| • | | • | the abilityextent to which our business, the medical community and the global economy will continue to be materially and adversely impacted by the effects of Alimera Sciences, Inc.the COVID-19 pandemic (the “Pandemic”), or Alimera,by other pandemics, epidemics or outbreaks; |
| • | the potential for EYP-1901, as a twice-yearly sustained delivery intravitreal anti-VEGF treatment targeting wet age-related macular degeneration (“wet AMD”), with potential in diabetic retinopathy (“DR”) and retinal vein occlusion (“RVO”); |
| • | our expectations regarding the timing and outcome of our Phase 1 clinical trial for EYP-1901 for the treatment of wet AMD; |
| • | our expectations to obtain regulatory approvalavoid the toxicity seen in the prior clinical trials of orally delivered vorolanib, a tyrosine kinase inhibitor (“TKI”) by delivering vorolanib locally using a bioerodible Durasert® technology as EYP-1901 at a significantly lower total dose; |
| • | our expectations regarding the timing and commercialize ILUVIENclinical development of our product candidates, including EYP-1901 and YUTIQ50; |
| • | the potential advantages of YUTIQ® and DEXYCU® for the treatment ofnon-infectious posterior segment uveitis, in Europe, the Middle East and Africa; eye diseases; |
the implication of results frompre-clinical and clinical trials and our other research activities;
| • | our cash flow expectations from commercial sales of YUTIQ and DEXYCU; |
| • | | • | our intentionsability to manufacture YUTIQ and DEXYCU, or any future products or product candidates, in sufficient quantities and quality; |
| • | our belief that our cash and cash equivalents of $44.9 million at December 31, 2020, combined with the approximately $108.0 million of net proceeds from the February 2021public offering of shares of our common stock and anticipated net cash inflows from product sales will fund our operating plan through the second quarter of 2022, under current expectations regarding (i) the timing and outcomes of our Phase 1 clinical trial for EYP-1901 for the treatment of wet AMD, and (ii) initiation of our Phase 2 clinical trials for EYP-1901 for the treatment of wet AMD; |
| • | our ability to obtain additional capital in sufficient amounts and on terms acceptable to us, and the consequences of failing to do so; |
| • | our future expenses and capital expenditures; |
| • | our expectations regarding the timing and results of the subpoena from the Division of Enforcement of the U.S. Securities and Exchange Commission (“SEC”) seeking production of certain documents and information on topics including product sales and demand, revenue recognition and accounting in relation to product sales, product sales and cash projections, and related financial reporting, disclosure and compliance matters (the “SEC” investigation”); |
| • | our expectations regarding our research into other usesability to obtain and applicationsadequately maintain sufficient intellectual property protection for EYP-1901, YUTIQ, DEXYCU and YUTIQ50 and any future products or product candidates, and to avoid claims of our Durasert™ and Verisome® technology platforms;infringement of third-party intellectual property rights; |
our expectations regarding our ability to obtain and adequately maintain sufficient intellectual property protection for DEXYCU, YUTIQ and our other product candidates, and to avoid claims of infringement of third party intellectual property rights;
| • | our expectation that we will continue to incur significant expenses and that our operating losses and our net cash outflows to fund operations will continue for the foreseeable future; |
our expectation that we will continue to incur significant expenses and that our operating losses and our net cash outflows to fund operations will continue for the foreseeable future;
| • | our expectations regarding our partnership with ImprimisRx; |
the scope and duration of intellectual property protection; and
| • | our expectation regarding the potential for our Paycheck Protection Program Loan (the “PPP Loan”) to be forgiven in full; and |
the effect of legal and regulatory developments.
| • | the effect of legal and regulatory developments. |
Forward-looking statements also include statements other than statements of current or historical fact, including, without limitation, all statements related to any expectations of revenues, expenses, cash flows, earnings or losses from operations, cash required to maintain current and planned operations, capital or other financial items; any statements of the plans, strategies and objectives of management for future operations; any plans or expectations with respect to product research, development and commercialization, including regulatory approvals; any other statements of expectations, plans, intentions or beliefs; and any statements of assumptions underlying any of the foregoing. We often, although not always, identify forward-looking statements by using words or phrases such as “likely”, “expect”, “intend”, “anticipate”, “believe”, “estimate”, “plan”, “project”, “forecast” and “outlook”.
The following are some of the factors that could cause actual results to differ materially from the anticipated results or other expectations expressed, anticipated or implied in our forward-looking statements: uncertainties with respect to: our ability to achieve profitable operations and access to needed capital; fluctuations in our operating results; the number of clinical trials, including clinical trials conducted outside the United States, or U.S., and data required for marketing approval for YUTIQ in the U.S.; our ability to use data in promotion for YUTIQ; our ability to successfully produce commercial supply of DEXYCU and successfully commercialize DEXYCU in the U.S.; our ability to successfully build a commercial infrastructure and enter into and maintain commercial agreements for the launch of DEXYCU and, if approved, YUTIQ; our ability to successfully commercialize YUTIQ, if approved, in the U.S.; potentialoff-label sales of ILUVIEN for uveitis; consequences of fluocinolone acetonide side effects; successful commercialization of, and receipt of revenues from, ILUVIEN for diabetic macular edema, or DME, which depends on Alimera’s ability to continue as a going concern; Alimera’s ability to obtain additional marketing approvals and the effect of pricing and reimbursement decisions on sales of ILUVIEN for DME; Alimera’s ability to obtain marketing approval for ILUVIEN in its licensed territories fornon-infectious posterior segment uveitis; the development of our next-generation Durasert short-acting treatment for uveitis; potential declines in Retisert® royalties; our ability to market and sell products; the success of current and future license agreements, including our agreement with Alimera; termination or breach of current license agreements, including our agreement with Alimera; our dependence on contract research organizations, contract sales organizations, vendors and investigators; effects of competition and other developments affecting sales of products; market acceptance of products; effects of guidelines, recommendations and studies; protection of intellectual property and avoiding intellectual property infringement; retention of key personnel; product liability; industry consolidation; compliance with environmental laws; manufacturing risks; risks and costs of international business operations; effects of the potential exit of the United Kingdom from the European Union; legislative or regulatory changes; volatility of stock price; possible dilution; absence of dividends; and other factors described in our filings with the Securities and Exchange Commission. | • | the extent to which the Pandemic impacts our business, the medical community and the global economy; |
| • | the effectiveness and timeliness of our preclinical studies and clinical trials, and the usefulness of the data; |
| • | uncertainties with respect to the duration, scope and outcome of the SEC investigation and its impact on our financial condition, results of operations and cash flows; |
| • | our ability to achieve profitable operations and access to needed capital; |
| • | fluctuations in our operating results; |
| • | our ability to successfully produce sufficient commercial quantities of YUTIQ and DEXYCU and to successfully commercialize YUTIQ and DEXYCU in the U.S.; |
| • | our ability to sustain and enhance an effective commercial infrastructure and enter into and maintain commercial agreements for the commercialization of YUTIQ and DEXYCU; |
| • | consequences of fluocinolone acetonide side effects for YUTIQ; |
| • | consequences of dexamethasone side effects for DEXYCU; |
| • | the success of current and future license and collaboration agreements, including our agreements with Ocumension Therapeutics (“Ocumension”) and Equinox Science, LLC (“Equinox”); |
| • | our dependence on contract research organizations, contract sales organizations, vendors and investigators; |
| • | effects of competition and other developments affecting sales of products; |
| • | market acceptance of our products; |
| • | protection of intellectual property and avoiding intellectual property infringement; |
| • | other factors described in our filings with the SEC. |
We cannot guarantee that the results and other expectations expressed, anticipated or implied in any forward-looking statement will be realized. The risks set forth under Item 1A of this Annual Report on Form10-K describe major risks to our business, and you should read and interpret any forward-looking statements together with these risks. A variety of factors, including these risks, could cause our actual results and other expectations to differ materially from the anticipated results or other expectations expressed, anticipated or implied in our forward-looking statements. Should known or unknown risks materialize, or should underlying assumptions prove inaccurate, actual results could differ materially from past results and those anticipated, estimated or projected in the forward-looking statements. You should bear this in mind as you consider any forward-looking statements. Our forward-looking statements speak only as of the dates on which they are made. We do not undertake any obligation to publicly update or revise our forward-looking statements even if experience or future changes makes it clear that any projected results expressed or implied in such statements will not be realized. DEXYCU®, YUTIQ®, and Durasert® are our trademarks. Retisert® and Vitrasert® are Bausch & Lomb’s trademarks. ILUVIEN® is Alimera Sciences Inc.’s trademark. Verisome® is a trademark owned by Ramscor, Inc. and exclusively licensed to us. The reports we file or furnish with the SEC, including this Annual Report on Form 10-K, also contain trademarks, trade names and service marks of other companies, which are the property of their respective owners. Risk Factor Summary The risk factors summarized below could materially harm our business, operating results and/or financial condition, impair our future prospects and/or cause the price of our common stock to decline. For more information, see “Item 1A. Risk Factors” in this Annual Report on Form 10-K for the year ended December 31, 2020. Material risks that may affect our business, operating results and financial condition include, but are not necessarily limited to, the following: Risks Related To Our Financial Position and our Capital Resources | • | We will likely need additional capital to fund our operations. If we are unable to obtain sufficient capital, we will need to curtail and reduce our operations and costs and modify our business strategy. |
| • | We have incurred significant losses since our inception and anticipate that we will continue to incur losses for the foreseeable future. |
| • | We may never achieve profitability from future operations. |
| • | The ongoing novel coronavirus (COVID-19) pandemic has had and will likely continue to have a material and adverse impact on our business. |
| • | We received a subpoena from the SEC Enforcement Division requesting documents and information in an investigation relating to product sales and demand, revenue recognition and accounting. If the SEC commences an enforcement action against us, the resolution of such an enforcement action could have a material adverse effect on our business, financial condition, results of operations and cash flows. In addition, we have expended and expect to continue to expend significant financial and managerial resources responding to the SEC subpoena, which could also have a material adverse effect on our business, financial condition, results of operations and cash flows. |
| • | We will need to raise additional capital in the future, which may not be available on favorable terms and may be dilutive to stockholders or impose operational restrictions. |
| • | We must maintain compliance with the terms of our CRG loan or receive a waiver for any non-compliance. Our failure to comply with the covenants or other terms of the loan, including as a result of events beyond our control, could result in a default under the loan agreement that would materially and adversely affect the ongoing viability of our business. |
| • | Our Loan Agreement contains restrictions that limit our flexibility in operating our business. |
| • | Certain potential payments to the Lenders could impede a sale of our company. |
| • | To service our indebtedness, we will require a significant amount of cash and our ability to generate cash depends on many factors beyond our control. |
Risks Related To The Regulatory Approval And Clinical Development Of Our Product Candidates | • | We are substantially dependent on the success of our lead product candidate, EYP-1901, which is in the early stages of development and must go through clinical trials, which are very expensive, time-consuming and difficult to design and implement. The outcomes of clinical trials are uncertain, and delays in the completion of or the termination of any clinical trial of EYP-1901 or our other product candidates could harm our business, financial condition and prospects. |
| • | Clinical trial results may fail to support approval of EYP-1901 or our other product candidates. |
| • | We may expend significant resources to pursue our lead product candidate, EYP-1901 for the potential treatment of wet AMD, and fail to capitalize on the potential of EYP-1901, or our other product candidates, for the potential treatment of other indications that may be more profitable or for which there is a greater likelihood of success. |
| • | Initial results from a clinical trial do not ensure that the trial will be successful and success in early stage clinical trials does not ensure success in later-stage clinical trials. |
| • | We face risks related to health epidemics and outbreaks, including the Pandemic, which could significantly disrupt our preclinical studies and clinical trials. |
| • | We may find it difficult to enroll patients in our clinical trials, which could delay or prevent clinical trials of our product candidates. |
| • | We are largely dependent on the future commercial success of our lead product candidate, EYP-1901. |
Risks Related To The Commercialization Of Our Products And Product Candidates | • | Our current business strategy relies on our ability to successfully commercialize YUTIQ and DEXYCU and in the U.S. Our approved products may not achieve market acceptance or be commercially successful. |
| • | Our products may become subject to unfavorable pricing regulations, third-party reimbursement practices or healthcare reform initiatives, which could harm our business. |
| • | If we fail to comply with reporting and payment obligations under the Medicaid Drug Rebate program or other governmental pricing programs, we could be subject to additional reimbursement requirements, penalties, sanctions, and fines which could have a material adverse effect on our business, financial condition, results of operations and growth prospects. |
| • | Even though regulatory approvals for YUTIQ and DEXYCU have been obtained in the U.S., we will still face extensive FDA regulatory requirements and may face future regulatory difficulties. |
| • | Our relationships with physicians, patients and payors in the U.S. are subject to applicable anti-kickback, fraud and abuse laws and regulations. In addition, we are subject to patient privacy regulation by both the federal government and the states in which we conduct our business. Our failure to comply with these laws could expose us to criminal, civil and administrative sanctions, reputational harm, and could harm our results of operations and financial conditions. |
| • | If the market opportunities for our products and product candidates, including EYP-1901, are smaller than we believe they are, our results of operations may be adversely affected and our business may suffer. |
| • | If any of our products have newly discovered or developed safety problems, our business would be seriously harmed. |
| • | The Affordable Care Act and any changes in healthcare laws may increase the difficulty and cost for us to commercialize DEXYCU and YUTIQ in the U.S. and affect the prices we may obtain. |
| • | Patient assistance programs for pharmaceutical products have come under increasing scrutiny by governments, legislative bodies and enforcement agencies. These activities may result in actions that have the effect of reducing prices or demand for our products, harming our business or reputation, or subjecting us to fines or penalties. |
| • | If competitive products are more effective, have fewer side effects, are more effectively marketed and/or cost less than our products or product candidates, or receive regulatory approval or reach the market earlier, our product candidates may not be approved, and our products or product candidates may not achieve the sales we anticipate and could be rendered noncompetitive or obsolete. |
| • | If the FDA or other applicable regulatory authorities approve generic products that compete with any of our products or product candidates, it could reduce our sales of those products or product candidates. |
Risks Related To Our Intellectual Property | • | If we are unable to protect our intellectual property rights or if our intellectual property rights are inadequate to protect our product candidates, our competitors could develop and commercialize technology and products similar to ours, and our competitive position could be harmed. |
| • | We may become involved in lawsuits to protect or enforce our patents or the patents of our licensors, which could be expensive, time consuming and unsuccessful. |
| • | We may not be able to protect our intellectual property rights throughout the world. |
| • | Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements. |
| • | Third parties may initiate legal proceedings alleging that we are infringing their intellectual property rights, the outcome of which could be uncertain and could harm our business. |
| • | Our competitors may be able to circumvent our patents by developing similar or alternative technologies or products in a non-infringing manner. |
Risks Related To Our Reliance On Third Parties | • | The development and commercialization of our lead product candidate, EYP-1901, is dependent on intellectual property we license from Equinox Science. If we breach our agreement with Equinox or the agreement is terminated, we could lose license rights that are important to our business. |
| • | If we are unable to maintain our agreement with ImprimisRx to co-promote DEXYCU, we may be unable to generate significant revenue from this product. |
| • | If we encounter issues with our CMOs or suppliers, we may need to qualify alternative manufacturers or suppliers, which could impair our ability to sufficiently and timely manufacture and supply DEXYCU. |
| • | We use our own facility for the manufacturing of YUTIQ, which requires significant resources, and which could adversely affect its commercial viability. |
| • | Our YUTIQ manufacturing operations depend on our Watertown, MA facility. If this facility is destroyed or is out of operation for a substantial period of time, our business may be adversely impacted. |
| • | If third-party manufacturers, wholesalers and distributors fail to devote sufficient time and resources to DEXYCU or their performance is substandard, our product supply may be impacted. |
Risks Related To Ownership Of Our Common Stock | • | The trading price of the shares of our common stock has been highly volatile, and purchasers of our common stock could incur substantial losses. |
| • | EW Healthcare and Ocumension own a substantial amount of our common stock and can exert significant control over matters subject to stockholder approval, which would prevent new investors from influencing significant corporate decisions. |
| • | Certain covenants related to our share purchase agreement with Ocumension may restrict our ability to obtain future financing and cause additional dilution for our stockholders. |
Introduction
Our BusinessOverview
We are a specialty biopharmaceuticalpharmaceutical company committed to developing and commercializing innovative ophthalmictherapeutics to help improve the lives of patients with serious eye disorders. Our pipeline leverages our proprietary Durasert® technology for extended intraocular drug delivery including EYP-1901, a potential twice-yearly sustained delivery intravitreal anti-VEGF treatment initially targeting wet age-related macular degeneration (“wet AMD”), the leading cause of vision loss among people 50 years of age and older in the United States. Our product candidate pipeline also includes YUTIQ50, a potential twice-yearly treatment for non-infectious uveitis affecting the posterior segment of the eye, one of the leading causes of blindness. We also have two commercial products: YUTIQ®, a once every three-year treatment for chronic non-infectious uveitis affecting the posterior segment of the eye, and DEXYCU®, a single dose treatment for postoperative inflammation following ocular surgery. Local drug delivery for treating ocular diseases is a significant challenge due to the effectiveness of the blood-eye barrier. This barrier makes it difficult for systemically-administered drugs to reach the eye in sufficient quantities to have a beneficial effect without causing unacceptable adverse side effects to other organs. Our validated Durasert technology, which has already been included in four products approved for marketing by the U.S. Food and Drug Administration (“FDA”), is designed to provide consistent, sustained delivery of small molecule drugs over a period of months to years through a single intravitreal injection. Our lead product candidate, EYP-1901, combines a bioerodible formulation of our proprietary Durasert sustained-release technology with vorolanib, a tyrosine kinase inhibitor (“TKI”). We are currently evaluating EYP-1901 in a Phase 1 clinical trial as a potential twice-yearly sustained delivery intravitreal treatment for wet AMD. Current approved treatments for wet AMD require monthly or bi-monthly eye injections in a physician’s office, which can cause inconvenience and discomfort and often lead to reduced compliance and poor outcomes. In two prior clinical trials of vorolanib as an orally delivered therapyconducted by a third party, vorolanib had a strong clinical signal with no significant ocular adverse events. We expect initial data from the Phase 1 clinical trial in the second half of 2021. YUTIQ® (fluocinolone acetonide intravitreal implant) 0.18 mg for intravitreal injection, is a non-erodible intravitreal implant containing fluocinolone acetonide (“FA”) lasting for up to 36 months and is indicated for the treatment of eye diseases. Our lead product, DEXYCU™chronic non-infectious uveitis affecting the posterior segment of the eye. This disease affects between 60,000 to 100,000 people each year in the U.S., causes approximately 30,000 new cases of blindness every year and is the third leading cause of blindness. YUTIQ utilizes our proprietary Durasert® sustained-release drug delivery technology platform. DEXYCU® (dexamethasone intraocular suspension) 9%, approved by the United States, or U.S., Food and Drug Administration, or FDA, in February 2018,for intraocular administration, is administered as a single dose at the end of ocular surgery and is the first long-acting intraocular product approved by the FDAindicated for the treatment of post-operative ocular inflammation.inflammation, with our primary focus on its use immediately following cataract surgery as a single dose treatment. DEXYCU utilizes our proprietary Verisome® drug-delivery platform, which allowstechnology. We are also developing YUTIQ50 as a potential six-month intravitreal treatment for chronic non-infectious uveitis affecting the posterior segment of the eye. We have consulted with the FDA and identified a clinical pathway for a single intraocular injectionsupplemental new drug application (“sNDA”) filing that releases dexamethasone,we expect will involve a corticosteroid, over time. Thereclinical trial of a small population. We are approximately four million cataract surgeries performed annuallycurrently evaluating the timeline and investment requirements for the initiation of this trial. We are also seeking to enhance our long-term growth potential by expanding EYP-1901 beyond wet AMD into diabetic retinopathy (“DR”) and retinal vein occlusion (“RVO”), both large and growing ocular disease areas. We also plan to potentially identify and advance additional product candidates through clinical and regulatory development. This may be accomplished through internal discovery efforts, potential research collaborations and/or in-licensing arrangements with partner molecules and potential acquisitions of additional ophthalmic products, product candidates or technologies that complement our current product portfolio. The novel coronavirus (COVID-19) pandemic (the “Pandemic”) has had, and will likely continue to have, a material and adverse impact on our business, including as a result of preventive and precautionary measures that we, other businesses, and governments have and will likely continue to take. This includes a significant impact on cash flows from expected revenues due to the closure of ambulatory surgery centers for DEXYCU and a significant reduction in physician office visits impacting YUTIQ. These closures precipitated the restructuring of our commercial organization that was announced on April 1, 2020 along with a reduction in planned spending for calendar year 2020 and into calendar year 2021. Due to the continued Pandemic, these factors continued to have an adverse impact on our revenues, financial condition and cash flows in the U.S.,fourth quarter of 2020 and we planinto the first quarter of 2021. We have experienced and may continue to launch DEXYCUexperience significant and unpredictable reductions in the U.S.demand for our products as customers have shut down their facilities and non-essential surgical procedures have been postponed in the first half of calendar year 2019 with a primary focus on its use following cataract surgery. Our lead product candidate is YUTIQ™, a three-yearnon-erodible fluocinolone acetonide, or FA, insert foran effort to promote social
distancing and to redirect medical resources and priorities towards the treatment ofnon-infectious posterior segment uveitis, or NIPU. Injected into COVID-19. We are monitoring the eye in an office visit, YUTIQ is a tiny micro-insert that delivers a micro-dose of a corticosteroid to the back of the eye on a sustained constant (zero order release) basis for approximately three years. On March 19, 2018, the FDA accepted our New Drug Application, or NDA, for YUTIQPandemic and set an FDA Prescription Drug User Fee Act, or PDUFA, action date of November 5, 2018. YUTIQ is basedits potential effect on our proprietary Durasert™ sustained-release drug delivery technology platform, which can deliver drugs for predetermined periodsfinancial position, results of time ranging from monthsoperations and cash flows.
Our Pipeline and Commercial Products The following table describes the stage of each of our programs: 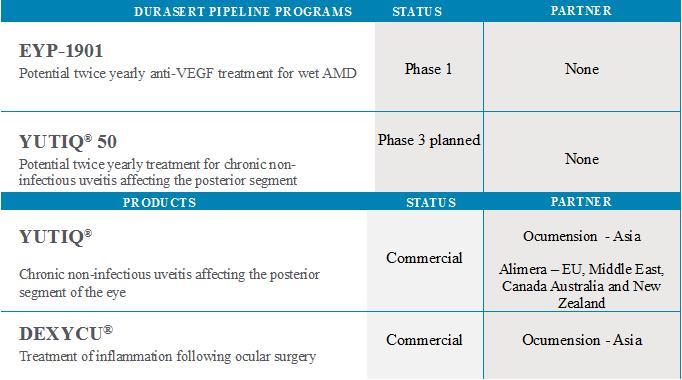
Strategy Our strategy is to years. Posterior segment uveitis isbecome a leading pharmaceutical company commercializing innovative therapeutics to help improve the third leading causelives of blindness in the U.S. and affects between 55,000 to 120,000 people in the U.S. If approved in November 2018, we expect to launch YUTIQ in the U.S. in the first halfpatients with serious eye disorders. The key elements of calendar year 2019.our strategy include: | • | Advance EYP-1901 through clinical development for wet AMD. |
| • | Advance EYP-1901 through clinical development in additional indications, including DR and RVO after completion of a positive Phase 1 clinical trial in wet AMD. |
| • | Advance YUTIQ50 into clinical development for a potential sNDA filing as a twice-yearly sustained delivery treatment for chronic non-infectious uveitis affecting the posterior segment of the eye. |
| • | Identify and in-license, partner or acquire additional transformative ophthalmology products to build long-term stockholder value targeting programs that can utilize our Durasert technologies. |
| • | Grow commercial product revenues for both YUTIQ and DEXYCU in the U.S. |
| • | Leverage our Durasert and Verisome technologies through research collaborations and out-licenses with other pharmaceutical and biopharmaceutical companies, institutions and other organizations. We believe these technologies can provide sustained, targeted delivery of therapeutic agents, resulting in improved therapeutic effectiveness, safer administration and better patient compliance and convenience, with reduced product development risk and cost. |
The Unmet Need in the Treatment of Eye Disease The human eye is an organ which reacts to light to provide sight. The eye has two principal anatomical segments: the anterior segment and the posterior segment. The anterior segment consists of the cornea, iris, pupil, lens and aqueous humor, while the posterior segment consists of the retina, choroid, vitreous humor and the optic nerve.
The tissues and structures in the anterior and posterior segment of the eye work in concert to produce sight. Light from an object or scene enters the eye through the anterior chamber, beginning with the cornea. The cornea bends the light such that it passes freely through the pupil, which is the opening in the center of the iris. The iris works like a shutter in a camera, enlarging or shrinking dependingWe are primarily focused on how much light is entering the eye. After passing through the iris, the light rays pass through the eye’s natural crystalline lens. This clear, flexible structure works like the lens in a camera, shortening and lengthening its width in order to focus light rays properly. Light rays then pass from the anterior segment intodiseases affecting the posterior segment of the eye starting with a dense, transparentgel-like substance, called the vitreous. The vitreous fills the globe of the eyeball, which bathes the eye in nutrients and helps the eye hold its spherical shape. In a normal eye, the light rays come to a sharp focusing point on the retina. The retina functions much like the film in a camera, capturing the light rays, processing them into light impulses through millions of tiny nerve endings and then sending these light impulses through over a million nerve fibers to the optic nerve. Because the process of producing sight requires the precise coordination of the tissues and structures in both the anterior and posterior segments of the eye, if disease affects any one of these components, vision can be impaired or potentially blinded.
Diseases of the anterior chamber of the eye include ocular inflammation, cataracts, dry eye, infection, and refractive disorders. Glaucoma, which is a disease that damages the optic nerve, can also be caused by inflammation in the anterior chamber (inflammatory or uveitic glaucoma). Because the anterior segment is readily accessible, physicians typically treat these diseases with topically-applied eye drops. However, there are several limitations of eye drops. First, the eye often eliminates topically applied medications via tear elimination, limiting the penetration of drugs into the ocular tissue. Second, eye drops are often administered by patients themselves, which often leads to misuse ornon-compliance by patients due to complicated and arduous eye drop regimens.
eye. Diseases of the posterior segment of the eye include conditions such asage-related macular degeneration, or wet AMD, diabetic retinopathy, diabetic macular edema, or DME,DR, RVO and chronic non-infectious uveitis affecting the posterior segment uveitis.of the eye. These diseases frequently result in damage to the vasculature of the eye, leading to poor visual function, and often to proliferation of new, abnormal and leaky blood vessels in the back of the eye. These conditions can lead to retinal damage, scarring and irreversible loss of vision. Because the posterior chambersegment is not readily accessible, physicians typically treat these diseases with intravitreal injections. However, there are several limitations of frequent intravitreal injections. First, these injections can be painful to the eye and often cause swelling or bleeding. Second, repeated intravitreal injections can cause scarring of the eye sclera. The sclera, also known as the white of the eye, is the opaque, fibrous, protective, outer layer of the human eye containing mainly collagen and some elastic fiber. Many patients
with retinal diseases require lifelong treatment and over time, these chronic intravitreal injections can cause significant sclera scarring, increased risk of intraocular infection and vitreous hemorrhage. Further, most ocular drugs are delivered via a bolus injection that requires monthly or bi-monthly re-injections. Each time the patient or the physician lengthens the treatment interval due to either missed appointments, cost to the patient, or lack of reimbursement, the patient’s disease can reactivate, which leads to incremental and cumulative damage to the retina. Over time this may lead to permanent loss of vision. Thus, monthly or bi-monthly injections are not an effective means of delivering a steady state dose to the site of disease. Drug delivery for treating ophthalmic diseases in both Finally, the anterior and posterior segments of the eye is a significant challenge. Due to the effectiveness of theblood-eye barrier, it is difficult for systemically (orally or
intravenously) administered drugs to reach the retina in sufficient quantities to have a beneficial effect without causing adverse side effects to other parts of the body. Injecting drugs in solution directly into the back of the eye can achieve effective, but often transient, dosage levels in the eye, requiring repeated injections. In addition to the issues of inconvenience, cost and noncompliance, repeated intravitreal injections have medical risks, including intraocular infection, perforated sclera and vitreous hemorrhage.
Ophthalmic drugs, whether drops, injections or oral dosage forms, are often not administered on the optimal schedule or at all, because patients do not self-administer as prescribed or do not get medical professional administration as required. The risk of patientnon-compliance increases when treatment involves multiple products or complex or painful dosing regimens, as patients age or suffer cognitive impairment or serious illness, or when the treatment is lengthy or expensive.
Drug delivery for treating ophthalmic diseases in posterior segments of the eye is a significant challenge. Due to the effectiveness of the blood-eye barrier, it is difficult for systemically (orally or intravenously) administered drugs to reach the retina in sufficient quantities to have a beneficial effect without causing adverse side effects to other parts of the body. Due to the drawbacks of traditionalfrequent intravitreal injections, eye drops and oral or systemic injectable delivery, we believe the development of methods to deliver drugs to patients in a more precise, micro dose zero order release, controlled fashion over sustainedlonger periods of time satisfies anwith Durasert can satisfy a large patient and physician unmet medical need by assuring complianceneed. In addition, with less frequent injections, or daily eye drops, we believe patients will be able to thecomply better with their prescribed treatment regimen. Our products, DEXYCU and, if approved, YUTIQ are intended to address diseases of both the anterior and posterior segments of the eye, respectively, through long-acting and sustained delivery technologies. Our Products and Product Candidates
The following table describes the stage of each of our programs:
| | | | | | | Product
| | Disease
| | Approved Products
| | Partner
| DEXYCU
| | Occular post-surgical inflammation | | FDA-approved | | None | ILUVIEN
| | DME | | Approved in the U.S. and 17 EU countries; direct commercialization in the U.S., U.K., Germany, Portugal, Ireland and Austria; distribution through sublicense partners in Spain, Italy, France and various countries in the Middle East | | Alimera | RETISERT
| | Posterior segment uveitis | | FDA-approved; commercialized in the U.S. since 2005 | | Bausch & Lomb | VITRASERT
| | CMV retinitis | | FDA-approved; commercialized from 1996 through 2012 (patent expiration) | | Bausch & Lomb | | | | | Product Candidate
| | Disease
| | Stage of Development
| | Partner
| YUTIQ
| | Posterior segment uveitis | | NDA accepted with PDUFA action date of November 5, 2018
Type II variation accepted for review in the 17 EU countries previously approved for ILUVIEN for DME
| | For U.S.: to commercialize directly pending NDA approval
For EMEA: regulatory, reimbursement and distribution licensed to Alimera under ILUVIEN
| YUTIQshorter-acting uveitis
| | Posterior segment uveitis | | Bioequivalence and animal safety studies | | None | Durasert TKI for Wet AMD
| | Wet AMD | | Pre-clinical | | None |
DEXYCU
DEXYCU is the first long-acting intraocular product approved by the FDA for the treatment of post-operative ocular inflammation. Cataract surgery is one of the most frequent surgical procedures performed in the U.S., with approximately four million procedures performed annually. However, patients can experience post-operative ocular inflammation. Under the current standard of care for inflammation associated with cataract surgery, patients, many of whom are elderly, must self-administer medicated eye drops several times a day over a period of several weeks. DEXYCU, administered as a single intraocular injection at the conclusion of surgery, utilizes our Verisome technology to dispense a biodegradable extended-release formulation of dexamethasone, a corticosteroid, in the posterior chamber directly behind the iris. We believe that a single administration of a corticosteroid at the site of inflammation may benefit patients by eliminatingnon-compliance and dosing errors associated with the current practice of dispensing multiple daily self-administered eye drops following cataract surgery over a period of several weeks.
The efficacy of DEXYCU was demonstrated in a double-masked randomized Phase 3 clinical trial of 394 patients. In the clinical trial, patients received an intraocular dose of 517 micrograms, or mcg, of DEXYCU, 342 mcg of DEXYCU, or placebo administered by a physician at the end of cataract surgery. The primary efficacy outcome in the clinical trial was anterior chamber cell clearing in the study eye on the eighth day following surgery. The percentage of patients meeting the primary efficacy outcome was 20% in the placebo group while 57% and 60% met the primary efficacy outcome in the 342 and 517 mcg DEXYCU treatment groups, respectively. In addition, the percentage of patients receiving rescue medication of ocular steroid or a nonsteroidal anti-inflammatory drug was significantly lower at day one, three, eight, 15 and 30 in the 342 and 517 mcg treatment groups versus placebo. The most common adverse reactions (5 – 15%) reported with DEXYCU were increased intraocular pressure, or IOP, corneal edema and iritis. Other adverse reactions occurring in 1 – 5% of subjects included corneal endothelial cell loss, blepharitis, eye pain, cystoid macular edema, dry eye, ocular inflammation, posterior capsule opacification, blurred vision, reduced visual acuity, vitreous floaters, foreign body sensation, photophobia and vitreous detachment. Warnings and precautions included on the label for DEXYCU include increases in IOP, delayed healing, exacerbation of infection and cataract progression which are side effects generally associated with intraocular steroids.
TheFDA-approved dosage of DEXYCU is 0.005 milliliters, or mL, of dexamethasone 9% (equivalent to 517 mcg), administered as a single dose intraocularly in the posterior chamber, directly behind the iris, at the end of surgery. DEXYCU will be available as a 9% intraocular suspension equivalent to dexamethasone 103.4 mg/mL in a single-dose vial provided in a kit. The drug is encapsulated in the fully bioerodible Verisome technology, which provides a steady release of dexamethasone for up to 22 days post-injection.
We acquired the rights to DEXYCU on March 28, 2018 through the acquisition of Icon Bioscience, Inc., or Icon. We paid Icon’s security holders approximately $15 million at the closing of the transaction, and are obligated to pay certain post-closing contingent cash payments upon the achievement of specified milestones based upon certain net sales and partnering revenue standards, in each case subject to the terms and conditions set forth in that certain Agreement and Plan of Merger, dated March 28, 2018, by and among us, Oculus Merger Sub, Inc., or Merger Sub, Icon and Shareholder Representative Services LLC, solely in its capacityregimen as the representativeburden of Icon’s securityholders, which we referhaving to as the Merger Agreement. These include, but are not limited to, (i) aone-time development milestone of $15.0 million payable in cash upon the first commercial sale of DEXYCU in the U.S., (ii) sales milestone payments totaling up to $95.0 million upon the achievement of certain sales thresholds and subject to certain Centers for Medicare & Medicaid Services, or CMS, reimbursement conditions set forth in the Merger Agreement, (iii) quarterlyearn-out payments equal to 12% on net sales of DEXYCU in a given year, whichearn-out payments will increase to 16% of net sales of DEXYCU in such year beginning in the calendar quarter for such year to the extent aggregate annual consideration of DEXYCU exceeds $200.0 million in such year, (iv) quarterlyearn-out payments equal to 20% of partnering revenue received by us for DEXYCU sales outside of the U.S., and (v) single-digit percentage quarterlyearn-out payments with respect to net sales and/or partnering income, if any, resulting from future clinical development,
regulatory approval and commercialization of any other product candidates we acquired in connection with the acquisition of Icon.
We plan to launch DEXYCU in the U.S. in the first half of calendar year 2019 following the successful scale up of commercial infrastructure and commercial supplies. We plan to commercialize DEXYCU ourselves in the U.S. through a contract sales organization, or CSO, whereby the sales leadership (National Sales Director and Regional Managers) are hired by us, and the key account managers, or KAMs, and sales representatives are hired by the CSO with the option for us to hire these sales reps after a period of time. We believe this flexible sales model provides less execution risk to us as CSOs can leverage costs across multiple clients, and thus are able to cost-effectively build the necessary infrastructure to support sales activities using varied, industry-wide experience to provide the most impactful solutions.
We believe that approximately four million cataract surgeries are performed annually in the U.S. The current standard of care in the U.S. for treating post-operative inflammation is primarily a combination of steroid, antibiotic andnon-steroidal eye drops on a tapered treatment regimen that can last up to four weeks. This eye drop treatment regimen is complicated and can result in up to 100 eye drops being administered over time. Steroid eye drops are the most complicated medication to administer in this regimen, requiring up to 70 eye drops over3-4 weeks on a tapered dosing schedule. Further, cataract surgery patients are often elderly and can have compromised cognitive function, osteoarthritis in their hands and poor eyesight due to the cataract surgery. These complexities can lead to poor compliance due to failing to administer eye drops according to the prescribed schedule, or administering an eye drop but failing to have itfrequently go into the eye, and/or not finishing the treatment regimen. In addition, patients often call their physician’s office multiple times to have themre-explain the treatment regimen. We believe DEXYCU addresses many of these issues and potentially eliminates the need for post-surgical steroid eye drops by providing one injection immediately post-surgery into the same incision site where the new intraocular lens has been placed. We believe physicians will react positively to this single injection because the full steroid dose will be placed at the surgical site where inflammation can occur post-surgery.
Approximately 40% of patients who undergo cataract surgery are covered by Medicare Part B. New drugs approved by the FDA that are part of cataract surgery performed in a hospital outpatient department or ambulatory surgical center may receive an additional transitional pass-through payment under Medicare provided it meets certain criteria, including a “not insignificant” cost criterion. This “pass-through payment” consists of Medicare reimbursement for the drug based on a defined formula for calculating the minimum fee that a manufacturer may charge for the drug.
DEXYCU qualified for Medicare transitional pass-through payment and we received a pass-throughC-code from CMS, which will be effective on October 1, 2018. We have not yet determined final pricing, other than that it will be modestly higher than $485 to ensure we will continue to qualify for pass-through status after including normal industry discounts and rebates given to providers or commercial payors.
Under current CMS regulations, pass-through status applies for a period of three years, measured from the date Medicare makes its first pass-through payment for DEXYCU, following which DEXYCU would be incorporated into the cataract bundled payment system, which will significantly reduce the pricing for DEXYCU. We are working with outside consultants to potentially gain an extension to the transitional payment system, or to separate the drug payment from the bundled cataract surgery payment after the three-year transitional payment ends and continue to be reimbursed separately for a longer period of time, potentially through patent life.
Our DEXYCU U.S. patent portfolio includes two issued patents under an exclusive license from Ramscor, Inc. for all ophthalmic conditions. These two issued patents contain composition claims for delivering biologically active substances using citric acid esters. In addition, two more U.S. applications pertaining to DEXYCU have issued as patents. These patents, one with method of use claims and the other with device claims, will provide further protection for DEXYCU through May 2034.
The drug delivery technology used in DEXYCU is called Verisome. The basic technology can be formulated into numerous products, as a biodegradable solid, gel, or liquid substance that provides drug release in a controlled mannerinjections, usually over a period of weeks to several monthslifetime after diagnosis, presents issues for ocular, systemic, or topical applications. Ophthalmic applications are focused on the ability of this system to create an injectable liquid or slightly viscous gel. Verisome-based products can be injected into the aqueous or vitreous humor as a liquid via a small gauge needle. When the drug is injected into an ocular chamber, it coalesces into a single spherical dose that settles in the lower portion of the chamber. The system is biodegradable and versatile for administering different drugs; furthermore, duration of use can be tailored. Shrinkage of the Verisome sphere over time reflects simultaneous degradation of the delivery system and release of the active agent. In ophthalmology, this mode of delivery offers advantages because the physician can easily assess the status of therapy by observing the drug-containing system within the eye. When the sphere is no longer visible, the entire drug has been released, and no inactive ingredient remains in the eye. Potential applications could include intraocular products to treat inflammation, ocular hypertension and glaucoma.patients.
Durasert Technology Platform Our Durasert technology platform uses proprietary sustained polymerrelease technology to deliver drugs over periods of up to treat chronic diseases, especially those affecting the hard to access posterior segment of the eye.three years through a single intravitreal injection. To date, threefour products utilizing successive generations of the Durasert technology have been approved by the FDA. TheseIn addition to our own YUTIQ, these products include ILUVIEN® (FA intravitreal implant) 0.19 mg, licensed to Alimera Sciences Inc. (“Alimera”), or Alimera, and Retisert® (FA intravitreal implant) 0.59 mg and Vitrasert® (ganciclovir) 4.5 mg, which are both licensed to Bausch & Lomb. Currently,Although the earlier ophthalmic products that utilize the Durasert technology, Retisert and Vitrasert, are surgically implanted, ILUVIEN and YUTIQ were designed to be injected during a physician office visit. The Durasert technology platform utilizescreates a miniaturized, injectable, sustained-releasesustained release insert forof a small moleculesmolecule compound that can deliver a drug for periods of up to three years. This insertThe current FDA-approved products utilize the non-erodible formulation of Durasert. For these products, the drug core matrix is only 3.5 mm in lengthcoated with an external diameterone or more polymer layers, and the permeability of just 0.37 mm.those layers and other design aspects control the rate and duration of drug release. By changing elements of the design, we can alter both the rate and duration of release to meet different therapeutic needs. EYP-1901 utilizes a bioerodible formulation of the Durasert technology. The insert can be administered in an office setting through a needle as small as25-gauge.bioerodible formulation eliminates the non-erodible polymer coating allowing the body to absorb the drug core matrix. Our Durasert technology platform is designed to address the issue ofprovide sustained delivery for ophthalmic diseases and other product candidates. Specifically, our Durasert platformconditions with the following features: | • | | • | Extended Delivery. The delivery of drugs for predetermined periods of time ranging from months to years. We believe that uninterrupted, sustained delivery offers the opportunity to develop products that reduce the need for repeated applications, thereby reducing the risks of patient noncompliance and adverse effects from repeated administrations. |
| • | | • | Controlled Release Rate. The release of therapeutics at a zero order kinetics controlled rate. We believe that this feature allows us to develop products that deliver optimal concentrations of therapeutics over time and eliminate excessive variability in dosing during treatment. |
| • | | • | Localized Delivery. The delivery of therapeutics directly to a target site. We believe this administration can allow the natural barriers of the body to isolate and assist in maintaining appropriate concentrations at the target site in an effort to achieve the maximum therapeutic effect while minimizing unwanted systemic effects. |
Our Durasert technology platform providesProduct Candidates EYP-1901 for wet AMD EYP-1901 is a potential twice-yearly sustained localized delivery intravitreal anti-VEGF treatment initially targeting wet AMD. Wet AMD is when new, abnormal blood vessels grow under the retina. These vessels may leak blood or other fluids, causing scarring of small molecule drugsthe macula. This form of AMD is less common but much more serious. AMD is one of the major causes of vision loss (accounts for 8.7%) of the total vision impairment globally. Age is the greatest risk factor for developing AMD and individuals aged 50+ are more prone to the posterior segmentdisease. Among all AMD patients in the United States, wet AMD accounts for only 10% of cases, yet it alone accounts for 90% of legal blindness.
EYP-1901 Market Opportunity There are several effective and safe treatments for wet AMD available on the eyemarket, including anti-VEGF intravitreal injectable drugs marketed under the brands names Lucentis, Eylea, Beovu and is utilized in threeFDA-approved products and in our product candidates, including YUTIQ and other shorter duration product candidates. InAvastin (off label use). However, these products and product candidates, a drug core is surrounded with one or more polymer layers, and the permeability of those layers and other design aspects of the product or product candidate control the rate and duration of drug release. By changing elements of the design, we can alter both the rate and duration of release to meet different therapeutic needs. Although the earlier ophthalmic products that utilize the Durasert technology, Retisert and Vitrasert, are surgically implanted, ILUVIEN, YUTIQ and our other ophthalmic product candidates are designed totreatments must be injected at the target site in ana physician’s office visit. either monthly, bi-monthly or every three months, which can cause inconvenience and discomfort and often lead to reduced compliance and poor outcomes.YUTIQ
YUTIQ, our lead product candidate, is based on our Durasert technology platform and consists of an injectable, sustained-release micro-insert designed to treat chronic NIPU, intermediate uveitis and panuveitis affecting the posterior segment of the eye. YUTIQ is designed to provide sustained daily release of a total of 0.18 mg of theoff-patent corticosteroid FA at a controlled rate directly to the back of the eye over approximately three years from a single administration performed in an office visit. It is injected with our proprietary inserterSeparate published studies using a25-gauge needle. We are developing YUTIQ independently and have licensed regulatory, reimbursement and distribution rights to Alimera for Europe, the Middle East and Africa, or the EMEA, under its ILUVIEN tradename. Subject to NDA approval by the FDA, we plan to independently commercialize YUTIQreal world data-one study in the U.S. Further,and another that includes Canada, France, Germany, Ireland, Italy, the Netherlands, UK and Venezuela-indicate that despite initial efficacy, approved wet AMD treatments still result in conjunctionvision loss over time.
We believe that EYP-1901, with its possibility for twice yearly injections, has the commercializationpotential to offer wet AMD sufferers a convenient and effective treatment option, if approved. Pre-Clinical and Clinical Development Vorolanib, the active drug candidate in EYP-1901, is a small molecule TKI that blocks all 3 isoforms of DEXYCU,VEGFR, the main driver of the proliferation of blood vessels that are the hallmark of wet AMD. Vorolanib has been previously studied in Phase 1 and 2 clinical trials by Tyrogenix, Inc. (“Tyrogenix”) as an orally delivered therapy for the treatment of wet AMD and data from these trials demonstrated a positive clinical signal. Although the Phase 2 clinical trial was discontinued due to systemic toxicity, no significant ocular adverse events were observed in either clinical trial. The Phase 1 clinical trial of orally delivered vorolanib demonstrated: | • | Best-corrected visual acuity (“BCVA”) was maintained to within 4 letters of baseline at the 24-week endpoint, or improved in all but 1 participant |
| • | 60% (15/25) of patients required no rescue injection while on oral vorolanib therapy |
| • | Mean ocular coherence tomography (“OCT”) thickness in completers was reduced by -50 +/- 97 µm; and |
| • | Mean OCT thickness in treatment-naïve patients was reduced by ~80 µm |
The Phase 2 clinical trial of orally delivered vorolanib demonstrated less anti-VEGF rescue versus placebo for all doses with no ocular toxicity despite a strict pre-defined recue criteria. By delivering vorolanib locally, in the vitreous humor, as EYP-1901 using a bioerodible Durasert formulation at significantly lower doses, we expect to spreadavoid the systemic toxicities seen in the prior clinical trials of vorolanib and typically associated with orally delivered TKIs. This concept has been supported by initial pharmacokinetic (“PK”), safety and GLP toxicology studies including a non-GLP PK and safety study that demonstrated drug levels in the vitreous and retina/choroid above the IC50 for VEGFR. These studies have been conducted in support of the EYP-1901 investigational new drug (“IND”) application filed with the FDA. The IND application for EYP-1901 was filed with the FDA in December 2020 in support of initiation of a Phase 1 clinical trial in wet AMD patients. We enrolled the first patient in the Phase 1 clinical trial in January 2021. The Phase 1 clinical trial is a dose escalation trial of three ascending doses with a total planned enrollment of 13 wet AMD patients. The primary endpoint of the trial is safety, and key secondary endpoints are BCVA and central subfield thickness. Top level data from this clinical trial is anticipated in the second half of 2021, assuming that patient enrollment is not impacted by the Pandemic. Intellectual Property In February 2020, we entered into an Exclusive License Agreement with Equinox Science, LLC (“Equinox”), pursuant to which Equinox granted us an exclusive, sublicensable, royalty-bearing right and license to certain patents and other Equinox intellectual property to research, develop, make, have made, use, sell, offer for sale and import the compound vorolanib and any pharmaceutical products comprising the compound for the prevention or treatment of age-related macular degeneration, diabetic retinopathy and retinal vein occlusion using our commercial, medical, legal, corporateproprietary localized delivery technologies, in each case, throughout the world except China, Hong Kong, Taiwan and Macau (the “Territory”). In consideration for the rights granted by Equinox, we (i) made a one time, non-refundable, non-creditable upfront cash payment of $1.0 million to Equinox in February 2020, and (ii) agreed to pay milestone payments totaling up to $50 million upon the achievement of certain development and regulatory infrastructure over two products.milestones, consisting of (a) completion of a Phase 2 clinical trial for the compound or a licensed product, (b) the filing of a new drug application or foreign equivalent for the compound or a licensed product in the United States, European Union or United Kingdom and (c) regulatory approval of the compound or a licensed product in the United States, European Union or United Kingdom. Posterior segment uveitisWe also agreed to pay Equinox tiered royalties based upon annual net sales of licensed products in the Territory. The royalties are payable with respect to a licensed product in a particular country in the Territory on a country-by-country and licensed product-by-licensed product basis until the later of (i) twelve years after the first commercial sale of such licensed product in such country and (ii)
the first day of the month following the month in which a generic product corresponding to such licensed product is launched in such country (collectively, the “Royalty Term”). The royalty rates range from the high-single digits to low-double digits depending on the level of annual net sales. The royalty rates are subject to reduction during certain periods when there is no valid patent claim that covers a licensed product in a particular country. YUTIQ50 YUTIQ50 is a potential twice-yearly sustained delivery treatment for chronicnon-infectious inflammatory disease uveitis affecting the posterior segment of the eye, often involving the retina, and is a leading cause of blindness in developed countries. It afflicts people of all ages, producing swelling and destroying eye tissues, which can lead to severe vision loss and blindness. In the U.S., posterior segment uveitis is estimated to affect approximately55,000-120,000 people in the U.S., resulting in approximately 30,000 cases of blindness and making it the third leading cause of blindness in the U.S. Patients with posterior segment uveitis are typically treated with ocular injected steroids and systemic steroids, but frequently develop serious side effects from systemic steroids over time that can limit effective dosing. Patients who do not tolerate systemic steroids then are offered—as the last line of treatment—therapy with systemic immunosuppressants or biologics, which themselves can cause severe side effects. YUTIQ Phase 3 Trials
In our two Phase 3 trials to assess the safety and efficacy of YUTIQ, we achieved the primary efficacy endpoint of prevention of recurrence of uveitis through six months with statistical significance (p value of < 0.001 in each study). These studies are randomized, sham injection-controlled, double-masked trials with the primary endpoint of both trials defined as recurrence of uveitis at six months, with patients followed for three years. Our first Phase 3 trial enrolled 129 patients in 16 centers in the U.S. and 17 centers outside the U.S, with 87 eyes treated with YUTIQ and 42 eyes receiving sham injections. Our second Phase 3 trial enrolled 153 patients in 15 centers in India with 101 eyes treated with YUTIQ and 52 eyes receiving sham injections. The36-month patientfollow-up was recently completed in the first of the two Phase 3 trials.
Our first Phase 3 trial met its primary efficacy endpoint of prevention of recurrence of disease at 6 months with statistical significance (p < 0.001, intent to treat analysis; recurrence of 18.4% for YUTIQ versus 78.6% for control). The trial yielded similar efficacy through 12 months of follow up (p < 0.0001, intent to treat analysis; recurrence of 27.6% for YUTIQ versus 85.7% for control). YUTIQ was generally well tolerated through12-months offollow-up. The incremental risk of elevated IOP for YUTIQ-treated eyes compared to control eyes was lower through 12 months than through six months for elevation over 21 mmHg (6.1% versus 10.9%) as well as for the more serious elevation over 25 mmHg (7.6% versus 11.3%). Elevated IOP was generally well treated with eye drops. Through 12 months, the percentage of eyes requiring filtration surgery was low and similar between YUTIQ-treated and control eyes (3.4% versus 2.4%). Of the 63 study eyes with a natural lens at baseline, 33.3% of YUTIQ-treated eyes compared to 4.8% of control eyes required cataract surgery through 12 months. Cataracts are both a side effect of treatment with steroids and a natural consequence of uveitis.
Our second Phase 3 trial also met its primary efficacy endpoint of prevention of recurrence of disease at 6 months with statistical significance (p < 0.001, intent to treat analysis; recurrence of 21.8% for YUTIQ versus 53.8% for control). As in the first Phase 3 trial, YUTIQ was generally well tolerated through 6 months, and12-monthfollow-up efficacy and safety data was consistent with the12-month data from our first Phase 3 trial. The36-month patientfollow-up is expected to be completed in October 2019.
We also conducted a multi-center, randomized, controlled, single-masked study of the safety and utilization of two different inserters for YUTIQ. We enrolled 26 subjects (38 eyes) in this study in 6 centers in the U.S. The utilization and safety results of this study have been included in our NDA filing for YUTIQ.
YUTIQ Regulatory Strategy
On March 19, 2018, the FDA accepted our NDA for YUTIQ and set a PDUFA action date of November 5, 2018. If approved, we plan to launch YUTIQ in the U.S. in the first half of calendar year 2019.
We haveout-licensed the rights for YUTIQ for the treatment of NIPU to Alimera for the EMEA as an extension of our original license agreement with Alimera. Pursuant to the original agreement, we granted worldwide license rights to ILUVIEN for DME and other potentialback-of-the-eye diseases (other than uveitis) utilizing a corticosteroid with our Durasert technology. In the European Economic Area, or EEA, Alimera has submitted our previously-filed YUTIQ data as a Type II variation in each of the 17 countries in which it previously obtained regulatory approval for ILUVIEN for DME. According to Alimera’s public filings, Alimera plans to submit follow-up data supporting its Type II variation application in the fourth quarter of calendar year 2018 and expects it will obtain approval for its application in the first half of calendar 2019.
YUTIQ Marketing Strategy
Subject to approval by the FDA, we intend to commercialize YUTIQ ourselves in the U.S. We believe that the uveitis market in the U.S. is relatively modest in size, with an estimated patient prevalence for NIPU of approximately 55,000 to 120,000 patients. Consequently, the number of retinal physicians who treat the majority of this patient population is estimated to be fewer than 500. As a result, we believe the commercial footprint and cost to market for YUTIQ will be less than a typical pharmaceutical product launch with a larger physician call population. Members of our leadership team have extensive commercialization experience and we believe that commercializing YUTIQ ourselves in the U.S. will maximize the value of YUTIQ to us. Outside of the U.S., we expanded Alimera’s license agreement to include uveitis, including NIPU, in the EMEA. This additional license right was part of the July 10, 2017 amended and restated collaboration agreement with Alimera, or the Amended Alimera Agreement. Alimera has reported that it plans to commercialize the NIPU EMEA indication under its ILUVIEN trademark. We plan to seekout-license partner arrangements in other territories.
Development Product: Shorter Duration YUTIQ
We are developing a next-generation, shorter-duration treatment for posterior segment uveitis, using the same non-erodible Durasert technologyformulation and drugsteroid (FA) as in YUTIQ. This program is designed to offer an intravitreal micro insert with a shorter delivery period, thus providing physicians with flexibility for multiple dosing intervals. Our market research has indicated a strong preference amongst those physicians surveyed for both a six to nine-month drug delivery product and ain addition to the three-year drug delivery option.option provided by YUTIQ. Although we believe many patients would likely opt for a longer-acting treatment option, some doctors may prefer to initially treat their uveitis patients over shorter time periods. We have consulted with the FDA and identified a potential clinical pathway for an sNDA filing that involves a clinical trial of approximately 60 patients, randomized 2:1. We are currently evaluating the timeline and investment requirements for the initiation of this trial.
Development Product: Tyrosine Kinase Inhibitor InsertEYP-1901 for Wet AMDDR and RVO
We are investigatingalso seeking to enhance our long-term growth potential by expanding beyond wet AMD for EYP-1901 and into DR and RVO, both large and growing ocular disease areas.DR is a frequent complication of diabetes mellitus. Slow but progressive changes in the developmentsmall blood vessels of an injectable, bioerodible, sustained-release Durasert insert deliveringthe retina may cause no symptoms or only mild vision problems in early stages. As the disease progresses, retina bleeding and fluid accumulation can eventually lead to blindness. RVO is a tyrosine kinase inhibitor, or TKI, for treatment of wet AMD. AMD, the leadingcommon cause of vision loss in peopleolder individuals with over 65,90% of cases occurring in patients over the age of 55 years. It is the second most commonly treated with intravitreal injectionscommon retinal vascular disease after DR. As in wet AMD, the hypoxic retinal tissue in RVO releases VEGF and inflammatory mediators, thereby inducing the complication of biologics that block vascular endothelial growth factor, or VEGF.FDA-approved Lucentis®macular edema, a cause of significant visual acuity loss. We intend to advance EYP-1901 through clinical development in DR and Eylea® andoff-label useRVO after completion of anti-cancer drug Avastin® are the leading treatments fora successful Phase 1 clinical trial in wet AMD. These biologics must be injected into Our Commercial Products YUTIQ YUTIQ (fluocinolone acetonide intravitreal implant or “FA” 0.18 mg) for intravitreal injection, was approved by the FDA in October 2018 and we commercially launched YUTIQ in the U.S. in February 2019. YUTIQ is indicated for the treatment of chronic non-infectious uveitis affecting the posterior segment of the eye. YUTIQ is a once every three-year treatment utilizing a nonerodable formulation of our proprietary Durasert technology that is administered during a physician office visit. In addition to commercialization of YUTIQ in the U.S., we have (i) licensed regulatory, reimbursement and distribution rights to the product to Alimera for Europe, Middle East, and Africa (“EMEA”)under its ILUVIEN tradename and (ii) licensed clinical development, regulatory, reimbursement and distribution rights to Durasert FA to Ocumension Therapeutics (“Ocumension”) for Mainland China, Hong Kong, Macau, Taiwan, South Korea and other jurisdictions across Southeast Asia. Market Opportunity Chronic non-infectious uveitis affecting the posterior segment of the eye as frequently as monthlyis an inflammatory disease that afflicts people of all ages, producing swelling and typicallydestroying eye tissues, which can lose efficacy over time, resulting inlead to severe vision loss and returnblindness. This disease affects between 60,000 to 100,000 people each year in the U.S. and causes approximately 30,000 new cases of blindness every year. The standard of care treatment for this disease typically involves the use of short-acting corticosteroids to reduce uveitic flares followed by additional treatments of sustained release, lower dose steroids to minimize the risk of further flares. Recent Clinical Development Highlights In March 2020, we announced positive topline 36-month follow-up data from a second Phase 3 trial of YUTIQ for the treatment of chronic non-infectious uveitis affecting the posterior segment of the disease.eye. This second double-masked, randomized Phase 3 trial of YUTIQ enrolled 153 patients in 15 clinical centers in India, with 101 eyes treated with YUTIQ and 52 eyes receiving sham injections. At 36-months, the recurrence rate in YUTIQ randomized eyes was significantly lower than in sham treated eyes (46.5% vs. 75.0%, respectively; p=0.001). Visual acuity gains or losses of 3-lines or more were both similar between treatment groups. Safety data showed no unanticipated side effects at each follow-up timepoint at 12, 24 and 36-months. These positive results were consistent with the findings from the first Phase 3 study of YUTIQ and provide further validation of its long-term ability to reduce uveitic flares.
In November 2020, positive data for YUTIQ® was featured in a presentation at the American Academy of Ophthalmology (AAO) 2020 Virtual Annual Meeting. This presentation demonstrated statistically significant efficacy results from the second Phase 3 trial of YUTIQ. Intellectual Property We own the rights for YUTIQ in the U.S. and all foreign jurisdictions and have licensed these rights in EMEA and Mainland China, Hong Kong, Macau and Taiwan. In August 2020, we expanded the out-license agreement to include South Korea and other jurisdictions across Southeast Asia. We have patent rights for YUTIQ in the U.S. through at least August 2027 and internationally through dates ranging from October 2024 to May 2027. Sales and Marketing YUTIQ was granted a permanent and specific J-code by the Centers for Medicare & Medicaid Services (“CMS”), effective October 1, 2019. Approximately sixteen Key Account Managers (“KAMs”) are dedicated to calling on uveitis and retinal specialists across the U.S. In cancer therapy, TKIs2020, the retinal and uveitis markets were impacted by the Pandemic as most teaching hospitals and many independent practices significantly reduced the patient access and flow into the clinics. As a result, many patients were unable to receive the treatments needed to control the inflammatory disease in a timely manner. We started to see customer demand return in the third and fourth quarter of 2020. DEXYCU DEXYCU (dexamethasone intraocular suspension) 9%, for intraocular administration, was approved by the FDA in February 2018 for the treatment of post-operative ocular inflammation and commercially launched in the U.S. in March 2019 with a primary focus on its use immediately following cataract surgery. DEXYCU is administered as a single dose directly into the surgical site at the end of ocular surgery and is the first long-acting intraocular product approved by the FDA for the treatment of post-operative inflammation. DEXYCU utilizes our proprietary Verisome® drug-delivery technology, which allows for a single intraocular injection that releases dexamethasone, a corticosteroid, for up to 22 days. Market Opportunity DEXYCU is approved for ocular post-surgical inflammation. The initial market we have focused on for DEXYCU is post-operative inflammation associated with cataract surgery as there were approximately 3.8 million cataract surgeries performed in 2018 in the U.S. Prior to the launch of DEXYCU, the standard of care for post-operative reduction of inflammation and pain in cataract surgery had been a combination of steroid, antibiotic and non-steroidal eye drops administered multiple times each day over a period of several weeks. Recent Clinical Development Highlights Positive retrospective case study data supporting DEXYCU was highlighted in an oral presentation at the 2020 Caribbean Eye Meeting in an oral session entitled, “Drug Delivery: Real-World Experience With Dexamethasone Intraocular Suspension”. The ongoing retrospective study is designed to provide large-scale, real-world data on early experiences with DEXYCU from surgeons. Interim results presented are taken orally butfrom 154 patients administered DEXYCU with each time point of data based on patient chart data and frequency of measurement by participating physicians. The proportion of patients with complete anterior chamber cell clearing (cell score=0) was 47.5%, 50.0%, 84.1% and 87.5% at postoperative day 1, 8, 14 and 30, respectively. The proportion of patients with no anterior chamber flares (flare score=0), another measurement of inflammation, was 77.7%, 98.5%, 98.8% and 99.1% at postoperative day 1, 8, 14 and 30, respectively. Mean intraocular pressure at postoperative day 1 was 17.6mmHg, with levels decreasing through to postoperative day 30. In November 2020, positive data DEXYCU was featured in three presentations at the American Academy of Ophthalmology (AAO) 2020 Virtual Annual Meeting. This included positive results from a post-cataract surgery inflammatory reduction data from a multicenter retrospective study of real-world usage of DEXYCU. Intellectual Property We own the worldwide rights to all indications for DEXYCU and in January 2020 we out-licensed clinical development, regulatory, reimbursement and distribution rights for the product in Mainland China, Hong Kong, Macau and Taiwan. In August 2020, we expanded the out-license agreement to include South Korea and other jurisdictions across Southeast Asia.
Sales and Marketing In October 2018, DEXYCU was granted “pass through status” by the CMS that provides for reimbursement of DEXYCU separate from the cataract procedure payment bundle for a 3-year period. The 3-year period commenced in April 2019, the quarter that the first claim for reimbursement for DEXYCU was made with CMS and will expire in March 2022. In addition, in November 2018, CMS assigned a specific and permanent J-code for DEXYCU, effective January 1, 2019, that enabled reimbursement across all types of payers. We have 10 KAMS selling DEXYCU, supplemented by a marketing alliance with ImprimisRx that was signed in August of 2020. ImprimisRx is utilizing their toxicity preventsinternal sales representatives and their systemicnumerous indirect representatives to promote DEXYCU to their existing cataract surgery customers. The KAMs are dedicated to calling on cataract surgeons and ASCs and are supported by our market access, marketing and commercial sales management teams. The impact of the Pandemic has been significant on the cataract market as elective surgeries were completely eliminated or vastly reduced in many parts of the country for extended periods of time in 2020. We started to see customer demand return in the third and fourth quarter of 2020. At this time it is unknown how long the impact of the Pandemic will continue to impact patient access to these procedures. Manufacturing The FDA regulates the quality of pharmaceuticals very carefully. The main regulatory standard for ensuring pharmaceutical quality is the Current Good Manufacturing Practice (“cGMPs”) regulation for human pharmaceuticals. Manufacturing of our clinical trial materials (“CTM”) and of our commercial products is subject to these cGMPs which govern record-keeping, manufacturing processes and controls, personnel, quality control and quality assurance, among other activities. Incoming raw materials and components from suppliers are inspected upon arrival according to pre-specified criteria prior to use in the CTM or the commercial product. During product manufacture, in-process tests are conducted on intermediate products according to treat AMD. Usingpre-specified criteria; testing is finally conducted on the finished product prior to its release. Our systems and our contractors are required to comply with cGMP requirements, and we assess compliance regularly through performance monitoring and audits. EYP-1901 Production, assembly, and packaging of EYP-1901 CTM is done in the Class 10,000 clean room located at our Watertown, MA facility. We source the active pharmaceutical ingredient (“API”) vorolanib from Equinox and various raw materials and components for both EYP-1901 and its injector from third-party vendors. Our agreements with Equinox and these third parties include confidentiality and intellectual property provisions to protect our proprietary rights related to EYP-1901. YUTIQ50 Production, assembly, and packaging of YUTIQ50 CTM is done in the Class 10,000 clean room located at our Watertown, MA facility. We utilize the same vendors for YUTIQ50 materials and components as for YUTIQ, as described below. YUTIQ Production, assembly and packaging of YUTIQ is done in the Class 10,000 clean room located at our Watertown, MA facility. We source the API and various raw materials and components for YUTIQ from third-party vendors. Our agreements with these third parties include confidentiality and intellectual property provisions to protect our proprietary rights related to YUTIQ. DEXYCU We currently use a contract manufacturer for the commercial supply of DEXYCU. A separate contract manufacturer provides kitting and packaging of the finished product, and other vendors provide sterilization, testing and storage services. Our agreements with these third parties include confidentiality and intellectual property provisions to protect our proprietary rights related to DEXYCU. We require our contract manufacturers to operate in accordance with cGMPs and all other applicable laws and regulations. We employ personnel with extensive technical, manufacturing, analytical and quality experience to oversee contract manufacturing and testing activities, and to compile manufacturing and quality information for our regulatory submissions. U.S. Sales and Marketing We launched YUTIQ and DEXYCU in the U.S. during the first quarter of 2019 utilizing a contract sales organization (“CSO”) model. This model involved the hiring of sales and marketing leadership professionals providing oversight and leadership to the CSO teams. We believe this flexible sales model provided less execution risk as CSOs leverage costs across multiple clients allowing us to cost-effectively build the necessary infrastructure to support sales activities. In addition, we are able to utilize CSO installed systems and processes for, inter alia, regulatory filings, data tracking, field incentive compensation, training, hiring of KAMS, territory sizing / alignment, sample tracking, and customer relationship management systems.
Members of our sales and marketing leadership team have extensive commercialization experience with ophthalmic products at previous companies. We have 16 KAMS selling YUTIQ and we have 10 KAMS selling DEXYCU. In addition, we signed a marketing alliance with ImprimisRx in August of 2020 which adds sales efforts for DEXYCU by its 12 sales representatives and its numerous indirect representatives. The KAMs have extensive sales experience, with most having prior ophthalmological or pharmaceutical sales experience. In January 2020, the YUTIQ KAMs were converted to full-time employees from our CSO, and in October of 2020 the DEXYCU KAMs were converted to full-time employees. U.S. Market Access and Payer Reimbursement In 2018 we recruited a team of highly experienced personnel to form our market access team. The team is comprised of our VP of Market Access and Government Affairs, Assoc. Director of Patient Access, Director of National Accounts (“NAD”), and Field Reimbursement Managers (“FRMs”) who handle the reimbursement for both YUTIQ and DEXYCU. Their roles include the discussions with payers regarding the costs and benefits of our products for their members; assisting with the addition of our products to the medical policy of payers; and providing the market with assistance regarding reimbursement queries. We have initiated a patient assistance platform called EyePoint AssistSM to provide co-pay and coinsurance relief for eligible commercial patients. Reimbursement for YUTIQ is obtained using a permanent J code, established October 1, 2019, which enables reimbursement from both Medicare and commercial payers. DEXYCU has three-year pass through status with Medicare whereby it is routinely reimbursed for Medicare Part B patients. The issuance of a specific and permanent J code for DEXYCU in November 2018 has enabled our market access team to work with non-Medicare payers with regard to adding DEXYCU to their medical policies. We believe that products that are reimbursable using a specific J code (as opposed to a C code or miscellaneous J code) are simpler for payers to process and therefore have a greater likelihood of reimbursement. U.S. Product Distribution Channel We have established a distribution channel in the United States for the commercialization of YUTIQ and DEXYCU that provides physicians with several options for ordering our products. This includes agreements with a nationally recognized third-party logistics provider (“3PL”), several distributors and a specialty pharmacy provider for physicians who prefer to use a traditional buy-and-bill model. The 3PL provides fee-based services related to logistics, warehousing, order fulfilment, invoicing, returns and accounts receivable management. Research Agreements From time to time we enter into research agreements funded by third parties to evaluate our Durasert technology we plan to develop an implant to deliver a TKI directly toplatform for the back of the eye with a total dose that is significantly lower than what is customarily used in a course of cancer therapy. Our development goal is to provide sustained treatment of wet AMDophthalmic and other diseases. We intend to continue this activity with partner compounds that could be successfully delivered with our Durasert and, potentially, Verisome technology platforms on a fee-for-service basis with the potential for six months with a single injection of aTKI-based product, targeting VEGF while avoiding or reducing the toxic systemic side effects of TKIsfuture clinical and the frequent injections of current wet AMD anti-VEGF biologics. Using a model TKI (that is not patentable), we have generatedpre-clinical data that demonstrate that a TKI delivered by a sustained release insert was comparably efficaciouscommercial milestones and royalties.
FDA Approved Products Licensed to a commercially available biologic indicated for wet AMD delivered by injection, both in preventing choroidal neovascularization and in reducing vascular leakage. On the basis of these data, we are currently evaluating other, potentially patentable TKIs for sustained release over several months and with comparable therapeutic effects.Others Approved Durasert Technology Product: ILUVIEN for DME
ILUVIEN is an injectable, sustained-release micro-insert based on our Durasert technology platform and delivers 0.19 mg of FA to the back of the eye for treatment of DME. The ILUVIEN micro-insert is substantially the same micro-insert as YUTIQ. ILUVIEN is injected in an office visit using a25-gauge inserter, and delivers approximately 36 months of continuous,low-dose corticosteroid therapy with a single injection. ILUVIEN is approved in the U.S. for the treatment of DME in patients who have been previously treated with a course of corticosteroids and did not have a clinically significant rise in IOP. In the 17 EU countries where ILUVIEN has been approved, it is indicated for the treatment of vision impairment associated with chronic DME considered insufficiently responsive to available therapies. DME is a disease suffered by diabetics where leaking capillaries cause swelling in the macula, the most sensitive part of the retina. DME is a leading cause of blindness in theworking-age population in most developed countries. The ILUVIEN micro-insert is substantially the same micro-insert as YUTIQ. We originally licensed our Durasert proprietary insert technology to Alimera for use in ILUVIEN for the treatment of all ocular diseases (excluding uveitis). Alimera has sold ILUVIEN for DME in the U.K. and Germany since 2013, in Portugal and the U.S. since 2015 and in Austria and Ireland since 2017. ILUVIEN also has marketing approvals in 12 other European countries. In addition, Alimera has entered into various agreements under which distributors will provide regulatory, reimbursement and/or sales and marketing support for commercialization or future commercialization of ILUVIEN in several countries in the Middle East, as well as in France, Italy, Spain, Australia, New Zealand and Canada. On July 10, 2017, we entered into the Amended Alimera Agreement, pursuant to which we (i) expanded the license to Alimera to our proprietary Durasert sustained-release drug delivery technology platform to include uveitis, including NIPU,chronic non-infectious uveitis affecting the posterior segment of the eye, in the EMEA and (ii) converted the net profit share arrangement for each licensed product (including ILUVIEN) under the original collaboration agreement with Alimera or the Prior(the “Prior Alimera Agreement,Agreement”) to a sales-based royalty on a calendar quarter basis commencing July 1, 2017, with payments from Alimera due 60 days following the end of each calendar quarter. Sales-based royalties startstarted at the rate of 2%. Commencing January 1, 2019 (or earlier under certain circumstances), the sales-based royalty will increase and increased, commencing December 12, 2018, to 6% on aggregate calendar year net sales up to $75 million and 8% in excess of $75 million. Alimera’s share of contingently recoverable accumulated ILUVIEN commercialization losses under the Prior Alimera Agreement, capped at $25 million, are to be reduced as follows: (i) $10.0 million
was cancelled in lieu of an upfront license fee on the effective date of the Amended Alimera Agreement; (ii) for calendar years 2019 and 2020, 50% of earned sales-based royalties in excess of 2% will be offset against the quarterly royalty payments otherwise due from Alimera; (iii) on January 1,in March 2020, another $5 million will bewas cancelled provided, however, that such dateupon Alimera’s receipt of cancellation may be extended under certain circumstances related to Alimera’s regulatory approval process for ILUVIEN for the treatment of NIPU, with such extension, if any, subject to mutual agreement by the parties;uveitis indication; and (iv) commencing in calendar year 2021, 20% of earned sales-based royalties in excess of 2% will be offset against the quarterly royalty payments due from Alimera until such time as the balance of the original $25 million of recoverable commercialization losses has been fully recouped. On December 17, 2020, we sold our interest in royalties payable to us under our license agreement with Alimera in connection with Alimera’s sales of ILUVIEN® to SWK Funding, LLC (“SWK”) in exchange for a one-time $16.5 million payment from SWK. FollowingRetisert for chronic non-infectious uveitis affecting the completionposterior segment of the Amended Alimera Agreement, we withdrew our previously filed EU marketing approval application and our EU orphan drug designation for YUTIQ, and Alimera was responsible
for filing a Type II variation for ILUVIEN for the treatment of NIPU. In January 2018, Alimera received validation of a Type II variation submitted in December 2017 in all seventeen European countries in which it previously received regulatory approval for ILUVIEN for DME. According to Alimera’s public filings, Alimera plans to submitfollow-up data supporting its Type II variation application in the fourth quarter of calendar year 2018 and expects it will obtain approval for its application in the first half of calendar 2019. If the variation is approved, Alimera has reported that it plans to commercialize the three-year uveitis indication under its ILUVIEN trademark.
Approved Durasert Technology Product: Retisert for Posterior Segment Uveitiseye
Retisert is a sustained-release non-erodible implant based on our Durasert technology platform for the treatment of chronic non-infectious uveitis affecting the posterior segment uveitis.of the eye. Surgically implanted, it delivers 0.59 mg of FA to the back of the eye for approximately 30 months. Retisert is licensed to Bausch & Lomb, with which weco-developed the product. ApprovedRetisert is approved in the U.S., Bausch & Lomb sells the product and payspaid sales-based royalties to us. Approved Durasert Technology Product: Vitrasert for CMV Retinitis
Vitrasert The patent with which Retisert is a sustained-release implant based onmarketed expired in March 2019. As such, pursuant to our Durasert technology platform for the treatment of cytomegalovirus retinitis, a blinding eye disease that occurs in individualsagreement with advanced acquired immune deficiency syndrome. Surgically implanted, Vitrasert provided sustained delivery of the anti-viral drug ganciclovir for six to eight months. Approved in the U.S. and EU, Vitrasert was licensed to Bausch & Lomb, which discontinued sales in fiscal 2013payment of sales-based royalties concluded at the end of March 2019 following patent expiration.
Development Product: Severe Knee Osteoarthritis Implant
We have developed an implant for the treatment of pain associated with severe knee osteoarthritis, or OA, in collaboration with Hospital for Special Surgery pursuant to an Investigatory-Initiated Research Agreement. This implant was evaluated in an investigator-sponsored pilot study of six patients, which has been completed. The implant is composed of a specially manufactured surgical screw containing a Durasert system that delivers dexamethasone directly to the joint on a sustained basis. Dexamethasone is anoff-patent corticosteroid that is frequently used for the treatment of OA. Implanted in thenon-articulating area of the knee in an outpatient procedure, the implant is designed to provide long-term pain relief and thereby delay the need for knee replacement surgery. This implant represents the first use of our Durasert technology outside of ophthalmology. We believe this design, if successful, could be adapted for severe OA in other large joints. We plan toout-license our rights to this product.
Feasibility Study Agreements
From time to time we have entered into feasibility study agreements funded by third parties to evaluate our Durasert technology platform for the treatment of ophthalmic and other diseases. We presently are engaged in one such collaboration for a back of the eye disease. We intend to continue to identify other companies with compounds that could be successfully delivered with our Durasert and Verisome technology platforms and, through appropriate agreements, seek to generatenon-dilutive operating capital from such agreements.
We have received Notices of Allowance from the U.S. Patent and Trademark Office, or the USPTO, for trademarks DEXYCU™, YUTIQ™, DELIVERING INNOVATION TO THE EYE™ and Durasert™ in the U.S. Retisert® and Vitrasert® are Bausch & Lomb’s trademarks. ILUVIEN® is Alimera’s trademark. The reports we file or furnish with the Securities and Exchange Commission, or the SEC, including this Annual Report on Form10-K, also contain trademarks, trade names and service marks of other companies, which are the property of their respective owners.
Information with respect to ILUVIEN, including regulatory and marketing information, and Alimera’s plans and intentions, reflects information publicly disclosed by Alimera.
Fiscal 2018, fiscal 2017 and fiscal 2016 mean the twelve months ended June 30, 2018, 2017 and 2016, respectively, and fiscal 2019 means the twelve months ending June 30, 2019.
Strategy
Our strategy is to market multiple drugs for use in diseases of the eye. In addition to DEXYCU and YUTIQ, we plan to (i) acquire orin-license ophthalmology approved products or product candidates developed by third parties, (ii) independently developnon-proprietary drugs in combination with our Durasert or Verisome technology platforms and (iii) continue to leverage our Durasert and Verisome technology platforms through collaborations and license agreements, as appropriate. We plan to execute our strategy as follows:
Launch and maximize the commercial potential of DEXYCU for post-operative inflammation.In February 2018, the FDA approved DEXYCU for the treatment of postoperative inflammation following ocular surgery. DEXYCU is the first long-acting intraocular product approved by the FDA for the treatment of postoperative inflammation. We plan to launch DEXYCU in the U.S. in the first half of calendar year 2019 following the successful scale up of commercial infrastructure and supplies.
Obtain regulatory approval for, and maximize the commercial potential of, YUTIQ. On March 19, 2018, the FDA accepted our NDA for YUTIQ and set a PDUFA action date of November 5, 2018. Posterior segment uveitis is a high unmet need area with limited treatment options and the third leading cause of blindness in the U.S. If approved, we expect to launch YUTIQ in the U.S. in the first half of calendar year 2019.
Acquire orin-license ophthalmology products or product candidates developed by third parties.We plan to expand our commercial portfolio of treatments for eye disease by evaluating for acquisition and/orin-licensing approved products or product candidates in late stage clinical development.
Leverage our Durasert and Verisome technologies.We plan to use our proprietary Durasert and Verisome drug delivery technology platforms to independently develop new drug delivery products that use already-approved drugs to treat ophthalmic and other diseases, while continuing to leverage our technology platforms through collaborations and licenses with other pharmaceutical and biopharmaceutical companies, institutions and other organizations. We believe our technologies can provide sustained, targeted delivery of therapeutic agents, resulting in improved therapeutic effectiveness, safer administration and better patient compliance and convenience, with reduced product development risk and cost. We believe that our proven track record of four approved products, all providing sustained release of previously approved drugs, reflects the benefits of this strategy.
Develop Sustained Delivery ofOff-Patent Drugs.Many drugs are now, or will soon be,off-patent. It is estimated that over the next several years, patent coverage will end on products with world-wide sales aggregating billions of dollars annually. We are using our technology platforms to evaluate potential product candidates that deliveroff-patent and generic drugs, primarily focused on ocular diseases with significant market opportunities, where less frequent dosing through sustained delivery and/or targeted delivery at the treatment site would materially improve the effectiveness, safety or convenience of the original drug. By focusing on delivery of already-approved drugs, particularly those requiring potentially shorter clinical development programs, we believe we may be able to reduce the substantial risks and financial investment required for product approval.
Continue Partnering with Leading Biopharmaceutical and Pharmaceutical Companies. We intend to continue to partner with leading biopharmaceutical and pharmaceutical companies, institutions and others, where patent protection, development and regulatory costs, expertise and/or other factors make it desirable for us to have a partner. For example, drugs that might be more effectively delivered by our technology platforms or may have extended patent protection could make collaborations with the patent holders attractive. We may also seek to partner the development of product candidates that could materially benefit from sustained delivery but would require expensive clinical trials or are in treatment areas outside of our technical expertise. We may also seek to partner
| with companies with drugs coming off patent where our drug delivery technologies could offer an improved product and effectively extend patent protection.
|
Strategic Collaborations We have entered into a number of collaboration/license agreements to develop and commercialize our product candidates and technologies. In all of these collaboration agreements, we have retained the right to use and develop the underlying technologies outside of the scope of the exclusive licenses granted. Alimera
In February 2005, as amended The license and restated in March 2008, we granted Alimera an exclusive worldwidecollaboration arrangements typically include, among other terms and conditions, non-refundable upfront license to manufacture, develop, marketfees, milestone payments and sell ILUVIEN for the treatmentroyalty and/or profit sharing obligations.See Note 3, "Product Revenue Reserves and prevention of human eye diseases other than uveitis pursuantAllowances-License and Collaboration Agreements" to the Prior Alimera Agreement. We also granted Alimera a worldwidenon-exclusive license to manufacture, develop, marketConsolidated Financial Statements included under Item 15, "Exhibits and sell certain additional Durasert-based products (1) to deliver a corticosteroid and no other active ingredient by a direct delivery method to the back of the eye solely for the treatment and prevention of eye diseases in humans other than uveitis and (2) to treat DME in humans by delivering a compound by a direct delivery method through an incision no smaller than that required for a25-gauge or larger needle. Thenon-exclusive license is limited to those products that, among other things, (i) have a drug core within a polymer layer (with certain limitations regarding chemically bonded combinations of active agents) and (ii) are approved, or designed to be approved, to deliver a corticosteroid and no other active ingredient by a direct delivery to the posterior portion of the eye, or to treat DME by delivering a compound by a direct delivery through an incision required for a25-gauge or larger needle. We are not permitted to use or grant a license to any third party to use, the licensed technologies to make or sell any products that are or would be subject to thenon-exclusive license granted to Alimera.Financial Statement Schedules."
In October 2014, Alimera paid us a $25.0 million milestone upon FDA approval of ILUVIEN as provided in in the Prior Alimera Agreement.
In July 2017, we entered into the Amended Alimera Agreement to (i) expand the license to Alimera for our proprietary Durasert sustained-release drug delivery technology platform to include uveitis, including NIPU, in the EMEA and (ii) convert the previous net profit share arrangement on acountry-by-country basis to sales-based royalties for ILUVIEN for DME, NIPU and any other ILUVIEN indications that obtain regulatory approval in various jurisdictions in the future, provided that certain amounts of Alimera’s previous ILUVIEN net commercialization losses can be offset against earned sales-based royalties (as described below). We are entitled to receive a 2% sales-based royalty within 60 days following the end of each quarterly period through calendar year 2018. Commencing January 1, 2019 (or earlier under certain circumstances) the sales-based royalty will increase to 6% on aggregate calendar year net sales up to $75 million and 8% on any calendar year sales in excess of $75 million. Alimera’s share of accumulated ILUVIEN commercialization losses under the original net profit share arrangement (as set forth in the Prior Alimera Agreement), is capped at $25 million. Under the Amended Alimera Agreement, these recoverable losses will be reduced as follows: (i) $10 million was cancelled in lieu of any upfront license fee on the effective date of the Amended Alimera Agreement; (ii) for calendar years 2019 and 2020, 50% of earned sales-based royalties in excess of 2% of net sales will be offset against the quarterly royalty payments due from Alimera; (iii) on January 1, 2020 (or earlier under certain circumstances), another $5 million of the accumulated commercialization losses will be cancelled, provided, however, that such date of cancellation may be extended further under certain circumstances related to Alimera’s regulatory approval process for ILUVIEN for the treatment of NIPU, with such extension, if any, subject to mutual agreement by the parties; and (iv) commencing in calendar year 2021, 20% of earned sales-based royalties in excess of 2% of net sales will be offset against the quarterly royalty payments due from Alimera until such time as the remaining balance of the original $25 million of commercialization losses has been recouped by Alimera.
Bausch & Lomb
Under a 2003 amended license agreement, Bausch & Lomb has a worldwide exclusive license to make and sell Retisert and other first-generation products defined in the agreement in return for royalties based on sales. This agreement also covered Vitrasert prior to patent expiration. Bausch & Lomb can terminate its agreement with us without penalty at any time upon 90 days’ written notice.
Pfizer
In June 2011, we entered into an Amended and Restated Collaborative Research and License Agreement with Pfizer, Inc., or Pfizer, which we refer to as the Restated Pfizer Agreement, to focus solely on the development of a sustained-release bioerodible micro-insert injected into the subconjunctiva designed to deliver latanoprost for human ophthalmic disease or conditions other than uveitis, or the Latanoprost Product. Pfizer made an upfront payment of $2.3 million and we agreed to provide Pfizer options under various circumstances for an exclusive, worldwide license to develop and commercialize the Latanoprost Product. On October 25, 2016, we notified Pfizer that we had discontinued development of the Latanoprost Product, which provided Pfizer a60-day option to acquire a worldwide license in return for a $10.0 million payment and potential sales-based royalties and development, regulatory and sales performance milestone payments. Pfizer did not exercise its option and the Restated Pfizer Agreement automatically terminated on December 26, 2016. Per the terms of the Restated Pfizer Agreement, we retained the right thereafter to develop and commercialize the Latanoprost Product on our own or with a partner.
OncoSil Medical Ltd.
Our December 2012 license agreement, amended and restated in March 2013, with Enigma Therapeutics Limited, a wholly-owned subsidiary of OncoSil Medical Ltd, or OncoSil Medical, provides OncoSil Medical with an exclusive, worldwide, royalty-bearing license for the development of BrachySil (now named OncoSil™), a product candidate for the treatment of pancreatic and other cancers. We received an upfront fee of $100,000 and are entitled to an 8% sales-based royalty, 20% of sublicense consideration and milestones based on aggregate product sales. To date, OncoSil Medical has not received regulatory approval for OncoSil in any jurisdiction. OncoSil Medical is obligated to pay an annual license maintenance fee of $100,000, creditable during each ensuing twelve-month period against reimbursable patent maintenance costs and sales-based royalties. Annual license maintenance fees of $100,000 were paid in respect of each calendar year from 2013 through 2017. OncoSil Medical has the right to terminate this license upon 60 days’ prior written notice.
Research and Development
Our clinical andpre-clinical research programs primarily focus on ophthalmic applications of our technology platforms. Our research and development expenses totaled $16.2 million in fiscal 2018, $14.9 million in fiscal 2017 and $14.4 million in fiscal 2016. Of these amounts, $13.9 million in fiscal 2018, $13.0 million in fiscal 2017 and $12.8 million in fiscal 2016 were incurred for costs of research and development personnel, clinical andpre-clinical studies, contract services, testing and laboratory facilities. The remaining expense of $2.3 million in fiscal 2018, $1.9 million in fiscal 2017 and $1.6 million in fiscal 2016 consisted ofnon-cash charges for amortization of intangible assets, depreciation of property and equipment and stock-based compensation expense specifically allocated to research and development personnel.
During the first quarter of fiscal 2017, we consolidated all of our research and development operations in our facility in Watertown, Massachusetts. We closed our research facility in Malvern, U.K. and terminated the employment of all our employees in that location.
Intellectual Property We own or license patents in the U.S. and other countries. Our patents generally cover the design, formulation, manufacturing methods and use of our sustained release therapeutics, devices and technologies. Patents for individual products extend for varying periods according to the date of patent filing or grant and legal term of patents in the various countries where patent protection is obtained. The actual protection afforded by a patent, which can vary from country to country, depends upon the type of patent, the scope of its coverage and the availability of legal remedies in the country. Patent term extension may be available in various countries to compensate for a patent office delay or a regulatory delay in approval of the product. The U.S. patent with whichpatents that were previously listed in the USFDA Orange Book for Retisert is marked expiresexpired in March 2019. The lastlatest expiring patent covering Retisert expireslisted in April 2020. The latest expiring patentthe USFDA Orange Book covering ILUVIEN and YUTIQ expires in August 2027 in the U.S. and in October 2024 in the EU, although extensions have been obtained or applied for through May 2027 in various EU countries. The last of the previously issued patents covering DEXYCU expire in July 2023, but two additional patents have issued in the U.S. that will cover DEXYCU until at least 2034. The last expiring patent covering the vorolanib compound licensed to us by Equinox Science and used in EYP-1901 expires in September 2027, but additional patents have been applied for which, if issued, will extend coverage of EYP-1901 until at least 2037. The following table provides general details relating to our owned and licensed patents (including both patents that have been issued and applications that have been accepted for issuance) and patent applications as of August 31, 2018:February 28, 2021: | Technology | | United States
Patents | | | United States
Applications | | | Foreign
Patents | | | Foreign
Applications | | | Patent
Families | | | United States Patents | | | United States Applications | | | Foreign Patents | | | Foreign Applications | | | Patent Families | | Durasert | | | 11 | | | | 7 | | | | 72 | | | | 12 | | | | 12 | | | | 4 | | | | 3 | | | | 128 | | | | 10 | | | | 6 | | Verisome | | | 9 | | | | 4 | | | | 31 | | | | 26 | | | | 8 | | | Verisome (Ocular) | | | | 12 | | | | 5 | | | | 121 | | | | 18 | | | | 7 | | Other | | | 15 | | | | 11 | | | | 39 | | | | 47 | | | | 15 | | | | 11 | | | | 4 | | | | 72 | | | | 12 | | | | 8 | | | | | | | | | | | | | | | | | | | Total | | | 35 | | | | 22 | | | | 142 | | | | 85 | | | | 35 | | | | 27 | | | | 12 | | | | 321 | | | | 40 | | | | 21 | | | | | | | | | | | | | | | | | | |
Employees
WeHuman Capital Resources
To achieve the goals and expectations of our Company, it is critical that we continue to attract and retain top talent. To facilitate talent attraction and retention we strive to make our company a safe and rewarding workplace, with opportunities for our
employees to grow and develop in their careers, supported by strong compensation, benefits and health and wellness programs, and by programs that build connections between our employees. As of February 28, 2021, we had 44 101 employees, as98 of August 31, 2018.whom were full-time employees in the United States. None of our employees are coveredrepresented by a collective bargaining agreement. During fiscal 2020 our voluntary turnover rate was 13%, which is consistent with the average voluntary turnover rates for Boston-area Biotech companies. ManufacturingThe success of our business is fundamentally connected to the well-being of our employees. Accordingly, we are committed to their health, safety, and wellness. We provide our employees and their families with access to a variety of innovative, flexible and convenient health and wellness programs, including benefits that provide protection and security so that they have peace of mind concerning events that may require time away from work, or that impact their financial well-being, that support their physical and mental health and providing tools and resources to help them improve or maintain their health status and encourage engagement in healthy behaviors, and that offer choice where possible so they can customize their benefits to meet their needs and the needs of their families. In response to the Pandemic we implemented significant changes that we determined were in the best interest of our employees, as well as the communities in which we operate, and which comply with government regulations. This includes having many of our non-laboratory employees work from home, while implementing additional safety measures for employees continuing on-site work.
We currently planprovide robust compensation and benefits programs to use a contract manufacturer for commercial suppliesmeet the needs of our DEXYCU approved product. Subjectemployees. In addition to FDA approval,competitive base salaries, these programs include annual discretionary bonuses, stock awards, a 401(k) plan and employer match, an employee stock purchase program, health, dental and vision insurance benefits, health savings and flexible spending accounts, paid time off, family leave and flexible work schedules, among others. Our broad-based equity programs include all employees with vesting conditions to facilitate the retention of employees with critical skills and experience and motivate employees to perform to the best of their abilities, while we plan to source raw materials and components necessary to manufacture YUTIQ through third-party vendors and assemble commercial supplies of finished product ourselvesachieve our objectives. As a company our success is rooted in our Watertown, MA facility. Allthe diversity of our otherpre-clinical studyteams and clinical trial supplies for product candidates that utilize our Durasert technology platform have been,commitment to inclusion. We value diversity at all levels and will continue to be, manufactured ourselves. Raw materialsfocus on extending our diversity and components are availableinclusion initiatives across our workforce – from multiple sources. The manufacture of each of Retisert and ILUVIEN is the responsibility of our licensees. Sales and Marketing
We currently are building out our U.S. marketing and sales staff. Members of our leadership team have extensive commercialization experience at previous companies. We plan to launch DEXYCU in the U.S. in the first half of calendar year 2019 following the successful scale up of commercial infrastructure and supplies and, if approved by FDA, we expect to also launch YUTIQ in the U.S. in the first half of calendar year 2019. We have invested substantially in our sales and marketing infrastructure since May 2018 and that will continue during the balance of calendar year 2018 in preparation for the aforementioned product launches. We have entered into an agreementworking with a CSO for the field-based sales representatives and key account managers to promote DEXYCU
and, if approved byrecruit diverse team members to the FDA, YUTIQ, to our defined audiences in the U.S. We plan toout-license DEXYCU for any territories outside the U.S. We have alreadyout-licensed YUTIQ to Alimera for the EMEA and would expect to seek furtherout-licenses for other territories outside the U.S. and EMEA.advancement of leaders from different backgrounds.
Competition The market for products treating eye diseases is highly competitive and is characterized by extensive research efforts and rapid technological progress. We face substantial competition for ourFDA-approved product products and our product candidates. Pharmaceutical, drug delivery and biotechnology companies, as well as research organizations, governmental entities, universities, hospitals, other nonprofit organizations and individual scientists, have developed and are seeking to develop drugs, therapies and novel delivery methods to treat diseases targeted by our products and product candidates. Most of our competitors and potential competitors are larger, better established, more experienced and have substantially more resources than we or our partners have. Competitors may reach the market earlier, may have obtained or could obtain patent protection that dominates or adversely affects our products and potential products, and may offer products with greater efficacy, lesser or fewer side effects and/or other competitive advantages. We believe that competition for treatments ofback-of-the-eye eye diseases is based upon the effectiveness of the treatment, side effects, time to market, reimbursement and price, reliability, ease of administration, dosing or injection frequency, patent position and other factors. Many companies have or are pursuing products to treat eye diseases that are or would be competitive with DEXYCU, ILUVIEN for DME or, if approved,EYP-1901, YUTIQ50, YUTIQ, and ILUVIEN for NIPU.DEXYCU. Some of these products and potential productsproduct candidates include the following: | • | | Inflammation following cataract surgery.There is a high unmet medical need among patients who undergo cataract surgery as the current standard of care to treat the inflammation post-surgery includes a schedule of up to 70 eye drops over a period of3- 4 weeks. In August 2018, Kala Pharmaceuticals, Inc. announced that the FDA approved INVELTYS™ (loteprednol etabonate ophthalmic suspension) 1% for the topical treatment of post-operative inflammation and pain following ocular surgery. INVELTYS is the first twice-daily ocular corticosteroid approved for this indication while all other available ocular steroid eyedrops are only approved forfour-times-a-day dosing. This product is expected to improve compliance and allow for less burdensome self-dosing with eyedrops for patients. Ocular Therapeutix Inc. is developing DEXTENZA®, which is a corticosteroid intracanalicular insert placed through the punctum, a natural opening in the eye lid, into the canaliculus, and is designed to deliver dexamethasone to the ocular surface for up to 30 days. Following treatment, DEXTENZA is intended to resorb and exit the nasolacrimal system without the need for removal. DEXTENZA has completed a Phase 3 clinical trial in the U.S. and is currently limited by U.S. law to investigational use only because the product has not been approved by the FDA. On June 29, 2018, Ocular Therapeutixre-filed its NDA with the FDA, which approved itsre-submission and set a PDUFA action date of December 28, 2018. Both INVELTYS and DEXTENZA deliver a steroid on the surface of the eye and therefore are dependent on penetration through the cornea to reach the intended target of the anti-inflammatory effect. On the other hand, since DEXYCU is delivered directly into the posterior chamber and bypasses that anatomical barrier, we believe that it can exert its anti-inflammatory effect upon dosing.
|
| • | | DME.Genentech USA Inc.’s LUCENTIS® (ranibizumab) and Regeneron Pharmaceutical’s EYLEA® (aflibercept) are approved in the U.S. and the EU for the treatment of DME. Roche’s lower-cost AVASTIN® is approved to treat various cancers, but is usedoff-label for treatment of diabetic retinopathy. These products are VEGF inhibitors which are considered first line therapy for DME due to their ability to block the VEGF protein, which at high levels can cause abnormal blood vessels to grow in the eye and leak fluid. Genentech is a wholly-owned member of the Roche Group. Novartis AG, or Novartis, has the right to market and sell LUCENTIS outside of the U.S. Regeneron maintains exclusive rights to EYLEA in the U.S., and Bayer HealthCare Pharmaceuticals LLC owns the
|
| exclusive marketing rights outside the U.S. LUCENTIS, EYLEA and AVASTINWet AMD, a common condition and a leading cause of vision loss for people age 50 and older, are all injected into the back of the eye on a monthly orbi-monthly basis. Allergan, Inc.’s, or Allergan, OZURDEX® (dexamethasone intravitreal implant), a bioerodible intravitreal implant, has been approved for the treatment of DME, retinal vein occlusion and posterior segment uveitis, and has a therapeutic duration of several months. As with ILUVIEN, OZURDEX delivers a corticosteroid (dexamethasone) to the back of the eye through an intravitreal injection. However, it only lasts for up to several months, resulting in frequent injections compared to ILUVIEN lasting for up to three years. Other companies, including Roche, are working on the development of product candidates and extended delivery systems for the potential treatment of DME. RG7716, being developed by Roche, is a bispecific antibody that simultaneously binds to and inactivates vascular endothelial growth factor A, orVEGF-A, andangiopoietin-2. In a Phase 2 clinical trial, RG7716 demonstrated clinically meaningful visual acuity gains from baseline, and statistically significant improvements in visual acuity compared with ranibizumab. Roche’s Phase 3 clinical trial of RG7716 in DME is expected to begin later in calendar year 2018.
|
| • | | Posterior Segment Uveitis.Periocular steroid injections and systemic delivery of corticosteroids are routinely used to treat posterior segment uveitis, which is a chronic, inflammatory condition of the eye. It is treated both aggressively and frequently by physicians in order to minimize the disease “flares”, which are the main cause of vision deterioration and potential blindness. OZURDEX, which is marketed by Allergan, is approved in the U.S. and EU for posterior segment uveitis through an intravitreal bioerodible implant that provides treatment which lasts for several months. As with DME, the several-month effectiveness of OZURDEX can result in frequent intravitreal injections of the implant. AbbVie, Inc. recently obtained FDA approval for HUMIRA® (adalimumab) for the treatment of all types ofnon-infectious uveitis (intermediate, posterior and panuveitis) and it is administered subcutaneously every other week for systemic delivery. HUMIRA is a biologic that blocks tumor necrosis factor alpha, a naturally occurring cytokine that is involved in normal inflammatory and immune responses. Humira’s retail price in the U.S. is approximately $50,000 per year. Other companies have ongoing trials of posterior segment uveitis treatments, including Santen Pharmaceutical Co. Ltd., which recently received a Complete Response Letter, or CRL, from the FDA for their filed NDA for sirolimus, which is administered through intravitreal injection every two months. Sirolimus is a mammalian target of rapamycin inhibitor and modulator of the immune system, and is being developed for NIPU. Clearside’sCLS-TA (triamcinolone acetonide, a steroid) for macular edema associated withnon-infectious uveitis is in Phase 3 trials and it is administered through a suprachoroidal injection administered every 12 weeks. Preliminary clinical data indicated that the suprachoroidal route may reduce the risk of increased IOP that is typically associated with intraocular injection of steroids. The results of the Phase 3 trial, presented in September 2018, indicated that while about 50% of patients experienced significant improvements in visual acuity through 24 weeks, adverse events of IOP increase were reported in about 12% of patients.
|
| • | | WetAge-Related Macular Degeneration.Wet AMD, the leading cause of vision loss in people over 65, is most commonly treated with intravitreal injections of biologics that block VEGF.FDA-approved Lucentis and EYLEA andoff-label use of the anti-cancer Avastin® are the leading treatments for wet AMD. These biologics must be injected into the eye frequently and typically can lose efficacy over time, resulting in vision loss and return of the disease. However, EYLEA was recently approved by the FDA for dosing every 12 weeks. Novartis is currently developing an antibody fragment, brolucizumab, with high affinity to all VEGF-A isoforms. In May 2018, Novartis announced that treatment with brolucizumab, which could be given every 12 weeks, showednon-inferior improvements in best corrected visual acuity (BCVA) when compared with EYLEA every 8 weeks in two Phase 3 trials. Novartis plans to apply for approval with the FDA by the end of calendar year 2018. Abicipar pegol is a monoDARPin (Designed Ankyrin Repeat Protein) that blocks all isoforms ofVEGF-A and is currently being developed by Allergan. Smaller molecular size (34 kDa) may lead to longer duration (12 weeks) than the currently availableanti-VEGF-A agents. Allergan is conducting Phase 3 trials to compare treatment arms of abicipar every 8 weeks, abicipar every 12 weeks and ranibizumab every
|
| four weeks. In July 2018, Allergan announced the release of two positive clinical trials, SEQUOIA and CEDAR for abicipar, demonstrating that both the8-week and12-week treatment regimens met thepre-specified primary endpoint ofnon-inferiority to ranibizumab. The port delivery system with ranibizumab, or PDS, is a refillable reservoir system being developed by Genentech and it is designed to gradually release Lucentis (ranibizumab). The drug is released using a diffusion-control mechanism and the port is placed under the conjunctiva, fixed to the pars plana, and no sutures are needed. The port is then refilled as anin-office procedure with the help of a refill needle system that simultaneously introduces the drug into the reservoir and removes any remaining contents. In July 2018, Roche announced positive Phase 2 results: the majority of PDS patients went 6 months or longer between the implant of the device and first required refill, and patients in the high dose PDS group achieved similar vision outcomes as monthly ranibizumab eye injections. Phase 3 clinical trials are expected to begin later in calendar year 2018 to evaluate PDS in wet AMD. In cancer therapy, TKIs are taken orally, but their toxicity prevents their systemic use to treat AMD. Graybug Vision, Inc.’s, or Graybug Vision, lead product,GB-102, is an intravitreal injectable depot formulation of a tyrosine kinase inhibitor, sunitinib malate, that blocks multiple angiogenesis pathways. In 2017, Graybug Vision launched the first clinical trial ofGB-102 given every 6 months in patients with wet AMD. This Phase 1/2 clinical trial is designed to evaluate patients being treated with available intravitreal anti-VEGF agents who are later switched over to justGB-102.
|
RevenuesFDA-approved LUCENTIS and EYLEA and off-label use of the cancer drug AVASTIN® are the leading treatments for wet AMD. These biologics must be injected into the eye frequently and typically can lose efficacy over time, resulting in vision loss and return of the disease. EYLEA was approved for dosing every 12 weeks after one year of effective therapy.
We operateFDA approved Beovu® brolucizumab injection on October 8, 2019 for a three-month dosing interval immediately after a three-month loading phase. Novartis launched a safety review of Beovu and confirmed 3 newly identified side effects that cause severe vision loss: Retinal vasculitis, retinal vascular occlusion (blocked blood flow to or from the retina) and a combination of retinal vasculitis and retinal vascular occlusion. As a result, Novartis updated the Beovu safety information to include these warnings.
Genentech is developing a refillable reservoir port delivery system (“PDS”) designed to gradually release LUCENTIS (ranibizumab) using a diffusion-control mechanism. The port is placed under the conjunctiva, fixed to the pars plana, and no sutures are needed. The port is then refilled as an in-office procedure with the help of a refill needle system that simultaneously introduces the
drug into the reservoir and removes any remaining contents. The company had publicly indicated that it planned to file the NDA for the PDS for FDA approval in one business segment. The following table summarizes our revenues by type and by geographical location. Revenue is allocated geographicallywet AMD by the locationend of 2020 and launch in the U.S. in 2021. Kodiak Sciences is developing KSI-301, an anti-VEGF antibody biopolymer conjugate being developed for treatment-naïve wet age-related macular degeneration, diabetic macular edema and retinal vein occlusion. The Phase 2b/3 DAZZLE pivotal study in patients with treatment-naïve wet AMD completed enrollment in November 2020, and Kodiak initiated the Phase 3 GLEAM, GLIMMER, and BEACON pivotal studies of KSI-301 in diabetic macular edema and retinal vein occlusion in September 2020. These studies are anticipated to form the basis of the subsidiarycompany's initial BLA to support potential approval and commercialization. Graybug Vision, Inc.’s, lead product, GB-102, is an intravitreal injectable depot formulation of sunitinib malate, an anti-VEGF TKI, that earnsblocks multiple angiogenesis pathways. Graybug Vision’s Phase 2b trial of GB-102, initiated in October 2019. Ocular Therapeutix, Inc. is developing OTX-TKI, a bioresorbable hydrogel formulated with TKI particles in an injectable fiber that can be delivered through a small-gauge, sterile injection needle to the revenue.back of the eye. OTX-TKI is designed to deliver drug to the target tissues for a period of up to nine months. Ocular Therapeutix began recruiting for a phase 1 clinical trial outside the United States in 2018 evaluating safety, durability, and tolerability in individuals with wet AMD. Completion is anticipated in November 2021. AR-13503 (Aerie Pharmaceuticals), an inhibitor of rho kinase and protein kinase C (“PKC”), is a sustained-release implant being investigated for the treatment of wet AMD and DME. Suspended in a bioerodible polymer, AR-13503 provides controlled release of its active ingredients. In a September 2020 press release, Aerie reported results of a recently completed phase 2 multicenter study of 49 patients. The implant “demonstrated positive and sustained treatment effects with both formulations as shown by increases in best corrected visual acuity and reductions in macular edema,” according to the company. | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | Year Ended June 30, | | | | | 2018 | | | 2017 | | | 2016 | | | | | U.S. | | | U. K. | | | Total | | | U.S. | | | U. K. | | | Total | | | U.S. | | | U. K. | | | Total | | | | | (In thousands) | | Revenues: | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | Collaborative research and development | | $ | 1,243 | | | $ | 100 | | | $ | 1,343 | | | $ | 6,469 | | | $ | 100 | | | $ | 6,569 | | | $ | 298 | | | $ | 100 | | | $ | 398 | | Royalty income | | | 1,618 | | | | — | | | | 1,618 | | | | 970 | | | | — | | | | 970 | | | | 1,222 | | | | — | | | | 1,222 | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | $ | 2,861 | | | $ | 100 | | | $ | 2,961 | | | $ | 7,439 | | | $ | 100 | | | $ | 7,539 | | | $ | 1,520 | | | $ | 100 | | | $ | 1,620 | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
In January 2021, Clearside Biomedical announced that the first patients was enrolled in its Phase 1/2a clinical trial of CLS-AX (axitinib injectable suspension) in patients with neovascular age-related macular degeneration (wet AMD). Clinical sites, all based in the United States, are activated and currently screening wet AMD patients for this Phase 1/2a trial, known as OASIS, involving CLS-AX, a proprietary suspension of axitinib for suprachoroidal injection.With suprachoroidal administration of axitinib, there is the potential to achieve prolonged duration and targeted delivery to affected tissue layers. REGENXBIO Inc. and Adverum Biotechnologies, Inc. are developing gene therapy treatments for wet AMD. REGENXBIO is developing RGX-314, a gene therapy utilizing its NAV AAV8 vector containing a gene encoding for a monoclonal antibody fragment which inhibits VEGF. Adverum is developing ADVM-022, a gene therapy utilizing an AAV.7m8 vector containing a gene encoding for a protein that expresses aflibercept. YUTIQ and YUTIQ50 for Posterior Segment Uveitis Periocular and intravitreal steroid injections, and systemic delivery of corticosteroids are routinely used to treat posterior segment uveitis, which is a chronic, inflammatory condition of the eye. It is treated both aggressively and frequently by physicians in order to minimize the disease “flares”, which are the main cause of vision deterioration and potential blindness. OZURDEX®, marketed by Allergan, is approved in the U.S. and EU for posterior segment uveitis through an intravitreal bioerodible implant that provides treatment which lasts for several months. This limited duration effectiveness of OZURDEX can result in frequent intravitreal injections of the implant. AbbVie, Inc. has FDA approval for HUMIRA® (adalimumab) for the treatment of all types of non-infectious uveitis (intermediate, posterior and panuveitis) and it is administered subcutaneously every other week for systemic delivery. HUMIRA is a biologic that blocks tumor necrosis factor alpha, a naturally occurring cytokine that is involved in normal inflammatory and immune responses. Humira’s retail price in the U.S. is approximately $50,000 per year. Other companies have ongoing trials of posterior segment uveitis treatments, including Santen Pharmaceutical Co. Ltd., which received a Complete Response Letter, or CRL, in December 2017 from the FDA for its filed NDA for sirolimus, which is administered through intravitreal injection every two months. Sirolimus is a mammalian target of rapamycin inhibitor and modulator of the immune system and is being developed for chronic non-infectious uveitis affecting the posterior segment of the eye. Santen has since initiated a Phase 3 clinical trial of sirolimus in December 2018 in the U.S. Clearside Biomedical Inc.’s (“Clearside”) CLS-TA (triamcinolone acetonide, a steroid) for macular edema associated with non-infectious uveitis has been accepted by the FDA for review and it is administered through a suprachoroidal injection administered every 12 weeks. Preliminary clinical data indicated that the suprachoroidal route may reduce the risk of increased IOP that is typically associated with intraocular injection of steroids. The results of the Phase 3 trial, presented in September 2018, indicated that while about 50% of patients experienced significant improvements in visual acuity through 24 weeks, adverse events of IOP increase were reported in about 12% of patients. On December 19, 2018, Clearside submitted an NDA for XIPERE™ (CLS-TA) to the U.S. FDA for the treatment of macular edema associated with uveitis. On October 18, 2019, Clearside received a CRL from the FDA regarding its NDA for XIPERE.The CRL
included the FDA’s request for additional stability data, reinspection of the drug product manufacturer and additional data on clinical use of the final to-be-marketed SCS Microinjector™ delivery system.Clearside indicated that it expects to resubmit its New Drug Application for XIPERE to FDA for review in the first quarter of 2020. On October 23, 2019, Bausch Health Companies Inc. acquired an exclusive license for the commercialization and development of XIPERE in the United States and Canada. DEXYCU for Inflammation following cataract surgery. Kala Pharmaceuticals, Inc. (“Kala”) FDA approved INVELTYS™ (loteprednol etabonate ophthalmic suspension) 1% for the topical treatment of post-operative inflammation and pain following ocular surgery. INVELTYS is the first twice-daily ocular corticosteroid approved for this indication. In addition, there are various formulations of steroids that are produced by compounding pharmacies and that are in drop form or are injected into the eye following ocular surgery. Ocular Therapeutix™ Inc.’s (“Ocular”) FDA approved DEXTENZA® (dexamethasone ophthalmic insert) 0.4 mg, which is a corticosteroid intracanalicular insert placed through the punctum, a natural opening in the eye lid, into the canaliculus, and is designed to deliver dexamethasone to the ocular surface for up to 30 days. Bausch & Lomb’s FDA approved LOTEMAX®SM (loteprednol etabonate ophthalmic gel) 0.38%, a new gel formulation for the treatment of postoperative inflammation and pain following ocular surgery. LOTEMAX SM delivers a submicron particle size for faster drug dissolution in tears. Bausch & Lomb’s FDA approved LOTEMAX®SM (loteprednol etabonate ophthalmic gel) 0.38%, a new gel formulation for the treatment of postoperative inflammation and pain following ocular surgery. LOTEMAX SM delivers a submicron particle size for faster drug dissolution in tears. EYP-1901 for DR The central retina area that is located between the main branches (superior and inferior arcades) of the central retinal vessels in the eye is known as the “macular area”. The retina beyond this is considered “peripheral retina”. The central retinal area can develop abnormal findings in DR. These findings can be present in the non-proliferative or the proliferative forms of the disease. These changes in the macula include the presence of abnormally dilated small vessel outpouchings (called microaneurysms), retinal bleeding (retinal hemorrhages) and yellow lipid and protein deposits (hard exudates). The macula can get thicker than normal- referred to as macular edema (DME). Non-proliferative retinopathy (NPDR) can be classified into mild, moderate or severe stages based upon the presence or absence of retinal bleeding, abnormal venous beading of the vessel wall (venous beading) or abnormal vascular findings (intraretinal microvascular anomalies or IRMA). No treatment is usually done at this stage. Proliferative retinopathy (PDR) is progressive and requires treatment to prevent bleeding and scar tissue formation. Macular edema is a complication of DR and is a major cause of vision loss in a diabetic eye. Treatment of macular edema is usually needed in order to prevent loss of vision or to try to improve vision. Treatment includes the use of lasers or injection of anti-VEGF drugs that cause the retinal swelling/macular edema (from leaking blood vessels) to resolve. Patients are seen monthly if being injected or every 3 months post-laser for macular edema. Several studies indicate that anti-VEGF drugs are more effective than focal laser (DRCR, READ2, RIDE, RISE, DAVINCI). A recent study by the DRCR network has shown all three drugs – Avastin (bevacizumab), Lucentis (ranibizumab) and Eylea (aflibercept) are effective for macular edema therapy. Treatment of PDR is laser photocoagulation of the peripheral retina/panretinal photocoagulation (PRP). The laser is used to create scars on the peripheral retina. If successful, vitreous bleeding may be averted. Sometimes the proliferative disease is advanced and there is bleeding filling the eye (and preventing laser to be done) or scar tissue that wrinkles the retina or pulls it off the eyewall surface. In these situations, surgery is necessary (see vitrectomy for more information). In cases of abnormal blood vessel growth anti-VEGF injections into the eye can also be used. DRCR protocol S showed that anti-VEGF drug ranibizumab was noninferior to PRP in PDR. Anti-VEGF injections are sometimes used in concert with laser when blood vessels grow in the iris and neovascular glaucoma is present. Anti-VEGF are also given prior to vitrectomy surgery in selected cases. Follow-up is crucial for these patients. Thus, in a patient who is for any reason unlikely to return for follow-up, anti-VEGF is not the treatment of choice and PRP should be done. In addition to their efficacy at treating DME, anti-VEGF drugs such as Avastin, Lucentis and Eylea, have all been shown in a number of studies to have promise for halting and reversing DR. Looking towards the future, the treatment intervals and follow-up required to maintain improvements in DR and PDR will need to be determined, but long-acting anti-VEGF agents and small
molecules, such as TKIs, formulated in novel sustained delivery methods have the potential to transform the diabetic retinopathy treatment landscape. EYO-1901 for RVO RVO can cause retinal ischemia, neovascular complications such as glaucoma, vitreous hemorrhage and retinal traction, and macular edema. Patients often present with acute visual acuity loss. They may report a history of cardiovascular risk factors including a history of diabetes mellitus and hypertension. No treatment is available to reverse the retinal vein occlusions. However, the iris or retinal neovascularization or macular edema may be managed with anti-VEGF or steroid injections. Vein occlusion can affect the central retinal vein (CRVO) or a smaller branch (BRVO). Medical therapy can limit complications from retinal vein occlusions. Anti-VEGF intraocular injections can cause regression of iris neovascularization and macular edema. In addition, the SCORE study demonstrated the benefit of triamcinolone acetonide for macular edema secondary to central retinal vein occlusions but did not demonstrate benefit for branch retinal vein occlusions (vs. focal laser). The mainstay of therapy is now anti-VEGF therapy for macular edema with either CRVO or BRVO. Both Lucentis (ranibizumab, BRAVO and CRUISE studies) and Eylea (aflibercept, GALILEO/COPERNICUS and VIBRANT studies) have been shown to be efficacious in the treatment of macular edema. Significant gains in visual acuity results and the retinal edema subsides with therapy. Both drugs are recommended to be used monthly for the 6 treatments and then as needed. Avastin (bevacizumab) is also used off-label to treat macular edema. RVO physiopathology is highly dependent on VEGF levels resulting from retina ischemia and requires frequent intravitreal injections of current therapies. Therefore, long-acting anti-VEGF agents and small molecules, such as TKIs, formulated in novel sustained delivery methods and being developed for wAMD and DR have the potential to transform the treatment landscape in this condition as well. Government Regulation We are subject to extensive regulation by the FDA and other federal, state, and local regulatory agencies. The Federal Food, Drug and Cosmetic Act or the FD(the “FD&C Act,Act”), and FDA’s implementing regulations set forth, among other things, requirements for the testing, development, manufacture, quality control, safety, effectiveness, approval, labeling, storage,record-keeping, reporting, distribution, import, export, advertising and promotion of our products and product candidates. Although the discussion below focuses on regulation in the U.S., we currentlyout-license certain of our products and may seek approval for, and market, other products in other countries in the future. Generally, our activities in other countries will be subject to regulation that is similar in nature and scope asto that imposed in the U.S., although there can be important differences. Additionally, some significant aspects of regulation in the EU are addressed in a centralized way through the European Medicines Agency, or EMA, and the European Commission, but country specificcountry-specific regulation remains essential in many respects. The process of obtaining regulatory marketing approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources and may not be successful. Development and Approval Under the FD&C Act, FDA approval of an NDA is required before any new drug can be marketed in the U.S. NDAs require extensive studies and submission of a large amount of data by the applicant. Pre-clinical Testing.Before testing any compound in human patients in the U.S., a company must generate extensivepre-clinical data.Pre-clinical testing generally includes laboratory evaluation of product chemistry and formulation, as well as toxicological and pharmacological studies in several animal species to assess the qualitytoxicity and safetydosing of the product. Certain animal studies must be performed in compliance with the FDA’s Good Laboratory Practice, or GLP, regulations and the U.S. Department of Agriculture’s Animal Welfare Act. IND Application.Human clinical trials in the U.S. cannot commence until an investigational new drug, or IND, application is submitted and becomes effective. A company must submitpre-clinical testing results to the FDA as part of the IND, and the FDA must evaluate whether there is an adequate basis for testing the drug in initial clinical studies in human volunteers. Unless the FDA raises concerns, the IND becomes effective 30 days following its receipt by the FDA.FDA, and the clinical trial proposed in the IND may begin. Once human clinical trials have commenced, the FDA may stop a clinical trial by placing it on “clinical hold” because of concerns about the safety of the product being tested, or for other reasons. Clinical Trials.Clinical trials involve the administration of a drug to healthy human volunteers or to patients under the supervision of a qualified investigator. The conduct of clinical trials is subject to extensive regulation, including compliance with the FDA’s bioresearch monitoring regulations and Good Clinical Practice, or GCP, requirements, which establish standards for conducting, recording data from, and reporting the results of, clinical trials, and are intended to assure that the data and reported results are credible and accurate, and that the rights, safety, andwell-being of study participants are protected. Clinical trials must be
conducted under protocols that detail the study objectives, parameters for monitoring safety, and the efficacy criteria, if any, to be evaluated. Each protocol is reviewed by the FDA as part of the IND. In addition, each clinical trial must be reviewed and approved by, and conducted under the auspices of, an Institutional Review Board, or IRB.IRB, for each clinical site. Companies sponsoring the clinical trials, investigators, and IRBs also must comply with, as applicable, regulations and guidelines for obtaining informed consent from the study patients, following the protocol and investigational plan, adequately monitoring the clinical trial, and timely reporting of adverse events, or AEs. Foreign studies conducted under an IND must meet the same requirements that apply to studies being conducted in the U.S. Data from a foreign study not conducted under an IND may be submitted in support of an NDA if the study was conducted in accordance with GCP and the FDA is able to validate the data. A study sponsor is required to publicly post specified details about certain clinical trials and clinical trial results on government or independent websites (e.g., http://clinicaltrials.gov). Human clinical trials typically are conducted in three sequential phases, although the phases may overlap with one another: Phase 1 clinical trials involve the initial administration of the investigational drug to humans, typically to a small group of healthy human patients, but occasionally to a group of patients with the targeted disease or disorder. Phase 1 clinical trials generally are intended to determine the metabolism and pharmacologic actions of the drug, the side effects associated with increasing doses, and, if possible, to gain early evidence of effectiveness.be combined:
Phase 2 clinical trials generally are controlled studies that involve a relatively small sample of the intended patient population and are designed to develop initial data regarding the product’s effectiveness, to determine dose response and the optimal dose range, and to gather additional information relating to safety and potential AEs.
Phase 3 clinical trials are conducted after preliminary evidence of effectiveness has been obtained and are intended to gather the additional information about safety and effectiveness necessary to evaluate the drug’s overallrisk-benefit profile, and to provide a basis for physician labeling. Generally, Phase 3 clinical development programs consist of expanded,large-scale studies of patients with the target
| • | Phase 1 clinical trials involve the initial administration of the investigational drug to humans, typically to a small group of healthy human subjects, but occasionally to a group of patients with the targeted disease or disorder. Phase 1 clinical trials generally are intended to evaluate the safety, metabolism and pharmacologic actions of the drug, the side effects associated with increasing doses, and, if possible, to gain early evidence of effectiveness. |
| • | Phase 2 clinical trials generally are controlled studies that involve a relatively small sample of the intended patient population and are designed to develop initial data regarding the product’s effectiveness, to determine dose response and the optimal dose range, and to gather additional information relating to safety and potential AEs. |
| • | Phase 3 clinical trials are conducted after preliminary evidence of effectiveness has been obtained and are intended to gather the additional information about dosage, safety and effectiveness necessary to evaluate the drug’s overall risk-benefit profile, and to provide a basis for regulatory approval. Generally, Phase 3 clinical development programs consist of expanded, large-scale studies of patients with the target disease or disorder to obtain statistical evidence of the efficacy and safety of the drug at the proposed dosing regimen. |
The sponsoring company, the FDA, or the IRB may suspend or terminate a clinical trial at any time on various grounds, including a finding that the patients are being exposed to an unacceptable health risk. Further, success inearly-stage clinical trials does not assure success inlater-stage clinical trials. Data obtained from clinical activities are not always conclusive and may be subject to alternative interpretations that could delay, limit or prevent regulatory approval. NDA Submission and Review.The FD&C Act provides two pathways for the approval of new drugs through an NDA. An NDA under Section 505(b)(1) of the FD&C Act is a comprehensive application to support approval of a product candidate that includes, among other things, data and information to demonstrate that the proposed drug is safe and effective for its proposed uses, that production methods are adequate to ensure its identity, strength, quality, and purity of the drug, and that proposed labeling is appropriate and contains all necessary information. A 505(b)(1) NDA contains results of the full set ofpre-clinical studies and clinical trials conducted by or on behalf of the applicant to characterize and evaluate the product candidate. Section 505(b)(2) of the FD&C Act provides an alternate regulatory pathway to obtain FDA approval for new formulations or new uses of previously approved drug products. Specifically, Section 505(b)(2)that permits the filing of an NDA where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference. The applicant may rely to some extent upon the FDA’s findings of safety and effectiveness for an approved product that acts as the reference listed drug, or RLD, and submit its ownproduct-specific data— data — which may include data frompre-clinical studies or clinical trials conducted by or on behalf of the applicant—applicant — to address differences between the product candidate and the RLD.reference drug. The submission of an NDA under either Section 505(b)(1) or Section 505(b)(2) generally requires payment of a substantial user fee to the FDA.FDA, subject to certain limited deferrals, waivers and reductions. The FDA reviews applications to determine, among other things, whether a product is safe and effective for its intended use and whether the manufacturing controls are adequate to assure and preserve the product’s identity, strength, quality, and purity. For some NDAs, the FDA may convene an advisory committee to seek insights and recommendations on issues relevant to approval of the application. Although the FDA is not bound by the recommendation of an advisory committee, the agency usually considers such recommendations carefully when making decisions. Our products and product candidates include products that combine drug and device components in a manner that meet the definition of a "combination product" under FDA regulations. The FDA exercises significant discretion over the regulation of combination products, including the discretion to require separate marketing applications for the drug and device components in a combination product. For YUTIQ, FDA’s Center for Drug Evaluation and Research (CDER) had primary jurisdiction for review of the NDA, and both the drug and device were reviewed under one marketing application. For a drug-device combination product for which CDER has followed such recommendations.primary jurisdiction, CDER typically consults with the Center for Devices and Radiological Health in the NDA review process. The FDA may determine that a Risk Evaluation and Mitigation Strategy or REMS,(“REMS”), is necessary to ensure that the benefits of a new product outweigh its risks, and the product can therefore be approved. A REMS may include various elements, ranging from a medication guide or patient package insert to limitations on who may prescribe or dispense the drug, depending on what the FDA
considers necessary for the safe use of the drug. Under the Pediatric Research Equity Act or PREA,(“PREA”), certain applications for approval must also include an assessment, generally based on clinical study data, of the safety and effectiveness of the subject drug in relevant pediatric populations. Before approving an NDA, the FDA will inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with current Good Manufacturing Practice, or cGMP, requirements and adequate to assure consistent production of the product within required specifications. Once the FDA accepts an NDA submission for filing—filing — which occurs, if at all, within 60 days after submission of the NDA—NDA — the FDA’s goal for anon-priority review of an NDA is ten months. The review process can be and often is significantly extended, however, by FDA requests for additional information, studies, or clarification. After review of an NDA and the facilities where the product candidate is manufactured, the FDA may decide to not approve the applicationeither issues an approval letter or may issue a complete response letter or CRL,(“CRL”), outlining the deficiencies in the submission. The CRL also may requestrequire additional testing or information, including additionalpre-clinical or clinical data.data, for the FDA to reconsider the application. Even if such additional information and data are submitted, the FDA may decide that the NDA still does not meet the standards for approval. Data from clinical trials are not always conclusive and the FDA may interpret data differently than the sponsor. FDA approval of any application may include many delays or never be granted. If FDA grants approval, an approval letter authorizes commercial marketing of the product candidate with specific prescribing information for specific indications. Obtaining regulatory approval often takes a number of years, involves the expenditure of substantial resources, and depends on a number of factors, including the severity of the disease in question, the availability of alternative treatments, and the risks and benefits demonstrated in clinical trials. Additionally, as a condition of approval, the FDA may impose restrictions that could affect the commercial success of a drug or requirepost-approval commitments, including the completion within a specified time period of additional clinical studies, which often are referred to as “Phase 4” or“post-marketing” “post-marketing” studies. Post-approval modifications to the drug, such as changes in indications, labeling, or manufacturing processes or facilities, may require a sponsor to develop additional data or conduct additionalpre-clinical studies or clinical trials, to be submitted in a new or supplemental NDA, which would require FDA approval. Post-Approval Regulation Once approved, drug products are subject to continuing regulation by the FDA. If ongoing regulatory requirements are not met, or if safety or manufacturing problems occur after the product reaches the market, the FDA may at any time withdraw product approval or take actions that would limit or suspend marketing. Additionally, the FDA may requirepost-marketing studies or clinical trials, changes to a product’s approved labeling, including the addition of new warnings and contraindications, or the implementation of other risk management measures, including distribution-related restrictions, if there are new safety information developments. Good Manufacturing Practices.Companies engaged in manufacturing drug products or their components must comply with applicable cGMP requirements andproduct-specific regulations enforced by the FDA and other regulatory agencies. Compliance with cGMP includes adhering to requirements relating to organization and training of personnel, buildings and facilities, equipment, control of components and drug product containers and closures, production and process controls, quality control and quality assurance, packaging and labeling controls, holding and distribution, laboratory controls, and records and reports. The FDA regulates and inspects equipment, facilities, and processes used in manufacturing pharmaceutical products prior to approval. If, after receiving approval, a company makes a material change in manufacturing equipment, location, or process (all of which are, to some degree, incorporated in the NDA), additional regulatory review and approval may be required. The FDA also conducts regular, periodic visits tore-inspect equipment, facilities, and processes following the initial approval of a product. Failure to comply with applicable cGMP requirements and conditions of product approval may lead the FDA to take enforcement actions or seek sanctions, including fines, issuance of warning letters, civil penalties, injunctions, suspension of manufacturing operations, operating restrictions, withdrawal of FDA approval, seizure or recall of products, and criminal prosecution. Although we periodically monitor the FDA compliance of ourthird-party manufacturers, we cannot be certain that our present or futurethird-party manufacturers will consistently comply with cGMP and other applicable FDA regulatory requirements.
We also may need to comply with some of the FDA's manufacturing and safety reporting regulations for devices. In addition to cGMP, the FDA may require that our drug-device combination products comply with the Quality System Regulation (“QSR”), which sets forth the FDA’s manufacturing quality standards for medical devices. In addition to drug safety reporting requirements, the FDA may also require that we comply with some device safety reporting requirements for our drug-device combination product. Advertising and Promotion.The FDA and other federal regulatory agencies closely regulate the marketing and promotion of drugs through, among other things, standards and regulations fordirect-to-consumer advertising, advertising and promotion to healthcare professionals, communications regarding unapproved uses,industry-sponsored scientific and educational activities, and promotional activities involving the Internet. A product cannot be commercially promoted before it is approved. After approval, product promotion can include only those claims relating to safety and effectiveness that are consistent with the labeling approved by the FDA. Healthcare providers are permitted to prescribe drugs for“off-label” uses— “off-label” uses — that is, uses not approved by the FDA and not described in the product’s labeling—labeling — because the FDA does not regulate the practice of medicine. However, FDA regulations impose restrictions on manufacturers’ communications regardingoff-label uses. Broadly speaking, a manufacturer may not promote a drug foroff-label use, but under certain conditions may engage innon-promotional, balanced, scientific communication regardingoff-label use. Failure to comply with applicable FDA requirements and restrictions in this area may subject a company to adverse publicity and enforcement action by the FDA, the Department of Justice, or the Office of the Inspector General of the Department of Health and Human Services, as well as state authorities. This could subject a company to a range of penalties that could have a significant commercial impact, including civil and criminal fines and agreements that materially restrict the manner in which a company promotes or distributes a drug. Other Requirements.NDA holders must comply with other regulatory requirements, including submitting annual reports, reporting information about adverse drug experiences, and maintaining certain records. Hatch-Waxman Act The Drug Price Competition and Patent Term Restoration Act of 1984, or theHatch-Waxman Act, establishes two abbreviated approval pathways for pharmaceutical products that are in some wayfollow-on versions of already approved products. Generic Drugs.A generic version of an approved drug is approved by means of an abbreviated NDA, or ANDA, by which the sponsor demonstrates that the proposed product is the same as the approved,brand-name drug, which is referred to as the reference listed drug, or RLD. Generally, an ANDA must contain data and information showing that the proposed generic product and RLD (i) have the same active ingredient, in the same strength and dosage form, to be delivered via the same route of administration, (ii) are intended for the same uses, and (iii) are bioequivalent. This is instead of independently demonstrating the proposed product’s safety and effectiveness, which are inferred from the fact that the product is the same as the RLD, which the FDA previously found to be safe and effective. 505(b)(2) NDAs.As discussed above, if a product is similar, but not identical, to an already approved product, itpreviously, products may also be submitted for approval via an NDA under section 505(b)(2) of the FD&C Act. Unlike an ANDA, this does not excuse the sponsor from demonstrating the proposed product’s safety and effectiveness. Rather, the sponsor is permitted to rely to some degree on information from investigations that were not conducted by or for the FDA’s finding thatapplicant and for which the RLD is safe and effective,applicant has not obtained a right of reference, and must submit its ownproduct-specific data of safety and effectiveness to an extent necessary because of the differences between the products. An NDA approved under 505(b)(2) may in turn serve as an RLD for subsequent applications from other sponsors.
RLD Patents.In an NDA, a sponsor must identify patents that claim the drug substance or drug product or a method of using the drug. When the drug is approved, those patents are among the information about the product that is listed in the FDA publication,Approved Drug Products with Therapeutic Equivalence Evaluations, which is referred to as theOrange Book. The sponsor of an ANDA or 505(b)(2) application seeking to rely on an approved product as the RLD must make one of several certifications regarding each listed patent. A “Paragraph I” certification is the sponsor’s statement that patent information has not been filed for the RLD. A “Paragraph II” certification is the sponsor’s statement that the RLD’s patents have expired. A “Paragraph III” certification is the sponsor’s statement that it will wait for the patent to expire before obtaining approval for its product. A “Paragraph IV” certification is an assertion that the patent does not block approval of the later product, either because the patent is invalid or unenforceable or because the patent, even if valid, is not infringed by the new product. Regulatory Exclusivities.TheHatch-Waxman Act provides periods of regulatory exclusivity for products that would serve as RLDs for an ANDA or 505(b)(2) application. If a product is a “new chemical entity,” or NCE—NCE — generally meaning that the active moiety has never before been approved in any drug—drug — there is a period of five years from the product’s approval during which the FDA may not accept for filing any ANDA or 505(b)(2) application for a drug with the same active moiety. An ANDA or 505(b)(2) application may be submitted after four years, however, if the sponsor of the application makes a Paragraph IV certification. A product that is not an NCE may qualify for athree-year period of exclusivity if the NDA contains new clinical data (other than bioavailability studies), derived from studies conducted by or for the sponsor, that were necessary for approval. In that instance, the exclusivity period does not preclude filing or review of an ANDA or 505(b)(2) application; rather, the FDA is precluded from granting final approval to the ANDA or 505(b)(2) application until three years after approval of the RLD. Additionally, the exclusivity applies only to the conditions of approval that required submission of the clinical data.
Once the FDA accepts for filing an ANDA or 505(b)(2) application containing a Paragraph IV certification, the applicant must within 20 days provide notice to the RLD NDA holder and patent owner that the application has been submitted and provide the factual and legal basis for the applicant’s assertion that the patent is invalid or not infringed. If the NDA holder or patent owner files suit against the ANDA or 505(b)(2) applicant for patent infringement within 45 days of receiving the Paragraph IV notice, the FDA is prohibited from approving the ANDA or 505(b)(2) application for a period of 30 months or the resolution of the underlying suit, whichever is earlier. If the RLD has NCE exclusivity and the notice is given and suit filed during the fifth year of exclusivity, the30-month regulatory stay does not begin until fiveextends to 7.5 years after the RLD approval. The FDA may approve the proposed product before the expiration of the30-month regulatory stay if a court finds the patent invalid or not infringed or if the court shortens the period because the parties have failed to cooperate in expediting the litigation. Patent Term Restoration.A portion of the patent term lost during product development and FDA review of an NDA is restored if approval of the application is the first permitted commercial marketing of a drug containing the active ingredient. The patent term restoration period is generallyone-half the time between the effective date of the IND or the date of patent grant (whichever is later) and the date of submission of the NDA, plus the time between the date of submission of the NDA and the date of FDA approval of the product. The maximum period of restoration is five years, and the patent cannot be extended to more than 14 years from the date of FDA approval of the product. Only one patent claiming each approved product is eligible for restoration and the patent holder must apply for restoration within 60 days of approval. The USPTO, in consultation with the FDA, reviews and approves the application for patent term restoration. European and Other International Government Regulation In addition to regulations in the U.S., we are subject to a variety of regulations in other jurisdictions governing, among other things, clinical trials and any commercial sales and distribution of our products. Whether or not we obtain FDA approval for a product, we must obtain the requisite approvals from regulatory authorities in foreign countries prior to the commencement of clinical trials or marketing of the product in those countries. Some countries outside of the U.S. have a similar process that requires the submission of a clinical trial application, or CTA, much like the IND prior to the commencement of human clinical trials. In the EU, for example, similar to the FDA a CTA must be submitted for authorization to the competent national authority of each EU Member State in which the clinical trial is to be conducted. Furthermore, the applicant may only start a clinical trial at a specific study site after the competent ethics committee, much like the IRB, has issued a favorable opinion. Once the CTA is approved in accordance with the EU Clinical Trials Directive 2001/20/EC and the related national implementing provisions of the relevant individual EU Member States’ requirements, clinical trial development may proceed. In April 2014, the new Clinical Trials Regulation, (EU) No 536/2014, or Clinical Trials Regulation, was adopted. The Regulation is anticipated to enter into force in 2020.2021 or 2022. The Clinical Trials Regulation will be directly applicable in all the EU Member States, repealing the current Clinical Trials Directive 2001/20/EC. Conduct of all clinical trials performed in the EU will continue to be bound by currently applicable provisions until the new Clinical Trials Regulation becomes applicable. The extent to whichon-going clinical trials will be governed by the Clinical Trials Regulation will depend on when the Clinical Trials Regulation becomes applicable and on the duration of the individual clinical trial. If a clinical trial continues for more than three years from the day on which the Clinical Trials Regulation becomes applicable the Clinical Trials Regulation will at that time begin to apply to the clinical trial. The new Clinical Trials Regulation is intended to simplify and streamline the approval of clinical trials in the EU. The main characteristics of the regulation include: a streamlined application procedure through a single entry point, the “EU portal”; a single set of documents to be prepared and submitted for the application as well as simplified reporting procedures for clinical trial sponsors; and a harmonized procedure for the assessment of applications for clinical trials, which is divided in two parts. To obtain regulatory approval to commercialize a new drug under EU regulatory systems, we must submit a marketing authorization application, or MAA, to the competent regulatory authority. In the EU, marketing authorization for a medicinal product can be obtained through a centralized, mutual recognition, decentralized procedure, or the national procedure of an individual EU Member State. A marketing authorization, irrespective of its route to authorization, may be granted only to an applicant established in the EU. The centralized procedure provides for the grant of a single marketing authorization by the European Commission that is valid for all 2827 EU Member States and three of the four European Free Trade Association or EFTA States, Iceland, Liechtenstein and Norway. Under the centralized procedure, the Committee for Medicinal Products for Human Use, or the CHMP, established at the EMA is responsible for conducting the initial assessment of a product. The maximum timeframe for the evaluation of an MAA is 210 days. This period excludes clock stops during which additional information or written or oral explanation is to be provided by the applicant in response to questions posed by the CHMP. Accelerated evaluation might be granted by the CHMP in exceptional cases, when a medicinal product is expected to be of a major public health interest. A major public health interest defined by three cumulative criteria: (i) the seriousness of the disease (for example, heavy disabling or life-threatening diseases) to be treated, (ii) the absence or insufficiency of an appropriate alternative therapeutic approach, and (iii) anticipation of high therapeutic benefit. If the CHMP accepts to review a medicinal product as a major public health interest, the time limit of 210 days will be reduced to 150 days. It is, however, possible that the CHMP can revert to the standard time limit for the centralized procedure if it considers that it is no longer appropriate to conduct an accelerated assessment.
Irrespective of the related procedure, at the completion of the review period the CHMP will provide a scientific opinion concerning whether or not a marketing authorization should be granted in relation to a medicinal product. This opinion is based on a review of the quality, safety, and efficacy of the product. Within 15 days of the adoption, the EMA will forward its opinion to the European Commission for its decision. Following the opinion of the EMA, the European Commission makes a final decision to grant a centralized marketing authorization. The centralized procedure is mandatory for certain types of medicinal products, including orphan medicinal products, medicinal products derived from certain biotechnological processes, advanced therapy medicinal products and medicinal products containing a new active substance for the treatment of certain diseases. This route is optional for certain other products, including medicinal products that are of significant therapeutic, scientific or technical innovation, or whose authorization would be in the interest of public or animal health.health at EU level. Unlike the centralized authorization procedure, the decentralized marketing authorization procedure requires a separate application to, and leads to separate approval by, the competent authorities of each EU Member State in which the product is to be marketed. This application process is identical to the application that would be submitted to the EMA for authorization through the centralized procedure.procedure and must be completed within 210 days, excluding potential clock-stops, during which the applicant can respond to questions. The reference EU Member State prepares a draft assessment and drafts of the related materials within 120 days after receipt of a valid application.materials. The resulting assessment report is submitted to the concerned EU Member States who, within 90 days of receipt, must decide whether to approve the assessment report and related materials. If a concerned EU Member State cannot approve the assessment report and related materials due to concerns relating to a potential serious risk to public health, disputed elements may be referred to the European Commission, whose decision is binding on all EU Member States. The mutual recognition procedure is similarly based on the acceptance by the competent authorities of the EU Member States of the marketing authorization of a medicinal product by the competent authorities of other EU Member States. The holder of a national marketing authorization may submit an application to the competent authority of an EU Member State requesting that this authority recognize the marketing authorization delivered by the competent authority of another EU Member State. Marketing authorization holders are subject to comprehensive regulatory oversight by the EMA and the competent authorities of the individual EU Member States both before and after grant of marketing authorization. This includes control of compliance by the entities with EU cGMP rules, which govern quality control of the manufacturing process and require documentation policies and procedures. For other countries outside of the EU, such as countries in Eastern Europe, Latin America or Asia, the requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. Internationally, clinical trials are generally required to be conducted in accordance with GCP, applicable regulatory requirements of each jurisdiction and the medical ethics principles that have their origin in the Declaration of Helsinki. Compliance During all phases of development(pre- andpost-marketing), in the post-market setting, failure to comply with applicable regulatory requirements may result in administrative or judicial sanctions. These sanctions could include the FDA’s imposition of a clinical hold on trials, refusal to approve pending applications, withdrawal of an approval, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, product detention or refusal to permit the import or export of products, injunctions, fines, civil penalties or criminal prosecution. Third country authorities can impose equivalent penalties. Any agency or judicial enforcement action could have a material adverse effect on us. Other Exclusivities Pediatric Exclusivity.Section 505A of the FD&C Act provides for six months of additional exclusivity or patent protection if an NDA sponsor submits pediatric data that fairly respond to a written request from the FDA for such data. The data does not need to show that the product is effective in the pediatric population studied; rather, if the clinical trial is deemed to fairly respond to the FDA’s request, the additional protection is granted. If reports of requested pediatric studies are submitted to and accepted by FDA within the statutory time limits, whatever statutory or regulatory periods of exclusivity orOrange Book listed patent protection that cover the drug are extended by six months. This is not a patent term extension, but it effectively extends the regulatory period during which the FDA cannot approve an ANDA or 505(b)(2) application owing to regulatory exclusivity or listed patents. When any product is approved, we will evaluate seeking pediatric exclusivity as appropriate. In the EU, Regulation No 1901/2006, or the Pediatric Regulation, requires that prior to obtaining a marketing authorization in the EU, applicants demonstrate compliance with all measures included in an European Medicines Agency, or EMA, approved Pediatric Investigation Plan, or PIP. This PIP covers all subsets in a pediatric population, unless the EMA has granted either, a product-specific waiver, a class waiver, or a deferral for one or more of the measures included in the PIP. Where all measures provided in the agreed PIP are completed, a six monthssix-month extension period of qualifying Supplementary Protection Certificates or SPCs, is granted.
Orphan Drug Exclusivity.The Orphan Drug Act provides incentives for the development of drugs intended to treat rare diseases or conditions, which generally are diseases or conditions affecting less than 200,000 individuals in the U.S., or a disease or condition affecting
more than 200,000 individuals in the U.S. but there is no reasonable expectation that the cost of developing and making the drug product would be recovered from sales in the U.S. If a sponsor demonstrates that a drug is intended to treat a rare disease or condition,product qualifies for orphan drug designation, the FDA may grant orphan drug designation to the product for that use. The benefits of orphan drug designation include research and development tax credits and exemption from user fees. A drug that is approved for the orphan drug designated indication generally is granted seven years of orphan drug exclusivity. During that period, the FDA generally may not approve any other application for the same product for the same indication, although there are exceptions, most notably when the later product is shown to be clinically superior to the product with exclusivity. The FDA can revoke a product’s orphan drug exclusivity under certain circumstances, including when the product sponsor is unable to assure the availability of sufficient quantities of the product to meet patient needs. Orphan drug exclusivity does not prevent the FDA from approving a different drug for the same disease or condition, or the same biologic for a different disease or condition. In the EU, medicinal products: (a) that are used to diagnose, treat or prevent life-threatening or chronically debilitating conditions that affect no more than five in 10,000 people in the EU; or (b) that are used to treat or prevent life-threatening or chronically debilitating conditions and that, for economic reasons, would be unlikely to be developed without incentives; and (c) where no satisfactory method of diagnosis, prevention or treatment of the condition concerned exists, or, if such a method exists, the medicinal product would be of significant benefit to those affected by the condition, may be granted an orphan designation in the EU. The application for orphan designation must be submitted to the EMA’s Committee for Orphan Medicinal Products and approved by the European Commission before an application is made for marketing authorization for the product. Once authorized, orphan medicinal product designation entitles an applicant to financial incentives such as reduction of fees or fee waivers. In addition, orphan medicinal products are entitled to ten years of market exclusivity following authorization. During thisten-year period, with a limited number of exceptions, neither the competent authorities of the EU Member States, the EMA, or the European Commission are permitted to accept applications or grant marketing authorization for other similar medicinal products with the same therapeutic indication. However, marketing authorization may be granted to a similar medicinal product with the same orphan indication during theten-year period with the consent of the marketing authorization holder for the original orphan medicinal product or if the manufacturer of the original orphan medicinal product is unable to supply sufficient quantities. Marketing authorization may also be granted to a similar medicinal product with the same orphan indication if this latter product is safer, more effective or otherwise clinically superior to the original orphan medicinal product. The period of market exclusivity may, in addition, be reduced to six years if it can be demonstrated on the basis of available evidence that the original orphan medicinal product is sufficiently profitable not to justify maintenance of market exclusivity Data Exclusivity. In the EU, if a marketing authorization is granted for a medicinal product containing a new active substance, that product benefits from eight years of data exclusivity, during which generic marketing authorization applications referring to the data of that product may not be accepted by the regulatory authorities. The product also benefits from 10 years’ market exclusivity during which generic products, even if authorized, may not be placed on the market. The overallten-year period will be extended to a maximum of 11 years if, during the first eight years of those ten years, the marketing authorization holder obtains an authorization for one or more new therapeutic indications which, during the scientific evaluation prior to their authorization, are held to bring a significant clinical benefit in comparison with existing therapies. U.S. Healthcare Reform The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, which we refer to together as the Affordable Care Act, or ACA, is a sweeping measure intended to expand healthcare coverage within the U.S., primarily through the imposition of health insurance mandates on employers and individuals, the provision of subsidies to eligible individuals enrolled in plans offered on the health insurance exchanges, and expansion of the Medicaid program. This law substantially changeschanged the way healthcare is financed by both governmental and private insurers and has significantly impactsimpacted the pharmaceutical industry. Changes that may affect our business include those governing enrollment in federal healthcare programs, reimbursement changes, benefits for patients within a coverage gap in the Medicare Part D prescription drug program (commonly known as the “donut hole”), rules regarding prescription drug benefits under the health insurance exchanges, changes to the Medicaid Drug Rebate program, expansion of the Public Health Service’sService Act’s 340B drug pricing discount program, or 340B program, fraud and abuse, and enforcement. These changes have impacted and will continue to impact existing government healthcare programs and are resultinghave resulted in the development of new programs, including Medicare payment for performance initiatives and improvements to the physician quality reporting system and feedback program.initiatives. Some states have elected not to expand their Medicaid programs to individuals with an income of up to 133% of the federal poverty level, as is permitted under the Affordable Care Act. For each state that does not choose to expand its Medicaid program, there may be fewer insured patients overall, which could impact our sales of products and product candidates for which we receive regulatory approval, and our business and financial condition. Where new patients receive insurance coverage under any of the new Medicaid options made available through the Affordable Care Act, the possibility exists that manufacturers may be required to pay Medicaid rebates on drugs used under these circumstances, a decision that could impact manufacturer revenues. In addition, the federal government has announced delays in the implementation of key Certain provisions of the Affordable Care Act. Moreover, legislative changesAct have been subject to judicial challenges as well as efforts to repeal, replace, or regulatory changes under the Affordable Care Act remain possible in the 115th U.S. Congressotherwise modify them or to alter their interpretation and under the Trump Administration.implementation. For example, on December 22, 2017, the U.S.
government signed into law comprehensive tax legislation, referred to as the Tax Cuts and Jobs Act, or the Tax Act, which includes a provision repealing, effective January 1, 2019, thetax-based shared responsibility payment imposed by the Affordable Care Act on certain
individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate.” Further, the Bipartisan Budget Act of 2018, among other things, amended the Medicare statute to reduce the coverage gap in most Medicare drugs plans, commonly known as the “donut hole,” by raising the required manufacturer point-of-sale discount from 50% to 70% off the negotiated price effective as of January 1, 2019. Additional legislative changes, to and regulatory changes, underand judicial challenges related to the Affordable Care Act remain possible. We expectpossible, but the nature and extent of such potential changes or challenges are uncertain at this time. It is unclear how the Affordable Care Act and its implementation, as well as efforts to repeal, replace, or otherwise modify, or invalidate, the Affordable Care Act, or portions thereof, will affect our business, financial condition and results of operations. It is possible that the Affordable Care Act, as currently enacted or as it may be amended or replaced in the future, and other healthcare reform measures that may be adopted in the future could have a material adverse effect on our industry generally and on our ability to maintain or increase sales of our products or product candidates for which we receive regulatory approval or to successfully commercialize our products and product candidates, if approved.candidates. Coverage and Reimbursement Significant uncertainty exists as to the coverage and reimbursement status of any products for which we may obtain regulatory approval. Sales of any of our products and product candidates, if approved, will depend, in part, on the extent to which the costs of the products will be covered bythird-party payors, including government healthcare programs such as Medicare and Medicaid, and private payors, such as commercial health insurers and managed care organizations.Third-party payors determine which drugs they will cover and the amount of reimbursement they will provide for a covered drug. In the U.S., there is no uniform system among payors for making coverage and reimbursement decisions. In addition, the process for determining whether a payor will provide coverage for a product may be separate from the process for setting the price or reimbursement rate that the payor will pay for the product once coverage is approved.Third-party payors Payors may limit coverage to specific products on an approved list, or formulary, which might not include all of theFDA-approved products for a particular indication.
In order to secure coverage and reimbursement for our products, if approved for sale, we may need to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity andcost-effectiveness of the product, in addition to the costly studies required to obtain FDA or other comparable regulatory approvals. Even if we conduct pharmacoeconomic studies, our product candidates may not be considered medically necessary orcost-effective by payors. Further, a payor’s decision to provide coverage for a product does not imply that an adequate reimbursement rate will be approved.approved because HCPs negotiate their own reimbursement directly with commercial payors. In the past, payors have implemented reimbursement metrics and periodically revised those metrics as well as the methodologies used as the basis for reimbursement rates, such as average sales price, or ASP, average manufacturer price, or AMP, and actual acquisition cost. The existing data for reimbursement based on these metrics is relatively limited, although certain states have begun to survey acquisition cost data for the purpose of setting Medicaid reimbursement rates. The CMS surveys and publishes retail pharmacy acquisition cost information in the form of National Average Drug Acquisition Cost files to provide state Medicaid agencies with a basis of comparison for their own reimbursement and pricing methodologies and rates. We intend to participate in, and have certain price reporting obligations to, the Medicaid Drug Rebate program.Program. This program would requirerequires us to pay a rebate for each unit of drug reimbursed by Medicaid. The amount of the “basic” portion of the rebate for each product is set by law as the larger of: (i) 23.1% of quarterly AMP, or (ii) the difference between quarterly AMP and the quarterly best price available from us to any commercial ornon-governmental customer, or Best Price. AMP must be reported on a monthly and quarterly basis and Best Price is reported on a quarterly basis only. In addition, the rebate also includes the “additional” portion, which adjusts the overall rebate amount upward as an “inflation penalty” when the drug’s latest quarter’s AMP exceeds the drug’s AMP from the first full quarter of sales after launch, adjusted for increases in the Consumer PriceIndex-Urban. The upward adjustment in the rebate amount per unit is equal to the excess amount of the current AMP over theinflation-adjusted AMP from the first full quarter of sales. The rebate amount is recomputedcomputed each quarter based on our report to CMS of current quarterly AMP and Best Price for our drug. The terms of our participation in the program would impose a requirement for usWe are required to report revisions to AMP or Best Price within a period not to exceed 12 quarters from the quarter in which the data was originally due. Any such revisions could have the impact of increasing or decreasing our rebate liability for prior quarters, depending on the direction of the revision. The Affordable Care Act made significant changes to the Medicaid Drug Rebate Program, and CMS issued a final regulation, which became effective on April 1, 2016, to implement the changes to the Medicaid Drug Rebate program under the Affordable Care Act. On December 21, 2020, CMS issued a final regulation that modified existing Medicaid Drug Rebate Program regulations to permit reporting multiple Best Price figures with regard to value‑based purchasing arrangements (beginning in 2022); provide definitions for “line extension,” “new formulation,” and related terms with the practical effect of expanding the scope of drugs considered to be line extensions (beginning in 2022); and revise AMP and Best Price exclusions of manufacturer-sponsored patient benefit programs, specifically regarding inapplicability of such exclusions in the context of pharmacy benefit manager “accumulator” programs (beginning in 2023). It is currently unclear if the Biden administration will delay the effectiveness or take other actions with respect to this regulation. Federal law requires that any manufacturer that participates in the Medicaid Drug Rebate programProgram also participate in the Public Health Service’s 340B drug pricing program in order for federal funds to be available for the manufacturer’s drugs under Medicaid and Medicare Part B. The 340B program, which is administered by the Health Resources and Services Administration, or HRSA, requires participating manufacturers to agree to charge statutorily defined covered entities no more than the 340B “ceiling price” for
the manufacturer’s covered outpatient drugs. These 340B covered entities include a variety of community health clinics and other entities that receive health services grants from the Public Health Service, as well as hospitals that serve a disproportionate share oflow-income patients. The 340B ceiling price is calculated using a statutory formula, which is based on the AMP and rebate amount for the covered outpatient drug as calculated under the Medicaid Drug Rebate program.Program. Any changes to the definition of AMP and the Medicaid rebate amount under the Affordable Care Act or other legislation could affect our 340B ceiling price calculations and negatively impact our results of operations. HRSA issued a final regulation regarding the calculation of the 340B ceiling price and the imposition of civil monetary penalties on manufacturers that knowingly and intentionally overcharge covered entities, which became effective on January 1, 2019. It is currently unclear how HRSA will apply its enforcement authority under this regulation. HRSA has also implemented a ceiling price reporting requirement related to the 340B program under which we are required to report 340B ceiling prices to HRSA on a quarterly basis, and HRSA then publishes that information to covered entities. Moreover, under a final regulation effective January 13, 2021, HRSA newly established an administrative dispute resolution (“ADR”), process for claims by covered entities that a manufacturer has engaged in overcharging, and by manufacturers that a covered entity violated the prohibitions against diversion or duplicate discounts. Such claims are to be resolved through an ADR panel of government officials rendering a decision that could be appealed only in federal court. An ADR proceeding could subject us to onerous procedural requirements and could result in additional liability. In addition, legislation may be introduced that, if passed, would further expand the 340B program to additional covered entities or would require participating manufacturers to agree to provide 340B discounted pricing on drugs used in an inpatient setting. Federal law also requires that a company that participates in the Medicaid Drug Rebate program report ASP information each quarter to CMS for certain categories of drugs that are paid under the Medicare Part B program. Manufacturers calculate the ASP based on a statutorily defined formula as well as regulations and interpretations of the statute by CMS. CMS uses these submissions to determine payment rates for drugs under Medicare Part B. In a November 20, 2020 interim final rule, CMS established a “Most Favored Nation” demonstration model that would lower Medicare Part B reimbursement of certain drugs based on international reference prices. The rule has become subject to judicial challenges, and federal courts have enjoined the rule at this time. There is also proposed legislation pending that would establish an international reference price-based payment methodology. For more information about Medicare Part B, refer to the risk factor entitled “Our products may become subject to unfavorable pricing regulations, third-party reimbursement practices or healthcare reform initiatives, which could harm our business” set forth under the section titled “Risk Factors” in this Annual Report on Form 10-K. In the U.S. Medicare program, outpatient prescription drugs may be covered under Medicare Part D. Medicare Part D is a voluntary prescription drug benefit, through which Medicare beneficiaries may enroll in prescription drug plans offered by private entities for coverage of outpatient prescription drugs. Part D plans include bothstand-alone prescription drug benefit plans and prescription drug coverage as a supplement to Medicare Advantage plans provided for under Medicare Part C. Coverage and reimbursement for covered outpatient drugs under Part D are not standardized. Part D prescription drug plan sponsors are not required to pay for all covered Part D drugs, and each drug plan can develop its own drug formulary that identifies which drugs it will cover and at what tier or level. Any formulary used by a Part D prescription drug plan must be developed and reviewed by a pharmacy and therapeutic committee. Although Part D prescription drug formularies must include drugs within each therapeutic category and class of covered Part D drugs, they have some flexibility to establish those categories and classes and are not required to cover all of the drugs in each category or class. Medicare Part D prescription drug plans may use formularies to limit the number of drugs that will be covered in any therapeutic class and/or impose differential cost sharing or other utilization management techniques. The availability of coverage under Medicare Part D coverage is available for our products and may increase demandbe available for productsany future product candidates for which we receive marketing approval. However, in order for the products that we market to be included on the formularies of Part D prescription drug plans, we likely will have to offer pricing that is lower than the prices we might otherwise obtain. Changes to Medicare Part D that give plans more freedom to limit coverage or manage utilization, and other cost reduction initiatives in the program, could decrease the coverage and price that we receive for any approved products and could seriously harm our business.
In order to be eligible to have our products paid for with federal funds under the Medicaid and Medicare Part B programs and purchased by certain federal agencies and grantees, we expect tomust participate in the U.S. Department of Veterans Affairs, or VA,(“VA”), Federal Supply Schedule, or FSS,(“FSS”), pricing program. Under this program, we would beare obligated to make our “innovator” drugs available for procurement on an FSS contract and charge a price to four federal agencies—agencies — the VA, U.S. Department of Defense, or DoD,(“DoD”), Public Health Service and U.S. Coast Guard—Guard — that is no higher than the statutory Federal Ceiling Price, or FCP.(“FCP”). The FCP is based on thenon-federal average manufacturer price, orNon-FAMP,(“Non-FAMP”), which we calculate and report to the VA on a quarterly and annual basis. We also expect tomay participate in the Tricare Retail Pharmacy program, under which we would pay quarterly rebates on utilization of innovator products that are dispensed through the Tricare Retail Pharmacy network to Tricare beneficiaries. The rebates are calculated as the difference between the annualNon-FAMP and FCP. Federal law also requires that a company that participates in the Medicaid Drug Rebate program report ASP information each quarter to CMS for certain categories of drugs that are paid under the Medicare Part B program. Manufacturers calculate the average sales price based on a statutorily defined formula as well as regulations and interpretations of the statute by CMS. CMS uses these submissions to determine payment rates for drugs under Medicare Part B. For more information about Medicare Part B, refer to the risk factor entitled “Even if we are
able to commercialize our products, the products may become subject to unfavorable pricing regulations, third-party reimbursement practices or healthcare reform initiatives, which could harm our business” set forth under the section titled “Risk Factors” in this Annual Report on Form 10-K.
Pricing and rebate calculations vary across products and programs, are complex, and are often subject to interpretation by manufacturers,us, governmental or regulatory agencies, and the courts. Civil monetary penalties canWe could be appliedheld liable for errors associated with our submission of pricing data. In addition to retroactive Medicaid rebates and the potential for issuing 340B program refunds, if a manufacturer iswe are found to have knowingly submitted any false priceAMP, Best Price, or Non-FAMP information to the government, or failswe may be liable for significant civil monetary penalties per item of false information. If we are found to have made a misrepresentation in the reporting of our ASP, the Medicare statute provides for significant civil monetary penalties for each misrepresentation for each day in which the misrepresentation was applied. Our failure to submit the required pricemonthly/quarterly AMP and Best Price data on a timely basis.basis could result in a significant civil monetary penalty per day for each day the information is late beyond the due date . Such conduct also could be grounds for CMS to terminate the manufacturer’sour Medicaid drug rebate agreement, in which case federal payments may not be available under Medicaid or Medicare Part B for the manufacturer’sour covered outpatient drugs. Significant civil monetary penalties also could apply to late submissions of Non-FAMP information. Civil monetary penalties could also be applied if we are found to have charged 340B covered entities more than the statutorily mandated ceiling price. In addition, claims submitted tofederally-funded healthcare programs, such as Medicare and Medicaid, for drugs priced based on incorrect pricing data provided by a manufacturer can implicate the federal civil False Claims Act. The containment of healthcare costs has become a priority of federal, state and foreign governments, and the prices of drugs have been a focus in this effort. The U.S. government, state legislatures, and foreign governments have shown significant interest in implementingcost-containment programs to limit the growth ofgovernment-paid healthcare costs, including price controls, restrictions on reimbursement, and requirements for substitution of generic products for branded prescription drugs. For example, there have been several recent U.S. Congressional inquiries and proposed federal and state legislation designed to, among other things, bring more transparency to drug pricing, review the relationship between pricing and manufacturer patient programs, reduce the cost of drugs, and reform government program reimbursement methodologies for drug products. Beginning April 1, 2013, Medicare payments for all items and services, including drugs, were reduced by 2% under the sequestration (i.e., automatic spending reductions) required by the Budget Control Act of 2011, as amended by the American Taxpayer Relief Act of 2012. Subsequent legislation extended the 2% reduction, on average,generally to 2027.2030, with a pause during the Pandemic. If Congress does not take action in the future to modify these sequestrations, Medicare Part D plans could seek to reduce their negotiated prices for drugs. Other legislative or regulatory cost containment legislation could have a similar effect. Further, the Affordable Care Act may reduce the profitability of drug products. It expanded manufacturers’ rebate liability under the Medicaid program fromfee-for-service Medicaid utilization to include the utilization of Medicaid managed care organizations as well, and increased the minimum Medicaid rebate due for most innovator drugs, and capped the total rebate amount for innovator drugs at 100% of AMP.drugs. The Affordable Care Act and subsequent legislation also changed the definition of AMP. On February 1, 2016, CMS issued final regulations to implement the changes to the Medicaid drug rebate program under the Affordable Care Act. These regulations became effective on April 1, 2016. The Affordable Care Act requires pharmaceutical manufacturers of branded prescription drugs to pay a branded prescription drug fee to the federal government. Each such manufacturer pays a prorated share of the branded prescription drug fee of $4.1$2.8 billion in 2018,2019 and thereafter, based on the dollar value of its branded prescription drug sales to certain federal programs identified in the law. The Affordable Care Act also expanded the Public Health Service’sService Act’s 340B program to include additional types of covered entities. Substantial new provisions affecting compliance have also been enacted, which may affect our business practices with healthcare practitioners. It appears likely that the Affordable Care Act will continue the pressure on pharmaceutical pricing, especially under the Medicare and Medicaid programs, and may also increase our regulatory burdens and operating costs. LegislativeAdditional legislative changes, to and regulatory changes, underand judicial challenges related to the Affordable Care Act remain possible, in the 115th U.S. Congress and under the Trump Administration, as discussed above under the heading “U.S. Healthcare Reform.” In addition, there likely will continue to be proposals by legislators at both the federal and state levels, regulators, andthird-party payors to contain healthcare costs. Thus, even if we obtain favorable coverage and reimbursement status for our products and any productsproduct candidates for which we receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
Different pricing and reimbursement schemes exist in other countries. In the EU, each EU Member State can restrict the range of medicinal products for which its national health insurance system provides reimbursement and can control the prices of medicinal products for human use marketed on its territory. As a result, following receipt of marketing authorization in an EU Member State, through any application route, the applicant is required to engage in pricing discussions and negotiations with the competent pricing authority in the individual EU Member State. The governments of the EU Member States influence the price of pharmaceutical products through their pricing and reimbursement rules and control of national healthcare systems that fund a large part of the cost of those products to consumers. Some EU Member States operate positive and negative list systems under which products may only be marketed once a reimbursement price has been agreed upon. To obtain reimbursement or pricing approval, some of these countries may require the completion of clinical trials that compare thecost-effectiveness of a particular product candidate to currently available therapies. Other EU Member States allow companies to fix their own prices for medicines, but monitor and control company profits. Others adopt a system of reference pricing, basing the price or reimbursement level in their territories either on the pricing and reimbursement levels in other countries or on the pricing and reimbursement levels of medicinal products intended for the same
therapeutic indication. Further, some EU Member States approve a specific price for the medicinal product or may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. The downward pressure on healthcare costs in general, particularly prescription drugs, has become more intense. As a result, increasingly high barriers are being erected to the entry of new products. In addition, we may face competition for our product candidates fromlower-priced products in foreign countries that have placed price controls on pharmaceutical products. In addition, in some countries,cross-border imports fromlow-priced markets exert a commercial pressure on pricing within a country. Health Technology Assessment, or HTA, of medicinal products, however, is becoming an increasingly common part of the pricing and reimbursement procedures in some EU Member States. These EU Member States include the U.K., France, Germany, Ireland, Italy and Sweden. HTA is the procedure according to which the assessment of the public health impact, therapeutic impact and the economic and societal impact of use of a given medicinal product in the national healthcare systems of the individual country is conducted. HTA generally focuses on the clinical efficacy and effectiveness, safety, cost, and cost-effectiveness of individual medicinal products as well as their potential implications for the healthcare system. Those elements of medicinal products are compared with other treatment options available on the market. The outcome of HTA regarding specific medicinal products will often influence the pricing and reimbursement status granted to these medicinal products by the competent authorities of individual EU Member States. The extent to which pricing and reimbursement decisions are influenced by the HTA of the specific medicinal product varies between EU Member States. In addition, pursuant to Directive 2011/24/EU on the application of patients’ rights in cross-border healthcare, a voluntary network of national authorities or bodies responsible for HTA in the individual EU Member States was established. The purpose of the network is to facilitate and support the exchange of scientific information concerning HTAs. This may lead to harmonization of the criteria taken into account in the conduct of HTAs between EU Member States and in pricing and reimbursement decisions and may negatively affect price in at least some EU Member States. Healthcare Fraud and Abuse Laws In addition to FDA restrictions on marketing of pharmaceutical products, our business is subject to healthcare fraud and abuse regulation and enforcement by both the federal government and the states in which we conduct our business. These laws include, but are not limited to the following: The federalAnti-Kickback Statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving remuneration, directly or indirectly, in cash or in kind, to induce or in return for purchasing, leasing, ordering or arranging for or recommending the purchase, lease or order of any healthcare item or service reimbursable, in whole or in part, under Medicare, Medicaid or other federally financed healthcare programs. This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on one hand and prescribers, purchasers, and formulary managers on the other. A violation of theAnti-Kickback Statute may be established without proving actual knowledge of the statute or specific intent to violate it. The Affordable Care Act amended federal law to provide that the government may assert that a claim including items or services resulting from a violation of the federalAnti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act. Although there are a number of statutory exceptions and regulatory safe harbors protecting certain common activities from prosecution, the exceptions and safe harbors are drawn narrowly and practices that involve remuneration to those who prescribe, purchase, or recommend pharmaceuticals, including certain discounts, or engaging such individuals as consultants, speakers or advisors, may be subject to scrutiny if they do not fit squarely within the exception or safe harbor. In November 2020, the U.S. Department of Health and Human Services finalized a previously abandoned proposal to amend the discount safe harbor regulation of the Anti-Kickback Statute in a purported effort to create incentives to manufacturers to lower their list prices, and to lower federal program beneficiary out-of-pocket costs. The rule, which is currently slated to take full effect January 1, 2023, revises the Anti-Kickback Statute discount safe harbor to exclude manufacturer rebates to Medicare Part D plans, either directly or through pharmacy benefit managers (“PBMs”), creates a new safe harbor for point-of-sale price reductions that are set in advance and are available to the beneficiary at the point-of-sale, and creates a new safe harbor for service fees paid by manufacturers to PBMs for services rendered to the manufacturer. It is too early to know whether the Biden Administration will further delay, rewrite, or allow the rule to go into effect, and what effect the rule may have on negotiations for coverage for products with Medicare Part D plans or commercial insurers. Our practices may not in all cases meet all of the criteria for safe harbor protection fromanti-kickback liability. Moreover, there are no safe harbors for many common practices, such as educational and research grants, charitable donations, product support and patient assistance programs. Arrangements that implicate theAnti-Kickback Statute and do not fit within an exception or safe harbor are reviewed on acase-by-case basis to determine whether, based on the facts and circumstances, they violate the statute.
The federal civil False Claims Act prohibits any person from, among other things, knowingly presenting, or causing to be presented, a false or fraudulent claim for payment of government funds, or knowingly making, using, or causing to be made or used, a false record or statement material to an obligation to pay money to the government or knowingly concealing or knowingly and improperly avoiding, decreasing, or concealing an obligation to pay money to the federal government. Actions under the False Claims Act may be brought by private individuals known as qui tam relators in the name of the government.government and to share in any monetary recovery. In recent years, several pharmaceutical and other healthcare companies have faced enforcement actions under the False Claims Act for, among other things, providing free product to customers with the expectation that the customers would bill federal programs for the product, inflating prices reported to private price publication services which are used to set drug payment rates under government healthcare programs, and other interactions with prescribers and other customers including interactions that may have affected customers’ billing or coding practices on claims submitted to the federal government. Other companies have faced enforcement actions for causing false claims to be submitted because of the company’s marketing the product for unapproved, and thusnon-reimbursable, uses. Federal enforcement agencies also have shown increased interest in pharmaceutical companies’ product and patient assistance programs, including reimbursement andco-pay support services, and a number of investigations into these programs have resulted in significant civil and criminal settlements. The Health Insurance Portability and Accountability Act of 1996 as amended by the Health Information Technology for Economic and Clinical Health Act, which we refer to collectively as HIPAAits implementing regulations (collectively “HIPAA”) prohibits, among other things, knowingly and willfully executing a scheme to defraud any healthcare benefit program, including privatethird-party payors. HIPAA also prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement or representation, or making or using any false writing or document knowing the same to contain any materially false, fictitious or fraudulent statement or entry in connection with the delivery of or payment for healthcare benefits, items or services. We may obtain health information from third parties that are subject to privacy and security requirements under HIPAA and we could potentially be subject to criminal penalties if we, our affiliates, or our agents knowingly obtain or disclose individually identifiable health information maintained by a HIPAA-covered entity in a manner that is not authorized or permitted by HIPAA. The majority of states also have statutes or regulations similar to the federalanti-kickback and false claims laws, which apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor. Several states now require pharmaceutical companies to report expenses relating to the marketing and promotion of pharmaceutical products in those states and to report gifts and payments to individual health care providers in those states. Some of these states also prohibit certainmarketing-related activities including the provision of gifts, meals, or other items to certain health care providers. Other states have laws requiring pharmaceutical sales representatives to be registered or licensed, and still others impose limits onco-pay assistance that pharmaceutical companies can offer to patients. In addition, several states require pharmaceutical companies to implement compliance programs or marketing codes. The Physician Payments Sunshine Act, implemented as the Open Payments program, and its implementing regulations, require certainrequires manufacturers of drugs, devices, biologics and medical supplies that are reimbursablefor which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) to report annually to CMS information related to certaindirect or indirect payments made in the previous calendar year and other transfers of value to physicians and teaching hospitals, as well as ownership and investment interests held in the company by physicians and their immediate family members. Beginning in 2022, applicable manufacturers also will be required to report information regarding payments and transfers of value provided to physician assistants, nurse practitioners, clinical nurse specialists, certified nurse anesthetists and certified nurse-midwives. Compliance with such laws and regulations will require substantial resources. Because of the breadth of these various fraud and abuse laws, it is possible that some of our business activities could be subject to challenge under one or more of such laws. Such a challenge could have material adverse effects on our business, financial condition and results of operations. In the event governmental authorities conclude that our business practices do not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations, they may impose sanctions under these laws, which are potentially significant and may include civil monetary penalties, damages, exclusion of an entity or individual from participation in government health care programs, criminal fines and imprisonment, additional reporting requirements if we become subject to a corporate integrity agreement or other settlement to resolve allegations of violations of these laws, as well as the potential curtailment or restructuring of our operations. Even if we are not determined to have violated these laws, government investigations into these issues typically require the expenditure of significant resources and generate negative publicity.
Healthcare Privacy Laws We may be subject to federal, state and foreign laws and regulations coveringgoverning data privacy and security of health information, and the collection, use, disclosure, and protection ofhealth-related and other personal information. information, including state data breach notification laws, state health information and/or genetic privacy laws, and federal and state consumer protection laws, such as Section 5 of the FTC Act, many of which differ from each other in significant ways, thus complicating compliance efforts. Compliance with these laws is difficult, constantly evolving, and time consuming. Many of these state laws enable a state attorney general to bring actions and provide private rights of action to consumers as enforcement mechanisms. There is also heightened sensitivity around certain types of health information, such as sensitive condition information or the health information of minors, which may be subject
to additional protections. Federal regulators, state attorneys general, and plaintiffs’ attorneys, including class action attorneys, have been and will likely continue to be active in this space. The legislative and regulatory landscape for privacy and data protection continues to evolve, and there has been an increasing focus on privacy and data protection issues which may affect our business. Numerous federal and state laws and regulations, including state security breach notification laws, state health information privacy laws and federal and state consumer protection laws, govern the collection, use, disclosure, and protection of personal information. Failure to comply with suchthese laws and regulations could result in government enforcement actions and create liability for us (including the imposition of significant civil and/or criminal penalties), private litigation and/or adverse publicity that could negatively affect our business. In addition, healthcareWe may obtain health information from third parties, such as health care providers who prescribe our products and research institutions we collaborate with are subject to privacy and security requirements under HIPAA. Although we are not directly subject to HIPAA, other than potentially with respect to providing certain employee benefits, we could be subject to criminal penalties if we or our affiliates or agents knowingly obtain, use, or disclose individually identifiable health information maintained by a HIPAA-covered entity in a manner that is not authorized or permitted by HIPAA.
In California, the California Consumer Privacy Act (“CCPA”) took effect on January 1, 2020. The CCPA establishes certain requirements for data use and sharing transparency and provides California residents certain rights concerning the use, disclosure, and retention of their personal data. The CCPA and its implementing regulations have already been amended multiple times since their enactment. Similarly, there are a number of legislative proposals in the EU, the United States, at both the federal and state level, as well as other jurisdictions that could impose new obligations or limitations in areas affecting our business. In addition, some countries are considering or have passed legislation implementing data protection requirements or requiring local storage and processing of data or similar requirements that could increase the cost and complexity of delivering our services and research activities. These laws and regulations are evolving and subject to interpretation, and may impose limitations on our activities or otherwise adversely affect our business. The obligations to comply with the CCPA and evolving legislation may require us, among other things, to update our notices and develop new processes internally and with our partners. These laws and regulations, as well as any associated claims, inquiries, or investigations or any other government actions may lead to unfavorable outcomes including increased compliance costs, delays or impediments in the development of new products, negative publicity, increased operating costs, diversion of management time and attention, and remedies that harm our business, including fines or demands or orders that we modify or cease existing business practices. Outside the U.S., the legislative and regulatory landscape for privacy and data security continues to evolve. There has been increased attention to privacy and data security issues that could potentially affect our business, including the EU General Data Protection Regulation (“GDPR”), which entered into effect on May 25, 2018 and imposes penalties up to 4% of annual global turnover The GDPR regulates the processing of personal data and imposes strict obligations and restrictions on the ability to collect, analyze and transfer personal data from the EU to the US, including health data from clinical trials. Foreign Corrupt Practices Act In addition, the U.S. Foreign Corrupt Practices Act of 1977, as amended, or FCPA,(“FCPA”), prohibits corporations and individuals from engaging in certain activities to obtain or retain business or to influence a person working in an official capacity. It is illegal to pay, offer to pay or authorize the payment of anything of value to any official of another country, government staff member, political party or political candidate in an attempt to obtain or retain business or to otherwise influence a person working in that capacity. Corporate Information We were incorporated under the laws of the state of Delaware on March 19, 2008 under the name New pSivida, Inc.; our predecessor, pSivida Limited, was formed in December 2000 as an Australian company incorporated in Western Australia. We subsequently changed our name to pSivida Corp. in May 2008 and again to EyePoint Pharmaceuticals, Inc. in March 2018. Our principal executive office is located at 480 Pleasant Street, Suite B300, Watertown, Massachusetts 02472 and our telephone number is(617) 926-5000. Additional Information Our website address is http://www.eyepointpharma.com. Information contained on, or connected to, our website is not incorporated by reference into this Annual Report on Form10-K. Copies of this Annual Report on Form 10-K, and our annual reports on Form 10-K, proxy statements, quarterly reports on Form10-Q, current reports on Form8-K and, if applicable, amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended, or the Exchange Act, are available free of charge through our website under “Investors—“Investors – Financial Information—Information – SEC Filings” as soon as reasonably practicable after we electronically file these materials with, or otherwise furnish them to, the SEC. Further, a copy of this Annual Report on Form10-K is located at the SEC’s Public Reference Room at 100 F Street, NE, Washington, D.C. 20549. Information on the operation of the Public Reference Room can be obtained by calling the SEC at1-800-SEC-0330. The SEC maintains an Internet site that contains reports, proxy and information statements and other information regarding issuers that file electronically with the SEC atwww.sec.gov.
ITEM 1A.RISK FACTORS RISKS RELATED TO OUR FINANCIAL POSITION AND OUR CAPITAL RESOURCES We will likely need additional capital to fund our operations and continue as a going concern.operations. If we are unable to obtain sufficient capital, we will need to curtail and reduce our operations and costs and modify our business strategy. Our operations have consumed substantial amounts of cash. To date, we have financed our operations primarily through the sale and issuance of capital stock, and debtproceeds from term loan agreements and the receipt of license fees, milestone payments, research and development funding and royalty payments from our collaboration partners. In 2019, we commenced the U.S. launch of our first two commercial products, YUTIQ and DEXYCU, and, in the first quarter of 2021, we commenced the Phase 1 clinical trial for EYP-1901 for the treatment of wet AMD. However, we have no prior history of direct commercialization of our products and no expectation of revenues from our research and development programs, including EYP-1901, prior to the successful completion of clinical trials for such programs. Therefore, we have no sufficient historical evidence to assert that it is probable that we will receive sufficient revenues from our product sales to fund operations. As of June 30, 2018,December 31, 2020, our cash and cash equivalents totaled $38.8$44.9 million. These capital resources includedWe believe that our cash and cash equivalents of $44.9million at December 31, 2020, together with the net proceeds of approximately $108.0 million received in February 2021 from the issuance on June 25, 2018 of $25.5 million of units, which we refer to individually each as an Unit and collectively as the Units, with each Unit consisting of (i) one shareshares of our common stock and (ii) one warrant to purchase one share of our common stock, or the Second Tranche Warrants, andin an additional $5.0 million draw down on June 26, 2018 under our existing term loan. We refer to the issuance of the Units pursuant to the Second Tranche Securities Purchase Agreement as the Second Tranche Transaction. We believe that our existing capital resources, together with expected amounts to be received from existing collaboration agreements, shouldunderwritten public offering will enable us to fund our operations as currently planned intooperating plan through the firstsecond quarter of calendar year 2019. However, changing circumstances may cause us2022,under current expectations regarding (i) the timing and outcomes of our Phase 1 clinical trial for EYP-1901 for the treatment of wet AMD, and (ii) initiation of our Phase 2 clinical trials for EYP-1901 for the treatment of wet AMD. Due to consume capital faster than we currently anticipate,the difficulty and we may needuncertainty associated with the design and implementation of clinical trials, we will continue to spend more money than currently expected becauseassess our cash and cash equivalents and future funding requirements. Actual cash requirements could differ from our projections due to many factors, including the continued effect of such circumstances. Our ability to fundthe Pandemic on our planned operations beyond that time, including obtaining regulatory approvalbusiness and the medical community, the timing and results of our clinical trials for YUTIQ, commercializing DEXYCU and, if approved, YUTIQ, and continuing ourEYP-1901, additional investments in research and development programprograms, the success of commercialization for our other product candidates, will require additional capital. In addition, our current plan to commercializeYUTIQ and DEXYCU, the actual costs of these commercialization efforts, competing technological and YUTIQ ourselves inmarket developments and the U.S. will require significant operating cost investment related to product manufacturing, marketing, sales, distribution and other commercialization costs. To meet our further capital needs, we are considering multiple alternatives, including but not limited to, equity financings, additional debt financings, corporate collaborations, partnerships and othercosts of any strategic transactions and fundingacquisitions and/or development of complementary business opportunities. However, there can be no assurance that we will be able to complete any one or more of such other transactions on acceptable terms or otherwise. These factors raise substantial doubt about our ability to continue as a going concern. As a result, our independent registered public accounting firm has included an explanatory paragraph in its report on our audited consolidated financial statements for the fiscal year ended June 30, 2018 related to our ability to continue as a going concern.
If we are unable to raise additional capital in sufficient amounts or on terms acceptable to us, we will need to curtail and reduce our operations and costs, and modify our business strategy, which may require us to, among other things: significantly delay, scale back or discontinue the development or commercialization of one or more of our products or product candidates or one or more of our other research and development initiatives;
| • | significantly delay, scale back or discontinue the commercialization or development of one or more of our products or product candidates or one or more of our other research and development initiatives; |
| • | seek partners or collaborators for one or more of our products or product candidates at an earlier stage than otherwise would be desirable or on terms that are less favorable than might otherwise be available; |
| • | sell or license on unfavorable terms our rights to one or more of our technologies, products or product candidates that we otherwise would seek to develop or commercialize ourselves; and/or |
| • | seek to sell our company at an earlier stage than would otherwise be desirable or on terms that are less favorable than might otherwise be available. |
seek partners or collaborators for one or more of our products or product candidates at an earlier stage than otherwise would be desirable or on terms that are less favorable than might otherwise be available;
sell or license on unfavorable terms our rights to one or more of our technologies, products or product candidates that we otherwise would seek to develop or commercialize ourselves; or
seek to sell our company at an earlier stage than would otherwise be desirable or on terms that are less favorable than might otherwise be available.
We have incurred significant losses since our inception and anticipate that we will continue to incur losses for the foreseeable future. We have incurred significant losses since our inception, have not generated significant revenue from commercial sales of our products and, with the exception of fiscal year 2010 and fiscal year 2015, we have never been profitable. Investment in drug development is highly speculative because it entails substantial upfront operating expenses and significant risk that a product candidate will fail to successfully complete clinical trials, gain regulatory approval or become commercially viable. We continue to incur significant operating expenses due primarily to investments in clinical trials, sales and marketing infrastructure, research and development, and other expenses related to our ongoing operations, including development of YUTIQ and other early stage and/or potential product candidates, and the planned commercialization of DEXYCU.operations. For the fiscal yearyears ended June 30, 2018,December 31, 2020 and 2019, we had a losslosses from operations of $26.3$37.3 million and a$47.9 million, respectively, and net losslosses of $53.2$45.4 million and $56.8 million, respectively, and we had a total accumulated deficit of $364.0$510.7 million at June 30, 2018.December 31, 2020. We expect to continue to incur significant expenses and operating losses for the foreseeable future. We anticipate that our expenses will continue to be significant if, and as, we: begin to commercialize DEXYCU and, if approved, YUTIQ and further scale up our manufacturing and distribution capabilities to commercialize DEXYCU and, if approved, YUTIQ or any other product candidate for which we may obtain regulatory approval;
| • | continue the research and pre-clinical and clinical development of our product candidates, including EYP-1901 and YUTIQ50; |
| • | initiate additional pre-clinical studies, clinical trials or other studies or trials for EYP-1901 and our other product candidates, including YUTIQ50; |
| • | continue to commercialize YUTIQ and DEXYCU; |
| • | add additional operational, financial and management information systems and personnel, including personnel to support our development and commercialization efforts; |
| • | hire additional commercial, clinical, manufacturing and scientific personnel and engage third party commercial, clinical and manufacturing organizations; |
| • | further develop the manufacturing process for our product candidates; |
| • | change or add additional manufacturers or suppliers; |
| • | seek regulatory approvals for our product candidates that successfully complete clinical trials; |
| • | seek to identify and validate additional product candidates; |
| • | acquire or in-license other products, product candidates and technologies; |
| • | maintain, protect and expand our intellectual property portfolio; |
| • | create additional infrastructure to support our product development and planned future commercial sale efforts; and |
| • | experience any delays or encounter issues with any of the above. |
adapt our regulatory compliance efforts to incorporate requirements applicable to marketed drugs;
add operational, financial and management information systems and personnel, including personnel to support commercialization efforts;
hire additional commercial, clinical, manufacturing and scientific personnel and engage third party commercial, clinical, manufacturing organizations;
continue the research andpre-clinical and clinical development of our product candidates;
initiate additionalpre-clinical, clinical or other studies or trials for our product candidates;
further develop the manufacturing process for our product candidates;
change or add additional manufacturers or suppliers;
seek regulatory approvals for our product candidates that successfully complete clinical trials;
seek to identify and validate additional product candidates;
acquire orin-license other product candidates and technologies;
maintain, protect and expand our intellectual property portfolio;
attract and retain skilled personnel;
create additional infrastructure to support our product development and planned future commercialization efforts; and
experience any delays or encounter issues with any of the above.
The net losses we incur may fluctuate significantly from quarter to quarter and year to year, such that aperiod-to-period comparison of our results of operations may not be a good indication of our future performance. In any particular quarter or quarters, our operating results could be below the expectations of securities analysts or investors, which could cause our stock price to decline.
We may never achieve profitability from future operations. Our ability to generate revenue and achieve profitability depends on our ability, alone or with strategic collaboration partners, to successfully commercialize our current products and complete the development of, and obtain the regulatory approvals necessary for, the manufacture and commercialization of our productsproduct candidates, including EYP-1901 and product candidates. Our operations have consumed substantial amounts of cash. To date, we have financed our operations primarily through the sale and issuance of capital stock and debt and the receipt of license fees, milestone payments, research and development funding and royalty income from our collaboration partners.YUTIQ 50. To become and remain profitable, we and/or our licensees must succeed in developing and commercializing products that generate significant revenue. This will require us and/or our licensees to be successful in a range of challenging activities, including completingpre-clinical testing and clinical trials of our product candidates, discovering additional product candidates, obtaining regulatory approval for these product candidates, manufacturing, marketing and selling any products for which we or our licensees may obtain regulatory approval, satisfying any post-marketing requirements and obtaining reimbursement for our products from private insurance or government payors. To date, none of our approved licensed products, including Vitrasert, Retisert and ILUVIEN, has generated significant revenues to us from sales. We have not yet commercialized DEXYCU and YUTIQ has not yet obtained regulatory approval. We do not know whenthe extent to which YUTIQ or DEXYCU, or, if approved, YUTIQ, or any of our other product candidates, including EYP-1901, if approved, will generate significant revenue for us, if at all. We may never succeed in these activities and, even if we do, we may never generate revenues significant enough to achieve profitability. Because of the numerous risks and uncertainties associated with pharmaceutical product development and commercialization, we are unable to accurately project when or if we will be able to achieve profitability from operations. Even if we do so, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our failure to become and remain profitable would depress the value of our company and could impair our ability to raise capital, expand our business, maintain our research and development efforts, diversify our product offerings or even continue our operations. Our ability to generate revenue from our current or future products and product candidates will depend on a number of factors, including: our ability to successfully commercialize DEXYCU and, if approved, YUTIQ;
| • | our ability to successfully complete development activities, including the necessary clinical trials, with respect to EYP-1901 and our other product candidates; |
our ability to complete and submit applications to, and obtain regulatory approval from, foreign regulatory authorities, if we choose to commercialize DEXYCU outside the U.S. and, if approved, YUTIQ, in unpartnered jurisdictions outside the U.S.;
| • | our ability to successfully commercialize YUTIQ and DEXYCU; |
the size of the markets in the territories for which we gain regulatory approval;
| • | our ability to complete and submit applications to, and obtain regulatory approval from, foreign regulatory authorities, if we choose to commercialize YUTIQ and DEXYCU in unpartnered jurisdictions outside the U.S.; |
| • | the size of the markets in the territories for which we gain regulatory approval; |
| • | our ability to further develop our commercial organization capable of sales, marketing and distribution for YUTIQ and DEXYCU, and any of our other product candidates for which we may obtain marketing approval; |
| • | our ability to enter into and maintain commercially reasonable agreements with manufacturers, wholesalers, distributors and other third parties in our supply chain; |
| • | our success in establishing a commercially viable price for our products; |
| • | our ability to manufacture commercial quantities of our products at acceptable cost levels; and |
| • | our ability to obtain coverage and adequate reimbursement from third parties, including government payors |
The ongoing novel coronavirus (COVID-19) pandemic has had and will likely continue to have a material and adverse impact on our business. The Pandemic has had, and will likely continue to have, a material and adverse impact on our business, including as a result of preventive and precautionary measures that we, other businesses, and governments have and will likely continue to take. This includes a significant impact on cash flows from expected revenues due to the closure of ambulatory surgery centers for DEXYCU and a significant reduction in physician office visits impacting YUTIQ. These closures precipitated the restructuring of our commercial
organization that was announced on April 1, 2020 along with a reduction in planned spending for calendar year 2020 and into calendar year 2021. Due to the continued Pandemic, these factors continued to have an adverse impact on our revenues, financial condition and cash flows in the fourth quarter of 2020 and into the first quarter of 2021. We have experienced and may continue to experience significant and unpredictable reductions in the demand for our products as customers have shut down their facilities and non-essential surgical procedures have been postponed due to the Pandemic. Such reductions may have a material and adverse impact on our future revenues. While we cannot presently predict the future scope and severity of current or any potential business shutdowns or disruptions related to COVID-19, if approved, YUTIQ, andwe or any of our other product candidates for whichthe third parties with whom we may obtain marketing approval; our ability to enter into and maintain commercially reasonable agreements with wholesalers, distributorsengage, including the suppliers, manufacturers and other third parties in our global supply chain;
chain, clinical trial sites, clinical research organizations, patients who may be candidates for clinical trials, regulators, surgeons, ASCs, potential business development partners and other third parties with whom we conduct business, were to experience prolonged shutdowns or other business disruptions, including the imposition of restrictions on the export or import of our success in establishing a commercially viable price for our products; key supplies from countries outside of the United States, our ability to manufacture commercial quantities ofconduct our products at acceptable cost levels; business in the manner and on the timelines presently planned could be materially and negatively impacted. Further, any sustained disruption in the capital markets from the Pandemic could negatively impact our ability to obtain coverageraise capital. To the extent the Pandemic continues to adversely affect our business, results of operations, financial condition and adequate reimbursementcash flows, it may also heighten many of the other risks described herein as well as in any amendment or update to our risk factors reflected in subsequent filings with the SEC. The ultimate impact of the Pandemic on our business, results of operations, financial condition and cash flows is dependent on future developments, which are still highly uncertain and cannot be predicted with confidence, including the duration of the Pandemic, as well as the timing and phasing of business reopening, including the full resumption of the performance of elective surgical procedures such as cataract surgeries. We received a subpoena from third parties,the SEC Enforcement Division requesting documents and information in an investigation relating to product sales and demand, revenue recognition and accounting. If the SEC commences an enforcement action against us, the resolution of such an enforcement action could have a material adverse effect on our business, financial condition, results of operations and cash flows. In addition, we have expended and expect to continue to expend significant financial and managerial resources responding to the SEC subpoena, which could also have a material adverse effect on our business, financial condition, results of operations and cash flows.
In May 2020, we received a subpoena from the SEC Enforcement Division requesting documents and information on topics including government payors;product sales and our demand, revenue recognition and accounting in relation to product sales, sales and cash guidance, and related financial reporting, disclosure and compliance matters. We have cooperated and continue to cooperate with the SEC’s investigation. We cannot predict the outcome of the investigation, but there can be no assurance that the SEC will not commence an enforcement action against us, or as to what the ultimate outcome of any such investigation might be. Under applicable law, the SEC has the ability to successfully complete development activities,impose sanctions on companies which are found to have violated the provisions of applicable federal securities laws, including civil monetary penalties, cease and desist orders, and other remedies. The resolution of any such enforcement action, should there be one, could have a material adverse effect on our business, financial condition, results of operations and cash flows. We have expended and expect to continue to expend significant financial and managerial resources in connection with the necessary clinical trials, with respect toinvestigation, which also could have a material adverse effect on our other product candidates.business, financial condition, results of operations and cash flow.
We will need to raise additional fundscapital in the future, which may not be available on favorable terms and may be dilutive to stockholders or impose operational restrictions. We will need to raise additional capital in the future to help fund our commercialization of DEXYCU and, if approved, YUTIQ, and for the development and commercialization of EYP-1901 and our other product candidates.candidates, if approved, and the continued commercialization of YUTIQ and DEXYCU. The amount of additional capital we will require will be influenced by many factors, including, but not limited to: the success of our commercialization of DEXYCU for the treatment of postoperative ocular inflammation including, among other things, patient and physician acceptance of DEXYCU and our ability to obtain adequate coverage and reimbursement for DEXYCU;
| • | our clinical development plans for EYP-1901 and our other product candidates including YUTIQ 50; |
the cost and timing of commercialization activities for DEXYCU, including product manufacturing, marketing, sales and distribution;
| • | the outcome, timing and cost of the regulatory approval process for EYP-1901 and our other product candidates, including the potential for the FDA to require that we perform more studies and clinical trials than those we currently expect; |
the amount of revenues we earn from commercial sales of DEXYCU;
| • | product revenues received and cash flow generated from sales of YUTIQ and DEXYCU; |
| • | whether and to what extent we internally fund, whether and when we initiate, and how we conduct other product development programs; |
| • | whether and when we are able to enter into strategic arrangements for our products or product candidates and the nature of those arrangements; |
the timing, cost and success of our clinical development, regulatory approval and planned direct U.S. commercialization of YUTIQ;
| • | the costs involved in preparing, filing, and prosecuting patent applications, and maintaining, and enforcing our intellectual property rights; |
the amount of future revenues we receive with respect to the commercialization of ILUVIEN for DME and, if approved in the EMEA, ILUVIEN for NIPU;
| • | changes in our operating plan, resulting in increases or decreases in our need for capital; |
whether and to what extent we internally fund, whether and when we initiate, and how we conduct other product development programs;
the amount of Retisert royalties and other payments we receive under collaboration agreements;
| • | our views on the availability, timing and desirability of raising capital; and |
whether and when we are able to enter into strategic arrangements for our products or product candidates and the nature of those arrangements;
timely and successful development, regulatory approval and commercialization of other potential product candidates;
the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing any patent claims;
changes in our operating plan, resulting in increases or decreases in our need for capital;
our views on the availability, timing and desirability of raising capital; and
the costs of operating as a public company.
| • | the costs of operating as a public company. |
We do not know if additional capital will be available to us when needed or on terms favorable to us or our stockholders. Collaboration, licensing or other commercial agreements may not be available on favorable terms, or at all. We do not know the extent to which we will receive funds from the commercialization of DEXYCU, ILUVIEN, Retisert or, if approved, YUTIQ. If we seek to sell our equity securities under ourat-the-market or ATM, (“ATM”) program or in another offering, we do not know whether and to what extent we will be able to do so, or on what terms. Further, the rules and regulations of the Nasdaq Stock Market LLC, or Nasdaq,(“Nasdaq”), require us to obtain stockholder approval for sales of our equity securities under certain circumstances, which could delay or prevent us from raising additional capital from such sales. Also, the state of the economy and financial and credit markets at the time or times we seek any additional financing may make it more difficult or more expensive to obtain. If available, additional equity financing may be dilutive to stockholders, debt financing may involve restrictive covenants or other unfavorable terms and dilute our existing stockholders’ equity, and funding through collaboration, licensing or other commercial agreements may be on unfavorable terms, including requiring us to relinquish rights to certain of our technologies or products. If adequate financing is not available if and when needed, we may delay, reduce the scope of, or eliminate research or development programs, planned independent U.S. commercialization of DEXYCU, YUTIQ, if approved, or other new products, if any, postpone or cancel the pursuit of product candidates such as EYP-1901, includingpre-clinical and clinical trials and new business opportunities, independent U.S. commercialization of YUTIQ and DEXYCU, or other new products, if any, reduce staff and operating costs, or otherwise significantly curtail our operations to reduce our cash requirements and extend our capital. The anticipated benefits of the Icon Acquisition may not be fully realized and may take longer to realize than expected.
On March 28, 2018, or the Closing Date, we and our wholly-owned subsidiary, Merger Sub, entered into the Merger Agreement with Icon and the other signatories thereto, pursuant to which we acquired Icon through a reverse triangular merger, which we refer to as the Icon Acquisition. The Icon Acquisition was consummated on the Closing Date. The anticipated benefits of the Icon Acquisition may not be fully realized and may take longer to realize than expected. We have devoted and will continue to devote significant management attention and resources to the commercialization of DEXYCU and potential further development of product candidates and other programs utilizing the Verisome technology platform we acquired in the Icon Acquisition. Delays or unexpected difficulties in the development or commercialization process could adversely affect our business,
financial results and financial condition. We also may not realize the full achievement of the benefits of the Icon Acquisition within a reasonable period of time. In addition, we may have not yet discovered during the due diligence process unknown factors regarding Icon that could produce unintended and unexpected consequences for us. Undiscovered factors could cause us to incur potentially material financial liabilities and prevent us from achieving the expected benefits from the Icon Acquisition within our desired time frames, if at all.
Our profitability will be impaired by our obligations to make royalty and milestone payments to the former securityholders of Icon and other third-party collaborators.
In connectionmust maintain compliance with the Icon Acquisition, we madeterms of our CRG loan or receive a $15.0 million cash payment upon the closing of the Icon Acquisition and are obligated to pay certain post-closing contingent cash payments upon the achievement of specified milestones and based upon certain net sales and partnering revenue standards, in each case subject to the terms and conditions set forth in the Merger Agreement. These include but are not limited to (i) aone-time cash payment of $15.0 million payable within 30 days following the first commercial sale of DEXYCU in the U.S., (ii) sales milestone payments totaling up to $95.0 million beginning no earlier than three years after the effectiveness of a pass-through reimbursement code by CMS and upon the achievement of certain sales thresholds and subject to certain CMS reimbursement conditions set forth in the Merger Agreement, (iii) quarterlyearn-out payments equal to 12% on net sales of DEXYCU, whichearn-out payments will increase to 16% of net sales of DEXYCU in a given year beginning in the calendar quarterwaiver for a given year to the extent aggregate annual consideration of DEXYCU exceeds $200.0 million in such year, (iv) quarterlyearn-out payments equal to 20% of partnering revenue received by us for DEXYCU outside of the U.S., and (v) single-digit percentage quarterlyearn-out payments with respect to net sales and/or partnering income, if any resulting from future clinical development, regulatory approval and commercialization of any other product candidates we might develop utilizing the Verisome technology acquired in the Icon Acquisition.
Even if we are able to successfully launch DEXYCU, our profitability will be impacted by our obligations to make payments to the former securityholders of Icon. Although we believe, under such circumstances, that the increase in revenue will exceed the corresponding payments, our obligations to the former securityholders of Icon and other third-party collaborators could have a material adverse effect on our business, financial condition and results of operations if we are unable to manage our operating costs and expenses at profitable levels.
non-compliance. Our failure to comply with the covenants or other terms of the Credit Agreement,loan, including as a result of events beyond our control, could result in a default under the Credit Agreementloan agreement that couldwould materially and adversely affect the ongoing viability of our business. On theFebruary 13, 2019 (the “CRG Closing Date,Date”), we entered into a Credit Agreement, or the Credit Agreement, with SWK Fundingloan agreement (the “CRG Loan Agreement”) among us, as borrower, CRG Servicing LLC, as administrative agent or SWK,and collateral agent (“CRG”), and the lenders party thereto collectively referredfrom time to herein as the Lenders,time (“Lenders”), providing for a senior secured term loan of up to $20$60 million or the Loan.(the “CRG Loan”). On the CRG Closing Date, $15$35 million of the CRG Loan orwas advanced (the “CRG Initial Advance”). We utilized the proceeds from the CRG Initial Advance was advancedfor the repayment in full of all outstanding obligations under the credit agreement (the “SWK Credit Agreement”) with SWK Funding LLC (“SWK”). In April 2019, we exercised our option to borrow an additional $15 million under the CRG Loan Agreement (the “CRG Second Advance”). We did not draw any additional funds under the CRG Loan by the final draw deadline of March 31, 2020. On December 17, 2020, we and our wholly owned subsidiary, EyePoint Pharmaceuticals US, Inc., entered into a Royalty Purchase Agreement (the “RPA”) with SWK. Pursuant to the RPA, we sold our interest in royalties payable to us under our license agreement with Alimera in connection with Alimera’s sales of ILUVIEN. We received a one-time $16.5 million payment from SWK and, in return, SWK is entitled to receive future royalties payable to us under our existing license agreement with Alimera. The transaction closed on December 17, 2020. With CRG’s consent, we applied $15.0 million of net proceeds from the transaction with SWK against existing long-term debt obligations with CRG. As of December 31, 2020, our outstanding balance, including principal, interest and the remaining $5back end facility fee, under the CRG Loan was approximately $40.5 million, or the Additional Advance, was advanced on June 26, 2018 following the closingand consisted of approximately $38.3 million of carrying value (see Note 14), and $2.2 million of the Second Tranche Transaction.remaining balance of unamortized debt discount related to the CRG Loan. The CRG Loan is due and payable on March 27, 2023.December 31, 2023 (the “Maturity Date”). The proceeds of the Initial Advance were used to fund a portion of the Icon Acquisition and to pay fees and expenses related to the Credit Agreement and Icon Acquisition. We intend to use the net proceeds from the Additional Advance for working capital purposes and to fund the commercialization of DEXYCU and, if approved by the FDA, YUTIQ. TheCRG Loan bears interest at a per annum rate (subject to increase during an event of default) equal to 12.5%, of which 2.5% may be paid in-kind at our election, so long as no default or event of default under the three-month London Interbank Offered Rate, or LIBOR, subject to a 1.5% floor, plus 10.50%. The CreditCRG Loan Agreement permits us to pay interest only on the principal amount loaned thereunder for the first eight payments (paymentshas occurred and is continuing. We are due on a quarterly basis). Following the interest-only period, we will be required to make quarterly, interest only payments until the Maturity Date. In addition, we are required to pay an upfront fee of interest, plus repayments1.5% of the principal amount of the CRG Loan (excluding any paid-in-kind amounts), which is payable as amounts are advanced under the Credit Agreement in an aggregate amount of up to $1,667,000 per quarter.CRG Loan. Upon repayment of the CRG Loan, we are also required to pay an exit fee equal to 6% of the aggregate principal amountamounts advanced under the CreditCRG Loan Agreement.
The CRG Loan Agreement contains affirmative and negative covenants customary for financings of this type, including limitations on our and our subsidiaries’ abilities, among other things, to incur additional debt, grant or permit additional liens, make investments and acquisitions, merge or consolidate with others, dispose of assets, pay dividends and distributions and enter into affiliate transactions, in each case, subject to certain exceptions. In addition, the CRG Loan Agreement contains the following financial covenants requiring us and the guarantors under the CRG Loan Agreement to maintain: | • | liquidity in an amount which shall exceed the greater of (i) $5 million and (ii) to the extent we have incurred certain permitted debt, the minimum cash balance, if any, required of us by the creditors of such permitted debt; and |
| • | annual minimum product revenue from YUTIQ and DEXYCU: (i) for the twelve-month period beginning on January 1, 2019 and ending on December 31, 2019, of at least $15 million, (ii) for the twelve-month period beginning on January 1, 2020 and ending on December 31, 2020, of at least $45 million, (iii) for the twelve-month period beginning on January 1, 2021 and ending on December 31, 2021, of at least $80 million and (iv) for the twelve-month period beginning on January 1, 2022 and ending on December 31, 2022, of at least $90 million. |
In November 2019, CRG waived the financial covenant associated with our revenue derived from sales of our products, YUTIQ and DEXYCU, for the twelve-month period ending December 31, 2019. In October 2020, CRG (i) waived the financial covenant associated with our revenue derived from sales of our products, YUTIQ and DEXYCU, for the twelve-month period ending December 31, 2020 and (ii) amended the financial covenant associated with our minimum product revenue to $45 million from $80 million, for the twelve-month period ending December 31, 2021. Due to the effects on our business of the Pandemic, we may not meet the financial covenants associated with our revenue derived from sales of YUTIQ and DEXYCU for the twelve-month period ending December 31, 2021. If we do not maintain compliance with all of the continuing covenants and other terms and conditions of the CRG Loan or secure a waiver for any non-compliance, then the Lenders may choose to declare an event of default and require that we immediately repay all amounts outstanding, plus penalties and interest, including an exit fee and any prepayment fees, and foreclose on the collateral granted to them to secure such indebtedness. Such repayment would have a material adverse effect on our business, operating results and financial condition.In addition, the repayment of all unpaid principal and accrued interest under the CRG Loan may be accelerated upon consummation of a specified change of control transaction or the occurrence of certain other events of default (as specified in the CreditCRG Loan Agreement), including, among other things: our default in a payment obligation under the Credit Agreement;
| • | our default in a payment obligation under the CRG Loan Agreement; |
| • | our default in a payment obligation under any of our other debt agreements evidencing indebtedness in an aggregate principal amount in excess of $500,000; |
| • | our breach of the negative covenants or, subject to specified cure periods, other terms of the CRG Loan Agreement; |
| • | invalidity of the loan documents, including CRG ceasing to have a first priority, perfected security interest on any material portion of the collateral; |
| • | the occurrence of a material adverse effect (as specified in the CRG Loan Agreement); |
| • | certain specified insolvency and bankruptcy-related events; and |
| • | an injunction lasting more than 90 days or a mandatory recall or voluntary withdrawal of any product that results in liability in excess of the greater of $4,000,000 and 7.5% of our last twelve months’ revenue. |
our default in a payment obligation under any of our other debt agreements evidencing indebtedness in an aggregate principal amount in excess of $250,000;
our breach of the negative covenants or, subject to specified cure periods, other terms of the Credit Agreement;
invalidity of the Loan documents, including SWK ceasing to have a first priority, perfected security interest on any material portion of the collateral;
the occurrence of a material adverse effect (as specified in the Credit Agreement); and
certain specified insolvency and bankruptcy-related events.
Subject to any applicable cure period set forth in the CreditCRG Loan Agreement, upon the occurrence of a bankruptcy-related event of default, all amounts outstanding with respect to the CRG Loan (principal, accrued interest, exit fee and any prepayment fees) would become due and payable immediately, and, upon the occurrence of any other event of default, SWK may, and upon the written request of the majority Lenders shall,may accelerate all or any amounts outstanding with respect to the CRG Loan. Our assets or cash flow may not be sufficient to fully repay our obligations under the CreditCRG Loan Agreement if the obligations thereunder are accelerated upon an event of default. Further, if we are unable to repay, refinance or restructure our obligations under the CreditCRG Loan Agreement, the CRG Lenders could proceed to protect and enforce their rights under the CreditCRG Loan Agreement by exercising such remedies as are available to the CRG Lenders thereunder and in respect thereof under applicable law, either by suit in equity or by action at law, or both, whether for specific performance of any covenant or other agreement contained in the CreditCRG Loan Agreement or in aid of the exercise of any power granted in the CreditCRG Loan Agreement. The foregoing would materially and adversely affect the ongoing viability of our business. Our CreditLoan Agreement contains restrictions that limit our flexibility in operating our business. The CreditCRG Loan Agreement contains various covenants that limit our ability to engage in specified types of transactions without the Lenders’ prior consent. These covenants limit our ability to, among other things: sell, transfer, lease or dispose of our assets;
| • | sell, transfer, lease or dispose of our assets; |
create, incur or assume additional indebtedness;
encumber or permit liens on certain of our assets;
| • | create, incur or assume additional indebtedness; |
make restricted payments, including paying dividends on, repurchasing or making distributions with respect to, our common stock;
| • | encumber or permit liens on certain of our assets; |
make specified investments (including loans and advances);
| • | make restricted payments, including paying dividends on, repurchasing or making distributions with respect to, our common stock; |
consolidate, merge, sell or otherwise dispose of all or substantially all of our assets;
| • | make specified investments (including loans and advances); |
enter into certain transactions with our affiliates;
| • | consolidate, merge, sell or otherwise dispose of all or substantially all of our assets; |
permit our cash and cash equivalents held in certain deposit accounts to be less than $4,000,000 at any month end; and
| • | enter into certain transactions with our affiliates; |
permit our aggregate revenue and earnings before interest, taxes, depreciation, and amortization to fall below certain agreed projection levels.
| • | permit our cash and cash equivalents held in certain deposit accounts to be less than the greater of (i) $5,000,000 and (ii) to the extent we have incurred certain permitted debt, the minimum cash balance, if any, required of us by the creditors of such permitted debt at any time; and |
| • | permit our annual product revenue from YUTIQ and DEXYCU to fall below certain agreed projection levels. |
The covenants in our CreditLoan Agreement may limit our ability to take certain actions that may be in our long-term best interests. In the event that we breach one or more covenants, the Lenders may choose to declare an event of default and require that we immediately repay all amounts outstanding, plus penalties and interest, including the exit fee and any prepayment fees, terminate their commitments to extend further credit and foreclose on the collateral granted to them to secure such indebtedness. Such repayment could have a material adverse effect on our business, operating results and financial condition. Certain potential payments to the Lenders could impede a sale of our company. Subject to certain exceptions, we are required to make mandatory prepayments of the CRG Loan with the proceeds derived from asset sales and insurance proceeds. In addition, we may make a voluntary prepayment of the CRG Loan, in whole but notor in part, at any time on or after the first anniversary of the Closing Date.time. All mandatory and voluntary prepayments of the CRG Loan are subject to the payment of prepayment premiums as follows: (i) in the case of mandatory prepayments, if prepayment occurs after December 31, 2020 and on or prior to the first anniversary of the Closing Date, a customary make-whole amount equal to the amount of interest that would have accrued on the principal amount so prepaid had it remained outstanding through the first anniversary of the Closing Date, (ii) if prepayment occurs on or after the first anniversary of the Closing Date but prior to the second anniversary of the Closing Date, 6% of the aggregate amount of the principal prepaid and (iii) if prepayment occurs on or after the second anniversary of the Closing Date but prior to the third anniversary of the Closing Date,December 31, 2021, an amount equal to 1%3% of the aggregate outstanding principal amount of the CRG Loan being prepaid. No prepayment premium is due on any principal prepaid on or after the third anniversary of the Closing Date.December 31, 2021. These provisions may make it more costly for a potential acquirer to engage in a business combination transaction with us. Provisions that have the effect of discouraging, delaying or preventing a change in control could discourage a third party from attempting to acquire us, limit the opportunity for our stockholders to receive a premium for their shares of our common stock and could also affect the price that some investors are willing to pay for our common stock. To service our indebtedness, we will require a significant amount of cash and our ability to generate cash depends on many factors beyond our control. Our ability to make cash payments on our indebtedness will depend on our ability to generate significant operating cash flow in the future. This ability is, to a significant extent, subject to general economic, financial, competitive, legislative, regulatory and other factors, that will be beyond our control. In addition, our business may not generate sufficient cash flow from operations to enable us to pay our indebtedness or to fund our other liquidity needs. In any such circumstance, we may need to refinance all or a portion of our indebtedness, on or before maturity. We may not be able to refinance any indebtedness on commercially reasonable terms or at all. If we cannot service our indebtedness, we may have to take actions such as selling assets, seeking additional equity or reducing or delaying capital expenditures, strategic acquisitions and investments. Any such action, if necessary, may not be effected on commercially reasonable terms or at all. The instruments governing our indebtedness may restrict our ability to sell assets and our use of the proceeds from such sales.
Our loan under the Paycheck Protection Program may not be forgiven or may subject us to challenges and investigations regarding qualification for the loan.
On April 22, 2020, we received a PPP Loan, which was established under the CARES Act, in the principal amount of $2.0 million. Pursuant to Section 1106 of the CARES Act, on October 7, 2020, we applied for forgiveness of the entire PPP Loan, and we are awaiting the SBA’s response to our application. Such forgiveness will be determined, subject to limitations, based on the SBA’s evaluation of our use of the PPP loan proceeds, and whether the SBA agrees that our use of the PPP loan proceeds satisfied CARES Act criteria for qualifying expenses, which include payroll costs, rent, and utility costs over the allowable measurement period following our receipt of the loan proceeds.
Given the evolving nature of the SBA’s guidance regarding the PPP Loan application and forgiveness process, we cannot give any assurance that our PPP Loan will be forgiven in whole or in part.
Additionally, the PPP Loan application required us to certify that the economic uncertainty at the time we applied for the PPP Loan made the PPP Loan request necessary to support our ongoing operations. While we made this certification in good faith after analyzing, among other things, our financial situation and access to alternative forms of capital, and believe that we satisfied all eligibility criteria for the PPP Loan and that our receipt of the PPP Loan was consistent with the broad objectives of the Paycheck Protection Program of the CARES Act, the certification described above does not contain any objective criteria and is subject to interpretation. In addition, the SBA has stated that it is unlikely that a public company with substantial market value and
access to capital markets will be able to make the required certification in good faith. The lack of clarity regarding loan eligibility under the program has resulted in significant media coverage and controversy with respect to public companies applying for and receiving loans. If, despite our good faith belief that we satisfied all eligibility requirements for the PPP Loan, we are found to have been ineligible to receive the PPP Loan or in violation of any of the laws or regulations that apply to us in connection with the PPP Loan, including the False Claims Act, we may be subject to penalties, including significant civil, criminal and administrative penalties and would be required to repay the PPP Loan. Because we are seeking forgiveness of the PPP Loan, we were required to make certain certifications which will be subject to audit and review by governmental entities and could subject us to significant penalties and liabilities if found to be inaccurate, including being required to repay the PPP loan. In addition, our receipt of the PPP Loan may result in adverse publicity and damage to our reputation, and a review or audit by the SBA or other government entity or claims under the False Claims Act could consume significant financial and management resources. Any of these events could materially harm our business, results of operations and financial condition. Our profitability will be impacted by our obligations to make royalty and milestone payments to the former securityholders of Icon Bioscience, Inc. and other third-party collaborators. In connection with our acquisition of Icon Bioscience, Inc. (“Icon”) in March 2018 (the “Icon Acquisition”), we are obligated to pay certain post-closing contingent cash payments upon the achievement of specified milestones and based upon certain net sales and partnering revenue standards, in each case subject to the terms and conditions set forth in the Merger Agreement, dated March 28, 2018 (the “Merger Agreement”). These future obligations include (i) sales milestone payments totaling up to $95.0 million, beginning no earlier than three years after the October 1, 2018 effective date of the pass-through reimbursement code approved by CMS, upon the achievement of certain sales thresholds and subject to certain CMS reimbursement conditions set forth in the Merger Agreement, (ii) quarterly earn-out payments equal to 12% on net sales of DEXYCU, which earn-out payments will increase to 16% of net sales of DEXYCU in a given year beginning in the calendar quarter for a given year to the extent aggregate annual consideration of DEXYCU exceeds $200.0 million in such year, (iii) quarterly earn-out payments equal to 20% of partnering revenue received by us for DEXYCU outside of the U.S., and (iv) single-digit percentage quarterly earn-out payments with respect to net sales and/or partnering income, if any, resulting from future clinical development, regulatory approval and commercialization of any other product candidates we might develop utilizing the Verisome technology acquired in the Icon Acquisition. As of December 31, 2020, we made DEXYCU product revenue-based royalty payments totaling $2.1 million, of which $1.3 million were related to the partnering income in connection with the Icon Acquisition. Our profitability with respect to DEXYCU is impacted by our obligations to make payments to the former securityholders of Icon. Our obligations to the former securityholders of Icon and other third-party collaborators could have a material adverse effect on our business, financial condition and results of operations if we are unable to manage our operating costs and expenses at profitable levels. Our ability to use our net operating loss carryforwards and other tax attributes may be limited. As of June 30, 2018,December 31, 2020, including pre-acquisition amounts related to the Icon acquisition,, we had U.S. net operating loss or NOL,(“NOL”) carryforwards of approximately $165.1$269.1 million for U.S. federal income tax and approximately $123.5$196.2 million for state income tax purposes available to offset future taxable income and U.S. federal and state research and development tax credits of approximately $2.9$4.7 million, prior to consideration of annual limitations that may be imposed under Section 382 of the Internal Revenue Code of 1986, as amended or (“Section 382.382”). Our U.S. NOL carryforwards begin to expire in 2023 if not utilized. Our U.S. NOL and tax credit carryforwards could expire unused and be unavailable to offset future income tax liabilities. Under Section 382, and corresponding provisions of U.S. state law, if a corporation undergoes an “ownership change,” generally defined as a greater than 50% change, by value, in its equity ownership over a three-year period, the corporation’s ability to use itspre-change U.S. NOLs and otherpre-change tax attributes, such as research and development tax credits, to offset its post-change income may be limited. Our most recent analysesThe latest analysis performed under Section 382, were performed through 2017, with a preliminary update through JuneSeptember 30, 2018, and we cannot forecast or otherwise determineconfirmed that the exercise of certain warrants in late September 2018 resulted in a greater than 50% cumulative ownership change, which will cause annual limitations on the use of our ability to derive benefit from our various federal or statethen existing NOL balances and other pre-change tax attribute carryforwards.attributes. As a result, if we earn net taxable income in future periods, our ability to use ourpre-change U.S. NOL carryforwards to offset U.S. federal taxable income maywill be subject to limitations, which could potentially result in increased future tax liabilities to us. In addition, at the state level, there may be periods during which the use of U.S. NOLs is suspended or otherwise limited, which could accelerate or permanently increase state taxes owed.
In addition, we may experience additional ownership changes in the future as a result of subsequent shifts in our stock ownership, including through completed or contemplated financings, some of which may be outside of our control. If we determine that ana future ownership change has occurred and our ability to use our historical net operating loss and tax credit carryforwards is materially limited, it would harm our future operating results by effectively increasing our future tax obligations.
RISKS RELATED TO THE REGULATORY APPROVAL AND CLINICAL DEVELOPMENT OF OUR PRODUCT CANDIDATES We are substantially dependent on the success of our lead product candidate, EYP-1901, which is in the early stages of development and must go through clinical trials, which are very expensive, time-consuming and difficult to design and implement. The outcomes of clinical trials are uncertain, and delays in the completion of or the termination of any clinical trial of EYP-1901 or our other product candidates could harm our business, financial condition and prospects. Our most recent Section 382 analysis,research and development program for our lead product candidate, EYP-1901, and certain of our other product candidates, are at an early stage of development. We must demonstrate EYP-1901’s and our other product candidates’ safety and efficacy in humans through extensive clinical testing. Such testing is expensive and time-consuming and requires specialized knowledge and expertise. Clinical trials are expensive and difficult to design and implement, in part because they are subject to rigorous regulatory requirements. The clinical trial process is also time-consuming, and the outcome is not certain. We estimate that clinical trials of our product candidates will take multiple years to complete. Failure can occur at any stage of a clinical trial, and we could encounter problems that cause us to abandon or repeat clinical trials. The commencement and completion of clinical trials may be delayed or precluded by a number of factors, including: | • | decisions not to pursue development of product candidates due to pre-clinical or clinical trial results or market factors; |
| • | lack of sufficient funding; |
| • | delays or inability to attract clinical investigators for trials; |
| • | clinical sites dropping out of a clinical trial; |
| • | time required to add new clinical sites; |
| • | any shelter-in-place orders from local, state or federal governments or clinical trial site policies resulting from the COVID-19 pandemic that determine essential and non-essential functions and staff, which may impact the ability of site staff to conduct assessments, or result in delays to the conduct of the assessments, as part of our clinical trial protocols, or the ability to enter assessment results into clinical trial databases in a timely manner; |
| • | delays or inability to recruit patients in sufficient numbers or at the expected rate; |
| • | decisions by licensees not to exercise options for products or not to pursue or promote products licensed to them; |
| • | failure of trials to demonstrate safety and efficacy; |
| • | failure to reach agreement with the FDA or other regulatory agency requirements for clinical trial design or scope of the development program; |
| • | patients’ delays or failure to complete participation in a clinical trial or inability to follow patients adequately after treatment; |
| • | changes in the design or manufacture of a product candidate; |
| • | failures by, changes in our (or our licensees’) relationship with, or other issues at, CROs, vendors and investigators responsible for pre-clinical testing and clinical trials; |
| • | imposition of a clinical hold following an inspection of our clinical trial operations or trial sites by the FDA or foreign regulatory authorities; |
| • | delays or failures in obtaining required IRB approval; |
| • | inability to obtain supplies and/or to manufacture sufficient quantities of materials for use in clinical trials; |
| • | stability issues with clinical materials; |
| • | failure to comply with GLP, GCP, cGMP or similar foreign regulatory requirements that affect the conduct of pre-clinical and clinical studies and the manufacturing of product candidates; |
| • | requests by regulatory authorities for additional data or clinical trials; |
| • | governmental or regulatory agency assessments of pre-clinical or clinical testing that differ from our (or our licensees’) interpretations or conclusions; |
| • | governmental or regulatory delays, or changes in approval policies or regulations; and |
| • | developments, clinical trial results and other factors with respect to competitive products and treatments. |
We, the FDA, other regulatory authorities outside the United States, or an IRB may suspend a clinical trial at any time for various reasons, including a preliminary update through June 30, 2018, indicatesif it appears that the clinical trial is exposing participants to unacceptable health risks or if the Second Tranche Warrants are exercisedFDA or one or more other regulatory authorities outside the United States find deficiencies in full, this would resultour IND or similar application outside the United States or the conduct of the trial. If we experience delays in an ownership change resulting in annual limitations, not yet determinable, onthe completion of, or the termination of, any clinical trial of any of our product candidates, including EYP-1901, the commercial prospects of such product candidate will be harmed, and our ability to usegenerate product revenues from such product candidate will be delayed. In addition, any delays in completing ourpre-change NOLs clinical trials will increase our costs, slow down our product candidate development and otherpre-change tax attributes.approval process, and jeopardize our ability to commence product sales and generate revenues. Any of these occurrences may harm our business, financial condition, results of operations, cash
flows and prospects significantly. In addition, many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of our product candidates. Our operatingClinical trial results may fluctuate significantly from periodfail to period.support approval of EYP-1901 or our other product candidates.
Our operatingEven if our clinical trials are successfully completed as planned, the results have fluctuated significantly from periodmay not support approval of EYP-1901 or our other product candidates under the laws and regulations of the FDA or other regulatory authorities outside the United States. The clinical trial process may fail to perioddemonstrate that our product candidates are both safe and effective for their intended uses. Pre-clinical and clinical data and analyses are often able to be interpreted in the past anddifferent ways. Even if we view our results favorably, if a regulatory authority has a different view, we may continuestill fail to do so in the future due to many factors, including:
the costsobtain regulatory approval of our commercialization efforts;product candidates. This, in turn, would significantly adversely affect our business prospects.
costsWe may expend significant resources to pursue our lead product candidate, EYP-1901 for the potential treatment of internally fundedwet AMD, and fail to capitalize on the potential of EYP-1901, or our other product candidates, for the potential treatment of other indications that may be more profitable or for which there is a greater likelihood of success.
Because we have limited financial and managerial resources, we focus on research programs and product candidates for specific indications. Specifically, with regard to EYP-1901, we are initially focusing our efforts on the treatment of wet AMD. As a result, we may forego or delay pursuit of opportunities with EYP-1901 or other product candidates for the treatment of other indications that later prove to have greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities. Our spending on current and future research and development including contract research organizations, or CROs,programs and other costs relatedproduct candidates for specific indications may not yield any commercially viable products. Furthermore, until such time as we are able to clinical development and costs ofpre-clinical studies and research; build a broader product candidate pipeline, if ever, any adverse developments with respect to our products andleading product candidates, both licensed and independently developed, includingpre-clinical and clinical trial data and results, regulatory developments and marketing and sales results; timing, receipt and amountcandidate, EYP-1901, would have a more significant adverse effect on our overall business than if we maintained a broader portfolio of revenues, including receipt and recognition of collaborativeproduct candidates.
We have historically based our research and development licensing, milestone, royaltyefforts primarily on our proprietary technologies for the treatment of chronic eye diseases. As a result of pursuing the development of product candidates using our proprietary technologies, we may fail to develop product candidates or address indications based on other scientific approaches that may offer greater commercial potential or for which there is a greater likelihood of success. Research programs to identify new product candidates require substantial technical, financial and human resources. These research programs may initially show promise in identifying potential product candidates, yet fail to yield product candidates for clinical development. Initial results from a clinical trial do not ensure that the trial will be successful and success in early stage clinical trials does not ensure success in later-stage clinical trials. Results from pre-clinical testing, early clinical trials, prior clinical trials, investigator-sponsored studies and other payments;data and information often do not accurately predict final pivotal clinical trial results. EYP-1901 relies on vorolanib as its active pharmaceutical agent. Vorolanib is a small molecule TKI that has been previously studied by Tyrogenix in Phase 1 and 2 clinical trials as an orally delivered therapy for the treatment of wet AMD. The Phase 2 clinical trial was discontinued due to systemic toxicity. There can be no assurance that such systemic toxicities will not occur in our Phase 1 clinical trial for EYP-1901. In addition, data from one pivotal clinical trial may not be predictive of the results of other pivotal clinical trials for the same product candidate, even if the trial designs are the same or similar. Data obtained from pre-clinical studies and clinical trials are susceptible to varying interpretations, which may delay, limit or prevent regulatory approval. Adverse side effects may be observed in clinical trials that delay, limit or prevent regulatory approval, and even after a product candidate has received marketing approval, the emergence of adverse side effects in more widespread clinical practice may cause the product’s regulatory approval to be limited or even rescinded. Additional trials necessary for approval may not be undertaken or may ultimately fail to establish the safety and efficacy of our product candidates. In addition, while the clinical trials of our product candidates, including our lead product candidate, EYP-1901, are designed based on the available relevant information, in view of the uncertainties inherent in drug development, such clinical trials may not be designed with a focus on indications, patient populations, dosing regimens, safety or efficacy parameters or other variables that will provide the necessary safety and efficacy data to support regulatory approval to commercialize the product. In addition, the methods we select to assess particular safety or efficacy parameters may not yield statistically significant results regarding our product candidates’ effects on patients. Even if we believe the data collected from clinical trials of our product candidates are promising, these data may not be sufficient to support approval by the FDA or foreign regulatory authorities. Pre-clinical and clinical data can be interpreted in different ways. Accordingly, the FDA or foreign regulatory authorities could interpret these data in different ways from us or our partners, which could delay, limit or prevent regulatory approval.
We face risks related to health epidemics and outbreaks, including the Pandemic, which could significantly disrupt our preclinical studies and clinical trials. announcement, execution, amendmentWe are currently conducting Phase 1 clinical trials for EYP-1901 in multiple jurisdictions within the U.S. Enrollment of patients in these clinical trials and future clinical trials in these regions may be delayed due to the outbreak of the Pandemic. In addition, we rely on independent clinical investigators, contract research organizations and other third-party service providers to assist us in managing, monitoring and otherwise carrying out our preclinical studies and clinical trials, and the outbreak may affect their ability to devote sufficient time and resources to our programs. As a result, the expected timeline for data readouts of our preclinical studies and clinical trials and certain regulatory filings may be negatively impacted, which would adversely affect our business.
We may find it difficult to enroll patients in our clinical trials, which could delay or prevent clinical trials of our product candidates. Identifying and qualifying patients to participate in clinical trials of our product candidates, including EYP-1901, is critical to our success. The timing of our clinical trials depends in part on the speed at which we can recruit patients to participate in testing our product candidates. If patients are unwilling to participate in our trials because of the COVID-19 pandemic and restrictions on travel or healthcare institution policies, negative publicity from adverse events in the biotechnology industries, public perception of vaccine safety issues or for other reasons, including competitive clinical trials for similar patient populations, the timeline for recruiting patients, conducting studies and obtaining regulatory approval of potential products may be delayed. These delays could result in increased costs, delays in advancing our product development, delays in testing the effectiveness of our technology or termination of collaborationthe clinical trials altogether. We may not be able to identify, recruit and otherenroll a sufficient number of patients, or those with required or desired characteristics to achieve diversity in a clinical trial, or complete our clinical trials in a timely manner. Patient enrollment is affected by a variety factors including, among others: | • | severity of the disease under investigation; |
| • | design of the trial protocol and size of the patient population required for analysis of the trial’s primary endpoints; |
| • | size of the patient population; |
| • | eligibility criteria for the trial in question; |
| • | perceived risks and benefits of the product candidate being tested; |
| • | willingness or availability of patients to participate in our clinical trials (including due to the COVID-19 pandemic); |
| • | proximity and availability of clinical trial sites for prospective patients; |
| • | our ability to recruit clinical trial investigators with the appropriate competencies and experience; |
| • | availability of competing vaccines and/or therapies and related clinical trials; |
| • | efforts to facilitate timely enrollment in clinical trials; |
| • | our ability to obtain and maintain patient consents; |
| • | the risk that patients enrolled in clinical trials will drop out of the trials before completion; |
| • | patient referral practices of physicians; and |
| • | ability to monitor patients adequately during and after treatment. |
We may not be able to initiate or continue clinical trials if we cannot enroll a sufficient number of eligible patients to participate in the clinical trials required by regulatory agencies. Even if we enroll a sufficient number of eligible patients to initiate our clinical trials, we may be unable to maintain participation of these patients throughout the course of the clinical trial as required by the clinical trial protocol, in which event we may be unable to use the research results from those patients. If we have difficulty enrolling and maintaining the enrollment of a sufficient number of patients to conduct our clinical trials as planned, we may need to delay, limit or terminate ongoing or planned clinical trials, any of which would have an adverse effect on our business. We are largely dependent on the future commercial agreements; scope, duration and success of collaborationour lead product candidate, EYP-1901.
Our ability to generate revenues and otherbecome profitable will depend in large part on the future commercial agreements; general and industry-specific adverse economic conditions that may affect, among other things, our and our collaborators’ operations and financial results; and
changes in accounting estimates, policies or principles and intangible asset impairments.
Due to fluctuations in our operating results, quarterly comparisonssuccess of our financial resultslead product candidate, EYP-1901, if it is approved for marketing. If EYP-1901 or any other product that we commercialize in the future does not gain an adequate level of acceptance among physicians, patients and third parties, we may not necessarily be meaningful,generate significant product revenues or become profitable. Market acceptance by physicians, patients and investors should not rely upon such results as an indicationthird party payors of future performance. In addition, investorsEYP-1901 or other products we may react adversely if our reported operating results are less favorable than in a prior period or are less favorable than those anticipated by investorscommercialize in the financial community,future will depend on a number of factors, some of which are beyond our control, including:
| • | their efficacy, safety and other potential advantages in relation to alternative treatments; |
| • | their relative convenience and ease of administration; |
| • | the availability of adequate coverage or reimbursement by third parties, such as insurance companies and other healthcare payors, and by government healthcare programs, including Medicare and Medicaid; |
| • | the prevalence and severity of adverse events; |
| • | their cost of treatment in relation to alternative treatments, including generic products; |
| • | the extent and strength of our third party manufacturer and supplier support; |
| • | the extent and strength of marketing and distribution support; |
| • | the limitations or warnings contained in a product’s approved labeling; and |
| • | distribution and use restrictions imposed by the FDA or other regulatory authorities outside the United States. |
For example, even if EYP-1901 gains approval by the FDA, physicians and patients may result in decreases in our stock price.not immediately be receptive to it and may be slow to adopt it. If EYP-1901 does not achieve an adequate level of acceptance among physicians, patients and third party payors, we may not generate meaningful revenues from EYP-1901 and we may not become profitable. RISKS RELATED TO THE COMMERCIALIZATION OF OUR PRODUCTS AND PRODUCT CANDIDATES We have no history of commercializing products, which may make it difficult for you to evaluate the success of our business to date and to assess our future viability.
Our operations to date have been largely focused on raising capital and developing Retisert, ILUVIEN, YUTIQ and our other product candidates, including undertakingpre-clinical studies and conducting clinical
trials. Bausch & Lomb and Alimera were responsible for completing the clinical development of, obtaining regulatory approval for, and initiating the commercial launch of Retisert and ILUVIEN, respectively, under our license agreements with each of them. Icon completed the clinical development of, and obtained regulatory approval for, DEXYCU. To date, we have not yet demonstrated our ability to successfully complete clinical development through the submission and attainment of marketing approvals for any product candidate, manufacture at commercial scale, or, with the exception of Retisert and ILUVIEN, arrange for a third party to do so on our behalf, or conduct sales, marketing and distribution activities necessary for successful product commercialization. Consequently, any predictions about our future success or viability may not be as accurate as they could be if we had a longer history of successfully developing and commercializing products.
Our current business strategy relies heavily on our ability to successfully commercialize YUTIQ and DEXYCU and if approved, YUTIQ, in the U.S. Our approved products may not achieve market acceptance or be commercially successful. Our ability to successfully commercialize DEXYCUYUTIQ and if approved, YUTIQ,DEXYCU in the U.S. is criticalimportant to the execution of our business strategy. Neither DEXYCUYUTIQ nor YUTIQ, if approved,DEXYCU may achieve broad market acceptance among retinal specialists and other doctors, patients, government health administration authorities and other third-party payors, and may not be commercially successful in the U.S. The degree of market acceptance and commercial success of our approved products will depend on a number of factors, including the following: the acceptance of our products by patients and the medical community and the availability, perceived advantages and relative cost, safety and efficacy of alternative and competing treatments;
| • | the acceptance of our products by patients and the medical community and the availability, perceived advantages and relative cost, safety and efficacy of alternative and competing treatments; |
our ability to obtain reimbursement for our products from third party payors at levels sufficient to support commercial success;
| • | our ability to obtain reimbursement for our products from third party payors at levels sufficient to support commercial success; |
the cost effectiveness of our products;
| • | the cost effectiveness of our products; |
the effectiveness of our marketing, sales and distribution strategies and operations;
| • | the effectiveness of our marketing, sales and distribution strategies and operations; |
our ability and the ability of our contract manufacturing organizations, or CMOs, as applicable, to manufacture commercial supplies of our products, to remain in good standing with regulatory agencies, and to develop, validate and maintain commercially viable manufacturing processes that are, to the extent required, compliant with cGMP regulations;
| • | our ability and the ability of our contract manufacturing organizations, or CMOs, as applicable, to manufacture commercial supplies of our products, to remain in good standing with regulatory agencies, and to develop, validate and maintain commercially viable manufacturing processes that are, to the extent required, compliant with cGMP regulations; |
the degree to which the approved labeling supports promotional initiatives for commercial success;
| • | the degree to which the approved labeling supports promotional initiatives for commercial success; |
a continued acceptable safety profile of our products;
| • | a continued acceptable safety profile of our products; |
results from additional clinical trials of our products or further analysis of clinical data from completed clinical trials of our products by us or our competitors;
| • | results from additional clinical trials of our products or further analysis of clinical data from completed clinical trials of our products by us or our competitors; |
our ability to enforce our intellectual property rights;
| • | our ability to enforce our intellectual property rights; |
our products’ potential advantages over other therapies;
| • | our products’ potential advantages over other therapies; |
our ability to avoid third-party patent interference or patent infringement claims; and
| • | our ability to avoid third-party patent interference or patent infringement claims; and |
maintaining compliance with all applicable regulatory requirements.
| • | maintaining compliance with all applicable regulatory requirements. |
As many of these factors are beyond our control, we cannot assure you that we will ever be able to generate meaningful revenues through product sales. In particular, if governments, private insurers, governmental insurers and other third-party payors do not provide adequate and timely coverage and reimbursement levels for our products or limit the frequency of administration, the market acceptance of our products and product candidates will be limited. Governments, governmental insurers, private insurers and other third-party payors attempt to contain healthcare costs by limiting coverage and the level of reimbursement for products and, accordingly, they may challenge the price and cost-effectiveness of our products or refuse to provide coverage for our products. Any inability on our part to successfully commercialize DEXYCUYUTIQ and if approved, YUTIQ,DEXYCU, and our other product candidates in the U.S. or any foreign territories where they may be approved, or any significant delay in such approvals, could have a material adverse impact on our ability to execute upon our business strategy and our future business prospects.
If we are unable to maintain agreements with third parties to market and sell DEXYCU and, if approved, YUTIQ, we may be unable to generate any revenue from these products.
We currently have limited sales, marketing or distribution capabilities, our approved products are commercialized by others and, as a company, we have no experience in commercializing products. We have contracted to use an outsourced CSO to commercialize DEXYCU and, if approved, YUTIQ. Any CSO that we may use may not dedicate sufficient resources to the commercialization of our products or may otherwise fail in its commercialization due to factors beyond our control. Additionally, any CSO that we may use may fail to comply with applicable legal or regulatory requirements or may enter into agreements with other parties that have products and services that could compete with our products.
In the event that we fail to successfully launch and commercialize DEXYCU or, if approved, YUTIQ, through a CSO, we may also enter into a strategic collaboration with a third party. We face significant competition in seeking appropriate strategic collaborators, and these strategic collaborations can be intricate and time-consuming to negotiate and document. We may not be able to negotiate strategic collaborations on acceptable terms, or at all. We are unable to predict when, if ever, we will enter into any strategic collaborations because of the numerous risks and uncertainties associated with establishing strategic partnerships.
We do not know if we will decide to directly commercialize any other product candidates ourselves, if approved. If we decide to commercialize a product in one or more countries, there is no assurance we will be able to hire and manage a successful sales and marketing capability or have the financial resources necessary to fund independent commercialization of any products in any country.
Even if the FDA were to grant approval of YUTIQ, the terms of the approval may limit its commercial potential.
Even if we were to successfully obtain approval from the FDA for YUTIQ, any such approval might significantly limit the indications for use or patient populations, require that black box warnings, contraindications or warnings and precautions be included on the product labeling, require expensive and time-consuming post-approval clinical trials, a Risk Evaluation and Mitigation Strategy, or REMS, or surveillance as conditions of approval, or otherwise through product labeling limit the claims that we may make, any of which may impede the successful commercialization of YUTIQ. Depending on the extent of any REMS requirements, our costs to commercialize YUTIQ may increase significantly and distribution restrictions could limit sales. Further, if the approval of YUTIQ contains other significant product label limitations, our ability to address our full target market will be reduced and our ability to realize the full market potential of YUTIQ will be harmed and we may have to limit our sales and marketing efforts.
Even if we are able to commercialize our products, theOur products may become subject to unfavorable pricing regulations, third-party reimbursement practices or healthcare reform initiatives, which could harm our business.
The regulations that govern marketing approvals, pricing and reimbursement for new drug products vary widely from country to country. Some countries require approval of the sale price of a product before it can be marketed. In many countries, the pricing review period begins after marketing or product licensing approval is granted. In some foreign markets, prescription pharmaceutical pricing remains subject to continuing governmental control even after initial approval is granted. As a result, we might obtain marketing approval for a product in a particular country, but then be subject to price regulations that delay our commercial launch of the product, possibly for lengthy time periods, which could negatively impact the revenues we are able to generate from the sale of the product in that particular country. Adverse pricing limitations may hinder our ability to recoup our investment in one or more of our products. Our ability to commercialize any products successfully willsuccess also dependdepends in part on the extent to which coverage and reimbursement for these products and related treatments will be available from government health administration authorities, private health insurers and other organizations. Government authorities and third-party payors, such as private health insurers and health maintenance organizations, determine which medications they will cover and establish reimbursement levels. A primary trend in the U.S. healthcare industry and elsewhere is cost containment. Government authorities and third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medications. Increasingly, third-party payors are requiring that drug companies provide them with predetermined discounts from list prices and are challenging the prices charged for medical products. Third-party payors also may seek additional clinical evidence, beyond the data required to obtain marketing approval, demonstrating clinical benefits and value in specific patient populations, before covering our products for those patients. We cannot be sure that coverage and reimbursement will be available for any product that we commercialize and, if reimbursement is available, what the level of reimbursement will be. Coverage and reimbursement may impact the demand for, or the price of, any product candidate for which we obtain marketing approval. If reimbursement is not available or is available only to limited levels, we may not be able to successfully commercialize any product candidate for which we obtain marketing approval. For example, under current Medicare Part B policy, payment to hospital outpatient departments and ambulatory surgical centers for products furnished to patients during a procedure is typically packaged into the payment for the associated procedure and thus not paid separately. Products granted pass-through status are excluded from this payment packaging policy and currently receive separate payment from the associated procedure for a period of three years. While DEXYCU has been granted pass-through status and will receive separate payment in these settings from Medicare for a period of three years (measured fromon the basis of the date Medicare makesreceives its first pass-through paymentclaim for reimbursement for DEXYCU), at the end of that three year period or any future extension of the three year period, or if such three-year period is shortened by a change in law, regulation or administrativeAdministrative interpretation, payment for DEXYCU may be packaged into the payment for the associated procedure and no longer be paid separately, which we expect would materially decrease our revenues from sales of DEXYCU and correspondingly have a material adverse effect on our results of operations and financial condition. There may be significant delays in obtaining coverage and reimbursement for newly approved drugs, and coverage may be more limited than the purposes for which the drug is approved by the FDA or comparable foreign regulatory authorities. Moreover, eligibility for coverage and reimbursement does not imply that any drug will be paid for in all cases or at a rate that covers our costs, including research, development, manufacturing, selling and distribution costs. Reimbursement rates may vary according to the use of the drug and the clinical setting in which it is used, may be based on reimbursement levels already set for lower cost drugs and may be incorporated into existing payments for other services. Net prices for products may be reduced by mandatory discounts or rebates required by government healthcare programs or private payors and by any future relaxation of laws that presently restrict imports of products from countries where they may be sold at lower prices than in the U.S. Third-party payors often rely upon Medicare coverage policy and payment limitations in setting their own reimbursement policies. Our inability to promptly obtain coverage and profitable reimbursement rates from both government-funded and private payors for any approved products that we develop could have a material adverse effect on our operating results, our ability to raise capital needed to commercialize products, and our overall financial condition. We intend to participate in, and have certain price reporting obligations to, the Medicaid Drug Rebate program. ParticipationProgram. This program requires us to pay a rebate for each unit of drug reimbursed by Medicaid. The amount of the “basic” portion of the rebate for each product is requiredset by law as the larger of: (i) 23.1% of quarterly average manufacturer price, or AMP, or (ii) the difference between quarterly AMP and the quarterly best price available from us to any commercial or non-governmental customer, or Best Price. AMP must be reported on a monthly and quarterly basis and Best Price is reported on a quarterly basis only. In addition, the rebate also includes the “additional” portion, which adjusts the overall rebate amount upward as an “inflation penalty” when the drug’s latest quarter’s AMP exceeds the drug’s AMP from the first full quarter of sales after launch, adjusted for federal fundsincreases in the Consumer Price Index-Urban. The upward adjustment in the rebate amount per unit is equal to be availablethe excess amount of the current AMP over the inflation-adjusted AMP from the first full quarter of sales. The rebate amount is computed each quarter based on our report to CMS of current quarterly AMP and Best Price for our covered outpatient drugs under Medicaid and Medicare Part B. Underdrug. We are required to report revisions to AMP or Best Price within a period not to exceed 12 quarters from the quarter in which the data was originally due. Any such revisions could have the impact of increasing or decreasing our rebate liability for prior quarters, depending on the direction of the revision. The Affordable Care Act made significant changes to the Medicaid Drug Rebate Program, we would be requiredand CMS issued a final regulation, which became effective on April 1, 2016, to pay a rebate to each state Medicaid program for our covered outpatient drugs that are dispensed to Medicaid beneficiaries and paid for by a state Medicaid program as a condition of having federal funds being made availableimplement the changes to the statesMedicaid Drug Rebate program under the Affordable Care Act. On December 21, 2020, CMS issued a final regulation that modified
existing Medicaid Drug Rebate Program regulations to permit reporting multiple Best Price figures with regard to value‑based purchasing arrangements (beginning in 2022); provide definitions for our“line extension,” “new formulation,” and related terms with the practical effect of expanding the scope of drugs under Medicaidconsidered to be line extensions (beginning in 2022); and Medicare Part B.revise AMP and Best Price exclusions of manufacturer-sponsored patient benefit programs, specifically regarding inapplicability of such exclusions in the context of pharmacy benefit manager “accumulator” programs (beginning in 2023). It is currently unclear if the Biden administration will delay the effectiveness or take other actions with respect to this regulation. Federal law also requires that any companymanufacturer that participates in the Medicaid Drug Rebate Program also participate in the Public Health Service’s 340B drug pricing program in order for federal funds to be available for the manufacturer’s drugs under Medicaid and Medicare Part B. The 340B drug pricing program, which is administered by the Health Resources and Services Administration, or HRSA, requires participating manufacturers to agree to charge statutorily-defined covered entities no more than the 340B “ceiling price” for the manufacturer’s covered outpatient drugs. These 340B covered entities include, but are not limited to, a variety of community health clinics and other entities that receive health services grants from the Public Health Service, as well as hospitals that serve a disproportionate share oflow-income patients. The 340B ceiling price is calculated using a statutory formula, which is based on the AMP and rebate amount for the covered outpatient drug as calculated under the Medicaid Drug Rebate Program. Any changes to the definition of AMP and the Medicaid rebate amount under the Affordable Care Act or other legislation could affect our 340B ceiling price calculations and negatively impact our results of operations. HRSA issued a final regulation regarding the calculation of the 340B ceiling price and the imposition of civil monetary penalties on manufacturers that knowingly and intentionally overcharge covered entities, which became effective on January 1, 2019. It is currently unclear how HRSA will apply its enforcement authority under this regulation. HRSA has also implemented a ceiling price reporting requirement related to the 340B program under which we are required to report 340B ceiling prices to HRSA on a quarterly basis, and HRSA then publishes that information to covered entities. Moreover, under a final regulation effective January 13, 2021, HRSA newly established an ADR process for claims by covered entities that a manufacturer has engaged in overcharging, and by manufacturers that a covered entity violated the prohibitions against diversion or duplicate discounts. Such claims are to be resolved through an ADR panel of government officials rendering a decision that could be appealed only in federal court. An ADR proceeding could subject us to onerous procedural requirements and could result in additional liability. In addition, legislation may be introduced that, if passed, would further expand the 340B program to additional covered entities or would require participating manufacturers to agree to provide 340B discounted pricing on drugs used in an inpatient setting. Federal law also requires that a company that participates in the Medicaid Drug Rebate program report average sales price, or ASP, information each quarter to CMS for certain categories of drugs that are paid under the Medicare Part B program. Manufacturers calculate the ASP based on a statutorily defined formula as well as regulations and interpretations of the statute by CMS. CMS uses these submissions to determine payment rates for drugs under Medicare Part B. In a November 20, 2020 interim final rule, CMS established a “Most Favored Nation” demonstration model that would lower Medicare Part B reimbursement of certain drugs based on international reference prices. The rule has become subject to judicial challenges, and federal courts have enjoined the rule at this time. There is also proposed legislation pending that would establish an international reference price-based payment methodology. In order to be eligible to have itsour products paid for with federal funds under the Medicaid and Medicare Part B programs and purchased by certain federal agencies and grantees, a manufacturer alsowe must participate in the U.S. Department of Veterans Affairs, or VA Federal Supply Schedule, or FSS pricing program. Under this program, the manufacturer iswe would be obligated to make its innovator and single source productsour “innovator” drugs available for procurement on an FSS contract and charge a price to four federal agencies—VA, U.S. Department of Defense, or DoD, Public Health Service and U.S. Coast Guard—that is no higher than the statutory Federal Ceiling Price. Moreover, pursuantFCP. The FCP is based on the Non-FAMP, which we calculate and report to regulations issued by the DoD Defense Health AgencyVA on a quarterly and annual basis. We also expect to implement Section 703 ofparticipate in the National Defense Authorization Act for Fiscal Year 2008, manufacturers are required to provideTricare Retail Pharmacy program, under which we would pay quarterly rebates on utilization of their innovator and single source products that are dispensed through the Tricare Retail Pharmacy network to TRICARE beneficiaries by TRICARE network retail pharmacies.beneficiaries. The rebates are calculated as the difference between the annual Non-FAMP and FCP. The requirements under the 340B, FSS, and TRICARE programs will impact ourgross-to-net revenue for our current products and any productsproduct candidates that are commercialized in the future and could adversely affect our business and operating results. We intend to shipare shipping YUTIQ if approved, directly to physician offices or clinics to be administered to patients. YUTIQ will beis being shipped to physician offices or clinics primarily through specialty pharmacies and distributors and billeddistributors. Most prefer to doctorsbuy the product directly through our select distributors under a “buy and bill” model. Physicians who may not be unwillingwilling to purchase our products especially giventhrough a specialty distributor because they do not prefer the relatively high per unit price we expectbuy and bill method may prefer to chargehave another entity called a specialty pharmacy ship them the product at no cost to the physician. The specialty pharmacy bills the health plan for YUTIQ.our product directly and then ships the product to the physician such that no costs are incurred by the physician. We also may not be able to obtainhave obtained a permanent “J” code for YUTIQ thereby limiting ourwhich assists physicians and hospitals in their ability to obtain reimbursement from Medicare and making it more difficultbill all payer types for us to obtain reimbursement from commercial or Medicare Advantage plans.the product. We intend to shipare shipping DEXYCU to ambulatory surgical centers, or ASCs, or to hospital outpatient surgical centers through specialty pharmacies.pharmacies and distributors. DEXYCU will initially beis being reimbursed for Medicare Part B patients in these settings through a transitional pass throughpass-through payment utilizing a “C”“J” code. After the initial3-year period (measured fromon the basis of the date Medicare makesreceives its first pass-through paymentclaim for reimbursement for DEXYCU), unless this 3-year period is extended, DEXYCU may not qualify for separate payment and, therefore, may be subject to cataract bundled payment rates, which would significantly limit our ability to gain utilization and subsequent revenues. In addition, ASCs and hospital outpatient surgical centers may find the administrative burden of submitting Medicare Part B claims for DEXYCU with aC-code too burdensome and may have their invoices rejected for payment due to billing errors, which may discourage them from utilizing DEXYCU. They may also be unable to efficiently delineate between patients who have Medicare Part B payment coverage, Medicare Advantage coverage and commercial coverage and thus no longer utilize DEXYCU due to the insurance payment differences between these different insurance types.
If we successfully commercialize any of our products and if we participate in the Medicaid drug rebate program or other governmental pricing programs, failurefail to comply with reporting and payment obligations under thesethe Medicaid Drug Rebate program or other governmental pricing programs, we could result inbe subject to additional reimbursement requirements, penalties, sanctions, and fines which could have a material adverse effect on our business, financial condition, results of operations and growth prospects. The Medicaid Drug Rebate ProgramOur price reporting and other governmental pricing programs require participating manufacturers to report pricing data to various government agencies. Pricing calculations vary among products and programs and include average manufacturer price and best price forobligations under the Medicaid Drug Rebate Program, average sales price for certain categories of drugs that are paid underMedicare Part B, of the Medicare340B program, andnon-federal average manufacturer price for the VA VA/FSS program are described in the risk factor entitled “Our products may become subject to unfavorable pricing program.regulations, third-party reimbursement practices or healthcare reform initiatives, which could harm our business.” Pricing and rebate calculations vary across products and programs, are complex, and are often subject to interpretation by the manufacturer,us, governmental or regulatory agencies, and the courts. In the case of Medicaid pricing data, we will report after joining the program, if we become aware that our reporting for a prior quarterperiod was incorrect or has changed as a result of a recalculation of the pricing data, we will beare obligated to resubmit the corrected data for up to three years
after those data were originally were due. Such restatements and recalculations will increase our costs for complying with the laws and regulations governing the Medicaid Drug Rebate program and could result in an overage or underage in our rebate liability for past quarters. IfPrice recalculations also may affect the ceiling price at which we successfully commercialize any ofare required to offer our products under the 340B program and participate in such governmental pricing programs, we will bemay require us to offer refunds to covered entities. We are liable for errors associated with our submission of pricing data. That liability could be significant. For example,In addition to retroactive Medicaid rebates and the potential for issuing 340B program refunds, if we are found to have knowingly submitted false average manufacturer price, average sales price, best price,AMP, Best Price, ornon-federal average manufacturer price Non-FAMP information to the government, or fail to timely submit such information, we may be liable for significant civil monetary penalties. The foregoingpenalties per item of false information. If we are found to have made a misrepresentation in the reporting of our ASP, the Medicare statute provides for significant civil monetary penalties for each misrepresentation for each day in which the misrepresentation was applied. Our failure to submit monthly/quarterly AMP and Best Brice data on a timely basis could result in a significant civil monetary penalty per day for each day the information is late beyond the due date. Such conduct also could be grounds for other sanctions,CMS to terminate our Medicaid drug rebate agreement, in which case federal payments may not be available under Medicaid or Medicare Part B for our covered outpatient drugs. Significant civil monetary penalties also could apply to late submissions of Non-FAMP information. Civil monetary penalties could also be applied if we are found to have charged 340B covered entities more than the statutorily mandated ceiling price. Moreover, under a final regulation effective January 13, 2021, HRSA newly established an ADR process that has jurisdiction over claims by covered entities that a manufacturer has engaged in overcharging. An ADR proceeding could subject us to onerous procedural requirements and could result in additional liability. In addition, claims submitted to federally-funded healthcare programs, such as termination fromMedicare and Medicaid, for drugs priced based on incorrect pricing data provided by a manufacturer can implicate the Medicaid Drug Rebate Program.federal civil False Claims Act. If we overcharge the government in connection with our FSS contract or our anticipated Tricare Agreement, whether due to a misstated FCP or otherwise, we are required to refund the difference to the government. Failure to make necessary disclosures and/or to identify contract overcharges can result in allegations against us under the False Claims Act and other laws and regulations. Unexpected refunds to the government, and responding to a government investigation or enforcement action, would be expensive and time-consuming, and could have a material adverse effect on our business, financial condition, results of operations and growth prospects. We cannot assure you that our submissions will not be found by CMS or another governmental agency to be incomplete or incorrect. Even though regulatory approvalapprovals for YUTIQ and DEXYCU hashave been obtained in the U.S., we will still face extensive FDA regulatory requirements and may face future regulatory difficulties. Even though regulatory approvalapprovals for YUTIQ and DEXYCU hashave been obtained in the U.S., the FDA and state regulatory authorities may still impose significant restrictions on the indicated uses or marketing of YUTIQ and DEXYCU, or impose ongoing requirements for potentially costly post-approval studies or post-marketing surveillance. For example, as part of its approval of DEXYCU for the treatment of postoperative ocular inflammation, the FDA required under the Pediatric Research Equity Act, or PREA, that a Phase 3/4 prospective, randomized, active treatment-controlled, parallel-design multicenter trial be conducted to evaluate the safety of DEXYCU for the treatment of inflammation following ocular surgery for childhood cataract. This pediatric study maywill likely require us to undergo a costly and time-consuming development process. If we do not meet our obligations under the PREA for this pediatric study, the FDA may issue anon-compliance letter and may also consider DEXYCU to be misbranded and subject to potential enforcement action. We submitted a pediatric study protocol to the FDA as required. We have identified clinical sites and are continuing study start-up activities that are expected to lead to dosing of a first patient in early 2022. We are also subject to ongoing FDA requirements governing the labeling, packaging, storage, distribution, safety surveillance, advertising, promotion, record-keeping and reporting of safety and other post-marketing information. The holder of an approved NDA is obligated to monitor and report adverse events and any failure of a product to meet the specifications in the NDA. The holder of an approved NDA must also submit new or supplemental applications and obtain FDA approval for certain changes to the approved product, product labeling or manufacturing process. Advertising and promotional materials must comply with FDA regulations and may be subject to other potentially applicable federal and state laws. The applicable regulations in countries outside the U.S. grant similar powers to the competent authorities and impose similar obligations on companies.
In addition, manufacturers of drug products and their facilities are subject to payment of substantial user fees and continual review and periodic inspections by the FDA and other regulatory authorities for compliance with cGMP regulations and adherence to commitments made in the NDA. We willmay also need to comply with some of the FDA’s manufacturing regulations for devices with respect to YUTIQ. We and our third-party providers are generally required to maintain compliance with cGMP and other stringent requirements and are subject to inspections by the micro-insert for YUTIQ. FDA and comparable agencies in other jurisdictions to confirm such compliance. Any delay, interruption or other issues that arise in the manufacture, fill-finish, packaging or storage of our products as a result of a failure of our facilities or the facilities or operations of third parties to pass any regulatory agency inspection could significantly impair our ability to develop and commercialize our products. Significant noncompliance could also result in the imposition of monetary penalties or other civil or criminal sanctions and damage our reputation. In addition to cGMP, the FDA requiresmay require that our micro-insertYUTIQ manufacturers comply with the Quality System Regulation, or QSR, which sets forth the FDA’s manufacturing quality standards for medical devices, and other applicable government regulations and corresponding foreign standards. If we, or a regulatory authority, discover previously unknown problems with YUTIQ or DEXYCU, such as adverse events of unanticipated severity or frequency, or problems with a facility where the product is manufactured, a regulatory authority may impose restrictions relative to YUTIQ, DEXYCU or thetheir respective manufacturing facility,facilities, including requiring recall or withdrawal of the product from the market, suspension of manufacturing, or other FDA action or other action by foreign regulatory authorities. If we fail to comply with applicable regulatory requirements for DEXYCUYUTIQ or if approved, YUTIQ,DEXYCU, a regulatory authority may: issue a warning letter asserting that we are in violation of the law;
| • | issue a warning letter asserting that we are in violation of the law; |
| • | seek an injunction or impose civil or criminal penalties or monetary fines; |
| • | suspend, modify or withdraw regulatory approval; |
| • | suspend any ongoing clinical trials; |
| • | refuse to approve a pending NDA or a pending application for marketing authorization or supplements to an NDA or to an application for marketing authorization submitted by us; |
| • | seize our product; and/or |
| • | refuse to allow us to enter into supply contracts, including government contracts. |
seek an injunction or impose civil or criminal penalties or monetary fines;
suspend, modify or withdraw regulatory approval;
suspend any ongoing clinical trials;
refuse to approve a pending NDA or a pending application for marketing authorization or supplements to an NDA or to an application for marketing authorization submitted by us;
seize our product; and/or
refuse to allow us to enter into supply contracts, including government contracts.
Our relationships with physicians, patients and payors in the U.S. are subject to applicable anti-kickback, fraud and abuse laws and regulations. In addition, we will beare subject to patient privacy regulation by both the federal government and the states in which we conduct our business. Our failure to comply with these laws could expose us to criminal, civil and administrative sanctions, reputational harm, and could harm our results of operations and financial conditions. Our current and future operations with respect to the commercialization of DEXYCUYUTIQ and if approved, YUTIQ,DEXYCU are subject to various U.S. federal and state healthcare laws and regulations. These laws impact, among other things, our proposed sales, marketing, support and education programs and constrain our business and financial arrangements and relationships with third-party payors, healthcare professionals and others who may prescribe, recommend, purchase or provide our products, and other parties through which we market, sell and distribute our products. Finally, our current and future operations are subject to additional healthcare-related statutory and regulatory requirements and enforcement by foreign regulatory authorities in jurisdictions in which we conduct our business. The laws include, but are not limited to, the following: The U.S. federal Anti-Kickback Statute prohibits persons or entities from, among other things, knowingly and willfully soliciting, offering, receiving or paying any remuneration, directly or indirectly, overtly or covertly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, lease, order, or arranging for or recommending the purchase, lease or order of, any good or service, for which payment may be made, in whole or in part, under federal healthcare programs such as Medicare and Medicaid. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. This statute has been interpreted to apply to arrangements between pharmaceutical companies on one hand and Medicare patients, prescribers, purchasers and formulary managers on the other. In addition, the government may assert that a claim including items or services resulting from a violation of the federal anti-kickback statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act. Although there are a number of statutory exemptions and regulatory safe harbors protecting certain common manufacturer business arrangements and activities from prosecution and administrative sanction, the exemptions and safe harbors are drawn narrowly, and practices or arrangements that involve remuneration may be subject to scrutiny if they do not qualify for an exemption or safe harbor. Our practices may not in all cases meet all of the criteria for safe harbor protection, and therefore would be subject to a facts and circumstances analysis to determine potential Anti-Kickback statute liability.
The federal civil False Claims Act (which can be enforced through “qui tam,” or whistleblower actions, by private citizens on behalf of the federal government) prohibits any person from, among other things, knowingly presenting, or causing to be presented false or fraudulent claims for payment of government funds or knowingly making, using or causing to be made or used, a false record or statement material to an obligation to pay money to the government or knowingly and improperly avoiding, decreasing or concealing an obligation to pay money to the U.S. federal government. Many pharmaceutical and other healthcare companies have been investigated or subject to lawsuits by whistleblowers and have reached substantial financial settlements with the federal government under the False Claims Act for a variety of alleged improper marketing activities, including providing free product to customers with the expectation that the customers would bill federal programs for the product; providing consulting fees, grants, free travel, and other benefits to physicians to induce them to prescribe the company’s products; and inflating
| • | The U.S. federal Anti-Kickback Statute prohibits persons or entities from, among other things, knowingly and willfully soliciting, offering, receiving or paying any remuneration, directly or indirectly, overtly or covertly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, lease, order, or arranging for or recommending the purchase, lease or order of, any good or service, for which payment may be made, in whole or in part, under federal healthcare programs such as Medicare and Medicaid. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. This statute has been interpreted to apply to arrangements between pharmaceutical companies on one hand and Medicare patients, prescribers, purchasers and formulary managers on the other. In addition, the government may assert that a claim including items or services resulting from a violation of the federal anti-kickback statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act. Although there are a number of statutory exemptions and regulatory safe harbors protecting certain common manufacturer business arrangements and activities from prosecution and administrative sanction, the exemptions and safe harbors are drawn narrowly, and practices or arrangements that involve remuneration may be subject to scrutiny if they do not qualify for an exemption or safe harbor. In November 2020, the U.S. Department of Health and Human Services finalized a previously abandoned proposal to amend the discount safe harbor regulation of the Anti-Kickback Statute in a purported effort to create incentives to manufacturers to lower their list prices, and to lower federal program beneficiary out-of-pocket costs. The rule, which is currently slated to take full effect January 1, 2023, revises the Anti-Kickback Statute discount safe harbor to exclude manufacturer rebates to Medicare Part D plans, either directly or through PBMs, creates a new safe harbor for point-of-sale price reductions that are set in advance and are available to the beneficiary at the |
| | point-of-sale, and creates a new safe harbor for service fees paid by manufacturers to PBMs for services rendered to the manufacturer. It is too early to know whether the Biden Administration will further delay, rewrite, or allow the rule to go into effect, and what effect the rule might may have on negotiations for coverage for products with Medicare Part D plans or commercial insurers. Our practices may not in all cases meet all of the criteria for safe harbor protection, and therefore would be subject to a facts and circumstances analysis to determine potential Anti-Kickback statute liability. |
| • | The federal civil False Claims Act (which can be enforced through “qui tam,” or whistleblower actions, by private citizens on behalf of the federal government) prohibits any person from, among other things, knowingly presenting, or causing to be presented false or fraudulent claims for payment of government funds or knowingly making, using or causing to be made or used, a false record or statement material to an obligation to pay money to the government or knowingly and improperly avoiding, decreasing or concealing an obligation to pay money to the U.S. federal government. Many pharmaceutical and other healthcare companies have been investigated or subject to lawsuits by whistleblowers and have reached substantial financial settlements with the federal government under the False Claims Act for a variety of alleged improper marketing activities, including providing free product to customers with the expectation that the customers would bill federal programs for the product; providing consulting fees, grants, free travel, and other benefits to physicians to induce them to prescribe the company’s products; and inflating prices reported to private price publication services, which are used to set drug reimbursement rates under government healthcare programs. In addition, the government and private whistleblowers have pursued False Claims Act cases against pharmaceutical companies for causing false claims to be submitted as a result of the marketing of their products for unapproved uses. Pharmaceutical and other healthcare companies also are subject to other federal false claim laws, including federal criminal healthcare fraud and false statement statutes that extend tonon-government health benefit programs. |
HIPAA imposes criminal and civil liability for, among other things, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, or knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statement, in connection with the delivery of, or payment for healthcare benefits, items or services by a healthcare benefit program, which includes both government and privately funded benefits programs; similar to the U.S. federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
| • | HIPAA imposes criminal and civil liability for, among other things, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, or knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statement, in connection with the delivery of, or payment for healthcare benefits, items or services by a healthcare benefit program, which includes both government and privately funded benefits programs; similar to the U.S. federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. |
HIPAA, as amended by the Health Information Technology and Clinical Health Act, or HITECH and its implementing regulations, impose certain obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information and impose notification obligations in the event of a breach of the privacy or security of individually identifiable health information.
| • | HIPAA, and its implementing regulations, impose certain obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information and impose notification obligations in the event of a breach of the privacy or security of individually identifiable health information. |
Numerous federal and state laws and regulations that address privacy and data security, including state data breach notification laws, state health information privacy laws, and federal and state consumer protection laws (e.g., Section 5 of the Federal Trade Commission Act, or FTC Act), govern the collection, use, disclosure and protection of health-related and other personal information, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts.
| • | Numerous federal and state laws and regulations that address privacy and data security, including state data breach notification laws, state health information and/or genetic privacy laws, and federal and state consumer protection laws (e.g., Section 5 of the Federal Trade Commission Act, or FTC Act), govern the collection, use, disclosure and protection of health-related and other personal information, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts. Compliance with these laws is difficult, constantly evolving, and time consuming, and companies that do not comply with these state laws may face civil penalties. |
The majority of states have adopted analogous laws and regulations, including state anti-kickback and false claims laws, that may apply to our business practices, including but not limited to, research, distribution, sales and marketing arrangements and claims involving healthcare items or services reimbursed by any third-party payer, including private insurers. Other states have adopted laws that, among other things, require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the U.S. federal government, or otherwise restrict payments that may be made to healthcare providers and other potential referral sources; and state laws and regulations that require drug manufacturers to file reports relating to pricing and marketing information, which requires tracking gifts and other remuneration and items of value provided to healthcare professionals and entities. In addition, some states have laws requiring pharmaceutical sales representatives to be registered or licensed, and still others impose limits onco-pay assistance that pharmaceutical companies can offer to patients.
| • | The majority of states have adopted analogous laws and regulations, including state anti-kickback and false claims laws, that may apply to our business practices, including but not limited to, research, distribution, sales and marketing arrangements and claims involving healthcare items or services reimbursed by any third-party payer, including private insurers. Other states have adopted laws that, among other things, require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the U.S. federal government, or otherwise restrict payments that may be made to healthcare providers and other potential referral sources; and state laws and regulations that require drug manufacturers to file reports relating to pricing and marketing information, which requires tracking gifts and other remuneration and items of value provided to healthcare professionals and entities. In addition, some states have laws requiring pharmaceutical sales representatives to be registered or licensed, and still others impose limits on co-pay assistance that pharmaceutical companies can offer to patients. |
The Physician Payments Sunshine Act, implemented as the Open Payments program, and its implementing regulations, require certain manufacturers of drugs, devices, biologics and medical supplies that are reimbursable under Medicare, Medicaid, or the Children’s Health Insurance Program to report annually to the CMS information related to certain payments made in the preceding calendar year and other transfers of value to physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members.
| • | The Physician Payments Sunshine Act, implemented as the Open Payments program, and its implementing regulations, require certain manufacturers of drugs, devices, biologics and medical supplies that are reimbursable under Medicare, Medicaid, or the Children’s Health Insurance Program to report annually to the CMS information related to certain payments made in the preceding calendar year and other transfers of value to physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. Beginning in 2022, applicable manufacturers also will be required to report information regarding payments and transfers of value provided to physician assistants, nurse practitioners, clinical nurse specialists, certified nurse anesthetists and certified nurse-midwives. |
The shifting commercial compliance environment and the need to build and maintain robust and expandable systems to comply with different compliance or reporting requirements in multiple jurisdictions increase the possibility that a healthcare or pharmaceutical company may fail to comply fully with one or more of these requirements. Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations may involve substantial costs. It is possible that governmental authorities will conclude that our business practices do not comply with applicable fraud and abuse or other healthcare laws and regulations or guidance. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, imprisonment, exclusion from government funded healthcare programs, such as Medicare and Medicaid, additional oversight
and reporting requirements if we become subject to a corporate integrity agreement to resolve allegations ofnon-compliance with these laws, and the curtailment or restructuring of our operations. If any of the physicians or other providers or entities with whom we expect to do business is found not to be in compliance with applicable laws, they may be subject to the same criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs. Even if we are not determined to have violated these laws, government investigations into these issues typically require the expenditure of significant resources and generate negative publicity, which could harm our financial condition and divert resources and the attention of our management from operating our business. The occurrence of any event or penalty described above may inhibit our ability to commercialize DEXYCUYUTIQ and if approved, YUTIQDEXYCU in the U.S. and generate revenues, which would have a material adverse effect on our business, financial condition and results of operations. If the market opportunities for our products and product candidates, including EYP-1901, are smaller than we believe they are, our results of operations may be adversely affected and our business may suffer. We focus our research and product development primarily on treatments offor eye diseases. Our projections of both the number of people who have these diseases, as well as the subset of people with these diseases who have the potential to benefit from treatment with our products and product candidates, such as our projections of the number of patients with wet AMD who may benefit from treatment with EYP-1901 if it is approved for use, are based on estimates. These estimates may prove to be incorrect and new studies or clinical trials may change the estimated incidence or prevalence of these diseases. The number of patients in the U.S. and elsewhere may turn out to be lower than expected, may not be otherwise amenable to treatment with our products, or new patients may become increasingly difficult to identify or gain access to, all of which would adversely affect our results of operations and our business. For example, we are developing our leading product candidate, EYP-1901, for the treatment of wet AMD. Although we believe wet AMD is a common condition and a leading cause of vision loss for people age 50 and older, our estimates of the potential market opportunity for EYP-1901 may be incorrect. If any of our products were to become the subject ofhave newly discovered or developed safety problems, related to their safety, our business would be seriously harmed. All of our approved products are and will be subject to continued oversight by the FDA or other foreign regulatory bodies, and we cannot assure you that newly discovered or developed safety issues will not arise. Although we have seenobserved no material safety issues to date, we cannot rule out that issues may arise in the future. For example, with the use of any newly marketed drug by a wider patient population, serious adverse events may occur from time to time that initially do not appear to relate to the drug itself. If such events are subsequently associated with the drug, or if any other safety issues emerge,issue emerges, we or our collaboration partners may voluntarily, or FDA or other regulatory authorities may require that we suspend or cease marketing of our approved products or modify how we or they market our approved products. In addition, newly discovered safety issues may subject us to substantial potential liabilities and adversely affect our financial condition and business. The Affordable Care Act and any changes in healthcare laws may increase the difficulty and cost for us to commercialize DEXYCU and obtain marketing approval of and commercialize YUTIQ in the U.S., and affect the prices we may obtain. The U.S. hasand state governments have enacted orand proposed legislative and regulatory changes affecting the healthcare system that could prevent or delay marketing of DEXYCU and if approved, YUTIQ, in the U.S., restrict or regulate post-approval activities and affect our ability to profitably sell YUTIQ and DEXYCU, prevent or delay marketing of our other product candidates, and if approved, YUTIQ.restrict or regulate post-approval activities. The U.S. government and state legislatures and agencies also have shown significant interest in implementing cost-containment programs to limit the growth of government-paid healthcare costs, including price controls, restrictions on reimbursement and requirements for substitution of generic products for branded prescription products. TheFor example, the Affordable Care Act was intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add transparency requirements for the
healthcare and health insurance industries, impose new taxes and fees on the health industry and impose additional health policy reforms. Among the provisions of the Affordable Care Act that have been implemented since enactment and are of importance to the commercialization of DEXYCUYUTIQ and if approved, YUTIQ,DEXYCU in the U.S. are the following: an annual, nondeductible fee on any entity that manufactures or imports specified branded prescription drugs or biologic agents;
| • | an annual, nondeductible fee on any entity that manufactures or imports specified branded prescription drugs or biologic agents; |
an increase in the statutory minimum rebates a manufacturer must pay under the Medicaid Drug Rebate Program;
| • | an increase in the statutory minimum rebates a manufacturer must pay under the Medicaid Drug Rebate Program; |
expansion of healthcare fraud and abuse laws, including the U.S. civil False Claims Act and the Anti-Kickback Statute, new government investigative powers, and enhanced penalties for noncompliance;
| • | expansion of healthcare fraud and abuse laws, including the U.S. civil False Claims Act and the Anti-Kickback Statute, new government investigative powers, and enhanced penalties for noncompliance; |
a Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50%point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for a manufacturer’s outpatient drugs to be covered under Medicare Part D (such manufacturer discounts will increase from 50% to 70% beginning in 2019 as required by the Bipartisan Budget Act of 2018);
| • | a Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50% point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for |
extension of manufacturers’ Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations;
price reporting requirements for drugs that are inhaled, infused, instilled, implanted, or injected;
| | a manufacturer’s outpatient drugs to be covered under Medicare Part D (such manufacturer discounts were increased from 50% to 70% effective as of January 1, 2019 as required by the Bipartisan Budget Act of 2018); |
expansion of eligibility criteria for Medicaid programs;
| • | extension of manufacturers’ Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations; |
addition of entity types eligible for participation in the Public Health Service’s 340B drug pricing program;
| • | price reporting requirements for drugs that are inhaled, infused, instilled, implanted, or injected; |
a requirement to annually report certain information regarding drug samples that manufacturers and distributors provide to physicians; and
| • | expansion of eligibility criteria for Medicaid programs; |
a Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research.
| • | addition of entity types eligible for participation in the Public Health Service Act’s 340B drug pricing program; |
| • | a requirement to annually report certain information regarding drug samples that manufacturers and distributors provide to physicians; and |
| • | a Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research. |
Certain legislative changes to and regulatory changes underprovisions of the Affordable Care Act have occurred in the 115th U.S. Congress and under the Trump Administration.been subject to judicial challenges as well as efforts to repeal, replace, or otherwise modify them or to alter their interpretation or implementation. For example, on December 22, 2017, the U.S. government signed into law comprehensive tax legislation, referred to as the Tax Cuts and Jobs Act, or the Tax Act, which includes a provision repealing, effective January 1, 2019, thetax-based shared responsibility payment imposed by the Affordable Care Act on certain individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate.” Further, the Bipartisan Budget Act of 2018, among other things, amended the Medicare statute to reduce the coverage gap in most Medicare drugs plans, commonly known as the “donut hole,” by raising the required manufacturer point-of-sale discount from 50% to 70% off the negotiated price effective as of January 1, 2019. Additional legislative changes, to and regulatory changes, underand judicial challenges related to the Affordable Care Act remain possible. We expectIt is unclear how the Affordable Care Act and its implementation, as well as efforts to repeal, replace, or otherwise modify, or invalidate, the Affordable Care Act, or portions thereof, will affect our business, financial condition and results of operations. It is possible that the Affordable Care Act, as currently enacted or as it may be amended in the future, and other healthcare reform measures that may be adopted in the future, could have a material adverse effect on our industry generally and on our ability to maintain or increase sales of DEXYCUYUTIQ and if approved, YUTIQ,DEXYCU in the U.S. or to successfully commercialize either product in the U.S. We also expect that the Affordable Care Act, as well as other healthcare reform measures that have been adopted and that may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on the price that we receive for DEXYCUYUTIQ and if approved, YUTIQ,DEXYCU in the U.S., and could seriously harm our future revenues. Any reduction in reimbursement from Medicare, Medicaid, or other government programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenues, attain profitability or successfully commercialize DEXYCUYUTIQ and if approved, YUTIQDEXYCU in the U.S. There has been increasing legislative, regulatory, and enforcement interest in the United States with respect to drug pricing practices. For example, the Trump Administration issued a rule updating the discount safe harbor at 42 CFR 1001.952(h) to explicitly exclude reductions in price offered by drug manufacturers to PBMs and Part D plans from the safe harbor's definition of a "discount." This rule also creates a new safe harbor designed specifically for price reductions on pharmaceutical products, but only those that are reflected in the price charged to the patient at the pharmacy counter. As another example, on November 20, 2020, CMS issued an interim final rule to implement a “Most Favored Nation” demonstration project to test Medicare Part B reimbursement of certain separately payable drugs and biologicals based on international reference prices. The rule has become subject to judicial challenges, and federal courts have enjoined the rule at this time. If the rule survives judicial scrutiny, the Most Favored Nation model will subject certain drugs or biologicals identified by CMS as having the highest annual Medicare Part B spending to an alternative payment methodology based on international reference prices, with the list of products to be updated annually to add more products and products not to be removed absent limited circumstances. There has also been legislation that would establish an international reference price-based Medicare Part B drug and biological payment methodology. Patient assistance programs for pharmaceutical products have come under increasing scrutiny by governments, legislative bodies and enforcement agencies. These activities may result in actions that have the effect of reducing prices or demand for our products, harming our business or reputation, or subjecting us to fines or penalties.We sponsor patient assistance programs, which are available to qualified patients for our products, including insurance premium and copay assistance programs. We also make donations to third-party charities that provide such assistance. Recently, there has been enhanced scrutiny of such company-sponsored programs and services. If we, our vendors or donation recipients, are deemed to have failed to comply with relevant laws, regulations or government guidance in any of these areas, we could be subject to criminal and civil sanctions, including significant fines, civil monetary penalties and exclusion from participation in government healthcare programs, including Medicare and Medicaid, and burdensome remediation measures. Actions could also be brought against executives overseeing our business or other employees. It is possible that any actions taken by the Department of Justice (DOJ) as a result of this industry-wide inquiry could reduce demand for our products and/or reduce coverage of our products, including by federal and state health care programs such as Medicare and Medicaid. If any or all of these events occur, our business, prospects and stock price could be materially and adversely affected.
If competitive products receive regulatory approval or reach the market earlier, are more effective, have fewer side effects, are more effectively marketed and/or cost less than our products or product candidates, or receive regulatory approval or reach the market earlier, our products or product candidates may not be approved, and our products or product candidates may not achieve the sales we anticipate and could be rendered noncompetitive or obsolete. We believe that pharmaceutical, drug delivery and biotechnology companies, research organizations, governmental entities, universities, hospitals, other nonprofit organizations and individual scientists are seeking to develop drugs, therapies, products, approaches or methods to treat our targeted diseases or their underlying causes. For our targeted diseases, competitors have alternate therapies that are already commercialized or are in various stages of development, ranging from discovery to advanced clinical trials. Any of these drugs, therapies, products, approaches or methods may receive government approval or gain market acceptance more rapidly than our products and product candidates, may offer therapeutic or cost advantages, or may more effectively treat our targeted diseases or their underlying causes, which could result in our product candidates not being approved, reduce demand for our products and product candidates or render them noncompetitive or obsolete. Many of our competitors and potential competitors for our leading product candidate, EYP-1901, and our commercialized products, YUTIQ and DEXYCU, have substantially greater financial, technological, research and development, marketing and personnel resources than we do. Our competitors may succeed in developing alternate technologies and products that, in comparison to the products or product candidates we have and are seeking to develop: are more effective and easier to use;
| • | are more effective and easier to use; |
have fewer side effects;
| • | have fewer side effects; |
offer other benefits; or
| • | offer other benefits; or |
may otherwise render our products less competitive or obsolete.
| • | may otherwise render our products less competitive or obsolete. |
Many of these competitors have greater experience in developing products, conducting clinical trials, obtaining regulatory approvals or clearances and manufacturing and marketing products than we do. Guidelines, recommendations and studies published by various organizations could reduce the use of our products and potential use of product candidates.
Government agencies, professional societies, practice management groups, private health and science foundations and organizations focused on various diseases may publish guidelines, recommendations or studies that affect our or our competitors’ products and product candidates. Any such guidelines, recommendations or studies that reflect negatively on our products or product candidates, either directly or relative to our competitive products, could result in current or potential decreased use, sales of, and revenues from one or more of our products and product candidates. Furthermore, our success depends in part on our and our partners’ ability to educate healthcare providers and patients about our products and product candidates, and these education efforts could be rendered ineffective by, among other things, third-parties’ guidelines, recommendations or studies.
The micro-insert for ILUVIEN and YUTIQ delivers FA, a corticosteroid that is associated with certain adverse side effects in the eye, which may affect the success of this micro-insert for treatment of DME and posterior segment uveitis.
The micro-insert for both ILUVIEN and YUTIQ delivers thenon-proprietary corticosteroid FA, which is associated with cataract formation and elevated IOP and may increase the risk of glaucoma and related surgery to manage those side effects. These side effects shown in the Phase 3 trials for ILUVIEN resulted in limitations to the approved indications of ILUVIEN, and sales of ILUVIEN may be adversely affected by the potential side effects from FA relative to other treatments for DME. The extent of ILUVIEN’s long-term side-effect profile beyond month 36 is not yet known. Alimera is conducting a five-year post-authorization, open label registry
study of the safety of ILUVIEN in 800 patients treated with the European labeled indication, which was a condition of European approval. In July 2017, Alimera announced that the Medicines and Healthcare Products Regulatory Agency gave final approval for Alimera to cap total enrollment at 550 patients, with the last three-year patientfollow-up visit anticipated in January 2020. Data from this study or other commercial experience could result in the withdrawal of ILUVIEN’s marketing approval in one or more jurisdictions. Further, delay in the commercial launch of ILUVIEN could result in the withdrawal of marketing or regulatory authorization for ILUVIEN in jurisdictions where ILUVIEN has already received marketing authorization. In addition, the perception by physicians of this benefit of efficacy versus the side-effect profile could adversely affect sales of ILUVIEN.
YUTIQ has achieved encouraging safety results through the lastfollow-up visit at month 12 in each of its two Phase 3 trials. However, there is no assurance that encouraging safety results will continue in these trials. There is also no assurance that the overall risk-benefit profile for YUTIQ will be favorable or that it will be determined to be safe for the treatment of posterior segment uveitis in light of potential side effects from FA. These side effects may limit the population for which marketing authorization is granted or for which reimbursement is provided in one or more jurisdictions and/or adversely affect sales of YUTIQ, if approved. In addition, because the micro-insert for ILUVIEN and YUTIQ are substantially the same, any safety issues that arise with respect to the ILUVIEN micro-insert could raise concerns about the YUTIQ micro-insert, which could result in delays in the approval process, prevent the FDA from approving YUTIQ and, even if approved, cause us to suspend marketing of YUTIQ or subject us to substantial liability, which would adversely affect our financial condition and business.
DEXYCU is an intraocular suspension that delivers dexamethasone, a corticosteroid that is associated with certain adverse side effects in the eye, which may affect the success of DEXYCU for the treatment of post-operative inflammation. DEXYCU is an intraocular suspension that delivers dexamethasone, a corticosteroid, which is associated with certain adverse side effects in the eye. The safety analyses from DEXYCU’s clinical trials revealed that the most commonly reported adverse reactions were increases in IOP, corneal edema and iritis, a type of uveitis affecting the front of the eye. These side effects may limit commercial use in the population for which marketing authorization was granted or for which reimbursement is provided in one or more jurisdictions and/or adversely affect sales of DEXYCU. If the FDA or other applicable regulatory authorities approve generic products that compete with any of our products or product candidates, it could reduce our sales of those products or product candidates. In the U.S., after an NDA is approved, the product generally becomes a “listed drug” which can, in turn, be relied upon by potential competitors in support of approval of an ANDA. The Federal Food, Drug, and Cosmetic Act, FDA regulations and other applicable regulations and policies provide incentives to manufacturers to create generic,non-infringing versions of a drug to facilitate the approval of an ANDA. These manufacturers might show that their product has the same active ingredients, dosage form, strength, route of administration, conditions of use, and labeling as our product candidate and tomight conduct a relatively inexpensive study to demonstrate that the generic product is absorbed in the body at the same rate and to the same extent as, or is bioequivalent to, our product. These generic equivalents would be significantly less costly than ours to bring to market, and companies that produce generic equivalents are generally able to offer their products at lower prices. Thus, after the introduction of a generic competitor, a significant percentage of the sales of any branded product are typically lost to the generic product. Accordingly, competition from generic equivalents to our products would substantially limit our ability to generate revenues and therefore to obtain a return on the investments we have made in our products. Product liability lawsuits against us could cause us to incur substantial liabilities and to limit commercialization of DEXYCUYUTIQ and if approved, YUTIQ,DEXYCU, and any other product candidates that we may develop and commercialize.commercialize, including EYP-1901. We currently face an inherentthe risk of product liability exposure related to the testing of our product candidates in human clinical trials, and this risk will increase significantly as we commercialize DEXYCUYUTIQ and if approved, YUTIQ,DEXYCU, and other product candidates that we may develop and commercialize. We also may face product liability claims regardless of FDA approval for commercial manufacturing and sale as product liability claims may be brought against us byfrom patients who have used these products andare treated with any of our product candidates in any of our clinical trials, future patients, healthcare providers or others using, administering or selling our products, if and when approved.trials. If we cannot successfully defend ourselves against claims that our products or product candidates caused injuries, we could incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in: decreased demand for our products;
| • | decreased demand for our products; |
injury to our reputation and significant negative media attention;
| • | injury to our reputation and significant negative media attention; |
termination of clinical trial sites or entire trial programs that we conduct in the future relating to DEXYCU, YUTIQ or our other product candidates;
withdrawal of clinical trial participants from any future clinical trial relating to DEXYCU, YUTIQ or our other product candidates;
| • | termination of clinical trial sites or entire trial programs that we conduct in the future relating to YUTIQ, DEXYCU, EYP-1901 or our other product candidates; |
significant costs to defend the related litigation;
| • | withdrawal of clinical trial participants from any future clinical trial relating to YUTIQ, DEXYCU, EYP-1901 or our other product candidates; |
substantial money awards to patients;
| • | significant costs to defend the related litigation; |
loss of revenue;
| • | substantial money awards to patients; |
diversion of management and scientific resources from our business operations; and
an increase in product liability insurance premiums or an inability to maintain product liability insurance coverage.
| • | diversion of management and scientific resources from our business operations; and |
| • | an increase in product liability insurance premiums or an inability to maintain product liability insurance coverage. |
We currently carry product liability insurance with coverage up to $10.0$30.0 million in the aggregate, with a per incident limit of $10.0$30.0 million, which may not be adequate to cover all liabilities that we may incur. Further, we may not be able to maintain insurance coverage at a reasonable cost or in an amount adequate to satisfy any liability that may arise. Our inability to maintain sufficient product liability insurance at an acceptable cost could prevent or inhibit the commercialization of DEXYCUYUTIQ and if approved, YUTIQ,DEXYCU, or the development and commercialization of our other product candidates.candidates, including EYP-1901. Additionally, any agreements we may enter into in the future with collaborators in connection with the development or commercialization of YUTIQ, DEXYCU, YUTIQEYP-1901 or any of our other product candidates may entitle us to indemnification against product liability losses, but such indemnification may not be available or adequate should any claim arise. In addition, several of our agreements require us to indemnify third parties and these indemnificationsindemnification obligations may exceed the coverage under our product liability insurance policy. Regulatory approval for any approved product is limited by the FDA to those specific indications and conditions for which clinical safety and efficacy have been demonstrated.
Our promotional materials, statements and training methods must comply with applicable laws and regulations, including FDA’s prohibition of the promotion of unapproved, oroff-label, use. Physicians may use our productsoff-label, as the FDA does not restrict or regulate a physician’s independent choice of treatment within the practice of medicine. If the FDA determines that our promotional materials, statements or activities constitute promotion of anoff-label use, we could be required to modify our promotional materials, statements or training methods or subject us to regulatory or enforcement actions, such as the issuance of an untitled letter, a
warning letter, injunction, seizure, civil fine, disgorgement of money, operating restrictions or criminal penalties. We may also be subject to actions by other governmental entities or private parties, such as the U.S. civil False Claims Act, civil whistleblower or “qui tam” actions. It is also possible that other federal, state or foreign enforcement authorities might take action if they consider our promotional, materials or activities to constitute promotion of anoff-label use, which could result in significant fines or penalties under other statutory authorities. In that event, our reputation could be damaged and market adoption of our approved products could be impaired.
Even though FDA approval for DEXYCU has been obtained in the U.S., we may never obtain approval for or successfully commercialize it outside of the U.S., which would limit our ability to realize its full market potential.
In order to market DEXYCU outside of the U.S., we must obtain marketing authorizations and comply with numerous and varying regulatory requirements of other countries regarding quality, safety and efficacy. Clinical trials conducted in one country may not be accepted by foreign regulatory authorities, and regulatory approval in one country does not mean that regulatory approval will be obtained in any other country. Approval processes vary among countries and can involve additional product testing and validation and additional administrative review periods. Seeking foreign regulatory approval could result in difficulties and costs for us and require additionalnon-clinical studies or clinical trials, which could be costly and time consuming. Regulatory requirements can vary widely from country to country and could delay or prevent the introduction of DEXYCU in those countries. While our management team has experience in obtaining foreign regulatory approvals at other companies, we, as a company, do not have experience in obtaining regulatory approval in international markets. If we fail to comply with regulatory requirements in international markets or to obtain and maintain required approvals, or if regulatory approval in international markets is delayed, we would not be able to realize the full market potential of DEXYCU.
RISKS RELATED TO THE REGULATORY APPROVAL AND CLINICAL DEVELOPMENT OF OUR PRODUCT CANDIDATES
We are substantially dependent on the success of our lead product candidate, YUTIQ, which is in a later stage of development than our other product candidates. To the extent regulatory approval of YUTIQ is delayed or not granted, our business, financial condition and results of operations may be materially adversely affected and the price of our common stock may decline.
We are focusing a significant portion of our activities and resources on our lead product candidate, YUTIQ, and we believe our prospects are highly dependent on, and a significant portion of the value of our company relates to, our ability to successfully develop, obtain regulatory approval for, and commercialize YUTIQ in the U.S. The U.S. regulatory approval of YUTIQ is subject to many risks, including the risks discussed in other risk factors set forth in this report, and YUTIQ may not receive marketing approval from the FDA or such marketing approval may be delayed. If the results or timing of the regulatory process, regulatory developments, any additional clinical trials orpre-clinical studies, or other activities, actions or decisions related to YUTIQ do not meet our or others’ expectations, the market price of our common stock could decline significantly.
We submitted an NDA for YUTIQ in January 2018 and have a PDUFA action date of November 5, 2018. Although we have discussed our clinical development plans with the FDA, the agency may ultimately determine that our Phase 3 clinical trials or other aspects of our NDA are not sufficient for regulatory approval and may issue a CRL instead of approval. If we receive a CRL, the FDA would outline deficiencies in our NDA and may request the submission of additional information, including clinical data.
The facilities used by us to manufacture our products and any product candidates that we may develop are subject to inspections, includingpre-approval inspections following our submission of any NDAs to the FDA for any product candidates that we may develop. For example, the FDA conducted apre-approval inspection related to our NDA for YUTIQ from March 26, 2018 to April 4, 2018. Most inspections by the FDA, including the FDA
inspection related to our NDA for YUTIQ, result in an Establishment Inspection Report as well as a Summary of Observations immediately following the inspection. As required, the Company has responded to these observations in a timely manner. In any event, our commercialization of YUTIQ in the U.S. may be delayed and we may incur additional costs and be required to devote additional resources to address the FDA’s concerns. If we are unable to timely address the FDA’s summary of observations from such inspection or if the FDA requires us to conduct additional clinical trials or studies, or requires our manufacturers to improve or change their practices, our timeline for commercialization of YUTIQ in the U.S. will be delayed and we will incur additional costs. Further, there can be no assurance that we will complete such studies or clinical trials or address manufacturing issues in a manner that is acceptable to the FDA.
In addition, one of our collaborators, Alimera, holds an exclusive license to our proprietary Durasert insert for the treatment of uveitis, including NIPU, in the EMEA. Alimera was responsible for filing a Type II variation for ILUVIEN for the treatment of NIPU. In December 2017, Alimera submitted a Type II variation for ILUVIEN to add the indication of recurrent and persistent NIPU to the ILUVIEN label. In January 2018, Alimera received validation of this Type II variation submission in all 17 European countries in which Alimera had previously received regulatory approval for ILUVIEN for DME. According to Alimera’s public filings, Alimera plans to submitfollow-up data supporting its Type II variation application in the fourth quarter of calendar year 2018 and expects it will obtain approval for its application in the first half of calendar 2019. Obtaining regulatory approval for such a variation is uncertain and Alimera may fail to obtain the approval. The MAA variation review processes and the processes of other regulatory authorities, are extensive, lengthy, expensive, and uncertain, and such regulatory authorities may delay, limit, or deny approval of ILUVIEN for NIPU.
Any delay or setback in the development or regulatory approval of YUTIQ will adversely affect our business and could cause our stock price to decline.
The regulatory approval processes of the FDA or other foreign regulatory authorities are lengthy, time consuming and inherently unpredictable, and if we are ultimately unable to obtain regulatory approval for our product candidates, our business may be substantially harmed.
The time required to obtain approval by the FDA or other foreign regulatory authorities is unpredictable, but typically takes many years following the commencement of clinical trials and depends upon numerous factors, including the substantial discretion of the regulatory agency. In addition, approval policies, regulations, or the type and amount of clinical data necessary to gain approval may change during the course of a product candidate’s clinical development. It is possible that none of our existing product candidates or any product candidates we may seek to develop in the future will ever obtain regulatory approval.
Our product candidates could fail to receive regulatory approval for many reasons, including the following:
the regulatory authority may not accept our application for filing;
the regulatory authority may disagree with the design, scope or implementation of our clinical trials;
we may be unable to demonstrate to the satisfaction of the regulatory authority that a product candidate is safe and effective for its proposed indication and/or that its clinical and other benefits outweigh its safety risks;
the regulatory authority may disagree with our interpretation of data frompre-clinical studies or clinical trials;
the data collected from clinical trials of our product candidates may not be sufficient to support the approval of a drug application or marketing authorization application;
the regulatory authority may fail to approve our or our third-party manufacturers’ manufacturing processes or facilities for clinical and commercial supplies; and
the approval policies or regulations of the regulatory authority may change in a manner rendering our clinical data insufficient for approval.
We cannot be certain that any of our current product candidates will receive regulatory approval. If we do not receive regulatory approval for our product candidates, our business may be substantially harmed.
Delays in clinical trials are common and have many causes, and any delay could result in increased costs to us and jeopardize or delay our ability to obtain regulatory approval and commence product sales.
Other than YUTIQ, all of our product development is at earlier stages. Product development at all stages involves a high degree of risk, and only a small proportion of research and development programs result in product candidates that advance to pivotal clinical trials or result in approved products. There is no assurance that any feasibility study agreements we have, or enter into, with third parties, or our own research and development programs and collaborations will result in any new product candidates, or that we or any licensees will commence clinical trials for any new product candidates or continue clinical trials once commenced. If clinical trials conducted by or for us or any licensees for any product candidates do not provide the necessary evidence of safety and efficacy, those product candidates will not receive the necessary regulatory approvals, cannot be sold, and will not generate revenues for us.
We may also experience delays in clinical trials of our product candidates or the time required to complete clinical trials for our product candidates may be longer than anticipated. Our future clinical trials may not begin on time, have an effective design, enroll a sufficient number of patients, or be completed on schedule, if at all. Our clinical trials can be delayed, or even terminated, for a variety of reasons, including, but not limited to:
decisions not to pursue development of product candidates due topre-clinical or clinical trial results or market factors;
lack of sufficient funding;
inability to attract clinical investigators for trials;
inability to recruit patients in sufficient numbers or at the expected rate;
decisions by licensees not to exercise options for products or not to pursue or promote products licensed to them;
failure of trials to demonstrate safety and efficacy;
failure to meet FDA or other regulatory agency requirements for clinical trial design, or inadequate clinical trial design;
inability to follow patients adequately after treatment;
changes in the design or manufacture of a product candidate;
failures by, changes in our (or our licensees’) relationship with, or other issues at, CROs, vendors and investigators responsible forpre-clinical testing and clinical trials;
imposition of a clinical hold following an inspection of our clinical trial operations or trial sites by the FDA or foreign regulatory authorities;
inability to obtain supplies and/or to manufacture sufficient quantities of materials for use in clinical trials;
stability issues with clinical materials;
failure to comply with GLP, GCP, cGMP or similar foreign regulatory requirements that affect the conduct ofpre-clinical and clinical studies and the manufacturing of product candidates;
requests by regulatory authorities for additional data or clinical trials;
governmental or regulatory agency assessments ofpre-clinical or clinical testing that differ from our (or our licensees’) interpretations or conclusions;
governmental or regulatory delays, or changes in approval policies or regulations; and
developments, clinical trial results and other factors with respect to competitive products and treatments.
If clinical trials for our product candidates are delayed or terminated for any of the above reasons or other reasons, our development costs may increase, our approval process could be delayed and our ability to commercialize our product candidates could be materially harmed.
We may expend our limited resources to pursue a particular product candidate or indication and fail to capitalize on product candidates or indications that may be more profitable or for which there is a greater likelihood of success.
Because we have limited financial and managerial resources, we focus on research programs and product candidates for specific indications. As a result, we may forego or delay pursuit of opportunities with other product candidates or for other indications that later prove to have greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities. Our spending on current and future research and development programs and product candidates for specific indications may not yield any commercially viable products.
We have historically based our research and development efforts primarily on our Durasert technology platform to develop proprietary sustained-release pharmaceutical products for the treatment of posterior uveitis and other chronic eye diseases. As a result of pursuing the development of product candidates using our proprietary technologies, we may fail to develop product candidates or address indications based on other scientific approaches that may offer greater commercial potential or for which there is a greater likelihood of success. Research programs to identify new product candidates require substantial technical, financial and human resources. These research programs may initially show promise in identifying potential product candidates, yet fail to yield product candidates for clinical development.
Initial results from a clinical trial do not ensure that the trial will be successful and success in early stage clinical trials does not ensure success in later-stage clinical trials.
Results frompre-clinical testing, early clinical trials, investigator-sponsored studies and other data and information often do not accurately predict final pivotal clinical trial results. In addition, data from one pivotal clinical trial may not be predictive of the results of other pivotal clinical trials for the same product candidate, even if the trial designs are the same or similar. Data obtained frompre-clinical studies and clinical trials are susceptible to varying interpretations, which may delay, limit or prevent regulatory approval. Adverse side effects may be observed in clinical trials that delay, limit or prevent the regulatory approval, and even after a product candidate has received marketing approval, the emergence of adverse side effects in more widespread clinical practice may cause the product’s regulatory approval to be limited or even rescinded. Additional trials necessary for approval may not be undertaken or may ultimately fail to establish the safety and efficacy of our product candidates.
In addition, while the clinical trials of our product candidates are designed based on the available relevant information, in view of the uncertainties inherent in drug development, such clinical trials may not be designed with a focus on indications, patient populations, dosing regimens, safety or efficacy parameters or other variables that will provide the necessary safety and efficacy data to support regulatory approval to commercialize the product. In addition, the methods we select to assess particular safety or efficacy parameters may not yield
statistically significant results regarding our product candidates’ effects on patients. Even if we believe the data collected from clinical trials of our product candidates are promising, these data may not be sufficient to support approval by the FDA or foreign regulatory authorities.Pre-clinical and clinical data can be interpreted in different ways. Accordingly, the FDA or foreign regulatory authorities could interpret these data in different ways from us or our partners, which could delay, limit or prevent regulatory approval.
Changes in regulatory requirements and guidance may occur and we may need to amend clinical trial protocols submitted to applicable regulatory authorities to reflect these changes. Amendments may require us to resubmit clinical trial protocols to institutional review boards or ethics committees forre-examination, which may impact the costs, timing or successful completion of a clinical trial.
The FDA’s and other regulatory authorities’ policies may change, and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the U.S. or abroad. If we are required to conduct additional clinical trials or other studies with respect to our product candidates beyond those that we currently contemplate, or if we are unable to successfully complete our clinical trials or other studies, we may be delayed in obtaining regulatory approval of any of our product candidates, we may not be able to obtain regulatory approval at all or we may obtain approval of indications that are not as broad as intended. Our product development costs will also increase if we experience delays in testing or approvals, and we may not have sufficient funding to complete the testing and approval process for our product candidates. Significant clinical trial delays could allow our competitors to bring products to market before we do and impair our ability to commercialize our products if and when approved. If any of this occurs, our business would be harmed.
RISKS RELATED TO OUR INTELLECTUAL PROPERTY If we are unable to protect our intellectual property rights or if our intellectual property rights are inadequate to protect our product candidates, our competitors could develop and commercialize technology and products similar to ours, and our competitive position could be harmed. Our commercial success will depend in large part on our ability to obtain and maintain patent and other intellectual property protection in the U.S. and other countries with respect to our proprietary technology and products. We rely on trade secret, patent, copyright and trademark laws, and confidentiality and other agreements with employees and third parties, all of which offer only limited protection. We seek patent protection for many different aspects of our product candidates, including their compositions, their methods of use, processes for their manufacture, and any other aspects that we deem to be commercially important to the development of our business. The patent prosecution process is expensive and time-consuming, and we and any licensors and licensees may not be able to apply for or prosecute patents on certain aspects of our product candidates or delivery technologies at a reasonable cost, in a timely fashion, or at all. For technology licensed to third parties, we may not have the right to control the preparation, filing and/or prosecution of the corresponding patent applications, or to maintain patent rights corresponding to such technology. Therefore, these patents and applications may not be prosecuted and enforced in a manner consistent with the best interests of our business. It is also possible that we, or any licensors or licensees, will fail to identify patentable aspects of inventions made in the course of development and commercialization activities before it is too late to obtain patent protection on them. It is possible that defects of form in the preparation or filing of our patents or patent applications may exist, or may arise in the future, such as with respect to proper priority claims, inventorship, claim scope or patent term adjustments. If any licensors or licensees are not fully cooperative or disagree with us as to the prosecution, maintenance, or enforcement of any patent rights, such patent rights could be compromised, and we might not be able to prevent third parties from making, using, and selling competing products. If there are material defects in the form or preparation of our patents or patent applications, such patents or applications may be invalid or unenforceable. Moreover, our competitors may independently develop equivalent knowledge, methods,and know-how. Any of these outcomes could impair our ability to prevent competition from third parties, which may have an adverse impact on our business, financial condition, and operating results. The patent positions of pharmaceutical companies generally are highly uncertain, involve complex legal and factual questions and have in recent years been the subject of much litigation. As a result, the issuance, scope, validity, enforceability and commercial value of any patents that issue, are highly uncertain. For example, recent changes to the patent laws of the U.S. provide additional procedures for third parties to challenge the validity of issued patents. Under the Leahy-Smith America Invents Act, or AIA, which was signed into law on September 16, 2011, patents issued from applications with an effective filing date after March 15, 2013 may be challenged by third parties using the post-grant review procedure which allows challenges for a number of reasons, including prior art, sufficiency of disclosure, and subject matter eligibility. Under the AIA, patents may also be challenged under the inter partes review procedure. Inter partes review provides a mechanism by which any third party may challenge the validity of any issued U.S. Patent in the USPTO on the basis of prior art. Because of a lower evidentiary standard necessary to invalidate a patent claim in USPTO proceedings as compared to the evidentiary standard relied on in U.S. federal court, a third party could potentially provide evidence in a USPTO proceeding sufficient for the USPTO to hold a claim invalid even though the same evidence would be insufficient to
invalidate the claim if first presented in a district court action. Accordingly, a third party may attempt to use the USPTO procedures to invalidate our patent claims that would not have been invalidated if first challenged by the third party as a defendant in a district court action. With respect to foreign jurisdictions, the laws of foreign countries may not protect our rights to the same extent as the laws of the U.S. or vice versa. For example, European patent law restricts the patentability of methods of treatment of the human body more than U.S. law does. Also, patents granted by the European Patent Office may be opposed by any person within nine months from the publication of their grant. Our patents and patent applications, even if unchallenged by a third party, may not adequately protect our intellectual property or prevent others from designing around our claims. The steps we have taken to protect our proprietary rights may not be adequate to preclude misappropriation of our proprietary information or infringement of our intellectual property rights, both inside and outside the U.S. Further, the examination process may require us to narrow the claims of pending patent applications, which may limit the scope of patent protection that may be obtained if these applications issue. The rights that may be granted under future issued patents may not provide us with the proprietary protection or competitive advantages we are seeking. If we are unable to obtain and maintain patent protection for our technology and products, or if the scope of the patent protection obtained is not sufficient, our competitors could develop and commercialize technology and products similar or superior to ours, and our ability to successfully commercialize our technology and products may be impaired. As of August 31, 2018,March 1, 2021, we had 177348 patents or granted applications and 10752 pending patent applications, including patents and pending applications covering our Durasert, DEXYCU, Verisome and other technologies. With respect to these patent rights, we do not know whether any of our patent applications will result in issued patents or, if any of our patent applications do issue, whether such patents will protect our technology in whole or in part, or whether such patents will effectively prevent others from commercializing competitive technologies and products. There is no guarantee that any of our issued or granted patents will not later be found invalid or unenforceable. Furthermore, since patent applications in the U.S. and most other countries are confidential for a period of time after filing, we cannot be certain that we were the first to either (i) file any patent application related to our product candidates or (ii) invent any of the inventions claimed in our patents or patent applications. For applications with an effective filing date before March 16, 2013, or patents issuing from such applications, an interference proceeding can be provoked by a third party or instituted by the USPTO to determine who was the first to invent any of the subject matter covered by the patent claims of our applications and patents. As of March 16, 2013, the U.S. transitioned to a“first-to-file” “first-to-file” system for deciding which party should be granted a patent when two or more patent applications are filed by different parties claiming the same invention. A third party that files a patent application in the USPTO before us could therefore be awarded a patent covering an invention of ours even if we had made the invention before it was made by the third party. This will require us to be cognizant going forward of the time from invention to filing of a patent application. The change to“first-to-file” “first-to-file” from“first-to-invent” “first-to-invent” is one of the changes to the patent laws of the U.S. resulting from the AIA. Publications of discoveries in the scientific literature often lag behind the actual discoveries, and patent applications in the U.S. and other jurisdictions are typically not published until 18 months after filing or in some cases not at all, until they are issued as a patent. Therefore, we cannot be certain that we were the first to make the inventions claimed in our pending patent applications, that we were the first to file for patent protection of such inventions, or that we have found all of the potentially relevant prior art relating to our patents and patent applications that could invalidate one or more of our patents or prevent one or more of our patent applications from issuing. Even if patents do successfully issue and even if such patents cover our product candidates, third parties may initiate oppositions,interferences, re-examinations, post-grant reviews, inter partes reviews, nullification or derivation actions in court or before patent offices or similar proceedings challenging the validity, enforceability, or scope of such patents, which may result in the patent claims being narrowed or invalidated. Furthermore, even if they are unchallenged, our patents and patent applications may not adequately protect our intellectual property, provide exclusivity for our product candidates, or prevent others from designing around our claims. Any of these outcomes could impair our ability to prevent competition from third parties. Furthermore, the issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability, and our owned and licensed patents may be challenged in the courts or patent offices in the U.S. and abroad. Such challenges may result in loss of exclusivity or freedom to operate or in patent claims being narrowed, invalidated or held unenforceable, in whole or in part, which could limit our ability to stop others from using or commercializing similar or identical technology and products, or limit the duration of the patent protection of our technology and products. Given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such product candidates might expire before or shortly after such product candidates are commercialized. As a result, our owned and licensed patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours.
We may become involved in lawsuits to protect or enforce our patents or the patents of our licensors, which could be expensive, time consuming and unsuccessful. Competitors may infringe our patents or the patents of any party from whom we may license patents from in the future. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time-consuming. In a patent litigation in the U.S., defendant counterclaims alleging invalidity or unenforceability are commonplace. Grounds for a validity challenge could be an alleged failure to meet any of several statutory requirements, for example, lack of novelty, obviousnessor non-enablement. Grounds for an unenforceability assertion could be an allegation that someone connected with prosecution of the patent withheld relevant information from the USPTO or made a misleading statement during prosecution. The outcome following legal assertions of invalidity and unenforceability during patent litigation is unpredictable. A court may decide that a patent of ours or of any of our future licensors is not valid, or is unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. In addition, to the extent that we have to file patent litigation in a federal court against a U.S. patent holder, we would be required to initiate the proceeding in the state of incorporation or residency of such entity. With respect to the validity question, for example, we cannot be certain that no invalidating prior art exists. An adverse result in any litigation or defense proceedings could put one or more of our patents at risk of being invalidated, found unenforceable, or interpreted narrowly, and it could put our patent applications at risk of not issuing. Defense of these claims, regardless of their merit, would involve substantial litigation expense and would be a substantial diversion of employee resources from our business. If a defendant were to prevail on a legal assertion of invalidity or unenforceability, we would lose at least part, and perhaps all, of the patent protection on one or more of our products. Such a loss of patent protection could compromise our ability to pursue our business strategy. As noted above, interference proceedings brought by the USPTO for applications with an effective filing date before March 16, 2013, or for patents issuing from such applications may be necessary to determine the priority of inventions with respect to our patents and patent applications or those of our collaborators or licensors. An unfavorable outcome could require us to cease using the technology or to attempt to license rights to it from the prevailing party. Our business could be harmed if a prevailing party does not offer us a license on terms that are acceptable to us. Litigation or interference proceedings may fail and, even if successful, may result in substantial costs and distraction of our management and other employees. We may not be able to prevent, alone or with any of our future licensors, misappropriation of our proprietary rights, particularly in countries where the laws may not protect those rights as fully as in the U.S. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our common stock. Moreover, we may be subject toa third-party pre-issuance submission of prior art to the USPTO or other foreign patent offices, or become involved in opposition, derivation, reexamination, inter partes review, post-grant review or interference proceedings challenging our patent rights or the patent rights of others. An adverse determination in any such submission, proceeding or litigation could invalidate or reduce the scope of, our patent rights, allow third parties to commercialize our technology or drugs and compete directly with us, without payment to us, or result in our inability to manufacture or commercialize drugs without infringing third-party patent rights. In addition, if the breadth or strength of protection provided by our patents and patent applications is threatened, it could dissuade companies from collaborating with us to license, develop, or commercialize current or future product candidates. We may not be able to protect our intellectual property rights throughout the world. Filing, prosecuting and defending patents on our product candidates throughout the world would be prohibitively expensive, and our intellectual property rights in some countries outside the U.S. may be less extensive than those in the U.S. In addition, the laws and practices of some foreign countries do not protect intellectual property rights, especially those relating to life sciences, to the same extent as federal and state laws in the U.S. For example, novel formulations of drugs and manufacturing processes may not be patentable in certain jurisdictions, and the requirements for patentability may differ in certain countries, particularly developing countries. Also, some foreign countries, including EU countries, India, Japan and China, have compulsory licensing laws under which a patent owner may be compelled under certain circumstances to grant licenses to third parties. Consequently, we may have limited remedies if patents are infringed or if we are compelled to grant a license to a third party, and we may not be able to prevent third parties from practicing our inventions in all countries outside the U.S., or from selling or importing products made using our inventions into or within the U.S. or other jurisdictions. This could limit our potential revenue opportunities. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products, and may export otherwise infringing products to territories where we have patent protection, but where enforcement is not as strong as that in the U.S. These products may compete with our products in jurisdictions where we do not have any issued patents and our patent claims or other intellectual property rights may not be effective or sufficient to prevent them from competing with us in these jurisdictions. Accordingly, our efforts to enforce intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from our intellectual property. We may not prevail in any lawsuits that we initiate in these foreign countries and the damages or other remedies awarded, if any, may not be commercially meaningful.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminatedfor non-compliance with these requirements. Periodic maintenance fees, renewal fees, annuity fees and various other governmental fees on patents and applications are required to be paid to the USPTO and various governmental patent agencies outside of the U.S. in several stages over the lifetime of the patents and applications. The USPTO and various non-U.S. governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process and after a patent has issued. There are situations inwhich non-compliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Third parties may initiate legal proceedings alleging that we are infringing their intellectual property rights, the outcome of which could be uncertain and could harm our business. Our commercial success depends upon our ability, and the ability of our partners and collaborators, to develop, manufacture, market and sell our products and product candidates, if approved, and use our proprietary technologies without infringing the proprietary rights of third parties. While many of our product candidates are inpre-clinical studies and clinical trials, we believe that the use of our product candidates in thesepre-clinical studies and clinical trials falls within the scope of the exemptions provided by 35 U.S.C. Section 271(e) in the U.S., which exempts from patent infringement liability activities reasonably related to the development and submission of information to the FDA. As our other product candidates progress toward commercialization, the possibility of a patent infringement claim against us increases. Accordingly, we may invest significant time and expense in the development of our product candidates only to be subject to significant delay and expensive and time-consuming patent litigation before our product candidates may be commercialized. There can be no assurance that our products or product candidates do not infringe other parties’ patents or other proprietary rights, and competitors or other parties may assert that we infringe their proprietary rights in any event. There is considerable intellectual property litigation in the biotechnology and pharmaceutical industries. We may become party to, or threatened with, future adversarial proceedings or litigation regarding intellectual property rights with respect to our product candidates, including interference or derivation proceedings before the USPTO. Numerous U.S. and foreign issued patents and pending patent applications owned by third parties exist in the fields in which we are developing our product candidates. Third parties may assert infringement claims against us based on existing patents or patents that may be granted in the future. If we are found to infringe a third party’s intellectual property rights, we could be required to obtain a license from such third party to continue commercializing our products or product candidates. However, we may not be able to obtain any required license on commercially reasonable terms or at all. Even if a license can be obtained on acceptable terms, the rights maybe non-exclusive, which could give our competitors access to the same technology or intellectual property rights licensed to us. If we fail to obtain a required license, we may be unable to effectively market products or product candidates based on our technology, which could limit our ability to generate revenues or achieve profitability and possibly prevent us from generating revenues sufficient to sustain our operations. Alternatively, we may need to redesign our infringing products, which may be impossible or require substantial time and monetary expenditure. Under certain circumstances, we could be forced, including by court order, to cease commercializing our products or product candidates. In addition, in any such proceeding or litigation, we could be found liable for substantial monetary damages, potentially including treble damages and attorneys’ fees, if we are found to have willfully infringed. A finding of infringement could prevent us from commercializing our products or product candidates or force us to cease some of our business operations, which could harm our business. Any claims by third parties that we have misappropriated their confidential information or trade secrets could have a similar negative impact on our business. The cost to us in defending or initiating any litigation or other proceeding relating to patent or other proprietary rights, even if resolved in our favor, could be substantial, and litigation would divert our management’s attention. Some of our competitors may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could compromise our commercialization efforts, delay our research and development efforts and limit our ability to continue our operations. There could also be public announcements of the results of the hearing, motions, or other interim proceedings or developments. If securities analysts or investors perceive those results to be negative, it could cause the price of shares of our common stock to decline. Our competitors may be able to circumvent our patents by developing similar or alternative technologies or products ina non-infringing manner. Our competitors may seek approval to market their own products that are the same as, similar to or otherwise competitive with our products or product candidates. In these circumstances, we may need to defend or assert our patents by various means, including filing lawsuits alleging patent infringement requiring us to engage in complex, lengthy and costly litigation or other proceedings. In any of these types of proceedings, a court or government agency with jurisdiction may find our patents invalid, unenforceable or not infringed. We may also fail to identify patentable aspects of our research and development before it is too late to obtain patent protection. Even if we have valid and enforceable patents, these patents still may not provide protection against competing products or processes sufficient to achieve our business objectives.
Changes in either U.S. or foreign patent law or interpretation of such laws could diminish the value of patents in general, thereby impairing our ability to protect our products or product candidates. As is the case with other pharmaceutical companies, our success is heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the pharmaceutical industry involve both technological and legal complexity, and it therefore is costly, time-consuming and inherently uncertain. As noted above, the AIA has significantly changed U.S. patent law. In addition to transitioning from a“first-to-invent” “first-to-invent” to“first-to-file” “first-to-file” system, the AIA also limits where a patentee may file a patent infringement suit and provides opportunities for third parties to challenge issued patents in the USPTO via post-grant review or inter partes review, for example. All of our U.S. patents, even those issued before March 16, 2013, may be challenged by a third party seeking to institute inter partes review. Depending on decisions by the U.S. Congress, the federal courts, the USPTO, or similar authorities in foreign jurisdictions, the laws and regulations governing patents could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce our existing patents and patents that we might obtain in the future. We may be subject to claims asserting that our employees, consultants, independent contractors and advisors have wrongfully used or disclosed confidential information and/or alleged trade secrets of their current or former employers or claims asserting ownership of what we regard as our own intellectual property. Although we try to ensure that our employees, consultants, independent contractors and advisors do not use the proprietary informationor know-how of others in their work for us, we may be subject to claims that these individuals or we have inadvertently or otherwise used or disclosed confidential information and/or intellectual property, including trade secrets or other proprietary information, of the companies that any such individual currently or formerly worked for or provided services to. Litigation may be necessary to defend against these claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to our business. In addition, while we require our employees and contractors who may be involved in the conception or development of intellectual property to execute agreements assigning such intellectual property to us, we may be unsuccessful in executing such an agreement with each party who, in fact, conceives or develops intellectual property that we regard as our own. The assignment of intellectual property rights may not be self-executing or the assignment agreements may be breached, and we may be forced to bring claims against third parties, or defend claims that they may bring against us, to determine the ownership of what we regard as our intellectual property. Intellectual property rights do not prevent all potential threats to competitive advantages we may have. The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations, and intellectual property rights may not adequately protect our business or permit us to maintain our competitive advantage. The following examples are illustrative: others may be able to make drug and device components that are the same as or similar to our product candidates but that are not covered by the claims of the patents that we own or have exclusively licensed;
| • | others may be able to make drug and device components that are the same as or similar to our product candidates but that are not covered by the claims of the patents that we own or have exclusively licensed; |
we or any of our licensors or collaborators might not have been the first to make the inventions covered by the issued patent or pending patent application that we own or have exclusively licensed;
| • | we or any of our licensors or collaborators might not have been the first to make the inventions covered by the issued patent or pending patent application that we own or have exclusively licensed; |
we or any of our licensors or collaborators might not have been the first to file patent applications covering certain of our inventions;
| • | we or any of our licensors or collaborators might not have been the first to file patent applications covering certain of our inventions; |
others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our intellectual property rights;
| • | others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our intellectual property rights; |
the prosecution of our pending patent applications may not result in granted patents;
| • | the prosecution of our pending patent applications may not result in granted patents; |
granted patents that we own or have licensed may not cover our products or may be held not infringed, invalid or unenforceable, as a result of legal challenges by our competitors;
| • | granted patents that we own or have licensed may not cover our products or may be held not infringed, invalid or unenforceable, as a result of legal challenges by our competitors; |
with respect to granted patents that we own or have licensed, especially patents that we either acquireor in-license, if certain information was withheld from or misrepresented to the patent examiner, such patents might be held to be unenforceable;
| • | with respect to granted patents that we own or have licensed, especially patents that we either acquire or in-license, if certain information was withheld from or misrepresented to the patent examiner, such patents might be held to be unenforceable; |
patent protection on our product candidates may expire before we are able to develop and commercialize the product, or before we are able to recover our investment in the product;
| • | patent protection on our product candidates may expire before we are able to develop and commercialize the product, or before we are able to recover our investment in the product; |
our competitors might conduct research and development activities in the U.S. and other countries that provide a safe harbor from patent infringement claims for such activities, as well as in countries in which we do not have patent rights, and may then use the information learned from such activities to develop competitive products for sale in markets where we intend to market our product candidates;
we may not develop additional proprietary technologies that are patentable;
| • | our competitors might conduct research and development activities in the U.S. and other countries that provide a safe harbor from patent infringement claims for such activities, as well as in countries in which we do not have patent rights, and may then use the information learned from such activities to develop competitive products for sale in markets where we intend to market our product candidates; |
the patents of others may have an adverse effect on our business; and
| • | we may not develop additional proprietary technologies that are patentable; |
we may choose not to file a patent application for certain technologies, trade secretsor know-how, and a third party may subsequently file a patent covering such intellectual property.
| • | the patents of others may have an adverse effect on our business; and |
| • | we may choose not to file a patent application for certain technologies, trade secrets or know-how, and a third party may subsequently file a patent covering such intellectual property. |
Should any of these events occur, they could significantly harm our business, financial condition, results of operations and prospects. If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed. In addition to seeking patent protection for certain aspects of our product candidates and technologies, we also consider trade secrets, including confidential andunpatented know-how, important to the maintenance of our competitive position. We protect trade secrets and confidential andunpatented know-how, in part, by customarily enteringinto non-disclosure and confidentiality agreements with parties who have access to such knowledge, such as our employees, outside scientific and commercial collaborators, CROs, CMOs, consultants, advisors and other third parties. We also enter into confidentiality and invention or patent assignment agreements with our employees and consultants that obligate them to maintain confidentiality and assign their inventions to us. Despite these efforts, any of these parties may breach the agreements and disclose our proprietary information, including our trade secrets, and we may not be able to obtain adequate remedies for such breaches. In addition, our trade secrets may otherwise become known, including through a potential cybersecurity breach, or may be independently developed by competitors. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive and time-consuming, and the outcome is unpredictable. In addition, some courts in the U.S. and certain foreign jurisdictions are less willing or unwilling to protect trade secrets. If any of our trade secrets were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent them from using that technology or information to compete with us. If any of our trade secrets were to be disclosed to or independently developed by a competitor, our competitive position would be harmed. If our trademarks are not adequately protected, then we may not be able to build name recognition in our markets of interest and our business may be adversely affected. We expect to rely on trademarks as one means to distinguish any of our approved products from the products of our competitors. We have received Notices of Allowanceregistrations for DEXYCU™YUTIQ®, YUTIQ™DEXYCU®, DELIVERING INNOVATION TO THE EYE™ and Durasert™. ILUVIEN® is Alimera’s trademark. RetisertEYE® and VitrasertDURASERT®. The Verisome® are Bausch & Lomb’s trademarks.technology is exclusively licensed to us by Ramscor, Inc and the Verisome® mark is owned by Ramscor, Inc. Our and our licensees’ trademarks may be challenged, infringed, circumvented or declared generic or determined to be infringing on other marks. For instance, Sun Pharma has filed an extension of time to file an opposition to our trademark application for Durasert. Wetrademarks, we have negotiatedentered into aco-existence agreement with Sun Pharma.Pharma and a settlement agreement with Merck allowing continued, though somewhat limited, use of two of our marks. If we are unable to establish name recognition based on our trademarks, we may not be able to compete effectively. ILUVIEN® is Alimera’s trademark. Retisert® and Vitrasert® are Bausch & Lomb’s trademarks. RISKS RELATED TO OUR RELIANCE ON THIRD PARTIES We do not control theThe development orand commercialization of YUTIQ inour lead product candidate, EYP-1901, is dependent on intellectual property we license from Equinox Science. If we breach our agreement with Equinox or the EMEA, whichagreement is licensedterminated, we could lose license rights that are material to Alimera, and as a resultour business.
Pursuant to our license agreement with Equinox, we may not realize the full market potential of YUTIQ. Under the Amended Alimera Agreement, we granted Alimeraacquired exclusive rights to use our proprietary drug delivery platformpatents, patent applications and know-how owned or controlled by Equinox relating to the compound vorolanib, a tyrosine kinase inhibitor, for the treatment of uveitis,wet AMD, diabetic retinopathy, and retinal vein occlusion. Our lead product candidate, EYP-1901, utilizes vorolanib in combination with our proprietary Durasert sustained release technology. Our license agreement with Equinox imposes various development, regulatory, commercial, financial and other obligations on us. If we fail to comply with our obligations under the agreement with Equinox, or otherwise materially breach the agreement with Equinox, and fail to remedy such failure or cure such breach within 90 days, Equinox will have the right to terminate the agreement. If our agreement with Equinox is terminated by Equinox for our uncured material breach, we would lose our license and all rights to the use of vorolanib for EYP-1901. The loss of the license from Equinox would prevent us from developing and commercializing EYP-1901 and could subject us to claims of breach of contract and patent infringement from Equinox if any continued research, development, manufacture or commercialization of EYP-1901 is covered by the affected patents. Accordingly, the loss of our license from Equinox would materially harm our business.
If we are unable to maintain our agreement with ImprimisRx to co-promote DEXYCU, we may be unable to generate significant revenue from this product. When we launched YUTIQ and DEXYCU in 2019, we contracted with an outsourced CSO to commercialize the products. We terminated our relationship with the CSO and converted the CSO’s remaining YUTIQ KAMs to full-time employees as of January 2020, and we converted the CSO’s remaining DEXYCU KAMs to full-time employees as of October 2020. In August 2020, to complement and augment the efforts of our internal sales team for DEXYCU, we signed a Commercial Alliance Agreement with ImprimisRx for the sale of DEXYCU to its customers. To a significant degree, we are relying on our strategic collaboration with ImprimisRx to grow our sales of DEXYCU. As a result of our agreement with ImprimisRx, ImprimisRx now executes a large part of the sales efforts for DEXYCU and those efforts may be affected by ImprimisRx’s organization, operations, activities and events both related and unrelated to DEXYCU. Our co-promotion efforts with ImprimisRx have encountered and continue to encounter a number of difficulties, uncertainties and challenges, including NIPU,curtailments in the EMEA (underperformance of cataract surgeries due to the ILUVIEN trademark)Pandemic, which have impacted DEXYCU sales growth. Any failure to fully optimize this co-promotion arrangement with ImprimisRx, by either partner, could also cause DEXYCU sales and subsequently withdrew our financial results to be disappointing and hurt the price of our common stock. Any disputes with ImprimisRx over these or other issues could harm the promotion and sales of DEXYCU and could result in substantial costs to us. If we encounter issues with our CMOs or suppliers, we may need to qualify alternative manufacturers or suppliers, which could impair our ability to sufficiently and timely manufacture and supply DEXYCU. We currently depend on CMOs and suppliers for DEXYCU. Although we could obtain the drug product and other components for DEXYCU from other CMOs and suppliers, we would need to qualify and obtain FDA approval for such CMOs or suppliers as alternative sources, which could be costly and cause significant delays. In addition, the manufacturer of the drug product in DEXYCU conducts its manufacturing operations for us at a single facility. Unless and until we qualify additional facilities, we may face limitations in our ability to respond to manufacturing issues. For example, if regulatory, manufacturing or other problems require this manufacturer to discontinue production at its facility, or if the equipment used for the production of the drug product in this facility is significantly damaged or destroyed by fire, flood, earthquake, power loss or similar events, the ability of such manufacturer to manufacture DEXYCU may be significantly impaired. In the event that this party suffers a temporary or protracted loss of its materials, facility or equipment, we would still be required to obtain FDA approval to qualify a new manufacturer as an alternate manufacturer for the drug product before any drug product manufactured by such manufacturer could be sold or used. Any production shortfall that impairs the supply of DEXYCU could adversely affect our ability to satisfy demand for DEXYCU, which could have a material adverse effect on our product sales, results of operations and financial condition. The Pandemic may also have an adverse impact on our CMOs or suppliers as a result of employees or other key personnel becoming infected, preventive and precautionary measures that governments or such third parties are taking, such as social distancing, quarantines, and other restrictions, and shortages of supplies necessary for the manufacture of DEXYCU. Any of these circumstances could adversely impact the ability of third parties on which we rely to manufacture and distribute adequate volumes of DEXYCU. We use our own facility for the manufacturing of YUTIQ, MAAwhich requires significant resources, and orphan drug designation for posterior segment uveitis. Alimerawhich could adversely affect its commercial viability. We currently manufacture commercial supplies of YUTIQ ourselves at our Watertown, MA facility. We have, and will continue, to perform extensive audits of our suppliers, vendors and contract laboratories. The cGMP requirements govern, among other things, recordkeeping, production processes and controls, personnel and quality control. To ensure that we continue to meet these requirements, we have and will continue to expend significant time, money and effort. The commercial manufacture of medical products is now responsible for obtaining all regulatory approvalscomplex and requires significant expertise and capital investment, including the development of advanced manufacturing techniques and process controls. Manufacturers of medical products often encounter difficulties in production, particularly in scaling out and validating initial production and ensuring the absence of contamination. These problems include difficulties with production costs and yields, quality control, including stability of the product, quality assurance testing, operator error, shortages of qualified personnel, as well as compliance with strictly enforced federal, state and foreign regulations. We cannot assure you that any issue relating to the manufacture of YUTIQ will not occur in the EMEA. Underfuture. The FDA also may, at any time following approval of a product for sale, audit our manufacturing facilities. If any such inspection or audit identifies a failure to comply with applicable regulations or if a violation of our product specifications or applicable regulation occurs independent of such an inspection or audit, FDA may issue a Form FDA-483 and/or an untitled or warning letter, or we or the FDA may require remedial measures that may be costly and/or time consuming for us to implement and that may include the
temporary or permanent suspension of commercial sales, recalls, market withdrawals, seizures or the temporary or permanent closure of a facility. Any such remedial measures imposed upon us could materially harm our business. In addition, although we could contract with other third parties to manufacture YUTIQ, we would need to qualify and obtain FDA approval for a contract manufacturer or supplier as an alternative source for YUTIQ, which could be costly and cause significant delays. Our YUTIQ manufacturing operations depend on our Watertown, MA facility. If this agreement,facility is destroyed or is out of operation for a substantial period of time, our business may be adversely impacted. We currently conduct our manufacturing operations related to YUTIQ in our facility located in Watertown, MA. If regulatory, manufacturing or other problems, including Pandemic-related impacts on our employees, require us to suspend or discontinue production at our Watertown, MA facility, we will not be able to have noor maintain adequate commercial supply of YUTIQ, which would adversely impact our business. If the facility or the equipment in it is significantly damaged or destroyed by fire, flood, power loss or similar events, we may not be able to quickly or inexpensively replace our facility. In the event of a temporary or protracted loss of either facility or equipment, we might not be able to transfer manufacturing to a third party. Even if we could transfer manufacturing to a third party, the shift would likely be expensive and time-consuming, particularly since the new facility would need to comply with necessary regulatory requirements. The Pandemic may also have an adverse impact on our manufacturing activities for YUTIQ as a result of employees or other key personnel becoming infected, preventive and precautionary measures that governments or such third parties are taking, such as social distancing, quarantines, and other restrictions, and shortages of supplies necessary for the manufacture of YUTIQ. Any of these circumstances could adversely impact our ability to manufacture and distribute adequate volumes of YUTIQ. If third-party manufacturers, wholesalers and distributors fail to devote sufficient time and resources to DEXYCU or their performance is substandard, our product supply may be impacted. Our reliance on a limited number of manufacturers, wholesalers and distributors exposes us to the following risks, any of which could limit commercial supply of our products, result in higher costs, or deprive us of potential product revenues: | • | our CMOs, or other third parties we rely on, may encounter difficulties in achieving the volume of production needed to satisfy commercial demand, may experience technical issues that impact quality or compliance with applicable and strictly enforced regulations governing the manufacture of pharmaceutical products, and may experience shortages of qualified personnel to adequately staff production operations; |
| • | our wholesalers and distributors could become unable to sell and deliver DEXYCU for regulatory, compliance and other reasons; |
| • | our CMOs, wholesalers and distributors could default on their agreements with us to meet our requirements for commercial supply of DEXYCU; |
| • | our CMOs, wholesalers and distributors may not perform as agreed or may not remain in business for the time required to successfully produce, store, sell and distribute DEXYCU and we may incur additional cost; and |
| • | if our CMOs, wholesalers and distributors were to terminate our arrangements or fail to meet their contractual obligations, we may be forced to delay the commercialization of DEXYCU. |
Our reliance on third parties reduces our control over Alimera’s regulatoryour development and commercialization activities in the EMEA (with the exception of the completionbut does not relieve us of our ongoing Phase 3 uveitis clinical trials), includingresponsibility to ensure compliance with all required legal, regulatory approvals, and no direct control over commercialization efforts for YUTIQ inscientific standards. For example, the EMEA. Alimera has only limited experience in filingFDA and supporting the applications necessary to obtain marketing approvals for product candidates. Obtaining approval of an MAA by the EMA is uncertain and Alimera may fail to obtain the approval. The MAA review processes, and the processes of other regulatory authorities are extensive, lengthy, expensive,require that our product candidates and uncertain,any products that we may eventually commercialize be manufactured according to cGMP and such regulatory authorities maysimilar foreign standards. Any failure by our third-party manufacturers to comply with cGMP or failure to scale up manufacturing processes, including any failure to deliver sufficient quantities of product candidates in a timely manner, could lead to a delay limit,in, or deny approval of YUTIQ. Further, Alimera may abandon further development of YUTIQ in the EMEA. Because the full market potential of YUTIQ is contingent upon the successful development and commercialization of YUTIQ in the EMEA, we will be dependent on Alimerafailure to achieve the full market potential of YUTIQ. If Alimera does not succeed in obtainingobtain, regulatory approval of YUTIQ inany of our product candidates or supply our commercial volume of DEXYCU. In addition, such failure could be the EMEAbasis for any reason,the FDA to issue a warning or does not succeed in securing market acceptanceuntitled letter, withdraw approvals for products previously granted to us, or take other regulatory or legal action, including recall or seizure, total or partial suspension of YUTIQ inproduction, suspension of ongoing clinical trials, refusal to approve pending applications or supplemental applications, detention or product, refusal to permit the EMEA,import or elects for any reason to discontinue developmentexport of YUTIQ, we will be unable to realize the full market potential of YUTIQ.products, injunction, imposing civil penalties or pursuing criminal prosecution.
If our CROs, vendors and investigators do not successfully carry out their responsibilities or if we lose our relationships with them, our development efforts with respect to our product candidates could be delayed. We are dependent on CROs, vendors and investigators forpre-clinical testing and clinical trials related to our product development programs.programs, including for EYP-1901. These parties are not our employees, and we cannot control the amount or timing of resources that they devote to our programs. If they do not timely fulfill their responsibilities or if their performance is inadequate, the development and commercialization of our product candidates could be delayed. The parties with which we contract for execution of clinical trials play a significant role in the conduct of the trials and the subsequent collection and analysis of data. Their failure to meet their obligations could adversely affect clinical development of our product candidates. In addition, if we or our CROs fail to comply with applicable current Good Clinical Practices or cGCPs,(“GCP”), the clinical data generated in our clinical trials may be deemed unreliable and the FDA may require us to perform additional clinical trials before approving any marketing applications. Upon inspection, the FDA may determine that our clinical trials did not comply with cGCPs.GCP. Switching or adding additional CROs involves additional cost and requires management time and focus. Identifying, qualifying and managing performance of third-party service providers can be difficult, time-consuming and cause delays in our development programs. In addition, there is a natural transition period when a new CRO commences work and the new CRO may not provide the same type or level of services as the original provider. Though we carefully manage our relationships with our CROs, there can be no assurance that we will not encounter challenges or delays in the future or that these delays or challenges will not have a material adverse impact on our business, financial condition and prospects. If any of our relationships with our CROs terminate, we may not be able to enter into arrangements with alternative CROs or to do so on commercially reasonable terms. As a result, delays may occur, which can materially impact our ability to meet our desired clinical development timelines. Because we have relied on third parties, our internal capacity to perform these functions is limited. Outsourcing these functions involves risks that third parties may not perform to our standards, may not produce results in a timely manner or may fail to perform at all. In addition, the use of third-party service providers requires us to disclose our proprietary information to these parties, which could increase the risk that this information will be misappropriated. We currently have a small number of employees, which limits the internal resources we have available to identify and monitor our third-party providers. To the extent we are unable to identify and successfully manage the performance of third-party service providers in the future, our ability to advance our product candidates through clinical trials will be compromised. Though we carefully manage our relationships with our CROs, there can be no assurance that we will not encounter similar challenges or delays in the future or that these delays or challenges will not have a material adverse impact on our business, financial condition and prospects. If we encounter difficulties in negotiating commercial manufacturing and supply agreements with our third-party manufacturers and suppliers of DEXYCU, our ability to commercialize DEXYCU would be impaired.
We currently rely, and expect to continue to rely, on a limited number of CMOs and suppliers who assist in the production, assembly, test, supply, storage and distribution of DEXYCU in our FDA registration, and we control only some of the aspects of their activities. We may not be able to obtain terms that are favorable to us or enter into commercial manufacturing and supply agreements at all with each of the necessary third parties. If we are unable to enter into such agreements on commercially reasonable terms, our ability to commercialize DEXYCU would be impaired, and our business, financial condition and results of operations would be materially adversely affected.
If we encounter issues with our contract manufacturers or suppliers, we may need to qualify alternative manufacturers or suppliers, which could impair our ability to sufficiently and timely manufacture and supply DEXYCU.
We currently depend on contract manufacturers and suppliers for DEXYCU. We expect to obtain the drug product for commercial supply of DEXYCU from one CMO. Although we could obtain the drug product for
DEXYCU from other third-party suppliers, we would need to qualify and obtain FDA approval for another contract manufacturer or supplier as an alternative source for the drug product, which could be costly and cause significant delays. In addition, the manufacturer of the drug product in DEXYCU conducts its manufacturing operations for us at a single facility. Unless and until we qualify additional facilities, we may face limitations in our ability to respond to manufacturing and supply issues. For example, if regulatory, manufacturing or other problems require this manufacturer to discontinue production at its facility, or if the equipment used for the production of the drug product in this facility is significantly damaged or destroyed by fire, flood, earthquake, power loss or similar events, the ability of such manufacturer to manufacture DEXYCU may be significantly impaired. In the event that this party suffers a temporary or protracted loss of its materials, facility or equipment, we would still be required to obtain FDA approval to qualify a new manufacturer or supplier, as applicable, as an alternate manufacturer or source for the drug product before any drug product manufactured by such manufacturer or by such supplier could be sold or used.
Any production shortfall that impairs the supply of DEXYCU could have a material adverse effect on our business, financial condition and results of operations and adversely affect our ability to satisfy demand for DEXYCU, which could adversely affect our product sales and operating results materially.
If we or our licensees encounter problems with product manufacturing, there could be delays in product development or commercialization, which would adversely affect our future profitability.
Our ability and that of our licensees to conduct timelypre-clinical and clinical research and development programs, obtain regulatory approvals, and develop and commercialize our product candidates will depend, in part, upon our and our licensees’ ability to manufacture our products and product candidates, either directly or through third parties, in accordance with FDA and other regulatory requirements. The manufacture, packaging and testing of our products and product candidates are regulated by the FDA and similar foreign regulatory entities and must be conducted in accordance with applicable cGMP and comparable foreign requirements. Any change in a manufacturing process or procedure used for one of our products or product candidates, including a change in the location at which a product or product candidate is being manufactured or in the third-party manufacturer being used, may require the FDA’s and similar foreign regulatory entities’ prior review and/or approval in accordance with applicable cGMP or other regulations. Additionally, the FDA and similar foreign regulatory entities may implement new standards, or change their interpretation and enforcement of existing standards, for the manufacture, packaging and testing of products at any time.
There are a limited number of manufacturers that operate under cGMP and other foreign regulations that are both capable of manufacturing our products and product candidates and are willing to do so. Alimera has contracted with individual third-party manufacturers for the manufacture of ILUVIEN and its components. If any of Alimera’s third-party manufacturers breach their agreements or are unable or unwilling to perform for any reason or fail to comply with cGMP and comparable foreign requirements, Alimera may not be able to locate alternative acceptable manufacturers, enter into favorable agreements with them or get them approved by the applicable regulatory authorities in a timely manner. Delays in the commercial production of ILUVIEN could delay or impair Alimera’s marketing of ILUVIEN, which, in turn, could adversely affect Alimera’s generation of sales-based royalties to us. Failure by us, our collaborative partners, or our or their third-party manufacturers, to comply with applicable manufacturing requirements could result in sanctions being imposed on us or our collaborative partners, including fines, injunctions, civil penalties, failure of regulatory authorities to grant marketing approval of our product candidates, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of product, operating restrictions and criminal prosecutions. If we or our collaborative partners are unable to develop our own manufacturing facilities or to obtain or retain third-party manufacturing on acceptable terms, we may not be able to conduct certain futurepre-clinical and clinical testing of our product candidates and our collaborative partners may not be able to conduct certain futurepre-clinical and clinical testing or to supply commercial quantities of our products. We manufacture supplies in connection withpre-clinical or clinical studies conducted by us and our licensees. Our licensees have the exclusive rights to
manufacture commercial quantities of products, once approved for marketing. Our licensees’ reliance on third-party manufacturers entails risks, including:
failure of third parties to comply with cGMP and other applicable U.S. and foreign regulations and to employ adequate quality assurance practices;
inability to obtain the materials necessary to produce a product or to formulate the active pharmaceutical ingredient on commercially reasonable terms, if at all;
supply disruption, deterioration in product quality or breach of a manufacturing or license agreement by the third party because of factors beyond our or our licensees’ control;
termination ornon-renewal of a manufacturing or licensing agreement with a third party at a time that is costly or difficult; and
inability to identify or qualify an alternative manufacturer in a timely manner, even if contractually permitted to do so.
If third-party manufacturers, wholesalers and distributors fail to devote sufficient time and resources to DEXYCU or their performance is substandard, our product supply may be impacted.
Our reliance on a limited number of manufacturers, wholesalers and distributors exposes us to the following risks, any of which could limit commercial supply of our products, result in higher costs, or deprive us of potential product revenues:
our CMOs, or other third parties we rely on, may encounter difficulties in achieving the volume of production needed to satisfy commercial demand, may experience technical issues that impact quality or compliance with applicable and strictly enforced regulations governing the manufacture of pharmaceutical products, and may experience shortages of qualified personnel to adequately staff production operations;
our wholesalers and distributors could become unable to sell and deliver DEXYCU for regulatory, compliance and other reasons;
our CMOs, wholesalers and distributors could default on their agreements with us to meet our requirements for commercial supply of DEXYCU;
our CMOs, wholesalers and distributors may not perform as agreed or may not remain in business for the time required to successfully produce, store, sell and distribute DEXYCU and we may incur additional cost; and
if our CMOs, wholesalers and distributors were to terminate our arrangements or fail to meet their contractual obligations, we may be forced to delay the commercialization of DEXYCU.
Our reliance on third parties reduces our control over our development and commercialization activities but does not relieve us of our responsibility to ensure compliance with all required legal, regulatory and scientific standards. For example, the FDA and other regulatory authorities require that our product candidates and any products that we may eventually commercialize be manufactured according to cGMP and similar foreign standards. Any failure by our third-party manufacturers to comply with cGMP or failure to scale up manufacturing processes, including any failure to deliver sufficient quantities of product candidates in a timely manner, could lead to a delay in, or failure to obtain, regulatory approval of any of our product candidates or supply our commercial volume of DEXYCU. In addition, such failure could be the basis for the FDA to issue a warning or untitled letter, withdraw approvals for products previously granted to us, or take other regulatory or legal action, including recall or seizure, total or partial suspension of production, suspension of ongoing clinical trials, refusal to approve pending applications or supplemental applications, detention or product, refusal to permit the import or export of products, injunction, imposing civil penalties or pursuing criminal prosecution.
Manufacturing issues may arise that could result in delays or suspension of the commercialization of DEXYCU.
As we further scale up manufacturing of DEXYCU and conduct required stability testing, issues may arise involving, among other things, the sensitivity of the tests to confirm product stability, the performance of production personnel, product-packaging or third-party equipment malfunctions. These issues may require refinement or resolution in order to initiate or continue the commercial marketing of DEXYCU. In addition, quality issues may arise duringscale-up and of commercial manufacturing processes. By way of example, we are still in the process of validating the manufacturing process for the commercial supply of DEXYCU. Any issues in our product or delivery devices could result in delays in the commercialization of DEXYCU or the suspension of commercialization of DEXYCU.
We intend to use our own facility for the manufacturing of YUTIQ, which will require significant resources, which could adversely affect its commercial viability.
If approved by the FDA, we plan to manufacture commercial supplies of YUTIQ ourselves at our Watertown MA facility. We currently manufacture products only for clinical and testing purposes in this facility and we do not manufacture products for commercial use. We must obtain FDA approval of our manufacturing process before we can commercially manufacture YUTIQ in the U.S. In addition, we must pass apre-approval inspection of our manufacturing facility before we can obtain marketing approval for YUTIQ. In order to obtain approval, all of our manufacturing methods, equipment and processes must comply with the FDA’s cGMP requirements. We will also need to perform extensive audits of our suppliers, vendors and contract laboratories. The cGMP requirements govern, among other things, recordkeeping, production processes and controls, personnel and quality control. To ensure that we meet these requirements, we will expend significant time, money and effort. Due to the unique nature of our Durasert technology platform, we cannot predict the likelihood that the FDA will approve our facility as compliant with cGMP requirements even if we believe that we have taken the steps necessary to achieve compliance.
The FDA, in its regulatory discretion, may require us to undergo additional clinical trials with respect to any new or improved manufacturing process we may develop or utilize in the future. This could delay or prevent approval of YUTIQ or any of our other product candidates. If we fail to comply with cGMP requirements, pass an FDApre-approval inspection or obtain FDA approval of our manufacturing process, we would not receive FDA approval and would be subject to possible regulatory action, including recall or withdrawal of the product from the market or suspension of manufacturing. The failure to successfully implement our manufacturing process may delay or prevent our ability to commercialize YUTIQ.
If we do obtain FDA approval for YUTIQ, including satisfying the FDA with regard to a validated manufacturing process, we may still be unable to commercially manufacture YUTIQ. The commercial manufacture of medical products is complex and requires significant expertise and capital investment, including the development of advanced manufacturing techniques and process controls. Manufacturers of medical products often encounter difficulties in production, particularly in scaling out and validating initial production and ensuring the absence of contamination. These problems include difficulties with production costs and yields, quality control, including stability of the product, quality assurance testing, operator error, shortages of qualified personnel, as well as compliance with strictly enforced federal, state and foreign regulations. We cannot assure you that any stability or other issues relating to the manufacture of YUTIQ, if approved, will not occur in the future.
The FDA also may, at any time following approval of a product for sale, audit our manufacturing facilities. If any such inspection or audit identifies a failure to comply with applicable regulations or if a violation of our product specifications or applicable regulation occurs independent of such an inspection or audit, FDA may issue a FormFDA-483 and/or an untitled or warning letter, or we or the FDA may require remedial measures that may be costly and/or time consuming for us to implement and that may include the temporary or permanent
suspension of commercial sales, recalls, market withdrawals, seizures or the temporary or permanent closure of a facility. Any such remedial measures imposed upon us could materially harm our business.
Our employees, collaborators, service providers, independent contractors, principal investigators, consultants, CSOs,co-promotion partners, vendors and CROs may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements. We are exposed to the risk that our employees, collaborators, independent contractors, principal investigators, consultants, CSOs,co-promotion partners, vendors and CROs may engage in fraudulent or other illegal activity with respect to our business. Misconduct by these employees could include intentional, reckless and/or negligent conduct or unauthorized activity that violates: FDA regulations, including those laws requiring the reporting of true, complete and accurate information to the FDA;
| • | FDA regulations, including those laws requiring the reporting of true, complete and accurate information to the FDA; |
manufacturing standards;
| • | manufacturing standards; |
federal and state healthcare fraud and abuse laws and regulations; or
| • | federal and state healthcare fraud and abuse laws and regulations; or |
laws that require the true, complete and accurate reporting of financial information or data.
| • | laws that require the true, complete and accurate reporting of financial information or data. |
In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Misconduct by these parties could also involve individually identifiable information, including, without limitation, the improper use of information obtained in the course of clinical trials, or illegal misappropriation of drug product, which could result in regulatory sanctions and serious harm to our reputation. Any incidents or any other conduct that leads to an employee receiving an FDA debarment could result in a loss of business from third parties and severe reputational harm. Although we have adopted a Code of Business Conduct to govern and deter such behaviors, it is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of civil, criminal and administrative penalties, damages, monetary fines, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, contractual damages, reputational harm, diminished profits and future earnings, additional reporting requirements and oversight if we become subject to a corporate integrity agreement or similar agreement to resolve allegations ofnon-compliance with these laws, and curtailment of our operations. The success of our current and possible future collaborative and licensing arrangements depends and will depend heavily on the experience, resources, efforts and activities of our licensees, and if they are not successful in developing and marketing our products or product candidates, as applicable, it will adversely affect our revenues, if any, from those products.
Our business strategy includes continuing to leverage our technology platform by entering into collaborative and licensing arrangements for the development and commercialization of our products and product candidates, where appropriate. The success of current and future collaborative and licensing arrangements do and will depend heavily on the experience, resources, skill, efforts and activities of our licensees. Our licensees have had, and are expected to have, significant discretion in making decisions related to the development of product
candidates and the commercialization of products under these collaboration agreements. Risks that we face in connection with our collaboration and licensing strategy include the following:
our collaborative and licensing arrangements are, and are expected to be, subject to termination under various circumstances, including on short notice and without cause;
we are required, and expect to be required, under our collaborative and licensing arrangements, not to conduct specified types of research and development in the field that is the subject of the arrangement or not to sell products in such field, limiting the areas of research, development and commercialization that we can pursue;
our licensees may develop and commercialize, either alone or with others, products that are similar to or competitive with our products;
our licensees may change the focus of their development and commercialization efforts or decrease or fail to increase spending related to our products or product candidates, thereby limiting the ability of these products to reach their potential;
our licensees may lack the funding, personnel or experience to develop and commercialize our products successfully or may otherwise fail to do so; and
our licensees may not perform their obligations, in whole or in part.
We currently have collaboration and licensing arrangements with various companies, most significantly Alimera and Bausch & Lomb. While Bausch & Lomb has significant experience in the ophthalmic field and substantial resources, there is no assurance whether, and to what extent, that experience and those resources will be devoted to Retisert, and we do not expect revenues from Retisert to increase, and they may decline further. Although we believe potential revenues from ILUVIEN are important to our future results of operations and financial condition, Alimera has limited experience and limited financial resources, and ILUVIEN for DME is currently Alimera’s first and only commercial product. Alimera has reported that its negative cash flows from operations and accumulated deficit may raise substantial doubt about its ability to continue as a going concern. Further, due to the limited revenue generated by Alimera to date, Alimera may not be able to maintain compliance with covenants under its loan agreement and, in the event of a default, we do not know whether Alimera will be able to obtain amendments or waivers of those covenants. We do not know if Alimera will be able to raise additional financing if and when required.
If our current and future licensees are not successful in developing and marketing our products, it will adversely affect our revenues, if any, from those products.
Our current licensees may terminate their agreements with us at any time or fail to fulfill their obligations under those agreements, and, if they do, we will lose the benefits of those agreements.
Our licensees have rights of termination under our agreements with them and could terminate those agreements without cause on short notice. Further, our licensees may fail to fulfill their obligations under their agreements, or we may disagree with them over the rights and obligations under those agreements, which could result in breach of the agreements and/or termination. Exercise of termination rights by one or more of our licensees or by us may leave us without the financial benefits and development, marketing or sales resources provided under the terminated agreement. It could be necessary for us to replace, or seek to provide ourselves, the services provided by the licensee, and there is no assurance we would be successful in doing so. It could delay, impair or stop the development or commercialization of products or product candidates licensed to them or require significant additional capital investment by us, which we may not have the resources to fund. If any of our licensees do not perform their obligations under our agreements or if any of those agreements are terminated, it could have an adverse effect on our business, financial condition and results of operations.
Off-label sales of ILUVIEN to treat posterior segment uveitis may adversely affect sales of YUTIQ, if approved.
The micro-inserts that comprise ILUVIEN and YUTIQ have substantially the same design, polymers and release rate, and both deliver the corticosteroid FA. Although YUTIQ delivers a somewhat lower dose of FA than ILUVIEN, ILUVIEN is already approved and marketed. It is possible that physicians will prescribe ILUVIEN for the treatment of posterior segment uveitis on anoff-label basis, which could adversely affect the sales of YUTIQ, if approved.
There is no assurance that Alimera will successfully commercialize ILUVIEN for DME or that we will receive significant revenues from the commercialization of ILUVIEN.
We are entitled to royalties on acountry-by-country andquarter-by-quarter basis on net sales of ILUVIEN where Alimera markets ILUVIEN directly and to a percentage of product revenues, royalties andnon-royalty consideration where Alimera sublicenses the marketing of ILUVIEN. The commercialization of ILUVIEN for DME is a significant undertaking by Alimera, and ILUVIEN for DME is Alimera’s first and only commercial product. Alimera’s sales of ILUVIEN have not been significant to date, Alimera has continued to incur operating losses, and it has violated, and in the future may violate, the financial covenants of its loan agreement. We do not know if, when, or to what extent Alimera’s ILUVIEN net revenues will increase significantly, which would generate royalties to us from the commercialization of ILUVIEN for DME. The amount and timing of any revenues we receive will be affected by many factors including:
Alimera’s and its distributors’ and sublicensees’ ability to effectively market and sell ILUVIEN in each country where sold;
the manner of sale, whether directly by Alimera or by sublicensees or distributors, and the terms of sublicensing and distribution agreements;
the amount and timing of sales of ILUVIEN in each country;
regulatory approvals, appropriate labeling, and desirable pricing, insurance coverage and reimbursement;
commencement of marketing in additional countries; and
Alimera’s ability to raise adequate capital as needed to fund its operations, to maintain compliance with its loan agreement and to achieve profitability from its operations.
If Alimera is not successful in commercializing ILUVIEN for DME, it would adversely affect our business, operating results and financial condition.
Alimera may need alternative financing to replace its $40.0 million debt facility or additional capital to maintain compliance with the financial covenants under its loan agreement, which Alimera may be unable to obtain, and Alimera’s continued losses and financial condition may cast doubt on its ability to continue to operate as a going concern.
Although Alimera commercially markets ILUVIEN for DME in the U.S. and in certain countries in Europe, Alimera had a net loss from operations of $16.5 million for its fiscal year ended December 31, 2017, a net loss of $11.6 million for the interimsix-month period ended June 30, 2018 and had an accumulated deficit of $410.8 million as of June 30, 2018. Alimera has not generated revenues that cover its actual or anticipated expenses and cannot project the extent of its future losses. Alimera may continue to incur operating losses, and as a result, it is uncertain when or if it will achieve or sustain profitability. Alimera’s ability to generate royalty payments to us is dependent on its ability to successfully market and sell ILUVIEN for DME and, if approved, ILUVIEN for NIPU.
Alimera failed to meet a revenue threshold in January 2016 and a liquidity threshold as of June 30, 2016 under the financial covenants of its former loan agreement. While these failures were subsequently waived by its former lender, Alimera was required to pay substantial amounts and grant concessions in connection with these waivers. In January 2018, Alimera refinanced its debt with a $40 million loan agreement with a new lender. If Alimera defaults on its obligations under its new loan agreement, its new lender may call the loan, which could require Alimera to pay back the entire amount owed and pay an early termination fee, or if the lender does not call the loan, Alimera may have to pay an increased rate of interest, pay additional monetary amounts in exchange for a waiver or modification of the loan agreement, or grant equity or warrant coverage and agree to further restrictions on its operations that could hinder it in the future. Alimera’s failure to comply with the covenants under its new loan agreement could create substantial doubt about Alimera’s ability to continue as a going concern and to market and sell ILUVIEN. The termination provisions of our agreement with Alimera include various bankruptcy events.
Further, due to the limited revenue generated by ILUVIEN to date, even if Alimera is able to maintain compliance with its debt covenants, Alimera may need to raise additional capital to fund the continued commercialization of ILUVIEN. If Alimera is unable to raise sufficient additional financing, it may need to adjust its commercial plans, which likely would adversely affect Alimera’s ability to market ILUVIEN and make any potential royalty payments to us.
There is no assurance our Retisert royalty income will continue at current levels or at all.
Our Retisert royalty income, which had ranged between $1.2 million and $1.4 million from fiscal 2012 through fiscal 2016, decreased to $970,000 for fiscal 2017 and totaled approximately $1.0 million for fiscal 2018. We do not expect Retisert royalty income to increase materially, if at all, and it may decline further or cease. Bausch & Lomb’s obligation to pay a royalty terminates on a licensed product by licensed product basis and country by country basis upon the date that the last to expire patent expires. The patent with which Retisert is marketed expires in March 2019. The latest patent covering Retisert expires in April 2020, and we will not receive any Retisert royalty income after that time. Bausch & Lomb previously ceased selling Vitrasert, our product previously licensed to Bausch & Lomb, on its patent expiration.
Sales of ILUVIEN for DME may be materially adversely affected by pricing and reimbursement decisions of regulatory bodies, insurers and others.
Prices, coverage and reimbursement to consumers of ILUVIEN for DME, like other products, are generally regulated by third-party payors, such as government health administration authorities and plans, private health insurers and other organizations and affect ILUVIEN’s sales. The timing and complexity of those reimbursements also affect sales. Prices in the EU are generally lower and coverage and access to products more limited than in the U.S. For example, in the U.K. and Scotland, National Health Service coverage is limited to the treatment of the eyes of chronic DME patients unresponsive to existing therapies that have undergone cataract surgery, subject to simple patient access schemes. Alimera may not achieve satisfactory agreements with statutory or other insurers. We do not know what levels of pricing will be approved or reimbursed for ILUVIEN, or what restrictions will be placed on its use or reuse in countries where ILUVIEN is not currently sold. In the U.S., Alimera has offered extended customer payment terms. Future net sales of ILUVIEN and, accordingly, the amount of royalties that we may receive from such net sales, may be adversely affected by pricing and reimbursement decisions, and such effects may be material.
If we or our licensees fail to comply with environmental laws and regulations, our or their ability to manufacture and commercialize products may be adversely affected.
Medical and biopharmaceutical research and development involves the controlled use of hazardous materials, such as radioactive compounds and chemical solvents. We and our licensees are subject to federal, state and local laws and regulations in the U.S. and abroad governing the use, manufacture, storage, handling and
disposal of such materials and waste products. We and they could be subject to both criminal liability and civil damages in the event of an improper or unauthorized release of, or exposure of individuals to, hazardous materials. In addition, claimants may sue us or them for resulting injury or contamination, and the liability may exceed our or their ability to pay. Compliance with environmental laws and regulations is expensive, and current or future environmental regulations may impair the research, development or production efforts of our company or our licensees and harm our operating results.
RISKS RELATED TO OUR INDUSTRY, STRATEGY AND OPERATIONS
If we fail to retain key personnel, our business could suffer.
We are dependent upon the principal members of our management and scientific staff. In addition, we believe that our future success in developing and marketing our products will depend on whether we can attract and retain additional qualified management and scientific personnel as well as a sales and marketing staff. There is strong competition for qualified personnel within the industry in which we operate, and we may not be able to attract and retain such personnel. As we have a small number of employees and we believe our products and product candidates are unique and highly specialized, the loss of the services of one or more of the principal members of our management or scientific staff, or the inability to attract and retain additional personnel and develop expertise as needed, could have a material adverse effect on our results of operations and financial condition.
We will need to grow the size of our organization, and we may experience difficulties in managing this growth.
Implementation of our development and commercialization strategies will require additional managerial, operational, sales, marketing, financial and other resources. Our current management, personnel and systems may not be adequate to effectively manage the expansion of our operations, which may result in weaknesses in our infrastructure, give rise to operational mistakes, loss of business opportunities, employee turnover and reduced productivity. Future growth could require significant capital expenditures and may divert financial resources from other projects, such as the development of our existing or future product candidates. Future growth would impose significant added responsibilities on members of management, including:
managing the commercialization of DEXYCU and, if approved, YUTIQ;
overseeing ourpre-clinical studies and clinical trials effectively;
identifying, recruiting, maintaining, motivating and integrating additional employees, including any sales and marketing personnel engaged in connection with the commercialization of DEXYCU and, if approved, YUTIQ;
engaging and managing our relationship with any contract sales organizations; and
managing our internal development efforts effectively while complying with our contractual obligations to licensors, licensees, contractors and other third parties; and improving our managerial, development, operational and financial systems and procedures.
As our operations expand, we will need to manage additional relationships with various strategic collaborators, suppliers and other third parties. Our future financial performance and our ability to commercialize our product candidates and to compete effectively will depend, in part, on our ability to manage any future growth effectively. To that end, we must be able to manage our development efforts and clinical trials effectively and hire, train and integrate additional management, administrative and sales and marketing personnel. Failure to accomplish any of these activities could prevent us from successfully growing our company.
Consolidation in the pharmaceutical and biotechnology industries may adversely affect us.
There has been consolidation in the pharmaceutical and biotechnology industries. Consolidation could result in the remaining companies having greater financial resources and technological capabilities, thus intensifying
competition, and fewer potential collaboration partners or licensees for our product candidates. In addition, if a consolidating company is already doing business with any of our competitors, we could lose existing or potential future licensees or collaboration partners as a result of such consolidation.
Economic conditions and regulatory changes leading up to and following the U.K.’s likely exit from the EU could have a material adverse effect on our business and results of operations.
The U.K. held a referendum on June 23, 2016 in which a majority voted for the U.K.’s withdrawal from the EU, which we refer to as Brexit. As a result of this vote, negotiations have commenced to determine the terms of the U.K.’s withdrawal from the EU as well as its relationship with the EU going forward, including the terms of trade between the U.K. and the EU. The effects of Brexit have been and are expected to continue to befar-reaching. Brexit and the perceptions as to its impact may adversely affect business activity and economic conditions in Europe and globally and could continue to contribute to instability in global financial and foreign exchange markets. Brexit could also have the effect of disrupting the free movement of goods, services and people between the U.K. and the EU; however, the full effects of Brexit are uncertain and will depend on any agreements the U.K. may make to retain access to EU markets.
In addition, we expect that Brexit could lead to legal uncertainty and potentially divergent national laws and regulations as the U.K. determines which EU laws to replicate or replace. If the U.K. were to significantly alter its regulations affecting our industry, we could face significant new costs. It may also be time-consuming and expensive for us to alter our internal operations in order to comply with new regulations. Altered regulations could also add time and expense to the process by which our products or product candidates or those of our licensees or collaborators receive regulatory approval in the U.K. and EU. Similarly, it is unclear at this time what Brexit’s impact will have on our intellectual property rights and the process for obtaining, maintaining and defending such rights. It is possible that certain intellectual property rights, such as trademarks, granted by the EU will cease being enforceable in the U.K. absent special arrangements to the contrary, and we are required to refile our trademarks and other intellectual property applications domestically in the U.K.
Lastly, as a result of the Brexit, other European countries may seek to conduct referenda with respect to their continuing membership in the EU. Given these possibilities and others we may not anticipate, as well as the lack of comparable precedent, the full extent to which our business, results of operations and financial condition could be adversely affected by Brexit is uncertain.
Our business and operations would suffer in the event of computer system failures, cyberattacks or a deficiency in our cybersecurity.
Despite the implementation of security measures, our internal computer systems and those of our contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures, cyberattacks or cyber-intrusions over the Internet, attachments to emails, persons inside our organization, or persons with access to systems inside our organization. The risk of a security breach or disruption, particularly through cyber-attacks or cyber intrusion, including by computer hackers, foreign governments, and cyber terrorists, has generally increased as the number, intensity and sophistication of attempted attacks and intrusions from around the world have increased. Such an event could cause interruption of our operations. As part of our business, we and our vendors maintain large amounts of confidential information, includingnon-public personal information on patients and our employees Breaches in security could result in the loss or misuse of this information, which could, in turn, result in potential regulatory actions or litigation, including material claims for damages, interruption to our operations, damage to our reputation or otherwise have a material adverse effect on our business, financial condition and operating results. We expect to have appropriate information security policies and systems in place in order to prevent unauthorized use or disclosure of confidential information, includingnon-public personal information, there can be no assurance that such use or disclosure will not occur.
If we fail to comply with data protection laws and regulations, we could be subject to government enforcement actions, which could include civil or criminal penalties, as well as private litigation and/or adverse publicity, any of which could negatively affect our operating results and business.
We may be subject to laws and regulations that address privacy and data security of patients who use our products or product candidates in the U.S. and in states in which we conduct our business. In the U.S., numerous federal and state laws and regulations, including state data breach notification laws, state health information privacy laws, and federal and state consumer protection laws (e.g., Section 5 of the FTC Act) govern the collection, use, disclosure, and protection of health-related and other personal information. For instance, HIPAA imposes certain obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information and imposes notification obligations in the event of a breach of the privacy or security of individually identifiable health information on entities subject to HIPAA and their business associates that perform certain activities that involve the use or disclosure of protected health information on their behalf. Failure to comply with applicable data protection laws and regulations could result in government enforcement actions and create liability for us, which could include civil and/or criminal penalties, as well as private litigation and/or adverse publicity that could negatively affect our operating results and business.
We may be exposed to liabilities under the FCPA and other U.S. and foreign anti-corruption anti-money laundering, export control, sanctions, and other trade laws and regulations, and any determination that we violated these laws could have a material adverse effect on our business.
We are subject to export control and import laws and regulations, including the U.S. Export Administration Regulations, U.S. Customs regulations, and various economic and trade sanctions regulations administered by the U.S. Treasury Department’s Office of Foreign Assets Control. We are also subject to the FCPA, the U.S. domestic bribery statute contained in 18 U.S.C. § 201, the U.S. Travel Act, the USA PATRIOT Act, the United Kingdom Bribery Act 2010, the Proceeds of Crime Act 2002, and possibly other anti-bribery and anti-money laundering laws in countries outside of the U.S. in which we conduct our activities. Anti-corruption laws are interpreted broadly and prohibit companies and their employees and third-party intermediaries from authorizing, promising, offering, providing, soliciting, or accepting, directly or indirectly, improper payments or benefits to or from any person whether in the public or private sector. As we commercialize DEXYCU, if approved, YUTIQ, and any of other product candidates that we may develop, we may engage with third-party manufacturers and collaborators who operate abroad and are required to obtain certain necessary permits, licenses and other regulatory approvals with respect to our business. Our activities abroad create the risk of unauthorized payments or offers of payments by employees, consultants, sales agents or distributors, even though they may not always be subject to our control. It is our policy to implement safeguards to discourage these practices by our employees, consultants, sales agents and distributors. However, our existing safeguards and any future improvements may prove to be less than effective, and the employees, consultants, sales agents, or distributors of our company may engage in conduct for which we might be held responsible, even if we do not explicitly authorize such activities.
Noncompliance with anti-corruption, anti-money laundering, export control, sanctions, and other trade laws could subject us to whistleblower complaints, investigations, sanctions, settlements, prosecution, other enforcement actions, disgorgement of profits, significant fines, damages, other civil and criminal penalties or injunctions, suspension and/or debarment from contracting with certain persons, the loss of export privileges, reputational harm, adverse media coverage and other collateral consequences. If any subpoenas or investigations are launched, or governmental or other sanctions are imposed, or if we do not prevail in any possible civil or criminal litigation, our business, results of operations and financial condition could be materially harmed. Responding to any action will likely result in a materially significant diversion of management’s attention and resources and significant defense and compliance costs and other professional fees. In addition, the U.S. government may seek to hold us liable for successor liability FCPA violations committed by companies in which we invest or that we acquire. As a general matter, enforcement actions and sanctions could harm our business, results of operations, and financial condition.
RISKS RELATED TO OWNERSHIP OF OUR COMMON STOCK The trading price of the shares of our common stock has been highly volatile, and purchasers of our common stock could incur substantial losses. The price of our common stock is highly volatile and may be affected by developments directly affecting our business, as well as by developments out of our control or not specific to us. The closing price of our common stock has ranged from $3.72 to $20.50 per share during the period from January 2, 2020 to March 5, 2021. The pharmaceutical and biotechnology industries, in particular, and the stock market generally, are vulnerable to abrupt changes in investor sentiment. Prices of securities and trading volumes of companies in the pharmaceutical and biotechnology industries, including ours, can swing dramatically in ways unrelated to, or that bear a disproportionate relationship to, our performance. The price of our common stock and their trading volumes may fluctuate based on a number of factors including, but not limited to: the timing, costs and progress of our commercialization efforts;
| • | clinical trials and their results, and other product and technological developments and innovations; |
clinical trials and their results, and other product and technological developments and innovations;
| • | the timing, costs and progress of our commercialization efforts; |
FDA and other domestic and international governmental regulatory actions, receipt and timing of approvals of our product candidates, and any denials and withdrawal of approvals;
| • | FDA and other domestic and international governmental regulatory actions, receipt and timing of approvals of our product candidates, and any denials and withdrawal of approvals; |
competitive factors, including the commercialization of new products in our markets by our competitors;
| • | competitive factors, including the commercialization of new products in our markets by our competitors; |
advancements with respect to treatment of the diseases targeted by our products or product candidates;
| • | advancements with respect to treatment of the diseases targeted by our products or product candidates; |
developments relating to, and actions by, our collaborative partners, including execution, amendment and termination of agreements, achievement of milestones and receipt of payments;
| • | developments relating to, and actions by, our collaborative partners, including execution, amendment and termination of agreements, achievement of milestones and receipt of payments; |
the success of our collaborative partners in marketing any approved products and the amount and timing of payments to us;
| • | the success of our collaborative partners in marketing any approved products and the amount and timing of payments to us; |
availability and cost of capital and our financial and operating results;
| • | availability and cost of capital and our financial and operating results; |
actions with respect to pricing, reimbursement and coverage, and changes in reimbursement policies or other practices relating to our products or the pharmaceutical or biotechnology industries generally;
| • | actions with respect to pricing, reimbursement and coverage, and changes in reimbursement policies or other practices relating to our products or the pharmaceutical or biotechnology industries generally; |
meeting, exceeding or failing to meet analysts’ or investors’ expectations, and changes in evaluations and recommendations by securities analysts;
| • | meeting, exceeding or failing to meet analysts’ or investors’ expectations, and changes in evaluations and recommendations by securities analysts; |
economic, industry and market conditions, changes or trends; and
| • | the use of social media platforms by customers or investors; |
other factors unrelated to us or the pharmaceutical and biotechnology industries.
| • | the issuance of additional shares upon the exercise of currently outstanding options or warrants or upon the settlement of stock units; |
| • | future sales of substantial amounts of shares of our common stock in the market; |
| • | economic, industry and market conditions, changes or trends; and |
| • | other factors unrelated to us or the pharmaceutical and biotechnology industries. |
In addition, low trading volume in our common stock may increase their price volatility. Holders of our common stock may not be able to liquidate their positions at the desired time or price. Finally, we will need to continue to meet the listing requirements of Nasdaq including the minimum stock price, for our stock to continue to be traded on Nasdaq. Additional shares that may be issued upon the exercise of currently outstanding options or warrants or upon the settlement of restricted, performance or deferred stock units would dilute the voting power of our currently outstanding common stock and could cause our stock price to decline.
As of August 31, 2018, we had outstanding options to acquire approximately 8.2 million shares of our common stock, outstanding restricted stock units to acquire 898,129 shares of our common stock, outstanding performance stock units to acquire approximately 1.2 million shares of our common stock, outstanding deferred stock units to acquire 35,001 shares of our common stock, lender warrants to acquire 409,091 and 77,721 shares of our common stock at exercise prices of $1.10 and $1.93, respectively, and the Second Tranche Warrants to acquire 20,184,224 shares of our common stock, or approximately 29.4% of our shares on a fully diluted basis. The issuance of shares of our common stock upon exercise of the stock options or warrants or settlement of the restricted, performance or deferred stock units could result in dilution to the interests of other holders of our common stock and could adversely affect our stock price.
EW Healthcare ownsand Ocumension own a substantial amount of our common stock and can exert significant control over matters subject to stockholder approval, which would prevent new investors from influencing significant corporate decisions. Following the closing of the Second Tranche Transaction on June 25, 2018, EW Healthcare and Ocumension, our largest stockholder,stockholders, beneficially owns approximately 34.2%own 4,190,921 and 3,010,722 shares of our common stock, respectively, or 14.6% and 10.5% of our total outstanding common stock, respectively, as of September 13, 2018. Based upon 74,512,048 outstanding shares of common stock at September 13, 2018 and assuming the exercise of the Second Tranche Warrants byMarch 5, 2021. EW Healthcare and the other Second Tranche Investors, EW Healthcare would beneficially own approximately 44.7% % of our common stock. Upon such exercise in full of the Second Tranche Warrants, EW Healthcare hasOcumension each have the ability to significantly influence the outcome of matters submitted to our stockholders for approval, including the election and removal of directors and any merger, consolidation or sale of all or substantially all of our assets. EW Healthcare and Ocumension have agreed that, for so long as such investor owns a number of shares equal to at least 75% of the shares of common stock it owns as of December 31, 2020, at any meeting of our stockholders, however called, or at any adjournment thereof, or in any other circumstances in which EW Healthcare or Ocumension, as applicable, are entitled to vote, consent or give any other approval, except as otherwise agreed to in writing in advance by us, EW Healthcare and Ocumension shall (a) appear at each such meeting or otherwise cause the shares of our common stock owned by such investor or their respective affiliates to be counted as present thereat for purposes of calculating a quorum; and (b) vote (or cause to be voted), in person or by proxy, all such shares of our common stock that are beneficially owned by such investor or as to which such investor has, directly or indirectly, the right to vote or direct the voting, (i) in favor of any proposals recommended by our board of directors for approval; and (ii) against any proposals that our board of directors recommends our stockholders vote against; provided, however, that the foregoing does not apply to meetings or proposals that are inconsistent with the investor’s rights and obligations under certain agreements between the applicable investor and us.
In addition, thisthe concentration of voting power in EW Healthcare and Ocumension may: (i) delay, defer or prevent a change in control; (ii) entrench our management and Board; or (iii) delay or prevent a merger, consolidation, takeover or other business combination involving us on terms that other stockholders may desire. Comprehensive tax reform legislation could adversely affectIn addition, each of EW Healthcare and Ocumension currently have the right to nominate one or more individuals to our businessboard of directors. While the directors appointed by EW Healthcare and financial condition.Ocumension are obligated to act in accordance with their fiduciary duties under Delaware law, they may have equity or other interests in EW Healthcare or Ocumension and, accordingly, their personal interests may be aligned with EW Healthcare’s or Ocumension’s interests, which may not always coincide with our corporate interests or the interests of our other stockholders. The directors are required to disclose any potential material conflicts of interest. The current EW Healthcare nominated directors are Dr. Göran Ando and Ron Eastman. The current Ocumension nominated director is Ye Liu.
. The Tax Act, which was passed on December 22, 2017, introduced significant changesCertain covenants related to the U.S. tax laws. The Tax Act, among other things, contains significant changes to corporate taxation, including but not limited to the reduction of the corporate tax rate from a top rate of 35% to a flat rate of 21%, limitation of the tax deduction for interest expense to 30% of adjusted earnings (except for certain small businesses), limitation of the deduction for net operating losses to 80% of current year taxable income in respect of losses arising in taxable years beginning after 2017 and elimination of net operating loss carrybacks, one time taxation of offshore earnings at reduced rates regardless of whether they are repatriated, immediate deductions for certain new investments instead of deductions for depreciation expense over time, and modifying or repealing many business deductions and credits (including reducing the business tax credit for certain clinical testing expenses incurred in the testing of certain drugs for rare diseases or conditions generally referred to as “orphan drugs”). Any federal net operating loss carryovers for taxable years beginning after 2017 will be carried forward indefinitely pursuant to the Tax Act. The Tax Act also limits deductions for compensation in excess of $1 million, which could impactour share purchase agreement with Ocumension may restrict our ability to deduct such corporate expenses. We continue to examine the impact the Tax Act may have onobtain future financing and cause additional dilution for our business. Notwithstanding the reductionstockholders.
On December 31, 2020 we entered into a Share Purchase Agreement (the “Share Purchase Agreement”) with Ocumension Therapeutics, incorporated in the federal corporate income tax rate,Cayman Islands with limited liability (“Ocumension”), pursuant to which we offered and sold to the overall impact of the Tax Act is uncertain and our business and financial condition could be adversely affected. Future salesOcumension 3,010,722 shares of our common stock may depress our stock price.
The marketat a purchase price of $5.2163 per share, which was the five-day volume weighted average price of our common stock could decline as of the close of trading on December 29, 2020 (the “Ocumension Transaction”). Pursuant to the Share Purchase Agreement, for so long as Ocumension owns a resultnumber of sales of substantial amountsshares of our common stock equal to at least 75% of the shares of our common stock it acquired at the closing of the Ocumension Transaction, Ocumension is entitled to participate in subsequent issuances of our equity securities in order to maintain its ownership percentage, subject to certain exceptions for, among other things, the issuance of equity awards pursuant to equity incentive plans, inducement awards and/or employee stock purchase plans and the issuance of shares of our common stock pursuant to “at-the-market” equity offering programs. Any participation rights granted to Ocumension in the public market,Share Purchase Agreement would be effected via a separate private placement. These participation rights could severely impact our ability to engage investment bankers to structure a financing transaction and raise additional financing on favorable terms. Furthermore, negotiating and obtaining a waiver to these participation rights may either not be possible or as a resultmay be costly to us. If Ocumension exercises its participation rights, our existing stockholders would be further diluted to the extent of the perception that these sales could occur, which could occur if we issue a large number of shares of common stock (or securities convertible into our common stock) in connection with a future financing, as our common stock is trading at low levels. These factors could make it more difficult for usOcumension acquires to raise funds through future offerings of common stock or other equity securities.maintain its ownership percentage.
Provisions in our charter documents could prevent or delay stockholders’ attempts takeover our company. Our board of directors is authorized to issue “blank check” preferred stock, with designations, rights and preferences as they may determine. Accordingly, our board of directors may in the future, without stockholder approval, issue shares of preferred stock with dividend, liquidation, conversion, voting or other rights that could adversely affect the voting power or other rights of the holders of our common stock. This type of preferred stock could also be issued to discourage, delay or prevent a change in our control. The ability to issue “blank check” preferred stock is a traditional anti-takeover measure. This provision in our charter documents makes it difficult for a majority stockholder to gain control of our company. Provisions like this may be beneficial to our management and our board of directors in a hostile tender offer and may have an adverse impact on stockholders who may want to participate in such a tender offer. Provisions in our bylaws provide for indemnification of officers and directors, which could require us to direct funds away from our business and the development of our product candidates. Our bylaws provide for the indemnification of our officers and directors. We may in the future be required to advance costs incurred by an officer or director and to pay judgments, fines and expenses incurred by an officer or director, including reasonable attorneys’ fees, as a result of actions or proceedings in which our officers and directors are involved by reason of being or having been an officer or director of our company. Funds paid in satisfaction of judgments, fines and expenses may be funds we need for the operation of our business and the development of our product candidates, thereby affecting our ability to attain profitability.
GENERAL RISK FACTORS We do not currently intendwill need to pay dividends on our common stock, and any return to investors is expected to come, if at all, only from potential increases ingrow the pricesize of our common stock.organization, and we may experience difficulties in managing this growth. We have never declared or paid cash dividends onImplementation of our capital stock,development and you shouldcommercialization of product strategies will require additional managerial, operational, sales, marketing, financial and other resources. Our current management, personnel and systems may not rely on an investmentbe adequate to effectively manage the expansion of our operations, which may result in weaknesses in our common stockinfrastructure, give rise to provide dividend income. We currently intend to retain alloperational mistakes, loss of our future earnings, if any, to financebusiness opportunities, employee turnover and reduced productivity. Future growth could require significant capital expenditures and may divert financial resources from other projects, such as the growth and development of our businessexisting or future product candidates. Future growth would impose significant added responsibilities on members of management, including:
| • | overseeing our clinical trials for EYP-1901 effectively; |
| • | managing the commercialization of YUTIQ and DEXYCU; |
| • | identifying, recruiting, maintaining, motivating and integrating additional employees, including any research and development personnel engaged in our clinical trials for EYP-1901, as well as sales and marketing personnel engaged in connection with the commercialization of YUTIQ and DEXYCU; |
| • | engaging and managing our relationship with any co-promotion partners or contract sales organizations; and |
| • | managing our internal development efforts effectively while complying with our contractual obligations to licensors, licensees, contractors and other third parties; and improving our managerial, development, operational and financial systems and procedures. |
As our operations expand, we will need to manage additional relationships with various strategic collaborators, suppliers and do not anticipate declaring or paying any cash dividends for the foreseeable future. In addition, the Credit Agreement contains certain covenants that limitother third parties. Our future financial performance and our ability to pay or make any dividendcommercialize our product candidates and the terms ofto compete effectively will depend, in part, on our ability to manage any future debt agreements may further precludegrowth effectively. To that end, we must be able to manage our development efforts and clinical trials effectively and hire, train and integrate additional management, administrative and sales and marketing personnel. Failure to accomplish any of these activities could prevent us from paying dividends.successfully growing our company. Our business and operations would suffer in the event of computer system failures, cyberattacks or a deficiency in our cybersecurity. Despite the implementation of security measures, our internal computer systems and those of our contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures, cyberattacks or cyber-intrusions over the Internet, attachments to emails, persons inside our organization, or persons with access to systems inside our organization. The risk of a security breach or disruption, particularly through cyber-attacks or cyber intrusion, including by computer hackers, foreign governments, and cyber terrorists, has generally increased as the number, intensity and sophistication of attempted attacks and intrusions from around the world have increased. Such an event could cause interruption of our operations. As part of our business, we and our vendors maintain large amounts of confidential information, including non-public personal information on patients and our employees Breaches in security could result in the loss or misuse of this information, which could, in turn, result in potential regulatory actions or litigation, including material claims for damages, interruption to our operations, damage to our reputation or otherwise have a result, capital appreciation, ifmaterial adverse effect on our business, financial condition and operating results. We expect to have appropriate information security policies and systems in place in order to prevent unauthorized use or disclosure of confidential information, including non-public personal information, there can be no assurance that such use or disclosure will not occur. If we fail to comply with data protection laws and regulations, we could be subject to government enforcement actions, which could include civil or criminal penalties, as well as private litigation and/or adverse publicity, any of which could negatively affect our common stock will be your sole source of gain for the foreseeable future.operating results and business.
We may be subject to securities litigation, which is expensivelaws and could divert our management’s attention. As we operateregulations that address privacy and data security in the pharmaceuticalU.S. and biotechnology industries,in states in which we conduct our business. The legislative and regulatory landscape for privacy and data protection continues to evolve, and there has been an increasing focus on privacy and data protection issues which may be especially vulnerable to volatility in the market price ofaffect our common stock, especially to the extent that various factors affect the common stock of companies in our industry.business. In the past, companies thatU.S., numerous federal and state laws and regulations govern the collection, use, disclosure, and protection of health-related and other personal information, including state data breach notification laws, state health information privacy laws, state genetic privacy laws, and federal and state consumer protection and privacy laws (including, for example, Section 5 of the FTC Act and the CCPA). Compliance with these laws is difficult, constantly evolving, and time consuming. In addition, state laws govern the privacy and security of health, research and genetic information in specified circumstances, many of which differ from each other in significant ways and may not have experienced volatility in the market price of their stock have been subjectsame effect, thus complicating compliance efforts. Failure to securities class action litigation. We may be the target of this type of litigation in the future. Securities litigation against uscomply with these laws and regulations could result in substantial costsgovernment enforcement
actions and divert our management’s attention from other business concerns,create liability for us, which could seriouslyinclude civil and/or criminal penalties, as well as private litigation and/or adverse publicity that could negatively affect our operating results and business. For instance, HIPAA imposes certain obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information and imposes notification obligations in the event of a breach of the privacy or security of individually identifiable health information on entities subject to HIPAA and their business associates that perform certain activities that involve the use or disclosure of protected health information on their behalf. We may obtain health information from third parties (e.g., research institutions from which we obtain clinical trial data) that are subject to privacy and security requirements under HIPAA. Although we are not directly subject to HIPAA – other than potentially with respect to providing certain employee benefits – we could potentially be subject to criminal penalties if we, our affiliates, or our agents knowingly obtain, use, or disclose individually identifiable health information maintained by a HIPAA-covered entity in a manner that is not authorized or permitted by HIPAA. In addition, the CCPA became effective on January 1, 2020 and establishes certain requirements for data use and sharing transparency, and provides California residents certain rights concerning the use, disclosure, and retention of their personal data. The CCPA and its implementing regulations have already been amended multiple times since their enactment. Similarly, there are a number of legislative proposals in the United States, at both the federal and state level, that could impose new obligations or limitations in areas affecting our business. These laws and regulations are evolving and subject to interpretation, and may impose limitations on our activities or otherwise adversely affect our business. The obligations to comply with the CCPA and evolving legislation may require us, among other things, to update our notices and develop new processes internally and with our partners. If we, our agents, or our third party partners fail to comply or are alleged to have failed to comply with these or other applicable data protection and privacy laws and regulations, or if we were to experience a data breach involving personal information, we could be subject to government enforcement actions or private lawsuits. Any associated claims, inquiries, or investigations or other government actions could lead to unfavorable outcomes that have a material impact on our business including through significant penalties or fines, monetary judgments or settlements including criminal and civil liability for us and our officers and directors, increased compliance costs, delays or impediments in the development of new products, negative publicity, increased operating costs, diversion of management time and attention, or other remedies that harm our business, including orders that we modify or cease existing business practices. Outside the U.S., the legislative and regulatory landscape for privacy and data security continues to evolve. There has been increased attention to privacy and data security issues that could also require us to make substantial payments to satisfy judgments or to settle litigation. If securities or industry analysts do not publish research or publish inaccurate or unfavorable research aboutpotentially affect our business, our stock price and trading volume could decline.
The trading market for our common stock depends in partincluding the EU General Data Protection Regulation (“GDPR”), which imposes strict obligations on the research and reports that securities or industry analysts publish about us or our business. We do not have any control over these analysts.processing of personal data, including relating to the transfer of personal data from the European Economic Area to third countries such as the US. If one or morewe act in violation of the analysts who cover us downgrade our stockGDPR we may face significant penalties of up to 10,000,000 Euros or publish inaccurate or unfavorable research about our business, our stock price would likely decline. If one or more of these analysts cease coverageup to 2% of our companytotal worldwide annual turnover for certain violations, or failup to publish reports on us regularly, demand20,000,000 Euros or up to 4% of our total worldwide annual turnover for our stock could decrease, which might cause our stock price and trading volume to decline.more serious violations.
None. ITEM 2. | ITEM 2. PROPERTIES |
We do not own any real property. On May 17, 2018, we amended our lease, dated November 1, 2013, to extend our Watertown, Massachusetts lease term from April 2019 through approximately AprilMay 2025 and to add an additional 6,590 square feet of rentable area for a resulting total of 20,240 square feet. Followingbuild-out of the additional space, for which the landlord provided a construction allowance of $671,000, we took occupancy on September 10, 2018. We are entitled to base rent abatement for the aggregate space for the four months ending December 31, 2018. The aggregate leased space consists of 1,750 square feet of laboratory space, 1,000 square feet of Class 10,000 clean room space and 17,490 square feet of office space. We have an option to extend the term of the lease for one additional five-year period at market rates.
We lease 3,000 square feet of office space in Liberty Corner, New Jersey under a lease agreement that expires in June 2022. On June 11, 2018, we subleased an additional 1,381 square feet of office space in this building through May 2022. We believe our leased facilities are adequate for our present and anticipated needs.
ITEM 3. | ITEM 3. LEGAL PROCEEDINGS |
We are subject to various routine legal proceedings and claims incidental to our business, which management believes will not have a material effect on our financial position, results of operations or cash flows. On May 14, 2020, we received a subpoena from the Division of Enforcement of the SEC seeking production of certain documents and information on topics including product sales and demand, revenue recognition and accounting in relation to product sales, product sales and cash projections, and related financial reporting, disclosure and compliance matters. We are cooperating fully in connection with this investigation. Based on procedures performed to date in relation to our revenue recognition practices, we have not identified any accounting items that are not in accordance with GAAP. In addition, we believe our public statements regarding our business, including with respect to product sales and demand, have been and are in compliance with federal securities laws. At this time, we are unable to predict the duration, scope or outcome of this matter or whether it could have a material impact on our financial condition, results of operations or cash flow. ITEM 4. |
ITEM 4. MINE SAFETY DISCLOSURES |
Not applicable.
PART II ITEM 5. | ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
Market Information, Holders and Dividends
Our common stock is traded on the Nasdaq Global Market under the trading symbol “EYPT”. The following table sets forth the high and low selling prices per share of our common stock as reported on the Nasdaq Global Market for the periods indicated: | | | | | | | | | | | | High | | | Low | | Fiscal year ended June 30, 2018: | | | | | | | | | First Quarter | | $ | 1.76 | | | $ | 1.03 | | Second Quarter | | | 1.44 | | | | 1.02 | | Third Quarter | | | 1.35 | | | | 0.93 | | Fourth Quarter | | | 2.88 | | | | 1.07 | | Fiscal year ended June 30, 2017: | | | | | | | | | First Quarter | | $ | 4.25 | | | $ | 2.85 | | Second Quarter | | | 3.22 | | | | 1.50 | | Third Quarter | | | 2.16 | | | | 1.63 | | Fourth Quarter | | | 2.45 | | | | 1.57 | |
On September 13, 2018, the last reported sale price of our common stock on the Nasdaq Global Market was $2.21. As of that date,March 5, 2021, we had approximately 11480 holders of record of our common stock and, according to our estimates, approximately 8,500stock. This number does not include beneficial owners of our common stock. Ourwhose shares of common stock previously tradedare held by nominees in the form of CHESS Depositary Interests on the Australian Securities Exchange under the symbol “PVA” until our delisting from the ASX, which occurred effective as of May 7, 2018.
We have never paid cash dividends, and we do not anticipate paying cash dividends in the foreseeable future. In addition, the Credit Agreement contains negative covenants which limit our ability to pay cash dividends on our equity securities.street name.
Equity Compensation Plan Information The following table provides information about the securities authorized for issuance under the Company’sInformation required by Item 5 of Form 10-K regarding our equity compensation plans asis incorporated herein by reference to Item 12 of June 30, 2018:Part III of this Annual Report on Form 10-K
| | | | | | | | | | | | | Plan category | | Number of securities
to be issued upon
exercise of
outstanding options,
warrants and rights
(a) | | | Weighted-average
exercise price of
outstanding options,
warrants and rights
(b) | | | Number of securities
remaining available
for future issuance
under equity
compensation plans
(excluding securities
reflected in Column a)
(c) | | Equity Compensation plans approved by security holders | | | 7,310,244 | (1) | | $ | 2.89 | | | | 1,497,528 | | Equity Compensation plans not approved by security holders (2) | | | 440,000 | (3) | | | 1.95 | | | | — | | | | | | | | | | | | | | | Total | | | 7,750,244 | | | $ | 2.89 | | | | 1,497,528 | | | | | | | | | | | | | | |
(1) | Consists of outstanding stock options, performance-based stock units, restricted stock units and deferred stock units to purchase 7,310,244 awards pursuant to our 2008 Equity Incentive Plan, as amended, and our 2016 Long Term Incentive Plan, as amended, or the 2016 LTIP.
|
(2) | Our Board has not established any specific number of shares that could be issued without stockholder approval. Inducement grants to new key employees are determined on acase-by-case basis. Other than possible inducement grants, we expect all equity awards will be made under stockholder-approved plans.
|
(3) | In May 2018, we issued 440,000 inducement equity awards outside of the 2016 LTIP in accordance with Nasdaq Listing Rule 5635(c)(4). The inducement equity awards were, upon the recommendation of the Compensation Committee of our Board, approved by our Board and were made as an inducement material to Leonard Blum’s acceptance of employment with us in accordance with Nasdaq Listing Rule 5635(c)(4). Mr. Blum is our Executive Vice President and General Manager, U.S. Mr. Blum’s inducement awards consist of twonon-qualified stock option awards to purchase a total of 440,000 shares of common stock and performance stock units, or PSUs, entitling Mr. Blum to receive up to 225,000 shares of common stock based on the achievement of prescribed performance metrics determined by the Compensation Committee. The stock option to purchase 375,000 shares of common stock will vest ratably on each of the first, second and third anniversaries of the date of grant, subject to the terms of grant. The stock option to purchase 65,000 shares of common stock will vest on the first anniversary of the date of grant. The performance metrics associated with the PSU award will be subject to measurement following the3-year period ending June 30, 2021. Effective September 26, 2018, Mr. Blum will cease to be employed by us. Per the terms of his employment agreement with us and the applicable stock option award agreements, upon the cessation of Mr. Blum’s employment with us “without Cause” (as such term is defined in Mr. Blum’s employment agreement), any unvested portion of his stock option awards that would have vested had Mr. Blum continued his employment through the first anniversary of his employment will vest immediately prior to his cessation of employment and will remain exercisable until 5:00 P.M. Eastern Time on the last day of the three-month period commencing on the date of Mr. Blum’s cessation of employment. In addition, Mr. Blum’s employment agreement and our form of performance stock unit award agreement provides that Mr. Blum will forfeit the unvested portion of his PSUs upon the cessation of his employment. Accordingly, (i) 125,000 shares of common stock underlying Mr. Blum’s firstnon-qualified stock option award and (ii) all 65,000 shares of common stock underlying Mr. Blum’s secondnon-qualified stock option award will vest immediately prior to Mr. Blum’s cessation of employment, become immediately exercisable and remain exercisable until December 26, 2018. All of the shares of common stock underlying Mr. Blum’s PSUs will be forfeited upon the cessation of his employment.
|
Recent Sales of Unregistered Securities Other than as previously disclosed on our Current Reports on Form8-K or Quarterly Reports on Form 10-Q filed with the SEC, we did not issue any unregistered equity securities during the yeartwelve months ended June 30, 2018.December 31, 2020. Issuer RepurchasesPurchases of Equity Securities by the Issuer and Affiliated Purchasers
None. ITEM 6. | SELECTED FINANCIAL DATA
|
The selected historical financial data set forth belowITEM 6. SELECTED FINANCIAL DATA
Because we are a smaller reporting company as of June 30, 2018, 2017, 2016, 2015 and 2014 and for eachdefined by Rule 12b-2 of the years then ended have been derived from our audited consolidated financial statements, of which the financial statements as of June 30, 2018 and 2017 and for the years ended June 30, 2018, 2017 and 2016 are included elsewhere inExchange Act, with respect to this Annual Report on Form10-K. The 10-K, we are not required to provide the information set forth below should be read in conjunction with Item 7, “Management’s Discussion and Analysis of Financial Condition and Results of Operations”, and the audited Consolidated Financial Statements, and the Notes thereto, and other financial information included elsewhere herein. Our historical financial information may not be indicative of our future results of operations or financial position.under this Item.
| | | | | | | | | | | | | | | | | | | | | | | | Year Ended June 30, | | | | | 2018 | | | 2017 | | | 2016 | | | 2015 | | | 2014 | | | | | (In thousands except per share data) | | Consolidated Statements of Operations Data: | | | | | | | | | | | | | | | | | | | | | Revenues: | | | | | | | | | | | | | | | | | | | | | Collaborative research and development (1) | | $ | 1,343 | | | $ | 6,569 | | | $ | 398 | | | $ | 25,411 | | | $ | 2,155 | | Royalty income (2) | | | 1,618 | | | | 970 | | | | 1,222 | | | | 1,154 | | | | 1,318 | | | | | | | | | | | | | | | | | | | | | | | Total revenues | | | 2,961 | | | | 7,539 | | | | 1,620 | | | | 26,565 | | | | 3,473 | | | | | | | | | | | | | | | | | | | | | | | Operating expenses: | | | | | | | | | | | | | | | | | | | | | Research and development | | | 16,178 | | | | 14,880 | | | | 14,381 | | | | 12,088 | | | | 9,573 | | Sales and marketing | | | 1,512 | | | | — | | | | — | | | | — | | | | — | | General and administrative | | | 11,545 | | | | 11,235 | | | | 9,013 | | | | 8,056 | | | | 7,468 | | Gain on sale of property and equipment | | | — | | | | — | | | | — | | | | — | | | | (78 | ) | | | | | | | | | | | | | | | | | | | | | | Total operating expenses | | | 29,235 | | | | 26,115 | | | | 23,394 | | | | 20,144 | | | | 16,963 | | | | | | | | | | | | | | | | | | | | | | | Operating (loss) income | | | (26,274 | ) | | | (18,576 | ) | | | (21,774 | ) | | | 6,421 | | | | (13,490 | ) | Interest and other income, net | | | 101 | | | | 91 | | | | 72 | | | | 22 | | | | 5 | | Interest expense | | | (720 | ) | | | — | | | | — | | | | — | | | | — | | Change in fair value of derivative liability | | | (26,278 | ) | | | — | | | | — | | | | — | | | | — | | | | | | | | | | | | | | | | | | | | | | | (Loss) income before income taxes | | | (53,171 | ) | | | (18,485 | ) | | | (21,702 | ) | | | 6,443 | | | | (13,485 | ) | Income tax benefit (expense) | | | — | | | | — | | | | 155 | | | | (96 | ) | | | 130 | | | | | | | | | | | | | | | | | | | | | | | Net (loss) income | | $ | (53,171 | ) | | $ | (18,485 | ) | | $ | (21,547 | ) | | $ | 6,347 | | | $ | (13,355 | ) | | | | | | | | | | | | | | | | | | | | | | Net (loss) income per share: | | | | | | | | | | | | | | | | | | | | | Basic | | $ | (1.15 | ) | | $ | (0.52 | ) | | $ | (0.68 | ) | | $ | 0.22 | | | $ | (0.49 | ) | | | | | | | | | | | | | | | | | | | | | | Diluted | | $ | (1.15 | ) | | $ | (0.52 | ) | | $ | (0.68 | ) | | $ | 0.21 | | | $ | (0.49 | ) | | | | | | | | | | | | | | | | | | | | | | Weighted average common shares outstanding: | | | | | | | | | | | | | | | | | | | | | Basic | | | 46,226 | | | | 35,344 | | | | 31,623 | | | | 29,378 | | | | 27,444 | | | | | | | | | | | | | | | | | | | | | | | Diluted | | | 46,226 | | | | 35,344 | | | | 31,623 | | | | 30,584 | | | | 27,444 | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | As of June 30, | | | | | 2018 | | | 2017 | | | 2016 | | | 2015 | | | 2014 | | | | | (In thousands) | | Consolidated Balance Sheet Data: | | | | | | | | | | | | | | | | | | | | | Cash and cash equivalents | | $ | 38,776 | | | $ | 16,898 | | | $ | 15,313 | | | $ | 19,121 | | | $ | 15,334 | | Marketable securities | | | — | | | | — | | | | 13,679 | | | | 9,414 | | | | 2,944 | | Intangible assets, net | | | 31,358 | | | | 364 | | | | 1,102 | | | | 1,925 | | | | 2,765 | | Total assets | | | 71,670 | | | | 18,677 | | | | 31,619 | | | | 32,367 | | | | 22,671 | | Long-term debt | | | 17,309 | | | | — | | | | — | | | | — | | | | — | | Derivative liability | | | 19,780 | | | | — | | | | — | | | | — | | | | — | | Total deferred revenue—current and long-term | | | — | | | | 50 | | | | 5,732 | | | | 5,629 | | | | 5,722 | | Total stockholders’ equity | | | 11,687 | | | | 13,336 | | | | 20,881 | | | | 23,368 | | | | 14,924 | |
(1) | Includes the following: from our Prior Alimera Agreement (including patent reimbursement costs): $148,000 in fiscal 2018, $659,000 in fiscal 2017, $233,000 in fiscal 2016, $25.1 million in fiscal 2015 and
|
| $114,000 in fiscal 2014; from our Restated Pfizer Agreement: $5.6 million in fiscal 2017 and $7,000 in fiscal 2014; from feasibility study agreements: $1.1 million in fiscal 2018, $211,000 in fiscal 2017, $33,000 in fiscal 2016, $144,000 in fiscal 2015 and $1.9 million in fiscal 2014; from our license agreement with OncoSil Medical: $100,000 in fiscal 2018, $100,000 in fiscal 2017, $100,000 in fiscal 2016, $100,000 in fiscal 2015 and $102,000 in fiscal 2014. See Note 4 in the accompanying Notes to the Consolidated Financial Statements contained in this Annual Report on Form10-K for additional information. |
(2) | Includes the following: from our Amended Alimera Agreement; $575,000 in fiscal 2018; from our license agreement with Bausch & Lomb: $1.0 million in fiscal 2018, $970,000 in fiscal 2017, $1.2 million in fiscal 2016, $1.2 million in fiscal 2015 and $1.3 million in fiscal 2014.
|
ITEM 7. | ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
|
The following discussion and analysis of financial condition and results of operations should be read in conjunction with our audited Consolidated Financial Statements and related Notes beginning on pageF-1 of this Annual Report on Form10-K. This discussion contains forward-looking statements, based on current expectations and related to future events and our future financial performance, that involve risks and uncertainties. Our actual results may differ significantly from those anticipated or implied in these forward-looking statements as a result of many important factors, including, but not limited to, those set forth under Item 1A, “Risk Factors”, and elsewhere in this report. The following Management’s Discussion and Analysis (“MD&A”) provides a narrative of our results of operations for the year ended December 31, 2020 and the comparable period ended December 31, 2019, respectively, and our financial position as of December 31, 2020 and 2019, respectively. The MD&A should be read together with our consolidated financial statements and related notes included on pages F-1 through F-34 of this Annual Report on Form 10-K. Overview We are a specialty biopharmaceuticalpharmaceutical company committed to developing and commercializing innovative ophthalmic productstherapeutics to help improve the lives of patients with serious eye disorders. Our pipeline leverages our proprietary Durasert® technology for extended intraocular drug delivery including EYP-1901, a potential twice-yearly sustained delivery intravitreal anti-VEGF treatment initially targeting wet age-related macular degeneration (“wet AMD”), the leading cause of vision loss among people 50 years of age and older in the United States. Our product candidate pipeline also includes YUTIQ50, a potential twice-yearly treatment for non-infectious uveitis affecting the posterior segment of the eye, diseases. Our lead product,one of the leading causes of blindness. We also have two commercial products: YUTIQ®, a once every three-year treatment for chronic non-infectious uveitis affecting the posterior segment of the eye, and DEXYCU (dexamethasone intraocular suspension) 9%®, approved by the FDA in February 2018, is administered as a single dose at the end oftreatment for postoperative inflammation following ocular surgerysurgery. Fiscal 2020 Overview and is the first long-acting intraocular product approved by the FDA for the treatment of post-operative inflammation. DEXYCU utilizes our proprietary Verisome drug-delivery platform, which allows for a single intraocular injection that releases dexamethasone, a corticosteroid, over time. There are approximately four million cataract surgeries performed annually in the U.S. and we plan to launch DEXYCU in the U.S. in the first half of calendar year 2019 with a primary focus on its use following cataract surgery. Our lead product candidate is YUTIQ, a three-yearnon-erodible FA insert for the treatment of NIPU. Injected into the eye in an office visit, YUTIQ is a tiny micro-insert that delivers a micro-dose of a corticosteroid to the back of the eye on a sustained constant (zero order release) basis for approximately three years. On March 19, 2018, the FDA accepted our NDA for YUTIQ and set a PDUFA action date of November 5, 2018. YUTIQ is based on our proprietary Durasert sustained-release drug delivery technology platform, which can deliver drugs for predetermined periods of time ranging from months to years. Posterior segment uveitis is the third leading cause of blindness in the U.S. and affects between 55,000 to 120,000 people in the U.S. If approved in November 2018, we expect to launch YUTIQ in the U.S. in the first half of calendar year 2019.COVID-19 Impact Both Phase 3 clinical trials investigating YUTIQ met their primary efficacy endpoint of prevention of recurrence of disease through six months with statistical significance (p < 0.001, intent to treat analysis) and with safety data consistent with the known effects of ocular corticosteroid use. Statistical significance for efficacy and encouraging safety results were maintained through 12 months offollow-up in both Phase 3 clinical trials. In Europe, we filed a MAA in June 2017 and subsequently withdrew the application after expanding the license for our proprietary Durasert sustained-release drug delivery technology platform to Alimera to include uveitis, including NIPU. In January 2018, Alimera received validation of a Type II variation submitted in all seventeen European countries in which it previously received regulatory approval for ILUVIEN for DME. According to Alimera’s public filings, Alimera plans to submitfollow-up data supporting its Type II variation application in the fourth quarter of calendar year 2018 and expects it will obtain approval for its application in the first half of calendar 2019. If the variation is approved, Alimera plans to commercialize the uveitis indication under its ILUVIEN trademark.
ILUVIEN is an injectable, sustained-release micro-insert that provides three years of treatment of DME from a single injection. ILUVIEN and YUTIQ are based on the same technology and deliver the same corticosteroid, FA. ILUVIEN was developed in collaboration with, and is licensed to and sold by Alimera. Pursuant the Prior Alimera Agreement, ILUVIEN has been sold directly in the U.K. and Germany since 2013, in the U.S. and Portugal since 2015 and in Austria and Ireland since 2017. ILUVIEN also has marketing approvals in 12 other European countries. In addition, Alimera has entered into various agreements under which distributors will provide regulatory, reimbursement or sales and marketing support for commercialization or future commercialization of ILUVIEN in several countries in the Middle East, as well as France, Italy, Spain, Australia, New Zealand and Canada. In July 2017, we entered into the Amended Alimera Agreement pursuant to
which we (i) expanded the license to Alimera for our proprietary Durasert drug delivery technology platform to include uveitis, including NIPU, in the EMEA and (ii) converted the net profit share arrangement for each licensed product (including ILUVIEN) under the Prior Alimera Agreement to a sales-based royalty on a calendar quarter basis commencing July 1, 2017, with payments from Alimera due 60 days following the end of each quarter.
Our development programs are focused primarily on developing sustained release drug products using our proven Durasert technology platform to deliver small molecule drugs to treat uveitis, wetage-related macular degeneration, glaucoma and other diseases.
Fiscal Year 2018 Overview
The fiscal year ended June 30, 2018December 31, 2020 was highlighted by the following events: | • | Underlying customer demand and Distributor purchases by specialty distributors and specialty pharmacies (collectively, the “Distributors”) of both YUTIQ and DEXYCU was negatively impacted beginning in the first and especially the second quarter of 2020 due to shutdowns associated with the Pandemic in the U.S. Although a modest return of customer demand began in June 2020 and produced sequential product sales growth into the third and fourth quarters, we expect these reduced demand levels to continue through the duration of the Pandemic until various restrictions on elective surgeries and office visits are fully removed. During the Pandemic, our sales organization continued to call on such offices, though at a reduced frequency.There have been no disruptions to the supply chains for YUTIQ and DEXYCU during the Pandemic and we continue to produce finished product for commercial sale. |
| • | In January 2020, we entered into an exclusive license agreement with Ocumension Therapeutics for the development and commercialization in Mainland China, Hong Kong, Macau and Taiwan of DEXYCU for the treatment of post-operative inflammation following ocular surgery. Under the terms of the license agreement, we received an upfront payment of $2 million and will be eligible to receive up to an additional $12.0 million if certain future prespecified development, regulatory and commercial sales milestones are achieved by Ocumension. In addition, we are entitled to receive a mid-single digit sales-based royalty. In exchange, Ocumension will receive exclusive rights to develop and commercialize the product in the greater China territory, at its own cost and expense with us supplying product for clinical trials and commercial sale. |
| • | In February 2020, we completed an underwritten public offering of 1,500,000 shares of our common stock at a public offering price of $14.50 per share. The gross proceeds of the offering were $21,750,000, before deducting the underwriting discounts and commissions and other transaction expenses. This offering closed on February 25, 2020. |
| • | In April 2020, we announced a reorganization of our commercial operations and the cancellation or deferral of planned spending to conserve cash due to the impact of the Pandemic, particularly the postponement of nearly all elective surgeries including cataract surgery. This reorganization was primarily focused on a reduction in the external contract sales organization for DEXYCU. We allocated our remaining DEXYCU commercial resources to high-volume ASCs in key U.S. regions. The reorganization was expected to result in annual savings of approximately $7 million and one-time savings of approximately $10 million from other planned expenditure cancellations and deferrals. |
| • | In April 2020, we received a $2.0 million Paycheck Protection Program (PPP) loan through the Small Business Administration’s Paycheck Protection Program (PPP) under the Coronavirus Aid, Relief and Economic Security Act of |
| | 2020 (the CARES Act). The PPP loan enabled us to retain key commercial infrastructure and employees and avoid furloughs as product demand and revenues remained significantly reduced due to ASC and physician office closures necessitated by the Pandemic. We used the proceeds of the PPP loan to cover payroll costs, rent and utilities in accordance with the CARES Act and anticipate the loan will be fully forgiven. |
| • | In August 2020, we announced the signing of a commercial alliance for the joint promotion of DEXYCU with ImprimisRx (Harrow Health). Through this agreement, we are able to access the established and complementary ImprimisRx commercial operations in cataract surgery to include DEXYCU as a prioritized product in its existing portfolio of product offerings. |
| • | In August 2020, we received $9.5 million from Ocumension Therapeutics under the Memorandum of Understanding (“2020 MOU”). This expanded agreement grants Ocumension the rights to commercialize both YUTIQ and DEXYCU under their own brand names in South Korea and other jurisdictions across Southeast Asia and acts as the full and final prepayment of all remaining development, regulatory, and commercial sale milestone payments under the original license agreements. We remain entitled to receive royalties on future sales of these products by Ocumension. |
| • | In October 2020, we announced an amendment to our existing debt facility with CRG Servicing LLC (CRG). Under the terms of the amendment, CRG waived the covenant associated with our net product revenue of YUTIQ and DEXYCU for the twelve-month period ending on December 31, 2020. The parties also agreed to a reduction of the December 31, 2021 net product revenue covenant to $45 million from $80 million based on the promising recovery and return in customer demand for both products at that time following COVID-19-related closures. There were no additional costs incurred by us for the waiver. |
| • | In December 2020, we announced a 1-for-10 reverse stock split which enabled us to regain compliance with the $1.00 minimum closing bid price required for continued listing on the Nasdaq Global Market. |
| • | In December 2020, we announced a $16.5 million monetization of our ILUVIEN Royalty (Alimera) with SWK Holdings Corporation. $15 million of the net proceeds was applied against existing debt obligations with CRG Servicing LLC. |
| • | In December 2020, we announced a $15.7 million equity investment by Ocumension Therapeutics, our partner in Asia. |
On January 5, 2018, we filed our NDA for YUTIQ. On March 19, 2018,
Recent Developments Recent developments and ongoing activities regarding the FDA accepted our NDA forcommercialization of YUTIQ include: | • | Customer demand in Q4, represented as units purchased by physicians from our distributors, was up 10% over Q3, driven by underlying growth. |
Recent developments and set a PDUFA action dateongoing activities regarding the commercialization of November 5, 2018;DEXYCU include: | • | Customer demand in Q4, represented as units purchased by ambulatory surgical centers, was up 30% over Q3, driven by increases in cataract surgeries and some re-opening of ASC’s. |
| • | DEXYCU co-promotion partner, ImprimisRx®, began driving volume through its experienced cataract surgery field force, materially adding to Q4 customer demand. |
On March 28, 2018, we completed the Icon Acquisition pursuant to a merger agreement, dated March 28, 2018, by and among us, Merger Sub (our wholly-owned subsidiary), Icon and the other signatories thereto. Under the terms of the Merger Agreement, Merger Sub was merged with and into Icon, with Icon being the surviving corporation and our wholly-owned subsidiary
| • | In February 2021, we sold 10,465,000 shares of Common Stock in an underwritten public offering at a price of $11.00 per share, including the exercise in full by the underwriters of their option to purchase up to 1,365,000 additional shares of our common stock. The gross proceeds of the offering are approximately $115.1 million. Underwriter discounts and commissions and other share issue costs totaled approximately $7.1 million. |
On March 28, 2018, in connection with the Icon Acquisition, we entered into the Credit Agreement with SWK Funding, a wholly-owned subsidiary of SWK Holdings Corporation, pursuant to which the Lenders provided to us anon-dilutive term loan in the principal amount of $15.0 million, which was increased by an additional $5.0 million drawdown at our request on June 26, 2018 following the closing of the Second Tranche Transaction on June 25, 2018. We may draw down an additional $10.0 million Loan, subject to (i) SWK Funding obtaining additional loan commitments and (ii) the satisfaction of certain conditions (as set forth in the Credit Agreement);R&D Highlights
On March 28, 2018, in connection with the Initial Advance, we issued a warrant, or the SWK Warrant, to SWK Funding to purchase (i) 409,091 shares of our common stock, or the Initial Advance Warrant Shares, and (ii) 77,721 shares of our common stock equal to 3% of the Additional Advance, or the Additional Advance Warrant Shares. The SWK Warrant was exercisable with respect to the Initial Advance Warrant Shares upon issuance of the SWK Warrant at an exercise price of $1.10, which was the consolidated closing bid price of a share of our common stock on the Nasdaq Global Market immediately preceding the issuance of Initial Advance Warrant Shares. The SWK Warrant became exercisable with respect to the Additional Advance Warrant Shares on June 26, 2018 at an exercise price of $1.93 per share, which was the consolidated closing bid price of a share of our common stock on the Nasdaq Global Market immediately preceding the issuance of the Additional Advance Warrant Shares. The SWK Warrant will remain exercisable (x) until the close of business on March 28, 2025 with respect to the Initial Advance Warrant Shares and (y) until the close of business on June 26, 2025 with respect to the Additional Advance Warrant Shares;
| • | In February 2020, we signed an exclusive license agreement with Equinox Science, LLC, to develop vorolanib, a tyrosine kinase inhibitor, for the treatment of wet age-related macular degeneration, retinal vein occlusion and diabetic retinopathy. Vorolanib is being developed as EYP-1901 utilizing a bioerodible formulation of our Durasert technology. |
On March 28, 2018, in connection with the Icon Acquisition, we entered into a securities purchase agreement, or the First Tranche Securities Purchase Agreement, with EW Healthcare Partners, L.P. and EWHealthcare Partners-A, L.P, collectively the First Tranche Investors, pursuant to which we sold an aggregate of 8,606,324 shares of our common stock at a purchase price of $1.10 per share, or the First Tranche Purchase Price, for gross proceeds of $9.5 million, which we refer to as the First Tranche Transaction; and
On March 28, 2018, in connection with the Icon Acquisition and the execution of the First Tranche Securities Purchase Agreement, we entered into the Second Tranche Securities Purchase Agreement, with the First Tranche Investors and certain other accredited investors signatory thereto, or the Second Tranche Investors, pursuant to which we agreed to offer and sell, subject to the approval of our
| stockholders, an aggregate•
| In March 2020, we announced positive topline 36-month follow-up data from the second Phase 3 trial of up to approximately $25.5 millionYUTIQ for the treatment of Units. Our stockholders approvedchronic non-infectious uveitis affecting the sale and issuanceposterior segment of the Unitseye. This second double-masked, randomized Phase 3 trial of YUTIQ enrolled 153 patients in 15 clinical centers in India, with 101 eyes treated with YUTIQ and 52 eyes receiving sham injections. At 36-months, the recurrence rate in YUTIQ randomized eyes was significantly lower than in sham treated eyes (46.5% vs. 75.0%, respectively; p=0.001). Visual acuity gains or losses of 3-lines or more were both similar between treatment groups. Safety data showed no unanticipated side effects at each follow-up timepoint at 12, 24 and 36-months. These positive results were consistent with the findings from the first Phase 3 study of YUTIQ and provide further validation of its long-term ability to reduce uveitic flares. |
| • | Positive retrospective case study data supporting DEXYCU was highlighted in an oral presentation at the 2020 Caribbean Eye Meeting in an oral session entitled, “Drug Delivery: Real-World Experience With Dexamethasone Intraocular |
| | Suspension”. The ongoing retrospective study is designed to provide large-scale, real-world data on June 22, 2018early experiences with DEXYCU from surgeons. Interim results presented are from 154 patients administered DEXYCU with each time point of data based on patient chart data and frequency of measurement by participating physicians. The proportion of patients with complete anterior chamber cell clearing (cell score=0) was 47.5%, 50.0%, 84.1% and 87.5% at postoperative day 1, 8, 14 and 30, respectively. The proportion of patients with no anterior chamber flares (flare score=0), another measurement of inflammation, was 77.7%, 98.5%, 98.8% and 99.1% at postoperative day 1, 8, 14 and 30, respectively. Mean intraocular pressure at postoperative day 1 was 17.6mmHg, with levels decreasing through to postoperative day 30. |
| • | In July 2020, we sold 20,184,224 Units toannounced the Second Tranche Investors uponappointment of Dr. Jay Duker as our Chief Strategic Scientific Officer. Dr. Duker brings more than thirty years of ophthalmology experience with roles held in the closingclinical, research, business, and academic settings. Dr. Duker is also the Director of the Second Tranche Transaction on June 25, 2018.New England Eye Center. He is also Professor and Chair of Ophthalmology at Tufts Medical Center and Tufts University School of Medicine, as well as the Chairman of the Board of Sesen Bio, Inc., a publicly traded clinical stage biopharmaceutical company. |
| • | In December 2020, we reported positive results from our GLP Toxicology Study of EYP-1901, a potential twice-yearly sustained-delivery intravitreal anti-VEGF treatment targeting wet age-related macular degeneration, or wet AMD. |
| • | In December 2020, we filed an IND application with the FDA for a Phase 1 trial for EYP-1901, and subsequently dosed the first patient in the Phase 1 clinical trial in January 2021. |
ILUVIEN, an injectable, sustained-release micro-insert delivering 0.19mg of FA to the back of the eye for the treatment of DME, was licensed to and developed with Alimera under the Prior Alimera Agreement. In July 2017, we entered into the Amended Alimera Agreement pursuant to which we (i) expanded the license to Alimera for our proprietary Durasert drug delivery technology platform to include uveitis, including NIPU, in the EMEA and (ii) converted the net profit share arrangement for each licensed product (including ILUVIEN) under the Prior Alimera Agreement to a sales-based royalty on a calendar quarter basis. Sales-based royalties start at the rate of 2% effective as of July 1, 2017. Commencing January 1, 2019 (or earlier under certain circumstances), the sales-based royalty will increase to 6% (8% on total ILUVIEN net sales in excess of $75 million on a calendar year basis). Alimera’s share of contingently recoverable accumulated ILUVIEN commercialization losses under the original net profit share arrangement (as set forth in the Prior Alimera Agreement), was capped at $25 million. Under the Amended Alimera Agreement those recoverable losses will be reduced as follows: (i) $10.0 million was cancelled in lieu of an upfront license fee on the effective date of the Amended Alimera Agreement; (ii) for calendar years 2019 and 2020, 50% of earned sales-based royalties in excess of 2% of net sales will be offset against the quarterly royalty payments otherwise due from Alimera; (iii) on January 1, 2020 (or earlier under certain circumstances), another $5 million will be cancelled, provided, however, that such date of cancellation may be extended under certain circumstances related to Alimera’s regulatory approval process for ILUVIEN for NIPU, with such extension, if any, subject to mutual agreement by the parties; and (iv) commencing in calendar year 2021, 20% of earned sales-based royalties in excess of 2% of net sales will be offset against the quarterly royalty payments due from Alimera until such time as the remaining balance of the original $25 million of recoverable commercialization losses has been fully recouped.
We believe that the terms of the Amended Alimera Agreement for ILUVIEN for DME have standardized and simplified the agreement and improved the potential total value of the agreement for us. ILUVIEN for DME has been sold by Alimera in the U.S. since 2015, where it is indicated for the treatment of DME in patients previously treated with a course of corticosteroids without a clinically significant rise in IOP. ILUVIEN for DME has been sold by Alimera in the U.K. and Germany since 2013, in Portugal since 2015 and in Austria and Ireland since 2017. ILUVIEN also has marketing approvals in 12 other European countries. In addition, Alimera has entered into various agreements under which distributors will provide regulatory, reimbursement or sales and marketing support for commercialization or future commercialization of ILUVIEN in several countries in the Middle East, as well as France, Italy, Spain, Australia, New Zealand and Canada. ILUVIEN for DME has marketing approvals in a total of 17 European countries, where it is indicated for the treatment of chronic DME considered insufficiently responsive to available therapies.
FDA-approved Retisert is an implant that provides sustained treatment of posterior segment uveitis for 30 months that wasco-developed with and licensed to Bausch & Lomb. Implanted in a surgical procedure, Retisert delivers the same corticosteroid as Durasert but in a larger dose. We receive royalties from Retisert sales.
We are also using our Durasert technology platform to identify potential product candidates that provide sustained treatment of wet and dry AMD, glaucoma and other diseases. We are also conductingpre-clinical evaluations of various TKI candidates for wet AMD. We are also developing a next-generation short-duration treatment for posterior segment uveitis, using the same Durasert technology and drug (FA) as in YUTIQ.
Summary of Critical Accounting Policies and Estimates The discussion and analysis of our financial condition and results of operations is based upon our consolidated financial statements, which have been prepared in accordance with U.S. generally accepted accounting principles, or (“U.S. GAAP.GAAP”). The preparation of these financial statements requires that we make certain estimates, judgments and assumptions that affect the reported amounts of assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting periods. We base our estimates on historical experience, anticipated results and trends and various other factors believed to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily available from other sources. By their nature, these estimates, judgments and assumptions are subject to an inherent degree of uncertainty, and management evaluates them on an ongoing basis for changes in facts and circumstances. Changes in estimates are recorded in the period in which they become known. Actual results may differ from our estimates under different assumptions or conditions. While our significant accounting policies are more fully described in Note 2 in the accompanying Notes to the accompanying consolidated financial statements,Consolidated Financial Statements contained in this Annual Report on Form 10-K, we believe that the following accounting policies are critical to understanding the judgments and estimates used in the preparation of our financial statements. It is important that the discussion of our operating results that follows be read in conjunction with the critical accounting policies discussed below. Revenue Recognition Revenue is recognized when a customer obtains control of promised goods or services, in an amount that reflects the consideration which the entity expects to receive in exchange for those goods or services. To determine revenue recognition for arrangements that an entity determines are within the scope of ASC 606, Revenue from Contracts with Customers (“ASC 606”), we perform the following five steps: (i) identify the contract(s) with a customer; (ii) identify the performance obligations in the contract; (iii) determine the transaction price; (iv) allocate the transaction price to the performance obligations in the contract; and (v) recognize revenue when (or as) the entity satisfies a performance obligation. We only apply the five-step model to contracts when it is probable that the entity will collect the consideration it is entitled to in exchange for the goods or services it transfers to the customer. At contract inception, once the contract is determined to be within the scope of ASC 606, we assess the goods or services promised within each contract, determines those that are performance obligations and assesses whether each promised good or service is distinct. We then recognize as revenue the amount of the transaction price that is allocated to the respective performance obligation when (or as) the performance obligation is satisfied. Sales, value add, and other taxes collected on behalf of third parties are excluded from revenue. Product sales, net — We sell YUTIQ and DEXYCU to a limited number of specialty distributors and specialty pharmacies (collectively the “Distributors”) in the U.S., with whom we have entered into formal agreements, for delivery to physician practices for YUTIQ and to hospital outpatient departments and ambulatory surgical centers for DEXYCU. We recognize revenue on sales of our products when Distributors obtain control of the products, which occurs at a point in time, typically upon delivery. In addition to agreements with Distributors, we also enter into arrangements with healthcare providers, ambulatory surgical centers, and payors that provide for government mandated and/or privately negotiated rebates, chargebacks, and discounts with respect to their purchase of our products from Distributors.
Reserves for variable consideration — Product sales are recorded at the wholesale acquisition costs, net of applicable reserves for variable consideration. Components of variable consideration include trade discounts and allowances, provider chargebacks and discounts, payor rebates, product returns, and other allowances that are offered within contracts between us and our Distributors, payors, and other contracted purchasers relating to our product sales. These reserves, as detailed below, are based on the amounts earned, or to be claimed on the related sales, and are classified either as reductions of product revenue and accounts receivable or a current liability, depending on how the amount is to be settled. Overall, these reserves reflect our best estimates of the amount of consideration to which it is entitled based on the terms of the respective underlying contracts.Actual amounts of consideration ultimately received may differ from our estimates. If actual results in the future vary from the estimates, we adjust these estimates, which would affect product revenue and earnings in the period such variances become known. Distribution fees — We compensate our Distributors for services explicitly stated in our contracts and they are recorded as a reduction of revenue in the period the related product sale is recognized. Provider chargebacks and discounts — Chargebacks are discounts that represent the estimated obligations resulting from contractual commitments to sell products at prices lower than the list prices charged to our Distributors. These Distributors charge us for the difference between what they pay for the product and our contracted selling price. These reserves are established in the same period that the related revenue is recognized, resulting in a reduction of product revenue and the establishment of a current liability. Reserves for chargebacks consist of amounts that we expect to pay for units that remain in the distribution channel inventories at each reporting period-end that we expect will be sold under a contracted selling price, and chargebacks that Distributors have claimed, but for which we have not yet settled. Government rebates — We are subject to discount obligations under state Medicaid programs and Medicare. These reserves are recorded in the same period the related revenue is recognized, resulting in a reduction of product revenue and the establishment of a current liability which is included in accrued expenses and other current liabilities on the condensed consolidated balance sheets. Our business strategyliability for these rebates consists of invoices received for claims from prior quarters that have not been paid or for which an invoice has not yet been received, estimates of claims for the current quarter, and estimated future claims that will be made for product that has been recognized as revenue, but which remains in the distribution channel inventories at the end of each reporting period. Payor rebates — We contract with certain private payor organizations, primarily insurance companies, for the payment of rebates with respect to utilization of our products. We estimate these rebates and records such estimates in the same period the related revenue is recognized, resulting in a reduction of product revenue and the establishment of a current liability. Co-Payment assistance — We offer co-payment assistance to commercially insured patients meeting certain eligibility requirements. The calculation of the accrual for co-pay assistance is based on an estimate of claims and the cost per claim that we expect to receive associated with product that has been recognized as revenue. Product returns — We generally offer a limited right of return based on our returned goods policy, which includes entering into collaborativedamaged product and remaining shelf life. We estimate the amount of our product sales that may be returned and record this estimate as a reduction of revenue in the period the related product revenue is recognized, as well as reductions to trade receivables, net on the condensed consolidated balance sheets. License and collaboration agreement revenue — We analyze each element of our license and development agreements forcollaboration arrangements to determine the development and commercialization of product candidates utilizing our technology system.appropriate revenue recognition. The terms of the license agreement may include payment to us of non-refundable up-front license fees, milestone payments if specified objectives are achieved, and/or royalties on product sales. We recognize revenue from upfront payments at a point in time, typically upon fulfilling the delivery of the associated intellectual property to the customer. If the contract contains a single performance obligation, the entire transaction price is allocated to the single performance obligation. Contracts that contain multiple performance obligations require an allocation of the transaction price based on the estimated relative standalone selling prices of the promised products or services underlying each performance obligation. We determine standalone selling prices based on the price at which the performance obligation is sold separately. If the standalone selling price is not observable through past transactions, we estimate the standalone selling price taking into account available information such as market conditions and internally approved pricing guidelines related to the performance obligations. We recognize sales-based milestone payments as revenue upon the achievement of the cumulative sales amount specified in the contract in accordance with ASC 606-10-55-65. For those milestone payments which are contingent on the occurrence of particular future events, we determine that these arrangementsneed to be considered for inclusion in the calculation of total consideration from the contract as a component of variable consideration using the most-likely amount method. As such, we assess each milestone to determine the probability and substance behind achieving each milestone. Given the inherent uncertainty associated with these future events, we will not recognize revenue from such milestones until there is a high probability of occurrence, which typically include multiple deliverables by us (suchoccurs near or upon achievement of the event.
When determining the transaction price of a contract, an adjustment is made if payment from a customer occurs either significantly before or significantly after performance, resulting in a significant financing component. Applying the practical expedient in paragraph 606-10-32-18, we do not assess whether a significant financing component exists if the period between when we perform our obligations under the contract and when the customer pays is one year or less. None of our contracts contained a significant financing component as granting of license rights, providingDecember 31, 2020. Reimbursement of costs — We may provide research and development services manufacturingand incur maintenance costs of clinical materialslicensed patents under collaboration arrangements to assist in advancing the development of licensed products. We act primarily as a principal in these transactions and, participatingaccordingly, reimbursement amounts received are classified as a component of revenue to be recognized consistent with the revenue recognition policy summarized above. We record the expenses incurred and reimbursed on jointa gross basis. Royalties— We recognize revenue from license arrangements with our commercial partners’ net sales of products. Such revenues are included as royalty income. In accordance with ASC 606-10-55-65, royalties are recognized when the subsequent sale of the commercial partner’s products occurs. Our commercial partners are obligated to report their net product sales and the resulting royalty due to us typically within 60 days from the end of each quarter. Based on historical product sales, royalty receipts and other relevant information, we recognize royalty income each quarter and subsequently determine a true-up when we receive royalty reports and payment from our commercial partners. Historically, these true-up adjustments have been immaterial. Sale of Future Royalties — We have sold our rights to receive certain royalties on product sales. In the circumstance where we have sold our rights to future royalties under a royalty purchase agreement and also maintains limited continuing involvement in the arrangement (but not significant continuing involvement in the generation of the cash flows that are due to the purchaser), we defer recognition of the proceeds we receive for the sale of royalty streams and recognizes such unearned revenue as revenue under the units-of-revenue method over the life of the underlying license agreement. Under the units-of-revenue method, amortization for a reporting period is calculated by computing a ratio of the proceeds received from the purchaser to the total payments expected to be made to the purchaser over the term of the agreement, and then applying that ratio to the period’s cash payment. Estimating the total payments expected to be received by the purchaser over the term of such arrangements requires management to use subjective estimates and assumptions. Changes to our estimate of the payments expected to be made to the purchaser over the term of such arrangements could have a material effect on the amount of revenues recognized in any particular period. Research Collaborations — We recognize revenue over the term of the statements of work under any funded research committees) in exchangecollaborations. Revenue recognition for consideration, if any, related to us of some combination of one or more ofnon-refundablea license fees, funding of research and development activities, payments based upon achievement of clinical development, regulatory and sales milestones and/or royalties in the form of a designated percentage of product sales or participation in profits. Revenue arrangements with multiple deliverables are divided into separate units of accounting if certain criteria are met, including whether the delivered element has stand-alone value to the collaborative partner andoption right is assessed based on the selling priceterms of any such future license agreement or is otherwise recognized at the completion of the deliverables. When deliverables are separable, consideration received is allocatedresearch collaborations. Please refer to the separate units of accounting basedNote 3 for further details on the relative selling price method using management’s best estimatelicense and collaboration agreements into which we have entered and corresponding amounts of revenue recognized during the standalone selling price of deliverables when vendor-specific objective evidence or third-party evidence of selling price is not available. Allocated consideration is recognized as revenue upon application ofcurrent and prior year periods.
Deferred Revenue Amounts received prior to satisfying the appropriateabove revenue recognition principles to each unit. The assessment of multiple deliverable arrangements requires judgment in order to determine the appropriate units of accounting, the estimated selling price of each unit of accounting, and the points in time that, or periods over which,criteria are recorded as deferred revenue should be recognized.
For the years ended June 30, 2018 and 2017, we reported $1.3 million and $6.6 million, respectively, of collaborative research and development revenue. Of the total for fiscal 2017, $5.6 million representednon-cash revenue recognized upon the termination of our Restated Pfizer Agreement (see Note 4 in the accompanying Notesconsolidated balance sheets. Amounts not expected to be recognized within one year following the Consolidated Financial Statements contained in this Annual Reportbalance sheet date are classified as non-current deferred revenue.
Please refer to Note 4 for further details on Form10-Kthe license and collaboration agreements into which we have entered and corresponding amounts of revenue recognized for more information). Revenue is recognized when there is persuasive evidence that an arrangement exists, delivery has occurred, the price is fixedyear ended December 31, 2020 and determinable and collection is reasonably assured.2019. Recognition of Expense in Outsourced Clinical Trial Agreements We recognize research and development expense with respect to outsourced agreements for clinical trials with CROscontract research organizations (“CROs”) as the services are provided, based on our assessment of the services performed. We make our assessments of the services performed based on various factors, including evaluation by the third-party CROs and our own internal review of the work performed during the period, measurements of progress by us or by the third-party CROs, data analysis with respect to work completed and our management’s judgment. We have had agreements with two CROs to conduct the Phase 3 clinical trial program for YUTIQ. Our financial obligations underYUTIQ, which we completed in the agreements are determined by the services that we request from time to time under the agreements. The actual amounts owed under the agreements and the timingfirst half of those obligations will depend on various factors, including changes to the protocols and/or services requested, the number of patients to be enrolled and the rate of patient enrollment, achievement ofpre-defined direct cost milestone events and other factors relating to the clinical trials.2020. As of June 30, 2018, ourDecember 31, 2020, there were no material obligations remaining under those agreements. In August 2020, we entered into an agreement with a CRO agreements providedto conduct a Phase 1 study for two Phase 3 clinical trials at an aggregate remainingcostEYP-1901, which we started in the first quarter of approximately $4.5 million, excluding any potential remaining contract change orders. We can terminate the agreements at any time without penalty, and if terminated, we would be liable only for services through the termination date plusnon-cancellable CRO obligations to third parties.2021. During fiscal 2018,2020 and 2019, we recognized approximately $4.6$91,000 and $1.2 million, respectively, of research and development expense attributable to our YUTIQ Phase 3 clinical trial program. Changes in our estimates or differences between the actual level of services performed and our estimates may result in changes to our research and development expenses in future periods.
Results of Operations Years Ended June 30, 2018December 31, 2020 and 20172019 | | | | | | | | | | Year Ended December 31, | | | Change | | | | | Year Ended June 30, | | Change | | | 2020 | | | 2019 | | | Amounts | | | % | | | | | 2018 | | 2017 | | Amounts | | % | | | | | | | | | | | | | | | | | | | | | (In thousands except percentages) | | | (In thousands except percentages) | | Revenues: | | | | | | | | | | | | | | | | | | | | | | | | | Collaborative research and development | | $ | 1,343 | | | $ | 6,569 | | | $ | (5,226 | ) | | (80 | )% | | Product sales, net | | | $ | 20,831 | | | $ | 16,824 | | | $ | 4,007 | | | | 24 | % | License and collaboration agreements (including licensing fees from a related party of $11,500 and $1,000 for the years ended December 31, 2020 and 2019, respectively) | | | | 11,942 | | | | 1,361 | | | | 10,581 | | | | 777 | % | Royalty income | | | 1,618 | | | 970 | | | 648 | | | 67 | % | | | 1,664 | | | | 2,180 | | | | (516 | ) | | | (24 | )% | | | | | | | | | | | | | | | Total revenues | | | 2,961 | | | 7,539 | | | (4,578 | ) | | (61 | )% | | | 34,437 | | | | 20,365 | | | | 14,072 | | | | 69 | % | | | | | | | | | | | | | | | Operating expenses: | | | | | | | | | | | | | | | | | | | | | | | | | Cost of sales, excluding amortization of acquired intangible assets | | | | 5,824 | | | | 2,687 | | | | 3,137 | | | | 117 | % | Research and development | | | 16,178 | | | 14,880 | | | 1,298 | | | 9 | % | | | 17,424 | | | | 15,368 | | | | 2,056 | | | | 13 | % | Sales and marketing | | | 1,512 | | | | — | | | 1,512 | | | na | | | | 25,293 | | | | 29,772 | | | | (4,479 | ) | | | (15 | )% | General and administrative | | | 11,545 | | | 11,235 | | | 310 | | | 3 | % | | | 20,726 | | | | 17,939 | | | | 2,787 | | | | 16 | % | | | | | | | | | | | | | | | Amortization of acquired intangible assets | | | | 2,460 | | | | 2,460 | | | | — | | | N/A | | Total operating expenses | | | 29,235 | | | 26,115 | | | 3,120 | | | 12 | % | | | 71,727 | | | | 68,226 | | | | 3,501 | | | | 5 | % | | | | | | | | | | | | | | | Operating loss | | | (26,274 | ) | | (18,576 | ) | | (7,698 | ) | | (41 | )% | | Loss from operations | | | | (37,290 | ) | | | (47,861 | ) | | | 10,571 | | | | (22 | )% | Other income (expense) | | | | | | | | | | | | | | | | | | Interest income and other, net | | | 101 | | | 91 | | | 10 | | | 11 | % | | | 58 | | | | 1,054 | | | | (996 | ) | | | (94 | )% | Interest expense | | | (720 | ) | | | — | | | (720 | ) | | na | | | | (7,257 | ) | | | (6,176 | ) | | | (1,081 | ) | | | (18 | )% | Change in fair value of derivative liability | | | (26,278 | ) | | | — | | | (26,278 | ) | | na | | | | | | | | | | | | | | | | | Loss on extinguishment of debt | | | | (905 | ) | | | (3,810 | ) | | | 2,905 | | | | 76 | % | Other expense, net | | | | (8,104 | ) | | | (8,932 | ) | | | 828 | | | | 9 | % | Net loss | | $ | (53,171 | ) | | $ | (18,485 | ) | | $ | (34,686 | ) | | (188 | )% | | $ | (45,394 | ) | | $ | (56,793 | ) | | $ | 11,399 | | | | 20 | % | | | | | | | | | | | | | | |
Revenues
Collaborative researchProduct Sales, net
Product sales, net represents the gross sales of YUTIQ and development revenue totaled $1.3DEXYCU less provisions for product sales allowances. Product sales, net increased by $4.0 million to $20.8 million for 2020 compared to $16.8 million in fiscal 2018,the prior year. We commenced U.S. commercial sales of YUTIQ in February 2019 and DEXYCU in March 2019. Product sales during 2020 were negatively impacted due to shutdowns associated with the Pandemic in the U.S. Although we did see a decreasemodest return of $5.2 million, or 80%, comparedcustomer demand for both products in the third and fourth quarters, we expect demand to $6.6 million in fiscal 2017. This decrease was attributable primarily to $5.6 million of revenue recognized uponcontinue at decreased levels through the terminationduration of the Restated Pfizer Agreement in December 2016Pandemic until restrictions on elective surgeries and a $535,000 reduction of revenues attributable tooffice visits are removed. Please see the Prior Alimera Agreement, which included $136,000 of revenue recognized from a May 2017 arbitration settlement of Alimera’s calendar year 2014 reporting of ILUVIEN net profits, partially offset by an $879,000 increase in revenues derived from feasibility study agreements. In July 2017, we entered into the Amended Alimera Agreement, pursuant to which we (i) expanded the license to Alimera for our proprietary Durasert drug delivery technology platform to include uveitis, including NIPU, in the EMEA and (ii) converted the net profit share arrangement for each licensed product (including
ILUVIEN) under the Prior Alimera Agreement to a sales-based royalty on a calendar quarter basis. We expect this conversion to result in increased revenues from Alimera over time, as well as better predictability and consistency of revenues to be recognized from Alimera. Based on60-day payment terms from Alimera following the end of each calendar quarter, sales-based royalties earned from Alimera are being recognized as revenues one quarter in arrears. Commencing in calendar 2019, the royalty rate on net sales of ILUVIEN will increase from 2% to 4% (which represents a 6% royalty rate less a 2% offset for Alimera’s recovery of accumulated commercialization losses under the Prior Alimera Agreement). See Note 4 in the accompanying Notes to the Consolidated Financial Statements contained in this Annual Report on Form10-KRecent Development section for more information related toon the Alimeraimpact of the Pandemic on, among other things, our product sales.
License and collaboration agreement License and collaboration agreement andrevenues increased by $10.6 million to the settlement of a dispute relating to the computation of ILUVIEN net profits for calendar year 2014 Royalty income totaled $1.6$11.9 million for fiscal 2018, an increase of $648,000, or 67%,in 2020 compared to $970,000 in fiscal 2017. Royalty income attributable to the Amended Alimera Agreement totaled $575,000 in fiscal 2018. Retisert royalty income increased by $73,000, or 8%, to approximately $1.0$1.4 million in fiscal 2018 compared to $970,000 in fiscal 2017. We expect Retisert royalty income to remain flat or to decline somewhat in the next fiscal year.
Research and Development
Research and development expenses totaled $16.2 million in fiscal 2018, an increase of $1.3 million, or 9%, compared to $14.9 million in fiscal 2017.2019. This increase was attributable primarily to (i)the recognition of $9.5 million under our Ocumension 2020 MOU entered into August 2020 (see Note 3) as well as approximately $2.0 million from Ocumension upon signing a $1.8license agreement for DEXYCU in China, compared with $1.0 million increase of professional services related primarilyrecognized in 2019.
Royalty Income Royalty income decreased by $516,000 to our YUTIQ three-year uveitis Phase 3 clinical development program and regulatory filings, (ii) a $774,000 increase$1.7 million in U.S. personnel and related costs, including incentive compensation and (iii) a $404,000 increase inpre-clinical studies, related primarily2020. The decrease was attributable to our short-acting YUTIQ product candidate, and other third-party research costs; (iv) a $257,000 increase in amortization of intangible assets, attributable primarily to $615,000 of amortizationthe impact of the DEXYCU / Icon intangible asset offset byroyalty monetization agreement with SWK Holdings that grants to SWK all future royalty payments under the completed amortizationAmended Alimera Agreement beginning with Q4 2020 for a one-time payment of our previous patent technology intangible assets as of December 2017; and (v) a $116,000 increase in U.S. stock-based compensation, partially offset by decreases of (i) $1.6 million of CRO costs for our YUTIQ three-year uveitis clinical development, (ii) $472,000 of direct U.K. restructuring costs in fiscal 2017. In anticipation of the commercial launch of DEXYCU and, if approved, YUTIQ in the first half of calendar 2019, we expect fiscal 2019 research and development expense to increase significantly compared to fiscal 2018, due primarily to increased personnel and third-party costs for medical and regulatory affairs, pharmacovigilance, quality assurance and operations and full year amortization of the DEXYCU intangible asset, partially offset by anticipated reductions in CRO costs for the YUTIQ Phase 3 clinical trials and regulatory professional services related$16.5 million. Due to the YUTIQ NDA filing. Salesaccounting treatment for this agreement (see Revenue Recognition section), we did not record any accrued royalty revenue under the Amended Alimera Agreement in Q4-2020 and Marketing
In anticipation of the commercial launch of DEXYCU and, if approved by the FDA, YUTIQ, we commenced thebuild-out of our commercial infrastructure and marketing activities in the fourth quarter of fiscal 2018. Sales and marketing expense totaled $1.5 million in fiscal 2018 and consisted primarily of $1.0 million of marketing program and agency costs and approximately $450,000 of personnel and related costs, including travel and stock-based compensation. We expect major increases in sales and marketing costs in fiscal 2019, including additional headcount, implementation of our contract sales organization agreement, participation in numerous ophthalmology conferences and congresses and managed markets and sales operations activities.will
General and Administrative
General and administrative expenses totaled $11.5 million in fiscal 2018, an increase of $310,000, or 3%, compared to $11.2 million in fiscal 2017. The increase was attributable primarily to (i) approximately $900,000 of consulting services, which included interim CFO services, business development and the effect ofrecognize a $218,000 fiscal 2017 credit for audit costs in connection with the May 2017 Alimera arbitration settlement; (ii) approximately $204,000 of facility and office expense, which included our New Jersey office that opened in July 2017; and (iii) approximately $116,000 of stockholder meeting and stock exchange costs, including our delisting
from the Australian Securities Exchange, partially offset by net decreases of (i) approximately $813,000 for personnel and related costs largely due to the absence of $1.2 million of fiscal 2017 severance compensation to our former CEO and former Vice President, Corporate Affairs and General Counsel, and (ii) approximately $230,000 of legal, audit and other professional fees that resulted from a combination of lower patent legal fees and the absence of approximately $605,000 of legal fees associated with our CEO transition and the arbitration proceedings and the restructuring of our Alimera collaboration agreement, which were partially offset by fiscal 2018 legal fees associated with potential financing and business development transactions and our ASX delisting. In anticipation of the commercial launch of DEXYCU and, if approved, YUTIQ in the first half of calendar year 2019, we expect fiscal 2019 general and administrative expenses to increase significantly, primarily for headcount additions and associated personnel and related costs.
Interest (Expense) Income and Other
On March 28, 2018, we borrowed $15.0 million under a term loan facility in connection with the Icon Acquisition. Following consummation of the Second Tranche Transaction on June 25, 2018, we borrowed an additional $5.0 million under that term loan facility. For fiscal 2018 we incurred $511,000 of interest expense on the term loan and amortized $209,000non-cash portion of deferred debt issue costs and debt discount. Assuming that the LIBOR rate remains constant at the current rate 2.34%, we expectrevenue as Alimera pays royalties to incur interest expense of approximately $2.6 million during fiscal 2019. In addition,non-cash amortization of the balance of debt discount at June 30, 2018 utilizing the effective interest methodSWK beginning in Q1 2021 (see Note 3). Accordingly, revenue recognized for Alimera Royalty Income is expected to be approximately $652,000 duringlower in future periods based on this methodology.
Separately, the quarter ended March 31, 2019 was the last period we recognized the Retisert royalty as the licensee, Bausch and Lomb informed us in early 2019 that they consider this agreement to have ended due to the expiration of certain patents. Cost of Sales, Excluding Amortization of Acquired Intangible Assets Cost of sales, excluding amortization of acquired intangible assets, increased by $3.1 million to $5.84 million for fiscal 2019. Interest income and other increased to $101,000 in fiscal 2018 compared to $91,000 in fiscal 2017, due primarily to higher interest rates on amounts invested in an institutional money market fund.
Change in Fair Value of Derivative Liability
Change in fair value of derivative liability totaled $26.32020 from $2.7 million in fiscal 2018,the prior year. This increase was primarily attributable to the classification(i) approximately $1.3 million of the Second Tranche transaction as a liability (see Notes 10 and 12 to the accompanying consolidated financial statements). The Unitsroyalty expense associated with the Second Tranche Transaction were settled to equity upon$9.5 million received from Ocumension under the Second Tranche Transaction closing on June 25, 2018. The Second Tranche Warrants were also liability classified and were revalued at June 30, 2018. The resulting $19.82020 MOU as well as $2 million derivative liability balance at June 30, 2018 will be revalued immediately prior toreceived from the exercise or the September 28, 2018 expirationOcumension DEXYCU signing payment received, (ii) $897,000 from a one-time write-down of the Second Tranche Warrants.
Years Ended June 30, 2017 and 2016
| | | | | | | | | | | | | | | | | | | | Year Ended June 30, | | | Change | | | | | 2017 | | | 2016 | | | Amounts | | | % | | | | | (In thousands except percentages) | | Revenues: | | | | | | | | | | | | | | | | | Collaborative research and development | | $ | 6,569 | | | $ | 398 | | | $ | 6,171 | | | | 1,551 | % | Royalty income | | | 970 | | | | 1,222 | | | | (252 | ) | | | (21 | )% | | | | | | | | | | | | | | | | | | Total revenues | | | 7,539 | | | | 1,620 | | | | 5,919 | | | | 365 | % | | | | | | | | | | | | | | | | | | Operating expenses: | | | | | | | | | | | | | | | | | Research and development | | | 14,880 | | | | 14,381 | | | | 499 | | | | 3 | % | General and administrative | | | 11,235 | | | | 9,013 | | | | 2,222 | | | | 25 | % | | | | | | | | | | | | | | | | | | Total operating expenses | | | 26,115 | | | | 23,394 | | | | 2,721 | | | | 12 | % | | | | | | | | | | | | | | | | | | Operating loss | | | (18,576 | ) | | | (21,774 | ) | | | 3,198 | | | | 15 | % | Interest and other income, net | | | 91 | | | | 72 | | | | 19 | | | | 26 | % | | | | | | | | | | | | | | | | | | Loss before income taxes | | | (18,485 | ) | | | (21,702 | ) | | | 3,217 | | | | 15 | % | Income tax benefit | | | — | | | | 155 | | | | (155 | ) | | | (100 | )% | | | | | | | | | | | | | | | | | | Net loss | | $ | (18,485 | ) | | $ | (21,547 | ) | | $ | 3,062 | | | | 14 | % | | | | | | | | | | | | | | | | | |
Revenues
Collaborative research and development revenue totaled $6.6 million in fiscal 2017, an increaseout of $6.2 million, or 1,551%, compared to $398,000 in fiscal 2016. This increase was attributable primarily to $5.6 million of revenue recognized upon the termination of the Restated Pfizer Agreement in December 2016. In addition, revenues derivedspecification DEXYCU production batches from our collaboration agreementthird-party manufacturer and (iii) increased costs associated with Alimera increased by $426,000, which included $136,000 of revenue recognized from a May 2017 arbitration settlement of Alimera’s calendar year 2014 reporting of ILUVIEN net profits.higher product sales, primarily product cost and distribution fees.
In July 2017, we restructured the Alimera collaboration agreement to (a) license Durasert three-year uveitis in the EMEA to Alimera and (b) to convert the net profit share arrangement to a sales-based royalty for all ILUVIEN licensed indications. See Notes 4 in the accompanying Notes to Consolidated Financial Statements contained in this Annual Report on Form10-K for more information related to the Alimera collaboration agreement and to the settlement of a dispute relating to the computation of ILUVIEN net profits for calendar year 2014.
Retisert royalty income decreased by $252,000, or 21%, to $970,000 in fiscal 2017 compared to $1.22 million in fiscal 2016.
Research and Development Research and development expenses totaled $14.9increased by $2.1 million to $17.4 million for 2020 from $15.4 million in fiscal 2017, an increase of $499,000, or 3%, compared to $14.4 million in fiscal 2016. This increase was attributable primarily to (i) a $1.4 million increase of professional services related primarily to our Durasert three-year uveitis Phase 3 clinical development program and completed MAA filing and planned NDA filing, (ii) an $879,000 increase in U.S. personnel and related costs, including incentive compensation and the August 2016 hire of our Chief Medical Officer and (iii) a $596,000 increase in U.S. stock-based compensation, partially offset by decreases of (i) $1.1 million of CRO costs for our Durasert three-year uveitis clinical development, (ii) $1.0 million of U.K. costs primarily related to the effect of the U.K. restructuring, which included a $147,000 foreign exchange impact of a stronger US$ currency, and (iii) $268,000 of U.S.pre-clinical studies and other third-party research costs related primarily to prior year studies of potential TKI compounds and purchases of lab and clinical supplies for our Durasert three-year uveitis clinical development program. General and Administrative
General and administrative expenses totaled $11.2 million in fiscal 2017, an increase of $2.2 million, or 24%, compared to $9.0 million in fiscal 2016.year. This increase was attributable primarily to (i) approximately $1.5$1.4 million of expense for EYP-1901 related studies, (ii) a $1.0 million payment for the licensing of Vorolanib (EYP-1901), (iii) approximately $650,000 in lab and non-clinical expenses primarily for support of the EYP-1901 initiatives and (iv) approximately $422,000 of expense for DEXYCU related studies, partially offset by approximate comparative decreases of (i) $1.1 million in YUTIQ Phase 3 expense as the trial ended in Q2 2020 and (ii) $385,000 in stability and other testing.
Sales and Marketing Sales and marketing expenses decreased by approximately $4.5 million to $25.3 million for fiscal 2020 from $29.8 million in the prior year. This decrease was primarily attributable to (i) approximately $4.0 million of net contract sales organization (CSO) expenses due to the reduction in DEXYCU KAMs as per our previously announced restructuring plan, including severance and (ii) $1.6 million in marketing and related expenses in conjunction with our restructuring plan, partially offset by (i) an approximate $900,000 increase in personnel (other than KAMs converted to employees) and related expenses, primarily from the full year to date impact of prior year additions. General and Administrative General and administrative expenses increased by $2.8 million to $20.7 million for 2020 from $17.9 million in the prior year. This increase was attributable primarily to (i) $1.0 million in legal and other professional fees, (ii) $666,000 in insurance expense, due primarily to D&O policy costs, (iii) $597,000 in consulting expenses and (iv) $432,000 in personnel and related costs, including annual incentive compensation,expenses. Amortization of Acquired Intangible Assets Amortization of acquired intangible assets totaled $2.5 million for both 2020 and 2019. This amount was attributable to the DEXYCU product intangible asset that resulted from the Icon Acquisition (see Note 5). Interest (Expense) Income Interest expense totaled $7.3 million for 2020, which $1.2included $745,000 of amortization of debt discount and $977,000 of non-cash payment-in-kind interest expense all related to the CRG Debt. Interest expense in 2019 was $6.2 million represented severance compensation to our former CEOwhich included $596,000 of amortization of debt discount and former Vice President, Corporate Affairs and General Counsel, (ii) approximately $1.3$1.1 million of legal fees, which included approximately $175,000non-cash payment-in-kind interest expense. During the prior year period, we extinguished the SWK Loan and established a new term loan facility with CRG (see Note 9). Interest income from amounts invested in an institutional money market fund decreased to $58,000 for fiscal 2020 compared to $1.1 million, due primarily to reduced interest-bearing assets and lower money market interest rates versus 2019. Loss on Extinguishment of legal feesDebt In Q4 2020, we paid down $13.7 million to partially reduce principal on the CRG term loan. We recorded a $905,000 loss on partial extinguishment of debt associated with the CEO transition and severance arrangements, $430,000write-off of legal feesthe balance of unamortized debt discount related to this partial prepayment.
Repayment of the Alimera arbitration proceedings and agreement restructuring and $253,000SWK Loan in February 2019 resulted in a $3.8 million loss on extinguishment of patent legal fees, partially offset by a $619,000 decrease in consulting services costs,debt, which consisted primarily of prior year uveitis market assessment analyses and the $218,000 fiscal 2017 cancellation of previously accrued audit costs in connection with the Alimera arbitration settlement. Interest and Other Income
Interest and other income increased to $91,000 in fiscal 2017, an increase of $19,000, or 23%, compared to $72,000 in fiscal 2016, due primarily to higher money market interest rates.
Income Tax Benefit
Income tax benefit was $0 in fiscal 2017 compared to an income tax benefit of $155,000 in fiscal 2016. We incurred $4,000 in fiscal 2016 of federal alternative minimum tax expense based on U.S. taxable income for
calendar year 2014 attributable primarily to the $25.0(i) a $2.3 million ILUVIENFDA-approval milestone. Refundable foreign research and development tax credits were not available for fiscal 2017 as a resultwrite-off of the consolidationremaining balance of our researchunamortized debt discount; (ii) a $1.2 million prepayment penalty; and development activities in(iii) a $306,000 make-whole interest payment covering the U.S. duringperiod from the quarter ended September 30, 2016.date of the loan repayment to what would have been the first anniversary of the original loan closing date, or March 28, 2019.
Inflation and Seasonality
Our management believes inflation has not had a material impact on our operations or financial condition and that our operations are not currently subject to seasonal influences.
Recently Adopted and Recently Issued Accounting Pronouncements New accounting pronouncements are issued periodically by the Financial Accounting Standards Board or FASB,(“FASB”), and are adopted by us as of the specified effective dates. Unless otherwise disclosed below, we believe that the impact of recently issued and adopted pronouncements will not have a material impact on our financial position, results of operations and cash flows or do not apply to our operations. In May 2014, the FASB issued Accounting Standards UpdateNo. 2014-09,Revenue from Contracts with Customers(Topic 606), which we refer to as ASU2014-09, which requires an entity to recognize revenue in an amount that reflects the consideration to which the entity expects to be entitled in exchange for the transfer of promised goods or services to customers. The standard will replace most existing revenue recognition guidance in U.S. GAAP. In August 2015, the FASB issued ASU2015-14, which officially deferred the effective date of ASU2014-09 by one year, while also permitting early adoption. As a result, ASU2014-09 becomes effective on July 1, 2018. The Company has initiated an assessment of the potential changes from adopting ASU2014-09 and two revenue streams are expected to be applicable under the standard. We have adopted the new standard effective July 1, 2018 using the modified retrospective method. The adoption did not have an impact on the recorded amounts in the current balance sheet and income statement based on our existing collaboration agreements. Additional disclosures will be made in future periods related to the recognition of amounts as a result of adopting the new standard.
In February 2016, the FASB issued ASUNo. 2016-02,Leases (Topic 842) (“ASU 2016-02”). In July 2018, the FASB issued ASU No. 2018-11, Leases (Topic 842), Targeted Improvements, which contains certain amendments to ASU 2016-02 intended to provide relief in implementing the new standard. The new standard establishes aright-of-use or ROU, (“ROU”) model that requires a lessee to record a ROU asset and a lease liability on the balance sheet for all operating leases, with terms longer than 12 months. Leases will be classified as either financean exception provided for leases with a duration of one year or operating,less. We adopted ASU 2016-02 on January 1, 2019 using the modified retrospective transition approach which, pursuant to ASU 2018-11, allows companies to recognize existing leases at the adoption date without requiring comparable period presentation. Comparative periods are presented in accordance with classification affecting the patternprevious guidance in Accounting Standards Codification (“ASC”) 840, Leases. In June 2016, the FASB issued ASU No. 2016-13, Financial Instruments - Credit Losses (Topic 326) (“ASU 2016-13”): Measurement of expense recognition inCredit Losses on Financial Instruments, to replace the income statement. The new standardcurrent incurred loss impairment methodology for financial assets measured at amortized cost with a methodology that reflects expected credit losses and requires consideration of a broader range of reasonable and supportable information, including forecasted information, to develop credit loss estimates. ASU 2016-13 is effective for fiscal years beginning after December 15, 2018, including interim periods within those fiscal years. As a result, ASU2016-02 will become effective on July 1, 2019. A modified retrospective transition approach is required for lessees for capital and operating leases existing at, or entered into after, the beginning of the earliest comparative period presented in the financial statements, with certain practical expedients available. We are currently evaluating the impact of the pending adoption of the new standard on our consolidated financial statements. In November 2016, the FASB issued ASUNo. 2016-18,Restricted Cash. The standard addresses the classification and presentation of restricted cash and restricted cash equivalents within the statement of cash flows. The ASU is effective for fiscal years beginning after December 15, 2017, with early adoption permitted. We adopted this standard during the quarter ended June 30, 2018. As a result of the adoption of this standard, we combined restricted cash balances of $150,000 at each of June 30, 2018, 2017 and 2016 with cash and cash equivalents when reconciling the beginning and ending balances within the consolidated statements of cash flows for fiscal 2018, 2017 and 2016. As of June 30, 2018, cash, cash equivalents and restricted cash of $38.9 million, as reported within the consolidated statements of cash flows, included $38.8 million of cash and cash equivalents and $150,000 of restricted cash, as reported within the consolidated balance sheets.
In January 2017, the FASB issued ASU No.2017-01, Business Combinations (Topic 805): Clarifying the Definition of a Business, which we refer to as ASU2017-01, to clarify the definition of a business by adding guidance to assist entities with evaluating whether transactions should be accounted for as acquisitions or
disposals of assets versus businesses. ASU2017-01 is effective for fiscal years beginning after December 15, 2017, and interim periods within those fiscal years. Early adoption is permitted. We adopted this standard early to account for the Icon Acquisition (see Note 3 in the accompanying Notes to Consolidated Financial Statements contained in this Annual Report on Form10-K).
In June 2018, the FASB issued ASU2018-07,Improvements to Nonemployee Share-Based Payment Accounting. The standard aligns the measurement and classification guidance for share-based payments to nonemployees with the guidance for share-based payments to employees, with certain exceptions. Under the new guidance, the measurement of equity-classified nonemployee awards will be fixed at the grant date. The ASU is effective for fiscal years beginning after December 15, 2018,2019, and interim periods within those fiscal years. Early adoption is permitted for fiscal years beginning after December 15, 2018. We adopted ASU 2016-13 on January 1, 2020. The adoption of this standard did not have a material impact on our consolidated financial statements.
In December 2019, the FASB issued ASU No. 2019-12, Income Taxes (Topic 740) (“ASU 2019-12”): Simplifying the Accounting for Income Taxes. The amendments simplify the accounting for income taxes by removing certain exceptions for recognizing deferred taxes for investments, performing intraperiod allocation and calculating income taxes in interim periods. The ASU also adds guidance to reduce complexity in certain areas, including recognizing deferred taxes for tax goodwill and allocating taxes to members of a consolidated group ASU 2019-12 is effective for fiscal years beginning after December 15, 2020, and interim periods within those fiscal years. Early adoption is permitted, including adoption in interim or annual periods for which financial statements have not yet been issued. This standard will be effective for us in the first quarter of our fiscal year ending December 31, 2021. We are currently evaluating the impact the adoption of this update will have on our consolidated financial statements, but do not before an entity adoptsbelieve the adoption of the new revenue guidance. We do not expect ASU2018-07 tostandard will have a significantmaterial impact on our consolidated financial statements. Liquidity and Capital Resources We have had a history of operating losses and an absence of significant recurring cash inflows from revenue, and at December 31, 2020 we had a total accumulated deficit of $510.7 million. Our operations have been financed primarily from sales of our equity securities, issuance of debt and a combination of license fees, milestone payments, royalty income and other fees received from collaboration partners. In the first quarter of 2019, we commenced the U.S. launch of our first two commercial products, YUTIQ and DEXYCU. However, we have not received sufficient revenues from our product sales to fund operations and we do not expect revenues from our product sales to generate sufficient funding to sustain our operations in the near-term. Financing Activities Our operations for fiscal 2018 operations2020 were financed primarily from existing capital resources at June 30, 2017, gross proceeds of $7.3 million from sales of 5,900,000 shares of common stock under our existing ATM program and the First Tranche Transaction and the Loan that resulted in gross proceeds of $24.5 million, $15.0$22.2 million of which was used to fund the closing payment to acquire Icon and approximately $3.5 million was used to pay certain financing and transaction costs. Following approval of our stockholders at a special meeting held on June 22, 2018, we closed the Second Tranche Transaction resulting in the issuance of 20,184,224 Units, which consisted of (i) the sale of 20,184,224 shares of our common stock at a per share price of $1.265 for gross proceeds of $25.5 million and (ii) the issuance of 20,184,224 warrants, which we collectively refer to as the Second Tranche Warrants, to purchase shares of our common stock. The Second Tranche Warrants are exercisable at any time until the close of business on September 28, 2018. The exercise price of each Second Tranche Warrant will be an amount equal to the lower of (a) $1.43 (a 30% premium to the First Tranche Purchase Price) and (b) a 20% discount to the VWAP of the shares of the Company’s common stock on Nasdaq for the 20 trading days immediately prior to the exercise of a Second Tranche Warrant; provided, however, that the exercise price cannot be lower than $0.88, which is a 20% discount to the First Tranche Purchase Price. In connection with the Second Tranche Transaction, a $5.0 million draw down of the Additional Advance was consummated on June 26, 2018. At June 30, 2018, our principal sources of liquidity were cash and cash equivalents that totaled $38.8 million. As of June 30, 2018, our debt consists ofat December 31, 2019, approximately $20.0 million which amount representsof gross proceeds from the amount outstandingFebruary 2020 underwritten stock offering and approximately $14.2 million from sales under our at-the-market facility. In addition, on April 8, 2020, we submitted an application through Silicon Valley Bank for the Paycheck Protection Program Loan (the “PPP Loan”) pursuant to the Credit Agreement. Coronavirus Aid, Relief and Economic Security Actadministered by the U.S. Small Business Administration (the “SBA”). On April 22, 2020, we received PPP Loan proceeds of $2.0 million. We also received $15.7 million in December 2020 from an equity investment by Ocumension. In addition to our cash and cash equivalents of $44.9 million at December 31, 2020, we received incremental financing cash flows of approximately $108 million net proceeds from our February 2021 stock offering.
The CRG Loan is due and payable on March 27, 2023.December 31, 2023 (the “Maturity Date”). The CRG Loan bears interest at a per annum rate (subject to increase during an event of default) equal to 12.5%, of which 2.5% may be paid in-kind at our election, so long as no
default or event of default under the three-month LIBOR, subjectCRG Loan Agreement has occurred and is continuing. We are required to a 1.5% floor, plus 10.50%. The Credit Agreement permits us to paymake interest only on the principal amount loaned thereunder for the first eight payments (payments are due on a quarterly basis). Followingbasis until the interest-only period, weMaturity Date. We will also be required to make quarterly payments of interest, plus repayments of the principal amount loaned under the Credit Agreement in an aggregate amount of up to $1,667,000 per quarter, or the Quarterly Principal Repayment Cap. Subject to the Quarterly Principal Repayment Cap, the amount of any quarterly principal payments during any fiscal year is based on (x) a percentage ofour year-to-date net revenue through the end of such quarter less (y) any prior quarterly principal and interest payments made during such fiscal year. In addition, we paid an upfront fee of 1.5% of the aggregate principal amount of the Loan. We are also required to pay an exit fee equal to 6% of the aggregate principal amountamounts advanced (including any paid-in-kind amounts) under the CreditCRG Loan Agreement. Subject to certain exceptions, we are required to make mandatory prepayments of the CRG Loan with the proceeds of assets sales and insurance proceeds.in the event of a change of control of our Company. In addition, we may make a voluntary prepayment of the CRG Loan, in whole but notor in part, at any time on or after the first anniversary of March 28, 2018.time. All mandatory and voluntary prepayments of the CRG Loan are subject to the payment of prepayment premiums as follows: (i) in the case of mandatory prepayments, if prepayment occurs after December 31, 2019 and on or prior to December 31, 2020, 5% of the first anniversary of March 28, 2018, a customary make-whole amount equal to theaggregate outstanding principal amount of interest that would have accrued on the principal amount soCRG Loan being prepaid
had it remained outstanding through the first anniversary of March 28, 2018, and (ii) if prepayment occurs after December 31, 2020 and on or after the first anniversary of March 28, 2018 but prior to the second anniversary of March 28, 2018, 6% of the aggregate amount of the principal prepaid and (iii) if prepayment occurs on or after the second anniversary of March 28, 2018 but prior to the third anniversary of March 28, 2018,December 31, 2021, an amount equal to 1%3% of the aggregate outstanding principal amount of the CRG Loan being prepaid. No prepayment premium is due on any principal prepaid after December 31, 2021.
Certain of our existing and future subsidiaries, including the Guarantors, are guaranteeing the obligations of ours under the Loan Agreement. Our obligations under the Loan Agreement and the guarantee of such obligations are secured by a pledge of substantially all of our and the Guarantors’ assets. The CRG Loan Agreement contains affirmative and negative covenants customary for financings of this type, including limitations on our and our subsidiaries’ abilities, among other things, to incur additional debt, grant or permit additional liens, make investments and acquisitions, merge or consolidate with others, dispose of assets, pay dividends and distributions and enter into affiliate transactions, in each case, subject to certain exceptions. In addition, the CRG Loan Agreement contains the following financial covenants requiring us and the Guarantors to maintain: | • | liquidity in an amount which shall exceed the greater of (i) $5 million and (ii) to the extent we have incurred certain permitted debt, the minimum cash balance, if any, required of us by the creditors of such permitted debt; and |
| • | annual minimum product revenue from YUTIQ and DEXYCU: (i) for the twelve-month period beginning on January 1, 2021 and ending on December 31, 2021, of at least $45 million and (ii) for the twelve-month period beginning on January 1, 2022 and ending on December 31, 2022, of at least $90 million. |
On October 8, 2020, we entered into a Waiver to the CRG Loan Agreement (the “Waiver”) pursuant to which CRG waived the financial covenant associated with our revenue derived from sales of YUTIQ and DEXYCU for the twelve-month period ended December 31, 2020 and reduced the revenue covenant for the twelve-month period ending December 31, 2021 from $80 million to $45 million. On November 19, 2019, we entered into a Waiver to the CRG Loan Agreement (the “Waiver”) pursuant to which CRG waived the financial covenant associated with our revenue derived from sales of YUTIQ and DEXYCU for the twelve-month period ended December 31, 2019. If we do not maintain compliance with all of the continuing covenants and other terms and conditions of the CRG Loan or secure a waiver for any non-compliance, then the Lenders may choose to declare an event of default and require that we immediately repay all amounts outstanding, plus penalties and interest, including an exit fee and any prepayment fees, and foreclose on the collateral granted to them to secure such indebtedness. Such repayment would have a material adverse effect on our business and financial condition. On December 17, 2020, we paid $15.0 million against the CRG Loan obligations in connection with the consummation of the RPA agreement (see Note 3). In addition to repayment of the $13.8 million principal portion of the CRG Loan, we paid (i) the $828,000 Exit Fee, and (ii) accrued and unpaid interest of $378,000 through that date. CRG waived the financial covenant in the CRG Loan associated with the payment of prepayment premiums if prepayment occurred after December 31, 2019 and on or afterprior to December 31, 2020.As of December 31, 2020, our outstanding balance, including principal and the third anniversaryexit fee, under the CRG Loan was approximately $40.5 million, and consisted of March 28, 2018. With the exceptionapproximately $38.3 million of net income for the fiscal year ended June 30, 2015 resulting from our receiptcarrying value (see Note 14), and $2.2 million of the $25.0 million ILUVIENFDA-approval milestone, we have predominantly incurred operating losses since inception, and at June 30, 2018remaining balance of unamortized debt discount related to the CRG Loan.
Future Funding Requirements At December 31, 2020, we had a total accumulated deficitcash and cash equivalents of $364.0$44.9 million. We do not currently have any significant assured sources of future revenue, and our anticipated recurring use of cash to fund operations in combination with no probable source of additional capital raises substantial doubt about our ability to continue as a going concern for one year from the issuance of our financial statements included in this Annual Report on Form10-K. We have historically financed our operations primarily from the proceeds of sales of our equity securities and receipt of license fees, milestone payments, research and development funding and royalty income from our collaboration partners. We believeexpect that our cash and cash equivalents combined with the $108 million of $38.8 million at June 30, 2018net proceeds from the February 2021 stock offering and expectedanticipated net cash inflows under existing collaboration agreementsfrom product sales will enable us to fund our operating plan through the second quarter of 2022,under current expectations regarding (i) the timing and planned operations (including continuationoutcomes of our two Phase 31 clinical trial for EYP-1901 for the treatment of wet AMD, and (ii) initiation of our Phase 2 clinical trials for YUTIQEYP-1901 for the treatment of wet AMD. Due to the difficulty and plans for commercial launchuncertainty associated with the design and implementation of DEXYCUclinical trials, we will continue to assess our cash and if approved, YUTIQ) into the first quarter of calendar year 2019. In order to extend our ability to fund our operations beyond then, including our planned U.S. commercial launch of DEXYCUcash equivalents and if approved, YUTIQ, we may receive proceeds from the exercise of the Second Tranche Warrants. Therefuture funding requirements. However, there is no assurance that additional funding will be achieved and that we will receivesucceed in our future operations. Actual cash requirements could differ from management’s projections due to many factors, including cash generation from sales of YUTIQ and DEXYCU, additional investments in research and development programs, clinical trial expenses for EYP-1901, competing technological and market developments and the costs of any strategic acquisitions and/or development of complementary business opportunities. In addition, the Pandemic has had, and will likely continue to have, a material and adverse impact on our
business, including as a result of preventive and precautionary measures that we, other businesses, and governments are taking. Due to these impacts and measures, we have experienced and will likely continue to experience significant revenues fromand unpredictable reductions in the planned commercializationdemand for our commercial products as customers have shut down their facilities and non-essential surgical procedures have been postponed in an effort to promote social distancing and to redirect medical resources and priorities towards the treatment of DEXYCU or, if approved, YUTIQ, or from license revenues from ILUVIEN or be able to obtain financing from any other sources.COVID-19. The amount of additional capital we will require will be influenced by many factors, including, but not limited to: whether the Second Tranche Warrants are exercised;
| • | the potential for EYP-1901, as a twice-yearly sustained delivery intravitreal anti-VEGF treatment targeting wet age-related macular degeneration (“wet AMD”), with potential in diabetic retinopathy (“DR”) and retinal vein occlusion (“RVO”); |
the success and timing of our commercialization efforts with respect to DEXYCU;
| • | our expectations regarding the timing and clinical development of our product candidates, including EYP-1901 and YUTIQ50; |
whether we are required to make additional milestone andearn-out payments to the former security-holders of Icon;
| • | the success of our U.S. direct commercialization of YUTIQ for the treatment of chronic non-infectious uveitis affecting the posterior segment of the eye including, among other things, patient and physician acceptance of YUTIQ and our ability to obtain adequate coverage and reimbursement for YUTIQ; |
the amount of future revenues we receive with respect to our planned commercialization of DEXYCU and, if approved by the FDA, YUTIQ and license revenues from ILUVIEN for DME and, if and when approved in the EMEA, of ILUVIEN for NIPU;
| • | the success of our U.S. direct commercialization of DEXYCU for the treatment of postoperative ocular inflammation including, among other things, patient and physician acceptance of DEXYCU and our ability to obtain adequate coverage and reimbursement for DEXYCU; |
the timing, cost and success of our clinical development, regulatory approval and planned direct U.S. commercialization of YUTIQ;
| • | the cost of commercialization activities for YUTIQ and DEXYCU, including product manufacturing, marketing, sales and distribution; |
whether and to what extent we internally fund, whether and when we initiate, and how we conduct other product development programs;
| • | whether and to what extent we internally fund, whether and when we initiate, and how we conduct other product development programs; |
the amount of Retisert royalties and other payments we receive under collaboration agreements;
| • | payments we receive under any new collaboration agreements; |
whether and when we are able to enter into strategic arrangements for our products or product candidates and the nature of those arrangements;
| • | whether and when we are able to enter into strategic arrangements for our products or product candidates and the nature of those arrangements; |
timely and successful development, regulatory approval and commercialization of our products and product candidates;
| • | the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing any patent claims; |
the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing any patent claims;
| • | changes in our operating plan, resulting in increases or decreases in our need for capital; |
changes in our operating plan, resulting in increases or decreases in our need for capital; and
| • | the duration, scope and outcome of the SEC investigation and its impact on our financial condition, results of operations or cash flows |
our views on the availability, timing and desirability of raising capital.
| • | the forgiveness of the $2.0M PPP Loan by the U.S. Small Business Administration |
| • | our views on the availability, timing and desirability of raising capital; and |
| • | the extent to which our business could be adversely impacted by the effects of the Pandemic or by other pandemics, epidemics or outbreaks. |
We do not know if additional capital will be available when needed or on terms favorable to us or our stockholders. Collaboration, licensing or other agreements may not be available on favorable terms, or at all. We do not know the extent to which we will be able to establish commercial and other capabilities to successfully launch DEXYCU or, if approved, YUTIQ. Although we expect that our restructured Alimera collaboration agreement will provide a more consistent flow of royalty income, we do not knowreceive funds from the extent to which Alimera will achieve increasing revenues from its commercialization of ILUVIEN for DME and, if approved in the EMEA, for NIPU.YUTIQ or DEXYCU. If we seek to sell shares under our ATM program or in another offering,equity securities, we do not know whether and to what extent we will be able to do so, or on what terms. Further, the rules and regulations of the Nasdaq Global Market require us to obtain stockholder approval for sales of our equity securities under certain circumstances, which could delay or prevent us from raising additional capital from such sales. Also, the state of the economy and financial and credit markets at the time or times we seek any additional financing may make it more difficult or expensive to obtain. If available, additional equity financing may be dilutive to stockholders, debt financing may involve restrictive covenants or other unfavorable terms and dilute our existing stockholders’ equity, and funding through collaboration, licensing or other commercial agreements may be on unfavorable terms, including requiring us to relinquish rights to certain of our technologies or products. If adequate financing is not available if and when needed, we may delay, reduce the scope of, or eliminate research or development programs, potential independent commercialization of DEXYCUYUTIQ and if approved, YUTIQDEXYCU, or other new products, if any, and postpone or cancel the pursuit of product candidates, includingpre-clinical and clinical trials and new business opportunities, reduce staff and operating costs, or otherwise significantly curtail our operations to reduce our cash requirements and extend our capital.
Our consolidated statements of historical cash flows are summarized as follows:follows (in thousands): | | | | Year Ended June 30, | | | Year ended December 31, | | | | | | | | | | 2018 | | 2017 | | 2016 | | | 2020 | | | 2019 | | | | | | | | | (In thousands) | | | | | | | | | | | | | | Net loss: | | $ | (53,171 | ) | | $ | (18,485 | ) | | $ | (21,547 | ) | | $ | (45,394 | ) | | $ | (56,793 | ) | | | | | Changes in operating assets and liabilities | | | 924 | | | 318 | | | 2,073 | | | | 20,136 | | | | (12,536 | ) | | | | | Other adjustments to reconcile net loss to cash flows from operating activities | | | 30,340 | | | (2,323 | ) | | 3,158 | | | | 10,823 | | | | 12,630 | | | | | | | | | | | | | | | | | Cash flows used in operating activities | | $ | (21,907 | ) | | $ | (20,490 | ) | | $ | (16,316 | ) | | $ | (14,435 | ) | | $ | (56,699 | ) | | | | | | | | | | | | | | | | Cash flows (used in) provided by investing activities | | $ | (16,888 | ) | | $ | 13,577 | | | $ | (4,462 | ) | | $ | (362 | ) | | $ | (213 | ) | | | | | | | | | | | | | | | | Cash flows provided by financing activities | | $ | 60,671 | | | $ | 8,503 | | | $ | 16,990 | | | $ | 37,492 | | | $ | 33,864 | | | | | | | | | | | | | | | | |
Sources and uses of operating cash flows for the years ended June 30, 2018, 2017 and 2016 are summarized as follows:
| | | | | | | | | | | | | | | | Year Ended June 30, | | | | | 2018 | | | 2017 | | | 2016 | | | | | (In thousands) | | Operating cash inflows: | | | | | | | | | | | | | License and collaboration agreements | | $ | 1,296 | | | $ | 891 | | | $ | 507 | | Royalty income | | | 1,556 | | | | 1,008 | | | | 1,298 | | Foreign R&D tax credits | | | — | | | | 132 | | | | 163 | | Investment interest received, net | | | 106 | | | | 120 | | | | 176 | | | | | | | | | | | | | | | | | | 2,958 | | | | 2,151 | | | | 2,144 | | | | | | | | | | | | | | | Operating cash outflows: | | | | | | | | | | | | | Personnel costs | | | (7,220 | ) | | | (7,229 | ) | | | (5,133 | ) | Professional fees | | | (8,013 | ) | | | (6,074 | ) | | | (3,610 | ) | Clinical development and third-party R&D | | | (6,788 | ) | | | (7,115 | ) | | | (7,615 | ) | Loan interest payments | | | (258 | ) | | | — | | | | — | | All other operating cash outflows, net | | | (2,586 | ) | | | (2,223 | ) | | | (2,102 | ) | | | | | | | | | | | | | | | | | (24,865 | ) | | | (22,641 | ) | | | (18,460 | ) | | | | | | | | | | | | | | Cash flows used in operating activities | | $ | (21,907 | ) | | $ | (20,490 | ) | | $ | (16,316 | ) | | | | | | | | | | | | | |
Operating cash inflows for each year consisted primarily of payments received pursuant to license and collaboration agreements and royalty income. As a percentage of total license and collaboration and royalty income cash inflows, amounts attributable to Alimera represented 25.4% in fiscal 2018, 58.6% in fiscal 2017 and 72.4% in fiscal 2016, amounts attributable to Bausch & Lomb represented 34.7% in fiscal 2018, 54.1% in fiscal 2017 and 74.2% in fiscal 2016, amounts attributable to OncoSil Medical represented 3.5% in fiscal 2018, 5.3% in fiscal 2017, 5.4% in fiscal 2016 and amounts attributable to various feasibility study agreements represented 36.5% in fiscal 2018 and 13.2% in fiscal 2017.
Operating cash outflows increasedfor the year ended December 31, 2020 totaled $14.4 million, primarily due to our net loss of $45.4 million, reduced by approximately $2.2$10.8 million or 9.8%,of non-cash expenses, which included $5.5 million of stock-based compensation and $2.5 million of amortization of the DEXYCU finite-lived intangible asset. Further adjustments of cash in operating activities resulted from fiscal 2017an increase of $16.5 million in deferred revenue primarily related to fiscal 2018, asthe SWK Royalty Payment Agreement and a result$1.9 million decrease in accounts receivable from product sales. Operating cash outflows for the year ended December 31, 2019 totaled $56.7 million, primarily due to our net loss of (a)$56.8 million, reduced by $12.6 million of non-cash expenses, which included $4.7 million of stock-based compensation, a $3.8M loss on extinguishment of our SWK debt and $2.5 million amortization of the DEXYCU finite-lived intangible asset. Further use of cash in operating activities resulted from primarily an increase of $11.1 million in accounts receivable from our launch of our two commercial products, a $4.6 million increase in prepaid expenses primarily related to commercialization activities and a $1.9 million increase in professional fees, which consisted primarily of YUTIQ regulatory and clinical consulting fees and DEXYCU and YUTIQ marketing and managed markets consulting services, partially offset by lower patent legal fees; (b) $258,000 of term loan interest payments; (c) a $352,000 increase in third-party research and development costs; (d) a $135,000 increase in travel and facility costs; and (e) a $98,000 increase in stockholder meeting and statutory filing costs, partially offset by a $679,000 decrease in CRO costs for the YUTIQ Phase 3 development program. Personnel and related costs were flat on a year-over-year basis, reflecting a significant net increase in employee headcount and higher incentive compensation offset by $1.2 million of severance compensation in fiscal 2017. Operating cash outflows increased by $4.2 million, or 22.7%, from fiscal 2016 to fiscal 2017, as a result primarily of (a) $2.5 million of professional fees, which consisted primarily of YUTIQ three-year uveitis clinical and regulatory consulting fees and general legal fees associated with the CEO transition and costs of the Alimera arbitration and (b) $2.1 million of personnel and benefit costs, which included $1.2 million of severance compensation and in fiscal 2017, the additions of a Chief Medical Officer and EVP of Corporate and Commercial Development and an approximate $390,000 increase in fiscal 2016 incentive compensation awards (paid in fiscal 2017) compared to the prior year, partially offset by a $208,000 decrease in CRO costs for YUTIQ clinical development.product inventory. Cash flows fromused in investing activities in fiscal 2018 consisted principally of $16.8 million of cash used, net of cash acquired plus transaction costs, for the Icon acquisition. Cash flows from investing activities in fiscal 2017years ended December 31, 2020 and 2016 were attributable primarily to maturities2019 consisted of marketable securities, net of purchases of $13.7 million for fiscal 2017 and purchases of marketable securities, net of maturities, of $4.3 million for fiscal 2016. Purchases of property and equipment totaled $108,000 in fiscal 2018, $147,000 in fiscal 2017totaling $362,000 and $113,000 in fiscal 2016. Cash flows fromNet cash provided by financing activities infor fiscal 2018 were related primarily to the Icon acquisition2020 totaled $37.50 million and to support investments in commercial infrastructure, sales, marketing and medical affairs in preparation for the launch of DEXYCU and, if approved by the FDA, YUTIQ. These financing cash flows included approximately (i) $35.0 million of aggregate gross proceeds from the sale of 8,606,324 shares of common stock in the First Tranche Transaction and the sale of 20,184,224 Units in the Second Tranche Transaction (see Note 10consisted of the accompanying Notes to Consolidated Financial Statements contained in this Annual Report on Form10-K)following:
| (i) | $20.0 million of net proceeds from the issuance of 1,500,000 shares of our Common Stock; and |
| (ii) | $2.0 million of net proceeds from the PPP Loan; and |
| (iii) | $294,000 of proceeds from stock issued our employee stock purchase plan; |
| (iv) | $14.2 million of net proceeds from the issuance of 2,590,093 shares of our Common Stock sold utilizing our ATM; and |
| (v) | $15.7 million of net proceeds from the issuance of 3,010,722 shares of our Common Stock to Ocumension Therapeutics; partially offset by |
| (vi) | $14.6 million partial repayment of the CRG Term Loan, which included $13.7M of principal and $828,000 in Exit Fee. |
Net cash provided by financing activities for fiscal 2019 totaled $33.9 million and (ii) $20.0 million of gross proceeds from a term loan agreement (see Note 9consisted of the accompanying Notes to Consolidated Financial Statements contained in this Annual Report on Form10-K). Share issue and debt issue costs totaled approximately $1.9 million in connection with these financing transactions. Cash flows from financing activities in fiscal 2018 also included the sale of 5,900,000 shares of common stock under our ATM program for gross proceeds of $7.3 million, net of $239,000 of share issue costs. Cash flows from financing activities in fiscal 2017 were related predominately to the sale of shares of our common stock under our ATM program for gross proceeds of $8.9 million, less $233,000 of program adoption costs and $244,000 of share issue costs. Cash flows from financing activities in fiscal 2016 were attributable primarily to an underwritten public offering in January 2016 for gross proceeds of $17.8 million, net of $1.3 million of share issue costs. In addition, cash flows from financing activities included proceeds from the exercise of stock options totaling $503,000 in fiscal 2018, $99,000 in fiscal 2017 and $490,000 in fiscal 2016.following:
| (i) | $33.8 million of net proceeds from the initial drawdown under the CRG Loan Agreement, net of debt issue costs; and |
| (ii) | $18.3 million of net proceeds from the issuance of 10,526,500 shares of our common stock (“Common Stock”); and |
| (iii) | $14.8 million of net proceeds from our second drawdown under the CRG Loan Agreement offset by payment of a $15.0 million development milestone that was due to the former Icon security holders following the first commercial sale of DEXYCU; |
| (iv) | $398,000 of proceeds from the exercise of stock options; and |
| (v) | $4.3 million of net proceeds from the issuance of 2,998,877 shares of our Common Stock sold utilizing our ATM.; partially offset by |
| (vi) | $22.7 million repayment of the SWK Loan, which included principal of $20.0 million, a $1.2 million prepayment penalty, a $1.2 million Exit Fee and $306,000 of make whole interest. |
Off-Balance Sheet Arrangements We do not have anyoff-balance sheet arrangements that have, or are reasonably likely to have, a current or future effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources that would be material to investors. Tabular Disclosure of Contractual Obligations
The following table summarizes our minimum contractual obligations as of June 30, 2018:
| | | | | | | | | | | | | | | | | | | | | | | | Payments Due by Period | | Contractual Obligations | | Total | | | Less than
1 year | | | 1-3 years | | | 3-5 years | | | More than
5 years | | | | | (In thousands) | | Operating Lease Obligations | | $ | 5,520 | | | $ | 548 | | | $ | 1,756 | | | $ | 1,698 | | | $ | 1,518 | | Purchase Obligations | | | 1,125 | | | | 1,125 | | | | — | | | | — | | | | — | | | | | | | | | | | | | | | | | | | | | | | Total | | $ | 6,645 | | | $ | 1,673 | | | $ | 1,756 | | | $ | 1,698 | | | $ | 1,518 | | | | | | | | | | | | | | | | | | | | | | |
Our operating lease obligations consist predominantly of office and lab space in Watertown, Massachusetts and office space in Liberty Corner, New Jersey. Our purchase obligations consist of contractual agreements primarily for medical affairs and commercialization activities, as well asnon-cancellable purchase orders for supplies and services.
We have agreements with two CROs to conduct the clinical development program for YUTIQ. Our financial obligations under the agreements are determined by the services that we request from time to time under the agreements. The actual amounts owed under the agreements and the timing of those obligations will depend on various factors, including the number of patients and rate of patient enrollment, any protocol amendments and other factors relating to the clinical trials. We can change the services requested and thereby increase or decrease our obligations under the agreements from time to time. As of June 30, 2018, our CRO agreements provided for two Phase 3clinical trials at an aggregate remainingcost of approximately $4.5 million, excluding any potential additional contract change notifications. We can terminate the agreements at any time without penalty.
We also have employment agreements with four executive officers that would require us to make severance payments to them if we terminate their employment without cause or the executives resign for good cause. These
payments are contingent upon the occurrence of various future events, and the amounts payable under these provisions depend upon the level of compensation at the time of termination of employment, which are therefore not calculable at this time, and, as a result, we have not included any such amounts in the table above.
ITEM 7A. | ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK |
We are exposed to various market risks, which may result in potential losses arising from adverse changes in market rates, sucha smaller reporting company as interest rates. We do not enter into derivatives or other financial instruments for trading or speculative purposes and do not believe we are exposed to material market risk with respect to our cash and cash equivalents. As of June 30, 2018, we had cash and cash equivalents of $38.8 million. We do not engage in any hedging activities against changes in interest rates. Becausedefined by Rule 12b-2 of the short-term maturitiesSecurities Exchange Act of our cash1934 and cash equivalents, we doare not believe that an immediate 10% increase in interest rates would have a significant impact onrequired to provide the realized value of our investments.
The interest rate on our Loaninformation under the Credit Agreement is variable based on the three-month LIBOR, subject to a 1.5% floor, plus 10.50%. Accordingly, such interest rate is affected by changes in market interest rates. As of June 30, 2018, we had $20.0 million of aggregate principal amount outstanding under the Credit Agreement. As of June 30, 2018, the three-month LIBOR was 2.34%. A hypothetical 1% increase in the three-month LIBOR would result in $200,000 in incremental annual interest expense under the Loan.this Item.
ITEM 8. | ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA |
The information required by this item may be found on pagesF-1 throughF-36 F-34 of this Annual Report.Report on Form 10-K. ITEM 9. | ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
None. ITEM 9A. | ITEM 9A. CONTROLS AND PROCEDURES |
Evaluation of Disclosure Controls and Procedures Our management, with the participation of our principal executive officer and principal financial officer, evaluated the effectiveness of our disclosure controls and procedures as of June 30, 2018.December 31, 2020. The term “disclosure controls and procedures”, as defined in Rules13a-15(e) and15d-15(e) under the Exchange Act, means controls and other procedures of a company that are designed to ensure that information required to be disclosed by us in the reports that we file or submit under the Exchange Act, is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms, and that such information is accumulated and communicated to our management, including our principal executive officer and principal financial officer, as appropriate, to allow timely decisions regarding required disclosure. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their desired objectives, and our management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on the evaluation of our disclosure controls and procedures as of June 30, 2018,December 31, 2020, our principal executive officer and principal financial officer concluded that, as of such date, our disclosure controls and procedures were effective at the reasonable assurance level. (a) | Management’s Report on Internal Control over Financial Reporting |
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as defined in Rule13a-15(f) or15d-15(f) of the Exchange Act, as a process designed by, or under the supervision of, our principal executive and principal financial officers and effected by our board of directors, management and other personnel to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles in the U.S., and includes those policies and procedures that: | • | pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets; |
| • | provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures are being made only in accordance with authorizations of management and our directors; and |
| • | provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements. |
All internal control systems, no matter how well designed, have inherent limitations and may not prevent or detect misstatements. Projections of any evaluations of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate. Our management assessed the effectiveness of our internal control over financial reporting as of June 30, 2018.December 31, 2020. In making this assessment, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission inInternal Control—Integrated Framework (2013). Based on this assessment, our management concluded that, as of such date, our internal control over financial reporting was effective based on those criteria.
(b) | Changes in Internal Control over Financial Reporting |
There were no changes in our internal control over financial reporting during the last quarter of the fiscal yearperiod covered by this Annual Report on Form10-K that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting. Due to the continued Pandemic, certain employees of the Company continued working remotely through the year ended December 31, 2020. As a result of the continued remote working environment the Company has not identified any material changes in the Company’s internal control over financial reporting. The Company is continually monitoring and assessing the Pandemic situation to determine any potential impacts on the design and operating effectiveness of our internal controls over financial reporting.ITEM 9B. | ITEM 9B.OTHER INFORMATION |
None.
PART III Certain information required by Part III is omitted from this Annual Report on Form10-K and is incorporated herein by reference from our definitive proxy statement relating to our 20182021 annual meeting of stockholders, pursuant to Regulation 14A of the Exchange Act of 1934, also referred to in this Annual Report on Form10-K as our 20182021 Proxy Statement, which we expect to file with the SEC no later than October 29, 2018.April 30, 2021. ITEM 10. | ITEM 10. DIRECTORS, EXECUTIVE OFFICERS, AND CORPORATE GOVERNANCE |
Corporate Governance We have adopted a written Code of Business Conduct that applies to all of our employees, officers and directors. This Code of Business Conduct is designed to ensure that our business is conducted with integrity and in compliance with SEC regulations and Nasdaq listing standards. The Code of Business Conduct covers adherence to laws and regulations as well as professional conduct, including employment policies, conflicts of interest and the protection of confidential information. The Code of Business Conduct is available under “Governance Overview” within the “Investors – Corporate Governance” section of our website at www.eyepointpharma.com. We intend to disclose any future amendments to, or waivers from, the Code of Business Conduct that affect our directors or senior financial and executive officers within four business days of the amendment or waiver by posting such information on the website address and location specified above. Other Information The other information required to be disclosed in Item 10 is hereby incorporated by reference to our 20182021 Proxy Statement. ITEM 11. | ITEM 11. EXECUTIVE COMPENSATION |
The information required to be disclosed in Item 11 is hereby incorporated by reference to our 20182021 Proxy Statement. ITEM 12. | ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
The information required to be disclosed in Item 12 is hereby incorporated by reference to our 20182021 Proxy Statement. ITEM 13. | ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE |
The information required to be disclosed in Item 13 is hereby incorporated by reference to our 20182021 Proxy Statement. ITEM 14. | ITEM 14. PRINCIPAL ACCOUNTING FEES AND SERVICES |
The information required to be disclosed in Item 14 is hereby incorporated by reference to our 20182021 Proxy Statement.
PART IV ITEM 15. | ITEM 15. EXHIBITS AND FINANCIAL STATEMENTS |
(a)(1) Financial Statements The financial statements filed as part of this report are listed on the Index to Consolidated Financial Statements onpage F-1. (a)(2) Financial Statement Schedules Schedules have been omitted because of the absence of conditions under which they are required or because the required information is included in our Consolidated Financial Statements or Notes thereto. ITEM 16. FORM 10-K SUMMARY Not applicable. (a)(3) Exhibits. | | | | | | | | | | | | | Incorporated by Reference to SEC Filing | Exhibit No. | | Exhibit Description | | Incorporated by Reference to SEC Filing | | Exhibit No. | | Exhibit Description | | Form | | SEC Filing Date | | Exhibit No. | | | Form | | SEC Filing
Date | | | Exhibit
No. | | | | | | | | | | | | | | | | Articles of Incorporation andBy-Laws | | | | | | | | | Articles of Incorporation and By-Laws | | | | | | | | | | | | | | | | | | | | 3.1 | | Certificate of Incorporation of pSivida Corp. | | 8-K12G3 | | | 06/19/08 | | | | 3.1 | | 3.1 | | Certificate of Incorporation of pSivida Corp. | | 8-K12G3 | | 06/19/08 | | 3.1 | | | | | | | | | | | | | | 3.2 | | Certificate of Amendment of the Certificate of Incorporation of pSivida Corp. | | 10-K | | | 09/13/17 | | | | 3.2 | | 3.2 | | Certificate of Amendment of the Certificate of Incorporation of pSivida Corp. | | 10-K | | 09/13/17 | | 3.2 | | | | | | | | | | | | | | 3.3 | | Certificate of Amendment of the Certificate of Incorporation of pSivida Corp. | | 8-K | | | 04/02/18 | | | | 3.1 | | 3.3 | | Certificate of Correction to Certificate of Amendment of the Certificate of Incorporation of pSivida Corp. | | 8-K | | 04/02/18 | | 3.1 | | | | | | | | | | | | | | 3.4 | | Certificate of Amendment of Certificate of Incorporation, as amended of EyePoint Pharmaceuticals, Inc. | | 8-K | | | 06/27/18 | | | | 3.1 | | 3.4 | | Certificate of Amendment of Certificate of Incorporation, as amended of EyePoint Pharmaceuticals, Inc. | | 8-K | | 06/27/18 | | 3.1 | | | | | | | | | | | | | 3.5(a) | | By-Laws of EyePoint Pharmaceuticals, Inc. | | | | | | | | By-Laws of EyePoint Pharmaceuticals, Inc. | | 10-K | | 09/18/18 | | 3.5 | | | | | | | | | | | | | 3.6 | | 3.6 | | Amendment No. 1 to the By-Laws of EyePoint Pharmaceuticals, Inc. | | 8-K | | 11/06/18 | | 3.1 | | | | | | | | | | | | 3.7 | | 3.7 | | Certificate of Amendment of the Certificate of Incorporation, as amended, of EyePoint Pharmaceuticals, Inc. | | 8-K | | 06/23/20 | | 3.1 | | | | | | | | | | | | 3.8 | | 3.8 | | Certificate of Amendment of the Certificate of Incorporation, as amended, of EyePoint Pharmaceuticals, Inc. | | 8-K | | 12/08/20 | | 3.1 | | | | | | | | | | | | | | Instruments Defining the Rights of Security Holders | | | | | | | | | Instruments Defining the Rights of Security Holders | | | | | | | | | | | | | | | | | | | | 4.1 | | Form of Specimen Stock Certificate for Common Stock | | 8-K12G3 | | | 06/19/08 | | | | 4.1 | | 4.1 | | Form of Specimen Stock Certificate for Common Stock | | 8-K12G3 | | 06/19/08 | | 4.1 | | | | | | | | | | | | | | 4.2 | | Warrant to Purchase Common Stock of pSivida Corp., issued March 28, 2018, to SWK Funding, LLC | | 8-K | | | 03/29/18 | | | | 4.1 | | 4.2 | | Warrant to Purchase Common Stock of pSivida Corp., issued March 28, 2018, to SWK Funding, LLC | | 8-K | | 03/29/18 | | 4.1 | | | | | | | | | | | | | | 4.3 | | Form of Warrant to Purchase Common Stock of EyePoint Pharmaceuticals, Inc. | | 8-K | | | 06/27/18 | | | | 4.1 | | 4.3 | | Registration Rights Agreement, dated as of March 28, 2018, by and among pSivida Corp. and EW Healthcare Partners, L.P. and EW Healthcare Partners-A, L.P. | | 8-K | | 03/29/18 | | 10.3 | | | | | | | | | | | | | | 4.4 | | Registration Rights Agreement, dated as of March 28, 2018, by and among pSivida Corp. and EW Healthcare Partners, L.P. and EW HealthcarePartners-A, L.P. | | 8-K | | | 03/29/18 | | | | 10.3 | | 4.4 | | Second Registration Rights Agreement, dated as of June 25, 2018, by and among EyePoint Pharmaceuticals, Inc. and EW Healthcare Partners, L.P. and EW Healthcare Partners-A, L.P. and each other person identified on the signature pages thereto | | 8-K | | 06/27/18 | | 10.1 | | | | | | | | | | | | | | 4.5 | | Second Registration Rights Agreement, dated as of June 25, 2018, by and among EyePoint Pharmaceuticals, Inc. and EW Healthcare Partners, L.P. and EW HealthcarePartners-A, L.P. and each other person identified on the signature pages thereto | | 8-K | | | 06/27/18 | | | | 10.1 | | | 4.5(a) | | 4.5(a) | | Description of Securities of EyePoint Pharmaceuticals, Inc. | | | | | | | | | | | | | | | | | | | | | Material Contracts—Management Contracts and Compensatory Plans | | | | | | | | | Material Contracts - Management Contracts and Compensatory Plans | | | | | | | | | 10.1 | | Employment Agreement between pSivida Corp. and Nancy Lurker, dated September 15, 2016 | | 10-Q | | | 11/08/16 | | | | 10.1 | | | | | 10.2 | | Amended and Restated Performance-Based Restricted Stock Unit Award Agreement, dated December 21, 2016, by and between pSivida Corp. and Nancy Lurker | | 8-K | | | 12/23/16 | | | | 10.1 | | | | | 10.3 | | Nonstatutory Stock Option Inducement Award granted to Nancy Lurker, subject to shareholder approval, with effect from September 15, 2016 | | 10-Q | | | 11/08/16 | | | | 10.3 | | | | | 10.4 | | Employment Agreement between pSivida Corp. and Deb Jorn, dated November 2, 2016 | | 10-Q | | | 11/08/16 | | | | 10.4 | | |
| | | | | | | | | | | | | Exhibit No. | | Exhibit Description | | Incorporated by Reference to SEC Filing | | | | Form | | SEC Filing
Date | | | Exhibit
No. | | 10.5 | | Amended and Restated Performance-Based Restricted Stock Unit Award Agreement, dated December 21, 2016, by and between pSivida Corp. and Deb Jorn | | 8-K | | | 12/23/16 | | | | 10.2 | | | | | | | | 10.6 | | Nonstatutory Stock Option granted to Deb Jorn on November 2, 2016 | | 10-Q | | | 11/08/16 | | | | 10.6 | | | | | | | | 10.7 | | Employment Agreement, between pSivida Corp. and Leonard S. Ross, dated December 17, 2010 | | 8-K | | | 12/21/10 | | | | 10.1 | | | | | | | | 10.8 | | Option Amendment Agreement, between pSivida Corp. and Leonard S. Ross, dated December 17, 2010 | | 8-K | | | 12/21/10 | | | | 10.2 | | | | | | | | 10.9 | | Retention Bonus Letter, dated January 5, 2017, by and between pSivida Corp. and Leonard Ross | | 8-K | | | 01/10/17 | | | | 10.1 | | | | | | | | 10.10 | | Employment Agreement, between EyePoint Pharmaceuticals, Inc. and Dario Paggiarino, dated March 27, 2018 | | 10-Q | | | 05/10/18 | | | | 10.7 | | | | | | | | 10.11 | | Employment Agreement, dated August 1, 2018, by and between EyePoint Pharmaceuticals, Inc. and David Price | | 8-K | | | 08/03/18 | | | | 10.1 | | | | | | | | 10.12(a) | | Employment Agreement, dated May 11, 2018, by and between EyePoint Pharmaceuticals, Inc. and Leonard Blum | | | | | | | | | | | | | | | | | 10.13 + | | Form of Stock Option Certificate for grants to executive officers under the EyePoint Pharmaceuticals, Inc. 2016 Long Term Incentive Plan, as amended | | 10-Q | | | 02/08/18 | | | | 10.1 | | | | | | | | 10.14 + | | Form of Deferred Stock Unit Award for grants tonon-executive directors under the EyePoint Pharmaceuticals, Inc. 2016 Long Term Incentive Plan, as amended | | 10-Q | | | 02/08/18 | | | | 10.2 | | | | | | | | 10.15+(a) | | Form of Stock Option Award Agreement for Inducement grants to executive officers | | | | | | | | | | | | | | | | | | Material Contracts—Management Contracts and Compensatory Plans (continued) | | | | | | | | | | | | | | | | | 10.16 | | 2008 Equity Incentive Plan, as amended on November 19, 2009 | | 10-K | | | 09/10/15 | | | | 10.6 | | | | | | | | 10.17 + | | Form of Stock Option Certificate for grants to executive officers under the pSivida Corp. 2008 Incentive Plan | | 8-K | | | 09/10/08 | | | | 10.1 | | | | | | | | 10.18 | | pSivida Corp. 2016 Long Term Incentive Plan, as amended | | 10-Q | | | 02/09/17 | | | | 4.1 | | | | | | | | 10.19+(a) | | Form of Indemnification Agreement between EyePoint Pharmaceuticals, Inc. and its officers and directors | | | | | | | | | | | | | | | | | 10.20(a) | | EyePoint Pharmaceutical Short Term Incentive Plan | | | | | | | | | | | 10.21 + | | Form of Restricted Stock Unit Award for grants to executive officers under the pSivida Corp. 2016 Long Term Incentive Plan, as amended | | 10-K | | | 09/13/17 | | | | 10.18 | |
| | | | | | | | | | | | | Exhibit No. | | Exhibit Description | | Incorporated by Reference to SEC Filing | | | | Form | | SEC Filing
Date | | | Exhibit
No. | | | | | | | | 10.22 + | | Form of Performance-Based Stock Unit Award for grants under the pSivida Corp. 2016 Long Term Incentive Plan, as amended | | 10-K | | | 09/13/17 | | | | 10.19 | | | | | | | | | Material Contracts—Leases | | | | | | | | | | | | | | | | | 10.23 | | Lease Agreement between pSivida Corp. and Farley White Aetna Mills, LLC dated November 1, 2013 | | 10-Q | | | 11/13/13 | | | | 10.1 | | | | | | | | 10.24(a) | | First Amendment of Lease, dated February 6, 2014, between Farley White Aetna Mills, LLC and pSivida Corp. | | | | | | | | | | | | | | | | | 10.25(a) | | Second Amendment of Lease, dated May 14, 2018, between Whetstone Riverworks Holdings, LLC and EyePoint Pharmaceuticals, Inc. | | | | | | | | | | | | | | | | | | Material Contracts—License and Collaboration Agreements | | | | | | | | | | | | | | | | | 10.26# | | Amended and Restated License Agreement between Control Delivery Systems, Inc. and Bausch & Lomb Incorporated dated December 9, 2003, as amended on June 28, 2005 | | 20-F | | | 01/18/06 | | | | 4.12 | | | | | | | | 10.27# | | Second Amendment to Amended and Restated License Agreement between pSivda US, Inc. and Bausch & Lomb dated August 1, 2009 | | 10-K | | | 09/25/09 | | | | 10.13 | | | | | | | | 10.28# | | Second Amended and Restated Collaboration Agreement by and between pSivida US, Inc. and Alimera Sciences, Inc. dated July 10, 2017 | | 10-K | | | 09/13/17 | | | | 10.23 | | | | | | | | 10.29# | | Amended and Restated Collaborative Research and License Agreement, dated as of June 14, 2011, by and among pSivida Corp, pSivida US, Inc., pSiMedica Limited and Pfizer, Inc. | | 10-K/A | | | 12/27/11 | | | | 10.13 | | | | | | | | 10.30 | | Agreement, dated April 11, 2017, by and between pSivida Corp., pSiMedica Limited and Pfizer, Inc. | | 10-K | | | 09/13/17 | | | | 10.25 | | | | | | | | | Material Contracts—Other Agreements | | | | | | | | | | | | | | | | | 10.31 | | At Market Issuance Sales Agreement, dated February 8, 2017, by and between pSivida Corp. and FBR Capital Markets & Co. | | 8-K | | | 02/08/17 | | | | 10.1 | | | | | | | | 10.32 | | Securities Purchase Agreement, dated as of March 28, 2018, by and among pSivida Corp. and EW Healthcare Partners, L.P. and EW HealthcarePartners-A, L.P. | | 8-K | | | 03/29/18 | | | | 10.1 | | | | | | | | 10.33 | | Second Securities Purchase Agreement, dated as of March 28, 2018, by and among pSivida Corp. and EW Healthcare Partners, L.P. and EW HealthcarePartners-A, L.P. and each other person identified on the signature pages thereto | | 8-K | | | 03/29/18 | | | | 10.2 | |
| | | | Incorporated by Reference to SEC Filing | Exhibit No. | | Exhibit Description | | Form | | SEC Filing Date | | Exhibit No. | 10.1 | | Employment Agreement between pSivida Corp. and Nancy Lurker, dated September 15, 2016 | | 10-Q | | 11/08/16 | | 10.1 | | | | | | | | | | | | | | | | | | | 10.2 | | Nonstatutory Stock Option Inducement Award granted to Nancy Lurker, subject to shareholder approval, with effect from September 15, 2016 | | 10-Q | | 11/08/16 | | 10.3 | | | | | | | | | | 10.3 | | Employment Agreement, between EyePoint Pharmaceuticals, Inc. and Dario Paggiarino, dated March 27, 2018 | | 10-Q | | 05/10/18 | | 10.7 | | | | | | | | | | 10.4 | | Employment Agreement between EyePoint Pharmaceuticals, Inc. Scott Jones, dated May 30, 2019 | | 10-Q | | 08/07/19 | | 10.4 | | | | | | | | | | 10.5 | | Employment Agreement, dated November 14, 2019, by and between EyePoint Pharmaceuticals, Inc. and George Elston | | 8-K | | 11/19/19 | | 10.1 | | | | | | | | | | 10.6+ | | Form of Stock Option Certificate for grants to executive officers under the pSivida Corp. 2008 Incentive Plan | | 8-K | | 09/10/08 | | 10.1 | | | | | | | | | | 10.7+ | | Form of Stock Option Certificate for grants to executive officers under the EyePoint Pharmaceuticals, Inc. 2016 Long Term Incentive Plan, as amended | | 10-Q | | 02/08/18 | | 10.1 | | | | | | | | | | 10.8+ | | Form of Deferred Stock Unit Award for grants to non-executive directors under the EyePoint Pharmaceuticals, Inc. 2016 Long Term Incentive Plan, as amended | | 10-Q | | 02/08/18 | | 10.2 | | | | | | | | | | 10.9+ | | Form of Stock Option Award Agreement for Inducement grants to executive officers | | 10-K | | 09/18/18 | | 10.15 | | | | | | | | | | 10.10 | | 2008 Equity Incentive Plan, as amended on November 19, 2009 | | 10-K | | 09/10/15 | | 10.6 | | | | | | | | | | | | | | | | | | | 10.11 | | pSivida Corp. 2016 Long Term Incentive Plan, as amended | | 10-Q | | 02/09/17 | | 4.1 | | | | | | | | | | | | | | | | | | | 10.12+ | | Form of Restricted Stock Unit Award for grants to executive officers under the pSivida Corp. 2016 Long Term Incentive Plan, as amended | | 10-K | | 09/13/17 | | 10.18 | | | | | | | | | | 10.13+ | | Form of Performance-Based Stock Unit Award for grants under the pSivida Corp. 2016 Long Term Incentive Plan, as amended | | 10-K | | 09/13/17 | | 10.19 | | | | | | | | | | | | | | | | | | | 10.14 | | Stock Option Award Agreement, dated August 14, 2018, by and between EyePoint Pharmaceuticals, Inc. and John Weet | | 10-Q | | 11/09/18 | | 10.5 | | | | | | | | | | | | | | | | | | | 10.15 | | Stock Option Award Agreement, dated November 26, 2018, by and between EyePoint Pharmaceuticals, Inc. and Ron Honig | | 10-K | | 03/18/19 | | 10.25 | | | | | | | | | | 10.16 | | EyePoint Pharmaceuticals, Inc. 2016 Long Term Incentive Plan | | 8-K | | 06/28/19 | | 10.1 | | | | | | | | | | 10.17 | | Amendment No. 1 to EyePoint Pharmaceuticals, Inc. 2016 Long Term Incentive Plan | | 8-K | | 06/28/19 | | 10.2 | | | | | | | | | | 10.18 | | EyePoint Pharmaceuticals, Inc. 2019 Employee Stock Purchase Plan | | 8-K | | 06/28/19 | | 10.3 | | | | | | | | | | 10.19(a)+ | | Form of Indemnification Agreement between EyePoint Pharmaceuticals, Inc. and its officers and directors | | | | | | | | | | | | | | | | | | Material Contracts – Leases | | | | | | | | | | | | | | | | 10.20 | | Lease Agreement between pSivida Corp. and Farley White Aetna Mills, LLC dated November 1, 2013 | | 10-Q | | 11/13/13 | | 10.1 | | | | | | | | | | 10.21 | | First Amendment of Lease, dated February 6, 2014, between Farley White Aetna Mills and pSivida Corp. | | 10-K | | 09/18/18 | | 10.24 |
| | | | | | | | | | | | | Exhibit No. | | Exhibit Description | | Incorporated by Reference to SEC Filing | | | | Form | | SEC Filing
Date | | | Exhibit
No. | | 10.34 | | Agreement and Plan of Merger, dated March 28, 2018, by and among pSivida Corp., Oculus Merger Sub, Inc., Icon Bioscience, Inc. and Shareholder Representative Services LLC | | 8-K | | | 03/29/18 | | | | 10.5 | | | | | | | | 10.35 | | Credit Agreement, dated as of March 28, 2018, among pSivida Corp., SWK Funding LLC and the financial institutions party thereto from time to time as lenders | | 8-K | | | 03/29/18 | | | | 10.4 | | | | | | | | | Other Exhibits | | | | | | | | | | | | | | | | | 21.1(a) | | Subsidiaries of EyePoint Pharmaceuticals, Inc. | | | | | | | | | | | | | | | | | 23.1(a) | | Consent of Independent Registered Public Accounting Firm, Deloitte & Touche LLP | | | | | | | | | | | | | | | | | 31.1(a) | | Certification of Principal Executive Officer pursuant to Rule13a-14(a) and Rule15d-14(a) of the Securities Exchange Act, as amended | | | | | | | | | | | | | | | | | 31.2(a) | | Certification of Principal Financial Officer pursuant to Rule13a-14(a) and Rule15d-14(a) of the Securities Exchange Act, as amended | | | | | | | | | | | | | | | | | 32.1(a) | | Certification of Principal Executive Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | | | | | | | | | | | | | | | | | 32.2(a) | | Certification of Principal Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | | | | | | | | | | | | | | | | | 101 | | The following materials from EyePoint Pharmaceuticals’ Annual Report on Form10-K for the year ended June 30, 2018, formatted in eXtensible Business Reporting Language (XBRL): (i) Consolidated Balance Sheets at June 30, 2018 and 2017; (ii) Consolidated Statements of Comprehensive (Loss) Income for the years ended June 30, 2018, 2017 and 2016; (iii) Consolidated Statements of Stockholders’ Equity for the years ended June 30, 2018, 2017 and 2016; (iv) Consolidated Statements of Cash Flows for the years ended June 30, 2018, 2017 and 2016; and (v) Notes to Consolidated Financial Statements. | | | | | | | | | | |
| | | | Incorporated by Reference to SEC Filing | Exhibit No. | | Exhibit Description | | Form | | SEC Filing Date | | Exhibit No. | | | | | | | | | | 10.22 | | Second Amendment of Lease, dated May 14, 2018, between Whetstone Riverworks Holdings, LLC and EyePoint Pharmaceuticals, Inc. | | 10-K | | 09/18/18 | | 10.25 | | | | | | | | | | | | Material Contracts - License and Collaboration Agreements | | | | | | | | | | | | | | | | 10.23# | | Second Amended and Restated Collaboration Agreement by and between pSivida US, Inc. and Alimera Sciences, Inc. dated July 10, 2017 | | 10-K | | 09/13/17 | | 10.23 | | | | | | | | | | 10.24# | | Exclusive License Agreement between EyePoint Pharmaceuticals, Inc. and Equinox Science, LLC. | | 10-K | | 03/16/20 | | 10.32 | | | | | | | | | | | | | | | | | | | | | Material Contracts - Other Agreements | | | | | | | | | | | | | | | | 10.25 | | Securities Purchase Agreement, dated as of March 28, 2018, by and among pSivida Corp. and EW Healthcare Partners, L.P. and EW Healthcare Partners-A, L.P. | | 8-K | | 03/29/18 | | 10.1 | | | | | | | | | | 10.26 | | Second Securities Purchase Agreement, dated as of March 28, 2018, by and among pSivida Corp. and EW Healthcare Partners, L.P. and EW Healthcare Partners-A, L.P. and each other person identified on the signature pages thereto | | 8-K | | 03/29/18 | | 10.2 | 10.27 | | Agreement and Plan of Merger, dated March 28, 2018, by and among pSivida Corp., Oculus Merger Sub, Inc., Icon Bioscience, Inc. and Shareholder Representative Services LLC | | 8-K | | 03/29/18 | | 10.5 | | | | | | | | | | 10.28 | | Term Loan Agreement, dated February 13, 2019, among EyePoint Pharmaceuticals, Inc., as Borrower, EyePoint Pharmaceuticals US, Inc. and Icon Bioscience, Inc., as Subsidiary Guarantors, and CRG Servicing LLC, as Administrative Agent and Collateral Agent | | 8-K | | 02/19/19 | | 10.1 | | | | | | | | | | 10.29 | | Fee Letter, dated February 13, 2019, by and between EyePoint Pharmaceuticals, Inc. and CRG Servicing LLC | | 8-K | | 02/19/19 | | 10.2 | | | | | | | | | | 10.30 | | Waiver To Term Loan Agreement, dated November 19, 2019, among EyePoint Pharmaceuticals, as Borrower, EyePoint Pharmaceuticals US, Inc. and Icon Bioscience, Inc., as subsidiary guarantors and CRG Servicing LLC, as Administrative Agent and Collateral Agent | | 8-K | | 11/22/19 | | 10.1 | | | | | | | | | | 10.31 | | Note dated April 21, 2020 between EyePoint Pharmaceuticals, Inc. and Silicon Valley Bank | | 8-K | | 04/28/20 | | 99.1 | | | | | | | | | | 10.32 | | Controlled Equity OfferingSM Sales Agreement, dated August 5, 2020, by and between EyePoint Pharmaceuticals, Inc. and Cantor Fitzgerald & Co. | | 8-K | | 08/05/20 | | 1.1 | | | | | | | | | | 10.33 | | Amendment No. 2 and Waiver To Term Loan Agreement, dated October 8, 2020, among EyePoint Pharmaceuticals, Inc. as Borrower, EyePoint Pharmaceuticals US, Inc. and Icon Bioscience, Inc., as subsidiary guarantors and CRG Servicing LLC, as Administrative Agent and Collateral Agent. | | 8-K | | 10/08/20 | | 10.1 | | | | | | | | | | 10.34# | | Commercial Alliance agreement, dated as of August 1, 2020 between EyePoint Pharmaceuticals, Inc. and ImprimisRx, LLC. | | 10-Q | | 11/06/20 | | 10.1 | | | | | | | | | | 10.35#(a) | | Amendment One to the Commercial Alliance Agreement dated November 12, 2020 between EyePoint Pharmaceuticals, Inc. and ImprimisRx, LLC | | | | | | | | | | | | | | | | 10.36#(a) | | Royalty Purchase Agreement, dated December 17, 2020, by and between EyePoint Pharmaceuticals, Inc. and SWK Funding, LLC | | | | | | | | | | | | | | | | 10.37 | | Share Purchase Agreement, dated December 31, 2020, by and between EyePoint Pharmaceuticals, Inc. and Ocumension Therapeutics. | | 8-K | | 01/04/21 | | 10.1 | | | | | | | | | |
| | | | Incorporated by Reference to SEC Filing | Exhibit No. | | Exhibit Description | | Form | | SEC Filing Date | | Exhibit No. | 10.38 | | Voting and Investor Rights Agreement, dated December 31, 2020, by and among EyePoint Pharmaceuticals, Inc., Ocumension Therapeutics, and EW Healthcare Partners, L.P. and EW Healthcare Partners-A,L.P. | | 8-K | | 01/04/21 | | 10.2 | | | | | | | | | | 10.39 | | First Amendment to Share Purchase Agreement, dated February 1, 2021, by and between EyePoint Pharmaceuticals, Inc. and Ocumension Therapeutics | | 8-K | | 02/03/21 | | 10.1 | | | | | | | | | | 21.1(a) | | Subsidiaries of EyePoint Pharmaceuticals, Inc. | | | | | | | | | | | | | | | | 23.1(a) | | Consent of Independent Registered Public Accounting Firm, Deloitte & Touche LLP | | | | | | | | | | | | | | | | 31.1(a) | | Certification of Principal Executive Officer pursuant to Rule 13a-14(a) and Rule 15d-14(a) of the Securities Exchange Act, as amended | | | | | | | | | | | | | | | | 31.2(a) | | Certification of Principal Financial Officer pursuant to Rule 13a-14(a) and Rule 15d-14(a) of the Securities Exchange Act, as amended | | | | | | | | | | | | | | | | 32.1(b) | | Certification of Principal Executive Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | | | | | | | | | | | | | | | | 32.2(b) | | Certification of Principal Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | | | | | | | | | | | | | | | | | | | | | | | | | 101.INS | | Inline XBRL Instance Document – the instance document does not appear in the Interactive Data File because XBRL tags are embedded within the Inline XBRL document. | | | | 101.SCH | | Inline XBRL Taxonomy Extension Schema Document | | | | 101.CAL | | Inline XBRL Taxonomy Extension Calculation Linkbase Document | | | | 101.DEF | | Inline XBRL Taxonomy Extension Definition Linkbase Document | | | | 101.LAB | | Inline XBRL Taxonomy Extension Label Linkbase Document | | | | 101.PRE | | Inline XBRL Taxonomy Extension Presentation Linkbase Document | | | | | | | | | | | | | 104 | | Cover Page Interactive Data File (Embedded within the Inline XBRL document and included in Exhibit 101). | | | | | | | | | | | | | | | |
# | Confidential treatment has been granted for portionsPortions of this exhibit have been omitted in compliance with Item 601 of Regulation S-K.
|
† | Confidential Treatment has been requested with respect to certain portions of this exhibit. Omitted portions have been filed separately with the U.S. Securities and Exchange Commission.+
|
+ | The final versions of documents denoted as “form of” have been omitted pursuant to Rule12b-31. Such final versions are substantially identical in all material respects to the filed versions of such documents, provided that the name of the investor, and the investor’s and/or the Company’s signatures are included in the final versions. |
SIGNATURES Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized. | | | EYEPOINT PHARMACEUTICALS, INC. | | | By: | | /S/ NANCY LURKER
| | | Nancy Lurker | | | By: | | /s/ Nancy Lurker | | | | Nancy Lurker | | | | President and Chief Executive Officer | | | | Date: | | September 18, 2018Date: March 12, 2021 |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant in the capacities and on the dates indicated. | | | | | Name | | Title | | Date | /S/ GÖRAN ANDO
Göran Ando
| | | | | /S/ GÖRAN ANDO | | Chairman of the Board of Directors | | September 18, 2018March 12, 2021 | Göran Ando | | | | | | | | | | /S/ NANCY LURKER Nancy LurkerNANCY LURKER
| | President, Chief Executive Officer
and Director (Principal
| | March 12, 2021 | Nancy Lurker | | (Principal Executive Officer) | | September 18, 2018 | | | | | | /S/ DAVID PRICE David PriceGEORGE O. ELSTON
| | Chief Financial Officer (Principal
Financial Officer and Principal Accounting Officer) | | September 18, 2018March 12, 2021 | George O. Elston | | | | | | | | | | /S/ LEONARD S. ROSS Leonard S. RossWENDY DICICCO
| | VP, Finance and Chief Accounting
Officer (Principal Accounting
Officer)
| | September 18, 2018 | | | | /S/ DAVID J. MAZZO
David J. Mazzo
| | Director | | September 18, 2018March 12, 2021 | Wendy DiCicco | | | | | | | | | | /S/ MICHAEL ROGERS Michael RogersDOUGLAS GODSHALL
| | Director | | September 18, 2018March 12, 2021 | | | | /S/ DOUGLAS GODSHALL
Douglas Godshall | | | | | | | | | | /S/ YE LIU | | Director | | September 18, 2018March 12, 2021 | Ye Liu | | | | | | | | | | /S/ JAY DUKER Jay DukerRONALD W. EASTMAN
| | Director | | September 18, 2018March 12, 2021 | | | | /S/ KRISTINE PETERSON
Kristine Peterson
| | Director
| | September 18, 2018 | | | | /S/ RONALD W. EASTMAN
Ronald W. Eastman | | | | | | | | | | /S/ JOHN LANDIS | | Director | | September 18, 2018March 12, 2021 | John Landis | | | | | | | | | | /S/ DAVID R. GUYER | | Director | | March 12, 2021 | David R. Guyer | | | | |
EYEPOINT PHARMACEUTICALS, INC. AND SUBSIDIARIES INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM To the stockholdersStockholders and the Board of Directors of EyePoint Pharmaceuticals, Inc. Opinion on the Financial Statements We have audited the accompanying consolidated balance sheets of EyePoint Pharmaceuticals, Inc. (formerly pSivida Corp.) and subsidiaries (the “Company”"Company") as of June 30, 2018December 31, 2020 and 2017,December 31, 2019, the related consolidated statements of comprehensive loss, stockholders’stockholders' equity, and cash flows, for each of the three years in the period ended June 30, 2018,December 31, 2020, and 2019, and the related notes (collectively referred to as the “financial statements”"financial statements"). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of June 30, 2018December 31, 2020, and 2017,2019, and the results of its operations and its cash flows for each of the threetwo years in the period ended June 30, 2018,December 31, 2020, in conformity withaccountingwith accounting principles generally accepted in the United States of America. Basis for Opinion These financial statements are the responsibility of the Company’sCompany's management. Our responsibility is to express an opinion on the Company’sCompany's financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB. We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion. Going ConcernCritical Audit Matter
The accompanyingcritical audit matter communicated below is a matter arising from the current-period audit of the financial statements have been prepared assuming that was communicated or required to be communicated to the Company will continue as a going concern. As discussed in Note 1audit committee and that (1) relates to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the accounts or disclosures to which it relates. Revenue Transaction Price and Variable Consideration – Provisions for Chargebacks — Refer to Note 2 and 3 to the financial statements. Critical Audit Matter Description Revenue is stated net of certain sales deductions, including provisions for chargebacks. Provisions for chargebacks are deducted from gross revenues at the time revenues are recognized and are included in accrued rebates, returns and discounts in the Company’s anticipated recurring useConsolidated Balance Sheets. The estimated provision for product chargebacks of cash to fund operationsthe Company’s products requires significant judgment by management. Management’s model for estimating chargebacks is based on historical activity while also considering levels of inventory in combination with no probable sourcethe channel. We identified the estimated provision for chargebacks of additional capital raises substantial doubt about its ability to continuethe Company’s products as a going concern. Management’s planscritical audit matter because of the significant estimates and assumptions relating to the limited history of production of the Company’s products for which a provision for chargebacks was applied, coupled with the level of estimation uncertainty involved, required a high degree of auditor judgment and an increased extent of effort. How the Critical Audit Matter Was Addressed in regardthe Audit Our audit procedures related to these matters are also described in Note 1. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.Revenue Sales Deductions – Provisions for Chargebacks included the following, among others: • | We tested the mathematical accuracy of management’s calculation of the provisions for chargebacks. |
• | We evaluated the Company’s methodology and significant assumptions made in developing the provision for chargebacks, including auditing the completeness and accuracy of the underlying data used by management in their estimates. |
• | We evaluated the reasonableness of management’s provision for chargebacks by comparing the assumptions used in the projections to external market sources, information produced by the entity and inquiries with management. |
• | We evaluated management’s ability to accurately forecast product chargeback activity by comparing product chargeback reserves in earlier periods during the year to actual chargebacks. |
/s/ Deloitte & Touche LLP Boston, Massachusetts September 18, 2018March 12, 2021
We have served as the Company’sCompany's auditor since 2008.
EYEPOINT PHARMACEUTICALS, INC. AND SUBSIDIARIES CONSOLIDATED BALANCE SHEETS (In thousands except share amounts) | | | | June 30, | | | December 31, | | | December 31, | | | | | 2018 | | 2017 | | | 2020 | | | 2019 | | Assets | | | | | | | | | | | | | Current assets: | | | | | | | | | | | | | Cash and cash equivalents | | $ | 38,776 | | | $ | 16,898 | | | $ | 44,909 | | | $ | 22,214 | | Accounts and other receivables | | | 353 | | | 251 | | | Accounts and other receivables, net (including due from a related party of $104 and $0 at December 31, 2020 and 2019, respectively) | | | | 9,453 | | | | 11,368 | | Prepaid expenses and other current assets | | | 780 | | | 591 | | | | 3,419 | | | | 5,997 | | | | | | | | | | Inventory | | | | 5,337 | | | | 2,138 | | Total current assets | | | 39,909 | | | 17,740 | | | | 63,118 | | | | 41,717 | | Property and equipment, net | | | 253 | | | 313 | | | | 630 | | | | 357 | | Operating lease right-of-use assets | | | | 2,610 | | | | 3,078 | | Intangible assets, net | | | 31,358 | | | 364 | | | | 25,209 | | | | 27,669 | | Other assets | | | — | | | 110 | | | Restricted cash | | | 150 | | | 150 | | | | 150 | | | | 150 | | | | | | | | | | Total assets | | $ | 71,670 | | | $ | 18,677 | | | $ | 91,717 | | | $ | 72,971 | | | | | | | | | | Liabilities and stockholders’ equity | | | | | | | | | | | | | | Current liabilities: | | | | | | | | | | | | | Accounts payable | | $ | 2,940 | | | $ | 1,016 | | | $ | 4,811 | | | $ | 4,192 | | Accrued expenses | | | 3,723 | | | 4,224 | | | | 8,445 | | | | 6,832 | | Accrued development milestone | | | 15,000 | | | | — | | | Deferred revenue | | | — | | | 50 | | | | 945 | | | | 15 | | | | | | | | | | Other current liabilities | | | | 687 | | | | 481 | | Total current liabilities | | | 21,663 | | | 5,290 | | | | 14,888 | | | | 11,520 | | Long-term debt | | | 17,309 | | | | — | | | | 37,977 | | | | 47,223 | | Derivative liability | | | 19,780 | | | | — | | | Deferred revenue - noncurrent | | | | 15,616 | | | | — | | Operating lease liabilities - noncurrent | | | | 2,330 | | | | 2,898 | | Other long-term liabilities | | | 1,231 | | | 51 | | | | 2,365 | | | | 3,000 | | | | | | | | | | Total liabilities | | | 59,983 | | | 5,341 | | | | 73,176 | | | | 64,641 | | | | | | | | | | Commitments and contingencies (Note 15) | | | | | | | Contingencies (Note 17) | | | | | | | | | | Stockholders’ equity: | | | | | | | | | | | | | Preferred stock, $.001 par value, 5,000,000 shares authorized, no shares issued and outstanding | | | — | | | | — | | | Common stock, $.001 par value, 150,000,000 and 120,000,000 shares authorized, 74,512,048 and 39,356,999 shares issued and outstanding, each at June 30, 2018 and 2017, respectively | | | 74 | | | 39 | | | Preferred stock, $.001 par value, 5,000,000 shares authorized, 0 shares issued and outstanding | | | | — | | | | — | | Common stock, $.001 par value, 300,000,000 and 150,000,000 shares authorized at December 31, 2020 and 2019, respectively; 18,139,981 and 10,941,659 shares issued and outstanding at December 31, 2020 and 2019, respectively | | | | 18 | | | | 11 | | Additionalpaid-in capital | | | 374,766 | | | 323,284 | | | | 528,362 | | | | 472,765 | | Accumulated deficit | | | (363,991 | ) | | (310,820 | ) | | | (510,680 | ) | | | (465,286 | ) | Accumulated other comprehensive income | | | 838 | | | 833 | | | | 841 | | | | 840 | | | | | | | | | | Total stockholders’ equity | | | 11,687 | | | 13,336 | | | | 18,541 | | | | 8,330 | | | | | | | | | | Total liabilities and stockholders’ equity | | $ | 71,670 | | | $ | 18,677 | | | $ | 91,717 | | | $ | 72,971 | | | | | | | | | |
See notes to consolidated financial statements
EYEPOINT PHARMACEUTICALS, INC. AND SUBSIDIARIES CONSOLIDATED STATEMENTS OF COMPREHENSIVE LOSS (In thousands except per share data) | | | | Year Ended June 30, | | | Year Ended December 31, | | | Year Ended December 31, | | | | | 2018 | | 2017 | | 2016 | | | 2020 | | | 2019 | | Revenues: | | | | | | | | | | | | | | | Collaborative research and development | | $ | 1,343 | | | $ | 6,569 | | | $ | 398 | | | Product sales, net | | | $ | 20,831 | | | $ | 16,824 | | License and collaboration agreements (including licensing fees from a related party of $11,500 and $1,030 for the years ended December 31, 2020 and 2019, respectively) | | | | 11,942 | | | | 1,361 | | Royalty income | | | 1,618 | | | 970 | | | 1,222 | | | | 1,664 | | | | 2,180 | | | | | | | | | | | | | Total revenues | | | 2,961 | | | 7,539 | | | 1,620 | | | | 34,437 | | | | 20,365 | | | | | | | | | | | | | Operating expenses: | | | | | | | | | | | | | | | Cost of sales, excluding amortization of acquired intangible assets | | | | 5,824 | | | | 2,687 | | Research and development | | | 16,178 | | | 14,880 | | | 14,381 | | | | 17,424 | | | | 15,368 | | Sales and marketing | | | 1,512 | | | | — | | | | — | | | | 25,293 | | | | 29,772 | | General and administrative | | | 11,545 | | | 11,235 | | | 9,013 | | | | 20,726 | | | | 17,939 | | Amortization of acquired intangible assets | | | | 2,460 | | | | 2,460 | | Total operating expenses | | | | 71,727 | | | | 68,226 | | Loss from operations | | | | (37,290 | ) | | | (47,861 | ) | | | | | | | | | | | | | | | | | | | Total operating expenses | | | 29,235 | | | 26,115 | | | 23,394 | | | | | | | | | | | | | | Operating loss | | | (26,274 | ) | | (18,576 | ) | | (21,774 | ) | | Other income (expense): | | | | | | | | | | Interest and other income, net | | | 101 | | | 91 | | | 72 | | | | 58 | | | | 1,054 | | Interest expense | | | (720 | ) | | | — | | | | — | | | | (7,257 | ) | | | (6,176 | ) | Change in fair value of derivative liability | | | (26,278 | ) | | | — | | | | — | | | | | | | | | | | | | | Loss before income taxes | | | (53,171 | ) | | (18,485 | ) | | (21,702 | ) | | Income tax benefit | | | — | | | | — | | | 155 | | | | | | | | | | | | | | Loss on extinguishment of debt | | | | (905 | ) | | | (3,810 | ) | Total other expense, net | | | | (8,104 | ) | | | (8,932 | ) | Net loss | | $ | (53,171 | ) | | $ | (18,485 | ) | | $ | (21,547 | ) | | $ | (45,394 | ) | | $ | (56,793 | ) | | | | | | | | | | | | Net loss per share: | | | | | | | | | | | | | | | Basic and diluted | | $ | (1.15 | ) | | $ | (0.52 | ) | | $ | (0.68 | ) | | $ | (3.54 | ) | | $ | (5.44 | ) | | | | | | | | | | | | Weighted average common shares outstanding: | | | | | | | | | | | | | | | Basic and diluted | | | 46,226 | | | 35,344 | | | 31,623 | | | | 12,836 | | | | 10,431 | | | | | | | | | | | | | | Net loss | | $ | (53,171 | ) | | $ | (18,485 | ) | | $ | (21,547 | ) | | $ | (45,394 | ) | | $ | (56,793 | ) | | | | | | | | | | | | Other comprehensive income (loss): | | | | | | | | | | | | | | | Foreign currency translation adjustments | | | 5 | | | (21 | ) | | (96 | ) | | | 1 | | | | 1 | | Net unrealized gain on marketable securities | | | — | | | 2 | | | 3 | | | | | | | | | | | | | | Other comprehensive income (loss) | | | 5 | | | (19 | ) | | (93 | ) | | | 1 | | | | 1 | | | | | | | | | | | | | Comprehensive loss | | $ | (53,166 | ) | | $ | (18,504 | ) | | $ | (21,640 | ) | | $ | (45,393 | ) | | $ | (56,792 | ) | | | | | | | | | | | |
See notes to consolidated financial statements
EYEPOINT PHARMACEUTICALS, INC. AND SUBSIDIARIES CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY (In thousands except share data) | | | | | | | | | | | | | | Common Stock | | | Additional | | | | | | | Accumulated Other | | | Total | | | | | Common Stock | | | Additional
Paid-In
Capital | | Accumulated
Deficit | | Accumulated
Other
Comprehensive
Income | | Total
Stockholders’
Equity | | | Number of Shares | | | Par Value Amount | | | Paid-In Capital | | | Accumulated Deficit | | | Comprehensive Income | | | Stockholders’ Equity | | | | | Number of
Shares | | Par Value
Amount | | | Balance at July 1, 2015 | | 29,412,365 | | | $ | 29 | | | $ | 293,060 | | | $ | (270,666 | ) | | $ | 945 | | | $ | 23,368 | | | Net loss | | | — | | | | — | | | | — | | | (21,547 | ) | | | — | | | (21,547 | ) | | Other comprehensive loss | | | — | | | | — | | | | — | | | | — | | | (93 | ) | | (93 | ) | | Issuance of stock, net of issue costs | | 4,440,000 | | | 5 | | | 16,495 | | | | — | | | | — | | | 16,500 | | | Exercise of stock options | | 320,554 | | | | — | | | 490 | | | | — | | | | — | | | 490 | | | Stock-based compensation | | | — | | | | — | | | 2,163 | | | | — | | | | — | | | 2,163 | | | | | | | | | | | | | | | | | | | | | | | Balance at June 30, 2016 | | 34,172,919 | | | 34 | | | 312,208 | | | (292,213 | ) | | 852 | | | 20,881 | | | Cumulative effect of change in accounting principle (Note 2) | | | — | | | | — | | | 122 | | | (122 | ) | | | — | | | | — | | | Net loss | | | — | | | | — | | | | — | | | (18,485 | ) | | | — | | | (18,485 | ) | | Other comprehensive loss | | | — | | | | — | | | | — | | | | — | | | (19 | ) | | (19 | ) | | Issuance of stock, net of issue costs | | 5,100,000 | | | 5 | | | 8,399 | | | | — | | | | — | | | 8,404 | | | Exercise of stock options | | 84,080 | | | | — | | | 99 | | | | — | | | | — | | | 99 | | | Stock-based compensation | | | — | | | | — | | | 2,456 | | | | — | | | | — | | | 2,456 | | | | | | | | | | | | | | | | | | | | | | | Balance at June 30, 2017 | | 39,356,999 | | | 39 | | | 323,284 | | | (310,820 | ) | | 833 | | | 13,336 | | | Balance at December 31, 2018 | | | | 9,537,151 | | | $ | 10 | | | $ | 445,277 | | | $ | (408,493 | ) | | $ | 839 | | | $ | 37,633 | | Net loss | | | — | | | | — | | | | — | | | (53,171 | ) | | | — | | | (53,171 | ) | | | — | | | | — | | | | — | | | | (56,793 | ) | | | — | | | | (56,793 | ) | Other comprehensive income | | | — | | | | — | | | | — | | | | — | | | 5 | | | 5 | | | | — | | | | — | | | | — | | | | — | | | | 1 | | | | 1 | | Issuance of stock, net of issue costs | | 34,690,548 | | | 35 | | | 47,947 | | | | — | | | | — | | | 47,982 | | | | 1,352,538 | | | | 1 | | | | 22,626 | | | | — | | | | — | | | | 22,627 | | Fair value of warrants issued | | | — | | | | — | | | 355 | | | | — | | | | — | | | 355 | | | Exercise of stock options | | 310,900 | | | | — | | | 503 | | | | — | | | | — | | | 503 | | | | 22,342 | | | | — | | | | 414 | | | | — | | | | — | | | | 414 | | Vesting of stock units | | 153,601 | | | | — | | | (27 | ) | | | — | | | | — | | | (27 | ) | | | 29,628 | | | | — | | | | (120 | ) | | | — | | | | — | | | | (120 | ) | Stock-based compensation | | | — | | | | — | | | 2,704 | | | | — | | | | — | | | 2,704 | | | | — | | | | — | | | | 4,568 | | | | — | | | | — | | | | 4,568 | | | | | | | | | | | | | | | | | | | | | | Balance at June 30, 2018 | | 74,512,048 | | | $ | 74 | | | $ | 374,766 | | | $ | (363,991 | ) | | $ | 838 | | | $ | 11,687 | | | | | | | | | | | | | | | | | | | | | | | Balance at December 31, 2019 | | | | 10,941,659 | | | $ | 11 | | | $ | 472,765 | | | $ | (465,286 | ) | | $ | 840 | | | $ | 8,330 | | Net loss | | | | — | | | | — | | | | — | | | | (45,394 | ) | | | — | | | | (45,394 | ) | Other comprehensive income | | | | — | | | | — | | | | — | | | | — | | | | 1 | | | | 1 | | Issuance of stock, net of issue costs | | | | 7,100,815 | | | | 7 | | | | 49,846 | | | | — | | | | — | | | | 49,853 | | Employee stock purchase plan | | | | 33,697 | | | | — | | | | 294 | | | | — | | | | — | | | | 294 | | Vesting of stock units | | | | 63,810 | | | | — | | | | (90 | ) | | | — | | | | — | | | | (90 | ) | Stock-based compensation | | | | — | | | | — | | | | 5,547 | | | | — | | | | — | | | | 5,547 | | Balance at December 31, 2020 | | | | 18,139,981 | | | $ | 18 | | | $ | 528,362 | | | $ | (510,680 | ) | | $ | 841 | | | $ | 18,541 | |
See notes to consolidated financial statements
EYEPOINT PHARMACEUTICALS, INC. AND SUBSIDIARIES CONSOLIDATED STATEMENTS OF CASH FLOWS (In thousands) | | | | Year Ended June 30, | | | Year Ended December 31, | | | Year Ended December 31, | | | | | 2018 | | 2017 | | 2016 | | | 2020 | | | 2019 | | Cash flows from operating activities: | | | | | | | | | | | | | | | Net loss | | $ | (53,171 | ) | | $ | (18,485 | ) | | $ | (21,547 | ) | | $ | (45,394 | ) | | $ | (56,793 | ) | Adjustments to reconcile net loss to cash flows used in operating activities: | | | | | | | | | | | | | | | Amortization of intangible assets | | | 981 | | | 724 | | | 756 | | | | 2,460 | | | | 2,460 | | Depreciation of property and equipment | | | 167 | | | 91 | | | 152 | | | | 189 | | | | 144 | | Amortization of debt discount | | | 209 | | | | — | | | | — | | | | 745 | | | | 596 | | Amortization of bond (discount) premium on marketable securities | | | — | | | (9 | ) | | 87 | | | Amortization of noncurrent portion of deferred revenue | | | — | | | (5,585 | ) | | | — | | | Non-cash interest expense | | | | 977 | | | | 1,052 | | Loss on extinguishment of debt | | | | 905 | | | | 3,810 | | Stock-based compensation | | | 2,704 | | | 2,456 | | | 2,163 | | | | 5,547 | | | | 4,568 | | Change in fair value of derivative liability | | | 26,278 | | | | — | | | | — | | | Changes in operating assets and liabilities: | | | | | | | | | | | | | | | Accounts and other receivables | | | 7 | | | 219 | | | 116 | | | Prepaid expenses and other current assets | | | (174 | ) | | (99 | ) | | 187 | | | Accounts payable | | | 1,747 | | | (346 | ) | | 626 | | | Accrued expenses | | | (585 | ) | | 650 | | | 1,036 | | | Accounts receivable and other current assets | | | | 4,846 | | | | (15,304 | ) | Inventory | | | | (3,200 | ) | | | (1,859 | ) | Accounts payable and accrued expenses | | | | 1,872 | | | | 4,596 | | Right-of-use assets and operating lease liabilities | | | | 72 | | | | 46 | | Deferred revenue | | | (50 | ) | | (97 | ) | | 103 | | | | 16,546 | | | | (15 | ) | Deferred rent | | | (20 | ) | | (9 | ) | | 5 | | | | | | | | | | | | | | Net cash used in operating activities | | | (21,907 | ) | | (20,490 | ) | | (16,316 | ) | | | (14,435 | ) | | | (56,699 | ) | | | | | | | | | | | | Cash flows from investing activities: | | | | | | | | | | | | | | | Purchases of marketable securities | | | — | | | (5,052 | ) | | (17,517 | ) | | Maturities of marketable securities | | | — | | | 18,743 | | | 13,168 | | | Acquisition of Icon Bioscience Inc., net of cash acquired | | | (16,780 | ) | | | — | | | | — | | | Purchases of property and equipment | | | (108 | ) | | (147 | ) | | (113 | ) | | | (362 | ) | | | (213 | ) | Proceeds from sale of property and equipment | | | — | | | 33 | | | | — | | | | | | | | | | | | | | Net cash (used in) provided by investing activities | | | (16,888 | ) | | 13,577 | | | (4,462 | ) | | | | | | | | | | | | | Net cash used in investing activities | | | | (362 | ) | | | (213 | ) | Cash flows from financing activities: | | | | | | | | | | | | | | | Proceeds from issuance of stock, net of issuance costs | | | 41,515 | | | 8,404 | | | 16,500 | | | | 49,918 | | | | 22,627 | | Proceeds under paycheck protection program loan | | | | 2,041 | | | | — | | Proceeds from issuance of long-term debt | | | 20,000 | | | | — | | | | — | | | | — | | | | 50,000 | | Payment of debt issue costs | | | (1,347 | ) | | | — | | | | — | | | | — | | | | (1,341 | ) | Payment of long-term debt principal | | | | (13,794 | ) | | | (20,000 | ) | Payment of extinguishment of debt costs | | | | (828 | ) | | | (2,716 | ) | Net settlement of stock units to satisfy statutory tax withholding | | | | (90 | ) | | | (120 | ) | Proceeds from exercise of stock options | | | 503 | | | 99 | | | 490 | | | | 294 | | | | 414 | | | | | | | | | | | | | Payment of contingent development milestone | | | | — | | | | (15,000 | ) | Principal payments on finance lease obligations | | | | (49 | ) | | | — | | Net cash provided by financing activities | | | 60,671 | | | 8,503 | | | 16,990 | | | | 37,492 | | | | 33,864 | | | | | | | | | | | | | Effect of foreign exchange rate changes on cash and cash equivalents | | | 2 | | | (5 | ) | | (20 | ) | | | — | | | | 1 | | | | | | | | | | | | | Net increase (decrease) in cash, cash equivalents and restricted cash | | | 21,878 | | | 1,585 | | | (3,808 | ) | | | 22,695 | | | | (23,047 | ) | Cash, cash equivalents and restricted cash at beginning of year | | | 17,048 | | | 15,463 | | | 19,271 | | | | 22,364 | | | | 45,411 | | | | | | | | | | | | | Cash, cash equivalents and restricted cash at end of year | | $ | 38,926 | | | $ | 17,048 | | | $ | 15,463 | | | $ | 45,059 | | | $ | 22,364 | | | | | | | | | | | | | Supplemental cash flow information: | | | | | | | | | | | | | | | Cash interest paid | | $ | 258 | | | $ | — | | | $ | — | | | $ | 5,510 | | | $ | 4,870 | | Supplemental disclosure ofnon-investing and financing activities: | | | | | | | | Accrued development milestone | | | 15,000 | | | | — | | | | — | | | Supplemental disclosure of non-cash investing and financing activities: | | | | | | | | | | Accrued term loan exit fee | | | 1,200 | | | | — | | | | — | | | | 122 | | | | 3,000 | | Fair value of second tranche purchase liability | | | 4,734 | | | | — | | | | — | | | Fair value of warrants issued with debt | | | 355 | | | | — | | | | — | | | Fair value of second tranche warrants | | | 18,165 | | | | — | | | | — | | |
See notes to consolidated financial statements
EYEPOINT PHARMACEUTICALS, INC. AND SUBSIDIARIES NOTES TO CONSOLIDATED FINANCIAL STATEMENTS EyePoint Pharmaceuticals, Inc. (together with its subsidiaries, the “Company”), incorporated in Delaware, is a specialty biopharmaceuticalpharmaceutical company committed to developing and commercializing innovative ophthalmic products fortherapeutics to help improve the treatmentlives of patients with serious eye diseases.disorders. The Company’s leadpipeline leverages its proprietary Durasert® technology for extended intraocular drug delivery including EYP-1901, a potential twice-yearly sustained delivery intravitreal anti-VEGF treatment initially targeting wet age-related macular degeneration (“wet AMD”), the leading cause of vision loss among people 50 years of age and older in the United States. The Company’s product DEXYCU™ (dexamethasone intraocular suspension) 9%candidate pipeline also includes YUTIQ50, a potential twice-yearly treatment for non-infectious uveitis affecting the posterior segment of the eye, one of the leading causes of blindness. The Company also has 2 commercial products: YUTIQ®, a once every three-year treatment for chronic non-infectious uveitis affecting the posterior segment of the eye, and DEXYCU®, a single dose treatment for postoperative inflammation following ocular surgery. Local drug delivery for treating ocular diseases is a significant challenge due to the effectiveness of the blood-eye barrier. This barrier makes it difficult for systemically-administered drugs to reach the eye in sufficient quantities to have a beneficial effect without causing unacceptable adverse side effects to other organs. The Company’s validated Durasert technology, which has already been included in 4 products approved for marketing by the United States (“U.S.”) Food and Drug Administration (“FDA”), is designed to provide consistent, sustained delivery of small molecule drugs over a period of months to years through a single intravitreal injection. The Company’s lead product candidate, EYP-1901, combines a bioerodible formulation of its proprietary Durasert sustained-release technology with vorolanib, a tyrosine kinase inhibitor (“TKI”). The Company is currently evaluating EYP-1901 in February 2018a Phase 1 clinical trial as a potential twice-yearly sustained delivery intravitreal treatment for wet AMD. Current approved treatments for wet AMD require monthly or bi-monthly eye injections in a physician’s office, which can cause inconvenience and discomfort and often lead to reduced compliance and poor outcomes. In two prior clinical trials of vorolanib as an orally delivered therapy, vorolanib had a strong clinical signal with no significant ocular adverse events. The Company expects initial data from the Phase 1 clinical trial in the second half of 2021. YUTIQ® (fluocinolone acetonide intravitreal implant) 0.18 mg for intravitreal injection, is a non-erodible intravitreal implant containing fluocinolone acetonide (“FA”) lasting for up to 36 months and is indicated for the treatment of chronic non-infectious uveitis affecting the posterior segment of the eye. This disease affects between 60,000 to 100,000 people each year in the U.S. causes approximately 30,000 new cases of blindness every year and is the third leading cause of blindness. YUTIQ utilizes the Company’s proprietary Durasert® sustained-release drug delivery technology platform. DEXYCU® (dexamethasone intraocular suspension) 9%, for intraocular administration, is indicated for the treatment of post-operative ocular inflammation, is administeredwith the Company’s primary focus on its use immediately following cataract surgery as a single dose at the end of ocular surgery and is the first long-acting intraocular product approved by the FDA for the treatment of post-operative inflammation.treatment. DEXYCU utilizes the Company’s proprietary Verisome® drug-delivery platform, which allowstechnology. The Company is also developing YUTIQ50 as a potential six-month intravitreal treatment for chronic non-infectious uveitis affecting the posterior segment of the eye. The Company has consulted with the FDA and identified a clinical pathway for a single intraocular injectionsupplemental new drug application (“sNDA”) filing that releases dexamethasone, a corticosteroid, over time. There are approximately four million cataract surgeries performed annually in the U.S. and the Company expects will involve a clinical trial of a small population. The Company is currently evaluating the timeline and investment requirements for the initiation of this trial. The Company is also seeking to enhance its long-term growth potential by expanding EYP-1901 beyond wet AMD into diabetic retinopathy (“DR”) and retinal vein occlusion (“RVO”), both large and growing ocular disease areas. The Company also plans to launch DEXYCU in the U.S. in the first half of calendar year 2019 with a primary focus on its use following cataract surgery.potentially identify and advance additional product candidates through clinical and regulatory development. The Company acquired DEXYCU in connectionexpects to utilize its internal discovery efforts, potential research collaborations and/or in-licensing arrangements with itspartner molecules and potential acquisition of Icon Bioscience, Inc. (“Icon”additional ophthalmic products, product candidates or technologies that complement its current product portfolio. Effects of the COVID-19 Coronavirus Pandemic The outbreak of the COVID-19 coronavirus pandemic (the “Pandemic”) in March 2018. The Company’s lead product candidate is YUTIQ™,2020 has had and will likely continue to have, a three-yearnon-erodible fluocinolone acetonide insert for the treatment ofnon-infectious posterior uveitis (“NIPU”). Injected into the eye in an office visit, YUTIQ is a tiny micro-insert that delivers a micro-dose of a corticosteroid to the back of the eye on a sustained constant (zero order release) basis for approximately three years. On March 19, 2018, the FDA accepted the Company’s New Drug Application (“NDA”) for YUTIQmaterial and set an FDA Prescription Drug User Fee Act (“PDUFA”) action date of November 5, 2018. YUTIQ is basedadverse impact on the Company’s proprietary Durasert™ sustained-release drug delivery technology platform, which can deliver drugs for predetermined periodsbusiness, including as a result of time ranging from months to years. Posterior segment uveitis is the third leading cause of blindness in the U.S. and affects between 55,000 to 120,000 people. If approved in November 2018,measures that the Company, expectsother businesses, and government have taken and will likely continue to launch YUTIQtake. This includes a significant impact on cash flows from expected revenues due to the closure of ambulatory surgery centers for DEXYCU and a significant reduction in physician office visits impacting YUTIQ. These closures precipitated the first half of calendar year 2019. ILUVIEN® for diabetic macular edema (“DME”), the Company’s lead licensed product, is sold directly in the U.S. and several European Union (“EU”) countries by Alimera Sciences, Inc. (“Alimera”). Retisert®, onerestructuring of the Company’s earlier generation products,commercial organization that was approvedannounced on April 1, 2020 along with a reduction in 2005 by the FDAplanned spending for the treatment of posterior segment uveitiscalendar year. Due to the continued Pandemic, these factors have had an adverse impact on the Company’s revenues, financial condition and is soldcash flows in 2020 and will continue into 2021. Although customer demand for the Company’s products demonstrated sequential growth in the U.S. by Bausch & Lomb Incorporated (“Bausch & Lomb”).third and fourth quarter of 2020, the extent and duration of the impact on the Company’s business is uncertain at this time. The Company is monitoring the Pandemic and its potential effect on the
Company’s development programs are focused primarilyfinancial position, results of operations and cash flows. This uncertainty could have an impact in future periods on developing sustained release products that utilize its Durasertcertain estimates used in the preparation of the Company’s periodic financial results, including reserves for variable consideration related to product sales, realizability of certain receivables, assessment for excess or obsolete inventory, and Verisome technology platforms to deliver approved drugs to treat chronic diseases. The Company’s strategy includes developing products independently while continuing to leverage its technology platforms through collaborationsimpairment of long-lived assets. Uncertainty around the extent and license agreements.duration of the Pandemic, and any future related financial impact cannot be reasonably estimated at this time. Liquidity The Company has financed its operations primarilyhad cash and cash equivalents of $44.9million at December 31, 2020. On February 4, 2021, the Company received net proceeds of approximately $108.0 million from salesthe issuance of equity securities, debt andshares of the receipt of license fees, milestone payments, research and development funding and royalty income from its collaboration partners.Company’s common stock (“Common Stock”) in an underwritten public offering (see Note 19). The Company has a history of operating losses and to date, has not had significant recurring cash inflows from revenue. The Company’s anticipated recurring useoperations have been financed primarily from sales of cashits equity securities, issuance of debt and a combination of license fees, milestone payments, royalty income and other fees received from its collaboration partners. In the first quarter of 2019, the Company commenced the U.S. launch of its first two commercial products, YUTIQ and DEXYCU. However, the Company has not received sufficient revenues from its product sales to fund operations and the Company does not expect revenues from its product sales to generate sufficient funding to sustain its operations in combination with no probable sourcethe near-term. The Company expects to continue fulfilling its funding needs through cash inflows from revenue of YUTIQ and DEXYCU product sales, licensing and research collaboration transactions, additional equity capital raises substantial doubt about its ability to continue as a going concern for one year from the issuance of its financial statements. During June 2018, the Company closed two financing transactions that consisted of (i) $25.3 million of net proceeds from the sale of 20,184,224 units (each a “Unit”, and collectively, the “Units”), with each Unit consisting of one share of the Company’s common stock (“Common Stock”) and one warrant to purchase one share of Common Stock (each a “Second Tranche Warrant”, and collectively, the “Second Tranche Warrants”), to certain accredited investors on June 25, 2018 (see Note 10) and (ii) $4.9 million of net proceeds from an additional advance under its term loan facility on June 26, 2018 (see Note 9). The Company had cash and cash equivalents of $38.8 million at June 30, 2018.other arrangements. The Company believes that its cash and cash equivalents of $44.9 million at June 30, 2018December 31, 2020 and the net proceeds of approximately $108.0 million received in February 2021 from the issuance of Common Stock, coupled with expected proceedscash inflows from existing collaboration agreementsits product sales will enable the Company to maintainfund its current and planned operations (including continuationfor at least the next twelve months from the date these consolidated financial statements were issuedand therefore the conditions raising substantial doubt raised in prior periods has been alleviated. Actual cash requirements could differ from management’s projections due to many factors, including the continued effect of its two Phase 3the Pandemic on the Company’s business and the medical community, the timing and results of the Company’s clinical trials for YUTIQ) intoEYP-1901, additional investments in research and development programs, the first quartersuccess of calendar year 2019. In order to extendcommercialization for YUTIQ and DEXYCU, the Company’s ability to fund its operations beyond then, including its planned U.S. commercial launch of DEXYCU and, if approved, YUTIQ, management’s plans include obtaining additional equity financing from the potential exercise of the Warrants, from the sale of equity securities through an underwritten public offering or the sale of Common Stock through anat-the-market (“ATM”) program or from other sources and/or additional debt financing and/or, as applicable, reducing or deferring operating expenses. The timing and extent of the Company’s implementationactual costs of these plans is expected to depend oncommercialization efforts, competing technological and market developments and the amount and timingcosts of cash receipts from existing or any future collaborations or other agreementsstrategic acquisitions and/or proceeds from any financing transactions. There is no assurance that the Company will receive significant revenues from its planned commercializationdevelopment of DEXYCU or, if approved, YUTIQ, or from its product license revenues under existing collaboration agreements, or that it will receive proceeds from the exercise of the Warrants.complementary business opportunities.
2. | Significant Accounting Policies |
Basis of Presentation The consolidated financial statements are presented in U.S. dollars in accordance with generally accepted accounting principles in the U.S. (“U.S. GAAP”) and include the accounts of EyePoint Pharmaceuticals, Inc. and its wholly-owned subsidiaries. All intercompany accounts and transactions have been eliminated. The Company’s fiscal year ends on June 30 of each year. The years ended June 30, 2018, 2017 and 2016 may be referred to herein as fiscal 2018, fiscal 2017 and fiscal 2016, respectively. Use of Estimates The preparation of consolidated financial statements in conformity with U.S. GAAP requires management to make certain estimates and assumptions that affect the reported amounts and disclosure of assets and liabilities at the date of the consolidated financial statements and the reported amounts and disclosure of revenues and expenses during the reporting periods. Significant management estimates and assumptions include, among others, those related to reserves for variable consideration related to product sales, revenue recognition for multiple-deliverable arrangements, recognition of expense in outsourced clinical trial agreements, and realization of deferred tax assets and the valuation of stock option and other equity awards.assets. Actual results could differ from these and other estimates. Foreign Currency The functional currency of the Company and each of its subsidiaries is the currency of the primary economic environment in which each such entity operates - operates—the U.S. dollar or the Pound Sterling. Assets and liabilities of the Company’s foreign subsidiary are translated atperiod-end exchange rates. Amounts included in the consolidated statements of comprehensive loss and cash flows are translated at the weighted average exchange rates for the period. Gains and losses from currency translation are included in accumulated other comprehensive income as a separate component of stockholders’ equity in the consolidated balance sheets. The balance of accumulated other comprehensive income attributable to foreign currency translation was $838,000$841,000 and $840,000 at June 30, 2018December 31, 2020 and $833,000 at June 30, 2017.2019, respectively. Foreign currency gains or losses arising from transactions denominated in foreign currencies, whether realized or unrealized, are recorded in interest and other income, net in the consolidated statements of comprehensive loss and were not significantmaterial for all periods presented. Cash Equivalents Cash equivalents represent highly liquid investments with maturities of three months or less at the date of purchase, principally consisting of institutional money market funds. Marketable Securities
Marketable securities consist of investments with an original or remaining maturity of greater than three months at the date of purchase. The Company has historically classified its marketable securities as
available-for-sale. Accordingly, the Company records these investments at fair value, with unrealized gains and losses excluded from earnings and reported, net of tax, in accumulated other comprehensive income, which is a component of stockholders’ equity. If the Company determines that a decline of any investment is other-than-temporary, the investment is written down to fair value. As of June 30, 2018 and 2017, there were no investments in marketable securities. During fiscal 2017, $5.1 million of marketable securities were purchased and $18.7 million matured.
The fair value of marketable securities is determined based on quoted market prices at the balance sheet date of the same or similar instruments. The amortized cost of debt securities is adjusted for amortization of premiums and accretion of discounts through to the earlier of sale or maturity. Such amortization and accretion amounts are included in interest and other income, net in the consolidated statements of comprehensive loss. The cost of marketable securities sold is determined by the specific identification method.
Concentrations of Credit Risk Financial instruments that potentially subject the Company to concentrations of credit risk consist principally of cash, cash equivalents and marketable securities. At June 30, 2018,December 31, 2020, a total of $28.8$23.5 million, representing all of the Company’s interest-bearing cash equivalent balances, were concentrated in one U.S. Government institutional money market fund that had investments consisting primarily of U.S. Government Agency debt, U.S. Treasury debt, U.S. Treasury Repurchase Agreements and U.S. Government Agency Repurchase Agreements. Generally, these deposits may be redeemed upon demand and, therefore, the Company believes they have minimal risk. The Company had no0 investments in marketable securities at June 30, 2018December 31, 2020 and 2017.2019, respectively. The Company’s investment policy, approved by the Company’s Board of Directors, includes guidelines relative to diversification and maturities designed to preserve principal and liquidity. RevenuesAs of December 31, 2020, accounts receivable from AlimeraASD Specialty Healthcare LLC and McKesson Specialty Care Distribution LLC accounted for $723,000, or 24% of total revenues in fiscal 2018, $659,000, or 9% of total revenues in fiscal 201756.0% and $233,000, or 14% of total revenues in fiscal 2016. Revenues from Pfizer accounted for $5.6 million, or 74% of total revenues in fiscal 2017 and were inconsequential in fiscal 2018 and fiscal 2016. Revenues from Bausch & Lomb accounted for approximately $1.0 million, or 35% of total revenues in fiscal 2018, $984,000, or 13% of total revenues in fiscal 2017 and $1.3 million, or 77% of total revenues in fiscal 2016. Revenues from feasibility study agreements accounted for approximately $1.1 million, or 37% of total revenues in fiscal 2018, $211,000, or 3%, of total revenues in fiscal 2017 and were inconsequential in fiscal 2016.
Accounts receivable from Bausch & Lomb accounted for $306,000, or 87%37.0% of total accounts receivable, at June 30, 2018respectively. For the year ended December 31, 2020, revenues from ASD Specialty Healthcare LLC, Ocumension Therapeutics, and $246,000, or 98%McKesson Specialty Care Distribution LLC accounted for 39.0%, 33.0%, and 18.0% of total revenues, respectively.
As of December 31, 2019, accounts receivable from the Specialty Distributor, an affiliate of the Third-party Logistics Provider (the “3PL”), ASD Specialty Healthcare LLC, FFF Enterprises, Inc., and McKesson Specialty Care Distribution LLC accounted for 37.0%, 34.0%, 15.0%, and 12.0% of total accounts receivable, at June 30, 2017.respectively. For the year ended December 31, 2019, revenues from the Specialty Distributor, an affiliate of the 3PL, ASD Specialty Healthcare LLC, and Alimera Sciences accounted for 56.0%, 15.0%, and 10.0% of total revenues, respectively. Fair Value Measurements The Company accounts for certain assets and liabilities at fair value. The hierarchy below lists three levels of fair value based on the extent to which inputs used in measuring fair value are observable in the market. The Company categorizes each of its fair value measurements in one of these three levels based on the lowest level input that is significant to the fair value measurement in its entirety. These levels are: Level 1 – Inputs are quoted prices (unadjusted) in active markets that are accessible at the measurement date for identical assets and liabilities.
| • | Level 1 – Inputs are quoted prices (unadjusted) in active markets that are accessible at the measurement date for identical assets and liabilities. |
Level 2 – Inputs are directly or indirectly observable in the marketplace, such as quoted prices for similar assets or liabilities in active markets or quoted prices for identical assets or liabilities with insufficient volume or infrequent transaction (less active markets).
| • | Level 2 – Inputs are directly or indirectly observable in the marketplace, such as quoted prices for similar assets or liabilities in active markets or quoted prices for identical assets or liabilities with insufficient volume or infrequent transaction (less active markets). |
Level 3 – Inputs are unobservable estimates that are supported by little or no market activity and require the Company to develop its own assumptions about how market participants would price the assets or liabilities.
| • | Level 3 – Inputs are unobservable estimates that are supported by little or no market activity and require the Company to develop its own assumptions about how market participants would price the assets or liabilities. |
The carrying amounts of cash equivalents, accounts receivable, accounts payable and accrued expenses approximate fair value because of their short-term maturity. Accounts and Other Receivables, Net Receivables consistarise primarily from the Company’s products sold in the U.S. The balance in accounts and other receivables, net consists primarily of quarterly royalties earned underamounts due from customers, net of applicable revenue reserves. The majority of the Company’s accounts receivable have standard payment terms that require payment within 120-157 days. The Company performs ongoing credit evaluations of its customers’ financial condition and continuously monitor collections and payments from its customers and analyzes accounts that are past due for collectability. The allowance for credit losses is estimated based on the Company’s analysis of trends in overall receivables aging, specific identification of certain receivables that are at risk of not being paid, past collection experience and current economic trends. Given the nature and limited history of collectability of the Company’s accounts receivable, the Company recorded 0 allowance for credit losses as of December 31, 2020 and 2019. Inventory Inventory is stated at the lower of cost or net realizable value, net on a license agreementfirst-in, first-out (“FIFO”) basis. The inventory costs for YUTIQ include purchases of various components and the active pharmaceutical ingredient (“API”) and internal labor and overhead for the product manufactured in the Company’s Watertown, MA facility. The inventory costs for DEXYCU include purchased components, the API and third-party manufacturing and assembly.
Capitalization of inventory costs begins after FDA approval of the product. Prior thereto, inventory costs of products and product candidates are recorded as research and development expense, even if this inventory may later be sold as commercial product. The Company assesses the recoverability of inventory and it writes down any excess and obsolete inventories to their estimated realizable value in the period in which the impairment is first identified. Write-downs are based on the age of the inventory, lower of cost or market, along with Bausch & Lomb.significant management judgments concerning future demands for the inventory. Such impairment charges, should they occur, are recorded within cost of sales, excluding amortization of acquired intangible assets. The determination of whether inventory costs will be realizable requires estimates by management. If actual market conditions are less favorable than projected by management, additional write-downs of inventory might be recorded in future periods. Cost of sales, excluding amortization of acquired intangible assets, consist of costs associated with the manufacture of YUTIQ and DEXYCU, certain period costs for DEXYCU product revenue, product shipping and, as applicable, royalty expense. The inventory costs for YUTIQ include purchases of various components, the active pharmaceutical ingredient (“API”) and direct labor and overhead for the product manufactured in the Company’s Watertown, MA facility. The inventory costs for DEXYCU include purchased components, the API and third-party manufacturing and assembly. Capitalization of inventory costs begins after FDA approval of a product. Prior thereto, inventory costs of products and product candidates are recorded as research and development expense, even if this inventory may later be sold as commercial product. The Company accrued DEXYCU product revenue-based royalty expense of $2.3 million and $781,000 for the year ended December 31, 2020 and 2019, respectively, as a component of cost of sales, of which $1.3 million and $0 of accrued revenue-based royalty expense were related to the partnering income equal to 20% of DEXYCU share of the Accelerated Milestone Payment received in August 2020 and upfront payment received in February 2020 from Ocumension, in connection with the acquisition of Icon Bioscience, Inc. in March 2018 for the year ended December 31, 2020 and 2019, respectively. Debt and Equity Instruments Debt and equity instruments are classified as either liabilities or equity in accordance with the substance of the contractual arrangement. Derivative Instruments Derivative financial liabilities are recorded at fair value, with gains and losses arising from changes in fair value recognized in change in fair value of derivative liability within the consolidated statements of comprehensive loss at each period end while such instruments are outstanding. The Company’s derivative liabilities are beingfrom certain financing transactions were primarily valued using Monte Carlo simulation models. Refer to Notes 10 and 12 for additional information. Property and Equipment Property and equipment are recorded at cost and depreciated over their estimated useful lives (generally three to five years) using the straight-line method. Leasehold improvements are amortized on a straight-line basis over the shorter of the remainingnon-cancellable lease term or their estimated useful lives. Repair and maintenance costs are expensed as incurred. When assets are retired or sold, the assets and accumulated depreciation are derecognized from the respective accounts and any gain or loss is recognized. Leases The Company leases real estate and office equipment under operating leases. Its primary real estate lease contains rent holiday and rent escalation clauses. The Company recognizesadopted Accounting Standards Codification No. 842, Leases (“ASC 842”), as of January 1, 2019. The adoption of ASC 842 represents a change in accounting principle that establishes a right-of-use (“ROU”) model that requires a lessee to record a ROU asset and a lease liability on the balance sheet for all operating leases, with an exception provided for leases with a duration of one year or less. The Company applied ASC 842 using the modified retrospective transition approach which, allows companies to recognize existing leases at the adoption date without requiring comparable period presentation. Comparative periods are presented in accordance with the previous guidance in Accounting Standards Codification (“ASC”) 840, Leases.
In adopting the new standard, the Company elected to utilize the available package of practical expedients permitted under the transition guidance within the new standard, which does not require the reassessment of the following: (i) whether existing or expired arrangements are or contain a lease, (ii) the lease classification of existing or expired leases, and (iii) whether previous initial direct costs would qualify for capitalization under the new lease standard. Additionally, the Company elected to combine lease and non-lease components and to exclude leases with a term of 12 months or less. The adoption of this accounting standard resulted in recording operating lease ROU assets for 3 real estate operating lease arrangements and corresponding operating lease liabilities of $3.5 million and $3.7 million, respectively, as of January 1, 2019. The operating lease assets at adoption were lower than the operating lease liabilities because the balance of the Company’s deferred rent holidayliabilities at December 31, 2018, which represented lease incentives, was reclassified into operating lease assets. The adoption of the standard did not have a material effect on the Company’s consolidated statements of operations or consolidated statements of cash flows. Under Topic 842, the Company determines whether the arrangement is or contains a lease at inception. Operating leases are recognized on the consolidated balance sheets as ROU assets, current portion of lease liabilities and scheduled rent increaseslong-term lease liabilities. ROU assets represent the Company’s right to use an underlying asset for the lease term and lease liabilities represent the Company’s obligation to make lease payments arising from the lease. Operating lease liabilities and their corresponding ROU assets are recorded based on the present value of lease payments over the expected remaining lease term. For this purpose, the Company considers only payments that are fixed and determinable at the time of commencement. The operating lease ROU assets also include any lease payments made and adjustments for prepayments and lease incentives. The interest rate implicit in lease contracts is typically not readily determinable. As a result, the Company utilized its incremental borrowing rate, which is the rate incurred to borrow on a collateralized basis over a similar term an amount equal to the lease payments in a similar economic environment. Lease terms may include options to extend or terminate the lease when it is reasonably certain that the Company will exercise that option. Lease expense for lease payments is recognized on a straight-line basis over the lease term, with the excess of cumulative rent expense over cash payments recorded as a deferred rent liability.term. Impairment of Intangible Assets The Company’s finite life intangible assets which have historically included its Durasert and Tethadur™ patented technologies, also include the DEXYCU product (utilizing the Verisome technology) following the March 2018 acquisition of Icon. The previous intangible assets were amortized on a straight-line basis over twelve years and were fully amortized as of December 31, 2017. The DEXYCU intangible asset is being amortized on a straight-line basis over its estimated useful life of thirteen years. The intangible asset lives were determined based upon the anticipated period that the Company would derive future cash flows from the intangible assets, considering the effects of legal, regulatory, contractual, competitive and other economic factors. The Company continually monitors whether events or circumstances have occurred that indicate that the remaining estimated useful life of its intangible assets may warrant revision. The Company assesses potential impairments to its intangible assets when there is evidence that events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. An impairment loss is recognized when the future undiscounted net cash flows expected to result from the use of an asset are less than its carrying value. If the Company considers an asset to be impaired, the impairment charge to be recognized is measured as the amount by which the carrying value of the asset exceeds its estimated fair value. Revenue Recognition Collaborative Research and Development and Multiple-Deliverable Arrangements
The Company enters into collaborative arrangementsadopted Accounting Standards Codification No. 606, Revenue from Contracts with strategic partners forCustomers (“ASC 606”), as of July 1, 2018. The adoption of ASC 606 represents a change in accounting principle that more closely aligns revenue recognition with the development and commercializationdelivery of products and product candidates utilizing the Company’s technologies.services. The termsCompany applied ASC 606 using the modified retrospective method. The cumulative effect of these agreements have typically included multiple deliverables byinitially applying the Company (for example, license rights, research and development services and manufacturing of clinical materials)new revenue standard resulted in exchange for considerationa $218,000 reduction to the Companyopening balance of some combination ofnon-refundable license fees, research and development funding, payments based upon achievement of clinical development or other milestones and royalties in the form of a designated percentage of product sales.accumulated deficit at July 1, 2018. Revenue is recognized when there is persuasive evidencea customer obtains control of promised goods or services, in an amount that reflects the consideration which the entity expects to receive in exchange for those goods or services. To determine revenue recognition for arrangements that an arrangement exists, delivery has occurred,entity determines are within the scope of ASC 606, Revenue from Contracts with Customers (“ASC 606”), the Company performs the following five steps: (i) identify the contract(s) with a customer; (ii) identify the performance obligations in the contract; (iii) determine the transaction price; (iv) allocate the transaction price to the performance obligations in the contract; and (v) recognize revenue when (or as) the entity satisfies a performance obligation. The Company only applies the five-step model to contracts when it is fixedprobable that the entity will collect the consideration it is entitled to in exchange for the goods or services it transfers to the customer. At contract inception, once the contract is determined to be within the scope of ASC 606, the Company assesses the goods or services promised within each contract, determines those that are performance obligations and determinable, and collectionassesses whether each promised good or service is reasonably assured. Multiple-deliverable arrangements, suchdistinct. The Company then recognizes as license and development agreements, are analyzed to determine whetherrevenue the deliverables can be separated or whether they must be accounted for as a single unitamount of accounting. When deliverables are separable, consideration receivedthe transaction price that is allocated to the separate unitsrespective performance obligation when (or as) the performance obligation is satisfied. Sales, value add, and other taxes collected on behalf of accountingthird parties are excluded from revenue. Product sales, net — The Company sells YUTIQ and DEXYCU to a limited number of specialty distributors and specialty pharmacies (collectively the “Distributors”) in the U.S., with whom the Company has entered into formal agreements, for delivery to physician practices for YUTIQ and to hospital outpatient departments and ambulatory surgical centers for DEXYCU. The Company
recognizes revenue on sales of its products when Distributors obtain control of the products, which occurs at a point in time, typically upon delivery. In addition to agreements with Distributors, the Company also enters into arrangements with healthcare providers, ambulatory surgical centers, and payors that provide for government mandated and/or privately negotiated rebates, chargebacks, and discounts with respect to the purchase of the Company’s products from Distributors. Reserves for variable consideration — Product sales are recorded at the wholesale acquisition costs, net of applicable reserves for variable consideration. Components of variable consideration include trade discounts and allowances, provider chargebacks and discounts, payor rebates, product returns, and other allowances that are offered within contracts between the Company and its Distributors, payors, and other contracted purchasers relating to the Company’s product sales. These reserves, as detailed below, are based on the relativeamounts earned, or to be claimed on the related sales, and are classified either as reductions of product revenue and accounts receivable or a current liability, depending on how the amount is to be settled. Overall, these reserves reflect the Company’s best estimates of the amount of consideration to which it is entitled based on the terms of the respective underlying contracts.Actual amounts of consideration ultimately received may differ from the Company’s estimates. If actual results in the future vary from the estimates, the Company adjusts these estimates, which would affect product revenue and earnings in the period such variances become known. Distribution fees — The Company compensates its Distributors for services explicitly stated in the Company’s contracts and are recorded as a reduction of revenue in the period the related product sale is recognized. Provider chargebacks and discounts — Chargebacks are discounts that represent the estimated obligations resulting from contractual commitments to sell products at prices lower than the list prices charged to the Company’s Distributors. These Distributors charge the Company for the difference between what they pay for the product and the Company’s contracted selling price. These reserves are established in the same period that the related revenue is recognized, resulting in a reduction of product revenue and the establishment of a current liability. Reserves for chargebacks consist of amounts that the Company expects to pay for units that remain in the distribution channel inventories at each reporting period-end that the Company expects will be sold under a contracted selling price, method using management’s bestand chargebacks that Distributors have claimed, but for which the Company has not yet settled. Government rebates — The Company is subject to discount obligations under state Medicaid programs and Medicare. These reserves are recorded in the same period the related revenue is recognized, resulting in a reduction of product revenue and the establishment of a current liability which is included in accrued expenses and other current liabilities on the condensed consolidated balance sheets. The Company’s liability for these rebates consists of invoices received for claims from prior quarters that have not been paid or for which an invoice has not yet been received, estimates of claims for the current quarter, and estimated future claims that will be made for product that has been recognized as revenue, but which remains in the distribution channel inventories at the end of each reporting period. Payor rebates — The Company contracts with certain private payor organizations, primarily insurance companies, for the payment of rebates with respect to utilization of its products. The Company estimates these rebates and records such estimates in the same period the related revenue is recognized, resulting in a reduction of product revenue and the establishment of a current liability. Co-Payment assistance — The Company offers co-payment assistance to commercially insured patients meeting certain eligibility requirements. The calculation of the accrual for co-pay assistance is based on an estimate of claims and the cost per claim that the Company expects to receive associated with product that has been recognized as revenue. Product returns — The Company generally offers a limited right of return based on its returned goods policy, which includes damaged product and remaining shelf life. The Company estimates the amount of its product sales that may be returned and records this estimate as a reduction of revenue in the period the related product revenue is recognized, as well as reductions to trade receivables, net on the condensed consolidated balance sheets. License and collaboration agreement revenue — The Company analyzes each element of its license and collaboration arrangements to determine the appropriate revenue recognition. The terms of the license agreement may include payment to the Company of non-refundable up-front license fees, milestone payments if specified objectives are achieved, and/or royalties on product sales. The Company recognizes revenue from upfront payments at a point in time, typically upon fulfilling the delivery of the associated intellectual property to the customer.
If the contract contains a single performance obligation, the entire transaction price is allocated to the single performance obligation. Contracts that contain multiple performance obligations require an allocation of the transaction price based on the estimated relative standalone selling prices of the promised products or services underlying each performance obligation. The Company determines standalone selling prices based on the price at which the performance obligation is sold separately. If the standalone selling price of deliverables when vendor-specific objective evidence or third-party evidence ofis not observable through past transactions, the Company estimates the standalone selling price is not available. Allocated consideration is recognizedtaking into account available information such as market conditions and internally approved pricing guidelines related to the performance obligations. The Company recognizes sales-based milestone payments as revenue upon applicationthe achievement of the appropriate revenue recognition principles to each unit. Whencumulative sales amount specified in the contract in accordance with ASC 606-10-55-65. For those milestone payments which are contingent on the occurrence of particular future events, the Company determines that an arrangement shouldthese need to be accountedconsidered for inclusion in the calculation of total consideration from the contract as a single unitcomponent of accounting, it must determinevariable consideration using the period over whichmost-likely amount method. As such, the performance obligations will be performed and revenue will be recognized. The Company estimates its performance period used for revenue recognition based on the specific terms ofassesses each agreement, and adjusts the performance periods, if appropriate, based on the applicable facts and circumstances. Significant management judgment may be requiredmilestone to determine the levelprobability and substance behind achieving each milestone. Given the inherent uncertainty associated with these future events, the Company will not recognize revenue from such milestones until there is a high probability of effort required underoccurrence, which typically occurs near or upon achievement of the event.
When determining the transaction price of a contract, an arrangement andadjustment is made if payment from a customer occurs either significantly before or significantly after performance, resulting in a significant financing component. Applying the practical expedient in paragraph 606-10-32-18, the Company does not assess whether a significant financing component exists if the period over whichbetween when the Company is expected to completeperforms its performance obligations under the arrangement. Ifcontract and when the Company cannot reasonably estimate when its performance obligations either are completedcustomer pays is one year or become inconsequential, then revenue recognition is deferred until the Company can reasonably make such estimates. Revenue is then recognized over the remaining estimated period of performance using the cumulativecatch-up method. Royalties
Royalty income is recognized upon the saleless. None of the related products, provided that the royalty amounts are fixed or determinable, collectionCompany’s contracts contained a significant financing component as of the related receivable is reasonably assured and theDecember 31, 2020.
Royalties — The Company has no remaining performance obligations under the arrangement.recognizes revenue from license arrangements with its commercial partners’ net sales of products. Such revenues are included as royalty income. If In accordance with ASC 606-10-55-65, royalties are receivedrecognized when the subsequent sale of the commercial partner’s products occurs. The Company’s commercial partners are obligated to report their net product sales and the resulting royalty due to the Company typically within 60 days from the end of each quarter. Based on historical product sales, royalty receipts and other relevant information, the Company recognizes royalty income each quarter and subsequently determines a true-up when it receives royalty reports and payment from its commercial partners. Historically, these true-up adjustments have been immaterial.
Sale of Future Royalties — The Company has sold its rights to receive certain royalties on product sales. In the circumstance where the Company has remaining performance obligations,sold its rights to future royalties under a royalty purchase agreement and also maintains limited continuing involvement in the royalty payments would be attributedarrangement (but not significant continuing involvement in the generation of the cash flows that are due to the services being providedpurchaser), the Company defers recognition of the proceeds it receives for the sale of royalty streams and recognizes such unearned revenue as revenue under the arrangementunits-of-revenue method over the life of the underlying license agreement. Under the units-of-revenue method, amortization for a reporting period is calculated by computing a ratio of the proceeds received from the purchaser to the total payments expected to be made to the purchaser over the term of the agreement, and therefore revenue wouldthen applying that ratio to the period’s cash payment. Estimating the total payments expected to be received by the purchaser over the term of such arrangements requires management to use subjective estimates and assumptions. Changes to the Company’s estimate of the payments expected to be made to the purchaser over the term of such arrangements could have a material effect on the amount of revenues recognized as such performance obligations are performed. Any such revenues are included as collaborative research and development revenues. Reimbursement of Costs
in any particular period.
Research Collaborations — The Company may providerecognizes revenue over the term of the statements of work under any funded research collaborations (including feasibility study agreements). Revenue recognition for consideration, if any, related to a license option right is assessed based on the terms of any such future license agreement or is otherwise recognized at the completion of the research collaborations (including feasibility study agreements). Please refer to Note 3 for further details on the license and development servicescollaboration agreements into which the Company has entered and incur maintenance costs of licensed patents under collaboration arrangements to assist in advancing the development of licensed products. The Company acts primarily as a principal in these transactions and, accordingly, reimbursementcorresponding amounts received are classified as a component of revenue to be recognized consistent withduring the revenue recognition policy summarized above. The Company records the expenses incurredcurrent and reimbursed on a gross basis. Deferred Revenue Amounts received prior to satisfying the above revenue recognition criteria are recorded as deferred revenue in the accompanying consolidated balance sheets. Amounts not expected to be recognized within one year following the balance sheet date are classified asnon-current deferred revenue.
Research and Development Research and development costs are charged to operations as incurred. These costs include all direct costs, including cash compensation,and stock-based compensation and benefits for research, clinical development, quality assurance, quality control, operations and developmentmedical affairs personnel, amortization of intangible assets, third-party costs and services for clinical trials, clinical materials,pre-clinical programs, regulatory and medical affairs, external consultants, and other operational costs related to the Company’s research and development of its product candidates. Reverse Stock Split On December 8, 2020, the Company’s stockholders approved areverse stock split (the “reverse split”) of the Company’s issued and outstanding shares of Common Stock.As a result of the reverse split, every 10 shares of the Company’s Common Stock issued and outstanding were converted into one share of Common Stock. No fractional shares were issued in connection with the reverse split. Stockholders who would otherwise be entitled to a fractional share of Common Stock instead received cash in lieu of fractional shares based on the closing sales price of the Company’s Common Stock as quoted on the Nasdaq Global Market on December 8, 2020. The reverse split did not reduce the number of authorized shares of the Common Stock or preferred stock (the “Preferred Stock”), or change the par values of the Company’s Common Stock or Preferred Stock. The reverse split affected all stockholders uniformly and did not affect any stockholder's ownership percentage of the Company's shares of Common Stock (except to the extent that the reverse split would result in some of the stockholders receiving cash in lieu of fractional shares). All outstanding options, warrants, restricted stock units and deferred stock units entitling their holders to receive or purchase shares of the Company’s Common Stock have been adjusted as a result of the reverse split, as required by the terms of each security. The number of shares reserved for future issuance pursuant to the Company’s 2016 Long-Term Incentive Plan and the number of shares reserved for future issuance pursuant to the Company’s 2019 Employee Stock Purchase Plan have also been appropriately adjusted. Stock-Based Compensation The Company may award stock options and other equity-based instruments to its employees, directors and consultants pursuant to stockholder-approved plans. In the fourth quarter of fiscal 2017, the Company early adopted Accounting Standards Update (“ASU”) No. 2016-09 (“ASU2016-09,Compensation— 2016-09”), Compensation – Stock Compensation (Topic 718): Improvements to Employee Share-Based Payment Accounting, pursuant to which it elected to account for forfeitures as they occur. As a result, the Company recorded an adjustment of $122,000 to accumulated deficit and additionalpaid-in capital as of July 1, 2016. Prior to the adoption of ASU2016-09, the Company recognized compensation expense for only the portion of share-based payment awards that were expected to vest. Based on historical trends, the Company applied estimated forfeiture rates to determine the number of awards that were expected to vest. Additional expense was recorded if the actual forfeiture rate for each tranche of option grants was lower than estimated, and a recovery of prior expense was recorded if the actual forfeiture rate was higher than estimated. Compensation cost related to such share-based payment awards is based on the fair value of the instrument on the grant date and is recognized on a graded vesting basis over the requisite service period for each separately vesting tranche of the awards. The Company may also grant share-based payment awards that are subject to objectively measurable performance and service criteria. Compensation expense for performance-based awards begins at such time as it becomes probable that the respective performance conditions will be achieved. The Company continues to recognize the grant date fair value of performance-based awards through the vesting date of the respective awards so long as it remains probable that the related performance conditions will be satisfied. The Company estimates the fair value of stock option awards using the Black-Scholes option valuation model and the fair value of performance stock units, restricted stock units and deferred stock units based on the observed grant date fair value of the underlying Common Stock. Net Loss per Share Basic net loss per share is computed by dividing net loss by the weighted-average number of common shares outstanding during the period. For periods in which the Company reports net income, diluted net income per share is determined by adding to the weighted-average number of common shares outstanding the average number of dilutive common equivalent shares using the treasury stock method, unless the effect is anti-dilutive. The numbers of shares in the following table reflect the Company’s one-for-10 reverse split of its issued and outstanding shares of Common Stock on December 8, 2020. As a result of the reverse split, every 10 shares of Common Stock issued and outstanding were converted into 1 share of Common Stock.
PotentialOutstanding potential Common Stock equivalents excluded from the calculation of diluted earnings per share because the effect would have been anti-dilutive were as follows:
| | | | | | | | | | | | | | | | Year Ended June 30, | | | | | 2018 | | | 2017 | | | 2016 | | Options outstanding | | | 7,750,244 | | | | 6,895,685 | | | | 4,981,421 | | Warrants outstanding | | | 20,671,036 | | | | 623,605 | | | | 623,605 | | Restricted stock units outstanding | | | 1,398,129 | | | | 948,500 | | | | — | | Performance stock units outstanding | | | 466,668 | | | | 210,000 | | | | — | | Deferred stock units | | | 35,001 | | | | — | | | | — | | | | | | | | | | | | | | | | | | 30,321,078 | | | | 8,677,790 | | | | 5,605,026 | | | | | | | | | | | | | | |
| | Year Ended December 31, | | | Year Ended December 31, | | | | | 2020 | | | 2019 | | | Stock options | | | 1,338,880 | | | | 1,090,973 | | | ESPP | | | 27,713 | | | | 13,737 | | | Warrants | | | 48,683 | | | | 48,683 | | | Restricted stock units | | | 149,004 | | | | 78,684 | | | | | | 1,564,280 | | | | 1,232,077 | | |
Comprehensive Loss Comprehensive loss is comprised of net loss, foreign currency translation adjustments and unrealized gains and losses onavailable-for-sale marketable securities. Income Tax The Company accounts for income taxes under the asset and liability method. Deferred income tax assets and liabilities are computed for the expected future impact of differences between the financial reporting and income tax bases of assets and liabilities and for the expected future benefit to be derived from tax credits and loss carry forwards. Such deferred income tax computations are measured based on enacted tax laws and rates applicable to the years in which these temporary differences are expected to be recovered or settled. A valuation allowance is provided against net deferred tax assets if, based on the available evidence, it is more likely than not that some or all of the net deferred tax assets will not be realized. The Company determines whether it is more likely than not that a tax position will be sustained upon examination. If it is not more likely than not that a position will be sustained, none of the benefit attributable to the position is recognized. The tax benefit to be recognized for any tax position that meets the more likely than not recognition threshold is calculated as the largest amount that is more than 50% likely of being realized upon resolution of the uncertainty. The Company accounts for interest and penalties related to uncertain tax positions as part of its income tax benefit. Recently Adopted and Recently Issued Accounting Pronouncements New accounting pronouncements are issued periodically by the Financial Accounting Standards Board (“FASB”) and are adopted by the Company as of the specified effective dates. Unless otherwise disclosed below, the Company believes that the impact of recently issued and adopted pronouncements will not have a material impact on the Company’s financial position, results of operations and cash flows or do not apply to the Company’s operations. In May 2014, the FASB issued Accounting Standards UpdateNo. 2014-09,Revenue from Contracts with Customers(Topic 606) (“ASU2014-09”), which requires an entity to recognize revenue in an amount that reflects the consideration to which the entity expects to be entitled in exchange for the transfer of promised goods or services to customers. The standard will replace most existing revenue recognition guidance in U.S. GAAP. In August 2015, the FASB issued ASU2015-14, which officially deferred the effective date of ASU2014-09 by one year, while also permitting early adoption. As a result, ASU2014-09 became effective on July 1, 2018. The Company has initiated an assessment of the potential changes from adopting ASU2014-09 and two revenue streams are expected to be applicable under the standard. The Company adopted the new standard effective July 1, 2018 using the modified retrospective method. The adoption did not have an impact on the recorded amounts in
the current balance sheet and income statement based on its existing collaboration agreements. Additional disclosures will be made in future periods related to the recognition of amounts as a result of adopting the new standard.
In February 2016, the FASB issued ASUNo. 2016-02,Leases (Topic 842) (“ASU 2016-02”). In July 2018, the FASB issued ASU No. 2018-11, Leases (Topic 842), Targeted Improvements, which contains certain amendments to ASU 2016-02 intended to provide relief in implementing the new standard. The new standard establishes aright-of-use (“ROU”) model that requires a lessee to record a ROU asset and a lease liability on the balance sheet for all operating leases, with terms longer than 12 months. Leases will be classified as either financean exception provided for leases with a duration of one year or operating,less. The Company adopted ASU 2016-02 on January 1, 2019 using the modified retrospective transition approach which, pursuant to ASU 2018-11, allows companies to recognize existing leases at the adoption date without requiring comparable period presentation. Comparative periods are presented in accordance with classification affecting the patternprevious guidance in Accounting Standards Codification (“ASC”) 840, Leases. In June 2016, the FASB issued ASU No. 2016-13, Financial Instruments - Credit Losses (Topic 326) (“ASU 2016-13”): Measurement of expense recognition inCredit Losses on Financial Instruments, to replace the income statement. The new standardcurrent incurred loss impairment methodology for financial assets measured at amortized cost with a methodology that reflects expected credit losses and requires consideration of a broader range of reasonable and supportable information, including forecasted information, to develop credit loss estimates. ASU 2016-13 is effective for fiscal years beginning after December 15, 2018, including interim periods within those fiscal years. As a result, ASU2016-02 will become effective on July 1, 2019. A modified retrospective transition approach is required for lessees for capital and operating leases existing at, or entered into after, the beginning of the earliest comparative period presented in the financial statements, with certain practical expedients available. The Company is evaluating the impact the adoption of this standard will have on its consolidated financial statements. In November 2016, the FASB issued ASUNo. 2016-18,Restricted Cash. The standard addresses the classification and presentation of restricted cash and restricted cash equivalents within the statement of cash flows. The ASU is effective for fiscal years beginning after December 15, 2017, with early adoption permitted. The Company adopted this standard during the quarter ended June 30, 2018. As a result of the adoption of this standard, the Company combined restricted cash balances of $150,000 at each of June 30, 2018, 2017 and 2016 with cash and cash equivalents when reconciling the beginning and ending balances within the consolidated statements of cash flows for fiscal 2018, 2017 and 2016. As of June 30, 2018, cash, cash equivalents and restricted cash of $38.9 million, as reported within the consolidated statements of cash flows, included $38.8 million of cash and cash equivalents and $150,000 of restricted cash, as reported within the consolidated balance sheets.
In January 2017, the FASB issued ASU No.2017-01, Business Combinations (Topic 805): Clarifying the Definition of a Business (“ASU2017-01”), to clarify the definition of a business by adding guidance to assist entities with evaluating whether transactions should be accounted for as acquisitions or disposals of assets versus businesses. ASU2017-01 is effective for fiscal years beginning after December 15, 2017, and interim periods within those fiscal years. Early adoption is permitted. The Company adopted this standard early to account for the acquisition of Icon (see Note 3).
In June 2018, the FASB issued ASU2018-07,Improvements to Nonemployee Share-Based Payment Accounting. The standard aligns the measurement and classification guidance for share-based payments to nonemployees with the guidance for share-based payments to employees, with certain exceptions. Under the new guidance, the measurement of equity-classified nonemployee awards will be fixed at the grant date. The ASU is effective for fiscal years beginning after December 15, 2018,2019, and interim periods within those fiscal years. Early adoption is permitted but not before an entity adopts the new revenue guidance. for fiscal years beginning after December 15, 2018. The Company doesadopted ASU 2016-13 on January 1, 2020. The adoption of this standard did not expect ASU2018-07 to have a significantmaterial impact on its consolidated financial statements.
In December 2019, the FASB issued ASU No. 2019-12, Income Taxes (Topic 740) (“ASU 2019-12”): Simplifying the Accounting for Income Taxes. The amendments simplify the accounting for income taxes by removing certain exceptions for recognizing deferred taxes for investments, performing intraperiod allocation and calculating income taxes in interim periods. The ASU also adds guidance to reduce complexity in certain areas, including recognizing deferred taxes for tax goodwill and allocating
taxes to members of a consolidated group ASU 2019-12 is effective for fiscal years beginning after December 15, 2020, and interim periods within those fiscal years. Early adoption is permitted, including adoption in interim or annual periods for which financial statements have not yet been issued. This standard will be effective for the Company in the first quarter of its fiscal year ending December 31, 2021. The Company is currently evaluating the impact the adoption of this update will have on its consolidated financial statements, but does not believe the adoption of the new standard will have a material impact on the Company’s consolidated financial statements. 3. | Acquisition of Icon Bioscience, Inc.Product Revenue Reserves and Allowances
|
On March 28, 2018, the Company and its newly-created wholly-owned subsidiary, Oculus Merger Sub, Inc., acquired Icon, a specialty biopharmaceutical company, through a reverse triangular merger (the “Icon Acquisition”) pursuant to an Agreement and PlanThe Company’s product revenues have been primarily from sales of Merger (the “Merger Agreement”) among the Company, Icon,YUTIQ and Shareholder Representative Services LLC (“SRS”), solely in its capacity as representative of Icon’s securityholders. The Icon Acquisition has been accounted for as an asset acquisition because substantially all of the fair value of the gross assets acquired were deemed to be concentrated in a group of similar identifiable assets related to Icon’s lead product, DEXYCU. A portion of the Icon Acquisition was funded by an equity financing and a debt financing, both of which closed concurrently with the Icon Acquisition (see Notes 10 and 9, respectively).
Pursuant to the Merger Agreement, the Company made a closing payment of $15.0 million to SRS, net of an estimated $127,000 working capital adjustment, and is obligated to pay certain post-closing contingent cash payments upon the achievement of specified milestones and based upon certain net sales and partnering revenue standards, in each case subject to the terms and conditions set forth in the Merger Agreement. These include but are not limited to (i) aone-time development milestone of $15.0 million payable in cash upon the first commercial sale of DEXYCU in the U.S., (ii) sales milestone payments totaling upwhich it began shipping to $95.0 million upon the achievement of certain sales thresholdsits customers in February 2019 and subject to certain Centers for Medicare & Medicaid Services (“CMS”) reimbursement conditions set forth in the Merger Agreement, (iii) quarterlyearn-out payments equal to 12% on net sales of DEXYCU in a given year, whichearn-out payments will increase to 16% of net sales of DEXYCU in such year beginning in the calendar quarter for such year to the extent aggregate annual consideration of DEXYCU exceeds $200.0 million in such year, (iv) quarterlyearn-out payments equal to 20% of partnering revenue receivedMarch 2019, respectively.
Net product revenues by the Company for DEXYCU outside of the U.S., and (v) single-digit percentage quarterlyearn-out payments with respect to net sales and/or partnering income, if any, resulting from future clinical development, regulatory approval and commercialization of any other product candidates the Company acquired in the Icon Acquisition. The purchase price on the date of the Icon Acquisition was $32.0 million, comprised of the closing consideration of $15.0 million, including the assumption of an estimated $127,000 of net current liabilities of Icon, the contingent development milestone payment of $15.0 million and transaction costs of approximately $2.0 million. Through June 30, 2018, cash paid for the acquisition totaled $16.8 million, which consisted of $14.9 million at closing and approximately $1.9 million of transaction costs, net of $38,000 of cash acquired. Given the stage of development of DEXYCU, the Company has determined these payments do not represent research and development costs. The contingent consideration in the form of sales milestones will be capitalized as additional intangible assets when any such consideration becomes probable and can be reasonably estimated. Sales-based royalty payments will be expensed as incurred.
The $32.0 million purchase price was allocated to a single finite-lived intangible asset with an expected amortization life of approximately 13 years (see Note 5). The acquisition did not have a net tax impact due to a full valuation allowance against the acquired net deferred tax assets.
4. | License and Collaboration Agreements
|
Alimera
Under a collaboration agreement with Alimera, as amended in March 2008 (the “Prior Alimera Agreement”), the Company licensed to Alimera the rights to develop, market and sell certain product candidates, including ILUVIEN for DME, and Alimera assumed all financial responsibility for the development of licensed products. In addition, the Company was entitled to receive 20% of any net profits (as defined) on sales of each licensed product (including ILUVIEN) by Alimera, measured on aquarter-by-quarter andcountry-by-country basis. Alimera was entitled to recover 20% of previously incurred and unapplied net losses (as defined) for commercialization of each product in a country, but only by an offset of up to 4% of the net profits earned in that country each quarter, reducing the Company’s net profit share to 16% in each country until those net losses were recouped. In the event that Alimera sublicensed commercialization in any country, the Company was entitled to 20% of royalties and 33% ofnon-royalty consideration received by Alimera, less certain permitted deductions. The Company was also entitled to reimbursement of certain patent maintenance costs with respect to the patents licensed to Alimera.
Because the Company has no remaining performance obligations under the Prior Alimera Agreement, all amounts received from Alimera were generally recognized as revenue upon receipt or at such earlier date, if applicable, on which any such amounts were both fixed and determinable and reasonably assured of collectability. In instances when payments were received and subject to a contingency, revenue was deferred until such contingency was resolved.
Revenue under the Prior Alimera Agreement, including all patent reimbursements, totaled $148,000 for fiscal 2018, $659,000 for fiscal 2017 and $233,000 for fiscal 2016, which are included in collaborative research and development revenues in the accompanying consolidated statements of comprehensive loss. These revenues included (i) $50,000 and $585,000 of net profit share earned in fiscal 2018 and 2017, respectively, of which fiscal 2017 included $136,000 recognized in connection with an arbitration settlement related to calendar year 2014 reporting by Alimera; and (ii) $157,000 ofnon-royalty sublicense consideration earned in fiscal 2016. The remainder of Alimera revenues included in collaborative research and development for each year consisted principally of patent fee reimbursements.
On July 10, 2017, the Company entered into a further amended and restated collaboration agreement (the “Amended Alimera Agreement”), pursuant to which the Company (i) expanded the license to Alimera to the Company’s proprietary Durasert sustained-release drug delivery technology platform to include uveitis, including NIPU, in Europe, the Middle East and Africa and (ii) converted the net profit share arrangement for each licensed product (including ILUVIEN) under the Prior Alimera Agreement to a sales-based royalty on a calendar quarter basis commencing July 1, 2017, with payments from Alimera due 60 days following the end of each quarter.
Sales-based royalties started at the rate of 2%. Commencing January 1, 2019 (or earlier under certain circumstances), the sales-based royalty will increase to 6% on aggregate calendar year net sales up to $75 million and to 8% on any calendar years sales in excess of $75 million. Alimera’s share of contingently recoverable accumulated ILUVIEN commercialization losses under the original net profit share arrangement, capped at $25 million, are to be reduced as follows: (i) $10.0 million was cancelled in lieu of an upfront license fee on the effective date of the Amended Alimera Agreement; (ii) for calendar years 2019 and 2020, 50% of earned sales-based royalties in excess of 2% will be offset against the quarterly royalty payments otherwise due from Alimera; (iii) on January 1, 2020, another $5 million will be cancelled, provided, however, that such date of cancellation may be extended under certain circumstances related to Alimera’s regulatory approval process for ILUVIEN for posterior uveitis, with such extension, if any, subject to mutual agreement by the parties; and (iv) commencing in calendar year 2021, 20% of earned sales-based royalties in excess of 2% will be offset against the quarterly royalty payments due from Alimera until such time as the balance of the original $25 million of recoverable commercialization losses has been fully recouped.
Revenue under the Amended Alimera Agreement totaled $575,000 in fiscal 2018 and was recorded as royalty income in the accompanying consolidated statements of comprehensive loss.
Following consummation of the Amended Alimera Agreement, the Company withdrew its previously filed EU marketing approval application (“MAA”) and its orphan drug designation for posterior uveitis, and Alimera became responsible for filing a Type II variation for ILUVIEN for the treatment of NIPU in the 17 EU countries where ILUVIEN is currently approved for the treatment of DME. In January 2018, Alimera received validation of a Type II variation submitted in December 2017 in all seventeen European countries in which it previously received regulatory approval for ILUVIEN for DME. Alimera has reported that it expects to receive regulatory approval for the Type II variation in the first half of calendar year 2019. If the variation is approved, Alimera plans to commercialize the three-year NIPU indication under its ILUVIEN trademark.
Pfizer
In June 2011, the Company and Pfizer entered into an Amended and Restated Collaborative Research and License agreement (the “Restated Pfizer Agreement”) to focus solely on the development of a sustained-release bioerodible micro-insert injected into the subconjunctiva designed to deliver latanoprost for human ophthalmic disease or conditions other than uveitis (the “Latanoprost Product”). Pfizer made an upfront payment of $2.3 million and the Company agreed to provide Pfizer options under various circumstances for an exclusive, worldwide license to develop and commercialize the Latanoprost Product.
The estimated selling price of the combined deliverables under the Restated Pfizer Agreement of $6.7 million was partially recognized as collaborative research and development revenue over the expected
performance period using the proportional performance method with costs associated with developing the Latanoprost Product reflected in operating expenses in the period in which they were incurred. No collaborative research and development revenue was recorded during each of fiscal 2017 and fiscal 2016.
On October 25, 2016, the Company notified Pfizer that it had discontinued development of the Latanoprost Product, which provided Pfizer a60-day option to acquire a worldwide license in return for a $10.0 million payment and potential sales-based royalties and development, regulatory and sales performance milestone payments. Pfizer did not exercise its option and the Restated Pfizer Agreement automatically terminated on December 26, 2016. The remaining deferred revenue balance of $5.6 million was recognized as revenue in the three-month period ended December 31, 2016. Per the terms2020 and 2019 were as follows (in thousands):
| | Year Ended | | | Year Ended | | | | December 31, 2020 | | | December 31, 2019 | | YUTIQ (A) | | $ | 13,878 | | | $ | 12,046 | | DEXYCU (B) | | | 6,953 | | | | 4,778 | | Total product sales, net | | $ | 20,831 | | | $ | 16,824 | |
| (A) | The Company recognized approximately $205,000 and $91,000 of revenue from YUTIQ product sales to Ocumension for the years ended December 31, 2020 and 2019, respectively. |
| (B) | The Company recognized approximately $8,000 and $0 of revenue from DEXYCU product sales to Ocumension for the years ended December 31, 2020 and 2019, respectively. |
The following table summarizes activity in each of the Restricted Pfizer Agreement,product revenue allowance and reserve categories for the Company has retainedyears ended December 31, 2020 and 2019 (in thousands): | | Chargebacks, Discounts and Fees | | | Government and Other Rebates | | | Returns | | | Total | | Beginning balance at January 1, 2020 | | $ | 1,618 | | | $ | 271 | | | $ | 352 | | | $ | 2,241 | | Provision related to sales in the current year | | | 2,141 | | | | 1,056 | | | | 978 | | | | 4,175 | | Adjustments related to prior period sales | | | (387 | ) | | | — | | | | 50 | | | | (337 | ) | Deductions applied and payments made | | | (2,798 | ) | | | (792 | ) | | | (777 | ) | | | (4,367 | ) | Ending balance at December 31, 2020 | | $ | 574 | | | $ | 535 | | | $ | 603 | | | $ | 1,712 | | | | | | | | | | | | | | | | | | | | | Chargebacks, Discounts and Fees | | | Government and Other Rebates | | | Returns | | | Total | | Beginning balance at January 1, 2019 | | $ | 0 | | | $ | 0 | | | $ | 0 | | | $ | 0 | | Provision related to sales in the current year | | | 2,301 | | | | 429 | | | | 732 | | | | 3,462 | | Adjustments related to prior period sales | | | 0 | | | | 0 | | | | 0 | | | | 0 | | Deductions applied and payments made | | | (683 | ) | | | (158 | ) | | | (380 | ) | | | (1,221 | ) | Ending balance at December 31, 2019 | | $ | 1,618 | | | $ | 271 | | | $ | 352 | | | $ | 2,241 | |
Returns are recorded as a reduction of accounts receivable on the right to developcondensed consolidated balance sheets. Chargebacks, discounts and commercializefees and rebates are recorded as a component of accrued expenses on the Latanoprost Product on its own or with a partner.condensed consolidated balance sheets (See Note 7). Bausch & LombLicense and Collaboration Agreements and Royalty Income
Alimera Pursuant to a licensing and development agreement, as amended, Bausch & LombAlimera Sciences, Inc. has a worldwide exclusive license to develop, make, market and sell RetisertILUVIEN in return for royalties based on sales. Royaltysales and patent fee reimbursements. Total revenue was $1.7 million and $2.1 million for the years ended December 31, 2020 and 2019, respectively. In addition to patent fee reimbursements in
those periods, the Company recorded royalty income totaled approximately $1.0$1.7 million and $2.0 million for the years ended December 31, 2020 and 2019, respectively. SWK Royalty Purchase Agreement On December 17, 2020, the Company entered into a royalty purchase agreement (the “RPA”) with SWK Funding LLC (“SWK”). Under the RPA, the Company sold its right to receive royalty payments on future sales of products subject to the Amended Alimera Agreement for an upfront cash payment of $16.5 million. Except for the rights to the royalties, the Company retains all rights and obligations under the Amended Alimera Agreement, pursuant to which, Alimera owns worldwide rights to the Company’s Durasert technology in fiscal 2018, $970,000ILUVIEN for DME and rights for ILUVIEN (currently marketed by the Company as YUTIQ in fiscal 2017the U.S.) for non-infectious posterior uveitis in the EMEA. Alimera has the sole rights to utilize the intellectual property developed under the Amended Alimera Agreement. There has been no intellectual property developed jointly by Alimera and approximately $1.2the Company as part of the Amended Alimera Agreement. The Company cannot utilize the intellectual property for the indication licensed to Alimera in order to manufacture and sell ILUVIEN. The Company’s ongoing efforts under the Amended Alimera Agreement will consist of continuing to maintain and enforce its patents as well as providing safety data and regulatory support as necessary. None of these obligations require significant efforts on the part of the Company with respect to the generation of sales in the market. The Company will only be required to expend more extensive efforts if litigation were to arise that requires the Company to protect its patents rights pursuant to the terms of the Amended Alimera Agreement. Historically, such a defense has not been required. Similarly, regulatory support and safety data is only provided on an ad-hoc basis depending on the regulatory requests, which has been minimal historically. It remains Alimera’s sole responsibility to manufacture, actively market and promote the products under the Amended Alimera Agreement to generate the sales, which ultimately generate the royalties to be paid to SWK. The Company classified the proceeds received from SWK as deferred revenue, to be recognized as revenue under the units-of-revenue method over the life of the RPA because of the Company’s limited continuing involvement in the Amended Alimera Agreement. SWK has no recourse and the Company assumes no credit risk in event that Alimera fails to make a royalty payment. The Company must only forward all material correspondence from Alimera to SWK, including royalty reports, notices and any other correspondence with respect to royalties to SWK. SWK has the right to audit and inspect the books and records pertaining to net sales and royalties under the Amended Alimera Agreement. Neither the Company nor SWK has the unilateral ability to cancel the transaction. There is no cap or limitation on the royalties to be received by SWK in the future and its return will reflect all royalties paid by Alimera. Because the transaction was structured as a non-cancellable sale, the Company does not have significant continuing involvement in the generation of the cash flows due to SWK and there is no limitation on the rates of return to SWK, the Company recorded the total proceeds of $16.5 million in fiscal 2016. Accounts receivableas deferred revenue under royalty sale agreement. The deferred revenue is being recognized as revenue over the life of the RPA under the "units-of-revenue" method. Under this method, amortization for a reporting period is calculated by computing a ratio of the proceeds received from Bausch & Lomb totaled $306,000 at June 30, 2018SWK to the payments expected to be made by Alimera to SWK over the term of the Amended Alimera Agreement, and $246,000 at June 30, 2017.then applying that ratio to the period’s cash payment. As of December 31, 2020, the Company classified $885,000 and $15.6 million as current and non-current deferred revenue recognized under royalty sale agreement, respectively. As 0 royalty payments have yet been paid to SWK, the Company has 0t recognized any revenue related to the RPA for the period ended December 31, 2020. OncoSil MedicalMedical The Company entered into an exclusive, worldwide royalty-bearing license agreement in December 2012, amended and restated in March 2013, with OncoSil Medical UK Limited (f/k/a Enigma Therapeutics Limited), a wholly-owned subsidiary of OncoSil Medical Ltd (“OncoSil”) for the development of BrachySil, the Company’s previously developedprevious product candidate for the treatment of pancreatic and other types of cancer. The Company received an upfront fee of $100,000 and is entitled to 8% sales-based royalties, 20% of sublicense consideration and milestone payments based on aggregate product sales. OncoSil is obligated to pay an annual license maintenance fee of $100,000 by the end of each calendar year, the most recent of which was received in December 2017.2020. For each calendar year commencing with 2014, the Company is entitled to receive reimbursement of any patent maintenance costs, sales-based royalties andsub-licensee sales-based royalties earned, but only to the extent such amounts, in the aggregate, exceed the $100,000 annual license maintenance fee. In March 2020, the U.S. Food and Drug Administration granted Breakthrough Device Designation for the OncoSil™ device for treatment of unresectable locally advanced pancreatic cancer (LAPC) in combination with chemotherapy. In April 2020, the British Standards Institute (BSI) grants European CE marking for the OncoSil™ System and designates OncoSil™ a breakthrough device for the treatment of locally advanced pancreatic cancer (LAPC) in combination with chemotherapy. As of June 30, 2018,December 31, 2020, OncoSil has not received regulatory approval in any jurisdiction, although an application for CE Mark approval in Europe continues to be pending. the EU, United Kingdom, Switzerland, Singapore, Malaysia and New Zealand. The Company has no0 consequential performance obligations under the OncoSil license agreement. For the years ended December 31, 2020 and 2019, revenue of $100,000 and $100,000 was recorded for this agreement, respectively.
Ocumension Therapeutics In November 2018, the Company entered into an exclusive license agreement with Ocumension Therapeutics (“Ocumension”) for the development and accordingly, any amountscommercialization of its three-year micro insert using the Durasert technology for the treatment of chronic non-infectious uveitis affecting the posterior segment of the eye (YUTIQ in the U.S.) in Mainland China, Hong Kong, Macau and Taiwan. The Company received a one-time upfront payment of $1.75 million from Ocumension and is eligible to receive up to (i) $7.25 million upon the achievement by Ocumension of certain prescribed development and regulatory milestones, and (ii) $3.0 million commercial sales-based milestones. In addition, the Company is entitled to receive mid-single digit sales-based royalties. Ocumension has also received a special approval by the Hainan Province People's Government to market this product for chronic, non-infectious posterior segment uveitis in the Hainan Bo Ao Lecheng International Medical Tourism Pilot Zone (“Hainan Pilot Zone”). In March 2019, the Company entered into a Memorandum of Understanding (“2019 MOU”), pursuant to which, the Company will supply product for the clinical trials and Hainan Pilot Zone use. Paralleling to Ocumension’s normal registration process of the product with the Chinese Regulatory Authorities, the 2019 MOU modified the Company’s entitlement to the development and regulatory milestones of up to $7.25 million under the license agreement to product supply milestones or development milestones, whichever comes first, totaling up to $7.25 million. In August 2019, the Company began shipping this product to Ocumension. The Company was required to provide a fixed number of hours of technical assistance support to Ocumension at no cost, which support has been completed and no future performance obligation exists. Ocumension is responsible for all development, regulatory and commercial costs, including any additional technical assistance requested. Ocumension has a first right of negotiation for an additional exclusive license to the Company’s shorter-duration line extension candidate for this indication. In August 2019, the Company received a $1.0 million development milestone payment from Ocumension triggered by the approval of its Investigational New Drug (“IND”) in China for this program. The IND allows the importation of finished product into China for use in a clinical trial to support regulatory filing. In January 2020, the Company entered into an exclusive license agreement with Ocumension for the development and commercialization in Mainland China, Hong Kong, Macau and Taiwan of DEXYCU for the treatment of post-operative inflammation following ocular surgery. Pursuant to the terms of the license agreement, the Company received upfront payments of $2.0 million from Ocumension in February 2020 and will be eligible to receive up to (i) $6.0 million upon the achievement by Ocumension of certain prescribed development and regulatory milestones, and (ii) $6.0 million commercial sales-based milestones. In addition, the Company is entitled to receive mid-single digit sales-based royalties. In exchange, Ocumension will receive exclusive rights to develop and commercialize DEXYCU in Mainland China, Hong Kong, Macau and Taiwan, at its own cost and expense with the Company supplying product for clinical trials and commercial sale. In addition, Ocumension will receive a fixed number of hours of technical assistance support from the Company at no cost. In August 2020, the Company entered into a Memorandum of Understanding (“2020 MOU”), pursuant to which, the Company received a one-time non-refundable payment of $9.5 million (the “Accelerated Milestone Payment”) from Ocumension as a full and final payment of the combined remaining development, regulatory and sales milestone payments under the Company’s license agreements with Ocumension for the treatment of chronic non-infectious uveitis affecting the posterior segment of the eye and for the treatment of post-operative inflammation following ocular surgery, respectively. Upon payment of the Accelerated Milestone Payment, the remaining $11.75 million in combined remaining development and sales milestone payments under the Company’s original license agreement are recognized as revenue onwith Ocumension upon the earlierachievement by Ocumension of receipt or when collectability is reasonably assured. Revenue related(i) remaining development and regulatory milestones of $6.25 million and commercial sales-based milestones of $3.0 million for the development and commercialization of its three-year micro insert using the Durasert technology for the treatment of chronic non-infectious uveitis affecting the posterior segment of the eye; and (ii) $6.0 million upon the achievement by Ocumension of certain prescribed development and regulatory milestones, and $6.0 million commercial sales-based milestones for the development and commercialization in Mainland China, Hong Kong, Macau and Taiwan of DEXYCU for the treatment of post-operative inflammation following ocular surgery, totaling up to $21.25 million, were permanently extinguished and will no longer be due and owed to the OncoSil agreement totaled $100,000Company. In exchange, Ocumension also received exclusive rights to develop and commercialize YUTIQ and DEXYCU products under its own brand names in eachSouth Korea and other jurisdictions across Southeast Asia in Brunei, Burma (Myanmar), Cambodia, Timor-Leste, Indonesia, Laos, Malaysia, the Philippines, Singapore, Thailand and Vietnam, at its own cost and expense with the Company supplying product for clinical trials and commercial sale. The Company continues to be entitled to royalties on future product sales by Ocumension. Other than a fixed number of fiscal 2018, fiscal 2017hours of technical assistance support to be provided at no cost by the Company, Ocumension is responsible for all development, regulatory and fiscal 2016. At June 30, 2018, nocommercial costs, including any additional technical assistance requested. All technical assistance was provided during 2020. The Chief Executive Officer of Ocumension became a director of the Company starting December 31, 2020 (See Note 10), at which time, Ocumension became a related party of the Company.
During the years ended December 31, 2020 and 2019, the Company recognized approximately $11.5 million and $1.0 million of license and collaboration revenue, respectively, in addition to $213,000 and $91,000 of revenue from product sales, respectively. As of December 31, 2020 and 2019, 0 deferred revenue was recorded for this agreement.agreement, respectively. Feasibility Study AgreementsThe Company recorded sales-based royalty expense of $1.3 million during the year ended December 31, 2020, with respect to partnering income equal to 20% of DEXYCU share of the Accelerated Milestone Payment received in August 2020 and upfront payment received in February 2020 from Ocumension, in connection with the Icon acquisition in March 2018. NaN sales-based royalty expense was recorded during the year ended December 31, 2019.
Research Collaborations The Company from time to time enters into funded agreements to evaluate the potential use of its technology systems for sustained release of third partythird-party drug candidates in the treatment of various diseases. Consideration received is generally recognized as revenue over the term of the research collaborations (including feasibility study agreement.agreements). Revenue recognition for consideration, if any, related to a license option right is assessed based on the terms of any such future license agreement or is otherwise recognized at the completion of the research collaborations (including feasibility study agreement.agreements). Revenues under research collaborations (including feasibility study agreementsagreements) totaled $1.1 million in fiscal 2018, $211,000 in fiscal 2017$255,000 and $33,000 in fiscal 2016.$135,000 for the years ended December 31, 2020 and 2019, respectively. At December 31, 2020 and 2019, $60,000 and $15,000 deferred revenue was recorded for the research collaborations (including feasibility study agreements), respectively. Inventory consisted of the following (in thousands): | | December 31, 2020 | | | December 31, 2019 | | Raw materials | | $ | 2,664 | | | $ | 1,476 | | Work in process | | | 747 | | | | 346 | | Finished goods | | | 1,926 | | | | 316 | | Total inventory | | $ | 5,337 | | | $ | 2,138 | |
The reconciliation of intangible assets for the years ended June 30, 2018December 31, 2020 and 2017 was as follows2019 (in thousands): | | | | | | | | | | | | June 30, | | | | | 2018 | | | 2017 | | Patented technologies | | | | | | | | | Gross carrying amount at beginning of year | | $ | 35,610 | | | $ | 36,196 | | Acquisition of Icon Bioscience Inc. | | | 31,973 | | | | — | | Foreign currency translation adjustments | | | 739 | | | | (586 | ) | | | | | | | | | | Gross carrying amount at end of year | | | 68,322 | | | | 35,610 | | | | | | | | | | | Accumulated amortization at beginning of year | | | (35,246 | ) | | | (35,094 | ) | Amortization expense | | | (981 | ) | | | (724 | ) | Foreign currency translation adjustments | | | (737 | ) | | | 572 | | | | | | | | | | | Accumulated amortization at end of year | | | (36,964 | ) | | | (35,246 | ) | | | | | | | | | | Net book value at end of year | | $ | 31,358 | | | $ | 364 | | | | | | | | | | |
| | Year Ended | | | Year Ended | | | | December 31, | | | December 31, | | | | 2020 | | | 2019 | | Patented technologies | | | | | | | | | Gross carrying amount at beginning of period | | $ | 68,322 | | | $ | 68,322 | | Gross carrying amount at end of period | | | 68,322 | | | | 68,322 | | Accumulated amortization at beginning of period | | | (40,653 | ) | | | (38,193 | ) | Amortization expense | | | (2,460 | ) | | | (2,460 | ) | Accumulated amortization at end of period | | | (43,113 | ) | | | (40,653 | ) | Net book value at end of period | | $ | 25,209 | | | $ | 27,669 | |
The net book value of the Company’s intangible assets at June 30, 2018December 31, 2020 and 20172019 is summarized as follows (in thousands): | | | | | | | | | | | Estimated | | | | | | | | | | | | | Remaining | | | | | | | | | | | | | Useful Life at | | | | | | | | | | December 31, | | | December 31, | | | December 31, | | | | | June 30, | | | Estimated
Remaining
Useful Life at
June 30, 2018 | | | 2020 | | | 2019 | | | 2020 | | | | | 2018 | | | 2017 | | | (Years) | | | | | | | | | | | (Years) | | Patented technologies | | | | | | | | | | | | | | | | | | | DEXYCU / Verisome | | $ | 31,358 | | | | — | | | | 12.75 | | | $ | 25,209 | | | $ | 27,669 | | | | 10.25 | | Durasert | | | — | | | | 265 | | | | — | | | Tethadur | | | — | | | | 99 | | | | — | | | | | | | | | | | | | $ | 25,209 | | | $ | 27,669 | | | | | | | | $ | 31,358 | | | $ | 364 | | | | | | | | | | | | | | |
The Company amortizes its intangible assets with finite lives on a straight-line basis over their respective estimated useful lives. Amortization expense for intangible assets totaled $981,000$2.5 million in fiscal 2018, $724,000 in fiscal 2017each of the two years ended December 31, 2020 and $756,000 in fiscal 2016.2019, respectively. In connection with the Icon Acquisition, (see Note 3), the initial purchase price of $32.0 million was attributed to the DEXYCU product intangible asset. This finite-lived intangible asset is being amortized on a straight-line basis over its expected remaining useful life estimated to be 13of 10.25 years at the rate of approximately $2.5 million per year. Amortization expense was reported as a component of cost of sales for the year ended December 31, 2020 and 2019, respectively. 6. | Property and Equipment, Net |
Property and equipment, net consisted of the following (in thousands): | | | | June 30, | | | December 31, | | | December 31, | | | | | 2018 | | 2017 | | | 2020 | | | 2019 | | Property and equipment | | $ | 805 | | | $ | 698 | | | $ | 1,403 | | | $ | 1,095 | | Leasehold improvements | | | 101 | | | 101 | | | | 255 | | | | 101 | | | | | | | | | | Gross property and equipment | | | 906 | | | 799 | | | | 1,658 | | | | 1,196 | | Accumulated depreciation and amortization | | | (653 | ) | | (486 | ) | | | (1,028 | ) | | | (839 | ) | | | | | | | | | $ | 630 | | | $ | 357 | | | | $ | 253 | | | $ | 313 | | | | | | | | | | |
Depreciation expense totaled $167,000$189,000 and $144,000 in fiscal 2018, $91,000 in fiscal 2017the years ended December 31, 2020 and $152,000 in fiscal 2016.2019, respectively. Accrued expenses consisted of the following (in thousands): | | | | | | December 31, | | | December 31, | | | | | June 30, | | | 2020 | | | 2019 | | | | | 2018 | | | 2017 | | | Personnel costs | | | $ | 5,686 | | | $ | 3,263 | | Clinical trial costs | | $ | 742 | | | $ | 1,984 | | | | 0 | | | | 345 | | Personnel costs | | | 1,763 | | | | 1,632 | | | Professional fees | | | 926 | | | | 590 | | | | 647 | | | | 700 | | Interest | | | 254 | | | | — | | | Sales chargebacks, rebates and other revenue reserves | | | | 1,109 | | | | 1,889 | | Other | | | 38 | | | | 18 | | | | 1,003 | | | | 635 | | | | | | | | | | $ | 8,445 | | | $ | 6,832 | | | | $ | 3,723 | | | $ | 4,224 | | | | | | | | | | |
In January 2017,
On May 17, 2018, the Company entered into retention bonus agreements with five employees. Under these agreements (a) cash payments totaling $319,000 were made on December 22, 2017amended the lease for its headquarters in Watertown, Massachusetts. The original five-year lease for approximately 13,650 square feet of combined office and (b) subject to continuing employment, a total of 305,616 restricted stock units (“RSUs”) of an equal value were granted at that date based on a closing share price of $1.045 per share with aone-year vesting period. Included in personnel costs in the above table is $160,000 at June 30, 2017, representing pro rata accrual of the cash bonus component. In July 2016, the Company announced its plan to consolidate all research and development activities in its U.S. facility. Following employee consultations under local U.K. law, the Company determined to close its U.K. research facility and terminated the employment of its U.K. employees. The U.K. facility lease,laboratory space was set to expire in April 2019. Under the amendment, the Company leased an additional 6,590 square feet of rentable area of the building, with a commencement date of September 10, 2018. The amendment extended the term of the lease for the combined space through May 31, 2025. The landlord agreed to provide the Company a construction allowance of up to $670,750 to be applied toward the aggregate work completed on August 31, 2016,the total space. The Company has an option to further extend the term of the lease for one additional five-year period. Per the terms of the lease agreement, the Company does not have a residual value guarantee. The Company previously provided a cash-collateralized $150,000 irrevocable standby letter of credit as security for the Company’s obligations under the lease, which was extended through November 30, 2016the period that is four months beyond the expiration date of the amended lease. The Company will also be required to facilitatepay its proportionate share of certain operating costs and property taxes applicable to the leased premises in excess of new base year amounts.
In July 2017, the Company leased approximately 3,000 square feet of office space in Basking Ridge, New Jersey under a lease term extending through June 2022, with 2 five-year renewal options at 95% of the then-prevailing market rates. In addition to base rent, the Company is obligated to pay its proportionate share of building operating expenses and real estate taxes in excess of base year amounts. In June 2018, the Company subleased an orderly transitionadditional 1,381 square feet of adjoining space from Caladrius Biosciences, Inc. (“Caladrius”) through May 2022. The Chief Executive Officer of Caladrius was a director of the Company through June 2020. Per the terms of the lease and sublease agreements, the Company does not have any residual value guarantees. The Company identified and assessed the following significant assumptions in recognizing its right-of-use (“ROU”) assets and corresponding lease liabilities: | • | As the Company’s leases do not provide an implicit rate, the Company estimated the incremental borrowing rate in calculating the present value of the lease payments. The Company utilized the borrowing rate under its existing 5-year term loan facility (see Note 9) as the discount rate. |
| • | Since the Company elected to account for each lease component and its associated non-lease components as a single combined component, all contract consideration was allocated to the combined lease component. |
| • | The expected lease terms include noncancelable lease periods. Renewal option periods have not been included in the determination of the lease terms as they are not deemed reasonably certain of exercise. |
| • | Variable lease payments, such as common area maintenance, real estate taxes and property insurance are not included in the determination of the lease’s ROU asset or lease liability. |
As of December 31, 2020, the weighted average remaining term of the Company’s operating leases was 4.3 years and the required restorationlease liabilities arising from obtaining ROU assets reflect a weighted average discount rate of 12.5%. Supplemental balance sheet information related to operating leases as of December 31, 2020 and 2019, respectively are as follows (in thousands): | December 31, | | | December 31, | | | 2020 | | | 2019 | | Other current liabilities - operating lease current portion | $ | 568 | | | $ | 481 | | Operating lease liabilities – noncurrent portion | | 2,330 | | | | 2,898 | | Total operating lease liabilities | $ | 2,898 | | | $ | 3,379 | |
Operating lease expense recognized related to ROU assets was $852,000 and$852,000, excluding $36,000 and $36,000 of variable lease costs, during each of the premises. A summary reconciliationyears ended December 31, 2020 and 2019, respectively, and were included in general and administrative expense in the Company’s statement of comprehensive loss. Cash paid for amounts included in the restructuring costsmeasurement of operating lease liabilities was $867,000 and $808,000 for the years ended December 31, 2020 and 2019, respectively. The Company is a party to 2 finance leases for laboratory equipment. The equipment leases expire in December 2021 and December 2022, respectively.
Supplemental balance sheet information related to the finance lease as of December 31, 2020 is as follows (in thousands): | | | | | | | | | | | | | | | | | | | | Balance at
June 30, 2016 | | | Charged to
Expense | | | Payments | | | Balance at
June 30, 2017 | | Termination benefits | | $ | 118 | | | $ | 273 | | | $ | (391 | ) | | $ | — | | Facility closure | | | 40 | | | | 73 | | | | (113 | ) | | | — | | Other | | | 29 | | | | 126 | | | | (155 | ) | | | — | | | | | | | | | | | | | | | | | | | | | $ | 187 | | | $ | 472 | | | $ | (659 | ) | | $ | — | | | | | | | | | | | | | | | | | | |
| December 31, | | | 2020 | | Property and equipment, at cost | $ | 239 | | Accumulated amortization | | (52 | ) | Property and equipment, net | $ | 187 | | | | | | Other current liabilities – finance lease current portion | $ | 119 | | Other long-term liabilities | | 71 | | Total finance lease liabilities | $ | 190 | |
The components of finance lease expense recognized during the year ended December 31, 2020 related to ROU assets was $52,000. Interest on lease liabilities was $9,000 during the year ended December 31, 2020. Cash paid for amounts included in the measurement of finance lease liabilities was operating cash flows of $9,000 and financing cash flows of $49,000 during the year ended December 31, 2020, respectively. The Company had 0 finance lease in 2019. As of December 31, 2020, the weighted average remaining term of the Company’s finance lease was 1.7 years and the lease liabilities arising from obtaining ROU assets reflect a weighted average discount rate of 12.5%. The Company’s total future minimum lease payments under non-cancellable leases at December 31, 2020 were as follows (in thousands): | Operating Leases | | | Finance Leases | | 2021 | | 889 | | | | 135 | | 2022 | | 849 | | | | 75 | | 2023 | | 815 | | | | — | | 2024 | | 830 | | | | — | | 2025 | 346 | | | 0 | | Total lease payments | $ | 3,729 | | | $ | 210 | | Less imputed interest | | (831 | ) | | | (20 | ) | Total | $ | 2,898 | | | $ | 190 | |
Paycheck Protection Program Loan On April 8, 2020, the Company applied to Silicon Valley Bank (the “SVB”) for a Paycheck Protection Program Loan (the “PPP Loan”) of $2.0 million that is administered by the U.S. Small Business Administration (the “SBA”), under the Coronavirus Aid, Relief and Economic Security Act (the “CARES Act”). On April 22, 2020, the PPP Loan was approved and the Company received the PPP Loan proceeds. The PPP Loan bears interest at a fixed rate of 1.0% per annum and has a two-year term that matures on April 21, 2022. Monthly principal and interest payments will commence on November 21, 2020. The PPP Loan may be forgiven partially or fully if the PPP Loan proceeds are used for covered payroll costs, rent and utility costs and the maintenance of employee and compensation levels. The Paycheck Protection Program Flexibility Act of 2020 (the “PPP Flexibility Act”), enacted on June 5, 2020, amended the Paycheck Protection Program, among others, as follows: (i) extended the covered period from 8 weeks to the earlier of 24 weeks from the date the PPP Loan is originated and December 31, 2020, during which PPP funds needed to be expended in order to be forgiven. A borrower may submit a loan forgiveness application any time on or before the maturity date of the loan – including before the end of the covered period – if the borrower has used all of the loan proceeds for which the borrower is requesting forgiveness; (ii) at least 60% of PPP funds must be spent on payroll costs, with the remaining 40% available to spend on other eligible expenses; (iii) payments are deferred until the date on which the amount of forgiveness determined is remitted to the lender. If a borrower fails to seek forgiveness within 10 months after the last day of its covered period, then payments will begin on the date that is 10 months after
the last day of the covered period. In addition, the PPP Flexibility Act modified the CARES Act by increasing the maturity date for loans made after the effective date from two years to a minimum maturity of five years from the date on which the borrower applies for loan forgiveness. Existing PPP loans made before the new legislation retain their original two-year term, but may be renegotiated between a lender and a borrower to match the 5-year term permitted under the PPP Flexibility Act. The Company recorded approximately $472,000 of restructuring costs during fiscal 2017 to research and development expense. These costs consisted of (i) $273,000 of additional employee severance for discretionary termination benefits upon notificationused all of the affected employeesloan proceeds from the PPP Loan to pay expenses during the covered period that the Company believes were for eligible purposes. On September 25, 2020, the Company submitted an application to SVB for full loan forgiveness. No assurance is provided that the Company will obtain forgiveness of the PPP Loan in whole or in part. As of the date of this filing, the application for the PPP Loan forgiveness is still pending review. The PPP Loan proceeds of $2.0 million were recorded as a loan in accordance with ASC 420,Exit or Disposal Cost Obligations;470, Debt, and (ii) $199,000included in long-term debt in the Company’s balance sheet as of professional fees, travel and lease extension costs. In addition,December 31, 2020. Accrued interest expense based on the stated interest rate of 1% per annum was $14,000 for the first quarter of fiscal 2017, the Company recorded $99,000 ofnon-cash stock-based compensation expense in connection with the extension, through June 30, 2017, of the exercise period for all vested stock options held by the U.K. employees at Julyyear ended December 31, 2016 and a $133,000 credit to stock-based compensation expense to account for forfeitures of allnon-vested stock options at that date.2020.
The Company paid all of the restructuring costs associated with the plan of consolidation as of March 31, 2017.
9. | CRG Term Loan Agreement |
On March 28, 2018February 13, 2019 (the “Closing“CRG Closing Date”), the Company entered into a Creditthe CRG Loan Agreement (the “Credit Agreement”) among the Company, as borrower, SWK FundingCRG Servicing LLC, as administrative agent and collateral agent (the “Agent”),and the lenders party thereto from time to time (the “Lenders”), providing for a senior secured term loan of up to $20$60 million (the “Loan”“CRG Loan”). On the CRG Closing Date, $35 million of the CRG Loan was advanced (the “CRG Initial Advance”). The Company utilized the proceeds from the CRG Initial Advance for the repayment in full of all outstanding obligations under its prior credit agreement (the “SWK Credit Agreement”) with SWK Funding LLC (“SWK”). In April 2019, the Company exercised its option to borrow an additional $15 million of the CRG Loan was advanced (the “Initial“CRG Second Advance”). The remaining $5 million of the Loan was advanced on June 26, 2018 following satisfaction of the Minimum Capital Raise (as defined in the Credit Agreement) (the “Additional Advance”). The LoanCompany may be increased bydraw up to an additional $10 million, upon the request of the Company, subject to the Agent obtaining additional loan commitmentsachievement of prescribed three-month trailing product revenues of YUTIQ and satisfaction of certain conditions in the Credit Agreement.DEXYCU on or before March 31, 2020. The CRG Loan is due and payable on March 27,December 31, 2023 (the “Maturity Date”). The CRG Loan bears interest at a fixed rate of 12.5% per annum rate of the three-month LIBOR rate (subject to a 1.5% floor) plus 10.50%. The Credit Agreement permits the Company to pay interest onlypayable in arrears on the principal amount for the first eight payments (payments are due on a quarterly basis commencing May 15, 2018). Following the interest-only period, thelast business day of each calendar quarter. The Company will beis required to make quarterly, interest only payments ofuntil the Maturity Date. So long as no default has occurred and is continuing, the Company may elect on each applicable interest plus repaymentspayment date to pay 2.5% of the principal in an aggregate12.5% per annum interest as Paid In-Kind (“PIK”), whereby such PIK amount of up to $1.67 million per quarter (the “Quarterly Principal Repayment Cap”). Subjectwould be added to the Quarterly Principal Repayment Cap,aggregate principal amount and accrue interest at 12.5% per annum. Through December 31, 2020, total PIK amounts of $2.0 million have been added to the amount of any quarterly principal payments during any fiscal yearbalance of the Company is based on (x) a percentage of theyear-to-date net revenue of the Company through the end of such quarter less (y) any prior quarterly principal and interest payments made during such fiscal year.CRG Loan. In addition, the Company paidis required to pay an upfront fee of 1.5% of amounts borrowed under the aggregate principal amount ofCRG Loan (excluding any paid-in-kind amounts), which is payable as amounts are advanced under the CRG Loan. The Company iswill also be required to pay an exit fee equal to 6% of (i) the aggregate principal amountamounts advanced and (ii) PIK amounts issued, under the Credit Agreement (the “Exit Fee”), which amount is included in other long-term liabilities inCRG Loan Agreement. In connection with the accompanying consolidated balance sheet.CRG Initial Advance, a 1.5% financing fee of $525,000 and an expense reimbursement of $350,000 were deducted from the net borrowing proceeds. In connection with the CRG Second Advance, a 1.5% financing fee of $225,000 was deducted from the net borrowing proceeds. Upon the occurrence of a bankruptcy-related event of default, all amounts outstanding with respect to the CRG Loan become due and payable immediately, and upon the occurrence of any other Event of Default (as defined in the CreditCRG Loan Agreement), all or any amounts outstanding with respect to the CRG Loan may become due and payable upon request of the Agent or majority Lenders. Additionally, subjectSubject to certain exceptions, the Company is required to make mandatory prepayments of the CRG Loan with the proceeds of assetassets sales and insurance proceeds. Thein the event of a change of control of the Company. In addition, the Company may make a voluntary prepayment of the CRG Loan, in whole but notor in part, at any time on or after the first anniversary of the Closing Date.time. All mandatory and voluntary prepayments of the CRG Loan are subject to the payment of prepayment premiums as follows: (i) in the case of mandatory prepayments, if prepayment occurs prior to the first anniversary of the Closing Date, a customary make-whole amount equal to the amount of interest that would have accrued on the principal amount so prepaid had it remained outstanding through the first anniversary of the Closing Date, (ii) if prepayment occurs on or after the first anniversary of the Closing Date, but prior to the second anniversary of the Closing Date, 6%December 31, 2019, an amount equal to 10% of the aggregate outstanding principal amount of the CRG Loan being prepaid, (ii) if prepayment occurs after December 31, 2019 and on or prior to December 31, 2020, 5% of the aggregate outstanding principal amount of the CRG Loan being prepaid, which was waived on December 17, 2020 when the Company paid $15.0 million against the CRG Loan obligations in connection with the consummation of the RPA agreement (see Note 3), and (iii) if prepayment occurs after December 31, 2020 and on or after the second anniversary of the Closing Date, but prior to the third anniversary of the Closing Date,December 31, 2021, an amount equal to 1%3% of the aggregate outstanding principal amount of the Loan being prepaid. NoNaN prepayment premium is due on any principal prepaid on or after the third anniversaryDecember 31, 2021. Certain of the Company’s existing and future subsidiaries are guaranteeing the obligations of the Company under the CRG Loan Agreement. The obligations of the Company under the CRG Loan Agreement and the guarantee of such obligations are secured by a pledge of substantially all of the Company’s and the guarantors’ assets.
The CRG Loan Agreement contains affirmative and negative covenants customary for financings of this type, including limitations on our and our subsidiaries’ abilities, among other things, to incur additional debt, grant or permit additional liens, make investments and acquisitions, merge or consolidate with others, dispose of assets, pay dividends and distributions and enter into affiliate transactions, in each case, subject to certain exceptions. In addition, the CRG Loan Agreement contains the following financial covenants requiring the Company and the Guarantors to maintain: | • | liquidity in an amount which shall exceed the greater of (i) $5 million and (ii) to the extent the Company has incurred certain permitted debt, the minimum cash balance, if any, required of the Company by the creditors of such permitted debt; and |
| • | annual minimum product revenue from YUTIQ and DEXYCU: (i) for the twelve-month period beginning on January 1, 2019 and ending on December 31, 2019, of at least $15 million, (ii) for the twelve-month period beginning on January 1, 2020 and ending on December 31, 2020, of at least $45 million, (iii) for the twelve-month period beginning on January 1, 2021 and ending on December 31, 2021, of at least $80 million and (iv) for the twelve-month period beginning on January 1, 2022 and ending on December 31, 2022, of at least $90 million. |
In November 2019, CRG waived the financial covenant associated with the Company’s revenue derived from sales of its products, DEXYCU and YUTIQ, for the twelve-month period ending December 31, 2019. In October 2020, CRG (i) waived the financial covenant associated with the Company’s revenue derived from sales of its products, DEXYCU and YUTIQ, for the twelve-month period ending December 31, 2020 and (ii) amended the financial covenant associated with the Company’s minimum product revenue to $45 million from $80 million, for the twelve-month period ending December 31, 2021. There were no other material changes to the Loan Agreement and the Company incurred 0 incremental charges for the issuance of the waivers. The total debt discount related to the CRG Initial Advance was approximately $3.2 million and consisted of (i) the accrual of a $2.1 million exit fee; (ii) the $525,000 upfront fee; and (iii) $591,000 of legal and other transaction costs. This amount is being amortized as additional interest expense over the term of the Loan using the effective interest rate method. The total debt discount related to the CRG Second Advance was approximately $1.1 million and consisted of (i) the accrual of a $900,000 exit fee; and (ii) the $225,000 upfront fee. This amount is being amortized as additional interest expense over the term of the Loan using the effective interest rate method. On December 17, 2020, the Company paid $15.0 million against the CRG Loan obligations in connection with the consummation of the RPA agreement (see Note 3). This payment included (i) a $13.8 million principal portion of the CRG Loan (ii) the $828,000 Exit Fee, and (iii) accrued and unpaid interest of $378,000 through that date. In connection with the partial prepayment of the CRG Loan, the Company recorded a loss on partial extinguishment of debt of $905,000 in the year ended December 31, 2020, associated with the write-off of the remaining balance of unamortized debt discount related to the partial prepayment of the CRG Loan. Amortization of debt discount under the CRG Loan totaled $745,000 and $512,000 for the years ended December 31, 2020 and 2019, respectively. SWK Credit Agreement On March 28, 2018 (the “SWK Closing Date.Date”), the Company entered into the SWK Credit Agreement among the Company, as borrower, SWK, as agent, and the lenders party thereto from time to time, providing for a senior secured term loan of up to $20 million (the “SWK Loan”). On the SWK Closing Date, $15 million of the SWK Loan was advanced (the “SWK Initial Advance”). The remaining $5 million of the SWK Loan was advanced on June 26, 2018 (the “SWK Additional Advance”). In connection with the SWK Loan, the Company issued a warrant (the “SWK Warrant”) to the Agent to purchase (a) 409,09140,910 shares of Common Stock (the “Initial Advance Warrant Shares”) at an exercise price equal of $1.10 per share and (b) 77,7217,773 shares of Common Stock (the “Additional Advance Warrant Shares”) at an exercise price of $1.93 per share.share (see Note 10). The SWK Warrant is exercisable (i) with respect to the Initial Advance Warrant Shares, any time on or after the SWK Closing Date until the close of business on the7-year anniversary of the SWK Initial Advance and (ii) with respect to the Additional Advance Warrant Shares, any time on or after the closing of the SWK Additional Advance until the close of business on the7-year anniversary of the SWK Additional Advance. The Agent may exercise the SWK Warrant on a cashless basis at any time. In the event the Agent exercises the SWK Warrant on a cashless basis, the Company will not receive any proceeds. The Additional Advance Warrant Shares were recorded as a liability at the Closing Date and were remeasured at fair value at each reporting period until the date of the SWK Additional Advance. The aggregate fair value of the Additional Advance Warrant Shares at the Closing Date was $69,000. The Initial Advance Warrant Shares were recorded as equity on the Company’s balance sheet at their relative fair value of $284,000. The remaining $14.6 million of the proceeds received were allocated to the SWK Initial Advance term loan. Upon the closing of the SWK Additional Advance in June 2018, the Additional Advance Warrant Shares werere-valued at $87,000 and reclassified to equity.
The total debt discount related to the SWK Initial Advance was $2.1 million and was comprised of (1) $1.8 million, which included thea 1.5% upfront fee, the Exit Feea 6% exit fee (the “Exit Fee”) and legal and other transaction costs, which were ratably allocated to each of the two2 tranches of the SWK Loan based upon the total principal amount available to the Company under each tranche and (2) $353,000 related to the aggregate fair value of the Initial Advance Warrant Shares and the Additional Advance Warrant Shares. This amount iswas being amortized as additional interest expense over the term of the SWK Loan using the effective interest rate method. The total debt issue costs related to the SWK Additional Advance was $299,000 and was comprised of the allocated portions of the 1.5% upfront fee and the Exit Fee. This amount was recorded as a prepaid expense to be amortized ratably from the SWK Closing Date through December 31, 2018. Through the date of the SWK Additional Advance, $97,000 was amortized and the remaining balance of $202,000 was reclassified to debt discount.discount in June 2018. Together with the 6% Exit Fee on the SWK Additional Advance and other transaction costs, total debt discount of $652,000 associated with the SWK Additional Advance willwas to be amortized over the remaining life of the SWK Additional Advance portion of the SWK Loan using the effective interest rate method. The SWK Loan was originally scheduled to mature on March 27, 2023 and bore interest at a per annum rate of the three-month LIBOR rate (subject to a 1.5% floor) plus 10.50%. On February 13, 2019, the Company repaid the SWK Loan in connection with the consummation of the CRG Loan Agreement. In addition to repayment of the $20 million principal balance, the Company paid (i) a $1.2 million prepayment penalty, (ii) the $1.2 million Exit Fee, (iii) accrued and unpaid interest of $664,000 through that date and (iv) an additional make-whole interest payment of $306,000 covering the additional period through what would have been the first anniversary of the SWK Loan. In connection with the prepayment of the SWK Loan, the Company recorded a loss on extinguishment of debt of $3.8 million in the three months ended March 31, 2019. In addition to the prepayment penalty and make-whole interest payment amounts, the loss on extinguishment of debt included the write-off of the remaining balance of unamortized debt discount of approximately $2.3 million. Amortization of debt discount under the SWK Loan totaled $84,000 in the first quarter of 2019 through the SWK loan extinguishment date. 20182020 Equity Financing
On the Closing Date,ATM Facility
In August 2020, the Company entered into an at-the-market facility (the “ATM Facility”) with Cantor Fitzgerald & Co (“Cantor”). Pursuant to the ATM Facility, under a SecuritiesForm S-3 shelf registration statement that was declared effective by the SEC in December 2018, the Company may, at its option, offer and sell shares of its Common Stock from time to time for an aggregate offering price of up to $25.0 million. The Company will pay Cantor a commission of 3.0% of the gross proceeds from any future sales of such shares. During the year ended December 31, 2020, the Company sold 2,590,093 shares of its Common Stock at a weighted average price of $5.74 per share for gross proceeds of approximately $14.9 million. Share issue costs, including sales agent commissions, totaled $646,000 during the reporting period. Share Offering On December 31, 2020, the Company entered into a Share Purchase Agreement (the “First Tranche Securities“Share Purchase Agreement”) with EW Healthcare Partners, L.P. and EW HealthcarePartners-A, L.P. (collectively, the “First Tranche Investors”),Ocumension, pursuant to which the Company offered and sold to the First Tranche Investors an aggregate of 8,606,324Ocumension 3,010,722 shares of Common Stock, at a purchase price of $1.10approximately $5.22 per share, (the “First Tranche Purchase Price”) for aggregate gross proceeds of approximately $9.5 million (the “First Tranche Transaction”). Onwhich was the Closing Date, the Company entered into a Second Securities Purchase Agreement (the “Second Tranche Securities Purchase Agreement” and together with the First Tranche Securities Purchase Agreement, the “Securities Purchase Agreements”) with the First Tranche Investors and certain other accredited investors (collectively, the “Second Tranche Investors”), pursuant to which the Company, subject to the approval of the Company’s stockholders, would offer and sell to the Second Tranche Investors an aggregate of approximately $25.5 million of Units, subject to a maximum of 27,250,000 Units, with each Unit consisting of (a) one share of Common Stock and (b) one warrant to purchase a share of Common Stock (the “Second Tranche Transaction” and together with the First Tranche Transaction, the “Equity Transactions”).
The purchase price for each share of Common Stock issuable in the Second Tranche Transaction was defined as the lower of (a) $1.265 (which is a 15% premium to the First Tranche Purchase Price) and (b) a 20% discount to thefive-day volume weighted average price (“VWAP”) of the Common Stock as of the close of trading on December 29, 2020. The aggregate gross proceeds from the Transaction were approximately $15.7 million. Share issue costs totaled approximately $0.1 million.
In February 2020, the Company sold 1,500,000 shares of Common Stock onin an underwritten public offering at a price of $14.5 per share for gross proceeds of $21.75 million. Underwriter discounts and commissions and other share issue costs totaled approximately $1.8 million. At the Nasdaq Stock Market for the 20 trading days immediately prior to the closingAnnual Meeting of the Second Tranche Transaction; provided, however, that the purchase price could not be lower than $0.88, which is a 20% discount to the First Tranche Purchase Price. At a special meeting of stockholdersStockholders (the “Annual Meeting”) held on June 22, 2018,23, 2020, the Company’s stockholders approved the Second Tranche Transaction, following which, on June 25, 2018, the Company sold to the Second Tranche Investors an aggregate of 20,184,224 Units at a purchase price of $1.265 per Unit for gross proceeds of
approximately $25.5 million, not including any proceeds that would be received from an exercise of the warrants. In addition, the stockholders approved the adoption of an amendment to the Company’s Certificate of Incorporation, as amended, to increase the number of authorized shares of its Common Stock from 120,000,000150,000,000 shares to 150,000,000300,000,000 shares.
The Second Tranche Warrants are exercisable any time until on or prior to the close of business on the 15th business day following the date on which the holders of the Second Tranche Warrants receive written notice from the Company that CMS has announced that a newC-Code has been established for DEXYCU and will be effective at the start of the first calendar quarter after such notice. CMS has approved transitional pass-through status and reimbursement through aC-code with an effective date of October 1, 2018. Following written notice of such approval to the holders of the Second Tranche Warrants on September 7, 2018, the Second Tranche Warrants are exercisable until the close of business on September 28, 2018. The exercise price of each Second Tranche Warrant issued in the Second Tranche Transaction is an amount equal to the lower of (a) $1.43 (a 30% premium to the First Tranche Purchase Price) and (b) a 20% discount to the VWAP of the shares of Common Stock on the Nasdaq Stock Market for the 20 trading days immediately prior to the exercise of a Second Tranche Warrant; provided, however, that the exercise price cannot be lower than $0.88, which is a 20% discount to the First Tranche Purchase Price.
The Company determined thatfiled the sharesCertificate of Common Stock issued in the First Tranche Transaction and the future obligation to issue Units in the Second Tranche Transaction were freestanding instruments. The Common Stock issued in the First Tranche Transaction was recorded as equity on the Company’s Balance Sheet. The future obligation to issue Units in the Second Tranche Transaction was recorded as a liability on the Company’s Balance Sheet, subject to remeasurement at fair value at each reporting period until settled. The Company determined that the First Tranche Transaction and the Second Tranche Transaction should be accounted for as a single transaction. Accordingly, the total consideration received on the Closing Date of $9.5 million was first allocated to the future obligation to issue Units in the Second Tranche Transaction at fair value as of the Closing Date, with the residual amount allocated to the Common Stock issued in the First Tranche Transaction. Further, issuance costs of $343,000 were allocated to each of the freestanding instruments on the basis of relative fair value. A net amount of approximately $4.6 million was allocated to the Common Stock issued in the First Tranche Transaction and the future obligation to issue Units in the Second Tranche Transaction, respectively, as of the Closing Date. As of March 31, 2018, the fair value of the Second Tranche Transaction liability was approximately $6.9 million and the Company recorded the $2.2 million change in fair value for the quarter ended March 31, 2018.
The future obligation to issue Units in the second tranche transaction was revalued immediately prior to the Second Tranche Transaction and resulted in a change in fair value of approximately $22.2 million. Upon consummation of the Second Tranche Transaction, the resulting derivative liability balance of approximately $29.1 million was reclassified to equity.
The Company determined that the Second Tranche Warrants are considered puttable warrants that represent an obligation that is indexed to repurchasing the Company’s shares and may require a transfer of assets that require classification as liabilities. The initial valuation of the Second Tranche WarrantsAmendment on June 25, 2018 of approximately $18.2 million wasre-measured at the balance sheet date, resulting in a change in fair value of derivative of approximately $1.6 million and a derivative liability balance of $19.8 million at June 30, 2018.23, 2020.
2019 Equity Financing ATM Facility In February 2017,January 2019, the Company entered into an at-the-market program (the “ATM Program”). Pursuant to the ATM program pursuant to which,Program, under itsa FormS-3 shelf registration statement that was declared effective by the SEC in December 2018, the Company may, at its option, offer and sell shares of its Common Stock from time to time for an aggregate offering price of up to $20.0 million. The Company payswill pay the sales agent a commission of up to 3.0% of the gross proceeds from the saleany future sales of such shares. The Company incurred approximately $223,000 of legal, accounting and other costs to establish and activate During the ATM program. Prior to the Company’s delisting from the Australian Securities Exchange (“ASX”) in May 2018, the Company’s ability to sell shares under the ATM program was subject to ASX listing rules, as defined, limiting the number of shares the Company could issue in any12-month period without stockholder approval, as well as other applicable rules and regulations of the ASX and Nasdaq Stock Market. During fiscal 2017 (from March 2017 through May 9, 2017),year ended December 31, 2019, the Company sold 5,100,000299,888 shares of its Common Stock under the ATM program (“ATM Shares Sold”) at a weighted average price of $1.74$15.03 per share for gross proceeds of approximately $8.9$4.5 million. Share issue costs, including sales agent commissions, totaled $244,000$221,000 during fiscal 2017. At a special meeting of stockholders held on June 27, 2017, the Company’s stockholders ratified the ATM Shares Sold, thereby refreshing the Company’s capacity to issue shares of Common Stock without prior stockholder approval under the ASX listing rules. In addition, the stockholders approved the adoption of an amendment to the Company’s Certificate of Incorporation, as amended, to increase the number of authorized shares of Common Stock from 60,000,000 shares to 120,000,000 shares.
During fiscal 2018 (from July 2017 through November 7, 2017), the Company sold 5,900,000 shares of Common Stock under the ATM program at a weighted average price of $1.23 per share for gross proceeds of approximately $7.3 million. Share issue costs, including sales agent commissions, totaled $239,000 during fiscal 2018.reporting period.
Share Offering In January 2016,April 2019, the Company sold 4,440,0001,052,650 shares of Common Stock in an underwritten public offering at a price of $4.00$19.00 per share for gross proceeds of $17.8$20.0 million. Underwriter discounts and commissions and other share issue costs totaled approximately $1.3$1.7 million. Warrants to Purchase Common Shares In connection with the sale of the Units in the Second Tranche Transaction, the Company issued the Second Tranche Warrants on June 25, 2018 to the Second Tranche Investors. The Second Tranche Warrants are exercisable at any time until on or prior to the close of business on the 15th business day following the date on which the holders of the Second Tranche Warrants receive written notice from the Company that CMS has announced that a newC-code has been established for DEXYCU and will be effective at the start of the first calendar quarter after such notice. CMS has approved transitional pass-through status and reimbursement through aC-code with an effective date of October 1, 2018. Following written notice of such approval to the holders of the Second Tranche Warrants on September 7, 2018, the Second Tranche Warrants are exercisable until the close of business on September 28, 2018. The exercise price of each Second Tranche Warrant to be issued in the Second Tranche Transaction will be an amount equal to the lower of (a) $1.43 (a 30% premium to the First Tranche Purchase Price) and (b) a 20% discount to the VWAP of the shares of the Common Stock on Nasdaq for the 20 trading days immediately prior to the exercise of a Second Tranche Warrant; provided, however, that the exercise price cannot be lower than $0.88, which is a 20% discount to the First Tranche Purchase Price.
Based on the above price formula, if exercised in full the aggregate gross proceeds from the Second Tranche Warrants will range from a minimum of approximately $17.8 million to a maximum of approximately $28.9 million.
The following table provides a reconciliation of fixed price warrants to purchase shares of the Company’s Common Stock for the years ended June 30, 2018December 31, 2020 and 2017, which excludes the Second Tranche Warrants that are subject to a variable exercise price:2019: | | | | | | | | | | | | | | | | | | | | Year Ended June 30, | | | | | 2018 | | | 2017 | | | | | Number
of
Warrants | | | Weighted
Average
Exercise
Price | | | Number
of
Warrants | | | Weighted
Average
Exercise
Price | | Balance at beginning of year | | | 623,605 | | | $ | 2.50 | | | | 623,605 | | | $ | 2.50 | | Issued | | | 486,812 | | | | 1.23 | | | | — | | | | — | | Expired | | | (623,605 | ) | | | 2.50 | | | | — | | | | — | | | | | | | | | | | | | | | | | | | Balance and exercisable at end of year | | | 486,812 | | | $ | 1.23 | | | | 623,605 | | | $ | 2.50 | | | | | | | | | | | | | | | | | | |
| | Year Ended | | | Year Ended | | | | December 31, | | | December 31, | | | | 2020 | | | 2019 | | | | Number of Warrants | | | Weighted Average Exercise Price | | | Number of Warrants | | | Weighted Average Exercise Price | | Balance at beginning of period | | | 48,683 | | | $ | 12.33 | | | | 48,683 | | | $ | 12.33 | | Expired | | | | | | | | | | | 0 | | | | 0 | | Balance and exercisable at end of period | | | 48,683 | | | $ | 12.33 | | | | 48,683 | | | $ | 12.33 | |
In connection with the LoanSWK Credit Agreement (see Note 9), the Company issued warrants (i)the SWK Warrant to purchase 409,091 shares of Common Stock(i) 40,910 Initial Advance Warrant Shares on March 28, 2018 at an exercise price of $1.10$11.00 per share with a seven-year term and (ii) to purchase 77,721 shares of Common Stock7,773 Additional Advance Warrant Shares on June 26, 2018 at an exercise price of $1.93$19.30 per share with a seven-year term. On August 7, 2017, previously issued5-year investor At December 31, 2020, the weighted average remaining life of the warrants expired unexercised.was approximately 4.28 years.
11. | Share-Based Payment Awards |
Equity Incentive Plans The 2016 Long-Term Incentive Plan (the “2016 Plan”), approved by the Company’s stockholders on December 12, 2016 (the “Adoption Date”), provides for the issuance of up to 3,000,000300,000 shares of the Company’s Common Stock reserved for issuance under the 2016 Plan plus any additional shares of the Company’s Common Stock that were available for grant under the 2008 Incentive Plan (the “2008 Plan”) at the Adoption Date or would otherwise become available for grant under the 2008 Plan as a result of subsequent termination or forfeiture of awards under the 2008 Plan. At the Company’s Annual Meeting of Stockholders held on June 30, 2018,25, 2019, the Company’s stockholders approved an amendment to the 2016 Plan to increase the number of shares authorized for issuance by 1,100,000 shares. At December 31, 2020, a total of 1,497,528603,000 shares were available for new awards. Certain inducement awards, although not awarded under the 2016 Plan or the 2008 Plan, are subject to and governed by the terms and conditions of the 2016 Plan or 2008 Plan, as applicable. Stock Options The following table provides a reconciliation of stock option activity under the Company’s equity incentive plans and for inducement awards for the year ended June 30, 2018:December 31, 2020: | | | | | | | | | | | | | | | | | | | | Number of
options | | | Weighted
Average
Exercise
Price | | | Weighted
Average
Remaining
Contractual
Life | | | Aggregate
Intrinsic
Value | | | | | | | | | | | (in years) | | | (in thousands) | | Outstanding at July 1, 2017 | | | 6,045,685 | | | $ | 3.34 | | | | | | | | | | Granted | | | 2,609,814 | | | | 1.90 | | | | | | | | | | Exercised | | | (310,900 | ) | | | 1.62 | | | | | | | | | | Forefeited | | | (1,884,355 | ) | | | 3.54 | | | | | | | | | | | | | | | | | | | | | | | | | | | Outstanding at June 30, 2018 | | | 6,460,244 | | | $ | 2.79 | | | | 7.28 | | | $ | 700 | | | | | | | | | | | | | | | | | | | Exercisable at June 30, 2018 | | | 2,906,334 | | | $ | 3.23 | | | | 4.91 | | | $ | 321 | | | | | | | | | | | | | | | | | | |
| | Number of options | | | Weighted Average Exercise Price | | | Weighted Average Remaining Contractual Life | | | Aggregate Intrinsic Value | | | | | | | | | | | | (in years) | | | (in thousands) | | Outstanding at January 1, 2020 | | | 1,090,973 | | | $ | 25.21 | | | | | | | | | | Granted | | | 415,339 | | | | 12.09 | | | | | | | | | | Exercised | | | — | | | | — | | | | | | | | | | Forfeited | | | (99,031 | ) | | | 23.27 | | | | | | | | | | Expired | | | (68,401 | ) | | | 33.58 | | | | | | | | | | Outstanding at December 31, 2020 | | | 1,338,880 | | | $ | 20.86 | | | | 7.55 | | | $ | 36 | | Exercisable at December 31, 2020 | | | 716,764 | | | $ | 25.05 | | | | 6.50 | | | $ | — | |
During the year ended June 30, 2018,
In January 2019, the Company granted 2,109,813 optionsexpanded the terms of its annual stock option grants to employeesinclude vesting ratable monthly over four years, or with 25% vesting after one year followed by ratable monthly vesting over three years. Previously, the Company’s option grants generally had ratable annual vesting over 3three years, 240,001 options tonon-executive directors withor 1-year cliff vesting, 120,000 options to two newly appointednon-executive directors with ratable annual vesting over 3 years, 40,000 additional options to onenon-executive director appointed the previous year in connection with such director’s offer letter with ratable annual vesting over 2 years and 100,000 options to an external consultant with 6.5 months cliff vesting at June 30, 2018. In accordance with ASX Listing Rules, all equityvesting. Nonemployee awards authorized by the Compensation Committee of the Boardare granted similar to the Company’s executive andnon-executive directors were subject to stockholder approval prior to the Company’s delisting from ASX, with the grant date fair value measured at the stockholder approval date and vesting measured from the Compensation Committee authorization date.employee awards. All option grants have a10-year term. Options to purchase a total of 413,000 shares of the Company’s Common Stock vested during the year ended December 31, 2020.
In determining the grant date fair value of option awards, under the equity incentive plans, the Company applied the Black-Scholes option pricing model. Based upon limited option exercise history, the Company has generally used the “simplified” method outlined in SEC Staff Accounting Bulletin No. 110 to estimate the expected life of stock option grants. Management believes that the historical volatility of the Company’s stock price on Nasdaq best represents the expected volatility over the estimated life of the option. The risk-free interest rate is based upon published U.S. Treasury yield curve rates at the date of grant corresponding to the expected life of the stock option. An assumed dividend yield of zero reflects the fact that the Company has never paid cash dividends and has no intentions to pay dividends in the foreseeable future. The key assumptions used to apply the Black-Scholes option pricing model for options granted under the 2016 Plan during the years ended June 30, 2018, 2017December 31, 2020 and 20162019 were as follows:
| | | | | | | | Year Ended December 31, | | | Year Ended December 31, | | | | | 2018 | | 2017 | | 2016 | | 2020 | | | 2019 | | Option life (in years) | | 5.50 - 6.00 | | 5.50 - 6.25 | | 5.50 - 6.25 | | 5.50 - 6.10 | | | 5.50 - 6.08 | | Stock volatility | | 59% - 64% | | 70% - 72% | | 76% - 80% | | 64% - 70% | | | 60% - 65% | | Risk-free interest rate | | 2.18% - 2.89% | | 1.23% - 2.08% | | 1.47% - 1.97% | | 0.32% - 1.76% | | | 1.37% - 2.63% | | Expected dividends | | 0.0% | | 0.0% | | 0.0% | | 0.0% | | | 0.0% | |
The following table summarizes information about employee, consultant and director stock options under the Company’s equity incentive plans for the years ended June 30, 2018, 2017December 31, 2020 and 20162019 (in thousands except per share amounts): | | | | | | | | Year Ended December 31, | | | Year Ended December 31, | | | | | 2018 | | | 2017 | | | 2016 | | | 2020 | | | 2019 | | Weighted-average grant date fair value per share | | $ | 1.06 | | | $ | 1.95 | | | $ | 2.74 | | | $ | 7.07 | | | $ | 9.35 | | Total cash received from exercise of stock options | | | 503 | | | | 99 | | | | 490 | | | | — | | | | 414 | | Total intrinsic value of stock options exercised | | | 152 | | | | 53 | | | | 967 | | | | — | | | | 84 | |
Time-Vested Restricted Stock Units Time-vested RSUsrestricted stock units (“RSUs”) issued to date under the 2016 Plan generally vest on a ratable annual basis over 3 years. The related stock-based compensation expense is recorded over the requisite service period, which is the vesting period. The fair value of all time-vested RSUs is based on the closing share price of the Common Stock on the date of grant. In connection with retention bonus agreements entered into in January 2017 (see Note 7), a total of 305,616 RSUs were issued on December 22, 2017 subject toone-year cliff vesting.
The following table provides a reconciliation of RSU activity under the 2016 Plan for the year ended June 30, 2018:December 31, 2020: | | | | | | Number of Restricted Stock Units | | | Weighted Average Grant Date Fair Value | | | | | Number of
Restricted
Stock Units | | Weighted
Average
Grant Date
Fair Value | | | Nonvested at July 1, 2017 | | | 248,500 | | | $ | 1.77 | | | Nonvested at January 1, 2020 | | | | 78,684 | | | $ | 18.34 | | Granted | | | 802,459 | | | 1.52 | | | | 152,575 | | | | 12.42 | | Vested | | | (107,830 | ) | | 1.53 | | | | (71,657 | ) | | | 15.25 | | Forfeited | | | (45,000 | ) | | 1.77 | | | | (10,598 | ) | | | 17.17 | | | | | | | | | | Nonvested at June 30, 2018 | | | 898,129 | | | $ | 1.58 | | | | | | | | | | | Nonvested at December 31, 2020 | | | | 149,004 | | | $ | 13.85 | |
The weighted-average remaining vesting term of the RSUs at June 30, 2018December 31, 2020 was 1.341.01 years. Performance-Based Stock Units
Performance Stock Units (“PSUs”) have been awarded to certain employees. The performance conditions associated with the PSU awards are as follows: (a) for one third of the PSUs, upon an FDA acceptance of the Company’s NDA submission of YUTIQ for review on or before March 31, 2018 and (b) fortwo-thirds of the PSUs, upon an FDA approval of YUTIQ on or before March 31, 2019. For each performance criteria that is achieved, 50% of the PSUs that are associated with that performance condition vest at the achievement date and 50% vest on the first anniversary of such date, in each case subject to continued employment through such date. As a result of the achievement of the first performance condition on March 19, 2018, 48,332 PSUs vested at that date and the other 48,332 PSUs became subject to a service-based condition with a vesting date of March 19, 2019. As of June 30, 2018, the second performance condition associated with the PSUs was not yet deemed probable of achievement and, accordingly, stock-based compensation has not been recorded for that portion of the PSUs during the year ended June 30, 2018.
The following table provides a reconciliation of PSU activity under the 2016 Plan for the year ended June 30, 2018:
| | | | | | | | | | | | Number of
Performance
Stock Units | | | Weighted
Average
Grant Date
Fair Value | | Outstanding at July 1, 2017 | | | 210,000 | | | $ | 1.77 | | Granted | | | 115,000 | | | | 1.13 | | Vested | | | (48,332 | ) | | | 1.52 | | Forfeited | | | (35,000 | ) | | | 1.77 | | | | | | | | | | | Outstanding at June 30, 2018 | | | 241,668 | | | $ | 1.52 | | | | | | | | | | |
The weighted-average remaining vesting term of the PSUs associated with the first performance condition at June 30, 2018 was approximately 8.6 months.
Deferred Stock Units A total of 67,500There were 0 non-vested deferred stock units (“DSUs”) were issued and outstanding to incumbentthe Company’s non-executive directors at each of December 31, 2020 and ratified at2019, respectively. Each DSU vests one year from the December 15, 2017 annual meeting of stockholders with a vesting date of June 27, 2018 and a grant date fair value of $1.13 per share. A total of 35,001 DSUs were issued to incumbent directors on June 21, 2018 withone-year cliff vesting and a grant date fair value of $1.95 per share.grant. Subsequent to vesting, the DSUs will be settled in shares of the Company’s Common Stock upon the earliest to occur of (i) each director’s termination of service
on the Company’s Board of Directors and (ii) the occurrence of a change of control as defined in the award agreement. Upon Dr. James Barry’s terminationAt December 31, 2020, there were 1,916 vested DSUs that have not been settled in shares of service on the Company’s Board of Directors on May 7, 2018, all of Dr. Barry’s 12,500 DSUs vested in full and were settled in shares. The following table provides a reconciliation of DSU activity under the 2016 Plan for the year ended June 30, 2018:Common Stock.
| | | | | | | | | | | | Number of
Deferred
Stock Units | | | Weighted
Average
Grant Date
Fair Value | | Outstanding at July 1, 2017 | | | — | | | $ | — | | Granted | | | 102,501 | | | | 1.41 | | Vested | | | (67,500 | ) | | | 1.13 | | | | | | | | | | | Outstanding at June 30, 2018 | | | 35,001 | | | $ | 1.95 | | | | | | | | | | |
AtEmployee Stock Purchase Plan
On June 30, 2018,25, 2019, the weighted-average remaining vesting termCompany’s stockholders approved the adoption of the DSUs was approximately 11.7 months. Market-Based RestrictedEyePoint Pharmaceuticals, Inc. 2019 Employee Stock Units
Purchase Plan (the “ESPP”) and authorized up to 110,000 shares of Common Stock reserved for issuance to participating employees. The following table provides a reconciliation of market-based restricted stock units for the year ended June 30, 2018: | | | | | | | | | | | | Number of
Market-Based
Restricted
Stock Units | | | Weighted
Average
Grant Date
Fair Value | | Outstanding at July 1, 2017 | | | 700,000 | | | $ | 1.35 | | Forfeited | | | (200,000 | ) | | | 1.09 | | | | | | | | | | | Outstanding at June 30, 2018 | | | 500,000 | | | $ | 1.45 | | | | | | | | | | |
At June 30, 2017, there were 700,000 market-based Restricted Stock Units (“market-based RSUs”) outstanding, which included 500,000 issued as an inducement awardESPP allows qualified participants to purchase the Company’s PresidentCommon Stock twice a year at 85% of the lesser of the average of the high and CEO and 200,000 issued to another employee under the 2008 Plan. At June 30, 2018, there were 500,000 market-based RSUs outstanding due to a forfeiture of 200,000 market-based RSUs upon the other employee’s resignation. Subject to a service condition, the number of shares underlying the one remaining market-based RSU vests based upon a relative percentile rank of the3-year change in the closinglow sales price of the Company’s Common Stock compared to thaton (i) the first trading day of the companies that make uprelevant offering period and (ii) the Nasdaq Biotechnology Index through September 15, 2019. last trading day of the relevant offering period. The number of shares of the Company’s Common Stock each employee may purchase under this plan, when combined with all other employee stock purchase plans, is limited to the lower of an aggregate fair market value of $25,000 during each calendar year, or 5,000 shares of the Company’s Common Stock in any one offering period. The first six month offering period under the ESPP began on August 1, 2019 and ended on January 31, 2020. As of December 31, 2020, 33,697 shares of the Company’s Common Stock were issued pursuant to the ESPP.
The Company estimated the fair value of the market-based RSUsoption component of the ESPP shares at the date of grant using a Monte CarloBlack-Scholes valuation model on the respective dates of grant. Other Inducement Award Grants
In connection with the September 15, 2016 hire of the Company’s President and CEO, the Company granted 850,000 options to purchase Common Stock with ratable annual vesting over 4 years, an exercise price of $3.63 per share and a10-year term. Although the stock options were not awarded under the 2008 Plan, the stock options are subject to and governed by the terms and conditions of the 2008 Plan.
In connection with the May 14, 2018 hire of the Company’s Executive Vice President and General Manager, US, the Company granted 375,000 options to purchase Common Stock with ratable annual vesting over 3 years, 65,000 options with1-year cliff vesting and 225,000 PSUs. The options have an exercise price of
$1.95 per share and a10-year term and, although not awarded under the 2016 Plan, are subject to and governed by the terms and conditions of the 2016 Plan. The PSUs are subject to proportional vesting based on cumulative measurement for the3-year period ending June 30, 2021, withtwo-thirds of the award based upon defined amounts of the Company’s product revenues andone-third upon measurement of the net present value of certain business development transactions that are consummated by the Company.
The following table provides a reconciliation of the Company’s inducement stock option awards formodel. During the year ended June 30, 2018:December 31, 2020, the compensation expense from ESPP shares was immaterial.
| | | | | | | | | | | | | | | | | | | | Number of
options | | | Weighted
Average
Exercise
Price | | | Weighted
Average
Remaining
Contractual
Life | | | Aggregate
Intrinsic
Value | | | | | | | | | | | (in years) | | | (in thousands) | | Outstanding at July 1, 2017 | | | 850,000 | | | $ | 3.63 | | | | | | | | | | Granted | | | 440,000 | | | | 1.95 | | | | | | | | | | | | | | | | | | | | | | | | | | | Outstanding at June 30, 2018 | | | 1,290,000 | | | $ | 3.06 | | | | 8.78 | | | $ | 57 | | | | | | | | | | | | | | | | | | | Exercisable at June 30, 2018 | | | 212,500 | | | $ | 3.63 | | | | 8.22 | | | $ | — | | | | | | | | | | | | | | | | | | |
Stock-Based Compensation Expense The Company’s statements of comprehensive loss included total compensation expense from stock-based payment awards as follows (in thousands): | | | | Year Ended June 30, | | | Year Ended December 31, | | | Year Ended December 31, | | | | | 2018 | | | 2017 | | | 2016 | | | 2020 | | | 2019 | | Compensation expense included in: | | | | | | | | | | | | | | | Research and development | | $ | 1,252 | | | $ | 1,109 | | | $ | 702 | | | $ | 1,411 | | | $ | 1,073 | | Sales and marketing | | | 50 | | | | — | | | | — | | | | 907 | | | | 715 | | General and administrative | | | 1,402 | | | | 1,347 | | | | 1,461 | | | | 3,229 | | | | 2,780 | | | | | | | | | | | | | $ | 5,547 | | | $ | 4,568 | | | | $ | 2,704 | | | $ | 2,456 | | | $ | 2,163 | | | | | | | | | | | | | |
In connection with termination benefits provided to the Company’s former Chief Executive Officer, the vesting of certain options was accelerated in accordance with the terms of the options, the exercise period for all vested options was extended through September 14, 2017, and all remainingnon-vested options were forfeited. Additionally, in connection with the U.K. restructuring, the exercise period of all vested options held by the former U.K. employees was extended through June 30, 2017 and allnon-vested options were forfeited. These option modifications and forfeitures were accounted for in the quarter ended September 30, 2016, the net effect of which resulted in an approximate $274,000 increase of stock-based compensation expense included in general and administrative expense and an approximate $35,000 reduction of stock-based compensation expense included in research and development expense for the year ended June 30, 2017 in the table above.
In connection with termination benefits provided to the Company’s former Vice President, Corporate Affairs and General Counsel, the vesting of certain options was accelerated in accordance with the terms of the options, the exercise period for all vested options was extended through June 26, 2018 and all remainingnon-vested options were forfeited. The option modification and forfeitures were accounted for in the quarter endedAt December 31, 2016, the net effect of which resulted in an approximate $104,000 reduction of stock-based compensation expense included in general and administrative expense for the year ended June 30, 2017 in the table above.
At June 30, 2018,2020, there was approximately $5.4$2.9 million of unrecognized compensation expense related to outstanding equity awards under the 2016 Plan, the 2008 Plan, andThe inducement awards and the ESPP that is expected to be recognized as expense over a weighted-average period of approximately 1.71.49 years.
12.In-License Agreement Equinox Science, LLC In February 2020, the Company entered into an Exclusive License Agreement with Equinox Science, LLC (“Equinox”), pursuant to which Equinox granted us an exclusive, sublicensable, royalty-bearing right and license to certain patents and other Equinox intellectual property to research, develop, make, have made, use, sell, offer for sale and import the compound vorolanib and any pharmaceutical products comprising the compound for the prevention or treatment of age-related macular degeneration, diabetic retinopathy and retinal vein occlusion using our proprietary localized delivery technologies, in each case, throughout the world except China, Hong Kong, Taiwan and Macau (the “Territory”). In consideration for the rights granted by Equinox, the Company (i) made a one time, non-refundable, non-creditable upfront cash payment of $1.0 million to Equinox in February 2020, and (ii) agreed to pay milestone payments totaling up to $50 million upon the achievement of certain development and regulatory milestones, consisting of (a) completion of a Phase II clinical trial for the compound or a licensed product, (b) the filing of a new drug application or foreign equivalent for the compound or a licensed product in the United States, European Union or United Kingdom and (c) regulatory approval of the compound or a licensed product in the United States, European Union or United Kingdom. The Company also agreed to pay Equinox tiered royalties based upon annual net sales of licensed products in the Territory. The royalties are payable with respect to a licensed product in a particular country in the Territory on a country-by-country and licensed product-by-licensed product basis until the later of (i) twelve years after the first commercial sale of such licensed product in such country and (ii) the first day of the month following the month in which a generic product corresponding to such licensed product is launched in such country (collectively, the “Royalty Term”). The royalty rates range from the high-single digits to low-double digits depending on the level of annual net sales. The royalty rates are subject to reduction during certain periods when there is no valid patent claim that covers a licensed product in a particular country. The Company recorded $1.0 million of R&D expense during the year ended December 31, 2020 for this license. 13.Restructuring Charges Fiscal Year 2020 Restructuring Plan On April 1, 2020, the Company committed to and announced a restructuring plan (the “Plan”) with regard to its commercial operations. The Plan is a result of decline in product demand associated with shut-downs of customer facilities and postponements of elective surgical procedures in response to the Pandemic. In connection with the Plan, the Company, among other things, downsized its current workforce, with reductions coming primarily from its external DEXYCU sales force and supporting commercial operations, as cataract surgery is considered a non-essential procedure due to the Pandemic. The Company recorded approximately $590,000 of external DEXYCU sales force personnel and employee severance for discretionary termination benefits during the year ended December 31, 2020, upon notification of the affected external DEXYCU sales force personnel and employees in accordance with ASC 420, Exit or Disposal Cost Obligations. The charges of $590,000 were recognized in the Company’s operating results, of which $542,000 and $48,000 were included in sales and marketing expense and general and administrative expense, respectively. The Plan was completed during the fourth quarter of fiscal 2020. | Employee Severance and Benefits | | | Total | | Beginning balance at January 1, 2020 | $ | 0 | | | $ | 0 | | Restructuring charge | | | 590 | | | | | 590 | | Cash payments | | | (590 | ) | | | | (590 | ) | Ending balance at December 31, 2020 | $ | | 0 | | | $ | | 0 | |
12.14. | Fair Value Measurements |
The following tables summarize the Company’s assets and liabilities carried at fair value measured on a recurring basis at June 30, 2018December 31, 2020 and 20172019, respectively, by valuation hierarchy (in thousands): | | | | June 30, 2018 | | | December 31, 2020 | Description | | Total Carrying
Value | | | Quoted prices in
active markets
(Level 1) | | | Significant other
observable inputs
(Level 2) | | | Significant
unobservable inputs
(Level 3) | | | Total Carrying Value | | | Quoted prices in active markets (Level 1) | | | Significant other observable inputs (Level 2) | | Significant unobservable inputs (Level 3) | Assets: | | | | | | | | | | | | | | | | | | | | | Cash equivalents | | $ | 28,826 | | | $ | 28,826 | | | $ | — | | | $ | — | | | $ | 23,538 | | | $ | 23,538 | | | | | | | | | | | | | | | | | | | | $ | 23,538 | | | $ | 23,538 | | | | | | | | $ | 28,826 | | | $ | 28,826 | | | $ | — | | | $ | — | | | | | | | | | | | | | | | | | Liabilities: | | | | | | | | | | Derivative liabilities | | $ | 19,780 | | | $ | — | | | $ | — | | | $ | 19,780 | | | | | | | | | | | | | | | | | | | $ | 19,780 | | | $ | — | | | $ | — | | | $ | 19,780 | | | | | | | | | | | | | | | | |
| | | | June 30, 2017 | | | December 31, 2019 | | Description | | Total Carrying
Value | | | Quoted prices in
active markets
(Level 1) | | | Significant other
observable inputs
(Level 2) | | | Significant
unobservable inputs
(Level 3) | | | Total Carrying Value | | | Quoted prices in active markets (Level 1) | | | Significant other observable inputs (Level 2) | | | Significant unobservable inputs (Level 3) | | Assets: | | | | | | | | | | | | | | | | | | | | | | | | | Cash equivalents | | $ | 13,521 | | | $ | 13,521 | | | $ | — | | | $ | — | | | $ | 19,976 | | | $ | 19,976 | | | $ | 0 | | | $ | 0 | | | | | | | | | | | | | | | | $ | 19,976 | | | $ | 19,976 | | | $ | 0 | | | $ | 0 | | | | $ | 13,521 | | | $ | 13,521 | | | $ | — | | | $ | — | | | | | | | | | | | | | | | | |
Financial instruments that potentially subject the Company to concentrations of credit risk have historically consisted principally of cash and cash equivalents. At June 30, 2018December 31, 2020 and June 30, 2017,2019, respectively, substantially all of the Company’s interest-bearing cash equivalent balances were concentrated in one U.S. Government money market fund that has investments consisting primarily of U.S. Government Agency debt, U.S. Treasury debt, U.S. Treasury Repurchase Agreements and U.S. Government Agency Repurchase Agreements. These deposits may be redeemed upon demand and, therefore, generally have minimal risk. The Company’s cash equivalents are classified within Level 1 on the basis of valuations using quoted market prices. The Second Tranche Transaction was determined to be liability classified (see Note 10), which required that the liability be measured atcarrying amounts of accounts receivable, accounts payable and accrued expenses approximate fair value each period with changes inbecause of their short-term maturity. The fair value recorded as a component ofnon-operating expense in the consolidated statement of comprehensive loss. This valuation wasCompany’s CRG Loan is determined to be a level 3 valuation because it includes unobservable inputs. The Second Tranche Transaction liability was valued using a Monte Carlo simulation valuation model. This model incorporated several inputs, including the Common Stock pricediscounted cash flow analysis based on the date of valuation, the historical volatilitymarket rates for observable similar instruments as of the price of Common Stock, the risk-free interest rate and management’s assessment of the probability and timing of the issuance of the Units occurring. A significant fluctuation in the Company’s stock price or the Company’s estimate of the number of Units to be issued could result in a material increase or decrease incondensed consolidated balance sheet dates. Accordingly, the fair value of the Second Tranche liability.CRG Loan is categorized as Level 2 within the fair value hierarchy. The Second Tranche Transaction liability was settled upon the closingcarrying value of the Second Tranche TransactionCRG Loan at December 31, 2020 was approximately $38.3million, and consisted of $36.0million of its carrying amount as reported in June 2018. The Company remeasured the Second Tranche Transaction liability to fair value immediately prior to settlement. This valuation at settlement was calculatedlong-term debt, and $2.3 million of debt exit fee as the excessreported in other long-term liabilities of the sum of (i) thecondensed consolidated balance sheet, respectively. The fair value of the Second Tranche Warrants and (ii) theCRG Loan was approximately $38.0 million at December 31, 2020. The fair value of the shares of Common Stock issued to settle the liability over the cash proceeds received by the Company for the Units. Significant assumptions used toCRG Loan approximated its carrying value this liability were as follows:at December 31, 2019. | | | | | | | | | | | | March 28, 2018
(Date of Issuance) | | | June 25, 2018
(Date of Settlement) | | Volatility | | | 54.20 | % | | | N/A | | Risk free interest rate | | | 1.70 | % | | | N/A | | Estimated date of stockholder approval | | | June 2018 | | | | N/A | | Estimated number of units issuable | | | 26,900,000 | | | | 20,184,224 | | Valuation date stock price | | $ | 1.07 | | | $ | 1.93 | |
The Additional Advance Warrants were initially determined to be liability classified (see Note 9), which required that the liability be measured at fair value each period with changes in fair value being recorded as a component of non-operating expense in the consolidated statement of comprehensive loss. This valuation was determined to be a level 3 valuation because it includes unobservable inputs. The Additional Advance Warrant liability was valued using a Monte Carlo simulation valuation model. This model incorporated several inputs including the Common Stock price on the date of valuation, the historical volatility of the price of the Common Stock, the risk-free interest rate and management’s assessments of the probability of the Additional Advance being drawn upon. Upon the closing of the Second Tranche Transaction in June 2018, the Additional Advance Warrants no longer met the criteria to be classified as a liability. The Company remeasured the Additional Advance Warrants immediately prior to the close of the Second Tranche Transaction and reclassified the liability balance to equity. Significant assumptions used to value this liability were as follows: | | | | | | | | | | | | March 28, 2018
(Date of Issuance) | | | June 25, 2018
(Date of
Reclassification to
Equity) | | Volatility | | | 55.20 | % | | | 55.10 | % | Risk free interest rate | | | 1.70 | % | | | 2.80 | % | Term (in years) | | | 7 | | | | 7 | | Dividend rate | | | 0 | % | | | 0 | % | Valuation date stock price | | $ | 1.07 | | | $ | 1.93 | | Probability of issuance | | | 80 | % | | | 100 | % |
Upon the closing of the Second Tranche Transaction, the Company issued the Second Tranche Warrants, which were determined to be liability classified, which requires that the liability be measured at fair value each period with changes in fair value being recorded as a component of non-operating expense in the consolidated statement of comprehensive loss. This valuation was determined to be a level 3 valuation because it includes unobservable inputs. The Second Tranche Warrants were valued using a Monte Carlo simulation valuation model. This model incorporated several inputs, including the Common Stock price on the date of valuation, the historical volatility of the price of the Common Stock and the risk-free interest rate. Significant assumptions used to value this liability are as follows:
| | | | | | | | | | | | June 25, 2018
(Date of issuance) | | | June 30, 2018 | | Volatility | | | 81.00 | % | | | 85.40 | % | Risk free interest rate | | | 2.10 | % | | | 2.10 | % | Term (in years) | | | 0.5 | | | | 0.5 | | Dividend rate | | | 0 | % | | | 0 | % | Valuation date stock price | | $ | 2.00 | | | $ | 2.08 | | Probability of issuance | | | 100 | % | | | 100 | % |
The following table sets forth a summary of changes in the fair value of the Company’s derivative liabilities for which fairPPP Loan approximated its carrying value is determined by Level 3 inputs (in thousands):
| | | | | | | | | | | | | | | | | | | | Second Tranche
Liability | | | Additional
Advance
Warrant
Liability | | | Second
Tranche
Warrants | | | Total | | Balance at June 30, 2017 | | $ | — | | | $ | — | | | $ | — | | | $ | — | | Initial fair value of derivative liability | | | 4,734 | | | | 69 | | | | 18,165 | | | | 22,968 | | Change in fair value | | | 24,319 | | | | 18 | | | | 1,615 | | | | 25,952 | | Reclassification to equity | | | — | | | | (87 | ) | | | — | | | | (87 | ) | Settlement | | | (29,053 | ) | | | — | | | | — | | | | (29,053 | ) | | | | | | | | | | | | | | | | | | Balance at June 30, 2018 | | $ | — | | | $ | — | | | $ | 19,780 | | | $ | 19,780 | | | | | | | | | | | | | | | | | | |
Also included in the change in fair value was $326,000 of transaction costs that were expensed in connection with the issuance of the derivative liabilities.$2.0 million at December 31, 2020.
The Company operates a defined contribution plan intended to qualify under Section 401(k) of the U.S. Internal Revenue Code. Participating U.S. employees may contribute a portion of theirpre-tax compensation, as defined, subject to statutory maximums. The Company matches employee contributions up to 5% of eligible compensation, subject to a stated calendar year Internal Revenue Service maximum. The Company operated a defined contribution pension plan for U.K. employees pursuant to which the Company made contributions on behalf of employees plus a matching percentage of elective employee contributions. This pension plan was terminated in the quarter ending September 30, 2016 following termination of employment of all U.K. employees. The Company contributed a total of $220,000$690,000 and $619,000 for fiscal 2018, $193,000 for fiscal 2017the year ended December 31, 2020 and $209,000 for fiscal 20162019, respectively, in connection with these retirement plans.
The components of income tax benefit are as follows (in thousands):
| | | | | | | | | | | | | | | | Year Ended June 30, | | | | | 2018 | | | 2017 | | | 2016 | | U.S. operations: | | | | | | | | | | | | | Current income tax expense | | $ | — | | | $ | — | | | $ | 4 | | Deferred income tax benefit | | | — | | | | — | | | | — | | | | | | | | | | | | | | | | | | — | | | | — | | | | 4 | | | | | | | | | | | | | | | Non-U.S. operations: | | | | | | | | | | | | | Current income tax benefit | | | — | | | | — | | | | (159 | ) | Deferred income tax benefit | | | — | | | | — | | | | — | | | | | | | | | | | | | | | | | | — | | | | — | | | | (159 | ) | | | | | | | | | | | | | | Income tax benefit | | $ | — | | | $ | — | | | $ | (155 | ) | | | | | | | | | | | | | |
During the fiscal year ended June 30, 2016, the Company recognized a current income tax benefit of $159,000 related to foreign research and development tax credits earned by its U.K. subsidiary.
The components of loss before income taxes are as follows (in thousands): | | | | Year Ended June 30, | | | Year Ended December 31, | | | Year Ended December 31, | | | | | 2018 | | 2017 | | 2016 | | | 2020 | | | 2019 | | U.S. operations | | $ | (53,000 | ) | | $ | (17,566 | ) | | $ | (19,780 | ) | | $ | (45,492 | ) | | $ | (56,866 | ) | Non-U.S. operations | | | (171 | ) | | (919 | ) | | (1,922 | ) | | | 98 | | | | 73 | | | | | | | | | | | | | Loss before income taxes | | $ | (53,171 | ) | | $ | (18,485 | ) | | $ | (21,702 | ) | | $ | (45,394 | ) | | $ | (56,793 | ) | | | | | | | | | | | |
On December 22, 2017, theTax Cuts and Jobs Act(the “Tax Act”) was signed into law, making significant changes to the federal tax law. Amongst other things, the Tax Act reduces the federal corporate tax rate from 34% to 21% effective for tax years beginning after December 31, 2017 and has resulted in a remeasurement of the Company’s deferred tax assets included in the Company’s fiscal 2018 rate reconciliation. The difference between the Company’s expected income tax benefit, as computed by applying the blended statutory U.S. federal tax rate of 27.5%21% for fiscal 2018the year ended December 31, 2020 and 34%21% for each of fiscal 2017 and fiscal 2016the year ended December 31, 2019 to loss before income taxes, and actual income tax benefit is reconciled in the following table (in thousands): | | | | Year Ended June 30, | | | Year Ended December 31, | | | Year Ended December 31, | | | | | 2018 | | 2017 | | 2016 | | | 2020 | | | 2019 | | Income tax benefit at statutory rate | | $ | (14,622 | ) | | $ | (6,284 | ) | | $ | (7,379 | ) | | $ | (9,533 | ) | | $ | (11,927 | ) | State income taxes, net of federal benefit | | | (1,552 | ) | | (928 | ) | | (1,044 | ) | | | (2,760 | ) | | | (3,685 | ) | Non-U.S. income tax rate differential | | | (66 | ) | | (121 | ) | | 778 | | | | (8 | ) | | | 374 | | Change in fair value of derivative | | | 7,227 | | | | — | | | | — | | | 0 | | | | 0 | | Change in federal tax rate | | | 14,673 | | | | — | | | | — | | | 0 | | | | 0 | | Research and development tax credits | | | (284 | ) | | (242 | ) | | (397 | ) | | | (403 | ) | | | (150 | ) | Permanent items | | | (15 | ) | | (9 | ) | | 216 | | | | 288 | | | | 55 | | Changes in valuation allowance | | | (5,385 | ) | | 7,489 | | | 6,789 | | | | 13,068 | | | | 15,608 | | Other, net | | | 24 | | | 95 | | | 882 | | | | (652 | ) | | | (275 | ) | | | | | | | | | | | | Income tax benefit | | $ | — | | | $ | — | | | $ | (155 | ) | | $ | 0 | | | $ | 0 | | | | | | | | | | | | |
In addition to the $5.4 million change in valuation allowance in the above table, the Company recorded a deferred tax asset of $6.2 million and a valuation allowance of the same amount in connection with the Icon acquisition.
The significant components of deferred income taxes are as follows (in thousands): | | | | June 30, | | | December 31, | | | December 31, | | | | | 2018 | | | 2017 | | | 2020 | | | 2019 | | Deferred tax assets: | | | | | | | | | | | | | Net operating loss carryforwards | | $ | 47,774 | | | $ | 39,439 | | | $ | 74,876 | | | $ | 66,400 | | Deferred revenue | | | — | | | | 20 | | | | 150 | | | | 8 | | Lease liability | | | | 806 | | | | 923 | | Stock-based compensation | | | 4,241 | | | | 5,107 | | | | 6,847 | | | | 5,805 | | Tax credits | | | 3,463 | | | | 1,727 | | | | 4,503 | | | | 3,687 | | Other | | | 185 | | | | 186 | | | | 2,514 | | | | 1,473 | | | | | | | | | | Total deferred tax assets | | | 55,663 | | | | 46,479 | | | | 89,696 | | | | 78,296 | | | | | | | | | | Deferred tax liabilities: | | | | | | | | | | | | | Intangible assets | | | 8,542 | | | | 123 | | | | 6,087 | | | | 7,559 | | | | | | | | | | Right-of-use assets | | | | 713 | | | | 841 | | Total deferred tax liabilities | | | | 6,800 | | | | 8,400 | | Deferred tax assets, net | | | 47,121 | | | | 46,356 | | | | 82,896 | | | | 69,896 | | Valuation allowance | | | 47,121 | | | | 46,356 | | | | 82,896 | | | | 69,896 | | | | | | | | | | Total deferred tax liability | | $ | — | | | $ | — | | | $ | 0 | | | $ | 0 | | | | | | | | | |
The valuation allowance generally reflects limitations on the Company’s ability to use the tax attributes and reduces the value of such attributes to themore-likely-than-not realizable amount. Management assessed the available positive and negative evidence to estimate if sufficient taxable income will be generated to use the existing net deferred tax assets. Based on a weighting of the
objectively verifiable negative evidence in the form of cumulative operating losses over the three-year period ended June 30, 2018, management believes that it is not more likely than not that the deferred tax assets will be realized and, accordingly, a full valuation allowance has been established. The valuation allowance increased $765,000, $7.5$13.1 million and $6.8$15.6 million duringfor the fiscal years ended June 30, 2018, 2017December 31, 2020 and 2016,2019, respectively, with such increases attributed to there-measurement of the net deferred tax assets at theyear-end dates. The valuation allowance decreased by $5.4 million from current year activity, including the impact of the 2017 Tax Act, offset by an increase of $6.2 million related to the Icon acquisition. The Company has tax net operating loss and tax credit carry forwards in its individual tax jurisdictions. Including approximately $49.3 million related to the Icon acquisition, at June 30, 2018December 31, 2020 the Company had U.S. federal net operating loss carry forwards of approximately $165.1 million,$269.1 million. The net operating losses consist of $151.8, which expire at various dates between calendar years 2023 and 2038. The utilization of certain of these loss and tax credit carry forwards may be limited by Sections 382 and 383 of the Internal Revenue Code as a result of historical or future changes in the Company’s ownership. At June 30, 2018,December 31, 2020, the Company had state net operating loss carry forwards of approximately $123.5$196.2 million, which expire between 2033 and 2038, as well as U.S. federal and state research and development tax credit carry forwards of approximately $2.9$4.7 million, which expire at various dates between calendar years 20182021 and 2038. In addition, at June 30, 2018December 31, 2020 the Company had net operating loss carry forwards in the U.K. of £21.1£20.9 million (approximately $27.9$27.6 million), which are not subject to any expiration dates. The Company’s U.S. federal income tax returns for calendar years 2003 through 2017 remain subject to examination by the Internal Revenue Service. The Company’s U.K. tax returns for fiscal years 2006 through 20172019 remain subject to examination. Through June 30, 2018,December 31, 2020, the Company had no0 unrecognized tax benefits in its consolidated statements of comprehensive loss and no0 unrecognized tax benefits in its consolidated balance sheets as of June 30, 2018 or 2017.December 31, 2020 and 2019, respectively. As of June 30, 2018December 31, 2020 and 2017,2019, the Company had no0 accrued penalties or interest related to uncertain tax positions. 15.17. | Commitments and Contingencies
|
Operating Leases
On May 17, 2018, the Company entered into a Second Amendment (the “Second Amendment”) to its Lease, dated November 1, 2013, as amended (the “Lease”), with Whetstone Riverworks Holdings, LLC (assuccessor-in-interest to Farley White Aetna Mills, LLC) (the “Landlord”). The Lease was initially for approximately 13,650 square feet of rentable area (the “Existing Space”) of the building located at 480 Pleasant Street, Watertown, MA 02472 (the “Premises”) and was set to expire in April 2019. Under the Second Amendment, the Company will lease an additional 6,590 square feet of rentable area (the “Additional Space”, and together with the Existing Space, the “Total Space”) on the Premises, commencing on September 10, 2018 (the “Additional Space Effective Time”). The Landlord has agreed to provide the Company a construction allowance of up to $670,750 to be applied toward the aggregate work to be conducted on the Total Space. The Second Amendment also extended the term of the Lease, which will now expire 80 calendar months after the Additional Space Effective Time; provided, however, that the base rent for the Total Space will be abated during the first four months following the Additional Space Effective Time. The Company also has an option to extend the term of the Lease for one additional five-year period. The Company will also be required to pay its proportionate share of certain operating costs and property taxes applicable to the leased premises.
Commencing July 1, 2017, the Company leases approximately 3,000 square feet of office space in Liberty Corner, New Jersey under a lease term extending through June 2022, with two five-year renewal options at 95% of the then-prevailing market rates. In addition to base rent, the Company is obligated to pay its proportionate share of building operating expenses and real estate taxes in excess of base year amounts. In June 2018, the Company subleased an additional 1,381 square feet of adjoining space from Caladrius Biosciences, Inc. (“Caladrius”) through May 2022. The Chief Executive Officer of Caladrius is a director of the Company.
At June 30, 2018, the Company’s total future minimum lease payments undernon-cancellable operating leases were as follows (in thousands):
| | | | | Fiscal Year: | | | | 2019 | | $ | 548 | | 2020 | | | 870 | | 2021 | | | 886 | | 2022 | | | 892 | | 2023 and beyond | | | 2,324 | | | | | | | | | $ | 5,520 | | | | | | |
Rent expense related to the Company’s real estate and other operating leases charged to operations was approximately $508,000 for fiscal 2018, $442,000 for fiscal 2017 and $485,000 for fiscal 2016.
Legal Proceedings The Company is subject to various other routine legal proceedings and claims incidental to its business, which management believes will not have a material effect on the Company’s financial position, results of operations or cash flows. U.S. Securities and Exchange Commission Subpoena On May 14, 2020, the Company received a subpoena from the Division of Enforcement of the SEC seeking production of certain documents and information on topics including product sales and demand, revenue recognition and accounting in relation to product sales, product sales and cash projections, and related financial reporting, disclosure and compliance matters. The Company is cooperating fully in connection with this investigation. Based on procedures performed to date in relation to the Company’s revenue recognition practices, the Company has not identified any accounting items that are not in accordance with GAAP. At this time, the Company is unable to predict the duration, scope or outcome of this matter or whether it could have a material impact on the Company’s financial condition, results of operations or cash flow.16.18. | Segment and Geographic Area Information |
Business Segment The Company operates in only one1 business segment, beingwhich is the biotechnology sector.business of developing and commercializing innovative ophthalmic products for the treatment of eye diseases. Operating segments are identified as components of an enterprise about which separate discrete financial information is available for evaluation by the chief operating decision maker in making decisions regarding resource allocation and assessing performance. The chief operating decision maker made such decisions and assessed performance at the company level, as one segment.
Geographic Area Information The following table summarizes the Company’s revenues and long-lived assets, net by geographic area (in thousands): | | | | | | | | | | | | Revenues | | | Long-lived assets, net | | | | | Revenues | | | Long-lived assets, net | | | Twelve Months Ended December 31, | | | Twelve Months Ended December 31, | | | At December 31, | | | At December 31, | | | | | 2018 | | | 2017 | | | 2016 | | | 2018 | | | 2017 | | | 2020 | | | 2019 | | | 2020 | | | 2019 | | U.S. | | $ | 2,861 | | | $ | 7,439 | | | $ | 1,520 | | | $ | 253 | | | $ | 313 | | | $ | 22,624 | | | $ | 19,144 | | | $ | 630 | | | $ | 357 | | China | | | $ | 11,713 | | | $ | 1,121 | | | | — | | | | — | | U.K. | | | 100 | | | | 100 | | | | 100 | | | | — | | | | — | | | | 100 | | | | 100 | | | | — | | | | — | | | | | | | | | | | | | | | | | | | Consolidated | | $ | 2,961 | | | $ | 7,539 | | | $ | 1,620 | | | $ | 253 | | | $ | 313 | | | $ | 34,437 | | | $ | 20,365 | | | $ | 630 | | | $ | 357 | | | | | | | | | | | | | | | | | | |
17.19. | Quarterly Financial Data (unaudited)Subsequent Events
|
The following table summarizesIn February 2021, the quarterly resultsCompany sold 10,465,000 shares of operations for the years ended June 30, 2018 and 2017 (in thousands exceptCommon Stock in an underwritten public offering at a price of $11.0 per share, amounts):including the exercise in full by the underwriters of their option to purchase up to 1,365,000 additional shares of Common Stock. The gross proceeds of the offering to the Company are approximately $115.1 million. Underwriter discounts and commissions and other share issue costs totaled approximately $7.1 million.
| | | | | | | | | | | | | | | | | | | | | | | | Fiscal Year 2018 | | | | | First Quarter
Ended
September 30,
2017 | | | Second Quarter
Ended
December 31,
2017 | | | Third Quarter
Ended
March 31,
2018 | | | Fourth Quarter
Ended
June 30,
2018 | | | Year Ended
June 30,
2018 | | | | | | | | | | | (1) | | | (1) | | | | | Total revenues | | $ | 385 | | | $ | 933 | | | $ | 928 | | | $ | 715 | | | $ | 2,961 | | Operating loss | | | (6,006 | ) | | | (5,808 | ) | | | (4,678 | ) | | | (9,782 | ) | | | (26,274 | ) | Net loss | | | (5,983 | ) | | | (5,782 | ) | | | (6,978 | ) | | | (34,428 | ) | | | (53,171 | ) | | | | | | | | | | | | | | | | | | | | | | Net loss per share—basic and diluted | | $ | (0.15 | ) | | $ | (0.13 | ) | | $ | (0.15 | ) | | $ | (0.62 | ) | | $ | (1.15 | ) | | | | | | | | | | | | | | | | | | | | | | Weighted average common shares—basic and diluted | | | 39,430 | | | | 44,530 | | | | 45,644 | | | | 55,387 | | | | 46,226 | | | | | | | | | | | | | | | | | | | | | | |
F-34 | | | | | | | | | | | | | | | | | | | | | | | | Fiscal Year 2017 | | | | | First Quarter
Ended
September 30,
2016 | | | Second Quarter
Ended
December 31,
2016 | | | Third Quarter
Ended
March 31,
2017 | | | Fourth Quarter
Ended
June 30,
2017 | | | Year Ended
June 30,
2017 | | | | | | | | (2) | | | | | | | | | | | Total revenues | | $ | 277 | | | $ | 5,971 | | | $ | 590 | | | $ | 701 | | | $ | 7,539 | | Operating loss | | | (7,186 | ) | | | (94 | ) | | | (5,160 | ) | | | (6,136 | ) | | | (18,576 | ) | Net loss | | | (7,162 | ) | | | (67 | ) | | | (5,140 | ) | | | (6,116 | ) | | | (18,485 | ) | | | | | | | | | | | | | | | | | | | | | | Net loss per share—basic and diluted | | $ | (0.21 | ) | | $ | — | | | $ | (0.15 | ) | | $ | (0.16 | ) | | $ | (0.52 | ) | | | | | | | | | | | | | | | | | | | | | | Weighted average common shares—basic and diluted | | | 34,175 | | | | 34,177 | | | | 34,366 | | | | 38,673 | | | | 35,344 | | | | | | | | | | | | | | | | | | | | | | |
(1) | Results for the third and fourth quarters of fiscal 2018 included $2.3 million and $24.0 million, respectively, of change in fair value of derivative liability in connection with the Second Tranche Transaction (see Notes 10 and 12). The fourth quarter of fiscal 2018 includes an out-of-period expense of $1.2 million reflecting the increase in the fair value of the Company’s derivative liability which occurred, but was not recorded, in the third quarter of fiscal 2018.
|
(2) | Results for the second quarter of fiscal 2017 included $5.6 million of revenue recognized as a result of the December 2016 termination of the Company’s Restated Pfizer Agreement (see Note 4).
|
F-36
|
|
|
|