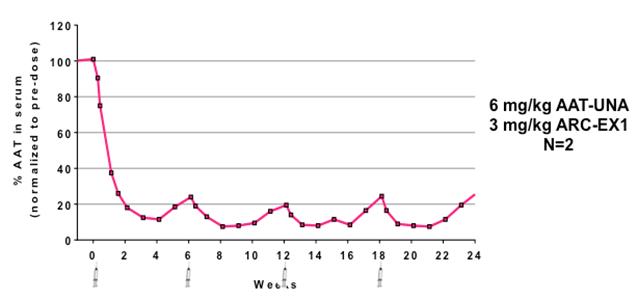
Figure 12: Long term reduction of AAT in NHPs following repeat dosing with ARC-AAT (ARC-EX1 is a DPC™ delivery system) 14
The Alpha-1 Project Arrowhead has an agreement with The Alpha-1 Project (TAP), the venture philanthropy subsidiary of the Alpha-1 Foundation. TAP’s mission is to support organizations in pursuit of cures and therapies for lung and liver disease caused by Alpha-1 Antitrypsin Deficiency. Under the terms of the agreement, TAP will partially fund the development of ARC-AAT. In addition to the funding, TAP will make its scientific advisors available to Arrowhead, assist with patient recruitment for clinical trials with its Alpha-1 Foundation Patient Research Registry, and engage in other collaborative efforts that support the development of ARC-AAT. Phase 1 Arrowhead is conducting a single dose Phase 1 clinical study in 2013. Adipotide (Formerly Prohibitin-TP01) – Obesity and Metabolic Disorders
Obesityof ARC-AAT. The study is a global health threatmulti-center, randomized, placebo-controlled, double-blind, single dose-escalation, first-in-human study to evaluate the safety, tolerability and onepharmacokinetics of ARC-AAT and the leading causeseffect on circulating AAT levels. The study has been enrolling dose cohorts of preventable deaths in the United States. Arrowhead’s anti-obesity drug candidate, Adipotide, was designedsix participants each, with participants randomized at a ratio of 2:1 (active:placebo) to selectively disrupt the blood supply that supports unhealthy fat by the targeted inductionreceive a single intravenous injection of apoptosis (cell death) in the vasculature of adipose tissue.either ARC-AAT or placebo (normal saline). The Adipotide peptidestudy consists of two functional domains. The homing domain targetsparts; Part A in healthy volunteers and part B currently being conducted in patients with PiZZ genotype AATD. Dosing in patients (part B) began at a membrane associated protein, Prohibitin, on adipose vascular endothelial cells. The membrane disrupting domain causes apoptosis by disrupting mitochondrial membranes inside the cells.
An Investigational New Drug Application (IND) for Adipotide was filed with the FDA, and we began enrolling patients in 2012 as part of a Phase 1 clinical trial to test the safety of the compound in human patients. Our collaborator, MD Anderson Cancer Center in Houston, plans to enroll up to 39 obese prostate cancer patients in the Phase 1 study and has agreed to bear all direct costs of this trial. Up to five dose levels of the drug candidate will be tested in the trial. Three participants will be enrolled at each dose level with the first group of participants receiving the lowestused in healthy volunteers that met pre-defined criteria for AAT knockdown and then continued dose level by injectionescalation may proceed under the skin once per dayprotocol. The study evaluates participants for 28 days and each new group receiving a higher dose than the group before it,following dosing, with additional follow-up if no intolerable side effects are seen. This will continueneeded every 2 weeks until the highest tolerable dose is found or the study terminates.AAT levels return to baseline.
Potential Advantages of Adipotide:
Shown to promote weight loss of 11% to 30% of total body mass in preclinical studies using rodents and spontaneously obese rhesus monkeys after just 28 days of treatment;
Shown to reduce abnormalities in blood chemistry associated with diabetes;
Novel mechanism of action compared to other known therapeutics on the market or in clinical trials;
No modulation of neurotransmitters seen in pre-clinical studies, thus unlikely to have psychological side effects;
No amphetamine-like mechanism of action and thus unlikely to yield GI side effects.
Adipotide is based on Arrowhead’s Homing Peptide™ library developed at MD Anderson Cancer Center. White adipose (fat) tissue is highly vascularized and both the expansion and maintenance of adipose tissue depend on a continued ability to build supporting vasculature. This peptide targeting library provides a map of the unique cell receptors on the vasculature that varies in different tissues. Targeting vasculature based on this variation allows for specific delivery of drug payloads to specific target cells, while avoiding collateral injury to other healthy/non-targeted cells. Using this technique, peptide sequences that target receptors specific to white adipose tissue were identified. Adipotide has been developed by our majority-owned subsidiary, Ablaris Therapeutics, Inc. (“Ablaris”). Arrowhead owns 64% of the fully diluted shares of Ablaris.
CALAA-01 – Solid Tumors
CALAA-01 is a combination of our RONDEL delivery technology and a patented siRNA targeting the M2 subunit of ribonucleotide reductase, a clinically-validated cancer target. Ribonucleotide reductase catalyzes the conversion of ribonucleosides to deoxyribonucleosides and is necessary for DNA synthesis and replication, and thus tumor growth. The internally developed siRNA demonstrates potent anti-proliferative activity across multiple types of cancer cells. CALAA-01 was the first siRNA therapeutic candidate to target cancer in a human clinical study and we believe was also the first successful systemic delivery of an siRNA therapeutic candidate.
In August 2012 enrollment into the Phase 1 clinical trial was completed. Adverse events observed coincided with an increase in certain cytokine levels. Elevation in cytokines is consistent with an acute immune response to the natural siRNA used in CALAA-01. These reactions also appeared to be transient, such that if a patient stayed on CALAA-01, the cytokine responses often subsided. Based on these results, a Phase 1b trial was initiated using a modified dosing schedule in which patients were pretreated with a lower dose to assess whether this strategy can increase patient safety and further increase the maximum tolerated dose. Patient enrollment was completed in August 2012 and analysis of final study data is being prepared.
Interim clinical results were presented at the 2010 American Society of Clinical Oncology meeting (ASCO). Data from 15 patients accrued to 5 dose levels (3, 9, 18, 24, 30 mg/m2) showed that treatment-related adverse events were mostly mild to moderate with fatigue, fever/chills, allergic, or gastrointestinal-related adverse events most frequently observed. Importantly, no changes in coagulation, liver function tests, or kidney function were observed.
4
Analysis of tumor biopsies from three melanoma patients showed the presence of intracellular nanoparticles in amounts that correlated with dose. Additionally, a reduction was found in both the RRM2 messenger RNA and protein levels when compared to pre-dosing tissue. Furthermore, the presence of siRNA-mediated mRNA cleavage products was confirmed by 5’-RACE, demonstrating that siRNA-mediated mRNA cleavage occurred specifically at the site predicted for an RNAi mechanism. These results were published in March 2010 in the scientific journalNature, citing these interim data from our Phase 1 trial as the first evidence of systemic delivery of siRNA, and the successful “silencing” of a widely recognized cancer gene via RNA interference in humans.
Partnered Programs
Cyclosert and CRLX-101 (formerly IT-101)
The linear cyclodextrin-based drug delivery platform, Cyclosert, was designed for the delivery of small molecule drugs. In December 2008, we completed a Phase 1 trial with IT-101, a conjugate of the linear cyclodextrin polymer and Camptothecin, a potent anti-cancer drug, with a positive safety profile and indications of efficacy.Orphan Drug Designation
In June 2009, we entered into a transaction2015, Arrowhead was granted the orphan drug designation for ARC-AAT by the U.S. Food and Drug Administration (FDA). Orphan drug designation provides incentives for sponsors to develop products for rare diseases. These incentives include increased engagement with Cerulean Pharmaceuticals, Inc., a privately-held Boston, Massachusetts based company. Cerulean licensed rightsFDA on drug development activities, exemption from all future product-specific regulatory fees, the opportunity to further researchapply for R&D funding, tax credits, an increased chance of priority review, and commercialize IT-101 (now known as “CRLX-101”), and the Cyclosert platform for all products except for nucleic acids, tubulysin, cytolysin and second generation epothilones. In connection with the transaction, we assigned certain patents to Cerulean and Cerulean granted back to us rights necessary to research and commercialize the excluded products. As such, we retain the rights to the RONDEL siRNA delivery platform, as well as CALAA-01. We received an initial payment7 years of $2.4 million, and may receive development and sales milestones, and royalty payments if CRLX-101 or other products based on the Cyclosert platform are successfully developed. Should Cerulean sublicense CRLX-101 to a third party, we are entitle to receive a percentage of any sublicensing income at rates between 10% and 40%, depending on the stage of the drug’s developmentorphan exclusivity at the time of sublicensing.New Drug Application (NDA).
Tubulin InhibitorPreclinical Programs
ArrowheadIn addition to our clinical candidates, we are actively engaged in the discovery and development of additional pre-clinical stage products for intravenous and subcutaneous administered therapeutics targeting the liver, as well as programs targeting extra-hepatic tissues. We focus on disease targets that are well suited for intervention with targeted RNAi therapeutics using our DPC™ delivery platform.
ARC-521 ARC-521 is a new RNAi-based therapeutic in Arrowhead’s HBV portfolio that was developed to target both cccDNA derived mRNA transcripts, like ARC-520, as well as those from HBV DNA that has integrated into the host DNA. It is intended to address specific patient populations that are predicted to have higher ratios of integrated DNA versus cccDNA. The Company plans to begin clinical studies of ARC-521 in mid-2016. ARC-F12 ARC-F12 is an RNAi-based therapeutic designed to reduce the production of factor 12 with the goal of providing a licenseprophylactic treatment for hereditary angioedema (HAE) and joint development agreement with Vienna, Austria based biotech Tube Pharmaceuticals GmbH, which grants Tube Pharma the right to develop Cyclosert enabled tubulin inhibitors. Tubulysins are a novel tubulin-targeted class of natural compounds with potent anti-proliferative activity against multiple cancer types. Tube Pharmathromboembolic diseases. Arrowhead is conducting preclinical studies.relevant disease models, and is considering other potential studies to support advancement of ARC-F12 into clinical trials. ARC-HIF2 ARC-HIF2 is an RNAi-based therapeutic designed to reduce the production of hypoxia-inducible factor 2α (HIF-2α) to treat clear cell renal cell carcinoma. It is the first drug candidate using a new DPC™ delivery vehicle designed to target tissues outside of the liver. Arrowhead is eligibleconducting tumor models and is working on manufacturing scale up for the new delivery vehicle to receive milestones and royalties on sales.support advancement of ARC-HIF2 into IND-enabling studies. ARC-LPA ARC-LPA is an RNAi-based therapeutic designed to reduce production of apolipoprotein A (apo(a)), a key component of lipoprotein(a) (Lp(a)), which has been genetically linked with increased risk of cardiovascular diseases. ARC-LPA employs Arrowhead’s new hepatic delivery format being developed for subcutaneous administration. This program has not yet been officially designated by Arrowhead as entering pre-IND development. Partner-based Programs Alnylam Pharmaceuticals 15
In January 2012, Arrowhead granted Alnylam Pharmaceuticals, Inc., (“Alnylam”) a license to utilize the Dynamic PolyconjugateDPC™ delivery technology for a single RNAi therapeutic product. Alnylam is collaborating with Arrowhead to develop this technology for an undisclosed target in its “Alnylam 5x15” pipeline, which is focused on genetically defined targets and diseases.target. Alnylam has not publically disclosed what progress, if any, it may have made with respect to this target. Arrowhead is eligible to receive milestone payments and royalties on sales from Alnylam.worldwide sales. Shire In December 2012, Arrowhead signed a research collaboration and license agreement with Shire AG (“Shire”) to develop and commercialize targeted peptide-drug conjugates (PDCs) utilizing Arrowhead’s human-derived Homing Peptide platform and Shire’s therapeutic payloads. Arrowhead may receive research funding and could be eligible for development, regulatory, and commercialization milestone payments of up to $32.8 million for each development candidate, plus additional milestone payments for a second indication, and royalties on worldwide sales. Preclinical Programs
In addition to our clinical candidates and our partner-based programs, we are actively engaged in the discovery and development of additional pre-clinical stage products. We focus on disease targets that are well suited for intervention with guided therapeutics like our PDCs and targeted RNAi therapeutics using our DPC delivery platform. These may include liver disease, oncology, and other therapeutic areas.
RNAI DELIVERY PROGRAM
In October 2011, Arrowhead acquired Roche’s RNAi business, including its RNA therapeutic assets, related intellectual property and research facility in Madison, Wisconsin. We believe that these assets position Arrowhead as one of the most advanced and broadest RNAi therapeutics companies in the world. Arrowhead now possesses the following siRNA assets:
Non-exclusive license from Alnylam to use canonical siRNAs in oncology, respiratory diseases, metabolic diseases and certain liver diseases. This includes a sub-license from Isis Pharmaceuticals giving Arrowhead license for siRNA chemical modifications for these specific disease areas.
Non-exclusive license from City of Hope Comprehensive Cancer Center to Dicer substrate and Meroduplex siRNAs. The Dicer technology may provide advantages over canonical siRNAs in certain circumstances. In addition, different siRNA formats may trigger RNAi more or less efficiently on a target-by-target basis.
Patent estate covering the Dynamic Polyconjugate siRNA delivery system.
Access to certain patents on targeting siRNA drugs with antibodies and small molecules.
5
State-of-the-art laboratory facilities in Madison, Wisconsin, managed by long term leaders in oligonucleotide therapeutics and delivery, including a small animal research facility and an offsite primate colony.
Intellectual property covering Roche’s internally developed liposomal nanoparticle drug delivery technology.
RONDEL siRNA delivery system which has demonstrated gene knockdown in humans in the CALAA-01 clinical trial.
Minority ownership position in Leonardo Biosystems, Inc., a private company developing a multi-stage silicon-based delivery system.
CALAA-01 Phase 1 oncology drug candidate.
We believe this represents one of the broadest siRNA drug technology and delivery portfolios in the world.
RNA Interference & the Benefits of siRNARNAi Therapeutics RNA interference (RNAi) is a mechanism present in living cells that inhibits the expression of a specific gene, thereby affecting the production of a specific protein. Deemed to be one of the most important recent discoveries in life science with the potential to transform medicine, the discoverers of RNAi were awarded a Nobel Prize in 2006 for their work. Mediated by small interfering RNAs (siRNA), a class of ribonucleic acid (RNA) molecules, 20-25 nucleotides in length, RNAi-based therapeutics canmay leverage this natural pathway of gene silencing to potentially target and shut down specific disease causing genes. 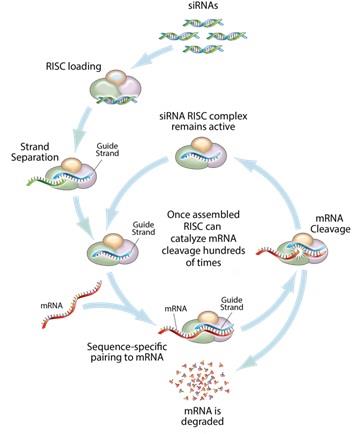
Figure 13: Mechanism of RNA interference Small molecule and antibody drugs have proven effective at inhibiting certain cell surface, intracellular, and extracellular targets. However, certainother drug targets such as intranuclear genes and some proteins have proven difficult to inhibit with traditional drug-based and biologic therapeutics. Developing effective drugs for these targets would have the potential to address large underserved markets for the treatment of many diseases. Using the ability to specifically silence any gene, siRNARNAi therapeutics may be able to address previously “undruggable” targets, unlocking the market potential of such targets. Mechanism of RNA interference
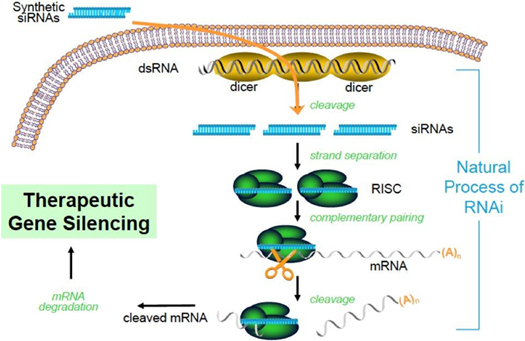
Advantages of RNAi as a Therapeutic Modality | · | Silences the expression of disease causing genes; |
16
Potential to address any target in the transcriptome including previously “undruggable” targets;
| · | Potential to address any target in the transcriptome including previously "undruggable" targets; |
| · | Rapid lead identification; |
| · | Opportunity to use multiple RNA sequences in one drug product for synergistic silencing of related targets; and |
| · | RNAi therapeutics are uniquely suited for personalized medicine through target and cell specific delivery and gene knockdown. |
Rapid lead identification;
Opportunity to use multiple RNA sequences in one drug product for synergistic silencing of related targets; and
siRNAs are uniquely suited for personalized medicine through target and cell specific delivery and knockdown.
6
Addressing the siRNARNAi Delivery Challenge To date, the primary challenge to the development of siRNARNAi therapeutics has been delivering the fragile, often immunogenic and otherwise rapidly cleared siRNARNAi trigger molecules, into the cytoplasm of the cell, where RNAi activity occurs. This hurdle has prevented siRNAmany RNAi therapeutics from reaching their full potential. Many companies have attempted to overcome the delivery challenge. Most early systems involved cholesterol conjugates orvarious carriers such as liposomes. However, development in humans has been limited due to toxicity and immunogenicity of these approaches when studied in clinical trials. To address the delivery challenge, Arrowhead has a leading team of researchers with extensive siRNA therapeutic know-how and two validated delivery platforms:
The Dynamic Polyconjugate system is an amphipathic polymer to which shielding agents and targeting ligands are reversibly attached. | • | | The RONDELTM delivery system utilizes targeted cyclodextrin polymers to deliver siRNA and other oligonucleotides to tumors. Humanin vivo gene knockdown has been demonstrated in a Phase 1 cancer trial, establishing human proof of concept for the RONDEL system.
|
These are both modular systems that may be optimized on a target-by-target basis. Importantly, they also may be targeted to address a variety of tissues.
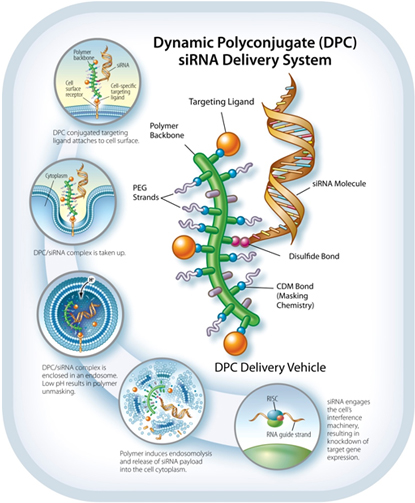
The Dynamic Polyconjugate siRNAPolyconjugate™ RNAi Delivery System
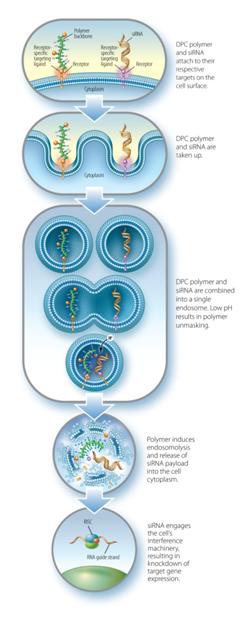
The DPCDPC™ delivery system represents an innovative solution to the siRNARNAi delivery problem, specifically designed to overcome barriers to systemic administration of siRNA.RNAi trigger molecules. Developed by ourArrowhead scientists, in Madison, Wisconsin, the inspiration for DPCDPC™ technology came from the physical characteristics of viruses, nature’s own nanoparticles for nucleic acid delivery. Viruses are efficient at finding their target cells and delivering their nucleic acid payload to the proper cellular compartment. Key features of viruses are their small size, their overall negative surface charge, their specificity for particular cell types based on receptors unique to that cell, and their ability to disassemble and release their nucleic acid cargo to the proper cell compartment in response to cellular triggers. All of these features are incorporated into DPCDPC™ technology. DPCsPrototypical DPC delivery systems are small, nanoparticles,generally 5-20 nanometers (nm) in size, with an amphipathic polymer backbone. Arrowhead has a library of polymers, that may be employed with the system, enabling optimization based on factors such as preferred mode of administration, pharmacokinetics, and target tissue. Shielding agents such as polyethylene glycol and targeting ligands aremay be reversibly attached to the polymer backbone. In some constructs, the siRNARNAi trigger payload is attached to the DPC,DPC™ delivery system, while in other constructs, the siRNARNAi trigger circulates attached to a different carrier. When attached, the DPCDPC™ delivery system construct protects the siRNARNAi trigger payload while allowing the polymer to circulate in the blood without creating undue toxicity. The targeting ligand guides the nanoparticlesit to the cell type of interest where, together with the siRNA,RNAi trigger, it is taken up into a membrane-enclosed cellular compartment known as an endosome. The polymer is selected for its ability to disrupt the endosomal membrane, which releasesallows the siRNARNAi trigger to be released into the cytoplasm. There, it engages the cell’s RNAi machinery, ultimately resulting in knockdown of target gene expression. This lyticmembrane active chemistry of the DPCDPC™ polymeric backbone is modified, or “masked”, using proprietary chemistry.chemistry until it reaches the endosome where pH changes cause the masking chemistries to fall off. Masking of the polymer’s lyticmembrane active chemistry accomplishes two interrelated objectives that are critical toin vivo siRNA RNAi delivery:
Reduction of toxicity by controlling when the membrane lytic property of the polymer is activated.
| · | Reduction of toxicity by controlling when the membrane disruptive property of the polymer is activated; and |
| · | Inhibition of non-specific interactions with blood components and non-targeted cell types. |
Inhibition17
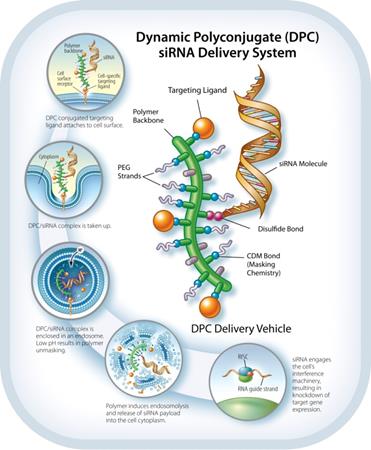
Figure 14: Single molecule DPCs™ 18
Co-injection of non-specific interactions with blood componentsDPCs™ and non-targeted cell types. 7
Arrowhead has developed multiple forms of the prototypical DPCDPC™ delivery system. Our ARC-520 clinical candidate utilizesand ARC-AAT use a DPC™ formulation wherewhereby the siRNARNAi trigger is conjugated to cholesterol and is not attached to the DPC.DPC™. Pre-clinical studies have shown co-injection of the liver-targeted DPCDPC™ polymer together with siRNAthe RNAi trigger conjugated to a lipophilic moiety, such as cholesterol, results in a >500-foldgreater than 500-fold increase in the potency when compared to the siRNA-cholesterolRNAi trigger with cholesterol alone. This formulation retains the potent endosomal escape capabilities of Arrowhead’s DPCArrowhead's DPC™ platform, simplifies drug manufacturing, and creates new targeting opportunities. 8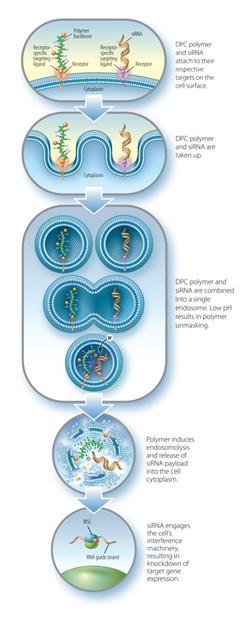
Figure 15: Mechanism of action for co-injected DPCs 19
DPCs using Co-injection StrategySubcutaneous Administration of RNAi Therapeutics
A DPC formulationArrowhead has also developed multiple delivery formulations for subcutaneous administration has also been developed using Arrowhead’s latest proprietary polymer masking technology.of RNAi therapeutics where the RNAi triggers are conjugated to the delivery vehicle. Using DPCs to deliver siRNA,these vehicles, high-level target gene knockdown is observed at low siRNA doses with limited toxicity in rodents and non-human primates. Arrowhead studies have shown knockdown of 99% in monkeys after a single injection of 1 mg/kg, >90% at 0.5 mg/kg, and 80% in mice at 0.05 mg/kg, which represents greater knockdown at lower doses than reported results of other clinical candidates. PK and biodistribution studies indicate that the new masking technology is highly stable, allowing for maximal bioavailability and long circulation times. Arrowhead is developing this formulation for use in multiple therapeutic areas including oncology.
RONDEL Delivery System
For this delivery system, polymers form the foundation for a three-part RNAi/Oligonucleotide Nanoparticle Delivery (RONDEL) technology. The first component is the positively charged polymer that, when mixed with siRNA, binds to the negatively charged “backbone” of the siRNA. The polymer and siRNA self-assemble into nanoparticles less than 100 nm diameter that are designed to protect the siRNA from nuclease degradation in serum. The cyclodextrin in the polymer enables the surface of the particles to be decorated by stabilizing agents and targeting ligands. These surface modifications are formed by proprietary methods involving the cyclodextrins. The surface-modifying agents have terminal adamantane groups that form inclusion complexes with the cyclodextrin and contain polyethylene glycol (PEG) to endow the particles with properties that prevent aggregation, enhance stability and enable systemic administration. Targeting molecules can be covalently attached to the adamantane-PEG modifier, enabling the siRNA-containing particles to be targeted to tissues of interest.
9
RONDEL Nanoparticle
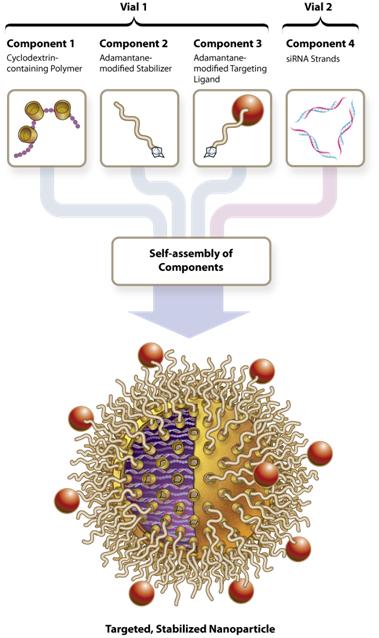
Based on a novel polymeric sugar (linear cyclodextrin) molecule, RONDEL has been applied thus farobserved in animal studies. Arrowhead intends to the delivery of two classes of therapeutics: siRNAutilize a subcutaneous formulation in its ARC-LPA program and small molecule drugs. The polymer is combined with the drug molecule to form a drug containing nanoparticle between 10 nanometers and 100 nanometers in size. We believe that this particle size is important because drug molecules below 10 nanometers are quickly cleared from the body in the urine while nanoparticles larger than 100 nanometers are not always able to escape the tumor vasculature to reach tumor cells. Nanoparticles between 10 and 100 nanometers can lead to preferential accumulation in tumor tissue,additional programs where the drug can take effect, leaving other tissues less affected. The drug delivery system has the added benefits of increasing solubility and allowing targeting of the nanoparticles.
chronic dosing may be required.Intellectual Property 10
The RONDEL delivery system offers the following advantages: | • | | Generalized delivery system—Binds to and self-assembles with the siRNA to form uniform colloidal-sized particles. Analysis has shown that these particles are spherical and between 10 nm and 100 nm in diameter.
|
Any siRNA sequence can be easily substituted—Because RONDEL binds to the siRNA backbone, other siRNAs sequences can be easily incorporated to form a new drug product.
| • | | Safety—The RONDEL technology has been shown to have a positive safety profilein vitro testing with human cell cultures, and the fully formulated polymer/siRNA particles exhibit a significant therapeutic window of safety in animals, even when repeated doses (up to eight doses over a four week period) are used.
|
| • | | Effective targeted delivery—We have demonstrated successful delivery of functional siRNA therapeutics to tumor cells and to hepatocytes by systemic administration and confirmed sequence-specific gene inhibition.
|
| • | | Human proof of concept—CALAA-01, the first clinical candidate developed using the RONDEL system, has established several important “firsts” in human testing of an siRNA therapeutic including first to show systemic siRNA delivery, first to show dose dependent accumulation in target cells and first to show RNAi mediated mRNA and protein knockdown.
|
CALAA-01 and RONDEL have been developed by Arrowhead’s majority-owned subsidiary, Calando Pharmaceuticals, Inc. (“Calando’). Arrowhead owns 74% of the fully diluted shares of Calando.
HOMING PEPTIDE PROGRAM
In April 2012, Arrowhead acquired Alvos Therapeutics, Inc. (“Alvos”). Alvos licensed a discovery platform and large library of proprietary human-derived Homing Peptides from the MD Anderson Cancer Center. This discovery platform is designed to identify targeting agents, such as peptides, that selectively accumulate in primary and metastatic tumors, associated vasculature, and to 30 healthy tissue types. Such targeting agents are of interest for drug development because they hold the promise of shepherding drugs into specific cells while sparing others. This new platform was acquired because it fit well into our existing business. One of the key advantages of our RNA delivery systems is their ability to be targeted. With a vast proprietary targeting library of our own, we believe that we can enhance the value of our RNAi programs and differentiate our capabilities from those of our competitors. In addition, we believe that we can apply the homing peptide sequences to non-RNA therapeutics and present attractive value to potential partners. The platform has the potential to allow Arrowhead to:
Develop therapeutic agents that hunt down and destroy known tumors, as well as distant unidentified metastases;
Convert cancer therapeutics that generally interact with most cells in the body to “smart” drugs that accumulate primarily at tumor sites and affect cancer cells preferentially, thereby improving the toxicity and side effects of currently used cancer drugs; and
Selectively target non-cancer therapeutics to virtually any tissue type in the body where they can have the desired pharmacologic effect.
This platform is potentially powerful in the specificity of the targeting sequences, the large number of unique sequences and their origin from human screening. In addition, because of the human-based identification process, there is lower risk that animal model data will not translate. Our proprietary library of 42,000 unique targeting sequences can be used with our own delivery platforms, as well as with small molecule drugs. This platform has achieved clinical proof of concept in targeting metastatic prostate cancer with the first sequence tested in humans.
Drs. Renata Pasqualini and Wadih Arap, who developed this technology, run a large laboratory at MD Anderson Cancer Center. They focus on discovering novel cell-surface receptors and validated receptors on tumor sites and identifying peptide sequences that will bind to those receptors. Importantly, their method identifies peptides that are rapidly internalized into cells. These peptide-receptor pairs hold the promise of shuttling therapeutic payloads preferentially and directly into those cells. The ability to target and deliver cytotoxins would address some of the problems with current cancer therapeutics by limiting side effects and increasing efficacy.
In order to discover these receptors and sequences that target them, Drs. Pasqualini and Arap used a technique calledin vivophage display. Over the past several years they have applied phage display screening to end-stage cancer patients with primary and metastatic tumors under rigorous ethical standards. To our knowledge, they are the only group in the world that is generating this type of human-derived data. Direct screening in human cancer patients has the potential to eliminate some of the uncertainty that has plagued current discovery methods with animal models. This strategy sought to map the human vasculature into “zip codes” and has discovered a large number of novel receptors that are expressed only on the cell surface of tumor sites and nowhere else. The library can be further increased by continuing to work with MD Anderson to screen additional patients.
11
Arrowhead is working to apply this technology to targeting our proprietary siRNA delivery vehicles. Our two primary delivery platforms, DPCs and RONDEL, are highly attractive in part because they have been shown to be well tolerated, effective, capable of delivering RNAs to multiple organ systems, and they are targetable. The Homing Peptide library provides our targeted therapeutic program with a powerful new source of flexibility. The library is also valuable in creating a new class of therapeutics, Peptide-Drug Conjugates, or PDCs. By linking the Homing Peptides to traditional small molecule drugs we can potentially transform a therapeutic that interacts with most cells in the body into one that interacts preferentially with the cell of choice. We believe that this transition from untargeted to targeted drugs is a paradigm shift for cancer therapeutics and that our new library puts us at the forefront of this transformation. We intend to build our own pipeline and work with partners to apply our targeting sequences to their drugs. We believe that this specific targeting will enable us to make existing generics safer and more effective and we intend to work with partners to help make their proprietary drugs better. Given the large number of approved APIs for oncology and the thousands of Homing Peptide sequences that we now have, there are many potential combinations of targeting sequence and drug molecules.
PDCs share the promise of the original class of guided therapeutics, antibody-drug conjugates or ADCs, in that they could increase efficacy and decrease toxicity relative to current standard of care oncology products. Benefits of PDCs as a class are as follows:
They are potentially faster, cheaper, and simpler to make than ADCs, making them attractive development projects for biopharmaceutical companies;
Their targets are expressed on a high percentage of multiple tumor types, giving them a larger potential commercial market than genetically targeted agents that are efficacious in only a small subset of patient populations; and
The use of Homing Peptides that were discovered in human cancer patients as the targeting moieties for PDCs potentially increases clinical probability of success.
We believe this unique mix of benefits will be attractive to potential partners in the biopharmaceutical industry. This technology has the potential to facilitate the rapid development of multiple new product candidates, each of which could meet a critical unmet medical need. In addition, screening in man has broad applicability in other therapeutic areas of interest to the biopharmaceutical industry.
Intellectual Property
We controlCompany controls approximately 154306 issued patents (including 66 for DPCs; 25 for hydrodynamic gene delivery; 26 for pH labile molecules; 7 for polyampholyte; 5 for template polymerization; 12 for delivery polymers; 133 for RNAi trigger molecules; 1 for targeting molecules; 1 for liver expression vector; and 29 for Homing Peptides), including European validations)validations, and 292247 patent applications.applications (108 applications in 13 families for DPCs; 140 applications in 23 families for RNAi trigger targets; and 8 for Homing Peptides). The pending applications have been filed throughout the world, including, in the United States, Argentina, ARIPO (Africa Regional Intellectual Property Organization), Australia, Brazil, Canada, Chile, China, Eurasian Patent Organization, Europe, the Arab States of the Gulf,Hong Kong, Israel, India, Indonesia, Iraq, Jordan, Japan, Republic of Korea, Mexico, New Zealand, OAPI (African Intellectual Property Organization), Peru, Philippines, Russian Federation, Saudi Arabia, Singapore, Thailand, Taiwan, Venezuela, Vietnam, and Venezuela.South Africa.
RONDELRNAi Triggers
Calando controls an intellectual property portfolio ofThe Company owns patents directed to certain linear cyclodextrin polymers and related technology (the “Linear Cyclodextrin System”). The portfolio is directedRNAi triggers targeted to both RONDEL and Cyclosert. In June 2009, Calando sold and assigned to Cerulean certain patents (“Cerulean Assigned Patents”) directed toward linear cyclodextrin polymers conjugated to drugs. Additionally, Calando granted Cerulean an exclusive license under its rights toreduce expression of hepatitis B viral proteins as well the Linear Cyclodextrin System to develop and commercialize CRLX-101 and Cerulean Products. Calando retained rights to use the Linear Cyclodextrin System to develop drugs in which a therapeutic agent is (i) a nucleic acid (e.g., siRNA), (ii) a second generation epothilone, (iii) tubulysin or (iv) cytolysin (collectively, the “Calando Products”).RRM2 gene.
| | Patent Group | Estimated Year of Expiration | RNAi Triggers | Patent directed to HBV RNAi triggers | 2032 | Patent directed to RRM2 RNAi triggers | 2031 | α-ENaC | 2028 | β-ENaC | 2031 | β-Catenin | 2033 | KRAS | 2033 | HSF1 | 2030, 2032 | APOC3 | 2034 | Cx43 | 2029 | HIF1A | 2026 | FRP-1 | 2026 | HCV | 2023 | PDtype4 | 2026 | PI4Kinase | 2028 | HRH1 | 2027 | SYK | 2027 | TNF-α | 2027 |
20
Dynamic Polyconjugates The issued patents include approximately 55 patents directed to the RONDEL and CYLCOSERT drug delivery platforms. Included in these 55 patents are approximately 34 patents covering linear cyclodextrin copolymers utilized in RONDEL and CYCLOSERT, issued in the United States, Europe (Austria, Belgium, Switzerland, Germany, Denmark, Spain, Finland, France, the United Kingdom, Greece, Ireland, Israel, Italy, Luxembourg, Monaco, Netherlands, Portugal, Sweden), Australia, Brazil, Canada, China, Cyprus, Japan, Republic of Korea, Mexico, Russian Federation, Singapore and South Africa. Approximately 14 patents are directed to inclusion complexes and drug-cyclodextrin complexes utilized in the RONDEL and CYLCOSERT platforms. These patents have issued in the United States, Australia, China, Israel, Japan, Republic of Korea, Russian Federation, Singapore, Taiwan and South Africa. Approximately seven additional patents issued in the United States and Europe (Austria, Switzerland, Germany, France and the United Kingdom) are directed to supramolecular complexes containing therapeutic agents. Calando also owns a U.S. issued patent (in addition to 14 patents in Europe, i.e., Austria, Belgium, Switzerland, Germany, Denmark, Spain, Finland, France, the United Kingdom, Hungary, Italy, Netherlands, Poland and Sweden) directed to the siRNAs targeting the gene targeted by the active ingredient in CALAA-01, as well as a U.S. patent directed to the siRNA active ingredient of CALAA-02.
12
HOMING PEPTIDES
We also control 18 patentsDPC related to our Homing Peptide platform, related to Adipotide, our drug candidate for the treatment for obesity and related metabolic disorders. Approximately five of these patents are United States patents and the remaining patents are validated in Belgium, Switzerland, Germany, Spain, France, the United Kingdom, Ireland, Greece, Italy, Netherlands, Portugal, Sweden and Turkey.
DPC’S
In addition, we control eleven patents related to our Dynamic Polyconjugate drug delivery platform. These patents have issued in the United States, Australia, Canada, Europe, France, Germany, Italy, Spain, Switzerland, United Kingdom, India, Japan, Mexico, New Zealand, Philippines, Russia, South Korea, Singapore, and South Africa. WeThe Company also control approximately 41controls a number of patents directed to hydrodynamic nucleic acid delivery, which issued in the United States, Australia and Europe (Austria,(validated in Austria, Belgium, Switzerland, Germany, Denmark, Spain, Finland, France, the United Kingdom, Hungary, Ireland, Italy, Netherlands and Sweden).
Thirteen additional patents are directed to various precursors to our DPC delivery platform, and other membrane active polymers, as well as additional drug and gene delivery methodologies and carriers (e.g., lipid- and micelle-based systems).
The approximate year of expiration for each of these various groups of patents are set forth below: | | | Patent Group | | Estimated Year of Expiration | Dynamic PolyconjugatesRONDEL™ and CYCLOSERT™TM (DPCTM) | Linear cyclodextrin copolymersDynamic Polyconjugates
| | 2018 | Inclusion complexes
| | 2021 | Drug-cyclodextrin complexes
| | 2024 | Supramolecular complexes containing therapeutic agents
| | 2019 | CALAA-012027
| Patent directed to RRM2 siRNAsDynamic Polyconjugates – second iteration
| | 2028 | CALAA-02
| Patent directed to HIF-2 alpha (EPAS1) siRNAs
| | 2030 | Adipotide®
| Targeting moieties and conjugates
| | 2021 | Targeted Pharmaceutical Compositions
| | 2021 | Dynamic Polyconjugates® (DPC®)2031
| Membrane Active Polymers | | 2027 | Membrane Active Polymers—Polymers – Additional Iterations | 2024 | Membrane Active Polymers – Additional Iterations | 20242032 | Copolymer Systems | | 2024 | Polynucleotide-Polymer Composition | | 2024 | Polynucleotide-Polymer Composition—Additional IterationsARC-520 Polymer
| 2031 | ARC-521 Composition | 2035 | Masking Chemistry | 2031 | Masking Chemistry | 2035 | Polyampholyte Delivery | | 2017 | pH Labile Molecules | | 2020 | Endosomolytic Polymers | | 2020 | Hydrodynamic delivery
| Various iterations
| | 2015 | Homing PeptidesHydrodynamic delivery
| EphA5 Targeting PeptidesFirst iterations
| | 20272015 | IL-11R Targeting PeptidesSecond iteration
| 2020 | Third iteration | 20222024 |
13
Calando has licensed patents from Alnylam relevant to siRNA therapeutics for both CALAA-01 and CALAA-02. Calando has out-licensed to Tube Pharmaceuticals GmbH, the use of the linear cyclodextrin system for delivering second generation synthetic epothilone drugs. Calando has also out-licensed to Tube Pharmaceuticals GmbH, the use of the linear cyclodextrin system for delivering tubulysin and cytolysin.
The RNAi and nanoparticle drug delivery patent landscapes are complex and rapidly evolving. As such, we may need to obtain additional patent licenses prior to commercialization of our candidates. You should review the factors identified in “Risk Factors” in Part I, Item 1A of this Annual Report on Form 10-K. License AgreementsHoming Peptides
Cerulean License
The linear cyclodextrin-basedWe also control patents related to our Homing Peptide platforms, related to Adipotide, our drug delivery platform, Cyclosert, was designedcandidate for the deliverytreatment for obesity and related metabolic disorders. Approximately seven of small molecule drugs. Cyclosert provides many ofthese patents are United States patents and the same benefits asremaining patents are validated in Belgium, Switzerland, Germany, Spain, France, Japan, the RONDEL system. In December 2008, we completed a Phase 1 trial with IT-101, a conjugate of Calando’s linear cyclodextrin polymerUnited Kingdom, Ireland, Greece, Italy, Netherlands, Portugal, Sweden and Camptothecin, a potent anti-cancer drug, with a positive safety profileTurkey.
Patent Group | Estimated Year of Expiration | Adipotide® | Targeting moieties and conjugates | 2021 | Targeted Pharmaceutical Compositions | 2021 | Homing Peptides | EphA5 Targeting Peptides | 2027 | IL-11R Targeting Peptides | 2022 |
21
Non-Exclusively Licensed Patent Rights obtained from Roche Roche and indications of efficacy. On June 23, 2009, wethe Company entered into a transactionStock and Asset Purchase Agreement on October 21, 2011 pursuant to which Roche assigned to Arrowhead its entire rights under certain licenses including: the License and Collaboration Agreement between Roche and Alnylam dated July 8, 2007 (the “Alnylam License”); the Non-Exclusive Patent License Agreement between Roche and MDRNA, Inc. dated February 12, 2009 (“MDRNA License”); and the Non-Exclusive License Agreement between Roche and City of Hope dated September 19, 2011 (the “COH License”) (Collectively the “RNAi Licenses”). The RNAi Licenses provide the Company with Cerulean related to Cyclosertnon-exclusive, worldwide, perpetual, irrevocable, royalty-bearing rights and IT-101 (the “Cerulean Transaction”). In the Cerulean Transaction, we granted Cerulean an irrevocable, perpetual, royalty bearing worldwide license with the right to sublicense a broad portfolio of intellectual property relating to the discovery, development, manufacture, characterization, and use of therapeutic products that function through the mechanism of RNA interference for specified targets.
Terms of the Alnylam License The Alnylam License provides us with a non-exclusive, worldwide, perpetual, irrevocable, royalty-bearing right and sublicensable license under Alnylam’s rights in certain patent rightsintellectual property existing as of its effective date, to engage in discovery, development, commercialization and know-how in the field of human diseases solely in order to: (a) conduct research and development on the Linear Cyclodextrin System,manufacturing activities, including making improvements thereto, in order to research and commercialize our clinical asset IT-101 (now known as “CRLX-101”), as well as certain other products in which no therapeutic agent is specifically defined (the “Cerulean Products”); (b) research, develop, make, have made, use, market, offer to sell, distribute,for sale, sell and import CRLX-101 and Cerulean Products; and (c) use, copy, modify and distribute certain know-how for those purposes. We retained all rights with respect tolicensed products in whichcertain fields. The fields include the treatment or prophylaxis of indications comprising an RNAi compound complementary to, and function in mediating the RNAi of, a target known or believed to be primarily implicated in one or more primary therapeutic agent is a (i) tubulysin, (ii) cytolysin, (iii) second generation epothilone or (iv) nucleic acid (hereinafter “Calando Products”).areas. The primary therapeutic areas are cancer, hepatic, metabolic disease and pulmonary disease. The hepatic therapeutic area specifically excludes targets of infectious pathogen. The Cerulean Transaction also involvedAlnylam license excludes access to intellectual property specifically related to “Blocked Targets.” “Blocked Targets” are those targets that are subject to a contractual obligation of a pre-existing agreement between Alnylam and its alliance partners. Under the sale and assignment by us of certain patents directed to Cyclosert and CRLX-101 (the “Cerulean Assigned Patents”) to Cerulean. Cerulean then granted back to us an exclusive, irrevocable, perpetual, royalty free, worldwide license, with the right to grant sublicenses, under the Cerulean Assigned Patents solely to the extent necessary to research and commercialize products in which each therapeutic agent is a cytolysin, tubulysin, second generation epothilone or any nucleic acid. As such,Alnylam License, we retain the rights to the RONDEL siRNA delivery platform, as well as the siRNA-based products, CALAA-01 and CALAA-02. The Cerulean Transaction resulted in an initial payment to Calando of $2.4 million. Cerulean ismay be obligated to pay development and sales milestone payments of up to $2.75 million if CRLX-101the mid to upper double digit millions of dollars for each licensed product that progresses through clinical trials andin a particular indication, receives marketing approval. If approved,approval for that indication and is the subject of a first commercial sale. Additionally, we are also entitledmay be obligated to receive uppay mid to an additional $30 million in sales milestone payments, plushigh single digit percentage royalties on net sales. Should Cerulean sublicense CRLX-101sales of such products.
Core Patents relating to RNAi The RNAi Licenses include patents relating to the general structure, architecture, and design of double-stranded oligonucleotide molecules, which engage RNA interference mechanisms in a third party,cell. These rights include the “Tuschl II” patents, including issued U.S. Patent Nos. 7,056,704; 7,078,196; 7,078,196; 8,329,463; 8,362,231; 8,372,968; and 8,445,327; “Tuschl I” patents, including U.S. Patent Nos. 8,394,628 and 8,420,391; and allowed “Tuschl I” patent application, U.S. Publication No. 2011024446; “City of Hope” patents, including U.S. Patent No. 8,084,599; and “Kreutzer-Limmer” patents assigned to Alnylam, including U.S. Patent Nos. 7,829,693; 8,101,594; 8,119,608; 8,202,980; and 8,168,776. Thomas Tuschl is the first named inventor on “Tuschl I” and “Tuschl II.” “Tuschl I” refers to the patents arising from the patent application entitled “The Uses of 21-23 Sequence-Specific Mediators of Double-Stranded RNA Interference as a Tool to Study Gene Function and as a Gene-Specific Therapeutic.” “Tuschl II” patents refer to the patents and patent applications arising from the patent application entitled “RNA Interference Mediating Small RNA Molecules.” “City of Hope” is the first named assignee of certain core RNAi trigger patents. The second named assignee of these patents is Integrated DNA Technologies, Inc. Kreutzer-Limmer patents refer to the Alnylam patents and patent applications, relating to core siRNA IP, which includes inventors, Roland Kreutzer and Stefan Limmer. Chemical modifications of double-stranded oligonucleotides The RNAi Licenses also include patents related to modifications of double-stranded oligonucleotides, including modifications to the base, sugar, or internucleoside linkage, nucleotide mimetics, and end modifications, which do not abolish the RNAi activity of the double-stranded oligonucleotides. Also included are patents relating to modified double-stranded oligonucleotides, such as meroduplexes described in in U.S. Publication No. 20100209487 assigned to Marina Biotech (f/k/a MDRNA, Inc.), and microRNAs described in U.S. Patent Nos. 7,582,744; 7,674,778, and 7,772,387 assigned to Alnylam as well as U.S. Patent No. 8,314,227 related to unlocked nucleic acids (UNA). The ‘227 patent was assigned by Marina Biotech to Arcturus Therapeutics, Inc. but remains part of the MDRNA License. The RNAi Licenses also include rights from INEX/Tekmira relating to lipid-nucleic acid particles, and oligonucleotide modifications to improve pharmacokinetic activity including resistance to degradation, increased stability, and more specific targeting of cells from Alnylam and ISIS Pharmaceuticals, Inc. 22
Manufacturing techniques for the double-stranded oligonucleotide molecules or chemical modifications The RNAi Licenses also include patents relating to the synthesis and manufacture of double-stranded oligonucleotide molecules for use in RNA interference, as well as chemical modifications of such molecules, as described above. These include methods of synthesizing the double-stranded oligonucleotide molecules such as in the core “Tuschl I” allowed U.S. Application No. 12/897,749, the core “Tuschl II” U.S. Patent Nos. 7,056,704; 7,078,196; and 8,445,327; and Alnylam’s U.S. Patent Nos. 8,168,776, as well as methods of making chemical modifications of the double-stranded oligonucleotides such as described in Alnylam’s U.S. Patent No. 7,723,509 and INEX’s U.S. Patent Nos. 5,976,567; 6,858,224; and 8,484,282. Patent applications are currently pending that further cover manufacturing techniques for double-stranded oligonucleotide molecules or chemical modifications. Uses and Applications of Double-Stranded Oligonucleotide Molecules or Chemical modifications The RNAi Licenses also include patents related to uses of the double-stranded oligonucleotides that function through the mechanism of RNA interference. These include for example, the core “Tuschl I” U.S. Patent No. 8,394,628 and “Tuschl II” U.S. Patent No. 8,329,463; Alnylam’s U.S. Patent Nos. 7,763,590; 8,101,594, and 8,119,608, and City of Hope‘s U.S. Patent No. 8,084,599. Other more specific uses have been acquired and patent applications are currently pending that cover additional end uses and applications of double-stranded oligonucleotides functioning through RNA interference. License from Alnylam In January 2012, we shall receiveobtained a percentagelicense from Alnylam under its rights in certain RNAi intellectual property to develop and commercialize RNAi-based products targeting RNAs encoded by the genome of HBV. Alnylam granted us a worldwide non-exclusive sublicensable royalty bearing license under Alnylam’s general RNAi intellectual property estate to research, develop and commercialize RNAi-based products targeting HBV RNAs in combination with DPC™ technology. Alnylam further granted us a worldwide sublicensable exclusive royalty bearing license under its target-specific RNAi patent rights to research, develop and commercialize RNAi-based products targeting HBV RNAs in combination with DPC™ technology. Alnylam further agreed to forego the development of any sublicensing income at rates between 10% and 40%, depending onRNAi-based products targeting HBV RNAs in combination with DPC™ technology. Under the stage of the drug’s development at the time of sublicensing. Cerulean islicense from Alnylam, we may be obligated to further pay development and sales milestone payments of up to $3 millionthe low double digit millions of dollars for each Cerulean Productlicensed product that progresses through clinical trials, and receives marketing approval. If Cerulean Products are approved,approval and is the subject of a first commercial sale. Additionally, we are entitledmay be obligated to receive uppay low single digit percentage royalties on sales of such products.
License to an additional $15 millionAlnylam In consideration for the licenses from Alnylam, in January 2012 we granted Alnylam a worldwide non-exclusive, sublicensable royalty bearing license under our broad and target-specific DPC™ intellectual property rights to research, develop and commercialize RNAi-based products against a single undisclosed target in combination with DPC™ technology. Under the license to Alnylam, Alnylam may be obligated to pay us development and sales milestone payments plus single digit royalties on net sales. Should Cerulean sublicense a Cerulean Product to a third party, we shall receive a percentage of any sublicensing income at a rate in the tens. The terms of the agreements of the Cerulean Transactions are tiedup to the expirationlow double digit millions of certain controlled patent rightsdollars for each licensed product that progresses through clinical trials, receives marketing approval and Cerulean Assigned Patents. Ceruleanis the subject of a first commercial sale. Additionally, Alnylam may terminate the agreements on thirty (30) days’ notice and unless there is a drug safety concern, would be obligated to re-assign the CRLX-101 IND back topay us and provide us with an exclusive license thereto under the Cerulean Assigned Patents. We are responsible for the costs associated with prosecutionlow single digit percentage royalties on sales of the patents we control and have licensed to Cerulean.
such products.14
University of Texas MD Anderson Cancer Center License In December 2010, we obtained an exclusive world-wideworldwide license from at the University of Texas MD Anderson Cancer Center in Houston, Texas (“UTMDACC”) related to Adipotide technology (the “UTMDACC License”). The UTMDACC License granted us a royalty-bearing, exclusive right (with the right to sublicense) under certain UTMDACC patents to develop and commercialize certain products in the fields of: 1) therapeutics, diagnostics and research services that both (i) incorporate peptides that specifically target adipose tissue, and (ii) are used to treat, diagnose or research solely either (a) obesity, overweight and/or (b) metabolic conditions related to, caused by and/or associated with obesity and overweight, e.g., diabetes; and 2) cancer therapies, diagnostics and research products associated with a specific targeting moiety. We also have rights to certain improvements to the UTMDACC technology arising in the lab of Drs. Wadih Arap and Renata Pasqualini (“UTMDACC Improvements”).technology. In consideration for the license, we paid UTMDACC an upfront fee of $2 million and are obligated to pay annual fees initially equal to $50,000 increasing up to a maximum of $100,000, with such annual fees creditable against milestone payments. We may be obligated to pay development milestone payments of up to $8.3 million for each UTMDCC licensed product that progresses through clinical trials and receives U.S. marketing approval are required.approval. Additional EU and Japanese approval milestone payments are in the low single digit million dollar range. If a commercial drug is developed and approved, royalty payments on net sales of UTMDACC licensed products are in the low single digit range. ShouldIf we sublicense or partner a UTMDACC licensed product, 23
UTMDACC would receive partnering fee percentages in the range of single digits to the twenties, depending on the stage of development of the partnered UTMDACC licensed product. The term of the UTMDACC License is linked to the last to expire patents licensed therein or 15 years if a licensed product contains only licensed know-how. We are obligated to actively and effectively attempt to commercialize the UTMDACC Technology and submit to UTMDACC a Phase 2 clinical trial protocol within two years of obtaining an approved IND. We are also obligated to commence a Phase 2 clinical trial within four years and a Phase 3 clinical trial within seven years of approval of an IND. However, we may obtain yearly extensions of time upon the payment of an increasing fee in the range of tens of thousands of dollars up to several hundred thousand dollars. We also have diligence obligations with respect to any UTMDACC Improvements later added to the license. Acquisition of Assets from Novartis On March 3, 2015, the Company entered into an Asset Purchase and Exclusive License Agreement (the “RNAi Purchase Agreement”) with Novartis pursuant to which the Company acquired Novartis’ RNAi assets and rights thereunder. Pursuant to the RNAi Purchase Agreement, the Company acquired or licensed certain patents and patent applications owned or controlled by Novartis related to RNAi therapeutics, assignment of Novartis’s rights under a license from Alnylam (the “Alnylam-Novartis License”), rights to three pre-clinical RNAi candidates, and a license to certain Novartis assets (the “Licensed Novartis Assets”). The UTMDACCpatents acquired from Novartis include multiple patent families covering delivery technologies and RNAi-trigger design rules and modifications. The Licensed Novartis Assets include an exclusive, worldwide right and license, shall automatically terminate if we file for bankruptcysolely in the RNAi field, with the right to grant sublicenses through multiple tiers under or are unablewith respect to pay our bills as they come due.certain patent rights and know how relating to delivery technologies and RNAi-trigger design rules and modifications. Under the assigned Alnylam-Novartis License, the Company has acquired a worldwide, royalty-bearing, exclusive license with limited sublicensing rights to existing and future Alnylam intellectual property (coming under Alnylam’s control on or before March 31, 2016), excluding intellectual property concerning delivery technology, to research, develop and commercialize 30 undisclosed gene targets. Research and Development Research and Development Facility Arrowhead operates aArrowhead’s research and development facilityoperations are located in Madison, Wisconsin. ThisWisconsin and are housed in two facilities. The main facility was built and equipped by Roche and was part of our 2011 acquisition of theirRoche’s RNA therapeutics business. We have integratedA second research and development facility, located in a nearby Madison suburb, was opened in 2015 as operations into that facility, including work onexpanded. Substantially all of the Company’s assets are located either in these facilities or in our platforms RONDEL, DPCs, Homing Peptides, and our clinical candidates CALAA-01, Adipotide, and ARC-520.corporate headquarters in Pasadena. A summary of the facilityour research and development facilities is provided below:
Approximately 40
| · | Approximately 87 scientists; |
| · | State-of-the-art laboratories in two buildings: main building has 27,000 total sq. ft. (16,000 sq. ft. lab, 11,000 sq. ft. office); second building has 2,900 total sq. ft. (1,700 sq. ft. lab, 1,200 sq. ft. office); |
| · | Complete small animal facility; |
| · | Primate colony housed at the Wisconsin National Primate Research Center, an affiliate of the University of Wisconsin; |
| · | In-house histopathology capabilities; |
| · | Animal models for metabolic, viral, and oncologic diseases; |
| · | Animal efficacy and safety assessment; |
| · | Polymer, peptide, oligonucleotide and small molecule synthesis and analytics capabilities (HPLC, NMR, MS, etc.); |
| · | Polymer, peptide and oligonucleotide PK, biodistribution, clearance methodologies; and |
| · | Conventional and confocal microscopy, flow cytometry, Luminex platform, qRT-PCR, clinical chemistry analytics. |
State-of-the-art laboratories: 24,000 total sq. ft. of lab space;
Complete small animal facility with capacity for 10,000 rodents in 2012;
Primate colony housed at University of Wisconsin;
In-house histopathology capabilities;
Animals models for metabolic, viral, and oncologic diseases;
Animal efficacy and safety assessment;
Peptide synthesis and analytics capabilities;
Polymer and small molecule synthesis and analytics capabilities (NMR, mass spec, etc.);
Polymer and siRNA PK, biodistribution, clearance methodologies; and
Confocal microscopy, flow cytometry, Luminex platform, clinical chemistry analytics.
Research and Development Expenses Research and development (R&D) expenses consist of costs incurred in identifying,discovering, developing and testing our product programs. Theseclinical candidates and platform technologies. R&D expenses consist primarilyalso include costs related to clinical trials, including costs of salaries and related expenses for personnel, license fees, consulting fees, contract research organizations to recruit patients and manage clinical trials. Other costs associated with clinical trials include manufacturing of clinical supplies, as well as good laboratory practice (“GLP”) toxicology studies necessary to support clinical trials, both of which are 24
outsourced to cGMP-compliant manufacturers and the costs of laboratory equipment and facilities. ResearchGLP-compliant laboratories. Total research and development expense for 2012fiscal 2015 was $8.7$47.3 million, an increase from $3.5$23.1 million in 2011,2014 and $8.7 million in 2013. We employ approximately 87 employees in an R&D function, primarily due toworking from our facility in Madison, Wisconsin. These employees are engaged in various areas of research on Arrowhead candidate and platform development including synthesis and analytics, PK/biodistribution, formulation, CMC and analytics, tumor and extra-hepatic targeting, bioassays, live animal research, toxicology/histopathology, clinical and regulatory operations, and other areas. Salaries and payroll-related expenses for our R&D activities were $11.6 million in fiscal 2015, $7.8 million in fiscal 2014, and $4.1 million in fiscal 2013. Laboratory supplies including animal-related costs for in-vivo studies were $3.1 million, $2.3 million, and $1.3 million in fiscal 2015, 2014, and 2013, respectively. Costs related to manufacture of clinical supplies, GLP toxicology studies and other outsourced lab studies, as well as clinical trial costs were $41.8 million, $18.8 million, and $6.1 million in fiscal 2015, 2014, and 2013 respectively. Facility-related costs, primarily rental costs for our leased laboratory in Madison, Wisconsin were $1.0 million, $0.9 million, and $0.7 million in fiscal 2015, 2014, and 2013, respectively. Other research and development expenses were $1.4 million, $1.2 million, and $0.6 million in fiscal 2015, 2014 and 2013, respectively. These expenses are primarily related to milestone payments, which can vary from period to period depending on the acquisitionnature of our various license agreements, and the Madison facility.timing of reaching various development milestones requiring payment. Government Regulation GovernmentalGovernment authorities in the U.S.United States, at the federal state, and local levels, and in other countries and jurisdictions, including the European Union, extensively regulate, among other things, the research, development, testing, product approval, manufacture, quality control, manufacturing changes, packaging, storage, recordkeeping, labeling, promotion, advertising, sales, distribution, marketing, and marketing, among other things,import and export of drugs and biologic products. All of our foreseeable product candidates are expected to be regulated as drug products.
drugs. The processes for obtaining regulatory approval in the U.S. and in foreign countries and jurisdictions, along with compliance with applicable statutes and regulations and other regulatory authorities both pre- and post-commercialization, are a significant factor in the production and marketing of our products and our R&D activities and require the expenditure of substantial time and financial resources.Review and Approval of Drugs in the United States 15
In the U.S., the FDA regulates drug productsand other government entities regulate drugs under the Federal Food, Drug, and Cosmetic Act (the “FDCA”), and other laws within the Public Health Service Act.Act, and the regulations promulgated under those statutes, as well as other federal and state statutes and regulations. Failure to comply with applicable legal and regulatory requirements in the U.S. requirements, both before andat any time during the product development process, approval process, or after approval, may leadsubject us to a variety of administrative andor judicial sanctions, such as a delay in approving or refusal by the FDA to approve pending applications, withdrawal of approvals, delay or suspension of clinical trials, issuance of warning letters and other types of regulatory letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil monetary penalties, refusals of or debarment from government contracts, exclusion from the federal healthcare programs, restitution, disgorgement of profits, civil or criminal investigations by the FDA, U.S. Department of Justice, State Attorneys General, and/or other agencies, False Claims Act suits and/or other litigation, and/or criminal prosecutions. Before An applicant seeking approval to market and distribute a new drug products are marketed theyin the U.S. must be approved bytypically undertake the FDA. The steps required before a novel drug product is approved by the FDA include: following: (1) completion of pre-clinical laboratory tests, animal studies, and formulation tests; studies in compliance with the FDA’s GLP regulations; (2) submission to the FDA of an Investigational New Drug Application (“IND”) for human clinical testing, which must become effective without FDA objection before human clinical trials may begin; (3) approval by an independent institutional review board (“IRB”), representing each clinical site before each clinical trial may be initiated; (4) performance of adequate and well-controlled human clinical trials in accordance with the FDA’s current good clinical practice (“cGCP”) regulations, to establish the safety and effectiveness of the proposed drug product for each indication for which approval is sought; (4) (5) preparation and submission to the FDA of a New Drug Application (“NDA”); (5) 25
(6) satisfactory review of the NDA by an FDA advisory committee, where appropriate or if applicable, (7) satisfactory completion of aone or more FDA inspectioninspections of the manufacturing facility or facilities at which the drug product, isand the active pharmaceutical ingredient or ingredients thereof, are produced to assess compliance with cGMP;current good manufacturing practice (“cGMP”) regulations and FDA reviewto assure that the facilities, methods, and finally (6)controls are adequate to ensure the product’s identity, strength, quality, and purity; (8) payment of user fees, as applicable, and securing FDA approval of the NDA; and (9) compliance with any post-approval requirements, such as any Risk Evaluation and Mitigation Strategies (“REMS”) or post-approval studies required by the FDA. Preclinical Studies and an NDA.IND Pre-clinical testsPreclinical studies can include in vitro and animal studies to assess the potential for adverse events and, in some cases, to establish a rationale for therapeutic use. The conduct of preclinical studies is subject to federal regulations and requirements, including GLP regulations. Other studies include laboratory evaluationsevaluation of product chemistry, toxicitythe purity, stability and formulation, as well as animal studies. Thephysical form of the manufactured drug substance or active pharmaceutical ingredient and the physical properties, stability and reproducibility of the formulated drug or drug product. An IND sponsor must submit the results of the pre-clinicalpreclinical tests, together with manufacturing information, and analytical data, are submittedany available clinical data or literature and plans for clinical studies, among other things, to the FDA as part of an IND. Some preclinical testing, such as longer-term toxicity testing, animal tests of reproductive adverse events and carcinogenicity, may continue after the IND which must become effective before human clinical trials may begin.is submitted. An IND will automatically becomebecomes effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions such asrelated to a proposed clinical trial and places the conduct of the trials as outlined in the IND.trial on clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding FDA concerns or questions before the clinical trialstrial can proceed. There can be no assurance thatbegin. As a result, submission of an IND willmay not result in the FDA authorizationallowing clinical trials to commencecommence.
Following commencement of a clinical trials. Oncetrial under an IND, the FDA may place a clinical hold on that trial. A clinical hold is in effect, eachan order issued by the FDA to the sponsor to delay a proposed clinical trialinvestigation or to be conductedsuspend an ongoing investigation. A partial clinical hold is a delay or suspension of only part of the clinical work requested under the IND must be submittedIND. For example, a specific protocol or part of a protocol is not allowed to proceed, while other protocols may do so. No more than 30 days after imposition of a clinical hold or partial clinical hold, the FDA whichwill provide the sponsor a written explanation of the basis for the hold. Following issuance of a clinical hold or partial clinical hold, an investigation may only resume after the FDA has notified the sponsor that the investigation may proceed. The FDA will base that determination on information provided by the sponsor correcting the deficiencies previously cited or may not allowotherwise satisfying the trial toFDA that the investigation can proceed. Human Clinical Studies in Support of an NDA Clinical trials involve the administration of the investigational drugproduct to human subjects under the supervision of qualified physician-investigatorsinvestigators in accordance with cGCP requirements, which include, among other things, the requirement that all research subjects provide their informed consent in writing before their participation in any clinical trial. Clinical trials are conducted under written study protocols detailing, among other things, the objectives of the study, the parameters to be used in monitoring safety and healthcare personnel. Clinicalthe effectiveness criteria to be evaluated. A protocol for each clinical trial and any subsequent protocol amendments must be submitted to the FDA as part of the IND. In addition, an IRB representing each institution participating in the clinical trial must review and approve the plan for any clinical trial before it commences at that institution, and the IRB must conduct continuing review and reapprove the study at least annually. The IRB must review and approve, among other things, the study protocol and informed consent information to be provided to study subjects. An IRB must operate in compliance with FDA regulations. Information about certain clinical trials must be submitted within specific timeframes to the NIH for public dissemination on its ClinicalTrials.gov website. Human clinical trials are typically conducted in three definedsequential phases, but the phaseswhich may overlap or be combined. combined: Phase 1 usually involves1: The product candidate is initially introduced into healthy human subjects or patients with the initial administration of the investigational drugtarget disease or biologic product to healthy individuals to evaluate itscondition and tested for safety, dosage tolerance, absorption, metabolism, distribution, excretion and, pharmacodynamics. if possible, to gain an early indication of its effectiveness. Phase 2 usually involves trials in2: The product candidate is administered to a limited patient population withto identify possible adverse effects and safety risks, to preliminarily evaluate the disease or conditionefficacy of the product for which the test material is being developed,specific targeted diseases and to evaluatedetermine dosage tolerance and appropriate dosage; identify possible adverse side effects and safety risks; and preliminarily evaluate the effectiveness of the drug or biologic for specific indications. optimal dosage. Phase 3 trials usually further evaluate effectiveness and test further for safety by administering the drug or biologic3: The product candidate in its final form inis administered to an expanded patient population. Ourpopulation, generally at geographically dispersed clinical trial sites, in well-controlled clinical trials to generate enough data to statistically evaluate the efficacy and safety of the product development partners,for 26
approval, to establish the overall risk-benefit profile of the product, and to provide adequate information for the labeling of the product. Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and more frequently if serious adverse events occur. Phase 1, Phase 2, and Phase 3 clinical trials may not be completed successfully within any specified period, or at all. Furthermore, the FDA or wethe sponsor may suspend or terminate a clinical trialstrial at any time on various grounds, including any situation where we believea finding that patientsthe research subjects are being exposed to an unacceptable health riskrisk. Similarly, an IRB can suspend or are obtaining no medical benefit fromterminate approval of a clinical trial at its institution, or an institution it represents, if the test material.clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients. The FDA will typically inspect one or more clinical sites in late-stage clinical trials to assure compliance with cGCP and the integrity of the clinical data submitted. Submission of an NDA to the FDA Assuming successful completion of the required clinical testing and other requirements, the results of the pre-clinical trialspreclinical and the clinical trials,studies, together with other detailed information including information onrelating to the product’s chemistry, manufacture, controls and composition of the product,proposed labeling, among other things, are submitted to the FDA in the formas part of an NDA requesting approval to market the drug product for one or more indications. Under federal law, the submission of most NDAs is additionally subject to an application user fee, currently exceeding $2.3 million for fiscal year 2016, and the sponsor of an approved NDA is also subject to annual product and establishment user fees, currently exceeding $114,450 per product and $585,200 per establishment for fiscal year 2016. These fees are typically increased annually. Under certain circumstances, the FDA will waive the application fee for the first human drug application that a small business, defined as a company with less than 500 employees, or its affiliate, submits for review. An affiliate is defined as a business entity that has a relationship with a second business entity if one business entity controls, or has the power to control, the other business entity, or a third party controls, or has the power to control, both entities. In addition, an application to market a prescription drug product that has received orphan designation is not subject to a prescription drug user fee unless the application includes an indication for other than the rare disease or condition for which the drug was designated. The FDA conducts a preliminary review of an NDA within 60 days of its receipt and informs the sponsor by the 74th day after the FDA’s receipt of the submission to determine whether the application is sufficiently complete to permit substantive review. The FDA may request additional information rather than accept an NDA for filing. In this event, the application must be resubmitted with the additional information. The resubmitted application is also subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review. The FDA has agreed to specified performance goals in the review process of NDAs. Most such applications are meant to be reviewed within ten months from the date of filing, and most applications for “priority review” products are meant to be reviewed within six months of filing. The review process may be extended by the FDA for three additional months to consider new information or clarification provided by the applicant to address an outstanding deficiency identified by the FDA following the original submission. Before approving an application,NDA, the FDA typically will usually inspect the facility or facilities where the product is manufactured, andmanufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product unless cGMP compliance is satisfactory. Ifwithin required specifications. Additionally, before approving an NDA, the FDA determineswill typically inspect one or more clinical sites to assure compliance with cGCP. The FDA also may require submission of an REMS plan to mitigate any identified or suspected serious risks. The REMS plan could include medication guides, physician communication plans, assessment plans, and elements to assure safe use, such as restricted distribution methods, patient registries, or other risk minimization tools. The FDA is required to refer an application for a novel drug to an advisory committee or explain why such referral was not made. Typically, an advisory committee is a panel of independent experts, including clinicians and other scientific experts, that reviews, evaluates and provides a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions. 27
The FDA’s Decision on an NDA On the basis of the FDA’s evaluation of the NDA is not acceptable,and accompanying information, including the results of the inspection of the manufacturing facilities, the FDA may outlineissue an approval letter or a complete response letter. An approval letter authorizes commercial marketing of the product with specific prescribing information for specific indications. A complete response letter generally outlines the deficiencies in the submission and may require substantial additional testing or information in order for the FDA to reconsider the application. If and when those deficiencies have been addressed to the FDA’s satisfaction in a resubmission of the NDA, and oftenthe FDA will requestissue an approval letter. The FDA has committed to reviewing such resubmissions in two or six months depending on the type of information included. Even with submission of this additional information. information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval. If the FDA approves a product, it may limit the NDA, certainapproved indications for use for the product, require that contraindications, warnings or precautions be included in the product labeling, require that post-approval studies, including Phase 4 clinical trials, be conducted to further assess the drug’s safety after approval, require testing and surveillance programs to monitor the product after commercialization, or impose other conditions, including distribution restrictions or other risk management mechanisms, including REMS, which can materially affect the potential market and profitability of the product. The FDA may prevent or limit further marketing of a product based on the results of post-market studies or surveillance programs. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes orand additional labeling claims, are subject to further testing requirements and FDA review and approval. The testingproduct may also be subject to official lot release, meaning that the manufacturer is required to perform certain tests on each lot of the product before it is released for distribution. If the product is subject to official release, the manufacturer must submit samples of each lot, together with a release protocol showing a summary of the history of manufacture of the lot and approval process requires substantial time, effortthe results of all of the manufacturer’s tests performed on the lot, to the FDA. The FDA may in addition perform certain confirmatory tests on lots of some products before releasing the lots for distribution. Finally, the FDA will conduct laboratory research related to the safety and financial resources, and approval on a timely basis, if at all, cannot be guaranteed.effectiveness of drug products. Under the Orphan Drug Act, the FDA may grant orphan drug designation to a drug intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the U.S. and for which there is no reasonable expectation that the cost of developing and making available in the U.S. a drug for this type of disease or condition will be recovered from sales in the U.S. for that drug. Orphan drug designation must be requested before submitting an NDA. NDA, and both the drug and the disease or condition must meet certain criteria specified in the Orphan Drug Act and FDA’s implementing regulations at 21 C.F.R. Part 316. The granting of an orphan drug designation does not alter the standard regulatory requirements and process for obtaining marketing approval. Safety and effectiveness of a drug must be established through adequate and well-controlled studies. After the FDA grants orphan drug designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. If a product that has orphan drug designation subsequently receives the first FDA approval for the disease for which it has such designation, the product is entitled to orphan product exclusivity, which means that the FDA may not approve any other application to market the same drug for the same indication, except in very limited circumstances, for seven years. Post-Approval Requirements Drugs manufactured or distributed pursuant to FDA approvals are subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to recordkeeping, periodic reporting, product sampling and distribution, advertising and promotion and reporting of adverse experiences with the product. After approval, most changes to the approved product, such as adding new indications or other labeling claims, are subject to prior FDA review and approval. There also are continuing, annual user fee requirements for any marketed products and the establishments at which such products are manufactured, as well as new application fees for supplemental applications with clinical data. In addition, regardlessdrug manufacturers and other entities involved in the manufacture and distribution of the type of approval, we and our partnersapproved drugs are required to complyregister their establishments with a number of FDA requirements both before and after approval. For example, drug makers are required to report certain adverse reactions and production problems, if any, to the FDA and state agencies, and are subject to comply with certain requirements concerning advertising and promotion for our products. In addition, quality control and manufacturing procedures must continue to conform to cGMP after approval, andperiodic unannounced inspections by the FDA periodically inspects manufacturing facilities to assessand these state agencies for compliance with cGMP.cGMP requirements. Changes to the manufacturing process are strictly regulated and often require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP and impose reporting and documentation requirements upon the sponsor and any third-party manufacturers that the sponsor may decide to use. Accordingly, manufacturers must continue to expend time, money, and effort in all areasthe area of regulatory compliance, including production and quality control to maintain cGMP compliance. 28
Once an approval is granted, the FDA may withdraw the approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events or problems with manufacturing processes of unanticipated severity or frequency, or failure to comply with cGMP. In addition, discovery of problems, such as safety problems,regulatory requirements, may result in revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical trials to assess new safety risks; or imposition of distribution or other restrictions under a REMS program. Other potential consequences include, among other things: | · | restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls; |
| · | fines, warning letters or holds on post-approval clinical trials; |
| · | refusal of the FDA to approve pending NDAs or supplements to approved NDAs, or suspension or revocation of product license approvals; |
| · | product seizure or detention, or refusal to permit the import or export of products; or |
| · | injunctions or the imposition of civil or criminal penalties. |
The FDA strictly regulates marketing, labeling, advertising and promotion of products that are placed on the market. Drugs may be promoted only for the approved indications and in accordance with the provisions of the approved label. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability. In addition, the distribution of prescription pharmaceutical products is subject to the Prescription Drug Marketing Act (PDMA), which regulates the distribution of drugs and drug samples at the federal level, and sets minimum standards for the registration and regulation of drug distributors by the states. Both the PDMA and state laws limit the distribution of prescription pharmaceutical product samples and impose requirements to ensure accountability in distribution. Abbreviated New Drug Applications for Generic Drugs In 1984, with passage of the Drug Price Competition and Patent Term Restoration Act of 1984 (commonly referred to as the “Hatch-Waxman Amendments”) amending the FDCA, Congress authorized the FDA to approve generic drugs that are the same as drugs previously approved by the FDA under the NDA provisions of the statute. To obtain approval of a generic drug, an applicant must submit an abbreviated new drug application (ANDA) to the agency. In support of such applications, a generic manufacturer may rely on the preclinical and clinical testing previously conducted for a drug product previously approved under an NDA, known as the reference listed drug, or RLD. To reference that information, however, the ANDA applicant must demonstrate, and the FDA must conclude, that the generic drug does, in fact, perform in the same way as the RLD it purports to copy. Specifically, in order for an ANDA to be approved, the FDA must find that the generic version is identical to the RLD with respect to the active ingredients, the route of administration, the dosage form, and the strength of the drug. At the same time, the FDA must also determine that the generic drug is “bioequivalent” to the innovator drug. Under the statute, a generic drug is bioequivalent to a RLD if “the rate and extent of absorption of the generic drug do not show a significant difference from the rate and extent of absorption of the RLD.” Upon approval of an ANDA, the FDA indicates that the generic product is “therapeutically equivalent” to the RLD and it assigns a therapeutic equivalence rating to the approved generic drug in its publication “Approved Drug Products with Therapeutic Equivalence Evaluations,” also referred to as the “Orange Book.” Physicians and pharmacists consider the therapeutic equivalence rating to mean that a generic drug is fully substitutable for the RLD. In addition, by operation of certain state laws and numerous health insurance programs, the FDA’s designation of a therapeutic equivalence rating often results in substitution of the generic drug without the knowledge or consent of either the prescribing physician or patient. Under the Hatch-Waxman Amendments, the FDA may not approve an ANDA until any applicable period of nonpatent exclusivity for the RLD has expired. The FDCA provides a period of five years of data exclusivity for new drug containing a new chemical entity. In cases where such exclusivity has been granted, an ANDA may not be filed with the FDA until the expiration of five years unless the submission is accompanied by a Paragraph IV certification, in which case the applicant may submit its application four years following the original product approval. The FDCA also provides for a period of three years of exclusivity if the NDA includes reports of one or more new clinical investigations, other than bioavailability or bioequivalence studies, that were conducted by or for the applicant and are essential to the approval of the application. This three-year exclusivity period often protects changes to a previously approved drug product, such as a new dosage form, route of administration, combination or indication. 29
Hatch-Waxman Patent Certification and the 30 Month Stay Upon approval of an NDA or a supplement thereto, NDA sponsors are required to list with the FDA each patent with claims that cover the applicant’s product or a method of using the product. Each of the patents listed by the NDA sponsor is published in labelingthe Orange Book. When an ANDA applicant files its application with the FDA, the applicant is required to certify to the FDA concerning any patents listed for the reference product in the Orange Book, except for patents covering methods of use for which the ANDA applicant is not seeking approval. Specifically, the applicant must certify with respect to each patent that: | · | the required patent information has not been filed; |
| · | the listed patent has expired; |
| · | the listed patent has not expired, but will expire on a particular date and approval is sought after patent expiration; or |
| · | the listed patent is invalid, unenforceable or will not be infringed by the new product. |
A certification that the new product will not infringe the already approved product’s listed patents or restrictionsthat such patents are invalid or unenforceable is called a Paragraph IV certification. If the applicant does not challenge the listed patents or indicate that it is not seeking approval of a patented method of use, the ANDA application will not be approved until all the listed patents claiming the referenced product have expired. If the ANDA applicant has provided a Paragraph IV certification to the FDA, the applicant must also send notice of the Paragraph IV certification to the NDA and patent holders once the ANDA has been accepted for filing by the FDA. The NDA and patent holders may then initiate a patent infringement lawsuit in response to the notice of the Paragraph IV certification. The filing of a patent infringement lawsuit within 45 days after the receipt of a Paragraph IV certification automatically prevents the FDA from approving the ANDA until the earlier of 30 months, expiration of the patent, settlement of the lawsuit or a decision in the infringement case that is favorable to the ANDA applicant. To the extent that a Section 505(b)(2) applicant is relying on studies conducted for an already approved product, the applicant is required to certify to the FDA concerning any patents listed for the approved product in the Orange Book to the same extent that an ANDA applicant would. As a result, approval of a 505(b)(2) NDA can be stalled until all the listed patents claiming the referenced product manufacturerhave expired, until any non-patent exclusivity, such as exclusivity for obtaining approval of a new chemical entity, listed in the Orange Book for the referenced product has expired, and, in the case of a Paragraph IV certification and subsequent patent infringement suit, until the earlier of 30 months, settlement of the lawsuit or a decision in the infringement case that is favorable to the Section 505(b)(2) applicant. Pediatric Studies and Exclusivity Under the Pediatric Research Equity Act of 2003, an NDA holder,or supplement thereto must contain data that are adequate to assess the safety and effectiveness of the drug product for the claimed indications in all relevant pediatric subpopulations, and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. With enactment of the Food and Drug Administration Safety and Innovation Act, or FDASIA, in 2012, sponsors must also submit pediatric study plans prior to the assessment data. Those plans must contain an outline of the proposed pediatric study or studies the applicant plans to conduct, including removalstudy objectives and design, any deferral or waiver requests, and other information required by regulation. The applicant, the FDA, and the FDA’s internal review committee must then review the information submitted, consult with each other, and agree upon a final plan. The FDA or the applicant may request an amendment to the plan at any time. The FDA may, on its own initiative or at the request of the applicant, grant deferrals for submission of some or all pediatric data until after approval of the product for use in adults, or full or partial waivers from the market. 16
Corporate Information
pediatric data requirements. Additional requirements and procedures relating to deferral requests and requests for extension of deferrals are contained in FDASIA. Unless otherwise noted, (1)required by regulation, the pediatric data requirements do not apply to products with orphan designation. Pediatric exclusivity is another type of non-patent marketing exclusivity in the United States and, if granted, provides for the attachment of an additional six months of marketing protection to the term “Arrowhead” refersof any existing regulatory exclusivity, including the non-patent and orphan exclusivity. This six-month exclusivity may be granted if an NDA sponsor submits pediatric data that fairly respond to Arrowhead Research Corporation, a Delaware corporation, (2)written request from the termsFDA for such data. The data do not need to show the “Company,” “we,” “us,” and “our,” referproduct to be effective in the pediatric population studied; rather, if the clinical trial is deemed to fairly respond to the ongoing business operationsFDA’s request, the additional protection is granted. If reports of Arrowheadrequested pediatric studies are submitted to and its Subsidiaries, whether conductedaccepted by the FDA within the statutory time limits, whatever statutory or regulatory periods of exclusivity or patent protection cover the product are extended by six months. This is not a patent term extension, but it effectively extends the regulatory period during which the FDA cannot accept or approve another application. 30
Patent Term Restoration and Extension A patent claiming a new drug product may be eligible for a limited patent term extension under the Hatch-Waxman Amendments. Those Amendments permit a patent restoration of up to five years for patent term lost during product development and the FDA regulatory review. The restoration period granted is typically one-half the time between the effective date of an IND and the submission date of a NDA, plus the time between the submission date of a NDA and ultimate approval. Patent term restoration cannot be used to extend the remaining term of a patent past a total of 14 years from the product’s approval date. Only one patent applicable to an approved drug product is eligible for the extension, and the application for the extension must be submitted prior to the expiration of the patent in question. The U.S. Patent and Trademark Office reviews and approves the application for any patent term extension or restoration in consultation with the FDA. Review and Approval of Drug Products in the European Union In order to market any product outside of the United States, a company must also comply with numerous and varying regulatory requirements of other countries and jurisdictions regarding quality, safety and efficacy and governing, among other things, clinical trials, marketing authorization, commercial sales and distribution of our products. Whether or not it obtains FDA approval for a product, the company would need to obtain the necessary approvals by the comparable foreign regulatory authorities before it can commence clinical trials or marketing of the product in those countries or jurisdictions. The approval process ultimately varies between countries and jurisdictions and can involve additional product testing and additional administrative review periods. The time required to obtain approval in other countries and jurisdictions might differ from and be longer than that required to obtain FDA approval. Regulatory approval in one country or jurisdiction does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country or jurisdiction may negatively impact the regulatory process in others. Pursuant to the European Clinical Trials Directive, a system for the approval of clinical trials in the European Union has been implemented through Arrowhead ornational legislation of the member states. Under this system, an applicant must obtain approval from the competent national authority of a subsidiary of Arrowhead, (3) the term “Subsidiaries” refers collectively to Arrowhead Madison Inc. (“Madison”, formerly known as Roche Madison, Inc.), Alvos Therapeutics, Inc. (“Alvos”), Calando Pharmaceuticals, Inc. (“Calando”), Ablaris Therapeutics, Inc. (“Ablaris”), Agonn Systems, Inc. (“Agonn”,) and Tego Biosciences Corporation (“Tego”) as well as our former subsidiary, Unidym, Inc. (“Unidym”), which was divested in January 2011, (4) the term “Minority Investments” refers collectively to Nanotope, Inc. (“Nanotope”) and Leonardo Biosystems, Inc. (“Leonardo”)European Union member state in which the company holdsclinical trial is to be conducted. Furthermore, the applicant may only start a less than majority ownership position,clinical trial after a competent ethics committee has issued a favorable opinion. Clinical trial applications must be accompanied by an investigational medicinal product dossier with supporting information prescribed by the European Clinical Trials Directive and (5)corresponding national laws of the term “Common Stock” refersmember states and further detailed in applicable guidance documents. To obtain marketing approval of a drug under European Union regulatory systems, an applicant must submit a marketing authorization application (MAA) either under a centralized or decentralized procedure. The centralized procedure provides for the grant of a single marketing authorization by the European Commission that is valid for all European Union member states. The centralized procedure is compulsory for specific products, including for medicines produced by certain biotechnological processes, products designated as orphan medicinal products, advanced therapy products and products with a new active substance indicated for the treatment of certain diseases. For products with a new active substance indicated for the treatment of other diseases and products that are highly innovative or for which a centralized process is in the interest of patients, the centralized procedure may be optional. Under the centralized procedure, the Committee for Medicinal Products for Human Use (CHMP) established at the European Medicines Agency (EMA) is responsible for conducting the initial assessment of a drug. The CHMP is also responsible for several post-authorization and maintenance activities, such as the assessment of modifications or extensions to Arrowhead’s Common Stockan existing marketing authorization. Under the centralized procedure in the European Union, the maximum timeframe for the evaluation of an MAA is 210 days, excluding clock stops, when additional information or written or oral explanation is to be provided by the applicant in response to questions of the CHMP. Accelerated evaluation might be granted by the CHMP in exceptional cases, when a medicinal product is of major interest from the point of view of public health and in particular from the viewpoint of therapeutic innovation. In this circumstance, the EMA ensures that the opinion of the CHMP is given within 150 days. The decentralized procedure is available to applicants who wish to market a product in various European Union member states where such product has not received marketing approval in any European Union member states before. The decentralized procedure provides for approval by one or more other, or concerned, member states of an assessment of an application performed by one member state designated by the applicant, known as the reference member state. Under this procedure, an applicant submits an application based on identical dossiers and related materials, including a draft summary of product characteristics, and draft labeling and package leaflet, to the reference member state and concerned member states. The reference member state prepares a draft assessment report and drafts of the related materials within 120 days after receipt of a valid application. Within 90 days of receiving the reference member state’s assessment report and related materials, each concerned member state must decide whether to approve the assessment report and related materials. 31
If a member state cannot approve the assessment report and related materials on the grounds of potential serious risk to public health, the disputed points are subject to a dispute resolution mechanism and may eventually be referred to the European Commission, whose decision is binding on all member states. Data and Market Exclusivity in the European Union In the European Union, new chemical entities qualify for eight years of data exclusivity upon marketing authorization and an additional two years of market exclusivity. This data exclusivity, if granted, prevents regulatory authorities in the European Union from referencing the innovator’s data to assess a generic (abbreviated) application for eight years, after which generic marketing authorization can be submitted, and the term “stockholder(s)” refersinnovator’s data may be referenced, but not approved for two years. The overall ten-year period will be extended to a maximum of eleven years if, during the first eight years of those ten years, the marketing authorization holder obtains an authorization for one or more new therapeutic indications which, during the scientific evaluation prior to their authorization, are held to bring a significant clinical benefit in comparison with existing therapies. Even if a compound is considered to be a new chemical entity and the sponsor is able to gain the prescribed period of data exclusivity, another company nevertheless could also market another version of the drug if such company can complete a full MAA with a complete database of pharmaceutical test, preclinical tests and clinical trials and obtain marketing approval of its product. Pharmaceutical Coverage, Pricing and Reimbursement Significant uncertainty exists as to the holderscoverage and reimbursement status of Arrowhead Common Stock.products approved by the FDA and other government authorities. Sales of products will depend, in part, on the extent to which the costs of the products will be covered by third-party payors, including government health programs such as, in the United States, Medicare and Medicaid, commercial health insurers and managed care organizations. The process for determining whether a payor will provide coverage for a product may be separate from the process for setting the price or reimbursement rate that the payor will pay for the product once coverage is approved. Third-party payors may limit coverage to specific products on an approved list, or formulary, which might not include all of the approved products for a particular indication. In order to secure coverage and reimbursement for any product that might be approved for sale, a company may need to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and cost-effectiveness of the product, in addition to the costs required to obtain FDA or other comparable regulatory approvals. A payor’s decision to provide coverage for a drug product does not necessarily imply that an adequate reimbursement rate will be approved. Third-party reimbursement may not be sufficient to maintain price levels high enough to realize an appropriate return on our investment in product development. The containment of healthcare costs has become a priority of federal, state and foreign governments, and the prices of drugs have been a focus in this effort. Third-party payors are increasingly challenging the prices charged for medical products and services and examining the medical necessity and cost-effectiveness of medical products and services, in addition to their safety and efficacy. If these third-party payors do not consider a product to be cost effective compared to other available therapies, they may not cover the product after approval as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow a company to sell its products at a profit. The U.S. government, state legislatures and foreign governments have shown significant interest in implementing cost containment programs to limit the growth of government-paid health care costs, including price controls, risk sharing, restrictions on reimbursement and requirements for substitution of generic products for branded prescription drugs. Adoption of such controls and measures, and tightening of restrictive policies in jurisdictions with existing controls and measures, could limit payments for pharmaceuticals. As a result, the marketability of any product which receives regulatory approval for commercial sale may suffer if the government and third-party payors fail to provide adequate coverage and reimbursement. In addition, an increasing emphasis on managed care in the United States has increased and will continue to increase the pressure on drug pricing. Coverage policies, third-party reimbursement rates and drug pricing regulation may change at any time. In particular, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, contains provisions that may reduce the profitability of drug products, including, for example, increased rebates for drugs sold to Medicaid programs, extension of Medicaid rebates to Medicaid managed care plans, mandatory discounts for certain Medicare Part D beneficiaries and annual fees based on pharmaceutical companies’ share of sales to federal health care programs. Even if favorable coverage and reimbursement status is attained for one or more products that receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future. 32
In the European Union, pricing and reimbursement schemes vary widely from country to country. Some countries provide that drug products may be marketed only after a reimbursement price has been agreed. Some countries may require the completion of additional studies that compare the cost-effectiveness of a particular product candidate to currently available therapies. For example, the European Union provides options for its member states to restrict the range of drug products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. European Union member states may approve a specific price for a drug product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the drug product on the market. Other member states allow companies to fix their own prices for drug products, but monitor and control company profits. The downward pressure on health care costs in general, particularly prescription drugs, has become intense. As a result, increasingly high barriers are being erected to the entry of new products. In addition, in some countries, cross-border imports from low-priced markets exert competitive pressure that may reduce pricing within a country. Any country that has price controls or reimbursement limitations for drug products may not allow favorable reimbursement and pricing arrangements for any of our products. Healthcare Laws and Regulation Healthcare providers, physicians and third-party payors will play a primary role in the recommendation and prescription of drug products that are granted marketing approval. Arrangements with third-party payors and customers are subject to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we market, sell and distribute our products for which we obtain marketing approval. Restrictions under applicable federal and state healthcare laws and regulations, include the following: | · | the federal healthcare Anti-Kickback Statute prohibits, among other things, persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made, in whole or in part, under a federal healthcare program such as Medicare and Medicaid; |
| · | the federal Foreign Corrupt Practices Act (FCPA) prohibits, among other things, U.S. corporations and their persons acting on their behalf from offering, promising, authorizing or making payments to any foreign government official, including certain healthcare professionals in many countries, in an attempt to obtain or retain business or otherwise seek preferential treatment abroad; |
| · | the federal False Claims Act imposes civil penalties, and provides for civil whistleblower or qui tam actions, against individuals or entities for knowingly presenting, or causing to be presented, to the federal government, claims for payment that are false or fraudulent, making a false statement material to a false or fraudulent claim, or improperly avoiding, decreasing, or concealing an obligation to pay money to the federal government; |
| · | the federal Health Insurance Portability and Accountability Act of 1996 (HIPAA) imposes criminal and civil liability for executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters; |
| · | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act and its implementing regulations, also imposes obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information; |
| · | the federal false statements statute prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statement in connection with the delivery of or payment for healthcare benefits, items or services; |
| · | the federal Physician Payment Sunshine Act requires manufacturers of drugs, devices, biologics and medical supplies to report to the Department of Health and Human Services information related to payments and other transfers of value to physicians and teaching hospitals and physician ownership and investment interests; and |
| · | analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws, may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by nongovernmental third-party payors, including private insurers. |
Some state laws require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government in addition to requiring drug manufacturers to report information related to payments to physicians and other health care providers or marketing expenditures. State and foreign laws also govern the privacy and security of health information in some circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts. Certain Financial Information The financial information required in this Item 1 is included in Part II, Item 6 and Part IV, Item 15 of this Annual Report Form 10-K. 33
Corporate Information Arrowhead was originally incorporated in South Dakota in 1989, and was reincorporated in Delaware in 2000. The Company’s principal executive offices are located at 225 South Lake Avenue, Suite 1050, Pasadena, California 91101, and its telephone number is (626) 304-3400. We also operate a 24,000 square foot research and development facility in Madison, Wisconsin. As of September 30, 2012,2015, Arrowhead had 52104 full-time employees. Other Business InterestsInvestor Information
Leonardo Biosystems, Inc.Our internet website address is http://www.arrowheadresearch.com Our reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended, including our annual reports on Form 10-K, our quarterly reports on Form 10-Q and our current reports on Form 8-K, and amendments to those reports, are accessible through our website, free of charge, as soon as reasonably practicable after these reports are filed electronically with, or otherwise furnished to, the SEC. These SEC reports can be accessed through the “Investors” section of our website.
LeonardoYou may read and copy any materials we file with the SEC at the SEC’s Public Reference Room at 100 F Street, NE, Washington, DC 20549. You may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. The SEC also maintains an Internet website that contains reports, proxy and information statements, and other information regarding Arrowhead and other issuers that file electronically with the SEC. The SEC’s Internet website address is a drug delivery company that employs a novel multi-stage drug delivery mechanism aimed at dramatically increasing targeting efficiency of pharmaceuticals. Arrowhead has an approximately 3% ownership interest in Leonardo. Leonardo’s silicon micro-particulate technology involves transporting a therapeutic agent past multiple biological barriers using multiple carriers, each optimized for a specific barrier. Leonardo’s proprietary primary vehicles are designed to preferentially accumulate at tumor vasculature. Secondary carriers are then released from the primary carriers that are designed to accumulate around tumor cells and release their therapeutic payloads. Pre-clinical testing in animal disease models suggests that Leonardo’s platform enables significantly increased targeting of tumors and also provides sustained release of cancer therapies. Further development of Leonardo’s technology is dependent on cash resources available to Leonardo.
Nanotope, Inc.
Nanotope is a regenerative medicine company with license to a suite of nanotechnology-based products customized to regenerate specific tissues: including neuronal, bone and cartilaginous tissues. During 2012, Nanotope closed its R&D facility and ceased internal development of its technology. Development is continuing at Northwestern University in the lab of Sam Stupp, Nanotope’s scientific founder. Arrowhead has an approximately 23% ownership interest in Nanotope.
Unidym, Inc.
In January 2011, Arrowhead sold Unidym, Inc. to Wisepower Co., Ltd., a publicly-traded, Seoul, Korea-based electronics company (KOSDAQ: 040670)http://www.sec.gov. Unidym was a majority-owned subsidiary that developed nanotechnology-enabled materials to be used in the manufacturing of certain electronics components. Upfront consideration consisted of stock and convertible bonds valued at $5,000,000 with certain restrictions as to timing of stock sales. Additional cash earn-out payments of up to US $140 million are possible based on cumulative sales and licensing milestones, and up to 40% of licensing revenue.
34
You should carefully consider the risks discussed below and all of the other information contained in this report in evaluating us and an investment in our securities. If any of the following risks and uncertainties should occur, they could have a material adverse effect on our business, financial condition or results of operations. In that case, the trading price of our Common Stock could decline. Additionally, we note that we are a development stage company and we have accrued net losses annually since inception.inception given the stage of our drug development. We urge you to consider our likelihood of success and prospects in light of the risks, expenses and difficulties frequently encountered by entities at similar stages of development. Risks Related to Our Company Drug development is time consuming, expensive and risky. We are focused on technology related to new and improved pharmaceutical candidates. Product candidates that appear promising in the early phases of development, such as in animal and early human clinical trials, often fail to reach the market for a number of reasons, such as: ● | Clinical trial results may be unacceptable, even though preclinical trial results were promising; |
● | Inefficacy and/or harmful side effects in humans or animals; |
● | The necessary regulatory bodies, such as the U.S. Food and Drug Administration, may not approve our potential product for the intended use, or at all; and |
● | Manufacturing and distribution may be uneconomical. |
For example, the positive pre-clinical results in animals for ARC-520, ARC-AAT, and our other programs may not be replicated in human clinical studies. These programs may be also found to be unsafe in humans, particularly at higher doses needed to achieve the desired levels of efficacy. Also, the positive safety results for ARC-520 from single dose human clinical studies may not be replicated in other human studies, including multiple dose studies. Clinical trial results are frequently susceptible to varying interpretations by scientists, medical personnel, regulatory personnel, statisticians and others, which often delays, limits, or prevents further clinical development or regulatory approvals of potential products. Clinical trials can take many years to complete, including the process of study design, clinical site selection and the recruitment of patients. As a result, we can experience significant delays in completing clinical studies, which can increase the cost of developing a drug candidate and shorten the time that an approved product may be protected by patents. If our drug candidates are not successful in human clinical trials, we may be forced to curtail or abandon certain development programs. If we experience significant delays in commencing or completing our clinical studies, we could suffer from significant cost overruns, which could negatively affect our capital resources and our ability to complete these studies. There are substantial risks inherent in attempting to commercialize new drugs, and, as a result, we may not be able to successfully develop products for commercial use. Our research and development efforts involve therapeutics based on RNA interference and our delivery systems, which are largely unproven technologies. Our scientists and engineers are working on developing technology in the early stages. However, such technology’s commercial feasibility and acceptance are unknown. Scientific research and development requires significant amounts of capital and takes a long time to reach commercial viability, if it can be achieved at all. To date, our research and development projects have not produced commercially viable drugs, and may never do so. During the research and development process, we may experience technological barriers that we may be unable to overcome. Further, certain underlying premises in our development programs, including ARC-520, are not proven. For instance, the theory that knockdown of S-antigen in chronic hepatitis B patients will result in a functional cure is unproven. Thus, even if ARC-520 is successful at reducing S-antigen levels in patients, it may not be a commercially viable drug if there is not a corresponding medical benefit related to the underlying Hepatitis B infection. Similarly, if ARC-AAT successfully reduces the production of mutant alpha-1 antitrypsin in the liver, it may not lead to a reduction of globules in the liver, and even if it leads to a reduction in such globules, this may not lead to other beneficial hepatic changes. It is also unknown at this time what changes in the liver may be required to gain regulatory approval and/or favorable reimbursement,. Because of these and similar uncertainties, it is possible that no commercial products will be successfully developed. If we are unable to successfully develop commercial products, we will be unable to generate revenue or build a sustainable or profitable business. Our drug candidates are in the early stages of development and because we have a short development history with both RNA interference and our delivery technologies, there is a limited amount of information about us upon which you can evaluate our business and prospects. We have no approved drugs and thus have not begun to market or generate revenues from the commercialization of any products. We have only a limited history upon which one can evaluate our RNAi therapeutic business as our drug candidates are still at an early stage of development. Thus, we have limited experience and have not yet demonstrated an ability to successfully overcome 35
many of the risks and uncertainties frequently encountered by companies in new and rapidly evolving fields, particularly in the biopharmaceutical area. For example, to execute our business plan, we will need to successfully: ● | Execute product development activities using unproven technologies; |
● | Build, maintain and protect a strong intellectual property portfolio; |
● | Demonstrate safety and efficacy of our drug candidates in multiple human clinical studies; |
● | Receive FDA approval and approval from similar foreign regulatory bodies; | ● | Gain market acceptance for the development and commercialization of any drugs we develop; |
● | Ensure our products are reimbursed by commercial and/or government payors at a rate that permits commercial viability; | ● | Develop and maintain successful strategic relationships with suppliers, distributors, and commercial licensing partners; and |
● | Manage our spending and cash requirements as our expenses will increase in the near term if we add programs and additional preclinical and clinical trials. |
If we are unsuccessful in accomplishing these objectives, we may not be able to develop products, raise capital, expand our business or continue our operations. We may be unable to attract revenue-generating collaborations with other pharmaceutical and biotech companies to advance our drug candidates. Our business strategy includes obtaining collaborations with other pharmaceutical and biotech companies to support the development of our therapeutic siRNA and other drug candidates. We may not be able to attract such partners, and even if we are able to enter into such partnerships, the terms may be less favorable than anticipated. Further, entering into partnership agreements may limit our commercialization options and/or require us to share revenues and profits with our partners. If we are unable to enter into any of these agreements on commercially attractive terms, we may be unable to develop certain programs due to a limited availability of resources, and we may need to raise additional capital, which could be dilutive to our existing investors. We will need to achieve commercial acceptance of our drug candidates to generate revenues and achieve profitability. Even if our research and development efforts yield technologically feasible applications, we may not successfully develop commercial products. Drug development takes years of study in human clinical trials prior to regulatory approval, and, even if we are successful, it may not be on a timely basis. During our drug development period, superior competitive technologies may be introduced which could diminish or extinguish the potential commercial uses for our drug candidates. Additionally, the degree to which the medical community and consumers will adopt any product we develop is uncertain. The rate and degree of market acceptance of our products will depend on a number of factors, including the establishment and demonstration in the medical community of the clinical efficacy and safety of our products, their potential advantage over alternative treatments, and the costs to patients and third-party payors, including insurance companies and Medicare. Recent efforts in the United States and abroad to reduce overall healthcare spending has put significant pressure on the price of prescription drugs and certain companies have been publicly criticized for the relatively high cost of their therapies. These pressures may force us to sell any approved drugs at a lower price than we or analysts may anticipate, or may result in lower levels of reimbursement and coverage from third parties. We cannot predict whether significant commercial market acceptance for our products, if approved, will ever develop, and we cannot reliably estimate the projected size of any such potential market. Our revenue growth and achievement of profitability will depend substantially on our ability to introduce products that will be accepted by the medical community. If we are unable to cost-effectively achieve acceptance of our technology among the medical establishment and patients, or if the associated products do not achieve wide market acceptance, our business will be materially and adversely affected. Risks Related to Our Financial Condition Our independent auditors have issued a report questioning our ability to continue as a going concern.
The report of our independent auditors contained in our financial statements explains that we have not yet established an ongoing source of revenue sufficient to cover operating costs and allow us to continue as a going concern. Our ability to continue as a going concern is dependent on obtaining adequate capital to fund operating losses until we become profitable. If we are unable to obtain adequate capital, we may have to delay, scale back, or discontinue the development and/or commercialization of one or more product candidates, or relinquish or otherwise dispose of rights to technologies, product candidates, or products that we would otherwise seek to develop or commercialize ourselves and/or cease operations.
17
We have a history of net losses, and we expect to continue to incur net losses and may not achieve or maintain profitability. We have incurred net losses since our inception, including net losses of $22.1$91.9 million for the year ended September 30, 2012 and a cumulative net loss since inception of approximately $153.7 million.2015. We expect that our operating losses will continue for the foreseeable future as we fundcontinue our drug development and discovery efforts. To achieve profitability, we must, either directly or through licensing and/or partnering relationships, meet certain milestones, successfully develop and obtain regulatory approval for aone or more drug candidatecandidates and effectively manufacture, market and sell any drugs we successfully develop. Even if we successfully commercialize drug candidates that receive regulatory approval, we may not be able to realize revenues at a level that would allow us to achieve or sustain profitability. 36
Accordingly, we may never generate significant revenue and, even if we do generate significant revenue, we may never achieve profitability. We have limited cash resources.will require substantial additional funds to complete our research and development activities. Our business currently does not generate the cash that is necessary to finance our operations. We incurred net losses of approximately $22.1 million in 2012 and $153.7 million since our inception. Subject to the success of the research and development programs of our company and our partners, and potential licensing or partnering transactions, we will likely need to raise significant additional capital in the immediate future to: Fund research and development activities relating to our development of our product candidates, including clinical and pre-clinical trials;
● | Fund research and development infrastructure and activities relating to the development of our drug candidates, including pre-clinical and clinical trials and manufacturing to support these efforts; |
● | Fund our general and administrative infrastructure and activities; |
Pursue licensing
● | Pursue business development opportunities for our technologies; |
Protect
● | Add to and protect our intellectual property; and |
● | Retain our management and technical staff. |
Our future capital needs depend on many factors, including: · | The scope, duration and expenditures associated with our research and development; |
Continued scientific progress in these programs;
· | Regulatory requirements for our clinical trials; |
The outcome of potential partnering or licensing transactions, if any;
· | The extent to which our R&D and clinical efforts are successful; |
Competing technological developments;
· | The outcome of potential partnering or licensing transactions, if any, and the extent to which our business development efforts result in the acquisition of new programs or technologies; |
Our proprietary patent position, if any, in our products; and
· | Competing technological developments; |
· | Our intellectual property positions, if any, in our products; and |
The regulatory approval process for our products.
· | The regulatory approval process and regulatory standards for our drug candidates. |
We will need to raise substantial additional funds through public or private equity offerings, debt financings or additional strategic alliances and licensing arrangements in the immediate future to continue our operations. We may not be able to obtain additional financing on terms favorable to us, if at all. General market conditions as well as market conditions for companies that are facing financial distress, may make it very difficult for us to seek financing from the capital markets, and the terms of any financing may adversely affect the holdings or the rights of our stockholders. For example, if we raise additional funds by issuing equity securities, further dilution to our stockholders will result, which may substantially dilute the value of your investment. In addition, as a condition to providing additional funds to us, future investors may demand, and may be granted, rights superior to those of existing stockholders. Debt financing, if available, may involve restrictive covenants that could limit our flexibility in conducting future business activities and, in the event of insolvency, would be paid before holders of equity securities received any distribution of corporate assets. WeIn order to raise additional funds through alliance, joint venture or licensing arrangements, we may be required to relinquish rights to our technologies or drug candidates, or grant licenses on terms that are not favorable to us, in order to raise additional funds through alliance, joint venture or licensing arrangements.us. If adequate funds are not available, we may have to further delay, reduce or eliminate one or more of our planned activities. These actions would likely reduce the market price of our common stock. The currentIf the estimates we make, or the assumptions on which we rely, in preparing our consolidated financial market conditionsstatements prove inaccurate, our actual results may exacerbate certain risks affectingvary from those reflected in our business.accruals.
We do not yet generate substantial revenue, and our operations and research and development activitiesOur consolidated financial statements have been primarily fundedprepared in accordance with accounting principles generally accepted in the United States of America (GAAP). The preparation of these consolidated financial statements requires us to date throughmake estimates and judgments that affect the sale of Company securities and securitiesreported amounts of our Subsidiaries. assets, liabilities, revenues and expenses, the amounts of charges accrued by us and related disclosure of contingent assets and liabilities. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances. We cannot assure you, however, that our estimates, or the assumptions underlying them, will be correct.
The global financial markets are volatile and those market conditions may impair our ability to raise the capital we require. If we are unable to secure additional cash resources from the sale of securities or other sources, it could become necessary to slow or suspend development efforts. In addition, we may have to reduce expenses, which could impair our ability to manage our business. Even if investment capital is available to us, the terms may be onerous. Because we have not generated significant revenues to cover our operating expenses, we are dependent on raising additional capital from investors or lenders.
To date, we have only generated a small amount of revenue. Given our strategy of financing new and unproven technology research, there can be no assurance we will ever generate significant revenue. Our revenue-producing opportunities depend on our ability to attract collaborations or out-licenses with other companies, receive milestone and royalty payments from prior divestitures, and/or generate income from the sales of products. These sources of revenue are uncertain as to the amount and timing of potential revenue. Accordingly, our revenue prospects are uncertain and we must plan to finance our operations through the sales of equity securities or debt financing. If we are unable to continue raising operating capital from these sources, we may be forced to curtail or cease our operations.
18
We will need to achieve commercial acceptance of our applicationscash, cash equivalents and fixed income marketable securities is subject to generate revenuesrisks which may cause losses and achieve profitability.affect the liquidity of these investments.
Even ifAt September 30, 2015, we had $17.5 million in fixed income marketable securities. These investments are in corporate bonds nearing maturity, but our researchinvestments may also include commercial paper, securities issued by the U.S. government obligations, certificates of deposit and development efforts yield technologically feasible applications, wemoney market funds meeting the criteria of our investment policy, which is focused on the preservation of
37
our capital. These investments are subject to general credit, liquidity, and market and interest rate risks, particularly in the current economic environment. We may not successfully develop commercial productsrealize losses in the fair value of these investments or a complete loss of these investments, which would take years to study in human clinical trials prior to regulatory approval, and, even if successfully developed, we may not do so onhave a timely basis. During this development period, superior competitive technologies may be introduced which could diminish or extinguish the potential commercial uses for our drug candidates. Additionally, the degree to which patients and consumers will adopt any product we develop is uncertain. We cannot predict whether significant commercial market acceptance for our products, if approved, will ever develop, and we cannot reliably estimate the projected size of any such potential market. Our revenue growth and achievement of profitability will depend substantially on our ability to introduce new technological applications to manufacturers for products accepted by customers. If we are unable to cost-effectively achieve acceptance of our technology among the medical establishment and patients, or if the associated products do not achieve wide market acceptance, our business will be materially and adversely affected. We have debtnegative effect on our consolidated balance sheet throughfinancial statements. In addition, should our subsidiary, Calando, which couldinvestments cease paying or reduce the amount of interest paid to us, our interest income would suffer. The market risks associated with our investment portfolio may have negative consequences if we were unable to repay the principal or interest due.
Calando has a $500,000 unsecured convertible promissory note outstanding. The note bears 10% interest accrued annually, and matures in November 2013. The note is payable at two times face value at maturity and upon the occurrencean adverse effect on our results of certain events, including, the license of Calando’s siRNA delivery system. If Calando is unable to meet its obligations to the bearer of the note, Arrowhead may not be in a position to lend Calando sufficient cash to pay such demand note. Unless other sources of financing become available, this could result in Calando’s insolvency.
Our Subsidiaries are party to technology license agreements with third parties that require us to satisfy obligations to keep them effective and, if these agreements are terminated, our technology and our business would be seriously and adversely affected.
Through our Subsidiaries, we are party into exclusive, long-term license agreements with University of Texas MD Anderson Cancer Center, California Institute of Technology, Alnylam Pharmaceuticals, Inc. and other entities to incorporate their proprietary technologies into our proposed products. These license agreements require us to pay royalties and satisfy other conditions, including conditions in some cases related to the commercialization of the licensed technology. We may not be able to successfully incorporate these technologies into marketable products or, if we do, whether sales will be sufficient to recover the amounts that we are obligated to pay to the licensors. If we fail to satisfy our obligations under these agreements the terms of the licenses may be materially modified, such as by rendering the licenses non-exclusive, or may give our licensors the right to terminate their respective agreement with us, which would limit our ability to implement our current business plan and harm our businessoperations, liquidity and financial condition.
Risks Related to Our Company
Drug development is time consuming, expensive and risky.
We are focused on technology related to new and improved pharmaceutical candidates. Product candidates that appear promising in the early phases of development, such as in animal and early human clinical trials, often fail to reach the market for a number of reasons, such as:
Clinical trial results may be unacceptable, even though preclinical trial results were promising;
Inefficacy and/or harmful side effects in humans or animals;
The necessary regulatory bodies, such as the U.S. Food and Drug Administration, may not approve our potential product for the intended use; and
Manufacturing and distribution may be uneconomical.
For example, the positive pre-clinical results for Adipotide in animals may not be replicated in human clinical studies or it may be found to be unsafe in humans. Additionally, clinical trial results are frequently susceptible to varying interpretations by scientists, medical personnel, regulatory personnel, statisticians and others, which often delays, limits, or prevents further clinical development or regulatory approvals of potential products. Clinical trials can take many years to complete, including the process of study design, clinical site selection and the enrollment of patients. As a result, we can experience significant delays in completing clinical studies, which can increase the cost of developing a drug candidate. If our drug candidates are not successful in human clinical trials, we may be forced to curtail or abandon certain development programs. If we experience significant delays in commencing or completing our clinical studies, we could suffer from significant cost overruns, which could negatively affect our capital resources and our ability to complete these studies.
19
We may be unable to attract revenue-generating collaborations with other pharmaceutical and biotech companies to advance our drug candidates.
Our business strategy includes obtaining collaborations with other pharmaceutical and biotech companies to support the development of our therapeutic siRNA and other drug candidates. We may not be able to attract such partners, and even if we are able to enter into such partnerships, the terms may be less favorable than anticipated. Further, entering into partnership agreements may limit our commercialization options and/or require us to share revenues and profits with our partners.
Our products are in the early stages of our development and because we have a short development history with both DPCs and Homing Peptides, there is a limited amount of information about us upon which you can evaluate our business and prospects.
We have not begun to market or generate revenues from the commercialization of any products. We have only a limited history upon which one can evaluate our targeted therapeutic business and prospects as our therapeutic products are still at an early stage of development. Thus, we have limited experience and have not yet demonstrated an ability to successfully overcome many of the risks and uncertainties frequently encountered by companies in new and rapidly evolving fields, particularly in the biopharmaceutical area. For example, to execute our business plan, we will need to successfully:
Execute product development activities using an unproven technology;
Build, maintain and protect a strong intellectual property portfolio;
Gain acceptance for the development and commercialization of any product we develop;
Develop and maintain successful strategic relationships; and
Manage our spending and cash requirements as our expenses are expected to increase in the near term due to preclinical and clinical trials.
If we are unsuccessful in accomplishing these objectives, we may not be able to develop products, raise capital, expand our business or continue our operations.
We may lose a considerable amount of control over our intellectual property and may not receive anticipated revenues in strategic transactions, particularly where the consideration is contingent on the achievement of development or sales milestones.
Our business model has been to develop new technologies and to exploit the intellectual property created through the research and development process to develop commercially successful products. Calando has licensed a portion of its technology to Cerulean Pharma, Inc. and we intend to pursue licensing arrangements with other companies. A significant portion of the potential value from these licenses is tied to the achievement of the development and sales milestones, which we cannot control. Similarly, the majority of the consideration, up to $140 million, potentially payable by Wisepower in connection with our sale of Unidym is tied to the achievement of commercialization milestones, which we cannot control. Although Wisepower and Cerulean are required to use certain minimum efforts to achieve the post-closing milestones, we cannot control whether they actually achieve these milestones. If the acquirers fail to achieve performance milestones, we may not receive a significant portion of the total value of any sale, license or other strategic transaction.
There are substantial risks inherent in attempting to commercialize new technological applications, and, as a result, we may not be able to successfully develop products for commercial use.
Our research and development efforts involve therapeutics based on nanotechnology and RNA interference, which are largely unproven technologies. Our scientists and engineers are working on developing technology in various stages. However, such technology’s commercial feasibility and acceptance are unknown. Scientific research and development requires significant amounts of capital and takes a long time to reach commercial viability, if at all. To date, our research and development projects have not produced commercially viable applications, and may never do so. During the research and development process, we may experience technological barriers that we may be unable to overcome. Because of these uncertainties, it is possible that none of our potential applications will be successfully developed. If we are unable to successfully develop applications of our technology for commercial use, we will be unable to generate revenue or build a sustainable or profitable business.
We will need to establish additional relationships with strategic and development partners to fully develop and market our products.
We do not possess all of the financial and development resources necessary to develop and commercialize products that may result from our technologies on a mass scale. Unless we expand our product development capacity and enhance our internal marketing capability, we will need to make appropriate arrangements with strategic partners to develop and commercialize current and future products. If we do not find appropriate partners, or if our existing arrangements or future agreements are not successful, our ability to develop and commercialize products could be adversely affected. Even if we are able to find collaborative partners, the overall success of the development and commercialization of product candidates in those programs will depend largely on the efforts of other parties and is beyond our control. In addition, in the event we pursue our commercialization strategy through collaboration, there are a variety of technical, business and legal risks, including:
A development partner would likely gain access to our proprietary information, potentially enabling the partner to develop products without us or design around our intellectual property;
20
We may not be able to control the amount and timing of resources that our collaborators may be willing or able to devote to the development or commercialization of our product candidates or to their marketing and distribution; and
Disputes may arise between us and our collaborators that result in the delay or termination of the research, development or commercialization of our product candidates or that result in costly litigation or arbitration that diverts our management’s resources.
The occurrence of any of the above events or other related events not foreseen by us could impair our ability to generate revenues and harm our business and financial condition.
We may not be able to effectively secure first-tier technologies when competing against other companies or investors. Our future success may require that we acquire patent rights and know-how to new or complimentary technologies. However, we compete with a substantial number of other companies that may also compete for technologies we desire. In addition, many venture capital firms and other institutional investors, as well as other pharmaceutical and biotech companies, invest in companies seeking to commercialize various types of emerging technologies. Many of these companies have greater financial, scientific and commercial resources than us. Therefore, we may not be able to secure the technologies we desire. Furthermore, should any commercial undertaking by us prove to be successful, there can be no assurance competitors with greater financial resources will not offer competitive products and/or technologies. Risks Associated with Reliance on Third Parties We will need to establish additional relationships with strategic and development partners to fully develop our drug candidates and market any approved products. We do not possess all of the financial and development resources necessary to develop and commercialize products that may result from our technologies. Unless we expand our product development capacity and enhance our internal marketing capability, we may need to make appropriate arrangements with strategic partners to develop and commercialize any drug candidates that may be approved. If we do not find appropriate partners, or if our existing arrangements or future agreements are not successful, our ability to develop and commercialize products could be adversely affected. Even if we are able to find collaborative partners, the overall success of the development and commercialization of product candidates in those programs will depend largely on the efforts of other parties and will be beyond our control. In addition, in the event we pursue our commercialization strategy through collaboration or licenses to third parties, there are a variety of technical, business and legal risks, including: ● | We may not be able to control the amount and timing of resources that our collaborators may be willing or able to devote to the development or commercialization of our drug candidates or to their marketing and distribution; and |
● | Disputes may arise between us and our collaborators that result in the delay or termination of the research, development or commercialization of our drug candidates or that result in costly litigation or arbitration that diverts our management’s resources. |
The occurrence of any of the above events or other related events could impair our ability to generate revenues and harm our business and financial condition. We may lose a considerable amount of control over our intellectual property and may not receive anticipated revenues in strategic transactions, particularly where the consideration is contingent on the achievement of development or sales milestones. Our business model has been to develop new technologies and to utilize the intellectual property created through the research and development process to develop commercially successful products. If the acquirers of our technologies fail to achieve performance milestones, we may not receive a significant portion of the total value of any sale, license or other strategic transaction. We rely on outside sources for various components and processes for our products. We rely on third parties for various components and processes for our products. While we try to have at least two sources for each component and process, weproduct candidates. We may not be able to achieve multiple sourcing because there may be no acceptable second source, other companies may choose not to work with us, or the component or process sought may be so new that a second source does not exist, or does not exist on acceptable terms. There may be a disruption or delay in the performance of our third-party contractors, suppliers or collaborators which is beyond our control. If such third parties are unable to satisfy their commitments to us, the development of our businessproducts would be adversely affected. Therefore, it is possible that our businessdevelopment plans will have to be slowed down or stopped completely at times due to our inability to obtain required raw materials, components and outsourced processes at an acceptable cost, if at all, or to get a timely response from vendors. We must overcome the many obstacles associated with integrating and operating varying development programs.
Our model to integrate and oversee research and development projects presents many risks, including:
The difficulty of integrating operationsWe have limited manufacturing capability and personnel;must rely on third-party manufacturers to manufacture our clinical supplies and
The diversion commercial products, if and when approved, and if they fail to meet their obligations, the development and commercialization of our management’s attentionproducts could be adversely affected.
We have limited manufacturing capabilities and experience. ARC-520 and our other drug candidates are composed of multiple components and require specialized formulations for which scale-up and manufacturing could be difficult. We have limited experience 38
in such scale-up and manufacturing requiring us to depend on a limited number of third parties, who may not be able to deliver in a timely manner, or at all. In order to develop products, apply for regulatory approvals and commercialize our products, we will need to develop, contract for, or otherwise arrange for the necessary manufacturing capabilities. Our internal manufacturing capabilities are limited to small-scale production of material for use in in vitro and in vivo experiments that is not required to be produced under cGMP standards. There are a limited number of manufacturers that supply synthetic siRNAs. There are risks inherent in pharmaceutical manufacturing that could affect the ability of our contract manufacturers to meet our delivery time requirements or provide adequate amounts of material to meet our needs. Included in these risks are synthesis and purification failures and contamination during the manufacturing process, which could result in unusable product and cause delays in our development process, as well as additional expense to us. Additionally, our product candidates have not yet been manufactured for commercial use. If any of our product candidates become approved for commercial sale, we will need to establish third-party manufacturing capacity. A third-party manufacturing partner may require us to fund capital improvements to support the scale-up of manufacturing and related activities. The third-party manufacturer may not be able to establish scaled manufacturing capacity for an approved product in a resulttimely or economic manner, if at all. If a manufacturer is unable to provide commercial quantities of evaluating, negotiatingsuch an approved product, we will have to successfully transfer manufacturing technology to a different manufacturer. Engaging a new manufacturer for such an approved product could require us to conduct comparative studies or utilize other means to determine bioequivalence of the new and integrating acquisitionsprior manufacturers’ products, which could delay or new business ventures. prevent our ability to commercialize such an approved product. If any of these manufacturers is unable or unwilling to increase its manufacturing capacity or if we are unable to establish alternative arrangements on a timely basis or on acceptable terms, the development and efficiently designcommercialization of such an approved product may be delayed or there may be a shortage in supply. Any inability to manufacture our product candidates or future approved drugs in sufficient quantities when needed would seriously harm our business. Manufacturers of our approved products, if any, must comply with cGMP requirements enforced by the FDA and integrate administrativeforeign health authorities through facilities inspection programs. These requirements include quality control, quality assurance, and operational support forthe maintenance of records and documentation. Manufacturers of our Subsidiaries, weapproved products, if any, may be unable to manage projects effectively,comply with these cGMP requirements and with other FDA, state, and foreign regulatory requirements. We have little control over our manufacturers’ compliance with these regulations and standards. A failure to comply with these requirements may result in fines and civil penalties, suspension of production, suspension or delay in product approval, product seizure or recall, or withdrawal of product approval. If the safety of any quantities supplied is compromised due to our manufacturer’s failure to adhere to applicable laws or for other reasons, we may not be able to obtain regulatory approval for or successfully commercialize our products, which would seriously harm our business. We rely on third parties to conduct our clinical trials, and if they fail to fulfill their obligations, our development plans may be adversely affected. We rely on independent clinical investigators, contract research organizations and other third-party service providers to assist us in managing, monitoring and otherwise carrying out our clinical trials. We have contracted with, and we plan to continue to contract with, certain third-parties to provide certain services, including site selection, enrollment, monitoring and data management services. Although we depend heavily on these parties, we do not control them and therefore, we cannot be assured that these third-parties will adequately perform all of their contractual obligations to us. If our third-party service providers cannot adequately and timely fulfill their obligations to us, or if the quality and accuracy of our clinical trial data is compromised due to failure by such third-parties to adhere to our protocols or regulatory requirements or if such third-parties otherwise fail to meet deadlines, our development plans may be delayed or terminated. Further, if clinical study results are compromised, then we may need to repeat the affected studies, which could adversely affect our ability to meet our business objectives and the value of an investmentresult in the Company could decline. In addition, consummating acquisitions and strategic relationships could adversely impact our cash position, and dilute stockholder interests, for many reasons, including:
Collaboration terms that decrease future cash flows from products in exchange for near term benefits;
Changes to our income to reflect the amortization of acquired intangible assets, including goodwill;
Interestsignificant additional costs and debt service requirements for any debt incurreddelays to fundus.
Risks related to managing our growth strategy; andoperations Any issuance of securities to fund our operations or growth, which dilutes or lessens the rights of current stockholders.
Our success depends on the attraction and retention of senior management and scientists with relevant expertise. Our future success depends to a significant extent on the continued services of our key employees, including Dr. Anzalone, our Presidentsenior scientific, technical and Chief Executive Officer, Dr. Bruce Given, our Chief Operating Officer, and Ken Myszkowski, our Chief Financial Officer.managerial personnel. We do not maintain key man life insurance for any of our executives. Ourexecutives and we do not maintain employment agreements with most senior officers or employees. Competition for qualified employees in the pharmaceutical industry is high, and our ability to execute our strategy also will depend on our ability to continue to attract and retain qualified scientists and management. If we are unable to find, hire and retain qualified individuals, we couldwill have difficulty implementing our business plan in a timely manner, or at all. 21
Members of our senior management team and BoardWe may have difficulty expanding our operations successfully as we evolve from a conflictcompany primarily involved in discovery and pre-clinical testing into one that develops and commercializes drugs.
39
We expect that as we increase the number of interest inproduct candidates we are developing we will also serving as officers and/need to expand our operations. This expected growth may place a strain on our administrative and operational infrastructure. As product candidates we develop enter and advance through clinical trials, we will need to expand our development, regulatory, manufacturing, marketing and sales capabilities or directors ofcontract with other organizations to provide these capabilities for us. As our Subsidiaries. Whileoperations expand due to our development progress, we expect that we will need to manage additional relationships with various collaborators, suppliers and other organizations. Our ability to manage our officersoperations and directors who also serve as officers and/future growth will require us to continue to improve our operational, financial and management controls, reporting systems and procedures. We may not be able to implement improvements to our management information and control systems in an efficient or directorstimely manner and may discover deficiencies in existing systems and controls.
Our business and operations could suffer in the event of information technology system failures. Our internal computer systems and those of our Subsidiaries will comply with their fiduciary duties owedcontractors and consultants are vulnerable to our stockholders, they may have conflicting fiduciary obligations to our stockholdersdamage from computer viruses, unauthorized access, natural disasters, terrorism, war, and the minority stockholderstelecommunication and electrical failures. Such events could cause interruption of our Subsidiaries. Specifically, Dr. Anzalone,operations and loss of intellectual property. For example, the loss of pre-clinical trial data or data from completed or ongoing clinical trials for our Presidentproduct candidates could result in delays in our regulatory filings and CEO, is the founder, CEOdevelopment efforts and a board member of Nanotope, a regenerative medicine company in which the Company owns a 23% interest. Further, Dr. Anzalone as well as Dr. Mauro Ferrari, an Arrowhead board member, are board members of Leonardo, a drug delivery company in which Arrowhead owns a 3% interest. Dr. Anzalone owns a noncontrolling interest in the stock of Nanotope. Drs. Anzalone and Ferrari own a noncontrolling interest in Leonardo. Douglass Given, a member ofsignificantly increase our board of directors, is the brother of Bruce Given.costs. To the extent that any disruption or security breach were to result in a loss of or damage to our data, or inappropriate disclosure of confidential, proprietary or private information, we could incur liability, we could lose valuable trade secret rights, and the development of our directors chooseproduct candidates could be delayed. Risks related to recuse themselves from particular Board actions to avoid a conflict of interest, the other membersdevelopment and regulatory approval of our Boardproduct candidates The manufacture and sale of Directorshuman therapeutic products are governed by a variety of statutes and regulations. There can be no assurance that our product candidates will have a greater influence on such decisions.obtain regulatory approval. We face uncertainty related to healthcare reform, pricing and reimbursement, which could reduce our revenue.
In the United States, President Obama signed in March 2010 the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act (collectively, “PPACA”), which is expected to substantially change the way health care is financed by both governmental and private payers. PPACA provides for changes to extend medical benefits to those who currently lack insurance coverage, encourages improvementsThe sale of human therapeutic products in the qualityU.S. and foreign jurisdictions is subject to extensive and time consuming regulatory approval which requires:
· | controlled research and human clinical testing; |
· | establishment of the safety and efficacy of the product; |
· | government review and approval of a submission containing manufacturing, pre-clinical and clinical data; and |
· | adherence to cGMP regulations during production and storage. |
The product candidates we currently have under development will require significant development, pre-clinical and clinical testing and investment of health care itemssignificant funds before their commercialization. Some of our product candidates, if approved, will require the completion of post-market studies. There can be no assurance that any of our products will be further developed and services,approved. The process of completing clinical testing and significantly impacts the U.S. pharmaceutical industry inobtaining required approvals will take a number of ways, further listed below. By extending coverageyears and require the use of substantial resources. Further, there can be no assurance that product candidates employing a new technology will be shown to be safe and effective in clinical trials or receive applicable regulatory approvals. If we fail to obtain regulatory approvals for any or all of our products, we will not be able to market such product and our operations may be adversely affected. If testing of a larger population, PPACAparticular product candidate does not yield successful results, then we will be unable to commercialize that product candidate. We must demonstrate our product candidates’ safety and efficacy in humans through extensive clinical testing. Our research and development programs are at an early stage of development. We may substantially change the structureexperience numerous unforeseen events during, or as a result of, the health insurance systemtesting process that could delay or prevent commercialization of any products, including the following: · | the results of pre-clinical studies may be inconclusive, or they may not be indicative of results that will be obtained in human clinical trials; |
· | safety and efficacy results attained in early human clinical trials may not be indicative of results that are obtained in later clinical trials; |
· | after reviewing test results, we may abandon projects that we might previously have believed to be promising; |
· | we or our regulators, may suspend or terminate clinical trials because the participating subjects or patients are being exposed to unacceptable health risks; and |
· | our product candidates may not have the desired effects or may include undesirable side effects or other characteristics that preclude regulatory approval or limit their commercial use if approved. |
40
Clinical testing is very expensive, takes many years, and the methodologyoutcomes are uncertain. The data collected from our clinical trials may not be sufficient to support approval of our product candidates by the regulatory authorities. The clinical trials of our product candidates may not be completed on schedule, and the regulatory authorities may not ultimately approve any of our product candidates for reimbursing medical services, drugscommercial sale. If we fail to adequately demonstrate the safety and devices. These structural changes,efficacy of a product candidate, it would prevent regulatory approval of the product candidate, which could prevent us from achieving profitability. It may take us longer than we are currently projecting to complete our clinical trials, and we may not be able to complete them at all. Although for planning purposes, we project the commencement, continuation and completion of our clinical trials, a number of factors, including scheduling conflicts with participating clinicians and clinical institutions, and difficulties in identifying or enrolling patients who meet trial eligibility criteria, may cause significant delays. We may not commence or complete clinical trials involving any of our product candidates as well as other changes thatprojected or may not conduct them successfully. Even if our clinical studies are successful and we achieve regulatory approval, the approved product label may be mademore limited than we or analysts anticipate, which could limit the commercial opportunity for our product candidates. At the time drugs are approved for commercialization, they are given a “product label” from the FDA or other regulatory body. In most countries this label sets forth the approved indication for marketing, and identifies potential safety concerns for prescribing physicians and patients. While we intend to seek as part of deficit and debt reduction efforts in Congress, could entail modifications to the existing system of private payers and government programs,broad a product label as possible for our product candidates, we may receive a narrower label than is expected by either us or third parties, such as Medicare, Medicaidstockholders and State Children’s Health Insurance Program,securities analysts. For example, any approved products may only be indicated to treat refractory patients (i.e., those who have failed some other first-line therapy). Similarly, it is possible that only a specific sub-set of patients safely responds to our drug candidates. As a result, our product candidates, even if successful in clinical trials, could be approved only for a subset of patients. Additionally, safety considerations may result in contraindications that could further limit the scope of an approved product label. Any of these or other safety and efficacy considerations could limit the commercial opportunity for our product candidates. Even if our product candidates are approved for commercialization, future regulatory reviews or inspections may result in the suspension or withdrawal of one or more of our products, closure of a facility or enforcement of substantial fines. If regulatory approval to sell any of our product candidates is received, regulatory agencies will subject any marketed product(s), as well as the creation ofmanufacturing facilities, to continual review and periodic inspection. If previously unknown problems with a government-sponsored healthcare insurance source,product or some combination of both. Such restructuringmanufacturing and laboratory facility are discovered, or we fail to comply with applicable regulatory approval requirements, a regulatory agency may impose restrictions on that product or on us. The agency may require the withdrawal of the coverageproduct from the market, closure of medical care in the United States could impact the extentfacility or enforcement of reimbursementsubstantial fines. We face potential product liability exposure, and if successful claims are brought against us, we may incur substantial liability for prescribed drugs, includinga product candidate and may have to limit its commercialization. The use of our product candidates biopharmaceuticals,in clinical trials and medical devices. Somethe sale of any products for which we obtain marketing approval expose us to the risk of product liability claims. Product liability claims might be brought against us by clinical trial participants, consumers, health-care providers, pharmaceutical companies, or others selling our products. If we cannot successfully defend ourselves against these claims, we may incur substantial liabilities. Regardless of merit or eventual outcomes of such claims, product liability claims may result in: · | decreased demand for our product candidates; |
· | impairment of our business reputation; |
· | withdrawal of clinical trial participants; |
· | substantial monetary awards to patients or other claimants; |
· | the inability to commercialize our product candidates. |
Although we currently have liability insurance for our clinical trials, our insurance coverage may not be sufficient to reimburse us for all expenses or losses we may suffer. Moreover, insurance coverage is becoming increasingly expensive and, in the future, we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses. 41
If a natural or man-made disaster strikes our research and development facility or otherwise affects our business, it could delay our progress developing our product candidates. We conduct research and development in a facility in Madison, Wisconsin. The facilities and the equipment we use are costly to replace and require substantial lead time to repair or replace. Our facilities may be harmed by natural or man-made disasters, including, without limitation, earthquakes, floods, fires and acts of terrorism; and if our facilities are affected by a disaster, our development efforts would be delayed. Significant delays in our development efforts could materially impact our ability to obtain regulatory approval and to commercialize our products. Any insurance we maintain against damage to our property and the disruption of our business due to disaster may not be sufficient to cover all of our potential losses and may not continue to be available to us on acceptable terms, or at all. In addition, our development activities could be harmed or delayed by a shutdown of the specific PPACA provisions, among other things:U.S. government, including the FDA. Establish annual, non-deductible fees on any entity that manufactures or imports certain branded prescription drugs and biologics, beginning in 2011;
Increase minimum Medicaid rebates owed by manufacturers under the Medicaid Drug Rebate Program;
Extend manufacturers’ Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations;
Establish a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in and conduct comparative clinical effectiveness research;
Require manufacturers to participate in a coverage gap discount program, under which they must agree to offer 50 percent point-of-sale discounts off negotiated pricesThe successful commercialization of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D, beginning in 2011; and
Increase the number of entities eligible for discounts under the Public Health Service pharmaceutical pricing program, effective January 2010.
If future reimbursement for approvedour product candidates, if any, is substantially less than we project, or rebate obligations associated with them are substantially increased, our business could be materiallyapproved, will depend in part on the extent to which government authorities and adversely impacted.health insurers establish adequate reimbursement levels and pricing policies.
Sales of any approved drug candidate will depend in part on the availability of coverage and reimbursement from third-party payers such as government insurance programs, including Medicare and Medicaid, private health insurers, health maintenance organizations and other health care related organizations.organizations, who are increasingly challenging the price of medical products and services. Accordingly, coverage and reimbursement may be uncertain. Adoption of any drug candidate by the medical community may be limited if third-party payers will not offer coverage. Additionally, significant uncertainty exists as to the reimbursement status of newly approved drugs. Cost control initiatives may decrease coverage and payment levels for any new drug and, in turn, the price that we will be able to charge.charge and/or the volume of our sales. We are unable to predict all changes to the coverage or reimbursement methodologies that will be applied by private or government payers. Any denial of private or government payer coverage or inadequate reimbursement could harm our business and reduce our revenue. If we partner with third parties with respect to any of our product candidates, we may be reliant on that partner to obtain reimbursement from government and private payors for the drug, if approved, and any failure of that partner to establish adequate reimbursement could have a negative impact on our revenues and profitability. In addition, both the federal and state governments in the United States and foreign governments continue to propose and pass new legislation, regulations, and policies affecting coverage and reimbursement policies,rates, which are designed to contain or reduce the cost of health care, as well as hold public hearings on these matters, which has resulted in certain private companies dropping the prices of their drugs.care. Further federal and state proposals and healthcare reforms are likely, which could limit the prices that can be charged for the product candidates that we develop and may further limit our commercial opportunity. There may be future changes that result in reductions in currentpotential coverage and reimbursement levels for our product candidates, if approved and commercialized, and we cannot predict the scope of any future changes or the impact that those changes would have on our operations. 22
There may be a difference in the investment valuations thatIf future reimbursement for approved product candidates, if any, is substantially less than we used when making initialproject, or rebate obligations associated with them are substantially greater than we expect, our net revenue and subsequent investments in our Subsidiaries and minority investments and actual market values.
Our investments in our Subsidiaries and noncontrolling interests were the result of negotiation with subsidiary management and equity holders, and the investment valuations may not always have been independently verified. Traditional methods used by independent valuation analysts include a discounted cash flow analysis and a comparable company analysis. We have not generated a positive cash flow to date and do not expect to generate significant cash flow in the near future. Additionally, we believe that few comparable public companies exist to provide meaningful valuation comparisons. Accordingly, we have not always sought independent valuation analysis in connection with our investments and may have invested in our various holdings at higher or lower valuations than an independent source would have recommended. There mayprofitability could be no correlation between the investment valuations that we used over the years for our investments and the actual market values. If we should eventually sell all or a part of any of our consolidated business or that of a subsidiary, the ultimate sale price may be for a value substantially different than previously determined by us, which could materially and adversely impairimpacted.
We may not enjoy the valuemarket exclusivity benefits of our Common Stock.orphan drug designation. Although we have obtained an orphan designation for ARC-AAT in the treatment of alpha-1 antitrypsin deficiency, ARC-AAT may not be the first product to receive approval for that indication. Under the Orphan Drug Act, the first product with an orphan designation receives market exclusivity, which prohibits FDA from approving the “same” drug for the same indication. The FDA has stated that drugs can be the “same” even when they are not identical, but has not provided guidance with respect to how it will determine “sameness” for RNAi drugs. It is possible that another RNAi drug could be approved for the treatment of alpha-1 antitrypsin deficiency before ARC-AAT, which means that we may not obtain orphan drug exclusivity and could also potentially be blocked from approval until the first product’s orphan drug exclusivity period expires or we demonstrate, if we can, that ARC-AAT is superior. Further, even if we obtain orphan drug exclusivity for a product, that exclusivity may not effectively protect the product from competition because different drugs can be approved for the same condition. Even after an orphan drug is approved and granted orphan drug exclusivity, the FDA can subsequently approve the same drug for the same condition if the FDA concludes that the later drug is safer, more effective or makes a major contribution to patient care. Risks Related to Our Intellectual Property Rights Our ability to protect our patents and other proprietary rights is uncertain, exposing us to the possible loss of competitive advantage. We have licensed rights to pending patents and have filed and expect to continue to file patent applications. Researchers sponsored by us may also file patent applications that we choosemay need to license. Such patent applications may not be available for licensing or may not be economically feasible to license. Certain of our patents may not be granted or may not contain claims of the necessary breadth because, for example, prior patents exist. If a particular patent is not granted, the value of the invention described in the patent would be diminished. Further, even if these patents are granted, they may be difficult to enforce. Even if ultimately 42
successful, efforts to enforce our patent rights could be expensive, distracting for management, cause our patents to be invalidated or held unenforceable, and thus frustrate commercialization of products. Additionally, evenEven if patents are issued and are enforceable, others may independently develop similar, superior or parallel technologies to any technology developed by us orand not infringe on our patents. Our technology may prove to infringe upon patents or rights owned by others. Finally, patentPatent prosecution and maintenance is expensive, and we may be forced to curtail prosecution or maintenance if our cash resources are limited. Thus, the patents held by or licensed to us may not afford us any meaningful competitive advantage. If we are unable to derive value from our licensed or owned intellectual property, the value of your investment may decline. We are party to technology license agreements with third parties that require us to satisfy obligations to keep them effective and, if these agreements are terminated, our technology and our business would be seriously and adversely affected. We are party to license agreements with Novartis, Alnylam, City of Hope, and other entities to incorporate their proprietary technologies into our drug products under development. These license agreements require us to pay royalties and satisfy other conditions, including conditions in some cases related to the commercialization of the licensed technology. We may not be able to successfully incorporate these technologies into marketable products or, if we do, sales may not be sufficient to recover the amounts that we are obligated to pay to the licensors. If we fail to satisfy our obligations under these agreements, the terms of the licenses may be materially modified, such as by rendering currently exclusive licenses non-exclusive, or it may give our licensors the right to terminate their respective agreement with us, which would limit our ability to implement our current business plan and harm our business and financial condition. We may be subject to patent infringement claims, which could result in substantial costs and liability and prevent us from commercializing our potential products. Because the intellectual property landscape in the fields in which we participate is rapidly evolving and interdisciplinary, it is difficult to conclusively assess our freedom to operate without infringing on third party rights. However, we are currently aware of certain patent rights held by third parties that, if found to be valid and enforceable, could be alleged to render one or more of our business lines infringing. If a claim should be brought and is successful, we may be required to pay substantial damages, be forced to abandon any affected business lines and/or seek a license from the patent holder. In addition, any patent infringement claims brought against us, whether or not successful, may cause us to incur significant expenses and divert the attention of our management and key personnel from other business concerns. These could negatively affect our results of operations and prospects. We cannot be certain that patents owned or licensed by us or our Subsidiaries will not be challenged, potentially successfully, by others. In addition, if our potential productsproduct candidates infringe the intellectual property rights of third parties, these third parties may assert infringement claims against our customers, licensees, and other parties with whom we have business relationships and we may be required to indemnify our customersthose parties for any damages they suffer as a result of these claims. The claims may require us to initiate or defend protracted and costly litigation on behalf of customers, licensees, and other parties regardless of the merits of these claims. If any of these claims succeed, we may be forced to pay damages on behalf of our customersthose parties or may be required to obtain licenses for the products they use. If we cannot obtain all necessary licenses on commercially reasonable terms, we may be unable to continue selling such products. We license patent rights from third-party owners and we rely on such owners to obtain, maintain and enforce the patents underlying such licenses. We are a party to a number of licenses that give us rights to third-party intellectual property that is necessary or useful for our business. In particular, we have obtained licenses from, among others, Novartis and Alnylam. We also expect to enter into additional licenses to third-party intellectual property in the future. Our success will depend in part on the ability of our licensors to obtain, maintain and enforce patent protection for our licensed intellectual property, in particular, those patents to which we have secured exclusive rights. Our licensors may not successfully prosecute the patent applications to which we are licensed. Even if patents are issued in respect of these patent applications, our licensors may fail to maintain these patents, may determine not to pursue litigation against other companies that are infringing these patents, or may pursue such litigation less aggressively than we would. Without protection for the intellectual property we license, other companies might be able to offer substantially identical products for sale, which could adversely affect our competitive business position and harm our business prospects. Our technology licensed from various third parties may be subject to government rights and retained rights of the originating research institutions.rights. We license technology from the University of Texas MD Anderson Cancer Center, Caltech, and other universities and companies. Our licensors may have obligations to government agencies or universities. Under their agreements, a government agency or university may obtain certain rights over the technology that we have developed and licensed, including the right to require that a compulsory license be granted to one or more third parties selected by the government agency.
In addition, our licensors often retain certain rights under their agreements with us, including the right to use the underlying technology for noncommercial academic and research use, to publish general scientific findings from research related to the technology, and to make customary scientific and scholarly disclosures of information relating to the technology. It is difficult to monitor whether our licensors
43
limit their use of the technology to these uses, and we could incur substantial expenses to enforce our rights to our licensed technology in the event of misuse. Confidentiality agreements with employees and others may not adequately prevent disclosure of trade secrets and other proprietary information. In order to protect our proprietary technology and processes, we rely in part on confidentiality agreements with our collaborators, employees, consultants, outside scientific collaborators and sponsored researchers, and other advisors. These agreements may not effectively prevent disclosure of confidential information and may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. In addition, others may independently discover trade secrets and proprietary information, and in such cases we could not assert any trade secret rights against such party. Costly and time-consuming litigation could be necessary to enforce and determine the scope of our proprietary rights, and failure to obtain or maintain trade secret protection could adversely affect our competitive business position. 23
Risks Related to Regulation of Our Products
Our corporate compliance program cannot guarantee that we are in compliance with all applicable federal and state regulations.
Our operations, including our research and development and our commercialization efforts, such as clinical trials, manufacturing and distribution, are subject to extensive federal and state regulation. While we have developed and instituted a corporate compliance program, we cannot be assured that the Company or our employees are, or will be in compliance with all potentially applicable federal and state regulations or laws. If we fail to comply with any of these regulations or laws, a range of actions could result, including, but not limited to, the termination of clinical trials, the failure to approve a commercialized product, significant fines, sanctions, or litigation, any of which could harm our business and financial condition.
Risks Related to our Stock Stockholder equity interest may be substantially diluted in any additional financing. Our certificate of incorporation authorizes the issuance of 145,000,000 shares of Common Stock and 5,000,000 shares of Preferred Stock, on such terms and at such prices as our Board of Directors may determine. Adjusted forThe following serves as a summary of share issuance activity during the 1 for 10 stock split that was implemented on November 17, 2011,fiscal year ended September 30, 2015: | · | Issued 250,143 shares of Common Stock pursuant to the exercise of stock options, warrants and exchange rights and the vesting of restricted stock units; |
| · | Issued 3,321,383 shares of Common Stock in consideration for the assets acquired as part of the Novartis asset acquisition in March 2015; and |
| · | Issued 1,316,215 shares of Common Stock pursuant to the conversion of certain Preferred Stock held by the Company’s shareholders. |
As of September 30, 2012,2015, we had 13,579,18559,544,677 shares of Common Stock issued and outstanding. The issuance of additional securities in financing transactions by us or through the exercise of options or warrants will dilute the equity interests of our existing stockholders, perhaps substantially, and mightcould result in dilution in the tangible net book value of a share of our Common Stock, depending upon the price and other terms on which the additional shares are issued. Our Common Stock price has fluctuated significantly over the last several years and may continue to do so in the future, without regard to our results of operations and prospects. Because we are aearly in the stage of our drug development, stage company, there are few objective metrics by which our progress may be measured. Consequently, we expect that the market price of our Common Stock will likely continue to fluctuate significantly. We may not generate substantial revenue from the license or sale of our technology for several years, if at all. In the absence of product revenue as a measure of our operating performance, we anticipate that investors and market analysts will assess our performance by considering factors such as: ● | Announcements of developments related to our business; |
● | Our ability to enter into or extend investigation phase, development phase, commercialization phase and other agreements with new and/or existing partners; |
Announcements regarding the status of any or all of our collaborations or products;
● | Announcements regarding the status of any or all of our collaborations or products, including clinical trial results; |
● | Market perception and/or investor sentiment regarding our technology; |
Announcements regarding developments in the RNA interference or biotechnology fields in general;
● | Announcements of actions taken by regulatory authorities; such as the U.S. Food and Drug Administration; |
Market perception and/or announcements regarding other companies developing products in the field of RNA interference;
● | Announcements regarding developments in the RNA interference or biotechnology fields in general; |
The issuance of competitive patents or disallowance or loss of our patent rights; and
● | Announcements regarding clinical trial results with our products or competitors’ products; |
● | Market perception and/or announcements regarding other companies developing products in the field of biotechnology generally or specifically RNA interference; | ● | The issuance of competitive patents or disallowance or loss of our patent rights; |
44
● | The addition or departure of key executives; and |
● | Variations in our operating results. |
We will not have control over many of these factors but expect that they may influence our stock price. As a result, our stock price may be volatile and such volatility could result in the loss of all or part of your investment. Additionally, Litigation claims may result in the past, whenfinancial losses or harm our reputation and may divert management resources. When the market price of a stock has beenis volatile, holders of that stock have often initiated securities class action litigation against the company that issued the stock. If anyCertain of our stockholders have recently brought a lawsuitsuch lawsuits against us, pursuant to which we could incur substantial costscosts. We cannot predict with certainty the eventual outcome of this or any other litigation, arbitration or third-party inquiry. We may not be successful in defending ourselves or asserting our rights in current or future lawsuits, investigations, or claims that have been or may be brought against us and, as a result, our business could be materially harmed. These lawsuits, arbitrations, investigations or claims may result in large judgments or settlements against us, any of which could have a negative effect on our financial performance and business. Additionally, lawsuits, arbitrations and investigations can be expensive to defend, whether or not the lawsuit. The lawsuit, could alsoarbitration or investigation has merit, and the defense of these actions may divert the time and attention of our management.management and other resources that would otherwise be engaged in running our business. The market for purchases and sales of our Common Stock may be very limited, and the sale of a limited number of shares could cause the price to fall sharply. Although our Common Stock is listed for trading on the NASDAQ CapitalGlobal Select Market, historicallyat various times our securities have been relatively thinly traded. Investor trading patterns could serve to exacerbate the volatility of the price of theour stock. For example, mandatory sales of our Common Stock by institutional holders could be triggered if an investment in our Common Stock no longer satisfies their investment standards and guidelines. Accordingly, itIt may be difficult to sell shares of our Common Stock quickly without significantly depressing the value of the stock. Unless we are successful in developing continued investor interest in our stock, sales of our stock could continue to result in major fluctuations in the price of the stock. 24
If securities or industry analysts do not publish research reports about our business or if they make adverse recommendations regarding an investment in our stock, our stock price and trading volume may decline. The trading market for our Common Stock can be influenced by the research and reports that industry or securities analysts publish about our business. We do not currently have and may never obtain researchCurrently, coverage of our Company by industry orand securities analysts.analysts is limited. Investors have many investment opportunities and may limit their investments to companies that receive greater coverage from analysts. If noadditional industry or securities analysts do not commence coverage of the Company, the trading price of our stock could be negatively impacted. In the event we obtain industry or security analyst coverage, ifIf one or more of the analysts downgrade our stock or comment negatively on our prospects, our stock price may decline. If one or more of these analysts cease to cover our industry or us or failsfail to publish reports about the Company regularly, our Common Stock could lose visibility in the financial markets, which could also cause our stock price or trading volume to decline. The market Further, incorrect judgments, estimates or assumptions made by research analysts may adversely affect our stock price, particularly if subsequent performance falls below the levels that were projected by the research analyst(s), even if we did not set or endorse such expectations. Any of these events could cause further volatility in our stock price and could result in substantial declines in the value of our Common Stock may be adversely affected by the sale of shares by our management or founding stockholders.stock.
Sales of our Common Stock by our officers, directors and founding stockholders could adversely and unpredictably affect the price of those securities. Additionally, the price of our Common Stock could be affected even by the potential for sales by these persons. We cannot predict the effect that any future sales of our Common Stock, or the potential for those sales, will have on our share price. Furthermore, due to relatively low trading volume of our stock, should one or more large stockholders seek to sell a significant portion of their stock in a short period of time, the price of our stock may decline.
We do not intend to declare cash dividends on our Common Stock. We will not distribute cash to our stockholders unless and until we can develop sufficient funds from operations to meet our ongoing needs and implement our business plan. The time frame for that is unpredictable and investors should not expect dividends in the near future, if at all. Our Board of Directors has the authority to issue shares of “blank check” preferred stock, which may make an acquisition of the Company by another company more difficult. We have adopted and may in the future adopt certain measures that may have the effect of delaying, deferring or preventing a takeover or other change in control of the Company that a holder of our Common Stock might consider in its best interest. Specifically, our Board of Directors, without further action by our stockholders, currently has the authority to issue up to 5,000,000 shares of preferred stock and to fix the rights (including voting rights), preferences and privileges of these shares (“blank check” preferred). Such preferred stock may have rights, including economic rights, senior to our Common Stock.
ITEM 1B. | UNRESOLVED STAFF COMMENTS |
None. At September 30, 2012, we had leases for our corporate headquarters, located in Pasadena, California, and our research facility in Madison, Wisconsin. The Company does not own any real property. The following table summarizes the company’s leased facilities:facilities as of September 30, 2015. The Company does not own any real property.
| | | | | | | | | | | | | | | | | | | | Office
Space | | | Monthly
Rent | | | Lease
Commencement | | | Lease Term | | Pasadena, CA | | | 5,300 sq. ft. | | | $ | 13,000 | | | | August 16, 2011 | | | | 5.5 years | | Madison, WI | | | 24,000 sq. ft. | | | $ | 56,500 | | | | February 16, 2009 | | | | 10 Years | |
| Office
Space | | | Monthly
Rent | | | Lease
Commencement | | | Lease Term | | Pasadena, California | | 8,500 sq. ft. | | | $ | 24,000 | | | | August, 16, 2012 | | | | 7 years | | Madison, Wisconsin | | 27,000 sq. ft. | | | $ | 79,000 | | | | February, 16, 2009 | | | | 10 years | | Middleton, Wisconsin | | 2,900 sq. ft | | | $ | 6,000 | | | | May, 18, 2015 | | | | 1 year | |
None.
Legal Proceedings are set forth in our financial statement schedules in Part IV, Item 15 of this Annual Report and are incorporated herein by reference. See Note 7 — Commitments and Contingencies of Notes to Consolidated Financial Statements of Part IV, Item 15. Exhibits and Financial Statement Schedules.
ITEM 4. | MINE SAFETY DISCLOSURES |
Not applicable. PART II 25
PART II
ITEM 5. | MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
Price Range of Common Stock Our Common Stock is traded on the NASDAQ StockGlobal Select Market under the symbol “ARWR”. The following table sets forth the high and low sales prices for a share of the Company’s Common Stock during each period indicated. On November 17, 2011, the Company effected a 1 for 10 reverse stock split. The share prices in the table below are shown on a post-split basis. | | | | Fiscal Year Ended September 30, | | Fiscal Year Ended September 30, | | | | | 2012 | | | 2011 | | 2015 | | | 2014 | | | | | High | | | Low | | | High | | | Low | | High | | | Low | | | High | | | Low | | 1st Quarter | | $ | 7.50 | | | $ | 3.60 | | | $ | 11.00 | | | $ | 8.30 | | $ | 14.03 | | | $ | 5.00 | | | $ | 11.07 | | | $ | 5.86 | | 2nd Quarter | | | 6.38 | | | | 4.13 | | | | 10.00 | | | | 6.00 | | | 9.06 | | | | 6.15 | | | | 26.57 | | | | 9.92 | | 3rd Quarter | | | 7.14 | | | | 3.12 | | | | 7.00 | | | | 4.30 | | | 8.00 | | | | 6.08 | | | | 18.28 | | | | 9.79 | | 4th Quarter | | | 3.84 | | | | 2.60 | | | | 5.90 | | | | 3.70 | | | 7.62 | | | | 5.01 | | | | 16.96 | | | | 10.72 | |
Shares Outstanding At December 19, 2012, an aggregate of 15,719,07911, 2015, 59,554,677 shares of the Company’s Common Stock were issued and outstanding, and were owned by 293135 stockholders of record, based on information provided by the Company’s transfer agent. Dividends The Company has never paid dividends on its Common Stock and does not anticipate that it will do so in the foreseeable future. Securities Authorized for Issuance Under the Equity Compensation Plans The disclosure required under this item related to equity compensation plans is incorporated by reference from Item 12 under the caption “Equity Compensation Plan Information” inof Part III of this Annual Report on Form 10-K. 46
Sales of Unregistered Securities All information under this Item has been previously reported on our Current Reports on Form 8-K. Repurchases of Equity Securities We did not repurchase any shares of our Common Stock during fiscal 2012 or fiscal 2011.the years ended September 30, 2015, 2014 and 2013. Performance Graph The following performance graph shall not be deemed “soliciting material” or to be “filed” with the SEC, nor shall such information be incorporated by reference into any future filing under the Securities Act of 1933 or Securities Exchange Act of 1934, each as amended, except to the extent that we specifically incorporate it by reference into such filing. The graph compares the cumulative 5-year total return to shareholders on our Common Stock relative to the cumulative total returns of the NASDAQ Composite Index and the NASDAQ Biotechnology Index. We selected the NASDAQ Biotechnology Index because we believe the index reflects the market conditions within the industry in which we primarily operate. The comparison of total return on investment, defined as the change in year-end stock price plus reinvested dividends, for each of the periods assumes that $100 was invested on September 30, 2010, in each of our Common Stock, the NASDAQ Composite Index and the NASDAQ Biotechnology Index, with investment weighted on the basis of market capitalization. The comparisons in the following graph are based on historical data and are not intended to forecast the possible future performance of our Common Stock. 
47
ITEM 6. | SELECTED FINANCIAL DATA |
As a “Smaller Reporting Company,
The following selected financial data has been derived from our audited consolidated financial statements and should be read in conjunction with the consolidated financial statements, the related notes thereto and the independent auditors’ report thereon, and “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” wewhich are not required to provideincluded elsewhere in this information. Form 10-K and in previously filed annual reports on Form 10-K of Arrowhead Research Corporation. | | Year Ended September | | | | 2015 | | | 2014 | | | 2013 | | | 2012 | | | 2011 | | OPERATING SUMMARY | | | | | | | | | | | | | | | | | | | | | REVENUE | | $ | 382,000 | | | $ | 175,000 | | | $ | 290,266 | | | $ | 146,875 | | | $ | 296,139 | | OPERATING EXPENSES | | | | | | | | | | | | | | | | | | | | | Research and development | | | 47,267,361 | | | | 23,138,050 | | | | 8,705,627 | | | | 5,391,463 | | | | 3,277,760 | | Acquired in-process research and development | | | 10,142,786 | | | | - | | | | - | | | | - | | | | - | | Salaries and payroll-related costs | | | 16,554,008 | | | | 12,829,355 | | | | 6,667,669 | | | | 6,414,921 | | | | 1,408,366 | | General and administrative expenses | | | 7,931,184 | | | | 5,894,008 | | | | 3,488,864 | | | | 6,439,323 | | | | 3,821,550 | | Stock-based compensation | | | 10,232,897 | | | | 5,696,173 | | | | 1,536,271 | | | | 1,241,404 | | | | 1,376,921 | | Depreciation and amortization | | | 2,336,207 | | | | 1,345,655 | | | | 1,751,412 | | | | 1,748,975 | | | | 241,808 | | Impairment expense | | | - | | | | 2,172,387 | | | | 1,308,047 | | | | - | | | | - | | Contingent consideration - fair value adjustments | | | 1,891,533 | | | | 2,375,658 | | | | 1,421,652 | | | | - | | | | - | | TOTAL OPERATING EXPENSES (a) | | | 96,355,976 | | | | 53,451,286 | | | | 24,879,542 | | | | 21,236,086 | | | | 10,126,405 | | OPERATING LOSS | | | (95,973,976 | ) | | | (53,276,286 | ) | | | (24,589,276 | ) | | | (21,089,211 | ) | | | (9,830,266 | ) | LOSS FROM CONTINUING OPERATIONS | | | (91,940,882 | ) | | | (58,725,412 | ) | | | (31,703,079 | ) | | | (22,110,643 | ) | | | (8,785,008 | ) | Income (loss) from discontinued operations | | | - | | | | - | | | | (354 | ) | | | (80 | ) | | | 1,373,396 | | Gain on disposal of discontinued operations | | | - | | | | - | | | | - | | | | - | | | | 3,919,213 | | NET LOSS | | | (91,940,882 | ) | | | (58,725,412 | ) | | | (31,703,433 | ) | | | (22,110,723 | ) | | | (3,492,399 | ) | Net (gain) loss attributable to non-controlling interests | | | - | | | | 95,222 | | | | 560,144 | | | | 984,795 | | | | 363,514 | | NET LOSS ATTRIBUTABLE TO ARROWHEAD | | $ | (91,940,882 | ) | | $ | (58,630,190 | ) | | $ | (31,143,289 | ) | | $ | (21,125,928 | ) | | $ | (3,128,885 | ) | EARNINGS PER SHARE (BASIC): | | | | | | | | | | | | | | | | | | | | | Income (loss) from continuing operations attributable to Arrowhead common shareholders | | $ | (1.60 | ) | | $ | (1.25 | ) | | $ | (1.30 | ) | | $ | (1.90 | ) | | $ | (1.18 | ) | Income (loss) from discontinued operations attributable to Arrowhead common shareholders | | $ | - | | | $ | - | | | $ | - | | | $ | - | | | $ | 0.74 | | Net Income (Loss) attributable to Arrowhead common shareholders | | $ | (1.60 | ) | | $ | (1.25 | ) | | $ | (1.30 | ) | | $ | (1.90 | ) | | $ | (0.44 | ) | | | | | | | | | | | | | | | | | | | | | | EARNINGS PER SHARE (DILUTED): | | | | | | | | | | | | | | | | | | | | | Income (loss) from continuing operations attributable to Arrowhead common shareholders | | $ | (1.60 | ) | | $ | (1.25 | ) | | $ | (1.30 | ) | | $ | (1.90 | ) | | $ | (1.08 | ) | Income (loss) from discontinued operations attributable to Arrowhead common shareholders | | $ | - | | | $ | - | | | $ | - | | | $ | - | | | $ | 0.68 | | Net Income (Loss) attributable to Arrowhead common shareholders | | $ | (1.60 | ) | | $ | (1.25 | ) | | $ | (1.30 | ) | | $ | (1.90 | ) | | $ | (0.40 | ) | | | | | | | | | | | | | | | | | | | | | | Weighted average shares outstanding - basic | | | 57,358,442 | | | | 46,933,030 | | | | 24,002,224 | | | | 11,129,766 | | | | 7,181,121 | | Weighted average shares outstanding - diluted | | | 57,358,442 | | | | 46,933,030 | | | | 24,002,224 | | | | 11,129,766 | | | | 7,830,407 | | | | | | | | | | | | | | | | | | | | | | | CASH DIVDEND PAID PER COMMON SHARE | | $ | - | | | $ | - | | | $ | - | | | $ | - | | | $ | - | | | | September 30, | | | | 2015 | | | 2014 | | | 2013 | | | 2012 | | | 2011 | | FINANCIAL POSITION SUMMARY | | | | | | | | | | | | | | | | | | | | | CASH AND CASH EQUIVALENTS (b) | | $ | 81,214,354 | | | $ | 132,510,610 | | | $ | 19,114,444 | | | $ | 3,377,288 | | | $ | 7,507,389 | | SHORT- AND LONG-TERM INVESTMENTS (b) | | | 17,539,902 | | | | 44,741,378 | | | | 10,732,414 | | | | 106,500 | | | | 634,585 | | TOTAL ASSETS (b) | | | 132,267,914 | | | | 182,816,756 | | | | 37,329,631 | | | | 16,527,818 | | | | 15,888,563 | | CAPITAL LEASE OBLIGATIONS | | | 758,340 | | | | 972,331 | | | | 1,282,458 | | | | 1,497,259 | | | | - | | OTHER LONG-TERM OBLIGATIONS | | | 6,204,917 | | | | 4,226,137 | | | | 1,808,709 | | | | 1,108,563 | | | | 742,446 | |
| (a) | The increase in our Total Operating Expenses during the year ended September 30, 2015 is primarily due to the progress achieved on our lead clinical candidate for HBV, ARC-520. The primary costs incurred during fiscal year 2015 related to manufacturing of clinical supplies for the ARC-520 phase 2b clinical trial, toxicology studies, and the cost associated with the administration of the clinical trials. Additionally, the Company incurred a $10.1 million expense related to the acquisition of in-process research and development from the Novartis asset acquisition in March 2015. |
| (b) | The decrease in Cash and Cash Equivalents, Short- and Long-Term Investments, and Total Assets as of September 30, 2015 as compared to September 30, 2014, primarily relates to expenditures discussed above. |
48
ITEM 7. | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
Description of Business Unless otherwise noted, (1) the term “Arrowhead” refers to Arrowhead Research Corporation, a Delaware corporation, (2) the terms the “Company,” “we,” “us,” and “our,” refer to the ongoing business operations of Arrowhead and its Subsidiaries, whether conducted through Arrowhead or a subsidiary of Arrowhead, (3) the term “Subsidiaries” refers collectively to Arrowhead Madison Inc. (“Arrowhead Madison”), formerly known as “Roche Madison, Inc.”, Alvos Therapeutics, Inc.Arrowhead Australia Pty Ltd (“Alvos”Arrowhead Australia”), Calando Pharmaceuticals, Inc. (“Calando”),and Ablaris Therapeutics, Inc. (“Ablaris”), Agonn Systems, Inc. (“Agonn”), and Tego Biosciences Corporation (“Tego”) as well as our former subsidiary, Unidym, Inc. (“Unidym”), which was divested in January 2011, (4) the term “Minority Investments” refers collectively to Nanotope, Inc. (“Nanotope”) and Leonardo Biosystems, Inc. (“Leonardo”) in which the company holds a less than majority ownership position, and (5) the term “Common Stock” refers to Arrowhead’s Common Stock, (5) the term “Preferred Stock” refers to Arrowhead’s Preferred Stock and the term “stockholder(s)“Stockholder(s)” refers to the holders of Arrowhead Common Stock. All Arrowhead share and per share data have been adjusted to reflect a one for ten reverse stock split effected on November 17, 2011. 26
Overview Arrowhead Research Corporationdevelops novel drugs to treat intractable diseases by silencing the genes that cause them. Using the industry’s broadest portfolio of RNA chemistries and efficient modes of delivery, Arrowhead therapies trigger the RNA interference mechanism to induce rapid, deep and durable knockdown of target genes. RNA interference (RNAi) is a mechanism present in living cells that inhibits the expression of a specific gene, thereby affecting the production of a specific protein. Deemed to be one of the most important recent discoveries in life science with the potential to transform medicine, the discoverers of RNAi were awarded a Nobel Prize in 2006 for their work. Arrowhead’s RNAi-based therapeutics leverage this natural pathway of gene silencing to target and shut down specific disease causing genes. Arrowhead operates lab facilities in Madison and Middleton, Wisconsin, where the Company’s research and development activities, including the development of RNAi therapeutics, are based. The Company’s principal executive offices are located in Pasadena, California. During fiscal year 2015, the Company continued to develop its lead clinical stage targeted therapeutics company with development programs in oncology, obesity, andcandidate, ARC-520, for the treatment of chronic hepatitis B virus infection. Arrowhead is focusedas well as its second clinical candidate, ARC-AAT, an RNAi therapeutic designed to treat liver disease associated with Alpha-1 antitrypsin deficiency (AATD). The Company continued its Phase 2 studies in ARC-520, with no dose-limiting toxicities or serious adverse events having been observed to date. The Company submitted an Investigational New Drug application to the FDA in December 2014 for ARC-520 to initiate phase 2b multi-dose studies to determine the depth of hepatitis B surface antigen (HBsAg) reduction following ARC-520 injection. The Company received feedback from the FDA, and based on creating new therapeutics that are preferentially taken up by target tissuesfeedback the Company adjusted the protocol in order to maximizebegin the trial. In April 2015, the application was approved by the FDA. In June 2015, the Company received regulatory clearance in Germany for two additional Phase 2b multiple-dose studies of ARC-520 to be conducted in parallel, and also expects to file with additional Australian, Asian and European agencies to begin additional phase 2 studies. In May 2015, the Company completed protocol-required dosing of healthy volunteers in an on-going phase 1 study of ARC-AAT, and in July 2015, initiated dosing of patients in Part B of that same study. The study recently received regulatory clearance in the United Kingdom, Germany and New Zealand, and is currently recruiting patients at several sites in those countries. In June 2015, ARC-AAT was granted orphan drug designation by the FDA. During fiscal year 2015, the Company expanded its candidate pipeline by nominating several additional pre-clinical candidates including: ARC-F12, a drug’s efficacytreatment for factor 12 (F12) mediated angioedemic and potentially limit side effectsthromboembolic diseases, ARC-HIF2, a treatment for clear cell renal cell carcinoma (ccRCC), and ARC-521, a complementary candidate to ARC-520 for the treatment of chronic hepatitis B infection. In March 2015, the Company completed the acquisition of Novartis’ entire RNAi research and development portfolio and associated with exposureassets. The acquisition included assignment of certain patents and patent applications owned or controlled by Novartis related to healthy cells. Arrowhead has assembled a broad set of technologies and licenses to enable targeted RNAi therapeutics, capablean exclusive license in the RNAi field to other patents and patent applications owned or controlled by Novartis, assignment of silencing specifica third party license, three pre-clinical RNAi candidates, and other related assets. This acquisition is expected to significantly expand the Company’s RNAi capabilities as the assets are integrated. The Company continues to develop other clinical candidates for future clinical trials, including intravenously-administered therapeutics targeting gene productsknockdown in specific cells. Arrowhead has also assembled a proprietary targeting library that may be used with its RNAi platformsthe liver, as well as formulations for administering RNAi-based therapeutics by subcutaneous administration. Clinical candidates are tested internally and through GLP toxicology studies at outside laboratories, and drug materials for such studies, and for clinical trials, are contracted to third-party manufactures when cGMP production is required. The Company engages third-party contract research organizations (CROs) to manage clinical trials and works cooperatively with small molecule or peptide drugs.such organizations on all aspects of clinical trial management, including plan design, patient recruiting, and follow up. These platformsoutside costs, relating to the preparation for and administration of clinical trials, are referred to as program costs, and if the clinical candidates progress through human testing, program costs will increase. 49
Net losses were $91.9 million, $58.7 million and $31.7 million during the years ended September 30, 2015, 2014 and 2013, respectively. Diluted losses per share were $1.60, $1.25 and $1.30 during the years ended September 30, 2015, 2014 and 2013, respectively. The Company strengthened its liquidity and financial position through two securities offerings completed in October 2013 and February 2014 which generated approximately $172.6 million of cash proceeds for the Company. These cash proceeds secured the funding needed to advance both ARC-520 and ARC-AAT into clinical trials and will also assist as the Company expands its pipeline of other clinical candidates. The Company had $81.2 million of cash and cash equivalents and $132.3 million of total assets as of September 30, 2015 as compared to $132.5 million and $182.8 million as of September 30, 2014, respectively. The increased operating expenses and net losses and decrease in cash and cash equivalents and total assets reflects expenditures associated with the Company’s research and development efforts for its clinical candidates and pipeline, as well as cash paid for the Novartis RNAi assets acquired. Based upon the Company’s current cash resources and operating plan, the Company expects to have yielded several drug candidates under both internal and partnered development.sufficient liquidity to fund operations for at least the next twelve months. Critical Accounting Policies and Estimates Management makes certain judgments and uses certain estimates and assumptions when applying accounting principles generally accepted in the United StatesGAAP in the preparation of our Consolidated Financial Statements. We evaluate our estimates and judgments on an ongoing basis and base our estimates on historical experience and on assumptions that we believe to be reasonable under the circumstances. Our experience and assumptions form the basis for our judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may vary from what we anticipate and different assumptions or estimates about the future could change our reported results. We believe the following accounting policies are the most critical to us, in that they are important to the portrayal of our consolidated financial statements and require our most difficult, subjective or complex judgments in the preparation of our consolidated financial statements. For further information, seeNote 1, Organization and Significant Accounting Policies, to our Consolidated Financial Statements, which outlines our application of significant accounting policies and new accounting standards.policies. Revenue Recognition Revenue from product sales areis recorded when persuasive evidence of an arrangement exists, title has passed and delivery has occurred, a price is fixed and determinable, and collection is reasonably assured. We may generate revenue from technology licenses, collaborative research and development arrangements, research grants and product sales. Revenue under technology licenses and collaborative agreements typically consists of nonrefundable and/or guaranteed technology license fees, collaborative research funding, and various milestone and future product royalty or profit-sharing payments. Revenue associated with research and development funding payments under collaborative agreements is recognized ratably over the relevant periods specified in the agreement, generally the research and development period. Revenue from up-front license fees, milestones and product royalties are recognized as earned based on the completion of the milestones and product sales, as defined in the respective agreements. Payments received in advance of recognition as revenue are recorded as deferred revenue. Business CombinationsAsset Acquisition
In October 2011, weOn March 3, 2015, the Company entered into an Asset Purchase and Exclusive License Agreement (the “RNAi Purchase Agreement”) with Novartis Institutes for BioMedical Research, Inc., a Delaware corporation (“Novartis”), pursuant to which the Company acquired allNovartis’ RNAi assets and rights thereunder. Pursuant to the Agreement, the Company acquired or licensed certain patents and patent applications owned or controlled by Novartis related to RNAi therapeutics, assignment of a third-party license, rights to three pre-clinical RNAi candidates, and other related assets (collectively, the “Purchased Assets”). The acquisition of the outstanding common stock of Roche Madison, Inc. and certain related intellectual property assets for a $50,000 promissory note and 1,288,158 shares of Arrowhead Common Stock, an estimated consideration value of $5.1 millionPurchased Assets closed on the dateMarch 3, 2015, concurrent with execution of the acquisition. We assigned the valueAgreement (the “Closing”). The Company accounted for this transaction as an acquisition of the consideration to the tangibleRNAi assets, including patents, a third-party license and identifiable intangible assetsin process research and the liabilities assumed on the basis of their fair values on the date of acquisition. The excess of net assets over the consideration was recorded as a nonoperating gain.
In April 2012, we acquired all of the outstanding common stock of Alvos Therapeutics, Inc. in exchangedevelopment for the issuance of 315,457 shares of Arrowhead Common Stock, valued at $2.0 million at the time of acquisition. The consideration was assigned to its tangible and intangible assets, and liabilities based on estimated fair values at the time of acquisition.
pre-clinical candidates. The allocation of the purchase price to each asset was determined by estimating the relative fair value of each asset acquired and applying that to certain items, including propertythe total cost of the acquisition for the Company. The Company capitalized the patents and equipment, intangible assetslicense acquired as Intangible Assets as they require no future development and certain liabilitieswill have alternative future uses as the Company expands its RNAi capabilities. The Company expensed the portion of the purchase consideration allocated to the pre-clinical candidates as they will require management judgment,future development in order to be commercialized. This expense is recorded in the “Acquired in-process research and is based upondevelopment” line item of the information available at the timeConsolidated Statements of acquisition.Operations. 50
Impairment of Long-lived Assets We review long-lived assets for impairment whenever events or changes in business circumstances indicate that the carrying amount of assets may not be fully recoverable or that our assumptions about the useful lives of these assets are no longer appropriate. If impairment is indicated, recoverability is measured by a comparison of the carrying amount of an asset to the estimated undiscounted future cash flows expected to be generated by the asset. If the carrying amount of an asset exceeds its estimated future cash flows, an impairment charge is recognized in the amount by which the carrying amount of the asset exceeds the fair value of the asset. 27
Impairment of Intangible assets Intangible assets consist of in-process research and development, patentslicense agreements and license agreementspatents acquired in conjunction with a business or asset acquisition. Intangible assets are monitored for potential impairment whenever events or circumstances indicate that the carrying amount may not be recoverable, and are also reviewed annually to determine whether any impairment is necessary. Based on early adoption of ASU 2012-02, the annual review of intangible assets is performed via a two-step process. First, a qualitative assessment is performed to determine if it is more likely than not that the intangible asset is impaired. If required, a quantitative assessment is performed and, if necessary, impairment is recorded. Stock-Based Compensation We recognize stock-based compensation expense for stock options based on the grant date fair value using the Black-Scholes options pricing model, which requires us to make assumptions regarding certain variables including the risk-free interest rate, expected stock price volatility, assumed forfeitures, and the expected life of the award. The grant date fair value of restricted stock units granted is based upon the quoted closing market price per share on the date of grant, adjusted for assumed forfeitures. For performance-based stock awards, the value of the award is measured at the grant date. Expense for stock options and restricted stock units is recognized over the requisite service period. The assumptions used in calculating stock-based compensation expense represent management’s best estimates, but these estimates involve inherent uncertainties, and if factors change or the Company used different assumptions, its stock-based compensation expense could be materially different in the future. Derivative Assets and Liabilities We account for warrants and other derivative financial instruments as either equity or assets/liabilities based upon the characteristics and provisions of each instrument. Warrants classified as equity are recorded as additional paid-in capital on our consolidated balance sheetConsolidated Balance Sheet and no further adjustments to their valuation are made. Some of our warrants were determined to be ineligible for equity classification because of provisions that may result in an adjustment to their exercise price. Warrants classified as derivative liabilities and other derivative financial instruments that require separate accounting as assets or liabilities are recorded on our consolidated balance sheetConsolidated Balance Sheet at their fair value on the date of issuance and are revalued on each subsequent balance sheet date until such instruments are exercised or expire, with any changes in the fair value between reporting periods recorded as other income or expense. We estimate the fair value of these assets/liabilities using option pricing models that are based on the individual characteristics of the warrants or instruments on the valuation date, as well as assumptions for expected volatility, expected life and risk-free interest rate. Changes in the assumptions used could have a material impact on the resulting fair value. The primary input affecting the value of our derivatives liabilities is the Company’s stock price. Contingent Consideration The consideration for our acquisitions often includes future payments that are contingent upon the occurrence of a particular event. For example, milestone payments might be based on progress of clinical development, the achievement of various regulatory approvals or future sales milestones, and royalty payments might be based on drug product sales levels. The Company records a contingent consideration obligation for such contingent payments at September 30, 2012, a 50% changefair value on the acquisition date. The Company estimates the fair value of contingent consideration obligations through valuation models designed to estimate the probability of the occurrence of such contingent payments based on various assumptions and incorporating estimated success rates. Estimated payments are discounted using present value techniques to arrive at estimated fair value at the balance sheet date. Changes in the fair value of our contingent consideration obligations are recognized within our Consolidated Statements of Operations. Changes in the fair value of the Company’s stock price would affect the value of the derivative liability by approximately $0.3 millioncontingent consideration obligations can result from changes to $0.4 million, depending on other inputs. Reverse Stock Split
As of November 17, 2011, the Company effected a 1 for 10 reverse stock split (the “reverse stock split”). As a result of the reverse stock split, each ten shares of the Company’s Common Stock issued and outstanding immediately priorone or multiple inputs, including adjustments to the reverse split was combined into one sharediscount rates, changes in the amount or timing of Common Stock. Also, as a resultexpected expenditures associated with product development, changes in the amount or timing of cash flows from products upon commercialization, changes in the Reverse Stock Split,assumed achievement or timing of any development milestones, changes in the per share exercise priceprobability of certain clinical events and changes in the number of shares of Common Stock underlying outstanding Company stock options, warrants, Series A Preferred and any Common Stock based equity grants outstanding immediately prior to the reverse stock split was proportionally adjusted,assumed probability associated with regulatory approval. These fair value measurements are based on significant inputs not observable in the one-for-ten split ratio,market. Substantial judgment is employed in accordance withdetermining the termsappropriateness of such options, warrants or other Common Stock based equity grantsthese assumptions as the case may be. No fractional shares of Common Stock were issued in connection with the reverse stock split. Stockholders instead received cash payment in lieu of any fractional shares. Unless otherwise noted, all share and per share amounts in these have been retrospectively adjusted to reflect the reverse stock split.
Full Year Review
On October 21, 2011, the Company acquired Roche Madison, Inc. and other intangible assets from Roche. The acquisition included a laboratory research facility in Madison, Wisconsin comprising over 24,000 square feet. Roche Madison Inc. employed 41 employees at the time of the acquisition. Due to the significant new costs associated with the facility, its people and research programs, salary costs, general and administrative costs, and research and development costs increased significantly relative to prior periods. Going forward, we expect this increased cost structure to continue as research and development efforts are accelerated.
On April 11, 2012, the Company acquired Alvos Therapeutics, Inc., a targeted therapeutics company. Prior to the acquisition, Alvos licensed a large platform proprietary human-derived Homing Peptides and the method for their discovery from MD Anderson Cancer Center. The company hired one employee as a result of the acquisition date and for each subsequent period. Accordingly, changes in assumptions could have a material impact on the operationsamount of Alvos are being integrated into our research facilitycontingent consideration expense the Company records in Madison, Wisconsin.any given period.
51
Results of Operations The following data summarizes our results of operations for the following periods indicated: | | Year ended September 30, | | | | 2015 | | | 2014 | | | 2013 | | Revenue | | $ | 382,000 | | | $ | 175,000 | | | $ | 290,266 | | Operating Loss | | | (95,973,976 | ) | | | (53,276,286 | ) | | | (24,589,276 | ) | Loss from Continuing Operations | | | (91,940,882 | ) | | | (58,725,412 | ) | | | (31,703,079 | ) | Net Loss | | | (91,940,882 | ) | | | (58,725,412 | ) | | | (31,703,433 | ) | Earnings per Share (Basic and Diluted) | | $ | (1.60 | ) | | $ | (1.25 | ) | | $ | (1.30 | ) |
The increase in our Total Operating Expenses during the year ended September 30, 2015 is primarily due to the progress achieved on our lead clinical candidate for HBV, ARC-520. The primary costs incurred during fiscal year 2015 related to manufacturing of clinical supplies for the ARC-520 phase 2b clinical trial, toxicology studies, and the cost associated with the administration of the clinical trials. Additionally, the Company hadincurred a net loss$10.1 million expense related to the acquisition of $22.1 millionin-process research and development from Novartis in March 2015. Results of Operations Comparison for 2015 and 2014 Revenues Total revenue was $382,000 for the year ended September 30, 2012, compared to a net loss of $3.5 million2015 and $175,000 for the year ended September 30, 2011, an increase of $18.6 million. 28
The change in the net loss was the result of a number of factors. During the year ended September 30, 2011,2014. Revenue is primarily related to licensed technology. In addition, the Company recognized income from discontinued operations of $5.4 million related to the gain on disposal of Unidym, which was not repeated in the current period. In fiscal 2012, the Company recorded an impairment charges and recorded as reserve against a receivable from its unconsolidated affiliates, in the amount of $4.1 million. In fiscal 2012, the company recorded a loss on disposal of equipment of $1.1 million, related to non-strategic equipment obtained in conjunction with the acquisition of Roche Madison, and subsequently sold. These losses were partially offset by a gain recorded on the acquisition of Roche Madison of $1.6 million. All of these items are non-operating, one-time occurrences. However, research and development costs increased significantly in the current fiscal year due to the acquisition of Roche Madison, its facility costs, personnel costs, and program costs. Details of the results of operations are presented below.
Revenues
The Company generatedhad collaboration revenue of $147,000$160,000 during the year ended September 30, 2012, due to license agreements obtained in conjunction with the acquisition of Roche Madison, as compared to revenue of $296,000 during the year ended September 30, 2011. The revenue in 2011 was primarily related to a qualifying therapeutic discovery grant received by Calando.2015.
Operating Expenses The analysis below details the operating expenses and discusses the expenditures of the Company within the major expense categories. Certain reclassifications have been made to prior-period operating expense categories to conform to the current period presentation. For purposes of comparison, the amounts for the years ended September 30, 20122015 and 20112014 are shown in the tabletables below. Research and Development Expenses R&D expenses are related to the Company’s on-going research and development efforts, primarily related to program costs, composed primarily of outsourced costs related to the manufacturing of clinical supplies, toxicity/efficacy studies and clinical trial expenses. Internal costs primarily relate to operations at our research and development facility in Madison, Wisconsin, including facility costs and laboratory-related expenses. The following table provides details of research and development expense for the periods indicated: (in thousands) | | Twelve | | | | | | | Twelve | | | | | | | | | | | Months Ended | | | | | | | Months Ended | | | | | | | | | | | September 30, 2015 | | | % of Total | | | September 30, 2014 | | | % of Total | | | Increase (Decrease) | | Laboratory supplies & services | | $ | 2,531 | | | | 5 | % | | $ | 2,071 | | | | 9 | % | | $ | 460 | | | | 22 | % | In vivo studies | | | 556 | | | | 1 | % | | | 224 | | | | 1 | % | | | 332 | | | | 148 | % | Outside labs & contract services | | | 489 | | | | 1 | % | | | 691 | | | | 3 | % | | | (202 | ) | | | -29 | % | Toxicity/efficacy studies | | | 6,572 | | | | 14 | % | | | 6,306 | | | | 27 | % | | | 266 | | | | 4 | % | Drug manufacturing | | | 21,431 | | | | 46 | % | | | 9,142 | | | | 40 | % | | | 12,289 | | | | 134 | % | Clinical trials | | | 13,329 | | | | 28 | % | | | 2,627 | | | | 11 | % | | | 10,702 | | | | 407 | % | License, royalty & milestones | | | 1,065 | | | | 2 | % | | | 1,096 | | | | 5 | % | | | (31 | ) | | | -3 | % | Facilities and related | | | 977 | | | | 2 | % | | | 881 | | | | 4 | % | | | 96 | | | | 11 | % | Other research expenses | | | 317 | | | | 1 | % | | | 100 | | | | 0 | % | | | 217 | | | | 217 | % | Total | | $ | 47,267 | | | | 100 | % | | $ | 23,138 | | | | 100 | % | | $ | 24,129 | | | | 104 | % |
Laboratory supplies and services expense increased $460,000 from $2,071,000 during the year ended September 30, 2014 to $2,531,000 during the current period. The increase in laboratory supplies and services is a result of additional supplies necessary to 52
support increased efforts in pre-clinical research as the Company supports ongoing clinical efforts and accelerates efforts to identify new clinical candidates. In vivo studies expense increased $332,000 from $224,000 during the year ended September 30, 2014 to $556,000 during the current period. In vivo expense can vary depending on the stage of preclinical candidates, the nature and amount of testing required and the varying costs of different in vivo testing models. The expense in both periods relates to studies in connection with the development of new clinical candidates. During fiscal year 2015, the Company expanded its candidate pipeline which resulted in additional studies conducted. Outside labs and contract services expense decreased $202,000 from $691,000 during the year ended September 30, 2014 to $489,000 during the current period. The decrease in the current period primarily relates to a one-time fee paid in the prior year related to access to a certain animal study. Toxicity/efficacy studies expense increased $266,000 from $6,306,000 during the year ended September 30, 2014 to $6,572,000 during the current period. This category includes IND-enabling toxicology studies, post-IND toxicology studies, such as long-term toxicology studies, and other efficacy studies. The full-year expense primarily relates to IND-enabling toxicology studies related to ARC-AAT, on-going long-term toxicology studies to support later clinical trials, and studies related to ARC-520 to support our anticipated phase 2b clinical trial. These amounts can vary quarter to quarter based on stage of development. Drug manufacturing expense increased $12,289,000 from $9,142,000 during the year ended September 30, 2014 to $21,431,000 during the current period. The current period expense primarily relates to drug manufacturing to supply toxicology studies for our ARC-520 Phase 2b clinical trials and clinical supplies for the ARC-520 Phase 2b clinical trial. The manufacturing campaign for the Phase 2b clinical trial for ARC-520 is largely complete, however, as other clinical candidates are nominated, and as other clinical trials advance, further expenditures will be incurred. Clinical trials expense increased $10,702,000 from $2,627,000 during the year ended September 30, 2014 to $13,329,000 during the current period. The increase is primarily driven by costs incurred in preparation for our anticipated phase 2b clinical trial for ARC-520. We are also incurring costs in fiscal 2015 related to our clinical trial for our second clinical candidate ARC-AAT. We expect clinical trial expenses to increase further in fiscal year 2016 as enrollment in our clinical trials increases. License, royalty and milestones expense was consistent at $1,096,000 during the year ended September 30, 2014 and $1,065,000 during the current period. This category can include milestone payments which can vary from period to period depending on the nature of our various license agreements, and the timing of reaching various development milestones requiring payment. We reached milestones related to our clinical candidates that required a $1 million payment in each period. Facilities expense increased $96,000 from $881,000 during the year ended September 30, 2014 to $977,000 during the current period. The increase relates to higher rent in our Madison facility somewhat offset by lower repairs and maintenance costs on our lab equipment. Other research expense increased $217,000 from $100,000 during the year ended September 30, 2014 to $317,000 during the current period. The increase in the current period primarily relates to costs associated with a collaboration agreement to identify muscle targeting peptide molecules in fiscal 2015, for which the Company has been reimbursed from its collaboration partner. Salary & Wage Expenses—Fiscal 2012 compared to Fiscal 2011and Payroll-Related Expenses The Company employs management,scientific, technical and administrative and scientific and technical staff at its corporate offices and its research facility.facilities. Salaries and wagespayroll-related expense consists of salary, bonuses, payroll taxes and related benefits. Salary and benefitspayroll-related expenses include two major categories: general and administrative (G&A) compensation expense, and research and development (R&D) compensation expense, based on the primary activities of each employee. The following table provides detail of salary and related benefitspayroll-related expenses for the years ended September 30, 2012 and 2011.periods indicated: (in thousands) | | | | | | | | | | | | | | | | | | | | | | | | | | | | Twelve months
Ended | | | % of
Expense | | | Twelve months
Ended | | | % of
Expense | | | Increase (Decrease) | | | | | September 30, 2012 | | | Category | | | September 30, 2011 | | | Category | | | $ | | | % | | G&A—compensation-related | | $ | 3,107 | | | | 48 | % | | $ | 1,144 | | | | 81 | % | | $ | 1,963 | | | | 172 | % | R&D—compensation-related | | | 3,308 | | | | 52 | % | | | 264 | | | | 19 | % | | | 3,044 | | | | 1153 | % | | | | | | | | | | | | | | | | | | | | | | | | | | Total | | $ | 6,415 | | | | 100 | % | | $ | 1,408 | | | | 100 | % | | $ | 5,007 | | | | 356 | % | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Twelve months | | | | | | | Twelve months | | | | | | | | | | | | | | | | Ended | | | | | | | Ended | | | | | | | | | | | September 30, 2015 | | | % of Total | | | September 30, 2014 | | | % of Total | | | Increase (Decrease) | | R&D - compensation-related | | $ | 11,605 | | | | 70 | % | | $ | 7,828 | | | | 61 | % | | $ | 3,777 | | | | 48 | % | G&A - compensation-related | | | 4,949 | | | | 30 | % | | | 5,001 | | | | 39 | % | | | (52 | ) | | | -1 | % | Total | | $ | 16,554 | | | | 100 | % | | $ | 12,829 | | | | 100 | % | | $ | 3,725 | | | | 29 | % |
During
53
R&D compensation expense increased $3,777,000 from $7,828,000 during the year ended September 30, 2012, 2014 to $11,605,000 during the current period. An increase in headcount accounted for the majority of the change in compensation-related expense. G&A compensation expense increased $1,963,000. During the fiscal year, upon the acquisition of Roche Madison, the Company expanded its senior management team. Its G&A headcount also increased due to several Madison employees classified as G&A. Duringwas consistent at $5,001,000 during the year ended September 30, 2012, R&D compensation expense increased $3,044,000. This increase was due to employees hired upon2014 and $4,949,000 during the acquisition of Roche Madison.current period. G&A headcount has remained consistent during the periods. General & Administrative Expenses—Fiscal 2012 compared to Fiscal 2011Expenses The following table provides details of our general and administrative expenses for the fiscal years 2012 and 2011.periods indicated: (in thousands) | | | | | | | | | | | | | | Twelve | | | | | | | Twelve | | | | | | | | | | | | Twelve months
Ended | | | % of
Expense | | Twelve months
Ended | | | % of
Expense | | Increase (Decrease) | | | Months Ended | | | | | | | Months Ended | | | | | | | | | | | | September 30, 2012 | | | Category | | September 30, 2011 | | | Category | | $ | | % | | | September 30, 2015 | | | % of Total | | | September 30, 2014 | | | % of Total | | | Increase (Decrease) | | Professional/outside services | | $ | 1,800 | | | | 28 | % | | $ | 2,383 | | | | 63 | % | | $ | (583 | ) | | | -24 | % | | $ | 3,989 | | | | 50 | % | | $ | 2,465 | | | | 42 | % | | $ | 1,524 | | | | 62 | % | Patent expense | | | 1,024 | | | | 16 | % | | | 604 | | | | 16 | % | | | 420 | | | | 70 | % | | | 950 | | | | 12 | % | | | 632 | | | | 11 | % | | | 318 | | | | 50 | % | Facilities and related | | | 120 | | | | 2 | % | | | 168 | | | | 4 | % | | | (48 | ) | | | -29 | % | | | 308 | | | | 4 | % | | | 193 | | | | 3 | % | | | 115 | | | | 60 | % | Travel | | | 369 | | | | 6 | % | | | 201 | | | | 5 | % | | | 168 | | | | 84 | % | | | 841 | | | | 11 | % | | | 654 | | | | 11 | % | | | 187 | | | | 29 | % | Business insurance | | | 202 | | | | 3 | % | | | 194 | | | | 5 | % | | | 8 | | | | 4 | % | | | 523 | | | | 7 | % | | | 302 | | | | 5 | % | | | 221 | | | | 73 | % | Communication and Technology | | | 196 | | | | 3 | % | | | 96 | | | | 3 | % | | | 100 | | | | 104 | % | | | 691 | | | | 9 | % | | | 388 | | | | 7 | % | | | 303 | | | | 78 | % | Office expenses | | | 91 | | | | 1 | % | | | 54 | | | | 1 | % | | | 37 | | | | 69 | % | | | 270 | | | | 3 | % | | | 380 | | | | 6 | % | | | (110 | ) | | | -29 | % | Other | | | 2,637 | | | | 41 | % | | | 95 | | | | 3 | % | | | 2,542 | | | | NM | | | | 359 | | | | 4 | % | | | 880 | | | | 15 | % | | | (521 | ) | | | -59 | % | | | | | | | | | | | | | | | | | | | | | Total | | $ | 6,439 | | | | 100 | % | | $ | 3,795 | | | | 100 | % | | $ | 2,644 | | | | 70 | % | | $ | 7,931 | | | | 100 | % | | $ | 5,894 | | | | 100 | % | | $ | 2,037 | | | | 35 | % | | | | | | | | | | | | | | | | | | | | |
Professional/outside services include legal, accounting, consulting and other outside services retained by Arrowhead and its subsidiaries.the Company. All periods include normally occurringrecurring legal and accountingaudit expenses related to SEC compliance and other corporate matters. Professional/outside services expense was $1,800,000increased $1,524,000 from $2,465,000 during the year ended September 30, 2012, compared2014 to $2,383,000$3,989,000 during the current period. The increase in the comparable prior period. In the prior period,professional fees primarily related to higher legal fees related to recent litigation events. See Note 7 – Commitments and Contingencies for further discussion. Additionally, the Company recorded expenses of $663,000 relatedincurred higher recruiting fees to stock issued for financing commitments in association with the September 2011 financing in conjunction with the acquisition of Roche Madison, Inc.fill new positions. 29
Patent expense was $1,024,000increased $318,000 from $632,000 during the year ended September 30, 2012, compared2014 to $604,000 in$950,000 during the comparable priorcurrent period. DuringPatent expense costs increase due to additional prosecution requirements associated with new patents acquired through the year ended September 30, 2012, approximately half of the patent expense was related to fees paid to patent counsel for the maintenance of newly acquired intellectual property in conjunction with the acquisition of Roche Madison. The balance of patent expense primarily relates to Calando’s intellectual property portfolio, and to a lesser extent the intellectual property acquired in conjunction with the Alvos acquisition and the Ablaris patent portfolio.Novartis asset acquisition. The Company expects to continuecontinues to invest in patent protection as thefor its DPC™ technology, related product candidates and other RNAi technology through patent filings in multiple countries. The Company extendsexpects to extend and maintainsmaintain protection for its current portfolios, as appropriate, and filesfile new patent applications as its product applicationstechnologies are developed and improved. Expenses can vary from period to period as patents proceed through their prosecution life cycle. Facilities and relatedFacilities-related expense was $120,000increased $115,000 from $193,000 during the year ended September 30, 2012, compared2014 to $168,000$308,000 in the comparable priorcurrent period. Facilities and related expense within general and administrative expenses primarily relateincreased due to rental costs associated with the Company’sexpansion of our corporate headquarters in Pasadena, California. Facilities expense decreased due to reduction in the company’s rental expense because its lease for its corporate headquarters expired. During most of fiscal 2012, the Company occupied smaller and less expensive office space. In August 2012, the Company moved into a new facility. Its rental costs in fiscal 2013 are expected to increase relative to the temporary space occupied in 2012.Pasadena.
Travel expense was $369,000increased $187,000 from $654,000 during the year ended September 30, 2012, compared2014 to $201,000 in$841,000 during the comparable priorcurrent period. Travel expense increased due to travel associated with the acquisitionin support of Roche Madison Inc.,our R&D function, primarily our GMP manufacturing campaign and our clinical trials, as well as additional travel costsother corporate and business development related to Madison-based employees. During fiscal 2012, the Company hired a Chief Operating Officer and a Chief Business Officer, whose job functions require travel. Also, travel costs are expected to increase in the future due to increased travel between the Madison and Pasadena locations. Travel expense includes costs related to travel by Company personnel for operational business meetings at other company locations, business initiatives and collaborations throughout the world with other companies, marketing, investor relations, fund raising and public relations purposes. Travel expenses can fluctuate from quarter to quarter and from year to year depending on current projects and activities. Business insurance expense was $202,000 during the year ended September 30, 2012, compared to $194,000 in the comparable prior period. The company experienced favorable rate decreases in its Directors and Officers insurance coverage, which was offset by additional insurance costs associated with Madison. Communication and technology expense was $196,000 during the year ended September 30, 2012, compared to $96,000 in the comparable prior period. The increase was related to software maintenance costs at Madison, primarily desk top software and license renewal fees on software related to the operation of laboratory equipment.
Office expenses are administrative costs to facilitate the operations of the Company’s office facilities in Pasadena and Madison, and include office supplies, copier/printing costs, postage/delivery, professional dues/memberships, books/subscriptions, staff amenities, and professional training. Office expenses were $91,000 during the year ended September 30, 2012, compared to $54,000 in the comparable prior period. The increase in office expenses was related to costs incurred at its newly acquired Madison facility.
Other expense was $2.6 million during the year ended September 30, 2012 compared to $95,000 in the comparable prior period. During the year ended September 30, 2012, the Company recorded reserves against receivableincreased $221,000 from its unconsolidated affiliates, Nanotope and Leonardo in the amount of $2.5 million.
Research and Development Expenses—Fiscal 2012 compared to Fiscal 2011
R&D expenses are related to the Company’s on-going research and development efforts, primarily related to its laboratory research facility in Madison, Wisconsin, and also include outsourced R&D services. The following table provides detail of research and development expense for the years ended September 30, 2012 and 2011.
30
(in thousands)
| | | | | | | | | | | | | | | | | | | | | | | | | | | | Twelve months
Ended | | | % of
Expense | | | Twelve months
Ended | | | % of
Expense | | | Increase (Decrease) | | | | | September 30, 2012 | | | Category | | | September 30, 2011 | | | Category | | | $ | | | % | | Outside labs & contract services | | $ | 1,096 | | | | 20 | % | | $ | 605 | | | | 19 | % | | $ | 491 | | | | 81 | % | In vivo studies | | | 302 | | | | 6 | % | | | 29 | | | | 1 | % | | | 273 | | | | 941 | % | Drug Manufacturing | | | 1,256 | | | | 23 | % | | | 68 | | | | 2 | % | | | 1,188 | | | | 1747 | % | Consulting | | | 655 | | | | 12 | % | | | 440 | | | | 13 | % | | | 215 | | | | 49 | % | License, royalty & milestones | | | 274 | | | | 5 | % | | | 2,045 | | | | 63 | % | | | (1,771 | ) | | | -87 | % | Laboratory supplies & services | | | 793 | | | | 15 | % | | | 2 | | | | 0 | % | | | 791 | | | | NM | | Facilities and related | | | 787 | | | | 15 | % | | | 8 | | | | 0 | % | | | 779 | | | | NM | | Sponsored research | | | 185 | | | | 3 | % | | | 75 | | | | 2 | % | | | 110 | | | | 147 | % | Other research expenses | | | 43 | | | | 1 | % | | | 6 | | | | 0 | % | | | 37 | | | | 617 | % | | | | | | | | | | | | | | | | | | | | | | | | | | Total | | $ | 5,391 | | | | 100 | % | | $ | 3,278 | | | | 100 | % | | $ | 2,113 | | | | 64 | % | | | | | | | | | | | | | | | | | | | | | | | | | |
Outside lab and services expense was $1,096,000 during the year ended September 30, 2012, compared to $605,000 in the comparable prior period. The increase is due to outside services contracted to complement the research performed at our Madison facility, which was acquired in October 2012, and not part of the prior period expenses.
In vivo studies expense was $302,000 during the year ended September 30, 2012, compared2014 to 29,000 in$523,000 during the comparable priorcurrent period. The current period expense relatesBusiness insurance costs increased primarily due to preclinical animal studies at our Madison research facility, and we expect this increased level of expense for such studies to continue at an elevated level as the company accelerates its product development efforts. The prior period expenseadded coverage related to certain limited outsourcedthe Company’s clinical trials, as well as increases in vivo studies related to Calando.other corporate liability insurance.
Drug manufacturingCommunication and technology expense was $1,256,000increased $303,000 from $388,000 during the year ended September 30, 2012, compared2014 to $68,000 in$691,000 during the comparable priorcurrent period. Approximately half of the drug manufacturing expenseThe increase was related to raw materials, specifically, polymer components for RONDEL. Prior year costs for this program were $68,000. The other half of the drug manufacturing costs relateequipment purchases to our manufacturing campaignreplace outdated equipment and new equipment purchases related to the Company’s Hepatitis B Virus (HBV) program, which began in the fourth quarter of fiscal 2012, for use in upcoming GLP toxicity studies planned in the first half of fiscal 2013. The Company is utilizing outside manufacturers to produce these components; these costs will continue until the manufacturing campaign is completed in 2013.new employees.
ConsultingOffice expense was $655,000decreased $110,000 from $380,000 during the year ended September 30, 2012, compared2014 to $440,000$270,000 during the current period. These expenses relate to conferences/training, office supplies, miscellaneous administrative expenses, and expenses related to office expansions at our R&D facility in Madison and our corporate headquarters in Pasadena. During fiscal year 2014, the Company incurred certain charges related to its Madison office expansion which was not repeated in the comparable prior period. The increase in consultingcurrent year.
54
Other expense was primarily related to fees paid to our consultants monitoring our clinical trial at Calando, as well as clinical consulting costs for a planned clinical trial in HBV, as well as higher costs associated with the scientific advisory board at Ablaris. License, royalty & milestone expense was $274,000decreased $521,000 from $880,000 during the year ended September 30, 2012, compared2014 to $2,045,000 in the comparable prior period. The licensing fees, royalty and milestones expenses$359,000 during the prior year reflect a one-time to $2 million in licensingcurrent period. This category consists primarily of conference attendance fees, paid to University of Texas M.D. Anderson Cancer Center for the anti-obesity compound licensed by Ablaris.franchise and property tax expenses and marketing expenses. The current year expense also relates to Ablaris anddecrease was payable to the University of Texas M.D. Anderson Cancer Center related to a milestone achieved by dosing its first patientreduction in the provision for franchise taxes as well as an obesity/prostate cancer clinical trial.allowance recorded for certain other receivables during fiscal year 2014 that was not repeated in 2015.
Acquired in-process research and development – Novartis pre-clinical candidates Acquired in-process research and development expense for the Novartis pre-clinical candidates was $10,142,786 for fiscal year 2015 and zero for the previous periods. This expense pertains to the acquisition of the Novartis RNAi assets discussed above. The pre-clinical candidates were expensed during the period, while certain other patents and a third-party license were capitalized as intangible assets. Stock-based compensation expense Stock-based compensation expense, a noncash expense, was $1,241,000increased $4,536,724 from $5,696,173 during the year ended September 30, 2012, compared2014 to $1,376,000$10,232,897 during the comparable priorcurrent period. Stock-based compensation expense is based upon the valuation of stock options and restricted stock units granted to employees, directors, and certain consultants. Many variables affect the amount expensed, including the Company’s stock price on the date of the grant, as well as other assumptions. Based on the completion of vesting of a number ofadditional options and restricted stock options during the second half of fiscal 2011, compensation expense related to those awards ended. This was mostly offset by additional optionsunits granted to new and existing employees in fiscal 2012.2015, compensation expense has increased from the prior year. Depreciation and amortization expense Depreciation and amortization expense, a noncash expense, was $1,749,000increased $990,552 from $1,345,655 during the year ended September 30, 2012, compared2014 to $268,000$2,336,207 during the comparable priorcurrent period. The majority of depreciation and amortization expense relates to depreciation on lab equipment obtained as part of the acquisition of Roche’s RNAi business in 2011. In addition, the Company records depreciation on leasehold improvements at its Madison research facility and its Pasadena corporate headquarters. The increase in depreciation and amortization expense is primarily due to the amortization of the intangible assets acquired in the Novartis asset acquisition. Impairment expense Impairment expense, a noncash expense, decreased $2,172,387 from $2,172,387 during the year ended September 30, 2014 to $0 during the current period. During the year ended September 30, 2014, the Company determined that the carrying value of the Alvos in-process research and development (IPR&D) may not be recoverable and should be fully impaired. During the current period, the Company did not find any of its intangible or long-lived assets to be impaired. Contingent Consideration – Fair Value Adjustments Contingent Consideration – Fair Value Adjustments increased $484,125 from $2,375,658 during the year ended September 30, 2014 to $1,891,533 during the current period. Contingent consideration resulting from the acquisition of Roche’s RNAi business is calculated by modeling research and development activities for clinical candidates, forecasting timelines to market, and using “peak sales” estimate modeling, cash flows and potential milestone and royalty payments. The modeling assumes certain success rates, and discount factors related to riskiness of projects and the time value of money to calculate a net present value of future consideration payments to Roche. Each reporting period, the Company re-evaluates its contingent consideration, and if material, makes adjustments to the recorded liability. The Company increased this liability by $1.9 million and $2.4 million during the years ended September 30, 2015 and 2014, respectively, for a total liability of $5.9 million, which is recorded on the Company’s Consolidated Balance Sheets. Other Income / Expense Other income / expense was expense of $5,443,826 during the year ended September 30, 2014 as compared to income of $4,035,494 during the current period. The largest component of this line item is related to the change in the value of derivative liabilities related to certain warrants with a price adjustment feature, which requires derivative accounting. The change in value of derivative liabilities was a reduction of approximately $2.9 million in 2015 and an increase of approximately $6.0 million in 2014. The fluctuations in each period were primarily driven by changes in the Company’s stock price, which had a corresponding impact to the valuation of the underlying warrants. 55
Results of Operations Comparison for 2014 and 2013 Revenues Total revenue was $175,000 for the year ended September 30, 2014 and $290,000 for the year ended September 30, 2013. Revenue is primarily related to licensed technology. In addition, the Company had collaboration revenue of $115,000 during the year ended September 30, 2013. Operating Expenses The analysis below details the operating expenses and discusses the expenditures of the Company within the major expense categories. Certain reclassifications have been made to prior period operating expense categories to conform to the current period presentation. For purposes of comparison, the amounts for the years ended September 30, 2014 and 2013 are shown in the tables below. Research and Development Expenses R&D expenses are related to the Company’s on-going research and development efforts, primarily related to program costs, composed primarily of outsourced costs related to the manufacturing of clinical supplies, toxicity/efficacy studies and clinical trial expenses. Internal costs primarily relate to operations at our research facility in Madison, Wisconsin, including facility costs and laboratory-related expenses. The following table provides details of research and development expense for the periods indicated: (in thousands) | | Twelve | | | | | | | Twelve | | | | | | | | | | | Months Ended | | | | | | Months Ended | | | | | | | | | | September 30, 2014 | | | % of Total | | | September 30, 2013 | | | % of Total | | | Increase (Decrease) | | Laboratory supplies & services | | $ | 2,071 | | | | 9 | % | | $ | 1,058 | | | | 12 | % | | $ | 1,013 | | | | 96 | % | In vivo studies | | | 224 | | | | 1 | % | | | 232 | | | | 3 | % | | | (8 | ) | | | -3 | % | Outside labs & contract services | | | 691 | | | | 3 | % | | | 533 | | | | 6 | % | | | 158 | | | | 30 | % | Toxicity/efficacy studies | | | 6,306 | | | | 27 | % | | | 1,658 | | | | 19 | % | | | 4,648 | | | | 280 | % | Drug manufacturing | | | 9,142 | | | | 40 | % | | | 2,625 | | | | 30 | % | | | 6,517 | | | | 248 | % | Clinical trials | | | 2,627 | | | | 11 | % | | | 1,306 | | | | 15 | % | | | 1,321 | | | | 101 | % | License, royalty & milestones | | | 1,096 | | | | 5 | % | | | 322 | | | | 4 | % | | | 774 | | | | 240 | % | Facilities and related | | | 881 | | | | 4 | % | | | 732 | | | | 8 | % | | | 149 | | | | 20 | % | Other research expenses | | | 100 | | | | 0 | % | | | 240 | | | | 3 | % | | | (140 | ) | | | -58 | % | Total | | $ | 23,138 | | | | 100 | % | | $ | 8,706 | | | | 100 | % | | $ | 14,432 | | | | 166 | % |
Laboratory supplies and services expense increased $1,013,000 from $1,058,000 during the year ended September 30, 2013 to $2,071,000 during the year ended September 30, 2014. The Company expanded its laboratory facility and increased its R&D headcount during 2014. The increase in laboratory supplies and services is a result of additional supplies necessary to support increased efforts in pre-clinical research as the Company supports ongoing clinical efforts and accelerates efforts to identify new clinical candidates. In vivo studies expense decreased $8,000 from $232,000 during the year ended September 30, 2013 to $224,000 during the year ended September 30, 2014. In vivo expense can vary depending on the stage of preclinical candidates, the nature and amount of testing required and based on the varying costs of different in vivo testing models. The expense in both periods relates to studies related to development of new clinical candidates, including ARC-AAT. Outside labs and contract services expense increased $158,000 from $533,000 during the year ended September 30, 2013 to $691,000 during the year ended September 30, 2014. The increase was primarily related to oligonucleotide synthesis related to development of new clinical candidates. Toxicity/efficacy studies expense increased $4,648,000 from $1,658,000 during the year ended September 30, 2013 to $6,306,000 during the year ended September 30, 2014. This category includes IND-enabling toxicology studies as well as post-clinical toxicology studies, such as long-term toxicology studies, and other efficacy studies. The expense during the year ended September 30, 2014 primarily relates to toxicology studies related to ARC-520, our clinical candidate for HBV, specifically toxicology studies to support our anticipated phase 2b clinical trial. Additionally, during 2014 we began incurring costs related to IND-enabling toxicology studies related to ARC-AAT. 56
Drug manufacturing expense increased $6,517,000 from $2,625,000 during the year ended September 30, 2013 to $9,142,000 during the year ended September 30, 2014. The expense during the year ended September 30, 2014 relates to drug manufacturing to supply toxicology studies for our HBV Phase 2b clinical trial, clinical supplies for the HBV Phase 2b clinical trial, as well as clinical supplies for our clinical trial for ARC-AAT. Clinical trials expense increased $1,321,000 from $1,306,000 during the year ended September 30, 2013 to $2,627,000 during the year ended September 30, 2014. Clinical trial expenses increased as the Company advanced ARC-520 into its Phase 1 and Phase 2a clinical trial during 2014. License, royalty and milestones expense increased $774,000 from $322,000 during year ended September 30, 2013 to $1,096,000 during the year ended September 30, 2014. The increase during the year ended September 30, 2014 relates to a $1 million milestone payment due pursuant to a license agreement based upon a milestone reached on one of its clinical candidates. Milestone payments can vary from period to period depending on the nature of our various license agreements, and the timing of reaching various development milestones requiring payment. Facilities expense increased $149,000 from $732,000 during the year ended September 30, 2013 to $881,000 during the year ended September 30, 2014. Facilities expenses were higher during the year ended September 30, 2014 primarily due to repairs and maintenance costs on lab equipment. Other research expense decreased $140,000 from $240,000 during the year ended September 30, 2013 to $100,000 during the year ended September 30, 2014. Other research expense in the prior period relates to work at the University of Cincinnati related to our obesity program, which studies have been completed, and no further studies are planned. The expenses during the year ended September 30, 2014 primarily relate to medical and hazardous waste removal costs. Salary and Payroll-Related Expenses The following table provides detail of salary and payroll-related expenses for the periods indicated: (in thousands) | | Twelve months | | | | | | Twelve months | | | | | | | | | | | Ended | | | | | | Ended | | | | | | | | | | | September 30, 2014 | | | % of Total | | | September 30, 2013 | | | % of Total | | | Increase (Decrease) | | | R&D - compensation-related | | $ | 7,828 | | | | 61 | % | | $ | 4,127 | | | | 62 | % | | $ | 3,701 | | | | 90 | % | | G&A - compensation-related | | | 5,001 | | | | 39 | % | | | 2,541 | | | | 38 | % | | | 2,460 | | | | 97 | % | | Total | | $ | 12,829 | | | | 100 | % | | $ | 6,668 | | | | 100 | % | | $ | 6,161 | | | | 92 | % | |
R&D compensation expense increased $3,701,000 from $4,127,000 during the year ended September 30, 2013 to $7,828,000 during the year ended September 30, 2014. Increased headcount and salary increases accounted for the majority of the change in compensation-related expense. Additionally, expenses related to annual performance bonuses were accrued during the year ended September 30, 2014 totaling $1,602,000 in expense; no performance bonuses were paid in the prior period. G&A compensation expense increased $2,460,000 from $2,541,000 during the year ended September 30, 2013 to $5,001,000 during the year ended September 30, 2014. The majority of this change relates to expense recorded for annual performance bonuses in the year ended September 30, 2014. Additionally, a portion of the increase is due to salary increases. G&A headcount remained fairly consistent during the past twelve months. 57
General & Administrative Expenses The following table provides details of our general and administrative expenses for the periods indicated: (in thousands) | | Twelve | | | | | | | Twelve | | | | | | | | | | | Months Ended | | | | | | Months Ended | | | | | | | | | | September 30, 2014 | | | % of Total | | | September 30, 2013 | | | % of Total | | | Increase (Decrease) | | Professional/outside services | | $ | 2,465 | | | | 42 | % | | $ | 1,319 | | | | 38 | % | | $ | 1,146 | | | | 87 | % | Patent expense | | | 632 | | | | 11 | % | | | 941 | | | | 27 | % | | | (309 | ) | | | -33 | % | Facilities and related | | | 193 | | | | 3 | % | | | 170 | | | | 5 | % | | | 23 | | | | 14 | % | Travel | | | 654 | | | | 11 | % | | | 440 | | | | 13 | % | | | 214 | | | | 49 | % | Business insurance | | | 302 | | | | 5 | % | | | 216 | | | | 6 | % | | | 86 | | | | 40 | % | Communication and Technology | | | 388 | | | | 7 | % | | | 184 | | | | 5 | % | | | 204 | | | | 111 | % | Office expenses | | | 380 | | | | 6 | % | | | 110 | | | | 3 | % | | | 270 | | | | 245 | % | Other | | | 880 | | | | 15 | % | | | 109 | | | | 3 | % | | | 771 | | | | 707 | % | Total | | $ | 5,894 | | | | 100 | % | | $ | 3,489 | | | | 100 | % | | $ | 2,406 | | | | 69 | % |
Professional/outside services include legal, accounting, consulting and other outside services retained by the Company. All periods include normally recurring legal and audit expenses related to SEC compliance and other corporate matters. Professional/outside services expense increased $1,146,000 from $1,319,000 during the year ended September 30, 2013 to $2,465,000 during the year ended September 30, 2014. The increase in professional fees primarily related to professional recruiting fees for the hiring of additional R&D personnel to support and expand its clinical pipeline. Additionally, the Company incurred higher SEC filing fees associated with financing in February 2014 and higher NASDAQ fees based on a higher number of shares outstanding. Patent expense decreased $309,000 from $941,000 during the year ended September 30, 2013 to $632,000 during the year ended September 30, 2014. Patent expenses related to Calando declined by $215,000 in the current period as Calando terminated its license agreement with Caltech in August 2013, which had obligated Calando to pay certain related patent costs, and by curtailing prosecution of other non-strategic patents. During the year ended September 30, 2014, Arrowhead deconsolidated Calando, thus no further Calando patent expense will be incurred. Facilities-related expense increased $23,000 from $170,000 during the year ended September 30, 2013 to $193,000 in the year ended September 30, 2014. Facilities expense increased due to the expansion of our corporate headquarters in Pasadena as well as routine increases in ancillary lease charges. Travel expense increased $214,000 from $440,000 during the year ended September 30, 2013 to $654,000 during the year ended September 30, 2014. Travel expense increased due to travel in support of our R&D function, primarily our GMP manufacturing campaign and our clinical trials. Business insurance expense increased $86,000 from $216,000 during the year ended September 30, 2013 to $302,000 during the year ended September 30, 2014. Business insurance costs increased primarily related to added coverage related to the Company’s clinical trials. Communication and technology expense increased $204,000 from $184,000 during the year ended September 30, 2013 to $388,000 during the year ended September 30, 2014. The increase was related to equipment purchases to replace outdated equipment and to cost associated with new equipment related to new employees. Office expense increased $270,000 from $110,000 during the year ended September 30, 2013 to $380,000 during the year ended September 30, 2014. The increase was related to conferences/training, office supplies, miscellaneous administrative expenses, and expenses related to an office expansions at our R&D facility in Madison and our corporate headquarters in Pasadena. Other expense increased $771,000 from $109,000 during the year ended September 30, 2013 to $880,000 during the year ended September 30, 2014. The increase was related to an increase in estimated franchise taxes accrued as well as an allowance recorded for certain other receivables. 58
Stock-based compensation expense Stock-based compensation expense, a noncash expense, increased $4,160,000 from $1,536,000 during the year ended September 30, 2013 to $5,696,000 during the year ended September 30, 2014. Based on the additional options and restricted stock units granted to new and existing employees in fiscal 2014, compensation expense increased from the prior year. Depreciation and amortization expense Depreciation and amortization expense, a noncash expense, decreased $405,000 from $1,751,000 during the year ended September 30, 2013, to $1,346,000 during the year ended September 30, 2014. The majority of depreciation and amortization expense relates to depreciation on lab equipment obtained as part of the acquisition of the Roche Madison.RNAi business n in 2011. In addition, the Company records depreciation on leasehold improvements at its Madison research facility. The Madison facility was acquiredAmortization expense decreased in October 2011; therefore,2014 as compared to 2013 as capitalized patents related to Calando were fully written off in 2013, accordingly there was no related depreciationexpense recorded in 2014. Impairment expense Impairment expense, a noncash expense, increased $864,000 from $1,308,000 during the prior year. Finally, certain patents acquired previously have been capitalized and amortized overyear ended September 30, 2013 to $2,172,000 during the remaining useful livesyear ended September 30, 2014. During the year ended September 30, 2014, the Company determined that the carrying value of the respective patents. Alvos IPR&D may not be recoverable and should be fully impaired. The Company recorded an impairment charge of $2.2 million. During the year ended September 30, 2013, management determined that the value of the Calando patents was impaired, and the Company recorded a $1.3 million impairment charge.Contingent Consideration – Fair Value Adjustments 31Contingent Consideration – Fair Value Adjustments increased $954,000 from $1,422,000 during the year ended September 30, 2013 to $2,376,000 during the year ended September 30, 2014. Contingent consideration resulting from the acquisition of Roche’s RNAi business is calculated by modeling research and development activities for clinical candidates, forecasting timelines to market, and using “peak sales” estimate modeling, cash flows and potential milestone and royalty payments. The modeling assumes certain success rates, and discount factors related to riskiness of projects and the time value of money to calculate a net present value of future consideration payments to Roche. Each reporting period, the Company re-evaluates its contingent consideration, and if material, makes adjustments to the recorded liability. The Company increased this liability by $2.5 million and $1.4 million during the years ended September 30, 2014 and 2013, respectively, for a total liability of $4.0 million, which is recorded on the Company’s Consolidated Balance Sheets.
Other Income / Expense Other income / expense changeddecreased $1,670,000 from income of $1,045,000 in fiscal 2011 to other expense of $1,021,000$7,114,000 during the year ended September 30, 2013 to expense of $5,444,000 during the year ended September 30, 2014. The decrease in fiscal 2012. During fiscal 2012,expense is primarily due to a $1.0 million write down of a note receivable related to the sale of our former subsidiary, Unidym, in 2013, which was not repeated in the current year. Additionally, the Company recorded several nonrecurring items: Impairment ofreceived increased interest income during 2014 as it has invested its investmentexcess cash balances in its unconsolidated affiliate, Nanotope of $1.4 million, a loss on the disposal of fixed assets of $1.1 million,short and a gain recorded upon the acquisition of Roche Madison of $1.6 million, and an impairment of its investment in Leonardo of $0.2 million. Otherlong-term corporate bonds. The largest component of other income/expense wasthis line item is related to the change in the value of derivative liabilities related to certain warrants with a price adjustment feature, which requires derivative accounting. The change in value of derivatives, whichderivative liabilities was $387,000 in fiscal 2012, compared to $1.1approximately $6.0 million in fiscal 2011.2014 and $5.3 million in 2013. Liquidity and Cash Resources As a development stage company, Arrowhead has historically financed its operations through the sale of securities of Arrowhead and its Subsidiaries. Research and development activities have required significant capital investment since the Company’s inception, and are expected to continue to require significant cash investmentexpenditure in fiscal 2012.year 2016, and beyond.
At September 30, 2012,2015, the Company had cash on hand of approximately $3.4 million. In addition, the Company had subscriptions receivable from previous financings of $1.0$81.2 million and a short term note receivable of approximately $2.4 million. Cash and cash equivalents decreased $4.1 million during fiscal 2012 from $7.5as compared to $132.5 million at September 30, 2011 to $3.42014. Excess cash invested in fixed income securities was $17.5 million at September 30, 2012.2015, compared to $44.7 million at September 30, 2014. The Company believes its current financial resources are sufficient to fund its operations through at least the next twelve months. Cash
59
A summary of cash flows for the years ended September 30, 2015, 2014, and 2013 is as follows: | | Year ended September 30, | | | | 2015 | | | 2014 | | | 2013 | | Cash Flow from Continuing Operations: | | | | | | | | | | | | | Operating Activities | | $ | (65,707,615 | ) | | $ | (35,416,373 | ) | | $ | (19,032,826 | ) | Investing Activities | | | 14,120,838 | | | | (36,481,770 | ) | | | (9,520,867 | ) | Financing Activities | | | 290,521 | | | | 185,294,309 | | | | 44,291,203 | | Cash Flow from Discontinued Operations: | | | | | | | | | | | | | Operating Activities | | | - | | | | - | | | | (354 | ) | Investing Activities | | | - | | | | - | | | | - | | Financing Activities | | | - | | | | - | | | | - | | Net increase (decrease) in cash | | | (51,296,256 | ) | | | 113,396,166 | | | | 15,737,156 | | Cash at beginning of period | | | 132,510,610 | | | | 19,114,444 | | | | 3,377,288 | | Cash at end of period | | $ | 81,214,354 | | | $ | 132,510,610 | | | $ | 19,114,444 | |
During the year ended September 30, 2015, the Company used $65.7 million in cash from operating activities, was $16.0 million, which represents the on-going expenses of its research and development programs and corporate overhead. Cash outlays were primarily composed of the following: research and development costs were $41.2 million, salary and payroll-related costs was $6.5expenses were $16.6 million, and general and administrative costs were $4.0 million, research and development costs were $4.8$7.9 million. $0.9 million was used to fund operating expenses at Arrowhead’s two minority interest companies, Nanotope and Leonardo. Cash expenses were somewhat offset by cash received from revenues of $0.2 million. Cash provided by investing activities was $0.4$14.1 million, primarily related to cash received from the salematurity of investments of $0.5 million, proceeds from the disposal of fixed assets of $0.3 million,certain marketable securities partially offset by capital$10.0 million of cash used to acquire the Novartis assets discussed above. Capital expenditures of $0.5were $2.0 million. Cash provided by financing activities of $10.8$0.3 million primarily includes the exercise of warrants and stock options during the year ended September 30, 2015. During the year ended September 30, 2014, the Company used $35.4 million in cash from operating activities, which represents the on-going expenses of its research and development programs and corporate overhead. Cash outlays were primarily composed of the following: research and development costs were $21.7 million, salary and payroll-related expenses were $9.1 million, and general and administrative costs were $4.6 million. Cash used by investing activities was $36.5 million, primarily related to net cash investments in fixed income securities of $34.8 million. Capital expenditures were $1.7 million. Cash provided by financing activities of $185.3 million includes $11.0$172.6 million received related toof cash received from equity financings by the saleCompany in October 2013 and February 2014. The exercise of Common Stock, offset by principal payments on capital leaseswarrants and stock options during fiscal 2014 resulted in additional cash inflow of $0.2$12.9 million. These matters raise substantial doubt aboutDuring the Company’s ability to continue as a going concern. These financial statements were prepared under the assumption thatyear ended September 30, 2013, the Company used $19.0 million in cash from operating activities, which represents the on-going expenses of its research and development programs and corporate overhead. Cash outlays were primarily composed of the following: research and development costs were $8.7 million, salary and payroll-related expenses were $6.7 million, and general and administrative costs were $3.5 million. Cash used by investing activities was $9.5 million, primarily related to net cash investments in fixed income securities of $9.3 million. Capital expenditures were $0.3 million. Cash provided by financing activities of $44.3 million includes $42.4 million of cash received from equity financings by the Company during 2013. The exercise of warrants and stock options during fiscal 2013 resulted in additional cash inflow of $2.1 million.
Contractual Obligations In the table below, we set forth our enforceable and legally binding obligations and future commitments at September 30, 2015 for the categories shown, as well as obligations related to contracts in such categories that we are likely to continue. Some of the figures that we include in this table are based on management’s estimates and assumptions about these obligations, including their duration, the possibility of renewal, anticipated actions by third parties and other factors. Because these estimates and assumptions are necessarily subjective, the obligations we will continue as a going concern and doactually pay in future periods may vary from those reflected in the table. The following table does not include any adjustments that might result from the outcome of that uncertainty. Recent Financing Activity / Sources of Capital:
In December 2012, the Company sold 1.9 million units at a price of $2.26 per unit in a public offering. Each unit consisted of one share of Common Stock and a warrant to purchase 0.5 share of Common Stock, exercisable at $2.20. Gross proceeds from the offering were $4.3 million, which included a $500,000 promissory note due February 1, 2013. Commissions and other offering expenses are expected to be approximately $300,000.
On August 10, 2012, the Company sold 2.3 million units at a price of $2.76 per unit in a registered offering to institutional and individual investors. Each unit consisted of one share of Common Stock and a warrant to purchase 0.75 share of Common Stock exercisable at $3.25 per share. Gross proceeds from the offering were approximately $6.2 million, with net proceeds of approximately $5.8 million after deducting commissions and other offering expenses.
On September 30, 2011, the Company sold 1,458,917 shares of Common Stock at a price of $3.80 per share. Cash proceeds received in fiscal 2011 were $4.6 million, cash proceeds in the first six months of fiscal 2012 were $0.4 million, and the balance is expected to be received in 2012. On October 4, 2011, the Company completed a second closing to the offering in which the Company sold 138,158 shares of Common Stock at a price of $3.80 per share. Cash proceeds were $525,000.
On October 20, 2011, the Company and Lincoln Park Capital Fund, LLC, an Illinois limited liability company (“LPC”) entered into a $15 million purchase agreement, together with a registration rights agreement, whereby LPC agreed to purchase up to $15 million of Common Stock, subject to certain limitations, from time to time during the three-year term of the agreement. Additionally, the Company filed a registration statement with the U.S. Securities & Exchange Commission covering the resale of the sharesfuture obligations that may be issued to LPCowed under the agreement. On January 30, 2012, the SEC declared the registration statement effective for the resale of such shares. The Company has the right, in its sole discretion, over a 36-month period to sell up to $15 million of Common Stock (subject to certain limitations) to LPC, depending on certain conditions as set forth in the agreement. As of September 30, 2012, the Company had drawn $1 million from the facility.
Although the Company has sources of liquidity, as described above, the Company anticipates that further equity financings, and/or asset sales andexisting license agreements, will be necessary to continue to fund operations inas the future.
certainty of achieving the relevant milestones that would trigger these payments is currently unknown. | | Payments due by Period | | | | Total | | | Less than 1 Year | | | 1-3 Years | | | 3-5 Years | | | More than 5 Years | | Debt | | | - | | | | - | | | | - | | | | - | | | | - | | Capital Leases | | | 758,340 | | | | 217,548 | | | | 446,007 | | | | 94,785 | | | | - | | Operating Leases | | | 2,343,075 | | | | 631,881 | | | | 1,251,561 | | | | 459,633 | | | | - | | Purchase Obligations | | | 49,300,000 | | | | 35,500,000 | | | | 13,800,000 | | | | - | | | | - | | Other Long-Term Liabilities | | | 6,204,917 | | | | - | | | | - | | | | 342,453 | | | | 5,862,464 | | Total | | $ | 58,606,332 | | | $ | 36,349,429 | | | $ | 15,497,568 | | | $ | 896,871 | | | $ | 5,862,464 | |
32
Off-Balance Sheet Arrangements As of September 30, 2012,2015, we did not have any off-balance sheet arrangements, as defined in Item 303(a)(4)(ii) of SEC Regulation S-K. Recent Accounting Pronouncements See Note 1 to our Consolidated Financial Statements of this annual report on Form 10-K for a description of recent accounting pronouncements applicable to our business.
ITEM 7A. | QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK |
We are exposed to market risk related to changes in interest rates, which could adversely affect the value of our interest rate-sensitive assets and liabilities. We do not hold any instruments for trading purposes and investment criteria are governed by the Company’s Investment Policy. As of September 30, 2015 and 2014, we had cash and cash equivalents of $81.2 million and $132.5 million, respectively. We invest our cash reserves in corporate bonds typically with maturities of less than 2 years, and we classify these investments as held-to-maturity. Due to the relatively short-term nature of the investments that we hold, we do not believe that the results of operations or cash flows would be affected to any significant degree by a “Smaller Reporting Company,” we aresudden change in market interest rates relative to our investment portfolio. Our liability instrument sensitive to changes in interest rates is our derivative liability with its fair value determined using an option pricing model, which uses interest rate as an input. However, any change associated with this valuation would result in a noncash expense and would not required to provide this information.significantly impact our operations. ITEM 8. | FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA |
The information required by this item is included in Item 15 of this Annual Report Form 10-K. ITEM 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE. |
None. ITEM 9A. | CONTROLS AND PROCEDURES. |
Our Chief Executive Officer and our Chief Financial Officer, after evaluating our “disclosure controls and procedures” (as defined in Securities Exchange Act of 1934 (the “Exchange Act”) Rules 13a-15(e) and 15d-15(e)) as of the end of the period covered by this Annual Report on Form 10-K (the “Evaluation Date”) have concluded that as of the Evaluation Date, our disclosure controls and procedures are effective to ensure that information we are required to disclose in reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in Securities and Exchange Commission rules and forms, and to ensure that information required to be disclosed by us in such reports is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, where appropriate, to allow timely decisions regarding required disclosure. No change in the Company’s internal controls over financial reporting (as defined in Rule 13a-15(f) and 15d-15(f) of the Exchange Act) occurred during the Company’s most recent fiscal quarter that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting. Management’s Annual Report on Internal Control over Financial Reporting Internal Control over Financial Reporting Our management is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act. Our internal control over financial reporting is designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles in the United States. This process includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of our assets; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures are being made only in accordance with authorizations of our management and directors; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on our financial statements. Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of the internal control over financial reporting to future periods are subject to risk that the internal control may become inadequate because of changes in conditions, or that the degree of compliance with policies or procedures may deteriorate. Management’s Assessment of the Effectiveness of our Internal Control over Financial Reporting Management has evaluated the effectiveness of ourThe Company’s management is responsible for establishing and maintaining adequate internal control over financial reporting as(as defined in Rule 13a-15(f) under the Exchange Act). Management conducted an assessment of September 30, 2012. In conducting its evaluation, management used the frameworkeffectiveness of the Company’s internal control over financial reporting based on the criteria set forth in Internal Control—Control – Integrated Framework issued by the
62
Committee of Sponsoring Organizations of the Treadway Commission (COSO)(2013 framework). Based on our evaluation under such framework,the Company’s assessment, management has concluded that ourits internal control over financial reporting was effective as of September 30, 2012.2015 to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements in accordance with GAAP. The Company’s independent registered public accounting firm, Rose, Snyder and Jacobs LLP, has issued an audit report on the Company’s internal control over financial reporting, which appears in Item 15 of this Form 10-K. Changes in Internal Control Over Financial Reporting There was no change in our internal control over financial reporting that occurred during the fourth quarter of the year ended September 30, 2012,2015 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting. ITEM 9B. | OTHER INFORMATION |
None
None. 33
PART III ITEM 10. | DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE. |
BoardThe information called for by this Item will be incorporated by reference from our Definitive Proxy Statement to be filed for our 2016 Annual Meeting of Directors:
The names and ages of our directors serving as of December 14, 2012 are provided below. Directors are elected annually for a one year term. Biographical information regarding these officers is set forth under the following table.Stockholders, which proxy statement will be filed no later than January 28, 2016.
| | | | | | | | | | ITEM 11. | NameEXECUTIVE COMPENSATION
| | Age
| | Position with Arrowhead
| | | | | Christopher Anzalone
| | 43 | | Chief Executive Officer & President and Director | | | | | | | | | | Douglass Given
| | 60 | | Chairman of the Board | | | | | | | | | | Mauro Ferrari
| | 53 | | Director | | | | | | | | | | Edward W. Frykman
| | 76 | | Director | | | | | | | | | | Charles P. McKenney
| | 74 | | Director | | | | | | | | | | Michael S. Perry
| | 53 | | Director | | |
Dr. Christopher Anzalone has been President, Chief Executive Officer and DirectorThe information called for by this Item will be incorporated by reference from our Definitive Proxy Statement to be filed for our 2016 Annual Meeting of the Company since December 1, 2007. In 2005, Dr. Anzalone formed and served as CEO of the Benet Group LLC, private equity firm focused on creating and building new nano-biotechnology companies from university-generated science. While at The Benet Group, Dr. Anzalone was founding CEO in two portfolio companies, Nanotope Inc., a tissue regeneration company, and Leonardo Biosystems Inc., a cancer drug delivery company. Dr. Anzalone remains CEO and director of Nanotope. Dr. Anzalone is a director of Arrowhead’s wholly-owned subsidiary, Arrowhead Madison Inc., majority-owned subsidiaries, Calando Pharmaceuticals, Inc., Ablaris Therapeutics, Inc., and Tego Biosciences Corporation and minority investment, Leonardo Biosystems, Inc. Prior to his tenure at Benet Group, from 1999 until 2003, he was a partner at the Washington, DC-based private equity firm Galway Partners, LLC, where he was in charge of sourcing, structuring, and building new business ventures and was founding CEO of NanoInk, Inc., a leading nanolithography company. Dr. Anzalone holds a Ph.D. in Biology from UCLA and a B.A. in Government from Lawrence University. We believe Dr. Anzalone’s qualifications to serve on the Board include his deep understanding of the business through his role as Chief Executive Officer; in addition Dr. Anzalone has extensive experience in nanotechnology, biotechnology, company-building and venture capital.
Dr. Mauro Ferrariwas appointed to the Arrowhead Board of Directors in 2010. Dr. Ferrari is the President and CEO of The Methodist Hospital Research Institute (TMHRI). He is also the President of The Alliance for NanoHealth. Dr. Ferrari is a director of Arrowhead’s minority investment, Leonardo Biosystems, Inc. Dr. Ferrari is an internationally recognized expert in nanomedicine and biomedical nanotechnology. Prior to assuming leadership of TMHRI, Dr. Ferrari was Professor and Chairman of The Department of NanoMedicine and Biomedical Engineering at The University of Texas Health Science Center at Houston, Professor of Experimental Therapeutics at the MD Anderson Cancer Center, Adjunct Professor of Bioengineering at Rice University, and Adjoint Professor of Biomedical Engineering at the University of Texas in Austin. His previous academic appointments include professorships at UC Berkeley and Ohio State University.
From 2003 to 2005, he served as Special Expert on Nanotechnology and Eminent Scholar at The National Cancer Institute, where he led in the development of the NCI’s program in Nanotechnology,Stockholders, which remains the largest program in NanoMedicine in the world. Dr. Ferrari has been serving as the Editor-in-Chief for “Biomedical Microdevices: BioMEMS and Biomedical Nanotechnology” since 1997. We believe Dr. Ferrari’s qualifications to serve on the Board include his extensive training and experience in the fields of nanotechnology, biotechnology and biomedical applications. Dr. Ferrari has significant technical training, several academic appointments and numerous published articles and patents. Additionally, Dr. Ferrari has extensive experience in developmental stage organizations having founded several startup companies.
Edward W. Frykman has been a director of the Company sinceproxy statement will be filed no later than January 2004. Mr. Frykman was an Account Executive with Crowell, Weedon & Co., a position he held from 1992 until 2008 when he retired. Before his service at Crowell, Weedon & Co., Mr. Frykman served as Senior Vice President of L.H. Friend & Co. Both Crowell Weedon & Co. and L.H. Friend & Co. are investment brokerage firms located in Southern California. In addition, Mr. Frykman was a Senior Account Executive with Shearson Lehman Hutton, where he served as the Manager of the Los Angeles Regional Retail Office of E. F. Hutton & Co. Mr. Frykman was a director in Arrowhead’s predecessor company since its inception in May 2003 until January 2004, when he became a director of the Company. Mr. Frykman is also a director of Acacia Research Corporation, a publicly-held corporation based in Newport Beach, California. Mr. Frykman is a director of Arrowhead’s majority-owned subsidiaries Calando Pharmaceuticals, Inc., Ablaris Therapeutics, Inc., and Tego Biosciences Corporation. We believe Mr. Frykman’s qualifications to serve on the Board include his long tenure as a member of the Board which enabled Mr. Frykman to gain a deep understanding of the company’s operations, strategy and finances. Mr. Frykman also has extensive experience in the fields of finance and public company oversight.
28, 2016. 34
Dr. Douglass Givenhas been a director of the company since November 2010. He is an Investment Partner at Bay City Capital and has been with the firm since October 2000. He was formerly Chief Executive Officer and a director of NeoRx, Corporate Sr. Vice President and Chief Technical Officer of Mallinckrodt, and Chief Executive Officer and a director of Progenitor and Mercator Genetics. He held positions as Vice President at Schering Plough Research Institute, Vice President at Monsanto/G.D. Searle Research Laboratories, and Medical Advisor at Lilly Research Laboratories. Dr. Given is the Chairman of VIA Pharmaceuticals, and Chairman of Vivaldi Biosciences. He is Chairman of the Visiting Committee to the Division of Biological Sciences and the Pritzker School of Medicine at the University of Chicago, a member of the Johns Hopkins Bloomberg School of Public Health Advisory Board, and a member of the Harvard School of Public Health AIDS Initiative International Advisory Council.
Dr. Given holds an MD with honors and a PhD from the University of Chicago, and an MBA from the Wharton School, University of Pennsylvania. He was a fellow in Internal Medicine and Infectious Diseases at Harvard Medical School and Massachusetts General Hospital. We believe Dr. Given’s qualifications to serve on the Board include his extensive experience in finance and business transactions, particularly investments in the life sciences industry as well as directorship roles in start-up biotechnology companies. Dr. Given also has significant leadership roles, including CEO and Senior Vice President, at several large pharmaceutical companies. Dr. Douglass Given is a brother of Dr. Bruce Given, our chief operating officer.
Charles P. McKenneyhas been a director of the Company since April 2004. Mr. McKenney has maintained a government affairs law practice in Pasadena, California since 1989, representing businesses and organizations in their relations with state and local government regarding their obligations under state and local land use and trade practices laws. From 1973 through 1989, he served as Attorney for Corporate Government Affairs for Sears, Roebuck and Co., helping organize and carry out Sears’s western state and local government relations programs. Mr. McKenney has served two terms on the Pasadena, California, City Council as well as on several city boards and committees, including three city Charter Reform Task Forces. Mr. McKenney is a director of Arrowhead’s majority-owned subsidiaries Calando Pharmaceuticals, Inc., Ablaris Therapeutics, Inc., and Tego Biosciences Corporation. We believe Mr. McKenney’s qualifications to serve on the Board include his long tenure as a member of the Board resulting in a deep understanding of the Company’s operations, strategy and finances. Mr. McKenney also has extensive experience providing strategic legal and advisory services to developmental stage organizations.
Dr. Michael S. Perry joined Arrowhead’s Board of Directors in December 2011. Dr. Perry is Global Head, Cell Therapy at Novartis Pharma. Prior to his appointment at Novartis, Dr. Perry was a Venture Partner with Bay City Capital LLP from 2005 until November 2012 and President and Chief Medical Officer of Poniard Pharmaceuticals from 2010 to November 2012. He also currently serves as a member of the board of directors of AmpliPhi Biosciences Corporation (APHB.PK). He was Chief Development Officer at VIA Pharmaceuticals, Inc., a publicly held drug development company, from April 2005 until May 2009. Prior thereto, he served as Chairman and Chief Executive Officer of Extropy Pharmaceuticals, Inc., a privately held pediatric specialty pharmaceutical company, from June 2003 to April 2005. From 2002 to 2003, Dr. Perry served as President and Chief Executive Officer of Pharsight Corporation, a publicly held software and consulting services firm. From 2000 to 2002, Dr. Perry served as Global Head of Research and Development for Baxter BioScience. From 1997 to 2000, Dr. Perry was President and Chief Executive Officer of both SyStemix Inc. and Genetic Therapy Inc., two wholly owned subsidiaries of Novartis Corp. and from 1994 to 1997, he was Vice President of Regulatory Affairs for Novartis Pharma (previously Sandoz Pharmaceuticals). Prior to 1994, Dr. Perry held various management positions with Syntex Corporation, Schering-Plough Corporation and BioResearch Laboratories, Inc. Dr. Perry holds a Doctor of Veterinary Medicine, a Ph.D. in Biomedical Pharmacology and a B.Sc. in Physics from the University of Guelph, Ontario, Canada. He is a graduate of the International Management Program at Harvard Business School. We believe Dr. Perry’s qualifications to serve on the board include his medical expertise and his extensive experience in preclinical and clinical drug development, including executive level leadership roles in several publicly held biotech companies.
Executive Officers:
The names and ages of our executive officers and the positions held by each as of December 19, 2012 are as follows:
| | | | | | | | | | ITEM 12. | Name
| | Age
| | Position with Arrowhead
| | | | | Christopher Anzalone
| | 43 | | Chief Executive Officer & President and Director | | | | | Kenneth A. Myszkowski
| | 46 | | Chief Financial Officer | | | | | Bruce Given
| | 58 | | Chief Operating Officer | | |
Dr. Christopher Anzalone(see Board of Directors)
Kenneth A. Myszkowski, Chief Financial Officer, joined Arrowhead in 2009. Prior to joining Arrowhead, Mr. Myszkowski served as the corporate controller for Broadwind Energy, a public energy company which provides products and services to the wind energy industry. Previous to his position at Broadwind, Mr. Myszkowski was controller for Epcor USA, the U.S. headquarters for Epcor Utilities, Inc., a public energy company. Prior to Epcor, Mr. Myszkowski was controller for two start-up ventures: NanoInk, specializing in Dip Pen Nanolithography, a nanofabrication technology, and Delphion, which provided on-line tools for intellectual
35
property research. Mr. Myszkowski also held several corporate roles at FMC Corporation, and Premark International, both Fortune 500 conglomerates. He began his career in the audit practice of Arthur Andersen & Co. in Chicago, Illinois. Mr. Myszkowski received his undergraduate degree from the University of Illinois, and his MBA from the University of Chicago Booth School of Business. He is a certified public accountant.
Dr. Bruce Given, Chief Operating Officer, joined Arrowhead in 2011. Since October 1, 2009, Dr. Given has been a director of the Company’s subsidiary, Calando Pharmaceuticals, Inc., and since February 1, 2010, Dr. Given has been Chief Executive Officer of Leonardo Biosystems, Inc., a company in which Arrowhead holds a minority equity interest. Dr. Given has been a member of the Board of Directors for ICON, plc. since 2007 and Chairman of the Board of Directors since 2010. Dr. Given served as the President and Chief Executive Officer, and as a member of the Board of Directors of Encysive Pharmaceuticals, an R&D-based commercial pharmaceutical company, roles he held from 2002 through 2007. Subsequent to his tenure at Encysive until present, Dr. Given has been President of Bruce Given Consulting, a firm that provides consulting services to biotech companies. Prior to his tenure at Encysive, Dr. Given held several senior executive roles at Johnson and Johnson, Sandoz Pharmaceuticals, and Schering-Plough. Dr. Given obtained his bachelor of sciences degree from Colorado State University, graduating Phi Beta Kappa. He received his M.D. degree with honors from the University of Chicago, Pritzker School of Medicine and completed his medical training at the University of Chicago and at Brigham and Women’s Hospital in Boston, where he was a Clinical Fellow at Harvard Medical School. He is board certified in internal medicine and endocrinology and metabolism and has authored 33 scientific publications. Dr. Bruce Given is a brother of Dr. Douglass Given, a director of the company.
Section 16(a) Beneficial Ownership Reporting Compliance
Under Section 16(a) of the Securities Exchange Act of 1934, the Company’s directors and officers and its significant stockholders (defined by statute as stockholders beneficially owning more than ten percent (10%) of the Common Stock) are required to file with the SEC and the Company reports of ownership, and changes in ownership, of common stock. Based solely on a review of the reports received by it, the Company believes that, during the fiscal year ended September 30, 2012, all of its officers, directors and significant stockholders complied with all applicable filing requirements under Section 16(a).
Code of Ethics
We have adopted a code of conduct that applies to our Chief Executive Officer, Chief Financial Officer, and to all of our other officers, directors and employees. The code of conduct is available at the Corporate Governance section of the Investor Relations page on our website at www.arrowheadresearch.com. Any waivers from or amendments to the code of conduct, if any, will be posted on our website.
Corporate Governance
The Audit Committee of the Board is currently comprised of three directors and operates under a written charter adopted by the Board. The members of the Audit Committee are Edward W. Frykman, Charles P. McKenney and Mike Perry. All members of the Audit Committee are “independent,” as defined in Rule 10A-3 under the Exchange Act and Rule 5605 of the NASDAQ Marketplace Rules, and financially literate. The Board has determined that Mr. Frykman is an “audit committee financial expert” in accordance with the applicable regulations.
ITEM 11. | EXECUTIVE COMPENSATION. |
Executive Officers
Summary Compensation Table
The following table summarizes compensation paid, awarded or earned for services rendered during fiscal 2012 and fiscal 2011 by our Chief Executive Officer, our Chief Financial Officer, and our other executive officer serving the Company as of September 30, 2012. We refer to those persons collectively as our “Named Executive Officers”.
36
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | Name and Principal Position | | Year | | | Salary
($) | | | Bonus
($) | | | Stock
Awards
($) | | | Option
Awards
($)(1) | | | All Other
Compensation
($)(2) | | | Total
($) | | Christopher Anzalone | | | | | | | | | | | | | | | | | | | | | | | | | | | | | President & Chief | | | 2012 | | | | 473,000 | | | | 200,000 | | | | — | | | | 811,685 | | | | — | | | | 1,484,685 | | Executive Officer | | | 2011 | | | | 400,000 | | | | 25,000 | | | | — | | | | — | | | | — | | | | 425,000 | | | | | | | | | | Ken Myszkowski | | | | | | | | | | | | | | | | | | | | | | | | | | | | | Chief Financial | | | 2012 | | | | 262,000 | | | | 60,000 | | | | — | | | | 425,672 | | | | 9,731 | | | | 757,403 | | Officer | | | 2011 | | | | 225,000 | | | | 7,500 | | | | — | | | | — | | | | 9,000 | | | | 241,500 | | | | | | | | | | Bruce Given | | | | | | | | | | | | | | | | | | | | | | | | | | | | | Chief Operating | | | | | | | | | | | | | | | | | | | | | | | | | | | | | Officer (3) | | | 2012 | | | | 317,000 | | | | — | | | | — | | | | 605,445 | | | | 9,459 | | | | 931,904 | |
(1) | This column represents the total grant date fair value, computed in accordance with ASC 718, of stock options granted during fiscal year 2012. No stock options were granted during fiscal 2011. The assumptions used to calculate the value of the stock underlying the option awards are set forth in Note 9 of the Notes to the Consolidated Financial Statements attached hereto. |
(2) | Amounts consist of 401(k) matching contributions. |
(3) | Bruce Given was hired on October 26, 2012, thus compensation reflected is for a partial year of approximately eleven months. |
Outstanding Equity Awards at Fiscal Year-End
The following table provides information, with respect to the Named Executive Officers, concerning the Outstanding Equity Awards of the Company’s stock as of September 30, 2012. Note that the information in Item 11 reflects the adjustment related to the 1- for-10 reverse stock split which occurred on November 17, 2011.
| | | | | | | | | | | | | | | | | | | | Option Awards (1) | | | | | Number of
Securities
Underlying
Unexercised
Options (#)
(Exercisable) | | | Number of
Securities
Underlying
Unexercised
Options (#)
(Unexercisable) | | | Option
Exercise Price
($) | | | Option
Expiration
Date | | Christopher Anzalone | | | 2,500 | | | | — | | | | 21.30 | | | | 6/11/2018 | | | | | 112,650 | | | | — | | | | 5.10 | | | | 10/8/2019 | | | | | 56,325 | | | | — | | | | 5.20 | | | | 3/4/2020 | | | | | 26,042 | | | | 23,958 | | | | 9.90 | | | | 8/16/2020 | | | | | 3,438 | | | | 11,562 | | | | 4.60 | | | | 10/21/2021 | | | | | 24,792 | | | | 145,208 | | | | 5.19 | | | | 2/16/2022 | | | | | — | | | | 50,000 | | | | 2.62 | | | | 9/28/2022 | | Ken Myszkowski | | | 16,927 | | | | 8,073 | | | | 7.00 | | | | 11/16/19 | | | | | 8,000 | | | | — | | | | 5.20 | | | | 3/4/2020 | | | | | 6,250 | | | | 5,750 | | | | 9.90 | | | | 8/16/2020 | | | | | 3,438 | | | | 11,562 | | | | 4.60 | | | | 10/21/2021 | | | | | 12,104 | | | | 70,896 | | | | 5.19 | | | | 2/16/2022 | | | | | — | | | | 25,000 | | | | 2.62 | | | | 9/28/2022 | | Bruce Given | | | 771 | | | | 229 | | | | 6.20 | | | | 9/28/19 | | | | | 6,875 | | | | 23,125 | | | | 5.20 | | | | 10/26/2021 | | | | | 16,042 | | | | 93,958 | | | | 5.19 | | | | 2/16/2022 | | | | | — | | | | 35,000 | | | | 2.62 | | | | 9/28/2022 | |
(1) | Except for 30,000 options granted to Bruce Given as an inducement grant upon his hire, all option awards were granted under the 2000 Stock Option Plan or the 2004 Equity Incentive Plan of the Company. Options are priced at the market closing price on the day of the award. Options have various vesting parameters, but generally vest within 48 months or less after the award is granted. |
37
Director Compensation
Directors who are also employees of the Company receive no separate compensation from the Company for their service as members of the Board. Non-employee directors currently receive a cash retainer of $37,500 per year. Additionally, non-employee directors received periodic grants of stock options based on recommendation of the compensation committee. Based on the policy of his current employer, Dr. Ferrari has waived his right to receive cash compensation and has waived his right to received stock option grants during fiscal 2012. Dr. Given declined to accept cash compensation during fiscal 2012. The following table sets forth the total compensation paid to our directors in fiscal 2012.
| | | | | | | | | | | | | Name | | Fee Earned
or
Paid in Cash
($) (1) | | | Option
Awards
($) (2) (3) | | | Total ($) | | Douglass Given | | $ | — | | | $ | 130,210 | | | $ | 130,210 | | Edward Frykman | | $ | 33,125 | | | $ | 74,500 | | | $ | 107,625 | | Charles McKenney | | $ | 33,125 | | | $ | 74,500 | | | $ | 107,625 | | Mike Perry | | $ | 28,125 | | | $ | 158,300 | | | $ | 186,425 | | Mauro Ferrari | | $ | — | | | $ | — | | | $ | — | |
(1) | Beginning in February 2012, quarterly compensation to non-employee directors was increased from $5,000 to $9,375. There are no additional payments for being a member of a committee. Dr. Ferrari and Dr. Given have declined to receive cash compensation at this time. |
(2) | This column represents the total grant date fair value, computed in accordance with ASC 718, of stock options granted during fiscal year 2012. The assumptions used to calculate the value of option awards are set forth under Note 7 to the Consolidated Financial Statements attached hereto. |
(3) | Option grants to non-employee directors generally vest one year from date of grant. At September 30, 2012, Mr. Frykman had outstanding option grants to purchase 55,500 shares at prices ranging from $2.62 to $20.20; Mr. McKenney had outstanding option grants to purchase 53,000 shares at prices ranging from $2.62 to $20.20; Mr. Perry had outstanding option grants to purchase 50,000 shares at prices ranging from $2.62 to $5.19; Dr. Given had outstanding option grants to purchase 40,000 shares at prices ranging from $2.62 to $5.19; and Dr. Ferrari had outstanding option grants to purchase 24,843 shares at prices ranging from $9.60 to $28.70. |
Risk Management and Mitigation
In reviewing the compensation structure, the Compensation Committee has considered how the Company’s compensation policies may affect the Company’s risk profile and how compensation policies may be used to mitigate risks facing the Company. In considering these issues, the Compensation Committee determined that the use of performance-based bonuses and long-term equity awards did not appear to create undue risks for the Company or encourage excessive risk-taking behavior on the part of employees.
With respect to bonus awards, the amount of an individual’s award depends principally on overall Company performance, which reduces the ability and incentive for an individual to take undue risks in an effort to increase the amount of his or her bonus award for a particular year. The Company’s performance goals are reviewed and approved by the Compensation Committee at the beginning of each fiscal year and are considered to be generally of the nature that would not encourage or reward excessive risk taking. Additionally, the Compensation Committee monitors Company performance throughout the year and has the ability to intervene in instances where actions by the Company vis-à-vis Company performance goal attainment would be considered unduly risky to prevent or penalize such actions.
With respect to equity awards, these awards typically vest over several years, meaning that long-term value creation, contrasted with short-term gain, presents the best opportunity for employees to profit from these awards. To the extent that performance-based equity awards are used, the events that trigger vesting are expected to be realized several years in the future. The Company has not historically used claw-back provisions or imposed holding periods for vested awards, although the Compensation Committee will consider whether such mechanisms might be appropriate in the future to mitigate risk as the Company transitions from a drug development company to a fully integrated specialty pharmaceutical company with commercial operations. Additionally, the use of financial-based performance metrics to determine employee compensation may subject those payouts to claw-back penalties under the Dodd-Frank Act, to the extent that there is a subsequent restatement of the financial measure that was used to determine a payout.
Compensation Committee Interlocks and Insider Participation
During the last completed fiscal year, no member of the Compensation Committee was a current or former officer or employee of the Company. None of our executive officers served as a member of the compensation committee (or board of directors serving the compensation function) of another entity where such entity’s executive officers served on our Compensation Committee. Moreover, none of our executive officers served as a member of the compensation committee (or board of directors serving the compensation function) of another entity where such entity’s executive officers served on our Board.
38
ITEM 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS. |
The following table sets forth the beneficial ownershipinformation called for by this Item will be incorporated by reference from our Definitive Proxy Statement to be filed for our 2016 Annual Meeting of the Company’s Common Stock as of November 30, 2012, by (i) each of the named executive officers named in the table under Executive Compensation – Summary Compensation Table,” (ii) each director, (iii) all current directors and executive officers as a group, and (iv) the holders of greaterStockholders, which proxy statement will be filed no later than 5% of our total shares outstanding known to us. Unless otherwise specified in the footnotes to the table below, the persons and entities named in the table have sole voting and investment power with respect to all shares beneficially owned, subject to community property laws, where applicable and the address of each stockholder, unless otherwise indicated below, is c/o Arrowhead Research Corporation, 225 South Lake Avenue, Suite 1050, Pasadena, California 91101. All information in Item 12 has been adjusted to reflect the 1 for 10 reverse stock split which was effected on November 17, 2011. January 28, 2016. 39
| | | | | | | | | | | | Number and Percentage of Shares | | | | | Beneficially Owned (1) | | | | | Shares | | | Percentage | | 5% Beneficial Owners | | | | | | | | | M. Robert Ching (2) | | | 1,344,339 | | | | 9.9 | % | Galloway Ltd. (3) | | | 1,270,981 | | | | 9.4 | % | Roche Finance Ltd. (5) | | | 1,141,596 | | | | 8.4 | % | Sabby Healthcare Volatility Master Fund, Ltd. (4) | | | 905,796 | | | | 6.7 | % | Vermogensverwaltungs—Gesellschaft Zurich (6) | | | 675,000 | | | | 5.0 | % | Executive Officers and Directors | | | | | | | | | Chris Anzalone (7) | | | 315,844 | | | | 2.3 | % | Kenneth Myszkowski (8) | | | 61,552 | | | | * | | Bruce Given (9) | | | 38,355 | | | | * | | Edward Frykman (10) | | | 37,500 | | | | * | | Charles McKenney (11) | | | 29,020 | | | | * | | Mauro Ferrari (12) | | | 26,796 | | | | * | | Mike Perry (13) | | | 6,771 | | | | * | | Douglass Given | | | — | | | | — | | | | | Executive officers and directors as a group (8 persons) (14) | | | 515,838 | | | | 3.8 | % |
(1) | Based on 13,579,184 common shares issued and outstanding as of November 30, 2012. Shares not outstanding but deemed beneficially owned by virtue of the right of a person to acquire them as of November 30, 2012, or within sixty days of such date, are treated as outstanding only when determining the percentage owned by such individual and when determining the percentage owned by a group. |
(2) | Includes 793,611 shares of common stock and 1,102,232 shares of common stock issuable upon the exercise of common stock purchase warrants, of which 371,687 shares of common stock, and 262,805 shares of common stock issuable upon the exercise of common stock purchase warrants are held by BBB Assets for which M. Robert Ching holds investment and voting control. Certain of the warrants are subject to a contractual blocker whereby the right to exercise such warrant is limited such that Dr. Ching will not have greater than 9.99% beneficial ownership of the outstanding common stock. Warrants to purchase 306,939 shares are currently not exercisable due to this limitation. |
(3) | Denham Eke holds voting and investment control with respect to the shares owned by Galloway, Ltd. The address for Galloway, Ltd. is Viking House, Nelson Street, Douglas, Isle of Man, IM1 2AH |
(4) | Hal Mintz holds voting and investment control with respect to shares owned by Sabby Healthcare Volatility Master Fund, Ltd., the address for which is 89 Nexus Way, Camana Bay, Grand Cayman KY1-9007, Cayman Islands |
(5) | Carole Nuechterlein, Head of Roche Venture Fund, holds voting and investment control with respect to the shares owned by Roche Finance, Ltd. The address for Roche Finance Ltd. is Grenzacherstrasse 124, 4058 Basel Switzerland. |
(6) | Markus Winkler holds voting and investment control with respect to the shares owned by Vermogensverwaltungs—Gesellschaft Zurich (VGZ), the address for VGZ is Mainaustrasse 30, CH - 8034 Zurich Switzerland |
(7) | Includes 249,498 shares issuable upon the exercise of stock options, and 32,173 shares issuable upon the exercise of common stock purchase warrants that are exercisable within 60 days of November 30, 2012. |
(8) | Includes 60,052 shares issuable upon the exercise of stock options that are exercisable within 60 days of November 30, 2012. |
(9) | Includes 38,355 shares issuable upon the exercise of stock options that are exercisable within 60 days of November 30, 2012. |
(10) | Includes 30,500 shares issuable upon the exercise of stock options that are exercisable within 60 days of November 30, 2012. |
(11) | Includes 28,000 shares issuable upon the exercise of stock options that are exercisable within 60 days of November 30, 2012. |
(12) | Includes 24,845 shares issuable upon the exercise of stock options that are exercisable within 60 days of November 30, 2012. |
(13) | Includes 6,771 shares issuable upon the exercise of stock options that are exercisable within 60 days of November 30, 2012. |
(14) | Includes 438,031 shares issuable upon the exercise of stock options, and 32,173 shares issuable upon the exercise of common stock purchase warrants that are exercisable within 60 days of November 30, 2012. |
40
EQUITY COMPENSATION PLAN INFORMATION
The following table provides information as of September 30, 2012 with respect to shares of our Common Stock that may be issued under our equity compensation plans. On November 17, 2011, the Company effected a 1 for 10 reverse stock split. The share data in the table below are listed on a post-split basis.
| | | | | | | | | | | | | | | | Equity Compensation Plan Information | | Plan Category | | Number of
securities to be issued
upon exercise of
outstanding options,
warrants and rights | | | Weighted
average
exercise price
of outstanding
options,
warrants and
rights | | | Number of
securities
remaining
available for
future issuance
under equity
compensation
plans (excluding
securities
reflected in
column (a)) | | Equity compensation plans approved by security holders(1) | | | 1,659,594 | | | $ | 6.20 | | | | 459,166 | | Equity compensation plans not approved by security holders (2) | | | 251,200 | | | $ | 4.70 | | | | — | | | | | | | | | | | | | | | Total | | | 1,910,794 | | | $ | 6.01 | | | | 459,166 | |
(1) | Includes options outstanding representing 1,506,694 shares subject to the 2004 Equity Incentive Plan and 152,900 shares subject to the 2000 Option Plan. No shares are available for issuance under the 2000 option plan. |
(2) | Includes inducement options issued to newly hired employees upon the acquisition of Roche Madison in October 2011. |
| ITEM 13. | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE. |
As of September 30, 2012, a majority of the members of the Board are independent directors, as definedThe information called for by the NASDAQ Marketplace Rules. The Board has determined that all of the Company’s directors are independent, except Dr. Anzalone, the Company’s Chief Executive Officer, and Dr. Doug Given, the brother of the Company’s Chief Operating Officer. Non-employee directors do not receive consulting, legal or other feesthis Item will be incorporated by reference from the Company, other than Board compensation.
Nanotope and Leonardo were co-founded by the Company’s President and Chief Executive Officer, Dr. Christopher Anzalone, who beneficially owns approximately 14.2% and 9.4% of the outstanding voting securities of Nanotope and Leonardo, respectively. Dr. Anzalone does not hold options, warrants or any other rights to acquire securities of Nanotope or Leonardo. Dr. Anzalone has the right to appoint a representative to the Board of Directors of each Nanotope and Leonardo. Dr. Anzalone is serving as the President and Chief Executive Officer of Nanotope. Dr. Anzalone has not received any compensation for his work on behalf of Nanotope or Leonardo since joining the Company on December 1, 2007.
During fiscal 2012, a portion of Arrowhead employee salary costs, including Dr. Anzalone’s salary and administrative overhead, was charged to Nanotope and Leonardo for management and administrative services provided by Arrowhead to Nanotope and Leonardo. During fiscal 2012, the charge for services provided to Nanotope and Leonardo were $198,269 and $239,783, respectively. In addition, during fiscal 2012, Arrowhead made cash advances to Nanotope of $475,000 and cash advances to Leonardo of $56,000. The amounts due from Nanotope have been fully reserved, and the operations of Nanotope have been suspended. Accordingly, future cash advances are expectedour Definitive Proxy Statement to be minimal. The majorityfiled for our 2016 Annual Meeting of the balance due Arrowhead from Leonardo is expected toStockholders, which proxy statement will be repaid in cash or converted to equity in fiscal 2013. In addition, Dr. Bruce Given, the Company’s Chief Operating Officer, and CEO of Leonardo is the brother of Dr. Doug Given, a member of Arrowhead’s Board of Directors. Dr. Doug Given hasfiled no financial interest in Leonardo.
In August 2010, the Company retained Mr. Vincent Anzalone, the brother of Arrowhead’s Chief Executive Officer, as a consultant for the Company, focusing on business development and market analysis. Mr. Vincent Anzalone was paid $120,000 during the fiscal year ended September 30, 2011, and $120,000 during the fiscal year ended September 30, 2012. Mr. Vincent Anzalone joined the Company as an employee on September 24, 2012 at an annual salary of $130,000.
later than January 28, 2016. 41
ITEM 14. | PRINCIPAL ACCOUNTANT FEES AND SERVICES |
The Audit Committee regularly reviews and determines whether specific projects or expenditures withinformation called for by this Item will be incorporated by reference from our independent auditors, Rose, Snyder & Jacobs (RS&J), may potentially affect their independence. The Audit Committee’s policy is to pre-approve all audit and permissible non-audit services provided by RS&J. Pre-approval is generally provided by the Audit Committee for up to one year, detailed to the particular service or category of servicesDefinitive Proxy Statement to be rendered and is generally subject to a specific budget. The Audit Committee may also pre-approve additional servicesfiled for our 2016 Annual Meeting of specific engagements on a case-by-case basis. All engagements of our independent registered public accounting firm in 2012 and 2011 were pre-approved by the audit committee. The following table sets forth the aggregate fees invoiced by RS&J for the fiscal years ended September 30, 2012, and September 30, 2011:Stockholders, which proxy statement will be filed no later than January 28, 2016.
| | | | | | | | | | | | Year Ended September 30, | | | | | 2012 | | | 2011 | | Audit fees (1) | | $ | 159,065 | | | $ | 116,200 | | Audit-related fees (2) | | | 42,459 | | | | 16,250 | | Tax fees (3) | | | — | | | | 20,085 | | | | | | | | | | | Total | | $ | 201,524 | | | $ | 152,435 | | | | | | | | | | |
(1) | Fees invoiced by RS&J include year-end audit and quarterly reviews of Form 10-Q. |
(2) | Fees invoiced by RS&J related to Arrowhead Comfort Letter and Consents, and other agreed-upon procedures. |
(3) | This category consists of professional services rendered by RS&J for tax return preparation and consulting. The Company has retained another public accounting firm for tax preparation and consulting. |
PART IV
PART IV ITEM 15. | EXHIBITS AND FINANCIAL STATEMENT SCHEDULES |
The following documents are filed as part of this Annual Report on Form 10-K: See Index to Financial Statements and Schedule on page F-1. | (2) | Financial Statement Schedules. |
See Index to Financial Statements and Schedule on page F-1. All other schedules are omitted as the required information is not present or is not present in amounts sufficient to require submission of the schedule, or because the information required is included in the consolidated financial statements or notes thereto. 63 42
The following exhibits are filed (or incorporated by reference herein) as part of this Annual Report on Form 10-K: | | | | | | | | | Exhibit
Number | | | | | | Description | | Incorporated by Reference Herein | | Description
| Form | Form
| | Date | 2.1 | | | | | 2.1 | | | Stock and Asset Purchase Agreement between Arrowhead Research Corporation and Roche entities, dated October 21, 2011† | | Annual Report on Form 10-K for the fiscal year ended September 30, 2011, as Exhibit 2.1 | | December 20, 2011 | | | | | | | | 2.2 | 3.1 | Asset Purchase and Exclusive License Agreement between Arrowhead Research Corporation and Novartis Institutes for BioMedical Research, Inc., dated March 3, 2015† | | Quarterly Report on Form 10-Q, as Exhibit 2.1 | | May 11, 2015 | | | | | | | | 3.1 | | Certificate of Incorporation of InterActive Group, Inc., a Delaware corporation, datedfiled with the Secretary of State of the State of Delaware on February 13, 2001 | | Schedule 14C, as Exhibit A | | December 22, 2000 | | | | | | | | 3.2 | 3.2 | | | Certificate of Amendment to Certificate of Incorporation of InterActive Group, Inc. (effecting, among other things a change in the corporation’s name to “Arrowhead Research Corporation”), filed with the Secretary of State of the State of Delaware on January 12, 2004 | | Schedule 14C, as Exhibit 1 | | December 22, 2003 | | | | | | | | 3.3 | 3.3 | | | Certificate of Amendment to Certificate of Incorporation of Arrowhead Research Corporation, datedfiled with the Secretary of State of the State of Delaware on January 25,26, 2005 | | Form 10-QSB for the quarter ended December 31, 2004, as Exhibit 3.4 | | February 11, 2005 | | | | | | | | 3.4 | 3.4 | | | Certificate of Amendment to Certificate of Incorporation of Arrowhead Research Corporation, filed with the Secretary of State of the State of Delaware on dated October 13,14, 2009 | | Annual Report on Form 10-K for the fiscal year ended September 30, 2009, as Exhibit 3.4 | | December 22, 2009 | | | | | | | | 3.5 | 3.5 | | | Series A Certificate of Designations, datedfiled with the Secretary of State of the State of Delaware on October 25, 2011 | | Current Report on Form 8-K, as Exhibit 3.1 | | October 26, 2011 | | | | | | | | 3.6 | 3.6 | | | Certificate of Amendment to Certificate of Incorporation of Arrowhead Research Corporation, datedfiled with the Secretary of State of the State of Delaware on November 17,15, 2011 | | Current Report on Form 8-K, as Exhibit 3.1 | | November 17, 2011 | | | | | | | | 3.7 | 3.7 | Bylaws | | Bylaws | | Schedule 14C, as Exhibit B | | December 22, 2000 | | | | | | | | 3.8 | 3.8 | | | Amendment No. 1 to the Bylaws of Arrowhead Research Corporation | | Current Report on Form 8-K, as Exhibit 3.1 | | April 27, 2010 | | | | | | | | 3.9 | 4.1 | Series B Certificate of Designations, filed with the Secretary of State of the State of Delaware on May 1, 2013 | | Form of Registration Rights Agreement, July and August 2009 | | Current Report on Form 8-K, as Exhibit 10.23.1 | | July 17, 2009May 1, 2013 | | | | | | | | 3.10 | 4.2 | Series C Certificate of Designations, filed with the Secretary of State of the State of Delaware on October 10, 2013 | | Form of Registration Rights Agreement, dated December 11, 2009 | | Annual Report on Form 10-K for the fiscal year ended September 30, 2009, as Exhibit 4.2 | | December 22, 2009 | | | | | | 4.3 | | | Form of Warrant to Purchase Shares of Common Stock expiring in July and August 2013 | | Current Report on Form 8-K, as Exhibit 10.23.1 | | August 26, 2008October 10, 2013 | | | | | | | | 4.1 | 4.4 | | | Form of Common Stock Warrant expiring in September 2013 | | Current Report on Form 8-K, as Exhibit 10.2 | | September 11, 2008 | | | | | | 4.5 | | | Form of Warrant to Purchase Capital Stock expiring June 2014 | | Current Report on Form 8-K, as Exhibit 4.1 | | July 17, 2009 | | | | | | | | 4.2 | 4.6 | | | Form of Warrant to Purchase Capital Stock expiring December 2014 | | Annual Report on Form 10-K for the fiscal year ended September 30, 2009, as Exhibit 4.7 | | December 22, 2009 | | | | | | | | 4.3 | 4.7 | | | Form of Warrant to Purchase Common Stock expiring May 2017 | | Current Report on Form 8-K, as Exhibit 4.1 | | May 30, 2007 |
43
| | | | | | | Exhibit
Number | | | | Incorporated by Reference Herein
| | Description
| | Form
| | Date
| | 4.84.4 | | Form of Warrant to Purchase Common Stock dated June 2010expiring December 2015 | | Current Report on Form 8-K, as Exhibit 4.1 | | June 18, 2010 |
64
Exhibit
Number | | Description | | Incorporated by Reference Herein | | | Form | | Date | 4.9
| | Form of Registration Rights Agreement between Arrowhead Research Corporation and Lincoln Park Capital Fund, LLC, dated October 20, 2011 | | Current Report on Form 8-K, as Exhibit 10.2 | | October 26, 2011 | 4.5 | | | | 4.10 | | Form of Registration Rights Agreement between Arrowhead Research Corporation and Roche entities, dated October 21, 2011 | | Annual Report on Form 10-K for the fiscal year ended September 30, 2011, as Exhibit 4.10 | | December 20, 2011 | | | | | 4.11 | | Form of Warrant to Purchase Shares of Capital Stock of Arrowhead Research Corporation expiring September 16, 2015 | | Current Report on Form 8-K, as Exhibit 4.1 | | September 22, 2010 | | | | | | | | 4.124.6 | | Form of Warrant to Purchase Shares of Common Stock Expiring August 13, 2016 | | Current Report on Form 8-K, as Exhibit 4.1 | | August 13, 2012 | | | | | | | | 4.134.7 | | Form of Common Stock Certificate | | Amendment No. 2 to Registration Statement on Form S-1, as Exhibit 4.7 | | September 11, 2009 | | | | | | | | 4.144.8 | | Form of Series AB Preferred Stock Certificate | | Annual Report on Form 10-K for the fiscal year ended September 30, 2011,2013, as Exhibit 4.134.15 | | December 20, 201118, 2013 | | | | | | | | 4.9 | | Form of Warrant to Purchase Shares of Common Stock expiring August 17, 2016 | | Current Report on Form 8-K, as exhibit 4.2 | | August 13, 2012 | | | | | | | | 4.10 | | Form of Warrant to Purchase Shares of Common Stock expiring December 12, 2017 | | Current Report on Form 8-K, as exhibit 4.2 | | December 12, 2012 | | | | | | | | 4.11 | | Form of Warrant to Purchase Shares of Common Stock expiring January 30, 2018 | | Current Report on Form 8-K, as exhibit 4.2 | | January 30, 2013 | | | | | | | | 4.12 | | Form of Series C Preferred Stock Certificate | | Annual Report on Form 10-K for the fiscal year ended September 30, 2013, as Exhibit 4.15 | | December 18, 2013 | | | | | | | | 10.1** | | Arrowhead Research Corporation (fka InterActive, Inc.) 2000 Stock Option Plan | | Schedule 14C, as Exhibit D | | December 22, 2000 | | | | | | | | 10.2** | | Arrowhead Research Corporation 2004 Equity Incentive Plan, as amended | | Schedule 14C, as Annex A | | January 12, 2012 | | | | | | | | 10.3** | | Arrowhead Research Corporation 2013 Incentive Plan | | Schedule 14C, as Annex A | | December 20, 2013 | | | | | | | | 10.4** | | Form of Stock Option Agreement for use with the 2013 Incentive Plan | | Current Report on Form 8-K, as Exhibit 10.1 | | February 12, 2014 | | | | | | | | 10.5** | | Form of Restricted Stock Unit Agreement for use with the 2013 Incentive Plan | | Current Report on Form 8-K, as Exhibit 10.2 | | February 12, 2014 | | | | | | | | 10.6** | | Executive Incentive Plan, adopted December 12, 2006 | | Annual Report on Form 10-K for the fiscal year ended September 30, 2006, as Exhibit 10.11 | | December 14, 2006 | | | | | | | | 10.4*10.7** | | Compensation Policy for Non-Employee Directors, as amended | | Annual Report on Form 10-K for the fiscal year ended September 30, 2006, as Exhibit 10.12 | | December 14, 2006 | | | | | 10.5** | | Employment Agreement between Arrowhead and Dr. Christopher Anzalone, dated June 11, 2008 | | Current Report on Form 8-K, as Exhibit 10.1 | | June 13, 2008 | | | | | | | | 10.6*10.8** | | Amendment to Employment Agreement between Arrowhead and Dr. Christopher Anzalone, effective May 12, 2009 | | Annual Report on Form 10-K for the fiscal year ended September 30, 2009, as Exhibit 10.8 | | December 22, 2009 | | | | | 10.7 | | Insert Therapeutics, Inc. Amended and Restated Investors’ Rights Agreement, dated April 17, 2008 | | Current Report on Form 8-K, as Exhibit 10.3 | | April 23, 2008 | | | | | 10.8 | | Form of Unsecured Convertible Promissory Note Agreement, dated November 26, 2008 | | Current Report on Form 8-K, as Exhibit 10.1 | | December 3, 2008 | | | | | 10.9 | | Platform Agreement by and between Calando Pharmaceuticals, Inc and Cerulean Pharma Inc., dated as of June 23, 2009 † | | Form 10-Q for the quarter ended June 30, 2009, as Exhibit 10.1 | | August 10, 2009 | | | | | 10.10 | | IT-101 Agreement by and between Calando Pharmaceuticals, Inc and Cerulean Pharma, Inc., dated as of June 23, 2009† | | Form 10-Q for the quarter ended June 30, 2009, as Exhibit 10.2 | | August 10, 2009 | | | | | 10.11 | | License and Enforcement Agreement between Unidym, Inc. and Samsung Electronics Co., Ltd., dated December 2010 | | Form 10-Q for the quarter ended December 31, 2010, as Exhibit 10.1 | | February 10, 2011 |
44
| | | | | | | Exhibit
Number | | | | Incorporated by Reference Herein
| | Description
| | Form
| | Date
| | 10.12 | | CNT Production Patent License Agreement between Unidym, Inc. and Samsung Electronics Co., Ltd., dated December 2010 | | Form 10-Q for the quarter ended December 31, 2010, as Exhibit 10.2 | | February 10, 2011 | 10.9 | | | | 10.13 | | Intellectual Property Purchase and Business Cooperation Agreement between Unidym, Inc. and Samsung Electronics Co., Ltd., dated December 2010 | | Form 10-Q for the quarter ended December 31, 2010, as Exhibit 10.3 | | February 10, 2011 | | | | | 10.14 | | Patent and Technology License Agreement between Arrowhead Research Corporation and the Board of Regents of The University of Texas System, dated December 14, 2010 | | Form 10-Q for the quarter ended December 31, 2010, as Exhibit 10.4 | | February 10, 2011 | | | | | 10.15 | | Form of Series A Preferred Stock Purchase Agreement among Ablaris Therapeutics Inc. and certain investors, dated January 2011 | | Form 10-Q for the quarter ended March 31, 2011, as Exhibit 10.2 | | May 12, 2011 | | | | | 10.16 | | Stock Purchase Agreement between Arrowhead Research Corporation and Calando Pharmaceuticals, Inc., dated January 10, 2011 | | Form 10-Q for the quarter ended March 31, 2011, as Exhibit 10.1 | | May 12, 2011 | | | | | 10.17 | | Agreement and Plan of Merger among Wisepower Co., Ltd., Unicycle Acquisition Corp, Unidym, Inc. and Arrowhead Research Corporation, dated January 17, 2011 | | Current Report on Form 8-K, as Exhibit 10.1 | | January 21, 2011 | | | | | 10.18 | | Stock Purchase Agreement between Wisepower Co., Ltd. and Arrowhead Research Corporation, dated January 17, 2011 | | Current Report on Form 8-K, as Exhibit 10.2 | | January 21, 2011 | | | | | 10.19 | | Bond Purchase Agreement between Wisepower Co., Ltd. and Arrowhead Research Corporation, dated January 17, 2011 | | Current Report on Form 8-K, as Exhibit 10.3 | | January 21, 2011 | | | | | 10.20 | | Form of Subscription Agreement between Arrowhead Research Corporation and certain Investors, dated September 2011 | | Current Report on Form 8-K, as Exhibit 10.1 | | October 6, 2011 | | | | | 10.21 | | Form of Series A Subscription Agreement between Arrowhead Research Corporation and certain investors | | Current Report on Form 8-K, as Exhibit 10.3 | | October 26, 2011 | | | | | 10.22 | | Form of Purchase Agreement between Arrowhead Research Corporation and Lincoln Park Capital Fund, LLC, dated October 20, 2011 | | Current Report on Form 8-K, as Exhibit 10.1 | | October 26, 2011 | | | | | 10.23 | | Form of Common Stock Subscription Agreement between Arrowhead Research Corporation certain Investors, dated October 21, 2011 | | Current Report on Form 8-K, as Exhibit 10.4 | | October 26, 2011 | | | | | 10.24 | | Non-Exclusive License Agreement between Arrowhead Research Corporation and Roche entities, dated October 21, 2011† | | Annual Report on Form 10-K for the fiscal year ended September 30, 2011, as Exhibit 10.33 | | December 20, 2011 | | | | | 10.25 | | Form of Investor Subscription Agreement between Arrowhead Research Corporation and Lincoln Park Capital Fund, LLC, dated October 24, 2011 | | Current Report on Form 8-K, as Exhibit 10.1 | | October 27, 2011 |
65 45
| | | | | | | Exhibit
Number | | | | | Description | | Incorporated by Reference Herein | | Description
| Form | Form
| | Date |
| | | | | | | 10.2610.10 | | License and Collaboration Agreement, dated July 8, 2007 † | | Annual Report on Form 10-K for the fiscal year ended September 30, 2011, as Exhibit 10.35 | | December 20, 2011 | | | | | | | | 10.2710.11 | | Collaboration Agreement by and among Alnylam Pharmaceuticals, Inc. and F. Hofmann-LaHoffmann-La Roche Ltd and Hoffman-La Roche Inc., dated October 29, 2009 † | | Annual Report on Form 10-K for the fiscal year ended September 30, 2011, as Exhibit 10.36 | | December 20, 2011 | | | | | | | | 10.2810.12 | | Office Lease Agreement between South Lake Avenue Investors LLC and the Company, dated April 12, 2012 | | Quarterly Report on Form 10-Q, as Exhibit 10.1 | | May 8, 2012 | | | | | | | | 10.2910.13 | | Form of Subscription Agreement between Arrowhead Research Corporation and certain Investors | | Current Report on Form 8-K, as Exhibit 99.1 | | December 12, 2012 | | | | | | | | 10.14 | | Form of Subscription Agreement between Arrowhead Research Corporation and certain Investors | | Current Report on Form 8-K, as Exhibit 99.1 | | January 30, 2013 | | | | | | | | 10.15 | | Form of Securities Purchase Agreement between Arrowhead Research Corporation and certain Investors | | Current Report on Form 8-K, as Exhibit 99.110.1 | | August 10, 2012April 30, 2013 | | | | | | | | 21.110.16 | | Securities Purchase Agreement between the Company and the purchasers listed thereon. | | Current Report on Form 8-K, as Exhibit 10.1 | | October 10, 2013 | | | | | | | | 10.17 | | License Agreement by and between Alnylam Pharmaceuticals, Inc., Arrowhead Research Corporation and Arrowhead Madison, Inc.† | | Quarterly Report on Form 10-Q, as Exhibit 10.1 | | August 12, 2014 | | | | | | | | 21.1 | | List of Subsidiaries* | | | | | | | | | | | | 23.1 | | Consent of Independent Public Registered Accounting Firm* | | | | | | | | | | | | 24.1 | | Power of Attorney (contained on signature page) | | | | | | | | | | | | 31.1 | | Certification of Chief Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002* | | | | | | | | | | | | 31.2 | | Certification of Chief Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002* | | | | | | | | | | | | 32.1 | | Certification by Chief Executive Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002*** | | | | | | | | | | | | 32.2 | | Certification by Chief Financial Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002*** | | | | | | | | | | | | 101.INS | | XBRL Instance Document* | | | | | | | | | | | | 101.SCH | | XBRL Schema Document* | | | | | | | | | | | | 101.CAL | | XBRL Calculation Linkbase Document* | | | | | | | | | | | | 101.LAB | | XBRL Label Linkbase Document* | | | | | | | | | | | | 101.PRE | | XBRL Presentation Linkbase Document* | | | | | | | | | | | | 101.DEF | | XBRL Definition Linkbase Document* | | | | |
Pursuant to Rule 406T of Regulation S-T, the Interactive Data Files included in Exhibit 101 hereto are deemed not filed or a part of a registration statement or prospectus for purposes of Sections 11 or 12 of the Securities Act of 1933, as amended, are deemed not filed for purposes of Section 18 of the Securities and Exchange Act of 1934, as amended, and otherwise are not subject to liability under those sections.
** | Indicates compensation plan, contract or arrangement. |
† | Confidential treatment has been requested with respect to certain portions of this exhibit. Omitted portions have been filed separately with the Securities and Exchange Commission. |
46
SIGNATURESSIGNATURE
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this Report on Form 10-K to be signed on its behalf by the undersigned, thereunto duly authorized, on this 28th14th day of December 2012.2015. Dated: December 14, 2015 | | | ARROWHEAD RESEARCH CORPORATION | | | | By: | | /s/ CHRISTOPHER ANZALONE Christopher Anzalone | | | Christopher Anzalone | | | Chief Executive Officer |
Pursuant to the requirements of the Securities Exchange Act of 1934, this Report on Form 10-K has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated: Signature | | Title | | Date | Signature
| | Title
| | Date
| | | | /s/ CHRISTOPHER ANZALONEChristopher Anzalone | | Chief Executive Officer, President and | | December 28, 201214, 2015 | Christopher Anzalone | | Director (Principal Executive Officer) | | | | | | | | | /s/ KENNETHKenneth A. MYSZKOWSKIMyszkowski | | Chief Financial Officer (Principal | | December 28, 201214, 2015 | Kenneth A. Myszkowski | | Financial and Accounting Officer) | | | | | | | | | /s/ DOUGLASS GIVENDouglass Given | | Director, Chairman of the Board of Directors | | December 28, 201214, 2015 | Douglass Given | | | | | | | | | | | /s/ MAURO FERRARIMauro Ferrari | | Director | | December 28, 201214, 2015 | Mauro Ferrari | | | | | | | | /s/ EDWARD W. FRYKMAN
| | Director | | December 28, 2012
| /s/ Edward W. Frykman | | Director | | | | | December 14, 2015 | /s/ CHARLES P. MCKENNEYEdward W. Frykman
| | Director | | December 28, 2012
| | | Charles P. McKenney | | | | | | | | /s/ MICHAEL S. PERRY | | Director | | December 28, 2012
| | Michael S. Perry | | Director | | December 14, 2015 | Michael S. Perry | | | | |
4767
INDEX TO FINANCIAL STATEMENTS AND SCHEDULE | | | | | ReportReports of Independent Registered Public Accounting Firm
| | F-2 | F-2 | | Consolidated Balance Sheets of Arrowhead Research Corporation, and Subsidiaries, September 30, 20122015 and 20112014 | | F-4 | F-3 | | Consolidated Statements of Operations of Arrowhead Research Corporation and Subsidiaries for the years ended September 30, 20122015, 2014 and 2011 and the period from May 7, 2003 (inception) through September 30, 20122013 | | F-5 | F-4 | | Consolidated Statement of Stockholders’ Equity of Arrowhead Research Corporation and Subsidiaries for the period from May 7, 2003 (inception) throughyears ended September 30, 20122015, 2014, and 2013 | | F-6 | F-5 | | Consolidated Statements of Cash Flows of Arrowhead Research Corporation and Subsidiaries for the years ended September 30, 20122015, 2014 and 2011 and the period from May 7, 2003 (inception) through September 30, 20122013 | | F-7 | F-6 | | Notes to Consolidated Financial Statements of Arrowhead Research Corporation and Subsidiaries | | F-8 | F-9 | |
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM To the Board of Directors and Stockholders of Arrowhead Research Corporation We have audited the accompanying consolidated balance sheets of Arrowhead Research Corporation (a Delaware corporation) and Subsidiaries (the “Company”) as of September 30, 20122015 and 20112014, and the related consolidated statements of operations and comprehensive loss, stockholders’ equity and cash flows for each of the three years in the period ended September 30, 2012, and 2011 and for the period from May 7, 2003 (inception) through September 30, 2012.2015. These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits. We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the consolidated financial statements,statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall consolidated financial statement presentation. We believe that our audits provide a reasonable basis for our opinion. In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the consolidated financial position of Arrowhead Research Corporation and Subsidiaries as ofat September 30, 20122015 and 2011,2014, and the consolidated results of their operations and their cash flows for each of the three years in the period ended September 30, 2012 and 2011, and for the period from May 7, 2003 (inception) through September 30, 20122015, in conformity with accounting principlesU.S. generally accepted accounting principles. We also have audited, in accordance with the United Statesstandards of America.the Public Company Accounting Oversight Board (United States), Arrowhead Research Corporation and Subsidiaries’ internal control over financial reporting as of September 30, 2015, based on criteria established in Internal Control – Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) and our report dated December 11, 2015 expressed an unqualified opinion thereon. The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the consolidated financial statements, the Company has incurred recurring net losses. This condition raises substantial doubt about the Company’s ability to continue as a going concern. Management’s plans regarding those matters also are described in Note 1. The accompanying consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/
Rose, Snyder & Jacobs LLP Encino, California December 27, 201211, 2015 F-2
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM To the Board of Directors and Stockholders of Arrowhead Research Corporation We have audited Arrowhead Research Corporation and Subsidiaries’ (the “Company”) internal control over financial reporting as of September 30, 2015, based on criteria established in Internal Control – Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (“the COSO criteria”). Arrowhead Research Corporation’s management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Annual Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion. A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements. Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate. In our opinion, Arrowhead Research Corporation and Subsidiaries maintained, in all material respects, effective internal control over financial reporting as of September 30, 2015, based on the COSO criteria. (A Development Stage Company)We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the 2015 consolidated financial statements of Arrowhead Research Corporation and Subsidiaries and our report dated December 11, 2015 expressed an unqualified opinion thereon.
Rose, Snyder & Jacobs LLP Encino, California December 11, 2015
PART I. FINANCIAL INFORMATION ITEM 1. | FINANCIAL STATEMENTS |
Arrowhead Research Corporation Consolidated Balance Sheets | | | | | | | | | | | | September 30, 2012 | | | September 30, 2011 | | ASSETS | | | | | | | | | CURRENT ASSETS | | | | | | | | | Cash and cash equivalents | | $ | 3,377,288 | | | $ | 7,507,389 | | Trade receivable | | | 9,375 | | | | — | | Other receivables | | | 9,930 | | | | 1,608,382 | | Prepaid expenses and other current assets | | | 618,130 | | | | 110,818 | | Marketable securities | | | 106,500 | | | | 634,585 | | Note receivable, net | | | 2,446,113 | | | | — | | | | | | | | | | | TOTAL CURRENT ASSETS | | | 6,567,336 | | | | 9,861,174 | | PROPERTY AND EQUIPMENT | | | | | | | | | Computers, office equipment and furniture | | | 323,376 | | | | 285,266 | | Research equipment | | | 3,319,027 | | | | 3,515 | | Software | | | 69,623 | | | | 77,020 | | Leasehold improvements | | | 2,749,409 | | | | — | | | | | | | | | | | | | | 6,461,435 | | | | 365,801 | | Less: Accumulated depreciation and amortization | | | (1,565,783 | ) | | | (340,364 | ) | | | | | | | | | | PROPERTY AND EQUIPMENT, NET | | | 4,895,652 | | | | 25,437 | | OTHER ASSETS | | | | | | | | | Patents and other intangible assets, net | | | 4,784,569 | | | | 1,731,211 | | Note receivable, net | | | — | | | | 2,272,868 | | Investment in Nanotope Inc., equity basis | | | — | | | | 1,649,748 | | Investment in Leonardo Biosystems Inc., at cost | | | — | | | | 187,000 | | Derivative asset and other non-current assets | | | 280,261 | | | | 161,125 | | | | | | | | | | | TOTAL OTHER ASSETS | | | 5,064,830 | | | | 6,001,952 | | | | | | | | | | | TOTAL ASSETS | | $ | 16,527,818 | | | $ | 15,888,563 | | | | | | | | | | | LIABILITIES AND STOCKHOLDERS’ EQUITY | | | | | | | | | CURRENT LIABILITIES | | | | | | | | | Accounts payable | | $ | 877,986 | | | $ | 576,809 | | Accrued expenses | | | 730,775 | | | | 864,511 | | Accrued payroll and benefits | | | 1,127,219 | | | | 195,649 | | Deferred revenue | | | 37,500 | | | | — | | Derivative liabilities | | | 647,213 | | | | 944,980 | | Capital lease obligation | | | 214,801 | | | | — | | Other current liabilities | | | 1,692,394 | | | | — | | | | | | | | | | | TOTAL CURRENT LIABILITIES | | | 5,327,888 | | | | 2,581,949 | | | | | | | | | | | LONG-TERM LIABILITIES | | | | | | | | | Note payable, net of current portion | | | 839,421 | | | | 606,786 | | Capital lease obligation, net of current portion | | | 1,282,458 | | | | — | | Other non-current liabilities | | | 269,142 | | | | 135,660 | | | | | | | | | | | TOTAL LONG-TERM LIABILITIES | | | 2,391,021 | | | | 742,446 | | | | | | | | | | | Commitments and contingencies | | | | | | | | | STOCKHOLDERS’ EQUITY | | | | | | | | | Arrowhead Research Corporation stockholders’ equity: | | | | | | | | | Preferred stock, $0.001 par value; 5,000,000 shares authorized; no shares issued or outstanding | | | — | | | | — | | Common stock, $ 0.001 par value; 145,000,000 shares authorized; 13,579,185 and 8,642,286 shares issued and outstanding as of September 30, 2012 and September 30, 2011, respectively | | | 108,354 | | | | 86,423 | | Additional paid-in capital | | | 145,917,968 | | | | 127,476,435 | | Subscription receivable | | | (1,016,000 | ) | | | (900,000 | ) | Accumulated deficit during the development stage | | | (134,997,680 | ) | | | (113,871,752 | ) | | | | | | | | | | Total Arrowhead Research Corporation stockholders’ equity | | | 10,012,642 | | | | 12,791,106 | | Noncontrolling interest | | | (1,203,733 | ) | | | (226,938 | ) | | | | | | | | | | TOTAL STOCKHOLDERS’ EQUITY | | | 8,808,909 | | | | 12,564,168 | | | | | | | | | | | TOTAL LIABILITIES AND STOCKHOLDERS’ EQUITY | | $ | 16,527,818 | | | $ | 15,888,563 | | | | | | | | | | |
| | | | | | | | | September 30, 2015 | | | September 30, 2014 | | ASSETS | | | | | | | | CURRENT ASSETS | | | | | | | | Cash and cash equivalents | $ | 81,214,354 | | | $ | 132,510,610 | | Prepaid expenses | | 3,293,285 | | | | 588,626 | | Other current assets | | 823,620 | | | | 48,502 | | Short term investments | | 17,539,902 | | | | 21,653,032 | | TOTAL CURRENT ASSETS | | 102,871,161 | | | | 154,800,770 | | Property and equipment, net | | 4,526,848 | | | | 3,872,753 | | Intangible assets, net | | 24,824,116 | | | | 1,013,473 | | Investments | | - | | | | 23,088,346 | | Other assets | | 45,789 | | | | 41,414 | | TOTAL ASSETS | $ | 132,267,914 | | | $ | 182,816,756 | | LIABILITIES AND STOCKHOLDERS’ EQUITY | | | | | | | | CURRENT LIABILITIES | | | | | | | | Accounts payable | $ | 5,031,706 | | | $ | 2,579,478 | | Accrued expenses | | 5,376,119 | | | | 1,399,486 | | Accrued payroll and benefits | | 3,824,062 | | | | 3,268,506 | | Deferred revenue | | 103,125 | | | | 103,125 | | Derivative liabilities | | 1,301,604 | | | | 4,173,943 | | Capital lease obligation | | 217,548 | | | | 213,991 | | Notes payable | | - | | | | 50,000 | | Other current liabilities | | 46,407 | | | | 58,495 | | TOTAL CURRENT LIABILITIES | | 15,900,571 | | | | 11,847,024 | | LONG-TERM LIABILITIES | | | | | | | | Capital lease obligation, net of current portion | | 540,792 | | | | 758,340 | | Contingent consideration obligations | | 5,862,464 | | | | 3,970,931 | | Other non-current liabilities | | 342,453 | | | | 255,206 | | TOTAL LONG-TERM LIABILITIES | | 6,745,709 | | | | 4,984,477 | | Commitments and contingencies | | | | | | | | STOCKHOLDERS’ EQUITY | | | | | | | | Arrowhead Research Corporation stockholders' equity: | | | | | | | | Preferred stock, $0.001 par value; 5,000,000 shares authorized; 15,652 and 18,300 shares issued and outstanding as of September 30, 2015 and September 30, 2014, respectively | | 16 | | | | 18 | | Common stock, $0.001 par value; 145,000,000 shares authorized; 59,544,677 and 54,656,936 shares issued and outstanding as of September 30, 2015 and September 30, 2014, respectively | | 151,914 | | | | 147,026 | | Additional paid-in capital | | 426,873,358 | | | | 391,164,558 | | Accumulated other comprehensive income (loss) | | (136,425 | ) | | | - | | Accumulated deficit | | (316,712,041 | ) | | | (224,771,159 | ) | Total Arrowhead Research Corporation stockholders' equity | | 110,176,822 | | | | 166,540,443 | | Noncontrolling interest | | (555,188 | ) | | | (555,188 | ) | TOTAL STOCKHOLDERS’ EQUITY | | 109,621,634 | | | | 165,985,255 | | TOTAL LIABILITIES AND STOCKHOLDERS’ EQUITY | $ | 132,267,914 | | | $ | 182,816,756 | |
The accompanying notes are an integral part of these consolidated financial statements. F-3
F-4
Arrowhead Research Corporation and Subsidiaries (A Development Stage Company)
Consolidated Statements of Operations and Comprehensive Loss | | | | Year Ended September 30, | | May 7, 2003 (Inception) to | | | Year Ended September 30, | | | | 2012 | | 2011 | | September 30, 2012 | | | 2015 | | | 2014 | | | 2013 | | | REVENUE | | $ | 146,875 | | | $ | 296,139 | | | $ | 4,138,834 | | | $ | 382,000 | | | $ | 175,000 | | | $ | 290,266 | | | OPERATING EXPENSES | | | | | | | | | | | | | | | | | | | | Research and development | | | | 47,267,361 | | | | 23,138,050 | | | | 8,705,627 | | | Acquired in-process research and development | | | | 10,142,786 | | | | - | | | | - | | | Salaries and payroll-related costs | | | 6,414,921 | | | | 1,408,366 | | | | 26,392,130 | | | | 16,554,008 | | | | 12,829,355 | | | | 6,667,669 | | | General and administrative expenses | | | 6,439,323 | | | | 3,795,380 | | | | 31,353,215 | | | | 7,931,184 | | | | 5,894,008 | | | | 3,488,864 | | | Research and development | | | 5,391,463 | | | | 3,277,760 | | | | 42,618,748 | | | Stock-based compensation | | | 1,241,404 | | | | 1,376,921 | | | | 13,581,468 | | | | 10,232,897 | | | | 5,696,173 | | | | 1,536,271 | | | Depreciation and amortization | | | 1,748,975 | | | | 267,978 | | | | 7,409,286 | | | | 2,336,207 | | | | 1,345,655 | | | | 1,751,412 | | | | | | | | | | | | | | Impairment expense | | | | - | | | | 2,172,387 | | | | 1,308,047 | | | Contingent consideration - Fair Value Adjustments | | | | 1,891,533 | | | | 2,375,658 | | | | 1,421,652 | | | TOTAL OPERATING EXPENSES | | | 21,236,086 | | | | 10,126,405 | | | | 121,354,847 | | | | 96,355,976 | | | | 53,451,286 | | | | 24,879,542 | | | | | | | | | | | | | | OPERATING LOSS | | | (21,089,211 | ) | | | (9,830,266 | ) | | | (117,216,013 | ) | | | (95,973,976 | ) | | | (53,276,286 | ) | | | (24,589,276 | ) | | OTHER INCOME (EXPENSE) | | | | | | | | | | | | | | | | | | | | Equity in income (loss) of unconsolidated affiliates | | | (240,154 | ) | | | (163,180 | ) | | | (963,407 | ) | | | - | | | | (78,874 | ) | | | (641,141 | ) | | Impairment of investment in unconsolidated affiliates | | | (1,642,775 | ) | | | — | | | | (1,642,775 | ) | | Gain on purchase of Roche Madison | | | 1,576,107 | | | | — | | | | 1,576,107 | | | Loss on sale of property and equipment, net | | | (1,079,377 | ) | | | — | | | | (1,206,465 | ) | | Realized and unrealized gain (loss) on marketable securities | | | (58,091 | ) | | | (261,219 | ) | | | 62,954 | | | Gain (loss) on sale of fixed assets, net | | | | 19,195 | | | | (58,878 | ) | | | (76,388 | ) | | Interest income (expense), net | | | 35,966 | | | | 86,530 | | | | 2,750,444 | | | | 729,158 | | | | 645,493 | | | | (97,910 | ) | | Change in value of derivatives | | | 386,892 | | | | 1,133,127 | | | | 3,281,404 | | | | 2,869,267 | | | | (6,033,659 | ) | | | (5,300,389 | ) | | Gain on sale of stock in subsidiary | | | — | | | | — | | | | 2,292,800 | | | Other income | | | — | | | | 250,000 | | | | 250,000 | | | | | | | | | | | | | | Other income (expense) | | | | 417,874 | | | | 82,092 | | | | (997,975 | ) | | TOTAL OTHER INCOME (EXPENSE) | | | (1,021,432 | ) | | | 1,045,258 | | | | 6,401,062 | | | | 4,035,494 | | | | (5,443,826 | ) | | | (7,113,803 | ) | | | | | | | | | | | | | LOSS FROM CONTINUING OPERATIONS BEFORE INCOME TAXES | | | (22,110,643 | ) | | | (8,785,008 | ) | | | (110,814,951 | ) | | LOSS BEFORE INCOME TAXES | | | | (91,938,482 | ) | | | (58,720,112 | ) | | | (31,703,079 | ) | | Provision for income taxes | | | — | | | | — | | | | — | | | | (2,400 | ) | | | (5,300 | ) | | | - | | | | | | | | | | | | | | LOSS FROM CONTINUING OPERATIONS | | | (22,110,643 | ) | | | (8,785,008 | ) | | | (110,814,951 | ) | | | (91,940,882 | ) | | | (58,725,412 | ) | | | (31,703,079 | ) | | Income (loss) from discontinued operations | | | (80 | ) | | | 1,373,396 | | | | (47,546,642 | ) | | Gain on disposal of discontinued operations | | | — | | | | 3,919,213 | | | | 4,708,588 | | | | | | | | | | | | | | NET INCOME (LOSS) FROM DISCONTINUED OPERATIONS | | | (80 | ) | | | 5,292,609 | | | | (42,838,054 | ) | | | | | | | | | | | | | Loss from discontinued operations | | | | - | | | | - | | | | (354 | ) | | NET LOSS FROM DISCONTINUED OPERATIONS | | | | - | | | | - | | | | (354 | ) | | NET LOSS | | | (22,110,723 | ) | | | (3,492,399 | ) | | | (153,653,005 | ) | | | (91,940,882 | ) | | | (58,725,412 | ) | | | (31,703,433 | ) | | Net (income) loss attributable to noncontrolling interests | | | 984,795 | | | | 363,514 | | | | 18,819,285 | | | | | | | | | | | | | | Net loss attributable to non-controlling interests | | | | - | | | | 95,222 | | | | 560,144 | | | NET LOSS ATTRIBUTABLE TO ARROWHEAD | | $ | (21,125,928 | ) | | $ | (3,128,885 | ) | | $ | (134,833,720 | ) | | $ | (91,940,882 | ) | | $ | (58,630,190 | ) | | $ | (31,143,289 | ) | | | | | | | | | | | | | Earnings per share—basic and diluted: | | | | | | | | Loss from continuing operations attributable to Arrowhead common shareholders | | $ | (1.90 | ) | | $ | (1.18 | ) | | | | Loss from discontinued operations attributable to Arrowhead common shareholders | | | — | | | | 0.74 | | | | | | | | | | | | | | | Net loss attributable to Arrowhead shareholders | | $ | (1.90 | ) | | $ | (0.44 | ) | | | | | | | | | | | | | | Weighted average shares outstanding | | | 11,129,766 | | | | 7,181,121 | | | | | | | | | | | | | | | NET LOSS PER SHARE ATTRIBUTABLE TO ARROWHEAD | | | | | | | | | | | | | | | SHAREHOLDERS - BASIC & DILUTED: | | | $ | (1.60 | ) | | $ | (1.25 | ) | | $ | (1.30 | ) | | Weighted average shares outstanding - basic and diluted | | | | 57,358,442 | | | | 46,933,030 | | | | 24,002,224 | | | OTHER COMPREHENSIVE INCOME (LOSS), NET OF TAX | | | | | | | | | | | | | | | Foreign Currency Translation Adjustments | | | | (136,425 | ) | | | - | | | | - | | | COMPREHENSIVE LOSS ATTRIBUTABLE TO ARROWHEAD | | | $ | (92,077,307 | ) | | $ | (58,630,190 | ) | | $ | (31,143,289 | ) | |
The accompanying notes are an integral part of these consolidated financial statements. F-4
F-5
Arrowhead Research Corporation and Subsidiaries (A Development Stage Company) -
Consolidated Statement of Stockholders’ Equity From inception to September 30, 2012
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | Common Stock | | | Preferred Stock | | | Additional Paid-in | | | Subscription | | | Accumulated Deficit during the | | | Noncontrolling | | | | | | | | Shares | | | Amount | | | Shares | | | Amount | | | Capital | | | Receivable | | | Development Stage | | | interest | | | Totals | | Initial Issuance of Stock: | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | Common stock & warrants issued for cash @ $0.01 per unit | | | 300,000 | | | | 3,000 | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | 3,000 | | Common stock & warrants issued for cash @ $10.00 per unit | | | 168,000 | | | | 1,680 | | | | — | | | | — | | | | 1,678,320 | | | | — | | | | — | | | | — | | | | 1,680,000 | | Stock issuance cost charged to additional paid-in capital | | | — | | | | — | | | | — | | | | — | | | | (168,000 | ) | | | — | | | | — | | | | — | | | | (168,000 | ) | Net loss for period from inception to September 30, 2003 | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | (95,238 | ) | | | — | | | | (95,238 | ) | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | Balance at September 30, 2003 | | | 468,000 | | | | 4,680 | | | | — | | | | — | | | | 1,510,320 | | | | — | | | | (95,238 | ) | | | — | | | | 1,419,762 | | Exercise of stock options | | | 7,500 | | | | 75 | | | | — | | | | — | | | | 14,925 | | | | — | | | | — | | | | — | | | | 15,000 | | Common stock & warrants issued for cash @ $10.00 per unit | | | 47,500 | | | | 475 | | | | — | | | | — | | | | 474,525 | | | | — | | | | — | | | | — | | | | 475,000 | | Common stock & warrants issued for marketable securities @ $10.00 per unit | | | 50,000 | | | | 500 | | | | — | | | | — | | | | 499,500 | | | | — | | | | — | | | | — | | | | 500,000 | | Stock issuance cost charged to additional paid-in capital | | | — | | | | — | | | | — | | | | — | | | | (96,500 | ) | | | — | | | | — | | | | — | | | | (96,500 | ) | Common stock and warrants issued for cash @ $15.00 per unit | | | 660,879 | | | | 6,609 | | | | — | | | | — | | | | 9,906,573 | | | | — | | | | — | | | | — | | | | 9,913,182 | | Common stock issued in reverse acquisition | | | 70,553 | | | | 706 | | | | — | | | | — | | | | (151,175 | ) | | | — | | | | — | | | | — | | | | (150,469 | ) | Common stock issued as a gift for $10.90 per share | | | 15,000 | | | | 163 | | | | — | | | | — | | | | 162,587 | | | | — | | | | — | | | | — | | | | 162,750 | | Common stock and warrants issued as stock issuance cost @ $15.00 per unit | | | 35,623 | | | | 356 | | | | — | | | | — | | | | 533,988 | | | | — | | | | — | | | | — | | | | 534,344 | | Stock issuance cost charged to additional paid-in capital | | | — | | | | — | | | | — | | | | — | | | | (991,318 | ) | | | — | | | | — | | | | — | | | | (991,318 | ) | Exercise of stock option @ $2.00 per share | | | 7,500 | | | | 75 | | | | — | | | | — | | | | 14,925 | | | | — | | | | — | | | | — | | | | 15,000 | | Exercise of stock options @ $10.00 per share | | | 600 | | | | 6 | | | | — | | | | — | | | | 5,994 | | | | — | | | | — | | | | — | | | | 6,000 | | Stock-based compensation | | | — | | | | — | | | | — | | | | — | | | | 175,653 | | | | — | | | | — | | | | — | | | | 175,653 | | Net loss for the year ended September 30, 2004 | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | (2,528,954 | ) | | | 1,777,699 | | | | (751,255 | ) | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | Balance at September 30, 2004 | | | 1,363,155 | | | | 13,645 | | | | — | | | | — | | | | 12,059,997 | | | | — | | | | (2,624,192 | ) | | | 1,777,699 | | | | 11,227,149 | | Exercise of warrants @ $15.00 per share | | | 1,381,289 | | | | 13,813 | | | | — | | | | — | | | | 20,705,522 | | | | — | | | | — | | | | — | | | | 20,719,335 | | Exercise of stock options @ $10.00 per share | | | 2,500 | | | | 25 | | | | — | | | | — | | | | 24,975 | | | | — | | | | — | | | | — | | | | 25,000 | | Common stock issued to purchase Insert Therapeutics share @ $39.80 per share | | | 50,226 | | | | 502 | | | | — | | | | — | | | | 1,999,498 | | | | — | | | | — | | | | | | | | 2,000,000 | | Common stock issued for services | | | 1,250 | | | | 12 | | | | — | | | | — | | | | 49,988 | | | | — | | | | — | | | | — | | | | 50,000 | | Stock-based compensation | | | — | | | | — | | | | — | | | | — | | | | 508,513 | | | | — | | | | — | | | | — | | | | 508,513 | | Change in percentage of ownership in subsidiary | | | — | | | | — | | | | — | | | | — | | | | 230,087 | | | | — | | | | — | | | | — | | | | 230,087 | | Net loss for the year ended September 30, 2005 | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | (6,854,918 | ) | | | 121,491 | | | | (6,733,427 | ) | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | Balance at September 30, 2005 | | | 2,798,419 | | | | 27,997 | | | | — | | | | — | | | | 35,578,580 | | | | — | | | | (9,479,110 | ) | | | 1,899,190 | | | | 28,026,657 | | Exercise of stock options | | | 11,579 | | | | 116 | | | | — | | | | — | | | | 341,421 | | | | — | | | | — | | | | — | | | | 341,537 | | Common stock issued @ $48.80 per share | | | 20,485 | | | | 205 | | | | — | | | | — | | | | 999,795 | | | | — | | | | — | | | | — | | | | 1,000,000 | | Common stock issued @ $38.40 per share | | | 1,500 | | | | 15 | | | | — | | | | — | | | | 57,585 | | | | — | | | | — | | | | — | | | | 57,600 | | Common stock issued @ $35.00 per share | | | 559,000 | | | | 5,590 | | | | — | | | | — | | | | 19,539,410 | | | | — | | | | — | | | | — | | | | 19,545,000 | | Common stock issued @ $59.10 per share | | | 2,536 | | | | 25 | | | | — | | | | — | | | | 149,975 | | | | — | | | | — | | | | — | | | | 150,000 | | Common stock issued to purchase Calando Pharmaceuticals, Inc. @ $51.70 per share | | | 20,838 | | | | 208 | | | | — | | | | — | | | | 1,077,125 | | | | — | | | | — | | | | — | | | | 1,077,333 | | Stock-based compensation | | | — | | | | — | | | | — | | | | — | | | | 1,369,478 | | | | — | | | | — | | | | — | | | | 1,369,478 | | Net loss for the year ended September 30, 2006 | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | (18,997,209 | ) | | | (964,752 | ) | | | (19,961,961 | ) | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | Balance at September 30, 2006 | | | 3,414,359 | | | | 34,156 | | | | — | | | | — | | | | 59,113,369 | | | | — | | | | (28,476,319 | ) | | | 934,438 | | | | 31,605,644 | | Exercise of stock options | | | 18,616 | | | | 186 | | | | — | | | | — | | | | 434,541 | | | | — | | | | — | | | | — | | | | 434,727 | | Common stock issued @ $57.80 per share, net | | | 284,945 | | | | 2,849 | | | | — | | | | — | | | | 15,149,366 | | | | — | | | | — | | | | — | | | | 15,152,215 | | Arrowhead’s increase in proportionate share of Insert Therapeutics’ equity | | | — | | | | — | | | | — | | | | — | | | | 2,401,394 | | | | — | | | | — | | | | — | | | | 2,401,394 | | Common stock issued for purchase of Carbon Nanotechnologies, Inc. @ $37.70 per share | | | 143,122 | | | | 1,431 | | | | — | | | | — | | | | 5,398,569 | | | | — | | | | — | | | | — | | | | 5,400,000 | | Stock-based compensation | | | — | | | | — | | | | — | | | | — | | | | 2,175,544 | | | | — | | | | — | | | | — | | | | 2,175,544 | | Net loss for the year ended September 30, 2007 | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | (29,931,118 | ) | | | (781,829 | ) | | | (30,712,947 | ) | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | Balance at September 30, 2007 | | | 3,861,042 | | | | 38,622 | | | | — | | | | — | | | | 84,672,783 | | | | — | | | | (58,407,437 | ) | | | 152,609 | | | | 26,456,577 | | Exercise of stock options | | | 10,536 | | | | 106 | | | | — | | | | — | | | | 289,921 | | | | — | | | | — | | | | — | | | | 290,027 | | Common stock issued at approximately $18.00 per share, net | | | 386,399 | | | | 3,867 | | | | — | | | | — | | | | 6,956,718 | | | | — | | | | — | | | | — | | | | 6,960,585 | | Arrowhead’s increase in proportionate share of Unidym’s equity | | | — | | | | — | | | | — | | | | — | | | | 1,720,962 | | | | — | | | | — | | | | — | | | | 1,720,962 | | Common stock issued @ $27.20 per share to Rice University | | | 5,000 | | | | 50 | | | | — | | | | — | | | | 135,950 | | | | — | | | | — | | | | — | | | | 136,000 | | Common stock issued @ $28.30 per share to purchase shares of Unidym, Inc. | | | 7,055 | | | | 71 | | | | — | | | | — | | | | 199,929 | | | | — | | | | — | | | | — | | | | 200,000 | | Common stock issued @ $29.50per share to purchase MASA Energy, LLC | | | 10,505 | | | | 105 | | | | — | | | | — | | | | 309,895 | | | | — | | | | — | | | | — | | | | 310,000 | | Common stock issued @ $21.90 per share to Unidym for the acquisition of Nanoconduction | | | 11,416 | | | | 114 | | | | — | | | | — | | | | 249,886 | | | | — | | | | — | | | | — | | | | 250,000 | | Common stock issued @ $21.80 per share | | | 1,500 | | | | 15 | | | | — | | | | — | | | | 32,685 | | | | — | | | | — | | | | — | | | | 32,700 | | Stock-based compensation | | | — | | | | — | | | | — | | | | — | | | | 3,187,397 | | | | — | | | | — | | | | — | | | | 3,187,397 | | Net loss for the year ended September 30, 2008 | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | (27,089,030 | ) | | | (152,609 | ) | | | (27,241,639 | ) | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | Balance at September 30, 2008 | | | 4,293,452 | | | | 42,950 | | | | — | | | | — | | | | 97,756,126 | | | | — | | | | (85,496,467 | ) | | | — | | | | 12,302,609 | | Common Stock issued @ $5.50 per share to Unidym stockholder in exchange for Unidym’s shares | | | 205,839 | | | | 2,059 | | | | — | | | | — | | | | 1,131,617 | | | | — | | | | — | | | | — | | | | 1,133,676 | | Common Stock issued @ $5.20 per share to TEL Ventures in exchange for Unidym’s shares | | | 222,222 | | | | 2,222 | | | | — | | | | — | | | | 1,156,111 | | | | — | | | | — | | | | — | | | | 1,158,333 | | Reclassification of former Unidym mezzanine debt to equity | | | — | | | | — | | | | — | | | | — | | | | 2,000,000 | | | | — | | | | — | | | | — | | | | 2,000,000 | | Arrowhead’s increase in proportionate share of Calando’s equity | | | — | | | | — | | | | — | | | | — | | | | 2,120,250 | | | | — | | | | — | | | | — | | | | 2,120,250 | | Common stock issued @ $3.00 per share | | | 919,664 | | | | 9,197 | | | | — | | | | — | | | | 2,749,796 | | | | — | | | | — | | | | — | | | | 2,758,993 | | Change in percentage ownership in subsidiary | | | — | | | | — | | | | — | | | | — | | | | 16,297 | | | | — | | | | — | | | | — | | | | 16,297 | | Stock-based compensation | | | — | | | | — | | | | — | | | | — | | | | 2,676,170 | | | | — | | | | — | | | | — | | | | 2,676,170 | | Issuance of Preferred Stock for Subscription in Unidym | | | — | | | | — | | | | — | | | | — | | | | 300,000 | | | | (300,000 | ) | | | — | | | | — | | | | — | | Amortization of discount on Unidym Series D Preferred Stock | | | — | | | | — | | | | — | | | | — | | | | 163,960 | | | | — | | | | (163,960 | ) | | | — | | | | — | | Net loss for the year ended September 30, 2009 | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | (19,308,392 | ) | | | — | | | | (19,308,392 | ) | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | Balance at September 30, 2009 | | | 5,641,177 | | | | 56,428 | | | | — | | | | — | | | | 110,070,327 | | | | (300,000 | ) | | | (104,968,819 | ) | | | — | | | | 4,857,937 | | Exercise of stock options | | | 688 | | | | 7 | | | | — | | | | — | | | | 7,624 | | | | — | | | | — | | | | — | | | | 7,631 | | Issuance of Preferred Stock for Subscription in Unidym | | | — | | | | — | | | | — | | | | — | | | | — | | | | 300,000 | | | | — | | | | — | | | | 300,000 | | Issuance of Unidym’s common stock to minority shareholders | | | — | | | | — | | | | — | | | | — | | | | 245,345 | | | | — | | | | — | | | | 54,655 | | | | 300,000 | | Common stock issued @ $6.30 per share | | | 508,343 | | | | 5,083 | | | | — | | | | — | | | | 3,217,813 | | | | — | | | | — | | | | — | | | | 3,222,896 | | Common stock issued @ $13.12 per share | | | 659,299 | | | | 6,593 | | | | — | | | | — | | | | 3,692,078 | | | | — | | | | — | | | | — | | | | 3,698,671 | | Common Stock issued to Calando stockholders in exchange for Calando’s shares | | | 122,000 | | | | 1,220 | | | | — | | | | — | | | | (160,667 | ) | | | — | | | | — | | | | 159,447 | | | | — | | Common Stock issued to Unidym stockholders in exchange for Unidym’s shares | | | 15,318 | | | | 153 | | | | — | | | | — | | | | (1,435 | ) | | | — | | | | — | | | | 1,282 | | | | — | | Stock-based compensation | | | — | | | | — | | | | — | | | | — | | | | 1,582,149 | | | | — | | | | — | | | | — | | | | 1,582,149 | | Exercise of warrants | | | 225,189 | | | | 2,250 | | | | — | | | | — | | | | 1,063,600 | | | | — | | | | — | | | | 200 | | | | 1,066,050 | | Net loss for the year ended September 30, 2010 | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | (5,774,048 | ) | | | (1,182,990 | ) | | | (6,957,038 | ) | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | Balance at September 30, 2010 | | | 7,172,014 | | | | 71,734 | | | | — | | | | — | | | | 119,716,834 | | | | — | | | | (110,742,867 | ) | | | (967,406 | ) | | | 8,078,295 | | Exercise of warrants | | | 8,656 | | | | 87 | | | | — | | | | — | | | | 43,192 | | | | — | | | | — | | | | — | | | | 43,279 | | Exercise of stock options | | | 2,700 | | | | 27 | | | | — | | | | — | | | | 13,857 | | | | — | | | | — | | | | — | | | | 13,884 | | Divestiture of Unidym | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | 254,275 | | | | 254,275 | | Issuance of preferred stock in subsidiary | | | — | | | | — | | | | — | | | | — | | | | 1,618,509 | | | | — | | | | — | | | | — | | | | 1,618,509 | | Change in percentage of ownership in subsidiary | | | — | | | | — | | | | — | | | | — | | | | (849,707 | ) | | | — | | | | — | | | | 849,707 | | | | — | | Stock-based compensation | | | — | | | | — | | | | — | | | | — | | | | 1,404,640 | | | | — | | | | — | | | | — | | | | 1,404,640 | | Common stock issued @ $3.80 per share | | | 1,458,917 | | | | 14,574 | | | | — | | | | — | | | | 4,629,110 | | | | — | | | | — | | | | — | | | | 4,643,684 | | Issuance of Common Stock for Subscription | | | | | | | | | | | — | | | | — | | | | 900,000 | | | | (900,000 | ) | | | | | | | | | | | — | | Net loss for the year ended September 30, 2011 | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | (3,128,885 | ) | | | (363,514 | ) | | | (3,492,399 | ) | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | Balance at September 30, 2011 | | | 8,642,286 | | | $ | 86,422 | | | | — | | | $ | — | | | $ | 127,476,435 | | | $ | (900,000 | ) | | $ | (113,871,752 | ) | | $ | (226,938 | ) | | | 12,564,167 | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | Exercise of stock options | | | 4,583 | | | | 45 | | | | — | | | | — | | | | 23,788 | | | | — | | | | — | | | | — | | | | 23,833 | | Stock-based compensation | | | — | | | | — | | | | — | | | | — | | | | 1,241,404 | | | | — | | | | — | | | | — | | | | 1,241,404 | | Common stock issued @ $3.80 per share | | | 138,158 | | | | 1,382 | | | | — | | | | — | | | | 523,618 | | | | (100,000 | ) | | | — | | | | — | | | | 425,000 | | Common stock issued @ $3.70 per share | | | 675,000 | | | | 6,750 | | | | — | | | | — | | | | 2,241,000 | | | | | | | | — | | | | — | | | | 2,247,750 | | Common stock issued @ $4.00 per share | | | 100,000 | | | | 1,000 | | | | — | | | | — | | | | 399,000 | | | | — | | | | — | | | | — | | | | 400,000 | | Common stock issued @ $6.23 per share | | | 83,211 | | | | 83 | | | | — | | | | — | | | | 499,918 | | | | — | | | | — | | | | — | | | | 500,001 | | Common stock issued @ $5.11 per share | | | 97,831 | | | | 98 | | | | — | | | | — | | | | 499,904 | | | | — | | | | — | | | | — | | | | 500,002 | | Common stock issued @ $2.76 per share | | | 2,260,869 | | | | 2,261 | | | | — | | | | — | | | | 5,809,979 | | | | (16,000 | ) | | | | | | | | | | | 5,796,240 | | Common stock issued under Committed Capital Agreement | | | 68,926 | | | | 689 | | | | — | | | | — | | | | (689 | ) | | | — | | | | — | | | | — | | | | — | | Common stock issued in acquisitions | | | 1,217,159 | | | | 9,332 | | | | — | | | | — | | | | 6,138,498 | | | | — | | | | — | | | | — | | | | 6,147,830 | | Fractional shares redeemed in reverse stock split | | | (131 | ) | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | Preferred stock issued @ $1,000 per share | | | — | | | | — | | | | 1,015 | | | | 1 | | | | 1,014,999 | | | | — | | | | — | | | | — | | | | 1,015,000 | | Preferred stock converted to common stock | | | 275,782 | | | | 276 | | | | (1,015 | ) | | | (1 | ) | | | (275 | ) | | | — | | | | — | | | | — | | | | — | | Exercise of Calando stock options | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | 8,000 | | | | 8,000 | | Exercise of warrants | | | 15,511 | | | | 16 | | | | — | | | | — | | | | 50,390 | | | | — | | | | — | | | | — | | | | 50,406 | | Net loss for the year ended September 30, 2012 | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | (21,125,928 | ) | | | (984,795 | ) | | | (22,110,723 | ) | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | Balance at Septemeber 30, 2012 | | | 13,579,185 | | | $ | 108,354 | | | | — | | | $ | — | | | $ | 145,917,968 | | | $ | (1,016,000 | ) | | $ | (134,997,680 | ) | | $ | (1,203,733 | ) | | $ | 8,808,910 | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Preferred Stock | | | Amount ($) | | | Common Stock | | | Amount ($) | | | Additional Paid-In Capital | | | Subscription Receivable | | | Accumulated Other Comprehensive Income (loss) | | | Accumulated Deficit | | | Non-controlling Interest | | | Totals | | Balance at September 30, 2012 | | - | | | $ | - | | | | 13,579,185 | | | $ | 108,354 | | | $ | 145,917,968 | | | $ | (1,016,000 | ) | | $ | - | | | $ | (134,997,680 | ) | | $ | (1,203,733 | ) | | $ | 8,808,909 | | Exercise of warrants | | - | | | | - | | | | 1,182,451 | | | | 1,183 | | | | 2,053,416 | | | | - | | | | - | | | | - | | | | - | | | | 2,054,599 | | Exercise of stock options | | - | | | | - | | | | 675 | | | | 1 | | | | 2,578 | | | | - | | | | - | | | | - | | | | - | | | | 2,579 | | Stock-based compensation | | - | | | | - | | | | - | | | | - | | | | 1,536,271 | | | | - | | | | - | | | | - | | | | - | | | | 1,536,271 | | Subscription payment | | - | | | | - | | | | - | | | | - | | | | - | | | | 16,000 | | | | - | | | | - | | | | - | | | | 16,000 | | Subscription reversal | | - | | | | - | | | | (267,444 | ) | | | (2,674 | ) | | | (997,326 | ) | | | 1,000,000 | | | | - | | | | - | | | | - | | | | - | | Common stock issued @ $4.49 to Roche | | - | | | | - | | | | 239,894 | | | | 240 | | | | 985,809 | | | | - | | | | - | | | | - | | | | - | | | | 986,049 | | Common stock and warrants issued @ $2.26 | | - | | | | - | | | | 1,825,079 | | | | 1,825 | | | | 3,814,643 | | | | - | | | | - | | | | - | | | | - | | | | 3,816,468 | | Common stock and warrants issued @ $2.12 | | - | | | | - | | | | 1,667,051 | | | | 1,667 | | | | 3,255,192 | | | | - | | | | - | | | | - | | | | - | | | | 3,256,859 | | Common stock and warrants issued @ $1.83 | | - | | | | - | | | | 14,262,553 | | | | 14,263 | | | | 25,445,236 | | | | - | | | | - | | | | - | | | | - | | | | 25,459,499 | | Establish and settlements related to derivative liability | | - | | | | - | | | | - | | | | - | | | | 1,600,989 | | | | - | | | | - | | | | - | | | | - | | | | 1,600,989 | | Preferred stock issued @ 1,000 per share | | 9,900 | | | | 10 | | | | - | | | | - | | | | 9,899,990 | | | | - | | | | - | | | | - | | | | - | | | | 9,900,000 | | Net loss for the year ended September 30, 2013 | | - | | | | - | | | | - | | | | - | | | | - | | | | - | | | | - | | | | (31,143,289 | ) | | | (560,144 | ) | | | (31,703,433 | ) | Balance at September 30, 2013 | | 9,900 | | | $ | 10 | | | | 32,489,444 | | | $ | 124,859 | | | $ | 193,514,766 | | | $ | - | | | $ | - | | | $ | (166,140,969 | ) | | $ | (1,763,877 | ) | | $ | 25,734,789 | | Exercise of warrants | | - | | | | - | | | | 2,911,919 | | | | 2,911 | | | | 10,145,133 | | | | - | | | | - | | | | - | | | | - | | | | 10,148,044 | | Exercise of stock options | | - | | | | - | | | | 454,863 | | | | 455 | | | | 2,729,545 | | | | - | | | | - | | | | - | | | | - | | | | 2,730,000 | | Stock-based compensation | | - | | | | - | | | | - | | | | - | | | | 5,696,173 | | | | - | | | | - | | | | - | | | | - | | | | 5,696,173 | | Common stock issued @ $5.86 | | - | | | | - | | | | 3,071,672 | | | | 3,072 | | | | 14,057,040 | | | | - | | | | - | | | | - | | | | - | | | | 14,060,112 | | Common stock issued @ $18.95 | | - | | | | - | | | | 6,325,000 | | | | 6,325 | | | | 112,575,234 | | | | - | | | | - | | | | - | | | | - | | | | 112,581,559 | | Preferred stock issued @ $1,000 per share | | 46,000 | | | | 46 | | | | - | | | | - | | | | 45,999,954 | | | | - | | | | - | | | | - | | | | - | | | | 46,000,000 | | Common stock issued to Galloway | | - | | | | - | | | | 131,579 | | | | 132 | | | | 499,868 | | | | - | | | | - | | | | - | | | | - | | | | 500,000 | | Settlements related to derivative liability | | - | | | | - | | | | - | | | | - | | | | 5,956,079 | | | | - | | | | - | | | | - | | | | - | | | | 5,956,079 | | Preferred stock converted to common stock | | (37,600 | ) | | | (38 | ) | | | 9,272,459 | | | | 9,272 | | | | (9,234 | ) | | | - | | | | - | | | | - | | | | - | | | | - | | Deconsolidation of Calando Pharmaceuticals, Inc. | | - | | | | - | | | | - | | | | - | | | | - | | | | - | | | | - | | | | - | | | | 1,303,911 | | | | 1,303,911 | | Net loss for the year ended September 30, 2014 | | - | | | | - | | | | - | | | | - | | | | - | | | | - | | | | - | | | | (58,630,190 | ) | | | (95,222 | ) | | | (58,725,412 | ) | Balance at September 30, 2014 | | 18,300 | | | $ | 18 | | | | 54,656,936 | | | $ | 147,026 | | | $ | 391,164,558 | | | $ | - | | | $ | - | | | $ | (224,771,159 | ) | | $ | (555,188 | ) | | $ | 165,985,255 | | Exercise of warrants | | - | | | | - | | | | 79,828 | | | | 81 | | | | 401,795 | | | | - | | | | - | | | | - | | | | - | | | | 401,876 | | Exercise of stock options | | - | | | | - | | | | 28,758 | | | | 29 | | | | 101,841 | | | | - | | | | - | | | | - | | | | - | | | | 101,870 | | Stock-based compensation | | - | | | | - | | | | - | | | | - | | | | 10,232,897 | | | | - | | | | - | | | | - | | | | - | | | | 10,232,897 | | Exercise of exchange rights | | - | | | | - | | | | 5,250 | | | | 5 | | | | 3,067 | | | | - | | | | - | | | | - | | | | - | | | | 3,072 | | Preferred stock converted to common stock | | (2,648 | ) | | | (2 | ) | | | 1,316,215 | | | | 1,316 | | | | (1,314 | ) | | | - | | | | - | | | | - | | | | - | | | | - | | Common stock- Restricted Stock Unit vesting | | - | | | | - | | | | 136,307 | | | | 136 | | | | (26,165 | ) | | | - | | | | - | | | | - | | | | - | | | | (26,029 | ) | Common stock issued to Novartis @ $7.53 | | - | | | | - | | | | 3,321,383 | | | | 3,321 | | | | 24,996,679 | | | | - | | | | - | | | | - | | | | - | | | | 25,000,000 | | Foreign currency translation adjustments | | - | | | | - | | | | - | | | | - | | | | - | | | | - | | | | (136,425 | ) | | | - | | | | - | | | | (136,425 | ) | Net loss for the year ended September 30, 2015 | | - | | | | - | | | | - | | | | - | | | | - | | | | - | | | | - | | | | (91,940,882 | ) | | | - | | | | (91,940,882 | ) | Balance at September 30, 2015 | | 15,652 | | | $ | 16 | | | | 59,544,677 | | | $ | 151,914 | | | $ | 426,873,358 | | | $ | - | | | $ | (136,425 | ) | | $ | (316,712,041 | ) | | $ | (555,188 | ) | | $ | 109,621,634 | |
The accompanying notes are an integral part of these consolidated financial statements. F-5F-6
Arrowhead Research Corporation and Subsidiaries ( A Development Stage Company )
Consolidated Statements of Cash Flows | | | | | | | | | | | | | | | | Year ended September 30, | | | May 7, 2003 (Date of inception) to September 30, | | | | | 2012 | | | 2011 | | | 2012 | | CASH FLOWS FROM OPERATING ACTIVITIES: | | | | | | | | | | | | | Net income (loss) | | $ | (22,110,723 | ) | | $ | (3,492,399 | ) | | $ | (153,653,005 | ) | Net (income) loss attributable to noncontrolling interests | | | 984,795 | | | | 363,514 | | | | 18,819,285 | | | | | | | | | | | | | | | Net income (loss) attributable to Arrowhead | | | (21,125,928 | ) | | | (3,128,885 | ) | | | (134,833,720 | ) | (Income) loss from discontinued operations | | | 80 | | | | (5,292,609 | ) | | | 42,838,054 | | Realized and unrealized (gain) loss on investments | | | 58,091 | | | | 261,218 | | | | (762,954 | ) | Charge for bad debt allowance | | | 2,497,300 | | | | — | | | | 2,497,300 | | (Gain) loss from sale of subsidiary | | | — | | | | — | | | | (306,344 | ) | (Gain) loss on purchase of Roche Madison | | | (1,576,107 | ) | | | — | | | | (1,576,107 | ) | Loss on disposal of fixed assets | | | 1,079,377 | | | | — | | | | 1,206,465 | | Stock issued for professional services | | | — | | | | 193,885 | | | | 741,632 | | Change in value of derivatives | | | (386,892 | ) | | | (1,133,127 | ) | | | (3,281,404 | ) | Purchased in-process research and development | | | — | | | | — | | | | 15,851,555 | | Stock-based compensation | | | 1,241,404 | | | | 1,376,921 | | | | 13,581,468 | | Depreciation and amortization | | | 1,748,975 | | | | 267,978 | | | | 7,409,286 | | Amortization (accretion) of note discounts, net | | | 9,390 | | | | (7,938 | ) | | | 1,452 | | Gain on sale of stock in subsidiary | | | — | | | | — | | | | (2,292,800 | ) | Non-cash impairment expense | | | 1,642,775 | | | | — | | | | 1,642,775 | | Equity in (income)loss of unconsolidated affiliates | | | 240,154 | | | | 163,180 | | | | 963,407 | | Noncontrolling interest | | | (984,795 | ) | | | (363,514 | ) | | | (18,819,285 | ) | Changes in operating assets and liabilities: | | | | | | | | | | | | | Receivables | | | 162,855 | | | | — | | | | 100,040 | | Other receivables | | | (938,179 | ) | | | (736,253 | ) | | | (2,543,142 | ) | Prepaid expenses | | | (338,531 | ) | | | 99,005 | | | | (481,099 | ) | Other current assets | | | (6,853 | ) | | | 18,473 | | | | (103,213 | ) | Deposits | | | (23,747 | ) | | | — | | | | (60,542 | ) | Accounts payable | | | 291,876 | | | | 157,079 | | | | 498,310 | | Accrued expenses | | | 186,369 | | | | 452,843 | | | | 720,909 | | Other liabilities | | | 882,296 | | | | 15,751 | | | | 1,130,161 | | | | | | | | | | | | | | | NET CASH USED IN OPERATING ACTIVITIES OF CONTINUING OPERATIONS | | | (15,340,090 | ) | | | (7,655,993 | ) | | | (75,877,796 | ) | CASH FLOWS FROM INVESTING ACTIVITIES OF CONTINUING OPERATIONS: | | | | | | | | | | | | | Purchase of property and equipment | | | (479,710 | ) | | | (9,674 | ) | | | (4,045,309 | ) | Proceeds from sale of investments | | | 509,009 | | | | 1,534,687 | | | | 3,313,609 | | Proceeds from sale of fixed assets | | | 290,312 | | | | — | | | | 432,687 | | Cash transferred in acquisition/divestitures | | | 121,033 | | | | (1,700,398 | ) | | | (1,579,365 | ) | Purchase of marketable securities—US Treasury Bills | | | — | | | | — | | | | (18,575,915 | ) | Purchase of MASA Energy, LLC | | | — | | | | — | | | | (250,000 | ) | Minority equity investment | | | — | | | | — | | | | (2,000,000 | ) | Cash paid for interest in Insert | | | — | | | | — | | | | (10,150,000 | ) | Cash obtained from interest in Insert | | | — | | | | — | | | | 10,529,594 | | Proceeds from sale of marketable securities—US Treasury Bills | | | — | | | | — | | | | 18,888,265 | | Proceeds from sale of subsidiaries | | | — | | | | — | | | | 359,375 | | Payment for patents | | | — | | | | — | | | | (303,440 | ) | Restricted cash | | | — | | | | — | | | | 50,773 | | | | | | | | | | | | | | | NET CASH PROVIDED BY (USED IN) INVESTING ACTIVITIES OF CONTINUING OPERATIONS | | | 440,644 | | | | (175,385 | ) | | | (3,329,726 | ) | CASH FLOWS FROM FINANCING ACTIVITIES OF CONTINUING OPERATIONS: | | | | | | | | | | | | | Principal payments on capital leases | | | (196,606 | ) | | | — | | | | (196,606 | ) | Proceeds from issuance of Calando debt | | | — | | | | — | | | | 2,516,467 | | Proceeds from issuance of stock in subsidiary | | | 8,000 | | | | 1,718,932 | | | | 20,902,100 | | Proceeds from issuance of common stock and warrants, net | | | 10,958,231 | | | | 4,507,389 | | | | 106,253,011 | | | | | | | | | | | | | | | NET CASH PROVIDED BY FINANCING ACTIVITIES OF CONTINUING OPERATIONS | | | 10,769,625 | | | | 6,226,321 | | | | 129,474,972 | | | | | | | | | | | | | | | Cash flows from discontinued operations: | | | | | | | | | | | | | Operating cash flows | | | (280 | ) | | | 2,265,284 | | | | (46,003,787 | ) | Investing cash flows | | | — | | | | — | | | | 790,625 | | Financing cash flows | | | — | | | | — | | | | (1,677,000 | ) | | | | | | | | | | | | | | Net cash provided by (used in) discontinued operations: | | | (280 | ) | | | 2,265,284 | | | | (46,890,162 | ) | | | | | | | | | | | | | | NET INCREASE(DECREASE) IN CASH | | | (4,130,101 | ) | | | 660,227 | | | | 3,377,288 | | CASH AT BEGINNING OF PERIOD | | | 7,507,389 | | | | 6,847,162 | | | | — | | | | | | | | | | | | | | | CASH AT END OF PERIOD | | $ | 3,377,288 | | | $ | 7,507,389 | | | $ | 3,377,288 | | | | | | | | | | | | | | | Supplementary disclosures: | | | | | | | | | | | | | Interest paid | | $ | 46,269 | | | $ | 105,000 | | | $ | 280,688 | | Taxes paid | | $ | — | | | $ | 742,500 | | | $ | 742,500 | |
| Year ended September 30, | | | 2015 | | | 2014 | | | 2013 | | CASH FLOWS FROM OPERATING ACTIVITIES: | | | | | | | | | | | | Net loss | $ | (91,940,882 | ) | | $ | (58,725,412 | ) | | $ | (31,703,433 | ) | Net loss attributable to non-controlling interests | | - | | | | 95,222 | | | | 560,144 | | Net loss attributable to Arrowhead | | (91,940,882 | ) | | | (58,630,190 | ) | | | (31,143,289 | ) | Loss from discontinued operations | | - | | | | - | | | | 354 | | (Gain) loss on disposal of fixed assets | | (19,195 | ) | | | 58,878 | | | | 76,388 | | Change in value of derivatives | | (2,869,267 | ) | | | 6,033,659 | | | | 5,300,389 | | Contingent consideration - fair value adjustments | | 1,891,533 | | | | 2,375,658 | | | | 1,421,652 | | Acquired in-process research and development | | 10,142,786 | | | | - | | | | - | | Stock-based compensation | | 10,232,897 | | | | 5,696,173 | | | | 1,536,271 | | Depreciation and amortization | | 2,336,207 | | | | 1,345,655 | | | | 1,751,412 | | Amortization of note premiums | | 1,110,524 | | | | 793,887 | | | | 128,406 | | Gain on debt extinguishment | | - | | | | (84,721 | ) | | | - | | Noncash gain in equity investment | | - | | | | (87,197 | ) | | | - | | Noncash impairment expense | | - | | | | 2,172,387 | | | | 2,315,721 | | Non-controlling interest | | - | | | | (95,222 | ) | | | (560,144 | ) | Changes in operating assets and liabilities: | | | | | | | | | | | | Receivables | | - | | | | 75,000 | | | | (65,625 | ) | Other receivables | | (789,090 | ) | | | (25,867 | ) | | | 1,080 | | Prepaid expenses | | (2,693,839 | ) | | | (15,812 | ) | | | 44,713 | | Other current assets | | (2,492 | ) | | | (13,287 | ) | | | (1,811 | ) | Accounts payable | | 2,497,804 | | | | 1,412,275 | | | | 321,647 | | Accrued expenses | | 4,435,784 | | | | 3,478,094 | | | | 27,920 | | Other | | (40,385 | ) | | | 94,257 | | | | (187,910 | ) | NET CASH USED IN OPERATING ACTIVITIES | | (65,707,615 | ) | | | (35,416,373 | ) | | | (19,032,826 | ) | CASH FLOWS FROM INVESTING ACTIVITIES: | | | | | | | | | | | | Cash paid for acquisitions | | (10,000,000 | ) | | | - | | | | - | | Purchases of property and equipment | | (1,970,612 | ) | | | (1,717,362 | ) | | | (296,880 | ) | Proceeds from sale of fixed assets | | 500 | | | | 10,000 | | | | 89,505 | | Purchase of marketable securities | | - | | | | (46,365,528 | ) | | | (10,732,571 | ) | Proceeds from sale of marketable securities | | 26,090,950 | | | | 11,591,120 | | | | 1,419,079 | | NET CASH PROVIDED BY (USED IN) INVESTING ACTIVITIES | | 14,120,838 | | | | (36,481,770 | ) | | | (9,520,867 | ) | CASH FLOWS FROM FINANCING ACTIVITIES: | | | | | | | | �� | | | | Principal payments on capital leases | | (213,991 | ) | | | (225,406 | ) | | | (214,801 | ) | Proceeds from issuance of common stock and preferred stock, net | | - | | | | 172,641,671 | | | | 42,448,826 | | Proceeds from the exercise of warrants and stock options | | 504,512 | | | | 12,878,044 | | | | 2,057,178 | | NET CASH PROVIDED BY FINANCING ACTIVITIES | | 290,521 | | | | 185,294,309 | | | | 44,291,203 | | Cash flows from discontinued operations: | | | | | | | | | | | | Operating cash flows | | - | | | | - | | | | (354 | ) | Investing cash flows | | - | | | | - | | | | - | | Financing cash flows | | - | | | | - | | | | - | | Net cash used in discontinued operations: | | - | | | | - | | | | (354 | ) | NET INCREASE (DECREASE) IN CASH | | (51,296,256 | ) | | | 113,396,166 | | | | 15,737,156 | | CASH AT BEGINNING OF PERIOD | | 132,510,610 | | | | 19,114,444 | | | | 3,377,288 | | CASH AT END OF PERIOD | $ | 81,214,354 | | | $ | 132,510,610 | | | $ | 19,114,444 | | Supplementary disclosures: | | | | | | | | | | | | Interest paid | $ | 14,429 | | | $ | 25,635 | | | $ | 42,044 | | 3,321,383 shares of Common stock issued to Novartis for asset acquisition | $ | 25,000,000 | | | $ | - | | | $ | - | | 131,579 shares of Common stock issued to Galloway Limited in settlement of services agreement | $ | - | | | $ | 500,000 | | | $ | - | | 239,894 shares of Common stock issued to Roche for stock and asset acquisition | $ | - | | | $ | - | | | $ | 986,049 | |
The accompanying notes are an integral part of these consolidated financial statements. F-6
SUPPLEMENTARY NON CASH TRANSACTIONS
All Arrowhead share amounts have been adjusted to reflect the 1 for 10 reverse stock split effected on November 17, 2011.
On March 23, 2005, Arrowhead purchased 7,375,000 shares of Insert Therapeutics, Inc. common stock from two minority stockholders of Insert for 50,226 newly issued shares of Arrowhead Common Stock valued at $2,000,000 based on the closing market price of Arrowhead Common Stock on NASDAQ on the date of the closing.
On March 31, 2006, Arrowhead purchased 964,000 shares of Calando Pharmaceuticals, Inc. common stock from minority stockholders of Calando for $1,928,000 consisting of 20,838 newly issued shares of Arrowhead Common Stock valued at $1,077,333 plus $850,667 in cash. The 20,838 shares of Arrowhead Common Stock were valued based on the average closing price of Arrowhead’s Common Stock on NASDAQ the ten trading days immediately prior to the date of the closing.
On April 20, 2007, Arrowhead purchased the Series E Preferred Stock of Carbon Nanotechnologies, Inc. in exchange for 143,122 shares of Arrowhead Common Stock with an estimated fair market value of $5,400,000 based on the average closing price of Arrowhead’s Common Stock on NASDAQ the ten trading days immediately prior to March 24, 2007, as set forth in the Agreement and Plan of Merger among Unidym, Carbon Nanotechnologies, Inc., Arrowhead, and others.
On April 23, 2008, Arrowhead purchased 200,000 shares of the Common Stock of Unidym Inc., in exchange for 7,054 shares of Arrowhead Common Stock with an estimated fair market value of $200,000 based on the average closing price of Arrowhead’s Common Stock on NASDAQ the ten trading days immediately prior to the date of the closing.
On April 29, 2008, Arrowhead purchased all of the membership units of MASA Energy, LLC for $560,000. The purchase price consisted of 10,504 shares of Arrowhead Common Stock with an estimated fair market value of $310,000 based on the average closing price of Arrowhead’s Common Stock on NASDAQ the ten trading days immediately prior to the date of the closing, plus $250,000 in cash.
On August 8, 2008, Unidym acquired all of the outstanding stock of Nanoconduction, Inc. in exchange for 11,411 shares of Arrowhead stock with an estimated fair market value of $250,000.
On June 11, 2009, Arrowhead issued 132,462 shares of Common Stock with an estimated fair market value of $688,802 in exchange for an equal number of Series A Preferred Stock of Unidym, with minority stockholders of Unidym.
On June 25, 2009, Arrowhead issued 194,444 shares of Common Stock with an estimated fair market value of $972,222 in exchange for an equal number of Series C Preferred Stock of Unidym, with a minority stockholder of Unidym.
On September 22, 2009, Arrowhead issued 9,149 shares of Common Stock with an estimated fair market value of $46,662 in exchange for an equal number of Series A Preferred Stock of Unidym with a minority stockholder of Unidym.
On September 28, 2009, Arrowhead issued 64,227 shares of Common Stock with an estimated fair market value of $398,209 in exchange for 5,574 shares of Series A Preferred Stock and 636,699 shares of Series C Preferred Stock of Unidym, with several minority stockholders of Unidym.
On September 30, 2009, Arrowhead issued 27,777 shares of Common Stock with an estimated fair market value of $186,111 in exchange for an equal number of Series C-1 Preferred Stock of Unidym, with a minority stockholder of Unidym.
In October and November 2009, Arrowhead issued 15,317 shares of Common Stock with an estimated fair market value of $47,485 in exchange for an equal number of shares of Series C Preferred Stock of Unidym, with several minority stockholders of Unidym.
In October and November 2009, Arrowhead issued 114,000 shares of Common Stock with an estimated fair market value of $706,800 in exchange for 2,850,000 shares of Calando’s common stock, with several minority stockholders of Calando. In conjunction with the exchange, Arrowhead also issued 24,000 Warrants to purchase Arrowhead Common Stock in exchange for 600,000 Warrants to purchase Calando common stock.
In February 2010, Arrowhead issued 8,000 shares of Common Stock and 2,400 warrants to purchase Arrowhead Common Stock, at an exercise price of $5.00, to several Calando shareholders, in exchange for 200,000 shares of Calando common stock and 60,000 warrants to purchase Calando common stock.
In March 2010, a warrant holder exercised 24,788 warrants to purchase Arrowhead Common Stock, in a cashless exercise, whereby Arrowhead issued to the warrant holder 12,870 shares of Arrowhead Common Stock.
In September 2010, Arrowhead issued warrants to purchase 390,625 shares of Arrowhead Common Stock, at an exercise price of $5.00, to two Calando shareholders, in exchange for 1,562.5 shares of Series A Preferred Stock of Calando Pharmaceuticals, Inc.
On October 21, 2011, Arrowhead entered into a Stock and Asset Purchase Agreement whereby in acquired all of the outstanding common stock of Roche Madison Inc. and certain intellectual property rights in exchange for 1,288,158 shares of Arrowhead Common Stock, a promissory note of $50,000, and potential contingent consideration based on the achievement of certain regulatory milestones, and sales milestones and royalty payments after drug approval.
On April 5, 2012, Arrowhead entered into a Stock Purchase Agreement whereby it acquired all of the outstanding common stock of Alvos Therapeutics, Inc. for 315,457 shares of Arrowhead Common Stock and potential contingent consideration based on the achievement of certain clinical, regulatory and sales milestones.
F-7
Arrowhead Research Corporation (A Development Stage Company)
Notes to Consolidated Financial Statements September 30, 2012
Unless otherwise noted, (1) the term “Arrowhead” refers to Arrowhead Research Corporation, a Delaware corporation, (2) the terms the “Company,” “we,” “us,” and “our,” refer to the ongoing business operations of Arrowhead and its Subsidiaries, whether conducted through Arrowhead or a subsidiary of Arrowhead, (3) the term “Subsidiaries” refers collectively to Arrowhead Madison Inc. (“Arrowhead Madison”) , Alvos Therapeutics, Inc. (“Alvos”), Calando Pharmaceuticals, Inc.Arrowhead Australia Pty Ltd (“Calando”Arrowhead Australia”), and Ablaris Therapeutics, Inc. (“Ablaris”), Agonn Systems, Inc. (“Agonn”), and Tego Biosciences Corporation (“Tego”) as well as our former subsidiary, Unidym, Inc. (“Unidym”), which was divested in January 2011, (4) the term “Minority Investments” refers collectively to Nanotope, Inc. (“Nanotope”) and Leonardo Biosystems, Inc. (“Leonardo”) in which the company holds a less than majority ownership position, and (5) the term “Common Stock” refers to Arrowhead’s Common Stock, (5) the term “Preferred Stock” refers to Arrowhead’s Preferred Stock and the term “stockholder(s)“Stockholder(s)” refers to the holders of Arrowhead Common Stock. All Arrowhead share and per share data have been adjusted to reflect a one for ten reverse stock split effected on November 17, 2011.
NOTE 1.ORGANIZATION AND SIGNIFICANT ACCOUNTING POLICIES Nature of Business and Going Concern Arrowhead Research Corporationdevelops novel drugs to treat intractable diseases by silencing the genes that cause them. Using the industry’s broadest portfolio of RNA chemistries and efficient modes of delivery, Arrowhead therapies trigger the RNA interference mechanism to induce rapid, deep and durable knockdown of target genes. RNA interference (RNAi) is a clinical stage targeted therapeutics companymechanism present in living cells that inhibits the expression of a specific gene, thereby affecting the production of a specific protein. Deemed to be one of the most important recent discoveries in life science with development programs in oncology, obesity, and chronic hepatitis B virus infection. Our novel delivery technologies have the potential to enable revolutionary new classestransform medicine, the discoverers of drugs, such as RNAi interferencewere awarded a Nobel Prize in 2006 for their work. Arrowhead’s RNAi-based therapeutics leverage this natural pathway of gene silencing to target and peptide drug conjugates, for a broad rangeshut down specific disease causing genes. Liquidity Historically, the Company’s primary source of unmet medical needs. Our pipeline of targeted therapeutics are designed to have increased effectiveness through guided delivery and decreased toxicity through reduction of side effects associated with unwanted exposure in healthy cells and tissues. Liquidity
Arrowheadfinancing has historically financed its operationsbeen through the sale of securities of Arrowheadits securities. Research and its Subsidiaries. Developmentdevelopment activities have required significant capital investment since the Company’s inception and weinception. We expect our current portfolio companiesoperations to continue to require cash investment in fiscal 2013to pursue our research and beyonddevelopment goals, including clinical trials and related drug manufacturing. Based upon the Company’s current cash resources and operating plan, the Company expects to continue development.have sufficient liquidity to fund operations for at least the next twelve months.
At September 30, 2012,2015, the Company had $3.4$81.2 million in cash to fund operations. In addition to its cash resources, the Company has invested excess cash in investment grade commercial bonds maturing in less than 12 months. These bonds provide a source of liquidity, though the Company plans to hold them until maturity. At September 30, 2015, the Company had invested $17.5 million in bonds. During the year ended September 30, 2012,2015, the Company’s cash position decreased by $4.1 million. The Company received$51.3 million which was primarily the result of cash from the issuanceoutflows related to operating activities of equity of $11.0$65.7 million, cash from the sale of its holdings of stock in Wisepower Co. Ltd of $0.5 million, and cash collections from licensing revenue of $0.2 million. The company had cash outflow of $16.0 million related to its continuing operating activities and capital expenditures of $0.5 million. As a result of the sale of the Company’s subsidiary, Unidym, in January 2011, the Company received $2.5 million in stock of the acquirer, Wisepower Co. Ltd. (“Wisepower”) and a $2.5 million convertible bond from Wisepower, of which approximately $200,000 is owed to a third party who was a minority investor in Unidym. Following the divestiture, through the quarter ended December 31, 2011, the Company liquidated its position in Wisepower stock. The convertible bond has a face value of $2.5 million and can be redeemedpaid for cash on January 17, 2013, and at which time could represent an additional source of liquidity for the company. In September 2011, the Company entered into an equity line facility whereby it has the ability to draw capital up to $15 million, subject to certain provisions, including maintaining a minimum stock price of $2.00 per share. Through September 30, 2012, the Company has drawn $1 million from this facility. In December 2012, the Company sold 1.9 million units at a price of $2.26 per unit in a public offering. Each unit consisted of one share of Common Stock and a warrant to purchase 0.5 share of Common Stock, exercisable at $2.20. Gross proceeds from the offering were $4.3 million, which included a $500,000 promissory note due February 1, 2013.
On October 21, 2011, Arrowhead completed the acquisition of certain RNAi assets from Hoffmann-La Roche Inc.Novartis of $10.0 million (see footnote 2) and F. Hoffmann-La Roche Ltd., including intellectual propertycapital expenditures of $2.0 million, partially offset by maturities of fixed income investments totaling $26.1 million and a research and development facility in Madison, Wisconsin. At the time of the acquisition, the facility had 41 employees. Due to the costs associated with maintaining and operating the facility, including personnel costs, rent, research and development expenses, and other costs, cash expenses have increased, and it is expected that the Company will incur higher cash expenses during the remainder of 2012 and beyond, relative to periods prior to the acquisition, as the Company accelerates its preclinical and clinical development efforts.
Going Concern
The accompanying consolidated financial statements have been prepared on a going concern basis, which contemplates the realization of assets and the satisfaction of liabilities and commitments in the normal course of business. The Company has experienced negative cash flows from operations since inception and has an accumulated deficit of approximately $135 million. The Company has funded its activities to date almost exclusively from equity financings.
F-8
The Company plans to fund its development activities as cash resources allow. However, the company is dependent on additional equity financing and/or the signing of collaboration/partnership arrangements to supply cash for future development. The Company cannot be certain that such funding will be available on acceptable terms or available at all. To the extent that the Company raises additional funds by issuing equity securities, its stockholders may experience significant dilution. If the Company is unable to raise funds when required or on acceptable terms, it may have to delay, scale back, or discontinue the development and/or commercialization of one or more product candidates, or relinquish or otherwise dispose of rights to technologies, product candidates, or products that it would otherwise seek to develop or commercialize itself and possibly cease operations.
In addition to the normal risks associated with a new business venture, there can be no assurance that the Company’s research and development will be successfully completed or that any product will be approved or commercially viable. The Company is subject to risks common to companies in the biotechnology industry including, but not limited to, dependence on collaborative arrangements, development by the Company or its competitors of new technological innovations, dependence on key personnel, protection of proprietary technology, and compliance with Food and Drug Administration (“FDA”) and other governmental regulations and approval requirements.
These matters raise substantial doubt about the Company’s ability to continue as a going concern. These financial statements were prepared under the assumption that the Company will continue as a going concern and do not include any adjustments that might resultproceeds from the outcomeexercise of that uncertainty.warrants and options of $0.5 million.
Although the Company has sources of liquidity, as described above, the Company anticipates that further equity financings, and/or asset sales and license agreements will be necessary to continue to fund operations in the future.
Summary of Significant Accounting Policies Principles of Consolidation—The consolidated financial statements include the accounts of Arrowhead and its Subsidiaries,Subsidiaries. Arrowhead’s primary operating subsidiary is Arrowhead Madison, Alvos, Calando, Ablaris, Tego, Agonn,which is located in Madison, Wisconsin, where the Company’s research and until its disposition in January 2011, Unidym. Prior to April 2008, Arrowhead’s Subsidiaries included Insert Therapeutics, Inc. (“Insert”), which was merged with Calando in April 2008. The merged entity is majority-owned by Arrowhead and continues to operate under the name of Calando. In January 2011, Arrowhead sold its interests in Unidym to Wisepower, and in December 2009, Tego completed a sale of its assets to Luna Innovations, Inc. Unidym and Tego resultsdevelopment facilities are included in the Income (Loss) from Discontinued Operations. Income (Loss) from Discontinued Operations also includes Aonex Technologies, Inc., sold in May 2008, and Nanotechnica, Inc., dissolved in June 2005.located. All significant intercompany accounts and transactions are eliminated in consolidation, and noncontrolling interests are accounted for in the Company’s financial statements.consolidation. Basis of Presentation and Use of Estimates—The preparation of financial statements in conformity with accounting principles generally accepted accounting principlesin the United States requires management to make estimates and assumptions that affect the reported amounts reported in the accompanying financial statements. Significant estimates made in preparing these financial statements include valuing the stock of the Subsidiaries, assumptions to calculate stock-based compensation expense, allowance for doubtful accounts, deferred tax asset valuation allowance, derivative assets and liabilities noncontrolling interest and useful livesdisclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. In the opinion of management, all adjustments, consisting of normal recurring accruals, considered necessary for depreciable and amortizable assets.a fair presentation have been included. Actual results could materially differ from those estimates. Additionally, certain reclassifications have been made to prior period financial statements to conform to the current period presentation. In the opinion of management, all adjustments, including normal recurring accruals considered necessary for a fair presentation, have been included. Cash and Cash Equivalents—The Company considers all liquid debt instruments purchased with a maturity of three months or less to be cash equivalents. The Company had no restricted cash at September 30, 2015 and September 30, 2014. Concentration of Credit Risk—The Company maintains checkingseveral bank accounts for Arrowhead and separate accounts for each Subsidiaryits operations at any of threetwo financial institutions. These accounts are insured by the Federal Deposit Insurance Corporation (FDIC) for up to $250,000 per account.institution. Management believes the Company is not exposed to significant credit risk due to the financial position of the depository institutioninstitutions in which these deposits are held. F-8
Investments—The Company may invest excess cash balances in short-term and long-term marketable debt securities. Investments may consist of certificates of deposits, money market accounts, government-sponsored enterprise securities, corporate bonds and/or commercial paper. The Company accounts for its investment in marketable securities in accordance with FASB ASC 320, Investments – Debt and Equity Securities. This statement requires certain securities to be classified into three categories: Held-to-maturity—Debt securities that the entity has the positive intent and ability to hold to maturity are reported at amortized cost. Trading Securities—Debt and equity securities that are bought and held primarily for the purpose of selling in the near term are reported at fair value, with unrealized gains and losses included in earnings. Available-for-Sale—Debt and equity securities not classified as either securities held-to-maturity or trading securities are reported at fair value with unrealized gains or losses excluded from earnings and reported as a separate component of shareholders’ equity. The Company classifies its investments in marketable debt securities based on the facts and circumstances present at the time of purchase of the securities. At September 30, 2015, the Company classified all of its investments as held-to-maturity. Held-to-maturity investments are measured and recorded at amortized cost on the Company’s Consolidated Balance Sheet. Discounts and premiums to par value of the debt securities are amortized to interest income/expense over the term of the security. No gains or losses on investment securities are realized until they are sold or a decline in fair value is determined to be other-than-temporary. Property and Equipment—Property and equipment are recorded at cost, which may equal fair market value in the case of property and equipment acquired in conjunction with a business acquisition. Depreciation of property and equipment is recorded using the straight-line method over the respective useful lives of the assets ranging from three to seven years. Leasehold improvements are amortized over the lesser of the expected useful life or the remaining lease term. Long-lived assets, including property and equipment are reviewed for impairment whenever events or circumstances indicate that the carrying amount of these assets may not be recoverable. F-9
Intangible Assets subjectSubject to amortization—Amortization—At September 30, 2012,2015, intangible assets subject to amortization includedinclude certain patents and certain license agreements. Intangible assets subject to amortization are reviewed for impairment whenever events or circumstances indicate that the carrying amount of these assets may not be recoverable. In-Process Research & Development (IPR&D) – —IPR&D assets represent capitalized on-going research projects that Arrowheadwere acquired through business combinations. Such assets are initially measured at their acquisition date fair values. The amounts capitalized are being accounted for as indefinite-lived intangible assets, subject to impairment testing until completion or abandonment of R&D efforts associated with the project. Upon successful completion of a project, Arrowhead will make a determination as to the then remaining useful life of the intangible asset and begin amortization. Based on early adoption of ASU 2012-02, Arrowhead tests its indefinite-lived assets for impairment at least annually, through a two-step process. The first step is a qualitative assessment to determine if it is more likely than not that the indefinite lived assets are impaired. Arrowhead considers relevant events and circumstances that could affect the inputs used to determine the fair value of the intangible assets. If the qualitative assessment indicates that it is more likely than not that the intangible assets isare impaired, a second step is performed which is a quantitative test to determine the fair value of the intangible asset. If the carrying amount of the intangible assets exceeds its fair value, an impairment loss is recorded in the amount of that excess. If circumstances determine that it is appropriate, the Company may also elect to bypass step one, and proceed directly to the second step. Equity Investments—Arrowhead has a noncontrolling equity investment in Nanotope, a privately held biotechnology company, which is recorded in Other Assets. Historically, this investment was carried at cost less Arrowhead’s proportionate share of Nanotope’s operating lossF-9
Contingent Consideration—The consideration for the period since investment. BasedCompany’s acquisitions often includes future payments that are contingent upon the occurrence of a particular event. For example, milestone payments might be based on the lackachievement of significant progress in Nanotope’s researchvarious regulatory approvals or future sales milestones, and development efforts,royalty payments might be based on drug product sales levels. The Company records a contingent consideration obligation for such contingent payments at fair value on the investment has been fully reserved and is carried at a net bookacquisition date. The Company estimates the fair value of zero. Arrowhead utilizescontingent consideration obligations through valuation models designed to estimate the equity methodprobability of accounting as it owns more than 20% of the voting equitysuch contingent payments based on various assumptions and has the abilityincorporating estimated success rates. Estimated payments are discounted using present value techniques to exercise significant influence over this company. Minority Equity Investments—The Company’s minority equity investment in Leonardo, a privately held biotechnology company, has been recorded in Other Assets, however, an impairment charge has been recorded in 2012, and its net bookarrive at estimated fair value at September 30, 2012 is zero. This investment has been accounted for under the cost method of accounting.
Noncontrolling Interests in Majority-Owned Subsidiaries—Operating losses applicable to majority-owned Calando, Ablaris and, prior to its disposal, Unidym have periodically exceeded the noncontrolling interestsbalance sheet date. Changes in the equity capital of either Subsidiary. Such excess losses applicable to the noncontrolling interests have been and are borne by the Company as there is no obligation of the noncontrolling interests to fund any losses in excess of their original investment. There is also no obligation or commitment on the part of the Company to fund operating losses of any Subsidiary whether wholly-owned or majority-owned. The Company allocates the noncontrolling interest’s share of net loss in excess of the noncontrolling interest’s initial investment in accordance with FASB ASC 810-10, which was effective for the Company on October 1, 2009.
When there is a change in the Company’s proportionate share of a development-stage Subsidiary resulting from additional equity transactions in a Subsidiary, the change is accounted for as an equity transaction in consolidation. To the extent that the increase in the calculatedfair value of the contingent consideration obligations are recognized within the Company’s interestConsolidated Statements of Operations. Changes in the equityfair value of the Subsidiary exceedscontingent consideration obligations can result from changes to one or multiple inputs, including adjustments to the Company’s investmentdiscount rates, changes in the offering, that increaseamount or timing of expected expenditures associated with product development, changes in the amount or timing of cash flows from products upon commercialization, changes in the assumed achievement or timing of any development milestones, changes in the probability of certain clinical events and changes in the assumed probability associated with regulatory approval. These fair value measurements are based on significant inputs not observable in the market. Substantial judgment is referred toemployed in determining the appropriateness of these assumptions as the Company’s “increase in its proportionate share of the Subsidiary’s equity”acquisition date and for each subsequent period. Accordingly, changes in assumptions could have a material impact on the amount is recorded as an increaseof contingent consideration expense the Company records in the Company’s Additional Paid-in Capital.any given period.
Revenue Recognition—Revenue from license fees are recorded when persuasive evidence of an arrangement exists, title has passed or services have been rendered, a price is fixed and determinable, and collection is reasonably assured. WeThe Company may generate revenue from product sales, technology licenses, collaborative research and development arrangements, and research grants. Revenue under technology licenses and collaborative agreements typically consists of nonrefundable and/or guaranteed technology license fees, collaborative research funding and various milestone and future product royalty or profit-sharing payments. Revenue associated withPayments under collaborative research and development funding payments under collaborative agreements isare recognized as revenue ratably over the relevant periods specified in the agreement, generally the period during which research and development period.is conducted. Revenue from up-front license fees, milestones and product royalties are recognized as earned based on the completion of the milestones and product sales, as defined in the respective agreements. Payments received in advance of recognition as revenue are recorded as deferred revenue.
Allowance for Doubtful Accounts—The Company accrues an allowance for doubtful accounts based on estimates of uncollectible revenues by analyzing historical collections, accounts receivable aging and other factors. Accounts receivable are written off when all collection attempts have failed. Research and Development—Costs and expenses that can be clearly identified as research and development are charged to expense as incurred in accordance with FASB ASC 730-10. Included in research and development costs are operating costs, facilities, supplies, external services, clinical trial and manufacturing costs, overhead directly related to the Company’s research and development operations, and costs to acquire technology licenses. Earnings (Loss) per Share—Basic earnings (loss) per share is computed using the weighted-average number of common shares outstanding during the period. Diluted earnings (loss) per share are computed using the weighted-average number of common shares and dilutive potential common shares outstanding during the period. Dilutive potential common shares primarily consist of stock options and restricted stock units issued to employees and consultants and warrants to purchase Common Stock of the Company. All outstanding stock options, restricted stock units and warrants for the years ended September 30, 2015, 2014 and 2013 have been excluded from the calculation of Diluted earnings (loss) per share due to their anti-dilutive effect. F-10
Stock-Based Compensation—The Company accounts for share-based compensation arrangements in accordance with FASB ASC 718, which requires the measurement and recognition of compensation expense for all share-based payment awards to be based on estimated fair values. We useThe Company uses the Black-Scholes option valuation model to estimate the fair value of ourits stock options at the date of grant. The Black-Scholes option valuation model requires the input of subjective assumptions to calculate the value of stock options. We useFor restricted stock units, the value of the award of based on the Company’s stock price at the grant date. For performance-based stock awards, the value of the award is measured at the grant date. The Company uses historical data amongand other information to estimate the expected price volatility and the expected forfeiture rate. Expense is recognized over the vesting period, commencing at the time the Company determines the achievement of performance conditions is probable. This determination requires significant judgment by management. F-10
Derivative Assets and Liabilities—The Company accounts for warrants and other derivative financial instruments as either equity or assets/liabilities based upon the characteristics and provisions of each instrument. Warrants classified as equity are recorded as additional paid-in capital on the Company’s Consolidated Balance Sheet. Some of the Company’s warrants were determined to be ineligible for equity classification because of provisions that may result in an adjustment to their exercise price. Warrants classified as derivative liabilities and other derivative financial instruments that require separate accounting as assets or liabilities are recorded on the Company’s Consolidated Balance Sheet at their fair value on the date of issuance and are revalued on each subsequent balance sheet date until such instruments are exercised or expire, with any changes in the fair value between reporting periods recorded as other income or expense. The Company estimates the fair value of these assets/liabilities using option pricing models that are based on the individual characteristics of the warrants or instruments on the valuation date, as well as assumptions for expected volatility, expected life and risk-free interest rate. Income Taxes—The Company accounts for income taxes under the liability method, which requires the recognition of deferred income tax assets and liabilities for the expected future tax consequences of events that have been included in the financial statements or tax returns. Under this method, deferred income taxes are recognized for the tax consequences in future years of differences between the tax bases of assets and liabilities and their financial reporting amounts at each period end based on enacted tax laws and statutory tax rates applicable to the periods in which the differences are expected to affect taxable income. Valuation allowances are established, when necessary, to reduce deferred income tax assets to the amount expected to be realized. The provision for income taxes, if any, represents the tax payable for the period and the change in deferred income tax assets and liabilities during the period. Recently IssuedRecent Accounting StandardsPronouncements
In July 2012,November 2015, the FASB issued ASU 2012-02,Testing Indefinite-Lived Intangible Assets2015-17, Balance Sheet Classification of Deferred Taxes, which simplifies the presentation of deferred income taxes by eliminating the need for Impairment,entities to separate deferred income tax liabilities and assets into current and noncurrent amounts in a classified statement of financial position. The adoption of this update is not expected to have a material effect on our financial statements. In August 2014, the FASB issued ASU 2014-15, Presentation of Financial Statements – Going Concern (Topic 915): Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern, which amended the guidancestates that in ASU 2011-08 to simplify the testing of indefinite-lived intangible assets other than goodwillconnection with preparing financial statements for impairment. ASU 2012-02 becomes effective foreach annual and interim impairment tests performed for fiscal years beginning onreporting period, an entity’s management should evaluate whether there are conditions or after September 15, 2012 and earlier adoption is permitted. We adopted this standardevents, considered in the third quarteraggregate, that raise substantial doubt about the entity’s ability to continue as a going concern within one year after the date that the financial statements are issued (or within one year after the date that the financial statements are available to be issued when applicable). The adoption of fiscal year 2012. We believe adoptionthis update did not have a material effect on our financial statements. In September 2011, the Financial Accounting Standards Board (FASB) issued Accounting Standards Update (ASU) No. 2011-08, Intangibles – Goodwill and Other (Topic 350) – Testing Goodwill for Impairment (ASU 2011-08), to allow entities to use a qualitative approach to determine whether it is more likely than not that the fair value of a reporting unit is less than its carrying value. If after performing the qualitative assessment an entity determines it is not more likely than not that the fair value of a reporting unit is less than its carrying amount, then performing the two-step goodwill impairment test is unnecessary. However, if an entity concludes otherwise, then it is required to perform the first step of the two-step goodwill impairment test. ASU 2011-08 is effective for us in fiscal 2013 and earlier adoption is permitted. The adoption of ASU 2011-08 is not anticipated to have any impact on our financial position, results of operations or cash flows.
In May 2011, the Financial Accounting Standards Board (“FASB”) issued ASU 2011-04, Fair Value Measurement (“ASU 2011-04”), which amended ASC 820, Fair Value Measurements (“ASC 820”), providing a consistent definition and measurement of fair value, as well as similar disclosure requirements between U.S. GAAP and International Financial Reporting Standards. ASU 2011-04 changes certain fair value measurement principles, clarifies the application of existing fair value measurement and expands the disclosure requirements. ASU 2011-04 was effective for us beginning January 1, 2012. The adoption of ASU 2011-04 did not have a material effect on our consolidated financial statements or disclosures. In June 2010,2014, the FASB issued ASU No. 2010-17,2014-09 Revenue Recognition—Milestone Methodfrom Contracts with Customers (Topic 605): Milestone Method of Revenue Recognition. This606), which will supersede nearly all existing revenue recognition guidance under GAAP. ASU codifies the consensus reached in EITF Issue No. 08-9, “Milestone Method of Revenue Recognition.” The amendments to the Codification provide guidance on defining a milestone and determining2014-09 provides that an entity recognize revenue when it maytransfers promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be appropriate to applyentitled in exchange for those goods or services. This update also requires additional disclosure about the milestone methodnature, amount, timing and uncertainty of revenue recognitionand cash flows arising from customer contracts, including significant judgments and changes in judgments, and assets recognized from costs incurred to obtain or fulfill a contract. ASU No. 2014-09 allows for researcheither full retrospective or development transactions. Consideration that is contingent on achievement of a milestone in its entirety may be recognized as revenuemodified retrospective adoption and will become effective for the Company in the period in whichfirst quarter of 2018. The Company is evaluating the milestone is achieved only ifpotential effects of the milestone is judged to meet certain criteria to be considered substantive. Milestones should be considered substantive in their entirety and may not be bifurcated. An arrangement may contain both substantive and nonsubstantive milestones, and each milestone should be evaluated individually to determine if it is substantive. This guidance was adopted effective October 1, 2010. The adoption of this guidance did not have a material impactupdate on our consolidatedits financial statements.
NOTE 2. ACQUISITIONS Roche Madison
On October 21, 2011,March 3, 2015, the Company entered into a Stockan Asset Purchase and Asset PurchaseExclusive License Agreement (the “RNAi Purchase Agreement”) with Hoffmann-La RocheNovartis Institutes for BioMedical Research, Inc. and F Hoffmann-La Roche Ltd (collectively, “Roche”, a Delaware corporation (“Novartis”), pursuant to which the Company purchased from Roche (i) all of the outstanding common stock of Roche Madison Inc. (“Roche Madison”) and (ii) the intellectual property rights then held by Roche related to its RNAi business and identified in the RNAi Purchase Agreement (the “Transaction”). In consideration for the purchase of Roche Madison and the Rocheacquired Novartis’ RNAi assets the Company issuedand rights thereunder. Pursuant to Roche a promissory note with a principal value of $50,000 and 901,702 shares of Common Stock (as adjusted for the 1-for-10 reverse stock split on November 17, 2011). Subsequently, as required by the RNAi Purchase Agreement, the Company issued an additional 386,456 shares of Common Stock. The acquisition provides additional technology, particularlyacquired or licensed certain patents and patent applications owned or controlled by Novartis related to RNAi delivery,therapeutics, assignment of a third-party license, rights to three pre-clinical RNAi candidates, and providedother related assets (collectively, the “Purchased Assets”). The acquisition of the Purchased Assets closed on March 3, 2015, concurrent with execution of the RNAi Purchase Agreement (the “Closing”). In consideration for the Purchased Assets, the Company made certain payments to Novartis, including: (a) a state-of-the-art lab facility. payment of $10,000,000 in cash, and 3,321,383 shares of the Company’s common stock (the “Shares”); (b) escalating royalties in the single digits based upon annual net sales thresholds for certain RNAi products sold by the Company; and (c) milestone payments tied to the achievement of certain development and sales milestones for each target being developed by the Company. F-11
Pursuant to the RNAi Purchase Agreement, Rocheprior to initiation of a phase 2 Clinical Trial (as defined, along with all other italicized terms in this section, in the RNAi Purchase Agreement) for a given RNAi Product or Arrowhead RNAi Product directed to an Initial Target, Novartis has an exclusive right to negotiate a license under any Intellectual Property Rights owned or exclusively licensed to the Company to make, sell or otherwise commercially exploit such RNAi Product or Arrowhead RNAi Product. After initiation of a phase 2 Clinical Trial for a given Arrowhead RNAi Product (“ROFN Candidate”), Novartis shall have a right of first negotiation on certain product candidatesthe ROFN Candidate developed by the Company and its affiliates relating to the purchased assets. If the Company proposes to out-license, or enters into substantive negotiations to out-license, any ClinicalROFN Candidate, or Existing Candidate (as such terms are defined in the RNAi Purchase Agreement), the Company must give notice of the ROFN Candidate it proposes to out-license and negotiate exclusively and in good faith with RocheNovartis for 90 daysa period of time regarding the applicable out-license. This right of first negotiation applies to all Existing Candidates (as defined in the RNAi Purchase Agreement) and the first five Clinical Candidates for which the Company delivers notice to Roche and subsequently enters into an out-license. In addition to the consideration paid by the Company as perat the closing terms,of the Transaction, the Company is obligated to make certain royalty and milestone payments to RocheNovartis upon the occurrence of certain events. For certain product candidates that are developed bysales of any RNAi Products for which Novartis and the Company or its affiliates and that are covered by a valid claim by the patent rights transferred in the Transaction for which the Company and Roche do not enter into a licensing arrangement, the Company will be obligated to pay a 3% royalty on Net Sales (as definedrates ranging in the low to mid-single digits on Net Sales depending upon the type of RNAi Purchase Agreement),Product provided that the royalty rate may be reduced or offset in certain circumstances. The obligation to pay royalties on such candidates will last until the later of (i) the expiration of the last to expire patent right related toValid Claim Covering such product candidate that was transferredRNAi Product in the Transactionsuch country and (ii) ten11 years after the first commercial sale of such product candidate.RNAi Product (as such italicized terms are defined in the RNAi Purchase Agreement). The Company will also be obligated to make cash payments to RocheNovartis upon the achievement of various milestones includingfor any RNAi Products for which Novartis and the firstCompany do not enter into a licensing arrangement. These milestones include the initiation of phase 2 and 3 clinical trials, U.S. and other regulatory approval of an Existing Candidate in certain jurisdictionsapprovals, and upon certain annual sales milestones for Existing Candidates that may receive regulatory approval. The potentialmilestones. These milestone payments range from $2,500,000could total an amount in the mid to $6,000,000 per milestone. Based on the Company’s estimateupper double digit millions of future payments, a net present value of $84,935 was calculated as contingent consideration, and is recorded as a part of other noncurrent liabilities.dollars. The following table summarizes the estimated relative fair values of the assets acquired at the date of acquisition: | | | | | Current assets | | $ | 432,709 | | Property and equipment | | | 7,215,206 | | Intangible assets | | | 1,174,935 | | Other noncurrent assets | | | 6,264 | | Current liabilities | | | (414,122 | ) | Noncurrent liabilities | | | (1,570,072 | ) | Gain on purchase | | | (1,576,106 | ) | | | | | | Total purchase consideration | | $ | 5,268,814 | | | | | | |
Intangible assets - patents | | $ | 21,728,334 | | Intangible assets – license | | | 3,128,880 | | Acquired in-process research and development - Pre-Clinical Candidates | | | 10,142,786 | | Total purchase consideration | | $ | 35,000,000 | |
The purchase consideration was composed of the following: | | | | | Promissory note due Roche | | $ | 50,000 | | Contingent consideration | | | 84,935 | | Shares issued to Roche | | | 5,133,879 | | | | | | | Total purchase consideration | | $ | 5,268,814 | | | | | | |
| | | | | Cash Paid | | $ | 10,000,000 | | Value of Shares Issued | | | 25,000,000 | | Total purchase consideration | | $ | 35,000,000 | |
We estimatedThe Company accounted for this transaction as an acquisition of RNAi assets, including patents, a third-party license and in process research and development for the fair value of the assets and liabilities acquired through various valuation techniques including a market approach and an income approach. Because the net identifiable tangible and intangible assets and liabilities were in excesspre-clinical candidates. The allocation of the purchase price a gain onto each asset was determined by estimating the relative fair value of each asset acquired and applying that to the total cost of the acquisition for the Company. The Company capitalized the patents and license acquired as Intangible Assets as they require no future development and will have alternative future uses as the Company expands its RNAi capabilities (see footnote 5 for additional discussion of the useful lives and amortization of these Intangible Assets). The Company expensed the portion of the purchase of $1.6 million was recorded. The most significant assets capitalized were research equipment and certainconsideration allocated to the pre-clinical candidates as they will require future development in order to be commercialized. This expense is recorded in the “Acquired in-process research and development. We believe that we were able to acquire these assets at a reasonable purchase price and generate a gain on the transaction due in part from the seller’s desire to exit the relatively early stagedevelopment” line item of the RNAi business, as compared to the seller’s other business operations, as well as the seller’s desire to disposeConsolidated Statements of certain on-going costs associated with the facility, primarily lease costs and personnel costs, which were synergistic to the Company’s strategy to establish a research facility to advance development efforts for its drug product candidates.
Alvos Therapeutics
On April 5, 2012, the Company entered into a Stock Purchase Agreement to purchase all of the outstanding shares of Alvos Therapeutics, Inc., (“Alvos”), a privately held company that licensed a large platform of proprietary human-derived Homing Peptides, and the method for their discovery, from MD Anderson Cancer Center. In conjunction with the acquisition, Arrowhead hired one employee from Alvos, and retained one employee on a consulting basis. In exchange for all of the outstanding shares of Alvos, Arrowhead issued an upfront payment of 315,467 shares of Common Stock. The former Alvos stockholders are also eligible to receive additional issuances of stock valued at up to $23.5 million at the time of issuance based on the future achievement of clinical, regulatory and sales milestones. Based on the Company’s estimate of future payments, a net present value of $88,686 was calculated as contingent consideration, and is recorded as a part of other noncurrent liabilities. The Alvos acquisition provided key technology in targeted therapeutics.
Operations. F-12NOTE 3. PROPERTY AND EQUIPMENT
The following table summarizes the estimated fair values at the dateour major classes of acquisition:property and equipment: | | | | | Current assets | | $ | 29,332 | | In-process R&D | | | 2,172,387 | | Current liabilities | | | (113,033 | ) | | | | | | Total purchase consideration | | $ | 2,088,686 | | | | | | |
| | | | September 30, 2015 | | | September 30, 2014 | | Computers, office equipment and furniture | $ | 404,964 | | | $ | 334,162 | | Research equipment | | 6,354,584 | | | | 4,614,176 | | Software | | 110,428 | | | | 69,623 | | Leasehold improvements | | 3,117,537 | | | | 3,045,022 | | Total gross fixed assets | | 9,987,513 | | | | 8,062,983 | | Less: Accumulated depreciation and amortization | | (5,460,665 | ) | | | (4,190,230 | ) | Property and equipment, net | $ | 4,526,848 | | | $ | 3,872,753 | |
F-12
NOTE 4. INVESTMENTS The purchase consideration was comprised solelyCompany invests a portion of sharesits excess cash balances in short-term and long-term debt securities. Investments at September 30, 2015 consisted of Arrowhead Common Stock issued tocorporate bonds with maturities remaining of less than one year. The Company may also invest excess cash balances in certificates of deposit, money market accounts, U.S. Treasuries, U.S. government agency obligations, corporate debt securities, and/or commercial paper. The Company accounts for its investments in accordance with FASB ASC 320, Investments – Debt and Equity Securities. At September 30, 2015, all investments were classified as held-to-maturity securities. The following tables summarize the former shareholdersCompany’s short- and long-term investments as of Alvos Therapeutics, Inc.September 30, 2015, and September 30, 2014: | As of September 30, 2015 | | | Amortized Cost | | | Gross Unrealized Gains | | | Gross Unrealized Losses | | | Fair Value | | Commercial notes (due within one year) | $ | 17,539,902 | | | $ | — | | | $ | (304,942 | ) | | $ | 17,234,960 | | Commercial notes (due after one year through two years) | $ | — | | | | — | | | $ | — | | | $ | — | | Total | $ | 17,539,902 | | | $ | — | | | $ | (304,942 | ) | | $ | 17,234,960 | |
| As of September 30, 2014 | | | Amortized Cost | | | Gross Unrealized Gains | | | Gross Unrealized Losses | | | Fair Value | | Commercial notes (due within one year) | $ | 21,653,032 | | | $ | — | | | $ | (189,830 | ) | | $ | 21,463,202 | | Commercial notes (due after one year through two years) | $ | 23,088,346 | | | | — | | | $ | (217,693 | ) | | $ | 22,870,653 | | Total | $ | 44,741,378 | | | $ | — | | | $ | (407,523 | ) | | $ | 44,333,855 | |
| | | | | Shares issued | | | 315,457 | | Price per share | | $ | 6.34 | | | | | | | Share consideration | | $ | 2,000,000 | | Contingent consideration | | | 88,686 | | | | | | | Total purchase consideration | | $ | 2,088,686 | | | | | | |
NOTE 3. 5. INTANGIBLE ASSETS Intangible assets consist of in-process research and development (IPR&D)(“IPR&D”) not subject to amortization, and patents and other intangible assetslicense agreements subject to amortization, which were capitalized as a part of aan asset acquisition or business combination. IPR&D represents projects that have not yet received regulatory approval and are required to be classified as indefinite assets until the successful completion or the abandonment of the associated R&D efforts. Accordingly, during the development period after the date of acquisition, these assets will not be amortized until approval is obtained in one or more jurisdictions which, individually or combined, are expected to generate a significant portion of the total revenue expected to be earned by an IPR&D project. At that time, wethe Company will determine the useful life of the asset, reclassify the asset out of IPR&D and begin amortization. If the associated R&D effort is abandoned the related IPR&D assets will likely be written off and wethe Company would record an impairment loss. Intangible assets not subject to amortization include patents capitalized as part of a business combination as well as license agreementsIPR&D capitalized as part of a business combination from the acquisition of the Roche Madison.RNAi business. Intangible assets subject to amortization include patents and a license agreement capitalized as part of the Novartis RNAi asset acquisition and license agreements capitalized from the acquisition of the Roche RNAi business. The license agreement associated with the Novartis RNAi asset acquisition is being amortized over the estimated life remaining at the time of acquisition which was 21 years, and the accumulated amortization of the asset is approximately $86,570. The license agreements associated with the acquisition of the Roche RNAi business are being amortized over the estimated life remaining at the time of acquisition, which was 4 years. Patentsyears, and the accumulated amortization of the assets is approximately $216,116. The patents associated with the Novartis RNAi asset acquisition are being amortized over a periodthe estimated life remaining at the time of threeacquisition, which was 14 years, to twenty years. The weighted average originaland the accumulated amortization periodof the assets is twelve years.approximately $905,347. Amortization of license agreementsexpense for the years ended September 30, 2015, 2014 and patents2013 was $1,046,571, $54,653 and $236,009, respectively. Amortization expense is expected to be approximately $300,000 for fiscal years 2013, 2014, and 2015, $250,000$1,714,313 in 2016, $240,000$1,700,429 in 2017, $1,700,429 in 2018, $1,700,429 in 2019, $1,700,429 in 2020, $1,700,429 in 2021 and $280,000$13,662,723 thereafter. We review amounts capitalized as in-process research and development for impairment at least annually in the fourth quarter, and whenever events or changes in circumstances indicate that the carrying value of an asset may not be recoverable. In the event the carrying value of the assets is not expected to be recovered, the assets are written down to their estimated fair values. We continue to test our indefinite-lived IPR&D assets for potential impairment until the projects are completed or abandoned.F-13
The belowfollowing table provides details on ourthe Company’s intangible asset balances: | | | | | | | | | | | | | | | | Intangible assets
not subject to
amortization | | | Intangible assets
subject to
amortization | | | Total
Intangible
assets | | Balance at September 30, 2010 | | $ | 0 | | | $ | 1,973,019 | | | $ | 1,973,019 | | Amortization | | | 0 | | | | (241,808 | ) | | | (241,808 | ) | | | | | | | | | | | | | | Balance at September 30, 2011 | | $ | 0 | | | $ | 1,731,211 | | | $ | 1,731,211 | | Additions – Madison acquisition | | | 944,935 | | | | 230,000 | | | | 1,174,935 | | Additions – Alvos acquisition | | | 2,172,387 | | | | 0 | | | | 2,172,387 | | Amortization | | | 0 | | | | (293,964 | ) | | | (293,964 | ) | | | | | | | | | | | | | | Balance at September 30, 2012 | | $ | 3,117,322 | | | $ | 1,667.247 | | | $ | 4,784,569 | | | | | | | | | | | | | | |
| Intangible assets
not subject to
amortization | | | Intangible assets
subject to
amortization | | | Total
Intangible assets | | Balance at September 30, 2013 | $ | 3,117,322 | | | $ | 123,191 | | | $ | 3,240,513 | | Impairment | | (2,172,387 | ) | | | - | | | | (2,172,387 | ) | Amortization | | - | | | | (54,653 | ) | | | (54,653 | ) | Balance at September 30, 2014 | $ | 944,935 | | | $ | 68,538 | | | $ | 1,013,473 | | Acquisition of Novartis RNAi Assets | | - | | | | 24,857,214 | | | | 24,857,214 | | Amortization | | - | | | | (1,046,571 | ) | | | (1,046,571 | ) | Balance at September 30, 2015 | $ | 944,935 | | | $ | 23,879,181 | | | $ | 24,824,116 | |
F-13
NOTE 4.INVESTMENT IN SUBSIDIARIES
In addition to 100% ownership interest in Arrowhead Madison, Inc. and Alvos Therapeutics, Inc., Arrowhead also maintains majority ownership in Calando Pharmaceuticals, Ablaris Therapeutics, Inc., and minority investments in Nanotope, Inc. and Leonardo Biosystems, Inc.
Calando Pharmaceuticals, Inc.
Calando is a clinical stage RNAi therapeutics company. On April 17, 2008, Calando merged with and into Insert, with Insert as the surviving company. Prior to the merger, Arrowhead invested an aggregate of $23.2 million in Calando through equity and debt financings. As a condition of the merger, the Preferred Stock of each of Calando and Insert was converted into common stock and the loans were converted to equity. As a result of the merger, shares of Insert common stock were issued to the stockholders of the former Calando, and Insert changed its name to Calando Pharmaceuticals, Inc.
On November 26, 2008, Calando entered into Unsecured Convertible Promissory Note Agreements (“Notes”) for $2.5 million with accredited investors and Arrowhead, which invested $200,000 in the Notes offering. Arrowhead subsequently invested an additional $600,000 in the same offering. Except for one Note in the principal amount of $500,000, all Notes and accrued interest were converted into a total of 2,950 shares of Calando Series A Preferred Stock on June 23, 2009. The remaining Note is due November 26, 2013; see Note 4 for further information.
In fiscal 2010, Arrowhead issued 122,000 shares of its Common Stock in exchange for shares of Calando common stock, with several minority stockholders of Calando. In conjunction with this exchange, Arrowhead also issued 26,400 warrants to purchase Arrowhead Common Stock in exchange for warrants to purchase Calando common stock.
In January 2011, Arrowhead invested $9.1 million, through a cash investment of $1.0 million and the conversion of $8.1 million intercompany debt, acquiring newly issued Calando Series B and Series C preferred stock.
As of September 30, 2012, Calando owed to Arrowhead $3.1 million under a series of 8% simple interest notes and advances. It is expected that these loans will either be repaid or converted to equity in the future. The balance of the notes and advances is eliminated in consolidation.
As of September 30, 2012, Arrowhead owned 79% of the outstanding shares of Calando and 76% on a fully diluted basis.
Ablaris Therapeutics, Inc.
Ablaris was formed and began operations in the first quarter of fiscal 2011, based on the license of certain anti-obesity technology developed at the MD Anderson Cancer Center at the University of Texas. During the year ended September 30, 2011, Ablaris raised $2.9 million in cash, of which $1.3 million was invested by Arrowhead and $1.6 million was invested by outside investors, through the issuance of Ablaris Series A Preferred stock.
As of September 30, 2012, Arrowhead owned 64% of the outstanding shares of Ablaris and 64% on a fully diluted basis.
Nanotope, Inc.
Nanotope’s primary areas of focus include development of treatments for spinal cord injuries, cartilage regeneration and wound healing. As of September 30, 2012, Arrowhead owned 23% of the outstanding shares of Nanotope, and 19% on a fully diluted basis. Arrowhead accounts for its investment in Nanotope using the equity method of accounting.
During the quarter ended June 30, 2012, Nanotope closed its R&D operations at its facility in Skokie, Illinois, and moved R&D operations to the laboratories of Northwestern University under the direction of Dr. Samuel Stupp, the Company’s founder. Development work will be performed by personnel in Dr. Stupp’s lab, and Nanotope terminated the employment of its staff. The company’s research equipment was sold to Arrowhead, to be utilized at its research facility in Madison, Wisconsin. Certain components of Nanotope’s intellectual property portfolio continue to be maintained at Arrowhead’s expense, potentially allowing Nanotope to benefit from successful developments. These circumstances required Arrowhead to examine its investment in Nanotope, as well as its receivable from Nanotope, for impairment. Upon review, it was determined that the remaining book value of its investment exceeded its fair value as determined by discounted future cash flows. As a result, upon completion of the assessment, management recorded a non-cash impairment charge of $1.4 million during the year ended September 30, 2012 to reduce the carrying value of the investment to $0. Additionally, the company recorded a full reserve against its receivable from Nanotope in the amount of the $1.9 million.
Summarized financial information for Nanotope, Inc. is as follows:
| | | | | | | | | | | | September 30, 2012 | | | September 30, 2011 | | Current assets | | $ | 31,000 | | | $ | 21,000 | | Non-current assets | | | 0 | | | | 85,000 | | Liabilities | | | 2,070,000 | | | | 1,255,000 | | Equity | | | (2,039,000 | ) | | | (1,149,000 | ) |
F-14
| | | | | | | | | | | | For the year ended
September 30, 2012 | | | For the year ended
September 30, 2011 | | Revenue | | $ | 0 | | | $ | 515,000 | | Operating expenses | | | 808,000 | | | | 1,161,000 | | Net Loss | | $ | (890,000 | ) | | $ | (709,000 | ) | | | | | | | For the year ended
September 30, 2012 | | | For the year ended
September 30, 2011 | | Cash flows used in operating activities | | $ | (831,000 | ) | | $ | (705,000 | ) | Cash flows provided by (used in) investing activities | | | 61,000 | | | | (31,000 | ) | Cash flows provided by financing activities | | | 784,000 | | | | 746,000 | |
Leonardo Biosystems, Inc.
Leonardo is developing a drug-delivery platform technology based on novel methods of designing porous silicon microparticles that selectively accumulate in tumor vasculature. Arrowhead accounts for its investment in Leonardo using the cost method of accounting. As of September 30, 2012, Leonardo owed to Arrowhead $547,000, included in other receivables. Although it is expected to be repaid or converted to equity, the Company has provided a full reserve against the receivable from Leonardo. As of September 30, 2012, Arrowhead’s ownership interest in Leonardo was 3%.
NOTE 5.DISCONTINUED OPERATIONS
Unidym, Inc.
Founded by Arrowhead in 2005, Unidym is developing electronic applications of carbon nanotubes. Arrowhead sold its ownership interest in Unidym to Wisepower, a Korean corporation, in January 2011. The consideration included $2.5 million in Wisepower stock, a convertible bond with a face value of $2.5 million, a percentage of certain revenue streams, as well as contingent payments up to $140 million based on revenue milestones over a ten-year period. The consideration received in the form of Wisepower stock has been liquidated. The consideration received in the form of a bond is currently held as a current asset. The bond which bears no interest, became convertible on January 17, 2012, is redeemable in cash on January 17, 2013, and becomes due on January 17, 2014.
In conjunction with the disposition of Unidym, the gain on the sale and the results of historical operations are recorded as discontinued operations in the Company’s Statements of Operations. Additionally, the cash flows from Unidym are reflected separately as cash flows from discontinued operations in the Company’s Consolidated Statement of Cash Flows. Any future cash flows as discussed above will also be reflected as a part of cash flows from discontinued operations.
Tego Biosciences, Inc.
On April 20, 2007, Tego, a wholly-owned subsidiary of Arrowhead, acquired the assets of C Sixty, Inc., a Texas-based company developing protective products based on the anti-oxidant properties of fullerenes.
In December 2009, Tego completed the sale of all of its intellectual property assets to Luna Innovations, Inc. The consideration included an upfront purchase price of $350,000 and reimbursements of patent and license expenses of $80,000, as well as contingent payments based on milestones and royalties for each fullerene product developed by Luna and covered by Tego intellectual property. Due to the sale of substantially all of Tego’s assets, the operations of Tego ceased and the gain on the sale and the results of historical operations are recorded as discontinued operation in the Company’s Statements of Operations. Additionally, the cash flows from Tego are reflected separately as cash flows from discontinued operations. Any future cash flows as discussed above will be reflected as a part of cash flows from discontinued operations in the Company’s Consolidated Statements of Cash Flows.
NOTE 6.NOTES PAYABLE On November 26, 2008, Calando entered into Unsecured Convertible Promissory Note Agreements (“Notes”) for $2.5 million with accredited investors and Arrowhead, which invested $200,000 in the Notes offering. Arrowhead subsequently invested an additional $600,000 in the same offering. Except for one Note in the principal amount of $500,000, all Notes and accrued interest were converted into a total of 2,950 shares of Calando Series A Preferred Stock on June 23, 2009. The remaining Note had a 10% interest rate, matured on November 26, 2010, and was renegotiated and extended until November 26, 2013. The terms of the new note include a 10% interest rate and require two times principal payment upon certain events as defined in the note and at maturity. At September 30, 2012, The Note is reflected on the balance sheet at the maturity amount of $1,000,000 less a discount of $210,579.
F-15
NOTE 7.STOCKHOLDERS’ EQUITY
At September 30, 2012,2015, the Company had a total of 150,000,000 shares of capital stock authorized for issuance, consisting of 145,000,000 shares of Common Stock, par value $0.001 per share, and 5,000,000 shares of Preferred Stock, par value $0.001.$0.001 per share. At September 30, 2012, 13,579,1842015, 59,544,677 shares of Common Stock were outstanding; nooutstanding. Additionally, 15,652 shares of Series C Preferred Stock were outstanding.outstanding, which are convertible into 2,670,990 shares of Common Stock. At September 30, 2012, 153,2002015, 8,099,777 shares and 1,965,860 sharesof Common Stock were reserved for issuance upon exercise of options and vesting of restricted stock units granted or available for grant under Arrowhead’s 2000 Stock Option Plan and 2004 Equity Incentive Plan respectively.and 2013 Incentive Plan, as well as for inducement grants made to new employees. The Preferred Stock is convertible to Common Stock by its holder at its stated conversion price, though it is not convertible to the extent the holder would beneficially own more than 9.99% of the number of shares of outstanding Common Stock immediately after the conversion. The holders of Preferred Stock are eligible to vote with the Common Stock of the Company on an as-converted basis, but only to the extent they are eligible for conversion without exceeding the 9.99% ownership limitation. The Preferred Stock does not carry a coupon, but it is entitled to receive dividends on a pari passu basis with Common Stock, when and if declared. In any liquidation or dissolution of the Company, the holders of Preferred Stock are entitled to participate in the distribution of the assets, to the extent legally available for distribution, on a pari passu basis with the Common Stock. On September 30, 2011,October 11, 2013, the Company sold 1,458,9173,071,672 shares of Common Stock, at a price of $3.80$5.86 per share. Cash proceeds received in fiscal 2011 were $4.5 million; the balance of $1 million is expected to be received in 2012. On October 4, 2011, the Company completed a second closing of the offering in which the Company sold 138,158 shares of Common Stock at a price of $3.80 per share. Cash proceeds were $525,000. On October 20, 2011, the Companyshare, and Lincoln Park Capital Fund, LLC, an Illinois limited liability company (“LPC”) entered into a $15 million purchase agreement (the “Purchase Agreement”), together with a registration rights agreement, whereby LPC agreed to purchase up to $15 million of Common Stock, subject to certain limitations, from time to time during the three-year term of the Purchase Agreement. Additionally, the Company filed a registration statement with the U.S. Securities & Exchange Commission covering the resale of the shares that may be issued to LPC under the Purchase Agreement. On January 30, 2012, the SEC declared the registration statement effective for the resale of such shares. The Company has the right, in its sole discretion, over a 36-month period to sell up to $15 million of Common Stock (subject to certain limitations) to LPC, depending on certain conditions as set forth in the Purchase Agreement. As of September 30, 2012, the Company had drawn $1 million from the facility.
On October 21, 2011 and October 24, 2011, the Company entered into Subscription Agreements with certain accredited investors (the “Series A Purchasers”), pursuant to which the Company issued and sold an aggregate of 1,01546,000 shares of Series AC Preferred Convertible Stock, $0.001 par value per share, at a purchase price of $1,000 per share. The aggregate purchase price paid for thePreferred Stock is convertible into shares of Series A Preferred was $1,015,000. On February 16, 2012, upon approval by the Company’s shareholders, 1,015 shares of Arrowhead Series A Preferred Convertible Stock, $0.001 par value per share, were converted to 275,782 shares of Common Stock.
On October 21, 2011, the Company entered into a Subscription Agreement with an accredited investor, pursuant to which the Company issued and sold an aggregate of 675,000 shares of Common Stock, $0.001 par value per share,common stock at a purchaseconversion price of $3.70 per share.$5.86. The aggregate purchase price paid by the purchaserpurchasers for the Common Stock and Series C Preferred Stock was $64,000,000 and the Company received net proceeds of approximately $60,000,000, after advisory fees and offering expenses.
On February 24, 2014, the Company sold 6,325,000 shares of Common Stock, is $2,497,500. On August 10, 2012 the Company sold 2,260,869 units at a public offering price of $2.76$18.95 per unit. Each unit consisted of one share of common stockshare. Net proceeds were approximately $112.6 million after underwriting commissions and a warrant to purchase 0.75 shares of common stock at an exercise price of $3.25. Gross proceeds from thediscounts and other offering were $6.2 million excluding offering fees and expenses.
As of November 17, 2011, the Company effected a 1 for 10 reverse stock split. As a result of the reverse stock split, each ten shares of the Company’s Common Stock issued and outstanding immediately prior to the reverse stock split was combined into one share of Common Stock. Also, as a result of the reverse stock split, the per share exercise price, and the number of shares of Common Stock underlying Company stock options, warrants, and any Common Stock based equity grants outstanding immediately prior to the reverse stock split was proportionally adjusted, based on the one-for-ten split ratio, in accordance with the terms of such options, warrants or other Common Stock based equity grants as the case may be. No fractional shares of Common Stock were issued in connection with the reverse split. Stockholders received a cash payment in lieu of any fractional shares. All share and per share amounts in these financial statements have been retrospectively adjusted to reflect the reverse stock split.
The following table summarizes information about warrants outstanding at September 30, 2012:2015: | | | | | | | | | Exercise prices | | Number of Warrants | | | Remaining
Life in Years | | $70.60 | | | 94,897 | | | | 4.6 | | $20.00 | | | 386,400 | | | | 0.9 | | $5.00 | | | 1,155,023 | | | | 2.2 | | $5.09 | | | 461,024 | | | | 2.2 | | $1.38 | | | 322,150 | | | | 3.2 | | $4.16 | | | 1,000 | | | | 4.2 | | $3.25 | | | 1,695,654 | | | | 3.9 | | | | | | | | | | | Total warrants outstanding | | | 4,116,147 | | | | | | | | | | | | | | |
Exercise prices | | Number of
Warrants | | | Remaining
Life in Years | | $ | 70.60 | | | | 94,897 | | | | 1.6 | | $ | 5.00 | | | | 364,375 | | | | 0.8 | | $ | 5.09 | | | | 239,534 | | | | 0.8 | | $ | 1.38 | | | | 24,324 | | | | 0.2 | | $ | 4.16 | | | | 1,000 | | | | 1.2 | | $ | 3.25 | | | | 334,347 | | | | 0.9 | | $ | 2.12 | | | | 75,000 | | | | 2.2 | | $ | 1.83 | | | | 277,284 | | | | 2.2 | | $ | 7.14 | | | | 80,000 | | | | 2.7 | | Total warrants outstanding | | | 1,490,761 | | | | | |
F-16F-14
NOTE 8.LEASES7. COMMITMENTS AND CONTINGENCIES In May 2012, theLeases
The Company signed a lease for newleases office space for its corporate headquarters in Pasadena, California. In March 2014, the Company signed a lease addendum to expand its corporate headquarters, and moved into the new locationspace became available in August 2012.September 2014. The lease has a five-year term; rentalleases for the expansion space and the current space will expire in September 2019. Rental costs, including the expansion space, are approximately $13,000$24,000 per month, increasing approximately 3% annually. The Company’s research facility in Madison, Wisconsin is leased through February 28, 2019. Monthly rental expense is approximately $22,000.$26,000. Other monthly rental expenses include common area maintenance and real estate taxes totaling approximately $13,000$18,000 per month. Utilities costs are approximately $16,000 per month. IncludingTotal monthly costs are approximately $79,000 per month, including monthly payments recorded under a capital lease of approximately $21,000, total$19,000. In May 2015, the Company signed a lease for additional research facility space in Middleton, Wisconsin, and this space is leased through May 2016. Monthly rental expense for the additional space is approximately $4,000. Other monthly costs arerental expenses include common area maintenance and real estate taxes totaling approximately $72,000$2,000 per month. Facility and equipment rent expense related to continuing operations, for the yearyears ended September 30, 20122015, 2014 and 20112013 was $480,000$744,000, $554,000 and $162,000,$534,000, respectively. From inception to date, rent expense was $4,125,485. Rent expense related to Unidym, until its disposal in January 2011, is included as a part of income/loss from discontinued operations. As of September 30, 2012,2015, future minimum lease payments due in fiscal years under capitalized leases are as follows: | 2013 | | $ | 256,846 | | | 2014 | | | 256,846 | | | 2015 | | | 256,846 | | | 2016 | | | 256,846 | | $ | 228,420 | | 2017 | | | 256,846 | | | 2018 and thereafter | | | 363,864 | | | 2017 | | | 228,420 | | 2018 | | | 228,420 | | 2019 | | | 95,175 | | 2020 | | | - | | 2021 and thereafter | | | - | | Less interest | | | (150,835 | ) | | (22,095 | ) | | | | | | Principal | | | 1,497,259 | | | 758,340 | | Less current portion | | | (214,801 | ) | | (217,548 | ) | | | | | | Noncurrent portion | | $ | 1,282,458 | | $ | 540,792 | | | | | | |
As of September 30, 2012,2015, future minimum lease payments due in fiscal years under operating leases are as follows: | | | | | 2013 | | $ | 356,672 | | 2014 | | | 434,229 | | 2015 | | | 445,921 | | 2016 | | | 457,961 | | 2017 | | | 470,154 | | 2018 and thereafter | | | 484,785 | | | | | | | Total | | $ | 2,649,723 | | | | | | |
2016 | $ | 631,881 | | 2017 | | 613,664 | | 2018 | | 637,897 | | 2019 | | 459,633 | | 2020 | | - | | 2021 and thereafter | | - | | Total | $ | 2,343,075 | |
Litigation The Company and certain of its officers and directors have been named as defendants in a consolidated class action pending before the United States District Court for the Central District of California regarding certain public statements in connection with the Company’s hepatitis B drug research. The consolidated class action, initially filed as Wang v. Arrowhead Research Corp., et al., No. 2:14-cv-07890 (C.D. Cal., filed Oct. 10, 2014), and Eskinazi v. Arrowhead Research Corp., et al., No. 2:14-cv-07911 (C.D. Cal., filed Oct. 13, 2014), asserts claims under Sections 10(b) and 20(a) of the Securities Exchange Act of 1934 and seeks damages in an unspecified amount. Additionally, three putative stockholder derivative actions captioned Weisman v. Anzalone et al., No. 2:14-cv-08982 (C.D. Cal., filed Nov. 20, 2014), Bernstein (Backus) v. Anzalone, et al., No. 2:14-cv-09247 (C.D. Cal., filed Dec. 2, 2014); and Johnson v. Anzalone, et al., No. 2:15-cv-00446 (C.D. Cal., filed Jan. 22, 2015), were filed in the United States District Court for the Central District of California, alleging breach of fiduciary duty by the Company’s Board of Directors in connection with the facts underlying the securities claims. An additional consolidated derivative action asserting similar claims is pending in Los Angeles County Superior Court, initially filed as Bacchus v. Anzalone, et al., (L.A. Super., filed Mar. 5, 2015); and Jackson v. Anzalone, et al. (L.A. Super., filed Mar. 16, 2015). Each of these suits seeks damages in unspecified amounts and some seek various forms of injunctive relief. F-15
The Company and two of its former executives have been named as defendants in a complaint filed on November 11, 2014 and captioned William Marsh Rice University vs. Unidym, Inc. and Arrowhead Research Corporation, No. 2014-66088, currently pending in the United States District Court for the Southern District of Texas relating to alleged breaches of a license agreement between Rice University and the Company’s former subsidiary, Unidym, Inc. The plaintiff has alleged that the Company and its former executives acted fraudulently with respect to Unidym’s license from Rice University and seeks injunctive relief, damages, including unspecified compensatory and punitive damages, and attorneys’ fees. The Company believes it has meritorious defenses and intends to vigorously defend itself in each of the above matters. The Company makes provisions for liabilities when it is both probable that a liability has been incurred and the amount can be reasonably estimated. No such liability has been recorded related to these matters. The Company does not expect these matters to have any material effect on its Consolidated Financial Statements. With regard to legal fees, such as attorney fees related to these matters or any other legal matters, the Company’s accounting policy is to recognize such costs as incurred. Purchase Commitments In the normal course of business, we enter into various purchase commitments for the manufacture of drug components, toxicology studies, and for clinical studies. As of September 30, 2015, these future commitments were approximately $49.3 million, of which approximately $35.5 million is expected to be incurred in fiscal 2016, and $13.8 million is expected to be incurred beyond fiscal 2016. Technology License Commitments The Company has licensed from third parties the rights to use certain technologies that it uses in its research and development activities, as well as in any products the Company may develop using these licensed technologies. These agreements and other similar agreements often require milestone and royalty payments. Milestone payments, for example, may be required as the research and development process progresses through various stages of development, such as when clinical candidates enter or progress through clinical trials, upon NDA and upon certain sales level milestones. These milestone payments could amount to the mid to upper double digit millions of dollars. In certain agreements, the Company may be required to make mid to high single digit percentage royalty payments based on a percentage of the sales of the relevant products. NOTE 9.8. STOCK-BASED COMPENSATION Arrowhead has two plans that provide for equity-based compensation. Under the 2000 Stock OptionArrowhead’s 2004 Equity Incentive Plan 153,200and 2013 Incentive Plan, as of September 30, 2015, 2,537,018 and 5,088,971 shares, respectively, of Arrowhead’s Common Stock are reserved for issuance upon exercise of non-qualified stock options. No further grants can be made under the 2000 Stock Option Plan. The 2004 Equity Incentive Plan reserves 1,965,860 shares for the grant of stock options, stock appreciation rights, restricted stock awards and performance unit/share awards by the Board of Directorsaward to employees, consultants and others. No further grants may be made under the 2004 Equity Incentive Plan. As of September 30, 2012,2015, there were options granted and outstanding to purchase 153,2002,537,018 and 1,767,8942,354,000 shares of Common Stock under the 2000 Stock Option Plan and the 2004 Equity Incentive Plan respectively.and the 2013 Incentive Plan, respectively, and there were 877,500 restricted stock units granted and outstanding under the 2013 Incentive Plan. Also, as of September 30, 2015, there were 544,622 shares reserved for options and 56,667 restricted stock units issued as inducement grants to new employees outside of equity compensation plans. During the year ended September 30, 2012, 988,3002015, no options or restricted stock units were granted under the 2004 Equity Incentive Plan, 1,609,000 options and 251,200 options675,000 restricted stock units were granted outside of Equityunder the 2013 Incentive plansPlan, and 120,000 options and 30,000 restricted stock units were granted as inducement stock optionsawards to new employees hired in conjunction withoutside of equity incentive plans. Additionally, the Company’s acquisition of its Madison research facility in October 2011. All share2000 Stock Option Plan and per share data in this footnote has been adjusted to reflect38,000 stock options that were outstanding under the 1 for 10 reverse stock split effected on November 17, 2011.2000 Stock Option Plan expired during the year ended September 30, 2015. The following tables summarize information about stock options: | | | | | | | | | Number of
Options
Outstanding | | | Weighted-
Average
Exercise
Price
Per Share | | | Weighted-
Average
Remaining
Contractual
Term | | | Aggregate
Intrinsic
Value | | | | | Number of
Options
Outstanding | | Weighted-
Average
Exercise
Price
Per Share | | | Weighted-
Average
Remaining
Contractual
Term | | Aggregate
Intrinsic
Value | | Balance At September 30, 2010 | | | 812,334 | | | $ | 10.62 | | | | | | | Balance At September 30, 2014 | | | 3,850,840 | | | | 6.99 | | | | | | | | | Granted | | | 20,000 | | | | 5.86 | | | | | | | 1,729,000 | | | | 6.35 | | | | | | | | | Cancelled | | | (100,539 | ) | | | 21.32 | | | | | | | (115,442) | | | | 11.38 | | | | | | | | | Exercised | | | (2,699 | ) | | | 5.10 | | | | | | | (28,758) | | | | 3.54 | | | | | | | | | | | | | | | | | | | | | | Balance At September 30, 2015 | | | 5,435,640 | | | $ | 6.71 | | | | 7.9 years | | | $ | 5,520,448 | | Exercisable At September 30, 2015 | | | 2,683,856 | | | $ | 6.07 | | | | 7.0 years | | | $ | 3,554,184 | |
F-16 F-17
| | | | | | | | | | | | | | | | | | | | Number of
Options
Outstanding | | | Weighted-
Average
Exercise
Price
Per Share | | | Weighted-
Average
Remaining
Contractual
Term | | | Aggregate
Intrinsic
Value | | Balance At September 30, 2011 | | | 729,096 | | | | 9.03 | | | | | | | | | | Granted | | | 1,229,500 | | | | 4.40 | | | | | | | | | | Cancelled | | | (42,919 | ) | | | 11.77 | | | | | | | | | | Exercised | | | (4,883 | ) | | | 5.20 | | | | | | | | | | | | | | | | | | | | | | | | | | | Balance At September 30, 2012 | | | 1,910,794 | | | $ | 6.01 | | | | 8.2 years | | | $ | 0 | | | | | | | | | | | | | | | | | | | Exercisable At September 30, 2012 | | | 811,214 | | | $ | 7.96 | | | | 6.6 years | | | $ | 0 | | | | | | | | | | | | | | | | | | |
Stock-based compensation expense related to stock options for the year ended September 30, 2012 and 2011 was $1,241,404 and $1,376,921, respectively. For the years ended September 30, 20122015, 2014 and 2011, $02013 was $4,760,831, $3,144,776, and $27,519, respectively, of this expense is included in discontinued operations. There is no$1,536,271, respectively. The Company does not recognize an income tax benefit as the companyCompany is currently operating at a loss and an actual income tax benefit may not be realized. TheFor non-qualified stock options, the loss creates a timing difference, resulting in a deferred tax asset, which is fully reserved by a valuation allowance. The fair value of the options granted by Arrowhead for the years ended September 30, 20122015, 2014 and 2011 is estimated at $4,091,1172013 was $7,338,395, $9,267,048, and $93,004, respectively. The aggregate fair value of options granted by Calando during the years ended September 30, 2012 and 2011 is estimated at $33,690 and $33,870,$2,843,575, respectively. The intrinsic value of the options exercised during the years ended September 30, 20122015, 2014 and 20112013 was $0$128,391, $4,360,850 and $3,666,$554, respectively. As of September 30, 2012,2015, the pre-tax compensation expense for all outstanding unvested stock options at Arrowhead in the amount of approximately $3,738,217$11,961,288 will be recognized in ourthe Company’s results of operations over a weighted average period of 3.1 years. As of September 30, 2012, the pre-tax compensation expense for all unvested stock options at Calando in the amount of approximately $63,250 will be recognized in our results of operations over a weighted average period of 2.6 2.7 years. The fair value of each stock option award is estimated on the date of grant using the Black-Scholes option pricing model. The Black-Scholes option valuation model was developed for use in estimating the fair value of traded options, which do not have vesting restrictions and are fully transferable. The determination of the fair value of each stock option is affected by our stock price on the date of grant, as well as assumptions regarding a number of highly complex and subjective variables. Because the Company’s employee stock options have characteristics significantly different from those of traded options, and because changes in the subjective input assumptions can materially affect the fair value estimate, in management’s opinion, the existing models do not necessarily provide a reliable single measure of the fair value of its employee stock options. The assumptions used to value stock options are as follows: | | | | Year ended September 30, | | Year ended September 30, | | | | 2012 | | 2011 | | 2015 | | 2014 | | 2013 | Dividend yield | | — | | — | | — | | — | | — | Risk-free interest rate | | 0.9% to 1.7% | | 1.11% to 2.90% | | 1.46 – 1.89% | | 1.8 – 2.4% | | 0.7 – 2.3% | Volatility | | 90% -100% | | 100% | | 75% | | 69% | | 69% | Expected life (in years) | | 5.5 to 6.25 | | 5.5 to 6.25 | | 6 - 6.25 | | 6.25 – 9.47 | | 5.5 – 6.25 | Weighted average grant date fair value per share of options granted | | $3.32 | | $4.70 | | $4.24 | | $8.92 | | $1.88 |
The dividend yield is zero as the Company currently does not pay a dividend. The risk-free interest rate is based on that of the U.S. Treasury bond. Volatility is estimated based on volatility average of the Company’s Common Stock price. Restricted Stock Units Restricted Stock Units (RSUs) were granted in fiscal year 2015 under the Company’s 2013 Incentive Plan and as inducement grants granted outside of the Plan. Of the restricted stock units granted during the years ended September 30, 2015 , 2014 and, 2013, 30,000, 40,000 and 0 shares, respectively, were granted outside of the Plan as inducement grants to new employees. At vesting, each RSU will be exchanged for one share of the Company’s Common Stock. Restricted stock unit awards generally vest subject to the satisfaction of service requirements or the satisfaction of both service requirements and achievement of certain performance targets. The following table summarizes the activity of the Company’s Restricted Stock Units: | Number of
RSUs | | | Weighted-
Average
Grant
Date
Fair Value | | Unvested at September 30, 2014 | | 510,000 | | | $ | 14.58 | | Granted | | 705,000 | | | | 7.41 | | Vested | | (280,833 | ) | | | 14.56 | | Forfeited | | — | | | | — | | Unvested at September 30, 2015 | | 934,167 | | | $ | 9.18 | |
F-17
The Company recorded $4,489,931, $2,551,397 and $0 of expense relating to restricted stock units during the years ended September 30, 2015, 2014, and 2013, respectively. Such expense is included in stock-based compensation expense in the Company’s Consolidated Statement of Operations and Comprehensive Loss. As of September 30, 2015, the pre-tax compensation expense for all unvested restricted stock units in the amount of approximately $3,739,892 will be recognized in the Company’s results of operations over a weighted average period of 1.6 years. NOTE 10.9. FAIR VALUE MEASUREMENTS & DERIVATIVE INSTRUMENTS The Company measures its financial assets and liabilities at fair value. Fair value is defined as the price that would be received to sell an asset or paid to transfer a liability (i.e., exit price) in an orderly transaction between market participants at the measurement date. Additionally, the Company is required to provide disclosure and categorize assets and liabilities measured at fair value into one of three different levels depending on the assumptions (i.e., inputs) used in the valuation. Level 1 provides the most reliable measure of fair value while Level 3 generally requires significant management judgment. Financial assets and liabilities are classified in their entirety based on the lowest level of input significant to the fair value measurement. The fair value hierarchy is defined as follows: Level 1—Valuations are based on unadjusted quoted prices in active markets for identical assets or liabilities. F-18
Level 2—Valuations are based on quoted prices for similar assets or liabilities in active markets, or quoted prices in markets that are not active for which significant inputs are observable, either directly or indirectly. Level 3—Valuations are based on prices or valuation techniques that require inputs that are both unobservable and significant to the overall fair value measurement. Inputs reflect management’s best estimate of what market participants would use in valuing the asset or liability at the measurement date. The following table summarizes fair value measurements at September 30, 20122015 and September 30, 20112014 for assets and liabilities measured at fair value on a recurring basis: September 30, 2012:2015: | | | | | | | | | | | | | | | | | | | | Level 1 | | | Level 2 | | | Level 3 | | | Total | | Cash and cash equivalents | | $ | 3,377,288 | | | $ | — | | | $ | — | | | $ | 3,377,288 | | Marketable securities | | $ | 106,500 | | | $ | — | | | $ | — | | | $ | 106,500 | | Derivative assets | | $ | — | | | $ | — | | | $ | 250,250 | | | $ | 250,250 | | Derivative liabilities | | $ | — | | | $ | — | | | $ | 647,213 | | | $ | 647,213 | | Contingent consideration | | $ | — | | | $ | — | | | $ | 173,621 | | | $ | 173,621 | | | | | | | September 30, 2011: | | | | | | | | | | | | | | | | | | | | | | | | | Level 1 | | | Level 2 | | | Level 3 | | | Total | | Cash and cash equivalents | | $ | 7,507,389 | | | $ | — | | | $ | — | | | $ | 7,507,389 | | Marketable securities | | $ | 634,585 | | | $ | — | | | $ | — | | | $ | 634,585 | | Derivative assets | | $ | — | | | $ | — | | | $ | 161,125 | | | $ | 161,125 | | Derivative liabilities | | $ | — | | | $ | — | | | $ | 944,980 | | | $ | 944,980 | | Contingent consideration | | $ | — | | | $ | — | | | $ | — | | | $ | — | |
| Level 1 | | | Level 2 | | | Level 3 | | | Total | | Cash and cash equivalents | $ | 81,214,354 | | | $ | — | | | $ | — | | | $ | 81,214,354 | | Derivative liabilities | $ | — | | | $ | — | | | $ | 1,301,604 | | | $ | 1,301,604 | | Acquisition-related contingent consideration obligations | $ | — | | | $ | — | | | $ | 5,862,464 | | | $ | 5,862,464 | |
As partSeptember 30, 2014:
| Level 1 | | | Level 2 | | | Level 3 | | | Total | | Cash and cash equivalents | $ | 132,510,610 | | | $ | — | | | $ | — | | | $ | 132,510,610 | | Derivative liabilities | $ | — | | | $ | — | | | $ | 4,173,943 | | | $ | 4,173,943 | | Acquisition-related contingent consideration obligations | $ | — | | | $ | — | | | $ | 3,970,931 | | | $ | 3,970,931 | |
The Company invests its excess cash balances in short- and long-term corporate bonds, generally with remaining maturities of less than two years. At September 30, 2015, the saleCompany had short-term investments of proceeds from the sale of Unidym in January 2011, Arrowhead received a bond from Wisepower in the face amount of $2.5 million. The bond is convertible to Wisepower common stock at a price of $2.00 per share. The conversion feature is subject to derivative accounting as prescribed under ASC 815. Accordingly, the fair value of the conversion feature on the date of issuance was estimated using an option pricing model and recorded on the Company’s consolidated balance sheet as a derivative asset.$17,539,902. The fair value of its investment at September 30, 2015 was $17,234,960. The Company expects to hold such investments until maturity, and thus unrealized gains and losses from the conversion feature is estimated at the end of each reporting period and the changefluctuations in the fair value of the conversion feature is recorded as a nonoperating gain/loss as change in value of derivatives in Company’s Consolidated Statement of Operations. A portion of the bond is owedsecurities are not likely to a third party, as such the company records a derivative asset for the entire conversion feature and records a derivative liability for the portion related to the third party. The original fair value of the derivative relating to the third party was $26,310; the fair value at September 30, 2011 was $6,854, and the fair value at September 30, 2012 was $10,645. The gain from the change in value of the derivative asset, net of the derivative liability, for the year ended September 30, 2012 was $85,334, and is reflected in the change in value of derivatives in the Company’s consolidated statement of operations.be realized. During the year ended September 30, 2012, the Company recorded a gain from the change in fair value of the derivative asset of $89,125. The assumptions used in valuing the derivative asset as of September 30, 2012 and 2011 were as follows:F-18
| | | | | | | | September 30, 2012 | | September 30, 2011 | Risk free interest rate | | 0.23% | | 0.4% | Expected life | | 1.3 Years | | 2.3 Years | Dividend yield | | none | | none | Volatility | | 72% | | 72% |
The following is a reconciliation of the derivative asset for the years ended September 30, 2012 and 2011:
| | | | | Value at September 30, 2010 | | $ | — | | Receipt of instruments | | | 618,500 | | Change in value | | | (457,375 | ) | | | | | | Value at October 1, 2011 | | | 161,125 | | Receipt of instruments | | | — | | Increase in value | | | 89,125 | | Net settlements | | | — | | | | | | | Value at September 30, 2012 | | $ | 250,250 | | | | | | |
F-19
As part of thean equity financing onin June 17, 2010, Arrowhead issued warrants to acquirepurchase up to 329,649 shares of Common Stock (the “2010 Warrants”), of which 24,324 warrants were outstanding at September 30, 2015. Similarly, as part of a financing in December 2012, Arrowhead issued warrants to purchase up to 912,543 shares of Common Stock (the “2012 Warrants”) of which 265,161 warrants were outstanding at September 30, 2015. Further, as part of a financing in January 2013, Arrowhead issued warrants to purchase up to 833,530 shares of Common Stock (the “2013 Warrants” and, together with the 2010 Warrants and the 2012 Warrants, the “Warrants”) of which contain12,123 warrants were outstanding at September 30, 2015. Each of the Warrants contains a mechanism to adjust the strike price upon the issuance of certain dilutive equity securities. If during the termterms of the Warrants, the Company issues Common Stock at a price lower than the exercise price offor the Warrants, the exercise price of the Warrants would be reduced to the amount equal to the issuance price of the Common Stock. As a result of this feature,these features, the Warrants are subject to derivative accounting as prescribed under ASC 815. Accordingly, the fair value of the Warrants on the date of issuance was estimated using an option pricing model and recorded on the Company’s consolidated balance sheetConsolidated Balance Sheet as a derivative liability. The fair value of the Warrants is estimated at the end of each reporting period and the change in the fair value of the Warrants is recorded as a nonoperatingnon-operating gain or loss as change in value of derivatives in the Company’s consolidated statementConsolidated Statement of operations.Operations and Comprehensive Loss. During the yearyears ended September 30, 2012,2015, 2014 and 2013, the Company recorded a non-cash lossgain/(loss) from the change in fair value of the derivative liability of $281,038. $2,684,712, $(5,821,796), and $5,066,591, respectively. The assumptions used in valuing the derivative liability as of September 30, 2012 and 2011 were as follows: | | | | September 30, 2012 | | September 30, 2011 | | Risk free interest rate | | 0.31% | | 0.9% | | 2010 Warrants | | | September 30, 2015 | | | September 30, 2014 | | September 30, 2013 | Risk-free interest rate | | | 0.1% | | | 0.13% | | 0.33% | Expected life | | 3.2 Years | | 4.2 Years | | 0.2 Years | | | 1.2 Years | | 2.2 Years | Dividend yield | | none | | none | | - | | | - | | - | Volatility | | 100% | | 100% | | 75% | | | 69% | | 69% | | | | | | | | | | 2012 Warrants | | | September 30, 2015 | | | September 30, 2014 | | September 30, 2013 | Risk-free interest rate | | | 0.6% | | | 1.07% | | 1.39% | Expected life | | | 2.2 Years | | | 3.2 Years | | 4.2 Years | Dividend yield | | | - | | | - | | - | Volatility | | | 75% | | | 69% | | 69% | | | | | | | | | | 2013 Warrants | | | September 30, 2015 | | | September 30, 2014 | | September 30, 2013 | Risk-free interest rate | | | 0.6% | | | 1.07% | | 1.39% | Expected life | | | 2.3 Years | | | 3.3 Years | | 4.3 Years | Dividend yield | | | - | | | - | | - | Volatility | | | 75% | | | 69% | | 69% |
The following is a reconciliation of the derivative liability related to these warrants for the years ended September 30, 2012 and 2011:warrants: | | | | | Value at September 30, 2010 | | $ | 2,408,522 | | Receipt of instruments | | | — | | Change in value | | | (1,501,289 | ) | | | | | | Value at September 30, 2011 | | $ | 907,233 | | Receipt of instruments | | | — | | Change in value | | | (281,038 | ) | Net settlements | | | — | | | | | | | Value at September 30, 2012 | | $ | 626,195 | | | | | | |
Value at September 30, 2013 | $ | 4,091,797 | | Issuance of instruments | | — | | Change in value | | 5,821,796 | | Net settlements | | (5,956,079 | ) | Value at September 30, 2014 | $ | 3,957,514 | | Issuance of instruments | | — | | Change in value | | (2,684,712) | | Net settlements | | — | | Value at September 30, 2015 | $ | 1,272,802 | | | | | |
F-19
In conjunction with the financing of Ablaris during the year ended September 30,in fiscal 2011, Arrowhead sold exchange rights to certain investors whereby the investors have the right to exchange their shares of Ablaris for a prescribed number of Arrowhead shares of Common Stock based upon a predefined ratio. The exchange rights have a seven-year term. During the first year, the exchange right allows the holder to exchange one Ablaris share for 0.06 Arrowhead shares (as adjusted for a subsequent reverse stock split).shares. This ratio declines to 0.04 in the second year, 0.03 in the third year and 0.02 in the fourth year. In the fifth year and beyond the exchange ratio is 0.01. Exchange rights for 675,000 Ablaris shares were sold during the year ended September 30,in fiscal 2011, and 500,000 remain outstanding at September 30, 2012.2015. The exchange rights are subject to derivative accounting as prescribed under ASC 815. Accordingly, the fair value of the exchange rights on the date of issuance was estimated using an option pricing model and recorded on the Company’s consolidated balance sheetConsolidated Balance Sheet as a derivative liability. The fair value of the exchange rights is estimated at the end of each reporting period and the change in the fair value of the exchange rights is recorded as a nonoperatingnon-operating gain or loss in the Company’s Consolidated Statement of Operations.Operations and Comprehensive Loss. During the yearyears ended September 30, 2012,2015, 2014 and 2013, the Company recorded a non-cash lossgain/(loss) from the change in fair value of the derivative liability of $20,520. $184,555, $(211,860) and $5,806, respectively. The assumptions used in valuing the derivative liability as of September 30, 2012 and 2011 were as follows: | | | | | September 30, 2015 | | September 30, 2014 | September 30, 2013 | | | | September 30, 2012 | | September 30, 2011 | | Risk free interest rate | | 0.62% | | 1.3% | | Risk-free interest rate | | 1.00% | | 1.07% | 1.39% | Expected life | | 5.3 Years | | 6.3 Years | 2.5 Years | | 3.3 Years | 4.3 Years | Dividend yield | | none | | none | - | | - | - | Volatility | | 100% | | 100% | 75% | | 100% | 100% |
The following is a reconciliation of the derivative liability related to these exchange rightsrights: Value at September 30, 2013 | $ | 4,569 | | Issuance of instruments | | — | | Change in value | | 211,860 | | Net settlements | | — | | Value at September 30, 2014 | $ | 216,429 | | Issuance of instruments | | — | | Change in value | | (184,555 | ) | Net settlements | | (3,072) | | Value at September 30, 2015 | $ | 28,802 | | | | | |
The derivative assets/liabilities are estimated using option pricing models that are based on the individual characteristics of the warrants or instruments on the valuation date, as well as assumptions for expected volatility, expected life and risk-free interest rate. Changes in the years endedassumptions used could have a material impact on the resulting fair value. The primary input affecting the value of our derivatives liabilities is the Company’s stock price. Other inputs have a comparatively insignificant effect. As of September 30, 2012 and 2011: | | | | | Value at September 30, 2010 | | $ | — | | Issuance of instruments | | | 100,650 | | Change in value | | | (69,758 | ) | | | | | | Value at September 30, 2011 | | $ | 30,892 | | Issuance of instruments | | | — | | Change in value | | | (20,520 | ) | Net settlements | | | — | | | | | | | Value at September 30, 2012 | | $ | 10,372 | | | | | | |
F-20
During2015, the year ended September 30, 2012,Company has liabilities for contingent consideration was recorded uponrelated to its acquisition of the acquisitions of Roche Madison, Inc.RNAi business and Alvos Therapeutics, Inc., totaling $173,621.the Novartis asset acquisition discussed in footnote 2. The fair value measurement of the contingent consideration obligations is determined using Level 3 inputs. The fair value of contingent consideration obligations is based on a discounted cash flow model using a probability-weighted income approach. The measurement is based upon unobservable inputs supported by little or no market activity based on our ownthe Company’s assumptions and experience. Estimating timing to complete the development and obtain approval of products is difficult, and there are inherent uncertainties in developing a product candidate, such as obtaining U.S. Food and Drug Administration (FDA) and other regulatory approvals. In determining the probability of regulatory approval and commercial success, we utilizethe Company utilizes data regarding similar milestone events from several sources, including industry studies and ourits own experience. These fair value measurements represent Level 3 measurements as they are based on significant inputs not observable in the market. Significant judgment is employed in determining the appropriateness of these assumptions as of the acquisition date and for each subsequent period. Accordingly, changes in assumptions could have a material impact on the amount of contingent consideration expense we recordthe Company records in any given period. Changes in the fair value of the contingent consideration obligations are recorded in our consolidated statementthe Company’s Consolidated Statement of operations. There were no changes inOperations and Comprehensive Loss.
F-20
The following is a reconciliation of contingent consideration fair value as of September 30, 2012.value: | Value at September 30, 2011 | | $ | — | | | Value at September 30, 2013 | | $ | 1,595,273 | | Purchase price contingent consideration | | | 173,621 | | | — | | Contingent consideration payments | | | — | | | — | | Change in fair value of contingent consideration | | | — | | | 2,375,658 | | | | | | | Value at September 30, 2012 | | $ | 173,621 | | | | | | | | Value at September 30, 2014 | | $ | 3,970,931 | | Purchase price contingent consideration | | | — | | Contingent consideration payments | | | — | | Change in fair value of contingent consideration | | | 1,891,533 | | Value at September 30, 2015 | | $ | 5,862,464 | |
The fair value of contingent consideration obligations is estimated through valuation models designed to estimate the probability of such contingent payments based on various assumptions and incorporating estimated success rates. Estimated payments are discounted using present value techniques to arrive at estimated fair value at the balance sheet date. Changes in the fair value of the contingent consideration obligations can result from changes to one or multiple inputs, including adjustments to the discount rates, changes in the amount or timing of expected expenditures associated with product development, changes in the amount or timing of cash flows from products upon commercialization, changes in the assumed achievement or timing of any development milestones, changes in the probability of certain clinical events and changes in the assumed probability associated with regulatory approval. Each of these assumptions can have a significant impact on the calculation of contingent consideration. The carrying amounts of the Company’s other financial instruments, which include accounts receivable, accounts payable, and accrued expenses approximate their respective fair values due to the relatively short-term nature of these instruments. The carrying value of the Company’s debt obligations approximates fair value based on market interest rates.
NOTE 11.10. - INCOME TAXES The Company utilizes the guidance issued by the FASB for accounting for income taxes which requires the recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have been included in the financial statements or tax returns. Under this method, deferred income taxes are recognized for the tax consequences in future years of differences between the tax bases of assets and liabilities and their financial reporting amounts at each year-end based on enacted tax laws and statutory tax rates applicable to the periods in which the differences are expected to affect taxable income. Valuation allowances are established, when necessary, to reduce deferred tax assets to the amount expected to be realized. The provision for income taxes represents the tax payable for the period and the change during the period in deferred tax assets and liabilities. ForComponents of the years endednet deferred tax asset (liability) at September 30, 20122015 and 2011, the Company had consolidated net2014 are as follows:
| | 2015 | | | 2014 | | Deferred tax assets: | | | | | | | | Reserve for other receivables | $ | - | | | $ | 233,014 | | Accrued compensation | | 1,513,021 | | | | 1,313,354 | | Stock compensation | | 6,571,774 | | | | 3,011,369 | | Capitalized research and development | | - | | | | 13,536,745 | | Fair value adjustments | | 2,850,125 | | | | 1,864,364 | | Net operating losses | | 88,965,968 | | | | 42,268,526 | | Intangible Assets | | 5,551,705 | | | | - | | Total deferred tax assets | | 105,452,593 | | | | 62,227,372 | | Valuation allowance | | (95,085,045 | ) | | | (55,224,802 | ) | Deferred tax liabilities: | | | | | | | | State taxes | | (10,282,834 | ) | | | (6,277,587 | ) | Equity investments | | - | | | | (7,675 | ) | Intangible assets | | - | | | | (475,829 | ) | Fixed assets | | (84,714 | ) | | | (241,479 | ) | Total deferred tax liability | | (10,367,548 | ) | | | (7,002,570 | ) | Net deferred tax assets | $ | — | | | $ | — | |
The Company’s book losses of $22.4 million and $3.5 million, respectively. The lossesother timing differences result in a net deferred income tax benefit which is offset by a deferred tax provision for the valuation allowance for a net deferred provisionasset of zero. SinceThe Company has concluded, in accordance with the applicable accounting F-21
standards, that it is more likely than not that the Company is a development stage company,may not realize the benefit of all of its deferred tax assets. Accordingly, management has provided a 100% valuation allowance against its deferred tax assets until such time as management believes that its projections of future profits as well as expected future tax rates make the realization of these deferred tax assets more-likely-than-not. Significant judgment is required in the evaluation of deferred tax benefits and differences in future results from our estimates could result in material differences in the realization of these assets. As of September 30, 2012, the Company has available gross2015 and 2014, federal net operating loss (NOL) carry forwards of $107.2 million and gross state NOL carry forwards of $75.9 million which expire at various dates through 2032. As of September 30, 2012, the deferred tax assets were $40.9 million.$105.5 million and $62.2 million, respectively. The Company has recorded a full valuation allowance of $40.9 million related to federal and state net operating loss carry forwards.all of its deferred tax assets. The Company has performed an assessment of positive and negative evidence regarding the realization of the net deferred tax asset in accordance with FASB ASC 740-10, “Accounting for Income Taxes.” This assessment included the evaluation of scheduled reversals of deferred tax liabilities, the availability of carry forwards and estimates of projected future taxable income.
As of September 30, 2014, the Company had available gross federal net operating loss (NOL) carry forwards of $115.8 million and gross state NOL carry forwards of $170.8 million which expire at various dates through 2034. Gross federal NOL carry forwards for 2015 are estimated at $70.5 million, and gross state NOL carry forwards for 2015 are estimated at $124.1 million. The provision for income taxes for the years ended September 30, 2015 and 2014 are as follows: | 2015 | | | 2014 | | Federal: | | | | | | | | Current | | — | | | | — | | Deferred | | — | | | | — | | Total Federal | | — | | | | — | | State: | | | | | | | | Current | $ | 2,400 | | | | 5,300 | | Deferred | | — | | | | — | | Total State | $ | 2,400 | | | | 5,300 | | Provision from income taxes | $ | 2,400 | | | | 5,300 | |
F-21
The Company’s effective income tax rate differs from the statutory federal income tax rate as follows for the years ended September 30, 2015 and 2014: | | 2015 | | | 2014 | | At U.S. federal statutory rate | | 34.0 | % | | | 34.0 | % | State taxes, net of federal effect | | 9.3 | | | | 7.6 | | Stock compensation | | (0.7 | ) | | | (0.1 | ) | Mark-to-market adjustments | | 0.7 | | | | (3.5 | | Write-off of net operating losses | | 0.0 | | | | (32.7 | ) | Valuation allowance | | (43.4 | ) | | | (5.2 | ) | Other | | 0.1 | | | | (0.1 | ) | Effective income tax rate | | 0.0 | % | | | 0.0 | % |
The Company has adopted guidance issued by the FASB that clarifies the accounting for uncertainty in income taxes recognized in an enterprise’s financial statements and prescribes a recognition threshold of more likely than not and a measurement process for financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return. In making this assessment, a company must determine whether it is more likely than not that a tax position will be sustained upon examination, based solely on the technical merits of the position and must assume that the tax position will be examined by taxing authorities. OurThe Company’s policy is to include interest and penalties related to unrecognized tax benefits in income tax expense. InterestThe Company has not recognized any unrecognized tax benefits and does not have any interest or penalties totaled $0 for the years endedrelated to uncertain tax positions as of September 30, 20122015 and 2011, respectively, and $0 for the period from May 7, 2003 (date of inception) through September 30, 2012. 2014. The Company files income tax returns with the Internal Revenue Service (“IRS”), the state of California and certain other taxing jurisdictions. For jurisdictions in which tax filings are prepared, theThe Company is no longer subject to income tax examinations by the IRS and by state tax authorities for years through fiscal 2007, and byuntil the IRS for the years through fiscal 2008. Our review of prior year tax positions using the criteria and provisions presented by the FASB did not result in a material impact on the Company’s financial position or results of operations.net operating losses are settled. The provision for income taxes differs from the federal statutory rate due to state income taxes and changes in the valuation allowance for deferred income tax assets.
F-22
NOTE 13.RELATED PARTY TRANSACTIONS11. Christopher Anzalone, Arrowhead’s President and CEO, owns 1,395,900 shares of Nanotope, Inc. common stock or approximately 14.2% of Nanotope’s outstanding voting securities. Dr. Anzalone does not hold options, warrants or any other rights to acquire securities of Nanotope. Dr. Anzalone has the right to appoint a representative to the board of directors of Nanotope. Dr. Anzalone currently serves on the Nanotope board in a seat reserved for Nanotope’s CEO, and another individual holds the seat designated by Dr. Anzalone. Dr. Anzalone has served as President and Chief Executive Officer of Nanotope since its formation and continues to serve in these capacities. Dr. Anzalone has not received any compensation for his work on behalf of Nanotope since joining the Company on December 1, 2007. Dr. Anzalone has also waived his right to any unpaid compensation accrued for work done on behalf of Nanotope before he joined the Company.
In August 2010, the Company retained Mr. Vincent Anzalone, the brother of Arrowhead’s Chief Executive Officer, as a consultant for the Company, focusing on business development and market analysis, with a monthly remuneration of $10,000 per month. Mr. Vincent Anzalone was paid $120,000 during the fiscal years ended September 30, 2012, and 2011.
NOTE 14.EMPLOYEE BENEFIT PLANS
In January 2005, the Company began sponsoringadopted a defined contribution 401(k) retirement savings plan covering substantially all of its employees. The Plan wasis administered under the “safe harbor” provision of ERISA. Under the terms of the plan, an eligible employee may elect to contribute a portion of their salary on a pre-tax basis, subject to federal statutory limitations. The plan allowedallows for a discretionary match in an amount up to 100% of each participant’s first 3% of compensation contributed plus 50% of each participant’s next 2% of compensation contributed. For the years ended September 30, 20122015, 2014, and 2011,2013, we recorded expenses under these plans of approximately $162,000$407,603, $264,193, and $43,000, respectively and $616,000 since inception of the Company.$191,947, respectively. In addition to the employee benefit plans described above, the Company participates inprovides certain customary employee benefitsbenefit plans, including those which provide health and life insurance benefits to employees. NOTE 15.SUBSEQUENT EVENTS
In December 2012, the Company sold 1.9 million units at a price of $2.26 per unit in a public offering. Each unit consisted of one share of Common Stock and a warrant to purchase 0.5 share of Common Stock, exercisable at $2.20. Gross proceeds from the offering were $4.3 million, which included a $500,000 promissory note due February 1, 2013. Commissions and other offering expenses are expected to be approximately $300,000.
F-22NOTE 12. UNAUDITED QUARTERLY FINANCIAL DATA
The following table presents selected unaudited quarterly financial data for each full quarterly period of the years ended September 30, 2015 and 2014: | | First | | Second | | Third | | Fourth | | Year ended September 30, 2015 | | Quarter | | Quarter | | Quarter | | Quarter | | Revenues | | $ | 170,750 | | $ | 43,750 | | $ | 123,750 | | $ | 43,750 | | Operating Losses | | $ | (25,115,276) | | $ | (29,632,743) | | $ | (15,993,706) | | $ | (25,232,251) | | Net Loss | | $ | (22,575,282) | | $ | (28,683,993) | | $ | (15,936,053) | | $ | (24,745,554) | | Net Loss Attributable to Arrowhead | | $ | (22,575,282) | | $ | (28,683,993) | | $ | (15,936,053) | | $ | (24,745,554) | | Loss per share (Basic and Diluted) | | $ | (0.41) | | $ | (0.51) | | $ | (0.27) | | $ | (0.42) | |
| | First | | Second | | Third | | Fourth | | Year ended September 30, 2014 | | Quarter | | Quarter | | Quarter | | Quarter | | Revenues | | $ | 43,750 | | $ | 43,750 | | $ | 43,750 | | $ | 43,750 | | Operating Losses | | $ | (7,009,382) | | $ | (11,212,498) | | $ | (12,700,100) | | $ | (22,354,306) | | Net Loss | | $ | (10,685,372) | | $ | (13,982,700) | | $ | (11,626,451) | | $ | (22,430,889) | | Net Loss Attributable to Arrowhead | | $ | (10,628,312) | | $ | (13,942,521) | | $ | (11,626,919) | | $ | (22,432,438) | | Loss per share (Basic and Diluted) | | $ | (0.28) | | $ | (0.31) | | $ | (0.22) | | $ | (0.42) | |
F-23 |
|
|
|