This Annual Report on Form 10-K, or this Annual Report, includes our trademarks, trade names and service marks, such as Liquidia, the Liquidia logo, YUTREPIA and PRINT, or Particle Replication In Non-wetting Templates, which are protected under applicable intellectual property laws and are the property of Liquidia Corporation.Technologies, Inc. This Annual Report also contains trademarks, trade names and service marks of other companies, which are the property of their respective owners. Solely for convenience, trademarks, trade names and service marks referred to in this Annual Report on Form 10-K may appear without the ®, ™ or SM symbols, but such references are not intended to indicate, in any way, that we will not assert, to the fullest extent under applicable law, our rights or the right of the applicable licensor to these trademarks, trade names and service marks. We do not intend our use or display of other parties’ trademarks, trade names or service marks to imply, and such use or display should not be construed to imply, a relationship with, or endorsement or sponsorship of us by, these other parties.
Manufacturers and certain other entities involved in the manufacturing and distribution of approved products are required to register their establishments with the FDA and certain state agencies and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP and other laws. The cGMP requirements apply to all stages of the manufacturing process, including the production, processing, sterilization, packaging, labeling, storage and shipment of the product. Manufacturers must establish validated systems to ensure that products meet specifications and regulatory standards and test each product batch or lot prior to its release. Combination products are subject to FDA regulation to ensure the quality of both the constituent parts and the finished product.
Changes to the manufacturing process are strictly regulated and often require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP and impose reporting and documentation requirements upon the sponsor and any third-party manufacturers that the sponsor may decide to use. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain cGMP compliance.
The FDA may impose a number of post-approval requirements as a condition of approval of an application. For example, the FDA may require post-marketing testing, including Phase 4 clinical trials, and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization.
The FDA may withdraw a product approval if compliance with regulatory requirements is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, problems with manufacturing processes, or failure to comply with regulatory requirements, may result in restrictions on the product or even complete withdrawal of the product from the market.
Potential implications include required revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical trials to assess new safety risks; or imposition of distribution or other restrictions under a REMS program. Other potential consequences include, among other things:
The FDA strictly regulates marketing, labeling, advertising, and promotion of products that are placed on the market. Drugs may be promoted only for the approved indications and in accordance with the provisions of the approved label. As a compliance best practice and risk mitigation measure, pharmaceutical companies typically train their sales force regarding the limitations on promotion of products relative to their approved indications for use and concerns regarding potential “off-label promotion.” However, a physician may use products off-label when, in the physician’s independent professional medical judgment, he or she deems it appropriate. Recent court decisions have impacted FDA’s enforcement activity regarding off-label promotion in the light of First Amendment considerations; however, there are still significant risks in this area in part due to the potential for False Claims Act exposure. Further, the FDA as not materially changed its position on off-label promotion following legal setbacks on First Amendment grounds and the U.S. Department of Justice has consistently asserted in False Claims Act briefings that “speech serves as a conduit for violations of the law is not constitutionally protected.”
A primary trend in the U.S. healthcare industry and elsewhere is cost containment. There have been a number of federal and state proposals during the last few years regarding the pricing of pharmaceutical and biopharmaceutical products, limiting coverage and reimbursement for drugs and other medical products, government control and other changes to the healthcare system in the United States including the Patient Protection and Affordable Care Act (ACA).
In the future, there may continue to be additional proposals relating to the reform of the U.S. healthcare system, some of which could further limit the prices we will be able to charge for our product candidates, or the amounts of reimbursement available for our product candidates. If future legislation were to impose direct governmental price controls or access restrictions, it could have a significant adverse impact on our business. Managed care organizations, as well as Medicaid and other government agencies, continue to seek price discounts. Some states have implemented, and other states are considering, measures to reduce costs of the Medicaid program, and some states are considering implementing measures that would apply to broader segments of their populations that are not Medicaid-eligible. Due to the volatility in the current economic and market dynamics, we are unable to predict the impact of any unforeseen or unknown legislative, regulatory, payor or policy actions, which may include cost containment and healthcare reform measures. Such policy actions could have a material adverse impact on our profitability.
These and other healthcare reform initiatives may result in additional reductions in Medicare and other healthcare funding, which could have a material adverse effect on our financial operations. We expect that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for our product candidates or additional pricing pressures.
In order to market any product outside of the United States, we will need to comply with numerous and varying regulatory requirements of other countries and jurisdictions regarding development, approval, commercial sales and distribution of our products, and governing, among other things, clinical trials, marketing authorization, commercial sales and distribution of our products, if approved. Whether or not we obtain FDA approval for a product, we must obtain the necessary approvals by the comparable regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those countries. The approval process varies between countries and jurisdictions and can involve additional product testing and additional administrative review periods. The time required to obtain approval in other countries and jurisdictions might differ from and be longer than that required to obtain FDA approval. Regulatory approval in one country or jurisdiction does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country or jurisdiction may negatively impact the regulatory process in others.
Item 1A. Risk Factors
The following is a summary of the principal risk factors described in this section:
Since our incorporation, we have invested heavily in the development of our product candidates and technologies, as well as in recruiting management and scientific personnel. To date, we have not commenced the commercialization of our product candidates and all of our pre-acquisition revenue has been derived from up-front fees and milestone payments made to us in connection with licensing and collaboration arrangements we have entered into.into and the Promotion Agreement, under which we share in the profit derived from the sale of Treprostinil Injection in the United States. These up-front fees and milestone payments have been, and combined with revenue generated from the ProductTreprostinil Injection may continue to be, insufficient to match our operating expenses. We expect to continue to devote substantial financial and other resources to the clinical development of our product candidates and, as a result, must generate significant revenue to achieve and maintain profitability or raise additional capital to fund clinical development. We may continue to incur losses and negative cash flow and may never transition to profitability or positive cash flow.
Remote work policies, quarantines, shelter-in-place and similar government orders, shutdowns or other restrictions on the conduct of business operations related to the COVID-19 pandemic may negatively impact productivity and our research and development activities, the magnitude of which will depend, in part, on the length and severity of the restrictions and other limitations on our ability to conduct our business in the ordinary course. In addition, although our employees are accustomed to working remotely, changes in internal controls due to remote work arrangements may result in control deficiencies in the preparation of our financial reports, which could be material. Currently, most of our employees are working remotely, with only essential personnel working on site as needed to produce LIQ861 and prepare for a pre-approval inspection by the FDA.
The spread of COVID-19, which has caused a broad impact globally, may materially affect us economically. While the potential economic impact brought by, and the duration of, COVID-19 may be difficult to assess or predict, a widespread pandemic could result in significant disruption of global financial markets, reducing our ability to access capital, which could in the future negatively affect our liquidity. In addition, a recession or market correction resulting from the spread of COVID-19 could materially affect our business and the value of our common stock.
Our common stock has been listed on the Nasdaq Capital Market under the symbol “LQDA” since November 19, 2020. Between July 26, 2018 and November 18, 2019,2020, the common stock of Liquidia Technologies, our wholly owned subsidiary and predecessor-in-interest for SEC reporting purposes, was listed on the Nasdaq Capital Market under the symbol “LQDA.” Prior to July 26, 2018, there was no established public trading market for our common stock. As of March 15, 2021, the closing price of our common stock was $2.90 per share.
We have never paid any cash dividends on our capital stock. We anticipate that we will retain earnings, if any, to support operations and to finance the growth and development of our business. In addition, the terms of our LSARIFA with SVBHCR precludes us from paying cash dividends, except in certain prescribed circumstances, without the prior written consent of SVB.HCR. Therefore, we do not expect to pay cash dividends for the foreseeable future.
Information regarding equity compensation plans is set forth in Item 12 of this Annual Report on Form 10-K and is incorporated herein by reference.
Not applicable.
Item 6. Selected Financial Data.[Reserved].
Our primary uses of capital are, and we expect will continue to be, compensation and related personnel expenses, clinical costs, manufacturing process development costs, external research and development services, laboratory and related supplies, legal and other regulatory expenses, legal costs, administrative and overhead costs and debt service.repayments under the RIFA. We also expect to incur significant commercialization expenses related to product manufacturing, sales, marketing and distribution as we prepare to potentially receive regulatory approval for YUTREPIA. Our future funding requirements will be heavily determined by the timing of the potential commercialization of YUTREPIA and the resources needed to support development of our product candidates. Additionally, as a publicly traded company we incur significant legal, accounting and other expenses. In addition,If the Sarbanes-Oxley Act, as well as rules adoptedCompany is unable to access the contingent Investment Amounts from the RIFA or generate substantial YUTREPIA product revenue by the SEC and Nasdaq Stock Market LLC (“Nasdaq”)second quarter of 2024, the Company will require public companies to implement specified corporate governance practices.
We may raise additional capital through licensing activities, other business arrangements or the sale of equity or convertible debt securities. In such an event, the ownership of our existing shareholders will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect the rights associated with holdings of our common stock.
Because of the numerous risks and uncertainties associated with research, development and commercialization of pharmaceuticals, we are unable to estimate the exact amount of our working capital requirements. Our future funding requirements will depend on many factors, including:
PART IV
PARTIV
Item15. Exhibits and Financial Statement Schedules.
Financial Statement Schedules
| (a) | The following documents are filed as part of this Annual Report on Form 10-K: |
| (2) | Financial Statement Schedules. |
All schedules are omitted as the information required is inapplicable or the information is presented in the consolidated financial statements or the related notes.
| (3) | Required information is included in the notes to the financial statements.
|
See Exhibit Index below.
| (b) | The following exhibits are filed as part of this Annual Report on Form 10-K. |
Exhibit
No.
| | Description | 2.1 | | Agreement and Plan of Merger, dated as of June 29, 2020, by and among the Company, Liquidia Technologies, Inc., RareGen, LLC, Gemini Merger Sub I, Inc., Gemini Merger Sub II, LLC and PBM RG Holdings, LLC (incorporated by reference to Exhibit 2.1 of the Company’s Registration Statement on Form S-4, filed with the SEC on August 5, 2020). | 2.2 | | Limited Waiver and Modification to Agreement and Plan of Merger, dated as of August 3, 2020, by and among the Company, Liquidia Technologies, Inc., RareGen, LLC, Gemini Merger Sub I, Inc., Gemini Merger Sub II, LLC and PBM RG Holdings, LLC (incorporated by reference to Exhibit 2.2 of the Company’s Registration Statement on Form S-4, filed with the SEC on August 5, 2020). | 3.1 | | Certificate of Incorporation of Liquidia Corporation (incorporated by reference to Exhibit 3.1 of the Company’s Registration Statement on Form S-4, filed with the SEC on August 5, 2020). | 3.2 | | Bylaws of Liquidia Corporation (incorporated by reference to Exhibit 3.2 of the Company’s Registration Statement on Form S-4, filed with the SEC on August 5, 2020). | 4.1 | | Form of Specimen Common Stock Certificate of Liquidia Corporation (incorporated by reference to Exhibit 4.1 of the Company’s Registration Statement on Form S-4, filed with the SEC on August 5, 2020). | 4.2 | | Form of Warrant to Purchase Shares of Preferred Stock, issued by Liquidia Technologies, Inc. in January 2017 and February 2017 (incorporated herein by reference to Exhibit 4.4 to Liquidia Technologies, Inc.’s’s Registration Statement on Form S-1, filed with the SEC on June 28, 2018). | 4.3 | | Seventh Amended and Restated Investors’Investors’ Rights Agreement, dated as of February 2, 2018, by and among the Company, the Investors party thereto and the Common Holders party thereto (incorporated herein by reference to Exhibit 4.5 to Liquidia Technologies, Inc.’s’s Registration Statement on Form S-1, filed with the SEC on June 28, 2018). |
4.4 | | Warrant to Purchase Stock, issued February 26, 2021, by Liquidia Corporation to Silicon Valley Bank (incorporated herein by reference to Exhibit 4.1 to the Company’s Current Report on Form 8-K, filed with the SEC on March 3, 2021). | 4.5*4.5
| | Warrant to Purchase Stock, dated as of January 7, 2022, by and between Liquidia Corporation and Silicon Valley Bank (incorporated herein by reference to Exhibit 4.1 to the Company’s Current Report on Form 8-K, filed with the SEC on January 11, 2022). | 4.6 | | Warrant to Purchase Stock, dated as of January 7, 2022, by and between Liquidia Corporation and SVB Innovation Credit Fund VIII, L.P. (incorporated herein by reference to Exhibit 4.2 to the Company’s Current Report on Form 8-K, filed with the SEC on January 11, 2022). | 4.7 | | Warrant to Purchase Stock, dated as of January 7, 2022, by and between Liquidia Corporation and Innovation Credit Fund VIII-A L.P. (incorporated herein by reference to Exhibit 4.3 to the Company’s Current Report on Form 8-K, filed with the SEC on January 11, 2022). | 4.8 | | Description of Securities of the Company.Company (incorporated herein by reference to Exhibit 4.5 to the Company’s Annual Report on Form 10-K, filed with SEC on March 25, 2021). | 10.1# | | Liquidia Technologies, Inc. Stock Option Plan (2004), as amended, and forms of award agreements thereunder (incorporated herein by reference to Exhibit 10.1 to Liquidia Technologies, Inc.’s’s Annual Report on Form 10-K, filed with the SEC on February 26, 2019). | 10.2# | | Liquidia Technologies, Inc. 2016 Equity Incentive Plan, as amended, and forms of award agreements thereunder (incorporated herein by reference to Exhibit 10.2 to Liquidia Technologies, Inc.’s’s Registration Statement on Form S-1, filed with the SEC on June 28, 2018). | 10.3# | | Liquidia Technologies, Inc. 2018 Long-Term Incentive Plan, and forms of award agreements thereunder (incorporated herein by reference to Exhibit 99.3 to Liquidia Technologies, Inc.’s’s Registration Statement on Form S-8, filed with the SEC on July 26, 2018). |
10.4#*10.4#
| | Liquidia Corporation 2020 Long-Term Incentive Plan, and forms of award agreements thereunder.thereunder (incorporated by reference to Exhibit 10.4 to the Company’s Annual Report on Form 10-K, filed with the SEC on March 25, 2021). | 10.5# | | Amendment to the Liquidia Corporation 2020 Long-Term Incentive Plan (incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K, filed with the SEC on June 17, 2022). | 10.6# | | Liquidia Corporation 2022 Inducement Plan (incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K, filed with the SEC on January 31, 2022). | 10.7# | | Form of Stock Option Grant Notice and Stock Option Agreement under the 2022 Inducement Plan (incorporated herein by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K, filed with the SEC on January 31, 2022). | 10.8# | | Form of Indemnification Agreement with the Company’sCompany’s executive officers and directors (incorporated herein by reference to Exhibit 10.2 to the Company’sCompany’s Current Report on 8-K12B, filed with the SEC on November 18, 2020). | 10.610.9
| | Litigation Funding and Indemnification Agreement, dated as of November 17, 2020, by and between RareGen, LLC and PBM RG Holdings, LLC (incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K12B, filed with the SEC on November 18, 2020). | 10.710.10
| | Form of Lock-Up Agreement by and among the Company, Liquidia Technologies, Inc. and each of the RareGen members party thereto (incorporated herein by reference to Exhibit 10.7 to the Company’s Registration Statement on Form S-4, filed with the SEC on August 5, 2020). | 10.810.11++*
| | Loan and SecurityRevenue Interest Financing Agreement, dated as of February 26, 2021,January 9, 2023, by and among Silicon Valley Bank, Liquidia Corporation, Liquidia Technologies, Inc., Healthcare Royalty Partners IV, L.P., and Liquidia PAH, LLC (incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K, filed with the SEC on March 3, 2021).HCR Collateral Management, LLC.
| 10.9+10.12+
| | Inhaled Collaboration and Option Agreement, dated as of June 15, 2012, by and between Liquidia Technologies, Inc. and Glaxo Group Limited (incorporated herein by reference to Exhibit 10.14 to Liquidia Technologies, Inc.’s’s Registration Statement on Form S-1, filed with the SEC on June 28, 2018). | 10.10+10.13+
| | Amendment No. 1 to the Inhaled Collaboration and Option Agreement, dated as of May 13, 2015, by and between Liquidia Technologies, Inc. and Glaxo Group Limited (incorporated herein by reference to Exhibit 10.15 to Liquidia Technologies, Inc.’s’s Registration Statement on Form S-1, filed with the SEC on June 28, 2018). | 10.11+10.14+
| | Second Amendment to the Inhaled Collaboration and Option Agreement, dated as of November 19, 2015, by and between Liquidia Technologies, Inc. and Glaxo Group Limited (incorporated herein by reference to |
| | Exhibit 10.16 to Liquidia Technologies, Inc.’s’s Registration Statement on Form S-1, filed with the SEC on June 28, 2018). | 10.12+10.15++
| | Amendment No. 3 to the Inhaled Collaboration and Option Agreement, effective as of June 24, 2019, by and between Liquidia Technologies, Inc. and Glaxo Group Limited (incorporated herein by reference to Exhibit 10.1 to Liquidia Technologies, Inc.’s’s Current Report on Form 8-K, filed with the SEC on June 28, 2019). | 10.13+10.16+
| | Amended and Restated License Agreement, dated as of December 15, 2008, by and between Liquidia Technologies, Inc. and The University of North Carolina at Chapel Hill (incorporated herein by reference to Exhibit 10.17 to Liquidia Technologies, Inc.’s’s Registration Statement on Form S-1, filed with the SEC on June 28, 2018). | 10.14+10.17+
| | First Amendment to Amended and Restated License Agreement, dated as of June 8, 2009, by and between Liquidia Technologies, Inc. and The University of North Carolina at Chapel Hill (incorporated herein by reference to Exhibit 10.18 to Liquidia Technologies, Inc.’s’s Registration Statement on Form S-1, filed with the SEC on June 28, 2018). | 10.1510.18
| | 6th Amendment to Amended and Restated License Agreement, dated as of June 10, 2016, by and between Liquidia Technologies, Inc. and The University of North Carolina at Chapel Hill (incorporated herein by reference to Exhibit 10.19 to Liquidia Technologies, Inc.’s’s Registration Statement on Form S 1,S-1, filed with the SEC on June 28, 2018). | 10.16+10.19+
| | Manufacturing Development and Scale-up Agreement, dated as of March 19, 2012, by and between Liquidia Technologies, Inc. and Chasm Technologies, Inc. (incorporated herein by reference to Exhibit 10.20 to Liquidia Technologies, Inc.’s’s Registration Statement on Form S-1, filed with the SEC on June 28, 2018). | 10.17+10.20+
| | 1st Amendment to Manufacturing Development and Scale up Agreement, dated as of May 25, 2017, by and between Liquidia Technologies, Inc. and Chasm Technologies, Inc. (incorporated herein by reference to Exhibit 10.21 to Liquidia Technologies, Inc.’s’s Registration Statement on Form S-1, filed with the SEC on June 28, 2018). | 10.18#10.21#
| | Severance Agreement and General Release, dated as of January 13, 2021, by and between Liquidia Technologies, Inc. and Neal Fowler (incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K, filed with the SEC on January 14, 2021).
| 10.19#
| | Executive Employment Agreement, dated as of December 14, 2020, by and between Liquidia Technologies, Inc. and Damian deGoa (incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K, filed with the SEC on December 16, 2020).
| 10.20#
| | Nonstatutory Stock Option Inducement Award Agreement, dated as of December 15, 2020, by and between the Company and Damian deGoa (incorporated herein by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K, filed with the SEC on December 16, 2020). | 10.21#10.22#
| | Separation Agreement and General Release, dated as of January 31, 2022, by and between Liquidia Technologies, Inc. and Damian deGoa (incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K, filed with the SEC on February 4, 2022). | 10.23# | | Executive Employment Agreement, dated as of January 3, 2022, by and between Liquidia Corporation and Roger A. Jeffs, Ph.D. (incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K, filed with the SEC on January 4, 2022). | 10.24# | | Executive Employment Agreement, dated as of November 30, 2020, by and between Liquidia Technologies, Inc. and Michael Kaseta (incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K, filed with the SEC on December 1, 2020). | 10.22#10.25#
| | Amended and Restated Executive Employment Agreement, dated as of July 25, 2018, by and between Liquidia Technologies, Inc. and Robert Lippe (incorporated herein by reference to Exhibit 10.2 to Liquidia Technologies, Inc.’s’s Current Report on Form 8-K, filed with the SEC on July 30, 2018). | 10.23#*10.26#
| | Executive EmploymentSeverance Agreement and General Release, dated as of May 18, 2020,June 28, 2022, by and between Liquidia Technologies, Inc. and Tushar Shah.Shah, M.D (incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K, filed with the SEC on July 1, 2022).
|
10.2410.27#
| | Executive Employment Agreement, dated as of June 13, 2022, by and between Liquidia Technologies, Inc. and Rajeev Saggar (incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K, filed with the SEC on June 22, 2022). | 10.28 | | Cooperation Agreement by and among the Company, Liquidia Technologies, Inc., PBM Capital Finance, LLC and PD Joint Holdings, LLC Series 2016-A, dated as of June 29, 2020 (incorporated by reference to Exhibit 10.5 of the Company’s Registration Statement on Form S-4, filed with the SEC on August 5, 2020). | 10.2510.29
| | Cooperation Agreement by and among the Company, Liquidia Technologies, Inc. and Serendipity BioPharma LLC, dated as of June 29, 2020 (incorporated by reference to Exhibit 10.6 of the Company’s Registration Statement on Form S-4, filed with the SEC on August 5, 2020). |
10.30 | | Standstill Agreement, dated as of April 12, 2021, by and among Liquidia Corporation and the Purchasers party thereto (incorporated herein by reference to Exhibit 10.3 to the Company’s Current Report on Form 8-K, filed with the SEC on April 13, 2021). | 10.26#10.31#
| | Liquidia Corporation 2020 Employee Stock Purchase Plan (incorporated herein by reference to Exhibit 10.12 to the Company’s Registration Statement on Form S-4, filed with the SEC on August 5, 2020). | 10.27#10.32#
| | Amendment No. 1 to the Liquidia Corporation 2020 Employee Stock Purchase Plan (incorporated herein by reference to Exhibit 10.36 to the Company’s Annual Report on Form 10-K, filed with the SEC on March 17, 2022). | 10.33# | | Liquidia Corporation Annual Cash Bonus Plan (incorporated herein by reference to Exhibit 10.32 to the Company’s Registration Statement on Form S-4, filed with the SEC on August 5, 2020). | 10.28#*10.34#
| | Liquidia Corporation Executive Severance and Change in Control Plan.Plan (incorporated herein by reference to Exhibit 10.28 to the Company’s Annual Report on Form 10-K, filed with the SEC on March 25, 2021). | 10.2910.35
| | Lease Agreement, dated as of June 29, 2007, by and between Liquidia Technologies, Inc. and Durham KTP Tech 4, LLC, as amended (incorporated herein by reference to Exhibit 10.34 to the Company’sCompany’s Registration Statement on Form S-4, filed with the SEC on August 5, 2020). | 10.30+10.36++
| | Promotion Agreement, dated as of August 1, 2018, by and between RareGen, LLC and Sandoz Inc. (incorporated herein by reference to Exhibit 10.36 to the Company’s Registration Statement on Form S-4, filed with the SEC on August 5, 2020). | 10.31+10.37++
| | First Amendment to Promotion Agreement, dated as of May 8, 2020, by and between RareGen, LLC and Sandoz Inc. (incorporated herein by reference to Exhibit 10.37 to the Company’s Registration Statement on Form S-4, filed with the SEC on August 5, 2020). | 10.3210.38
| | Second Amendment to Promotion Agreement, dated as of September 4, 2020, by and between RareGen, LLC and Sandoz Inc. (incorporated herein by reference to Exhibit 10.38 to Amendment No. 1 to the Company’s Registration Statement on Form S-4, filed on September 4, 2020). | 10.3310.39++*
| | Third Amendment to Promotion Agreement, dated as of November 18, 2022 by and between Liquidia PAH, LLC and Sandoz Inc. | 10.40 | | Joint Development Agreement, dated May 3, 2019, between RareGen, LLC and Carelife USA Inc. (incorporated herein by reference to Exhibit 10.4010.39 to the Company’s Registration Statement on Form S-4, filed with the SEC on August 5, 2020). | 10.41++ | | LIQ861 API Supply Agreement, dated as of January 10, 2020, by and among LGM Pharma LLC, Yonsung Fine Chemicals Co. Ltd. and Liquidia Technologies, Inc. (incorporated herein by reference to Exhibit 10.44 to the Company’s Annual Report on Form 10-K, filed with the SEC on March 17, 2022). | 10.42++ | | Commercial Manufacturing Services and Supply Agreement, dated November 12, 2020, by and between Liquidia Technologies, Inc. and Xcelience, LLC (now Lonza Tampa, LLC) (incorporated herein by reference to Exhibit 10.45 to the Company’s Annual Report on Form 10-K, filed with the SEC on March 17, 2022). | 10.43++* | | Device Development and Supply Agreement, dated as of December 1, 2022, by and among Mainbridge Health Partners, LLC, Sandoz Inc. and Liquidia PAH, LLC. | 21.1* | | Subsidiaries of Liquidia Corporation. | 23.1* | | Consent of PricewaterhouseCoopers LLP, independent Registered Public Accounting Firm. | 31.1* | | Certification of Principal Executive Officer pursuant to Rules 13a-14(a) and 15d-14(a), as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | 31.2* | | Certification of Principal Financial Officer pursuant to Rules 13a-14(a) and 15d-14(a), as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | 32.1** | | Certification of Principal Executive Officer pursuant to 18 U.S.C Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | 32.2** | | Certification of Principal Financial Officer pursuant to 18 U.S.C Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | 101*101.INS*
| | The following materials from Liquidia Corporation’s Annual Report on Form 10-K for the year ended December 31, 2020, formatted in Inline eXtensible Business Reporting Language (iXBRL): (i) Consolidated Balance Sheets as of December 31, 2020 and 2019, (ii) Consolidated Statements of Operations and Comprehensive Loss for the years ended December 31, 2020 and 2019 (iii) Consolidated Statements of Stockholders’ Equity for the years ended December 31, 2020 and 2019, (iv) Consolidated Statements of Cash Flows for the years ended December 31, 2020 and 2019 and (v) Notes to Consolidated Financial Statements.XBRL Instance Document
| 104*101.SCH* | | Inline XBRL Taxonomy Extension Schema Document |
101.CAL* | | Inline XBRL Taxonomy Extension Calculation Linkbase Document | 101.DEF* | | Inline XBRL Taxonomy Extension Definition Linkbase Document | 101.LAB* | | Inline XBRL Taxonomy Extension Label Linkbase Document | 104* | | Cover Page Interactive Data File (formatted as Inline XBRL and Contained in Exhibit 101). |
+ Confidential treatment has been granted with respect as to certain portions of this exhibit. Such portions have been redacted and submitted separately to the SEC. ++ Portions of this exhibit have been redacted in compliance with Regulation S-K Item 601(b)(10). The omitted information is not material and would likely cause competitive harm to the Company if publicly disclosed. ** | FiledFurnished herewith.
|
**#
| Furnished herewith.Indicates management contract or compensatory plan.
|
#
| Indicates management contract or compensatory plan.
|
Item16. Form 10-K Summary. None. SIGNATURES Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized. | Liquidia Corporation | | | Date: March 25, 202120, 2023 | By: | /s/ Damian deGoaRoger A. Jeffs, Ph.D. | | Name: | Damian deGoaRoger A. Jeffs, Ph.D.
| | Title:
| Chief Executive OfficerTitle:
| Chief Executive Officer |
Pursuant to the requirements of the Securities Exchange Act of 1934, report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated: Name | | Position |
| Date | | | | | | /s/ Damian deGoaRoger A. Jeffs, Ph.D. | | Director and Chief Executive Officer | | March 25, 202120, 2023 | Damian deGoaRoger A. Jeffs, Ph.D.
| | (Principal Executive Officer) | | | | | | | | /s/ Michael Kaseta | | Chief Financial Officer | | March 25, 202120, 2023 | Michael Kaseta | | (Principal Financial and Accounting Officer) | | | | | | | | /s/ Dr. Stephen Bloch | | Chairman of the Board of Directors | | March 25, 202120, 2023 | Dr. Stephen Bloch | | | | | | | | | | /s/ Damian deGoa | | Director | | March 20, 2023 | Damian deGoa | | | | | | | | | | /s/ Katherine Rielly-Gauvin | | Director | | March 25, 202120, 2023 | Katherine Rielly-Gauvin | | | | | | | | | | /s/ Dr. Joanna Horobin | | Director | | March 25, 202120, 2023 | Dr. Joanna Horobin | | | | | | | | | | /s/ Roger A. Jeffs, Ph.D.David Johnson | | Director | | March 25, 202120, 2023 | Roger A. Jeffs, Ph.D.David Johnson
| | | | | | | | | | /s/ Arthur Kirsch | | Director | | March 25, 202120, 2023 | Arthur Kirsch | | | | | | | | | | /s/Paul B. Manning | | Director | | March 25, 202120, 2023 | Paul B. Manning | | | | | | | | | | /s/ Raman Singh | | Director | | March 25, 202120, 2023 | Raman Singh | | | |
| | | |
LIQUIDIA CORPORATION FINANCIAL STATEMENTS TABLE OF CONTENTS
Report of Independent Registered Public Accounting Firm To the Board of Directors and Stockholders of Liquidia Corporation Opinion on the Financial Statements We have audited the accompanying consolidated balance sheets of Liquidia Corporation and its subsidiaries (the “Company”) as of December 31, 20202022 and 2019,2021, and the related consolidated statements of operations and comprehensive loss, of stockholders’ equity and of cash flows for the years then ended, including the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 20202022 and 2019,2021, and the results of its operations and its cash flows for the years then ended in conformity with accounting principles generally accepted in the United States of America. Change in Accounting Principle
As discussed in Note 2 to the financial statements, the Company changed the manner in which it accounts for leases in 2019.
Basis for Opinion These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB. We conducted our audits of these consolidated financial statements in accordance with the standards of the PCAOB. Those standards require that we plan and perform the auditsaudit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion. Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion. Emphasis of Matter As discussed in Note 1 to the consolidated financial statements, the Company willmay require additional financing to fund future operations. Management’s evaluation of the events and conditions and plans to mitigate this matter are also described in Note 1. /s/ PricewaterhouseCoopers LLP Raleigh, North Carolina March 25, 202120, 2023 We have served as the Company’s auditor since 2014. Liquidia Corporation Consolidated Balance Sheets (in thousands, except share and per share data) | | | December 31, 2020 | | | December 31, 2019 | | Assets | | | | | | | | | Current assets: | | | | | | | | | Cash | | $ | 65,316,481 | | | $ | 55,796,378 | | Prepaid expenses and other current assets | | | 752,447 | | | | 590,251 | | Total current assets | | | 66,068,928 | | | | 56,386,629 | | Property, plant and equipment, net | | | 6,805,570 | | | | 9,253,965 | | Operating lease right-of-use assets, net | | | 2,649,328 | | | | 2,823,430 | | Indemnification asset, related party | | | 1,387,275 | | | | 0 | | Contract acquisition costs, net | | | 12,792,491 | | | | 0 | | Intangible asset, net | | | 5,534,843 | | | | 0 | | Goodwill | | | 3,903,282 | | | | 0 | | Other assets | | | 390,043 | | | | 378,043 | | Total assets | | $ | 99,531,760 | | | $ | 68,842,067 | | Liabilities and stockholders’ equity | | | | | | | | | Current liabilities: | | | | | | | | | Accounts payable | | $ | 3,734,227 | | | $ | 3,498,043 | | Accrued compensation | | | 3,259,515 | | | | 3,164,687 | | Accrued stock offering costs | | | 0 | | | | 1,289,413 | | Other accrued expenses | | | 1,386,880 | | | | 1,525,919 | | Refund liability | | | 1,768,864 | | | | 0 | | Current portion of operating lease liabilities | | | 664,670 | | | | 566,390 | | Current portion of finance lease liabilities | | | 923,218 | | | | 1,244,229 | | Current portion of long-term debt | | | 0 | | | | 5,585,637 | | Total current liabilities | | | 11,737,374 | | | | 16,874,318 | | Litigation finance payable | | | 1,154,360 | | | | 0 | | Long-term operating lease liabilities | | | 5,006,301 | | | | 5,670,971 | | Long-term finance lease liabilities | | | 255,402 | | | | 1,056,747 | | Long-term debt | | | 10,292,485 | | | | 10,292,484 | | Total liabilities | | | 28,445,922 | | | | 33,894,520 | | Commitments and contingencies | | | | | | | | | Stockholders’ equity: | | | | | | | | | Preferred stock — 10,000,000 shares authorized as of December 31, 2020 and December 31, 2019, 0 shares issued and outstanding as of December 31, 2020 and December 31, 2019 | | | 0 | | | | 0 | | Common stock — $0.001 par value, 80,000,000 and 40,000,000 shares authorized as of December 31, 2020 and December 31, 2019, respectively, 43,336,277 and 28,231,267 shares issued and outstanding as of December 31, 2020 and December 31, 2019, respectively | | | 43,336 | | | | 28,231 | | Additional paid-in capital | | | 346,044,721 | | | | 250,158,766 | | Accumulated deficit | | | (275,002,219 | ) | | | (215,239,450 | ) | Total stockholders’ equity | | | 71,085,838 | | | | 34,947,547 | | Total liabilities and stockholders’ equity | | $ | 99,531,760 | | | $ | 68,842,067 | |
| | | | | | | | | | December 31, | | December 31, | | | | 2022 | | 2021 | | Assets | | | | | | | | Current assets: | | | | | | | | Cash and cash equivalents | | $ | 93,283 | | $ | 57,494 | | Accounts receivable, net | | | 5,017 | | | 2,990 | | Prepaid expenses and other current assets | | | 1,511 | | | 792 | | Total current assets | | | 99,811 | | | 61,276 | | Property, plant and equipment, net | | | 4,151 | | | 5,017 | | Operating lease right-of-use assets, net | | | 2,101 | | | 2,412 | | Indemnification asset, related party | | | 6,595 | | | 6,282 | | Contract acquisition costs, net | | | 8,604 | | | 10,138 | | Intangible asset, net | | | 3,726 | | | 4,390 | | Goodwill | | | 3,903 | | | 3,903 | | Other assets | | | 307 | | | 311 | | Total assets | | $ | 129,198 | | $ | 93,729 | | Liabilities and stockholders’ equity | | | | | | | | Current liabilities: | | | | | | | | Accounts payable | | $ | 2,197 | | $ | 1,070 | | Accrued expenses and other current liabilities | | | 5,522 | | | 5,171 | | Current portion of operating lease liabilities | | | 900 | | | 775 | | Current portion of finance lease liabilities | | | 181 | | | 311 | | Total current liabilities | | | 8,800 | | | 7,327 | | Litigation finance payable | | | 6,594 | | | 6,143 | | Long-term operating lease liabilities | | | 3,332 | | | 4,232 | | Long-term finance lease liabilities | | | 171 | | | 352 | | Long-term debt | | | 19,879 | | | 10,410 | | Total liabilities | | | 38,776 | | | 28,464 | | Commitments and contingencies (Note 15) | | | | | | | | Stockholders’ equity: | | | | | | | | Preferred stock — 10,000,000 shares authorized, none outstanding | | | — | | | — | | Common stock — $0.001 par value, 80,000,000 shares authorized, 64,517,912 and 52,287,737 shares issued and outstanding as of December 31, 2022 and December 31, 2021, respectively | | | 64 | | | 52 | | Additional paid-in capital | | | 440,954 | | | 374,794 | | Accumulated deficit | | | (350,596) | | | (309,581) | | Total stockholders’ equity | | | 90,422 | | | 65,265 | | Total liabilities and stockholders’ equity | | $ | 129,198 | | $ | 93,729 | |
The accompanying notes are an integral part of these consolidated financial statements. Liquidia Corporation Consolidated Statements of Operations and Comprehensive Loss (in thousands, except share and per share data) | | | Year Ended December 31, | | | | | 2020 | | | 2019 | | Net service revenue | | $ | 739,628 | | | $ | 0 | | Collaboration revenue | | | 0 | | | | 8,072,120 | | Total revenue | | | 739,628 | | | | 8,072,120 | | Costs and expenses: | | | | | | | | | Cost of net service revenue | | | 237,712 | | | | 0 | | Cost of collaboration revenue | | | 0 | | | | 807,192 | | Research and development | | | 32,222,393 | | | | 40,491,358 | | General and administrative | | | 27,368,653 | | | | 13,597,119 | | Total costs and expenses | | | 59,828,758 | | | | 54,895,669 | | Loss from operations | | | (59,089,130 | ) | | | (46,823,549 | ) | Other income (expense): | | | | | | | | | Interest income | | | 184,359 | | | | 613,716 | | Interest expense | | | (857,998 | ) | | | (1,373,622 | ) | Total other expense, net | | | (673,639 | ) | | | (759,906 | ) | Net loss and comprehensive loss | | $ | (59,762,769 | ) | | $ | (47,583,455 | ) | | | | | | | | | | | Net loss per share attributable to common stockholders, basic and diluted | | $ | (1.76 | ) | | $ | (2.57 | ) | Weighted average common shares outstanding, basic and diluted | | | 33,888,434 | | | | 18,482,455 | |
| | | | | | | | | Year Ended December 31, | | | 2022 | | 2021 | Revenue | | $ | 15,935 | | $ | 12,853 | Costs and expenses: | | | | | | | Cost of revenue | | | 2,859 | | | 3,023 | Research and development | | | 19,435 | | | 20,517 | General and administrative | | | 32,411 | | | 23,110 | Total costs and expenses | | | 54,705 | | | 46,650 | Loss from operations | | | (38,770) | | | (33,797) | Other income (expense): | | | | | | | Interest income | | | 1,090 | | | 33 | Interest expense | | | (2,338) | | | (762) | Loss on extinguishment of debt | | | (997) | | | (53) | Total other expense, net | | | (2,245) | | | (782) | Net loss and comprehensive loss | | $ | (41,015) | | $ | (34,579) | Net loss per common share, basic and diluted | | $ | (0.67) | | $ | (0.70) | Weighted average common shares outstanding, basic and diluted | | | 60,958,862 | | | 49,677,737 |
The accompanying notes are an integral part of these consolidated financial statements. Liquidia Corporation Consolidated Statements of Stockholders’Stockholders’ Equity Equity(in thousands, except share amounts)
For the Years Ended December 31, 2020 and 2019
| | | | | | | | | | | | | | | | | Common | | Common | | Additional | | | | | Total | | | Stock | | Stock | | Paid in | | Accumulated | | Stockholders’ | | | Shares | | Amount | | Capital | | Deficit | | Equity | Balance as of December 31, 2020 | | 43,336,277 | | $ | 43 | | $ | 346,045 | | $ | (275,002) | | $ | 71,086 | Issuance of common stock upon exercise of stock options | | 14,699 | | | — | | | 41 | | | — | | | 41 | Issuance of common stock under employee stock purchase plan | | 270,185 | | | — | | | — | | | — | | | — | Issuance of common stock upon vesting of restricted stock units | | 40,539 | | | — | | | — | | | — | | | — | Sale of common stock, net | | 8,626,037 | | | 9 | | | 21,701 | | | — | | | 21,710 | Issuance of warrants | | — | | | — | | | 261 | | | — | | | 261 | Stock-based compensation | | — | | | — | | | 6,746 | | | — | | | 6,746 | Net loss | | — | | | — | | | — | | | (34,579) | | | (34,579) | Balance as of December 31, 2021 | | 52,287,737 | | $ | 52 | | $ | 374,794 | | $ | (309,581) | | $ | 65,265 | Issuance of common stock upon exercise of stock options | | 232,877 | | | — | | | 838 | | | — | | | 838 | Issuance of common stock upon vesting of restricted stock units | | 54,181 | | | — | | | — | | | — | | | — | Issuance of common stock under employee stock purchase plan | | 51,941 | | | — | | | 258 | | | — | | | 258 | Issuance of warrants | | — | | | — | | | 1,317 | | | — | | | 1,317 | Equity consideration for acquisition | | 616,666 | | | 1 | | | (1) | | | — | | | — | Sale of common stock, net | | 11,274,510 | | | 11 | | | 54,450 | | | — | | | 54,461 | Stock-based compensation | | — | | | — | | | 9,298 | | | — | | | 9,298 | Net loss | | — | | | — | | | — | | | (41,015) | | | (41,015) | Balance as of December 31, 2022 | | 64,517,912 | | $ | 64 | | $ | 440,954 | | $ | (350,596) | | $ | 90,422 |
| | | Common Stock
Shares | | | Common Stock
Amount | | | Additional
Paid in
Capital | | | Accumulated
Deficit | | | Total
Stockholders’
Equity | | Balance as of December 31, 2018 | | | 15,519,469 | | | $ | 15,520 | | | $ | 185,726,048 | | | $ | (167,053,897 | ) | | $ | 18,687,671 | | Cumulative adjustment - adoption of ASC 842 | | | — | | | | 0 | | | | 0 | | | | (602,098 | ) | | | (602,098 | ) | Issuance of common stock upon exercise of stock options | | | 32,325 | | | | 32 | | | | 141,295 | | | | 0 | | | | 141,327 | | Issuance of common stock upon exercise of common stock warrants | | | 64,629 | | | | 64 | | | | 649 | | | | 0 | | | | 713 | | Issuance of common stock upon vesting of restricted stock units | | | 40,954 | | | | 41 | | | | (41 | ) | | | 0 | | | | 0 | | Sale of common stock, net | | | 12,573,890 | | | | 12,574 | | | | 60,914,510 | | | | 0 | | | | 60,927,084 | | Share-based compensation | | | — | | | | 0 | | | | 3,376,305 | | | | 0 | | | | 3,376,305 | | Net loss | | | — | | | | 0 | | | | 0 | | | | (47,583,455 | ) | | | (47,583,455 | ) | Balance as of December 31, 2019 | | | 28,231,267 | | | $ | 28,231 | | | $ | 250,158,766 | | | $ | (215,239,450 | ) | | $ | 34,947,547 | | Issuance of common stock upon exercise of stock options | | | 40,685 | | | | 40 | | | | 67,876 | | | | 0 | | | | 67,916 | | Issuance of common stock under employee stock purchase plan | | | 5,090 | | | | 5 | | | | 19,410 | | | | 0 | | | | 19,415 | | Issuance of common stock upon vesting of restricted stock units | | | 2,810 | | | | 4 | | | | (4 | ) | | | 0 | | | | 0 | | Sale of common stock, net | | | 9,506,425 | | | | 9,506 | | | | 71,006,892 | | | | 0 | | | | 71,016,398 | | Equity consideration for acquisition | | | 5,550,000 | | | | 5,550 | | | | 20,837,781 | | | | 0 | | | | 20,843,331 | | Share-based compensation | | | — | | | | 0 | | | | 3,954,000 | | | | 0 | | | | 3,954,000 | | Net loss | | | — | | | | 0 | | | | 0 | | | | (59,762,769 | ) | | | (59,762,769 | ) | Balance as of December 31, 2020 | | | 43,336,277 | | | $ | 43,336 | | | $ | 346,044,721 | | | $ | (275,002,219 | ) | | $ | 71,085,838 | |
The accompanying notes are an integral part of these consolidated financial statements. Liquidia Corporation Consolidated Statements of Cash Flows (in thousands) | | | Year Ended December 31, | | | | | 2020 | | | 2019 | | Operating activities | | | | | | | | | Net loss | | $ | (59,762,769 | ) | | $ | (47,583,455 | ) | Adjustments to reconcile net loss to net cash used in operating activities: | | | | | | | | | Share-based compensation | | | 3,954,000 | | | | 3,376,305 | | Depreciation and amortization | | | 3,129,579 | | | | 2,567,742 | | Non-cash lease expense | | | 174,102 | | | | 225,537 | | Amortization of discount and debt issuance costs on long-term debt | | | 61,424 | | | | 75,364 | | Loss on disposal of property and equipment | | | 10,802 | | | | 6,587 | | Changes in operating assets and liabilities, net of business acquired: | | | | | | | | | Accounts receivable, net | | | 0 | | | | 272,557 | | Prepaid expenses and other current assets | | | (132,007 | ) | | | (371,194 | ) | Other non-current assets | | | (12,000 | ) | | | 807,192 | | Accounts payable | | | (297,160 | ) | | | 294,514 | | Accrued compensation | | | 42,312 | | | | 649,168 | | Other accrued expenses | | | 180,736 | | | | (108,707 | ) | Refund liability | | | (927,136 | ) | | | 0 | | Operating lease liabilities | | | (566,390 | ) | | | (422,364 | ) | Deferred revenue | | | 0 | | | | (8,071,920 | ) | Net cash used in operating activities | | | (54,144,507 | ) | | | (48,282,674 | ) | Investing activities | | | | | | | | | Cash acquired from acquisition of business | | | 1,000,000 | | | | 0 | | Purchases of property, plant and equipment | | | (752,086 | ) | | | (1,850,099 | ) | Net cash provided by (used in) investing activities | | | 247,914 | | | | (1,850,099 | ) | Financing activities | | | | | | | | | Principal payments on finance leases | | | (1,122,356 | ) | | | (998,687 | ) | Payments for finance lease deposits | | | 0 | | | | (34,649 | ) | Proceeds from issuance of long-term debt | | | 0 | | | | 5,000,000 | | Principal payments on long-term debt | | | (5,647,060 | ) | | | 0 | | Proceeds from sale of common stock, net of underwriting fees and commissions | | | 71,225,398 | | | | 63,039,490 | | Payments for offering costs | | | (1,634,467 | ) | | | (754,028 | ) | Receipts from litigation financing | | | 507,849 | | | | 0 | | Proceeds from issuance of common stock under stock incentive plans | | | 87,332 | | | | 141,327 | | Proceeds from exercise of warrants | | | 0 | | | | 713 | | Net cash provided by financing activities | | | 63,416,696 | | | | 66,394,166 | | Net increase (decrease) in cash | | | 9,520,103 | | | | 16,261,393 | | Cash, beginning of period | | | 55,796,378 | | | | 39,534,985 | | Cash, end of period | | $ | 65,316,481 | | | $ | 55,796,378 | | Supplemental disclosure of cash flow information | | | | | | | | | Cash paid for interest | | $ | 820,889 | | | $ | 887,038 | | Cash paid for operating lease liabilities | | $ | 1,172,759 | | | $ | 1,081,582 | | Right of use assets obtained with lease liabilities | | $ | 0 | | | $ | 834,693 | | Changes in purchases of property and equipment in accounts payable and accrued expenses | | $ | 412,096 | | | $ | 184,424 | | Noncash acquisition of business, net of acquired cash | | $ | 19,843,331 | | | | 0 | | Accrued tenant improvements and receivable from landlord | | $ | 0 | | | $ | 936,104 | | Deferred offering costs incurred but not paid | | $ | 0 | | | $ | 1,358,378 | |
| | | | | | | | | Year Ended December 31, | | | 2022 | | 2021 | Operating activities | | | | | | | Net loss | | $ | (41,015) | | $ | (34,579) | Adjustments to reconcile net loss to net cash used in operating activities: | | | | | | | Stock-based compensation | | | 9,298 | | | 6,746 | Depreciation and amortization | | | 3,647 | | | 5,612 | Non-cash lease expense | | | 311 | | | 237 | Loss on disposal of property and equipment | | | 4 | | | 44 | Loss on extinguishment of debt | | | 997 | | | 53 | Non-cash interest expense | | | 328 | | | 232 | Changes in operating assets and liabilities: | | | | | | | Accounts receivable, net | | | (2,027) | | | (2,990) | Prepaid expenses and other current assets | | | (719) | | | (40) | Other non-current assets | | | 4 | | | 80 | Accounts payable | | | 814 | | | (7,559) | Accrued expenses and other current liabilities | | | 545 | | | 563 | Refund liability | | | — | | | (1,769) | Operating lease liabilities | | | (775) | | | (665) | Net cash used in operating activities | | | (28,588) | | | (34,035) | Investing activities | | | | | | | Purchases of property, plant and equipment | | | (592) | | | (107) | Proceeds from the sale of property, plant and equipment | | | 5 | | | — | Net cash used in investing activities | | | (587) | | | (107) | Financing activities | | | | | | | Principal payments on finance leases | | | (311) | | | (477) | Principal payments on long-term debt | | | (10,500) | | | (10,353) | Proceeds from issuance of long-term debt with warrants, net | | | 19,767 | | | 10,410 | Receipts from litigation financing | | | 451 | | | 4,989 | Proceeds from sale of common stock, net of underwriting fees and commissions | | | 54,461 | | | 21,710 | Proceeds from issuance of common stock under stock incentive plans | | | 1,096 | | | 41 | Net cash provided by financing activities | | | 64,964 | | | 26,320 | Net increase (decrease) in cash and cash equivalents | | | 35,789 | | | (7,822) | Cash and cash equivalents, beginning of period | | | 57,494 | | | 65,316 | Cash and cash equivalents, end of period | | $ | 93,283 | | $ | 57,494 | Supplemental disclosure of cash flow information | | | | | | | Cash paid for interest | | $ | 1,626 | | $ | 423 | Cash paid for operating lease liabilities | | $ | 1,244 | | $ | 1,208 | Reduction of lease liability and right-of-use asset from lease modification | | $ | — | | $ | 39 | Non-cash increase in property, plant and equipment through accounts payable | | $ | 139 | | $ | — | Non-cash increase in indemnification asset through accounts payable | | $ | 313 | | $ | 4,895 |
The accompanying notes are an integral part of these consolidated financial statements. Liquidia Corporation Notes to Consolidated Financial Statements 1. Business(tabular dollars in thousands)
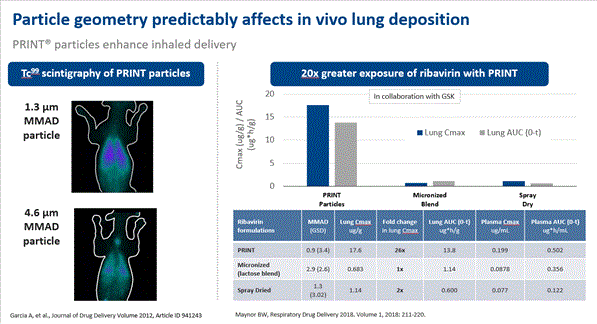
1. Business Description of the Business Liquidia Corporation (“Liquidia” or the “Company”) is a biopharmaceutical company focused on the development, manufacture, and commercialization of products that address unmet patient needs, with current focus directed towards the treatment of pulmonary arterial hypertension (“PAH”PH”). Liquidia Corporation operates through the company’sits wholly owned operating subsidiaries, Liquidia Technologies, Inc. (“Liquidia Technologies”) and Liquidia PAH, LLC (“Liquidia PAH”), formerly known as RareGen, LLC (“RareGen”). The Company conducts research, development and manufacturing of novel products by applying its proprietary PRINT® technology, a particle engineering platform, to enable precise production of uniform drug particles designed to improve the safety, efficacy and performance of a wide range of therapies. The Company is currently developing two product candidates for which it holds worldwide commercial rights: LIQ861 to treat PAH, and LIQ865 to treat post-operative pain.
The Company’s most advanced product, LIQ861, is an inhaled dry powder formulation of treprostinil designed to improve the therapeutic profile of treprostinil by enhancing deep lung delivery and achieving higher dose levels than current inhaled therapies. The Company submitted the New Drug Application (“NDA”) for LIQ861 in January 2020 and is actively preparing its reply to the Complete Response Letter (“CRL”) issued by the Food and Drug Administration (“FDA”) in November 2020.
The Company’s second product candidate, LIQ865, is designed to deliver sustained release of bupivacaine, a non-opioid anesthetic, to treat local post-operative pain for three to five days through a single administration. The Company has completed two Phase 1 clinical trials and additional toxicology studies to help enable continued clinical development in Phase 2 studies of LIQ865.
The Company generates revenue primarily pursuant to thea promotion agreement between Liquidia PAH and Sandoz Inc. (“Sandoz”), dated as of August 1, 2018, as amended (the “Promotion Agreement”), sharing profit derived from the sale of the first-to-file fullySandoz’s substitutable generic treprostinil injection (“Treprostinil Injection”) in the United States. Liquidia PAH has the exclusive rights to conduct commercial activities to encourage the appropriate use of Treprostinil Injection. The Company employs a small, targeted sales force calling on physicians involvedand hospital pharmacies in the treatment of PAH in the United States,pulmonary arterial hypertension (“PAH”), as well as key stakeholders involved in the distribution and reimbursement of Treprostinil Injection. Strategically, the Company believes that its commercial presence in the field will enable an efficient base to expand from for the launch of LIQ861YUTREPIA upon final approval, leveraging existing relationships and further validating ourits reputation as a company committed to supporting PAH patients. The Company conducts research, development and manufacturing of novel products by applying its subject matter expertise in cardiopulmonary diseases and our proprietary PRINT® technology, a particle engineering platform that enables precise production of uniform drug particles designed to improve the safety, efficacy, and performance of a wide range of therapies. Through development of the Company’s own products and research with third parties, the Company has experience applying PRINT across multiple routes of administration and drug payloads including inhaled therapies, vaccines, biologics, nucleic acids and ophthalmic implants, among others. The Company’s lead product candidate, for which it holds worldwide commercial rights, is YUTREPIA for the treatment of PAH. YUTREPIA is an inhaled dry powder formulation of treprostinil designed with PRINT to improve the therapeutic profile of treprostinil by enhancing deep lung delivery while using a convenient, low resistance dry-powder inhaler (“DPI”) and by achieving higher dose levels than the labelled dose of current inhaled therapies. The Company’s New Drug Application (“NDA”) for YUTREPIA was tentatively approved by the U.S. Food and Drug Administration (“FDA”) for the treatment of PAH in November 2021. The FDA also confirmed that the clinical data in the NDA would support the Company’s pursuit of a supplemental NDA to treat patients with pulmonary hypertension and interstitial lung disease (PH-ILD) upon the expiration of regulatory exclusivity for the nebulized form of treprostinil in March 2024. Recent Developments On January 9, 2023, the Company entered into a Revenue Interest Financing Agreement (the “RIFA”) with HealthCare Royalty Partners IV, L.P. (“HCR”) and HealthCare Royalty Management, LLC. Pursuant to the RIFA and subject to customary closing conditions, HCR has agreed to pay the Company an aggregate investment amount of up to $100.0 million (the “Investment Amount”). Under the terms of the RIFA, $32.5 million of the Investment Amount was funded on January 27, 2023 (the “Initial Investment Amount”), $22.4 million of which was used to satisfy in full and retire the Company’s indebtedness under the Amended and Restated Loan and Security Agreement with Silicon Valley Bank, with the excess proceeds less transaction costs of approximately $0.7 million funded to the Company. Under the RIFA, an additional $35.0 million of the Investment Amount will be funded fifteen business days after the earlier of regulatory approval of YUTREPIA or a favorable determination relating to the asserted patents in the ongoing patent litigation with United Therapeutics Corporation and $25.0 million of the Investment Amount will be funded fifteen business days after the mutual agreement of HCR and the Company to fund such amount. See Note 16 for further information. Risks and Uncertainties The Company is subject to risks and uncertainties common to early-stage companies in the biotechnology industry, including, but not limited to, development by competitors of new technological innovations, dependence on third parties and key personnel, protection of proprietary technology, compliance with government regulations, the impact of the COVID-19 coronavirus, and the ability to secure additional capital to fund operations. The current global macro-economic environment is volatile, which may result in supply chain constraints and elevated rates of inflation. In addition, the Company operates in a dynamic and highly competitive industry and believes that changes in any of the following areas could have a material adverse effect on the Company’s future financial position, results of operations, or cash flows: the ability to obtain future financing; advances and trends in new technologies and industry standards; results of clinical trials; regulatory approval and market acceptance of the Company’s products; development of sales channels; certain strategic relationships; litigation or claims against the Company related to intellectual property, product, regulatory, or other matters; and the Company’s ability to attract and retain employees necessary to support its growth. Product candidates developed by the Company require approval from the FDA and/or other international regulatory agencies prior to commercial sales. There can be no assurance that the Company's product candidates will receive the necessary approvals. If the Company is denied approval, approval is delayed, or the Company is unable to maintain approval, it could have a material adverse impact on the Company. The Company relies on single source manufacturers and suppliers for the supply of its product candidates. This adds to the manufacturing risks faced by the Company, which could be left without backup facilities in the event of any failure by a supplier. Any disruption from these manufacturers or suppliers could have a negative impact on the Company’s business, financial position and results of operations. Liquidity The Company expects to incur significant expenses and operating losses for the foreseeable future as it advances product candidates through clinical trials, seeks regulatory approval and pursuesprepares for commercialization of any approved product candidates. In addition, if the Company obtains marketing approval for any of its product candidates, it would incur significant commercialization expenses related to product manufacturing, marketing, sales and distribution. These efforts require significant amounts of additional capital, adequate personnel and infrastructure, and extensive compliance-reporting capabilities. Even if the Company's development efforts are successful, it is uncertain when, if ever, the Company will realize significant revenue from product sales. The Company will seekmay require additional funding through public or private financings, debt financing or collaboration.capital in advance of a potential commercial launch of YUTREPIA. If the Company is unable to access the contingent Investment Amounts from the RIFA or generate substantial YUTREPIA product revenue by the second quarter of 2024, the Company will require additional capital. The Company may also require additional capital to pursue in-licenses or acquisitions of other product candidates. If the Company concludes it requires but is unable to obtain funding, the Company could be required to delay, reduce, or eliminate research and development programs, product portfolio expansion, or future commercialization efforts, which could adversely affect its business prospects. In accordance with Accounting Standards Update (“ASU”) 2014-15,2014-15,Disclosure of Uncertainties about an Entity’sEntity’s Ability to Continue as a Going Concern (Subtopic 205-40)205-40), the Company has evaluated whether there are conditions and events, considered in the aggregate, that raise substantial doubt about the Company’s ability to continue as a going concern within one year after the date that the consolidated financial statements are issued. We haveThe Company has financed ourits growth and operations through a combination of funds generated from revenues, the issuance of convertible preferred stock and common stock, finance leases, bank borrowings, bank borrowings with warrants and the issuance of convertible notes and warrants.warrants, and revenue interest financing. Since inception, the Company has incurred recurring losses, including net lossesloss of $59.8$41.0 million for the year ended December 31,2020 2022 and the Company had an accumulated deficit of $275.0$350.6 million as of December 31, 2020. The2022. Although the Company expects to continue to generate operating losses for the foreseeable future. Asfuture, management believes that based on its current operating plan, excluding any potential contingent Investment Amounts from the issuance date of the consolidated financial statements for the year ended December 31,2020, the Company expects thatRIFA and future YUTREPIA product revenue, its cash and cash equivalents will be sufficient to fund its operating expensesoperations and capital expenditure requirements and allow it to remain in compliance with its minimum cash covenants pursuant to the RIFA for at least 12twelve months from the issuance date of the annualthese consolidated financial statements. The future viability ofIf the Company is dependent on itsunable to access additional Investment Amounts from the RIFA, there could be substantial doubt about the Company’s ability to raise additional capital to finance its future operations. Accordingly, the accompanying consolidated financial statements have been prepared on the basis of continuity of operations, realization of assets, and the satisfaction of liabilities and commitments in the ordinary course of business.
Recent Developments
Acquisition of RareGen, LLC (now Liquidia PAH, LLC)
On November 18, 2020 (the “Closing Date”), the Company completed the previously announced acquisition contemplated by the Agreement and Plan of Merger, datedcontinue as a going concern as of June 29, 2020, as amended by a Limited Waiver and Modification to the Merger Agreement, dated as of August 3, 2020 (the “Merger Agreement”), by and among Liquidia Technologies, the Company, RareGen, Gemini Merger Sub I, Inc., a Delaware corporation (“Liquidia Merger Sub”), Gemini Merger Sub II, LLC, a Delaware limited liability company (“RareGen Merger Sub”), and PBM RG Holdings, LLC, a Delaware limited liability company (“PBM”). Pursuant to the Merger Agreement, Liquidia Merger Sub, a former wholly owned subsidiarydate of the Company, merged with and into Liquidia Technologies (the “Liquidia Technologies Merger”), and RareGen Merger Sub, a former wholly owned subsidiaryissuance of the Company’s second quarter 2023 financial statements. The Company merged withhas based these estimates on assumptions that may differ from actual results, and into RareGen (the “RareGen Merger” and, together with the Liquidia Technologies Merger, the “Merger Transaction”). Upon consummation of the Merger Transaction, the separate corporate existences of Liquidia Merger Sub and RareGen Merger Sub ceased and Liquidia Technologies and RareGen (now Liquidia PAH) continue as wholly owned subsidiaries of Liquidia Corporation.it could use its available resources sooner than expected.
On the Closing Date, an aggregate of 5,550,000 shares of common stock, $0.001 par value per share (“Liquidia Corporation Common Stock”), were issued to RareGen members in exchange for 10,000 RareGen common units, representing all of the issued and outstanding RareGen equity. Additionally, on the Closing Date, an aggregate of 616,666 shares of Liquidia Corporation Common Stock were withheld from RareGen members to secure the indemnification obligations of RareGen members. Additionally, RareGen members received a pro rata portion of the RareGen cash at closing in excess of $1 million. RareGen members are also entitled to receive a pro rata portion of up to an additional 2,708,333 shares of Liquidia Corporation Common Stock in the aggregate in 2022, based on the amount of 2021 net sales of the generic treprostinil product (“Net Sales Earnout Shares”) owned by Sandoz, which RareGen markets pursuant to the Promotion Agreement. The fair value of the purchase consideration or the purchase price was approximately $20.8 million. See Note 3 below for further details.
2. Basis of Presentation and Summary of Significant Accounting Policies Basis of Presentation The Company has prepared the accompanying financial statements in conformity with generally accepted accounting principles in the United States of America (“GAAP”). Such financial statements reflect all adjustments that are, in management’s opinion, necessary to present fairly, in all material respects, the Company’s financial position, results of operations and cash flows and are presented in U.S. Dollars. Consolidation The accompanying consolidated financial statements include the Company’s wholly owned subsidiaries, Liquidia Technologies and Liquidia PAH. All intercompany accounts and transactions have been eliminated. Use of Estimates The preparation of financial statements in accordance with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, and the disclosure of contingent assets and liabilities, at the date of the financial statements, as well as the reported amounts of revenues and expenses during the period. These estimates are based on historical experience and various other assumptions believed reasonable under the circumstances. The Company evaluates its estimates on an ongoing basis, including those related to the valuation of stock-based awards, certain accruals, and intangible and contract acquisition cost amortization, and makes changes to the estimates and related disclosures as experience develops or new information becomes known. Actual results will most likely differ from those estimates. Revision of Previously Issued Financial Statements
During the three months ended June 30, 2020, the Company identified an error in the matter in which it calculated diluted weighted common shares outstanding and diluted net loss per common share. While the Company has included common stock warrants whose exercise price is de minimis in the calculation of basic weighted average common shares outstanding and basic net loss per common share, these warrants were inappropriately excluded from the calculation of diluted weighted common shares outstanding and diluted net loss per common share, which resulted in an error in those previously reported amounts for the year ended December 31, 2019. The Company has evaluated this error and determined that this presentation error was not material to any prior annual or interim periods. However, the Company is revising the previously presented December 31, 2019 diluted weighted common shares outstanding and diluted net loss per common share as follows:
| | | Year Ended | | | | | December 31, 2019 | | | | | As Presented | | | As Revised | | | | | | | | | | | | Net loss per common share: Diluted | | $ | (2.59 | ) | | $ | (2.57 | ) | | | | | | | | | | | Diluted weighted average shares outstanding | | | 18,371,083 | | | | 18,482,455 | |
Summary of Significant Accounting Policies Cash Cash
The Company considers all highly liquid investments with a maturity of three months or less when purchased,at the date of purchase to be cash equivalents. The Company had 0 cash equivalents as of December 31,2020 and 2019. Accounts Receivable
Accounts receivable are stated at net realizable value including an allowance for doubtful accounts as of each balance sheet date, if applicable. The Company has not recorded an allowance for doubtful accounts during the years ended December 31, 2020 and 2019.
Concentration of Credit Risk
Financial instruments that potentially subject the Company to concentrations of credit risk consist of cash and accounts receivable.cash equivalents. The Company is exposed to credit risk, subject to federal deposit insurance, in the event of default by the financial institutions holding its cash and cash equivalents to the extent of amounts recorded on the consolidated balance sheet. With regard to cash, 99%As of December 31, 2022 all of the Company’s cash isand cash equivalents were held on deposit with Pacific WesternSilicon Valley Bank (“Pacific Western”SVB”). With regardFollowing the March 10, 2023 closure of SVB, substantially all of the Company’s cash and cash equivalents were moved to revenuesa different accredited financial institution. The Company has not experienced any losses on such accounts and concentration ofdoes not believe that it is subject to unusual credit risk GlaxoSmithKline plc (“GSK”beyond the normal credit risk associated with commercial banking relationships. Such deposits have and “GSK Inhaled”)will continue to exceed federally insured limits. Accounts Receivable Accounts receivable are stated at net realizable value and net of an allowance for credit losses as of each balance sheet date, if applicable. One customer accounted for $099% and $8.1 million98% of our revenue during the years ended December 31,2020 and 2019, respectively, or 0% and 100%, respectively, of our total revenue. Sandoz accounted for $0.7 million and $0 of our revenue during the years ended accounts receivable at December 31, 2020 2022 and 2019, respectively, or 100%2021, respectively. As of December 31, 2022 and 0%, respectively, of our total revenue. Leases
In February 2016, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) 2016-02,Leases, as amended (Topic 842) (“ASU 2016-02”). The Company adopted Topic 842, as amended, as of January 1, 2019, using the modified retrospective approach. The modified retrospective approach provided a method for recording existing leases at adoption that approximates the results of a full retrospective approach in the year of adoption. In addition,2021, the Company elected the package of practical expedients permitted under the transition guidance within the new standard, which among others, allowed the Company to carry forward the historical lease classification. Adoption of ASU 2016-02 resulted in the recording of net lease assets and lease liabilities of approximately $6.4 million and $9.1 million respectively, as of January 1, 2019. The net impact of applying Topic 842 washas not recorded as an adjustment to increase the accumulated deficit by $0.6 million as of January 1, 2019. Adoption of ASU 2016-02 had no impact on the Statement of Cash Flows.allowance for credit losses.
Leases The provisions of ASU 2016-02 setASC 842 Leases sets out the principles for the recognition, measurement, presentation and disclosure of leases for both lessees and lessors. The new standard requires lessees to apply a dual approach, classifying leases as either finance or operating leases based on the principle of whether or not the lease is effectively a financed purchase by the lessee. This classification will determine whether lease expense is recognized based on an effective interest method or on a straight-line basis over the term of the lease. A lessee is also required to record a right-of-use asset and a lease liability for all leases with a term of greater than 12 months regardless of their classification. The Company has elected to account for leases with a term of 12 months or less in a similar manner as under existing guidance for operating leases. For operating leases, the asset and liability is expensed over the lease term on a straight-line basis, with all cash flows classified as an operating activity in the Statement of Cash Flows. For finance leases, interest on the lease liability is recognized separately from the amortization of the right-of-use asset in the Statement of Operations and Comprehensive Loss and the repayment of the principal portion of the lease liability is classified as a financing activity, while the interest component is classified as an operating activity in the Statement of Cash Flows.
Property, Plant and Equipment Property, plant and equipment are stated at cost. Depreciation of property, plant and equipment is computed using the straight-line method over the estimated useful lives of the assets beginning when the assets are placed in service. Estimated useful lives for the major asset categories are: Lab and build-to-suit equipment (years) | | | 5 | - | 7 | | Office equipment (years) | | | | 5 | | | Furniture and fixtures (years) | | | | 10 | | | Computer equipment (years) | | | | 3 | | | Leasehold improvements | | Lesser of life of the asset or remaining lease term | |
| | | Lab and build-to-suit equipment (years) | | 5 - 7 | Office equipment (years) | | 5 | Furniture and fixtures (years) | | 10 | Computer equipment (years) | | 3 | Leasehold improvements | | Lesser of life of the asset or remaining lease term |
Major renewals and improvements are capitalized to the extent that they increase the useful economic life or increase the expected economic benefit of the underlying asset. Maintenance and repairs are charged to operations as incurred. When items of property, plant and equipment are sold or retired, the related cost and accumulated depreciation or amortization is removed from the accounts, and any gain or loss is included in operating expenses in the accompanying Statements of Operations and Comprehensive Loss. Business Combination
In a business combination, the acquisition method of accounting requires that the assets acquired and liabilities assumed be recorded as of the date of the acquisition at their respective fair values with limited exceptions. Assets acquired and liabilities assumed in a business combination that arise from contingencies are generally recognized at fair value. If fair value cannot be determined, the asset or liability is recognized if probable and reasonably estimable; if these criteria are not met, no asset or liability is recognized. Transaction costs and costs to restructure the acquired company are expensed as incurred. The operating results of the acquired business are reflected in the Company’s consolidated financial statements after the date of the acquisition.
Long-Lived Assets The Company reviews long-lived assets, including definite-life intangible assets for realizability on an ongoing basis. Changes in depreciation and amortization, generally accelerated depreciation and variable amortization, are determined and recorded when estimates of the remaining useful lives or residual values of long-term assets change. The Company also reviews for impairment when conditions exist that indicate the carrying amount of the assets may not be fully recoverable. In those circumstances, the Company performs undiscounted operating cash flow analyses to determine if an impairment exists. When testing for asset impairment, the Company groups assets and liabilities at the lowest level for which cash flows are separately identifiable. Any impairment loss is calculated as the excess of the asset’s carrying value over its estimated fair value. Fair value is estimated based on the discounted cash flows for the asset group over the remaining useful life or based on the expected cash proceeds for the asset less costs of disposal. Any impairment losses would be recorded in the consolidated statements of operations. To date, no such impairments have occurred. Goodwill Goodwill
The Company acquired goodwill on its balance sheet during the fourth quarter of 2020 from the Merger Transaction. The Company assesses goodwill for impairment at least annually as of July 1 or whenever events or changes in circumstances indicate that the carrying amount of such assets may not be recoverable. For example, significant and unanticipated changes or our inability to obtain or maintain regulatory approvals for our product candidates, including the NDA for YUTREPIA, could trigger testing of our goodwill for impairment. The Company has 1one reporting unit. The Company has the option to first assess qualitative factors to determine whether events or circumstances indicate it is more likely than not that the fair value of a reporting unit is greater than its carrying amount, in which case a quantitative impairment test is not required.
Per ASU 2017-04ASC 350 Intangibles-Goodwill and Other the quantitative goodwill impairment test is performed by comparing the fair value of the reporting unit with its carrying amount, including goodwill. If the fair value of the reporting unit exceeds its carrying amount, goodwill is not impaired. An impairment loss is recognized for any excess of the carrying amount of the reporting unit’s goodwill over the fair value up to the amount of goodwill allocated to the reporting unit. Income tax effects from any tax-deductible goodwill on the carrying amount of the reporting unit are considered when measuring the goodwill impairment loss, if applicable. AsThe Company completed its annual goodwill impairment test as of December 31, 2020 the Company concluded there were July 1, 2022. There have been no significant events or changes in circumstances that indicated thataffecting the carrying amountvaluation of goodwill was not recoverable.subsequent to the assessment.
Revenue Recognition from Promotion Agreements The Company recognizes revenue in accordance with Accounting Standards Update (ASU) 2014-09,ASU 2014-09, Revenue from Contracts with Customers (Topic 606)606). The core principle of Topic 606 is that a company should recognize revenue to depict the transfer of promised goods or services to customers in an amount that reflects the consideration to which the company expects to be entitled in exchange for those goods or services. The following five steps are applied to achieve that core principle: | ● | Step 1: Identify the contract with the customer |
| ● | Step 2: Identify the performance obligations in the contract |
| ● | Step 3: Determine the transaction price |
| ● | Step 4: Allocate the transaction price to the performance obligations in the contract |
| ● | Step 5: Recognize revenue when the company satisfies a performance obligation |
In order to identify the performance obligations in a contract with a customer, the Company assesses the promised goods or services in the contract and identifies each promised good or service that is distinct. If a good or service is not distinct, the good or service is combined with other promised goods or services until a bundle of goods or services is identified that is distinct. The transaction price is the amount of consideration to which an entity expects to be entitled in exchange for transferring promised goods or services to a customer. The consideration promised in a contract with a customer may include fixed amounts, variable amounts, or both. Variable consideration is included in the transaction price only to the extent that it is probable that a significant reversal in the amount of cumulative revenue recognized will not occur when the uncertainty associated with the variable consideration is subsequently resolved. The Company evaluates any non-cash consideration, consideration payable to the customer, potential returns and refunds, and whether consideration contains a significant financing element in determining the transaction price. Revenue is measured based on consideration specified in a contract with a customer. The Company recognizes revenue when it satisfies a performance obligation by transferring control over a service to a customer. The amount of revenue recognized reflects estimates for refunds and returns, which are presented as a reduction of Accounts receivable where the right of setoff exists. On August 1,2018, the Company partnered with Sandoz in the Promotion Agreement to launch the first-to-filefirst-to-file generic of Treprostinil Injection for the treatment of patients with PAH. Under the Promotion Agreement, the Company provides certain promotional and nonpromotional activities on an exclusive basis for the product in the United States of America for the treatment of PAH, in exchange for a share of Sandoz’s net profits, as defined within the Promotion Agreement. In addition, the Company paid Sandoz $20$20.0 million at the inception of the Promotion Agreement, in consideration for the right to conduct the promotional activities for the product. In exchange for its services, the Company is entitled to receive a portion of net profits based on specified profit levels associated with the product. The Company determined that certain activities within the contract are within the scope of ASC 808, Collaborative Arrangements. The commercialization of the product is a joint operating activity where the Company will provide promotional activities for Sandoz’s intellectual property and Sandoz will be responsible for items such as supply of the product, distribution to customers, managing sales, processing returns, and regulatory matters, and protection of patents. Both parties will be active participants, each carrying out its assigned responsibilities, and participating in the joint operating activity and will share in the risks and rewards of the commercialization through the profit-sharing arrangement. In addition, the Company determined that the services provided under the Promotion Agreement fall within the scope of Topic 606. The promotional activities the Company performs are one of the services the Company expects to provide as part of its ordinary activities, and it is receiving consideration for this service from Sandoz in the form of a share of “Net Profits” (as defined in the Promotion Agreement). The Company has one combined performance obligation under the Promotion Agreement, which is to perform promotional and non-promotional activities to encourage the appropriate use of the product in accordance with the product labeling and applicable law. As such, and in accordance with ASU 2018-18:2018-18: Clarifying the Interaction between Topic 808 and Topic 606, the Company will account for the entire Promotion Agreement under Topic 606. Revenue Recognition from Research and Development Services
The Company derives collaboration revenue primarily from licensing its proprietary PRINT technology and from performing research and development services. Collaboration revenues are recognized as services are performed in an amount that reflects the consideration the Company expects to be entitled to in exchange for those services and technology.
The Company’s research, development and licensing agreements provide for multiple promised goods and services to be satisfied by the Company and include a license to the Company’s technology in a particular field of study, participation in collaboration committees, performance of certain research and development services and obligations for certain manufacturing services.
The transaction price for these contracts includes non-refundable fees and fees for research and development services. Non-refundable up-front fees which may include, for example, an initial payment upon effectiveness of the contractual relationship or payment to secure a right for a future license, are recorded as deferred revenue and recognized into revenue over time as the Company provides the research services under the contract required to advance the products to the point where the Company is able to transfer control of the licensed technology to the customer (“Technology Transfer”). The contract consideration may also include additional non-refundable payments due to the Company based on the achievement of research, development, regulatory or commercialization milestone events. In agreements involving multiple goods or services promised to be transferred to customers, the Company must assess, at the inception of the contract, whether each promise represents a separate performance obligation (i.e., is “distinct”), or whether such promises should be combined as a single performance obligation. As these goods and services are considered to be highly interrelated, they may be considered to represent a single, combined performance obligation. The Company includes an estimate of the probable amount of milestone payments to which it will be entitled in the transaction price. The estimate requires evaluation of factors which are outside of the Company’s control and significantly limit the Company’s ability to achieve the remaining milestone payments. Therefore, the Company has not included any future milestone payments in the transaction price allocated to research, development and licensing agreements as of December 31,2020 or December 31, 2019. The Company revises the transaction price to include milestone payments once the specific milestone achievement is not considered to be subject to a significant reversal of revenue. At that time, the estimated transaction price is adjusted and a cumulative catch-up adjustment is recorded to adjust the amount of revenue to be recognized from the license inception to the date the milestone was deemed probable of achievement. The milestone is included with other non-refundable up-front fees and recognized into revenue over time as the Company continues to provide services under the contract through the Company’s Technology Transfer. The amount of revenue recognized is based on the proportion of total research services performed to date to the expected services to be provided through the Technology Transfer.
The estimate of the research services to be provided through the Technology Transfer requires significant judgment to evaluate assumptions regarding the level of effort required for the Company to have performed sufficient obligations for the customer to be able to utilize the licensed technology without requiring further services from the Company. If the estimated level of effort changes, the remaining deferred revenue is recognized over the revised period in which the expected research services and Technology Transfer are required. Changes in estimates occur for a variety of reasons, including but not limited to (i) research and development acceleration or delays, (ii) customer prioritization of research projects, or (iii) results of research and development activities. The Company recognizes the consideration it is entitled to receive for research and development services, which are primarily billed quarterly in arrears on a time and materials basis, as the services are performed (under a proportional performance model) and collection is reasonably assured. Additionally, any up-front or development milestone payments received are also recognized as revenues, over time, under this same proportional performance model.
Royalties related to product sales will be recognized as revenue when the sale occurs since payments relate directly to products that will have been fully developed and for which the Company will have satisfied all of its performance obligations.
Segment Information U.S. GAAP requires segmentation based on an entity’s internal organization and reporting of revenue and operating income based upon internal accounting methods commonly referred to as the “management approach.” Operating segments are defined as components of an enterprise about which separate financial information is available that is evaluated regularly by the chief operating decision maker (CODM), or decision making group, in deciding how to allocate resources and in assessing performance. The Company’s CODM is its Chief Executive Officer. The Company has determined that it has one operating and reporting segment. Research and Development Expense Research and development costs are expensed as incurred and include direct costs incurred to third parties related to the salaries of, and share-basedstock-based compensation for, personnel involved in research and development activities, contractor fees, grant expenses, administrative expenses and allocations of research-related overhead costs. Administrative expenses and research-related overhead costs included in research and development expense consist of allocations of facility and equipment lease charges, depreciation and amortization of assets and insurance directly related to research and development activities. Patent Maintenance The Company is responsible for all patent costs, past and future, associated with the preparation, filing, prosecution, issuance, maintenance, enforcement and defense of United States patent applications. Such costs are recorded as general and administrative expenses as incurred. To the extent that the Company’s licensees share these costs, such benefit is recorded as a reduction of the related expenses. Share-BasedStock-Based Compensation
The Company estimates the grant date fair value of its share-basedstock-based awards and amortizes this fair value to compensation expense over the requisite service period or vesting term (see Note 5)8).
Net Loss Per Share Basic net loss per share is calculated by dividing net loss attributable to common stockholders by the weighted average shares outstanding during the period, without consideration of common stock equivalents. Diluted net loss per share is calculated by adjusting weighted average shares outstanding for the dilutive effect of common stock equivalents outstanding for the period, determined using the treasury-stock method. For purposes of the diluted net loss per share calculation, stock options and warrants are considered to be common stock equivalents but are excluded from the calculation of diluted net loss per share because their effect would be anti-dilutive. Due to their anti-dilutive effect, the calculation of diluted net loss per share for the years ended December 31,2020 and 2019 does not includeexcludes the following common stock equivalent shares: | | | Year Ended December 31, | | | | | 2020 | | | 2019 | | Stock options | | | 2,652,525 | | | | 1,979,411 | | Restricted stock units | | | 98,705 | | | | 0 | | Total | | | 2,751,230 | | | | 1,979,411 | |
| | | | | | | Year Ended | | | December 31, | | | 2022 | | 2021 | Stock Options | | 7,757,017 | | 5,234,582 | Restricted Stock Units | | 399,349 | | 259,705 | Warrants | | 445,205 | | 168,767 | Total | | 8,601,571 | | 5,663,054 |
Certain common stock warrants are included in the calculation of basic and diluted net loss per share since their exercise price is de minimis. Fair Value of Financial Instruments The carrying values ofamounts reflected in the Company's consolidated balance sheets for cash, accounts receivable, prepaid expenses and other current assets, accounts payable at December 31,2020and 2019 approximatedaccrued expenses and other liabilities approximate their fair valuevalues due to the short maturity of these instruments.their short-term nature. The Company’s valuation of financial instruments is based on a three-tieredthree-tiered approach, which requires that fair value measurements be classified and disclosed in one of three tiers. The fair value hierarchy defines a three-levelthree-level valuation hierarchy for disclosure of fair value measurements as follows: Level 1 — Quoted prices in active markets for identical assets or liabilities; Level 2 — Other than quoted prices included in Level 1 inputs that are observable for the asset or liability, either directly or indirectly; and Level 3 — Unobservable inputs for the asset and liability used to measure fair value, to the extent that observable inputs are not available. The categorization of a financial instrument within the valuation hierarchy is based upon the lowest level of input that is significant to the fair value measurement. The following tables present the placement in the fair value hierarchy of financial liabilities measured at fair value: | | | | | | | | | | | | | | | Quoted | | Significant | | | | | | | | | Prices in | | Other | | Significant | | | | | | Active | | Observable | | Unobservable | | | | | | Markets | | Inputs | | Inputs | | Carrying | December 31, 2022 | | (Level 1) | | (Level 2) | | (Level 3) | | Value | Assets | | | | | | | | | | | | | Money market mutual funds (cash equivalents) | | $ | 92,283 | | $ | — | | $ | — | | $ | 92,283 | Liabilities | | | | | | | | | | | | | A&R Silicon Valley Bank term loan | | $ | — | | $ | 18,853 | | $ | — | | $ | 19,879 |
| | | | | | | | | | | | | | | Quoted | | Significant | | | | | | | | | Prices in | | Other | | Significant | | | | | | Active | | Observable | | Unobservable | | | | | | Markets | | Inputs | | Inputs | | Carrying | December 31, 2021 | | (Level 1) | | (Level 2) | | (Level 3) | | Value | Assets | | | | | | | | | | | | | Money market mutual funds (cash equivalents) | | $ | 56,494 | | $ | — | | $ | — | | $ | 56,494 | Liabilities | | | | | | | | | | | | | Silicon Valley Bank term loan | | $ | — | | $ | 10,021 | | $ | — | | $ | 10,410 |
Money market mutual funds are included in cash and cash equivalents on the Company's consolidated balance sheets. They are valued using quoted market prices and therefore are classified within Level 1 of the fair value as of December 31, 2020 and 2019: December 31, 2020 | | Quoted Prices in Active Markets (Level 1) | | | Significant Other Observable Inputs (Level 2) | | | Significant Unobservable Inputs (Level 3) | | | Carrying Value | | Pacific Western Bank note - A&R LSA | | $ | 0 | | | $ | 9,842,069 | | | $ | 0 | | | $ | 10,292,485 | |
December 31, 2019 | | Quoted Prices in Active Markets (Level 1) | | | Significant Other Observable Inputs (Level 2) | | | Significant Unobservable Inputs (Level 3) | | | Carrying Value | | Pacific Western Bank note - A&R LSA | | $ | 0 | | | $ | 14,094,792 | | | $ | 0 | | | $ | 15,878,121 | |
hierarchy. The fair value of debt is measured in accordance with ASU 2016-01,ASC 820, Financial Instruments–Overall: Recognition and Measurement of Financial Assets and Financial Liabilities. The fair value is determined based on the remaining years to maturity, interest and principal payments, as well as an interest rate consistent with the Company’s current estimated cost of debt.
Deferred Offering Costs
The Company capitalizes certain legal, professional accounting and other third-party fees that are directly associated with in-process equity financings as deferred offering costs until such equity financings are consummated. After consummation of the equity financing, these costs are recorded in stockholders’ equity as a reduction of proceeds generated as a result of the offering.
Income Taxes The asset and liability method is used in the Company’s accounting for income taxes. Under this method, deferred tax assets and liabilities are determined based on differences between financial reporting and tax bases of assets and liabilities and are measured using the enacted tax rates and laws that are expected to be in effect when the differences are expected to reverse. The Company records a valuation allowance against deferred tax assets when realization of the tax benefit is uncertain. A valuation allowance is recorded, if necessary, to reduce net deferred taxes to their realizable values if management believes it is more likely than not that the net deferred tax assets will not be realized. The Company may recognize the tax benefit from an uncertain tax position only if it is more likely than not that the tax position will be sustained on examination by the taxing authorities based on the technical merits of the position. The tax benefits recognized in the financial statements from such a position are measured based on the largest benefit that has a greater than 50% likelihood of being realized upon ultimate settlement. Recent Accounting Pronouncements In October 2018, August 2020, the FASB issued ASU 2018-18,2020-06, Collaborative ArrangementsAccounting for Convertible Instruments and Contracts in an Entity’s Own Equity (Topic 808): Clarifying. This guidance simplifies the Interaction between Topic 808accounting for certain financial instruments with characteristics of liabilities and Topic 606 ("ASU 2018-18"). The provisions of ASU 2018-18 clarify when certain transactions between collaborative arrangement participants should be accounted for under ASC 606equity, including convertible instruments and incorporates unit-of-account guidance consistent with ASC 606 to aidcontracts in this determination. The guidance is effective for annual periods and interim periods within those annual periods beginning after December 15, 2019, with early adoption permitted. Thean entity’s own equity. Effective January 1, 2022, the Company adopted ASU 2018-18 effective January 1, 2020 and it did not have an effect2020-06, which had no impact on the Company's consolidatedCompany’s financial statements for the year ended December 31, 2020.and related disclosures. In January 2017,May 2021, the FASB issued ASU No.2017-04,2021-04, SimplifyingIssuer’s Accounting for Certain Modifications or Exchanges of Freestanding Equity-Classified Written Call Options. This guidance clarifies and reduces diversity in the Testaccounting for Goodwill Impairment,modifications or exchanges of freestanding equity-classified written call options (for example warrants) that remain equity classified after modification or exchange. Effective January 1, 2022, the Company adopted ASU 2021-04, which eliminated the requirement to calculate the implied fair value of goodwill. An entity should perform its annual, or interim, goodwill impairment test by comparing the fair value of a reporting unit with its carrying amount. An entity should recognize an impairment charge for the amount by which the carrying amount exceeds the reporting unit’s fair value; however, the loss recognized should not exceed the total amount of goodwill allocated to that reporting unit. The update also eliminated the requirements for any reporting unit with a zero or negative carrying amount to perform a qualitative assessment and, if it fails that qualitative test, to perform Step 2 of the goodwill impairment test. An entity is required to disclose the amount of goodwill allocated to each reporting unit with a zero or negative carrying amount of net assets. The guidance was effective for the Company’s interim and annual reporting periods during the year ending December 31, 2020. The adoption of this accounting guidance did not have a materialhad no impact on the Company’s consolidated financial statements.statements and related disclosures. 3. Property, Plant and Equipment In June 2016, Property, plant and equipment consisted of the FASB issued ASU 2016-13,Financial Instruments – Credit Losses, which provides new guidance on the accounting for credit losses on financial instruments. The new guidance introduces an expected loss model for estimating credit losses, replacing the incurred loss model. The new guidance also changes the impairment model for available-for-sale debt securities, requiring the use of an allowance to record estimated credit losses (and subsequent recoveries). The Company adopted the new guidance effective January 1, 2020. The adoption of this accounting guidance had no impact on the Company’s consolidated financial statements.following:
| | | | | | | | | December 31, | | December 31, | | | 2022 | | 2021 | Lab and build-to-suit equipment | | $ | 6,257 | | $ | 6,600 | Office equipment | | | 19 | | | 19 | Furniture and fixtures | | | 134 | | | 177 | Computer equipment | | | 291 | | | 347 | Leasehold improvements | | | 11,409 | | | 11,457 | Construction-in-progress | | | 155 | | | — | Total property, plant and equipment | | | 18,265 | | | 18,600 | Accumulated depreciation and amortization | | | (14,114) | | | (13,583) | Property, plant and equipment, net | | $ | 4,151 | | $ | 5,017 |
3. Acquisition of RareGen LLC (now Liquidia PAH, LLC)
On November 18, 2020 (the “Closing Date”), the Company completed the previously announced acquisition of RareGen, LLC (now Liquidia PAH) (See Note 1).
Reasons for the Acquisition and Merger
The Company acquired Liquidia PAH to improve financial strengthrecorded depreciation and operational efficiencies including the generationamortization expense of cash flow through sales of a generic version of Remodulin, which is a parenteral formulation of treprostinil,$1.4 million and $1.8 million for the treatment of PAH. Strategically, the Company believes that its commercial presence in the field will enable an efficient launch of LIQ861 upon approval, leveraging existing relationships and further validating its reputation as a company committed to supporting PAH patients.
Merger Consideration
The fair value of the purchase consideration or the purchase price, was approximately $20.8 million. The purchase consideration consisted of the 6,166,666 shares of Liquidia Corporation Common Stock based on a per share price of $3.38, which represented the closing price of Liquidia Technologies Common Stock on the Closing Date. 5,550,000 of the shares were issued as of years ended December 31, 2020 2022 and 2021, respectively. Maintenance and repairs are expensed as incurred and were $0.3 million and $0.1 million for the remaining 616,666 shares were withheld from RareGen members to secure their indemnification obligations pursuant to the Merger Agreement.years ended December 31, 2022 and 2021, respectively.
4. Contract Acquisition Costs and Intangible Asset, and Goodwill The total purchase priceContract acquisition costs and allocated purchase price isintangible asset are summarized as follows:
Number of common shares to be issued to RareGen’s members | | | 6,166,666 | | Multiplied by the fair value per share of Liquidia Technologies common stock | | $ | 3.38 | | Total estimated purchase price | | $ | 20,843,331 | |
| | | | | | | | | | | | | | | | | | | | | December 31, 2022 | | December 31, 2021 | | | Gross Carrying Amount | | Accumulated Amortization | | Net Carrying Amount | | Gross Carrying Amount | | Accumulated Amortization | | Net Carrying Amount | Contract acquisition costs | | $ | 12,980 | | $ | (4,376) | | $ | 8,604 | | $ | 12,980 | | $ | (2,842) | | $ | 10,138 | Intangible asset | | $ | 5,620 | | $ | (1,894) | | | 3,726 | | $ | 5,620 | | $ | (1,230) | | | 4,390 |
Accounting for the Acquisition
The acquisition of Liquidia PAH was accounted for as a business combination and reflects the application of acquisition accounting in accordance with Accounting Standards Codification (ASC) 805, Business Combinations. The acquired Liquidia PAH assets, including identifiable intangible assets and liabilities assumed, have been recorded at their estimated fair values with the excess purchase price assigned to goodwill. A preliminary purchase price allocation has been performed and the recorded amounts for intangible assets, other assets, indemnification asset, goodwill, litigation finance payable, deferred tax liability and other liabilities are subject to change pending finalization of valuation efforts and review of tax matters. The amounts recognized will be finalized as the information necessary to complete the analysis is obtained, but no later than one year after the Closing Date.
Purchase Price Allocation
The preliminary purchase price allocation resulted in the following amounts being allocated to the assets acquired and liabilities assumed as of the Closing Date of November 18, 2020 based on their respective preliminary fair values summarized below:
Cash | | $ | 1,000,000 | | Property and equipment | | | 79,330 | | Prepaid and other current assets | | | 30,190 | | Intangible asset | | | 5,620,000 | | Contract acquisition costs | | | 12,980,000 | | Indemnification asset | | | 1,065,538 | | Goodwill | | | 3,903,282 | | Less other current liabilities | | | (492,499 | ) | Less refund liability | | | (2,696,000 | ) | Less litigation finance payable, current | | | (646,510 | ) | Total estimated purchase price | | $ | 20,843,331 | |
Contract Acquisition Costs, Intangible Asset and Goodwill Acquired
Prior to the Merger Transaction, the Company did not have contract acquisition costs, intangible assets or goodwill on its Balance Sheet. Contract acquisition costs and Intangible asset of $12,980,000 and $5,620,000, respectively, were acquired in the Merger Transaction relating to and consisting of the Promotion Agreement. The Company is amortizing the value of the Promotion Agreement contract acquisition costs and customer relationship intangible asset on a pro-rata basis based on the estimated total revenue or net profits to be recognized over the period from November 18, 2020 through December 2032, the termination date of the Merger Transaction through May 2027 (seePromotion Agreement (see Note 2 for Revenue Recognition accounting policy)2-Revenue Recognition). Amortization of contract acquisition costs is recorded as a reduction of revenue and amortization of the intangible asset is recorded as cost of revenue.
The Company recorded amortization related to the contract acquisition costs of $1.5 million and $2.7 million for the years ended December 31, 2022 and 2021, respectively. The Company recorded amortization related to the intangible asset of $0.7 million and $1.1 million for the years ended December 31, 2022 and 2021, respectively. Annual amortization over the next five years is expected to be lower than prior years primarily due to an amendment to the Sandoz Agreement entered into during the fourth quarter of 2022, which extended the term of the Agreement by five years. During the year ended December 31, 2020 the Company recorded total amortizationgoodwill of $187,508 from the contract acquisition costs as a reduction in net service revenue. Net contract acquisition costs totaled $12,792491 as of December 31, 2020. During the year ended December 31, 2020, the Company recorded total amortization of $85,157 from the intangible asset as an increase to cost of revenue. The net intangible asset totaled $5,534,843 as of December 31, 2020. The Company acquired goodwill in the Merger Transaction of $3,903,282$3.9 million, which primarily represents the Liquidia PAH assembled workforce and the residual value of the purchase consideration and assumed liabilities that exceeded the assets acquired (see Note 2 for Goodwill accounting policy). None of the goodwill recognized is expected to be deductible for income tax purposes.
Refund Liability
In accordance with the Promotion Agreement, Liquidia PAH receives consideration from Sandoz in the form of a share of Net Profits for the promotional activities it performs. The share of Net Profits received is subject to adjustments from Sandoz for items such as distributor chargebacks, rebates, inventory returns, inventory write-offs and other adjustments (the “Net Profits Adjustment”)2-Goodwill). As of the date of the Merger Transaction,December 31, 2022, the Company identified approximately $2,696,000concluded that there were no events or changes in circumstances that indicated that the carrying amount of Net Profits Adjustment that are expected to be refunded to Sandoz during 2021 for items that had been incurred prior to the Merger Transaction. The Company has recorded a refund liability as partgoodwill was not recoverable.
5. Indemnification Asset with Related Party and Litigation Finance Payable Prior to the Closing Date of the Merger Transaction,On June 3, 2020, Liquidia PAH entered into a litigation financing arrangement (the “Financing Agreement”) with Henderson SPV, LLC (“Henderson”). Liquidia PAH, along with Sandoz (collectively the “Plaintiffs”), are pursuing litigation against United Therapeutics Corporation (“United Therapeutics”) and, prior to entering into a binding settlement term sheet with Smiths Medical ASC (see Note 13).in November 2020, were pursuing litigation against Smiths Medical. Under the Financing Agreement, Henderson will fund Liquidia PAH’s legal fees and/orand litigation expenses (referred to as “Deployments”) up to a total commitment of $10 million, in exchange for a share of certain litigation or settlement proceeds.
Deployments received from Henderson are recorded as a Litigation finance payable. Litigation proceeds will be split equally between Liquidia PAH and Sandoz. LitigationUnless there is an event of default by Henderson, litigation proceeds received by Liquidia PAH must be applied first to repayment of total Deployments received. Litigation proceeds in excess of Deployments received are split between Liquidia PAH and Henderson according to a formula. ProceedsUnless there is an event of default by PBM, all proceeds received by Liquidia PAH are due to PBM as described further below. In connection with the Merger Transaction,On November 17, 2020, Liquidia PAH entered into a Litigation Funding and Indemnification Agreement (“Indemnification Agreement”) with PBM. PBM is considered to be a related party as it is controlled by a major stockholder (which beneficially owns approximately 11 percent9.3% of Liquidia Corporation Common Stock as of the date of these financial statements) andMarch 1, 2023) who is also a member of the Company’s Board of Directors.
Prior to the Merger Transaction, Liquidia PAH was actively managing the litigation, and had sole decision-making authority over the litigation. Under the terms of the Indemnification Agreement, PBM now controls the litigation, with Liquidia PAH’s only requirementprimarily responsibility being to cooperate to support the litigation proceedings as needed. The Indemnification Agreement also provides that Liquidia PAH and its affiliates will not be entitled to any proceeds resulting from, or bear any financial or other liability for, the United Therapeutics and Smiths Medical ASC litigation.litigation unless there is an event of default by PBM. Any Liquidia PAH litigation expenses not reimbursed by Henderson under the Financing Agreement will be reimbursed by PBM. Any proceeds received which Henderson is not entitled to under the Financing Agreement will be due to PBM. As of the Closing Date of the Merger Transaction, Liquidia PAH recorded as an
The Indemnification Asset $1,065,538. Legalis increased as the Company records third party legal and litigation expenses incurred forrelated to the United Therapeutics and Smiths Medical ASC litigation are recorded as an increase to the Indemnification Asset. Subsequent Deployments received from Henderson are recorded as an increase to the Litigation Finance Payable.
litigation. As of December 31, 2020, 2022, the Indemnification Asset and Litigation Finance Payable were classified as long-term assets and liabilities, respectively as it is considered unlikely that the litigation wouldwill conclude during the year 2021. Liquidia PAH Results of Operations
Liquidia PAH’s results of operations and cash flows are included in the Company’s consolidated financial statements for the period subsequentprior to November 18, 2020 through December 31, 2020, 2023.
6. Accrued Expenses and Liquidia PAH’s assetsOther Current Liabilities Accrued expenses and other current liabilities were recorded at their estimated fair values in the Company’s Consolidated Balance Sheets as of December 31, 2020. Liquidia PAH’s actual results for the period from the Closing Date November 18, 2020 through December 31, 2020, which are included in the Consolidated Statement of Operations and Comprehensive Loss for the year ended December 31, 2020 were as follows: Revenue | | $ | 739,628 | | Costs and expenses: | | | | | Cost of revenue | | | 237,712 | | Research and development | | | 35,919 | | General and administrative | | | 216,787 | | Total costs and expenses | | | 490,418 | | Total operating income and net income | | $ | 249,210 | |
Transaction Costs
In connection with the Merger Transaction, the Company incurred significant one-time expenses in the year ended December 31, 2020 primarily including transaction costs (e.g., bankers’ fees, legal fees, consultant fees, etc.). Total transaction costs recorded in general and administrative expense totaled $4.8 million for the year ended December 31, 2020.
Supplemental Pro Forma Financial Information
The following unaudited pro forma financial information assumes the companies were combined as of January 1, 2019. The unaudited pro forma financial information as presented below is for informational purposes only and is based on estimates and assumptions that have been made solely for purposes of developing such pro forma information. This is not necessarily indicativeconsisted of the resultsfollowing:
| | | | | | | | | December 31, | | December 31, | | | 2022 | | 2021 | Accrued compensation | | $ | 2,862 | | $ | 3,157 | Accrued research and development expenses | | | 1,757 | | | 344 | Accrued other expenses | | | 903 | | | 1,670 | Total accrued expenses and other current liabilities | | $ | 5,522 | | $ | 5,171 |
| | | Years Ended | | | | | December 31, | | | | | 2020 | | | 2019 | | Revenue | | $ | 4,756,625 | | | $ | 18,160,476 | | Net loss | | $ | (57,406,967 | ) | | $ | (52,876,681 | ) | Net loss per common share, basic and diluted | | $ | (1.48 | ) | | $ | (2.15 | ) |
4. Stockholders’7. Stockholders’ Equity
Authorized Capital As of December 31,2020, 2022, the authorized capital of the Company consists of 90,000,000 shares of capital stock, $0.001 par value per share, of which 80,000,000 shares are designated as common stock and 10,000,000 shares are designated as preferred stock. Common Stock Upon any voluntary or involuntary liquidation, dissolution or winding up of the affairs of the Company, the holders of the common stock shall be entitled to receive that portion of the remaining funds to be distributed to the stockholders, subject to the liquidation preferences of any outstanding preferred stock, if any. Such funds shall be paid to the holders of common stock on the basis of the number of shares so held by each of them. Issuance of Common Stock on July 2, 2020 April 18, 2022 from an Underwritten Public Offering On June 29, 2020, Liquidia Technologies entered into an underwriting agreement (the “Underwriting Agreement”) with Jefferies LLC, as representativeApril 12, 2022, the Company sold 11,274,510 shares of the several underwriters named therein (collectively, the “Underwriters”), pursuant to which 9,375,000 shares of Liquidia TechnologiesCompany’s common stock were sold in an underwritten registered public offering at an offering price of $8.00$5.10 per Shareshare (the “Offering”). The Offering closed on July 2, 2020. The grossApril 18, 2022, and the Company received net proceeds of approximately $54.5 million from the offering were $75.0 million and net proceeds were $70.3 million,sale of the shares, after deducting the underwriting discounts and commissions and other offering expenses. The Company intends to use Caligan Partners LP (“Caligan”), the net proceeds from thisCompany’s largest stockholder, and Paul B. Manning, a member of the Company’s board of directors, participated in the Offering and purchased shares of common stock in an aggregate amount of $11.0 million at the public offering price per share and on the same terms as the other purchasers in the Offering. Caligan purchased 1,764,705 shares of common stock in the Offering for ongoing commercial developmentan aggregate purchase price of LIQ861,$9.0 million and Paul B. Manning purchased 392,156 shares of common stock in the Offering for continued developmentan aggregate purchase price of LIQ865 and for general corporate purposes. The Company’s management will retain broad discretion over the allocation of the net proceeds.$2.0 million. Issuance of Common Stock on March 31, 2022 from Merger Transaction On November 18, 2020 (the “Closing Date”), the Company completed the acquisition of RareGen as contemplated by that certain Agreement and Plan of Merger, dated as of June 29, 2020, as amended by a Limited Waiver and Modification to the Merger Agreement, dated as of August 3, 2020 (the “Merger Agreement”). On the Closing Date, an aggregate of 5,550,000 shares of the Company’s common stock, were issued to RareGen members in exchange for all of the issued and outstanding RareGen equity. On March 31, 2022, an aggregate of 616,666 shares of the Company’s common stock, which were held back on the Closing Date for indemnification purposes, were issued to RareGen members. Issuance of Common Stock on April 13, 2021 from a Private Placement in December 2019 On December 23, 2019, Liquidia TechnologiesApril 12, 2021, the Company entered into a Common Stock Purchase Agreement (the “Purchase Agreement”) with a fund and account managed by Caligan Partners LP and certain institutionalother accredited investors (the “Purchasers”) for the sale by Liquidia Technologiesthe Company in a private placement (the “Private Placement”) of an aggregate of 7,164,5348,626,037 shares (the “Private Placement Shares”) of common stock,the Company’s Common Stock at a purchase price of $3.13$2.52 per share. The Private Placement Share. The closing ofclosed on April 13, 2021 and the Private Placement occurred on December 27, 2019. Liquidia Technologies granted the Purchasers indemnification rights with respect to its representations, warranties, covenants, and agreements under the Purchase Agreement. The gross proceeds from the sale of the Private Placement Shares were $22.4 million and net proceeds were $21.0 million, after deducting placement agent fees and offering expenses. Issuance of Common Stock from the ATM Agreement Commencing in August 2019
Liquidia Technologies entered into a sales agreement (the “ATM Agreement”) with Jefferies LLC (“Jefferies”) to issue and sell shares of Liquidia Technologies common stock, having an aggregate offering price of up to $40.0 million, from time to time during the term of the ATM Agreement, through an “at-the-market” equity offering program at Liquidia Technologies’ sole discretion, under which Jefferies acted as Liquidia Technologies’ agent and/or principal. Liquidia Technologies paid Jefferies a commission equal to 3.0% of theCompany received gross proceeds of any common stock sold through Jefferies under the ATM Agreement. During the year ended December 31, 2020, Liquidia Technologies sold 131,425 sharesapproximately $21.7 million.
Issuance of Common Stock from an Underwritten Public Offering in March 2019
In March 2019, Liquidia Technologies closed an underwritten offering of 3,000,000 shares of its common stock at a public offering price of $11.50 per share. The gross proceeds from the offering were $34.5 million and net proceeds were $31.8 million, after deducting underwriting discounts and commissions and other offering expenses.
Warrants During the year ended December 31, 2020, 02022, no warrants to purchase shares of common stock were exercised. During the year ended December 31,2019, 64,629 2021, 40,702 warrants to purchase shares of common stock were exercised. As of December 31, 2020 and 2019, there were2022, outstanding warrants to purchase 106,274 sharesconsisted of common stock with an exercise price of $0.0168 per share. The warrants expire on December 31, 2026.the following: | | | | | | | | | | Number of | | | | | | | | warrants | | Exercise Price | | Expiration Date | A&R SVB Warrant - Initial Tranche (see Note 12) | | 250,000 | | $ | 5.14 | | January 6, 2032 | SVB Warrant - Initial Tranche (see Note 12) | | 100,000 | | $ | 3.05 | | February 26, 2031 | SVB Warrant - Term B and Term C Tranches (see Note 12) | | 100,000 | | $ | n/a | | February 26, 2031 | Other warrants | | 65,572 | | $ | 0.02 | | December 31, 2026 |
5. Share-Based Compensation
8. Stock-Based Compensation 2020 Long-Term Incentive Plan The Company’s 2020 Long-Term Incentive Plan (the “2020“2020 Plan”) was approved by stockholders in November 2020. The 2020 Plan replaced the 2018 Plan as the Company’s primary long-term incentive program. In addition to stock options, the 2020 Plan provides for the granting of stock options, appreciation rights, stock awards, stock units, and other share-based awards. The 2020 Plan providesstock-based awards and for accelerated vesting under certain change of control transactions. A totalThe number of 1,700,000 shares of the Company’s common stock was initially authorized and reservedavailable for issuance under the 2020 Plan. This reserve Plan will automatically increase each subsequent anniversary of on January 1 of each year through 2030, by an amount equal to the smaller of (a) 4% of the number of shares of common stock issued and outstanding on the immediately preceding December 31, or (b) an amount determined by the Board of Directors (the “Evergreen Provision”). On January 1, 2021, 2023, the number of shares of common stock available for issuance under the 2020 Plan automatically increased by 1,733,4322,580,716 shares to 2,955,432 shares from 1,222,000 pursuant to the Evergreen Provision. The Company’s 2018 Long-Term Incentive Plan (the “2018 Plan”) was approved by stockholders in July 2018. In addition to stock options, the 2018 Plan provided for the granting of stock appreciation rights, stock awards, stock units, and other share-based awards. A total of 1,600,000 shares of the Company’s common stock was initially authorized and reserved for issuance under the 2018 Plan.
On January 1, 2019, the number of shares of common stock available for issuance under the 2018 Plan automatically increased by 620,778 shares to 2,220,778 shares from 1,600,0002,851,611 shares pursuant to the Evergreen Provision. On January 1, 2020, the numberAs of December 31, 2022, there were 270,895 shares of common stock available for issuancefuture grants under the 2018 Plan automatically increased by 1,129,250 shares to 2,416,811 shares from 1,287,561 shares pursuant to the Evergreen Provision. The 2018 Plan provides for accelerated vesting under certain change of control transactions. The 2018 Plan was discontinued following stockholder approval of the 2020 Plan, but the outstanding awards under the 2018 Plan will continue to remain in effect in accordance with its terms. Shares that are returned under the 2018 Plan upon cancellation, termination or otherwise of awards outstanding under the 2018 Plan will not be available for grant under the 2020 Plan. As of December 31, 2020 the Company had reserved for issuance 1,455,465 shares of common stock under the 2018 Plan, representing the remaining outstanding options and restricted stock units granted under the 2018Plan.
The 20182020 Plan replaced the 2016Company’s prior equity award plans and 2004 Plans as the Company’s primary long-term incentive program. The 2016 and 2004 Plans weresuch plans have been discontinued, following stockholder approval of the 2018 Plan, buthowever, the outstanding awards under the 2016 and 2004 Plans will continue to remain in effect in accordance with their terms. Shares that are returned under the 2016 and 2004 Plansthese prior plans upon cancellation, termination or otherwiseexpiration of awards outstanding under the 2016 and 2004 Plans will not be available for grant under the 20182020 Plan. As of December 31, 2020, 2022, the Company had reserved for issuance 580,847a total of 673,880 shares of common stock reserved for issuance related to the remaining outstanding equity awards granted under the 2016prior plans. 2022 Inducement Plan On January 25, 2022, the Board approved the adoption of the Company’s 2022 Inducement Plan (the “2022 Inducement Plan”). The 2022 Inducement Plan was recommended for approval by the Compensation Committee of the Board (the “Compensation Committee”), and subsequently approved and adopted by the Board without stockholder approval pursuant to Rule 5635(c)(4) of the rules and regulations of The Nasdaq Stock Market, LLC (the “Nasdaq Listing Rules”). The Company reserved 310,000 shares of the Company's common stock for issuance pursuant to equity awards granted under the 2022 Inducement Plan, and 265,890the 2022 Inducement Plan will be administered by the Compensation Committee. In accordance with Rule 5635(c)(4) of the Nasdaq Listing Rules, equity awards under the 2022 Inducement Plan may only be made to an employee who has not previously been an employee or member of the Board (or any subsidiary of the Company), or following a bona fide period of non-employment by the Company (or a subsidiary of the Company), if he or she is granted such equity awards in connection with his or her commencement of employment with the Company or a subsidiary and such grant is an inducement material to his or her entering into employment with the Company or such subsidiary. As of December 31, 2022, the Company had a total of 12,800 shares available to issue under the 2022 Inducement Plan. Employee Stock Purchase Plan In November 2020, stockholders approved the Liquidia Corporation 2020 Employee Stock Purchase Plan (the “ESPP”). The ESPP allows eligible employees to purchase shares of the Company’s common stock at a discount through payroll deductions, subject to plan limitations. Unless otherwise determined by the administrator, the Company’s common stock will be purchased for the accounts of employees participating in the ESPP at a price per share that is 85% of the lesser of the fair market value of the Company’s common stock on the first and last trading day of the offering period. During the year ended December 31, 2022, 51,941 shares of common stock were issued under the 2004 Plan, representingESPP. As of December 31, 2022, a total of 548,059 shares of the remaining outstanding options grantedCompany’s common stock are reserved for issuance under the 2016 and 2004 Plans.ESPP. On January 1, 2023, in connection with an evergreen provision contained in the ESPP, an additional 150,000 shares of the Company’s common stock were reserved for issuance under the ESPP. CEO Options During December 2020, the Company issued a stock option grant to its then new chief executive officer (the “CEO”)Chief Executive Officer, Damian deGoa, to purchase up to 2,000,000 shares of the Company’s common stock (the “CEO Option”) at the exercise price on the grant date of $3.00 per share. The CEO Option was issued outside of the 2020 Plan and 1,375,000 options vested in the fourth quarter of 2021 upon achievement of certain milestones and the passage of time, and ceased vesting upon the termination of Mr. deGoa’s employment on January 31, 2022. However, the CEO Option will remain exercisable so long as Mr. deGoa remains a director of the Company in accordance with his Separation Agreement. This change to vesting terms was treated as a modification of the original award resulting in a stock-based compensation charge of $2.9 million during the year ended December 31, 2022. On June 16, 2022, pursuant to Roger Jeffs’s executive employment agreement dated January 3, 2022 (the “Jeffs Employment Agreement”), the Company granted Dr. Jeffs 931,745 nonstatutory stock options (the “Second Tranche Option”), with an exercise price per share equal to the closing price of a share of common stock on the date of grant. The Second Tranche Option is subject to the following vesting schedule;schedule: 25% of the CEO Optiongrant vested and became exercisable on January 3, 2023, and the remaining portion of the grant will become vested and exercisable, on the first anniversary of December 14, 2020 and the balance will become vested and exercisableas applicable, in equal monthly installments over the following thirty-six months, subject to the CEO’sDr. Jeffs’ continuous employment with the Company. However,Company on each such vesting date. Notwithstanding the CEO Option is subject toforegoing, in the following accelerated vesting; (i) ifevent of a Change in Control (as defined in the Company receives tentative approval by the U.S. Food and Drug Administration (the “FDA”)2020 Plan), 100% of the Company’s New Drug Application for LIQ861 prior to June 30, 2022, and the CEO is actively employed by the Company on such date, then 25% of the then-unvestedunvested portion of the CEO OptionOptions shall become vested and exercisable as of the closing date of the FDA’s approval; and (ii) if the Company achieves commercial availability of the subcutaneous Treprostinil product with cartridge supplies sufficient to support the market for one year by December 31, 2021, and the CEOsuch Change in Control, provided that Dr. Jeffs is actively employed bywith the Company on such date, then 25% of the then-unvested portion of the CEO Option shall become vested and exercisable as of the date the Company can document by competent proof to the Board of the achievement of such milestone. In addition, the CEO Option will become 100% vested upon certain change of control transactions.date.
Share-BasedStock-Based Compensation Valuation and Expense
Total stock-based compensation expense recognized for employees and non-employees was as follows: | | | | | | | | | | Year Ended | | | | December 31, | | By Expense Category: | | 2022 | | 2021 | | Research and development | | $ | 1,409 | | $ | 1,923 | | General and administrative | | | 7,889 | | | 4,823 | | Total stock-based compensation expense | | $ | 9,298 | | $ | 6,746 | |
The following table summarizes the unamortized compensation expense and the remaining years over which such expense would be expected to be recognized, on a weighted-average basis, by type of award: | | | | | | | | As of December 31, 2022 | | | | | | Weighted | | | | | | Average | | | | | | Remaining | | | | | | Recognition | | | Unamortized | | Period | | | Expense | | (Years) | Stock options | | $ | 15,532 | | 2.9 | Restricted stock units | | $ | 1,723 | | 2.9 |
The Company accounts for its employee share-basedstock-based compensation plans using the fair value method. The fair value method requires the Company to estimate the grant-date fair value of its share-basedstock-based awards and amortize this fair value to compensation expense over the requisite service period or vesting term. The Company uses the Black-Scholes option-pricing model to estimate the fair value of each option grant is estimated using a Black-Scholes option-pricing model. stock options granted and purchase rights issued under the ESPP. For restricted stock units (“RSUs”), the grant-date fair value is based upon the market price of the Company’s common stock on the date of the grant. This fair value is then amortized to compensation expense over the requisite service period or vesting term. The Company recorded the following share-based compensation expense:
| | | Year Ended December 31, | | By Expense Category: | | 2020 | | | 2019 | | Research and development | | $ | 1,099,000 | | | $ | 1,119,382 | | General and administrative | | | 2,855,000 | | | | 2,256,923 | | Total | | $ | 3,954,000 | | | $ | 3,376,305 | |
| | | Year Ended December 31, | | By Type of Award: | | 2020 | | | 2019 | | Stock Options | | $ | 3,817,000 | | | $ | 3,240,376 | | Restricted Stock Units | | | 137,000 | | | | 135,929 | | Total | | $ | 3,954,000 | | | $ | 3,376,305 | |
The following table summarizes the unamortized compensation expense and the remaining years over which such expense would be expected to be recognized, on a weighted-average basis, by type of award:
| | | As of December 31, 2020 | | | | | Unamortized Expense | | | Weighted Average Remaining Recognition Period (Years) | | Stock Options | | $ | 10,977,000 | | | | 3.5 | | Restricted Stock Units | | $ | 359,000 | | | | 3.1 | |
Stock Options
The following table summarizes the assumptions used for estimating the fair value of stock options granted under the Black-Scholes option-pricing model during:during the years ended December 31, 2022 and 2021: | | | Year Ended | | | | | | December 31, | | | | | | 2020 | | | 2019 | | | | | | | | | | | | Year Ended | | | | December 31, | | | | 2022 | | 2021 | Expected dividend yield | | | —% | | | | —% | | | | — | | — | Risk-free interest rate | | 0.40% | - | 1.60% | | | 1.40% | - | 2.40% | | | 1.46% - 3.96% | | 0.62% - 1.67% | Expected Volatility | | 87% | - | 94% | | | 83% | - | 88% | | | Expected volatility | | | 90% - 95% | | 91% - 96% | Expected life (years) | | 5.8 | - | 6.2 | | | | 6.04 | | | | 5.8 - 6.1 | | 5.2 - 6.1 |
As a result of using these
The following table summarizes the assumptions inused for estimating the fair value purchase rights granted to employees under the ESPP under the Black-Scholes option-pricing model the weighted average fair value for options granted during the yearsyear ended December 31, 2020 and 2019 was $2.78 and $8.00 per share, respectively.2022: | | | | | Year Ended | | | December 31, | | | 2022 | Expected dividend yield | | — | Risk-free interest rate | | 0.69% - 3.92% | Expected volatility | | 80% - 129% | Expected life (years) | | 0.50 |
The following describes each of these assumptions and the Company’s methodology for determining each assumption: Expected Dividend YieldYield: The dividend yield percentage is 0zero because the Company neither currently payshas not historically paid dividends nor intendsand does not expect to do so duringfor the expected option term.foreseeable future.
Risk-Free Interest RateRate: The risk-free interest rate is based on the U.S. Treasury yield curve approximating the term of the expected life of the award in effect on the date of grant. Expected VolatilityVolatility: Expected stock price volatility is based on a weighted average of several peer public companies and the historical volatility of the Company’s common stock during the period for which it has traded since the initial public offering. For purposes of identifying peer companies, the Company considered characteristics such as industry, length of trading history and similar vesting terms. Expected LifeLife: The expected life represents the period the awards are expected to be outstanding. The Company’s historical share option exercise experience does not provide a reasonable basis upon which to estimate an expected term because of a lack of sufficient data. Therefore, the Company estimates the expected term by using the simplified method. Stock Options The following table summarizes the Company’s stock option activity including the CEO Option, during the year ended December 31, 2020:2022: | | | Number of
Shares | | | Weighted
Average Exercise
Price | | | Weighted
Average
Contractual
Term
(in years) | | | Aggregate
Intrinsic
Value | | Outstanding as of December 31, 2019 | | | 2,052,976 | | | $ | 9.33 | | | | | | | | | | Granted | | | 3,481,191 | | | $ | 3.75 | | | | | | | | | | Exercised | | | (91,413 | ) | | $ | 2.28 | | | | | | | | | | Cancelled | | | (750,683 | ) | | $ | 8.22 | | | | | | | | | | Outstanding as of December 31, 2020 | | | 4,692,071 | | | $ | 5.51 | | | | 7.2 | | | $ | 37,996 | | Exercisable as of December 31, 2020 | | | 1,167,357 | | | $ | 8.77 | | | | 2.9 | | | $ | 1,196 | | Vested and expected to vest as of December 31, 2020 | | | 4,572,623 | | | $ | 5.47 | | | | 7.2 | | | $ | 37,996 | |
| | | | | | | | | | | | | | | | | | Weighted | | | | | | | | Weighted | | Average | | | | | | | | Average | | Contractual | | Aggregate | | | Number of | | Exercise | | Term | | Intrinsic | | | Shares | | Price | | (in years) | | Value | Outstanding as of December 31, 2021 | | 5,598,009 | | $ | 4.19 | | | | | | Granted | | 4,489,277 | | | 5.22 | | | | | | Exercised | | (233,356) | | | 3.60 | | | | | | Cancelled | | (1,455,668) | | | 5.73 | | | | | | Outstanding as of December 31, 2022 | | 8,398,262 | | $ | 4.49 | | 8.5 | | $ | 17,628 | Exercisable as of December 31, 2022 | | 3,327,055 | | $ | 4.03 | | 7.8 | | $ | 9,442 | Vested and expected to vest as of December 31, 2022 | | 7,914,670 | | $ | 4.47 | | 8.5 | | $ | 16,849 |
The weighted average fair value for options granted during the years ended December 31, 2022 and 2021 was $3.94 and $2.23 per share, respectively. The aggregate intrinsic value of stock options in the table above represents the difference between the $2.95$6.37 closing price of the Company’s common stock as of December 31,2020 2022 and the exercise price of outstanding, exercisable, and vested and expected to vest in-the-money stock options. The following table summarizes information about the Company’s stock options as of December 31,2020:
Exercise Price or Range of Exercise Price | | | Options Outstanding | | | Weighted Average
Contractual Life
(Years) | | | Options Exercisable | | | $1.85 | to | $2.79 | | | | 233,771 | | | | 9.8 | | | | 3,771 | | | | $3.00 | | | | | 2,000,000 | | | | 9.9 | | | | 0 | | | $3.14 | to | $3.40 | | | | 595,233 | | | | 6.0 | | | | 78,174 | | | $3.64 | to | $7.95 | | | | 670,899 | | | | 5.8 | | | | 308,379 | | | $8.00 | to | $9.01 | | | | 8,780 | | | | 4.9 | | | | 6,213 | | | | $9.31 | | | | | 515,990 | | | | 2.8 | | | | 394,976 | | | $10.04 | to | $21.36 | | | | 667,398 | | | | 3.9 | | | | 375,844 | | | | | | | | | 4,492,071 | | | | 7.2 | | | | 1,167,357 | |
Additional information related to our stock options is summarized below: | | | Year Ended December 31, | | | | | | 2020 | | | 2019 | | | Intrinsic value of options exercised | | $ | 176,433 | | | $ | 266,040 | | | | | | | | | | | | | | December 31, | | | | 2022 | | 2021 | Cash proceeds from options exercised | | | $ | 837 | | $ | 41 | Aggregate intrinsic value of options exercised | | | $ | 553 | | $ | 21 | Fair value of options vested | | $ | 4,178,659 | | | $ | 4,131,982 | | | $ | 4,427 | | $ | 6,169 |
During the years ended December 31, 2020 and 2019, 91,413 and 32,325 stock options were exercised for the purchase
Restricted Stock Unit AwardsUnits During the year ended December 31, 2020, the Board of Directors approved grants of an aggregate of 138,614 non-performance-based RSUs to employees. RSUsRestricted Stock Units (“RSUs”) represent the right to receive shares of common stock of the Company at the end of a specified time period. The RSUs vest overperiod or upon the achievement of a four-year period similar to stock options granted to employees.specific milestone. RSUs can only be settled in shares of the Company’s common stock.
A summary of nonvested RSU awards outstanding as of During the year ended December 31, 2020 2022, the Board of Directors approved grants of an aggregate of 503,403 time-based RSUs to employees. 93,834 of these RSUs were issued to Dr. Rajeev Saggar, the Company’s Chief Medical Officer since July 2022, pursuant to his employment agreement of which 50% will vest on the first anniversary of his start date with the balance to vest quarterly through July 2025. 63,230 of these RSUs were issued to Dr. Jeffs pursuant to his employment agreement and changesvest quarterly through January 2023. The remaining 346,339 RSUs vest over a four-year period similar to stock options granted to employees.
The following table summarizes the Company’s RSU activity during the year then ended is as follows: | | | Number of RSUs | | | Weighted
Average Grant-Date Fair Value (per RSU) | | Nonvested as of December 31, 2019 | | | 7,493 | | | $ | 28.87 | | Granted | | | 138,614 | | | | 3.32 | | Vested | | | (2,810 | ) | | | 28.87 | | Forfeited | | | (55,166 | ) | | | 3.31 | | Nonvested as of December 31, 2020 | | | 88,131 | | | $ | 4.68 | |
Employee Stock Purchase Plan
In May 2019, stockholders approved the Liquidia Technologies, Inc. 2019 Employee Stock Purchase Plan (the “2019 ESPP”). A total of 300,000 shares of Liquidia Technologies common stock had been reserved for issuance under the 2019 ESPP Plan. The offering periods were six months each and begin in March and September of each year, with the initial offering period having commenced on September 3, 2019. Under the 2019 ESPP, during Liquidia Technologies’ first offering period from September 1, 2019 to February 28, 2020, based upon 85% of the closing price of $4.13 on February 28, 2020, 3,269 shares were purchased based upon employee withholdings. During Liquidia Technologies’ second offering period from March 1, 2020 to August 31, 2020, based upon 85% of the closing price of $5.12 on August 31, 2020, 1,821 shares were purchased based upon employee withholdings.
In November 2020, stockholders approved the Liquidia Corporation 2020 Employee Stock Purchase Plan (the “2020 ESPP”). Upon adoption of the 2020 ESPP, the 2019 ESPP was terminated. As of December 31, 2020, a total of 300,000 shares of the Company’s common stock are reserved for issuance under the 2020 ESPP. Subject to any plan limitations, the 2020 ESPP allows eligible employees to contribute through payroll deductions up to $25,000 per year of their earnings for the purchase of the Company’s common stock at a discounted price per share. The offering periods are typically six months each and begin in March and September of each year, with the initial three-month offering period to commence on or about June 1, 2021, followed by successive six-month offering periods thereafter. Unless otherwise determined by the administrator, the Company’s common stock will be purchased for the accounts of employees participating in the 2020 ESPP at a price per share that is 85% of the fair market value of the Company’s common stock on the last trading day of the offering period.2022:
F- 21
| | | | | | | | | | Weighted | | | | | Average | | | | | Grant-Date | | | Number of | | Fair Value | | | RSUs | | (per RSU) | Unvested as of December 31, 2021 | | 15,204 | | $ | 3.31 | Granted | | 503,403 | | | 5.64 | Vested | | (54,181) | | | 4.91 | Forfeited | | (56,700) | | | 6.25 | Unvested as of December 31, 2022 | | 407,726 | | $ | 5.57 |
6. License Agreements
The Company performs research under a license agreement with The University of North Carolina at Chapel Hill (“UNC”) as amended to date (the “UNC Letter Agreement”). As part of the UNC Letter Agreement, the Company holds an exclusive license to certain research and development technologies and processes in various stages of patent pursuit, for use in its research and development and commercial activities, with a term until the expiration date of the last to expire patent subject to the UNC Letter Agreement, subject to industry standard contractual compliance. Under the UNC Letter Agreement, the Company is obligated to pay UNC royalties equal to a low single digit percentage of all net sales of drug products whose manufacture, use or sale includes any use of the technology or patent rights covered by the UNC Letter Agreement. The Company may grant sublicenses of UNC licensed intellectual property in return for specified payments based on a percentage of any fee, royalty or other consideration received.
7.9. Revenue From Contracts With Customers
TheOn August 1, 2018, the Company derived its revenue during the year ended December 31, 2020 frompartnered with Sandoz in the Promotion Agreement. DuringAgreement to launch the year ended December 31, 2019, first-to-file generic of Treprostinil Injection for the Company derived its revenue primarily from licensing its proprietary PRINT technology and from performing research and development services. Revenue was recognized as services were performed in an amount that reflectedtreatment of patients with PAH. Under the consideration the Company expected to be entitled to in exchange for those services and technology.
The Company’s research, development and licensing agreements provide for multiple promised goods and services to be satisfied by the Company and include a license to the Company’s technology in a particular field of study, participation in collaboration committees, performance of certain research and development services and obligations for certain manufacturing services.
The transaction price for these contracts includes non-refundable fees and fees for research and development services. Non-refundable up-front fees which may include, for example, an initial payment upon effectiveness of the contractual relationship or payment to secure a right for a future license, are recorded as deferred revenue and recognized into revenue over time asPromotion Agreement, the Company provides certain promotional and nonpromotional activities on an exclusive basis for the research services underproduct in the contract required to advanceUnited States of America for the products to the point where thetreatment of PAH. The Company is able to transfer control of the licensed technology to the customer (“Technology Transfer”). The contract consideration may also include additional non-refundable payments due to the Company based on the achievement of research, development, regulatory or commercialization milestone events. In agreements involving multiple goods or services promised to be transferred to customers, the Company must assess,paid Sandoz $20.0 million at the inception of the contract, whether each promise represents a separate performance obligation (i.e., is “distinct”), or whether such promises should be combined as a single performance obligation. As these goods and services are considered to be highly interrelated, they were considered to represent a single, combined performance obligation. The Company includes an estimate of the probable amount of milestone payments to which it will be entitledPromotion Agreement, in the transaction price. The estimate requires evaluation of factors which are outside of the Company’s control and significantly limit the Company’s ability to achieve the remaining milestone payments. Therefore, the Company has not included any future milestone payments in the transaction price allocated to research, development, and licensing agreements as of December 31,2020. The Company revises the transaction price to include milestone payments once the specific milestone achievement is not considered to be subject to a significant reversal of revenue. At that time, the estimated transaction price is adjusted and a cumulative catch-up adjustment is recorded to adjust the amount of revenue to be recognized from the license inception to the date the milestone was deemed probable of achievement. The milestone is included with other non-refundable up-front fees and recognized into revenue over time as the Company continues to provide services under the contract through the Company’s Technology Transfer. The amount of revenue recognized is based on the proportion of total research services performed to date to the expected services to be provided through the Technology Transfer.
The estimate of the research services to be provided through the Technology Transfer requires significant judgment to evaluate assumptions regarding the level of effort requiredconsideration for the Companyright to have performed sufficient obligationsconduct the promotional and nonpromotional activities for the customer to be able to utilizeproduct. In exchange for its services, the licensed technology without requiring further services from the Company. If the estimated level of effort changes, the remaining deferred revenue is recognized over the revised period in which the expected research services and Technology Transfer are required. Changes in estimates occur for a variety of reasons, including but not limited to (i) research and development acceleration or delays, (ii) customer prioritization of research projects, or (iii) results of research and development activities. The Company recognizes the consideration it is entitled to receive a portion of net profits, as defined within the Promotion Agreement, based on specified profit levels associated with the product. See Note 2 for researchRevenue Recognition accounting policy.
In accordance with the Promotion Agreement, Liquidia PAH receives consideration from Sandoz in the form of a share of Net Profits for the promotional activities it performs. The share of Net Profits received is subject to adjustments from Sandoz for items such as distributor chargebacks, rebates, inventory returns, inventory write-offs and development services, which are primarily billed quarterly in arrears onother adjustments (the “Net Profits Adjustment”). The Company expects to refund certain amounts to Sandoz through a time and materials basis, asreduction of the services are performed (under a proportional performance model) and collection is reasonably assured. Additionally, any up-front or development milestone paymentscash received are also recognized as revenue, over time,from future Net Profits generated under this same proportional performance model. NaN royalties have been recognized during the years ended Promotion Agreement. As of December 31, 2020 or 2019.2022 and 2021, a $0.5 million refund liability is offset against accounts receivable from Sandoz.
In September 2015, GSK Inhaled exercised the option to permanently license the technology for a non-refundable payment to theThe Company derived approximately 98% and 99% of $15.0 million. Pursuant to the license provisions of the collaboration agreement, GSK Inhaled is potentially required to pay the Company for certain milestones reached in addition to tiered royalties on the worldwide sales of the licensed products at percentages rangingits revenue from the mid-single digits to low-single digits depending on the total number of products developed and other royalty step-down events with a fixed low-single digit royalty floor. In February 2016, GSK Inhaled paid the Company a $3.0 million milestone payment pursuant to the collaboration agreement. The combined $18 million in up-front and milestone payments was subject to deferral pursuant to the adoption of ASC 606 and the revenue policy described herein.
In July 2018, GSK notified the Company of its plans to discontinue development of the inhaled antiviral for viral exacerbations in chronic obstructive pulmonary disease under the GSK Inhaled collaboration agreement after completion of the related Phase 1 clinical trial. In June 2019, the Company and GSK executed the third amendment to the collaboration agreement providing the Company rights to develop and commercialize additional inhaled programs at the Company’s sole cost and development. This amendment granted the Company the right to develop three additional molecular entities for application in inhaled programs using the Company’s PRINT technology and a mechanism to acquire further molecular entities for inhaled applications. New inhaled programs developed under this amendment would carry milestone and royalty payments due to GSK upon initiation of Phase 3 studies and subsequent commercialization, respectively. This amendment, among other factors including the lack of continued performance anticipated by the Company and GSK under the original agreement, led the Company to the belief that no further research and development services will be provided to GSK under the collaboration agreement and the earnings process related to the up-front and development milestone payments previously received under the collaboration agreement is completed under the proportional performance model. Therefore, the remaining deferred revenue of $8.1 million was recognized as revenue during the year ended December 31,2019. If GSK were to request additional services under the original agreement, which the Company believes is a remote likelihood, the Company does not expect the value of any incremental efforts that the Company might agree to perform to be material. Any potential milestone or royalty payments from the Company to GSK associated with this amendment will be recorded as operating expenses.
The following tables represent a disaggregation of revenue by each significant research, development and licensing agreement and payment typePromotion Agreement for the years ended December 31,2020 and 2019:
| | | Revenue for the Year Ended December 31, | | | | | 2020 | | | 2019 | | Sandoz Promotion Agreement | | $ | 739,628 | | | | 0 | | GSK Inhaled revenue: | | | | | | | | | Non-refundable milestones | | | 0 | | | | 1,345,320 | | Non-refundable upfront payments | | | 0 | | | | 6,726,600 | | Total GSK Inhaled revenue | | | 0 | | | | 8,071,920 | | Other research and development services | | | 0 | | | | 200 | | Total Revenue | | $ | 739,628 | | | | 8,072,120 | |
Deferred Sublicense Payments
Sublicense payments to UNC are considered direct and incremental fulfillment costs of the Company’s research, development and licensing agreements as the PRINT technology resources used by the Company are continually researched by UNC. These costs are deferred and then amortized into Cost of Sales over the same estimated period of benefit as the period of the underlying revenue recognition. In conjunction with the June 2019 amendment to the GSK collaboration agreement, the balance of deferred sublicense payments was expensed to Cost of revenue during the year ended December 31, 2019. 2022 and 2021, respectively.
8. Property, Plant and Equipment
Property, plant and equipment consisted
| | | December 31, | | | December 31, | | | | | 2020 | | | 2019 | | Lab and build-to-suit equipment | | $ | 7,499,645 | | | $ | 7,562,263 | | Office equipment | | | 31,205 | | | | 128,669 | | Furniture and fixtures | | | 257,774 | | | | 237,951 | | Computer equipment and software | | | 404,558 | | | | 804,046 | | Leasehold improvements | | | 11,524,738 | | | | 11,762,351 | | Construction-in-progress | | | 65,820 | | | | 91,797 | | Total property, plant and equipment | | | 19,783,740 | | | | 20,587,077 | | Accumulated depreciation and amortization | | | (12,978,170 | ) | | | (11,333,112 | ) | Property, plant and equipment, net | | $ | 6,805,570 | | | $ | 9,253,965 | |
The Company recorded depreciation and amortization expense of $2,856,914 and $2,567,742 for the years ended December 31,2020 and 2019, respectively. Maintenance and repairs are expensed as incurred and were $194,192 and $220,658, respectively, for the years ended December 31,2020 and 2019.
The following table details the activity of Construction in Progress (“CIP”) in 2020 and 2019 and the associated transfer to Leasehold Improvements, Laboratory Equipment and Computer Software when the assets were placed in service:
| | | Leasehold | | | Build-to-suit | | | Lab | | | | | | | | | Improvements | | | Equipment | | | Equipment | | | Total | | Balance as of December 31, 2018 | | $ | 63,351 | | | $ | 0 | | | $ | 91,797 | | | $ | 155,148 | | Add: Purchases related to CIP | | | 2,820,640 | | | | 0 | | | | 0 | | | | 2,820,640 | | Less: Transfer due to being placed in service | | | (2,883,991 | ) | | | 0 | | | | 0 | | | | (2,883,991 | ) | Balance as of December 31, 2019 | | | 0 | | | | 0 | | | | 91,797 | | | | 91,797 | | Add: Acquired in Merger Transaction | | | 0 | | | | 65,820 | | | | 0 | | | | 65,820 | | Less: Transfer due to being placed in service | | | 0 | | | | 0 | | | | (91,797 | ) | | | (91,797 | ) | Balance as of December 31, 2020 | | $ | 0 | | | $ | 65,820 | | | $ | 0 | | | $ | 65,820 | |
9.10. Income Taxes
NaNNo provision for federal and state income tax expense has been recorded for the years ended December 31,2020 2022 and 20192021 due to the valuation allowance recorded against the net deferred tax asset and recurring losses.
Deferred income taxes reflect the net tax effect of temporary differences between the carrying amount of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of the Company’s deferred tax assets and liabilities are as follows as of December 31,2020 2022 and 2019:2021: | | | 2020 | | | 2019 | | | | | | | | | | | | | | 2022 | | 2021 | Deferred income tax assets: | | | | | | | | Tax loss carryforwards | | $ | 54,844,147 | | | $ | 42,107,372 | | | $ | 59,241 | | $ | 57,302 | Research and development credits | | 3,995,782 | | | 3,016,889 | | | | 3,942 | | | 4,204 | R&D section 174 costs | | | | 4,584 | | | — | Share-based compensation | | 1,501,172 | | | 973,905 | | | | 4,637 | | | 3,213 | Lease liability | | 1,576,326 | | | 1,508,645 | | | | 1,157 | | | 1,627 | Compensation | | 193,490 | | | 564,572 | | | | 621 | | | 800 | Fixed assets | | 17,421 | | | 0 | | | | 369 | | | 250 | Refund liability | | 620,442 | | | 0 | | | Patent amortization | | 76,017 | | | 86,985 | | | | 476 | | | 325 | Accrued litigation costs | | | | 1,546 | | | 1,641 | Settlement reserve | | | | 123 | | | 141 | Other | | 267,422 | | | 55,972 | | | | 2 | | | 1 | Valuation allowance | | | (61,595,499 | ) | | | (47,505,967 | ) | | | (74,549) | | | (66,987) | Total deferred income tax assets | | 1,496,720 | | | 808,373 | | | | 2,149 | | | 2,517 | Deferred income tax liabilities: | | | | | | | | Fixed assets | | 0 | | | 158,787 | | | Section 481(a) adjustment | | 62,420 | | | 0 | | | | 21 | | | 48 | Intangible assets | | 675,866 | | | 0 | | | | 1,546 | | | 1,679 | Right of use asset | | | 758,434 | | | | 649,586 | | | | 582 | | | 790 | Total deferred income tax liabilities | | | 1,496,720 | | | | 808,373 | | | | 2,149 | | | 2,517 | Total net deferred tax | | $ | 0 | | | $ | 0 | | | $ | — | | $ | — |
As of December 31,2020 2022 and 2019,2021, the Company has established a full valuation allowance against its net deferred tax assets since, at the time, the Company could not assert that it was more likely than not that its deferred tax assets would be realized. As a result, there was an increase in the valuation allowance in 20202022 of approximately $13,980,000.$7.6 million.
As of December 31,2020, 2022, the Company had federal and state income tax loss carryforwards of $238,509,075$278.7 million and $239,195,945,$304.1 million, respectively, which begin to expire in 2024 for federal purposes and in 20212023 for state purposes. In addition, the Company has tax credit carryforwards for federal tax purposes of approximately $4,399,000$4.3 million as of December 31,2020, 2022, which begin to expire in 2026. The utilization of net operating loss and tax credit carryforwards to reduce future income taxes will depend on the Company’s ability to generate sufficient taxable income prior to the expiration of the loss carryforwards. The Internal Revenue Code of 1986, as amended, contains provisions which limit the ability to utilize the net operating loss carryforwards in the case of certain events, including significant changes in ownership interests. If the Company’s net operating loss carryforwards are limited, and the Company has taxable income which exceeds the permissible yearly net operating loss carryforwards, the Company would incur a federal income tax liability even though net operating loss carryforwards would be available in future years. The reasons for the difference between actual income tax expense for the years ended December 31,2020 2022 and 20192021 and the amount computed by applying the statutory federal income tax rate to income before income tax are as follows: | | | 2020 | | | 2019 | | | | | | | | | | % of Pretax | | | | | | % of Pretax | | | | | | Amount | | | Earnings | | | Amount | | | Earnings | | | | | | | | | | | | | | | | | | | 2022 | | 2021 | | | | | | | | % of | | | | | % of | | | | | | | | Pretax | | | | | Pretax | | | | | Amount | | Earnings | | Amount | | Earnings | | Income tax benefit at statutory rate | | $ | (12,550,174 | ) | | 21.0 | % | | $ | (10,019,974 | ) | | 21.0 | % | | $ | (8,613) | | 21.0 | % | $ | (7,261) | | 21.0 | % | State income taxes, net of federal tax benefit | | (1,203,286 | ) | | 2.0 | | | (957,616 | ) | | 2.0 | | | | (1,787) | | 4.4 | | | (2,626) | | 7.6 | | Non-deductible expenses | | 2,929 | | | 0 | | | 94,903 | | | (0.2 | ) | | | 1 | | — | | | — | | — | | Share-based compensation | | 247,011 | | | (0.4 | ) | | 258,338 | | | (0.5 | ) | | Transaction costs | | 573,494 | | | (1.0 | ) | | 0 | | | 0 | | | Stock-based compensation | | | | 310 | | (0.8) | | | 286 | | (0.8) | | Credits | | (978,793 | ) | | 1.6 | | | (634,842 | ) | | 1.3 | | | | — | | — | | | (262) | | 0.8 | | Deferred tax true-up | | | | 1,159 | | (2.9) | | | — | | — | | Change in state rate | | (667 | ) | | 0 | | | 3,887 | | | 0 | | | | 1,368 | | (3.3) | | | 4,454 | | (12.9) | | Other | | (70,721 | ) | | 0.1 | | | 77,009 | | | (0.1 | ) | | | — | | — | | | 18 | | (0.1) | | Change in valuation allowance | | | 13,980,207 | | | | (23.3 | ) | | | 11,178,295 | | | | (23.5 | ) | | | 7,562 | | (18.4) | | | 5,391 | | (15.6) | | Provision for income taxes | | $ | 0 | | | | 0.0 | % | | $ | 0 | | | | 0.0 | % | | $ | — | | — | % | $ | — | | — | % |
The Company has determined that there may be a future limitation on the Company’s ability to utilize its entire federal R&D credit carryover. Therefore, the Company recognized an uncertain tax benefit associated with the federal R&D credit carryover during the years ended December 31,2020 2022 and 2019,2021, as follows: Balance at December 31, 2018 | | $ | 0 | | Increases related to 2019 | | | 158,710 | | Increases related to prior periods | | | 0 | | Balance at December 31, 2019 | | | 158,710 | | Increases related to 2020 | | | 244,698 | | Increases related to prior periods | | | 0 | | Balance at December 31, 2020 | | $ | 403,408 | |
| | | | Balance at December 31, 2020 | | $ | 403 | Increases related to 2021 | | | 52 | Balance at December 31, 2021 | | | 455 | Decreases related to 2022 | | | (65) | Balance at December 31, 2022 | | $ | 390 |
The Company recognizes the tax benefit from an uncertain tax position only if it is more likely than not that the tax position will be sustained on examination by the taxing authorities based on the technical merits of the position. The Company has determined that it had no other material uncertain tax benefits for the year ended December 31,2020. 2022. The Company’s policy for recording interest and penalties related to uncertain tax provisions is to record them as a component of the provision for income taxes. The Company did not have any accrued interest or penalties associated with any unrecognized tax positions as of December 31,2020 2022 and 2019,2021, and there were no such interest or penalties recognized during the years ended December 31,2020 2022 and 2019.2021. On November 18, 2021, North Carolina enacted the 2021 Appropriations Act, which included a gradual corporate income tax rate decrease from the current 2.5% to 0% by 2030. The Company is in a cumulative loss position and does not have significant deferred tax liabilities that can be utilized as a source of taxable income in the future. Therefore, in 2021, the Company reduced its deferred tax asset related to North Carolina NOLs to zero, as no benefit is expected to be realized from these deferred tax assets prior to 2030 when there would be no income tax in North Carolina. The reduction in the value of the deferred tax assets resulted in $5.7 million of cumulative tax expense, which is fully offset by the reduction in the corresponding valuation allowance. If the Company becomes profitable prior to 2030, the Company will recognize an income tax benefit related to the portion of its deferred tax asset related to North Carolina NOLs utilized. The Company has all tax years open to examination by federal tax and state tax jurisdictions. No income tax returns are currently under examination by taxing authorities.
10.11. Leases Commitments and Contingencies
Leases
The Company leases certain laboratory space, office space, and equipment. Leases with an initial term of 12 months or less are not recorded on the Balance Sheet;balance sheet; the Company recognizes lease expense for these leases on a straight-line basis over the lease term. For lease agreements entered into or reassessed after the adoption of Topic 842, the Company combines lease and non-lease components, if any. Most leases include one or more options to renew. The exercise of lease renewal options is at the Company’s sole discretion. Certain leases also include options to purchase the leased property. Consistent with past practice and current intent, the Company has recognized all such purchase options as part of its right-of-use assets and lease liabilities. The depreciable life of assets and leasehold improvements are limited by the expected lease term unless there is a transfer of title or purchase option reasonably certain of exercise. The Company’s lease agreements do not contain any material residual value guarantees or material restrictive covenants. The Company conducts its operations from leased facilities in Morrisville, North Carolina. As of December 31, 2020, the Company’s amended leases for its primary building is for usage of approximately 45,000 square feet in Morrisville, North Carolina with a lease expiration date of space expiring October 31, 2026. The leases are for general office, laboratory, research and development and light manufacturing space. The lease agreements requireIn addition, the Company to pay property taxes, insurance, common area expenses and maintenance costs. In November 2018, the Company amended the lease of its primary building to expand by 8,264 additional square footage expiring October 31, 2026 in exchange for terminating the Company’s other lease with the same landlord for 4,400 noncontiguous square feet. A tenant allowance of approximately $1.0 million was also made available for use to help fund the build out related to the expansion of the primary building lease. The incremental rent over the terminated lease for the first12 months of this lease expansion amounts to $0.1 million, subject to lease escalation in subsequent periods. In June 2019, the Company signed a commitment to incur construction costs of up to $3.1 million related to the leasehold improvements for this lease expansion, against which the tenant allowance will be applied. The leasehold improvements were substantially completed in 2019 and the Company took occupancy of the additional square footage in 2019. The total construction costs incurred in 2019 approximated $2.8 million. The Company leases specialized laboratory equipment under finance leases. The related right-of-use assets are amortized on a straight-line basis over the lesser of the lease term or the estimated useful life of the asset.
The Company does not have access to certain inputs used by its lessors to calculate the rate implicit in its finance leases. As such, the Company utilizesutilized its estimated incremental borrowing rate for the discount rate applied to its finance leases. The original incremental borrowing rate used on finance leases was 7.5%. During February 2021, the Company exercised the lease purchase option for certain finance leases that had expired and entered into a lease modification agreement with its existing lessor for certain other finance leases. The modification resulted in an increase in the remaining lease term of between 24 and 48 months as well as a decrease in the monthly payments associated with the respective modified leases. The incremental borrowing rate used on the modified leases was 6.5%. The lease modification had an immaterial impact on the Company’s 2021 consolidated financial statements. The Company’s lease cost is reflected in the accompanying Statements of Operations and Comprehensive Loss as follows: | | | Year Ended December 31, | | | | | Classification | | 2020 | | | 2019 | | | Operating lease cost | General and administrative | | $ | 780,470 | | | $ | 884,597 | | | | | | | | | | | | | | | | | | Year Ended December 31, | | | | Classification | | 2022 | | 2021 | Operating lease cost: | | | | | | | | | | Fixed lease cost | | | Research and development | | $ | 702 | | $ | 702 | Fixed lease cost | | | General and administrative | | | 78 | | | 78 | Finance lease cost: | | | | | | | | | | Amortization of lease assets | General and administrative | | 1,356,307 | | | 1,316,924 | | | Research and development | | | 135 | | | 267 | Interest on lease liabilities | Interest expense | | | 125,659 | | | 190,687 | | | Interest expense | | | 32 | | | 43 | Total Lease Cost | Total Lease Cost | | $ | 2,262,436 | | | $ | 2,392,208 | | | | | $ | 947 | | $ | 1,090 |
The weighted average remaining lease term and discount rates as of December 31,2020 2022 were as follows: | | | | Weighted average remaining lease term (years): | | | | | Operating leases | | 3.8 | 5.8 | | Finance leases | | 1.9 | 0.9 | | Weighted average discount rate: | | | | | Operating leases | | | 10.3 | % | Finance leases | | | 7.3 | %6.5
| % |
The discount rate for operating leases was estimated based upon market rates of collateralized loan obligations of comparable companies on comparable terms. The future minimum lease payments as of December 31,2020 2022 were as follows: | | | Operating | | Finance | | | | | | | | | | | | | | | | | | | | Operating | | Finance | | | | Year ending December 31: | | Leases | | Leases | | Total | | | Leases | | Leases | | Total | 2021 | | $ | 1,207,708 | | | $ | 957,193 | | | $ | 2,164,901 | | | 2022 | | 1,243,934 | | | 260,857 | | | 1,504,791 | | | 2023 | | 1,283,253 | | | 0 | | | 1,283,253 | | | $ | 1,283 | | $ | 195 | | $ | 1,478 | 2024 | | 1,316,540 | | | 0 | | | 1,316,540 | | | | 1,317 | | | 115 | | | 1,432 | 2025 | | 1,355,923 | | | 0 | | | 1,355,923 | | | | 1,356 | | | 64 | | | 1,420 | Thereafter | | | 1,157,807 | | | | 0 | | | | 1,157,807 | | | 2026 | | | | 1,158 | | | — | | | 1,158 | Total minimum lease payments | | 7,565,165 | | | 1,218,050 | | | 8,783,215 | | | | 5,114 | | | 374 | | | 5,488 | Less: Interest | | | (1,894,194 | ) | | | (39,430 | ) | | | (1,933,624 | ) | | | (882) | | | (22) | | | (904) | Present value of lease liabilities | | $ | 5,670,971 | | | $ | 1,178,620 | | | $ | 6,849,591 | | | $ | 4,232 | | $ | 352 | | $ | 4,584 |
Other Commitments and Contingencies
In connection with the Merger Transaction, we agreed to issue additional consideration of up to 2,708,333 additional shares of common stock to the former equity holders of Raregen (now Liquidia PAH) contingent on achievement of certain Liquidia PAH revenue targets during the year ending December 31, 2021. As of December 31, 2020, the fair value of this contingent consideration was deemed to be immaterial.
In March 2012, the Company entered into an agreement, as amended, with Chasm Technologies, Inc. for manufacturing consulting services related to the Company’s manufacturing capabilities during the term of the agreement. The Company agreed to pay future contingent royalties on net sales totaling no more than $1,500,000, none of which were earned as of December 31, 2020.
We enter into contracts in the normal course of business with contract service providers to assist in the performance of our research and development and manufacturing activities. Subject to required notice periods and our obligations under binding purchase orders, we can elect to discontinue the work under these agreements at any time. In addition, we have entered into a multi-year agreement with LGM Pharma, LLC (LGM) to produce active pharmaceutical ingredients for LIQ861. Under our manufacturing agreement with LGM, we are required to provide rolling forecasts, a portion of which will be considered a binding, firm order, subject to an annual minimum purchase commitment of $3,050,000 for the term of the agreement. The agreement expires five years from the first marketing authorization approval of LIQ861. This minimum commitment was waived for the year ending December 31, 2021.
The Company from time-to-time is subject to claims and litigation in the normal course of business, none of which the Company believes represent a risk of material loss or exposure. See Note 13 for further discussion of pending legal proceedings.
We also have employment agreements with certain employees which require the funding of a specific level of payments, if certain events, such as a change in control or termination without cause, occur.
11.12. Long-Term Debt
Long-term debt consisted of the following asfollowing: | | | | | | | | | | | | | December 31, | | December 31, | | | Maturity Date | | 2022 | | 2021 | A&R Silicon Valley Bank term loan | | December 1, 2025 | | $ | 19,879 | | $ | — | Silicon Valley Bank term loan | | September 1, 2024 | | | — | | | 10,410 | Long-term debt | | | | $ | 19,879 | | $ | 10,410 |
On January 9, 2023, the Company entered into a Revenue Interest Financing Agreement (the “RIFA”) with HealthCare Royalty Partners IV, L.P. (“HCR”) and HealthCare Royalty Management, LLC, pursuant to which and subject to the terms and conditions contained therein, the HCR agreed to pay the Company an aggregate investment amount of December 31, 2020 and 2019: | | | | December 31, | | | December 31, | | | | Maturity Date | | 2020 | | | 2019 | | Pacific Western Bank note | October 25, 2022 | | $ | 10,292,485 | | | $ | 15,878,121 | | Less current portion | | | 0 | | | | (5,585,637 | ) | Long-term debt, less current portion | | $ | 10,292,485 | | | $ | 10,292,484 | |
up to $100.0 million (the “Investment Amount”). $32.5 million of the Investment Amount was funded on January 27, 2023, $22.4 million of which was used to satisfy the Company’s existing obligations under the A&R SVB LSA (defined below), with the excess proceeds funded to the Company. Note 16. Subsequent Events for more information. Pacific Western BankAmended and Restated Loan and Security Agreement dated January 7, 2022
In October 2018, Liquidia Technologies and Pacific WesternOn January 7, 2022 (the “A&R SVB LSA Effective Date”), the Company entered into an Amended and Restated Loan and Security Agreement with SVB and SVB Innovation Credit Fund VIII, L.P. (“AInnovation”) (the “A&R SVB LSA”) in which Liquidia Technologies received an initial tranche of $11.0 million to extinguish its existing debt of $8.0 million under the LSA, repay in full the $1.8 million in outstanding indebtedness under a promissory note and for general corporate purposes. The indebtedness under the A&R LSA bore interest at the greater of the Prime rate or 5% and had a four-year term maturing in October 2022. . The A&R SVB LSA provided for access toestablished a second trancheterm loan facility in the aggregate principal amount of up to $5.0$40.0 million available in three tranches.$20.0 million was funded on the A&R SVB LSA Effective Date, $10.5 million of which was used to be drawn at Liquidia Technologies’ option through June 30, 2019. satisfy its existing obligations under the SVB LSA (see below). The second tranche became accessible as a resultCompany accounted for the repayment of the full enrollmentSVB LSA in accordance with ASC 405-20, Extinguishments of the LIQ861 INSPIRE clinical trial, without observing any materially adverse data through the two-week endpoint. The entire second tranche of $5.0 million was drawn by the CompanyLiabilities, which resulted in May 2019 bringing the total amount outstanding to $16.0 million. Both tranches required payments of interest-only through December 31, 2019.
The A&R LSA carried a one-time success fee of $375,000 and a prepayment penalty of 1% if the drawn tranche was prepaid prior to October 27, 2020. The success fee was triggered in December 2019 by the sale of common stock and was recorded as interest expense of $375,000loss on extinguishment during the year ended December 2019. Accrued interest was included in Other accrued expenses in the Balance Sheets as of December 31, 2020 2022 of $1.0 million.
The A&R SVB LSA was to mature on December 1, 2025 and 2019.consisted of interest-only payments through December 31, 2023. The minimum cash covenant was $8.5 million. Pacific Western maintainedoutstanding principal amount of the term loans accrued interest at a blanket lien on all assets excludingfloating rate per year equal to the greater of 7.25% and the prime rate of interest plus 4.0%. The A&R SVB LSA contains customary affirmative and negative covenants, including but not limited to certain financial covenants, protection of intellectual property for which it was provided a negative pledge. Pursuant torights, the A&R LSA, the Company was also obligated to comply with various other customary covenants, including, among others, restrictions on its ability to disposedisposition of certain assets, replace or suffer the departure of the CEO or CFO without delivering ten days’ prior written notification to Pacific Western, suffer a change on the Board of Directors which would result in the failure of at least one partner of Canaan Partners or their respective affiliates to serve as a voting member in each case without having used best efforts to deliver at least 15 days’ prior written notification to Pacific Western, make acquisitions, be acquired, incur indebtedness, grant liens, make distributions to its stockholders, make investments, enter into certain transactions with affiliates or pay down subordinated debt, subject to specified exceptions.
In May 2019, the Company and Pacific Western entered into a First Amendment to A&R LSA to provide for a limit of $2.5 million of Liquidia Technologies’ capital expenditures during the year ended December 31, 2019. As of December 31, 2020, thematerial adverse changes. The Company was in compliance with all such covenants underat December 31, 2022.
As an inducement to enter into the A&R LSA. On July 3, 2020, SVB LSA, the Company Liquidia Technologies, Liquidia Merger Subissued SVB, Innovation, and RareGen Merger Sub entered intoInnovation Credit Fund VIII-A L.P. (“Innovation Credit”) certain warrants to purchase shares of the Company’s common stock pursuant to the Warrant to Purchase Stock agreements by and between the Company and each recipient (collectively, the “A&R SVB Warrants”). The respective A&R SVB Warrants provided recipients the right to obtain a Joindertotal of 250,000 shares of the Company’s stock at an exercise price of $5.14 per share. The A&R SVB Warrants provide an option for a cashless exercise.
In accordance with ASC 470, Debt, the value of the A&R SVB Warrants and Second AmendmentA&R SVB LSA was allocated using a relative fair value allocation. The fair value of the A&R SVB Warrants was determined to be $1.3 million and included in additional paid-in-capital, of which $0.7 million was recognized as a component of the loss on extinguishment and $0.6 million as a debt discount. The remaining $19.4 million was allocated to the A&R LSA (the “Second Amendment”), with Pacific Western pursuant to which, among other things, (i)SVB LSA. In addition, the Company Liquidia Merger Subincurred fees of less than $0.1 million, which were recorded as debt issuance costs. The debt discount and RareGen Merger Sub executed a joinderdebt issuance costs are being amortized to interest expense and the Final Payment Fee is being accreted using the effective interest method over the term of the A&R LSA forSVB LSA. The Company evaluated the purposefeatures of acknowledging that it shall be a “Borrower” along with Liquidia Technologies thereunder and, in connection therewith, collaterally assigned and transferred to Pacific Western, and grant the Bank a continuing security interest in, all of such parties’ now owned and existing or hereafter acquired and arising assets of “Collateral” (as defined in the A&R LSA)SVB LSA and A&R SVB Warrants in accordance with ASC 480, Distinguishing Liabilities from Equity and ASC 815, (ii)Derivatives and Hedging. The Company determined that the Borrowers shall A&R SVB LSA and A&R SVB Warrants did not make contain any payments pursuant tofeatures that certain Litigation Fundingwould qualify as a derivative or embedded derivative. In addition, the Company determined that the A&R SVB Warrants should be classified as equity. The estimated fair value of the A&R SVB Warrant was calculated using the Black-Scholes Option Pricing Model based on the following inputs: | | | Expected dividend yield | | — | Risk-free interest rate | | 1.76% | Expected volatility | | 97.2% | Expected life (years) | | 10.0 |
Loan and IndemnificationSecurity Agreement entered into by and between RareGen and the Members’ Representative prior to closing the Merger Transaction, without Pacific Western’s consent unless such payments are reimbursed within 30 days and do not exceed $250,000 at any time and (iii) the definition of “Permitted Indebtedness” was amended to accommodate the transactions contemplated by the Litigation Funding and Indemnification Agreement. On dated February 26, 2021 (the “Effective Date”), the
The Company and its two wholly owned subsidiaries, Liquidia Technologies and Liquidia PAH, entered into a Loan and Security Agreement with SVB on February 26, 2021 (the “Loan Agreement”“Effective Date”) and a First Loan Modification Agreement with Silicon Valley Bank, a California corporation, as lender (“SVB”SVB on August 26, 2021 (the “SVB LSA”). The Loan AgreementSVB LSA established a term loan facility in the aggregate principal amount of up to $20.5 million, (the “Term Loan Facility”). An initialof which $10.5 million (the “Term A Loan”) was funded on March 1, 2021 and was used to satisfy the Company’s existing obligations under the A&R LSA, consisting of approximately $9.4 million, in outstanding principal and interest, and such obligations are considered fully repaid and terminated as of that date, with the excess proceeds funded to the Company. As a resultThe Company accounted for the repayment of the refinance, the entire balance of outstanding debt related to the A&R LSA at December 31, 2020 has been presented as noncurrentloan obligation in accordance with ASC 470-10-45-14. See Note 14 for additional details regarding405-20, Extinguishments of Liabilities, which resulted in a loss on extinguishment during the nine months ended September 30, 2021 of less than $0.1 million. In connection with the Loan Agreement.Agreement, the Company issued to SVB a warrant, dated as of the Effective Date to purchase 200,000 shares of common stock (the “SVB Warrant”), of which 100,000 shares vested on the Effective Date, with an exercise price per share equal to $3.05 (the “Initial Tranche”). The remaining 100,000 shares did not vest as additional amounts were not funded under the SVB LSA (the “Term B and C Tranches”). The Company evaluated the features of the SVB LSA and SVB Warrant in accordance with ASC 480, Distinguishing Liabilities from Equity and ASC 815, Derivatives and Hedging. The Company determined that the Loan Agreement and Warrant did not contain any features that would qualify as a derivative or embedded derivative. In addition, the Company determined that the SVB Warrant should be classified as equity. The estimated fair value of the SVB Warrant of was calculated using the Black-Scholes Option Pricing Model based on the following inputs: | | | Expected dividend yield | | — | Risk-free interest rate | | 1.43% | Expected volatility | | 90.8% | Expected life (years) | | 10.0 |
13. Defined Contribution Retirement Plan The Company maintains a defined contribution 401(k)401(k) retirement plan for its employees, pursuant to which employees who have completed sixty days of service may elect to contribute a portion of their compensation on a tax-deferred basisbasis. The Company matches 100% of eligible employee contributions up to 4% of an employee’s salary, subject to the maximum amount permitted by the Internal Revenue Code. The Company provides a 4% matching contribution to eligible employee contributions. Matching contributions are paid subsequent to the year to which they relate. The Company’s matching contributions were $0.4 million and $0.3 million for the years ended December 31, 2020 2022 and 2019 were $416,345 and $400,378, respectively, and such amounts were included in Other Accrued Expenses on the Consolidated Balance Sheets as of December 31,2020 and 2019,2021, respectively. 13.14. Legal Proceedings
YUTREPIA-Related Litigation LIQ861-Related Litigation
On In June 4, 2020, United Therapeutics filed a complaint for patent infringement against usthe Company in the U.S. District Court for the District of Delaware (Case No.1:20-cv-00755-UNA)20-cv-00755-RGA) (the “Hatch-Waxman Litigation”), asserting infringement by usthe Company of U.S. Patent Nos. 9,604,901, entitled “Process to Prepare Treprostinil, the Active Ingredient in Remodulin®” (the “’901“‘901 Patent”), and 9,593,066, entitled “Process to Prepare Treprostinil, the Active Ingredient in Remodulin®” (the “’066“‘066 Patent”), relating to United Therapeutics’ Tyvaso®, a nebulized treprostinil solution for the treatment of PAH. On July 16, 2020, we filed an answer to United Therapeutics’ complaint and also included counterclaims of invalidity, non-infringement, and Orange Book de-listing of the ’901 Patent and ’066 Patent. United Therapeutics seeks a judgment that the asserted patents are infringed and an injunction of FDA final approval and subsequent commercial launch of LIQ861 product until after the latest to expire asserted patent. United Therapeutics’ complaint iswas in response to the Company’s NDA for LIQ861,YUTREPIA, filed with the FDA, requesting approval to market LIQ861,YUTREPIA, a dry powder inhalation of treprostinil for the treatment of PAH. The LIQ861YUTREPIA NDA was filed under the 505(b)(2)505(b)(2) regulatory pathway with Tyvaso® as the reference listed drug. Under the Hatch-Waxman Act, the FDA is automatically precluded from approving the LIQ861 NDA for up to 30 months, absent an earlier judgment unfavorable to United Therapeutics by the court. Although the Company believes its LIQ861 dry powder inhaler for the treatment of PAH is highly differentiated from Tyvaso®, since the Company is seeking approval of the LIQ861 NDA under the 505(b)(2) regulatory pathway, the LIQ861 NDA is subject to the provisions of the Hatch-Waxman Act.
On In July 21, 2020, the U.S. Patent and Trademark Office (the “USPTO”) issued U.S. Patent No.10,716,793 (the ““‘793 Patent”), entitled “Treprostinil Administration by Inhalation”, to United Therapeutics. On In July 22, 2020, United Therapeutics filed an amended complaint in the Hatch-Waxman Litigation asserting infringement of the ‘793‘793 Patent by the practice of LIQ861. The infringement allegationYUTREPIA.
In June 2021, the Court held a claim construction hearing. Based on the Court’s construction of the ‘793 Patent is separate from the 30-month regulatory stay on final approvalclaim terms, United Therapeutics filed a stipulation of the NDA for LIQ861, which is only associated with the infringement allegations of the ‘901 Patent and the ‘066 Patent. The Company intends to make a certificationpartial judgment with respect to the ‘793‘901 Patent in its future resubmissionDecember 2021 under which United Therapeutics agreed to the entry of judgment of the NDA for LIQ861.Company’s non-infringement of the ’901 Patent. United Therapeutics’ motionTherapeutics preserved its appellate rights with respect to dismiss the Company’s invalidity defenses and counterclaims concerning‘901 Patent in the ‘793 Patent was denied byevent the U.S. District Court forCourt’s construction of those terms is reversed. Trial proceedings in the District of Delaware on November 3, 2020. On July 30, 2020, Hatch-Waxman Litigation were held in March 2022. In August 2022, Judge Andrews, who was presiding over the Hatch-Waxman Litigation, conductedissued an opinion that claims 1, 2, 3, 6 and 9 of the ‘066 Patent were invalid, that the remaining asserted claims of the ‘066 Patent were not infringed by the Company, and that all of the asserted claims of the ‘793 Patent were both valid and infringed by the Company, based on the arguments presented by the Company in the Hatch-Waxman Litigation. In September 2022, Judge Andrews entered a scheduling conferencefinal judgment in the Hatch-Waxman Litigation that incorporated the findings from his opinion and setordered that the effective date of any final approval by the FDA of YUTREPIA shall be a claim construction hearingdate which is not earlier than the expiration date of the ’793 Patent, which will be in May 2021 2027. Both the Company and setUnited Therapeutics have appealed Judge Andrews’ decision to the trial to begin in March 2022.United States Court of Appeals for the Federal Circuit. The appeal remains pending.
On March 30, 2020, In September of 2022, following entry of final judgment, the Company filed a motion requesting that Judge Andrews stay enforcement of the order delaying the effective date of any final approval by the FDA of YUTREPIA until the expiration of the ’793 Patent. Briefing on the motion for stay of enforcement is complete, and the motion remains pending with the Court.
In March 2020, the Company filed two petitions for inter partes review with the Patent Trial and Appeal Board (the “PTAB”) of the USPTO. One petition was for inter partes review of the ‘901‘901 Patent and sought a determination that the claims in the ‘901‘901 Patent are invalid, and a second petition was for inter partes review of the ‘066‘066 Patent and sought a determination that the claims in the ‘066‘066 Patent are invalid. Both the ‘901 Patent and ‘066 Patent are owned by United Therapeutics, and both patents are related to U.S. Patent No.8,497,393 which was granted to United Therapeutics and subsequently invalidated by the USPTO in an inter partes review instituted in 2016 by SteadyMed Ltd. On In October 13, 2020, the PTAB instituted an inter partes review of the ‘901‘901 Patent and concurrently denied institution on the ‘066‘066 Patent, stating that the ‘066‘066 petition has not established a reasonable likelihood that it would prevail in showing that at least one of the challenged claims is unpatentable. AIn October 2021, the PTAB issued a final written decision determining the validityconcluding that seven of the challenged claims in the ‘901 patent were unpatentable, leaving only the narrower dependent claims 6 and 7, both of which require actual storage at ambient temperature of treprostinil sodium. In November 2021, United Therapeutics submitted a rehearing request with respect to the PTAB’s decision in the inter partes review of the ‘901‘901 Patent. The rehearing request was denied in June 2022. In August 2022, United Therapeutics appealed the decision of the PTAB with respect to the ‘901 Patent is expected within 12 months from institution.to the United States Court of Appeals for the Federal Circuit. The appeal remains pending. On In January 7, 2021, the Company filed a petition for inter partes review with the PTAB relating to the ‘793 patent,‘793 Patent, seeking a determination that the claims in the ‘793 patent‘793 Patent are invalid. A determinationIn August 2021, the PTAB instituted an inter partes review of the ‘793 Patent, finding that the Company had demonstrated a reasonable likelihood that it would prevail with respect to showing that at least one challenged claim of the ‘793 patent is unpatentable as obvious over the combination of certain prior art cited by the Company in its petition to the PTAB. In July 2022, the PTAB to institute the petition is expectedruled in the third quarter of 2021, and a final written decision determiningCompany’s favor, concluding that based on the validitypreponderance of the challengedevidence, all the claims of the ‘793 patent, if’793 Patent have been shown to be unpatentable. In August 2022, United Therapeutics submitted a rehearing request with respect to the petition is instituted byPTAB’s decision in the inter partes review of the ‘793 Patent. The rehearing request was denied in February 2023. United Therapeutics has publicly stated that it will appeal the PTAB’s decision with respect to the ‘793 Patent. The PTAB’s decision with respect to the ‘793 Patent will not override Judge Andrews’ order in the Hatch-Waxman Litigation that YUTREPIA may not be approved due to infringement of the ‘793 Patent unless and until the decision of the PTAB is expected within 12 months from institution.affirmed on appeal.
Trade Secret Litigation Liquidia PAH-RelatedIn December 2021, United Therapeutics filed a complaint in the Superior Court in Durham County, North Carolina, alleging that the Company and a former United Therapeutics employee, who later joined the Company as an employee many years after terminating his employment with United Therapeutics, conspired to misappropriate certain trade secrets of United Therapeutics and engaged in unfair or deceptive trade practices. In January 2022, the Company’s co-defendant in the lawsuit removed the lawsuit to the United States District Court for the Middle District of North Carolina. Subsequently, in January 2022, United Therapeutics filed an amended complaint eliminating their claim under the federal Defend Trade Secrets Act and a motion seeking to have the case remanded to North Carolina state court. In April 2022, the Court granted United Therapeutics’ motion to have the case remanded to North Carolina state court. In May 2022, the Company filed a motion to dismiss all of the claims made by United Therapeutics in the lawsuit. The motion was denied by the Court in October 2022. Discovery in the case is ongoing.
RareGen Litigation On In April 16, 2019, Sandoz and Liquidia PAH (then known as RareGen) filed a complaint against United Therapeutics and Smiths Medical in the District Court of New Jersey (Case No.No.3:19-cv-10170)19-cv-10170), (the “UTC/Smiths Medical litigation”“RareGen Litigation”), alleging that United Therapeutics and Smiths Medical violated the Sherman Antitrust Act of 1890, state law antitrust statutes and unfair competition statutes by engaging in anticompetitive acts regarding the drug treprostinil for the treatment of PAH. On In March 20, 2020, Sandoz and Liquidia PAH filed a first amended complaint adding a claim that United Therapeutics breached a settlement agreement that was entered into in 2015, in which United Therapeutics agreed to not interfere with Sandoz’s efforts to launch its generic treprostinil, by taking calculated steps to restrict and interfere with the launch of Sandoz’s competing generic product. United Therapeutics developed treprostinil under the brand name Remodulin® and Smiths Medical manufactured a pump and cartridges that are used to inject treprostinil into patients continuously throughout the day. Sandoz and Liquidia PAH allege that United Therapeutics and Smiths Medical entered into anticompetitive agreements (i) whereby Smiths Medical placed restrictions on the cartridges such that they can only be used with United Therapeutics’ branded Remodulin® product and (ii) requiring Smiths Medical to enter into agreements with specialty pharmacies to sell the cartridges only for use with Remodulin®.
On January 29,In November 2020, the court denied Liquidia PAH’s and Sandoz’s motion for a preliminary injunction and United Therapeutics’ and Smiths Medicals’ motion to dismiss. On November 6, 2020, Sandoz and Liquidia PAH entered into a binding term sheet (the “Term Sheet”) with Smiths Medical in order to resolve the outstanding UTC/Smiths Medical litigationRareGen Litigation solely with respect to disputes between Smiths Medical, Liquidia PAH and Sandoz. In April 2021, Liquidia PAH and Sandoz entered into a Long Form Settlement Agreement (the “Term Sheet”“Settlement Agreement”). In accordance with Smiths Medical to further detail the terms of the settlement among such parties as reflected in the Term Sheet. Pursuant to the Term Sheet and the Settlement Agreement, the former RareGen members and Sandoz received a payment of $4.25 million or the Settlement Proceeds, whichthat was evenly split between the parties. In addition, pursuant to the
Term Sheet and Settlement Agreement, Smiths Medical disclosed and made available to Sandoz and Liquidia PAH certain specifications and other information related to the cartridge that Smiths Medical developed and manufactures for use with the CADD-MS 3 Infusion infusion pump or the CADD-MS (the “CADD-MS 3 Cartridge. Cartridge”). Pursuant to the Term Sheet,Settlement Agreement, Smiths Medical also granted Liquidia PAH and Sandoz a non-exclusive, royalty-free license in the United States to Smiths Medical’s patents and copyrights associated with the CADD-MS 3 Cartridge and certain other information for use of the CADD-MS 3 pump and the CADD-MS 3 Cartridges. AsSmiths also agreed in the Settlement Agreement to provide information and assistance in support of Liquidia PAH’s efforts to receive FDA clearance for the RG 3ml Medication Cartridge (the “RG Cartridge”) and to continue to service certain CADD-MS 3 pumps that are available for use with the Treprostinil Injection through January 1, 2025. Liquidia PAH and Sandoz agreed, among other things, to indemnify Smiths from certain liabilities related to the RG Cartridge. In September 2021, United Therapeutics filed a motion for summary judgment with respect to all of the dateclaims brought by Sandoz and Liquidia PAH against United Therapeutics. At the same time, Sandoz filed a motion for summary judgment with respect to the breach of this Annual Report on Form 10-K,contract claim. In March 2022, the UTC/Smiths MedicalCourt issued an order granting partial summary judgment to United Therapeutics with respect to the antitrust and unfair competition claims, denying summary judgment to United Therapeutics with respect to the breach of contract claim, and granting partial summary judgment to Sandoz with respect to the breach of contract claim. The RareGen Litigation will now proceed to a trial to determine the amount of damages due from United Therapeutics to Sandoz with respect to the breach of contract claim. The Court has ordered that a three-day bench trial will be scheduled for summer of 2023. Under the Promotion Agreement, all proceeds from the litigation was still in process.will be divided evenly between Sandoz and Liquidia PAH. Under the litigation finance agreements that Liquidia PAH has entered into with Henderson and PBM, any net proceeds received by Liquidia PAH with respect to the RareGen Litigation will be divided between Henderson and PBM. F- 29
14. Subsequent Event15. Commitments and Contingencies
Mainbridge Health Care Device Development and Supply Agreement On February 26, 2021 (December 1, 2022, the “Effective Date”Company entered into a Device Development and Supply Agreement (the “Pump Development Agreement”), with Mainbridge Health Partners, LLC (“Mainbridge”) and Sandoz Inc. (“Sandoz”). The Pump Development Agreement provides for the cooperation between the Company, Sandoz and Mainbridge to develop a new pump that is suitable for the subcutaneous administration of Treprostinil Injection. Mainbridge will perform all development, validation and testing activities required for the pump and related consumables in anticipation of submitting a 510(k) clearance application for the pump to the FDA in 2023. In connection with the Pump Development Agreement, the Company and its two wholly owned subsidiaries, Liquidia TechnologiesSandoz have agreed to pay Mainbridge certain future contingent milestone payments in accordance with the terms and Liquidia PAH entered into a Loan and Security Agreement (the “Loan Agreement”) with Silicon Valley Bank, a California corporation, as lender (“Lender”).conditions set forth therein. UNC License Agreement The Loan Agreement establishedCompany performs research under a term loan facility in the aggregate principal amountlicense agreement with The University of upNorth Carolina at Chapel Hill (“UNC”) as amended to $20.5 milliondate (the “Term Loan Facility”“UNC License Agreement”). An initial $10.5 million (the “Term A Loan”) was funded to the Company on the Effective Date. Availability of $5.0 million under the second trancheAs part of the Term Loan Facility (the “Term B Loan”) is conditioned upon Liquidia having received tentative U.S. Food and Drug Administration (FDA) approval for LIQ861 by June 30, 2022, and availability of $5.0 million under the third tranche of the Term Loan Facility (the “Term C Loan” and, collectively with the Term A Loan and Term B Loan, the “Term Loans”) is conditioned upon Liquidia having received final and unconditional FDA approval for LIQ861 by December 31, 2022. The entire Term A Loan was used to satisfy the Company’s existing obligations under its previously disclosed Amended and Restated Loan and Security Agreement, dated as of October 26, 2018, as amended, by and between Liquidia and Pacific Western Bank, consisting of approximately $9.4 million in outstanding principal and interest, and such obligations are considered fully repaid and terminated. As security for its obligations under the Loan Agreement, Liquidia granted Lender a continuing security interest in substantially all of the assets of Liquidia, other than intellectual property.
The Term Loans made under the Term Loan Facility mature on September 1, 2024 (the “Maturity Date”) and have an interest-only monthly payment period through March 31, 2023 (the “Interest-Only Period”). Following the Interest-Only Period, the Company will begin making monthly payments of principal and interest until the Maturity Date. Interest will accrue on the unpaid principal balance of the outstanding Term Loans at a floating per annum rate equal to the greater of (i) the Wall Street Journal prime rate plus 0.75% and (ii) four percent (4.0%). Furthermore, on the earliest to occur of (x) the Maturity Date, (y) the date the Term Loans are repaid in full or (z) the date of termination of the LoanUNC License Agreement, the Company shall payholds an exclusive license to Lender five percent (5.0%)certain research and development technologies and processes in various stages of the aggregate original principal amount of all Term Loans made by the Lender (the “Final Payment”).
In the event that Liquidia elects to terminate the Term Loan Facilitypatent pursuit, for use in its entirety, it may do so at any time by payingresearch and development and commercial activities, with a term until the outstanding principal balance, unpaid accrued interest, the Final Payment and a prepayment fee equal to (i) five percent (5.0%) of the outstanding principal balance, if such prepayment is made during the Interest-Only Period or (ii) zero, if such prepayment is made after the Interest-Only Period and before the Maturity Date.
Subject to certain exceptions, the Loan Agreement contains covenants prohibiting the Company from, among other things, and subject to certain limited exceptions: (a) conveying, selling, leasing, transferring or otherwise disposing of its properties or assets; (b) liquidating or dissolving; (c) engaging in any business other than the business currently engaged in or reasonably related thereto by it or any of its subsidiaries; (d) engaging in mergers or acquisitions; (e) incurrence of additional indebtedness; (f) allowing any lien or encumbrance on any of its property; (g) paying any dividends; (h) repurchasing its equity; and (i) making payment on subordinated debt. In addition, the Loan Agreement requires Liquidia to maintain an unrestricted and unencumbered “Minimum Cash Balance” (as defined therein) equal to at least (i) $30.0 million during the period commencing on the Effective Date and including the date immediately prior to the fundingexpiration date of the Term B Loan (the “Term B Loan Funding Date”) and (ii) during the period commencing on the Term B Loan Funding Date through and including the date immediately priorlast to expire patent subject to the funding dateUNC License Agreement, subject to industry standard contractual compliance. Under the UNC License Agreement, the Company is obligated to pay UNC royalties equal to a low single digit percentage of all net sales of drug products whose manufacture, use or sale includes any use of the Term C Loan (the “Term C Loan Funding Date”), $35.0 million. Moreover,technology or patent rights covered by the UNC License Agreement, including YUTREPIA. The Company may grant sublicenses of UNC licensed intellectual property in return for specified payments based on a percentage of any fee, royalty or other consideration received.
Chasm Technologies In March 2012, the eventCompany entered into an agreement, as amended, with Chasm Technologies, Inc. for manufacturing consulting services related to the Minimum Cash Balance is not achieved during any calendar quarterCompany’s manufacturing capabilities during the term of the Loan Agreement,agreement. The Company agreed to pay future contingent milestones and royalties on net sales totaling no more than $1.5 million, none of which has been earned as of December 31, 2022. Employment Agreements The Company has agreements with certain employees which require the Loan Agreement requires Liquidia to maintain cumulative “Cash Burn” (as definedfunding of a specific level or payments if certain events, such as a change in control or termination without cause, occur. Purchase Obligations The Company enters into contracts in the Loan Agreement)normal course of business with contract service providers to assist in the performance of research and development and manufacturing activities. Subject to required notice periods and obligations under binding purchase orders, the Company can elect to discontinue the work under these agreements at any time. As of December 31, 2022, the Company has non-cancelable commitments for product manufacturing costs of approximately $3.7 million for the periodsyear ending March 31, 2021, June 30, 2021, September 30, 2021, December 31, 2021, March 31, 2022 and June 30, 2022 and2023. In addition, the Company has entered into a multi-year supply agreement with LGM Pharma, LLC (LGM) to produce active pharmaceutical ingredients for each calendar quarter thereafter equalYUTREPIA. Under the supply agreement with LGM, the Company is required to $10.5provide rolling forecasts, a portion of which will be considered a binding, firm order, subject to an annual minimum purchase commitment of $2.7 million $17.0 million, $23.0 million, $28.5 million, $33.5 million and $38.0 million, respectively; provided, however, thatfor the above amounts shall be increased by an amount equal to 75%term of the agreement. The agreement expires five years from the first marketing authorization approval of YUTREPIA. Other Contingencies and Commitments The Company from time-to-time is subject to claims and litigation in the normal course of business, none of which the Company believes represent a risk of material loss or exposure. See Note 14 for further discussion of pending legal proceedings. In addition to the commitments described above, the Company is party to other commitments, including non-cancelable leases and long-term debt, which are described elsewhere in these financial statements. 16. Subsequent Events Revenue Interest Financing Agreement On January 9, 2023, the Company entered into a Revenue Interest Financing Agreement (the “RIFA”) with HealthCare Royalty Partners IV, L.P. (“HCR”) and HealthCare Royalty Management, LLC. Pursuant to the RIFA and subject to customary closing conditions, HCR has agreed to pay the Company an aggregate net cashinvestment amount of up to $100.0 million (the “Investment Amount”). Under the terms of the RIFA, $32.5 million of the Investment Amount was funded on January 27, 2023 (the “Initial Investment Amount”), $22.4 million of which was used to satisfy in full and retire the Company’s indebtedness under the A&R SVB LSA, with the excess proceeds receivedless transaction costs of approximately $0.7 million funded to the Company. An additional $7.5 million of the Investment Amount will be funded fifteen business days after a request made by the Company to HCR to fund acquisition of rights, whether in the form of an acquisition, license, joint venture or similar transaction, to a clinical stage or commercial stage biopharmaceutical product to diagnose, prevent, or treat pulmonary hypertension, an additional $35.0 million of the Investment Amount will be funded fifteen business days after the earlier of regulatory approval of YUTREPIA or a favorable determination relating to the asserted patents in the ongoing patent litigation with United Therapeutics, and the remaining $25.0 million of the Investment Amount will be funded fifteen business days after the mutual agreement of HCR and the Company to fund such amount (the “Fourth Investment Amount”). As consideration for the Investment Amount and pursuant to the RIFA, the Company has agreed to pay HCR a tiered royalty on annual net revenue of the Company after the first commercial sale of YUTREPIA (the “Revenue Interests”). Except as may otherwise be mutually agreed to in connection with the funding of the Fourth Investment Amount, the applicable tiered percentage will range from 3.60% to 10.00% on the first $250 million on annual net revenue, 1.44% to 4.00% on the next $250 million in annual net revenue, and 0.36% to 1.00% on all annual net revenue in excess of $500 million. The specific royalty rate within such ranges will depend upon the total amount advanced by the HCR and the Company’s achievement of a certain annual net revenue threshold for the calendar year 2025. The Company will also make certain fixed quarterly payments to HCR, plus an additional amount on a ratable basis to reflect the funding of additional amounts by HCR under the RIFA. The Company will be required to make additional payments to HCR in the event that the first commercial sale of YUTREPIA does not occur by June 30, 2025 and certain minimum quarterly royalty payments beginning in 2026. If HCR has not received cumulative minimum payments from the saleCompany equal to 60% of the Company’s equity securitiesamount funded to date by December 31, 2026 or 100% of the amount funded to date by December 31, 2028, the Company must make a cash payment immediately following each applicable date to HCR sufficient to gross HCR up to such minimum amounts after giving full consideration of the cumulative amounts paid to HCR by the Company through each date. The net sale thresholds described above are not to be interpreted as financial guidance or projections for future net sales of the Company. HCR’s rights to receive the Revenue Interests will terminate on or after the Effective Date butdate on or priorwhich HCR has received payments equal to 175% of funded portion of the Investment Amount less the aggregate amount of all payments made to HCR as of such date (the “Hard Cap”), plus an amount, if any, that HCR would need to receive to yield an internal rate of return on the funded Investment Amount equal to 18% (the “IRR True-Up Payment”), unless the RIFA is earlier terminated. If a change of control of the Company occurs, HCR may accelerate payments due under the RIFA up to the last day of such calendar quarter; provided, further, that uponHard Cap, plus the Term C Loan Funding Date, the Cash Burn covenant shall no longer apply. The Loan Agreement also contains customary events of default, including amongIRR True-Up Payment, plus any other things, the Company’s failure to make any principal or interest payments when due, the occurrence of certain bankruptcy or insolvency events or the Company’s breach of the covenantsobligations payable under the Loan Agreement, or other material adverse changes relating to Liquidia. Furthermore, per the Loan Agreement, an event of default shall occur upon any formal court ruling against Liquidia that the Lender determines in its good faith busines judgment is reasonably likely to prohibit its ability to obtain final approval from the FDA with respect to its New Drug Application for LIQ861 or impair or delay Liquidia’s ability to commercialize LIQ861 as currently contemplated.RIFA. Upon the occurrence of an event of default, SVB HCR may accelerate payments due under the RIFA up to the Hard Cap, plus the IRR True-Up Payment, plus any other obligations payable under the RIFA.
The RIFA contains customary affirmative and negative covenants and customary events of default and other events that would cause acceleration, including, among other things, accelerate Liquidia’s obligations under the Loan Agreement. In connection withoccurrence of certain material adverse events or the Loan Agreement,material breach of certain representations and warranties and specified covenants, in which event HCR may elect to terminate the RIFA and require the Company issued to the Lender a warrant, dated as of the Effective Date (the “Warrant”)make payments to purchase up to 200,000 shares of the Company’s common stock, $0.001 par value per share (the “Common Stock”), of which (x) 100,000 shares vested on the Effective Date, with an exercise price per share equal to $3.05, and (y) 50,000 shares shall vest on each of the Term B Loan Funding Date and Term C Loan Funding Date, with an exercise price per shareHCR equal to the lower of (i) the trailing 10-day average pricelesser of the Common StockHard Cap, plus any other obligations payable under the RIFA, or the funded portion of the Investment Amount, minus payments received by HCR in respect of the Revenue Interests, plus the IRR True-Up Payment. If the FDA grants final approval to an inhaled treprostinil product therapeutically equivalent to YUTREPIA and HCR has not received 100% of the amount funded by HCR to date, then the Company will be required to make payments to HCR equal to 100% of the amount funded by HCR to date, minus payments received by HCR in respect of the Revenue Interests.
In addition, the RIFA contains a financial covenant that requires us to maintain cash and cash equivalents in an amount at least equal to $7.5 million during the calendar year beginning on January 1, 2024 and at least equal to $15.0 million for the applicable funding date and (ii)remainder of the closing price per sharepayment term after the calendar year ended December 31, 2024. As of Common Stock on the trading day prior to applicable funding date. The Warrant is exercisable for ten (10) years from thefiling date of issuance, and will be exercised automaticallythis Annual Report on a net issuance basis if not exercised prior toForm 10-K, the expiration date and if the then-current fair market valueCompany was not aware of one shareany breach of Common Stock is greater than the exercise price then in effect.covenants, occurrence of material adverse event, nor had it received any notice of event of default from HCR. |