As of March 23, 2012 was 84,608,826.6, 2015, there were outstanding 171,715,551 shares of the registrant’s common stock, par value $0.001 per share.
Documents Incorporated by Reference
Portions of the registrant’s definitive Proxy Statement for its 2015 Annual Meeting of Stockholders are incorporated by reference into Part III of this Annual Report on Form 10- K where indicated. Such Proxy Statement will be filed with the Securities and Exchange Commission no later than 120 days after the end of the registrant’s fiscal year ended December 31, 2014.
THERAPEUTICSMD, INC.
ANNUAL REPORT ON FORM 10-K
Fiscal Year Ended December 31, 2014
TABLE OF CONTENTS
vitaMedMD®, TherapeuticsMD®, and BocaGreenMD® are registered trademarks of our company. This Annual Report also contains trademarks and trade names of other companies.
This Annual Report includes market and industry data that we obtained from periodic industry publications, third-party studies and surveys, government agency sources, filings of public companies in our industry, and internal company surveys. Industry publications and surveys generally state that the information contained therein has been obtained from sources believed to be reliable. Although we believe the foregoing industry and market data to be reliable at the date of the report, this information could provide to be inaccurate as a result of a variety of matters.
Statement Regarding Forward-Looking Information
This Annual Report on Form 10-K (the “Report”)contains forward-looking statements that involve substantial risks and uncertainties. For example, statements regarding our operations, financial position, business strategy, product development, and other plans and objectives for future operations, and assumptions and predictions about future product development and demand, research and development, marketing, expenses and sales are all forward-looking statements. These statements may be found in the items of this Annual Report entitled “Business” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” as well as in this Annual Report generally. These statements are generally accompanied by words such as “intend,” “anticipate,” “believe,” “estimate,” “potential(ly),” “continue,” “forecast,” “predict,” “plan,” “may,” “will,” “could,” “would,” “should,” “expect,” or the negative of such terms or other comparable terminology.
We have based these forward-looking statements on our current expectations and projections about future events. We believe that the assumptions and expectations reflected in such forward-looking statements are reasonable, based on information available to us on the date hereof, but we cannot assure you that these assumptions and expectations will prove to have been correct or that we will take any action that we may presently be planning. These forward-looking statements are inherently subject to known and unknown risks and uncertainties. Actual results or experience may differ materially from those expected or anticipated in the forward-looking statements. Factors that could cause or contribute to such differences include, but are not limited to, research and product development uncertainties, regulatory policies and approval requirements, competition from other similar businesses, market and general economic factors, and the other risks discussed in Item 1A of this Annual Report. This discussion should be read in conjunction with the consolidated financial statements and notes thereto included in this Annual Report.
We have identified some of the important factors that could cause future events to differ from our current expectations and they are described in this Annual Report in the section entitled “Risk Factors” that you should review carefully. Please consider our forward-looking statements in light of those risks as you read this Annual Report. If one or more of these or other risks or uncertainties materialize, or if our underlying assumptions prove to be incorrect, actual results may vary materially from what we project. We do not undertake to update any forward-looking statements or to publicly announce the results of any revisions to any statements to reflect new information or future events or developments.
| Item 1. | Business |
Overview
Our Company
We are a women’s health care product company focused on creating and commercializing products targeted exclusively for women. Currently, we are focused on conducting the clinical trials necessary for regulatory approval and commercialization of advanced hormone therapy pharmaceutical products. The current drug candidates used in our clinical trials are designed to alleviate the symptoms of and reduce the health risks resulting from menopause-related hormone deficiencies, including hot flashes, osteoporosis, and vaginal discomfort. We are developing these hormone therapy drug candidates, which contain estradiol and progesterone alone or in combination, with the aim of demonstrating equivalent clinical efficacy at lower doses, thereby enabling an enhanced side effect profile compared with competing products. Our drug candidates are created from a platform of hormone technology that enables the administration of hormones with high bioavailability alone or in combination. In addition, we manufacture and distribute branded and generic prescription prenatal vitamins, as well as over-the-counter, or OTC, vitamins.
We have obtained U.S. Food and Drug Administration, or FDA, acceptance of our Investigational New Drug, or IND, applications to conduct clinical trials for four of our hormone therapy drug candidates: TX-001HR, our oral combination of progesterone and estradiol; TX-002HR, our oral progesterone alone; TX-003HR, our oral estradiol alone; and TX-004HR, our vaginal suppository estradiol alone. We are currently conducting phase 3 clinical trials for TX-001HR and TX-004HR. In July 2014, we temporarily suspended enrollment in the phase 3 clinical trial for TX-002HR, and in October 2014 we temporarily stopped the trial in order to update the phase 3 protocol based on discussions with the FDA. We have no current plans to conduct clinical trials for TX-003HR.
Throughout this Annual Report, the terms “we,” “us,” “our,” “Therapeutics,“TherapeuticsMD,” or “our Company” referscompany” refer to TherapeuticsMD™,TherapeuticsMD, Inc., a Nevada corporation, together with itsand unless specified otherwise, include our wholly owned subsidiary, vitaMedMD®, LLC, a Delaware limited liability company (“VitaMed”). Unless otherwise stated or unless the context otherwise requires, the description of our business set forth below is provided on a combined basis, taking into account our subsidiary, VitaMed.
Hormone Therapy Market
The menopause hormone therapy market includes two major components: an FDA-approved drug market and a non-FDA approved drug market supplied by compounding pharmacies. On November 27, 2013, the Drug Quality and Security Act became law and the FDA was given additional oversight over compounding pharmacies. We believe FDA-approved products are easily measured and monitored, while non-FDA approved hormone therapy drug products, typically referred to as bioidenticals, when produced and sold by compounding pharmacies are not easily measured or monitored. We estimate the non-FDA approved compounded bioidentical hormone therapy sales of estradiol and progesterone products by compounding pharmacies approximate $1.5 billion per year and the FDA-approved market approximates $4.2 billion per year. Our phase 3 clinical trials are intended to establish an indication of the safety and efficacy of our bioidentical drug candidates at specific dosage levels. We intend our hormone therapy drug candidates, if approved, to provide hormone therapies with well characterized safety and efficacy profiles that can be consistently manufactured to target specifications. This would provide an alternative to the non-FDA approved compounded bioidentical market. This is based on our belief that our drug candidates will offer advantages in terms of demonstrated safety and efficacy consistency in the hormone dose, lower patient cost as a result of insurance coverage, and improved access as a result of availability from major retail pharmacy chains than custom order or formulation by individual compounders.
Pipeline of our Hormone Therapy Drug Candidates
TX-001HR
TX-001HR, our combination estradiol and progesterone drug candidate, is undergoing clinical trials for the treatment of moderate to severe vasomotor symptoms due to menopause, including hot flashes, night sweats, sleep disturbances, and vaginal discomfort, for post-menopausal women with an intact uterus. The hormone therapy drug candidate is chemically identical to the hormones that naturally occur in a woman’s body, namely estradiol and progesterone, and is being studied as a continuous-combined regimen, in which the combination of estrogen and progesterone are taken together in one product daily. If approved by the FDA, we believe this would represent the first time a combination product of estradiol and progesterone biologically identical, or bioidentical, to the estradiol and progesterone produced by the ovaries, would be approved for use in a single combined product. According to Symphony Health Solutions, the total FDA-approved market for menopause-related combination estrogen/progestin was approximately $659 million in U.S. sales for the 12 months ended December 31, 2011, Company management determined2014.
TX-002HR
TX-002HR is a natural progesterone formulation for the treatment of secondary amenorrhea without the potentially allergenic component of peanut oil. The hormone therapy drug candidate is chemically identical to the hormones that VitaMed would becomenaturally occur in a woman’s body. If approved by the sole focusFDA, we believe TX-002HR will be similarly effective to traditional treatments, but may be effective at lower dosages. According to Symphony Health Solutions, the total FDA-approved market for oral progestin was approximately $400 million in U.S. sales for the 12 months ended December 31, 2014.
TX-003HR
TX-003HR is a natural estradiol formulation. This hormone therapy drug candidate is chemically identical to the hormones that naturally occur in a woman’s body. We currently do not have plans to further develop this hormone therapy drug candidate. According to Symphony Health Solutions, the total FDA-approved market for oral estradiol was approximately $131 million in U.S. sales for the 12 months ended December 31, 2014.
TX-004HR
TX-004HR is a vaginal suppository estradiol drug candidate for the treatment of vulvar and vaginal atrophy, or VVA, in post-menopausal women with vaginal linings that do not receive enough estrogen. We believe that our drug candidate will be at least as effective as the traditional treatments for VVA because of an early onset of action with less systemic exposure, inferring a greater probability of dose administration to the target tissue, and it will have an added advantage of being a simple, easier to use dosage form versus traditional VVA treatments. According to Symphony Health Solutions, the total FDA-approved market for VVA treatment was approximately $1.3 billion in U.S. sales for the 12 months ended December 31, 2014.
Preclinical Development
Based upon leveraging our hormone platform technology, we have seven preclinical projects that include development of our proposed progesterone-alone and combination estradiol and progesterone products in a topical cream and transdermal patch form, which we refer to as TX-005HR and TX-006HR, respectively, and transdermal patch form, which we refer to as TX-007HR and TX-008HR, respectively. We recently completed a pilot pharmacokinetic, or PK, study of TX-005HR in eight patients that tested the topical administration on the upper arm of 50 mcg of estradiol and 25 mg of progesterone. The results of the CompanyPK study suggest that topical formulations of estradiol and services previously performed relativeprogesterone may be possible using our proprietary solubilized forms of the compounds. We intend to file an IND with respect to TX-005HR and TX-006HR during the second half of 2015 and then commence phase 1 clinical trials. We may in the future engage with a financing partner to advance our topical cream and transdermal patch projects. We are also evaluating various other indications for our hormone technology, including oral contraception, treatment of preterm birth, and premature ovarian failure. According to Symphony Health Solutions, the total FDA-approved menopause-related market for estrogen alone and in combination was approximately $3.7 billion in U.S. sales for the 12 months ended December 31, 2014.
Current Products
As we continue the clinical development of our hormone therapy drug candidates, we continue to manufacture and distribute our prescription and OTC product lines, consisting of prenatal vitamins, iron supplements, vitamin D supplements, and natural menopause relief products under our vitaMedMD® brand name and duplicate formulations of some of our prescription prenatal vitamin products, also referred to as “generic” formulations, under our BocaGreenMD® Prena1™ name. All of our prenatal vitamins are gluten-, sugar-, and lactose-free. We believe our product attributes result in greater consumer acceptance and satisfaction than competitive products while offering the highest quality and patented ingredients.
Industry and Market
Health Care and Pharmaceutical Market
According to EvaluatePharma® World Preview report, the pharmaceutical industry experienced an unprecedented decline in worldwide prescription drug sales in 2012. Worldwide prescription drug sales fell by 1.6% to $714 billion in 2012, with the United States representing about 36% of the market. Loss of patent protection on a number of blockbuster brands and fiscal austerity affecting Eurozone countries (compounded by a weak euro compared to the aforementioned Licensing AgreementU.S. Dollar) contributed to this unprecedented contraction. In total, $38 billion of sales were discontinued. Unless otherwise statedlost as a result of expired patent protection, including drugs such as Lipitor and Plavix. According to the report, this anomaly is not expected to continue and sustained sales growth should start returning at the end of 2013 at an average rate of 3.8% per annum between 2012 and 2018.
In terms of numbers of new drug approvals in the United States, 2012 was the best year since 1997 when the Pfizer drug, Lipitor, was approved. But perhaps more important than the large number of approvals in 2012 (45 versus 35 in 2011), quality was also significantly better than in previous years, as judged by analysts’ consensus expectations of sales five-year post launch.
Women’s Health Care Market
According to the GBI Research (a provider of industry-leading business intelligence solutions on a global basis) report “Women’s Health Therapeutic Market through 2018,” the women’s health therapeutics market is one of the most attractive markets in the global pharmaceutical industry. Hormone therapy, gynecological disorders, and musculoskeletal disorders in women are the prime areas of focus in the women’s health therapeutics market. The women’s health therapeutics market in the United States was valued at $12.5 billion in 2011. Revenues are projected to increase to $15.1 billion in 2018 at a compound and growth rate of 2.7%. This can be attributed to the launch of new drug molecules.
Hormone Therapy Market
Menopause is the spontaneous and permanent cessation of menstruation, which naturally occurs in most women between the ages of 40 and 58. It is defined as the final menstrual period and is confirmed when a woman has not had her period for 12 consecutive months. These symptoms are caused by the reduced levels of circulating estrogen as ovarian production shuts down. The symptoms include hot flashes, night sweats, sleep disturbances, and vaginal dryness. According to Symphony Health Solutions, prescriptions for hormone therapy products for the treatment of menopause symptoms or unlessprevention of osteoporosis generated total sales of over $4.2 billion on over 35 million prescriptions for the context otherwise requires,12 months ended December 31, 2014. Oral hormone therapy accounted for $2.0 billion on 23 million prescriptions over the descriptionsame time period.
Prescriptions for menopausal hormone therapy in the United States dropped significantly following the Women’s Health Initiative, or WHI, study in 2002, which found that subjects using estrogen plus synthetic progestin had, among other things, a greater incidence of coronary heart disease, breast cancer, stroke, and pulmonary embolism. A number of additional studies regarding the benefits and risks of hormone therapy have been conducted over the last decade since the WHI results were first published. In general, recommendations for hormone therapy use are to be judged on an individual basis, and the FDA recommends that women with moderate to severe menopausal symptoms who want to try menopausal hormone therapy for relief use it for the shortest time needed and at the lowest effective dose.
There were approximately 41.7 million women in the United States between the ages of 45 and 64 in 2010, projected to increase slightly (2.8%) to 42.9 million in 2015 and to approximately 44.3 million in 2040, according to the 2010 National Census population figures. These women are the target market for hormone therapy to treat menopausal related symptoms.
Hormone Therapy Products
Estrogen (with or without a progestin) is the most effective treatment of vasomotor symptoms due to menopause according to the North American Menopause Society, or NAMS. According to Symphony Health Solutions, sales of total FDA-approved oral, transdermal, and suppository estrogen (with and without a progestin) hormone therapy products were approximately $3.7 billion for the 12 months ended December 31, 2014, an approximately 7% increase over the prior year. The three primary hormone therapy products are estrogen, progestin, and combination of estrogen and progestin, which are produced in a variety of forms, including oral tablets or capsules, skin patches, gels, emulsion, or vaginal suppositories and creams.
Estrogen-Only Therapies
Estrogen therapies are used to treat vasomotor symptoms due to menopause that are a direct result of the decline in estrogen levels associated with ovarian shutdown at menopause. Estrogen therapy has been used to manage these symptoms for more than 50 years. Estrogen is a generic term for any substance, natural or synthetic, that exerts biological effects characteristic of estrogenic hormones, such as estradiol. Based upon the age demographic for all women receiving prescriptions for estrogen therapy and the average age range during which women experience vasomotor symptoms, we believe that estrogen is primarily used for the treatment of vasomotor symptoms, but also prescribed for the prevention of osteoporosis.
Estrogen-only therapy, or ET, is used primarily in women who have had a hysterectomy and have undergone a surgical menopause, as those women do not require a progestin to protect the uterine endometrium from proliferation. Approximately 433,000 women undergo a hysterectomy each year in the United States according to the United States Centers for Disease Control and Prevention. Sales of FDA-approved ET were approximately $3.0 billion for the 12 months ended December 31, 2014, according to Symphony Health Solutions.
ET is also used for the treatment of VVA, which has a variety of indications, including vaginal dryness, vaginal itching and irritation, painful intercourse, painful urination, and other symptoms. Sales of FDA-approved ET for VVA were approximately $1.3 billion for the 12 months ended December 31, 2014, according to Symphony Health Solutions.
ET is also approved for the prevention of osteoporosis. Multiple studies conducted on various estrogen compositions, including studies published in the Journal of the American Medical Association in 2002, Osteoporosis International in 2000, The Lancet in 2002, Maturitas in 2008, and Climacteric in 2005, demonstrated efficacy based on increases in bone mineral density. Epidemiological and some fracture prevention studies, such as the study published in the New England Journal of Medicine in 1980, also have demonstrated a decrease in bone fractures as a result of ET.
Progestin-Only Therapies
Progestins include the naturally occurring hormone progesterone and a number of synthetic progestin compounds that have progestational activity. These agents are used for a variety of indications and conditions, but most often, progestins are used either alone or in combination with an estrogen for hormonal contraception and to prevent endometrial hyperplasia from unopposed estrogen in hormone therapy. Progestins alone are also used to treat women with secondary amenorrhea in order to create withdrawal bleeding in these women who have not had regular menses. Progestins are also used to treat dysfunctional uterine bleeding and endometriosis. Progesterone has also been used to prevent threatened or recurrent pregnancy loss and for the prevention of preterm birth. Progestins have also been used in fertility treatments. Progestins have also been used as a palliative measure for metastatic endometrial carcinoma and in the treatment of renal and breast carcinoma.
Estrogen/Progestin Combination Products
Progestins are used in combination with estrogen in post-menopausal women with uteruses to avoid an increase in the incidence of endometrial hyperplasia, which is a condition caused by chronic use of estrogen alone by a woman with a uterus and is associated with an increased incidence of uterine, or endometrial, cancer. Studies have shown that, after one year, the incidence of endometrial hyperplasia is less than 1% in women taking estrogen/progestin combinations, in contrast to up to 20% in women taking estrogen alone. In accordance with FDA recommendations, doctors typically recommend that a menopausal or post-menopausal woman who has a uterus take estrogen plus a progestin, either as a combination drug or as two separate drugs. Symphony Health Solutions estimates that sales of FDA-approved estrogen/progestin combinations were approximately $659 million in the United States for the 12 months ended December 31, 2014.
Limitations of Existing Estrogen/Progestin Therapies
The most commonly prescribed progestin is a synthetic progestin (medroxyprogesterone acetate), which can cause some women to experience painful vaginal bleeding, breast tenderness, and bloating and may reduce cardio-protective benefits potentially associated with estrogen therapy by limiting the estrogen’s ability to raise high-density lipoprotein cholesterol, or good cholesterol, and low-density lipoprotein, or bad cholesterol.
A widely prescribed naturally occurring progesterone is known as Prometrium® (progesterone USP), sold by AbbVie Inc. Natural progesterone is used in combination with estrogen for hormone therapy; however, we believe there are currently no FDA-approved hormone therapy combination products with natural progesterone.
Prenatal Vitamin Market
According to the American Pregnancy Association, approximately six million women become pregnant each year, resulting in approximately four million births. Of these women, over 75% receive prenatal care during the first trimester, and most doctors encourage taking a prenatal vitamin as the recommended standard of care. Prenatal vitamins are usually dietary supplements intended to be taken before and during pregnancy and during postnatal lactation that provide nutrients recognized by various health organizations as helpful for a healthy pregnancy outcome.
There are hundreds of prenatal vitamins available, with both prescription and OTC choices. According to Symphony Health Solutions’ PHAST 2.0 Report, during the 12 months ended November 30, 2014, approximately 8.2 million prescriptions for prenatal vitamins were issued in the United States resulting in total sales of approximately $399 million, with sales between branded and generic products split nearly evenly. According to the 2012 Gallup Target Market Report on Prenatal Vitamins, supplement use has been fairly constant overall between 2008 and 2011. However, shifts have occurred in terms of types used, with the trend toward OTC prenatal vitamins and away from prescription prenatal vitamins. During this same period, the use of OTC products surpassed the use of prescription products, largely driven by increased use among women currently pregnant.
Our Business Model
We are a women’s health care product company focused on creating and commercializing products targeted exclusively for women, including products specifically for pregnancy, childbirth, nursing, pre-menopause, menopause, and post-menopause. We intend to use our current prescription and OTC product lines, consisting of prenatal vitamins, iron supplements, vitamin D supplements, and natural menopause relief products, as the foundation of our business set forth below is providedplatform. If approved and commercialized, our hormone therapy drug candidates will allow us to enter the $4.2 billion hormone therapy market, based on a combined basis, taking into account our newly-acquired wholly owned subsidiary, VitaMed.
Our current product line is marketed and sold shares of its Common Stock (not including any shares issued upon the exercise of options and/or warrants or upon the conversion of any convertible securities) or the five-day average closing bid price immediately preceding the date of conversion. At December 31, 2011, the outstanding principle balance of the Secured Notes was $500,000 each.
Our sales model focuses on the “4Ps”: patient, provider, pharmacist, and payor. We market both over-the-counter (“OTC”) and sell our current products primarily through a direct national sales force of approximately 34 full-time professionals that calls on health care providers in the OB/GYN market space, as well as through our website directly to consumers. In addition, our products allow health care providers to offer an alternative to patients to meet their individual nutritional and financial requirements related to co-payment and cost-of-care considerations and help patients realize cost savings over competing products. We also believe that our combination of branded, generic, and OTC lines offers physicians, women, and payors cost-effective alternatives for top-quality care. We supply our prescription nutritional supplements, drugs, medical foodsproducts to consumers through retail pharmacies. We market our OTC products either directly to consumers via our website and other medicalphone sales followed by direct shipment to their homes or offices or through physicians who then re-sell them to their patients. Our fully staffed customer care center uses current customer relationship management software to respond to health care providers, pharmacies, and consumers via incoming and outgoing telephone calls, e-mails, and live-chat. We also facilitate repeat customer orders for our non-prescription products through pharmacies and our web-site with the recommendation of physicians by creating a unique value propositions for patients, physician/providers and insurance payors.
As health market. As we continue our product development efforts for both new products and refinements to existing products, we are also seeking proprietary ingredients and formulations that can be exclusively licensed or patented for use in women’s healthcare that will further differentiate our products from the competition.
We prenatal vitamins are gluten-, sugar-, and lactose-free. | ||
Our Growth Strategy
Our goal is to become the women’s health care company recommended by health care providers to all patients by becoming the new standard in women’s health with a complete line of products, all under one quality brand. Key elements of our strategy to achieve this goal are as follows:
Exclusive Focus on Women’s Health Issues. We plan to focus exclusively on women’s health issues to enable us to build long-term relationships with women as they move through their life cycles of birth control, pregnancy, child birth, and pre- and post- menopause.
Focus on Hormone Therapy Products. We plan to focus on the development, clinical trials, and commercialization of hormone therapy products designed to (1) alleviate the systems of and reduce the health effects resulting from menopause-related hormone deficiencies, including hot flashes, osteoporosis, and vaginal dryness, and (2) demonstrate equivalent clinical efficacy at lower doses, enabling an enhanced side effect profile compared with competing products.
Penetrate Bioidentical Market with FDA-approved Products. As we are not aware of any current FDA-approved bioidentical hormone therapy combination products, we believe that our hormone therapy drug candidate for combined estradiol and progesterone, if approved by the FDA, will provide a safer and more effective alternative to non-FDA approved compounded bioidentical hormone therapy products, at a lower price to patients due to insurance coverage.
Marketing Emphasis. We plan to maintain an emphasis on large group OB/GYN practices that provide opportunities to reach large patient bases and that are receptive to the data and savings we provide.
Multiple Distribution Channels. We are pursuing multiple distribution channels, including physicians and pharmacies, through our sales force and our website.
Geographical Expansion. We plan to expand our geographic market and sales team to cover the entire country by increasing our current 37 sales territories to 60 sales territories by the end of 2016.
Introducing New Products. We continue to develop our hormone therapy drug candidates which currently consist of a (1) bioidentical oral combination of progesterone and estradiol product, and (2) a suppository VVA estradiol product.
Our Current Product Lines
We offer a wide range of products targeted for women’s health specifically associated with pregnancy, child birth, nursing, post-child birth, and menopause, including prescription and OTC prenatal vitamins, iron supplements, vitamin D supplements, and natural menopause relief products under our vitaMedMD brand name and duplicate formulations of some of our prescription prenatal vitamin products, referred to as “generic” formulations, under our BocaGreenMD Prena1 name.
FortheyearsendedDecember 31,2014,2013and2012,approximately98%,98%,and97%, respectively, of our consolidated revenue was generated by our prenatal vitamin products. Our prenatal vitamin products are marketed as either OTC products or prescription products. Our OTC and prescription prenatal vitamin products are generally variations of the same product with modifications in formulation and marketing. The primary significant difference between our OTC and prescription prenatal vitamin products is the source of payment. Purchasers of our OTC prenatal vitamin products pay for the product directly while purchasers of our prescription prenatal vitamin products pay for the product via third-party payor. For both our OTC and prescription prenatal vitamin products, we employ the same sales force and marketing team to sell and market our products.
In March 2012, we launched our first prescription prenatal vitamin, vitaMedMD Plus Rx, with subsequent launches of our second prescription prenatal vitamin, vitaMedMD One Rx, in April 2012 and our third prescription prenatal vitamin, vitaMedMD RediChew™ Rx, in May 2012. In the fourth quarter of 2012, we launched our BocaGreenMD Prena1 line of prescription prenatal vitamins, which included three prescription prenatal vitamins that were generic formulations of our vitaMedMD-branded prescription prenatal vitamins. In the first quarter of 2014, we introduced a new prescription prenatal vitamin product under our branded vitaMedMD name as vitaPearl and under our generic Prena1 name as Prena1 Pearl, which features a unique, proprietary combination of FOLMAX™, FePlus™, and pur-DHA™. Our product line is detailed below.
vitaPearl™
vitaPearl is our newest prescription product and a complete prenatal vitamin in one tiny pearl. vitaPearl provides 40% more folic acid as Flowmax™, our proprietary modified-release folic acid than the leading prescription prenatal vitamin. vitaPearl delivers 14 key vitamins and minerals plus 200 mg of DHA, as par-DHA™, providing comprehensive support for a women and her body whether she is planning a pregnancy, pregnant, or nursing.
vitaMedMD Plus
vitaMedMD Plus is a once-daily, two pill combo pack that contains a complete multivitamin with 16 essential vitamins and minerals and 300 mg of plant based docosohexaenoic acid, or DHA, and is vegan and Kosher certified.
vitaMedMD One Prenatal Multivitamin
vitaMedMD One is a single-dose daily multivitamin that provides 14 vitamins and minerals and 200 mg of plant-based DHA. Each convenient, easy-to-swallow softgel features 975 mcg of folic acid.
vitaMedMD Plus Rx Prenatal Multivitamin
vitaMedMD Plus Rx is a once-daily, two pill combo prescription product containing one prenatal vitamin tablet with Quatrefolic®, the fourth generation folate, and one plant-based DHA 300 mg capsule. Quatrefolic® is a registered trademark of Gnosis S.P.A. All minerals, including iron, zinc, and copper, are chelated to improve absorption.
vitaMedMD One Rx Prenatal Multivitamin
vitaMedMDOne Rx is a prescription product with a single-dose daily multivitamin that provides 14 vitamins and minerals, Quatrefolic®, and 200 mg of plant-based DHA.
vitaMedMD RediChew®Rx Prenatal Multivitamin
vitaMedMDRediChew®Rx is a prescription, easy-to-chew, small, vanilla-flavored tablet containing Quatrefolic®, vitamin D3, vitamin B2, vitamin B6 and vitamin B12. We believevitaMedMD RediChew Rx is an excellent option for women who have difficulty swallowing tablets or softgels, or are experiencing nausea and morning sickness.
vitaMedMD Iron 21/7
vitaMedMD Iron 21/7 is an iron replacement supplement with a three weeks-on/one week-off dosing schedule intended to maximize absorption and enhance tolerability. It is formulated with 150 mg of chelated iron to help improve tolerability and limit typical side effects associated with iron replacements. Each easy-to-swallow single tablet serving also includes 800 mcg of folic acid, plus vitamins C and B12, and succinic acid to aid in absorption.
vitaMedMD Menopause Relief
vitaMedMD Menopause Reliefoffers a natural treatment for hot flashes, night sweats, and mood disturbances. Each single tablet dosage delivers 120 mg of Lifenol®, a well-studied female hops extract recognized for its potency and support in alleviating hot flashes, plus plant phytoestrogens. It also includes calcium and vitamin D3 for added bone support.
vitaMedMD Vitamin D3 50,000 IU and Vitamin D3 2,000 IU
vitaMedMD Vitamin D3 50,000 IU and Vitamin D3 2,000 IU are dietary supplements provided in a small, easy-to-swallow gel capsule that help replenish and maintain beneficial levels of vitamin D in the body. Sustaining adequate levels of vitamin D in the body is essential to bone health, enhancing the absorption of calcium and phosphorus. Vitamin D3, also known as cholecalciferol, is considered the most preferred form of vitamin D as it is the most active form of the nutrient. We believevitaMedMDVitamin D3 50,000 IU andVitamin D3 2,000 IU are ideal for pregnant, breastfeeding, and menopausal women to sustain adequate levels of vitamin D.
| 8 |
BocaGreenMD Prena1 Pearl
BocaGreenMD Prena1 Pearl is our newest prescription product and a complete prenatal vitamin in one tiny pearl. vitaPearl provides 40% more folic acid than the leading prescription prenatal vitamin. vitaPearl delivers 14 key vitamins and minerals plus 200 mg of DHA, providing comprehensive support for a women and her body whether she is planning a pregnancy, pregnant, or nursing.
BocaGreenMD Prena1 Chew
BocaGreenMD Prena1 Chew is a prescription, single daily easy-to-chew, vanilla-flavored tablet well-suited for women planning a pregnancy and those with difficulty swallowing tablets or capsules or when nausea or morning sickness make taking tablets or capsules difficult.
Our Hormone Therapy Drug Candidates
We have obtained FDA acceptance of our IND applications to conduct clinical trials for four of our proposed products: TX-001HR, our oral combination of progesterone and estradiol; TX-002HR, our oral progesterone alone; TX-003HR, our oral estradiol alone; and TX-004HR, our estradiol alone vaginal suppository.
TX-001HR
TX-001HR, our combination estradiol and progesterone drug candidate, is undergoing clinical trials for the treatment of moderate to severe vasomotor symptoms due to menopause, including hot flashes, night sweats, sleep disturbances, and vaginal dryness, for post-menopausal women with an intact uterus. The hormone therapy drug candidate is chemically identical to the hormones that naturally occur in a woman’s body, namely estradiol and progesterone, and is being studied as a continuous-combined regimen, in which the combination of estrogen and progesterone are taken together in one product daily. If approved by the FDA, we believe this would represent the first time a combination product of estradiol and progesterone bioidentical to the estradiol and progesterone produced by the ovaries, would be approved for use in a single combined product.
On September 5, 2013, we initiated the REPLENISH trial, a multicenter, double-blind, placebo-controlled, phase 3 study of TX-001HR in postmenopausal women with an intact uterus. The study is designed to evaluate the efficacy of TX-001HR for the treatment of moderate to severe vasomotor symptoms due to menopause and the endometrial safety of TX-001HR. Patients are assigned to one of five treatment arms, four active and one placebo, and receive study medication for 12 months. The primary endpoint for the reduction of endometrial hyperplasia is an incidence of endometrial hyperplasia of less than 1% at 12 months, as determined by endometrial biopsy. The primary endpoint for the treatment of moderate to severe vasomotor symptoms is the mean change of frequency and severity of moderate to severe vasomotor symptoms at weeks four and 12 compared to placebo, as measured by the number and severity of hot flushes. Only subjects experiencing a minimum daily frequency of seven moderate to severe hot flushes at screening are included in the vasomotor symptoms analysis, while all subjects are included in the endometrial hyperplasia analysis. The secondary endpoints include reduction in sleep disturbances and improvement in quality of life measures, night sweats and vaginal dryness, measured at 12 weeks, six months and 12 months. We intend to enroll approximately 1,750 patients at approximately 100 sites. We currently anticipate that enrollment in the REPLENISH Trial will be completed in the first-half of 2015 and that results of the trial will be reported in the middle of 2016. Based on such timeline and successful reports of the trial, we would anticipate filing a New Drug Application, or NDA, for TX-001HR during the first-half of 2016 and that such NDA would be approved by the FDA during the first-half of 2017.
We previously conducted a PK study of TX-001HR to demonstrate that our drug candidate is bioequivalent to the reference listed drug based on the criterion that the 90% confidence interval on the test-to-reference ratio is contained entirely within the interval 80% to 125%. The study compared our combined capsule TX-001HR of 2 mg estradiol and 200 mg of progesterone to 2 mg of Estrace®and 200 mg of Prometrium®.
The study compared the mean plasma concentrations for free estradiol between TX-001HR and Estrace® in 62 female test subjects. When the results of a single dose-fed study were compared over 48 hours by the test drug versus reference drug, the ratio was 0.93 with the standard deviation within the subject being 0.409 for an upper 95% confidence bound of -0.089. The maximum plasma concentration levels of free estradiol showed that the drug -versus -reference drug ratio was 0.88 with the standard deviation within the subject being 0.344 for an upper 95% confidence bound of -0.040 over 48 hours.
The study also compared the mean plasma concentrations for progesterone between TX-001HR and Prometrium® in 62 female test subjects. When the results were compared over 48 hours of the test that the drug-versus-reference drug, the ratio was 1.05 with the standard deviation within the subject being 0.956 for an upper 95% confidence bound of -0.542. The maximum plasma concentration levels of progesterone showed drug versus reference drug ratio as 1.16 with the standard deviation within the subject being 1.179 for an upper 95% confidence bound of -0.785 over 48 hours.
We believe these data are sufficient to demonstrate the bioequivalence of TX-001HR to Estrace® and Prometrium® based on the criteria for demonstrating bioequivalence established in connection with the study. On September 5, 2013, we began enrollment of the REPLENISH trial, a multicenter, double-blind, placebo-controlled, phase 3 study of TX-001HR in postmenopausal women with an intact uterus. The study is designed to evaluate the efficacy of TX-001HR for the treatment of moderate to severe vasomotor symptoms due to menopause and the endometrial safety of TX-001HR. Patients are assigned to one of five treatment arms, four active and one placebo, and receive study medication for 12 months. The primary endpoint for the reduction of endometrial hyperplasia is an incidence of endometrial hyperplasia of less than 1% at 12 months, as determined by endometrial biopsy. The primary endpoint for the treatment of moderate to severe vasomotor symptoms is the mean change of frequency and severity of moderate to severe vasomotor symptoms at weeks four and 12 compared to placebo, as measured by the number and severity of hot flushes. Only subjects experiencing a minimum daily frequency of seven moderate to severe hot flushes at screening are included in the vasomotor symptoms analysis, while all subjects are included in the endometrial hyperplasia analysis. The secondary endpoints include reduction in sleep disturbances and improvement in quality of life measures, night sweats and vaginal dryness, measured at 12 weeks, six months and 12 months. We intend to enroll approximately 1,750 patients at approximately 100 sites. We currently anticipate that enrollment in the REPLENISH Trial will be complete in 2015 and that results of the trial will be reported in 2016.
TX-002HR
TX-002HR is a natural progesterone formulation for the treatment of secondary amenorrhea without the potentially allergenic component of peanut oil. The hormone therapy drug candidate is chemically identical to the hormones that naturally occur in a woman’s body. We believe it will be similarly effective to traditional treatments, but may demonstrate efficacy at lower dosages.
In January 2014, we began recruitment of patients in the SPRY trial, a phase 3 clinical trial designed to measure the safety and effectiveness of TX-002HR in the treatment of secondary amenorrhea. During the first two quarters of 2014, the SPRY trial encountered enrollment challenges because of Institutional Review Board, or IRB, approved clinical trial protocols and FDA inclusion and exclusion criteria. In July 2014, we temporarily suspended enrollment and in October 2014 we stopped the SPRY trial in order to update the phase 3 protocol based on discussions with the FDA. We intend to update the phase 3 protocol to, among other things, target only those women with secondary amenorrhea due to polycystic ovarian syndrome and to amend the primary endpoint of the trial. We believe that the updated phase 3 protocol, if approved by the FDA, will allow us to mitigate the enrollment challenges in, and shorten the duration of, the SPRY trial. However, there can be no assurance that the FDA will approve the updated phase 3 protocol that we intend to propose.
TX-003HR
TX-003HR is a natural estradiol formulation. This hormone therapy drug candidate would be chemically identical to the hormones that naturally occur in a woman’s body. We currently do not have plans to further develop this hormone therapy drug candidate.
TX-004HR
TX-004HR is a vaginal suppository estradiol drug candidate for the treatment of VVA in post-menopausal women with vaginal linings that do not receive enough estrogen. We believe that our drug candidate will be at least as effective as the traditional treatments for VVA because of an early onset of action with less systemic exposure, inferring a greater probability of dose administration to the target tissue, and it will have an added advantage of being a simple, easier to use dosage form versus traditional VVA treatments.
We initiated the REJOICE trial, a multicenter, double-blind, placebo-controlled phase 3 clinical trial during the third quarter of 2014 to assess the safety and efficacy of TX-004HR for the treatment of moderate to severe dyspareunia, or painful intercourse, as a symptom of VVA due to menopause. We are conducting a single 12 week study, evaluating three different doses of estradiol: 4 mcg, 10 mcg and 25 mcg versus placebo. The FDA has to date noted that in order to approve a drug based on a single trial, the trial would need to show statistical significance at a 0.01 level. The study has been designed to include four primary endpoints: the reduction of vaginal pH levels to less than 5.0, an increase in superficial cells, a decrease in parabasal cells and the improvement of dyspareunia. If approved, the 4 mcg formulation would represent a lower effective dose than the currently available VVA therapies approved by the FDA. The trial is designed to enroll approximately 700 patients across approximately 100 sites. We currently anticipate that enrollment in the REJOICE trial will be complete during the second quarter of 2015 and that results of the trial will be reported during the third quarter of 2015. Based on such timeline and successful reports of the trial, we would anticipate filing an NDA for TX-004HR during the fourth quarter of 2015 and that such NDA would be approved by the FDA during the fourth quarter of 2016.
In August 2013, we initiated a phase 2 clinical trial for VVA, designed to measure the effect of TX-004HR on certain clinical endpoints, including a study candidate’s pH levels, vaginal cytology, and most bothersome symptom of VVA, out of the symptoms identified in FDA guidance. Based upon our phase 2 results, we believe we have a rapidly acting product that differs substantially from the reference listed drug Vagifem® sold by Novo Nordisk.
Preclinical Development
Based upon leveraging our hormone platform technology, we have seven preclinical projects that include development of our proposed progesterone-alone and combination estradiol and progesterone products in a topical cream and transdermal patch form, which we refer to as TX-005HR and TX-006HR, respectively, and transdermal patch form, which we refer to as TX-007HR and TX-008HR, respectively. We recently completed a pilot pharmacokinetic, or PK, study of TX-005HR in eight patients that tested the topical administration on the upper arm of 50 mcg of estradiol and 25 mg of progesterone. The results of the PK study suggest that topical formulations of estradiol and progesterone may be possible using our proprietary solubilized forms of the compounds. We intend to file an IND with respect to TX-005HR and TX-006HR during the second half of 2015 and then commence phase 1 clinical trials. We may in the future engage with a financing partner to advance our topical cream and transdermal patch projects. We are also evaluating various other indications for our hormone technology, including oral contraception, treatment of preterm birth, and premature ovarian failure. According to Symphony Health Solutions, the total FDA-approved menopause-related market for estrogen alone and in combination was approximately $3.7 billion in U.S. sales for the 12 months ended December 31, 2014.
Sales Strategy
Although our direct national sales force is similar to that of a traditional pharmaceutical company in that sales representatives are callingcall on OB/GYN practices to provide education and sampling, we believe our sales representatives are more customer centric in their sales approach. Our sales representatives offerapproach by offering physicians more than just differences in our products from the competition; they are also able to offer an array of partnering opportunities to promote efficiency and cost savings. Our OPERA technology allows us to collect and analyze critical data from various inputs allowing us to provide significant value to patients, providers and payors.
Our national rollout strategy ishas been to focus first on the largest metropolitan areas in the United States. In order to accelerate the sales rampramp-up in a new territory, we employ a national sales/large practice sales effort to identify key practices in new or expanding markets. Concurrent with our provider sales effort, we work with both commercial insurance and Medicaid insurance payors for partnerships in which the payor can support the prescription and/or recommendation of our products for the benefit of the patient, physician, and payor, with an end result of providing better outcomes for all three constituents.
At the forefront of our sales approach is the philosophy that the physician should recommend or prescribe products based only on what is best for theirthe patient. In general, a better outcome is achieved by providing patients with the best products and care at the best value. HavingWe believe having an assortment of high qualityhigh-quality product options that can be recommended or prescribed by both the physician and payor is the foundation of providing valuable options to the patient.
We believe our sales force has developed strong relationships and partnerships in the OB/GYN market to sell our current products. We have also established relationships with some of the largest OB/GYN practices in their respective markets. By delivering additional products through the same sales channel, we believe we can leverage our already deployed assets to increase our sales and achieve profitability. We intend to leverage and grow our current marketing and sales organization to commercialize our drug candidates in the United States assuming the successful completion of the FDA regulatory process.
Online Commerce
A vast majority of our OTC product sales are completed online. The Internet has continued to increase its influence over communication, content, and commerce. We believe several factors will contribute to this continuing increase, including convenience, expanded range of available products targeted forand services, improved security and electronic payment technology, increased access to broadband Internet connections and widespread consumer confidence and acceptance of the Internet as a means of commerce.
Retail Commerce
The vast majority of our prescription product sales are completed through the traditional pharmacy distribution network. Although online and mail order pharmacy commerce continues to grow, the majority of products are still purchased directly by the consumer locally at traditional stores. As this division of our business expands, we will continue to employ strategies that help us reduce inefficiencies in this channel and develop relationships that allow our products to be differentiated from the competition.
Competition
Pharmaceutical Industry
The pharmaceutical industry is subject to intense competition and is characterized by extensive research efforts and rapid technological change. Competition in our industry occurs in a variety of areas, including developing and bringing new products to market before others, developing new technologies to improve existing products, developing new products to provide the same benefits as existing products at lower cost, and developing new products to provide benefits superior to those of existing products. Most major pharmaceutical companies, as well as numerous specialty pharmaceutical companies, sell products in the women’s health sector of the pharmaceutical industry, which is comprised of products designed for post-pubescent females and associatedis generally considered very fragmented. There are many companies focused on the women’s health sector of the pharmaceutical industry that have significantly greater financial and other resources than we do, including generic manufacturers, drug compounding pharmacies, and large pharmaceutical companies. In addition, academic and other research institutions could be engaged in research and development efforts for the indications targeted by our products.
Hormone Therapy Market
The menopause hormone therapy market includes two major components: an FDA-approved drug market and a non-FDA approved drug market supplied by compounding pharmacies. On November 27, 2013, the Drug Quality and Security Act became law and the FDA was given additional oversight over compounding pharmacies. We believe FDA-approved products are easily measured and monitored, while non-FDA approved hormone therapy drug products, typically referred to as bioidenticals, when produced and sold by compounding pharmacies, are not easily measured or monitored. Our phase 3 clinical trials are intended to establish an indication of the safety and efficacy of our bioidentical drug candidates at specific dosage levels. We intend our hormone therapy drug candidates, if approved by the FDA, to provide hormone therapies with pregnancy, child birth, nursing, post birthwell characterized safety and efficacy profiles that can be consistently manufactured to target specifications. This would provide an alternative to the non-FDA approved compounded bioidentical market. This aim is based on our belief that our drug candidates will offer advantages in terms of demonstrated safety and efficacy consistency in the hormone dose, lower patient cost as a result of insurance coverage and improved access as a result of availability from major retail pharmacy chains rather than custom order or formulation by individual compounders.
TX-001HR, our combination estradiol and progesterone drug candidate, is undergoing clinical trials for the treatment of moderate to severe vasomotor symptoms due to menopause. OurThe combination of estradiol and progesterone for the treatment of moderate to severe vasomotor symptoms due to menopause for postmenopausal women with an intact uterus is comprised of two components: the FDA-approved drug market and the non-FDA-approved compounded drug market. According to the PHAST Prescription Monthly by Symphony Health Solutions, the U.S. FDA-approved market for menopause-related combination estradiol and progesterone was approximately $659 million for the 12 months ended December 31, 2014. The largest competitors in the FDA-approved market are PREMARIN cream (Pfizer), generic estradiol and progestins (TEVA), FemHRT (Warner Chilcott), Angeliq (Bayer), and Activella (Novo Nordisk), with sales of PREMARIN cream constituting a majority of such sales. None of the current FDA-approved drugs for the treatment of moderate to severe vasomotor symptoms due to menopause is bioidentical to the estradiol and progesterone produced by the ovaries. Based on various reports, included data recently presented at the North American Menopause Society, including Knowledge, Use, and Prescribing of Custom-Compounded Bioidentical Hormones for Menopausal Women: It’s Not What You Think, by JoAnn V. Pinkerton, et al., we estimate that U.S. sales of non-FDA -approved compounded combination estradiol and progesterone products approximate $1.5 billion per year. The market for non-FDA -approved compounded hormone therapy products is generally considered very fragmented because the products are prepared and sold by individual compounding pharmacies. We believe that TX-001HR, if approved by the FDA, would represent the first time a bioidentical combination product of estradiol and progesterone would be approved for use in a single combined product.
TX-002HR, our progesterone only drug candidate, is for the treatment of secondary amenorrhea. According to PHAST Prescription Monthly by Symphony Health Solutions, the U.S. progesterone alone oral market for 2014 was approximately $400 million. The largest competitors in the progestin alone oral market are Aygestin tablets (TEVA), Provera tablets (Pfizer), and Prometrium capsules (AbbVie). We believe that TX-002HR, if approved by the FDA, would provide for treatment of secondary amenorrhea without the potentially allergenic component of peanut oil found in existing products.
TX-004HR, our vaginal suppository estradiol drug candidate, is undergoing clinical trials for the treatment of VVA in post-menopausal women with vaginal linings that do not receive enough estrogen. According to the PHAST Prescription Monthly by Symphony Health Solutions, the U.S. market for vaginal suppository estradiol for the treatment of VVA in post-menopausal women was approximately $1.3 billion for the 12 months ended December 31, 2014. Approximately $1.15 billion of such sales were by three products currently on the market: PREMARIN cream (Pfizer), ESTRACE cream (Warner Chilcott), and Vagifem tablets (Novo Nordisk). We believe that TX-004HR, if approved by the FDA, will be at least as effective as the existing treatments for VVA because of an early onset of action with less systemic exposure inferring a greater probability of dose administration to the target tissue, and it will have an added advantage of being a simple, easier to use dosage form versus traditional VVA treatments.
Prenatal Vitamin Market
The prenatal vitamin market is highly fragmented, with dozens of companies selling hundreds of competitive products. Prenatal vitamin products are marketed as either OTC product line availableproducts or prescription products, with many companies marketing their products through our website includesboth channels. According to Symphony Health Solutions’ PHAST 2.0 Report, during the 12 months ended December 31, 2014, approximately 8.2 million prescriptions for prenatal vitamins DHA, iron supplements, calcium supplements, Vitamin D supplements, women’s multivitamins, natural (non-hormonal) menopause relief,were issued in the United States resulting in total sales of approximately $399 million. According to the 2012 Gallup Target Market Report on Prenatal Vitamins, supplement use has been fairly constant overall between 2008 and scar reduction creams. In March 2012, we launched our first prescription-only2011. However, shifts have occurred in terms of types used, with the trend toward OTC prenatal vitamins and away from prescription prenatal vitamins. We estimate that the U.S. OTC prenatal vitamin vitaMedMD™ Plus Rx, and planmarket is approximately $800 million a year.
Seasonality
The specialty pharmaceutical industry is not subject to launchseasonal sales fluctuation.
Products in Development
We introduced our second prescription-only prenatal vitamin, vitaMedMD™ One Rx, in April 2012. Our product line is detailed below.
Raw Materials for Our Products
We acquire all raw materials and ingredients for our proprietary products are purchased from a group of third-party suppliers specializing in raw material manufacturing, processing, and specialty distribution. Our manufacturers maintainprimary manufacturer maintains multiple supply and purchasing relationships throughout the raw materials marketplace to provide an uninterrupted supply of product to meet our manufacturing requirements.
Availability of and Dependence Upon Suppliers
We currently obtain over 90%approximately 97% of our vitaMedMD and BocaGreenMD products from Lang; therefore,Lang Pharma Nutrition, or Lang, a full-service, private label and corporate brand manufacturer specializing in premium health benefit driven products, including medical foods, nutritional supplements, beverages, bars, and functional foods in the dietary supplement category. As a result, we are dependent upon themon Lang and its subcontractors for the manufacture of most of our products. We believe the terms of our agreements with Lang are competitive with other suppliers and manufacturers. Although we anticipate continuing our relationship with Lang, we believe that we could obtain similar terms with other suppliers to provide the same services. We have experienced no difficulties in obtaining the products we need in the amounts we require and do not anticipate those issues in the future.
Manufacturing of Our Products
Our vitamin products are manufactured and regulated byin accordance with the same FDA quality standards (Controls UsedFDA’s current Good Manufacturing Practice, or cGMPs, for Manufacturing, Processing, Packing, or Holding Dietary Supplements for FDA 21 CFR Part 110/111 CGMP Regulations (“CFR 111”)) and current good manufacturing practices (“cGMP”) as prescription nutritional therapies.dietary supplements. In addition, we conduct two additional un-required certificates of analysis on every lot to ensure quality and we employ an outside third party to enforce rigorous quality audits.
All of our manufacturing is performed by third partythird-party manufacturers. Over 90%In addition to manufacturing substantially all of our manufacturing is handled byproducts, Lang a full-service, private label and corporate brand manufacturer specializing in premium health benefit driven™ products, including medical foods, nutritional supplements, beverages, bars, and functional foods in the dietary supplement category. Langalso provides a variety of additional services to us, including development processes, prototype development, raw materials sourcing, regulatory review, and packaging production. At present, we believe our relationship with Lang is excellent, and we intend to continue to use themLang as our third partythird-party manufacturer for most of our products. In the event our relationship with Lang terminates for any reason, there are a number of other manufacturers available to us; accordingly, management is of the opinionus. Accordingly, we do not believe that such termination would not have a material adverse effect on our business.
We use third-party manufacturers to source key raw materials and manufacture and package our products. The FDA must approve the manufacturing facility for compliance with the FDA’s cGMP regulations before an NDA for a new drug is approved. Accordingly, we intend to engage only those third-party contract manufacturers that have consistently shown the ability to satisfy these requirements for our hormone therapy drug candidates.
Quality Control for Ourour Products
A quality assurance team establishes process controls and documents and tests every stage of the manufacturing process to ensure we meet product specifications and that our finished dietary supplementsproducts contain the correct ingredients, purity, strength, and composition in compliance with FDA regulations. We test incoming raw materials and finished goods to ensure they meet or exceed FDA and U.S. Pharmacopeia standards, including quantitative and qualitative assay and microbial and heavy metal contamination.
Our manufacturers’ quality and production standards are designed to meet or exceed the latestcurrent FDA regulations. To ensure the highest quality, our manufacturing operations are audited by AIB International, Inc. (“AIB”), or AIB, among others, for independent cGMP certification. AIB is an independent, not-for-profit organization that offers programs and services to augment and support the work of regulatory officials around the country, including standards development, product testing and certification, and onsite audits and inspections. The manufacturing facilities we primarily use are also ISO 9001 certified, which is a family of standards related to quality management systems and are designed to help organizations ensure they meet the needs of customers. In addition, our manufacturers are hazard analysis critical control point (“HACCP”) certified which is a systematic preventive approach for food and pharmaceutical safety that addresses physical, chemical and biological hazards as a means of prevention rather than finished product inspection.
Distribution of our Products
We use a variety of distribution channels dependent upon product type. OTC products are sold directlyWe sell our prescription prenatal vitamins to consumer via the Internet and phone sales and the products are shipped directly from the Company to the consumer’s home. In a few instances, the Company sells product to physicians who then sell the product directly to their patients. Our prescription products are sold to the patient directlypatients through their pharmacy.pharmacies. Since the launch of our prescription products, in addition to third-party logistics providers, we use some of the same majornational and regional distributors as other pharmaceutical companies, including Cardinal, McKesson, AmerisourceBergen, H.D. Smith, and AmerisourceBergen.
Customer Service
Our goal is 100% customer satisfaction by consistently delivering superior customer experiences;experiences before, during, and after the sale. To achieve this goal, we maintain a fully staffed-staffed customer care center for both inbound and outboundthat uses current customer service using the most current technologiesrelationship management software to respond to customershealth care providers, pharmacies, and consumers and accept orders for non-prescription products via incoming and outgoing telephone calls, e-mails, and live-chat. We believe our customer service initiatives allow us to establish and maintain long-term customer relationships and facilitate repeat visits and purchases.
Our representatives receive regular training so that they can effectively and efficiently field questions from current and prospective customers and are also trained not to answer questions that should be directed to a customer’s physician. Having a quality customer care center allows our representatives to provide an array of valuable data in the areas of sales, market research, quality assurance, lead generation, and customer retention.
Our Return Policy
We sell our prescription products through third-party logistics providers, major distributors, and pharmacies, all of whom may return a product within six months prior to or up to 12 months after the expiration date of the product. Once customers buy a product from the pharmacy, the product may not be returned. Customers may return or exchange our OTCnon-prescription products for any reason by returning the product within thirty (30)30 days of receipt. We will refund the entire purchase price, less shipping. The customer is responsible for the cost of returning the products to us, except in cases wherein which the product is being returned because of a defect or an error made in our order fulfillment. If the purchased product exceeded a thirty-day30-day supply, the unused product must be returned to receive the full refund. All unopened OTC products may be exchanged for different products; the customer will be responsible for the difference in price if the replacement product is more expensive or we will refund the difference if the replacement product is less expensive.
Our Quality Guarantee
We proudly stand behind the quality of our products. OurWe believe our guarantee makes it easy, convenient, and safe for customers to purchase our products. Under our quality guarantee, we:
help health care providers and payors by delivering information on patient compliance and satisfaction;
ensure a safe, secure online shopping experience through our encrypted website.
We value frequent communication with and feedback from our customers in order to continue to improve our offerings and services.
Research and Development
Our product development programs are concentrated in the area of advanced hormone therapy pharmaceutical products. We engage in programs to provide alternatives to both the FDA-approved and the non-FDA -approved compounded bioidentical market for hormone therapy. Our programs seek to bring new products to market in unique delivery systems or formats that enhance the effectiveness, safety, and reliability of existing hormone therapy alternatives.
We intend our hormone therapy drug candidates, if approved, to provide an alternative to the non-FDA -approved compounded bioidentical market based on our belief that our drug candidates will offer advantages in terms of proven safety, efficacy, and stability, lower patient cost as a result of insurance coverage, and improved access as a result of availability from major retail pharmacy chains rather than custom order or formulation by individual compounders.
Our research and development expenses were $43.2 million in 2014, $13.6 million in 2013, and $4.5 million in 2012.
Intellectual Property
Our success depends, in part, on our ability to obtain patents, maintain trade secret protection, and operate without infringing the proprietary rights of others. Our intellectual property portfolio is one of the means by which we attempt to protect our competitive position. We rely primarily on a combination of know-how, trade secrets, patents, trademarks, and contractual restrictions to protect our products and to maintain our competitive position. We are constantlydiligently seeking ways to protect our intellectual property through registrationsvarious legal mechanisms in relevant jurisdictions.
We have severaldeveloped hormone products using our SYMBODA® platform, which is our advanced hormone therapy platform to enable delivery of bio-identical hormones through a variety of dosage forms and administrative routes.
In addition to numerous pending patent applications, as of December 31, 2014, we had four issued patents, including:
| • | one method patent that relates to our OPERA® information technology platform, which is owned by us and is a U.S. jurisdiction patent with an expiration date in 2029; and |
Subsequent to December 31, 2014, one additional patent was issued related to our combination progesterone and estradiol formulations, which are owned by us and are U.S. jurisdiction patents with an expiration date in 2032.
As of December 31, 2014, we had filed 24 non-provisional and 13 provisional patent applications with the U.S. Patent and Trademark Office, (the “USPTO”). We intendor the USPTO, with respect to file additionalour hormone therapy drug candidates, and 55 international patent applications when appropriate; however, we may not file any such applications or, if filed, the patents may not be issued. with respect to our hormone therapy drug candidates.
We hold numerousmultiple U.S. trademark registrations and have numerous pending trademark applications. Issuance of a federally registered trademark creates a rebuttable presumption of ownership of the mark; however, it is subject to challenge by others claiming first use in the mark in some or all of the areas in which it is used. Federally registered trademarks have a perpetual life as long as they are maintained and renewed on a timely basis and used properly as trademarks, subject to the rights of third parties to seek cancellation of the trademarks if they claim priority or confusion of usage. We believe our patents and trademarks are valuable and provide us certain benefits in marketing our products.
We intend to actively protect our intellectual property with patents, trademarks, trade secrets, andor other legal avenues for the protection of intellectual property.
OPERA® is our patent-pendingpatented information technology platform used in our business. TheWe believe the deployment of OPERAOPERA® and the further development and deployment of related technology creates a sustainable competitive advantage that has led to our market share growth. We are currently developing additional intellectual property in the following areas:
As we continue to develop proprietary intellectual property, we will expand our protection by applying for additional patents around the business process for OPERA and patents on future technologies, including developing mobile applications to more effectively communicate with patients.technologies. As we examine our current product offerings and new product pipeline, we are in the process of modifying and developing new formulations that will enable us to gain patent protection for these products.
While we seek broad coverage under our patent applications, there is always a risk that an alteration to the process may provide sufficient basis for a competitor to avoid infringement claims. In addition, patents expire and we cannot provide any assurance that any patents will be issued from our pending application or that any potentially issued patents will adequately protect our intellectual property.
Government Regulation
In the United States, the FDA regulates pharmaceuticals, dietary supplements, and cosmetics under the Federal Food, Drug, and Cosmetic Act, or FDCA, and its influence over communication, content and commerce. According to Forrester Research, an independent research company providing advice to global leaders in business and technology, U.S. online retail sales increased 12.2% from 2010. Forrester projects online retail sales to grow at a 10% CAGR to $278.9 billion by 2015. We believe several factors will contribute to this increase including convenience, expanded range of available products and services, improved security and electronic payment technology, increased access to broadband Internet connections and widespread consumer confidence and acceptance of the Internet as a means of commerce.
Pharmaceutical Regulation
The process required by the FDA before a new drug product may be marketed in the United States generally involves the following:
An IND is a request for authorization from the FDA to administer an investigational drug product to humans. We currently have effective INDs for all of our business.
Clinical trials involve the administration of the investigational drug to human subjects under the supervision of qualified investigators in accordance with current Good Clinical Practices, or cGCPs, which include the requirement that all research subjects provide their informed consent for their participation in any clinical trial. A protocol for each clinical trial and any subsequent protocol amendments must be submitted to the FDA as part of the IND. Additionally, approval must also be obtained from each clinical trial site’s IRB before the trials may be initiated, and the IRB must monitor the study until completed. There are also requirements governing the reporting of ongoing clinical trials and clinical trial results to public registries.
Clinical trials are usually conducted in three phases. Phase 1 clinical trials are normally conducted in small groups of healthy volunteers to assess safety and find the potential dosing range. After a safe dose has been established, the drug is currently great uncertaintyadministered to small populations of sick patients (phase 2) to look for initial signs of efficacy in many statestreating the targeted disease or condition and to continue to assess safety. Phase 3 clinical trials are usually multi-center, double-blind controlled trials in hundreds or even thousands of subjects at various sites to assess as fully as possible both the safety and effectiveness of the drug.
The FDA, the IRB, or the clinical trial sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects are being exposed to an unacceptable health risk. Additionally, some clinical trials are overseen by an independent group of qualified experts organized by the clinical trial sponsor, known as a data safety monitoring board or committee, or DSMB. This group reviews unblinded data from clinical trials and provides authorization for whether or how existing laws governing issuesnot a trial may move forward at designated check points based on access to certain data from the study. We may also suspend or terminate a clinical trial based on evolving business objectives and/or competitive climate.
Assuming successful completion of all required testing in accordance with all applicable regulatory requirements, detailed investigational drug product information is submitted to the FDA in the form of an NDA requesting approval to market the product for one or more indications. The application includes all relevant data available from pertinent preclinical and clinical trials, including negative or ambiguous results as well as positive findings, together with detailed information relating to the product’s chemistry, manufacturing, controls and proposed labeling, among other things.
Once the NDA submission has been accepted for filing, the FDA’s goal is to review applications within ten months of filing. However, the review process is often significantly extended by FDA requests for additional information or clarification. The FDA may refer the application to an advisory committee for review, evaluation, and recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee, but it typically follows such recommendations.
After the FDA evaluates the NDA and conducts inspections of manufacturing facilities in which the drug product will be formulated and its active pharmaceutical ingredient, or API, will be produced, it may issue an approval letter or, instead, a Complete Response Letter. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications. A Complete Response Letter indicates that the review cycle of the application is complete and the application is not ready for approval. A Complete Response Letter may require additional clinical data and/or an additional pivotal phase 3 clinical trial(s), and/or other significant, expensive and time-consuming requirements related to clinical trials, preclinical studies or manufacturing. Even if such additional information is submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. The FDA could also approve the NDA with a REMS plan to mitigate risks, which could include medication guides, physician communication plans, or elements to assure safe use, such as property ownership, salesrestricted distribution methods, patient registries and other taxes,risk minimization tools. The FDA also may condition approval on, among other things, changes to proposed labeling, development of adequate controls and libelspecifications, or a commitment to conduct one or more post-market studies or clinical trials. Such post-market testing may include phase 4 clinical trials and personal privacy applysurveillance to further assess and monitor the Internetproduct’s safety and commercial online retailers. These issues may take yearseffectiveness after commercialization.
After regulatory approval of a drug product is obtained, we are required to resolve. For example, tax authorities incomply with a number of post-approval requirements. As a holder of an approved NDA, we would be required to report, among other things, certain adverse reactions and production problems to the FDA, to provide updated safety and efficacy information, and to comply with requirements concerning advertising and promotional labeling for any of our products. Also, quality control and manufacturing procedures must continue to conform to cGMP after approval to ensure and preserve the long-term stability of the drug product. The FDA periodically inspects manufacturing facilities to assess compliance with cGMP, which imposes extensive procedural, substantive, and record keeping requirements. In addition, changes to the manufacturing process are strictly regulated, and, depending on the significance of the change, may require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP and impose reporting and documentation requirements upon us and any third-party manufacturers that we may decide to use. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with cGMP and other aspects of regulatory compliance.
We rely, and expect to continue to rely, on third parties for the production of clinical and commercial quantities of our drug candidates. Future FDA and state inspections may identify compliance issues at our facilities or at the facilities of our contract manufacturers that may disrupt production or distribution, or require substantial resources to correct. In addition, discovery of previously unknown problems with a product or the failure to comply with applicable requirements may result in restrictions on a product, manufacturer, or holder of an approved NDA, including withdrawal or recall of the product from the market or other voluntary, FDA-initiated or judicial action that could delay or prohibit further marketing. Newly discovered or developed safety or effectiveness data may require changes to a product’s approved labeling, including the addition of new warnings and contraindications, and also may require the implementation of other risk management measures. Also, new government requirements, including those resulting from new legislation, may be established, or the FDA’s policies may change, which could delay or prevent regulatory approval of our products under development.
Our hormone therapy drug candidates may compete with unapproved hormone therapy products supplied by compounding pharmacies. Pharmacy compounding is a practice in which a licensed pharmacist combines, mixes, or alters ingredients in response to a prescription to create a medication tailored to the medical needs of an individual patient. The medications created by the compounding pharmacy are technically “new drugs” subject to the new drug approval requirements of the FDCA. However, FDA’s 2002 Compliance Policy Guide 460.200 states that FDA will exercise enforcement discretion to exclude compounded drugs from the new drug approval requirements except where compounding pharmacies act more akin to traditional drug manufacturers. The FDA does not exercise the same authority to regulate compounding pharmacies as pharmaceutical manufacturers. For example, compounding pharmacies are not required to report adverse events associated with compounded drugs, while commercial drug manufacturers are subject to stringent regulatory reporting requirements.
505(b)(2) Applications
We intend to submit NDAs for our hormone therapy drug candidates, assuming that the clinical data justify submission, under section 505(b)(2) of the FDCA, or Section 505(b)(2). Section 505(b)(2) permits the filing of an NDA when at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference. The applicant may rely upon published literature and the FDA’s findings of safety and effectiveness based on certain pre-clinical or clinical studies conducted for an approved product. The FDA may also require companies to perform additional studies or measurements to support the change from the approved product. The FDA may then approve the new drug candidate for all or some of the label indications for which the referenced product has been approved, as well as a Congressional advisory commission,for any new indication sought by the Section 505(b)(2) applicant. In regards to TX-001HR, we are currently reviewing the appropriate tax treatmentrequired to conduct phase 3 studies for vasomotor symptoms versus placebo and an endometrial protection study.
Phase 3 clinical trials for secondary amenorrhea versus placebo will be required for TX-002HR. TX-003HR would be required to undergo phase 3 studies of companies engaged in online commerce and new state tax regulations may subject usvasomotor symptoms compared to additional state sales and income taxes. New legislation or regulation, the application of laws and regulations from jurisdictions whose lawsplacebo, though we currently do not currently applyhave plans to continue development of this drug candidate.
As part of our business,submission, we intend to certify that all of the patents for approved products referenced in the NDA for each of the hormone therapy drug candidates as listed in the FDA’s Orange Book have expired and that we will not be compelled to certify that any patent is invalid, unenforceable, or will not be infringed by the new product. If, in fact, this assessment is incorrect, it can have a change in application of existing lawsserious and regulations to the Internet and commercial online services could result in significant additional taxes on our business. These taxes could have an adverse effect on our resultsability to obtain FDA approval or market our new product. If we are compelled to certify that a patent is invalid, unenforceable, or not infringed, then the holder of operations.
Marketing Exclusivity
A 505(b)(2) NDA applicant may be eligible for its own regulatory exclusivity period, such as three-year exclusivity. The first approved 505(b)(2) NDA applicant for a particular condition of ourapproval, or change to a marketed product, such as a new extended release formulation for a previously approved product, may be granted three-year Hatch-Waxman exclusivity if one or more clinical studies, other than bioavailability or bioequivalence studies, was essential to the approval of the application and was conducted/sponsored by the applicant. Should this occur, the FDA would be precluded from making effective any other application for the same condition of use or for a change to the drug product that was granted exclusivity until after that three-year exclusivity period has run. Additional exclusivities may also apply.
Additionally, the 505(b)(2) NDA applicant may have relevant patents in the Orange Book, and if it does, it can initiate patent infringement litigation against those applicants that challenge such patents, which could result in a 30-month stay delaying those applicants.
Dietary Supplement Regulation
Our currently marketed products are regulated as dietary supplements. The processing, formulation, safety, manufacturing, packaging, labeling, advertising, and distribution of these products are subject to extensive regulation inby one or more federal agencies, including the U.S. The FDA enforces the Federal Food, Drug and Cosmetic Act (FDCA) and related regulations which govern the identity, purity, quality, strength, and composition of dietary supplements and regulate the formulation, manufacture, packaging, labeling, holding, sale, and distribution of dietary supplements, foods and OTC and prescription drugs, and prohibit the sale of misbranded and adulterated dietary supplements and dietary supplements that by the intention of the manufacturer or distributor or label or labeling claims are unapproved new drugs.
Generally, our nutritional product formulations are proprietary in that in designing them, we attempt to blend an optimal combination of nutrients that appear to have beneficial impact based upon scientific literature and advertising actsinput from physicians; however, we are generally prohibited from making disease treatment and practices associated withprevention claims in the promotion and sale of these products. The U.S. Postal Inspection Service enforces federal laws governing fraudulent use of the mail. Regulation of certain aspects of the dietary supplement business at the federal level is also governed by the Consumer Product Safety Commission (CPSC) (e.g., concerning the presence of adulterated substances, such as toxic levels of lead or iron, that render products unsafe for consumption and require an ordered recall), the Department of Agriculture (e.g., forour products that are intended for ingestion as dietary supplements for animals) and the Environmental Protection Agency (e.g., in the methods of disposal used for certain dietary ingredients, such as colloidal silver). Federal and state anti-kickback statutes, the Ethics in Patient Referrals Act, false claims statutes and HIPPA also apply to our business.
The FDCA has been amended several times affecting provisions that concern dietary ingredients and dietary supplements, including by the Dietary Supplement Health and Education Act of 1994, (DSHEA).or DSHEA, formally defined what may be sold asamended the FDCA to establish a dietary supplement, defined statementsnew framework governing the composition, safety, labeling, manufacturing, and marketing of nutritional support and the conditions under which they may lawfully be used, and included provisions that permit the FDA to regulate manufacturing practices and labeling claims peculiar to dietary supplements. “Dietary supplements” are defined as vitamins, minerals, herbs, other botanicals, amino acids and other dietary substances that are used to supplement the diet, as well as concentrates, constituents, extracts, metabolites, or combinations of such dietary ingredients. Generally, under DSHEA,the FDCA, dietary ingredients that were onmarketed in the market beforeUnited States prior to October 15, 1994 may be used in dietary supplements without notifying the FDA. However, a “new”“New” dietary ingredient (i.e.ingredients (i.e., a dietary ingredientingredients that was notwere “not marketed in the U.S.United States before October 15, 1994)1994”) must be the subject of a new dietary ingredient notification submitted to the FDA unless the ingredient has been “present in the food supply as an article used for food” without having beenbeing “chemically altered.” A new dietary ingredient notification must provide the FDA with evidence of a “history of use or other evidence of safety” which establishesestablishing that use of the dietary ingredient “will reasonably be expected to be safe.” A new dietary ingredient notification must be submitted to the FDA at least 75 days before the initial marketing of the new dietary ingredient. The FDA may determine that a new dietary ingredient cannotification does not provide an adequate basis to conclude that a dietary ingredient is reasonably expected to be marketed. There can be no assurance that the FDA will accept evidence purporting to establish the safety of any new dietary ingredients that we may want to market and the FDA’s refusal to accept such evidencesafe. Such a determination could prevent the marketing of such dietary ingredients.
The FDA or other agencies could leadtake actions against products or product ingredients that in its determination present an unreasonable health risk to consumers that would make it illegal for us to sell such products. In addition, the FDA could issue consumer warnings with respect to challenge dietarythe products or ingredients alreadyin such products. Such actions or warnings could be based on information received through FDCA-mandated reporting of serious adverse events. The FDCA requires that reports of serious adverse events be submitted to the market as “illegal” underFDA, and based in part on such reports, the FDCA because of the failureFDA has issued public warnings to file a new dietary ingredient notification or because the substance may be one foundconsumers to be the subject of an investigational new drug application for which clinical trials have commenced and been publicized.
The FDA prohibits disease treatment claims entirely when made for a dietary supplement; however,FDCA permits “statements of nutritional support,” including so-called “structure/function claims” are permittedsupport” to be included in labeling for dietary supplements without premarket approval. Such statements must be submitted to the FDA pre-approval.within 30 days of marketing. Such statements may describe how a particular dietary ingredient affects the structure, function, or general well-being of the body, or the mechanism of action by which a dietary ingredient may affect thebody structure, function, or well-being, of the body, but such statements may not stateexpressly or implicitly represent that a dietary supplement will reduce the riskdiagnose, cure, mitigate, treat, or incidence ofprevent a disease unless such claim has been reviewed and approved by the FDA.disease. A company that uses a statement of nutritional support in labeling must possess scientific evidence substantiating that the statement is truthful and not misleading. Such statements must be submitted toIf the FDA no later than thirty days after first marketing the product with the statement and must be accompanied by the following FDA mandated label disclaimer: “This statement has not been evaluated by the Food and Drug Administration. This product is not intended to diagnose, treat, cure or prevent any disease.” There can be no assurance that the FDA will not determinedetermines that a particular statement of nutritional support that we want to use is an unacceptable diseasedrug claim, conventional food claim, or an unauthorized nutrient-disease relationship claim otherwise permitted with FDA approval asversion of a “health claim.claim,” Suchor, if the FDA determines that a determination might preventparticular claim is not adequately supported by existing scientific data or is false or misleading, we would be prevented from using the use of such a claim.
In addition, DSHEA provides that certainso-called “third-party literature,” such as a reprint of a peer-reviewed scientific publication linking a particular dietary ingredient with health benefits, may be used “in connection with the sale of a dietary supplement to consumers” without the literature being subject to regulation as labeling. The literature : (1) must not be exemptfalse or misleading; (2) may not “promote” a particular manufacturer or brand dietary supplement; (3) must present a balanced view of the available scientific information on the subject matter; (4) if displayed in establishment, must be physically separate from labeling regulation. However, the FDA has adopted an “intentdietary supplements; and (5) should not have appended to use” doctrine wherebyit any information by sticker or another method. If the literature fails to satisfy each of these requirements, we may be prevented from disseminating such literature even if exempt from labeling, may nonetheless formwith our products, and any dissemination could subject our product to regulatory action as an illegal drug.
In June 2007, pursuant to the basis for an agency determination that the literature in context reveals a company’s intent to sell a dietary ingredient or dietary supplement as a drug, thereby rendering the supplement an unlawful, unapproved new drug. Because the “intent to use” doctrine is predicated on a subjective assessment of all facts and circumstances associated with the promotion and sale of a dietary supplement, we cannot know whether any particular piece of literature otherwise exempt from labeling will be deemed by the FDA unlawful for use in association with the sale of the dietary ingredient or dietary supplement.
The FDA has broad authority to enforce the provisions of the FDCA concerning medical foods,federal law applicable to dietary supplements, and drugs, including powers to issue a public “warning letter”Warning Letters or Untitled Letters to a company, to quarantine and prohibit the sale of products deemed adulterated or misbranded, to publicize information about illegal products, todetain products intended for import, require the reporting of serious adverse events, request a voluntary recall of illegal or unsafe products from the market, toand request that the Department of Justice initiate a seizure action, an injunction action, or a criminal prosecution in the U.S. courts,courts. The FSMA expands the reach and to seek disgorgement from a federal courtregulatory powers of all proceeds received from the sale of products deemed misbranded or adulterated. For instance, the FDA recently announced that any unapproved new drug introduced after September 19, 2011 will be subject to immediate enforcement action, without prior notice and without regardwith respect to the enforcement priorities set outproduction and importation of food, including dietary supplements. The expanded reach and regulatory powers include the FDA’s ability to order mandatory recalls, administratively detain domestic products, require certification of compliance with domestic requirements for imported foods associated with safety issues and administratively revoke manufacturing facility registrations, effectively enjoining manufacturing of dietary ingredients and dietary supplements without judicial process. The regulation of dietary supplements may increase or become more restrictive in CPG 440.100. The FDA will continue to apply the enforcement priorities established in 2006. These give a higher priority to enforcement actions involving drugs in certain high-risk categories, such as drugs that pose a potential safety risk or lack evidence of effectiveness.
The FTC exercises jurisdiction over the advertising of medical foods, dietary supplements and drugs. cosmetics. In recent years, the FTC has instituted numerous enforcement actions against companies for failure to have adequate substantiation for claims made in advertising or for the use of false or misleading advertising claims.
In recent years, the FTC has instituted numerous enforcement actions against dietary supplement companies for making false or misleading advertising claims and for failing to adequately substantiate claims made in advertising. These enforcement actions have often resulted in consent decrees and the payment of civil penalties and/or restitution by the companies involved. The FTC also regulates other aspects of consumer purchases, including but not limited to, promotional offers of savings compared policies, telemarketing, continuity plans, and “free” offers.
We are also subject to regulation under various state, local, and international laws that include provisions governing, among other things, the formulation, manufacturing, packaging, labeling, advertising, and distribution of dietary supplements and drugs. For example, Proposition 65 in the Statestate of California is a list of substances deemed to pose a risk of carcinogenicity or birth defects at or above certain levels. If any such ingredient exceeds the permissible levels in a dietary supplement, cosmetic, or drug, the product may be lawfully sold in California only if accompanied by a prominent warning label alerting consumers that the product contains an ingredient linked to cancer or birth defect risk. Private attorney general actions as well as California attorney general actions may be brought against non-compliant parties and can result in substantial costs and fines.
Other U.S. Health Care Laws and Compliance Requirements
We are also subject to additional health care regulation and enforcement by the federal government and the states in which we conduct our business. Applicable federal and state healthcarehealth care laws and regulations include butthe following:
Health Insurance Portability and Accountability Act of 1996, as amended, or HIPAA, imposes criminal and civil liability for executing a scheme to defraud any health care benefit program and also imposes obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security, and transmission of individually identifiable health information.
Efforts to ensure that our business arrangements with third parties comply with applicable healthcarehealth care laws and regulations could be costly. Although we believe that our regulatory counsel has assisted us in establishing business practices are structured to be compliant with applicable laws, it is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations, or case law involving applicable fraud and abuse or other healthcarehealth care laws and regulations. If our past or present operations, including activities conducted by our sales team or agents, are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal, and administrative penalties, damages, fines, exclusion from third party payor programs, such as Medicare and Medicaid, and the curtailment or restructuring of our operations. If any of the physicians, providers, or entities with whom we do business isare found to be not in compliance with applicable laws, they may be subject to criminal, civil, or administrative sanctions, including exclusionsexclusion from government funded healthcarehealth care programs.
Many aspects of these laws have not been definitively interpreted by the regulatory authorities or the courts, and their provisions are open to a variety of subjective interpretations whichthat increases the risk of potential violations. In addition, these laws and their interpretations are subject to change. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses, divert our management’s attention from the operation of our business, and damage our reputation.
In addition, from time to time in the future, we may become subject to additional laws or regulations administered by the FDA, the FTC, or by other federal, state, local, or foreign regulatory authorities, to the repeal of laws or regulations that we generally consider favorable, such as DSHEA, or to more stringent interpretations of current laws or regulations. We are not able to predict the nature of such future laws, regulations, repeals, or interpretations, and we cannot predict what effect additional governmental regulation, if and when it occurs, would have on our business in the future. Such developments could, however, require reformulation of certain products to meet new standards, recalls or discontinuance of certain products not able to be reformulated, additional record-keeping requirements, increased documentation of the properties of certain products, additional or different labeling, additional scientific substantiation, additional personnel, or other new requirements. Any such developments could have a material adverse effect on our business.
The growth and demand for eCommerce could result in more stringent consumer protection laws that impose additional compliance burdens on online retailers. These consumer protection laws could result in substantial compliance costs and could interfere with the Past Five Years
There is currently great uncertainty in many states whether or how existing laws governing issues such as property ownership, sales and do not directly handle, storeother taxes, and libel and personal privacy apply to the Internet and commercial online retailers. These issues may take years to resolve. For example, tax authorities in a number of states, as well as a Congressional advisory commission, are currently reviewing the appropriate tax treatment of companies engaged in online commerce and new state tax regulations may subject us to additional state sales and income taxes. New legislation or transport hazardous materials or waste products. We depend on these third parties to abide by all applicable federal, state and localregulation, the application of laws and regulations governing the use, manufacture, storage, handling and disposal of hazardous materials and waste products. Wefrom jurisdictions whose laws do not anticipate the costcurrently apply to our business, or a change in application of complying with theseexisting laws and regulations to be material.
Employees
As of December 31, 2011,2014, we had 51100 full-time employees, four (4)five of whom are executive officers. Additionally, from time to time, we hire temporary contract employees. None of our employees are covered by a collective bargaining agreement, and we are unaware of any union organizing efforts. We have never experienced a major work stoppage, strike, or dispute. We consider our relationship with our employees to be good.
| 23 |
Our corporate headquartersHistory
On October 3, 2011, we changed our name to TherapeuticsMD, Inc. On October 4, 2011, we closed a reverse merger with VitaMedMD, LLC, pursuant to which (1) all outstanding membership units of VitaMed were exchanged for shares of our common stock, (2) all outstanding VitaMed options and warrants were exchanged and converted into options and warrants to purchase shares of our common stock, and (3) VitaMed became our wholly owned subsidiary. As of December 31, 2011, we determined that VitaMed would become the sole focus of our company and services previously performed relative to the licensing agreement discussed in the following paragraph were discontinued.
We were incorporated in Utah in 1907 under the name Croff Mining Company. Prior to 2008, Croff’s operations consisted entirely of oil and natural gas leases. Due to a spin-off of its operations in December 2007, Croff had no business operations or revenue source and had reduced its operations to a minimal level although it continued to file reports required under the Securities Exchange Act of 1934, or the Exchange Act. As a result of the spin-off, Croff was a “shell company” under the rules of the Securities and Exchange Commission, or the SEC. In July 2009, Croff (i) closed a transaction to acquire America’s Minority Health Network, Inc. as a wholly owned subsidiary, (ii) ceased being a shell company, and (iii) experienced a change in control in which the former stockholders of America’s Minority Health Network, Inc. acquired control of our company. On June 11, 2010, we closed a transaction to acquire Spectrum Health Network, Inc. as a wholly owned subsidiary. On July 20, 2010, we filed Articles of Conversion and Articles of Incorporation to redomicile in the state of Nevada. On July 31, 2010, we transferred the assets of America’s Minority Health Network, Inc. to a secured noteholder in exchange for the satisfaction of certain associated debt. On February 15, 2011, we transferred the assets of Spectrum Health Network, Inc. to a secured noteholder in exchange for the satisfaction of associated debt and in exchange for a licensing agreement under which we subsequently sold subscription services and advertising on the Spectrum Health Network for commissions.
Available Information
We are locateda Nevada corporation. We maintain our principal executive offices at 9516800 Broken Sound Parkway NW, Suite 320,Third Floor, Boca Raton, FLFlorida 33487. Our telephone number is 561-961-1911(561) 961-1900. We maintain websites atwww.therapeuticsmd.com,www.vitamedmd.com,www.vitamedmdrx.com, andwww.bocagreenmd.com. The information contained on our fax numberwebsites or that can be accessed through our websites is 561-431-3389.
We are subject tofile reports with the requirements of Section 13(a) under the Exchange Act which requires us to file annual reportsSEC, including Annual Reports on Form 10-K, quarterly reportsQuarterly Reports on Form 10-Q, and current reportsCurrent Reports on Form 8-K, and any other filings required by the SEC. Through our website, we are required to comply with all other obligations of the Exchange Act applicable to issuers filing registration statements pursuant to Section 12(g) of the Exchange Act.
The public may read and copy any materials we file with, or furnish to, the SEC at the SEC’s Public Reference Room at 100 F Street, NE, Washington, DC 20549. The public may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. The SEC maintains an Internet site (www.sec.gov) that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC.
| Item 1A. | Risk Factors |
Investing in our common stock involves a high degree of risk. You should carefully consider the following risk factors, together with all of the information included in this Annual Report before you decide to purchase shares of our common stock. We believe the risks and uncertainties described below are the most significant we face. Additional risks and uncertainties of which we are unaware, or that we currently deem immaterial, also may become important factors that affect us. If any of the following risks occur, our business, financial condition, or results of operations could be materially and adversely affected. In that case, the trading price of our common stock could decline, and you may lose all or part of your investment.
Risks Related to Our Business
We have incurred significant operating losses since inception and anticipate that we will incur continued losses for the foreseeable future.
We have incurred recurring net losses, including net losses of $54 million, $28 million, and $35 million for the years ended December 31, 2014, 2013, and 2012, respectively. As of December 31, 2014, we had an accumulated deficit of approximately $135 million. We have generated limited revenue and have funded our operations to date primarily from public and private sales of equity and private sales of debt securities. We expect to incur substantial additional losses over the next several years as our research, development, and clinical trial activities increase, especially those related to our hormone therapy drug candidates. As a result, we may never achieve or maintain profitability unless we successfully commercialize our products, in particular, our hormone therapy drug candidates. If we are unable to make required payments under any of our obligations for any reason, our creditors may take actions to collect their debts, including foreclosing on our property that collateralizes our obligations. If we continue to incur substantial losses and are unable to secure additional financing, we could be forced to discontinue or curtail our business operations, sell assets at unfavorable prices, refinance existing debt obligations on terms unfavorable to us, or merge, consolidate, or combine with a company with greater financial resources in a transaction that might be unfavorable to us.
We currently derive all of our revenue from sales of our women’s health care products, and our failure to maintain or increase sales of these products would have a material adverse effect on our business, financial condition, results of operations, and growth prospects.
We currently derive all of our revenue from sales of women’s health care products, including prenatal and women’s multi-vitamins, iron supplements, vitamin D supplements, and natural menopause relief. While sales of our vitamin products grew from 2010 through 2014, we cannot assure you that such sales will continue to grow. In addition to other risks described herein, our ability to maintain or increase existing product sales is subject to a number of risks and uncertainties, including the following:
If revenue from sales of our existing prescription and OTC prenatal vitamins does not continue or increase, we may be required to reduce our operating expenses or to seek to raise additional funds, which could have a material adverse effect on our business, financial condition, results of operations, and growth prospects, or we may not be able to commence or continue clinical trials to seek approval for and commercialize our hormone therapy drug candidates or any other products we may choose to develop in the future.
If our products do not have the effects intended or cause undesirable side effects, our business may suffer.
Although many of the ingredients in our current dietary supplement products are vitamins, minerals, and other substances for which there is a long history of human consumption, they also contain innovative ingredients or combinations of ingredients. Although we believe all of these products and the combinations of ingredients in them are safe when taken as directed, the products could have certain undesirable side effects if not taken as directed or if taken by a consumer who has certain medical conditions, such as the potential effect of high doses of folic acid masking pernicious anemia. In addition, these products may not have the effect intended if they are not taken in accordance with certain instructions, which include certain dietary restrictions. Furthermore, there can be no assurance that any of the products, even when used as directed, will have the effects intended or will not have harmful side effects in an unforeseen way or on an unforeseen cohort. If any of our products or products we develop or commercialize in the future are shown to be harmful or generate negative publicity from perceived harmful effects, our business, financial condition, results of operations, and prospects would be harmed significantly.
Our future success will depend in large part on our ability to commercialize our hormone therapy drug candidates designed to alleviate the symptoms of and reduce the health risks resulting from menopause, including hot flashes, osteoporosis, and vaginal dryness.
Our future success will depend in large part on our ability to successfully develop and commercialize our hormone therapy drug candidates designed to alleviate the symptoms of and reduce the health risks resulting from menopause, including hot flashes, osteoporosis, and vaginal dryness. We have submitted IND applications for our four hormone therapy drug candidates, which the FDA has made effective and which permit us to conduct clinical testing on these proposed products. We currently intend to clinically test three of those drug candidates. However, we may not be able to complete the development of these drug candidates, the results of the clinical trials may not be sufficient to support NDA for any of them, and even if we believe the results of our clinical trials are sufficient to support any NDA that we submit, the FDA may disagree and may not approve our NDA. In addition, even if the FDA approves one or more of our NDAs, it may do so with restrictions on the intended uses that may make commercialization of the product or products financially untenable. The failure to commercialize or obtain necessary approval for any one or more of these products would substantially harm our prospects and our business.
We may not be able to complete the development and commercialization of our hormone therapy drug candidates if we fail to obtain additional financing.
We need substantial amounts of cash to complete the clinical development of our hormone therapy drug candidates. Our existing cash and cash equivalents will not be sufficient to fund these requirements. In addition, changing circumstances may cause us to consume funds significantly faster than we currently anticipate, and we may need to spend more money than currently expected because of circumstances beyond our control. We do not currently have any committed external source of funds. We will attempt to raise additional capital from the issuance of equity or debt securities, collaborations with third parties, licensing of rights to these products, or other means, or a combination of any of the foregoing. Securing additional financing will require a substantial amount of time and attention from our management and may divert a disproportionate amount of their attention away from our day-to-day activities, which may adversely affect our ability to conduct our day-to-day operations. In addition, we cannot guarantee that future financing will be available in sufficient amounts or on terms acceptable to us, if at all. If we are unable to raise additional capital when required or on acceptable terms, we may be required to take one or more of the following actions:
Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures, or declaring dividends. To the extent that we raise additional capital through the sale of equity or convertible debt securities, the ownership interest of our existing stockholders will be diluted, and the terms of these new securities may include liquidation or other preferences that adversely affect the rights of our existing stockholders. If we raise additional funds through collaborations, strategic alliances, or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs, or proposed products or grant licenses on terms that may not be favorable to us.
If we are unable to raise additional capital in sufficient amounts or on terms acceptable to us, we will be prevented from pursuing discovery, development, and commercialization efforts, and our ability to generate revenue and achieve or sustain profitability will be substantially harmed.
We have no experience as a company in bringing a drug to regulatory approval.
We have never obtained regulatory approval for, or commercialized, a drug. It is possible that the FDA may refuse to accept any or all of our planned NDAs for substantive review or may conclude, after review of our data, that our applications are insufficient to obtain regulatory approval of any of our hormone therapy drug candidates. We have recently begun to conduct validation and scale up of the manufacturing processes for our proposed combination estradiol and progesterone drug candidate and our proposed suppository estradiol VVA product. The FDA may also require that we conduct additional clinical or manufacturing validation studies, which may be costly and time-consuming, and submit that data before it will reconsider our applications. Depending on the extent of these or any other FDA required studies, approval of any NDA that we submit may be significantly delayed, possibly for years, or may require us to expend more resources than we have available or can secure. Any delay or inability in obtaining regulatory approvals would delay or prevent us from commercializing our hormone therapy drug candidates, generating revenue from these proposed products, and achieving and sustaining profitability. It is also possible that additional studies, if performed and completed, may not be considered sufficient by the FDA to approve any NDA we submit. If any of these outcomes occur, we may be forced to abandon our planned NDAs for one or more of our hormone therapy drug candidates, which would materially adversely affect our business and could potentially cause us to cease operations.
Clinical trials involve a lengthy and expensive process with an uncertain outcome, and results of earlier studies and trials may not be predictive of future trial results.
Three hormone therapy drug candidates are currently in various stages of clinical testing. We have begun phase 3 clinical trial of our estradiol and progesterone combination drug candidate, our progesterone alone drug candidate and our vaginal suppository estradiol drug candidate. Clinical trials are expensive, can take many years to complete and have highly uncertain outcomes. For example, we recently temporarily suspended enrollment in and subsequently stopped the SPRY trial for our progesterone alone drug candidate in order to update the phase 3 protocol based on discussions with the FDA. Failure can occur at any time during the clinical trial process as a result of inadequate performance of a drug, inadequate adherence by patients or investigators to clinical trial protocols, or other factors. New drugs in later stages of clinical trials may fail to show the desired safety and efficacy traits despite having progressed through earlier clinical trials. A number of companies in the biopharmaceutical industry have suffered significant setbacks in advanced clinical trials as a result of a lack of efficacy or adverse safety profiles, despite promising results in earlier trials. Our future clinical trials may not be successful or may be more expensive or time-consuming than we currently expect. Prior to approving a new drug, the FDA generally requires that the safety and efficacy of the drug be demonstrated in two adequate and well-controlled clinical trials. In some situations the FDA approves drugs on the basis of a single well-controlled clinical trial. We believe we may be required to conduct only a single phase 3 clinical trial of each of our estradiol and progesterone combination drug candidate, our progesterone alone drug candidate and our vaginal suppository estradiol drug candidate for the treatment of VVA. However, in connection with our VVA drug candidate, the FDA has to date noted that in order to approve a drug based on a single trial, the trial would need to show statistical significance at a 0.01 level, and that a trial that is merely statistically significant may not provide sufficient evidence to support an NDA filing or approval of a drug candidate where the NDA relies on a single clinical trial. If clinical trials for any of our hormone therapy drug candidates fail to demonstrate safety or efficacy to the satisfaction of the FDA, the FDA will not approve that drug and we would not be able to commercialize it, which will have a material adverse effect on our business, financial condition, results of operations, and prospects.
Delays in clinical trials are common for many reasons, and any such delays could result in increased costs to us and jeopardize or delay our ability to obtain regulatory approval and commence product sales as currently contemplated.
We may experience delays in clinical trials for our hormone therapy drug candidates. Our planned clinical trials might not begin on time; may be interrupted, delayed, suspended, or terminated once commenced; might need to be redesigned; might not enroll a sufficient number of patients; or might not be completed on schedule, if at all. Clinical trials can be delayed for a variety of reasons, including the following:
Patient enrollment, a significant factor in the timing of clinical trials, is affected by many factors, including the size and nature of the patient population, the proximity of patients to clinical sites, the eligibility criteria for the trial, the design of the clinical trial, competing clinical trials, and clinicians’ and patients’ perceptions as to the potential advantages of the drug being studied in relation to other available therapies, including any new drugs that may be approved for the indications we are investigating. Any of these delays in completing our clinical trials could increase our costs, slow down our product development and approval process, and jeopardize our ability to commence product sales and generate revenue.
We may be required to suspend or discontinue clinical trials because of adverse side effects or other safety risks that could preclude approval of our hormone therapy drug candidates.
Our clinical trials may be suspended or terminated at any time for a number of reasons. A clinical trial may be suspended or terminated by us, our collaborators, the FDA, or other regulatory authorities because of a failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols, presentation of unforeseen safety issues or adverse side effects, failure to demonstrate a benefit from using the investigational drug, changes in governmental regulations or administrative actions, lack of adequate funding to continue the clinical trial, or negative or equivocal findings of the DSMB or the IRB for a clinical trial. An IRB may also suspend or terminate our clinical trials for failure to protect patient safety or patient rights. We may voluntarily suspend or terminate our clinical trials if at any time we believe that they present an unacceptable risk to participants. In addition, regulatory agencies may order the temporary or permanent discontinuation of our clinical trials at any time if they believe the clinical trials are not being conducted in accordance with applicable regulatory requirements or present an unacceptable safety risk to participants. If we elect or are forced to suspend or terminate any clinical trial of any proposed product that we develop, the commercial prospects of such proposed product will be harmed and our ability to generate product revenue from any of these proposed products will be delayed or eliminated. Any of these occurrences may harm our business, financial condition, results of operations, and prospects significantly.
We rely on third parties to conduct our research and development activities, including our clinical trials, and we may experience delays in obtaining or may be unsuccessful in obtaining regulatory approval for, or in commercializing, our hormone therapy drug candidates if these third parties do not successfully carry out their contractual duties or meet expected deadlines.
We do not have the resources to independently conduct research and development activities. Therefore, we have relied, and plan to continue to rely, on various third-party CROs to conduct our research and development activities and to recruit patients and monitor and manage data for our on-going clinical programs for our hormone therapy drug candidates, as well as for the execution of our clinical studies. Although we control only certain aspects of our CROs’ activities, we are responsible for ensuring that each of our studies is conducted in accordance with the applicable protocol, legal, regulatory, and scientific standards and our reliance on the CROs does not relieve us of our regulatory responsibilities. We cannot assure you that the CROs will conduct the research properly or in a timely manner, or that the results will be reproducible. We and our CROs are required to comply with the FDA’s cGCPs, which are regulations and guidelines enforced by the FDA for all of our products in clinical development. The FDA enforces these cGCPs through periodic inspections of trial sponsors, principal investigators, and clinical trial sites. If we or our CROs fail to comply with applicable cGCPs, the clinical data generated in our clinical trials may be deemed unreliable or invalid, and the FDA may require us to perform additional clinical trials before approving our proposed products. We cannot assure you that, upon inspection, the FDA will determine that any of our clinical trials comply with cGCPs. In addition, to evaluate the safety and effectiveness compared to placebo of our hormone therapy drug candidates to a statistically significant degree, our clinical trials will require an adequately large number of test subjects. Any clinical trial that a CRO conducts abroad on our behalf is subject to similar regulation. Accordingly, if our CROs fail to comply with these regulations or recruit a sufficient number of patients, we may be required to repeat clinical trials, which would delay the regulatory approval process.
In addition, we do not employ the personnel of our CROs, and, except for remedies available to us under our agreements with such organizations, we cannot control whether or not they will devote sufficient time and resources to our on-going clinical and pre-clinical programs. Our CROs may also have relationships with other commercial entities, including one or more of our competitors, for which they may also be conducting clinical studies or other drug development activities, which could impede their ability to devote appropriate time to our clinical programs. If our CROs do not successfully carry out their contractual duties or obligations or meet expected deadlines, if they need to be replaced, or if the quality or accuracy of the clinical data they obtain is compromised because of the failure to adhere to our clinical protocols or regulatory requirements, or for other reasons, our clinical trials may be extended, delayed, or terminated, and we may not be able to obtain regulatory approval for or successfully commercialize our hormone therapy drug candidates that we seek to develop. As a result, our financial results and the commercial prospects for our hormone therapy drug candidates that we seek to develop would be harmed, our costs could increase, and our ability to generate revenue could be delayed or ended.
We typically engage one or more CROs on a project-by-project basis for each study or trial. While we have developed and plan to maintain our relationships with CROs that we have previously engaged, we also expect to enter into agreements with other CROs to obtain additional resources and expertise in an attempt to accelerate our progress with regard to on-going clinical programs and, specifically, the compilation of clinical trial data for submission with an NDA for each of our hormone therapy drug candidates. If any of our relationships with these third parties terminate, we may not be able to enter into arrangements with alternative CROs or do so on commercially reasonable terms. Switching or entering into new relationships with CROs involves substantial cost and requires extensive management time and focus. In addition, there is a natural transition period when a new CRO commences work. As a result, delays occur, which can materially affect our ability to meet our desired clinical development timelines and can increase our costs significantly. Although we try to carefully manage our relationships with our CROs, there can be no assurance that we will not encounter challenges or delays in the future or that these delays or challenges will not have a material adverse impact on our business, financial condition, results of operations, or prospects.
| 29 |
Future legislation, regulations, and policies adopted by the FDA or other regulatory authorities may increase the time and cost required for us to conduct and complete clinical trials for our hormone therapy drug candidates.
The FDA has established regulations, guidelines, and policies to govern the drug development and approval process, as have foreign regulatory authorities. Any change in regulatory requirements resulting from the adoption of new legislation, regulations, or policies may require us to amend existing clinical trial protocols or add new clinical trials to comply with these changes. Such amendments to existing protocols or clinical trial applications or the need for new ones, may significantly and adversely affect the cost, timing, and completion of the clinical trials for our hormone therapy drug candidates.
In addition, the FDA’s policies may change and additional government regulations may be issued that could prevent, limit, or delay regulatory approval of our drug candidates, or impose more stringent product labeling and post-marketing testing and other requirements. For example, in the past the FDA has indicated it would regulate prenatal vitamins containing greater than 0.8 mg of folic acid as a drug under the FDCA. More recently the FDA indicated that there is no specified upper limit on the amount of folic acid permitted in a dietary supplement. If the FDA were to seek to regulate products with higher amounts of folic acid as drugs, it may require us to stop selling certain of our dietary supplement products and otherwise adversely affect our business. If we are slow or unable to adapt to any such changes, our business, prospects, and ability to achieve or sustain profitability would be adversely affected.
Even if we obtain regulatory approval for our hormone therapy drug candidates, we will still face extensive, ongoing regulatory requirements and review, and our products may face future development and regulatory difficulties.
Even if we obtain regulatory approval for one or more of our hormone therapy drug candidates in the United States, the FDA may still impose significant restrictions on a product’s indicated uses or marketing or to the conditions for approval, or impose ongoing requirements for potentially costly post-approval studies, including phase 4 clinical trials or post-market surveillance. As a condition to granting marketing approval of a product, the FDA may require a company to conduct additional clinical trials. The results generated in these post-approval clinical trials could result in loss of marketing approval, changes in product labeling, or new or increased concerns about side effects or efficacy of a product. For example, the labeling for our hormone therapy drug candidates, if approved, may include restrictions on use or warnings. The Food and Drug Administration Amendments Act of 2007, or FDAAA, gives the FDA enhanced post-market authority, including the explicit authority to require post-market studies and clinical trials, labeling changes based on new safety information, furtherand compliance with FDA-approved Risk Evaluation and Mitigation Strategies, or REMS, programs. If approved, our hormone therapy drug candidates will also be subject to ongoing FDA requirements governing the manufacturing, labeling, packaging, storage, distribution, safety surveillance, advertising, promotion, record keeping, and reporting of safety and other post-market information. The FDA’s exercise of its authority could result in delays or increased costs during product development, clinical trials and regulatory review, increased costs to comply with additional post-approval regulatory requirements, and potential restrictions on sales of approved products. Foreign regulatory agencies often have similar authority and may impose comparable costs. Post-marketing studies, whether conducted by us or by others and whether mandated by regulatory agencies or voluntary, and other emerging data about marketed products, such as adverse event reports, may also adversely affect sales of our hormone therapy drug candidates once approved, and potentially our other marketed products. Further, the discovery of significant problems with a product similar to one of our products that implicate (or are perceived to implicate) an entire class of products could have an adverse effect on sales of our approved products. Accordingly, new data about our products could negatively affect demand because of real or perceived side effects or uncertainty regarding efficacy and, in some cases, could result in product withdrawal or recall. Furthermore, new data and information, including information about product misuse, may lead government agencies, professional societies, and practice management groups or organizations involved with various diseases to publish guidelines or recommendations related to the Companyuse of our products or the use of related therapies or place restrictions on sales. Such guidelines or recommendations may lead to lower sales of our products.
The holder of an approved NDA also is subject to obligations to monitor and report adverse events and instances of the failure of a product to meet the specifications in the NDA. Application holders must submit new or supplemental applications and obtain FDA approval for certain changes to the approved product, product labeling, or manufacturing process. Application holders must also submit advertising and other promotional material to the FDA and report on ongoing clinical trials. Legal requirements have also been enacted to require disclosure of clinical trial results on publicly available databases.
In addition, manufacturers of drug products and their facilities are subject to continual review and periodic inspections by the FDA and other regulatory authorities for compliance with the FDA’s cGMPs regulations. If we or a regulatory agency discovers previously unknown problems with a product, such as adverse events of unanticipated severity or frequency or problems with the facility where the product is manufactured, a regulatory agency may impose restrictions on that product, the manufacturing facility, or us, including requiring recall or withdrawal of the product from the market or suspension of manufacturing, requiring new warnings or other labeling changes to limit use of the drug, requiring that we conduct additional clinical trials, imposing new monitoring requirements, or requiring that we establish a REMS. Advertising and promotional materials must comply with FDA rules in addition to other potentially applicable federal and state laws. The distribution of product samples to physicians must comply with the requirements of the Prescription Drug Marketing Act. Sales, marketing, and scientific/educational grant programs must comply with the anti-fraud and abuse provisions of the Social Security Act, the False Claims Act, and similar state laws. Pricing and rebate programs must comply with the Medicaid rebate requirements of the Omnibus Budget Reconciliation Act of 1990 and the Veterans Healthcare Act of 1992. If products are made available to authorized users of the Federal Supply Schedule of the General Services Administration, additional laws and requirements apply. All of these activities are also potentially subject to federal and state consumer protection and unfair competition laws. If we or our third-party collaborators fail to comply with applicable regulatory requirements, a regulatory agency may take any of the following actions:
The occurrence of any of the foregoing events or penalties may force us to expend significant amounts of time and money and may significantly inhibit our ability to bring to market or continue to market our products and generate revenue. Similar regulations apply in foreign jurisdictions.
Our dependence upon third parties for the manufacture and supply of our existing women’s health care products and our hormone therapy drug candidates may cause delays in, or prevent us from, successfully developing, commercializing, and marketing our products.
We do not currently have nor do we plan to build the infrastructure or capability internally to manufacture our existing women’s health care products. For example, we depend on Lang, a full-service, private label and corporate brand manufacturer specializing in premium health benefit driven products, including medical foods, nutritional supplements, beverages, bars and functional foods in the dietary supplement category, to supply approximately 97% of our vitaMedMD and BocaGreen products. In certain circumstances, including our failure to satisfy our production forecasts to Lang, we may be obligated to reimburse Lang for the costs of excess raw materials purchased by Lang that it cannot use in another product category that it then sells. We also rely on third-party contract manufacturing organizations, or CMOs to supply our hormone therapy drug candidates for use in the conduct of our clinical trials. We rely on these third parties to manufacture these products in accordance with our specifications and in compliance with applicable regulatory requirements. We do not have long-term contracts for the commercial supply of our products or our hormone therapy drug candidates. We intend to pursue long-term manufacturing agreements, but we may not be able to negotiate such agreements on acceptable terms, if at http://www.therapeuticsmd.com. Youall.
In addition, regulatory requirements could pose barriers to the manufacture of our products, including our hormone therapy drug candidates. Our third-party manufacturers are required to comply with cGMP regulations. As a result, the facilities used by any of our current or future manufacturers must be approved by the FDA. Holders of NDAs, or other forms of FDA approvals or clearances, or those distributing a regulated product under their own name, are responsible for manufacturing even though that manufacturing is conducted by a third-party CMO. All of our existing products are, and our hormone therapy drug candidates, if approved, will be, manufactured by CMOs. These CMOs are required by the terms of our contracts to manufacture our products in compliance with the applicable regulatory requirements. If our manufacturers cannot successfully manufacture material that conforms to our specifications and the strict regulatory requirements of the FDA and any applicable foreign regulatory authority, they will not be able to secure the applicable approval for their manufacturing facilities. If these facilities are not approved for the commercial manufacture of our existing products or our hormone therapy drug candidates, we may need to find alternative manufacturing facilities, which would result in disruptions of our sales and significant delays of up to several years in obtaining approval for our hormone therapy drug candidates. In addition, our manufacturers will be subject to ongoing periodic unannounced inspections by the FDA and corresponding state and foreign agencies for compliance with cGMPs and similar regulatory requirements. Failure by any of our manufacturers to comply with applicable cGMP regulations could result in sanctions being imposed on us, including fines, injunctions, civil penalties, delays, suspensions or withdrawals of approvals, operating restrictions, interruptions in supply, recalls, withdrawals, issuance of safety alerts, and criminal prosecutions, any of which could have a material adverse impact on our business, financial condition, results of operations, and prospects. Finally, we also could experience manufacturing delays if our CMOs give greater priority to the supply of other products over our products and proposed products or otherwise do not satisfactorily perform according to the terms of their agreements with us.
If any supplier of the product for our hormone therapy drug candidates experiences any significant difficulties in its respective manufacturing processes, does not comply with the terms of the agreement between us, or does not devote sufficient time, energy, and care to providing our manufacturing needs, we could experience significant interruptions in the supply of our hormone therapy drug candidates, which could impair our ability to supply our hormone therapy drug candidates at the levels required for our clinical trials and commercialization and prevent or delay their successful development and commercialization.
The commercial success of our existing products and our hormone therapy drug candidates that we develop, if approved in the future, will depend upon gaining and retaining significant market acceptance of these products among physicians and payors.
Physicians may not prescribe our products, including any of our hormone therapy drug candidates, if approved by the appropriate regulatory authorities for marketing and sale, which would prevent us from generating revenue or becoming profitable. Market acceptance of our products, including our hormone therapy drug candidates, by physicians, patients, and payors, will depend on a number of factors, many of which are beyond our control, including the following:
Even if the medical community accepts that our products are safe and efficacious for their approved indications, physicians may not immediately be receptive to the use or may be slow to adopt our products as an accepted treatment for the symptoms for which they are intended. We cannot assure you that any labeling approved by the FDA will permit us to promote our products as being superior to competing products. If our products, including, in particular our hormone therapy drug candidates, if approved, do not achieve an adequate level of acceptance by physicians and payors, we may not generate sufficient or any revenue from these products and we may not become profitable. In addition, our efforts to educate the medical community and third-party payors on the benefits of our products may require significant resources and may never be successful.
Our products, including our hormone therapy drug candidates if approved, face significant competition from branded and generic products, and our operating results will suffer if we fail to compete effectively.
Development and awareness of our brand will depend largely upon our success in increasing our customer base. The dietary supplement and pharmaceutical industries are intensely competitive and subject to rapid and significant technological change. Our products, including any hormone therapy drug candidates that are approved, face intense competition, including from major multinational pharmaceutical and dietary supplement companies, established biotechnology companies, specialty pharmaceutical, and generic drug companies. A new non-hormonal product, Brisdelle, produced by Noven Pharmaceuticals, was approved by the FDA for treatment of vasomotor symptoms in June 2013. Many of these companies have greater financial and other resources, such as larger research and development staffs and more experienced marketing and manufacturing organizations. As a result, these companies may obtain regulatory approval more rapidly and may be more effective in selling and marketing their products. They also may invest heavily to accelerate discovery and development of novel compounds or to in-license novel compounds that could make the products that we sell or develop obsolete. As a result, our competitors may succeed in commercializing products before we do. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large, established companies. If we are unable to economically promote or maintain our brand, our business, results of operations and financial condition could be severely harmed. In addition, our efforts to provide an alternative to the non FDA-approved compound bioidentical market for estradiol and progesterone products sold by compounding pharmacies may not be successful.
Coverage and reimbursement may not be available for our products, which could make it difficult for us to sell our products profitably.
Market acceptance and sales of our products, including any hormone therapy drug candidates, will depend on coverage and reimbursement policies and may be affected by health care reform measures. Government authorities and third-party payors, such as private health insurers and health maintenance organizations, decide which products they will pay for and establish reimbursement levels. Third-party payors generally do not cover OTC products, and coverage for vitamins and dietary supplements varies. We cannot be sure that coverage and reimbursement will be available for our products, including any hormone therapy drug candidates, if approved. We also cannot be sure that the amount of reimbursement available, if any, will not reduce the demand for, or the price of, our products. If reimbursement is not available or is available only at limited levels, we may not be able to successfully compete through sales of our existing dietary supplement products or successfully commercialize our hormone therapy drug candidates.
Specifically, in both the United States and some foreign jurisdictions, there have been a number of legislative and regulatory proposals to change the health care system in ways that could affect our ability to sell our products profitably. In the United States, the Medicare Prescription Drug, Improvement, and Modernization Act of 2003, also called the Medicare Modernization Act, or MMA, changed the way Medicare covers and pays for pharmaceutical products. The legislation expanded Medicare coverage for drug purchases by the elderly and certain others and introduced a new reimbursement methodology based on average sales prices for physician-administered drugs. In addition, this legislation provided authority for limiting the number of certain outpatient drugs that will be covered in any therapeutic class. As a result of this legislation and the expansion of federal coverage of drug products, we expect that there will be additional pressure to contain and reduce costs. These and future cost-reduction initiatives could decrease the coverage and price that we receive for our products, including our hormone therapy drug candidates, if approved, and could seriously harm our business. While the MMA applies only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policies and payment limitations in setting their own reimbursement rates, and any reduction in reimbursement under Medicare may result in a similar reduction in payments from private payors.
In March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, or collectively, PPACA, became law in the United States. The goal of PPACA is to reduce the cost of health care and substantially change the way health care is financed by both governmental and private insurers. Among other measures, PPACA imposes increased rebates on manufacturers for certain covered drug products reimbursed by state Medicaid programs. While we cannot predict the full effect PPACA will have on federal reimbursement policies in general or on our business specifically, the PPACA may result in downward pressure on drug reimbursement, which could negatively affect market acceptance of our products. In addition, we cannot predict whether new proposals will be made or adopted, when they may be adopted, or what impact they may have on us if they are adopted.
On March 4, 2015, the U.S. Supreme Court heard oral arguments inKing v. Burwell where the Supreme Court has been asked to decide the validity an IRS regulation paying subsidies, in the form of federal tax credits, to individuals who meet certain income guidelines, but who reside in states that have elected to use the federal government’s insurance exchange rather than establishing one of their own. Those challenging the IRS regulation have argued that federal subsidies are only available to those residing in states that have established their own insurance exchanges. Should the Supreme Court rule against the government, that could have adverse implications for most health care providers, including companies manufacturing prescription drugs and other products covered by health insurance under the PPACA.
The availability of generic products at lower prices than branded products may also substantially reduce the likelihood of reimbursement for branded products, such as our hormone therapy drug candidates, if approved. We expect to experience pricing pressures in connection with the sale of our products generally due to the trend toward managed health care, the increasing influence of health maintenance organizations, and additional legislative proposals. If we fail to successfully secure and maintain adequate coverage and reimbursement for our products or are significantly delayed in doing so, we will have difficulty achieving market acceptance of our products and our business will be harmed.
Product liability lawsuits could divert our resources, result in substantial liabilities and reduce the commercial potential of our products.
We face an inherent risk of product liability claims as a result of the marketing of our current products and the clinical testing of our hormone therapy drug candidates despite obtaining appropriate informed consents from our clinical trial participants, and, in light of the history of product liability claims related to other hormone replacement therapy products, we will face an even greater risk if we obtain FDA approval and commercialize our hormone therapy drug candidates in the United States or other additional jurisdictions or if we engage in the clinical testing of proposed new products or commercialize any additional products. For example, we may be sued if any product we develop allegedly causes injury or is found to be otherwise unsuitable during clinical testing, manufacturing, marketing, or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, failures to warn of dangers inherent in the product, negligence, strict liability, or breaches of warranties. Claims could also be asserted under state consumer protection acts. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our existing products or hormone therapy drug candidates, if approved. Even successful defense would require significant financial and management resources. Regardless of the merits or eventual outcome, product liability claims may result in any of the following:
Although we maintain general liability insurance of up to $10 million in the aggregate and clinical trial liability insurance of $10 million in the aggregate for our hormone therapy drug candidates, this insurance may not fully cover potential liabilities. The cost of any product liability litigation or other proceeding, even if resolved in our favor, could be substantial. In addition, our inability to obtain or maintain sufficient insurance coverage at an acceptable cost or to otherwise protect against potential product liability claims could prevent or inhibit the development and commercial production and sale of our products, which could adversely affect our business, financial condition, results of operations, and prospects.
Our business may be affected by unfavorable publicity or lack of consumer acceptance.
We are highly dependent upon consumer acceptance of the safety and quality of our products, as well as similar products distributed by other companies. Consumer acceptance of a product can be significantly influenced by scientific research or findings, national media attention, and other publicity about product use. A product may be received favorably, resulting in high sales associated with that product that may not be sustainable as consumer preferences change. Future scientific research or publicity could be unfavorable to our industry or any of our particular products and may not be consistent with earlier favorable research or publicity. A future research report or publicity that is perceived by our consumers as less than favorable or that may question earlier favorable research or publicity could have a material adverse effect on our ability to generate revenue. Adverse publicity in the form of published scientific research, statements by regulatory authorities or otherwise, whether or not accurate, that associates consumption of our product or any other similar product with illness or other adverse effects, or that questions the benefits of our product or a similar product, or that claims that such products do not have the effect intended could have a material adverse effect on our business, reputation, financial condition, or results of operations.
If we use hazardous and biological materials in a manner that causes injury or violates applicable law, we may be liable for damages.
Our research and development activities involve the controlled use of potentially hazardous substances, including chemical, biological, and radioactive materials. In addition, our operations produce hazardous waste products. Federal, state, and local laws and regulations in the United States govern the use, manufacture, storage, handling, and disposal of hazardous materials. Although we believe that our procedures for use, handling, storing, and disposing of these materials (all of which only occur at third-party sites operated by our contractors) comply with legally prescribed standards, we may incur significant additional costs to comply with applicable laws in the future. We also cannot predict the impact on our business of new or amended environmental laws or regulations or any changes in the way existing and future laws and regulations are interpreted or enforced. Also, even if we are in compliance with applicable laws, we cannot completely eliminate the risk of contamination or injury resulting from hazardous materials, and we may incur liability as a result of any such contamination or injury. In the event of an accident, we could be held liable for damages or penalized with fines, and the liability could exceed our resources, and we do not carry liability insurance covering the use of hazardous materials. If we fail to comply with applicable requirements, we could incur substantial costs, including civil or criminal fines and penalties, clean-up costs, or capital expenditures for control equipment or operational changes necessary to achieve or maintain compliance. Compliance with applicable environmental laws and regulations is expensive, and current or future environmental regulations may impair our research, development and production efforts, which adversely affect our business, financial condition, results of operations, and prospects.
We are subject to extensive and costly government regulation.
The products we currently market, including the vitamins, and the pharmaceutical products we are developing and planning to develop in the future, are subject to extensive and rigorous domestic government regulation, including regulation by the FDA, the Centers for Medicare & Medicaid Services, or CMS, other divisions of the U.S. Department of Health and Human Services, including its Office of Inspector General, the U.S. Department of Justice, the Departments of Defense and Veterans Affairs, to the extent our products are paid for directly or indirectly by those departments, state and local governments, and their respective foreign equivalents. The FDA regulates dietary supplements, cosmetics, and drugs under different regulatory schemes. For example, the FDA regulates the processing, formulation, safety, manufacturing, packaging, labeling, and distribution of dietary supplements and cosmetics under its dietary supplement and cosmetic authority, respectively. The FDA also regulates the research, development, pre-clinical and clinical testing, manufacture, safety, effectiveness, record keeping, reporting, labeling, storage, approval, advertising, promotion, sale, distribution, import, and export of pharmaceutical products under various regulatory provisions. If any drug products we develop are tested or marketed abroad, they will also be subject to extensive regulation by foreign governments, whether or not we have obtained FDA approval for a given product and its uses. Such foreign regulation may be equally or more demanding than corresponding U.S. regulation.
Government regulation substantially increases the cost and risk of researching, developing, manufacturing, and selling products. Our failure to comply with these regulations could result in, by way of example, significant fines, criminal and civil liability, product seizures, recalls, withdrawals, withdrawals of approvals, and exclusion and debarment from government programs. Any of these actions, including the inability of our hormone therapy drug candidates to obtain and maintain regulatory approval, would have a materially adverse effect on our business, financial condition, results of operations, and prospects.
We are subject to additional federal and state laws and regulations relating to our business, and our failure to comply with those laws could have a material adverse effect on our results of operations and financial conditions.
We are subject to additional health care regulation and enforcement by the federal government and the states in which we conduct our business. The laws that may affect our ability to operate include the following:
Further, the recently enacted PPACA, among other things, amends the intent requirement of the federal anti-kickback and criminal health care fraud statutes. A person or entity can now be found guilty of fraud or false claims under PPACA without actual knowledge of the statute or specific intent to violate it. In addition, PPACA provides that the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the false claims statutes. Possible sanctions for violation of these anti-kickback laws include monetary fines, civil and criminal penalties, exclusion from Medicare, Medicaid and other government programs and forfeiture of amounts collected in violation of such prohibitions. Any violations of these laws, or any action against us for violation of these laws, even if we successfully defend against it, could result in a material adverse effect on our reputation, business, results of operations, and financial condition.
PPACA also imposes new reporting requirements on device and pharmaceutical manufacturers to make annual public disclosures of payments to health care providers and ownership of their stock by health care providers. Failure to submit required information may result in civil monetary penalties of up to an aggregate of $150,000 per year (or up to an aggregate of $1 million per year for “knowing failures”), for all payments, transfers of value, or ownership or investment interests that are not reported. Manufacturers were required to begin data collection on August 1, 2013 and were required to report such data to CMS by March 31, 2014.
In addition, there has been a recent trend of increased federal and state regulation of payments made to physicians for marketing. Some states, such as California, Massachusetts and Vermont, mandate implementation of corporate compliance programs, along with the tracking and reporting of gifts, compensation, and other remuneration to physicians.
The scope and enforcement of these laws is uncertain and subject to change in the current environment of health care reform, especially in light of the lack of applicable precedent and regulations. We cannot predict the impact on our business of any changes in these laws. Federal or state regulatory authorities may challenge our current or future activities under these laws. Any such challenge could have a material adverse effect on our reputation, business, results of operations, and financial condition. Any state or federal regulatory review of us, regardless of the outcome, would be costly and time-consuming.
If we are not successful in attracting and retaining highly qualified personnel, we may not be able to successfully implement our business strategy.
Our ability to compete in the highly competitive pharmaceutical industry depends in large part on our ability to attract and retain highly qualified managerial, scientific, and medical personnel. In order to induce valuable employees to remain with us, we have, among other things, provided stock-based compensation that vests over time. The value to employees of stock-based compensation will be significantly affected by movements in our stock price that we cannot control and may at any time be insufficient to counteract more lucrative offers from other companies. Despite our efforts to retain valuable employees, members of our management, scientific, and medical teams may terminate their employment with us on short notice. We do not have employment agreements with a number of our key employees. As a result, most employees are employed on an at-will basis, which means that any of these employees could leave our employment at any time, with or without notice, and may go to work for a competitor. The loss of the services of any of our executive officers or other key employees could potentially harm our business, operating results, and financial condition. Our success also depends on our ability to continue to attract, retain, and motivate highly skilled scientific and medical personnel.
Any failure to adequately expand a direct sales force will impede our growth.
We expect to be substantially dependent on a direct sales force to attract new business and to manage customer relationships. We plan to expand our direct sales force and believe that there is significant competition for qualified, productive direct sales personnel with advanced sales skills and technical knowledge. Our ability to achieve significant growth in revenue in the future will depend, in large part, on our success in recruiting, training, and retaining sufficient direct sales personnel. New and future hires may not become as productive as expected, and we may be unable to hire sufficient numbers of qualified individuals in the future in the markets in which we do business. While there presently exists a high rate of unemployment, if we are unable to hire and develop sufficient numbers of productive sales personnel our business prospects could suffer.
Other pharmaceutical companies with which we compete for qualified personnel have greater financial and other resources, different risk profiles, and longer histories than we do. They also may provide more diverse opportunities and better chances for career advancement. Some of these characteristics may be more appealing to high-quality candidates than what we offer. If we are unable to continue to attract and retain high-quality personnel, our ability to commercialize drug candidates will be limited.
Our success is tied to our distribution channels.
We sell our prescription prenatal vitamin products to wholesale distributors, specialty pharmacies, specialty distributors, and chain drug stores that generally sell products to retail pharmacies, hospitals, and other institutional customers. However, over 75% of our product shipments since inception were to only four customers. Our business would be harmed if any of these customers refused to distribute our products or refused to purchase our products on commercially favorable terms to us.
A failure to maintain optimal inventory levels to meet commercial demand for our products could harm our reputation and subject us to financial losses.
Our ability to maintain optimal inventory levels to meet commercial demand depends on the performance of third-party contract manufacturers. In some instances, our products have unique ingredients used under license arrangements. If our manufacturers are unsuccessful in obtaining raw materials, if we are unable to manufacture and release inventory on a timely and consistent basis, if we fail to maintain an adequate level of product inventory, if inventory is destroyed or damaged, or if our inventory reaches its expiration date, patients might not have access to our products, our reputation and brands could be harmed, and physicians may be less likely to recommend our products in the future, each of which could have a material adverse effect on our business, financial condition, results of operations, and cash flows.
Our ability to utilize net operating loss carryforwards may be limited.
As of December 31, 2014, we had net operating loss carryforwards, or NOLs, of approximately $105.5 million available to offset future taxable income through 2034. These NOLs may be used to offset future taxable income, to the extent we generate any taxable income, and thereby reduce or eliminate our future federal income taxes otherwise payable. Section 382 of the Internal Revenue Code of 1986, as amended, imposes limitations on a corporation’s ability to utilize NOLs if it experiences an ownership change as defined in Section 382. In general terms, an ownership change may result from transactions increasing the ownership of certain stockholders in the stock of a corporation by more than 50 percent over a three-year period. In the event that an ownership change has occurred, or were to occur, utilization of our NOLs would be subject to an annual limitation under Section 382 determined by multiplying the value of our stock at the time of the ownership change by the applicable long-term tax-exempt rate. Any unused annual limitation may be carried over to later years. We may be found to have experienced an ownership change under Section 382 as a result of events in the past or the issuance of shares of our common stock in the future. If so, the use of our NOLs, or a portion thereof, against our future taxable income may be subject to an annual limitation under Section 382, which may result in expiration of a portion of our NOLs before utilization.
Our success depends on how efficiently we respond to changing consumer preferences and demand.
Our success depends, in part, on our ability to anticipate and respond to changing consumer trends and preferences. We may not be able to respond in a timely or commercially appropriate manner to these changes. Our failure to accurately predict these trends could negatively impact our inventory levels, sales, and consumer opinion of us as a source for the latest product. The success of our new product offerings depends upon a number of factors, including our ability to achieve the following:
If we do not introduce new products, make enhancements to existing products, or maintain the appropriate inventory levels to meet customers’ demand in a timely manner, our business, results of operations, and financial condition could be materially and adversely affected.
We may initiate product recalls or withdrawals, or may be subject to regulatory enforcement actions that could negatively affect our business.
We may be subject to product recalls, withdrawals, or seizures if any of the products we formulate, manufacture, or sell are believed to cause injury or illness or if we are alleged to have violated governmental regulations in the manufacture, labeling, promotion, sale, or distribution of any of our products. A recall, withdrawal, or seizure of any of our products could materially and adversely affect consumer confidence in our brands and lead to decreased demand for our products. In addition, a recall, withdrawal, or seizure of any of our products would require significant management attention, would likely result in substantial and unexpected expenditures, and could materially and adversely affect our business, financial condition, and results of operations.
We will need to grow our organization, and we may experience difficulties in managing this growth, which could disrupt our operations.
As of December 31, 2014, we had 100 employees. As our development and commercialization plans and strategies develop, we expect to expand our employee base for managerial, operational, financial, and other resources and, depending on our commercialization strategy, we may further expand our employee base for sales and marketing resources. Future growth would impose significant added responsibilities on members of management, including the need to identify, recruit, maintain, motivate, and integrate additional employees. Also, our management may need to divert a disproportionate amount of its attention away from their day-to-day activities and devote a substantial amount of time to managing these growth activities. We may not be able to effectively manage the expansion of our operations, which may result in weaknesses in our infrastructure, give rise to operational mistakes, loss of business opportunities, loss of employees and reduced productivity among remaining employees. Our growth could require significant capital expenditures and may divert financial resources from other projects, such as the development of additional drug candidates. If we are unable to effectively manage our expected growth, our expenses may increase more than expected, our ability to increase revenue could be reduced and we may not be able to implement our business strategy. Our future financial performance and our ability to commercialize our hormone therapy drug candidates, if approved, and compete effectively will depend, in part, on our ability to effectively manage any future growth in our organization.
Our employees may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements and insider trading.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include intentional failures to comply with FDA regulations, to provide accurate information about Vitamedto the FDA, to comply with federal and state health care fraud and abuse laws and regulations, to report financial information or data accurately, or to disclose unauthorized activities to us. In particular, sales, marketing, and business arrangements in the health care industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing, and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs, and other business arrangements. Employee misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. We have adopted a Code of Conduct and Ethics, but it is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with these laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
Risks Related to our Intellectual Property
Another party could develop hormone therapy products and obtain FDA regulatory exclusivity in the United States before we do, potentially preventing our ability to commercialize our hormone therapy drug candidates and other products in development.
We plan to seek to obtain market exclusivity for our hormone therapy drug candidates and any other drug candidates we develop in the future. To the extent that patent protection is not available or has expired, FDA marketing exclusivity may be the only available form of exclusivity available for these proposed products. Marketing exclusivity can delay the submission or the approval of certain marketing applications. Potentially competitive products may also be seeking marketing exclusivity and may be in various stages of development, including some more advanced than us. We cannot predict with certainty the timing of FDA approval or whether FDA approval will be granted, nor can we predict with certainty the timing of FDA approval for competing products or whether such approval will be granted. It is possible that competing products may obtain FDA approval with marketing exclusivity before we do, which could delay our ability to submit a marketing application or obtain necessary regulatory approvals, result in lost market opportunities with respect to our hormone therapy drug candidates, and materially adversely affect our business, financial condition, and results of operations.
If our efforts to protect the proprietary nature of the intellectual property covering our hormone therapy drug candidates and other products are not adequate, we may not be able to compete effectively in our market.
Our commercial success will depend in part on our ability to obtain additional patents and protect our existing patent positions as well as our ability to maintain adequate protection of other intellectual property for our hormone therapy drug candidates and other products. If we do not adequately protect our intellectual property, competitors may be able to use our technologies and erode or negate any competitive advantage we may have, which could harm our business and ability to achieve profitability. The patent positions of pharmaceutical companies are highly uncertain. The legal principles applicable to patents are in transition due to changing court precedent and legislative action, and we cannot be certain that the historical legal standards surrounding questions of validity will continue to be applied or that current defenses relating to issued patents in these fields will be sufficient in the future. Changes in patent laws in the United States, such as the America Invents Act of 2011, may affect the scope, strength, and enforceability of our patent rights or the nature of proceedings that may be brought by us related to our patent rights. In addition, the laws of some foreign countries do not protect proprietary rights to the same extent as the laws of the United States, and we may encounter significant problems in protecting our proprietary rights in these countries. We will be able to protect our proprietary rights from unauthorized use by third parties only to the extent that our proprietary technologies are covered by valid and enforceable patents or are effectively maintained as trade secrets.
These risks include the possibility of the following:
While we apply for patents covering our technologies and products, as we deem appropriate, many third parties may already have filed patent applications or have received patents in our areas of product development. These entities’ applications, patents, and other intellectual property rights may conflict with patent applications to which we have rights and could prevent us from obtaining patents or could call into question the validity of any of our patents, if issued, or could otherwise adversely affect our ability to develop, manufacture, or commercialize our hormone therapy drug candidates. In addition, if third parties file patent applications in the technologies that also claim technology to which we have rights, we may have to participate in interference, derivation, or other proceedings with the USPTO or foreign patent regulatory authorities to determine our rights in the technologies, which may be time-consuming and expensive. Moreover, issued patents may be challenged during in the courts or in post-grant proceedings at http://www.vitamedmd.com,the USPTO, or in similar proceedings in foreign countries. These proceedings may result in loss of patent claims or adverse changes to the scope of the claims.
If we, our licensors, or our strategic partners fail to obtain and http://www.vitamedmdrx.com.
In addition, mechanisms exist in much of the world permitting some form of challenge by generic drug marketers to our patents prior to, or immediately following, the expiration of any regulatory exclusivity, and generic companies are increasingly employing aggressive strategies, such as “at risk” launches to challenge relevant patent rights.
Our business also may rely on unpatented proprietary technology, know-how, and trade secrets. If the confidentiality of this intellectual property is breached, it could adversely impact our business.
If we are sued for infringing intellectual property rights of third parties, litigation will be costly and time consuming and could prevent or delay us from developing or commercializing our drug candidates.
Our commercial success depends, in part, on our not infringing the patents and proprietary rights of other parties and not breaching any collaboration or other agreements we have entered into with regard to our technologies and products. We are aware of numerous third-party U.S. and non-U.S. issued patents and pending applications that exist in the areas of hormone therapy, including compounds, formulations, treatment methods, and synthetic processes, which may be applied towards the synthesis of hormones. Patent applications are confidential when filed and remain confidential until publication, approximately 18 months after initial filing, while some patent applications remain unpublished until issuance. As such, there may be other third-party patents and pending applications of which we are currently unaware with claims directed towards composition of matter, formulations, methods of manufacture, or methods for treatment related to the use or manufacture of our products or drug candidates. Therefore, we cannot ever know with certainty the nature or existence of every third-party patent filing. We cannot provide assurances that we or our partners will be free to manufacture or market our drug candidates as planned or that we or our licensors’ and partners’ patents will not be opposed or litigated by third parties. If any third-party patent was held by a court of competent jurisdiction to cover aspects of our materials, formulations, methods of manufacture, or methods of treatment related to the use or manufacture of any of our drug candidates, the holders of any such patent may be able to block our ability to develop and commercialize the applicable drug candidate unless we obtained a license or until such patent expires or is finally determined to be held invalid or unenforceable. There can be no assurances that we will be able to obtain a license to such patent on favorable terms or at all. Failure to obtain such license may have a material adverse effect on our business.
There is a smaller reporting company,substantial amount of litigation involving intellectual property in the pharmaceutical industry generally. If a third party asserts that we infringe its patents or other proprietary rights, we could face a number of risks that could adversely affect our business, financial condition, results of operations, and as such,prospects, including the following:
We are party from time to time to legal proceedings relating to our intellectual property, and third parties in the future may file claims asserting that our technologies, processes, or products infringe on their intellectual property. We cannot predict whether third parties will assert these claims against us or our strategic partners or against the licensors of technology licensed to us, or whether those claims will harm our business. In addition, the outcome of intellectual property litigation is subject to uncertainties that cannot be adequately quantified in advance. If we or our partners were to face infringement claims or challenges by third parties relating to our drug candidates, an adverse outcome could subject us to significant liabilities to such third parties, and force us or our partners to curtail or cease the development of some or all of our drug candidates, which could adversely affect our business, financial condition, results of operations, and prospects.
We intend to submit NDAs for our hormone therapy drug candidates, assuming that the clinical data justify submission, under Section 505(b)(2), which was enacted as part of the Drug Price Competition and Patent Term Restoration Act of 1984, otherwise known as the Hatch-Waxman Act. Section 505(b)(2) permits the filing of an NDA when at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference. To the extent that a Section 505(b)(2) NDA relies on clinical trials conducted for a previously approved drug product or the FDA’s prior findings of safety and effectiveness for a previously approved drug product, the Section 505(b)(2) applicant must submit patent certifications in its Section 505(b)(2) NDA with respect to any patents for the approved product on which the application relies that are listed in the FDA’s publication, Approved Drug Products with Therapeutic Equivalence Evaluations, commonly referred to as the Orange Book. Specifically, the applicant must certify for each listed patent that (i) the required patent information has not been filed; (ii) the listed patent has expired; (iii) the listed patent has not expired, but will expire on a particular date and approval is not sought until after patent expiration; or (iv) the listed patent is invalid, unenforceable or will not be infringed by the proposed new product. A certification that the new product will not infringe the previously approved product’s listed patent or that such patent is invalid or unenforceable is known as a Paragraph IV certification.
If the Section 505(b)(2) NDA applicant has provided a Paragraph IV certification to the FDA, the applicant must also send notice of the Paragraph IV certification to the owner of the referenced NDA for the previously approved product and relevant patent holders within 20 days after the Section 505(b)(2) NDA has been accepted for filing by the FDA. The NDA and patent holders may then initiate a patent infringement suit against the Section 505(b)(2) applicant. Under the FDCA, the filing of a patent infringement lawsuit within 45 days of receipt of the notification regarding a Paragraph IV certification automatically prevents the FDA from approving the Section 505(b)(2) NDA for 30 months beginning on the date the patent holder receives notice, or until a court deems the patent unenforceable, invalid or not infringed, whichever is earlier. The court also has the ability to shorten or lengthen the 30 month period if either party is found not to be reasonably cooperating in expediting the litigation. Thus, the Section 505(b)(2) applicant may invest a significant amount of time and expense in the development of its product only to be subject to significant delay and patent litigation before its product may be commercialized. Alternatively, if the NDA or relevant patent holder does not file a patent infringement lawsuit within the specified 45 day period, the FDA may approve the Section 505(b)(2) application at any time.
If we cannot certify that all of the patents listed in the Orange Book for the approved products referenced in the NDAs for each of our hormone therapy drug candidates have expired, we will be compelled to include a Paragraph IV certification in the NDA for such drug candidate. Our inability to certify that all of the patents listed in the FDA’s Orange Book for approved products referenced in the NDAs for each of our hormone therapy drug candidates could have a serious and significant adverse effect on the timing for obtaining approval of our hormone therapy drug candidates. For example, at least one approved product that may be referenced in our 505(b)(2) application as a reference product for our vaginal suppository estradiol product currently lists unexpired patents in the Orange Book.
We may be required to file lawsuits or take other actions to protect or enforce our patents or the patents of our licensors, which could be expensive and time-consuming.
Competitors may infringe our patents or the patents of our licensors. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time-consuming. Moreover, there can be no assurance that we will have sufficient financial or other resources to file and pursue such infringement claims, which typically last for years before they are concluded. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection, particularly those relating to pharmaceuticals, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally.
In addition, in an infringement proceeding, a court may decide that a patent of ours or our licensors is not valid or is unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that our patents, or those of our licensors, do not cover the technology in question or on other grounds. An adverse result in any litigation or defense proceedings could put one or more of our patents, or those of our licensors, at risk of being invalidated, held unenforceable, or interpreted narrowly and could put our patent applications, or those of our licensors, at risk of not issuing. Moreover, we may not be able to prevent, alone or with our licensors, misappropriation of our proprietary rights, particularly in countries in which the laws may not protect those rights as fully as in the United States or in those countries in which we do not file national phase patent applications. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, if securities analysts or investors perceive public announcements of the results of hearings, motions, or other interim proceedings or developments to be negative, the price of our common stock could be adversely affected. The occurrence of any of the above could adversely affect our business, financial condition, results of operations, and prospects.
If we are unable to protect the confidentiality of certain information, the value of our products and technology could be materially adversely affected.
We also rely on trade secrets, know-how, and continuing technological advancement to develop and maintain our competitive position. To protect this competitive position, we regularly enter into confidentiality and proprietary information agreements with third parties, including employees, independent contractors, suppliers, and collaborators. We cannot, however, ensure that these protective arrangements will be honored by third parties, and we may not have adequate remedies if these arrangements are breached. In addition, enforcement of claims that a third party has illegally obtained and is using trade secrets, know-how, or technological advancements is expensive, time-consuming, and uncertain. Non-U.S. courts are sometimes less willing than U.S. courts to protect this information. Moreover, our trade secrets, know-how, and technological advancements may otherwise become known or be independently developed by competitors in a manner providing us with no practical recourse against the competing parties. If any such events were to occur, they could adversely affect our business, financial condition, results of operations, and prospects.
We may be subject to claims that our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
As is common in the pharmaceutical industry, we employ individuals who were previously employed at other biotechnology or pharmaceutical companies, including our competitors or potential competitors. We may be subject to claims that these employees, or we, have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Such claims may lead to material costs for us, or an inability to protect or use valuable intellectual property rights, which could adversely affect our business, financial condition, results of operations, and prospects.
Risks Related to Ownership of Our Common Stock
The market price of our common stock may be highly volatile, and you could lose all or part of your investment.
The trading price of our common stock on NYSE MKT is likely to be volatile. This volatility may prevent you from being able to sell your shares at or above the price you paid for your shares. Our stock price could be subject to wide fluctuations in response to a variety of factors, which include the following:
In addition, the stock market in general and the stock of biotechnology companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies. Broad market and industry factors may negatively affect the market price of our common stock, regardless of our actual operating performance.
Our principal stockholders and management own a significant percentage of our stock and will be able to exert significant control over matters subject to stockholder approval.
At December 31, 2014, our executive officers, directors, holders of 5% or more of our stock, and their affiliates beneficially owned approximately 61.1% of our common stock on an as converted basis. These stockholders may be able to determine the outcome of all matters requiring stockholder approval. For example, these stockholders may be able to control elections of directors, amendments of our organizational documents, or approval of any merger, sale of assets, or other major corporate transaction. This may prevent or discourage unsolicited acquisition proposals or offers for our common stock that you may feel are in your best interest as one of our stockholders. In addition, pursuant to this item.a Securities Purchase Agreement dated September 26, 2012, we granted certain of our stockholders the right, expiring in October 2015, if they elect, to purchase on the same terms as in any offering of our common stock, a number of shares of common stock that is sufficient to maintain their respective pro rata ownership percentage of our common stock.
If we fail to maintain proper internal controls, our ability to produce accurate financial statements or comply with applicable regulations could be impaired.
Pursuant to Section 404 of the Sarbanes-Oxley Act, our management is required annually to deliver a report that assesses the effectiveness of our internal control over financial reporting and our independent registered public accounting firm is required annually to deliver an attestation report on the effectiveness of our internal control over financial reporting. If we are unable to maintain effective internal control over financial reporting or if our independent auditors are unwilling or unable to provide us with an attestation report on the effectiveness of internal control over financial reporting for future periods as required by Section 404 of the Sarbanes-Oxley Act, we may not be able to produce accurate financial statements, and investors may therefore lose confidence in our operating results, our stock price could decline and we may be subject to litigation or regulatory enforcement actions.
If securities or industry analysts do not publish research or publish inaccurate or unfavorable research about our business, our stock price and trading volume could decline.
The trading market for our common stock will depend in part on the research and reports that securities or industry analysts publish about us or our business. If one or more of the analysts who cover us downgrade our stock or publish inaccurate or unfavorable research about our business, our stock price would likely decline. If one or more of these analysts cease coverage of us or fail to publish reports on us regularly, we could lose visibility in the financial markets, which might cause our stock price and trading volume to decline.
We do not intend to pay dividends on our common stock so any returns will be limited to the value of our stock.
We have never declared or paid any cash dividends on our common stock. We currently anticipate that we will retain any future earnings for the development, operation, and expansion of our business and do not anticipate declaring or paying any cash dividends for the foreseeable future. Any return to stockholders will be limited to the value of their stock.
Some provisions of our charter documents and Nevada law may have anti-takeover effects that could discourage an acquisition of us by others, even if an acquisition would be beneficial to our stockholders and may prevent attempts by our stockholders to replace or remove our current management.
Provisions in our articles of incorporation and bylaws, as well as certain provisions of Nevada law, could make it more difficult for a third party to acquire us or increase the cost of acquiring us, even if an acquisition would benefit our stockholders, and could also make it more difficult to remove our current management. These provisions in our articles of incorporation and bylaws include the following:
prohibiting cumulative voting in the election of directors, which would otherwise allow less than a majority of stockholders to elect director candidates; and
In addition, we are subject to Nevada’s Combination with Interested Stockholders statute (Nevada Revised Statute Sections 78.411 - 78.444), which prohibits an “interested stockholder” from entering into a “combination” with a company, unless certain conditions are met. An “interested stockholder” is a person who, together with affiliates and associates, beneficially owns (or within the prior two years, did beneficially own) 10% or more of the corporation’s capital stock entitled to vote.
|
None.
|
Our corporate headquarters is located at 951 Broken Sound Parkway NW, Suite 320,in Boca Raton, FL 33487. On July 9, 2009, VitaMed entered into a 45-monthFlorida, where we lease for approximately 7,13017,686 square feet of office space (the “Lease”). Overpursuant to a 63 month non-cancelable operating lease that commenced on July 1, 2013, was amended on February 18, 2015, and expires on September 30, 2018. The primary functions performed at this location are executive, administrative, accounting, treasury, marketing, and human resources.
We believe that our current facility is in good working order and is capable of supporting our operations for the term of the Lease, the Company will pay an average monthly cost of $9,352 which includes base rent, common area fees, taxes and insurance. Terms of the Lease provide for an extension for an additional two-year period. The Company’s management believes that the leased premises are suitable and adequateforeseeable future.
| Legal Proceedings |
From time to meet its needs.
|
Not applicable.
Market Information
Since April 23, 2013, our common stock has been listed on the OTCQBNYSE MKT under the symbol “TXMD.” Prior to that time, our common stock was quoted on the OTCQB. The following table showssets forth for the price rangeperiods indicated the high and low bid or sales prices of our Common Stock for each quarter duringcommon stock on the years ended December 31, 2011OTCQB and 2010.the NYSE MKT, as applicable. The below quotations reflect inter-dealer prices, without retail mark-up, mark-down or commission, and may not represent actual transactions. Prices listed are historic prices
| High | Low | |||||||
| 2014 | ||||||||
| Fourth Quarter | $ | 4.95 | $ | 3.44 | ||||
| Third Quarter | $ | 6.29 | $ | 3.88 | ||||
| Second Quarter | $ | 6.35 | $ | 3.42 | ||||
| First Quarter | $ | 9.01 | $ | 4.86 | ||||
| 2013 | ||||||||
| Fourth Quarter | $ | 5.50 | $ | 2.86 | ||||
| Third Quarter | $ | 3.18 | $ | 2.03 | ||||
| Second Quarter | $ | 3.23 | $ | 1.73 | ||||
| First Quarter | $ | 3.70 | $ | 1.65 | ||||
On March 6, 2015, there were approximately 277 shareholders of record and on February 23, 2015, there were not adjusted to reflect the 1:100 Reverse Split that was effective on October 3, 2011.
| Quarter Ended | High | Low | ||||||
| Fiscal Year 2011 | ||||||||
| Fourth Quarter | $ | 1.70 | $ | 0.01 | ||||
| Third Quarter | $ | 0.04 | $ | 0.01 | ||||
| Second Quarter | $ | 0.07 | $ | 0.01 | ||||
| First Quarter | $ | 0.10 | $ | 0.02 | ||||
| Fiscal Year 2010 | ||||||||
| Fourth Quarter | $ | 0.15 | $ | 0.03 | ||||
| Third Quarter | $ | 0.90 | $ | 0.06 | ||||
| Second Quarter | $ | 1.34 | $ | 0.25 | ||||
| First Quarter | $ | 1.60 | $ | 0.10 | ||||
Dividends
Historically, we had 84,608,826 outstanding shares of Common Stock.
Performance Graph
The following line graph compares cumulative total shareholder return for the five years ended December 31, 2014 for (i) our common stock; (ii) NASDAQ Stock Market (US Companies); (iii) NASDAQ Pharmaceutical Index; and (iv) Peer Group (includes: Acorda Therapeutics, Inc., AMAG Pharmaceuticals, Inc., Arena Pharmaceuticals, Inc., Dendreon Corporation, Dyax Corporation, Exelixis, Inc., Halozyme Therapeutics, Inc., Orexigen Therapeutics, Ind., Spectrium Pharmaceuticals, Inc., and VIVUS Inc.) does not include the following companies that our Boardwere included in the prior year's performance graph: Amarillo Biosciences Inc., which filed for bankruptcy protection in October 2013, Avanir Pharmaceuticals, Inc., which entered into an agreement and plan of Directors deems relevant.
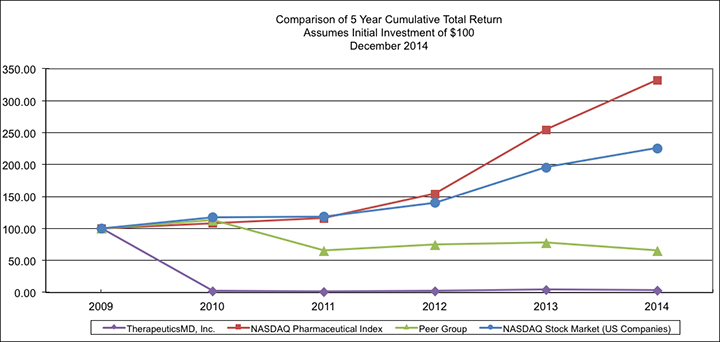
The following line graph compares cumulative total shareholder return for the period beginning when our common stock became listed on the NYSE MKT exchange (April 23, 2013) and consultantsended December 31, 2014 for (i) our common stock; (ii) NASDAQ Stock Market (US Companies); (iii) NASDAQ Pharmaceutical Index; and (iv) our Peer Group (includes: Acorda Therapeutics, Inc., AMAG Pharmaceuticals, Inc., Arena Pharmaceuticals, Inc., Dendreon Corporation, Dyax Corporation, Exelixis, Inc., Halozyme Therapeutics, Inc., Orexigen Therapeutics, Ind., Spectrium Pharmaceuticals, Inc., and VIVUS Inc.) does not include the following companies that were included in the prior year’s performance graph: Amarillo Biosciences Inc., which filed for bankruptcy protection in October 2013, Avanir Pharmaceuticals, Inc., which entered into an agreement and plan of merger to be acquired in December 2014 and was acquired in January 2015, and Cadence Pharmaceuticals Inc., which was acquired in March 2014. The graph assumes $100 invested on April 23, 2013 and includes reinvestment of dividends. Measurement points are April 23, 2013 and the last trading day of the Company who are ablefiscal years ended December 31, 2014 and 2013 and each of the following quarters ended therein beginning with the quarter ended June 30, 2013. The stock price performance on the following graph is not necessarily indicative of future stock price performance.
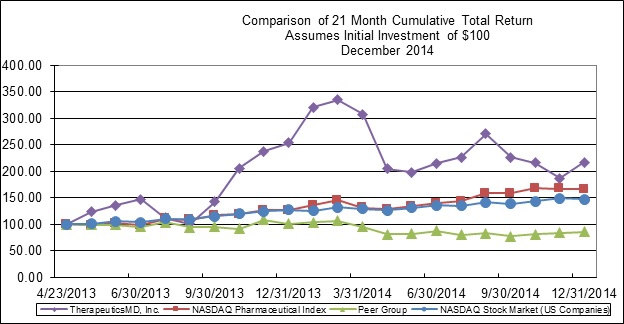
The performance graphs shall not be deemed “filed” for purposes of Section 18 of the Exchange Act or otherwise subject to contribute towards the creationliability of that section. The performance graphs will not be deemed incorporated by reference into any filing of our company under the Exchange Act or who have created stockholder value by providing them stock optionsthe Securities Act.
| Item 6. | Selected Financial Data |
The following table sets forth selected consolidated financial and other stockdata as of and cash incentives (the “Awards”). The Awards available underfor the LTIP consistperiods indicated. You should read the following information together with the more detailed information contained in “Management’s Discussion and Analysis of stock options, stock appreciation rights, restricted stock, restricted stock units, performance stock, performance units, EVA awards,Financial Condition and other stock or cash awards as described in the LTIP. There are 25,000,000 shares authorized for issuance thereunder. The LTIP is administered by the Company’s BoardResults of Directors, who determine: (i) the persons to be granted stock options under the Plan; (ii) the number of shares subject to each optionOperations” and our consolidated financial statements and the exercise pricerelated notes included elsewhere in this Annual Report. The consolidated statements of each option; (iii) whetheroperations for the stock option will be exercisable at any time duringyears ended December 31, 2014, 2013, and 2012, and the option period of ten (10) years or whether it shall be exercisable in installments or by vesting only.
| Plan Category | Number of Securities to be issued upon exercise of outstanding options (a) | Weighted-average exercise price of outstanding options (b) | Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a) (c) | |||||||||
| Equity compensation plans not approved by security holders | -0- | $ | -0- | -0- | ||||||||
| Equity compensation plan approved by security holders: LTIP | 10,536,161 | $ | 0.16 | 14,317,782 | ||||||||
| Total | 10,536,161 | $ | 0.16 | 14,317,782 | ||||||||
THERAPEUTICSMD, INC. AND SUBSIDIARIES
(in thousands, except per share data)
| Year Ended December 31, | ||||||||||||||||||||
| 2014 | 2013 | 2012 | 2011 | 2010 | ||||||||||||||||
| (Restated) | ||||||||||||||||||||
| Consolidated Statements of Operations Data: | ||||||||||||||||||||
| Revenue, net | $ | 15,026 | $ | 8,776 | $ | 3,818 | $ | 2,088 | $ | 1,242 | ||||||||||
| Cost of goods sold | 3,672 | 1,960 | 1,348 | 947 | 556 | |||||||||||||||
| Gross profit | 11,354 | 6,816 | 2,470 | 1,141 | 686 | |||||||||||||||
| Operating expenses: | ||||||||||||||||||||
| Sales, general, and administration | 22,124 | 19,015 | 14,070 | 6,406 | 3,465 | |||||||||||||||
| Research and development | 43,219 | 13,551 | 4,492 | 107 | 65 | |||||||||||||||
| Depreciation and amortization | 52 | 58 | 56 | 55 | 23 | |||||||||||||||
| Total operating expense | 65,395 | 32,624 | 18,618 | 6,568 | 3,553 | |||||||||||||||
| Operating loss | (54,041 | ) | (25,808 | ) | (16,148 | ) | (5,427 | ) | (2,867 | ) | ||||||||||
| Other (expense) income | (176 | ) | (2,611 | ) | (18,972 | ) | (7,486 | ) | ||||||||||||
| Net loss | $ | (54,217 | ) | $ | (28,419 | ) | $ | (35,120 | ) | $ | (12,913 | ) | $ | (2,867 | ) | |||||
| Net loss per share, basic and diluted | $ | (0.36 | ) | $ | (0.22 | ) | $ | (0.38 | ) | $ | (0.21 | ) | $ | (0.07 | ) | |||||
| Weighted average number of common shares outstanding | 149,727 | 127,570 | 91,630 | 62,516 | 38,289 | |||||||||||||||
| Consolidated Balance Sheet Data (at end of period) | ||||||||||||||||||||
| Total assets | $ | 59,079 | $ | 62,016 | $ | 5,926 | $ | 1,439 | $ | 1,197 | ||||||||||
| Total liabilities | $ | 10,690 | $ | 7,318 | $ | 7,359 | $ | 3,151 | $ | 233 | ||||||||||
| Total stockholder' surplus (deficit) | $ | 48,389 | $ | 54,698 | $ | (1,433 | ) | $ | (1,712 | ) | $ | 964 | ||||||||
| Other Data: | ||||||||||||||||||||
| Capital expenditures | $ | 617 | $ | 480 | $ | 273 | $ | 38 | $ | 27 | ||||||||||
| Working Capital (deficit)(end of period) | $ | 45,545 | $ | 52,085 | $ | 1,015 | $ | (1,914 | ) | $ | 826 | |||||||||
Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
You should read the following discussion and capital expenditures were $5.0 millionanalysis in conjunction with the information set forth under “Selected Consolidated Financial and $2.9 millionOther Data” and our consolidated financial statements and the notes to those financial statements included elsewhere in this Annual Report. This discussion contains forward-looking statements based upon current expectations that involve risks and uncertainties. See “Special Note Regarding Forward-Looking Statements.” Our actual results may differ materially from those contained in or implied by any forward-looking statements as a result of various factors, including the risks and uncertainties described under “Risk Factors” elsewhere in this Annual Report.
Company Overview
We are a women’s health care product company focused on creating and commercializing products targeted exclusively for women. Currently, we are focused on conducting the clinical trials necessary for regulatory approval and commercialization of advanced hormone therapy pharmaceutical products. The current drug candidates used in our clinical trials are designed to alleviate the symptoms of and reduce the health risks resulting from menopause-related hormone deficiencies, including hot flashes, osteoporosis, and vaginal dryness. We are developing these hormone therapy drug candidates, which contain estradiol and progesterone alone or in combination, with the aim of demonstrating equivalent clinical efficacy at lower doses, thereby enabling an enhanced side effect profile compared with competing products. Our drug candidates are created from a platform of hormone technology that enables the administration of hormones with high bioavailability alone or in combination. In addition, we manufacture and distribute branded and generic prescription prenatal vitamins, as well as or OTC vitamins.
Research and Development – Overview
We have obtained FDA acceptance of our IND applications to conduct clinical trials for four of our hormone therapy drug candidates: TX-001HR, our oral combination of progesterone and estradiol; TX-002HR, our oral progesterone alone; TX-003HR, our oral estradiol alone; and TX-004HR, our vaginal suppository estradiol alone.
We are currently conducting phase 3 clinical trials for TX-001HR and TX-004HR. In July 2014, we suspended enrollment in the phase 3 clinical trial for TX-002HR and in October 2014 we temporarily stopped the trial in order to update the phase 3 protocol based on discussions with the FDA. We have no current plans to conduct clinical trials for TX-003HR.
TX-001HR, our combination estradiol and progesterone drug candidate, is undergoing clinical trials for the yearstreatment of moderate to severe vasomotor symptoms due to menopause, including hot flashes, night sweats, sleep disturbances, and vaginal dryness for post-menopausal women with an intact uterus. The hormone therapy drug candidate is chemically identical to the hormones that naturally occur in a woman’s body, namely estradiol and progesterone, and is being studied as a continuous-combined regimen, in which the combination of estrogen and progesterone are taken together in one product daily. If approved by the FDA, we believe this would represent the first time a combination product of estradiol and progesterone (bioidentical to the estradiol and progesterone produced by the ovaries) would be approved for use in a single combined product.
On September 5, 2013, we initiated the REPLENISH trial, a multicenter, double-blind, placebo-controlled, phase 3 study of TX-001HR in postmenopausal women with an intact uterus. The study is designed to evaluate the efficacy of TX-001HR for the treatment of moderate to severe vasomotor symptoms due to menopause and the endometrial safety of TX-001HR. Patients are assigned to one of five treatment arms, four active and one placebo, and receive study medication for 12 months. The primary endpoint for the reduction of endometrial hyperplasia is an incidence of endometrial hyperplasia of less than 1% at 12 months, as determined by endometrial biopsy. The primary endpoint for the treatment of moderate to severe vasomotor symptoms is the mean change of frequency and severity of moderate to severe vasomotor symptoms at weeks four and 12 compared to placebo, as measured by the number and severity of hot flushes. Only subjects experiencing a minimum daily frequency of seven moderate to severe hot flushes at screening are included in the vasomotor symptoms analysis, while all subjects are included in the endometrial hyperplasia analysis. The secondary endpoints include reduction in sleep disturbances and improvement in quality of life measures, night sweats and vaginal dryness, measured at 12 weeks, six months and 12 months. We intend to enroll approximately 1,750 patients at approximately 100 sites. We currently anticipate that enrollment in the REPLENISH Trial will be completed in the first half of 2015 and that results of the trial will be reported in the first half of 2016.
Based on such timeline and successful reports of the trial, we would anticipate filing an NDA for TX-001HR during the first-half of 2016 and that such NDA would be approved by the FDA during the first-half of 2017.
TX-002HR is a natural progesterone formulation for the treatment of secondary amenorrhea without the potentially allergenic component of peanut oil. The hormone therapy drug candidate is chemically identical to the hormones that naturally occur in a woman's body. We believe it will be similarly effective to traditional treatments, but may demonstrate efficacy at lower dosages. In January 2014, we began recruitment of patients in the SPRY trial, a phase 3 clinical trial designed to measure the safety and effectiveness of TX-002HR in the treatment of secondary amenorrhea. During the first two quarters of 2014, the SPRY trial encountered enrollment challenges because of IRB approved clinical trial protocols and FDA inclusion and exclusion criteria. In July 2014, we temporarily suspended enrollment, and in October 2014 we stopped the SPRY trial in order to update the phase 3 protocol based on discussions with the FDA. We intend to update the phase 3 protocol to, among other things, target only those women with secondary amenorrhea due to polycystic ovarian syndrome and to amend the primary endpoint of the trial. We believe that the updated phase 3 protocol, if approved by the FDA, will allow us to mitigate the enrollment challenges in, and shorten the duration of, the SPRY trial. However, there can be no assurance that the FDA will approve the updated phase 3 protocol that we intend to propose.
TX-004HR is a vaginal suppository estradiol drug candidate for the treatment of VVA in post-menopausal women with vaginal linings that do not receive enough estrogen. We believe that our drug candidate will be at least as effective as the traditional treatments for VVA because of an early onset of action with less systemic exposure inferring a greater probability of dose administration to the target tissue, and it will have an added advantage of being a simple, easier to use dosage form versus traditional VVA treatments. We initiated the REJOICE trial, a multicenter, double-blind, placebo-controlled phase 3 clinical trial during the third quarter of 2014 to assess the safety and efficacy of TX-004HR for the treatment of moderate to severe dyspareunia, or painful intercourse, as a symptom of VVA due to menopause. We are conducting a single 12 week study, evaluating three different doses of estradiol: 4 mcg, 10 mcg and 25 mcg versus placebo. The FDA has to date noted that in order to approve a drug based on a single trial, the trial would need to show statistical significance at a 0.01 level. The study has been designed to include four primary endpoints: the reduction of vaginal pH levels to less than 5.0, an increase in superficial cells, a decrease in parabasal cells and the improvement of dyspareunia. If approved, the 4 mcg formulation would represent a lower effective dose than the currently available VVA therapies approved by the FDA. The trial is designed to enroll approximately 700 patients across approximately 100 sites. We currently anticipate that enrollment in the REJOICE trial will be complete during the second quarter of 2015 and that results of the trial will be reported during the third quarter of 2015. Based on such timeline and successful reports of the trial, we would anticipate filing an NDA for TX-004HR during the fourth quarter of 2015 and that such NDA would be approved by the FDA during the fourth quarter of 2016.
Research and Development Expenses
A significant portion of our operating expenses to date have been incurred in research and development activities. Research and development expenses relate primarily to the discovery and development of our drug products. Our business model is dependent upon our company continuing to conduct a significant amount of research and development. Until one of our drug products receives IND approval from the FDA, products costs are listed as “Other Research and Development” costs in the accompanying condensed consolidated financial statements. Our research and development expenses consist primarily of expenses incurred under agreements with CROs, investigative sites, and consultants that conduct our clinical trials and a substantial portion of our preclinical studies; employee-related expenses, which include salaries and benefits, and non-cash share-based compensation; the cost of developing our chemistry, manufacturing and controls capabilities, and acquiring clinical trial materials; and costs associated with other research activities and regulatory approvals.
We make payments to the CROs based on agreed upon terms that may include payments in advance of a study starting date. Nonrefundable advance payments for goods and services that will be used in future research and development activities are expensed when the activity has been performed or when the goods have been received rather than when the payment is made. Advance payments to be expensed in future research and development activities were $1,175,082 and $2,606,405, at December 31, 2014 and December 31, 2013, respectively.
The following table indicates our research and development expense by project/category for the periods indicated (in thousands):
| Year Ended December 31, (000s) | ||||||||
| 2014 | 2013 | |||||||
| TX-001HR | 26,123 | 4,809 | ||||||
| TX-002HR | 1,443 | 1,059 | ||||||
| TX-004HR | 3,984 | — | ||||||
| Other research and development | 11,669 | 7,683 | ||||||
| Total research and development | $ | 43,219 | $ | 13,551 | ||||
Research and development expenditures will continue to be significant as we continue development of our drug candidates and advance the development of our proprietary pipeline of novel drug candidates. We expect to incur significant research and development costs as we develop our drug pipeline, complete the ongoing clinical trials of our drug candidates, conduct our planned phase 3 clinical trials, subject to receiving input from regulatory authorities, and prepare regulatory submissions.
The costs of clinical trials may vary significantly over the life of a project owing to factors that include, but are not limited to, the following: per patient trial costs, the number of patients that participate in the trials; the number of sites included in the trials; the length of time each patient is enrolled in the trial; the number of doses that patients receive; the drop-out or discontinuation rates of patients; the amount of time required to recruit patients for the trial, the duration of patient follow-up; and the efficacy and safety profile of the drug candidate. We base our expenses related to clinical trials on estimates that are based on our experience and estimates from CROs and other third parties.
Results of Operations
Comparison of Years Ended December 31, 2014, 2013, and 2012:
Year ended December 31, 2011 and 2010, respectively.
| Year Ended December 31, | ||||||||||||
| 2014 | 2013 | Change | ||||||||||
| (000s) | ||||||||||||
| Revenue | $ | 15,026 | $ | 8,776 | 6,250 | |||||||
| Cost of goods sold | 3,672 | 1,960 | 1,712 | |||||||||
| Operating expenses | 65,395 | 32,624 | 32,771 | |||||||||
| Operating loss | (54,041 | ) | (25,808 | ) | (28,233 | ) | ||||||
| Financing Costs | (260 | ) | (1,504 | ) | 1,244 | |||||||
| Interest expense | — | (1,166 | ) | 1,166 | ||||||||
| Other income (expense) net | 84 | 59 | 25 | |||||||||
| Net loss | $ | (54,217 | ) | $ | (28,419 | ) | $ | (25,798 | ) | |||
Revenue
Revenue for year ended December 31, 2014 increased by approximately $6,250,000, or 71%, to approximately $15,026,000, compared with approximately $8,776,000 for the operationyear ended December 31, 2013. Of this $6,250,000, approximately $2,003,494, or 32%, was attributable to an increase in the average sales price of our current business planexisting products, and approximately $4,247,127, or 68%, was attributable to sales of new products introduced during the year ended December 31, 2014.
Cost of Goods Sold
Cost of goods sold increased by approximately $1,712,000, or 87%, to approximately $3,672,000 for the year ended December 31, 2014, compared with approximately $1,960,000 for the year ended December 31, 2013. Our gross margins decreased to approximately 76% for the year ended December 31, 2014, compared to approximately 78% for the year ended December 31, 2013. The gross margin change was primarily attributable to increases in June 2008distribution related costs.
Operating Expenses
Our principal operating costs included the following items as a percentage of total operating expenses.
| Year Ended December 31, | ||||||||
| 2014 | 2013 | |||||||
| Human resource related costs | 16 | % | 33 | % | ||||
| Sales and marketing costs, excluding human resource costs | 9 | % | 15 | % | ||||
| Product research and development costs | 66 | % | 41 | % | ||||
| Professional fees and consulting costs | 4 | % | 4 | % | ||||
| Other operating expenses | 5 | % | 7 | % | ||||
Operating expenses increased by approximately $32,771,000, or 100%, to approximately $65,395,000 for the year ended December 31, 2014, compared with approximately $32,624,000 for year ended December 31, 2013, as a result of the following items:
| (000s) | |||
| Increase in product research and development costs | $ | 29,668 | |
| Increase in human resource related costs | 509 | ||
| Increase in sales and marketing, excluding human resource costs | 702 | ||
| Increase in professional and consulting costs | 852 | ||
| Increase in all other operating expenses | 1,040 | ||
| $ | 32,771 |
Research and development costs for the year ended December 31, 2014 increased by approximately $29,668,000, or 219%, to approximately $43,219,000, primarily as a result of the continuing phase 3 clinical trial of TX-001HR, the partial year phase 3 clinical trial for TX-002HR, and phase 1 and partial year phase 3 clinical trial for TX-004HR. Research and developments costs during the year ended December 31, 2014 included the following research and development projects:
During the year ended December 31, 2014 and the period February 2013 (project inception) through December 31, 2014, we have not yet attainedincurred approximately $26,123,000 and $30,932,000, respectively, in research and development costs with respect to TX-001HR, our combination estradiol and progesterone drug candidate.
During the year ended December 31, 2014 and the period April 2013 (project inception) through December 31, 2014, we have incurred approximately $1,443,000 and $2,502,000, respectively, in research and development costs with respect to TX-002HR, our progesterone only drug candidate.
During the year ended December 31, 2014 and since the project’s inception in August 2014, we have incurred approximately $3,984,000 in research and development costs with respect to TX-004HR, our vaginal suppository estradiol drug candidate.
For a leveldiscussion of revenuethe nature of efforts and steps necessary to allow uscomplete these projects, see “Item 1. Business — Research and Development.” For a discussion of the risks and uncertainties associated with completing development of our products, see “Item 1A. Risk Factors — Risks Related to meetOur Business.” For a discussion of the extent and nature of additional resources that we may need to obtain if our current overhead. Basedliquidity is not expected to be sufficient to complete these projects, see “— Liquidity and Capital Resources.” For a discussion as to whether a future milestone such as completion of a development phase, date of filing an NDA with a regulatory agency or approval from a regulatory agency can be reliably determined, see “Item 1. Business — Our Hormone Therapy Drug Candidates,” “Item 1. Business — Products in Development” and “Item 1. Business — Pharmaceutical Regulation.” Future milestones, including NDA submission dates, are not easily determinable as such milestones are dependent on various factors related to our currentclinical trials, including the timing of ongoing patient recruitment efforts to find eligible subjects for the applicable trials.
Human resource related costs, including salaries and benefits, increased by approximately $509,000, or 5%, to approximately $10,870,000, primarily as a result of an increase in salary and wages and related cost of $1,060,000 associated with additional employees required for our clinical trials, partially offset by a decrease in amortization of non-cash compensation totaling approximately $551,000 related to employee stock options issued during 2014 as compared to 2013.
Sales and marketing plancosts increased approximately $702,000, or 14%, to approximately $5,717,000, primarily as a result of expanded marketing, advertising, education, and expectedtraining. Many of these incurred added costs were associated with our new prenatal products introduced in 2014.
Professional and consulting costs increased approximately $852,000, or 67%, to approximately $2,368,000 as a result of additional costs incurred for legal, consulting, and regulatory compliance.
All other costs increased approximately $1,040,000, or 48%, to approximately $3,221,000 as a result of additional costs incurred for rent, travel, corporate communications, insurance, and contract labor.
Operating Loss
As a result of the foregoing, our operating loss increased approximately $28,233,000, or 109%, to approximately $54,041,000 for the year ended December 31, 2014, compared with approximately $25,808,000 for the year ended December 31, 2013, primarily as a result increased research and development costs associated with our continued development of our hormone therapy drug candidates, partially offset by increased revenue from sales demand,of our prenatal vitamin products.
As a result of the continued development of our hormone therapy drug candidates, we do not contemplate attaining profitable operationsanticipate that we will continue to have operating losses for the near future until 2013,our hormone therapy drug candidates are approved by the FDA and brought to market, although there is no assurance that we will attain such approvals or that any marketing of our hormone therapy drug candidates, if approved, will be successful.
Financing Costs
Financing costs decreased approximately $1,244,000, or 83%, to approximately $260,000 for the year ended December 31, 2014, compared with approximately $1,504,000 for the year ended December 31, 2013, primarily as a result of the amortization of the costs associated with warrants granted in 2013 in connection with a $10,000,000 revolving line of credit.
Interest Expense
We did not incur interest expense for the year ended December 31, 2014, compared with approximately $1,166,000 in interest expense incurred for the year ended December 31, 2013, as a result of the retirement of our debt during 2013.
Net Loss
As a result of the net effects of the foregoing, net loss increased approximately $25,798,000, or 91%, to approximately $54,217,000 for the year ended December 31, 2014, compared with approximately $28,419,000 for the year ended December 31, 2013. Net loss per share of common stock, basic and diluted, was ($0.36) for the year ended December 31, 2014, compared with ($0.22) per share of common stock for the year ended December 31, 2013. Net loss per share of common stock was positively affected by our issuance of shares of common stock in an underwritten public offering in August 2014.
Year ended December 31, 2013 compared with year ended December 31, 2012
| Year Ended December 31, | ||||||||||||
| 2013 | 2012 | Change | ||||||||||
| (000s) | ||||||||||||
| Revenue | $ | 8,776 | $ | 3,818 | $ | 4,958 | ||||||
| Cost of goods sold | 1,960 | 1,348 | 612 | |||||||||
| Operating expenses | 32,624 | 18,618 | 14,006 | |||||||||
| Operating loss | (25,808 | ) | (16,148 | ) | (9,660 | ) | ||||||
| Financing Costs | (1,504 | ) | — | (1,504 | ) | |||||||
| Interest expense | (1,166 | ) | (1,905 | ) | 739 | |||||||
| Other income (expense) net | 59 | (42 | ) | 101 | ||||||||
| Loss on extinguishment of debt | — | (10,308 | ) | 10,308 | ||||||||
| Beneficial conversion feature | — | (6,717 | ) | 6,717 | ||||||||
| Net loss | $ | (28,419 | ) | $ | (35,120 | ) | $ | (6,701 | ) | |||
Revenue
Revenue for the year ended December 31, 2013 increased by approximately $4,958,000, or 130%, to $8,776,000, compared with the year ended December 31, 2012. Of this $4,958,000, approximately $713,000, or 14%, was attributable to an increase in the average sales price of our existing products, and approximately $4,245,000, or 86%, was attributable to sales of new products introduced in 2012.
Cost of Goods Sold
Cost of goods sold increased by approximately $612,000, or 45%, to $1,960,000 for the year ended December 31, 2013, compared with the year ended December 31, 2012. Our gross margins increased to 78% in 2013 compared to 65% in 2012. The gross margin change was primarily attributable to the increase in average sales price of products sold and product mix of prescription and OTC products.
Operating Expenses
Our principal operating levelcosts included the following items as a percentage of total operating expenses.
| Year Ended December 31, | ||||||||
| 2013 | 2012 | |||||||
| Human resource related costs | 33 | % | 39 | % | ||||
| Sales and marketing, excluding human resource costs | 15 | % | 24 | % | ||||
| Production design and development costs | 41 | % | 24 | % | ||||
| Professional fees and consulting costs | 4 | % | 6 | % | ||||
| Other operating expenses | 7 | % | 7 | % | ||||
Operating expenses increased by approximately $14,006,000, or 75%, for the year ended December 31, 2013 from year ended December 31, 2012 as a result of the following items:
| (000s) | |||
| Increase in product research and development costs | $ | 9,059 | |
| Increase in human resource related costs | 3,333 | ||
| Increase in sales and marketing costs, excluding human resource costs | 582 | ||
| Increase in professional and consulting costs | 79 | ||
| Increase in all other operating expenses | 953 | ||
| $ | 14,006 |
Research and development costs increased by approximately $9,059,000, or 202%, to approximately $13,551,000, primarily as a result of the commencement of phase 3 clinical trials for TX-001HR as well as the preparation of phase 3 clinical trials for TX-002HR, and phase 3 clinical trials for TX-004HR. Research and developments costs during the year ended December 31, 2013 included the following research and development projects:
TX-001HR. Since the project’s inception in February 2013, we have incurred approximately $4,809,000 in research and development costs with respect to TX-001HR, our combination estradiol and progesterone drug candidate.
TX-002HR. Since the project’s inception in April 2013, we have incurred approximately $1,059,000 in research and development costs with respect to TX-002HR, our progesterone only drug candidate.
For a discussion of the nature of efforts and steps necessary to complete these projects, see “Item 1. Business — Research and Development.” For a discussion of the risks and uncertainties associated with completing development of our products, see “Item 1A. Risk Factors — Risks Related to Our Business.” For a discussion of the extent and nature of additional resources that we may need to obtain if our current liquidity is not expected to be sufficient to complete these projects, see “— Liquidity and Capital Resources.” For a discussion as to whether a future milestone such as completion of a development phase, date of filing an NDA with a regulatory agency or approval from a regulatory agency can ever be achieved. Wereliably determined, see “Item 1. Business — Our Hormone Therapy Drug Candidates,” “Item 1. Business — Products in Development” and “Item 1. Business — Pharmaceutical Regulation.” Future milestones, including NDA submission dates, are not easily determinable as such milestones are dependent upon obtaining additional financingon various factors related to our clinical trials, including the timing of ongoing patient recruitment efforts to find eligible subjects for the applicable trials.
Human resource related costs, including salaries and benefits, increased by approximately $3,333,000, or 46%, to approximately $10,611,000, primarily as a result of an increase in orderamortization of non-cash compensation totaling approximately $3,152,000 related to adequately fund working capital, infrastructure, manufacturing expensesemployee stock options issued during 2013 and significant marketing/investor related expenditures2012.
Sales and marketing costs increased by approximately $582,000, or 13%, to gain market recognition, so thatapproximately $5,015,000, primarily as a result of expanded marketing, advertising, education, and training. In addition, we can achieveincreased spending in the areas of travel, product samples, and commissions. We also incurred added costs associated with our new product distribution channels introduced in 2013.
Operating Loss
As a levelresult of the foregoing, our operating loss increased approximately $9,660,000, or 60%, to approximately $25,808,000 for the year ended December 31, 2013, compared with approximately $16,148,000 for the year ended December 31, 2012, primarily as a result increased research and development costs associated with our continued development of our hormone therapy drug candidates, partially offset by increased revenue adequate to supportfrom sales of our cost structure, noneprenatal vitamin products.
As a result of which can be assured. Management believes it will be able to raise the capital required to execute the Company’s business plan and become profitable.
Financing Costs
Financing costs increased from $0, to approximately $1,504,000, resulting from the amortization of costs associated with warrants granted in 2013 in connection with a $10,000,000 revolving line of credit.
Interest Expense
Interest expense decreased approximately $739,000, or 39%, to approximately $1,166,000, primarily as a result of the retirement of debt issued during 2012.
Net Loss
As a result of the net effects of the foregoing, including a lack of charges in 2013 for loss on extinguishment of debt and a beneficial conversion feature of debt that were included in our 2012 results, net loss decreased approximately $6,701,000, or 19%, to approximately $28,419,000 for the year ended December 31, 2013, compared with approximately $35,120,000 for the year ended December 31, 2012. Net loss per share of common stock, basic and diluted, was ($0.22) for the year ended December 31, 2013, compared with ($0.38) per share of common stock for the year ended December 31, 2012. Net loss per share of common stock was positively affected by our issuance of shares of common stock in underwritten public offerings in March and September 2013.
Liquidity and Capital Resources
We have funded our operations primarily through the private placement of equity and debt securities, and public offerings of our common stock. For the three year period ending December 31, 2014, we cannot provide assurance asreceived $9 million in net proceeds from the issuance of debt securities and $130 million in net proceeds from the issuance of shares of our common stock. As of December 31, 2014, we had cash totaling approximately $51.4 million: however, changing circumstances may cause us to how muchconsume funds significantly faster than we willcurrently anticipate, and we may need to spend in ordermore money than currently expected because of circumstances beyond our control.
We believe that our existing cash will allow us to develop, manufacture,fund our operating plan through at least the next 12 months. If our available cash and market new products and technologies incash equivalents is insufficient to satisfy our liquidity requirements, we may seek to sell additional equity or debt securities or obtain a credit facility. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures, or declaring dividends. To the future. We are currently working to bring additional products to market (some of which would require FDA approval). We expect to spend approximately $1.2 million on research and development in 2012. Asextent that we increase the market penetration of our current products and we expand our product base to include prescription products, the need for increased inventory levels will become a necessity. This increase is estimated to be approximately $0.8 million.
We need substantial amounts of cash to complete the clinical development of our hormone therapy drug candidates. The following table sets forth the primary sources and uses of cash for each of the periods set forth below:
Summary of (Uses) and Sources of Cash
| Year Ended December 31, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
| Net cash flows used in operating activities | $ | (45,520,996 | ) | $ | (20,768,069 | ) | $ | (12,737,326 | ) | |||
| Net cash flows used in investing activities | $ | (606,756 | ) | $ | (583,561 | ) | $ | (272,506 | ) | |||
| Net cash flows provided by financing activities | $ | 43,298,099 | $ | 73,989,416 | $ | 14,436,885 | ||||||
| 56 |
Operating Activities
The use of cash in further dilution. Our failure to raise capital when needed would adversely affectall periods resulted primarily from our business, financial conditionnet loss adjusted for non-cash charges and resultschanges in components of operations, and could force us to reduce or cease our operations.
| (000’s) | ||||
| At December 31, 2010 | $ | 423 | ||
| At December 31, 2011 | 126 | |||
| Decrease in cash and cash equivalents | $ | (297 | ) | |
| (000’s) | ||||
| At December 31, 2009 | $ | 123 | ||
| At December 31, 2010 | 423 | |||
| Increase in cash and cash equivalents | $ | 300 | ||
The increase (decrease)of approximately $25 million in cash and cash equivalents is comprised of the following componentsused in operating activities for the years ended December 31:
| 2011 | 2010 | |||||||
| Proceeds from notes payable and line of credit | $ | 3,084 | $ | -0- | ||||
| Proceeds from issuance of equity securities | 1,707 | 3,171 | ||||||
| Proceeds from exercise of stock options | 17 | -0- | ||||||
| Sources of cash and cash equivalents | 4,808 | 3,171 | ||||||
| Cash used in operating activities | 4,967 | 2,844 | ||||||
| Cash used to purchase equipment | 29 | 27 | ||||||
| Repayment of debt | 101 | -0- | ||||||
| Cash used in other investing activities | 8 | -0- | ||||||
| Uses of cash and cash equivalents | 5,105 | 2,871 | ||||||
| Increase (decrease) in cash and cash equivalents | $ | (297 | ) | $ | 300 | |||
| December 31, | ||||||||||||
| 2011 | 2010 | Change | ||||||||||
(000’s) | ||||||||||||
| Current assets | $ | 1,237 | $ | 1,059 | $ | 178 | ||||||
| Current liabilities | 3,151 | 233 | 2,918 | |||||||||
| Working capital | $ | (1,914 | ) | $ | 826 | $ | (2,740 | ) | ||||
| Year Ended December 31, | ||||||||||||
| 2011 | 2010 | Change | ||||||||||
(000’s) | ||||||||||||
| Revenue | $ | 2,088 | $ | 1,242 | �� | $ | 846 | |||||
| Cost of goods sold | 947 | 556 | 391 | |||||||||
| Operating expenses | 6,568 | 3,553 | 3,015 | |||||||||
| Operating loss | (5,427 | ) | (2,867 | ) | (2,560 | ) | ||||||
| Settlement of debt | (7,390 | ) | -0- | (7,390 | ) | |||||||
| Other expense, net | (96 | ) | -0- | (96 | ) | |||||||
| Net loss | $ | (12,913 | ) | $ | (2,867 | ) | $ | (10,046 | ) | |||
The Company’s costs of individual products did not change for year ended December 31, 2011 as compared to 2010.
Year Ended December 31, | ||||||||
| 2011 | 2010 | |||||||
| Human resource costs, including benefits | 52 | % | 52 | % | ||||
| Sales and marketing | 14 | % | 9 | % | ||||
| Product design and development costs | 2 | % | 2 | % | ||||
| Travel and entertainment | 6 | % | 5 | % | ||||
| Professional fees for legal, accounting and consulting | 7 | % | 8 | % | ||||
| Rent and other occupancy costs | 5 | % | 5 | % | ||||
| Non-cash costs | 3 | % | 5 | % | ||||
| Other | 11 | % | 14 | % | ||||
| (000’s) | ||||
| Increase in human resource costs | $ | 1,551 | ||
| Increase in sales and marketing | 560 | |||
| Increase in product design and development costs | 424 | |||
| Increase in travel and entertainment | 233 | |||
| Increase in professional and consulting | 318 | |||
| Increase in rent and other occupancy costs | 23 | |||
| Increase in non-cash compensation | 19 | |||
| Increase in all other | 270 | |||
| $ | 3,015 | |||
Investing Activities
The increase of approximately $23,000 in cash used in investing activities for the additionyear ended December 31, 2014 compared with the prior year was due to patent development costs and purchase of interest expense not incurredproperty and equipment.
The increase of approximately $311,000 in cash used in investing activities for the year ended December 31, 2013 compared with the prior year was due to patent development costs and purchase of property and equipment.
Financing Activities
Financing activities represent the principal source of our cash flow.
Our financing activities for the year ended December 31, 2014 provided net cash of approximately $43,298,000.
On July 29, 2014, we entered into an underwriting agreement relating to the issuance and sale by us of 8,565,310 shares of our common stock. Under the terms of the underwriting agreement, we granted the underwriters a 30-day option to purchase up to an additional 1,284,796 shares of our common stock, which was exercised in full on July 30, 2014. The offering closed on August 4, 2014. The net proceeds to us from this offering were approximately $42.8 million, after deducting underwriting discounts and commissions and other offering expenses payable by us.
Our financing activities for the year ended December 31, 2013 provided net cash of approximately $73,989,000.
On March 14, 2013, we entered into an underwriting agreement relating to the issuance and sale by us of 29,411,765 shares of our common stock. Under the terms of the underwriting agreement, we granted the underwriters a 30-day option to purchase up to an additional 4,411,765 shares of our common stock. On April 12, 2013, the underwriters exercised their option to purchase 1,954,587 shares of our common stock. The net proceeds to us from this offering were approximately $48 million, after deducting underwriting discounts and commissions and other offering expenses.
On September 25, 2013, we entered into an underwriting agreement relating to the issuance and sale by us of 13,750,000 shares of our common stock. The net proceeds to us from this offering were approximately $30 million, after deducting underwriting discounts and commissions and other offering expenses payable by us.
In March 2013, we repaid approximately $5 million in notes and credit lines.
Our financing activities for the year ended December 31, 2012 provided net cash of approximately $14,437,000.
In September 2012, we entered into a securities purchase agreement with multiple investors, relating to the issuance and sale of our common stock in a private placement that provided us with approximately $8 million in net proceeds. During 2012, we issued notes in the aggregate principal amount of approximately $9 million to multiple parties, of which approximately $2 million was repaid during 2010.
Critical Accounting Estimates and New Accounting Pronouncements
Critical Accounting Estimates
The preparation of financial statements in accordance with accounting principles generally accepted in the U.S.United States, or GAAP, requires managementus to make estimates and assumptions that affect reported amounts and related disclosures in the financial statements. Management considersWe consider an accounting estimate to be critical if:if
changes in the estimate or different estimates that could have been selected could have a material impact on our results of operations or financial condition.
We base our estimates and judgments on our experience, our current knowledge, our beliefs of what could occur in the future, our observation of trends in the industry, information provided by our customers, and information available from other sources. Actual results may differ from these estimates under different assumptions or conditions. We have identified the following accounting policies and estimates as those that we believe are most critical to our financial condition and results of operations and that require management’sour most subjective and complex judgments in estimating the effect of inherent uncertainties: share-based compensation expense and income taxes,taxes.
Revenue Recognition. We recognize revenue on arrangements in accordance with ASC 605, Revenue Recognition. We recognize revenue only when the price is fixed or determinable, persuasive evidence of an arrangement exists, the service is performed, and derivative financial instruments.
Our OTC and prescription prenatal vitamin products are generally variations of the same product with slight modifications in formulation and marketing. The primary difference between our OTC and prescription prenatal vitamin products is the source of payment. Purchasers of our OTC prenatal vitamin products pay for the product directly while purchasers of our prescription prenatal vitamin products pay for the product via third-party payers. Both OTC and prescription prenatal vitamin products share the same marketing support team utilizing similar marketing techniques.
Over-the-Counter Products
We generate OTC revenue from product sales primarily to retail consumers. We recognize revenue from product sales upon shipment, when the rights of ownership and risk of loss have passed to the consumer. We include outbound shipping and handling fees in sales and bill them upon shipment. We include shipping expenses in cost of sales. A majority of our customers pay for our products with credit cards, and we usually receive the cash settlement in two to three banking days. Credit card sales minimize accounts receivable balances relative to sales. We provide an unconditional 30-day money-back return policy under which we accept product returns from our retail and eCommerce customers. We recognize our revenue from OTC sales, net of returns, sales discounts, and eCommerce fees.
Prescription Products
We sell our name brand and generic prescription products primarily through drug wholesalers and retail pharmacies. We recognize revenue from prescription product sales, net of sales discounts, chargebacks, and rebates.
We accept returns of unsalable product from customers within a return period of six months prior to and up to 12 months following product expiration. Our prescription products currently have a shelf life of 24 months from the date of manufacture. Given the limited history of our prescription products, we currently cannot reliably estimate expected returns of the prescription products at the time of shipment. Accordingly, we defer recognition of revenue on prescription products until the right of return no longer exists, which occurs at the earlier of the time the prescription products are dispensed through patient prescriptions or expiration of the right of return.
We maintain various rebate programs in an effort to maintain a competitive position in the marketplace and to promote sales and customer loyalty. The consumer rebate program is designed to enable the end user to return a coupon to us. If the coupon qualifies, we send a rebate check to the end user. We estimate the allowance for consumer rebates based on our experience and industry averages, which is reviewed, and adjusted if necessary, on a quarterly basis.
Research and Development Expense. We rely on the services of external CRO’s to facilitate our clinical studies. Certain of these CRO’s require us to make payments based on agreed-upon terms, which may include payments in advance of a study starting date. We capitalize these advance payments into prepaid expense when paid. We expense these nonrefundable advance payments for goods and services that will be used in future research and development activities when the activity has been performed rather than when the payment is made. As a result, we amortize certain of these amounts based on factors relating to the progress of our clinical studies. These factors include successful enrollment of patients, expected duration of studies, and completion of clinical trial milestones. On a quarterly basis we re-assess the factors by which these advanced payments are expensed. If these factors change we adjust these prepaid balances accordingly.
Share-Based Compensation Expense
Income Taxes
The determination of our provision for income taxes requires significant judgment, the use of estimates, and the interpretation and application of complex tax laws. In the ordinary course of our business, there are transactions and calculations for which the ultimate tax determination is uncertain. In spite of our belief that we have appropriate support for all the positions taken on our tax returns, we acknowledge that certain positions may be successfully challenged by the taxing authorities. We determine the tax benefits more likely than not to be recognized with respect to uncertain tax positions. Although we believe our recorded tax assets and liabilities are reasonable, tax laws and regulations are subject to interpretation and inherent uncertainty; therefore, our assessments can involve both a series of complex judgments about future events and rely on estimates and assumptions. Although we believe these estimates and assumptions are reasonable, the final determination could be materially different than that which is reflected in our provision for income taxes and recorded tax assets and liabilities.
Segment Reporting.We are managed and operated as one business, which is focused on creating and commercializing products targeted exclusively for women. Our business operations are managed by a single management team that reports to our president. We do not operate separate lines of business with respect to any of our products and we do not prepare discrete financial information with respect to separate products. All product sales are derived from sales in the United States. Accordingly, we view our business as one reportable operating segment.
New Accounting Pronouncements
In December 2011,August 2014, the FASB issued Accounting Standards Update, (“ASU”) 2011-11, Balance Sheet - Offsetting. This guidanceor ASU, No. 2014-05, Presentation of Financial Statements-Going Concern (Subtopic 205-40): Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern. ASU 2014-15 requires disclosuresmanagement to evaluate whether there are conditions and events that raise substantial doubt about offsettingthe entity’s ability to continue as a going concern within one year after the financial statements are issued (or available to be issued when applicable) and, related arrangements for recognized financial instruments and derivative instruments. The standardif so, disclose that fact. ASU 2014-15 is effective for us asannual periods ending after December 15, 2016 and interim periods within annual periods beginning after December 15, 2016. Early adoption is permitted for annual or interim reporting periods for which the financial statements have not previously been issued. We do not expect the adoption of January 1,the ASU 2014-15 to have a material effect on our consolidated financial statements and disclosures.
In May 2014, the FASB and the International Accounting Standards Board (IASB) issued ASU, No. 2014-09, Revenue from Contracts with Customers (Topic 606). The standard’s core principle is that a company will recognize revenue when it transfers promised goods or services to customers in an amount that reflects the consideration to which the company expects to be entitled in exchange for those goods or services. In doing so, companies will need to use more judgment and make more estimates than under today’s guidance. These may include identifying performance obligations in the contract, estimating the amount of variable consideration to include in the transaction price and allocating the transaction price to each separate performance obligations. ASU 2014-09 is effective for public business entities, certain not-for-profit entities and certain employee benefit plans, for annual periods beginning after December 15, 2016, including interim periods within that period. Early adoption is not permitted under GAAP. We are currently evaluating the impact of ASU 2014-09 on our consolidated financial statements and disclosures.
In July 2013, and will not materially impact our financial statement disclosures.
In MayDecember 2011, the FASB issued ASU 2011-04, Fair Value Measurement: AmendmentsNo. 2011-11, Balance Sheet (Topic 210): Disclosures about Offsetting Assets and Liabilities, or ASU 2011-11. ASU 2011-11 enhances current disclosures about financial instruments and derivative instruments that are either offset on the statement of financial position or subject to Achieve Common Fair Value Measurementan enforceable master netting arrangement or similar agreement, irrespective of whether they are offset on the statement of financial position. Entities are required to provide both net and Disclosure Requirementsgross information for these assets and liabilities in U.S.order to facilitate comparability between financial statements prepared in conformity with GAAP and IFRSs. Thisfinancial statements prepared on the basis of International Financial Reporting Standards. ASU represents the converged guidance of the FASB and the IASB (the “Boards”) on fair value measurement, and results in common requirements2011-11 is effective for measuring fair value and for disclosing information about fair value measurements, including a consistent meaning of the term “fair value.” These amendments change some of the terminology used to describe many of the existing requirements in U.S. GAAP for measuring fair value and for disclosing information about fair value measurements. The amendments should be applied prospectively, and they are effective during interim and annual reporting periods beginning on or after December 15, 2011. Early application by public entities isJanuary 1, 2013, and interim periods within those years. ASU 2011-11 did not permitted. The adoption is not expected to have a material impact on the Company’s results of operations,our financial position or cash flows.
We do not believe there would behave been a material effect on the accompanying consolidated financial statements had any other recently issued, but not yet effective, accounting standards been adopted in the current period.
Off-Balance Sheet Arrangements
As of December 31, 2011,2014, 2013, and 2012, we had no material off-balance sheet arrangements.
In the ordinary course of business, we enter into agreements with third parties that include indemnification provisions, which, in our judgment, are normal and customary for companies in our industry sector. These agreements are typically with business partners, clinical sites, and suppliers. Pursuant to these agreements, we generally agree to indemnify, hold harmless, and reimburse indemnified parties for losses suffered or incurred by the indemnified parties with respect to our productdrug candidates, use of such productdrug candidates, or other actions taken or omitted by us. The maximum potential amount of future payments we could be required to make under these indemnification provisions is unlimited. We have not incurred material costs to defend lawsuits or settle claims related to these indemnification provisions. As a result, the estimated fair value of liabilities relating to these provisions is minimal. Accordingly, we have no liabilities recorded for these provisions as of December 31, 2011.
In the normal course of business, we may be confronted with issues or events that may result in a contingent liability. These generally relate to lawsuits, claims, environmental actions or the actions of various regulatory agencies. We consult with counsel and other appropriate experts to assess the claim. If, in our opinion, we have incurred a probable loss as set forth by accounting principles generally accepted in the U.S.,GAAP, an estimate is made of the loss and the appropriate accounting entries are reflected in our financial statements.
Effects of Inflation
For each of the periods for which financial information is presented, the Company’sfiscal years ended December 31, 2014, 2013, and 2012, our business and operations have not been materially affected by inflation.
Contractual Obligations
A summary of contractual cash obligations as of December 31, 2014 is as follows:
| Payments Due By Period | |||||||||||||||
| (in thousands) | |||||||||||||||
| Total | Less than 1 Year | 1-3 Years | 4-5 Years | ||||||||||||
| Operating Lease Obligations | $ | 1,450 | $ | 371 | $ | 1,079 | $ | 0 | |||||||
Seasonality
The specialty pharmaceutical industry component of women’s health segmentis not subject to seasonal sales fluctuation.
Item 7A. | Quantitative and Qualitative Disclosures about Market Risk |
We had cash and cash equivalents totaling $51.4 million as of December 31, 2014.We hold our cash in money market funds and the health care market. Weprimary objective of our investment policy is to preserve principal and maintain proper liquidity to meet operating needs.Our investment policy specifies credit quality standards for our investments and limits the amount of credit exposure to any single issue, issuer or type of investment.Our primary exposure to market risk is interest rate sensitivity, which is affected by changes in the general level of U.S. interest rates.To minimize this risk, we intend to sellmaintain a portfolio that may include cash, cash equivalents and investment securities available-for-sale in a variety of products including medical foods, nutritional supplements, prescription productssecurities which may include money market funds, government and ancillary productsnon-government debt securities and commercial paper, all with various maturity dates.Due to the low risk profile of our investments, an immediate 100 basis point change in interest rates would not have a material effect on the fair market value of our portfolio.
We do not hold or issue derivatives, derivative commodity instruments or other financial instruments for speculative trading purposes. Further, we do not believe our cash equivalents and investment securities have significant risk of default or illiquidity. We made this determination based on discussions with our investment advisors and a review of our holdings. While we believe our cash equivalents and investment securities do not contain excessive risk, we cannot provide absolute assurance that address women’s health needs. We plan to sell our products online and through physician’s offices and pharmacies. We anticipate the demand for our products to grow as we add territories, sales personnel and products. Our plan is to offer a complete line of products in the women’s healthfuture our investments will not be subject to adverse changes in market and we believe as we expand our product line and sales territories that our revenues will increase. Our sales team markets our products by detailing physicians who specialize in women’s health about the features and benefitsvalue. All of our products. We believe our sales and marketing strategy will enable usinvestments are held at fair value.
Item 8. | Financial Statements and Supplementary Data |
Reference is made to increase demand for our products thereby allowing our revenues to grow in upcoming quarters.
Item 9. |
We maintain disclosure controls and procedures (as defined under Rule 13a-15(e) under the Exchange Act) as of the end of the period covered by this report.
Our management, with the material weaknesses described below.
Management’s Annual Report on Internal Control Overover Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as defined inunder Exchange Act Rules 13a-15(f) and 15d-15(f) under the Exchange Act. The Company’s. Our internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with GAAP. Internal control over financial reporting includes those policies and procedures that:
Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2014. In making this assessment, our management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control—Integrated Framework (2013). Management’s assessment included an evaluation of the design of our internal control over financial reporting and testing of the operational effectiveness of its internal control over financial reporting. Based on management’s assessment, we believe that our internal controls over financial reporting were effective as of December 31, 2014.
Rosenberg Rich Baker Berman & Company, an independent registered public accounting firm, has audited the consolidated financial statements included in this Annual Report; and, as part of its audit, has issued an attestation report, included herein, on the effectiveness of our internal control over financial reporting.
Changes in Internal Control over Financial Reporting
There was no change in our internal control over financial reporting during our most recent fiscal quarter that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Inherent Limitations on Effectiveness of Controls
Our management, including our Chief Executive Officer and Chief Financial Officer, does not expect that our disclosure controls and procedures or our internal controls will prevent all error and all fraud. A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefit of controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues, misstatements, errors, and instances of fraud, if any, within our company have been or will be prevented or detected. Further, internal controls may become inadequate as a result of changes in conditions, or through the deterioration of the degree of compliance with policies or procedures.
Item 9B. | Other Information |
None.
Item 10. | Directors, Executive Officers, and Corporate Governance |
The information required by this Item relating to our directors and corporate governance is incorporated herein by reference to the definitive Proxy Statement to be filed pursuant to Regulation 14A of the Exchange Act for our 2015 Annual Meeting of Stockholders.
Item 11. | Executive Compensation |
The information required by this Item is incorporated herein by reference to the definitive Proxy Statement to be filed pursuant to Regulation 14A of the Exchange Act for our 2015 Annual Meeting of Stockholders.
Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
The information required by this Item is incorporated by reference to the definitive Proxy Statement to be filed pursuant to Regulations 14A of the Exchange Act for our 2015 Annual Meeting of Stockholders.
Item 13. | Certain Relationships and Related Transactions, and Director Independence |
The information required by this Item is incorporated herein by reference to the definitive Proxy Statements to be filed pursuant to Regulation 14A of the Exchange Act for our 2015 Annual Meeting of Stockholders.
Item 14. | Principal Accountant Fees and Services |
The information required by this Item is incorporated herein by reference to the definitive Proxy Statement to be filed pursuant to Regular 14A of the Exchange Act for our 2015 Annual Meeting of Stockholders.
Item 15. | Exhibits and Financial Statement Schedules |
(a)Financial Statements and Financial Statements Schedules
| (1) | Financial Statements are listed in the Index to Consolidated Financial Statements on page F-1 of this Annual Report. |
| (2) | No financial statement schedules are included because such schedules are not applicable, are not required, or because required information is included in the consolidated financial statements or notes thereto. |
(b)Exhibits
Exhibit | Date | Description |
| 2.1 | July 6, 2009 | Agreement and Plan of Reorganization among Croff Enterprises, Inc., AMHN Acquisition Corp., America’s Minority Health Network, Inc., and the Major Shareholders(1) |
| 2.2 | June 11, 2010 | Agreement and Plan of Reorganization among AMHN, Inc., SHN Acquisition Corp., Spectrum Health Network, Inc., and the Sole Shareholder of Spectrum Health Network, Inc.(2) |
| 2.3 | October 25, 2007 | Croff Enterprises, Inc. Plan of Corporate Division and Reorganization(3) |
| 2.4 | July 18, 2011 | Agreement and Plan of Merger among VitaMedMD, LLC, AMHN, Inc., and VitaMed Acquisition, LLC(4) |
| 3.1 | September 15, 2009 | Articles of Amendment to Articles of Incorporation (to change name to AMHN, Inc.)(5) |
| 3.2 | July 27, 2009 | Certificate of Merger of AMHN Acquisition Corp., with and into America’s Minority Health Network, Inc.(6) |
| 3.3 | December 27, 2007 | Articles of Amendment to Articles of Incorporation of Croff Enterprises, Inc. (to increase authorized common shares from 20,000,000 to 50,000,000)(3) |
| 3.4 | July 20, 2010 | Articles of Conversion of AMHN, Inc. filed in the State of Nevada(7) |
| 3.5 | July 20, 2010 | Articles of Incorporation of AMHN, Inc. filed in the State of Nevada(7) |
| 3.6 | August 29, 2011 | Certificate of Amendment and Restatement of Articles of Incorporation of AMHN, Inc. (to change name and increase authorized shares)(8) |
| 3.7 | n/a | Bylaws of AMHN, Inc.(9) |
| 4.1 | September 26, 2012 | Form of Securities Purchase Agreement (10) |
| 4.2 | n/a | Form of Certificate of Common Stock(11) |
| 10.1 | November 9, 2010 | Demand Promissory Note to Philip M. Cohen for $210,000(12) |
| 10.2 | April 18, 2011 | Convertible Promissory Note to First Conquest Investment Group, L.L.C. for $105,000(12) |
| 10.3 | April 18, 2011 | Convertible Promissory Note to Energy Capital, LLC for $105,000(12) |
| 10.4 | May 7, 2011 | Sales Representative Agreement between AMHN, Inc. and Mann Equity, LLC(12) |
| 10.5 | July 9, 2009 | Lease Agreement between Liberty Property Limited Partnership and VitaMedMD, LLC(13) |
| 10.6 | September 8, 2011 | Stock Purchase Agreement between AMHN, Inc. and Pernix Therapeutics, LLC(14) |
| 10.7 | September 8, 2011 | Lock-Up Agreement between AMHN, Inc. and Pernix Therapeutics, LLC(14) |
| 10.8 | n/a | Form of Common Stock Purchase Warrant (13) |
| 10.9* | n/a | Form of Non-Qualified Stock Option Agreement (13) |
| 10.10 | September 2011 | Form of Convertible Promissory Note(15) |
| 10.11 | September 20, 2011 | Financing Agreement between Lang Naturals, Inc. and VitaMedMD, LLC(16) |
| 10.12 | October 18, 2011 | Debt Conversion Agreement between the Company and Energy Capital, LLC(17) |
| 10.13 | October 18, 2011 | Debt Conversion Agreement between the Company and First Conquest Investment Group, LLC(17) |
| 10.14 | October 23, 2011 | Consulting Agreement among VitaMedMD, LLC, the Company, and Lang Naturals, Inc.(17) |
| 10.15 | October 23, 2011 | Common Stock Purchase Warrant to Lang Naturals, Inc.(17) |
Exhibit | Date | Description |
| 10.16 | October 23, 2011 | Lock-Up Agreement between the Company and Lang Naturals, Inc.(17) |
| 10.17 | November 3, 2011 | Software License Agreement between vitaMedMD, LLC and Pernix Therapeutics, LLC(18) |
| 10.18 | November 2011 | Form of Promissory Note (19) |
| 10.19 | February 24, 2012 | Note Purchase Agreement among the Company, Plato & Associates, Inc., and Steven G. Johnson(20) |
| 10.20 | February 24, 2012 | Form of Secured Promissory Note(20) |
| 10.21 | February 24, 2012 | Security Agreement among the Company, Plato & Associates, Inc., and Steven G. Johnson(20) |
| 10.22 | February 24, 2012 | Form of Common Stock Purchase Warrant(20) |
| 10.26 | April 17, 2012 | Master Services Agreement between the Company and Sancilio and Company, Inc.(21) |
| 10.27** | May 17, 2012 | Consulting Agreement between the Company and Sancilio and Company, Inc. (21) |
| 10.28* | November 8, 2012 | Form of Employment Agreement(22) |
| 10.29 | January 31, 2013 | Multiple Advance Revolving Credit Note, issued to Plato & Associates, LLC(23) |
| 10.30 | January 31, 2013 | Common Stock Purchase Warrant, issued to Plato & Associates, LLC(23) |
| 10.31* | May 8, 2013 | Agreement to Forfeit Non-Qualified Stock Options between the Company and Robert G. Finizio(24) |
| 10.32 | May 7, 2013 | Consulting Agreement between the Company and Sancilio and Company, Inc. (24) |
| 10.33 | May 16, 2013 | Lease between the Company and 6800 Broken Sound LLC(25) |
| 10.34* | n/a | Amended and Restated 2012 Stock Incentive Plan(26) |
| 10.35* | n/a | 2009 Long Term Incentive Compensation Plan, as amended(27) |
| 10.36† | February 18, 2015 | First Amendment to Lease between the Company and 6800 Broken Sound, LLC |
| 21.1 | December 31, 2012 | Subsidiaries of the Company(28) |
| 23.1† | March 12, 2015 | Consent of Rosenberg Rich Baker Berman & Company |
| 31.1† | March 12, 2015 | Certification of Chief Executive Officer pursuant to Rule 13a-14(a) and Rule 15d-14(a), promulgated under the Securities Exchange Act of 1934, as amended |
31.2† | March 12, 2015 | Certification of Chief Financial Officer pursuant to Rule 13a-14(a) and Rule 15d-14(a), promulgated under the Securities Exchange Act of 1934, as amended |
| 32.1† | March 12, 2015 | Certification pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 |
| 32.2† | March 12, 2015 | Certification pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 |
| 101.INS† | n/a | XBRL Instance Document |
| 101.SCH† | n/a | XBRL Taxonomy Extension Schema Document |
| 101.CAL† | n/a | XBRL Taxonomy Extension Calculation Linkbase Document |
| 101.DEF† | n/a | XBRL Taxonomy Extension Definition Linkbase Instance Document |
| 101.LAB† | n/a | XBRL Taxonomy Extension Label Linkbase Instance Document |
| 101.PRE† | n/a | XBRL Taxonomy Extension Presentation Linkbase Instance Document |
____________________________
| * | Indicates a contract with management or compensatory plan or arrangement. |
| ** | Certain information in this exhibit has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been requested with respect to the omitted portions. |
| † | Filed herewith. |
| (1) | Filed as an exhibit to Form 8-K filed with the Commission on July 10, 2009 and incorporated herein by reference (SEC File No. 000-16731). |
| (2) | Filed as an exhibit to Form 8-K filed with the Commission on June 14, 2010 and incorporated herein by reference (SEC File No. 000-16731). |
| (3) | Filed as an exhibit to Form 10-K for the year ended December 31, 2007 filed with the Commission on May 1, 2008 and incorporated herein by reference (SEC File No. 000-16731). |
| (4) | Filed as an exhibit to Form 8-K filed with the Commission on July 21, 2011 and incorporated herein by reference (SEC File No. 000-16731). |
| (5) | Filed as an exhibit to Form 10-Q for quarter ended September 30, 2009 filed with the Commission on November 16, 2009 and incorporated herein by reference (SEC File No. 000-16731). |
| (6) | Filed as an exhibit to Form 10-K for the year ended December 31, 2009 filed with the Commission on March 17, 2010 and incorporated herein by reference (SEC File No. 000-16731). |
| (7) | Filed as an exhibit to Form 10-Q for quarter ended June 30, 2010 filed with the Commission on August 3, 2010 and incorporated herein by reference (SEC File No. 000-16731). |
| (8) | Filed as an exhibit to Definitive 14C Information Statement filed with the Commission on September 12, 2011 and incorporated herein by reference (SEC File No. 000-16731). |
| (9) | Filed as an exhibit to Definitive 14C Information Statement filed with the Commission on June 29, 2010 and incorporated herein by reference (SEC File No. 000-16731). |
| (10) | Filed as an exhibit to Form 8-K filed with the Commission on October 2, 2012 and incorporated herein by reference (SEC File No. 000-16731). |
| (11) | Filed as an exhibit to Form S-3 filed with the Commission on January 25, 2013 and incorporated hereby by reference (SEC File No. 333-186189). |
| (12) | Filed as an exhibit to Form 10-Q for quarter ended March 31, 2011 filed with the Commission on May 19, 2011 and incorporated herein by reference (SEC File No. 000-16731). |
| (13) | Filed as an exhibit to Form 8-K filed with the Commission on October 11, 2011 and incorporated herein by reference (SEC File No. 000-16731). |
| (14) | Filed as an exhibit to Form 8-K filed with the Commission on September 14, 2011 and incorporated herein by reference (SEC File No. 000-16731). |
| (15) | Filed as an exhibit to Form 8-K/A filed with the Commission on November 22, 2011 and incorporated herein by reference (SEC File No. 000-16731). |
| (16) | Filed as an exhibit to Form 8-K/A filed with the Commission on February 2, 2012 and incorporated herein by reference (SEC File No. 000-16731). |
| (17) | Filed as an exhibit to Form 8-K filed with the Commission on October 24, 2011 and incorporated herein by reference (SEC File No. 000-16731). |
| (18) | Filed as an exhibit to Form 10-Q for quarter ended September 30, 2011 filed with the Commission on November 7, 2011 and incorporated herein by reference (SEC File No. 000-16731). |
| (19) | Filed as an exhibit to Form 8-K filed with the Commission on November 23, 2011 and incorporated herein by reference (SEC File No. 000-16731). |
| (20) | Filed as an exhibit to Form 8-K filed with the Commission on February 24, 2012 and incorporated herein by reference (SEC File No. 000-16731). |
| (21) | Filed as an exhibit to Form 10-Q for quarter ended June 30, 2012 filed with the Commission on August 9, 2012 and incorporated herein by reference (SEC File No. 000-16731). |
| (22) | Filed as an exhibit to Form 10-Q for quarter ended September 30, 2012 filed with the Commission on November 13, 2012 and incorporated herein by reference (SEC File No. 000-16731). |
| (23) | Filed as an exhibit to Form 8-K filed with the Commission on February 6, 2013 and incorporated herein by reference (SEC File No. 000-16731). |
| (24) | Filed as an exhibit to Form 10-Q for quarter ended March 31, 2013 filed with the Commission on May 10, 2013 and incorporated herein by reference (SEC File No. 001-00100). |
| (25) | Filed as an exhibit to Form 10-Q for quarter ended June 30, 2013 filed with the Commission on August 7, 2013 and incorporated herein by reference (SEC File No. 001-00100). |
| (26) | Filed as an exhibit to Form 8-K filed with the Commission on August 22, 2013 and incorporated herein by reference (SEC File No. 001-00100). |
| (27) | Filed as an exhibit to Registration Statement on Form S-8 filed with the Commission on October 15, 2013 and incorporated herein by reference (SEC File No. 333-191730). |
| (28) | Filed as an exhibit to Form 10-K for the year ended December 31, 2012 filed with the Commission on March 12, 2013 and incorporated herein by reference (SEC File No. 000-16731). |
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| Date: March 12, 2015 | THERAPEUTICSMD, INC. | |
| /s/ Robert G. Finizio | ||
| Robert G. Finizio | ||
| Chief Executive Officer |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the date indicated.
| Signature | Capacity | Date |
| /s/ Robert G. Finizio | Chief Executive Officer, Director (Principal Executive Officer) | March 12, 2015 |
| Robert G. Finizio | ||
| /s/ John C.K. Milligan, IV | President, Secretary, Director | March 12, 2015 |
| John C.K. Milligan, IV | ||
| /s/ Daniel A. Cartwright | Chief Financial Officer, Treasurer (Principal Financial and Accounting Officer) | March 12, 2015 |
| Daniel A. Cartwright | ||
| /s/ Tommy G. Thompson | Chairman | |
| Tommy G. Thompson | March 12, 2015 | |
| /s/ Brian Bernick | Director | March 12, 2015 |
| Brian Bernick | ||
| /s/ Cooper C. Collins | Director | March 12, 2015 |
| Cooper C. Collins | ||
| /s/ Robert V. LaPenta, Jr. | Director | March 12, 2015 |
| Robert V. LaPenta, Jr. | ||
| /s/ Nicholas Segal | Director | March 12, 2015 |
| Nicholas Segal |
| /s/ Jules Musing | Director | March 12, 2015 |
| Jules Musing | ||
| /s/ Randall Stanicky | Director | March 12, 2015 |
| Randall Stanicky |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and
Stockholders of TherapeuticsMD, Inc.
We have audited the accompanying balance sheets of TherapeuticsMD, Inc. as of December 31, 2014 and 2013, and the related statements of operations, stockholders’ equity, and cash flows for each of the years in the three year period ended December 31, 2014. We also have audited TherapeuticsMD’s internal control over financial reporting as of December 31, 2014, based on criteria established inInternal Control—Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). TherapeuticsMD’s management is responsible for these financial statements, for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Annual Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on these financial statements and an opinion on the company’s internal control over financial reporting based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement and whether effective internal control over financial reporting was maintained in all material respects. Our audits of the financial statements included examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted in the United States of America (“GAAP”). The Company’saccounting principles. A company’s internal control over financial reporting includes those policies and procedures that:
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
| Name and Principal Position | Year | Salary ($) | Bonus ($) | Stock Awards ($) | Option Awards ($)(1) | Nonequity Incentive Plan Compensation ($) | Non-qualified deferred compensation earnings ($) | All other compensation ($) | Total ($) | ||||||||||||||||||
| Robert G. Finizio | 2011 | 156,000 | -0- | -0- | -0- | -0- | -0- | 15,986 | 171,986 | ||||||||||||||||||
Chief Exec. Officer(2) | 2010 | 140,282 | -0- | -0- | -0- | -0- | -0- | 2,250 | 142,532 | ||||||||||||||||||
| John C.K. Milligan | 2011 | 156,000 | -0- | -0- | -0- | -0- | -0- | 25,329 | 181,329 | ||||||||||||||||||
President/Secretary(3) | 2010 | 144,787 | -0- | -0- | -0- | -0- | -0- | 9,554 | 154,341 | ||||||||||||||||||
| Daniel A. Cartwright | 2011 | 79,615 | -0- | -0- | 46,216 | -0- | -0- | 730 | 126,561 | ||||||||||||||||||
CFO/Treasurer(4) | 2010 | -0- | -0- | -0- | -0- | -0- | -0- | -0- | -0- | ||||||||||||||||||
| Mitchell L. Krassan | 2011 | 110,000 | -0- | -0- | -0- | -0- | -0- | -0- | 110,000 | ||||||||||||||||||
Chief Strategy Officer(5) | 2010 | 15,096 | -0- | -0- | 62,301 | -0- | -0- | -0- | 77,397 | ||||||||||||||||||
| Option Awards | Stock Awards | ||||||||||||||||||||||||||||||||
| Name | Number of Securities Underlying Unexercised Options (#) Exercisable | Number of Securities Underlying Unexercised Options (#) Unexercisable | Equity Incentive Plan Awards: Number of Securities Underlying Unexercised Unearned Options (#) | Option Exercise Price ($) | Option Expiry Date | Number of Shares or Units of Stock That Have Not Vested (#) | Market Value of Shares or Units of Stock That Have Not Vested ($) | Equity Incentive Plan Awards: Number of Unearned Shares, Units or Other Rights That Have Not Vested (#) | Equity Incentive Plan Awards Market or Payout Value of Unearned Shares, Units or Other Rights That Have Not Vested ($) | ||||||||||||||||||||||||
| Robert G. Finizio, CEO | 1,431,987 | (1) | 40,914 | (1) | -0- | $ | 0.10 | 01/01/19 | -0- | -0- | -0- | -0- | |||||||||||||||||||||
| John CK Milligan, IV, Pres./Sec. | 1,995,248 | (1) | 57,007 | (1) | -0- | $ | 0.10 | 01/01/19 | -0- | -0- | -0- | -0- | |||||||||||||||||||||
| Daniel A. Cartwright, CFO/Treas. | -0- | 300,000 | (2) | -0- | $ | 0.38 | 10/21/21 | -0- | -0- | -0- | -0- | ||||||||||||||||||||||
| Mitchell Krassan, Exec. VP | 73,646 | (3) | -0- | -0- | $ | 0.19 | 05/01/20 | -0- | -0- | -0- | -0- | ||||||||||||||||||||||
| 23,015 | (4) | 69,042 | (4) | -0- | $ | 0.19 | 05/10/10 | -0- | -0- | -0- | -0- | ||||||||||||||||||||||
| 265,943 | (5) | 470,512 | (5) | -0- | $ | 0.20 | 09/01/20 | -0- | -0- | -0- | -0- | ||||||||||||||||||||||
| Brian Bernick, Director | 1,391,082 | (1) | 81,828 | (1) | -0- | $ | 0.10 | 01/01/19 | -0- | -0- | -0- | -0- | |||||||||||||||||||||
| Name (a) | Fees earned or paid in cash ($) (b) | Stock awards ($) (c) | Option awards ($) (d) | Non-equity incentive plan compensation ($) (e) | Nonqualified deferred compensation earnings ($) (f) | All other compensation ($) (g) | Total ($) (h) | |||||||||||||||||||||
Robert G. Finizio(1) | -0- | -0- | $ | -0- | -0- | -0- | -0- | $ | -0- | |||||||||||||||||||
John C.K. Milligan IV(2) | -0- | -0- | $ | -0- | -0- | -0- | -0- | $ | -0- | |||||||||||||||||||
Brian Bernick, MD(3) | -0- | -0- | $ | -0- | -0- | -0- | -0- | $ | -0- | |||||||||||||||||||
| Name and Address of Beneficial Owner | Title of Class | Number of Shares Beneficially Owned(1) | Percent of Class | |||||||
| Robert G. Finizio Chairman and Chief Executive Officer | Common Stock | 23,813,493 | (2) | 27.61 | % | |||||
| John C.K. Milligan, IV President, Secretary and Director | Common Stock | 8,660,642 | (3) | 9.97 | % | |||||
| Daniel A. Cartwright Chief Financial Officer, Vice Pres. Finance, Treasurer | Common Stock | 163,632 | (4) | 0.19 | % | |||||
| Mitchell L. Krassan Executive Vice President, Chief Strategy Officer | Common Stock | 677,128 | (5) | 0.79 | % | |||||
| Brian Bernick, M.D. Director | Common Stock | 10,654,049 | (6) | 12.37 | % | |||||
| Samuel A. Greco Director | Common Stock | 400,000 | (7) | 0.47 | % | |||||
| Cooper C. Collins Director | Common Stock | 2,631,579 | (8) | 3.11 | % | |||||
| Robert V. LaPenta, Jr. Director | Common Stock | 5,000 | (9) | 0.01 | % | |||||
| Nicholas Segal Director | Common Stock | 3,948,719 | (10) | 4.66 | % | |||||
| All directors and executive officers as a group (9 persons) | Common Stock | 50,95,242 | (11) | 55.94 | % | |||||
| Steven G. Johnson, Shareholder 804 Tree Haven Ct., Highland Village, TX 75077 | Common Stock | 8,318,283 | (12) | 9.38 | % | |||||
| Robert J. Smith, Shareholder 13650 Fiddlesticks Blvd., #202-324; Ft. Myers, FL 33912 | Common Stock | 8,304,334 | (13) | 9.36 | % | |||||
| Wellington Management Company, LLP 280 Congress St., Boston, MA 02210 | Common Stock | 5,000,000 | (14) | 5.90 | % | |||||
Parks & Company, LLC
Certified Public Accountants & Consultants
1761 W. Hillsboro Boulevard, Suite 326
Deerfield Beach, FL 33442 Phone (954) 719-7569
www.parkscpas.com Fax (954) 719-3704
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors andMembers’of VitamedMD, LLC
We have audited the accompanying balance sheet of VitamedMD, LLC as of December 31, 2010, and the related statements of operations, changes in members’ equity and cash flows for the year ended December 31, 2010. VitamedMD, LLC’s management is responsible for these financial statements. Our responsibility is to express an opinion on these financial statements based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free ofall material misstatement. The company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audit included consideration ofrespects, effective internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audit provides a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of VitamedMD, LLC as of December 31, 2010, and2014, based on criteria established inInternal Control—Integrated Framework (2013) issued by the resultsCommittee of its operations and its cash flows forSponsoring Organizations of the year ended December 31, 2010 in conformity with accounting principles generally accepted in the United States of America.
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note C to the financial statements, the Company has not yet established profitable operations and has incurred significant losses since inception. These factors raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans regarding these matters are also described in Note C. The accompanying financial statements do not include any adjustments that might result from the outcome of this uncertainty.
As discussed in Note N, the Company restated its 2010 financial statements to correct errors related to the valuation of compensation and consultant expense using the Black-Scholes option-pricing model.Treadway Commission (COSO).
Parks/s/ Rosenberg Rich Baker Berman & Company LLC
Somerset, New Jersey
March 12, 2015
Deerfield Beach, Florida
February 28, 2012
| F-6 |
| December 31, | ||||||||
| 2011 | 2010 | |||||||
| ASSETS | (Restated) | |||||||
| Current Assets: | ||||||||
| Cash | $ | 126,421 | $ | 422,939 | ||||
| Accounts receivable, net of allowance for doubtful accounts of $1,500 and $0, respectively | 26,720 | 11,812 | ||||||
| Inventory | 588,073 | 618,069 | ||||||
| Other current assets | 496,060 | 6,292 | ||||||
| Total current assets | 1,237,274 | 1,059,112 | ||||||
| Fixed Assets: | ||||||||
| Property and equipment, net of accumulated depreciation of $81,500 and $26,655, respectively | 70,113 | 96,192 | ||||||
| Other Assets: | ||||||||
| Security deposit | 31,949 | 31,949 | ||||||
| Patent costs | 18,870 | 10,000 | ||||||
| Other assets | 80,515 | - | ||||||
| 131,334 | 41,949 | |||||||
| Total assets | $ | 1,438,721 | $ | 1,197,253 | ||||
| LIABILITIES AND STOCKHOLDERS' EQUITY | ||||||||
| Current Liabilities: | ||||||||
| Notes payable | $ | 2,150,000 | $ | - | ||||
| Accounts payable | 306,511 | 117,636 | ||||||
| Notes payable, related parties | 200,000 | - | ||||||
| Accrued interest | 28,321 | - | ||||||
| Other current liabilities | 465,747 | 115,206 | ||||||
| Total current liabilities | 3,150,579 | 232,842 | ||||||
| Commitments and Contingencies | ||||||||
| Stockholders’ Equity: | ||||||||
| Preferred stock - par value $0.001; 10,000,000 shares authorized; no shares issued and outstanding | - | - | ||||||
| Common stock - par value $0.001; 250,000,000 shares authorized; 82,978,804 and 55,487,321 issued and outstanding, respectively | 82,979 | 55,487 | ||||||
| Additional paid in capital | 15,198,241 | 4,988,637 | ||||||
| Accumulated deficit | (16,993,078 | ) | (4,079,713 | ) | ||||
| Total stockholders' equity (deficit) | (1,711,858 | ) | 964,411 | |||||
| Total liabilities and stockholders' equity | $ | 1,438,721 | $ | 1,197,253 | ||||
| Year Ended December 31, | ||||||||
| 2011 | 2010 | |||||||
| (Restated) | ||||||||
| Revenues, net | $ | 2,088,177 | $ | 1,241,921 | ||||
| Cost of goods sold | 947,112 | 556,390 | ||||||
| Gross profit | 1,141,065 | 685,531 | ||||||
| Operating expenses: | ||||||||
| Sales, general, and administration | 6,406,197 | 3,464,810 | ||||||
| Research and development | 107,241 | 65,402 | ||||||
| Depreciation and amortization | 54,845 | 22,783 | ||||||
| Total operating expense | 6,568,283 | 3,552,995 | ||||||
| Operating loss | (5,427,218 | ) | (2,867,464 | ) | ||||
| Other income and (expense) | ||||||||
| Settlement of debt | (7,390,000 | ) | - | |||||
| Interest expense | (64,380 | ) | - | |||||
| Loan guaranty costs | (38,159 | ) | - | |||||
| Other income | 6,392 | - | ||||||
| Total other income (expense) | (7,486,147 | ) | - | |||||
| Loss before taxes | (12,913,365 | ) | (2,867,464 | ) | ||||
| Provision for income taxes | - | - | ||||||
| Net loss | $ | (12,913,365 | ) | $ | (2,867,464 | ) | ||
| Loss per share, basic and diluted: | ||||||||
| Net loss per share, basic and diluted | $ | (0.21 | ) | $ | (0.07 | ) | ||
| Weighted average number of common shares outstanding | 62,516,461 | 38,289,463 | ||||||
| Additional | ||||||||||||||||||||
| Common Stock | Paid in | Accumulated | ||||||||||||||||||
| Shares | Amount | Capital | Deficit | Total | ||||||||||||||||
| Balance, December 31, 2009 | 39,516,450 | $ | 39,516 | $ | 1,656,364 | $ | (1,212,249 | ) | $ | 483,631 | ||||||||||
| Shares issued in private placement | 15,970,871 | 15,971 | 3,154,672 | - | 3,170,643 | |||||||||||||||
| Options issued as compensation | - | - | 177,601 | - | 177,601 | |||||||||||||||
| Net loss | - | - | - | (2,867,464 | ) | (2,867,464 | ) | |||||||||||||
| Balance, December 31, 2010 (Restated) | 55,487,321 | 55,487 | 4,988,637 | (4,079,713 | ) | 964,411 | ||||||||||||||
Effect of merger and recapitalization pursuant to execution of Security Exchange Agreement | 165,879 | 166 | (255,919 | ) | - | (255,753 | ) | |||||||||||||
| Shares issued in private placement | 5,551,589 | 5,552 | 1,701,448 | - | 1,707,000 | |||||||||||||||
| Shares issued in exchange for debt | 21,681,958 | 21,682 | 8,217,455 | - | 8,239,137 | |||||||||||||||
| Shares issued in exercise of warrants | 92,057 | 92 | 17,158 | - | 17,250 | |||||||||||||||
| Options issued as compensation | - | - | 183,355 | - | 183,355 | |||||||||||||||
| Warrants issued for services | - | - | 190,280 | - | 190,280 | |||||||||||||||
| Warrants issued for loan guaranty costs-related parties | - | - | 93,969 | - | 93,969 | |||||||||||||||
| Warrants issued for financing costs | - | - | 45,362 | - | 45,362 | |||||||||||||||
| Warrants issued as financing costs-related parties | 9,338 | - | 9,338 | |||||||||||||||||
| Warrants issued as compensation-related party | - | - | 7,158 | - | 7,158 | |||||||||||||||
| Net loss | - | - | - | (12,913,365 | ) | (12,913,365 | ) | |||||||||||||
| Balance, December 31, 2011 | 82,978,804 | $ | 82,979 | $ | 15,198,241 | $ | (16,993,078 | ) | $ | (1,711,858 | ) | |||||||||
| Year Ended December, 31, | ||||||||
| 2011 | 2010 | |||||||
| (Restated) | ||||||||
| CASH FLOWS FROM OPERATING ACTIVITES | ||||||||
| Net loss | $ | (12,913,365 | ) | $ | (2,867,463 | ) | ||
| Adjustments to reconcile net loss to net cash flows used in operating activities: | ||||||||
| Effect of merger and recapitalization pursuant to execution of Security Exchange Agreement | (255,753 | ) | - | |||||
| Depreciation | 54,845 | 22,783 | ||||||
| Allowance for doubtful accounts | 1,500 | - | ||||||
| Amortization of debt discount | 28,719 | - | ||||||
| Stock based debt settlement | 7,600,000 | - | ||||||
| Stock based compensation | 190,513 | 177,601 | ||||||
| Warrants issued for services | 22,630 | - | ||||||
| Non-cash financing costs | 25,980 | - | ||||||
| Loan guaranty costs | 38,159 | - | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Accounts receivable | (16,409 | ) | (6,008 | ) | ||||
| Inventory | 29,996 | (454,683 | ) | |||||
| Other current assets | (346,822 | ) | 152,916 | |||||
| Accounts payable | 188,876 | 95,034 | ||||||
| Accrued interest | 33,994 | - | ||||||
| Accrued expenses and other current liabilities | 350,541 | 36,033 | ||||||
| Net cash flows used in operating activities | (4,966,596 | ) | (2,843,787 | ) | ||||
| CASH FLOWS FROM INVESTING ACTIVITIES | ||||||||
| Purchase of property and equipment | (28,766 | ) | (27,348 | ) | ||||
| Patent costs, net of abandoned costs | (8,870 | ) | - | |||||
| Net cash flows used in investing activities | (37,636 | ) | (27,348 | ) | ||||
| CASH FLOWS FROM FINANCING ACTIVITIES | ||||||||
| Proceeds from notes and loans payable | 2,484,160 | - | ||||||
| Proceeds from sale of common stock | 1,000,000 | - | ||||||
| Proceeds from sale of membeship units, net of expenses | 707,000 | 3,170,645 | ||||||
| Proceeds bank line of credit | 300,000 | - | ||||||
| Proceeds from notes and loans payable-related parties | 300,000 | - | ||||||
| Proceeds from exercise of options | 17,250 | - | ||||||
| Repayment of notes payable-related party | (100,696 | ) | - | |||||
| Net cash flows provided by financing activities | 4,707,714 | 3,170,645 | ||||||
| Increase (decrease) in cash | (296,518 | ) | 299,510 | |||||
| Cash, beginning of period | 422,939 | 123,429 | ||||||
| Cash, end of period | $ | 126,421 | $ | 422,939 | ||||
| SUPPLEMENTAL DISCLOSURES OF CASH FLOW INFORMATION: | ||||||||
| Cash paid for interest | $ | 696 | $ | - | ||||
| Cash paid for income taxes | $ | - | $ | - | ||||
| SUPPLEMENTAL SCHEDULE OF NON-CASH FINANCING ACTIVITIES: | ||||||||
| Warrants issued for financing | $ | 148,668 | $ | - | ||||
| Warrants issued for services | $ | 190,280 | $ | - | ||||
| Conversion of notes payable and accrued interest into common stock | $ | 849,137 | $ | - | ||||
NOTE A1 – THE COMPANY
TherapeuticsMD, Inc., a Nevada corporation, (“Therapeutics”or TherapeuticsMD or the “Company”) was incorporated in Utah in 1907 under the name Croff Mining Company. The Company, changed its name to Croff Oil Company in 1952 and in 1996 changed its name to Croff Enterprises, Inc. In the twenty (20) years prior to 2008, Croff’s operations consisted entirely of oil and natural gas leases. Due to a spin-off of its operations in December 2007, Croff had no business operations or revenue source and had reduced its operations to a minimal level although it continued to file reports required under the Exchange Act. As a result of the spin-off, Croff was a “shell company” under the rules of the Commission. In July 2009, the Company (i) closed a transaction to acquire America’s Minority Health Network, Inc. as ahas two wholly owned subsidiary, (ii) ceased being a shell company, and (iii) experienced a change in control in which the former shareholders of America’s Minority Health Network, Inc. acquired control of the Company. On September 14, 2009, the Company changed its name to AMHN, Inc. On June 11, 2010, the Company closed a transaction to acquire Spectrum Health Network, Inc. as a wholly owned subsidiary. On July 20, 2010, the Company filed Articles of Conversion and Articles of Incorporation to redomicile in the State of Nevada and changed the par value of its shares of capital stock to $0.001 per share. On July 31, 2010, the Company transferred the assets of America’s Minority Health Network, Inc. to a secured noteholder in exchange for the satisfaction of debt associated therewith. On February 15, 2011, the Company transferred the assets of Spectrum Health Network, Inc. to a secured noteholder in exchange for the satisfaction of debt associated therewith and in exchange for an Exclusive Licensing, Distribution and Advertising Sales Agreement (“Licensing Agreement”) under which the Company could sell subscription services and advertising on the Spectrum Health Network for commissions. On August 3, 2011 (with an effective date of October 3, 2011), in anticipation of closing the Merger (as defined and described below), the Company filed Amended and Restated Articles of Incorporation to change its name to TherapeuticsMD, Inc. and to increase the shares of Common Stock authorized for issuance to 250,000,000. On October 4, 2011, the Company closed the Merger withsubsidiaries, vitaMedMD, LLC, a Delaware limited liability company, (“VitaMed”). As of December 31, 2011, Company management determined thator VitaMed, would become the sole focus of the Company and services performed relative to the Licensing Agreement were discontinued.BocaGreenMD, Inc., a Nevada corporation, or BocaGreen. Unless otherwise stated or unless the context otherwise requires, TherapeuticsMD, VitaMed, and BocaGreen collectively are sometimes referred to as “our company,” “we,” “our,” or “us.”
Nature of Business
We are a women’s health care product company focused on creating and commercializing products targeted exclusively for women. As of the descriptiondate of these consolidated financial statements, we are focused on conducting the clinical trials necessary for regulatory approval and commercialization of our business set forth below is provided on a combined basis, taking into accountadvanced hormone therapy pharmaceutical products. The drug candidates used in our newly-acquired wholly owned subsidiary, VitaMed.
DECEMBER 31, 2011 AND 2010
Principles of Consolidation
The accompanying consolidated financial statements include the accounts of the Companyour company and itsour wholly owned subsidiary.subsidiaries, VitaMed and BocaGreen. All material intercompany balances and transactions have been eliminated in consolidation.
Cash and Cash Equivalents
We maintain cash at financial institutions and,that at times balances may exceed the federally insured limits. The Company haslimit of $250,000 per financial institution. We have never experienced any losses related to these balances. All of our non-interest bearing cash balances were fully insured at December 31, 2011 and 2010 due to a temporary federal program in effect from December 31, 2010 through December 31, 2012. Under the program, there is no limit to the amount of insurance for eligible accounts. Beginning 2013, insurance coverage will revert to $250,000 per depositor at each financial institution, and our non-interest bearing cash balances may again exceed federally insured limits. The Company had no interest-bearing amounts on deposit in excess of federally insured limits at December 31, 2011 and 2010.
Trade Accounts Receivable and Allowance for Doubtful Accounts
Trade accounts receivable are customer obligations due under normal trade terms. The Company reviews theWe review accounts receivable for uncollectible accounts and credit card charge-backs and providesprovide an allowance for doubtful accounts, which is based upon a review of outstanding receivables, historical collection information, and existing economic conditions. TradeWe consider trade accounts receivable past due for more than 90 days are consideredto be delinquent. DelinquentWe write off delinquent receivables are written off to bad debt expenseagainst our allowance for doubtful accounts based on individual credit evaluations, the results of collection efforts, and specific circumstances of the customer. Recoveriescustomers. We record recoveries of accounts previously written off are recorded as reductions of bad debt expenseincrease in allowance for doubtful accounts when received.Historically, our bad debt expense has been limited becauseTo the majority of our trade receivables are paid via credit card. Dataextent data we use to calculate these estimates does not accurately reflect bad debts; adjustments to these reserves may be required.At December 31, 2011 and 2010, the Company recorded an allowance for doubtful accounts of $1,500 and $0, respectively.
THERAPEUTICSMD, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 2 – SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
Inventories
Inventories represent packaged vitamins, nutritional products and supplements and raw materials, which are valued at the lower of cost or market using the average costaverage-cost method.
Pre-Launch Inventory
Inventory costs associated with product candidates that have not yet received regulatory approval are capitalized if we believe there is probable future commercial use and future economic benefit. If the probability of future commercial use and future economic benefit cannot be reasonably determined, then pre-launch inventory costs associated with such product candidates are expensed as research and development expenses during the period the costs are incurred.
Fixed Assets
Equipment
We state equipment is stated at cost, net of accumulated depreciation. MaintenanceWe charge maintenance costs, which do not significantly extend the useful lives of the respective assets, and repair costs are charged to operating expense as incurred. Depreciation is computedWe compute depreciation using the straight-line method over the estimated useful lives of the related assets, which range from 3three to 7seven years. Depreciation expense totaled $25,686 and $5,105 for
Leasehold Improvements
We state improvements at cost, net of accumulated depreciation. We compute depreciation using the years ended December 31, 2011 and 2010, respectively.
IntangibleAssets
Patent and $17,678 for the years ended December 31, 2011 and 2010, respectively.Trademarks
The Company has
We have adopted the provisions ofFinancial Accounting Standards Board, (“FASB”)or FASB, Accounting Standards Codification, or ASC,350,Intangible-Goodwill and Other,or(“ASC 350”)350.
THERAPEUTICSMD, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2011 AND 2010
Impairment of Long-Lived Assets
We review the carrying values of property and equipment and finite-livedlong-lived intangible assets are reviewed for impairment whenever events or changes in circumstances indicate that their carrying values may not be recoverable. Such events or circumstances include but are not limited to:the following:
• | ||
significant adverse changes in the business climate that could impact an asset’s value, including adverse actions or assessments by regulators;
If impairment indicators are present, the Company determineswe determine whether an impairment loss should be recognized by testing the applicable asset or asset group’s carrying value for recoverability. This test requires long-lived assets to be grouped at the lowest level for which identifiable cash flows are largely independent of the cash flows of other assets and liabilities, the determination of which requires judgment. The Company estimatesWe estimate the undiscounted future cash flows expected to be generated from the use and eventual disposal of the assets and comparescompare that estimate to the respective carrying values in order to determine if such carrying values are recoverable. This assessment requires the exercise of judgment in assessing the future use of and projected value to be derived from the eventual disposal of the assets to be held and used. AssessmentsIn our assessments, we also consider changes in asset utilization, including the temporary idling of capacity and the expected timing for placing this capacity back into production. If the carrying value of the assets is not recoverable, then we record a loss is recorded for the difference between the assets’ fair value and respective carrying value. Thevalues. We determine the fair value of the assets is determined using an “income approach” based upon a forecast of all the expected discounted future net cash flows associated with the subject assets. Some of the more significant estimates and assumptions include:include market size and growth, market share, projected selling prices, manufacturing cost, and discount rate. The Company’sWe base estimates are based upon its historical experience, itsour commercial relationships, market conditions, and available external information about future trends. The Company believes itsWe believe our current assumptions and estimates are reasonable and appropriate; however, unanticipatedappropriate. Unanticipated events and changes in market conditions, however, could affect such estimates, resulting in the need for an impairment charge in future periods.
Fair Value of Financial Instruments
Our financial instruments consist primarily of receivables,accounts receivable, accounts payable and accrued expenses and short-term debt.expenses. The carrying amount of receivables,accounts receivable, accounts payable and accrued expenses approximates itstheir fair value because of the short-term maturity of such instruments. Interest rates thatinstruments, which are currently available to the Company for issuance of short-term debt with similar terms and remaining maturities are used to estimateconsidered Level 1 assets under the fair value of the Company’s short-term debt.
THERAPEUTICSMD, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE B2 – SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
Fair Value of Financial Instruments (continued)
We categorize our assets and liabilities that are valued at fair value on a recurring basis into a three-level fair value hierarchy as defined by ASC 820,“Fair Value Measurements and Disclosures” (“ASC 820”). Measurements.The fair value hierarchy gives the highest priority to quoted prices in active markets for identical assets and liabilities (Level 1) and lowest priority to unobservable inputs (Level 3).
| Level 1 |
| Level 2 |
| Level 3 |
At December 31, 20112014, and 2010, the Company2013, we had no assets or liabilities that arewere valued at fair value on a recurring basis.
The fair value of indefinite-lived assets is measured on a non-recurring basis using significant unobservable inputs (Level 3) in connection with our impairment test. There was no impairment of intangible assets during the years ended December 31, 2014, 2013, and 2012.
Income Taxes
Based upon a change in our business model, deferred income taxes are accounteddetermined by calculating the loss from operations of our company starting October 4, 2011.
We account for income taxes under the asset and liability method. DeferredWe recognize deferred tax assets and liabilities are recognized for the estimated future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax basis. DeferredWe measure deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which the related temporary differences are expected to be recovered or settled. TheWe recognize the effect on deferred tax assets and liabilities of a change in tax rates is recognized when the rate change is enacted. Valuation allowances are recorded to reduce deferred tax assets to the amount that will more likely than not be realized.
In accordance with ASC 740,Income Taxes, the Company recognizeswe recognize the effect of uncertain income tax positions only if the positions are more likely than not of being sustained in an audit, based on the technical merits of the position. RecognizedWe measure recognized uncertain income tax positions are measured atusing the largest amount that has a likelihood of being realized that is greater than 50% likely of being realized.. Changes in recognition or measurement are reflected in the period in which those changes in judgment occur. The Company recognizesAt December 31, 2014, 2013, and 2012 we had no uncertain income tax positions.
We recognize both interest and penalties related to uncertain tax positions as part of the income tax provision. As ofAt December 31, 20112014 and 2010, the Company has2013, we had no tax positions relating to open tax returns that were considered to be uncertain.
THERAPEUTICSMD, INC. AND SUBSIDIARIES
In December 2004,NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 2 – SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
Income Taxes (continued)
Our tax returns are subject to review by the FASB issued ASC 718, Compensation – Stock Compensation (“ASC 718”). ASC 718 companiesInternal Revenue Service three years after they are requiredfiled. Currently, years filed after 2011 are subject to review.
Share-Based Compensation
We measure the compensation costs of unit-basedshare-based compensation arrangements based on the grant-date fair value and recognize the costs in the financial statements over the period during which employees are required to provide services. Unit-basedShare-based compensation arrangements include unit options, restricted share plans,stock, restricted stock units, performance-based awards, share appreciation rights, and employee
Equity instruments (“instruments”) issued to other than employeesnon-employees are recorded on the basis of the fair value of the instruments, as required by ASC 718. FASB ASC 505,Equity Based Payments to Non-Employeesdefines,or ASC 505.ASC 505defines the measurement date and recognition period for such instruments. In general, the measurement date is when either (a) a (a) performance commitment, as defined, is reached or (b) the earlier of (i) the non-employee performance is complete or (ii) the instruments are vested. The measured value related to the instruments is recognized over a period based on the facts and circumstances of each particular grant as defined in ASC 505.
We recognize the ASC.
Debt Discounts
Costs incurred withfrom parties whothat are providing long-term financing, which include warrants issued in connection with the underlying debt, are reflected as a debt discount based on the relative fair value of the debt and warrants to the total proceeds. These We generally amortize discounts are generally amortized over the life of the related debt using the effective interest rate method. In connection with debt issued during the years ended December 31, 2011 and 2010, the Company recorded debt discounts totaling $28,719 and $0, respectively. Amortization expense related to debt discounts totaled $28,719 and $0 for the years ended December 31, 2011 and 2010, respectively, and is included in interest expense on the accompanying consolidated financial statements. Debt discount was fully amortized at December 31, 2011.
Revenue Recognition
We recognize revenue on arrangements in accordance with ASC 605, “Revenue Recognition” (“ASC 605”)Recognition. Revenue is recognizedWe recognize revenue only when the price is fixed or determinable, persuasive evidence of an arrangement exists, the service is performed, and collectability is reasonably assured.
THERAPEUTICSMD, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 2 – SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
Revenue Recognition (continued)
Our OTC and prescription prenatal vitamin products are generally variations of the same product with slight modifications in formulation and marketing. The Company generatesprimary difference between our OTC and prescription prenatal vitamin products is the source of payment. Purchasers of our OTC prenatal vitamin products pay for the product directly while purchasers of our prescription prenatal vitamin products pay for the product via third-party payers. Both OTC and prescription prenatal vitamin products share the same marketing support team utilizing similar marketing techniques. The revenue that is generated by us from major external customers is all generated from sales of our prescription prenatal vitamin products which is disclosed in Note 13. There are no major external customers for our OTC prenatal vitamin or other products.
Over-the-Counter Products
We generate OTC revenue from product sales primarily to retail consumers. The Company’s policy is toWe recognize revenue from product sales upon shipment, when the rights of ownership and risk of loss have passed to the consumer. OutboundWe include outbound shipping and handling fees are included in sales and are billedbill them upon shipment. ShippingWe include shipping expenses are included in cost of sales. TheA majority of the Company’s sales are paidour customers pay for our products with credit cards, and the Companywe usually receivesreceive the cash settlement in two to three banking days. Credit card sales minimize accounts receivable balances relative to sales.We provide an unconditional thirty-day30-day money-back return policy wherebyunder which we accept product returns from our retail wholesale and eCommerce customers.We recognize our revenue from OTC sales, net of returns, sales discounts, and eCommerce fees.
Prescription Products
We sell our name brand and generic prescription productsprimarily through drug wholesalers and retail pharmacies. We recognize revenue from prescription product sales, net of sales discounts, chargebacks, and rebates.
We accept returns of unsalable product from customers within a return period of six months prior to and up to twelve months following product expiration. Our prescription products currently have a shelf life of 24 months from the date of manufacture. Given the limited history of our prescription products, we currently cannot reliably estimate expected returns of the prescription products at the time of shipment. Accordingly, we defer recognition of revenue on prescription products until the right of return no longer exists, which occurs at the earlier of the time the prescription products are dispensed through patient prescriptions or expiration of the right of return.
We maintain various rebate programs in an effort to maintaina competitive position in the marketplace and to promote sales and customer loyalty. The consumer rebate program is designed to enable the end user to return a coupon to us. If the coupon qualifies, we send a rebate check to the end user. We estimate the allowance for consumer rebates based on our experience and industry averages, which is reviewed, and adjusted if necessary, on a quarterly basis.
| F-12 |
THERAPEUTICSMD, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE B2 – SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
Revenue Recognition (continued)
Segment Reporting
We are managed and operated as one business, which is focused on creating and commercializing products targeted exclusively for women. Our business operations are managed by a single management team that reports to the President of our Company. We do not operate separate lines of business with respect to any of our products and we have experienced returns (monitored on a daily basis) equaldo not prepare discrete financial information with respect to approximatelyseparate products. All product sales are derived from sales in the United States. Accordingly, we view our business as one percent of sales.
Shipping and Handling Costs
We expense all shipping and handling costs as incurred. TheseWe include these costs are included in cost of sales on the accompanying consolidated financial statements.
Advertising Costs
We expense advertising costs when incurred. Advertising expenses totaled $19,408costs were $698,871, $11,739 and $25,698$65,944 during the years ended December 31, 20112014, 2013 and 2010,2012, respectively.
Research and Development Expenses
Research and development, expenditures, which areor R&D, expenses include internal R&D activities, services of external contract research organizations, or CROs, costs of their clinical research sites, and other activities. Internal R&D activity expenses include laboratory supplies, salaries, benefits, and non-cash share-based compensation expenses. Advance payments to be expensed as incurred, totaled $107,241in future R&D activities were $1,175,082 and $65,402 during$2,606,405 for the years ended December 31, 20112014 and 2010,2013, respectively.
THERAPEUTICSMD, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 2 – SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
Earnings Per Share
We calculate earnings per share, (“EPS”)or EPS, in accordance with ASC 260, “Earnings Per Share,” which requires the computation and disclosure of two EPS amounts,amounts: basic and diluted. BasicWe compute basic EPS is computed based on the weighted averageweighted-average number of shares of common stock, par value $0.001 per share, or Common Stock, outstanding during the period. DilutedWe compute diluted EPS is computed based on the weighted averageweighted-average number of common shares of our Common Stock outstanding plus all potentially dilutive common shares of our Common Stock outstanding during the period. Such potentialpotentially dilutive common shares of our Common Stock consist of stock options and warrants. Potential common shares totaling 96,618,626warrants and 165,752 (Reverse Split shares) at December 31, 2011 and 2010, respectively, have beenwere excluded from the calculation of diluted earnings per share calculation as they arebecause their effect would have been anti-dilutive due to the net loss reported by us.
The table below presents the Company.
| As of December 31, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
| Stock options | 16,792,443 | 15,632,742 | 13,733,488 | |||||||||
| Warrants | 13,927,916 | 14,293,499 | 12,193,499 | |||||||||
| 30,720,359 | 29,926,241 | 25,926,987 | ||||||||||
Use of Estimates
Our consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America.America, or GAAP. The preparation of these financial statements requires us to make significant estimates and judgments that affect the reported amounts of assets, liabilities, revenues,revenue, expenses, and related disclosure of contingent assets and liabilities. We evaluate our estimates, including those related to contingencies, on an ongoing basis. We base our estimates on historical experience and on various other assumptions that are believedwe believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ, at times in material amounts, from these estimates under different assumptions or conditions.
Recently Issued Accounting Pronouncements
In August 2014, the FASB issued Accounting Standards Update, or ASU, No. 2014-05, Presentation of Financial Statements-Going Concern (Subtopic 205-40): Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern. ASU 2014-15 requires management to evaluate whether there are conditions and events that raise substantial doubt aboutthe entity’s ability to continue as a going concern within one year after the financial statements are issued (or available to be issued when applicable) and, if so, disclose that fact. ASU 2014-15 is effective for annual periods ending after December 15, 2016 and interim periods within annual periods beginning after December 15, 2016. Early adoption is permitted for annual or interim reporting periods for which the financial statements have not previously been issued. We do not expect the adoption of the ASU 2014-15 to have a material effect on our consolidated financial statements and disclosures.
THERAPEUTICSMD, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE B2 – SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
Recently Issued Accounting Pronouncements
In December 2011,May 2014, the FASB issuedand the International Accounting Standards Update (“ASU”) 2011-11, Balance Sheet - Offsetting. This guidance requires disclosures about offsettingBoard (IASB) issued ASU, No. 2014-09, Revenue from Contracts with Customers (Topic 606). The standard’s core principle is that a company will recognize revenue when it transfers promised goods or services to customers in an amount that reflects the consideration to which the company expects to be entitled in exchange for those goods or services. In doing so, companies will need to use more judgment and related arrangements for recognized financial instrumentsmake more estimates than under previous guidance. These may include identifying performance obligations in the contract, estimating the amount of variable consideration to include in the transaction price and derivative instruments. The standardallocating the transaction price to each separate performance obligations. ASU 2014-09 is effective for us aspublic business entities, certain not-for-profit entities and certain employee benefit plans, for annual periods beginning after December 15, 2016, including interim periods within that period. Early adoption is not permitted under GAAP. We are currently evaluating the impact of January 1,ASU 2014-09 on our consolidated financial statements and disclosures.
In July 2013, and will not materially impact our financial statement disclosures.
In MayDecember 2011, the FASB issued ASU 2011-04, Fair Value Measurement: AmendmentsNo. 2011-11, Balance Sheet (Topic 210): Disclosures about Offsetting Assets and Liabilities, or ASU 2011-11. ASU 2011-11 enhances current disclosures about financial instruments and derivative instruments that are either offset on the statement of financial position or subject to Achieve Common Fair Value Measurementan enforceable master netting arrangement or similar agreement, irrespective of whether they are offset on the statement of financial position. Entities are required to provide both net and Disclosure Requirementsgross information for these assets and liabilities in U.S.order to facilitate comparability between financial statements prepared in conformity with GAAP and IFRSs. Thisfinancial statements prepared on the basis of International Financial Reporting Standards. ASU represents the converged guidance of the FASB and the IASB (the “Boards”) on fair value measurement, and results in common requirements2011-11 is effective for measuring fair value and for disclosing information about fair value measurements, including a consistent meaning of the term “fair value.” These amendments change some of the terminology used to describe many of the existing requirements in U.S. GAAP for measuring fair value and for disclosing information about fair value measurements. The amendments should be applied prospectively, and they are effective during interim and annual reporting periods beginning on or after December 15, 2011. Early application by public entities isJanuary 1, 2013, and interim periods within those years. ASU 2011-11 did not permitted. The adoption is not expected to have a material impact on the Company’s results of operations,our financial position or cash flows.
We do not believe there would behave been a material effect on the accompanying consolidated financial statements had any other recently issued, but not yet effective, accounting standards been adopted in the current period.
Reclassifications
Certain 2013 and 2012 amounts have been reclassified to conform to current year presentation.
| F-15 |
THERAPEUTICSMD, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 3 – INVENTORY
Inventory consists of the following:
| December 31, | ||||||||
| 2014 | 2013 | |||||||
| Finished product | $ | 874,294 | $ | 621,679 | ||||
| Raw material | 155,341 | 250,943 | ||||||
| Deferred costs | 152,478 | 170,996 | ||||||
| TOTAL INVENTORY | $ | 1,182,113 | $ | 1,043,618 | ||||
NOTE 4 – OTHER CURRENT ASSETS
Other current assets consist of the following:
| December 31, | ||||||||
| 2014 | 2013 | |||||||
| Prepaid consulting | $ | 411,864 | $ | 530,596 | ||||
| Prepaid insurance | 394,878 | 145,722 | ||||||
| Prepaid research and development costs | 299,498 | 1,267,588 | ||||||
| Other receivables-related party (Note 12) | 249,981 | 249,981 | ||||||
| Other prepaid costs | 181,186 | 23,806 | ||||||
| Deferred financing costs | — | 260,022 | ||||||
| TOTAL OTHER CURRENT ASSETS | $ | 1,537,407 | $ | 2,477,715 | ||||
NOTE 5 – FIXED ASSETS
Fixed assets consist of the following:
| December 31, | ||||||||
| 2014 | 2013 | |||||||
| Equipment | $ | 132,150 | $ | 108,458 | ||||
| Furniture and fixtures | 53,895 | 46,625 | ||||||
| 186,045 | 155,083 | |||||||
| Accumulated depreciation | (122,752 | ) | (93,765 | ) | ||||
| TOTAL FIXED ASSETS | $ | 63,293 | $ | 61,318 | ||||
Depreciation expense for the years ended December 31, 2014, 2013, and 2012 was $28,987, $47,883, and $27,484, respectively. In December 2013, accumulated depreciation was reduced by $11,980 associated with leasehold improvements of our previously leased office property.
| F-16 |
THERAPEUTICSMD, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 6 –PREPAID EXPENSE
Prepaid expense consists of the following:
| December 31, | ||||||||
| 2014 | 2013 | |||||||
| Prepaid manufacturing costs | $ | 899,000 | $ | 899,000 | ||||
| Prepaid research and development costs | 463,720 | 824,221 | ||||||
| Accreted prepaid costs | 64,543 | 27,234 | ||||||
| TOTAL PREPAID EXPENSE | $ | 1,427,263 | $ | 1,750,455 | ||||
NOTE 7 – INTANGIBLE ASSETS
The following table sets forth the gross carrying amount and accumulated amortization of our intangible assets as of December 31, 20112014 and December 31, 2013:
| December 31, 2014 | ||||||||||||||||
| Gross Carrying Amount | Accumulated Amortization | Net Amount | Weighted- Average Remaining Amortization Period (yrs.) | |||||||||||||
| Amortizing intangible assets: | ||||||||||||||||
| OPERA® software patent | $ | 31,951 | $ | (2,496 | ) | $ | 29,455 | 14.75 | ||||||||
| Development costs of corporate website | 91,743 | (91,743 | ) | — | n/a | |||||||||||
| Approved hormone therapy drug candidate patents | 439,184 | (19,401 | ) | 419,783 | 18 | |||||||||||
| Non-amortizing intangible assets: | ||||||||||||||||
| Hormone therapy drug candidate patents (pending) | 675,982 | — | 675,982 | n/a | ||||||||||||
| Multiple trademarks for vitamins/supplements | 103,368 | — | 103,368 | n/a | ||||||||||||
| Total | $ | 1,342,228 | $ | (113,640 | ) | $ | 1,228,588 | |||||||||
THERAPEUTICSMD, INC. AND 2010
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE C7 – GOING CONCERN
| December 31, 2013 | ||||||||||||||||
| Gross Carrying Amount | Accumulated Amortization | Net Amount | Weighted- Average Remaining Amortization Period (yrs.) | |||||||||||||
| Amortizing intangible assets: | ||||||||||||||||
| OPERA® software patent | $ | 31,951 | $ | (499 | ) | $ | 31,452 | 15.8 | ||||||||
| Development costs of corporate website | 91,743 | (89,661 | ) | 2,082 | 0.3 | |||||||||||
| Non-amortizing intangible assets: | ||||||||||||||||
| Hormone therapy drug candidate patents (pending) | 572,726 | — | 572,726 | n/a | ||||||||||||
| Multiple trademarks for vitamins/supplements | 59,328 | — | 59,328 | n/a | ||||||||||||
| Total | $ | 755,748 | $ | (90,160 | ) | $ | 665,588 | |||||||||
We amortized the Company will continue as a going concern. The Company incurred a loss from operationsintangible asset related to development costs for corporate website over 36 months, which is the prescribed life for software and website development costs. We amortize the intangible asset related to OPERA® using the straight-line method over the estimated useful life of approximately $5,400,000, had negative cash flow from operations20 years, which is the life of the intellectual property patents. We amortize the approved hormone therapy drug candidate patents using straight-line method over the estimated useful life of approximately $5,000,000 and had an accumulated deficit of approximately $17,000,000 at20 years. During the years ended December 31, 2011. These matters raise substantial doubt about2014 and 2013, there was no impairment recognized.
In addition to numerous pending patent applications, as of December 31, 2014, we had 4 issued patents, including:
• | 3 utility patents that relate to our combination progesterone and estradiol formulations, which are owned by us and are U.S. jurisdiction patents with expiration dates in 2032. We have pending patent applications with respect to certain of these patents in Argentina, Australia, Canada, the European Union, Mexico, Brazil, Japan, Russia, South Africa and South Korea. |
Subsequent to December 31, 2014, 1 additional patent was issued related to our combination progesterone and estradiol formulations.
THERAPEUTICSMD, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 7 – INTANGIBLE ASSETS
Amortization expense was $23,480, $10,262 and $28,776 for the Company’s abilityyears ended December 31, 2014, 2013, and 2012, respectively. As of December 31, 2014, the estimated amortization expense for the next five years is as follows:
| Year Ending December 31, | Estimated Amortization | |||||
| 2015 | $ | 25,138 | ||||
| 2016 | $ | 25,138 | ||||
| 2017 | $ | 25,138 | ||||
| 2018 | $ | 25,138 | ||||
| 2019 | $ | 25,138 | ||||
NOTE 8 – OTHER CURRENT LIABILITIES
Other current liabilities consist of the following:
| December 31, | ||||||||
| 2014 | 2013 | |||||||
| Accrued clinical trial costs | $ | 1,706,542 | $ | 665,782 | ||||
| Accrued payroll, bonuses and commission costs | 814,205 | 941,313 | ||||||
| Accrued vacation costs | 442,430 | 256,920 | ||||||
| Accrued legal and accounting expense | 276,470 | 224,550 | ||||||
| Other accrued expenses(1) | 185,965 | 177,900 | ||||||
| Allowance for wholesale distributor fees | 160,503 | 306,303 | ||||||
| Accrued royalties | 72,710 | 52,188 | ||||||
| Allowance for coupons and returns | 90,446 | 126,233 | ||||||
| Accrued rent | 91,368 | — | ||||||
| Accrued financing costs | — | 850,000 | ||||||
| TOTAL OTHER CURRENT LIABILITIES | $ | 3,840,639 | $ | 3,601,189 | ||||
_________________
(1) In June 2008, we declared and paid a special dividend of $0.40 per share of our Common Stock to continueall stockholders of record as of June 10, 2008, of which $41,359 remained unclaimed by certain stockholders at both December 31, 2014 and 2013.
NOTE 9 – NOTES PAYABLE
Issuance and Payment of Multiple Advance Revolving Credit Note
On January 31, 2013, we entered into a going concern. Management’s plans include raisingbusiness loan agreement with Plato and Associates, LLC, or Plato, for a Multiple Advance Revolving Credit Note, or the Revolving Credit Note. The Revolving Credit Note allowed us to draw down funding up to a $10,000,000 maximum principal amount, at a stated interest rate of 6% per annum. Plato was able to make advances to us from time to time under the Revolving Credit Note at our request, which advances were of a revolving nature and were able to be made, repaid, and made from time to time. Interest payments were due and payable on the tenth day following the end of each calendar quarter in which any interest was accrued and unpaid, commencing on April 10, 2013, and the principal balance outstanding under the Revolving Credit Note, together with all accrued interest and other amounts payable under the Revolving Credit Note, if any, was due and payable on February 24, 2014. The Revolving Credit Note was secured by substantially all of our assets. On each of February 25 and March 13, 2013, $200,000 was drawn against the Revolving Credit Note. On March 21, 2013, we repaid $401,085, which included accrued interest, and there was no balance outstanding under the Revolving Credit Note as of December 31, 2013 and February 24, 2014 when it expired. As additional proceeds from debtconsideration for the Revolving Credit Note, we granted to Plato a warrant to purchase 1,250,000 shares of our Common Stock at an exercise price of $3.20 per share (See Note 10).
THERAPEUTICSMD, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Borrowing under Business Loan Agreement and equity transactionsPromissory Note
In March 2011, VitaMed entered into a business loan agreement with First United Bank for a $300,000 bank line of credit for which personal guarantees and cash collateral were required. Personal guarantees and cash collateral limited to continue to increase its sales$100,000 each were provided by Robert Finizio and marketing activities; however, there are no assurances that management will be successful in their efforts.John Milligan, officers of our Company, and by Reich Family Limited Partnership, an entity controlled by Mitchell Krassan, also an officer of our Company. The financial statements do not include adjustments relatingbank line of credit accrued interest at the rate of 3.02% per annum based on a year of 360 days and was due on March 1, 2012. We negotiated a one-year extension with First United to the recoverabilitybank line of credit, which was executed on March 19, 2012. Under the extension, borrowings bore interest at a rate of 2.35% and realizationwere due on March 1, 2013. On November 13, 2012, the outstanding balance of assets$299,220 was repaid in full, and classificationwe amended the line of liabilitiescredit to reflect a $100,000 bank line of credit. In accordance with the amended line of credit, the personal guarantee and cash collateral limited to $100,000 provided by the Reich Family Limited Partnership remained in place, while the personal guarantees and cash collateral were removed for Mr. Finizio and Mr. Milligan. In February 2013, we borrowed $100,000 from First United Bank under the amended bank line of credit. On April 25, 2013, we re-paid $100,735, which represented the principal and interest that might be necessary shouldwas due under the Company be unableamended bank line of credit. On May 1, 2013, the amended bank line of credit expired and was not renewed. Accordingly, the personal guarantee was canceled, and the cash collateral was refunded to continuethe Reich Family Limited Partnership. During the years ended December 31, 2013 and 2012, we paid $735 and $7,366, respectively, of interest expense, which are included in operation.
Issuance of the Company’s Common Stock. January and February 2012 Promissory Notes
In addition, all VitaMed OptionsJanuary and VitaMed Warrants were exchanged and converted into Company Options and Company Warrants. All Units , VitaMed Options and VitaMed Warrants were exchanged on a pro-rata basis for shares of the Company’s Common Stock whichFebruary 2012, we issued 6% promissory notes in the aggregate totaled 70,000,000principal amount of $900,000, due March 1, 2012, which were subsequently increased to an aggregate principal amount of $1,700,000. As discussed below in Issuance and Settlement of February 2012 Notes, these promissory notes were modified on February 24, 2012 through the issuance of secured promissory notes.
THERAPEUTICSMD, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 9 – NOTES PAYABLE (Continued)
Issuance and Settlement of February 2012 Promissory Notes
On February 24, 2012, we issued promissory notes, the February 2012 Notes, to an individual and an entity, both of which were stockholders of our company, in the principal amount of $1,358,014 and $1,357,110, respectively, and granted warrants to purchase an aggregate of 9,000,000 shares of our Common Stock pursuant to the terms of a Note Purchase Agreement, dated February 24, 2012. We received an aggregate of $1,000,000 of new funding upon issuance of the February 2012 Notes and related warrants and surrender by the holders of certain promissory notes, which we previously issued in the aggregate amount of $1,700,000 plus the aggregate accrued interest of $15,124 (collectively, the “Prior Notes”). Under the February 2012 Notes, as amended, we borrowed an additional $3,000,000 during March, April, and May 2012.
We granted warrants to purchase an aggregate of 5,685,300 shares of Common Stock in consideration of the modification of the Prior Notes and warrants to purchase an aggregate of 3,314,700 shares of Common Stock in connection with the issuance of the February 2012 Notes. We determined that the resulting modification of the Prior Notes was substantial in accordance with ASC 470-50, Modifications and Extinguishments. As such the modification was accounted for as an extinguishment and restructuring of the debt, and the 5,685,300 warrants issued in consideration of the modification were expensed (see Warrant Activity during 2012 in NOTE 10 for more details). The fair value of the Prior Notes was estimated by calculating the present value of the future cash flows discounted at a market rate of return for comparable debt instruments to be $1,517,741, resulting in a conversion ratio calculated bydebt discount of $197,383 and recognized a loss on extinguishment of debt of $10,307,864, which represented the sum of all Units, VitaMed Options and VitaMed Warrants divided by 70,000,000 (the “Conversion Ratio”). Pursuant to the Conversion Ratio, the Company issued 58,407,331 sharesfair value of the Company’s Common Stock5,685,300 warrants net of the difference between the carrying amount of the Prior Notes and their fair value as of the date of the modification on the accompanying consolidated financial statements.
On June 19, 2012, we settled an aggregate amount of $3,102,000 of principal and accrued interest of the February 2012 Notes in exchange for the Units, reservedexercise of warrants to purchase 8,145,486 shares of our Common Stock. As discussed below in Issuance and Payment of June 2012 Notes, the remaining balance of $2,691,847 of the February 2012 Notes was modified on June 19, 2012 through the issuance of the June 2012 Notes (as defined below) (see NOTE 10 for issuancemore details).
Issuance and Payment of June 2012 Promissory Notes
On June 19, 2012, we issued secured promissory notes, or the June 2012 Notes, to the holders of the February 2012 Notes in the principal amounts of $2,347,128 and $2,344,719, pursuant to the terms of a Note Purchase Agreement. In connection with the June 2012 Notes, the holders of the February 2012 Notes surrendered the remaining balance of such notes in the aggregate amount of $1,347,128 and $1,344,719, which sums included principal and accrued interest through June 19, 2012, and we received an aggregate of 10,119,796 shares issuable upon$2,000,000 of new funding, or the exerciseJune Funding, from the holders of the Company OptionsFebruary 2012 Notes. The principal amount of each of the June 2012 Notes, plus any additional advance made to us thereafter, together with accrued interest at the annual rate of 6%, was due in one lump sum payment on February 24, 2014. As security for our obligations under the June 2012 Notes, we entered into a security agreement and reserved for issuancepledged all of our assets, tangible and intangible, as further described therein. We also granted warrants to purchase an aggregate of 1,472,9167,000,000 shares of our Common Stock in connection with the June Funding. On March 21, 2013, we repaid $4,882,019 of the June 2012 Notes, including accrued interest, leaving a balance of $21,595 in accrued interest as of March 31, 2013. On April 25, 2013, the balance of accrued interest was paid in full.
THERAPEUTICSMD, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 9 – NOTES PAYABLE (Continued)
Issuance and Payment of Additional Promissory Notes in 2012
In August and September 2012, we issued 6% promissory notes in the amount of $1,600,000 due on October 1, 2012, which due date was subsequently extended. These notes were paid in full in October 2012.
In September 2012, we issued a 6% promissory note for $200,000 due on October 15, 2012. This note was paid in full in October 2012.
Issuance and Payment of Additional Promissory Notes in 2011
In December 2011, we issued 4% promissory notes to Mr. Finizio and Mr. Milligan for an aggregate of $100,000 due March 1, 2012. These promissory notes were subsequently extended by mutual agreement to June 1, 2012. In June 2012, we paid the promissory note held by Mr. Finizio in full, including $888 in accrued interest. Mr. Milligan’s promissory note was extended to October 15, 2012. On October 4, 2012, we paid Mr. Milligan’s promissory note in full, including $1,519 in accrued interest.
Conversion of July 2011 Secured Notes
In July 2011, VitaMed issued two senior secured promissory notes, or the Secured Notes, each in the amount of $500,000 and also entered into a security agreement under which VitaMed pledged all of its assets to secure the obligation. The Secured Notes were assumed by us upon the merger with VitaMed in October 2011 and bore interest at the rate of 6% per annum, were due on the one year anniversary thereof, and were convertible into shares of our Common Stock at our option. We were permitted to satisfy the obligations underlying the Secured Notes by delivering such number of shares of our Common Stock calculated by dividing the then-outstanding principal balance by the Share Price. For purposes of the Secured Notes, the “Share Price” meant the lower of the most recent price at which we offered and sold shares of our Common Stock (not including any shares of our Common Stock issued upon the exercise of options and/or warrants or upon the Company Warrants.
Issuance, Payment and Conversion of VitaMed Promissory Notes
In June 2011, VitaMed issued promissory notes, or the VitaMed Notes, in the aggregate principal amount of $500,000. In connection with the VitaMed Notes, VitaMed granted warrants to purchase equity interests in VitaMed that were equivalent to an aggregate of 613,718 shares of our Common Stock when the VitaMed Notes were assumed by us upon the merger with VitaMed. The VitaMed Notes bore interest at a rate of 4% per annum and were due at the earlier of (i) the six month anniversary of the date of issuance and (ii) such time as VitaMed received the proceeds of a promissory note(s) issued in an amount of not less than $1,000,000, or the Funding. Upon the closing of the Funding in July 2011, as more fully described above in Conversion of July 2011 Secured Notes, two of the VitaMed Notes in the aggregate principal amount of $200,000 were paid in full. By mutual agreement, the remaining VitaMed Notes in the aggregate principal amount of $300,000 were extended. In October 2011, one of the VitaMed Notes in the aggregate principal amount of $50,000 was paid in full, and, by mutual agreement, certain of the VitaMed Notes in the aggregate principal amount of $100,000 were converted into 266,822 shares of our Common Stock at $0.38 per share, which represented the fair value of our Common Stock on the date of conversion. In June 2012, a VitaMed Note held by an unaffiliated individual was paid in full, including $2,160 in accrued interest. The remaining VitaMed Notes, held by Mr. Milligan and by BF Investment Enterprises, Ltd., which is owned by Brian Bernick, a director of our company, in the aggregate principal amount of $100,000, were extended to October 15, 2012. On October 4, 2012, we re-paid the outstanding VitaMed Notes in full, including $5,341 in accrued interest.
THERAPEUTICSMD, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 9 – NOTES PAYABLE (Continued)
Issuance, Payment and Conversion of VitaMed Promissory Notes (continued)
In September and October 2011, VitaMed issued convertible notes, or the VitaMed Convertible Notes, in the aggregate amount of $534,160. The VitaMed Convertible Notes bore interest at the rate of 4% per annum and were due December 1, 2011. On November 18, 2011, we entered into Debt Conversion Agreements with the holders of the VitaMed Convertible Notes, pursuant to which we converted the principal and accrued interest of the VitaMed Convertible Notes into 1,415,136 shares of our Common Stock at $0.38 per share, which represented the fair value of the shares of our Common Stock on the date of conversion.
For the year ended December 31, 2012, we recorded an aggregate of $6,344 as interest expense on the accompanying consolidated financial statements.
NOTE 10 – STOCKHOLDERS’ EQUITY
Preferred Stock
At December 31, 2011, the Company2014, we had 10,000,000 shares of Preferred Stock, par value $0.001, authorized and none outstanding,for issuance, of which no shares can be designated by the Company’s Board of Directors.
Common Stock
At December 31, 2011, the Company2014, we had250,000,000 shares of Common Stock, $0.001 par value $0.001,authorized for issuance, of which156,097,019 shares of our Common Stock were issued and outstanding.
Issuances During 2014
On July 29, 2014, we entered into an underwriting agreement with 82,978,781Goldman Sachs & Co, as the representative of the underwriters named therein, or the Goldman Sachs Underwriters, relating to an underwritten public offering of 8,565,310 shares of our Common Stock. The price to the public in the offering was $4.67 per share. Under the terms of the underwriting agreement, we granted the Goldman Sachs Underwriters a 30-day option to purchase up to an additional 1,284,796 shares of our Common Stock. On July 30, 2014, the Goldman Sachs Underwriters exercised their option to purchase the additional 1,284,796 shares of our Common Stock. Net proceeds from this offering were approximately $42.8 million, after deducting underwriting discounts and commissions and other offering expenses payable by us. The offering closed on August 4, 2014.
THERAPEUTICSMD, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 10 – STOCKHOLDERS’ EQUITY (Continued)
During the year ended December 31, 2014, certain individuals exercised stock options to purchase 860,800 shares of our Common Stock. Stock options to purchase shares of our Common Stock were exercised as follows: (i) 724,193 options for $345,746 in cash and (ii)136,607 options, pursuant to the stock options’ cashless provision, wherein 130,380 shares of Common Stock were issued. In addition, during 2014, we issued 50,000 shares of Common Stock to an employee upon the vesting of restricted stock units that were granted in December 2013.
During the year ended December 31, 2014, certain individuals exercised warrants to purchase 365,583 shares of our Common Stock for $181,000 in cash.
Issuances During 2013
On March 14, 2013, we entered into an underwriting agreement with Jefferies LLC as the representative of the underwriters named therein, or the Jefferies Underwriters, relating to the issuance and outstanding.
On September 25, 2013, we entered into an underwriting agreement with Stifel, Nicolaus & Company, Incorporated, as the representative of the underwriters named therein, or the Stifel Underwriters, relating to the issuance and sale of 13,750,000 shares of our Common Stock. The price to the public in the offering was $2.40 per share. The net proceeds to us from this offering were approximately $30.2 million, after deducting underwriting discounts and commissions and other offering expenses payable by us. The offering closed on September 30, 2013.
During 2013 certain individuals exercised stock options to purchase an aggregate of 75,423 shares of our Common Stock for approximately $31,000.
Issuances During 2012
During June 2012, we settled $3,102,000 in principal and accrued interest of the February 2012 Notes in exchange for the holders’ exercise of a portion of the related warrants for an aggregate of 8,145,486 shares of Common Stock. During June 2012, we and May 2011, VitaMedthe holders also agreed to convert a portion of the February 2012 Notes, and principal and accrued interest through June 19, 2012 totaling $1,054,647, into 2,775,415 shares of Common Stock at $0.38 per share.
THERAPEUTICSMD, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 10 – STOCKHOLDERS’ EQUITY (Continued)
Issuances During 2012 (continued)
In September 2012, we entered into a Securities Purchase Agreement with multiple investors, relating to the issuance and sale of Common Stock in a private placement. The private placement closed on October 2, 2012, through which we sold 2,892,630 Unitsan aggregate of 3,953,489 shares of Common Stock at $2.15 per share, for an aggregate purchase price of $707,000.
During the year ended December 31, 2012, certain individuals exercised stock options to purchase 2,631,5791,958,216 shares of the Company’s Common Stock. Stock (the “Shares”) at aoptions to purchase price of $0.38 per share for a total purchase price of $1,000,000 (“Purchase Price”). In connection with the Stock Purchase Agreement, the Company and Pernix entered into a Lock-Up Agreement which, among other things, restricts the sale, assignment, transfer, encumbrance and other disposition of the Shares issued to Pernix. Pursuant to the terms of the Lock-Up Agreement, Pernix agreed
Warrants to Purchase Common Stock
As of December 2011, a former director of VitaMed, exercised Company Options31, 2014, we had warrants outstanding to purchase 92,057an aggregate of 13,927,916 shares of the Company’sour Common Stock at an aggregatewith a weighted-average contractual remaining life of 3.0 years, and exercise prices ranging from $0.24 to $3.20 per share, resulting in a weighted average exercise price of $17,250.
The valuation methodology used to determine the fair value of Common Stock purchaseour warrants is the Black-Scholes-Merton option-pricing model (“Black-Scholes Model”), an acceptable model in accordance with ASC 718-10.Model. The Black-Scholes Model requires the use of a number of assumptions, including volatility of the stock price, the risk-free interest rate and the term of the Common Stock purchase warrant.
Warrant Activity During 2014
During the year ended December 31, 2011,2014, we did not issue any warrants. As of December 31, 2014, unamortized costs associated with the Sancilio & Company, Inc., or SCI warrants issued in 2013 and 2012 totaled approximately $875,600.
Warrant Activity During 2013
In January 2013, we issued warrants to purchase 1,250,000 shares of our Common Stock in connection with the following:
| Purpose | Number of Shares Under Company Warrants | Exercise Price | Exercise Term in Years | Fair Value | ||||||||||||
| Loan guaranty | 613,713 | $0.24 | 10 | $ | 93,969 | |||||||||||
| Loan consideration | 613,718 | $0.41 | 5 | 30,993 | ||||||||||||
| Product consulting | 1,045,485 | $0.38-$0.41 | 5-10 | 189,942 | ||||||||||||
| Services | 784,711 | $0.38-$1.50 | 5-10 | 159,363 | ||||||||||||
| 3,057,627 | $ | 474,267 | ||||||||||||||
THERAPEUTICSMD, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 10 – STOCKHOLDERS’ EQUITY (Continued)
Warrant Activity During 2013 (continued)
In May 2013, we entered into a consulting agreement with SCI, to develop drug platforms to be used in our hormone replacement drug candidates. These services include support of our efforts to successfully obtain U.S. Food and Drug Administration, or the FDA, approval for our drug candidates, including a vaginal capsule for the treatment of vulvar and vaginal atrophy, or VVA. In connection with the agreement, SCI agreed to forfeit its rights to receive warrants to purchase 833,000 shares of our Common Stock that were to be granted pursuant to the terms of a prior consulting agreement dated May 17, 2012. As consideration under the agreement, we agreed to issue to SCI a warrant to purchase 850,000 shares of our Common Stock at $2.01 per share that has vested or will vest, as applicable, as follows:
| 1. | 283,333 shares were earned on May 11, 2013 upon acceptance of an Investigational New Drug application by the FDA for an estradiol-based drug candidate in a softgel vaginal capsule for the treatment of VVA; however, pursuant to the terms of the consulting agreement, the shares did not vest until June 30, 2013. The fair value of $405,066 for the shares vested on June 30, 2013 was determined by using the Black-Scholes Model on the date of vesting using a term of 5 years; a volatility of 45.89%; risk free rate of 1.12%; and a dividend yield of 0%. We recorded the entire $405,066 as non-cash compensation as of June 30, 2013; |
| 2. | 283,333 shares vested on June 30, 2013. The fair value of $462,196 for these shares was determined by using the Black-Scholes Model on the date of the vesting using a term of 5 years; a volatility of 45.84%; risk free rate of 1.41%; and a dividend yield of 0%. As of December 31, 2014, we recorded $154,068 as prepaid expense-short term and $77,026 as prepaid expense-long term in the accompanying consolidated financial statements. During the years ended December 31 2014 and 2013, we recorded $154,068 and $77,034, respectively, as non-cash compensation in the accompanying consolidated financial statements; and |
| 3. | 283,334 shares will vest upon the receipt by us of any final FDA approval of a drug candidate that SCI helped us design. It is anticipated that this event will not occur before December 2015. |
Warrant Activity During 2012
In February 2012, we issued warrants for the purchase of an aggregate of 5,685,300 shares of Common Stock in connection with the modification of certain existing promissory notes, or the Modification Warrants, and warrants for the purchase of an aggregate of 3,314,700 shares of our Common Stock in connection with the issuance of the February 2012 Notes, or the February 2012 Warrants (see NOTE 9). Both the Modification Warrants and the February 2012 Warrants are exercisable at $0.38 per share. The Modification Warrants have a fair value of $10,505,247, and the February 2012 Warrants have a fair value of $6,124,873. Fair value was determined on the date of the issuance using a term of five years; a volatility of 44.5%; risk free rate of 0.89%; and a dividend yield of 0%. We recorded the fair value of the Modification Warrants as part of the loss on extinguishment of debt in the accompanying consolidated financial statements. The relative fair value of the February 2012 Warrants of $859,647 was recorded as debt discount. As a result of the surrender of the February 2012 Notes on June 19, 2012, we expensed the remaining unamortized debt discount.
THERAPEUTICSMD, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 10 – STOCKHOLDERS’ EQUITY (Continued)
Warrant Activity During 2012
In March 2012, we issued warrants to purchase an aggregate of 31,000 shares of Common Stock to five unaffiliated individuals for services rendered. These warrants were valued on the date of the issuance using a term of five years; a volatility of 44.81%; risk free rate of 1.04%; and a dividend yield of 0%; we recorded $29,736 as consulting expense in the accompanying consolidated financial statements.
In May 2012, we issued warrants to purchase an aggregate of 1,300,000 shares of Common Stock to an unaffiliated entity for services to be rendered over approximately five years beginning in May 2012. Services provided are to include (a) services in support of our drug development efforts, including services in support our ongoing and future drug development and commercialization efforts, regulatory approval efforts, third-party investment and financing efforts, marketing efforts, chemistry, manufacturing and controls efforts, drug launch and post-approval activities, and other intellectual property and know-how transfer associated therewith; (b) services in support of our efforts to successfully obtain New Drug Approval; and (c) other consulting services as mutually agreed upon from time to time in relation to new drug development opportunities. The warrants were valued at $1,532,228 on the date of the issuance using an exercise price of $2.57; a term of five years; a volatility of 44.71%; risk free rate of 0.74%; and a dividend yield of 0%. At December 31, 2014, we had $257,796 reported as prepaid expense-short term and $386,694 recorded as prepaid expense-long term. During the years ended December 31, 2014, 2013 and 2012, we recorded $309,165, $360,528 and $218,045, respectively as non-cash compensation with respect to these warrants in the accompanying consolidated financial statements. The contract will expire upon the commercial manufacture of a drug product. We have determined that the process will take approximately five years. As a result, we are amortizing the $1,532,228 over five years.
In June 2012, we issued warrant to purchase aggregate of 7,000,000 shares of Common Stock in connection with the issuance of the June 2012 Notes, or the June 2012 Warrants, (see NOTE 9). Of the June 2012 Warrants issued, 6,000,000 are exercisable at $2.00 per share and 1,000,000 are exercisable at $3.00 per share. The fair value of the June 2012 Warrants of $9,424,982 was determined on the date of the issuance using a term of five years; a volatility of 44.64%; risk free rate of 0.75%; and a dividend yield of 0%. The Company Warrant vested immediately. Of the $12,548 fair value, $5,612 was recorded as
In June 2012, we issued warrants to purchase an aggregate of 1,500 shares of Common Stock to three unaffiliated individuals for services performed. rendered. The Company Warrant waswarrants were valued on the date of the grantissuance using a term of 10five years; a volatility of 45.94%44.78%; risk free rate of 2.23%0.72%; and a dividend yield of 0%. The Company Warrant vests over a 44-month period beginning on November 21, 2011 (or 13,636 shares for months 1-43 and 13,652 shares for month 44). Of the $133,045 fair value, $7,158A total of $1,656 was recorded as non-cash compensation on consulting expense in the accompanying consolidated financial statementsstatements.
. The remaining $125,887 will be expense to non-cash compensation equitably over the remaining 42 months.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE D10 – STOCKHOLDERS’ EQUITY (Continued)
Summary of our Warrant activity and related information for 184,211 shares with a fair value of $25,980 to an unrelated entity for consulting services covered under a two (2) month agreement. The Company Warrant was valued on the date of the grant using a term of five (5) years; volatility of 41.04%; risk free rate of 1.08%; and a dividend yield of 0%. The $25,980 fair value was recorded as
| Number of Shares Under Warrants | Weighted Average Exercise Price | Weighted Average Remaining Contractual Life in Years | Aggregate Intrinsic Value | |||||||||||||
| Balance at December 31, 2011 | 3,057,627 | $ | 0.36 | 7.9 | $ | 3,483,691 | ||||||||||
| Issued | 17,332,500 | $ | 1.26 | |||||||||||||
| Exercised | (8,145,486 | ) | $ | 0.38 | ||||||||||||
| Expired | — | |||||||||||||||
| Cancelled | (51,142 | ) | $ | 0.24 | ||||||||||||
| Balance at December 31, 2012 | 12,193,499 | $ | 1.63 | 4.8 | $ | 17,971,994 | ||||||||||
| Issued | 2,100,000 | $ | 2.72 | |||||||||||||
| Exercised | — | |||||||||||||||
| Expired | — | |||||||||||||||
| Cancelled | — | |||||||||||||||
| Balance at December 31, 2013 | 14,293,499 | $ | 1.79 | 3.9 | $ | 48,932,777 | ||||||||||
| Issued | — | |||||||||||||||
| Exercised | (365,583 | ) | $ | 0.50 | ||||||||||||
| Expired | — | |||||||||||||||
| Cancelled | — | |||||||||||||||
| Balance at December 31, 2014 | 13,927,916 | $ | 1.82 | 3.0 | $ | 36,623,875 | ||||||||||
| Vested and Exercisable at December 31, 2014 | 13,562,764 | $ | 1.83 | 2.9 | $ | 35,599,540 | ||||||||||
The weighted average fair value per share of Company Warrants grantedwarrants issued and the assumptions used in the Black-Scholes Model during the years ended December 31, 20112014, 2013 and 2012 are set forth in the table below.
| 2014 | 2013 | 2012 | ||||||||||
| Weighted average fair value | $ | — | $ | 2.83 | $ | 2.05 | ||||||
| Risk-free interest rate | — | % | 0.88-1.12 | % | 0.72-1.04 | % | ||||||
| Volatility | — | % | 44.29-45.89 | % | 44.64-44.81 | % | ||||||
| Term (in years) | — | 5-6 | 5 | |||||||||
| Dividend yield | 0.00 | % | 0.00 | % | 0.00 | % | ||||||
The risk-free interest rate assumption is based upon observed interest rates on zero coupon U.S. Treasury bonds whose maturity period is appropriate for the term.
Estimated volatility is a measure of the amount by which the Company’sour stock price is expected to fluctuate each year during the term of the award. The Company’sOur estimated volatility is an average of the historical volatility of the stock prices of itsour peer entities whose stock prices were publicly available. The Company’sOur calculation of estimated volatility is based on historical stock prices over a period equal to the term of the awards. The CompanyWe used the historical volatility of peer entities due to the lack of sufficient historical data of itsour stock price during 2001-2011.
THERAPEUTICSMD, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE D10 – STOCKHOLDERS’ EQUITY (Continued)
Options to Purchase Common Stock purchase warrants during the year ended December 31, 2010.
| Number of Shares Under Company Warrant | Weighted Average Exercise Price | Weighted Average Remaining Contractual Life | Aggregate Intrinsic Value | |||||||||||||
| Balance at December 31, 2010 | -0- | |||||||||||||||
| Granted | 3,057,627 | $ | 0.36 | 8.7 | $ | 3,483,691 | ||||||||||
| Exercised | -0- | |||||||||||||||
| Expired | -0- | |||||||||||||||
| Cancelled | -0- | |||||||||||||||
| Balance at December 31, 2011 | 3,057,627 | $ | 0.36 | 8.7 | $ | 3,483,691 | ||||||||||
| Vested and Exercisable at December 31, 2011 | 2,254,758 | $ | 0.37 | 5.6 | $ | 2,361,339 | ||||||||||
In 2009, the Companywe adopted the 2009 Long Term Incentive Compensation Plan, (the “LTIP”)or the 2009 Plan, to provide financial incentives to employees, members of the Board, anddirectors, advisers, and consultants of the Companyour company who are able to contribute towards the creation of or who have created stockholder value by providing them stock options and other stock and cash incentives, (the “Awards”).or the Awards. The Awards available under the LTIP2009 Plan consist of stock options, stock appreciation rights, restricted stock, restricted stock units, performance stock, performance units, EVA awards, and other stock or cash awards as described in the LTIP.2009 Plan. There are 25,000,000 shares authorized for issuance thereunder. Prior to the Merger,merger with VitaMed, no awardsAwards had been issued under the LTIP.
| Number of Shares Under Company Option | Weighted Average Exercise Price | Weighted Average Remaining Contractual Life | Aggregate Intrinsic Value | |||||||||||||
| Balance at December 31, 2010 | -0- | |||||||||||||||
Granted (1) | 10,682,218 | $ | 0.16 | 7.6 | $ | 14,188,484 | ||||||||||
| Exercised | (92,057 | ) | $ | 0.19 | ||||||||||||
| Expired | -0- | |||||||||||||||
| Cancelled | -0- | |||||||||||||||
| Balance at December 31, 2011 | 10,590,161 | $ | 0.16 | 7.6 | $ | 14,067,649 | ||||||||||
| Vested and Exercisable at December 31, 2011 | 6,581,049 | $ | 0.13 | 7.5 | $ | 9,038,719 | ||||||||||
On February 23, 2012, we adopted the 2012 Stock Incentive Plan, or the 2012 Plan, a non-qualified plan that was amended in August 2013. The 2012 Plan was designed to serve as an aggregateincentive for retaining qualified and competent key employees, officers, directors, and certain consultants and advisors of 10,590,161our company. There are 10,000,000 shares with a weighted average contractual life of 7.6 years and exercise prices ranging from $0.10 to $1.22 per share resulting in a weighted average exercise priceour Common Stock authorized for issuance thereunder. As of $0.16 per share.
The valuation methodology used to determine the fair value of Company Optionsstock options is the Black-Scholes-Merton option-pricing model (“Black-Scholes Model”), an acceptable model in accordance with ASC 718-10.Model. The Black-Scholes Model requires the use of a number of assumptions including volatility of the stock price, the risk-free interest rate, and the expected life.
| 2011 | 2010 | |||
| Risk-free interest rate | 0.91-2.54% | 1.27-3.12% | ||
| Volatility | 37.92-40.48% | 36.34-42.46% | ||
| Expected life (in years) | 5.5-6.25 | 5-6.25 | ||
| Dividend yield | 0.00% | 0.00% |
| 2014 | 2013 | 2012 | ||||||||||
| Risk-free interest rate | 0.07-1.77 | % | 0.65-1.71 | % | 0.61-2.23 | % | ||||||
| Volatility | 68.05-82.29 | % | 33.35-45.76 | % | 40.77-46.01 | % | ||||||
| Term (in years) | 0.5-6.25 | 5-6.25 | 5-6.25 | |||||||||
| Dividend yield | 0.00 | % | 0.00 | % | 0.00 | % | ||||||
The risk-free interest rate assumption is based upon observed interest rates on zero coupon U.S. Treasury bonds whose maturity period is appropriate for the expected life. Estimated volatility is a measure of the amount by which the Company’s stock price of Common Stock is expected to fluctuate each year during the term of the award. The Company’sOur estimated volatility is an average of the historical volatility of the stock prices of itsour peer entities whose stock prices were publicly available. The Company’sOur calculation of estimated volatility is based on historical stock prices over a period equal to the term of the awards. The CompanyWe used the historical volatility of our peer entities due to the lack of sufficient historical data of itson our stock price during 2001-2011.price. The average expected life is based on the contractual term of the option using the simplified method.
THERAPEUTICSMD, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 10 – STOCKHOLDERS’ EQUITY (Continued)
Options to Purchase Common Stock of the Company (continued)
A summary of activity under the 2009 and 2012 Plans and related information for 2012-2014 follows:
| Number of Shares Under Options | Weighted Average Exercise Price | Weighted Average Remaining Contractual Life in Years | Aggregate Intrinsic Value | |||||||||||||
| Balance at December 31, 2011 | 10,590,161 | $ | 0.16 | 7.6 | $ | 14,067,649 | ||||||||||
| Granted | 5,121,250 | $ | 2.80 | |||||||||||||
| Exercised | (1,931,788 | ) | ||||||||||||||
| Expired | — | |||||||||||||||
| Cancelled | (46,135 | ) | ||||||||||||||
| Balance at December 31, 2012 | 13,733,488 | $ | 1.16 | 7.7 | $ | 26,804,117 | ||||||||||
| Granted | 2,583,677 | $ | 3.31 | |||||||||||||
| Exercised | (75,423 | ) | ||||||||||||||
| Expired | (250 | ) | ||||||||||||||
| Cancelled | (608,750 | ) | ||||||||||||||
| Balance at December 31, 2013 | 15,632,742 | $ | 1.44 | 7.2 | $ | 58,878,132 | ||||||||||
| Granted | 2,442,000 | $ | 4.71 | |||||||||||||
| Exercised | (854,573 | ) | ||||||||||||||
| Expired | (250 | ) | ||||||||||||||
| Cancelled | (427,476 | ) | ||||||||||||||
| Balance at December 31, 2014 | 16,792,443 | $ | 1.88 | 6.9 | $ | 43,996,311 | ||||||||||
| Vested and Exercisable at December 31, 2014 | 13,276,462 | $ | 1.39 | 5.5 | $ | 40,720,977 | ||||||||||
At December 31, 2014, our outstanding options had exercise prices ranging from $0.10 to $5.21 per share.
Share-based compensation expense for Company Optionsoptions recognized in our results for the years ended December 31, 20112014, 2013, and 20102012 ($180,0874,393,455, $3,200,655 and $177,601$1,832,061, respectively) is based on awards vested and we estimated no forfeitures. ASC 718-10 requires forfeitures to be estimated at the time of grant and revised in subsequent periods if actual forfeitures differ from the estimates.
At December 31, 2011 and 2010,2014, total unrecognized estimated compensation expense related to non-vested Company Optionsunvested options granted prior to that date was approximately $244,000 and $206,000, respectively,$5,160,000, which is expected to be recognized over a weighted-average period of 3.32.9 years. No tax benefit was realized due to a continued pattern of operating losses.
In December 2013, we granted a restricted stock unit, or the RSU, under our 2012 Plan to an employee of 50,000 shares of our Common Stock having a fair value of $233,500. During the years ended December 31, 2014 and 2013, we recorded $53,428 and $180,072, respectively, of non-cash compensation related to the RSU on the accompanying consolidated financial statements. The RSU vested and the shares of Common Stock underlying the RSU were issued in June 2014.
THERAPEUTICSMD, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE E–11 – INCOME TAXES
For the advent of the Merger,
At December 31, 2011, the Company had2014, we have a federal net operating loss carry forward of approximately $2.1 million,$105,529,416 available to offset future taxable income through 2031. The Company established valuation allowances equal2034.
A reconciliation between taxes computed at the federal statutory rate and the consolidated effective tax rate is as follows:
| 2014 | 2013 | 2012 | ||||||||||
| Federal statutory tax rate | 34.0 | % | 35.0 | % | 35.0 | % | ||||||
| State tax rate, net of federal tax benefit | 5.8 | % | 5.8 | % | 5.5 | % | ||||||
| Adjustment in valuation allowances | (50.9 | )% | (32.4 | )% | (18.2 | )% | ||||||
| Permanent and other differences | 11.1 | % | (8.4 | )% | (22.3 | )% | ||||||
| Provision (Benefit) for Income Taxes | — | % | — | % | — | % | ||||||
Our deferred tax asset and liability as presented in the accompanying consolidated financial statements consist of the following:
| 2014 | 2013 | 2012 | ||||||||||
| Deferred Income Tax Assets: | ||||||||||||
| Net operating losses | $ | 43,091,437 | $ | 14,773,537 | $ | 5,920,861 | ||||||
| R&D Credit | 0 | 547,511 | 186,346 | |||||||||
| Total deferred income tax asset | 43,091,437 | 15,321,048 | 6,107,207 | |||||||||
| Valuation allowance | (43,091,437 | ) | (15,321,048 | ) | (6,107,207 | ) | ||||||
| Deferred Income Tax Assets, net | $ | — | $ | — | $ | — | ||||||
We believe that it is more likely than not that we will not generate sufficient future taxable income to realize the full amount oftax benefits related to the deferred tax assets due toon our balance sheet and as such, a valuation allowance has been established against the uncertainty of the utilization of the operating losses in future periods. The Company periodically assesses the likelihood that it will be able to recover its deferred tax assets. The Company considers all available evidence, both positive and negative, including historical levels of income, expectations and risks associated with estimates of future taxable income.
| Expected income tax benefit at statutory rate | $ | (4,519,678 | ) | |
| Non-deductible expenses: | ||||
| Debt settlement | 2,586,500 | |||
| VitaMed pre-merger loss | 1,164,629 | |||
| Other non-deductible expenses | 22,912 | |||
| Change in valuation account | 745,637 | |||
| Income tax expense (benefit) | $ | -0- |
| December 31, | ||||||||
| 2011 | 2010 | |||||||
| Deposits with vendors | $ | 300,503 | $ | -0- | ||||
| Prepaid consulting | 95,962 | -0- | ||||||
| Prepaid insurance | 52,611 | 6,292 | ||||||
| Prepaid guaranty costs | 46,984 | -0- | ||||||
| TOTAL OTHER CURRENT ASSETS | $ | 496,060 | $ | 6,292 | ||||
| December 31, | ||||||||
| 2011 | 2010 | |||||||
| Website | $ | 91,743 | $ | 65,791 | ||||
| Equipment | 33,651 | 30,837 | ||||||
| Furniture and fixtures | 26,219 | 26,219 | ||||||
| 151,613 | 122,847 | |||||||
| Accumulated depreciation | (81,500 | ) | (26,655 | ) | ||||
| TOTAL FIXED ASSETS | $ | 70,113 | $ | 96,192 | ||||
Unrecognized Tax Benefits
As of the following:
| December 31, | ||||||||
| 2011 | 2010 | |||||||
| Prepaid consulting | $ | 71,689 | $ | -0- | ||||
| Prepaid guaranty costs | 8,826 | -0- | ||||||
| TOTAL OTHER ASSETS | $ | 80,515 | $ | -0- | ||||
NOTE I12 – NOTES PAYABLE
Loan Guaranty
In March 1, 2011, the Company entered into a Demand Promissory Note with the Company’s then majority shareholder wherein the Company could periodically borrow funds to satisfy its operational requirements. Interest accrued at 20% per annum. On October 4, 2011, this Demand Promissory Note plus accrued interest totaling $170,152 was forgiven. The forgiveness of this related party debt was included in additional paid in capital on the accompanying financial statements.
THERAPEUTICSMD, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 12 – RELATED PARTIES (Continued)
Loans from Affiliates
In June 2011, VitaMed issued the VitaMed Notes in the aggregate principal amount of $500,000, of which $100,000 was sold to affiliates. In June 2012, the affiliate notes were extended to October 15, 2012 (one held by Mitchell Krassan, also an officer of VitaMed. The Bank LOC accrued interest at the rate of 3.020% per annum based onMr. Milligan for $50,000 and one for $50,000 held by BF Investments, LLC, which is owned by Brian Bernick, a year of 360 days and was due on March 1, 2012. The bank and VitaMed negotiated a one-year extension to the Bank LOC which was executed on March 19, 2012 (the “Bank LOC Extension”). The Bank LOC Extension accrues interest at the rate of 2.35% and is due on March 1, 2013. At December 31, 2011, the outstanding principle balancemember of the Bank LOC was $300,000. board of directors of our company. On October 4, 2012 these VitaMed Notes were paid in full including $5,341 in accrued interest.
In consideration for the personal guaranteesDecember 2011, we issued 4% promissory notes to Mr. Finizio and cash collateral, VitaMed issued VitaMed WarrantsMr. Milligan and for an aggregate of 499,998 Units (or Company Warrants for an aggregate of 613,713 shares pursuant to the
| December 31, | ||||||||
| 2011 | 2010 | |||||||
| Accrued payroll | $ | 227.477 | $ | -0- | ||||
| Accrued vacation | 68,438 | 24,208 | ||||||
| Other accrued expenses | 128,473 | 90,998 | ||||||
Dividends payable(1) | 41,359 | -0- | ||||||
| TOTAL OTHER CURRENT LIABILITIES | $ | 465,747 | $ | 115,206 | ||||
Purchases by mutual agreement to April 14, 2012.
During 2011 and 2010, the Company2012, we sold itsour products to Dr. Brian Bernick, a director of the Company,our company, in the amountsamount of $20,669$2,632, and $25,269, respectively. At$1,272 of receivables related thereto remained outstanding at December 31, 20112012. No products were sold to Dr. Bernick during 2014 and 2010, $0 and $79, respectively, remained outstanding.
Agreements with Pernix Therapeutics, LLC
On February 29, 2012, Cooper C. Collins, president and largest shareholder of Pernix Therapeutics, LLC, or Pernix, was elected to serve on October 4, 2011.our board of directors. From time to time, the Company has, and will continue to, enterwe have entered into agreements with Pernix in the normal course of business, whichbusiness. All such agreements are reviewed by independent directors of our company or a committee consisting of independent directors of our company. During the years ended December 31, 2014, 2013, and will be negotiated2012, we made purchases of $0, $0 and $404,000, respectively, from Pernix. At December 31, 2014, 2013, and 2012, there were amounts due Pernix of approximately $46,000, $46,000, and $308,000 outstanding, respectively.
Additionally, there were amounts due to us from Pernix for legal fee reimbursement relating to a litigation matter pursuant to a license and supply agreement in arms-length transactions. The Presidentthe amount of $249,981 for each of the years ended December 31, 2014 and largest shareholder2013.
Warrants assigned to Related Party
In June 2012, a warrant to purchase 100,000 shares of Pernix, Cooper C. Collins,our Common Stock was recently electedassigned by a non-affiliated third party to serve on the Company’s Boardson of Directors.
NOTE L13 - BUSINESS CONCENTRATIONS
We purchase our products from several suppliers with approximately 95%82%, 98%, and 93% coming76% of our purchases supplied from one suppliervendor for the years ended ending December 31, 20112014, 2013, and 2010,2012, respectively.
THERAPEUTICSMD, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE M13 - BUSINESS CONCENTRATIONS (Continued)
We sell our vitamins, prenatal dietary supplement products to wholesale distributors, specialty pharmacies, specialty distributors, and chain drug stores that generally sell products to retail pharmacies, hospitals, other institutions and directly to retail customers. Revenue generated from four customers accounted for approximately 75%, 79%, and 28% of our recognized revenue and 97%, 97%, and 98% of our deferred revenue for the years ended December 31, 2014, 2013, and 2012, respectively.
For the years ended December 31, 2014, 2013 and 2012, we had four customers that generated more than 10% of our sales – these customers are designated as customers “A”, “B”, “C” and “D”. Customers A, B, C and D generated $1,609,950, $1,586,903, $1,804,018 and $4,053,838, respectively, in sales in 2014; $1,221,212, $1,711,417, $2,588,626, and $1,312,192, respectively, in sales in 2013; and $67,599, $490,092, $830,902, and $0, respectively, in sales in 2012.
NOTE 14 – COMMITMENTS AND CONTINGENCIES
Operating Lease
We lease administrative and distribution facilitiesoffice space in Boca Raton, Florida pursuant to a forty-five63 month non-cancelable operating lease expiring in 2013.that commenced on July 1, 2013 and expires on September 30, 2018. The lease stipulates, among other things, average base monthly rents of $5,443 plus the Company’s share$30,149 (inclusive of monthly estimated operating expenses of $3,500expenses) and sales tax. tax, for a total future minimum payments over the life of the lease of $1,899,414.
The lease contains one renewal option for an additional two-year period.
| Years Ending December 31, | ||||
| 2012 | $ | 111,725 | ||
| 2013 | 56,601 | |||
| 2014 | -0- | |||
| 2015 | -0- | |||
| Thereafter | -0- | |||
| Total | $ | 168,326 | ||
As of December 31, 20102014, future minimum rental payments under our office lease are as follows:
| Years Ending December 31, | ||||||
| 2015 | $ | 371,240 | ||||
| 2016 | 382,377 | |||||
| 2017 | 393,848 | |||||
| 2018 | 302,748 | |||||
| 2019 | — | |||||
| Total minimum lease payments | ||||||
| Non-cancellable sub-lease income | — | |||||
| Net minimum lease payments | $ | 1,450,213 | ||||
Legal Proceedings
From time to time, we are involved in litigation and proceedings in the $90,238 reduction for the period from Inception throughordinary course of business. We are not currently involved in any legal proceeding that we believe would have a material effect on our consolidated financial condition, results of operations, or cash flows.
Off-Balance Sheet Arrangements
As of December 31, 2009.2014, 2013 and 2012, we had no off-balance sheet arrangements that have had or are reasonably likely to have current or future effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources that are material to investors.
| F-33 |
| As of | ||||||||
| December 31, 2010 | ||||||||
| As Reported | As Restated | |||||||
| Additional paid in capital | $ | 537,561 | $ | 261,174 | ||||
| Accumulated deficit | $ | (4,356,100 | ) | $ | (4,079,713 | ) | ||
For the Year Ended December 31, 2010 | ||||||||
| As Reported | As Restated | |||||||
| Sales, general and administration | $ | 3,650,959 | $ | 3,464,810 | ||||
| Total operating expense | $ | 3,739,144 | $ | 3,552,994 | ||||
| Operating loss | $ | (3,053,613 | ) | $ | (2,867,464 | ) | ||
| Net loss | $ | (3,053,613 | ) | $ | (2,867,464 | ) | ||
On February 24, 2012, TherapeuticsMD, Inc. (the “Company”) sold and issued Secured Promissory Notes (the “Notes”) to Steven G. Johnson (“Johnson”) and Plato & Associates, LLC (“Plato”) in the principal base amount of $1,358,014 and $1,357,110 respectively (the “Principal Base Amount(s)”) pursuant to the terms of that certain Note Purchase Agreement (the “Note Purchase Agreement”) of even date therewith. As consideration for the Notes, Johnson and Plato surrendered certain promissory notes previously issued by the Company in the aggregate amount of $858,014 and $857,110 respectively (which sums include principle and interest through February 24, 2011) (collectively known as the “Prior Notes”). As a result of the foregoing the Company received an aggregate of $1,000,000 of new funding from Johnson and Plato. On March 23, 2012, each of Johnson and Plato loaned the Company an additional $500,000 under the Notes for an aggregate of $1,000,000.
THERAPEUTICSMD, INC. AND SUBSIDIARYSUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE O15 – SELECTED QUARTERLY FINANCIAL DATA (UNAUDITED)
Summarized quarterly financial data for fiscal years 2014, 2013, and 2012 is as follows:
| 2014 Quarters | ||||||||||||||||
| (In thousands, except per share) | 1st | 2nd | 3rd | 4th | ||||||||||||
| Revenues | $ | 2,831 | $ | 3,752 | $ | 4,186 | $ | 4,257 | ||||||||
| Gross profit | $ | 2,000 | $ | 2,859 | $ | 3,118 | $ | 3,377 | ||||||||
| Net loss | $ | (9,183 | ) | $ | (10,899 | ) | $ | (17,832 | ) | $ | (16,303 | ) | ||||
| Loss per common share, basic and diluted | $ | (0.06 | ) | $ | (0.07 | ) | $ | (0.12 | ) | $ | (0.10 | ) | ||||
| 2013 Quarters | ||||||||||||||||
| (In thousands, except per share) | 1st | 2nd | 3rd | 4th | ||||||||||||
| Revenues | $ | 1,537 | $ | 2,081 | $ | 2,295 | $ | 2,863 | ||||||||
| Gross profit | $ | 1,157 | $ | 1,617 | $ | 1,646 | $ | 2,396 | ||||||||
| Net loss | $ | (6,376 | ) | $ | (6,009 | ) | $ | (7,673 | ) | $ | (8,361 | ) | ||||
| Loss per common share, basic and diluted | $ | (0.06 | ) | $ | (0.05 | ) | $ | (0.06 | ) | $ | (0.06 | ) | ||||
| 2012 Quarters | ||||||||||||||||
| (In thousands, except per share) | 1st | 2nd | 3rd | 4th | ||||||||||||
| Revenues | $ | 722 | $ | 819 | $ | 1,036 | $ | 1,241 | ||||||||
| Gross profit | $ | 386 | $ | 447 | $ | 729 | $ | 908 | ||||||||
| Net loss | $ | (13,290 | ) | $ | (11,850 | ) | $ | (4,253 | ) | $ | (5,727 | ) | ||||
| Loss per common share, basic and diluted | $ | (0.16 | ) | $ | (0.14 | ) | $ | (0.04 | ) | $ | (0.06 | ) | ||||
NOTE 16 – SUBSEQUENT EVENTS (Continued)
Election of Additional Directors
On February 29, 2012,10, 2015, we entered into an underwriting agreement, or the Company’s BoardCowen Agreement, with Cowen and Company, LLC, as the representative of Directors elected fourthe several underwriters, or the Cowen Underwriters, relating to an underwritten public offering of 13,580,246 shares of the our Common Stock, at a public offering price of $4.05 per share. Under the terms of the Cowen Agreement, we granted the Cowen Underwriters a 30-day option to purchase up to an aggregate of 2,037,036 additional individualsshares of Common Stock, which option was exercised in full. The net proceeds to serve as members of its Board of Directors, including: Samuel A. Greco, Cooper Collins, Robert V. LaPenta, Jr.us from the offering were approximately $59.1 million, after deducting underwriting discounts and Nicholas Segal.
Issuance of Company Optionscommissions and other estimated offering expenses payable by us. The offering closed on February 17, 2015.
On February 27, 2012,18, 2015, we entered into an agreement to lease administrative office space in Boca Raton, Florida, pursuant to an addendum to our existing 63 month non-cancelable operating lease commencing on July 1, 2013 and expiring on September 30, 2018. This addendum will be effective beginning April 1, 2015 and will expire with the Company issued Company Options to Robert G. Finizio and John Milligan, officers and directors of the Company. The ten-year Company Options are for 300,000 shares each and have an exercise price of $2.20 per share. The Company Options vest in fulloriginal lease term on February 27, 2013.
Approval of Committee Charters and Committee Appointments
On February 29, 2012, the Company’s Board of Directors (i) approved charters for each of the Audit Committee, Compensation Committee and Corporate Governance Committee, (ii) appointed members to each committee and (iii) named a Chair of each committee.
Members of the Audit Committee include Robert V. LaPenta, Jr., Samuel A. Greco and Nicholas Segal. Mr. LaPenta, Jr. will serve as Chair.
Members of the Compensation Committee include Cooper Collins, Robert G. Finizio and Nicholas Segal. Mr. Collins will serve as Chair.
Members of the Corporate Governance Committee include John C.K. Milligan, IV, Brian Bernick and Robert LaPenta, Jr. Mr. Milligan will serve as Chair.September 30, 2018.
| F-34 |
On March 1, 2012, the Company launched its first prescription prenatal vitamin,vitaMedMD™ Plus Rx,a single-dose product containing one prenatal vitamin tablet and one life’s DHA™ capsule.