_________________________________
2022 Delaware 34-1940305 7555 Innovation Way, Mason, OH 45040 (Address of principal executive offices) (Zip Code) Title of each class Trading Symbol(s) Name of each exchange on which registered Common Stock, $.001 par value ATRC NASDAQ Global Market Class Outstanding February 17, 2023 Common Stock, $.001 par value SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS globally. manner that will cure them. In addition, Afib is thought to be responsible for approximately 15% to 20% of the estimated 800,000 strokes that occur annually in the United States. According to the American Heart Association, the risk of stroke is five times higher in people with Afib. Studies have also suggested that 90% of clots that cause strokes in patients who have Afib originate from within the LAA. Recently, a very large independent international randomized trial, Left Atrial Appendage Occlusion Study (LAAOS) III, demonstrated a significant reduction in strokes when the LAA was managed during cardiac surgery. Afib accounts for billions of dollars in hospitalization-related and office visit costs in the United States each year. Indirect costs, such as the management of Afib-related strokes, are also believed to be significant. Because of the risk of stroke and the significant cost burden on the healthcare system, more and more surgeons are routinely addressing the LAA, both in patients who have Afib, but also in those who do not have Afib but may be at increased risk of developing the disease in the future. We believe that our AtriClip system is safer, more effective and easier to use than other products and techniques for excluding the LAA during cardiac surgery. Therefore, we believe that the market for our ablation products and the AtriClip system represent a significant growth opportunity. addiction to prescription narcotics, making alternative, non-opioid pain management modalities, such as other benefits. AtriClip System. The AtriClip® reusable cardiac surgery instruments. In the United States, a significant risk device requires the prior submission of an application for an Investigational Device Exemption (IDE) to FDA for approval before initiating a clinical trial. Clinical trials are required to support a pre-market approval (PMA) and are sometimes required for 510(k) clearance. Some trials require a feasibility study followed by a pivotal trial. In April 2022, FDA approved the protocol for the Left Atrial Appendage Exclusion for Prophylactic Stroke Reduction (LeAAPS) IDE clinical trial. The trial is designed to evaluate the effectiveness of prophylactic LAA exclusion using the AtriClip LAA Exclusion System for the prevention of ischemic stroke or systemic arterial embolism in cardiac surgery patients without pre-operative AF diagnosis who are at risk for these events. This prospective, multicenter, randomized trial evaluates safety at 30 days post-procedure to demonstrate no increased risk with LAA exclusion during cardiac surgery and effectiveness with a minimum follow-up of five years post procedure for all subjects. The trial provides for enrollment of up to 6,500 subjects at up to 250 sites worldwide. Site initiation and enrollment is ongoing. DEEP AF Pivotal clinical trials. Sales, Marketing and Medical Education a similar therapy. Physicians are reimbursed for their services separately under the Medicare Part B physician fee schedule. When performing a surgical cardiac ablation with and without a concomitant open-heart procedure, surgeons report Current Procedural Terminology (CPT) codes to receive a professional fee payment. Multiple CPT codes may be reported by a physician during a procedure if multiple procedures are performed. There are category one CPT codes for both concomitant and standalone surgical Afib Pervasive and Continuing Regulation. There are numerous regulatory requirements that apply after a product is cleared or approved by removals. Our manufacturing facilities and processes are also subject to FDA inspections to ensure compliance with QSR. professionals. Afib and other diseases and conditions. components, specific supplier requirements and current market demand for the components and subassemblies. To date, we have not experienced significant product availability or delay issues directly related to obtaining any of our components. harmed, and we may not achieve or sustain profitability. results. products to compete directly with ours. Companies also compete with us to attract qualified scientific and technical personnel as well as funding. Most of our competitors and potential competitors have greater financial, manufacturing, marketing and research and development capabilities than we have, and may obtain FDA approval or clearance for their Our clinical trials are expensive to conduct, typically products to be marketed specifically for those indications. Some payors may deny coverage or payment for the use of our products for indications not specifically approved or cleared by FDA. Often, these denials can be overcome through an appeals process, but there is no guarantee of success in these cases. We rely upon single and limited source third-party suppliers and third-party operations. materially adversely impacted. emission or disposal of hazardous substances that could cause us to incur substantial liabilities, including costs for investigation and remediation. 73. 2022. Effective December 31, 2022, we have ceased use of the NASDAQ Medical Equipment Index and transitioned to use of the NASDAQ Health Care Index for the comparison. We believe the NASDAQ Health Care Index to be a more appropriate index for this comparison, as it is more accessible to stockholders than the NASDAQ Medical Equipment Index and is widely recognized and used. 12/31/2015 12/31/2016 12/31/2017 12/31/2018 12/31/2019 AtriCure, Inc. $ 112.42 $ 98.05 $ 91.38 $ 153.31 $ 162.88 NASDAQ Composite $ 106.96 $ 116.45 $ 150.96 $ 146.67 $ 200.49 NASDAQ Medical Equipment $ 111.06 $ 116.87 $ 166.41 $ 187.88 $ 227.84 Year Ended December 31, 2019 (1) 2018 (2) 2017 2016 2015 (3) (in thousands, except per share data) Operating Results: Revenue $ 230,807 $ 201,630 $ 174,716 $ 155,109 $ 129,755 Gross profit $ 170,335 $ 147,120 $ 126,163 $ 111,101 $ 92,875 Gross margin 73.8% 73.0% 72.2% 71.6% 71.6% Net loss $ (35,194) $ (21,137) $ (26,892) $ (33,338) $ (27,212) Basic and diluted net loss per share $ (0.94) $ (0.62) $ (0.83) $ (1.05) $ (0.97) Weighted average shares outstanding 37,589 34,087 32,387 31,609 28,058 Financial Position: Cash, cash equivalents and investments $ 94,476 $ 124,402 $ 34,451 $ 47,009 $ 42,284 Working capital 93,244 134,457 50,355 56,889 43,164 Total assets 557,880 356,759 267,704 276,421 273,092 Long-term debt and leases 74,204 47,743 36,861 37,205 13,710 Stockholders’ equity 247,343 249,381 161,166 168,442 186,685 2021 Year Ended December 31, 2019 2018 % of % of Amount Revenue Amount Revenue (dollars in thousands) Revenue $ 230,807 100.0 % $ 201,630 100.0 % Cost of revenue 60,472 26.2 54,510 27.0 Gross profit 170,335 73.8 147,120 73.0 Operating expenses: Research and development expenses 41,230 17.9 34,723 17.2 Selling, general and administrative expenses 162,227 70.3 129,524 64.2 Total operating expenses 203,457 88.2 164,247 81.5 Loss from operations (33,122) (14.4) (17,127) (8.5) Other income (expense): Interest expense (4,111) (1.8) (4,607) (2.3) Interest income 2,398 1.0 1,006 0.5 Other (160) (0.1) (183) (0.1) Other expense (1,873) (0.8) (3,784) (1.9) Loss before income tax expense (34,995) (15.2) (20,911) (10.4) Income tax expense 199 0.1 226 — Net loss $ (35,194) (15.2) % $ (21,137) (10.5) % Revenue. Selling, general and administrative expenses. Selling, general and administrative expenses increased The following table Less than More than Contractual Obligations Total 1 year 1-3 years 3-5 years 5 years Long-term debt(1) $ 60,000 $ — $ 32,195 $ 27,805 $ — Finance leases(2) 17,937 1,597 3,225 3,316 9,799 Operating leases(3) 4,688 1,465 2,515 708 — Royalty obligations(4) 2,895 2,895 — — — Restricted grants 726 726 — — — Total contractual obligations $ 86,246 $ 6,683 $ 37,935 $ 31,829 $ 9,799 Inflation has impacted our operating costs throughout 2022. Continued increases in our cost of revenue may effect our ability to maintain our gross margin if the selling prices of our products do not increase commensurately, while continued increases in our operating expenses may adversely effect our operating results and the ability to make discretionary investments. We will continue to monitor the impact of inflation on our cost of revenue and operating expenses. on historical experience, current conditions and other reasonable factors. Actual results could differ from those estimates under different assumptions or conditions. We account for revenue in accordance with FASB ASC 606, “Revenue from Contracts with Customers”. Significant judgments and estimates involved in the Company’s recognition of revenue include the Share-Based Employee Compensation— We estimate the fair value of future periods. more information regarding recent accounting pronouncements. Page Financial Statements: /s/ Deloitte & Touche LLP 22, 2023 2021 2019 2018 Assets Current assets: Cash and cash equivalents $ 28,483 $ 32,231 Short-term investments 53,318 92,171 Accounts receivable, less allowance for doubtful accounts of $1,124 and $547 28,046 25,195 Inventories 29,414 22,484 Prepaid and other current assets 3,899 2,592 Total current assets 143,160 174,673 Property and equipment, net 32,646 27,080 Operating lease right-of-use assets 4,032 — Long-term investments 12,675 — Intangible assets, net 129,881 49,254 Goodwill 234,781 105,257 Other noncurrent assets 705 495 Total Assets $ 557,880 $ 356,759 Liabilities and Stockholders’ Equity Current liabilities: Accounts payable $ 14,948 $ 9,659 Accrued liabilities 32,750 25,840 Other current liabilities and current maturities of debt and leases 2,218 4,717 Total current liabilities 49,916 40,216 Long-term debt 59,634 35,571 Finance lease liabilities 11,774 12,172 Operating lease liabilities 2,796 — Contingent consideration and other noncurrent liabilities 186,417 19,419 Total Liabilities 310,537 107,378 Commitments and contingencies (Note 12) Stockholders’ Equity: Common stock, $0.001 par value, 90,000 shares authorized; 39,655 and 38,604 issued and outstanding 40 39 Additional paid-in capital 529,658 496,544 Accumulated other comprehensive loss (158) (199) Accumulated deficit (282,197) (247,003) Total Stockholders’ Equity 247,343 249,381 Total Liabilities and Stockholders’ Equity $ 557,880 $ 356,759 (LOSS) INCOME 2020 2019 2018 2017 Revenue $ 230,807 $ 201,630 $ 174,716 Cost of revenue 60,472 54,510 48,553 Gross profit 170,335 147,120 126,163 Operating expenses: Research and development expenses 41,230 34,723 34,144 Selling, general and administrative expenses 162,227 129,524 116,998 Total operating expenses 203,457 164,247 151,142 Loss from operations (33,122) (17,127) (24,979) Other income (expense): Interest expense (4,111) (4,607) (2,264) Interest income 2,398 1,006 227 Other (160) (183) 138 Loss before income tax expense (34,995) (20,911) (26,878) Income tax expense 199 226 14 Net loss $ (35,194) $ (21,137) $ (26,892) Basic and diluted net loss per share $ (0.94) $ (0.62) $ (0.83) Weighted average shares outstanding – basic and diluted 37,589 34,087 32,387 Comprehensive loss: Unrealized gain (loss) on investments $ 137 $ (31) $ 15 Foreign currency translation adjustment (96) (202) 487 Other comprehensive income (loss) 41 (233) 502 Net loss (35,194) (21,137) (26,892) Comprehensive loss, net of tax $ (35,153) $ (21,370) $ (26,390) 2020 Accumulated Additional Other Total Common Stock Paid-in Accumulated Comprehensive Stockholders’ Shares Amount Capital Deficit Income (Loss) Equity Balance—December 31, 2016 33,342 $ 33 $ 367,851 $ (198,974) $ (468) $ 168,442 Issuance of common stock under equity incentive plans 1,112 2 2,387 — — 2,389 Issuance of common stock under employee stock purchase plan 132 — 2,110 — — 2,110 Share-based employee compensation expense — — 14,615 — — 14,615 Other comprehensive income — — — — 502 502 Net loss — — — (26,892) — (26,892) Balance—December 31, 2017 34,586 $ 35 $ 386,963 $ (225,866) $ 34 $ 161,166 Issuance of common stock through public offering 2,875 3 82,870 — — 82,873 Issuance of common stock for settlement of contingent consideration 232 — 6,279 — — 6,279 Issuance of common stock under equity incentive plans 781 1 1,554 — — 1,555 Issuance of common stock under employee stock purchase plan 130 — 2,383 — — 2,383 Share-based employee compensation expense — — 16,495 — — 16,495 Other comprehensive loss — — — — (233) (233) Net loss — — — (21,137) — (21,137) Balance—December 31, 2018 38,604 $ 39 $ 496,544 $ (247,003) $ (199) $ 249,381 Issuance of common stock for SentreHEART acquisition 699 1 20,306 — — 20,307 Issuance of common stock under equity incentive plans 248 — (7,831) — — (7,831) Issuance of common stock under employee stock purchase plan 104 — 2,662 — — 2,662 Share-based employee compensation expense — — 17,977 — — 17,977 Other comprehensive income — — — — 41 41 Net loss — — — (35,194) — (35,194) Balance—December 31, 2019 39,655 $ 40 $ 529,658 $ (282,197) $ (158) $ 247,343 2020 2019 2018 2017 Cash flows from operating activities: Net loss $ (35,194) $ (21,137) $ (26,892) Adjustments to reconcile net loss to net cash used in operating activities: Share-based compensation expense 17,977 16,495 14,615 Depreciation 7,423 7,244 7,761 Amortization of intangible assets 1,943 1,510 1,367 Amortization of deferred financing costs 375 515 264 Non-cash lease expense 751 — — Loss on disposal of property and equipment and impairment of assets 604 323 336 Realized loss (gain) from foreign exchange on intercompany transactions 181 165 (173) (Accretion) amortization of investments (922) (362) 30 Provision for doubtful accounts 582 598 (172) Change in fair value of contingent consideration (4,916) (10,825) (4,078) Payment of contingent consideration in excess of purchase accounting amount — (96) — Changes in operating assets and liabilities, net of amounts acquired: Accounts receivable (3,201) (2,837) (1,464) Inventories (5,151) (146) (4,477) Other current assets (1,199) (367) 829 Accounts payable 2,790 (2,398) 1,290 Accrued liabilities 3,108 7,016 2,228 Other noncurrent assets and liabilities (962) 131 (408) Net cash used in operating activities (15,811) (4,171) (8,944) Cash flows from investing activities: Purchases of available-for-sale securities (73,249) (106,588) (16,455) Sales and maturities of available-for-sale securities 100,485 27,389 26,600 Purchases of property and equipment (12,182) (6,211) (6,384) Proceeds from sale of property and equipment 39 6 — Cash paid for business combination (17,240) — — Net cash (used in) provided by investing activities (2,147) (85,404) 3,761 Cash flows from financing activities: Proceeds from sale of stock, net of offering costs of $229 — 82,873 — Proceeds from debt borrowings 20,000 17,381 — Payments on debt and finance leases (629) (1,755) (1,689) Payment of debt fees (329) (1,136) (50) Proceeds from stock option exercises 1,202 6,012 4,402 Shares repurchased for payment of taxes on stock awards (9,033) (4,457) (2,013) Proceeds from issuance of common stock under employee stock purchase plan 2,662 2,383 2,110 Payment of contingent consideration liability previously established in purchase accounting — (1,125) — Proceeds from economic incentive loan 500 — — Net cash provided by financing activities 14,373 100,176 2,760 Effect of exchange rate changes on cash and cash equivalents (163) (179) 24 Net (decrease) increase in cash and cash equivalents (3,748) 10,422 (2,399) Cash and cash equivalents—beginning of period 32,231 21,809 24,208 Cash and cash equivalents—end of period $ 28,483 $ 32,231 $ 21,809 Supplemental cash flow information: Cash paid for interest $ 3,719 $ 3,870 $ 2,002 Cash paid for income taxes 259 65 37 Non-cash investing and financing activities: Contingent consideration in business combinations 171,300 — — Stock issuance in business combinations 20,307 — — Share-settled portion of contingent consideration — 6,279 — Accrued purchases of property and equipment 1,053 348 650 Assets obtained in exchange for finance lease obligations 270 24 2 Finance lease early termination — (6) — year ended December 31, 2021 excludes 404 shares because the effect would be anti-dilutive. 2019 2018 2017 Total accumulated other comprehensive (loss) income at beginning of period $ (199) $ 34 $ (468) Unrealized gains (losses) on investments Balance at beginning of period $ (37) $ (6) $ (21) Other comprehensive income (loss) before reclassifications 137 (31) 15 Amounts reclassified from accumulated other comprehensive (loss) income to other income — — — Balance at end of period $ 100 $ (37) $ (6) Foreign currency translation adjustment Balance at beginning of period $ (162) $ 40 $ (447) Other comprehensive (loss) income before reclassifications (277) (367) 660 Amounts reclassified from accumulated other comprehensive (loss) income to other income 181 165 (173) Balance at end of period $ (258) $ (162) $ 40 Total accumulated other comprehensive (loss) income at end of period $ (158) $ (199) $ 34 Research and Development Costs—Research and development If such targets are not met or service is not rendered for the requisite service period, no compensation cost is recognized, and any recognized compensation cost in prior periods will be reversed. For PSAs with a market condition, a Monte Carlo simulation is performed to estimate the fair value on the date of grant, and compensation cost is recognized over the requisite service period as the employee renders service, even if the market condition is not satisfied. The Company’s determination of the fair value is affected by the Company and market index stock performance, as defined by the award agreement, at the beginning of the service period and grant date; the expected volatility of the Company and market index stock performance over the performance period and the correlation coefficient of the daily returns for the Company and market index over the performance period. The Company’s long-lived assets are located in the United States, except for $1,616 as of December 31, 2022 and $1,399 as of December 31, 2021 located primarily in Europe. Quoted Prices in Active Significant Significant Markets for Other Other Identical Observable Unobservable Assets Inputs Inputs (Level 1) (Level 2) (Level 3) Total Assets: Money market funds $ — $ 14,502 $ — $ 14,502 Repurchase agreements — 10,000 — 10,000 Commercial paper — 13,755 — 13,755 U.S. government agencies and securities 8,539 — — 8,539 Corporate bonds — 24,852 — 24,852 Asset-backed securities — 18,847 — 18,847 Total assets $ 8,539 $ 81,956 $ — $ 90,495 Liabilities: Acquisition-related contingent consideration $ — $ — $ 185,157 $ 185,157 Total liabilities $ — $ — $ 185,157 $ 185,157 The following table represents the Company’s fair value hierarchy for its financial assets and liabilities measured at fair value on a recurring basis as of December 31, Quoted Prices in Active Significant Significant Markets for Other Other Identical Observable Unobservable Assets Inputs Inputs (Level 1) (Level 2) (Level 3) Total Assets: Money market funds $ — $ 16,193 $ — $ 16,193 Commercial paper — 40,731 — 40,731 U.S. government agencies and securities 6,734 — — 6,734 Corporate bonds — 30,195 — 30,195 Asset-backed securities — 14,511 — 14,511 Total assets $ 6,734 $ 101,630 $ — $ 108,364 Liabilities: Acquisition-related contingent consideration — — $ 18,773 $ 18,773 Total liabilities $ — $ — $ 18,773 $ 18,773 During 2021, the Company was informed that data from the aMAZE clinical trial did not achieve statistical superiority, and the Company assessed the projected probability of payment to be remote. The Weighted average Fair Value Valuation Technique Input Range by relative fair value Discount rate 5.56 % 5.56 % Regulatory & Commercialization-based milestones $ 185,157 Probability-weighted scenario approach Projected month and year of payment June 2020 - n/a Probability of payment 77.40 - 85.00 % 81.75 % 2019 2018 2017 Beginning Balance – January 1 $ 18,773 $ 37,098 $ 41,176 Amounts acquired 171,300 — — Settlement of trial enrollment milestone — (7,500) — Changes in fair value included in selling, general and administrative expenses (4,916) (10,825) (4,078) Ending Balance – December 31 $ 185,157 $ 18,773 $ 37,098 Unrealized Gains Cost Basis (Losses) Fair Value Corporate bonds $ 24,796 $ 56 $ 24,852 U.S. government agencies and securities 8,529 10 8,539 Commercial paper 13,755 — 13,755 Asset-backed securities 18,813 34 18,847 Total $ 65,893 $ 100 $ 65,993 Unrealized Gains Cost Basis (Losses) Fair Value Corporate bonds $ 30,223 $ (28) $ 30,195 U.S. government agencies and securities 6,734 — 6,734 Commercial paper 40,731 — 40,731 Asset-backed securities 14,520 (9) 14,511 Total $ 92,208 $ (37) $ 92,171 August 13, 2019 Inventories $ 1,848 Current assets 328 Operating lease right-of-use asset 2,929 Property and equipment 94 Intangible assets 82,570 Other assets 202 Total identifiable assets $ 87,971 Current liabilities $ 5,719 Operating lease liability 2,929 Total liabilities assumed $ 8,648 Net identifiable assets acquired $ 79,323 Goodwill 129,524 Total consideration $ 208,847 Amortization Term Valuation (in years) Developed technology $ 270 15 IPR&D 82,300 Indefinite Total $ 82,570 Year Ended December 31, (unaudited) 2019 2018 Revenue $ 232,768 $ 205,725 Net loss (40,970) (42,959) Basic and diluted net loss per share $ (1.09) $ (1.23) 2019 2018 Estimated Accumulated Accumulated Useful Life Cost Amortization Cost Amortization Technology 3-15 years $ 11,691 $ 8,131 $ 12,250 $ 7,017 IPR&D 126,321 — 44,021 — Total $ 138,012 $ 8,131 $ 56,271 $ 7,017 Future amortization expense is projected as follows: 2020 $ 1,822 2021 1,511 2022 18 2023 18 2024 18 2025 and thereafter 173 Total $ 3,560 2019 2018 Raw materials $ 11,126 $ 9,100 Work in process 1,260 1,232 Finished goods 17,028 12,152 Inventories $ 29,414 $ 22,484 Estimated Useful Life 2019 2018 Generators and other capital equipment 1-3 years $ 20,167 $ 18,158 Building under finance lease 15 years 14,250 14,250 Computer and other office equipment 3 years 7,606 6,360 Machinery, equipment and vehicles 3-7 years 5,905 4,859 Furniture and fixtures 3-7 years 5,009 4,702 Leasehold improvements 5-15 years 6,078 3,943 Construction in progress N/A 5,708 1,868 Land N/A 502 — Equipment under finance leases 3-5 years 483 213 Total 65,708 54,353 Less accumulated depreciation (33,062) (27,273) Property and equipment, net $ 32,646 $ 27,080 $3,637. 2019 2018 Accrued bonus $ 10,840 $ 9,100 Accrued commissions 8,734 8,065 Accrued payroll and employee-related expenses 6,748 4,512 Sales returns and allowances 3,979 1,410 Other accrued liabilities 59 1,205 Accrued taxes and value-added taxes payable 1,658 886 Accrued royalties 732 662 Total $ 32,750 $ 25,840 2020 $ — 2021 14,634 2022 17,561 2023 17,561 2024 10,244 Total long-term debt, of which $60,000 is noncurrent $ 60,000 2022. Twelve Months Ended December 31, 2019 Cash paid for amounts included in the measurement of lease liabilities: Operating cash flows from operating leases $ 1,026 Operating cash flows from finance leases 872 Financing cash flows from finance leases 629 Right-of-use assets obtained in exchange for lease obligations: Operating Leases 1,884 Finance Leases 270 Operating lease right-of-use asset obtained in business combination 2,929 Supplemental balance sheet information related to leases Operating Leases Finance Leases 2020 $ 1,465 $ 1,597 2021 1,337 1,602 2022 1,178 1,623 2023 708 1,646 2024 — 1,670 2025 and thereafter — 9,799 Total payments $ 4,688 $ 17,937 Less imputed interest (427) (5,410) Total $ 4,261 $ 12,527 2019 2018 2017 Current Tax Expense Federal $ (26) $ (51) $ — State 34 28 44 Foreign 165 198 72 Total current tax expense 173 175 116 Deferred Tax Expense Federal $ (7,655) $ (3,048) $ 18,485 State (1,368) 178 (1,337) Foreign (1,690) 45 (2,241) Change in valuation allowance 10,739 2,876 (15,009) Total deferred tax expense 26 51 (102) Total tax expense $ 199 $ 226 $ 14 2019 2018 Deferred tax assets (liabilities): Net operating loss carryforward $ 111,000 $ 68,563 Research and development and AMT credit carryforwards, net 8,193 6,206 Deferred interest 909 774 Equity compensation 8,233 4,750 Accruals and reserves 3,513 802 Inventories 1,007 726 Intangible assets (30,996) (11,448) Property and equipment, net (1,482) (608) Finance and operating lease liabilities 4,016 — Right-of-use assets (3,476) — Other, net 287 135 Subtotal 101,204 69,900 Less valuation allowance (101,178) (69,849) Total $ 26 $ 51 or fifteen years, depending on where research is conducted. The Company has federal net operating loss carryforwards of 2019 2018 2017 Federal tax at statutory rate 21.00 % $ (6,950) 21.00 % $ (4,391) 34.00 % $ (9,139) Federal and Foreign tax rate change 1.40 (462) (6.84) 1,430 (109.68) 29,480 Federal R&D credit 2.53 (837) 4.39 (918) (0.40) 107 Federal deferred adjustment 3.28 (1,085) (10.77) 2,253 — — Federal NOL adjustment for ASU — — — — 10.48 (2,816) Valuation allowance (32.45) 10,739 (13.75) 2,876 55.84 (15,009) State income taxes 4.02 (1,334) (0.99) 206 4.81 (1,292) Foreign NOL rate change (1.17) 388 (1.22) 256 1.30 (348) Foreign tax rate differential (0.38) 126 (0.60) 125 (2.45) 658 Permanent differences and other 1.17 (386) 7.70 (1,611) 6.05 (1,627) Effective tax rate (0.60) % $ 199 (1.08) % $ 226 (0.05) % $ 14 2020. beginning in 2019 2018 2017 Balance at the beginning of the year $ 1,157 $ 1,157 $ 3,175 Increases (decreases) for prior year tax positions 620 — (2,018) Increases (decreases) for current year tax positions — — — Increases (decreases) related to settlements — — — Decreases related to statute lapse — — — Balance at the end of the year $ 1,777 $ 1,157 $ 1,157 Stock Incentive Plan Stock options, restricted stock awards and restricted stock units granted generally vest at a rate of 33.3% on the first, second and third anniversaries of the grant date. Stock options Weighted Weighted Average Number of Average Remaining Aggregate Shares Exercise Contractual Intrinsic Time-Based Stock Options Outstanding Price Term Value Outstanding at January 1, 2019 1,582 $ 13.83 Granted 42 28.77 Exercised (110) 10.91 Cancelled (7) 30.48 Outstanding at December 31, 2019 1,507 $ 14.38 4.25 $ 27,340 Vested and expected to vest 1,503 $ 14.35 4.24 $ 27,319 Exercisable at December 31, 2019 1,392 $ 13.55 3.90 $ 26,398 Weighted Weighted RSA Average PSA Average Shares Grant Date Shares Grant Date Restricted Stock Awards and Performance Share Awards Outstanding Fair Value Outstanding Fair Value Outstanding at January 1, 2019 1,746 $ 18.19 90 $ 17.71 Awarded 435 30.12 174 30.77 Released (776) 18.44 — — Forfeited (3) 18.02 — — Outstanding at December 31, 2019 1,402 $ 21.76 264 $ 26.34 PSAs provide that each PSA that vests represents the right to receive one share of the Company’s common stock at the end of the performance period. With respect to the PSAs, the number of shares that vest and are issued to the recipient is based upon the Company’s performance with respect to specified targets at the end of the three year performance period. PSAs granted in 2020 have performance targets based on the Company’s compound annual revenue growth rate (CAGR) over the three year performance period, and payout opportunities range from 0% to 100% of the target amount. PSAs awarded subsequent to 2020 have two weighted performance targets: (i) the Company’s CAGR and (ii) relative total shareholder return (TSR). TSR is measured against the Nasdaq Health Care Index constituents and the 20-trading-day average stock price prior to the start and end of the performance period. PSAs granted in 2021 have payout opportunities ranging from 0% to 200% of the target amount, based on equally weighting of the performance targets. Awards granted in 2022 have payout opportunities ranging from 0% to 300% of the target amount and are weighted 60% on the CAGR performance target and 40% on the TSR performance target. These ranges are used to determine the number of shares that will be issuable when the award vests. The performance and market condition payouts will be determined independently and accumulated to determine the total payout for the three year performance period, subject to the maximum payout defined in the PSA agreements. All or a portion of the PSAs may vest following a change of control or a termination of service by reason of death or disability. Weighted Weighted Average Number of Average Remaining Aggregate Shares Exercise Contractual Intrinsic Performance Stock Options Outstanding Price Term Value Outstanding at January 1, 2019 450 $ 13.48 Granted — — Exercised — — Cancelled — — Outstanding at December 31, 2019 450 $ 13.48 3.45 $ 8,566 Exercisable at December 31, 2019 350 $ 13.48 3.45 $ 6,662 Activity under the plans during Weighted Weighted Average Number of Average Remaining Aggregate Shares Exercise Contractual Intrinsic Time-Based Stock Options Outstanding Price Term Value Outstanding at January 1, 2018 2,026 $ 13.30 Granted 52 26.05 Exercised (474) 12.70 Cancelled (22) 18.14 Outstanding at December 31, 2018 1,582 $ 13.83 5.02 $ 26,587 Vested and expected to vest 1,574 $ 13.78 5.00 $ 26,525 Exercisable at December 31, 2018 1,419 $ 12.99 4.63 $ 24,991 Weighted Weighted RSA Average PSA Average Shares Grant Date Shares Grant Date Restricted Stock Awards and Performance Share Awards Outstanding Fair Value Outstanding Fair Value Outstanding at January 1, 2018 1,845 $ 18.22 — $ — Awarded 630 18.71 90 17.71 Released (638) 18.87 — — Forfeited (91) 17.97 — — Outstanding at December 31, 2018 1,746 $ 18.19 90 $ 17.71 Weighted Weighted Average Number of Average Remaining Aggregate Shares Exercise Contractual Intrinsic Performance Stock Options Outstanding Price Term Value Outstanding at January 1, 2018 450 $ 13.48 Granted — — Exercised — — Cancelled — — Outstanding at December 31, 2018 450 $ 13.48 4.45 $ 5,555 Exercisable at December 31, 2018 350 $ 13.48 4.45 $ 4,321 The total intrinsic value of options exercised during the years ended December 31, and restricted stock award grants. 2019 2018 2017 Cost of revenue $ 917 $ 1,545 $ 610 Research and development expenses 2,374 1,987 2,052 Selling, general and administrative expenses 14,686 12,963 11,953 Total $ 17,977 $ 16,495 $ 14,615 2019 2018 2017 Restricted Stock Awards & Time-Based Stock Options $ 13,922 $ 15,032 $ 13,908 Performance Share Awards 3,254 766 — Performance Stock Options — — 43 ESPP 801 697 664 Total $ 17,977 $ 16,495 $ 14,615 2019 2018 2017 Range of risk-free interest rate 1.43-2.64 % 2.31 - 3.01 % 1.75 - 2.12 % Range of expected life of stock options (years) 5.13 to 5.69 5.14 to 5.71 5.21 to 5.76 Range of expected volatility of stock 40.00 - 42.00 % 41.00 - 42.00 % 43.00 - 48.00 % Weighted-average volatility 40.87 % 41.51 % 44.50 % Dividend yield 0.00 % 0.00 % 0.00 % The fair value of performance share awards with a market condition is estimated on the grant date using a Monte Carlo simulation and includes the following assumptions: 2019 2018 2017 Stock options $ 11.56 $ 10.97 $ 8.60 Restricted stock awards 30.12 18.71 19.38 Performance share awards 30.77 17.71 — tax: 2019 2018 2017 United States $ 185,829 $ 162,146 $ 138,387 Europe 27,929 25,912 21,901 Asia 15,976 12,687 13,616 Other international 1,073 885 812 Total international 44,978 39,484 36,329 Total revenue $ 230,807 $ 201,630 $ 174,716 2019 2018 2017 Open ablation $ 80,205 $ 72,250 $ 64,517 Minimally invasive ablation 34,842 35,053 34,421 Appendage management 68,166 52,891 37,281 Total ablation and appendage management 183,213 160,194 136,219 Valve tools 2,616 1,952 2,168 Total United States $ 185,829 $ 162,146 $ 138,387 2019 2018 2017 Open ablation $ 24,945 $ 21,118 $ 20,718 Minimally invasive ablation 8,349 9,176 8,007 Appendage management 11,476 8,988 7,251 Total ablation and appendage management 44,770 39,282 35,976 Valve tools 208 202 353 Total international $ 44,978 $ 39,484 $ 36,329 For the Three Months Ended March 31, June 30, September 30, December 31, 2019 2018 2019 2018 2019 2018 2019 2018 Operating Results: Revenue $ 53,966 $ 46,994 $ 58,906 $ 51,802 $ 56,614 $ 49,941 $ 61,321 $ 52,893 Gross profit 39,871 34,503 43,893 38,079 41,797 35,948 44,774 38,590 Loss from operations (5,320) (9,430) (3,839) 958 (8,637) (6,048) (15,326) (2,607) Net loss (5,635) (10,134) (4,101) (338) (9,362) (7,235) (16,096) (3,430) Net loss per share (basic and diluted) $ (0.15) $ (0.31) $ (0.11) $ (0.01) $ (0.25) $ (0.22) $ (0.42) $ (0.09) Beginning Additions Ending Balance Expenses Other (1) Deductions Balance Reserve for sales returns and allowances Year ended December 31, 2019 $ 1,410 $ 369 $ 2,240 $ 40 $ 3,979 Year ended December 31, 2018 1,169 $ 312 $ — $ 71 $ 1,410 Year ended December 31, 2017 834 $ 441 $ — $ 106 $ 1,169 Allowance for inventory valuation Year ended December 31, 2019 $ 1,029 $ 848 $ — $ 360 $ 1,517 Year ended December 31, 2018 889 $ 718 $ — $ 578 $ 1,029 Year ended December 31, 2017 1,080 $ 1,004 $ — $ 1,195 $ 889 Valuation allowance for deferred tax assets Year ended December 31, 2019 $ 69,849 $ 10,739 $ 20,590 $ — $ 101,178 Year ended December 31, 2018 66,973 $ 2,876 $ — $ — $ 69,849 Year ended December 31, 2017 81,982 $ — $ — $ 15,009 $ 66,973 REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM is incorporated herein by reference. Number of securities Weighted-average Number of securities remaining Plan Category (a) (b) (c) Equity compensation plans approved by security holders (3) 3,623,588 $ 14 1,577,687 Equity compensation plans not approved by security holders — — — Total 3,623,588 $ 14 1,577,687 reference. Exhibit No. Description 3.1 3.2 4.1 10.1# 10.2# 10.3# 10.4# 10.5 10.6 10.7# 10.8# 10.9# 10.10# 10.11 10.16§ 23.1 31.1 31.2 32.1 32.2 101.INS XBRL Instance Document 101.SCH XBRL Taxonomy Extension Schema Document 101.CAL XBRL Taxonomy Extension Calculation Linkbase Document 101.DEF XBRL Taxonomy Definition Linkbase Document 101.LAB XBRL Taxonomy Extension Label Linkbase Document 101.PRE XBRL Taxonomy Extension Presentation Linkbase Document 104 Cover Page Interactive Data File _________________________ SIGNATURES AtriCure, Inc. (REGISTRANT) Date: February /s/ Michael H. Carrel Michael H. Carrel President and Chief Executive Officer (Principal Executive Officer) Date: February /s/ Chief Financial Officer (Principal Accounting and Financial Officer) Signature Title(s) /s/ /s/ Michael H. Carrel Michael H. Carrel Michael H. Carrel Director, President and Chief Executive Officer (Principal Executive Officer) /s/ Mark A. Collar Mark A. Collar Mark A. Collar Director /s/ Regina E. Groves Regina E. Groves Regina E. Groves Director /s/ Karen N. Prange Karen N. Prange Karen N. Prange Director /s/ Deborah H. Telman Sven A. Wehrwein Sven A. Wehrwein Director /s/ Robert S. White Robert S. White Robert S. White Director_________________________________ ANNUAL REPORT UNDER SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 19342019 TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
Delaware34-1940305None_________________________________ No No No No ☐☐ No 2019,2022, the last business day of the registrant’s most recently completed second fiscal quarter as reported on the NASDAQ Global Market, was $1,110.6approximately $1,851.6 million.ClassOutstanding February 20, 202040,048,97246,568,044_________________________________
TABLETABLE OF CONTENTS“Risk Factors,”“Quantitative and Qualitative Disclosures about Market Risk” contains forward-looking statements regarding our future performance. All forward-looking information is inherently uncertain and actual results may differ materially from assumptions, estimates or expectations reflected or contained in the forward-looking statements as a result of various factors, including those set forth under “Risk Factors” and elsewhere in this Form 10-K. There may be additional risks of which we are not presently aware or that we currently believe are immaterial which could have an adverse impact on our business. Forward-looking statements address our expected future business, financial performance, financial condition and results of operations, and often contain words such as “intends,” “estimates,” “anticipates,” “hopes,” “projects,” “plans,” “expects,” “seek,” “believes,” “see,” “should,” “will,” “would,” “opportunity,” “could,” “can,” “may,” “future,” “predicts,” “target,” “potential,” and similar expressions and the negative versions thereof.of those words, and may be identified by the context in which they are used. Such statements are based only upon current expectations of AtriCure. Any forward-looking statement speaks only as of the date made. Reliance should not be placed on forward-looking statements because they involve known and unknown risks, uncertainties and other factors which may cause actual results, performance or achievements to differ materially from those expressed or implied. Forward-looking statements include statements that address activities, events, circumstances or developments that AtriCure expects, believes or anticipates will or may occur in the future. Forward-looking statements are based on AtriCure’s experience and perception of current conditions, trends, expected future developments and other factors it believes are appropriate under the circumstances and are subject to numerous risks and uncertainties, many of which are beyond AtriCure’s control. With respect to the forward-looking statements, we claim the protection of the safe harbor for forward-looking statements contained in the Private Securities Litigation Reform Act of 1995. These forward-looking statements speak only as of the date of this Form 10-K. We undertake no and hereby disclaim any and all, obligation to publicly update or revise any forward-looking statements to reflect new information or future events or otherwise unless required by law.PAWe own or have the rights to use various trademarks referred to in this Annual Report on Form 10-K, including Isolator® RTSynergy TM clamp, EPi-Sense® coagulation device, AtriClip® Flex·V®,, and cryoSPHERE® probe, among others, and their respective logos. Solely for convenience, we may refer to trademarks in this Annual Report on Form 10-K without the TM and ® symbols. Such references are not intended to indicate, in any way, that we will not assert, to the fullest extent permitted by law, our rights to our trademarks.ITEM and, left atrial appendage (LAA) management.management and post-operative pain. Afib is an irregular heartbeat, or arrhythmia, which affects approximately 1% of the populationover 37 million people worldwide, including more than eight million people in the United States.States, and is a growing epidemic. It is the most common cardiac arrhythmia or irregular heartbeat, encountered in clinical practice and accountsresults in high utilization of healthcare services. Patients often progress from being in Afib intermittently (paroxysmal) to being in Afib continuously. The continuous Afib patient population includes early persistent Afib, which lasts seven days to 6 months, persistent Afib, which lasts 6 months to one year, and long-standing persistent Afib, which lasts longer than one year. It is estimated that 3.5 million people in the United States suffer from long-standing persistent Afib. Afib often occurs in conjunction with other cardiovascular diseases, including hypertension, congestive heart failure, left ventricular dysfunction, coronary artery disease and valvular disease.more doctor visitsother emergent heart conditions, and hospital days than anyour products are used in conjunction with (or “concomitant” to) such a procedure. Minimally invasive procedures are performed on a standalone basis, and often include multi-disciplinary or “hybrid” approaches, combining surgical procedures using AtriCure ablation and AtriCure LAAM products with catheter ablation performed by an electrophysiologist.arrhythmia.arrhythmia, with approximately 1.2 million diagnoses annually in the United States. Afib is an under-diagnosed condition due in large part to the fact that patients with Afib often have mild or no symptoms, and their Afib is often only diagnosed when they seek treatment for an associated condition, such as a structural heart disease or stroke. Symptoms of Afib may include heart palpitations, dizziness, fatigue and shortness of breath, and these symptoms may be debilitating and life threatening in some cases. When a patient is in Afib, abnormal electrical impulses cause the atria, or upper chambers of the heart, to fibrillate, or beat rapidly, irregularly and in an uncoordinated fashion. As a result, blood in the atria may be in stasis, increasing the risk that a blood clot will form and cause a stroke or other serious complications. In patients with Afib, a significant percentage of those clots can form inside of the LAA. Symptoms of Afib may include heart palpitations, dizziness, fatigue and shortness of breath, and these symptoms may be debilitating and life threatening in some cases. Patients often progress from being in Afib intermittently (paroxysmal) to being in Afib continuously (persistent and long standing persistent). Afib often occurs in conjunction with other cardiovascular diseases, including hypertension, congestive heart failure, left ventricular dysfunction, coronary artery disease and valvular disease.Our products are used by physicians during both open-heart and minimally invasive procedures, either in conjunction with heart surgery for other conditions (“concomitant” to such a procedure), or on a standalone basis. We have several product lines for the ablation of cardiac tissue, including our Isolator® Synergy™ Ablation System, the first and only surgical device approved by the United States Food and Drug Administration (FDA) for the treatment of persistent and long-standing persistent forms of Afib in patients undergoing certain open concomitant procedures. We also offer a variety of minimally invasive ablation devices and access tools to facilitate less invasive cardiac and thoracic procedures. Our cryoICE® cryosurgery product line offers a variety of cryoablation devices for use in multiple types of cardiothoracic surgery. Our AtriClip® LAA Exclusion System is a device specifically designed to exclude the heart’s left atrial appendage.We believe that we are currently the market leader in the surgical treatment of Afib. Our Isolator Synergy System is approved by FDA for the treatment of persistent and long-standing persistent Afib concomitant to other open-heart surgical procedures. All of our other ablation devices are cleared for sale in the United States under FDA 510(k) clearances, including our other RF and cryoablation products, which are indicated for the ablation of cardiac tissue and/or the treatment of cardiac arrhythmias. In addition, certain of our cryoablation probes are cleared for managing pain by temporarily ablating peripheral nerves. Our AtriClip products are 510(k)-cleared with an indication for the exclusion of the LAA, performed under direct visualization and in conjunction with other cardiac surgical procedures. Direct visualization, in this context, requires that the surgeon is able to see the heart directly, with or without assistance from a camera, endoscope or other appropriate viewing technologies. The LARIAT® system is cleared for soft tissue ligation and is currently being studied to support an indication of exclusion of the LAA in patients with persistent and long-standing persistent Afib also undergoing a pulmonary vein isolation. We also offer reusable surgical instruments typically used in cardiac valve replacement or repair. Our Isolator Synergy clamps, Isolator Synergy pens, Coolrail® linear pen, cryosurgery devices, certain products of the AtriClip LAA Exclusion System, COBRA Fusion® Ablation System, NumerisTM System, the EPi-Sense® Guided Coagulation System with VisiTrax® technology, and LARIAT Suture Delivery Device bear the CE mark and may be commercially distributed throughout the member states of the European Union and other countries that comply with or mirror the Medical Device Directive. Our Isolator Synergy clamps, Isolator Synergy pens, Coolrail linear pen, cryosurgery devices, and certainproducts of the AtriClip LAA Exclusion System are available in select Asia-Pacific countries. We anticipate that substantially all of our revenue for the foreseeable future will relate to products we currently sell, or are in the process of developing.We sell our products to medical centers through our direct sales force in the United States and in certain international markets, such as Germany, France, the United Kingdom and the Benelux region. We also sell our products to distributors who in turn sell our products to medical centers in other international markets. Our business is primarily transacted in U.S. Dollars with the exception of transactions with our European customers, which are transacted primarily in the Euro or the British Pound.Market OverviewAfib is the most commonly diagnosed sustained cardiac arrhythmia, and affects approximately 33 million people worldwide, including six millionin the United States. It is estimated that the incidence of Afib doubles with each decade of an adult’s life. At age 40, remaining lifetime risk for Afib is 26% for men and 23% for women. Afib is an under-diagnosed condition due in large part to the fact that patients with Afib often have mild or no symptoms, and their Afib is only diagnosed when they seek treatment for an associated condition, such as a structural heart disease or stroke. We believe that increasing awareness of Afib and improved diagnostic screening will result in an increased number of patients diagnosed with Afib. Recently, there have been several new diagnostic technologies introduced in the United States that allow for less invasive screening options, which should assist patients with more proactive identification of Afib.Afib over time. Also, since the prevalence of Afib increases with age, there will likely be an increase in the number of diagnosed Afib patients in the United States as the population ages. We believe that the samethese trends in the United States also apply globally, as the incidence of Afib is increasing as the population ages in many geographies.which are typically classified as “persistent”or persistent and “long-standing persistent”long-standing persistent Afib. Over the past two decades, technology advancements have made surgical ablation more effective, repeatable and available to cardiac surgeons and electrophysiologists around the world. Societal guideline changes from the Society of Thoracic Surgeons (STS), Heart Rhythm Society (HRS), and American AssociatedAssociation of Thoracic Surgery (AATS) now have Class I recommendations for concomitant surgical ablation, meaning that it is a “recommended” treatment no longer just “reasonable”, for patients who have structural heart disease and Afib. In addition, guidelines for the treatment of more serious forms of Afib for patients without structural heart disease have also been introduced in the past several years. These societal guidelinesall structural heart patients who also have Afib.250,000300,000 are potential candidates for surgical ablation using our products. Today, we estimate that approximately 25% to 35%less than 20% of those candidates are being treated, but we believe many are not treated properly or fully. Of the population diagnosed with Afib,in a large percentage of patients are symptomatic and do not respond to pharmacological therapy. Additionally, there is a large population of patients who have no other underlying cardiac disease but who suffer from serious forms of Afib. Many of these patients fail traditional therapies, and thus we believe could benefit from a minimally invasive or multi-disciplinary (“hybrid”) Afib treatment using our products.infor the cardiac surgery market.Company.times result in significant post-operative pain and longer hospital recovery times as patients refrain from mobilizing their chest near the incision site. Most cardiothoracic surgeons will employ a multi-modal pain management protocol that includes global and local pain management techniques. Global techniques include epidural delivery of medication directly around the spinal cord, intravenous, or oral delivery of opioid and non-opioid pain medications. Local, more focused, techniques include syringe injections between vertebrates and cryo nerve block, the use of cryo-energy to temporarily ablate peripheral nerves. Cryo nerve block can be delivered using our cryoICE cryoSPHERE® probe, which is specifically designed for cryo nerve block, as well as our cryoICE CRYO2 probe, one of the same probes used to treat cardiac arrhythmias. Depending on the degree of invasiveness of the cardiothoracic surgery, physicians and their nursing staff will take advantage of multiple modes of pain management. It is estimated that each year roughly 140,000approximately 150,0000 cardiac and thoracic procedures are performed in the United States through thoracotomy access.States. Hospital recovery times can vary from two to eight days depending on the procedure, operative complications associated with the procedure, pain management protocol and other factors. Most surgeons will employ a multi-modal pain management protocol that includes various pain management techniques, including techniques such as epidural delivery of medication directly around the spinal cord, intravenous or oral delivery of opioid and non-opioid pain medications, or other strategies. More focused, local techniques include syringe injections between vertebrates, and Cryo Nerve Block which uses cryothermic energy to ablate peripheral nerves, temporarily stopping the transmission of pain signals coming from the chest wall during surgery. The nerve “scaffolds” remain intact, allowing axons to regenerate and restore nerve function over time. Cryo Nerve Block can be delivered using our cryoICE cryoSPHERE® probe, which is specifically designed for Cryo Nerve Block. Depending on the degree of invasiveness, physicians and their nursing staff will take advantage of multiple ways of managing pain for their patients. In recent years, prescription narcotics, or opioids, have come under heavy scrutiny due to their potential for long-term dependency, overdose and possible death. The Center for Disease Control has reported over 42,000 deaths involving opioids in the United States in a single year, and bothBoth federal and local governments in the United States have proposed and implemented new regulations to curb the opioid overdose epidemic. It is also estimated that one in seven cardiothoracic surgical patients develops an unhealthy post-proceduralcryo nerve block,Cryo Nerve Block, an increasingly important.important part of how physicians manage post-operative pain.The SolutionSolutions and Productsand catheter-based ablation devices currently marketed by our competition are not ideal for safely, rapidly and reliably creating lesions that completely and permanently block the abnormal electrical impulses that cause Afib, particularly for patients with more severe formstreatment of Afib and pioneers of the application of Cryo Nerve Block in thoracic and cardiothoracic procedures. We anticipate that substantially all our revenue for the foreseeable future will relate to products we currently sell or patients who have failed single or multiple catheter ablations.are in the process of developing. Our products enable cardiothoracic surgeons to mimic all or portions of the cut and sew MazeCOX-MAZE procedure with faster, less invasive and less technically challenging approaches. We have completed, and continue to invest in, clinical studies for the use of ourAfib.Afib and reduce stroke. Leading cardiothoracic surgeons and electrophysiologists, including those who serve or who have served as consultants to us, have published results of initialpreclinical and clinical studies utilizing our devices. The results of these studies have assessed efficacy, ease of use and safety endpoints.We also offer product lines for left atrial appendage management and open and minimally invasive cardiac tissue ablation. are characterized as eitherinclude those that heatcreate scar tissue using Radio Frequencyradio frequency (RF) energy to create the tissue effects or those that cool tissue using cryo-thermal heat transfer to create the tissue effects.cryothermic modalities. Our ablation products fall intoare part of platforms each consisting of disposable handpieceshand pieces which connect to compacteither a RF power generation sourcesgenerator or the cryoICE Box generator that wea cryothermic generator. We generally place this capital equipment with our direct customers and sell to our distributors.•Isolator Synergy Clamps. Our Isolator Synergy System represents our primary product line and currently generates the majority of our RF ablation-related revenue. All of our clamps are single-use disposables and havedisposable RF products with jaws that close in a parallel fashion. We sell multiple configurations of our Isolator Synergy clamps. The various configurations provide the user with options to address patient specific procedure requirements or anatomy; however, all the clamps withprovide consistent performance using the primary difference being the form of the clamping jaws.same core technology. The parallel closure compresses tissue and evacuates the blood and fluids from the energy pathway in order to make the ablation more effective.is currently being evaluated underhas been studied in multiple FDA approved clinical trials, including the previously completed ABLATE clinical trial which supported a pre-market approval (PMA) in 2011, as well as the ongoing DEEP AF IDE pivotal trial and HEAL-IST clinical trial.® clamp, following 510(k) clearance for ablation of cardiac tissue during cardiac surgery in July 2021. The EnCompass clamp was cleared through the FDA 510(k) process and is indicated for cardiac soft tissue ablation. The configuration is designed to make concomitant surgical ablations more efficient and is expected to drive deeper penetration of cardiac surgery procedures.configurations which have different contact lengths and are powered by an RF generator.configurations. The MAX Pen devices enable surgeons to evaluate cardiac arrhythmias, perform temporary cardiac pacing, sensing and stimulation and ablate cardiac tissue with the same device. Surgeons are able tocan readily toggle back and forth between these functions. The device comes in multiple configurations that have unique tissue contacting and shaft lengths. The Coolrail® device enables the user to make longer linear lines of ablation. Surgeons generally use one or more of our pen and linear devices in combination with Isolator Synergy clamps.•cryoICE Cryoablation System. The cryoICE cryoablation system is used in both open ablation procedures and cryoanalgesia. The system consists of the cryoICE BoxBOX generator along with a variety of single-use disposable probe.probes. The primary differences between these cryoablation probes is the form of the tissue contacting distal end. The cryoICE devices enablesenable the user to make linear ablations of varied lengths. Surgeons may utilize the cryoICE devices in combination with Isolator Synergy clamps or independently.systemprobe is used to apply cryo-energycryothermic energy to targeted intercostal peripheral nerves in the ribcage in order to provide temporary pain relief. This technique, called cryo nerve block,Cryo Nerve Block, is applied intra-operativelyintraoperatively by the cardiothoracic surgeonor thoracic surgeons and results in temporary pain relief for up to 90 days after the procedure. Sensation typically returns to the affected region of the chest after this period. Studies are ongoing to characterizecryo nerve blockCryo Nerve Block has generally shown a significant reduction in prescription of opioids, significantly reduced length of stay for patients in the hospital and further refine the procedure.Products for minimally invasive ablation:EPi-Sense Guided Coagulation System with VisiTraxTechnology. The EPi-Sense Guided Coagulation System with VisiTrax technology utilizes monopolar energy for the coagulation of tissue. The Epi-Sense device is a single-use disposable which is also capable of intra-operative cardiac signal sensing and recording when connected to an external recording device. We are conducting the CONVERGE IDE clinical trial to evaluate the safety and efficacy of the EPi-Sense Guided Coagulation System with VisiTrax technology to treat symptomatic persistent and long-standing persistent Afib patients who are refractory or intolerant to at least one Class I and/or III anti-arrhythmic drug.The AtriClipLAA Exclusion System includes various combinations of an implantable device (AtriClip) coupled to a single-use disposable applier. The AtriClip is designed to exclude the left atrial appendage by mechanically clamping the appendage from the outside of the heart, eliminatingheart. The left atrial appendage has been shown to be a source of arrhythmias. The exclusion of the LAA eliminates blood flow between the left atrial appendage and the atrium while avoiding contact with circulating blood.blood and provides electrical isolation benefits after placement. We believe that the AtriClip system is potentially safer, more effective and easier to use than otheravailable products and techniques for permanently excluding the left atrial appendage. These benefits compared to other techniques include permanent exclusion and electrical isolation of the appendage. The AtriClip device comes in two geometries (a rectangular configuration which encircles the targeted tissue and “V” shape which allows for an alternative lateral access) and a variety of lengths, allowing the userwhich are matched to select a configuration specific to the patient and in two geometries (rectangular and “V” shape).each patient's anatomy. The appliers come in multiple forms tailored to specific procedural needs depending on the type of surgery and how the surgeon is accessing the heart.different deployment mechanisms. an indication for the exclusion of the LAA, performed under direct visualization and in conjunction with other cardiac surgical procedures. Direct visualization, in this context, requires that the surgeon can see the heart directly, with or without assistance from a camera, endoscope or other appropriate viewing technologies. Certain products of our AtriClip LAA Exclusion System bear the CE mark for commercial distribution throughout the member states of the European Union and other countries that comply with or mirror the Medical Device Directive. These products are available for sale in a number of other countries globally.includes various combinations of AtriClipsis currently being evaluated under the Left Atrial Appendage Exclusion for Prophylactic Stroke Reduction (LeAAPS™) IDE clinical trial.appliers.LARIAT System.enabling technologies that hold 510(k) approvals and/or bear the CE mark. The LARIAT® System is a suture-based solution for soft-tissue closure and is compatible with a wide range of anatomical shapes. The Lariat Systemthat includes a suture loop coupled to a single-use disposable applier. The loop is designed to occlude the left atrial appendage by mechanically cinching the appendage from the outside of the heart, eliminating blood flow between the left atrial appendage and the atrium while avoiding contact with circulating blood. The product is currently being studied in the aMAZE clinical trial, the aim of which is to determine if the combination of two percutaneous procedures, including using the LARIAT to exclude the left atrial appendage along with pulmonary vein isolation using an endocardial catheter ablation device, may treat persistent or long-standing persistent atrial fibrillation more effectively than pulmonary vein isolation alone.In addition to the above product lines we also sell enabling technologies including our Lumitip™ dissectors, COBRA Fusion Surgical Ablation System, the Fusion Magnetic Retriever System and a line of reusable cardiac surgery (valve) instruments. The Lumitip dissector is used by surgeons to separate tissues to provide access to key anatomical structures that areThe COBRA Fusion Surgical Ablation System’s Versapolar technology combines bipolar temperature-controlled RF energy with monopolar energy and incorporates a unique suction design to draw tissue in to assure stable contact and optimize ablation performance. The Fusion Magnetic Retriever System™ allows access around key anatomical structures and facilitates positioning of the Cobra Fusion Surgical Ablation System™. Cardiac surgery instruments are used during certain surgical proceduresOther enabling technologies include our Glidepath™ guides for repair or replacement of heart valves.Current Afib Treatment AlternativesPhysicians usually begin treating Afib patients with a variety of drugs intended to prevent blood clots, control heart rate or restore the heart to normal sinus rhythm. If a patient’s Afib cannot be adequately controlled with drug therapy, doctors may perform one of several open-heart or minimally-invasive procedures that vary depending on the severity of the Afib symptoms and whether or not the patient suffers from other forms of heart disease.Alternative treatments to open-heart and minimally invasive procedures include:Drugs. Pharmaceutical options called anti-arrhythmics are available to treat Afib. Depending on a patient’s severity of the disease and heart condition, physicians typically administer these medications in a hospital setting with continuous monitoring. If the patient goes back into a normal rhythm, the physician will often prescribe a similar anti-arrhythmic drug to try to prevent a recurrence of Afib. The effectiveness of drug therapy varies based on the patient population and the drug being prescribed, among other factors. Often, pharmaceuticals to thin the blood (anti-coagulants) are prescribed due to the increased risk of stroke for patients who also have Afib.Implantable Devices. Implantable devices, such as defibrillators and pacemakers, can be effective in reducing the symptoms of Afib episodes, but neither device is intended to treat Afib. Patients may continue to experience the adverse effects of Afib as well as some of the symptoms and complications, including dizziness, fatigue, palpitations and stroke because the Afib continues.Catheter Ablation. Catheter ablation is a procedure that is typically performed by an electrophysiologist. The ablations are made from the inside of the heart using a flexible catheter. The heart is reached via a blood vessel, most commonly through the femoral vein. In proportion to the prevalence of Afib, only a small number of catheter-based Afib treatments are performed each year in the United States.We do not promote our products specifically for Afib treatment in the United States, except for the Isolator Synergy System, which may be promoted according to its FDA-approved indication for patients with persistent and long-standing persistent Afib undergoing certain open concomitant procedures. During elective open-heart surgical procedures, such as bypass or valve surgery, cardiothoracic surgeons use our ablation systems to treat patients with a pre-existing history of Afib. Surgeons use our products to perform cardiac procedures that may vary depending on the length of time a patient has been diagnosed with Afib and whether the patient’s Afib is intermittent, known as paroxysmal, or more continuous (non-paroxysmal), which is typically further classified as persistent, long-standing persistent or permanent. Patients who have been diagnosed with Afib for a longer duration and have non-paroxysmal forms of Afib generally receive more extensive ablation procedures than patients who have been diagnosed with Afib for a shorter duration or who have paroxysmal Afib. Additionally, during an open-heart procedure, physicians may use our AtriClip system to exclude the left atrial appendage.For those patients with Afib who do not require a concomitant open-heart surgical procedure, surgeons have used our products for minimally invasive Afib treatment procedures. These procedures have generally been performed through minimally invasiveincisions without the need to place patients on a heart-lung bypass machine. We do not currently have any products with FDA-approved indications for the standalone treatment of Afib.Certain physicians are combining various minimally invasive stand-alone epicardial ablation procedures (surgical ablation on the outside of the heart) with endocardial ablation and mapping techniques (catheter ablation from the inside of the heart). These combination procedures are often referred to as “hybrid” or “multi-disciplinary” approaches, in that both surgical ablation and catheter ablations are performed. Sometimes, both procedures are performed on the same day or in the same hospital stay, where other times they are performed days or weeks apart. Patient health condition, physician preference, hospital logistics and procedural room availability influence the decision whether to perform hybrid ablations in a single or a staged setting. Physicians are reporting that they are performing these procedures utilizing certainplacement of our productsclamps, Subtle™ Cannula’s to primarily treat patients who have non-paroxysmal formssupport access for our EPi-Sense catheters and a line of Afib.reducing the global Afib epidemic and healing the lives of those affected.patients affected by Afib and pain after surgery. Our strategy is to expand the treatment options for patients who suffer from Afib, or have a high risk of stroke, or who suffer from post-operative pain, through the continued development of our technologies and expansion of our product offerings, global commercial expansion and clinical science investments. The key elements of our strategy include:market opportunitiesmarkets or expand our growth in existing markets. trials, including the CONVERGE, aMAZE and ICE-AFIB IDE trials to validate the long-term results of procedures using our products and to support applications to regulatory agencies for expanded indications. We also make clinical research grants to support our product development efforts and expand the body of clinical evidence.evaluatedevelop and developevaluate our products. Additionally, we have formedregularly form advisory boards made up of key opinion leaders in multiple specialties to overseeprovide input to our training and clinical programs. We are building these relationships along with extended care professionals such as nurse practitioners and advanced practice providers, to provide insight regarding treatment trends, input on future product direction and education for providers involved in treating the disease.threefive years, both the Society for Thoracic Surgeons and the Heart Rhythm Society have released new guidelines on the surgical treatment of Afib in both open-heart and minimally-invasive settings.for the treatment of non-paroxysmal Afib, we instituted a program to train providers on the use of the Isolator Synergy System to treat persistent and long-standing persistent Afib in patients undergoing open-heart surgery. With the approval of the EPi-Sense System, we began programs to train physicians on the use of the EPi-Sense system in a hybrid approach to treating patients with long-standing persistent Afib. We believe thisthese training and education program hasprograms have increased awareness about the surgical treatment of Afib, during open-heart procedures, and we will continue to make investments to serve our physician customers. As a result of the educational process, we believe that awareness of our technologies is growing and will result in the increased use of our products. Minimally Invasive Products. We believe that the catalysts for expanded adoption of our minimally invasive products include completing clinical trials, including the CONVERGE and DEEP AF IDE clinical trials, procedural advancements, such as the hybrid or multi-disciplinary procedure for treatment of long-standing persistent Afib, continued innovation and product development, training and education of new customers, and the publication of additional scientific evidence supporting the safety and efficacy of hybrid treatments for persistent and long-standing persistent Afib.evidence. We believe these efforts will help validate the successful, long-term use of our products for patients with persistent and long-standing persistent Afib. Wealso believe that ongoing research activities, including prospective clinical trials, new procedural techniques and anticipated presentations and publications will create an increased demand for our minimally invasive products.investment in clinical science, product innovation, clinical differentiation and other strategic and financial considerations.Clinical TrialsAn IDE supplement is a means of obtaining approval to initiate a pivotal trial following the conclusion of a feasibility trial. We are conducting several clinical trials to validate the long-term results of procedures using our productsare conductingconducted the CONVERGE IDE clinical trial to evaluate the safety and efficacy of the EPi-Sense Guided Coagulation System with VisiTrax technology to treat symptomatic persistent and long-standing persistent Afib patients who are refractory or intolerant to at least one Class I and/or III anti-arrhythmic drug. In April 2021, we announced the PMA approval of the EPi-Sense System for treatment of symptomatic, drug-refractory, long-standing persistent atrial fibrillation, when augmented with an endocardial ablation catheter. We believe the Convergent procedure, or Hybrid AF therapy, provides the only compelling treatment option for a large and vastly underpenetrated population of Afib patients. The CONVERGE trial providesdemonstrated superiority in the hybrid therapy arm compared to endocardial catheter ablation alone. In patients diagnosed with long-standing persistent Afib, the therapy arm showed a 29% absolute difference in efficacy at 12 months (78% relative improvement) and an absolute difference of 35% at 18 months (110% relative improvement). There was also a 33% absolute difference in Afib burden reduction in favor of the Hybrid AF therapy at 12 months, which increased to 37% at 18 months. In April 2021, we also received approval from FDA to conduct the CONVERGE Post Approval Study (PAS). This study allows for enrollment of325 patients to be enrolled at up to 153 patients at 27 domestic medical centers and three international medical centers. Enrollment began in 2014 and was completed in August 2018.50 sites. The study protocol requiresfirst patient follow-up for twelve months post procedure for the primary effectiveness endpoint assessment and long-term follow-up through five years. The final PMA module was submitted in late 2019. In September 2019, we received approval for the Continued Access Protocol (CAP) for the CONVERGE study. The CAP is a single arm study of patients undergoing hybrid ablation only and currently provides for initial enrollment of up to 30 patients at 27 domestic sites, with the possibly of expanding the CAP study to enroll additional patients by submitting an IDE Supplement to FDA. Enrollment in the CONVERGE CAPtrial occurred in June 2022; site initiation and enrollment is expected to begin in early 2020.ongoing.aMAZE. In connection with our acquisition of SentreHEART, which we describe below, we are conducting the aMAZE IDE clinical trial. aMAZE is an FDA-approved, prospective, multicenter, randomized controlled trial evaluating the LARIAT Suture Delivery Device for LAA closure adjunctive to Pulmonary Vein Isolation (PVI) catheter ablation for the treatment of persistent and long-standing persistent Afib. The objective of the aMAZE Trial is to demonstrate that using the LARIAT device for LAA closure, plus a PVI ablation, will lead to a reduced incidence of recurrent Afib compared to PVI alone, with a favorable safety profile. The aMAZE Trial provides enrollment of up to 600 patients at 65 sites with one-year follow up. Primary endpoint measures are freedom from episodes of Afib greater than 30 seconds at one-year post treatment. Enrollment of 600 patients across 53 sites was completed in December 2019. In January 2020, we received approval for a CAP for the aMAZE study. The aMAZE CAP provides for additional patient enrollment of up to 85 patients at existing aMAZE trial sites, with the opportunity to further expand to 250 patients while the pre-market application is under review. We acquired SentreHEART, a privately held developer of percutaneous left atrial appendage management solutions sponsoring the aMAZE trial, in a merger on August 13, 2019.We received IDE approval from FDA to proceed with the ICE-AFIB trial in November 2018. Enrollment began in FebruaryJanuary 2019 and remains ongoing.ATLAS. The ATLAS study is a non-IDE randomized pilot study evaluating outcomes of patients with risk factors for developing postoperative Afib as well as risk of bleeding on oral anticoagulation. There are two types of patients subject to this study: those with a postoperative Afib diagnosis and receiving prophylactic exclusion of the left atrial appendage with the AtriClip device concomitant to cardiac surgery and those with a postoperative Afib diagnosis who are medically managed. Enrollment began in February 2016 and ended in March 2018. Preliminary data was presented at the Heart Rhythm Society meeting in May 2019, and a manuscript of the final results is being drafted for publication.FROST.conducted a cryo nerve block study, which was a non-IDE randomized pilot study evaluating intraoperative intercostal cryoanalgesia.invested in other clinical trials to validate the long-term results of procedures using our products and to support applications to regulatory agencies for expanded indications. The study involves treatment arm patients who received intercostal cryoanalgesiaCompany is in conjunction with standard post-operative pain management and control arm patients who receive standard post-operative pain management only. The study providedthe process of analyzing data for enrollmentpublication, future development activities, or possible evaluation of up to 100 patients at five medical centers. Enrollment began in June 2016 and an interim data analysis was completed when a total of 80 patients were enrolled in 2019. Enrollment was stopped followinglabel expansions. These trials include the interim analysis due to early achievement of statistical significance. Results from the trial were presented at the Society of Thoracic Surgeons podium in January 2020 and will be published in The Annals of Thoracic Surgery.Study. The DEEPStudy, CEASE AF and aMAZE IDE pivotal trial evaluates the safety and efficacy of the Isolator Synergy System when used in a staged approach where a minimally invasive surgical ablation procedure is first performed and the patient undergoes the intracardiac catheter procedure approximately 90-120 days later. The study began in 2014 and was paused during 2016-2017 due to our work to mitigate the risk related to esophageal injury during the procedure. We are committed to patient safety, and we worked collaboratively with FDA and obtained approval to resume enrollment in the trial in 2018. A total of 70 patients have been treated, and we have FDA approval to enroll an additional 18 patients. We plan to seek approval to enroll the full cohort of 220 patients, pending FDA’s review of additional safety data.CEASE AF. We are also pursuing a non-IDE trial in Europe to compare staged hybrid ablation treatment (minimally invasive surgical ablation procedure is first performed and the patient undergoes the intracardiac catheter procedure approximately 91-180 days later) versus catheter ablation alone. We expect the study to have an enrollment of approximately 210 patients at twelve sites. Enrollment began in November 2015 and remains ongoing.technicalclinical benefits. We only promote our products for uses described in their labeling as cleared or approved by the relevant regulatory agencies. Weagencies, and train our sales force on the use of our products to the extent the products are cleared or approved.160 employees supporting approximately 53 sales territories.260 employees. We select our sales personnel based on their expertise, sales experience and reputation in the medical device industry and their knowledge of cardiac and thoracic surgery procedures and technologies.marketscountries outside of the United States through a combination of independent distributors and direct sales personnel. Our international sales team includes sales representativesapproximately 50 employees focused on our direct markets, such as Germany, France, the United Kingdom, Australia and the Benelux region. We also maintain a network of distributors who market and sell our products in Asia, South America and Canada, as well as certain countries in Europe, who market and sell our products.Europe. We continue to evaluate opportunities for further expansion into markets outside of the United States.MostWe compete with other companies and divisions of our competitors have greater financial and human resources than we do and have established reputations with our target customers, as well as worldwide distribution channelscompanies that are more established and developed than ours.sell a single or limited number of competitive product lines or in certain geographies. Our primary competitor in the cardiac surgery market is Medtronic, plc, who provides similar surgical ablation products to ours that have been adopted by physicians for the treatment of Afib and related conditions. AtriCure has the only medical devices that are approved by FDA for treating long-standing persistent Afib: the Isolator Synergy Ablation, the first medical device to receive FDA approval for the treatment of persistent Afib in a concomitant setting, and the EPi-Sense System, which received FDA approval for standalone treatment of Afib with Hybrid AF Therapy. Several other companies offer intracardiac catheter devices that are commonly used by electrophysiologists to treat Afib. These catheter devices are FDA-approved to treat the paroxysmal formand persistent forms of Afib, but they are not FDA indicated to treat persistent or long-standing persistent Afib. AtriCure’s Isolator Synergy System is the only medical deviceIn particular, because Hybrid AF Therapy involves both epicardial and endocardial techniques, these catheters are complementary to our business, not competitive. We believe that is FDA approved to treatour products improve treatment outcomes for patients with non-paroxysmal forms of Afib in a surgical setting, and the only medical device approved to treat persistent or long-standing persistent Afib in a concomitant setting. when combined with intracardiac catheter devices.Afib. We believe thatAfib, although we are not aware of any ongoing FDA trials by other companies to study ablation of long-standing persistent Afib patients. New product introductions, technological advances and regulatory clearances from competitors may impact the use of our products compare favorably against competing products during both open-heart and minimally invasive procedures, and that our products compare favorably to intracardiac catheter devices when used to treat non-paroxysmal forms of Afib.To compete effectively, we strive to demonstrate that our products are an attractive alternative to other treatments by differentiating our products on the basis of safety, efficacy, performance, ease of use, reputation, service and price.in cardiac procedures. In addition to the cardiac surgery market, we invest heavilyalso consider competition within the post-operative pain market. Currently, we are not aware of other companies in training and education to ensure that our customers understand availablethe United States who are pursuing cryothermic nerve block therapies for thoracic surgery. There are other companies outside of the United States who market their devices techniques, and approaches for optimal treatment. We have encountered and expect to continue to encounter potential customers who prefer products offered by our competitors.Paymentpatienthealth care services in the United States is generally made by third-party payors. These payors include private insurers and government insurance programs, such as Medicare and Medicaid. The Medicare program, the largest single payor in the United States, is a federal health benefit program administered by the Centers for Medicare and Medicaid Services (CMS) and covers certain medical care items and services for eligible beneficiaries, such asprimarily individuals over 65 years old, as well as chronically disabled individuals. Because Medicare beneficiaries comprise a large percentage of the populations for which our products are used, and private insurers may follow the coverage and payment policies for Medicare, Medicare’s coding, coverage and payment policies for cardiothoracic surgical procedures are significant to our business.LAA exclusionLAAM device (LAAM) is used during a concomitant open-heart procedure, Medicare’s hospital reimbursement is based upon the patient’s primary structural heart surgical procedure. Therefore, any additional procedure concomitant to the primary procedure would not receive incremental hospital payment. In contrast, sole therapy minimally invasive ablation or surgical LAAM procedures typically are reimbursed under a general cardiac surgery or intracardiac procedure MS-DRG. We believe hospital reimbursement rates for sole therapy and concomitant therapy cardiac surgical ablation or surgical LAAM are adequate to cover the cost of our products even when multiple procedures are performed. Similar to surgical ablation for Afib or surgical LAAM, cryoablation performed for post-operative pain management is reimbursed as part of the primary procedure, open thoracic or cardiac surgery, MS-DRG. We believe hospital reimbursement rates are adequate in these situations.treatment. At this time, there are no category one CPT codes for the physician to reporttreatment, as well as surgical LAAM. However, some providers utilize unlisted CPT codes to obtain reimbursement in these situations. a third-party reimbursement consultantconsultants that providesprovide support to our customers in the event of a coverage denial.eveneven though a new medical device may have been approved for commercial distribution, we may find limited demand for the device until coverage and sufficient reimbursement levels have been obtained from governmental and private third-party payors. In addition, some private third-party payors require that certain procedures or the use of certain products be authorized in advance as a condition of reimbursement. In some countries, cost containment initiatives and health care reforms include initiatives like governmental reviews of reimbursement rate benchmarks, which may significantly reduce reimbursement for procedures using our medical devices or deny coverage for those procedures altogether. We are actively working to pursue market access initiatives in certain geographies, which includes applying for new reimbursement for therapies in which our devices are being used or pursuing specific reimbursement for utilization of our devices.thatwhich are performed on our behalf, to ensure that medical products distributed domestically or exported internationally are safe and effective for their intended uses. The FDA regulates the total product lifecycle from early design, development and testing, to support submissions for clearance or approval, tomanufacturing and commercialization activities, as well as post-market surveillance and reporting, including corrective actions, removals and recalls. Unless an exemption applies, most medical devices distributed in the United States require either 510(k) clearance or PMA from FDA.new PMA or PMA supplement is required for significant modification tochanges affecting the safety or effectiveness of a PMA-approved device, including indicatedbut not limited to new indications for use, a different manufacturing facility, or changes in the manufacturing process, labeling, andor design specifications or components of a device that is approved through the premarket approval process.device.human subject protection regulations.Accountability Act (HIPAA).manufacturers.manufacturers and prohibits the promotion by manufacturers of uses that are not within the approved or cleared labeling of the device. FDA does not regulate the practice of medicine or the conduct or content of medical education conducted by third parties.parties, which may include uses that are not within approved or cleared device labeling. Manufacturers may provide unrestricted financial support for suchindependent third-party medical education programs in the form of educational grants intended to offset the cost of such programs. If the manufacturer controls or unduly influences the content of such programs, FDA considers those programs to be promotional activities by the manufacturer and thus subject to FDA regulation including promotional restrictions. We seek to ensure that the activities we support pursuant to our educational grants program areis conducted in accordance with FDA criteria for independent educational activities. However, we cannot provide an assurance that FDA or other government authorities would view the third-party programs we have supported as being independent.FDA’s Quality System Regulation (QSR),FDA, including, but not limited to: annual establishment registration and product listing; currentchangeany changes to devices,a device; monitoring and reporting ofserious and adverse events and certain device modifications when necessary, monitoring the safety of the product,malfunctions; and performingreporting certain device corrections and removals when necessary.Federalfederal health care program. The Federalfederal False Claims Act (FCA) imposes civil liability on any person or entity that submits, or causes the submission of, a false or fraudulent claim to the United States government. Damages under the FCA consist of the imposition of fines and penalties and can be significant. The FCA also allows a private individual or entity with knowledge of past or present fraud against the federal government to sue on behalf of the government to recover the civil penalties and treble damages.Progressively, many countries that did not previously have medical device regulations have adopted them, and minimal requirements previously in place are becoming more stringent worldwide. While some harmonization of global regulations has taken hold, requirements continue to differ significantly. In China, for example, the product must first have approval in the country of origin. In Japan and China, successful results from local product safety testing precedes submission of documentation to obtain approval. In addition, regulatory agencies and authorities can halt distribution within the country or otherwise take action in accordance with local laws.2020 for all devices seeking CE mark after that date.2021. The method for assessing conformity varies depending on the type and class of the product, but typically involves a combination of self-assessment by the manufacturerquality system assessment and a third-partyproduct conformity assessment by a third-party notified body, an independent and neutral institution appointed by a country to conduct the conformity assessment. This third-party assessment includes a review of documentation related to the device that ismay be as extensive as the documentation requirements that the USUnited States FDA requires for higher risk products. The notified body also audits the manufacturer’s quality system and performs a detailed review of the testing of the manufacturer’s device. Successful completion of a conformity assessment procedure allows a manufacturer to issue a declaration of conformity with the requirements of the relevant directive and affix the CE mark to the device. Devices that bear the CE mark may be commercially distributed throughout the member states of the European Union and other countries that comply with or mirror the medical device directives.directives or medical device regulations.Quality Systemquality system and individual device certificates on a periodic basis.Intellectual PropertyProtection of our intellectual property is a priority for ourethical business practices and we relyhave put in place strict guidelines to advise medical technology manufacturers on a combination of patent, copyright, trademark and trade secret lawshow to protect our interests. Our ability to protect and use our intellectual property rightscollaborate ethically with healthcare professionals (HCPs). These guidelines are set out in the continued development and commercializationMedTech Europe Code of our technologies and products, operate without infringingEthical Business Practice (MedTech Code), which regulates all aspects of the proprietary rights of others, and prevent others from infringing our proprietary rights is important to our continued success. We will be able to protect our products and technologies from unauthorized use by third parties only to the extent that they are covered by valid and enforceable patents, trademarks or copyrights, or are effectively maintained as trade secrets, know-how or other proprietary information.We hold numerous issued United States and international patents. We also have multiple pending United States and international patent applications. We seek patent protection relating to technologies and products we develop in both the United States and in selected foreign countries. While we own much of our intellectual property, including patents, patent applications, trademarks, trade secrets, know-how and proprietary information, we also license patents and related technology of importance to the commercialization of our products. To continue developing and commercializing our current and future products, we may license intellectual property from commercial or academic entities to obtain the rights to technology that is required for our research, development and commercialization activities.All of our employees and technical consultants are required to execute confidentiality agreements in connection with their employment and consultingindustry's relationships with us. WeHCPs and healthcare organizations (HCOs). It covers medical education and research and development. It also generally require them to agree to discloseintroduces an independent enforcement mechanism and assign to us all inventions conceived in connection with their relationship with us. We devote significant resources to obtaining patentstransparency obligations. The Code sets clear and other intellectual property and protecting our other proprietary information. If valid and enforceable, these patents may give us a means of blocking competitors from using infringing technology to compete directly with our products. We also have proprietary information that may not be patentable. With respect to proprietary information that is not patentable, we have chosen to rely on trade secret protection and confidentiality agreements to protect our interests.ManufacturingWe assemble, inspect, test and package the majority of our products at our facilities in Ohio and California, and our products are sterilized by third parties. Purchased components are generally sourced from a single supplier, but alternatives to these suppliers are available in the event this would be needed.To minimize supply chain risks, we maintain inventory levels of components and raw materials specific to the respective part or device. We assess tooling and equipment on an ongoing basis. Order quantities and lead times for components purchased from outside suppliers are based on our forecasts derived from historical demand and anticipated future demand. Lead times may vary significantly depending on the size of the order, time required to fabricate and test the components, specific supplier requirements and current market demandtransparent rules for the componentsindustry's relationships with HCPs and subassemblies. To date, weHCOs, including company events, third party organized events, arrangements with consultants, gifts, research and financial support to medical education. We have not experienced significant delaysadopted the MedTech Code and incorporated its principles in obtaining any of our components.We regularly audit our suppliers for compliancestandard operating procedures, employee training programs and relationships with our quality system requirements, the QSR and/or applicable ISO standards. We are an FDA-registered medical device manufacturer and certified to ISO 13485:2016. In addition, we have successfully participated in the Medical Device Single Audit Program (MDSAP) and have been certified accordingly. The MDSAP program is recognized in Australia, Brazil, Canada, Europe, Japan and the United States.We are subject to numerous federal, state and local laws relating to such matters as laboratory practices, the experimental use of animals, the use and disposal of hazardous or potentially hazardous substances, safe working conditions, manufacturing practices, environmental protection and fire hazard controlAfib.Codes.Code of Ethical Business Practice. As such, they provide for payment of a fair market value fee only for legitimate services rendered to us. We do not expect or require the consultant to utilize or promote our products, and consultants are required to disclose their relationship with us as appropriate, such as when publishing an article in which one of our products is discussed. Amounts paid to physicians in the United States are disclosed by us in annual reports submitted to CMS under the federal “Open Payments” law. Amounts paid to physicians in certain other countries are also disclosed by us in reports submitted to various governmental agencies in those countries, in accordance with the laws of the jurisdictions where those physicians reside or practice, or where the payments are made.Employees730 full-time1,050 employees as of January 31, 2020.2023. None of the employees were represented by a labor union, or covered by a collective bargaining agreement. Weand we have never experienced any employment-related work stoppages andstoppages. We consider our employee relations to be in good standing. At AtriCure, the employee experience is crucial to the ongoing success of the company. We work to provide a culture that augments the intrinsic rewards of our mission – one where employees feel valued and supported every day. We strive to engage with our employees across every level of the organization, celebrate their personal milestones and cultivate a sense of trust and transparency. Our culture provides opportunities for employees to feel a part of a community, and specific benefits such as paid leave for volunteering and individual recognition with “Heart of AtriCure” awards highlight our commitment to this culture. Our employees have voted us as a Top Workplace seven times in the past eight years, and our culture is regularly cited in our internal engagement surveys as a leading positive attribute of the Company. Our culture is a central asset to our Company.Risks Relating To •Competition from existing and new products and procedures may decrease our market share.Businesssuccess depends, in part, on the adoption of the EPi-Sense device for the treatment of Afib following 2021 FDA pre-market approval of this product.Industryprivate payors to contain or reduce healthcare spending, including for procedures that utilize our products.ablation and left atrial appendage management products as our primary sources of revenue. If we aredo not successfulachieve widespread market acceptance in selling these productsthe United States, our operating results will be harmed.our left atrial appendage management productsLAAM product sales in the United States generate the majority of our revenue. We expect that sales of these products will continue to account for a majority of our revenue for the foreseeable future and that our future revenue will depend on the increasing acceptance by the medical community of our products as a standard treatmentsof care for treating Afib, managing the LAA and managing the LAA. Since we believe that physicians are using our ablation products largely for surgical treatment of Afib, if physicians do not use our products to treat Afib, we would lose substantially all of our revenue. We may not be able to maintain or increase market acceptance of our products for a number of reasons, including those set forth elsewhere in this “Risk Factors” section.If our products do not achieve widespread market acceptance in the United States, our operating results will be harmed, and we may not achieve or sustain profitability.Our success will depend, in large part, on the medical community’s acceptance of our principal products in the United States, which is the largest revenue market in the world for medical devices.pain with Cryo Nerve Block therapy. The U.S. medical community’s acceptance of our products will depend upon our ability to demonstrate the safety and efficacy, advantages, long-term clinical performance and cost-effectiveness of our products. In addition, acceptance of products for the treatment of Afib is dependent upon, among other factors, the level of screening for Afib general awareness and education of the medical community about the surgical treatment of Afib and the existence, effectiveness and safety of our products. Market acceptance and adoption of our products for the treatment of Afib also depends on the level of health insurer (including Medicare) reimbursement to physicians and hospitals for the use ofprocedures using our products.We cannot predict whether the U.S. medical community will accept our products or, if accepted, the extent of their use. Negative publicity resulting from incidents involving our products, otheror similar products related to those we sell or products or procedures subject to our clinical trials could have a significant adverse effect on the overall acceptance of our products. If we encounter difficulties growing the market for our products in the U.S., we may not be able to increase our revenue enough to achieve or sustain profitability, and our business and operating results will be seriously harmed.resultsactivities of its participants.activities. There is no assurance that our products will compete effectively against drugs, catheter-based ablation, implantable devices, other surgical ablation systems,devices, other products or techniques to occlude the left atrial appendage or other surgical Afib treatments, which may be more well-established among physiciansproducts and hospitals.techniques to manage post-operative pain. Our products may become obsolete prior to the end of their anticipated useful lives, or we may introduce new products or next-generation products prior to the end of the useful life of a prior generation,our current products, either of which may require us to dispose of existing inventory and related capital equipment and/or write off their value or accelerate their depreciation. In addition, such other products or techniques may be sold or implemented at lower prices. Due to the size of the Afib and LAA exclusionour markets, and the unmet need for an Afib cure, we anticipate that new or existing competitors may develop competing products, procedures and/or clinical solutions. There are few barriers to prevent new entrants or existing competitors from developingproducts before we do.products. The introduction of new products, procedures or clinical solutions, or of our competitors obtaining FDA approvals or clearances, may result in price reductions, reduced margins, loss of market share, or may render our products obsolete, which could adversely affect our revenue and future profitability.Worldwide economic conditions may reduce demand for procedures using or otherwise result in adverse implications on our business, operating results and financial condition.General worldwide economic conditions may deteriorate due to the effects of, among other developments, general credit market crises, collateral effects on the finance and banking industries, concerns about inflation, slower economic activity which may be caused by many factors, including natural disasters or other catastrophes, decreased consumer confidence, reduced corporate profits and capital spending, adverse business conditions and liquidity concerns. We are unable to predict the extent to which current or future worldwide economic conditions may impact our business. Specifically, because many procedures using our products are elective, they can be deferred by patients. In addition, patients may not be as willing under current or future economic conditions to take time off from work or spend their money on deductibles and co-payments often required in connection with the procedures that use our products.Beyond patient demand, any current or future deterioration in worldwide economic conditions, including in particular their effects on the credit and capital markets, may have other adverse implications for our business. For example, our customers’ ability to borrow money from their existing lenders or to obtain credit from other sources to purchase our products may be impaired, resulting in a decrease in sales. Although we maintain allowances for estimated losses resulting from the inability of our customers to make required payments, we cannot guarantee that we will accurately predict the loss rates we will experience, especially given any continuing turmoil in the worldwide economy. A significant change in the liquidity or financial condition of our customers could cause unfavorable trends in our receivable collections and additional allowances may be required, which could adversely affect our operating results. Further, given the economic and political challenges facing Eurozone countries, concerns have been raised regarding the stability and suitability of the Euro as a single currency. The failure of the Euro as a single currency could adversely affect our operating results. See “The United Kingdom’s withdrawal from the European Union may have a negative effect on global economic conditions, financial markets and out business.”Healthcare costs have risen significantly over the past decade. There have been and may continue to be proposals by legislators, regulators and third-party payors to keep, contain or reduce healthcare costs.The continuing efforts of governments, insurance companies and other payors of healthcare costs to contain or reduce these costs, combined with closer scrutiny of such costs, could lead to patients being unable to obtain approval for payment from these third-party payors. The cost containment measures that healthcare providers are instituting both in the U.S. and internationally could harm our business. Some healthcare providers in the U.S. have adopted or are considering a managed care system in which the providers contract to provide comprehensive healthcare for a fixed cost per person. Healthcare providers may attempt to control costs by authorizing fewer elective surgical procedures or by requiring the use of the least expensive devices possible, which could adversely affect the demand for our products or the price at which we can sell our products. Some healthcare providers have sought to consolidate and create new companies with greater market power, including hospitals. As the healthcare industry consolidates, competition to provide products and services has become and will continue to become more intense. This has resulted and likely will continue to result in greater pricing pressures and the exclusion of certain suppliers from important marketing segments.We face significant uncertainty in the industry due to government healthcare reform.The U.S. Patient Protection and Affordable Care Act (PPACA), as amended, and other healthcare reform have a significant impact on our business. The impact of the PPACA on the healthcare industry is extensive and includes, among other things, the federal government assuming a larger role in the healthcare system, expanding healthcare coverage of United States citizens and mandating basic healthcare benefits. The PPACA impacted our business by requiring an excise tax on all U.S. medical device sales beginning in January 2013 until suspended January 1, 2016 ; this excise tax was permanently repealed in December 2019. When in effect, the increased tax burden from the PPACA impacted our results of operations and cash flows.It is possible that legislation will be introduced and passed by Congress repealing the PPACA in whole or in part and signed into law. Because of the continued uncertainty about the implementation or continued effectiveness of the PPACA, including the potential for further legal challenges or repeal of that legislation, we cannot quantify or predict with any certainty the likely impact of the PPACA or its repeal on our business model, prospects, financial condition or results of operations.Any healthcare reforms enacted in the future may, like the PPACA, be phased in over a number of years but, if enacted, could reduce our revenue, increase our costs or require us to revise the ways in which we conduct business or put us at risk for loss of business. In addition, our results of operations, financial position and cash flows could be materially adversely affected by changes under the PPACA and changes under any federal or state legislation adopted in the future.We sell our products outside of the United States, and we are subject to various regulatory and other risks relating to international operations, which could harm our revenue and profitability.Doing business outside of the United States exposes us to risks distinct from those we face in our domestic operations. For example, our operations outside of the United States are subject to different regulatory requirements in each jurisdiction where we operate or have sales. Our failure, or the failure of our distributors, to comply with current or future foreign regulatory requirements, or the assertion by foreign authorities that we or our distributors have failed to comply, could result in adverse consequences, including enforcement actions, fines and penalties, recalls, cessation of sales, civil and criminal prosecution, and the consequences could be disproportionate to the relative contribution of our international operations to our results of operations. Moreover, if political or economic conditions deteriorate in these countries, or if any of these countries are affected by a natural disaster or other catastrophe, our ability to conduct our international operations or collect on international accounts receivable could be limitedpositive, and our costs could be increased, which could negatively affect our operating results. Engaging in business outside of the United States inherently involves a number of other difficultiescurrent and risks, including, butplanned clinical trials may not limited to:export restrictions and controls relating to technology;pricing pressure that we may experience internationally;difficulties in enforcing agreements and collecting receivables through certain foreign legal systems;political and economic instability;consequences arising from natural disasters and other similar catastrophes, such as hurricanes, tornados, earthquakes, floods and tsunamis;potentially adverse tax consequences, tariffs and other trade barriers;the need to hire additional personnel to promote our products outside of the United States;international terrorism and anti-American sentiment;fluctuations in exchange rates for future sales denominated in foreign currency, which represent a portion of our sales outside of the United States; anddifficulty in obtaining and enforcing intellectual property rights.In addition, our business practices in foreign countries must comply with U.S. laws, including the Foreign Corrupt Practices Act (FCPA). We have a compliance program in place designed to reduce the likelihood of potential violations of the FCPA and other U.S. and foreign anti-bribery and anti-corruption laws. If violations were to occur, they could subject us to fines and other penalties as well as increased compliance costs.Our exposure to each of these risks may increase our costs and require significant management attention. We cannot assure you that one or more of these factors will not harm our business.Compliance with developing European Union medical device regulation may limit our ability to maintain sales of our products in European markets or to introduce new products into European markets.Many foreign countries which we market or may market our products have regulatory bodies and restrictions similar to those of FDA. International sales are subject to foreign government regulation,satisfy the requirements of which vary substantially from country to country. The time required to obtain approval by a foreign country may be longer or shorter than that required forthe FDA clearance and the requirements may differ. In particular, marketing of medical devices in the EU is subject to compliance with the Medical Device Directive 93/92/EEC (MDD). A medical device may be placed on the market within the EU only if it conforms to certain “essential requirements” and bears the CE Mark. The most fundamental and essential requirement is that a medical device must be designed and manufactured in such a way that it will not compromise the clinical condition or safety of patients, or the safety and health of users and others. In addition, the device must achieve the essential performance intended by the manufacturer and be designed, manufactured and packaged in a suitable manner.Manufacturers must demonstrate that their devices conform to the relevant essential requirements through a conformity assessment procedure. The nature of the assessment depends upon the classification of the device. The classification rules are mainly based on three criteria: the length of time the device is in contact with the body, the degree of invasiveness and the extent to which the device affects the anatomy. Conformity assessment procedures for all but the lowest risk classification of device involve a notified body. Notified bodies are often private entities and are authorized or licensed to perform such assessments by government authorities. Manufacturers usually have some flexibility to select a notified body for conformity assessment procedures for a particular class of device and to reflect their circumstances, e.g., the likelihood that the manufacturer will make frequent modifications to its products. Conformity assessment procedures require an assessment of available clinical evidence, literature data for the product and post-market experience in respect of similar products already marketed. Notified bodies also may review the manufacturer’s quality systems. If satisfied that the product conforms to the relevant essential requirements, the notified body issues a certificate of conformity, which allows the general commercializing of a product in the EU. The product can also be subjected to local registration requirementsdepending on the country. We maintain CE Marking on all of our products that require such markings as well as local registrations as required.In May 2017, the EU adopted a new Medical Device Regulation (EU) 2017/745 (MDR), which will repeal and replace the MDD with effect from May 26, 2020. The MDR clearly envisages, among other things, stricter controls of medical devices, including strengthening of the conformity assessment procedures, increased expectations with respect to clinical data for devices and pre-market regulatory review of high-risk devices. The MDR also envisages greater control over notified bodies and their standards, increased transparency, more robust device vigilance requirements and clarification of the rules for clinical investigations. Under transitional provisions, medical devices with notified body certificates issued under the MDD prior to May 26, 2020 may continue to be placed on the market for the remaining validity of the certificate, until May 27, 2024 at the latest. After the expiry of any applicable transitional period, only devices that have been CE marked under the MDR may be placed on the market in the EU. If we fail to comply with the new MDR, we may not be able to continue to sell existing products in the EU or introduce new products for sale in the EU, either of which could materially harm our results of operations and financial condition. See also, “The United Kingdom’s withdrawal from the European Union may have a negative effect on global economic conditions, financial markets and our business.”Our quarterly financial results are likely to fluctuate significantly because our sales prospects are uncertain.Due to current worldwide economic conditions, natural disasters and other factors discussed in this “Risk Factors” section which may impact our sales results, our quarterly operating results are difficult to predict and may fluctuate significantly from quarter to quarter or from prior year to current year periods. These fluctuations may also affect our annual operating results and may cause those results to fluctuate unexpectedly from year to year.Surgeons may not commit enough time to sufficiently learn our products, and restrictions in our ability to train surgeons in the use of our products could reduce the market acceptance of our products and in turn could reduce our revenue or result in injuries to patients or other adverse events that could possibly lead to litigation that could harm us.It is critical to the success of our sales efforts to ensure that there are a sufficient number of surgeons familiar with, trained on and proficient in the use of our products. In order for surgeons to learn to use our products, they must attend structured training sessions in order to familiarize themselves with the products, and they must be committed to learning the technology. Further, surgeons must utilize the technology on a regular basis to ensure they maintain the skill set necessary to use the products. Continued market acceptance could be delayed by lack of surgeon willingness to attend training sessions, by the time required to complete this training or by state or institutional restrictions on our ability to provide training.While we train providers in the safe and effective use of our products, we do not train them to use any of our products specifically to treat Afib unless the product is FDA-approved specifically for the treatment of Afib. Our Isolator Synergy System is approved for the treatment of persistent and long-standing persistent forms of Afib concomitant to open-heart bypass graft or valve replacement surgery. The procedure using our Isolator Synergy System in this manner is known as the MAZE IV procedure. Following FDA approval, we instituted a program to train all new and existing users of the Isolator Synergy System in the MAZE IV procedure. We also make available training on the safe and effective use of our other products consistent with their FDA approved or cleared indications. We cannot assure that we will be able to maintain a consistent level of funding for these training programs or a sufficient number of surgeons will become aware of training programs. An inability to train a sufficient number of surgeons to generate adequate demand for our products could have a material adverse impact on our financial condition.Our marketing strategy is dependent on collaboration with physician “thought leaders”.Our research and development efforts and our marketing strategy depend heavily on obtaining support, physician training assistance and collaboration from highly-regarded physicians at leading commercial and research hospitals, particularly in the U.S. and Europe. If we are unable to gain and/or maintain such support, training services and collaboration, or if the reputation or standing of these physicians is impaired or otherwise adversely affected, our ability to market our products and, as a result, our financial condition, results of operations and cash flow, could be materially and adversely affected.Unless and until we obtain additional FDA approval for our products, we will not be able to promote most of them to treat Afib or to prevent stroke, and our ability to maintain and grow our business could be harmed.Although our Isolator Synergy System received FDA approval for the treatment of some forms of Afib in certain procedures, we have not received FDA clearance or approval to promote our other products for the treatment of Afib or the prevention of stroke. See “Business—Government Regulation”. Unless and until we obtain FDA clearance or approval for the use of our products to treat Afib or prevent stroke, we, and others acting on our behalf, may not claim in the U.S. that our products are safe and effective for such uses or otherwise promote them for such uses. Similar restrictions exist outside of the U.S. There is no assurance that future clearances or approvals of our products will be granted or that current or future clearances or approvals will not be withdrawn. Failure to obtain a clearance or approval or loss of an existing clearance or approval, could hurt our ability to maintain and grow our business.In order to obtain additional FDA approvals to promote our products for the treatment of Afib or reduction in stroke risk, we will need to demonstrate in clinical trials that our products are safe and effective for such use. Development of sufficient and appropriate clinical protocols to demonstrate quality, safety and efficacy may be required and we may not adequately develop suchprotocols to support approval. We cannot assure you that any of our clinical trials will be completed in a timely manner or successfully or that the results obtained will be acceptable to FDA. We, FDA or the IRB may suspend a clinical trial at any time for various reasons, including a belief that the risks to study subjects outweigh the anticipated benefits. In addition, if the results obtained from our clinical trials, any other clinical studies, or clinical or commercial experience indicate that any of our products are not safe or effective, or not as safe or effective as other treatment options, FDA may not approve our products for the treatment of Afib or reduction in stroke risk, and the adoption of the use of our products may suffer and our business would be harmed.time consuming, expensivetaking many years to complete and the outcome uncertain.have uncertain outcomes. Delays in patient enrollment or failure of patients to consent or continue to participate in a clinical trial may cause an increase in costs and delays in the approval and attempted commercialization of our products or result in the failure of the clinical trial. Conducting successful clinical studies may require the enrollment of large numbers of clinical sites and patients, and suitable patients may be difficult to identify and recruit. Patient enrollment in clinical trials and completion of patient participation and follow-up depends on many factors, including the size of the patient population; the nature of the trial protocol; the attractiveness of, or the discomforts and risks associated with, the treatments received by enrolled subjects; the availability of appropriate clinical trial investigators, support staff, and proximity of patients to clinical sites; and the ability to comply with the eligibility and exclusion criteria for participation in the clinical trial and patient compliance. For example,may be discouragedthat experience Afib, stroke, or continued arrhythmias such as IST, following treatment with our products and the number of patients that have serious complications resulting from enrolling inablations or LAA exclusion using our products. We cannot provide any assurance that the data collected during our clinical trials ifwill be compelling to the medical community because it may not be scientifically meaningful, may identify unexpected safety concerns, and may not demonstrate that procedures utilizing ourprotocol requires themexperience should not be relied upon as evidence that any of our products will gain market acceptance or that they will satisfy regulatory requirements for product approval. There can be no assurance that the results of studies conducted by collaborators or other third parties will be viewed favorably or are indicative of our own future study results. We may be required to undergo extensive post-treatment proceduresdemonstrate with substantial evidence through well-controlled clinical trials that our product candidates are either (i) safe and effective for use in a diverse population for their intended uses or follow-up(ii) are substantially equivalent to assesspredicate devices under section 510(k) of the Food, Drug and Cosmetic Act (FDCA). Success in early clinical trials does not mean that future clinical trials will be successful because product candidates in later-stage clinical trials may fail to demonstrate sufficient safety and efficacy to the satisfaction of the FDA and other regulatory authorities despite having progressed through initial clinical trials.if they determine thatprocedures for the treatments received undertreatment of Afib, including but not limited to the trial protocolsEPi-Sense System, also depends on the level of health insurer (including Medicare) reimbursement to physicians and hospitals for procedures using our products. Negative publicity resulting from incidents involving our products, or similar products, could have a significant adverse effect on the overall acceptance of our products. Market acceptance could be delayed by lack of physician willingness to attend training sessions by the time required to complete this training, or by restrictions on our ability to provide training. If we are unable to gain and/or maintain such support, training services and collaboration, our ability to grow the market for our products may be impacted and we may not attractivebe able to increase our revenue enough to achieve or involve unacceptable riskssustain profitability, and our business and operating results may be seriously harmed.discomforts. Patients may also not participate in our clinical trials if they chooseunwillingness of surgeons to participate in contemporaneoussuch training, could reduce the market acceptance of our products.competitivehospitals serving as our clinical trial sites. Key clinical trial activities, such as clinical trial site monitoring, subject visits and study procedures, may be interrupted. We may also encounter interruption or delays in the operations of FDA or other regulatory authorities, which may impact review and approval timelines. Geopolitical issues around the world have impacted the global supply chain and could materially adversely affect global economic growth, disrupt discretionary spending habits and generally decrease demand for our products and services. Our customers’ ability to borrow money from their existing lenders or to obtain credit from other sources to purchase our products may be impaired, resulting in a decrease in sales. We are unable to predict the extent to which current or future worldwide economic conditions may impact our business.can obtaindeem it medically appropriate, such as for the treatment without participatingof Afib or the reduction in stroke risk, even though FDA may not have approved or cleared our trial.andor our industry. This publicity could have a negative impact on our ability to attract and retain customers, our sales, clinical studies involving our products, our reputation and our stock price.certain of our consultants who are involved with clinical studies and the publication of articles concerning our products.consultants. We believe that such publicity would potentially have a negative impact on our clinical studies, business, results of operations and financial condition, or cause other adverse effects, including a decline in the price of our stock.We may be subject to fines, penalties, injunctions and other sanctions if we are deemed to be promoting the use of our products for unapproved, or off-label, uses.Our business and future growth depend on the continued use of our products for the treatment of Afib or prevention of stroke. Unless the products are approved or cleared by FDA specifically for the treatment of Afib or prevention of stroke, we may not make claims about the safety or effectiveness of our products for such uses.These limitations present a material risk that FDA or other federal or state law enforcement authorities could determine that the nature and scope of our sales, marketing and/or support activities, though designed to comply with all FDA requirements, constitute the promotion of our products for an unapproved use in violation of the Federal Food, Drug and Cosmetic Act (FDCA). We also face the risk that FDA or other governmental authorities might pursue enforcement based on past activities that we have discontinued or changed, including sales activities, arrangements with institutions and doctors, educational and training programs and other activities. Investigations concerning the promotion of unapproved uses and related issues, are typically expensive, disruptive and burdensome and generate negative publicity. If our promotional activities are found to be in violation of the law, we may face significant fines and penalties and may be required to substantially change our sales, promotion, grant and educational activities. There is also a possibility that we could be enjoined from selling some or all of our products for any unapproved use. In addition, as a result of an enforcement action against us or our executive officers, we could be excluded from participation in government healthcare programs such as Medicare and Medicaid.We are currently under investigation by the United States Department of Justice, and any adverse finding, allegation, or exercise of enforcement or regulatory discretion by the DOJ could materially and adversely affect our business, financial condition or results of operations.As previously disclosed, on December 11, 2017, the Company received a Civil Investigative Demand (CID) from the U.S. Department of Justice (DOJ) stating that it is investigating the Company to determine whether the Company has violated the False Claims Act, relating to the promotion of certain medical devices related to the treatment of atrial fibrillation for off-label use and submitted or caused to be submitted false claims to certain federal and state health care programs for medically unnecessary healthcare services related to the treatment of Afib. The CID covers the period from January 2010 to December 2017 and requires the production of documents and answers to written interrogatories. The Company had no knowledge of the investigation prior to receipt of the CID. The Company maintains rigorous policies and procedures to promote compliance with the False Claims Act and other applicable regulatory requirements. The Company provided the DOJ with documents and answers to the written interrogatories and iscooperating with the investigation. However, the Company cannot predict when the investigation will be resolved, the outcome of the investigation or its potential impact on the Company. While the Company believes its practices are lawful, there can be no assurance that the DOJ’s ongoing investigation or future exercise of its enforcement, regulatory, discretionary or other powers will not result in findings or alleged violations of federal laws that could lead to enforcement actions, proceedings or litigation and the imposition of damages, fines, penalties, restitution, other monetary liabilities, sanctions, settlements or changes to the Company’s business practices or operations that could have a material adverse effect on the Company’s business, financial condition or results of operations or eliminate altogether the Company’s ability to operate its business or on terms substantially similar to those on which it currently operates.The use of products we sell may result in injuries or other adverse events that lead to product liability suits, which could be costly to our business or our customers’ businesses.The use of products we sell may result in a variety of serious complications, including damage to the heart, internal bleeding, death or other adverse events, potentially leading to product liability claims. Serious complications are commonly encountered in connection with surgical procedures. If products we sell are defectively designed, manufactured or labeled, contain inadequate warnings, contain defective components, are misused or are associated with serious injuries or deaths, we may become subject to costly litigation by our customers or their patients. We carry product liability insurance that is limited in scope and amount and may not be adequate to fully protect us against product liability claims. We could be required to pay damages that exceed our insurance coverage, and such amounts could be significant. Any product liability claim, with or without merit, could also result in an increase in our product insurance rates or our inability to secure coverage on reasonable terms, if at all. Even in the absence of a claim, our insurance rates may rise in the future. Any product liability claim, even a meritless or unsuccessful one, would be time-consuming and expensive to defend and could result in the diversion of our management’s attention from our business and result in adverse publicity, withdrawal of clinical trial participants, injury to our reputation and loss of revenue. Any of these events could negatively affect our financial condition.Our intellectual property rights may not provide meaningful commercial protection for our products, which could enable third parties to use our technology or methods, or very similar technology or methods, and could reduce our ability to compete.Our success depends significantly on our ability to protect our proprietary rights to the technologies used in our products. We rely on patent protection, as well as a combination of copyright, trade secret and trademark laws and nondisclosure, confidentiality and other contractual restrictions to protect our proprietary technology. However, these legal means afford only limited protection and may not adequately protect our rights or permit us to gain or keep any competitive advantage. Our patent applications may not issue as patents at all or in a form that will be advantageous to us. Our issued patents and those that may be issued in the future may be challenged, invalidated or circumvented, which could limit our ability to stop competitors from marketing related products. Although we have taken steps to protect our intellectual property and proprietary technology, we cannot assure you that third parties will not be able to design around our patents or, if they do infringe upon our technology, that we will be successful in or will have sufficient resources to pursue a claim of infringement against those third parties. We believe that third parties may have developed or are developing products that could infringe upon our patent rights. Any pursuit of an infringement claim by us may involve substantial expense or diversion of management attention. In addition, although we have generally entered into confidentiality agreements and intellectual property assignment agreements with our employees, consultants, investigators and advisors, such agreements may be breached, may not be enforceable or may not provide meaningful protection for our trade secrets or other proprietary information in the event of unauthorized use or disclosure or other breaches of the agreements. Additionally, as is common in the medical device industry, some of these individuals were previously employed at other medical equipment or biotechnology companies, including our competitors. Although no claims are currently pending against us, we may be subject to claims that these individuals or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers.The laws of foreign countries may not protect our intellectual property rights to the same extent as the laws of the United States. Foreign countries generally do not allow patents to cover methods for performing surgical procedures. If our intellectual property does not provide significant protection against foreign or domestic competition, our competitors could compete more directly with us, which could result in a decrease in our revenue and market share. All of these factors may harm our competitive position.The medical device industry is characterized by extensive litigation and administrative proceedings over patent and other intellectual property rights and any litigation or claim against us may cause us to incur substantial costs, could place a significant strain on our financial resources, divert the attention of management from our business and harm our reputation.Despite measures taken to protect our intellectual property, unauthorized parties might copy aspects of our products or obtain and use information that we regard as proprietary. Whether a product infringes a patent involves complex legal and factual issues, the determination of which is often uncertain. Any patent dispute, even one without merit or an unsuccessful one, would be time-consuming and expensive to defend and could result in the diversion of our management’s attention from our business and result in adverse publicity, the disruption of development and marketing efforts, injury to our reputation and loss of revenue. Litigation also puts our patent applications at risk of being rejected and our patents at risk of being invalidated or interpreted narrowly and may provoke third parties to assert claims against us. Any of these events could negatively affect our financial condition.In the event of a patent dispute, if a third party’s patents were upheld as valid and enforceable and we were found to be infringing, or found to be inducing infringement by others, we could be prevented from selling our products unless we were able to obtain a license to use technology or ideas covered by such patent or are able to redesign our system to avoid infringement, or we may be ordered to pay substantial damages to the patent holders. A license may not be available at all or on terms acceptable to us, and we may not be able to redesign our products to avoid any infringement. Modification of our products or development of new products could require us to conduct additional clinical trials and to revise our filings with FDA and other regulatory bodies, which would be time-consuming and expensive. If we are not successful in obtaining a license or redesigning our products, we may be unable to sell our products and our business could suffer.The increase in cost of medical malpractice premiums to physicians and hospitals or the lack of malpractice insurance coverage due to the use of our products by physicians for an off-label indication may cause certain physicians or hospitals to decide not to use our products and may damage our ability to maintain or grow the market for our products.Insurance carriers have been raising premiums charged for medical malpractice insurance due, at least in part, to increased risks associated with off-label procedures, including higher damage awards for successful plaintiffs. Insurance carriers may continue to raise premiums or they may deny malpractice coverage for procedures performed using products such as ours on an off-label basis. If this trend continues or worsens, our revenue may fall as physicians or hospitals decide against purchasing our products due to the cost or unavailability of insurance coverage.We have a history of net losses, and we may never become profitable.We have incurred net losses each year since our inception, including, most recently, net losses of $35,194 in 2019, $21,137 in 2018 and $26,892 in 2017. As of December 31, 2019, we had an accumulated deficit of $282,197.Our net losses have resulted principally from costs and expenses relating to sales, training and promotional efforts, research and development, clinical trials, seeking regulatory clearances and approvals and general operating expenses. We expect to continue to incur substantial expenditures and to potentially incur additional operating losses in the future as we further develop and commercialize our products. If sales of our products do not continue to grow as we anticipate, we will not be able to achieve profitability. Our expansion efforts may prove to be more expensive than we currently anticipate, and we may not succeed in increasing our revenue sufficiently to offset these higher expenses. Our losses have had, and are expected to continue to have, an adverse impact on our working capital, total assets and accumulated deficit.Our capital needs after the next twelve months are uncertain, and we may need to raise additional funds in the future and such funds may not be available on acceptable terms, if at all.We believe that our current cash, cash equivalents and investments, along with the cash we expect to generate or use for operations or access via our term loan and revolving line of credit will be sufficient to meet our projected capital requirements for at least the next 12 months. Our Loan and Security Agreement with Silicon Valley Bank (SVB), as amended and restated effective February 23, 2018 and as further amended December 28, 2018, and August 12, 2019 (the “Loan Agreement”), provides for a $60,000 term loan and $20,000 revolving line of credit. The term loan and revolving credit facility both mature in August 2024. According to the Loan Agreement, principal payments on the term loan are to be made ratably commencing March 1, 2021 through the loan’s maturity date of August 1, 2024. If we meet certain conditions, as specified by the agreement, the commencement of term loan principal payments may be deferred by an additional six months. The term loan accrues interest at the greater of the Prime Rate and 5.00% plus 0.75%. As of December 31, 2019, we had outstanding borrowings under the term loan of $60,000. Borrowing availability under the revolving credit facility is based on the lesser of $20,000 or a borrowing base calculation as defined by the Loan Agreement. We may not draw on the revolving line of credit without SVB’s consent if the obligations in the aggregate under the Loan Agreement would exceed $70,000 after the incurrence of such requested withdraw. The applicable interest rate on advances outstanding under the revolving credit facility is the greater of the Prime Rate and 5.00%. The Loan Agreement also provides for certain prepayment and early termination fees, as well as establishes a minimum liquidity covenant and includes other customary terms and conditions. As of December 31, 2019, we had no borrowings under the revolving credit facility, and we had borrowing availability of $8,750.The nContact acquisition also provided for contingent consideration to be paid upon attaining specified regulatory approval in the coming year. Subject to the terms and conditions of the nContact merger agreement, such contingent consideration is paid in AtriCure common stock and cash, with a requirement to make payments in AtriCure common stock first, up to a specified maximum number of shares. Over the next twelve months, we do not expect our cash requirements to include significant payments of contingent consideration based on terms of the acquisition agreement and related milestones. Significant changes to the estimated consideration to be paid could result in a substantial increase in liabilities for contingent consideration and our accumulated deficit and reduce our net income or increase our net loss for the year in which the changes occur, which could contribute to difficulty in raising additional funds. The issuance of our stock to nContact shareholders to settle contingent consideration obligations would dilute the holdings of our existing stockholders.We believe we have adhered to the nContact contract provisions that provide for contingent consideration if the conditions described above are met. nContact representatives have disputed, and in the future may dispute our adherence to the contract andpursue a claim for non-adherence which could involve complex legal and factual issues, the determination of which is often uncertain. Any such claim, even one without merit or an unsuccessful one, would be time-consuming and expensive to defend and could result in the diversion of our management’s attention from our business, adverse publicity, the disruption of development and marketing efforts, injury to our reputation and adversely impact our financial condition.The SentreHEART acquisition provided for contingent consideration to be paid upon attaining specified clinical and reimbursement milestones on or before December 31, 2026. All contingent consideration will be payable in a combination of cash and stock, with the maximum number of shares that may be issued pursuant to the SentreHEART merger agreement limited to 19.9% of AtriCure’s total shares outstanding as of the date of merger. The maximum contingent consideration payable by us will not exceed $260,000. Over the next twelve months, we do not expect our cash requirements to include significant payments of contingent consideration based on terms of the acquisition agreement and related milestones. Significant changes to the estimated consideration to be paid could result in a substantial increase in liabilities for contingent consideration and our accumulated deficit and reduce our net income or increase our net loss for the year in which the changes occur, which could contribute to difficulty in raising additional funds. The issuance of our stock to SentreHEART shareholders to settle contingent consideration obligations would dilute the holdings of our existing stockholders.If we need to raise additional funds for any reason, we cannot be certain that such funds will be available to us on acceptable terms, if at all. Furthermore, if we issue equity securities to raise additional funds, our existing stockholders will experience dilution, and if we issue equity or debt securities, such securities may have rights, preferences and privileges senior to those of our existing stockholders. In addition, if we raise additional funds through collaboration, licensing or other similar arrangements, it may be necessary to relinquish potentially valuable rights to our future products or proprietary technologies or grant licenses on terms that are not favorable to us. If we cannot raise funds on acceptable terms, we may not be able to expand our operations, develop new products, take advantage of future opportunities or respond to competitive pressures or unanticipated customer requirements.We may be unable to comply with the covenants of our Loan Agreement.Our Loan Agreement with SVB contains a minimum liquidity covenant and other customary terms and conditions. The occurrence of an event of default could result in an increase to the applicable interest rate by 3.0%, an acceleration of all obligations, an obligation to repay all obligations in full and a right by SVB to exercise all remedies available to them. If we are unable to pay those amounts, SVB could proceed against the collateral granted to it pursuant to the Loan Agreement, and we may in turn lose access to our current source of borrowing availability.Our federal tax net operating loss (NOL) and general business credit carryforwards generated or acquired may expire or will be limited because we experienced an ownership change of more than 50 percent, which could result in greater future income tax expense and adversely impact future cash flows.On June 30, 2001, we experienced an ownership change as defined by Section 382 of the Internal Revenue Code of 1986. Section 382 imposes limitations (Section 382 limitation) on a company’s ability to use net operating loss and general business credit carryforwards if a company experiences a more-than-50-percent ownership change over a three-year testing period. Additionally, in connection with acquisitions, additional acquired NOLs are also subject to Section 382 limitation. The Section 382 limitations could limit the availability of our net operating loss and general business credit carryforwards to offset any future taxable income, which may increase our future income tax expense and adversely impact future cash flows. Net operating losses generated prior to 2018 are also subject to expiration under current IRS regulations. We have total federal income tax net operating loss and research and development credit carryforwards that, if not used to reduce our taxable income, will begin to expire in 2021. We have generated or acquired available net operating loss and research and development credit carryforwards of $340,079 and $8,168.If our goodwill or other intangible assets become impaired, it could materially reduce the value of our assets and reduce our net income or increase our net loss for the year in which the impairment occurs.As of December 31, 2019, we had $234,781 in goodwill related to acquisitions, which represents the purchase price we paid in excess of the fair value of the net assets we acquired. The Financial Accounting Standards Board’s (FASB) Accounting Standards Codification (ASC) 350, “Goodwill and Other Intangible Assets” requires that goodwill be tested for impairment at least annually (absent any impairment indicators). The testing includes comparing the fair value of each reporting unit with its carrying value. We estimate fair value using several valuation methods, including discounted cash flows, market multiples and market capitalization. Impairment adjustments, if any, are required to be recognized as operating expenses. We may have future impairment adjustments to our recorded goodwill. Any finding that the value of our goodwill has been impaired would require us to record an impairment charge which could materially reduce the value of our assets and reduce our net income or increase our net loss for the year in which the impairment charge occurs and increase our accumulated deficit.In Process Research and Development (IPR&D) valued at $126,321 was recorded as an intangible asset in connection with the nContact and SentreHEART acquisitions. If we do not obtain the regulatory approvals that would confirm the technological feasibility of the respective IPR&D projects, or if the IPR&D projects are abandoned for any other reason, we could have an impairment adjustment of this asset that could require us to write off a portion or all of the recorded asset value. Additionally, and similar togoodwill, if the IPR&D asset is deemed to be impaired (as a result of the estimated fair value being less than carrying value), we would be required to write off the impaired portion of the IPR&D asset. This would materially reduce the value of our assets and reduce our net income or increase our net loss for the year in which the write off occurs and increase our accumulated deficit.An inability to forecast future revenue or estimate life cycles of products may result in inventory-related charges that would negatively affect our gross margins and results of operations.To mitigate the risk of supply interruptions, we may choose to maintain additional inventory of our products or component parts. Managing our inventory levels is important to our cash position and results of operations and is challenging in the current economic environment. As we grow and expand our product offerings, managing our inventory levels becomes more difficult, particularly as we expand into new product areas and bring product enhancements to market. While we rely on our personnel and information technology systems for inventory management to effectively manage accounting and financial functions, our personnel and information technology systems may fail to adequately perform these functions or may experience an interruption. An excessive amount of inventory reduces our cash available for operations and may result in excess or obsolete materials. Conversely, inadequate inventory levels may make it difficult for us to meet customer product demand, resulting in decreased revenue. An inability to forecast future revenue or estimated life cycles of products may result in inventory-related charges that would negatively affect our gross margins and results of operations and increase our accumulated deficit, any of which could contribute to difficulty in raising additional funds.logisticsservice providers, making us vulnerable to supply problems and price fluctuations which could harm our business.Guided Coagulation System with VisiTrax technology and related RF generator. It would be a time consuming and lengthy process to secure these products from an alternative supplier. We have significant concentrations with a limited number of vendors. Additionally, our devices are sterilized prior to use using ethylene oxide at third-party sterilizers. Recently, certain sterilization facilities have experienced mandated temporary closures due to concerns over the impact of emissions of ethylene oxide from such facilities, and the Environmental Protection Agency has proposed regulations aimed at reducing hazardous air pollutants. We also rely on a third partyparties to handle our warehousing and logistics functions for European and Middle Easternseveral international markets on our behalf.•we may not be able to obtain adequate supply in a timely manner or on commercially reasonable terms;•we may have difficulty timely locating and qualifying alternative suppliers;suppliers or sterilizers;manufacture or sell our products;•future regulatory actions to modify sterilization processes may cause sterilizers to close, even on a temporary basis, or require new regulatory approvals for us to use, creating lost sterilization capacity and delays;•our suppliers may encounter financial hardships unrelated to our demand for components, which could inhibit their ability to fulfill our orders and meet our requirements.a replacement of warehousing and logistics provider,providers, if required, may not be accomplished quickly and could involve significant additional costs. Any interruption or delay in the supply of components, materials, sterilization or warehousing and logistics, or our inability to obtain components or materials from alternate sources at acceptable prices in a timely manner, could impair our ability to meet the demand of our customers and cause them to cancel orders or switch to competitive products and could therefore have a material adverse effect on our business, financial condition and results of operations.Ifortake precautions and are in process of qualifying a second building on our third-party vendors fail to comply with extensive FDA regulations relating toOhio campus, we do not maintain a backup manufacturing facility, making us dependent on the manufacturingcurrent facility and production workers for the continued operation of our productsbusiness. A natural or any component part, we may be subject to fines, injunctions and penalties, and our ability to commercially distribute and sell our products may be hurt.Our manufacturing facilities and the manufacturing facilities of any of our third-party component manufacturers, critical suppliersother disaster could damage or third-party sterilization facility are required to comply with FDA’s QSR which sets forth minimum standards for the procedures, execution and documentation of the design, testing, production, control, quality assurance, labeling, packaging, sterilization, storage and shipping of the products we sell. FDA may evaluate our compliance with the QSR, among other ways, through periodic announced or unannounced inspections which could disrupt our operations and interrupt our manufacturing. If in conducting an inspection ofdestroy our manufacturing facilities or the manufacturing facilities of any of our third-party component manufacturers, critical suppliers or third-party sterilization facility, an FDA investigator observes conditions or practices believed to violate the QSR, the investigator may document their observations on a Form FDA-483 that is issued at the conclusion of the inspection. A manufacturer that receives an FDA-483 may respondequipment and cause substantial delays in writing and explain any corrective actions taken in response to the inspectional observations. FDA will typically review the facility’s written response and may re-inspect to determine the facility’scompliance with the QSR and other applicable regulatory requirements. Failure to take adequate and timely corrective actions to remedy objectionable conditions listed on an FDA-483 could result in FDA taking administrative or enforcement actions. Among these may be FDA’s issuance of a Warning Letter to a manufacturer, which informs the manufacturer that FDA considers the observed violations to be of “regulatory significance” that, if not corrected, could result in further enforcement action. FDA enforcement actions, which include seizure, injunction and criminal prosecution, could result in total or partial suspension of a facility’s production and/or distribution, product recalls, fines, suspension of FDA’s review of product applications and FDA’s issuance of adverse publicity. Thus, an adverse inspection could force a shutdown of our manufacturing operations, or a recall of our products. Adverse inspections could also delay FDA approval of our products and could have an adverse effect on our production, sales and financial condition.We and any of our third-party vendors may also encounter other problems during manufacturing including failure to follow specific protocols and procedures, equipment malfunction and environmental factors, any of which could delaylead to additional expense and decreased revenue due to lack of supply. The insurance we maintain may not be adequate to cover our losses. With or impedewithout insurance, damage to our abilityfacility or our other property due to meet demand. The manufacture of our product also subjects us to risks thata natural disaster or casualty event could harm our business, including problems relating to the sterilization of our products or facilities and errors in manufacturing components that could negatively affect the efficacy or safety of our products or cause delays in shipment of our products. Any interruption or delay in the manufacture of the product or any of its components could impair our ability to meet the demand of our customers and cause them to cancel orders or switch to competitive products and could, therefore, have a material adverse effect on our business, financial condition and results of operations.If we fail to comply with the extensive FDA regulations relating to our business, webe subject to fines, injunctions and penalties, and our ability to commercially distribute and promote our products may be hurt.Our products are classified by FDA as medical devices and, as such, are subject to extensive regulationenter into significant acquisitions in the United States by FDAfuture. Acquisitions have inherent uncertainties and numerous other federal, stateinvolve risks and foreign governmental authorities. FDA regulations, guidance, notices and other issuances specific to medical devices are broad and regulate numerous aspects ofdifficulties in integrating that may adversely affect our business.Compliance with FDA, state and other regulations can be complex, expensive and time-consuming. FDA and other authorities have broad enforcement powers. Furthermore, changes in the applicable governmental regulations could prevent further commercialization of our products and technologies and could materially harm our business.If a serious failure to comply with applicable regulatory requirements was determined, it could result in enforcement action by FDA or other state or federal agencies, including the DOJ, which may include any of the following sanctions, among others:warning letters, fines, injunctions, consent decrees and civil penalties;repair, replacement, refunds, recall or seizure of our products;operating restrictions, partial suspension or total shutdown of production;suspension or termination of our clinical trials;refusing or delaying our pending requests for 510(k) clearance or PMAs, new intended uses or modifications to existing products;withdrawing 510(k) clearance or PMAs that have already been granted; andcriminal prosecution.If any of these events were to occur, we could lose customers and our production, product sales, business, results of operations and financial condition wouldcondition.harmed.We are also subjectno assurance that we will be able to medical device reporting regulations thatlocate suitable acquisition candidates, acquire possible acquisition candidates, acquire such candidates on commercially reasonable terms, or integrate acquired businesses successfully in the future. In addition, any governmental review or investigation of our proposed acquisitions, such as by the Federal Trade Commission, may impede, limit or prevent us from proceeding with an acquisition. Future acquisitions may require us to file reportsincur additional debt and contingent liabilities, which may adversely affect our business, results of operations and financial condition. The process of integrating acquired businesses into our existing operations may result in operating, contract and supply chain difficulties, such as the failure to retain customers or management personnel. Such difficulties may divert significant financial, operational and managerial resources from our existing operations and make it more difficult to achieve our operating and strategic objectives.FDA ifproduction yields and quality control, component supply and shortages of qualified personnel. These problems could result in delays in product availability and increases in expenses. Any such delay or increased expense could adversely affect our productsability to generate revenues and adversely impact our operating results.caused or contributed to a death or serious injury or,any insurance in the event of product malfunction, that if such malfunction were to recur, would likelythe death or disability of key personnel. Our officers and key employees, with the exception of our President and Chief Executive Officer, do not have employment agreements, and they may terminate their employment and work elsewhere without notice and without cause or contribute to a death or serious injury. There have been incidents, including patient deaths, which have occurred during or following procedures using our products thatgood reason. Currently we have not,non-compete agreements with our officers and believe were not required to be, reported to FDA because we determined that our products did not cause or contributeother employees. Due to the outcomes in these incidents. If FDA disagrees with us, however, and determines that we should have submitted reports for these adverse events, we could be subject to significant regulatory fines or other penalties. In addition, the numberspecialized knowledge of medical device reports we make, or the magnitude of the problems reported, could cause us or FDA to terminate or modify our clinical trials or recall or cease the saleeach of our products, and could hurt commercial acceptance of our products and harm our reputationofficers with customers.Modifications to our products may require new clearances or approvals or may require us to cease promoting or to recall the modified products until such clearances or approvals are obtained and FDA may not agree with our conclusions regarding whether new clearances or approvals were required.Any modification to a 510(k)-cleared device that would constitute a change in its intended use, design or manufacture could require a new or supplemental 510(k) clearance or, possibly, submission and FDA approval of a PMA. FDA requires every medical device company to make the determination as to whether a 510(k) must be filed, but FDA may review any medical device company’sdecision. We have made modificationsrespect to our products and concluded that such modifications did notour operations and the limited pool of people with relevant experience in the medical device field, the loss of service of one or more of these individuals could significantly affect our ability to operate and manage our business. The announcement of the loss of one or more of our key personnel could negatively affect our stock price.submitattract and retain additional management and technical personnel. We rely primarily on direct sales employees to sell our products in the United States and in Europe, and failure to adequately train them in the use and benefits of our products will prevent us from achieving our market share and revenue growth goals. We have key relationships with physicians that involve procedure, product, market and clinical development and training. If any of these physicians end their relationship with us, our business could be negatively impacted. We cannot assure you that we will be able to attract and retain the personnel and physician relationships necessary to grow and expand our business and operations. If we fail to identify, attract, retain and motivate these highly skilled personnel and physicians, we may be unable to continue our development and sales activities.510(k). FDAregular basis. Despite our security measures, including employee training, our information technology and infrastructure are vulnerable to cyber-attacks, malicious intrusions, breakdowns, destruction, loss of data privacy, breaches due to employee error, malfeasance or other disruptions. Cyber-attacks are becoming more sophisticated and frequent, and our systems could be the target of malware, ransomware and other cyber-attacks. We have invested in our systems and the protection of our data to reduce the risk of an intrusion or interruption, and we monitor our systems on an ongoing basis for any current or potential threats. We canagreecover our indemnification obligations and other liabilities associated with our decisions regarding whether submissions were required.FDA wereour insurance is not adequate or available to disagreepay liabilities associated with us and require usour operations, or if we are unable to submit a 510(k), PMApurchase adequate insurance at reasonable rates in the future, our business, financial condition, results of operations or a PMA supplement for then-existing modifications, we couldcash flows may be required to cease promoting or to recall the modified product until we obtain clearance or approval. In addition, we could be subject to significant regulatory fines or other penalties. Furthermore, our products could be subject to recall if FDA determines, for any reason, that our products are not safe or effective or that appropriate regulatory submissions were not made. Delays in receipt or failure to receive clearances or approvals, the loss of previously received clearances or approvals or the failure to comply with existing or future regulatory requirements could reduce our sales, profitability and future growth prospects.are unable todo not fully comply with such regulations, we could face substantial penalties.state consumer protection, fraud and business practice laws, including the California Consumer Privacy Act (“CCPA”), which became effective on January 1, 2020, which among other things, requires new disclosures to California consumers and provides consumers new abilities to opt out of certain sales of personal information;•the Federal Anti-Kickback Statute, which prohibits persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce either the referral of an individual, or furnishing or arranging for a good or service, for which payment may be made under federal healthcare programs such as the Medicare and Medicaid Programs;•the Federal False Claims Act, which prohibits submitting a false claim or causing of the submission of a false claim to the government;•Medicare laws and regulations that prescribe the requirements for coverage and payment, including the amount of such payment, and laws prohibiting false claims for reimbursement under Medicare and Medicaid;•state consumer protection, fraud and business practice laws, including the California Consumer Privacy Act (“CCPA”), which among other things, requires disclosures to California consumers and provides consumers new abilities to opt out of certain sales of personal information;•federal and state healthcare fraud and abuse laws or laws protecting the privacy of patient medical information, including the Health Insurance Portability and Accountability Act (HIPAA) which protects medical records and other personal health information by limiting their use and disclosure, giving individuals the right to access, amend and seek accounting reasonably necessary to accomplish the intended purpose;•laws and regulations, with respect tosuch as the General Data Protection Regulation in the European Union, that govern collection, use, disclosure, transfer and storage of personal data that we may collect from our employees, consultants or in conjunction with clinical trials such as the General Data Protection Regulation in the European Union;trials;•similar and other regulations outside the United States. For example, if we were found to be in violation of the Federal False Claims Act, we would likely face significant fines and penalties and would likely be required to change substantially our sales, promotion, grant and educational activities. There is also a possibility that we could face an injunction that would prohibit in whole or in part our current business activities, and, as a result of enforcement actions against us or our senior officers, we could be excluded from participation in government healthcare programs such as Medicare and Medicaid. If there is a change in law, regulation or administrative or judicial interpretations, we may have to change our business practices or our existing business practices could be challenged as unlawful, which could have a material adverse effect on our business, financial condition and results of operations.completely eliminate the risk of accidental contamination or injury to third parties from the use, storage, handling or disposal of these materials. In the event of contamination or injury, we could be held liable for any resulting damages, and any liability could exceed any applicable insurance coverage we may have. Our manufacturing operations may result in the release, discharge,Any clinical data that is generated regardingnot be positive or consistent, which would affect the rate at which ourhurt.adoptedclassified by FDA as medical devices and, as such, are subject to extensive regulation by FDA and numerous other federal, state and foreign governmental authorities. FDA regulations, guidance, notices and other issuances specific to medical devices are broad and regulate numerous aspects of our business. Compliance with FDA, state and other regulations can be complex, expensive and time-consuming. FDA and other authorities have broad enforcement powers. Furthermore, changes in the medical community.Important factors upon which the efficacyapplicable governmental regulations could prevent further commercialization of our products willand technologies and could materially harm our business.measured include data onharmed.patients that experience Afibmedical device reports we make, or stroke following treatment withthe magnitude of the problems reported, could cause us or FDA to terminate or modify our clinical trials or recall or cease the sale of our products, and the number of patients that have serious complications resulting from ablations or LAA occlusion using our products. While we believe we are now well-positioned to provide sufficient data regarding the safety and efficacy of our products, such data could identify unexpected safety issues. We cannot provide any assurance that the data collected during our clinical trials will be compelling to the medical community because it may not be scientifically meaningful and may not demonstrate that procedures utilizing our products are an attractive option when compared against data from alternative procedures and products. Negative data would affect the usehurt commercial acceptance of our products and harm our businessreputation with customers.prospects.Adverse changes in payors’ policies toward coverageuntil we obtain additional FDA approval for our products, we will not be able to promote most of them to prevent stroke, and reimbursement for surgical procedures would harm our ability to promotemaintain and sellgrow our products.Third-party payorsbusiness could be harmed. We may be subject to fines, penalties, injunctions and other sanctions if we are increasingly exerting pressure on medical device companiesdeemed to reduce their prices. Even to the extent thatbe promoting the use of our products is reimbursed by private payorsfor unapproved, or off-label, uses.governmental payors, adverse changes in payors’ policies toward coverage and reimbursement for surgical procedures would also harm our ability to promote and sell our products. Payors continue to review their policies and can, without notice, deny coverage for treatments that includefuture growth depend on the continued use of our products. Because each third-party payor individually approves coverageproducts for the treatment of Afib. Unless the products are approved or cleared by FDA specifically for the treatment of Afib or prevention of stroke, we may not make claims about the safety or effectiveness of our products for such uses. In order to obtain additional FDA approvals to promote our products for the treatment of Afib or reduction in stroke risk, we will need to demonstrate in clinical trials that our products are safe and reimbursement, obtaining these approvalseffective for such use. Development of sufficient and appropriate clinical protocols to demonstrate quality, safety and efficacy may be time-consumingrequired and costly. In addition, third-party payorswe may require usnot adequately develop such protocols to provide scientificsupport approval. We cannot assure you that any of our clinical trials will be completed in a timely manner or successfully or that the results obtained will be acceptable to FDA. We, FDA or the IRB may suspend a clinical trial at any time for various reasons, including a belief that the risks to study subjects outweigh the anticipated benefits.clinicalscope of our sales, marketing and/or support activities, though designed to comply with all FDA requirements, constitute the promotion of our products for an unapproved use in violation of the FDCA. We also face the risk that FDA or other governmental authorities might pursue enforcement based on past activities that we have discontinued or changed, including sales activities, arrangements with institutions and doctors, educational and training programs and other activities. Investigations concerning the promotion of unapproved uses and related issues, are typically expensive, disruptive and burdensome and generate negative publicity. If our promotional activities are found to be in violation of the law, we may face significant fines and penalties and may be required to substantially change our sales, promotion, grant and educational activities. There is also a possibility that we could be enjoined from selling some or all of our products for any unapproved use.usetreatment of some forms of Afib in certain procedures, we have not received FDA clearance or approval to promote our products. Adverse changes in coverage and reimbursement for surgical procedures could harm our business and reduce our revenue.FDA does not regulate the practice of medicine. Physicians may use ourother products in circumstances where they deem it medically appropriate, such as for the treatment of Afib or the reduction in stroke risk, even thoughprevention of stroke. Unless and until we obtain FDA may not have approvedclearance or cleared our products to be marketed specificallyapproval for those indications. Some payors may deem the use of our other products to treat Afib or prevent stroke, we, and others acting on our behalf, may not claim in the U.S. that such products are safe and effective for indications not specifically approvedsuch uses or cleared by FDA to be experimental and, asotherwise promote them for such may deny coverage or payment. Often, these denials can be overcome through an appeals process, but there is no guarantee of success in these cases.If coverage and adequate levels of reimbursement from governmental and third-party payorsuses. Similar restrictions also exist outside of the United States are not obtained and maintained, salesU.S. There is no assurance that future clearances or approvals of our products outsidewill be granted or that current or future clearances or approvals will not be withdrawn. Failure to obtain a clearance or approval or loss of an existing clearance or approval, could hurt our ability to maintain and grow our business.United Statesmodified products until such clearances or approvals are obtained and FDA may decrease,not agree with our conclusions regarding whether new clearances or approvals were required.failbe subject to achieve or maintain significant sales outside of the United States.Our revenue generated from sales outside of the United States is also dependent upon the availability of coveragefines, injunctions and reimbursement within prevailing foreign healthcare payment systems. Foreign healthcare payors generally do not provide the same level of reimbursement for sole-therapy minimally invasive procedures utilizing ablation devicespenalties, and related products as payors in the United States. In addition, healthcare cost containment efforts similarour ability to those we face in the United States are prevalent in many of the other countries in which wecommercially distribute and sell our products may be hurt.these efforts are expected to continue. To the extent that the usemanufacturing facilities of any of our devices has historically received reimbursement under a foreign healthcare payment system, such reimbursement, if any, has typically been significantly less thanthird-party component manufacturers, critical suppliers or third-party sterilization facilities are required to comply with FDA’s QSR, which sets forth minimum standards for the reimbursement provided in the United States. If coverageprocedures, execution and adequate levels of reimbursement from governmental and third-party payors outsidedocumentation of the United States aredesign, testing, production, control, quality assurance, labeling, packaging, sterilization, storage and shipping of the products we sell. FDA may evaluate our compliance with the QSR, among other ways, through periodic announced or unannounced inspections which could disrupt our operations and interrupt our manufacturing. If in conducting an inspection of our manufacturing facilities or the manufacturing facilities of any of our third-party component manufacturers, critical suppliers or third-party sterilization facilities, an FDA investigator observes conditions or practices believed to violate the QSR, the investigator may document their observations on a Form FDA-483 that is issued at the conclusion of the inspection. A manufacturer that receives an FDA-483 may respond in writing and explain any corrective actions taken in response to the inspection observations. FDA will typically review the facility’s written response and may re-inspect to determine the facility’s compliance with the QSR and other applicable regulatory requirements. Failure to take adequate and timely corrective actions to remedy objectionable conditions listed on an FDA-483 could result in FDA taking administrative or enforcement actions. Among these may be FDA’s issuance of a Warning Letter to a manufacturer, which informs the manufacturer that FDA considers the observed violations to be of “regulatory significance” that, if not obtainedcorrected, could result in further enforcement action. FDA enforcement actions, which include seizure, injunction and maintained, salescriminal prosecution, could result in total or partial suspension of a facility’s production and/or distribution, product recalls, fines, suspension of FDA’s review of product applications and FDA’s issuance of adverse publicity. Thus, an adverse inspection could force a shutdown of our manufacturing operations or a recall of our products. Adverse inspections could also delay FDA approval of our products outsideand could have an adverse effect on our production, sales and financial condition.United States may decrease, and we may failproduct or any of its components could impair our ability to achieve or maintain significant sales outside ofmeet the United States.Fluctuations in foreign currency exchange rates could result in declines in our reported sales and results of operations.Because somedemand of our international sales are denominated in local currencies and not in U.S. Dollars, our reported sales and earnings are subject to fluctuations in foreign currency exchange rates, primarily the Euro and British Pound. We translate results of transactions denominated in local currencies into U.S. Dollars using market conversion rates applicable to the period in which the transaction is reported. As a result, changes in exchange rates during a period can unpredictably and adversely affect our consolidated operating results and our asset and liability balances, even if the underlying value of the item in its original currency has not changed. At present, we do not hedge our exposure to foreign currency fluctuations. As a result, sales and expenses occurring in the future thatare denominated in foreign currencies may be translated into U.S. Dollars at less favorable rates, resulting in reduced revenues and earnings.Our manufacturing operations are primarily conducted at a single location, and any disruption at our manufacturing facility could increase our expenses and decrease our revenue.Our manufacturing operations are primarily conducted at a single location in Ohio, while select products are manufactured in California. While we take precautions at the Ohio location, we do not maintain a backup manufacturing facility, making us dependent on the current facility for the continued operation of our business. A natural or other disaster could damage or destroy our manufacturing equipmentcustomers and cause substantial delays in our manufacturing operations, whichthem to cancel orders or switch to competitive products and could, lead to additional expense and decreased revenue due to lack of supply. The insurance we maintain may not be adequate to cover our losses in any particular case. With or without insurance, damage to our facility or our other property due to a natural disaster or casualty event couldtherefore, have a material adverse effect on our business, financial condition and results of operations.relyare currently defending against a lawsuit brought under the False Claims Act, and any adverse finding, judgement, or enforcement action could materially and adversely affect our business, financial condition or results of operations.independent distributorsDecember 11, 2017, the Company received a Civil Investigative Demand (CID) from the USDOJ stating that it was investigating the Company to marketdetermine whether the Company has violated the False Claims Act, relating to the promotion of certain medical devices related to the treatment of atrial fibrillation for off-label use and sell our products insubmitted or caused to be submitted false claims to certain markets outsidefederal and state health care programs for medically unnecessary healthcare services related to the treatment of Afib. The Company provided the USDOJ with documents and answers to the written interrogatories, and cooperated with the investigation. In 2021, USDOJ informed the Company that the investigation resulted from a lawsuit by a private individual, or "relator", brought on behalf of the United States and various state and local governments under the qui tam provisions of the federal and similar state and local laws. Although the USDOJ and all of the state and local governments declined to intervene, the relator continues to pursue the lawsuit. During the third quarter of 2022, the relator filed a failureFourth Amended Complaint, which dropped allegations of off-label promotion and now alleges that the Company paid illegal kickbacks to healthcare providers in exchange for using or referring the Company’s products, in violation of the federal Anti-Kickback Statute and various comparable state and localindependent distributorsproducts may result in a variety of serious complications, including damage to successfully marketthe heart, nerves, internal bleeding, death, paralysis or other adverse events. Serious complications are commonly encountered in connection with surgical procedures. If products we sell are defectively designed, manufactured or labeled, contain inadequate warnings, contain defective components, are misused or are associated with serious injuries or deaths, we may become subject to costly litigation by our customers or their patients. We carry product liability insurance that is limited in scope and amount and may not be adequate to fully protect us against product liability claims. We could be required to pay damages that exceed our insurance coverage, and such amounts could be significant. Any product liability claim, with or without merit, could also result in an increase in our insurance rates or our inability to secure coverage on reasonable terms, if at all. Even in the absence of a claim, our insurance rates may rise in the future. Any product liability claim, even a meritless or unsuccessful one, would be time-consuming and expensive to defend and could result in the diversion of our management’s attention from our business and result in adverse publicity, withdrawal of clinical trial participants, injury to our reputation and loss of revenue. Any of these events could negatively affect our financial condition.any disruption in theirmethods, or very similar technology or methods, and could reduce our ability to compete.soinfringe upon our technology, that we will be successful in or will have sufficient resources to pursue a claim of infringement against those third parties. We believe that third parties may adversely impacthave developed or are developing products that could infringe upon our sales.We depend on third-party distributorspatent rights. Any pursuit of an infringement claim by us may involve substantial expense or diversion of management attention. In addition, although we have generally entered into confidentiality agreements and intellectual property assignment agreements with our employees, consultants, investigators and advisors, such agreements may be breached, may not be enforceable or may not provide meaningful protection for our trade secrets or other proprietary information in the event of unauthorized use or disclosure or other breaches of the agreements. Additionally, as is common in the medical device industry, some of these individuals were previously employed at other medical equipment or biotechnology companies, including our competitors. Although no claims are currently pending against us, we may be subject to sellclaims that these individuals have used or disclosed trade secrets or other proprietary information of their former employers.products in certain markets outsideintellectual property rights to the same extent as the laws of the United States, and if these distributorsStates. Foreign countries generally do not perform, we may be unableallow patents to maintaincover methods for performing surgical procedures. If our intellectual property does not provide significant protection against foreign or increase our level of international revenue. We intend to continue to grow our business outside of the United States, and to do so, we will need to attract additional distributorsdomestic competition, any current or hire direct sales personnel to expand the territories in which we sell our products. Independent distributors may terminate their relationshipfuture competitors could compete more directly with us, or devote insufficient sales efforts towhich could result in a decrease in our products. We are not able to control our independent distributors,revenue and they may not be successful in implementing our marketing plans. In addition, many of our independent distributors outside of the United States initially obtain and maintain foreign regulatory approval for sale of our products in their respective countries. Our failure to maintain our relationships with our independent distributors outside of the United States, or our failure to recruit and retain additional skilled independent distributors in these locations, could have an adverse effect on our operations. Turnover among our independent distributors, even if replaced, may adversely affect our short-term financial results while we transition to new independent distributors or direct sales personnel. The abilitymarket share. All of these third-party distributorsfactors may harm our competitive position.market and sell our products could also be adversely affected by unexpected events, including, but not limited to, power failures, nuclear events, natural or other disasters and war or terrorist activities. In addition, in light of the worldwide economic crisis, the ability of our distributors to borrow money from their existing lenders or to obtain credit from other sources to purchase our products may be impaired or our distributors could experience a significant change in their liquidity or financial condition, all of which could impair their ability to distribute our products and eventually lead to distributor turnover, and may adversely impact our sales.If we fail to properly manage our anticipated growth, our business could suffer.We may experience periods of rapid growth and expansion, whichincur substantial costs, could place a significant strain on our personnel,financial resources, divert the attention of management from our business and harm our reputation.technology systemsthat we regard as proprietary. Whether a product infringes a patent involves complex legal and other resources. In particular,factual issues, the increase in our direct sales force requires significant managementdetermination of which is often uncertain. Any patent dispute, even one without merit or an unsuccessful one, would be time-consuming and other supporting resources. Any failure by usexpensive to manage our growth effectively could have an adverse effect on our ability to achieve our developmentdefend and commercialization goals.To achieve our revenue goals, we must successfully increase production output as required by customer demand. In the future, we may experience difficulties in increasing production, including problems with production yields and quality control, component supply and shortages of qualified personnel. These problems could result in delaysthe diversion of our management’s attention from our business and result in product availabilityadverse publicity, the disruption of development and increases in expenses.marketing efforts, injury to our reputation and loss of revenue. Litigation also puts our patent applications at risk of being rejected and our patents at risk of being invalidated or interpreted narrowly and may provoke third parties to assert claims against us. Any such delay or increased expenseof these events could adverselynegatively affect our abilityfinancial condition.generate revenues.Future growth will also impose significant added responsibilities on management, including the needbe infringing, or found to identify, recruit, train and integrate additional employees. In addition, rapid and significant growth will placebe inducing infringement by others, we could be prevented from selling our products unless we were able to obtain a strain onlicense to use technology or ideas covered by such patent or are able to redesign our administrative and operational infrastructure. In ordersystem to manage our operations and growth,avoid infringement, or we will need to continue to improve our operational and management controls, reporting and information technology systems and financial internal control procedures. If we are unable to manage our growth effectively, it may be difficult forordered to pay substantial damages to the patent holders. A license may not be available at all or on terms acceptable to us, and we may not be able to redesign our products to avoid any infringement. Modification of our products or development of new products could require us to executeconduct additional clinical trials and to revise our business strategy and our operating results and business could suffer.We depend on our officersfilings with FDA and other skilledregulatory bodies, which would be time-consuming and experienced personnel to operate our business effectively.expensive. If we are not ablesuccessful in obtaining a license or redesigning our products, we may be unable to retain our current employees or recruit additional qualified personnel, our business will suffer and our future revenue and profitability will be impaired.We are highly dependent on the skills and experience of our President and Chief Executive Officer, Michael H. Carrel, and certain other officers and key employees. We do not have any insurance in the event of the death or disability of key personnel. Our officers and key employees, with the exception of our President and Chief Executive Officer, do not have employment agreements, and they may terminate their employment and work elsewhere without notice and without cause or good reason. Currently we have non-compete agreements with our officers and other employees. Due to the specialized knowledge of each of our officers with respect tosell our products and our business could suffer.pooland our costs could be increased, which could negatively affect our operating results. Engaging in business outside of people with relevantthe United States inherently involves a number of other difficulties and risks, including, but not limited to:medical device field,need to hire additional personnel to promote our products outside of the lossUnited States;service ofthese risks may increase our costs and require significant management attention. We cannot assure you that one or more of these individuals could significantly affectfactors will not harm our ability to operate and manage our business. The announcementthe loss of one or more of our key personnel could negatively affect our stock price.ContentsWe depend on our scientific and technical personnel for successful product development and innovation, which are criticalsuccess of our business. In addition, to succeed in the implementation of our business strategy, our management team must rapidly execute our sales strategy, obtain expanded FDA clearances and approvals, achieve market acceptance for our products and further develop products, while managing anticipated growth by implementing effective planning, manufacturing and operating processes. Managing this growth will require us to attract and retain additional management and technical personnel. We rely primarily on direct sales employees to sell our products in the United States and failure to adequately train them in the use and benefits of our products will prevent us from achieving our market share and revenue growth goals. We have key relationships with physicians that involve procedure, product, market and clinical development. If any of these physicians end their relationship with us, our business could be negatively impacted. We cannot assure you that we will be able to attract and retain the personnel and physician relationships necessary to grow and expand our business and operations. If we fail to identify, attract, retain and motivate these highly skilled personnel and physicians, we may be unable to continue our development and sales activities.Our business growth strategy involves the potential for significant acquisitions, which involve risks and difficulties in integrating potential acquisitions and may adversely affect our business, results of operations and financial condition.All acquisitions involve inherent uncertainties, which may include, among other things, our ability to:successfully identify targets for acquisition;negotiate reasonable terms;properly perform due diligence and determine significant risks associated with a particular acquisition;properly evaluate target company management capabilities; andsuccessfully transition and integrate the acquired company into our business and achieve the desired performance.We may acquire businesses with unknown liabilities, contingent liabilities or internal control deficiencies. We have plans and procedures in place to conduct reviews of potential acquisition candidates for compliance with applicable regulations and laws prior to acquisition. Despite these efforts, realization of any of these liabilities or deficiencies may increase our expenses, adversely affect our financial position through the initiation, pendency or outcome of litigation or otherwise, or cause us to fail to meet our public financial reporting obligations.We have consummated three significant acquisitions since 2013 and in the future may continue to invest a substantial amount of capital in acquisitions. We continue to evaluate potential acquisition opportunities to support, strengthen and grow our business. There can be no assurance that we will be able to locate suitable acquisition candidates, acquire possible acquisition candidates, acquire such candidates on commercially reasonable terms, or integrate acquired businesses successfully in the future. In addition, any governmental review or investigation of our proposed acquisitions, such as by the Federal Trade Commission, may impede, limit or prevent us from proceeding with an acquisition. Future acquisitions may require us to incur additional debt and contingent liabilities, which may adversely affect our business, results of operations and financial condition. The process of integrating acquired businesses into our existing operations may result in operating, contract and supply chain difficulties, such as the failure to retain customers or management personnel. Such difficulties may divert significant financial, operational and managerial resources from our existing operations and make it more difficult to achieve our operating and strategic objectives.Disruptions of critical information systems or material breaches in the security of our systems could harm our business, customer relations and financial condition.In the ordinary courseglobal nature of our business, we collect and store sensitive data, including intellectual property, our proprietary business information and that of our customers, suppliers and business partners, and personally identifiable information of our customers and employees in our data centers and on our networks. The secure processing, maintenance and transmission of this information is critical to our operations and business strategy. Despite our security measures, our information technology and infrastructure may be vulnerableexposed to attacks by hackers or breached due to employee error, malfeasance orliabilities under the Foreign Corrupt Practices Act and various other disruptions. Any such breach could compromise our networks and the information stored there could be accessed, publicly disclosed, lost or stolen. Any such access, disclosure or other loss of information could result in legal claims or proceedings, liability under laws that protect the privacy of personal information, regulatory penalties, disrupt our operations and the services we provide to customers, and damage our reputation and cause a loss of confidence in our products and services, which could adversely affect our business, operating margins, revenues and competitive position.We also rely in part on information technology to store information, interface with customers, maintain financial accuracy, secure our data and accurately produce our financial statements. If our information technology systems do not effectively and securely collect, store, process and report relevant data for the operation of our business, whether due to equipment malfunction or constraints, software deficiencies, human error or cyber incident, our ability to effectively plan, forecast and execute our business plan and complywith applicableanti-corruption laws, and regulations would be materially impaired. Any such impairmentany allegation or determination that we violated these laws could have a material adverse effect on our business.conditioncondition.timelinessfuture as we further develop and commercialize our products. If sales of our products do not continue to grow as we anticipate, we may not be able to achieve profitability. Our expansion efforts may prove to be more expensive than we currently anticipate, and we may not succeed in increasing our revenue sufficiently to offset these higher expenses. Our losses have had, and are expected to continue to have, an adverse impact on our working capital, total assets and accumulated deficit.reportoperate may also adopt some form of these guidelines. In such case, we may need to change our approach to intercompany transfer pricing in order to maintain compliance under the new rules. Our effective tax rate may increase or decrease, including changes in minimum taxation, depending on the current location of global operations at the time of the change. Finally, we might not always be in compliance with all applicable customs, exchange control, value added tax and transfer pricing laws despite our efforts to be aware of and to comply with such laws. In such case, we may need to adjust our operating results.procedures and our business could be adversely affected.tointo countries outside the United States that may experience economic turmoil.The United Kingdom’s withdrawal from the European Union may have a negative effect on global economic conditions, financial markets and our business.In June 2016, a majority of voters in the United Kingdom elected to withdraw from the EU, in a national referendum, commonly referred to as Brexit. In March 2017, the United Kingdom formally notified the EU of its intention to withdraw pursuant to Article 50 of the Lisbon Treaty. The United Kingdom left the EU on January 31, 2020, which has initiated an eleven-month transition period by which the United Kingdom is to leave the single market and customs union. The withdrawal has created significant uncertainty about the future relationship between the United Kingdom and the EU, including with respect to the laws and regulations that will apply as the United Kingdom determines which EU laws to replace or replicate to facilitate the withdrawal. From a regulatory perspective, the United Kingdom’s withdrawal gives rise to significant complexity and risks. Since the medical device regulatory framework in the United Kingdom is derived from the EU Medical Devices Directive, the United Kingdom’s withdrawal could materially impact the continued marketing of EU medical devices in the United Kingdom. Further, the withdrawal may also significantly delay the transport of our products into the United Kingdom, which could adversely impact our sales.Because of the continued uncertainty about the effects, implementation, or potential repeal of Brexit, we cannot quantify or predict with any certainty the likely impact of Brexit or related legislation on our business model, prospects, financial condition or results of operations. In the absence of a future trade deal, the United Kingdom’s trade with the EU and the rest of the world would be subject to tariffs and duties set by the World Trade Organization. Additionally, the movement of goods and personnel between the United Kingdom and the remaining member states of the EU will be subject to additional inspections and documentation checks, leading to possible delays at ports of entry and departure. The withdrawal may result in significant changes to the trading relationship between the United Kingdom and the EU. These changes to the trading relationship between the United Kingdom and EU would likely result in increased cost of goods imported into and exported from the United Kingdom and may decrease the profitability of our operations. Additional currency volatility could drive a weaker British pound, which could increase the cost of goods imported into the United Kingdom and may decrease the profitability of our operations. A weaker British pound versus the U.S. dollar may also cause local currency results of our operations to be translated into fewer U.S. dollars during a reporting period.Additionally, we may face new regulations regarding trade, aviation, tax, security and employees, among others, in the United Kingdom. Compliance with such regulations could be costly, negatively impacting our business, results of operations and financial condition. Brexit could also adversely affect European and worldwide economic and market conditions and could contribute to instability in global financial markets. With a range of outcomes still possible, the impact from Brexit remains uncertain and will depend, in part, on the final outcome of tariff, trade, regulatory and other negotiations.Our effective income tax rate may fluctuate, which may adversely affect our operations, earnings and earnings per share.Our effective income tax rate is influenced by our projected profitability in the various taxing jurisdictions in which we operate. The global nature of our business increases our tax risks. In addition, revenue authorities in many of the jurisdictions in which we operate are known to have become more active in their tax collection activities. Changes in the distribution of profits and losses among taxing jurisdictions may have a significant impact on our effective income tax rate, which in turn could have an adverse effect on our net income and earnings per share. The application of tax laws in various taxing jurisdictions, including the United States, is subject to interpretation, and tax authorities in various jurisdictions may have diverging and sometimes conflicting interpretations of the application of tax laws. Changes in tax laws or tax rulings, in the United States or other tax jurisdictions in which we operate, could materially impact our effective tax rate.Factors that may affect our effective income tax rate include, but are not limited to:the requirement to exclude from our quarterly worldwide effective income tax calculations losses in jurisdictions where no income tax benefit can be recognized;actual and projected full year pre-tax income, including differences between actual and anticipated income before taxes in various jurisdictions;changes in tax laws, or in the interpretation or application of tax laws, in various taxing jurisdictions;changes in the relative mix and staffing levels in various tax jurisdictions;audits or other challenges by taxing authorities; andthe establishment of valuation allowances against a portion or all of certain deferred income tax assets if we determined that it is more likely than not that future income tax benefits will not be realized.These changes may cause fluctuations in our effective income tax rate that could adversely affect our results of operations and cause fluctuations in our earnings and earnings per share.Governmental authorities may question our intercompany transfer pricing policies or change their laws in a manner that could increase our effective tax rate or otherwise harm our business.As a U.S. company doing business in international markets through subsidiaries, we are subject to foreign tax and intercompany pricing laws, including those relating to the flow of funds between the parent and subsidiaries. Tax authorities in the United States and in foreign markets closely monitor our corporate structure and how we account for intercompany fund transfers. If tax authorities challenge our corporate structure, transfer pricing mechanisms or intercompany transfers, our operationsWe may be negatively impacted and our effective tax rate may increase. Tax rates vary from country to country and if regulators determine that our profits in one jurisdiction should be increased, we might not be able to fully utilize all foreign tax credits that are generated, which would increase our effective tax rate. Additionally, the Organization for Economic Cooperation and Development, or OECD, has issued certain proposed guidelines regarding base erosion and profit sharing. Once these guidelines are formally adopted by the OECD, it is possible that separate taxing jurisdictions may also adopt some form of these guidelines. In such case, we may need to change our approach to intercompany transfer pricing in order to maintain compliance under the new rules. Our effective tax rate may increase or decrease depending on the current location of global operations at the time of the change. Finally, we might not always be in compliance with all applicable customs, exchange control, Value Added Tax and transfer pricing laws despite our efforts to be aware of and to comply with such laws. In such case, we may need to adjust our operating procedures and our business could be adversely affected.Due to the global nature of our business, we may be exposed to liabilities under the Foreign Corrupt Practices Act and various other anti-corruption laws, and any allegation or determination that we violated these laws could have a material adverse effect on our business.We are requiredunable to comply with the FCPA, UK Bribery Actcovenants of 2010our Loan Agreement.U.S.customary terms and foreign anti-corruption laws,conditions. The occurrence of an event of default could result in an increase to the applicable interest rate by 3.0%, an acceleration of all obligations, an obligation to repay all obligations in full and a right by SVB to exercise all remedies available to them. If we are unable to pay those amounts, SVB could proceed against the collateral granted to it pursuant to the Loan Agreement, and we may in turn lose access to our current source of borrowing availability.prohibit companiescould cause a decline in our stock price.engaging in bribery including corruptly or improperly offering, promising, or providing money or anything else of value to foreign officialscompetitive devices and certain other recipients. In addition, the FCPA imposes certain books, recordstherapies, and accounting control obligations on public companies and other issuers. We operate in partsstability of the worldmacro-economic environment in our key markets. Furthermore, analysts and investors may develop and publish their own projections of our business, which corruption can be common and compliance with anti-bribery laws may conflict with local customs and practices.form a consensus about our future performance. Our global operations face the riskbusiness results may vary significantly from such guidance or that consensus due to a number of unauthorized payments or offers being made by employees, consultants, sales agents and other business partnersfactors, many of which are outside of our control, or without our authorization. It is our policy to implement safeguards (including mandatory training) to prohibit these practices by our employees and business partners with respect to our operations. However, irrespective of these safeguards, or as a result of monitoring compliance with such safeguards, it is possible that we or certain other parties may discover or receive information at some point that certain employees, consultants, sales agents, or other business partners may have engaged in corrupt conduct for which we might be held responsible. Violations of the FCPA or other foreign anti-corruption laws may result in restatements of, or irregularities in, our financial statements as well as severe criminal or civil sanctions, and we may be subject to other liabilities, which could negatively affect our business, operating results and financial condition. In some cases, companies that violate the FCPA may be debarred by the U.S. government and/or lose their U.S. export privileges. Changes in anti-corruption laws or enforcement priorities could also result in increased compliance requirements and related costs which could adversely affect our business, financial conditionoperations and operating results. Furthermore, if we make downward revisions of our previously announced guidance, or if our publicly announced guidance of future operating results fails to meet expectations of operations. In addition, the U.S.securities analysts, investors, or other governments may seek to hold us liable for successor liability FCPA violations or violations of other anti-corruption laws committed by companies in which we invest or that we acquired or will acquire.The impact of restrictive trade policies in the United States and the potential corresponding actions by other countries could adversely affect our financial performance.The U.S. federal government has recently implemented tariffs on certain products imported into the United States from China, and the Chinese government has responded with retaliatory tariffs on certain products, including medical devices, exported from the United States to China. We cannot predict whether the United States will implement additional trade restrictions with respect to China or other countries and how such countries may respond to such trade restrictions. If these tariffs continue or are expanded, they may make it more difficult to sell our products in China or other markets outside of the United States. Restrictive trade policies may also harm the United States and global economies generally, which would adversely affect our business in a variety of ways, includingreducing the market for our products, causing a downturn in the trading price of our common stock, and restricting access to credit if we seek it for future growth.Our insurance may not cover all of our indemnification obligations and other liabilities associated with our operations.We maintain insurance designed to provide coverage for ordinary risks associated with our operations and our ordinary indemnification obligations, which we believe to be customary for our industry. The coverage provided by such insurance may not be adequate for all claims we may make or may be contested by our insurance carriers. If our insurance is not adequate or available to pay liabilities associated with our operations, or if we are unable to purchase adequate insurance at reasonable rates in the future, our business, financial condition, results of operations or cash flows may be materially adversely impacted.Shutdowns of the U.S. federal government could materially impair our business and financial condition.Regulatory approval may be delayed for reasons beyond our control. For example, over the last several years the U.S. government has shut down several times and certain regulatory agencies, such as the FDA, CMS and the SEC, have had to furlough their government employees and stop critical activities. If a prolonged government shutdown or budget sequestration occurs, it could significantly impact the ability of the FDA to timely review and process our regulatory submissions, which could have a material adverse effect on our business. Further, in our operations as a public company, future government shutdowns could impact our ability to communicate with the SEC on various topics, such as shareholder proposals, or to have our registration statements declared effective, which could affect our ability to access the capital markets quickly.An epidemic of the coronavirus disease is ongoing in China and other parts of the world and may adversely affect our operations and financial condition.An epidemic of the coronavirus disease is ongoing in China and other parts of the world. As the outbreak is still evolving, much of its impact remains unknown. It is impossible to predict the effect and potential spread of the coronavirus in China and globally. Should the coronavirus continue to spread or not be contained in China or other parts of the world, our business operations could be delayed or interrupted. China has implemented travel bans to contain the coronavirus, and several countries have expanded screenings of travelers. If bans are implemented and extended to other countries, our business operations could be adversely affected.Risks Relating To Our Common StockThe price and trading volume of our common stock may experience extreme fluctuations and our stockholders could lose some or all of their investment.Because we operate within the medical device segment of the healthcare industry, our stock price is likely to be volatile. The market price of our common stock may have and has had a history of substantial fluctuation due to a variety of factors, including, but not limited to:variations in our quarterly financial and operating results;physician and patient acceptance of the surgical treatment of Afib or exclusion of the LAA using our products;adverse regulatory developments with respect to our products, such as recalls, new regulatory requirements, changes in regulatory requirements or guidance and timing of regulatory clearances and approvals for new products;coverage and reimbursement determinations for our products and the related procedures;the timing of orders received;delays or interruptions in manufacturing or shipping of our products;pricing of our products;clinical trial results;media reports, publications or announcements about products or new innovations that could compete with our products or about the medical device product segment in general;investigations, claims or allegations by regulatory agencies, such as the Department of Justice and Financial Industry Regulatory Authority;market conditions or trends related to the medical device and healthcare industries or the market in general;additions to or departures of our key personnel;disputes, litigation or other developments relating to proprietary rights, including patents, and our ability to obtain patent protection for our technologies;changes in financial estimates, investors’ perceptions or recommendations by securities analysts;failure to achieve or maintain an effective healthcare compliance environment;changes in accounting principles; andfailure to achieve and maintain an effective internal control environment.These factors, some of which are not within our control, may cause the price of our stock to fluctuate substantially. We believe the quarterly and annual comparisons of our financial results are not necessarily meaningful and should not be relied upon as an indication of our future performance.The market prices of the securities of medical device companies, particularly companies like ours without consistent revenue and earnings, have been highly volatile and are likely to remain highly volatile in the future. This volatility has often been unrelated to the operating performance of these particular companies. In the past, companies that experience volatility in the market price of their securities have often faced securities class action litigation. Whether or not meritorious, litigation brought against us could result in substantial costs, divert our management’s attention and resources and harm our ability to grow our business.We may be obligated to issue additional shares of our common stock to the former stockholders of nContact and SentreHEART as a result of our satisfaction of certain milestones set forth in the respective merger agreements, resulting in dilution of our current stock ownership.Under the terms of each of the nContact and SentreHEART merger agreements, we could issue additional shares of our common stock, or make payments in cash, to the former stockholders of nContact and SentreHEART as contingent consideration upon our satisfaction of milestones described in the merger agreements. The nContact merger agreement limits the total number of shares of AtriCure common stock issued in connection with the acquisition to 5,660, of which 3,757 shares were issued at the closing of the nContact acquisition on October 13, 2015 and 232 shares were issued and delivered to the former shareholders of nContact on September 20, 2018 for satisfaction of the trial enrollment milestone. The SentreHEART merger agreement limits the total number of shares of AtriCure common stock issued in connection with the acquisition to 7,021, of which 699 shares were issued at the closing of the SentreHEART acquisition on August 13, 2019. Issuing additional shares of our common stock in satisfaction of contingent consideration dilutes the ownership interests of holders of our common stock on the dates of such issuances. If we are unable to realize the strategic, operational and financial benefits anticipated from our acquisitions of nContact and SentreHEART, our stockholders may experience dilution of their ownership interests in our company upon any such future issuances of shares of our common stock without receiving any commensurate benefit.The sale of material amounts of common stock could encourage short sales by thirdinterested parties, and depress the price of our common stock. As a result, our stockholders may lose all or part of their investment.The downward pressure on our stock price caused by the sale of a significant number of shares of our common stock or the perception that such sales could occur by any of our significant stockholders could cause our stock price to decline, thus allowing short sellers of our stock an opportunity to take advantage of any decrease in the value of our stock. The presence of short sellers in our common stock may further depress the price of our common stock. Some of our directors and executive officers have entered into, or may enter into, Rule 10b5-1 trading plans pursuant to which they may sell shares of our stock from time to time in the future. Actual or potential sales by these insiders, including those under a pre-arranged Rule 10b5-1 trading plan, could be interpreted by the market as an indication that the insider has lost confidence in our stock and adversely impact the market price of our stock.Sales of common stock by us in a capital raising transaction may dilute stockholder ownership of common stock and cause a decline in the market price of our common stock.We may need to raise capital in the future to fund our operations or new initiatives or reduce or pay in full our indebtedness. If we raise funds by issuing equity securities, our stock price may decline and our existing stockholders may experience significant dilution. Furthermore, we may enter into capital raising transactions at prices that represent a substantial discount to market price. A negative reaction by investors and securities analysts to any sale of our equity securities could result in a decline in the trading price of our common stock.Anti-takeover provisions in our amended and restated certificate of incorporation and amended and restated bylaws and under Delaware law could inhibit a change in control or a change in management that stockholders consider favorable.Provisions in our certificate of incorporation and bylaws could delay or prevent a change of control or change in management that would provide a premium to the market price of common stock. These provisions include those:authorizing the issuance without further approval of “blank check” preferred stock that could be issued by our board of directors to increase the number of outstanding shares and thwart a takeover attempt;prohibiting cumulative voting in the election of directors, which would otherwise allow less than a majority of stockholders to elect director candidates;limiting the ability of stockholders to call special meetings of stockholders;prohibiting stockholder action by written consent, thereby requiring all stockholder actions to be taken at a meeting of stockholders; andestablishing advance notice requirements for nominations for election to the board of directors or for proposing matters that can be acted upon by stockholders at stockholder meetings.In addition, Section 203 of the Delaware General Corporation Law limits business combination transactions with 15% stockholders that have not been approved by our board of directors. These provisions and others could make it difficult for a third party to acquire us, or for members of our board of directors to be replaced, even if doing so would be beneficial to our stockholders. Because our board of directors is responsible for appointing the members of our management team, these provisions could, in turn, affect any attempt to replace the current management team. If a change of control or change in management is delayed or prevented, stockholders may lose an opportunity to realize a premium on shares of common stock or the market price of our common stock could decline.We do not expect to pay dividends in the foreseeable future. As a result, stockholders must rely on stock appreciation for any return on investment.We do not anticipate paying cash dividends on our common stock in the foreseeable future. Any paymentTable of cash dividends will also depend on our financial condition, results of operations, capital requirements and other factors and will be at the discretion of our board of directors. Accordingly, stockholders will have to rely on capital appreciation, if any, to earn a return on investment in our common stock. Furthermore, pursuant to our credit facility, we are currently subject to restrictions on our ability to pay dividends and we may in the future become subject to other contractual restrictions on, or prohibitions against, the payment of dividends.Contentsbusiness.markets. If sufficient securities analysts do not cover our common stock, the lack of research coverage may adversely affect the market price of our common stock. It may be difficult for companies such as ours, with a smaller market capitalizations,capitalization, to attract and maintain sufficient independent financial analysts that will cover our common stock. This could have a negative effect on the market price of our stock.faillose all or part of their investment.meetdecline, thus allowing short sellers of our publicly announced guidancestock an opportunity to take advantage of any decrease in the value of our stock. The presence of short sellers in our common stock may further depress the price of our common stock. Some of our directors and executive officers have entered into, or other expectations aboutmay enter into, Rule 10b5-1 trading plans pursuant to which they may sell shares of our businessstock from time to time in the future. Actual or potential sales by these insiders, including those under a prearranged Rule 10b5-1 trading plan, could be interpreted by the market as an indication that the insider has lost confidence in our stock and future operating results, which couldadversely impact the market price of our stock.financial guidance abouta premium to the market price of common stock. These provisions include those:future operating results. In developing this guidance,others could make it difficult for a third party to acquire us, or for members of our board of directors to be replaced, even if doing so would be beneficial to our stockholders. Because our board of directors is responsible for appointing the members of our management makes certain assumptions and judgments about our future operating performance, including projected hiringteam, these provisions could, in turn, affect any attempt to replace the current management team. If a change of sales professionals, continued increasecontrol or change in management is delayed or prevented, stockholders may lose an opportunity to realize a premium on shares of our market share, and continued stability of the macro-economic environment in our key markets. Furthermore, analysts and investors may develop and publish their own projections of our business, which may form a consensus about our future performance. Our business results may vary significantly from such guidancecommon stock or that consensus due to a number of factors, many of which are outside of our control, and which could adversely affect our operations and operating results. Furthermore, if we make downward revisions of our previously announced guidance, or if our publicly announced guidance of future operating results fails to meet expectations of securities analysts, investors, or other interested parties, the market price of our common stock could decline.The requirements of being a public company may strain our resources and distract management.public company, we are subject to the reporting requirements of the Securities Exchange Act of 1934, as amended (Exchange Act), and the Sarbanes-Oxley Act of 2002 (Sarbanes-Oxley Act). result, stockholders must rely on stock appreciation for any return on investment.are also subject to certain provisions of the Dodd-Frank Wall Street Reform and Consumer Protection Act of 2010 (Dodd-Frank Act). The Exchange Act requires that we file annual, quarterly and current reports with respect to our business and financial condition. The Dodd-Frank Act requires the SEC to adopt certain rules and regulations relating to our public disclosures, corporate governance and executive compensation, among other things, and such rules and regulations require significant attention from management. Compliance with all of these laws, rules and regulations may from time to time divert management’s attention from other business concerns, which could have a material adverse effectdo not anticipate paying cash dividends on our business,common stock in the foreseeable future. Any payment of cash dividends will also depend on our financial condition, results of operations, capital requirements and cash flows.The Sarbanes-Oxley Act requires that we maintain effective disclosure controlsother factors and procedures and internal controls over financial reporting and management is required to evaluatewill be at the effectivenessdiscretion of our internal control over financial reporting asboard of the end of each fiscal year. In orderdirectors. Accordingly, stockholders will have to maintain the effectiveness ofrely on capital appreciation, if any, to earn a return on investment in our disclosure controls and procedures and internal control over financial reporting, significant resources and management oversight is required. Ifcommon stock. Furthermore, pursuant to our credit facility, we are not successful in maintaining effective internal control over financial reporting, there could be inaccuracies or omissionscurrently subject to restrictions on our ability to pay dividends and we may in the consolidated financial information we are requiredfuture become subject to file withother contractual restrictions on, or prohibitions against, the Securities and Exchange Commission. Additionally, even if there are no inaccuracies or omissions, we will be required to publicly disclose the conclusionpayment of our management that our internal control over financial reporting or disclosure controlsand procedures are not effective. These events could cause investors to lose confidence in our reported financial information, adversely impact our stock price, result in increased costs to remediate any deficiencies, or attract regulatory scrutiny or lawsuits that could be costly to resolve and distract management’s attention.The SEC has adopted rules regarding the disclosure of the use of conflict minerals (commonly referred to as tantalum, tin, tungsten and gold) which are mined from the Democratic Republic of the Congo (DRC) and neighboring countries. Under the rules, we are required to disclose the procedures we employ to determine the sourcing of such minerals and metals produced from those minerals. The requirements require due diligence efforts and could affect the sourcing of components used in our products. If the conflict minerals included in our products are found to be sourced from the DRC or surrounding countries, we may take actions to change materials or product designs to reduce the possibility that our purchase of conflict minerals may fund armed groups in the region. These actions could add engineering and other costs to the manufacture of our products. We expect to continue to incur costs in the investigation of the origin of the conflict minerals used in our products and in the reporting of the findings of our investigation. Our reputation may suffer if we have included conflict minerals in our products that are found to have funded armed groups in the DRC region.ITEMITEM 1B. UNRESOLVED STAFF COMMENTSITEMmaintains its headquartersoperates in the following principal locations:in a leasedincluding our global headquarters facility totaling approximately 92,000 square feet. The facilitythat contains the Company’sCompany's administrative, clinical, regulatory, engineering, product development, distribution and manufacturing functions. The Company also leases the following principal locations:headquarters facility is approximately 92,000 square feet. The Mason Ohio – This locationSouth facility is primarily used for warehousing and distribution activities. The facilityactivities and is approximately 40,000 square feet. The Mason Manufacturing Building, opened during 2022, is approximately 37,000 square feet and when qualified will be used for manufacturing and engineering activities.•Minneapolis,Minnetonka, Minnesota – This location includes both administrative, clinical, regulatory and product development space. The office is approximately 27,50032,000 square feet.•Redwood City,Pleasanton, California – This location is primarily used for product development research and development and manufacturing activities for the LARIAT System and is approximately 19,5006,000 square feet.•Amsterdam, Netherlands – This location is primarilyhouses administrative functions for the administration of our European subsidiaries andinternational operations. The space is approximately 9,000 square feet.ITEMThe Company is not party to any material pending or threatened litigation. 1210 – Commitments and Contingencies to our Consolidated Financial Statements.ITEMPARTPART IIITEM20, 2020,17, 2023, the closing price of our common stock on the NASDAQ Global Market was $40.70$40.34 per share, and the number of stockholders of record was 91.January 1, 2015December 31, 2017 and ending on December 31, 2019.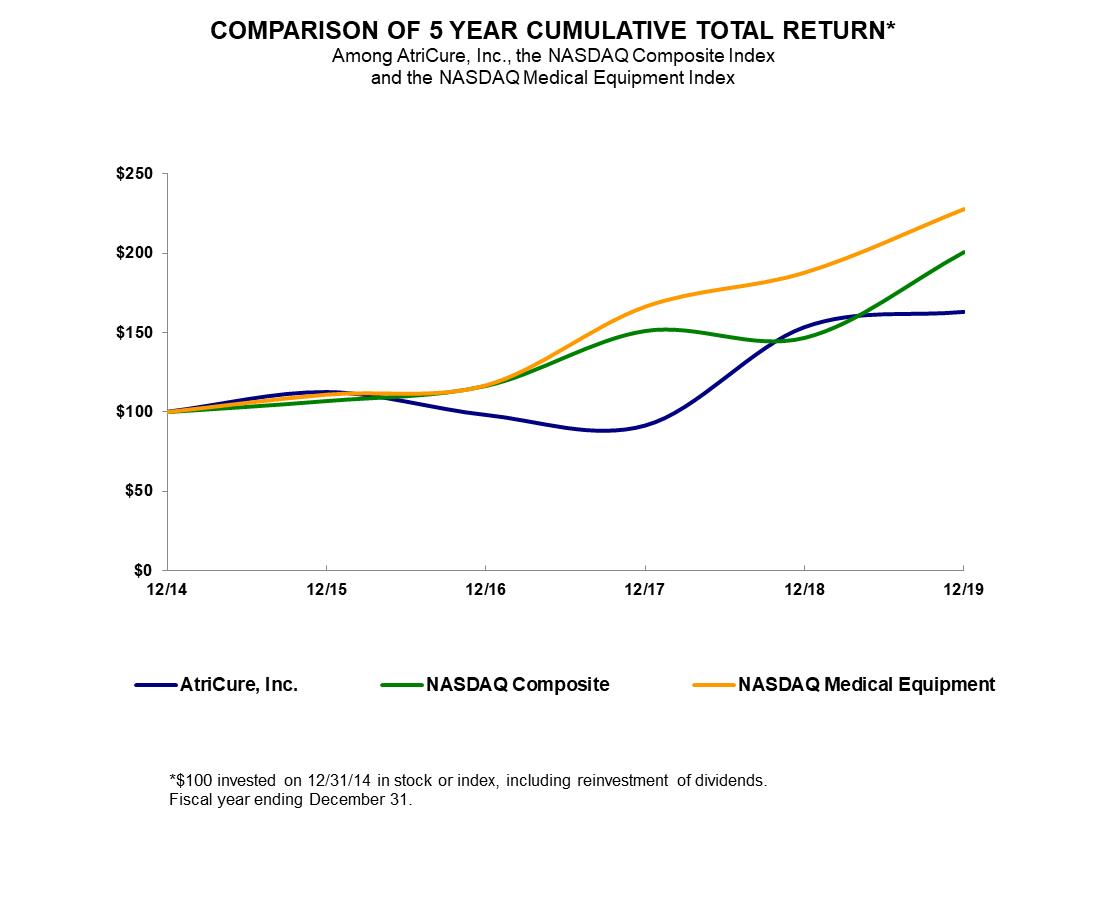
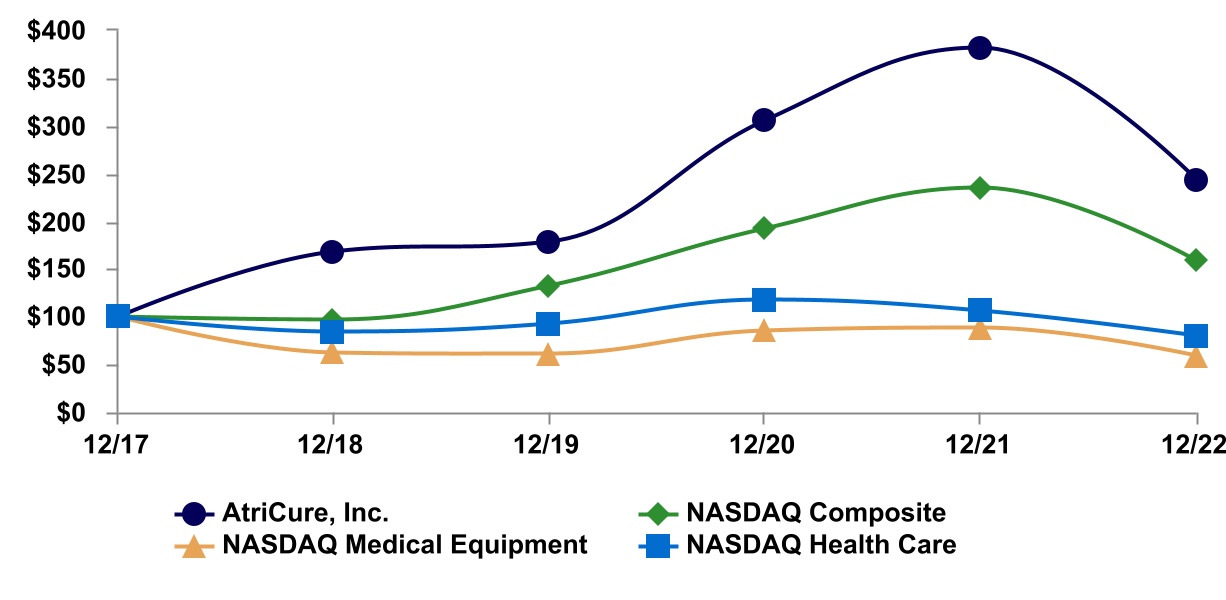
20142017 in our common stock, the NASDAQ Composite Index, and the NASDAQ Medical Equipment Index, and the NASDAQ Health Care Index, and that all dividends are reinvested. No dividends have been declared or paid on our common stock. Stock performance shown in the above chart for our common stock is historical and should not be considered indicative of future price performance. 12/31/2018 12/31/2019 12/31/2020 12/31/2021 12/31/2022 AtriCure, Inc. $ 167.76 $ 178.23 $ 305.21 $ 381.20 $ 243.31 NASDAQ Composite $ 97.16 $ 132.81 $ 192.47 $ 235.15 $ 158.65 NASDAQ Health Care $ 83.86 $ 92.88 $ 118.12 $ 106.27 $ 79.91 NASDAQ Medical Equipment $ 62.72 $ 61.17 $ 85.34 $ 88.20 $ 59.54 ITEMITEM 6. SELECTED FINANCIAL DATAThe following table reflects selected financial data derived from our Consolidated Financial Statements for each of the last five years. The operating results data for the years ended December 31, 2019, 2018 and 2017 and the financial position data as of December 31, 2019 and 2018 are derived from our audited financial statements included in this Form 10-K. The operating results data for the years ended December 31, 2016 and 2015 and the financial position data as of December 31, 2017, 2016 and 2015 are derived from our audited financial statements not included in this Form 10-K. Historical results are not necessarily indicative of future results. The selected financial data set forth below should be read in conjunction with our financial statements, the related notes and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included elsewhere in this Form 10-K._________________________(1)We acquired SentreHEART for $208,847 on August 13, 2019. The acquisition is included in our Consolidated Balance Sheets beginning August 13, 2019, and the results of operations are included in our Consolidated Statements of Operations and Comprehensive Loss beginning with the period August 14, 2019 through December 31, 2019.We adopted FASB ASC 842, “Leases” using the transition method provided by Accounting Standard Update (ASU ) 2018-11, “Leases (Topic 842): Targeted Improvements” on January 1, 2019. Under this method, we applied the new requirements to leases that existed as of January 1, 2019. As a result of the adoption, the Company recorded operating right-of-use assets and operating lease liabilities of approximately $1,884 and $2,189 as of January 1, 2019.(2)We adopted FASB ASC 606, “Revenue from Contracts with Customers” using the modified retrospective method effective January 1, 2018. The adoption of ASC 606 did not have a material impact on the amount and timing of revenue recognized in the Consolidated Financial Statements.(3)We acquired nContact for $116,842 on October 13, 2015. The acquisition is included in our Consolidated Balance Sheets beginning October 13, 2015, and the results of operations are included in our Consolidated Statements of Operations and Comprehensive Loss beginning with the period October 14, 2015 through December 31, 2015.
ITEMITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONSconsolidated financial statementsConsolidated Financial Statements and notes thereto contained in Item 8, “Financial Statements and Supplementary Data,” to provide an understanding of our results of operations, financial condition and cash flows. This section of this Form 10-K generally discusses 2022 and 2021 items and year-to-year comparisons between 2022 and 2021. This discussion and analysis contains forward-looking statements that involve risks, uncertainties and assumptions. The actual results may differ from those anticipated in these forward-looking statements as a result of many factors, including but not limited to those set forth under Item 1A “Risk Factors,” the cautionary statement regarding forward-looking statements at the beginning of Part I and elsewhere in this Form 10-K.(LAA) management. Afib affects approximately 1% of the population in the United States. It is the most common cardiac arrhythmia, or irregular heartbeat, encountered in clinical practicemanagement (LAAM) products are used by physicians during both open-heart and accountsminimally invasive procedures. In open-heart procedures, physicians are typically performing heart surgery for more doctor visitsother conditions, and hospital days than any other cardiac arrhythmia. When a patient is in Afib, abnormal electrical impulses cause the atria, or upper chambers of the heart, to fibrillate, or beat rapidly, irregularly, and in an uncoordinated fashion. As a result, blood in the atria may be in stasis, increasing the risk that a blood clot will form and cause a stroke or other serious complications. In patients with Afib, a significant percentage of those clots can form inside of the LAA. Symptoms of Afib may include heart palpitations, dizziness, fatigue and shortness of breath, and these symptoms may be debilitating and life threatening in some cases. Patients often progress from being in Afib intermittently (paroxysmal) to being in Afib continuously (persistent and long standing persistent). Afib often occursour products are used in conjunction with other cardiovascular diseases, including hypertension, congestive heart failure, left ventricular dysfunction, coronary artery disease(or “concomitant” to) such a procedure. Minimally invasive procedures are performed on a standalone basis, and valvular disease.We believe that we are currently the market leader in theoften include multi-disciplinary or “hybrid” approaches, combining surgical treatment of Afib.procedures using AtriCure ablation and AtriCure LAAM products with catheter ablation procedures performed by an electrophysiologist. Our Isolator Synergy Systempain management device is approvedused by FDA for the treatment of persistent and long-standing persistent Afib concomitantphysicians to other open-heartfreeze nerves during cardiothoracic or thoracic surgical procedures. All of our other ablation devices are cleared for sale in the United States under FDA 510(k) clearances, including our other RF and cryoablation products, which are indicated for the ablation of cardiac tissue and/or the treatment of cardiac arrhythmias. In addition, certain of our cryoablation probes are cleared for managing pain by temporarily ablating peripheral nerves. Our AtriClip products are 510(k)-cleared with an indication for the exclusion of the LAA, performed under direct visualization and in conjunction with other cardiac surgical procedures. Direct visualization, in this context, requires that the surgeon is able to see the heart directly, with or without assistance from a camera, endoscope or other appropriate viewing technologies. The LARIAT® system is cleared for soft tissue ligation and is currently being studied to support an indication of exclusion of the LAA in patients with persistent and long-standing persistent Afib also undergoing a pulmonary vein isolation. We also offer reusable surgical instruments typically used in cardiac valve replacement or repair. Our Isolator Synergy clamps, Isolator Synergy pens, Coolrail® linear pen, cryosurgery devices, certain products of the AtriClip LAA Exclusion System, COBRA Fusion® Ablation System, NumerisTM System, the EPi-Sense® Guided Coagulation System with VisiTrax® technology, and LARIAT Suture Delivery Device bear the CE mark and may be commercially distributed throughout the member states of the European Union and other countries that comply with or mirror the Medical Device Directive. Our Isolator Synergy clamps, Isolator Synergy pens, Coolrail linear pen, cryosurgery devices, and certain products of the AtriClip LAA Exclusion System are available in select Asia-Pacific countries. We anticipate that substantially all of our revenue for the foreseeable future will relate to products we currently sell or are in the process of developing.Dollars withDollars; direct sales transactions outside the exception of transactions with our European customers, whichUnited States are transacted in the EuroEuros, British Pounds or British Pound.Throughout 2019,Australian Dollars.sustainexperience variability in demand for our revenue growthproducts as non-emergent procedures were deferred in order to preserve resources for COVID-19 patients and caregivers, and hospital staffing was impacted by the pandemic and related factors. Beginning in the second quarter, many regions began to stabilize with overall improvements in procedure volume. We expect some variability to continue as we operate in many geographic regions with diverse restrictions that are impacted as new variants of the virus emerge and hospital staffing constraints continue to impact allocation of resources. Despite the challenging environment resulting from the pandemic, we reported annual revenues of $330,379 for the year ended December 31, 2022, an increase of 20.4% when compared to our prior year as a result of growing adoption across key product lines. We continue to build on our strategic initiatives of product innovation, investing in clinical science and providing superior training and education.August 2019,April 2022, we acquired SentreHEART with up-front paymentlaunched our EnCompass® clamp, following the July 2021 510(k) clearance for ablation of approximately $40,000, plus additional considerationcardiac tissue during cardiac surgery. The EnCompass clamp marks innovation in our core open ablation market, and is expected to drive deeper penetration of cardiac surgery procedures. During September 2022, we received final labeling approval for the next generation EPi-Sense ST device and began a limited launch evaluation in the fourth quarter.$260,000 contingent on the achievement of clinical and reimbursement milestones. The acquisition of SentreHEART significantly expands our addressable markets with a product designed for electrophysiologists, broadens our LAA management portfolio and augments our commitment142 patients at up to clinical science with the aMAZE trial. Enrollment in the aMAZE trial was completed in December 2019, and in early January 2020, we received approval for the Continued Access Protocol (CAP) for the aMAZE study. In addition to the SentreHEART acquisition in 2019, we continued to focus our efforts on the regulatory submissions for the CONVERGE trial and also received approval in September 2019 for a CAP for CONVERGE.For the year ended December 31, 2019 we reported annual revenues of $230,807, increasing 14.5% over the prior year, and surpassed 200,000 AtriClip devices sold to date. In February 2019, we launched the cryoICE cryoSPHERE probe40 sites in the United States, United Kingdom and continuedEuropean Union. The first patient enrollment in the trial occurred in June 2022; site initiation and enrollment is ongoing.build a dedicated teamevaluate the effectiveness of prophylactic LAA exclusion using the AtriClip LAA Exclusion System for the prevention of ischemic stroke or systemic arterial embolism in cardiac surgery patients without pre-operative AF diagnosis who are at risk for these events. This prospective, multicenter, randomized trial evaluates safety at 30 days post-procedure to demonstrate our commitmentno increased risk with LAA exclusion during cardiac surgery, and efficacy over a minimum follow-up of five years post procedure. The trial provides for enrollment of up to innovation6,500 subjects at up to 250 sites worldwide. In January 2023, we announced first patient enrollment in Cryo Nerve Block Therapy.the trial; site initiation and enrollment is ongoing.net lossprofessional education and marketing teams conduct virtual, in-person and mobile training for fiscal year 2019 was $35,194 as compared to $21,137 for fiscal year 2018, primarily as a result of increased operating expenses relatedto personnel costs due to increased headcountphysicians and variable compensation, incremental investments in clinical trials, product development and physician training,healthcare professionals, as well as SentreHEART acquisition expenses, offset partially by improved gross margin. See the “Results of Operations” section below for additional analysisour sales teams. Our training methods ensure invaluable access to continuing education and awareness of our 2019 results.products and related procedures. The 2021 FDA approval of the EPi-Sense System has enabled us to educate and train physicians on the benefits of Hybrid AF therapy in treating long-standing persistent Afib patients. Our Advanced Hybrid Ablation Training Courses are co-sponsored by the Heart Rhythm Society (HRS).20192022 compared to December 31, 2018Year Ended December 31, 2022 2021 % of % of Amount Revenue Amount Revenue Revenue $ 330,379 100.0 % 274,329 100.0 % Cost of revenue 84,439 25.6 68,469 25.0 Gross profit 245,940 74.4 205,860 75.0 Operating expense (benefit): Research and development expenses 57,337 17.4 48,506 17.7 Selling, general and administrative expenses 231,272 70.0 204,649 74.6 Change in fair value of contingent consideration — — (184,800) (67.4) Intangible asset impairment — — 82,300 30.0 Total operating expenses 288,609 87.4 150,655 54.9 (Loss) income from operations (42,669) (12.9) 55,205 20.1 Other expense, net (3,529) (1.1) (4,818) (1.8) (Loss) income before income tax expense (46,198) (14.0) 50,387 18.4 Income tax expense 268 0.1 188 0.1 Net (loss) income $ (46,466) (14.1) % $ 50,199 18.3 % TotalThe following table sets forth, for the periods indicated, our revenue by product type and geography expressed as dollar amounts and the corresponding change in such revenues between periods, in both dollars and percentages:Year Ended December 31, Change 2022 2021 Amount % Open ablation $ 86,119 $ 72,396 $ 13,723 19.0 % Minimally invasive ablation 38,553 39,380 (827) (2.1) % Pain management 39,974 22,787 17,187 75.4 % Appendage management 112,555 94,568 17,987 19.0 % Total United States $ 277,201 $ 229,131 $ 48,070 21.0 % Total International 53,178 45,198 7,980 17.7 % Total Revenue $ 330,379 $ 274,329 $ 56,050 20.4 % 14.5% (15.2%20.4% (21.8% on a constant currency basis). Revenue from customers inThroughout the United States market, cardiac surgery volumes recovered and product adoption continued. Our Isolator Synergy System continued to generate the majority of our ablation-related revenue. Key drivers of growth included the AtriClip® Flex-V® device within the appendage management franchise, the cryoSPHERE® probe for pain management, and the 2022 launch of the EnCompass clamp in open ablation. Minimally invasive ablation sales decreased as declines in legacy product sales outpaced growth in Hybrid AF therapy procedures using the EPi-Sense system. International revenue increased $23,683, or 14.6%, and revenue from international customers increased $5,494, or 13.9% (17.6%17.7% (25.7% on a constant currency basis). Sales in throughout our major European and Asia markets. Similar to the UnitedUnites States, grew across several key product categories. OpenInternational revenue growth was driven by appendage management, open ablation and pain management products, while minimally invasive ablation sales increased $7,955, or 11.0% primarilydeclined due to reduction in revenues from legacy products exceeding the positive impact ofgrowth in Hybrid AF therapy procedures using the CryoSPHERE device launch and continued volume increases for cardiac ablation devices. Minimally invasive (MIS) ablation sales decreased $211, or 0.6%, reflecting a decline in Fusion product sales, partially offset by increases in legacy RF ablation devices. Appendage management sales increased $15,275, or 28.9%, due to continued growth of the AtriClip Flex·VEPi-Sense system.® LAA Exclusion System, volume increases of the minimally invasive LAA Exclusion system and LARIAT System sales. International growth results from increased volume in AtriClip and open ablation product sales, as well as increasing Epi-sense device sales which partially offset a decline in all other minimally invasive RF ablation products. International revenue grew primarily in China, the United Kingdom, Germany, Australia and Japan.$5,962 and$15,970 primarily reflecting revenue growth. The gross margin increased 0.8%decrease of approximately 60 basis points was driven by inflationary and supply chain pressures and a shift in product mix to 73.8% in 2019. Improvements in grosslower margin are reflective of operational improvements and lower production costs at our headquarters. Additionally, there was a $628 decrease in share-based compensation in 2019 primarily due toproducts, partially offsetting the acceleration of vesting of restricted stock awards in 2018.benefit from higher volume.$6,507,$8,831, or 18.7%18.2%. The increaseWe expanded our product development, regulatory and clinical teams throughout 2022, resulting in research and development expense is comprised of $2,015 ofadditional $4,551 personnel costs resulting fromincluding variable compensation, travel and share-based compensation. Product development project spend increased headcount$1,053 as we continue to evolve our product pipeline. Clinical activities, regulatory submissions and $1,059 in clinical trialconsulting expenses, driven by the aMAZE clinical trial enrollment. Other expense drivers include $1,353 higher consulting, product development and regulatory expenses, $687 higher grant and research expenses, $433 increase inincluding compliance with EU MDR, drove $1,944 incremental costs, while amortization expense $387 higher share-based compensation, and $573 increase in various operating costs.increased $820 following the April 2021 PMA of the CONVERGE IDE clinical trial. See Note 4 of the Consolidated Financial Statements for further discussion.$32,703,$26,623, or 25.2%, primarily due to higher personnel expense of $14,902 resulting from increased13.0%. Higher headcount and variable compensation, $3,978rising travel expenses contributed $18,575 increase in personnel costs. Our commitment to physician training and return to in-person meetings, trade shows and marketing activities drove a $5,007 increase in expenses as compared to the prior year. Other administrative and operating expenses increased $3,114, largely for legal activity and information technology costs.acquisition-relatedcontingent consideration. The credit to operating expenses during the year ended December 31, 2021 reflects a change in the forecasted timing and $1,723probability of share-based compensation. Additionally, there was a $5,909 lower reductionachievement of the regulatory and reimbursement milestones related to the contingent consideration liability as compared to prior year (seeaMAZE clinical trial. See Note 3 – Fair Value in2 of the Consolidated Financial Statements). Other expense drivers include a $2,805 rise in operating costs, including organization meetings, facility expenses, and dues and subscriptions, $1,749 increase in marketing, training and tradeshow activities, $758 incremental legal, consulting and professional fees, and $879 increase in software agreements and other information technology expenses.Statements for further discussion.Net interest expense. Table of ContentsNet interest expense was $1,713 During the year ended December 31, 2021, the Company recorded an impairment charge for 2019 and $3,601 for 2018. Interest expense is for outstanding amountsthe IPR&D asset associated with our term loan and finance lease obligations, as well as the amortizationaMAZE PMA. See Note 4 of financing costs. Interest income reflects returns on our investments, including gains and losses on investments sold during the period. The decrease in net interest expense was driven by $1,392 higher interest income as a result of a higher investment balance throughout 2019 as compared to 2018 and $496 decrease in interest expense reflecting a lower interest rate on the term loan in 2019.Consolidated Financial Statements for further discussion.Year Ended December 31, 2018 compared to December 31, 2017For a comparison of our results of operations Net interest expense was $2,992 for the fiscal years ended December 31, 20182022 and December 31, 2017, see “Part II, Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations” of our annual report$4,452 for 2021. The decrease in net interest expense was driven by higher interest income from funds received for interest on Form 10-K for the fiscal year ended December 31, 2018, filed with the SEC on March 1, 2019.past due trade receivables.2019, the Company2022, we had cash, cash equivalents and investments of $94,476$172,622 and outstanding debt of $60,000. We had unused borrowing capacity of $8,750 under our revolving credit facility. Mostapproximately $28,750. All cash equivalents and investments and most of our operating cash and all cash equivalents and investments are held byin United States financial institutions. A minor portion of our cash is held in foreign banks to support our international operations. We had net working capital of $93,244$156,822 and an accumulated deficit of $282,197$326,619 as of December 31, 2019.2022.Cash flows used Our executive officers and Board of Directors review our funding sources and future capital requirements in connection with our annual operating activities. Net cash used inplan and periodic updates to the plan. Our future capital requirements depend on a number of factors, including, without limitation: market acceptance of our current and future products; costs to develop and support our products, including clinical evidence needs; future expenses to expand and support our sales, training and marketing efforts; operating activities was $15,811 during 2019. The primary net uses of cash for operating activities were as follows:the net loss of $35,194, which contains $23,998 of non-cash expenses including$17,977 in share-based compensation,$9,366 of depreciation and amortization,$1,355 of noncash lease expense and loss on disposal and impairment of assets,offset by a decrease in fair value of contingent consideration of $4,916; anda net decrease in cash used relatedfiling costs relating to changes in operating assetsregulatory policies or laws; costs for clinical trials and liabilitiesto secure regulatory approval for new products; legal defense costs; costs to prosecute, defend and enforce our intellectual property rights; and possible acquisitions and joint ventures, including potential business integration costs. Our principal cash requirements include costs of $4,615, due primarily to the following:a $5,151 increase in all categories of inventories in anticipation of future growth;a $3,201 increase in accounts receivable due primarily to increased sales;a $2,790 increase in accounts payable from increased operating costs; anda $3,108 increase in accrued liabilities reflecting increased accrued variable compensationoperations, capital expenditures, debt service costs and employee costs.other contractual obligations.Cash flows used in investing activities. Net cash used in investing activities was $2,147 during 2019. The primary uses of cash were $17,240 cash paid in the acquisition of SentreHEART and $12,182 purchases of property and equipment, including the expansion of our corporate headquarters and placement of generators with our customers. These uses of cash were offset by $27,236 of net sales and maturities of available-for-sale securities.Cash flows provided by financing activities. Net cash provided by financing activities during 2019 was $14,373, which was primarily proceeds from debt borrowings of $20,000, issuance of common stock under our employee stock purchase plan of $2,662, and proceeds from stock option exercises of $1,202. These were partially offset by shares repurchased for payment of taxes on stock awards of $9,033, and finance lease payments of $629.The Company’sOur Loan and Security Agreement with Silicon Valley Bank (SVB), as amended, August 12, 2019 (Loan Agreement), provides for a $60,000 term loan, with an option to make available an additional $30,000 in term loan borrowings, and a $20,000$30,000 revolving line of credit. The term loan and revolving credit facility both mature or expire, as applicable, on August 1, 2024. Principal payments on the term loan are to be made ratably commencing March 1, 2021 through the loan’s maturity date. If the Company meets certain conditions, as specified in the Loan Agreement thecommencement of thehas a five year term loan principal payments may be deferred by an additional six months.and expires November 2026. The term loan accrues interest at the greater of the Prime Rate or 5.00%, plus 0.75%1.25% and is subject to an additional 3.00% fee on the $60,000 term loan principal amount payable at maturity or upon acceleration or prepaymentmaturity. Principal payments are to be made ratably commencing 24 months after the inception of the loan through the loan's maturity date. At the option of the Company, the commencement of term loan. Borrowing availability under the revolving credit facility is based on the lesser of $20,000 or a borrowing base calculation as defined by the Loan Agreement. Borrowing availability under the revolving credit facility is further limited by a cap on total debt outstanding under the Loan Agreement, including outstanding letters of credit, of $70,000.loan principal payments may be extended an additional twelve months. As of December 31, 2019, we2022, our outstanding debt was $60,000, of which $3,333 is classified as current and $56,667 is classified as noncurrent. We had no borrowingsunused borrowing capacity of approximately $28,750 under theour revolving credit facility, and we had borrowing availability of $8,750. The revolving line of credit is subject to an annual facility fee of 0.15% offacility. For additional information on the revolving line of credit, and any borrowings bear interest at the greater of the Prime Rate or 5.00%. The Loan Agreement also provides for certain prepayment and early termination fees only if the term loan is repaid before August 2024 and establishes a minimum liquidity ratio and dividend restrictions, along with other customary terms and conditions. Specified assets have been pledgedconditions, as collateral. We are in compliance with the covenants of the Loan Agreementwell as of December 31, 2019.applicable interest and fee payments, see Note 8 - Indebtedness.In connection with the terms of ourin the amount of $1,250 was issued to the landlord in October 2015. The letter of credit is renewedwhich renews annually and remains outstanding as of December 31, 2019.2022.UsesWe incur capital expenditures on an ongoing basis to continue investment in our growth and our ability to better serve our customers. Throughout 2021 and 2022, we expanded our manufacturing operations as we completed the renovation of liquidity and capital resources.an additional facility of our Mason, Ohio campus.capital requirements depend on a number of factors, including the rate of market acceptance of ourobligations include both current and future products;long-term obligations. In December 2022, the resourcesCompany entered into a clinical trial management agreement for the LeAAPS clinical trial. The terms of the agreement require we devote to developingmake milestone payments upon achievement of various enrollment and supporting our products; future expenses to expand and support our sales and marketing efforts;project milestones over the estimated ten year term, yet the agreement may be terminated early for any reason. Furthermore, we will incur additional variable costs, relating to changes in regulatory policies or laws that affect our operations andincluding pass through costs of filings; costs associated withfrom clinical trials and securing regulatory approval for new products; costs associated with acquiring and integrating businesses; costs associated with prosecuting, defending and enforcing our intellectual property rights; payments made for acquisition-related earnouts; costs to expand our facilities to support continued growth; and possible future acquisitions and joint ventures. Global economic turmoil may adversely impact our revenue, access to the capital markets or future demand for our products.We have on file with the SEC a shelf registration statement which allows us to sell any combination of senior or subordinated debt securities, common stock, preferred stock, warrants, depositary shares and units in one or more offerings should we choose to do so in the future.trial sites. We expect to disburse between $6,000 and $9,000 of fixed and variable costs based on estimated achievement of milestone payments, site initiation and trial enrollment within the next twelve months. We have operating and finance leases primarily for our corporate offices, manufacturing and warehouse facilities, as well as computer equipment. Our finance leases consist primarily of principal and interest payments related to our Mason, Ohio headquarters. As of December 31, 2022, we have current finance lease obligations of $992 and long-term obligations of $9,147. Our operating leases for office and warehouse space includes current obligations of $1,147 and long-term obligations of $3,095. For additional information, see Note 9 - Leases. We additionally maintain a license agreement with terms that require royalty payments of 5% of specified product sales. See Note 10 - Commitments and Contingencies for information about the effectivenessterms. We have contractual obligations for contingent consideration payments related to the SentreHEART acquisition. Subject to the terms and conditions of this shelf registration statement for the foreseeable future.SentreHEART merger agreement, such contingent consideration would be paid in AtriCure common stock and cash, up to a specified maximum number of shares. The SentreHEART milestones expire on December 31, 2023 and December 31, 2026. As of December 31, 2022, we believe the likelihood of payment is remote, and the estimated fair value of the contingent consideration is $0. See Note 2 – Fair Value.The nContact transaction provides for contingent considerationHowever, we have on file with the SEC a shelf registration statement which allows us to be paid upon attaining specified regulatory approvals before January 2021. The SentreHEART acquisition provides for contingent considerationsell any combination of debt securities, common stock, preferred stock, warrants, depository shares and units in one or more offerings should we choose to be paid upon PMA approval before December 2023 and CPT reimbursement before December 2026. Subjectdo so in the future. We expect to maintain the terms and conditionseffectiveness of the nContact and SentreHEART merger agreements, such contingent consideration will be paid in AtriCure common stock and cash, up to a specified maximum number of shares. Overshelf registration statement for the next twelve months, we do not expect our cash requirements to include significant payments of contingent consideration based on terms of the respective acquisition agreements and progress towards achievement of the related milestones. See the heading “Legal” in Note 12 for a description of an earnout objection statement received from the nContact shareholder representative.foreseeable future.couldwould have rights senior to those associated with our common stock and could contain covenants that would restrict our operations. Finally, our term loan agreement and revolving line of credit require compliance with certain financial and other covenants. If we are unable to maintain these financing arrangements, we may be required to reduce the scope of our planned research and development, clinical activities and selling, training, education and marketing efforts.Contractual Obligations and Commitmentssets forthsummarizes our approximate aggregate obligations at December 31, 2019consolidated cash flow activities:Years Ended December 31, 2022 2021 Change Net cash used in operating activities $ (22,141) $ (13,780) $ 8,361 Net cash provided by investing activities 44,006 23,504 20,502 Net cash used in financing activities (7,059) (7,642) (583) future payments under contractsworking capital and other contingent commitments:assets and liabilities of $14,034. Working capital fluctuations are primarily due to the $11,237 reduction in accrued liabilities from higher annual variable compensation payments in 2022 due to improved operating performance in 2021 versus 2020, as well as an increase of $3,031 from our investment in inventories._________________________(1)Cash flows provided by investing activities. Long-term debt represents principal repayments relatedNet cash provided by investing activities increased by $20,502 in 2022 compared to our term loan. Principal payments under2021, reflecting higher net sales and maturities of available-for-sale securities of $27,630, offset by an increase of $7,128 for the term loan commence on March 1, 2021purchase of property and are made ratably until maturity in August 1, 2024. Interest on the term loan accrues at the greater of the Prime Rate or 5.00%, plus 0.75% and is payable monthly over the term of the loan. In addition, the term loan is subject to an additional 3.00% fee on the term loan principal, or $1,800, that is payable at maturity or upon acceleration or prepayment of the term loan. Finally, we have a contractual obligation to pay interest on amounts drawn on the revolving credit facility.(2)Finance leases consist of principal and interest payments related to our Mason, Ohio headquarters and computer equipment. See Note 11 – Leases.(3)Represents lease commitments under various operating leases,equipment primarily for officethe expansion of our manufacturing facilities.warehouse space. See Note 11 – Leases.(4)Represents obligationsfewer shares repurchased for royalty agreements ranging from 3% to 5%payment of specified product sales estimated using 2019 sales. Royalty obligations beyond one yeartaxes for stock awards offset with slight increases in employee stock purchase plan activity.not been included as payments are based on specified product sales and not estimable at this time. See Note 12 – Commitments and Contingencies to our Consolidated Financial Statements.We have contractual obligations for contingent consideration payments related to the nContact and SentreHEART acquisitions. Subject to the terms and conditions of the nContact and SentreHEART merger agreements, such contingent consideration will be paid in AtriCure common stock and cash, up to a specified maximum number of shares. The nContact contingent consideration expires on December 31, 2020. The SentreHEART milestones expire on December 31, 2023 and December 31, 2026. See Note 3 – Fair Value.Off-Balance-Sheet ArrangementsWe are not a party to any off-balance sheet arrangements that have, or are reasonably likely to have, a current or future material effect on our financial condition, changes in financial condition, sales or expenses, results of operations, liquidity, capital expenditures or capital resources.InflationInflation has not had a significant impact on our historical operations, and we do not expect it to have a significantan adverse impact on our results of operations or financial condition in the foreseeable future.consolidated financial statements,Consolidated Financial Statements, which have been prepared in accordance with accounting principles generally accepted in the United States. The preparation of consolidated financial statements requires management to make estimates and judgments that affect the reported amounts of assets and liabilities, revenue and expenses, and disclosures of contingent assets and liabilities at the date of the financial statements. On a periodic basis, we evaluate our estimates, using authoritative pronouncements, historical experience and other assumptions as the basis for making estimates. Actual results could differ from those estimates under different assumptions or conditions. We have described our significant accounting policies in Note 1 – Description of Business and Summary of Significant Accounting Policies to our consolidated financial statementsConsolidated Financial Statements included in this Form 10-K.affectinvolve a significant level of estimation uncertainty and judgments that are reasonably likely to have a material impact on our more significant Consolidated Financial Statements. We base ourused in the preparation of our consolidated financial statements. primarily from the sale of medical devices. The Company recognizesWe recognize revenue in an amount that reflects the consideration the Company expectswe expect to be entitled to in exchange for those devices when control of promised devices is transferred to customers. At contract inception, the Company assesses the products promised in its contracts with customers and identifies a performance obligation for each promise to transfer to the customer a product that is distinct. TheCompany’s devices are distinct and represent performance obligations. These performance obligations are satisfied and revenue is recognized at a point in time upon shipment or delivery of products. Sales of devices are categorized as follows: open ablation, minimally invasive ablation, appendage management and valve tools. Shipping and handling activities performed after control over products transfers to customers are considered activities to fulfill the promise to transfer the products rather than as separate promises to customers. Products are sold primarily through a direct sales force and through distributors in certain international markets. Terms of sale are generally consistent for both end-users and distributors, except that payment terms are generally net 30 days for end-users and net 60 days for distributors, with limited exceptions. The Company does not maintain any post-shipping obligations to customers. No installation, calibration or testing of products is performed by the Company subsequent to shipment in order to render products operational.determination of the timing of transfer of control of products to customers and the estimation of a provision for returns. The Company considersWe estimate the following indicators when determining when control of the product transfers to customers: (i) the Company has a right to payment in accordance with the shipping terms set forth in its contracts with customers; (ii) customers have legal title to products in accordance with shipping terms; (iii) the Company transfers physical possession of products either when the Company presents the products to a third party carrier for delivery to a customer (FOB shipping point) or when a customer receives the delivered goods (FOB destination); (iv) customers have the significant risks and rewards of ownership of products; and (v) customers have accepted products in connection with contractual shipping terms.We maintain a provision for sales returns and allowances to account for expected returns of defective or damaged products, products shipped in error and invoice adjustments. We adjust the provision using the expected value method based on historical experience.Allowance for Doubtful Accounts Receivable—We evaluate the collectability of accounts receivable to determine the appropriate reserve for doubtful accounts. In determining the amount of the reserve, we consider the aging of account balances, customer-specific informationexperience and other relevant factors. We review accounts receivablefactors that we believe could impact our expected returns, including defective or damaged products and adjustinvoice adjustments. In the allowance based on current circumstancesnormal course of business, we generally do not accept product returns unless a product is defective as manufactured, and charge off uncollectible receivables againstwe do not provide customers with the allowance when all attemptsright to collect the receivable have failed. If circumstances change, our estimates of the collectability of amounts could be changed by a material amount. Our history of write-offs against the allowance has not been significant.refund.Inventories—Inventories—Our inventories are stated at the lower of cost or net realizable value based on the first-in, first-out cost method (FIFO) and consist of raw materials, work in process and finished goods. Our industry is characterized by rapid product development and frequent new product introductions. We estimate reserves for excess, expired and obsolete inventory on a quarterly basis which are impacted by expiration of products, uncertainUncertain timing of product approvals, variability in product launch strategies and variation in product use.Propertyuse all impact inventory reserves for excess, obsolete and Equipment—We state property and equipment at cost less accumulated depreciation. Depreciation is computed using the straight-line method for financial reporting purposes and applied over the estimated useful lives of the assets. Includedexpired products. An increase to inventory reserves results in property and equipment are generators and other capital equipment (such as our RF and cryo generators) that are placed with direct customers that use our disposable products. These generators and other capital equipment are depreciated over a period of one to three years, which approximates their useful lives, and such depreciation is includedcorresponding increase in cost of revenue. We estimateInventories are written off against the useful lives of this equipment based on anticipated usage by our customers and the timing and impact of our expected new technology rollouts. To the extent we experience changes in the usage of this equipment or the introductions of new technologies, the estimated useful lives of this equipment may change in a future period.IPR&D Intangible Asset—In Process Research and Development (IPR&D) represents the value of acquired technology which has not yet reached technological feasibility. The primary basis for determining the technological feasibility is obtaining specific regulatory approvals. IPR&D is accounted for as an indefinite-lived intangible asset until completion or abandonment of the IPR&D project. Upon completion of the development project, the IPR&D will be amortized over its estimated useful life. The IPR&D asset represents an estimate of the fair value of the PMA that may result from the CONVERGE IDE and aMAZE IDE clinical trials. We review intangible assets for impairment annually on October 1, or more often if impairment indicatorsreserve when they are present, using our best estimates based on reasonable and supportable assumptions and projections of expected future cash flows. If the IPR&D project is abandoned or regulatory approvals are not obtained, we may have a full or partial impairment charge related to the IPR&D, calculated as the excess carrying value of the IPR&D assets over the estimated fair value.physically disposed.Goodwill— Goodwill represents the excess of purchase price over the fair value of the net assets acquired in business combinations. Our goodwill is accounted for in a single reporting unit representing the Company as a whole. We test goodwill for impairment annually on October 1, or more often if impairment indicators are present. The impairment test requires a comparison of the estimated fair value of the reporting unit to the carrying value of the assets and liabilities of that reporting unit. If the carrying value of the reporting unit exceeds the fair value of the reporting unit, the carrying value of the reporting unit’s goodwill is reduced toits fair value through an impairment charge to adjust the goodwill balance. The estimates of fair value and the determination of reporting units requires management judgment.We account for share-based compensation for all share-based payment awards, including stock options, restricted stock awards, restricted stock units, performance share awards, and stock purchases related to an employee stock purchase plan, based on their estimated fair values. We estimate the fair value of time-based options on the date of grant using the Black-Scholes option pricing model (Black-Scholes model). Our determination of fair value of share-based payment awards is affected by our stock price, as well as assumptions regarding a number of subjective variables. These variables include but are not limited to our expected stock price volatility over the term of the awards and actual and projected employee stock option exercise behaviors. The value of the portion of the awards that is ultimately expected to vest is recognized as expense over the requisite service periods in our Consolidated Statements of Operations and Comprehensive Loss.restricted stock awards, restricted stock units and performance share awards with a performance condition initially based uponon the closing stock price on the date of grant date closing market price of our common stock. The estimated fair value ofassuming the performance goal will be achieved. Such performance share awards have specified performance targets based on the compound annual growth rate (CAGR) of our revenue over a three-year performance period. With respect to these performance share awards, the number of shares that vest and are issued to the recipient is based upon revenue performance over the performance period. We may be adjustedadjust the expense over the performance period based on changes to estimates of performance target achievement.We also have an employee stock purchase plan (ESPP) which If such goals are not met or service is available to all eligible employees as defined bynot rendered for the plan document. Under the ESPP, shares of our common stock may be purchased at a discount. We estimate the number of shares to be purchased under the ESPP at the beginning of the purchaserequisite service period, and calculate estimatedno compensation expense using the Black-Scholes model based upon the fair value of the stock at the beginning of the purchase period. Compensation expensecost is recognized, over each purchase period, and expense is adjusted at the time of stock purchase.any recognized compensation cost from prior periods will be reversed.Acquisition-Related Contingent Consideration—Contingent consideration arrangements obligate the Company to pay former shareholders of acquired companies certain amounts if specified future events occur or conditions are met, such as the achievement of certain regulatory milestones or reimbursement milestones. We measure such liabilities using unobservable inputs by applying the probability-weighted scenario method. Various key assumptions, such as the probability and timing of achievement of the agreed milestones and the discount rate, are used in the determination of fair value of contingent consideration arrangements and are not observable in the market. Subsequent revisions to key assumptions, which impact the estimated fair value of contingent consideration liabilities, are reflected in selling, general and administrative expenses.We measure deferredDeferred income tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred income tax assets and liabilities from changes in tax rates is recognized in the period that includes the enactment date. us to make significant estimates and judgments about our future operating results. Deferred income tax assets are reduced by valuation allowances if, based on the consideration of all available evidence, it is more-likely-than-not that some portion of thea deferred tax asset will not be realized. Significant weight is given to evidence that can be objectively verified. We evaluate deferred income tax assets on an annual basis to determine if valuation allowances are required by considering all available evidence. Deferred income tax assets are realized by having sufficient future taxable income to allow the related tax benefits to reduce taxes otherwise payable. The sources of taxable income that may be available to realize the benefit of deferred tax assets are future taxable income, future reversals of existing taxable temporary differences, future taxable income exclusive of reversing temporary differences and carryforwardsin prior carryforward years and tax planning strategies that are both prudent and feasible. In evaluating whether to recordthe need for a valuation allowance, the applicable accounting standards deem that the existence of cumulative losses in recent years is a significant piece of objectively verifiable negative evidence that must be overcome by objectively verifiableobjectively-verifiable positive evidence to avoid the need to recordfor a valuation allowance.We believe our critical accounting policies regarding revenue recognition, Our valuation allowance for doubtful accounts receivable, inventories, property and equipment, IPR&D intangible asset, goodwill, share-based employee compensation, acquisition-related contingent consideration andoffsets substantially all net deferred income taxes affect our more significant judgments and estimates usedtax assets as it is more-likely-than-not that the benefit of such deferred income tax assets will not be recognized in the preparation of our consolidated financial statements. We base our judgments and estimates on historical experience, current conditions and other reasonable factors.21 – RecentDescription of Business and Summary of Significant Accounting PronouncementsPolicies to ourthe Consolidated Financial Statements in Item 8 of Part II for further information.
ITEMITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISKonRate Riskfacility accruefacility. The term loan accrues interest at a variable rate based on the Prime Rate plus 1.25% and any borrowings under the revolving credit facility bear interest at the Prime Rate.11.7%9.0% and 12.5%9.9% of the Company’s total revenue for the years ended December 31, 20192022 and 2018. Since such revenue was primarily denominated in Euros or British Pounds,2021. Accordingly, the Company is exposed to exchange rate fluctuations between the Euro and the U.S. Dollar and between the British Pound and the Euro. For the years ended December 31, 20192022 and 2018,2021, foreign currency transaction gains (losses) of $180$559 and $(183)$387 were recorded primarily in connection with settlements of the intercompany balances and invoices transacted in British Pounds. For revenue denominated in Euros, if there is an increase in the rate at which Euros are exchanged for U.S. Dollars, it will require more Euros to equal a specified amount of U.S. Dollars than before the rate increase. In such cases, and if products are priced in Euros, the Company will receive less in U.S. Dollars than was received before the rate increase went into effect. The Euro to U.S. Dollar conversion rate fluctuations may impact our reported revenue and expenses.other international markets,December 2022, we entered into a clinical trial management agreement for the Company denominates salesLeAAPS clinical trial. The terms of the agreement require we make fixed milestone payments upon achievement of various enrollment and project milestones over the estimated ten year term. Additional variable costs, including pass through costs incurred at clinical trial sites, will be billed to us by the contracted party. Fixed milestone payments are denominated in U.S. Dollars. If products are pricedCanadian Dollars, while variable pass-through fees incurred at clinical trial sites outside the United States may be billed in U.S. Dollars and competitors price their productsor other local currencies. Fluctuations in the local currency, an increase in the relative strengthconversation rates of the U.S. Dollar could result into the Company’s price not being competitive in a market where business is not transacted in U.S. Dollars.The Company invests its cash primarily in money market accounts, repurchase agreements, U.S. government agenciesCanadian Dollar and securities, corporate bonds, asset-backed securities and commercial paper. Although the Company believes its cash to be invested in a conservative manner, with cash preservation being the primary investment objective, the valuelocal currencies of the securities held will fluctuate with changes in the financial markets including, among other things, changes in interest rates, credit quality and general volatility. This risk is managed by investing in high quality investment grade securities with short-term maturities.Financial instruments that potentially subject the Company to credit risk consist of cash and cash equivalents balances and investments in corporate bonds. Certain of AtriCure’s cash and cash equivalents balances exceed FDIC insured limits or are invested in money market accounts with investment banks that are not FDIC-insured. The Company places its cash and cash equivalents in what it believes to be credit-worthy financial institutions. As of December 31, 2019, $28,096 ofinternational trial sites may impact the cash outlay required for future milestone payments and cash equivalents balance was in excess of FDIC limits.
ITEMITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATAPage4243Financial Statement Schedule:72REPORTMason, Ohio20192022 and 2018,2021, the related consolidated statements of operations and comprehensive loss,(loss) income, stockholders’ equity, and cash flows, for each of the three years in the period ended December 31, 2019,2022, and the related notes and the schedule listed in the Index at Item 15 (collectively referred to as the "financial statements"). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 20192022 and 2018,2021, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2019,2022, in conformity withaccounting principles generally accepted in the United States of America.2019,2022, based on criteria established in Internal Control — Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated February 24, 2020,22, 2023, expressed an unqualified opinion on the Company's internal control over financial reporting.Matters Matter
mattersmatter communicated below are mattersis a matter arising from the current-period audit of the financial statements that werewas communicated or required to be communicated to the audit committee and that (1) relaterelates to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the financial statements, taken as a whole, and we are not, by communicating the critical audit mattersmatter below, providing a separate opinionsopinion on the critical audit mattersmatter or on the accounts or disclosures to which they relate.it relates.Acquisition-Related Contingent Consideration and In Process Research and Development Pursuant to the SentreHEART Merger AgreementPerformance Share Awards with a Market Condition - Refer to Note 515 to the financial statementsThe Company acquired 100%outstanding equity intereststhree-year performance period: (i) the Company's revenue compound annual growth rate, a performance condition; and (ii) relative total shareholder return (TSR), a market condition. The performance and market condition payouts are determined independently.SentreHEART on August 13, 2019 for an aggregate purchase price of $208.8 million. The transaction was accounted for using the acquisition method of accounting for business combinations.Auditing the Company’s accounting for the SentreHEART acquisition was complex duePSAs with a market condition that vest and are issued to the estimation uncertainty and key assumptions involved in determining opening balance sheet fair values recorded forrecipient is based upon the acquisition-related contingent consideration liabilityCompany's TSR relative to the TSR of $171.3 million and the in process research and development (IPR&D) intangible assetselected market index at the end of $82.3 million.The Company measured the liability associated withthree-year performance period. A Monte Carlo simulation was performed to estimate the acquisition-related contingent consideration at fair value using unobservable inputs by applyingon the probability-weighted scenario method. Various key assumptions, includingdate of grant, with associated share-based compensation expense recognized over the probability and timing of achievement of regulatory or reimbursement milestones (“key assumptions”), were used inrequisite service period as the employee renders service.opening balance sheetfair value on the date of grant is affected by the stock price of the Company and the market index, as defined by the award agreement, at the beginning of the service period and grant date, the expected stock price volatility of the Company and the market index over the performance period and the correlation coefficient of the daily returns for the Company and the market index over the performance period.acquisition-related contingent consideration and are not observable in thePSAs with a market thus representing Level 3 measurements within the fair value hierarchy.The Company measured the IPR&D intangible asset at fair value, using the excess earnings method and key cash flow assumptions, such as revenue growth rates, related profit margins and obsolescence rates (“cash flow assumptions”).Given that the valuation of the acquisition-related contingent consideration is based on unobservable inputs and is sensitive to changes in key assumptions, and the IPR&D asset required management to make significant estimates related to key cash flows assumptions,condition, audit procedures required a high degree of auditor judgment and an increased extent of effort, including the need to involve our fair value specialists.assumptions impactingCompany's determination of the grant date fair value calculation of the acquisition-related contingent consideration and IPR&DPSAs with a market condition included the following, among others:Related to the valuation of the acquisition-related contingent consideration liability:•We inquired of management of the key valuation assumptions and the Company’s clinical research personnel to understand each milestone and key assumption, including current progress and any clinical results received to date.Monte Carlo simulation methodology used in the determination of the grant date fair value of the PSAs.•We tested the design and operating effectiveness of the Company’sCompany's internal controls over the valuationdetermination of the acquisition-related contingent consideration liability, including management’s projections of key assumptions used in the valuation.We evaluated management’s ability to accurately project the key assumptions by comparing actual progress to management’s historical projections.We evaluated the reasonablenessgrant date fair value of the key assumptions by comparing them to (1) internal communications to management and the Board of Directors and (2) information included in the Company’s external communications.PSAs.We examined regulatory trends to consider the impact of changes in the regulatory environment on the key assumptions.We independently corroborated the reasonableness of the key assumptions by verifying the process and timing necessary to achieve each milestone.Related to the valuation of the IPR&D intangible asset:We inquired of management and the Company’s commercial personnel to understand the key cash flow assumptions.•We tested the design and operating effectivenessaccuracy of the Company’s internal controls over the valuation of the IPR&D intangible asset, including management’s controls over estimatesdata used in determiningmeasuring the cash flow assumptions.We evaluated whetherawards by agreeing the cash flow assumptions used were reasonable by considering industry dataunderlying inputs, such as grant date, share price, and current market forecasts, and whethervesting conditions, among others, back to source documents, such assumptions were consistent with evidence obtained in other areas of the audit.as compensation committee minutes or PSA agreements.We evaluated management’s ability to accurately project the key cash flow assumptions by comparing actual progress to management’s historical projections.•With the assistance of our fair value specialists, we evaluated the reasonablenessmanagement's valuation of the significant valuation assumptions and calculationsPSAs with a market condition by:▪Evaluating the excess earnings method,TestingMonte Carlo simulation methodology and the reasonableness of the valuation assumptions, utilized, including the discountrisk-free interest rate, expected volatility, and the correlation coefficients.▪Testing the mathematical accuracy of the discounted cash flows used to determine the estimatedIndependently calculating a fair value of the IPR&D intangible asset.Valuation of Acquisition-Related Contingent Consideration — Refer to Note 3 to the financial statementsCritical Audit Matter DescriptionThe Company has acquisition-related contingent consideration arrangements totaling $185.2 million as of December 31, 2019 arising from the nContact and SentreHEART acquisitions which obligate the Company to pay former shareholders of acquired companies certain amounts if specified future events occur or conditions are met, such as the achievement of certain regulatory or reimbursement milestones (“milestones”).The Company measures the liability associated with these acquisition-related contingent consideration arrangements at fair value, using unobservable inputs by applying the probability-weighted scenario method. Various key assumptions, including the probability and timing of achievement of the milestones (“key assumptions”), are used in the determination of fair value of acquisition-related contingent consideration arrangements and are not observable inestimate for the market thus representing Level 3 measurements withincondition PSAs using the fair value hierarchy.Given that theunderlying PSA agreement and independently calculated valuation of the acquisition-related contingent consideration arrangements is based on unobservable inputs and is sensitive to changes in the probability and timing of achievement of the milestones, auditing these key assumptions required a high degree of auditor judgment and an increased extent of effort.inputs.How the Critical Audit Matter Was Addressed in the AuditOur audit procedures related to the Company’s key assumptions used in the determination of the fair value of acquisition-related contingent consideration arrangements included the following, among others:We inquired of management and the Company’s clinical research personnel to understand each milestone and key assumption, including current progress and any clinical results received to date.We tested the design and operating effectiveness of the Company’s internal controls over management’s estimates of key assumptions used in the valuation of the acquisition-related contingent consideration arrangements.We evaluated management’s ability to accurately project the key assumptions by comparing actual progress to management’s historical projections.We evaluated the reasonableness of the key assumptions by comparing them to (1) internal communications to management and the Board of Directors and (2) information included in the Company’s external communications.We examined regulatory trends to consider the impact of changes in the regulatory environment on the key assumptions.We independently corroborated the reasonableness of the key assumptions by verifying the process and timing necessary to achieve each milestone.24, 2020ATRICURE,ATRICURE, INC. AND SUBSIDIARIES20192022 and 20182022 2021 Assets Current assets: Cash and cash equivalents $ 58,099 $ 43,654 Short-term investments 63,014 75,436 Accounts receivable, less allowance for credit losses of $230 and $1,096 42,693 33,021 Inventories 45,931 38,964 Prepaid and other current assets 5,477 5,001 Total current assets 215,214 196,076 Long-term investments 51,509 104,338 Property and equipment, net 38,833 31,409 Operating lease right-of-use assets 3,787 4,761 Intangible assets, net 39,339 42,992 Goodwill 234,781 234,781 Other noncurrent assets 1,985 955 Total Assets $ 585,448 $ 615,312 Liabilities and Stockholders’ Equity Current liabilities: Accounts payable $ 19,898 $ 18,597 Accrued liabilities 33,022 36,092 Other current liabilities and current maturities of debt and leases 5,472 1,756 Total current liabilities 58,392 56,445 Long-term debt 56,834 59,741 Finance lease liabilities 9,147 10,082 Operating lease liabilities 3,095 4,068 Other noncurrent liabilities 1,226 1,220 Total Liabilities 128,694 131,556 Commitments and contingencies (Note 10) Stockholders’ Equity: Common stock, $0.001 par value, 90,000 shares authorized; 46,563 and 46,016 issued and outstanding 47 46 Additional paid-in capital 787,422 764,811 Accumulated other comprehensive loss (4,096) (948) Accumulated deficit (326,619) (280,153) Total Stockholders’ Equity 456,754 483,756 Total Liabilities and Stockholders’ Equity $ 585,448 $ 615,312
ATRICURE,ATRICURE, INC. AND SUBSIDIARIESLOSS2019, 20182022, 2021 and 20172022 2021 2020 Revenue $ 330,379 $ 274,329 $ 206,531 Cost of revenue 84,439 68,469 57,222 Gross profit 245,940 205,860 149,309 Operating expenses (benefit): Research and development expenses 57,337 48,506 43,070 Selling, general and administrative expenses 231,272 204,649 150,829 Change in fair value of contingent consideration (Note 2) — (184,800) (357) Intangible asset impairment (Note 4) — 82,300 — Total operating expenses 288,609 150,655 193,542 (Loss) income from operations (42,669) 55,205 (44,233) Other income (expense): Interest expense (4,986) (4,918) (4,885) Interest income 1,994 466 1,101 Other (537) (366) (24) (Loss) income before income tax expense (46,198) 50,387 (48,041) Income tax expense 268 188 114 Net (loss) income $ (46,466) $ 50,199 $ (48,155) Net (loss) income per share: Basic net (loss) income per share $ (1.02) $ 1.11 $ (1.14) Diluted net (loss) income per share $ (1.02) $ 1.09 $ (1.14) Weighted average shares outstanding: Basic 45,740 45,066 42,125 Diluted 45,740 46,039 42,125 Comprehensive (loss) income: Unrealized loss on investments $ (2,811) $ (941) $ (46) Foreign currency translation adjustment (337) (319) 516 Other comprehensive (loss) income (3,148) (1,260) 470 Net (loss) income (46,466) 50,199 (48,155) Comprehensive (loss) income, net of tax $ (49,614) $ 48,939 $ (47,685)
ATRICURE,ATRICURE, INC. AND SUBSIDIARIES2019, 2018,2022, 2021, and 2017Balance—December 31, 2019 39,655 $ 40 $ 529,658 $ (282,197) $ (158) $ 247,343 Issuance of common stock through public offering 4,574 5 188,953 — — 188,958 Issuance of common stock under equity incentive plans 1,013 — (2,194) — — (2,194) Issuance of common stock under employee stock purchase plan 104 — 3,330 — — 3,330 Share-based employee compensation expense — — 22,642 — — 22,642 Other comprehensive income — — — — 470 470 Net loss — — — (48,155) — (48,155) Balance—December 31, 2020 45,346 $ 45 $ 742,389 $ (330,352) $ 312 $ 412,394 Issuance of common stock under equity incentive plans 589 1 (9,837) — — (9,836) Issuance of common stock under employee stock purchase plan 81 — 4,181 — — 4,181 Share-based employee compensation expense — — 28,078 — — 28,078 Other comprehensive loss — — — — (1,260) (1,260) Net income — — — 50,199 — 50,199 Balance—December 31, 2021 46,016 $ 46 $ 764,811 $ (280,153) $ (948) $ 483,756 Issuance of common stock under equity incentive plans 426 1 (10,385) — — (10,384) Issuance of common stock under employee stock purchase plan 121 — 4,225 — — 4,225 Share-based employee compensation expense — — 28,771 — — 28,771 Other comprehensive loss — — — — (3,148) (3,148) Net loss — — — (46,466) — (46,466) Balance—December 31, 2022 46,563 $ 47 $ 787,422 $ (326,619) $ (4,096) $ 456,754
ATRICURE,ATRICURE, INC. AND SUBSIDIARIES2019, 20182022, 2021 and 20172022 2021 2020 Cash flows from operating activities: Net (loss) income $ (46,466) $ 50,199 $ (48,155) Adjustments to reconcile net (loss) income to net cash used in operating activities: Share-based compensation expense 28,771 28,078 22,642 Depreciation 8,057 7,534 7,866 Amortization of intangible assets 3,653 2,907 1,682 Amortization of deferred financing costs 507 759 509 Amortization of investments 1,478 2,482 1,236 Change in fair value of contingent consideration — (184,800) (357) Intangible asset impairment — 82,300 — Other non-cash adjustments 739 1,607 1,347 Changes in operating assets and liabilities: Accounts receivable (8,989) (10,087) 5,087 Inventories (7,305) (4,274) (5,265) Other current assets (515) (700) (477) Accounts payable 2,677 4,710 (1,560) Accrued liabilities (2,966) 8,271 (4,908) Other noncurrent assets and liabilities (1,782) (2,766) 484 Net cash used in operating activities (22,141) (13,780) (19,869) Cash flows from investing activities: Purchases of available-for-sale securities (24,637) (173,105) (227,045) Sales and maturities of available-for-sale securities 85,524 206,362 75,306 Purchases of property and equipment (16,881) (9,753) (5,259) Proceeds from capital grant — — 800 Net cash provided by (used in) investing activities 44,006 23,504 (156,198) Cash flows from financing activities: Proceeds from sale of stock, net of offering costs of $218 — — 188,958 Proceeds from debt borrowings — 5,000 — Payments on debt and finance leases (899) (5,816) (667) Payment of debt fees — (1,171) (35) Proceeds from stock option exercises 1,816 8,175 10,835 Shares repurchased for payment of taxes on stock awards (12,201) (18,011) (13,029) Proceeds from issuance of common stock under employee stock purchase plan 4,225 4,181 3,330 Net cash (used in) provided by financing activities (7,059) (7,642) 189,392 Effect of exchange rate changes on cash and cash equivalents (361) (372) 136 Net increase in cash and cash equivalents 14,445 1,710 13,461 Cash and cash equivalents—beginning of period 43,654 41,944 28,483 Cash and cash equivalents—end of period $ 58,099 $ 43,654 $ 41,944 Supplemental cash flow information: Cash paid for interest $ 4,270 $ 4,223 $ 4,366 Cash paid for income taxes, net of refunds 192 190 217 Non-cash investing and financing activities: Accrued purchases of property and equipment 272 1,552 298 Assets obtained in exchange for finance lease obligations — — 22 DESCRIPTIONDESCRIPTION OF BUSINESS AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES and, left atrial appendage (LAA) management and post-operative pain management, and sells its products to medical centers globally through its direct sales force and distributors.ourits wholly-owned subsidiaries. All intercompany accounts and transactions have been eliminated in consolidation.certain financial institutions.makes investmentsinvests primarily in U.S. Government agenciesgovernment and securities,agency obligations, corporate bonds, commercial paper and asset-backed securities and classifies all investments as available-for-sale. Investments with maturities ofmaturing in less than one year are classified as short-term.short-term investments. Investments are recorded at fair value, with unrealized gains and losses recorded as accumulated other comprehensive income (loss). Gains and losses are recognized using the specific identification method when securities are sold and are included in interest income or expense.income.Recognition—Recognition—Revenue is generated primarily from the sale of medical devices. Sales of devices are categorized based on the type of product as follows: open ablation, minimally invasive ablation, pain management and appendage management. The Company recognizes revenue when control of promised goodsdevices is transferred to customers in an amount that reflects the consideration the Company expects to be entitled to in exchange for those goods. This generally occursdevices. Revenue is recognized at a point in time upon shipment or delivery of goodsproducts. Shipping and handling activities performed after control transfers to customers. See Note 13customers are considered activities to fulfill the promise to transfer the products. Revenue includes shipping and handling revenue of $1,496, $1,354 and $1,192 in the years ended December 31, 2022, 2021 and 2020.further discussionboth end-users and distributors, except that payment terms are generally net 30 days for end-users and net 60 days for distributors, with some exceptions. The Company does not maintain any post-shipping obligations to customers; no installation, calibration or testing of products is performed subsequent to shipment in order to render products operational. The Company expects to be entitled to the total consideration for the products ordered as product pricing is fixed and payment terms fall within one year to forgo adjustment for the effects of a significant financing component. The Company excludes taxes assessed by governmental authorities on revenue-producing transactions from the measurement of the transaction price. products shipped in error and invoice adjustments. The Company adjusts the provision using the expected value method based on historical experience. Increases to the provision result in a reduction ofreduce revenue, and the provision is included in accrued liabilities.DoubtfulCredit Losses on Accounts Receivable—Receivable—The Company evaluates the collectability ofexpected credit losses on accounts receivable, to determine the appropriate reserve for doubtful accounts. In determining the amount of the reserve, the Company considers aging of account balances,considering historical credit losses, current customer-specific information and other relevant factors.factors when determining the allowance. An increase to the allowance for doubtful accountscredit losses results in a corresponding increase in selling, general and administrative expenses. The Company charges off uncollectible receivables against the allowance when all attempts to collect the receivable have failed. The Company’s history of write-offs has not been significant. Recoveries are recognized when received as a reduction to the allowance for credit losses by decreasing bad debt expense. The followingYear Ended December 31, 2022 2021 2020 Beginning balance - January 1 $ 1,096 $ 1,096 $ 1,124 Adoption of ASU 2016-13 — — (28) Provision for expected credit losses 190 65 — Recovery (1,056) (65) — Ending balance - December 31 $ 230 $ 1,096 $ 1,096 Inventory reserves for excess, obsolete and expired products are impacted by uncertainUncertain timing of regulatory approvals, variability in product launch strategies and variation in product use.use all impact inventory reserves for excess, obsolete and expired products. An increase to inventory reserves results in a corresponding increase in cost of revenue. Inventories are written off against the reserve when they are physically disposed.computeddetermined using the straight-line method over the estimated useful life. The estimated useful life of leasehold improvements is the shorter of the estimated life or the lease term. The estimated useful lives of assets (see Note 8).buildings is 15 to 20 years, while furniture, fixtures, computers and office equipment are depreciated from three to seven years. The Company’s radiofrequency and cryothermic generators are generally placed with customers that use the Company’s disposable products. The estimated useful lives of generators are based on anticipated usage by customers and may change in future periods with changes in usage or introduction of new technology. Depreciation related to generators is recorded in cost of revenue over three years. Maintenance and repair costs are expensed as incurred. The Company reassessesassesses the useful lives of property and equipment at least annually and retires assets if they are no longer in service. Maintenance and repair costs are expensed as incurred.use.The Company’s RF and cryo generators are generally placed with customers served by our direct sales force. The estimated useful lives of this equipment are based on anticipated usage by customers and the timing and impact of expected new technology rollouts by the Company and may change in a future period if the Company experiences changes in the usage of the equipment or introduces new technologies. Depreciation related to generators and other capital equipment is recorded in cost of revenue.The Company reviews property and equipment for impairment at least annually using its best estimates based on reasonable and supportable assumptions and projections of expected future cash flows. Property and equipment impairments recorded by the Company have not been significant.periodsfifteen year period benefited. The Company reassesses the useful lives of intangible assets annually.Included in intangible assets is In Process Research and Development (IPR&D), representing the value of acquired technologies which have not yet reached technological feasibility. The primary basis for determining the technological feasibility is obtaining specific regulatory approvals. IPR&D is accounted for as an indefinite-lived intangible asset until completion or abandonment of the IPR&D project. Upon completion of the development project, the IPR&D will be amortized over its estimated useful life. The IPR&DATRICURE, INC. AND SUBSIDIARIESNOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)(In Thousands, Except Per Share Amounts)assets represent estimates of the fair value of the pre-market approval (PMA) that may result from the CONVERGE IDE and aMAZE IDE clinical trials. The Company reviews intangible assets at least annually for impairment using its best estimates based on reasonable and supportable assumptions and projections of expected future cash flows. The Company performs impairment testing annually on October 1. If the IPR&D project is abandoned or regulatory approvals are not obtained, we may have a full or partial impairment charge related to the IPR&D, calculated as the excess carrying value of the IPR&D assets over the estimated fair value.projections.tests goodwill forperforms impairment testing annually on October 1 or more often if impairment indicators are present.Contingent ConsiderationLong-lived Assets—The Company reviews property and equipment and intangible assets, excluding goodwill, for impairment whenever events or changes in circumstances indicate the carrying amount of an asset may not be recoverable. When such an event occurs, management determines whether there has been impairment by comparing the anticipated undiscounted future net cash flows to the related asset's carrying value.the contingent consideration recorded in business combinations, as well as deferred revenues,contractual obligations, including asset retirement obligations and other contractual obligations. The contingent consideration balance is included in noncurrent liabilities as such settlement is both required and expected to be made primarily in shares of the Company’s common stock pursuant to the nContact merger agreement and SentreHEART merger agreement.Pounds.Pounds and Australian Dollars. existing assets and liabilities and their respective tax bases and operating loss and tax credit carryforwards. Deferred income tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred income tax assets and liabilities from a change in tax rates is recognized in the period that includes the enactment date.thea deferred income tax asset will not be realized. Significant weight is given to evidence that can be objectively verified. The Company evaluates deferred income tax assets on an annual basis to determine if valuation allowances are required.required by considering all available evidence. Deferred income tax assets are realized by having sufficient future taxable income to allow the related tax benefits to reduce taxes otherwise payable. The sources of taxable income that may be available to realize the benefit of deferred income tax assets are future taxable income, future reversals of existing taxable temporary differences, future taxable income exclusive of reversing temporary differences and carryforwards,in prior carryforward years and tax planning strategies that are both prudent and feasible. In evaluating the need for a valuation allowance, the existence of cumulative losses in recent years is significant objectively verifiableobjectively-verifiable negative evidence that must be overcome by objectively verifiableobjectively-verifiable positive evidence to avoid the need to recordfor a valuation allowance. The Company has recorded a fullCompany's valuation allowance againstoffsets substantially all net deferred income tax assets as it is more-likely-than-not that the benefit of the deferred income tax assets will not be recognized in future periods. The Tax Cut and Jobs Act (Tax Reform Act) allows companies an election to reclassify theCompany has not reclassified income tax effects of the Tax ReformCuts and Jobs Act on items within accumulated other comprehensive (loss) income (loss) to retained earnings. The Company has not made this electionearnings due to its full valuation allowance.Net LossEarnings Per Share—Basic and diluted net lossearnings per share is computed by dividing net (loss) income available to common stockholders by the weighted average number of shares of common stock outstanding during the period. Diluted earnings per share reflects net lossincome available to common stockholders divided by the weighted average number of common shares outstanding during the period. Sinceperiod and any dilutive common share equivalents, including shares issuable upon the Company has experienced net losses for all periods presented, net loss per share excludes the effectvesting of 3,623, 3,869 and 4,321 stock options, restricted stock awards and restricted stock units, and performance share awardsexercise of stock options as ofwell as shares issuable under the Company's employee stock purchase plan (ESPP).Year Ended December 31, 2022 2021 2020 Net (loss) income available to common stockholders $ (46,466) $ 50,199 $ (48,155) Basic weighted average common shares outstanding 45,740 45,066 42,125 Effect of dilutive securities — 973 — Diluted weighted average common shares outstanding 45,740 46,039 42,125 Basic net (loss) income per common share $ (1.02) $ 1.11 $ (1.14) Diluted net (loss) income per common share $ (1.02) $ 1.09 $ (1.14) 2019, 20182022 and 2017 because they are anti-dilutive. Therefore,2020, the number of shares calculated for basic net loss per share is also used for the diluted net loss per share calculation.calculation, and net loss per share excludes the effect of 1,292 and 2,301 sharesComprehensive Income (Loss) and Accumulated Other Comprehensive Income (Loss)—In addition to net losses,because the comprehensive loss includes foreign currency translation adjustments and unrealized gains and losses on investments.Accumulated other comprehensive (loss) income consistedeffect would be anti-dilutive. The computation of diluted earnings per share in the following (net of tax): costs are expensed as incurred. These costs include compensation and other internal and external costs associated with the development of and research related toof new and existing products or concepts, preclinical studies, clinical trials healthcare compliance and related regulatory affairs.activities, as well as amortization of technology assets. Research and development costs are expensed as incurred. Clinical trial costs and other development costs incurred by third parties are expensed as contracted work is performed.$635, $785$1,233, $907 and $900$655 during the years ended December 31, 2019, 20182022, 2021 and 2017.2020.recordsrecognizes share-based compensation expense for all share-based payment awards, including stock options, restricted stock awards, restricted stock units, performance sharesshare awards (PSAs) and stock purchases related to an employee stock purchase plan, based on estimated fair values.The Company estimates the fair value of share-based payment awards on the date of grant. The value of the portion of thean award that is ultimately expected to vest is recognized as expense over the requisite service periods in the Company’s Consolidated Statements of Operations and Comprehensive Loss.period. The Company estimates forfeitures at the time of grant and revises them, ifas necessary, in subsequent periods ifas actual forfeitures differ from those estimates.assumptions regarding several subjective variables. These variables include, but are not limited to,assumptions, such as the Company’s expected stock price volatility over the term of the awards and actual and projected employee stock option exercise behaviors. The value of the portion of the awards that is ultimately expected to vest is recognized as expense over the requisite service periods in the Consolidated Statements of Operations and Comprehensive Loss. The Company estimates the fair value of restricted stock awards and restricted stock units and performance share awards based upon the grant date closing market price of the Company’s common stock.estimatedCompany estimates the fair value of PSAs with a performance share awardscondition based on the closing stock price on the date of grant assuming the performance target will be achieved and may be adjustedadjust expense over the performance period based on changes to estimates of performance target achievement.management to make estimates and assumptions that affect the reported amounts of assets and liabilities, disclosure ofincluding intangible assets, contingent assets and liabilities at the date of the financial statements and the reported amounts of revenue and expense during the reporting period. Estimates are based on historical experience, where applicable, and other assumptions believed to be reasonable by management. Actual results could differ from those estimates.agencies and securities,agency obligations, accounts receivable, short-term other current assets, and accounts payable and accrued liabilities as Level 1. The carrying amounts of these assets and liabilities approximate their fair value due to their relatively short-term nature. Cash equivalents and investments in corporate bonds, repurchase agreements, commercial paper and asset-backed securities are classified as Level 2 within the fair value hierarchy. The fair value of fixed term debt is estimated by calculating the net present value of future debt payments at current market interest rates and is classified as Level 2. The book value of the Company’s fixed term debt approximates its fair value because the interest rate varies with market rates. Significant unobservable inputs with respect to the fair value measurements of the Level 3 contingent consideration liabilities are developed using Company data. See Note 32 – Fair Value for further information on fair value measurements.2. RECENT ACCOUNTING PRONOUNCEMENTSIn June 2016, the FASB issued ASU 2016-13, “Financial Instruments - Credit Losses (Topic 326): Measurement of Credit Losses on Financial Instruments” (ASU 2016-13). This guidance requires—The Company has considered all recent accounting pronouncements and has concluded that financial assets measured at amortized costs, such as trade receivables and contract assets, be presented net of expected credit losses,there are no recent accounting pronouncements which may be estimated based on relevant information such as historical experience, current conditions and future expectations for each pool of similar financial assets. The new guidance requires enhanced disclosures related to the trade receivables and associated credit losses. The guidance is effective for interim and annual periods beginning within 2020. The adoption of this guidance is expected to increase the level of disclosures related to the Company’s trade receivables, however it is notare expected to have a material impacteffect on the Company’s consolidatedCompany's financial statements.In January 2017, the FASB issued ASU 2017-04, “Intangibles - Goodwill and Other (Topic 350): Simplifying the Accounting for Goodwill Impairment” (ASU 2017-04). The guidance removes the requirement to perform a hypothetical purchase price allocation to measure goodwill impairment. Under ASU 2017-04, a goodwill impairment will be the amount by which a reporting unit’s carrying value exceeds its fair value, not to exceed the carrying amount of goodwill. The guidance becomes effective for interim and annual periods beginning within 2020, with early adoption permitted, and applied prospectively. The Company does not expect the adoption of this standardcontinues to have a material impact on its consolidated financial statements.In August 2018, the FASBmonitor and evaluate recently issued ASU 2018-13, “Fair Value Measurement (Topic 820), Disclosure Framework – Changes to the Disclosure Requirementsaccounting guidance upon issuance for Fair Value Measurement” (ASU 2018-13). The amendments modify the disclosure requirements for fair value measurements and are effective for all entities for interim and annual reporting periods beginning within 2020. Early adoption of either the entire standard or only the provisions that eliminate or modify the requirements is permitted. The Company has elected to early adopt the guidance, and the fair value measurement disclosures herein reflect the adoption of the provisions of ASU 2018-13 as of December 31, 2019.any potential impact.In August 2018, the FASB issued ASU 2018-15, “Customer’s Accounting for Implementation Costs Incurred in a Cloud Computing Arrangement That Is a Service Contract” (ASU 2018-15). The amendments in this ASU align the requirements for capitalizing implementation costs incurred in a hosting arrangement that is a service contract with the requirements for capitalizing implementation costs incurred to develop or obtain internal-use software (and hosting arrangements that include an internal-use software license). The guidance in ASC 350-40 on internal-use software is applied when capitalizing implementation costs related to a hosting arrangement that is a service contract and expense the capitalized implementation costs related to a hosting arrangement that is a service contract over the hosting arrangement's term, presenting the expense in the same line item in the statement of operations and comprehensive loss as that in which the fee associated with the hosting arrangement is presented. The amendments are effective for interim and annual reporting periods beginning within 2020, with early adoption permitted. During 2019, the Company adopted this standard prospectively, and has deferred eligible costs related to implementation of hosting arrangements within other current and noncurrent assets. These costs are amortized in the same income statement line as the associated hosting subscription fees and operating expenses. The adoption of ASU 2018-15 did not have a material impact on the Company’s consolidated financial statements.In December 2019, the FASB issued ASU 2019-12, “Income Taxes (Topic 740): Simplifying the Accounting for Income Taxes” (2019-12). The amendment simplifies the accounting for income taxes by eliminating some exceptions to the general approach in ASC 740, Income Taxes. It also clarifies certain aspects of the existing guidance to promote more consistent application, among other things. The guidance is effective for interim and annual reporting periods beginning within 2021 with early adoption permitted. The Company has elected to early adopt the simplification guidance, and there is no impact on prior periods.ATRICURE, INC. AND SUBSIDIARIESNOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)(In Thousands, Except Per Share Amounts)3.2. FAIR VALUE (ASC 820), defines fair value as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. Valuation techniques used to measure fair value must maximize the use of observable inputs and minimize the use of unobservable inputs. The fair value hierarchy is based on three levels of inputs, of which the first two are considered observable and the last unobservable, that may be used to measure fair value:•Level 1—Quoted prices in active markets for identical assets or liabilities that the Company has the ability to access at the measurement date. An active market for the asset or liability is a market in which transactions for the asset or liability occur with sufficient frequency and volume to provide pricing information on an ongoing basis. The valuation under this approach does not entail a significant degree of judgment.•Level 2—Inputs other than Level 1 that are observable, either directly or indirectly, such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities. The valuation technique for the Company’s Level 2 assets is based on quoted market prices for similar assets from observable pricing sources at the reporting date.•Level 3—Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities. Unobservable inputs shall be used to measure fair value to the extent that observable inputs are not available, thereby allowing for situations in which there is little, if any, market activity for the asset or liability at the measurement date.2019:2022:Quoted Prices
in Active
Markets for
Identical
Assets
(Level 1)Significant
Other
Observable
Inputs
(Level 2)Significant
Other
Unobservable
Inputs
(Level 3)Total Assets: Money market funds $ — $ 54,414 $ — $ 54,414 Commercial paper — 11,935 — 11,935 Government and agency obligations 32,637 — — 32,637 Corporate bonds — 67,598 — 67,598 Asset-backed securities — 2,353 — 2,353 Total assets $ 32,637 $ 136,300 $ — $ 168,937 TableThere were no changes in the levels or methodology of ContentsATRICURE, INC. AND SUBSIDIARIESNOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)(In Thousands, Except Per Share Amounts)2018:2021:Quoted Prices
in Active
Markets for
Identical
Assets
(Level 1)Significant
Other
Observable
Inputs
(Level 2)Significant
Other
Unobservable
Inputs
(Level 3)Total Assets: Money market funds $ — $ 38,360 $ — $ 38,360 Commercial paper — 22,978 — 22,978 Government and agency obligations 32,690 — — 32,690 Corporate bonds — 95,845 — 95,845 Asset-backed securities — 28,261 — 28,261 Total assets $ 32,690 $ 185,444 $ — $ 218,134 There were no changes in the levels or methodology of measurement of financial assets and liabilities during the years ended December 31, 2019 and 2018.Acquisition-Related Contingent Consideration. The Company hasCompany's contingent consideration arrangements arising from the nContact and SentreHEART acquisitions. Contingent consideration arrangements under the nContact merger agreement obligate the Company to pay former shareholders of nContact for the following milestones, if achieved:Regulatory Milestone – up to $42,500 upon the completion of the CONVERGE IDE clinical trial and receiving a PMA from FDA for the EPi-Sense AF Guided Coagulation System and/or any other nContact product with an indication for symptomatic persistent Afib or similar or related indication. The full contingent consideration amount of $42,500 is only earned if such regulatory approvals are received on or before January 1, 2020. The potential contingent consideration is reduced by 8.33% (or one-twelfth) each month following January 2020 and is reduced to 0 if the regulatory milestone is achieved after December 31, 2020. Any payment of the regulatory milestone contingent consideration is due within 30 days following the receipt of the related PMA approval.Trial Enrollment Milestone – $7,500 upon completion of patient enrollment in the CONVERGE IDE clinical trial. The Company completed patient enrollment on August 21, 2018, and payment was made to former nContact shareholders on September 20, 2018.Commercial Milestone – for calendar years 2016 through 2019, nContact revenues in excess of specified target revenue amounts will result in contingent consideration equal to 1.5 times the revenues in excess of target. Payments of contingent consideration when the commercial milestone is achieved are due within 65 days of each calendar year end. NaN payments were made for calendar years 2016 through 2019 as revenues did not exceed the targets for these years.Subject to the terms and conditions of the merger agreement, all contingent consideration must be paid first in shares of AtriCure common stock. The merger agreement limits the total number of shares of AtriCure common stock issued in connection with the acquisition to 5,660, of which 3,757 shares were issued at closing of the nContact acquisition on October 13, 2015 and an additional 232 shares were issued upon completion of the trial enrollment milestone in 2018.Contingent consideration arrangements under the SentreHEART merger agreement obligate the Company to pay certain defined amounts to former shareholders of SentreHEART if specified milestones are met related to the aMAZE IDE clinical trial, including PMA approval and reimbursement for the therapy involving SentreHEART’sSentreHEART's devices. In connection with the acquisition of SentreHEART on August 13, 2019, preliminary fair value of $171,300 was recordedThe achievement periods for the SentreHEART contingent consideration. See Note 5 for more details regarding the SentreHEART acquisition-related contingent consideration. Subject to the termsPMA approval and conditions of the SentreHEART merger agreement, all contingent consideration would be paid in cashreimbursement milestones expire on December 31, 2023 and stock at the discretion of the Company, subject to certain limitations, with the maximum number of shares that may be issued after closing limited to 7,021, ofATRICURE, INC. AND SUBSIDIARIESNOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)(In Thousands, Except Per Share Amounts)which 699 shares were issued at closing of the SentreHEART acquisition on August 13, 2019.December 31, 2026, respectively. The maximum contingent consideration payable by AtriCure will not exceed $260,000.The Company measures contingent consideration liabilities are measured by applying the probability weighted scenario method using unobservable inputs, by applying an income approach, such as the discounted cash flow technique or the probability-weighted scenario method. Various key assumptions, such as the probability and timing of achievement of the agreed milestones and projected revenues, are used in the determination of fair value of contingent consideration arrangements and are not observable in the market, thus representing a Level 3 measurement within the fair value hierarchy.recurring Level 3Company recorded a credit to operating expenses of $184,800 reflecting the change in fair value measurements of the contingent consideration liabilities includeconsideration. The Company has assessed the following significant inputsprojected probability of payment during the contractual achievement periods to be remote, resulting in no fair value as of December 31, 2019:
September 2025Contingent consideration liabilities are periodically remeasured. Changes in the discount rate, time until payment2022 and probabilities2021.2022 2021 2020 Beginning Balance – January 1 $ — $ 184,800 $ 185,157 Amounts acquired — — — Changes in fair value of contingent consideration — (184,800) (357) Ending Balance – December 31 $ — $ — $ 184,800 Contingent consideration liabilities are classified as noncurrent liabilities as the Company expects to settle the majority of the milestone payments in stock. As of December 31, 2019, the Company estimates 15% of the nContact regulatory milestone, or approximately $1,839 will be paid in cash during 2020.4.20192022 consisted of the following: Cost Basis Unrealized
LossesFair Value Corporate bonds $ 69,832 $ (2,234) $ 67,598 Government and agency obligations Government and agency obligations 33,971 (1,334) 32,637 Commercial paper 11,935 — 11,935 Asset-backed securities 2,483 (130) 2,353 Total $ 118,221 $ (3,698) $ 114,523 Cost Basis Unrealized
LossesFair Value Corporate bonds $ 96,408 $ (563) $ 95,845 Government and agency obligations 32,953 (263) 32,690 Commercial paper 22,978 — 22,978 Asset-backed securities 28,322 (61) 28,261 Total $ 180,661 $ (887) $ 179,774 Available-for-sale Amortized Cost Fair Value Due in 1 year or less $ 63,596 $ 62,840 Due after 1 year through 5 years 52,142 49,330 Due after 5 years through 10 years — — Instruments not due at a single maturity date 2,483 2,353 Total $ 118,221 $ 114,523 Investments as of December 31, 2018 consisted of the following:The Company has 0t experienced any significant realized gains or losses on its investments in the years ended December 31, 2019, 2018 and 2017.5. BUSINESS COMBINATIONSOn August 13, 2019, the Company acquired 100% of the outstanding equity interests of SentreHEART. Founded in 2005 and based in Redwood City, California, SentreHEART developed innovative technology for remote delivery of a suture for closure of anatomic structures including the left atrial appendage (LAA). This technology is currently being studied in the aMAZE IDE clinical trial, an FDA-approved, prospective, multicenter, randomized controlled trial. The objective of the aMAZE Trial is to demonstrate that the LARIAT® device for LAA closure, plus a Pulmonary Vein Isolation (PVI) ablation, will lead to a reduced incidence of recurrent Afib compared to PVI alone. Management believes the acquisition of SentreHEART will significantly expand the Company’s addressable markets with a product designed for electrophysiologists, and the acquisition of SentreHEART deepens the Company’s commitment to provide the broadest possible offering of ablation and LAA management solutions to patients and customers.The total consideration paid to SentreHEART’s former shareholders at the acquisition date was $18,008 in cash and 699 shares of AtriCure common stock valued at approximately $20,307. The cash paid at acquisition was subject to adjustment for net working capital balances outside of a specified range, resulting in $768 adjustment paid to the Company in November 2019. The merger agreement also provides for the Company to pay contingent consideration, as follows:PMA Milestone – up to $140,000 upon receiving PMA from FDA for the LARIAT system with an approved indication allowing commercial distribution in the United States for the closure of the LAA for treatment of atrial fibrillation. The full contingent consideration amount is only received if PMA approval is received on or before December 31, 2022. The potential contingent consideration is reduced by 4.17% (or one-twenty-fourth) each month following December 2022 and is reduced to 0 if the milestone is achieved after December 31, 2023. Payment of $25,000 of the PMA milestone may be accelerated upon achievement of an Interim Success Milestone as defined by the merger agreement.CPT Reimbursement Milestone – up to $120,000 upon approval of a Medicare Category 1 Current Procedural Terminology (CPT) Code by the American Medical Association. The full contingent consideration amount is only received if approval of the CPT Code is received on or before December 31, 2025. The potential contingent consideration is reduced by 4.17% (or one-twenty-fourth) each month following December 2025 and is reduced to 0 if the milestone is achieved after December 31, 2026.Subject to the terms and conditions of the merger agreement, all contingent consideration would be paid in cash and stock at the discretion of the Company, subject to certain limitations, with the maximum number of shares that may be issued after closing limited to 7,021, of which 699 were paid at closing. The maximum contingent consideration payable by AtriCure will not exceed $260,000.The Company accounted for the acquisition in accordance with ASC 805, “Accounting for Business Combinations”. The assets acquired, liabilities assumed and the estimated contingent consideration obligations are recorded at their respective fair values as of the date of acquisition. The process of estimating fair values of identifiable assets, certain intangible assets and assumed liabilities requires significant assumptions and estimates. The judgments used to determine the estimated fair value assigned to each class ofATRICURE, INC. AND SUBSIDIARIESNOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)(In Thousands, Except Per Share Amounts)assets acquired and liabilities assumed, as well as asset lives, can materially impact the amounts recorded and the Company’s results of operations.The components of the aggregate purchase price for the SentreHEART acquisition are as follows:Fair value of AtriCure common stock issued at closing$20,307Cash17,240Fair value of contingent consideration liabilities171,300Total purchase price$208,847The fair value of the contingent consideration liabilities was determined by applying the probability-weighted scenario method. Key assumptions in the valuation of the contingent consideration liabilities are based on management’s judgment and estimates and include the probability of achievement of each of the milestones, timing of achievement and discount rates, reflecting the inherent risks of achieving the respective milestones. Some assumptions are not observable in the market, and thus represent a Level 3 measurement within the fair value hierarchy. See Note 3 for disclosure of unobservable inputs.The following table summarizes the estimated fair values of the assets acquired and the liabilities assumed based on the information that was available as of the acquisition date:During the measurement period, the Company recorded adjustments for the fair value of consideration transferred, including settlement of working capital, and the evaluation of certain tax attributes. As of December 31, 2019, the purchase price allocation has not yet been finalized as the Company evaluates certain tax attributes of SentreHEART. Net deferred tax assets of $20,590 and offsetting valuation allowances were also recognized at the acquisition date for the future tax consequences attributable to differences between the above financial statement carrying amounts of existing assets and liabilities and their respective tax bases and acquired operating loss and tax credit carryforwards of SentreHEART. At acquisition, SentreHEART had approximately $184,036 of federal and state net operating loss carryforwards, which begin to expire in 2026 and $37,906 of federal net operating loss carryforwards which have no expiration as a result of the Tax Reform Act. A portion of the net operating loss carryforwards are subject to certain limitations under Internal Revenue Code Section 382. The Company recorded a full valuation allowance against the net deferred tax assets at acquisition. The goodwill recorded is not deductible for tax purposes.The valuation of the intangible assets acquired and related amortization periods are as follows:The fair value of the LARIAT developed technology was estimated using the relief-from-royalty method, an income approach. The LARIAT developed technology asset is amortized on a straight-line basis over its estimated useful life. The IPR&D asset was estimated using the excess earnings method, also an income approach. The IPR&D asset represents an estimate of the fair value of theATRICURE, INC. AND SUBSIDIARIESNOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)(In Thousands, Except Per Share Amounts)PMA approval from the in-process aMAZE IDE clinical trial and is accounted for as an indefinite-lived intangible asset until completion or abandonment of the project.The Company recorded the excess of the aggregate purchase price over the estimated fair values of the identifiable net assets acquired as goodwill. Goodwill is primarily attributable to the benefits the Company expects to realize by enhancing its product offering and addressable markets, thereby contributing to an expanded revenue base. As discussed in Note 1, the Company accounts for goodwill in a single reporting unit representing the Company as a whole.The operating results of SentreHEART, including $1,280 of appendage management revenue and $8,505 of net loss, are included in the Consolidated Statements of Operations and Comprehensive Loss beginning August 14, 2019. The Consolidated Balance Sheet as of December 31, 2019 reflects the acquisition of SentreHEART. The Company recognized approximately $3,978 of acquisition-related costs in the year ended December 31, 2019, consisting of legal, audit, tax and other due diligence expenses. Acquisition-related costs are included in selling, general and administrative expenses.The following supplemental pro forma information presents the financial results of the Company for the twelve months ended December 31, 2019 and 2018 as if the acquisition of SentreHEART had occurred on January 1, 2018.Certain pro forma adjustments have been made when calculating the amounts above to reflect the impact of the purchase transaction, primarily consisting of the exclusion of SentreHEART’s interest expense incurred on debt paid off or converted to equity in the acquisition, exclusion of fair value adjustments for SentreHEART’s derivative liabilities and preferred warrants settled as part of the acquisition, adjustments for amortization of intangible assets with determinable lives and exclusion of contingent consideration remeasurement. The Company also eliminated transaction expenses incurred by both AtriCure and SentreHEART. The supplemental pro forma information has been prepared for comparative purposes and does not purport to be indicative of what would have occurred had the acquisition been made on January 1, 2018, nor is it indicative of any future results. The pro forma information does not include any adjustments for potential revenue enhancements, cost synergies or other operating efficiencies that could result from the acquisition.6.4. INTANGIBLE ASSETS AND GOODWILLAmortization expense related 2022 2021 Cost Accumulated Amortization Cost Accumulated Amortization Technology $ 46,470 $ 7,131 $ 55,712 $ 12,720 intangible assets with definite lives, which excludesreduce the carrying value of the aMAZE IPR&D asset to $0 as of December 31, 2022 as a result of data from the aMAZE clinical trial not achieving statistical superiority. This impairment charge was $1,943, $1,510reflected as a component of operating expenses. The $9,242 reduction in technology cost and $1,367accumulated amortization during 2022 is a result of a write-off fully-amortized asset no longer in use.2019, 20182022, 2021 and 2017. In 2018, the Company reduced the ten-year estimated useful life of the Fusion technology asset by two years based on changes in estimated periods benefited. This change in estimate was applied prospectively.2020.ATRICURE, INC. AND SUBSIDIARIESNOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)(In Thousands, Except Per Share Amounts)2023 $ 2,953 2024 2,953 2025 2,953 2026 2,953 2027 2,953 2028 and thereafter 24,574 Total $ 39,339 Net carrying amount as of December 31, 2017$105,257Additions—Net carrying amount as of December 31, 2018105,257Additions129,524Net carrying amount as of December 31, 2019$234,7817.Net carrying amount as of December 31, 2020 $ 234,781 Additions — Net carrying amount as of December 31, 2021 234,781 Additions — Net carrying amount as of December 31, 2022 $ 234,781 2022 2021 Raw materials $ 19,880 $ 12,653 Work in process 2,959 2,064 Finished goods 23,092 24,247 Inventories $ 45,931 $ 38,964 8. 2022 2021 Buildings and improvements $ 28,947 $ 22,977 Generators 21,354 20,175 Machinery and office equipment 20,184 14,758 Computer equipment and software 10,251 7,762 Construction in progress 3,909 5,999 Land 1,006 1,006 Total 85,651 72,677 Less accumulated depreciation (46,818) (41,268) Property and equipment, net $ 38,833 $ 31,409 $7,423, $7,244$8,057, $7,534 and $7,761$7,866 for the years ended December 31, 2019, 20182022, 2021 and 2017. Depreciation related to generators and other capital equipment was $2,910, $3,191 and $3,574 for the years ended 2019, 2018 and 2017.2020. As of December 31, 20192022 and 2018,2021, the net carrying value of generators and other capital equipment was $4,272$4,447 and $4,545.ATRICURE, INC. AND SUBSIDIARIESNOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)(In Thousands, Except Per Share Amounts)9.7. ACCRUED LIABILITIES 2022 2021 Accrued compensation and employee-related expenses $ 26,924 $ 30,990 Other accrued liabilities 3,301 2,686 Sales returns and allowances 2,797 2,416 Total $ 33,022 $ 36,092 10.originally effective February 23, 2018 and modified December 28, 2018 was further modified and amended on August 12, 2019 in connection with the SentreHEART acquisition. The Loan Agreement includes a $60,000 term loan, with an option to make available an additional $30,000 in term loan borrowings, and $20,000a $30,000 revolving line of credit; however the total combinedcredit. The Loan Agreement has a five year term, loan and revolving line of credit outstandingexpiring November 2026. cannot exceed $70,000 at any time prior to SVB’s consent. The term loan and revolving credit facility both mature or expire, as applicable, on August 1, 2024.Principal payments of the term loan are to be made ratably commencing March 1, 202124 months after inception through the loan’sloan's maturity date. IfAt the Company meets certain conditions, as specified byoption of the Loan Agreement,Company, the commencement of term loan principal payments may be deferred byextended an additional sixtwelve months. The term loan accrues interest at the greater of the Prime Rate or 5.00%, plus 0.75%1.25% and is subject to an additional 3.00% fee on the $60,000 term loan principal payableamount at maturity or upon acceleration or prepayment of the term loan.maturity. The Company is accruing the 3.00% fee over the term of the Loan Agreement, with $135 accrued$420 included in the outstanding loan balance as of December 31, 2019.2022. Additionally, the originalunamortized financing costs related to the term loan of $501$253 are netted against the outstanding loan balance in the Consolidated Balance Sheets and are amortized ratably over the term of the Loan Agreement. The August 2019 refinancing was treated as a debt modification.0.15%0.20% of the revolving line of credit, and any borrowings thereunder bear interest at the greater of the Prime Rate or 5.00%.Rate. Borrowing availability under the revolving credit facility is based on the lesser of $20,000$30,000 or a borrowing base calculation as defined by the Loan Agreement. The borrowing availability is also limited to allow total debt outstanding under the Loan Agreement to not exceed $70,000 at any time prior to SVB’s consent and further reduced by outstanding letters of credit (as specified). As of December 31, 2019,2022, the Company had 0no borrowings under the revolving credit facility and had borrowing availability of $8,750.approximately $28,750. Financing costs related to the revolving line of credit are included in other assets in the Consolidated Balance Sheets and amortized ratably over the twelve-month period of the annual fee.principal paymentsmaturities of long-term debt, excluding the term loan final fee, are projected as follows:11. LEASESThe Company adopted the new lease guidance on January 1, 2019 using the transition method provided by ASU 2018-11, “Leases (Topic 842): Targeted Improvements”. Under this method, the Company has applied the new requirements to leases that existed as of January 1, 2019, rather than at the earliest comparative period presented in the financial statements. Prior periods are presented under legacy ASC 840 lease guidance. As a result of the adoption, the Company recorded operating right-of-use assets andATRICURE, INC. AND SUBSIDIARIESNOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)(In Thousands, Except Per Share Amounts)2023 $ 3,333 2024 20,000 2025 20,000 2026 16,667 Total long-term debt, of which $3,333 is current and $56,667 is noncurrent $ 60,000 operating lease liabilities of approximately $1,884 and $2,189 as of January 1, 2019. The difference between the initial operating right-of-use asset and operating lease liability of $305 is accrued rent previously recognized under ASC 840. corporate offices, manufacturing and warehouse facilities and computer equipment. The Company has applied the practical expedient and does not separate lease components from nonlease components. The Company has applied the short-term lease recognition exemption and recognizes lease payments in profit or loss for leases that have a lease term of twelve months or less at commencement and do not include a renewal option whose exercise is reasonably certain. Short term lease expense is not significant during the twelve months ended December 31, 2019.The Company’s leases have remaining lease terms of one year to eleveneight years. Except for the operating lease acquired as part of the SentreHEART acquisition, optionsOptions to renew or extend leases beyond their initial term have been excluded from measurement of the ROU assets and lease liabilities for the majority of leases as exercise is not reasonably certain.for operating leases and finance leases is 3.5 years and 11.0 years as of December 31, 2019. The weighted averagethe discount rate used to measurefor the outstanding operating lease liabilities and finance lease liabilities is 5.9% and 7.0%reporting periods are as of December 31, 2019. In connection with the terms of the Company’s corporate headquarters lease, afollows:As of As of As of December 31, 2022 December 31, 2021 December 31, 2020 Operating Leases Weighted average remaining lease term (years) 4.4 3.6 3.2 Weighted average discount rate 4.60 % 4.69 % 5.68 % Finance Leases Weighted average remaining lease term (years) 7.6 8.6 9.7 Weighted average discount rate 6.92 % 6.91 % 6.91 % lessor in October 2015. The letter of credit2015, and is renewed annually and remains outstanding as of December 31, 2019. Year Ended Year Ended Year Ended December 31, 2022 December 31, 2021 December 31, 2020 Operating lease cost $ 1,133 $ 1,052 $ 1,237 Finance lease cost: Amortization of right-of-use assets 1,016 1,019 1,050 Interest on lease liabilities 735 792 844 Total finance lease cost $ 1,751 $ 1,811 $ 1,894
ATRICURE, INC. AND SUBSIDIARIESTwelve Months EndedDecember 31, 2019Operating lease cost$952Finance lease cost:Amortization of right-of-use assets998Interest on lease liabilities872Total finance lease cost$1,870iswas as follows:ATRICURE, INC. AND SUBSIDIARIESNOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)(In Thousands, Except Per Share Amounts)Year Ended Year Ended Year Ended December 31, 2022 December 31, 2021 December 31, 2020 Cash paid for amounts included in the measurement of lease liabilities: Operating cash flows for operating leases $ 845 $ 998 $ 1,236 Operating cash flows for finance leases 735 620 844 Financing cash flows for finance leases 899 792 664 Right-of-use assets obtained in exchange for lease obligations: Operating Leases — 3,752 1,421 Finance Leases 62 — 22 Early termination of operating lease — — 2,743 iswas as follows:December 31, 2019Operating LeasesOperating lease right-of-use assets$4,032Other current liabilities and current maturities of leases and long-term debt(1,465)Operating lease liabilities(2,796)Total operating lease liabilities$(4,261)Finance LeasesProperty and equipment, at cost$14,733Accumulated depreciation(4,197)Property and equipment, net$10,536Other current liabilities and current maturities of leases and long-term debt$(753)Finance lease liabilities(11,774)Total finance lease liabilities$(12,527)As of December 31, 2022 As of December 31, 2021 Operating Leases Operating lease right-of-use assets $ 3,787 $ 4,761 Other current liabilities and current maturities of debt and leases 1,147 861 Operating lease liabilities 3,095 4,068 Total operating lease liabilities $ 4,242 $ 4,929 Finance Leases Property and equipment, at cost $ 14,645 $ 14,607 Accumulated depreciation (7,109) (6,116) Property and equipment, net $ 7,536 $ 8,491 Other current liabilities and current maturities of debt and leases $ 992 $ 895 Finance lease liabilities 9,147 10,082 Total finance lease liabilities $ 10,139 $ 10,977 2019 are2022 were as follows:Operating Leases Finance Leases 2023 $ 1,160 $ 1,665 2024 1,164 1,689 2025 920 1,638 2026 592 1,671 2027 609 1,703 2028 and thereafter 259 4,824 Total payments $ 4,704 $ 13,190 Less imputed interest (462) (3,051) Total lease liabilities $ 4,242 $ 10,139 12.RoyaltyLicense Agreements. The Company has royalty agreements in place with termsa license agreement that include paymentrequires payments of royalties of 3% to 5% of specified product sales. One royaltyThe agreement remains in effect through 2023, while the other agreement remains in effectterminates the later of 20252023 or until expiration of the underlying patents or patent applications.applications, which is expected to occur after 2023. Parties to the royalty agreementslicense agreement have the right at any time to terminate the agreement immediately for cause. Royalty expense of $2,892, $2,715was $3,264, $3,124 and $2,323 was recorded as part of cost of revenue$2,596 for the years ended December 31, 2019, 20182022, 2021 and 2017.2020.certain vendorssuppliers in the ordinary course of business, generally with terms that allow cancellation.WhenA liability is established once management has assessed thatdetermines a loss is probable and an amount can be reasonably estimated, the Company records a liability in the Consolidated Financial Statements.estimated.requiresrequired the production of documents and answers to written interrogatories. The Company had no knowledge of the investigation prior to receipt of the CID. The Company maintains rigorous policies and procedures to promote compliance with the False Claims Act and other applicable regulatory requirements. The Company provided the USDOJ with documents and answers to the written interrogatoriesinterrogatories. In March 2021, USDOJ informed the Company that its investigation was based on a lawsuit brought on behalf of the United States and various state and local governments under the qui tam provisions of federal and certain state and local False Claims Acts. Although the USDOJ and all of the state and local governments declined to intervene, the relator continues to pursue the case. During the third quarter of 2022, the relator filed a Fourth Amended Complaint, which dropped allegations of off-label promotion and now alleges that the Company paid illegal kickbacks to healthcare providers in exchange for using or referring the Company’s products, in violation of the federal Anti-Kickback Statute and various comparable state and local laws. While the Company is contesting the case, it is not possible to predict when this matter may be resolved or what impact, if any, the outcome of this matter might have on our consolidated financial position, results of operations or cash flows.cooperating with its investigation. However, the Company cannot predict when the investigation will be resolved, the outcomesales of the investigation or its potential impact on the Company.cryoSPHEREThe Company acquired nContact Surgical, Inc. pursuant® product, was historically presented within open ablation revenue and is now a separately stated revenue product type. Valve revenue, historically presented as a separate product type revenue, is now included in open ablation revenue. Revenue amounts for comparative prior fiscal periods have been reclassified to a merger agreement dated October 4, 2015. The merger agreement provides for contingent consideration or “earnout” to be paid upon attaining specified regulatory approvals and clinical and revenue milestones. The merger agreement’s earnout provisions require the Company to deliver periodic earnout reports to a designated representative of former nContact stockholders. In responseconform to the reports delivered in and after February 2018, the Company received letters from the representative purporting to serve as “earnout objection statements” (as that term is defined in the merger agreement) and claim that for purposes of determining the commercial milestone payment, the Company should be including revenues of certain products that the Company has not included in its earnout statements. The representative is seeking indemnification under the merger agreement related to its claims. The Company has engaged with the representative regarding the earnout objection statements and disputes the basis of the representative’s claims.current period presentation.13. REVENUEThe Company adopted FASB ASC 606, “Revenue from Contracts with Customers” (ASC 606) using the modified retrospective method effective January 1, 2018. The adoption of ASC 606 did not have a material impact on the amount and timing of revenue recognized in the Consolidated Financial Statements.Revenue is generated primarily from the sale of medical devices. The Company recognizes revenue in an amount that reflects the consideration the Company expects to be entitled to in exchange for those devices when control of promised devices is transferred to customers. At contract inception, the Company assesses the products promised in its contracts with customers and identifies a performance obligation for each promise to transfer to the customer a product that is distinct. The Company’s devices are distinct and represent performance obligations. These performance obligations are satisfied and revenue is recognized at a point in time upon shipment or delivery of products. Sales of devices are categorized as follows: open ablation, minimally invasive ablation, appendage management and valve tools. Shipping and handling activities performed after control over products transfers to customers are considered activities to fulfill the promise to transfer the products rather than as separate promises to customers. Revenue includes shipping and handling revenue of $1,485, $1,236 and $1,090 in 2019, 2018 and 2017.Products are sold primarily through a direct sales force and through distributors in certain international markets. Terms of sale are generally consistent for both end-users and distributors, except that payment terms are generally net 30 days for end-users and net 60 days for distributors, with limited exceptions. The Company does not maintain any post-shipping obligations to customers. No installation, calibration or testing of products is performed by the Company subsequent to shipment in order to render products operational.Significant judgments and estimates involved in the Company’s recognition of revenue include the determination of the timing of transfer of control of products to customers and the estimation of a provision for returns. The Company considers the following indicators when determining when the control of products transfers to customers: (i) the Company has a right to payment in accordance with the shipping terms set forth in its contracts with customers; (ii) customers have legal title to products in accordance with shipping terms; (iii) the Company transfers physical possession of products either when the Company presents the products to a third party carrier for delivery to a customer (FOB shipping point) or when a customer receives the delivered goods (FOB destination); (iv) customers have the significant risks and rewards of ownership of products; and (v) customers have accepted products in connection with contractual shipping terms.In the normal course of business, the Company does not accept product returns unless a product is defective as manufactured. The Company establishes estimated provisions for returns based on the expected value method considering historical experience. The Company does not provide customers with the right to a refund. In connection with the acquisition of SentreHEART, the Company recognized an allowance for sales returns and refunds of $2,240 for transition to ASC 606 to reflect SentreHEART’s historical refund practices.The Company expects to be entitled to the total consideration for the products ordered by customers as product pricing is fixed according to the terms of customer contracts and payment terms are short. Payment terms fall within the one-year guidance for the practical expedient which allows the Company to forgo adjustment of the promised amount of consideration for the effects of a significant financing component. The Company excludes taxes assessed by governmental authorities on revenue-producing transactions from the measurement of the transaction price.Costs associated with product sales include commissions and royalties. Considering that product sales are performance obligations in contracts that are satisfied at a point in time, commission expense associated with product sales and royalties paid based on sales of certain products is incurred at that point in time rather than over time. Therefore, the Company applies the practicalATRICURE, INC. AND SUBSIDIARIESNOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)(In Thousands, Except Per Share Amounts)expedient and recognizes commissions and royalties as expense when incurred because the expense is incurred at a point in time and the amortization period is less than one year. Commissions are recorded as selling expense and royalties are recorded as cost of revenue.See Note 18 for disaggregatedUnited States revenue by geographic area andproduct type is as follows: 2022 2021 2020 Open ablation $ 86,119 $ 72,396 $ 65,301 Minimally invasive ablation 38,553 39,380 25,647 Pain management 39,974 22,787 11,315 Total ablation $ 164,646 $ 134,563 $ 102,263 Appendage management 112,555 94,568 66,981 Total United States $ 277,201 $ 229,131 $ 169,244 category.14.type is as follows: 2022 2021 2020 Open ablation $ 26,809 $ 23,194 $ 18,760 Minimally invasive ablation 5,986 6,409 6,171 Pain management 558 61 3 Total ablation $ 33,353 $ 29,664 $ 24,934 Appendage management 19,825 15,534 12,353 Total International $ 53,178 $ 45,198 $ 37,287 2022 2021 2020 United States $ 277,201 $ 229,131 $ 169,244 Europe 30,428 27,931 23,217 Asia 20,734 16,077 13,118 Other International 2,016 1,190 952 Total International 53,178 45,198 37,287 Total Revenue $ 330,379 $ 274,329 $ 206,531 local and foreign income tax returns in jurisdictions with varying statutes of limitations. Income taxes are computed usingThe Company uses the asset and liability method in accordance with FASB ASC 740, “Income Taxes”, under which deferred income taxes are provided for the temporary differences between the financial reporting basis and the tax basis of the Company’s assets and liabilities. Deferred taxes are measured using provisions of currently enacted tax laws. A valuation allowance against deferred tax assets is recorded when it is more likely than not that such assets will not be fully realized. The Company has recorded a fullCompany's valuation allowance againstoffsets substantially all its net deferred tax assets as it is more likely than not that the benefit of the deferred tax assets will not be recognized in future periods.ATRICURE, INC. AND SUBSIDIARIESNOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)(In Thousands, Except Per Share Amounts) 2022 2021 2020 Current tax expense Federal $ — $ — $ (26) State 142 42 78 Foreign 118 125 74 Total current tax expense 260 167 126 Deferred tax expense Federal $ (8,351) $ (30,925) $ (10,304) State (459) (4,803) (1,686) Foreign (1,636) (826) (3,071) Change in valuation allowance 10,454 36,575 15,049 Total deferred tax expense 8 21 (12) Total tax expense $ 268 $ 188 $ 114 2022 2021 Deferred tax assets: Net operating loss carryforwards $ 138,263 $ 137,920 Research and development credit carryforwards 13,205 11,269 Research and experimental expenditures 10,104 — Equity compensation 8,287 7,974 Finance and operating lease liabilities 3,395 3,700 Deferred interest 2,411 2,469 Inventories 1,896 2,434 Accruals and reserves 1,332 1,755 Other 506 587 Total deferred tax assets 179,399 168,108 Deferred tax liabilities: Intangible assets (9,278) (9,993) Right-of-use assets (2,626) (3,037) Property and equipment (2,568) (1,264) Total deferred tax liabilities (14,472) (14,294) Valuation allowance (164,918) (153,798) Net deferred tax assets $ 9 $ 16 The Company recorded $20,590Provisions enacted in the Tax Cut and Jobs Act of net deferred2017 related to the capitalization of research and experimental expenditures for tax assetspurposes became effective on January 1, 2022. These provisions require us to capitalize and offsetting valuation allowances as part of the SentreHEART acquisition.amortize research and experimental expenditures for tax purposes over five$340,079$331,169 which expire between 2023 and 2037 and $175,758 which have expirations between 2021 and 2038 and $80,101 which has no expiration as a result of the Tax Reform Act.expiration. The Company has state and local net operating loss carryforwards of $260,924 with varying expirations from 2020$322,819 which expire between 2023 to 2040.2042. A portion of the Company’s federal and state net operating loss carryforwards are subject to certain limitations under Internal Revenue Code Sections 382 and 383. The Company has federal research and development credit carryforwards of $8,168$13,205 which have expirationsexpire between 2023 and 2040.2042. Additionally, the Company has foreign net operating loss carryforwards of approximately $42,712$60,381 which have expirations between 2020 and 2028. On January 1, 2017, the Company adopted ASU 2016-09, “Improvements to Employee Share-Based Payment Accounting” and recognized $2,816no expiration.2019, 20182022, 2021 and 20172020 effective income tax rates differ from the federal statutory rate as follows: 2022 2021 2020 Federal tax at statutory rate 21.0 % $ (9,701) 21.0 % $ 10,580 21.0 % $ (10,088) Permanent differences (1.9) 876 (80.3) (40,439) (2.5) 1,214 Valuation allowance (22.6) 10,454 72.6 36,575 (31.3) 15,048 State income taxes 0.7 (317) (9.4) (4,760) 3.3 (1,607) Federal R&D credit 4.2 (1,936) (3.7) (1,878) 2.0 (985) Foreign income taxes (0.5) 215 0.7 344 4.5 (2,140) Federal deferred adjustments (1.5) 677 (0.5) (234) 2.8 (1,328) Effective tax rate (0.6) % $ 268 0.4 % $ 188 (0.2) % $ 114 loss(loss) income for domestic and international operations was $(28,002)$(38,008) and $(6,993)$(8,190) for 2019, $(13,443)2022, $55,666 and $(7,468)$(5,279) for 20182021 and $(19,409)$(43,218) and $(7,469)$(4,823) for 2017.$304$379 at December 31, 2019.2022. The Company does not consider these earnings as permanently reinvested and thus has recognized appropriate U.S.determined that no current and deferred taxes are required on such amounts.20162019 are open for examination. Generally, state and foreign income tax returns for periodsATRICURE, INC. AND SUBSIDIARIESNOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)(In Thousands, Except Per Share Amounts)20152018 are open for examination. However, taxing authorities have the ability to adjustaudit net operating loss and tax credit carryforwards from years prior to these periods. The Company has not recognized certain tax benefits because of the uncertainty of realizing the entire value of the tax position taken on income tax returns upon review by the taxing authorities.2019, 20182022, 2021 and 20172020 is presented below:The Internal Revenue Service completed its review of the Company’s 2014 federal income tax return in February 2017. In 2017, the Company also completed a detailed analysis of R&D credit carryforwards for the tax years 2008 through 2016. As a result of this analysis, as well as completion of the IRS audit of the 2014 credit, the Company has reduced both the R&D credit carryforward and related unrecognized tax benefits by $2,018. The Company’s increase for prior year tax positions relates to uncertain income tax benefits assumed pursuant to the SentreHEART acquisition. Historically, the Company did not have any interest and penalties accrued for unrecognized income tax benefits as a result of offsetting net operating losses. The Company has accrued interest and penalties associated with uncertain income tax benefits assumed pursuant to the SentreHEART acquisition as of December 31, 2019, and recognized interest and penalties within income tax expense. The amount is not significant.There are 0 amounts included in the balance of unrecognized tax benefits at December 31, 2018 and 2017 that, if recognized, would affect the effective tax rate. 2022 2021 2020 Balance at the beginning of the year $ 1,798 $ 1,798 $ 1,777 Increases (decreases) for prior year tax positions (36) — 21 Increases (decreases) for current year tax positions — — — Increases (decreases) related to settlements — — — Decreases related to statute lapse — — — Balance at the end of the year $ 1,762 $ 1,798 $ 1,798 20192022, 2021 and 2020 includes $1,777$1,762, $1,798 and 1,798 of tax benefits that, if recognized, would result in adjustments to other tax accounts, primarily deferred taxes and valuation allowance. The Company does not expect that its unrecognized tax benefits for research credits will significantly change within twelve months of December 31, 2019.15.2022.2019, 20182022, 2021 and 2017,2020, approximately 12.0%9.7%, 10.8%10.5% and 13.2%10.8% of the Company’s total net revenue was derived from its top 10ten customers. During 2019, 20182022, 2021 and 20172020 no individual customer accounted for more than 10% of the Company’s revenue.20192022 and 2018, 16.5%2021, 11.7% and 11.8%16.0% of the Company’s total accounts receivable balance was derived from its top ten customers. No individual customer accounted for more than 10% of the Company’s accounts receivable as of December 31, 20192022 and 2018.2021.2019, $28,0962022, $57,849 of the cash and cash equivalents balance was in excess of the FDIC limits.16.2019, 2018the year ended December 31, 2022, the Company made matching contributions of 50% on the first 8% of employee contributions to the 401(k) Plan. During the year ended December 31, 2021 and 20172020, the Company made matching contributions of 50% on the first 6% of employee contributions to the 401(k) Plan. The Company’s matching contributions expensed during 2019, 2018in 2022, 2021 and 20172020 were $1,915, $1,560$4,447, $2,651 and $1,367.$2,237. Additional amounts may be contributed to the 401(k) Plan at the discretion of the Company’s Board of Directors, however, 0no such discretionary contributions were made during 2019, 2018in 2022, 2021 or 2017.2020. The Company also provides retirement benefits for employees of AtriCure Europe B.V. and otherits foreign subsidiaries. Total contributions to foreign retirement plans for these employees were $248, $243$446, $349 and $205$244 in 2019, 20182022, 2021 and 2017.17.2020.ATRICURE, INC. AND SUBSIDIARIESNOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)(In Thousands, Except Per Share Amounts) may grant incentive stock options to Company employees and may grant restricted stock awards or restricted stock units (collectively RSAs), nonstatutory stock options, performance share awards (PSAs) or stock appreciation rights to Company employees, directors and consultants.consultants, and may grant incentive stock options to Company employees. The administrator (the Compensation Committee of the Board of Directors)Directors, as the administrator of the 2014 Plan, has the authority to determine the terms of any awards, including the number of shares subject to each award, the exercisability of the awards and the form of consideration. As of December 31, 2019, 11,9992022, 13,999 shares of common stock had been reserved for issuance under the 2014 Plan and 1,5782,183 shares were available for future grants.During 2019 and 2018, the Compensation Committee approved the grant of performance share awards to the Company’s named executive officers and certain other executive employees pursuant to the Company’s 2014 Plan. The form of award agreement for the PSAs (PSA Grant Form) provides, among other things, that (i) each PSA that vests represents the right to receive 1 share of the Company’s common stock; (ii) the PSAs vest based on the Company achieving specified performance measurements over a performance period of three years; (iii) the performance measurements include revenue CAGR as defined in the PSA Grant Form; (iv) threshold, target and maximum payout opportunities established for the PSAs will be used to calculate the number of shares that will be issuable when the award vests, which may range from 0% to 200% of the target amount; (v) any PSAs that are earned are scheduled to vest and be settled in shares of the Company’s common stock at the end of the performance period; and (vi) all or a portion of the PSAs may vest following a change of control or a termination of service by reason of death or disability (each as described in greater detail in the PSA Grant Form).With respect to the PSAs, the number of shares that vest and are issued to the recipient is based upon the Company’s performance as measured against the specified targets at the end of the three-year performance period as determined by the Compensation Committee. The Company estimated the fair value of the PSAs based on its closing stock price on the grant date and will adjust compensation expense over the performance period based on its estimate of performance target achievement.granted prior to 2018 under the 2014 Plan generally vest at a rate of 25% on the first anniversary date of the grant and ratably each month thereafter over the following three years. Restricted stock awards granted prior to 2018 generally vest between one year and four years from the date of grant. Stock options generally expire ten years from the date of grant.Activity underplans during 2019 was as follows:20182022 was as follows:Weighted Average Number of Remaining Shares Contractual Time-Based Stock Options Outstanding Term Outstanding at January 1, 2022 653 $ 25.53 Granted — — Exercised (159) 11.45 Forfeited (13) 55.33 Outstanding at December 31, 2022 481 $ 29.34 4.2 $ 9,437 Vested and expected to vest 479 $ 29.17 4.2 $ 9,436 Exercisable at December 31, 2022 411 $ 23.46 3.5 $ 9,352 2019, 20182022, 2021 and 20172020 was $1,985, $5,343$5,565, $27,318 and $5,121.$29,594. As a result of the Company’s full valuation allowance on its net deferred tax assets, 0no tax benefit was recognized related to the stock option exercises. The exercise price per share of each option is equal to the fair market value of the underlying share on the date of grant. For 2019, 20182022, 2021 and 2017, $1,202, $6,0122020, $1,816, $8,175 and $4,402$10,835 in cash proceeds from the exercise of stock options were included in the Company’s Consolidated Statements of Cash Flows as a result of the exercise of stock options. Flows.RSA PSA Shares Shares Restricted Stock Awards and Performance Share Awards Outstanding Outstanding Outstanding at January 1, 2022 628 $ 50.96 227 $ 64.27 Awarded 356 63.14 117 91.05 Released (362) 47.58 (116) 44.21 Forfeited (24) 57.45 (15) 55.17 Outstanding at December 31, 2022 598 $ 60.00 213 $ 90.70 2019, 20182022, 2021 and 20172020 was $23,479, $11,864$23,242, $40,510 and $6,235.$34,200. The total fair value of performance share awards vested during 2022, 2021 and 2020 was $5,185, $8,165 and $4,003. The Company issues registered shares of common stock to satisfy stock option exercisesATRICURE, INC. AND SUBSIDIARIESNOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)(In Thousands, Except Per Share Amounts)grants.The Company has awarded 450and performance options to its President and Chief Executive Officer. The options expire ten years from the date of grant and vest in increments of 25 shares when the volume adjusted weighted average closing price of the common stock of the Company as reported by NASDAQ (or any other exchange on which the common stock of the Company is listed) for 30 consecutive days equals or exceeds each of $10.00 per share, $12.50 per share, $15.00 per share, $17.50 per share, $20.00 per share, $25.00 per share, $30.00 per share, $35.00 per share and $40.00 per share. A Monte Carlo simulation was performed to estimate the fair values, vesting terms and vesting probabilities for each tranche of options. Expense calculated using these estimates was recognized over the estimated vesting terms. As of December 31, 2017, compensation costs related to non-vested performance options were fully recognized.The ESPP is available to eligible employees as defined in the plan document. (currently 15%(15%) ofto the lesser of the closing price of the Company’s common stock on the first trading day or the last trading day of the offering period. The offering period (currently six months) and the offering price are subject to change. Participants may not purchase a value of more than $25 of the Company’s common stock in a calendar year and may not purchase a value of more than 3 shares during an offering period. As of December 31, 2019,2022, there were 491184 shares available for future issuance under the ESPP.2019, 20182022, 2021 and 2017.2020. The expense was allocated as follows: 2022 2021 2020 Cost of revenue $ 1,868 $ 2,243 $ 1,425 Research and development expenses 4,544 4,206 3,530 Selling, general and administrative expenses 22,359 21,629 17,687 Total $ 28,771 $ 28,078 $ 22,642 2022 2021 2020 Restricted Stock Awards & Time-Based Stock Options $ 18,633 $ 18,727 $ 18,612 Performance Share Awards 8,731 8,095 2,921 ESPP 1,407 1,256 1,109 Total $ 28,771 $ 28,078 $ 22,642 20192022 there was $17,971$23,252 of unrecognized compensation costs related to non-vested stock options and restricted stock arrangements ($9971,160 relating to stock options and $16,974$22,092 relating to restricted stock). This cost is expected to be recognized over a weighted-average period of 1.81.4 years for stock options and 1.71.8 years for restricted stock. As of December 31, 20192022 there was $6,625$11,648 of unrecognized compensation costs related to non-vested performance share awards, and this cost is expected to be recognized over a weighted-average period of 1.61.7 years.calculatingdetermining compensation expense, the fair value of restricted stock awards, restricted stock units and performance share awards with a performance condition is based on the market value of the Company’s stock on the grant date of the awards.awards or subsequent modification (as applicable). The fair value of the options is estimated on the grant date using the Black-Scholes model includingmodel. No options were granted during 2022. Options granted in prior years included the following assumptions: 2021 2020 Range of risk-free interest rate 0.43-1.22% 0.30 - 1.73% Range of expected life of stock options (years) 5.3 to 5.7 5.2 to 5.7 Range of expected volatility of stock 40.00 - 43.00% 40.00 - 43.00% Weighted-average volatility 41.84% 41.54% Dividend yield 0.00% 0.00% trading historystock price over the expected option life. The risk-free interest rate assumption is based upon the U.S. treasury yield curve at the time of grant for the expected option life. The Company estimates the expected terms of options using historical employee exercise behavior.2022 2021 Stock price $39.94 - $69.59 $ 66.31 Expected term (years) 2.6 to 2.8 2.8 Company volatility 43.50 - 46.90% 42.10% Market index average volatility 90.30 - 92.00% 91.00% Market index average correlation 33.50 - 35.40% 31.50% Risk-free interest rate 1.40 - 2.70% 0.20% Dividend yield 0.00% 0.00% 2019, 20182022, 2021 and 20172020 was as follows:18. SEGMENT AND GEOGRAPHIC INFORMATIONThe Company evaluates reporting segments in accordance with FASB ASC 280, “Segment Reporting”. The Company develops, manufactures 2022 2021 2020 Stock options $ — $ 27.31 $ 15.25 Restricted stock awards 63.14 67.51 40.77 Performance share awards 91.05 89.36 38.42 sells devices designed primarily for the surgical ablation of cardiac tissue and systems designed for the exclusionunrealized losses on investments. Accumulated other comprehensive (loss) income consisted of the left atrial appendage. These devices are developed and marketed to a broad basefollowing, net of medical centers globally. Management considers all such sales to be part of a single operating segment. Revenue attributed to geographic areas is based on the location of the customers to whom products are sold.Revenue by geographic area was as follows:United States revenue by product type was as follows:International revenue by product type was as follows:The Company’s long-lived assets are located primarily in the United States, except for $1,228 as of December 31, 2019 and $1,296 as of December 31, 2018, which are located primarily in Europe.2022 2021 2020 Total accumulated other comprehensive (loss) income at beginning of period $ (948) $ 312 $ (158) Unrealized (losses) gains on investments Balance at beginning of period $ (887) $ 54 $ 100 Other comprehensive (loss) income before reclassifications (2,739) (941) (70) Amounts reclassified from accumulated other comprehensive (loss) income to interest income (72) — 24 Balance at end of period $ (3,698) $ (887) $ 54 Foreign currency translation adjustment Balance at beginning of period $ (61) $ 258 $ (258) Other comprehensive (loss) income before reclassifications (774) (768) 555 Amounts reclassified from accumulated other comprehensive (loss) income to other (expense) income 437 449 (39) Balance at end of period $ (398) $ (61) $ 258 Total accumulated other comprehensive (loss) income at end of period $ (4,096) $ (948) $ 312 ATRICURE, INC. AND SUBSIDIARIESNOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)(In Thousands, Except Per Share Amounts)19. SELECTED QUARTERLY FINANCIAL DATA (UNAUDITED)Amounts may not sum to consolidated totals for the full year due to rounding. Basic and diluted net loss per share is computed independently for each of the quarters presented. Therefore, the sum of the quarterly per share amounts will not necessarily equal the total for the year.SCHEDULE IIVALUATION AND QUALIFYING ACCOUNTS(1) In connection with the acquisition of SentreHEART, the Company recognized an allowance for sales returns and refunds of for transition to ASC 606 to reflect SentreHEART’s historical refund practices, and recorded an offsetting valuation allowance for deferred tax assets.
ITEMITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSUREITEM and Senior Vice President and Chief Financial Officer (the Principal Accounting and Financial Officer), has evaluated the effectiveness of the Company’s disclosure controls and procedures as defined in Rule 13(a) – 15(e) of the Securities Exchange Act of 1934 (Exchange Act), as of the end of the period covered by this report. Based on this evaluation, we concluded that, as of the end of the period covered by this report, our disclosure controls and procedures were effective in providing reasonable assurance that information required to be disclosed by us in the reports we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s forms and rules, and the material information relating to the Company is accumulated and communicated to management, including the President and Chief Executive Officer and the Senior Vice President and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosures.20192022 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.2019.2022. No matter how well designed, because of inherent limitations in all control systems, internal control over financial reporting may not prevent or detect misstatements should they occur. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the control procedures may deteriorate. In making this assessment, the Company’s management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission in Internal Control-Integrated Framework (2013). Based on such assessment, management has concluded that the Company’s internal control over financial reporting was effective as of December 31, 2019.2022.consolidated financial statementsConsolidated Financial Statements included in this Annual Report on Form 10-K and, as part of its audit, has issued an attestation report on the effectiveness of the Company’s internal control over financial reporting. The attestation report can be found on the following page as part of this Item 9A.Mason, Ohio2019,2022, based on criteria established in Internal Control — Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). In our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2019,2022, based on criteria established in Internal Control — Integrated Framework (2013) issued by COSO.2019,2022, of the Company and our report dated February 24, 2020,22, 2023, expressed an unqualified opinion on those financial statements.24, 2020
ITEMITEM 9B. OTHER INFORMATIONEffective February 21, 2020, the Company’s Board of Directors re-constituted its committees as follows:Audit: Sven A. Wehrwein (Chair), Mark R. Lanning, Daniel P. Florin, B. Kristine Johnson (Ms. Johnson’s service on the Audit Committee will end effective April 1, 2020.)Compensation: Mark R. Lanning (Chair), Mark A. Collar, B. Kristine Johnson, Karen N. PrangeCompliance, Quality and Risk: Regina E. Groves (Chair), Sven A. Wehrwein, Robert S. White, Daniel P. FlorinNominating and Corporate Governance: Mark A. Collar (Chair), Scott W. Drake, Robert S. White, Karen N. PrangeStrategy: Robert S. White (Chair), Regina E. Groves, B. Kristine JohnsonPARTITEMItemitem with respect to the Company’s Directors is incorporated by reference to thecontained in our definitive proxy statement (the “Proxy Statement”) for our 20202023 Annual Meeting of Stockholders to be filed withunder the Securitiesheading “Proposal One—Election of Directors” and Exchange Commission within 120 days after the end of 2019 (the “Proxy Statement”).ITEM 11. EXECUTIVE COMPENSATIONItemitem with respect to the Company’s Executive Officers is contained in the Proxy Statement under the heading “Management” and is incorporated herein by referencereference.Statement.Statement under the heading “Corporate Governance Guidelines—Code of Conduct” and is incorporated herein by reference.2019.2022.
to be issued upon
exercise of
outstanding options,
warrants and rights (1)
exercise price of
outstanding options,
warrants and rights (2)
available for future issuance
under equity compensation
plans (excluding securities
reflected in column (a)) Plan Category (a) (c) Equity compensation plans approved by security holders (3) 1,291,762 $ 29 2,183,428 Equity compensation plans not approved by security holders — — — Total 1,291,762 $ 29 2,183,428 performance stock options and performance shares as of December 31, 2019.2022. remaining information required by this Itemitem with respect to security ownership of certain beneficial owners and management is contained in the Proxy Statement under the heading “Security Ownership of Certain Beneficial Owners and Management” and is incorporated herein by reference to the Proxy Statement.ITEMreference.Itemitem with respect to director independence is contained in the Proxy Statement under the heading “Corporate Governance and Board Matters – Independence of the Board” and is incorporated herein by reference to the Proxy Statement.ITEMreference.Itemitem with respect to audit fees, tax fees and the Audit Committee’s pre-approval policies and procedures are contained in the Proxy Statement under the heading “Proposal Two-Ratification of Appointment of Independent Registered Public Accounting Firm” and is incorporated herein by reference to the Proxy Statement.PARTPART IVITEM(1) The financial statements required by Item 15(a) are filed in Item 8 of this Form 10-K.
10.1210.1310.1410.1210.1510.1310.16#10.141410.15Exhibit No. Description 10.17# 10.18# 10.19# 14 21
Exhibit No.DescriptionSIGNATURES24, 202024, 2020M. Andrew WadeAngela L. WirickM. Andrew WadeAngela L. WirickMENWOMEN AND WOMENMEN BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Michael H. Carrel and M. Andrew Wade,Angela L. Wirick, her or his attorney-in-fact, with the power of substitution, for her or him in any and all capacities, to sign any and all amendments to this Form 10-K, and to file the same, with exhibits thereto and other documents in connection therewith, with the U.S. Securities and Exchange Commission, granting unto said attorneys-in-fact, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done therewith, as fully to all intents and purposes as she or he might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact, and any of them or her or his substitute or substitutes, may do or cause to be done by virtue thereof.24, 2020.22, 2023.Scott W. DrakeB. Kristine JohnsonScott W. DrakeB. Kristine JohnsonScott W. DrakeB. Kristine JohnsonChairmanChair of the Board/s/ M. Andrew WadeM. Andrew WadeM. Andrew WadeChief Financial Officer(Principal Accounting and Financial Officer)/s/ Daniel P. FlorinDaniel P. FlorinDaniel P. FlorinDirector/s/ B. Kristine JohnsonB. Kristine JohnsonB. Kristine JohnsonDirector/s/ Mark R. LanningMark R. LanningMark R. LanningDirectorDeborah H. Telman Deborah H. Telman Director /s/ Sven A. Wehrwein /s/ Maggie Yuen Maggie Yuen Maggie Yuen Director