UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
(Mark One)
☒ ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the Fiscal Year Ended December 31, 20222023
or
☐ TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
Commission File Number:file number: 001-40064
VIRPAX PHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
| Delaware | 82-1510982 | |
| (State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification |
1055 Westlakes Drive, Suite 300 Berwyn, PA | 19312 | |
| (Address of principal executive offices) | (Zip |
(610) 727-4597
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of | Trading | Name of Registered | ||
| Common Stock, par value $0.00001 per share | VRPX | The Nasdaq Capital Market |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ☐ | Accelerated filer | ☐ |
| Non-accelerated filer | ☒ | Smaller reporting company | ☒ |
| Emerging growth company | ☒ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☐
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to Section 240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ☐ No ☒
As of June 30, 2022,2023, the last business day of the registrant’s most recently completed second fiscal quarter, the aggregate market value of the registrant’s common stock held by non-affiliates of the registrant was $14,614,844$9,398,700 based on the closing sale price on June 30, 2022,2023, as reported on the NASDAQNasdaq Capital Markets.
As of March 21, 202322, 2024 the number of outstanding shares of the registrant’s common stock, par value $0.00001 per share, was 11,714,284.1,171,233.
DOCUMENTS INCORPORATED BY REFERENCE
None.
VIRPAX PHARMACEUTICALS, INC.
ANNUAL REPORT ON FORM 10-K
FOR THE FISCAL YEAR ENDED DECEMBER 31, 20222023
TABLE OF CONTENTS
i
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This reportAnnual Report on Form 10-K contains forward-looking statements made pursuant to the safe harbor provisions of the Private Securities Litigation Reform Act of 1995 under Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. Our forward-looking statements include, but are not limited to, statements regarding our or our management team’s expectations, hopes, beliefs, intentions or strategies regarding the future. In addition, any statements that refer to projections, forecasts or other characterizations of future events or circumstances, including any underlying assumptions, are forward-looking statements. The words “anticipate,” “believe,” “contemplate,” “continue,” “could,” “estimate,” “expect,” “intends,” “may,” “might,” “plan,” “possible,” “potential,” “predict,” “project,” “should,” “will,” “would” and similar expressions may identify forward-looking statements, but the absence of these words does not mean that a statement is not forward-looking. Forward-looking statements in this report on Annual Report on Form 10-K may include, for example, statements about:
| ● | our lack of operating history; |
| ● | the expectation that we will incur significant operating losses for the foreseeable future and will need significant additional capital; | |
| ● | our current and future capital requirements to support our development and commercialization efforts for our product candidates and our ability to satisfy our capital needs; | |
| ● | the outcome of certain current litigation in which we and our former Chief Executive Officer are named as defendants (See Part 1-Item 1A-Risk Factors, Item 3-Legal Proceedings); |
| ● | our ability to raise additional capital; |
| ● | our dependence on our product candidates, which are still in preclinical or early stages of clinical development; |
| ● | our, or that of our third-party manufacturers, ability to manufacture current good manufacturing practice (“cGMP”) quantities of our product candidates as required for preclinical and clinical trials and, subsequently, our ability to manufacture commercial quantities of our product candidates; |
| ● | our ability to complete required clinical trials for our product candidates and obtain approval from the US Food and Drug Administration (“FDA”) or other regulatory agencies in different jurisdictions; |
| ● | our lack of a sales and marketing organization and our ability to commercialize our product candidates if we obtain regulatory approval; |
| ● | our dependence on third-parties to manufacture our product candidates; |
| ● | our reliance on third-party contract research organizations (“CROs”) to conduct our clinical trials; |
| ● | our ability to maintain or protect the validity of our intellectual property; |
| ● | our ability to internally develop new inventions and intellectual property; |
| ● | interpretations of current laws and the passages of future laws; |
| ● | acceptance of our business model by investors; |
| ● | the accuracy of our estimates regarding expenses and capital requirements; |
| ● | our ability to maintain our Nasdaq listing; and |
| ● | our ability to adequately support organizational and business |
The foregoing does not represent an exhaustive list of matters that may be covered by the forward-looking statements contained herein or risk factors that we are faced with that may cause our actual results to differ from those anticipated in such forward-looking statements. Please see “Part I—Item 1A—Risk Factors” for additional risks which could adversely impact our business and financial performance.
All forward-looking statements are expressly qualified in their entirety by this cautionary notice. You are cautioned not to place undue reliance on any forward-looking statements, which speak only as of the date of this report or the date of the document incorporated by reference into this report. We have no obligation, and expressly disclaims any obligation, to update, revise or correct any of the forward-looking statements, whether as a result of new information, future events or otherwise. We have expressed our expectations, beliefs and projections in good faith and believe they have a reasonable basis. However, we cannot assure you that our expectations, beliefs or projections will result or be achieved or accomplished.
ii
PART I
ITEM 1. BUSINESS
All referencesForward-Looking Statements
This Annual Report contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended (the “Securities Act”), and Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), that involve substantial risks and uncertainties. The forward-looking statements are contained principally in Part I, Item 1. “Business,” Part I, Item 1A. “Risk Factors,” and Part II, Item 7. “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” but are also contained elsewhere in this reportAnnual Report and in some cases you can identify forward-looking statements by terminology such as “may,” “should,” “potential,” “continue,” “expects,” “anticipates,” “intends,” “plans,” “believes,” “estimates,” and similar expressions. These statements are based on our current beliefs, expectations, and assumptions and are subject to “Virpax,”a number of risks and uncertainties, many of which are difficult to predict and generally beyond our control, that could cause actual results to differ materially from those expressed, projected or implied in or by the “Company,”forward-looking statements.
You should refer to Item 1A. “Risk Factors” section of this Annual Report for a discussion of important factors that may cause our actual results to differ materially from those expressed or implied by our forward-looking statements. As a result of these factors, we cannot assure you that the forward-looking statements in this Annual Report will prove to be accurate. Furthermore, if our forward-looking statements prove to be inaccurate, the inaccuracy may be material. In light of the significant uncertainties in these forward-looking statements, you should not regard these statements as a representation or warranty by us or any other person that we will achieve our objectives and plans in any specified time frame, or at all. We do not undertake any obligation to update any forward-looking statements. Unless the context requires otherwise, references to “we,” “us,” or “our” mean“our,” and “Virpax,” refer to Virpax Pharmaceuticals, Inc. and its subsidiaries unless we state otherwise or the context otherwise indicates.subsidiaries.
Our CompanySummary Risk Factors
Risks Related to Our Financial Position and Need for Additional Capital
| ● | We are a preclinical stage biopharmaceutical company with a limited operating history. |
| ● | We have incurred losses since inception and anticipate that we will continue to incur losses for the foreseeable future. We are not currently profitable, and we may never achieve or sustain profitability. |
| ● | The report of our independent registered public accounting firm for the fiscal years ended December 31, 2023 and 2022 contains an explanatory paragraph regarding substantial doubt about our ability to continue as a going concern. |
| ● | We will require additional capital to fund our operations, we may not be able to raise additional capital, and if we fail to obtain necessary financing, we will not be able to complete the development and commercialization of our drugs. |
| ● | Raising additional capital will cause dilution to our stockholders, restrict our operations or require us to relinquish rights to our technologies or product candidates. |
| ● | Our ability to use our net operating loss carryforwards to offset future taxable income will be subject to certain limitations. |
| ● | Our ability to further develop our product candidates will be adversely affected by the terms of the Settlement Agreement (See Part 1-Item 1A-Risk Factors, Item 3-Legal Proceedings and Recent Developments – Litigation). |
| ● | The Company has been affected by significant litigation which requires the Company to pay an additional $2.5M by July 1, 2024, as part of the Settlement Agreement, and may in the future be affected by new litigation and indemnification and contribution claims related to our recently settled litigation with Sorrento Therapeutics, Inc. and Scilex Pharmaceuticals, Inc., which such claims may be material, and potential estimated separation payments that we make to our former Chief Executive Officer, which may be material. In addition, our officers and directors may be subject to various types of litigation in the future, and our insurance may not be sufficient to cover damages related to those claims. See “Risk Factor- Our business, financial condition and results of operations could be adversely affected by indemnification and other claims related to the damages awarded in our recently settled litigation with Sorrento Therapeutics, Inc. and Scilex Pharmaceuticals, Inc”, (See Part 1-Item 1A-Risk Factors, and Item 3-Legal Proceedings and Recent Developments – Litigation). |
Risks Related to Development, Clinical Testing, Manufacturing and Regulatory Approval
| ● | Clinical trials are expensive, time-consuming and difficult to design and implement, and involve an uncertain outcome. |
| ● | Disruptions in the global economy and supply chains may have a material adverse effect on our business, financial condition and results of operations. |
| ● | Adverse global conditions, including economic uncertainty, may negatively impact our financial results. |
| ● | Our development activities for Probudur are conducted in Israel. The war in the Middle East, may affect our operations. |
| ● | The regulatory approval processes of the FDA and comparable foreign authorities are lengthy, time consuming and inherently unpredictable, and if we are ultimately unable to obtain regulatory approval for our Product Candidates or any other product candidates, our business will be substantially harmed. |
| ● | If we are unable to file for approval of Epoladerm and Probudur under Section 505(b)(2) of the FDCA or if we are required to generate additional data related to safety and efficacy in order to obtain approval under Section 505(b)(2), we will be unable to meet our anticipated development and commercialization timelines. |
| ● | Enrollment and retention of patients in clinical trials is an expensive and time-consuming process and could be made more difficult or rendered impossible by multiple factors outside our control. |
| ● | The COVID-19 pandemic has adversely affected our business and a resurgence of COVID-19 or another health epidemic or pandemic may have an adverse impact on our business in the future. |
| ● | Results of preclinical studies, early clinical trials or analyses may not be indicative of results obtained in later trials. |
| ● | Interim “top-line” and preliminary data from our clinical trials that we announce or publish from time to time may change as more patient data become available and are subject to audit and verification procedures that could result in material changes in the final data. |
| ● | Our product candidates may cause serious adverse events or undesirable side effects, which may delay or prevent marketing approval, or, if approved, require them to be taken off the market, require them to include safety warnings or otherwise limit their sales. |
| ● | The market opportunities for our Product Candidates, if approved, may be smaller than we anticipate. |
| ● | We have never obtained marketing approval for a product candidate and we may be unable to obtain, or may be delayed in obtaining, marketing approval for any of our product candidates. |
| ● | Even if we obtain FDA approval for our Product Candidates or any other product candidate in the United States, we may never obtain approval for or commercialize our Product Candidates or any other product candidate in any other jurisdiction, which would limit our ability to realize their full market potential. |
| ● | Even if we obtain regulatory approval for our Product Candidates or any product candidate, we will still face extensive and ongoing regulatory requirements and obligations and any product candidates, if approved, may face future development and regulatory difficulties. |
| ● | Potential product liability lawsuits against us could cause us to incur substantial liabilities and limit commercialization of any products that we may develop. |
Risks Related to Commercialization
| ● | We face significant competition from other biotechnology and pharmaceutical companies and our operating results will suffer if we fail to compete effectively. |
| ● | The successful commercialization of our Product Candidates and any other product candidate we develop will depend in part on the extent to which governmental authorities and health insurers establish adequate coverage, reimbursement levels, and pricing policies. Failure to obtain or maintain coverage and adequate reimbursement for our product candidates, if approved, could limit our ability to market those products and decrease our ability to generate revenue. |
| ● | Even if our Product Candidates or any product candidate we develop receives marketing approval, it may fail to achieve market acceptance by physicians, patients, third-party payors or others in the medical community necessary for commercial success. |
| ● | If we are unable to establish sales, marketing, and distribution capabilities either on our own or in collaboration with third parties, we may not be successful in commercializing our Product Candidates, if approved. |
| ● | A variety of risks associated with operating internationally could materially adversely affect our business. |
Risks Related to Our Dependence on Third Parties
| ● | Our employees and independent contractors, including principal investigators, CROs, consultants, vendors, and any third parties we may engage in connection with development and commercialization, may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements, which could have a material adverse effect on our business. |
| ● | We currently rely on third-party contract manufacturing organizations (“CMOs”) for the production of clinical supply of our Product Candidates and intend to rely on CMOs for the production of commercial supply of our Product Candidates, if approved. Our dependence on CMOs may impair the development and commercialization of the drug, which would adversely impact our business and financial position. |
| ● | We intend to rely on third parties to conduct, supervise and monitor our clinical trials. If those third parties do not successfully carry out their contractual duties, or if they perform in an unsatisfactory manner, it may harm our business. |
Risks Related to Healthcare Laws and Other Legal Compliance Matters
| ● | Enacted and future healthcare legislation may increase the difficulty and cost for us to obtain marketing approval of and commercialize our product candidates, if approved, and may affect the prices we may set. |
| ● | Our business operations and current and future relationships with investigators, healthcare professionals, consultants, third-party payors, patient organizations, and customers will be subject to applicable healthcare regulatory laws, which could expose us to penalties. |
| ● | Any clinical trial programs we conduct or research collaborations we enter into in the European Economic Area may subject us to the General Data Protection Regulation. |
| ● | We are subject to environmental, health and safety laws and regulations, and we may become exposed to liability and substantial expenses in connection with environmental compliance or remediation activities. |
Risks Related to Our Intellectual Property
| ● | If we fail to comply with our obligations under our existing intellectual property licenses, we risk losing the rights to our intellectual property. |
| ● | If we are unable to obtain and maintain patent protection for our technology, products, and product candidates or if the scope of the patent protection obtained is not sufficiently broad, we may not be able to compete effectively in our markets. |
| ● | We may become subject to third parties’ claims alleging infringement of their patents and proprietary rights, or we may need to become involved in lawsuits to protect or enforce our patents, which could be costly, time consuming, delay or prevent the development and commercialization of our products and product candidates or put our patents and other proprietary rights at risk. |
| ● | We may not identify relevant third-party patents or may incorrectly interpret the relevance, scope or expiration of a third-party patent, which might adversely affect our ability to develop, manufacture and market our products and product candidates. |
| ● | Changes in patent laws or patent jurisprudence could diminish the value of patents in general, thereby impairing our ability to protect our products and product candidates. |
| ● | Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements. |
| ● | We enjoy only limited geographical protection with respect to certain patents and we may not be able to protect our intellectual property rights throughout the world. |
| ● | If we do not obtain patent term extension in the United States under the Hatch-Waxman Act and in foreign countries under similar legislation, thereby potentially extending the term of marketing exclusivity for our products, our business may be materially harmed. |
| ● | Intellectual property rights do not address all potential threats to our competitive advantage. |
| ● | Our reliance on third parties requires us to share our trade secrets, which increases the possibility that our trade secrets will be misappropriated or disclosed, and confidentiality agreements with employees and third parties may not adequately prevent disclosure of trade secrets and protect other proprietary information. |
| ● | If our trademarks and trade names are not adequately protected, then we may not be able to build name recognition in our markets of interest and our business may be adversely affected. |
| ● | We may need to license certain intellectual property from third parties, and such licenses may not be available or may not be available on commercially reasonable terms. |
| ● | We may be subject to claims that our employees, consultants, collaborators contractors or advisors have wrongfully used or disclosed confidential information of their former employers or other third parties. |
| ● | Our proprietary information may be lost, or we may suffer security breaches. |
Risks Related to Our Employees, Managing Our Growth and Our Operations
| ● | We have experienced turnover in our senior management team, and the loss of one or more of our executive officers or key employees or an inability to attract and retain highly skilled employees could adversely affect our business. |
| ● | We expect to expand our development, regulatory, and sales and marketing capabilities, and as a result, we may encounter difficulties in managing our growth, which could disrupt our operations. |
| ● | We may engage in acquisitions that could disrupt our business, cause dilution to our stockholders or reduce our financial resources. |
| ● | Our business and operations would suffer in the event of system failures. |
| ● | We are increasingly dependent on information technology, and our systems and infrastructure face certain risks, including cybersecurity and data leakage risks. |
Risks Related to Our Common Stock
| ● | Our failure to meet the continued listing requirements of The Nasdaq Capital Market could result in a de-listing of our common stock. |
| ● | The market price of our common stock has been volatile and can fluctuate substantially, which could result in substantial losses for purchasers of our common stock. |
| ● | We could be subject to securities class action litigation. |
| ● | Our directors, executive officers and certain stockholders (one of which is an affiliate of our former Chief Executive Officer) will continue to own a significant percentage of our common stock and, if they choose to act together, will be able to exert significant control over matters subject to stockholder approval. |
| ● | If securities or industry analysts do not publish research or reports about our business, or if they issue an adverse or misleading opinion regarding our common stock, our stock price and trading volume could decline. |
| ● | Because we do not anticipate paying any cash dividends on our common stock in the foreseeable future, capital appreciation, if any, will be your sole source of gain. |
| ● | We have incurred and will continue to incur increased costs as a result of operating as a public company, and our management will be required to devote substantial time to new compliance initiatives and corporate governance practices. |
| ● | We are an “emerging growth company,” and the reduced reporting requirements applicable to emerging growth companies may make our common stock less attractive to investors. |
| ● | Anti-takeover provisions contained in our certificate of incorporation and bylaws, as well as provisions of Delaware law, could impair a takeover attempt. |
| ● | Our certificate of incorporation, as amended, designates the Court of Chancery of the State of Delaware as the sole and exclusive forum for certain types of actions and proceedings that may be initiated by our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers or other employees. |
Our Company
We are a preclinical-stage pharmaceutical company focused on developing novel and proprietary drug delivery systems across various pain indications in order to enhance compliance and optimize each product candidate in our pipeline. Our drug-delivery systems and drug-releasing technologies being developed are focused on advancing non-opioid and non-addictive pain management treatments and treatments for central nervous system (“CNS”) disorders to enhance patients’ quality of life.
We have exclusive global rights to the following proprietary patented technologies: (i) Molecular Envelope Technology (“MET”) that uses an intranasal device to deliver enkephalin for the management of acute and chronicsevere pain, including post cancer pain associated with cancer (Envelta™) and PTSD (PES200)post-traumatic stress disorder (“PTSD”), (ii) Injectable “local anesthetic” Liposomal Gel Technology for postoperative pain management (Probudur™), and (iii) Topical Spray Film Delivery Technology for osteoarthritis pain (Epoladerm™). We intend to utilize these delivery technologies to selectively develop a portfolio of patented 505(b)(2) and new chemical entity (“NCE”) candidates for commercialization. We also have exclusive global rights to develop and commercialize an investigationalInvestigational formulation delivered via the nasal route to enhance pharmaceutical-grade cannabidiol (“CBD”) transport to the brain (“NobrXiolTMNobrXiol™”, formerly VRP324) to potentially treat epileptic seizures associated with tuberous sclerosis complex (“TSC”), Lennox-Gastaut syndrome (LGS) and Dravet syndrome (DS) in pediatric patients one yeartwo years of age and older.
While we We are currently focused on advancing non-opioid and non-addictive pain management treatments and treatmentsalso exploring value creative opportunities for CNS disorders, we also plan on using our proprietary delivery technologies to develop our anti-viral therapy (AnQlar™)two nonprescription product candidates including seeking regulatory approval for commercialization of such products: AnQlarTM, which is being developed as a 24 hour prophylactic viral barrier to potentially preventinhibit viral infection by influenza or reduce the risk or the intensity of viral infections in humans, including, but not limitedSARS-CoV-2, and Epoladerm™, which is a topical diclofenac epolamine metered dosed spray film formulation being developed to influenza and SARS-CoV-2 (“COVID 19”).We have exclusive global rights for AnQlar pursuant to a collaboration and license agreement.manage pain associated with osteoarthritis.
Our PortfolioRecent Developments
Reverse Stock Split
On February 29, 2024, we filed a certificate of amendment to our Amended and Restated Certificate of Incorporation for purposes of effecting a 1-for-10 reverse stock split (the “Reverse Split”) of our outstanding shares of common stock such that, effective upon March 1, 2024, the day after the filing thereof, every 10 issued and outstanding shares of our common stock were subdivided and reclassified into one validly issued, fully paid and non-assessable share of our common stock.
Litigation
On February 29, 2024, Sorrento Therapeutics, Inc. (“Sorrento”), and Scilex Pharmaceuticals Inc. (“Scilex” and together with Sorrento, the “Plaintiffs”) and the Company entered into a Settlement Agreement and Mutual Release (the “Settlement Agreement”) to fully resolve all issues related to the litigation with Plaintiffs captioned Sorrento Therapeutics, Inc. and Scilex Pharmaceuticals Inc. v. Anthony Mack and Virpax Pharmaceuticals, Inc., Case No. 2021-0210-PAF (the “Action”), subject to the entry by the United States Bankruptcy Court for the Southern District of Texas, which is handling the Sorrento bankruptcy filing (the “Bankruptcy Court”), of an order approving the Settlement Agreement (the “Settlement Order”). On March 1, 2024, the Plaintiffs filed a motion to approve the Settlement Agreement and grant the related relief with the Bankruptcy Court. On March 14, 2024, the Bankruptcy Court entered an order approving the Settlement Agreement and on March 20th the Plaintiffs filed a Stipulation of Dismissal with the Chancery Court dismissing the Action. See “Part I—Item 3—Legal Proceedings” for additional information regarding the litigation with the Plaintiffs.
As settlement consideration, the Company agreed to pay Sorrento and Scilex a total cash payment of $6 million, of which $3.5 million was paid two business days after the date that the Settlement Order was entered by the Bankruptcy Court (the “Effective Date”), which payment was made on March 18, 2024 and the remaining $2.5 million is to be paid on or before July 1, 2024. Additionally, the Company agreed to pay to Plaintiffs royalties of 6% of annual net sales of products developed from drug candidates Epoladerm, Probudur and Envelta until the earlier of the expiration of the last-to-expire valid patent claim of such product and the expiration of any period of regulatory exclusivity for such product.
Pursuant to the Settlement Agreement, each of the Plaintiffs and the Company provided mutual releases of all claims as of the Effective Date, whether known or unknown, arising from any allegations set forth in the Action. Plaintiffs’ release relates to claims against the Company only. Plaintiffs’ release as to the Company was effective upon the Company’s initial payment of $3.5 million, and the Company’s release of the Plaintiffs was effective on the Effective Date.
Our Portfolio
Our portfolio currently consists of multiple preclinical stage product candidates: Epoladerm, Probudur, Envelta, PES200, AnQlar and NobrXiol. In the accompanying section we will describe each product candidate, its benefits, and our market strategy for each product candidate. The dates reflected in the below table are estimates only, and there can be no assurances that the events included in the table will be completed on the anticipated timeline presented, or at all.


We are also developing Envelta for a second indication, PES200, which utilizes the same delivery mechanism as Envelta. PES200 enables the delivery of a metabolically labile peptide drug (Enkephalin) into the brain for post-traumatic stress disorder.
1
Long-acting Bupivacaine Liposomal-gel 3.0% (LBL100 or ProbudurTM™)
Probudur is a drug product candidate based on a unique liposomal delivery system utilizing large multi-vesicularmulti-lamellar vesicles (“LMVVs”MLV”) encapsulating a high dose of the local anesthetic bupivacaine. These drug-loaded liposomes are composed of lecithin and cholesterol which are GRAS by the FDA. These LMVVs are embedded in hydrogel beads to form a Lipogel. The system delivers a local analgesic medicine from the Lipogel. Early non-clinicalpreclinical animal studies produced data which suggestsdemonstrated that Probudur may be able to provideprovided significantly improved onset and duration and peak performance propertiesof analgesic effect as compared to a similar product on the market. The animal studies were conducted by administering Probudur byinfiltrating the surgical/wound site with Probudur. Probudur’s prolonged effectiveness is due to the formulation’s ability to keep the local infiltration of the surgical site which resulted in keeping the active ingredient localizedanesthetic at the surgicalsurgical/wound site for a longeran extended period of time.time (at least 96 hours). Four nonclinical trials were conducted using three animal models. Data from these animal studies showed that after treatment with Probudur (50 mg/kg), statistically demonstrated significant analgesic activity (measured as threshold pressure at animal’s withdrawal of the treated extremity) was observed in comparison to control (vehicle), for as long as 96 hours post-treatment (22.33±3.67g vs 5.00±0.58g; p<0.05), which is 24 hours longer than the leading product on the market.
If we are able to demonstrate a successful Phase III clinical trial, we believe Probudur may represent the first long actinglong-acting local anesthetic with an opioid sparing label. The slow release of the drug from the liposomal depot reduces the peak plasma levels, reducing toxicity while also potentially providing longer-lasting post-operativepostoperative pain control. We believe this property may permit administration of higher bupivacaine doses (3% versus 1.3% in leading market product); however, there can be no assurances, based on these animal studies, that Probudur will be safe and effective as these determinations are solely within the authority of the FDA. Further, there can be no assurance that Probudur will receive FDA approval.
Image 2 below illustrates the results of the early animal studies of Probudur:

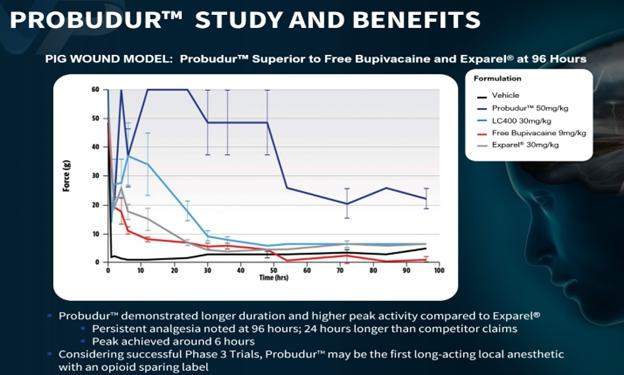
Image 2
In September 2023, we announced results of two pre-clinical Probudur dose escalation studies, where superior efficacy was demonstrated in head-to-head animal studies against Exparel®. The first study compared Probudur to Exparel utilizing a planar incision model. Two doses of Probudur, at 3 mg and 6 mg, were administered to rats. The results demonstrated three times longer efficacy for Probudur than Exparel. In the second study, two different formulations at the same dose of Probudur were compared to Exparel in rat incision models. In this study, Probudur demonstrated a four to five times longer effect than the comparable product.
In the studies, Probudur demonstrated longer duration and higher peak activity compared to Exparel. Persistent analgesia (sustained pain control) with Probudur was noted at 96 hours, which is 24 hours longer than claims of our competitors. Probudur also demonstrated biphasic release for fast onset and duration and peak activity at 6 hours with a single injection. Additional studies for efficacy, toxicity, and pharmacokinetics are ongoing. Assuming the success of our Phase 3 trials, we believe that Probudur has the potential to receive Opioid Sparing Label.
2
We plan to market Probudur to general surgeons, anesthesiologists, and orthopedic surgeons within the $35 billion post operativepostoperative pain management market local anesthetic post-surgical market. If the product candidate isBased on head-to-head preclinical studies compared to an approved liposomal bupivacaine formulation, if used appropriately, we believe this product candidate could potentiallyProbudur has the potential to eliminate or significantly reduce the need forto prescribe opioids for post-operativepostoperative pain relief. As a result of our IND review,pre-IND meeting, the FDA has indicated that it is reasonable for us to pursue a 505(b)(2) accelerated NDA for Probudur. There can be no assurance that we will be successful in securing regulatory approval under the 505(b)(2) pathway or that we will be successful in mitigating risks associated with the clinical development of this product candidate.
Image 3, below, displays the planned delivery of Probudur at the wound site:
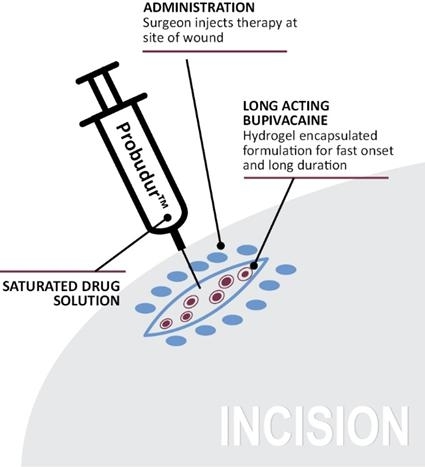
Image 3
Charles River Laboratories was engaged to perform seven preclinical animal studies during the second half of 2021, including method, dosage, and toxicity as part The development of the required FDA enabling trials for an IND filing for Probudur. However, we elected to strategically delay these trialsProbudur formulation was successfully completed in order to enhance the formulationthird quarter of Probudur to increase stability for manufacturing purposes and to possibly extend the lifetime of a relevant patent.2023. We anticipate this relevant provisional patentpatents will be filed at some point duringin the first half of 2023.2024. Lipocure RX, Ltd. (“Lipocure”) is currently in the process of working through the scale up of Probudur to a larger batch size. If Lipocure is able to achieve scale up of the enhanced formula, we anticipate IND enabling studies have started. The FDA minutes indicated that we are to commence towardsinitiate our clinical studies in targeted patient populations following the endcompletion of 2023.our nonclinical toxicity studies. We anticipate filing an IND in 2024; however, we may need to adjust this timeline if Lipocure, a company based in Israel, becomes unable to continue development work due to the war in the Middle East.
Image 3, below, displays the planned delivery of Probudur at the wound site:

Image 3
Current Development Status
Our research and development activities for our product candidates are performed for us by third parties that we contract with.
On June 30, 2021, we entered into an Agreement for Rendering of Research Services with Yissum Research Development Company of the Hebrew University of Jerusalem Ltd (“Yissum”) (the “June 2021 Yissum Research Agreement”). Under the June 2021 Yissum Research Agreement, we provided funding for research and development studies performed by researchers at Hebrew University related to the optimization of the Liposomal Bupivacaine formulation of Probudur and to increase stability for manufacturing purposes. In consideration for the research services, we paid research service fees of $337,500 in six equal quarterly installments. All services provided$337,500. On January 31, 2023, we entered into an Agreement for Rendering of Research Services with Yissum (the “January 2023 Yissum Research Agreement”) on substantially similar terms and conditions as detailed above under the June 2021 Yissum Research Agreement. In consideration for the research services, we paid research service fees of $326,000. On January 1, 2024, we entered into an Agreement initiatedfor Rendering of Research Services with Yissum (the “January 2024 Yissum Research Agreement”) for additional work on July 1, 2021formulation, method development, animal studies and were substantially complete bypatent related work. In consideration for the endresearch services, we will pay research service fees of 2022.$343,467 in four equal quarterly installments. We may terminate the agreement at any time and will only be responsible to pay Yissum for work performed through the date of termination.
3
On June 29, 2021, we entered into an Agreement for Rendering of Research Services with Lipocure RX, Ltd. (the “June 2021 Lipocure Research Agreement”). Under the June 2021 Lipocure Research Agreement, we shall provideprovided funding for research and development related to the optimization of the Liposomal Bupivacaine formulation of Probudur and eventual manufacturing of preclinical batches including for stability testing, animal studies, toxicology, and toxicologypatent related work. This will also include work associated with the potential filing of additional provisional patent applications. We may terminate the agreement at any time upon 30 days written notice and shall be only responsible to pay Lipocure for work performed through the date of such notice. In consideration for the research services, we agreed to paypaid research service fees of $200,000 upon execution, as well as $400,000 in July 2021, $270,000 in both September 2021 and January 2022, and three additional payments$1,890,000. On February 1, 2023, we entered into an Agreement for Rendering of $270,000 during 2022. We also agreed to pay $250,000 to Lipocure upon successful completion of Chemistry, Manufacturing and Controls (“CMC”) filing with the FDA. All services to be provided under the June 2021Research Services (the “February 2023 Lipocure Research Agreement initiatedAgreement”) with Lipocure on July 1, 2021similar terms and were substantially complete byconditions and for similar services - optimization of the endLiposomal Bupivacaine formulation, manufacture of 2022. We recorded $990,000pre-clinical batches including batches for stability testing, animal studies and $870,000 in research and development expensetoxicology work. In consideration for the yearsresearch services, we paid research service fees of $1,453,000 for the year ended December 31, 2022 and 2021, respectively, associated with this agreement.2023.
On April 28, 2022, we entered into a cooperative research and development agreement (“CRADA”) with the U.S. Army Institute of Surgical Research (“USAISR”) to evaluate Probudur as well as thea potential novel analgesics for our other potential product candidates.battlefield injury-induced pain solution. The current research project will evaluate the analgesic effectiveness and physiologic effects of Probudur. ThisThe initial term of the agreement will automaticallywas to expire on September 30, 2023 unless it iswas revised by mutual written agreement. The CRADA was modified and signed on October 10, 2023, and extended the terms of the agreement until September 2024. No funding is being provided by either party to the other party under the agreement. Each party is responsible for funding its own work performed and other activities undertaken for the research project under this agreement. The parties may elect to terminate this agreement, or portions thereof, at any time by mutual consent. Either party may unilaterally terminate this entire agreement at any time by giving the other party written notice, not less than thirty (30) days prior to the desired termination date.
On January 31, 2023, we entered into an Agreement for Rendering of Research Services with Yissum (the “January 2023 Yissum Research Agreement”) on substantially similar terms and conditions as detailed above under the June 2021 Yissum Research Agreement. Under the January 2023 Yissum Research Agreement, we will provide funding for research and development studies to be performed by researchers at Hebrew University related to the optimization of the Liposomal Bupivacaine formulation and to increase stability for manufacturing purposes. We may terminate the agreement at any time and will only be responsible to pay Yissum for work performed through the date of termination. In consideration for the research services, we agreed to pay aggregate research service fees of $326,000 in four equal quarterly installments ($81,500 per calendar quarter). All services to be provided under the January 2023 Yissum Research Agreement initiated on January 1, 2023, and are anticipated to be completed by the end 2023.
On February 1, 2023, we entered into an Agreement for Rendering of Research Services (the “January 2023 Lipocure Research Agreement”) with Lipocure. Under the January 2023 Lipocure Research Agreement, we shall provide funding for research and development related to the optimization of the Liposomal Bupivacaine formulation and eventual manufacture of pre-clinical batches including batches for stability testing, animal studies and toxicology work. This will also include work associated with the potential filing of additional provisional patent applications. We may terminate the agreement at any time upon 30 days written notice and shall be only responsible to pay Lipocure for work performed through the date of such notice. In consideration for the research services, we agreed to pay research service fees of $1,286,000 in four equal quarterly installments ($321,500 per calendar quarter). All services to be provided under the January 2023 Lipocure Research Agreement initiated on January 1, 2023, and are anticipated to be completed towards the end 2023.
Molecular Envelope Technology Enkephalin Intranasal Spray (EnveltaTM™)
EnveltaTM™ is a nanotechnology-based intranasal spray drug product candidate which enables the delivery of a metabolically labile peptide drug (Enkephalin) into the brain. It is manufactured using high pressure homogenization and spray drying. There is pharmacological evidence of activity of MET enabled enkephalin in morphine-tolerant animals. Preclinical studies were conducted in animals for between 6 and 28 days through intravenous, oral and intranasal dosing. Twelve studies were conducted using three animal models whereby the animal studies were aimed at determining safety pharmacology and genetic toxicology. The preliminary data from these early animal studies of Envelta have shown that Envelta exhibited pain control in morphine tolerant animals, without the development of tolerance itself. These animal models tested the anti-hyperalgesic effects in rats against evoked stimuli in a model of chronic inflammatory pain and against ongoing neuropathic pain in a conditioned placement preference model with spinal nerve ligation. Envelta and morphine were compared at the same dose level of 7.5 mg kg-1in this model and Envelta was determined to have a similar analgesic effect. With respect to respiratory depression, delta opioid receptor agonists may actually reverse the respiratory depression caused by morphine agonists, meaning that we believe Envelta will be unlikely to cause respiratory depression. However, there can be no assurances, based on these preclinical animal studies, that Envelta will be safe and effective. Further, there can be no assurance that Envelta will receive FDA approval.
We believe we have identified a large unmet need and market opportunity for current prescribers of opioids, including pain and hospice treatment centers. Currently, these prescribers may be using morphine-like opioids, which target three opioid receptors: mu, delta and kappa. Most analgesics used clinically target mu receptor, however, this receptor is also responsible for the majority of undesirable side effects associated with opioids. Currently, enkephalins are limited in their therapeutic potential by their pharmacokinetic profiles due to their inability to cross the blood-brain barrier to reach opioid receptors located in the central nervous system. However, we believe Envelta’s novel nasally delivered formulation, based on early animal studies, enhances enkephalin transport to the brain by protecting the drug in a molecular envelope (MET),MET, facilitating its crossing of the blood-brain barrier. Enkephalins, which are naturally occurring analgesics that are quickly metabolized in the body and lack an endogenous mechanism to allow them to cross the blood-brain barrier and reach their target delta-receptor, bind predominantly to the delta-receptor which is typically not associated with the dangers associated with opioids. Envelta has many competitive advantages including naturally occurring analgesics in our body, delta receptor agonist, the absence of liver first pass effect, and a comparable efficacy to morphine. We believe Envelta may have analgesic potential without opioid tolerance, and has not exhibited any indications of withdrawal, respiratory depression, euphoria, or addiction in the early animal studies. A study published in Proceedings of the National Academy of Sciences (PNAS) indicates, “Delta opioid receptors have a built-in mechanism for pain relief and can be precisely targeted with drug-delivering nanoparticles, making them a promising target for treating chronic inflammatory pain with fewer side effects.” There can be no assurances, based on these preclinical animal studies, that Envelta will be safe and effective in human trials.
4
Additionally, we believe Envelta may significantly reduce constipation and early animal clinical trials have not demonstrated any opioid dependence, drug seeking or respiratory depression. We plan to use the endogenous NCE regulatory pathway to bring this product candidate to market. We plan to target our marketing and selling efforts to pain specialists, anesthesiologists, orthopedics, surgeons, PCPs, Nurse Practitioners (“NPs”), oncologists, and neurologists within the $20 billiontreating patients with severe pain, including post cancer pain, and non-cancer pain market.which market is valued at approximately $20 billion.
Envelta is a neuroactive peptide drug product (enkephalin) with a proprietary composition formulated for administration by all routes except the topical route. A preassembled device and cartridge would be used to propel the enkephalin formulation through the nose to the brain via the olfactory nerve/bulb route of transmission. A MET will encapsulate the drug product, protecting it from degradation, and help to carry the drug across the blood-brain barrier to promptly suppress pain.
Image 4 and Image 5, below, display the planned delivery of enkephalin peptide nanoparticles to the brain via the olfactory route:
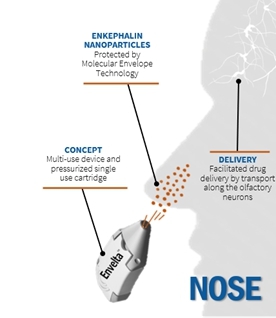

Image 4

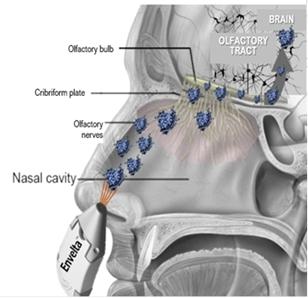
Image 5
5
Image 6 below illustrates the concept Envelta delivery device:


Image 6
We may also develop Envelta for a second indication utilizing the same delivery mechanism as Envelta, which we refer to as PES200. PES200 enables the delivery of a metabolically labile peptide drug (Enkephalin) into the brain and is also covered under the same intellectual property listed elsewhere in this Annual Report on Form 10-K for Envelta. We believe PES200’s capabilities and attributes are the same as Envelta and we may be able to reference the Envelta PreIND and possible Phase I studies to open a potential PES200 IND followed by potential Phase II trials. Our plan is to validate proof-of-concept followed by IND-enabling studies for the development of a novel enkephalin-based formulation to treat Post-Traumatic Stress Disorder.
Current Development Status
On August 25, 2020, we entered into the CRADA with NCATS. This collaboration is for the continued development of our product candidate, Envelta, an intranasal peptide, for the management of acute and chronic non-cancerto control severe pain, including post cancer pain. The term of the CRADA is for a period of four years from the effective date of the agreement and can be terminated by both parties at any time by mutual written consent. In addition, either party may unilaterally terminate the CRADA at any time by providing written notice of at least sixty (60) days before the desired termination date. The agreement provides for studies that are focused on the pre-clinical characterization of Envelta as a novel analgesic for acute and chronic non-cancerto control severe pain, including post cancer pain., and for studies to further develop Envelta through IND enabling studies. There are certain development “Go/No Go” provisions within the agreement whereby, if certain events occur, or do not occur, NCATS may terminate the CRADA. These “No GO” provisions include: i) lack of efficacy in all animal pain models, ii) no reliable and sensitive bioanalytical method can be developed, iii) manufacturing failure due to inherent process scalability issues, iv) unacceptable toxicity or safety profile to enable clinical dosing, and v) inability to manufacture the Envelta dosage form. As of March 25, 2024, we have not received any Go/No Go notifications from NCATS.
With respect to NCATS rights to any invention made solely by an NCATS employee(s) or made jointly by an NCATS employee(s) and our employee(s), the CRADA grants to us an exclusive option to elect an exclusive or nonexclusive commercialization license. For inventions owned solely by NCATS or jointly by NCATS and us, and licensed pursuant to our option, we must grant to NCATS a nonexclusive, nontransferable, irrevocable, paid-up license to practice the invention or have the invention practiced throughout the world by or on behalf of the United States government. For inventions made solely by an employee of ours, we grant to the United States government a nonexclusive, nontransferable, irrevocable, paid-up license to practice the invention or have the invention practiced throughout the world by or on behalf of the United States government for research or other government purposes.
6
We believe Envelta and PES200 could support the current effort amongmay provide prescribers, regulators, and patients to seekalternative non-addictive treatment options for pain.to control severe pain, including post cancer pain and potentially manage symptoms related to PTSD. We plan to utilize these delivery technologies to selectively develop a portfolio of patented 505(b)(2) and NCE candidates for commercialization. We intend to use these studies as a source for INDs for two additional potential indications, cancer pain and post-traumatic stress disorder. To date, all fourFour planned in vitro studies have beenwere successfully completed as well as the in vivo acute efficacy studies. In February 2022, we completed a 14-day intranasal dose range finding toxicity study of Envelta in rats with a 14-day recovery period which showed no adverse related findings in hematology, coagulation and serum chemistry data, with no treatment related toxicology findings or mortality noted. A 14-day intranasal dose range finding toxicity study of Envelta in dogs with a 14-day recovery period was also conducted and showed no adverse toxicologic findings. Bioavailability studies are anticipated to be completed by end of March 2023 and the in vivo chronic efficacy studies are being planned and are estimated to be completed sometime during the second half of 2023.
The preclinical manufacturing work being performedstudies under the CRADA necessaryare expected to filecontinue over the IND is ongoing. NCATS is working with its manufacturer on scaling up production of Envelta and anticipates completion of this task in the second half of 2023.next nine months. We currently anticipate filing an IND with Envelta duringin 2024. However, the second quarterIND timing is subject to risks in manufacturing of 2024.the MET/LENK, COA for GMP material and filling of cartridges for a 28-day dog bridging study and may be extended into 2025.
NobrXiol
Cannabidiol Nanoparticles with Molecular Envelope Technology (NobrXiol™)
NobrXiol
NobrXiol™ is being developed by Nanomerics Ltd. (‘Nanomerics”) as an investigational formulation delivered via the nasal route that uses MET as its delivery system to enhance Cannabidiol (“CBD”) transport to the brain. CBD acts on CB receptors of the endocannabinoid system in the brain, which regulates neuronal excitability response relevant to the pathophysiology of epilepsy. NobrXiol uses a proprietary preassembled delivery device that holds single-use cartridges that are sealed in inert gas and pressurized for easy activation that can be self-administered. Activation of the cartridge system to propelpropels the CBD powder formulation into the nose and to the brain via the olfactory nerve/bulb. This product candidate will be formulated to potentially treat epileptic seizures associated with Lennox-Gastaut syndrome (LGS) and Dravet syndrome (DS) in pediatric patients two years of age and older. Lennox-Gastaut syndrome (LGS) and Dravet syndrome (DS) are rare central nervous system diseases considered serious epileptic encephalopathies that cause different types of epileptic seizures, as well as cognitive and behavioral changes, and are generally resistant to treatment. The FDA previously granted Orphan Drug Designation for another drug for the treatment of the same diseases. Therefore, NobrXiol may also be able to receive Orphan Drug Designation for the treatment of Lennox-Gastaut syndrome (LGS) and Dravet syndrome (DS) in pediatric patients. NobrXiol has many potential competitive advantages including fast onset of action, reduced peripheral side effects, no liver first-pass metabolism, avoidance of drug to drug interactions, no gastrointestinal interaction, and the potential to eliminate enzymatic deactivation. On September 17, 2021, we entered into a collaboration and license agreement with Nanomerics (the “Nanomerics License Agreement – NobrXiol”) for the exclusive worldwide license to develop and commercialize the product candidate. We plan to target our marketing and selling efforts to healthcare practitioners specializing in pediatric epilepsy within the $16.5$16.915 billion market for managing epilepsy in pediatrics and adults.pediatrics. We have engaged Destum Partners to search for a Global Animal Healthcare sublicensing partner.
On April 21, 2022, we notified Nanomerics that the study aim of demonstrating the ability of Nanomerics platform technology delivering CBD to the brain via nasal administration in an animal model was met. Pursuant to the Nanomerics License Agreement - NobrXiol, we paid a milestone payment of $500,000 upon meeting this study aim in April 2022. We submitted the pre-IND Briefing Book with the FDA in October 2022 and received comments back from the FDA in December 2022. Upon our review of the FDA response letter,minutes, we now believe we have the appropriate guidance from the FDA to move forward with our overall development plan for this new product candidate and the ability to identify any need for further data prior to submitting the IND. Our current plan is to utilize potential grant awards to fund the development of NobrXiol through to an IND filing whilefiling. In April 2023, we focusentered into a participation agreement with the National Institute of Neurological Disorders and Stroke (“NINDS”), a part of NIH, to supply our cash resources on more immediate needs with regardproduct candidate compounds to the NINDS’s Epilepsy Therapy Screening Program (“ETSP”). NINDS ETSP will test our other product candidates.compounds in epilepsy animal models to determine whether our compounds have activity against resistant epilepsy and related disorders.
Current Development Status
An acute seizure model study, Maximal Electroshock Seizures (MES) in rats, is currently ongoing at NINDS ETSP. Once that study is complete, NINDS plans to explore behavioral tolerability screens, and chronic seizure models. There are also plans to examine the differentiation with various models for acute and sub-chronic dosing and to explore ancillary drug resistant epilepsy models.
Diclofenac Epolamine Spray Film (Epoladerm)(Epoladerm™)
We believe the Topical Spray Film Delivery Technology, which we refer to as Epoladerm,Epoladerm™, could provide a pathway for additional proprietary spray formulations with strong adhesion and accessibility properties upon application, especially around active joints and curvedcontoured body surfaces to manage pain associated with osteoarthritis. Our belief is based on, in part, research done to assess the effectiveness and safety of different preparations and doses of non-steroidal anti-inflammatory drugs (“NSAIDs”), opioids, and paracetamol for knee and hip osteoarthritis pain and physical function to enable effective and safe use of these drugs at their lowest possible dose. Osteoarthritis, which we believe to be a bettersignificant global market opportunity for us, is a painful condition that results in reduced physical function and quality of life and increased risk of all-cause mortality. A recent large meta-analysisOn June 6, 2017, the Company entered into a license agreement, as amended on pharmacologic treatmentsSeptember 2, 2017 and October 31, 2017 (the “MedPharm License Agreement”), with MedPharm Limited (“MedPharm”) for kneethe exclusive global rights to discover, develop, make, sell, market, and hip osteoarthritis indicatedotherwise commercialize any pharmaceutical composition or preparation (in any and all dosage forms) in final form containing one or more compounds, including Diclofenac Epolamine (“Epoladerm”), that topical diclofenac had the largest effect on painwas developed, manufactured or commercialized utilizing MedPharm’s spray formulation technology (“MedPharm Product”), to be used for any and physical function with a better safety profile than oral diclofenac. We believe these agents should be considered as a first-line pharmacological treatment for osteoarthritis.all uses in humans (including all diagnostic, therapeutic and preventative uses).
7
We believe Epoladerm’s proprietary spray film technology may lead to adhesion capabilities superior to those of currently available transdermal patches (e.g. Epoladerm does not require any tape reinforcement), while maintaining comparable skin absorption capabilities to transdermal patches currently on the market. Specifically, because the Epoladerm technology does not require a patch to deliver the drug through the skin, we believe Epoladerm may have better adhesion to the skin and may have better accessibility, particularly around joints and other curved body surfaces. Additionally, because Epoladerm is a spray, we believe it will be more aesthetically appealing to patients than transdermal patches. As a spray, Epoladerm was studied in ex-vivo skin studies (skin from abdominoplasty) and recent studies indicate drying times of approximately 120 seconds for chronic osteoarthritis of the knee. Unlike other topical NSAIDs, Epoladerm does not require physical handling of the actual drug and enables dosing that provides an accurate amount of active ingredient per spray application.
Pursuant to a Research and Option Agreement with MedPharm Limited (the “MedPharm Research and Option Agreement”), MedPharm will conduct certain research and development activities of proprietary formulations incorporating certain MedPharm technologies and certain of our proprietary molecules. Under the agreement, we were granted an option to obtain an exclusive, world-wide, sub-licensable, royalty bearing, irrevocable license to research, develop, market, use, commercialize, and sell any product utilizing MedPharm’s spray formulation technology.
Image 1,7, below, displays the expected delivery system of Epoladerm for the treatment of chronic osteoarthritis:

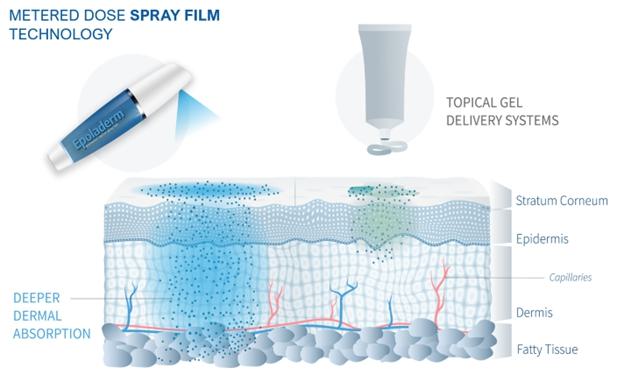
Image 1
Image 7
When discussing nonopioid treatments for chronic pain, the Centers for Disease Control (“CDC”) notes clinicians should consider topical agents as alternative first-line analgesics, thought to be safer than systemic medications. In an August 18, 2020 article appearing in the Annals of Internal Medicine, the American College of Physicians and the American Academy of Family Physicians announced a joint clinical guideline, “Nonpharmacologic and Pharmacologic Management of Acute Pain from Non-Low Back, Musculoskeletal Injuries in Adults,” whereby they recommend topical NSAIDs as first-line therapy for patients experiencing pain from non-low back, musculoskeletal injuries. The clinical guideline also recommends that clinicians not prescribe opioids for these injuries except in cases of severe injury or if patients cannot tolerate first-line therapeutic options. A recent large meta-analysis research report published in October 2021 on pharmacologic treatments for knee and hip osteoarthritis indicated that topical diclofenac had the largest effect on pain and physical function with a better safety profile than oral diclofenac. Based on this meta-analysis it was recommended that topical diclofenac should be considered as a first-line pharmacological treatment for knee osteoarthritis.
8
As a result of our pre-investigational new drug (“IND”) review,meeting, we believe it is reasonable for us to pursue a 505(b)(2) or OTC accelerated new drug application (“NDA”) for Epoladerm. There can be no assurance that we will be successful in securing regulatory approval under the 505(b)(2) pathway or that we will be successful in mitigating risks associated with the clinical development of this product candidate.
Current Development Status
In December 2021, we completed a single dose pharmacokinetic study of dermal administration of Epoladerm in minipigs as part of the required IND enabling trials. Single-dose transdermal delivery of Epoladerm was well-tolerated in all minipigs and no treatment-related clinical observations, changes in body weight, or dermal irritation were observed. All Epoladerm treated animals had plasma levels of Epoladerm confirming transdermal absorption. The maximum plasma concentration (“Cmax”) was reached at 4 hours post-dose, and plasma Epoladerm remained at 24-hour post-dose for all animals.
In January 2022, we reported positive results of four preclinical dermal safety studies for Epoladerm. Researchers concluded that once daily dermal administration of Epoladerm for 28 days was well-tolerated with no serious adverse findings. The studies were performed by Charles River Laboratories, a well-known clinical research organization. The studies included a skin irritation study in rabbits; a dermal sensitization assessment in guinea pigs; and a phototoxicity assay in mouse fibroblasts. Epoladerm was well tolerated in each of the studies and no reportable dermal irritation, dermal sensitization or phototoxicity was observed.
In June 2022, we announced our intention to pursue a direct to Over-the-Counter (“OTC”) regulatory pathway for Epoladerm. The direct to OTC, non-prescription regulatory pathway is expected to provide a faster drug development timeline and faster global approval track than the prescription pathway we had originally pursued for Epoladerm. To support the OTC application, we plan to submit Epoladerm’s completed dermal toxicity, sensitization, irritation, phototoxicity studies and its pharmacokinetic characteristics will need to be submitted to the FDA. In addition, we anticipate that we will have to complete a consumer preference assessment and a pivotal study required by the FDA’s Office of Non-prescription Drugs. The originally scheduled preclinical toxicology studies for an osteoarthritis of the knee indication that were to run in parallel with our anticipated pilot study for Epoladerm will not be required for our direct to OTC regulatory pathway.
We made the determination to delay our First-in-Human study investigating Epoladerm for pain associated with chronic osteoarthritis oduedue to: (i) a delay in procuring the active pharmaceutical ingredient necessary for the drug product candidate, (ii) delays related to supply chain disruptions, and (iii) an extensive review of the formulation and potential degradants resulting in MedPharm replacing the potential degradant. This additionalAdditional formulation work and in-vitro permeation testing may enable the patent coverage of this asset to be extended until at least 2042.2042 and provide an Over the Counter (“OTC”) pathway. MedPharm is anticipated to complete the formulation work and permeation testing at the end of third quarter 2023. If this nonclinical testing is successful, we plan to file an IND with Epoladerm in the fourth quarter 2023. In addition, MedPharm will initiate manufacturing batches that will be utilized for the First-in-Human study that is anticipated to begin first quarterhalf of 2024.
We may seekare seeking to license out or partner this asset as we continue to focus our efforts on our prescription drug pipeline.
High-Density Molecular Masking Spray Formulation for the Prevention of Respiratory Viruses (AnQlar)(AnQlar™)
AnQlar is a high-density molecular masking spray we plan to develop as an anti-viral barrier to potentially prevent, or reduce the risk or the intensity of, viral infections in humans. We intend for this formulation to be delivered using a preassembled device and cartridgemetered dose nasal spray to propel the high-density molecular spray formulation into the nose. We intend for AnQlar to be used as a nasal powder spray tonose and potentially prevent viral binding to epithelial cells in the nasal cavity and the upper respiratory tract, potentially reducing respiratory related infections.
9
As an addition to standard personal protective equipment, we believe AnQlar may offer an additional layer of protection, in the form of a molecular mask, to protect healthcare workers and those at risk of serious disease from viral infections. AnQlar has completed IND-enabling toxicology studies and ex-vivo studies which demonstrated a reduction in infectivity from respiratory viruses like influenza and SARS-CoV-2. During the ex-vivo study, in the presence of AnQlar, SARS-CoV-2 viral replication inhibition was observed in a bronchial epithelial model reconstituted from healthy human donor cells. Virus replication was evaluated using RT-qPCR. The data was presented as a number of viral copies per ml. A lower viral yield was detected in the cultures treated with AnQlar than in the control with phosphate buffered saline (“PBS”) after 72 hours of infection. AnQlar could potentially be marketed to first responders, healthcare workers, clinics, military forces, transplant and other immune compromised or at risk patients within the $13 billion (as of 2019) anti-viral market.
Image 78 below illustrates the results of the AnQlar IND-enabling toxicology studies ex-vivo:
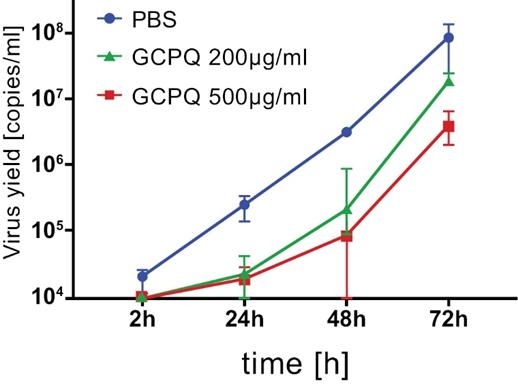
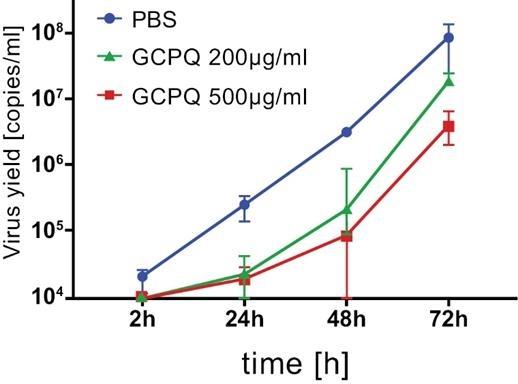
Image 78
A lower viral yield was detected in the cultures treated with AnQlar than in the control with Phosphate Buffered Saline (“PBS”) after 72 hours of infection.
Current Development Status
We submitted and received a written pre-investigational new drug (“pre-IND”) response from the FDA for AnQlar. In its pre-IND response, the FDA provided guidance on our pathway to pursue prophylactic treatment against SARS-CoV-2 and influenza for daily use as an OTC product. We believe the results of the pre-IND response support further research on AnQlar as a once daily intranasal prophylactic treatment of viral infections. The FDA has indicated that, upon successful completion of all necessary preclinical and clinical trials, we may pursue an NDA drug approval with the Office of Non-Prescription Drugs.
In August 2021, we engaged Syneos Health to assist with the design of the optimal clinical trial to facilitate an efficient regulatory and development timeline for AnQlar.
On August 25, 2021, we entered into a commercial manufacturing and supply agreement with Seqens, an integrated global leader in pharmaceutical solutions with 24 manufacturing sites worldwide and seven research and development facilities throughout the U.S. and Europe. The agreement with Seqens provides for both the supply material for our clinical studies as well as the long-term commercial supply of AnQlar. Seqens has initiated and is conducting process development and validation of additional large scale commercial quantities of AnQlar at its facilities in Devens and Newburyport, Massachusetts.
10
On September 29, 2021, we engaged a research and development firm to conduct a series of IND enabling toxicity studies for AnQlar which are expected to beAnQlar. These studies have been completed by the fourth quarter 2023. This was slightly delayed due to certain issues with finalizing the bioanalytical method developmentwithout an analysis of the product candidate. Upon successful completion of these studies, we intend to run a process using an investment banker to assist with sublicensing AnQlar to a global commercial partner in the respiratory space.pharmacokinetics (PK) data.
On July 5, 2022, we announced our intention to pursue an OTC Intranasal Medical Device Consumer regulatory pathway for AnQlar. To supportThe FDA has indicated that, upon successful completion of all necessary preclinical and clinical trials, we may pursue an NDA drug approval with the OTC Medical Device application, we plan to submit AnQlar’s completed in-vitro study, ex-vivo study using human mucosal cells, in-vivo study in rats, toxicology study and its pharmacokinetics (PK) characteristics studies to the FDA. Additionally, we will include AnQlar’s completed safety-pharmacology studies, drug-drug interaction studies and its virology studies evaluating AnQlar’s viral barrier properties against two variantsOffice of SARS-CoV-2 in a SARS-CoV-2 mouse model to the FDA. For the OTC medical device application, we anticipate that we will have to complete stability testing, human factors testing for medical devices, safety studies and supplementary in-vitro studies.Non-Prescription Drugs.
We recentlyOn July 27, 2022, Virpax conducted an initial review of the results from a preclinical virology study conducted by one of our CROs where we were evaluating the viral barrier properties of AnQlar™ versus two variants of the SARS CoV-2 virus. This review conducted by our external consultants indicates that the test article (AnQlar) shows an appropriate level of virus deactivation for a prophylactic viral barrier product candidate which was the outcome we were expecting. We have completed a validated bioanalytical method for the samples on February 12, 2024.
We are seeking to license out or partner this asset as we continue to seek opportunities to exploitfocus our product portfolio through licensing and other strategic transactions to further developefforts on our prescription drug product candidates. This includes seeking potential partners in further developing our drug product candidates and responding to inquiries of interest we have received concerning our product portfolio.pipeline.
Intellectual Property
We strive to protect and enhance the proprietary technologies, inventions and improvements that we believe are important to our business, including seeking, maintaining and defending patent rights, whether developed internally or licensed from third parties. Our policy is to seek to protect our proprietary position by, among other methods, pursuing and obtaining patent protection in the United States and in jurisdictions outside of the United States related to our proprietary technology, inventions, improvements, platforms and our product candidates that are important to the development and implementation of our business.
As of February 10, 2023March 15, 2024 our portfolio of owned and licensed patents and pending patent applications consisting of 1027 issued patents of which 11 are issued U.S. patents, 213 pending patent applications, of which 3 are pending U.S. patent applications, 1 pending Patent Cooperation Treaty (“PCT”) application, 26 issued patents 4 pending foreign applications and 12 provisional patent application.applications. These patent rights include issued US Patent Nos. 7,741,474, 8,470,371, 10,213,474, 8,278,277, 8,920,819, 9,713,591, 8,349,297 8,695,592, 10.213.474, 10,842,745 and 10,842,74511,839685 as well as patents and patent applications in Europe (Germany, France, UK), Canada, Japan, China, Israel, Australia, New Zealand, and South Korea. Below is a breakdown of patents by product:product, with unextended patent expiration dates indicated:
Epoladerm
The product candidate is covered by US Patent No. 8,349,297 (expires December 4, 2028) as well as issued patents in South Africa, Russian Federation, New Zealand, Norway, Mexico, Republic of Korea, Japan, China, Canada, Australia, Turkey, Slovakia, Slovenia, Sweden, Portugal, Poland, Netherlands, Latvia, Luxembourg, Lithuania, Italy, Ireland, Hungary, Greece, United Kingdom, France, Finland, Spain, Denmark, Germany, Czech Republic, Switzerland, Belgium and Austria (all of which expire September 14, 2026). The patents contain broad composition claims to a platform of pharmaceutical formulations which form a film on spray administration where the active agent is present at least 80% saturation and there is no undissolved active agent in the formulation. The claims also include a method of treatment and an aerosol dispenser containing the formulation. There is one pending provisional patent application that relates to specific spray formulations of diclofenac.
11
Probudur
The product candidate is covered by US Patent No. 9,713,591 (expires July 24, 2030) and US Patent No. 10,842,745 and 11,839685 (expires October 10, 2029) as well as a European patent (expires October 11, 2029) and a Chinese patent (expires October 11, 2029). There is also a pending US application which is a continuation of US Patent No. 10,842,745..11,839685. The patents containscontain composition claims to pharmaceutical compositions having an external storage solution containing an active pharmaceutical ingredient and particles of liposomes embedded in a polymeric matrix contained within the storage solution.
Envelta
The product candidate is covered by the following patent families which protect the chemistry of the MET polymer and its use in pharmaceutical products: Patent Family 1 includes US Patent No. 7,741,474 (expires March 18, 2026), and a Japanese Patent, Canadian Patent and European Patent (all which expire September 22, 2023). This patent family covers carbohydrate polymers with hydrophobic and hydrophilic side-groups suitable for solubilizing, for example, hydrophobic drugs.
Patent Family 2 includes US Patent No. 8,470,371 (expires July 29, 2029) and a Japanese Patent (expires August 8, 2027) and a European Patent (which expireexpires August 8, 2027). This family covers both composition and method claims for amphiphilic carbohydrate polymers which are capable of self-assembling to form micellar clusters in which the carbohydrate amphiphiles aggregate into hierarchically organized micellar clusters of individual aggregates. These micellar clusters formed by the aggregation of individual micelles may be transformed into stable nanoparticles with drugs, especially hydrophobic drugs that have poor aqueous solubility. This provides molar polymer/drug ratios that are greater than the ratios observed with block copolymers and improve the transfer of hydrophobic drugs across biological barriers.
Patent Family 3 includes US Patent Nos. 8,278,277 (expires August 16, 2030) and 8,920,819 (expires April 29, 2029), a Canadian patent (expires March 1, 2030) and a pending European application (which would expirepatent (expires March 1, 2030). This family covers lipophilic derivatives of hydrophilic drugs comprising a hydrophilic drug and a cleavable linker as well as methods of treatment using these compositions. In particular, the patents relate to compositions of a lipophilic derivative of the hydrophilic neuropeptide Leucine [5]-Enkephalin and an amphiphile compound, where the derivative includes a lipophilic linker attached to the side chain oxygen of the tyrosine in the Leucine [5]-Enkephalin, and where the amphiphile compound is quaternary palmitoyl glycol chitosan (GCPQ).
Patent Family 4 includes US Patent No. 10,213,474 (expires November 3, 2034), a Japanese patent (expires November 3, 2034), a European patent (expires November 3, 2034) and a Canadian patent ( expires(expires November 3, 2034). The patents cover methods for treating pain, comprising intranasally administering to a human or animal a composition comprising a therapeutically effective amount of a hydrophilic neuroactive peptide and an amphiphilic quaternary ammonium palmitoyl glycol chitosan (GCPQ); wherein the amphiphilic GCPQ is capable of self-assembly in aqueous media into particles having a mean particle size between 20-500 nm; where intranasally administering the composition delivers the hydrophilic neuroactive peptide to the brain of the human or animal.
Patent Family 5 includes national phase filings of WO 2021/1234413 which includes patent applications in the United States, Canada, Europe, Japan, and China. These patent applications cover acetylated amphiphilic carbohydrate compounds of average molecular weight 1-50 kDa which is based on a glycol chitosan, wherein the levels of acetylation can be modified. The compound can be formulated with hydrophobic compounds where the degree of acetylation of the carbohydrate compound is optimized to maximize solubilization of the compounds.
Patent Family 6 comprises US Provisional Patent Application No. 63/524764 which covers delivering leucine-5-enkephalin encapsulated in GCPQ intranasally via a drug or medicine dispersion and delivery system. Patents from this provisional application, if issued, would expire in 2044.
In addition to the patent families which protect the chemistry of the MET polymer and its use in pharmaceutical products there is a patent family which covers the delivery device that can be used to administer the pharmaceutical compositions including US Patent No. 8,695,592 (expires October 11, 2029), and a European patent (expires October 11, 2029). This family covers a capsule for use in dispensing a drug which has a pressurized container for a fluid, a chamber for containing a particulate, at least one channel running between the container and the chamber to provide fluidic communication and at least two distinct concave surfaces which impart rotational motion to a fluid flow so that within the chamber a rotationally turbulent flow of fluid is produced in order to engage with the particulate and to produce a mobile fluid comprising the particulate.
12
AnQlar
This product candidate is specifically covered by a PCT application WO 2022/043678A1 by a family of patent applications (United States, Canada, Japan, China, Europe and Australia) which describes a molecular masking spray for use as an anti-viral barrier to potentially prevent, or reduce the risk or the intensity of, viral infections in humans. Depending on the formulation and mode of administration of AnQlar, the product candidate may also be covered by many of the patents and patent application which cover Envelta.
NobrXiol
This product candidate is specifically covered by a provisionalPCT patent application which describes compositions containing an amphiphilic carbohydrate compound and a cannabinoid which may be intranasally delivered. Depending on the formulation and of NobrXiol, the product candidate may also be covered by many of the patents and patent application which cover EnveltaEnvelta.
Individual patents extend for varying periods depending on the date of filing of the patent application and the legal term of patents in the countries in which they are obtained. Generally, patents issued for regularly filed applications in the United States are granted a term of 20 years from the earliest effective non-provisional filing date. In addition, in certain instances, a patent term can be extended to recapture a portion of the U.S. Patent and Trademark Office, or USPTO, delay in issuing the patent as well as a portion of the term effectively lost as a result of the FDA regulatory review period. However, as to the FDA component, the restoration period cannot be longer than five years and the total patent term including the restoration period must not exceed 14 years following FDA approval. The duration of foreign patents varies in accordance with provisions of applicable local law, but typically is also 20 years from the earliest effective filing date. Many foreign jurisdictions also offer patent term extensions based on regulatory delays. However, the actual protection afforded by a patent varies on a product by product basis, from country to country and depends upon many factors, including the type of patent, the scope of its coverage, the availability of regulatory-related extensions, the availability of legal remedies in a particular country and the validity and enforceability of the patent.
Furthermore, we rely upon trade secrets and know-how and continuing technological innovation to develop and maintain our competitive position. We seek to protect our proprietary information, in part, using confidentiality agreements with our collaborators, employees, contractors, consultants and advisors and invention assignment agreements with our employees. We also have confidentiality agreements or invention assignment agreements with our collaborators, contractors and selected consultants. These agreements are designed to protect our proprietary information and, in the case of the invention assignment agreements, to grant us ownership of technologies that are developed through a relationship with a third party. These agreements may be breached, and we may not have adequate remedies for any breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors. To the extent that our collaborators, employees, contractors and consultants use intellectual property owned by others in their work for us, disputes may arise as to the rights in related or resulting know-how and inventions.
Our commercial success will also depend in part on not infringing upon the proprietary rights of third parties. It is uncertain whether the issuance of any third-party patent would require us to alter our development or commercial strategies, or our drugs or processes, or to obtain licenses or cease certain activities. Our breach of any license agreements or failure to obtain a license to proprietary rights that we may require to develop or commercialize our future drugs may have an adverse impact on us. If third parties have prepared and filed patent applications prior to the date of our earliest filed patent applications in the United States that also claim technology to which we have rights, we may have to participate in interference proceedings in the USPTO, to determine priority of invention. For more information, please see “Risk Factors — Risks Related to Our Intellectual Property.”
Material Agreements
MedPharm Limited
Research and Option Agreement
On April 11, 2017, we entered into a research and option agreement, as amended on May 30, 2018 (the “MedPharm Research and Option Agreement”), with MedPharm Limited, a company organized and existing under the laws of the United Kingdom (“MedPharm”), pursuant to which MedPharm granted us an option to obtain an exclusive, world-wide, royalty bearing license to use certain technology developed by MedPharm. Pursuant to the agreement, MedPharm will conduct certain research and development of proprietary formulations incorporating certain MedPharm technologies and certain of our proprietary molecules.
13
Under the MedPharm Research and Option Agreement, MedPharm granted us an option (the “MedPharm Option”) to obtain an exclusive (even to MedPharm), worldwide, sub-licensable (through multiple tiers), royalty bearing, irrevocable license to research, develop, market, commercialize, and sell any product utilizing MedPharm’s spray formulation technology which is the result of the activities performed under the MedPharm Research and Option Agreement, subject to our entry into a definitive license agreement with MedPharm. In order to exercise the MedPharm Option, we must provide MedPharm with written notice of such exercise before the end of the Option Period (as defined in the MedPharm Research and Option Agreement). The Option Period is subject to extension upon our mutual agreement with MedPharm.
Pursuant to the MedPharm Research and Option Agreement, we have a right of first refusal with respect to any license or commercial arrangement involving any Licensed Intellectual Property (as defined in the MedPharm Research and Option Agreement) in combination with any Virpax Molecule (as defined in the MedPharm Research and Option Agreement). In the event that MedPharm reaches an agreement with respect to a license or other commercial arrangement that involves technology or molecules covered by the right of first refusal, we have ten business days from the date of notice to notify MedPharm of our intention to exercise the right of first refusal and our intention to match the financial terms of the other license or commercial arrangement.
License Agreement (Epoladerm)
On June 6, 2017, as a result of ourthe Company’s exercise of the MedPharm Option under the MedPharm Research and Option Agreement, wethe Company entered into a license agreement, as amended on September 2, 2017 and October 9,31, 2017 (the “MedPharm License Agreement”), with MedPharm for the exclusive global rights to discover, develop, make, sell, market, and otherwise commercialize any pharmaceutical composition or preparation (in any and all dosage forms) in final form containing one or more compounds, including Epoladerm and OSF200,Diclofenac Epolamine (“Epoladerm”), that was developed, manufactured or commercialized utilizing MedPharm’s spray formulation technology (“MedPharm Product”), to be used for any and all uses in humans (including all diagnostic, therapeutic and preventative uses). Under the MedPharm License Agreement, we arethe Company is required to make future milestone and royalty payments to MedPharm. We areThe Company is obligated to make aggregate milestone payments to MedPharm of up to £1.15GBP 1.150 million upon the achievement of specified development milestones (payable in Great British Pounds). Additional milestone payments are due upon the achievement of certain development and commercial milestones achieved outside the United States, payable on a country-by-country basis. Royalty payments must be paid to MedPharm in an amount equal to a single-digit percentage of net sales of all MedPharm Product sold by us during the royalty term in the territory. Royalties shall be payable, on a country-by-country basis, during the period of time commencing on the first commercial sale and ending upon the expiration of the last-to-expire patent claim on the licensed product, which is set to expire on December 4, 2028. Each party has the right to terminate the agreement in its entirety upon written notice to the other party if (a) such other party is in material breach of the agreement and has not cured such breach within ninety (90) days after notice from the terminating party indicating the nature of such breach.breach or (b) if the other party is dissolved or liquidated or takes any corporate action for such purpose; makes a general assignment for the benefit of creditors; or has a receiver, trustee, custodian or similar agent appointed by order of any court of competent jurisdiction to take charge of or sell any material portion of its property or business. We have the right to terminate the agreement on a country by country basis for any reason or for no reason at any time upon ninety (90) days’ prior written notice to MedPharm and MedPharm has the right to terminate the agreement if we commence any interference or opposition proceeding with respect to, challenge the validity or enforceability of, or oppose any extension of term or the MedPharm Patent Rights (as such term is defined in the agreement) or we acquire or develop a Generic Version(as such term is defined in the agreement) of a MedPharm Product.
LipoCureRX, Ltd. (Probudur)
On March 19, 2018, we entered into a license and sublicense agreement (the “LipoCure Agreement”) with LipoCureRX, Ltd., a company organized and existing under the laws of Israel (“LipoCure”), for the sole and exclusive global license and sub-license rights to discover, develop, make, sell, market, and otherwise commercialize bupivacaine liposome, in injectable gel or suspension (“Licensed Compound”) or any pharmaceutical composition or preparation (in any and all dosage forms) in final form, including any combination product, containing a Licensed Compound (“Licensed Product”), including Probudur. Under the LipoCure Agreement, we were required to pay an upfront fee upon signing of $150,000 and are required to make future milestone and royalty payments to LipoCure. We are obligated to make aggregate milestone payments of up to $19.8 million upon the achievement of specified development and commercial milestones. Royalty payments must be paid in an amount equal to a single digit to low double-digit percentage of annual net sales of royalty qualifying products, subject to certain adjustments. Royalties shall be payable during the period of time, on a country-by-country basis, commencing on the first commercial sale and ending upon the expiration of the last-to-expire patent claim on the licensed product, which is set to expire on July 24, 2030. Each party has the right to terminate the agreement in its entirety upon written notice to the other party (a) if such other party is in material breach of the agreement and has not cured such breach within ninety (90) days after notice from the terminating party indicating the nature of such breach.breach or (b) with immediate effect if such other party passes a resolution for voluntary winding up or a winding up application is made against it and not set aside within 60 days; or (ii) a receiver of a liquidator is appointed for the other party; or (iii) the other party enters into winding up or insolvency or bankruptcy proceedings; or (iv) the other party enters into a scheme or arrangement in contemplation of the foregoing with its creditors.
14
Nanomerics Ltd.
Nanomerics Collaboration Agreement (Envelta)
On April 11, 2019, we entered into an exclusive collaboration and license agreement, as amended (the “Nanomerics Collaboration Agreement”), with Nanomerics Ltd. (“Nanomerics”), a company organized and existing under the laws of United Kingdom, for the exclusive world-wide license to develop and commercialize products, including Envelta, which contain hydrophilic neuropeptide Leucin5-Enkephalin and an amphiphile compound which is quaternary ammonium palmitoyl glycol chitosan, and to engage in a collaborative program utilizing Nanomerics’ knowledge, skills and expertise in the clinical development of products and in attracting external funding for such development. The Nanomerics Collaboration Agreement was also amended to include a program for the pre-clinical development of a product for post-traumatic stress disorder.
Under the Nanomerics Collaboration Agreement, we are required to make royalty payments equal to a single digit percentage of annual net sales of royalty qualifying products. We are also required to make aggregate milestone payments of up to $103 million upon the achievement of specified development and commercial milestones, and sublicense fees for any sublicense relationships we enter into subsequent to the Nanomerics Collaboration Agreement. Our obligation to pay royalties, on a country-by-country basis, shall commence on the date of first commercial sale of our licensed products and shall expire with respect to each separate licensed product, on the latest to occur of (a) the tenth (10th) anniversary of the first commercial sale of the first licensed product; (b) the expiration date of the last to expire of any valid claim (patent is set to expire on November 3, 2034); and, (c) the date upon which a generic product has been on the market for a period of no fewer than ninety (90) days. We have the right to terminate the agreement upon 180 days’ prior written notice to Nanomerics. Upon termination, we shall assign to Nanomerics all its right title and interest in all results other than results specific to (a) the Device (as defined in the Nanomerics Collaboration Agreement), its manufacture or use; and (b) the Technology, but excluding any clinical results relating to the Compound or Licensed Products (all terms as defined in the Nanomerics Collaboration Agreement).
Nanomerics License Agreement (AnQlar)
On August 7, 2020, we entered into a collaboration and license agreement with Nanomerics (the “Nanomerics License Agreement”) for the exclusive North American license to develop and commercialize a High-Density Molecular Masking Spray (AnQlar) as an anti-viral barrier to prevent or reduce the risk or the intensity of viral infections in humans. Under the Nanomerics License Agreement, we were required to make royalty payments and milestone payments upon the achievement of specified development and commercial milestones, and sublicense fees for any sublicense relationships we enter into subsequent to the Nanomerics License Agreement (any patent that issues from the currently filed provisional patent application would expire on August 24, 2041).
On March 9, 2022, we entered into an Amended and Restated Collaboration and License Agreement with Nanomerics (the “Amended Nanomerics License Agreement”) which amended and restated the August 7, 2020, Nanomerics License Agreement and expanded our North American rights for AnQlar to include exclusive global rights to develop and commercialize AnQlar as an anti-viral barrier to prevent or reduce the risk or the intensity of viral infections. The Amended Nanomerics License Agreement provides for payments up to $5.5 million upon the achievement of specified development milestones and profit share payments equal to between 30% to 40% of certain profits (as set forth in the Amended Nanomerics License Agreement), payable to Nanomerics upon the achievement of specified commercial milestones. The profit share payments are triggered upon determination by the FDA that AnQlar may be marketed as an Over-the-Counter product in the United States. In the event the profit share payments are not triggered as defined above, we would be obligated to pay royalties within a range of 5% to 15% of annual net sales of royalty qualifying products and commercial milestones on a worldwide basis amounting to aggregate milestone payments of up to $112.5 million upon the achievement of these commercial milestones. The Amended Nanomerics License Agreement also provides for additional aggregate milestone payments totaling $999,999 upon first receipt of regulatory approval for a licensed product in the European Union, Asia/Pacific region and South America/Middle East region. The Company’s obligation to pay royalties, on a country-by-country basis, shall commence on the date of first commercial sale of its licensed products and shall expire with respect to each separate licensed product, on the latest to occur of (a) the tenth (10th) anniversary of the first commercial sale of the first licensed product; (b) the expiration date of the last to expire of any valid claim; and, (c) the date upon which a generic product has been on the market for a period of no fewer than ninety (90) days. The Company has the right to terminate the Nanomerics License Agreement upon sixty (60) days’ prior written notice to Nanomerics. Each party has the right to terminate the agreement in its entirety upon written notice to the other party (a) if such other party is in material breach of the agreement and has not cured such breach within ninety (90) days after notice from the terminating party indicating the nature of such breach or (b) in the event that the other party shall file in any court or agency, pursuant to any statute or regulation of any state or country, a petition in bankruptcy or insolvency or for reorganization or for an the appointment of a receiver or trustee of such other Party or of its assets, or if the other Party proposes a written agreement of composition or extension of its debts of its debts, or if the other Party shall be served with an involuntary petition against it, filed in any insolvency proceeding, or if the other Party shall propose or be a party to any dissolution or liquidation, or if the other Party shall make an assignment for the benefit of its creditors. Upon termination, the Company shall assign to Nanomerics all its rights, title and interest in all of its results. Nanomerics has the right to terminate the agreement upon sixty (60) days’ prior written notice. In consideration for entering into this Amended Nanomerics License Agreement, the Company paid Nanomerics $1,500,000.$1,500,000 during the year ended December 31, 2022.
15
Nanomerics License Agreement (NobrXiol)
On September 17, 2021, we entered into a collaboration and license agreement with Nanomerics (the “Nanomerics License Agreement - NobrXiol”) for the exclusive worldwide license to develop and commercialize an investigational formulation delivered via the nasal route to enhance pharmaceutical-grade cannabidiol (“CBD”) transport to the brain to potentially treat seizures associated with tuberous sclerosis complex (“TSC”), Lennox-Gastaut syndrome and Dravet syndrome in patients one year of age and older. Under the Nanomerics License Agreement – NobrXiol, we are required to make royalty payments within a range of 5% to 15% of annual net sales of royalty qualifying products. Our obligation to pay royalties, on a country-by-country basis, shall commence on the date of first commercial sale of licensed products (as defined in the Nanomerics License Agreement – NobrXiol) and shall expire with respect to each separate licensed product, on the latest to occur of (a) the fifteen (15th) anniversary of the first commercial sale of the first licensed product; (b) the expiration date of the last to expire of any valid claim; and, (c) the date upon which a generic product has been on the market for a period of no fewer than ninety (90) days. We paid an upfront milestone payment upon signing of $200,000 and are required to make future milestone and royalty payments of up to $41 million upon the achievement of specified development and commercial milestones, and sublicense fees for any sublicense relationships we enter into subsequent to the Nanomerics License Agreement – NobrXiol (any patent that issues from the currently filed provisionalPCT patent application would expire on August 24, 2041)September 9, 2043). We have the right to terminate the Nanomerics License Agreement – NobrXiol upon one hundred and eighty (180) days’ prior written notice to Nanomerics. Upon termination, we shall assign to Nanomerics all its rights, title and interest in all of its results. Nanomerics has the right to terminate the agreement upon thirty (30) days’ prior written notice if we conclude in writing to Nanomerics that the study aim has not been achieved or we notify Nanomerics that we have decided against proceeding with a Phase III3 Clinical trial.
On April 21, 2022, the Company notified Nanomerics that the study aim of demonstrating the ability of Nanomerics platform technology delivering CBD to the brain via nasal administration in an animal model was met. Pursuant to the Nanomerics License Agreement - NobrXiol, the Company paid and incurred a milestone payment of $500,000 upon meeting this study aim in April 2022.
Sales and Marketing
If Epoladerm, Probudur, Envelta, PES200, AnQlar and/or NobrXiol are approved, we plan to enter into sales and marketing agreements with one or several pharmaceutical companies to sell to pain management clinics and specialists, general and orthopedic surgeons, anesthesiologists, primary care physicians (“PCPs”), Nurse Practitioners (“NPs”), oncologists, and neurologists.
On August 30, 2018, we entered into a Master Service Agreement (the “MSA”) with INC Research, LLC, a Syneos Health™ group company (“Syneos Health”) to operate as our Contract Sales Organization (“CSO”). Services provided by Syneos Health include clinical research services, bioanalytical analysis, statistics, validations, pharmacokinetics, and/or consulting, advertising, and public relations (communications), field team sales and education recruiting and deployment, and patient adherence services.
Manufacturing
Envelta is an intranasal spray and weWe rely on third-party contractors for manufacturing clinical supplies and plan to do so for commercial amounts also.
On August 25, 2021, we entered into a commercial manufacturing and supply agreement with Seqens, an integrated global leader in pharmaceutical solutions with 24 manufacturing sites worldwide and seven research and development facilities throughout the U.S. and Europe. The agreement with Seqens provides for both the supply material for our clinical studies as well as the long-term commercial supply of AnQlar. Seqens will conduct process development and validation of additional large scale commercial quantities of AnQlar at its facilities in Devens and Newburyport, Massachusetts.
16
We continue to explore manufacturing sources, in order to ensure that we have access to sufficient manufacturing capacity in order to meet potential demand for any of our product candidates in a cost-efficient manner. We plan to secure supply sources and contract with these or other parties to manufacture commercial quantities of any products we successfully develop. Among the conditions for FDA approval of a pharmaceutical product is the requirement that the manufacturer’s quality control and manufacturing procedures conform to cGMP, which must be followed at all times. The FDA typically inspects manufacturing facilities on an ongoing basis. In complying with Current Good Manufacturing Practice (“cGMP”) regulations, pharmaceutical manufacturers must expend resources and time to ensure compliance with product specifications as well as production, record keeping, quality control, reporting, and other requirements.
Competition
Competition
The pharmaceutical industry is intensely competitive and subject to rapid and significant technological change. We will continue to face competition from various global pharmaceutical, biotechnology, specialty pharmaceutical and generic drug companies that engage in drug development activities. Our key competitors include Pacira Biosciences, Inc. (which developed EXPAREL®) and Horizon (which developed Pennsaid®). Many of our competitors have similar products that focus on the same diseases and conditions that our current and future pipeline product candidates address. Many of our competitors have greater financial flexibility to deploy capital in certain areas as well as more commercial and other resources, marketing and manufacturing organizations, and larger research and development staff. As a result, these companies may be able to pursue strategies or approvals that we are not able to finance or otherwise pursue and may receive FDA, or other applicable regulatory approvals more efficiently or rapidly than us. Also, our competitors may have more experience in marketing and selling their products post-approval and gaining market acceptance more quickly. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large, established companies. Our product candidates could become less competitive if our competitors are able to license or acquire technology that is more effective or less costly and thereby offer an improved or a cheaper alternative to our product candidates.
We expect any product candidates that we develop and commercialize will compete on the basis of, among other things, efficacy, safety, convenience of administration and delivery, price and the availability of reimbursement from government and other third-party payors. We also expect to face competition in our efforts to identify appropriate collaborators or partners to help commercialize our product candidate portfolio in our target commercial markets.
Government Regulation and Approval Process
Government authorities in the United States at the federal, state and local level, including the FDA, the FTC and the DEA, extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, recordkeeping, promotion, advertising, distribution, marketing and export and import of products such as those we market. For both currently marketed and future products, failure to comply with applicable regulatory requirements can, among other things, result in suspension of regulatory approval and possible civil and criminal sanctions. Regulations, enforcement positions, statutes and legal interpretations applicable to the pharmaceutical industry are constantly evolving and are not always clear. Significant changes in regulations, enforcement positions, statutes and legal interpretations could have a material adverse effect on our financial condition and results of operations.
Additionally, future healthcare legislation or other legislative proposals at the federal and state levels could bring about major changes in the affected health care systems, including statutory restrictions on the means that can be employed by brand and generic pharmaceutical companies to settle Paragraph IV patent litigations. We cannot predict the outcome of such initiatives, but such initiatives, if passed, could result in significant costs to us in terms of costs of compliance and penalties associated with failure to comply.
With the support of President Trump and the HHS, the NIH launched the initiative Helping to End Addiction Long-term (“HEAL”), to provide solutions to the national opioid crisis. With the focus on ending the opioid crisis in America, HEAL, and new FDA guidance has made it more efficient to bring novel non-opioid medication to market.
17
The Office of National Drug Control Policy (“ONDCP”) is a component of the Executive Office of the President which works to reduce drug use and its consequences by leading and coordinating the development, implementation, and assessment of U.S. drug policy. In addition to its vital ongoing work, ONDCP also provided administrative and financial support to the President’s Commission on Combating Drug Addiction and the Opioid Crisis, established by Executive Order on March 29, 2017 by President Donald J. Trump. This commission was created to make recommendations to the President on how to best combat opioid addiction and abuse. In August 2017, the commission issued a preliminary report calling on President Trump to officially declare the crisis of opioid abuse a national emergency. On October 26, 2017, President Trump declared the opioid crisis a “national public health emergency.” The commission’s final report was released in early November 2017. In July 2017, the Pharmaceutical Care Management Association, a trade association representing pharmacy benefit managers, wrote a letter to the commissioner of FDA in which it expressed support for, among other things, the CDC guidelines and a seven-day limit on the supply of opioids for acute pain. In September 2018, CVS Pharmacy announced that it would only fill first time opioid prescriptions for acute pain for a seven-day supply.
Law enforcement and regulatory agencies may apply policies that seek to limit the availability of opioids. This creates the potential for aggressive enforcement, unfavorable publicity regarding the use or misuse of opioid drugs or the limitations of abuse-deterrent formulations, litigation, public inquiries or investigations related to the abuse, sales, marketing, distribution or storage of opioid products.
In addition, efforts by the FDA and other regulatory bodies to combat the abuse of opioids will positively impact the market for our novel non-opioid and non-addictive product candidates. We expect that the FDA will continue to evaluate the impact of abuse-deterrent opioids in the future, and this could impose further restrictions to opioid products currently on the market, which may include changing labeling, imposing additional prescribing restrictions, or seeking a product’s removal from the market.
Coinciding with HEAL,Helping to End Addiction Long Term (HEAL), a NIH initiative, there is new FDA guidance that streamlines and broadensare part of a larger effort to reduce the rangeuse of novel non-opioid medications.opioid analgesic drugs. The first guidance, will addressissued in June 2019, focuses on assessing the benefits and risks of developing new opioid pain drugs, including an updated framework for evaluating the risks associated with intentional or illicit misuse or abuse of these substances. In connection with the SUPPORT for Patients and Communities Act (SUPPORT Act), the purpose of this guidance is to spur the development of alternatives to opioids for the management of acute pain by providing information about product development-related issues, “opioid-sparing” claims, and expedited programs. The second guidance, issued in February 2022, addresses medications that can reduce the use of opioids in the treatment of acute pain, including how sponsors can demonstrate a clinically meaningful reduction in the use of opioid pain medications in the acute setting. The second guidance will focus on assessing the benefits and risks of developing new opioid pain drugs, including drafting an updated framework for evaluating the risks associated with intentional or illicit misuse or abuse of these substances. The third guidance, will “outlineissued March 2023, outlines a path for developing extended-release local anesthetics,” including clinical pharmacology, proper evaluation of safety and efficacy, and the types of studies that may support approval of these product candidates. Finally, FDA will issue guidance on the development of “new non-opioid pain medications for acute and chronic pain that can provide therapeutic alternatives to the use of opioids.”
Pharmaceutical Regulation in the United States
In the United States, the FDA regulates drugs under the FDCA and its implementing regulations. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval may subject an applicant to administrative or judicial sanctions. These sanctions could include the FDA’s refusal to approve pending applications, withdrawal of an approval, a clinical hold, Warning Letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement or civil or criminal penalties. Any agency or judicial enforcement action could have a material adverse effect on us.
FDA approval is required before any new unapproved drug or dosage form, including a new use of a previously approved drug or a generic version of a previously approved drug, can be marketed in the United States.
The process required by the FDA before a new drug may be marketed in the United States generally involves:
| ● | Completion of preclinical laboratory and animal testing and formulation studies in compliance with the FDA’s current GLP regulations; |
| ● | Submission to the FDA of an IND for human clinical testing, which must become effective before human clinical trials may begin in the United States; |
18
| ● | Approval by an IRB at each clinical site before each trial may be initiated; |
| ● | Performance of adequate and well-controlled human clinical trials in accordance with the FDA to establish the safety and efficacy of the proposed drug product for each intended use; |
| ● | Satisfactory completion of an FDA pre-approval inspection of the facility or facilities at which the product is manufactured to assess compliance with the FDA’s cGMP regulations to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity; |
| ● | Submission to the FDA of an NDA; |
| ● | Satisfactory completion of a potential review by an FDA advisory committee, if applicable; and |
| ● | FDA review and approval of the NDA. |
Preclinical Studies
When developing a branded product and bringing it to market, the first step in proceeding to clinical studies is preclinical testing. Preclinical tests are intended to provide a laboratory or animal study evaluation of the product to determine its chemistry, formulation and stability. Toxicology studies are also performed to assess the potential safety of the product. The conduct of the preclinical tests must comply with federal regulations and requirements, including GLPs. The results of these studies are submitted to the FDA as part of an IND application along with other information, including information about product chemistry, manufacturing and controls and a proposed clinical trial protocol. Long-term preclinical tests, such as animal tests of reproductive toxicity and carcinogenicity, may continue after the IND application is submitted.
Clinical Trials
Clinical trials involve the administration of the investigational new drug to human subjects under the supervision of qualified investigators in accordance with GCP requirements, which include the requirement that all research subjects provide their informed consent in writing for their participation in any clinical trial. Clinical trials are conducted under protocols detailing, among other things, the objectives of the trial, the parameters to be used in monitoring safety, and the effectiveness criteria to be evaluated. A protocol for each clinical trial and any subsequent protocol amendments must be submitted to the FDA as part of the IND. In addition, an IRB at each institution participating in the clinical trial must review and approve the plan for any clinical trial before it initiates at that institution. Information about certain clinical trials must be submitted within specific timeframes to the NIH for public dissemination on their website, www.ClinicalTrials.gov.
Human clinical trials are typically conducted in three sequential phases, which may overlap or be combined:
| ● | Phase I: The drug is initially introduced into healthy human subjects or patients with the target disease or condition and tested for safety, dosage tolerance, absorption, metabolism, distribution, excretion and, if possible, to gain an early indication of its effectiveness. |
| ● | Phase II: The drug is administered to a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage. |
| ● | Phase III: The drug is administered to an expanded patient population, generally at geographically dispersed clinical trial sites, in well-controlled clinical trials to generate enough data to statistically evaluate the efficacy and safety of the product for approval, to establish the overall risk-benefit profile of the product, and to provide adequate information for the labeling of the product. |
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and more frequently if serious adverse events occur. Phase 1, Phase 2, and Phase 3 trials may not be completed successfully within any specified period, or at all. Furthermore, the FDA or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients.
19
Disclosure of Clinical Trial Information
Sponsors of certain clinical trials of FDA-regulated products, including drugs, are required to register and disclose certain clinical trial information on www.ClinicalTrials.gov. Information related to the product, subject population, phase of investigation, study sites and investigators, and other aspects of the clinical trial is then made public as part of the registration. Sponsors are also obligated to discuss certain results of their clinical trials after completion. Disclosure of the results of these trials can be delayed until the new product or new indication being studied has been approved. Competitors may use this publicly available information to gain knowledge regarding the progress of development programs.
Marketing Approval
After completion of the required clinical testing, an NDA is prepared and submitted to the FDA. FDA approval of the NDA is required before marketing of the product may begin in the United States. The NDA must include, among other things, the results of all preclinical, clinical and other testing and a compilation of data relating to the product’s pharmacology, chemistry, manufacture and controls. Under federal law, the submission of most NDAs is subject to a substantial application user fee, and the manufacturer or sponsor under an approved NDA is also subject to annual program fees. The FDA has 60 days from its receipt of an NDA to determine whether the application will be accepted for filing based on the agency’s threshold determination that it is sufficiently complete to permit substantive review. The FDA may request additional information rather than accept an NDA for filing. In this event, the NDA must be resubmitted with the additional information and is subject to payment of additional user fees. The resubmitted application is also subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review. Under the Prescription Drug User Fee Act, as amended, the FDA has agreed to certain performance goals in the review of NDAs through a two-tiered classification system, Standard Review and Priority Review. Priority Review designation is given to drugs that are intended to treat a serious condition and, if approved, would provide a significant improvement in safety or effectiveness over existing therapies. The FDA endeavors to review most applications subject to Standard Review within ten to twelve months whereas the FDA’s goal is to review most Priority Review applications within six to eight months, depending on whether the drug is a new molecular entity.
The FDA may refer applications for novel drug products or drug products which present difficult questions of safety or efficacy to an advisory committee for review, evaluation and recommendation as to whether the application should be approved and under what conditions. Before approving an NDA, the FDA may inspect one or more clinical sites to assure compliance with GCP requirements. Additionally, the FDA will inspect the facility or the facilities at which the drug is manufactured. The FDA will not approve the NDA unless it determines that the manufacturing process and facilities are in compliance with cGMP requirements and are adequate to assure consistent production of the product within required specifications and the NDA contains data that provide substantial evidence that the drug is safe and effective for the labeled indication.
After the FDA evaluates the NDA and the manufacturing facilities, it issues either an approval letter or a complete response letter to indicate that the application is not ready for approval. A complete response letter generally outlines the deficiencies in the submission and may require substantial additional testing, or information, in order for the FDA to reconsider the application. Even with submission of this additional information, the FDA may ultimately decide that an application does not satisfy the regulatory criteria for approval. If, or when, the deficiencies have been addressed to the FDA’s satisfaction in a resubmission of the NDA, the FDA will issue an approval letter. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications.
As a condition of NDA approval, the FDA may require a REMS to help ensure that the benefits of the drug outweigh the potential risks. If the FDA determines a REMS is necessary during review of the application, the drug sponsor must agree to the REMS plan at the time of approval. A REMS may be required to include various elements, such as a medication guide or patient package insert, a communication plan to educate healthcare providers of the drug’s risks, limitations on who may prescribe or dispense the drug, or other elements to assure safe use, such as special training or certification for prescribing or dispensing, dispensing only under certain circumstances, special monitoring and the use of patient registries. In addition, the REMS must include a timetable to periodically assess the strategy. The requirement for a REMS can materially affect the potential market and profitability of a drug.
20
Moreover, as a condition of product approval, the FDA may require substantial post-approval testing, known as Phase IV testing, and/or surveillance to monitor the drug’s safety or efficacy, and the FDA has the authority to prevent or limit further marketing of a product based on the results of these post-marketing programs. Once granted, product approvals may be withdrawn if compliance with regulatory standards is not maintained or certain problems are identified following initial marketing. Drugs may be marketed only for the approved indications and in accordance with the provisions of the approved labeling, and, even if the FDA approves a product, it may limit the approved indications for use for the product or impose other conditions, including labeling or distribution restrictions or other risk-management mechanisms.
Further changes to some of the conditions established in an approved application, including changes in indications, labeling, or manufacturing processes or facilities, require submission and FDA approval of a new NDA or NDA supplement before the change can be implemented, which may require us to develop additional data or conduct additional preclinical studies and clinical trials. An NDA supplement for a new indication typically requires clinical data similar to that in the original application, and the FDA uses the similar procedures in reviewing NDA supplements as it does in reviewing NDAs.
Post-Approval Requirements
Once an NDA is approved, a product will be subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to drug listing and registration, recordkeeping, periodic safety reporting, product sampling and distribution, adverse event reporting and advertising, marketing and promotion, including standards and regulations for direct-to-consumer advertising, off-label promotion, industry-sponsored scientific and educational activities and promotional activities involving the Internet. Drugs may be marketed only for the approved indications and in a manner consistent with the provisions of the approved labeling. While physicians may prescribe for off-label uses, manufacturers may only promote for the approved indications and in accordance with the provisions of the approved labeling. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability. There also are extensive DEA regulations applicable to controlled substances.
Adverse event reporting and submission of periodic reports is also required following FDA approval of an NDA. Additionally, the FDA may place conditions on an approval, in addition to REMS programs of Phase IV testing, that could restrict the distribution or use of the product. Drug manufacturers and certain of their subcontractors are required to register their establishments and list their marketed products with the FDA and certain state agencies. Registration with the FDA subjects entities to periodic unannounced inspections by the FDA, during which the agency inspects manufacturing facilities to assess compliance with cGMPs. Accordingly, manufacturers must continue to expend time, money, and effort in the areas of production and quality-control to maintain compliance with cGMPs, including quality control and manufacturing processes. Regulatory authorities may withdraw product approvals or request product recalls if a company fails to comply with regulatory standards, if it encounters problems following initial marketing or if previously unrecognized problems are subsequently discovered. The FDA may also impose a REMS requirement on a drug already on the market if the FDA determines, based on new safety information, that a REMS is necessary to ensure that the drug’s benefits outweigh its risks. In addition, regulatory authorities may take other enforcement action, including, among other things, Warning Letters, the seizure of products, injunctions, consent decrees placing significant restrictions on or suspending manufacturing operations, refusal to approve pending applications or supplements to approved applications, civil penalties and criminal prosecution.
The Hatch-Waxman Amendments
505(b)(2) NDAs
The FDA is authorized to approve an alternative type of NDA under Section 505(b)(2) of the FDCA. Section 505(b)(2) permits the filing of an NDA where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference from the data owner. The applicant may rely upon the FDA’s findings of safety and efficacy for an approved product that acts as the “listed drug.” The FDA may also require 505(b)(2) applicants to perform additional studies or measurements to support the change from the listed drug. The FDA may then approve the new product candidate for all, or some, of the conditions of use for which the branded reference drug has been approved, or for a new condition of use sought by the 505(b)(2) applicant.
21
Abbreviated New Drug Applications (“ANDAs”)
The Hatch-Waxman amendments to the FDCA established a statutory procedure for submission and FDA review and approval of ANDAs for generic versions of listed drugs. An ANDA is a comprehensive submission that contains, among other things, data and information pertaining to the active pharmaceutical ingredient (“API\I”), drug product formulation, specifications and stability of the generic drug, as well as analytical methods, manufacturing process validation data and quality control procedures. Premarket applications for generic drugs are termed abbreviated because they generally do not include clinical data to demonstrate safety and effectiveness. However, a generic manufacturer is typically required to conduct bioequivalence studies of its test product against the listed drug. Bioequivalence is established when there is an absence of a significant difference in the rate and extent for absorption of the generic product and the reference listed drug. For some drugs, other means of demonstrating bioequivalence may be required by the FDA, especially where rate or extent of absorption are difficult or impossible to measure. The FDA will approve an ANDA application if it finds that the generic product does not raise new questions of safety and effectiveness as compared to the reference listed drug. A product is not eligible for ANDA approval if the FDA determines that it is not bioequivalent to the reference listed drug if it is intended for a different use or if it is not subject to, and requires, an approved Suitability Petition.
Patent Exclusivity and Orange Book Listing
In seeking approval for a drug through an NDA, including a 505(b)(2) NDA, applicants are required to list with the FDA certain patents whose claims cover the applicant’s product. Upon approval of an NDA, each of the patents listed in the application for the drug is then published in the Orange Book. Any applicant who files an ANDA seeking approval of a generic equivalent version of a drug listed in the Orange Book or a 505(b)(2) NDA referencing a drug listed in the Orange Book must certify to the FDA (i) that there is no patent listed with the FDA as covering the relevant branded product, (ii) that any patent listed as covering the branded product has expired, (iii) that the patent listed as covering the branded product will expire prior to the marketing of the generic product, in which case the ANDA will not be finally approved by the FDA until the expiration of such patent or (iv) that any patent listed as covering the branded drug is invalid or will not be infringed by the manufacture, sale or use of the generic product for which the ANDA is submitted. A notice of the Paragraph IV certification must be provided to each owner of the patent that is the subject of the certification and to the holder of the approved NDA to which the ANDA or 505(b)(2) application refers. The applicant may also elect to submit a “section viii” statement certifying that its proposed label does not contain (or carves out) any language regarding the patented method-of-use rather than certify to a listed method-of-use patent.
If the reference NDA holder and patent owners assert a patent challenge directed to one of the Orange Book listed patents within 45 days of the receipt of the Paragraph IV certification notice, the FDA is prohibited from approving the application until the earlier of 30 months from the receipt of the Paragraph IV certification, expiration of the patent, settlement of the lawsuit or a decision in the infringement case that is favorable to the applicant. The ANDA or 505(b)(2) application also will not be approved until any applicable non-patent exclusivity listed in the Orange Book for the branded reference drug has expired as described in further detail below.
Non-Patent Exclusivity
In addition to patent exclusivity, the holder of the NDA for the listed drug may be entitled to a period of non-patent exclusivity, during which the FDA cannot approve an ANDA or 505(b)(2) application that relies on the listed drug.
For example, a drug that is considered new chemical entity (NCE) at the time of approval may be awarded a five-year period of marketing exclusivity, starting at the time of product approval. An ANDA or 505(b)(2) application referencing that drug may not be approved until the five-year period expires. Also, an ANDA or 505(b)(2) application referencing that drug may not filed with the FDA until the expiration of five years, unless the submission is accompanied by a Paragraph IV certification, in which case the applicant may submit its application four years following the original product approval.
22
A drug, including one approved under Section 505(b)(2), may obtain a three-year period of exclusivity for a particular condition of approval, or change to a marketed product, such as a new formulation for a previously approved product, if one or more new clinical studies (other than bioavailability or bioequivalence studies) was essential to the approval of the application and was conducted/sponsored by the applicant.
DEA Regulation
Our product candidates may be regulated as “controlled substances” as defined in the Controlled Substances Act of 1970, as amended, which establishes registration, security, recordkeeping, reporting, storage, distribution and other requirements administered by the U.S. Drug Enforcement Agency (the “DEA”). The DEA is concerned with, among other things, the control of handlers of controlled substances and with the equipment and raw materials used in their manufacture and packaging, in order to prevent loss and diversion into illicit channels of commerce.
The DEA regulates controlled substances as Schedule I, II, III, IV or V substances. A pharmaceutical product may be listed as Schedule II, III, IV or V, with Schedule II substances considered to present the highest risk of abuse and Schedule V substances the lowest relative risk of abuse among such substances. The manufacture, shipment, storage, sale and use of Schedule II drugs are subject to a high degree of regulation. For example, Schedule XII drug prescriptions generally must be signed by a physician and may not be refilled without a new prescription. Substances in Schedule IV are considered to have a lower potential for abuse relative to substances in Schedule II. A prescription for controlled substances in Schedule III and IV may be issued by a practitioner through oral communication, in writing or by facsimile to the pharmacist and may be refilled if so, authorized on the prescription or by call-in. In the future, our other potential products may also be listed by the DEA as controlled substances.
Annual registration is required for any facility that manufactures, distributes, dispenses, imports or exports any controlled substance. The registration is specific to the particular location, activity and controlled substance schedule. For example, separate registrations are needed for import and manufacturing, and each registration will specify which schedules of controlled substances are authorized.
The DEA typically inspects a facility to review its security measures prior to issuing a registration. Security requirements vary by controlled substance schedule, with the most stringent requirements applying to Schedule I and Schedule II substances. Required security measures include background checks on employees and physical control of inventory through measures such as cages, surveillance cameras and inventory reconciliations. Records must be maintained for the handling of all controlled substances and periodic reports must be made to the DEA, including, for example, distribution reports for Schedule II controlled substances, Schedule III substances that are narcotics and other designated substances. Reports must also be made for thefts or losses of any controlled substance and authorization must be obtained to destroy any controlled substance. In addition, special authorization and notification requirements apply to imports and exports.
In addition, a DEA quota system controls and limits the availability and production of controlled substances in Schedule II. Distributions of any Schedule II controlled substance must also be accompanied by special order forms, with copies provided to the DEA. The DEA establishes annually an aggregate quota for how much of a Schedule II substance may be produced in total in the United States based on the DEA’s estimate of the quantity needed to meet legitimate scientific and medicinal needs. This limited aggregate amount of any particular Schedule II substance that the DEA allows to be produced in the United States each year is allocated among individual companies, who must submit applications annually to the DEA for individual production and procurement quotas. We and our contract manufacturers must receive an annual quota from the DEA in order to produce or procure any Schedule II substance for use in manufacturing. The DEA may adjust aggregate production quotas and individual production and procurement quotas from time to time during the year, although the DEA has substantial discretion in whether or not to make such adjustments. Our and our contract manufacturers’ quota of an active ingredient may not be sufficient to meet commercial demand or complete clinical trials. Any delay or refusal by the DEA in establishing our and our contract manufacturers’ quota for controlled substances could delay or stop our clinical trials or product launches, which could have a material adverse effect on our business, financial position and results of operations.
23
To meet its responsibilities, the DEA conducts periodic inspections of registered establishments that handle controlled substances. Failure to maintain compliance with applicable requirements, particularly as manifested in loss or diversion, can result in enforcement action that could have a material adverse effect on our business, results of operations and financial condition. The DEA may seek civil penalties, refuse to renew necessary registrations or initiate proceedings to revoke those registrations. In certain circumstances, violations could result in criminal proceedings.
Individual states also regulate controlled substances, and we and our contract manufacturers will be subject to state regulation on distribution of these products.
Pricing and Reimbursement
Successful commercialization of our products depends, in part, on the availability of governmental and third-party payor reimbursement for the cost of our products. Government authorities and third-party payors increasingly are challenging the price of medical products and services. On the government side, there is a heightened focus, at both the federal and state levels, on decreasing costs and reimbursement rates for Medicaid, Medicare and other government insurance programs. This has led to an increase in federal and state legislative initiatives related to drug prices, which could significantly influence the purchase of pharmaceutical products, resulting in lower prices and changes in product demand. If enacted, these changes could lead to reduced payments to pharmaceutical manufacturers. Many states have also created preferred drug lists and include drugs on those lists only when the manufacturers agree to pay a supplemental rebate. If our current products or future drug candidates are not included on these preferred drug lists, physicians may not be inclined to prescribe them to their Medicaid patients, thereby diminishing the potential market for our products.
In addition, third-party payors have been imposing additional requirements and restrictions on coverage and limiting reimbursement levels for pharmaceutical products. Third-party payors may require manufacturers to provide them with predetermined discounts from list prices and limit coverage to specific pharmaceutical products on an approved list, or formulary, which might not include all of the FDA-approved pharmaceutical products for particular indications. Third-party payors may challenge the price and examine the medical necessity and cost-effectiveness of pharmaceutical products in addition to their safety and efficacy. Manufacturers may need to conduct expensive pharmaco-economic studies in order to demonstrate the medical necessity and cost-effectiveness of pharmaceutical products in addition to the costs required to obtain the FDA approvals. Adequate third-party reimbursement may not be available to enable manufacturers to maintain price levels sufficient to realize an appropriate return on their investment in drug development.
Healthcare Reform
In the United States, there have been a number of federal and state proposals during the last several years regarding the pricing of pharmaceutical products, government control and other changes to the healthcare system of the United States. It is uncertain what other legislative proposals may be adopted or what actions federal, state, or private payors may take in response to any healthcare reform proposals or legislation. We cannot predict the effect such reforms may have on our business, and no assurance can be given that any such reforms will not have a material adverse effect.
By way of example, in March 2010, the ACA was signed into law, which, among other things, includes changes to the coverage and payment for drug products under government health care programs. The law includes measures that (i) significantly increase Medicaid rebates through both the expansion of the program and significant increases in rebates, (ii) substantially expand the Public Health System (340B) program to allow other entities to purchase prescription drugs at substantial discounts, (iii) extend the Medicaid rebate rate to a significant portion of Managed Medicaid enrollees, (iv) assess a rebate on Medicaid Part D spending in the coverage gap for branded and authorized generic prescription drugs, and (v) levy a significant excise tax on the industry to fund the healthcare reform.
24
Additionally, there has been heightened governmental scrutiny in the United States of pharmaceutical pricing practices in light of the rising cost of prescription drugs and biologics. On September 9, 2021, the Biden Administration published a wide-ranging list of policy proposals, most of which would need to be carried out by Congress, to reduce drug prices and drug payment. The United States Department of Health and Human Services (“HHS”) plan includes, among other reform measures, proposals to lower prescription drug prices, including by allowing Medicare to negotiate prices and disincentivizing price increases, and to support market changes that strengthen supply chains, promote biosimilars and generic drugs, and increase price transparency. These initiatives recently culminated in the enactment of the Inflation Reduction Act (the “IRA”) in August 2022, which will, among other things, allow the HHS to negotiate the selling price of certain drugs and biologics that the Centers for Medicare & Medicaid Services (“CMS”) reimburses under Medicare Part B and Part D, although only high-expenditure single-source drugs that have been approved for at least 7 years (11 years for biologics) can be selected by CMS for negotiation, with the negotiated price taking effect two years after the selection year. The negotiated prices, which will first become effective in 2026, will be capped at a statutory ceiling price beginning in October 2023, penalize drug manufacturers that increase prices of Medicare Part B and Part D drugs at a rate greater than the rate of inflation. The IRA permits the Secretary of HHS to implement many of these provisions through guidance, as opposed to regulation, for the initial years. Manufacturers that fail to comply with the IRA may be subject to various penalties, including civil monetary penalties. The IRA also extends enhanced subsidies for individuals purchasing health insurance coverage in ACA marketplaces through plan year 2025. These provisions will take effect progressively starting in 2023, although they may be subject to legal challenges.
Healthcare Regulations
Pharmaceutical companies are subject to various federal and state laws that are intended to combat health care fraud and abuse and that govern certain of our business practices, especially our interactions with third-party payors, healthcare providers, patients, customers and potential customers through sales and marketing or research and development activities. These include anti-kickback laws, false claims laws, sunshine laws, privacy laws and FDA regulation of advertising and promotion of pharmaceutical products.
Anti-kickback laws, including the federal Anti-Kickback Statute, make it a criminal offense knowingly and willfully to offer, pay, solicit, or receive any remuneration to induce or reward referral of an individual for, or the purchase, order or recommendation of, any good or service reimbursable by, a federal health care program (including our products). The federal Anti-Kickback Statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on the one hand and prescribers, purchasers and formulary managers on the other. Although there are several statutory exceptions and regulatory safe harbors protecting certain common activities from prosecution, the exceptions and safe harbors are drawn narrowly, and practices that involve remuneration intended to induce prescribing, purchasing or recommending may be subject to scrutiny if they do not qualify for an exception or safe harbor. In addition, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it to have committed a violation. Moreover, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act. The penalties for violating the federal Anti-Kickback Statute include administrative civil money penalties, imprisonment for up to five years, fines of up to $25,000 per violation and possible exclusion from federal healthcare programs such as Medicare and Medicaid.
The federal civil and criminal false claims laws, including the civil False Claims Act, prohibit knowingly presenting, or causing to be presented, claims for payment to the federal government (including Medicare and Medicaid) that are false or fraudulent (and, under the Federal False Claims Act, a claim is deemed false or fraudulent if it is made pursuant to an illegal kickback). Manufacturers can be held liable under these laws if they are deemed to “cause” the submission of false or fraudulent claims by, for example, providing inaccurate billing or coding information to customers or promoting a product off-label. Actions under the False Claims Act may be brought by the Attorney General or as a qui tam action by a private individual in the name of the government. Violations of the False Claims Act can result in significant monetary penalties, including fines ranging from $11,181 to $22,363 for each false claim, and treble damages. The federal government is using the False Claims Act, and the accompanying threat of significant liability, in its investigation and prosecution of pharmaceutical companies throughout the country, for example, in connection with the promotion of products for unapproved uses and other improper sales and marketing practices. The government has obtained multi-million and multi-billion dollar settlements under the False Claims Act in addition to individual criminal convictions under applicable criminal statutes. In addition, companies have been forced to implement extensive corrective action plans and have often become subject to consent decrees or corporate integrity agreements, severely restricting the manner in which they conduct their business. Given the significant size of actual and potential settlements, it is expected that the government will continue to devote substantial resources to investigating healthcare providers’ and manufacturers’ compliance with applicable fraud and abuse laws.
25
The Federal Civil Monetary Penalties Law prohibits, among other things, the offering or transferring of remuneration to a Medicare or Medicaid beneficiary that the person knows or should know is likely to influence the beneficiary’s selection of a particular supplier of Medicare or Medicaid payable items or services. Noncompliance can result in civil money penalties of up to $15,270 for each wrongful act, assessment of three times the amount claimed for each item or service and exclusion from the federal healthcare programs.
Federal criminal statutes prohibit, among other actions, knowingly and willfully executing or attempting to execute a scheme to defraud any healthcare benefit program, including private third-party payors, knowingly and willfully embezzling or stealing from a healthcare benefit program, willfully obstructing a criminal investigation of a healthcare offense, and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Like the federal Anti-Kickback Statute, the ACA amended the intent standard for certain healthcare fraud statutes under HIPAA such that a person or entity no longer needs to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
Analogous state and foreign laws and regulations, including state anti-kickback and false claims laws, may apply to products and services reimbursed by non-governmental third-party payors, including commercial payors. Additionally, there are state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government or that otherwise restrict payments that may be made to healthcare providers as well as state and foreign laws that require drug manufacturers to report marketing expenditures or pricing information.
Sunshine laws, including the Federal Open Payments law enacted as part of the ACA, require pharmaceutical manufacturers to disclose payments and other transfers of value to physicians and certain other health care providers or professionals, and in the case of some state sunshine laws, restrict or prohibit certain such payments. Pharmaceutical manufacturers are required to submit reports to the government by the 90th day of each calendar year. Failure to submit the required information may result in civil monetary penalties of up to an aggregate of $165,786 per year (or up to an aggregate of $1.105 million per year for “knowing failures”) for all payments, transfers of value or ownership or investment interests not reported in an annual submission and may result in liability under other federal laws or regulations. Certain states and foreign governments require the tracking and reporting of gifts, compensation and other remuneration to physicians.
Privacy laws, such as the privacy regulations implemented under HIPAA, restrict covered entities from using or disclosing protected health information. Covered entities commonly include physicians, hospitals and health insurers from which we may seek to acquire data to aid in our research, development, sales and marketing activities. Although pharmaceutical manufacturers are not covered entities under HIPAA, our ability to acquire or use protected health information from covered entities may be affected by privacy laws. Specifically, HIPAA, as amended by HITECH, and their respective implementing regulations, including the final omnibus rule published on January 25, 2013, imposes specified requirements relating to the privacy, security and transmission of individually identifiable health information.
Among other things, HITECH makes HIPAA’s privacy and security standards directly applicable to “business associates,” defined as independent contractors or agents of covered entities that create, receive, maintain or transmit protected health information in connection with providing a service for or on behalf of a covered entity. HITECH also increased the civil and criminal penalties that may be imposed against covered entities, business associates and possibly other persons, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorney’s fees and costs associated with pursuing federal civil actions. In addition, state laws govern the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways, thus complicating compliance efforts.
The FDA regulates the sale and marketing of prescription drug products and, among other things, prohibits pharmaceutical manufacturers from making false or misleading statements and from promoting products for unapproved uses. There has been an increase in government enforcement efforts at both the federal and state level. Numerous cases have been brought against pharmaceutical manufacturers under the Federal False Claims Act, alleging, among other things, that certain sales or marketing-related practices violate the Anti-Kickback Statute or the FDA’s regulations, and many of these cases have resulted in settlement agreements under which the companies were required to change certain practices, pay substantial fines and operate under the supervision of a federally appointed monitor for a period of years. Due to the breadth of these laws and their implementing regulations and the absence of guidance in some cases, it is possible that our practices might be challenged by government authorities. Violations of fraud and abuse laws may be punishable by civil and criminal sanctions including fines, civil monetary penalties, as well as the possibility of exclusion of our products from payment by federal health care programs.
26
Government Price Reporting
Government regulations regarding reporting and payment obligations are complex, and we are continually evaluating the methods we use to calculate and report the amounts owed with respect to Medicaid and other government pricing programs. Our calculations are subject to review and challenge by various government agencies and authorities, and it is possible that any such review could result either in material changes to the method used for calculating the amounts owed to such agency or the amounts themselves. Because the process for making these calculations, and our judgments supporting these calculations, involve subjective decisions, these calculations are subject to audit. In the event that a government authority challenges or finds ambiguity with regard to our report of payments, such authority may impose civil and criminal sanctions, which could have a material adverse effect on our business. From time to time we conduct routine reviews of our government pricing calculations. These reviews may have an impact on government price reporting and rebate calculations used to comply with various government regulations regarding reporting and payment obligations.
Many governments and third-party payors reimburse the purchase of certain prescription drugs based on a drug’s AWP. In the past several years, state and federal government agencies have conducted ongoing investigations of manufacturers’ reporting practices with respect to AWP, which they have suggested have led to excessive payments by state and federal government agencies for prescription drugs. We and numerous other pharmaceutical companies have been named as defendants in various state and federal court actions alleging improper or fraudulent practices related to the reporting of AWP.
Drug Pedigree Laws
State and federal governments have proposed or passed various drug pedigree laws which can require the tracking of all transactions involving prescription drugs from the manufacturer to the pharmacy (or other dispensing) level. Companies are required to maintain records documenting the chain of custody of prescription drug products beginning with the purchase of such products from the manufacturer. Compliance with these pedigree laws requires implementation of extensive tracking systems as well as heightened documentation and coordination with customers and manufacturers. While we fully intend to comply with these laws, there is uncertainty about future changes in legislation and government enforcement of these laws. Failure to comply could result in fines or penalties, as well as loss of business that could have a material adverse effect on our financial results.
Federal Regulation of Patent Litigation Settlements and Authorized Generic Arrangements
As part of the Medicare Prescription Drug Improvement and Modernization Act of 2003, companies are required to file with the U.S. Federal Trade Commission (“FTC”) and the U.S. Department of Justice (the “DOJ”) certain types of agreements entered into between brand and generic pharmaceutical companies related to the settlement of patent litigation or manufacture, marketing and sale of generic versions of branded drugs. This requirement could affect the manner in which generic drug manufacturers resolve intellectual property litigation and other disputes with brand pharmaceutical companies and could result generally in an increase in private-party litigation against pharmaceutical companies or additional investigations or proceedings by the FTC or other governmental authorities.
Other
The U.S. federal government, various states and localities have laws regulating the manufacture and distribution of pharmaceuticals, as well as regulations dealing with the substitution of generic drugs for branded drugs. Our operations are also subject to regulation, licensing requirements and inspection by the states and localities in which our operations are located or in which we conduct business.
Certain of our activities are also subject to FTC enforcement actions. The FTC also enforces a variety of antitrust and consumer protection laws designed to ensure that the nation’s markets function competitively, are vigorous, efficient and free of undue restrictions. Federal, state, local and foreign laws of general applicability, such as laws regulating working conditions, also govern us.
27
In addition, we are subject to numerous and increasingly stringent federal, state and local environmental laws and regulations concerning, among other things, the generation, handling, storage, transportation, treatment and disposal of toxic and hazardous substances, the discharge of pollutants into the air and water and the cleanup of contamination. We are required to maintain and comply with environmental permits and controls for some of our operations, and these permits are subject to modification, renewal and revocation by the issuing authorities. Our environmental capital expenditures and costs for environmental compliance may increase in the future as a result of changes in environmental laws and regulations or increased manufacturing activities at any of our facilities. We could incur significant costs or liabilities as a result of any failure to comply with environmental laws, including fines, penalties, third-party claims and the costs of undertaking a clean-up at a current or former site or at a site to which our wastes were transported. In addition, we have grown in part by acquisition, and our diligence may not have identified environmental impacts from historical operations at sites we have acquired in the past or may acquire in the future.
Human Capital Resources
As of March 21, 2023,15, 2024, we have a total of 7 full time employees and one contractor.employees. We have no collective bargaining agreements with our employees, and none are represented by labor unions. We consider our current relations with our employees to be good.
We believe that our future success will depend, in part, on our continued ability to attract, hire and retain qualified personnel. In particular, we depend on the skills, experience and performance of our senior management and research personnel. We compete for qualified personnel with other medical pharmaceutical, and healthcare companies, as well as universities and non-profit research institutions.
We provide competitive compensation and benefits programs to help meet the needs of our employees. In addition to salaries, these programs (which vary by country/region and employment classification) include incentive compensation plans, healthcare and insurance benefits, retirement investments, paid time off, and family leave, among others. We also use targeted equity-based grants with vesting conditions to facilitate retention of personnel, particularly for our key employees.
The success of our business is fundamentally connected to the well-being of our people. Accordingly, we are committed to the health and safety of our employees. In response to the COVID-19 pandemic, we implemented significant changes that we determined were in the best interest of our employees, as well as the communities in which we operate, and which comply with government regulations.
Facilities
Our principal address is 1055 Westlakes Drive, Suite 300, Berwyn, PAPennsylvania 19312. We believe our facilities are adequate to meet our current needs, although we may seek to negotiate new leases or evaluate additional or alternate space for our operations. We believe appropriate alternative space would be readily available on commercially reasonable terms.
Our telephone number is (610) 727-4597 and our website address is www.virpaxpharma.com. The information on our website is not incorporated by reference into this Annual Report on Form 10-K.
28
Item 1A. RISK FACTORS
An investment in our common stock is speculative and illiquid and involves a high degree of risk including the risk of a loss of your entire investment. You should carefully consider the risks and uncertainties described below and the other information contained in this report and our other reports filed with the Securities and Exchange Commission. The risks set forth below are not the only ones facing us. Additional unanticipated or unknown risks and uncertainties may exist that could also adversely affect our business, operations and financial condition in ways that are unknown to us or unpredictable. If any of the following risks actually materialize, our business, financial condition and/or operations could suffer. In such event, the value of our common stock could decline, and you could lose all or a substantial portion of the money that you pay for our common stock.
Summary of Risks Associated with Our Business
Our business and an investment in our company is subject to numerous risks and uncertainties, including those highlighted in the section titled “Risk Factors” immediately following this prospectus summary. Some of these risks include:
29
Risks Related to Our Financial Position and Need for Additional Capital
We are a preclinical stage biopharmaceutical company with a limited operating history.
We were established and began operations in 2017. Our operations to date have been limited to financing and staffing our company, licensing product candidates, conducting preclinical trialsstudies of Epoladerm for chronic osteoarthritis of the knee, Probudur for postoperative hip and knee replacement pain management, Envelta for the management of acute and chronicto control severe pain, including post cancer pain, associated with cancer, AnQlar as an anti-viral barrier to potentially prevent or reduce the risk or the intensity of viral infections in humans, including, but not limited to, influenza and SARS-CoV-2 (COVID 19), and NobrXiol to potentially treat epileptic seizures associated with, TSC, Lennox-Gastaut syndrome and Dravet syndrome in pediatric patients one yeartwo years of age and older. We have not yet demonstrated the ability to successfully complete a large-scale, pivotal clinical trial, obtain marketing approval, manufacture a commercial scale product, arrange for a third party to do so on our behalf, or conduct sales and marketing activities necessary for successful product commercialization. Consequently, predictions about our future success or viability may not be as accurate as they could be if we had a history of successfully developing and commercializing pharmaceutical products.
Accordingly, you should consider our prospects in light of the costs, uncertainties, delays and difficulties frequently encountered by companies in the early stages of development, especially preclinical stage pharmaceutical companies such as ours. Potential investors should carefully consider the risks and uncertainties that a company with a limited operating history will face. In particular, potential investors should consider that we cannot assure you that we will be able to, among other things:
| ● | successfully implement or execute our current business plan, and we cannot assure you that our business plan is sound; |
| ● | successfully manufacture our clinical product candidates and establish commercial supply; |
| ● | successfully complete the clinical trials necessary to obtain regulatory approval for the marketing of our product candidates; |
| ● | secure market exclusivity and/or adequate intellectual property protection for our product candidates; |
| ● | attract and retain an experienced management and advisory team; |
| ● | secure acceptance of our product candidates in the medical community and with third-party payors and consumers; |
| ● | raise sufficient funds in the capital markets or otherwise to effectuate our business plan; and |
| ● | utilize the funds that we do have and/or raise in the future to efficiently execute our business strategy. |
If we cannot successfully execute any one of the foregoing, our business may fail and your investment will be adversely affected.
30
We have incurred losses since inception and anticipate that we will continue to incur losses for the foreseeable future. We are not currently profitable, and we may never achieve or sustain profitability.
We are a preclinical stage biopharmaceutical company with a limited operating history and have incurred losses since our formation. We incurred net losses of approximately $21.7$15.2 million and $12.1$21.7 million for the years ended December 31, 20222023 and 2021,2022, respectively. As of December 31, 2022,2023, we had an accumulated deficit of approximately $44.4$59.5 million. We have not commercialized any product candidates and have never generated revenue from the commercialization of any product. To date, we have devoted most of our financial resources to research and development, including our preclinical work, general and administrative expenses including, but not limited to,which have primarily consisted of legal defense costs, legal settlement, and general corporate purposes, as well as to intellectual property.purposes.
We expect to incur significant additional operating losses for the next several years, at least, as we advance Epoladerm, Probudur, Envelta, AnQlar and EnveltaNobrXiol through preclinical development, complete clinical trials, seek regulatory approval and commercialize Epoladerm, Probudur, Envelta, AnQlar and NobrXiol (collectively, “Product Candidates"Candidates”), if approved. The costs of advancing product candidates into each clinical phase tend to increase substantially over the duration of the clinical development process. Therefore, the total costs to advance any of our product candidates to marketing approval in even a single jurisdiction will be substantial. Because of the numerous risks and uncertainties associated with pharmaceutical product development, we are unable to accurately predict the timing or amount of increased expenses or when, or if, we will be able to begin generating revenue from the commercialization of any products or achieve or maintain profitability. Our costs and expenses will also increase substantially if and as we:
| ● | Fund the remaining portion $2.5 million pursuant to the Settlement Agreement, make related indemnification and/or contribution payments, which payment, if any, may be material, or estimated separation payments we agree to make to our former Chief Executive Officer, which may be material (See Item 3-Legal Proceedings); |
| ● | are required by the FDA, to complete Phase 2 trials to support an NDA for our Product Candidates; |
| ● | are required by the FDA to complete Phase 3 trials to support NDAs for our Product Candidates; |
| ● | establish a sales, marketing and distribution infrastructure to commercialize our drugs, if approved, and for any other product candidates for which we may obtain marketing approval; |
| ● | maintain, expand and protect our intellectual property portfolio; |
| ● | hire additional clinical, scientific and commercial personnel; |
| ● | add operational, financial and management information systems and personnel, including personnel to support our product development and planned future commercialization efforts, as well as to support our transition to a public reporting company; and |
| ● | acquire or in-license or invent other product candidates or technologies. |
Furthermore, our ability to successfully develop, commercialize and license any product candidates and generate product revenue is subject to substantial additional risks and uncertainties, as described under “Risks Related to Development, Clinical Testing, Manufacturing and Regulatory Approval” and “Risks Related to Commercialization.” As a result, we expect to continue to incur net losses and negative cash flows for the foreseeable future. These net losses and negative cash flows have had, and will continue to have, an adverse effect on our stockholders’ equity and working capital. The amount of our future net losses will depend, in part, on the rate of future growth of our expenses and our ability to generate revenues. If we are unable to develop and commercialize one or more product candidates, either alone or through collaborations, or if revenues from any product that receives marketing approval are insufficient, we will not achieve profitability. Even if we do achieve profitability, we may not be able to sustain profitability or meet outside expectations for our profitability. If we are unable to achieve or sustain profitability or to meet outside expectations for our profitability, the value of our common stock will be materially and adversely affected.
The report of our independent registered public accounting firm for the fiscal years ended December 31, 2023 and 2022 contains an explanatory paragraph regarding substantial doubt about our ability to continue as a going concern.
31
The report of our independent registered public accounting firm on our financial statements as of and for the years ended December 31, 2023 and December 31, 2022 includes an explanatory paragraph indicating that there is substantial doubt about our ability to continue as a going concern. Due to our continuing losses and the anticipated significant decrease in our cash position from the payments to be made to the Plaintiffs pursuant to the Settlement Agreement, there exists substantial doubt about our ability to continue as a going concern. Pursuant to the Settlement Agreement, we have paid the Plaintiffs $3.5 million on March 18, 2024, and agreed to make an additional $2.5 million payment on or before July 1, 2024. The financial statements do not include any adjustments to the carrying amounts and classification of assets, liabilities, and reported expenses that may be necessary if we were unable to continue as a going concern.
We will require additional financings to fund our operations, including completing clinical development of and to commercially develop all of our product candidates and/or to fund litigation costs and expenses, including the required payment of the $2.5 million on or before July 1, 2024, related indemnification and/or contribution payments, if any, and which such payments may be material, as well as other potential estimated separation payments to our former Chief Executive Officer, which also may be material. There is no assurance that such financing will be available when needed or on acceptable terms. We also may be forced to curtail spending in research and development activities in order to conserve cash.
We will require additional capital to fund our operations, and if we fail to obtain necessary financing, we maywill not be able to complete the development and commercialization of our drugs.
Our operations have consumed substantial amounts of cash since inception. In addition, due to the $3.5 million payment that has been made, and the $2.5 million payment that will be required to be made on or before July 1, 2024, to the Plaintiffs pursuant to the Settlement Agreement our cash position has been and will be significantly decreased. The payment of the royalties to the Plaintiffs pursuant to the terms of the Settlement Agreement, will significantly impact our future revenue and may make it more difficult to engage in collaborations, licenses or the acquisition of certain product candidates, and may result in us ceasing to develop certain product candidates or all of our product candidates if we determine that it will not be financially profitable to do so. In addition, litigation related indemnification and/or contribution payments, if any, and which may be material, and any cash estimated separation payments, which may be material, that we make to our former Chief Executive Officer will further reduce our cash position. We expect to continue to spend substantial amounts to advance the clinical development of and launch and commercialize our product candidates if we receive regulatory approval. In addition, the damages that we are required to pay in connection with our litigation have and will impact the amount of cash available for clinical development of our product candidates. Our current cash position is not sufficient to enable us to fund our operations, including making the second payment under the Settlement Agreement. If we are unable to raise additional capital in the next few months, of which there can be no certainty, we may be forced to liquidate assets or initiate bankruptcy proceedings.
We will require additional capital for the further development and potential commercialization of our Product Candidates and may also need to raise additional funds sooner to pursue a more accelerated development of our Product Candidates. If we are unable to raise capital when needed or on attractive terms, we could be forced to delay, reduce or eliminate our research and development programs or any future commercialization efforts. In addition, our strategy for AnQlar and Epoladerm is to license out or partner these assets as we continue to focus our efforts on our prescription drug pipeline. If we are unsuccessful in our partnering activities and/or financing activities, we may be unable to develop AnQlar and Epoladerm.
At December 31, 2022,2023, we had cash of approximately $19.0$9.1 million. On March 18, 2024, we paid $3.5 million to the Plaintiffs and we have agreed to pay the Plaintiffs an additional $2.5 million on or before July 1, 2024. In addition, litigation related indemnification and/or contribution payments, if any, and which may be material, and any cash estimated separation payments that we make to our former Chief Executive Officer, which may be material, will further reduce our cash position. See Note 5 to the Notes to Financial Statements included in this Annual Report on Form 10-K for additional information regarding these payments. We have incurred continuing losses since inception, including a loss of $21.7$15.2 million for the year ended December 31, 2022, and are currently involved in litigation and while we believe we have meritorious defenses, the ultimate resolution of the action could result in a material loss.2023. Our future funding requirements, both near and long-term, will depend on many factors, including, but not limited to the:
| ● | costs associated with litigation, adverse remedy judgments and/or |
| ● | initiation, progress, timing, costs and results of preclinical studies and clinical trials, including patient enrollment in such trials, for our Product Candidates or any other future product candidates; |
| ● | clinical development plans we establish for our Product Candidates and any other future product candidates; |
| ● | obligation to make royalty and non-royalty sublicense receipt payments to third-party licensors, if any, under our licensing agreements; |
| ● | number and characteristics of product candidates that we discover or in-license and develop; |
| ● | outcome, timing and cost of regulatory review by the FDA and comparable foreign regulatory authorities, including the potential for the FDA or comparable foreign regulatory authorities to require that we perform more studies than those that we currently expect; |
| ● | costs of filing, prosecuting, defending and enforcing any patent claims and maintaining and enforcing other intellectual property rights; |
| ● | effects of competing technological and market developments; |
| ● | costs and timing of the implementation of commercial-scale manufacturing activities; |
| ● | costs and timing of establishing sales, marketing and distribution capabilities for any product candidates for which we may receive regulatory approval; and |
| ● | cost associated with being a public company. |
If we are unable to expand our operations or otherwise capitalize on our business opportunities due to a lack of capital, our ability to become profitable or survive will be compromised.
Our business, financial condition and results of operations could be adversely affected by indemnification and other claims related to the damages awarded in our recently settled litigation with Sorrento Therapeutics, Inc. and Scilex Pharmaceuticals, Inc.
Per the Settlement Agreement, the Plaintiff’s have released all claims against us. The Plaintiffs, however, can still pursue claims against Mr. Mack. Our Amended and Restated Bylaws dated November 18, 2020 (“Bylaws”) require us to “indemnify any person who was or is a party or is threatened to be made a party to any threatened, pending or completed action, suit or proceeding, whether civil, criminal, administrative or investigative (other than an action by or in the right of the Corporation) by reason of the fact that such person is or was a director or officer of the Corporation, or, while a director or officer of the Corporation….” Such indemnification, however, is limited to circumstances where the covered person “acted in good faith and in a manner such person reasonably believed to be in or not opposed to the best interests of the Corporation….” Mr. Mack may attempt to claim he is entitled to indemnification, should the Chancery Court find him liable for damages in the Action. Given the findings in the Memorandum Opinion issued in the Action, we believe we have a strong position that Mr. Mack would not be entitled to indemnification. There is a risk, however, that a court could find he is entitled to such indemnification. Additionally, per Section 7.6 of the Bylaws, we have been advancing Mr. Mack’s attorneys’ fees and costs for the Action. It is likely Mr. Mack will contend he is still entitled to advancement of any fees and/or costs for the Action going forward and may seek judicial intervention. However, as per the Bylaws, Mr. Mack is only entitled to advancement of expenses for indemnifiable actions. As noted above, given the Memorandum Opinion in the Action, we believe that we have a strong position that Mr. Mack is not entitled to indemnification, and therefore, not entitled to advancement of expenses. However, there is a risk that a court could find that Mr. Mack is entitled to such advancement. Further, Mr. Mack may attempt to seek damages from us based on the Chancery Court’s final judgment on damages under the theory of joint and several liability and seek contribution from us for any monetary judgment.
The Chancery Court is aware that Plaintiffs have settled with us and that the Settlement Agreement fully releases us from any claims or damages the Plaintiffs have against us related to the Action. Given the Settlement Agreement does not release Mr. Mack from liability related to the Action, the Chancery Court has requested supplemental briefing as to whether the Chancery Court can dismiss us from the lawsuit, as well as any claims Mr. Mack has against us arising from the Action. While we believe that any damages assessed may be awarded against Mr. Mack alone, Plaintiffs cannot seek additional damages from Virpax. However, there is a risk that Mr. Mack will still seek contribution from us for any damages claim arising from the Action. And, there is a risk that the Chancery Court will rule in Mr. Mack’s favor.
No further reimbursements are permitted from our insurance policy with respect to the litigation. Accordingly, if Mr. Mack was successful in seeking indemnification from us, we would have to pay such amounts in cash which would further reduce our cash position.
Raising additional capital may cause dilution to our stockholders, restrict our operations or require us to relinquish rights to our technologies or product candidates.
Until such time, if ever, as we can generate substantial revenue, we may finance our cash needs through a combination of equity offerings, debt financings, marketing and distribution arrangements and other collaborations, strategic alliances and licensing arrangements or other sources. We do not currently have any committed external source of funds. In addition, we may seek additional capital due to favorable market conditions or strategic considerations, even if we believe that we have sufficient funds for our current or future operating plans.
32
To the extent that we raise additional capital through the sale of equity or convertible debt securities, your ownership interest will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect your rights as a common stockholder. Debt financing and preferred equity financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. If we raise additional funds through collaborations, strategic alliances or marketing, distribution or licensing arrangements with third parties, we may be required to relinquish valuable rights to our technologies, intellectual property, future revenue streams or product candidates or grant licenses on terms that may not be favorable to us. If we are unable to raise additional funds through equity or debt financingsfinancing when needed, we may be required to delay, limit, reduce or terminate product candidate development or future commercialization efforts.
Changes in U.S. tax law may materially adversely affectIn addition, our financial condition, results of operationsstrategy for AnQlar and cash flows.
On March 27, 2020, the Coronavirus Aid, Relief, and Economic Security Act,Epoladerm is to license out or the CARES Act, was signed into law to address the COVID-19 crisis. The CARES Act is an approximately $2 trillion emergency economic stimulus package that includes numerous U.S. federal income tax provisions, including the modification of: (i) net operating loss rules (as discussed below), (ii) the alternative minimum tax refund and (iii) business interest deduction limitations under Section 163(j) of the Internal Revenue Code of 1986,partner this asset as amended, or the Code.
On December 22, 2017, President Trump signed into law federal tax legislation commonly referred to as the TCJA (defined below), which also significantly changed the U.S. federal income taxation of U.S. corporations. TCJA remains unclear in some respects and has been, and maywe continue to be, subject to amendments and technical corrections, as well as interpretations and implementing regulations by the Treasury and Internal Revenue Service, or the IRS, any offocus our efforts on our prescription drug pipeline, which could lessen or increase certain adverse impacts of TCJA.
TJCA (P.L. 115-97) modified the section 174 rules and beginningmay result in 2022, taxpayers may no longer currently deduct R&D expenditures but instead must amortize specified R&D expenditures ratably over five years (or 15 years for foreign expenditures).
While some of these U.S. federal income tax changes may adversely affect us in one or more reporting periods and prospectively, other changes may be beneficial on a going-forward basis. We continue to work with our tax advisors and auditors to determine the full impact TCJA will have on us. We urge our investors to consult with their legal and tax advisors with respect to TCJA and the potential tax consequences of investing in our common stock.shared revenue.
Our ability to use our net operating loss carryforwards to offset future taxable income may be subject to certain limitations.
Our net operating loss carryforwards (“NOLs”), and certain other tax attributes could expire unused and be unavailable to offset future income tax liabilities because of their limited duration or because of restrictions under U.S. tax law. As of December 31, 2022,2023, we had NOLs of approximately $28,397,000$35.2 million for federal and $37.1 million for state income tax purposes. Our federal NOL of $28,397,000$35.2 million includes $326,000 which expires in 2037, and the remaining NOL has an indefinite carryover period subject to limitation, and our state NOL’s of $28,397,000$37.1 million expire from 2037 through 2042.2043. Additionally, we have $546,000$654,000 of R&D credits which have a 20 year20-year carryforward period, which will expire from 2038 to 2042.2043.
33
Under TCJA (defined below), federal NOLs generated in tax years ending after December 31, 2017 may be carried forward indefinitely. Under the CARES Act, NOL carryforwards arising in tax years beginning after December 31, 2017 and before January 1, 2021 may be carried back to each of the five tax years preceding the tax year of such loss. Due to our cumulative losses through December 31, 2022,2023, we do not anticipate that such provision of the CARES Act will be relevant to us. The deductibility of federal NOLs, particularly for tax years beginning after December 31, 2022, may be limited. It is uncertain if and to what extent various states will conform to TCJA or the CARES Act.
In addition, our NOLs are subject to review and possible adjustment by the IRS, and state tax authorities. In general, under Sections 382 and 383 of the Code, a corporation that undergoes an “ownership change” is subject to limitations on its ability to use its pre-change NOLs and research & development credit to offset future taxable income. Due to previous ownership changes, or if we undergo an ownership change, our ability to use our NOLs and research & development credit could be limited by Section 382 of the Code. Future changes in our stock ownership, inclusive of a public offering and some of which are outside of our control, could result in an ownership change under Sections 382 and 383 of the Code. Furthermore, our ability to use NOLs and research & development credit of companies that we may acquire in the future may be subject to limitations. For these reasons, we may not be able to use a material portion of the NOLs and research & development credit, even if we attain profitability.
We examined the application of Section 382 and Section 383 with respect to ownership changes that took place during 2021, as well as the limitation on the application of net operating loss carry forwards. We determined that a more than 50% ownership change occurred on September 16, 2021. We also determined that the recent change in ownership limits our usage of net operating loss, other carry forwards and tax credits as of the change in ownership date to an annual amount of $4.1 million. Our net carryforwards and tax credits may be further limited in the future if additional ownership changes occur.
Our business, financial condition and results of operationsability to further develop our product candidates may be adversely affected if we are unsuccessful in our current litigation with Sorrento Therapeutics, Inc. and Scilex Pharmaceuticals, Inc.by the terms of the Settlement Agreement.
On March 12, 2021,The cash payments as well as the Company and itsroyalty payments that we have agreed to pay to the Plaintiffs pursuant to the Settlement Agreement as well as any payments we may be required to make to our former Chief Executive Officer, Anthony P. Mack (the “Defendants”), were named as defendantsmay result in a complaint (the “Complaint”) filed by Sorrento Therapeutics, Inc. (“Sorrento”), and Scilex Pharmaceuticals Inc. (“Scilex” and together with Sorrento, the “Plaintiffs”) in the Court of Chancery of the State of Delaware. In the Complaint, Plaintiffs alleged (i) Mr. Mack breached a Restrictive Covenants Agreement, dated as of November 8, 2016, between himself and Sorrento (the “Restrictive Covenants Agreement”), (ii) the Company tortiously interfered with the Restrictive Covenants Agreement, and (iii) the Company tortiously interfered with Scilex’s relationship with Mr. Mack. On May 7, 2021 Plaintiffs filed an Amended Complaint asserting the same three causes of action. On September 28, 2021, Plaintiffs filed a Second Amended Complaint asserting the same three causes of action as the prior complaints, as well as claims in which Plaintiffs alleged (i) Mr. Mack breached an Employment, Proprietary Information and Inventions Agreement, dated as of October 25, 2016, between himself and Sorrento (the “Employment Agreement”), (ii) the Company tortiously interfered with the Employment Agreement, (iii) Mr. Mack breached his fiduciary dutiesus ceasing to Scilex, and (iv) the Company aided and abetted Mr. Mack’s alleged breach of fiduciary duties to Scilex. On April 1, 2022, Plaintiffs filed a Third Amended Complaint. The Third Amended Complaint asserts the same causes of action as the Second Amended Complaint, as well as claims for (i) misappropriation of trade secrets by Defendants under Delaware law, and (ii) misappropriation of trade secrets by Defendants under California law. On April 18, 2022, Defendants filed answers to the Third Amended Complaint. Trial was held before Vice Chancellor Paul Fiorvanti from September 12 through September 14, 2022. Post-trial briefing was completed by December 12, 2022, and post-trial argument was held on January 20, 2023. The Company intends to vigorously defend the action. However, the Company is unable to predict the ultimate outcome of the lawsuit. Plaintiffs asserted alternative damages theories that would imply potential damages up to approximately $35.0 million. The Company countered that actual damages, even if Plaintiffs establish liability, could be zero because, among other things, Plaintiffs’ calculations use various unsupported assumptions, any alleged damages are speculative in nature, and there is a possibility the Company’sdevelop certain product candidates never reach market. To date,or all of our product candidates if we determine that it will not be financially profitable to do so. The payment of these royalties will significantly impact our future revenue and may make it more difficult to engage in collaborations, licenses or the parties have not had successful settlement negotiations. Based on the foregoing, the Company accrued $2 million in respectacquisition of the litigation. While the Company believes it has meritorious defenses, the ultimate resolution of the action could result in a material loss.certain product candidates.
Defending this lawsuit may cause us to incur significant legal expenses. Furthermore, in the event that we are unsuccessful in the defense of this lawsuit, we could be required to pay damages which could have a material adverse effect on our business, financial condition and results of operations.
34
The Company and our officers and directors may be subject to various types of litigation, and our insurance may not cover or be sufficient to cover damages related to those claims.
From time-to-time we may be involved in lawsuits or other claims arising in the ordinary course of business, including those related to product liability, consumer protection, employment, intellectual property, tort, privacy and data protection, and other matters. Although we maintain insurance in accordance with customary practice of companies of similar size and stage of development, our insurance may only cover us against some, and not all, of these potential claims. We may incur losses relating to claims filed against us or our directors and officers, including costs associated with defending against such claims, and there is risk that any such claims or liabilities will exceed our insurance coverage, or affect our ability to retain adequate liability insurance in the future. We may elect not to obtain insurance if we believe that the cost of available insurance is excessive relative to the risks presented. The levels of insurance we maintain may not be adequate to fully cover any and all losses or liabilities. Further, we may not be able to maintain insurance at commercially acceptable premium levels or at all. If any significant judgment, claim (or a series of claims), a settlement or other event is not fully insured or indemnified against, it could have a material adverse impact on our business, financial condition and results of operations. There can be no assurance as to the actual amount of these liabilities or the timing thereof. We cannot be certain that the outcome of current or future litigation will not have a material adverse impact on our business and results of operations.
The report of our independent registered public accounting firm for the fiscal years ended December 31, 2022 and 2021 contains an explanatory paragraph regarding substantial doubt about our ability to continue as a going concern.
The report of our independent registered public accounting firm on our financial statements as of and for the years ended December 31, 2022 and December 31, 2021 includes an explanatory paragraph indicating that there is substantial doubt about our ability to continue as a going concern. We are currently involved in litigation and have established an estimated litigation liability of $2.0 million. While we believe we have meritorious defenses, the ultimate resolution of the action could result in a material loss. Due to our continuing losses, the uncertainty regarding the outcome of this ongoing litigation and any potential claims related thereto, there exists substantial doubt about our ability to continue as a going concern. The financial statements do not include any adjustments to the carrying amounts and classification of assets, liabilities, and reported expenses that may be necessary if we were unable to continue as a going concern.
We will require additional financings to fund our operations, including litigation costs, and to complete clinical development of and to commercially develop all of our product candidates. There is no assurance that such financing will be available when needed or on acceptable terms. We also may be forced to curtail spending in research and development activities in order to conserve cash.
Risks Related to Development, Clinical Testing, Manufacturing and Regulatory Approval
Clinical trials are expensive, time-consuming and difficult to design and implement, and involve an uncertain outcome.
Clinical testing is expensive and can take many years to complete, and its outcome is inherently uncertain. Failure can occur at any time during the clinical trial process. Because the results of preclinical studies and early clinical trials are not necessarily predictive of future results, our Product Candidates may not have favorable results in later preclinical and clinical studies or receive regulatory approval. We may experience delays in initiating and completing any clinical trials that we intend to conduct, and we do not know whether planned clinical trials will begin on time, need to be redesigned, enroll patients on time or be completed on schedule, or at all. Clinical trials can be delayed for a variety of reasons, including delays related to:
| ● | the FDA or comparable foreign regulatory authorities disagreeing as to the design or implementation of our clinical studies; |
| ● | obtaining regulatory approval to commence a trial; |
| ● | reaching an agreement on acceptable terms with prospective CROs, and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; |
| ● | obtaining Institutional Review Board (“IRB”), approval at each site, or Independent Ethics Committee (“IEC”), approval at sites outside the United States; |
| ● | recruiting suitable patients to participate in a trial in a timely manner and in sufficient numbers; |
| ● | having patients complete a trial or return for post-treatment follow-up; |
| ● | imposition of a clinical hold by regulatory authorities, including as a result of unforeseen safety issues or side effects or failure of trial sites to adhere to regulatory requirements or follow trial protocols; |
| ● | clinical sites deviating from trial protocol or dropping out of a trial; |
| ● | addressing patient safety concerns that arise during the course of a trial; |
| ● | adding a sufficient number of clinical trial sites; or |
| ● | manufacturing sufficient quantities of product candidate for use in clinical trials. |
35
We could also encounter delays if a clinical trial is suspended or terminated by us, the IRBs or IECs of the institutions in which such trials are being conducted, the Data Safety Monitoring Board (“DSMB”) for such trial or the FDA or other regulatory authorities. Such authorities may impose such a suspension or termination due to a number of factors, including failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols, inspection of the clinical trial operations or trial site by the FDA or other regulatory authorities resulting in the imposition of a clinical hold, unforeseen safety issues or adverse side effects, failure to demonstrate a benefit from using a drug, changes in governmental regulations or administrative actions or lack of adequate funding to continue the clinical trial. Furthermore, we rely on CROs and clinical trial sites to ensure the proper and timely conduct of our clinical trials and, while we have agreements governing their committed activities, we have limited influence over their actual performance, as described in “Risks Related to Our Dependence on Third Parties”.
Disruptions in the global economy and supply chains may have a material adverse effect on our business, financial condition and results of operations.
The disruptions to the global economy which began in 2020 have impeded global supply chains, resulting in longer lead times and also increased critical component costs and freight expenses. We have encountered disruptions in our supply of various materials. We have taken and may have to take steps to minimize the impact of these disruptions in lead times and increased costs by working closely with our suppliers and other third parties on whom we rely for the conduct of our business. Despite the actions we have undertaken or may have to undertake to minimize the impacts from disruptions to the global economy, there can be no assurances that unforeseen future events in the global supply chain will not have a material adverse effect on our business, financial condition and results of operations.
We rely on, and expect to continue to rely on, third-party providers for the supply of materials and for research. The circumstances relating to the the Russian invasion of Ukraine, the war in the Middle East, as well as other global conditions, have caused significant shortages in the supply chain. We are continuously evaluating alternative and secondary source suppliers in order to ensure that we are able to source sufficient materials. In the event we are unable source sufficient materials from our current suppliers, or to develop relationships with additional suppliers, our business operations could suffer. To the extent our current suppliers, or any suppliers that we engage in the future, are unable to meet our requirements in a timely and cost-effective manner, including as a result of circumstances relating to the the Russian invasion of Ukraine, the war in the Middle East, we may not be able to obtain an adequate supply of materials. Any shortage of materials caused by any disruption or unavailability of supply could harm our business operations, delay the development of our product candidates, or increase our costs and decrease our revenue. Any such impacts or delays could adversely affect our financial condition and our business may be adversely affected. Our efforts to mitigate supply chain weaknesses may not be successful or may have unfavorable effects.
Furthermore, inflation can adversely affect us by increasing the costs of clinical trials, the research and development of our product candidates, as well as administration and other costs of doing business. We may experience increases in the prices of labor and other costs of doing business. In an inflationary environment, cost increases may outpace our expectations, causing us to use our cash and other liquid assets faster than forecasted. If this happens, we may need to raise additional capital to fund our operations, which may not be available in sufficient amounts or on reasonable terms, if at all, sooner than expected.
Adverse global conditions, including economic uncertainty, may negatively impact our financial results.
Global conditions, dislocations in the financial markets, any negative financial impacts affecting United States as a result of tax reform or changes to existing trade agreements or tax conventions, may adversely impact our business.
In addition, the global macroeconomic environment could be negatively affected by, among other things, resurgence of COVID-19 or other pandemics or epidemics, instability in global economic markets, increased U.S. trade tariffs and trade disputes with other countries, instability in the global credit markets, supply chain weaknesses, instability in the geopolitical environment as a result of the withdrawal of the United Kingdom from the European Union, the Russian invasion of Ukraine, andthe war in the Middle East, other political tensions, and foreign governmental debt concerns. Such challenges have caused, and may continue to cause, uncertainty and instability in local economies and in global financial markets.
Our development activities for Probudur are conducted in Israel. The war in the Middle East, may affect our operations.
Lipocure, the company performing all of the development work for Probudur, is located in Israel. If Lipocure were to be unable to continue to perform development work for us or were to be delayed in its performance of development work due to the war in the Middle East, our development timelines will be adversely impacted and we may not be able to develop Probudur within the timeline anticipated, if at all. There can be no assurance that we will be able to find alternative developers at favorable prices.
36
The regulatory approval processes of the FDA and comparable foreign authorities are lengthy, time consuming and inherently unpredictable, and if we are ultimately unable to obtain regulatory approval for our Product Candidates or any other product candidates, our business will be substantially harmed.
The time required to obtain approval by the FDA and comparable foreign authorities is unpredictable but typically takes many years following the commencement of clinical trials and depends upon numerous factors, including the substantial discretion of the regulatory authorities. In addition, approval policies, regulations or the type and amount of clinical data necessary to gain regulatory approval may change during the course of a product candidate’s clinical development and may vary among jurisdictions. We have not obtained regulatory approval for any product candidate and it is possible that we will never obtain regulatory approval for our Product Candidates or any other product candidate. We are not permitted to market any of our product candidates in the United States until we receive regulatory approval of an NDA from the FDA. Our product candidates could fail to receive regulatory approval for many reasons, including the following:
| ● | we may be unable to demonstrate to the satisfaction of the FDA or comparable foreign regulatory authorities that a product candidate is safe and effective for its proposed indication; |
| ● | serious and unexpected drug-related side effects experienced by participants in our clinical trials or by individuals using drugs similar to our product candidates, or other products containing the active ingredient in our product candidates; |
| ● | negative or ambiguous results from our clinical trials or results that may not meet the level of statistical significance required by the FDA or comparable foreign regulatory authorities for approval; |
| ● | we may be unable to demonstrate that a product candidate’s clinical and other benefits outweigh its safety risks; |
| ● | the FDA or comparable foreign regulatory authorities may disagree with our interpretation of data from preclinical studies or clinical trials; |
| ● | the data collected from clinical trials of our product candidates may not be acceptable or sufficient to support the submission of an NDA or other submission or to obtain regulatory approval in the United States or elsewhere, and we may be required to conduct additional clinical trials; |
| ● | the FDA or comparable foreign authorities may disagree regarding the formulation, labeling and/or the specifications of our product candidates; |
| ● | the FDA or comparable foreign regulatory authorities may fail to approve or find deficiencies with the manufacturing processes or facilities of third-party manufacturers with which we contract for clinical and commercial supplies; and |
| ● | the approval policies or regulations of the FDA or comparable foreign regulatory authorities may significantly change in a manner rendering our clinical data insufficient for approval. |
Prior to obtaining approval to commercialize a product candidate in the United States or abroad, we must demonstrate with substantial evidence from well-controlled clinical trials, and to the satisfaction of the FDA or foreign regulatory agencies, that such product candidates are safe and effective for their intended uses. Results from preclinical studies and clinical trials can be interpreted in different ways. Even if we believe the preclinical or clinical data for our product candidates are promising, such data may not be sufficient to support approval by the FDA and other regulatory authorities.
The FDA or any foreign regulatory bodies can delay, limit or deny approval of our product candidates or require us to conduct additional preclinical or clinical testing or abandon a program for many reasons, including:
| ● | the FDA or comparable foreign regulatory authorities may disagree with the design or implementation of our clinical trials; |
| ● | the FDA or comparable foreign regulatory authorities may disagree with our safety interpretation of our product candidate; |
| ● | the FDA or comparable foreign regulatory authorities may disagree with our efficacy interpretation of our product candidate; |
| ● | the FDA or comparable foreign regulatory authorities may regard our CMC package as inadequate. |
Of the large number of drugs in development, only a small percentage successfully complete the regulatory approval processes and are commercialized. This lengthy approval process, as well as the unpredictability of future clinical trial results, may result in our failing to obtain regulatory approval to market our Product Candidates or another product candidate, which would significantly harm our business, results of operations and prospects.
37
In addition, the FDA or the applicable foreign regulatory agency also may approve a product candidate for a more limited indication or patient population than we originally requested, and the FDA or applicable foreign regulatory agency may approve a product candidate with a label that does not include the labeling claims necessary or desirable for the successful commercialization of that product candidate. Any of the foregoing scenarios could materially harm the commercial prospects for our product candidates.
If we are unable to file for approval of Epoladerm Probudur and NobrXiolProbudur under Section 505(b)(2) of the FDCA or if we are required to generate additional data related to safety and efficacy in order to obtain approval under Section 505(b)(2), we may be unable to meet our anticipated development and commercialization timelines.
Our current plans for filing NDAs for Epoladerm Probudur and NobrXiolProbudur include efforts to minimize the data we will be required to generate in order to obtain marketing approval and therefore reduce the development time. We intend to file Section 505(b)(2) NDAs for Epoladerm Probudur and NobrXiolProbudur that might, if accepted by the FDA, save time and expense in the development and testing of these indications.
The timeline for filing and review of our NDAs for EpoladermProbudur and NobrXiolProbudur is based on our plan to submit each of the NDAs under Section 505(b)(2) of the FDCA, which would enable us to rely in part on data in the public domain or elsewhere. We have not yet filed an NDA under Section 505(b)(2) for any of our product candidates. Depending on the data that may be required by the FDA for approval, some of the data may be related to products already approved by the FDA. If the data relied upon is related to products already approved by the FDA and covered by third-party patents, we would be required to certify that we do not infringe the listed patents or that such patents are invalid or unenforceable. As a result of the certification, the third-party would have 45 days from notification of our certification to initiate an action against us. In the event that an action is brought in response to such a certification, the approval of our NDA could be subject to a stay of up to 30 months or more while we defend against such a suit. Approval of our product candidates under Section 505(b)(2) may therefore be delayed until patent exclusivity expires or until we successfully challenge the applicability of those patents to our product candidates. Alternatively, we may elect to generate sufficient additional clinical data so that we no longer rely on data which triggers a potential stay of the approval of our product candidates. Even if no exclusivity periods apply to our applications under Section 505(b)(2), the FDA has broad discretion to require us to generate additional data on the safety and efficacy of our product candidates to supplement third-party data on which we may be permitted to rely. In either event, we could be required, before obtaining marketing approval for any of our product candidates, to conduct substantial new research and development activities beyond those we currently plan to engage in order to obtain approval of our product candidates. Such additional new research and development activities would be costly and time consuming.
We may not be able to realize a shortened development timeline for Epoladerm Probudur and NobrXiol,Probudur, and the FDA may not approve either of our NDAs based on their review of the submitted data. Moreover, if products containing the reference drug are withdrawn from the market by the FDA for any safety reason, we may not be able to reference such products to support a 505(b)(2) NDA for our product candidates, and we may need to fulfill the more extensive requirements of Section 505(b)(1). If we are required to generate additional data to support approval, we may be unable to meet our anticipated development and commercialization timelines, may be unable to generate the additional data at a reasonable cost, or at all, and may be unable to obtain marketing approval of our lead product candidate.
Enrollment and retention of patients in clinical trials is an expensive and time-consuming process and could be made more difficult or rendered impossible by multiple factors outside our control.
The timely completion of clinical trials in accordance with their protocols depends, among other things, on our ability to enroll a sufficient number of patients who remain in the study until its conclusion. We may encounter delays in enrolling, or be unable to enroll, a sufficient number of patients to complete any of our clinical trials, and even once enrolled, we may be unable to retain a sufficient number of patients to complete any of our trials. Patient enrollment and retention in clinical trials depends on many factors, including:
| ● | the patient eligibility criteria defined in the protocol; |
| ● | the size of the patient population required for analysis of the trial’s primary endpoints; |
| ● | the nature of the trial protocol; |
38
| ● | the existing body of safety and efficacy data with respect to the product candidate; |
| ● | the proximity of patients to clinical sites; |
| ● | our ability to recruit clinical trial investigators with the appropriate competencies and experience; |
| ● | clinicians’ and patients’ perceptions as to the potential advantages of the product candidate being studied in relation to other available therapies, including any new drugs that may be approved for the indications we are investigating; |
| ● | competing clinical trials being conducted by other companies or institutions; |
| ● | our ability to maintain patient consents; and |
| ● | the risk that patients enrolled in clinical trials will drop out of the trials before completion. |
We face risks related toThe resurgence of COVID-19 pandemic could adversely affect our business or another health epidemicsepidemic or pandemic may have an adverse impact on our business in the future.
Our business, including our workforce, supply chain and outbreaks, including COVID-19, which could significantly disruptdisruption of our preclinical studies and clinical trials.
The duration and the geographic impact of the business disruption and related financial impact resulting from the COVID-19 pandemic cannot be reasonably estimated at this time and our businesstrials, could be adversely impactedaffected by its effects. In an effort to halt the outbreaka resurgence of COVID-19 or another health epidemic or pandemic may adversely affect us in the United States has, at times, placed significant restrictions on travel and many businesses have announced extended closures which couldfuture. It is possible that a resurgence of COVID-19 or another epidemic or pandemic will adversely impactaffect our operations. Enrollment of patients inbusiness, our clinical trials andworkforce, our planned and ongoingsupply chains, preclinical studies and clinical trials may be delayed dueor otherwise impact our ability to COVID-19. The impact ofconduct business in the COVID-19 pandemic on the operations of the FDA and other health authorities may delay potential approvals of our clinical trial protocols.future. In addition, we rely on independent clinical investigators, contract research organizations and other third-party service providers to assist us in managing, monitoring and otherwise carrying out our preclinical studies and clinical trials, and thea resurgence of COVID-19 or another epidemic pandemic may affect their ability to devote sufficient time and resources to our programs. We also rely on third party suppliers and contract manufacturers to produce the drug product we utilize in our clinical trials, and to the pandemic mayextent their businesses are adversely affected by such occurrences, they might cause delays in the delivery of raw materials and drug product.product or impact our ability to meet development timelines, which could adversely affect our results of operations. Temporary closure of facilities at which our clinical or preclinical trials are conducted, or restrictions on the ability of our employees, clinicians or patients enrolled in our trials to travel could adversely affect our operations and our ability to conduct and complete our preclinical and clinical trials. In addition, the COVID-19 pandemic, including insufficient vaccinationThe effects of the general population and the emergence of new variants, including the delta variant and the omicron variant, could affect theongoing or future health and availability of our workforce as well as those of the third parties on whom we rely. If new, more infectious or severe variants emerge, it is possible that the impact of the pandemicepidemics on our business may increase or lengthen in duration.remain uncertain and subject to change.
As a result of the foregoing factors, the expected timeline for data readouts of our preclinical studies and clinical trials and certain regulatory filings may be negatively impacted, which would adversely affect our business.
Results of preclinical studies, early clinical trials or analyses may not be indicative of results obtained in later trials.
The results of preclinical studies, early clinical trials or analyses of our product candidates may not be predictive of the results of later-stage clinical trials. Product candidates in later stages of clinical trials may fail to show the desired safety and efficacy traits despite having progressed through preclinical studies and initial clinical trials. A number of companies in the biopharmaceutical industry have suffered significant setbacks in advanced clinical trials due to lack of efficacy or adverse safety profiles, notwithstanding promising results in earlier trials. In addition, conclusions based on promising data from analyses of clinical results may be shown to be incorrect when implemented in prospective clinical trials. Even if our clinical trials for our Product Candidates are completed as planned, we cannot be certain that their results will support the safety and efficacy sufficient to obtain regulatory approval.
39
Interim “top-line” and preliminary data from our clinical trials that we announce or publish from time to time may change as more patient data become available and are subject to audit and verification procedures that could result in material changes in the final data.
From time to time, we may publish interim “top-line” or preliminary data from our clinical studies. Interim data from clinical trials that we may complete are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment continues and more patient data become available. Preliminary or “top-line” data also remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary data we previously published. As a result, interim and preliminary data should be viewed with caution until the final data are available. Adverse differences between preliminary or interim data and final data could significantly harm our business prospects.
Our product candidates may cause serious adverse events or undesirable side effects, which may delay or prevent marketing approval, or, if approved, require them to be taken off the market, require them to include safety warnings or otherwise limit their sales.
Serious adverse events or undesirable side effects caused by our Product Candidates or any other product candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA or other comparable foreign authorities. Results of any clinical trial we conduct could reveal a high and unacceptable severity and prevalence of side effects or unexpected characteristics. Any clinical trials for our drug product candidates which include our Product Candidates to date may fail to demonstrate acceptable levels of safety and efficacy which could prevent or significantly delay their regulatory approval or result in a more restrictive label by the FDA or other comparable foreign authorities.
If unacceptable side effects arise in the development of our product candidates, we, the FDA or the IRBs at the institutions in which our studies are conducted, or the DSMB, if constituted for our clinical trials, could recommend a suspension or termination of our clinical trials, or the FDA or comparable foreign regulatory authorities could order us to cease further development of or deny approval of a product candidate for any or all targeted indications. In addition, drug-related side effects could affect patient recruitment or the ability of enrolled patients to complete a trial or result in potential product liability claims. In addition, these side effects may not be appropriately recognized or managed by the treating medical staff. We expect to have to train medical personnel using our product candidates to understand the side effect profiles for our clinical trials and upon any commercialization of any of our product candidates. Inadequate training in recognizing or managing the potential side effects of our product candidates could result in patient injury or death. Any of these occurrences may harm our business, financial condition and prospects significantly.
Additionally, if one or more of our product candidates receives marketing approval, and we or others later identify undesirable side effects caused by such products, a number of potentially significant negative consequences could result, including:
| ● | regulatory authorities may withdraw approvals of such product; |
| ● | regulatory authorities may require additional warnings on the label, such as a “black box” warning or contraindication; |
| ● | additional restrictions may be imposed on the marketing of the particular product or the manufacturing processes for the product or any component thereof; |
| ● | we may be required to implement a Risk Evaluation and Mitigation Strategy (“REMS”) or create a medication guide outlining the risks of such side effects for distribution to patients; |
| ● | we could be sued and held liable for harm caused to patients; |
| ● | the product may become less competitive; and |
| ● | our reputation may suffer. |
Any of these events could prevent us from achieving or maintaining market acceptance of a product candidate, if approved, and could significantly harm our business, results of operations and prospects.
40
The market opportunities for our Product Candidates, if approved, may be smaller than we anticipate.
We expect to initially seek approval for Epoladerm for Osteoarthritis pain, Probudur for postoperative pain management, Envelta for the management for acute and chronicsevere pain including post cancer pain, NobrXiol to potentially treat seizures associated with cancer,Lennox-Gastaut syndrome and Dravet syndrome in pediatric patients two years of age and older in the United States, AnQlar as an antiviral barrier to potentially prevent or reduce the risk or the intensity of viral infections in humans, including, but not limited to, influenza and SARS-CoV-2 (COVID 19), and NobrXiol to potentially treat seizures associated with tuberous sclerosis complex (TSC), Lennox-Gastaut syndrome and Dravet syndrome in patients one year of age and older in the United States.Epoladerm for Osteoarthritis pain. Our estimates of market potential have been derived from a variety of sources, including scientific literature, patient foundations, and market research, and may prove to be incorrect. Even if we obtain significant market share for any product candidate, if approved, if the potential target populations are smaller than we anticipate, we may never achieve profitability without obtaining marketing approval for additional indications. In addition, our strategy for AnQlar and Epoladerm is to license out or partner this asset as we continue to focus our efforts on our prescription drug pipeline, which may result in shared revenue.
We have never obtained marketing approval for a product candidate and we may be unable to obtain, or may be delayed in obtaining, marketing approval for any of our product candidates.
We have never obtained marketing approval for a product candidate. It is possible that the FDA may refuse to accept for substantive review any NDAs that we submit for our product candidates or may conclude after review of our data that our application is insufficient to obtain marketing approval of our product candidates. If the FDA does not accept or approve our NDAs for our product candidates, it may require that we conduct additional clinical, preclinical, or manufacturing validation studies and submit that data before it will reconsider our applications. Depending on the extent of these or any other FDA-required studies, approval of any NDA that we submit may be delayed or may require us to expend more resources than we have available. It is also possible that additional studies, if performed and completed, may not be considered sufficient by the FDA to approve our NDAs.
Any delay in obtaining, or an inability to obtain, marketing approvals would prevent us from commercializing our product candidates, generating revenues, and achieving and sustaining profitability. If any of these outcomes occur, we may be forced to abandon our development efforts for our product candidates, which could significantly harm our business.
Even if we obtain FDA approval for our Product Candidates or any other product candidate in the United States, we may never obtain approval for or commercialize our Product Candidates or any other product candidate in any other jurisdiction, which would limit our ability to realize their full market potential.
In order to market any products in any particular jurisdiction, we must establish and comply with numerous and varying regulatory requirements on a country-by-country basis regarding safety and efficacy. Approval by the FDA in the United States does not ensure approval by regulatory authorities in other countries or jurisdictions. However, the failure to obtain approval in one jurisdiction may negatively impact our ability to obtain approval elsewhere. In addition, clinical trials conducted in one country may not be accepted by regulatory authorities in other countries, and regulatory approval in one country does not guarantee regulatory approval in any other country.
Approval processes vary among countries and can involve additional product testing and validation and additional administrative review periods. Seeking foreign regulatory approval could result in difficulties and increased costs for us and require additional preclinical studies or clinical trials which could be costly and time consuming. Regulatory requirements can vary widely from country to country and could delay or prevent the introduction of our products in those countries. We do not have any product candidates approved for sale in any jurisdiction, including in international markets, and we do not have experience in obtaining regulatory approval in international markets. If we fail to comply with regulatory requirements in international markets or to obtain and maintain required approvals, or if regulatory approvals in international markets are delayed, our target market will be reduced and our ability to realize the full market potential of any product we develop will be unrealized.
41
Even if we obtain regulatory approval for our Product Candidates or any product candidate, we will still face extensive and ongoing regulatory requirements and obligations and any product candidates, if approved, may face future development and regulatory difficulties.
Any product candidate for which we obtain marketing approval, along with the manufacturing processes, post-approval clinical data, labeling, packaging, distribution, adverse event reporting, storage, recordkeeping, export, import, advertising and promotional activities for such product, among other things, will be subject to extensive and ongoing requirements of and review by the FDA and other regulatory authorities. These requirements include submissions of safety and other post-marketing information and reports, establishment registration and drug listing requirements, continued compliance with cGMP requirements relating to manufacturing, quality control, quality assurance and corresponding maintenance of records and documents, requirements regarding the distribution of samples to physicians and recordkeeping and Good Clinical Practice (“GCP”) requirements for any clinical trials that we conduct post-approval.
Even if marketing approval of a product candidate is granted, the approval may be subject to limitations on the indicated uses for which the product candidate may be marketed or to the conditions of approval, including a requirement to implement a REMS. If any of our product candidates receive marketing approval, the accompanying label may limit the approved indicated use of the product candidate, which could limit sales of the product candidate. The FDA may also impose requirements for costly post-marketing studies or clinical trials and surveillance to monitor the safety or efficacy of a product. The FDA closely regulates the post-approval marketing and promotion of drugs to ensure drugs are marketed only for the approved indications and in accordance with the provisions of the approved labeling. The FDA imposes stringent restrictions on manufacturers’ communications regarding off-label use, and if we market our products for uses beyond their approved indications, we may be subject to enforcement action for off-label marketing. Violations of the FDCA relating to the promotion of prescription drugs may lead to FDA enforcement actions and investigations alleging violations of federal and state healthcare fraud and abuse laws, as well as state consumer protection laws.
In addition, later discovery of previously unknown adverse events or other problems with our products, manufacturers or manufacturing processes or failure to comply with regulatory requirements, may yield various results, including:
| ● | restrictions on manufacturing such products; |
| ● | restrictions on the labeling or marketing of products; |
| ● | restrictions on product distribution or use; |
| ● | requirements to conduct post-marketing studies or clinical trials; |
| ● | warning letters or untitled letters; |
| ● | withdrawal of the products from the market; |
| ● | refusal to approve pending applications or supplements to approved applications that we submit; |
| ● | recall of products; |
| ● | fines, restitution or disgorgement of profits or revenues; |
| ● | suspension or withdrawal of marketing approvals; |
| ● | refusal to permit the import or export of our products; |
| ● | product seizure; or |
| ● | injunctions or the imposition of civil or criminal penalties. |
42
Further, the FDA’s policies may change, and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained, which would adversely affect our business, prospects and ability to achieve or sustain profitability.
We also cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative or executive action, either in the United States or abroad. For example, certain policies of the current presidential administration may impact our business and industry. Namely, the current presidential administration has taken several executive actions, including the issuance of a number of Executive Orders, that could impose significant burdens on, or otherwise materially delay, the FDA’s ability to engage in routine regulatory and oversight activities such as implementing statutes through rulemaking, issuance of guidance, and review and approval of marketing applications. It is difficult to predict how these executive actions will be implemented, and the extent to which they will impact the FDA’s ability to exercise its regulatory authority. If these executive actions impose constraints on FDA’s ability to engage in oversight and implementation activities in the normal course, our business may be negatively impacted.
Potential product liability lawsuits against us could cause us to incur substantial liabilities and limit commercialization of any products that we may develop.
The use of our Product Candidates or any other product candidates we may develop in clinical trials and the sale of any products for which we obtain marketing approval exposes us to the risk of product liability claims. Product liability claims might be brought against us by patients, healthcare providers, pharmaceutical companies or others selling or otherwise coming into contact with our products. On occasion, large judgments have been awarded in class action lawsuits based on drugs that had unanticipated adverse effects. If we cannot successfully defend against product liability claims, we could incur substantial liability and costs. In addition, regardless of merit or eventual outcome, product liability claims may result in:
| ● | impairment of our business reputation and significant negative media attention; |
| ● | withdrawal of participants from our clinical trials; |
| ● | significant costs to defend the litigation; |
| ● | distraction of management’s attention from our primary business; |
| ● | substantial monetary awards to patients or other claimants; |
| ● | inability to commercialize our Product Candidates or any other product candidate; |
| ● | product recalls, withdrawals, or labeling, marketing, or promotional restrictions; |
| ● | decreased market demand for any product; and |
| ● | loss of revenue. |
Risks Related to Commercialization
We face significant competition from other biotechnology and pharmaceutical companies and our operating results will suffer if we fail to compete effectively.
The biopharmaceutical and pharmaceutical industries are highly competitive and subject to significant and rapid technological change. Our success is highly dependent on our ability to acquire, develop, and obtain marketing approval for new products on a cost-effective basis and to market them successfully. If any of our Product Candidates are approved, we will face intense competition from a variety of businesses, including large, fully integrated pharmaceutical companies, specialty pharmaceutical companies and biopharmaceutical companies in the United States and other jurisdictions. These organizations may have significantly greater resources than we do and may conduct similar research; seek patent protection; and establish collaborative arrangements for research, development, manufacturing and marketing of products that may compete with us.
43
Our competitors may, among other things:
| ● | have significantly greater name recognition, financial, manufacturing, marketing, drug development, technical, and human resources than we do, and future mergers and acquisitions in the biotechnology and pharmaceutical industries may result in even more resources being concentrated in our competitors; |
| ● | develop and commercialize products that are safer, more effective, less expensive, more convenient, or easier to administer, or have fewer or less severe effects; |
| ● | obtain quicker regulatory approval; |
| ● | implement more effective approaches to sales and marketing; or |
| ● | form more advantageous strategic alliances. |
Smaller and other early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These third parties compete with us in recruiting and retaining qualified scientific and management personnel; establishing clinical trial sites and patient registration; and in acquiring technologies complementary to, or necessary for, our programs. Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are more effective, have fewer or less severe side effects, or are more convenient or are less expensive than our Product Candidates. Our competitors may also obtain FDA or other regulatory approval for their product candidates more rapidly than we may obtain approval for our Product Candidates, which could result in our competitors establishing or strengthening their market position before we are able to enter the market.
The successful commercialization of our Product Candidates and any other product candidate we develop will depend in part on the extent to which governmental authorities and health insurers establish adequate coverage, reimbursement levels, and pricing policies. Failure to obtain or maintain coverage and adequate reimbursement for our product candidates, if approved, could limit our ability to market those products and decrease our ability to generate revenue.
The availability and adequacy of coverage and reimbursement by governmental healthcare programs such as Medicare and Medicaid, private health insurers and other third-party payors are essential for most patients to be able to afford prescription medications such as our Product Candidates, if approved. Our ability to achieve acceptable levels of coverage and reimbursement for products by governmental authorities, private health insurers and other organizations will have an effect on our ability to successfully commercialize our drug and any other product candidates we develop. Assuming we obtain coverage for our product candidates by a third-party payor, the resulting reimbursement payment rates may not be adequate or may require co-payments that patients find unacceptably high. We cannot be sure that coverage and reimbursement in the United States or elsewhere will be available for our product candidates or any product that we may develop, and any reimbursement that may become available may be decreased or eliminated in the future.
Third-party payors increasingly are challenging prices charged for pharmaceutical products and services, and many third-party payors may refuse to provide coverage and reimbursement for particular drugs or biologics when an equivalent generic drug, biosimilar, or a less expensive therapy is available. It is possible that a third-party payor may consider our product candidates as substitutable and offer to reimburse patients only for the less expensive product. Even if we show improved efficacy or improved convenience of administration with our product candidates, pricing of existing drugs may limit the amount we will be able to charge for our product candidates. These payors may deny or revoke the reimbursement status of a given product or establish prices for new or existing marketed products at levels that are too low to enable us to realize an appropriate return on our investment in our product candidates. If reimbursement is not available or is available only at limited levels, we may not be able to successfully commercialize our product candidates and may not be able to obtain a satisfactory financial return on our product candidates.
44
There is significant uncertainty related to the insurance coverage and reimbursement of newly approved products. In the United States, third-party payors, including private and governmental payors, such as the Medicare and Medicaid programs, play an important role in determining the extent to which new drugs and biologics will be covered. The Medicare and Medicaid programs increasingly are used as models in the United States for how private payors and other governmental payors develop their coverage and reimbursement policies for drugs and biologics. Some third-party payors may require pre-approval of coverage for new or innovative devices or drug therapies before they will reimburse healthcare providers who use such therapies. It is difficult to predict at this time what third-party payors will decide with respect to the coverage and reimbursement for our product candidates.
No uniform policy for coverage and reimbursement for products exists among third-party payors in the United States. Therefore, coverage and reimbursement for products can differ significantly from payor to payor. As a result, the coverage determination process is often a time-consuming and costly process that will require us to provide scientific and clinical support for the use of our product candidates to each payor separately, with no assurance that coverage and adequate reimbursement will be applied consistently or obtained in the first instance. Furthermore, rules and regulations regarding reimbursement change frequently, in some cases at short notice, and we believe that changes in these rules and regulations are likely.
We may also be subject to extensive governmental price controls and other market regulations outside of the United States, and we believe the increasing emphasis on cost-containment initiatives in other countries have and will continue to put pressure on the pricing and usage of medical products. In many countries, the prices of medical products are subject to varying price control mechanisms as part of national health systems. Other countries allow companies to fix their own prices for medical products but monitor and control company profits.
Additional foreign price controls or other changes in pricing regulation could restrict the amount that we are able to charge for our product candidates. Accordingly, in markets outside the United States, the reimbursement for our product candidates may be reduced compared with the United States and may be insufficient to generate commercially reasonable revenue and profits.
Moreover, increasing efforts by governmental and third-party payors in the United States to cap or reduce healthcare costs may cause such organizations to limit both coverage and the level of reimbursement for newly approved products and, as a result, they may not cover or provide adequate payment for our product candidates. We expect to experience pricing pressures in connection with the sale of our product candidates due to the trend toward managed health care, the increasing influence of health maintenance organizations and additional legislative changes. The downward pressure on healthcare costs in general, particularly prescription drugs and biologics and surgical procedures and other treatments, has become intense. As a result, increasingly high barriers are being erected to the entry of new products.
Even if our Product Candidates or any product candidate we develop receives marketing approval, it may fail to achieve market acceptance by physicians, patients, third-party payors or others in the medical community necessary for commercial success.
If our Product Candidates or any product candidate we develop receives marketing approval, it may nonetheless fail to gain sufficient market acceptance by physicians, patients, third-party payors, and others in the medical community. If it does not achieve an adequate level of acceptance, we may not generate significant product revenues or become profitable. The degree of market acceptance of our product candidates, if approved, will depend on a number of factors, including but not limited to:
| ● | the efficacy and potential advantages compared to alternative treatments; |
| ● | effectiveness of sales and marketing efforts; |
| ● | the cost of treatment in relation to alternative treatments, including any similar generic treatments; |
| ● | our ability to offer our products for sale at competitive prices; |
| ● | the convenience and ease of administration compared to alternative treatments; |
45
| ● | the willingness of the target patient population to try new therapies and of physicians to prescribe these therapies; |
| ● | the strength of marketing and distribution support; |
| ● | the availability of third-party coverage and adequate reimbursement; |
| ● | the prevalence and severity of any side effects; and |
| ● | any restrictions on the use of our product together with other medications. |
Because we expect sales of our product candidates, if approved, to generate substantially all of our revenues for the foreseeable future, the failure of our product candidates to find market acceptance would harm our business and could require us to seek additional financing.
If we are unable to establish sales, marketing, and distribution capabilities either on our own or in collaboration with third parties, we may not be successful in commercializing our Product Candidates, if approved.
We do not have any infrastructure for the sales, marketing, or distribution of our Product Candidates, and the cost of establishing and maintaining such an organization may exceed the cost-effectiveness of doing so. In order to market and successfully commercialize our drug or any product candidate we develop, if approved, we must build our sales, distribution, marketing, managerial and other non-technical capabilities or make arrangements with third parties to perform these services. We expect to build a focused sales distribution and marketing infrastructure to market our Product Candidates, if approved, in the United States and Europe. There are significant expenses and risks involved with establishing our own sales, marketing and distribution capabilities, including our ability to hire, retain and appropriately incentivize qualified individuals, generate sufficient sales leads, provide adequate training to sales and marketing personnel and effectively manage a geographically dispersed sales and marketing team. Any failure or delay in the development of our internal sales, marketing and distribution capabilities could delay any product launch, which would adversely impact the commercialization of that product. For example, if the commercial launch of our Product Candidates for which we recruit a sales force and establish marketing capabilities is delayed or does not occur for any reason, we would have prematurely or unnecessarily incurred these commercialization expenses. This may be costly, and our investment would be lost if we cannot retain or reposition our sales and marketing personnel.
Factors that may inhibit our efforts to commercialize our product candidates on our own include:
| ● | our inability to recruit, train and retain adequate numbers of effective sales and marketing personnel; |
| ● | the inability of sales personnel to obtain access to physicians or attain adequate numbers of physicians to prescribe our products; and |
| ● | unforeseen costs and expenses associated with creating an independent sales and marketing organization. |
We do not anticipate having the resources in the foreseeable future to allocate to the sales and marketing of our product candidates, if approved, in certain markets overseas. Therefore, our future success will depend, in part, on our ability to enter into and maintain collaborative relationships for such capabilities, the collaborator’s strategic interest in a product and such collaborator’s ability to successfully market and sell the product. We intend to pursue collaborative arrangements regarding the sale and marketing of our Product Candidates, if approved, for certain markets overseas; however, we cannot assure you that we will be able to establish or maintain such collaborative arrangements, or if able to do so, that they will have effective sales forces. To the extent that we depend on third parties for marketing and distribution, any revenues we receive will depend upon the efforts of such third parties, and there can be no assurance that such efforts will be successful.
46
If we are unable to build our own sales force or negotiate a collaborative relationship for the commercialization of our Product Candidates, we may be forced to delay the potential commercialization of the drug or reduce the scope of our sales or marketing activities. If we need to increase our expenditures to fund commercialization activities for our Product Candidates we will need to obtain additional capital, which may not be available to us on acceptable terms, or at all. We may also have to enter into collaborative arrangements for our Product Candidates at an earlier stage than otherwise would be ideal and we may be required to relinquish rights to it or otherwise agree to terms unfavorable to us. Any of these occurrences may have an adverse effect on our business, operating results, and prospects.
If we are unable to establish adequate sales, marketing, and distribution capabilities, either on our own or in collaboration with third parties, we will not be successful in commercializing our product candidates and may never become profitable. We will be competing with many companies that currently have extensive and well-funded marketing and sales operations. Without an internal team or the support of a third party to perform marketing and sales functions, we may be unable to compete successfully against these more established companies.
A variety of risks associated with operating internationally could materially adversely affect our business.
We currently have no international operations, aside from the development of our product candidates by companies that operate internationally, but our business strategy includes potentially expanding internationally if any of our product candidates receive regulatory approval. Doing business internationally involves a number of risks, including but not limited to:
| ● | multiple, conflicting and changing laws and regulations, such as privacy regulations, tax laws, export and import restrictions, employment laws, regulatory requirements and other governmental approvals, permits and licenses; |
| ● | failure by us to obtain and maintain regulatory approvals for the use of our products in various countries; |
| ● | additional potentially relevant third-party patent rights; |
| ● | complexities and difficulties in obtaining protection and enforcing our intellectual property; |
| ● | difficulties in staffing and managing foreign operations; |
| ● | complexities associated with managing multiple payor reimbursement regimes, government payors or patient self-pay systems; |
| ● | limits in our ability to penetrate international markets; |
| ● | financial risks, such as longer payment cycles, difficulty collecting accounts receivable, the impact of local and regional financial crises on demand and payment for our products and exposure to foreign currency exchange rate fluctuations; |
| ● | natural disasters, political and economic instability, including wars, terrorism and political unrest, outbreak of disease, boycotts, curtailment of trade and other business restrictions; |
| ● | certain expenses including, among others, expenses for travel, translation and insurance; and |
| ● | regulatory and compliance risks that relate to maintaining accurate information and control over sales and activities that may fall within the purview of the U.S. Foreign Corrupt Practices Act, its books and records provisions, or its anti-bribery provisions. |
Any of these factors could significantly harm any future international expansion and operations and, consequently, our results of operations.
47
Risks Related to Our Dependence on Third Parties
Our employees and independent contractors, including principal investigators, CROs, consultants, vendors, and any third parties we may engage in connection with development and commercialization, may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements, which could have a material adverse effect on our business.
Our employees and independent contractors, including principal investigators, consultants, vendors and any third parties we may engage in connection with development and commercialization of our product candidates, could engage in misconduct, including intentional, reckless or negligent conduct or unauthorized activities that violate: the laws and regulations of the FDA or other similar regulatory requirements of other authorities, including those laws that require the reporting of true, complete and accurate information to such authorities; manufacturing standards; data privacy, security, fraud and abuse and other healthcare laws and regulations; or laws that require the reporting of true, complete and accurate financial information and data. Specifically, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Activities subject to these laws could also involve the improper use or misrepresentation of information obtained in the course of clinical trials, creation of fraudulent data in preclinical studies or clinical trials or illegal misappropriation of drug product, which could result in regulatory sanctions and cause serious harm to our reputation. It is not always possible to identify and deter misconduct by employees and other third parties, and the precautions weWe take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with such laws or regulations. Additionally, we are subject to the risk that a person or government could allege such fraud or other misconduct, even if none occurred. If any such actions are instituted against us and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business and results of operations, including the imposition of significant civil, criminal and administrative penalties, damages, monetary fines, disgorgements, possible exclusion from participation in Medicare, Medicaid, other U.S. federal healthcare programs or healthcare programs in other jurisdictions, individual imprisonment, other sanctions, contractual damages, reputational harm, diminished profits and future earnings, and curtailment of our operations.
We currently rely on third-party contract manufacturing organizations (“CMOs”) for the production of clinical supply of our Product Candidates and intend to rely on CMOs for the production of commercial supply of our Product Candidates, if approved. Our dependence on CMOs may impair the development and commercialization of the drug, which would adversely impact our business and financial position.
We have limited personnel with experience in manufacturing, and we do not own facilities for manufacturing. Instead, we rely on and expect to continue to rely on CMOs for the supply of cGMP grade clinical trial materials and commercial quantities of our Product Candidates and any product candidates we develop, if approved. Reliance on CMOs may expose us to more risk than if we were to manufacture our product candidates ourselves. We intend to have manufactured a sufficient clinical supply of our Product Candidates drug substance to enable us to complete our clinical trials, and we have engaged or intend to engage a CMO to provide clinical and commercial supplies of the drug products.
The facilities used to manufacture our product candidates must be inspected by the FDA and comparable foreign authorities. While we provide oversight of manufacturing activities, we do not and will not control the execution of manufacturing activities by, and are or will be essentially dependent on, our CMOs for compliance with cGMP requirements for the manufacture of our product candidates. As a result, we are subject to the risk that our product candidates may have manufacturing defects that we have limited ability to prevent. If a CMO cannot successfully manufacture material that conforms to our specifications and the regulatory requirements, we will not be able to secure or maintain regulatory approval for the use of our product candidates in clinical trials, or for commercial distribution of our product candidates, if approved. In addition, we have limited control over the ability of our CMOs to maintain adequate quality control, quality assurance and qualified personnel. If the FDA or comparable foreign regulatory authority finds deficiencies with or does not approve these facilities for the manufacture of our product candidates or if it withdraws any such approval or finds deficiencies in the future, we may need to find alternative manufacturing facilities, which would delay our development program and significantly impact our ability to develop, obtain regulatory approval for or commercialize our product candidates, if approved. In addition, any failure to achieve and maintain compliance with these laws, regulations and standards could subject us to the risk that we may have to suspend the manufacture of our product candidates or that obtained approvals could be revoked. Furthermore, CMOs may breach existing agreements they have with us because of factors beyond our control. They may also terminate or refuse to renew their agreement at a time that is costly or otherwise inconvenient for us. If we were unable to find an adequate CMO or another acceptable solution in time, our clinical trials could be delayed, or our commercial activities could be harmed.
48
We rely on and will continue to rely on CMOs to purchase from third-party suppliers the raw materials necessary to produce our product candidates. We do not and will not have control over the process or timing of the acquisition of these raw materials by our CMOs. Moreover, we currently only have one agreement for the production of these raw materials (for AnQlar). Supplies of raw material could be interrupted from time to time and we cannot be certain that alternative supplies could be obtained within a reasonable timeframe, at an acceptable cost, or at all. In addition, a disruption in the supply of raw materials could delay the commercial launch of our product candidates, if approved, or result in a shortage in supply, which would impair our ability to generate revenues from the sale of our product candidates. Growth in the costs and expenses of raw materials may also impair our ability to cost effectively manufacture our product candidates. There are a limited number of suppliers for the raw materials that we may use to manufacture our product candidates and we may need to assess alternative suppliers to prevent a possible disruption of the manufacture of our product candidates.
Finding new CMOs or third-party suppliers involves additional cost and requires our management’s time and focus. In addition, there is typically a transition period when a new CMO commences work. Although we generally have not, and do not intend to, begin a clinical trial unless we believe we have on hand, or will be able to obtain, a sufficient supply of our product candidates to complete the clinical trial, any significant delay in the supply of our product candidates or the raw materials needed to produce our product candidates, could considerably delay conducting our clinical trials and potential regulatory approval of our product candidates.
As part of their manufacture of our product candidates, our CMOs and third-party suppliers are expected to comply with and respect the proprietary rights of others. If a CMO or third-party supplier fails to acquire the proper licenses or otherwise infringes the proprietary rights of others in the course of providing services to us, we may have to find alternative CMOs or third-party suppliers or defend against claims of infringement, either of which would significantly impact our ability to develop, obtain regulatory approval for or commercialize our product candidates, if approved.
We intend to rely on third parties to conduct, supervise and monitor our clinical trials. If those third parties do not successfully carry out their contractual duties, or if they perform in an unsatisfactory manner, it may harm our business.
We rely, and will continue to rely, on CROs, CRO-contracted vendors and clinical trial sites to ensure the proper and timely conduct of our clinical trials. Our reliance on CROs for clinical development activities limits our control over these activities, but we remain responsible for ensuring that each of our trials is conducted in accordance with the applicable protocol and legal, regulatory and scientific standards.
We and our CROs will be required to comply with the Good Laboratory Practice (“GLP”) requirements for our preclinical studies and GCP requirements for our clinical trials, which are regulations and guidelines enforced by the FDA and are also required by comparable foreign regulatory authorities. Regulatory authorities enforce GCP requirements through periodic inspections of trial sponsors, principal investigators and clinical trial sites. If we or our CROs fail to comply with GCP requirements, the clinical data generated in our clinical trials may be deemed unreliable and the FDA or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. We cannot assure you that upon inspection by a given regulatory authority, such regulatory authority will determine that any of our clinical trials comply with GCP requirements. In addition, our clinical trials must be conducted with product produced under cGMP requirements. Accordingly, if our CROs fail to comply with these requirements, we may be required to repeat clinical trials, which would delay the regulatory approval process.
Our CROs are not our employees, and we do not control whether or not they devote sufficient time and resources to our clinical trials. Our CROs may also have relationships with other commercial entities, including our competitors, for whom they may also be conducting clinical trials, or other drug development activities, which could harm our competitive position. We face the risk of potential unauthorized disclosure or misappropriation of our intellectual property by CROs, which may reduce our trade secret protection and allow our potential competitors to access and exploit our proprietary technology. If our CROs do not successfully carry out their contractual duties or obligations, fail to meet expected deadlines, or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols or regulatory requirements or for any other reason, our clinical trials may be extended, delayed or terminated, and we may not be able to obtain regulatory approval for, or successfully commercialize any product candidate that we develop. As a result, our financial results and the commercial prospects for any product candidate that we develop would be harmed, our costs could increase, and our ability to generate revenue could be delayed.
49
If our relationship with any CROs terminate, we may not be able to enter into arrangements with alternative CROs or do so on commercially reasonable terms. Switching or adding additional CROs involves substantial cost and requires management’s time and focus. In addition, there is a natural transition period when a new CRO commences work. As a result, delays occur, which can materially impact our ability to meet our desired clinical development timelines. Though we intend to carefully manage our relationships with our CROs, there can be no assurance that we will not encounter challenges or delays in the future or that these delays or challenges will not have an adverse impact on our business, financial condition and prospects.
| ● | the number and type of our collaborations could adversely affect our attractiveness to future collaborators or acquirers; and |
| ● | the loss of, or a disruption in our relationship with, any one or more collaborators could harm our business. |
If any collaborations do not result in the successful development and commercialization of products or if one of our collaborators terminates its agreement with us, we may not receive any future research and development funding or milestone or royalty payments under such collaborations. If we do not receive the funding we expect under these agreements, our continued development of our product candidates could be delayed, and we may need additional resources to develop additional product candidates. All of the risks relating to product development, regulatory approval and commercialization described in this Annual Report on Form 10-K also apply to the activities of any collaborators and there can be no assurance that our collaborations will produce positive results or successful products on a timely basis or at all.
In addition, subject to its contractual obligations to us, if one of our collaborators is involved in a business combination or otherwise changes its business priorities, the collaborator might deemphasize or terminate the development or commercialization of our product candidates. If a collaborator terminates its agreement with us, we may find it more difficult to attract new collaborators and the perception of our business and our stock price could be adversely affected.
We may in the future collaborate with additional pharmaceutical and biotechnology companies for development and potential commercialization of therapeutic products. We face significant competition in seeking appropriate collaborators. Our ability to reach a definitive agreement for a collaboration will depend, among other things, upon our assessment of the collaborator’s resources and expertise, the terms and conditions of the proposed collaboration and the proposed collaborator’s evaluation of a number of factors. If we are unable to reach agreements with suitable collaborators on a timely basis, on acceptable terms, or at all, we may have to curtail the development of a product candidate, reduce or delay its development program or one or more of our other development programs, delay its potential commercialization or reduce the scope of any sales or marketing activities, or increase our expenditures and undertake development or commercialization activities at our own expense. If we elect to fund and undertake development or commercialization activities on our own, we may need to obtain additional expertise and additional capital, which may not be available to us on acceptable terms or at all. If we fail to enter into collaborations and do not have sufficient funds or expertise to undertake the necessary development and commercialization activities, we may not be able to further develop our product candidates or bring them to market or continue to develop our programs, and our business may be materially and adversely affected.
Risks Related to Healthcare Laws and Other Legal Compliance Matters
Enacted and future healthcare legislation may increase the difficulty and cost for us to obtain marketing approval of and commercialize our product candidates, if approved, and may affect the prices we may set.
In the United States and other jurisdictions, there have been, and we expect there will continue to be, a number of legislative and regulatory changes and proposed changes to the healthcare system that could affect our future results of operations. In particular, there have been and continue to be a number of initiatives at the U.S. federal and state levels that seek to reduce healthcare costs and improve the quality of healthcare. For example, in March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act (collectively the “ACA”) was enacted, which substantially changed the way healthcare is financed by both governmental and private insurers. Among the provisions of the ACA, those of greatest importance to the pharmaceutical and biotechnology industries include the following:
| ● | an annual, non-deductible fee payable by any entity that manufactures or imports certain branded prescription drugs and biologic agents (other than those designated as orphan drugs), which is apportioned among these entities according to their market share in certain government healthcare programs; |
50
| ● | a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D; |
| ● | new requirements to report certain financial arrangements with physicians and teaching hospitals, including reporting “transfers of value” made or distributed to prescribers and other healthcare providers and reporting investment interests held by physicians and their immediate family members; |
| ● | an increase in the statutory minimum rebates a manufacturer must pay under the Medicaid Drug Rebate Program; |
| ● | a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs and biologics that are inhaled, infused, instilled, implanted, or injected; |
| ● | extension of a manufacturer’s Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations; | |
| ● | expansion of eligibility criteria for Medicaid programs thereby potentially increasing a manufacturer’s Medicaid rebate liability; |
| ● | a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research; |
| ● | establishment of a Center for Medicare Innovation at the Centers for Medicare & Medicaid Services, or CMS, to test innovative payment and service delivery models to lower Medicare and Medicaid spending, potentially including prescription drug spending; |
| ● | expansion of the entities eligible for discounts under the Public Health Service program; and |
| ● | a licensure framework for follow on biologic products. |
Other legislative changes have been proposed and adopted in the United States since the ACA was enacted. For example, the Budget Control Act of 2011, resulted in aggregate reductions of Medicare payments to providers of 2% per fiscal year. These reductions went into effect in April 2013 and, due to subsequent legislative amendments to the statute, will remain in effect through 2027 unless additional action is taken by Congress. In January 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, further reduced Medicare payments to several types of providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. Additionally, the orphan drug tax credit was reduced as part of a broader tax reform. These new laws or any other similar laws introduced in the future may result in additional reductions in Medicare and other health care funding, which could negatively affect our customers and accordingly, our financial operations.
51
Additionally, there has been heightened governmental scrutiny in the United States of pharmaceutical pricing practices in light of the rising cost of prescription drugs and biologics. On September 9, 2021, the Biden Administration published a wide-ranging list of policy proposals, most of which would need to be carried out by Congress, to reduce drug prices and drug payment. The United States Department of Health and Human Services (“HHS”) plan includes, among other reform measures, proposals to lower prescription drug prices, including by allowing Medicare to negotiate prices and disincentivizing price increases, and to support market changes that strengthen supply chains, promote biosimilars and generic drugs, and increase price transparency. These initiatives recently culminated in the enactment of the Inflation Reduction Act (the “IRA”) in August 2022, which will, among other things, allow the HHS to negotiate the selling price of certain drugs and biologics that the Centers for Medicare & Medicaid Services (“CMS”) reimburses under Medicare Part B and Part D, although only high-expenditure single-source drugs that have been approved for at least 7 years (11 years for biologics) can be selected by CMS for negotiation, with the negotiated price taking effect two years after the selection year. The negotiated prices, which will first become effective in 2026, will be capped at a statutory ceiling price beginning in October 2023, penalize drug manufacturers that increase prices of Medicare Part B and Part D drugs at a rate greater than the rate of inflation. The IRA permits the Secretary of HHS to implement many of these provisions through guidance, as opposed to regulation, for the initial years. Manufacturers that fail to comply with the IRA may be subject to various penalties, including civil monetary penalties. The IRA also extends enhanced subsidies for individuals purchasing health insurance coverage in ACA marketplaces through plan year 2025. These provisions will take effect progressively starting in 2023, although they may be subject to legal challenges.
Individual states in the United States have also become increasingly aggressive in passing legislation and implementing regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. Legally mandated price controls on payment amounts by third-party payors or other restrictions could harm our business, results of operations, financial condition and prospects. In addition, regional healthcare authorities and individual hospitals are increasingly using bidding procedures to determine what pharmaceutical products and which suppliers will be included in their prescription drug and other healthcare programs. This could reduce the ultimate demand for our product candidates or put pressure on our product pricing.
In markets outside of the United States, reimbursement and healthcare payment systems vary significantly by country, and many countries have instituted price ceilings on specific products and therapies. We cannot predict the likelihood, nature, or extent of government regulation that may arise from future legislation or administrative action in the United States or any other jurisdiction. If we or any third parties we may engage are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we or such third parties are not able to maintain regulatory compliance, our product candidates may lose any regulatory approval that may have been obtained and we may not achieve or sustain profitability.
Our business operations and current and future relationships with investigators, healthcare professionals, consultants, third-party payors, patient organizations, and customers will be subject to applicable healthcare regulatory laws, which could expose us to penalties.
Our business operations and current and future arrangements with investigators, healthcare professionals, consultants, third-party payors, patient organizations, and customers, may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations. These laws may constrain the business or financial arrangements and relationships through which we conduct our operations, including how we research, market, sell and distribute our product candidates, if approved. Such laws include:
| ● | the U.S. federal Anti-Kickback Statute, which prohibits, among other things, persons or entities from knowingly and willfully soliciting, offering, receiving, or providing any remuneration (including any kickback, bribe, or certain rebate), directly or indirectly, overtly or covertly, in cash or in kind, to induce or reward, or in return for, either the referral of an individual for, or the purchase, lease, order, or recommendation of, any good, facility, item, or service, for which payment may be made, in whole or in part, under U.S. federal and state healthcare programs such as Medicare and Medicaid. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. The U.S. federal Anti-Kickback Statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on the one hand and prescribers, purchasers and formulary managers on the other hand; |
52
| ● | the U.S. federal false claims and civil monetary penalties laws, including the civil False Claims Act (the “FCA”) which, among other things, impose criminal and civil penalties, including through civil whistleblower or qui tam actions, against individuals or entities for knowingly presenting, or causing to be presented, to the U.S. federal government, claims for payment or approval that are false or fraudulent, knowingly making, using or causing to be made or used, a false record or statement material to a false or fraudulent claim, or from knowingly making a false statement to avoid, decrease or conceal an obligation to pay money to the U.S. federal government. In addition, the government may assert that a claim including items and services resulting from a violation of the U.S. federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the FCA. A claim includes “any request or demand” for money or property presented to the federal government. In addition, manufacturers can be held liable under the FCA even when they do not submit claims directly to government payors if they are deemed to “cause” the submission of false or fraudulent claims; |
| ● | the U.S. federal Health Insurance Portability and Accountability Act of 1996 (“HIPAA”), which imposes criminal and civil liability for, among other things, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program or obtain, by means of false or fraudulent pretenses, representations, or promises, any of the money or property owned by, or under the custody or control of, any healthcare benefit program, regardless of the payor (e.g., public or private) and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statement, in connection with the delivery of, or payment for, healthcare benefits, items or services. Similar to the U.S. federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation; |
| ● | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009 (“HITECH”), and their respective implementing regulations, which impose, among other things, specified requirements relating to the privacy, security and transmission of individually identifiable health information without appropriate authorization by covered entities subject to the rule, such as health plans, healthcare clearinghouses and healthcare providers as well as their business associates that perform certain services involving the use or disclosure of individually identifiable health information. HITECH also created new tiers of civil monetary penalties, amended HIPAA to make civil and criminal penalties directly applicable to business associates and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorneys’ fees and costs associated with pursuing federal civil actions; |
| ● | the FDCA, which prohibits, among other things, the adulteration or misbranding of drugs, biologics and medical devices; |
| ● | the U.S. federal legislation commonly referred to as the Physician Payments Sunshine Act, enacted as part of the ACA, and its implementing regulations, which requires certain manufacturers of drugs, devices, biologics, and medical supplies that are reimbursable under Medicare, Medicaid, or the Children’s Health Insurance Program to report annually to the government information related to certain payments and other transfers of value to physicians and teaching hospitals, as well as ownership and investment interests held by the physicians described above and their immediate family members; and |
| ● | analogous U.S. state laws and regulations, including: state anti-kickback and false claims laws, which may apply to our business practices, including but not limited to, research, distribution, sales, and marketing arrangements and claims involving healthcare items or services reimbursed by any third-party payor, including private insurers; state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the U.S. federal government, or otherwise restrict payments that may be made to healthcare providers and other potential referral sources; state laws and regulations that require drug manufacturers to file reports relating to pricing and marketing information, which requires tracking gifts and other remuneration and items of value provided to healthcare professionals and entities; and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts. |
53
Because of the breadth of these laws and the narrowness of the statutory exceptions and regulatory safe harbors available under such laws, it is possible that some of our business activities, including our consulting agreements and other relationships with physicians and other healthcare providers, some of whom receive stock or stock options as compensation for their services, could be subject to challenge under one or more of such laws. Ensuring that our current and future internal operations and business arrangements with third parties comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices do not comply with current or future statutes, regulations, agency guidance or case law involving applicable fraud and abuse or other healthcare laws and regulations.
If our operations are found to be in violation of any of the laws described above or any other governmental laws and regulations that may apply to us, we may be subject to the imposition of civil, criminal and administrative penalties, damages, disgorgement, monetary fines, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, individual imprisonment, contractual damages, reputational harm, diminished profits and future earnings, additional reporting requirements or oversight if we become subject to a corporate integrity agreement or similar agreement to resolve allegations of non-compliance with these laws, and curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations. If any of the physicians or other providers or entities with whom we expect to do business are found to not be in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs and imprisonment, which could affect our ability to operate our business. Further, defending against any such actions can be costly, time-consuming and may require significant personnel resources. Therefore, even if we are successful in defending against any such actions that may be brought against us, our business may be impaired.
Any clinical trial programs we conduct or research collaborations we enter into in the European Economic Area may subject us to the General Data Protection Regulation.
If we conduct clinical trial programs or enter into research collaborations in the European Economic Area, we may be subject to the General Data Protection regulation (“GDPR”). The GDPR applies extraterritorially and implements stringent operational requirements for processors and controllers of personal data, including, for example, high standards for obtaining consent from individuals to process their personal data, robust disclosures to individuals, a comprehensive individual data rights regime, data export restrictions governing transfers of data from the European Union (the “EU”) to other jurisdictions, short timelines for data breach notifications, limitations on retention of information, increased requirements pertaining to health data, other special categories of personal data and coded data and additional obligations if we contract third-party processors in connection with the processing of personal data. The GDPR provides that EU member states may establish their own laws and regulations limiting the processing of personal data, including genetic, biometric or health data, which could limit our ability to use and share personal data or could cause our costs to increase. If our or our partners’ or service providers’ privacy or data security measures fail to comply with the GDPR requirements, we may be subject to litigation, regulatory investigations, enforcement notices requiring us to change the way we use personal data and/or fines of up to 20 million Euros or up to 4% of the total worldwide annual turnover of the preceding financial year, whichever is higher, as well as compensation claims by affected individuals, negative publicity, reputational harm and a potential loss of business and goodwill.
We are subject to environmental, health and safety laws and regulations, and we may become exposed to liability and substantial expenses in connection with environmental compliance or remediation activities.
Our operations, including our development, testing and manufacturing activities, are subject to numerous environmental, health and safety laws and regulations. These laws and regulations govern, among other things, the controlled use, handling, release and disposal of and the maintenance of a registry for, hazardous materials and biological materials, such as chemical solvents, human cells, carcinogenic compounds, mutagenic compounds and compounds that have a toxic effect on reproduction, laboratory procedures and exposure to blood-borne pathogens. If we fail to comply with such laws and regulations, we could be subject to fines or other sanctions.
As with other companies engaged in activities similar to ours, we face a risk of environmental liability inherent in our current and historical activities, including liability relating to releases of or exposure to hazardous or biological materials. Environmental, health and safety laws and regulations are becoming more stringent. We may be required to incur substantial expenses in connection with future environmental compliance or remediation activities, in which case, the production efforts of our third-party manufacturers or our development efforts may be interrupted or delayed.
54
Risks Related to Our Intellectual Property
If we fail to comply with our obligations under our existing intellectual property license, we risk losing the rights to our intellectual property.
Each of the material license agreements in which we have engaged, including the license agreements with MedPharm Limited, LipoCureRx Ltd. andAnd Nanomerics Ltd. has provisions by which each of those companies could terminate the license agreements thereby terminating our access to the intellectual property licensed under those agreements. Termination of these license agreements would prevent the commercialization of the products we are developing.
If we are unable to obtain and maintain patent protection for our technology, products, and product candidates or if the scope of the patent protection obtained is not sufficiently broad, we may not be able to compete effectively in our markets.
We rely upon a combination of patents, trade secret protection and confidentiality agreements to protect the intellectual property related to our drug development programs and product candidates. Our success depends in large part on our ability to obtain and maintain patent protection in the United States and other countries with respect to our Product Candidates and any future products and product candidates. We seek to protect our proprietary position by filing patent applications in the United States and abroad related to our development programs, and product candidates. The patent prosecution process is expensive and time-consuming, and we may not be able to file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner.
If the patent applications we own or license with respect to our development programs and product candidates fail to issue, if their breadth or strength of protection is threatened, or if they fail to provide meaningful exclusivity for our Product Candidates or any future product candidates, it could dissuade companies from collaborating with us to develop product candidates and threaten our ability to commercialize future product candidates. Any such outcome could have a materially adverse effect on our business.
The patent position of biotechnology and pharmaceutical companies generally is highly uncertain, involves complex legal and factual questions and has in recent years been the subject of much litigation. In addition, the laws of foreign countries may not protect our patent rights to the same extent as the laws of the United States. For example, European patent law restricts the patentability of methods of treatment of the human body more than U.S. law does. However, in certain instances, the laws of the United States are more restrictive than those of foreign countries. For example, a recent series of Supreme Court Cases has narrowed the types of subject matter considered eligible for patenting.
Accordingly, certain diagnostic methods are considered ineligible for patenting in the U.S. because they are directed to a “law of nature”. Further, publications of discoveries in scientific literature often lag behind the actual discoveries, and patent applications in the United States and other jurisdictions are typically not published until 18 months after filing, or in some cases not at all. Therefore, we cannot know with certainty whether we were the first to make the inventions claimed in our owned or licensed patents or pending patent applications, or that we were the first to file for patent protection of such inventions. As a result, the issuance, scope, validity, enforceability and commercial value of our patent rights are highly uncertain. Our pending and future patent applications may not result in patents being issued which protect our technology, products, or product candidates, in whole or in part, or patents being issued which effectively prevent others from commercializing competitive technologies and products. Changes in either the patent laws or interpretation of the patent laws in the United States and other countries may diminish the value of our patents or narrow the scope of our patent protection.
The issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability, and our owned and licensed patents may be challenged in the courts or patent offices in the United States and abroad. Such challenges may result in patent claims being narrowed, invalidated, held unenforceable, in whole or in part, or reduced patent term. Such a result could limit our ability to stop others from using or commercializing similar or identical technologies and products to ours. Moreover, patents have a limited lifespan. In the United States, the natural expiration of a patent is generally 20 years after it is filed.from the earliest effective non-provisional filing date. While various extensions may be available, the life of a patent is limited. Without patent protection for our current or future products, we may be open to competition from generic versions of such products. Given the amount of time required for the development, testing and regulatory review of new products, patents protecting such candidates might expire before or shortly after such candidates are commercialized. As a result, our owned and licensed patent portfolio may not provide us with sufficient rights to exclude others from using or commercializing technologies or products similar or identical to ours.
55
We may become subject to third parties’ claims alleging infringement of their patents and proprietary rights, or we may need to become involved in lawsuits to protect or enforce our patents, which could be costly, time consuming, delay or prevent the development and commercialization of our products and product candidates or put our patents and other proprietary rights at risk.
Our commercial success depends, in part, upon our ability to develop, manufacture, market and sell our products and product candidates without alleged or actual infringement, misappropriation or other violation of the patents and proprietary rights of third parties. Litigation relating to infringement or misappropriation of patent and other intellectual property rights in the pharmaceutical and biotechnology industries is common, including patent infringement lawsuits, interferences, oppositions, reexamination, derivation and post-grant proceedings before the U.S. Patent and Trademark Office (“USPTO”), and corresponding foreign patent offices.
The various markets in which we plan to operate are subject to frequent and extensive litigation regarding patents and other intellectual property rights. In addition, many companies in intellectual property-dependent industries, including the biotechnology and pharmaceutical industries, have employed intellectual property litigation as a means to gain an advantage over their competitors. Numerous U.S., European and other foreign issued patents and pending patent applications, which are owned by third parties, exist in the fields in which we are developing products and product candidates. Some claimants may have substantially greater resources than we do and may be able to sustain the costs of complex intellectual property litigation to a greater degree and for longer periods of time than we could. In addition, patent holding companies that focus solely on extracting royalties and settlements by enforcing patent rights may target us. As the biotechnology and pharmaceutical industries expand and more patents are issued, the risk increases that our products and product candidates may be subject to claims of infringement of the intellectual property rights of third parties.
We may be subject to third-party claims including infringement, interference or derivation proceedings, post-grant review and inter partes review before the USPTO or similar adversarial proceedings or litigation in other jurisdictions. Even if we believe such claims are without merit, a court of competent jurisdiction could hold that these third-party patents are valid, enforceable and infringed, and the holders of any such patents may be able to block our ability to commercialize the applicable product candidate unless we obtained a license under the applicable patents, or until such patents expire or are finally determined to be invalid or unenforceable. These proceedings may also result in our patent claims being invalidated, held unenforceable or narrowed in scope. Similarly, if ours or our licensors’ patents or patent applications are challenged during interference or derivation proceedings, a court may hold that a third-party is entitled to certain patent ownership rights instead of us. Further, if any third-party patents were held by a court of competent jurisdiction to cover aspects of our compositions, formulations, methods of manufacture, or methods of treatment, prevention or use, the holders of any such patents may be able to block our ability to develop and commercialize the applicable products and product candidates unless we obtained a license or until such patents expire or are finally determined to be invalid or unenforceable. In addition, defending such claims would cause us to incur substantial expenses and, if successful, could cause us to pay substantial damages, if we are found to be infringing a third party’s patent rights. If we are found to have infringed such rights willfully, the damages may be enhanced and may include attorneys’ fees. Further, if a patent infringement suit is brought against us or our third-party service providers, our development, manufacturing or sales activities relating to the product or product candidate that is the subject of the suit may be delayed or terminated. As a result of patent infringement claims, or in order to avoid potential infringement claims, we may choose to seek, or be required to seek, a license from the third party, which may require us to pay license fees or royalties or both. These licenses may not be available on acceptable terms, or at all. Even if a license can be obtained on acceptable terms, the rights may be nonexclusive, which could give our competitors access to the same intellectual property rights. If we are unable to enter into a license on acceptable terms, we could be prevented from commercializing one or more of our products and product candidates, forced to modify such products and product candidates, or to cease some aspect of our business operations, which could harm our business significantly. Modifying our products and product candidates to design around third-party intellectual property rights may result in significant cost or delay to us and could prove to be technically infeasible. Any of these events, even if we were ultimately to prevail, could require us to divert substantial financial and management resources that we would otherwise be able to devote to our business. In addition, if the breadth or strength of protection provided the patents and patent applications we own or in-license is threatened, it could dissuade companies from collaborating with us to license, develop or commercialize current or future products and product candidates.
56
If we were to initiate legal proceedings against a third party to enforce a patent covering one of our products and product candidates, the defendant could counterclaim that our patent is invalid or unenforceable. In patent litigation in the United States and in Europe, defendant counterclaims alleging invalidity or unenforceability are commonplace. Grounds for a validity challenge could be an alleged failure to meet any of several statutory requirements, for example, lack of eligibility, lack of written description, lack of novelty, obviousness or non-enablement. Third parties might allege unenforceability of our patents because someone connected with prosecution of the patent withheld relevant information, or made a misleading statement, during patent prosecution. The outcome of proceedings involving assertions of invalidity and unenforceability during patent litigation is unpredictable. With respect to the validity of patents, for example, we cannot be certain that there is no invalidating prior art of which we and the patent examiner were unaware during prosecution. If a defendant were to prevail on a legal assertion of invalidity or unenforceability, we would lose at least part, and perhaps all, of the patent protection on our products and product candidates. Furthermore, our patents and other intellectual property rights also will not protect our technology if competitors design around our protected technology without infringing our patents or other intellectual property rights.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. There could also be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors view these announcements in a negative light, the price of common stock could be adversely affected.
Finally, even if resolved in our favor, litigation or other legal proceedings relating to intellectual property claims may cause us to incur significant expenses and could distract our technical and management personnel from their normal responsibilities. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments and if securities analysts or investors view these announcements in a negative light, the price of our common stock could be adversely affected. Such litigation or proceedings could substantially increase our operating losses and reduce our resources available for development activities. We may not have sufficient financial or other resources to adequately conduct such litigation or proceedings. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their substantially greater financial resources. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could have an adverse effect on our ability to compete in the marketplace.
We may not identify relevant third-party patents or may incorrectly interpret the relevance, scope or expiration of a third-party patent, which might adversely affect our ability to develop, manufacture and market our products and product candidates.
We cannot guarantee that any of our or our licensors’ patent searches or analyses, including but not limited to the identification of relevant patents, the scope of patent claims or the expiration of relevant patents, are complete or thorough, nor can we be certain that we have identified each and every third-party patent and pending application in the United States, Europe and elsewhere that is relevant to or necessary for the commercialization of our products and product candidates in any jurisdiction. For example, in the United States, applications filed before November 29, 2000 and certain applications filed after that date that will not be filed outside the United States remain confidential until patents issue. Patent applications in the United States, Europe and elsewhere are published approximately 18 months after the earliest filing for which priority is claimed, with such earliest filing date being commonly referred to as the priority date. Therefore, patent applications covering our future products and product candidates, or their manufacture or use may currently be unpublished. Additionally, pending patent applications that have been published can, subject to certain limitations, be later amended in a manner that could cover our products and product candidates or the use thereof. The scope of a patent claim is determined by an interpretation of the law, the written disclosure in a patent and the patent’s prosecution history. Our interpretation of the relevance or the scope of a patent or a pending application may be incorrect, which may negatively impact our ability to market our products and product candidates. We may incorrectly determine that our products and product candidates are not covered by a third-party patent or may incorrectly predict whether a third party’s pending application will issue with claims of relevant scope. Our determination of the expiration date of any patent in the United States, Europe or elsewhere that we consider relevant may be incorrect, which may negatively impact our ability to develop and market our products and product candidates. Our failure to identify and correctly interpret relevant patents may negatively impact our ability to develop and market our products and product candidates.
57
From time to time we may identify patents or applications in the same general area as our products and product candidates. We may determine these third-party patents are irrelevant to our business based on various factors including our interpretation of the scope of the patent claims and our interpretation of when those patents expire. If the patents are asserted against us, however, a court may disagree with our determinations. Further, while we may determine that the scope of claims that will issue from a patent application does not present a risk, it is difficult to accurately predict the scope of claims that will issue from a patent application, our determination may be incorrect, and the issuing patent may be asserted against us. We cannot guarantee that we will be able to successfully settle or otherwise resolve such infringement claims. If we fail in any such dispute, in addition to being forced to pay monetary damages, we may be temporarily or permanently prohibited from commercializing our products and product candidates. We might, if possible, also be forced to redesign our products and product candidates so that we no longer infringe the third-party intellectual property rights. Any of these events, even if we were ultimately to prevail, could require us to divert substantial financial and management resources that we would otherwise be able to devote to our business.
Changes in patent laws or patent jurisprudence could diminish the value of patents in general, thereby impairing our ability to protect our products and product candidates.
As is the case with other biopharmaceutical and pharmaceutical companies, our success is heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the biopharmaceutical and pharmaceutical industries involve both technological complexity and legal complexity. Therefore, obtaining and enforcing biopharmaceutical and pharmaceutical patents is costly, time-consuming and inherently uncertain. In addition, the America Invents Act, or the AIA, which was passed in September 2011, resulted in significant changes to the U.S. patent system.
An important change introduced by the AIA is that, as of March 16, 2013, the United States transitioned to a “first-to-file”“first-inventor-to-file” system for deciding which party should be granted a patent when two or more patent applications are filed by different parties claiming the same invention. A third party that files a patent application in the USPTO after that date but before us could therefore be awarded a patent covering an invention of ours even if we made the invention before it was made by the third party. This will require us to be cognizant going forward of the time from invention to filing of a patent application, but circumstances could prevent us from promptly filing patent applications on our inventions.
Among some of the other changes introduced by the AIA are changes that limit where a patentee may file a patent infringement suit and providing opportunities for third parties to challenge any issued patent before the USPTO. This applies to all of our U.S. patents, even those effectively filed before March 16, 2013. Because of a lower evidentiary standard in USPTO proceedings compared to the evidentiary standard in U.S. federal courts necessary to invalidate a patent claim, a third party could potentially provide evidence in a USPTO proceeding sufficient for the USPTO to hold a claim invalid even though the same evidence would be insufficient to invalidate the claim if first presented in a district court action.
Accordingly, a third party may attempt to use the USPTO procedures to invalidate our patent claims that would not have been invalidated if first challenged by the third party as a defendant in a district court action. It is not clear what, if any, impact the AIA will have on the operation of our business. However, the AIA and its implementation could increase the uncertainties and costs surrounding the prosecution of our owned and in-licensed patent applications and the enforcement or defense of our owned and in-licensed patents.
Additionally, the U.S. Supreme Court has ruled on several patent cases in recent years either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. In addition to increasing uncertainty with regard to our ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on decisions by Congress, the federal courts and the USPTO, the laws and regulations governing patents could change in unpredictable ways that could weaken our ability to obtain new patents or to enforce our existing patents and patents that we might obtain in the future. Similarly, the complexity and uncertainty of European patent laws has also increased in recent years. In addition, the European patent system is relatively stringent in the type of amendments that are allowed during prosecution. Complying with these laws and regulations could limit our ability to obtain new patents in the future that may be important for our business.
58
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance and annuity fees on any issued patent are due to be paid to the USPTO, European Patent Office (“EPO”) and other foreign patent offices over the lifetime of a patent. In addition, the USPTO, EPO and other foreign patent offices require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. While an inadvertent failure to make payment of such fees or to comply with such provisions can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which such noncompliance will result in the abandonment or lapse of the patent or patent application, and the partial or complete loss of patent rights in the relevant jurisdiction. Non-compliance events that could result in abandonment or lapse of a patent or patent application include failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents within prescribed time limits. If we or our licensors fail to maintain the patents and patent applications covering our products and product candidates or if we or our licensors otherwise allow our owned or licensed patents or patent applications to be abandoned or lapse, our competitors might be able to enter the market, which would hurt our competitive position and could impair our ability to successfully commercialize our products and product candidates in any indication for which they are approved.
We enjoy only limited geographical protection with respect to certain patents and we may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting and defending patents covering our products and product candidates in all countries throughout the world would be prohibitively expensive. Competitors may use our owned and in-licensed technologies in jurisdictions where we have not obtained patent protection to develop their own products and, further, may export otherwise infringing products to territories where we and our licensors have patent protection, but enforcement is not as strong as that in the United States or the Europe. These products may compete with our products and product candidates, and our owned or in-licensed patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
In addition, we may decide to abandon national and regional patent applications before grant. The grant proceeding of each national or regional patent is an independent proceeding which may lead to situations in which applications might in some jurisdictions be refused by the relevant patent offices, while granted by others. For example, unlike other countries, China has a heightened requirement for patentability, and specifically requires a detailed description of medical uses of a claimed drug. Furthermore, generic drug manufacturers or other competitors may challenge the scope, validity or enforceability of our owned and in-licensed patents, requiring us or our licensors to engage in complex, lengthy and costly litigation or other proceedings. Generic drug manufacturers may develop, seek approval for and launch generic versions of our products. It is also quite common that depending on the country, the scope of patent protection may vary for the same product candidate or technology.
The laws of some jurisdictions do not protect intellectual property rights to the same extent as the laws or rules and regulations in the United States and Europe, and many companies have encountered significant difficulties in protecting and defending such rights in such jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents, trade secrets and other intellectual property protection, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally. Proceedings to enforce our patent rights in other jurisdictions, whether or not successful, could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly and our patent applications at risk of not issuing, and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate, and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license. Furthermore, while we intend to protect our intellectual property rights in our expected significant markets, we cannot ensure that we will be able to initiate or maintain similar efforts in all jurisdictions in which we may wish to market our product candidates. Accordingly, our efforts to protect our intellectual property rights in such countries may be inadequate, which may have an adverse effect on our ability to successfully commercialize our product candidates in all of our expected significant foreign markets. If we or our licensors encounter difficulties in protecting, or are otherwise precluded from effectively protecting, the intellectual property rights important for our business in such jurisdictions, the value of these rights may be diminished, and we may face additional competition from others in those jurisdictions.
59
Some countries also have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties. In addition, some countries limit the enforceability of patents against government agencies or government contractors. In these countries, the patent owner may have limited remedies, which could materially diminish the value of such patent. If we or any of our licensors is forced to grant a license to third parties with respect to any patents relevant to our business, our competitive position may be impaired.
If we do not obtain patent term extension in the United States under the Hatch-Waxman Act and in foreign countries under similar legislation, thereby potentially extending the term of marketing exclusivity for our products, our business may be materially harmed.
Patents have a limited lifespan. In the United States, if all maintenance fees are timely paid, the natural expiration of a patent is generally 20 years from its earliest U.S. non-provisional filing date. Various extensions may be available, but the life of a patent, and the protection it affords, is limited. Even if patents covering our products are obtained, once the patent life has expired for a product, we may be open to competition from competitive medications, including generic medications. Given the amount of time required for the development, testing and regulatory review of new products, patents protecting such candidates might expire before or shortly after such candidates are commercialized. As a result, our owned and licensed patent portfolio may not provide us with sufficient rights to exclude others from using or commercializing technologies or products similar or identical to ours.
Depending upon the timing, duration and conditions of FDA marketing approval of our product candidates, we may be able to extend the term of a patent covering each product candidate under the Drug Price Competition and Patent Term Restoration Act of 1984, referred to as the Hatch-Waxman Amendments and similar legislation in the EU. The Hatch-Waxman Amendments permit a patent term extension of up to five years for a patent covering an approved product as compensation for effective patent term lost during product development and the FDA regulatory review process. Patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval, and only one patent that is applicable to and covers an approved drug may be extended. Similar provisions are available in Europe, such as supplementary protection certificates, and in certain other non-United States jurisdictions to extend the term of a patent that covers an approved drug. However, we may not receive an extension if we fail to apply within applicable deadlines, fail to apply prior to expiration of relevant patents or otherwise fail to satisfy applicable requirements. Moreover, the length of a patent term extension could be less than we request. If we are unable to obtain patent term extension or the term of any such extension is less than we request, the period during which we can enforce our patent rights for that product will be shortened and our competitors may obtain approval to market competing products sooner. As a result, our revenue from applicable products could be reduced, possibly materially.
Further, under certain circumstances, the term of a patent covering our products may be extended for time spent during the pendency of the corresponding patent application in the USPTO (referred to as Patent Term Adjustment, or PTA). The laws and regulations underlying how the USPTO calculates the PTA is subject to change and any such PTA granted by the USPTO could be challenged by a third-party. If we do not prevail under such a challenge, the PTA may be reduced or eliminated, resulting in a shorter patent term, which may negatively impact our ability to exclude competitors.
Because PTA added to the term of patents covering pharmaceutical products has particular value, our business may be adversely affected if the PTA is successfully challenged by a third party and our ability to exclude competitors is reduced or eliminated.
60
Intellectual property rights do not address all potential threats to our competitive advantage.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations, and may not adequately protect our business, or permit us to maintain our competitive advantage. The following examples are illustrative:
| ● | others may be able to make products that are similar to our Product Candidates or our future products or product candidates but that are not covered by the claims of the patents that we own or license from others; |
| ● | others may independently develop similar or alternative technologies or otherwise circumvent any of our technologies without infringing our intellectual property rights; |
| ● | we or any of our collaborators might not have been the first to conceive and reduce to practice the inventions covered by the patents or patent applications that we own, license or will own or license; |
| ● | we or any of our collaborators might not have been the first to file patent applications covering certain technologies we or they own or have obtained a license, or will own or obtain a license; |
| ● | it is possible that our owned and in-licensed pending patent applications will not lead to issued patents; |
| ● | issued patents that we own and in-licensed may not provide us with any competitive advantage, or may be held invalid or unenforceable, as a result of legal challenges by our competitors; |
| ● | our competitors might conduct research and development activities in countries where we do not have patent rights, or in countries where research and development safe harbor laws exist, and then use the information learned from such activities to develop competitive products for sale in our major commercial markets; |
| ● | ownership and inventorship of our owned and in-licensed patents or patent applications may be challenged by third parties; and |
| ● | patents of third parties, or pending or future applications of third parties, if issued, may have an adverse effect on our business. |
Our reliance on third parties requires us to share our trade secrets, which increases the possibility that our trade secrets will be misappropriated or disclosed, and confidentiality agreements with employees and third parties may not adequately prevent disclosure of trade secrets and protect other proprietary information.
We consider proprietary trade secrets or confidential know-how and unpatented know-how to be important to our business. We may rely on trade secrets or confidential know-how to protect our technology, especially where patent protection is believed by us to be of limited value. Because we expect to rely on third parties to manufacture our Product Candidates and any future products and product candidates, and we expect to collaborate with third parties on the development of our Product Candidates and any future products and product candidates, we must, at times, share trade secrets with them. We also conduct joint research and development programs that may require us to share trade secrets under the terms of our research and development partnerships or similar agreements. However, trade secrets or confidential know-how can be difficult to maintain as confidential.
To protect this type of information against disclosure or appropriation by competitors, our policy is to require our employees, consultants, collaborators, contractors and advisors to enter into confidentiality agreements and, if applicable, material transfer agreements, consulting agreements or other similar agreements with us prior to beginning research or disclosing proprietary information. These agreements typically limit the rights of the third parties to use or disclose our confidential information, including our trade secrets. However, current or former employees, consultants, collaborators, contractors and advisors may unintentionally or willfully disclose our confidential information to competitors, and confidentiality agreements may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. The need to share trade secrets and other confidential information increases the risk that such trade secrets become known by our competitors, are inadvertently incorporated into the technology of others, or are disclosed or used in violation of these agreements. Given that our proprietary position is based, in part, on our know-how and trade secrets, a competitor’s discovery of our trade secrets or other unauthorized use or disclosure would impair our competitive position and may have an adverse effect on our business and results of operations. Enforcing a claim that a third party obtained illegally and is using trade secrets or confidential know-how is expensive, time consuming and unpredictable. The enforceability of confidentiality agreements may vary from jurisdiction to jurisdiction.
61
In addition, these agreements typically restrict the ability of our employees, consultants, collaborators, contractors and advisors to publish data potentially relating to our trade secrets, although our agreements may contain certain limited publication rights. Despite our efforts to protect our trade secrets, our competitors may discover our trade secrets, either through breach of our agreements with third parties, independent development or publication of information by any of our third-party collaborators. A competitor’s discovery of our trade secrets would impair our competitive position and have an adverse impact on our business.
If our trademarks and trade names are not adequately protected, then we may not be able to build name recognition in our markets of interest and our business may be adversely affected.
Our unregistered trademarks or trade names may be challenged, infringed, circumvented or declared generic or determined to be infringing on other marks. We may not be able to protect our rights to these trademarks and trade names, which we need to build name recognition among potential collaborators or customers in our markets of interest. At times, competitors may adopt trade names or trademarks similar to ours, thereby impeding our ability to build brand identity and possibly leading to market confusion. In addition, there could be potential trade name or trademark infringement claims brought by owners of other registered trademarks or trademarks that incorporate variations of our unregistered trademarks or trade names. Over the long term, if we are unable to successfully register our trademarks and trade names and establish name recognition based on our trademarks and trade names, then we may not be able to compete effectively, and our business may be adversely affected. Our efforts to enforce or protect our proprietary rights related to trademarks, trade secrets, domain names, copyrights or other intellectual property may be ineffective and could result in substantial costs and diversion of resources and could adversely impact our financial condition or results of operations.
We may need to license certain intellectual property from third parties, and such licenses may not be available or may not be available on commercially reasonable terms.
A third party may hold intellectual property, including patent rights that are important or necessary for the development or commercialization of our Product Candidates or our future products or product candidates. It may be necessary for us to use the patented or proprietary technology of third parties to commercialize our Product Candidates, in which case we would be required to obtain a license from these third parties. Such a license may not be available on commercially reasonable terms, or at all, which could materially harm our business. At this time, we are unaware of any intellectual property that interferes with ours or is complementary and needed to commercialize our Product Candidates.
We may be subject to claims that our employees, consultants, collaborators contractors or advisors have wrongfully used or disclosed confidential information of their former employers or other third parties.
We employ individuals who were previously employed at other biotechnology or pharmaceutical companies. Although we seek to protect our ownership of intellectual property rights by ensuring that our agreements with our employees, consultants, collaborators, contractors, advisors and other third parties with whom we do business include provisions requiring such parties to assign rights in inventions to us, we may be subject to claims that we or our employees, consultants, collaborators, contractors and advisors have inadvertently or otherwise used or disclosed confidential information of their former employers or other third parties. We have recently been involved in litigation involving claims of misappropriation of trade secrets by our former chief executive officer. See “Part 1 Business – Recent Developments – Litigation,” We may also be subject to claims that the former employers or other third parties have an ownership interest in our patents. Litigation may be necessary to defend against these claims. There is no guarantee of success in defending these claims, and if we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights, such as exclusive ownership of, or right to use, valuable intellectual property. Even if we are successful, litigation could result in substantial cost and be a distraction to our management and other employees.
Our proprietary information may be lost, or we may suffer security breaches.
In the ordinary course of our business, we collect and store sensitive data, including intellectual property, clinical trial data, proprietary business information, personal data and personally identifiable information of our clinical trial subjects and employees, in our data centers and on our networks. The secure processing, maintenance and transmission of this information is critical to our operations. Despite our security measures, our information technology and infrastructure may be vulnerable to attacks by hackers or breached due to employee error, malfeasance or other disruptions. Although, to our knowledge, we have not experienced any such material security breach to date, any such breach could compromise our networks and the information stored there could be accessed, publicly disclosed, lost or stolen. Any such access, disclosure or other loss of information could result in legal claims or proceedings, liability under laws that protect the privacy of personal information, significant regulatory penalties, disrupt our operations, damage our reputation and cause a loss of confidence in us and our ability to conduct clinical trials, which could adversely affect our reputation and delay our clinical development of our product candidates.
62
Risks Related to Our Employees, Managing Our Growth and Our Operations
Our future success depends onWe have experienced turnover in our ability to retainsenior management team, and the loss of one or more of our executive officers or key personnel andemployees or an inability to attract and retain and motivate qualified personnel.highly skilled employees could adversely affect our business.
We are highly dependent on the development, regulatory, commercialization and business development expertise of Anthony Mack, our Chief Executive Officer, as well as the other principal members of our management, scientific and clinical teams. Although we have employment agreements, offer letters or consulting agreements with our executive officers, these agreements do not prevent them from terminating their services at any time. We have in the past and may in the future experience changes in our executive management team resulting from the departure of executives, which may be disruptive to our business.
If we lose one or more of our executive officers or key employees, our ability to implement our business strategy successfully could be seriously harmed. We have in the past and may in the future experience changes in our executive management team resulting from the departure of executives, which may be disruptive to our business. To continue to develop our pipeline and execute our strategy, we must attract and retain highly skilled personnel in our industry. Furthermore, replacing executive officers and key employees may be difficult and may take an extended period of time because of the limited number of individuals in our industry with the breadth of skills and experience required to successfully develop product candidates, gain regulatory approval, and commercialize new products. Competition to hire from this limited pool is intense, and we may be unable to hire, train, retain or motivate these additional key personnel on acceptable terms given the competition among numerous pharmaceutical and biotechnology companies for similar personnel. We also experience competition for the hiring of scientific and clinical personnel from universities and research institutions. In addition, we rely on consultants and advisors, including scientific and clinical advisors, to assist us in formulating our research and development and commercialization strategy. Our consultants and advisors may be engaged by entities other than us and may have commitments under consulting or advisory contracts with other entities that may limit their availability to us. If we are unable to continue to attract and retain highly qualified personnel, our ability to develop and commercialize product candidates will be limited.
We expect to expand our development, regulatory, and sales and marketing capabilities, and as a result, we may encounter difficulties in managing our growth, which could disrupt our operations.
We expect to experience significant growth in the number of our employees and the scope of our operations, particularly in the areas of development, regulatory affairs and sales and marketing. To manage our anticipated future growth, we must continue to implement and improve our managerial, operational and financial systems, expand our facilities or acquire new facilities and continue to recruit and train additional qualified personnel. Due to our limited financial resources and the limited experience of our management team in managing a company with such anticipated growth, we may not be able to effectively manage the expansion of our operations or recruit and train additional qualified personnel. The expansion of our operations may lead to significant costs and may divert our management and business development resources. Any inability to manage growth could delay the execution of our business plans or disrupt our operations.
We may engage in acquisitions that could disrupt our business, cause dilution to our stockholders or reduce our financial resources.
In the future, we may enter into transactions to acquire other businesses, products or technologies. If we do identify suitable candidates, we may not be able to make such acquisitions on favorable terms, or at all. Any acquisitions we make may not strengthen our competitive position, and these transactions may be viewed negatively by customers or investors. We may decide to incur debt in connection with an acquisition or issue our common stock or other equity securities to the stockholders of the acquired company, which would reduce the percentage ownership of our existing stockholders. We could incur losses resulting from undiscovered liabilities of the acquired business that are not covered by the indemnification we may obtain from the seller. In addition, we may not be able to successfully integrate the acquired personnel, technologies and operations into our existing business in an effective, timely and nondisruptive manner. Acquisitions may also divert management attention from day-to-day responsibilities, increase our expenses and reduce our cash available for operations and other uses. We cannot predict the number, timing or size of future acquisitions or the effect that any such transactions might have on our operating results.
Our business and operations would suffer in the event of system failures.
Our computer systems, as well as those of our CROs and other contractors and consultants, are vulnerable to damage from computer viruses, unauthorized access, natural disasters (including hurricanes), terrorism, war and telecommunication and electrical failures. If such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our development programs. For example, the loss of preclinical or clinical trial data from completed, ongoing or planned trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach were to result in a loss of or damage to our data or applications, or inappropriate disclosure of personal, confidential or proprietary information, we could incur liability and the further development of our Product Candidates or any other product candidate could be delayed.
We are increasingly dependent on information technology, and our systems and infrastructure face certain risks, including cybersecurity and data leakage risks.
Significant disruptions to our information technology systems or breaches of information security could adversely affect our business. In the ordinary course of business, we collect, store and transmit large amounts of confidential information, and it is critical that we do so in a secure manner to maintain the confidentiality and integrity of such confidential information. The size and complexity of our information technology systems, and those of our third-party vendors with whom we contract, make such systems potentially vulnerable to service interruptions and security breaches from inadvertent or intentional actions by our employees, partners or vendors, from attacks by malicious third parties, or from intentional or accidental physical damage to our systems infrastructure maintained by us or by third parties. Maintaining the secrecy of this confidential, proprietary, or trade secret information is important to our competitive business position. While we have taken steps to protect such information and invested in information technology, there can be no assurance that our efforts will prevent service interruptions or security breaches in our systems or the unauthorized or inadvertent wrongful use or disclosure of confidential information that could adversely affect our business operations or result in the loss, dissemination, or misuse of critical or sensitive information. A breach of our security measures or the accidental loss, inadvertent disclosure, unapproved dissemination, misappropriation or misuse of trade secrets, proprietary information, or other confidential information, whether as a result of theft, hacking, fraud, trickery or other forms of deception, or for any other reason, could enable others to produce competing products, use our proprietary technology or information, or adversely affect our business or financial condition. Further, any such interruption, security breach, loss or disclosure of confidential information, could result in financial, legal, business, and reputational harm to us and could have a material adverse effect on our business, financial position, results of operations or cash flow.
63
Risks Related to Our Common Stock
The market price of our common stock has been volatile and can fluctuate substantially, which could result in substantial losses for purchasers of our common stock.
The market price of our common stock is highly volatile and for the year ended December 31, 2022,2023, the market price of our common stock has ranged from $0.61$2.40 to $3.67$12.00 per share.share, as adjusted for the 1-10 reverse stock split effected March 1, 2024. The recent fluctuations in our trading price and future trading in our common stock may be subject to wide fluctuations in response to a variety of factors, including the following:
| ● | any delay in the commencement, enrollment and ultimate completion of our clinical trials; |
| ● | any delay in submitting an NDA and any adverse development or perceived adverse development with respect to the FDA’s review of that NDA; |
| ● | failure to successfully develop and commercialize our Product Candidates or any future product candidate; |
| ● | results of ongoing litigation as previously described as well as any new litigation, to which we are party; |
| ● | inability to obtain additional funding; |
| ● | regulatory or legal developments in the United States and other countries applicable to our Product Candidates or any other product candidate; |
| ● | adverse regulatory decisions; |
| ● | changes in the structure of healthcare payment systems; |
| ● | inability to obtain adequate product supply for our Product Candidates or any other product candidate, or the inability to do so at acceptable prices; |
| ● | introduction of new products, services or technologies by our competitors; |
| ● | failure to meet or exceed financial projections we provide to the public; |
| ● | failure to meet or exceed the estimates and projections of the investment community; |
| ● | changes in the market valuations of companies similar to ours; |
| ● | market conditions in the pharmaceutical and biotechnology sectors, and the issuance of new or changed securities analysts’ reports or recommendations; |
| ● | announcements of significant acquisitions, strategic collaborations, joint ventures or capital commitments by us or our competitors; |
| ● | significant lawsuits, including patent or stockholder litigation, and disputes or other developments relating to our proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our technologies; |
| ● | additions or departures of key scientific or management personnel; |
| ● | sales of our common stock by us or our stockholders in the future; |
| ● | trading volume of our common stock; |
| ● | general economic, industry and market conditions; |
| ● | health epidemics and outbreaks, including COVID-19, which could significantly disrupt our preclinical studies and clinical trials, and therefore our receipt of necessary regulatory approvals could be delayed or prevented; and |
| ● | the other factors described in this “Risk Factors” section. |
64
In addition, the stock markets have experienced extreme price and volume fluctuations that have affected and continue to affect the market prices of equity securities of many companies. These fluctuations have often been unrelated or disproportionate to the operating performance of those companies. Broad market and industry factors, as well as general economic, political, regulatory and market conditions, may negatively affect the market price of our common stock, regardless of our actual operating performance. In particular, stock markets have experienced extreme volatility in 2020 due to the ongoing COVID-19 pandemic and investor concerns and uncertainty related to the impact of the pandemic on the economies of countries worldwide.
We could be subject to securities class action litigation.
In the past, securities class action litigation has often been brought against companies following a decline in the market price of their securities. This risk is especially relevant for us because biotechnology companies have experienced significant share price volatility in recent years. If we face such litigation, it could result in substantial costs and a diversion of management’s attention and resources, which could harm our business.
Our directors, executive officers and certain stockholders (one of which is an affiliate of our Chief Executive Officer) will continue to own a significant percentage of our common stock and, if they choose to act together, will be able to exert significant control over matters subject to stockholder approval.
Our directors, executive officers, and stockholders affiliated with our directors and executive officers beneficially own approximately 30% of the voting power of our outstanding common stock. In particular, Virpax Pharmaceuticals, LLC, which is controlled by Anthony Mack, our Chief Executive Officer, and of which Jeffrey Gudin, our Chief Medical Officer, is a member, beneficially own shares representing approximately 23.3% of our outstanding capital stock. As a result, such entities and individuals have the ability, acting together, to control the election of our directors and the outcome of corporate actions requiring stockholder approval, such as: (i) a merger or a sale of our company, (ii) a sale of all or substantially all of our assets, and (iii) amendments to our certificate of incorporation and bylaws. This concentration of voting power and control could have a significant effect in delaying, deferring or preventing an action that might otherwise be beneficial to our other stockholders and be disadvantageous to our stockholders with interests different from those entities and individuals. These individuals also have significant control over our business, policies and affairs as officers and directors of our company. Therefore, you should not invest in reliance on your ability to have any control over our company.
If securities or industry analysts do not publish research or reports about our business, or if they issue an adverse or misleading opinion regarding our common stock, our stock price and trading volume could decline.
The trading market for our common stock will depend, in part, on the research and reports that securities or industry analysts may publish about us or our business. We do not have any control over these analysts. If our financial performance fails to meet analyst estimates or one or more of the analysts who cover us downgrade our common stock or change their opinion of our common stock, our share price would likely decline. If one or more of these analysts cease coverage of us or fail to regularly publish reports on us, we could lose visibility in the financial markets, which could cause our share price or trading volume to decline.
Because we do not anticipate paying any cash dividends on our common stock in the foreseeable future, capital appreciation, if any, will be your sole source of gain.
We have never declared or paid any cash dividends on our common stock. We currently anticipate that we will retain future earnings for the development, operation and expansion of our business and do not anticipate declaring or paying any cash dividends for the foreseeable future. As a result, capital appreciation, if any, of our common stock would be your sole source of gain on an investment in our common stock for the foreseeable future. See “Dividend Policy” for additional information.
We have incurred and will continue to incur increased costs as a result of operating as a public company, and our management will be required to devote substantial time to new compliance initiatives and corporate governance practices.
As a public company, and particularly after we no longer qualify as an emerging growth company, we have incurred and will continue to incur significant legal, accounting and other.other costs. The Sarbanes-Oxley Act of 2002 (“SOX”), the Dodd-Frank Wall Street Reform and Consumer Protection Act, the listing requirements of Nasdaq, and other applicable securities rules and regulations impose various requirements on U.S. reporting public companies, including the establishment and maintenance of effective disclosure and financial controls and corporate governance practices. Our management and other personnel devote a substantial amount of time to these compliance initiatives. Moreover, these rules and regulations increase our legal and financial compliance costs and make some activities more time-consuming and costly. For example, our status as a public company makes it more difficult and more expensive for us to obtain director and officer liability insurance and we may be required to incur substantial costs to maintain the level of coverage that we believe is appropriate for a public Company. This could make it more difficult for us to attract and retain qualified senior management personnel or members for our board of directors (the “Board of Directors”). In addition, these rules and regulations are often subject to varying interpretations, and, as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies. This could result in continuing uncertainty regarding compliance matters and higher costs necessitated by ongoing revisions to disclosure and governance practices.
65
While we remain an emerging growth company, we will not be required to include an attestation report on internal control over financial reporting issued by our independent registered public accounting firm. To prepare for eventual compliance with Section 404, once we no longer qualify as an emerging growth company, we will be engaged in a process to document and evaluate our internal control over financial reporting, which is both costly and challenging. In this regard, we will need to continue to dedicate internal resources, potentially engage outside consultants and adopt a detailed work plan to assess and document the adequacy of internal control over financial reporting, continue steps to improve control processes as appropriate, validate through testing that controls are functioning as documented and implement a continuous reporting and improvement process for internal control over financial reporting. Despite our efforts, there is a risk that we will not be able to conclude, within the prescribed timeframe or at all, that our internal control over financial reporting is effective as required by Section 404. If we identify one or more material weaknesses, it could result in an adverse reaction in the financial markets due to a loss of confidence in the reliability of our financial statements.
We are an “emerging growth company,” and the reduced reporting requirements applicable to emerging growth companies may make our common stock less attractive to investors.
On April 5, 2012, the Jumpstart Our Business Startups Act of 2012 (“JOBS Act”) was signed into law. The JOBS Act contains provisions that, among other things, reduce certain reporting requirements for an “emerging growth company”. As an “emerging growth company,” we are electing to take advantage of the extended transition period afforded by the JOBS Act for the implementation of new or revised accounting standards, and as a result, we will comply with new or revised accounting standards on the relevant dates on which adoption of such standards is required for non-emerging growth companies. Section 107 of the JOBS Act provides that our decision not to take advantage of the extended transition period is irrevocable. Subject to certain conditions set forth in the JOBS Act, as an “emerging growth company,” we are not required to, among other things, (i) provide an auditor’s attestation report on our system of internal controls over financial reporting pursuant to Section 404, (ii) provide all of the compensation disclosure that may be required of non-emerging growth public companies under the Dodd-Frank Wall Street Reform and Consumer Protection Act, (iii) comply with any requirement that may be adopted by the Public Company Accounting Oversight Board regarding reporting and critical audit matters, and (iv) disclose certain executive compensation-related items such as the correlation between executive compensation and performance and comparisons of the chief executive officer’s compensation to median employee compensation. These exemptions will apply until the last day of the fiscal year including the fifth anniversary of the completion of our initial public offering or until we no longer meet the requirements for being an “emerging growth company,” whichever occurs first.
In addition, under the JOBS Act, emerging growth companies may delay adopting new or revised accounting standards until such time as those standards apply to private companies. Although we have not done so, we may elect not to avail ourselves of this exemption from new or revised accounting standards and, therefore, may be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies.
We cannot predict if investors will find our common stock less attractive because we may rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our share price may be more volatile.
Anti-takeover provisions contained in our certificate of incorporation and bylaws, as well as provisions of Delaware law, could impair a takeover attempt.
Our certificate of incorporation, bylaws and Delaware law contain provisions which could have the effect of rendering more difficult, delaying or preventing an acquisition deemed undesirable by our Board of Directors. Our corporate governance documents include provisions:
| ● | classifying our Board of Directors into three classes; |
66
| ● | authorizing “blank check” preferred stock, which could be issued by our Board of Directors without stockholder approval and may contain voting, liquidation, dividend, and other rights superior to our common stock; |
| ● | limiting the liability of, and providing indemnification to, our directors and officers; |
| ● | limiting the ability of our stockholders to call and bring business before special meetings; |
| ● | requiring advance notice of stockholder proposals for business to be conducted at meetings of our stockholders and for nominations of candidates for election to our Board of Directors; |
| ● | controlling the procedures for the conduct and scheduling of Board of Directors and stockholder meetings; and |
| ● | providing our Board of Directors with the express power to postpone previously scheduled annual meetings and to cancel previously scheduled special meetings. |
These provisions, alone or together, could delay or prevent hostile takeovers and changes in control or changes in our management.
As a Delaware corporation, we are also subject to provisions of Delaware law, including Section 203 of the Delaware General Corporation law, which prevents some stockholders holding more than 15% of our outstanding common stock from engaging in certain business combinations without approval of the holders of substantially all of our outstanding common stock.
Any provision of our certificate of incorporation, bylaws or Delaware law that has the effect of delaying or deterring a change in control could limit the opportunity for our stockholders to receive a premium for their shares of our common stock and could also affect the price that some investors are willing to pay for our common stock.
Our certificate of incorporation, as amended, designates the Court of Chancery of the State of Delaware as the sole and exclusive forum for certain types of actions and proceedings that may be initiated by our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers or other employees.
Our certificate of incorporation requires that, unless we consent in writing to the selection of an alternative forum, the Court of Chancery of the State of Delaware will, to the fullest extent permitted by law, be the sole and exclusive forum for each of the following:
| ● | any derivative action or proceeding brought on our behalf; |
| ● | any action asserting a claim for breach of any fiduciary duty owed by any director, officer or other employee of ours to the Company or our stockholders, creditors or other constituents; |
| ● | any action asserting a claim against us or any director or officer of ours arising pursuant to, or a claim against us or any of our directors or officers, with respect to the interpretation or application of any provision of, the DGCL, our certificate of incorporation or bylaws; or |
| ● | any action asserting a claim governed by the internal affairs doctrine; |
provided, that, if and only if the Court of Chancery of the State of Delaware dismisses any of the foregoing actions for lack of subject matter jurisdiction, any such action or actions may be brought in another state court sitting in the State of Delaware.
The exclusive forum provision is limited to the extent permitted by law, and it will not apply to claims arising under the Securities Exchange Act of 1934, as amended (the “Exchange Act”), or for any other federal securities laws which provide for exclusive federal jurisdiction.
Furthermore, Section 22 of the Securities Act creates concurrent jurisdiction for federal and state courts over all such Securities Act actions. Accordingly, both state and federal courts have jurisdiction to entertain such claims. To prevent having to litigate claims in multiple jurisdictions and the threat of inconsistent or contrary rulings by different courts, among other considerations, our amended and restated certificate of incorporation provides that the federal district courts of the United States of America will be the exclusive forum for resolving any complaint asserting a cause of action arising under the Securities Act. While the Delaware courts have determined that such choice of forum provisions are facially valid, a stockholder may nevertheless seek to bring such a claim arising under the Securities Act against us, our directors, officers, or other employees in a venue other than in the federal district courts of the United States of America. In such instance, we would expect to vigorously assert the validity and enforceability of the exclusive forum provisions of our amended and restated certificate of incorporation.
Our failure to meet the continued listing requirements of The Nasdaq Capital Market could result in a de-listing of our common stock.
Our shares of common stock are listed for trading on The Nasdaq Capital Market under the symbol “VRPX.” If we fail to satisfy the continued listing requirements of The Nasdaq Capital Market such as the corporate governance requirements, the stockholder’s equity requirement or the minimum closing bid price requirement, The Nasdaq Capital Market may take steps to de-list our common stock or warrants.
On April 10, 2023, we received a written notice (the “Notice”) from the Listing Qualifications Department of The Nasdaq Stock Market (“Nasdaq”) indicating that we are not in compliance with the $1.00 Minimum Bid Price requirement set forth in Nasdaq Listing Rule 5550(a)(2) for continued listing on The Nasdaq Capital Market (the “Bid Price Requirement”). On February 29, 2024, we filed a Certificate of Amendment to the Amended and Restated Certificate of Incorporation (the “Amendment”) with the Secretary of State of the State of Delaware to effect a 1-for-10 reverse stock split, which was effective on March 1, 2024. Although, we have regained compliance with the $1.00 Minimum Bid Price requirement set forth in Nasdaq Listing Rule 5550(a)(2) by effecting a reverse stock split there can be no assurance that we will continue to maintain compliance with the Nasdaq continued listing requirements. Any perception that we may not regain compliance for future noncompliance or a delisting of our common stock by Nasdaq could adversely affect our ability to attract new investors, decrease the liquidity of the outstanding shares of our common stock, reduce the price at which such shares trade and increase the transaction costs inherent in trading such shares with overall negative effects for our stockholder. In addition, delisting of our common stock from Nasdaq could deter broker-dealers from making a market in or otherwise seeking or generating interest in our common stock and might deter certain institutions and persons from investing in our common stock.
67
In the event of a de-listing, we would take actions to restore our compliance with The Nasdaq Capital Market’s listing requirements, but we can provide no assurance that any such action taken by us would allow our common stock become listed again, stabilize the market price or improve the liquidity of our common stock, prevent our common stock from dropping below The Nasdaq Capital Market, minimum bid price requirement or prevent future non-compliance with The Nasdaq Capital Market’s listing requirements.
The National Securities Markets Improvement Act of 1996, which is a federal statute, prevents or preempts the states from regulating the sale of certain securities, which are referred to as “covered securities.” Because our common stock is listed on The Nasdaq Capital Market, our common stock is covered securities. Although the states are preempted from regulating the sale of covered securities, the federal statute does allow the states to investigate companies if there is a suspicion of fraud, and, if there is a finding of fraudulent activity, then the states can regulate or bar the sale of covered securities in a particular case. Further, if we were to be delisted from The Nasdaq Capital Market, our common stock would cease to be recognized as covered securities and we would be subject to regulation in each state in which we offer our securities.
Although we believe this provision benefits us by providing increased consistency in the application of Delaware law in the types of lawsuits to which it applies, this provision may limit or discourage a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, officers or other employees, which may discourage such lawsuits against us and our directors, officers and other employees. Alternatively, if a court were to find the choice of forum provision contained in our certificate of incorporation to be inapplicable or unenforceable in an action, we may incur additional costs associated with resolving such action in other jurisdictions, which could adversely affect our business and financial condition.
We note that there is uncertainty as to whether a court would enforce the provision and that investors cannot waive compliance with the federal securities laws and the rules and regulations thereunder. Although we believe this provision benefits us by providing increased consistency in the application of Delaware law in the types of lawsuits to which it applies, the provision may have the effect of discouraging lawsuits against our directors and officers.
ITEM 1B. UNRESOLVED STAFF COMMENTS
Not applicable.
ITEM 1C. CYBERSECURITY
Risk Management and Strategy
We are a pre-clinical-stage biopharmaceutical company, focused on developing novel and proprietary drug delivery systems across various pain indications and treatments for CNS disorders. We have conducted a cyber security risk assessment performed by a third-party consultant and are in the process of developing a formal cybersecurity risk management program designed to identify, assess, manage, mitigate, and respond to cybersecurity threats. The risk assessment was performed against the National Institute of Standards and Technology (“NIST”) Cybersecurity Framework (“CSF”) standards.
We have implemented third-party risk management processes to manage the risks associated with reliance on vendors, critical service providers, and other third-parties that may lead to a service disruption or an adverse cybersecurity incident. This includes an assessment of vendors during the selection/onboarding process and a review of SOC 1 reports on an annual basis.
In addition, we maintain policies over areas such as information security, access on/offboarding, and access and account management, to help govern the processes put in place by management designed to protect our IT assets, data, and services from threats and vulnerabilities. We partner with industry recognized IT providers leveraging third-party technology and expertise. These third-party service providers are a key part of our current cybersecurity risk management and provide services including, maintenance of an IT assets inventory, periodic vulnerability scanning, identity access management controls including restricted access of privileged accounts, network integrity safeguarded by employing web-based software, including endpoint protection, endpoint detection and response, and remote monitoring management on all devices, industry-standard encryption protocols and critical data backups. Our outsourced information technology consultant conducts proactive patching and monitoring of all of our existing systems and has implemented systems and procedures to mitigate cybersecurity risks that we believe are appropriate for a company of our size, stage of growth and financial condition. In addition, we carry insurance with coverage for cyber events that we believe is suitable for a company of our size, stage of growth and financial condition.
As of the date of this Annual Report on Form 10-K, we are not aware of any cybersecurity threats, and have not experienced any cybersecurity incidents, that have materially affected us, including our business strategy, results of operations or financial condition.
For additional information concerning risks related to cybersecurity, see Item lA. Risk Factors: We are increasingly dependent on information technology, and our systems and infrastructure face certain risks, including cybersecurity and data leakage risks.
Governance
Management is responsible for the day-to-day management of the risks we face, while our Board of Directors has responsibility for the oversight of risk management, including as to risks from cybersecurity threats. In its risk oversight role, our Board of Directors has the responsibility to satisfy itself that the risk management processes designed and implemented by management are appropriate and functioning as designed. The Board of Directors has delegated to the Audit Committee of the Board of Directors the responsibility for the oversight of information technology, including cybersecurity risks. Member(s) of management assigned with cybersecurity oversight responsibility and/or third-party consultants providing cyber risk services brief the Audit Committee on cyber vulnerabilities identified through the risk management process, emerging threat landscape and new cyber risks, and provide updates on our processes to prevent, detect, and mitigate cybersecurity incidents.
ITEM 2. PROPERTIES
Our principal address is 1055 Westlakes Drive, Suite 300, Berwyn, PA 19312. We believe our facilities are adequate to meet our current needs, although we may seek to negotiate new leases or evaluate additional or alternate space for our operations. We believe appropriate alternative space would be readily available on commercially reasonable terms.
ITEM 3. LEGAL PROCEEDINGS
On March 12, 2021, the Company and the Company’sits former Chief Executive Officer, Anthony P. Mack (the(together, the “Defendants”), were named as defendants in a complaint (the “Complaint”) filed by Sorrento Therapeutics, Inc. (“Sorrento”), and Scilex Pharmaceuticals Inc. (“Scilex” and together with Sorrento, the “Plaintiffs”) in the Court of Chancery of the State of Delaware.Delaware captioned Sorrento Therapeutics, Inc. and Scilex Pharmaceuticals Inc. v. Anthony Mack and Virpax Pharmaceuticals, Inc., Case No. 2021-0210-PAF (the “Action”). In the Complaint, Plaintiffs alleged (i) Mr. Mack breached a Restrictive Covenants Agreement, dated as of November 8, 2016, between himself and Sorrento (the “Restrictive Covenants Agreement”), (ii) the Company tortiously interfered with the Restrictive Covenants Agreement, and (iii) the Company tortiously interfered with Scilex’s relationship with Mr. Mack. On May 7, 2021, Plaintiffs filed an Amended Complaint asserting the same three causes of action. On September 28, 2021, Plaintiffs filed a Second Amended Complaint asserting the same three causes of action as the prior complaints, as well as claims in which Plaintiffs alleged (i) Mr. Mack breached an Employment, Proprietary Information and Inventions Agreement, dated as of October 25, 2016, between himself and Sorrento (the “Employment Agreement”), (ii) the Company tortiously interfered with the Employment Agreement, (iii) Mr. Mack breached his fiduciary duties to Scilex, and (iv) the Company aided and abetted Mr. Mack’s alleged breach of fiduciary duties to Scilex. On April 1, 2022, Plaintiffs filed a Third Amended Complaint. The Third Amended Complaint asserts the same causes of action as the Second Amended Complaint, as well as claims for (i) misappropriation of trade secrets by Defendants under Delaware law, and (ii) misappropriation of trade secrets by Defendants under California law. On April 18, 2022, Defendants filed answers to the Third Amended Complaint. Trial was held before Vice Chancellor Paul Fiorvanti from September 12 through September 14, 2022. Post-trial briefing was completed
In March 2023, the Company collected $1,250,000 in reimbursement of legal costs pursuant to the Company’s directors’ and officers’ insurance policy, and recorded it as a reduction of general and administrative expense on the consolidated statements of operations. No further reimbursements are permitted from the insurance policy with respect to the litigation.
On September 1, 2023, the Chancery Court issued a memorandum opinion addressing liability in the Action and found in favor of Plaintiffs on all but three counts, which the Court found were waived. The Chancery Court found it proper to attribute Mr. Mack’s knowledge and actions to the Company, which Mr. Mack used to effectuate the tortious interference and breach of fiduciary duty. The Chancery Court found that Mr. Mack breached the Restrictive Covenants Agreement he entered into with Sorrento by December 12, 2022, and post-trial argument was held on January 20, 2023. The Company intends to vigorously defend the action. However,developing EpoladermTM; the Company is unableliable for tortious interference with contract; Plaintiffs were deemed to predicthave waived their claims for breach of Mr. Mack’s Employment Agreement and for tortious interference with prospective economic advantage; Mr. Mack breached his fiduciary duty of loyalty to Scilex; the ultimate outcomeCompany aided and abetted Mr. Mack’s breach of fiduciary duty; and Mr. Mack misappropriated certain Scilex trade secrets. The Court, however, stated that the question of an appropriate remedy must await further briefing.
On October 18, 2023, in accordance with the Chancery Court’s supplemental briefing schedule, Plaintiffs filed their supplemental brief requesting the following relief: an injunction, in the first instance, enjoining Mr. Mack from having any relationship with Virpax for a period of 18 months and 27 days; enjoining Virpax from further developing or marketing Epoladerm for a period of 18 months and 27 days; alternatively, if these two injunction requests were not granted, Plaintiffs requested a judgement of joint and several liability against Mr. Mack and Virpax of $14,684,833. In addition to these requests for injunctive relief (or in, the alternative, damages), Plaintiffs sought a constructive trust over the revenues of Epoladerm, ProbudurTM and EnveltaTM, or, in the alternative to a constructive trust, a royalty of 5 per cent of net sales of Epoladerm, 8-11 percent of net sales of Probudur and 7.5 percent of net sales of Envelta. In addition to the requests for injunctive relief, imposition of a constructive trust and/or royalties, Plaintiffs also requested additional damages, jointly and severally, against Mr. Mack and Virpax as follows: $1.3 million for misuse of Scilex resources, $6.7 million for misappropriation of trade secrets, $13.4 million for exemplary damage (trade secrets damage x2) and attorney’s fees in an unspecified amount. Finally, Plaintiffs sought injunctive relief, enjoining Mr. Mack and Virpax from further accessing Scilex’s trade secrets; requiring Mr. Mack and Virpax to return Scilex’s trade secrets to Plaintiffs; and enjoining Mr. Mack and Virpax from marketing or selling any products derived from or incorporating Scilex’s trade secrets.
On November 29, 2023, in accordance with the Chancery Court’s supplemental briefing schedule, Defendants filed their supplement brief on damages rebutting Plaintiffs’ damages analysis. Throughout the brief, Defendants argued Plaintiffs failed to meet their burden to prove damages, and as such, should be precluded from any damages award. However, given the Court’s instruction, Defendants proffered a reasonable damages analysis as follows. As for the injunctive relief requested against Mr. Mack, the Company took no position, as the request was directed to Mr. Mack personally. Concerning Plaintiffs’ request for an injunction against further development of Epoladerm for a period of 18 months and 27 days, Defendants opposed this request, arguing lack of irreparable harm, given Plaintiffs’ request for money damages. Defendants also argued a constructive trust is inappropriate, given Plaintiffs failed to articulate the parameters of such relief and, additionally, the lack of sales for the drug candidates preclude such relief. In terms of the lawsuit.money damages related to the three drug candidates, Defendants proffered a reasonable royalty rate of 1-3% of the net profits of the drug candidates, as opposed to lump sum damages, as such rate would alleviate the speculative nature of the damages requested by Plaintiffs. As for the misappropriation of trade secrets request of $6.7 million, given the Court found only 5 of the proffered 1,182 documents were trade secrets, Defendants contend Plaintiffs should receive no monetary damages (given the reasonable royalty would encompass use of these documents and, alternatively, Defendants would return such documents). However, if the Court were to award damages, such damages should be pro rata for the documents, or roughly $28,382. And, finally, Defendants opposed the request for attorneys’ fees and exemplary damages.
On December 21, 2023, Plaintiffs filed their reply brief on damages, generally reasserting their prior arguments on damages and rebutting Defendants’ arguments. Plaintiffs also asserted alternativethey supported their damages theories that would imply potential damages upclaims with sufficient evidence.
On February 29, 2024, Plaintiffs and the Company entered into a Settlement Agreement to approximately $35.0 million. fully resolve all issues related to settlement of the litigation with Plaintiffs, subject to the entry by the United States Bankruptcy Court for the Southern District of Texas, which is handling the Sorrento bankruptcy filing, of an order approving the Settlement Agreement. On March 1, 2024, the Plaintiffs filed a motion to approve the Settlement Agreement and grant the related relief with the Bankruptcy Court. On March 14, 2024, the Bankruptcy Court entered an order approving the Settlement Agreement and on March 20th the Plaintiffs filed a Stipulation of Dismissal with the Chancery Court dismissing the Action.
As settlement consideration, the Company agreed to pay Sorrento and Scilex a total cash payment of $6 million, of which $3.5 million was paid on March 18, 2024, two business days after the Effective Date, and the remaining $2.5 million is to be paid on or before July 1, 2024. Additionally, the Company agreed to pay to Plaintiffs royalties of 6% of annual net sales of products developed from drug candidates Epoladerm, Probudur and Envelta until the earlier of the expiration of the last-to-expire valid patent claim of such product and the expiration of any period of regulatory exclusivity for such product.
Pursuant to the Settlement Agreement, each of the Plaintiffs and the Company provided mutual releases of all claims as of the Effective Date, whether known or unknown, arising from any allegations set forth in the Action. Plaintiffs’ release relates to claims against the Company only. Plaintiffs’ release as to the Company was effective upon the Company’s initial payment of $3.5 million, and the Company’s release of the Plaintiffs was effective upon the Effective Date.
The Plaintiffs can still pursue claims against Mr. Mack. The Company’s Bylaws require the Company countered that actual damages, even if Plaintiffs establish liability, could be zero because, among other things, Plaintiffs’ calculations use various unsupported assumptions,to “indemnify any alleged damages are speculative in nature, and thereperson who was or is a possibilityparty or is threatened to be made a party to any threatened, pending or completed action, suit or proceeding, whether civil, criminal, administrative or investigative (other than an action by or in the Company’s product candidates never reach market. To date, the parties have not had successful settlement negotiations. Based on the foregoing, the Company accrued $2 million in respectright of the litigation. WhileCorporation) by reason of the fact that such person is or was a director or officer of the Corporation, or, while a director or officer of the Corporation….” Such indemnification, however, is limited to circumstances where the covered person “acted in good faith and in a manner such person reasonably believed to be in or not opposed to the best interests of the Corporation….” Mr. Mack may attempt to claim he is entitled to indemnification, should the Court find him liable for damages in the Action. Given the findings in the Memorandum Opinion issued in the Action, the Company believes it has meritorious defenses, the ultimate resolutiona strong position that Mr. Mack would not be entitled to indemnification. There is a risk, however, that a Court could find he is entitled to such indemnification. Additionally, per Section 7.6 of the actionBylaws, the Company has been advancing Mr. Mack’s attorneys’ fees and costs for the Action. It is likely Mr. Mack will contend he is still entitled to advancement of any fees and/or costs for the Action going forward and may seek judicial intervention. However, as per the Bylaws, Mr. Mack is only entitled to advancement of expenses for indemnifiable actions. As noted above, given the Memorandum Opinion in the Action, the Company believes that it has a strong position that Mr. Mack is not entitled to indemnification, and therefore, not entitled to advancement of expenses. However, there is a risk that a Court could resultfind that Mr. Mack is entitled to such advancement. Further, Mr. Mack may attempt to seek damages from the Company based on the Court’s final judgment on damages under the theory of joint and several liability and seek contribution from the Company for any monetary judgment. (See Item 1-Business and Item 1A-Risk Factors)
The Court is aware that Plaintiffs have settled with the Company and that the Settlement Agreement fully releases the Company from any claims or damages, the Plaintiff has against the Company, related to the Action. Given the Settlement Agreement does not release Mr. Mack from liability related to the Action, the Court has requested supplemental briefing as to whether the Court can dismiss the Company from the lawsuit, as well as any claims Mr. Mack has against the Company arising from the Action. While the Company believes that any damages assessed may be awarded against Mr. Mack alone, Plaintiffs cannot seek additional damages from Virpax. However, there is a risk that Mr. Mack will still seek contribution from the Company for any damages claim arising from the Action. And, there is a risk that the Court will rule in a material loss.Mr. Mack’s favor.
No further reimbursements are permitted from our insurance policy with respect to the litigation. Accordingly, if Mr. Mack was successful in seeking indemnification from us, we would have to pay such amounts in cash which would further reduce our cash position.
From time to time we are subject to claims by third parties under various legal disputes. The defense of such claims, or any adverse outcome relating to any such claims, could have a material adverse effect on our liquidity, financial condition and cash flows.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
68
PART II
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information
Our Common Stock trades on Nasdaq under the symbolssymbol “VRPX” and began trading on February 17, 2021. Prior to that date, there was no public market for our stock. The last reported sale price of our common stock on Nasdaq on March 22, 2024 was $4.03 per share of common stock.
Holders
As of March 21, 2023,22, 2024, there were approximately 3938 holders of record of our Common Stock. This number does not include beneficial owners whose shares are held in street name. The actual number of holders of our Common Stock is greater than this number of record holders and includes stockholders who are beneficial owners, but whose shares are held in street name by brokers or held by other nominees.
Dividends
We have never declared or paid cash dividends on our Common Stock. We do not intend to declare or pay cash dividends on our common stock for the foreseeable future, but currently intend to retain any future earnings to fund the development and growth of our business. The payment of cash dividends if any, on the common stock will rest solely within the discretion of our Board of Directors and will depend, among other things, upon our earnings, capital requirements, financial condition, and other relevant factors.
Recent Sales of Unregistered Securities
None.
Issuer Purchases of Equity Securities
None.
Equity Compensation Plan Information
The following table provides information with respect to our compensation plans under which equity compensation was authorized as of December 31, 2023.
| Number of securities to be issued upon exercise of outstanding options, warrants and rights | Weighted average exercise price of outstanding options, warrants and rights | Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column a) | ||||||||||
| Plan category | (a) | (b) | (c) | |||||||||
| Equity compensation plans approved by security holders(1) | 175,686 | (2) | $ | 34.60 | 116,511 | (3)(4) | ||||||
| Equity compensation plans not approved by security holders | - | - | - | |||||||||
| Total | 175,686 | $ | 34.60 | 116,511 | ||||||||
| (1) | The amounts shown in this row include securities under the 2017 Plan and the 2022 Plan. |
| (2) | Includes 98,886 and 76,800 shares of common stock issuable upon exercise of outstanding options pursuant to the 2017 Plan and the 2022 Plan, respectively, as of December 31, 2023. |
| (3) | In accordance with the “evergreen” provision in the 2022 Plan, an additional 23,428 shares were automatically made available for issuance on the first day of 2023, which represents 2% of the number of shares outstanding on December 31, 2023; these shares are excluded from this calculation. |
| (4) | Includes 0 and 116,511 shares of common stock available for issuance under the 2017 Plan and the 2022 Plan, respectively, as of December 31, 2023. |
ITEM 6. RESERVED[RESERVED]
Not applicable.
69
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
You should read the following discussion and analysis of our financial condition and results of operations together with our financial statements and the related notes appearing at the end of this Annual Report on Form 10-K. Some of the information contained in this discussion and analysis or set forth elsewhere in this Annual Report on Form 10-K, including information with respect to our plans and strategy for our business and related financing, includes forward-looking statements that involve risks and uncertainties. You should read “Cautionary Note Regarding Forward-Looking Statements” and Item 1A. Risk Factors of this Annual Report on Form 10-K for a discussion of important factors that could cause actual results to differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis.
Overview
Company Overview
We are a preclinical-stage pharmaceutical company focused on developing novel and proprietary drug delivery systems across various pain indications in order to enhance compliance and optimize each product candidate in our pipeline. Our drug-delivery systems, and drug-releasing technologies being developed are focused on advancing non-opioid and non-addictive pain management treatments and treatments for central nervous system (“CNS”) disorders to enhance patients’ quality of life.
We have exclusive global rights to the following proprietary patented technologies: (i) Molecular Envelope Technology (“MET”) that uses an intranasal device to deliver enkephalin for the management of acute and chronicto control severe pain, including post cancer pain associated with cancer (Envelta™) and PTSD, (ii) Injectable “local anesthetic” Liposomal Gel Technology for postoperative pain management (Probudur™), and (iii) Topical Spray Film Delivery Technology for osteoarthritis pain (Epoladerm™). We intend to utilize these delivery technologies to selectively develop a portfolio of patented 505(b)(2) and new chemical entity (“NCE”) candidates for commercialization. We also have exclusive worldwide rights to develop and commercialize an investigationalInvestigational formulation delivered via the nasal route to enhance pharmaceutical-grade cannabidiol (“CBD”) transport to the brain (“NobrXiol™”, formerly VRP324) to potentially treat epileptic seizures associated with tuberous sclerosis complex (“TSC”), Lennox-Gastaut syndrome and Dravet syndrome in pediatric patients one yeartwo years of age and older. We are also exploring value creative opportunities for our two nonprescription product candidates including seeking regulatory approval for commercialization of such products: AnQlar, which is being developed as a 24 hour prophylactic viral barrier to inhibit viral infection by influenza or SARS-CoV-2, and Epoladerm™, which is a topical diclofenac epolamine metered dosed spray film formulation being developed to manage pain associated with osteoarthritis.
While we are currently focused on advancing non-opioid and non-addictive pain management treatments and treatments for CNS disorders, we also plan on using our proprietary delivery technologies to develop our anti-viral therapy (AnQlar™) as a viral barrier to potentially prevent or reduce the risk or the intensity of viral infections in humans, including, but not limited to, influenza and SARS-CoV-2 (“COVID 19”). We have exclusive global rights for AnQlar pursuant to a collaboration and license agreement.
Critical Accounting Policies and Use of Estimates
We have based our management’s discussion and analysis of financial condition and results of operations on our financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States. The preparation of these financial statements requires us to make estimates that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements as well as the reported revenues and expenses during the reporting periods. On an ongoing basis, we evaluate our estimates and judgments, including those related to clinical development expenses and stock-based compensation. We base our estimates on historical experience and on various other factors that we believe to be appropriate under the circumstances. Actual results may differ from these estimates under different assumptions or conditions.
While our significant accounting policies are more fully discussed in Note 2 to our audited financial statements contained within this Annual Report on Form 10-K, we believe that the following accounting policies are critical to the process of making significant judgments and estimates in the preparation of our financial statements.
Research and Development Expenses
We rely on third parties to conduct our preclinical studies and to provide services, including data management, statistical analysis and electronic compilation. We have initiated preclinical trials and at the end of each reporting period, we compare the payments made to each service provider to the estimated progress towards completion of the related project. Factors that we consider in preparing these estimates include the status of preclinical studies, milestones achieved and other criteria related to the efforts of our vendors. These estimates are subject to change as additional information becomes available. Depending on the timing of payments to vendors and estimated services provided, we record net prepaid or accrued expenses related to these costs.
70
Stock-Based Compensation
Stock-based compensation cost is measured at the grant date based on the fair value of the award and is recognized as expense over the requisite service period, which is generally the vesting period. Our policy permits the valuation of stock-based awards granted to non-employees to be measured at fair value at the grant date.
Determining the appropriate fair value of share-based awards requires the use of subjective assumptions, including the fair value of our common shares prior to becoming a public company, and for options, the exceptedexpected life of the option and expected share price volatility. We use the Black-Scholes option pricing model to value its option awards. The assumptions used in calculating the fair value of share-based awards representsrepresent management’s best estimates and involve inherent uncertainties and the application of management’s judgment. As a result, if factors change and management uses different assumptions, share-based compensation expense could be materially different for future awards. See Note 87 to notes to the consolidated financial statements contained herein.
Legal and Other Contingencies
The outcomes of legal proceedings and claims brought against us and other loss contingencies are subject to significant uncertainty. We accrue a charge against income when our management determines that it is probable that an asset has been impaired, or a liability has been incurred and the amount of loss can be reasonably estimated. In determining the appropriate accounting for loss contingencies, we consider the likelihood of loss or impairment of an asset or the incurrence of a liability, as well as our ability to reasonably estimate the amount of loss. We regularly evaluate current information available to us to determine whether an accrual should be established or adjusted. Estimating the probability that a loss will occur and estimating the amount of a loss or a range of loss involves significant judgment. As noted in Note 5 Commitments and Contingencies, the Company has recently settled litigation with the Plaintiffs and entered into a Settlement Agreement. As of December 31, 2022, the Company had accrued $2.0 million with respect to the litigation. Based on the facts of the litigation and the Settlement Agreement that was executed, the Company has recognized an accrual totaling $6.0 million with respect to the litigation as of December 31, 2023. The Company recognized $4.0 million and $2.0 million for the years ended December 31, 2023 and 2022, respectively, included in general and administrative expenses on the consolidated statements of operations. We have also accrued an estimated $711,000 for payments to be made to our former Chief Executive Officer with respect to his separation from employment with us. While the Company believes this estimated expense related to the separation agreement to be reasonably possible, actual results may materially vary from these estimates. As part of the consideration for the separation agreement, Mr. Mack will be expected to release, discharge and waive any rights to indemnification, and/or contribution related to the Action. The accrual does not include any amounts that we may be required to pay for indemnification claims or contribution that he may seek against us.
Results of Operations
Years Ended December 31, 20222023 and 20212022
Operating expenses:
| Year Ended December 31, | Change | Year Ended December 31, | Change | |||||||||||||||||||||||||||||
| 2022 | 2021 | Dollars | Percentage | 2023 | 2022 | Dollars | Percentage | |||||||||||||||||||||||||
| Operating expenses: | ||||||||||||||||||||||||||||||||
| General and administrative | $ | 11,082,463 | $ | 7,186,385 | $ | 3,896,078 | 54 | % | $ | 10,572,181 | $ | 11,082,463 | $ | (510,282 | ) | (5 | )% | |||||||||||||||
| Research and development | 10,762,670 | 4,839,115 | 5,923,555 | 122 | % | 5,117,608 | 10,762,670 | (5,645,062 | ) | (52 | )% | |||||||||||||||||||||
| Total operating expenses | $ | 21,845,133 | $ | 12,025,500 | $ | 9,819,633 | 82 | % | $ | 15,689,789 | $ | 21,845,133 | $ | (6,155,344 | ) | (28 | )% | |||||||||||||||
General and administrative expenses increaseddecreased by $3,896,078,$510,282, or 54%5%, to $10,572,181 for the year ended December 31, 2023 from $11,082,463 for the year ended December 31, 2022 from $7,186,385 for the year ended December 31, 2021. The primary reasons for the increase2022. This decrease was a result of a significant decrease of legal defense costs of $2.3 million with regard to litigation, including reimbursement of legal defense costs of $1.25 million in general and administrative costs were (i) an increase in legal costs associated with litigation defense efforts of $3,417,321, including a $2,000,000 estimated litigation liability, (ii) an increase in salaries and wages and employee benefits of $257,438, (iii) an increase in insurance costs related2023 pursuant to our directors’ and officers’ insurance of $216,134, (iv)policy, and an increase of $2 million in non-executive board compensation of $181,667,estimated litigation settlement expense ($2 million in 2022 and (v)$4 million in 2023); and partially offset by an increase in grant writingof $1.8 million related to salaries and grant consulting fees of $114,973. This was offset by a decrease in stock based compensation of $316,768.wages, severance and professional fees.
71
Research and development expenses increaseddecreased by $5,923,555,$5,645,062, or 122%52%, to $5,117,608 for the year ended December 31, 2023, from $10,762,670 for the year ended December 31, 2022, from $4,839,115 for the year ended December 31, 2021.2022. The increasedecrease was primarily attributable to (i) an increase ina one-time milestone paymentspayment of $500,000$1.5 million made to Nanomerics in 2022 related to expanding AnQlar’s territory to global rights and a decrease in AnQlar preclinical activities of approximately $3.8 million, (ii) an increasea decrease in preclinical activity of $3,826,920 related to AnQlar’s ongoing IND enabling studies and regulatory consulting, (iii) increases in preclinical and regulatory activityactivities related to Epoladerm, of $513,517,and (iii) a decrease in preclinical activities related to NobrXiol. This was offset by an increase in preclinical workof approximately $1.3 million related to Probudur of $487,573 related to ongoing formula optimization, and (iv) an increase in NobrXiol of $599,800 mainly due to a milestone payment of $500,000 paid to Nanomerics upon achieving the study aim contained within a pre-clinical animal study.preclinical activities, which is our lead asset.
The following table presents R&D expenses tracked on a program-by-program basis for the year ended December 31, 20222023 and 2021.2022.
| Year Ended December 31, | Year Ended December 31, | |||||||||||||||
| 2022 | 2021 | 2023 | 2022 | |||||||||||||
| Program Expenses: | ||||||||||||||||
| Program expenses: | ||||||||||||||||
| Envelta | $ | 286,793 | $ | 296,142 | $ | 268,868 | $ | 286,793 | ||||||||
| Probudur | 1,583,093 | 1,095,520 | 2,872,819 | 1,583,093 | ||||||||||||
| Epoladerm | 1,830,689 | 1,317,173 | 714,471 | 1,830,689 | ||||||||||||
| AnQlar | 6,115,916 | 1,788,996 | 836,629 | 6,115,916 | ||||||||||||
| NobrXiol | 799,800 | 200,000 | 215,415 | 799,800 | ||||||||||||
| Total program expenses | $ | 10,616,291 | $ | 4,697,831 | 4,908,202 | 10,616,291 | ||||||||||
| Unallocated Expenses: | ||||||||||||||||
| Unallocated expenses: | ||||||||||||||||
| Stock based compensation | 146,379 | 58,811 | 209,406 | 146,379 | ||||||||||||
| Other research and development expenses | - | 82,473 | ||||||||||||||
| Total other research and development expense | 146,379 | 141,284 | 209,406 | 146,379 | ||||||||||||
| Total research and development expenses | $ | 10,762,670 | $ | 4,839,115 | $ | 5,117,608 | $ | 10,762,670 | ||||||||
Other expenses:
| Year Ended December 31, | Change | |||||||||||||||
| 2022 | 2021 | Dollars | Percentage | |||||||||||||
| Other income (expense): | ||||||||||||||||
| Interest expense | $ | - | $ | (92,822 | ) | $ | 92,822 | 100 | % | |||||||
| Other income | 194,413 | 62,259 | 132,154 | 212 | % | |||||||||||
| Total other expenses: | $ | 194,413 | $ | (30,563 | ) | $ | 224,976 | 736 | % | |||||||
| Year Ended December 31, | Change | |||||||||||||||
| 2023 | 2022 | Dollars | Percentage | |||||||||||||
| Other income: | ||||||||||||||||
| Other income | $ | 500,281 | $ | 194,413 | $ | 305,868 | 157 | % | ||||||||
| Total other income: | $ | 500,281 | $ | 194,413 | $ | 305,868 | 157 | % | ||||||||
Interest expense decreased by $92,822 for the year ended December 31, 2022, as compared to the year ended December 31, 2021 as a result of the repayment of a convertible promissory note in February 2021 and related party notes payable in September 2021. Other income increased by $305,868 primarily due to interest income as well as exchange rate gains during the year ended December 31, 2022. Other income during the year ended December 31, 2021 comprised mostly of our PPP loan forgiveness.income.
72
Liquidity and Capital Resources
Years Ended December 31, 20222023 and 20212022
Capital Resources
| As of December 31, | As of December 31, | Change | ||||||||||||||||||||||
| 2022 | 2021 | 2023 | 2022 | Dollars | Percentage | |||||||||||||||||||
| Current assets | $ | 19,673,649 | $ | 39,572,436 | $ | 9,628,345 | $ | 19,673,649 | (10,045,304 | ) | (51 | )% | ||||||||||||
| Current liabilities | 3,094,590 | 2,087,691 | $ | 7,694,024 | $ | 3,094,590 | 4,599,434 | 149 | % | |||||||||||||||
| Working capital | $ | 16,579,059 | $ | 37,484,745 | $ | 1,934,321 | $ | 16,579,059 | (14,644,738 | ) | (88 | )% | ||||||||||||
As of December 31, 2022,2023, our principal source of liquidity was our cash, which totaled approximately $19.0$9.1 million. On March 18, 2024, we paid $3.5 million to the Plaintiffs pursuant to the terms of the Settlement Agreement and we are obligated to pay an additional $2.5 million to the Plaintiffs on July 1, 2024. We will need to raise additional capital to fund operations and make the $2.5 million payment. We accrued $0.7 million for estimated payments to be made to our former Chief Executive Officer with respect to his separation from employment with us, which does not include accrual for any potential indemnification or contribution claims that he may seek from us that are related to the Action. We have not generated revenues and have not yet achieved profitable operations, nor have we ever generated positive cash flow from operations. There is no assurance that profitable operations, if achieved, could be sustained on a continuing basis. We are subject to those risks associated with any preclinical stage pharmaceutical company that has substantial expenditures for research and development. There can be no assurance that our research and development projects will be successful, that products developed will obtain necessary regulatory approval, or that any approved product will be commercially viable.
To continue to grow our business over the longer term, we plan to commit substantial resources to research and development, pre-clinical and clinical trials of our product candidates, other operations and potential product acquisitions and in-licensing.
We have evaluated and expect to continue to evaluate a wide array of strategic transactions as part of our planexplore opportunities to acquire or in-license and develop additional products and product candidates to augment our internal development pipeline. Strategic transaction opportunities that we may pursue could materially affect our liquidity and capital resources and may require us to incur additional indebtedness, seek equity capital or both. In addition, we may pursue development, acquisition or in-licensing of approved or development products in new or existing therapeutic areas or continue the expansion of our existing operations. Accordingly, we expect to continue to opportunistically seek access to additional capital to license or acquire additional products, product candidates or companies to expand our operations, or for general corporate purposes. Strategic transactions may require us to raise additional capital through one or more public or private debt or equity financings or could be structured as a collaboration or partnering arrangement. Any equity financing would be dilutive to our stockholders. We have no arrangements, agreements, or understandings in place at the present time to enter into any acquisition, in-licensing or similar strategic business transaction.
Cash Flows
Years Ended December 31, 20222023 and 20212022
The following table summarizes our cash flows from operating and financing activities:
| Year Ended December 31, | ||||||||
| 2022 | 2021 | |||||||
| Statement of cash flow data: | ||||||||
| Total net cash provided by (used in): | ||||||||
| Operating activities | $ | (17,846,708 | ) | $ | (14,542,592 | ) | ||
| Financing activities | - | 51,329,788 | ||||||
| (Decrease) increase in cash | $ | (17,846,708 | ) | $ | 36,787,196 | |||
| For the Year Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| Statement of cash flow data: | ||||||||
| Net cash used in operating activities | $ | (9,853,772 | ) | $ | (17,846,708 | ) | ||
| Net change in cash | $ | (9,853,772 | ) | $ | (17,846,708 | ) | ||
Operating Activities
For the year ended December 31, 2022,2023, cash used in operations was $17,846,708$9,853,772 compared to $14,542,592$17,846,708 for the year ended December 31, 2021.2022. The increasedecrease in cash used in operations was primarily the result of the increasedecrease in net loss primarily attributable to the decrease in research and development expenses and an increase in accounts payable and accrued expense, offset by a smaller decrease in prepaid expensesinsurance other current assets. In March 2023, we collected $1,250,000 in reimbursement of legal costs pursuant to our directors’ and current assets as well as an increase in current liabilities.
Financing Activities
Cash provided by financing activities was $51,329,788officers’ insurance policy, which decreased our net loss during the year ended December 31, 2021, attributable primarilyperiod. No further reimbursements are permitted from the insurance policy with respect to net proceeds received from our initial public offering in February 2021 of $15,834,087 and the underwritten offering in September 2021 of $36,999,465, after deducting underwriting discounts and offering expenses. These proceeds were offset by the repayment in full of our RRD Note of $493,480 in February 2021 and repayments of our promissory notes and the unforgiven portion of the PPP Loan of an aggregate of $1,503,764. No financing activities took place during the year ended December 31, 2022.litigation.
73
Future Capital Requirements
It is difficult to predict our spending for our product candidates prior to obtaining FDA approval. Moreover, changing circumstances may cause us to expend cash significantly faster than we currently anticipate, and we may need to spend more cash than currently expected because of circumstances beyond our control.
Our expectations regarding future cash requirements do not reflect the potential impact of any future acquisitions, mergers, dispositions, joint ventures or investments that we make in the future. We have no current understandings, agreements or commitments for any material acquisitions or licenses of any products, businesses or technologies. We maywill most likely need to raise substantial additional capital in order to engage in any of these types of transactions.
We expect to continue to incur substantial additional operating losses for at least the next several years as we continue to develop our product candidates and seek marketing approval and, subject to obtaining such approval, the eventual commercialization of our product candidates. If we obtain marketing approval for our product candidates, we will incur significant sales, marketing and outsourced manufacturing expenses. In addition, we expect to incur additional expenses to add operational, financial and information systems and personnel, including personnel to support our planned product commercialization efforts. We also expect to continue to incur significant costs to comply with corporate governance, internal controls and similar requirements applicable to us as a public company.
Our future use of operating cash and capital requirements will depend on many forward-looking factors, including the following:
| ● | the |
| ● |
| ● | the clinical development plans we establish for each product candidate; |
| ● | the number and characteristics of product candidates that we develop or may in-license; |
| ● | the terms of any collaboration agreements we may choose to execute; |
| ● | the outcome, timing and cost of meeting regulatory requirements established by the U.S. Drug Enforcement Administration, the FDA, the European Medicines Agency or other comparable foreign regulatory authorities; |
| ● | the cost of filing, prosecuting, defending and enforcing our patent claims and other intellectual property rights; |
| ● | the cost of defending intellectual property disputes, including patent infringement actions brought by third parties against us; |
| ● | costs and timing of the implementation of commercial scale manufacturing activities; and |
| ● | the cost of establishing, or outsourcing, sales, marketing and distribution capabilities for any product candidates for which we may receive regulatory approval in regions where we choose to commercialize our products on our |
To the extent that ourOur capital resources are currently insufficient to meet our future operating and capital requirements, and therefore we must finance our cash needs through public or private equity offerings, debt financings, collaboration and licensing arrangements or other financing alternatives. We have no committed external sources of funds. Additional equity or debt financing or collaboration and licensing arrangements may not be available on acceptable terms, if at all.
If we raise additional funds by issuing equity securities, our stockholders will experience dilution. Debt financing, if available, would result in increased fixed payment obligations and may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. Any debt financing or additional equity that we raise may contain terms, such as liquidation and other preferences that are not favorable to us or our stockholders. If we raise additional funds through collaboration and licensing arrangements with third parties, it may be necessary to relinquish valuable rights to our technologies, future revenue streams or product candidates or to grant licenses on terms that may not be favorable to us.
74
Liquidity
Since inception, we have been engaged in organizational activities, including raising capital and research and development activities. We have not generated revenues and have not yet achieved profitable operations, nor have we ever generated positive cash flow from operations. There is no assurance that profitable operations, if achieved, could be sustained on a continuing basis. We are subject to those risks associated with any preclinical stage pharmaceutical company that has substantial expenditures for research and development. There can be no assurance that our research and development projects will be successful, that products developed will obtain necessary regulatory approval, or that any approved product will be commercially viable. In addition, we operate in an environment of rapid technological change and is largely dependent on the services of its employees and consultants. Further, our future operations are dependent on the success of our efforts to raise additional capital.
We incurred a net loss of $21,650,720$15.2 million and $12,056,063$21.7 million for the yearyears ended December 31, 20222023 and 2021,2022, respectively, and had an accumulated deficit of $44,354,627$59.5 million as of December 31, 2022.2023. We anticipate incurring additional losses until such time, if ever, that we can generate significant revenue from our product candidates currently in development. Our primary source of capital has been from the $15.8 million that we raised from the issuance of debt and equity securities. We closedsecurities in our initial public offering that closed in February 2021 and an underwritten publicthe $37.0 million that we raised from the issuance of securities in a follow on offering that closed in September 2021, raising $15,783,207 and $36,999,465, net of issuance costs, respectively.2021.
At December 31, 2022,2023, we had cash of approximately $19.0$9.1 million, on March 22, 2024, we had cash of approximately $2.5 million. We are currently involved in litigation and we have established an estimated litigation liability of $2.0 million. While we believe we have meritorious defenses,paid the ultimate resolutionPlaintiff $3.5 million on March 18, 2024 pursuant to the terms of the action could result in a material loss.Settlement Agreement, and are obligated to pay an additional $2.5 million on or before July 1, 2024. We accrued $0.7 million for estimated payments to be made to our former Chief Executive Officer with respect to his separation from employment with us. The accrual does not include any amounts that we may be required to pay for indemnification claims or contribution that he may seek against us. We will need to raise additional capital to fund operations and make the $2.5 million payment. Due to our continuing losses and the uncertainty regarding the outcome of this ongoing litigation and any potential claims,our cash position, there exists substantial doubt about our ability to continue as a going concern. Our auditorsAuditor’s report contains an emphasis of matter regarding our substantial doubt of continuing as a going concern. The accompanying financial statements do not include any adjustments to the carrying amounts and classification of assets, liabilities, and reported expenses that may be necessary if we were unable to continue as a going concern.
Our future operations are dependent on the success of our efforts to raise additional capital. We currently do not have sufficient capital to fund the commercialization of any of our product candidates. Additional financing will be needed by us to fund our operations, including making payment pursuant to the Settlement Agreement, and to complete clinical development of and to commercially develop our product candidates. There is no assurance that such financing will be available when needed or on acceptable terms. Our ability to raise capital to date has been impacted by the uncertainty of the amount of damages we may be required to pay and it is likely that we will be unable to raise capital, if at all, until all uncertainties are resolved. Further, our ability to raise additional capital may be adversely impacted by potential worsening of global economic conditions, potential future global pandemics or health crises, and the recent disruptions to, and volatility in, the credit, banking, and financial markets in the United States. We also may be forced to curtail spending in research and development activities in order to conserve cash.
Additional financings will be needed by us to fund our operations, including litigation costs,making the $2.5 million payment on or before July 1, 2024 pursuant to the Settlement Agreement or pursuant to any damages awarded by the Chancery Court, and to complete clinical development of and to commercially develop our product candidates. There is no assurance that such financing will be available when needed or on acceptable terms. We also may be forced to curtail spending in research and development activities in order to conserve cash.
Global Macroeconomic Environment
The global macroeconomic environment could be negatively affected by, among other things, resurgence of COVID-19 or other pandemics or epidemics, instability in global economic markets, increased U.S. trade tariffs and trade disputes with other countries, instability in the global credit markets, supply chain weaknesses, instability in the geopolitical environment as a result of the withdrawal of the United Kingdom from the European Union, the ongoing conflict between Russia and Ukraine, andthe war in the Middle East, other political tensions, and foreign governmental debt concerns. Such challenges have caused, and may continue to cause, uncertainty and instability in local economies and in global financial markets.
While expected to be temporary, these disruptions may negatively impact our results of operations, financial condition, and liquidity in 20232024 and potentially beyond.
Significant Contractual Obligations and Commitments
We lease our office facilities under a six-month operating lease.
Recently Issued Accounting Standards
No relevant recent accounting pronouncements noted.
75
JOBS Act
On April 5, 2012, the Jumpstart Our Business Startups Act of 2012 (“JOBS Act”) was signed into law. The JOBS Act contains provisions that, among other things, reduce certain reporting requirements for an “emerging growth company”. As an “emerging growth company,” we are electing to take advantage of the extended transition period afforded by the JOBS Act for the implementation of new or revised accounting standards, and as a result, we will comply with new or revised accounting standards on the relevant dates on which adoption of such standards is required for non-emerging growth companies. Section 107 of the JOBS Act provides that our decision not to take advantage of the extended transition period is irrevocable. Subject to certain conditions set forth in the JOBS Act, as an “emerging growth company,” we are not required to, among other things, (i) provide an auditor’s attestation report on our system of internal controls over financial reporting pursuant to Section 404, (ii) provide all of the compensation disclosure that may be required of non-emerging growth public companies under the Dodd-Frank Wall Street Reform and Consumer Protection Act, (iii) comply with any requirement that may be adopted by the Public Company Accounting Oversight Board regarding reporting and critical audit matters, and (iv) disclose certain executive compensation-related items such as the correlation between executive compensation and performance and comparisons of the chief executive officer’s compensation to median employee compensation. These exemptions will apply until the last day of the fiscal year including the fifth anniversary of the completion of our initial public offering or until we no longer meet the requirements for being an “emerging growth company,” whichever occurs first.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Not Applicable.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
The information required by this item appears in a separate section of this Annual Report on Form 10-K beginning on page F-1 and is incorporated herein by reference.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, evaluated the effectiveness of our disclosure controls and procedures as of December 31, 2022.2023. The term “disclosure controls and procedures,” as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act, means controls and other procedures of a company that are designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is accumulated and communicated to the company’s management, including its principal executive and principal financial officers, as appropriate to allow timely decisions regarding required disclosure. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on the evaluation of our disclosure controls and procedures as of December 31, 2022,2023, our Chief Executive Officer and Chief Financial Officer concluded that, as of such date, our disclosure controls and procedures were effective at the reasonable assurance level.
76
Management’s Annual Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over our financial reporting. Internal control over financial reporting is defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act as a process designed by, or under the supervision of, our Chief Executive Officer and Chief Financial Officer, and effected by our Board of Directors, management and other personnel, to provide reasonable assurance regarding the reliability of our financial reporting and the preparation of our financial statements for external purposes in accordance with generally accepted accounting principles, and includes those policies and procedures that:
| ● | Pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of assets; |
| ● | provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures are being made only in accordance with the authorizations of management and directors; and |
| ● | provide reasonable assurance regarding the prevention or timely detection of unauthorized acquisition, use or disposition of assets that could have a material effect on our financial statements. |
Under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework provided in Internal Control — Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on this evaluation, our management concluded that our internal control over financial reporting was effective as of December 31, 2022.2023.
Changes in Internal Control Over Financial Reporting
There were no changes in our internal controls over financial reporting identified in management’s evaluation pursuant to Rules 13a-15(d) and 15d-15(d) of the Exchange Act that occurred during the yearquarter ended December 31, 20222023 that materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Inherent Limitations on Effectiveness of Controls
Our management, including our Chief Executive Officer and Chief Financial Officer, believes that our disclosure controls and procedures and internal control over financial reporting are designed to provide reasonable assurance of achieving their objectives and are effective at the reasonable assurance level. However, our management does not expect that our disclosure controls and procedures or our internal control over financial reporting will prevent all errors and all fraud. A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, have been detected. These inherent limitations include the realities that judgments in decision-making can be faulty, and that breakdowns can occur because of a simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people or by management override of the controls. The design of any system of controls also is based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions; over time, controls may become inadequate because of changes in conditions, or the degree of compliance with policies or procedures may deteriorate. Because of the inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and not be detected.
ITEM 9B. OTHER INFORMATION
None.During the three months ended December 31, 2023, no director or officer of the Company adopted or terminated a “Rule 10b5-1 trading arrangement” or “non Rule 10b5-1 trading arrangement,” as each term is defined in Item 408(a) of Regulation S-K.
On March 25, 2024, we entered into our standard director and officer indemnification agreement with Vinay Shah, a copy of which has been filed as an exhibit to this Annual Report on Form 10-K. The Indemnification Agreement provides for indemnification against expenses, judgments, fines and penalties actually and reasonably incurred by an indemnitee in connection with threatened, pending or completed actions, suits or other proceedings, subject to certain limitations. The Indemnification Agreement also provides for the advancement of expenses in connection with a proceeding prior to a final, nonappealable judgment or other adjudication, provided that the indemnitee provides an undertaking to repay to us any amounts advanced if the indemnitee is ultimately found not to be entitled to indemnification by us. The Indemnification Agreement sets forth procedures for making and responding to a request for indemnification or advancement of expenses, as well as dispute resolution procedures that will apply to any dispute between us and an indemnitee arising under the Indemnification Agreement.
In addition, and subject to certain limitations, the Indemnity Agreement provides for the advancement of expenses incurred by or on behalf of the Indemnitee in connection with any proceeding not initiated by the Indemnitee, and the reimbursement to us of the amounts advanced (without interest) to the extent that it is ultimately determined that the Indemnitee is not entitled to be indemnified by us.
Item 9C. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections.
Not applicable.
77
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
Executive Officers and Directors
The following sets forth certain information with respect to our officers and directors.
| Name | Age | Position | ||
| Executive Officers | ||||
| Chief Executive Officer (Principal Executive Officer) and | ||||
| Jeffrey Gudin, MD | Executive Vice President, Chief Medical Officer, and Class III Director | |||
| Chief Financial Officer (Principal Financial and Accounting Officer) and Corporate Secretary | ||||
| Sheila A. Mathias, PhD, J.D., MBA | 56 | Chief Scientific Officer | ||
| Directors | ||||
| Eric Floyd, PhD | 61 | Independent Class III | ||
| Jerrold Sendrow, CFP | Independent Class II Director and Audit Committee Chair | |||
| Thani Jambulingam, PhD | Independent Class II Director and Corporate Governance Committee Chair | |||
| Vanila M. Singh, MD | Independent Class I Director | |||
| Michael F. Dubin, CPA | Independent Class II Director | |||
| Barbara A. Ruskin, PhD, J.D. | 63 | Independent Class I Director |
Executive Officers
Anthony Mack, MBA Gerald W. Brucebecame has served as our Chief Executive Officer since November 20, 2023, as a director since July 2021 and as our Executive Vice President and our ChairmanCommercial Operations Officer from August 2017 until his appointment as our Chief Executive Officer. Mr. Bruce has spent over 30 years, including 20 years in senior leadership roles, in the Pharmaceutical and Medical Nutrition industry. He started his career in May 1983 at Johnson & Johnson Inc. where he was an award-winning sales representative and held leadership positions of increasing responsibility in sales and marketing ending with his role as Group Product Director of Analgesics in September 1998. From September 1998 to November 2000, he served as Vice President of Sales at Bristol-Myers Squibb Co. where he led the Cardiovascular and Metabolic sales force. From November 2000 to January 2006, he served as Vice President of Managed Markets where he led the team responsible for the development and implementation of the reimbursement strategy for Bristol-Myers Squibb’s US portfolio. From January 2006 to June 2008, Mr. Bruce was the Senior Vice President of Commercial Operations at NitroMed, Inc. where he was responsible for building the commercial strategy and led the team responsible for the development and implementation of the commercial plan for the start-up company’s first product for the treatment of heart failure. From April 2009 to November 2018, Mr. Bruce served as Vice President of Sales for Nutricia North America, Danone Medical Nutrition Division. Mr. Bruce currently serves on the Board of DirectorsTrustees for Lincoln University and Chief Executive Officeris a Board member for the National Sales Network. He received his bachelor’s degree in May 2017. Anthony P. Mack is an entrepreneur who has founded three successfulbusiness administration from Lincoln University and a master’s degree in leadership from the McDonough School of Business at Georgetown University. Mr. Bruce was selected as a director due to his extensive experience at pharmaceutical companies served on executive boards and has more than 35 yearsknowledge of experience in the industry as well as in finance. Prior to founding Virpax, Mr. Mack founded Scilex Pharmaceuticals and served as President, CEO and Director until February 2018. Scilex was sold to a publicly traded company. Mr. Mack founded his first pharmaceutical company, ProSolus Pharmaceuticals, in 2009 where he served as President and Director before selling the company to Mission Pharmacal in 2015. Mr. Mack has led training, marketing, and commercial distribution for billion-dollar pain management products and has forged key strategic alliances through high-level management positions with Purdue Pharma, Endo Pharmaceuticals, Novartis, and EKR Therapeutics. Prior to his pharmaceutical career, Mr. Mack worked in the financial sector, starting as a Series 7 broker dealer then a Series 24 licensed Principal, allowing him to supervise and manage brokers at affiliated branches. Mr. Mack holds an Executive MBA in Pharmaceutical and Healthcare Marketing from Saint Joseph’s University.industry.
Jeffrey Gudin, MD, one of our co-founders, became an Executive Vice President, and our Chief Medical Officer in January 2017. Prior to joining us, Dr. Gudin, was Director of Pain Management and Palliative Care at Englewood Hospital and Medical Center in New Jersey for almost 20 years. He is a Clinical Associate Professor in Anesthesiologyon faculty at the Rutgers New Jersey Medical School.University of Miami, Miller School of Medicine. Dr. Gudin is Board Certified in Pain Medicine, Anesthesiology, Addiction Medicine and Hospice and Palliative Medicine. He is an active speaker in the field of pain management. His clinical and research focus includes pain management, opioid abuse and potential solutions, and increasing clinician awareness of pain assessment and risk management. Dr. Gudin completed a residency in anesthesiology at Yale University School of Medicine, in New Haven, Connecticut. He continued his training with an extended postdoctoral fellowship in pain medicine at the Yale Center for Pain Management, where he was actively involved in research and teaching. Dr. Gudin was selected as a director due to his extensive experience in the field of pain management, opioid abuse and palliative care.
78
Gerald W. BruceVinay Shah. See below under “Directors.” became our Chief Financial Officer in June 2023. Previously, Mr. Shah served as the Chief Financial Officer of Aravive, Inc. from October 2018 until June 2022. Mr. Shah also served as the Chief Financial Officer of Aravive Biologics, Inc. from 2010 until June 2022, initially as a consultant and from 2017 as an employee. Mr. Shah brings more than 20 years of financial management experience in the medical device and biopharmaceutical industries to our company. From 2008 until 2016, he served in various positions at Pacira Pharmaceuticals Inc., a specialty pharmaceutical company, including Executive Director of Finance and Executive Director of Strategy Analytics, initially as a consultant and since 2010 as an employee. Before Pacira Pharmaceuticals Inc., Mr. Shah worked for Cardinal Health’s medical device group in various finance management positions. The group was subsequently consolidated and spun off as CareFusion and then sold to Becton, Dickinson and Company. His prior work experience includes positions at Pricewaterhouse Coopers LLP and KPMG in India and the Middle East. Mr. Shah received a Bachelor of Commerce degree from Ranchi University in India. He is a Chartered Accountant from the Institute of Chartered Accountants in India and has an MBA from W.P. Carey School of Business at Arizona State University.
Christopher M. Chipman, CPASheila A. Mathias, PhD, JD became our Chief FinancialScientific Officer in May 2020. Mr. ChipmanApril 2021. Dr. Mathias has more than 20 years of publicleadership experience in the pharmaceutical industry accelerating drug development. She brings extensive global regulatory affairs strategic guidance and private accounting experience.clinical development experience having worked across a range of therapeutic areas, including pain management, addiction medicine, and dermatology. This experience has spanned across big pharma, mid-sized, to start-up biotechnology companies. Most recently, she held the position Senior Director Global Regulatory Affairs at Sun Pharma Advanced Research Company from 2018 to 2021. Since November 2000, Mr. Chipman2018 she has been a managing memberserved on the Advisory Board for Tennessee State University Department of Chipman & Chipman, LLC, a consulting firm that assists public companies withBiology. Dr. Mathias has held increasing roles of responsibility, entering the preparationpharmaceutical industry at Merck US Human Health in the position of periodic reports requiredMedical Science Liaison. Dr. Mathias transitioned into Regulatory Affairs at Aventis Pharmaceuticals and has successfully brought multiple products through regulatory approval. Her experience includes roles at Braeburn Pharmaceuticals from 2015 to be filed with the Securities2018, Otsuka Pharmaceuticals from 2013 to 2018, Actelion Clinical Research from 2012 to 2013, Cephalon from 2007 to 2011, Novartis Pharmaceuticals from 2005 to 2007 and Exchange Commission and compliance with Section 404 of the Sarbanes Oxley Act of 2002. Mr. Chipman is a CPA and was Chief Financial Officer and Secretary of Capital Gold CorporationAventis Pharmaceuticals from March 20062002 to June 2011. Capital Gold Corporation was a publicly held gold production and exploration company, until its acquisition by AuRico Gold, Inc. (formerly, Gammon Gold). From July 1996 to August 1998, Mr. Chipman was a senior accountant with the accounting firm of Grant Thornton LLP and from August 1998 to March 2000 he was a Senior Financial Analyst for GlaxoSmithKline. Prior to that, from July 1994 to July 1996, he was an Audit Examiner for Wells Fargo Corporation. Mr. Chipman2005. Dr. Mathias received a B.A.B.S in EconomicsZoology from UrsinusHoward University, a PhD in Neurophysiology from Meharry Medical College, in 1994.an executive MBA from Saint Joseph’s University, and a JD from Northwestern California University School of Law.
Directors
Eric Floyd, PhD became a director in January 2017. Dr. Floyd was appointed as the Chairman of our Board of Directors effective November 20, 2023. Dr. Floyd currently serves as Chief Regulatory Officer at Neurogene Inc. He has nearly 21 years of regulatory experience within the pharmaceutical industry. Most recently, from November 2018 to December 2019 he was Senior Vice President, Regulatory Affairs, for Axovant Sciences. Prior to that, he served as President of Compliance Services and Chief Scientific Officer at Dohmen Life Science Services, Inc. from June 2015, Senior Vice President, U.S. Regulatory Affairs and Clinical Quality Compliance at Lundbeck Inc. December 2011, Global Vice President of Regulatory Affairs at Hospira Inc. (later acquired by Pfizer Inc.) from January 2010, Vice President of Worldwide Regulatory Affairs and Quality Assurance at Cephalon Inc. (later acquired by Teva Pharmaceuticals Industries Ltd.) from January 2007 and VP and Global Head of Respiratory, Dermatology, and Tropical Medicines Drug Regulatory Affairs at Novartis AG from February 2005. Dr. Floyd has also held senior leadership roles at Bristol Myers Squibb Co., Aventis Pharma and Merck Research Laboratories (a division of Merck & Co.). Dr. Floyd received a Ph.D. in Neurophysiology from Meharry Medical College, Nashville, an executive MBA from St. Joseph’s University, Philadelphia, an MS from Tennessee State University, a BS from the University of Illinois and has served as an Assistant Professor at Harvard University School of Medicine. Dr. Floyd served as an outside director on the board of directors of Scilex Pharmaceuticals Inc. from April 2014 to November 2016. Dr. Floyd was selected as a director due to his extensive experience at pharmaceutical companies and knowledge of the pharmaceutical industry.
Jerrold Sendrow, CFP became a director in January 2017. Mr. Sendrow has been a Certified Financial Planner since 1986 and continues to maintain his practice. Mr. Sendrow also served as an outside Director on the board of directors of SCILEX Pharmaceuticals Inc. from April 2014 to November 2016. Prior to that, Mr. Sendrow was an accountant in the audit departments of Touche Ross & Co. and Peat Marwick Mitchell & Co after returning from military service of two tours in the Vietnam conflict. Mr. Sendrow holds business degrees from Bernard Baruch College of the City University of New York and Adelphi University. Mr. Sendrow was selected as a director due to his leadership experience at other growth-stage companies and his financial accounting experience.
Gerald W. Bruce became a director in July 2021 and an Executive Vice President and our Commercial Operations Officer in August 2017. Mr. Bruce has spent over 30 years, including 20 years in senior leadership roles, in the Pharmaceutical and Medical Nutrition industry. He started his career in May 1983 at Johnson & Johnson Inc. (NYSE: JNJ) where he was an award-winning sales representative and held leadership positions of increasing responsibility in sales and marketing ending with his role as Group Product Director of Analgesics in September 1998. From September 1998 to November 2000, he served as Vice President of Sales at Bristol-Myers Squibb Co. (NYSE: BMY) where he led the Cardiovascular and Metabolic sales force. From November 2000 to January 2006 he served as Vice President of Managed Markets where he led the team responsible for the development and implementation of the reimbursement strategy for Bristol-Myers Squibb’s US portfolio. From January 2006 to June 2008, Mr. Bruce was the Senior Vice President of Commercial Operations at NitroMed, Inc. where he was responsible for building the commercial strategy and led the team responsible for the development and implementation of the commercial plan for the start-up company’s first product for the treatment of heart failure. From April 2009 to November 2018, Mr. Bruce served as Vice President of Sales for Nutricia North America, Danone Medical Nutrition Division. Mr. Bruce currently serves on the Board of Trustees for Lincoln University and is a Board member for the National Sales Network. He received his bachelor’s degree in Business Administration from Lincoln University and a master’s degree in Leadership from the McDonough School of Business at Georgetown University. Mr. Bruce was selected as a director due to his extensive experience at pharmaceutical companies and knowledge of the pharmaceutical industry.
79
Thani Jambulingam, PhD became a director in January 2017. Dr. Jambulingam is a Pfizer Fellow and Professor in the Department of Pharmaceutical and Healthcare Marketing at St Joseph’s University, Erivan K. Haub School of Business, in Philadelphia, Pennsylvania. He teaches in the executive MBA program for biopharmaceutical, medical device and physician executives. Dr. Jambulingam served as the chair of the department for eight years, from June 2003 to June 2010. Dr. Jambulingam’s research is focused on pharmaceutical and healthcare strategy and innovation. His research is regularly published in marketing and management journals. Dr. Jambulingam has also served as a consultant and facilitated training sessions in innovation and strategy for senior leadership and/or brand teams within several small, mid and large pharma and healthcare firms including Alkermes Plc, Abbott Industries, AstraZeneca plc, Cardinal Health, FMC, IQVIA, Lancaster General Hospital, Inspira Health, Lehigh Valley Health Network, Leo Pharma, Merck & Co., Novo Nordisk, Pfizer Inc., Sanofi, Solvay and Procter & Gamble Inc. During his sabbatical from Saint Joseph’s University, from July 2011 to August 2012, he joined Pfizer Inc. (NYSE: PFE) with the Prevenar Global Commercial Team contributing to development of Prevenar franchise positioning, healthy aging platform development, vaccine business strategy for emerging markets, pediatric expanded age strategy (life cycle management) and conducted strategy sessions for executive leadership within the specialty care division of Pfizer. Over the years, Dr. Jambulingam has successfully mentored several entrepreneurs in the life sciences industry. Dr. Jambulingam is a pharmacist and obtained his Ph.D. from the University of Wisconsin-Madison. Dr. Jambulingam completed the case method of teaching at Harvard. He has been inducted to the Rho Chi, the honor society in pharmacy and Beta Gamma Sigma, the honor society for business. In 2021, Dr. Jambuligam received the Tangelmann Award for lifetime excellence in research and teaching. For the past seven years, Dr. Jambulingam has been a faculty member conducting Bio-Entrepreneurship Bootcamp at the annual meeting at the Biotechnology Industry Organization (BIO). Dr. Jambulingam is also a visiting professor in the Wharton MBA Global program and teaches healthcare courses in India. Dr. Jambulingam was selected as a director due to his leadership experience at other companiesexpertise in innovation and strategy and his extensive knowledge of the pharmaceutical industry.
Vanila M. Singh, MD became a director in June 2020. From June 2017 to July 2019, Dr. Singh is the former Chief Medical Officer of the U.S. Department of Health and Human Services, where she served as the Chairperson of the highly regarded HHS Pain and Opioid Task Force in conjunction with the Department of Defense and the Veterans Administration. Since November 2019, Dr. Singh has been a director of Biodelivery Sciences International, Inc. (NASDAQ: BDSI), and since June 2004, Dr. Singh has been a clinical associate professor of Anesthesiology, Pain and Peri-operative Medicine at Stanford University and is a teaching mentor at Walter Reed National Military Medical Center. For over ten years, Dr. Singh served on medical ethics as well as on scientific editorial boards, committees for the American Society of Regional Anesthesia, American Society of Interventional Pain Physicians, California Medical Association, and the Santa Clara County Medical Association. Dr. Singh, who is double board-certified in pain and anesthesiology, focuses her practice on regional anesthesia and peri-operative, subacute, and the development of chronic pain, with an appreciation for complimentary and traditional medicine approaches that emphasize an individualized patient-centered approach. Dr. Singh received her medical degree from George Washington University Medical School and her B.A. from U.C. Berkeley in Molecular and Cell Biology and Economics. Dr. Singh was selected as a director due to her leadership experience and her extensive knowledge of the pharmaceutical industry and the regulatory environment.
Michael F. Dubin, CPA became a director in July 2021. From 2001 to 2016, Mr. Dubin held the title of Managing Partner, PA/SNJ Offices, with RSMUS LLP (RSM), a professional services company. He was presented with RSM’s “National Achievement Award” in 2010 and was a finalist for the company’s “National Integrity Award.” Prior to 2001, Mr. Dubin served as an audit partner for a regional accounting firm and a national accounting firm. Mr. Dubin obtained a BS in Economics (magna cum laude) from the Wharton School of Business, University of Pennsylvania. He served as a Board Member for RSM for four years. Mr. Dubin was also a board member and the Audit Committee Chairman for a privately held business in Philadelphia engaged in supplying energy efficiency services and facilities, and is a board member and the Risk Management Committee Chairman for a commercial bank in Pennsylvania and an advisory board member for an accounting firm in Pennsylvania. Mr. Dubin is professionally affiliated with the PICPA and AICPA. He was also an adjunct faculty member and course teacher for the Wharton School of Business for two years and has also been a guest lecturer at the Wharton School of the University of Pennsylvania, Temple University, University of Scranton and Lehigh University. He also served as an expert witness/consultant for the Federal Deposit Insurance Corporation and the Resolution Trust Corporation. Mr. Dubin was selected as a director due to his long history of successextensive leadership experience in public accounting and risk management and exposure to manufacturing, distribution, financial services, business and professional services, pharma, technology, retail and various other industries.
80
Barbara A Ruskin, PhD, JD became a director in March 2023. Dr. Ruskin brings over 25 years of experience in life science intellectual property and corporate law. Since May 2019, she has served as SVP, General Counsel and Chief Patent Officer and since September 2022 as Chief Intellectual Property and Innovation Officer for Silence Therapeutics, a Nasdaq listed international biotechnology company. Prior to that, from September 2017 to February 2019 she served as General Counsel and Chief Patent Officer of Molecular Templates Inc. Prior to holding these corporate positions, Dr. Ruskin served as outside counsel for pharmaceutical and biotechnology companies and their investors, including as an IP Corporate Partner at Ropes & Gray LLP and as an associate at Fish & Neave LLP, both in New York City. She has advised, managed and prosecuted worldwide patent portfolios for a number of biotech and pharmaceutical industry clients and has also worked in IP litigation in both the U.S. and Europe. Dr. Ruskin has since April 2007 served as a member of the Board of Directors at St. Jude Children’s GMP LLC (Memphis, TN), and served as Chairman of that Board from March 2017-March 2022. From 2016-2019, Dr. Ruskin also served on the Board of Directors for the Burke Neurological Institute (White Plains, NY). Dr. Ruskin did her post-doctoral research at the Whitehead Institute for Biomedical Research (MIT) and at the Institute for Neuroscience (University of Oregon). She has published numerous scientific articles and given legal presentations that combine her knowledge of biochemistry and law. In addition to her Ph.D. in Biochemistry & Molecular Biology, received from Harvard University, Dr. Ruskin received a B.A. in Biochemistry from the University of California, Berkeley, and her J.D. from Fordham University School of Law. Dr. Ruskin was selected as a director due to her leadership experience in corporate and intellectual property law with publicly listed biotechnology companies and extensive knowledge of the biotech and pharmaceutical industry.
Corporate Governance
Our bylaws delegate the authority over our management and officers to our Board of Directors. The Board of Directors may then delegate management of the Company to committees of the Board of Directors, or such other persons based on its reasonable discretion. Regardless of any delegation, the Board of Directors will remain responsible for the proper management of our affairs. The Board of Directors may create new committees or change the responsibilities of existing committees from time to time.
Board Structure and Committee Composition
Our business and affairs are managed under the direction of our Board of Directors. Our Board of Directors currently consists of eight directors. The Certificate of Incorporation provides that our Board of Directors shall consist of at least one director but not more than nine directors and that the number of directors may be fixed from time to time by resolution of our Board of Directors.
In accordance with the terms of the Certificate of Incorporation and bylaws, our Board of Directors is divided into three classes, Class I, Class II and Class III, with each class serving staggered three-year terms. Upon the expiration of the term of a class of directors, directors in that class are eligible to be elected for a new three-year term at the annual meeting of stockholders in the year in which their term expires. Our directors are divided among the three classes as follows:
| ● | The Class I directors are Mr. Bruce, Dr. Singh and Dr. |
| ● | The Class II directors are Dr. Jambulingam, Mr. Sendrow and Mr. Dubin; their terms will expire at the |
| ● | The Class III directors are |
We expect that any additional directorships resulting from an increase in the number of directors will be distributed among the three classes so that, as nearly as possible, each class will consist of one-third of the directors. The division of our Board of Directors into three classes with staggered three-year terms may delay or prevent a change of our management or a change in control.
Under our Certificate of Incorporation, directors have the authority to appoint one or more directors to our Board of Directors, subject to the maximum number of directors allowed for in our Certificate of Incorporation. A vacancy on our Board of Directors may be filled by the remaining directors and any director so appointed will hold office until our next annual general meeting. During any vacancy on our Board of Directors, the remaining directors will have full power to act as the board.
We have an Audit Committee, a Compensation Committee, a Nominating and Corporate Governance Committee and a Science and Technology Committee with the composition and responsibilities described below. Each committee operates under a written charter that has been approved by our Board of Directors.Directors, the full text of which is available on our website at www.virpaxpharma.com. The members of each committee are appointed by the Board of Directors and serve until their successor is elected and qualified unless they are earlier removed or resign.resign earlier. In addition, from time to time, special committees may be established under the direction of the Board of Directors when necessary to address specific issues.
Audit Committee
Our Audit Committee is comprised of Mr. Dubin, Mr. Sendrow, Dr. Floyd, and Dr. Jambulingam, with Mr. Sendrow serving as Chairman of the audit committee. Our Board of Directors has determined that each member of the Audit Committee meets the independence requirements of Rule 10A-3 under the Exchange Act and the applicable rules of the Nasdaq Capital Market. Our Board of Directors has determined that Mr. Dubin is an “audit committee financial expert” within the meaning of SEC regulations and the applicable rules of the Nasdaq Capital Market. The Audit Committee’s responsibilities include:
| ● | appointing, approving the compensation of, and assessing the qualifications, performance and independence of our independent registered public accounting firm, and in particular the provision of additional services to each entity covered by the committee; |
81
| ● | pre-approving audit and permissible non-audit services, and the terms of such services, to be provided by our independent registered public accounting firm; |
| ● | reviewing and discussing with management and the independent registered public accounting firm our annual and quarterly financial statements and related disclosures as well as critical accounting policies and practices used by us; |
| ● | monitoring the audit of our financial statements; |
| ● | setting policies for our hiring of employees or former employees of our independent registered public accounting firm; |
| ● | reviewing our significant risks or exposures and assessing the steps that management has taken or should take to monitor and minimize such risks or exposures; |
| ● | reviewing the adequacy of our internal control over financial reporting, including information system controls and security; |
| ● | monitoring the effectiveness of our systems of internal control, internal audit and risk management for each entity covered by the committee; |
| ● | establishing policies and procedures for the receipt and retention of accounting-related complaints and concerns; |
| ● | recommending, based upon the audit committee’s review and discussions with management and the independent registered public accounting firm, whether our audited financial statements shall be included in our Annual Report on Form 10-K; |
| ● | monitoring our compliance with legal and regulatory requirements as they relate to our financial statements and accounting matters; |
| ● | preparing the audit committee report required by the rules of the SEC to be included in our annual proxy statement; |
| ● | reviewing all related party transactions for potential conflict of interest situations and approving all such transactions; and |
| ● | reviewing and discussing with management and our independent registered public accounting firm our earnings releases and scripts. |
Compensation Committee
Our Compensation Committee is composed of Dr. Floyd, Mr. Sendrow, and Dr. Jambulingam, with Dr. Floyd serving as Chairman of the committee. Our Board of Directors has determined that each director serving on the Compensation Committee is “independent” in accordance with Rule 10C-1 under the Exchange Act and as defined under the applicable listing standards of the Nasdaq Capital Market. Further, the Board of Directors has determined that the directors serving on the Compensation Committee are “non-employee directors” as defined in rule 16b-3 promulgated under the Exchange Act and are “outside directors” as that term is defined in Section 162(m) of the Internal Revenue Code of 1986, as amended. The compensation committee’s responsibilities include:
| ● | reviewing and approving corporate goals and objectives relevant to the compensation of our chief executive officer, the officers who report directly to the chief executive officer and all officers who are “insiders” subject to Section 16 of the Exchange Act; |
82
| ● | evaluating the performance of our chief executive officer and such other officers in light of such corporate goals and objectives and determining and approving, or recommending to our Board of Directors for approval, the compensation of our chief executive officer and such other officers; |
| ● | appointing, compensating and overseeing the work of any compensation consultant, legal counsel or other advisor retained by the compensation committee; |
| ● | conducting the independence assessment outlined in the listing standards of the Nasdaq Capital Market with respect to any compensation consultant, legal counsel or other advisor retained by the compensation committee; |
| ● | annually reviewing and reassessing the adequacy of the committee charter; |
| ● | reviewing and establishing our overall management compensation and our compensation philosophy and policy; |
| ● | overseeing and administering our equity compensation and other compensatory plans; |
| ● | reviewing and approving our equity and incentive policies and procedures for the grant of equity-based awards and approving the grant of such equity-based awards; |
| ● | reviewing and making recommendations to our Board of Directors with respect to non-employee director compensation; and |
| ● | producing a report, if required, on executive compensation to be included in our annual proxy statement or Annual Report on Form 10-K. |
Nominating and Corporate Governance Committee
Our nominating and corporate governance committee will beis composed of Dr. Jambulingam, Mr. Sendrow, and Dr. Floyd, with Dr. Jambulingam serving as Chairman of the committee. Our Board of Directors has determined that Dr. Jambulingameach director serving on the Nominating and Corporate Governance Committee is “independent” as defined in the applicable rules of the Nasdaq Capital Market. The nominating and corporate governance committee’s responsibilities include:
| ● | establishing procedures for identifying and evaluating board of director candidates, including nominees recommended by stockholder; |
| ● | identifying individuals qualified to become members of our Board of Directors; |
| ● | recommending to our Board of Directors the persons to be nominated for election as directors and to each of our board’s committees; |
| ● | developing and recommending to our Board of Directors a set of corporate governance principles; |
| ● | articulating to each director what is expected, including reference to the corporate governance principles and directors’ duties and responsibilities; |
| ● | reviewing and recommending to our Board of Directors’ practices and policies with respect to directors; |
| ● | reviewing and recommending to our Board of Directors the functions and duties relative to corporate governance and the composition of the committees of our Board of Directors; |
| ● | reviewing and assessing the adequacy of the committee charter and submitting any changes to our Board of Directors for approval; |
83
| ● | considering and reporting to our Board of Directors any questions of possible conflicts of interest of Board of Directors’ members; |
| ● | providing for new director orientation and continuing education for existing directors on a periodic basis; |
| ● | performing an evaluation of the performance of the committee; and |
| ● | overseeing the evaluation of our Board of Directors. |
Science and Technology Committee
Our Science and Technology Committee is comprised of Dr. Floyd, Dr. Jambulingam, Dr. Singh and Dr. Jambulingam,Ruskin, with Dr. Floyd serving as the Chairman of the committee. Our Science and Technology Committee is responsible for, among other things:
| ● | periodically examining management’s strategic direction and investment in our biopharmaceutical research and development and technology initiatives; |
| ● | identifying and discussing significant emerging science and technology issues and trends; |
| ● | evaluating the soundness/risks associated with the technologies in which we are investing our research and development efforts; and |
| ● | periodically reviewing our overall patent strategies. |
Special Litigation Committee
Our Special Litigation Committee is comprised of Dr. Floyd, Dr. Jambulingam, Dr. Singh, Dr. Ruskin, Dr. Gudin, Mr. Sendrow and Mr. Dubin with Dr. Floyd serving as the Chairman of the committee. Our Special Litigation Committee was formed for the purpose of making decisions related to the litigation, the details of which are more fully disclosed under the sections titled “Risk Factors” and “Business—Recent Developments” elsewhere in this prospectus.
Code of Business Conduct and Ethics
We have adopted written code of business conduct and ethics (“Code of Ethics”) that applies to our directors, officers and employees, including our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions. The Code of Ethics is available on our website at www.virpaxpharma.com/www.virpaxpharma.com. In addition, we intend to post on our website all disclosures that are required by law or the Nasdaq Capital Market rules concerning any amendments to, or waivers from, any provision of the Code of Ethics. The reference to our website address does not constitute incorporation by reference of the information contained at or available through our website, and you should not consider it to be a part of this prospectus.
Legal Proceedings
See the disclosure under Item 3: Legal Proceedings above for a discussion of the Complaint naming Mr. Mack and the Company as defendants.
ITEM 11. EXECUTIVE COMPENSATION
As an emerging growth company under the JOBS Act we have opted to comply with the executive compensation disclosure rules applicable to “smaller reporting companies,” which require compensation disclosure for each of our principal executive officer andofficers serving during the most recently completed fiscal year, the two most highly compensated executive officers (other than our principal executive officer) serving as executive officers at the end of our most recently completed fiscal year and up to two additional individuals for whom disclosure would have been provided but for the fact that the individual was not serving as an executive officer of the company at the end of the most recently completed fiscal year (collectively, our “Named Executive Officers”). for services rendered in all capacities to us for the years ended December 31, 2023 and December 31, 2022. This section describes the executive compensation program in place for our Named Executive Officers during the year ended December 31, 2022, who are the individuals who served as our principal executive officer and two most highly compensated executive officers.2023.
This section discusses the material components of the executive compensation program for our executive officers who are named in the “Summary Compensation Table” below and the non-employee members of our Board of Directors. In 2022,2023, our “Named Executive Officers” and their positions were:
| ● | Gerald Bruce, our Chief Executive Officer; | |
| ● | Anthony Mack, our Former Chief Executive Officer and Former Chairman of the Board of Directors; |
| ● |
| ● | Vinay Shah, our Chief Financial Officer; and | |
| ● | Christopher |
84
This discussion may contain forward-looking statements that are based on our current plans, considerations, expectations and determinations regarding future compensation programs.
2022
2023 Summary Compensation Table
The following table sets forth information concerning the compensation of our Named Executive Officers for the years ended December 31, 20222023 and 2021:2022:
| Name & Principal Position | Year | Salary | Salary/ Bonus | Stock Awards | Option Awards | Total | |||||||||||||||||
| Anthony Mack | 2022 | $ | 467,000 | $ | 238,000 | $ | — | $ | 79,000 | (1) | $ | 784,000 | |||||||||||
| Chief Executive Officer, Chairman | 2021 | $ | 375,000 | $ | 225,000 | $ | — | $ | 158,000 | (1) | $ | 758,000 | |||||||||||
| Jeffrey Gudin, MD | 2022 | $ | 157,000 | $ | 47,000 | $ | — | $ | 81,000 | (1) | $ | 285,000 | |||||||||||
| Executive VP, Chief Medical Officer | 2021 | $ | 100,000 | $ | 45,000 | $ | — | $ | 158,000 | (1) | $ | 303,000 | |||||||||||
| Christopher Chipman | 2022 | $ | 296,000 | $ | 90,000 | $ | — | $ | 81,000 | (1) | $ | 467,000 | |||||||||||
| Chief Financial Officer(2) | 2021 | $ | 236,000 | $ | 100,000 | $ | — | $ | 79,000 | (1) | $ | 415,000 | |||||||||||
| Name & Principal Position | Year | Salary | Bonus | Option Awards (1) | All Other Compensation | Total | |||||||||||||||||
| Gerald W. Bruce(2) | 2023 | $ | 38,000 | $ | 29,000 | $ | 61,000 | $ | - | $ | 128,000 | ||||||||||||
| Chief Executive Officer | |||||||||||||||||||||||
| Anthony Mack | 2023 | $ | 445,000 | $ | - | $ | 60,000 | $ | 711,000 | $ | 1,216,000 | ||||||||||||
| Former Chief Executive Officer, Chairman(3) | 2022 | $ | 467,000 | $ | 238,000 | $ | 79,000 | $ | - | $ | 784,000 | ||||||||||||
| Sheila A. Mathias PhD, JD | 2023 | $ | 308,000 | $ | 115,000 | $ | 75,000 | $ | - | $ | 498,000 | ||||||||||||
| Chief Scientific Officer | |||||||||||||||||||||||
| Vinay Shah(4) | 2023 | $ | 123,000 | $ | 115,000 | $ | 86,000 | $ | - | $ | 324,000 | ||||||||||||
| Chief Financial Officer | |||||||||||||||||||||||
| Christopher Chipman | 2023 | $ | 169,000 | $ | - | $ | 75,000 | $ | 247,000 | $ | 491,000 | ||||||||||||
| Former Chief Financial Officer(5) | 2022 | $ | 296,000 | $ | 90,000 | $ | 81,000 | $ | - | $ | 467,000 | ||||||||||||
| (1) | Amounts reflect the full grant date fair value of stock options granted during the years ended December 31, |
| (2) | Mr. Gerald Bruce was appointed as our Chief Executive Officer effective as of November 20, 2023. Amounts included in the salary and bonus columns reflect compensation received as Chief Executive Officer between November 20, 2023 and December 31, 2023. Prior to being appointed as Chief Executive Officer, Mr. Bruce received equity compensation as a consultant serving as our Executive Vice President of Commercial Operations. |
| (3) | Mr. Mack resigned as our Chief Executive Officer effective November 17, 2023. The amount reflected in all Other Compensation represents estimated separation expense of $711,000. Refer to Note 5- Commitments and Contingencies to our consolidated financial statements included in this Annual Report on Form 10-K. |
| (4) | Mr. Shah was appointed as our Chief Financial Officer effective as of June 20, 2023. Amount reflects compensation received as Chief Financial Officer between June 20, 2023 and December 31, 2023. |
| (5) | Mr. Chipman resigned as our Chief Financial Officer effective June 30, 2023. Amounts reflect |
Our Board of Directors, in consultation with our Compensation Committee, annually reviews the compensation paid to our Named Executive Officers to assess the adequacy of the compensation paid to our Named Executive Officers. These annual assessments are done in order to periodically align our compensation practices with what our Board of Directors believe to be compensation levels more commensurate with companies of similar size and development stage as the Company. Pursuant to these annual assessments, on January 29, 2024, the Compensation Committee approved cash bonus awards for their 2023 performance to Mr. Bruce of $29,000 and Mr. Shah and Dr. Mathias of $115,000 each and equity awards of an option to purchase 12,500 shares of our Common Stock to Mr. Bruce and an equity award of an option to purchase 11,200 shares of our Common Stock to each of Mr. Shah and Dr. Mathias. All of the grants were made under the 2022 Plan. The options have an exercise price of $3.18 per share, the fair market value of the Common Stock on the date of grant and vests annually over three years commencing one year after the date of grant, as adjusted pursuant to the 1 to 10 reverse stock split effective March 1, 2024.
On January 25, 2023, the Compensation Committee approved cash bonus awards and increases to the base salaries for each of our Named Executive Officers based upon the Company’s and management’s performance in 2022. Based on these assessments, Mr. Mack Mr. Chipman and Mr. GudinChipman were awarded bonuses of $238,000 $90,000, and $47,000,$90,000, respectively. In addition, Mr. Mack’s base salary was increased to $494,000 per year and Mr. Chipman’s base salary was increased to $312,000 per year, and Dr. Gudin’s base salary was increased to $164,000 per year.
On January 31, 2022, based upon the recommendation of the Compensation Committee, our Board of Directors approved increases to the base salaries paid to our Named Executive Officers and approved cash bonus awards for each of our Named Executive Officers based upon the Company’s and management’s performance in 2021 . Based on these assessments, Mr. Mack, Mr. Chipman and Mr. Gudin were awarded bonuses of $225,000, $100,000 and $45,000, respectively. In addition, pursuant to the employment agreements with each of our Named Executive Officers, Mr. Mack’s base salary was increased to $475,000 per year, Mr. Chipman’s base salary was increased to $300,000 per year, and Dr. Gudin’s base salary was increased to $157,500 per year.
85
Compensation Arrangements with our Named Executive Officers
Mr. Mack.Bruce
Effective November 20, 2023, Mr. Bruce was appointed to serve as our Chief Executive Officer.
On December 6, 2023, we entered into an employment agreement with Mr. Bruce (the “Bruce Employment Agreement”). The term of the Bruce Employment Agreement initiated upon the commencement of the agreement and terminates upon either death, Disability, for Cause, for Good Reason (as such terms are defined in the Bruce Employment Agreement), or for other reasons by us or Mr. Bruce. Under the Bruce Employment Agreement, Mr. Bruce will be paid an annual base salary of $500,000 subject to annual increases at the discretion of the Board and will be eligible for an annual bonus in an amount up to 50% of his base salary, pro-rated for 2023, which will be awarded by the Board in its sole discretion based on the achievement of Company and personal performance metrics established by the Board on an annual basis. To receive any bonus, Mr. Bruce must be employed by the Company at the time of payment.
If Mr. Bruce’s employment is terminated in the event of Disability or death, the Company would have no further obligations under the Bruce Employment Agreement, except for any Accrued Obligations (as defined in the Bruce Employment Agreement) and any portion of an earned annual bonus which remains unpaid at the time of termination. If the Company terminates Mr. Bruce’s employment for Cause, the Company would have no further obligation under the Bruce Employment Agreement, except for any Accrued Obligations due. If the Company’s terminates Mr. Bruce’s employment other than for Disability or for Cause, in addition to any Accrued Obligations due, subject to Mr. Bruce executing a release, Mr. Bruce would be entitled to receive (i) severance payments in an amount equal to Mr. Bruce’s base salary for a period of twelve months after the effective date of the termination; (ii) reimbursement of medical insurance premiums until the earlier of (1) twelve months or (2) the date Mr. Bruce becomes eligible for medical benefits through another employer, subject to certain conditions; (iii) if vesting shall not have accelerated under the equity awards then held by Mr. Bruce, the Company will accelerate the vesting of the number of shares subject to options that would have vested in the twelve (12) month period after his separation, such that, effective as immediately prior to the separation date, he will be considered to have vested in all options granted to him through, and no later than twelve (12) months following the date of the separation; and (iv) effective as immediately prior to the separation date, the Company shall extend the period of time for Mr. Bruce to exercise any vested shares subject to options until the earlier of (1) the expiration date of the applicable option, or (2) twelve (12) months after his separation date.
If Mr. Bruce terminates his employment for Good Reason, Mr. Bruce would be entitled to receive the same payments and benefits on the same terms and conditions as would be applicable upon termination by the Company other than for Disability or for Cause.
Notwithstanding the above description, if Mr. Bruce’s employment is terminated by Mr. Bruce for Good Reason or by the Company without Cause (other than on account of Mr. Bruce’s death or Disability), in each case within twelve months following a Change in Control (as defined in the Bruce Employment Agreement), Mr. Bruce will be entitled to receive any Accrued Obligations due and, subject to Mr. Bruce’s compliance with the terms of the Bruce Employment Agreement and Mr. Bruce’s execution of a release, the following: (i) a lump sum payment equal to two times the sum of Mr. Bruce’s base salary for the year in which the termination date occurs (or if greater, the year immediately preceding the year in which the Change in Control occurs), (ii) a lump sum payment equal to two times the sum of Mr. Bruce’s cash bonus for the calendar year in which the termination date occurs (or if greater, the year in which the Change in Control occurs), and (iii) accelerated vesting of any award granted to Mr. Bruce under the 2022 Plan.
Prior to his appointment as our Chief Executive Officer, Mr. Bruce served as our Executive Vice President, Commercial Operations pursuant to the terms of a consulting agreement that we entered into with him on April 25, 2023. For his services Mr. Bruce was issued stock options to purchase up to 10,000 shares of our Common Stock upon execution of the consulting agreement.
Mr. Shah
Effective June 20, 2023, we entered into an employment agreement with Vinay Shah (the “Shah Employment Agreement”). The term of the Shah Employment Agreement initiated upon the commencement of the agreement and terminates upon either death, Disability, for Cause, for Good Reason (as such terms are defined in the Shah Employment Agreement), or for other reasons by us or Mr. Shah. The Shah Employment Agreement provides for Mr. Shah to serve as the Company’s Chief Financial Officer reporting to the Company’s Chief Executive Officer and provides for an annual base salary of $312,000, subject to annual increases at the discretion of the Board of Directors. Under the Shah Employment Agreement, Mr. Shah is eligible for an annual bonus with a target amount equal to 30% of his base salary which will not be pro rated for the first year, which will be awarded by our Board in its sole discretion based on the achievement of the Company and Mr. Shah of corporate and personal performance metrics established by the Board on an annual basis. To receive any bonus, Mr. Shah must be employed by the Company at the time of payment. Mr. Shah may also receive, in the discretion of the Board, equity awards under the 2022 Plan, or any other equity incentive plan that the Company may adopt in the future. Mr. Shah is also eligible to participate in all vacation and other fringe benefit programs of the Company to the extent and on the same terms and conditions as are accorded to other senior management employees of the Company. On June 20, 2023, Mr. Shah was awarded an option to purchase up to 10,000 shares of the Company’s common stock, 25% vesting after 12 months of his continuous services and the remaining 75% vesting in equal monthly installments over the next 24 months.
The Company may terminate the Shah Employment Agreement upon written notice to Mr. Shah in the event of Disability (as defined in the Shah Employment Agreement), in which event the Company would have no further obligations under the Shah Employment Agreement, except for any Accrued Obligations (as defined in the Shah Employment Agreement) and any portion of an earned annual bonus which remains unpaid at the time of termination. The Company may also terminate the Shah Employment Agreement for Cause (as defined in the Shah Employment Agreement) immediately upon providing written notice of such termination to Mr. Shah. If the Company terminates the Shah Employment Agreement for Cause, the Company would have no further obligation under the Shah Employment Agreement, except for any Accrued Obligations due. The Company may terminate the Shah Employment Agreement other than with respect to a Disability or for Cause immediately upon written notice of termination to Mr. Shah and if it does so subject to the Company’s receipt of a release, in addition to any Accrued Obligations due, Mr. Shah is entitled to receive (i) severance payments in an amount equal to Mr. Shah’s base salary for a period of twelve months after the effective date of the termination and (ii) reimbursement of medical insurance premiums until the earlier of (1) twelve months or (2) the date Mr. Shah becomes eligible for medical benefits through another employer, subject to certain conditions.
Mr. Shah may terminate his agreement for Good Reason (as defined in the Shah Employment Agreement) upon providing written notice of such termination to us. If Mr. Shah terminates his employment for Good Reason, Mr. Shah will be entitled to receive the same payments and benefits on the same terms and conditions as would be applicable upon termination by the Company other than for Disability or for Cause.
If the Shah Employment Agreement is terminated by Mr. Shah for Good Reason or by us without Cause (other than on account of Mr. Shah’s death or Disability), subject to the Company’s receipt of a release in each case within twelve months following a Change in Control (as defined in the Shah Employment Agreement), Mr. Shah will be entitled to receive the Accrued Obligations and, subject to Mr. Shah’s compliance with the terms of the Shah Employment Agreement, Mr. Shah will be entitled to receive the following: (i) a lump sum payment equal to two times the sum of Mr. Shah’s base salary for the year in which the termination date occurs (or if greater, the year immediately preceding the year in which the Change in Control occurs), (ii) a lump sum payment equal to two times the sum of Mr. Shah’s cash bonus for the calendar year in which the termination date occurs (or if greater, the year in which the Change in Control occurs), and (iii) accelerated vesting of any award granted to Mr. Shah under the 2022 Plan.
In connection with his entry into the Shah Employment Agreement, Mr. Shah entered into a customary Confidential Disclosure Invention Assignment Agreements with the Company.
Dr. Mathias
On April 7, 2021, we entered into an employment agreement with Sheila Mathias, (the “Mathias Employment Agreement”). The Mathias Employment Agreement provides for Dr. Mathias to serve as the Company’s Chief Scientific Officer reporting to the Company’s Chief Executive Officer and provided for an initial annual base salary of $250,000 which was increased to $312,000 on January 25, 2023 and is subject to annual increases at the discretion of the Board. Under the Mathias Employment Agreement, Dr. Mathias is eligible for an annual bonus with a target amount equal to 30% of her base salary, awarded by our Board in its sole discretion based on the achievement of the Company and Dr. Mathias of corporate and personal performance metrics established by the Board on an annual basis. To receive any bonus, Dr. Mathias must be employed by the Company at the time of payment. Dr. Mathias is also eligible to receive, in the discretion of the Board, equity awards under the 2022 Plan, or any other equity incentive plan that the Company adopted. Dr. Mathias is also eligible to receive other customary benefits described in the Mathias Employment Agreement. The Mathias Employment Agreement shall automatically terminate effective as of the date of Dr. Mathias’ death, or immediately upon written notice to Dr. Mathias in the event of Disability (as defined in the Mathias Employment Agreement), in which event the Company would have no further obligations under the Mathias Employment Agreement, except for any Accrued Obligations (as defined in the Mathias Employment Agreement) and any portion of an earned annual bonus which remains unpaid at the time of termination. We also may terminate the Mathias Employment Agreement for Cause (as defined in the Mathias Employment Agreement) immediately upon providing written notice of such termination to Dr. Mathias. If we terminate the Mathias Employment Agreement for Cause, we would have no further obligation under the Mathias Employment Agreement, except for any Accrued Obligations due. We could terminate the Mathias Employment Agreement without Cause immediately upon written notice of termination to Dr. Mathias. If we terminate the Mathias Employment Agreement without Cause, in addition to any Accrued Obligations due, Dr. Mathias is entitled to receive (i) severance payments in an amount equal to Dr. Mathias’ base salary for a period of six months after the effective date of the termination and (ii) reimbursement of medical insurance premiums until the earlier of (1) six months or (2) the date Dr. Mathias becomes eligible for medical benefits through another employer, subject to certain conditions.
Dr. Mathias may terminate her agreement for Good Reason (as defined in the Mathias Employment Agreement) upon providing written notice of such termination to us. If Dr. Mathias terminates her employment for Good Reason, Dr. Mathias would be entitled to receive the same payments and benefits on the same terms and conditions as would have been applicable upon termination by us without Cause.
If the Mathias Employment Agreement is terminated by Dr. Mathias for Good Reason or by us without Cause (other than on account of Dr. Mathias’ death or Disability), in each case within twelve months following a Change in Control (as defined in the Mathias Employment Agreement), Dr. Mathias shall be entitled to receive the Accrued Obligations and, subject to Dr. Mathias’ compliance with the terms of the Mathias Employment Agreement, shall be entitled to receive the following: (i) a lump sum payment equal to two times the sum of Dr. Mathias’ base salary for the year in which the termination date occurs (or if greater, the year immediately preceding the year in which the Change in Control occurs), (ii) a lump sum payment equal to two times the sum of Dr. Mathias’ cash bonus for the calendar year in which the termination date occurs (or if greater, the year immediately preceding the year in which the Change in Control occurs), and (iii) accelerated vesting of any award granted to Dr. Mathias under the equity incentive plan.
The Mathias Employment Agreement has a term of three years from the effective date and will be extended upon the expiration. In connection with her entry into the Mathias Employment Agreement, Dr. Mathias entered into a customary Confidential Disclosure Invention Assignment Agreements with the Company.
Former Executive Officers
Mr. Mack.
On September 18, 2018, we entered into an employment agreement with Mr. Mack, as amended (the “Mack Employment Agreement”). The term of the Mack Employment Agreement initiated upon the commencement of the agreement and terminates upon either death, disability, for cause, for good reason, or for other reasons by us or Mr. Mack. Under the Mack Employment Agreement, as amended, Mr. Mack iswas initially paid an annual base salary of $375,000 subjectwhich was amended to annual increases at the discretion of the Board of Directors$494,000 on January 25, 2023, and was entitled to an annual performance bonus targeted at an amount equal to 50% of his base salary based on the achievement of our corporate objectives and Mr. Mack’s individual performance metrics, in each case as established by the Board of Directors in consultation with Mr. Mack. Upon the recommendation of the Compensation Committee and in consultation with Mr. Mack, the Board of Directors may awardcould have awarded Mr. Mack an annual bonus in excess of the targeted amount. Mr. Mack’s annual base salary was increased to $494,000 on January 25, 2023. The Mack Employment Agreement may becould have been terminated by us immediately upon written notice to Mr. Mack, or by Mr. Mack upon 30 days’ notice provided to us. Concurrent with the execution of his employment agreement, we and Mr. Mack agreed to an executive confidentiality agreement (the “Executive Confidentiality Agreement”) that contains standard non-disclosure and non-competition provisions. In the event we terminateterminated the Mack Employment Agreement other than for cause, or Mr. Mack terminatesterminated the employment agreement for good reason, we willwould have been required to pay him the then effective base salary for a period of twelve months following the effective date of the termination. However, in the event of such termination, payment of the effective base salary is subject to the execution of a release of claims and the compliance by Mr. Mack with such release and all terms and provisions of the employment agreement and Executive Confidentiality Agreement that survive the termination of Mr. Mack’s employment.
Mr. Mack elected to forgo his salary and defer his compensation through March 2021. Upon the closing of our underwritten public offering in September 2021, we paid to Mr. Mack $1,051,875 in deferred compensation due to him under the Mack Employment Agreement.
Dr. Gudin
On AprilEffective August 15, 2021,2023, we entered into an employment agreement with Dr. Gudin, as amended (the “Gudin Employment Agreement”). The Gudinamendment to the Mack Employment Agreement, provides for Dr. Gudin to continue to serve as our Executive Vice President and Chief Medical Officer reporting to our Chief Executive Officer and provides for an annual base salary of $150,000, subject to annual increases atwhich provided that if the discretion of the Board of Directors. Dr. Gudin’s annual base salary was increased to $164,000 on January 25, 2023. Under the GudinMack Employment Agreement Dr. Gudin is eligible for an annual bonus with a target amount of 30% of his base salary, which will be awarded by our Board of Directors (the “Board”) in its sole discretion based on the achievement of the Company and Dr. Gudin of corporate and personal performance metrics established by the Board on an annual basis. Upon the recommendation of the Compensation Committee and in consultation with Dr. Gudin, the Board of Directors may award Dr. Gudin an annual bonus in excess of the targeted amount. To receive any bonus, Dr. Gudin must be employed by the Company at the time of payment. Dr. Gudin may also receive, in the discretion of the Board, equity awards under the 2022 Plan or any other equity incentive plan that the Company may adopt in the future. Dr. Gudin will also be eligible to receive other customary benefits described in the Gudin Employment Agreement.
We may terminate the Gudin Employment Agreement upon written notice to Dr. Gudin in the event of Disability (as defined in the Gudin Employment Agreement), in which event we would have no further obligations under the Gudin Employment Agreement, except for any Accrued Obligations (as defined in the Gudin Employment Agreement) and any portion of an earned annual bonus which remains unpaid at the time of termination. We may also terminate the Gudin Employment Agreement for Cause (as defined in the Gudin Employment Agreement) immediately upon providing written notice of such termination to Dr. Gudin. If we terminate the Gudin Employment Agreement for Cause, we would have no further obligation under the Gudin Employment Agreement, except for any Accrued Obligations due. We may terminate the Gudin Employment Agreement without Cause immediately upon written notice of termination to Dr. Gudin. If we terminate the Gudin Employment Agreement without Cause, in addition to any Accrued Obligations due, Dr. Gudin is entitled to receive (i) severance payments in an amount equal to Dr. Gudin’s base salary for a period of six months after the effective date of the termination and (ii) reimbursement of medical insurance premiums until the earlier of (1) six months or (2) the date Dr. Gudin becomes eligible for medical benefits through another employer, subject to certain conditions.
Dr. Gudin may terminate his agreement for Good Reason (as defined in the Gudin Employment Agreement) upon providing written notice of such termination to us. If Dr. Gudin terminates his employment for Good Reason, Dr. Gudin will be entitled to receive the same payments and benefits on the same terms and conditions as would be applicable upon termination by us without Cause.
86
If the Gudin Employment Agreement iswas terminated by Dr. GudinMr. Mack for Good Reason or by us without Cause (other than on account of Dr. Gudin’sMr. Mack’s death or Disability), in each casedisability) within twelve months following a Change in Control (as defined in the GudinMack Employment Agreement), Dr. Gudin will besubject to the Company’s receipt of a release in each case, Mr. Mack would have been entitled to receive thehis Accrued Obligations (as defined in the Mack Employment Agreement) and, subject to Dr. Gudin’sMr. Mack’s compliance with the terms of the GudinMack Employment Agreement, Dr. Gudin will beMr. Mack would have been entitled to receive the following: (i) a lump sum payment equal to two times the sum of Dr. Gudin’sMr. Mack’s base salary for the year in which the termination date occurs (or if greater, the year immediately preceding the year in which the Change in Control occurs), (ii) a lump sum payment equal to two times the sum of Dr. Gudin’sMr. Mack’s cash bonus for the calendar year in which the termination date occurs (or if greater, the year in which the Change in Control occurs), and (iii) accelerated vesting of any award granted to Dr. GudinMr. Mack under theour 2022 Plan.
The Gudin Employment Agreement has a term of three years from the effective date. In connection with his entry into the Gudin Employment Agreement, Dr. Gudin entered into a customary Confidential Disclosure Invention Assignment Agreements with us.Effective November 17, 2023, Mr. Mack resigned as our Chief Executive Officer.
Mr. Chipman
On April 7, 2021, we entered into an employment agreement with Christopher Chipman, as amended (the “Chipman Employment Agreement”). The Chipman Employment Agreement providesprovided for Mr. Chipman to continue to serve as the Company’s Chief Financial Officer reporting to the Company’s Chief Executive Officer and providesprovided for an annual base salary of $250,000, subject to annual increases at the discretion of the Board of Directors. Mr. Chipman’s annual base salarywhich was increased to $312,000 on January 25, 2023. Under the Chipman Employment Agreement, Mr. Chipman iswas eligible for an annual bonus with a target amount equal to 30% of his base salary, which will be awarded by our Board of Directors (the “Board”) in its sole discretion based on the achievement of the Company and Mr. Chipman of corporate and personal performance metrics established by the Board on an annual basis. Upon the recommendation of the Compensation Committee and in consultation with Mr. Chipman the Board of Directors may award Mr. Chipman an annual bonus in excess of the targeted amount. To receive any bonus, Mr. Chipman must be employed by the Company at the time of payment. Mr. Chipman maywas also eligible to receive, in the discretion of the Board, equity awards under the 2022 Plan, or any other equity incentive plan that the Company may adopt in the future. Mr. Chipman will also be eligible to receivefuture and other customary benefits described in the Chipman Employment Agreement.
We mayhad the right to terminate the Chipman Employment Agreement upon written notice to Mr. Chipman in the event of Disability (as defined in the Chipman Employment Agreement), in which event the Companywe would have no further obligations under the Chipman Employment Agreement, except for any Accrued Obligations (as defined in the Chipman Employment Agreement) and any portion of an earned annual bonus which remains unpaid at the time of termination. We may also had the right to terminate the Chipman Employment Agreement for Cause (as defined in the Chipman Employment Agreement) immediately upon providing written notice of such termination to Mr. Chipman. If we terminate the Chipman Employment Agreement for Cause,and we would have no further obligation under the Chipman Employment Agreement, except for any Accrued Obligations due. We mayalso had the right to terminate the Chipman Employment Agreement without Cause immediately upon written notice of termination to Mr. Chipman. IfChipman and we terminate thewould be obligated to pay Mr. Chipman Employment Agreement without Cause, in addition to any Accrued Obligations due Mr. Chipman is entitled to receiveand (i) severance payments in an amount equal to Mr. Chipman’s base salary for a period of six months after the effective date of the termination and (ii) reimbursement of medical insurance premiums until the earlier of (1) six months or (2) the date Mr. Chipman becomes eligible for medical benefits through another employer, subject to certain conditions.
Mr. Chipman mayhad the right to terminate his agreement for Good Reason (as defined in the Chipman Employment Agreement) upon providing written notice of such termination to us. If Mr. Chipman terminates his employment for Good Reason, Mr. Chipman willus and he would be entitled to receive the same payments and benefits on the same terms and conditions as would be applicable upon termination by us without Cause.
If the Chipman Employment Agreement iswas terminated by Mr. Chipman for Good Reason or by us without Cause (other than on account of Mr. Chipman’s death or Disability), in each case within twelve months following a Change in Control (as defined in the Chipman Employment Agreement), Mr. Chipman will bewould have been entitled to receive the Accrued Obligations and, subject to Mr. Chipman’s compliance with the terms of the Chipman Employment Agreement, Mr. Chipman willwould be entitled to receive the following: (i) a lump sum payment equal to two times the sum of Mr. Chipman’s base salary for the year in which the termination date occurs (or if greater, the year immediately preceding the year in which the Change in Control occurs), (ii) a lump sum payment equal to two times the sum of Mr. Chipman’s cash bonus for the calendar year in which the termination date occurs (or if greater, the year in which the Change in Control occurs), and (iii) accelerated vesting of any award granted to Mr. Chipman under the 2022 Plan.
The Chipman Employment Agreement hashad a term of three years from the effective date. In connection with his entry into the Chipman Employment Agreement, Mr. Chipman entered into a customary Confidential Disclosure Invention Assignment Agreements with the Company.
On June 18, 2023, Mr. Chipman notified the Chairman of the Board of his decision to resign from his position as our Chief Financial Officer to pursue other opportunities. Mr. Chipman’s employment terminated on June 30, 2023. We entered into a separation agreement and release with Mr. Chipman (the “Separation Agreement”), effective as of June 30, 2023, providing for (i) the payment to Mr. Chipman of a total of $234,000, (the “Severance Amount”) in four equal monthly installments of $58,500; (ii) reimbursement of COBRA payments for four months; and (iii) the acceleration of the vesting of all shares subject to option awards, such options to be exercisable until the Severance Amount was fully paid. Any options that were not timely exercised were nullified. The Separation Agreement also contained mutual non-disparagement obligations and a mutual standard release of claims. As of December 31, 2023, all amounts due to Mr. Chipman have been paid and all of his stock options have been forfeited.
Severance subject to release of claims.claims
Our obligation to provide an executive with severance payments and other benefits under each executive’s employment or consulting agreement, as applicable, is conditioned on the executive signing (and not subsequently revoking) an effective release of claims in favor of us.
Clawback Policy
The Board has adopted a clawback policy which requires the clawback of erroneously awarded incentive-based compensation of past or current executive officers awarded during the three full fiscal years preceding the date on which the issuer is required to prepare an accounting restatement due to the material noncompliance of the Company with any financial reporting requirement under the federal securities laws. There is no fault or misconduct required to trigger a clawback.
The Compensation Committee shall determine, in its sole discretion, the timing and method for promptly recouping such erroneously awarded compensation, which may include without limitation: (a) seeking reimbursement of all or part of any cash or equity-based award, (b) cancelling prior cash or equity-based awards, whether vested or unvested or paid or unpaid, (c) cancelling or offsetting against any planned future cash or equity-based awards, (d) forfeiture of deferred compensation, subject to compliance with Section 409A of the Internal Revenue Code and the regulations promulgated thereunder, and (e) any other method authorized by applicable law or contract. Subject to compliance with any applicable law, the Compensation Committee may affect recovery under this policy from any amount otherwise payable to the executive officer, including amounts payable to such individual under any otherwise applicable Company plan or program, including base salary, bonuses or commissions and compensation previously deferred by the executive officer.
87
DIRECTOR COMPENSATIONEquity compensation
Outstanding equity awards at fiscal year-end table
The following table sets forth information concerning the outstanding equity awards held by each of our Named Executive Officers as of December 31, 2023:
| Option Awards | |||||||||||||||
| Name | Number of Securities Underlying Unexercised Options (#) Exercisable | Number of Securities Underlying Unexercised Options (#) Unexercisable | Option Exercise Price ($) | Option Expiration Date | |||||||||||
| Gerald Bruce | 1,516 | — | 98.90 | 3/11/2030 | |||||||||||
| Chief Executive Officer | 3,000 | — | 46.20 | 4/7/2031 | |||||||||||
| 6,000 | — | 17.60 | 4/25/2032 | ||||||||||||
| 10,000 | — | 7.30 | 4/24/2033 | ||||||||||||
| Anthony Mack(4) | 2,022 | — | 98.90 | 2/17/2024 | |||||||||||
| Former Chief Executive Officer | 4,045 | — | 98.90 | 2/17/2024 | |||||||||||
| 3,333 | — | 46.20 | 2/17/2024 | ||||||||||||
| 2,022 | — | 23.40 | 2/17/2024 | ||||||||||||
| Sheila A. Mathias, PhD, JD | 1,667 | 833 | (1) | 46.20 | 4/07/2031 | ||||||||||
| Chief Scientific Officer | 2,023 | 4,045 | (1) | 21.30 | 1/31/2032 | ||||||||||
| — | 11,200 | (1) | 7.88 | 1/25/2033 | |||||||||||
| Vinay Shah | — | 10,000 | (2) | 9.90 | 6/20/2033 | ||||||||||
| Chief Financial Officer | |||||||||||||||
| Christopher Chipman(3) | - | - | - | - | |||||||||||
| Former Chief Financial Officer | |||||||||||||||
| (1) | These options vest equally over 3 years starting one year after anniversary date. |
| (2) | These options vest 25% after 12 months from the hire date (June 20, 2023) and the remaining 75% will vest in equal monthly installments over the next 24 months. |
| (3) | All of Mr. Chipman’s option awards were forfeited in October of 2023. |
| (4) | All of Mr. Mack’s options awards were forfeited in February of 2024. |
Securities Authorized for Issuance Under Equity Compensation Plans
The following table provides information with respect to our compensation plans under which equity compensation was authorized as of December 31, 2023.
| Number of securities to be issued upon exercise of outstanding options, warrants and rights | Weighted average exercise price of outstanding options, warrants and rights | Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column a) | ||||||||||
| Plan category | (a) | (b) | (c) | |||||||||
| Equity compensation plans approved by security holders | 175,686 | (1) | $ | 34.60 | 116,511 | (2)(3) | ||||||
| Equity compensation plans not approved by security holders | - | - | - | |||||||||
| Total | 175,686 | $ | 34.60 | 116,511 | ||||||||
| (1) | Includes 98,886 and 76,800 shares of common stock issuable upon exercise of outstanding options pursuant to the 2017 Plan and the 2022 Plan, respectively, as of December 31, 2023. |
| (2) | In accordance with the “evergreen” provision in the 2022 Plan, an additional 23,428 shares were automatically made available for issuance on the first day of 2024, which represents 2% of the number of shares outstanding on December 31, 2023; these shares are excluded from this calculation. |
| (3) | Includes 0 and 116,511 shares of common stock available for issuance under the 2017 Plan and the 2022 Plan, respectively, as of December 31, 2023. |
Employee benefits plans
We currently provide broad-based health and welfare benefits that are available to all of our employees, including our Named Executive Officers, including medical, dental, vision, life and disability insurance.
Director Compensation
The following table sets forth information concerning the compensation paid to certain of our non-employee directors, as well as employee directors who are not Named Executive Officers, during the year ended December 31, 2022:2023:
| Name | Cash ($) | Option ($)(1) | Total ($) | Fees Earned or Paid in Cash ($) | Option ($)(1)(3) | Total ($) | ||||||||||||||||||
| Eric Floyd, PhD | $ | 40,000 | $ | 31,000 | $ | 71,000 | $ | 55,000 | $ | 23,850 | $ | 78,850 | ||||||||||||
| Jerrold Sendrow, CFP | $ | 40,000 | $ | 25,000 | $ | 65,000 | $ | 55,000 | $ | 18,550 | $ | 73,550 | ||||||||||||
| Thani Jambulingam, PhD | $ | 40,000 | $ | 29,000 | $ | 69,000 | $ | 55,000 | $ | 21,200 | $ | 76,200 | ||||||||||||
| Vanila M. Singh, MD | $ | 40,000 | $ | 25,000 | $ | 65,000 | $ | 55,000 | $ | 7,950 | $ | 62,950 | ||||||||||||
| Gary S. Jacob, PhD(6) | $ | 23,000 | $ | 25,000 | $ | 48,000 | ||||||||||||||||||
| Michael F. Dubin(7) | $ | 40,000 | $ | 25,000 | $ | 65,000 | ||||||||||||||||||
| Michael F. Dubin | $ | 55,000 | $ | 10,600 | $ | 65,600 | ||||||||||||||||||
| Barbara A. Ruskin, PhD, J.D. | $ | 46,000 | $ | 17,750 | $ | 63,750 | ||||||||||||||||||
| Jeffrey Gudin, MD (2) | $ | 158,000 | $ | 34,000 | $ | 192,000 | ||||||||||||||||||
| (1) | Amounts reflect the full grant date fair value of stock options granted during |
| (2) |
| (3) |
| Number of Shares Subject to Outstanding options as of December 31, 2023 | ||||
| Jeffrey Gudin, MD | 20,617 | |||
| Eric Floyd, PhD | 11,066 | |||
| Jerrold Sendrow, CFP | 9,054 | |||
| Thani Jambulingam, | 9,756 | |||
| Vanila M. Singh, MD | 5,549 | |||
| Michael F. Dubin | 5,231 | |||
| Barbara Ruskin, PhD, J.D. | 2,500 | |||
88
On June 14, 2022, the Company established the Virpax Pharmaceuticals, Inc. 2022 Equity Incentive Plan (the “2022 Plan”). Upon stockholder approval of the 2022 Plan, all new grants of awards will be made under the 2022 Plan and no new grants of awards will be made under the 2017 Plan. Non-Employee Director Compensation Policy
The 2022 Plan includes a director compensation policy which provides for:
| ● | on January 1 of each year, each non-employee director will be granted Stock Options under the 2022 Plan to purchase |
| ● | each new non-employee director will be granted Stock Options under the 2022 Plan to purchase up to |
| ● | on January 1, of each year, each then serving non-Chair member of the Audit Committee, the Compensation Committee, the Nominating and Corporate Governance Committee and the Science and Technology Committee shall automatically be granted Stock Options to purchase |
In addition, our non-employee directors receive a cash payment $60,000 per year.
On January 1, 2022, our Board of Directors approved the grant of options to each of our non-employee directors pursuant to the above policy. Options to purchase an aggregate of 77,601 shares of our Common Stock were made, with all grants being made under the 2017 Plan. The options have an exercise price of $3.43 per share, the fair market value of the Common Stock on January 31, 2022, the date of grant, will vest upon the one-year anniversary of the grant date and have a ten-year expiration date.
On January 1, 2023, options were granted to the Non-Employee Directors pursuant to the 2022 Plan to purchase an aggregate of 155,00015,500 shares of Common Stock, with all grants being made under the 2022 Plan. The options have an exercise price of $0.622$6.22 per share, the fair market value of the Common Stock on the date of grant. The options granted to the directors will vest upon the one-year anniversary of the grant date and have a ten-year expiration date.
Equity compensationOn January 29, 2024, options to purchase 4,500 shares of Common Stock were granted to Dr Floyd for service as Chair of the Board of Directors and each other the Non-Employee Directors was granted an option to purchase an aggregate of 2,500 shares of Common Stock, with all grants being made under the 2022 Plan. The options have an exercise price of $3.18 per share, the fair market value of the Common Stock on the date of grant. The options granted to the directors will vest upon the one-year anniversary of the grant date and have a ten-year expiration date.
Outstanding equity awards at fiscal year-end table
The following table sets forth information concerning the outstanding equity awards held by each of our Named Executive Officers as of December 31, 2022:
| Option Awards | ||||||||||||||
| Name | Number of Securities Underlying Unexercised Options (#) Exercisable | Number of Securities Underlying Unexercised Options (#) Unexercisable | Option Exercise Price ($) | Option Expiration Date | ||||||||||
| Anthony Mack | 20,225 | — | 9.89 | 5/18/2029 | ||||||||||
| Chief Executive Officer | 40.450 | — | 9.89 | 5/22/2030 | ||||||||||
| 16,667 | 33,333 | 4.62 | 4/07/2031 | |||||||||||
| — | 60,676 | 2.13 | 1/31/2032 | |||||||||||
| Jeffrey Gudin, MD | 15,169 | — | 9.89 | 5/18/2029 | ||||||||||
| Chief Medical Officer | 30,338 | — | 9.89 | 5/22/2030 | ||||||||||
| 16,667 | 33,333 | 4.62 | 4/07/2031 | |||||||||||
| — | 60,676 | 2.13 | 1/31/2032 | |||||||||||
| Christopher Chipman | 40,450 | — | 9.89 | 5/01/2030 | ||||||||||
| Chief Financial Officer | 8,333 | 16,667 | 4.62 | 4/07/2031 | ||||||||||
| — | 60,676 | 2.13 | 1/31/2032 | |||||||||||
89
Securities Authorized for Issuance Under Equity Compensation Plans
The following table provides information with respect to our compensation plans under which equity compensation was authorized as of December 31, 2022.
| Number of securities to be issued upon exercise of outstanding options, warrants and rights | Weighted average exercise price of outstanding options, warrants and rights | Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column a) | ||||||||||
| Plan category | (a) | (b) | (c) | |||||||||
| Equity compensation plans approved by security holders(1) | 1,172,281 | (2) | $ | 5.38 | 1,515,591 | (3)(4) | ||||||
| Equity compensation plans not approved by security holders | - | - | - | |||||||||
| Total | 1,172,281 | 5.38 | 1,515,591 | |||||||||
Employee benefits plans
We currently provide broad-based health and welfare benefits that are available to all of our employees, including our Named Executive Officers, including medical, dental, vision, life and disability insurance.
Limitation of Directors Liability and Indemnification
The Delaware General Corporation Law authorizes corporations to limit or eliminate, subject to certain conditions, the personal liability of directors to corporations and their stockholder for monetary damages for breach of their fiduciary duties. Our Certificate of Incorporation limits the liability of our directors to the fullest extent permitted by Delaware law. In addition, we have entered into indemnification agreements with all of our directors and named executive officers whereby we have agreed to indemnify those directors and officers to the fullest extent permitted by law, including indemnification against expenses and liabilities incurred in legal proceedings to which the director or officer was, or is threatened to be made, a party by reason of the fact that such director or officer is or was a director, officer, employee or agent of ours, provided that such director or officer acted in good faith and in a manner that the director or officer reasonably believed to be in, or not opposed to, our best interests.
90
We have director and officer liability insurance to cover certain liabilities our directors and officers may incur in connection with their services to us, including matters arising under the Securities Act. Our Certificate of Incorporation and bylaws provide that we will indemnify our directors and officers who, by reason of the fact that he or she is or was one of our officers or directors of our Company, is involved in any action, suit or proceeding, whether civil, criminal, administrative or investigative related to their board role with us.
There However, there is no pending litigationfurther director and officer insurance coverage available with respect to the Action or proceeding involving any of our directors, officers, employees or agents in which indemnification will be required or permitted and we are not aware of any threatened litigation or proceeding that may result in a claim for such indemnification other than what is disclosed inpotential claims by Mr. Mack related thereto as set forth under Part I. Item 3. Legal Matters.
Indemnification Agreements
Indemnification Agreements
We have entered into Indemnification Agreements with each of our current directors and executive officers. The Indemnification Agreements provide for indemnification against expenses, judgments, fines and penalties actually and reasonably incurred by an indemnitee in connection with threatened, pending or completed actions, suits or other proceedings, subject to certain limitations. The Indemnification Agreements also provide for the advancement of expenses in connection with a proceeding prior to a final, nonappealable judgment or other adjudication, provided that the indemnitee provides an undertaking to repay to us any amounts advanced if the indemnitee is ultimately found not to be entitled to indemnification by us. The Indemnification Agreement sets forth procedures for making and responding to a request for indemnification or advancement of expenses, as well as dispute resolution procedures that will apply to any dispute between us and an indemnitee arising under the Indemnification Agreements.
Incentive plans
2022 Incentive Plan
On June 14, 2022, the Company established the Virpax Pharmaceuticals, Inc. 2022 Equity Incentive Plan, (the “2022 Plan”)at which time all new grants of awards will bewere made under the 2022 Plan and no new grants of awards will behave been made under the 2017 Plan. The Company believes that offering ownership interests in the Company is a key factor in retaining and recruiting employees, officers, non-employee directors and other individual service providers, and aligning and increasing their interests in the Company’s success.
2017 Incentive Plan
On May 20, 2017, our Board of Directors adopted the 2017 Plan, and on May 21, 2018, our Board of Directors approved the Amended and Restated 2017 Plan, which was further amended on April 25, 2020. The purpose of the 2017 Plan is to encourage the participants to contribute materially to our growth as a company, thereby benefitting our stockholder,stockholders, and will alignaligning the economic interests of the participants with those of the stockholder.stockholders. As noted above, upon the approval of the 2022 Plan, all new grants of awards will beare made under the 2022 Plan and no new grants of awards will be made under the 2017 Plan.
91
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
Security Ownership of Certain Beneficial Owners and Management
The following table sets forth information with respect to the beneficial ownership of our common stock, as of March 22, 2023:2024:
| ● | each person or group of affiliated persons known by us to beneficially own more than 5% of our common stock; |
| ● | each of our executive officers; |
| ● | each of our directors; and |
| ● | all of our executive officers and directors as a group. |
The number of shares beneficially owned by each stockholder is determined under rules issued by the SEC. Under these rules, beneficial ownership includes any shares as to which the individual or entity has sole or shared voting power or investment power. Applicable percentage ownership is based on 11,714,2841,171,233 shares of common stock outstanding as of March 22, 2023.2024. In computing the number of shares beneficially owned by an individual or entity and the percentage ownership of that person, shares of common stock subject to the exercise of options, warrants or other rights held by such person that are currently exercisable or will become exercisable within 60 days of March 22, 20232024 are counted as outstanding. Unless noted otherwise, the address of all listed stockholder is 1055 Westlakes Drive, Suite 300, Berwyn, PA 19312. Each of the stockholder listed has sole voting and investment power with respect to the shares beneficially owned by the stockholder unless noted otherwise, subject to community property laws where applicable.
| Name of Beneficial Owner | Number of Shares Beneficially Owned | Percentage of Shares Beneficially Owned | Number of Shares Beneficially Owned | Percentage of Shares Beneficially Owned | ||||||||||||
| 5% or Greater Stockholders | ||||||||||||||||
| Virpax Pharmaceuticals, LLC | 2,730,438 | (1) | 23.3 | % | 273,043 | (3) | 23.3 | % | ||||||||
| Sabby Management, LLC | 988,165 | (13) | 8.4 | % | ||||||||||||
| Executive Officers and Directors Other Than 5% or Greater Stockholders | ||||||||||||||||
| Anthony Mack | 366,796 | (1)(2) | 3.1 | % | ||||||||||||
| Named Executive Officers and Directors Other Than 5% or Greater Stockholders | ||||||||||||||||
| Gerald Bruce | 20,920 | (1) | 1.8 | % | ||||||||||||
| Vinay Shah | — | (2) | — | |||||||||||||
| Anthony P. Mack | 25,255 | (3)(4) | 2.1 | % | ||||||||||||
| Sheila Mathias, PhD, J.D., MBA | 9,445 | (5) | * | |||||||||||||
| Jeffrey Gudin, MD | 106,650 | (3) | * | 14,352 | (6) | * | ||||||||||
| Gerald Bruce | 109,214 | (4) | * | |||||||||||||
| Christopher Chipman | 77,342 | (5)(6) | * | |||||||||||||
| Eric Floyd, PhD | 73,700 | (7) | * | 11,866 | (7) | * | ||||||||||
| Jerrold Sendrow, CFP | 64,571 | (8) | * | 9,954 | (8) | * | ||||||||||
| Thani Jambulingam, PhD | 57,688 | (9) | * | 9,856 | (9) | * | ||||||||||
| Vanila Singh, MD, MACM | 40,499 | (10) | * | 5,549 | (10) | * | ||||||||||
| Michael F. Dubin | 32,316 | (11) | * | 5,231 | (11) | * | ||||||||||
| Directors and Officers as a Group (9 persons) | 3,659,213 | 29.6 | % | |||||||||||||
| Barbara A. Ruskin, PhD, J.D. | 2,500 | (12) | * | |||||||||||||
| Christopher Chipman | — | (13) | — | |||||||||||||
| Directors and Officers as a Group (12 persons) | 114,927 | 9.1 | % | |||||||||||||
| * | Less than 1%. |
| (1) | Includes 404 shares of common stock and 20,516 shares of common stock issuable upon exercise of stock options that are exercisable within 60 days of March 22, 2024. |
| (2) | Does not include 10,000 shares of common stock issuable upon exercise of stock options that are not exercisable within 60 days of March 22, 2024. |
| (3) | Anthony Mack, our Former Chief Executive Officer, and Jeffrey Gudin, our Executive Vice President and Chief Medical Officer, are the members of Virpax Pharmaceuticals, LLC. Due to Mr. Mack’s ownership of 88.8888% of the outstanding member units of Virpax Pharmaceuticals, LLC, he may be deemed to have sole voting and dispositive control over the shares of our common stock held by Virpax Pharmaceuticals, LLC. As a result, Mr. Mack may be deemed to beneficially own the shares of our common stock held by Virpax Pharmaceuticals, LLC. Mr. Mack resigned as our Chief Executive Officer and Chair of the Board, effective November 17, 2023. |
92
| Includes | |
| (5) | Includes 9,445 shares of common stock |
| Includes | |
| Includes | |
| (8) | Includes 900 shares of common stock and 9,054 shares of common stock issuable upon exercise of stock options exercisable within 60 days of March 22, 2024. |
| (9) | Includes 100 shares of common stock and includes 9,756 shares of common stock issuable upon exercise of stock options exercisable within 60 days of March 22, 2024. |
| (10) | Includes 5,549 shares of common stock issuable upon exercise of stock options that are exercisable within 60 days of March 22, |
| Includes | |
| Includes | |
| (13) |
93
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The following includes a summary of transactions since January 1, 20212022 to which we have been a party in which the amount involved exceeded or will exceed the lesser of $120,000 or 1% of the average of our total assets as of December 31, 20222023 and 2021,2022, and in which any of our directors, executive officers or, to our knowledge, beneficial owners of more than 5% of our capital stock or any member of the immediate family of any of the foregoing persons had or will have a direct or indirect material interest, other than equity and other compensation, termination, change in control and other arrangements, which are described under “Executive and Director Compensation.” We also describe below certain other transactions with our directors, executive officers, and stockholder.
Promissory Notes with our Named Executive OfficersThere were no related party transactions during the years ended December 31, 2023 and 2022.
In January 2021, we issued notes with an aggregate principal amount of $75,000 and $25,000, respectively. These notes were issued to Anthony Mack for $75,000 and Christopher Chipman, our Chief Financial Officer, for $25,000. These notes were subsequently repaid with proceeds from the initial public offering in February 2021.
Indemnification Agreements
We have entered into indemnification agreements with our directors and executive officers whereby we have agreed to indemnify those directors and officers to the fullest extent permitted by law, including indemnification against expenses and liabilities incurred in legal proceedings to which the director or officer was, or is threatened to be made, a party by reason of the fact that such director or officer is or was a director, officer, employee or agent of our Company, provided that such director or officer acted in good faith and in a manner that the director or officer reasonably believed to be in, or not opposed to, the best interests of our Company.
Policies and Procedures for Related Party Transactions:Transactions
Our Board of Directors has adopted a written related person transaction policy setting forth the policies and procedures for the review and approval or ratification of related person transactions. This policy covers, with certain exceptions set forth in Item 404 of Regulation S-K under the Securities Act, any transaction, arrangement or relationship, or any series of similar transactions, arrangements or relationships, in which we were or are to be a participant, where the amount involved exceeds the lesser of $120,000 or 1% of the average of our total assets as of December 31, 20222023 and 20212022 and a related person had, has or will have a direct or indirect material interest, including without limitation, purchases of goods or services by or from the related person or entities in which the related person has a material interest, indebtedness, guarantees of indebtedness and employment by us of a related person. In reviewing and approving any such transactions, our audit committee is tasked to consider all relevant facts and circumstances, including, but not limited to (i) whether the transaction is on terms comparable to those that could be obtained in an arm’s length transaction with an unrelated party; (ii) the extent of the related person’s interest in the transaction; (iii) the benefits to the Company; (iv) the impact on a director’s independence in the event the related person is a director, an immediately family member of a director or an entity in which a director is a partner, stockholder or executive officer; (v) the availability of other sources for comparable products or services; (vi) the terms of the transaction; and (vii) the terms available to unrelated third parties.
Director Independence
Our Board of Directors undertook a review of its composition, the composition of its committees and the independence of each director. Based upon information requested from and provided by each director concerning his or her background, employment and affiliations, including family relationships, our Board of Directors has determined that Dr. Floyd, Mr. Sendrow, Dr. Jambulingam, Dr. Singh, and Mr. Dubin and Dr. Ruskin do not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director and that each of these directors is “independent” as that term is defined under the Rules of the Nasdaq Market and the SEC.
94
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
Fees Paid to the Independent Registered Public Accounting Firm
The following table summarizes the fees paid for professional services rendered by EisnerAmper LLP, our independent registered public accounting firm, for each of the last two fiscal years.
| Year ending December 31, | 2022 | 2021 | 2023 | 2022 | ||||||||||||
| Audit fees(1) | $ | 231,420 | $ | 252,309 | $ | 329,805 | $ | 231,420 | ||||||||
| Audit related fees | - | - | - | - | ||||||||||||
| Tax fees | - | - | - | - | ||||||||||||
| All other fees | - | - | - | - | ||||||||||||
| Total | $ | 231,420 | $ | 252,309 | $ | 329,805 | $ | 231,420 | ||||||||
| (1) | Audit fees consist of fees incurred for professional services rendered for the audit of our annual financial statements and review of the quarterly financial statements, assistance with registration statements filed with the SEC, and services that are normally provided by our independent registered public accounting firm in connection with regulatory filings or engagements. Audit fees includes fees of approximately $29,000 for consents |
Auditor Independence
In our fiscal year ended December 31, 2022,2023, there were no other professional services provided by EisnerAmper LLP, located in Philadelphia, Pennsylvania, Firm ID: 274, that would have required our audit committee to consider their compatibility with maintaining the independence of EisnerAmper LLP.
Audit Committee Policy on Pre-Approval of Audit and Permissible Non-Audit Services of Independent Registered Public Accounting Firm
Our audit committee has established a policy governing our use of the services of our independent registered public accounting firm. Under this policy, our audit committee is required to pre-approve all audit and non-audit services performed by our independent registered public accounting firm in order to ensure that the provision of such services does not impair the public accountants’ independence. All fees paid to EisnerAmper LLP for our fiscal years ended December 31, 20222023 and 20212022 were pre-approved by our audit committee.
95
PART IV
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
(a)(1) Financial Statements
The financial statements and related notes, together with the report of EisnerAmper LLP appear at pages F-1 through F-23F-21 following the Exhibit List as required by “Part II—Item 8—Financial Statements and Supplementary Data” of this Annual Report on Form 10-K.
(a)(2) Financial Statement Schedules
All schedules have been omitted because the information required to be set forth therein is not applicable or is shown in the financial statements or notes thereto.
(a)(3) Exhibits
The following exhibits are filed as part of, or incorporated by reference into, this Annual Report on Form 10-K.
96
| * | Filed |
| ** | Furnished, not filed. |
| † | Denotes management compensation plan or contract. |
| # | Certain portions of this exhibit have been omitted because the omitted information is (i) not material and (ii) would likely cause competitive harm to the Company if publicly disclosed. |
ITEM 16. FORM 10-K SUMMARY
None.
97
SIGNATURES
Pursuant to the requirements of Section 13 and 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
| VIRPAX PHARMACEUTICALS, INC. | |
| (Registrant) | |
| Date: March | |
| Chief Executive Officer | |
| (Principal Executive Officer) | |
| Date: March | /s/ |
| Chief Financial Officer | |
| (Principal Financial and Accounting Officer) |
Pursuant to the requirements of the Securities Exchange Act of 1934, as amended, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Signature | Title | Date | ||
| /s/ | Chief Executive Officer and Director | March | ||
| (Principal Executive Officer) | ||||
/s/ | Chief Financial Officer and Secretary | March | ||
| (Principal Financial and Accounting Officer) | ||||
| /s/ Eric Floyd, PhD | Director and Chairman | March | ||
| Eric Floyd, PhD | | |||
| /s/ Jeffrey Gudin, MD | Chief Medical Officer, Director | March | ||
| Jeffrey Gudin, MD | ||||
| /s/ Jerrold Sendrow, CFP | Director | March | ||
| Jerrold Sendrow, CFP | ||||
| /s/ Thani Jambulingam, PhD | Director | March | ||
| Thani Jambulingam, PhD | ||||
| /s/ Vanila M. Singh, MD | Director | March | ||
| Vanila M. Singh, MD | ||||
| /s/ Michael Dubin | Director | March | ||
| Michael Dubin | ||||
| /s/ Barbara Ruskin | Director | March 25, 2024 | ||
| Barbara Ruskin |
98
VIRPAX PHARMACEUTICALS, INC
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
F-1
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of
Virpax Pharmaceuticals, Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Virpax Pharmaceuticals, Inc. (the “Company”) as of December 31, 20222023 and 2021,2022, and the related consolidated statements of operations, stockholders’ equity, (deficit), and cash flows for each of the years then ended and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the consolidated financial position of the Company as of December 31, 2023 and 2022, and 2021, and the consolidated results of itstheir operations and itstheir cash flows for each of the years then ended, in conformity with accounting principles generally accepted in the United States of America.
Going Concern
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the financial statements, the Company has incurred continuing losses and has litigation that may requireobligations for significant cash payments in the next year that raise substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ EisnerAmper LLP
We have served as the Company’s auditor since 2020.
EISNERAMPER LLP
Philadelphia, Pennsylvania
March 22, 202325, 2024
F-2
VIRPAX Pharmaceuticals, Inc.
CONSOLIDATED BALANCE SHEETS
| December 31, 2022 | December 31, 2021 | December 31, 2023 | December 31, 2022 | |||||||||||||
| ASSETS | ||||||||||||||||
| Current assets | ||||||||||||||||
| Cash | $ | 18,995,284 | $ | 36,841,992 | $ | 9,141,512 | $ | 18,995,284 | ||||||||
| Prepaid expenses and other current assets | 678,365 | 2,730,444 | 486,833 | 678,365 | ||||||||||||
| Total current assets | 19,673,649 | 39,572,436 | 9,628,345 | 19,673,649 | ||||||||||||
| Total assets | $ | 19,673,649 | $ | 39,572,436 | $ | 9,628,345 | $ | 19,673,649 | ||||||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY (DEFICIT) | ||||||||||||||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY | ||||||||||||||||
| Current liabilities | ||||||||||||||||
| Accounts payable and accrued expenses | $ | 1,094,590 | $ | 2,087,691 | $ | 1,694,024 | $ | 1,094,590 | ||||||||
| Estimated litigation liability | 2,000,000 | — | ||||||||||||||
| Litigation liability | 6,000,000 | 2,000,000 | ||||||||||||||
| Total current liabilities | 3,094,590 | 2,087,691 | 7,694,024 | 3,094,590 | ||||||||||||
| Total liabilities | 3,094,590 | 2,087,691 | 7,694,024 | 3,094,590 | ||||||||||||
| Commitments and contingencies | ||||||||||||||||
| Stockholders’ equity | ||||||||||||||||
| Preferred stock, par value $0.00001, 10,000,000 shares authorized, no shares issued and outstanding | — | — | ||||||||||||||
| Common stock, $0.00001 par value; 100,000,000 shares authorized, 11,714,284 shares issued and outstanding as of December 31, 2022; 11,714,885 shares issued and outstanding as of December 31, 2021 | 117 | 117 | ||||||||||||||
| Preferred stock, par value $0.00001, 10,000,000 shares authorized; no shares issued and outstanding as of the years ended December 31, 2023 and 2022 | — | — | ||||||||||||||
| Common stock, $0.00001 par value; 100,000,000 shares authorized, 1,171,233 shares issued and outstanding as of the years ended December 31, 2023 and 2022 | 12 | 12 | ||||||||||||||
| Additional paid-in capital | 60,933,569 | 60,188,535 | 61,478,444 | 60,933,674 | ||||||||||||
| Accumulated deficit | (44,354,627 | ) | (22,703,907 | ) | (59,544,135 | ) | (44,354,627 | ) | ||||||||
| Total stockholders’ equity | 16,579,059 | 37,484,745 | 1,934,321 | 16,579,059 | ||||||||||||
| Total liabilities and stockholders’ equity | $ | 19,673,649 | $ | 39,572,436 | $ | 9,628,345 | $ | 19,673,649 | ||||||||
See Notes to the Consolidated Financial Statements
F-3
VIRPAX Pharmaceuticals, Inc.
CONSOLIDATED STATEMENTS OF OPERATIONS
| For the Year Ended December 31, | ||||||||||||||||
| For the Year Ended December 31, 2022 | For the Year Ended December 31, 2021 | 2023 | 2022 | |||||||||||||
| OPERATING EXPENSES | ||||||||||||||||
| General and administrative | $ | 11,082,463 | $ | 7,186,385 | ||||||||||||
| General and administrative (net of insurance reimbursement of $1,250,000 during the year ended December 31, 2023 – See Note 5) | $ | 10,572,181 | $ | 11,082,463 | ||||||||||||
| Research and development | 10,762,670 | 4,839,115 | 5,117,608 | 10,762,670 | ||||||||||||
| Total operating expenses | 21,845,133 | 12,025,500 | 15,689,789 | 21,845,133 | ||||||||||||
| Loss from operations | (21,845,133 | ) | (12,025,500 | ) | (15,689,789 | ) | (21,845,133 | ) | ||||||||
| OTHER INCOME (EXPENSE) | ||||||||||||||||
| Interest expense | - | (92,822 | ) | |||||||||||||
| OTHER INCOME | ||||||||||||||||
| Other income | 194,413 | 62,259 | 500,281 | 194,413 | ||||||||||||
| Other income (expense) | 194,413 | (30,563 | ) | |||||||||||||
| Loss before tax provision | (21,650,720 | ) | (12,056,063 | ) | ||||||||||||
| Benefit from income taxes | — | — | ||||||||||||||
| Loss before income taxes | (15,189,508 | ) | (21,650,720 | ) | ||||||||||||
| Income taxes | — | — | ||||||||||||||
| Net loss | $ | (21,650,720 | ) | $ | (12,056,063 | ) | $ | (15,189,508 | ) | $ | (21,650,720 | ) | ||||
| Basic and diluted net loss per share | $ | (1.85 | ) | $ | (1.81 | ) | $ | (12.97 | ) | $ | (18.49 | ) | ||||
| Basic and diluted weighted average common stock outstanding | 11,712,186 | 6,677,271 | 1,171,233 | 1,171,020 | ||||||||||||
See Notes to the Consolidated Financial Statements
F-4
VIRPAX Pharmaceuticals, Inc.
CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERS’ EQUITY (DEFICIT)
| Common stock | Additional paid-in | Accumulated | Total stockholders’ Equity | Common stock | Additional paid-in | Accumulated | Total stockholders’ | |||||||||||||||||||||||||||||||||
| Shares | Amount | capital | deficit | (deficit) | ||||||||||||||||||||||||||||||||||||
| Balance at January 1, 2021 | 3,145,153 | 31 | 6,431,715 | (10,647,844 | ) | (4,216,098 | ) | |||||||||||||||||||||||||||||||||
| Common stock issued pursuant to initial public offering, net of offering costs | 1,800,000 | 18 | 15,783,189 | — | 15,783,207 | |||||||||||||||||||||||||||||||||||
| Common stock issued pursuant to underwritten public offering, net of offering costs | 6,670,000 | 67 | 36,999,398 | — | 36,999,465 | |||||||||||||||||||||||||||||||||||
| Cashless exercise of stock options and warrants | 85,669 | — | — | — | — | |||||||||||||||||||||||||||||||||||
| Stock-based compensation | — | — | 974,234 | — | 974,234 | |||||||||||||||||||||||||||||||||||
| Restricted stock awards granted | 15,000 | 1 | (1 | ) | — | — | ||||||||||||||||||||||||||||||||||
| Restricted stock awards forfeited | (937 | ) | — | — | — | — | ||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | (12,056,063 | ) | (12,056,063 | ) | |||||||||||||||||||||||||||||||||
| Shares | Amount | capital | deficit | equity | ||||||||||||||||||||||||||||||||||||
| Balance at December 31, 2021 | 11,714,885 | $ | 117 | $ | 60,188,535 | $ | (22,703,907 | ) | $ | 37,484,745 | 1,171,293 | $ | 12 | $ | 60,188,640 | $ | (22,703,907 | ) | $ | 37,484,745 | ||||||||||||||||||||
| Stock-based compensation | — | — | 745,034 | — | 745,034 | — | — | 745,034 | — | 745,034 | ||||||||||||||||||||||||||||||
| Restricted stock awards forfeited | (601 | ) | — | — | — | — | (60 | ) | — | — | — | — | ||||||||||||||||||||||||||||
| Net loss | — | — | — | (21,650,720 | ) | (21,650,720 | ) | — | — | — | (21,650,720 | ) | (21,650,720 | ) | ||||||||||||||||||||||||||
| Balance at December 31, 2022 | 11,714,284 | $ | 117 | $ | 60,933,569 | $ | (44,354,627 | ) | $ | 16,579,059 | 1,171,233 | 12 | 60,933,674 | (44,354,627 | ) | 16,579,059 | ||||||||||||||||||||||||
| Stock-based compensation | — | — | 544,770 | — | 544,770 | |||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | (15,189,508 | ) | (15,189,508 | ) | |||||||||||||||||||||||||||||||||
| Balance at December 31, 2023 | 1,171,233 | $ | 12 | $ | 61,478,444 | $ | (59,544,135 | ) | $ | 1,934,321 | ||||||||||||||||||||||||||||||
See Notes to the Consolidated Financial Statements
F-5
VIRPAX Pharmaceuticals, Inc.
CONSOLIDATED STATEMENTS OF CASH FLOWS
| For the Year Ended December 31, 2022 | For the Year Ended December 31, 2021 | |||||||
| CASH FLOWS FROM OPERATING ACTIVITIES | ||||||||
| Net loss | $ | (21,650,720 | ) | $ | (12,056,063 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Forgiveness of PPP loan | — | (61,816 | ) | |||||
| Stock-based compensation | 745,034 | 974,234 | ||||||
| Change in operating assets and liabilities: | ||||||||
| Prepaid expenses and other current assets | 2,052,079 | (2,712,171 | ) | |||||
| Accounts payable and accrued expenses | (993,101 | ) | (686,776 | ) | ||||
| Estimated litigation liability | 2,000,000 | — | ||||||
| Net cash used in operating activities | (17,846,708 | ) | (14,542,592 | ) | ||||
| CASH FLOWS FROM FINANCING ACTIVITIES | ||||||||
| Repayment of notes payable | — | (503,764 | ) | |||||
| Proceeds from related party notes payable | — | 100,000 | ||||||
| Repayment of related party notes payable | — | (1,100,000 | ) | |||||
| Offering costs related to underwritten public offering | — | (3,020,535 | ) | |||||
| Proceeds from underwritten public offering of common stock | — | 40,020,000 | ||||||
| Offering costs related to initial public offering | — | (2,165,913 | ) | |||||
| Proceeds from initial public offering of common stock | — | 18,000,000 | ||||||
| Net cash provided by financing activities | — | 51,329,788 | ||||||
| Net change in cash | (17,846,708 | ) | 36,787,196 | |||||
| Cash, beginning of year | 36,841,992 | 54,796 | ||||||
| Cash, end of year | $ | 18,995,284 | $ | 36,841,992 | ||||
| Supplemental disclosure of cash and non-cash financing activities | ||||||||
| Cash paid for interest | $ | — | $ | 362,822 | ||||
| Cash paid for taxes | $ | — | $ | — | ||||
| For the Year Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| CASH FLOWS FROM OPERATING ACTIVITIES | ||||||||
| Net loss | $ | (15,189,508 | ) | $ | (21,650,720 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Stock-based compensation | 544,770 | 745,034 | ||||||
| Change in operating assets and liabilities: | ||||||||
| Prepaid expenses and other current assets | 191,532 | 2,052,079 | ||||||
| Accounts payable and accrued expenses | 599,434 | (993,101 | ) | |||||
| Litigation liability | 4,000,000 | 2,000,000 | ||||||
| Net cash used in operating activities | (9,853,772 | ) | (17,846,708 | ) | ||||
| Net change in cash | (9,853,772 | ) | (17,846,708 | ) | ||||
| Cash, beginning of year | 18,995,284 | 36,841,992 | ||||||
| Cash, end of year | $ | 9,141,512 | $ | 18,995,284 | ||||
See Notes to the Consolidated Financial Statements
F-6
VIRPAX Pharmaceuticals, Inc.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Note 1. Business, and Liquidity and Going Concern
Business
Business
Virpax Pharmaceuticals, Inc. (“Virpax” or the “Company”) was incorporated on May 12, 2017 in the state of Delaware. Virpax is a preclinical stage pharmaceutical company focused on developing novel and proprietary drug-delivery systems, and drug-releasing technologies focused on advancing non-opioid and non-addictive pain management treatments and treatments for central nervous system (“CNS”) disorders to enhance patients’ quality of life.
On July 26, 2023, the Company formed Novvae Pharmaceuticals, Inc., a wholly owned subsidiary of the Company, in the state of Delaware, for the purpose of developing over the counter products. No activities have occurred during the year ended December 31, 2023.
Liquidity and Going Concern
The Company, since inception, has been engaged in organizational activities, including raising capital and research and development activities. The Company has not generated revenues and has not yet achieved profitable operations, nor has it ever generated positive cash flow from operations. There is no assurance that profitable operations, if achieved, could be sustained on a continuing basis. The Company is subject to those risks associated with any preclinical stage pharmaceutical company that has substantial expenditures for research and development. There can be no assurance that the Company’s research and development projects will be successful, that products developed will obtain necessary regulatory approval, or that any approved product will be commercially viable. In addition, the Company operates in an environment of rapid technological change and is largely dependent on the services of its employees and consultants. Further, the Company’s future operations are dependent on the success of the Company’s efforts to raise additional capital.
The Company incurred a net loss of $21,650,720$15,189,508 and $12,056,063$21,650,720 for the yearyears ended December 31, 20222023 and 2021,2022, respectively, and had an accumulated deficit of $44,354,627$59,544,135 as of December 31, 2022.2023. The Company anticipates incurring additional losses until such time, if ever, that it can generate significant revenue from its product candidates currently in development. The Company’s primary source of capital has been the issuance of debt and equity securities.
As noted in Note 6.5. Commitments and Contingencies, the Company has paid $3.5 million to the Plaintiffs on the Effective Date pursuant to the terms of the Settlement Agreement and is currently involved in defending litigation and intendsobligated to vigorously defendpay an additional $2.5 million to the action.Plaintiffs on July 1, 2024. The Company has established an estimated litigation liability of $2.0will need to raise additional capital to fund operations, make the $2.5 million payment, and, in respect of the litigation. While the Company believes it has meritorious defenses, the ultimate resolution of the action could result in a material loss.addition, fund other required payments, if any, to its former Chief Executive Officer. Due to the Company’s continuing losses and the uncertainty regarding the outcome of this ongoing litigation and any potential claims,cash position, there exists substantial doubt about the Company’s ability to continue as a going concern. The accompanying financial statements do not include any adjustments to the carrying amounts and classification of assets, liabilities, and reported expenses that may be necessary if the Company were unable to continue as a going concern.
Additional financings will be needed by the Company to fund its operations, including litigation costs, and to complete clinical development of and to commercially develop all of its product candidates. There is no assurance that such financing will be available when needed or on acceptable terms. The Company also may be forced to curtail spending in research and development activities in order to conserve cash.
Note 2. Summary of Significant Accounting Policies
Basis of Presentation — The accompanying financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America (“U.S. GAAP”) and reflect the operations of the Company. Any reference in these notes to applicable guidance is meant to refer to U.S. GAAP as found in the Accounting Standards Codification (“ASC”) and Accounting Standards Update (“ASU”) of the Financial Accounting Standards Board (“FASB”).
F-7
Use of Estimates — The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, including disclosure of contingent assets and liabilities, at the date of the financial statements, and the reported amounts of expenses during the reporting period. Due to the uncertainty of factors surrounding the estimates or judgments used in the preparation of the financial statements, actual results may materially vary from these estimates.
Significant items subject to such estimates and assumptions include research and development accruals and prepaid expenses, estimated litigation liability, and the valuation of stock-based compensation. Future events and their effects cannot be predicted with certainty; accordingly, accounting estimates requireIt is at least reasonably possible that the exerciseestimate of judgment.the effect of a condition, situation or set of circumstances that existed at the date of the financial statements, which management considered in formulating its estimate, could change in the near term due to one or more future confirming events. Accordingly, the actual results could differ from those estimates. Accounting estimates used in the preparation of these financial statements change as new events occur, as more experience is acquired, as additional information is obtained and as the operating environment changes.
Basic and Diluted Loss per Share — Basic net loss per share is determined using the weighted average number of shares of common stock outstanding during each period. Diluted net loss per share includes the effect, if any, from the potential exercise or conversion of securities, such as stock options and warrants, which would result in the issuance of incremental shares of common stock. The computation of diluted net loss per shares does not include the conversion of securities that would have an antidilutive effect. Equivalent common shares are excluded from the calculation of diluted net loss per share since their effect is antidilutive due to the net loss of the Company which consisted of the following:
| Year Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| Equivalent common shares | ||||||||
| Stock options | 175,686 | 117,228 | ||||||
| Warrants | 1,843 | 1,843 | ||||||
| Unvested restricted stock awards | — | 23 | ||||||
| Year Ended December 31, 2022 | Year Ended December 31, 2021 | |||||||
| Equivalent common shares | ||||||||
| Stock options | 1,172,281 | 669,067 | ||||||
| Warrants | 18,436 | 18,436 | ||||||
| Unvested restricted stock awards | 237 | 6,196 | ||||||
Cash —At— At times, The Company deposits its cash with reputable financial institutions that are insured by the Federal Deposit Insurance Corporation (“FDIC”). At times, the Company’s cash balances may exceed the current insured amounts underprovided by the Federal Deposit Insurance Corporation. TotalFDIC. The Company’s cash was $18,995,284balances exceeded federally insured limits by approximately $8,900,000 and $36,841,992$18,700,000 as of December 31, 20222023 and December 31, 2021,2022, respectively.
Fair Value of Financial Instruments — The carrying amounts of the Company’s financial instruments, including cash and accounts payable approximate fair value due to the short-term nature of those instruments.
Research and Development — Research and development costs are expensed as incurred. These expenses include the costs of proprietary efforts, as well as costs incurred in connection with certain licensing arrangements and external research and development expenses incurred under arrangements with third parties, such as contract research organizations (“CROs”) and consultants. At the end of each reporting period, the Company compares the payments made to each service provider to the estimated progress towards completion of the related project. Factors that the Company considers in preparing these estimates include the status of preclinical studies, milestones achieved, and other criteria related to the efforts of its vendors. These estimates will be subject to change as additional information becomes available.
Stock-based Compensation — Stock-based compensation cost is measured at the grant date based on the fair value of the award and is recognized as expense over the requisite service period, which is generally the vesting period. The Company’s policy permits the valuation of stock-based awards granted to non-employees to be measured at fair value at the grant date.date and records forfeitures as they occur.
F-8
Determining the appropriate fair value of share-based awards requires the use of subjective assumptions, including the fair value of the Company’s common shares prior to its initial public offering, and for options, the expected life of the option and expected share price volatility. The Company uses the Black-Scholes option pricing model to value its option awards. The assumptions used in calculating the fair value of share-based awards represents management’s best estimates and involve inherent uncertainties and the application of management’s judgment. As a result, if factors change and management uses different assumptions, share-based compensation expense could be materially different for future awards.
The expected life of options was estimated using the simplified method, as the Company has no historical information to develop reasonable expectations about future exercise patterns and post-vesting employment.
Income Taxes — The Company accounts for income taxes using the asset-and-liability method in accordance with ASC 740, Income Taxes (“ASC 740”). Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases and operating loss and tax credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on the deferred tax assets and liabilities of a change in tax rate is recognized in the period that includes the enactment date. A valuation allowance is recorded if it is more-likely-than-not that some portion or all of the deferred tax assets will not be realized in future periods.
The Company follows the guidance in ASC 740-10 in assessing uncertain tax positions. The standard applies to all tax positions and clarifies the recognition of tax benefits in the financial statements by providing for a two-step approach of recognition and measurement. The first step involves assessing whether the tax position is more-likely-than-not to be sustained upon examination based upon its technical merits. The second step involves measurement of the amount to be recognized. Tax positions that meet the more-likely than-not threshold are measured at the largest amount of tax benefit that is greater than 50% likely of being realized upon ultimate finalization with the taxing authority. The Company recognizes the impact of an uncertain income tax position in the financial statements if it believes that the position is more likely than not to be sustained by the relevant taxing authority. The Company will recognize interest and penalties related to tax positions in income tax expense. As of December 31, 2022,2023, the Company had no uncertain income tax positions.
Note 3. Prepaid Expenses and Other Current Assets
Prepaid expenses and other current assets consistsconsist of the following:
| December 31, 2023 | December 31, 2022 | |||||||
| Prepaid insurance | $ | 136,241 | $ | 156,754 | ||||
| Prepaid research and development | 283,370 | 496,270 | ||||||
| Other prepaid expenses and current assets | 67,222 | 25,341 | ||||||
| $ | 486,833 | $ | 678,365 | |||||
| December 31, 2022 | December 31, 2021 | |||||||
| Prepaid insurance | $ | 156,754 | $ | 152,801 | ||||
| Research and development | 496,270 | 2,466,140 | ||||||
| Legal retainer | — | 103,050 | ||||||
| Other prepaid expenses and current assets | 25,341 | 8,453 | ||||||
| $ | 678,365 | $ | 2,730,444 | |||||
Note 4. Accounts Payable and Accrued Liabilities
Accounts payable and accrued liabilities consists of the following:
| December 31, 2023 | December 31, 2022 | |||||||
| Accrued payroll | $ | 493,780 | $ | 654,765 | ||||
| Estimated separation expense | 711,000 | — | ||||||
| Research and development expenses | 143,071 | 254,904 | ||||||
| Legal expenses | 97,089 | 147,277 | ||||||
| Professional fees | 230,627 | — | ||||||
| Other | 18,457 | 37,644 | ||||||
| $ | 1,694,024 | $ | 1,094,590 | |||||
| December 31, 2022 | December 31, 2021 | |||||||
| Accrued payroll | $ | 654,765 | $ | 627,281 | ||||
| Research and development expenses | 254,904 | 930,561 | ||||||
| Legal expenses | 147,277 | 510,008 | ||||||
| Other | 37,644 | 19,841 | ||||||
| $ | 1,094,590 | $ | 2,087,691 | |||||
F-9
Note 5. Notes Payable
On October 1, 2018, the Company entered into a promissory note, as amended, (the “2018 Promissory Note”), which promised to pay Anthony Mack, Chief Executive Officer and significant investor, the principal amount of $500,000, and bore interest at a rate of 11.19% per annum. The 2018 Promissory Note stated that the principal was due on January 15, 2021. During the year ended December 31, 2021, the Company fully repaid the balance due of $500,000 on this promissory note with accrued interest of $166,296.
On January 15, 2019, the Company entered into a promissory note, as amended, (the “2019 Promissory Note”), which promised to pay Anthony Mack the principal amount of $500,000 and bore interest at a rate of 11.19% per annum. The 2019 Promissory Note stated that the principal was due on January 15, 2021. During the year ended December 31, 2021, the Company fully repaid the balance due of $500,000 on this promissory note with accrued interest of $149,977.
In January 2021, the Company issued notes with an aggregate principal amount of $75,000 and $25,000, respectively. These notes were issued to Anthony Mack for $75,000 and Christopher Chipman, Chief Financial Officer, for $25,000. These notes bore interest at a rate of 1.35% per annum and were subsequently fully repaid with proceeds from the IPO.
On August 29, 2019, the Company entered into a service provider convertible note purchase agreement (the “RRD Note”) with RRD International, LLC (“RRD”). Under the RRD Note, the Company and RRD agreed to make certain compensation due to RRD payable in the form of a convertible promissory note. The RRD Note stated that a maximum principal balance of $400,000 could be applied for services provided by RRD to the Company, which could be converted into equity or cash (all or in part) upon a Qualified Financing (as defined in the RRD Note) or the Conversion Date of March 31, 2020. Borrowings under the RRD Note bore simple interest on the outstanding principal amount of the RRD Note until paid in full at the fixed rate of 10% per annum. During 2020, the RRD Note was amended to increase the maximum principal to $600,000 and to extend the maturity and conversion dates through to January 31, 2021. In February 2021, the Company fully paid the balance on its RRD Note of $493,480, with $34,544 of accrued interest, with proceeds from the Company’s IPO.
On May 4, 2020, the Company entered into a Promissory Note (the “PPP Note”) with PNC Bank as the lender (the “Lender”), pursuant to which the Lender agreed to make a loan to the Company under the Paycheck Protection Program (the “PPP Loan”) offered by the U.S. Small Business Administration (the “SBA”) in a principal amount of $72,100 pursuant to Title 1 of the Coronavirus Aid, Relief and Economic Security Act (the “CARES Act”). The PPP Loan proceeds were available to be used to pay for payroll costs, including salaries, commissions, and similar compensation, group health care benefits, and paid leaves; rent; utilities; and interest on certain other outstanding debt. The amount that was forgiven was to be calculated in part with reference to the Company’s full-time headcount during the period ending October 31, 2020. The interest rate on the PPP Note was a fixed rate of 1% per annum. The PPP Note matured in two years. On July 2, 2021, the SBA notified the Company that the forgiveness amount totaled $61,816 which was recorded as other income within the statement of operations in 2021. The remaining balance of $10,284 was fully repaid by the Company during the year ended December 31, 2021.
Note 6.5. Commitments and Contingencies
Litigation
From time to time the Company is subject to claims by third parties under various legal disputes. The defense of such claims, or any adverse outcome relating to any such claims, could have a material adverse effect on the Company’s liquidity, financial condition and cash flows.
On March 12, 2021, the Company and the Company’sits former Chief Executive Officer, Anthony P. Mack (the(together, the “Defendants”), were named as defendants in a complaint (the “Complaint”) filed by Sorrento Therapeutics, Inc. (“Sorrento”), and Scilex Pharmaceuticals Inc. (“Scilex” and together with Sorrento, the “Plaintiffs”) in the Court of Chancery of the State of Delaware.Delaware captioned Sorrento Therapeutics, Inc. and Scilex Pharmaceuticals Inc. v. Anthony Mack and Virpax Pharmaceuticals, Inc., Case No. 2021-0210-PAF (the “Action”). In the Complaint, Plaintiffs alleged (i) Mr. Mack breached a Restrictive Covenants Agreement, dated as of November 8, 2016, between himself and Sorrento (the “Restrictive Covenants Agreement”), (ii) the Company tortiously interfered with the Restrictive Covenants Agreement, and (iii) the Company tortiously interfered with Scilex’s relationship with Mr. Mack. On May 7, 2021, Plaintiffs filed an Amended Complaint asserting the same three causes of action. On September 28, 2021, Plaintiffs filed a Second Amended Complaint asserting the same three causes of action as the prior complaints, as well as claims in which Plaintiffs alleged (i) Mr. Mack breached an Employment, Proprietary Information and Inventions Agreement, dated as of October 25, 2016, between himself and Sorrento (the “Employment Agreement”), (ii) the Company tortiously interfered with the Employment Agreement, (iii) Mr. Mack breached his fiduciary duties to Scilex, and (iv) the Company aided and abetted Mr. Mack’s alleged breach of fiduciary duties to Scilex. On April 1, 2022, Plaintiffs filed a Third Amended Complaint. The Third Amended Complaint asserts the same causes of action as the Second Amended Complaint, as well as claims for (i) misappropriation of trade secrets by Defendants under Delaware law, and (ii) misappropriation of trade secrets by Defendants under California law. On April 18, 2022, Defendants filed answers to the Third Amended Complaint. Trial was held before Vice Chancellor Paul Fiorvanti from September 12 through September 14, 2022. Post-trial briefing was completed
In March 2023, the Company collected $1,250,000 in reimbursement of legal costs pursuant to the Company’s directors’ and officers’ insurance policy, and recorded it as a reduction of general and administrative expense on the consolidated statements of operations. No further reimbursements are permitted from the insurance policy with respect to the litigation.
On September 1, 2023, the Chancery Court issued a memorandum opinion addressing liability in the Action and found in favor of Plaintiffs on all but three counts, which the Court found were waived. The Chancery Court found it proper to attribute Mr. Mack’s knowledge and actions to the Company, which Mr. Mack used to effectuate the tortious interference and breach of fiduciary duty. The Chancery Court found that Mr. Mack breached the Restrictive Covenants Agreement he entered into with Sorrento by December 12, 2022, and post-trial argument was held on January 20, 2023. The Company intends to vigorously defend the action. However,developing EpoladermTM; the Company is unableliable for tortious interference with contract; Plaintiffs were deemed to predicthave waived their claims for breach of Mr. Mack’s Employment Agreement and for tortious interference with prospective economic advantage; Mr. Mack breached his fiduciary duty of loyalty to Scilex; the ultimate outcomeCompany aided and abetted Mr. Mack’s breach of fiduciary duty; and Mr. Mack misappropriated certain Scilex trade secrets. The Court, however, stated that the question of an appropriate remedy must await further briefing.
On October 18, 2023, in accordance with the Chancery Court’s supplemental briefing schedule, Plaintiffs filed their supplemental brief requesting the following relief: an injunction, in the first instance, enjoining Mr. Mack from having any relationship with Virpax for a period of 18 months and 27 days; enjoining Virpax from further developing or marketing Epoladerm for a period of 18 months and 27 days; alternatively, if these two injunction requests were not granted, Plaintiffs requested a judgement of joint and several liability against Mr. Mack and Virpax of $14,684,833. In addition to these requests for injunctive relief (or in, the alternative, damages), Plaintiffs sought a constructive trust over the revenues of Epoladerm, ProbudurTM and EnveltaTM, or, in the alternative to a constructive trust, a royalty of 5 per cent of net sales of Epoladerm, 8-11 percent of net sales of Probudur and 7.5 percent of net sales of Envelta. In addition to the requests for injunctive relief, imposition of a constructive trust and/or royalties, Plaintiffs also requested additional damages, jointly and severally, against Mr. Mack and Virpax as follows: $1.3 million for misuse of Scilex resources, $6.7 million for misappropriation of trade secrets, $13.4 million for exemplary damage (trade secrets damage x2) and attorney’s fees in an unspecified amount. Finally, Plaintiffs sought injunctive relief, enjoining Mr. Mack and Virpax from further accessing Scilex’s trade secrets; requiring Mr. Mack and Virpax to return Scilex’s trade secrets to Plaintiffs; and enjoining Mr. Mack and Virpax from marketing or selling any products derived from or incorporating Scilex’s trade secrets.
On November 29, 2023, in accordance with the Chancery Court’s supplemental briefing schedule, Defendants filed their supplement brief on damages rebutting Plaintiffs’ damages analysis. Throughout the brief, Defendants argued Plaintiffs failed to meet their burden to prove damages, and as such, should be precluded from any damages award. However, given the Court’s instruction, Defendants proffered a reasonable damages analysis as follows. As for the injunctive relief requested against Mr. Mack, the Company took no position, as the request was directed to Mr. Mack personally. Concerning Plaintiffs’ request for an injunction against further development of Epoladerm for a period of 18 months and 27 days, Defendants opposed this request, arguing lack of irreparable harm, given Plaintiffs’ request for money damages. Defendants also argued a constructive trust is inappropriate, given Plaintiffs failed to articulate the parameters of such relief and, additionally, the lack of sales for the drug candidates preclude such relief. In terms of the lawsuit.money damages related to the three drug candidates, Defendants proffered a reasonable royalty rate of 1-3% of the net profits of the drug candidates, as opposed to lump sum damages, as such rate would alleviate the speculative nature of the damages requested by Plaintiffs. As for the misappropriation of trade secrets request of $6.7 million, given the Court found only 5 of the proffered 1,182 documents were trade secrets, Defendants contend Plaintiffs should receive no monetary damages (given the reasonable royalty would encompass use of these documents and, alternatively, Defendants would return such documents). However, if the Court were to award damages, such damages should be pro rata for the documents, or roughly $28,382. And, finally, Defendants opposed the request for attorneys’ fees and exemplary damages.
On December 21, 2023, Plaintiffs filed their reply brief on damages, generally reasserting their prior arguments on damages and rebutting Defendants’ arguments. Plaintiffs also asserted alternativethey supported their damages theories that would imply potential damages upclaims with sufficient evidence.
On February 29, 2024, Plaintiffs and the Company entered into a Settlement Agreement and Mutual Release (the “Settlement Agreement”) to approximately $35.0 million.fully resolve all issues related to settlement of the litigation with Plaintiffs, subject to the entry by the United States Bankruptcy Court for the Southern District of Texas, which is handling the Sorrento bankruptcy filing (the “Bankruptcy Court”), of an order approving the Settlement Agreement (the “Settlement Order”). On March 1, 2024, the Plaintiffs filed a motion to approve the Settlement Agreement and grant the related relief with the Bankruptcy Court. On March 14, 2024, the Bankruptcy Court entered an order approving the Settlement Agreement and on March 20th the Plaintiffs filed a Stipulation of Dismissal with the Chancery Court dismissing the Action.
As settlement consideration, the Company agreed to pay Sorrento and Scilex a total cash payment of $6 million, of which $3.5 million was be paid two business days after the Effective Date, March 18, 2024, and the remaining $2.5 million is to be paid on or before July 1, 2024. The Effective Date is defined as the date the Settlement Order is entered into if there are no objections to Sorrento’s motion for approval of the Settlement Agreement; or the date the Settlement Order becomes non-appealable if there are objections to Sorrento’s motion for approval of the Settlement Agreement. Additionally, the Company countered that actual damages, even ifagreed to pay to Plaintiffs establish liability, could be zero because, among other things, Plaintiffs’ calculations use various unsupported assumptions,royalties of 6% of annual net sales of products developed from drug candidates Epoladerm, Probudur and Envelta until the earlier of the expiration of the last-to-expire valid patent claim of such product and the expiration of any alleged damages are speculative in nature,period of regulatory exclusivity for such product.
The Plaintiffs can still pursue claims against Mr. Mack. The Company’s Amended and thereRestated Bylaws dated November 18, 2020 (“Bylaws”) require the Company to “indemnify any person who was or is a possibilityparty or is threatened to be made a party to any threatened, pending or completed action, suit or proceeding, whether civil, criminal, administrative or investigative (other than an action by or in the Company’s product candidates never reach market. To date, the parties have not had successful settlement negotiations. Based on the foregoing, the Company accrued $2 million in respectright of the litigation. WhileCorporation) by reason of the fact that such person is or was a director or officer of the Corporation, or, while a director or officer of the Corporation….” Such indemnification, however, is limited to circumstances where the covered person “acted in good faith and in a manner such person reasonably believed to be in or not opposed to the best interests of the Corporation….” Mr. Mack may attempt to claim he is entitled to indemnification, should the Court find him liable for damages in the Action. Given the findings in the Memorandum Opinion issued in the Action, the Company believes it has meritorious defenses, the ultimate resolutiona strong position that Mr. Mack would not be entitled to indemnification. There is a risk, however, that a Court could find he is entitled to such indemnification and any such award may be material. Additionally, per Section 7.6 of the actionBylaws, the Company has been advancing Mr. Mack’s attorneys’ fees and costs for the Action. It is likely Mr. Mack will contend he is still entitled to advancement of any fees and/or costs for the Action going forward and may seek judicial intervention. However, as per the Bylaws, Mr. Mack is only entitled to advancement of expenses for indemnifiable actions. As noted above, given the Memorandum Opinion in the Action, the Company believes that it has a strong position that Mr. Mack is not entitled to indemnification, and therefore, not entitled to advancement of expenses. However, there is a risk that a Court could resultfind that Mr. Mack is entitled to such advancement and such amounts may be material. Further, Mr. Mack may attempt to seek damages from the Company based on the Court’s final judgment on damages under the theory of joint and several liability and seek contribution from the Company for any monetary judgment and such amounts may be material.
The Court is aware that Plaintiffs have settled with the Company and that the Settlement Agreement fully releases the Company from any claims or damages, the Plaintiff has against the Company, related to the Action. Given the Settlement Agreement does not release Mr. Mack from liability related to the Action, the Court has requested supplemental briefing as to whether the Court can dismiss the Company from the lawsuit, as well as any claims Mr. Mack has against the Company arising from the Action. While the Company believes that any damages assessed may be awarded against Mr. Mack alone, Plaintiffs cannot seek additional damages from Virpax. However, there is a risk that Mr. Mack will still seek contribution from the Company for any damages claim arising from the Action. And, there is a risk that the Court will rule in a material loss.Mr. Mack’s favor and such amounts may be material.
F-10
As of December 31, 2022, the Company had accrued $2.0 million with respect to the litigation. Based on the facts of the litigation and the Settlement Agreement, the Company has recognized an accrual totaling $6.0 million with respect to the litigation as of December 31, 2023. The Company recognized $4.0 million and $2.0 million for the years ended December 31, 2023 and 2022, respectively, included in general and administrative expenses on the consolidated statements of operations.
Anthony Mack Resignation
On November 15, 2023, the Company accepted the resignation of Anthony P. Mack as Chief Executive Officer (“CEO”) and Chair of the Board of Directors (the “Board”) of the Company effective November 17, 2023. The resignation was not related to any disagreement with the Company on any matter relating to its operations, policies or practices. The Company is negotiating a separation agreement with Mr. Mack and has recorded estimated separation compensation expense related to the separation agreement of $711,000 and included in general and administrative expenses for the year ended December 31, 2023 and in accounts payable and accrued expenses as of December 31, 2023 . While the Company believes this estimated expense related to the separation agreement to be reasonably possible, actual results may materially vary from these estimates. As part of the consideration for the separation agreement, Mr. Mack will be expected to release, discharge and waive any rights to indemnification, and/or contribution related to the Action. The accrual does not include any amounts that we may be required to pay for indemnification claims or contribution that he may seek against us.
Global Macroeconomic Environment
The global macroeconomic environment could be negatively affected by, among other things, resurgence of COVID-19 or other pandemics or epidemics, instability in global economic markets, increased U.S. trade tariffs and trade disputes with other countries, instability in the global credit markets, supply chain weaknesses, instability in the geopolitical environment as a result of the withdrawal of the United Kingdom from the European Union, the Russian invasion of the Ukraine and, the war in the Middle East, other political tensions, and foreign governmental debt concerns. Such challenges have caused, and may continue to cause, uncertainty and instability in local economies and in global financial markets. As a result, the Company and its third party CMOs, and CROs have and may in the future face disruptions in procuring items that are essential to the Company’s research and development activities, including, for example, medical and laboratory supplies used in the Company’s preclinical studies that are sourced from abroad or for which there are shortages, or potential difficulties recruiting patients, and may cause delays and difficulties with ongoing and planned preclinical and clinical trials. The extent to which the Company’s financial condition, liquidity or results of operations are impacted is uncertain, and may negatively impact the Company’s results of operations, financial condition, and liquidity in 2023 and potentially beyond.
Note 7.6. Stockholders’ Equity
Overview
Preferred Stock
The Company’s Certificate of Incorporation, filed on May 12, 2017, and amended and restated on February 16, 2021, authorizes the issuance of preferred stock. The total number of shares which the Company is authorized to issue is 10,000,000, with a par value of $0.00001 per share.
Common Stock
The Company’s Certificate of Incorporation, filed on May 12, 2017, and amended and restated on February 16, 2021, authorizes the issuance of common stock. The total number of shares which the Company is authorized to issue is 100,000,000, with a par value of $0.00001 per share. As of December 31, 2023 and 2022 there were 1,171,233 common shares issued or outstanding.
On February 19, 2021,29, 2024, the Company issued 1,800,000filed a certificate of amendment to the Company’s Amended and Restated Certificate of Incorporation for purposes of effecting a 1-for-10 reverse stock split (the “Reverse Split”) of the Company’s outstanding shares of common stock related tosuch that, effective upon March 1, 2024, the Company’s IPO, for net proceeds totaling $15,783,207,day after deducting underwriting discountsthe filing thereof, every 10 issued and offering expenses. On September 16, 2021, the Company issued 6,670,000 shares of common stock related to the Company’s Underwritten Public Offering, for net proceeds totaling $36,999,465, after deducting underwriting discounts and offering expenses.
The Company issued 45,448outstanding shares of the Company’s common stock upon the exercise of 87,751 options in a cashless exercise during the year ended December 31, 2021.
Warrants
In conjunction with the IPO, the Company granted the underwriters warrants to purchase 90,000 shareswere subdivided and reclassified into one validly issued, fully paid and non-assessable share of the Company’s common stock at an exercise price of $12.50stock.
All share and per share which is 125% ofamounts in the initial public offering price. The warrantsconsolidated financial statements have a five-year term. The fair value allocatedbeen retroactively adjusted for all periods presented to give effect to the warrantsReverse Split, including reclassifying $105 equal to the reduction in par value to additional paid-in capital.
The Reverse Split affected all issued and outstanding shares of $639,000 was accounted for as a component of stockholders’ equity during the year ended December 31, 2021. The fair value of the warrants was estimated using the Black-Scholes option-pricing model and is affected by the Company’s share fair valueCommon Stock, as well as assumptions regarding a numberCommon Stock underlying stock options and warrants outstanding immediately prior to the effectiveness of complex and subjective variables, including a term of 5 years, expected price volatility of 100%, and a risk-free interest rate of 0.6%.the Reverse Split.
Warrants
There were warrants to purchase 18,436exercisable for 1,843 shares of the Company’s common stock outstanding as of December 31, 2023 and 2022. There were no warrants granted, exercised, or forfeited during the yearyears ended December 31, 2023 and 2022. The Company issued 40,221Warrants exercisable for 505 shares have an exercise price of the Company’s common stock upon the$98.89 with expiration date of September 22, 2030. Warrants exercisable for 1,338 shares have an exercise price of 76,620 warrants in a cashless exercise during the year ended December 31, 2021.$125.00 with an expiration date of February 16, 2026.
F-11
Note 8.7. Stock-Based Compensation
On May 20, 2017, the Company established the Virpax Pharmaceuticals, Inc. Amended and Restated 2017 Equity Incentive Plan (the “2017 Plan”). The Company’s Board of Directors (the “Board”), acting through its Equity Incentive Plan Committee, has determined that it would be to the advantage and best interest of the Company and its stockholders to grant restricted stock awards to certain individuals as compensation to serve as an employee of the Company and as an incentive for increased efforts during such service.
Restricted Stock
As of December 31, 2022 and 2021, there were 237 and 6,196 of unvested restricted stock awards issued totaling $2,342 and $39,862, respectively, based on a fair value of the Company’s common stock on the respective date of grant.
During the year ended December 31, 2022 and 2021, there were 0 and 15,000 restricted stock awards granted during the periods, respectively. There were 601 and 937 of restricted stock awards forfeited during the years ended December 31, 2022 and 2021, respectively. The Company recognized $37,520 and $79,438 of stock based compensation for vested restricted shares during the years ended December 31, 2022 and 2021, respectively.
Stock Options
On June 14, 2022, the Company established the Virpax Pharmaceuticals, Inc. 2022 Equity Incentive Plan (the “2022 Plan”) and no new grants of awards will be made under the 2017 Plan and all new grants of awards will be made under the 2022 Plan. The 2022 Plan and 2017 Plan are administered by the Compensation Committee of the Board (the “Compensation Committee”); provided that the entire Board may act in lieu of the Compensation Committee on any matter. The 2022 Plan enables the Company to continue to provide equity and equity-based awards to eligible employees, officers, non-employee directors and other individual service providers by reserving 1,500,000150,000 shares of the Company’s common stock for issuance under the 2022 Plan, as adjusted per the Reverse Split, subject to a 2% annual increase (like(similar to the 2017 Plan) under the 2%pursuant to an “evergreen” provision ofin the 2022 Plan (discussed further below). The Company believes that offering ownership interests in the Company is a key factor in retaining and recruiting employees, officers, non-employee directors and other individual service providers, and aligning and increasing their interests in the Company’s success.success.
The 2022 Plan (which is summarized below) is substantially similar to the 2017 Plan, except for (i) the increase in shares of common stock reserved for issuance as discussed above, and (ii) the elimination of annual limitations on grants of awards to eligible individuals and certain other provisions which had been included in the 2017 Plan in order to satisfy (now repealed) provisions of Section 162(m) of the Internal Revenue Code of 1986, as amended.
The 2022 Plan reserves an aggregate of (i) 1,500,000150,000 shares of ourthe Company’s common stock for the issuance of awards under the 2022 Plan (all of which may be granted as an Incentive Stock Option, or ISOs) plus (ii) an additional number of shares of common stock subject to outstanding awards under the 2017 Plan that become forfeited or canceled without payment or which are surrendered in payment of the exercise price and/or withholding taxes (collectively, the “Share Limit”). Pursuant to the 2022 Plan’s “evergreen” provision, the Share Limit shall be cumulatively increased on January 1, 2023, and on each January 1 thereafter, by 2% of the number of shares of common stock issued and outstanding on the immediately preceding December 31 or such lesser number of shares as determined by ourthe Board. The 2022 Plan increased by 23,428 shares on January 1, 2023.
In applying the aggregate share limitation under the 2022 Plan, shares of common stock (i) subject to awards that are forfeited, cancelled, returned to the Company for failure to satisfy vesting requirements or otherwise forfeited, or terminated without payment being made thereunder and (ii) that are surrendered in payment or partial payment of the exercise price of an option or stock appreciation right or taxes required to be withheld with respect to the exercise of Stock Options or stock appreciation rights or in payment with respect to any other form of award are not counted and, therefore, may be made subject to new awards under the 2022 Plan.
F-12
The 2022 Plan provides that:
All such options will become exercisable on the one-year anniversary of the date of grant.
There was no stock option activity and no stock options outstandingare 116,511 shares available for future grant under the 2022 Plan for the year endedat December 31, 2022.2023.
The Company recognized stock based compensation related to stock options under the 2017 Plan of $707,514 and $894,796 for the years ended December 31, 2022 and 2021, respectively. Total stock based compensation, inclusive of restricted shares and stock options, consists of the following:
| For the Year Ended December 31, | For the Year Ended December 31, | |||||||||||||||
| 2022 | 2021 | 2023 | 2022 | |||||||||||||
| General and administrative expense | 598,655 | 915,423 | $ | 335,363 | $ | 598,655 | ||||||||||
| Research and development expense | 146,379 | 58,811 | 209,407 | 146,379 | ||||||||||||
| 745,034 | 974,234 | $ | 544,770 | $ | 745,034 | |||||||||||
The fair value of option awards is estimated using the Black-Scholes option-pricing model. Exercise price of each award is generally not less than the per share fair value in effect as of that award date. The determination of fair value using the Black-Scholes model is affected by the Company’s share fair value as well as assumptions regarding a number of complex and subjective variables, including expected price volatility, risk-free interest rate and projected employee share option exercise behaviors. Options granted or modified under the Plan2017 and 2022 Plans during the years ended December 31, 20222023 and 20212022 were valued using the Black-Scholes option-pricing model with the following weighted-average assumptions:
| For the Year Ended December 31, | For the Year Ended December 31, | |||||||||||||||
| 2022 | 2021 | 2023 | 2022 | |||||||||||||
| Expected term (years) | 5.65 | 5.74 | 5.46 | 5.65 | ||||||||||||
| Risk-free interest rate | 1.96 | % | 1.04 | % | 3.67 | % | 1.96 | % | ||||||||
| Expected volatility | 77.12 | % | 78.98 | % | 113.12 | % | 77.12 | % | ||||||||
| Expected dividend yield | 0.00 | % | 0.00 | % | 0 | % | 0 | % | ||||||||
The Company estimates its expected volatility by using a combination of historical share price volatilities of similar companies within our industry. The risk-free interest rate assumption is based on observed interest rates for the appropriate term of the Company’s options on a grant date. The expected option term assumption is estimated using the simplified method and is based on the mid-point between vest date and the remaining contractual term of the option, since the Company does not have sufficient exercise history to estimate expected term of its historical option awards.
2017 Plan
Restricted stock
As of December 31, 2023 and 2022, there were 0 and 23 of unvested restricted stock awards issued totaling $0 and $2,342, respectively, based on a fair value of the Company’s common stock on the respective date of grant. There were no restricted stock awards granted during the years ended December 31, 2023 and 2022. There were 0 and 60 of restricted stock awards forfeited during the years ended December 31, 2023 and 2022, respectively. The Company recognized $2,342 and $37,520 of stock based compensation for vested restricted shares during the years ended December 31, 2023 and 2022, respectively.
F-13
The following isAs of January 1, 2023, there were a summarytotal of stock option activity under the 2017 Plan for the years ended December 31, 2022 and 2021:
| Number of Shares | Weighted- Average Exercise Price | Weighted- Average Remaining Contractual Term (Years) | Aggregate Intrinsic Value | |||||||||||||
| Options outstanding at January 1, 2021 | 486,101 | $ | 9.89 | 8.68 | $ | - | ||||||||||
| Forfeited | (5,000 | ) | 4.62 | |||||||||||||
| Exercised | (87,751 | ) | 9.89 | |||||||||||||
| Granted | 275,717 | 4.62 | - | - | ||||||||||||
| Options outstanding at December 31, 2021 | 669,067 | 7.75 | 8.34 | - | ||||||||||||
| Forfeited | (15,591 | ) | 3.14 | |||||||||||||
| Cancelled | - | - | ||||||||||||||
| Granted | 518,805 | 2.24 | - | - | ||||||||||||
| Options outstanding at December 31, 2022 | 1,172,281 | $ | 5.38 | 8.11 | $ | - | ||||||||||
| Options exercisable at December 31, 2022 | 620,734 | $ | 7.71 | 7.37 | $ | - | ||||||||||
On January 31, 2022, our Board approved an equity compensation award for the Company’s officers and employees. The Board approved this award of117,210 options to purchase an aggregate of 321,204 shares of Common Stock pursuant to the 2017 Plan. The options, other than Mr. Mack’s, have anoutstanding with a weighted average exercise price of $2.13 per share, the fair market value of the Common Stock on the date of grant. Mr. Mack’s options have an exercise price of $2.34 per share, which represents 110% of the fair market value on the date of grant. The options granted to the officers and employees vest in three equal installments beginning on the one-year anniversary of the grant date and have a ten-year expiration date.
On January 1, 2022, options were granted to the Non-Employee Directors pursuant to the 2017 Plan to purchase an aggregate of 77,601 shares of Common Stock. The options have an exercise price of $3.43 per share, the fair market value of the Common Stock on the date of grant. The options granted to the directors will vest upon the one-year anniversary of the grant date and have a ten-year expiration date.
On April 25, 2022, options were granted to a consultant pursuant to the 2017 Plan to purchase an aggregate of 60,000 shares of Common Stock. The options have an exercise price of $1.76 per share, the fair market value of the Common Stock on the date of grant. The options granted to the consultant will vest upon the one-year anniversary of the grant date and have a ten-year expiration date. Options were also granted to the Company’s EVP Commercial Operations and director pursuant to the 2017 Plan to purchase an aggregate of 60,000 shares of Common Stock. The options have an exercise price of $1.76 per share, the fair market value of the Common Stock on the date of grant. The options granted to this individual vest immediately upon grant and have a ten-year expiration date.
The weighted-average grant-date fair value of stock options granted during$53.81. During the year ended December 31, 2022 was $1.98.
2023, there were a total of 18,324 options forfeited with a weighted average exercise price of $44.56. As of December 31, 2022,2023, there was $519,190were a total of 98,886 options outstanding with a weighted average exercise price of $55.52 and weighted average remaining contractual life of 7.1 years, of which 82,880 options are exercisable with a weighted average exercise price of $61.25 and weighted average remaining contractual life of 7.0 years. As of December 31, 2023, $121,000 of total time-based unrecognized compensation costs related to unvested stock options.options within the 2017 Plan. These costs are expected to be recognized over a weighted average period of 1.640.9 years.
2022 Plan
The following is a summary of stock option activity under the activity under the 2022 Plan for the year ended December 31, 2023:
| 2022 Plan: | Number of Shares (in thousands) | Weighted Average Exercise Price | Weighted- Average Remaining Contractual Term (Years) | Aggregate Intrinsic Value (in thousands) | ||||||||||||
| Options outstanding at January 1, 2023 | — | — | — | — | ||||||||||||
| Forfeited | (24,050 | ) | 8.29 | — | — | |||||||||||
| Exercised | — | — | — | — | ||||||||||||
| Granted | 100,850 | 7.80 | — | — | ||||||||||||
| Options outstanding at December 31, 2023 | 76,800 | $ | 7.67 | 9.3 | — | |||||||||||
| Options exercisable at December 31, 2023 | 10,000 | $ | 7.30 | 9.5 | — | |||||||||||
Under the 2022 Plan, the Company may grant equity-based awards to individuals who are employees, officers, directors, or consultants of the Company. Options issued under the 2022 Plan will generally expire ten years from the date of grant and vest over a one-year to three-year period.
In accordance with Mr. Chipman’s Separation Agreement with the Company, he received accelerated vesting of 23,812 of his outstanding stock options (“Accelerated Options”) as of his separation date of June 30, 2023. Additionally, these Accelerated Options could be exercised until severance was fully paid, which was on October 15, 2023. Accelerated Options were not exercised as of that date and were cancelled. The accelerated vesting and increase in the time to exercise option awards after termination was treated as a stock option modification under ASC 718, Compensation - Stock Compensation. The total incremental expense resulting from the modification was de minimis for the year ended December 31, 2023.
The weighted-average grant-date fair value of stock options granted during the years ended December 31, 2023 and 2022 was $6.34 and $19.80, respectively.
As of December 31, 2023, there was $231,000 of total time-based unrecognized compensation costs related to unvested stock options under the 2022 Plan. These costs are expected to be recognized over a weighted average period of 2 years.
Note 9. Income Taxes
Note 8. Income Taxes
Deferred income taxes reflect the net effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. The Company’s deferred tax assets relate primarily to its net operating loss carryforwards and other balance sheet basis differences. In accordance with ASC 740, the Company recorded a valuation allowance to fully offset the deferred tax asset, because it is not more likely than not that the Company will realize future benefits associated with these deferred tax assets at December 31, 20222023 and 2021.2022. During the years ended December 31, 20222023 and 2021,2022, there was no income tax expense.
F-14
Significant components of the Company’s deferred tax assets at December 31, 20222023 and 2021:2022:
| December 31, 2023 | December 31, 2022 | |||||||
| Deferred tax assets: | ||||||||
| Net-operating loss carryforwards | $ | 8,854,000 | $ | 8,206,000 | ||||
| Capitalized R&D costs | 3,562,000 | 2,868,000 | ||||||
| Stock-based compensation | 829,000 | 922,000 | ||||||
| Accrued expenses | 1,989,000 | 743,000 | ||||||
| R&D credit | 654,000 | 546,000 | ||||||
| Other | 5,000 | 6,000 | ||||||
| Total deferred tax assets | 15,893,000 | 13,291,000 | ||||||
| Valuation allowance | (15,893,000 | ) | (13,291,000 | ) | ||||
| Net deferred tax asset | $ | — | $ | — | ||||
| December 31, 2022 | December 31, 2021 | |||||||
| Deferred tax assets: | ||||||||
| Net-operating loss carryforwards | $ | 8,206,000 | $ | 5,771,000 | ||||
| Capitalized R&D costs | 2,868,000 | - | ||||||
| Stock-based compensation | 922,000 | 805,000 | ||||||
| Accrued expenses | 743,000 | - | ||||||
| R&D credit | 546,000 | 86,000 | ||||||
| Other | 6,000 | 6,000 | ||||||
| Total deferred tax assets | 13,291,000 | 6,668,000 | ||||||
| Valuation allowance | (13,291,000 | ) | (6,668,000 | ) | ||||
| Net deferred tax asset | $ | — | $ | — | ||||
The Tax Cuts and Jobs Act modified the section 174 rules and beginning in 2022, taxpayers may no longer currently deduct R&D expenditures but instead must amortize specified R&D expenditures ratably over five years (or 15 years for foreign expenditures). Gross capitalized R&D Costs for the yearyears ended December 31, 2023 and 2022 amounted to approximately $10,559,000.$5,118,000 and $10,559,000, respectively.
The change in the valuation allowance for the years ended December 31, 20222023 and 20212022 was an increase of approximately $6,623,000$2,602,000 and $3,460,000,$6,623,000, respectively.
The Company’s reconciliation of the federal statutory tax rate and the effective tax rates for the years ended December 31, 20222023 and 20212022 is as follows:
| December 31, 2023 | December 31, 2022 | |||||||
| Federal statutory rate | 21.0 | % | 21.0 | % | ||||
| Increase (decrease) in tax expense at federal statutory rate | ||||||||
| State income taxes | 5.0 | % | 7.9 | % | ||||
| Change in state income tax rate | (9.2 | )% | 0.0 | % | ||||
| Change in valuation allowance | (17.1 | )% | (31.0 | )% | ||||
| Other | 0.3 | % | 2.1 | % | ||||
| Effective tax rate | 0.0 | % | 0.0 | % | ||||
| December 31, 2022 | December 31, 2021 | |||||||
| Federal statutory rate | 21.0 | % | 21.0 | % | ||||
| Increase (decrease) in tax expense at federal statutory rate | ||||||||
| State income taxes | 7.9 | % | 7.9 | % | ||||
| Change in valuation allowance | (31.0 | )% | (28.7 | )% | ||||
| Other | 2.1 | % | 0.2 | % | ||||
| Effective tax rate | 0.0 | % | 0.0 | % | ||||
The Company had approximately $28,397,000$35,164,000 of gross net operating loss (“NOL”) carryforwards for both federal and apportioned state NOL’s of approximately $37,095,000 as of December 31, 2022.2023. The Company has an R&D tax credit carryforward of approximately $654,000 as of December 31, 2023.
The Company’s ability to use net operating loss, other carry forwards and tax credits is subject to limitation in subsequent periods under certain provisions of Section 382 and Section 383 of the Internal Revenue Code of 1986, as amended, upon a more than 50% change in ownership of the Company’s stock by a 5% or greater shareholder. The Company examined the application of Section 382 with respect to ownership changes that took place during 2021, as well as the limitation on the application of net operating loss carry forwards. The Company has determined that a more than 50% ownership change occurred on September 16, 2021. The Company has determined that the recent change in ownership limits the Company’s usage of net operating loss, other carry forwards and tax credits of approximately $19,417,000 as of the change in ownership date to an annual amount of approximately $4.1 million which will be released by December 31, 2028. The Company’s net carryforwards and tax credits may be further limited in the future if additional ownership changes occur.
From the total of the Company’s federal NOL of $28,397,000,$35,164,000, $326,000 expires in 2037, and the remaining NOL has an indefinite carryover period but its usage is limited to 80% of taxable income in any subsequent year. The Company’s state NOL’s of $28,397,000$37,095,000 expire from 2037 through 2042.2043. Additionally, the Company has $546,000$654,000 of R&D credits which have a 20 year20-year carryforward period, which will expire from 2038 to 2042.2043.
F-15
Note 10.9. Research and Development and License Agreements
MedPharm Limited
Research and Option Agreement
On April 11, 2017, the Company entered into a research and option agreement, as amended on May 30, 2018 (the “MedPharm Research and Option Agreement”), with MedPharm Limited, a company organized and existing under the laws of the United Kingdom (“MedPharm”), pursuant to which MedPharm granted the Company an option to obtain an exclusive, world-wide, royalty bearing license to use certain technology developed by MedPharm. Pursuant to the MedPharm Research and Option Agreement, MedPharm will conduct certain research and development of proprietary formulations incorporating certain MedPharm technologies and certain of the Company’s proprietary molecules.
Under the MedPharm Research and Option Agreement, MedPharm granted the Company an option (the “MedPharm Option”) to obtain an exclusive (even to MedPharm), worldwide, sub-licensable (through multiple tiers), royalty bearing, irrevocable license to research, develop, market, commercialize, and sell any product utilizing MedPharm’s spray formulation technology which is the result of the activities performed under the MedPharm Research and Option Agreement, subject to the Company’s entry into a definitive license agreement with MedPharm. In order to exercise the MedPharm Option, the Company must provide MedPharm with written notice of such exercise before the end of the Option Period (as defined in the MedPharm Research and Option Agreement). The Option Period is subject to extension upon mutual agreement with MedPharm.
Pursuant to the MedPharm Research and Option Agreement, the Company has a right of first refusal with respect to any license or commercial arrangement involving any Licensed Intellectual Property (as defined in the MedPharm Research and Option Agreement) in combination with any Virpax Molecule (as defined in the MedPharm Research and Option Agreement). In the event that MedPharm reaches an agreement with respect to a license or other commercial arrangement that involves technology or molecules covered by the right of first refusal, the Company has ten business days from the date of notice to notify MedPharm of its intention to exercise the right of first refusal and the Company’s intention to match the financial terms of the other license or commercial arrangement.
License Agreement
On June 6, 2017, as a result of the Company’s exercise of the MedPharm Option under the MedPharm Research and Option Agreement, the Company entered into a license agreement, as amended on September 2, 2017 and October 31, 2017 (the “MedPharm License Agreement”), with MedPharm for the exclusive global rights to discover, develop, make, sell, market, and otherwise commercialize any pharmaceutical composition or preparation (in any and all dosage forms) in final form containing one or more compounds, including Diclofenac Epolamine (“Epoladerm”), that was developed, manufactured or commercialized utilizing MedPharm’s spray formulation technology (“MedPharm Product”), to be used for any and all uses in humans (including all diagnostic, therapeutic and preventative uses). Under the MedPharm License Agreement, the Company is required to make future milestone and royalty payments to MedPharm. We are obligated to make aggregate milestone payments to MedPharm of up to GBP 1.150 million upon the achievement of specified development milestones (payable in Great British Pounds). Additional milestone payments are due upon the achievement of certain development and commercial milestones achieved outside the United States, payable on a country-by-country basis. Royalty payments must be paid to MedPharm in an amount equal to a single-digit percentage of net sales of all MedPharm Product sold by us during the royalty term in the territory. Royalties shall be payable, on a country-by-country basis, during the period of time commencing on the first commercial sale and ending upon the expiration of the last-to-expire patent claim on the licensed product, which is set to expire on December 4, 2028. Each party has the right to terminate the agreement in its entirety upon written notice to the other party if such other party is in material breach of the agreement and has not cured such breach within ninety (90) days after notice from the terminating party indicating the nature of such breach.
LipoCureRx, Ltd.
On March 19, 2018, the Company entered into a license and sublicense agreement (the “LipoCure“Lipocure Agreement”) with LipoCureRx,LipocureRx, Ltd., a company organized and existing under the laws of Israel (“LipoCure”Lipocure”), for the sole and exclusive global license and sub-license rights to discover, develop, make, sell, market, and otherwise commercialize bupivacaine liposome, in injectable gel or suspension (“Licensed Compound”) or any pharmaceutical composition or preparation (in any and all dosage forms) in final form, including any combination product, containing a Licensed Compound (“Licensed Product”), including Probudur. Under the LipoCureLipocure Agreement, the Company was required to pay an upfront fee upon signing of $150,000 and is required to make future milestone and royalty payments to LipoCure.Lipocure. The Company is obligated to make aggregate milestone payments of up to $19.8 million upon the achievement of specified development and commercial milestones. Lipocure met the development milestone of $300,000 in the third quarter of 2023 for successfully completing a formulation for the Licensed Product. The Company paid $150,000 in the third quarter of 2023 and paid the balance in the fourth quarter of 2023. Royalty payments must be paid in an amount equal to a single digit to low double-digit percentage of annual net sales of royalty qualifying products, subject to certain adjustments. Royalties shall be payable during the period of time, on a country-by-country basis, commencing on the first commercial sale and ending upon the expiration of the last-to-expire patent claim on the licensed product, which is set to expire on July 24, 2030. Each party has the right to terminate the agreement in its entirety upon written notice to the other party if such other party is in material breach of the agreement and has not cured such breach within ninety (90) days after notice from the terminating party indicating the nature of such breach.
F-16The Company incurred $300,000 and $0 in research and development expenses, respectively, for the years ended December 31, 2023 and 2022. associated with this Lipocure agreement.
Nanomerics Ltd.
Nanomerics Collaboration Agreement
On April 11, 2019, the Company entered into an exclusive collaboration and license agreement, as amended (the “Nanomerics Collaboration Agreement”), with Nanomerics Ltd., a company organized and existing under the laws of United Kingdom (“Nanomerics”), for the exclusive world-wide license to develop and commercialize products, including NES100,Envelta™, which contain hydrophilic neuropeptide Leucin5-Enkephalin and an amphiphile compound which is quaternary ammonium palmitoyl glycol chitosan, to engage in a collaborative program utilizing Nanomerics’ knowledge, skills and expertise in the clinical development of products and to attract external funding for such development. The Nanomerics Collaboration Agreement was also amended to include a program for the pre-clinical development of a product for post-traumatic stress disorder.disorder (“PTSD”).
Under the Nanomerics Collaboration Agreement, the Company is required to make royalty payments equal to a single digit percentage of annual net sales of royalty qualifying products. The Company is also required to make aggregate milestone payments of up to $103 million upon the achievement of specified development and commercial milestones, and sublicense fees for any sublicense relationships we enterit enters into subsequent to the Nanomerics Collaboration Agreement. The Company’s obligation to pay royalties, on a country-by-country basis, shall commence on the date of first commercial sale of its licensed products and shall expire with respect to each separate licensed product, on the latest to occur of (a) the tenth (10th) anniversary of the first commercial sale of the first licensed product; (b) the expiration date of the last to expire of any valid claim (patent is set to expire on November 3, 2034); and, (c) the date upon which a generic product has been on the market for a period of no fewer than ninety (90) days. The Company has the right to terminate the agreement upon 180 days’ prior written notice to Nanomerics. Upon termination, the Company shall assign to Nanomerics all its right title and interest in all results other than results specific to (a) the Device (as defined in the Nanomerics Collaboration Agreement), including its manufacture or use; and (b) the Technology, but excluding any clinical Results relating to the Compound or Licensed Products (all terms as defined in the Nanomerics Collaboration Agreement).
Nanomerics License Agreement (AnQlar)
On March 9, 2022, the Company entered into an Amended and Restated Collaboration and License Agreement with Nanomerics (the “Amended Nanomerics License Agreement”) which amended and restated the August 7, 2020, Nanomerics License Agreement and expanded the Company’s North American rights for AnQlar to include exclusive global rights to develop and commercialize AnQlar as a viral barrier to prevent or reduce the risk or the intensity of viral infections. The Amended Nanomerics License Agreement provides for payments up to $5.5 million upon the achievement of specified development milestones and profit share payments equal to between 30% to 40% of certain profits (as set forth in the Amended Nanomerics License Agreement), payable to Nanomerics upon the achievement of specified commercial milestones. The profit share payments are triggered upon determination by the FDA that AnQlar may be marketed as an Over-the-Counter product in the United States. In the event the profit share payments are not triggered as defined above, the Company’s would be obligated to pay royalties within a range of 5% to 15% of annual net sales of royalty qualifying products and commercial milestones on a worldwide basis amounting to aggregate milestone payments of up to $112.5 million upon the achievement of these commercial milestones. The Amended Nanomerics License Agreement also provides for additional aggregate milestone payments totaling $999,999 upon first receipt of regulatory approval for a licensed product in the European Union, Asia/Pacific region and South America/Middle East region. The Company’s obligation to pay royalties, on a country-by-country basis, shall commence on the date of first commercial sale of its licensed products and shall expire with respect to each separate licensed product, on the latest to occur of (a) the tenth (10th) anniversary of the first commercial sale of the first licensed product; (b) the expiration date of the last to expire of any valid claim; and, (c) the date upon which a generic product has been on the market for a period of no fewer than ninety (90) days. The Company has the right to terminate the Nanomerics License Agreement upon sixty (60) days’ prior written notice to Nanomerics. Upon termination, the Company shall assign to Nanomerics all its rights, title and interest in all of its results. Nanomerics has the right to terminate the agreement upon sixty (60) days’ prior written notice. In consideration for entering into this Amended Nanomerics License Agreement, the Company paid Nanomerics a nonrefundable fee of $1,500,000 in March 2022, which is included in research and development expenses during the year ended December 31, 2022.
F-17
Nanomerics License Agreement (NobrXiol, formerly VRP324)
On September 17, 2021, the Company entered into a collaboration and license agreement with Nanomerics (the “Nanomerics License Agreement - NobrXiol”) for the exclusive worldwide license to develop and commercialize an investigational formulation delivered via the nasal route to enhance pharmaceutical-grade cannabidiol (“CBD”) transport to the brain to potentially treat seizures associated with tuberous sclerosis complex (TSC), Lennox-Gastaut syndrome and Dravet syndrome in patients one year of age and older. Lennox-Gastaut syndrome and Dravet syndrome are rare central nervous system diseases considered serious epileptic encephalopathies that cause different types of epileptic seizures as well as cognitive and behavioral changes and are generally resistant to treatment. Under the Nanomerics License Agreement – NobrXiol, the Company is required to make royalty payments within a range of 5% to 15% of annual net sales of royalty qualifying products. The Company’s obligation to pay royalties, on a country-by-country basis, shall commence on the date of first commercial sale of its licensed products and shall expire with respect to each separate licensed product, on the latest to occur of (a) the fifteenth (15th) anniversary of the first commercial sale of the first licensed product; (b) the expiration date of the last to expire of any valid claim; and, (c) the date upon which a generic product has been on the market for a period of no fewer than ninety (90) days. The Company paid an upfront milestone payment upon signing of $200,000 and is required to make future milestone and royalty payments of up to $41 million upon the achievement of specified development and commercial milestones, and sublicense fees for any sublicense relationships the Company enters into subsequent to the Nanomerics License Agreement (any patent that is issued from the currently filed provisional patent application would expire on August 24, 2041). The Company has the right to terminate the Nanomerics License Agreement upon one hundred and eighty (180) days’ prior written notice to Nanomerics. Upon termination, the Company shall assign to Nanomerics all its rights, title and interest in all of its results. Nanomerics has the right to terminate the agreement upon thirty (30) days’ prior written notice if the Company concludes in writing to Nanomerics that the study aim has not been achieved or the Company notifies Nanomerics that the Company has decided against proceeding with a Phase III3 Clinical trial.
On April 21, 2022, we notified Nanomerics that the study aim of demonstrating the ability of Nanomerics platform technology delivering CBD to the brain via nasal administration in an animal model was met. Pursuant to the Nanomerics License Agreement - VRP324, we paid a milestone payment of $500,000 upon meeting this study aim in April 2022. We submitted the pre-IND Briefing Book with the FDA in October 2022.
Research Agreements
Yissum
On October 11, 2020, the Company entered into an Agreement for Rendering of Research Services with Yissum (the “October 2020 Yissum Research Agreement”). Under the October 2020 Yissum Research Agreement, the Company shall provide funding for research and development studies to be performed by researchers at Hebrew University related to the formulation of Liposomal Bupivacaine as well as efficacy and PK studies in animals. In consideration for the research services, the Company agreed to pay research service fees of $81,000 in six equal monthly installments. In connection with the completion of the Company’s IPO, the Company paid Yissum $40,500 towards the total consideration of $81,000. All services to be provided under the October 2020 Yissum Research Agreement were completed by June 30, 2021.
On June 30, 2021, the Company entered into an Agreement for Rendering of Research Services with Yissum Research Development Company of the Hebrew University of Jerusalem Ltd (“Yissum”) (the “June 2021 Yissum Research Agreement”). Under the June 2021 Yissum Research Agreement, the Company provided funding for research and development studies performed by researchers at Hebrew University related to the optimization of the Liposomal Bupivacaine formulation (Probudur) and to increase stability for manufacturing purposes. In consideration for the research services, the Company agreed to pay research service fees of $337,500 in six equal quarterly installments.
On January 31, 2023, the Company entered into an Agreement for Rendering of Research Services with Yissum (the “January 2023 Yissum Research Agreement”) on substantially similar terms and conditions as detailed above under the October 2020June 2021 Yissum Research Agreement. Under the June 2021January 2023 Yissum Research Agreement, the Company shallagreed to provide funding for research and development studies to be performed by researchers at Hebrew University related to the optimization of the Liposomal Bupivacaine formulation (Probudur) and to increase stability for manufacturing purposes. The Company may terminate the agreement at any time and shall be only responsible to pay Yissum for work performed through the date of termination. In consideration for the research services, the Company agreed to pay research service fees of $337,500$326,000 in sixfour equal quarterly installments. All services to be provided underinstallments ($81,500 per calendar quarter).
The Company incurred $326,000 and $225,000 in research and development expenses respectively for the June 2021years ended December 31, 2023 and 2022 associated with these Yissum Research Agreement initiated on July 1, 2021 and were substantially complete by the end of 2022. See also Note 10 - Subsequent Events for discussion of January 2023 Yissum Research Agreement, entered into on January 31, 2023.agreements.
F-18
Lipocure
On June 29, 2021, the Company entered into an Agreement for Rendering of Research Services (the “June 2021 Lipocure Research Agreement”) with Lipocure RX, Ltd. (“Lipocure”). Under the June 2021 Lipocure Research Agreement, the Company shallagreed to provide funding for research and development related to the optimization of the Liposomal Bupivacaine formulation and eventual manufacture of pre-clinical batches including batches for stability testing, animal studies, toxicology, and toxicologypatent related work. This will also include work associated with the potential filing of additional provisional patent applications. The Company may terminate the agreement at any time upon 30 days written notice and shall be only responsible to pay Lipocure for work performed through the date of such notice. In consideration for the research services, the Company agreed to pay research service fees of $200,000 upon execution, as well as $400,000 in July 2021, $270,000 in both September 2021 and January 2022, and three additional payments of $270,000 during 2022. The Company also agreed to pay $250,000 to Lipocure upon successful completion of a Chemistry, Manufacturing and Controls “CMC” filing with the U.S. Food and Drug Administration (the (“FDA”). All
On February 1, 2023, the Company entered into an Agreement for Rendering of Research Services with Lipocure on similar terms and conditions and for similar services - optimization of the Liposomal Bupivacaine formulation, manufacture of pre-clinical batches including batches for stability testing, animal studies, toxicology, and patent related work. In consideration for the research services, the Company agreed to be provided under the June 2021 Lipocure Research Agreement initiated on July 1, 2021 and were substantially complete by the endpay research service fees of 2022. $1,286,000 in four equal quarterly installments ($321,500 per calendar quarter), as well as reasonable pass-through expenses.
The Company recorded $990,000incurred $1,453,000 and $870,000$1,220,000 in research and development expenseexpenses, respectively, for the years ended December 31, 20222023 and 2021, respectively,2022 associated with this agreement. See also Note 10 - Subsequent Events for discussion of January 2023these Lipocure Research Agreement, entered into on February 1, 2023.agreements.
NCATS-NIH Cooperative Research and Development Agreement
On August 25, 2020, the Company entered into a Cooperative Research and Development Agreement (“CRADA”) with the National Center for Advancing Translational Science (“NCATS”). This collaboration is for the continued development of the Company’s product candidate, NES100,Envelta, an intranasal peptide, for the management of acute and chronic non-cancerto control severe pain, including post cancer pain. The term of the CRADA is for a period of four years from May 6, 2020 (the effective date of the agreement) and can be terminated by both parties at any time by mutual written consent. In addition, either party may unilaterally terminate the CRADA at any time by providing written notice of at least sixty (60) days before the desired termination date. The agreement provides for studies that are focused on the pre-clinical characterization of NES100Envelta as a novel analgesic for acute and chronic non-cancerto control severe pain, including post cancer pain, and for studies to further develop NES100Envelta through investigative new drug (“IND”)IND enabling studies. There are certain development “Go/No Go” provisions within the agreement whereby, if certain events occur, or do not occur, NCATS may terminate the CRADA. These “No GO” provisions include: i) lack of efficacy in all animal pain models, ii) no reliable and sensitive bioanalytical method can be developed, iii) manufacturing failure due to inherent process scalability issues, iv) unacceptable toxicity or safety profile to enable clinical dosing, and v) inability to manufacture the NES100Envelta dosage form. As of March 25, 2024, we have not received any Go/No Go notifications from NCATS.
With respect to NCATS rights to any invention made solely by an NCATS employee(s) or made jointly by an NCATS employee(s) and our employee(s), the CRADA grants to the Company an exclusive option to elect an exclusive or nonexclusive commercialization license. For inventions owned solely by NCATS or jointly by NCATS and the Company, and licensed pursuant to the Company’s option, the Company must grant to NCATS a nonexclusive, nontransferable, irrevocable, paid-up license to practice the invention or have the invention practiced throughout the world by or on behalf of the United States government. For inventions made solely by an employee of the Company, we grant to the United States government a nonexclusive, nontransferable, irrevocable, paid-up license to practice the invention or have the invention practiced throughout the world by or on behalf of the United States government for research or other government purposes.
U.S Army Institute of Surgical Research
On April 28, 2022, the Company entered into a CRADA with the U.S. Army Institute of Surgical Research (USAISR) to evaluate Probudur.Probudur as a potential novel analgesic for battlefield injury-induced pain solution. The research project will evaluate the analgesic effectiveness and physiologic effects of Probudur. ThisThe initial term of this agreement will automaticallywas to expire on September 30, 2023 unless it iswas revised by mutual written agreement. The CRADA was modified and signed on October 10, 2023, and extended the terms of the agreement until September 2024. No funding is being provided by either party to the other party under the agreement. Each party is responsible for funding its own work performed and other activities undertaken for the research project under this agreement. The parties may elect to terminate this agreement, or portions thereof, at any time by mutual consent. Either party may unilaterally terminate this entire agreement at any time by giving the other party written notice, not less than thirty (30) days prior to the desired termination date.
F-19
Note 11.10. Subsequent Events
The Company has evaluated subsequent events from the balance sheet date through March 22, 2022. The25, 2024. In addition to those disclosed in Note 5 and Note 6, the following are subsequent events:
On January 1, 2023, options were granted to the Non-Employee Directors pursuant to the 2022 Plan to purchase an aggregate of 155,000 shares of Common Stock. The options have an exercise price of $0.622 per share, the fair market value of the Common Stock on the date of grant. The options granted to the directors will vest upon the one-year anniversary of the grant date and have a ten-year expiration date.
On January 25, 2023, our Compensation Committee approved an equity compensation award for the Company’s officers and employees. The Committee approved this award of options to purchase an aggregate of 528,500 shares of Common Stock pursuant to the 2022 Plan. The options, other than Mr. Mack’s, have an exercise price of $0.788 per share, the fair market value of the Common Stock on the date of grant. Mr. Mack’s options have an exercise price of $0.867 per share, which represents 110% of the fair market value on the date of grant and a five year expiration date. The remaining options granted to the officers and employees vest in three equal installments beginning on the one-year anniversary of the grant date, and have a ten-year expiration date.
On January 31, 2023, the Company2024, we entered into an Agreement for Rendering of Research Services with Yissum (the “January 20232024 Yissum Research Agreement”) for additional work on substantially similar termsformulation, method development, animal studies and conditions as detailed above underpatent related work. In consideration for the June 2021 Yissum Research Agreement. Under the January 2023 Yissum Research Agreement, the Company shall provide funding for research and development studies to be performed by researchers at Hebrew University related to the optimizationservices, we will pay research service fees of the Liposomal Bupivacaine formulation and to increase stability for manufacturing purposes. The Company$343,467 in four equal quarterly installments. We may terminate the agreement at any time and shallwill only be only responsible to pay Yissum for work performed through the date of termination. In consideration for the research services, the Company agreed to pay research service fees of $326,000 in four equal quarterly installments ($81,500 per calendar quarter). All services to be provided under the January 2023 Yissum Research Agreement initiated in January 1, 2023 and are anticipated to be completed towards the end 2023.
On February 1, 2023, the Company entered into an Agreement for Rendering of Research Services (the “January 2023 Lipocure Research Agreement”) with Lipocure. Under the January 2023 Lipocure Research Agreement, the Company shall provide funding for research and development related to the optimization of the Liposomal Bupivacaine formulation and eventual manufacture of pre-clinical batches including batches for stability testing, animal studies and toxicology work. This will also include work associated with the potential filing of additional provisional patent applications. The Company may terminate the agreement at any time upon 30 days written notice and shall be only responsible to pay Lipocure for work performed through the date of such notice. In consideration for the research services, the Company agreed to pay research service fees of $1,286,000 in four equal quarterly installments ($321,500 per calendar quarter). All services to be provided under the January 2023 Lipocure Research Agreement initiated on January 1, 2023 and are anticipated to be completed towards the end 2023.
In March 2023, the Company received $1,250,000 in reimbursement of legal costs pursuant to the Company’s D&O policy.
F-21