PART I
Diffusion Pharmaceuticals: Enhancing Oxygen, Fueling Life
We are a clinical stage biotechnologybiopharmaceutical company developing novel therapies that enhance the body’s ability to deliver oxygen to the areas where it is needed most. Our lead product candidate, TSC, is being developed to enhance the diffusion of oxygen to tissues with low oxygen levels, also known as hypoxia, a serious complication of many of medicine’s most intractable and difficult-to-treat conditions.
Our Strategy
Our goal is to become a leading biopharmaceutical company focused on extending the life expectancydevelopment and commercialization of novel therapies that enhance the body’s ability to deliver oxygen to areas where it is needed most and improve treatment outcomes for patients suffering from conditions complicated by hypoxia. To achieve this strategy, we are focused on the following key objectives:
| • | Demonstrate the effects of TSC on the oxygenation pathway. The results of our Oxygenation Trials – a series of three short-term, pharmacodynamic studies to evaluate the effects of TSC on different components of the oxygenation pathway that we expect to complete by the middle of 2022 – will be used to further inform the identification of potential indications appropriate for TSC and will guide the design of clinical trials aimed at supporting the commercialization of TSC as a treatment for conditions complicated by hypoxia. For additional information, see "Business — Our Lead Product Candidate: Trans Sodium Crocetinate -- The TSC Oxygenation Trials: Demonstrating Proof of Concept, Dose Response Relationships, and Potential Indications for Future Development.” |
| • | Develop TSC as an adjunct treatment for hypoxic solid tumors. The prevalence of hypoxic regions across numerous primary solid tumor types is believed to directly contribute to treatment resistance, whether it be radiotherapy, chemotherapy, or immunotherapy, increasing the metastatic potential of these tumors and the probability of unsuccessful treatment. We believe TSC’s novel mechanism of action supports the objective of developing TSC as a treatment for hypoxic solid tumors because TSC’s ability to enhance the oxygenation of hypoxic tissues may increase the effectiveness of standard-of-care radiation therapy and chemotherapy. Accordingly, in November 2021, we announced our Hypoxic Solid Tumor Program and our intent to commence our Planned Phase 2 Hypoxic Solid Tumor Trial in the second half of 2022. The data obtained during 2021 and early 2022 from our Oxygenation Trials and COVID Trial regarding TSC’s effects on oxygenation, dose response characteristics, pharmacokinetics, and pharmacodynamics are being used to guide the design of this trial. Through the combination of this new data, our past experiences, and further analyses and discussions of all available data with our Scientific Advisory Board and other external advisors, we believe we now have the necessary information to design the Hypoxic Solid Tumor Program to be more efficient and increase our likelihood of success compared to the Company’s past efforts to develop TSC as a cancer treatment, which were terminated in 2019 due to financial constraints. For additional information, see "Business — Our Lead Product Candidate: Trans Sodium Crocetinate – Our Hypoxic Solid Tumor Program.” |
| • | Accelerate development of TSC as a treatment for non-cancer indications. Beyond cancer, hypoxia is a complicating factor in many other intractable and difficult-to-treat conditions, including cardiovascular diseases, cerebrovascular diseases, respiratory diseases, skin and soft tissue diseases, and neurodegenerative diseases. We have previously undertaken clinical studies of TSC in a variety of non-cancer indications, including interstitial lung disease, COVID-19, stroke, and peripheral artery disease, and have evaluated TSC in preclinical models in an even wider range of indications. Data that we expect to obtain from our ongoing Altitude and ILD-DLCO Trials may identify additional, non-oncology development opportunities. Although our primary near-term focus will be our Hypoxic Solid Tumor Program, we will continue to explore pathways to progress TSC’s development in non-oncology indications through our preclinical studies. For additional information, see "Business — Our Lead Product Candidate: Trans Sodium Crocetinate – TSC’s Potential to Treat Other Conditions Complicated by Hypoxia.” |
| • | Maximize the commercial value of our pipeline. We have invested, and plan to continue to invest, significant time, effort, and resources into the development and maintenance of our intellectual property portfolio as we seek to retain worldwide development and commercial rights to our product candidates. In parallel with advancing our development programs, we will evaluate the commercial landscape for TSC with the intent of improving patient access to maximize our chances of commercial success, should the product be approved. |
| • | Opportunistically acquire or in-license additional product candidates that complement TSC and our overall strategy. We believe we can leverage what we have learned from the development of TSC and the significant skills and experience of our team to opportunistically identify and acquire or in-license novel product candidates that complement TSC and further our efforts to improve patient outcomes by enhancing standard-of-care therapy for conditions complicated by hypoxia. We also believe diversifying our asset portfolio through an acquisition or in-licensing would reduce our Company’s overall risk profile as an investment. |
Our Lead Product Candidate: Trans Sodium Crocetinate
Our lead product candidate, TSC, is being developed to enhance the diffusion of oxygen to tissues with low oxygen levels, also known as hypoxia.
Hypoxia is a condition in which there is an insufficient supply of oxygen to meet the energy demands of the cells in a tissue (e.g., skeletal muscle). Hypoxia is associated with the pathophysiology of many of medicine’s most intractable and difficult-to-treat conditions, including cancers, cardiovascular diseases, cerebrovascular diseases, respiratory diseases, skin and soft tissue diseases, and neurodegenerative diseases.
All currently approved treatments for hypoxia work by augmenting or supplementing the systemic availability of oxygen. For example, respirators use pressure to push oxygen across the lungs into the blood to increase oxygen concentration, certain medications and devices such as external mechanical pumps are designed to improve cardiac output, and blood transfusions are used to increase the concentration of red blood cells and oxygen-carrying hemoglobin molecules.
While these treatments are effective in certain circumstances, there are significant associated risks such as lung damage resulting from the use of respirators, toxic side effects and drug-to-drug interactions of medications that increase cardiac output, infections related to blood transfusions, and the risk of creating excessive oxygen-related tissue toxicity, or hyperoxia, by providing a patient with excessive amounts of oxygen, among others. In addition, in certain other indications, these currently approved therapies are ineffective in treating the associated hypoxia. For example, in the treatment of cancer, re-oxygenating hypoxic, cancerous tissue cells cannot typically be accomplished simply by increasing the systemic availability of oxygen.
We believe TSC is the first therapy specifically designed to enhance the efficiency of the oxygen diffusion process. Furthermore, in animal models, TSC’s diffusion-enhancing mechanism of action has been observed to affect hypoxic tissue preferentially while avoiding excessive oxygen-related tissue toxicity.
By supporting normal, physiologic levels of oxygen diffusion at the uptake and delivery points of the circulatory system, we believe TSC presents a significant opportunity to improve the current standard-of-care treatment for conditions complicated by hypoxia.
TSC's Mechanism of Action
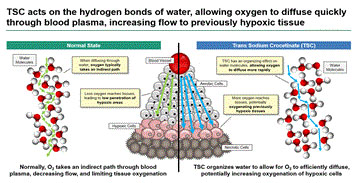
Blood plasma is approximately 90% water. The water molecules in the plasma are constantly moving, bound together in a loosely organized matrix by hydrogen bonds. TSC was designed to enhance the level of organization among the water molecules by increasing the amount of hydrogen bonding. This creates a less dense matrix of water molecules, reducing the resistance to oxygen diffusion across the concentration gradient.
Selected Preclinical Experiences
TSC has been evaluated in a variety of preclinical models intended to mimic relevant human conditions known to be complicated by hypoxia. In these studies, a variety of positive effects have been observed in connection with TSC administration, including:
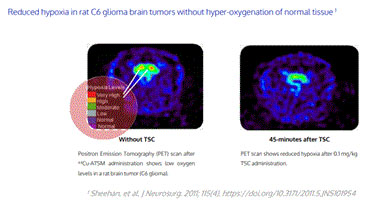
| • | Reducing hypoxia in rat brain tumors without hyper-oxygenation of normal tissue; |
| • | Improving survival in a rat brain tumor model when added to radiotherapy, whether alone or in combination with chemotherapy; |
| • | Improving tissue oxygenation without hyper-oxygenation of normal tissue and reducing infarct size in a rat ischemic stroke model; |
| • | Demonstrating a functional benefit in a rabbit ischemic stroke model, with or without tissue plasminogen activator at one-hour post-clot infection and with tissue plasminogen activator at three hours post-clot infection; and |
| • | Improving levels of arterial partial pressure of blood oxygen in a rat model of acute respiratory distress syndrome. |
Positron emission tomography scans showing reduction in hypoxia in a rat C6 glioma brain tumor model 45 min after TSC administration.
The TSC Oxygenation Trials: Demonstrating Proof of Concept, Dose Response Relationships, and Potential Indications for Future Development
While our clinical trials evaluating TSC conducted prior to 2021 provided certain preliminary signals regarding TSC’s mechanism of action and dose response characteristics, none of those studies were designed to demonstrate proof of concept by directly measuring TSC’s effects on tissue oxygenation and a variety of questions remained regarding TSC’s dose response characteristics, pharmacokinetics, and pharmacodynamics.
Given these identified gaps in our knowledge, in November 2020, we communicated our plan to design and execute our Oxygenation Trials, a series of short-term clinical studies using experimental models to evaluate the clinical effects of TSC on oxygenation, each designed to look at the effects of TSC on a different component of the oxygenation pathway. By quantifying the magnitude of effects of TSC on tissue oxygen levels and other direct clinical parameters assessing oxygen levels, and thereby demonstrating proof of concept of TSC’s effects on tissue oxygenation, as well as supplementing our knowledge of TSC’s dose response characteristics, pharmacokinetics and pharmacodynamics, we believe the data obtained from the Oxygenation Trials will allow us to significantly increase our clinical development strategy’s likelihood of success.
TCOM Trial: TSC's Effects on Peripheral Tissue Oxygenation
In March 2021, we initiated and completed enrollment in the first of the Oxygenation Trials, our TCOM Trial. In June 2021, we reported a positive trend in peripheral tissue oxygenation as compared to placebo among participants receiving TSC that persisted through the duration of the one-hour, post-dosing measurement period. Although the magnitude of this effect was not statistically significant due to limitations in the study design, the trends in the primary endpoint data indicated an improvement in peripheral oxygenation with TSC with no evidence of hyperoxygenation.
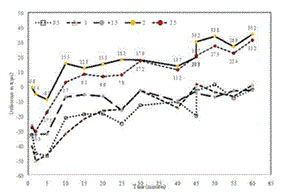
The figure below was created during a supplemental analysis of the TCOM Trial results by subtracting the median response observed in the TCOM Trial’s placebo group from the median response observed in each TSC dosage group at each of the measurement times during the one-hour period following dosing. These data highlight the persistent increase in peripheral tissue oxygenation relative to the placebo group through the duration of the one-hour measurement period following TSC administration, particularly at the two highest doses tested.
Effects of TSC on transcutaneous oxygen pressure.
We believe the TCOM Trial provides clinical evidence to support the hypothesis that TSC enhances the passive diffusion of oxygen from areas of high concentration to areas of low concentration without causing hyperoxygenation and that the data obtained from the study represent a positive and meaningful step towards the accomplishing of the Oxygenation Trials’ strategic objectives originally set forth in November 2020. These data have and will continue to guide the next steps of our development strategy, including the design of our Hypoxic Solid Tumor Program.
Altitude Trial: TSC's Effects Under Induced Hypoxic Conditions
On November 22, 2021, we announced dosing of the first participants in our second Oxygenation Trial, the Altitude Trial. This study is a double-blind, randomized, placebo-controlled crossover study designed to evaluate the effects of TSC on maximal oxygen consumption and partial pressure of blood oxygen in normal healthy volunteers subjected to incremental levels of physical exertion while exposed to “simulated altitude.” The study will enroll 30 healthy volunteers and give each volunteer a single dose of TSC at one of three different doses. Due to enrollment delays experienced during early 2022 related to the omicron variant wave of the COVID-19 pandemic, we now anticipate completing dosing in the Altitude Trial in the second quarter of 2022, with topline results reported within two months of study completion.
ILD-DLCO Trial: TSC's Effects on Oxygen Transfer Efficiency
On December 16, 2021, we announced dosing of the first patients in our third and final Oxygenation Trial, the ILD-DLCO Trial. This study is a double-blind, randomized, placebo-controlled, crossover study designed to evaluate the effects of TSC on the diffusion of carbon monoxide through the lungs, or DLCO, in patients with previously diagnosed ILD who have a baseline DLCO test result that is abnormal. DLCO is used in the study as a surrogate measure of oxygen transfer efficiency, or uptake, from the alveoli of the lungs through the plasma, and onto hemoglobin within red blood cells. The study will enroll 27 patients with confirmed ILD who will be randomized in a 2:1 ratio to a single dose of TSC or placebo via IV bolus. The study is statistically powered to evaluate the difference in effect of TSC versus placebo on improvement in DLCO measurements. In addition, patients will undergo a standard six-minute walk test intended to assess functional improvement in exercise capacity. Due to enrollment delays experienced during early 2022 related to the omicron variant wave of the COVID-19 pandemic, the effect of the omicron variant on patients with pulmonary diseases such as ILD, and staffing issues at the facilities where the ILD-DLCO Trial is being conducted, we now anticipate completing dosing in the ILD-DLCO Trial by improvingthe middle of 2022, with topline results reported within two months of study completion.
TSC's Demonstrated Clinical Safety Profile
TSC has been observed to be safe and well-tolerated at most doses tested in over 220 subjects included to-date in clinical studies at a variety of doses administered via intravenous infusion. Our clinical studies have included patients with a variety of medical conditions often complicated by hypoxia, including patients afflicted with GBM, peripheral artery disease with intermittent claudication, stroke, COVID-19, and interstitial lung disease. In each of these conditions and many others, hypoxia is a significant contributor to morbidity and mortality, and a considerable treatment obstacle for medical providers.
During 2021, we obtained additional data further demonstrating TSC's safety at higher doses and increased dosing frequencies from our COVID Trial and Oxygenation Trials. In the COVID Trial, completed in February 2021, TSC was administered on a different dosing regimen than any of our prior trials, with patients receiving an infusion multiple times per day for up to 15 days. In the TCOM Trial, based in part on a recommendation from the COVID Trial’s safety monitoring committee, arms evaluating even higher doses of TSC were included. No dose-limiting toxicities or serious adverse events were observed among any patients or participants in the either trial, including those who received the highest tested doses of TSC.
Our Hypoxic Solid Tumor Program
We believe TSC’s novel mechanism of action supports the objective of developing TSC as a treatment for hypoxic solid tumors because TSC’s ability to enhance the oxygenation of hypoxic tissues may increase the effectiveness of current standard-of-care treatments, including radiation therapy and chemotherapy. We are developing our lead product candidate,transcrocetinate sodium, also known astrans sodium crocetinate (“TSC”), for use in the many cancer types in which tumor oxygen deprivation (“hypoxia”) is known to diminish the effectiveness of current treatments. TSC is designed to target the cancer’s hypoxic micro-environment, re-oxygenating treatment-resistant tissue and making the cancer cells more susceptible to the therapeutic effects of standard-of-care radiation therapy and chemotherapy. Our lead development programs target TSC against cancers known to be inherently treatment-resistant, including brain
Solid tumors comprise approximately 90% of all adult cancers and, pancreatic cancer. A Phase 1/2 clinical trial of TSC combined with first-line radiation and chemotherapyaccording to the American Cancer Society, it was expected that roughly 1.9 million new cancer cases would be diagnosed in patients newly diagnosed with primary brain cancer (“glioblastoma” or “GBM”) was completed in 2015. This trial provided evidence of efficacy and safety in extending overall survival without the addition of toxicity. Based on these results, an End-of-Phase 2 meeting was held with the U.S. Food and Drug Administration (“FDA”) in August 2015, resulting in guidelines for the design of a single 400 patient pivotal Phase 3 registration study which, if successful, would be sufficient to support approval. The Companyduring 2021. It is also exploring the possibility of focusing on the subset of inoperable patientswell-documented in the GBM indication. Discussions withscientific literature that the FDA regarding extensionprevalence of the TSC development program from first line GBM into first-line pancreatic cancer treatment are currently underway. TSC has been granted Orphan Drug designation for the treatment of both GBM and brain metastases.
In addition to cancer, TSC also has potential applications in other indications involving hypoxia, such as hemorrhagic shock, stroke, peripheral artery disease, respiratory diseases and neurodegenerative diseases. In this regard, the Company is exploring the feasibility of testing TSC as a novel stroke treatment in an emergency medicine/ambulance setting.
Summary of Current Product Candidate Pipeline
The following table summarizes the targeted clinical indications for Diffusion’s lead molecule,trans sodium crocetinate:

In addition to the TSC programs depicted in the table, we are exploring alternatives regarding how best to capitalize upon our product candidate, RES-529, a novel PI3K/Akt/mTOR pathway inhibitor which has completed two Phase I clinical trials for age-related macular degeneration and is in preclinical development in oncology, specifically GBM.
Technology Overview
Our proprietary technology is targeted at overcoming treatment-resistance in solid cancerous tumors by combining our lead product candidate, TSC, with standard-of-care radiation and chemotherapy regimens, thus effecting a better patient survival outcome without the addition of harmful side effects.
Under normally oxygenated cellular conditions, radiation and chemotherapy generally have a powerful killing effect upon cancerous tumor tissue. However, in manyhypoxic regions across numerous primary solid tumor types cellular oxygen deprivation occurs asdirectly contributes to treatment resistance, whether it be radiotherapy, chemotherapy, or immunotherapy, increasing the resultmetastatic potential of rapid tumor growth, causing partsthese tumors and the probability of the tumor to outgrow its blood supply. When tumor tissue becomes hypoxic, it is up to three times more resistant to the cancer-killing power of the standard therapies (radiation and chemotherapy) currently used in the treatment of the vast majority of cancer patients. Cancerous tumor cells are known to thrive under hypoxic conditions, as the resultant changes in the tumor microenvironment confer “treatment-resistance” to radiation and chemotherapy within the cell.unsuccessful treatment.
Many solid cancerousof the challenges faced by practitioners in treating some of the most fatal and difficult-to-treat tumor types, are hypoxic and therefore subject to this treatment-resistance. TSC safely re-oxygenates treatment-resistant hypoxic tumor tissue via a novel mechanism of action, without affecting the oxygenation of normal tissue, thereby increasing therapeutic effectiveness. To date, no addition of serious harmful side effects, or exacerbation of the known side effects of standard-of-care treatments, has been observed in our clinical studies.
TSC’s distinctive re-oxygenation capabilities derive from its mechanism of action, which promotes enhanced diffusion of oxygen through blood plasma and into the hypoxic tumor microenvironment. Disruption of the treatment-resistance syndrome by re-oxygenation promotes enhanced cancer-killing power from radiation and chemotherapy, thereby safely extending patient survival. Because of the characteristics of this novel mechanism, oxygen levels of normal tissue remain unaffected, thereby avoiding side effects related to the syndrome referred to as “oxygen toxicity.” We believe this avoidance of oxygen toxicity confers a significant advantage to TSC’s diffusion-based approach over previous attempts to diminish treatment-resistance based on enhancing the oxygen concentration levels of the blood.
Our clinical development plan targets TSC at radiation and chemotherapy sensitization of hypoxic tumor types, with an initial focus on primary brain cancer,including GBM, pancreatic cancer, and brain metastases. We have been granted orphan drug designations by the FDA for the treatment of brain cancers based on the acknowledged unmet medical need and number of patients affected. Such orphan drug designations allow certain favorable treatments under FDA regulations in connection with exclusivity periods and the new drug approval process.
Tumor Hypoxia
We believe that our breakthrough small molecule approach to overcomingother solid tumor treatment-resistance by the reduction of cellular hypoxia may have significant implications for the improved treatment of cancer. Hypoxia is a deficiency in the supply of oxygen. It is well known that tumors, are especiallya product of those tumors’ highly hypoxic nature. Cancerous tumors are specifically susceptible to developing hypoxia driven bydue to a combination of rapid cellular growth, structural abnormalities of the tumor microvessels, and disturbed circulation within the tumor. There are a number of known treatment-resistanceHypoxic conditions within the tumor microenvironment have numerous negative consequences conferred by tumor hypoxia, including increases in:for patient outcomes and treatment, including:
| ●
• increased resistance to ionizing radiation, chemotherapy, immunotherapy and other treatment methods; • a more clinically aggressive phenotype; • increased potential for invasive growth and tumor progression; and • increased regional and distal tumor metastasis. | Resistance to ionizing radiation;
|
| ●
| Clinically aggressive phenotype;
|
| ●
| Potential for more invasive growth; and,
|
| ●
| Regional and distal tumor spreading.
|

In November 2021, based on the available preclinical and clinical data and the significant unmet medical need, we announced our intention to focus near-term efforts on developing TSC as an adjunct to standard of care therapy for hypoxic solid tumors. Through the combination of our past experiences, new knowledge gained from our Oxygenation Trials and COVID Trial regarding TSC’s effects on oxygenation, dose response characteristics, pharmacokinetics, and pharmacodynamics, and further analyses and discussions of all available data with our Scientific Advisory Board and other external advisors, we believe we now have the necessary information to design the Hypoxic Solid Tumor Program to be more efficient and increase our likelihood of success compared to the Company’s past efforts to develop TSC as a cancer treatment, which were terminated in 2019 due to financial constraints.
As part of the ongoing design of our Planned Phase 2 Hypoxic Tumor Trial – the first trial in our Hypoxic Solid Tumor Program which we currently expect to commence in the second half of 2022, subject to FDA feedback and the availability of clinical drug supply – we are currently drafting a trial protocol which we intend to file with the FDA. In parallel, we will continue our work to optimize the TSC manufacturing process and support the continued availability of high-quality drug product and undertake preclinical studies and other opportunities to continue developing data designed to demonstrate TSC’s potential uses in a broad spectrum of non-cancer indications.
TSC's Potential to Treat Other Conditions Complicated by Hypoxia
Beyond cancer, hypoxia is a complicating factor in many other intractable and difficult-to-treat conditions as well, including cardiovascular diseases, cerebrovascular diseases, respiratory diseases, skin and soft tissue diseases, and neurodegenerative diseases. In addition to our oncology programs past and present, we have previously conducted a variety of preclinical and clinical studies evaluating the effects of TSC in several of these other indications complicated by hypoxia, including COVID-19, stroke, and peripheral artery disease with intermittent claudication, and we intend to continue to pursue potential partnering opportunities in non-oncology indications.
Certain medical conditions complicated by hypoxia.
Our Phase 2 COVID-19 Trial
We believe TSC’s oxygen-enhancing mechanism of action could potentially provide benefit patients suffering from respiratory indications, such as COVID-19.
In September 2020, we announced the dosing of the first patients in our COVID Trial evaluating TSC in hospitalized COVID-19 patients at the National Institute of Infectious Diseases in Bucharest, Romania. The trial enrolled 24 patients divided into four sequential cohorts of six patients, with each patient in a cohort receiving the same intravenous doses of 0.25 mg/kg, 0.5 mg/kg, 1.0 mg/kg, or 1.5 mg/kg. All patients were administered intravenous doses of TSC every six hours for a minimum of five days and up to 15 days. On February 9, 2021, we completed dosing of the twenty-fourth and final patient in the trial.
The above-described phenomenon of hypoxia-related “treatment-resistance” has been known to the scientific and clinical communities for over half a century. The challenge has been to find an approach that can effectively mitigate treatment-resistance without the addition of toxic side effects or exacerbationprimary endpoint of the side effects associated with radiationCOVID Trial was to evaluate the safety and chemotherapy treatments. We believe thattolerability of TSC embodiesadministered every six hours for at least five and up to 15 days, a more frequent dosing regimen than had been used in our previous clinical studies. Secondary endpoints included pharmacokinetic measurement of TSC levels after dosing, relative improvements in blood oxygen levels, and certain other clinical parameters related to COVID-19, such an approach.as improvement in WHO ordinal scale by day 7 and through day 29, time on oxygen supplementation, and hospital length of stay.
Trans Sodium CrocetinateData from the trial, evaluated by the study’s independent safety monitoring committee, indicated TSC was safe and well-tolerated and that no dose-limiting toxicities or serious adverse events were observed among any patients in the study, including those who received the highest dose administered in the trial. Additionally, while the COVID Trial was not designed or powered to evaluate efficacy, the safety monitoring committee also observed that patients receiving the highest dose administered had improved outcomes in the trial’s secondary and exploratory endpoints compared to those receiving lower doses. Further, no patients that received TSC required dialysis or developed acute kidney injury, nor were there any reports of pulmonary embolism or deep vein thrombosis. Based on their observations, the safety monitoring committee also recommended the Company consider testing of higher TSC doses and a continuous intravenous infusion in future studies.
Product Development
Dr. John Gainer, our Chief Scientific Officer, one of our directorsResearch and Professor Emeritus of Chemical Engineering at the University of Virginia, was the first to propose the use of chemical compounds specifically to facilitate the diffusion of oxygen through blood plasma for the purpose of re-oxygenating hypoxic tissues. Dr. Gainer’s early laboratory work systematically examined various means to alter the diffusivity of oxygen through the use of small molecules that would affect the intermolecular forces existing in blood plasma. He originally identified crocetin, a natural carotenoid compound, as a molecule that could increase oxygen diffusion through the plasma, and crocetin was shown to be an effective treatment in a rabbit model of atherosclerosis and other indications. This work continued from the 1970s into the mid-1990s with various animal models, including radiation sensitization and hemorrhagic shock.
Because crocetin is an isomeric mixture, Dr. Gainer examined whether it was thetransDevelopment-isomer which was responsible for eliciting the therapeutic benefit. These experiments led to his development of a puretrans-isomer salt compound, which he namedtrans sodium crocetinate. (The USAN designated name istranscrocetinate sodium). TSC has been shown to be more effective than crocetin in a severe model of hemorrhagic shock in both rats and pigs. It also demonstrated safety and efficacy in animal models of stroke and myocardial infarction, as well as in enhancing the response of hypoxic tumors to the therapeutic effects of radiation and chemotherapy.
It is proposed that TSC works by altering the molecular arrangement of the water molecules in blood plasma (which is composed of 90% water), with the altered structure being less dense – and thus less resistant to oxygen diffusion – than untreated blood plasma. A water molecule is composed of two hydrogen atoms and one oxygen atom, with a net positive charge found on the hydrogen atoms and a net negative charge found on the oxygen atom. This results in the formation of hydrogen bonds, which are an attraction between the net-negatively charged oxygen of one water molecule and the net-positively charged hydrogen atoms of another water molecule. Theoretically, one water molecule can form four hydrogen bonds with neighboring water molecules. However, the literature on the subject indicates that a water molecule actually forms, on average, 2 to 3.6 hydrogen bonds. By promoting an increase in the average number of hydrogen bonds among the water molecules comprising the bulk of blood plasma, TSC enhances the ability of oxygen to diffuse more easily through the plasma and into hypoxic tissue.
In March 2017, we received a patent for bipolar trans carotenoid saltsrecent years, the majority of our research and their uses.This patent expands the coverage of the therapeutic use of TSC and other related compounds to five hypoxia-related conditions including congestive heart failure, chronic renal failure, acute lung injury (ALI), chronic obstructive pulmonary disease (COPD) and respiratory distress syndrome (RDS).
Trans Sodium Crocetinate Increases Oxygenation of Hypoxic Cancerous Tumors
While earlier studies focused on improved treatments for hemorrhagic shock, ischemia, and traumatic brain injury, the use of TSC as an agent to re-oxygenate hypoxic cancerous tumors became a central area of research for us following our founding in 2001. Because tumor hypoxia is a leading cause of solid tumor resistance to both radiation and chemotherapy, it was believed that an agent such as TSC – one that could safely increase the oxygenation of hypoxic tumor tissue – could prove effective in treatment-resistant cancers when combined with standard-of-care regimens of radiation and/or chemotherapy. This belief leddevelopment expenditures have been directed to the development of TSC. For example, during the year ended December 31, 2021, we incurred approximately $8.5 million in costs related to research and development of our products, a decrease of approximately $0.9 million compared to the year ended December 31, 2020. The majority of these costs were related to the development of TSC and related personnel, including costs associated with the Oxygenation Trials and the COVID Trial, which was completed in February 2021. We expect this trend to continue for the foreseeable future, including planned expenditures related to completion of the remaining Oxygenation Trials, the initiation of the Hypoxic Solid Tumor Program, and other costs associated with the conduct of additional, supportive preclinical studies and clinicalgeneral research and development programs targeted against treatment-resistanceactivities related to TSC and any other product candidates we may acquire or in-license in various cancers, withthe future.
As a focus on brain cancer types (both GBMdevelopment-stage company, we continuously evaluate opportunities to improve the value of our development pipeline, including potential acquisition and metastatic)in-licensing opportunities. Our efforts include ongoing evaluation and pancreatic cancer, allmodification of which are knownour development plans intended to maximize the probability of technical, developmental, and regulatory success while enhancing patient and stockholder value. These activities have, and we expect they will continue to be significantly hypoxic. The Company’s longer term goal isfor the foreseeable future, focused on opportunities that are synergistic with our overall corporate strategy to use TSC against treatment-resistance indevelop novel treatments for the entire rangetreatment of hypoxic cancers now treated with radiationhypoxia and chemotherapy.conditions complicated by hypoxia.

Glioblastoma ProgramIntellectual Property
Our lead programWe believe that a strong intellectual property portfolio is targetedcritical to our success. We are committed to obtaining and maintaining appropriate patent and other protections for our products candidates and other technologies, preserving and protecting our trade secrets and other confidential and proprietary information, and fiercely defending our intellectual property portfolio against newly diagnosed primary brain cancer, also known as glioblastoma (GBM). Glioblastoma is a grade IV brain tumor, characterizedany potential infringement by a heterogeneous cell population,third parties. We attempt to protect our intellectual property through among other things, the filing of applications for patent, trademark, and other appropriate intellectual property protections, the use of confidentiality agreements with a numberconsultants, contractors and other third parties, our employee policies regarding confidentiality, invention disclosure, and the assignment of negative attributes. GBM cells are typically genetically unstable (and thus prone to mutation), highly infiltrative, angiogenic, and resistant to radiation and chemotherapy. The mutations typically found in GBM allow the tumor to grow and thrive in a hypoxic environment. GBM is classified into two major subclasses, primary or secondary, depending upon the clinical propertiesinventions, as well as regular meetings of members of our internal development and legal teams, which contains key members of our management team. We are also committed to operating our business without infringing on the chromosomal and genetic alterations that are unique to each class. Primary GBM arisesde novo from normal glial cells and typically occurs in those over the ageintellectual property of 40, while secondary GBM arises from transformation of lower grade tumors and is usually seen in younger patients. Primary GBM is believed to account for approximately 95% of all GBM diagnoses.others.
While GBMIn general, patents extend for varying periods according to the date of patent filing or grant and the legal term of patents in various countries where patent protection is obtained, with term adjustments or extensions possible in certain cases based on patent office delays or pursuant to certain administrative and legislative exceptions. The actual protection afforded by a patent, which can vary from country to country, depends on the most common formtype of primary brain tumor involving glial cells it is still relatively rare, as approximately 12,000 peoplepatent, the scope of its coverage and the availability of legal remedies in the United States were diagnosed with GBM in 2014. The median age of GBM diagnosis is approximately 65 years, with the incidence of GBM in those over 65 increasing rapidly as shown by a doubling in incidence from 5.1 per 100,000 in the 1970s to 10.6 per 100,000 in the 1990s. Those diagnosed with the disease have a grim prognosis, with the median survival time of untreated patients being 4.5 months. Current standard-of-care treatment only provides 14-16 months of survival time after diagnosis.country.
Current Treatments for GBM
The standard-of-care for GBM tumors generally begins with surgical resection, unlessWe continue to invest significant time, effort, and resources into the tumor is deemed inoperable due to its location near vital centersdevelopment and maintenance of the brain. This surgery is performed both to alleviate the symptoms associated with the disease as well as to facilitate treatmentour patent portfolio. As of residual tumor cells. Even with advancesDecember 31, 2021, we owned 17 issued U.S. patents and more than 35 issued non-U.S. patents, and had numerous patent applications pending worldwide including issued patents and applications in surgical technique, complete removal of the tumor with clean margins is difficult to achieve,major markets such as the tumors are highly infiltrativeU.S., E.U., China, Japan, and typically extend intoIndia. The normal life (i.e. with no adjustments or extensions) of our key issued patents related to the composition of matter of TSC extends to 2026, with potential patent term extensions to 2031, and the normal brain parenchyma. Duelife of our patents related to this, almost all GBM patients have recurrencean oral formulation of the tumor,TSC extends to 2031, with 90%potential patent term extensions to 2036. The normal life of such recurrence occurring at the primary site.
Dueour key issued patents related to the invasive naturemethods of the tumors, surgical resection is promptly followed by radiotherapy coupled with the use of chemotherapeutic agents. Radiotherapy involves the administration of irradiationTSC extends to the whole brain. While nitrosoureas were historically a commonly used chemotherapeutic agent, temozolomide (“TMZ”) was approved in 20052037, not including potential patent term extensions. For additional information regarding patent term extensions, see "Business — Government Regulation— The Hatch-Waxman Amendments — Patent Term Restoration and is now a mainstay of the standard-of-care. This is based on a clinical trial that showed theMarketing Exclusivity. In addition, of TMZ to surgery and radiation increased median survival in newly diagnosed GBM patients to 14.6 months compared to 12.1 months for the surgery and radiation only group.
Most chemotherapeutic drugs have a limited ability to cross the blood brain barrier, thus a strategy to attempt to circumvent this was the development of Gliadel®, dissolvable chemotherapy wafers that could be placed in the tumor bed following surgical resection. Gliadel® contains the nitrosourea chemotherapeutic agent carmustine that is released for several weeks, in contrast to systemically administered carmustine that has a very short half-life. While Gliadel® wafers were shown to be safe, the drugs’ addition to radiation and TMZ did not result in a statistically significant increase in survival.
GBM tumors show increased expression of vascular endothelial growth factor (“VEGF”), and the anti-angiogenesis drug bevacizumabTSC has been approvedgranted Orphan Drug designation by the FDA for the treatment of recurrent GBM. A Phase 2 study found that bevacizumab treatment in patientsboth GBM and metastatic brain cancer, which may provide us with recurrent GBM increased six-month progression-free survival from a historical 9-15% to 25% with overall six-month survivalright of 54%. Another Phase 2 study showed that recurrent GBM patients treated with bevacizumab at a lower dose but a higher frequency had even higher six-month progression-free survival of 42.6%.
While bevacizumab has shown success in recurrent GBM, it is not utilized in newly diagnosed patients, our target patient population, as two separate clinical trials showed no difference in overall survival in patients treated with radiation, TMZ,exclusivity under certain FDA regulations. For additional information regarding orphan and bevacizumab compared to patients treated with only radiation and TMZ. Bevacizumab treatment did result in an increase in progression free survival in both studies; however, the reason why this increase in progression free survival did not translate to an increase in overall survival is unclear. In addition, certain studies have reported that patients treated with bevacizumab had an increased symptom burden, a worse quality of life, and a decline in neurocognitive function.
ultra-orphan designations, see "GBM Therapies Under DevelopmentBusiness
There are a number of companies developing GBM therapies. For example, a search on the website www.clinicaltrials.gov yields over 300 results for “glioblastoma multiforme” and “open trials.” Most of these trials focus on the recurrent patient population, whereas our target population is newly diagnosed patients. In addition to the therapeutics previously mentioned, current GBM trials include Northwest Therapeutics’ DCVax®-L, Bristol-Myers Squibb’s Nivolumab/Ipilimumab and AbbVie’s Veliparib. In addition, the medical device company Novocure has been developing a novel approach called Tumor Treating Fields (“TTFields”) using low intensity, alternating electric fields within the intermediate frequency range. TTFields are believed to disrupt cell division through physical interactions with key molecules during mitosis. This medical device approach has shown some success in the recurrent GBM patient population and more recently as an initial treatment.
—GBM is an Orphan Disease Government Regulation
GBM is diagnosed in approximately 12,000 individuals every year, making it an “orphan disease.”— Certain Other FDA Regulations — The Orphan Drug Act of 1983 was designed to provide financial incentives for, and to reduce the costs associated with, developing drugs for rare diseases and disorders. A “rare disease or disorder” is defined by the Orphan Drug Act of 1983 as affecting fewer than 200,000 Americans at the time of designation or one for which “there is no reasonable expectation that the cost of developing and making available in the United States…will be recovered from sales in the United States..” A sponsor must request that the FDA designate a drug currently under development for a “rare disease or condition” as an orphan drug, and if the FDA agrees that the drug and indication meet the criteria set forth in the Orphan Drug Act of 1983, certain financial and marketing incentives become available.
In July 2011, we announced that TSC was granted Orphan Drug Designation by the FDA for the treatment of GBM.Chemistry, Manufacturing, and Controls
Trans Sodium Crocetinate Phase 1/2 Clinical Trial in GBMTSC is currently formulated as a freeze-dried, injectable product, requiring a sophisticated manufacturing process.
We have evaluateddo not currently own or operate any facilities suitable for manufacturing TSC in 148 human subjects in various Phase 1or any of our other product candidates on a scale required to support either clinical development or commercialization. We currently use third-party CMOs to manufacture API, other starting materials, and Phase 2finished drug product for our preclinical studies and clinical trials and we intend to date,continue doing so for the foreseeable future. We anticipate this strategy will be scalable in a manner sufficient to support the production capacity required to support continued clinical development and ultimate commercialization. However, we do not have any formal agreements at this time with no serious adverse events attributableany CMO to TSC. Our Phase 1/2 clinical trialcover commercial production of TSC or any other product candidate. Although the use of third party CMOs to manufacture pharmaceutical products is common within the industry, this dependence on third-party CMOs exposes our business to certain risks, including those described under the heading, “Risk Factors – Risks Related to Our Dependence on Third Parties.”
Supply Chain Matters
Recently, many companies across a variety of sectors have reported disruptions, shortages, and other supply chain-related issues. In the biopharmaceutical sector, delays and interruptions in patients with newly diagnosed GBM completedthe supply chain were particularly pronounced as CMOs redirected resources under the Defense Production Act and to otherwise support the COVID-19 vaccine roll-out across the globe.
During 2021, we were able to effectively manage our supply of TSC and its component materials in 2015 is described in more detail below. TSC is targeted for testing against newly diagnosed GBM in an upcoming Phase 3a manner that avoided any significant interruptions to our clinical trial that, assumingprograms. We also took actions designed to increase the robustness of our drug supply and overall supply chain to mitigate risks related to the availability of funding resourcesour drug supply in the future. However, as constraints on the supply chain continue to impact the cost and general availability of manufacturing materials, related delays and shortages could affect the completioncost and timing of certain manufacturing and animal toxicology guidelines mandated by the FDA guidance, could begin within the next twelve (12) months.our available clinical study drug supply of TSC.
Our Phase 1/2 clinical trialCompetition
Currently, medical options to improve oxygenation without risk of hyper-oxygenation are limited and we believe that TSC's diffusion enhancing mechanism of action make it a first-in-class, novel, small molecule. However, there are several companies currently developing or marketing oxygen enhancing products, therapeutics, or devices that may nevertheless be competitive with TSC, if approved, including Hemoglobin Oxygen Therapeutics LLC, Hemotek Medical Inc., NuvOx Pharma LLC, Omniox, Inc., and VirTech Bio Inc. In addition, in GBM enrolled 59 newly diagnosed patients who received TSC in conjunctionthe first quarter of 2021, we became aware of a third party affiliated with radiation and TMZ. In the Phase I portiona former outside consultant of the trial, TSC was initially administered three times per week at half-dose to three patients prior to radiation. Subsequently, six additional patients received full-dose TSC for six weeks in combination with radiation. No dose-limiting toxicities were identified in the nine patients during the Phase I portion of the trial, nor were any serious adverse events relating to the drug observed. Fifty additional patients were enrolled in the Phase II trial and received full-dose TSC in combination with TMZ and radiation therapy. Four weeks after completion of radiation therapy, all patients underwent chemotherapy with higher doses of TMZ for five (5) days every four weeks, but no further TSC was administered.
We presented initial results from the trial at the 2015 American Society of Clinical Oncology (“ASCO”) Annual Meeting in June 2015,Company which discussed data from the 18 trial sites covering the first twenty-one months. Final results were published in the Journal of Neurosurgery online in May 2016. We compared results in relation to a historical control group from a 2005 study which showed that the addition of TMZ to standard-of-care (surgery plus radiation) increased overall survival from 12.1 months to 14.6 months. We reported that:
| ●
| TSC plus radiation and TMZ increased the patients’ chance of survival at two years by 37% compared to the historical control group. The overall survival at two years was 36.3% in the TSC group compared to 26.5% in the historical control group.
|
| ●
| In the subgroup of patients considered inoperable, the chance of survival at two years for those who received TSC was increased by 380%, (40% alive at two years for TSC group versus 10% for control).
|
| ●
| 71% of those treated with TSC were alive at one year compared to 61% of those in the historical control group. In 11 of 56 patients, tumors regressed to undetectable.
|
| ●
| No serious safety findings attributed to TSC were observed in the TSC study and adverse events were consistent with those seen in previous trials of GBM featuring radiation and TMZ.
|
End-of-Phase 2 FDA Meeting and Plans for TSC Phase 3 GBM Clinical Trial
Following the announcement of the results of the 2015 Phase 1/2 clinical trial in GBM, we held an end of Phase 2 meeting with the FDA in August 2015 to discuss planning for a Phase 3 clinical trial. At the meeting, guidance was received on a trial design for the Phase 3 study, including:
| ●
| A single, randomized trial of the agreed upon design, if successful, can serve as the basis for an application for approval.
|
| ●
| The trial will consist of 400 newly diagnosed GBM patients with half given TSC in conjunction with standard-of-care radiation and TMZ and half receiving standard-of-care radiation and TMZ only.
|
| ●
| Based on the Phase 1/2 safety results with supporting toxicology, TSC’s dosing exposure will be substantially increased, which means that TSC can now be used for both the radiation + chemotherapy and subsequent TMZ chemotherapy-only phase of GBM treatment, extending the TSC treatment duration from 6 weeks to 30 weeks.
|
| ●
| Diffusion will provide certain expanded information on animal toxicology, pharmacokinetics and manufacturing practices to the FDA before initiating the trial.
|
One of the major differences between the Phase 3 trial and the Phase 1/2 trial is the addition of TSC doses after the completion of the radiation/chemotherapy phase of treatment into the chemotherapy only phase. In the Phase 1/2 trial, TSC was only given prior to radiation (18 doses total over 6 weeks). In the Phase 3 study, we are planning to give the patients 36 total doses of TSC, 18 in conjunction with radiation/chemotherapy and 18 in conjunction with chemotherapy alone for a total TSC treatment duration of 30 weeks. The following figure gives a graphical representation of our planned Phase 3 trial, which, assuming the availability of financial resources and the completion of certain manufacturing and animal toxicology guidelines mandated by the FDA guidelines, we intend to commence enrollment in 2017, complete enrollment and potentially receive interim data in 2019 and complete conduct (enrollment and dosing) of the study in 2020. Data collection, analysis and regulatory interaction are projected to occur over the following 12 to 18 months.

Pancreatic Cancer Program
One of the most hypoxic of all the solid cancers, and therefore one of the most treatment-resistant, is pancreatic cancer. According to the American Cancer Society, pancreatic cancer is responsible for 7% of all cancer deaths in both men and women, making it the fourth leading cause of cancer death in the U.S. Estimates are that 40% of pancreatic cancer cases are sporadic in nature, 30% are related to smoking and 20% may be associated with dietary factors, with only 5-10% of cases hereditary in nature.
Pancreatic cancer is difficult to diagnose in early stages because initial symptoms are often nonspecific and subtle in nature, and include anorexia, malaise, nausea, fatigue, and back pain. Approximately 75% of all pancreatic carcinomas occur within the head or neck of the pancreas, 15-20% occur in the body of the pancreas, and 5-10% occur in the tail.
The only potential curative therapy for pancreatic cancer is complete surgical resection. Unfortunately, this is only possible for approximately 20% of cases, and even of those patients whose cancer is surgically resected, 80% will develop metastatic disease within two to three years following surgery. Patients with unresectable pancreatic cancer have a median overall survival of 10 to 14 months while patients diagnosed with Stage IV disease (indicative of metastases) have a 5-year overall survival of just 1%.
Multiple studies have confirmed that pancreatic cancers are highly hypoxic. A study reporting the direct measurement of oxygenation in human pancreatic tumors prior to surgery showed dramatic differences between tumors and normal tissue. The partial pressure of oxygen (“pO2”) ranged between 0-5.3 mmHg in tumors but in adjacent normal tissue it ranged from 9.3-92.7 mmHg. Hypoxic areas are also frequently found when examining tissue from mouse models of pancreatic cancer.
Current Treatment Options for Pancreatic Cancer
Surgery remains the primary mode of treatment for patients with pancreatic cancer. However, there is an important role for chemotherapy and/or radiation in an adjuvant setting (given to prevent recurrence) or neoadjuvant setting (given before surgery to shrink the tumor to make complete resection more probable), as well as in patients with unresectable disease.
Since its approval in 1996, gemcitabine has been partnered with approximately 30 different agents in late-stage clinical trials in an attempt to improve upon the effectiveness of gemcitabine alone in treating patients with metastatic pancreatic cancer. Only two of these trials have demonstrated improved efficacy, leading to FDA approvals – erlotinib (Tarceva®) and nab-paclitaxel (Abraxane®).
In patients with metastatic disease, the use of erlotinib with gemcitabine led to a significantly higher one-year survival rate than with the use of gemcitabine alone (23% vs. 17%,P= 0.023) as well as an increased median overall survival (6.24 months vs. 5.91 months,P = 0.038). A more recent study showed that the addition of nanoparticle albumin-bound (nab)-paclitaxel to gemcitabine significantly improved overall survival in treatment naïve patients with metastatic cancer, as overall survival was approximately two (2) months longer in patients treated with combination therapy (8.5 vs. 6.7 months).
The Folfirinox (leucovorin + 5-fluorouracil + oxaliplatin + irinotecan) regimen was shown to significantly improve overall survival compared to treatment with gemcitabine (11.1 months vs. 6.8 months). While dramatically improving overall survival, the Folfirinox treatment was accompanied by serious adverse events and thus is only recommended for patients with good performance status.
Other combinations of gemcitabine with cisplatin, oxaliplatin, irinotecan or docetaxel tested in Phase 3 trials have not been of superior benefit to gemcitabine alone. The combination therapy nab-Paclitaxel and gemcitabine was recently approved by the FDA as an additional standard-of-care for the treatment of patients with untreated pancreatic adenocarcinoma. However, the improvements were modest, and treatment of pancreatic cancer remains an area of intense research, with 92 products in all stages of clinical development with 14 of them in Phase 3 at this time according to clinicaltrials.gov.
Just recently, the FDA approved Onivyde® (irinotecan liposome injection) in combination with fluorouracil and leucovorin, to treat patients with metastatic pancreatic cancer who were previously treated with gemcitabine-based chemotherapy. In the pivotal clinical trial, patients treated with Onivyde® plus fluorouracil/leucovorin lived an average of 6.1 months, compared to 4.2 months for those treated with only fluorouracil/leucovorin.
Pancreatic Cancer Market Analysis
It is estimated that every year approximately 49,000 people are diagnosed with pancreatic cancer in the United States. More than half of these patients will be diagnosed with metastatic disease. The five-year survival rates for patients with pancreatic cancer are dismal (<14%) and are particularly bad for those with metastatic disease (~1%).
The current standard-of-care for patients with metastatic pancreatic cancer includes gemcitabine combined with either erlotinib or nab-paclitaxel. Gemzar® (gemcitabine) is now available as a generic, however prior to losing patent protection the drug generated peak revenues of approximately $700 million in the United States for Eli Lilly. Tarceva®(erlotinib), which is approved for the treatment of metastatic non-small cell lung cancer and metastatic pancreatic cancer, is marketed by Roche and Astellas and sales of the drug generated $1.2 billion in revenue in 2015. Abraxane® (nab-paclitaxel), which was approved for the treatment of breast cancer in 2005 and non-small cell lung cancer in 2012, was approved by the FDA in 2013 for the treatment of metastatic pancreatic cancer. Sales of Abraxane® totaled $848 million in 2014 for all indications.
TSC in Pancreatic Cancer
We believe that targeting hypoxia in pancreatic cancer with TSC, especially in combination with standard-of-care therapies involving gemcitabine and nab-paclitaxel, may be beneficial in the treatment of pancreatic cancer. Pancreatic cancer is one of the most hypoxic malignant tumors, making it one of the most resistant tumors to therapy. Patients with advanced pancreatic cancer of exocrine origin have few therapeutic options and, for patients with advanced cancers, the overall survival rate of all stages is less than 1% at 5 years, with most patients dying within 1 year. Gemcitabine remains to-date, the backbone of treatment of pancreatic cancer.
The antitumor efficacy of gemcitabine is knownclaims to be hindered byin early-stage development of a numberproduct candidate that purportedly may operate through a similar mechanism of hypoxia-related factors including, but not limited to:
| ●
| Limited Delivery of Gemcitabine to Intracellular Tumor Microenvironment. Hypoxia has been associated with resistance to nucleoside analogs such as gemcitabine by decreasing the expression of the human cross-cell membrane equilibrative nucleoside transporter 1 (hENT1), thereby decreasing transport of gemcitabine and other nucleoside analogues into tumor cells.
|
| ●
| Gemcitabine Intratumor Cell Inhibition of Ribonucleotide Reductase Compromised. Hypoxia has been associated with a decrease in intracellular tumor ribonucleotide reductase, an enzyme required for the antitumor effect of gemcitabine. Indeed, this decrease leads to cell cycle arrest in G1 or G2 phase, thereby allowing DNA repair before progression to S or M phase.
|
| ●
| Increased Breakdown of Gemcitabine. It has been also observed that under hypoxic conditions, intratumor cell levels of cytidine deaminase are substantially increased. Cytidine Deaminase is the main enzyme responsible for the breakdown of gemcitabine and similar nucleosides. As a result of this, the gemcitabine antitumor effect is substantially decreased under hypoxic conditions.
|
| ●
| Nab-paclitaxel Potentiates Gemcitabine by Decreasing Cytidine Deaminase. One of the ways nab-paclitaxel decreases cytidine deaminase has been observed to be through increasing reactive oxygen species (“ROSs”) which has been shown to deactivate cytidine deaminase.
|
Taken together, the four factors above are believed to explain, at least in part, the limitations of gemcitabine in hypoxic conditions and the efficacy observed with the gemcitabine plus nab-paclitaxel combination regimen for the treatment of pancreatic cancer. They also suggest that correction of hypoxia with an anti-hypoxia agent such as TSC may significantly improve the efficacy of the gemcitabine plus nab-paclitaxel combination regimen for the treatment of pancreatic cancer, including for the following reasons:
| ●
| TSC has been shown to improve the cytotoxic effect of gemcitabine in a pre-clinical rat model.
|
| ●
| By correcting hypoxia, TSC may improve delivery of gemcitabine to intracellular tumor microenvironment by increasing levels of hENT-1.
|
| ●
| By reversing hypoxia-induced cell cycle arrest via reactivation of ribonucleotide reductase, TSC may restore gemcitabine’s antitumor effect.
|
| ●
| By reversing the hypoxia-induced increase of cytidine deaminase, TSC may increase intratumoral gemcitabine levels. Addition of TSC to the gemcitabine plus nab-paclitaxel regimen could further improve the efficacy of the combination by further decreasing cytidine deaminase. Of note, hypoxia has also been implicated in conferring tumor resistance to taxane-based therapies.
|
Proposed Plans for TSC Phase 2 Clinical Trial in Pancreatic Cancer
The planned Phase 2 clinical trial for TSC in pancreatic cancer is based on preclinical safety and efficacy data, and findings from the Phase 1/2 clinical trial in GBM, as well as the facts noted above. Global experts in the field agree that pancreatic cancer is an appropriate target for expansion of the use of TSC, and a clinical advisory committee of these key opinion leaders has been assembled to facilitate our pancreatic cancer clinical development program. We are currently in discussions with recognized pancreatic cancer experts and the FDA regarding trial design, end-points, and patient numbers. Assuming the availability of financial resources, we anticipate beginning enrollment in this trial during 2017 and completing conduct of the study (enrollment and dosing) in 2019. Data collection, analysis and regulatory interaction are projected to occur over the following 9 to 12 months.
Brain Metastases Program
In contrast to the relative rarity of primary brain cancers, life-threatening cancers that metastasize to the brain are much more common and represent a serious complication in the treatment of many cancer types. Up to 30% of adult cancer patients will suffer from brain metastases. There are approximately 170,000 cases of metastatic brain cancer every year in the United States. Incidence of brain metastases varies depending upon the primary tumor type, although lung cancer appears to carry the greatest risk. The prognosis for patients with brain metastases is very grim, with current treatment options only resulting in median overall survival times of less than one year.
Treatment for brain metastases involves both controlling the symptoms associated with the condition as well as attacking the cancer directly. Brain metastases typically result in edema that can be controlled with the use of steroids; however, long-term use of steroids typically results in side effects that diminish a patient’s quality of life. Approximately 25-45% of patients will experience seizures and require the use of anti-epileptic drugs. Surgery is only utilized in patients with a solitary brain metastatic lesion. Radiation therapy remains the standard-of-care for the vast majority of patients with brain metastases. There is very limited evidence for the use of chemotherapy, as few clinical trials have been conducted. There are no medications currently approved for the treatment of brain metastases.
Plans for TSC Phase 2/3 Clinical Trial in Metastatic Brain Cancer
We are planning to conduct a Phase 2/3 clinical trial in metastatic brain cancer after further discussions with experts and the FDA regarding trial design, end-points, and patient numbers.
In December 2012, the FDA granted us Orphan Drug Designation for the use of TSC in brain metastases.
Description of Other Indications/Products
We have rights to and own technologies and potential products beyond those described above, including analog molecules as backupsaction to TSC. It is our strategy to focus at the current time on our TSC for oncology, specifically GBM and pancreatic cancer, as described herein. Beyond those described herein, we also intend to continue to review our technologies and potential products on a regular basis and consider internal development in the future and the potential to out-license portions of our technology and potential products to other biopharmaceutical companies with greater focus and resources than ours, or potentially in-license late stage products which are in or ready for human clinical trials.
As a result of the Merger, we acquired product candidates for ophthalmology, oncology and dermatology. One suchunclear if this Company’s product candidate is RES-529, a novel PI3K/Akt/mTOR pathway inhibitor which has completed two Phase I clinical trials for age-related macular degenerationwould, if developed and is in preclinical development in oncology, specifically GBM. The novel inhibition of the PI3K/Akt/mTOR pathway and targeting of the androgen receptor have also shown potential in a number of additional indications.
We are exploring partnering opportunities for our product candidates, such as strategic partnerships, alliances or licensing arrangements.
Competitionapproved, actually be competitive with TSC.
Our industry is highly competitive and subject to rapid and significant change. Potential competitors in the United States are numerous and include major pharmaceutical and specialty pharmaceutical companies, smaller biopharmaceutical companies, research universities, and other institutions. We generally divide ourothers. The biopharmaceutical and pharmaceutical industries are characterized by rapidly advancing technologies, intense competition, and a strong emphasis on developing proprietary therapeutics. Numerous companies are engaged in the pharmaceutical industry into four categories: (1) corporationsdevelopment, patenting, manufacturing, and marketing of health care products competitive with large research and developmental departmentsthose that develop and market products in many therapeutic areas; (2) companies that have moderate research and development capabilities and focus their product strategy on a small number of therapeutic areas; (3) small companies with limited development capabilities and only a few product offerings; and (4) university and other research institutions.we are developing. Many of our competitors have longer operating histories, greater name recognition, substantially greater financial resources, and larger research and development staffs than we do, as well as substantially greater experience than us in developing products, obtaining regulatory approvals, and manufacturing and marketing pharmaceutical products. AIn addition, a significant amount of research is carried out at academic and government institutions. These institutions are aware of the commercial value of their findings and are aggressive in pursuing patent protection and negotiating licensing arrangements to collect royalties for use of technology that they have developed.
There are several firms currently marketing One or developingmore of these companies or other entities may have one or more products under development that maywould be competitive with our products, including therapeutics and devices. We believe TSC is a first-in-class novel small molecule that re-oxygenates hypoxic tissue, enhancing efficacy of radiation and chemotherapy without harmful side effects.TSC.
MergersSales and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated among a smaller number of our competitors. Our commercial opportunity could be reduced or eliminated if our competitors develop or market products or other novel therapies that are more effective, safer or less costly than our current or future product candidates, or obtain regulatory approval for their products more rapidly than we may obtain approval for our product candidates. In addition, the first product to reach the market in a therapeutic or preventative area is often at a significant competitive advantage relative to later entrants in the market and may result in certain marketing exclusivity as per federal legislation. Acceptance by physicians and other health care providers, including managed care groups, also is critical to the success of a product versus competitor products. Our success will be based in part on our ability to identify, develop and manage a portfolio of product candidates that are safer and more effective than competing products.
Research and Product Development
We incurred approximately $7.2 million in 2016 and $3.9 million in 2015 on research and product development activities that related primarily to activities associated with the synthesis and formulations of our products then in development, additional preclinical studies and planning for Phase I/Phase II studies. We anticipate that our research and development expenses during 2017 will increase compared to 2016 and will consist primarily of expenses associated with the initiation of our glioblastoma and pancreatic cancer human clinical trials.
Intellectual Property
Our success depends and will continue to depend in part upon our ability to maintain proprietary protection for our products and technologies, to preserve our proprietary information, trademarks and trade secrets and to operate without infringing the proprietary rights of others. Our policy is to attempt to protect our technology by, among other things, filing patent applications on inventions that are important to the development and conduct of our business with the U.S. Patent and Trademark Office (“USPTO”), and its foreign counterparts or obtaining license rights for technology that we consider important to the development of our business.
As of December 31, 2016, we owned approximately 14 issued U.S. patents and 46 issued foreign patents, which include granted European, Japanese, Chinese and Indian patent rights, and over 50 pending patent applications worldwide, covering the product candidates we currently intend to develop. Our current patents expire between 2026 and 2031. TSC has been granted Orphan Drug Designation for the treatment of both GBM and metastatic brain cancer and an application is pending for pancreatic cancer. A formulation patent provides protection for the TSC oral drug product until 2031 with extensions possible.
Patents extend for varying periods according to the date of patent filing or grant and the legal term of patents in various countries where patent protection is obtained. The actual protection afforded by a patent, which can vary from country to country, depends on the type of patent, the scope of its coverage and the availability of legal remedies in the country.
In addition to patents, we use other forms of protection, such as trademark, copyright and trade secret protection, to protect our intellectual property, particularly where we do not believe patent protection is appropriate or obtainable. We aim to take advantage of all of the intellectual property rights that are available to us and believe that this comprehensive approach will provide us with proprietary positions for our product candidates, where available.Marketing
We also protect our proprietary information by requiring our employees, consultants, contractorscurrently have no marketed products and, accordingly, currently have no sales or marketing personnel.
Government Regulation
Pharmaceuticals like TSC and other advisors to execute nondisclosure and assignment of invention agreements upon commencement of their respective employment or engagement. Agreements with our employees also prevent them from bringingproduct candidates we may develop are highly regulated by governmental authorities in the proprietary rights of third parties to us. In addition, we also require confidentiality or service agreements from third parties that receive our confidential information or materials.
Government Regulation
FDA Drug Approval Process
In the United States, pharmaceutical products are subject to extensive regulation by the FDA. The Federal Food, Drug, and Cosmetic Act (the “FDC Act”)U.S. and other countries at the federal, state, and state statuteslocal levels. These regulations are numerous and regulations, govern,extensive in their scope, relating to, among other things, the research and development, testing, manufacture, storage, recordkeeping,quality control and testing, approval, labeling and packaging, promotion, marketing, and marketing,advertising, distribution, post-approval monitoring and reporting, samplingexport and import, and exportrecord keeping of pharmaceutical products. Failure
The FDA Drug Approval Process
Generally, before a new pharmaceutical product can be marketed, considerable data demonstrating its quality, safety and efficacy must be obtained, organized into a format specific for each applicable regulatory authority, submitted for review, and approved by the competent regulatory authority. In the United States, the competent regulatory authority is the FDA, which, pursuant to the FDC Act, is responsible for the review and approval of all data required to support a license to commercially market pharmaceuticals such as TSC.
The process of obtaining regulatory approvals and the subsequent compliance with FDA regulations requires the expenditure of substantial time and financial resources and failure to comply with the applicable U.S. requirements at any time during the product development process, approval process, or, if approved, following approval may subject a companyan applicant to a variety of administrative or judicial sanctions. These sanctions such as FDAcould include, among other actions, the FDA’s refusal to approve pending new drug applications, (“NDAs”),withdrawal of an approval, license revocation, a clinical hold, untitled or warning letters, voluntary or untitled letters,mandatory product recalls, market withdrawals, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement and civil or criminal penalties, and criminal prosecution.any of which could have a material adverse effect on our business, financial position, or results of operations.
Pharmaceutical product development for a new product or certain changes to an approved product inProcess Overview
The FDA drug approval process generally involves the United States typically involves preclinical laboratory and animal tests, the submission to the FDA of an investigational new drugfollowing steps:
| • | completion of extensive preclinical laboratory studies , including studies conducted in accordance with GLP requirements; |
| • | submission to the FDA of an IND application, (“IND”) which must become effective before clinical trials involving human subjects or patients may begin; |
| • | performance of adequate and well-controlled human clinical trials in accordance with applicable IND regulations, GCP requirements, and other clinical trial-related regulations to establish the safety and efficacy of the investigational product for each proposed indication, including approval by an IRB or independent ethics committee before each trial may be initiated; |
| • | submission to the FDA of an NDA; |
| • | a determination by the FDA within 60 days of its receipt of an NDA as to whether it will accept the filing for review; |
| • | satisfactory completion of one or more FDA pre-approval inspections of the manufacturing facility or facilities where the drug will be produced to assess compliance with cGMP requirements and to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity; |
| • | a potential FDA audit of the clinical trial sites that generated the data in support of the NDA; |
| • | payment of user fees for FDA review of the NDA; and |
| • | FDA review and approval of the NDA, including consideration of the views of any FDA advisory committee, prior to any commercial marketing or sale of the biologic or drug in the United States. |
The preclinical and clinical testing may commence, and adequateapproval process requires substantial time, effort, and well-controlled clinical trials to establish the safetyfinancial resources and effectiveness of the drug for each indication for which FDA approval is sought. Satisfactionsatisfaction of FDA pre-market approval requirements typically takes many years, andthough the actual time required may vary substantially based upon the type, complexity, and novelty of the applicable product or disease.indication to be treated. We cannot be certain that any approvals for TSC or any product candidates we attempt to develop in the future will be granted on a timely basis or at all.
Preclinical testsStudies
Preclinical studies include laboratory evaluation of product chemistry, formulation, and toxicity, as well as animal trials to assess the characteristics and potential safety and efficacy of the product. The conduct of the preclinical tests must comply with federal regulations and requirements, including good laboratory practices.drug. The results of preclinical testing are submitted to the FDA as part of an IND along with other information related to the drug, including information about product chemistry,regarding its chemical make-up, manufacturing process, and quality controls, andas well as a proposed clinical trial protocol. Long termLong-term preclinical tests, such as animal tests of reproductive toxicity and carcinogenicity, may continue after the IND is submitted.
Clinical Trials
AFollowing completion of preclinical studies and the submission on an IND to the FDA, a 30-day waiting period after the submission of each IND is required prior to the commencement of clinical testing in humans.required. If the FDA has neither commented on nor questioned the IND within this 30-day period, the clinical trial proposed in the IND may begin.
Clinical trials involve the administration of thean investigational new drug to healthy volunteers or patients under the supervision of a qualified investigator. ClinicalThese trials must be conducted: (1)conducted in compliance with federal regulations; (2) in compliance with good clinical practice (“regulations as well as GCP,”) an international standard meant to protect the rights and health of patients and to define the roles of clinical trial sponsors, administrators, and monitors; and (3)monitors. Each trial is conducted under protocolsa protocol detailing the objectives of the trial, the parameters to be used in monitoring safety, and the effectiveness criteria to be evaluated. Each protocol involving testing on U.S. patients and subsequent protocol amendments must be submitted to the FDA as part of the IND.
The FDA may order the temporary or permanent discontinuation of a clinical trial at any time, or impose other sanctions, if it believes that the clinical trial either is not being conducted in accordance with FDA requirements or presents an unacceptable risk to the clinical trial patients. The study protocol and informed consent information for patients in clinical trials must also be submitted to an institutional review board (“IRB”) for approval at each site at which the clinical trial will be conducted. An IRB may also require the clinical trial at the site to be halted, either temporarily or permanently, for failure to comply with the IRB’s requirements, or may impose other conditions.
Clinical trials to support NDAs for marketing approval are typically conducted in three sequential phases, but the phases may overlap, especially in cancer indications.certain indications such as cancer.
| • | Phase 1 - In Phase 1 trials, an investigational new drug is introduced into healthy human subjects and is evaluated to assess pharmacological actions, side effects associated with increasing doses and, in certain cases, early evidence on efficacy. |
| • | Phase 2 - In Phase 2 trials, the drug is introduced to a limited patient population in a particular indication to determine metabolism, pharmacokinetics, the effectiveness of the drug for the indication, dosage tolerance and optimum dosage, and to identify potential adverse effects and safety risks. |
| • | Phase 3 - In Phase 3 trials, if a drug has demonstrated evidence of effectiveness and an acceptable safety profile in prior Phase 2 trials, the drug is introduced to a larger patient population in the relevant indication to obtain the additional information about clinical efficacy and safety in a larger number of patients, typically at geographically dispersed clinical trial sites, to permit the FDA to evaluate the overall benefit-risk relationship of the drug, and to provide adequate information for the labeling of the drug, if approved. |
Not all drug development programs are required to follow the order and content of all three phases. For example, in August 2018, the FDA released a draft guidance entitled “Expansion Cohorts: Use in First-In-Human Clinical Trials to Expedite Development of Oncology Drugs and Biologics,” which outlines how drug developers can utilize an adaptive trial design commonly referred to as a seamless trial design in early stages of oncology drug development, i.e., the first-in-human clinical trial, to compress the traditional three phases of trials into one continuous trial called an expansion cohort trial. Information to support the design of individual expansion cohorts is included in IND applications and assessed by FDA. Expansion cohort trials can potentially bring efficiency to drug development and reduce developmental costs and time.
Post-approval trials, sometimes referred to as Phase 4 clinical trials, may be conducted after initial marketing approval. These trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication and are commonly intended to generate additional safety data regarding use of the product in a clinical setting. In certain instances, the FDA may mandate the performance of Phase 4 clinical trials as a condition of approval of an NDA.
Progress reports detailing the results of the clinical trials, among other information, must be submitted at least annually to the FDA and written IND safety reports must be submitted to the FDA and the investigators 15 calendar days after the trial sponsor determines the information qualifies for reporting for serious and unexpected suspected adverse events, findings from other studies or animal or in vitro testing that suggest a significant risk for human subjects and any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigator brochure. The sponsor also must notify the FDA of any unexpected fatal or life-threatening suspected adverse reaction as soon as possible but in no case later than seven calendar days after the sponsor’s initial receipt of the information.
Phase 1, the initial introduction of the drug into healthy human subjects or patients, the drug is tested to assess pharmacological actions, side effects associated with increasing doses and, if possible, early evidence on effectiveness. Phase 2, usually involves trials in a limited patient population to determine metabolism, pharmacokinetics, the effectiveness of the drug for a particular indication, dosage tolerance and optimum dosage, and to identify common adverse effects and safety risks. If a compound demonstrates evidence of effectiveness and an acceptable safety profile in Phase 2 evaluations, Phase 3, and other types of clinical trials are undertaken to obtainmay not be completed successfully within any specified period, if at all. The FDA or the additional information about clinical efficacy and safety insponsor may suspend or terminate a larger number of patients, typically at geographically dispersed clinical trial sites,at any time on various grounds, including a finding that the patients are being exposed to permit thean unacceptable health risk.
New Drug Application and FDA to evaluate the overall benefit-risk relationship of the drug and to provide adequate information for the labeling of the drug. In most cases the FDA requires two adequate and well-controlled Phase 3 clinical trials with statistically significant results to demonstrate the efficacy of the drug. With suitable FDA agreement, a single Phase 3 clinical trial with other confirmatory evidence may be sufficient. In those instances, the study is usually a large multicenter trial demonstrating internal consistency and a statistically persuasive finding of an effect on mortality, irreversible morbidity or prevention of a disease with a potentially serious outcome and confirmation of the result in a second trial would be practically or ethically impossible.Review Process
After completion of the required clinical testing, an NDA is prepared and submitted to the FDA.FDA containing data intended to provide substantial evidence that the drug is safe and effective in the relevant indication, and FDA approval of the NDA is required before commercial marketing of the product may begin in the United States. The NDA must include the results of all preclinical, clinical, and other testing and a compilation of data relating to the product’s pharmacology, chemistry, manufacturemanufacturing, and controls. The cost of preparing and submitting an NDA is substantial. Thesubstantial, and the submission of most NDAs is additionally subject to a substantial application user fee, and the manufacturer and sponsor under an approved new drug application are also subject to annual productsubstantial initial and establishment userongoing fees. These fees are typically increased annually.
The FDA has sixty (60)60 days from its receipt of an NDA to determine whether the applicationNDA will be accepted for filing based on the agency’s threshold determination that it is sufficiently complete to permit substantive review. Once the submission is accepted for filing, the FDA begins an in-depth review. The FDA has agreedreview subject to certain performance goals inagreed upon by the review of new drug applications.FDA. Priority review can be applied in certain instances, including with respect to drugs that the FDA determines offer major advances in treatment or provide a treatment where no adequate therapy exists. For certain drugs, priority review is further limited only for drugs intended to treat a serious or life-threatening disease relative to the currently approved products. The review process, for bothwhether standard andor priority, review may be extended by the FDA for three (3) additional months to consider certain late-submitted information or information intended to clarify information already provided in the submission.
The FDA may also refer applications for novel drug products, or drug products that present difficult questions of safety or efficacy, to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendations. Before approving an NDA, the FDA will typically inspect one or more clinical sites to assure compliance with GCP. Additionally, the FDA will inspect the facility or the facilities at which the drug is manufactured.manufactured to confirm compliance with cGMP. The FDA will not approvemay also refer applications for novel drug products, or drug products that present difficult questions of safety or efficacy, to an advisory committee. This advisory committee is typically a panel that includes clinicians and other experts in the product unless compliance with current good manufacturing practice (“cGMP”) is satisfactoryrelevant indication or subject matter who review and evaluate the NDA contains data thatand provide substantial evidence thata recommendation to the drugFDA as to whether the application should be approved. The FDA is safe and effective innot bound by the indication studied.recommendation of an advisory committee, but it generally follows such recommendations.
After the FDA evaluates the NDA, andthe clinical sites, the manufacturing facilities, and, as needed, receives a recommendation from the advisory committee, it issues either an approval letter or a complete response letter. A complete response letter generally outlines the deficiencies in the submission and may require substantial additional testing, or information, in order for the FDA to reconsider the application. If, or when, those deficiencies have been addressed to the FDA’s satisfaction in a resubmission of the NDA, the FDA will issue an approval letter. The FDA has committed to reviewing such resubmissions in two orto six (6) months depending on the type of new information included.
FDA Approval Letter
An FDA approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications. As a condition of NDA approval, the FDA may require a risk evaluation and mitigation strategy (“REMS”) to help ensure that the benefits of the drug outweigh the potential risks. REMS can include medication guides, communication plans for healthcare professionals and elements to assure safe use (“ETASU”). ETASU can include, but are not limited to, special training or certification for prescribing or dispensing, dispensing only under certain circumstances, special monitoring and the use of patient registries. The requirement for a REMS can materially affect the potential market and profitability of the drug. Moreover, product approval may require substantial post-approval testing and surveillance to monitor the drug’s safety or efficacy.efficacy including, in certain cases, REMS as described in more detail under the heading “—Certain Other FDA Regulations – Risk Evaluation and Mitigation Strategies.” Once granted, product approvals may be withdrawn if compliance with regulatory standards is not maintained or problems are identified following initial marketing.
ChangesFurther, changes to some of the conditions established in an approved application, including changes in indications, labeling, or manufacturing processes or facilities, require submission and FDA approval of a new NDA or NDA supplement before the change can be implemented. An NDA supplement for a new indication typically requires clinical data similar to that in the original application, and the FDA uses the samesimilar procedures and actions in reviewing NDA supplements as it does in reviewing NDAs.
The Hatch-Waxman ActAmendments
The Drug Price Competition and Patent Term Restoration Act, commonly referred to as the Hatch-Waxman Amendments, is a 1984 U.S. federal law which established the modern system of generic drug regulation in the U.S. The Hatch-Waxman Amendments were enacted to encourage the manufacture of generic drugs by outlining the process for generic pharmaceutical manufacturers to file an abbreviated new drug application and to provide certain related protections to drug development innovators, namely a new kind of market exclusivity period and the ability to potentially extend patent life by a portion of the time a drug is under regulatory review by the FDA.
Orange Book Listing and Abbreviated New Drug Applications
In seeking approval for a drug through an NDA, applicants are required to list with the FDA each patent whose claims cover the applicant’s product. Upon approval of a drug, each of the patents listed in the application for the drug is then published in the FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the Orange Book. Drugs listed in the Orange Book can, in turn, be cited by potential generic competitors in support of approval of an abbreviated new drug application (“ANDA”). ANDA.
An ANDA provides for marketing of a drug product that has the same active ingredients in the same strengths and dosage form as the listed drug and has been shown through bioequivalence testing to be therapeutically equivalent to the listed drug. Other than the requirement for bioequivalence testing, ANDA applicants are not required to conduct, or submit results of, preclinical or clinical tests to prove the safety or effectiveness of their drug product. Drugs approved in this way are commonly referred to as “generic equivalents” to the listed drug and can often be substituted by pharmacists under prescriptions written for the original listed drug.
TheAn ANDA applicant is required to certifymake certain certifications to the FDA concerning any such patents listed in the Orange Book for the approved product inreference drug intended to confirm that the FDA’s Orange Book. Specifically, the applicant must certify that: (1) the required patent information has not been filed; (2) the listed patent has expired; (3) the listed patent has not expired, but will expire on a particular date and approval is sought after patent expiration; or (4) the listed patent is invalid orproposed generic equivalent will not be infringed byinfringe on any intellectual property related to the new product.reference drug, commonly referred to as a Paragraph IV certification. The ANDA applicant may also electprocess gives the owner of the reference drug an opportunity to submitassert a section viii statement certifying thatpatent infringement claim if it believes its proposed ANDA label does not contain (or carves out) any language regardingintellectual property rights are being infringed upon following the patented method-of-use rather than certify tosubmission of a listed method-of-use patent. If the applicant does not challenge the listed patents, theParagraph IV certification.
An ANDA will not be approved until all the listed patents claiming the referenced product have expired.
A certification that the new product will not infringe the already approved product’s listed patents, or that such patents are invalid, is called a Paragraph IV certification. If the ANDA applicant has provided a Paragraph IV certification to the FDA, the applicant must also send notice of the Paragraph IV certification to the NDA and patent holders once the ANDA has been accepted for filing by the FDA. The NDA and patent holders may then commence a patent infringement lawsuit in response to the notice of the Paragraph IV certification. The filing of a patent infringement lawsuit within forty-five (45) days of the receipt of a Paragraph IV certification automatically prevents the FDA from approving the ANDA until the earlier of thirty (30) months, expiration of the patent, settlement of the lawsuit, or a decision in the infringement case that is favorable to the ANDA applicant.
The ANDA also will not be approved until any applicable non-patent exclusivity periods listed in the Orange Book for the referenced product hasreference drug have expired.
Patent Term Restoration and Marketing Exclusivity
UponCertain of our current and future product candidates, including TSC, may be eligible for patent term restoration and marketing exclusivity under the Hatch-Waxman Amendment.
The Hatch-Waxman Amendments permit restoration of the patent term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. Patent term restoration, however, cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent term restoration period is generally 50% of the amount of time between the effective date of an IND and the submission date of an NDA, plus the time between the submission date of an NDA and the approval of that application, except that the review period is reduced by any time during which the applicant failed to exercise due diligence. Only one patent applicable to an approved drug is eligible for such an extension and the application for the extension must be submitted prior to the expiration of the patent. The USPTO, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration.
Marketing exclusivity provisions under the FDC Act can also delay the submission or the approval of certain marketing applications. The FDC Act provides three years of marketing exclusivity for an NDA, or supplement to an existing NDA, if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example investigations related to new indications, dosages, or strengths of an existing drug. This three-year exclusivity covers only the modification for which the drug received approval on the basis of the new clinical investigations and does not prohibit the FDA from approving ANDAs for drugs containing the active agent for the original indication or condition of use. The FDC Act also provides a five-year period of non-patent marketing exclusivity within the United States to the first applicant to obtain approval of an NDA for a new chemical entity, (“NCE”)meaning the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance.
During the exclusivity period, the FDA may not accept for review an ANDA or a 505(b)(2) NDA submitted by another company for another drug based on the same active moiety, regardless of whether the drug is intended for the same indication as the original innovator drug or for another indication, where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement to one of the patents listed with the FDA by the innovator NDA holder. Three-year and five-year exclusivity will not delay the submission or approval of a full NDA. However, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the nonclinical studies and adequate and well-controlled clinical trials necessary to demonstrate safety and efficacy.
Certain Other FDA Regulations
The Orphan Drug Act of 1983
Under the Orphan Drug Act, the FDA may grant orphan designation to a drug product intended to treat a rare disease or condition, which is generally a disease or condition that containsaffects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the United States and for which there is no active moietyreasonable expectation that the cost of developing and making the product available in the United States for this type of disease or condition will be recovered from sales of the product. Our lead product candidate, TSC, has been approvedgranted Orphan Drug designations by the FDA for the treatment of both GBM and metastatic brain cancer.
Orphan drug designation must be requested before submitting an NDA. After the FDA grants orphan drug designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. For example, we previously announced that TSC was granted orphan drug designation by the FDA for the treatment of GBM and metastatic brain cancer in July 2011 and in December 2012, respectively. However, orphan drug designation on its own does not convey any advantage in or shorten the duration of the regulatory review and approval process but may result in certain financial and marketing incentives if approved.
If a product that has orphan designation subsequently receives the first FDA approval for the disease or condition for which it has such designation, the product is entitled to orphan drug exclusivity, which means that the FDA may not approve any other NDA, thatapplications to market the same drug receives fivefor the same indication for seven years from the date of marketingsuch approval, except in limited circumstances, such as a showing of clinical superiority to the product with orphan exclusivity during whichby means of greater effectiveness, greater safety, or providing a major contribution to patient care, or in instances of drug supply issues. Competitors, however, may receive approval of either a different product for the same indication or the same product for a different indication. In the latter case, because health care professionals are free to prescribe products for off-label uses, the competitor’s product could be used for the orphan indication despite our orphan exclusivity. Orphan drug exclusivity could also block the approval of one of our products for seven years if a competitor obtains approval before we do for the same drug and same indication, as defined by the FDA, cannot receive any ANDAfor which we are seeking approval, or if our product is determined to be contained within the scope of a generic version of that drug. Certain changes to athe competitor’s product for the same indication or disease. If we pursue marketing approval for an indication broader than the orphan drug such as the addition of a new indication to the package insert, are associated with a three-year period of exclusivity during which the FDA cannot approve an ANDA for a generic drug that includes the change.
An ANDA may be submitted one year before NCE exclusivity expires if a Paragraph IV certification is filed. If there is no listed patent in the Orange Book, theredesignation we have received, we may not be a Paragraph IV certificationentitled to orphan drug exclusivity. Orphan drug status in the E.U. has similar, but not identical, requirements and thus, no ANDA may be filed before the expiration of the exclusivity period.benefits.
REMSExpedited Development and Review Programs
The FDA has a fast track program that is intended to expedite or facilitate the authorityprocess for reviewing new drugs that meet certain criteria. Specifically, new drugs are eligible for fast track designation if they are intended to treat a serious or life-threatening condition and preclinical or clinical data demonstrate the potential to address unmet medical needs for the condition. Fast track designation applies to both the product and the specific indication for which it is being studied. The sponsor can request the FDA to designate the product for fast track status any time before receiving NDA approval, but ideally no later than the pre-NDA meeting. Any product submitted to the FDA for marketing, including under a fast track program, may be eligible for other types of FDA programs intended to expedite development and review, such as priority review and accelerated approval. Any product is eligible for priority review if it treats a serious or life-threatening condition and, if approved, would provide a significant improvement in safety and effectiveness compared to available therapies. The FDA will attempt to direct additional resources to the evaluation of an application for a new drug designated for priority review in an effort to facilitate the review.
A product may also be eligible for accelerated approval, if it treats a serious or life-threatening condition and generally provides a meaningful advantage over available therapies. In addition, it must demonstrate an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit or on a clinical endpoint that can be measured earlier than IMM that is reasonably likely to predict an effect on IMM or other clinical benefit. As a condition of approval, the FDA may require that a sponsor of a drug receiving accelerated approval perform adequate and well-controlled post-marketing clinical trials. If the FDA concludes that a drug shown to be effective can be safely used only if distribution or use is restricted, it will require such post-marketing restrictions, as it deems necessary to assure safe use of the product. If the FDA determines that the conditions of approval are not being met, the FDA can withdraw its accelerated approval for such drug.
Additionally, a drug may be eligible for designation as a breakthrough therapy if the product is intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening condition and preliminary clinical evidence indicates that the product may demonstrate substantial improvement over currently approved therapies on one or more clinically significant endpoints. The benefits of breakthrough therapy designation include the same benefits as fast track designation, plus intensive guidance from the FDA to ensure an efficient drug development program.
Even if a product qualifies for one or more of these programs, the FDA may later decide that the product no longer meets the conditions for qualification or the time period for FDA review or approval may not be shortened. Furthermore, fast track designation, priority review, accelerated approval, and breakthrough therapy designation do not change the standards for approval, but may expedite the development or approval process.
Rare Pediatric Disease Priority Review Voucher Program
In 2012, Congress authorized the FDA to award priority review vouchers to sponsors of certain rare pediatric disease product applications. This program is designed to encourage development of new drug and biological products for prevention and treatment of certain rare pediatric diseases. Specifically, under this program, a sponsor who receives an approval for a drug or biologic for a “rare pediatric disease” may qualify for a voucher that can be redeemed to receive a priority review of a subsequent marketing application for a different product. The sponsor of a rare pediatric disease drug product receiving a priority review voucher may transfer (including by sale) the voucher to another sponsor. The voucher may be further transferred any number of times before the voucher is used, as long as the sponsor making the transfer has not yet submitted the application. The FDA may also revoke any priority review voucher if the rare pediatric disease drug for which the voucher was awarded is not marketed in the U.S. within one year following the date of approval.
For purposes of this program, a “rare pediatric disease” is a (a) serious or life-threatening disease in which the serious or life-threatening manifestations primarily affect individuals aged from birth to 18 years, including age groups often called neonates, infants, children, and adolescents; and (b) a rare disease or condition within the meaning of the Orphan Drug Act. On December 27, 2020, the Rare Pediatric Disease Priority Review Voucher Program was extended. Under the current statutory sunset provisions, after September 30, 2024, FDA may only award a voucher for an approved rare pediatric disease product application if the sponsor has rare pediatric disease designation for the drug, and that designation was granted by September 30, 2024. After September 30, 2026, FDA may not award any Rare Pediatric Disease Priority Review Voucher.
Disclosure of Clinical Trial Information
Sponsors of clinical trials of FDA-regulated products are required to register and disclose to the public certain clinical trial information, including information related to the product, patient population, phase of investigation, study sites, investigators, and other aspects of the trial design. Sponsors are also obligated to discuss the results of their clinical trials after completion. However, disclosure of the results of these trials can be delayed until the new product or new indication being studied has been approved as competitors may otherwise use this or other publicly-available information to gain knowledge regarding the progress of development programs and gain a competitive advantage.
Risk Evaluation and Mitigation Strategies and Other Post-Approval Requirements
As a condition of NDA approval, the FDA may require a REMS to help ensure that the safe usebenefits of the drug.drug’s continued approval outweigh the potential risks. In determining whether a REMS is necessary, the FDA must consider the size of the population likely to use the drug, the seriousness of the disease or condition to be treated, the expected benefit of the drug, the duration of treatment, the seriousness of known or potential adverse events, and whether the drug is a new molecular entity. If the FDA determines a REMS is necessary, the drug sponsor must agree to the REMS plan at the time of approval. A REMS may be required to include various elements, such as a medication guide or patient package insert, a communication plan to educate healthcare providers of the drug’s risks, limitations on who may prescribe or dispense the drug, or other measures that the FDA deems necessary to assure the safe use of the drug. REMS can include medication guides, communication plans for healthcare professionals, and ETASU. ETASUelements to assure safe use. Elements to assure safe use can include, but are not limited to, special training or certification for prescribing or dispensing, dispensing only under certain circumstances, special monitoring requirements, and the use of patient registries. In addition, the REMS must include a timetable to periodically assess the strategy. The FDA may also impose a REMS requirement on a drug already on the market if the FDA determines, based on new safety information, that a REMS is necessary to ensure that the drug’s benefits outweigh its risks. The requirement for a REMS can materially affect the potential market and profitability of a drug.drug product.
Patent Term Extension
After NDA approval, owners of relevant drug patents may apply for up to a five-year patent extension. The allowable patent term extension is calculated as half of the drug’s testing phase, the time between IND application and NDA submission, and all of the review phase, the time between NDA submission and approval, up to a maximum of five years. The time can be shortenedEven if the FDA determines that the applicant diddoes not pursue approval with due diligence. The total patent term after the extension may not exceed 14 years.
For patents that might expire during the application phase, the patent owner may request an interim patent extension. An interim patent extension increases the patent term by one year and may be renewed up to four times. For each interim patent extension granted, the post-approval patent extension is reduced by one year. The director of the USPTO must determine that approval of the drug covered by the patent for whichrequire a patent extension is being sought is likely. Interim patent extensions are not available for a drug for which an NDA has not been submitted.
Post-Approval Requirements
OnceREMS, once an NDA is approved, a product will be subject to certain post-approval requirements.regulations. For instance, the FDA closely regulates the post-approval marketing and promotion of drugs, including standards and regulations for direct-to-consumer advertising, off-label promotion, industry-sponsored scientific and educational activities, and promotional activities involving the internet. Drugs may be marketed only for the approved indications and in accordance with the provisions of the approved labeling.
Adverse event reporting and the submission of periodic reports isare also required following FDA approval of an NDA. The FDA also may require post-marketing testing, known as Phase 4 testing, REMS, and surveillance to monitor the effects of an approved product, or the FDA may place conditions on an approval that could restrict the distribution or use of the product. In addition, quality-control, drug manufacture, packaging and labeling procedures must continue to conform to cGMPs after approval. Drug manufacturers and certain of their subcontractors are required to register their establishments with the FDA and certain state agencies. Registration with the FDA subjects entities to periodic unannounced inspections by the FDA, during which the agency inspects manufacturing facilities to assess compliance with cGMPs. Accordingly, manufacturers must continue to expend time, money and effort in the areas of production and quality-control to maintain compliance with cGMPs. Regulatory authorities may withdraw product approvals or request product recalls if a company fails to comply with regulatory standards, if it encounters problems following initial marketing, or if previously unrecognized problems are subsequently discovered.
Pediatric Information
Under the Pediatric Research Equity Act, NDAs or supplements to NDAs must contain data to assess the safetyDrug Approval Process and effectiveness of the drug for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the drug is safe and effective. The FDA may grant full or partial waivers, or deferrals, for submission of data.
The Best Pharmaceuticals for Children Act (“BPCA”) provides NDA holders a six-month extension of any exclusivity, patent or non-patent, for a drug if certain conditions are met. Conditions for exclusivity include the FDA’s determination that information relating to the use of a new drug in the pediatric population may produce health benefits in that population, the FDA making a written request for pediatric studies and the applicant agreeing to perform, and reporting on, the requested studies within the statutory timeframe. Applications under the BPCA are treated as priority applications, with all of the benefits that designation confers.
The Orphan Drug Act of 1983
The Orphan Drug Act of 1983 was designed to provide financial incentives for, and to reduce the costs associated with, developing drugs for rare diseases and disorders. A “rare disease or disorder” is defined by the Orphan Drug Act of 1983 as affecting fewer than 200,000 Americans at the time of designation or one for which “there is no reasonable expectation that the cost of developing and making available in the United States…will be recovered from sales in the United States.” A sponsor must request that the FDA designate a drug currently under development for a “rare disease or condition” as an orphan drug, and if the FDA agrees that the drug and indication meet the criteria set forth in the Orphan Drug Act of 1983, certain financial and marketing incentives become available. As mentioned previously, we have received Orphan Drug designations for GBM and metastatic brain cancer.
Disclosure of Clinical Trial Information
Sponsors of clinical trials of FDA-regulated products, including drugs, are required to register and disclose certain clinical trial information. Information related to the product, patient population, phase of investigation, study sites and investigators and other aspects of the clinical trial is then made public as part of the registration. Sponsors are also obligated to discuss the results of their clinical trials after completion. Disclosure of the results of these trials can be delayed until the new product or new indication being studied has been approved. Competitors may use this publicly-available information to gain knowledge regarding the progress of development programs.
RegulationOther Regulations Outside of the United StatesU.S.
In addition to regulations in the United States,U.S., we are and will be subject to the regulations of other countries governingin which we conduct any of our clinical trials andor engage in commercial sales andor other distribution of our product candidates.products, if approved. Whether or not we obtain FDA approval for conduct of a clinical trial or distribution of a product, we must obtain approval byfrom the comparablecompetent regulatory authority of any country or economic area in which we would seek to commence a clinical trial or market products. For example, conduct of the COVID Trial in Bucharest, Romania, required certain approvals from regulatory authorities of countries outside ofin Romania and the United States before we can commence clinical trials in such countries and approval of the regulators of such countries or economic areas, such as the European Union, before we may market products in those countries or areas.E.U. Certain countries outside of the United States have a process similar to the FDA’s thatIND process which requires the submission of a clinical trial application (“CTA”) much like the IND prior to the commencement of human clinical trials. In the European Union,E.U., for example, a CTA must be submitted to each country’s national health authority and an independent ethics committee, much like the FDA andwhich operates similar to an IRB respectively.under U.S. regulations. Once the CTA is approved in accordance with a country’s requirements, the clinical trial development may proceed.proceed in the applicable country. The approval process and requirements governing the conduct of clinical trials, product licensing, pricing, and reimbursement vary greatly from place to place, and the time may be longer or shorter than that required for FDA approval.
Under European Union regulatory systems,In particular, in the E.U. a company may submit marketing authorization applications (comparable to an NDA submission in the US to the FDA) under either under a centralized or decentralized procedure. The centralized procedure, which is compulsory for medicines produced by biotechnology or those medicines intended to treat AIDS, cancer, neurodegenerative disorders, or diabetes and is optional for those medicines which are highly innovative, provides for the grant of a single marketing authorization that is valid for all European UnionE.U. member states. The decentralized procedure provides for mutual recognition of national approval decisions. Under this decentralized procedure, the holder of a national marketing authorization in any E.U. member state may submit an application to the remaining member states. Within ninety (90) days of receiving the applications and assessments report, each member state must decide whether to recognize approval. If a member state does not recognize the marketing authorization, the disputed points are eventually referred to the European Commission, whose decision is binding on all member states.
Anti-Kickback and False Claims Laws
In addition The E.U. also has procedures similar to FDA restrictions on marketing of pharmaceutical products, several other types of state and federal laws have been applied to restrict certain marketing practices in the pharmaceutical industry. These laws include, among others, anti-kickback statutes, false claims statutes and other statutes pertaining to healthcare fraud and abuse. The federal healthcare program anti-kickback statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving remuneration to induce, or in return for, purchasing, leasing, ordering or arranging for the purchase, lease or order of any healthcare item or service reimbursable under Medicare, Medicaid or other federally financed healthcare programs. The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act of 2010, collectively referred to as the Affordable Care Act amended the intent elementthose of the federal anti-kickback statute so thatFDA pursuant to which a person company may obtain marketing exclusivity for a product for up to 11 years and/or entity no longer needsorphan drug designation and related exclusivity for up to have actual knowledge of the statute or specific intent to violate it. This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on the one hand and prescribers, purchasers and formulary managers on the other. Although there are a number of statutory exceptions and regulatory safe harbors protecting certain common activities from prosecution or other regulatory sanctions, the exceptions and safe harbors are drawn narrowly, and practices that involve remuneration intended to induce prescribing, purchases or recommendations may be subject to scrutiny if they do not qualify for an exception or safe harbor.
Federal false claims laws prohibit any person or entity from, among other things, knowingly presenting, or causing to be presented, a false claim for payment to the federal government, or knowingly making, or causing to be made, a false statement to have a false claim paid. This includes claims made to programs where the federal government reimburses, such as Medicaid,ten years, as well as programs where the federal government is a direct purchaser, such as when it purchases off the Federal Supply Schedule. Pharmaceutical and other healthcare companies have been prosecuted under these laws for, among other things, allegedly inflating drug prices they reportexpedited approval pathways available to pricing services, which in turn were used by the government to set Medicare and Medicaid reimbursement rates, and for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. In addition, certain marketing practices, including off-label promotion, may also violate federal false claims laws. Additionally, Affordable Care Act amended the federal healthcare program anti-kickback statute such that a violation of that statute can serve as a basis for liability under certain federal false claims laws.drugs.
The majority of states also have statutes or regulations similar to the federal anti-kickback law and false claims laws, which apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor.
Other federal statutes pertaining to healthcare fraud and abuse include the civil monetary penalties statute, which prohibits, among other things, the offer or payment of remuneration to a Medicaid or Medicare beneficiary that the offerer or payor knows or should know is likely to influence the beneficiary to order or receive a reimbursable item or service from a particular supplier, and the healthcare fraud and false statements statutes, which prohibit, among other things, knowingly and willfully executing or attempting to execute a scheme to defraud any healthcare benefit program or obtain by means of false or fraudulent pretenses, representations, or promises any money or property owned by or under the control of any healthcare benefit program in connection with the delivery of or payment for healthcare benefits, items, or services.
Violations of these federal healthcare fraud and abuse laws are punishable by imprisonment, criminal fines, civil monetary penalties and exclusion from participation in federal healthcare programs.
It is uncertain whether there will be any legislation that will replace or amend the Affordable Care Act. In addition, the Trump Administration has and will appoint many new secretaries, directors and the like into positions of authority in the U.S. Federal government dealing with the pharmaceutical and healthcare industries that may potentially have a negative impact on the prices and regulatory pathways for certain pharmaceuticals and healthcare products developed by the Company to market and sell its products in the U.S.
Other Federal and State Regulatory Requirements
The Centers for Medicare & Medicaid Services (“CMS”) has issued a final rule pursuant to Affordable Care Act that requires certain manufacturers of prescription drugs to annually collect and report information on payments or transfers of value to physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. Manufacturers were required to begin collecting information on August 1, 2013, with the first reports due March 31, 2014. The reported data is posted in searchable form on a public website. Failure to submit required information may result in civil monetary penalties.
In addition, several states now require prescription drug companies to report expenses relating to the marketing and promotion of drug products and to report gifts and payments to individual healthcare practitioners and entities in these states. Other states prohibit various other marketing-related activities. Still other states require the posting of information relating to clinical studies and their outcomes. In addition, California, Connecticut, Nevada and Massachusetts require pharmaceutical companies to implement compliance programs and marketing codes. Several additional states are considering similar proposals. Some of the state laws are broader in scope than federal laws. Compliance with these laws is difficult and time consuming, and companies that do not comply with these state laws face civil penalties.
Reimbursement
Sales of any of our product candidates that are approved will depend, in part, on the extent to which the costs of our approved products will be covered by third-party payors, such as government health programs, commercial insurance and managed healthcare organizations. These third-party payors are increasingly challenging the prices charged for medical products and services. Additionally, the containment of healthcare costs has become a priority of federal and state governments and the prices of drugs have been a focus in this effort. The U.S. government, state legislatures and foreign governments have shown significant interest in implementing cost-containment programs, including price controls, restrictions on reimbursement and requirements for substitution of generic products. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit our expected net revenue and results. If any of our products are approved and these third-party payors do not consider our approved products to be cost-effective compared to other therapies, they may not cover our approved products as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow us to sell our approved products on a profitable basis.
The Medicare Prescription Drug, Improvement, and Modernization Act of 2003 (“MMA”), imposed new requirements for the distribution and pricing of prescription drugs for Medicare beneficiaries. Under Part D, Medicare beneficiaries may enroll in prescription drug plans offered by private entities which will provide coverage of outpatient prescription drugs. Part D plans include both stand-alone prescription drug benefit plans and prescription drug coverage as a supplement to Medicare Advantage plans. Unlike Medicare Part A and B, Part D coverage is not standardized. Part D prescription drug plan sponsors are not required to pay for all covered Part D drugs, and each drug plan can develop its own drug formulary that identifies which drugs it will cover and at what tier or level. However, Part D prescription drug formularies must include drugs within each therapeutic category and class of covered Part D drugs, though not necessarily all the drugs in each category or class. Any formulary used by a Part D prescription drug plan must be developed and reviewed by a pharmacy and therapeutic committee. Government payment for some of the costs of prescription drugs may increase demand for our products for which we receive marketing approval. However, any negotiated prices for our approved products covered by a Part D prescription drug plan will likely be lower than the prices we might otherwise obtain. Moreover, while the MMA applies only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own payment rates. Any reduction in payment that results from the MMA may result in a similar reduction in payments from non-governmental payors.
The American Recovery and Reinvestment Act of 2009 provides funding for the federal government to compare the effectiveness of different treatments for the same illness. A plan for the research will be developed by the Department of Health and Human Services, the Agency for Healthcare Research and Quality and the National Institutes for Health, and periodic reports on the status of the research and related expenditures will be made to the U.S. Congress. Although the results of the comparative effectiveness studies are not intended to mandate coverage policies for public or private payors, it is not clear what effect, if any, the research will have on the sales of any approved product, if any such product or the condition that it is intended to treat is the subject of a study. It is also possible that comparative effectiveness research demonstrating benefits in a competitor’s product could adversely affect the sales of our product candidates. If third-party payors do not consider our approved products to be cost-effective compared to other available therapies, they may not cover our approved products as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow us to sell our approved products on a profitable basis.
The Affordable Care Act was enacted in March 2010, which includes measures to significantly change the way healthcare is financed by both governmental and private insurers. Among the provisions of the Affordable Care Act of importance to the pharmaceutical and biotechnology industry are the following:
| ●
| an annual, nondeductible fee on any entity that manufactures or imports certain branded prescription drugs and biologic agents, apportioned among these entities according to their market share in certain government healthcare programs, that began in 2011;
|
| ●
| an increase in the rebates a manufacturer must pay under the Medicaid Drug Rebate Program to 23.1% and 13% of the average manufacturer price for branded and generic drugs, respectively;
|
| ●
| a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50% point-of-sale discounts to negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D;
|
| ●
| extension of manufacturers’ Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations;
|
| ●
| expansion of eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to additional individuals and by adding new mandatory eligibility categories for certain individuals with income at or below 133% of the Federal Poverty Level, thereby potentially increasing manufacturers’ Medicaid rebate liability;
|
| ●
| expansion of the entities eligible for discounts under the Public Health Service pharmaceutical pricing program;
|
| ●
| expansion of healthcare fraud and abuse laws, including the False Claims Act and the Anti-Kickback Statute, new government investigative powers, and enhanced penalties for noncompliance;
|
| ●
| a licensure framework for follow-on biologic products;
|
| ●
| a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research;
|
| ●
| new requirements under the federal Open Payments program for drug manufacturers to report information related to payments and other transfers of value made to physicians and other healthcare providers as well as ownership or investment interests held by physicians and their immediate family members;
|
| ●
| a new requirement to annually report drug samples that manufacturers and distributors provide to physicians, effective April 1, 2012;
|
| ●
| creation of the Independent Payment Advisory Board which, beginning in 2014, will have authority to recommend certain changes to the Medicare program that could result in reduced payments for prescription drugs and those recommendations could have the effect of law even if Congress does not act on the recommendations; and
|
| ●
| establishment of a Center for Medicare Innovation at CMS to test innovative payment and service delivery models to lower Medicare and Medicaid spending, potentially including prescription drug spending that began on January 1, 2011.
|
In addition, in some non-U.S. jurisdictions, the proposed pricing for a product candidate must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country. For example, the European UnionE.U. provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. A member state may approve a specific price for the medicinal product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any of our product candidates. Historically, product candidates launched in the European UnionE.U. do not follow price structures of the United StatesU.S. and generally tend to be significantly lower.
It is uncertain whether thereCertain Other Legislation and Regulations
Current Healthcare Laws and Regulations
Healthcare providers, physicians, and third party payors, including governmental payors such as Medicare and Medicaid, will beplay a significant role in the recommendation and prescription of any products for which we obtain marketing approval. Our future arrangements with third party payors, healthcare providers, and physicians may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we market, sell, and distribute any drugs for which we obtain marketing approval.
These laws include, without limitation, state and federal anti-kickback, false claims, physician transparency, and patient data privacy and security laws and regulations, and other federal, state, and local regulations and legislation that will replace or amendimpacting the Affordable Care Act.pharmaceutical and biopharmaceutical industries, including but not limited to those described below.
| • | Health Insurance Portability and Accountability Act - HIPAA imposes criminal and civil liability for, among other things, knowingly and willfully executing a scheme, or attempting to execute a scheme, to defraud any healthcare benefit program, including private payors, or falsifying, concealing, or covering up a material fact or making any materially false statements in connection with the delivery of or payment for healthcare benefits, items or services. Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. HIPAA, as amended by HITECH and their respective implementing regulations, imposes, among other things, specified requirements on covered entities and their business associates relating to the privacy and security of individually identifiable health information, including mandatory contractual terms and required implementation of technical safeguards of such information. HITECH also created new tiers of civil monetary penalties, amended HIPAA to make civil and criminal penalties directly applicable to business associates, and gave state Attorneys General new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorneys’ fees and costs associated with pursuing federal civil actions. |
| • | Affordable Care Act - The ACA was enacted in March 2010 and included measures intended to significantly change the way healthcare is financed in the U.S. by both governmental and private insurers which have and may continue to impact the pharmaceutical and biopharmaceutical industries, including expanded Medicare and Medicaid benefits, expansion of healthcare fraud and abuse laws, establishment of the CMS, annual reporting requirements for manufacturers and distributors. Since its enactment, there have been numerous judicial, administrative, executive, and legislative challenges to certain aspects of the Affordable Care Act, and we expect there will be additional challenges and amendments to the ACA in the future. In addition, subsequent legislation, including the Budget Control Act of 2011, American Taxpayer Relief Act of 2012, and Coronavirus Aid, Relief, and Economic Security Act of 2020, has limited and supplemented various provisions of the Affordable Care Act. While we cannot predict what effect further changes to the Affordable Care Act would have on our business, the Affordable Care Act is likely to continue to impact the regulatory regime to which we are subject for the foreseeable future, and we cannot predict the ultimate content, timing, or effect of any healthcare reform legislation or the impact of potential legislation on us. |
21st Century Cures Act
| • | 21st Century Cures Act - The 21st Century Cures Act, which the U.S. House of Representatives passed in July 2015 and President Obama signed into law in December 2016, provides for a wide range of reforms that may impact our business, such as broadening the types of data required to support drug approval, extending protections from genetic competition, accelerating approval of breakthrough therapies, expanding the orphan drug product and compassionate use programs, and clarifying how manufacturers communicate about their products. |
| • | Anti-Kickback Laws - The federal Anti-Kickback Statute makes it illegal for any person, including a prescription drug manufacturer (or a party acting on its behalf), to knowingly and willfully solicit, receive, offer, or pay any remuneration, directly or indirectly, in cash or in kind, that is intended to induce or reward referrals, including the purchase, recommendation, order or prescription of a particular drug or any other good or service, for which payment may be made under a federal healthcare program, such as Medicare or Medicaid. Violations of this law are punishable by up to five years in prison, criminal fines, administrative civil money penalties, and exclusion from participation in federal healthcare programs. In addition, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it. |
| • | False Claims Laws - The federal False Claims Act imposes civil penalties, including through civil whistleblower or qui tam actions, against individuals or entities (including manufacturers) for, among other things, knowingly presenting, or causing to be presented, false or fraudulent claims for payment by a federal healthcare program or making a false statement or record material to payment of a false claim or avoiding, decreasing, or concealing an obligation to pay money to the federal government. The government may deem manufacturers to have “caused” the submission of false or fraudulent claims by, for example, providing inaccurate billing or coding information to customers or promoting a product off-label. |
| • | Medicare Prescription Drug, Improvement, and Modernization Act - The Medicare Prescription Drug, Improvement, and Modernization Act of 2003 imposes requirements on the distribution and pricing of prescription drugs for Medicare beneficiaries. Under Part D, Medicare beneficiaries may enroll in prescription drug plans offered by private entities which will provide coverage of outpatient prescription drugs. Part D plans include both stand-alone prescription drug benefit plans and prescription drug coverage as a supplement to Medicare Advantage plans, but plan sponsors are not required to pay for all covered Part D drugs, and each drug plan can develop its own drug formulary that identifies which drugs it will cover and at what tier or level. However, Part D prescription drug formularies must include drugs within each therapeutic category and class of covered Part D drugs, though not necessarily all the drugs in each category or class. Any reduction in payment that results from the these or similar regulations may result in a similar reduction in payments from non-governmental payors. |
| • | The Physician Payments Sunshine Act - The Sunshine Act, enacted as part of the Affordable Care Act,, imposed new annual reporting requirements for certain manufacturers of drugs, devices, biologics, and medical supplies for which payment is available under Medicare, Medicaid, or the Children’s Health Insurance Program, for certain payments and “transfers of value” provided to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. Effective January 1, 2022, these reporting obligations will extend to include transfers of value made to certain non-physician providers such as physician assistants and nurse practitioners. |
| • | State, Local, and Non-U.S. Legislation and Regulations - In addition, to the legislation summarized above, we may also be subject now or in the future to analogous state, local, and non-U.S. laws and regulations, such as state anti-kickback and false claims laws, which may be broader in scope and apply regardless of payor. Such laws are enforced by various state agencies and through private actions. For example, some state laws require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant federal government compliance guidance, require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers, and restrict marketing practices or require disclosure of marketing expenditures. Certain state and foreign laws also govern the privacy and security of health information in some circumstances and these data privacy and security laws may differ from both HIPAA and each other in significant ways, which would potentially increase our compliance burden. |
The 21st Century Cures Act,scope and enforcement of each of these laws is uncertain and subject to rapid change in the current environment of healthcare reform. Federal and state enforcement bodies have recently increased their scrutiny of interactions between healthcare companies and healthcare providers, which has led to a number of investigations, prosecutions, convictions, and settlements in the U.S. Househealthcare industry. If our operations are found to be in violation of Representatives passed in July 2015any of these laws or any other related governmental regulations that may apply to us, we may be subject to significant civil, criminal, and President Obama signed into law in December 2016, provides for a wide rangeadministrative penalties, damages, fines, imprisonment, disgorgement, exclusion of reforms,drugs from government funded healthcare programs, such as broadeningMedicare and Medicaid, reputational harm, additional oversight, and reporting obligations if we become subject to a corporate integrity agreement or similar settlement to resolve allegations of non-compliance with these laws and the typescurtailment or restructuring of data required to support drug approval, extending protections from genetic competition, accelerating approval of breakthrough therapies, expanding the orphan drug product program, and clarifying how manufacturers communicate about their products.our operations.
ManufacturingFuture Healthcare Laws and SupplyRegulations
We do notIn the United States and foreign jurisdictions, there have any facilities suitable for manufacturing onbeen a commercial scale anynumber of proposed changes regarding the healthcare system and its regulation that could prevent or delay marketing approval of our product candidates, nor do we haverestrict or regulate post-approval activities, and affect our ability to profitably sell any experience in volume manufacturing. We currently use third-party cGMP contract manufacturing organizations (“CMOs”) to manufacture our product candidates for our preclinical studies and clinical trials and intend to continue doing so in the future in accordance with FDA and other appropriate regulations. We anticipate that these CMOs will have capacity to support commercial scale, but we do not have any formal agreements at this time with any of these CMOs to cover commercial production. We also may elect to pursue other CMOs for manufacturing supplies for later-stage trials and for commercialization. We currently have no plans to establish a manufacturing capability, but rather plan to continue to rely on third-party cGMP manufacturers for any future trials and commercialization of our product candidates for which we retain manufacturing responsibility.obtain marketing approval. We expect that further implementation of current laws, as well as other healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on the price that we, or any strategic collaborators, may receive for any approved products. Further, there has recently been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products, which have resulted in several recent Congressional inquiries and proposed bills designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for products.
SalesU.S. Environmental, Health, and MarketingSafety Laws
We currently have no salesare subject to numerous environmental, health, and marketing personnelsafety laws and regulations. From time to sell anytime and in the future, our operations may involve the use of hazardous and flammable materials, including chemicals and biological materials, and may also produce hazardous waste products. Even if we contract with third parties for the disposal of these materials and waste products, we cannot completely eliminate the risk of contamination or injury resulting from these materials. In the event of contamination or injury resulting from the use or disposal of our product candidates on a commercial basis ifhazardous materials, we could be held liable for any resulting damages, and whenany liability could exceed our product candidates received required regulatory approvals. Ifresources. We also could incur significant costs associated with civil or criminal fines and whenpenalties for failure to comply with such laws and regulations.
In addition, we are readymay incur substantial costs in order to commercially launch a product, we will either contractcomply with current or hire qualified salesfuture environmental, health, and marketing personnelsafety laws and regulations. Current or seek a joint marketing partnerfuture environmental laws and regulations may impair our research, development or licenseeproduction efforts. In addition, failure to assist uscomply with this function.these laws and regulations may result in substantial fines, penalties or other sanctions.
EmployeesPublic Company Status
As a public company, we incur significant legal, accounting and other expenses to comply with the reporting requirements of the Exchange Act and applicable requirements of SOX and the Dodd-Frank Act, as well as rules and regulations subsequently implemented by the SEC and Nasdaq, including the establishment and maintenance of effective disclosure and financial controls, changes in corporate governance practices and required filing of annual, quarterly and current reports with respect to our business and operating results. These requirements increase our legal and financial compliance costs and make some activities more time-consuming and costly. In addition, our management and other personnel devote significant time and attention to these public company requirements.
Our People and Human Capital Resources
As of December 31, 2016,2021, we had 1016 full-time employees, including 4 in product development and 6 in management or administrative positions.up from 12 employees as of December 31, 2020. We also have engagedregularly work with several independent consultants and other contract organizations to support our organization.business and we regularly evaluate additional talent to help support our product manufacturing, development, financial, and other capabilities.
Diversity and Inclusion
We believe that an inclusive culture is required to understand and develop products that benefit all patients. By embracing differences, we aim to foster an environment of respect and trust in an effort to facilitate creativity, spark passion, and help us achieve better outcomes for all those who work at and with Diffusion. We are committed to creating and maintaining a workplace free from discrimination or harassment, including on the basis of any class protected by applicable law, and our recruitment, hiring, development, training, compensation, and advancement practices are based on qualifications, performance, skills, and experience without regard to gender, race, or ethnicity. Our management team and employees are expected to exhibit and promote honest, ethical, and respectful conduct in the workplace, including adhering to the standards for appropriate behavior set forth in our code of conduct.
Compensation and Benefits
We operate in a highly competitive environment for human capital, particularly as we seek to attract and retain talent with relevant experience in the biotechnology and pharmaceutical sectors. Therefore, we strive to provide a total rewards package to our employees that is competitive with our peer companies, including competitive pay, a comprehensive healthcare benefits package (including an 80% employer contribution to family medical coverage), 25 days of paid leave, a company-sponsored 401(k) savings plan, short-term and long-term disability, and other benefits, as well as remote working and flexible work schedules. We also offer every full-time employee the benefit of equity ownership in Diffusion through stock option grants. We believe these grants both help promote alignment between our employees and our stockholders and provide retention benefits, as the awards generally vest over a three-year period.
We do not have any employees that are represented by a labor union or that have entered into a collective bargaining agreement with the Company.
Safety and Wellness
At Diffusion, we believe that health matters to everyone, and the safety health, and wellness of our employees is one of our top priorities. We are committed to developing and fostering a work environment that is safe, professional, and promotes teamwork, diversity, and trust in order to afford all of our employees the opportunity to contribute to the best of their abilities.
Over the past two years, in response to the COVID-19 pandemic, we have taken certain measures and responded to changes in our operational needs, including actions designed to provide a safe work environment for our employees. These actions included investing in technology solutions to support increased work-from-home capabilities, shifting work schedules to reduce the number of people present in our offices, requiring mask wearing and social distancing, making hand sanitizer readily available, and other measures intended to comply with health and safety protocols as required by federal, state, and local governmental agencies, as well as guidance from the U.S. Centers for Disease Control and Prevention and similar public health authorities.
Employee Development and Training
We believe our people are among our most important assets. We focus on attracting, retaining, and cultivating talented individuals and believe in investing in our people to help them grow. Employees are encouraged to attend scientific, clinical, technological, and other relevant meetings and conferences and we strive to provide employees access to a broad set of internal resources intended to help them be successful, including a variety of training and educational materials. During 2021, we intend to implement a new, comprehensive employee evaluation program tied to the achievement of individual, team, and company goals to help further support, retain, and develop our people and further promote alignment of interests between our employees, future target customers, and our stockholders.
Directors and Executive Officers
The table below setsinformation set forth asin "Part III — Item 10 — Directors, Officers, and Corporate Governance," of March 15, 2017, certain information concerning our current directors and executive officers. No family relationships exist among any of our directors or executive officers.
Name
| | Age
| | Position with Diffusion
| |
David G. Kalergis
| | 68
| | Chairman and Chief Executive Officer
| |
John L. Gainer, Ph.D.
| | 78
| | Director and Chief Scientific Officer
| |
David R. Jones, M.D.
| | 53
| | Chief Medical Officer
| |
Ben L. Shealy
| | 58
| | Senior Vice President – Finance, Treasurer and Secretary
| |
Thomas Byrne
| | 59
| | General Counsel
| |
Isaac Blech
| | 67
| | Vice Chairman
| |
Robert Adams
| | 66
| | Director
| |
Mark T. Giles
| | 62
| | Director
| |
Alan Levin
| | 54
| | Director
| |
this Annual Report is incorporated herein by reference.
The following is a biographical summary of the experience of our directors and executive officers:Other Information About Our Company
David G. Kalergis – Mr. Kalergis has served as our Chairman of the Board and Chief Executive Officer since the completion of the Merger in January 2016. Mr. Kalergis, along with Dr. Gainer is the Company’s co-founder and has served as a director of Diffusion LLC since its inception in 2001 and as its Chief Executive Officer since 2004. Prior to joining the Company, Mr. Kalergis held positions with the University of Virginia; as the general counsel and director of business development for Pharmaceutical Research Associates, Inc., a pharmaceutical contract research organization; as an intelligence analyst for the U.S. Government; and with the law firm Dewey, Ballantine, Bushby, Palmer & Wood, practicing in the areas of corporate finance, public offerings and mergers and acquisitions. In addition, from July 1998 until May 2012, Mr. Kalergis served on the board of directors and audit committee of Virginia National Bank. Mr. Kalergis received a B.A. in psychology, as well as an M.B.A. and J.D., from the University of Virginia, and is a graduate of the Harvard Business School’s Leadership and Strategy in the Pharmaceutical and Biotechnology Industry program.
John L. Gainer, Ph.D. – Dr. Gainer has served as a director and as our Chief Scientific Officer since the completion of the Merger in January 2016. Dr. Gainer, along with Mr. Kalergis, is the Company’s co-founder and has served as one of Diffusions LLC’s directors and as its Chief Scientific Officer since its inception in 2001. From 1966 until his retirement in 2005, Dr. Gainer was a professor of chemical engineering at the University of Virginia. During his career, Dr. Gainer authored more than 100 scientific journal articles, including more than 30 published in medical journals, and spent two sabbaticals investigating drug actions and related research at Karolinska Institute in Stockholm and the laboratory of a major pharmaceutical company. He has been a member of the International Society for Oxygen Transport in Tissues since its inception in 1973. Dr. Gainer received a BSChE from West Virginia University, a MS in chemical engineering from the Massachusetts Institute of Technology, and a Ph.D. in chemical engineering from the University of Delaware.
David R. Jones, M.D. – Dr. Jones serves as our Chief Medical Officer. Dr. Jones was appointed Chief Medical Officer in connection with the completion of the Merger, and has served as Diffusion LLC’s Chief Medical Officer since September 2012. In addition to serving as the Company’s Chief Medical Officer, Dr. Jones is also the Fiona and Stanley Druckenmiller Endowed Professor for Lung Cancer Research and Chief of Thoracic Surgery at Memorial Sloan-Kettering Cancer Center in New York, NY, a position he has held since 2013. From 2007 to 2013, Dr. Jones was Professor of Surgery and Division Chief of Thoracic & Cardiovascular Surgery at the University of Virginia. In addition to his clinical practice, Dr. Jones has published more than 220 scientific articles, authored or co-authored over 35 book chapters, and served as Principal Investigator or Co-Investigator of over 30 clinical trials. Dr. Jones received an undergraduate degree in chemistry and an M.D. from West Virginia University, and completed his thoracic surgery residency and postdoctoral research fellowship in molecular oncology at the University of North Carolina - Chapel Hill.
Ben L. Shealy – Mr. Shealy serves as our Senior Vice President – Finance and Treasurer. Mr. Shealy was appointed Senior Vice President – Finance and Treasurer in connection with the completion of the Merger. Mr. Shealy has served as Diffusion LLC’s Senior Vice President – Finance and Treasurer, since December 2015, and prior to that had served as Diffusion LLC’s Chief Financial Officer since 2004. Prior to joining the Company, Mr. Shealy spent more than 20 years in the financial management industry focusing on private and public corporate financings, including serving as the Vice President of REBAR Inc. and positions with Donaldson, Lufkin & Jenrette, Prudential-Bache Capital Funding and the John Hancock Derivatives Group. Mr. Shealy received a B.S. in accounting from San Jose State University, an M.B.A. in finance from Columbia University and is a CFA Charter holder.
Thomas Byrne– Mr. Byrne was appointed as our General Counsel in connection with the completion of the Merger. Mr. Byrne served as a director of Diffusion LLC from 2001 to January 2016, as a director of the Company from January 2016 until April 2016 and has served as Diffusion LLC’s Secretary and Director of Patent Strategy since 2007. Prior to joining Diffusion LLC, Mr. Byrne served in in-house counsel positions at both Genentech Inc. and Amgen Inc., where he co-invented the erythropoiesis stimulating agent darbepoietin alpha (Aranesp®). From 1992 to 2000, he was a partner in the intellectual property law firm of Nixon and Vanderhye P.C. Mr. Byrne also currently acts as a consultant for start-up biotechnology companies on intellectual property, contract and business issues. He received a B.S. in chemical engineering and nuclear engineering, as well as a J.D., from the University of Virginia, and an M.S. in biochemical engineering from Yale University.
Isaac Blech –Mr. Blech has served as a director since August 2016. Mr. Blech’s current roles include serving as vice chairman of the board of directors of Edge Therapeutics, Inc., a clinical-stage biotechnology company, founder and director of Cerecor, Inc., a CNS company, director of ContraFect Corporation, an infectious disease company, director of Medgenics, Inc., a biotechnology company, and vice chairman of InspireMD, a stent company. He is vice chairman of the boards of directors of Centrexion Corporation, a private company which is developing new modalities of pain control, Regenovation, Inc., a private company developing new ways to regenerate human tissue, X4 Pharmaceuticals, a private cancer immunology company, Sapience Therapeutics, a private oncology company, Aridis Pharmaceuticals, a private company with a product to treat pneumonia, WaveGuide Corporation, a private company developing the world’s smallest NMR machine, SpendSmart Networks, Inc., a private electronic rewards company, and root9B Technologies, a private cyber security company. Over the past 35 years, Mr. Blech has founded and served on the boards of directors of companies which have produced major advances in a broad array of diseases, including the diagnosis of chlamydia, herpes, syphilis and HIV and the treatment of cystic fibrosis, sexual dysfunction, multiple myeloma and brain cancer. Mr. Blech was previously a member of the board of directors of the Company prior to the Merger.
Robert Adams – Mr. Adams has served as a director since the completion of the Merger in January 2016 and as a director of Diffusion LLC since 2002. Prior to his retirement in 2015, Mr. Adams was a partner in the intellectual property law firm of Nixon & Vanderhye P.C, where he had practiced for over 25 years, focusing on patent litigation and international patent licensing and negotiations. During that time period, Mr. Adams was lead litigation counsel in more than 50 major intellectual property lawsuits, where he directly handled, for example, all intellectual property valuations and settlements on behalf of his U.S. and foreign clients. Moreover, Mr. Adams served as the head negotiator for a well-known Japanese consumer products company for 15 years in various complicated licensing situations. Those negotiations typically involved the cross-licensing of up to hundreds of U.S. and foreign patent rights. His lead licensing activities on behalf of that client included, among other things, multi-year negotiations with Texas Instruments, Advanced Micro Devices and Freescale. Mr. Adams received a B.A. from the University of Maryland and a J.D. from George Washington University (with honors), and is a member of the Virginia State Bar.
Mark T. Giles – Mr. Giles has served as a director since the completion of the Merger in January 2016 and as a director of Diffusion LLC since 2008. Since July 2007, Mr. Giles has been the sole managing member of Panda Holdings, LLC, which engages in the investment and management of private capital. Prior to joining Panda Holdings, Mr. Giles served as the Chief Executive Officer of Virginia National Bank from July 1998 until June 2007 and thereafter continued to serve as the non-executive Chairman until December 2011. Prior to joining Virginia National Bank, Mr. Giles also served as the president of two publicly traded bank holding companies and subsidiary banks in Texas and practiced law with the banking group of a Houston law firm. He chairs the boards of Relay Foods, Inc. and Expedition Trust Company. He also serves on the boards of The Paramount Theater Foundation, The Paramount Theater Operating Company and the Computers4Kids Program. Mr. Giles received a B.S. from the McIntire School of Commerce at the University of Virginia and a J.D. from the University of Virginia School of Law.
Alan Levin – Mr. Levin has served as a director since the completion of the Merger in January 2016 and as a director of Diffusion LLC since June 2015. He previously served as Executive Vice President and Chief Financial Officer of Endo Health Solutions Inc. (“Endo”), a global specialty healthcare company, from June 2009 until his retirement in September 2013. Prior to joining Endo, Mr. Levin worked with Texas Pacific Group, a leading private equity firm, and one of their start-up investments. Before that, he was Senior Vice President & Chief Financial Officer of Pfizer, Inc. where he worked for 20 years in a variety of executive positions of increasing responsibility, including Treasurer and Senior Vice President of Finance & Strategic Management for the company’s research and development organization. Mr. Levin received a bachelor’s degree from Princeton University and a master’s degree from New York University’s Stern School of Business. Mr. Levin is a certified public accountant. He is a member of the board of directors of Aceto Corp, a NASDAQ-traded company specialized in generics and pharmaceutical intermediate products. He is also a member of the Advisory Board of Auven Therapeutics, a private equity fund; and the Critical Path Institute, a nonprofit collaboration between the Food and Drug Administration and pharmaceutical industry participants focused on streamlining and accelerating the development and regulatory pathways for innovative medicines.
Corporate Information and History
We were originally incorporated under the laws of the State of Nevada on January 10, 1995 and reincorporated under the laws of the State of Delaware on June 18, 2015 under the name, “RestorGenex Corporation.” On January 8, 2016, we completed the merger of our wholly owned subsidiary with and into Diffusion LLC, which was treated as a "reverse acquisition" under GAAP pursuant to which Diffusion LLC's historical results of operations replaced the Company's for all periods prior to the merger. Immediately following the closing of the merger, we changed our name from "RestorGenex Corporation" to "Diffusion Pharmaceuticals Inc."
On May 6, 2021, we received a written notice from the Nasdaq Staff indicating that we were not in compliance with the Bid Price Rule because the bid price for our common stock had closed below $1.00 per share for the previous 30 consecutive business days. On November 3, 2021, after failing to regain compliance with the Bid Price Rule within 180 days of our receipt of the first notice, we received an additional notice from the Staff providing that, although the Company had not regained compliance with the Bid Price Rule by the previously stated deadline, in accordance with Nasdaq Listing Rule 5810(c)(3)(A), the Company would be granted an additional 180 calendar days, or until May 2, 2022, to regain compliance with the Bid Price Rule. As part of our efforts to regain compliance with the Bid Price Rule, on February 28, 2022, we filed a definitive proxy statement related to a special meeting of our stockholders scheduled for Thursday, April 14, 2022 at which our stockholders will vote upon proposal, approved and recommended for stockholder approval by the Board, to amend our Certificate of Incorporation to effect a reverse stock split of our outstanding shares of common stock by a ratio of any whole number between 1-for-2 and 1-for-50 at any time prior to December 31, 2022, the timing and implementation of which will be subject to the discretion of the Board. To regain compliance, the bid price for the Company’s common stock must close at $1.00 per share or more for a minimum of 10 consecutive business days.
Our principal corporate headquarters areoffice is located at 2020 Avon Court, #4,300 East Main Street, Suite 201, Charlottesville, Virginia 22902, and our telephone number is (434) 220-0718,220-0718. Our website, www.diffusionpharma.com, including the Investor Relations section, investors.diffusionpharma.com, and our Internet web site addresssocial media channels – Facebook (www.facebook.com/diffusionpharmaceuticalsinc/), Twitter (www.twitter.com/diffusionpharma) and LinkedIn (https://www.linkedin.com/company/diffusion-pharmaceuticals/) -- contain a significant amount of information about the Company. We encourage investors to visit these websites and social media channels as information is www.diffusionpharma.com. Thefrequently updated and new information containedis shared. However, the information included on our web site or connected towebsite and available through our web sitesocial media channels is not incorporated by reference into, and should not be considered part of, this annual report on Form 10-K.Annual Report or any other filings we make with the SEC.
Merger with RestorGenex
On January 8, 2016, we completed the Merger with Diffusion LLC in accordance with the terms of the Merger Agreement. As a result of the Merger, Diffusion LLC, the surviving company in the Merger, became a wholly owned subsidiary of the Company.
At the effective time of the Merger (the “Effective Time”), each outstanding unit of membership interest of Diffusion LLC (the “Diffusion Units”) was converted into the right to receive 0.3652658 shares of our common stock, as determined pursuant to the Merger Agreement (the “Exchange Ratio”). Also at the Effective Time, the rights of the holders of each outstanding convertible promissory note convertible into Diffusion Units (the “Diffusion Convertible Notes”) to convert such Diffusion Convertible Notes into Diffusion Units was converted into the right to convert such securities into a number of shares of common stock equal to the number of Diffusion Units such Diffusion Convertible Note would be convertible into pursuant to its terms multiplied by the Exchange Ratio. In addition, at the Effective Time and as a result of the Merger, all outstanding options to purchase Diffusion Units were converted into and became options to purchase common stock on terms substantially identical to those in effect prior to the Effective Time, except for adjustments to the underlying number of shares and the exercise price based on the Exchange Ratio.
Immediately following the Merger, the former equity holders of Diffusion LLC owned approximately 84.1% of the Company’s common stock, and the stockholders of RestorGenex immediately prior to the Merger owned approximately 15.9% of the Company’s common stock, in each case, on a fully-diluted basis (subject to certain exceptions and adjustments) and calculated in accordance with the terms of the Merger Agreement. Also in connection with the Merger, the pre-Merger directors and officers of the Company tendered their resignations and the pre-Merger directors and officers of Diffusion LLC were appointed as the new directors and officers of the Company. Following the completion of the Merger, the Company changed its corporate name from “RestorGenex Corporation” to “Diffusion Pharmaceuticals Inc.” and changed the trading symbol of the Company’s common stock from “RESX” to “DFFN.” In November 2016, our common stock was approved for listing on the NASDAQ Capital Market and will continue to trade under the ticker symbol “DFFN.”
For accounting purposes, the Merger is treated as a “reverse acquisition” under generally acceptable accounting principles in the United States (“U.S. GAAP”) and Diffusion LLC is considered the accounting acquirer. Accordingly, Diffusion LLC’s historical results of operations have replaced the Company’s historical results of operations for all periods prior to the Merger.
Reverse Stock Split
On August 17, 2016, the Company filed a Certificate of Amendment to its Certificate of Incorporation with the Secretary of State of the State of Delaware to effect a 1-to-10 reverse stock split (the “Reverse Stock Split”) of the common stock. No fractional shares were issued in connection with the Reverse Stock Split. Stockholders who otherwise would have been entitled to receive fractional shares of common stock had their holdings rounded up to the next whole share. Proportional adjustments were made to the Company’s outstanding warrants, stock options and other equity securities and to the Company’s 2015 Equity Incentive Plan, as amended, to reflect the Reverse Stock Split, in each case, in accordance with the terms thereof. Unless the context otherwise requires, all share and per share amounts in this annual report on Form 10-K have been adjusted to reflect the Reverse Stock Split.
Private Placement
We are conducting a private placement (the “Private Placement”) of (i) shares of our Series A convertible preferred stock, $0.001 par value per share (the “Series A Convertible Preferred Stock”), each share of Series A Convertible Preferred Stock being convertible into one share of our common stock, subject to adjustment, and (ii) for each share of Series A Convertible Preferred Stock purchased in the Private Placement, a 5-year warrant to purchase one share of common stock (the “Warrant” and collectively with the Series A Convertible Preferred Stock, the “Securities”).
On March 14, 2017, we entered into Subscription Agreements with certain accredited investors and conducted an initial closing of the Private Placement pursuant to which we sold 7,837,023 shares of the Series A Convertible Preferred Stock at a purchase price of $2.02 per share (the “Initial Closing”). In addition, each investor received a Warrant to purchase one share of common stock for each share of Series A Convertible Preferred Stock purchased by such investor in the Private Placement at an exercise price equal to $2.22, subject to adjustment thereunder.
We received total net proceeds of approximately $14.1 million from the Initial Closing, after deducting $1.7 million in placement agent fees and expenses associated with the Initial Closing. We currently intend to use the anticipated proceeds of the Private Placement to fund research and development of TSC, including clinical trial activities, and for general corporate purposes. Pursuant to the Purchase Agreements, we may conduct one or more additional closings until we have sold up to an aggregate of $25.0 million of Securities.
The Securities are being offered and sold in a private placement only to accredited investors pursuant to exemptions from the registration requirements of the Securities Act of 1933, as amended (the “Securities Act”), afforded by Section 4(a)(2) and Rule 506 of Regulation D promulgated thereunder. To the extent that any shares of common stock are issued in connection with the conversion of the Series A Convertible Preferred Stock or the exercise of the Warrants, the common stock may not be offered, transferred or sold in the United States absent registration or the availability of an applicable exemption from the registration requirements of the Securities Act. The foregoing description of the Private Placement is neither an offer to sell, nor a solicitation of an offer to buy, any of the Securities and shall not constitute an offer, solicitation or sale in any state or jurisdiction in which such offer, solicitation or sale is unlawful.The Securities offered in the Private Placement have not been registered under the Securities Act or the securities laws of any state, and may not be offered or sold in the United States unless the Securities are registered under the Securities Act and all applicable state securities laws, or an exemption from such registration requirements is available. The Securities are being offered and sold in reliance on exemptions from the registration requirements of the Securities Act and applicable state securities laws.
For a more information on the Initial Closing and a description of the Securities please see our Current Report on Form 8-K filed with the Securities and Exchange Commission on March 15, 2017.
Available Information
We make available free of charge andon or through our Internet web site,website certain reports that we file with or furnish to the SEC in accordance with Exchange Act. These include our annual reportsAnnual Reports on Form 10-K, quarterly reportsour Quarterly Reports on Form 10-Q, current reportsand our Current Reports on Form 8-K, andas well as any amendments to any suchthose reports, filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange ActAct. We make this information available free of 1934, as amended,charge as soon as reasonably practicable after we electronically file such materialthe information with, or furnish it to, the SEC. The SEC also maintains a website, www.sec.gov, that contains reports, proxy and information statements, and other information regarding the Company and other issuers that file electronically with the SEC. We also make available, free of charge and through our Internet web site, to any shareholder who requests,website, the charters of our boardthe committees of the Board, our Corporate Governance Guidelines, and our Code of Business Conduct and Ethics. Requests for copies can be directed to Investor Relations at (434) 220-0718.
Cautionary Note Regarding Forward-Looking Statements
This Annual Report on Form 10-K (this “Annual Report”) includes forward-looking statements. We may, in some cases, use terms such as “believes,” “estimates,” “anticipates,” “expects,” “plans,” “intends,” “may,” “could,” “might,” “will,” “should,” “approximately” or other words that convey uncertainty of future events or outcomes to identify these forward-looking statements. Forward-looking statements appear in a number of places throughout this Annual Report and include statements regarding our intentions, beliefs, projections, outlook, analyses or current expectations concerning, among other things, our ongoing and planned preclinical development and clinical trials, the timing of and our ability to make regulatory filings and obtain and maintain regulatory approvals for our product candidates, our intellectual property position, the degree of clinical utility of our products, particularly in specific patient populations, our ability to develop commercial functions, expectations regarding clinical trial data, our results of operations, cash needs, financial condition, liquidity, prospects, growth and strategies, the industry in which we operate and the trends that may affect the industry or us.
By their nature, forward-looking statements involve risks and uncertainties because they relate to events, competitive dynamics and industry change, and depend on the economic circumstances that may or may not occur in the future or may occur on longer or shorter timelines than anticipated. Although we believe that we have a reasonable basis for each forward-looking statement contained or incorporated by reference in this Annual Report, we caution you that forward-looking statements are not guarantees of future performance and that our actual results of operations, financial condition and liquidity, and the development of the industry in which we operate may differ materially from the forward-looking statements contained or incorporated by reference in this Annual Report. In addition, even if our results of operations, financial condition and liquidity, and the development of the industry in which we operate are consistent with the forward-looking statements contained or incorporated by reference in this Annual Report, they may not be predictive of results or developments in future periods.
Actual results could differ materially from our forward-looking statements due to a number of factors, including risks related to:
| ●
| our estimates regarding expenses, capital requirements and needs for additional financing;
|
| ●
| the success and timing of our preclinical studies and clinical trials;
|
| ●
| our ability to obtain additional financing;
|
| ●
| the difficulties in obtaining and maintaining regulatory approval of our products and product candidates, and the labeling under any approval we may obtain;
|
| ●
| our plans and ability to develop and commercialize our product candidates;
|
| ●
| our failure to recruit or retain key scientific or management personnel or to retain our executive officers;
|
| ●
| the accuracy of our estimates of the size and characteristics of the potential markets for our product candidates and our ability to serve those markets;
|
| ●
| regulatory developments in the United States and foreign countries;
|
| ●
| the rate and degree of market acceptance of any of our product candidates;
|
| ●
| obtaining and maintaining intellectual property protection for our product candidates and our proprietary technology;
|
| ●
| our ability to operate our business without infringing the intellectual property rights of others;
|
| ●
| recently enacted and future legislation regarding the healthcare system;
|
| ●
| the success of competing products that are or become available; and
|
| ●
| the performance of third parties, including contract research organizations and manufacturers.
|
You should also read carefully the factors described in the “Risk Factors” section of this Annual Report and elsewhere to better understand the risks and uncertainties inherent in our business and underlying any forward-looking statements. As a result of these factors, we cannot assure you that the forward- looking statements contained or incorporated by reference in this Annual Report will prove to be accurate. Furthermore, if our forward-looking statements prove to be inaccurate, the inaccuracy may be material. In light of the significant uncertainties in these forward-looking statements, you should not regard these statements as a representation or warranty by us or any other person that we will achieve our objectives and plans in any specified timeframe, or at all.
Any forward-looking statements that we make contained or incorporated by reference in this Annual Report speak only as of the date of such statement, and, except as required by applicable law, we undertake no obligation to update such statements to reflect events or circumstances after the date of this Annual Report or to reflect the occurrence of unanticipated events. Comparisons of results for current and any prior periods are not intended to express any future trends or indications of future performance, unless expressed as such, and should only be viewed as historical data.
The followingInvesting in our common stock involves a high degree of risk. Set forth below are significant factorscertain material risks and uncertainties known to us that could materially harmadversely affect our business, operatingfinancial condition, or results or financial conditionof operations or could cause our actual results to differ materially from our anticipated results or other expectations including those expressed in any forward-looking statement madeelsewhere in this report:
Risks Related to Our Financial Needs
Wehave limited cash resources,have incurred significant losses since our inception and have a history of net losses and negative cash flow from operations.
We are a clinical-stage biotechnology company and, as a result, we have a limited operating history and there is little historical basis upon which to assess how we will respond to competitive or economic challenges or other challenges to our business. Our business and prospects must be considered in lightAnnual Report. The occurrence of the risks and uncertainties frequently encounteredevents contemplated by similarly situated companies.
We have limited cash resources, have generated substantial net losses and negative cash flow from operations since our inception, and we continue to incur significant research, development and other expenses related to our ongoing operations for other product candidates. We expect to incur losses and negative cash flow for the foreseeable future. Our ability to generate sufficient revenues from our other product candidates, if approved, will depend on numerous factors described in the following risk factors. Even if we achieve profitability in the future, we may not be able to sustain profitability in subsequent periods. Our prior losses and expected future losses have had and will continue to have an adverse effect on our stockholders’ equity.
We currently generate no revenue from the sale of products and may never become profitable.
To date, we have not generated any revenues from our product candidates. Even if we are able to successfully achieve regulatory approval for our product candidates, we do not know when any of these products will generate revenue for us, if at all. Our ability to generate revenue from our current products or future product candidates also depends on a number of additional factors, including our ability to:
| ●
| successfully complete development activities, including the necessary clinical trials;
|
| ●
| complete and submit NDAs to the FDA and obtain regulatory approval for indications for which there is a commercial market;
|
| ●
| complete and submit applications to, and obtain regulatory approval from, foreign regulatory authorities;
|
| ●
| develop a commercial organization capable of sales, marketing and distribution for the products we intend to sell ourselves in the markets in which we have retained commercialization rights; and
|
| ●
| find suitable distribution partners to help us market, sell and distribute our approved products in other markets.
|
In addition, because of the numerous risks and uncertainties associated with product development, including that our product candidates may not advance through development or achieve the endpoints of applicable clinical trials, we are unable to predict the timing or amount of increased expenses, or when or if we will be able to achieve or maintain profitability. Even if we are able to complete the process described above, we anticipate incurring significant costs associated with commercializing these products.
We will require additional capital to fund our operations and if we fail to obtain necessary financing, we may be unable to complete the development and commercialization of our product candidates.
Our operations have consumed substantial amounts of cash since inception. We expect to continue to spend substantial amounts to advance the clinical development of our product candidates and to commercialize any product candidates for which we receive regulatory approval. We expect our existing cash as of December 31, 2016, together with the proceeds from the Initial Closing of the Private Placement of our Series A Convertible Preferred Stock in March 2017, will enable us to fund our operating expenses and capital expenditure requirements into December of 2017. Even if we raise the maximum amount being offered in the Private Placement, we will require additional capital for the further development and commercialization of our product candidates and may also need to raise additional funds sooner in order to accelerate development of our product candidates. We may also require additional capital to repay our outstanding convertible indebtedness if the holders thereof do not elect to convert such indebtedness into equity prior to maturity.
We cannot be certain that additional funding will be available on acceptable terms, or at all. If we are unable to raise additional capital in sufficient amounts or on terms acceptable to us we may have to significantly delay, scale back or discontinue the development or commercialization of one or more of our product candidates or one or more of our other research and development initiatives. We also could be required to:
| ●
| seek collaborators for one or more of our current or future product candidates at an earlier stage than otherwise would be desirable or on terms that are less favorable than might otherwise be available; or
|
| ●
| relinquish or license on unfavorable terms our rights to technologies or product candidates that we otherwise would seek to develop or commercialize ourselves.
|
Our forecast of the period of time through which our financial resources will be adequate to support our operations is a forward-looking statement and involves risks and uncertainties, and actual results could vary as a result of a number of factors, including the factors discussed elsewhere in this “Risk Factors” section. We have based this estimate on assumptions that may prove to be wrong, and we describe below could utilize our available capital resources sooner than we currently expect. Our future funding requirements, both near and long-term, will depend on many factors, including, but not limited to:
| ●
| the initiation, progress, timing, costs and results of clinical trials for our product candidates and any future product candidates we may in-license;
|
| ●
| the clinical development plans we establish for these product candidates;
|
| ●
| the number and characteristics of product candidates that we in-license and develop;
|
| ●
| the outcome, timing and cost of regulatory approvals by the FDA and comparable foreign regulatory authorities, including the potential for the FDA or comparable foreign regulatory authorities to require that we perform more studies than those that we currently expect;
|
| ●
| the cost of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights;
|
| ●
| the effect of competing technological and market developments; and
|
| ●
| the cost and timing of completion of becoming a commercial organization.
|
Raising additional capital may cause dilution to our existing stockholders, restrict our operations or require us to relinquish rights to our technologies or product candidates.
We may seek additional capital through a combination of private and public equity offerings, debt financings, receivables or royalty financings, strategic partnerships and alliances and licensing arrangements. We do not currently have any committed external source of funds. To the extent that we raise additional capital through the sale of equity or convertible debt securities, your ownership interest will be diluted, and the terms may include liquidation or other preferences that adversely affect the rights of existing stockholders. Debt, receivables and royalty financings may be coupled with an equity component, such as warrants to purchase stock, which could also result in dilution of our existing stockholders’ ownership. The incurrence of indebtedness would result in increased fixed payment obligations and could also result in certain restrictive covenants, such as limitations on our ability to incur additional debt, limitations on our ability to acquire or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business and may result in liens being placed on our assets and intellectual property. If we were to default on such indebtedness, we could lose such assets and intellectual property. If we raise additional funds through strategic partnerships and alliances and licensing arrangements with third parties, we may have to relinquish valuable rights to our product candidates, or grant licenses on terms that are not favorable to us.
The issuance of shares ofSeries A ConvertiblePreferred Stock reduces the relative voting power of holders ofcommonstock, dilutes the ownership of such holders and may adversely affect the market price of ourcommonstock.
In the event of any vote or action by written consent, each holder of shares of Series A Convertible Preferred Stock shall be entitled to that number of votes equal to the whole number of shares of common stock into which the aggregate number of shares of Series A Convertible Preferred Stock held of record by such stockholder are convertible as of the close of business on the record date fixed for such vote or such written consent based on a conversion price, solely for such purpose, equal to the closing price of our common stock onto decline, resulting in the date such Series A Convertible Preferred Stock was issued. As holdersloss of Series A Convertible Preferred Stockall or part of any investment in our common stock. Furthermore, other risks that are entitledcurrently unknown to vote on an as-converted basisus or that we currently believe to be immaterial may also, nevertheless, adversely affect our business, financial condition, or results of operations in a way that is material.
Before investing in our common stock, you should carefully consider these risks and uncertainties, together with holders of common stock on all matters submitted to a vote of the holders of common stock, the issuance of the Series A Convertible Preferred Stock, and the subsequent payment of dividendsother information in shares of common stock, effectively reduces the relative voting power of the current holders of common stock.
We have a history of operating losses and expect to continue to incur losses in the forseeable future, which raises substantial doubt about our ability to continue as a going concern.
As discussed further in footnote 2 ofthis Annual Report, including our consolidated financial statements forand related notes, the year ended December 31, 2016, we have a historyinformation included in, Part II — Item 7 — Management’s Discussion and Analysis of operating lossesFinancial Condition and expect to continue to incur losses in the forseeable future, which raises substantial doubt regarding our ability to continue as a going concern.We currently have no sourcesResults of revenue and our ability to continue as a going concern is dependent on our ability to raise capital to fund our future business plans. Additionally, volatility in the capital markets and general economic conditions in the United States may be a significant obstacle to raising the required funds. These factors raise substantial doubt about our ability to continue as a going concern. The consolidated financial statements do not include any adjustments that might be necessary should the Company be unable to continue as a going concern. If the going concern basis were not appropriate for these financial statements, adjustments would be necessary in the carrying value of assets and liabilities, the reported expensesOperations, and the balance sheet classifications used.information incorporated herein by reference.
Risks Related to the Development, Regulatory Approval, and Commercialization of TSC and Our Other Product Candidates
Our businessThe success of Diffusion is dependent on the successful development, regulatory approval, and, ultimately, commercialization of TSC and our other product candidates, which include products primarilycandidates. However, the drug development process is expensive, time-consuming and uncertain. Our efforts to develop, obtain regulatory approval for, and commercialize TSC or any other product candidate could fail at any stage of the treatmentdevelopment process for a variety of glioblastoma multiforme, pancreatic cancerreasons, including the possibility that TSC fails to adequately demonstrate efficacy or proof of concept in the Oxygenation Trials. Furthermore, because the results of preclinical studies and brain metastases.early-stage clinical trials are not necessarily predictive of future results, even if we are able to advance TSC or another product candidate into additional clinical trials – for example, beyond our Planned Phase 2 Hypoxic Solid Tumor Trial – we may not continue to experience favorable results.
Our portfolio of product candidates includes TSC, which is a novel small molecule that re-oxygenates hypoxic tissue, enhancing the efficacy of radiation and chemotherapy without harmful side effects poised to enter Phase III clinical development for glioblastoma multiforme. In addition, TSC may have potential applications in pancreatic cancer and brain metastases. The success of our business,Diffusion, including our ability to finance our companyoperations and generate any revenue in the future, primarily will depend primarily on the successful development, regulatory approval, and, ultimately, commercialization of our product candidates. Currently, the majority of our product development resources are dedicated to our lead product candidate, TSC. In the future, we may also become dependent on oneseek to develop or more othercommercialize additional product candidates, or any futureincluding product candidates that we may in-license or acquire or develop.to supplement our internally developed portfolio.
The clinical and commercial success of our product candidates will depend on a number of factors, including the following:
| ●
| timely completion of our clinical trials, which may be significantly slower or cost more than we currently anticipate and will depend substantially upon the performance of third-party contractors;
|
| ●
| whether we are required by the FDA or similar foreign regulatory agencies to conduct additional clinical trials beyond those planned to support the approval and commercialization of our product candidates or any future product candidates;
|
| ●
| acceptance of our proposed indications and primary endpoint assessments relating to the proposed indications of our product candidates by the FDA and similar foreign regulatory authorities;
|
| ●
| our ability to demonstrate to the satisfaction of the FDA and similar foreign regulatory authorities, the safety and efficacy of our product candidates or any future product candidates;
|
| ●
| the prevalence, duration and severity of potential side effects experienced with our product candidates or future approved products, if any;
|
| ●
| the timely receipt of necessary marketing approvals from the FDA and similar foreign regulatory authorities;
|
| ●
| achieving and maintaining, and, where applicable, ensuring that our third-party contractors achieve and maintain, compliance with our contractual obligations and with all regulatory requirements applicable to our product candidates or any future product candidates or approved products, if any;
|
| ●
| the ability of third parties with whom we contract to manufacture clinical trial and commercial supplies of our product candidates or any future product candidates, remain in good standing with regulatory agencies and develop, validate and maintain commercially viable manufacturing processes that are compliant with cGMP;
|
| ●
| a continued acceptable safety profile during clinical development and following approval of our product candidates or any future product candidates;
|
| ●
| our ability to successfully commercialize our product candidates or any future product candidates in the United States and internationally, if approved for marketing, sale and distribution in such countries and territories, whether alone or in collaboration with others;
|
| ●
| acceptance by physicians and patients of the benefits, safety and efficacy of our product candidates or any future product candidates, if approved, including relative to alternative and competing treatments;
|
| ●
| our and our partners’ ability to establish and enforce intellectual property rights in and to our product candidates or any future product candidates;
|
| ●
| our and our partners’ ability to avoid third-party patent interference or intellectual property infringement claims; and
|
| ●
| our ability to in-license or acquire additional product candidates or commercial-stage products that we believe can be successfully developed and commercialized.
|
If we do not achieve one or more of these factors, many of which are beyond our control, in a timely manner or at all, we could experience significant delays or an inability to obtain regulatory approvals or commercialize our product candidates. Even if regulatory approvals are obtained, we may never be able to successfully commercialize any of our product candidates. Accordingly, we cannot assure you that we will be able to generate sufficient revenue through the sale of our product candidates or any future product candidates to continue our business.
Clinical drug development for our product candidates is very expensive, time-consuming and uncertain. Our clinical trials may fail to adequately demonstrate the safety and efficacy of our product candidates, which could prevent or delay regulatory approval and commercialization.
Clinical drug development for our product candidatesprocess is very expensive, time-consuming, difficult to design and implement, and its outcome is inherently uncertain. Before obtaining regulatory approval for the commercial sale of a product candidate, we must demonstrate through clinical trials that a product candidate is both safe and effective for use in the target indication. Most product candidates that commence clinical trials are never approved by regulatory authorities for commercialization. commercialization and success in early-stage clinical trials does not ensure that later clinical trials will demonstrate the efficacy and safety of an investigational drug in a manner adequate to support regulatory approval. Countless other companies, including many with greater resources and experience, have failed or suffered significant setbacks attempting to navigate the drug development process, including failed attempts to develop treatments for hypoxia and indications on the hypoxia-continuum that we may ultimately choose to target with TSC, and there can be no assurance that we will have success where others have failed.
Our product candidates, areincluding TSC, remain in early stages of development. Wethe development process and we expect that the additional clinical trials for these product candidatesnecessary to support an NDA will take several years but may take significantly longer than expected to complete. In addition,We do not know whether the clinical trials we may conduct will demonstrate adequate efficacy and safety or otherwise provide adequate information to result in regulatory approval to market any partner with which weof our product candidates in any particular jurisdiction. Furthermore, the timeline for our clinical trials may be delayed in the future collaborate,for a variety of reasons, including delays related to regulatory and IRB review and approval, slower than anticipated rates of enrollment in or early withdrawals from the FDA, an IRBtrial, third party performance issues beyond our control including any CRO engaged in the conduct of the trial, discovery of series or unexpected toxicities or side effects, or a lack of effectiveness.
Whether we are able to successfully develop TSC or any of our other regulatory authorities,product candidates will depend on a large number of factors, including state and local agencies and counterpart agencies in foreign countries, may suspend, delay, require modifications to or terminate our clinical trials at any time, for various reasons, including:the following:
| | ●•
| discovery of serious or unexpected toxicities or side effects experienced by study participants or other safety issues;our ability to complete our planned and future clinical trials in a timely manner and, with respect to any future trials beyond the ongoing Oxygenation Trials and our Planned Phase 2 Hypoxic Solid Tumor Trial, our ability to fund such trials;
|
| | ●•
| lackour ability to demonstrate safety and efficacy to the satisfaction of effectiveness ofthe FDA and similar foreign regulatory authorities, and whether we are required by any product candidate duringsuch body to conduct additional clinical trials or the failure of our product candidates to meet specified endpoints;support approval;
|
| | ●•
| slower than expected ratesthe receipt of subject recruitmentnecessary regulatory approvals, including acceptance of our proposed indications and enrollment rates in clinical trials resulting from numerous factors,primary endpoint assessments, marketing approvals, and labeling claims;
|
| • | a continued acceptable safety profile during development and following approval, including the prevalence, duration and severity of other companies’ clinical trials for their product candidates for the same indication, or clinical trials for indications for which patients do not as commonly seek treatment;potential side effects experienced; and |
| | ●•
| difficulty in retaining subjects who have initiated a clinical trial but may withdraw at any time dueour ability to adverse side effects from the therapy, insufficient efficacy, fatigue with the clinical trial process or for any other reason;
|
| ●
| difficulty in obtaining IRB approval for studies to be conducted at each site;
|
| ●
| delays in manufacturing or obtaining, or inability to manufacture or obtain, sufficient quantities of materials for use in clinical trials;
|
| ●
| inadequacy of or changes incommercialize successfully, including scaling our manufacturing processcapabilities, the development of sales and marketing capabilities internally or through a third party, acceptance by physicians and patients of the product formulation or method of delivery;
|
| ●
| changes in applicable laws, regulationsbenefits, safety and regulatory policies;
|
| ●
| delays or failure in reaching agreement on acceptable terms in clinical trial contracts or protocols with prospective clinical research organizations (“CROs”), clinical trial sites and other third-party contractors;
|
| ●
| inability to add a sufficient number of clinical trial sites;
|
| ●
| uncertainty regarding proper dosing;
|
| ●
| failureefficacy of our CROs or other third-party contractors to comply with contractual and regulatory requirements or to perform their services in a timely or acceptable manner;
|
| ●
| failure by us, our employees, our CROs or their employees or any partner with which we may collaborate or their employees to comply with applicable FDA or other regulatory requirements relating to the conduct of clinical trials or the handling, storage, security and recordkeeping for drug products;
|
| ●
| scheduling conflicts with participating clinicians and clinical institutions;
|
| ●
| failure to design appropriate clinical trial protocols;
|
| ●
| insufficient data to support regulatory approval;
|
| ●
| inability or unwillingness of medical investigators to follow our clinical protocols; or
|
| ●
| difficulty in maintaining contact with subjects during or after treatment, which may result in incomplete data.treatments.
|
WeAny of these factors, many of which are beyond our control, could result in significant delays or an inability to develop, obtain regulatory approvals for, or commercialize our product candidates, and we may ultimately be able to receive regulatory approval or generate revenue from the sale of TSC or any partner with which we may collaborate may suffer significant setbacks in our clinical trials similar to the experience of aother product candidate.
A number of other companies in the pharmaceutical and biotechnology industries,biopharmaceutical industry have suffered significant setbacks in later-stage Phase 3 clinical development even after receiving promising results in earlier preclinical studies or clinical trials. InIf later-stage clinical trials do not produce favorable results for TSC or any other product candidate, or we are unable to complete the eventnecessary clinical trials for any reason (including a lack of funding), our ability to achieve regulatory approval or successfully commercialize may be compromised. At any time, we may decide or be forced by circumstance to delay or discontinue the development or commercialization of TSC or any of our other product candidates, including as a result of unfavorable results in later-stage clinical trials, changes in our internal product, technology or indication focus, the appearance of new technologies that make our product candidate obsolete, competition from a competing product, or changes in (or failure to comply with) applicable regulatory requirements. If we or our potential partners abandondecide or are delayedforced to terminate the TSC development program or any future program in which we have invested significant resources, we will not receive any return on our investment despite the clinical development efforts related to our product candidates,allocation of significant resources, we may not be able to execute on our business plan effectively, and our business, financial condition, operating results of operations may be materially and prospects would be harmed.adversely affected.
WeEven if we are able to successfully complete the clinical trials and over development activities necessary to submit an NDA to the FDA or an application for marketing approval to an equivalent non-U.S. regulatory authority, we may be unable to obtain regulatory approval for TSC or any other product candidates we may attempt to develop in future, for the indications offor which we are seekinginitially seek approval or our other future product candidates under applicable regulatory requirements.at all. The FDA and foreignsimilar non-U.S. regulatory bodiesauthorities have substantialsignificant discretion in the approval process, including the ability to delay, limit, or deny approval of product candidates. The delay, limitation, or denial of any regulatory approval for any of our product candidates, and TSC in particular, would limit or restrict altogether our ability to commercialize the product and generate revenue, which could materially and adversely impact commercialization, our potential to generate revenue, our business, financial condition, and our operating results.results of operations.
We currently have no products approved for sale, and we may never obtain regulatory approval to commercialize TSC or any of our current or futureother product candidates.candidates we may attempt to develop in the future. The research, testing, manufacturing, safety surveillance, efficacy, quality control, recordkeeping, labeling, packaging, storage, approval, sale, marketing, distribution, import, export, and reporting of safety and other post-market information related to our drug products are subject to extensive regulation by the FDA and other regulatory authorities in the United States and in foreign countries, and such regulationsabroad, which often differ from country to country. We arewill not be permitted to market TSC or any of our other current product candidates in the United StatesU.S. until we receive approval of an NDA or other applicable regulatory filing from the FDA. We are alsoFDA, and we will not be permitted to market TSC or any of our other current product candidates in any foreignnon-U.S. countries until we receive the requisite approval from the applicable regulatory authorities of such countries.authorities.
To gain approval to market a new drug, such as TSC, the FDA and foreignsimilar non-U.S. regulatory authorities must receiverequire the submission of an NDA (or similar application) that contains preclinical and clinical data that adequately demonstratedemonstrating the safety, purity, potency, efficacy, and compliant manufacturing of the product for the intended indication applied for in an NDA or other applicable regulatory filing. The development and approval of new drug products involves a long, expensive and uncertain process, and delay or failure can occur at any stage. A number of companies in the pharmaceutical and biopharmaceutical industry have suffered significant setbacks in clinical trials, including in Phase 3 clinical development, even after promising results in earlier preclinical studies or clinical trials. These setbacks have been caused by, among other things, findings made while clinical trials were underway and safety or efficacy observations made in clinical trials, including previously unreported adverse events. Success in preclinical testing and early clinical trials does not ensure that later clinical trials will be successful, and the results of clinical trials by other parties may not be indicative of the results in trials we or our partners may conduct.
indication. The FDA and foreign regulatory bodiestheir non-U.S. counterparts have substantial discretion in the drug approval process, including the ability to delay, limit, or deny approval of product candidatesapplications for many reasons, including:
| | ●•
| the FDA or the applicable foreign regulatory body may disagreedeemed issues with the design or implementationexecution of one or more clinical trials;
|
| | ●•
| the FDA or the applicable foreign regulatory body may not deem a product candidate safe and effective for its proposed indication, or may deem a product candidate’s safety or other perceived risks to outweigh its clinical or other benefits;
|
| ●
| the FDA or the applicable foreign regulatory body may not find the data from preclinical studies and clinical trials sufficient to support approval, or the results of clinical trials may not meet the level of statistical or clinical significance required by the FDA or the applicable foreign regulatory body for approval;
|
| ●
| the FDA or the applicable foreign regulatory body may disagree with our interpretation of data from preclinical studies or clinical trials performed by us or third parties, or with the interpretation of any partner with which we may collaborate;
|
| ●
| the data collected from clinical trials may not be sufficient to support the submission of an NDA or other applicable regulatory filing;
|
| ●
| the FDA or the applicable foreign regulatory body may require additional preclinical studies or clinical trials;
|
| ●
| the FDA or the applicable foreign regulatory agency may identifydeemed deficiencies in the formulation, quality control, labeling, or specifications of our current or futurethe product candidates;candidate;
|
| | ●•
| the FDAdeemed issues in our manufacturing processes or the applicable foreign regulatory agency may require clinical trials in pediatric patients in order to establish pharmacokinetics or safety for this more drug-sensitive population;
|
| ●
| the FDA or the applicable foreign regulatory agency may grant approval contingent on the performance of costly additional post-approval clinical trials; |
| ●
| the FDA or the applicable foreign regulatory agency also may approve our current or any future product candidates for a more limited indication or a narrower patient population than we originally requested;
|
| ●
| the FDA or applicable foreign regulatory agency may not approve the labeling that we believe is necessary or desirable for the successful commercialization of our product candidates;
|
| ●
| the FDA or the applicable foreign regulatory body may not approve of the manufacturing processes, controls or facilities of third-party manufacturers or testing labs with which we contract; or
|
| | ●•
| a determination that the FDAdata from preclinical studies and clinical trials included in the application is not sufficient to support approval, or do not meet a required level of statistical or clinical significance, including as a result of a differing interpretation of the applicable foreign regulatory body may change its approval policies or adopt new regulationsdata than that presented by the Company in a manner rendering our clinical data or regulatory filings insufficient for approval.application; |
| • | a determination that the perceived risks of approving the product candidate outweigh the clinical and other benefits of approval; |
| • | a determination that additional preclinical studies or clinical trials are required, either prior to or as a contingency to approval, and, for certain target indications such as pediatric populations, in the targeted sub-population; |
| • | a determination that a product candidate may only be approved on a contingent basis or for a more limited indication or patient population than we request; |
| • | a determination that labeling we believe is necessary or desirable for successful commercialization cannot be approved; or |
| • | unanticipated future changes to the approval process and related regulations. |
OfHistorically, of the large number of drugs in development at any given time, only a small percentage successfully complete the FDA or other regulatory approval processes and are ultimately commercialized. Our product candidates may not be approved for sale and marketing by the FDA or applicable foreign regulatory agenciesany other governmental authority, even if they meet specified endpoints in our clinical trials. The FDA or applicable foreign regulatory agencies may ask us to conduct additional costly and time-consuming clinical trials in order to obtain marketing approval or approval to enter into an advanceda further phase of clinical development, or may change the requirements for approval even after such agency has reviewed and commented on the design for the clinical trials.
Any delay in obtaining, or inability to obtain, applicablethe regulatory approval for any ofapprovals necessary to market and sell our product candidates would delay or prevent commercialization of our product candidates and would harmmaterially and adversely affect our business, financial condition, operating results and prospects.
Even if our product candidates obtain regulatory approval, they may fail to achieve the broad degreeresult of physician and patient adoption and use necessary for commercial success.
The commercial success of any of our current or future product candidates, if approved, will depend significantly on the broad adoption and use of the resulting product by physicians and patients for approved indications, and may not be commercially successful. The degree and rate of physician and patient adoption of our current or future product candidates, if approved, will depend on a number of factors, including:
| ●
| the clinical indications for which the product is approved and patient demand for approved products that treat those indications;
|
| ●
| the effectiveness of our product as compared to other available therapies;
|
| ●
| the availability of coverage and adequate reimbursement from managed care plans and other healthcare payors for any of our product candidates that may be approved;
|
| ●
| the cost of treatment with our product candidates in relation to alternative treatments and willingness to pay for the product, if approved, on the part of patients;
|
| ●
| acceptance by physicians, major operators of clinics and patients of the product as a safe and effective treatment;
|
| ●
| physician and patient willingness to adopt a new therapy over other available therapies to treat approved indications;
|
| ●
| overcoming any biases physicians or patients may have toward particular therapies for the treatment of approved indications;
|
| ●
| proper training and administration of our product candidates by physicians and medical staff;
|
| ●
| patient satisfaction with the results and administration of our product candidates and overall treatment experience;
|
| ●
| the willingness of patients to pay for certain of our product candidates relative to other discretionary items, especially during economically challenging times;
|
| ●
| the revenue and profitability that our product candidate may offer a physician as compared to alternative therapies;
|
| ●
| the prevalence and severity of side effects;
|
| ●
| limitations or warnings contained in the FDA-approved labeling for our product candidates;
|
| ●
| any FDA requirement to undertake a REMS;
|
| ●
| the effectiveness of our sales, marketing and distribution efforts;
|
| ●
| adverse publicity about our product candidates or favorable publicity about competitive products; and
|
| ●
| potential product liability claims.
|
Our product candidates, if approved, will face significant competition and our failure to effectively compete may prevent us from achieving significant market penetration.
The pharmaceutical industry is characterized by rapidly advancing technologies, intense competition and a strong emphasis on developing proprietary therapeutics. Numerous companies are engaged in the development, patenting, manufacturing and marketing of health care products competitive with those that we are developing. We face competition from a number of sources, such as pharmaceutical companies, generic drug companies, biotechnology companies and academic and research institutions, many of which have greater financial resources, marketing capabilities, sales forces, manufacturing capabilities, research and development capabilities, clinical trial expertise, intellectual property portfolios, experience in obtaining patents and regulatory approvals for product candidates and other resources than us. Some of the companies that offer competing products also have a broad range of other product offerings, large direct sales forces and long-term customer relationships with our target physicians, which could inhibit our market penetration efforts.
Many pharmaceutical companies currently offer products, and continue to develop additional alternative product candidates and technologies, for indications similar to those targeted by our product candidates. We anticipate that,operations. Furthermore, if we obtain regulatory approval of our product candidates, we will face significant competition from other approved therapies. If approved, our product candidates may also compete with unregulated, unapproved and off-label treatments. Certain of our product candidates, if approved, will present novel therapeutic approaches for the approved indications and will have to compete with existing therapies, some of which are widely known and accepted by physicians and patients. To compete successfully in this market, we will have to demonstrate that the relative cost, safety and efficacy of our approved products, if any, provide an attractive alternative to existing and other new therapies. Such competition could lead to reduced market share for our product candidates and contribute to downward pressure on the pricing of our product candidates, which could harm our business, financial condition, operating results and prospects. For more information about the competition we face, see “Business—Competition.”
Due to less stringent regulatory requirements in certain foreign countries, there are many more products and procedures available for use in those international markets than are approved for use in the United States. In certain international markets, there are also fewer limitations on the claims that our competitors can make about the effectiveness of their products and the manner in which they can market them. As a result, we expect to face more competition in these markets than in the United States.
Any product candidates that we commercialize, or that any partner with which we may collaborate commercializes, will be subject to ongoing and continued regulatory review.
Even after we or our partners achieve U.S. regulatory approval for a product candidate, if any, we or our partners will be subject to continued regulatory review and compliance obligations. For example, with respect to our product candidates, the FDA may impose significant restrictions on the approved indicated uses for which the product may be marketed or on the conditions of approval. A product candidate’s approval may contain requirements for potentially costly post-approval studies and surveillance, including Phase 4 clinical trials or a REMS, to monitor the safety and efficacy of the product. We will also be subject to ongoing FDA obligations and continued regulatory review with respect to, among other things, the manufacturing, processing, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion and recordkeeping for our product candidates. These requirements include submissions of safety and other post-marketing information and reports, registration, as well as continued compliance with cGMP requirements and with the FDA’s GCP requirements and good laboratory practice (“GLP”) requirements, which are regulations and guidelines enforced by the FDA for all of our product candidates in clinical and preclinical development, and for any clinical trials that we conduct post-approval. To the extent that a product candidate is approved for sale in other countries, we may be subject to similar restrictions and requirements imposed by laws and government regulators in those countries.
In addition, manufacturers of drug products and their facilities are subject to continual review and periodic inspections by the FDA and other regulatory authorities for compliance with cGMP regulations. If we or a regulatory agency discovers previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facility where, or processes by which, the product is manufactured, a regulatory agency may impose restrictions on that product or us, including requesting that we initiate a product recall, or requiring notice to physicians, withdrawal of the product from the market or suspension of manufacturing.
If we, our product candidates or the manufacturing facilities for our product candidates fail to comply with applicable regulatory requirements, a regulatory agency may:
| ●
| impose restrictions on the marketing or manufacturing of the product, suspend or withdraw product approvals or revoke necessary licenses;
|
| ●
| mandate modifications to promotional materials or require us to provide corrective information to healthcare practitioners;
|
| ●
| require us or our partners to enter into a consent decree, which can include imposition of various fines, reimbursements for inspection costs, required due dates for specific actions and penalties for noncompliance;
|
| ●
| issue warning letters, show cause notices or untitled letters describing alleged violations, which may be publicly available;
|
| ●
| commence criminal investigations and prosecutions;
|
| ●
| impose injunctions, suspensions or revocations of necessary approvals or other licenses;
|
| ●
| impose other civil or criminal penalties;
|
| ●
| suspend any ongoing clinical trials;
|
| ●
| delay or refuse to approve pending applications or supplements to approved applications filed by us or our potential partners;
|
| ●
| refuse to permit drugs or precursor chemicals to be imported or exported to or from the United States;
|
| ●
| suspend or impose restrictions on operations, including costly new manufacturing requirements; or
|
| ●
| seize or detain products or require us or our partners to initiate a product recall.
|
The regulations, policies or guidance of the FDA and other applicable government agencies may change and new or additional statutes or government regulations may be enacted that could prevent or delay regulatory approval of our product candidates or further restrict or regulate post-approval activities. We cannot predict the likelihood, nature or extent of adverse government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we are not able to achieve and maintain regulatory compliance, we may not be permitted to market our product candidates, which would adversely affect our ability to generate revenue and achieve or maintain profitability.
We have conducted and maydetermine in the future conduct clinical trials for our product candidates outsidethat the United States and the FDA and applicable foreign regulatory authorities may not accept data from such trials.
We have conducted and may in the future choose to conduct one or more of our clinical trials outside the United States. Although the FDA or applicable foreign regulatory authority may accept data from clinical trials conducted outside the United States or the applicable jurisdiction, acceptance of such study data by the FDA or applicable foreign regulatory authority may be subject to certain conditions. Where data from foreign clinical trials are intended to serve as the basis for marketing approval in the United States, the FDA will not approve the application on the basis of foreign data alone unless those data are applicable to the U.S. population and U.S. medical practice; the studies were performed by clinical investigators of recognized competence; and the data are considered valid without the need for an on-site inspection by the FDA or, if the FDA considers such an inspection to be necessary, the FDA is able to validate the data through an on-site inspection or other appropriate means. Many foreign regulatory bodies have similar requirements. In addition, such foreign studies would be subject to the applicable local laws of the foreign jurisdictions where the studies are conducted. There can be no assurance the FDA or applicable foreign regulatory authority will accept data from trials conducted outside of the United States or the applicable jurisdiction. If the FDA or applicable foreign regulatory authority does not accept such data, it would likely result in the need for additional trials, which would be costly and time-consuming and delay aspects of our business plan.
Our product candidates may cause undesirable side effects or have other unexpected properties that could delay or prevent their regulatory approval, limit the commercial profile of an approved label or result in post-approval regulatory action.
Unforeseen side effects from any of our product candidates could arise either during clinical development, or, if approved, after the approved product has been marketed. Undesirable side effects caused by product candidates could cause us, any partners with which we may collaborate or regulatory authorities to interrupt, modify, delay or halt clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA or comparable foreign authorities. In such an event, trials could be suspended or terminated and the FDA or comparable foreign regulatory authorities could order us, or our potential partners, to cease further development of or deny approval of product candidates for any or all targeted indications. The drug-related side effects could affect patient recruitment or the ability of enrolled patients to complete the trial or result in product liability claims. Any of these occurrences may harm our business, financial condition, operating results and prospects.
Additionally, if we or others identify undesirable side effects, or other previously unknown problems, caused by our product candidates after obtaining U.S. or foreign regulatory approval, or other products with the same or related active ingredients, a numbercommercial prospects of potentially negative consequences could result, including:
| ●
| regulatory authorities may withdraw their approval of the product;
|
| ●
| regulatory authorities may require a recall of the product or we or our potential partners may voluntarily recall a product;
|
| ●
| regulatory authorities may require the addition of warnings or contraindications in the product labeling, narrowing of the indication in the product label or field alerts to physicians and pharmacies;
|
| ●
| we may be required to create a medication guide outlining the risks of such side effects for distribution to patients or institute a REMS;
|
| ●
| we may have limitations on how we promote the product;
|
| ●
| we may be required to change the way the product is administered or modify the product in some other way;
|
| ●
| the FDA or applicable foreign regulatory authority may require additional clinical trials or costly post-marketing testing and surveillance to monitor the safety or efficacy of the product;
|
| ●
| sales of the product may decrease significantly;
|
| ●
| we could be sued and held liable for harm caused to patients; and
|
| ●
| our brand and reputation may suffer.
|
Any of the above events resulting from undesirable side effects or other previously unknown problems could prevent us or our potential partners from achieving or maintaining market acceptance of the affected product candidate and could substantially increase the costs of commercializing our product candidates.
Because the results of preclinical studies and early-stage clinical trials are not necessarily predictive of future results, any product candidate we advance into additional clinical trials may not continueare insufficient to have favorable results or receive regulatory approval.
Success in preclinical studiesjustify our continued expenditure of the associated development and early clinical trials does not ensure that later clinical trials will generate adequate data to demonstrate the efficacy and safety of an investigational drug. Many companies in the pharmaceutical and biotechnology industries, including those with greater resources and experience, have suffered significant setbacks in clinical trials, even after reporting promising results in earlier clinical trials. We do not know whether the clinical trialsother costs, we may conduct will demonstrate adequate efficacy and safetychoose to delay, suspend, or otherwise provide adequate information to result in regulatory approval to market any ofabandon our product candidates in any particular jurisdiction. If later-stage clinical trials do not produce favorable results, our ability to achieve regulatory approval for any of our product candidates may be compromised.
We may face product liability exposure, and if successful claims are brought against us, we may incur substantial liability if our insurance coverage for those claims is inadequate.
We face an inherent risk of product liability as a result of the clinical testing of our product candidates and will face an even greater risk if we commercialize any products. This risk exists even if a product is approved for commercial sale by the FDA and manufactured in facilities licensed and regulated by the FDA or an applicable foreign regulatory authority. Our products and product candidates are designed to affect important bodily functions and processes. Any side effects, manufacturing defects, misuse or abuse associated with our product candidates could result in injury to a patient or even death. We cannot offer any assurance that we will not face product liability suits in the future, nor can we assure you that our insurance coverage will be sufficient to cover our liability under any such cases.
In addition, a liability claim may be brought against us even if our product candidates merely appear to have caused an injury. Product liability claims may be brought against us by consumers, health care providers, pharmaceutical companies or others selling or otherwise coming into contact with our product candidates, among others. If we cannot successfully defend ourselves against product liability claims we will incur substantial liabilities and reputational harm. In addition, regardless of merit or eventual outcome, product liability claims may result in:
| ●
| withdrawal of clinical trial participants;
|
| ●
| termination of clinical trial sites or entire trial programs;
|
| ●
| the inability to commercialize our product candidates;
|
| ●
| decreased demand for our product candidates;
|
| ●
| impairment of our business reputation;
|
| ●
| product recall or withdrawal from the market or labeling, marketing or promotional restrictions;
|
| ●
| substantial costs of any related litigation or similar disputes;
|
| ●
| distraction of management’s attention and other resources from our primary business;
|
| ●
| substantial monetary awards to patients or other claimants against us that may not be covered by insurance; or
|
We obtained product liability insurance coverage for our clinical trials. Large judgments have been awarded in class action or individual lawsuits based on drugs that had unanticipated side effects. Our insurance coverage may not be sufficient to cover all of our product liability related expenses or losses and may not cover us for any expenses or losses we may suffer. Moreover, insurance coverage is becoming increasingly expensive, and, in the future, we may not be able to maintain insurance coverage at a reasonable cost, in sufficient amounts or upon adequate terms to protect us against losses due to product liability. We will need to increase our product liability coverage if any of our product candidates receive regulatory approval, which will be costly, and we may be unable to obtain this increased product liability insurance on commercially reasonable terms, or at all. A successful product liability claim or series of claims brought against us could cause our stock price to decline and, if judgments exceed our insurance coverage, could decrease our cash and could harm our business, financial condition, operating results and prospects.
If any of our product candidates are approved for marketing and we are found to have improperly promoted off-label uses, or if physicians misuse our products or use our products off-label, we may become subject to prohibitions on the sale or marketing of our products, product liability claims and significant fines, penalties and sanctions, and our brand and reputation could be harmed.
The FDA and other regulatory agencies strictly regulate the marketing and promotional claims that are made about drug products. In particular, a product may not be promoted for uses or indications that are not approved by the FDA or such other regulatory agencies as reflected in the product’s approved labeling and comparative safety or efficacy claims cannot be made without direct comparative clinical data. If we are found to have promoted off-label uses of any of our product candidates, we may receive warning or untitled letters and become subject to significant liability, which would materially harm our business. Both federal and state governments have levied large civil and criminal fines against companies for alleged improper promotion and have enjoined several companies from engaging in off-label promotion. If we become the target of such an investigation or prosecution based on our marketing and promotional practices, we could face similar sanctions, which would materially harm our business. In addition, management’s attention could be diverted from our business operations, significant legal expenses could be incurred and our brand and reputation could be damaged. The FDA has also requested that companies enter into consent decrees or permanent injunctions under which specified promotional conduct is changed or curtailed. If we are deemed by the FDA to have engaged in the promotion of our products for off-label use, we could be subject to FDA regulatory or enforcement actions, including the issuance of an untitled letter, a warning letter, injunction, seizure, civil fine or criminal penalties. It is also possible that other federal, state or foreign enforcement authorities might take action if they consider our business activities constitute promotion of an off-label use, which could result in significant penalties, including criminal, civil or administrative penalties, damages, fines, disgorgement, exclusion from participation in government healthcare programs and the curtailment or restructuring of our operations.
We cannot, however, prevent a physician from using our product candidates outside of those indications for use when in the physician’s independent professional medical judgment he or she deems appropriate. Physicians may also misuse our product candidates or use improper techniques, potentially leading to adverse results, side effects or injury, which may lead to product liability claims. If our product candidates are misused or used with improper technique, we may become subject to costly litigation by physicians or their patients. Furthermore, the use of our product candidates for indications other than those cleared by the FDA may not effectively treat such conditions, which could harm our reputation among physicians and patients.
We may choose not to continue developing or commercializing any of our product candidates at any time during development or after approval,commercialization efforts with respect thereto, which would reduce or eliminate our potential return on investment for those product candidates.
AtOur ability to develop our product candidates depends, and, if TSC or any time,of our other product candidates are approved, our ability to successfully commercialize our products will depend, in part on our ability to successfully obtain sufficient quantities of the necessary APIs, other component substances and materials, and finished drug product for our product candidates. We are currently entirely dependent on third parties for the manufacture and supply of our product candidates and their component parts, including, with respect to TSC, a sole supplier. We may be unable to continue to develop or commercialize our product candidates or face significant delays in that process if we may decideare unable to discontinuesuccessfully obtain these materials or manufacture drug product in sufficient quantities.
Maintaining an adequate supply of TSC and our other product candidates to meet our needs is critical to the developmentsuccess of anyour business. However, manufacturing and supply of APIs, other substances and materials and finished drug products is a complex and technically challenging process, and changes beyond our direct control can impact the quality, volume, price, and successful delivery of our product candidates or not to continue commercializing oneimpede, delay, limit or moreprevent the successful development and commercialization of our approved product candidates for a varietycandidates. Mistakes and mishandling are not uncommon in the biopharmaceutical and pharmaceutical industry and can affect successful production and supply significantly.
As of reasons, including changes in our internal product, technology or indication focus, the appearancedate of new technologies that make our product obsolete, competition from a competing product or changes in or failure to comply with applicable regulatory requirements. If we terminate a program in whichthis Annual Report, we have invested significant resources,no internal manufacturing capabilities and therefore we willdo not receive any return onhave direct control over our investmentability to maintain drug supply sufficient to serve our needs for our ongoing and we will have missed the opportunity to have allocated those resources to potentially more productive uses.
Weplanned clinical trials or, our prospective partners may be subject to product recalls in the future that could harm our brand and reputation and could negatively affect our business.
We or our prospective partners may be subject to product recalls, withdrawals or seizures if any of our product candidates ifare approved, for marketing, fail to meet specifications or are believed to cause injury or illness or ifcommercialization. Although we are allegedultimately responsible for ensuring compliance with regulatory requirements such as cGMPs, we are dependent on our third party CMOs and other contract suppliers and manufacturers for the manufacture of our drug product, including both APIs and finished products, as well as day-to-day compliance with cGMPs and certain other manufacturing-related regulatory requirements. Facilities used by our contract suppliers and manufacturers to have violated governmental regulationsproduce the APIs and other substances and materials or finished products for commercial sale must pass inspection, provide regulators with certain technical information, and be approved by the FDA and other relevant regulatory authorities to confirm compliance with cGMP requirements and other regulatory requirements. If the safety of TSC or any of our other product candidates (or any component thereof) is found in the future to be compromised, we may not be able to successfully commercialize or obtain regulatory approval for the product candidate, and we may be held liable for injuries sustained as a result.
Any disruption in our relationship with these third parties or their ability to manufacture the APIs and finished drug product we need for our clinical trials and other development activities could result in significant delays in our anticipated development timelines and/or significant additional supply costs. Such a disruption could be the result of any number of reasons, including thosecontractual disputes with our partners, regulatory issues with our partners or at their facilities (whether or not related to Diffusion or our drug product), financial issues faced by our partners (including bankruptcy or insolvency), damages to our partners’ facilities or equipment, communication breakdowns, or acts of God. For example, during 2021 we faced certain delays in the manufacturing process for planned, new batches of TSC drug product due to the fact that, in connection with the U.S. federal government’s Operation Warp Speed initiative in response to the COVID-19 pandemic, the facility at which our former, primary CMO partner conducts significant portions of the TSC manufacturing process had been mandated to devote the majority of the facility’s available resources to the manufacture labeling, promotion, sale or distribution. Any recall, withdrawal or seizureof components of the COVID-19 vaccine.
Amplifying this risk is the fact that, notwithstanding the improvements made to our supply chain during 2021 described under, "Business - Product Development - Chemistry, Manufacturing, and Controls," we currently depend upon a sole source to manufacture our API for TSC and other aspects of our manufacturing process, limiting our available options to troubleshoot these issues. Although we actively manage this third-party relationship to ensure continuity, quality, and compliance with regulations and we intend to identify and develop alternative manufacturing and supply alternatives in the future, this process remains ongoing, will take time, and will involve significant costs. Even with these efforts, some events beyond our control, including global instability due to political unrest or from an outbreak of pandemic or contagious disease, such as COVID-19, could result in supply chain disruptions or the complete or partial failure of these manufacturing services. Any such failure or disruptions could materially adversely affect our business, financial condition, cash flows, and results of operations. Furthermore, due to the significant regulatory oversight of the pharmaceutical manufacturing process, any changes in the identity of our third-party partners or in our manufacturing processes – even if in the best interests of the Company and successful – could result in regulatory and other delays, as well as significant additional costs. In addition, if our current supplier terminated our arrangement or failed to meet our supply needs for any reason prior to the time we are able to identify sufficient alternative manufacturing capacity, we may be forced to delay our development plans significantly.
Our CMO and other manufacturing and supply partners are also engaged to supply and manufacture materials or products for other biopharmaceutical and pharmaceutical companies, exposing them to regulatory risks unrelated to the work they are doing for Diffusion but which may nevertheless impact their ability to meet their contractual requirements to us or otherwise impede their ability to supply us with sufficient quantities of drug product. Failure to meet the regulatory requirements for the production of those materials and products may also affect the regulatory clearance of a contract supplier’s or manufacturer’s facility. If the FDA or a comparable foreign regulatory agency does not approve these facilities for the supply or manufacture of our product candidates or if it withdraws its approval in the future, even if such lack of approval is unrelated to Diffusion or our product candidates, we may need to find alternative supply or manufacturing facilities.
In addition, to date we have only manufactured TSC and our other product candidates in relatively small quantities for preclinical studies and clinical trials. As we prepare for additional, later-stage clinical trials and potential commercialization, we will need to take steps to substantially increase the scale at which we are able to produce TSC, its API, and its other component parts. In order to meet these needs, our CMOs and suppliers will need to produce our API, other components, and finished product in larger quantities, more cost effectively and, in certain cases, at higher yields than they currently achieve. These third-party contractors may not be able to successfully increase the manufacturing capacity for any of such drug substance and product candidates in a timely or cost-effective manner or at all. Even if such a scale up is possible, it may require additional processes, technologies, and validation studies, which are costly, may not be successful, and which the FDA and foreign regulatory authorities would need to review and approve prior to any commercial sale of TSC or any other product candidate. In addition, quality issues may arise during those scale-up activities because of the inherent properties of a product candidate itself or in combination with other components added during the process of manufacturing, packaging, shipping, or storage.
Our reliance on contract manufacturers and suppliers further exposes us to the possibility that they, or third parties with access to their facilities, will have access to and may misappropriate our trade secrets or other proprietary information. In addition, the manufacturing facilities of certain of our suppliers are located outside of the United States. This may give rise to difficulties in importing our product candidates or their components into the United States or other countries as a result of, among other things, regulatory agency approval requirements or import inspections, incomplete or inaccurate import documentation or defective packaging.
Any of these factors could cause a delay or termination of preclinical studies, clinical trials, other development activities, regulatory submissions or approvals of our product candidates, or, if any of our product candidates is approved, commercial supply, and could result in significant, unanticipated costs or an inability to effectively develop our products candidates or commercialize our approved products on a timely basis, or at all, which could materially and adversely affect consumer confidence in our brands and lead to decreased demand for our approved products. In addition, a recall, withdrawal or seizure of any of our approved products would require significant management attention, would likely result in substantial and unexpected expenditures and would harm our business, financial condition, and operating results.results of operations.
If we or any partners with which we may collaborate are unable to achieve and maintain coverage and adequate levels of reimbursement for any of our product candidates for which we receive regulatory approval, or any future products we may seek to commercialize, their commercial success may be severely hindered.
For any of our product candidates that become available only by prescription, successful sales by us or by any partners with which we may collaborate depend on the availability of coverage and adequate reimbursement from third-party payors. Patients who are prescribed medicine for the treatment of their conditions generally rely on third-party payors to reimburse all or part of the costs associated with their prescription drugs. The availability of coverage and adequate reimbursement from governmental healthcare programs, such as Medicare and Medicaid, and private third-party payors is critical to new product acceptance. Coverage decisions may depend upon clinical and economic standards that disfavor new drug products when more established or lower cost therapeutic alternatives are already available or subsequently become available. If any of our product candidates do not demonstrate attractive efficacy profiles, they may not qualify for coverage and reimbursement. Even if we obtain coverage for a given product, the resulting reimbursement payment rates might not be adequate or may require co-payments that patients find unacceptably high. Patients are unlikely to use our products unless coverage is provided and reimbursement is adequate to cover a significant portion of the cost of our products.
In addition, the market for our product candidates will depend significantly on access to third-party payors’ drug formularies, or lists of medications for which third-party payors provide coverage and reimbursement. The industry competition to be included in such formularies often leads to downward pricing pressures on pharmaceutical companies. Also, third-party payors may refuse to include a particular branded drug in their formularies or otherwise restrict patient access to a branded drug when a less costly generic equivalent or other alternative is available.
Third-party payors, whether foreign or domestic, or governmental or commercial, are developing increasingly sophisticated methods of controlling healthcare costs. In addition, in the United States, although private third-party payors tend to follow Medicare, no uniform policy of coverage and reimbursement for drug products exists among third-party payors. Therefore, coverage and reimbursement for drug products can differ significantly from payor to payor. As a result, the coverage determination process is often a time-consuming and costly process that will require us to provide scientific and clinical support for the use of our product candidates to each payor separately, with no assurance that coverage and adequate reimbursement will be obtained.
Further, we believe that future coverage and reimbursement will likely be subject to increased restrictions both in the United States and in international markets. Third-party coverage and reimbursement for any of our product candidates for which we may receive regulatory approval may not be available or adequate in either the United States or international markets, which could harm our business, financial condition, operating results and prospects.
Healthcare reform measures could hinder or prevent the commercial success of our products and product candidates.
In the United States, there have been, and we expect there will continue to be, a number of legislative and regulatory changes to the healthcare system that could affect our future revenue and profitability and the future revenue and profitability of any partner with which we may collaborate. Federal and state lawmakers regularly propose and, at times, enact legislation that results in significant changes to the healthcare system, some of which are intended to contain or reduce the costs of medical products and services. For example, in March 2010, President Obama signed one of the most significant healthcare reform measures in decades the Affordable Care Act. It contains a number of provisions, including those governing enrollment in federal healthcare programs, reimbursement changes and fraud and abuse measures, all of which are expected to impact existing government healthcare programs and result in the development of new programs. The Affordable Care Act, among other things, (1) increased the minimum Medicaid rebates owed by most manufacturers under the Medicaid Drug Rebate Program and extended the rebate program to certain individuals enrolled in Medicaid managed care organizations, (2) established annual fees on manufacturers of certain branded prescription drugs and (3) enacted a Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50% point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D.
In addition, other legislative changes have been proposed and adopted in the United States since the Affordable Care Act was enacted. On August 2, 2011, the Budget Control Act of 2011, among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, was unable to reach required goals, thereby triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions of Medicare payments to providers up to 2% per fiscal year, which went into effect on April 1, 2013. On January 2, 2013, President Obama signed into law the American Taxpayer Relief Act of 2012, which, among other things, further reduced Medicare payments to several providers, including hospitals and imaging centers.
More recently, the Protecting Access to Medicare Act of 2014, signed into law in April 2014, provides for a 0.5% change from 2013 federal payment rates under the Medicare Physician Fee Schedule through 2014 and a 0% update from January 1 until April 1, 2015. Congressional failure to intervene to prevent these changes in payment rates may adversely affect our future revenue and operating results. The 21st Century Cures Act, which the U.S. House of Representatives passed in July 2015 and President Obama signed into law in December 2016, provides a wide range of reforms, such as broadening the types of data required to support drug approval, extending protections from genetic competition, accelerating approval of breakthrough therapies, expanding the orphan drug product program, and clarifying how manufacturers communicate about their products.
Additional state and federal healthcare reform measures may be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for our products once approved or additional pricing pressures.
It is also uncertain whether there will be any legislation that will replace or amend the Affordable Care Act. In addition, the Trump Administration has and will appoint and employ many new secretaries, directors and the like into positions of authority in the U.S. Federal government dealing with the pharmaceutical and healthcare industries that may potentially have a negative impact on the prices and the regulatory pathways for certain pharmaceuticals and healthcare products developed by the Company. Such changes in the regulatory pathways could adversely affect and or delay the ability of the Company to market and sell its products in the U.S.
We may also be subject to healthcare laws, regulation and enforcement and our failure to comply with those laws could adversely affect our business, operations and financial condition.
Certain federal and state healthcare laws and regulations pertaining to fraud and abuse and patients’ rights are and will be applicable to our business. We are subject to regulation by both the federal government and the states in which we or our partners conduct our business. The laws and regulations that may affect our ability to operate include:
| ●
| the federal Anti-Kickback Statute, which prohibits, among other things, any person or entity from knowingly and willfully offering, soliciting, receiving or providing any remuneration (including any kickback, bribe or rebate), directly or indirectly, overtly or covertly, in cash or in kind, to induce either the referral of an individual or in return for the purchase, lease, or order of any good, facility item or service, for which payment may be made, in whole or in part, under federal healthcare programs such as the Medicare and Medicaid programs;
|
| ●
| federal civil and criminal false claims laws and civil monetary penalty laws, including, for example, the federal civil False Claims Act, which impose criminal and civil penalties, including civil whistleblower or qui tam actions, against individuals or entities for, among other things, knowingly presenting, or causing to be presented, to the federal government, including the Medicare and Medicaid programs, claims for payment that are false or fraudulent or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government;
|
| ●
| the federal Health Insurance Portability and Accountability Act of 1996 (“HIPAA”), which created new federal criminal statutes that prohibit knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program or obtain, by means of false or fraudulent pretenses, representations or promises, any of the money or property owned by, or under the custody or control of, any healthcare benefit program, regardless of the payor (e.g., public or private), knowingly and willfully embezzling or stealing from a health care benefit program, willfully obstructing a criminal investigation of a health care offense and knowingly and willfully falsifying, concealing or covering up by any trick or device a material fact or making any materially false statements in connection with the delivery of, or payment for, healthcare benefits, items or services relating to healthcare matters;
|
| ●
| HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, and their implementing regulations, which impose obligations on covered entities, including healthcare providers, health plans, and healthcare clearinghouses, as well as their respective business associates that create, receive, maintain or transmit individually identifiable health information for or on behalf of a covered entity, with respect to safeguarding the privacy, security and transmission of individually identifiable health information;
|
| ●
| the federal physician sunshine requirements under the Affordable Care Act, which require manufacturers of drugs, devices, biologics and medical supplies to report annually to the Centers for Medicare & Medicaid Services information related to payments and other transfers of value provided to physicians and teaching hospitals, and ownership and investment interests held by physicians and their immediate family members; and
|
| ●
| state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws, which may apply to items or services reimbursed by any third-party payor, including commercial insurers; state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the applicable compliance guidance promulgated by the federal government, or otherwise restrict payments that may be provided to healthcare providers and other potential referral sources; state laws that require drug manufacturers to report information related to payments and other transfers of value to healthcare providers or marketing expenditures; and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts.
|
Recent health care reform legislation has strengthened these laws. For example, the recently enacted Affordable Care Act, among other things, amended the intent requirement of the federal Anti-Kickback Statute and certain criminal healthcare fraud statutes. A person or entity no longer needs to have actual knowledge of the statute or specific intent to violate it. In addition, the Affordable Care Act provided that the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act. It is uncertain whether there will be any legislation that will replace or amend the Affordable Care Act.
Achieving and sustaining compliance with these laws may prove costly. In addition, any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business. If our operations are found to be in violation of any of the laws described above or any other governmental laws or regulations that apply to us, we may be subject to penalties, including administrative, civil and criminal penalties, damages, fines, disgorgement, the exclusion from participation in federal and state healthcare programs, individual imprisonment or the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our financial results.
Our employees, independent contractors, principal investigators, consultants, vendors, CROs and any partners with which we may collaborate may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements.
We are exposed to the risk that our employees, independent contractors, principal investigators, consultants, vendors, CROs and any partners with which we may collaborate may engage in fraudulent or other illegal activity. Misconduct by these persons could include intentional, reckless or negligent conduct or unauthorized activity that violates: laws or regulations, including those laws requiring the reporting of true, complete and accurate information to the FDA or foreign regulatory authorities; manufacturing standards; federal, state and foreign healthcare fraud and abuse laws and data privacy; or laws that require the true, complete and accurate reporting of financial information or data. In particular, sales, marketing and other business arrangements in the healthcare industry are subject to extensive laws intended to prevent fraud, kickbacks, self-dealing and other abusive practices. These laws may restrict or prohibit a wide range of business activities, including research, manufacturing, distribution, pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Activities subject to these laws also involve the improper use of information obtained in the course of clinical trials, or illegal misappropriation of drug product, which could result in regulatory sanctions or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations, and serious harm to our reputation. In addition, federal procurement laws impose substantial penalties for misconduct in connection with government contracts and require certain contractors to maintain a code of business ethics and conduct. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of civil, criminal and administrative penalties, damages, monetary fines, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, contractual damages, reputational harm, diminished profits and future earnings, and curtailment of our operations, any of which could adversely affect our ability to operate our business and our operating results.
Risks Related to Our Dependence on Third Parties
We expect to rely on third-party CROs and other third parties to conduct and oversee our clinical trials and other aspects of our development process for TSC and our other product development.candidates. If these third parties do not meet our requirements or otherwise conduct the trials as required,or perform the other services for which they are engaged, we may not be able to satisfy our contractual obligations orsuccessfully develop, obtain regulatory approval for, or commercialize our product candidates when expected or at all. Furthermore, if we are not able to establish and maintain the necessary collaborative relationships with our CROs and other third party partners, we may have to alter our development and commercialization plans.
Conducting our clinical trials in a safe, compliant, and timely manner is critical to our success. We expect tocurrently rely on third-party CROs to conduct and oversee our clinical trials and other aspects of our product development. We also rely upondevelopment, as well as various medical institutions, clinical investigators, and contract laboratories, consultants, and other third parties to design and conduct our trials, to analyze the results therefrom, and to ensure that the trials are conducted in accordance with our clinical protocols and all applicable regulatory requirements, including the FDA’s regulations and GCPs, which are an international standard meant to protect the rights and health of patients and to define the roles of clinical trial sponsors, administrators and monitors, and state regulations governing the handling, storage, security and recordkeeping for drug products.GCPs. These CROs and other third parties play a significant role in the conduct of these trials and the subsequent collection and analysis of data from the clinical trials. We rely heavily on these parties for the execution of our clinical trials and preclinical studies, andtherefrom, as we control only certain aspects of their activities. Weactivities and rely heavily on them to execute our CROstrials in a safe, compliant, and other third-party contractors are requiredtimely manner. Although we intend to comply with GCPinternalize portions of some of these functions during 2021 and GLP requirements, which are regulations and guidelines enforced by the FDA and comparable foreign regulatory authorities for products in clinical development. Regulatory authorities enforce these GCP and GLP requirements through periodic inspections of trial sponsors, principal investigators and trial sites. Ifbeyond as our organization grows, we or any ofexpect to continue to rely on these third parties fail to comply with applicable GCP and GLP requirements,a significant degree in the clinical data generated in our clinical trials may be deemed unreliable and the FDA or other regulatory authority may require us to perform additional clinical trials before approving our or our partners’ marketing applications. We cannot assure you that upon inspection by a given regulatory authority, such regulatory authority will determine that any of our clinical or preclinical trials complies with applicable GCP and GLP requirements. In addition, our clinical trials must generally be conducted with product produced under cGMP regulations. Our failure to comply with these regulations and policies may require us to repeat clinical trials, which would delay the regulatory approval process.future.
If any of our CROs, or clinical trial sites, terminateor other third party partners terminates their involvement in one of our clinical trials (or with Diffusion entirely) for any reason, we may not be able to enter into alternative arrangements with alternative CROs or clinical trial sites, or do sosufficient to meet our needs, on a timely basis, on commercially reasonable terms.terms, or at all. In addition, if our relationship with clinical trial sites is terminated, we may incur significant additional costs or experience the loss of follow-up information on patients enrolled in our ongoing clinical trials, unless we are able to transfer the care of those patients to another qualified clinical trial site.
We, as well as the CROs and other third-party contractors acting on our behalf, are required to comply with GCP and GLP requirements in all of our clinical trials, which are enforced through periodic inspections of trial sponsors, principal investigators, and trial sites. If we or any of these third parties fail to comply with applicable GCP, GLP, or other regulatory requirements, the clinical data generated in our clinical trials may be deemed unreliable and we may be required to perform additional clinical trials to supplement or replace such data before receiving approval of a product candidate from the FDA foreign regulatory authority. Our clinical trials must also generally be conducted with product produced under cGMP regulations. Our and our partners’ compliance with these various regulations may be reviewed by regulatory inspections at any time, processes over which we will have very little control or immediate visibility, and a failure to comply with these regulations and policies by us, our CROs, or any of our other third party partners may result in significant delays in our development programs. In addition, principal investigators for our clinical trials may serve as scientific advisors or consultants to us from time to time and could receive cash or equity compensation in connection with such services. If these relationships and any related compensation result in perceived or actual conflicts of interest, the integrity of the data generated at the applicable clinical trial site may be questioned by the FDA.
We rely completely on third-party contractors to supply, manufacture and distribute clinical drug supplies for our product candidates, including certain sole-source suppliers and manufacturers, we intend to rely on third parties for commercial supply, manufacturing and distribution if any of our product candidates receive regulatory approval and we expect to rely on third parties for supply, manufacturing and distribution of preclinical, clinical and commercial supplies of any future product candidates. We do not currently have, nor do we plan to acquire, the infrastructure or capability to supply, manufacture or distribute preclinical, clinical or commercial quantities of drug substances or products.
Our ability to develop our product candidates depends and our ability to commercially supply our products will depend, in part, on our ability to successfully obtain theactive pharmaceutical ingredients (“APIs”) and other substances and materials used in our product candidates from third parties and to have finished products manufactured by third parties in accordance with regulatory requirements and in sufficient quantities for preclinical and clinical testing and commercialization. If we fail to develop and maintain supply relationships with these third parties, we may be unable to continue to develop or commercialize our product candidates.
We do not have direct control over the ability of our contract suppliers and manufacturers to maintain adequate capacity and capabilities to serve our needs, including quality control, quality assurance and qualified personnel. Although we are ultimately responsible for ensuring compliance with regulatory requirements such as cGMPs, we are dependent on our contract suppliers and manufacturers for day-to-day compliance with cGMPs for production of both APIs and finished products. Facilities used by our contract suppliers and manufacturers to produce the APIs and other substances and materials or finished products for commercial sale must pass inspection and be approved by the FDA and other relevant regulatory authorities. Our contract suppliers and manufacturers must comply with cGMP requirements enforced by the FDA through its facilities inspection program and review of submitted technical information. If the safety of any product or product candidate or component is compromised due to a failure to adhere to applicable laws or for other reasons, we may not be able to successfully commercialize or obtain regulatory approval for the affected product or product candidate, and we may be held liable for injuries sustained as a result. Any of these factors could cause a delay or termination of preclinical studies, clinical trials or regulatory submissions or approvals of our product candidates, and could entail higher costs or result in our being unable to effectively commercialize our approved products on a timely basis, or at all.
We also rely and will continue to rely on certain third parties as the sole source of the materials they supply or the finished products they manufacture. In the event an existing supplier fails to supply product on a timely basis or in the requested amount, supplies product that fails to meet regulatory requirements, becomes unavailable through business interruption or financial insolvency or loses its regulatory status as an approved source or if we or our manufacturers are unable to renew current supply agreements when such agreements expire and we do not have a second supplier, we likely would incur added costs and delays in identifying or qualifying replacement manufacturers and materials and there can be no assurance that replacements would be available to us on a timely basis, on acceptable terms or at all. In certain cases we may be required to get regulatory approval to use alternative suppliers, and this process of approval could delay production of our products or development of product candidates indefinitely. We and our manufacturers do not currently maintain inventory of these APIs and other substances and materials. Any interruption in the supply of an API or other substance or material or in the manufacture of a finished product could have a material adverse effect on our business, financial condition, operating results and prospects.
In addition, these contract manufacturers are engaged with other companies to supply and manufacture materials or products for such companies, which also exposes our suppliers and manufacturers to regulatory risks for the production of such materials and products. As a result, failure to meet the regulatory requirements for the production of those materials and products may also affect the regulatory clearance of a contract supplier’s or manufacturer’s facility. If the FDA or a comparable foreign regulatory agency does not approve these facilities for the supply or manufacture of our product candidates, or if it withdraws its approval in the future, we may need to find alternative supply or manufacturing facilities, which would negatively impact our ability to develop, obtain regulatory approval of or market our product candidates, if approved.
To date, our drug substances and product candidates have been manufactured in small quantities for preclinical studies and early-stage clinical trials. As we prepare for later-stage clinical trials and potential commercialization, we will need to take steps to increase the scale of production of our drug substances and product candidates, which may include transferring production to new third-party suppliers or manufacturers. In order to conduct larger or late-stage scale clinical trials for our product candidates and supply sufficient commercial quantities of the resulting drug product and its components, if that product candidate is approved for sale, our contract manufacturers and suppliers will need to produce our drug substances and product candidates in larger quantities, more cost effectively and, in certain cases, at higher yields than they currently achieve. These third-party contractors may not be able to successfully increase the manufacturing capacity for any of such drug substance and product candidates in a timely or cost-effective manner or at all. Significant scale up of manufacturing may require additional processes, technologies and validation studies, which are costly, may not be successful and which the FDA and foreign regulatory authorities must review and approve. In addition, quality issues may arise during those scale-up activities because of the inherent properties of a product candidate itself or of a product candidate in combination with other components added during the manufacturing and packaging process, or during shipping and storage of the APIs or the finished product. If our third-party contractors are unable to successfully scale up the manufacture of any of our product candidates in sufficient quality and quantity and at commercially reasonable prices, and we are unable to find one or more replacement suppliers or manufacturers capable of production at a substantially equivalent cost in substantially equivalent volumes and quality, and we are unable to successfully transfer the processes on a timely basis, the development of that product candidate and regulatory approval or commercial launch for any resulting products may be delayed, or there may be a shortage in supply, either of which could significantly harm our business, financial condition, operating results and prospects.
We expect to continue to depend on third-party contract suppliers and manufacturers for the foreseeable future. Our supply and manufacturing agreements, if any, do not guarantee that a contract supplier or manufacturer will provide services adequate for our needs. We and our contract suppliers and manufacturers continue to improve production processes, certain aspects of which are complex and unique, and we may encounter difficulties with new or existing processes. While we attempt to build in certain contractual obligations on such third-party suppliers and manufacturers, we may not be able to ensure that such third parties comply with these obligations. Depending on the extent of any difficulties encountered, we could experience an interruption in clinical or commercial supply, with the result that the development, regulatory approval or commercialization of our product candidates may be delayed or interrupted. In addition, third-party suppliers and manufacturers may have the ability to increase the price payable by us for the supply of the APIs and other substances and materials used in our product candidates, in some cases without our consent.
Additionally, any damage to or destruction of our third-party manufacturers’ or suppliers’ facilities or equipment may significantly impair our ability to have our product candidates manufactured on a timely basis. Furthermore, if a contract manufacturer or supplier becomes financially distressed or insolvent, or discontinues our relationship beyond the term of any existing agreement for any other reason, this could result in substantial management time and expense to identify, qualify and transfer processes to alternative manufacturers or suppliers, and could lead to an interruption in clinical or commercial supply.
Our reliance on contract manufacturers and suppliers further exposes us to the possibility that they, or third parties with access to their facilities, will have access to and may misappropriate our trade secrets or other proprietary information. In addition, the manufacturing facilities of certain of our suppliers are located outside of the United States. This may give rise to difficulties in importing our products or product candidates or their components into the United States or other countries as a result of, among other things, regulatory agency approval requirements or import inspections, incomplete or inaccurate import documentation or defective packaging. It is also uncertain what impact the election of Donald Trump as President will have on our third-party suppliers in light of his public statements.
Because we currently rely on a sole supplier to manufacture the active pharmaceutical ingredients of certain of our product candidates, any production problems with our supplier could adversely affect us.
We have relied upon supply agreements with third parties for the manufacture and supply of the bulk active pharmaceutical ingredients used in certain of our product candidates for purposes of preclinical testing and clinical trials. We presently depend upon a single source as the sole manufacturer of our supply of APIs for certain of our product candidates. Although we have identified alternate sources for these supplies, it would be time-consuming and costly to qualify these sources. Since we currently obtain our API from this manufacturer on a purchase-order basis, either we or the supplier may terminate our arrangement, without cause, at any time without notice. If our supplier was to terminate our arrangement or fail to meet our supply needs we might be forced to delay our development.
Manufacturing and supply of the APIs and other substances and materials used in our product candidates and finished drug products is a complex and technically challenging undertaking, and there is potential for failure at many points in the manufacturing, testing, quality assurance and distribution supply chain, as well as the potential for latent defects after products have been manufactured and distributed.
Manufacturing and supply of APIs, other substances and materials and finished drug products is technically challenging. Changes beyond our direct control can impact the quality, volume, price and successful delivery of our product candidates and can impede, delay, limit or prevent the successful development and commercialization of our product candidates. Mistakes and mishandling are not uncommon and can affect successful production and supply. Some of these risks include:
| ●
| failure of our manufacturers to follow cGMP requirements or mishandling of product while in production or in preparation for transit;
|
| ●
| inability of our contract suppliers and manufacturers to efficiently and cost-effectively increase and maintain high yields and batch quality, consistency and stability;
|
| ●
| difficulty in establishing optimal drug delivery substances and techniques, production and storage methods and packaging and shipment processes;
|
| ●
| transportation and import/export risk, particularly given the global nature of our supply chain;
|
| ●
| delays in analytical results or failure of analytical techniques that we depend on for quality control and release of product;
|
| ●
| natural disasters, labor disputes, financial distress, lack of raw material supply, issues with facilities and equipment or other forms of disruption to business operations of our contract manufacturers and suppliers; and
|
| ●
| latent defects that may become apparent after product has been released and which may result in recall and destruction of product.
|
Any of these factors could result in delays or higher costs in connection with our clinical trials, regulatory submissions, required approvals or commercialization of our products, which could harm our business, financial condition, operating results and prospects.
If we are not able to establish and maintain collaborations, we may have to alter our development and commercialization plans.
The development and potential commercialization of our product candidates will require substantial additional cash to fund expenses. In order to fund or otherwise further development of our current or future product candidates, we may collaborate with other pharmaceutical and biotechnology companies for theon their development and potential commercialization of those product candidates. We would face significant competition in seeking appropriate partners. Whetherpartners and whether we reach a definitive agreement for a collaboration will depend among other things, uponon many factors, including, our assessment of thea partner’s resources and experience, the terms and conditions of the proposed collaboration, and the proposed partner’s evaluation of a number of factors. Those factors may include the design or results of clinical trials; the likelihood of approval by the FDA or other regulatory authorities; the potential market for the subject product candidate; the costs and complexities of manufacturing and delivering such product candidate to patients; the potential of competing products; any uncertainty with respect to our ownership of our intellectual property; and industry and market conditions generally. The partner may also consider alternative product candidates or technologies for similar indications that may be available for collaboration and whether such a collaboration could be more attractive than the one with us for our product candidate. We may also be restricted under future license agreements from entering into agreements on certain terms with potential partners. CollaborationsThese types of collaborations are complex and time-consuming to negotiate and document. In addition, theredocument and could ultimately result in lower returns on investment for our stockholders than would have been achieved developing the product candidate without a significant number of recent business combinations among large pharmaceutical companies that have resulted in a reduced number of potential future partners. Collaborations typically impose detailed obligations on each party. Ifpartner. Further, if we were to breach our obligations under the agreements governing any such future collaboration, we may face substantial consequences, including potential termination of the collaboration, and our rights to our partners’ product candidates, in which we have invested substantial time and money, would be lost.
Any failure to successfully enter into and maintain the necessary relationships with CROs and our other current and future third party partners and collaborators could materially and adversely affect our business, financial condition, and results of operations.
WeGeneral Risks Related to the Development, Regulatory Approval, and Commercialization of TSC and Our Other Product Candidates
Our business, financial condition, or results of operations may notalso be successful in our effortsmaterially adversely affected by a number of general risks related to implement collaborations or other alternative arrangements for the development of our product candidates. When we partner with a third party for development and commercialization of a product candidate, we can expect to relinquish to the third party some or all of the control over the future success of that product candidate. Our collaboration partner may not devote sufficient resources to the commercializationregulatory approval of our product candidates or may otherwise fail in their commercialization. The terms of any collaboration or other arrangement that we establish mayare not be favorablespecific to us. In addition, any collaboration that we enter into may be unsuccessful in the development and commercialization of our product candidates. In some cases, we may be responsible for continuing preclinical and initial clinical development of a partnered product candidate or research program, and the payment we receive from our collaboration partner may be insufficient to cover the cost of this development.Company, including:
We may not be able to negotiate collaborations on a timely basis, on acceptable terms or at all. If• Our COVID Trial, which we are unable to do so,completed in February 2021, was conducted in Bucharest, Romania and we may have to curtail the development of a product candidate, reduce or delay its development program or one or more of our other development programs, delay its potential commercialization or increase our expenditures and undertake development or commercialization activities at our own expense. If we elect to increase our expenditures to fund development or commercialization activities on our own, we may need to obtain additional capital, which may not be available to us on acceptable terms or at all. If we do not have sufficient funds, we may not be able to further develop our product candidates or bring them to market and generate product revenue.
Risks Related to Our Business and Financial Operations
We will need to further increase the size and complexity of our organization in the future and weconduct additional clinical trials for TSC or our other product candidates outside the U.S. In connection with an application for marketing approval, the FDA may experience difficulties in executing our growth strategy and managing any growth.
Our management, personnel, systems and facilities currently in placedetermine not to accept data from clinical trials conducted outside of the U.S. if they determine the data presented therefrom cannot be considered valid without further inspection of the clinical trial site, are not adequateapplicable to support our business planthe U.S. population and future growth. We will need to further expand our scientific, clinical, managerial, operational, financial andU.S. medical practice, or as a result of certain other resources to support our planned research, development and commercialization activities, all of which will require additional capital resources.
Our need to manage our operations, growth and various projects effectively requires that we:
| ●
| continue to improve our operational, clinical, financial, management and regulatory compliance controls and reporting systems and procedures;
|
| ●
| attract and retain sufficient numbers of talented employees;
|
| ●
| manage our preclinical and clinical trials effectively;
|
| ●
| manage our third-party supply and manufacturing operations effectively and in a cost-effective manner, while increasing production capabilities for our current product candidates to commercial levels;
|
| ●
| manage our development efforts effectively while carrying out our contractual obligations to partners and other third parties;
|
| ●
| manage our commercialization activities for our product candidates effectively and in a cost-effective manner; and
|
| ●
| establish and maintain relationships with development and commercialization partners.
|
In addition, historically, we have utilized and continue to utilize the services of part-time outside consultants to perform a number of tasks for us, including tasks related to preclinical and clinical testing. Our growth strategy may also entail expanding our use of consultants to implement these and other tasks going forward. We rely on consultants for certain functions of our business and will need to effectively manage these consultants to ensure that they successfully carry out their contractual obligations and meet expected deadlines.factors. There can be no assurance that the FDA will accept any data we obtain from the COVID Trial or other trials we may conduct outside the U.S. in the future.
• We face a number of risks related to the potential for one or more of our future product candidates to cause undesirable side effects, have other unexpected properties, contain manufacturing defects, or be subject to misuse or abuse. The occurrence of one or more of these events with respect to a product candidate or product could delay or prevent its regulatory approval, limit its commercial potential, result in additional pre- or post-approval regulatory requirements, or subject us to product liability exposure to consumers, health care providers, or others. Product liability claims could be brought in the future even if a product candidate is ultimately approved for commercial sale and manufactured in facilities licensed and regulated by the appropriate governmental authorities, and if product liability claims brought against us in the future were to be successful, we could incur substantial liability if our insurance coverage for those claims proved to be inadequate.
• Our employees, independent contractors, principal investigators, consultants, vendors, CROs, and other third parties we work with in the course of our development activities may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements, during the course of their employment or other engagement with us. Any such misconduct or improper activities, whether intentional or negligent, could result in regulatory sanctions or other penalties against the Company, exclusion from federal healthcare programs such as Medicare and Medicaid, the incurrence of substantial defense costs, and serious harm to our reputation.
In addition, although we currently have no marketed products, in the event TSC or any of our other product candidates are approved for marketing and commercial sale by the FDA or any other regulatory authority, our business, financial condition, or results of operations may be materially adversely affected by a number of general risks related to the commercialization of such products that are not specific to our Company, including:
• Even if our product candidates obtain regulatory approval, they may fail to achieve the broad degree of physician and patient adoption and use necessary for commercial success. The degree and rate of physician and patient adoption will depend on a number of factors, including the clinical indications for which a product candidate is approved and its effectiveness compared to other therapies, cost and the availability of reimbursement and other coverage from third party payors, our ability to educate patients and healthcare providers regarding a new therapy, and the effectiveness of our sales and marketing efforts. Furthermore, we will face significant competition, often from products sold and marketed by companies with far greater resources than Diffusion, and our failure to effectively compete may prevent us from achieving significant market penetration.
• With respect to any such future products available only by prescription, if we are unable to achieve and maintain coverage and adequate levels of reimbursement from third party payors – including governmental health programs such as Medicare and Medicaid and private insurance companies – and access to such third party payors’ drug formularies, the commercial success of those products may be ableseverely hindered. If any such products do not demonstrate attractive efficacy profiles, they may not qualify for coverage and reimbursement and, even if we obtain coverage for a given product, the resulting reimbursement payment rates might not be adequate, may require co-payments that patients find unacceptably high, and may vary from payor to managepayor, and there is no assurance that coverage and reimbursement levels necessary to achieve commercial success will be obtained.
• Any such future products candidates that we commercialize will be subject to ongoing and continued regulatory review, including rules and regulations of the FDA and similar non-U.S. governmental authorities relating to advertising, marketing and labeling (including restrictions on the promotion of off-label use), potential REMS requirements, routine manufacturing and other review, and required compliance with GLP. If we or a regulatory agency discovers previously unknown problems with any such product, or any facility at or process by which it is manufactured, we may face restrictions on the sale or distribution of such product or on our existing consultantsCompany as a whole, including regulatory actions requiring us to modify marketing or findsales materials, suspend manufacturing or ongoing trials, initiate a recall or withdraw the product from the market entirely, enter into a consent decree, or submit to other competent outside consultants, as needed, on economically reasonable terms,civil or at all.criminal investigations and penalties. If we are not able to effectively manage our growthachieve and expand our organization by hiring new employees and expanding our use of consultants, we might be unable to implement successfully the tasks necessary to execute effectively on our planned research, development and commercialization activities and, accordingly, might not achieve our research, development and commercialization goals.
If we fail to attract and retain management and other key personnel, we may be unable to continue to successfully develop or commercialize our product candidates or otherwise implement our business plan.
Our ability to compete in the highly competitive pharmaceuticals industry depends upon our ability to attract and retain highly qualified managerial, clinical, scientific, medical, sales and marketing and other personnel. We are highly dependent on our management and scientific personnel, including our Chief Executive Officer, Chief Scientific Officer and our Senior Vice President – Finance, Treasurer and Secretary, certain consultants and members of our Board of Directors who are well known and respected in our industry. The loss of the services of any of these individuals could impede, delay or prevent the successful development of our product pipeline, completion of our planned clinical trials, commercialization of our product candidates or in-licensing or acquisition of new assets and could negatively impact our ability to successfully implement our business plan. If we lose the services of any of these individuals, we might not be able to find suitable replacements on a timely basis or at all, and our business could be harmed as a result. We do not maintain “key man” insurance policies on the lives of these individuals or the lives of any of our other employees. With the exception of our Chief Executive Officer, Chief Scientific Officer and our Senior Vice President – Finance and Treasurer, each of whom is subject to an employment agreement, we employ our executive officers and key personnel on an at-will basis and their employment can be terminated by us or them at any time, for any reason and without notice. In order to retain valuable employees at our company, in addition to salary and cash incentives, we provide stock options that vest over time. The value to employees of stock options that vest over time will be significantly affected by movements in our stock price that are beyond our control, and may at any time be insufficient to counteract offers from other companies.
We might not be able to attract or retain qualified management and other key personnel in the future due to the intense competition for qualified personnel among biotechnology, pharmaceutical and other businesses, particularly in the Charlottesville, Virginia area where we are headquartered. We could have difficulty attracting experienced personnel to our company and may be required to expend significant financial resources in our employee recruitment and retention efforts. Many of the other pharmaceutical companies with whom we compete for qualified personnel have greater financial and other resources, different risk profiles and longer histories in the industry than we do. They also may provide more diverse opportunities and better chances for career advancement. If we are not able to attract and retain the necessary personnel to accomplish our business objectives, we may experience constraints that will harm our ability to implement our business strategy and achieve our business objectives.
We currently have no sales and marketing personnel or capabilities. If we are unable to establish sales and marketing capabilities on our own or through third parties when we are ready to commercialize our product candidates, we will be unable to successfully commercialize our product candidates, if approved, or generate product revenue.
We currently have no sales and marketing personnel or capabilities. To commercialize our product candidates, if approved, in the United States, Canada, the European Union and other jurisdictions we may seek to enter, we must build our marketing, sales, distribution, managerial and other non-technical capabilities or make arrangements with third parties to perform these services, andregulatory compliance, we may not be successful in doing so. We as a company have no prior experience in the marketing, sale and distribution of pharmaceutical products and there are significant risks involved in building and managing a sales organization, includingpermitted to market our product candidates, which would adversely affect our ability to hire, retaingenerate revenue and incentivize qualified individuals, generate sufficient sales leads, provide adequate trainingachieve or maintain profitability.
• The biopharmaceutical and pharmaceutical industries are highly regulated and the potential for future legislative reform provides uncertainty and potential threats to salesour business and marketing personnelour potential future revenue and effectively manageprofitability of any such future products. In the U.S., there have been, and we expect there will continue to be, a geographically dispersed salesnumber of legislative and marketing team. Any failureregulatory changes to the healthcare system intended to contain or delayreduce the costs of medical products and medical services including those described under the heading Part I – Item 1. Business – Certain Other Legislation and Regulations – Current Healthcare Laws and Regulations. Additional state and federal healthcare reform measures may be adopted in the developmentfuture, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for our internal sales, marketing and distribution capabilities would adversely impactproducts once approved or additional pricing pressures. We cannot predict the commercializationlikelihood, nature, or extent of these products. Wegovernment regulation that may choose to collaborate with additional third parties that have direct sales forces and established distribution systems, either to augment our own sales force and distribution systemsarise from future legislation or administrative or executive action, whether in lieu of our own sales force and distribution systems. If we are unable to enter into such arrangements on acceptable termsthe U.S. or at all,other market territories we may not be able to successfully commercialize our product candidates. The inability to successfully commercialize our product candidates, either on our own or through collaborations with one or more third parties, would harm our business, financial condition, operating results and prospects.
Our failure to successfully in-license, acquire, develop and market additional product candidates or approved products would impair our ability to grow our business.pursue.
We are considering activities to in-license, acquire, develop and market additional products and product candidates. If we implement these activities, we may be dependent upon pharmaceutical companies, academic scientists and other researchers to sell or license products or technology to us. The success of this strategy depends partly upon our ability to identify and select promising pharmaceutical product candidates and products, negotiate licensing or acquisition agreements with their current owners and finance these arrangements.
The process of proposing, negotiating and implementing a license or acquisition of a product candidate or approved product is lengthy and complex. Other companies, including some with substantially greater financial, marketing, sales and other resources, may compete with us for the license or acquisition of product candidates and approved products. We have limited resources to identify and execute the acquisition or in-licensing of third-party products, businesses and technologies and integrate them into our current infrastructure. Moreover, we may devote resources to potential acquisitions or licensing opportunities that are never completed, or we may fail to realize the anticipated benefits of such efforts. We may not be able to acquire the rights to additional product candidates on terms that we find acceptable, or at all.
Further, any product candidate that we acquire may require additional development efforts prior to commercial sale, including preclinical or clinical testing and approval by the FDA and applicable foreign regulatory authorities. All product candidates are prone to risks of failure typical of pharmaceutical product development, including the possibility that a product candidate will not be shown to be sufficiently safe and effective for approval by regulatory authorities. In addition, we cannot provide assurance that any approved products that we acquire will be manufactured or sold profitably or achieve market acceptance.
If we implement activities to in-license and acquire product candidates and we in-license and acquire commercial-stage products or engage in other strategic transactions,it could impact our liquidity, increase our expenses and present significant distractions to our management.
If we implement a strategy to in-license and acquire product candidates, we may in-license and acquire commercial-stage products or engage in other strategic transactions. Additional potential transactions that we may consider include a variety of different business arrangements, including spin-offs, strategic partnerships, joint ventures, restructurings, divestitures, business combinations and investments. Any such transaction may require us to incur non-recurring or other charges, may increase our near- and long-term expenditures and may pose significant integration challenges or disrupt our management or business, which could adversely affect our operations and financial results. For example, these transactions entail numerous potential operational and financial risks, including:
| ●
| exposure to unknown liabilities;
|
| ●
| disruption of our business and diversion of our management’s time and attention in order to develop acquired products, product candidates or technologies;
|
| ●
| incurrence of substantial debt or dilutive issuances of equity securities to pay for acquisitions;
|
| ●
| substantial acquisition and integration costs;
|
| ●
| write-downs of assets or impairment charges;
|
| ●
| increased amortization expenses;
|
| ●
| difficulty and cost in combining the operations and personnel of any acquired businesses with our operations and personnel;
|
| ●
| impairment of relationships with key suppliers, partners or customers of any acquired businesses due to changes in management and ownership; and
|
| ●
| inability to retain our key employees or those of any acquired businesses.
|
Accordingly, there can be no assurance that we will undertake or successfully complete any transactions of the nature described above, and any transaction that we do complete could harm our business, financial condition, operating results and prospects. We have no current plan, commitment or obligation to enter into any other transaction described above.
Our operating results may fluctuate significantly, which makes our future operating results difficult to predict and could cause our operating results to fall below expectations.
Our near-term operations will be limited primarily to researching and developing our product candidates and undertaking preclinical studies and clinical trials of our product candidates. We have not yet obtained regulatory approvals for any of our product candidates. Consequently, any predictions you make about our future success or viability may not be as accurate as they could be if we had a longer operating history or approved products on the market. From time to time, we may enter into collaboration agreements and license agreements with other companies that include development funding and significant upfront and milestone expenditures and payments, and we expect that amounts earned from or paid pursuant to these agreements will be a significant source of our capital expenditures and an important source of our revenue. Accordingly, our revenue and profitability may depend on development funding and the achievement of development and clinical milestones under potential future collaboration and license agreements and sales of our products, if approved. These upfront and milestone payments may vary significantly from period to period and any such variance could cause a significant fluctuation in our operating results from one period to the next. In addition, we measure compensation cost for stock-based awards made to employees at the grant date of the award, based on the fair value of the award as determined by our Board, and recognize the cost as an expense over the employee’s requisite service period. As the variables that we use as a basis for valuing these awards change over time, including our underlying stock price and estimated volatility, the magnitude of the expense that we must recognize may vary significantly. Furthermore, our operating results may fluctuate due to a variety of other factors, many of which are outside of our control and may be difficult to predict, including the following:
| ●
| delays in the commencement, enrollment and the timing of clinical testing for our product candidates;
|
| ●
| the timing and success or failure of clinical trials for our product candidates or competing product candidates, or any other change in the competitive landscape of our industry, including consolidation among our competitors or partners;
|
| ●
| any delays in regulatory review and approval of product candidates in clinical development;
|
| ●
| the timing and cost of, and level of investment in, research and development activities relating to our product candidates, which may change from time to time;
|
| ●
| the cost of manufacturing our product candidates, which may vary depending on FDA guidelines and requirements, and the quantity of production;
|
| ●
| our ability to obtain additional funding to develop our product candidates;
|
| ●
| expenditures that we will or may incur to acquire or develop additional product candidates and technologies;
|
| ●
| the level of demand for our product candidates, should they receive approval, which may vary significantly;
|
| ●
| potential side effects of our product candidates that could delay or prevent commercialization or cause an approved drug to be taken off the market;
|
| ●
| the ability of patients or healthcare providers to obtain coverage of or sufficient reimbursement for our product candidates, if approved;
|
| ●
| our dependency on third-party manufacturers to supply or manufacture our product candidates;
|
| ●
| our ability to establish an effective sales, marketing and distribution infrastructure in a timely manner;
|
| ●
| market acceptance of our product candidates, if approved, and our ability to forecast demand for those product candidates;
|
| ●
| our ability to receive approval and commercialize our product candidates outside of the United States;
|
| ●
| our ability to establish and maintain collaborations, licensing or other arrangements;
|
| ●
| our ability and third parties’ abilities to protect intellectual property rights;
|
| ●
| costs related to and outcomes of potential litigation or other disputes;
|
| ●
| our ability to adequately support future growth;
|
| ●
| our ability to attract and retain key personnel to manage our business effectively;
|
| ●
| our ability to maintain adequate insurance policies; and
|
| ●
| future accounting pronouncements or changes in our accounting policies.
|
If we fail to comply with environmental, health and safety laws and regulations, we could become subject to fines or penalties or incur significant costs.
In connection with our research and development activities and our manufacture of materials and product candidates, we are subject to federal, state and local laws, rules, regulations and policies governing the use, generation, manufacture, storage, air emission, effluent discharge, handling and disposal of certain materials, biological specimens and wastes. Although we believe that we have complied with the applicable laws, regulations and policies in all material respects and have not been required to correct any material noncompliance, we may be required to incur significant costs to comply with environmental and health and safety regulations in the future. Current or future laws and regulations may impair our research, development or production efforts. Failure to comply with these laws and regulations also may result in substantial fines, penalties or other sanctions.
Our operating results and liquidity needs could be negatively affected by market fluctuations and economic downturn.
Our operating results and liquidity could be negatively affected by economic conditions generally, both in the United States and elsewhere around the world. Domestic and international equity and debt markets have experienced and may continue to experience heightened volatility and turmoil based on domestic and international economic conditions and concerns. In the event these economic conditions and concerns continue or worsen and the markets continue to remain volatile, our operating results and liquidity could be adversely affected by those factors in many ways, including weakening demand for certain of our product candidates and making it more difficult for us to raise funds if necessary, and our stock price may decline. Additionally, although we plan to market our products primarily in the United States, our partners may have extensive global operations, indirectly exposing us to risk.
Our ability to utilize our net operating loss (“NOL”) carryforwards and other deferred tax assets may be limited.
As of December 31, 2016, the Company had NOL carryforwards available to reduce future taxable income, if any, for income tax purposes. If not utilized, the federal and Virginia NOL carryforwards will begin expiring during the year ending December 31, 2034. Under Section 382 of Internal Revenue Code of 1986, as amended (the “Code”), if a corporation undergoes an “ownership change,” generally defined as a greater than 50% change (by value) in its equity ownership over a three-year period, the corporation’s ability to use its pre-change NOL carryforwards and other pre-change tax attributes (such as research tax credits) to offset its post-change income may be limited. We believe that our recent merger, and other transactions that have occurred, triggered an “ownership change” limitation that significantly limited our ability to utilize our NOL carryforwards. We may also experience ownership changes in the future as a result of the ongoing Private Placement and subsequent shifts in our stock ownership. As a result, if we earn net taxable income, our ability to use our pre-change NOL carryforwards and other deferred tax assets to offset U.S. federal taxable income may be subject to limitations, which could potentially result in increased future tax liability to us. In addition, similar limitations may apply at the state level and there may be periods during which the use of NOL carryforwards is suspended or otherwise limited, which could accelerate or permanently increase state taxes owed.
We may be adversely affected by natural disasters and other catastrophic events, and by man-made problems such as terrorism, that could disrupt our business operations and our business continuity and disaster recovery plans may not adequately protect us from a serious disaster.
Our corporate headquarters are located in Charlottesville, Virginia. If a disaster, power outage or other event occurred that prevented us from using all or a significant portion of our headquarters, that damaged critical infrastructure, such as enterprise financial systems, manufacturing resource planning or enterprise quality systems, or that otherwise disrupted operations, it may be difficult or, in certain cases, impossible for us to continue our business for a substantial period of time. Our contract manufacturers’ and suppliers’ facilities are located in multiple locations, where other natural disasters or similar events, such as blizzards, tornadoes, earthquakes, fires, explosions or large-scale accidents or power outages, could severely disrupt our operations and have a material adverse effect on our business, financial condition, operating results and prospects. In addition, acts of terrorism and other geo-political unrest could cause disruptions in our business or the businesses of our partners, manufacturers or the economy as a whole. All of the aforementioned risks may be further increased if we do not implement a disaster recovery plan or our partners’ or manufacturers’ disaster recovery plans prove to be inadequate. To the extent that any of the above should result in delays in the regulatory approval, manufacture, distribution or commercialization of our product candidates, our business, financial condition, operating results and prospects would suffer.
Our business and operations would suffer in the event of failures in our internal computer systems.
Despite the implementation of security measures, our internal computer systems and those of our current and any future partners, contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. While we have not experienced any such material system failure, accident or security breach to date, if such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our manufacturing activities, development programs and our business operations. For example, the loss of manufacturing records or clinical trial data from completed or future clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach were to result in a loss of, or damage to, our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and the further commercialization and development of our products and product candidates could be delayed.
Risks Related to Our Intellectual Property
We may not be able to obtain or enforce patent rights or other intellectual property rights that cover our product candidates and technologies that are of sufficient breadth to prevent third parties from competing against us.
Our successThe pharmaceutical industry is characterized by rapidly advancing technologies, intense competition, and a strong emphasis on developing proprietary therapeutics. Numerous companies are engaged in the development, patenting, manufacturing, and marketing of health care products competitive with respect to our product candidatesthose that we are developing, and technologies will depend in part onwe face competition from a number of sources, such as pharmaceutical companies, generic drug companies, biotechnology companies and academic and research institutions. Accordingly, our ability to obtain and maintain patent protection in both the United StatesU.S. and non-U.S. jurisdictions will be critical to our ability to successfully develop, obtain regulatory approval for, and, in particular, commercialize TSC and our other countries,product candidates. These protections are and will be essential to preservepreserving and protecting our novel inventions, proprietary developments, and trade secrets and to preventpreventing third parties from infringing upon them. In particular, our proprietary rights. Our ability to protect any of our product candidates from unauthorized or infringing use by third parties depends in substantial part on our ability to obtain and maintain valid and enforceable patents.patents in the U.S. and worldwide.
Our patent portfolio includes patents and patent applications in the United StatesU.S. and foreign jurisdictions where we believe there is a market opportunity forother major markets covering our products. The covered technology with varying scope, including issued U.S. patents related to composition of matter, formulation, methods of delivery, and methods of use and the scope of coverage vary from country to country. Although we believe that our intellectual property position is strong and are currently assessing our operations and existing portfolio for additional intellectual property opportunities, we do not have – and may be unable to obtain – patent protection for every aspect of our technology. For thoseaspects of our technology for which we do not have patent coverage, or in countries where we do not have granted patents, we may not have any ability to prevent the unauthorized use of our technologies. Anytechnologies or technologies substantially similar to ours, and any patents that we may obtain in the future may be narrow in scope and thus easily circumvented by competitors. Further, in countries where we do not have granted patents, third parties may be able to make, use or sell products identical to or substantially similar to, our product candidates.
The patent application process, also known as patent prosecution, is expensive and time-consuming, and we and our current or future licensors and licensees may not be able to prepare, file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner. It is also possible that we or our current licensors, or any future licensors or licensees, will fail to identify patentable aspects of inventions made in the course of development and commercialization activities before it is too late to obtain patent protection on them. Therefore, these and any of our patents and applications may not be prosecuted and enforced in a manner consistent with the best interests of our business. It is possible that defects of form in the preparation or filing of our patents or patent applications may exist, or may arise in the future, such as with respect to proper priority claims, inventorship, claim scope or patent term adjustments. If our current licensors, or any future licensors or licensees, are not fully cooperative or disagree with us as to the prosecution, maintenance or enforcement of any patent rights, such patent rights could be compromised and we might not be able to prevent third parties from making, using and selling competing products. If there are material defects in the form or preparation of our patents or patent applications, such patents or applications may be invalid and unenforceable. Moreover, our competitors may independently develop equivalent knowledge, methods and know-how. Any of these outcomes could impair our ability to prevent competition from third parties, which may have an adverse impact on our business, financial condition and operating results.
Due to legal standards relating to patentability, validity, enforceability and claim scope of patents covering pharmaceutical inventions, our ability to obtain, maintain and enforce patents is uncertain and involves complex legal and factual questions. Accordingly, rights underThe patent application process, also known as patent prosecution, is expensive and time-consuming, and we may not be able to prepare, file, and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner. It is also possible that we will fail to identify patentable aspects of inventions made in the course of development and commercialization activities before it is too late to obtain patent protection on them. While we are currently engaged in efforts to obtain additional intellectual property and patent protections for TSC, there is no assurance we will obtain such protections through our applications. Therefore, these and any existingof our patents and applications may not be prosecuted and enforced in an optimal manner. It is also possible that defects of form in the preparation or filing of our patents or patent applications may exist, or may arise in the future, such as with respect to inadvertent prior public disclosures, proper priority claims, inventorship, claim scope, or patent term adjustments. If our current or future third party development partners are not fully cooperative or disagree with us as to the prosecution, maintenance, or enforcement of any patent rights, those patent rights could be compromised and we might not be able to prevent third parties from making, using and selling competing products. Moreover, our competitors may independently develop equivalent knowledge, methods and know-how. Accordingly, we cannot guarantee that any patents will issue from any of our currently pending patent applications, which could impair our ability to prevent competition from third parties.
Even for aspects of our technology for which we mighthave obtained, or obtain or license may not cover our product candidates, orin the future, patent protection, the complexity of legal and factual questions underlying such claims means they may not provide us with sufficient protection for our product candidates to afford a commercial advantage against competitive products or processes, including those from branded and generic pharmaceutical companies. In addition, we cannot guarantee that any patents will issue from any pending or future patent applications owned by or licensed to us. Even if patents have issued or will issue, weWe cannot guarantee that the claims of these patents are or will be held valid or enforceable by the courts or will provide us with any significant protection against competitive products or otherwise be commercially valuable to us.
Competitors in the fields of oncology have created a substantial amount of prior art, including scientific publications, patents and patent applications. Our ability to obtain and maintain valid and enforceable patents depends on whether the differences between our technology and the prior art allow our technology to be patentable over the prior art. Although we believe that our technology includes certain inventions that are unique and not duplicative of any prior art, we do not have outstanding issued patents covering all of the recent developments in our technology and we are unsure of the patent protection that we will be successful in obtaining, if any. Even if the patents do successfully issue, third Third parties may design around or challenge the validity, enforceability or scope of such issued patents or any other issued patents we own or license, which may result in such patents being narrowed, invalidated or held unenforceable. If the breadth or strength of protection provided by the patents we hold or pursue with respect to our product candidates is challenged, it could dissuade companies from collaborating with us to develop, or threaten our ability to commercialize, our product candidates.
The laws of some foreign jurisdictions do not provide intellectual property rights to the same extent as in the United States and many companies have encountered significant difficulties in protecting and defending such rights in foreign jurisdictions. If we encounter such difficulties in protecting or are otherwise precluded from effectively protecting our intellectual property in foreign jurisdictions, our business prospects could be substantially harmed. The patent positions of pharmaceutical and biotechnology companies can be highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. Changes in either the patent laws or in the interpretations of patent laws in the United StatesU.S. and other countries may diminish the value of our intellectual property. Accordingly, we cannot predict the breadth of claims that may be allowed or enforced in our patents or in third-party patents.
The degree of future protection of our proprietary rights is uncertain. Patent protection may be unavailable or severely limited in some cases and may not adequately protect our rights or permit us to gain or keep our competitive advantage. For example:
| ●
| we might not have been the first to invent or the first to file the inventions covered by each of our pending patent applications and issued patents;
|
| ●
| others may independently develop similar or alternative technologies or duplicate any of our technologies;
|
| ●
| the patents of others may have an adverse effect on our business;
|
| ●
| any patents we obtain or our licensors’ issued patents may not encompass commercially viable products, may not provide us with any competitive advantages or may be challenged by third parties;
|
| ●
| any patents we obtain or our in-licensed issued patents may not be valid or enforceable; and
|
| ●
| we may not develop additional proprietary technologies that are patentable.
|
PatentsIn addition, patents have a limited lifespan.lifespan, presenting further challenges in effectively protecting our technologies and associated commercial position. In the United States,U.S., the natural expiration of a patent is generally 20 years after it is filed. Various extensions may be available; howeveravailable under a variety of legislative and regulatory avenues but often the life of a patent,afforded by these extensions and the protection it affords, is limited. Withoutprotections they afford are limited relative to full patent protection for our product candidates, we may be open to competition from generic versions of our product candidates. Further, theprotection. The extensive period of time between patent filing and regulatory approval for a product candidate limits the time during which we can market a product candidate under patent protection, which may particularly affect the profitability of our early-stage product candidates. Even if patents covering our products are obtained, once the patent life has expired, we may be open to competition from competitive products. If one of our products requires extended development, testing and/or regulatory review, patents protecting such products might expire before or shortly after such products are commercialized. As a result, our owned and licensed patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours, which could have a material adverse effect on our business, financial condition and results of operations.
To this point, we have been developing TSC since the founding of Diffusion LLC in the early 2000s and, as a result, portions of our patent portfolio, including certain patents related to TSC’s composition of matter, will expire beginning in 2023, which may be prior to the time that we are able to obtain regulatory approval for and, if approved, commercialize TSC. Moreover, the normal life (i.e., with no adjustments or extensions) of our key issued patents related to the composition of matter of TSC extends to 2026, with potential patent term extensions to 2031, and the normal life of our patents related to an oral formulation of TSC extends to 2031, with potential patent term extensions to 2036. While the Company is actively engaged in efforts to obtain additional patent protection covering our product candidates, there is no assurance that we will successfully obtain such patent protection.. As a result, our owned and licensed patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours. For example, we are aware of other companies that currently use different formulations of TSC, which could adversely affect the Company.
Furthermore, the laws of some foreign jurisdictions do not provide intellectual property rights to the same extent as in the United States and many companies have encountered significant difficulties in protecting and defending such rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection, particularly those relating to pharmaceuticals, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally in those countries. If we encounter such difficulties in protecting or are otherwise precluded from effectively protecting our intellectual property in foreign jurisdictions, our business prospects could be substantially harmed.
Proprietary trade secrets and unpatented know-how are also very important to our business. Although we have taken steps to protect our trade secrets and unpatented know-how by entering into confidentiality agreements with third parties, and intellectual property protection agreements with certain employees, consultants and advisors, third parties may still obtain this information or we may be unable to protect our rights. We also have limited control over the protection of trade secrets used by our suppliers, manufacturers and other third parties. There can be no assurance that binding agreements will not be breached, that we would have adequate remedies for any breach or that our trade secrets and unpatented know-how will not otherwise become known or be independently discovered by our competitors. If trade secrets are independently discovered, we would not be able to prevent their use. Enforcing a claim that a third party illegally obtained and is using our trade secrets or unpatented know-how is expensive and time-consuming, and the outcome is unpredictable. In addition, courts outside the United StatesU.S. may be less willing to protect trade secret information.
Changes in patent law could diminish the value of patents in general, thereby impairing our ability to protect our product candidates.
The United States has recently enacted and is currently implementing wide-ranging patent reform legislation. Further, recent U.S. Supreme Court rulings have either narrowed the scope of patent protection available in certain circumstancesIf we are unable to adequately obtain or weakened the rights of patent owners in certain situations. In addition to increasing uncertainty with regard to our ability to obtain patents in the future, this combination of events has created uncertainty with respect to the scope and value of patents, once obtained.
For our U.S. patent applications containing a priority claim after March 16, 2013, there is a greater level of uncertainty in the patent law. In September 2011, the Leahy-Smith America Invents Act, also known as the America Invents Act (“AIA”) was signed into law. The AIA includes a number of significant changes to U.S. patent law, including provisions that affect the way patent applications will be prosecuted and may also affect patent litigation. The USPTO is currently developing regulations and procedures to govern administration of the AIA, and many of the substantive changes to patent law associated with the AIA. It is not clear what other, if any, impact the AIA will have on the operation of our business. Moreover, the AIA and its implementation could increase the uncertainties and costs surrounding the prosecution ofenforce our patent applications and the enforcement or defenseother intellectual property rights for any reason, it could materially and adversely affect our business, financial condition, and results of our issued patents, all of which could have an adverse effect on our business. An important change introduced by the AIA is that, as of March 16, 2013, the United States transitioned to a “first-to-file” system for deciding which party should be granted a patent when two oroperations. For more patent applications are filed by different parties claiming the same invention. A third party that files a patent application in the USPTO after that date but before us could therefore be awarded a patent covering an invention of ours even if we had made the invention before it was made by the third party. This will require us to be cognizant going forward of the time from invention to filing of a patent application.
Among some of the other changes introduced by the AIA are changes that limit where a patentee may file a patent infringement suit and providing opportunities for third parties to challenge any issued patent in the USPTO. This applies to all of our U.S. patents, even those issued before March 16, 2013. Because of a lower evidentiary standard in USPTO proceedings compared to the evidentiary standard in United States federal court necessary to invalidate a patent claim, a third party could potentially provide evidence in a USPTO proceeding sufficient for the USPTO to hold a claim invalid even though the same evidence would be insufficient to invalidate the claim if first presented in a district court action. Accordingly, a third party may attempt to use the USPTO procedures to invalidate our patent claims that would not have been invalidated if first challenged by the third party as a defendant in a district court action.
Depending on decisions by the U.S. Congress, the U.S. federal courts, the USPTO or similar authorities in foreign jurisdictions, the laws and regulations governing patents could change in unpredictable ways that may weaken our and our licensors’ ability to obtain new patents or to enforce existing patents we and our licensors or partners may obtain in the future.
We may not be able to protectinformation about our intellectual property rights throughoutand our competition, see the world.information included under the heading, “Part I – Item 1. Business – Products, Product Development, and Our Competition – Our Intellectual Property” and “— Our Competition.”
Filing, prosecuting and defending patents on our product candidatesIf we become involved in all countries throughout the world would be prohibitively expensive. The requirements for patentability may differ in certain countries, particularly developing countries. In addition, the laws of some foreign countries do notlawsuits to protect intellectual property rights to the same extent as laws in the United States. Consequently, we may not be able to prevent third parties from practicing our inventions in all countries outside the United States. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and, further, may export otherwise infringing products to territories where we have patent protection, but enforcement on infringing activities is inadequate. These products may compete with our products, andor enforce our patents or other intellectual property, rights may not be effective or sufficient to prevent them from competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection, particularly those relating to pharmaceuticals, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly and our patent applications at risk of not issuing, and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded, if any, may not be commercially meaningful. In addition, certain countries in Europe and certain developing countries have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties. In those countries, we may have limited remedies if our patents are infringed or if we are compelled to grant a license to our patents to a third party, which could materially diminish the value of those patents. This could limit our potential revenue opportunities. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we own or license. Finally, our ability to protect and enforce our intellectual property rights may be adversely affected by unforeseen changes in foreign intellectual property laws.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance and annuity fees on any issued patent are due to be paid to the USPTO and foreign patent agencies in several stages over the lifetime of the patent. The USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. While an inadvertent lapse can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Non-compliance events that could result in abandonment or lapse of a patent or patent application include failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. If we or our licensors fail to maintain the patents and patent applications covering our product candidates, our competitors might be able to enter the market, which would have an adverse effect on our business.
If we are sued for infringing intellectual property rights of third parties, it will be costly and time-consuming, and an unfavorable outcome in that litigation could have a material adverse effect on our business.business, financial condition, or results of operations.
Our ultimate commercial success depends upon our ability to develop, manufacture, market, and sell our product candidates and use our proprietary technologies in the U.S. and non-U.S. markets. In order to do so, it is critical that we prevent third parties from infringing on our intellectual property rights and that we operate our business without infringing on the proprietaryintellectual property rights of third parties. We cannot assure you that marketing and selling such candidates and using such technologies will not infringe existing or future patents. Numerousothers.
However, numerous U.S. and foreignnon-U.S. issued patents and pending patent applications owned by third parties exist in the fields relating to TSC and our other product candidates. As the biotechnologycandidates, their potential methods of delivery, potential indications they may be used to treat, and pharmaceutical industries expandtheir other features, and, as more patents are issued over time, the risk increases that others may assert that our product candidates, technologies, or methods of delivery or use infringe their patent or other intellectual property rights, or that we discover a third party infringing on our rights. Moreover, it is not always clear to industry participants, including us, which patents cover various drugs, biologics, drug delivery systems, or their methods of use, and which of these patents may be valid and enforceable. Thus, because of the large number of patents issuedenforceable, and patent applications filed in our fields, therewhat inventions or technologies may be a risk that third parties may allege they haveclaimed by non-public patent rights encompassing our product candidates, technologies or methods.
In addition, there may be issued patents of third parties that are infringed or are alleged to be infringed by our product candidates or proprietary technologies. Because some patentapplications. Patent applications in the United States may be maintained in secrecy until the patents are issued, because patent applications in the United StatesU.S. and many foreign jurisdictions are typically not published until eighteen (18)18 months after their first non-provisional filing and because publications in the scientific literature often lag behind actual discoveries, meaning we cannot be certain thatwhether others, including our competitors, have not filed patent applications for technology covered by our own and in-licensed issued patents or our pending applications. Our competitors may have filed,applications and may in the future file, patent applications covering our product candidates or technology similar to ours. Anywhether any such patent application may havefiling has priority over our own and in-licensed patent applications or patents, which could further require us to obtain rights to issued patents covering such technologies. If another party has filed a U.S. patent application on inventions similar to those owned or in-licensed to us, we or, in the case of in-licensed technology, the licensor may have to participate, in the United States, in an interference proceeding to determine priority of invention.patents.
We may be exposed to, or threatened with, future litigation by third parties having patent or other intellectual property rights alleging that our product candidates or proprietary technologies infringe such third parties’ intellectual property rights, including litigation resulting from filing under Paragraph IV ofIn the Hatch-Waxman Act. These lawsuits could claim thatbiopharmaceutical and pharmaceutical industries in particular, there are existing patent rights for such drug and this type of litigation can be costly and could adversely affect our operating results and divert the attention of managerial and technical personnel, even if we do not infringe such patents or the patents asserted against us are ultimately established as invalid. There is a risk that a court would decide that we are infringing the third party’s patents and would order us to stop the activities covered by the patents. In addition, there is a risk that a court will order us to pay the other party damages for having violated the other party’s patents.
There is a substantial amount of litigation involving patent and other intellectual property rights inrights. This type of litigation may occur unexpectedly but may also be prompted by specific events, such as a patent application being made public by the biotechnologyUSPTO or a non-U.S. governmental authority or under Paragraph IV of the Hatch-Waxman Amendments. For more information regarding the Hatch-Waxman Amendments and pharmaceutical industries generally. ToParagraph IV thereunder, see the information included under the heading, “Part I – Item 1. Business – Government Regulation – The Hatch-Waxman Amendments.”
As of the date of this Annual Report, no litigation asserting infringement claims has been brought against us. Ifus, nor have we filed such a claim against any third partyparty. However, we cannot assure you that the development or future commercialization of any of our product candidates or other technologies will not result in claims that weour activities infringe itson the existing or future intellectual property rights we may face a number of issues, including:
| ●
| infringement and other intellectual property claims which, regardless of merit, may be expensive and time-consuming to litigate and may divert our management’s attention from our core business;
|
| ●
| substantial damages for infringement, which we may have to pay if a court decides that the product or technology at issue infringes or violates the third party’s rights, and if the court finds that the infringement was willful, we could be ordered to pay treble damages and the patent owner’s attorneys’ fees;
|
| ●
| a court prohibiting us from selling or licensing the product or using the technology unless the third party licenses its intellectual property rights to us, which it is not required to do;
|
| ●
| if a license is available from a third party, we may have to pay substantial royalties or upfront fees or grant cross-licenses to intellectual property rights for our products or technologies; and
|
| ●
| redesigning our products or processes so they do not infringe, which may not be possible or may require substantial monetary expenditures and time.
|
Some of ourthird parties. Furthermore, potential competitors may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources. In addition, any uncertainties resulting from the initiation and continuation of any litigation could harm our ability to raise additional funds or otherwise adversely affect our business, financial condition, operating results and prospects.
Because we may in the future rely on certain third-party licensors and partners, if one of those licensors or partners is sued for infringing a third party’s intellectual property rights, our business, financial condition, operating results and prospects could suffer in the same manner as if we were sued directly. In addition to facing litigation risks, we may agree in the future to indemnify certain third-party licensors and partners against claims of infringement caused by our proprietary technologies, and we may enter into cost-sharing agreements with some our licensors and partners that could require us to pay some of the costs of patent litigation brought against those third parties whether or not the alleged infringement is caused by our proprietary technologies. In certain instances, these cost-sharing agreements could also require us to assume greater responsibility for infringement damages than would be assumed just on the basis of our technology.
The occurrence of any of the foregoing could adversely affect our business, financial condition, operating results and prospects.
We may become involved in lawsuits to protect or enforce our patents or other intellectual property which could be expensive and time-consuming.
Competitors may infringe our intellectual property, including our patents. AsFor example, in the first quarter of 2021, we became aware of a result, wethird party affiliated with a former outside consultant of the Company which claims to be in early-stage development of a product candidate that purportedly may operate through a similar mechanism of action to TSC.
We may be required to file infringement claims to stop third-party infringement or unauthorized use.use or, if a third party claims we are infringing on their rights, respond to such claims. This process can be expensive particularly for a company of our size, and time-consuming. In addition,time consuming, and could result in an infringement proceeding, a court may decidedeciding that a patent of ours is not valid or is unenforceable, or may refusethat a third party is not required to stop using a technology we believe infringes on our rights, significant costs, or the other party from using the technology at issue on the grounds that our patent claims do not cover its technology or that the factors necessary to grant an injunction against an infringer are not satisfied.diversion of management’s time. An adverse determination ofin any litigation or other proceedings could put one or more of our patents at risk of being invalidated, interpreted narrowly, or amended such that they do not cover our product candidates. Moreover,candidates in a manner sufficient to support our development and commercialization needs or that such adverse determinations couldproduct candidate needs to be significantly redesigned, or put our pending patent applications at risk of not issuing, or issuing with limited and potentially inadequate scopescope. Further, some of our competitors may be able to cover our product candidates or to prevent others from marketing similar products.sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources.
Interference,In addition, interference, derivation, or other proceedings brought at the USPTO may be necessary to determine the priority or patentability of inventions with respect to our patents or patent applications or those of our licensors or potential partners.applications. Litigation or USPTO proceedings brought by us may fail or may be invoked against us by third parties. Even if we are successful in these proceedings, domestic or foreign litigation or USPTO or foreign patent office proceedings may result in substantial costs and distraction to our management.the diversion of management’s time. We may not be able alone or with our licensors or potential partners, to prevent all misappropriation of our proprietary rights, particularly in countries with a legal framework that offers limited intellectual property protections or where the laws may not protect such rights as fully as incosts of enforcement outweigh the United States.commercial and other benefits of maintaining intellectual property protections.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation or other proceedings, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation or other proceedings. In addition, during the courseproceedings, including as a result of this kind of litigation or proceedings, there could be public announcements of the results of hearings, motions or other interim proceedings or developments, or public access to related documents. If investors perceive these resultsThis type of disclosure could put us at a significant competitive disadvantage by disclosing important trade secrets or other proprietary information to be negative, the market price for common stock could be significantly harmed.our competitors and other third parties.
Any litigation or other challenge related to our intellectual property could materially and adversely affect our business, financial condition, and results of operations.
WeGeneral Risks Related to Our Intellectual Property
Our business, financial condition, or results of operations may also be materially adversely affected by a number of general risks related to our intellectual property that are not specific to our Company, including:
• As is common in the biopharmaceutical and pharmaceutical industries, some of our employees were formerly employed by companies in the industry, including our competitors or potential competitors, and some of our consultants actively work for other companies in the industry. As a result, although we have in place policies which prohibit the use of third-party confidential information in violation of any obligation to a former employer or otherwise, we may be subject to claims that our employees, consultants or independent contractors have wrongfully used or disclosed to us alleged trade secrets of their former employers or their former or current customers. In addition, if any of our current employees or consultants are engaged by a competitor in the future, it is possible that they may appropriate or otherwise improperly use our proprietary and confidential information. Any of the foregoing events could result in significant costs and the diversion of time and resources.
As is common• Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated as a result of any non-compliance with these requirements. We may also abandon certain intellectual property protections that we would otherwise maintain if we determine such protections are not expected to provide sufficient value relative to the cost of ongoing maintenance.
• Patent laws and other intellectual property protections available in the biotechnologyU.S., E.U., or other jurisdictions are subject to change. These changes may be unpredictable, weaken our overall intellectual property position, increase our costs related to maintenance and pharmaceutical industries, certainenforcement, or otherwise diminish the value of patents in general, thereby impairing our employees were formerly employed byability to protect our product candidates and maximize our return on investment thereon.
Risks Related to Our Business, Financial Position, Results of Operation, and Organizational Structure
We will require additional capital to fund our operations which may not be available on acceptable terms or at all. If we fail to obtain necessary financing, we may be forced to delay or curtail our clinical trials and other biotechnologydevelopment activities or pharmaceutical companies, including our competitors or potential competitors. Moreover, we engage the services of consultantsbe unable to assist us incomplete the development and commercialization of our product candidates manydue to a lack of whom were previously employedsufficient resources.
Although we expect that our existing cash resources will enable us to fund our operating expenses and capital expenditure requirements through 2023, we expect to continue to spend substantial amounts as we continue to develop TSC and our other product candidates. As a result, we will need to obtain additional financing in the future in order to complete the development of TSC and fund our other development and operational activities.
We cannot be certain that the additional funding we will require will be available on acceptable terms or at all. Investors may demand significant discounts to market prices or that we agree to restrictive covenants or other limitations on our ability to operate our business, and conditions in the capital markets may make equity and debt financing more difficult to obtain or negatively impact our ability to complete a financing transaction at all. If we are unable to raise additional capital in sufficient amounts or on terms acceptable to us, we may have previously beento significantly delay, scale back, discontinue the development or are currently providing consulting servicescommercialization of one or more of our product candidates, or seek alternative financing opportunities such as collaborations or licensing opportunities. For example, prior to other biotechnology or pharmaceutical companies, includingcompleting the May 2020 Offering and February 2021 Offering, we determined that we did not have adequate resources to fully support the randomized portion of the GBM Trial and commencement of enrollment in the trial was suspended following the run-in portion.
Furthermore, our competitors or potential competitors. We mayforecast of the period of time through which our financial resources will be subjectadequate to claims that these employeessupport our operations is a forward-looking statement and consultants or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers or their former or current customers.involves the associated risks and uncertainties. Although we have based this estimate on assumptions that we believe to be reasonable, they may prove to be wrong, we could utilize our available capital resources sooner than we currently expect, and actual results could vary greatly from our expectations expressed in this Annual Report as a result. The magnitude and timing of our future funding requirements, both near and long-term, will depend on many factors, including, but not limited to:
| • | the number, development stage, and other characteristics of product candidates that we choose to develop, including any product candidates that we may in-license or otherwise acquire in the future; |
| • | the clinical development plans we establish for these product candidates; |
| • | the magnitude of costs associated with filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights; |
| • | the initiation, progress, timing, costs, and results of clinical trials for such product candidates; |
| • | the outcome, timing and cost of regulatory approvals by the FDA and comparable foreign regulatory authorities, including the potential for the FDA or comparable foreign regulatory authorities to require that we perform more studies than those that we currently expect; |
| • | the cost and timing of completion of becoming a commercial organization; and |
| • | the effect of competing technological and market developments. |
We currently generate no knowledgerevenue from the sale of products, have incurred significant losses since our inception, have a history of net losses and negative cash flow from operations, expect to incur losses for the foreseeable future, and may never become profitable. In addition, our operating results may fluctuate significantly, which makes our future operating results difficult to predict and could cause our operating results to fall below expectations. As a result, any such claims being allegedinvestment in our common stock is speculative and risky.
We are a clinical stage biotechnology company and, as a result, we have a limited operating history from which to assess how we will respond to competitive, economic, or other challenges to our business, and our business and prospects must be considered in light of the risks and uncertainties frequently encountered by similarly situated companies.
We have limited cash resources, have generated substantial net losses and negative cash flow from operations since our inception, and we continue to incur significant research, development, and other expenses related to our ongoing operations, including the development of TSC and our other product candidates. To date, if such claims werewe have not yet obtained regulatory approvals for any of our product candidates and, accordingly, have not generated any revenues from the sale of products. We expect to arise, litigationcontinue to incur losses and negative cash flow for the foreseeable future. Furthermore, our future operating results may fluctuate due to a variety of other factors, many of which are outside of our control and may be necessarydifficult to defend againstpredict, including the delays in our product development programs including as a result of regulatory review, increased expenditures related to manufacturing or the enforcement of intellectual property rights, other litigation costs, changes in accounting policies, or other unanticipated events.
Our ability to generate sufficient revenues from TSC or any such claims.of our other product candidates, if approved, will depend on numerous factors described throughout this Annual Report. Even if we are able to successfully develop and receive regulatory approval for TSC or any of our other product candidates, we do not know if or when any such product will achieve commercial success or generate revenue for us, and we will incur significant costs associated with the commercialization that will need to be offset by revenue before achieving a profit. We may also in the future enter into collaboration agreements and license agreements with other companies that include milestone expenditures and payments, in which case our ability to generate revenue or achieve profitability may be dependent on the achievement of those milestones. Even if we achieve profitability in the future, we may not be able to sustain profitability in subsequent periods, and our prior losses and expected future losses have had and will continue to have an adverse effect on our stockholders’ equity. Furthermore, due to the uncertainty of the drug development process, we are often unable to predict the timing or amount of increased expenses, or when we will be able to achieve or maintain profitability, if at all.
We will need to further increase the size and complexity of our organization in the future including, if TSC or any of our other product candidates are approved for commercial sale, establishing sales and marketing capabilities. We may experience difficulties in executing our growth strategy or managing any growth that we do experience if we are unable to recruit and retain talented individuals in key positions.
Our ability to succeed in the highly competitive pharmaceuticals industry depends upon our ability to attract and retain highly qualified personnel. As of the date of this Annual Report, we have 14 full-time employees and no part-time employees. Particularly given our near-term plans for the TSC development program and our compliance requirements with the FDA, SEC, and other regulatory bodies, we believe that our current staffing levels are inadequate to support our future needs. We anticipate adding additional personnel to our team throughout the organization during 2022 in an effort to support the development of TSC, grow our business more generally, and optimize the size of our organization. In addition, assuming success in our ongoing and planned clinical trials, we expect to further supplement and grow our scientific, clinical, regulatory, financial, and other human resources to support our planned research, development, and commercialization plans for TSC and our other product candidates.
Our ability to effectively manage our anticipated growth will depend on multiple factors, including, among others, our ability to:
• effectively retain current talent and effectively recruit sufficient numbers of new talented employees;
• manage our third-party supply and manufacturing operations effectively and in a cost-effective manner,
• while increasing production capabilities for our current product candidates to commercial levels;
• establish and maintain relationships with development and commercialization partners;
• manage our development and commercialization efforts effectively and in a cost-effective manner; and
• continue to improve our operational, clinical, financial, management and regulatory compliance controls and reporting systems and procedures.
We are highly dependent on our management and scientific personnel, including our executive officers, certain other key employees and consultants, and the members of our Board. The loss of the services of any of these individuals could impede, delay, or prevent completion of our ongoing and planned clinical trials, regulatory approval, or commercialization of TSC or any of our other product candidates. If we lose the services of any of these individuals, we might not be able to find suitable replacements on a timely basis or at all, and our business could be harmed as a result. All of our employees, including our executive officers with whom we have employment agreements, are employed on an at-will basis and their employment can be terminated by us or them at any time. As part of our efforts to retain our valuable employees, we, among other things, provide a generous salary and benefits package, as described in more detail under the heading, "Part I — Item 1. Business – Our People and Human Capital Resources – Employees."
Nevertheless, we may be unable to attract or retain qualified management and other key personnel in the future due to the intense competition among biotechnology, pharmaceutical, and other businesses. There may be a limited number of persons with the requisite skills to serve in these positions, and we cannot assure you that we will be able to identify or employ qualified personnel for any such position on acceptable terms, if at all, and the high levels of competition within the industry may mean that we will be required to expend significant financial resources in our employee recruitment and retention efforts. Many of the other pharmaceutical companies with whom we compete for qualified personnel have greater financial and other resources, different risk profiles and longer histories in the industry than we do. They also may provide more diverse opportunities and better chances for career advancement. If we are not able to attract and retain the necessary personnel to accomplish our business objectives, we may experience constraints that will harm our ability to implement our business strategy and achieve our business objectives. Furthermore, as we currently have no marketed products, we currently have no sales or marketing personnel or capabilities. To commercialize TSC or any of our other product candidates, if approved, we will need to build our marketing, sales, distribution, and other related capabilities or arrange with third parties to perform these services, and we may not be successful in defending against anydoing so.
In addition, we have historically utilized the services of certain outside independent contractors to perform a number of critical functions for our company, including with respect to clinical development, regulatory matters, accounting, and human resources, a practice we expect to continue and may choose to expand in the future. We rely on these independent contractors and effectively managing our relationships with them is and will remain a priority. However, there can be no assurance that we will be able to manage these relationships effectively, that such claims, any such litigationcontractors will be able or choose to continue working with us in the future, or that we will be able to find additional or replacement services if and as needed, on economically reasonable terms or at all.
If we are not able to effectively manage our growth and expand our organization through a combination of effectively retaining our existing employees and third-party contractors and successfully recruiting new employees and contractors, we may be unable to effectively execute on our product development and other strategic plans, which may adversely affect our business, financial condition, or results of operations.
If we decide to in-license or acquire one or more additional product candidates or otherwise enter into a strategic transaction, it could be protracted, expensive, a distractionimpact our liquidity, increase our expenses, and present significant distractions to our management team, not viewed favorably by investorsteam.
We currently only have one product candidate under active development, TSC. We may in the future implement a strategy to in-license or acquire one or more additional product candidates to supplement our pipeline. We may also consider a variety of other strategic transactions, including spin-offs, partnerships, joint ventures, restructurings, divestitures, business combinations, and minority investments. Any such transaction would expose us to a number of risks and uncertainties, including the potential incurrence of recurring, non-recurring (including unknown liabilities), or other charges (including amortization expenses, write-downs, or other impairment charges), increase of short- and long-term expenditures, or dilution to our stockholders, as well as posing significant integration, implementation, or retention challenges and diverting our management team’s focus on other priorities, including the TSC development program. Any of the foregoing could have a material adverse effect on our business, financial condition, or results of operation. which could adversely affect our operations and financial results. There can be no assurance that we will undertake such a transaction or, if we do, that we will successfully complete the transaction or that the transaction will be additive to our business, financial condition, or results of operations.
Our ability to utilize our NOL carryforwards and other third partiesdeferred tax assets may be limited as a result of past and may potentially result in an unfavorable outcome.
Risks Related to Ownership of Our Common Stock and Other Securities
The trading volume of ourcommonstock has historically been very low, and recently has been extremely volatile, which may result in decreased periods of liquidity or large, short-term fluctuations in our stock price.
The number of shares of our common stock being traded on a daily basis has historically been very low and recently has been extremely volatile. Since January 8, 2016, the date we completed the Merger, through March 15, 2017, the daily trading volume for our Common Stock ranged from zero shares to 3,535,800 shares (after giving effect to the Reverse Stock Split). The quotation of our common stock on the NASDAQ Capital Market does not assure that a meaningful, consistent and liquid trading market currently exists.
Any holder of our common stock wishing to sell his, her or its shares may cause a significant fluctuation in the trading pricefuture issuances of our common stock. In addition, low trading volume
As of a stock increasesDecember 31, 2021, we had $23.4 million in federal and state NOL carryforwards available to reduce future taxable income, if any, for income tax purposes. If not utilized, the possibility that, despite rules against such activity,NOL carryforwards will begin expiring during the priceyear ending December 31, 2034. Under Section 382 of the stockTax Code, if a corporation undergoes an “ownership change,” generally defined as a greater than 50% change, measured by value, in its equity ownership over a three-year period, the corporation’s ability to use its pre-ownership change NOL carryforwards and other pre-ownership change tax attributes – such as research tax credits – to offset its post-ownership change income may be manipulated by persons acting in their own self-interest. We may not have adequate market makers and market making activity to prevent manipulation in our common stock. Such volatility may belimited. In the resultevent we issue additional shares of broad market and industry factors. In addition to market and industry factors, the price and trading volume for common stock to fund our future development efforts and those issuances result in additional ownership changes for purposes of Section 382 of the Tax Code, as we did in the May 2020 Offering and the February 2021 Offering, our NOL carryforwards could be further reduced.
General Risks Related to Our Business, Financial Condition, Results of Operations, and Organizational Structure
Our business, financial condition, or results of operations may also be highly volatile for factorsmaterially adversely affected by a number of general risks related thereto and to our organizational structure that are not specific to our own operations, many of which are beyond our control, including those described elsewhere in and incorporated into this annual report on Form 10-K and our other public filings.Company, including:
We may be at an increased risk of securities litigation, which is expensive and could divert management attention.
The market price of our common stock may be volatile, and in the past companies that have experienced volatility in the market price of their stock have been subject to securities class action litigation. We may be the target of this type of litigation in the future. Securities litigation against us could result in substantial costs and divert our management’s attention from other business concerns, which could seriously harm our business.
We consummated a “reverse merger” with a shell entity in 2008, which may result in certain limitations on the ability of our stockholders to use the Rule 144 safe harbor under the Securities Act, for resales ofcommonstock.
As we are a former shell company due to our “reverse merger” with a shell entity in 2008, the ability of our stockholders to resell shares of Common Stock under Rule 144 may be limited. The use of Rule 144 is one of the most common methods of selling restricted shares. Rule 144(i) pertains to shares issued by a former shell company. Under Rule 144(i), sales of shares may only be made under certain conditions, including that we are current with respect to certain filings required under the federal securities laws. As a result, permission to remove a restrictive legend on shares of Common Stock may be granted under more limited circumstances compared to an issuer that is not a former shell company.
• If we fail to maintain an effective system of internal control over financial reporting, we may not be able to accurately report our financial results or prevent fraud. Furthermore, our disclosure controls and procedures are subject to inherent limitations, human error, and other systematic breakdowns, and therefore may not prevent or detect all errors or acts of fraud. As a result, stockholders could lose confidence in our financial and other public reporting, which wouldcould harm our business, and the trading pricefinancial condition, or results ofcommonstock. operations.
Effective internal controls over financial reporting are necessary for us• As our Company, the industry in which we operate, and the world-at-large become increasingly virtual, our acquisition and implementation of additional information technology solutions and our compliance with global privacy and data security requirements could result in additional costs and liabilities or inhibit our ability to provide reliable financial reportscollect and together with adequate disclosure controls and procedures, are designed to prevent fraud. Anyprocess data globally. Furthermore, any failure to implement required newcomply with applicable requirements or improved controls,best practices – as well as other events outside of our control – could result in a security breach or difficulties encountered in their implementation, could cause us to fail to meet our reporting obligations. Ineffective internal controls could also cause investors to lose confidence in our reported financial information, which could have a negative effect on the trading price of common stock. In addition, any future testing by us conducted in connection with Section 404 of the Sarbanes-Oxley Act of 2002, as amended (“SOX”), or any required subsequent testing by our independent registered public accounting firm, may reveal deficiencies in our internal controls over financial reporting that are deemed to be material weaknesses or that may require prospective or retroactive changesother disruption to our consolidatedinformation technology systems, limit our capacity to effectively monitor and control our operations, compromise our or third parties’ confidential information, or otherwise adversely affect our business, financial statementscondition, or identify other areas for further attention or improvement.results of operations.
We are required, pursuant to Section 404 of SOX, to furnish a report by management on, among other things, the effectiveness of our internal control over financial reporting as of December 31, 2016. However, our independent registered public accounting firm is not required to attest to the effectiveness of our internal control over financial reporting pursuant to Section 404. Under the supervision and with the participation of our Chief Executive Officer and Senior Vice President - Finance our management conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework in Internal Control—Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on that evaluation, our management concluded that our internal control over financial reporting was effective as of December 31, 2016. An independent assessment of the effectiveness of our internal controls could detect problems that our management’s assessment might not. Undetected material weaknesses in our internal controls could lead to financial statement restatements and require us to incur the expense of remediation.
Our disclosure controls and procedures may not prevent or detect all errors or acts of fraud.
We are subject to the periodic reporting requirements of the Exchange Act. Our disclosure controls and procedures are designed to reasonably assure that information required to be disclosed by us in reports we file or submit under the Exchange Act is accumulated and communicated to management, recorded, processed, summarized and reported within the time periods specified in the rules and forms of the SEC. We believe that any disclosure controls and procedures or internal controls and procedures, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met.
These inherent limitations include the realities that judgments in decision-making can be faulty, and that breakdowns can occur because of simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people or by an unauthorized override of the controls. Accordingly, because of the inherent limitations in our control system, misstatements due to error or fraud may occur and not be detected.
• We incur significant costs as a result of our public company status and devote substantial management time to operating as a public company.company, including complying with the applicable requirements of the Securities Act, the Exchange Act, the Dodd-Frank Act, SOX, and the rules and regulations of Nasdaq. If, in the future, we are required to include in our annual report an attestation of our independent registered public accounting firm regarding internal control over financial reporting, the amount of these compliance costs would increase significantly.
• Although we have in place business continuity and disaster recovery plans, our business, financial condition, or results of operations could be negatively affected by volatility, disruptions, or other uncertainty caused by market fluctuations, economic downturns or unfavorable global economic conditions, pandemics, natural disasters or other catastrophic events, events of war, terrorism, or other man-made problems, or other geopolitical events outside of our control, including the COVID-19 pandemic and Brexit.
• If we fail to comply with applicable laws and regulations, including the healthcare laws and regulations described under the heading, Part I – Item 1. Business – Certain Other Legislation and Regulations – Current Healthcare Laws and Regulations and applicable environmental, health, and safety laws and regulations, we could become subject to fines, penalties, or other consequences.
Risks Related to Ownership of Our Common Stock
If we cannot continue to satisfy the NASDAQ Capital Market continued listing standards and other NASDAQ rules, our Common Stock could be delisted, which would harm our business, the trading price of our Common Stock, our ability to raise additional capital and the liquidity of the market for our Common Stock.
Our common stock is currently listed on the NASDAQ Capital Market. To maintain the listing of our common stock on the NASDAQ Capital Market, we are required to meet certain listing requirements, including, among others, either: (i) a minimum bid price of $1.00 per share, a market value of publicly held shares (excluding shares held by our executive officers, directors and 10% or more stockholders) of at least $1 million and stockholders’ equity of at least $2.5 million; or (ii) a minimum bid price of $1.00 per share, a market value of publicly held shares (excluding shares held by our executive officers, directors and 10% or more stockholders) of at least $1 million and a total market value of listed securities of at least $35 million. There is no assurance that we will regain and continue to meet the Bid Price Rule and Nasdaq’s other listing requirements.
As previously disclosed, on May 6, 2021, we received a public company,written notice from the Staff indicating that the Company was not in compliance with the Bid Price Rule because the bid price for the Company’s common stock had closed below $1.00 per share for the previous 30 consecutive business days. On November 3, 2021, after failing to regain compliance with the Bid Price Rule within 180 days of receipt of the first notice, we incurreceived an additional notice from the Staff providing that, although the Company had not regained compliance with the Bid Price Rule by the previously stated deadline, in accordance with Nasdaq Listing Rule 5810(c)(3)(A), the Company would be granted an additional 180 calendar days, or until May 2, 2022, to regain compliance with the Bid Price Rule.
To regain compliance with the Bid Price Rule, the bid price for our common stock must close at $1.00 per share or more for a minimum of 10 consecutive business days. NASDAQ’s written notice has no effect on the listing or trading of our common stock at this time, and we are currently evaluating our alternatives to resolve this listing deficiency. We have filed with the SEC a proxy statement relating to the Special Meeting, to be held on April 14, 2022, at which our stockholders will vote on a proposal to approve an amendment to our Certificate of Incorporation to effect the Reverse Stock Split, with the final decision of whether to proceed with the Reverse Stock Split, the effective time of the Reverse Stock Split, and the exact ratio of the Reverse Stock Split to be determined by the Board, in its discretion, at any time prior to December 31, 2022. However, there can be no assurance that the Reverse Stock Split will be approved at the Special Meeting or, if approved, will result in a sustained higher stock price, if effected, that will allow us to regain compliance with the Bid Price Rule and meet the NASDAQ stock price listing requirements, and there is no guarantee we will continue to satisfy the other NASDAQ Capital Market continued listing standards.
In the event that our common stock is delisted from NASDAQ and is not eligible for quotation or listing on another market or exchange, trading of our common stock could be conducted only in the over-the-counter market or on an electronic bulletin board established for unlisted securities such as the Pink Sheets or the OTC Bulletin Board. In such event, it could become more difficult for us to raise capital and for our stockholders to dispose of, or obtain accurate price quotations for, our common stock, and there would likely also be a reduction in our coverage by securities analysts and the news media, which could cause the price of our common stock to decline further.
Our stock price is volatile and any investment in our securities may suffer a decline in value. In addition, this volatility may subject our business to additional risks, such as an increased risk of securities litigation.
During the year ended December 31, 2021, the closing market price for our common stock as reported by Nasdaq varied between a high of $1.70 on February 16, 2021 and December 31, 2021 and a low of $0.31 on December 29-31, 2021. As a result of fluctuations in the price of our common stock, you may be unable to sell shares or our common stock at or above the price you paid for them, even if your holding period is relatively short. The market price of our common stock is likely to continue to be volatile and subject to significant legal, accountingprice and volume fluctuations in response to market, industry and other expenses to comply with the reporting requirements of the Exchange Act and applicable requirements of SOX and the Dodd-Frank Wall Street Reform and Consumer Protection Act, as well as rules and regulations subsequently implemented by the SEC and NASDAQ,factors, including the establishmentdegree of analyst coverage of our stock, their valuations and maintenance of effective disclosurerecommendations, and financial controls, changes in corporate governance practices and required filing of annual, quarterly and current reports with respect to our business and operating results. These requirements increase our legal and financial compliance costs and make some activities more time-consuming and costly. In addition, our management and other personnel devote significant time and attention to these public company requirements, which diverts their time attention from operational and other business matters.
If securities or industrywhether any such analysts do not publish research or publish inaccurate or unfavorable research about our business. If the results of our business do not meet these analysts’ forecasts, the expectations of investors or the financial guidance we provide to investors in any period, the market price of our common stock price and trading volume could decline.
The trading market for Furthermore, despite this volatility, due to the fact that we have never declared or paid cash dividends on our common stock will depend in part on the research and reports that securities or industry analysts publish about us or our business. We do not currently have and may never obtain research coverage by securities and industry analysts. If no securitiesanticipate declaring or industry analysts commence coveragepaying any cash dividends in the foreseeable future, we expect that only appreciation of the price of our company,common stock, if any, will provide a return to our stockholders for the foreseeable future.
Historically, the stock markets in general, and the markets for biotechnology stocks in particular, have experienced significant volatility that has at times been unrelated to the financial condition or results of operations of particular companies. These broad market fluctuations may adversely affect the trading price forof our common stock would be negatively impacted. If we obtain securities or industry analyst coverage and, if one or more ofconsequently, adversely affect the analysts who covers us downgrades our stock or publishes inaccurate or unfavorable research about our business, our stock price would likely decline. If one or more of these analysts ceases coverage of us or failsat which you are able to publish reports on us regularly, demand for common stock could decrease, which could cause our stock price and trading volume to decline.
Future sales ofsubstantial number ofsell any shares of our common stock that you own. In the past, following periods of volatility in the public market or other issuancessignificant price declines in individual securities or the market as a whole, securities class-action litigation has often been instituted against companies. Such litigation, if instituted against us, could result in substantial costs and diversion of management’s attention and resources, which could materially and adversely affect our business, financial condition, results of operations and growth prospects.
We have funded our operations to date through the issuance of securities, including common stock, or rightswarrants to purchase common stock, including pursuant to equity incentive plans,could resultconvertible preferred stock, and convertible debt securities, and we expect that in additional dilution of the percentage ownership of our stockholders and could cause our stock price to fall.
Wefuture we will need to raise substantialadditional capital through similar means to fund our continued development efforts for TSC and our other liquidity needs. Assuming funding is available on acceptable terms, any future issuance of common stock or securities convertible for or exchangeable into common stock will result in dilution to our existing stockholders and could depress the market price of our common stock. Furthermore, the terms of future financing transactions may contain provisions that restrict our operations or require us to relinquish certain rights to our product candidates or other technologies.
Although we believe we have sufficient cash resources to fund our other operating expenses and capital expenditure requirements through 2023, we will in the future need to raise additional funds through the Private Placement and otherwise, to continue our operations, fund additional clinical trials ofevaluating TSC and our other product candidates, and, potentially commercialize our product candidates.if approved, commercializing TSC. We plan to continue to finance our operations with a combination of equity issuances, debt arrangements, and, a possible partnershippotentially, licensing, or license of development and/or commercialization rightsother partnering relationships. In addition, our Board may determine at any time to our product candidates. Any issuance of convertible debt securities, preferred stock or common stock may be at a discount fromraise additional capital if it believes the then current trading price of our common stock. If we issue additional common or preferred stock or securities convertible into common stock, our stockholders will experience additional dilution, which may be significant. For example, the Securities soldterms are in the Private Placement are convertible into or exchangeable for sharesbest interests of our common stock. Furthermore, the Series A Convertible Preferred Stock is entitled to an 8.0% cumulative preferred dividend payable semi-annually in shares of common stock. Therefore, the Private Placement will result in substantial dilution to our stockholders.
In addition, pursuant to our 2015 Equity Incentive Plan, as amended, our compensation committee is authorized to grant equity-based incentive awards to our directors, executive officers and other employees and service providers, including officers, employees and service providersAccordingly, new issuances of our subsidiaries and affiliates. As of March 15, 2017, thea substantial number of shares of our common stock could occur at any time. For example, in February 2021, we have reserved for issuance under our 2015 Equity Incentive Plan is 356,148, and future option grants and issuances of common stock under our 2015 Equity Incentive Plan will dilute the percentage ownership of our investors and may adversely affect the market priceissued approximately 33.7 million shares of our common stock.
Further, future sales of a substantial number of shares of common stock or securities convertible into common stock in connection with the public market,completion of the February 2021 Offering. Any issuance or sale of shares, or the perception in the market thatof an intent to issue or sell shares in the near-term, by the Company or holders of a large number of shares intend to sell shares could reduce the market price of our common stock. We also cannot assure you that any such sale of common stock and impedeor other securities will be at a price per share that is equal to or greater than the price per share paid by you for our common stock. Furthermore, a depressed stock price could limit our ability to raise future capital.necessary capital through the sale of additional equity securities on terms that are acceptable.
We may also seek additional capital through other methods, either alone or in combination with the issuance of additional securities, including debt financings, receivables or royalty financings, strategic partnerships and alliances, and licensing arrangements, any of which could be coupled with an equity component, such as warrants to purchase stock. The conversionincurrence of indebtedness could result in increased fixed payment obligations, liens and other security interests being placed on certain of our assets, and certain restrictive covenants being imposed on the Series A Convertible Preferred Stockoperation of our business, such as limitations on our ability to incur additional debt or the exercise of the Warrants into common stock would dilute the ownership interest of existing holders of common stock, and any salesacquire intellectual property rights. We may also in the public marketfuture raise additional funds through strategic partnerships, alliances, and licensing arrangements with third parties, any of the common stock issuable upon conversionwhich could require us to relinquish valuable rights to TSC or our other product candidates. The restrictions imposed by any of the Series A Convertible Preferred Stockthese arrangements could materially decrease any potential returns on our investment in TSC or the exercise of the Warrants couldour other product candidates, or otherwise materially and adversely affect prevailing market pricesour business, financial condition, or results of operations.
Our organizational documents impose certain anti-takeover provisions and make the Delaware Chancery Court the exclusive forum for certain stockholder actions, which could depress the trading price of our common stock.
Our directors, executive officers and principal stockholders exert significant influence over us and could impede a change of corporate control.
As of March 15, 2017, our directors, executive officers and holders of more than five percent of common stock, together with their affiliates, beneficially owned, in the aggregate, 47% of our outstanding common stock (See “Security Ownership of Certain Beneficial Owners and Management”). As a result, these stockholders, acting together, would have the ability to exert significant influence on matters submitted to our stockholders for approval, including the election of directors and any merger, consolidation or sale of all or substantially all of our assets. In addition, these stockholders, acting together, would have the ability to significantly influence the management and affairs of our company. Accordingly, this concentration of ownership could harm the market price of common stock by:
| ●
| delaying, deferring or preventing a change of control of us;
|
| ●
| impeding a merger, consolidation, takeover or other business combination involving us; or
|
| ●
| discouraging a potential acquiror from making a tender offer or otherwise attempting to obtain control of us.
|
Delaware law and provisions in our restated articlescertificate of incorporation, as amended, and restated bylaws could make a merger, tender offer or proxy contest difficult, thereby depressing the trading price of Common Stock.
The anti-takeover provisions under Delaware corporate law may discourage, delay or prevent a change of control by prohibiting us from engaging in a business combination with stockholders owning in excess of 15 percent of our outstanding voting stock for a period of three years after the person becomes an interested stockholder, even if a change of control would be beneficial to our existing stockholders. In addition, our restated articles of incorporation and restated bylawsBylaws contain provisions that may make the acquisition of our company, a proxy contest, or the nomination of a director candidate by a stockholder more difficult than such actions would be in the absence of such provisions, including the following:that:
| | ●•
| provide that only our Board will havehas the right to fill a vacancy on the Board created by an expansion or by the expansionresignation, death, or removal of a director, which prevents stockholders from being able to fill vacancies on our Board or the resignation, death or removal of a director, which prevents stockholders from being able to fill vacancies on our Board;
|
| | ●•
| provide that only our Chairman of the Board, our Chief Executive Officer, or a majority of our Board will bedirectors are authorized to call a special meeting of stockholders;
|
| | ●•
| authorize the issuance ofwe may issue undesignated preferred stock, the terms of which may be established and shares of which may be issued without stockholder approval (notwithstanding any requirements imposed by the SEC or any exchange on which our common stock may now or in the future trade), and which may include rights superior to the rights of the holders of common stock;
|
| | ●•
| provide that our Board is expressly authorized to make, alteramend, restate, or repeal our bylaws;Bylaws; and
|
| | ●•
| establish advance notice requirements foris required with respect to any nominations for electionselection to our Board or for proposing matters that can be acted upon by stockholders at stockholder meetings; and
|
| ●
| certain priorities, voting rights and other rightsany meeting of the holders of our Series A Convertible Preferred Stockstockholders.
|
TheseIn addition our Bylaws provide that, unless we consent in writing to the selection of an alternative forum, the Court of Chancery of the State of Delaware is the sole and exclusive forum for certain actions, including derivative actions brought on the Company's behalf, stockholder actions claiming breaches of a fiduciary duty owed by any of our directors or officers, and claims arising under our organizational documents, in each case, subject to said Court of Chancery having personal jurisdiction over the indispensable parties named as defendants therein. Although this provision would not apply to any stockholder claims under the Exchange Act, there is uncertainty regarding whether a court would enforce such a forum selection provision as written to stockholder claims under the Securities Act. Nevertheless, this forum selection provision may limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers, employees, or agents, which may discourage lawsuits against us and such persons.
The limitations on certain stockholder rights imposed by these provisions could also discourage proxy contests and make it more difficult for our stockholders to elect directors of their choosing so as to cause us to take certain corporate actions our stockholders may desire to take.
We have never paid dividends on our capital stock, and we do not anticipate paying any cash dividends indepress the foreseeable future.
We have never declared nor paid cash dividends on our capital stock. We currently intend to retain any future earnings to finance the operation and expansion of our business, and we do not expect to declare or pay any dividends in the foreseeable future. Consequently, stockholders must rely on sales of their common stock after price appreciation, which may never occur, as the only way to realize any future gains on their investment.
We have broad discretion in the use of the net proceeds fromthe Private Placement and may not use them effectively.
On March 14, 2017, we completed the Initial Closing of the Private Placement. We received net proceeds of approximately $14.1 million from the sale, after deducting placement agent fees and expenses. In the future, we may conduct one or more additional closings in the Private Placement for up to aggregate gross proceeds of $25.0 million.
We plan to invest the net proceeds from the Private Placement to fund research and development of our lead product candidate, TSC, including clinical trial activities, and for general corporate purposes. The failure by our management to apply these funds effectively could result in financial losses that could have a material adverse effect on our business, cause the markettrading price of our common stock to decline and delay the development of our product candidates. Pending their use, we may invest the net proceeds from the offering in a manner that does not produce income or that loses value. If we do not invest the net proceeds from the offering in ways that enhance stockholder value, we may fail to achieve expected financial results, which could cause the price of our common stock to decline.
Any adjustments in the conversion price of ourSeries A ConvertiblePreferred Stock or the exercise price of our Warrants could have a depressive effect on our stock price and the market for our stock.
If we are required to adjust the conversion price for the Series A Convertible Preferred Stock or the Warrant exercise price pursuant to any of the adjustment provisions of the agreements related to the Private Placement, the adjustment or the perception that an adjustment may be required, may have a depressive effect on both our stock price and the market for common stock.
ITEM 1B. | UNRESOLVED STAFF COMMENTS |
None.
Our principal executiveOn November 8, 2021, the Company entered into a Deed of Lease Termination Agreement with the Carlton Landlord providing for the early termination of the Carlton Lease related to the Company's prior corporate headquarters. The Carlton Lease was previously scheduled to expire on April 30, 2022. In connection with the termination, the Company made a one-time payment to the Carlton Landlord of approximately $14,000, net of a security deposit subsequently returned in accordance with the Carlton Lease. In lieu of the fixed office is locatedand laboratory space previously available to us under the Carlton Lease, the Company has entered into short term agreements to utilize membership-based co-working space in a leased facility inboth Charlottesville, Virginia where we lease approximately 5,000 square feet of office space for approximately $5,500 per month. We lease this space on a month-to-month basis. In 2017, we plan to move into a different location with a similar cost profile, which we believe will be suitable and adequate for our current and immediately foreseeable needs.Philadelphia, Pennsylvania.
FromThe information in Note 6, Commitments and Contingencies — Legal Proceedings to our consolidated financial statements set forth in, Part II — Item 8 — Financial Statements of this Annual Report is incorporated herein by reference.
In addition, from time to time, we are subject to various pending or threatened legal actions and proceedings, including those that arise in the ordinary course of its business, which may include employment matters, breach of contract disputes and stockholder litigation. Such actions and proceedings are subject to many uncertainties and to outcomes that are not predictable with assurance and that may not be known for extended periods of time. We record a liability in our consolidated financial statements for costs related to claims, including future legal costs, settlements and judgments, when we have assessed that a loss is probable and an amount can be reasonably estimated. If the reasonable estimate of a probable loss is a range, we record the most probable estimate of the loss or the minimum amount when no amount within the range is a better estimate than any other amount. We disclose a contingent liability even if the liability is not probable or the amount is not estimable, or both, if there is a reasonable possibility that a material loss may have been incurred. In the opinion of management, as of the date hereof, the amount of liability, if any, with respect to these matters, individually or in the aggregate, will not materially affect our consolidated results of operations, financial position or cash flows.
On August 7, 2014, a complaint was filed in the Superior Court of Los Angeles County, California by Paul Feller, the Company’s former Chief Executive Officer under the captionPaul Feller v. RestorGenex Corporation, Pro Sports & Entertainment, Inc., ProElite, Inc. and Stratus Media Group, GmbH (Case No. BC553996). The complaint asserts various causes of action, including, among other things, promissory fraud, negligent misrepresentation, breach of contract, breach of employment agreement, breach of the covenant of good faith and fair dealing, violations of the California Labor Code and common counts. The plaintiff is seeking, among other things, compensatory damages in an undetermined amount, punitive damages, accrued interest and an award of attorneys’ fees and costs. On December 30, 2014, we filed a petition to compel arbitration and a motion to stay the action. On April 1, 2015, the plaintiff filed a petition in opposition to our petition to compel arbitration and a motion to stay the action. After a hearing for the petition and motion on April 14, 2015, the Court granted our petition to compel arbitration and a motion to stay the action. On January 8, 2016, the plaintiff filed an arbitration demand with the American Arbitration Association. No arbitration hearing has yet been scheduled. We believe this matter is without merit and we intend to defend the arbitration vigorously. Because this matter is in an early stage, we are unable to predict its outcome and the possible loss or range of loss, if any, associated with its resolution or any potential effect the matter may have on our financial position. Depending on the outcome or resolution of this matter, it could have a material effect on our financial position.
ITEM 4. | MINE SAFETY DISCLOSURES |
None.
PART II
| ITEM 5. | MARKET FOR REGISTRANT’SREGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
Market PriceInformation
Our common stock trades publicly on the NASDAQNasdaq Capital Market under the symbol “DFFN.” Prior to November 9, 2016, our common stock traded on the OTCQX over-the-counter bulletin board. The following table sets forth the high and low daily sale prices for our Common Stock, as quoted by the NASDAQ Capital Market or the OTCQX, as applicable, for each calendar quarter during 2016 and 2015, after giving effect to the Reverse Stock Split. The over-the-counter market quotations reflect inter-dealer prices, without retail mark-up, mark-down or commission and may not necessarily represent actual transactions.
2016 | High | Low |
| | | |
Fourth Quarter | $7.00 | $1.94 |
| | | |
Third Quarter | 9.25 | 5.27 |
| | | |
Second Quarter | 11.50 | 8.00 |
| | | |
First Quarter | 17.00 | 8.50 |
| | | |
2015 | High | Low |
| | | |
Fourth Quarter | $26.00 | $4.00 |
| | | |
Third Quarter | 17.50 | 8.00 |
| | | |
Second Quarter | 30.50 | 16.50 |
| | | |
First Quarter | 37.50 | 23.50 |
| | | |
| | | |
| | | |
| | | |
| | | |
In November 2016 our common stock was approved for listing, and commenced trading, on the NASDAQ Capital Market. Prior to that, our common stock traded on the OTCQX marketplace of the OTC Market Groups. In addition, prior to the Merger, our common stock was quoted on the OTCQX marketplace, under the symbol “RESX.” Effective January 25, 2016, our trading symbol changed to “DFFN.” On August 17, 2016, we implemented the Reverse Stock Split of our common stock on a 1:10 basis.
Number of Record Holders
As of March 15, 2017,2022, there were 466343 record holders of our common stock. This does not include beneficial owners of our common stock whose stock is held in nominee or “street name”.
Dividends
To date, we have not declared or paid any cash dividends on our common stock and do not intend to do so in the near future.
Securities Authorized for Issuance Under Equity Compensation Plans
For certainThe information concerning securities authorized for issuance under our equity compensation plan, seeset forth in, Part III — Item 12 — Security(Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.Matters) of this Annual Report is incorporated herein by reference to the extent required by Item 201(d) of Regulation S-K.
Recent Unregistered Sales of Unregistered Equity Securities and Use of Proceeds
During the fourth quarter ended December 31, 2016, we did not issue or sell any Diffusion equity securities without registration under the Securities Act.None
Issuer Purchases of Equity Securities by the Issuer and Affiliated Purchasers
During the fourth quarter ended December 31, 2016, we did not purchase any shares of Diffusion common stock or other Diffusion equity securities.None.
Our Board of Directors has not authorized any repurchase plan or program for the purchase of shares of our common stock or other securities on the open market or otherwise.
ITEM 6. | SELECTED FINANCIAL DATA |
The information required by Item 6 is not applicable to us as a smaller reporting company andof Form 10-K has been omitted.omitted from this Annual Report pursuant to the amendments to Regulation S-K adopted by the SEC on November 19, 2020.
ITEM 7. | MANAGEMENT’SMANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
|
Introduction
This Management’s Discussiondiscussion and Analysis provides materialanalysis contains information related to historical and prospective disclosuresevents intended to enable investors and other usersyou to assess our financial condition and results of operations. Statements that are not historical are forward-lookingThe information contained in this discussion and involve risks and uncertainties discussed under the headings “Part I. Item 1. Business—Cautionary Note Regarding Forward-Looking Statements” and “Part I. Item 1A. Risk Factors” of this report. The following discussion of our results of operations and financial conditionanalysis should be read in conjunction with our consolidated financial statements and the related notes thereto includedcontained elsewhere in this report. TheseAnnual Report, as well as the risks could cause our actual results to differ materially from any future performance suggested below.and uncertainties discussed under the heading, "Part 1A — Risk Factors."
Business OverviewDiffusion Pharmaceuticals: Enhancing Oxygen, Fueling Life
We are a clinical stage biotechnologybiopharmaceutical company focused on extendingdeveloping novel therapies that enhance the life expectancy of cancer patients by improvingbody’s ability to deliver oxygen to the effectiveness of current standard-of-care treatments, including radiation therapy and chemotherapy. We are developing ourareas where it is needed most. Our lead product candidate,transcrocetinate sodium, TSC, is being developed to enhance the diffusion of oxygen to tissues with low oxygen levels, also known astrans sodium crocetinateorTSC, for use in the hypoxia, a serious complication of many cancer types in which tumor oxygen deprivation, or hypoxia, is known to diminish the effectiveness of current treatments. TSC is designed to target the cancer’s hypoxic micro-environment, re-oxygenating treatment-resistant tissuemedicine’s most intractable and making the cancer cells more susceptible to the therapeutic effectsdifficult-to-treat conditions.
Highlights from Fourth Quarter of standard-of-care radiation therapy and chemotherapy.2021
| • | Announced Lead Development Program – In November 2021, we announced our intent to develop TSC as an adjunct to standard of care therapy for hypoxic solid tumors. |
| • | Initiated Final Two Oxygenation Trials – On November 22, 2021, we announced dosing of the first participants in the Altitude Trial and, on December 16, 2021, we announced dosing of the first patients in the ILD-DLCO Trial. We currently expect to complete dosing in the Altitude Trial in the first quarter of 2022 and to complete dosing in the ILD-DLCO Trial in the second quarter of 2022. |
| • | Enhanced Intellectual Property Portfolio – On November 30, 2021, the USPTO granted to the Company United States Patent No. 11,185,523, “Use of Bipolar Trans Carotenoids With Chemotherapy and Radiotherapy for Treatment of Cancer.” This patent includes new claims related to methods of treating cancerous tumors by administering TSC in combination with radiation therapy and chemotherapy. This new patent extends the normal life of our cancer-related intellectual property claims into 2037. |
| • | Implemented Cost-Saving Facility Changes – In November 2021, we terminated the Carlton Lease for our prior corporate headquarters, furthering our shift to a remote-work culture. During 2021, we also entered into short-term agreements to utilize membership-based co-working space in both Charlottesville, Virginia and Philadelphia, Pennsylvania, resulting in cost savings. |
Our lead development programs target TSC against cancers knownStrategic Priorities for 2022
In 2022, we plan to be inherently treatment-resistant, including brain cancerscontinue and pancreatic cancer. A Phase 1/2 clinical trial of TSC combined with first-line radiation and chemotherapy in patients newly diagnosed with primary brain cancer, glioblastoma, or GBM, was completed in 2015. This trial provided evidence of efficacy and safety in extending overall survival without the addition of toxicity. Based on these results, an End-of-Phase 2 meeting was held with the FDA in August 2015, resulting in agreementfurther our focus on the designdevelopment of a single 400 patient pivotal Phase 3 registration study which, if successful, would be sufficient to support approval. Discussions with the FDA regarding extension of the TSC development program from first line GBM into first-line pancreatic cancer treatment are currently underway. TSC has been granted Orphan Drug designations for the treatment of GBMhypoxia and metastatic brain cancer.as a platform to enhance standard-of-care treatment for conditions complicated by hypoxia with a particular near-term focus on hypoxic solid tumors.
As of March 15, 2022, TSC has been administered to more than 220 subjects across 11 clinical trials. Data from these clinical trials support our understanding of the safety, tolerability, pharmacokinetics and pharmacodynamic effects of TSC. In addition, post hoc analyses of two prior studies involving patients with PAD with claudication and unresected GBM tumors have provided preliminary evidence of TSC’s potential. However, the data available from these studies and the post hoc analyses of their outcomes are limited in several meaningful ways. For example, neither study was powered to cancer,formally demonstrate efficacy at a statistically significant level, there was no evaluation of whether TSC also has potential applicationsincreased oxygenation in other indications involving hypoxia, such as hemorrhagic shock, stroke, peripheral artery diseasethe target tissues in either study, and, neurodegenerative diseases.in the GBM Trial, no dose exploration was conducted.
On January 8, 2016,During 2021, to address these identified gaps in our TSC knowledge base, further inform our identification of potential indications appropriate for TSC, and guide the future of our TSC development program, we designed and executed on our Oxygenation Trials. The Oxygenation Trials are a series of three short-term, clinical studies designed to provide clinical evidence of the relationship between TSC dose and the effects on oxygenation, each specifically tailored to evaluate the effects of TSC on a different component of the oxygen delivery pathway. We completed the Mergerfirst of these studies, the TCOM Trial, in March 2021. The first participants and patients in the remaining two Oxygenation Trials, the Altitude Trial and the ILD-DLCO Trial, were dosed in November 2021 and December 2021, respectively. We currently expect to complete dosing in the Altitude Trial in the second quarter of 2022 and to complete dosing in the ILD-DLCO Trials in the middle of 2022 and, in each case, to report top-line results within two months of study completion.
In November 2021, based on the available preclinical and clinical data and the significant unmet medical need, we announced our intention to focus near-term efforts on developing TSC as an adjunct to standard of care therapy for hypoxic solid tumors. Through the combination of our past experiences, new knowledge gained from our Oxygenation Trials and COVID Trial regarding TSC’s effects on oxygenation, dose response characteristics, pharmacokinetics, and pharmacodynamics, and further analyses and discussions of all available data with Diffusion LLC. our Scientific Advisory Board and other external advisors, we believe we now have the necessary information to design the Hypoxic Solid Tumor Program to be more efficient and increase our likelihood of success compared to the Company’s past efforts to develop TSC as a cancer treatment, which were terminated in 2019 due to financial constraints.
As a resultpart of the Merger, Diffusion LLC,ongoing design of our Planned Phase 2 Hypoxic Tumor Trial – the surviving companyfirst trial in our Hypoxic Solid Tumor Program which we currently expect to commence in the Merger, becamesecond half of 2022, subject to FDA feedback and the availability of clinical drug supply – we are currently drafting a wholly-owned subsidiarytrial protocol which we intend to file with the FDA. In parallel, we will continue our work to optimize the TSC manufacturing process and support the continued availability of high-quality drug product and undertake preclinical studies and other opportunities to continue developing data designed to demonstrate TSC’s potential uses in a broad spectrum of non-cancer indications.
Financial Summary
As of December 31, 2021, we had a cash and cash equivalents balance of $37.3 million. We have incurred operating losses since inception, have not generated any product sales revenue, and have not achieved profitable operations. We incurred net losses of $24.1 million and $14.2 million for the years ended December 31, 2021 and 2020, respectively. To date, we have funded our operations and short-term liquidity needs primarily through the issuance and sale of common stock, warrants to purchase common stock, convertible debt, and convertible preferred stock. We expect to continue funding our operations through similar means for the foreseeable future, assuming the availability of additional capital, though we may enter into strategic partnerships or other alternative transactions in order to fund our ongoing capital requirements.
Our accumulated deficit as of December 31, 2021, was $130.0 million and we expect to continue to incur substantial losses in future periods for the foreseeable future. We also anticipate that our operating expenses will increase substantially as we continue to advance the development of TSC, including any costs related to:
| • | our ongoing and planned clinical trials, including the ongoing Oxygenation Trials and our Planned Phase 2 Hypoxic Solid Tumor Trial; |
| • | any additional studies we may undertake to evaluate TSC, including other preclinical and clinical studies to support the filing of any NDA with the FDA; |
| • | near-term investments intended to improve the quality and robustness of our supplier relationships and overall supply chain; |
| • | other research, development, and manufacturing activities designed to develop and optimize formulation, manufacturing processes, dosage, dose forms, and other characteristics prior to regulatory approval; |
| • | the maintenance, expansion, and protection our global intellectual property portfolio; |
| • | the hiring of additional clinical, manufacturing, scientific, sales, or other personnel; |
| • | research and development related to any other product candidates we may acquire or in-license in the future; and |
| • | investments in operational, financial, and management information systems. |
We intend to use our existing cash and cash equivalents for working capital and to fund the research and development of TSC. We expect that our cash and cash equivalents as of December 31, 2021 will enable us to fund our operating expenses and capital expenditure requirements through 2023.
Financial Operations Overview
Revenues
We have not yet generated any revenue from product sales. We do not expect to generate revenue from product sales for the foreseeable future.
Research and Development Expense
R&D expenses include, but are not limited to, third-party CRO arrangements and employee-related expenses, including salaries, benefits, stock-based compensation, and travel expense reimbursement. R&D activities are central to our business model. Product candidates in later stages of clinical development generally have higher development costs than those in earlier stages of clinical development, primarily due to the increased size and duration of later-stage clinical studies. As we advance our product candidates, we expect the amount of R&D costs will continue to increase for the foreseeable future. R&D costs are charged to expense as incurred.
Intangible Asset Impairment Charge
In the third quarter of 2021, the Company made a determination to no longer dedicate resources to the Company’s DFN-529 intangible asset and any future development efforts were abandoned. In connection with this decision, the Company concluded that DFN-529 was impaired in its entirety.
General and Administrative Expense
G&A expenses consist principally of salaries and related costs for executive and other personnel, including stock-based compensation, other employee benefit costs, expenses associated with investment bank and other financial advisory services, and travel expenses. Other G&A expenses include, facility-related costs, communication expenses and professional fees for legal, patent prosecution and maintenance, consulting, accounting, and other professional services.
Interest Income
Interest income consists of interest earned from our cash and cash equivalents.
Income Tax Benefit
The Company recorded an income tax benefit of $0.4 million during the year ended December 31, 2021. The income tax benefit was due to the tax effect of the Company. Immediately followingreduction in the Merger,deferred tax liability associated with the former equity holdersbasis difference from the IPR&D indefinite lived intangible asset. The Company maintains a full valuation allowance against its deferred tax assets due to the Company’s history of Diffusion LLC owned approximately 84.1%losses as of December 31, 2021.
Our NOLs and tax credit carryforwards are subject to review and possible adjustment by the Internal Revenue Service and state tax authorities. NOL and tax credit carryforwards may become subject to an annual limitation in the event of a greater than 50.0% cumulative change in the ownership interest of significant stockholders over a three year period, as defined under Sections 382 and 383 of the Internal Revenue Code as well as similar state provisions. The amount of the annual limitation is determined based on the Company’s common stock, and the stockholders of RestorGenexvalue immediately prior to the Merger owned approximately 15.9% ofownership change, and subsequent ownership changes may further affect the Company’s common stock,limitation in each case, on a fully-diluted basis and calculatedfuture years. We have not yet performed an analysis to determine whether or not ownership changes that have occurred in accordance with the terms ofyear ended December 31, 2020 or during the Merger Agreement. Also in connection with the Merger, the pre-Merger directors and officers of the Company tendered their resignations and the pre-Merger directors and officers of Diffusion LLC were appointed as the new directors and officers of the Company. Following the completion of the Merger, the Company changed its corporate name from “RestorGenex Corporation”year ended December 31, 2021 give rise to “Diffusion Pharmaceuticals Inc.” and changed the trading symbol of the Company’s Common Stock from “RESX” to “DFFN.” In November 2016, our common stock was approved for listing on the NASDAQ Capital Market and will continue to trade under the ticker symbol “DFFN”.any further limitations.
For accounting purposes, the Merger is treated as a “reverse acquisition” under U.S. GAAP and Diffusion LLC is considered the accounting acquirer. Accordingly, Diffusion LLC’s historical results of operations replaced the Company’s historical results of operations for all periods prior to the Merger. Unless otherwise stated, all comparisons in this Management’s Discussion and Analysis to prior year periods are to the results of Diffusion Pharmaceuticals LLC for such period on a stand-alone basis.
Recent Development
On March 14, 2017, we entered into Subscription Agreements with certain accredited investors and conducted the Initial Closing of a Private Placement pursuant to which we sold 7,837,023 shares of the Series A Convertible Preferred Stock at a purchase price of $2.02 per share. In addition, each investor received a Warrant to purchase one share of common stock for each share of Series A Convertible Preferred Stock purchased by such investor in the Private Placement at an exercise price equal to $2.22, subject to adjustment thereunder.
We received total net proceeds of approximately $14.1 million from the Initial Closing of the Private Placement, after deducting $1.7 million in placement agent fees and expenses associated with the Private Placement. The Company currently intends to use the proceeds of the Private Placement to fund research and development of TSC, including clinical trial activities, and for general corporate purposes. In the future, we may conduct one or more additional closings in the Private Placement for up to aggregate gross proceeds of $25.0 million.
Critical Accounting Policies and Estimates
Certain of our critical accounting policies require estimates requirethat involve the application of significant judgment by management in selecting the appropriate assumptions in determining the estimate. By their nature, these judgments are subject to an inherent degree of uncertainty. We develop these judgments based on our historical experience, terms of existing contracts, our observance of trends in the industry, and information available from other outside sources, as appropriate. Actual results may differ from these judgments under different assumptions or conditions. Different, reasonable estimates could have been used for the current period. Additionally, changes in accounting estimates are reasonably likely to occur from period to period. Both of these factors could have a material impact on the presentation of our financial condition, changes in financial condition, or results of operations. We believe the following accounting estimatespolicies described below are among the most critical to aid in fully understanding and evaluating our financial statements, as they require estimates which involve our most subjective or complex judgments:judgments.
GoodwillIntangible Assets
Goodwill is the excess of the cost of an acquired entity over the net amounts assigned to tangible and intangible assets acquired and liabilities assumed. We apply Accounting Standards Codification (ASC) 350 “Goodwill and Other Intangible Assets,” which requires testing goodwill for impairment on an annual basis. We assess goodwill for impairment as part of our annual reporting process on October 1 of each year. In between valuations, we conduct additional tests if circumstances indicate a need for testing. We evaluate goodwill on a consolidated basis as we are organized as a single reporting unit. We consider certain triggering events when evaluating whether an interim goodwill impairment analysis is warranted. Among these would be a significant long-term decrease in our market capitalization based on events specific to our operations. There was no impairment to our goodwill during the year ended December 31, 2016.
Intangible Assets
Our sole intangible asset as of December 31, 2020 consisted of DFN-529, which was acquired in 2016 consists ofpursuant to our merger with RestorGenex Corporation and was accounted for as an in-process research and development (“IPR&D”) intangible asset acquired as part of the Merger.asset. The fair value of the IPR&D assetsasset was determined as of the acquisition date using the cost approach, often referred to as current replacement cost, which establishes a value based on the cost of reproducing or replacing the asset, often referred to as current replacement costs.asset. The cost approach was chosen as we were not able to estimate an income stream attributable to the IPR&D assetsasset, given the fact that the related products havehad only completed limited preclinical and clinical trials and the timeline to commercial viability, if the FDA approval process iswere to be successful, is somewhatwas uncertain, and would take a number of years, and the costs would behave been significant. In August 2016,the third quarter of 2021, we abandonedmade a determination to no longer dedicate resources to our DFN-529 intangible asset and any future development efforts for the IPR&D asset associatedwere abandoned. In connection with our RES-440 product candidate and recorded an impairment charge equal to the acquired value of RES-440. As the development efforts for our remaining RES-529 IPR&D asset continue, based on the facts and circumstances at the time of a future valuation for the purposes of assessing impairment, it is possiblethis decision, we concluded that the value for RES-529 could be substantially reduced or eliminated, which could resultDFN-529 was impaired in a maximum pretax charge to operations equal to the current carrying value of our intangible asset of $8.6 million as of December 31, 2016. We tested the IPR&D intangible asset for impairment on October 1, which is our annual impairment testing date. We consider certain triggering events when evaluating whether an interim IPR&D impairment analysis is warranted. There was no impairment to RES-529 during the year ended December 31, 2016.
Research and development
Research and development costs are expensed as incurred and consist primarily of funds paid to third parties for the provision of services for product candidate development, clinical and preclinical development and related supply and manufacturing costs, and regulatory compliance costs. At the end of the reporting period, the Company compares payments made to third-party service providers to the estimated progress toward completion of the research or development objectives. Such estimates are subject to change as additional information becomes available. Depending on the timing of payments to the service providers and the progress that the Company estimates has been made as a result of the service provided, the Company may record net prepaid or accrued expense relating to these costs. Upfront payments made to third parties who perform research and development services on the Company’s behalf are expensed as services are rendered.
Stock-Based Compensation
We account for stock-based compensation based on the grant date fair value of the award. We recognize this cost as an expense over the requisite service period, which is generally the vesting period of the respective award. Forfeitures rates are used in stock-based compensation to adjust the recognized stock-based compensation expense to reflect the expected attrition of employees prior to their full vesting in stock-based compensation awards. We use the Black-Scholes option-pricing model to determine the estimated fair value of stock options. Inputs into the Black-Scholes option-pricing model include: the grant date fair value of our common stock; the option exercise price; the expected term of the option in years; the annualized volatility of the stock; and the risk-free interest rate. If any of the assumptions used in the Black-Scholes model changes significantly, stock-based compensation for future awards may differ materially compared with the awards granted previously. The inputs that create the most sensitivity in our option valuation are the volatility and expected term.
Given our limited history as a publicly traded company following the Merger in January 2016, we did not have sufficient trading data to calculate volatility based on our own common stock, and the expected volatility was calculated as of each grant date based on a peer group of publicly traded companies. The expected term of the stock options was determined based upon the simplified approach for employees, allowed under SEC Staff Accounting Bulletin No. 110, which assumes that the stock options will be exercised evenly from vesting to expiration. As data associated with future exercises is obtained, the expected term of future grants will be adjusted accordingly. For non-employee awards, we use the remaining contractual term.its entirety.
Financial Summary
At December 31, 2016, we had cash and cash equivalents balances of $1.6 million. We have incurred operating losses since inception, have not generated any product sales revenue and have not achieved profitable operations.We incurred a net loss of $18.0 million and $6.7 million for the years ended December 31, 2016 and 2015, respectively. Our accumulated deficit as of December 31, 2016, was $60.2 million, and we expect to continue to incur substantial losses in future periods.We anticipate that our operating expenses will increase substantially as we continue to advance our lead, clinical-stage product candidate, TSC. We anticipate that our expenses will substantially increase as we:
| ●
| complete regulatory and manufacturing activities and commence our planned Phase II and III clinical trials for TSC;
|
| ●
| continue the research, development and scale-up manufacturing capabilities to optimize products and dose forms for which we may obtain regulatory approval;
|
| ●
| conduct other preclinical and clinical studies to support the filing of a NDA for TSC with the FDA;
|
| ●
| maintain, expand and protect our global intellectual property portfolio;
|
| ●
| hire additional clinical, manufacturing, and scientific personnel; and
|
| ●
| add operational, financial and management information systems and personnel, including personnel to support our drug development and potential future commercialization efforts.
|
We intend to use our existing cash and cash equivalents for working capital and to fund the research and development of TSC. We expect that our existing cash as of December 31, 2016, together with the proceeds from the Initial Closing of the Private Placement of our Series A Convertible Preferred Stock in March 2017, will enable us to fund our operating expenses and capital expenditure requirements into December of 2017.
Financial Operations Overview
Revenues
We have not yet generated any revenue from product sales. We do not expect to generate revenue from product sales for the foreseeable future.
Research and Development Expense
Research and development costs include, but are not limited to, third-party contract research arrangements, employee-related expenses, including salaries, benefits, stock-based compensation and travel expense reimbursement, as well as impairment of our in-process research and development assets. Research and development activities are central to our business model. Product candidates in later stages of clinical development generally have higher development costs than those in earlier stages of clinical development, primarily due to the increased size and duration of later-stage clinical trials. As we advance our product candidates, we expect the amount of research and development costs will continue to increase for the foreseeable future. Research and development costs are charged to expense as incurred.
General and Administrative Expense
General and administrative expenses consist principally of salaries and related costs for executive and other personnel, including stock-based compensation, expenses associated with investment bank and other financial advisory services, and travel expenses. Other general and administrative expenses include costs associated with the Merger, professional fees that were incurred in connection with preparing to operate and operating as a public company, settlement of litigation matters, facility-related costs, communication expenses and professional fees for legal, patent prosecution and maintenance, and consulting and accounting services.
Interest Expense, Net
Interest expense, net for the year ended December 31, 2016 consisted principally of the interest expense recorded in connection with our convertible debt instruments offset by the interest earned from our cash and cash equivalents.
Income Tax Benefit
Since inception, we have incurred net losses and have not recorded any U.S. federal or state income tax benefits for the losses as they have been offset by valuation allowances. Indefinite lived intangibles, such as IPR&D, cannot be utilized for purposes of future realization of deferred tax assets. For the year ended December 31, 2016, we recognized an income tax benefit from the impairment charge related to our abandonment of the future development efforts for the RES-440 IPR&D asset.
Results of Operations for Year Ended December 31, 20162021 Compared to Year Ended December 31, 20152020
The following table sets forthsummarizes our results of operations for the years ended December 31, 20162021 and 2015:2020:
| | | Year ended December 31, | | | | | |
| | | 2016 | | | 2015 | | | Change | |
Operating expenses: | | | | | | | | | | | | |
Research and development | | $ | 7,252,241 | | | $ | 3,875,467 | | | $ | 3,376,774 | |
General and administrative | | | 11,094,146 | | | | 2,522,370 | | | | 8,571,776 | |
Depreciation | | | 25,342 | | | | 8,268 | | | | 17,074 | |
Loss from operations | | | 18,371,729 | | | | 6,406,105 | | | | 11,965,624 | |
Interest expense, net | | | 29,686 | | | | 313,117 | | | | (283,431 | ) |
Loss from operations before income tax benefit | | | (18,401,415 | ) | | | (6,719,222 | ) | | | (11,682,143 | ) |
Income tax benefit | | | (364,796 | ) | | | - | | | | (364,796 | ) |
Net loss | | $ | (18,036,619 | ) | | $ | (6,719,222 | ) | | $ | (11,317,347 | ) |
| | | Year ended December 31, | | | | | |
| | | 2021 | | | 2020 | | | Change | |
Operating expenses: | | | | | | | | | | | | |
Research and development | | $ | 8,499,414 | | | $ | 9,427,667 | | | $ | (928,253 | ) |
Intangible asset impairment charge | | | 8,639,000 | | | | — | | | $ | 8,639,000 | |
General and administrative | | | 7,445,277 | | | | 6,444,109 | | | | 1,001,168 | |
Depreciation | | | 93,416 | | | | 103,168 | | | | (9,752 | ) |
Loss from operations | | | (24,677,107 | ) | | | (15,974,944 | ) | | | 8,702,163 | |
Interest income | | | 137,487 | | | | 114,257 | | | | 23,230 | |
Loss from operations before income taxes | | | (24,539,620 | ) | | | (15,860,687 | ) | | | (8,678,933 | ) |
Income tax benefit (expense) | | | 443,893 | | | | 1,675,381 | | | | (1,231,488 | ) |
Net loss | | $ | (24,095,727 | ) | | $ | (14,185,306 | ) | | $ | (7,447,445 | ) |
Research and development expenses were $7.3$8.5 million during the year ended December 31, 20162021 compared to $3.9$9.4 million during the year ended December 31, 2015.2020, a decrease of 10%. This increasedecrease was primarilydue to lower project spending due to the completion and/or wind-down of our clinical studies evaluating TSC in Covid-19, GBM, and stroke.
In the third quarter of 2021, the Company made a result of an additional $1.2 million of expenses relateddetermination to animal toxicology studies as well as an increase of $1.0 million in active product ingredient manufacturing costs. We also incurred an additional $0.5 million in costs relatedno longer dedicate resources to our TSC pancreatic cancer programthe Company’s DFN-529 intangible asset and recognized a $1.0 million impairment charge upon our abandonment ofany future development efforts were abandoned. As a result, we recognized a nonrecurring $8.6 million non-cash impairment charge related to the write down of our RES-440DFN-529 IPR&D asset. Salaries and wages expense and stock compensation expense increased by $0.3 million and $0.4 million, respectively, during the year ended December 31, 2016 compared to the prior year, due to an increase in headcount. The overall increase in research and development expense was offset by a $0.9 million decrease in spend related to GBM trials. We expect that our research and development expenses will increase significantly in future periods compared to prior year periods due to our anticipated efforts to advance the research and development of our technologies and product candidates.
General and administrative expenses were $11.1$7.4 million during the year ended December 31, 20162021 compared to $2.5$6.4 million during the year ended December 31, 2015.2020, an increase of 16%. The increase was primarily attributabledue to increased headcount resulting in higher compensation expense and other costs associated with the hiring of new employees as well as an increase of $4.1 million in professional fees incurred in connection with preparing to operate as a public company, merger and transaction related fees and feesexpense related to investment bank advisoryconsulting services. Insurance expense also increased by $0.8 million during
For the year ended December 31, 2016 compared2021, we recognized an income tax benefit of $0.4 million due to the prior year, which was mainly attributable to directors and officer’s insurance. We also recognized a $2.5 million noncash charge upon the settlementtax effect of the Schmidt litigation matter duringreduction in the deferred tax liability associated with the basis differences from the DFN-529 IPR&D intangible asset that was written down in the third quarter of 2021. For the year ended December 31, 2016. Salaries and wages and stock compensation expense increased by $0.4 million and $0.4 million, respectively, during the year ended December 31, 2016 compared to the prior year due to our increase in headcount.
Interest expense decreased by $0.3 million in 2016 due to the conversion of debt into equity that occurred at the end of 2015 and in early 2016.
During the year ended December 31, 2016,2020, we recognized aan income tax benefit of $0.4$1.7 million in connection withto reflect the impairment charge associated with the write downutilization of indefinite deferred tax liabilities as a source of income against indefinite lived portions of our RES-440 IPR&D intangible asset.deferred tax assets. Prior to 2021, we recognized the full income tax benefit allowed by the 2017 Tax Act to utilize indefinite deferred tax liabilities as a source of income against indefinite lived portions of our deferred tax assets.
Liquidity and Capital Resources
Working Capital (Deficit)
The following table summarizes our working capital (deficit) as of December 31, 20162021 and 2015:2020:
| | | December 31, | |
| | | | | | | |
| | | 2016 | | | 2015 | |
| | | | | | | | | |
Cash and cash equivalents | | $ | 1,552,852 | | | $ | 1,997,192 | |
| | | | | | | | | |
Prepaid expenses, deposits and other assets | | | 50,844 | | | | 45,921 | |
| | | | | | | | | |
Total current liabilities | | | (4,438,422 | ) | | | (1,471,308 | ) |
| | | | | | | | | |
Working capital (deficit) | | $ | (2,834,726 | ) | | $ | 571,805 | |
| | | December 31, | |
| | | 2021 | | | 2020 | |
Cash and cash equivalents | | $ | 37,313,558 | | | $ | 18,515,595 | |
Prepaid expenses, deposits and other assets | | | 510,015 | | | | 260,825 | |
Total current liabilities | | | 2,927,684 | | | | 2,435,783 | |
Working capital | | $ | 34,895,889 | | | $ | 16,340,637 | |
We expect to continue to incur net losses for the foreseeable future. We intend to use our existing cash and cash equivalents for working capital and to fund the research and development of TSC and our other product candidates.candidates through 2023.
Cash Flows
The following table sets forth our cash flows for the years ended December 31, 20162021 and 2015:2020:
| | | December 31, | | |
| | | | | | | | | December 31, | |
Net cash (used in) provided by: | | 2016 | | | 2015 | | | 2021 | | | 2020 | |
| | | | | | | | | | |
Operating activities | | $ | (10,768,265 | ) | | $ | (5,185,328 | ) | | $ | (14,501,789 | ) | | $ | (13,552,629 | ) |
| | | | | | | | | | |
Investing activities | | | 8,498,271 | | | | 2,459,709 | | | 4,000 | | | — | |
| | | | | | | | | | |
Financing activities | | | 1,825,654 | | | | 2,386,292 | | | | 33,295,752 | | | | 17,890,875 | |
| | | | | | | | | | |
Net decrease in cash and cash equivalents | | $ | (444,340 | ) | | $ | (339,327 | ) | |
Net increase in cash and cash equivalents | | | $ | 18,797,963 | | | $ | 4,338,246 | |
Operating Activities
For the year ended December 31, 2021, net cash used in operating activities increased $0.9 million, or 7% compared to the year ended December 31, 2020.
Net cash used in operating activities of $10.8$14.5 million during the year ended December 31, 20162021 was primarily attributable to our net loss of $18.0$24.1 million and a $0.4 million change in deferred income taxes. These amounts were partially offset by $6.0a $8.6 million non cash impairment charge in connection with the write down of non-cash charges and $1.3 million for theour DFN-529 IPR&D asset, our net change in our operating assets and liabilities. Noncashliabilities of $0.4 million, and non-cash charges primarily consistedcomprised of $0.9 million of stock-based compensation expense, the loss on the disposal of $1.4property and equipment of $0.1 million the issuanceand depreciation expense of 148,073 shares of our common stock for advisory services at an estimated fair value of $1.4 million, a $1.0 million impairment charge in connection abandoning our future development efforts of RES-440 IPR&D asset, and $2.5 million noncash litigation settlement charge. The net change in our operating assets and liabilities is primarily attributable to the increase in our accounts payable and accrued expenses due to the timing in processing our payroll and payment to our vendors for professional services and costs associated with our clinical and preclinical activities.$0.1 million.
Net cash used in operating activities of $5.2$13.6 million during the year ended December 31, 20152020 was primarily attributable to our net loss of $6.7$14.2 million thatand a $1.7 million change in deferred income taxes. This amount was offset by $0.9 million of non-cash charges and $0.6 million for theour net change in our operating assets and liabilities. Noncashliabilities of $1.5 million, and non-cash charges primarily consistedcomprised of $0.7 million of stock-based compensation expense and non-cash interest related to our convertible debt.depreciation expense of $0.1 million.
Investing Activities
Net cash provided by investing activities was $8.5 million during the year ended December 31, 2016 compared to $2.5 million during the year ended December 31, 2015. We acquired $8.5 million as a result of the Merger during the year ended December 31, 2016. During the year ended December 31, 2015, certificates2021, we received $4,000 from the sale of deposit maturedproperty and proceeds of $2.5 million were received.equipment. For the year ended December 31, 2020, we had no cash flows from investing activities.
Financing Activities
For the year ended December 31, 2021, net cash provided by financing activities increased $15.4 million, or 86% compared to the year ended December 31, 2020.
Net cash provided by financing activities was $1.8of $33.3 million during the year ended December 31, 2016 and2021 was primarily attributable to net proceeds of $31.1 million received from the $1.9 million cash proceeds receivedsale of our common stock in connection with the issuanceFebruary 2021 Offering and $2.2 million in proceeds received from the exercise of 6.0% convertible notes in September 2016. previously issued common stock warrants.
Net cash provided by financing activities was primarily $2.4of $17.9 million during the year ended December 31, 2015, and2020 was primarily attributable to the issuance$10.8 million in proceeds (net of convertible debt.underwriting discounts and commissions payable by us) received in connection with the May 2020 Offering and $8.0 million in proceeds received in connection with the exercise of common stock warrants stock during the year. These cash inflows were offset in part by the payment of $1.0 million in additional financing costs.
Capital Requirements
We expect to continue to incur substantial expenses and generate significant operating losses as we continue to pursue our business strategy of developing our lead product candidate, TSC, for use in the treatmentTSC. Our operations have consumed substantial amounts of GBM, pancreatic cancercash since inception and brain metastases.
To date, we have primarily used equity and debt financings to fund our ongoing business operations and short-term liquidity needs. We expect to continue to spend substantial amounts of cash to advance the clinical development of TSC and any other product candidates we may in-license or acquire in the future. As of the date of this practiceAnnual Report, most of our cash resources for the foreseeable future.clinical development are dedicated to our ongoing and planned clinical trials. While we believe we have adequate cash resources to continue operations through 2023, we anticipate that we will need additional funding in order to complete development of TSC which, if available, could be obtained through additional capital raising transactions, entry into strategic partnerships or collaborations, or alternative financing arrangements.
In January 2016, we completed the Merger. For accounting purposes, Diffusion LLC is considered the acquiring entity in the Merger and, as a result, we acquired $8.5 million in cash. In September 2016, we issued convertible notes and received cash proceeds of $1.9 million. As of December 31, 2016,2021, we did not have any existing credit facilities in place under which we could borrow funds.
We expect our existing cash asfunds or any other sources of December 31, 2016, together withcommitted capital. In the proceeds from the Initial Closing of the Private Placement of our Series A Convertible Preferred Stock in March 2017, will enable us to fund our operating expenses and capital expenditure requirements into December of 2017. Wefuture, we may seek to raise additional funds through various sources, such as equity and debt financings, or through strategic collaborations and license agreements. Wesources. However, we can give no assurances that we will be able to secure additional sources of funds to support our operations, or if such funds are available to us, that such additional financing will be sufficient to meet our needs or be on terms acceptable to us. This risk may increase if economic and market conditions deteriorate. If we are unable to obtain additional financing when needed, we may need to terminate, significantly modify, or delay the development of TSC or our product candidates, and our operations, or we may need to obtain funds through collaboratorscollaborations or otherwise on terms that may require us to relinquish rights to our technologies or product candidates that we might otherwise seek to develop or commercialize independently. If we are unable to raise anyadequate additional capital as and when required in the near-term and/orfuture, we cannot significantly reduce our expenses and arecould be forced to cease development activities and terminate our operations, investors mayand you could experience a complete loss of theiryour investment.
To the extent that we raise additional capital in the future through the sale of our common stock or securities convertible or exchangeable for common stock such as common stock warrants, convertible preferred stock, or convertible debt instruments, the interests of our current stockholders may be diluted. If we issue preferred stockdiluted or convertible debt securities, it could affect the rights of our common stockholders or reduce the value of our common stock.otherwise impacted. In particular, specific rights granted to future holders of preferred stock or convertible debt securities may include voting rights, preferences as to dividends and liquidation, conversion and redemption rights, sinking fund provisions, and restrictions on our ability to merge with or sell our assets to a third party. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends.
Contractual Obligations
As of December 31, 2016, we had the following contractual commitments:
| | | Payments due by period | |
Contractual Obligations | | Total | | | Less than 1 year | | | 1-3 years | | | 3-5 years | | | More than 5 years | |
Principal on Convertible Notes (1) | | $ | 2,430,000 | | | $ | 1,880,000 | | | $ | 550,000 | | | $ | - | | | $ | - | |
Interest on Convertible Notes | | | 152,942 | | | | 112,800 | | | | 40,142 | | | | - | | | | - | |
Total | | $ | 2,582,942 | | | $ | 1,992,800 | | | $ | 590,142 | | | $ | - | | | $ | - | |
| (1)
| The Company issued convertible notes in March of 2011 and September of 2016, bearing interest at 1% and 6%, respectively. The convertible notes mature in September of 2017 and June of 2018, respectively, unless converted earlier into shares of common stock at the holders election or upon certain specified events.
|
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements, as defined by the rules and regulations of the SEC that have or are reasonably likely to have a material effect on our financial condition, changes in financial condition, revenue or expenses, results of operations, liquidity, capital expenditures or capital resources. As a result, we are not materially exposed to any financing, liquidity, market or credit risk that could arise if we had engaged in these arrangements.
Recently Issued Accounting Pronouncements
Recently issued accounting pronouncements are addressedThe information in Note 3, Basis of Presentation and Summary of Significant Accounting Policies to our consolidated financial statements set forth in, the Notes to Consolidated"Part II — Item 8 — Financial Statements.Statements" of this Annual Report is incorporated herein by reference.
ITEM 7A. | QUANTITATIVE AND QUALITATIVE DISCLOSURE ABOUT MARKET RISK |
As a "smaller reporting company" (as such term is defined in Rule 12b-2 of the Exchange Act), we are not required to provide the information described in Item 7A is not applicable to us as a smaller reporting company305 of Regulation S-K and, accordingly, the information required by Item 6 of Form 10-K has been omitted.omitted from this Annual Report.
ITEM 8. | FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA |
Description | Page |
| | |
Report of Independent Registered Public Accounting Firm (KPMG LLP, McLean, Virginia Auditor Firm ID: 185) | 6653
|
| | |
Consolidated Balance Sheets as of December 31, 20162021 and 20152020 | 6754
|
| | |
Consolidated Statements of Operations for the years ended December 31, 20162021 and 20152020 | 6855
|
| | |
Consolidated Statements of Changes in Stockholders’ Equity (Deficit) for the years ended December 31, 20162021 and 20152020 | 6956
|
| | |
Consolidated Statements of Cash Flows for the years ended December 31, 20162021 and 20152020 | 7057
|
| | |
Notes to the Consolidated Financial Statements for the years ended December 31, 20162021 and 20152020 | 7158
|
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRMReport of Independent Registered Public Accounting Firm
TheTo the Stockholders and Board of Directors
Diffusion Pharmaceuticals Inc.:
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheets of Diffusion Pharmaceuticals Inc. and subsidiaries (the Company) as of December 31, 20162021 and 2015, and2020, the related consolidated statements of operations, changes in stockholders’ equity, (deficit), and cash flows for the years then ended. ended, and the related notes (collectively, the consolidated financial statements). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2021 and 2020, and the results of its operations and its cash flows for each of the years then ended, in conformity with U.S. generally accepted accounting principles.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States).PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement. Anmisstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit includesof its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence supportingregarding the amounts and disclosures in the consolidated financial statements. An auditOur audits also includes assessingincluded evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statement presentation.statements. We believe that our audits provide a reasonable basis for our opinion.
In our opinion,Critical Audit Matters
Critical audit matters are matters arising from the current period audit of the consolidated financial statements referredthat were communicated or required to above present fairly, in allbe communicated to the audit committee and that: (1) relate to accounts or disclosures that are material respects, the financial position of Diffusion Pharmaceuticals Inc. and subsidiaries as of December 31, 2016 and 2015, and the results of their operations and their cash flows for the years then ended in conformity with U.S. generally accepted accounting principles.
The consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 2 to the consolidated financial statements the Company has suffered recurring losses from operations, has limited resources available to fund current research and development activities, and will require substantial additional financing to continue to fund its research and development activities. The conditions raise substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters(2) involved our especially challenging, subjective, or complex judgments. We determined that there are also described in Note 2. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
As discussed in Note 1 to the consolidated financial statements, on January 8, 2016, the Company completed a merger with RestorGenex Corporation under which a newly formed subsidiary of RestorGenex Corporation merged with and into the Company, with the Company surviving as a wholly-owned subsidiary of RestorGenex Corporation.
no critical audit matters.
/s/ KPMG LLP
We have served as the Company’s auditor since 2015.
McLean, Virginia
March 31, 201718, 2022
DIFFUSION PHARMACEUTICALS INC.
CONSOLIDATED BALANCE SHEETS
| | | December 31, | | | December 31, | |
| | | 2016 | | | 2015 | | | 2021 | | | 2020 | |
Assets | | | | | | | | | | | | |
Current assets: | | | | | | | | | |
Cash and cash equivalents | | $ | 1,552,852 | | | $ | 1,997,192 | | | $ | 37,313,558 | | | $ | 18,515,595 | |
Prepaid expenses, deposits and other current assets | | | 50,844 | | | | 45,921 | | | | 510,015 | | | | 260,825 | |
Total current assets | | | 1,603,696 | | | | 2,043,113 | | | 37,823,573 | | | 18,776,420 | |
Property and equipment, net | | | 79,755 | | | | 51,996 | | | 0 | | | 149,198 | |
Intangible asset | | | 8,639,000 | | | | - | | | 0 | | | 8,639,000 | |
Goodwill | | | 6,929,258 | | | | - | | |
Right of use asset | | | 0 | | | 149,162 | |
Other assets | | | 232,675 | | | | 181,487 | | | | 15,578 | | | | 15,771 | |
Total assets | | $ | 17,484,384 | | | $ | 2,276,596 | | | $ | 37,839,151 | | | $ | 27,729,551 | |
Liabilities and Stockholders’ Equity (Deficit) | | | | | | | | | |
Liabilities and Stockholders’ Equity | | | | | |
Current liabilities: | | | | | | | | | |
Current portion of convertible debt | | $ | 1,880,000 | | | $ | 424,964 | | |
Accounts payable | | | 1,684,158 | | | | 424,675 | | | $ | 947,495 | | | $ | 545,844 | |
Accrued expenses and other current liabilities | | | 874,264 | | | | 621,669 | | | 1,980,189 | | | 1,776,470 | |
Current operating lease liability | | | | 0 | | | | 113,469 | |
Total current liabilities | | | 4,438,422 | | | | 1,471,308 | | | 2,927,684 | | | 2,435,783 | |
Convertible debt, net of current portion | | | 550,000 | | | | 818,646 | | |
Deferred income taxes | | | 3,279,363 | | | | - | | | 0 | | | 443,893 | |
Other liabilities | | | 31,915 | | | | 28,265 | | |
Noncurrent operating lease liability | | | | 0 | | | | 35,693 | |
Total liabilities | | | 8,299,700 | | | | 2,318,219 | | | | 2,927,684 | | | | 2,915,369 | |
Commitments and Contingencies | | | | | | | | | |
Stockholders’ Equity (Deficit): | | | | | | | | | |
Common stock, $0.001 par value: | | | | | | | | | |
1,000,000,000 shares authorized; 10,345,637 and 8,118,939 shares issued and outstanding atDecember 31, 2016 and 2015, respectively | | | 10,346 | | | | 8,119 | | |
Commitments and Contingencies (Note 5) | | | | | | |
Stockholders’ Equity: | | |
Common stock, $0.001 par value: 1,000,000,000 shares authorized; 101,914,280 and 64,015,441 shares issued and outstanding at December 31, 2021 and 2020, respectively | | | 101,914 | | | 64,016 | |
Additional paid-in capital | | | 69,363,575 | | | | 42,102,876 | | | 164,814,664 | | | 130,659,550 | |
Accumulated deficit | | | (60,189,237 | ) | | | (42,152,618 | ) | | | (130,005,111 | ) | | | (105,909,384 | ) |
Total stockholders' equity (deficit) | | | 9,184,684 | | | | (41,623 | ) | |
Total liabilities and stockholders' equity (deficit) | | $ | 17,484,384 | | | $ | 2,276,596 | | |
Total stockholders' equity | | | | 34,911,467 | | | | 24,814,182 | |
Total liabilities and stockholders' equity | | | $ | 37,839,151 | | | $ | 27,729,551 | |
See accompanying notes to consolidated financial statements.
DIFFUSION PHARMACEUTICALS INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
| | | Year ended December 31, | | |
| | | 2016 | | | 2015 | | | Year Ended December 31, | |
| | | | | | | | | | | 2021 | | | 2020 | |
Operating expenses: | | | | | | | | | |
Research and development | | $ | 7,252,241 | | | $ | 3,875,467 | | | $ | 8,499,414 | | | $ | 9,427,667 | |
Intangible asset impairment charge | | | 8,639,000 | | | 0 | |
General and administrative | | | 11,094,146 | | | | 2,522,370 | | | 7,445,277 | | | 6,444,109 | |
Depreciation | | | 25,342 | | | | 8,268 | | | | 93,416 | | | | 103,168 | |
Loss from operations | | | 18,371,729 | | | | 6,406,105 | | | (24,677,107 | ) | | (15,974,944 | ) |
Interest expense, net | | | 29,686 | | | | 313,117 | | |
Loss from operations before income tax benefit | | | (18,401,415 | ) | | | (6,719,222 | ) | |
Other income: | | |
Interest income | | | | 137,487 | | | | 114,257 | |
Loss before income taxes | | | (24,539,620 | ) | | (15,860,687 | ) |
Income tax benefit | | | (364,796 | ) | | | - | | | | 443,893 | | | | 1,675,381 | |
Net loss | | $ | (18,036,619 | ) | | $ | (6,719,222 | ) | | $ | (24,095,727 | ) | | $ | (14,185,306 | ) |
Deemed dividend arising from warrant exchange | | | | 0 | | | | (1,950,378 | ) |
Net loss applicable to common stockholders | | | $ | (24,095,727 | ) | | $ | (16,135,684 | ) |
| | | | | | | | | | |
Per share information: | | | | | | | | | |
Share information: | | |
Net loss per share of common stock, basic and diluted | | $ | (1.76 | ) | | $ | (2.56 | ) | | $ | (0.25 | ) | | $ | (0.30 | ) |
Weighted average shares outstanding, basic and diluted | | | 10,232,791 | | | | 2,619,945 | | | | 97,626,748 | | | | 53,831,973 | |
See accompanying notes to consolidated financial statements.
DIFFUSION PHARMACEUTICALS INC.
CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERS’STOCKHOLDERS’ EQUITY (DEFICIT)
| | | | | | | | | | | Additional | | | | | | | Total | |
| | | Common Stock | | | Paid-in | | | Accumulated | | | Stockholders' | |
| | | Shares | | | Amount | | | Capital | | | Deficit | | | Equity (Deficit) | |
Balance at January 1, 2015 | | | 2,208,953 | | | $ | 2,209 | | | $ | 21,813,635 | | | $ | (35,433,396 | ) | | $ | (13,617,552 | ) |
Issuance of restricted stock | | | 18,412 | | | | 18 | | | | (18 | ) | | | - | | | | - | |
Conversion of convertible notes | | | 5,891,574 | | | | 5,892 | | | | 19,695,084 | | | | - | | | | 19,700,976 | |
Stock-based compensation expense | | | - | | | | - | | | | 594,175 | | | | - | | | | 594,175 | |
Net loss | | | - | | | | - | | | | - | | | | (6,719,222 | ) | | | (6,719,222 | ) |
Balance at December 31, 2015 | | | 8,118,939 | | | | 8,119 | | | | 42,102,876 | | | | (42,152,618 | ) | | | (41,623 | ) |
Fair value of RestorGenex shares | | | 1,861,503 | | | | 1,862 | | | | 19,544,138 | | | | - | | | | 19,546,000 | |
Estimated fair value of RestorGenex stock options outstanding | | | - | | | | - | | | | 1,321,000 | | | | - | | | | 1,321,000 | |
Estimated fair value of RestorGenex warrants outstanding | | | - | | | | - | | | | 384,000 | | | | - | | | | 384,000 | |
Common stock issued for advisory services | | | 148,073 | | | | 148 | | | | 1,409,215 | | | | - | | | | 1,409,363 | |
Conversion of convertible debt | | | 217,122 | | | | 217 | | | | 711,278 | | | | - | | | | 711,495 | |
Settlement of litigation matter upon issuance of convertible debt | | | - | | | | - | | | | 2,500,000 | | | | - | | | | 2,500,000 | |
Stock-based compensation expense | | | - | | | | - | | | | 1,391,068 | | | | - | | | | 1,391,068 | |
Net loss | | | - | | | | - | | | | - | | | | (18,036,619 | ) | | | (18,036,619 | ) |
Balance at December 31, 2016 | | | 10,345,637 | | | $ | 10,346 | | | $ | 69,363,575 | | | $ | (60,189,237 | ) | | $ | 9,184,684 | |
| | | Stockholders' Equity | |
| | | Common Stock | | | | | | | | | | | | | |
| | | Shares | | | Amount | | | Additional Paid-in Capital | | | Accumulated Deficit | | | Total Stockholders' Equity | |
Balance at December 31, 2019 | | | 33,480,365 | | | $ | 33,481 | | | $ | 111,824,859 | | | $ | (91,724,078 | ) | | $ | 20,134,262 | |
Issuance of common stock, pre-funded warrants and warrants, net of issuance costs | | | 11,428,572 | | | | 11,429 | | | | 10,330,202 | | | | 0 | | | | 10,341,631 | |
Issuance of common stock upon exercise of warrants | | | 19,106,504 | | | | 19,106 | | | | 7,768,370 | | | | 0 | | | | 7,787,476 | |
Stock-based compensation expense | | | — | | | | 0 | | | | 736,119 | | | | 0 | | | | 736,119 | |
Net loss | | | — | | | | 0 | | | | 0 | | | | (14,185,306 | ) | | | (14,185,306 | ) |
Balance at December 31, 2020 | | | 64,015,441 | | | | 64,016 | | | | 130,659,550 | | | | (105,909,384 | ) | | | 24,814,182 | |
Issuance of common stock and warrants, net of issuance costs | | | 33,658,538 | | | | 33,658 | | | | 31,060,644 | | | | 0 | | | | 31,094,302 | |
Issuance of common stock upon exercise of warrants | | | 4,230,000 | | | | 4,230 | | | | 2,197,220 | | | | 0 | | | | 2,201,450 | |
Vesting of restricted stock units | | | 10,301 | | | | 10 | | | | (10 | ) | | | 0 | | | | 0 | |
Stock-based compensation expense | | | — | | | | 0 | | | | 897,260 | | | | 0 | | | | 897,260 | |
Net loss | | | — | | | | 0 | | | | 0 | | | | (24,095,727 | ) | | | (24,095,727 | ) |
Balance at December 31, 2021 | | | 101,914,280 | | | $ | 101,914 | | | $ | 164,814,664 | | | $ | (130,005,111 | ) | | $ | 34,911,467 | |
See accompanying notes to consolidated financial statements.
DIFFUSION PHARMACEUTICALS INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
| | | Year Ended December 31, | |
| | | 2021 | | | 2020 | |
Operating activities: | | | | | | | | |
Net loss | | $ | (24,095,727 | ) | | $ | (14,185,306 | ) |
Adjustments to reconcile net loss to net cash used in operating activities: | | | | | | | | |
Depreciation and amortization | | | 93,416 | | | | 103,168 | |
Loss on disposal of property and equipment | | | 51,782 | | | | 0 | |
Stock-based compensation expense | | | 897,260 | | | | 736,119 | |
Abandonment of in-process research and development intangible asset | | | 8,639,000 | | | | 0 | |
Change in deferred income taxes | | | (443,893 | ) | | | (1,675,381 | ) |
Changes in operating assets and liabilities: | | | | | | | | |
Prepaid expenses, deposits and other assets | | | (248,997 | ) | | | 518,169 | |
Accounts payable, accrued expenses and other liabilities | | | 605,370 | | | | 950,602 | |
Net cash used in operating activities | | | (14,501,789 | ) | | | (13,552,629 | ) |
| | | | | | | | | |
Cash flows provided by investing activities: | | | | | | | | |
Cash received from sale of property and equipment | | | 4,000 | | | | 0 | |
Net cash provided by investing activities | | | 4,000 | | | | 0 | |
| | | | | | | | | |
Cash flows provided by financing activities: | | | | | | | | |
Proceeds from the sale of common stock, net of issuance costs | | | 31,094,302 | | | | 10,827,100 | |
Proceeds from the sale of common stock warrants | | | 2,201,450 | | | | 8,046,103 | |
Payment of offering costs | | | 0 | | | | (982,328 | ) |
Net cash provided by financing activities | | | 33,295,752 | | | | 17,890,875 | |
| | | | | | | | | |
Net increase in cash and cash equivalents | | | 18,797,963 | | | | 4,338,246 | |
| | | | | | | | | |
Cash and cash equivalents at beginning of year | | | 18,515,595 | | | | 14,177,349 | |
Cash and cash equivalents at end of year | | $ | 37,313,558 | | | $ | 18,515,595 | |
| | | | | | | | | |
Supplemental disclosure of non-cash investing and financing activities: | | | | | | | | |
Vesting of restricted stock units | | $ | 10 | | | $ | — | |
| | | Year ended December 31, | |
| | | 2016 | | | 2015 | |
Operating activities: | | | | | | | | |
Net loss | | $ | (18,036,619 | ) | | $ | (6,719,222 | ) |
Adjustments to reconcile net loss to net cash used in operating activities: | | | | | | | | |
Depreciation | | | 25,342 | | | | 8,268 | |
Loss on sale or disposal of assets | | | 6,761 | | | | 1,127 | |
Stock-based compensation expense | | | 1,391,068 | | | | 594,175 | |
Common stock issued for advisory services | | | 1,409,363 | | | | - | |
Abandonment of in-process research and development intangible asset | | | 951,000 | | | | - | |
Change in deferred income taxes | | | (364,796 | ) | | | - | |
Settlement of litigation matter | | | 2,500,000 | | | | - | |
Non-cash interest expense | | | 38,446 | | | | 165,329 | |
Amortization of debt issuance costs and debt discount | | | - | | | | 147,150 | |
Changes in operating assets and liabilities: | | | | | | | | |
Prepaid expenses, deposits and other assets | | | 319,545 | | | | (29,739 | ) |
Accounts payable, accrued expenses and other liabilities | | | 991,625 | | | | 647,584 | |
Net cash used in operating activities | | | (10,768,265 | ) | | | (5,185,328 | ) |
| | | | | | | | | |
Cash flows provided by investing activities: | | | | | | | | |
Purchases of property and equipment | | | (2,331 | ) | | | (40,291 | ) |
Maturities of certificates of deposit | | | - | | | | 2,500,000 | |
Cash received in reverse merger transaction | | | 8,500,602 | | | | - | |
Net cash provided by investing activities | | | 8,498,271 | | | | 2,459,709 | |
| | | | | | | | | |
Cash flows provided by financing activities: | | | | | | | | |
Proceeds from the issuance of convertible debt | | | 1,880,000 | | | | 2,401,602 | |
Payment of offering costs | | | (54,346 | ) | | | (15,310 | ) |
Net cash provided by financing activities | | | 1,825,654 | | | | 2,386,292 | |
| | | | | | | | | |
Net decrease in cash and cash equivalents | | | (444,340 | ) | | | (339,327 | ) |
| | | | | | | | | |
Cash and cash equivalents at beginning of period | | | 1,997,192 | | | | 2,336,519 | |
Cash and cash equivalents at end of period | | $ | 1,552,852 | | | $ | 1,997,192 | |
| | | | | | | | | |
Supplemental disclosure of non-cash investing and financing activities: | | | | | | | | |
Offering costs in accounts payable | | $ | 126,110 | | | $ | - | |
Conversion of convertible notes and related accrued interest into common stock | | | 711,495 | | | | 19,700,976 | |
Consideration in connection with RestorGenex Corporation merger transaction | | | 21,261,000 | | | | - | |
See accompanying notes to consolidated financial statements.
DIFFUSION PHARMACEUTICALS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
1. Organization and Description of Business
Diffusion Pharmaceuticals Inc. “Diffusion” or “the Company”("the Company"), a Delaware corporation, is a clinical-stage specialty pharmaceuticalbiopharmaceutical company developing new, small-molecule drugsnovel therapies that help regulateenhance the movement ofbody’s ability to deliver oxygen into tissue by a novel mechanism of action.to areas where it is needed most. The Company’s lead product candidate, trans sodium crocetinate (“TSC”), uses this novel mechanismTSC, is being developed to re-oxygenateenhance the microenvironmentdiffusion of solid cancerous tumors, thereby enhancing tumor cells’ responseoxygen to conventional treatment without additional side effects. TSC has received orphan drug designation for the treatment of glioblastoma multiforme (“GBM”), and the Company expects to enter a Phase III study in newly diagnosed GBM patients in the next twelve (12) months, assuming the availability of financial resources.
On December 15, 2015, the Company’s predecessor for accounting purposes, Diffusion Pharmaceuticals LLC (“Diffusion LLC”) entered into a definitive merger agreementtissues with the Company, thenlow oxygen levels, also known as “RestorGenex Corporation”hypoxia, a serious complication of many of medicine’s most intractable and traded on the over-the-counter stock exchange under the ticker symbol “RESX.” On January 8, 2016 the Company completed the merger with Diffusion LLC under which a newly formed subsidiary of the Company merged with and into Diffusion LLC in an all-stock transaction, with Diffusion LLC surviving as a wholly-owned subsidiary of the Company. Subsequent to the merger, the Company was renamed “Diffusion Pharmaceuticals Inc.” and the Company’s ticker symbol was changed to “DFFN.” On difficult-to-treat conditions. In November 9, 2016, the Company’s common stock (“Common Stock”) began trading on the NASDAQ Capital Market.
The merger transaction was accounted for as a reverse acquisition under the acquisition method of accounting. Because Diffusion LLC’s pre-transaction owners held an 84.1% economic and voting interest in the combined company immediately following the closing of the merger, Diffusion LLC is considered to be the acquirer of RestorGenex for accounting purposes (See Note 12). For purposes of these notes and the financial statements they accompany, unless the context otherwise requires, the terms “Diffusion” and “the Company” refer to Old Diffusion, and the term “RestorGenex” refers to the Company, in each case, for the periods prior to the merger transaction.
The Company’s members’ capital at December 31, 2015 has been recast as common stock and additional paid in capital. On August 17, 2016, the Company effected a 1-for-10 reverse split of its common stock. The accompanying consolidated financial statements and these notes give retroactive effect to this reverse stock split.
Each outstanding unit of membership interest of Diffusion LLC (the “Diffusion Units”) was converted into the right to receive 0.3652658 shares of Common Stock (the “Exchange Ratio”). Additionally, the right of holders of $1.1 million outstanding convertible notes of Diffusion LLC to convert such notes into Diffusion Units was converted into the right to convert such notes into a number of shares of Common Stock equal to the number of Diffusion Units into which such note would have been convertible under the original terms of the note multiplied by the Exchange Ratio. In addition, all outstanding options to purchase Diffusion Units were assumed by the Company and the right to exercise converted into 1,495,615 options to purchase Common Stock on terms substantially identical to those in effect prior to the merger transaction, except for adjustments to the underlying number of shares and the exercise price 2021, based on the Exchange Ratio.preclinical and clinical data accumulated to date and the significant unmet medical need, the Company announced that its near-term focus will be the design and execution of a clinical program to support the use of intravenously administered TSC as an adjunctive treatment for hypoxic solid tumors.
2. Liquidity
The Company has not generated any revenues from product sales and has funded operations primarily from the proceeds of public and private placementsofferings of its membership units (prior to the Merger)equity, convertible debt and convertible notes, as well as through the reverse merger.preferred stock. Substantial additional financing will be required by the Company to continue to fund its research and development activities. No assurance can be given that any such financing will be available when needed, or at all, or that the Company’s research and development efforts will be successful.
The Company regularly explores alternative means of financing its operations and seeks funding through various sources, including public and private securities offerings, collaborative arrangements with third parties and other strategic alliances and business transactions. The Company received net cash proceeds of approximately $14.1 million from the initial closing of a private placement (the “Private Placement”) of the Company’s Series A convertible preferred stock (the “Series A Convertible Preferred Stock”), after deducting $1.7 million in placement agent fees and expenses. The Company may periodically conduct one or more additional closings in the Private Placement for up to aggregate gross proceeds of $25.0 million.
DIFFUSION PHARMACEUTICALS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
CONTINUED
Other than as described in Note 17, the Company currently does not have any commitments to obtain additional funds and may be unable to obtain sufficient funding in the future on acceptable terms, if at all. If the Company cannot obtain the necessary funding, it will need to delay, scale back or eliminate some or all of its research and development programs or enter into collaborations with third parties to:to commercialize potential products or technologies that it might otherwise seek to develop or commercialize independently; consider other various strategic alternatives, including a merger or sale of the Company; or cease operations. If the Company engages in collaborations, it may receive lower consideration upon commercialization of such products than if it had not entered into such arrangements or if it entered into such arrangements at later stages in the product development process.
TheOperations of the Company has prepared its financial statements assuming that it will continue as a going concern, which contemplates realization of assetsare subject to certain risks and the satisfaction of liabilities in the normal course of business. The Company has incurred net losses since inception and it expects to generate losses from operations for the foreseeable future primarily due to research and development costs for its potential product candidates, which raises substantial doubt about the Company’s ability to continue as a going concern. Variousuncertainties including various internal and external factors that will affect whether and when the Company’s product candidates become approved drugs and how significant their market share will be, some of which are outside of the Company’s control. The length of time and cost of developing and commercializing these product candidates and/or failure of them at any stage of the drug approval process will materially affect the Company’s financial condition and future operations. The Company expects that its existing cash and cash equivalents as of December 31, 2016, together with the proceeds from the initial closing of the Private Placement of its Series A Convertible Preferred Stock in March 2017, 2021 will enable the Companyit to fund its operating expenses and capital expenditure requirements into December of 2017.through 2023.
3.
3. Basis of Presentation and Summary of Significant Accounting Policies
Basis of Presentation
The accompanying consolidated financial statements of the Company have been prepared in accordance with US GAAP. Any reference in these notes to applicable guidance is meant to refer to US GAAP as found in the Accounting Standards Codification (“ASC”) and Accounting Standards Updates (“ASU”) of the Financial Accounting Standards Board (“FASB”).Board.
Use of Estimates
The preparation of financial statements in conformity with US GAAP requires the Company to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of expenses during the reporting period.
On an ongoing basis, the Company evaluates its estimates using historical experience and other factors, including the current economic environment. Significant items subject to such estimates are assumptions used for purposes of determining stock-based compensation the fair value of the convertible notes, allocation of the purchase price and accounting for research and development activities. Management believes its estimates to be reasonable under the circumstances. Actual results could differ significantly from those estimates.
DIFFUSION PHARMACEUTICALS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Fair Value of Financial Instruments
The carrying amounts of the Company’s financial instruments, including cash equivalents and accounts payable and accrued expenses approximate fair value due to the short-term nature of those instruments. The carrying value of the contingent consideration liability is the estimated fair value of the liability (See Note 15).As of December 31, 2016, and December 31, 2015, the fair value of the Company’s outstanding convertible notes was approximately $2.6 million and $4.8 million, respectively.The fair value of the convertible notes is determined using a binomial lattice model that utilizes certain unobservable inputs that fall within Level 3 of the fair value hierarchy.
Concentration of Credit Risk
Financial instruments that potentially expose the Company to concentrations of credit risk consist principally of cash on deposit with multiple financial institutions, the balances of which frequently exceed federally insured limits.
Cash and Cash Equivalents
The Company considers any highly liquidhighly-liquid investments, such as money market funds, with an original maturity of three months or less to be cash and cash equivalents.
DIFFUSION PHARMACEUTICALS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
CONTINUED
Property and Equipment
The Company records property and equipment at cost less accumulated depreciation and amortization. Costs of renewals and improvements that extend the useful lives of the assets are capitalized. Maintenance and repairs are expensed as incurred. Depreciation is recognized on a straight-line basis over the estimated useful lives of the assets, which generally range from 2 to 15 years. The Company amortizes leasehold improvements over the shorter of the estimated useful life of the asset or the term of the related lease. Upon retirement or disposition of assets, the costs and related accumulated depreciation and amortization are removed from the accounts with the resulting gains or losses, if any, reflected in results of operations. In November 2021, the Company terminated the lease of its prior corporate headquarters and in connection with the termination the Company disposed of all of its related property and equipment.
Long-Lived AssetsIntangible Asset
Long-lived assets are reviewed for potential impairment whenever events indicate thatIn the carrying amountthird quarter of such assets may not be recoverable. The Company does2021, the Board of Directors made a determination to no longer dedicate financial resources to the Company's DFN-529 intangible asset and any future internal development efforts were abandoned. In connection with this by comparing the carrying value of the long-lived assets with the estimated future undiscounted cash flows expected to result from the use of the assets, including cash flows from disposition. If it is determined an impairment exists, the asset is written down to its estimated fair value. The Company has not recognized any impairment of long-lived assets during the years ended December 31, 2016 and 2015, respectively.
Intangible Assets
Intangible assets are comprised of identifiable in-process research and development (IPR&D) assets and are considered indefinite-lived intangible assets and are assessed for impairment annually on October 1 or more frequently if impairment indicators exist. If the associated research and development effort is abandoned, the related assets will be written-off anddecision, the Company will recordconcluded that DFN-529 was impaired in its entirety and as such, the Company recognized a non-cash impairment loss. For those compounds that reach commercialization,charge of $8.6 million during the IPR&D assets will be amortized over their estimated useful lives. In August 2016,third quarter of 2021. The abandonment also resulted in an income tax benefit of $0.4 million due to the Company abandoned the future development efforts with respect to its RES-440 product candidate, and the valuetax effect of the reduction in the deferred tax liability associated IPR&D asset was written down to zero (See Note 4).with the asset.
Goodwill
Goodwill is the excess of the cost of an acquired entity over the net amounts assigned to tangible and intangible assets acquired and liabilities assumed. Goodwill is not amortized, but is subject to an annual impairment test. The Company has a single reporting unit and all goodwill relates to that reporting unit.
The Company performs its annual goodwill impairment test on October 1 of each fiscal year or more frequently if changes in circumstances or the occurrence of events suggest that an impairment exists. If the fair value of the reporting unit is less than its carrying value, an impairment loss is recorded to the extent that the implied fair value of the reporting unit’s goodwill is less than the carrying value of the reporting unit’s goodwill. The Company did not recognize any impairment of goodwill during the year ended December 31, 2016.
Offering Costs
Offering costs consist principally of legal costs incurred through the balance sheet date related to our private placement financing (See Note 17) and are recognized in other assets on the consolidated balance sheet.
Research and Development
Major components of research and development costs include internal research and development (such as salaries and related employee benefits, equity-based compensation, supplies and allocated facility costs) and contracted services (research and development activities performed on the Company’s behalf). Costs incurred for research and development are expensed as incurred.
At the end of the reporting period, the Company compares payments made to third-partythird-party service providers to the estimated progress toward completion of the research or development objectives. Such estimates are subject to change as additional information becomes available. Depending on the timing of payments to the service providers and the progress that the Company estimates has been made as a result of the services provided, the Company may record net prepaid or accrued expenses relating to these costs.
DIFFUSION PHARMACEUTICALS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
CONTINUED
Upfront payments made to third parties who perform research and development services on the Company’s behalf are expensed as services are rendered.
Patent Costs
Patent costs, including related legal costs, are expensed as incurred and are recorded within general and administrative expenses in the consolidated statements of operations.
Income Taxes
Prior to the Merger, Diffusion LLC was treated as a partnership for federal and state income tax purposes. Diffusion LLC’s taxable income or loss, as well as certain other tax attributes, were passed through directly to its members and were reported in each member’s individual income tax return.
Upon completion of the Merger as discussed in Note 1, the Company converted from a partnership to a corporation for accounting purposes. As a corporation, the Company uses the asset and liability method of accounting for income taxes. Deferred tax assets and liabilities are recognized for the estimated future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases and operating loss and credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. A valuation allowance is provided when it is more likely than not that some portion or all of a deferred tax asset will not be realized. The Company recognizes the benefit of an uncertain tax position that it has taken or expects to take on its income tax return it files, if such a position is more likely than not to be sustained.
FASB ASC Subtopic 740-10, Debt Issuance CostsAccounting for Uncertainty of Income Taxes
Debt issuance costs incurred, (“ASC 740-10”) defines the criterion an individual tax position must meet for any part of the benefit of the tax position to be recognized in connectionfinancial statements prepared in conformity with financing arrangements are amortized to interest expense overGAAP. The Company may recognize the lifetax benefit from an uncertain tax position only if it is more likely than not such tax position will be sustained on examination by the taxing authorities, based solely on the technical merits of the respective financing arrangement usingtax position. The tax benefits recognized in the effectivefinancial statements from such a tax position should be measured based on the largest benefit having a greater than 50% likelihood of being realized upon ultimate settlement with the tax authority. In accordance with the disclosure requirements of ASC 740-10, the Company’s policy on income statement classification of interest method. Debt issuance costs, netand penalties related to income tax obligations is to include such items as part of related amortization, are deducted from the carrying amount of the related debt.total interest expense and other expense, respectively.
Stock-based Compensation
The Company measures employee and nonemployee stock-based awards at grant-date fair value and records compensation expense on a straight-line basis over the vesting period of the award. Stock-based awards issued to non-employees are revalued until the award vests. The Company uses the Black-Scholes option pricing modelModel to value its stock option awards. Estimating the fair value of stock option awards requires management to apply judgment and make estimates, including the volatility of the Company’s common stock, the expected term of the Company’s stock options, the expected dividend yield and the fair value of the Company’s common stock on the measurement date. As a result, if factors change and management uses different assumptions, stock-based compensation expense could be materially different for future awards.
TheFor certain stock option grants, the expected term of stock options was estimated using the “simplified method” for employee options as the Company has nolimited historical information to develop reasonable expectations about future exercise patterns and post vesting employment termination behavior for its stock option grants. The simplified method is based on the average of the vesting tranches and the contractual life of each grant. ForDuring the year ended December 31, 2020, it became apparent that the expected term of the Company's stock options granted to non-employees,was commensurate with the contractual life (i.e. 10 years) of the stock option and therefore the Company usesbegan to use the remaining contractual life. life as the expected term.
For stock price volatility, the Company uses a combination of its own historical stock price and comparable public companies as a basis for its expected volatility to calculate the fair value of option grants. The Company assumes no dividend yield because dividends are not expected to be paid in the near future, which is consistent with the Company’s history of not paying dividends.Thedividends. The risk-free interest rate is based on U.S. Treasury notes with a term approximating the expected lifeterm of the option. The Company accounts for forfeitures in the periods they occur.
DIFFUSION PHARMACEUTICALS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Net Loss Per Common Share
Basic net loss per share is computed by dividing net loss applicable to common stockholders by the weighted average number of shares of common stock outstanding during each period. Diluted net loss per share includes the effect, if any, from the potential exercise or conversion of securities, such as convertible debt, convertible preferred stock, common stock warrants, stock options and unvested restricted stock that would result in the issuance of incremental shares of common stock. In computing the basic and diluted net loss per share applicable to common stockholders, the weighted average number of shares remains the same for both calculations due to the fact that when a net loss exists, dilutive shares are not included in the calculation as the impact is anti-dilutive.
DIFFUSION PHARMACEUTICALS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
CONTINUED
The following potentially dilutive securities outstanding have been excluded from the computation of diluted weighted average shares outstanding, as they would be anti-dilutive:
| | | December 31, | | | December 31, | |
| | | 2016 | | | 2015 | | | 2021 | | | 2020 | |
Convertible debt | | | 757,909 | | | | 427,108 | | |
Common stock warrants | | | 460,721 | | | | — | | | 6,499,469 | | | 9,100,112 | |
Stock options | | | 2,207,409 | | | | 1,495,582 | | | 3,626,223 | | | 2,240,204 | |
Unvested restricted stock awards | | | 9,204 | | | | 15,341 | | |
Unvested restricted stock units | | | | 275,450 | | | | 153,000 | |
| | | | 3,435,243 | | | | 1,938,031 | | | | 10,401,142 | | | | 11,493,316 | |
Amounts in the table reflect the common stock equivalents of the noted instruments.
Recently Issued But Not Yet Adopted Accounting Pronouncements
Comprehensive Loss
The Company has no items of comprehensive income or loss other than net loss.
Recent Accounting Pronouncements
In AugustJune 2016, the FASB issued ASU 2016-15,2016-13, Financial Instruments—ClassificationCredit Losses, Measurement of Certain Cash ReceiptsCredit Losses on Financial Instruments (Topic 326). The standard amends the impairment model by requiring entities to use a forward-looking approach based on expected losses to estimate credit losses for most financial assets and Cash Payments,whichcertain other instruments that aren’t measured at fair value through net income. For available-for-sale debt securities, entities will reduce diversitybe required to recognize an allowance for credit losses rather than a reduction in practice on how certain cash receipts and cash payments are classified incarrying value of the statementasset. Entities will no longer be permitted to consider the length of cash flows. Thetime that fair value has been less than amortized cost when evaluating when credit losses should be recognized. This new guidance is applicable to public business entitieseffective for fiscal years beginning after December 15, 2017 and interim periods within those years. The ASU should be applied retrospectively and early adoption is permitted. the Company as of January 1, 2022. The Company is currently evaluating the impact of adopting this new guidance on its financial statements and related disclosures.
In March 2016, the FASB issued ASU 2016-09,Compensation – Improvements to Employee Share-Based Payment Accounting, which simplifies several aspectsbut does not expect that adoption of the accounting for employee share-based payment transactions including the accounting for income taxes, forfeitures, and statutory tax withholding requirements, as well as classification in the statement of cash flows. The guidance is applicable to public business entities for fiscal years beginning after December 15, 2016 and interim periods within those years. The Company is currently evaluating the effect that ASU 2016-09this standard will have a material impact on its consolidated financial statements and related disclosures.
DIFFUSION PHARMACEUTICALS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
CONTINUED
Recently Adopted Accounting Pronouncements
In February 2016, December 2019, the FASB issued ASU 2016-02,LeasesNo.2019-12, “Income Taxes (Topic 842)740): Simplifying the Accounting for Income Taxes. The FASB issued” This guidance applies to all entities and aims to reduce the update to require the recognitioncomplexity of lease assets and liabilities on the balance sheet of lessees. The standard will betax accounting standards while enhancing reporting disclosures. This guidance is effective for fiscal years beginning after December 15, 2018, including2020 and interim periods within such fiscal years. The ASU requires a modified retrospective transition method with the option to elect a package of practical expedients.therein. Early adoption is permitted. The Company is currently evaluating the potential impact of the adoption of this standard on its consolidated results of operations,permitted for any annual periods for which financial positionstatements have not been issued and cash flows and related disclosures.
In August 2014, the FASB issued ASU 2014-15,Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern. The amendments in this update will explicitly require a company’s management to assess an entity’s ability to continue as a going concern, and to provide related footnote disclosures in certain circumstances. The new standard is effective in the first annual period ending after December 15, 2016.interim periods therein. The Company adopted this standard as of December 31, 2016 in January 2021 and the adoption of this standard did not have a material impact on our financial statements.
Reclassification
Certain prior period balances have been reclassified to conform with the current period presentation.
4. Acquisitions
Merger with Diffusion Pharmaceuticals LLC
On December 15, 2015, the Company entered into a merger agreement (the “Merger Agreement”) with Diffusion LLC. On January 8, 2016, the Company completed the merger (the “Merger”), with Diffusion LLC surviving as a wholly-owned subsidiary of the Company. Subsequent to the Merger, the Company was renamed “Diffusion Pharmaceuticals Inc.” and the Company’s ticker symbol was changed from “RESX” to “DFFN.” In November 2016, the Company’s Common Stock was approved for listing on the NASDAQ Capital Market and continues to trade under the ticker symbol “DFFN”. The Company and Diffusion LLC entered into the Merger Agreement in an effort to provide improved access to the capital markets in order to obtain the resources necessary to accelerate development of TSC in multiple clinical programs and continue to build an oncology-focused company.
The Merger was accounted for as a reverse acquisition under the acquisition method of accounting. Because Diffusion LLC’s pre-transaction owners held an 84.1% economic and voting interest in the combined company immediately following the completion of the Merger, Diffusion LLC is considered to be the acquirer of the Company for accounting purposes.
Each Diffusion Unit was converted into the right to receive 0.3652658 shares of Common Stock based on the Exchange Ratio. Additionally, the rights of holders of Diffusion LLC’s outstanding convertible notes to convert such notes into Diffusion Units was converted into the right to convert such notes into a number of shares of Common Stock equal to the number of Diffusion Units into which such note would have been convertible under the terms of the note in effect immediately prior to the consummation of the Merger multiplied by the Exchange Ratio. In addition, all outstanding options to purchase Diffusion Units were assumed by the Company and the right to exercise such options for Diffusion Units converted into a right to exercise such options for Common Stock on terms substantially identical to those in effect prior to the Merger transaction, except for adjustments to the underlying number of shares and the exercise price based on the Exchange Ratio.
In connection with the Merger, the Company’s former Board of Directors authorized, declared and effected a distribution of contingent value rights (“CVRs”) to stockholders of the Company as of the close of business on January 7, 2016. Each CVR is a non-transferable right to potentially receive certain cash payments in the event the Company receives net cash payments during the five (5) year period after the Merger as a result of the sale, transfer, license or similar transaction or any other agreement to the extent relating to the development of the Company’s product currently known as RES-440, a “soft” anti-androgen. See below and Note 15 for additional fair value information.its related disclosures.
DIFFUSION PHARMACEUTICALS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
CONTINUED
The purchase consideration in a reverse acquisition is determined with reference to the value of equity that the accounting acquirer, in this case, Diffusion LLC, would have had to issue to the owners of the accounting acquiree, the Company, to give the pre-acquisition equity holders of the Company the same percentage interest in Diffusion LLC that such pre-acquisition equity holders held in the Company immediately following the Merger. The purchase price was calculated as follows:
Fair value of the Company’s pre-Merger shares outstanding | | $ | 19,546,000 | |
| | | | | |
Estimated fair value of the Company’s pre-Merger stock options outstanding | | | 1,321,000 | |
| | | | | |
Estimated fair value of the Company’s pre-Merger warrants outstanding | | | 384,000 | |
| | | | | |
CVRs – RES-440 product candidate | | | 10,000 | |
| | | | | |
Total purchase price | | $ | 21,261,000 | |
The Merger transaction has been accounted for using the acquisition method of accounting, which requires that assets acquired and liabilities assumed be recognized at their fair values as of the acquisition date. The valuation technique utilized to value the IPR&D was the cost approach.
The following table summarizes the allocation of the purchase price to the assets acquired and liabilities assumed as of the acquisition date:
Cash and cash equivalents | | $ | 8,500,602 | |
| | | | | |
Prepaid expenses and other assets | | | 195,200 | |
| | | | | |
Property and equipment | | | 57,531 | |
| | | | | |
Intangible assets | | | 9,600,000 | |
| | | | | |
Goodwill | | | 6,929,258 | |
| | | | | |
Accrued liabilities | | | (377,432 | ) |
| | | | | |
Deferred tax liability | | | (3,644,159 | ) |
| | | | | |
Net assets acquired | | $ | 21,261,000 | |
Qualitative factors supporting the recognition of goodwill due to the Merger include the Company’s anticipated enhanced ability to secure additional capital and gain access to capital market opportunities as a public company and the potential value created by having a more well-rounded clinical development portfolio by adding the earlier stage products acquired in the Merger to the Company’s later state product portfolio. The goodwill is not deductible for income tax purposes. The values allocated to IPR&D intangible assets are $8.6 million for the RES-529 compound and $1.0 million for the RES-440 compound, each with indefinite useful lives. See Note 6 for additional information.
Pro Forma Financial Information (Unaudited)
The following pro forma financial information reflects the consolidated results of operations of the Company as if the Merger had taken place on January 1, 2015. The pro forma financial information is not necessarily indicative of the results of operations as they would have been had the transactions been effected on the assumed date.
| | | Year ended December 31, | |
| | | | | | | |
| | | 2016 | | | 2015 | |
| | | | | | | |
Net revenues | | $ | — | | | $ | — | |
| | | | | | | | | |
Net loss | | $ | (16,471,398 | ) | | $ | (17,618,292 | ) |
| | | | | | | | | |
Basic and diluted loss per share | | $ | (1.60 | ) | | $ | (3.89 | ) |
DIFFUSION PHARMACEUTICALS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
CONTINUED
Non-recurring pro forma transaction costs directly attributable to the Merger were $1.6 million for the year ended December 31, 2016 and have been deducted from the net loss presented above. The costs deducted from the year ended December 31, 2016 period included a success fee and other transaction costs of $1.1 million and approximately 46,000 shares of common stock with a fair market value of $0.5 million paid to a financial advisor upon the closing of the Merger on January 8, 2016. Additionally, RestorGenex incurred approximately $3.0 million in severance costs as a result of resignations of executive officers immediately prior to the Merger and approximately $2.7 million in share based compensation expense as a result of the acceleration of vesting of stock options at the time of Merger. These costs are excluded from the pro forma financial information for the year ended December 31, 2016. The Company excluded an $11.1 million impairment charge incurred by RestorGenex as well as combined transaction costs from Diffusion and RestoGenex in the amount of $1.9 million from the pro forma financial information for the year ended December 31, 2015.
5. Property and Equipment
Property and equipment consists of the following:
| | | December 31, | |
| | | 2016 | | | 2015 | |
Laboratory equipment | | $ | 182,357 | | | $ | 182,357 | |
Furniture and office equipment | | | 137,290 | | | | 84,668 | |
Total property and equipment | | | 319,647 | | | | 267,025 | |
Less: accumulated depreciation | | | (239,892 | ) | | | (215,029 | ) |
Property and equipment, net | | $ | 79,755 | | | $ | 51,996 | |
Depreciation expense was approximately $25.0 thousand and $8.0 thousand for the year ended December 31, 2016 and 2015, respectively.
DIFFUSION PHARMACEUTICALS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
CONTINUED
6. Goodwill and Intangible Assets
The Company completes its annual impairment test on October 1 each year, or more frequently if triggering events indicate a possible impairment. The Company continually evaluates financial performance, economic conditions and other relevant developments in assessing if an interim period impairment test is necessary.
Changes in the carrying amount of goodwill for the period ended December 31, 2016 consisted of the following:
| | | Goodwill | |
Balance as of December 31, 2015 | | $ | — | |
Goodwill as a result of the Merger | | | 6,929,258 | |
Balance as of December 31, 2016 | | $ | 6,929,258 | |
Intangible assets consisted of the following:
| | | Acquisition Date Fair Value | | | Accumulated Amortization | | | Impairment Upon Abandonment | | | Carrying Value at December 31, 2016 | |
RES-529 | | $ | 8,639,000 | | | $ | - | | | $ | - | | | $ | 8,639,000 | |
RES-440 | | | 961,000 | | | | - | | | | (961,000 | ) | | $ | - | |
Total in-process research and development costs (IPR&D) | | $ | 9,600,000 | | | $ | - | | | $ | (961,000 | ) | | $ | 8,639,000 | |
The Company’s novel PI3K/Akt/mTOR pathway inhibitor, RES-529, is in preclinical development for oncology. Through a series of in vitro and in vivo animal models, RES-529 has been shown to have activity in several cancer types due to its ability to target and inhibit the PI3K/Akt/mTOR signal transduction pathway. RES-529 is a first-in-class inhibitor of both TORC1 and TORC2 that is mechanistically differentiated from other PI3K/Akt/mTOR pathway inhibitors currently in development. RES-529 has shown activity in both in vitro and in vivo glioblastoma animal models and has been demonstrated to be orally bioavailable and can cross the blood brain barrier.
In August 2016, the Board of Directors determined that RES-440, a “soft” anti-androgen compound for the treatment of acne vulgaris, was outside the Company’s core product focus, and was not a priority. Therefore, future development efforts were abandoned in the third quarter of 2016. In connection with its review of such abandonment, the Company concluded RES-440 was impaired in its entirety and the CVRs were remeasured and determined to have no value as of December 31, 2016. The abandonment resulted in a net impairment charge of $1.0 million and was recorded as a component of research and development expenses within the Company’s consolidated statement of operations for the year ended December 31, 2016. The abandonment also resulted in an income tax benefit of $0.4 million.
7. Other Assets
Other assets consist of the following:
| | | December 31, | |
| | | 2016 | | | 2015 | |
Offering costs | | $ | 180,456 | | | $ | — | |
Clinical studies deposits | | | — | | | | 160,400 | |
Other | | | 52,219 | | | | 21,087 | |
Total | | $ | 232,675 | | | $ | 181,487 | |
DIFFUSION PHARMACEUTICALS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
CONTINUED
8. 4.Accrued Expenses and Other Current Liabilities
Accrued expenses and other current liabilities consist of the following:
| | | December 31, | | | December 31, | |
| | | 2016 | | | 2015 | | | 2021 | | | 2020 | |
Accrued interest payable | | $ | 29,359 | | | $ | 14,009 | | |
Accrued payroll and payroll related expenses | | | 399,740 | | | | 56,947 | | | $ | 879,971 | | | $ | 653,899 | |
Accrued professional fees | | | 72,855 | | | | 327,950 | | | 247,704 | | | 31,809 | |
Accrued clinical studies expenses | | | 220,978 | | | | 184,737 | | | 786,579 | | | 1,055,398 | |
Other | | | 151,332 | | | | 38,026 | | | | 65,935 | | | | 35,364 | |
Total | | $ | 874,264 | | | $ | 621,669 | | | $ | 1,980,189 | | | $ | 1,776,470 | |
5.Commitments and Contingencies
9. Convertible DebtOffice Space Lease Commitment
On September 27, 2016, November 8, 2021, the Company issued and sold convertible promissory notes (“2016 Convertible Notes”) in an aggregate principal amountentered into a Deed of $1.9 million.Lease Termination Agreement with the Carlton Landlord providing for the early termination of the Carlton Lease related to the Company's prior corporate headquarters. The 2016 Convertible Notes were issuedCarlton Lease was previously scheduled to an investor and certain other parties inexpire on April 30, 2022. In connection with the settlementtermination, the Company made a one-time payment to the Carlton Landlord of approximately $14,000, net of a litigation matter (See Note 10). The 2016 Convertible Notes have a term of one (1) year and bear interest at a rate of 6.0% per annum with the principal and accrued interest due upon the earlier of the maturity date or conversion date. At any time prior to the maturity date, the holders may elect to convert, in whole or in part, the 2016 Convertible Notes (including any accrued but unpaid interest) into shares of the Company’s common stock at a conversion price of $3.50 per share. In the event of a Change of Control (as defined in the 2016 Convertible Notes), the holders of the 2016 Convertible Notes may declare the aggregate outstanding amount of the 2016 Convertible Notes to be immediately due and payable or may elect to convert the 2016 Convertible Notes and any accrued but unpaid interest as if such conversion took place on the maturity date.
The Company accounted for the issuance of the convertible promissory notessecurity deposit subsequently returned in accordance with ASC 470-10-25 and, at the time of issuance, recorded a litigation settlement expense of $2.5 million which represents the difference between the estimated fair valueCarlton Lease. In lieu of the 2016 Convertible Notes issued and the cash received from the noteholders.
From December 2009 through December 2015, the Company issued unsecured convertible promissory notes (the “Old Convertible Notes”) for aggregate gross proceeds of $22.4 million. The Old Convertible Notes bore interest at either 1% or 1.5% per annum, however, all Old Convertible Notes outstanding at December 31, 2016 bear interest at a rate of 1% per annum. The Old Convertible Notes accrue interest beginning on the date of issuance, with the principal and accrued interest due upon the earlier of the maturity date or conversion date. At any time prior to the maturity date, the holders may elect to convert, in whole or in part, the Old Convertible Notes and any related accrued but unpaid interest into common stock of the Company at a price per share equal to the conversion price.
In the event of a Change of Control or a Qualified Financing (each as defined below), the holders of the Old Convertible Notes may declare the aggregate outstanding amount of the Old Convertible Notes to be immediately due and payable or may elect to convert the Convertible Notes and any accrued but unpaid interest as if such conversion took place on the maturity date. A Change of Control is defined as: (i) a merger or consolidation in which the owners immediately prior to the transaction do not own, directly or indirectly, more than 50% of the surviving company; (ii) the acquisition of more than 50% of the Company’s outstanding shares by a single person, entity or group or persons or entities acting in concert or (iii) the sale or transfer of all or substantially all of the assets of the Company. A Qualified Financing is defined as a sale of shares or other transaction that results in gross proceeds to the Company of at least $50.0 million, including proceeds received in connection with the conversion of any Old Convertible Notes. Through the date the financial statements were available to be issued, there have been no Change of Control or Qualified Financing events.
The Company may prepay the Old Convertible Notes, in full or in part, at any time on a pari passu basis. Upon receipt of notice that the Company intends to prepay the Old Convertible Notes, holders will have the option to convert their notes in lieu of payment.
At the effective time of the Merger, $1.1 million in aggregate principal amount of Old Convertible Notes were outstanding and the rights of the holders of each such outstanding Old Convertible Note convertible into Diffusion Units were converted into the right to convert such securities into a number of shares of the Company’s common stock equal to the number of Diffusion Units such Old Convertible Note would be convertible into pursuant to its terms multiplied by the Exchange Ratio.
DIFFUSION PHARMACEUTICALS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
CONTINUED
The following table provides the details of the Convertible Notes outstanding at December 31, 2016:
ConvertibleNote Series | | Issue Date | | Maturity Date | | Conversion Price | | | Interest Rate | | | Total Principal | |
2016 Convertible Notes | | 9/27/2016 | | 9/27/2017 | | $ | 3.50 | | | | 6.00 | % | | $ | 1,880,000 | |
| | | | | | | | | | | | | | | | | |
Series B Note | | 3/15/2011 | | 6/30/2018 | | $ | 2.74 | | | | 1.00 | % | | | 550,000 | |
| | | | | | | | | | | | | | | | | |
| Total principal amount | | | | | | | | | | | | | | | 2,430,000 | |
| | | | | | | | | | | | | | | | | |
| Less current portion of convertible notes | | | | | | | | | | | | | | | (1,880,000 | ) |
| | | | | | | | | | | | | | | | | |
| Convertible notes, net of current portion | | | | | | | | | | | | | | $ | 550,000 | |
During the year ended December 31, 2016, Old Convertible Notes of $0.7 million and related accrued interest of $16.4 thousand were converted into 217,122shares of common stock:
The following table provides the details of the Convertible Notes outstanding at December 31, 2015:
ConvertibleNote Series | | Issue Date | | Maturity Date | | Conversion Price | | | Interest Rate | | | Total Principal | |
B | | 3/15/2011 | | 6/30/2018 | | $ | 2.74 | | | | 1.00 | % | | | 570,000 | |
| | | | | | | | | | | | | | | | | |
C | | 09/14/12 | | 6/30/2018 | | $ | 2.74 | | | | 1.00 | % | | | 425,000 | |
| | | | | | | | | | | | | | | | | |
E | | 06/30/2014 | | 06/30/2018 | | $ | 4.11 | | | | 1.00 | % | | | 50,000 | |
| | | | | | | | | | | | | | | | | |
F | | 12/04/2015 | | 12/04/2019 | | $ | 5.48 | | | | 1.00 | % | | | 200,000 | |
| | | | | | | | | | | | | | | | | |
Total principal amount | | | | | | | | | | | 1,245,000 | |
| | | | | | | | | | | | | |
Less: unamortized debt issuance costs | | | | | | | | | | | (1,390 | ) |
| | | | | | | | | | | | | |
Less current portion of convertible notes | | | | | | | | | | | (424,964 | ) |
| | | | | | | | | | | | | |
Convertible notes, net of current portion | | | | | | | | | | $ | 818,646 | |
DIFFUSION PHARMACEUTICALS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
CONTINUED
During the year ended December 31, 2015, Old Convertible Notes of $19.1 million and related accrued interest of $0.6 million were converted into 5,891,574 shares of common stock:
10. Commitments and Contingencies
Operating Lease
The Company leasesfixed office and laboratory facilitiesspace previously available to us under the Carlton Lease, the Company has entered into short term agreements to utilize membership-based co-working space in both Charlottesville, Virginia under a month-to-month cancelable operating lease. and Philadelphia, Pennsylvania.
Rent expense related to the Company's operating lease was $66.0 thousand for each ofboth the years ended December 31, 20162021 and 2015.2020 was approximately $0.1 million.
Research and Development Arrangements
In the course of normal business operations, the Company enters into agreements with universities and contract research organizations, or CROs, to assist in the performance of research and development activities and contract manufacturers to assist with chemistry, manufacturing, and controls related expenses. Expenditures to CROs represent a significant cost in clinical development for the Company. The Company could also enter into additional collaborative research, contract research, manufacturing, and supplier agreements in the future, which may require upfront payments and long-term commitments of cash.
Defined Contribution Retirement Plan
The Company has established a 401(k) defined contribution plan that covers all employees who qualify under the terms of the plan. Eligible employees may elect to contribute to the 401(k) Plan up to 90% of their compensation, limited by the IRS-imposed maximum. The Company provides a safe harbor match with a maximum amount of 4% of the participant’s compensation. The Company made matching contributions under the 401(k) Plan of approximately $75,000 and $68,000 for the years ended December 31,2021 and 2020, respectively.
DIFFUSION PHARMACEUTICALS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Legal Proceedings
On August 7, 2014, a complaint was filed in the Superior Court of Los Angeles County, California by Paul Feller, the Company’s former Chief Executive Officer of the Company’s legal predecessor under the captionPaul Feller v. RestorGenex Corporation, Pro Sports & Entertainment, Inc., ProElite, Inc. and Stratus Media Group, GmbH(Case (Case No. BC553996)BC553996). The complaint asserts various causes of action, including, among other things, promissory fraud, negligent misrepresentation, breach of contract, breach of employment agreement, breach of the covenant of good faith and fair dealing, violations of the California Labor Code and common counts. The plaintiff is seeking, among other things, compensatory damages in an undetermined amount, punitive damages, accrued interest and an award of attorneys’ fees and costs. On December 30, 2014, the Company filed a petition to compel arbitration and a motion to stay the action. On April 1, 2015, the plaintiff filed a petition in opposition to the Company’s petition to compel arbitration and a motion to stay the action. After a related hearing for the petition and motion on April 14, 2015, the Courtcourt granted the Company’s petition to compel arbitration and a motion to stay the action. On January 8, 2016, the plaintiff filed an arbitration demand with the American Arbitration Association. NoOn November 19, 2018 at an Order to Show Cause Re Dismissal Hearing, the court found sufficient grounds not to dismiss the case and an arbitration hearing has yet been scheduled. was scheduled, originally for November 2020 but later postponed due to the COVID-19 pandemic and related restrictions on gatherings in the State of California. In addition, following the November 2018 hearing, an automatic stay was placed on the arbitration in connection with the plaintiff filing for personal bankruptcy protection. On October 22, 2021, following a determination by the bankruptcy trustee not to pursue the claims and release them back to the plaintiff, the parties entered into a stipulation to abandon arbitration and return the matter to state court. A case management conference was held on February 23, 2022 at which a trial date of May 24, 2023 was set, and the parties have agreed to stipulate to mediation in advance of the trial.
The Company believes the claims in this matter isare without merit and intends to defend the arbitrationitself vigorously. BecauseHowever, at this matter is in an early stage, the Company is unable to predict itsthe outcome and the possible loss or range of loss, if any, associated with its resolution or any potential effect the matter may have on the Company’s financial position. Depending on the outcome or resolution of this matter, it could have a material effect on the Company’s financial position.position, results of operations and cash flows.
6.Stockholders' Equity and Common Stock Warrants
2021 Common Stock Offering
In February 2021, the Company completed the February 2021 Offering in which it offered and sold 33,658,538 shares of its common stock in an underwritten, public offering for a purchase price to the public of $1.025 per share, inclusive of shares offered and sold pursuant to the exercise in full by the underwriter of its 30-day option to purchase additional shares. The February 2021 Offering resulted in aggregate net proceeds to the Company of $31.1 million, after deducting underwriting commissions, discounts, and expenses. In addition, at the closings of the February 2021 Offering, the Company issued to designees of the underwriter of the transaction warrants to purchase up to an aggregate of 1,682,927 shares of common stock to designees. The underwriter warrants have an exercise price of $1.28125 per share and a term of five years from the date of issuance.
2020 Common Stock Offering
In May 2020, the Company completed the May 2020 Offering, a public offering of 11,428,572 shares of common stock for a purchase price of $1.05 per share for net proceeds of $10.3 million after deducting commissions, discounts, and other offering costs. In addition, at the closing of the May 2020 Offering, the Company issued warrants to purchase up to 571,429 shares of common stock to designees of the placement agent for the May 2020 Offering. The placement agent's warrants have an exercise price of $1.3125 per share and a term of five years from the date of issuance.
Additionally, also in May 2020, the Company entered into a warrant exercise agreement with an investor who held the Prior Warrant, a previously outstanding warrant to purchase up to an aggregate of 5,000,000 shares of our common stock at an exercise price of $0.35 per share. In consideration for the exercise of the Prior Warrant for cash and an additional $0.125 per each share of common stock in the Prior Warrant being exercised, the exercising investor received new unregistered warrants to purchase up to an aggregate of 5,000,000 shares of common stock in a private placement. The warrants are exercisable immediately at an exercise price of $0.5263 per share and exercisable until November 8, 2025. During the year ended December 31, 2020, the Company recognized a deemed dividend of $2.0 million to reflect the consideration given as an inducement for the investor to exercise the warrants. This deemed dividend was recorded in the Company's consolidated statement of operations during the year ended December 31, 2020 as an increase to the net loss applicable to common stockholders for purposes of computing net loss per share, basic and diluted.
DIFFUSION PHARMACEUTICALS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
CONTINUED
On September 21, 2015, David Schmidt, a former member of Diffusion LLC and current stockholder of the Company, filed suit (the “Original Complaint”) in the Circuit Court for Albemarle County, Virginia (Case. No. CL15-791, David G. Schmidt v. Diffusion Pharmaceuticals LLC), which Complaint was amended on April 14, 2016 (the “Amended Complaint”). The Original Complaint alleged that a breach of contract with respect to Mr. Schmidt’s previously converted convertible note, and requested relief of specific performance requiring Diffusion LLC to issue him additional units, representing the additional number of units he would have received had he converted at the renegotiated conversion price.
On September 27, 2016, the Company, Diffusion LLC, Mr. Schmidt and the other parties thereto entered into a Settlement Agreement (the “Settlement Agreement”) pursuant to which, among other things, (i) Mr. Schmidt and Diffusion each agreed to submit a consent order to the Court dismissing all claims set forth in the Amended Complaint with prejudice and without an admission of liability by any party, (ii) Mr. Schmidt and Diffusion each released, on behalf of such party and its heirs, assigns, representatives, affiliates and agents, all claims and causes of action of any nature against the other party existing as of the date of the Settlement Agreement and (iii) as consideration therefor, the Company agreed to issue and sell, to Mr. Schmidt and the other parties to the Settlement Agreement, the 2016 Convertible Notes in an aggregate principal amount of $1.9 million and are convertible into shares of Common Stock at an initial conversion price of $3.50 per share.
The Company recognized a litigation settlement charge of $2.5 million which represents the difference between the fair value of the 2016 Convertible Notes issued and the cash received from Mr. Schmidt and other parties. The settlement charge was recorded as a component of general and administrative expenses within the Company’s consolidated statement of operations for the year ended December 31, 2016.
11. Stockholder’s Equity
Common Stock
In connection with the Merger (See Note 4) the Company ascribed non-cash consideration of $0.4 million to 478,200 warrants outstanding prior to the Merger. During the year ended December 31, 2016,May 2020 Investor Warrant Exercise, the Company issued 2,226,698warrants to purchase up to 250,000 shares of its common stock to the placement agent with an exercise price of $0.5938 per share and otherwise have identical terms to the warrants issued to the investor.
Common Stock Warrants
During its evaluation of which 1,861,503 are shares held byequity classification for the pre-Merger stockholders ofCompany's common stock warrants issued in 2020 and 2019, the Company 45,643 shares were issuedconsidered the conditions as prescribed within ASC 815-40,Derivatives and Hedging, Contracts in an Entity’s own Equity. The conditions within ASC 815-40 are not subject to a probability assessment. The warrants do not fall under the liability criteria within ASC 480Distinguishing Liabilities from Equity as they are not puttable and do not represent an instrument that has a redeemable underlying security. The warrants do meet the definition of a derivative instrument under ASC 815, but are eligible for advisory services provided in connection with the Merger, 102,430 shares were issued for general financial advisory services providedscope exception as they are indexed to the Company’s own stock and would be classified in permanent equity if freestanding.
As of December 31,2021, the Company and 217,122 shares were issued pursuanthad the following warrants outstanding to conversions of convertible debt as discussed in Note 9. The Company did not purchase or retire anyacquire shares of its common stock.stock:
Warrants
| | | Outstanding | | | Range of exercise price per share | | Expiration dates |
Common stock warrants issued in 2017 related to Series A convertible preferred stock offering | | | 903,870 | | | | $33.30 | | | March 2022 |
Common stock warrants issued in 2018 related to the January 2018 common stock offering | | | 1,181,421 | | | $12.00 | - | $15.00 | | January 2023 |
Common stock warrants issued related to the May 2019 Offering | | | 1,382,913 | | | $5.00 | - | $6.11875 | | May and December 2024 |
Common stock warrants issued related to the November 2019 Offering | | | 213,570 | | | | $0.35 | | | May 2024 |
Common stock warrants issued related to the December 2019 Offering | | | 313,339 | | | | $0.6981 | | | December 2024 |
Common stock warrants issued related to the May 2020 Offering | | | 571,429 | | | | $1.3125 | | | March 2025 |
Common stock warrants issued related to the May 2020 Investor Warrant Exercise | | | 250,000 | | | | $0.5938 | | | November 2025 |
Common stock warrants issued related to the February 2021 Offering | | | 1,682,927 | | | | $1.28 | | | February 2026 |
| | | | 6,499,469 | | | | | | | |
During the year ended December 31, 2016, the Company did not grant any2021, 53,570 warrants to purchase shares of its common stockexpired and no4,230,000 warrants were exercised.exercised for proceeds of approximately $2.2 million. During the year ended December 31, 2016,2020, 0 warrants to purchase an aggregateexpired and 19,106,504 warrants were exercised for gross proceeds of 17,479 shares of common stock expired unexercised.$8.0 million.
DIFFUSION PHARMACEUTICALS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
7.Stock-Based Compensation
Warrants2015 Equity Plan
The 2015 Equity Plan provides for increases to purchase an aggregatethe number of 460,721shares reserved for issuance thereunder each January 1 equal to 4.0% of the total shares of the Company’s common stock were outstanding and exercisable as of the immediately preceding December 31, 2016, with per share exercise prices ranging from $20.00 to $750.00 andunless a weighted average exercise price of $54.27 per share.
12. Stock-Basedlesser amount is stipulated by the Compensation
Upon consummation Committee of the Merger, all outstanding options to purchase Diffusion UnitsCompany's board of directors. Accordingly, 4,076,571 shares were converted into stock options to purchase the Company’s common stock on terms substantially identical to those in effect prioradded to the reverse merger, except for adjustments to the underlying numberreserve as of January 1, 2022. All such shares and the exercise price based on the Exchange Ratio. At the time of the Merger, there were 301,156 stock options that were exercisable for shares of the Company’s common stock at a weighted average exercise price of $40.13 per share.
DIFFUSION PHARMACEUTICALS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
CONTINUED
2015 Equity Plan
The 2015 Equity Plan, as amended in July 2016, currently allows for the issuance of up to a maximum of 500,000 shares of common stockmay be issued in connection with the grant of stock-based awards, including stock options, restricted stock, restricted stock units, stock appreciation rights and other types of awards as deemed appropriate, not including shares subject to awards assumed in connectioneach case, in accordance with certain transactions, including the Merger.terms of the 2015 Equity Plan. As of December 31, 2016,2021, there were 40,507580,925 shares of common stock available for future issuance under the 2015 Equity Plan. In addition, beginning on January 1, 2017, on each January 1st throughGenerally, the options have a ten (10) year contractual term of the plan, up to 4.0% of the total shares of the Company’s common stock outstanding as of December 31st will be added to the plan reserve, unless a lesser amount is stipulated by the Compensation Committee of the Company’s Board of Directors. On January 1, 2017, the maximum number of shares of the 2015 Equity Plan increased by 413,825 shares, or 4.0% of the total shares of the Company’s common stock outstanding at December 31, 2016.and vest in equal monthly installments over three (3) years.
The Company recorded stock-based compensation expense in the following expense categories of its consolidated statements of operations for the periods indicated:
| | | Year ended December 31, | |
| | | 2016 | | | 2015 | |
| | | | | | | | | |
Research and development | | $ | 674,643 | | | $ | 309,579 | |
| | | | | | | | | |
General and administrative | | | 716,425 | | | | 284,596 | |
| | | | | | | | | |
Total stock-based compensation expense | | $ | 1,391,068 | | | $ | 594,175 | |
| | | Year ended December 31, | |
| | | 2021 | | | 2020 | |
Research and development | | $ | 154,041 | | | $ | 164,791 | |
General and administrative | | | 743,219 | | | | 571,328 | |
Total stock-based compensation expense | | $ | 897,260 | | | $ | 736,119 | |
The following table summarizes the activity related to all stock option grants to employees and non-employees for the year ended December 31, 2016:options:
| | | Number of Options | | | Weighted average exercise price per share | | | Weighted average remaining contractual life (in years) | | | Aggregate Intrinsic Value(A) | |
Balance at January 1, 2016 | | | 1,495,615 | | | $ | 3.92 | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
RestorGenex options outstanding | | | 301,156 | | | | 40.13 | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
Cancelled | | | (48,190 | ) | | | 61.30 | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
Granted | | | 458,828 | | | | 6.25 | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
Outstanding at December 31, 2016 | | | 2,207,409 | | | $ | 8.09 | | | | 7.8 | | | | $94,599 | |
| | | | | | | | | | | | | | | | | |
Exercisable at December 31, 2016 | | | 1,446,121 | | | $ | 9.36 | | | | 7.0 | | | | $94,599 | |
| | | | | | | | | | | | | | | | | |
Vested and expected to vest at December 31, 2016 | | | 2,203,011 | | | $ | 8.09 | | | | 7.8 | | | | $94,599 | |
| | |
| (A)
| The difference, if positive, between the stock option’s exercise price and the closing price of the Company’s common stock at December 31, 2016.
|
DIFFUSION PHARMACEUTICALS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
CONTINUED
Generally, the options have a ten (10) year contractual term and vest in equal monthly installments over three (3) years. In August 2016, the Company granted an option to purchase 204,907 shares of Common Stock to a director in connection with the director’s appointment to the board of directors. The options will vest in equal quarterly installments over ten (10) years and any options exercised are restricted from being sold until August 2021.
Non-employee Stock Options
Non-employee options are remeasured to fair value each period through operations using a Black-Scholes option-pricing model until the options vest. There were no stock options granted to non-employees during the year ended December 31, 2016. Key assumptions used to estimate the fair value of the non-employee stock options granted during the year ended December 31, 2015 included risk-free interest rates of 1.9% to 2.4%, an expected volatility of 125.8% to 127.4%, no expected dividend yield and an expected term equal to the remaining contractual option term. The total fair value of non-employee stock options vested during the years ended December 31, 2016 and 2015 was $0.8 million and $0.2 million, respectively. The weighted average stock price used to determine the fair value of non-employee stock options that vested during the years ended December 31, 2016 and 2015 was $8.27 and $1.90, respectively. At December 31, 2016, there was $75.0 thousand of unrecognized compensation cost related to 42,963 unvested stock options and subject to re-measurement until vested. The total unrecognized compensation expense will be recognized as expense over a weighted-average period of 1.0 year.
Employee Stock Options
| | | Number of Options | | | Weighted average exercise price per share | | | Weighted average remaining contractual life (in years) | | | Aggregate Intrinsic Value | |
Balance at January 1, 2020 | | | 309,276 | | | $ | 55.78 | | | | | | | | | |
Granted | | | 1,931,100 | | | | 0.68 | | | | | | | | | |
Expired | | | (172 | ) | | | 142.50 | | | | | | | | | |
Balance at December 31, 2020 | | | 2,240,204 | | | $ | 8.28 | | | | | | | $ | 289,067 | |
Granted | | | 1,815,767 | | | | 0.89 | | | | | | | | | |
Forfeited | | | (429,748 | ) | | | 1.37 | | | | | | | | | |
Outstanding at December 31, 2021 | | | 3,626,223 | | | | 5.40 | | | | 8.52 | | | | — | |
Exercisable at December 31, 2021 | | | 2,140,524 | | | $ | 8.58 | | | | 8.09 | | | | — | |
Vested and expected to vest at December 31, 2021 | | | 3,626,223 | | | $ | 5.40 | | | | 8.52 | | | | — | |
The weighted average grant date fair value of stock option awards granted to employees was $5.55$0.89 and $2.25$0.64 during the yearyears ended December 31, 20162021 and 2015,2020, respectively. The total fair value of options vested during the years ended December 31, 20162021 and 20152020 were $0.6$0.8 million and $0.3$0.7 million, respectively. NoNaN options were exercised during any of the periods presented. At December 31, 2016,2021, there was $2.9$1.1 million of unrecognized compensation cost related to unvested options that will be recognized as expense over a weighted-average period of 6.021.7 years. During the year ended December 31, 2021, the Company granted 385,267 performance-based stock options with an exercise price of $1.11 per share, subject to vesting based on the satisfaction of specified performance criteria. Compensation expense for the performance-based awards is recorded over the estimated service period for each milestone when the performance conditions are deemed probable of achievement. The Company recorded stock-based compensation expense of approximately $0.1 million year ended December 31, 2021, for service-based awards with performance conditions deemed probable of achievement and/or achieved. For performance-based awards containing performance conditions which were not deemed probable of achievement at December 31, 2021, no stock compensation expense was recognized and any previously recognized expense related to those awards originally deemed probable was reversed.
DIFFUSION PHARMACEUTICALS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
The grant date fair value of employee stock options is determined using the Black-Scholes model.Model. The following assumptions were used during the years ended December 31, 20162021 and 2015:2020:
| 2016 | | 2015 | | | 2021 | | | 2020 | |
Expected term (in years) | 5.76 | - | 7.48 | | | 5.76 | | | | | 10 | | | | 5.31 | — | 10 | |
Risk-free interest rate | 1.2% | - | 2.0% | | 2.1% | - | 2.3% | | | 1.3% | — | 1.7% | | | 0.4% | — | 1.7% | |
Expected volatility | 106.8% | - | 125.5% | | 125.6% | - | 125.8% | | | 122.6% | — | 125.8% | | | 113.4% | — | 124.8% | |
Dividend yield | | 0% | | | | 0% | | | | | 0% | | | | | 0% | | |
Restricted Stock Unit Awards
AsThe Company issues restricted stock ("RSU") to newly elected, non-executive members of December 31, 2016, there were 9,198 unvested sharesthe board of restricted stock. Duringdirectors that vest in six, tri-monthly installments beginning 18 months after the year ended December 31, 2016, 6,140 shares vested.respective grant date. The fair value asof a RSU is equal to the fair market value price of the respectiveCompany’s common stock on the date of grant. RSU expense is recorded on a straight-line basis over the service period.
The following table summarizes activity related to RSU stock-based payment awards:
| | | Number of Units | | | Weighted average grant date fair value | |
Balance at January 1, 2021 | | | 153,000 | | | $ | 0.65 | |
Granted | | | 138,800 | | | | 0.72 | |
Vested(1) | | | (16,350 | ) | | | 0.51 | |
Outstanding at December 31, 2021 | | | 275,450 | | | $ | 0.70 | |
(1) The Company withheld 6,049 shares of common stock to cover the tax liability associated with the vesting dates of the restricted stock awards was $12.0 thousandthese units in 2021.
The Company recognized approximately $54,000 and $6.0 thousand for 2016 and 2015, respectively. The grant date fair value of each restricted stock award granted$18,000 in expense related to these units during the yearyears ended December 31, 2015 was $1.97. There were no restricted stock awards granted in 2016.At 2021 and December 2020, respectively. At December 31, 2016,2021, there was $18.0 thousandapproximately $0.1 million of unrecognized compensation cost related to unvested restricted stock that will be recognized as expense over a weighted average period of 1.52.12 years.
13. Defined Contribution Retirement Plan
The Company has established a 401(k) defined contribution plan (the 401(k) Plan) that covers all employees who qualify under the terms of the plan. Eligible employees may elect to contribute to the 401(k) Plan up to 90% of their compensation, limited by the IRS-imposed maximum. The Company provides a safe harbor match with a maximum amount of 4% of the participant’s compensation. The Company made matching contributions under the 401(k) Plan of $28.0 thousand and $10.0 thousand for the years ended December 31, 2016 and 2015, respectively.
148.. Income Taxes
Prior to the Merger, the Company was treated as a partnership for federal income tax purposes.
DIFFUSION PHARMACEUTICALS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
CONTINUED
Deferred tax assets and liabilities are determined based on the differences between the financial statement carrying amounts and tax bases of assets and liabilities using enacted tax rates in effect for years in which differences are expected to reverse. A deferred tax liability of $3.6 million was recorded for the basis differences associated with indefinite-lived in-process R&D assets. In August 2016, the Company abandoned future development efforts for the IPR&D asset associated with RES-440 and recorded an impairment charge of $1.0 million which is equal to its acquired value. Upon recognizing the impairment, the Company recognized an income tax benefit of $0.4 million and reduced the carrying value of the related deferred tax liability as of December 31, 2016 (See Note 4).
Significant components of the Company's deferred tax assets for federal income taxes as of December 31, 2016 consisted of the following:
Deferred tax assets | | December 31, 2016 | |
Net operating loss carryforwards | | $ | 7,547,296 | |
| | | | | |
Stock option compensation | | | 2,720,186 | |
| | | | | |
Orphan Drug credits | | | 1,218,069 | |
| | | | | |
Capitalized start-up costs and other | | | 4,690,843 | |
| | | | | |
Valuation allowance | | | (16,176,394 | ) |
| | | | | |
Net deferred tax asset | | $ | — | |
| | | | | | | |
| | | | | | | |
Deferred tax liabilities | | | | |
| | | | | |
Intangible assets | | | (3,279,363 | ) |
| | | | | |
| Net deferred tax liability | | $ | (3,279,363 | ) |
Deferred tax assets | | December 31, 2021 | | | December 31, 2020 | |
Net operating loss carryforwards | | $ | 6,033,726 | | | $ | 3,864,189 | |
Stock option compensation | | | 1,641,354 | | | | 1,571,227 | |
Orphan Drug credits | | | 647,937 | | | | 541,384 | |
Lease liability | | | 0 | | | | 38,394 | |
Capitalized start-up costs and other | | | 12,403,925 | | | | 10,709,631 | |
Valuation allowance | | | (20,726,942 | ) | | | (14,906,646 | ) |
Deferred tax assets | | | 0 | | | | 1,818,179 | |
| | | | | | | | | |
Deferred tax liabilities | | | | | | | | |
Intangible assets | | | 0 | | | | (2,223,678 | ) |
Right of use asset | | | 0 | | | | (38,394 | ) |
Deferred tax liabilities | | | 0 | | | | (2,262,072 | ) |
| | | | | | | | | |
Net deferred tax liability | | $ | 0 | | | $ | (443,893 | ) |
The changes in the valuation allowance that occurred during 2016 were as follows:
Valuation allowance at beginning of year | | $ | — | |
| | | | | |
Changes recorded to income tax provision | | | 5,017,047 | |
| | | | | |
Changes recorded in purchase accounting | | | 11,159,347 | |
| | | | | |
Valuation allowance at end of year | | $ | 16,176,394 | |
The Company does not have unrecognized tax benefits as of December 31,2021 or December 31, 2016. 2020. The Company recognizes interest and penalties accrued on any unrecognized tax benefits as a component of income tax expense.
DIFFUSION PHARMACEUTICALS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
CONTINUED
The Company had net operating lossNOL carryforwards (“NOL”) for federal and state income tax purposes at December 31, 20162021 and 2020 of approximately:
Combined NOL Carryforwards: | | December 31, 2016 | |
| | | | |
Federal | | $ | 19,842,612 | |
| | | | | |
State | | $ | 20,222,492 | |
Upon completion of the Merger, the Company acquired the net operating losses for federal and state income tax purposes which RestorGenex has incurred since inception. The Tax Reform Act of 1986 (the “Act”) provides for limitation on the use of net operating loss following certain ownership changes (as defined in the Act) that could limit the Company’s ability to utilize these carryforwards. The Company experienced an ownership change as a result of the Merger.
Combined NOL Carryforwards: | | December 31, 2021 | | | December 31, 2020 | |
Federal | | $ | 23,442,045 | | | $ | 15,013,388 | |
State | | | 23,436,624 | | | | 15,007,966 | |
The pre-2018 net operating loss carryforwards begin expiring in 20342021 for both federal income tax purposes and for state income tax purposes. In January 2016, November 2019, the number of outstanding shares of RestorGenex common stockCompany increased in connection with the Merger discussed in note 4. This increase in the number of shares outstanding resultedresulting in a change of ownership, under the provisions of Internal Revenue Code Section 382 and similar state provisions. These provisions and limitslimit the Company’s ability to utilize these net operating loss carryforwards to offset future income. The amounts above reflect the amount of NOLs that the Company expects to be able to utilize as a result of the limitation. The Company recorded a 100% valuation allowance of the deferred tax assets as of December 31, 2016, 2021 because of the uncertainty of their realization.
DIFFUSION PHARMACEUTICALS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
A reconciliation of income tax benefit at the statutory federal income tax rate and income taxes as reflected in the consolidated financial statements as of December 31, 2016 is as follows:
Rate reconciliation: | | December 31, 2016
| |
Federal tax benefit at statutory rate
| | | (34.0 | ) % |
| | | | |
State tax, net of Federal benefit
| | | (2.3 | ) |
| | | | |
Litigation settlement charge
| | | 4.6 | |
| | | | |
Acquisition costs | | | 2.8 | |
| | | | |
Orphan drug credit
| | | (4.4 | ) |
| | | | |
Change in valuation allowance
| | | 27.2 | |
| | | | |
Stock compensation | | | 4.2 | |
| | | | |
Other
| | | (0.1 | ) |
| | | | |
Total provision
| | | (2.0 | ) % |
Rate reconciliation: | | December 31, 2021 | | | December 31, 2020 | |
Federal tax benefit at statutory rate | | | (21.0 | )% | | | (21.0 | )% |
State tax, net of Federal benefit | | | (4.7 | )% | | | (4.7 | )% |
Orphan drug credit | | | (0.4 | )% | | | (2.9 | )% |
Change in valuation allowance | | | 24.3 | % | | | 18.1 | % |
Total provision | | | (1.8 | )% | | | (10.5 | )% |
The Company files income tax returns in the U.S. federal jurisdiction and various state jurisdictions. The Company’s 20132018 to 20152021 tax years remain open and subject to examination. All net operating losses and credits remain subject to review until utilized.
DIFFUSION PHARMACEUTICALS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
CONTINUED
15. Fair Value Measurements
Certain assets and liabilities are carried at fair value under GAAP. Fair value is defined as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. Valuation techniques used to measure fair value must maximize the use of observable inputs and minimize the use of unobservable inputs. Financial assets and liabilities carried at fair value are to be classified and disclosed in one of the following three levels of the fair value hierarchy, of which the first two are considered observable and the last is considered unobservable:
| ●ITEM 9.
| Level 1—Quoted prices in active markets for identical assets or liabilities.
|
| ●
| Level 2—Observable inputs (other than Level 1 quoted prices), such as quoted prices in active markets for similar assets or liabilities, quoted prices in markets that are not active for identical or similar assets or liabilities, or other inputs that are observable or can be corroborated by observable market data.
|
| ●
| Level 3—Unobservable inputs that are supported by little or no market activity and that are significant to determining the fair value of the assets or liabilities, including pricing models, discounted cash flow methodologies and similar techniques.
|
The asset’s or liability’s fair value measurement level within the fair value hierarchy is based on the lowest level of any input that is significant to the fair value measurement.
The following table presents the Company’s assets and liabilities that are measured at fair value on a recurring basis:
| | | December 31, 2016 | |
| | | | | | | | | | |
| | | (Level 1) | | | (Level 2) | | | (Level 3) | |
Assets | | | | | | | | | | | | |
| | | | | | | | | | | | | |
Cash and cash equivalents | | $ | 1,552,852 | | | $ | — | | | $ | — | |
| | | | | | | | | | | | | |
Liabilities | | | | | | | | | | | | |
| | | | | | | | | | | | | |
Contingent value rights distribution | | $ | — | | | $ | — | | | $ | — | |
| | | December 31, 2015 | |
| | | | | | | | | | |
| | | (Level 1) | | | (Level 2) | | | (Level 3) | |
Assets | | | | | | | | | | | | |
| | | | | | | | | | | | | |
Cash and cash equivalents | | $ | 1,997,192 | | | $ | — | | | $ | — | |
| | | | | | | | | | | | | |
Liabilities | | | | | | | | | | | | |
| | | | | | | | | | | | | |
Contingent value rights distribution | | $ | — | | | $ | — | | | $ | — | |
Contingent Value Rights Distribution
Each CVR represents a non-transferable right (subject to certain limited exceptions) to potentially receive certain cash payments, not to exceed $50.0 million in the aggregate, in the event the Company receives net cash payments during the five (5) year period after the Merger as a result of the sale, transfer, license or similar transaction relating to RES-440. Any option or warrant holder of the Company as of the record date for the CVRs would, at the time of exercise, be entitled to receive one CVR for each share of the Company’s common stock issued upon the future exercise of the option or warrant, which would entitle the holder to a pro rata portion of any CVR payments made after the date of exercise.
DIFFUSION PHARMACEUTICALS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
CONTINUED
In connection with the abandonment of the RES-440 IPR&D asset (See Note 4), the Company remeasured the value of each CVR and as of December 31, 2016, determined the CVRs have no value.
The reconciliation of the contingent consideration liability measured at fair value on a recurring basis using significant unobservable inputs (Level 3) is as follows:
| | | Contingent Value Rights Distribution | |
Issued in connection with the Merger transaction | | $ | 10,000 | |
| | | | | |
Change in fair value upon abandonment of RES-440 | | | (10,000 | ) |
| | | | | |
Balance at December 31, 2016 | | $ | — | |
16. Related Party Transactions
The Company’s Director of Information Technologies is also the son of the Chief Executive Officer and he has held that position since December 2014.
17. Subsequent Events
The Company is conducting the Private Placement of (i) shares of its Series A Convertible Preferred Stock, each share of Series A Convertible Preferred Stock being convertible into one share of its common stock, subject to adjustment, and (ii) for each share of Series A Convertible Preferred Stock purchased in the Private Placement, a 5-year warrant to purchase one share of common stock (the “Warrant” and collectively with the Series A Convertible Preferred Stock, the “Securities”).
On March 14, 2017, the Company entered into Subscription Agreements with certain accredited investors and conducted an initial closing of the Private Placement pursuant to which the Company sold 7,837,023 shares of the Series A Convertible Preferred Stock at a purchase price of $2.02 per share (the “Initial Closing”). In addition, each investor received a Warrant to purchase one share of common stock for each share of Series A Convertible Preferred Stock purchased by such investor in the Private Placement at an exercise price of $2.22, subject to adjustment thereunder.
The Company received net cash proceeds of approximately $14.1 million from the Initial Closing, after deducting $1.7 million in placement agent fees and expenses associated with the Initial Closing. The Company may periodically conduct one or more additional closings up to aggregate gross proceeds of $25.0 million.
ITEM 9.
| CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
None.
ITEM 9A. | CONTROLS AND PROCEDURES |
Evaluation of Disclosure Controls and Procedures
The term “disclosure controls and procedures” means our controls and other procedures that are designed to ensure that information required to be disclosed by us in the reports that we file or submit under the Securities Exchange Act of 1934, as amended, is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by us in the reports that we file or submit under the Exchange Act is accumulated and communicated to our management, including our principal executive officer and principal financial officer, or persons performing similar functions, as appropriate to allow timely decisions regarding required disclosure.
We carried out an evaluation, under the supervision and with the participation of our management, including our principal executive officer and principal financial officer, of the effectiveness of our disclosure controls and procedures (as defined in Exchange Act Rules 13a – 15(e) and 15d – 15(e)). Based upon that evaluation, our principal executive officer and principal financial officer concluded that, as of the end of the period covered in this report, our disclosure controls and procedures were effective to ensure that information required to be disclosed in reports filed under the Exchange Act is recorded, processed, summarized and reported within the required time periods and is accumulated and communicated to our management, including our principal executive officer and principal financial officer, as appropriate to allow timely decisions regarding required disclosure.
Our principal executive officer and principal financial officer do not expect that our disclosure controls or internal controls will prevent all error and all fraud. Although our disclosure controls and procedures were designed to provide reasonable assurance of achieving their objectives, a control system, no matter how well conceived and operated, can provide only reasonable, not absolute assurance that the objectives of the system are met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within our company have been detected. These inherent limitations include the realities that judgments in decision-making can be faulty, and that breakdowns can occur because of a simple error or mistake. Additionally, controls can be circumvented if there exists in an individual a desire to do so. There can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions.
Management’sManagement’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rules 13a-15(f) and 15d-15(f). Under the supervision and with the participation of our Chief Executive Officer and Chief Financial Officer, our management conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework in Internal Control—Control—Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on that evaluation, our management concluded that our internal control over financial reporting was effective as of December 31, 2016.2021.
Based on the evaluation of our Chief Executive Officer and Senior Vice President, Finance for the period ended December 31, 2016, management concluded that our internal controls over financial reporting were operating effectively.
Attestation Report of the Independent Registered Public Accounting Firm
This report does not include an attestation report of our independent registered public accounting firm regarding internal control over financial reporting. Management’sAs a "smaller reporting company" (as such term is defined in Rule 12b-2 of the Exchange Act), pursuant to Section 989G of the Dodd-Frank Act, we are exempt from the requirement subjecting management’s report was not subject to attestation by our independent registered public accounting firm pursuant to Section 989G of the Dodd-Frank Wall Street Reform and Consumer Protection Act, which exempts smaller reporting companies from the auditor attestation requirement.firm.
Change in Internal Control Over Financial Reporting
There was no change in our internal control over financial reporting that occurred during our fourth quarter ended December 31, 20162021 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
ITEM 9B. | OTHER INFORMATION |
None.
PART III
ITEM 10. | DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE |
InformationCORPORATE GOVERNANCE
Introduction
Our common stock is currently listed for quotation on the Nasdaq Capital Market under the symbol “DFFN.” As required by the Listing Rules of the Nasdaq Capital Market, the Board has adopted certain governance standards, including its standard of independence.
Corporate Governance Guidelines
Our Board has adopted Corporate Governance Guidelines, a copy of which can be found on the Investor Relations—Corporate Governance section of our corporate website at www.diffusionpharma.com. Among the topics addressed in our Corporate Governance Guidelines are:
| • | Board size, composition and qualifications; |
| • | Retirement, term limits, and resignation policy; |
| • | Loans to directors and executive officers; |
| • | Chief Executive Officer evaluation; |
| • | Board and committee meetings; |
| • | Board and committee evaluations; |
| • | Executive sessions of outside directors; |
| • | Director continuing education; |
| • | Meeting attendance by directors and non-directors; |
| • | Appropriate information and access; |
| • | Related person transactions; |
| • | Ability to retain advisors; |
| • | Communication with directors; |
| • | Conflicts of interest and director independence; |
| • | Director attendance at annual meetings of stockholders; and |
| • | Board interaction with corporate constituencies; |
| • | Change of principal occupation and board memberships. |
| • | Stock ownership by directors and executive officers; |
Directors & Director Independence
The Board has determined that six of our seven current directors — Robert Adams, Eric Francois, Mark T. Giles, Jane H. Hollingsworth, Diana Lanchoney, and Alan Levin — are “independent directors” under the Listing Rules of the Nasdaq Capital Market.
Board Leadership Structure
The Board believes that our stockholders are best served if the Board retains the flexibility to adapt its leadership structure to applicable facts and circumstances, which necessarily change over time. Accordingly, under our Corporate Governance Guidelines, the office of Chairman of the Board and Chief Executive Officer may or may not be held by one person. The Board believes it is best not to have a fixed policy on this issue and that it should be free to make this determination based on what it believes is best under the circumstances.
Currently, Jane H. Hollingsworth serves as the Chair of the Board and Robert J. Cobuzzi, Jr. serves as our Chief Executive Officer. The Board believes that it is currently in the best interests of the Company’s stockholders to separate these offices. This separation allows for our Board Chair to act as a bridge between the Board and the operating organization, while our Chief Executive Officer focuses on running the Company’s business. The Board believes that this separation allows for a more effective utilization of the proven leadership capabilities, breadth of industry experience and business success of the individuals holding both positions, and that the Company and its stockholders are best currently served by this leadership structure.
Executive Sessions
Generally, at regular meetings of the Board, our independent directors meet in executive session with no company management present during a portion of the meeting. Ms. Hollingsworth typically presides over these executive sessions and serves as a liaison between the independent directors and our Chief Executive Officer.
Board Meetings and Attendance
During 2021, the Board held nine meetings, including one joint meeting with the Compensation Committee. Each of our directors attended 75 percent or more of the meetings of the Board and all committees on which he or she served during 2021. In addition, the Company’s directors are expected to participate in the annual meetings of stockholders, and all of the Company’s directors participated in the 2021 annual meeting of stockholders.
Board Committees
The Board has three standing committees: the Audit Committee, the Compensation Committee, and the Nominating and Corporate Governance Committee. Each of these committees has the composition and responsibilities described below. The Board, from time to time, may establish other committees to facilitate the management of the Company and may change the composition and the responsibilities of the existing committees. Each of the three standing committees has a charter which can be found on the Investor Relations—Corporate Governance section of our corporate website at www.diffusionpharma.com.
Audit Committee
Responsibilities
The primary responsibilities of the Audit Committee include:
• overseeing our accounting and financial reporting processes, systems of internal control over financial reporting and disclosure controls and procedures on behalf of the Board and reporting the results or findings of its oversight activities to the Board;
• having sole authority to appoint, retain and oversee the work of our independent registered public accounting firm and establishing the compensation to be paid to the independent registered public accounting firm;
• establishing procedures for the receipt, retention and treatment of complaints regarding accounting, internal accounting controls and/or auditing matters and for the confidential, anonymous submission by our employees of concerns regarding questionable accounting or auditing matters;
• reviewing and pre-approving all audit services and permissible non-audit services to be performed for us by our independent registered public accounting firm as provided under the federal securities laws and rules and regulations of the SEC; and
• overseeing our system to monitor and manage risk, and legal and ethical compliance programs, including the establishment and administration (including the grant of any waiver from) a written code of ethics applicable to each of our principal executive officer, principal financial officer, principal accounting officer or controller or persons performing similar functions.
The Audit Committee has the authority to engage the services of outside experts and advisors as it deems necessary or appropriate to carry out its duties and responsibilities.
Composition and Audit Committee Financial Expert
The current members of the Audit Committee are Messrs. Francois, Giles, and Levin and Ms. Hollingsworth. Mr. Levin is the chair of the Audit Committee.
Each current member of the Audit Committee qualifies as “independent” for purposes of membership on audit committees under the Listing Rules of the Nasdaq Capital Market and the rules and regulations of the SEC and is “financially literate” under the Listing Rules of the Nasdaq Capital Market. In addition, the Board has determined that Mr. Levin qualifies as an “audit committee financial expert” as defined by the rules and regulations of the SEC and meets the qualifications of “financial sophistication” under the Listing Rules of the Nasdaq Capital Market as a result of his experience in senior financial positions. Stockholders should understand that these designations related to the Audit Committee members’ experience and understanding with respect to this itemcertain accounting and auditing matters are disclosure requirements of the SEC and the Nasdaq Capital Market and do not impose upon any of them any duties, obligations or liabilities that are greater than those generally imposed on a member of the Audit Committee or of the Board.
Meetings
The Audit Committee met five times during 2021.
Processes and Procedures for Complaints
The Audit Committee has established procedures for the receipt, retention and treatment of complaints received by us regarding accounting, internal accounting controls, or auditing matters, and the submission by our employees, on a confidential and anonymous basis, of concerns regarding questionable accounting or auditing matters. Our personnel with such concerns are encouraged to discuss their concerns with their supervisor first, who in turn will be responsible for informing our Chief Executive Officer of any concerns raised. If an employee prefers not to discuss a particular matter with his or her own supervisor, the employee may instead discuss such matter with our Chief Executive Officer. If an individual prefers not to discuss a matter with the Chief Executive Officer or if the Chief Executive Officer is unavailable and the matter is urgent, the individual is encouraged to contact the Chair of the Audit Committee, Mr. Levin.
Compensation Committee
Responsibilities
The primary responsibilities of the Compensation Committee include:
• determining the annual salaries, incentive compensation, long-term incentive compensation, special or supplemental benefits or perquisites and any and all other compensation applicable to our Chief Executive Officer and other executive officers;
• determining any revisions to corporate goals and objectives with respect to compensation for our Chief Executive Officer and other executive officers and establishing and leading a process for the full Board to evaluate the performance of our Chief Executive Officer and other executive officers in light of those goals and objectives;
• administering our equity-based compensation plans, including determining specific grants of options and other awards for executive officers and other employees under our equity-based compensation plans; and
• establishing and leading a process for determination of the compensation applicable to the non-employee directors on the Board.
The Compensation Committee has the authority to engage the services of outside experts and advisors as it deems necessary or appropriate to carry out its duties and responsibilities.
Composition
The current members of the Compensation Committee are Messrs. Adams and Francois, Ms. Hollingsworth, and Dr. Lanchoney. Mr. Adams is the chair of the Compensation Committee. Each of the four current members of the Compensation Committee is an “independent director” under the Listing Rules of the Nasdaq Capital Market and a “non-employee director” within the meaning of Rule 16b-3 under the Exchange Act.
Meetings
The Compensation Committee met seven times during 2021, including one joint meeting with the Board.
Processes and Procedures for Consideration and Determination of Executive Compensation
The Compensation Committee has authority to determine all compensation applicable to our executive officers. In setting executive compensation for our executive officers, the Compensation Committee considers, among other things, the following primary factors: each executive’s position within the Company and the level of responsibility; the ability of the executive to affect key business initiatives; the executive’s individual experience and qualifications; compensation paid to executives of comparable positions by companies similar to our Company; Company and individual performance; and the executive’s current and historical compensation levels. The Compensation Committee has also from time to time – including during 2021 – retained the services of its independent consulting firm, Radford, to provide advice with respect to executive compensation, such as developing a group of comparable peer companies and reviewing executive and director compensation levels. In making decisions regarding the form and amount of compensation to be paid to our executives, the Compensation Committee may consider information gathered by, and the recommendations of, Radford, when necessary and appropriate.
In making decisions regarding the form and amount of compensation to be paid to our executive officers (other than our Chief Executive Officer), the Compensation Committee considers and gives weight to the recommendations of our Chief Executive Officer recognizing that due to his reporting and otherwise close relationship with each executive, the Chief Executive Officer often is in a better position than the Compensation Committee to evaluate the performance of each executive (other than himself). In making decisions regarding the form and amount of compensation to be paid to our Chief Executive Officer, the Compensation Committee considers the recommendation of the Chief Executive Officer with respect to his own compensation and the Compensation Committee’s own assessment of the Chief Executive Officer’s annual performance and input from other Board members. The Compensation Committee meets in executive session regularly and makes all executive compensation decisions about the Chief Executive Officer without the presence of the Chief Executive Officer or any executive or employee of our company.
Processes and Procedures for Consideration and Determination of Director Compensation
The Board has delegated to the Compensation Committee the responsibility, among other things, to establish and lead a process for determining compensation payable to our non-employee directors. The Compensation Committee makes recommendations regarding compensation payable to our non-employee directors to the entire Board, which then makes the final decision.
In making decisions regarding compensation to be paid to our non-employee directors, the Board considers factors such as its own views as to the form and amount of compensation to be paid, the current and anticipated time demands placed on non-employee directors and other factors that may be relevant, including the recommendations of Radford, when necessary and appropriate.
Nominating and Corporate Governance Committee
Responsibilities
The primary responsibilities of the Nominating and Corporate Governance Committee are:
• identifying individuals qualified to become Board members;
• recommending director nominees for each annual meeting of our stockholders and director nominees to fill any vacancies that may occur between meetings of stockholders;
• general management and director succession planning;
• being aware of best practices in corporate governance and developing and recommending to the Board a set forthof corporate governance standards to govern the Board, its committees, our company and our employees in the Proxy Statementconduct of our business and affairs;
• developing and overseeing a Board and Board committee evaluation process; and
• reviewing and discussing with our Chief Executive Officer and reporting periodically to the Board plans for executive officer development and succession plans for the 2016Chief Executive Officer and other key executive officers and employees; and
The Nominating and Corporate Governance Committee has the authority to engage the services of outside experts and advisors as it deems necessary or appropriate to carry out its duties and responsibilities.
Composition
The current members of the Nominating and Corporate Governance Committee are Messrs. Adams, Giles, and Levin and Dr. Lanchoney. Mr. Giles is the chair of the Nominating and Corporate Governance Committee. Each of the four current members of the Nominating and Corporate Governance Committee is an “independent director” within the meaning of the Listing Rules of the Nasdaq Capital Market.
Meetings
The Nominating and Corporate Governance Committee met three times during 2021.
Processes and Procedures for Consideration Director Nominations
In selecting nominees for the Board, the Nominating and Corporate Governance Committee first determines whether the incumbent directors are qualified to serve, and wish to continue to serve, on the Board. The Nominating and Corporate Governance Committee believes that our Company and stockholders benefit from the continued service of certain qualified incumbent directors because those directors have familiarity with and insight into our Company’s affairs that they have accumulated during their tenure with Diffusion. Appropriate continuity of Board membership also contributes to the Board’s ability to work as a collective body. Accordingly, it is the practice of the Nominating and Corporate Governance Committee, in general, to re-nominate an incumbent director at the upcoming annual meeting of stockholders if the director wishes to continue his or her service with the Board, the director continues to satisfy the Nominating and Corporate Governance Committee’s criteria for membership on the Board, the Nominating and Corporate Governance Committee believes the director continues to make important contributions to the Board and there are no special, countervailing considerations against re-nomination of the director.
In identifying and evaluating new candidates for election to the Board, the Nominating and Corporate Governance Committee intends to first solicit recommendations for nominees from persons whom the Nominating and Corporate Governance Committee believes are likely to be familiar qualified candidates having the qualifications, skills and characteristics required for Board nominees from time to time. Such persons may include members of the Board and senior management of Diffusion. In addition, the Nominating and Corporate Governance Committee may engage a search firm to assist it in identifying qualified candidates. The Nominating and Corporate Governance Committee then intends to review and evaluate each candidate whom it believes merits serious consideration, taking into account available information concerning the candidate, any qualifications or criteria for Board membership established by the Nominating and Corporate Governance Committee, the existing composition of the Board (including with respect to diversity), and other factors that it deems relevant. In conducting its review and evaluation, the Nominating and Corporate Governance Committee may solicit the views of our management, other Board members and any other individuals it believes may have insight into a candidate. The Nominating and Corporate Governance Committee may designate one or more of its members and/or other Board members to interview any proposed candidate.
The Nominating and Corporate Governance Committee will consider recommendations for the nomination of directors submitted by our stockholders. The Nominating and Corporate Governance Committee will evaluate candidates recommended by stockholders in the same manner as those recommended described above.
There are no formal requirements or minimum qualifications that a candidate must meet in order for the Nominating and Corporate Governance Committee to recommend the candidate to the Board. The Nominating and Corporate Governance Committee believes that each nominee should be evaluated based on his or her merits as an individual, taking into account the needs of the Company and the Board. However, in evaluating candidates, there are a number of criteria that the Nominating and Corporate Governance Committee generally views as relevant and is likely to consider. Some of these factors include:
| • | whether the candidate is an “independent director” under applicable independence tests under the federal securities laws and rules and regulations of the SEC; |
| • | whether the candidate is “financially sophisticated” and otherwise meets the requirements for serving as a member of an audit committee; |
| • | whether the candidate is an “audit committee financial expert” under the rules and regulations of the SEC for purposes of serving as a member of the Audit Committee; |
| • | the needs of the Company with respect to the particular talents and experience of our directors; |
| • | the personal and professional integrity and reputation of the candidate; |
| • | the candidate’s level of education and business experience; |
| • | the candidate’s business acumen; |
| • | the candidate’s level of understanding of our business and industry and other industries relevant to our business; |
| • | the candidate’s ability and willingness to devote adequate time to the work of the Board and its committees; |
| • | the fit of the candidate’s skills and personality with those of other directors and potential directors in building a board of directors that is effective, collegial and responsive to the needs of our company; |
| • | whether the candidate possesses strategic thinking and a willingness to share ideas; |
| • | the candidate’s diversity of experiences, expertise and background, in general and as compared to other directors on the Board; and |
| • | the candidate’s ability to represent the interests of all stockholders and not a particular interest group. |
While we do not have a stand-alone diversity policy, in considering whether to recommend any director nominee, including candidates recommended by stockholders, the Nominating and Corporate Governance Committee will consider the factors described above. The Nominating and Corporate Governance Committee seeks nominees with a broad diversity of experience, expertise, and backgrounds. The Nominating and Corporate Governance Committee does not assign specific weights to particular criteria and no particular criterion is necessarily applicable to all prospective nominees. We believe that the backgrounds and qualifications of the directors, considered as a group, should provide a significant mix of experience, knowledge and abilities that will allow the Board to fulfill its responsibilities.
Board Oversight of Risk
The Board as a whole has responsibility for risk oversight, with more in-depth reviews of certain areas of risk being conducted by the relevant Board committees that report on their deliberations to the full Board. The oversight responsibility of the Board and its committees is enabled by management reporting processes that are designed to provide information to the Board about the identification, assessment and management of critical risks and management’s risk mitigation strategies. The areas of risk that we focus on include regulatory, operational, financial (accounting, credit, liquidity and tax), legal, compensation, competitive, health, safety and environment, economic, political and reputational risks.
The committees of the Board oversee risks associated with their respective principal areas of focus. The Audit Committee’s role includes a particular focus on the qualitative aspects of financial reporting to stockholders, our processes for the management of business and financial risk, our financial reporting obligations, and our compliance with significant applicable legal, ethical and regulatory requirements. The Audit Committee, along with management, is also responsible for developing and participating in a process for review of important financial and operating topics that present potential significant risk to our company. The Compensation Committee is responsible for overseeing risks and exposures associated with our compensation programs and arrangements, including our executive and director compensation programs and arrangements. The Nominating and Corporate Governance Committee oversees risks relating to our corporate governance matters and policies and management and director succession planning.
We recognize that a fundamental part of risk management is understanding not only the risks a company faces and what steps management is taking to manage those risks, but also understanding what level of risk is appropriate for our Company. The involvement of the full Board in setting our business strategy is a key part of the Board’s assessment of management’s appetite for risk and also a determination of what constitutes an appropriate level of risk for our company.
We believe our current Board leadership structure is appropriate and helps ensure proper risk oversight for our company for a number of reasons, including: (1) general risk oversight by the full Board in connection with its role in reviewing our key long-term and short-term business strategies and monitoring on an on-going basis the implementation of our key business strategies; (2) more detailed oversight by our Board committees that are currently comprised of and chaired by our independent directors and (3) the focus of our Chairman of the Board on allocating appropriate Board agenda time for discussion regarding the implementation of our key business strategies and specifically risk management.
Code of Business Conduct and Ethics
Our Code of Business Conduct and Ethics applies to all of our directors, executive officers and other employees, and meets the requirements of the SEC. A copy of our Code of Business Conduct and Ethics is available on the Investor Relations—Corporate Governance—Code of Business Conduct and Ethics section of our corporate website at www.diffusionpharma.com.
Policy Regarding Director Attendance at Annual MeetingMeetings of Stockholders (“Proxy Statement”)
It is the policy of the Board that directors standing for re-election should attend our annual meeting of stockholders, if their schedules permit.
Process Regarding Stockholder Communications with Board
Stockholders may communicate with the Board or any one particular director by sending correspondence, to our General Counsel & Corporate Secretary via e-mail to info@diffusionpharma.com or via mail to 300 East Main Street, Suite 201, Charlottesville, Virginia 22902, with an amendmentinstruction to this Annual Reportforward the communication to the Board or one or more particular directors. Our General Counsel & Corporate Secretary will forward promptly all such stockholder communications to the Board or the one or more particular directors, with the exception of any advertisements, solicitations for periodical or other subscriptions and other similar communications.
DIRECTORS
Number of Directors
Our Bylaws provide that the Board will consist of at least one member, or such other number as may be determined by the Board or our stockholders. The Board has currently fixed the number of directors at seven.
Information About Our Directors
The table below sets forth, as of March 15, 2022, certain information that has been furnished to us by our current directors.
Name | Age | Director Since |
Robert Adams | 71 | 2016 |
Robert J. Cobuzzi, Jr., Ph.D. | 57 | 2020 |
Eric Francois | 47 | 2021 |
Mark T. Giles | 67 | 2016 |
Jane H. Hollingsworth | 63 | 2020 |
Diana Lanchoney, M.D. | 55 | 2021 |
Alan Levin | 59 | 2016 |
In addition, the paragraphs below provide further information about each current director, including all positions he or she holds, his or her principal occupation and business experience for the past five years, and the names of other publicly held companies of which he or she currently serves as a director or served as a director during the past five years. We believe that all of our directors and director nominees display personal and professional integrity; satisfactory levels of education and/or business experience; broad-based business acumen; an appropriate level of understanding of our business and its industry and other industries relevant to our business; the ability and willingness to devote adequate time to the work of the Board and its committees; a fit of skills and personality with those of our other directors that helps build a board of directors that is effective, collegial and responsive to the needs of our company; strategic thinking and a willingness to share ideas; a diversity of experiences, expertise and background; and the ability to represent the interests of all of our stockholders. The information presented below regarding each director and nominee for director also sets forth specific experience, qualifications, attributes and skills that led the Board to the conclusion that he or she should serve as a director in light of our business and structure.
Robert Adams — Mr. Adams has served as a director since January 2016 and as a director of Diffusion LLC since 2002. Prior to his retirement in 2015, Mr. Adams was a partner in the intellectual property law firm of Nixon & Vanderhye P.C, where he had practiced for over 25 years, focusing on Form 10-K (“Form 10-K/A”patent litigation and international patent licensing and negotiations. During that time period, Mr. Adams was lead litigation counsel in more than 50 major intellectual property lawsuits, where he directly handled, for example, all intellectual property valuations and settlements on behalf of his U.S. and foreign clients. Moreover, Mr. Adams served as the head negotiator for a well-known Japanese consumer products company for 15 years in various complicated licensing situations. Those negotiations typically involved the cross-licensing of up to hundreds of U.S. and foreign patent rights. His lead licensing activities on behalf of that client included, among other things, multi-year negotiations with Texas Instruments, Advanced Micro Devices and Freescale. Mr. Adams received a B.A. from the University of Maryland and a J.D. from George Washington University (with honors), and is a member of the Virginia State Bar.
The Board believes Mr. Adams’ perspective and experience as a director of Diffusion, as well as the depth and breadth of his intellectual property experience, provide him with the qualifications to serve as a director.
Robert J. Cobuzzi, Jr., Ph.D. – Dr. Cobuzzi has served as a director since January 2020 and as our President and Chief Executive Officer since September 2020. Cobuzzi also currently serves as a Venture Partner and Chairman of the Business Development Board for Sunstone Life Science Ventures, an early-stage, European-focused investor in human therapeutics. He also serves on behalf of Sunstone as a Board Observer for Synendos Therapeutics, a venture-backed, Swiss-based biopharmaceutical company focused on developing therapeutics for neuropsychiatric disorders. Previously, Dr. Cobuzzi served as an Advisor to the Mitochondrial Disease Research Program at the Children’s Hospital of Philadelphia, an internationally recognized hospital and research center devoted to children, from January 2019 to April 2020, and as President and Chief Executive Officer of MitoCUREia, Inc., an affiliated company, from July 2019 to July 2020. From 2005 to 2018, Dr. Cobuzzi served in various roles at Endo International PLC, a specialty branded and generic pharmaceuticals manufacturer, most recently serving as President of Endo Ventures Ltd. Dr. Cobuzzi received his Bachelor of Arts in Biochemistry and Art History from Colby College and his Ph.D. in Molecular and Cellular Biochemistry from Loyola University Chicago. He served as a Post-doctoral Fellow in Experimental Therapeutics at Roswell Park Cancer Institute.
The Board believes Dr. Cobuzzi’s experience and insight with drug development and business development and funding, both in the U.S. and abroad, as well as his experience and background as our Chief Executive Officer, provide him with the qualifications to serve as a director.
Eric Francois – Mr. Francois has served as a director since June 2021. Since November 2021, Mr. Francois has served as a Managing Director at the investment banking firm of Credit Suisse. From November 2015 until November 2021, Mr. Francois served as Chief Financial Officer of Scynexis, Inc., a pharmaceutical company pioneering innovative medicines to potentially help millions of patients worldwide in need of new options to overcome and prevent difficult-to-treat and drug-resistant infections. He previously served as co-founder and Chief Operating Officer of Topi, Inc., a technology startup, from July 2013 to October 2015, where he was responsible for all marketing, commercial and financial activities and helped grow the company from inception to over 250 clients worldwide. Previously, Mr. Francois served from September 2007 to July 2013 as a Director in the Equity Capital Markets Group at Lazard Ltd where he led capital raisings and advisory assignments for healthcare and biotechnology companies. He started his career in September 2000 at Cowen and Company in the Equity Capital Markets and Convertible Debt Groups. Mr. Francois holds a B.A. in Economics and Business Administration and a M.A. in Marketing from Pantheon-Sorbonne University, France.
The Board believes that the combination of Mr. Francois’s perspective and industry experience, his experience in financial reporting, corporate finance, and capital raising transactions, and his executive-level experience in the pharmaceutical industry all provide him with the qualifications and skills to serve as a director.
Mark T. Giles — Mr. Giles has served as a director since January 2016 and as a director of Diffusion LLC since 2008. Since July 2007, Mr. Giles has been the sole managing member of Panda Holdings, LLC, which engages in the investment and management of private capital. Since February 2015, Mr. Giles has been a general partner of Anchormark Holdings, LLC, which engages in the investment and management of private capital. Prior to joining Panda Holdings and Anchormark Holdings, Mr. Giles served as the Chief Executive Officer of Virginia National Bank from July 1998 until June 2007 and thereafter continued to serve as the non-executive Chairman until December 2011. Prior to joining Virginia National Bank, Mr. Giles also served as the president of two publicly traded bank holding companies and subsidiary banks in Texas and practiced law with the banking group of a Houston law firm. He chairs the board of Expedition Trust Company. Mr. Giles received a B.S. from the McIntire School of Commerce at the University of Virginia and a J.D. from the University of Virginia School of Law.
The Board believes Mr. Giles’ perspective and experience as a director of Diffusion, as well as the depth and breadth of his business and legal experience, provide him with the qualifications to serve as a director.
Jane H. Hollingsworth – Ms. Hollingsworth has served as a director since September 2020. She currently serves as the founding Managing Partner of Militia Hill Ventures, an organization that creates, builds, and invests in life sciences companies, a role she has held since 2013. While at Militia Hill, Jane co-founded and currently serves as Executive Chair of Eliksa Therapeutics, a regenerative medicine company, co-founded and served as Executive Chair of Spirovant Sciences, a gene therapy company sold to Sumitomo Dainippon Pharma, and served as Executive Chair and CEO of Immunome Inc., a cancer immunotherapy company. Prior to founding Militia Hill, Ms. Hollingsworth co-founded and served as Chief Executive Officer of NuPathe, Inc., a neuroscience focused biopharmaceutical company. . She also co-founded and served as EVP of Auxilium Pharmaceuticals, a urology and rare disease focused biopharmaceutical company. Ms. Hollingsworth also currently serves on the board of various industry and community organizations, including Afimmune Ltd., Ribonova, the University City Science Center, the Kimmel Center for the Performing Arts and Breatcancer.Org. Ms. Hollingsworth received her B.A. from Gettysburg College and her J.D. from Villanova University.
The Board believes Ms. Hollingsworth’s industry perspective and experience, including as chief executive officer and director of a publicly-traded biopharmaceutical company, as well as her depth of her other operating and senior management experience in our industry and educational background, provide her with the qualifications to serve as a director.
Diana Lanchoney, M.D. – Dr. Lanchoney has served as a director since June 2021. Since 2014, Dr. Lanchoney has served as a Vice President of CSL Behring, Inc., a global biopharmaceutical company manufacturing plasma-derived and recombinant therapeutic products, since October 2021 as Vice President, R&D Strategy Implementation, from January 2018 to October 2021 as Vice President, Clinical Pharmacology and Translational Development and prior to that as Vice President, R&D Project Management, from October 2014 to December 2017. Prior to joining CSL, Dr. Lanchoney served in positions of increasing responsibility with Merck & Co., a global pharmaceutical company, most recently as Associate Vice President, Corporate Strategy. Dr. Lanchoney received her B.A. in Economics and German Studies from Tufts University and her M.D. from the University of Pennsylvania.
The Board believes Dr. Lanchoney’s professional and academic background and experience provide her with the qualifications to serve as a director, including the depth and breadth of her experience with clinical development, corporate strategy, and pharmaceutical industry partnering.
Alan Levin — Mr. Levin has served as a director since January 2016 and as a director of Diffusion LLC since June 2015. He previously served as Executive Vice President and Chief Financial Officer of Endo Health Solutions Inc., a global specialty healthcare company, from June 2009 until his retirement in September 2013. Prior to joining Endo, Mr. Levin worked with Texas Pacific Group, a leading private equity firm, and one of their start-up investments. Before that, he was Senior Vice President & Chief Financial Officer of Pfizer, Inc. where he worked for 20 years in a variety of executive positions of increasing responsibility, including Treasurer and Senior Vice President of Finance & Strategic Management for the company’s research and development organization. Mr. Levin received a bachelor’s degree from Princeton University and a master’s degree from New York University’s Stern School of Business. Mr. Levin is a certified public accountant. Mr. Levin currently serves as a member of the board of directors of Biocryst Pharmaceuticals, Inc., a Nasdaq-traded biopharmaceuticals company. He is also a member of the Advisory Board of Auven Therapeutics, a private equity fund; and the Critical Path Institute, a nonprofit collaboration between the Food and Drug Administration and pharmaceutical industry participants focused on streamlining and accelerating the development and regulatory pathways for innovative medicines. From December 2013 to July 2019, he was a member of the board of directors of Aceto Corporation, a Nasdaq-traded company specialized in generics and pharmaceutical intermediate products.
The Board believes that the combination of Mr. Levin’s perspective and experience as a director of Diffusion; his experience in financial reporting, treasury and corporate finance (including his prior positions as chief financial officer of Endo and Pfizer, Inc.); and his executive-level experience in the pharmaceutical industry all provide him with the qualifications and skills to serve as a director.
Overview of Non-Employee Director Compensation Program
As described in more detail under the headings “Electionheading “Corporate Governance—Compensation Committee—Responsibilities,” the Board has delegated to the Compensation Committee the responsibility, among other things, to establish and lead a process for the determination of Directors,” “Executive Officers,” “Section 16(a) Beneficial Ownership Reporting Compliance”compensation payable to our non-employee directors. The Compensation Committee makes recommendations regarding compensation payable to our non-employee directors to the entire Board, which then makes final decisions regarding such compensation. Dr. Cobuzzi, our Chief Executive Officer, is not compensated separately for serving on the Board while also serving as an employee.
As further described below, the principal elements of our non-employee director compensation program for 2021 included cash compensation in the form of annual cash retainers and “Corporate Governance”long-term equity-based incentive compensation, in the form of stock options and restricted stock units.
Cash Compensation
The cash compensation paid to our non-employee members of the Board consists of the following cash retainers:
Description | | Annual Cash Retainer | |
Board Member | | $ | 40,000 | |
Chair of the Board | | $ | 25,000 | |
Audit Committee Chair | | $ | 15,000 | |
Compensation Committee Chair | | $ | 10,000 | |
Nominating and Corporate Governance Committee Chair | | $ | 8,000 | |
Audit Committee Member (other than Chair) | | $ | 7,500 | |
Compensation Committee Member (other than Chair) | | $ | 5,000 | |
Nominating and Corporate Governance Committee Member (other than Chair) | | $ | 4,000 | |
The annual cash retainers are paid in regular installments and otherwise in accordance with the Company’s standard payroll practices. The Compensation Committee has also reserved the right to make a portion of such payments in the form of equity rather than cash under certain conditions. During the fiscal year 2021, all retainers were paid in cash.
Long-Term Equity-Based Incentive Compensation
In addition to cash compensation, our non-employee directors receive long-term equity-based incentive compensation in the form of options to purchase shares of our common stock and restricted stock units. Upon a non-employee director’s initial appointment to the Board, he or she shall receive a stock option award to purchase a number of shares of common stock equal to 0.114% of our shares of common stock outstanding on the grant date, vesting in 18 equal monthly installments following his or her appointment to the Board. In addition, upon appointment he or she also receives a restricted stock unit award for an equivalent number of shares, vesting in six tri-monthly installments commencing on the 18-month anniversary of his or her appointment to the Board. Directors appointed prior to January 1, 2020 received the entirety of this initial appointment award in the form of an option.
In addition, each non-employee director annually receives a stock option award to purchase a number of shares of common stock equal to 0.114% of our shares of common stock outstanding on the grant date, vesting in equal monthly installments over one year, unless otherwise provided by the Compensation Committee.
All option awards granted to our non-employee directors have a ten-year term and an exercise price equal to the fair market value of our common stock on the grant date.
Director Compensation Table for 2021
The table below provides summary information concerning the compensation of each individual who served as a non-employee director of the Company during the year ended December 31, 2021:
| Name | | Fees Earned or Paid in Cash | | | Stock Awards (1) | | | Option Awards (2) | | | All Other Compensation | | | Total | |
Robert Adams | | $ | 57,616 | | | $ | - | | | $ | 57,084 | | | $ | - | | | $ | 114,700 | |
Robert J. Cobuzzi, Jr., Ph.D. (3) | | $ | 0 | | | $ | - | | | $ | 170,735 | (3) | | $ | 612,617 | (3) | | $ | 783,352 | |
Eric Francois (4) | | $ | 27,185 | | | $ | 49,968 | | | $ | 114,167 | | | $ | - | | | $ | 141,352 | |
John L. Gainer, Ph.D. (4) | | $ | 19,288 | | | $ | - | | | $ | - | | | $ | - | | | $ | 19,288 | |
Mark T. Giles | | $ | 57,911 | | | $ | - | | | $ | 57,084 | | | $ | - | | | $ | 114,995 | |
Jane H. Hollingsworth | | $ | 67,374 | | | $ | - | | | $ | 57,084 | | | $ | - | | | $ | 124,458 | |
David G. Kalergis (4) | | $ | 28,932 | | | $ | - | | | $ | - | | | $ | - | | | $ | 28,932 | |
Diana Lanchoney (4) | | $ | 25,373 | | | $ | 49,968 | | | $ | 114,167 | | | $ | - | | | $ | 139,540 | |
Alan Levin | | $ | 61,411 | | | $ | - | | | $ | 57,084 | | | $ | - | | | $ | 118,495 | |
| 1) | The amounts shown in this column reflect the grant date fair value of restricted stock unit awards granted during 2021 to the identified directors upon their respective initial appointments to the Board, calculated in accordance with the provisions of ASC Topic 718 and determined without regard to forfeitures. See the assumptions used in the Black-Scholes Model in Note 7 to the audited financial statements included in our Annual Report. As of December 31, 2021, the aggregate number of shares underlying restricted stock units granted to our directors were as follows: Dr. Cobuzzi – 98,100, 16,350 of which were vested; Mr. Francois – 69,400, none of which were vested; Ms. Hollingsworth – 54,900, none of which were vested; and Dr. Lanchoney – 69,400, none of which were vested. |
| 2) | The amounts shown in this column reflect the grant date fair value of option awards granted during 2021 to the identified directors and former directors, calculated in accordance with the provisions of ASC Topic 718 and determined without regard to forfeitures. See the assumptions used in the Black-Scholes Model in Note 7 to the audited financial statements included in our Annual Report. As of December 31, 2021, the aggregate number of shares subject to options awarded to our each of our directors and former directors were as follows: Mr. Adams – 177,745, of which 136,125 were vested; Dr. Cobuzzi – 761,726, of which 410,041 were vested; Dr. Gainer – 258,770, of which 258,770 were vested; Mr. Francois – 166,600, of which 69,417 were vested; Mr. Giles – 177,191, of which 135,541 were vested; Ms. Hollingsworth – 150,700, of which 101,562 were vested; Mr. Kalergis – 291,592, of which 291,592 were vested; Dr. Lanchoney – 166,600, of which 69,417 were vested; and Mr. Levin –176,557, of which 134,907 were vested. |
| 3) | Reflects compensation for Dr. Cobuzzi’s service as our President and Chief Executive Officer. See “Item 11. Executive Compensation -- Summary Compensation Table” for additional information. Dr. Cobuzzi does not receive any additional compensation for his service as a director. |
| 4) | Dr. Gainer and Mr. Kalergis did not stand for re-election to the Board at our 2021 annual meeting of stockholders and their service on the Board ended upon Mr. Francois’ and Dr. Lanchoney’s election to the Board at such meeting on June 25, 2021. |
EXECUTIVE OFFICERS
Information About Our Executive Officers
The table below sets forth, as of March 15, 2022, certain information concerning our executive officers. Biographical information for Dr. Cobuzzi is included above under the heading, “Directors — Information About Our Directors," and incorporated herein by reference.
Name | Age | Position with Diffusion |
Robert J. Cobuzzi, Jr., Ph.D. | 57 | President and Chief Executive Officer |
William K. Hornung | 53 | Chief Financial Officer |
Christopher D. Galloway, M.D. | 51 | Chief Medical Officer |
William R. Elder | 39 | General Counsel & Corporate Secretary |
In addition, the paragraphs below provide further information about each current director, including all positions he or she holds, his or her principal occupation and business experience for the past five years, and the names of other publicly held companies of which he or she currently serves as a director or served as a director during the past five years.
William K. Hornung – Mr. Hornung serves as our Chief Financial Officer, a position he has held since September 2018. Prior to his appointment as Chief Financial Officer, Mr. Hornung served as the Chief Business Officer at Diffusion from July 2017 through September 2018. Previously, Mr. Hornung served as Chief Financial Officer of Contravir Pharmaceuticals from June 2014 to November 2015 and helped the company up-list to Nasdaq and raise nearly $30 million. Prior to Contravir, from 2002 through 2014 Mr. Hornung held positions of increasing responsibility with PTC Therapeutics, most recently serving as Vice President of Finance from April 2012 to March 2014. While at PTC Therapeutics he oversaw the IPO process and raised more than $1 billion. From 1998 through 2002, Mr. Hornung was with Elan Pharmaceuticals (formerly The Proxy StatementLiposome Company) in various financial roles. At Liposome and Elan he was responsible for strategic planning and operations of the company's UK-based European headquarters. Earlier in his career Mr. Hornung worked for a clinical research organization where he was responsible for project management and nearly all financial aspects of the company. Mr. Hornung holds a Bachelor of Science in Accounting from the William Paterson State University of New Jersey.
Christopher D. Galloway, M.D. – Dr. Galloway has served as our Chief Medical Officer since October 2020. Prior to joining Diffusion, Dr. Galloway served as senior medical director in critical care for La Jolla Pharmaceutical Company from August 2018 to September 2020, where he chaired and oversaw La Jolla’s investigator-initiated and collaborative research programs and supported the commercial and medical teams in connection with the launch of GIAPREZA™(angiotensin II), which has been approved by the U.S. Food and Drug Administration as a vasoconstrictor to increase blood pressure in adults with septic or Form 10-K/Aother distributive shock. Prior to his time at La Jolla, Dr. Galloway served as medical director for global clinical development at Rakuten Medical Inc. (f/k/a Aspyrian Therapeutics, Inc.), a biotechnology company developing cell-targeting investigative immuno-oncology therapies, from December 2016 to July 2018 and as medical affairs director within Merck & Co., Inc.’s immunotherapy division from August 2015 to November 2016. Dr. Galloway received his doctor of medicine from the University of Texas Medical Branch at Galveston, completed his residency in emergency medicine at Carolinas Medical Center in Charlotte, NC, and received a B.A. in biology from the University of Texas at Austin. He is licensed to practice medicine in the State of Colorado and is a diplomate of the American Board of Emergency Medicine.
William R. Elder – Mr. Elder has served as our General Counsel & Corporate Secretary since September 2020. Prior to joining Diffusion, Mr. Elder principally served as president and chief executive officer of BillyVonElds, LLC, a season-long and daily fantasy sports company, where he managed all corporate, legal, and operational aspects of the business from April 2019 to September 2020. From July 2020 to September 2020, Mr. Elder also served as a part-time consultant to Diffusion. From 2011 to February 2019, Mr. Elder served as a corporate and securities associate for Dechert LLP, an international law-firm, where Mr. Elder’s practice focused primarily on counseling public companies on securities laws and regulatory requirements, corporate governance matters, and financial transactions in the equity and debt markets. He received his J.D. from the University of Pennsylvania Law School, an M.S. in finance from Villanova University, and a B.A. in economics from Tufts University.
ITEM 11. | EXECUTIVE COMPENSATION |
Summary Compensation Table
The table below provides summary compensation information concerning compensation awarded for service during the years ended December 31, 2021 and December 31, 2020 to the individuals that served as our named executive officers during the year ended December 31, 2021.
Name and Principal Position | Year | | Salary (1) | | | Bonus Compensation (2) | | | Stock Awards (3) | | | Option Awards (4) | | | All Other Compensation (5) | | | Total | |
| | | | | | | | | | | | | | | | | | | | | | | | | | |
Robert J. Cobuzzi, Jr., Ph.D. (6) | 2021 | | $ | 410,000 | | | $ | 164,000 | | | $ | - | | | $ | 170,735 | | | $ | 38,617 | | | $ | 783,352 | |
Chief Executive Officer | 2020 | | $ | 129,046 | | | $ | 67,650 | | | $ | 50,031 | | | $ | 512,233 | | | $ | 58,535 | | | $ | 817,495 | |
William K. Hornung | 2021 | | $ | 324,929 | | | $ | 96,667 | | | $ | - | | | $ | 84,838 | | | $ | 29,084 | | | $ | 535,518 | |
Chief Financial Officer | 2020 | | $ | 298,100 | | | $ | 93,902 | | | $ | - | | | $ | 130,083 | | | $ | 17,866 | | | $ | 539,951 | |
Christopher D. Galloway, M.D. (6) | 2021 | | $ | 375,000 | | | $ | 127,500 | | | $ | - | | | $ | 86,393 | | | $ | 37,092 | | | $ | 625,985 | |
Chief Medical Officer | 2020 | | $ | 66,827 | | | $ | 37,500 | | | $ | - | | | $ | 190,882 | | | $ | 4,599 | | | $ | 299,808 | |
William R. Elder (6) | 2021 | | $ | 256,250 | | | $ | 76,234 | | | $ | - | | | $ | 66,246 | | | $ | 5,877 | | | $ | 404,607 | |
General Counsel | 2020 | | $ | 68,270 | | | $ | 24,750 | | | $ | - | | | $ | 147,429 | | | $ | 25,266 | | | $ | 265,715 | |
| 1) | Represents cash portion of base salary as described below under “—Employment Agreements.” |
| 2) | Represents the annual cash incentive bonuses for service during the applicable year by our named executive officers as described further below under “—2021 Bonus Compensation”. |
| 3) | The amount shown in this column reflects the grant date fair value of a restricted stock unit award made to Dr. Cobuzzi in connection with his initial appointment to the Board in January 2020, calculated in accordance with the provisions of ASC Topic 718 and determined without regard to forfeitures. The assumptions used in the Black-Scholes model are disclosed in Note 7 to the audited financial statements included in this Annual Report. |
| 4) | The amounts shown in this column reflect the grant date fair value of option awards granted for service during the applicable year, calculated in accordance with the provisions of ASC Topic 718 and determined without regard to forfeitures. Amounts shown for 2020 include time-based awards for service during 2020 granted in March 2021 and (i) with respect to Dr. Cobuzzi, $49,971 granted in January 2020 in connection with his initial appointment to the Board, $50,867 granted in June 2020 in connection with his service as a non-employee director during 2020, and $354,483 granted in September 2020 in connection with his initial appointment as Chief Executive Officer; (ii) with respective to Dr. Galloway, $162,083 granted in October 2020 in connection with his initial appointment as Chief Medical Officer, and (iii) with respect to Mr. Elder, $54,523 granted in September 2020 in connection with his initial appointment as General Counsel. Amounts shown for 2021 include the full grant date fair value of milestone-based, performance awards also granted in March 2021. Pursuant to the terms of the corresponding award agreements, two-thirds of the underlying shares originally granted were automatically forfeited due to the first patient in the ILD-DLCO Trial not being dosed on or before September 30, 2021. The Company announced dosing of the first patients in the ILD-DLCO Trial on December 16, 2021. |
| 5) | The amounts reported in this column for 2020 represent (w) with respect to Dr. Cobuzzi, (i) $50,076 in fees for his service as a non-employee director from January 2020 to September 2020 prior to his appointment as Chief Executive Officer in September 2020 (including fees for committee service), (ii) $5,162 in 401(k) Plan matching contributions by the Company, and (iii) $3,297 in Company-paid health insurance premiums, (x) with respect to Mr. Hornung, (i) $6,459 in 401(k) Plan matching contributions by the Company and (ii) $11,407 in Company-paid health insurance premiums, (y) with respect to Dr. Galloway, (i) $2,500 in 401(k) Plan matching contributions by the Company and (ii) $2,099 in Company-paid health insurance premiums, and (z) with respect to Mr. Elder, (i) $24,775 in fees for his service as a consultant to the Company from July 2020 to September 2020 prior to his appointment as General Counsel & Corporate Secretary and (ii) $491 in Company-paid health insurance premiums. The amounts reported in this column for 2021 represent (w) with respect to Dr. Cobuzzi, (i) $11,600 in 401(k) Plan matching contributions by the Company and (iii) $27,017 in Company-paid health insurance premiums, (x) with respect to Mr. Hornung, (i) $11,600 in 401(k) Plan matching contributions by the Company and (ii) $17,484 in Company-paid health insurance premiums, (y) with respect to Dr. Galloway, (i) $11,600 in 401(k) Plan matching contributions by the Company and (ii) $25,492 in Company-paid health insurance premiums, and (z) with respect to Mr. Elder, $5,877 in Company-paid health insurance premiums. |
| 6) | Dr. Cobuzzi began his employment as President & Chief Executive Officer on September 8, 2020 and began his service as a director on January 7, 2020, Dr. Galloway began his employment as Chief Medical Officer on October 19, 2020, and Mr. Elder began his employment as General Counsel & Corporate Secretary on September 23, 2020. Accordingly, during 2020, each received pro-rated compensation in accordance with his respective term of service. |
Employment Agreements
Robert J. Cobuzzi, Jr., Ph.D., President & Chief Executive Officer
Effective September 8, 2020, we entered into an employment agreement with Dr. Cobuzzi pursuant to which he serves as our President & Chief Executive Officer. The employment agreement has an indefinite term. Dr. Cobuzzi is currently entitled to an initial annual base salary of $410,000, subject to increase at the discretion of the Board. Dr. Cobuzzi has the opportunity to earn a target annual bonus of 50 percent of his base salary. The Board may, in its discretion, pay a portion of Dr. Cobuzzi’ annual salary and annual bonus in the form of equity or equity-based compensation, provided that commencing with the year following the year in which a “change of control” (as defined in the employment agreement) occurs, Dr. Cobuzzi’ entire base salary and annual bonus will be filedpaid in cash. For 2021, Dr. Cobuzzi’ entire pro-rated base salary was paid in cash. The employment agreement contains certain severance and change of control provisions as described in more detail under the heading “—Post-Termination Severance and Change in Control Arrangements.” The employment agreement also contains certain non-competition and non-solicitation provisions (each applicable during employment and for 24 months thereafter), as well as confidentiality and non-disparagement provisions (each applicable during employment and at all times thereafter).
William K. Hornung, Chief Financial Officer
Effective September 21, 2018, we entered into an amended and restated employment agreement with Mr. Hornung pursuant to which he serves as our Chief Financial Officer. The employment agreement has an indefinite term. Mr. Hornung was entitled to an annual base salary of $298,100 during 2020, subject to increase at the discretion of the Board. Mr. Hornung has the opportunity to earn a target annual bonus of 35 percent of his base salary. The Board may, in its discretion, pay a portion of Mr. Hornung’s annual salary and annual bonus in the form of equity or equity-based compensation, provided that commencing with the SEC within 120 days afteryear following the endyear in which a “change of control” (as defined in the employment agreement) occurs, Mr. Hornung’s entire base salary and annual bonus will be paid in cash. For 2021, Mr. Hornung’s entire base salary was paid in cash. The employment agreement contains certain severance and change of control provisions as described in more detail under the heading “—Post-Termination Severance and Change in Control Arrangements.” The employment agreement also contains certain non-competition and non-solicitation provisions (each applicable during employment and for 18 months thereafter), as well as confidentiality and non-disparagement provisions (each applicable during employment and at all times thereafter).
Christopher D. Galloway, Chief Medical Officer
Effective October 19, 2020, we entered into an employment agreement with Dr. Galloway pursuant to which he serves as our Chief Medical Officer. The employment agreement has an indefinite term. Dr. Galloway is entitled to an initial annual base salary of $375,000, subject to increase at the discretion of the fiscal year covered by this Annual Report.Board. Dr. Galloway has the opportunity to earn a target annual bonus of 40 percent of his base salary. The employment agreement contains certain severance and change of control provisions as described in more detail under the heading “—Post-Termination Severance and Change in Control Arrangements.” The employment agreement also contains certain non-competition (applicable during employment and for 12 months thereafter) and non-solicitation provisions (applicable during employment and for 24 months thereafter), as well as confidentiality and non-disparagement provisions (each applicable during employment and at all times thereafter).
ITEM 11.
William R. Elder, General Counsel & Corporate Secretary | EXECUTIVE COMPENSATION
|
InformationEffective September 23, 2020, we entered into an employment agreement with Mr. Elder pursuant to which he serves as our General Counsel & Corporate Secretary. The employment agreement has an indefinite term. Mr. Elder is entitled to an initial annual base salary of $250,000, subject to increase at the discretion of the Board. Mr. Elder has the opportunity to earn a target annual bonus of 30 percent of his base salary. The Board may, in its discretion, pay a portion of Mr. Elder’s annual salary and annual bonus in the form of equity or equity-based compensation, provided that commencing with the year following the year in which a “change of control” (as defined in the employment agreement) occurs, Mr. Elder’s entire base salary and annual bonus will be paid in cash. For 2021, Mr. Elder’s entire pro-rated base salary was paid in cash. The employment agreement contains certain severance and change of control provisions as described in more detail under the heading “—Post-Termination Severance and Change in Control Arrangements.” The employment agreement also contains certain non-competition and non-solicitation provisions (each applicable during employment and for 24 months thereafter), as well as confidentiality and non-disparagement provisions (each applicable during employment and at all times thereafter).
Long-Term Equity Incentive Compensation and Other Compensatory Arrangements
The Compensation Committee administers the 2015 Equity Plan in which our named executive officers participate, the bonus payments made to our named executive officers provided for in the employment agreements described under the heading “—Employment Agreements,” and any other compensation-related matters as they otherwise determine in their discretion. The option grants made for service during 2020 to the named executive officers vest and become exercisable in equal (or as near equal as possible) installments over a 36-month period until fully vested, subject to their continued employment through the applicable vesting date.
During 2021, the Compensation Committee determined that 50% of any annual long-term equity incentive awards granted to our named executive officers for service during the year would be granted at the outset of the year in the form of performance-based options the vesting of which will be dependent on the achievement of specified performance metrics during the year of grant. The remaining 50 percent were granted in the form of option awards subject to time-based vesting early in the subsequent year (i.e. 2022) at the discretion of the Compensation Committee in accordance with past practice. In 2022, the Compensation Committee determined to return to the Company's historic practice of granting all such awards as options subject to time-based vesting.
2021 Bonus Compensation
Executive bonuses are determined by the Compensation Committee. The Compensation Committee determines whether bonuses are earned and the amounts of the bonus payout by considering a number of factors, the principal factor being based upon the performance goals developed by the Compensation Committee. Other important factors include clinical trial progress, business development activities, status of public filings, capital raising transactions, and stock price performance.
Outstanding Equity Awards at Fiscal Year End
Option Awards
The table below provides information regarding unexercised stock option awards held by each of our named executive officers that remained outstanding as of December 31, 2021. Unless otherwise indicated, each grant was awarded under our 2015 Equity Plan.
Name | Award Type | Grant Date | | Shares Underlying Unexercisable Options Exercisable | | | Shares Underlying Unexercisable Options Unexercisable | | | Exercise Price | | Expiration Date |
Robert J. Cobuzzi, Jr., Ph.D. | NQO | 1/7/2020 | | | 118,600 | | | | — | | | $ | 0.51 | | 1/7/2030 |
| | NQO | 6/17/2020 | | | 61,300 | | | | — | | | $ | 1.00 | | 6/17/2030 |
| | NQO | 9/8/2020 | | | 197,917 | | | | 277,083 | | | $ | 0.79 | | 9/8/2030 |
| | NQO | 3/1/2021 | | | 14,920 | | | | 38,793 | | | $ | 1.11 | | 3/1/2031 |
| | NQO** | 3/1/2021 | | | 17,904 | | | | 35,809 | | | $ | 1.11 | | 3/1/2031 |
William K. Hornung | NQO | 1/2/2018 | | | 6,000 | | | | — | | | $ | 17.70 | | 1/2/2028 |
| | NQO | 1/2/2019 | | | 16,334 | | | | — | | | $ | 2.10 | | 1/2/2029 |
| | NQO | 1/2/2020 | | | 35,067 | | | | 17,533 | | | $ | 0.46 | | 1/2/2030 |
| | NQO | 3/1/2021 | | | 34,103 | | | | 88,668 | | | $ | 1.11 | | 3/1/2031 |
| | NQO** | 3/1/2021 | | | 8,897 | | | | 17,793 | | | $ | 1.11 | | 3/1/2031 |
Christopher D. Galloway, M.D. | NQO* | 10/19/2020 | | | 77,778 | | | | 122,222 | | | $ | 0.85 | | 10/19/2030 |
| | NQO | 3/1/2021 | | | 7,550 | | | | 19,630 | | | $ | 1.11 | | 3/1/2031 |
| | NQO** | 3/1/2021 | | | 9,060 | | | | 18,119 | | | $ | 1.11 | | 3/1/2031 |
William R. Elder | NQO* | 9/22/2020 | | | 29,167 | | | | 40,833 | | | $ | 0.82 | | 9/22/2030 |
| | NQO | 3/1/2021 | | | 24,357 | | | | 63,327 | | | $ | 1.11 | | 3/1/2031 |
| | NQO** | 3/1/2021 | | | 6,947 | | | | 13,894 | | | $ | 1.11 | | 3/1/2031 |
* - Non-plan based equity award grant made as an inducement to the individual’s acceptance of employment with the Company in accordance with Nasdaq Listing Rule 5635(c)(4).
** - Pursuant to the terms of the corresponding award agreements, two-thirds of the underlying shares originally granted were automatically forfeited due to the first patient in the ILD-DLCO Trial not being dosed on or before September 30, 2021. The Company announced dosing of the first patients in the ILD-DLCO Trial on December 16, 2021.
Restricted Stock Unit Awards
The table below provides information regarding restricted stock unit awards held by each of our named executive officers that remained outstanding as of December 31, 2021, if any. Each grant was awarded under our 2015 Equity Plan.
Name | Award Type | Grant Date | | Number of Shares That Have Not Vested | | | Market Value of Shares That Have Not Vested* | |
Robert J. Cobuzzi, Jr., Ph.D. | RSU | 1/7/2020 | | | 81,750 | | | $ | 25,343 | |
* - Based on a price per share of $0.31, the closing price of our common stock on December 31, 2021 as reported by Nasdaq. The award was granted to Dr. Cobuzzi in connection with his appointment as a non-employee director in January 2020 and vests in six tri-monthly installments. The first such installment vested on October 31, 2021.
401(k) Retirement Plan
We maintain our 401(k) Plan pursuant to which all eligible employees are entitled to make pre-tax and after-tax contributions of their compensation. In addition, the Company makes discretionary matching contributions at a rate of 100% for contributions up to 3% of the participant’s eligible compensation and 50% for any additional contributions up to 5% of the participant’s eligible compensation. The matching contributions received by our named executive officers in 2021 and 2020 are reported in the “All Other Compensation” column of the Summary Compensation Table above.
Post-Termination Severance and Change in Control Arrangements
As described under the heading “—Employment Agreements,” we have entered into employment agreements with each of Drs. Cobuzzi and Galloway and Messrs. Hornung and Elder that provide for certain severance and change of control benefits, subject to the execution and non-revocation of a release of claims by the executive or his estate (as applicable).
Under Dr. Cobuzzi’s employment agreement, if his employment is terminated by us other than for “cause,” death or “disability,” or by Dr. Cobuzzi for “good reason” (as such terms are defined in the employment agreement), Dr. Cobuzzi will be entitled to any unpaid bonus earned in the year prior to the termination, a pro-rata portion of the bonus earned during the year of termination, continuation of base salary for 12 months, plus 12 months of COBRA premium reimbursement, provided that if such termination occurs within 60 days before or within 24 months following a “change of control” (as defined in the employment agreement), then Dr. Cobuzzi will be entitled to receive the same severance benefits as described above, except that he will receive (a) a payment equal to two times the sum of his base salary and the higher of his target annual bonus opportunity and the bonus payment he received for the year immediately preceding the year in which the termination occurred instead of 12 months of base salary continuation, and (b) a payment equal to 36 times the monthly COBRA premium for him and his eligible dependents instead of 12 months of COBRA reimbursements (the payments in clauses (a) and (b) are paid in a lump sum in some cases and partly in a lump sum and partly in installments over 12 months in other cases). In addition, if Dr. Cobuzzi’s employment is terminated by us without cause or by Dr. Cobuzzi for good reason, in either case, upon or within 24 months following a change of control, then Dr. Cobuzzi will be entitled to full vesting of all equity awards received by him from us (with any equity awards that are subject to the satisfaction of performance goals deemed earned at not less than target performance, and with any equity award that is in the form of a stock option or stock appreciation right to remain outstanding and exercisable for 24 months following the termination date (but in no event beyond the expiration date of the applicable option or stock appreciation right)).
Under the employment agreements for each of Dr. Galloway and Messrs. Hornung and Elder, in the event that his employment is terminated by us other than for “cause”, death or “disability” or upon his resignation for “good reason” (as such terms are defined in the applicable employment agreement), the applicable executive will be entitled to any unpaid bonus earned in the year prior to the termination, a pro-rata portion of the bonus earned during the year of termination, continuation of base salary for 9 months, plus 12 months of COBRA premium reimbursement, provided that if such termination occurs within 60 days before or within 24 months following a “change of control” (as defined in the applicable employment agreement), then he will be entitled to receive the same severance benefits as described above, except that he will receive (a) a payment equal to 1.5 times the sum of his base salary and the higher of his target annual bonus opportunity and the bonus payment he received for the year immediately preceding the year in which the termination occurred instead of 9 months of base salary continuation and (b) a payment equal to 18 times the monthly COBRA premium for him and his eligible dependents instead of 12 months of COBRA reimbursements (the payments in clauses (a) and (b) are paid in a lump sum in some cases and in installments over 9 or 12 months in other cases). In addition, if the applicable executive’s employment is terminated by the Company without cause or by the applicable executive for good reason, in either case, upon or within 24 months following a change of control, then the applicable executive will be entitled to full vesting of all equity awards received by him from us (with any equity awards that are subject to the satisfaction of performance goals deemed earned at not less than target performance, and with any equity award that is in the form of a stock option or stock appreciation right to remain outstanding and exercisable for 24 months following the termination date (but in no event beyond the expiration date of the applicable option or stock appreciation right)).
Under the employment agreements for each of our current named executive officers, in the event that the executive’s employment is terminated due to his death or disability, he (or his estate) will be entitled to any unpaid bonus earned in the year prior to the termination, a pro-rata portion of the bonus earned during the year of termination, 12 months of COBRA premium reimbursement and accelerated vesting of (a) all equity awards received in payment of base salary or an annual bonus and (b) with respect to this item will be set forthany other equity award, the greater of the portion of the unvested equity award that would have become vested within 12 months after the termination date had no termination occurred and the portion of the unvested equity award that is subject to accelerated vesting (if any) upon such termination under the applicable equity plan or award agreement (with performance goals deemed earned at not less than target performance, and with any equity award that is in the Proxy Statementform of a stock option or Form 10-K/Astock appreciation right to remain outstanding and exercisable for 12 months following the termination date or, if longer, such period as provided under the headings “Executive Compensation”applicable equity plan or award agreement (but in no event beyond the expiration date of the applicable option or stock appreciation right)).
Further, under the terms of the stock option agreements with our named executives officers, upon a completion of a “change of control” (as defined in the 2015 Equity Plan), options held by our named executive officers will become immediately vested and “Director Compensation,” and is incorporated herein by reference. The Proxy Statementremain exercisable through their expiration date regardless of whether the holder remains in the employment or Form 10-K/A will be filed withservice of the SEC within 120 daysCompany after the endchange of control. Alternatively, in connection with a change of control, the fiscal year covered by this Annual Report.Compensation Committee may, in its sole discretion, cash out the options.
ITEM 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
InformationBased on information available to us and filings with the SEC, the following table sets forth certain information regarding the beneficial ownership (as defined by Rule 13d-3 under the Exchange Act) of our outstanding common stock as of March 1, 2022 for (i) each person or group of affiliated persons known by us to be the beneficial owner of more than 5% of our common stock, if any, (ii) each of our current directors; (iii) each of our current executive officers (as defined in Item 402(a)(3) of Regulation S-K under the Exchange Act); and (iv) all of our current directors and named executive officers as a group. As of March 1, 2022, to our knowledge, no beneficial owner owned 5% or more of the shares of common stock then outstanding.
Beneficial ownership and percentage ownership are determined in accordance with the rules of the SEC and include voting or investment power with respect to this item will be set forth inshares of stock. This information does not necessarily indicate beneficial ownership for any other purpose. Under these rules, shares of common stock issuable under shares of preferred stock, stock options or warrants that are exercisable or convertible within 60 days of March 1, 2022 are deemed outstanding for the Proxy Statement or Form 10-K/Apurpose of computing the beneficial ownership percentage of the holder thereof, but are not deemed outstanding for the purpose of computing the beneficial ownership percentage of any other person. Ownership is based upon information provided by each respective director and is incorporated herein by reference. The Proxy Statement or Form 10-K/A will beofficer and public documents filed with the SEC, within 120 days after the endincluding Forms 3 and 4, Schedules 13D and 13G and certain other documents, which information may not be accurate as of the fiscalRecord Date.
Unless otherwise indicated and subject to applicable community property laws, to our knowledge, each stockholder named in the following table possesses sole voting and investment power over their shares of common stock, except for those jointly owned with that person’s spouse. Unless otherwise indicated below, the address of each person listed on the table is c/o Diffusion Pharmaceuticals Inc., 300 East Main Street, Suite 201, Charlottesville, Virginia 22902.
Name and Address of Beneficial Owner | | Shares of Common Stock Beneficially Owned (1) | | | Common Stock Beneficial Ownership Percentage (2) | |
Current Directors | | | | | | | | |
Robert Adams (3) | | | 166,515 | | | | 0.2 | % |
Robert J. Cobuzzi, Jr., Ph.D. (4) | | | 551,507 | | | | 0.5 | % |
Eric Francois (5) | | | 104,125 | | | | 0.1 | % |
Mark T. Giles (6) | | | 210,173 | | | | 0.2 | % |
Jane H. Hollingsworth (7) | | | 162,661 | | | | 0.2 | % |
Diana Lanchoney, M.D. (8) | | | 104,125 | | | | 0.1 | % |
Alan Levin (9) | | | 156,158 | | | | 0.2 | % |
Current Named Executive Officers | | | | | | | | |
William R. Elder (10) | | | 115,827 | | | | 0.1 | % |
Christopher D. Galloway, M.D. (11) | | | 171,063 | | | | 0.2 | % |
William K. Hornung (12) | | | 142,717 | | | | 0.1 | % |
All Current Directors, Director Nominees, and Named Executive Officers as a Group (ten persons) (13) | | | 1,884,871 | | | | 1.8 | % |
* Indicates less than 0.1%
1. Includes shares of common stock held as of March 1, 2022 plus shares of common stock that may be acquired upon exercise of options, warrants and other rights exercisable within 60 days of the March 1, 2022.
2. Based on 101,924,581 shares of common stock issued and outstanding as of March 1, 2022. The percentage ownership and voting power for each person (or all directors and executive officers as a group) is calculated by assuming (i) the exercise or conversion of all preferred stock, options, warrants and convertible securities exercisable or convertible within 60 days of March 1, 2022 held by such person and (ii) the non-exercise and non-conversion of all outstanding preferred stock, warrants, options and convertible securities held by all other persons (including our other directors and executive officers).
3. Consists of (a) 1,706 shares held directly by Mr. Adams, (b) 631 shares held jointly with Mr. Adams’ wife, (c) 1,260 shares held for the benefit of Mr. Adams in his 401(k) retirement account, and (d) 162,918 shares of common stock issuable upon the exercise of options exercisable within 60 days of March 1, 2022.
4. Consists of (a) 34,602 shares held directly by Dr. Cobuzzi and (b) 516,905 shares of common stock issuable upon the exercise of options exercisable within 60 days of March 1, 2022.
5. Consists of 104,125 shares of common stock issuable upon the exercise of options exercisable within 60 days of March 1, 2022.
6. Consists of (a) 294 shares held for the benefit of Mr. Giles in his individual retirement account, (b) 53,513 shares held by MTG Investment Holdings, LLC, and (c) 156,366 shares of common stock issuable upon the exercise of options exercisable within 60 days of March 1, 2022. Mr. Giles is the sole member of MTG Investment Holdings, LLC and may be deemed to be the beneficial owner of such securities. Mr. Giles disclaims beneficial ownership of such securities except to the extent of his pecuniary interest therein.
7. Consists of (a) 32,876 shares held directly by Ms. Hollingsworth and (b) 129,875 shares of common stock issuable upon the exercise of options exercisable within 60 days of March 1, 2022.
8. Consists of 104,125 shares of common stock issuable upon the exercise of options exercisable within 60 days of March 1, 2022.
9. Consists of (a) 1,654 shares held by Mr. Levin directly and (b) 154,504 shares of common stock issuable upon the exercise of options exercisable within 60 days of March 1, 2022.
10. Consists of (a) 15,000 shares held directly by Mr. Elder and (b) 100,827 shares of common stock issuable upon the exercise of options exercisable within 60 days of March 1, 2022.
11. Consists of (a) 20,000 shares held for the benefit of Dr. Galloway in his individual retirement account, (b) 10,000 shares held for the benefit of Dr. Galloway’s wife in her individual retirement account, and (c) 141,063 shares of common stock issuable upon the exercise of options exercisable within 60 days of March 1, 2022.
12. Consists of 142,717 shares of common stock issuable upon the exercise of options exercisable within 60 days of March 1, 2022.
13. Includes 1,713,425 shares of common stock issuable upon the exercise of options exercisable within 60 days of March 1, 2022.
Delinquent Section 16(a) Reports
Section 16(a) of the Exchange Act requires our directors and executive officers and all persons who beneficially own more than 10 percent of the outstanding shares of our common stock to file with the SEC initial reports of ownership and reports of changes in ownership of our common stock. Directors, executive officers and greater than 10 percent beneficial owners also are required to furnish us with copies of all Section 16(a) forms they file.
To our knowledge, based on a review of the copies of such reports and amendments to such reports furnished to us with respect to the year coveredended December 31, 2021, and based on written representations by this Annual Report.our directors and executive officers, all required Section 16 reports under the Exchange Act, for our directors, executive officers and beneficial owners of greater than 10 percent of our common stock were filed on a timely basis during the year ended December 31, 2021, except for the following, each of which were not timely filed: Forms 3 relating to Mr. Francois' and Dr. Lanchoney's respective elections to the Board on June 25, 2021, each filed on October 15, 2021; and a Form 4 relating to a July 1, 2021 grant of options and restricted stock units to Dr. Lanchoney in connection with her election to and service on the Board, filed on October 15, 2021.
ITEM 13. | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE |
Information with respect to this item will be set forth in the Proxy Statement or Form 10-K/A under the headings “CertainCertain Relationships and Related Party Transactions”Transactions
Our Audit Committee is charged with the responsibility of reviewing and “Corporate Governance”approving or ratifying all related person transactions in accordance with the Listing Rules of the Nasdaq Capital Market and other applicable law, rules and regulations and any related policies and procedures adopted by or on behalf of the Company and then in effect.
Since January 1, 2020 there has been no transaction to which we have been a party in which (i) the amount involved in the transaction exceeds $120,000 and (ii) any of our directors, executive officers or, to our knowledge, beneficial owners of more than 5% of our capital stock had or will have a direct or indirect material interest. Furthermore, no family relationships exist among any of our directors or executive officers.
Director Independence
The information required by Item 13 of Form 10-K with respect to director independence included above under the heading, "Item 10. Directors, Executive Officers and Corporate Governance — Corporate Governance," is incorporated herein by reference. The Proxy Statement or Form 10-K/A will be filed with the SEC within 120 days after the end of the fiscal year covered by this Annual Report.
ITEM 14. | PRINCIPAL ACCOUNTING FEES AND SERVICES |
Information with respect to this item will be set forth in the Proxy Statement or Form 10-K/A under the heading “Ratification of the Selection of Independent Registered Public Accounting Firm” and is incorporated herein by reference.
The Proxy Statement or Form 10-K/A will be filed with the SEC within 120 days after the end ofAudit Committee selected KPMG LLP as our independent registered public accounting firm for the fiscal year coveredending December 31, 2021. The Audit Committee's selection of KPMG LLP was ratified by this Annual Report.our stockholders at our 2021 annual meeting of stockholders.
Independent Registered Public Accounting Firm’s Fees
The table below presents fees billed to us for professional services rendered by KPMG LLP for the years ended December 31, 2021 and December 31, 2020.
| | | Aggregate Amount Billed | |
| | | 2021 | | | 2020 | |
Audit Fees | | $ | 365,000 | | | $ | 508,060 | |
Audit-Related Fees | | $ | — | | | $ | — | |
Tax Fees | | $ | — | | | $ | 80,000 | |
All Other Fees | | $ | — | | | $ | — | |
Total | | $ | 365,000 | | | $ | 588,060 | |
Tax fees billed in 2020 represent fees paid to KPMG LLP in connection with an analysis under Section 382 of the Tax Code.
Pre-Approval Policies and Procedures
The Audit Committee has adopted procedures pursuant to which all audit, audit-related, and tax services and all permissible non-audit services provided by our independent registered public accounting firm must be pre-approved by the Audit Committee. All services rendered by KPMG during 2021 and 2020 were permissible under applicable laws and regulations and were approved in advance by the Audit Committee in accordance with the rules adopted by the SEC in order to implement requirements of SOX.
PART IV
ITEM 15. | EXHIBITS, FINANCIAL STATEMENT SCHEDULES |
1. Our financial statements are included in Part II, Item 8 of Part II of this report.Annual Report.
2. All financial statement schedules have been omitted from this Item 15 as the required information is not applicable, is not present in amounts sufficient to require submission of such schedules, or because the information required is included in our financial statements or the related notes included in Part II, Item 8 of this Annual Report.
3. The exhibits set forth in the following "Index to Exhibits" are filed with, furnished with, and/or incorporated by reference into this report are listed on the Exhibit Index to this report.Annual Report, as set forth therein. A copy of any of the exhibits listedsuch exhibit will be furnished at a reasonable cost, upon receipt from any person of a written request for any such exhibit. Such request should be sent to Diffusion Pharmaceuticals Inc., 2020 Avon Court, #4,300 East Main Street, Suite 201, Charlottesville, Virginia 22902, Attention: Stockholder Information. The Exhibit Index indicates each management contract or compensatory plan or arrangement required to be filed as an exhibit to this report.General Counsel.
INDEX TO EXHIBITS
ITEM 16.Exhibit
No. | Description | Method of Filing |
3.1 | Certificate of Incorporation of Diffusion Pharmaceuticals Inc., as amended | Incorporated by reference to Exhibit 3.1 to the registrant’s annual report on Form 10-K for the year ended December 31, 2019 |
3.2 | Bylaws of Diffusion Pharmaceuticals Inc., as amended | Incorporated by reference to Exhibit 3.4 to the registrant’s annual report on Form 10-K for the year ended December 31, 2015 |
4.1 | Form of 2017 Private Placement Warrant | Incorporated by reference to Exhibit 4.1 to the registrant’s current report on Form 8-K filed on March 15, 2017 |
4.2 | Form of 2018 Common Stock Warrant | Incorporated by reference to Exhibit 4.1 to the registrant’s current report on Form 8-K filed on January 19, 2018 |
4.3 | Form of 2018 Underwriter's Warrant | Incorporated by reference to Exhibit 4.2 to the registrant’s current report on Form 8-K filed on January 22, 2018 |
4.4 | Form of May 2019 Common Stock Warrant | Incorporated by reference to Exhibit 4.1 to the registrant’s current report on Form 8-K filed on May 28, 2019 |
4.5 | Form of May 2019 Placement Agent’s Warrant | Incorporated by reference to Exhibit 4.2 to the registrant’s current report on Form 8-K filed on May 28, 2019 |
4.6 | Form of November 2019 Series I Common Stock Warrant | Incorporated by reference to Exhibit 4.1 to the registrant’s current report on Form 8-K filed on November 13, 2019 |
4.7 | Form of November 2019 Series II Common Stock Warrant | Incorporated by reference to Exhibit 4.2 to the registrant’s current report on Form 8-K filed on November 13, 2019 |
4.8 | Form of November 2019 Pre-Funded Common Stock Warrant | Incorporated by reference to Exhibit 4.3 to the registrant’s current report on Form 8-K filed on November 13, 2019 |
4.9 | Form of November 2019 Placement Agent’s Warrant | Incorporated by reference to Exhibit 4.4 to the registrant’s current report on Form 8-K filed on November 13, 2019 |
4.10 | Form of December 2019 Common Stock Warrant | Incorporated by reference to Exhibit 4.1 to the registrant’s current report on Form 8-K filed on December 13, 2019 |
4.11 | Form of December 2019 Placement Agent’s Warrant | Incorporated by reference to Exhibit 4.2 to the registrant’s current report on Form 8-K filed on December 13, 2019 |
4.12 | Form of May 2020 Common Stock Warrant | Incorporated by reference to Exhibit 4.1 to the registrant’s current report on Form 8-K filed on May 8, 2020 |
4.13 | Form of May 2020 Placement Agent's Warrant (In Respect of Exercise Transaction) | Incorporated by reference to Exhibit 4.2 to the registrant’s current report on Form 8-K filed on May 8, 2020 |
4.14 | Form of May 2020 Placement Agent's Warrant (In Respect of Offering Transaction) | Incorporated by reference to Exhibit 4.3 to the registrant’s current report on Form 8-K filed on May 20, 2020 |
4.15 | Form of February 2021 Underwriter's Warrant | Incorporated by reference to Exhibit 4.1 to the registrant’s current report on Form 8-K filed on February 18, 2021 |
4.16 | Description of Securities | Incorporated by reference to Exhibit 4.12 to the registrant's annual report on 10-K for the year ended December 31, 2019 |
10.1 | Employment Agreement dated as of September 8, 2020 by and between Robert J. Cobuzzi, Jr., Ph.D. and Diffusion Pharmaceuticals Inc.* | Incorporated by reference to Exhibit 10.2 to the registrant’s current report on Form 8-K filed on September 9, 2020 |
10.2 | Amended and Restated Employment Agreement, dated as of September 21, 2018, by and between William Karl Hornung and Diffusion Pharmaceuticals Inc. * | Incorporated by reference to Exhibit 10.2 to the registrant's Current Report on Form 8-K filed September 27, 2018 |
10.3 | Employment Agreement, dated as of October 19, 2020, by and between Christopher D. Galloway, M.D. and Diffusion Pharmaceuticals Inc. * | Incorporated by reference to Exhibit 10.1 to the registrant's Current Report on Form 8-K filed October 20, 2020 |
10.4 | Employment Agreement, dated as of September 23, 2020, by and between William Elder and Diffusion Pharmaceuticals Inc. * | Incorporated by reference to Exhibit 10.1 to the registrant's Current Report on Form 8-K filed September 25, 2020 |
10.5 | Diffusion Pharmaceuticals Inc. 2015 Equity Incentive Plan* | Incorporated by reference to Appendix C to the registrants definitive proxy statement on Schedule 14A filed on June 10, 2016 |
10.6 | Amendment No. 1 to Diffusion Pharmaceuticals Inc. 2015 Equity Incentive Plan* | Incorporated by reference to Appendix B to the registrants definitive proxy statement on Schedule 14A filed on June 10, 2016 |
10.7 | Form of Stock Option Award Agreement under 2015 Equity Incentive Plan* | Filed herewith |
10.8 | Form of Director RSU Agreement under 2015 Equity Incentive Plan* | Filed herewith |
10.9 | Form of Diffusion Pharmaceuticals LLC Stock Option Award Agreement* | Incorporated by reference to Exhibit 10.24 to the registrant’s annual report on Form 10-K for the year ended December 31, 2015 |
10.10 | Form of Indemnification Agreement between Diffusion Pharmaceuticals Inc. and each of its Directors and Officers* | Incorporated by reference to Exhibit 10.3 to the registrant’s annual report on Form 10-K for the year ended December 31, 2015 |
21.1 | Subsidiaries of Diffusion Pharmaceuticals Inc. | Filed herewith |
23.1 | Consent of KPMG LLP, independent registered public accounting firm | Filed herewith |
31.1 | Certification of Chief Executive Officer Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 and SEC Rule 13a-14(a) | Filed herewith |
31.2 | Certification of Principal Financial Officer Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 and SEC Rule 13a-14(a) | Filed herewith |
32.1 | Certification of Chief Executive Officer Pursuant to 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | Filed herewith |
101 | The following materials from the registrant’s annual report on Form 10-K for the year ended December 31, 2021, formatted in iXBRL (Inline Extensible Business Reporting Language): (i) Consolidated Balance Sheets, (ii) Consolidated Statements of Operations, (iii) Consolidated Statements of Cash Flows, and (iv) Notes to Consolidated Financial Statements | Filed herewith |
| 104 | Cover Page Interactive Data File (embedded within the Inline XBRL and contained in Exhibit 101) | |
* | Indicates a management contract or compensatory plan or arrangement. |
ITEM 16. | FORM 10-K SUMMARY |
None.
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, as amended, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
Dated: March 31, 201718, 2022 | DIFFUSION PHARMACEUTICALS INC. | |
| | By: | /s/ David G. KalergisRobert J. Cobuzzi, Jr., Ph.D. David G. KalergisRobert J. Cobuzzi, Jr., Ph.D.
Chairman andPresident, Chief Executive Officer, and Director
(Principal Executive Officer) | |
Pursuant to the requirements of the Securities Exchange Act of 1934, as amended, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Name and Signature | | Title | | Date |
| | | | |
/s/ David G. KalergisRobert J. Cobuzzi, Jr., Ph.D. | | Chairman of the Board andPresident, Chief Executive Officer, and Director
| | March 31, 201718, 2022 |
David G. KalergisRobert J. Cobuzzi, Jr., Ph.D.
| | (Principal Executive Officer) | | |
| | | | | |
/s/Ben Shealy William K. Hornung | | Senior Vice President, FinanceChief Financial Officer
| | March 31, 201718, 2022 |
Ben ShealyWilliam K. Hornung
| | (Principal Financial and Accounting Officer) | | |
| | | | | |
/s/ John L. Gainer, Ph.D.Jane H. Hollingsworth | | DirectorChair of the Board
| | March 31, 201718, 2022 |
John L. Gainer, Ph.D.
| | | | |
| | | | |
/s/ Isaac Blech
| | Vice Chairman
| | March 31, 2017
|
Isaac BlechJane H. Hollingsworth
| | | | |
| | | | | |
/s/ Robert Adams | | Director | | March 31, 201718, 2022 |
Robert Adams | | | | |
| | | | |
/s/ Eric Francois | | Director | | March 18, 2022 |
Eric Francois | | | | |
| | | | | |
/s/ Mark T. Giles | | Director | | March 31, 201718, 2022 |
Mark T. Giles | | | | |
| | | | |
/s/ Diana Lanchoney | | Director | | March 18, 2022 |
Diana Lanchoney | | | | |
| | | | | |
/s/ Alan Levin | | Director | | March 31, 201718, 2022 |
Alan Levin | | | | |
DIFFUSION PHARMACEUTICALS INC.
EXHIBIT INDEX TO ANNUAL REPORT ON FORM 10-K
FOR THE YEAR ENDED DECEMBER 31, 2016
Exhibit
No.
| | Description
| | Method of Filing
|
2.1
| | Plan of Conversion, dated June 18, 2015
| | Incorporated by reference to Exhibit 2.1 to the registrant’s current report on Form 8-K filed on June 18, 2015
|
| | | | |
2.2
| | Agreement and Plan of Merger dated as of December 15, 2015 among the Company, Arco Merger Sub, LLC and Diffusion Pharmaceuticals LLC *
| | Incorporated by reference to Exhibit 2.1 to the registrant’s current report on Form 8-K filed on December 15, 2015
|
| | | | |
3.1
| | Certificate of Incorporation of Diffusion Pharmaceuticals Inc., as amended
| | Filed herewith
|
| | | | |
3.2
| | Bylaws of Diffusion Pharmaceuticals Inc., as amended
| | Incorporated by reference to Exhibit 3.4 to the registrant’s annual report on Form 10-K for the year ended December 31, 2015
|
| | | | |
3.3
| | Certificate of Designation of Preferences, Rights and Limitations of the Series A Convertible Preferred Stock of Diffusion Pharmaceuticals Inc.
| | Incorporated by reference to Exhibit 3.1 to the registrant’s current report on Form 8-K filed on March 15, 2017
|
3.5
| | Certificate of Conversion, as filed with the Secretary of State of the State of Delaware on June 18, 2015
| | Incorporated by reference to Exhibit 3.2 to the registrant’s current report on Form 8-K filed on June 18, 2015
|
| | | | |
4.1
| | Form of Diffusion Pharmaceuticals Inc. Convertible Note Agreement
| | Incorporated by reference to Exhibit 4.1 to the registrant’s current report on Form 8-K filed on October 3, 2016
|
| | | | |
4.2
| | Form of Diffusion Pharmaceuticals LLC Convertible Note Agreement
| | Incorporated by reference to Exhibit 4.7 to the registrant’s annual report on Form 10-K for the year ended December 31, 2015
|
| | | | |
4.3
| | Form of Warrant issued to Investors in the 2017 Private Placement by Diffusion Pharmaceuticals Inc.
| | Incorporated by reference to Exhibit 4.1 to the registrant’s current report on Form 8-K filed on March 15, 2017
|
4.4
| | Form of Warrant issued to Investors in the 2014 Private Placement by the Company
| | Incorporated by reference to Exhibit 4.1 to the registrant’s current report on Form 8-K filed on April 29, 2014
|
| | | | |
4.5
| | Warrant, dated October 21, 2014, issued by the Company to Isaac Blech
| | Incorporated by reference to Exhibit 4.5 to the registrant’s annual report on Form 10-K for the fiscal year ended December 31, 2014
|
| | | | |
4.6
| | Warrant, dated April 29, 2014, issued by the Company to Sol J. Barer, Ph.D.
| | Incorporated by reference to Exhibit 4.4 to the registrant’s annual report on Form 10-K for the fiscal year ended December 31, 2014
|
| | | | |
4.7
| | Form of Registration Rights Agreement entered into by and among the Company and Investors in the 2014 Private Placement
| | Incorporated by reference to Exhibit 10.2 to the registrant’s current report on Form 8-K filed on April 29, 2014
|
| | | | |
4.8
| | Registration Rights Agreement dated November 18, 2013 between the Company and Certain Holders
| | Incorporated by reference to Exhibit 10.4 to the registrant’s current report on Form 8-K filed on November 22, 2013
|
| | | | |
10.1
| | Employment Agreement, dated as of September 6, 2016, by and between David G. Kalergis and Diffusion Pharmaceuticals Inc.**
| | Incorporated by reference to Exhibit 10.1 to the registrant’s current report on Form 8-K as filed on September 8, 2016
|
| | | | |
10.2
| | Employment Agreement, dated as of October 12, 2016, by and between Ben L. Shealy and Diffusion Pharmaceuticals Inc.**
| | Incorporated by reference to Exhibit 10.1 to the registrant’s current report on Form 8-K as filed on October 13, 2016
|
| | | | |
10.3
| | Diffusion Pharmaceuticals Inc. 2015 Equity Incentive Plan**
| | Incorporated by reference to Exhibit 10.2 to the registrant’s current report on Form 8-K as filed on June 18, 2015
|
| | | | |
10.4
| | Amendment No. 1 to Diffusion Pharmaceuticals Inc. 2015 Equity Incentive Plan**
| | Incorporated by reference to Appendix B to the registrant’s definitive proxy statement on Schedule 14A filed on June 10, 2016
|
| | | | |
10.5
| | Form of Diffusion Pharmaceuticals Inc. Stock Option Award Agreement**
| | Filed herewith
|
10.6
| | Form of Diffusion Pharmaceuticals LLC Stock Option Award Agreement**
| | Incorporated by reference to Exhibit 10.22 to the registrant’s annual report on Form 10-K for the year ended December 31, 2015
|
| | | | |
10.7
| | Form of 2015 Incentive Stock Option Agreement under the Diffusion Pharmaceuticals Inc. 2015 Equity Incentive Plan**
| | Incorporated by reference to Exhibit 10.3 to the registrant’s current report on Form 8-K filed on June 18, 2015
|
| | | | |
10.8
| | Form of 2015 Non-Statutory Stock Option Agreement under the Diffusion Pharmaceuticals Inc. 2015 Equity Incentive Plan**
| | Incorporated by reference to Exhibit 10.4 to the registrant’s current report on Form 8-K filed on June 18, 2015
|
| | | | |
10.9
| | Form of Stock Option Agreement between the Company and certain former Executive Officers**
| | Incorporated by reference to Exhibit 10.12 to the registrant’s annual report on Form 10-K for the fiscal year ended December 31, 2014
|
| | | | |
10.10
| | Form of Stock Option Agreement between the Company and certain former Directors**
| | Incorporated by reference to Exhibit 10.13 to the registrant’s annual report on Form 10-K for the fiscal year ended December 31, 2014
|
| | | | |
10.11
| | Form of Indemnification Agreement between Diffusion Pharmaceuticals Inc. and each of its Directors and Officers**
| | Incorporated by reference to Exhibit 10.3 to the registrant’s annual report on Form 10-K for the year ended December 31, 2015
|
| | | | |
10.12
| | Resignation Agreement, dated April 30, 2015, between the Company and Yael Schwartz, Ph.D.**
| | Incorporated by reference to Exhibit 10.1 to the registrant’s current report on Form 8-K filed on May 1, 2015
|
| | | | |
10.13
| | Placement Agency Agreement, dated January 27, 2017, by and between Diffusion Pharmaceuticals Inc. and Maxim Merchant Capital, a division of Maxim Group LLC
| | Incorporated by reference to Exhibit 10.1 to the registrant’s current report on Form 8-K filed on March 15, 2017
|
| | | | |
10.14
| | Form of 2017 Private Placement Subscription Agreement
| | Incorporated by reference to Exhibit 10.2 to the registrant’s current report on Form 8-K filed on March 15, 2017
|
| | | | |
10.15
| | Settlement Agreement, dated September 27, 2016, by and among Diffusion Pharmaceuticals Inc., Diffusion Pharmaceuticals LLC, David Schmidt and the other parties thereto
| | Incorporated by reference to Exhibit 10.1 to the registrant’s current report on Form 8-K filed on October 3, 2016
|
| | | | |
10.16
| | Contingent Value Rights Agreement, dated as of January 8, 2016, by and between Diffusion Pharmaceuticals Inc. and Computershare, Inc., as Rights Agent
| | Incorporated by reference to Exhibit 10.2 to the registrant’s current report on Form 8-K filed on January 8, 2016
|
| | | | |
10.17 | | Amendment to the Placement Agency Agreement, dated March 14, 2017, by and between Diffusion Pharmaceuticals Inc. and Maxim Merchant Capital, advisor of Maxim Group LLC | | Filed herewith |
21.1
| | Subsidiaries of Diffusion Pharmaceuticals Inc.
| | Filed herewith
|
| | | | |
23.1
| | Consent of KPMG LLP, independent registered public accounting firm
| | Filed herewith
|
| | | | |
31.1
| | Certification of Chief Executive Officer Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 and SEC Rule 13a-14(a)
| | Filed herewith
|
| | | | |
31.2
| | Certification of Principal Financial Officer Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 and SEC Rule 13a-14(a)
| | Filed herewith
|
| | | | |
32.1
| | Certification of Chief Executive Officer Pursuant to 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002
| | Furnished herewith
|
| | | | |
101
| | The following materials from the registrant’s annual report on Form 10-K for the year ended December 31, 2016, formatted in XBRL (Extensible Business Reporting Language): (i) Consolidated Balance Sheets, (ii) Consolidated Statements of Operations, (iii) Consolidated Statements of Cash Flows, and (iv) Notes to Consolidated Financial Statements
| | Filed herewith
|
*
| All exhibits and schedules to this agreement have been omitted pursuant to Item 601(b)(2) of Regulation S-K. The registrant will furnish the omitted exhibits and schedules to the SEC upon request by the SEC.
|
| |
**
| A management contract or compensatory plan or arrangement.
|
97