UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
☒ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 20202023
OR
☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 FOR THE TRANSITION PERIOD FROM TO |
Commission File Number 001-38244
Genprex, Inc.
(Exact Name of Registrant as Specified in Its Charter)
Delaware | 90-0772347 |
(State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification Number) |
|
|
|
|
Austin, Texas |
|
(Address of principal executive offices) | (Zip Code) |
Registrant’s telephone number, including area code: (877) 774-4679
Securities registered pursuant to Section 12(b) of the Act:
Title of each class |
| Trading Symbol(s) |
| Name of each exchange on which registered |
Common Stock, par value $0.001 per share |
| GNPX |
| The Nasdaq Capital Market |
Securities registered pursuant to 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically, every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer: |
| ☐ |
| Accelerated filer |
| ☐ |
Non-accelerated filer: |
| ☒ |
| Smaller reporting company |
| ☒ |
|
|
|
| Emerging growth company |
|
|
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☒☐
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to § 240.10D-1(b). ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. Yes ☐ No ☒
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ☐ No ☒
The aggregate market value of the voting and non-voting common equity held by non-affiliates of the registrant as of June 30, 20202023 was approximately $114$48.5 million, computed by reference to the closing price of the registrant’s common stock on June 30, 20202023 (the last business day of the registrant'sregistrant’s most recently completed second fiscal quarter) of $3.14 per share,, as reported by The Nasdaq Capital Market.
As of March 15, 2021,25, 2024, there were 43,276,7641,910,441 shares of the registrant’s common stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s definitive proxy statement to be filed with the Securities and Exchange Commission, or SEC, subsequent to the date hereof pursuant to Regulation 14A in connection with the registrant’s 20212024 annual meeting of stockholders, are incorporated by reference into Part III of this Annual Report on Form 10-K. Such proxy statement will be filed with the SEC not later than 120 days after the conclusion of the registrant’s fiscal year ended December 31, 2020.2023.
|
| Page |
| ||
Item 1. | ||
Item 1A. | ||
Item 1B. | ||
| Item 1C. | Cybersecurity | 76 |
Item 2. | ||
Item 3. | ||
Item 4. | ||
|
|
|
| ||
Item 5. | ||
Item 6. | ||
Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations | |
Item 7A. | ||
Item 8. | ||
Item 9. | Changes in and Disagreements With Accountants on Accounting and Financial Disclosure | |
Item 9A. | ||
Item 9B. | ||
| Item 9C. | Disclosure Regarding Foreign Jurisdictions that Prevent Inspections | 87 |
|
|
|
| ||
Item 10. | ||
Item 11. | ||
Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters | |
Item 13. | Certain Relationships and Related Transactions, and Director Independence | |
Item 14. | ||
|
|
|
| ||
Item 15. | ||
Item 16. | ||
|
|
|
|
| |
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K (this “Annual Report on Form 10-K” or this “Annual Report”) contains forward-looking statements that involve substantial risks and uncertainties. Unless the context requires otherwise, references to “Genprex,” the “Company,” “we,” “us” or “our” in this Annual Report refer to Genprex, Inc. Any statements in this Annual Report about our expectations, beliefs, plans, objectives, assumptions or future events or performance are not historical facts and are forward-looking statements. These statements are often, but not always, made through the use of words or phrases such as “believe,” “will,” “expect,” “anticipate,” “estimate,” “intend,” “plan” and “would.” For example, statements concerning financial condition, possible or assumed future results of operations, growth opportunities, industry ranking, plans and objectives of management, markets for our common stock and future management and organizational structure are all forward-looking statements. Forward-looking statements are not guarantees of performance. They involve known and unknown risks, uncertainties and assumptions that may cause actual results, levels of activity, performance or achievements to differ materially from any results, levels of activity, performance or achievements expressed or implied by any forward-looking statement.
Any forward-looking statements are qualified in their entirety by reference to the risk factors discussed throughout this Annual Report. Some of the risks, uncertainties and assumptions that could cause actual results to differ materially from estimates or projections contained in the forward-looking statements include but are not limited to:
Market conditions;
• | Market conditions; |
Our capital position;
• | Our capital position; |
Our ability to compete with larger better financed pharmaceutical companies;
• | Our ability to compete effectively and with larger and/or better-financed biotechnology and pharmaceutical companies; |
Our uncertainty of developing marketable products;
• | Our uncertainty of developing marketable products; |
Our ability to develop and commercialize our products;
• | Our ability to develop and commercialize our products; |
Our ability to obtain regulatory approvals;
• | Our ability to obtain regulatory approvals; |
Our ability and third parties’ ability to maintain and protect intellectual property rights;
• | Our ability and third parties’ ability to maintain and protect intellectual property rights; |
Our ability to raise additional future financing and possible lack of financial and other resources;
• | Our ability to raise additional future financing and possible lack of financial and other resources, our ability to maintain compliance with the continued listing requirements of The Nasdaq Capital Market, our ability to continue as a going concern, and to continue to support and fund our preclinical and clinical development programs and growth of our business; |
The success of our clinical trials through all phases of clinical development;
• | The effects and ultimate impact of public health crises such as the coronavirus pandemic, or any other health epidemic, on our business, our clinical trials, our research programs, healthcare systems or the global economy as a whole; |
Any delays in regulatory review and approval of our current and future product candidates;
• | The success of our clinical trials through all phases of clinical development, including the ability of our third-party suppliers or manufacturers to supply or manufacture our products on a timely, consistent basis in a manner sufficient and appropriate as is commensurate to meet our clinical trial timing, courses of treatment, and other requisite fulfillment considerations necessary to adequately advance our development programs; |
Our dependence on third-party manufacturers to supply or manufacture our products;
• | Our ability to conduct and complete our clinical trials in accordance with projected timelines; |
Our ability to control product development costs;
• | Any delays in regulatory review and approval of our current and future product candidates; |
Our ability to attract and retain key employees;
• | Our dependence on third-party suppliers or manufacturers to supply or manufacture our key ingredients and/or raw materials, products and/or product components and successfully carry out a sustainable, reproducible and scalable manufacturing process in accordance with specifications or applicable regulations; |
Our ability to compete effectively;
• | Our ability to control product development costs; |
Our ability to enter into new strategic collaborations, licensing or other arrangements;
• | Our ability to attract and retain key employees; |
Changes in government regulation affecting product candidates could increase our development costs;
• | Our ability to enter into new strategic collaborations, licensing or other arrangements; |
Our involvement in patent and other intellectual property litigation could be expensive and could divert management’s attention;
• | Changes in government regulation affecting product candidates that could increase our development costs; |
The possibility that there may be no market acceptance for our products; and
• | Our involvement in patent, trademark, and other intellectual property litigation that could be expensive and divert management’s attention; |
• | The possibility that there may be no market acceptance for our products; and |
Changes in third-party reimbursement policies which could adversely affect potential future sales of any of our products that are approved for marketing.
• | Changes in third-party reimbursement policies which could adversely affect potential future sales of any of our products that are approved for marketing. |
The foregoing list sets forth some, but not all, of the factors that could affect our ability to achieve results described in any forward-looking statements, which speak only as of the date of this Annual Report. Except as required by law, we assume no obligation and expressly disclaim any duty to update any forward-looking statement to reflect events or circumstances after the date of this Annual Report or to reflect the occurrence of unanticipated events. In addition, we cannot assess the impact of each factor on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements contained in this Annual Report. All forward-looking statements are expressly qualified in their entirety by the cautionary statements contained in this section.
RISK FACTOR SUMMARY
Our business is subject to significant risks and uncertainties that make an investment in us speculative and risky. Below we summarize what we believe are the principal risk factors but these risks are not the only ones we face, and you should carefully review and consider the full discussion of our risk factors in the section titled “Risk Factors,” together with the other information in this Annual Report on Form 10-K. If any of the following risks actually occurs (or if any of those listed elsewhere in this Annual Report on Form 10-K occur), our business, reputation, financial condition, results of operations, revenue, and future prospects could be seriously harmed. Additional risks and uncertainties that we are unaware of, or that we currently believe are not material, may also become important factors that adversely affect our business.
Risks Related to Our Financial Position and Need for Additional Capital
|
|
|
|
|
|
Risks Related to Development and Commercialization of Our Current and Future Product Candidates
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Risks Related to Regulatory Approval and Marketing of Our Current and Future Product Candidates and Other Legal Compliance Matters
|
|
|
|
|
|
|
|
|
|
Risks Related to Our Dependence on Third Parties
|
|
|
|
|
|
Risks Related to Our Intellectual Property
|
|
|
|
|
|
|
|
Risks Related to Employee Matters and Managing Growth
|
|
|
|
General Risk Factors
|
|
|
|
|
|
|
|
Overview
We are a clinical stage gene therapy company focused on developing life-changing treatmentspioneering the development of gene-based therapies for cancer and diabetes.large patient populations with unmet medical needs. Our lead cancer drug candidate, REQORSA™ Immunogene therapy drug (sometimes referred to as GPX-001), is being developed to treat non-small cell lung cancer ("NSCLC"). The active agent in REQORSA is a TUSC2 gene expressing plasmid that is encapsulatedoncology platform utilizes our systemic, non-viral ONCOPREX® Delivery System which uses lipid-based nanoparticles in a DOTAP cholesterol nanoparticle. TUSC2 is alipoplex form to deliver tumor suppressor gene which has both tumor killing (via apoptosis) and immunomodulatory effects. We utilize our novel proprietary ONCOPREX® Nanoparticle Delivery System to deliver the TUSC2 gene expressing plasmidgene-expressing plasmids to cancer cells. The TUSC2 geneproduct is one of a series of genes whose therapeutic useadministered intravenously, where it is coveredtaken up by our exclusive worldwide licenses from The University of Texas MD Anderson Cancer Center ("MD Anderson").
We are planning to initiate our Acclaim-1 and Acclaim-2 clinical trialstumor cells that then express tumor suppressor proteins that were deficient in 2021. Acclaim-1 is a Phase 1/2 clinical trial using a combination of REQORSA with AstraZeneca PLC’s Tagrisso® in patients with late-stage NSCLC with mutated epidermal growth factor receptors ("EGFRs") whose disease progressed after treatment with Tagrisso. In January 2020, we received Food and Drug Administration ("FDA") Fast Track Designation for the Acclaim-1 patient population. Acclaim-2 is a Phase 1/2 clinical trial using a combination of REQORSA with Merck & Co.’s Keytruda® in late-stage NSCLC patients who are low expressors (1% to 49%) of the protein programmed death-ligand 1 ("PD-L1").
Intumor. Our diabetes we are developing a gene therapy that is exclusively licensed from the University of Pittsburgh of the Commonwealth System of Higher Education ("University of Pittsburgh") for the treatment of Type 1 and Type 2 diabetes. This potential treatmenttechnology is designed to work in Type 1 diabetes by transforming alpha cells in the pancreas into functional beta-like cells, which can produce insulin but aremay be distinct enough from beta cells to evade the body’s immune system. OurIn Type 2 diabetes, product candidateour technology is currently being evaluated in preclinical studies.believed to work by replenishing and rejuvenating exhausted beta cells that make insulin.
Oncology Platform
Utilizing our non-viral ONCOPREX Nanoparticle Delivery System, we are developing cancer treatments that are designed to administer cancer fighting genes. We encapsulate the gene-expressing plasmids using ONCOPREX lipid nanoparticles, and administer them intravenously, where they are then taken up by tumor cells and express proteins that are missing or found in low quantities in the tumor cells. With ourOur lead oncology drug candidate, REQORSA® Immunogene Therapy (generic name: quaratusugene ozeplasmid), previously referred to as GPX-001, is initially being developed in combination with prominent, approved cancer drugs to treat Non-Small Cell Lung Cancer (“NSCLC”) and Small Cell Lung Cancer (“SCLC”). REQORSA there ishas a multimodal mechanism of action whereby REQORSAit interrupts cell signaling pathways that cause replication and proliferation of cancer cells, re-establishes pathways for apoptosis, or programmed cell death, in cancer cells, and modulates the immune response against cancer cells. In early studies, REQORSA has also been shown to block mechanismsbe complementary with targeted drugs and immunotherapies. Our strategy is to develop REQORSA in combination with current approved therapies and we believe REQORSA’s unique attributes position it to provide treatments that create drug resistance.improve on these current therapies for patients with NSCLC, SCLC, and possibly other cancers.
Acclaim – 1: We currently are enrolling and treating patients in the Phase 2a expansion portion of our Phase 1/2 Acclaim-1 clinical trial. The Acclaim-1 trial uses a combination of REQORSA and AstraZeneca’s Tagrisso® in patients with late-stage NSCLC that has activating epidermal growth factor receptor (“EGFR”) mutations and progression after treatment with Tagrisso. Following the May 2023 completion of the Phase 1 dose escalation portion of the study, the Acclaim-1 Safety Review Committee (“Acclaim-1 SRC”) approved advancement from the Phase 1 dose escalation portion to the Phase 2a expansion portion of the study. Based on a review of safety data which showed no dose limiting toxicities (“DLTs”), the Acclaim-1 SRC determined that the recommended Phase 2 dose (“RP2D”) of REQORSA will be 0.12 mg/kg. This was the highest dose level delivered in the Phase 1 portion of the study and is twice the highest dose level delivered in the Company’s prior clinical trial combining REQORSA with Tarceva® for the treatment of late-stage lung cancer. We opened the Phase 2a expansion portion of the study and enrolled and dosed the first patient in January 2024. The Phase 2a expansion portion of the trial is expected to enroll approximately 66 patients; half will be patients who received only prior Tagrisso treatment and the other half will be patients who received prior Tagrisso treatment and chemotherapy. The aim is to determine toxicity and efficacy profiles of patients with different eligibility criteria. There will be an interim analysis following the treatment of 19 patients in each cohort. We expect to complete the enrollment of 19 patients in each cohort of the Phase 2a expansion portion of the study by the end of 2024, and thus we expect the interim analyses in early 2025. The Food and Drug Administration (“FDA”) has granted Fast Track Designation for the Acclaim-1 treatment combination of REQORSA and Tagrisso in NSCLC patients who have progressed after Tagrisso treatment.
Acclaim – 2: We currently are enrolling and treating patients in the Phase 1 dose escalation portion of our Phase 1/2 Acclaim-2 clinical trial. The Acclaim-2 trial uses a combination of REQORSA and Merck & Co.’s Keytruda® in patients with late-stage NSCLC whose disease has progressed after treatment with Keytruda. Patients are currently being treated at the 0.06 mg/kg dose level in the first cohort of patients and, subject to Acclaim-2 Safety Review Committee (“Acclaim-2 SRC”) approval, will be treated at successive dose levels of 0.09 mg/kg and 0.12 mg/kg. In March 2023, we amended the Acclaim-2 protocol to include additional treatments in the control group with the goal of accelerating enrollment in the study by making the trial more attractive to a wider variety of investigators. We expect enrollment in the dose escalation portion of the study to be completed in the second half of 2024. We will then initiate and evaluate patients in the Phase 2a expansion portion of the study at the maximum tolerated dose (the “MTD”) or RP2D. The FDA has granted Fast Track Designation for the Acclaim-2 treatment combination of REQORSA and Keytruda in NSCLC patients who have progressed after Keytruda treatment.
The expansion portion of both the Acclaim-1 and Acclaim-2 trials are Phase 2 studies. The expansion portion of these studies provides us the advantage of early insight into drug effectiveness in defined and distinct patient populations at the MTD or RP2D in order to better evaluate efficacy and increase the likelihood of a successful randomized Phase 2 trial which will follow the expansion portion of each study.
Acclaim – 3: We currently are enrolling patients in the Phase 1 dose escalation portion of our Phase 1/2 Acclaim-3 clinical trial. The Acclaim-3 clinical trial uses a combination of REQORSA and Genentech, Inc.’s Tecentriq® as maintenance therapy in patients with extensive stage small cell lung cancer (“ES-SCLC”) who did not develop tumor progression after receiving Tecentriq and chemotherapy as initial standard treatment. Patients are treated with REQORSA and Tecentriq until disease progression or unacceptable toxicity is experienced. In January 2024, we opened the Phase 1 portion of the Acclaim-3 study for enrollment. We expect to complete the Phase 1 dose escalation portion of the study during the second half of 2024 and we expect to start the Phase 2 expansion portion of our Acclaim-3 study in the second half of 2024. In June 2023, the FDA granted Fast Track Designation for the Acclaim-3 treatment combination of REQORSA and Tecentriq as maintenance therapy in patients with ES-SCLC who did not develop tumor progression after receiving Tecentriq and chemotherapy as initial standard treatment. In August 2023, the FDA granted Orphan Drug Designation to REQORSA for the treatment of SCLC.
The TUSC2 gene, which is the key component of REQORSA and plays a vital role in cancer suppression and normal cell regulation, is one of a series of genes on the short arm of Chromosome 3 whose therapeutic use is covered by our exclusive worldwide licenses from The University of Texas MD Anderson Cancer Center (“MD Anderson”). We believe that our ONCOPREX Nanoparticle Delivery System could allowallows for delivery of a number of cancer-fighting genes, alone or in combination with other cancer therapies, to combat multiple types of cancer. We believe that REQORSA’s combinationcancer and we are in early stages of pan-kinase inhibition, direct inductiondiscovery programs to identify other cancer candidates. In August 2022, we entered into a three-year sponsored research agreement with MD Anderson to support further preclinical studies of apoptosis, anti-cancer immune modulationTUSC2 and complementary action with targeted drugs and immunotherapies is unique, and positions REQORSA to provide treatment for patients with NSCLC and possibly other cancers, who are not benefitting from current therapies.tumor suppressor genes.
Diabetes Gene Therapy
In diabetes, we have exclusively licensed from the University of Pittsburgh of the Commonwealth System of Higher Education (“University of Pittsburgh”) multiple technologies relating to the development of a gene therapy product for each of Type 1 and Type 2 diabetes. The same general novel approach is used in each of Type 1 and Type 2 diabetes whereby an adeno-associated virus (“AAV”) vector containing the Pdx1 and MafA genes is administered directly into the pancreatic duct. In humans, this can be done with a routine endoscopy procedure. Our diabetes gene therapy, also referred to as GPX-002, was developed by lead researcher Dr. George Gittes, at the Rangos Research Centerproduct candidates are currently being evaluated and optimized in preclinical studies at the University of Pittsburgh Medical Center ("UPMC") Children’s Hospital. This potentialPittsburgh. GPX-002 is being developed using the same construct for the treatment of both Type 1 diabetes and Type 2 diabetes. GPX-002 for Type 1 diabetes is designed to work by transforming alpha cells in the pancreas into functional beta-like cells, which can produce insulin but aremay be distinct enough from beta cells to evade the body’s immune system. In a similar approach, GPX-002 for Type 2 diabetes (formerly known as GPX-003), where autoimmunity is not at play, is believed to work by replenishing and rejuvenating exhausted beta cells that make insulin. We finalized the components of the diabetes construct to take forward for nonclinical studies and in December 2023, we submitted a request to meet with the FDA to obtain their guidance on the nonclinical studies needed to file an Investigational New Drug (“IND”) application and initiate first-in-human studies. As a result of the FDA’s response, the Company will continue with its planned additional nonclinical studies before requesting regulatory guidance in 2024 for the IND-enabling studies. In October 2023, we entered into a one-year extension to our August 2022 sponsored research agreement with the University of Pittsburgh for the use of GPX-002 in a non-human primate (“NHP”) model in Type 2 diabetes. The therapy utilizesextension includes a procedurerevised research plan to encompass our most recent technologies to which we acquired exclusive rights from the University of Pittsburgh in which an adeno-associated virus vector deliversJuly 2023. These include a MafB promoter to drive expression of the Pdx1 and MafA genestranscription factors that can potentially be used for both Type 1 and Type 2 diabetes. See also “Note 7 – Commitments and Contingencies” to our financial statements included in this Annual Report on Form 10-K. In February 2023, the pancreas.Company’s research collaborators at the University of Pittsburgh presented preclinical data in a NHP model of Type 1 diabetes highlighting the therapeutic potential of GPX-002 at the 16th International Conference on Advanced Technologies & Treatments for Diabetes (ATTD 2023) in Berlin, Germany. The statistically significant study results showed the treated animals had decreased insulin requirements, increased c-peptide levels, and improved glucose tolerance compared to baseline. In April 2023, the Company hosted a Key Opinion Leader virtual event entitled “Novel Gene Therapy to Treat Type 1 Diabetes,” which discussed preclinical data reported at ATTD 2023 supporting gene therapy to treat Type 1 diabetes.
Recent Developments
License Amendment with MD AndersonAt-the-Market Offering Program
On March 3, 2021,December 13, 2023, we entered into an amendmentAt The Market (“ATM”) Offering Agreement (the “MD License Amendment”“Agreement”) to the Patent and Technology License Agreement dated May 4, 2020 with MD Anderson. The MD License Amendment grants us a worldwide, exclusive, sublicensable licenseH.C. Wainwright & Co., LLC, serving as agent (the “Agent”) with respect to an additional portfolioATM offering program under which we may offer and sell through the Agent, from time to time at our sole discretion, up to such number or dollar amount of six patents and one patent application and related technology for methods for treating cancer by administrationshares (the “Shares”) of a TUSC2 therapy in conjunction with EGFR inhibitorsour common stock, par value $0.001 per share (the “Common Stock”), as registered on the prospectus supplement covering the ATM offering, as may be amended or other anti-cancer therapies in patients predictedsupplemented from time to be responsive to TUSC2 therapy. Pursuant to the MD License Amendment, wetime. We have agreed to (i) pay annual maintenance fees ranging from the mid five figuresAgent a commission equal to three percent (3%) of the low six figures, (ii) total milestone paymentsgross sales proceeds of $6,150,000, (iii) a one-time fee inany Shares sold through the mid five figuresAgent under the Agreement, and (iv) certain patent related expenses.
Financings
On February 11, 2021,also have provided the Agent with customary indemnification and contribution rights. As of December 31, 2023, we had not sold an aggregateany Shares through the Agent under the Agreement. From January 1, 2024 through the date of 4,000,000filing of this Annual Report on Form 10-K, we have sold 158,474 shares of our common stock for net proceeds to us totaling $881,946 through the Agent under the Agreement.
Reverse Stock Split
At our special meeting of stockholders held on December 14, 2023, the Company’s stockholders adopted and approved an amendment to our Amended and Restated Certificate of Incorporation to effect a purchase pricereverse stock split of $6.25our issued and outstanding shares of Common Stock, at a specific ratio, ranging from one-for-ten (1:10) to one-for-fifty (1:50), with such ratio to be determined by our Board in its discretion.
On January 31, 2024, we filed the amendment to our Amended and Restated Certificate of Incorporation with the Secretary of State of the State of Delaware to effect a reverse stock split of our Common Stock at a ratio of one-for-forty (1:40) (the “Reverse Stock Split”). The Reverse Stock Split became effective in accordance with the terms of the amendment at 12:01 AM Eastern Time on February 2, 2024 under a new CUSIP number, 372446-203. All share and per share amounts in this Annual Report on Form 10-K have been adjusted to reflect the Reverse Stock Split.
March 2024 Registered Direct Offering
On March 21, 2024, we completed a registered direct offering.offering, in which we sold to an institutional investor an aggregate of (i) 165,000 shares of our common stock, (ii) pre-funded warrants (the “March 2024 Pre-Funded Warrants”) exercisable for up to an aggregate of 1,377,112 shares of our common stock, and (iii) warrants (the “March 2024 Common Warrants”) exercisable for up to an aggregate of 1,542,112 shares of our common stock. The offering price for each share of common stock and accompanying March 2024 Common Warrant was $4.215, and the offering price for each March 2024 Pre-Funded Warrant and accompanying March 2024 Common Warrant was $4.2149. The March 2024 Pre-Funded Warrants are exercisable immediately upon issuance at an exercise price of $0.0001 per share and will expire when exercised in full. The March 2024 Common Warrants are exercisable immediately upon issuance at an exercise price of $4.09 per share and will expire in five years from the date of issuance. We received net proceeds of approximately $23.2$5.8 million after deducting estimatedcommissions and expenses, excluding any proceeds that may be received in the future from any exercise of the March 2024 Common Warrants. In connection with the offering, expenses.
we also amended certain existing warrants to purchase up to an aggregate of 194,248 shares of our common stock that were previously issued to investors in March 2023 and July 2023, with exercise prices of $44.00 and $35.40 per share and expiration dates of March 1, 2028 and July 21, 2028 for $0.125 per amended warrant, such that the amended warrants have a reduced exercise price of $4.09 per share and an expiration date of five years from the closing of this offering.
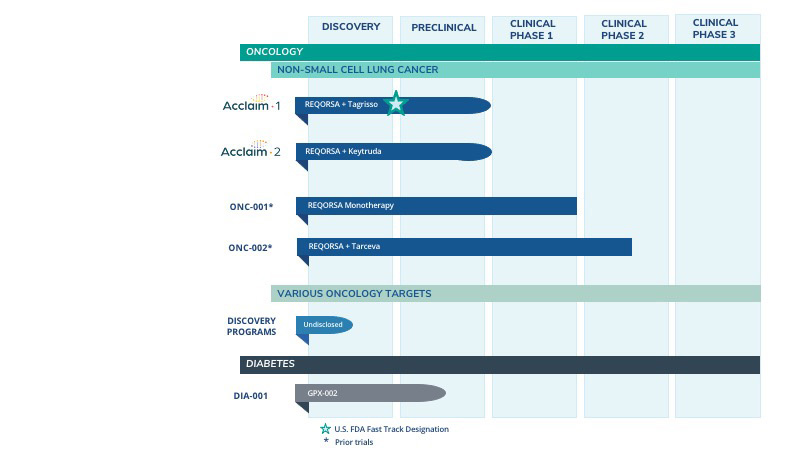
Our Pipeline
Our technologies are designed to administer disease-fighting genes to provide new treatment optionstherapies for large patient populations with cancer and diabetes who currently have limited treatment options. We are developing our lead oncology product candidate REQORSA to be administered with targeted therapies and with immunotherapies for NSCLC.NSCLC and SCLC. We continue to conduct preclinical research to explore how REQORSA may be administered with targeted therapies and immunotherapies in other solid tumors, such as ALK-positive NSCLC, and we are researching how other cancer fighting genes, such as NPRL2, can be appliedenhance our portfolio using our systemic, non-viral gene therapy platform, the ONCOPREX Nanoparticle Delivery System. WeDeliverySystem. Using a different gene therapy delivery system, we are also are developing our pre-clinicalpreclinical diabetes candidate GPX-002.GPX-002 using the same construct for both Type 1 diabetes and Type 2 diabetes (formerly known as GPX-003 when a different construct was being considered for Type 2 diabetes). The following table summarizes our product development pipeline.
Introduction – Cancer
Cancer and Genetic Mutations. Cancer results from genetic mutations. Mutations that lead to cancer are usually present in two major classes of genes: oncogenes, which are involved in functions such as signal transduction and transcription; and tumor suppressor genes, which play a rolemultiple roles in governing cell proliferation by regulating transcription.growth and proliferation. Transduction is the process by which chemical and physical signals are transmitted throughinto cells. Transcription is the process by which a cell’s DNA sequence is copied to make RNA molecules, which then play a role in protein expression. In normal cells, mutations in oncogenes are discovered and targeted for elimination by tumor suppressor genes. In cancer cells, the oncogene mutations may overwhelm the natural tumor suppression processes, or those tumor suppression processes may be impaired or absent. Functional alterations due to mutations in oncogenes or tumor suppressor genes may result in the abnormal and uncontrolled growth patterns characteristic of cancer. These genetic alterations facilitate such malignant growth by affecting signal transduction pathways and transcription, thussuch as inhibiting normal growth signaling in the cell, circumventing the natural process of apoptosis, evading the immune system’s response to cancer, and inducing angiogenesis, which is the formation of new blood vessels that supply cancer cells.
The most common genetic alterations present in NSCLClung cancer are in tumor suppressor genes, against which fewgenes. No targeted small molecule drugs historically considered the backbone of traditional medicine, have successfully been developed.developed against tumor suppressor gene mutations in NSCLC or SCLC.
Another genetic condition often associated with lung cancer is the overexpressionpresence of EGFRs and mutations of tyrosine kinases. KinasesTyrosine kinases are enzymes that play an important role in signal transduction through the modification of proteins by adding or taking away phosphate groups a process called dephosphorylation,(phosphorylation) onto the amino acid tyrosine, to change the proteins’ function. When an EGFR ligand binds to the EGFR, two EGFR transmembrane proteins are brought to proximityclose together on the cell membrane surface, or dimerize, either through a ligand, or binding molecule, that binds to the extracellular receptor, or through some other process,and the intracellular protein-kinasetyrosine kinase domains can autophosphorylate, and activate downstream processes, including cell signaling pathways that can lead to either cell cycle arrest or cell growth and proliferation. EGFRs and kinases can act similarly to a switch that turns “on” and “off” when phosphate groups are either added or taken away. Mutated kinases can have a malfunctioning on/off switch, causing the switch to be stuck in the “on” position or failing to turn the switch “off,” leading to the loss of control of cell control.growth.
Cancer and the Immune System. Cancer can also spread when the body’s natural immune functions are impaired, including by the cancer cells themselves. PD-1, or Programmed Death-1, is a receptor expressed on the surface of activated T cells, which are part of the body’s immune system. PD-L1 is a protein/receptorligand for PD-1 which is expressed on the surface of cancer and other cells. The binding of PD-1 to PD-L1 has been speculated to contribute to cancer cells’ ability to evade the body’s immune response. PD-1 and similar molecules are called immune checkpointscheckpoint inhibitors because they can impede the normal immune response, for example by blocking the T cells from attacking the cancer cells. In many cancers, PD-L1 receptors areis up-regulated. Substantial research is now beinghas been performed in the emerging field of immuno-oncology to discover drugs or antibodies that could block PD-L1PD-1 and similar receptors. It is believed that blocking the PD-1/PD-L1 interaction pathway and other similar checkpoints, such as cytotoxic T-lymphocyte-associated protein 4, or CTLA-4, with drugs called checkpoint inhibitors can prevent cancer cells from inactivating T cells.cells, leading to an attack of the immune system on the cancer.
Current Treatment of NSCLC. Chemotherapy is the standard treatment for the majority of NSCLC patients, as it is for many other cancer patients. Because it is a non-selective systemic treatment, rather than a targeted approach to treating cancer, chemotherapy also kills healthy cells and has a number of other undesirable side effects.
A subset of NSCLC patients carry one or both ofa mutation in EGFR, which makes their tumors sensitive to EGFR tyrosine kinase inhibitors (“EGFR TKIs”). The two EGFR mutations,most common are referred to as exon 19 deletion and exon 21 substitution, which make their tumors sensitive to tyrosine kinase inhibitors ("EGFR TKIs"). Because EGFR is frequently overexpressed in lung tumors, it has become a favored therapeutic target for pharmaceutical companies.substitution. Several pharmacological and biological approaches, including EGFR TKIs, have been developed specifically to block activated EGFR for cancer therapy. The class of drugs functioning as protein kinase inhibitors ("KIs") comprises the majority of targeted therapies for lung cancer, accounting for most sales and use. Of the KIs, the EGFR TKI drugs are the most common with drugs targeting EGFR kinases leading the sector growth.targeted therapies used in lung cancer. Several EGFR TKI therapies are marketed commercially including, but not limited to, Tagrisso, Tarceva, Iressa and Gilotrif.
Approximately 7%10% to 20% of NSCLC patients of North American and European descent and approximately 30%40% to 50% of NSCLC patients of Asian descent have activating EGFR mutations. This means that the majority of NSCLC patients do not have activating EGFR mutations and are therefore “EGFR negative” and not optimal candidates for EGFR TKIs.
However, while EGFR TKIs are most effective in patients who have an activating EGFR mutation and are therefore described as “EGFR positive,” they are significantly less effective in overall NSCLC populations and are generally not effective in patients without an activating EGFR mutation.
In addition, even among those patients who are EGFR positive and benefit from EGFR TKI therapy, nearly all eventually become resistant to and ultimately no longer respond to EGFR TKI therapy, resulting in eventual disease progression. For example, according to the FLAURA study, sponsored by AstraZeneca, the median time to tumor progression for lung cancer patients on Tagrisso monotherapy is approximately 18 months. Furthermore, although recent clinical trials have shown that combining EGFR TKIs with conventional chemotherapy does not increaseincreases progression free survival for lung cancer patients.patients with EGFR mutations compared to Tagrisso monotherapy, it is not yet clear whether the combination of Tagrisso and chemotherapy initially leads to longer overall survival than treatment with Tagrisso monotherapy (which has relatively few side effects) and then treatment with chemotherapy at the time of disease progression.
Current Treatment of SCLC. SCLC is staged as limited stage, in which the cancer is only on one side of the chest and can be treated with a single radiation therapy field, or as extensive stage (ES), which includes all other patients. Since SCLC is an aggressive disease, the vast majority of patients have extensive stage SCLC at the time of initial diagnosis. The standard treatment for ES-SCLC for many years was a combination chemotherapy with carboplatin and etoposide for 4 cycles of treatment, as treatment with chemotherapy for longer duration or treatment including other agents was not shown to be beneficial. In the last several years the addition of immune checkpoint inhibitors has been shown to have improved efficacy when added to 4 cycles of chemotherapy. Thus, standard treatment now consists of either Tecentriq or Imfinzi added to 4 cycles of carboplatin and etoposide, and then Tecentriq or Imfinzi are continued as maintenance therapy until disease progression.
However, treatment of ES-SCLC is not curative, and patients progress quickly. In patients receiving Tecentriq and chemotherapy, the progression free survival (“PFS”) after starting maintenance Tecentriq is only 2.6 months. Further improvements in the treatment of ES-SCLC are needed.
Epidemiology of Non-Small Cell Lung Cancer. According to the World Health Organization in 2019,2023, lung cancer was the leading cause of cancer deaths worldwide, causingworldwide. According to the American Cancer Society in 2023, lung cancer accounts for about one in five of all cancer deaths in the United States, and causes more deaths in the U.S. with more people dying of lung cancer in the U.S. than colorectal,of colon, breast liver or stomach cancers.and prostate cancers combined. In 2020, there were more than 2 million new lung cancer cases and approximately 1.8 million deaths from lung cancer worldwide. In the United States,U.S., according to the American Cancer Society, it is estimated that in 20212024 there will be more than 235,000234,000 new cases of lung cancer and more than 131,000125,000 deaths from this disease. The American Society of Clinical Oncology reports that NSCLC represents 84%about 82% of all lung cancers and has a 24%the five-year survival rate. However, according to the National Cancer Institute, 57%rate for patients with NSCLC with distant spread is 9 percent. SCLC represents about 14% of lung cancer diagnoses are distant, or have metastasized,patients and the five-year relative survival rate for Stage 4 (metastatic) NSCLCpatients with SCLC with distant spread is approximately 5%.3 percent. With limited benefit from current therapies, we believe there is a significant unmet medical need for new treatments for NSCLC and SCLC in the United StatesU.S. and globally, and we believe REQORSA may be suitable for athe majority of NSCLClung cancer patients.
REQORSA™®
Our REQORSA™REQORSA® immunogene therapy (generic name: quaratusugene ozeplasmid) is designed to (i) interrupt cell signaling pathways that cause replication and proliferation of cancer cells, (ii) to target and kill cancer cells, via receptor pathways, and (iii) to stimulate the natural immune responses against cancer. REQORSA is an immunogene therapy in that it combines features of gene therapy and immunotherapy. It up-regulates TUSC2 expression in the cell, and also increases the anti-tumor immune cell population and down-regulates PD-L1, receptors, thereby potentially boosting the immune response to cancer.
REQORSA consists of thea TUSC2 gene expressing plasmid encapsulated in non-viral lipid-based nanoparticles in a non-viral nanoparticle made from lipid moleculeslipoplex form (our ONCOPREX Nanoparticle Delivery System) with, which has a positive electrical charge. REQORSA is injected intravenously and specifically targets cancer cells. Cancer cells have elevated metabolism compared to healthy cells and as a result, are negatively charged compared to healthy cells, which are positively charged, or chargedgenerally charge neutral. REQORSA is designed to deliver the functioning TUSC2 gene to cancer cells while minimizing their uptake by normal tissue. Tumor biopsyLaboratory studies conducted at MD Anderson show that in three patients, the uptake of TUSC2 in tumor cells in vitro after REQORSA treatment was 10 to 33 times the uptake in normal cells. Biopsies in three patients with NSCLC treated with REQORSA show major increases in TUSC2 protein expression in the tumor cells one day after REQORSA administration. We believe that REQORSA unlike other gene therapies, which either need to be delivered directly into tumors or require cells to be removed from the body, re-engineered and then reinserted into the body, is the first systemic gene therapy to be used for cancer in humans. Many other gene therapies require complex procedures, such as removal of cells from a patient and modification of those cells which are then reinfused into the patient, and many, unlike REQORSA, lead to permanent changes in a patient’s DNA.
Many approved cancer therapeutics target only single molecules or a single specific genetic abnormality related to driving the proliferation and survival of cancer cells. In contrast, REQORSA has been shown to have a multimodal mechanism of action whereby it interrupts cell signaling pathways that cause replication and proliferation of cancer cells, re-establishes pathways for programmed cell death (apoptosis) in cancer cells, and modulates the immune response against cancer cells. REQORSA also has been shown to block mechanisms that create drug resistance.be complementary with targeted drugs and immunotherapies.
REQORSA is a pan-kinase inhibitor shown to simultaneously inhibit the EGFR and Protein kinase B, also known as AKT, oncogenic kinase pathways in vitro and in vivo. Once the cancer cell takes up the nanoparticles containing the TUSC2 expressing plasmid, it is reprogrammed to die. Resistance to targeted drugs and checkpoint inhibitors developoften develops through activation of alternate bypass pathways. For example, when PD-1 is blocked, the TIM-3 checkpoint is up-regulated. We believe that REQORSA’s multimodal activity will block emerging bypass pathways, thereby potentially reducing the probability that drug resistance develops.
Many approved cancer therapeutics target only single molecules or a single specific genetic abnormality related to driving the proliferation and survival of cancer cells. In contrast, REQORSA is designed to work by targeting several molecules within the cancer cell to interrupt cell signaling pathways that cause replication and proliferation of cancer cells, to target and kill cancer cells, to block mechanisms that create drug resistance and to stimulate the natural immune response.
Our preclinical and clinical data indicatesindicate that REQORSA is well tolerated and may be effective alone or in combination with targeted small molecule therapies. Preclinical data indicatesindicate that REQORSA may also be effective with immunotherapies, and in a three-drug combination with immunotherapy and chemotherapy. By facilitating the action of both drugs, we believe thereThese data suggest that REQORSA, when combined with other therapies, may be an expandedeffective in a large population of patients who may benefit from these advanced therapy regimens.lung cancer patients.
TUSC2, the Active AgentTumor Suppressor Gene in REQORSA™REQORSA®
TUSC2 is a multifunctional gene that plays a vital role in cancer suppression and normal cell regulation. Key TUSC2 anti-cancer mechanisms of action include the inactivation of multiple oncogenic kinases, the induction of apoptosis, the control of cell signaling and inflammation, and modulation of the immune system to fight cancer. REQORSA has also been shown to block mechanisms that create drug resistance.be complementary with targeted drugs and immunotherapies. Our preclinical data indicatesindicate that REQORSA in combination with both EGFR TKIs and with immunotherapies can achieve results more favorable than results achieved with either REQORSA or such other therapies alone, and may make those drugs effective for patients with drug resistance who would not otherwise benefit from them.
Normal TUSC2 function is often inactivated at the early onset ofin cancer development, making TUSC2 a potential target for all stages of cancer, including metastatic disease. The TUSC2 protein is reduced or absent in approximately 85%82% of lung cancers.NSCLCs and in 100% of SCLCs. In patients with NSCLC, the loss of TUSC2 expression has been associated with significantly worse overall survival than when TUSC2 expression is not impaired.decreased.
Studies show TUSC2 protein functions as a key mediator in the Apaf1-mediated mitochondrial apoptosis pathway by recruiting and directing cytoplasmic Apaf1 protein to a critical cellular location and activating it in situ,and by thereby up-regulating activity of other proapoptotic effectors. Normal TUSC2 function mediatesfunctions to mediate apoptosis in cancer cells through interaction with Apaf1 and also down-regulates multiple tyrosine kinases that control cell growth, including EGFR, AKT, platelet-derived growth factor receptor ("PDGFR"(“PDGFR”), c-Kit, and c-Abl. TUSC2 mediates apoptosis in cancer cells but not normal cells through its interaction with Apaf1 and down-regulates tyrosine kinases including EGFR, PDGFR, c-Kit, and c-Abl.
In normal cells, the proteins involved in the PI3K/AKTAKT/mTOR pathway (also called the mTOR pathway),play an important role in whichcellular function and cellular trafficking. In this pathway, PI3K, a kinase, generates messenger molecules required to translocate AKT, another protein kinase, to the cell’s plasma membrane where it is phosphorylated and activated, play an important role in cellular function and cellular trafficking.activated. These proteins are often found to be aberrantly active in cancers, causing cells to lose their ability to control cell growth, proliferation, and differentiation. Thus, mutations in PI3K (overexpression) and its upstream receptors,activators, such as EGFR, have been associated with many forms of cancers.
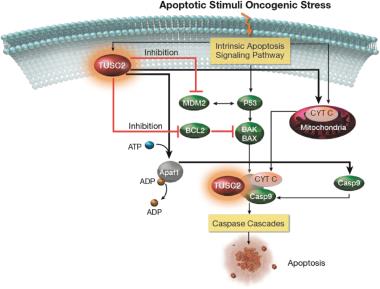
Similarly, proteins in the Ras/MAPK pathway, which is a signal transduction pathway that transduces signals to the cell nucleus where specific genes are activated for cell growth, division and differentiation, play a critical role in cellular responses to various stress stimuli, including osmotic stress, DNA damage, and proinflammatory factors.inflammation. As shown in the figures below, the TUSC2 protein, a potent pan-kinase inhibitor, blocks multiple cell signalingcell-signaling pathways downstream of the EGFR receptor (EGFR in the figures), leadingand leads to cell cycle interruption, and thereby preventing cancer cell proliferation and survival.
Under stress conditions, such as oncogenic stress, cells go through a regulated process of programmed cell death, also known as apoptosis,apoptosis. As illustrated in order to control cell development and replication. Thethe schematic below, the TUSC2 protein interacts via various apoptotic signaling pathways such as Apa1 to stimulate programmed cell death via the release of caspases, enzymes that play a significant role in apoptosis.

Pan-Kinase Inhibition by TUSC2
Stimulation of Apoptotic Signaling by TUSC2
Our clinical and preclinical data indicatesindicate that the combination of REQORSA with EGFR TKIs, may increase anti-tumor activity in cancers with or without the EGFR mutations and in cancers that have become resistant to EGFR TKI therapy, thus potentially expanding the number of patients who could benefit from those drugs.
TUSC2 and the Immune Response. In addition to its pro-apoptotic cytotoxicity and tyrosine kinase inhibitory activity, TUSC2 enhances the immune response to cancer. Data from preclinical studies at MD Anderson has shown a therapeutic benefit from the combination of TUSC2 and anti-PD-1 antibody and a key role for TUSC2 in regulating immune cell subpopulations including cytokines, natural killer ("NK"(“NK”) cells, and T lymphocytes. In addition, TUSC2 has been found to down-regulate PD-L1 receptors on the surface of cancer cells. ByAs a result, lymphocytes expressing the PD-1 receptor are more likely to recognize the cancer cell as an altered cell that should be destroyed. In addition, by inducing tumor cell apoptosis TUSC2 increases antigen release and presentation, thus promoting an enhanced antitumor response in the presence of other immune regulators.
NK cells, an important part of the innate immune system, have developed several mechanisms to distinguish healthy cells from target cells. These mechanisms allow NK cells to kill cells that are deemed dangerous to the host, including cancer cells. However, one of the consequences of malignant transformation is the ability of the cancer cell to evade the immune system. Cancer cells do so via the up-regulation and interplay of receptors, including checkpoint inhibitors such as PD-1 and PD-L1.
As shown in the illustration below, TUSC2 has been found to stimulate the release of interleukin-15, or IL-15, resulting in up-regulation of mature NK cells that circulate and target cancer cells.
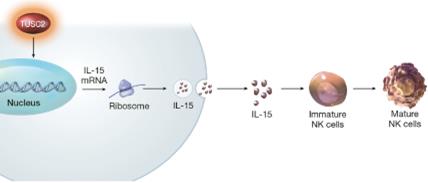
Modulation by TUSC2 of the Immune Response to Cancer
In work presented in an abstract for the 2024 American Association of Cancer Research (AACR) meeting, our clinical collaborators have shown that TUSC2 has metabolic effects both in lung cancers and in normal cells. TUSC2 is encoded by the nuclear DNA, but the TUSC2 protein resides in the inner membrane of the mitochondria. TUSC2 has been shown to play a critical role in mitochondrial respiration/energy metabolism, reactive oxygen species production, and in Ca2+ flux to and from mitochondria. This recent work demonstrated that TUSC2 re-introduction to TUSC2-deficient cancer cells consistently suppressed both glycolysis and mitochondrial ATP production, thus leaving cells without sufficient energy to support their vital functions. This suggests that TUSC2 protein introduced to cancer cells lacking TUSC2 can decrease the high metabolic rate that is characteristic of cancer cells, leading to marked decreases in cell growth. The data also showed that both glycolytic and mitochondrial metabolism of a normal epithelial cell line were strengthened after the introduction of TUSC2, suggesting a beneficial role of TUSC2 for the metabolic health of normal cells. This suggests that TUSC2 effects on metabolism in normal lymphocytes could lead to the increased immune response to lung cancers seen in animal models receiving REQORSA. The study further suggested that REQORSA may play an important role as a cancer treatment to target and disrupt the metabolism of cancer cells, leading to a decrease in the rate of glycolysis.
ONCOPREX®Nanoparticle Delivery System
Our immunogene therapyoncology platform consists of anti-cancer genes expressing DNA plasmids expressing tumor suppressor genes contained in non-viral lipidlipid-based nanoparticles in a lipoplex form (“lipoplexes”) delivered intravenously. Lipoplexes (see figure below) have lipid-based nanoparticles that clump together, thus protecting the DNA between them from being destroyed in the bloodstream. REQORSA our lead drug candidate, utilizes the ONCOPREX® Nanoparticle Delivery System to encapsulate the TUSC2 gene in positively charged nanoparticleslipoplexes that bindare attracted to actively replicating (and therefore negatively charged)charged cancer cells, and then enter the cancer cell through selective endocytosis, a process by which cells take in substances from outside the cell by engulfing them in a vesicle. The nanoparticles in our system differ significantly from liposomes historically used for drug delivery in that they are true particles encapsulating the therapeutic payload within a bilamellar lipid coat.
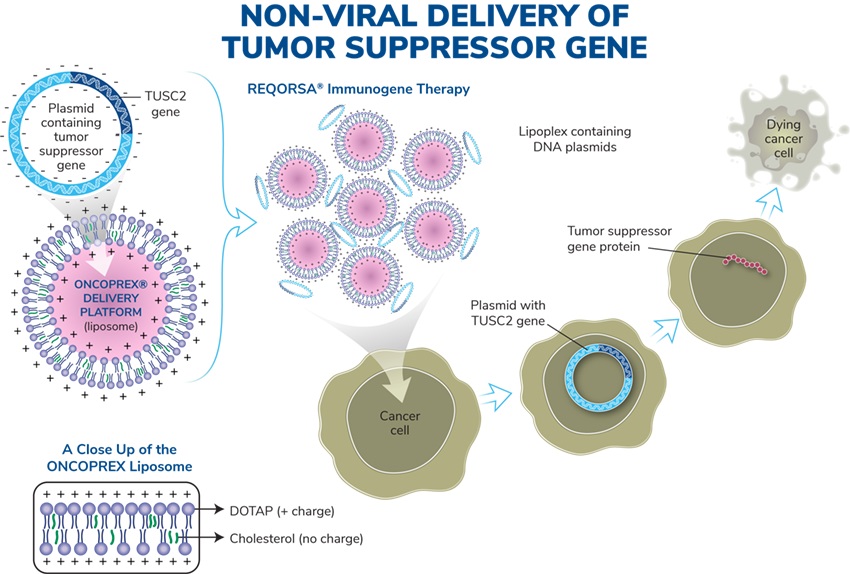
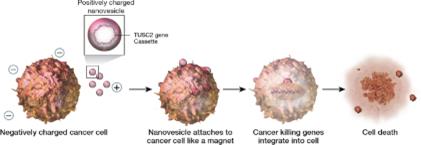
Operation of the ONCOPREX Nanoparticle Delivery System
The particle size is small enough to allow REQORSA to cross tight barriers in the lung, but large enough to avoid accumulation or clearance in the liver, spleen and kidney. The cationic (positive) charge of the nanoparticleslipoplexes helps to target cancer cells, and direct nanoparticle fusion avoids target cell endocytosis.which have a slight negative charge due to their high glycolytic rate. A Phase 1 clinical trial showed that intravenous REQORSA therapy selectively and preferentially targeted primary and metastatic tumor cells, resulting in anticancer activity. The nanoparticleslipoplexes are non-immunogenic, allowing repetitive therapeutic dosing and providing extended half-life in the circulation.
The ONCOPREX Nanoparticle Delivery System is based on a non-viral delivery system. MostMany gene therapies rely on viral based delivery systems. The benefit of the viral system is that viruses are skilled at penetrating cells. However, viruses can also affect more than one type of cell and it is possible that the virus may infect additional cells not justother than the targeted cells containing mutated genes. If this happens, healthy cells may be damaged causing other illness or diseases, such as cancer. WithOnce REQORSA once it is taken up into a cancer cell, the TUSC2 gene is expressed into aand TUSC2 protein that is capable of restoring certain defective functions arising in the cancer cell. REQORSA has been designed using the ONCOPREX Nanoparticle Delivery System to deliver the functioning TUSC2 gene to cancer cells while minimizing their uptake by normal tissue. Tumor biopsyLaboratory studies conducted at MD Anderson showed that in three patients, the uptake of TUSC2 in tumor cells after REQORSA treatment was 10 to 33 times the uptake in normal cells.cells, and studies in three NSCLC patients showed a major increase in TUSC2 expression in tumor tissue one day after REQORSA administration. REQORSA is also delivered systemically as opposed to many other gene therapies which are locally delivered.
REQORSA Origins, Development Rationale, and Strategy
TUSC2 was discovered through a lung cancer research consortium from MD Anderson and The University of Texas Southwestern Medical Center along with the National Cancer Institute. The TUSC2 discovery teams included Jack A. Roth, MD, FACS, chairman of our Scientific and Medical Advisory Board.
Our technology discoveries and research and development programs have been the subjects of numerous peer-reviewed publications and have been supported by Small Business Innovation Research ("SBIR") grants and grants from the National Institutes of Health, the United States Department of Treasury, and the State of Texas. We hold a worldwide, exclusive license from MD Anderson to patents covering the therapeutic use of TUSC2 and other genes that have been shown to have cancer fighting properties, as well as a number of related technologies, including 27 issued patents and 17 pending patent applications. The rights we have obtained pursuant to our license agreement with MD Anderson are made subject to the rights of the U.S. government to the extent that the technology covered by the licensed intellectual property was developed under a funding agreement between MD Anderson and the U.S. government.
REQORSA Development Rationale and Strategy
Our goal is to utilize our novel immunogenegene therapy platform to provide more effective treatments to large patient populations suffering from devastating illness with more effective treatments.illness.
REQORSA, our lead oncology product candidate, initially is being developed as a potential treatment for NSCLC.NSCLC and SCLC. Clinical and preclinical data indicatesindicate that REQORSA, when combined with EGFR TKIs such as Tagrisso, Tarceva and Iressa, provides a synergistic effect that could also benefit the larger population of NSCLC patients who are EGFR negative (which means they are not expected to benefit from EGFR TKI drugs alone).effect. Further, our data shows that REQORSA may re-sensitize EGFR positive patients who become resistant to, and therefore no longer benefit from, EGFR TKIs alone. Thus, REQORSA may both significantly expand the benefit of EGFR TKIs to the majority of patients who do not have EGFR activating mutations, and also extend the usefulness and benefit of EGFR TKIs for the population of NSCLC patients who are EGFR positive, but whose tumors progress on EGFR TKIs. Preclinical and clinical data support our belief that REQORSA may provide medical benefit in several subpopulations of NSCLC patients for which there is an unmet medical need, such as NSCLC patients with EGFR mutations, and ALK positive NSCLC patients. These data also served as the basis for the receipt from the FDA in January 2020 of aour first Fast Track Designation. In granting our Fast Track Designation, the FDA found that REQORSA may provide a benefit over existing therapies for patients whose tumors progress on Tagrisso. TheThis FDA Fast Track Designation is for use of the combination of REQORSA with TKI Tagrisso for the treatment of NSCLC patients with EGFR mutations whose tumors progressed after treatment with Tagrisso.
Pre-clinicalPreclinical data also hashave shown that REQORSA enhances the immune response to cancer. Data from preclinical studies at MD Anderson hashave shown a therapeutic benefit from the combination of TUSC2 and anti-PD-1 antibody or anti-PD-L1 antibody and a key role for TUSC2 in regulating immune cell subpopulations including cytokines, NK cells, and T lymphocytes. In addition, TUSC2 has been found to down-regulate PD-L1 receptors on the surface of cancer cells. These data, along with our previous preclinical and clinical data, provided the basis for the receipt from the FDA in December 2021 of our second Fast Track Designation. In granting this Fast Track Designation, the FDA found that REQORSA has the potential to provide a benefit over existing therapies for patients whose tumors progress on Keytruda. This FDA Fast Track Designation is for use of the combination of REQORSA with Keytruda for the treatment of NSCLC patients whose tumors progressed after treatment with Keytruda.
Pre-clinicalOur study in SCLC builds on the preclinical data showing that REQORSA enhances the immune response to cancer, and that the combination of REQORSA and immune checkpoint inhibitors demonstrates a therapeutic benefit over immune checkpoint inhibitors alone. Immune checkpoint inhibitors, such as Tecentriq, have recently been approved for use in ES-SCLC. Tecentriq, for instance, is used in combination with the chemotherapy drugs carboplatin and etoposide for 4 cycles of therapy, and then Tecentriq is administered alone as maintenance therapy until disease progression. Unfortunately, this is a relatively short time since the median PFS after starting maintenance therapy is 2.6 months. Our goal with combining REQORSA and Tecentriq as maintenance therapy is to prolong PFS and survival of ES-SCLC patients. In June 2023, the FDA granted our third Fast Track Designation. In granting this Fast Track Designation, the FDA found that REQORSA has the potential to provide a benefit over existing therapies for patients with ES-SCLC. This FDA Fast Track Designation is for the use of the combination of REQORSA with Tecentriq as maintenance therapy in patients with ES-SCLC who did not develop tumor progression after receiving Tecentriq and chemotherapy as initial standard treatment. In August 2023, the FDA also granted Orphan Drug Designation to REQORSA for the treatment of SCLC.
Preclinical studies by MD Anderson researchers have included combining REQORSA with:
● | the EGFR TKI gefitinib (marketed as Iressa® by AstraZeneca Pharmaceuticals) in animals and in human NSCLC cells; |
● | third generation EGFR TKIs such as osimertinib (marketed as Tagrisso® by AstraZeneca Pharmaceuticals); |
● | MK2206 in animals (MK2206 is an inhibitor of AKT kinases, which affect cell signaling pathways downstream from tyrosine kinases); |
● | the |
● |
|
● |
|
|
|
|
|
|
|
The manufacturers of the marketed drugs were not involved in any of our clinical or preclinical studies. In clinical studies involving marketed drugs, the drugs were administered concurrently with REQORSA without being modified in any way, and the antibodies used in our preclinical studies that did not use the marketed drugs were the non-humanized equivalent to marketed drugs.
Data from these clinical and preclinical studies indicates that combining REQORSA with these other therapies yields results more favorable than either these therapies or REQORSA alone, with minimal side effects relative to other lung cancer drugs, thereby potentially making REQORSA a therapy complementary to these cancer treatments.
Our Fast Track Designation, clinical and preclinical data have provided the basis for the Company’s development strategy for REQORSA and its use in treating NSCLC. Accordingly, we are planning to initiate, our Acclaim-1 and Acclaim-2 clinical trials in late-stage NSCLC in 2021.
Acclaim-1
InAs described above, in January 2020, we received Fast Track Designation from the FDA for use of REQORSA in combination with TKI Tagrisso for the treatment of NSCLC patients with EGFR mutations whose tumors progressed after treatment with Tagrisso. Given
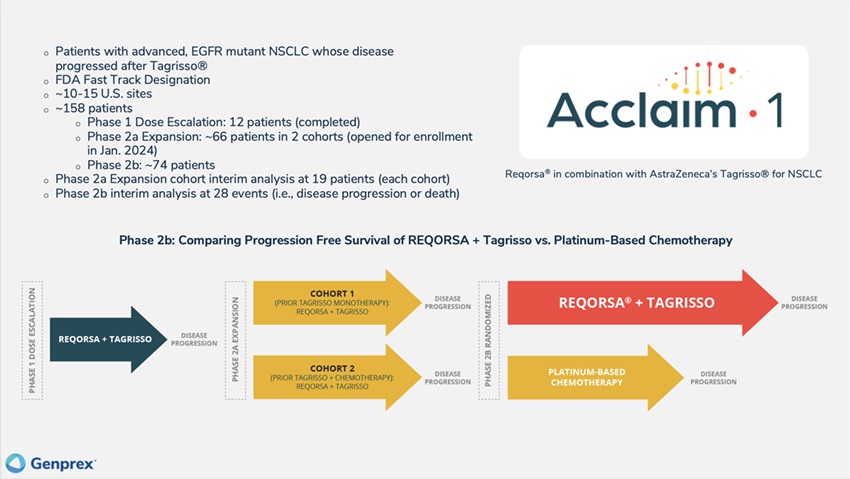
We currently are enrolling and treating patients in the poor prognosis for these patients andPhase 2a expansion portion of our FDA Fast Track Designation, we are prioritizing this drug combination and patient population and plan to initiate a Phase 1/2 Acclaim-1 clinical trial, in 2021.
The Acclaim-1 trial is a Phase 1/2 Open-Label, Dose-Escalationan open-label, dose-escalation and Clinical Response Studyclinical response study of REQORSA in Combinationcombination with Tagrisso in patients with advanced, EGFR-mutant, metastatic non-small-cell lung cancer who have progressed after treatment with Tagrisso. We anticipate enrolling patients at approximately 15 clinical sites and estimate that the Phase 1 portion of the Acclaim-1 trial will enroll up to 18 patients and that the Phase 2 portion will enroll approximately 74 patients (a 1:1 ratio of REQORSA and Tagrisso combination therapy versus Tagrisso monotherapy). Patients will be treated until a second progression event or unacceptable toxicity is experienced. Patients must have histologically confirmed unresectable stage III or IV EGFR-positive NSCLC (any histology) with:
● | radiological progression on Tagrisso (third generation EGFR-TKI); and |
● |
|
We enrolled 12 patients in the completed Phase 1 dose escalation portion of the Acclaim-1 study and estimate that the Phase 2a expansion portion will enroll approximately 66 patients in 2 cohorts, and the randomized Phase 2b portion will enroll approximately 74 patients. Starting with the Phase 2a expansion portion of the study, all patients receiving REQORSA in this study are required to submit an archival biopsy specimen that can be evaluated for TUSC2 expression. Half of the patients enrolled in the Phase 2a expansion portion of the study will be patients who received only prior Tagrisso treatment and the other half will be patients who received prior Tagrisso treatment and chemotherapy. The aim is to determine toxicity and efficacy profiles of patients with different eligibility criteria. There will be an interim analysis following the treatment of 19 patients in each cohort. In connection with the Phase 2 expansion portion of the trial, we are in the process of adding approximately 3-5 additional clinical sites and we expect to enroll patients at approximately 10-15 U.S. clinical sites for the Acclaim-1 study. We opened the Phase 2a expansion portion of the Acclaim-1 study and enrolled and dosed the first patient in January 2024. We expect to complete the enrollment of the 19 patients in each cohort of the Phase 2a expansion portion of the study by the end of 2024, and thus we expect the interim analyses in early 2025. Patients enrolled in the Phase 2b portion of the study will be randomized 1:1 to either REQORSA and Tagrisso combination therapy or to platinum-based chemotherapy. Patients will be treated until disease progression or unacceptable toxicity is experienced.
The primary endpoint of the Phase 1 portion isof the Acclaim-1 study was to determine a dose limiting toxicity ("DLT"), defined as ≥Grade 3 prolonged non-hematologicalwith DLT or, ≥Grade 4 prolonged hematological toxicity occurring duringif DLT was not experienced, to determine the first cyclerandomized RP2D. Since no DLTs were experienced in Phase 1, the 0.12 mg/kg dose of therapy and consideredREQORSA was determined to be possibly, probably, or definitely related to REQORSA and Tagrisso combination therapy.the RP2D. The primary endpoint of the Phase 22a expansion portion is overall response rate (ORR). The primary endpoint of the Phase 2b randomized portion of the trial is progression-free survivalPFS which is defined as time from randomization after first progression on Tagrisso, to first event (seconddisease progression) or death. Patients will be followed for upsurvival.
In October 2023, one of our clinical collaborators presented a poster presentation at the 2023 AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics detailing the Phase 1 results of the Acclaim-1 study. While the Phase 1 portion of the Acclaim-1 study was designed primarily to assess safety, we believe promising efficacy results were also observed. The reported results showed no DLTs, established a RP2D of 0.12 mg/kg (the highest dose level administered in the trial) and provided data showing early efficacy of REQORSA in combination with Tagrisso. Of the 12 patients treated with escalating doses of REQORSA and standard doses of Tagrisso, all of whom had progressed on Tagrisso containing regimens, three patients, as of data from January 2024, had experienced prolonged time to progression, including one with continuing partial response. Specifically, one patient at the 0.06 mg/kg dose level, previously treated with carboplatin, pemetrexed, and Tagrisso, had a partial remission by investigator evaluation and treatment is now ongoing in the trial after 28 cycles, which is approximately 19.5 months. A second patient at the 0.12 mg/kg dose level who was previously treated with cisplatin, pemetrexed, carboplatin, and Tagrisso has stable disease and is continuing to receive REQORSA after 14 cycles, or approximately 10 months. And a third patient who was at the 0.09 mg/kg dose level, previously treated with Tagrisso, had stable disease and received 14 cycles, over approximately 10 months followingbefore disease progression occurred. The extended PFS of each of these patients is consistent with long-term PFS seen in several patients in prior early stage clinical trials of REQORSA and is not expected with treatment with Tagrisso alone after progression on Tagrisso. REQORSA administration was generally well tolerated and there were no DLTs. The administration was associated with a delayed infusion-related reaction of muscle aches, fever and chills in some patients, which we believe is similar to reactions seen with the administration of their last doseantibodies routinely used in oncology treatment. This was managed with prophylactic steroids, acetaminophen and diphenhydramine, and symptoms decreased with repeat cycles. We believe this new mechanism and novel approach targeting lung cancer, which comes with a strong safety profile and early signs of Tagrisso. We have received centralized institutional review board ("IRB") approval for Acclaim-1.efficacy, is paving new ground in the fight against lung cancer.
Acclaim-2
In December 2021, we received Fast Track Designation from the FDA for use of REQORSA in combination with the checkpoint inhibitor Keytruda for the treatment of advanced NSCLC patients whose tumors progressed after treatment with Keytruda.
In 2019, preclinical data waswere presented by MD Anderson collaborators relating to the combination of TUSC2, the active agent in REQORSA, with Keytruda showing that TUSC2 combined with the checkpoint blockade mechanism of action of Keytruda was more effective than Keytruda alone in increasing the survival of mice with a human immune cellssystem (humanized mice) that had metastatic lung cancer with a human lung cancer. MD Anderson also presented preclinical data in 2019 for the combination of TUSC2, Keytruda and chemotherapy for the treatment of some of the most resistant metastatic lung cancers. This study found that the addition of TUSC2 demonstrates synergy with Keytruda and also with Keytruda combined with chemotherapy, and thus, may improve on the first-line standard of care for lung cancer. In May 2020, we entered into a worldwide, exclusive license agreement with The Board of Regents of the University of Texas System on behalf of MD Anderson for the use of TUSC2 in combination with immunotherapies, including Keytruda, and also for the use of TUSC2 in a three-drug combination of TUSC2, immunotherapy andcancer which includes chemotherapy.
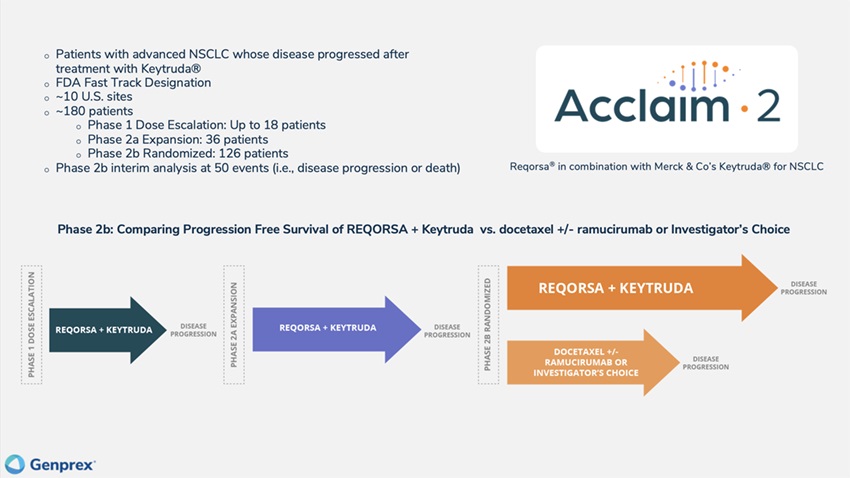
The Acclaim-2 trial is aWe currently are enrolling and treating patients in the Phase 1 dose escalation portion of our Phase 1/2 Acclaim-2 clinical trial, combiningan open-label, dose-escalation and clinical response study of REQORSA in combination with Keytruda in patients whose disease haswith advanced, metastatic non-small-cell lung cancer who have progressed on Keytrudaafter treatment with Keytruda. Patients must have histologically confirmed unresectable stage III or IV NSCLC (any histology) with:
| ● | radiological progression on Keytruda; and | |
| ● | an ECOG performance status of 0 to 1. |
Starting in NSCLC2024, all patients whoreceiving REQORSA in this study are low expressors (1%required to 49%)submit an archival biopsy specimen that can be evaluated for TUSC2 expression. We expect to enroll patients at approximately 10 U.S. clinical sites and estimate that the dose escalation portion of PD-L1. We plan to initiate the Acclaim-2 trial will enroll up to 18 patients, the Phase 2a expansion portion will enroll 36 patients, and the Phase 2b randomized portion will enroll approximately 126 patients. Patients enrolled in 2021.the Phase 2b randomized portion of the study will be randomized 2:1 to either REQORSA and Keytruda combination therapy in the experimental arm or to chemotherapy (docetaxel with or without ramucirumab), or to the investigator’s choice of therapy in the control arm. Patients will be treated until disease progression or unacceptable toxicity is experienced.
The primary endpoint of the dose escalation portion is DLT, defined as ≥Grade 3 prolonged non-hematological or ≥Grade 4 prolonged hematological toxicity occurring during the first cycle of therapy and considered to be possibly, probably, or definitely related to REQORSA and Keytruda combination therapy. The primary endpoint of the Phase 2a expansion portion and the Phase 2b randomized portion of the trial is progression-free survival which is defined as time from randomization to disease progression or death. Patients will be followed for survival.
Patients are currently being treated at the 0.06 mg/kg dose level and, subject to Acclaim-2 SRC approval, we will treat the two successive cohorts of patients at the 0.09 mg/kg and 0.12 mg/kg dose levels. In March 2023, we amended the Acclaim-2 protocol to include additional treatments in the control group with the goal of accelerating enrollment in the study by making the trial more attractive to a wider variety of investigators. We expect enrollment in the Phase 1 dose escalation portion of the Acclaim-2 trial will be completed in the second half of 2024. We will then initiate and evaluate patients in the Phase 2a expansion portion of the study at the MTD or RP2D to expand the safety and efficacy profile at that dose in order to better evaluate efficacy and to increase the likelihood of a successful Phase 2b randomized trial, which will follow the expansion portion.
Acclaim-3
In June 2023, the FDA granted Fast Track Designation for the Acclaim-3 treatment combination of REQORSA and Tecentriq as maintenance therapy in patients with ES-SCLC who did not develop tumor progression after receiving Tecentriq and chemotherapy as initial standard treatment. In August 2023, the FDA granted Orphan Drug Designation to REQORSA for the treatment of SCLC. We currently are enrolling patients in the Phase 1 dose escalation portion of our Phase 1/2 Acclaim-3 clinical trial. Patients in the study will be enrolled after receiving initial treatment with 3-4 cycles of carboplatin, etoposide, and Tecentriq, and achieving complete response, partial response or stable disease. They will then receive treatment with REQORSA and Tecentriq as maintenance therapy every 21 days until disease progression. In January 2024, we opened the Phase 1 portion of the Acclaim-3 study for enrollment and added multiple clinical sites through our collaboration with a large network of integrated, community-based oncology practices. We now anticipate enrolling patients at approximately 10 U.S. clinical sites and estimate that the Phase 1 dose escalation portion of the Acclaim-3 trial will enroll up to 12 patients, and the Phase 2 expansion portion will enroll approximately 50 patients at approximately 10 to 15 U.S clinical sites. Patients will be treated with REQORSA and Tecentriq until disease progression or unacceptable toxicity is experienced. The primary endpoint of the Phase 1 escalation portion is to determine the MTD or RP2D and the primary endpoint of the Phase 2 portion is to determine the 18-week progression-free survival rate from the time of the start of maintenance therapy with REQORSA and Tecentriq in patients with ES-SCLC. Patients will also be followed for survival. We expect to complete the Phase 1 portion of the study during the second half of 2024 and we expect to start the Phase 2 expansion portion of our Acclaim-3 study in the second half of 2024.
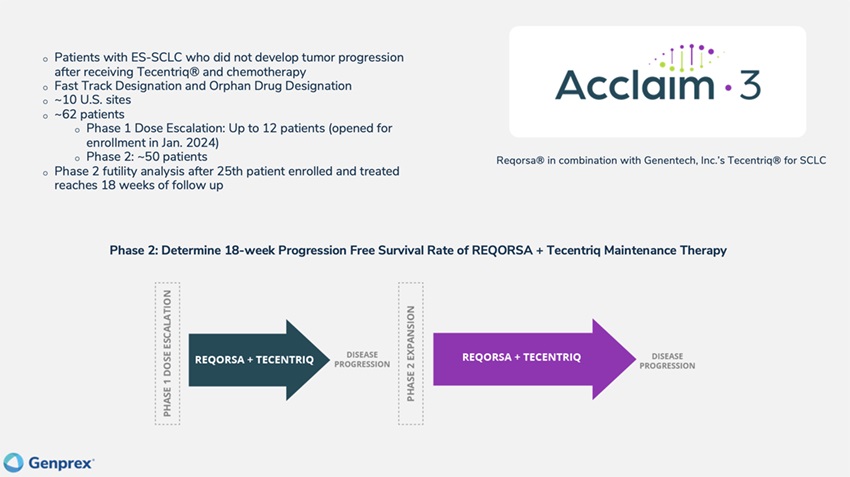
ONC-001: REQORSAREQORSA®™ Phase 1 Monotherapy Trial (completed)
In 2012, MD Anderson researchers completed a Phase 1 clinical trial of REQORSA as a monotherapy (the “REQORSA“Phase 1 Monotherapy Trial”). in patients with advanced NSCLC with disease progression at study entry. The primary objective of the REQORSA Monotherapy Trial was to assess the toxicity of REQORSA administered intravenously and to determine the maximum tolerated dose ("MTD")MTD and recommended Phase 2 doseRP2D of REQORSA alone. Secondary objectives were to assess the expression of TUSC2 following intravenous delivery of REQORSA in tumor biopsies and also to assess the anticancer activity of REQORSA. This trial showed that REQORSA was well tolerated and established the single-agent MTD and the therapeutic dosage for REQORSA at 0.06 mg/kg administered every 21 days. This MTD was established based on the occurrence of an asymptomatic, Grade 3 laboratory abnormality in 2 patients. Although this trial was not designed to show changes in outcomes, a halt in cancer growth was observed in a number of patients, and tumor regressions occurred in primary lung tumors and metastatic cancers in the liver, pancreas, and lymph nodes. In addition, pre- and post-treatment patient biopsies demonstrated that intravenous REQORSA selectively and preferentially targeted patients’patient’s cancer cells and suggested that clinical anti-cancer activity was mediated by TUSC2.increased expression of TUSC2 in the cancer cells.
In the Phase 1 Monotherapy Trial, REQORSA was injected intravenously in stage IV (metastatic) lung cancer patients who had received traditional platinum combination chemotherapy but still showedhad tumor progression at the time of entry into the study. 31Thirty-one subjects were treated at six dose levels. Seventy percent of subjects had received two or more prior chemotherapy regimens. The only serious adverse events defined as grade 3, 4 or 5 events under the Common Terminology Criteria for Adverse Events published by the U.S. Department of Health and Human Services, were grade 3 fever (experienced by three patients) and grade 3 hypotension (experienced by 1 patient). The only dose-limiting toxicities were two episodes of transient grade 3 hypophosphatemia (abnormally low levels of phosphate in the blood) resulting in an MTD of 0.06 mg/kg. Twenty-three subjects received two or more doses, of whom five subjects,Five patients, or 22% of the 23 subjects,evaluable patients, achieved disease control for periods ranging from 2.6 months to 10.8 months. The median disease control period for these subjectspatients was 5.0 months (95% CI: 2.0-7.6), while the other 18 subjects’ cancer progressed during the Phase 1 Monotherapy Trial. Disease control for cancer therapies is defined under the Response Evaluation Criteria in Solid Tumors ("RECIST") as Complete Response (CR) + Partial Response (PR) + Stable Disease (SD)>8weeks). Median survival for all subjects in the Phase 1 Monotherapy Trial was 8.3 months (95% CI 6.0-10.5 months) and mean survival time was 13.2 months (95%CI 8.9-7.5 months) with a range of two to 23+ months.
Two subjects had reductions in primary tumor size of 14% and 26%. One subject with stable disease, a 54-year-old female with a large cell neuroendocrine carcinoma who received 12 cycles of REQORSA therapy before having disease progression, had evidence of a durable metabolic response, which is a lasting reduction of metabolic activity in the tumor, as shown by positron emission tomography ("PET") imaging. The response was documented with PET scans performed after the second, fourth and sixth doses, all showing markedly decreased metabolic activity in the tumor with no changes in size or number of metastases by computed tomography ("CT") imaging. The illustration below is of the PET scan of this subject performed at baseline (Illustration A) and after the fourth dose.dose (Illustration B). This subject had received six prior chemotherapy regimens. Prior to entry in the Phase 1 Monotherapy Trial, two hepatic metastases were progressing on gemcitabine. The subject also had a metastasis in the head of the pancreas and a peripancreatic lymph node, shown by the arrows in the illustration below. Illustration A shows the pretreatment PET scan. The dose of Fluorodeoxyglucose (18F) was 8.8mCi. Illustration B shows the post treatment PET scan performed 20 days following the fourth dose of REQORSA. The dose of Fluorodeoxyglucose (18F) was 9.0mCi. All scans were performed within a 60 to 90 minute window after injection.
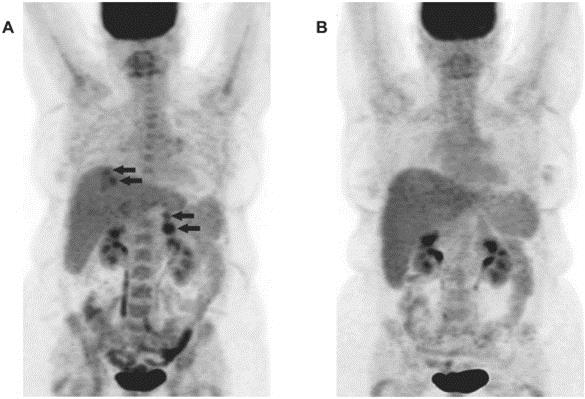
Metabolic Tumor Response in a Metastatic Lung Cancer Subject
This subject survived after subsequent therapy more than seven years after the final treatment with REQORSA, to our knowledge, without evidence of cancer progression in the responding sites.
ONC-002: Phase 1/2 - Trial Combining REQORSA with Tarceva (Phase 1 portion completed; Phase 2 portion no longer enrollingclosed in favor of conducting Acclaim-1)order to conduct Acclaim-1 instead)
Phase 1 Portion: The Phase 1 Monotherapy Trial showed that REQORSA is well tolerated, that high levels of TUSC2 expression are detected in the tumor post-treatment, and that there iswas evidence of tumor growth suppression. Based on the results from the Phase 1 Monotherapy Trial and substantial preclinical evidence that REQORSA is complementary with EGFR TKIs, we began a Phase 1/2 trial (the “Phase 1/2 Combination Tarceva Trial”) at MD Anderson combining REQORSA with Tarceva in patients with Stage IV (metastatic) or recurrent NSCLC that is not potentially curable by radiotherapy or surgery,surgery. Patients were enrolled whether or not they have received prior chemotherapy, and whether or not they havehad an activating EGFR mutation. Enrollment in the Phase 1 portion of the Phase 1/2 Combination Tarceva Trial commenced in 2014 at MD Anderson with Dr. Charles Lu as the Principal Investigator.
In the Phase 1 portion of the Phase 1/2 Combination Tarceva Trial, 18 subjects were treated with the following dose levels:
Dose Level | Drug Doses | |
1 | Tarceva (100 mg/day) + REQORSA | |
2 | Tarceva (100 mg/day) + REQORSA | |
3 | Tarceva (150 mg/day) + REQORSA | |
4 | Tarceva (150 mg/day) + REQORSA |
As in the Phase 1 Monotherapy Trial, subjects received a pre-treatment regimen of oral and intravenous dexamethasone and diphenhydramine to reduceprevent infusion reaction symptoms such as fever, along with an infusion of REQORSA every three weeks. Subjects received oral Tarceva daily during each three-week cycle during the treatment period.
The Phase 1 portion of the Phase 1/2 Combination Tarceva Trial was also a dose escalation study with the primary purpose of determining the MTD. DLT were defined as grade 3, 4, or 5 events during the first cycle of treatment that were considered to be treatment related. At dose level 1, one subject had grade 3 adverse events of fatigue, muscle weakness, and hyponatremia (low sodium level) considered to be related to the study treatment (Tarceva). Therefore, three additional subjects were treated at this dose level (six subjects total), none of whom sufferedhad a DLT. At dose level 2, there were no DLTs. At dose level 3, one subject had a grade 3 rash considered to be related to the study treatment (Tarceva); therefore, an additional three subjects were treated at this dose level (six subjects total). No additional subjects sufferedhad a DLT. At dose level 4, there were no DLTs; thus, dose level 4, as the highest dose evaluated, was determined to be the MTD.
Once the MTD for the study treatment combination was determined in the Phase 1 portion of the Phase 1/2 Combination Tarceva Trialdose to be Dose Level 4, accrual proceeded withused in the Phase 2 portion of the study.
Since the eligibility criteria, drug administration details (other than dose) and evaluation details were identical for the Phase 1 portion and the Phase 2 portion, the three subjects in the Phase 1 portion of the Phase 1/2 Combination Tarceva Trial who were treated at the MTDPhase 2 dose (0.06 mg/kg) were included in the analysis of the Phase 2 portion of the study.
Four patients in the Phase 1 portion of the study had stable disease ranging from 12 weeks to 36 weeks. The following observations from our preclinical studies and from the Phase 1 portion of the Phase 1/2 Combination Tarceva Trial provided the rationale for proceeding with the Phase 2 portion of the study:
|
|
|
|
|
|
|
|
Phase 2 Portion: The Phase 2 portion of the Phase 1/2 Combination Tarceva Trial was a Simon two-stage trial designed to include subjects treated with the combination of REQORSA and Tarceva at the MTDPhase 2 dose with the primary goal of measuring the response rate, and secondary endpoints of stable disease, time to progression and overall survival. The response rate for cancer therapies iswas defined under the RECIST as Complete Response (CR) + Partial Response (PR); disease control rate iswas defined under the RECIST criteria as Complete Response (CR) + Partial Response (PR) + Stable Disease (SD) > 8 weeks. Although this Simon two-stage trial was closed early to start the Acclaim-1 study instead, the trial had already met the required response rate to advance to the second stage and to complete the full enrollment.
Enrollment criteria for the Phase 2 portion were identical to those in the first phase. The first subject enrolled in the Phase 2 portion began Tarceva on Day 8, and subsequently every other enrolled subject began Tarceva on Day 8. The rationale for delaying Tarceva was to allow exploratory analyses of potential differential effects of REQORSA alone and in combination with Tarceva on downstream pathway activation and potential biomarkers of Tarceva resistance.1 portion. Subjects received three-week cycles of REQORSA in combination with Tarceva until the occurrence of progressive disease (PD), unacceptable toxicity, withdrawal of consent, or study treatment discontinuation for other reasons, whichever occurred first.
Of the 39 patients allowed in the protocolplanned for the Phase 2 portion of the trial, 10 were enrolled (three of whom were also subjects of the Phase 1 portion of the Phase 1/2 Combination Tarceva Trial) and nine were evaluable for response under the trial protocol, because they received two or more cycles of treatment.. None of the 10 subjects treated in the Phase 2 portion of the Phase 1/2 Combination Tarceva Trial sufferedhad a DLT. Interim resultsResults from the Phase 2 portion for the 9 evaluable10 patients show that:
● | One patient had a response |
● |
● |
|
| Disease control rate |
The patient with the CR, a 58-year-old female, upon enrollment in the study had metastatic NSCLC status following 6 cycles of pemetrexed and carboplatin and after two cycles of maintenance pemetrexed withhad cancer progression. The patient’s tumor hashad EGFR exon 18 and 20 missense mutations, which are not sensitive to Tarceva. As shown in the illustrations below, thisTarceva alone. This patient had disappearance of both the lung primary tumor and the lung, liver and lymph node metastases.
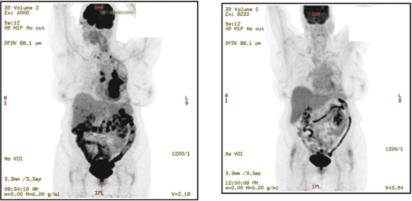
Subject with RECIST Complete Response
We are no longer enrolling the Phase 2 portion of the Phase 1/2 Combination Tarceva Trial in favor of conducting Acclaim-1.
The response rate and disease control rate observed in the Phase 2 portion of the Phase 1/2 Combination Tarceva Trial substantially exceeds the response rate of 7% (with no CRs) and disease control rate of 58% reported for a clinical trial of the EGFR TKI afatinib (marketed as Gilotrif® by Boehringer Ingelheim Pharmaceuticals, Inc.) in a study referred to as the LUX-Lung 1 clinical trial. A total of 585 patients were enrolled in that Phase 2b/3 clinical trial, whose primary endpoint was overall survival and whose secondary endpoints were progression-free survival, RECIST response, quality of life and safety. The LUX-Lung 1 clinical trial was a randomized, double blinded Phase 2b/3 clinical trial treating subjects with Stage IIIB or IV adenocarcinoma, a type of NSCLC. Patients in that trial had received one or two previous chemotherapy regimens and had disease progression after at least 12 weeks of treatment with EGFR inhibitors erlotinib or gefitinib. A total of 585 patients were enrolled in that Phase 2b/3 clinical trial, whose primary endpoint was overall survival and whose secondary endpoints included progression-free survival and RECIST response. The Phase 2 portion of our Phase 1/2 trial was not blinded and was designed to treat NSCLC subjects regardless of EGFR status.
The following table sets forth interimprovides data from the Phase 2 portion of the Phase 1/2 Combination Tarceva Trial for subjects with and without EGFR mutations.

Note that two patients with disease progression on Tarceva received 10 and 12 cycles of Tarceva, respectively, before disease progression and entry into the trial. With the combination of REQORSA and Tarceva, both of these patients had stable disease, suggesting that the combination therapy may be an effective treatment for patients whose disease is progressing after extensive Tarceva treatment. We are no longer enrolling the Phase 2 portion of the Phase 1/2 Combination Tarceva Trial in favor of conducting the Acclaim-1 trial, which combines REQORSA with Tagrisso, since Tagrisso has been shown to be more effective than Tarceva as initial therapy for patients with NSCLC with EGFR mutations.
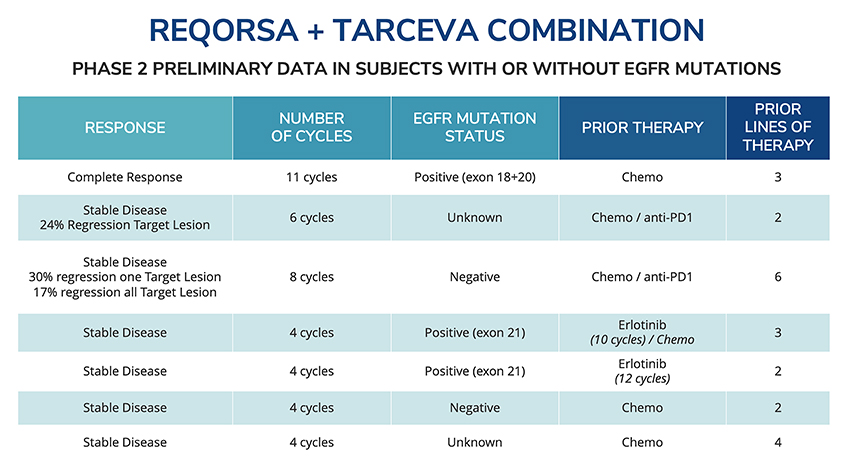
Preclinical Studies of REQORSA supporting our conductSupporting Our Conduct of Acclaim-1
REQORSA and Tyrosine Kinases. Investigators at MD Anderson conducted preclinical research showing that REQORSA alone blocked the activation of the c-Abl tyrosine kinase. A number of other studies at MD Anderson have shown the complementary effects of REQORSA combined with a variety of targeted kinase inhibitory agents, both marketed and in various stages of clinical development, including Tarceva, Iressa, Tagrisso, MK2206, and others. Researchers investigated the use of REQORSA combined with commercially available EGFR TKI drugs Tarceva and Iressa, and conducted preclinical in vitro and in vivo studies combining REQORSA with these drugs in a variety of human lung cancer cell lines, including cancers with activating EGFR mutations and EGFR mutation negative cancers. Lung cancers known to have intrinsic and acquired resistance to Tarceva therapy were also studied, as were Kras-related and other cancers. Notably, studies in xenograft animal models showed that REQORSA and either Tarceva or Iressa showed synergistic anti-cancer effects, superior to either agent used alone, in both EGFR mutation negative cancers (generally not candidates for Tarceva) and in EGFR mutation positive cancers (optimal candidates for Tarceva), including cancers known to be resistant to Tarceva therapy. The addition of REQORSA to either Tarceva or Iressa overcame drug-induced resistance by simultaneously inactivating EGFR and AKT signaling pathways and by inducing apoptosis in Tarceva- or Iressa-resistant cancers with EGFR mutations and with EGFR mutation-negative cancers.
In one study, MD Anderson researchers tested the combination of Tarceva and REQORSA against five human NSCLC cell lines: H1299, H322, A549, H460, and H1975, the latter of which has the L858R and T790M EGFR mutations and is highly resistant to Tarceva. The results showed that the combination of REQORSA and Tarceva significantly reduced NSCLC colony formation beyond the effect of Tarceva, REQORSA or controls alone (p<0.01 at both 1 and 2.3 uM concentrations for all cell lines). The cooperative interaction between Tarceva and REQORSA was confirmed in vivo using a lung colony formation metastases model in nu/nu mice with A549 human lung cancer cells injected in the tail vein. Mice were treated with the combination of REQORSA and Tarceva and various controls including empty nanovesicles, Tarceva alone, REQORSA alone, and other controls.
The greatest reduction in lung colonies occurred in the REQORSA with Tarceva combination (90% reduction) which was reduced compared to all control groups (p<0.0005). In terms of total tumor nodules, the cooperative effect is greater than 0.9999. This means that there is less than a 1 in 10,000 chance that the low dose Tarceva with TUSC2 combination does not have a cooperative effect and greater than 9,999 in 10,000 chance that the cooperative effect exists. P-value is the probability that the difference between two data sets was due to chance. The smaller the p-value, the more likely the differences are not due to chance alone. In general, if the p-value is less than or equal to 0.05, the outcome is considered statistically significant. The FDA’s evidentiary standard of efficacy generally relies on a p-value of less than or equal to 0.05.Tagrisso.
REQORSA and TUSC2 deficient and Tarceva or Iressa resistant cell lines. MD Anderson researchers also tested REQORSA in TUSC2-deficient and Tarceva- or Iressa-resistant NSCLC cell lines. Treatment of the NSCLC EGFR mutation negative cell lines H1299, H322, H358 and H460 cancer cell line showed that the REQORSA combination significantly sensitized (p<0.001) response of the cancer cell lines to both Tarceva or Iressa treatment and synergistically induced apoptosis in vitro. The findings were confirmed in vivo in an H322 orthotopic lung cancer mouse model. These studies included the Kras mutant cell line H460, which is significant because patients with Kras mutant tumors are generally unresponsive to Tarceva or Iressa. Synergistic induction of apoptosis was observed with the combination of REQORSA and concentrations of Tarceva or Iressa similar to steady-state serum concentrations achievable with oral dosing. The combination of REQORSA and either Tarceva or Iressa induced similar levels of tumor cell growth inhibition, apoptosis induction, and inactivation of oncogenic protein kinases.
Data from these and other MD Anderson studies suggest a combination of REQORSA with Iressa or Tarceva can promote synergistic tumor cell killing and overcome drug-induced resistance by simultaneously inactivating the EGFR and the AKT signaling pathways and by inducing apoptosis in resistant cells with nonmutated EGFR. These data suggest that NSCLC patients with an activating EGFR mutation, whose cancer progresses on Tarceva, may potentially benefit from REQORSA with Tarceva combination therapy. These data also suggest that NSCLC patients without an activating EGFR mutation (generally unresponsive to Tarceva) may potentially benefit from REQORSA with Tarceva combination therapy. These data provided strong support for the ONC-002 trial, which combined REQORSA with Tarceva.
REQORSA in Tagrisso resistant cell lines. Tagrisso (osimertinib), a third-generation EGFR inhibitor, shows robust clinical activity, yet patients inevitably develop secondary resistance. An osimertinib resistant H1975-OsiR isogenic cell line was developed through continuous exposure to osimertinib, and an osimertinib resistant clone was selected which showed 100-fold higher resistance to osimertinib compared with its parental counterpart (H1975-parental). Xenograft tumors from both H1975-parental and H1975-OsiR cells were developed in NSG mice and were treated with osimertinib. H1975-OsiR tumors were significantly less sensitive than its parental counterpart. Synergistic antitumor activity of TUSC2+osimertinib was found in H1975-OsiR tumors where both TUSC2+osimertinib (5mg/kg) and TUSC2+osimertinib (10mg/kg) combinations showed a robust antitumor effect compared with single agent treatment groups. No synergistic effect was observed for H1975-parental tumors. In conclusion, TUSC2 and Tumor Sensitivity. In another study, MD Anderson researchers analyzedtherapy in combination with osimertinib showed synergistic antitumor efficacy in EGFR mutant osimertinib resistant NSCLC tumors. These data provide a strong biologic rationale for the effectsAcclaim-1 clinical trial.
Preclinical Studies of TUSC2 re-expressionin the Immune Response to Cancer Supporting Our Conduct of Acclaim-2
Preclinical studies indicate that REQORSA is selectively taken up by tumor cells with a 10 to 33 fold differential favoring uptake by tumor cells, thus imparting a passive targeting property without the need to attach targeting ligands. REQORSA targeting is partly due to the attraction of opposite charges (REQORSA has a positive charge, normal cells no charge, and most cancer cells have a negative charge), and partly due to enhanced endocytosis by tumor cells, and is enhanced by the leakiness that is characteristic of tumor vasculature.
In experimental mouse xenograft models, the ONCOPREX delivery system was shown to efficiently deliver several therapeutic tumor suppressor genes (TP53, FHIT, TUSC2) to disseminated human cancer cells. Metastatic tumor growth was suppressed, and survival prolonged, after systemic administration of the genes via a nanovesicle vector. Human NSCLC A549 cells have virtually no TUSC2 protein. As shown in Figure 1, intratumoral administration of REQORSA (referred to as FUS1 in Figure 1) to subcutaneous NSCLC H1299 tumor xenografts resulted in inhibition of tumor growth.
Figure 1. Effect of REQORSA on the sensitivityGrowth of H1299 Subcutaneous Tumor Xenografts in Nude Mice
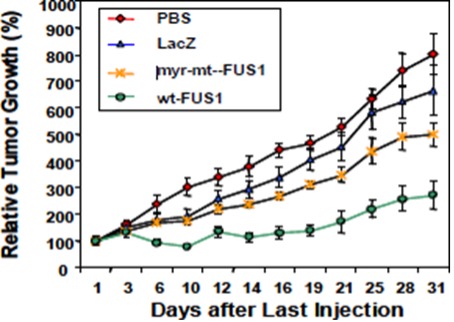
Moreover, intravenous injections of REQORSA into mice bearing experimental A549 lung metastases resulted in a decrease in the number of metastatic tumor cellsnodules. Lung tumor-bearing animals treated with REQORSA also had a significant increase (P=0.01) in survival time (median survival time: 80 days) compared with 48 to 51 days for control animals.
Analysis of TUSC2 expression by IHC following REQORSA treatment showed distribution of TUSC2 throughout the AKT inhibitor MK2206tumor in a high percentage of the tumor cells. These results demonstrate the potent tumor suppressing activity of the in vitroTUSC2 gene, supporting the feasibility of using nanovesicles for systemic plasmid delivery to metastases as well as to primary tumors, and in mice. The AKT pathway is an important intracellular, converging positive regulator of apoptosis. AKT stimulates apoptosisimplicating REQORSA as a promising therapeutic agent for primary and is frequently dysregulated in cancers, and this has been associated with reduced sensitivity to anti-tumor drugs. The study showed that the combination of TUSC2 transfection with MK2206 treatment suppressed tumor cell viability in vitro and effectively inhibited xenograft tumor growth in vivo more effectively than either agent alone.disseminated human lung cancer.
REQORSAPreclinical Studies of TUSC2 in the Immune Response to Cancer supporting our conduct of Acclaim-2 Synergizes with Pembrolizumab
Previous research hasIt was previously shown that TUSC2 regulates cytokine expressionthe combination of REQORSA and an anti-PD1 antibody inhibited tumor growth synergistically in subcutaneous and metastatic NSCLC in vitroKRAS. Cytokines are proteins that stimulate inflammation as part mutant syngeneic mouse models. To determine whether this synergy also applies to the KRAS/LKB1 mutant subtype of the immune response. Stable expression of TUSC2 in H1299human A549 NSCLC cells, altered expression ofhumanized mice harboring KRAS/LKB1 mutant A549 lung metastases were treated with REQORSA, pembrolizumab or the combination. These studies were performed with an improved humanized mouse model using fresh human umbilical cord blood derived CD34+ stem cells in irradiated NSG mice, in which mouse immune system cells have been largely destroyed. The reconstituted humanized mice have a wide spectrum of cytokines including IL2, IL7, IL8fully competent human immune system with major functional immune populations and 10, GM-CSFwere used here to evaluate synergy between REQORSA and PDGF-beta. TUSC2 is a positive regulator of innate immunity via regulation of IL-15 expression. IL-15 induces NK cell differentiation.pembrolizumab.
The systemic effect of thetreatment strategy is shown in Figure 2A. REQORSA (referred to as TUSC2 and anti-PD1 antibody combinationin Figure 2) was examined in two immunocompetent, syngeneic mouse models of Kras and p53 mutant lung cancer. C57BL/6 mice were subcutaneously injected with murine adenocarcinoma lung carcinoma CMT/167-luc cells (KrasG12V mutation). CMT/167 cells do not express TUSC2. Tumors from untreated mice, isotype antibody control, or those treated with anti-PD1 were used as controls. 344SQ (KrasG12D allele and a knock-in Trp53R172HÄG allele) adenocarcinomas which metastasize to the lung in 126S2 mice were also used. When tumors reached 50-100 mm3, mice were either injectedadministered intravenously with DOTAP:cholesterol (DC)-TUSC2 complex alone (at a dose of 25 “g of plasmid DNA and 10 nmol DC, every 48 hours for a total of three injections)injections, and pembrolizumab was administered every 3-4 days a total of three times. Bioluminescence imaging was performed to visualize the intensity of tumor burden for mice in different treatment groups both in humanized and non-humanized mice. Both REQORSA and pembrolizumab monotherapies reduced the tumor burden significantly, although pembrolizumab was moderately more effective. Importantly, REQORSA plus pembrolizumab inhibited tumor growth synergistically (*P< 0.05, **P< 0.005, ***, or (DC)-TUSC2 complex combinedP< 0.0005). (Figure 2B, C). There was no antitumor effect of pembrolizumab and reduced change with anti-PD1 antibody (250 “g for four injections) alone or combined with anti-CTLA4 (100ug for three injections). Tumor growth and developmentREQORSA in non-humanized mice, which was monitored by scoring ex-vivo luminescence using the IVIS Imaging System 200 Series. All tumor measurements were blinded to treatment and results were analyzed independently by biostatisticians.expected since these mice have no immune cells.
Preclinical Study Showing thatTo identify the TUSC2immunological features associated with efficacy of this combination, in depth immune profiling of the tumor microenvironment was performed. An increased number of reconstituted human CD3+ T cells was found in all groups, compared with the untreated control. CD8+ T cells were significantly upregulated by pembrolizumab and Anti-PD1 Combination Cooperatively Inhibits Growthits combination with REQORSA (Figure 2D). Levels of CMT/167 Lung Adenocarcinomas
Mouse experiments showed combined treatment with TUSC2 and anti-PD1 antibody superior to anti-PD1 alone in five independent experiments in two different tumor models. Results of a representative experiment are shownactivated CD8+ T cells (CD8+CD69+) were also significantly increased in the graph below. By week threecombination group and were slightly higher than the reductionpembrolizumab group (Figure 2D). There was no effect of pembrolizumab on NK/activated NK cells, whereas REQORSA alone enhanced their levels significantly, indicating REQORSA regulation of NK activation, which is consistent with the previous findings reported in tumor image intensity bysyngeneic mice. The combination had a similar effect as REQORSA monotherapy (Figure 2E). REQORSA, pembrolizumab, and the combination, were all associated with significant decrease of reconstituted human myeloid derived suppressor cells (“MDSCs”) (CD33+ve), (Figure 2F). Pembrolizumab and the combination had a profound stimulatory effect on HLA-DR+ dendritic cells (DCs), (Figure 2G). REQORSA alone enhanced HLA-DR+DC levels moderately. Taken together, these results show that the combination of TUSC2REQORSA and anti-PD1 and TUSC2, anti-PD1, plus anti-CTLA4 was greater thanpembrolizumab enhanced the reduction with TUSC2 alone, anti-PD1 combined with anti-CTLA4, or the isotype control. Spleens and blood were collected for immunological analysis profiling by multicolor flow cytometry. Immune profiling panels were designed to evaluateimmune response and major changes of specific regulatory innate and adaptive immune cells to TUSC2 or anti-PD1 treatment in peripheral blood and spleen.inhibited tumor growth synergistically (*P< 0.05, **P< 0.005, ***, P< 0.0005).
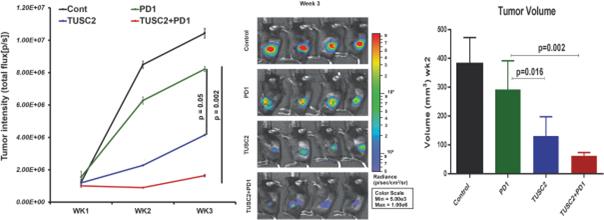
Preclinical Study Showing EffectREQORSA also showed synergistic antitumor activity with nivolumab in the same mouse model, highlighting the role of TUSC2 Anti-PD1 Combo on T LymphocytesREQORSA rendering KRAS/LKB1 mutant tumors more sensitive to immune checkpoint blockade. Thus, these data suggest that the synergy of REQORSA with immune checkpoint inhibitors is not limited to pembrolizumab.
Preclinical Study Showing theFigure 2. Synergistic Antitumor Effect of the TUSC2 and Anti-PD1 CombinationREQORSA Immunogene Therapy with Pembrolizumab on T Lymphocytes
The population of NK cells, cytotoxic lymphocytes critical to innate immune function, was assessed in peripheral blood mononuclear cells ("PBMCs") in tumor bearing mice treated with anti-PD1, TUSC2 alone and the combination. As shownKRAS/LKB1 Mutant Lung Metastases in the graph below, the NK cell population increased strongly in the TUSC2 alone and TUSC2+PD1 groups which correlated with tumor regression. Anti-PD1 alone had no effect on NK cell proliferation.
Tumor free mice without mutations that lead to metastasis were injected intravenously with TUSC2 which caused a threefold up-regulation of NK cells in the peripheral blood of TUSC2 injected mice as compared with non-injected mice. CD8 T cells, which are cytotoxic T cells ("CTL") for tumor killing, act as a prognostic marker of tumor regression. Increased numbers of CTL were found in the TUSC2 and TUSC2+PD1 groups as compared with that of the control group which directly correlated with the anti-tumor effect, as shown in the graph below. Lower levels of CD62L expression on T lymphocytes in TUSC2 treated mice suggests that TUSC2 regulates T cell activation. Moreover, TUSC2 induced down-regulation of regulatory T cells (Treg, CD4+CD25+). TUSC2 was shown to down-regulate checkpoint markers such as PD-1, CTLA-4, Tim-3, and LAG-3.Humanized Mouse Model
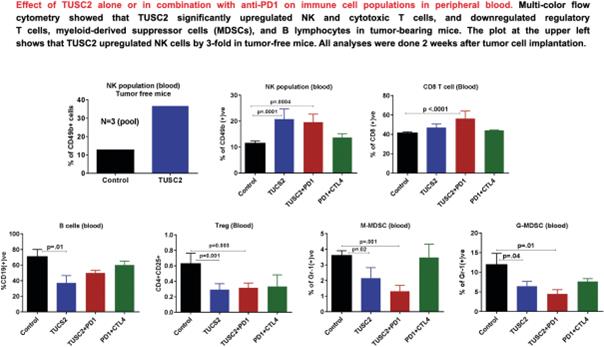
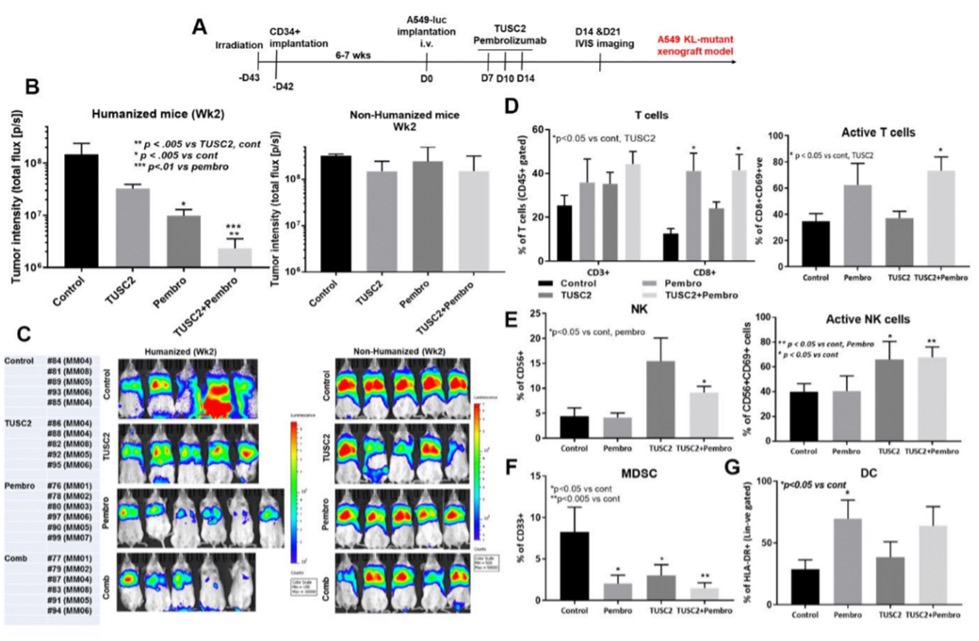
Effect of TUSC2 with Anti-PD1 on Immune Cell Populations in Peripheral Blood (Large)
Preclinical Study Showing thatStudies of TUSC2 Immunogene Therapy is Synergistic with Anti-PD1 in Lung Cancer Syngeneic Mouse ModelsSupporting Our Conduct of Acclaim-3
Based onTransfection of SCLC cells in vitro with TUSC2 showed growth inhibition and a marked suppression of colony formation compared to cells transfected with a control vector. These results demonstrate the prolonged responses that were observedpotential tumor suppression function of TUSC2 in TUSC2 clinical trials, whichSCLC cells and suggest that TUSC2-mediated gene therapy could be a useful therapeutic strategy for the treatment of SCLC.
Data presented at the October 2023 AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics from studies in humanized mouse models of SCLC that use human H841 cells have shown that the combination of quaratusugene ozeplasmid and atezolizumab provides significantly better control of tumor burden than either agent alone (Figure 3, Figure 4, and Figure 5). H841 is a human SCLC cell line that does not express TUSC2 may modulate theprotein, and which was labeled with luciferase for these experiments. When injected intravenously, H841 cells metastasize to both lung and liver. The reconstituted humanized mice have a fully competent human immune response, and on the fact thatsystem with major functional immune populations, which showed antigen specific T cell responses as well as antitumor activity with immune checkpoint blockade immunotherapy against PD1therapy and PD-L1 has yielded durable antitumor activitywas used here to evaluate synergy between quaratusugene ozeplasmid and atezolizumab.
In these studies, one million H841 cells/mouse were injected intravenously, and treatment began 12 days later, after the metastases were well established. Quaratusugene ozeplasmid was administered intravenously every other weekday at a dose of 25 μg/mouse of plasmid DNA: 10 nmol liposome solution in 100 μL of D5W for 5 doses. Atezolizumab was administered at a subsetdose of NSCLC patients, MD Anderson researchers conducted300 μg/mouse intraperitoneal injection twice a preclinical studyweek for 4 doses. Three weeks later, bioluminescence imaging was performed to investigatevisualize the intensity of tumor burden for mice in different treatment groups.
Humanized mice with H841 xenografts were treated with quaratusugene ozeplasmid, atezolizumab or the combination. With the combination of quaratusugene ozeplasmid and atezolizumab there was a highly significant reduction in tumor burden compared to the results with atezolizumab alone (p=0.002). There was approximately a 10-fold reduction in tumor burden in the quaratusugene ozeplasmid plus atezolizumab group compared to untreated control mice. Note that 3 out of 5 mice in the quaratusugene ozeplasmid plus atezolizumab group had complete or near-complete responses (Figure 3). Figure 4 provides a graph of the bioluminescence flux in the treatment groups.
Analysis of the tumor immune microenvironment in the H841 xenografts (Figure 5) shows that, compared to treatment with atezolizumab alone, the combination of quaratusugene ozeplasmid and atezolizumab leads to increased numbers of huCD8 T-cells, NK cells, and DC, and lower numbers of MDSCs. These changes indicate an increased immune response to TUSC2the xenograft and identify at least one mechanism responsible for the increased tumor response seen with quaratusugene ozeplasmid and atezolizumab in immune cell populations and the synergistic antitumor effect of TUSC2 in combination with anti-PD1 checkpoint blockade in syngeneic mouse NSCLC models.combination.
Two Kras-mutant syngeneic mouse models were usedIn summary, the data from these studies suggest that a combination treatment of quaratusugene ozeplasmid and atezolizumab can promote a significantly increased tumor cell killing effect in SCLC xenografts compared to explore the effectthat of TUSC2+anti-PD1 (+/- anti-CTLA-4) on immune cells infiltration into the tumor micro-environment. Activating Kras mutations are the most common driver mutations in lung adenocarcinomas. Lung cancer with mutant Kras has a poor prognosis, is often resistant to conventional therapy, and readily becomes resistant to targeted therapies with kinase inhibitors. Studies by researchers not at MD Anderson have found that PD1 expression was highly associated with the presence of Kras mutations and that PD-L1 expression was elevated in premalignant Kras-mutant cells, suggesting that Kras mutation may affect the function of the PD1/PD-L1 immune checkpoint pathway.atezolizumab alone.
The first syngeneic mouse model used a murine
Figure 3. Bioluminescence Flux after Quaratusugene Ozeplasmid and Atezolizumab as Single Agents
and in Combination in H841 SCLC Model in Humanized Mice
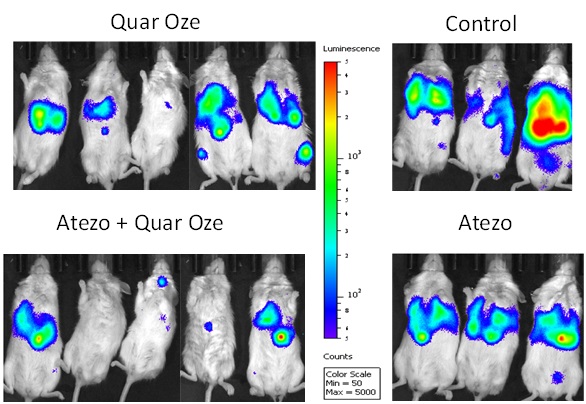
Abbreviations: Atezo = atezolizumab; max = maximum; min = minimum; Quar Oze = quaratusugene ozeplasmid; SCLC = small cell lung carcinoma cell line CMT/167-luc with a Kras G12V mutation and a low level of TUSC2 expression, implanted subcutaneously in C57BL/6 mice. The second syngeneic mouse model optimized an aggressive experimental metastatic lung cancer model using 129SvE mice injected with SQ344 lung cancer cells, which contained KrasG12D allele. The SQ344 tumor model was found to be less sensitive to anti-PD1 single agent treatment.
The image below shows the results of this preclinical study, in which anti-PD-1, TUSC2 and anti-CTLA-4 treatments were administered in the SQ344 metastatic lung tumor mouse model. The graph on the left shows the survival of the mice with the lung tumor cells treated with (a) no treatment, (b) a combination of anti-PD-1 and anti-CTLA-4, (c) TUSC2 alone, (d) a combination of TUSC2 and anti-PD-1, and (e) a combination of TUSC2, anti-PD-1 and anti-CTLA-4. The image on the right shows samples of untreated lung tissue and lung tissue treated with TUSC2.
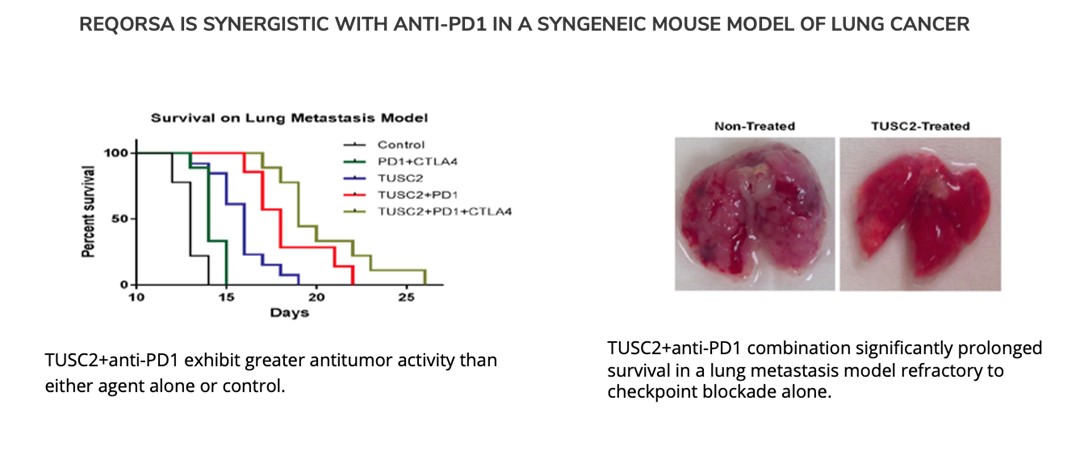
Preclinical Studies Showing that TUSC 2 Immunogene Therapy is Synergistic with Anti-PD1 in Lung Cancer Humanized Mouse Models
The results
Figure 4. Graph of a preclinical study combining anti-PD1 with TUSC2 immunogene therapy showed strong antitumor immune responses of anti-PD-1 against PDX (patient derived xenograft) tumors developedBioluminescence Flux after Quaratusugene Ozeplasmid and Atezolizumab as
Single Agents and in humanized mice. In addition, TUSC2 plus anti-PD1 plus a chemotherapy combination resultedCombination in metastasis regression significantly greater than either TUSC2 immunogene therapy alone or anti-PD1 plus chemotherapy. H841 SCLC Model in HumanizedMice
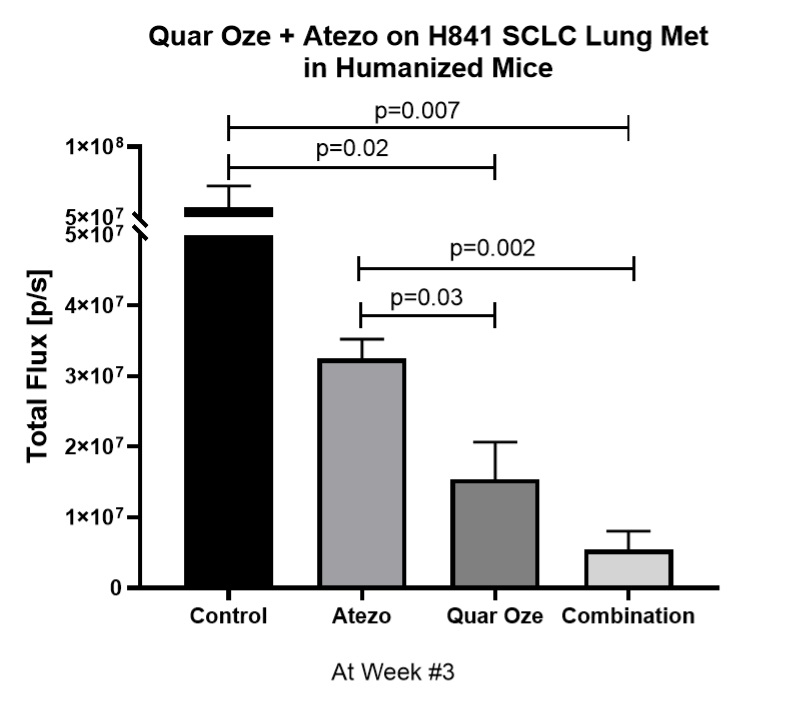
Abbreviations: Atezo = atezolizumab; Quar Oze = quaratusugene ozeplasmid; SCLC = small cell lung cancer
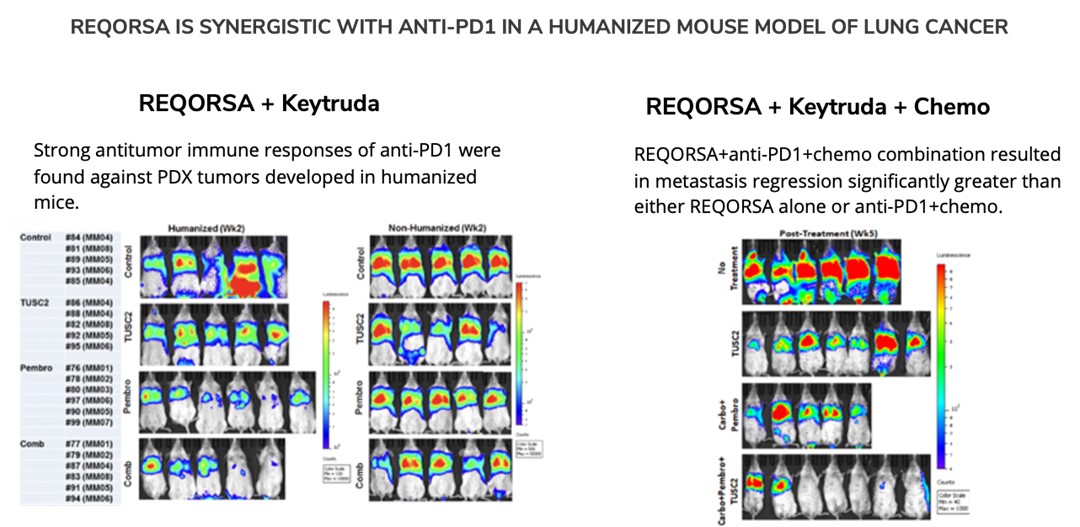
Discovery ProgramsFigure 5. Analysis of the Tumor Immune Microenvironment After Quaratusugene Ozeplasmid
ONCOPREX® Nanoparticle Delivery Systemand Atezolizumab as a platform. We believe that the ONCOPREX Nanoparticle Delivery System may be applicable to delivery of a range of therapeutic and prophylactic plasmid DNA and RNA interference constructs and show efficacy in cancers beyond NSCLC. The manufacturing methods we have developed for REQORSA have been optimized and we believe they may be useful for a wide array of disease treatments. Clinical data from the use of REQORSA has shown that the ONCOPREX Nanoparticle Delivery System is well tolerated in humans and can be delivered at high therapeutic doses.
Rights to other Tumor Suppressor Genes. We have licensed rights to a group of candidate tumor suppressor genes, including 101F6, NPRL2, CACNA2D2, PL6, BLU, RASSF1, HYAL 1 and HYAL2, in addition to tumor suppressor gene, TUSC2, all of which are located in a sub-region of human chromosome 3 known as 3p21.3. Using a number of techniques, MD Anderson researchers and their collaborators have identified these genes as potentially having cancer-fighting characteristics. MD Anderson researchers have subsequently conducted a number of preclinical studies on certain of these genes, particularly 101F6 and NPRL2, as well as TUSC2, both aloneSingle Agents and in combination with other compounds,Combination Using an H841 SCLC Model in order to assess their actual effects on NSCLC. Under our Sponsored Research Agreement with MD Anderson, we plan to continue to support continuing research into the cancer-fighting properties of these and other genes in the 3p21.3 sub-region. Humanized Mice
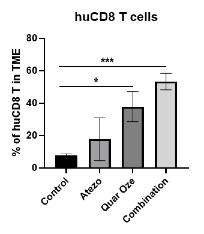
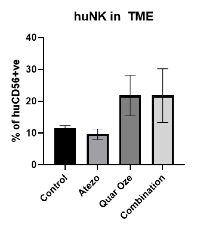
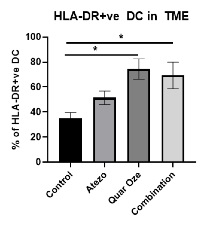
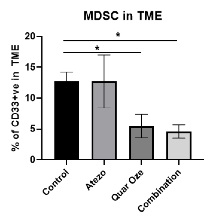
Abbreviations: Atezo = atezolizumab; DC = dendritic cells; HLA-DR = human leukocyte antigen – DR isotype; huCD8 T cells = human CD8 T cells; huNK = human natural killer cells; MDSC = myeloid derived suppressor cells; Quar Oze = quaratusugene ozeplasmid; TME = tumor microenvironment
Researchers at MD Anderson have collaborated with other researchers to identify other genes, such as those in the 3p21.3 chromosomal region, that may act as tumor suppressors or have other cancer fighting functions. We hold rights to certain of these genes under license agreements with MD Anderson. Data from preclinical studies performed by MD Anderson researchers and others suggest that TUSC2, the active agent in REQORSA, could be effective against other types of cancer, including glioblastoma, head and neck, breast (including triple-negative breast cancer), renal cell (kidney), thyroid, and soft tissue sarcoma, as well as NSCLC. Therefore, the ONCOPREX Nanoparticle Delivery System may allow delivery of a number of cancer fighting genes, alone or in combination with other cancer therapies, to combat multiple types of cancer.
Introduction – Diabetes
Diabetes Mellitus. Diabetes mellitus refers to a group of metabolic diseases that affect how the body produces and uses blood sugar (glucose). Glucose is vital to health because it is an important source of energy for the cells that make up the body’s muscles and tissues. It is also the brain'sbrain’s main source of fuel. Chronic diabetes conditions include Type 1 diabetes and Type 2 diabetes, both of which lead to excess sugarglucose in the blood and can cause serious health problems. Left untreated, high blood sugarglucose levels can damage the eyes, kidneys, nerves, and the heart, and can also lead to coma and death.
Epidemiology of Diabetes. According to the U.S. Center for Disease Control 34.2as of 2023, 38.4 million Americans, or approximately 10.5%11.6% of the U.S. population, have diabetes. It is also believed that more than 8897 million Americans aged 18 years or older have prediabetes, which representsprediabetes. In 2021, approximately 35%537 million adults (20-79 years) worldwide were living with diabetes, and the total number of people living with diabetes is projected to rise to 643 million by 2030 and 783 million in 2045. Also in 2021, diabetes caused more than 6.7 million deaths globally and diabetes resulted in approximately $966 billion dollars in health expenditures, a 316% increase over the U.S. population. The prevalence of this chronic disease is continuing to rise. preceding fifteen years.
The Role of Alpha Cells and Beta Cells. The two most abundant endocrine cell types in the pancreas, the beta and the alpha cells, are essential for the maintenance of blood glucose homeostasis whereby levels of glucose are maintained by the body within a narrow range. While the beta cell produces insulin, the only blood glucose-lowering hormone of the body, the alpha cell releases glucagon, which elevates blood glucose. While the release products of the beta cell inhibit alpha cell function, the alpha cell releases factors that are stimulatory for beta cell function and increase glucose-stimulated insulin secretion.
In people with Type 1 diabetes, however, beta cells are destroyed by the immune system and no longer secrete insulin, leading to an absolute deficit of insulin. Type 2 diabetes is due to "insulin“insulin resistance,"” an initial resistance of the body'sbody’s cells to obey the direction from insulin. To overcome this resistance, the beta cells secrete more insulin, and glucose is eventually forced into the cells. Glucose is maintained within normal limits, but at the expense of increased insulin secretion by the beta cells. After many years of such increased secretion, the beta cells become "tired"“tired” from working overtime, and the fatigue process begins. This fatigue tends to be progressive, and in time the compensation offor insulin resistance disappears. At that point, blood glucose levels start going up.
Current Treatments for Diabetes. Advances in new treatments have helped many people better manage the disease. However, despite patients'patients’ best attempts, managing diabetes remains a challenging, daily balancing act because exogenous insulin therapy simply cannot ideally mimic the body’s biological function.
Type 1 diabetes patients are treated with insulin, with most of the progress in therapy relating to enhanced delivery of the drug and improved methods for measuring blood glucose levels. A variety of drug release technologies have allowed for rapid-acting, intermediate-acting and long-acting insulin injections that provide drug anywhere from one to 24 hours. In addition, improvements in needles, continuous delivery ports, and inhalation technologies all have helped patients better manage their disease and may impact quality of life, but none of these advances are disease modifying.
Type 2 diabetes patients are advised to use diet and exercise to manage their condition. When these lifestyle changes alone do not control the disease, Type 2 diabetes patients may be prescribed a variety of medications that help alter how the body manages blood sugar levels. For example, biguanides such as Metformin®,metformin, reduce the amount of glucose produced in the liver. DPP-4 inhibitors, such as Januvia®, Onglyza®, and Tradjenta®, improve blood sugar levels and prevent them from dropping too low. Glucagon-like peptides, such as Byetta®, Trulicity® and Victoza®, change the way the body produces insulin. Drugs in the SGLT2 inhibitor class, such as Farxiga and Inokana,Invokana, release more glucose into the urine. Finally, insulin injections may be needed if these oral medications, along with diet and exercise, do not lower blood sugar levels enough. These medications, including insulin replacement therapy, while offering improvements for Type 2 diabetes patients, do not affect the underlying cause of the disease.
GPX-002
WeAs further described in the “Licenses and Research Collaborations” section below, we have exclusively licensed a pre-clinical gene therapy from the University of Pittsburgh multiple technologies relating to restore the functiondevelopment of beta cells that are destroyed bya gene therapy product for each of Type 1 and Type 2 diabetes. The same general novel approach is used in each of Type 1 and Type 2 diabetes whereby an AAV vector containing the immune system and overcome further destruction of insulin-producing cells. This technology, referred to us as GPX-002, infuses adeno-associated virus carrying Pdx1 and MafA gene expression cassettes throughgenes is administered directly into the pancreatic ductduct. In humans, we believe this can be done with a routine endoscopy procedure, called endoscopic retrograde cholangiopancreatography (ERCP). Our diabetes product candidates are currently being evaluated and optimized in preclinical studies at the University of Pittsburgh. GPX-002 is being developed using the same construct for the treatment of both Type 1 diabetes and Type 2 diabetes. GPX-002 for Type 1 diabetes is designed to reprogramwork by transforming alpha cells in the pancreas into functional beta-like cells, which can produce insulin but aremay be distinct enough from beta cells to evade the body’s immune system. In a similar approach, GPX-002 for Type 2 diabetes (formerly known as GPX-003), where autoimmunity is not at play, is believed to work by replenishing and rejuvenating exhausted beta cells that make insulin. We finalized the components of the diabetes construct to take forward for nonclinical studies and in December 2023, we submitted a request to meet with the FDA to obtain their guidance on the nonclinical studies needed to file an IND application and initiate first-in-human studies. As a result of the FDA’s response, the Company will continue with its planned additional nonclinical studies before requesting regulatory guidance in 2024 for the IND-enabling studies.
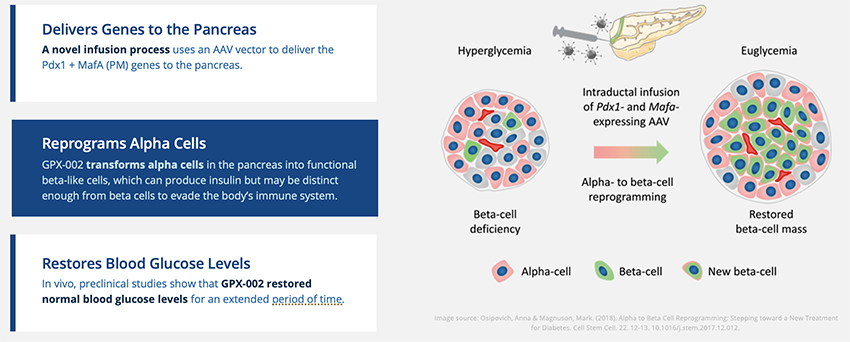
In October 2023, we entered into a one-year extension to our August 2022 sponsored research agreement with the University of Pittsburgh for the use of GPX-002 in a NHP model in Type 2 diabetes. The extension includes a revised research plan to encompass our most recent technologies to which we acquired exclusive rights from the University of Pittsburgh in July 2023. These include a MafB promoter to drive expression of the Pdx1 and MafA transcription factors that can potentially be used for both Type 1 and Type 2 diabetes.
Preclinical Studies
GPX-002This gene therapy approach has been tested in vivo in mice and nonhuman primates. NHPs using an earlier construct as described below.
Preclinical Mouse Studies
In studies in mice treated to destroy insulin producing beta cells and in non-obese diabetic (“NOD”) mice, a modelboth of which are models of Type 1 autoimmune diabetes, theour gene therapy approach restored normal blood glucose levels for an extended period of time, typically around four months. Accordingand markedly increased the mass of insulin producing beta cells.
The figures below show that starting approximately a week after injection of the engineered AAV construct (labeled AAV8-PM) into the pancreatic duct, the blood glucose level markedly improved in mice in which insulin producing cells had been destroyed by the drug alloxan (ALX). In addition, the mass of beta cells and beta-like cells producing insulin was significantly increased.
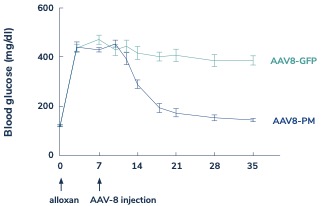
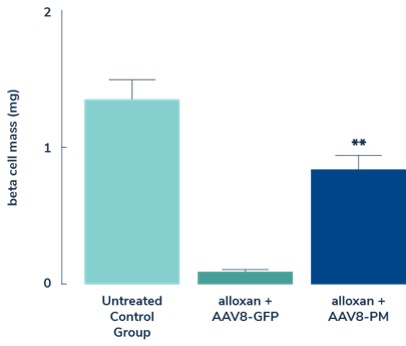
NOD mice develop diabetes due to an immune attack that destroys the insulin producing beta cells in the pancreas. The figures below show that starting approximately a week after injection of the engineered AAV construct (labeled AAV8-PM) into the pancreatic duct the blood glucose level markedly improved in NOD mice. In addition, there is a significant increase in the mass of beta cells and beta-like cells that produce insulin. The improvement in glucose level normalization lasted approximately 4 months, which, according to the researchers the duration of restored blood glucose levels in mice could potentially translate to decades in humans. If successful,
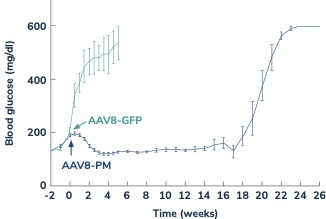
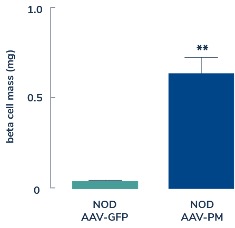
The researchers also carried out an experiment to determine if the same AAV engineered construct could be used to convert human alpha cells to beta-like cells that would produce insulin as shown in the figures below. Human pancreatic islets were treated with streptozotocin (STZ) to destroy beta cells, and then were treated with the AAV engineered construct. They were then transplanted into NOD mice that had been treated with alloxan (ALX), and also modified so they would not reject human cells. The NOD mice that received the AAV engineered construct had significantly lower blood glucose levels and higher mass of beta and beta-like cells that secrete insulin than did control mice. These data suggest that the same AAV engineered construct can convert human alpha cells into insulin secreting beta-like cells.
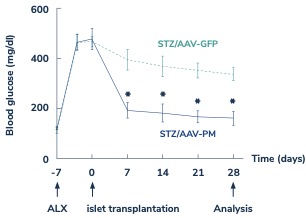
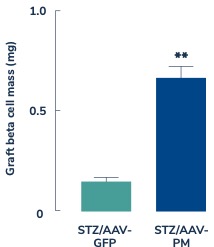
Preclinical Non-Human Primate Studies
In February 2023, the Company’s research collaborators at the University of Pittsburgh presented preclinical data in a NHP model of Type 1 diabetes highlighting the therapeutic potential of GPX-002 at the 16th International Conference on Advanced Technologies & Treatments for Diabetes (ATTD 2023) in Berlin, Germany. The statistically significant study results showed that after infusion of the AAV engineered construct all eight of the NHPs had:
● | Decreased insulin requirements (p<0.001); |
● | Increased c-peptide levels (p<0.05); | |
| ● | Improved glucose tolerance compared to baseline (p<0.05) with one demonstrating reestablished normoglycemia; and |
● | Insulin and glucagon staining in the same cells, suggesting the formation of insulin-producing cells. |
We believe these data in NHPs demonstrate the potential for this gene therapy couldtreatment to eliminate the need for insulin replacement therapy for Type 1 and Type 2 diabetic patients. In October 2023, we entered into a one-year extension to our August 2022 sponsored research agreement with the University of Pittsburgh for the use of GPX-002 in a NHP model of Type 2 diabetes. The extension includes a revised research plan to encompass our most recent technologies to which we acquired exclusive rights from the University of Pittsburgh in July 2023.
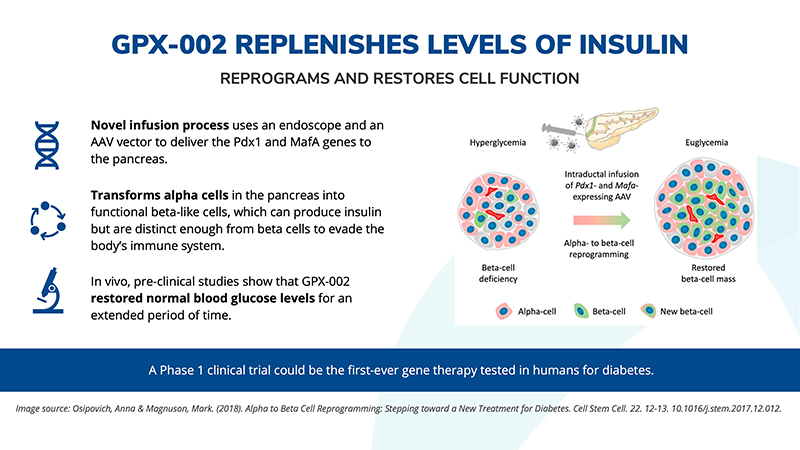
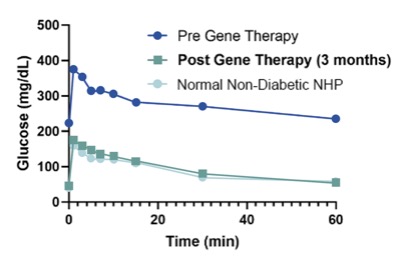
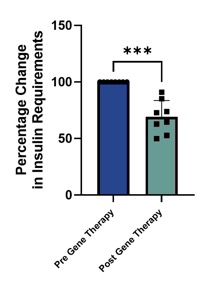
Discovery Programs
Oncology
ONCOPREX® Delivery System as a platform. We believe that the ONCOPREX Delivery System may be applicable to delivery of a range of therapeutic and prophylactic plasmid DNA and RNA interference constructs and shows efficacy in cancers beyond lung cancer. We also believe that the manufacturing methods we have developed for REQORSA may be useful for a wide array of disease treatments. Clinical data from the use of REQORSA has shown that the ONCOPREX Delivery System is well tolerated in humans and can be delivered at high therapeutic doses.
Rights to other Tumor Suppressor Genes. We have licensed rights to the tumor suppressor gene, TUSC2, which is located in a sub-region of human Chromosome 3 known as 3p21.3, on which multiple tumor suppressor genes are located, including for example, 101F6, NPRL2, CACNA2D2, PL6, BLU, RASSF1, HYAL 1 and HYAL2. Using a number of techniques, MD Anderson researchers and their collaborators have identified these genes as potentially having cancer-fighting characteristics. MD Anderson researchers have subsequently conducted a number of preclinical studies on certain of these genes, particularly NPRL2, as well as TUSC2, both alone and in combination with other compounds, in order to assess their actual effects on lung cancer. Under past and current sponsored research agreements with MD Anderson, we support continuing research into the cancer-fighting properties of these and other genes in the 3p21.3 sub-region.
Researchers at MD Anderson have collaborated with other researchers to identify other genes, such as those in the 3p21.3 chromosomal region, which may act as tumor suppressors or have other cancer fighting functions. We hold rights to certain of these genes under license agreements with MD Anderson. Data from preclinical studies performed by MD Anderson researchers and others suggest that TUSC2, the active agent in REQORSA, could be effective against other types of cancer, including glioblastoma, head and neck, breast (including triple-negative breast cancer), renal cell (kidney), thyroid, and soft tissue sarcoma, as well as NSCLC and SCLC. Therefore, the ONCOPREX Delivery System may allow delivery of a number of cancer fighting genes, alone or in combination with other cancer therapies, to combat multiple types of cancer.
Another abstract to be presented by our clinical collaborators at the 2024 AACR meeting supports further clinical study of REQORSA in Anaplastic Lymphoma Kinase (“ALK”)+ NSCLC, which are found in approximately 5% of patients with NSCLC. In this nonclinical study, TUSC2 expression in three ALK+ cell lines were evaluated before and after exposure to REQORSA, referred to as TUSC2 gene therapy in the abstract. Researchers in the University of Michigan Rogel Cancer Center Judith Tam ALK NSCLC research initiative found that overexpressing TUSC2 via REQORSA treatment in ALK+ lung cancer cell lines had the ability to inhibit colony formation by 50%, which is believed to indicate that REQORSA inhibits growth of ALK+ cells. Researchers documented a strong pro-apoptotic response to TUSC2 expression in ALK+ NSCLC. The study found that the use of REQORSA to overexpress TUSC2 in ALK+ NSCLC cell lines was effective in decreasing cell growth and proliferation. The findings suggest REQORSA therapy may be a potential therapy for ALK+ NSCLC and the researchers believe the results support further clinical study of REQORSA as an anti-ALK NSCLC treatment strategy.
Diabetes
In July 2023, we exclusively licensed from the University of Pittsburgh technology related to a gene therapy for both Type 1 and Type 2 diabetes using a MafB promoter to drive expression of the Pdx1 and MafA transcription factors. In October 2023, we entered into a one-year extension to our August 2022 sponsored research agreement with the University of Pittsburgh for the use of GPX-002 in a NHP model in Type 2 diabetes. The extension includes a revised research plan to encompass the technologies to which we acquired exclusive rights from the University of Pittsburgh in July 2023.
Process Development and Manufacturing
We have made substantial investment in manufacturing for our product candidates with the goal of mitigating the risks associated with the complex manufacturing required to deliver gene therapies. While we continue to use third-party contract development and manufacturing organizations (“CDMOs”) in the manufacture of our product candidates, we believe we have a competitive advantage in our field based on core competencies we have developed that we are leveraging in the manufacture of our product candidates. These core competencies include:
| ● | Extensive and diverse internal and external consulting expertise; | |
| ● | Customized chemistry, manufacturing and controls (“CMC”) strategy for accelerated development; | |
● | Risk assessment and remediation for FDA submissions; | |
| ● | Novel and proprietary manufacturing processes; | |
| ● | Integrated global Good Manufacturing Practices (“cGMP” or “GMP”) manufacturing network; and | |
| ● | Management of supply chain for business continuity. |
During 2023, we experienced a delay in the successful production of a new batch of REQORSA in connection with our transition to a third party CDMO with advanced and automated processes that we believe ultimately will allow us to more efficiently scale our product as we increase production for our trials. In December 2023, we completed the successful production of a new batch of REQORSA thereby securing REQORSA supply for our Acclaim clinical studies. In our oncology program, we are now focused on preparing for commercial readiness for REQORSA. To date we have developed a robust manufacturing process for REQORSA through years of process development.development activities that we continue to improve with the dramatic development and expansion of advanced technologies in the nascent gene therapy sector. REQORSA is an immunogene therapy with two main components. The active agent in REQORSA is a DNA plasmid encoding the TUSC2 protein. The plasmid is encapsulated by non-viral DOTAP cholesterol lipid nanoparticles.lipoplexes. This system of encapsulating DNA plasmid in non-viral lipid nanoparticlesusing lipoplexes to deliver the tumor suppressor gene-expressing plasmids to cancer cells is referred to by us as our systemic, non-viral ONCOPREX Nanoparticle Delivery System. Each of these two components is manufactured by separate third-party contract development and manufacturing organizations ("CDMOs") and then transported to another CDMO for final drug formulation. We do not currently have the internal infrastructure or facilities to manufacture REQORSA or any other product candidate for use in the conduct of our clinical trials or for commercial supply; however, our strategy could change in the future and we could choose to develop this infrastructure. Where other gene therapy agents need to be prepared individually for each patient or require viral vectors for gene delivery, REQORSA utilizes the ONCOPREX Nanoparticle Delivery System and has been shown to be scalable at current Good Manufacturing Practices (“cGMP”)cGMP and can be stored for at least one yearapproximately 12 to 18 months for later use. Successful tech transfer of REQORSA from MD Anderson, where it was developed and previously manufactured, to a CDMOCDMOs has been achieved as well as scale-up of our clinical grade manufacturing production in accordance with cGMP. TheAs noted above, the clinical grade production will bematerial is being used to supply our Acclaim-1, Acclaim-2 and Acclaim-2Acclaim-3 clinical trials.
For our diabetes program, which is an earlier stage program than our oncology program, we are working with our academic collaborators at University of Pittsburgh to transfer the technologies associated with the manufacture of our GPX-002 construct to an appropriate integrated network of CDMOs and other vendors. This also involves the investigation of novel advanced technologies to incorporate in these processes. GPX-002 involves the delivery of the Pdx1 and MafA genes into the pancreas via the pancreatic duct utilizing an AAV vector.
We manage our manufacturing arrangements with our CDMOCDMOs and other vendors through various agreements.
Intellectual Property
Patents and other proprietary rights such as trademarks and trade secrets are critical to our business and to our ability to successfully develop and commercialize our product candidates. Our goal is to obtain, maintain, enhance and enforce patent protection for our products, formulations, processes, methods and other proprietary technologies, preserve our trade secrets, and operate without infringing on the proprietary rights of other parties, both in the U.S. and in other countries. Our policy is to actively seek the broadest intellectual property protection possible for our products,product candidates, proprietary information, and proprietary technology through a combination of contractual arrangements, patents, trade secrets, trademarks, copyrights and patents,regulatory exclusivity both in the U.S. and elsewhere in the world. Patents provide a period of exclusivity intended to make it more difficult for competitors to make, use or sell competing technologies. We additionally rely on regulatory protection afforded through data exclusivity, market exclusivity, orphan drug designation and/or patent term extensions, where available. We have developed and/or in-licensed numerous patents and pending patent applications that relate to compositions-of-matter, methods-of-use and other technologies and possess substantial know-how and trade secrets relating to the development of gene therapy technologies.
WeAs further described in the “Licenses and Research Collaborations” section below, we hold a worldwide, exclusive license from MD Anderson to 27patents covering the therapeutic use of TUSC2 and other genes that have been shown to have cancer fighting properties, including 20 issued patents and 1712 pending patent applications for technologies developed at MD Anderson and The University of Texas Southwestern Medical Center. These patents comprise various therapeutic, diagnostic, technical and processing claims relating to REQORSA and our ONCOPREX Nanoparticle Delivery System. We expect these patents and patent applications, if issued, to expire from 2024 to 2038. The rights we have obtained pursuant to our license agreement with MD Anderson are made subject to the rights of the U.S. government to the extent that the technology covered by the licensed intellectual property was developed under a funding agreement between MD Anderson and the U.S. government. As further described in the “Licenses and Research Collaborations” section below, we also hold a worldwide, exclusive licenselicenses to an issued patent and 8 pending patent applications for diabetes technologies developed at the University of Pittsburgh. We expect these patents and patent applications, if issued, to expire from 2035 to 2043. We also are prosecuting four patent applications relating to various oncology targets in our discovery program. In addition, for certain of our product candidates we also expect to have further exclusivity in the form of data and marketing exclusivity under pharmaceutical regulatory laws, including for example, potentially up to 12 years of exclusivity from the date of first BLA approval of our product candidates. For a further description and discussion of these laws, exclusivities and their regulatory background, please see the “Business – Government Regulation” section below in this Part I, Item 1 of this Annual Report.
We also have received trademark registrations for the trademarks GENPREX, REQORSA, and ONCOPREX and have filedONCOPREX. For a discussion of the challenges we face in obtaining or maintaining patent, trademark application forand/or trade secret protection, please see the drug name REQORSA.risk factors under the heading “Risks Related to Our Intellectual Property” in Part I, Item 1A of this Annual Report.
Licenses and Research Collaborations
Agreements with MD Anderson
Our ONCOPREX and REQORSA technologies are exclusively licensed pursuant to a Patent and Technology License Agreement (“MD Anderson License Agreement”) dated July 20, 1994, with MD Anderson, as amended on September 1, 1996, August 11, 1997, July 31, 1994 and October 4, 2001 (collectively, the "1994 MD Anderson License Agreement"), between MD Anderson and Introgen Therapeutics, Inc. (f/k/a Intron Therapeutics, Inc.) (“Introgen”).
Pursuant to the 1994 MD Anderson License Agreement, we have rights to patents covering use of various genes, including the TUSC2 gene, for treatment of cancer, as well as know-how and related intellectual property.
The exclusive licenses under the 1994 MD Anderson License Agreement will continue until the expiration of all patents covered by such agreement. Upon the expiration of the exclusive licenses, we will have a non-exclusive, fully paid-up right and license to use and otherwise exploit the technology rights licensed under the agreement. MD Anderson may terminate the agreement for, among other things, a breach of the agreement by us which remains uncured.
Pursuant to a Technology Sublicense Agreement dated March 7, 2007 (“Sublicense Agreement”), Introgen sublicensed its rights under the 1994 MD Anderson License Agreement to Introgen Research Institute, Inc. (“IRI”). IRI is a Texas-based technology company formed and owned by Rodney Varner, our current President CEO and Chairman of the boardBoard and IRI’s sole officer. IRI is owned by trusts of directors.which Mr. Varner’s descendants are the sole beneficiaries.
Pursuant to an Assignment and Collaboration Agreement dated April 13, 2009 (“IRI Collaboration Agreement”) IRI assigned its rights under the Sublicense Agreement to us, and we granted to IRI a non-exclusive, royalty-free license to use and practice the licensed technology for non-commercial research purposes. As consideration for this assignment, we agreed to assume all of IRI’s obligations to MD Anderson under the 1994 MD Anderson License Agreement, including ongoing patent related expenses and royalty obligations.
The IRI Collaboration Agreement was amended by an Amended Collaboration and Assignment Agreement dated July 1, 2011 ("(“2011 IRI Collaboration Agreement"Agreement”). The 2011 Collaboration Agreement provided that IRI would provide additional licensing opportunities and services to us, in return for monthly payments and our obligation to pay to IRI a royalty of 1% on sales of products licensed to us under the 1994 MD Anderson License Agreement. In 2012, IRI’s obligation to provide those opportunities and services, and our obligation to make monthly payments to IRI, were terminated; however, we are required to pay a 1% royalty to IRI upon sales of products licensed to us under the 1994 MD Anderson License Agreement which royalty obligation continues for 21 years after the later of the termination of the 1994 MD Anderson License Agreement and the termination of the sublicense assigned by IRI to us.
Pursuant to a Technology Sublicense Agreement dated June 1, 2011, we granted to IRI a non-exclusive sublicense, for non-commercial purposes, to the rights under the Sublicense Agreement.
At the time that we entered into the 2011 IRI Collaboration Agreement, Mr. Varner was not an officer or director of Genprex, but he was deemed to be an “affiliate” of the Company due to his beneficial ownership of approximately 39% of our issued and outstanding shares. At the time we acquired the ONCOPREX and REQORSA technologies under the 2009 IRI Collaboration Agreement, they were the subject of the Phase 1 Monotherapy Trial. We completed the Phase 1 Monotherapy Trial and did substantial process development, manufacturing and regulatory work necessary to bring the technologies into a Phase 1/2 combination trial.
Pursuant to the 1994 MD Anderson License Agreement, the Sublicense Agreement and the 2009 IRI Collaboration Agreement, we are obligated to pay all fees, patent related expenses, royalties, and other amounts that become due with respect to the licensed patents, patent application and other technologies. We are also obligated to pay to MD Anderson royalties of 1.5% of net sales of the licensed products, as well as 1.5% of advance payments received by us (excluding amounts paid to us in reimbursement of development or other costs) from third parties pursuant to sublicense, marketing, distribution or franchise arrangements. Under the 2011 IRI Collaboration Agreement, we are obligated to pay to IRI a royalty of 1.0% of net sales of licensed products and 1.0% of certain other payments received by us. This royalty obligation continues for 21 years after the later of the termination of the 1994 MD Anderson License Agreement and the termination of the Sublicense Agreement. We have no other payment obligations to IRI under the 2009 IRI Collaboration Agreement or the 2011 IRI Collaboration Agreement. We were not required to make any up-front payments to MD Anderson or IRI when we entered into the 1994 MD Anderson License Agreement, the Sublicense Agreement or the 2009 IRI Collaboration Agreement.
On May 4, 2020 (the “MD Effective Date”), we entered into a Patent and Technology License Agreement with MD Anderson, as amended on March 3, 2021 (collectively, the “2020 License Agreement” and together with the 1994 MD Anderson License Agreement, collectively, the “MD Anderson License Agreements”). Pursuant to the 2020 License Agreement, MD Anderson granted us a worldwide, exclusive, sublicensable, royalty-bearing license to certain licensed intellectual property and technology, including, without limitation, use of chemotherapy in combination with TUSC2 therapy and methods for treating cancer by administration of a TUSC2 in conjunction with EGFR inhibitors or other anti-cancer therapies in patients that are expected to be responsive to TUSC2 therapy (collectively, the “Licensed IP”), to manufacture, use, commercialize, seller, offer for sale and import licensed products related to the treatment of cancer using TUSC2 therapy in combination with certain immunotherapies (the “Licensed Products”). In consideration for our use of the Licensed IP, we are required to make certain payments to MD Anderson, including, without limitation, an upfront license fee as well as a fee paid to amend the agreement, annual maintenance fees ranging from the low five figures to low six figures, milestone payments aggregating up to a maximum of $6,150,000, low single digit royalty payments to low double digits royalty payments with lower net sales being subject to lower royalty payments, and minimum annual royalties after the first sale in a low six figure amount. In addition, we shall be required to reimburse MD Anderson for certain patent expenses. The 2020 License Agreement shall expire on the later to occur of (a) the expiration of all patents issued under the Licensed IP and the cancellation, withdrawal, or express abandonment of all patent applications under the Licensed IP, or (b) 30 years after the MD Effective Date, unless earlier terminated pursuant to the terms thereof. In 2015
See also “Note 7 – Commitments and 2017, we entered into two option agreements with MD Anderson, paying MD Anderson $35,000 and $37,803, respectively, for the rightsContingencies” to negotiate exclusive rights to additional licensed intellectual property and technology from MD Anderson.our financial statements included in this Annual Report on Form 10-K.
License Agreement with P53, Inc.
On February 26, 2010, IRI and P53, Inc., which subsequently changed its name to MultiVir Inc., (“P53”) entered into a Technology License Agreement ("(“P53 License Agreement"Agreement”) pursuant to which IRI granted to P53 Inc. ("P53") a worldwide, exclusive license under certain patents related to the ONCOPREX Nanoparticle Delivery System that we are now using for the delivery of TUSC2, but only for P53’s use in gene therapy products in which the sole active genes are P53 and MDA-7. As a result of the 2009 IRI Collaboration Agreement, we are the licensor under the P53 License Agreement.
The P53 License Agreement authorizes P53 to develop, make and have made, use, offer for sale, sell, import and otherwise distribute the licensed products. As consideration for the P53 License Agreement, P53 agreed to pay IRI one-half of all amounts invoiced by MD Anderson to IRI, up to a maximum of $15,000 to be paid by P53, for patent prosecution expenses incurred prior to the effective date of the P53 License Agreement, as well as two-thirds of IRI’s ongoing patent prosecution expenses, in each case with respect to the licensed patents. Additionally, P53 agreed to pay all amounts that become due to IRI as a result of the P53 License Agreement or the sales, licensing, or other activities of P53 under the P53 License Agreement. Pursuant to the P53 License Agreement, P53 has granted to IRI a fully paid license with respect to improvements made by P53 to the technology licensed to P53 under the P53 License Agreement. The P53 License Agreement remains in effect until the expiration of the last of the patents licensed under the agreement. The last licensed patent under the P53 License Agreement will expire in April 2025. We may terminate the agreement for, among other things, P53’s breach of the agreement or if P53 challenges the validity or enforceability of any of the licensed patents. P53 may terminate the agreement upon 90 days’ written notice.
License Agreement with University of Pittsburgh - Of the Commonwealth System of Higher Education
On February 11, 2020, we entered into an exclusive license agreement, (the “UPas amended on August 17, 2022 and November 3, 2022 (collectively the “2020 UP License Agreement”), with the University of Pittsburgh - Of the Commonwealth System of Higher Education (“UP”University of Pittsburgh” or “UP”) pursuant to which UP granted us a worldwide, exclusive license to certain licensed technology, and a worldwide, non-exclusive license to use certain related know-how, all related to diabetes gene therapy. The 2020 UP License Agreement permits us to make, haveprevent others from making, having made, useusing, and sellselling certain licensed technology and to practice the patent rights in the field of diabetes therapy. We have agreed to sell the licensed technology to UP upon its request on terms and conditions as such products and/or processes are made available to our most favored customer. As consideration for the 2020 UP License Agreement, we agreed to pay UP an initial license fee, annual maintenance fees, running royalties minimum annual royalties beginning with the first commercial sale of the licensed technology pursuant to such agreement, a share of non-royalty sublicense income, and milestone payments in the aggregate amount of up to $3,975,000, as well as patent prosecution expenses incurred prior to and after the effective date of the 2020 UP License Agreement. The 2020 UP License Agreement shall remain in effect until the later of 20 years after the first commercial sale of the licensed technology or the expiration of the last Valid Claim (as defined in the 2020 UP License Agreement). UP may terminate the agreement if, among other things, (i) we fail to cure a default, (ii) if we fail to achieve the specified milestones within the specified time periods or (iii) our intentional practice of the licensed patent rights or know-how outside the field of diabetes therapy. We may terminate the 2020 UP License Agreement upon six months’ prior written notice to UP and payment of all amounts accrued or due to UP through the effective date of termination.
On November 22, 2022 and December 29, 2022, respectively, we entered into two separate exclusive license agreements with UP on substantially similar terms as the 2020 UP License Agreement as described in the preceding paragraph. Both the exclusive license agreement dated November 22, 2022 (the “November 2022 UP License Agreement”) and the exclusive license agreement dated December 29, 2022 (the “December 2022 UP License Agreement”, and together with the 2020 UP License Agreement and the November 2022 UP License Agreement, collectively, the “UP License Agreements”) relate to diabetes gene therapy. The November 2022 UP License Agreement relates in particular to the transformation of macrophages enabling them to reduce autoimmunity activity in Type 1 diabetes while the December 2022 UP License Agreement relates to gene therapy for Type 2 diabetes using an insulin promoter to drive expression of the Pdx1 and MafA transcription factors.
On July 14, 2023, we entered into an exclusive license agreement with UP (the “July 2023 UP License Agreement”) on substantially similar terms as the UP License Agreements as described in the preceding paragraphs. The July 2023 UP License Agreement related to a gene therapy for both Type 1 and Type 2 diabetes using a MafB promoter to drive expression of the Pdx1 and MafA transcription factors.
See also “Note 7 – Commitments and Contingencies” to our financial statements included in this Annual Report on Form 10-K.
Grants
WeOur technology discoveries and research and development programs have receivedbeen the subject of numerous peer-reviewed publications and have been supported by Small Business Innovation Research ("SBIR") grants and grants from the following entities: Texas Emerging Technology Fund, the SBIR program, the National Institutes of Health (“NIH”("NIH"), and the United States Department of Treasury, and the Treasury.State of Texas through its Texas Emerging Technology Fund. The rights we have obtained pursuant to our MD Anderson License Agreements are made subject to the rights of the U.S. government to the extent that the technology covered by the licensed intellectual property was developed under a funding agreement between MD Anderson and the U.S. government. Our collaborators at University of Pittsburgh have also received grants from the NIH in connection with pre-clinicalpreclinical work on GPX-002.GPX-002 and so, the rights we have obtained pursuant to our UP License Agreements are made subject to the rights of the U.S. government to the extent that the technology covered by the licensed intellectual property was developed under a funding agreement between the University of Pittsburgh and the U.S. government.
Competition
The biotechnology and pharmaceutical industries are intensely competitive and subject to rapid and significant technological change. There is also a strong emphasis on intellectual property and proprietary products. We have domestic and international competitors including major multinational pharmaceutical companies, established biotechnology companies, specialty pharmaceutical and generic drug companies and universities and other research institutions. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies.
Currently, there are a number of drugs approved and under development for treatment of lung cancer. Treatments competitive with our primary product candidates generally fall into the following categories: chemotherapies such as cisplatin, carboplatin, docetaxel and pemetrexed; targeted therapies such as Tarceva, Iressa, Gilotrif, and Tagrisso, and immunotherapies such as checkpoint inhibitors and CAR and CAR T cells, and oncolytic virus-based technology. Any such competing therapy may be more effective and/or cost-effective than ours.
Type 1 diabetes is an autoimmune disease that permanently destroys beta cells of the pancreatic islet leading to the body no longer having the ability to produce insulin. Type 2 diabetes, also known as adult onset diabetes, is a condition associated with developed resistance to insulin. There are a number of approved treatments and therapies to manage diabetes including insulin, insulin analogs, continuous glucose monitoring, novel approaches to administration such as insulin pens and insulin pumps, and preventative therapeutics. Any of these therapies may be more effective, cost-effective, or considered less invasive than ours.
Many of our competitors have greater financial and other resources, such as larger research and development staff and more experienced marketing and manufacturing organizations than we do. Large pharmaceutical companies, in particular, have extensive experience in clinical testing, obtaining regulatory approvals, recruiting patients and manufacturing pharmaceutical products. These companies also have significantly greater research, sales and marketing capabilities and collaborative arrangements in our target markets with leading companies and research institutions. Established pharmaceutical companies may also invest heavily to accelerate discovery and development of novel compounds or to in-license novel compounds that could make the product candidates that we develop obsolete. As a result of these factors, our competitors may succeed in obtaining patent protection and/or FDA approval or discovering, developing and commercializing drugs for the cancer indications that we are targeting before we do or may develop drugs that are deemed to be more effective or gain greater market acceptance than ours. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large, established companies. In addition, many universities and private and public research institutes may become active in our target disease areas. Our competitors may succeed in developing, acquiring or licensing on an exclusive basis, technologies and drug products that are more effective or less costly than any product candidates that we are currently developing or that we may develop, which could render our products obsolete or noncompetitive. Any product candidates that we successfully develop and commercialize may compete with existing and new therapies that may become available in the future. The availability of reimbursement from government and other third-party payers will also significantly affect the pricing and competitiveness of our products. For a further discussion of the challenges we face from competition, please see the "Risk Factors" section in Part I, Item 1A of this Annual Report.
Government Regulation
Government authorities in the U.S., at the federal, state and local level, and other countries extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, recordkeeping, promotion, advertising, distribution, post-approval monitoring and reporting, marketing and export and import of products such as those we are developing. The pharmaceutical drug product candidates that we develop must be approved by the FDA before they may be legally marketed.
In the United States, the FDA regulates pharmaceutical products under the Federal Food, Drug, and Cosmetic Act (“FDCA”) and implementing regulations and other federal, state and local statutes and regulations. In the case of biologics, the section of the FDCA that governs the approval of drugs via New Drug Applications (“NDAs”) does not apply to the approval of biologics. Rather, biologics, such as gene therapy products, are approved for marketing under provisions of the Public Health Service Act ("PHSA"(“PHSA”) via a Biologics License Application (“BLA”). However, the application process and requirements for approval of BLAs are very similar to those for NDAs. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, the approval process or after approval, may subject an applicant to administrative or judicial sanctions. FDA sanctions could include refusal to approve pending applications, withdrawal of an approval, a clinical hold, warning or untitled letters, product recalls, product seizures, total or partial suspension of production or distribution injunctions, fines, refusals of government contracts, restitution, disgorgement orand civil orand criminal penalties. Any agency or judicial enforcement action could have a material adverse effect on us.
Ethical, social, and legal concerns about gene therapy, genetic testing, and genetic research could result in additional regulations restricting or prohibiting the processes we may use. Federal and state agencies, congressional committees and foreign governments have expressed interest in further regulating biotechnology. More restrictive regulations or claims that our products are unsafe or pose a hazard could prevent us from commercializing any products. New government requirements may be established that could delay or prevent regulatory approval of our current and potential product candidates. It is impossible to predict whether legislative changes will be enacted, regulations, policies, or guidance changed, or interpretations by agencies or courts changed, or what the impact of such changes, if any, may be.
U.S. Biological Products Development Process
The process required by the FDA before a biological product, including our REQORSA, GPX-002, and potential future product candidate,candidates, may be marketed in the United States generally involves the following:
● | completion of |
● | submission to the FDA of an application for |
|
|
● | performance of adequate and well-controlled human clinical trials according to the FDA’s regulations, commonly referred to as Good Clinical Practices |
● |
|
● |
|
● |
|
● | potential FDA |
● | FDA review and approval, or licensure, of the BLA. |
Within the FDA, the Center for Biologics Evaluation and Research (“CBER”) regulates gene therapy products. CBER works closely with the NIH and its Novel and Exception Technology and Research Advisory Committee (“NExTRAC”) which makes recommendations to the NIH on gene therapy issues and engages in a public discussion of scientific, safety, ethical and societal issues related to proposed and ongoing gene therapy protocols. The FDA and the NIH have published guidance documents with respect to the development and submission of gene therapy protocols, including informed consent documents. The FDA also has published guidance documents related to, among other things, gene therapy products in general, their preclinical assessment, observing patients involved in gene therapy studies for delayed adverse events, potency testing, and chemistry, manufacturing and control information in gene therapy Investigational New Drugs (“INDs”).INDs.
Before testing any product candidate, including a gene therapy product, in humans, the product candidate enters the preclinical testing stage. Preclinical tests, also referred to aswhich are a subset of nonclinical studies, include laboratory evaluations of product chemistry, toxicity and formulation, as well as animal studies to assess the potential safety and activity of the product candidate. The conduct of certain preclinical tests must comply with federal regulations and requirements, including GLPs.
The clinical trial sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and a proposed clinical protocol, to the FDA as part of the IND. Some preclinicalnonclinical testing may continue even after the IND is submitted. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA places the clinical trial on a clinical hold within that 30-day time period. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. The FDA may also impose clinical holds on a product candidate at any time before or during clinical trials due to safety concerns or non-compliance. If the FDA imposes a clinical hold, trials may not recommence without FDA authorization and then only under terms authorized by the FDA.
The clinical trial sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and a proposed clinical protocol, to the FDA as part of the IND. Some preclinical testing may continue evenusually continues after the IND is submitted. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA places the clinical trial on a clinical hold within that 30-day time period. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. The FDA may also impose clinical holds on a product candidate at any time before or during clinical trials due to safety concerns or non-compliance. If the FDA imposes a clinical hold, trials may not recommence without FDA authorization and then only under terms authorized by the FDA.
Clinical trials involve the administration of the product candidate to volunteers or patients under the supervision of qualified investigators, generally physicians not employed by or under the clinical trial sponsor’s control. Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical trial, dosing procedures, patient selection and exclusion criteria, effectiveness criteria to be evaluated, and the parameters to be used to monitor patient safety, including stopping rules that assure a clinical trial will be stopped if certain adverse events occur. Each protocol and any amendments to the protocol must be submitted to the FDA as part of the IND. Clinical trials must be conducted and monitored in accordance with the FDA’s regulations comprising the GCP requirements, including the requirement that all patients provide informed consent. The FDA may order the temporary or permanent discontinuation of a clinical trial at any time, or impose other sanctions, if it believes that the clinical trial either is not being conducted in accordance with FDA regulations or presents an unacceptable risk to the clinical trial patients.
Further, each clinical trial must be reviewed and approved by an independent IRBinstitutional review board ("IRB") at or servicing each institution or site at which the clinical trial will be conducted. An IRB is charged with protecting the welfare and rights of clinical trial participants and considers such items as whether the risks to individuals participating in the clinical trials are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the form and content of the informed consent, which must be signed by each clinical trial patient or his or her legal representative, and must monitor the clinical trial until completed. Clinical trials involving biological product candidates also must be reviewed by an institutional biosafety committee ("IBC"(“IBC”), a local institutional committee that reviews and oversees basic and clinical research conducted at that institution. The IBC assesses the safety of the research and identifies any potential risk to public health or the environment.
Human clinical
Clinical trials to support BLAs for marketing approval are typically conducted in three sequential phases that may overlap or be combined:
● | Phase 1. The investigational product candidate is initially introduced into human patients and tested |
● | Phase 2. The investigational product candidate is evaluated in a limited patient population to identify possible common adverse effects and safety risks, to evaluate preliminarily the efficacy of the product candidate for specific targeted diseases, and |
● | Phase 3. Clinical trials are undertaken to evaluate further dosage, clinical efficacy, potency, and safety in an expanded patient population at geographically dispersed clinical trial sites. These clinical trials are intended to establish the overall risk/benefit ratio of the product candidate and provide an adequate basis for product labeling. |
In most cases, the FDA requires two adequate and well-controlled Phase 3 clinical trials to demonstrate the efficacy of the drug. A single Phase 3 trial may be sufficient in raresome instances, including (1) where the study is a large multicenter trial demonstrating internal consistency and a statistically very persuasive finding of a clinically meaningful effect on mortality, irreversible morbidity or prevention of a disease with a potentially serious outcome and confirmation of the result in a second trial would be practically or ethically impossible or (2) when in conjunction withthere is one adequate and well-controlled clinical investigation plus other confirmatory evidence. Typically, during the development of oncology therapies, all subjects enrolled in Phase 1 clinical trials are disease-affected patients and, as a result, considerably more information on clinical activity may be collected during such trials than during Phase 1 clinical trials for non-oncology therapies. A single pivotal trial may be sufficient in rare instances to provide substantial evidence of effectiveness (generally subject to the requirement of additional post-approval studies).
In addition, the manufacturer of an investigational biologic in a Phase 2 or Phase 3 clinical trial for a serious or life-threatening disease is required to make available, such as by posting on its website, its policy on evaluating and responding to requests for expanded access to such investigational biologic.
Post-approval clinical trials, sometimes referred to as Phase 4 clinical trials, may be conducted after initial marketing approval. These clinical trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication, particularly for long-term safety follow-up. The FDA recommends that sponsors observe patients for potential gene therapy-related delayed adverse events with agents such as those we are developing for a period of up to 15 years, including a minimum of five years of annual examinations followed by ten years of annual queries, either in person or by questionnaire, of clinical trial patients.
During all phases of clinical development, regulatory agencies require extensive monitoring and auditing of all clinical activities, clinical data, and clinical trial investigators. Annual progress reports detailing the results of the clinical trials must be submitted to the FDA. Written IND safety reports must be promptly submitted to the FDA, and the investigators for serious and unexpected adverse events, any findings from other trials, tests in laboratory animals or in vitro testing that suggest a significant risk for human patients, or any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigator brochure. The sponsor must submit an IND safety report within 15 calendar days after the sponsor determines that the information qualifies for expedited reporting. The sponsor must also notify the FDA of any unexpected fatal or life-threatening suspected adverse reaction within seven calendar days after the sponsor’s initial receipt of the information. Phase 1, Phase 2 and Phase 3 clinical trials may not be completed successfully within any specified period, if at all. The FDA, the sponsor, or its data safety monitoring board may suspend a clinical trial at any time on various grounds, including a finding that the patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the investigational product candidate has been associated with unexpected serious harm to patients.
There are also requirements governing the reporting of ongoing clinical trials and completed clinical trial results to public registries. Sponsors of clinical trials of FDA-regulated products, including biologics, are required to register and disclose certain clinical trial information, which is publicly available at www.clinicaltrials.gov. Information related to the product, patient population, phase of investigation, study sites and investigators, and other aspects of the clinical trial is then made public as part of the registration. Sponsors are also obligated to disclose the results of their clinical trials after completion. Disclosure of the results of these trials can be delayed in certain circumstances for up to two years after the date of completion of the trial.
Concurrently with clinical trials, companies usually complete additional animal studies and also develop additional information about the physical characteristics of the components of a product as well as finalize processes for manufacturing the components in commercial quantities in accordance with GMP requirements. To help reduce the risk of the introduction of adventitious agents with use of biological products, the PHSA, emphasizes the importance of manufacturing control for products whose attributes cannot be precisely defined. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, the sponsor must develop methods for testing the identity, strength, quality, potency and purity of the final product. Additionally, appropriate packaging must be selected and tested, and stability studies must be conducted to demonstrate that the components of a product candidate do not undergo unacceptable deterioration over their shelf life.
U.S. Review and Approval Processes
After the completion of clinical trials of an investigational biologic product, a BLA is prepared and submitted to the FDA. FDA approval of a BLA must be obtained before commercial marketing and distribution of the product may begin.begin in the United States. The BLA must include results of product development, laboratory, and animal studies, human trials, information on the manufacture and composition of the product, proposed labeling and other relevant information. The testing and approval processes require substantial time and effort and there can be no assurance that the FDA will file the BLA and, even if filed, that any approval will be granted on a timely basis, if at all.
Under the Prescription Drug User Fee Act, as amended (“PDUFA”), each BLA must be accompanied by a significant user fee. The FDA adjusts the PDUFA user fees on an annual basis. PDUFA also imposes annual program fees on prescription drugs, including biologics. Fee waivers or reductions are available in certain circumstances, including a waiver of the application fee for the first application filed by a small business. No user fees are assessed on BLAs for products designated as orphan drugs, unless the application also includes a non-orphan indication.
Within 60 days following submission, the FDA reviews the BLA to determine if it is substantially complete before the agency files it. The FDA may request additional information or may refuse to file any BLA that it deems incomplete or not properly reviewable at the time of submission and may request additional information.submission.
In this event, the BLA must be resubmitted with the additional information. The resubmitted application also is subject to an initial filing review before the FDA files it. Once the submission is filed, the FDA begins an in-depth substantive review of the BLA. Under PDUFA, FDA has agreed to performance goals to review 90% of original standard BLAs within 10 months of the 60-day filing date and 90% of original priority BLAs within six months of the 60-day filing date, whereupon a review decision is to be made. The FDA does not always meet its PDUFA goal dates for standard and priority BLAs. The review process and the PDUFA goal date may be extended by three months if the FDA requests or the BLA sponsor otherwise provides additional information or clarification regarding information already provided in the BLA submission. The FDA reviews the BLA to determine, among other things, whether the proposed product is safe and potent, or effective, for its intended use, and has an acceptable purity profile, and whether the product is being manufactured in accordance with GMP to assure and preserve the product’s identity, safety, strength, quality, potency and purity.
The FDA may refer applications for novel products or products that present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations when making decisions. During the product approval process, the FDA also will determine whether a Risk Evaluation and Mitigation Strategy (“REMS”) is necessary to assure that the benefits of the biologic outweigh the potential risks of the product to patients. A REMS can include medication guides, communication plans for healthcare professionals, and elements to assure a product’s safe use (“ETASU”). An ETASU can include, but is not limited to, special training or certification for prescribing or dispensing the product, dispensing the product only under certain circumstances, special monitoring, and the use of the product.patient-specific registries. If the FDA concludes that a REMS is needed, the sponsor of the BLA must submit a proposed REMS; the FDA will not approve the BLA without a REMS, if required.
Before approving a BLA, the FDA will typically inspect the facilities at which the product is manufactured. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in substantial compliance with GMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving a BLA, the FDA will typically inspect one or more clinical sites, to assure that the clinical trials were conducted in compliance with GCP requirements. In addition, the FDA may inspect one or more sites where animal studies were conducted to confirm compliance with GLP requirements. To assure GMP, GLPGCP and GCPGLP compliance, an applicant must incur significant expenditure of time, money, and effort in the areas of training, record keeping, production, and quality control.
Notwithstanding the submission of relevant data and information, the FDA may ultimately decide that the BLA does not satisfy its regulatory criteria for approval and deny approval. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently from how we interpret the same data. If the agency decides not to approve the BLA in its present form, the FDA will issue a complete response letter that usually describes all of the specific deficiencies in the BLA identified by the FDA. The deficiencies identified may be minor, for example, requiring labeling changes, or major, for example, requiring additional clinical trials. Additionally, the complete response letter may include recommended actions that the applicant might take to place the application in a condition for approval. If a complete response letter is issued, the applicant may resubmit the BLA, addressing all of the deficiencies identified in the letter, withdraw the application, or request a hearing.
If a product candidate receives regulatory approval, the FDA will issue an approval letter. The approval may be significantly limited to specific diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings, or precautions be included in the product labeling. The FDA may impose restrictions and conditions on product distribution, prescribing, or dispensing in the form of a risk management plan, or otherwise limit the scope of any approval. In addition, the FDA may require post marketing clinical trials, sometimes referred to as Phase 4 clinical trials, designed to assess further a biological product’s safety and effectiveness, and testing and surveillance programs to monitor the safety of approved products that have been commercialized.
One of the performance goals agreed to by the FDA under the PDUFA is to review 90% of original standard BLAs within 10 months of the 60-day filing date and 90% of original priority BLAs within six months of the 60-day filing date, whereupon a review decision is to be made. The FDA does not always meet its PDUFA goal dates for standard and priority BLAs. The review process and the PDUFA goal date may be extended by three months if the FDA requests or the BLA sponsor otherwise provides additional information or clarification regarding information already provided in the submission within the last three months before the PDUFA goal date.
Expedited Development and Review Programs
The FDA has four programs in place intended to facilitate and expedite development and review of new drugs and biologics intended to address unmet medical needs in the treatment of serious or life-threatening conditions. These are Fast Track Designation, Breakthrough Therapy Designation, Accelerated Approval Program, and Priority Review Designation.
The Fast Track program is intended to expedite or facilitate the process for reviewing a new product if it is intended for the treatment of a serious or life-threatening disease or condition, and it demonstrates the potential to address unmet medical needs for such a disease or condition. Fast Track Designation applies to the combination of the product and the specific indication for which it is being studied. The sponsor of a new drug or biologic may request the FDA to designate the drug or biologic as a Fast Track product at any time during the clinical development of the product. Unique to a Fast Track product, the FDA may consider for review sections of the marketing application on a rolling basis before the complete application is submitted, if the sponsor provides a schedule for the submission of the sections of the application, the FDA agrees to accept sections of the application and determines that the schedule is acceptable, and the sponsor pays any required user fees upon submission of the first section of the application.
A product can receive Breakthrough Therapy Designation if it is intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that it may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. A Breakthrough Therapy Designation conveys all of the features of Fast Track Designation in addition to more intensive FDA guidance on an efficient development program, organizational commitment involving senior managers, and eligibility for priority review. Specifically, the FDA intends to expedite the development and review of a Breakthrough Therapy by, where appropriate, intensively involving senior managers and experienced review staff in a proactive collaborative, cross-disciplinary review. Where appropriate, the FDA also intends to assign a cross-disciplinary project lead for the review team to facilitate an efficient review of the development program. The FDA notes that a compressed drug development program still must generate adequate data to demonstrate that the drug or biologic meets the statutory standard for approval. Omitting components of the development program that are necessary for such a determination can significantly delay, or even preclude, marketing approval.
Breakthrough Therapy Designation indicates that preliminary clinical evidence demonstrates the drug may have substantial improvement on one or more clinically significant endpoints over available therapy. Breakthrough Therapy Designation intensifies FDA involvement to ensure an efficient drug development program and is an organizational commitment from the FDA to involve its senior managers. A sponsor receiving Breakthrough Therapy Designation has up to six months after receiving the Breakthrough Therapy Designation to request an Initial Comprehensive Multidisciplinary meeting to discuss the drug development program. This initial meeting is a Type B meeting, used to discuss the overarching, high-level plan for drug development. These discussions include topics such as planned clinical trials and endpoints, any resizing or adaptations to the trials, plans for expediting the manufacturing development strategy and studies that potentially could be completed after approval. When Breakthrough Therapy Designation has been granted, the FDA is encouraged to meet regularly with the sponsor and subsequent meetings are considered Type B meetings and are established based on the needs of the program.
The FDA may grant accelerated approval under its Accelerated Approval Program to a product candidate for a serious or life-threatening condition upon a determination that the product candidate has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit, or an effect on a clinical endpoint that can be measured earlier than an effect on irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. Accelerated approval is contingent on a sponsor’s agreement to conduct at least one adequate and well-controlled additional post-approval trial to verify and describe the product’s clinical benefit. In addition, the FDA currently requires as a condition for accelerated approval pre-approval of promotional materials, which could adversely impact the timing of the commercial launch of the product.
Fast Track Designation, Breakthrough Therapy Designation, RMAT, and Accelerated Approval do not change the standards for approval but may expedite the development process. Additionally, Fast Track Designation or Breakthrough Therapy Designation may be withdrawn by the FDA if the FDA believes that the designation is no longer supported by data emerging in the clinical trial process, including considering any new drug or biologic approvals that address the unmet medical need.
An application for a product candidate may be eligible to obtain Priority Review Designation if it is intended to treat a serious condition and, if approved, would provide a significant improvement in safety or effectiveness. The FDA will attempt to direct additional resources to the evaluation of an application for a new product designated for priority review in an effort to facilitate the review. A Priority Review Designation means FDA’s goal is to take action on the marketing application within six months (compared to ten months under standard review) of the 60-day filing date. Priority Review Designation does not change the standards for approval but may expedite the review process.
Orphan Designation and Exclusivity
Under the Orphan Drug Act, the FDA may grant orphan designation to a drug or biologic intended to treat a rare disease or condition, defined as a disease or condition with a patient population of fewer than 200,000 individuals in the United States, or a patient population greater than 200,000 individuals in the United States and when there is no reasonable expectation that the cost of developing and making available the drug or biologic in the United States will be recovered from sales in the United States for that drug or biologic. Orphan drug designation must be requested before submitting a BLA or NDA. After the FDA grants orphan drug designation, the generic identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA.
If a product that has orphan drug designation subsequently receives the first FDA approval for a particular active ingredient for the disease for which it has such designation, the product is entitled to orphan product marketing exclusivity, which means that the FDA may not approve any other applications, including a full BLA, to market the same biologic for the same use or indication for seven years, except in limited circumstances, such as a showing of clinical superiority to the product with orphan drug exclusivity or if FDA finds that the holder of the orphan drug exclusivity has not shown that it can assure the availability of sufficient quantities of the orphan drug to meet the needs of patients with the disease or condition for which the drug was designated. Orphan drug exclusivity does not prevent the FDA from approving a different drug or biologic for the same disease or condition, or the same drug or biologic for a different disease or condition. Among the other benefits of orphan drug designation are tax credits for certain research and a waiver of the BLA or NDA application user fee.
A designated orphan drug may not receive orphan drug exclusivity if it is approved for a use that is broader than the indication for which it received orphan designation. In addition, orphan drug exclusive marketing rights in the United States may be lost if the FDA later determines that the request for designation was materially defective or, as noted above, if the second applicant demonstrates that its product is clinically superior to the approved product with orphan exclusivity or the manufacturer of the approved product is unable to assure sufficient quantities of the product to meet the needs of patients with the rare disease or condition.
Post-Approval Requirements
MaintainingOnce a BLA is approved, maintaining post-approval compliance with applicable federal, state, and local statutes and regulations requires the expenditure of substantial time and financial resources. Manufacturers and other entities involved in the manufacture and distribution of approved products are required to register the establishments where the approved products are made with the FDA and certain state agencies and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with GMP and other laws. Rigorous and extensive FDA regulation of products continues after approval, particularly with respect to GMP. We rely, and expect to continue to rely, on third parties for the production and distribution of clinical and commercial quantities of any products that we may commercialize. Manufacturers of our products are required to comply with applicable requirements in the GMP regulations, including quality control and quality assurance and maintenance of records and documentation. Other post-approval requirements include reporting of GMP deviations that may affect the identity, potency, purity and overall safety of a distributed product, record-keeping requirements, reporting of adverse effects, reporting updated safety and efficacy information, and complying with electronic record and signature requirements. After a BLA is approved, the product also may be subject to official lot release. As part of the manufacturing process, the manufacturer is required to perform certain tests on each lot of the product before it is released for distribution. If the product is subject to official release by the FDA, the manufacturer submits samples of each lot of product to the FDA together with a release protocol showing a summary of the history of manufacture of the lot and the results of all of the manufacturer’s tests performed on the lot. The FDA also may perform certain confirmatory tests on lots of some products before releasing the lots for distribution by the manufacturer. Accordingly, manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain GMP compliance. Discovery of problems with a product after approval may result in restrictions on a product, manufacturer, or holder of an approved BLA, including withdrawal of the product from the market. In addition, changes to the manufacturing process or facility generally require prior FDA approval before being implemented. Other types of changes to the approved product, such as adding new indications and additional labeling claims, are also subject to further FDA review and approval.
We also must comply with the FDA’s advertising and promotion requirements, such as those related to direct-to-consumer advertising, the prohibition on promoting products for uses or in patient populations that are not described in the product’s approved labeling (known as “off-label use”), industry-sponsored scientific and educational activities, and promotional activities involving the internet. Discovery of previously unknown problems or the failure to comply with the applicable regulatory requirements may result in restrictions on the marketing of a product or withdrawal of the product from the market as well as possible civil or criminal sanctions.
Manufacturers and other entities involved in the manufacture and distribution of approved products are required to register the establishments where the approved products are made with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with GMP and other laws. Accordingly, manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain GMP compliance. Discovery of problems with a product after approval may result in restrictions on a product, manufacturer, or holder of an approved BLA, including withdrawal of the product from the market. In addition, changes to the manufacturing process or facility generally require prior FDA approval before being implemented. Other types of changes to the approved product, such as adding new indications and additional labeling claims, are also subject to further FDA review and approval.
Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval may subject an applicant or manufacturer to administrative or judicial civil or criminal sanctions and adverse publicity. FDA sanctions could include refusal to approve pending applications, withdrawal of an approval, clinical hold, warning or untitled letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, mandated corrective advertising or communications with doctors, debarment, restitution, disgorgement of profits, or civil or criminal penalties. Any agency or judicial enforcement action could have a material adverse effect on us.
U.S. Patent Term Restoration
Depending upon the timing, duration, and specifics of the FDA approval of the use of our current and potential product candidates, some of our U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984 ("Hatch-Waxman Amendments"). The Hatch-Waxman Amendments permit a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent term restoration period is generally one-half the time between the effective date of an IND and the submission date of a BLA plus the time between the submission date of a BLA and the approval of that application. Only one patent applicable to an approved biological product is eligible for the extension and the application for the extension must be submitted prior to the expiration of the patent. The U.S. Patent and Trademark Office, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration.
Biosimilars and Exclusivity
The Biologics Price Competition and Innovation Act of 2009 ("BPCIA") created an abbreviated approval pathway for biological products shown to be highly similar to, or interchangeable with, an FDA-licensed reference biological product. The FDA has issued several guidance documents outlining an approach to review and approval of biosimilars.
Biosimilarity, which requires that there be no clinically meaningful differences between the biological product and the reference product in terms of safety, purity, and potency, can be shown through analytical studies, animal studies, and a clinical study or studies. Interchangeability requires that a product is biosimilar to the reference product and the product must demonstrate that it can be expected to produce the same clinical results as the reference product in any given patient and, for products that are administered multiple times to an individual, the biologic and the reference biologic may be alternated or switched after one has been previously administered without increasing safety risks or risks of diminished efficacy relative to exclusive use of the reference biologic. However, complexities associated with the larger, and often more complex, structures of biological products, as well as the processes by which such products are manufactured, pose significant hurdles to implementation of the abbreviated approval pathway that are still being worked out by the FDA.
The BPCIA includes, among other provisions:
● | A 12-year exclusivity period from the date of first licensure, or BLA approval, of the reference product, during which approval of a 351(k) application referencing that product may not be made effective; |
● | A four-year exclusivity period from the date of first licensure of the reference product, during which a 351(k) application referencing that product may not be |
|
|
● | An exclusivity period for certain biological products that have been approved through the 351(k) pathway as interchangeable biosimilars. |
The BPCIA also establishes procedures for identifying and resolving patent disputes involving applications submitted under section 351(k) of the PHSA.
The BPCIA is complex and its interpretation and implementation by the FDA remains unpredictable. In addition, government proposals have sought to reduce the 12-year reference product exclusivity period. Other aspects of the BPCIA, some of which may impact the BPCIA exclusivity provisions, have also been the subject of recent litigation. As a result, the ultimate effect, implementation, and meaning of the BPCIA is subject to uncertainty.
Disclosure of Clinical Trial Information
Sponsors of clinical trials of FDA-regulated products, including biological products, are required to register and disclose certain clinical trial information on the website www.clinicaltrials.gov. Information related to the product, patient population, phase of investigation, trial sites and investigators, and other aspects of a clinical trial are then made public as part of the registration. Sponsors are also obligated to disclose the results of their clinical trials after completion. Disclosure of the results of clinical trials can be delayed in certain circumstances for up to two years after the date of completion of the trial. Competitors may use this publicly available information to gain knowledge regarding the progress of clinical development programs as well as clinical trial design.
Pediatric Information
Under the Pediatric Research Equity Act (“PREA”), BLAs or supplements to BLAs must contain data to assess the safety and effectiveness of the biological product for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the biological product is safe and effective. The FDA may grant full or partial waivers, or deferrals, for submission of data. Unless otherwise required by regulation, PREA does not apply to any biological product with orphan product designation except a product with a new active ingredient that is a molecularly targeted cancer product intended for the treatment of an adult cancer and directed at a molecular target determined by FDA to be substantially relevant to the growth or progression of a pediatric cancer that is subject to a BLA submitted on or after August 18, 2020.
The Best Pharmaceuticals for Children Act (“BPCA”) provides a six-month extension of any non-patent exclusivity for a biologic if certain conditions are met. Conditions for exclusivity include the FDA’s determination that information relating to the use of a new drug or biologic in the pediatric population may produce health benefits in that population, the FDA making a written request for pediatric studies, and the applicant agreeing to perform, and reporting on, the requested studies within the statutory timeframe. Applications under the BPCA are treated as priority applications, with all of the benefits that designation confers.
Additional U.S. Regulation
In the United States, our activities are potentially subject to regulation by various federal, state and local authorities in addition to the FDA, including but not limited to, the Centers for Medicare and Medicaid Services ("CMS"(“CMS”), other divisions of the U.S. Department of Health and Human Services, for instance the Office of Inspector General, the U.S. Department of Justice ("DOJ"(“DOJ”), and individual U.S. Attorney offices within the DOJ, and state and local governments. For example, sales, marketing and scientific/educational grant programs must comply with the anti-fraud and abuse provisions of the Social Security Act, the false claims laws, the physician payment transparency laws, the privacy and security provisions of the Health Insurance Portability and Accountability Act ("HIPAA"(“HIPAA”), as amended by the Health Information Technology for Economic and Clinical Health Act ("HITECH"(“HITECH”) and similar state laws, each as amended.
In addition to the foregoing, state and federal laws regarding environmental protection and hazardous substances, including the Occupational Safety and Health Act, the Resource Conservancy and Recovery Act and the Toxic Substances Control Act, may affect our business. These and other laws govern our use, handling and disposal of various biological, chemical, and radioactive substances used in, and wastes generated by, our operations. If our operations result in contamination of the environment or expose individuals to hazardous substances, we could be liable for damages and governmental fines. We believe that we are in material compliance with applicable environmental laws and that continued compliance therewith is unlikely to have a material adverse effect on our business. We cannot predict, however, how changes in these laws may affect our future operations.
Federal and State Fraud and Abuse, Privacy and Transparency Laws
In the United States, our activities are potentially subject to regulation by various federal, state and local authorities in addition to the FDA, including but not limited to, CMS, other divisions of the U.S. Department of Health and Human Services, for instance, the Office of Inspector General, DOJ, and individual U.S. Attorney offices within the DOJ,Federal and state fraud and local governments. These federal and stateabuse laws, which generally will not be applicable to us or our current and potential product candidates unless and until we obtain FDA marketing approval for any of our current and potential product candidates, include, among others, anti-kickback statutes, the False Claims Act and related stated state and federal laws, the Stark Law and related state and federal laws, transparency laws, privacy and regulation regarding providing drug samples, sales and marketing activities and our relationships with customers and payors as follows.
The federal Anti-Kickback Statute prohibits, among other things, individuals and entities from knowingly and willfully offering, paying, soliciting, or receiving any remuneration, directly or indirectly, overtly or covertly, to induce or in return for purchasing, leasing, recommending, ordering, or arranging for the purchase, lease, recommendation or order of any health care item or service reimbursable, in whole or in part, under Medicare, Medicaid, or other federally financed healthcare programs. This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on one hand and prescribers, purchasers, and formulary managers on the other. Although there are a number of statutory exemptions and regulatory safe harbors protecting certain common activities from prosecution, the exemptions and safe harbors are drawn narrowly, and practices that involve remuneration that may be alleged to be intended to induce prescribing, purchases or recommendations may be subject to scrutiny if they do not qualify for an exemption or safe harbor. Additionally, the intent standard under the Anti-Kickback Statute was amended by the Affordable Care Act to a stricter standard such that a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. Further, the Affordable Care Act codified case law that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act.
HIPAA created additional federal criminal statutes that prohibit, among other actions, knowingly and willfully executing, or attempting to execute, a scheme to defraud or to obtain, by means of false or fraudulent pretenses, representations or promises, any money or property owned by, or under the control or custody of, any healthcare benefit program, including private third-party payers, willfully obstructing a criminal investigation of a healthcare offense, and knowingly and willfully falsifying, concealing or covering up by trick, scheme or device, a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
Federal false claims and civil monetary penalties laws, including the federal civil False Claims Act, prohibit, among other things, any person or entity from knowingly presenting, or causing to be presented, a false, fictitious or fraudulent claim for payment to, or approval by, the federal government, or knowingly making, using, or causing to be made or used, a false statement to get a false claim paid. Several pharmaceutical and other health care companies have been prosecuted under these laws for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. Other companies have been prosecuted for causing false claims to be submitted because of the company’s marketing of the product for unapproved, and thus non-reimbursable, uses.
The majority of states also have statutes or regulations similar to the federal Anti-Kickback Statute and false claims laws, which apply to items and services, reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payer.
We may also be subject to data privacy and security regulations by both the federal government and the states in which we conduct our business. HIPAA, as amended by HITECH, and their respective implementing regulations, including the final Omnibus Rule published in 2013, imposes requirements on certain types of entities, including mandatory contractual terms, relating to the privacy, security, and transmission of individually identifiable health information. Among other things, HITECH makes HIPAA’s security standards and certain privacy standards directly applicable to business associates, which are independent contractors or agents of covered entities that create, receive, maintain or transmit protected health information in connection with providing a service for or on behalf of a covered entity. HITECH also created four new tiers of civil monetary penalties, amended HIPAA to make civil and criminal penalties directly applicable to business associates, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorneys’ fees and costs associated with pursuing federal civil actions. In addition, many state laws govern the privacy and security of health information in specified circumstances, many of which differ from each other and from HIPAA in significant ways and may not have the same requirements, thus complicating compliance efforts.
Additionally, the federal Physician Payments Sunshine Act under the Affordable Care Act, and its implementing regulations require that certain manufacturers of drugs, devices, biological and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program, with certain exceptions, annually report to CMS information related to certain payments or other transfers of value made or distributed to physicians, physician assistants, certain types of advanced practice nurses and teaching hospitals, or to entities or individuals at the request of, or designated on behalf of, the physicians and teaching hospitals and to report annually certain ownership and investment interests held by physicians and their immediate family members. Failure to submit timely, accurately, and completely the required information may result in civil monetary penalties of up to an aggregate of $150,000 per year and up to an aggregate of $1 million per year for “knowing failures.” Certain states also mandate implementation of compliance programs, impose restrictions on pharmaceutical manufacturer marketing practices, and/or require the tracking and reporting of gifts, compensation, and other remuneration to healthcare providers and entities.
Because of the breadth of these laws and the narrowness of the exceptions and safe harbors, it is possible that some of our business activities could be subject to challenge under one or more of such laws. Such a challenge could have a material adverse effect on our business, financial condition, and results of operations. If our operations are found to be in violation of any of these or any other health regulatory laws that may apply to us, we may be subject to, without limitation, significant penalties, including the imposition of significant civil, criminal and administrative penalties, damages, monetary fines, disgorgement, individual imprisonment, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, contractual damages, reputational harm, diminished profits and future earnings, and curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations.
In addition, as part of the sales and marketing process, pharmaceutical companies frequently provide samples of approved products to physicians. This practice is regulated by the FDA and other governmental authorities, including, in particular, requirements concerning record keeping and control procedures. Any failure to comply with the regulations may result in significant criminal and civil penalties as well as damage to our credibility in the marketplace.
Coverage and Reimbursement
In many of the markets where we may do business in the future, the prices of pharmaceutical products are subject to direct price controls (by law) and to reimbursement programs with varying price control mechanisms. In the United States, significant uncertainty exists as to the coverage and reimbursement status of any product candidates for which we obtain product approval. Often private payers follow the coverage and reimbursement decisions of the Medicare program, and it is difficult to predict how CMS may decide to cover and reimburse approved products, especially novel products, and those determinations are subject to change.
Moreover, the process for determining whether a third-party payer will provide coverage for a drug product may be separate from the process for setting the price of a drug product or for establishing the reimbursement rate that such a payer will pay for the drug product. Third-party payers may limit coverage to specific products on an approved list, also known as a formulary, which might not include all of the FDA-approved drugs for a particular indication. A decision by a third-party payer not to cover our current and potential product candidates could reduce physician utilization of our products once approved. A payer’s decision to provide coverage for a drug product does not imply that an adequate reimbursement rate will be approved. Further, one payer’s determination to provide coverage for a drug product does not assure that other payers will also provide coverage for the drug product. Coverage and reimbursement for new products can differ significantly from payer to payer. As a result, the coverage determination process will require us to provide scientific and clinical support for the use of our products to each payer separately and will be a time-consuming process. Additionally, third-party reimbursement may not be available or may not be adequate to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development.
Federal, state and local governments in the United States and foreign governments continue to consider legislation to limit the growth of healthcare costs, including the cost of prescription drugs. Specifically, there have been several recent U.S. Congressional inquiries and proposed federal and state legislation designed to, among other things, bring more transparency to drug pricing, reduce the cost of prescription drugs under Medicare, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drugs. Future legislation could limit payments for pharmaceuticals such as the drug candidates that we are developing.
Different pricing and reimbursement schemes exist in other countries. In the EU, governments influence the price of drug products through their pricing and reimbursement rules and control of national health care systems that fund a large part of the cost of those products to consumers. Some jurisdictions operate systems under which products may be marketed only after a reimbursement price has been agreed. To obtain reimbursement or pricing approval, some of these countries may require the completion of studies or analyses of clinical trials that compare the cost-effectiveness of a particular product candidate to currently available therapies. Other member states allow companies to set their own prices for medicines, but exert cost controls in other ways, including but not limited to, placing revenue caps on product sales, providing reimbursement for only a subset of eligible patients, mandating price negotiations after a set period of time, or mandating that prices not exceed an average basket of prices in other countries. The downward pressure on health care costs in general, particularly treatments, has become very intense. As a result, increasingly high barriers are being erected to the entry of new products. In addition, European governments may periodically review and decrease prices based on factors, including, but not limited to, years-on-market, price in other countries, competitive entry, new clinical data, lack of supporting clinical data, or other factors.
The marketability of any product candidates for which we or our collaborators receive regulatory approval for commercial sale may suffer if the government and third-party payers fail to provide adequate coverage and reimbursement. In addition, an emphasis on cost containment measures in the United States has increased and we expect will continue to increase the pressure on pharmaceutical pricing. Third-party payers are increasingly challenging the prices charged for medical products and services, examining the medical necessity and reviewing the cost-effectiveness of drugs, medical devices and medical services, in addition to questioning safety and efficacy. If these third-party payers do not consider our products to be cost-effective compared to other available therapies, they may not cover our products after FDA approval or, if they do, the level of payment may not be sufficient to allow us to sell our products at a profit. Coverage policies and third-party reimbursement rates may change at any time. Even if favorable coverage and reimbursement status is attained for one or more products for which we or our collaborators receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
Legislative and Regulatory Changes, Including Health Care Reform
The laws and regulation that affect our business are subject to change from time to time, and entirely new laws and regulations are sometimes adopted. In March 2010, the Affordable Care Act was enacted, which affected, and may further affect, health care financing and delivery by both governmental and private insurers, and therefore the pharmaceutical and biotechnology industry. The Affordable Care Act has affected and may continue to affect existing governmentparticular, healthcare programs and may result in the development of new programs.
Among the Affordable Care Act’s provisions of importance to the pharmaceutical and biotechnology industries, in addition to those otherwise described above, are the following:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
There have been judicialadopted, and Congressional challenges to certain aspects of the Affordable Care Act. As a result, there have been delays in the implementation of, and action taken to repeal or replace, certain aspects of the Affordable Care Act. In January 2017, President Trump signed an Executive Order directing federal agencies with authorities and responsibilities under the Affordable Care Act to waive, defer, grant exemptions from or delay the implementation of any provision of the Affordable Care Act that would impose a fiscal or regulatory burden on states, individuals, healthcare providers, health insurers or manufacturers of pharmaceuticals or medical devices. In December 2018 a judge for the United States District Court for the Northern District of Texas ruled that the entire Affordable Care Act is unconstitutional. This ruling was appealed to the U.S. Court of Appeals for the Fifth Circuit, which held that the individual mandate is unconstitutional. This decision was appealed, and in November 2020, the United States Supreme Court held oral arguments. It is uncertain how the United States Supreme court will rule on this case or how healthcare measures of the Biden administration will impact the Affordable Care Act and our business. With respect to repeal or revision of the Affordable Care and its replacement with new or revised legislation, it is unclear when such legislation willmay be enacted, what it will provide and what impact it will have on the availability of healthcare and containing or lowering costs of healthcare.
The former Trump Administration proposed a number of initiatives to drive down prescription drug costs and health care spending. President Biden has indicated a desire to address prescription drug costs as well. However, it is unclear how any proposed or future initiatives may affect our business.
Other legislative changes have been proposed and adopted in the United States sincefuture, could result in further reductions in coverage and levels of reimbursement for pharmaceutical products, increases in rebates payable under U.S. government rebate programs and additional downward pressure on pharmaceutical product prices. In 2022, President Biden signed the Affordable CareInflation Reduction Act, was enacted. For example, in August 2011, the Budget Control Act of 2011, among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the fiscal year 2012 through 2021, was unable to reach required goals, thereby triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions of Medicare payments to providers of up to 2% per fiscal year, which went into effect in April 2013 and will remain in effect through 2030 unless additional Congressional action is taken. The CARES Act, which was signed into law in March 2020 and is designed to provide financial support and resources to individuals and businesses affected by the COVID-19 pandemic, suspended the 2% Medicare sequester from May 1, 2020 through December 31, 2020, and extended the sequester by one year, through 2030. The Consolidated Appropriations Act, 2021 extended the suspension of the 2% Medicare sequester through March 31, 2021. Further, in January 2013, President Obama signed into law the American Taxpayer Relief Act of 2012, which, among other things, further reducedcontains a provision that authorizes CMS to negotiate a "maximum fair price" for a limited number of high-cost, single-source drugs each year, and another provision that requires drug companies to pay rebates to Medicare paymentsif prices rise faster than inflation. It is unclear to several providers, including hospitals, imaging centerswhat extent these and cancer treatment centers,other statutory, regulatory, and increased the statute of limitations period for the government to recover overpayments to providers from three to fiveadministrative initiatives will be enacted and implemented in future years.
We anticipate that the Affordable Care Act and other legislative reforms will result in additional downward pressure on the price that we receive for any approved product, if covered, and could seriously harm our business. Any reduction in reimbursement from Medicare and other government programs may result in a similar reduction in payments from private payers. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability, or commercialize our products. In addition, it is possible that there will be further legislation or regulation that could harm our business, financial condition, and results of operations.
Environmental Regulation
In addition to the foregoing, state and federal laws regarding environmental protection and hazardous substances, including the Occupational Safety and Health Act, the Resource Conservancy and Recovery Act and the Toxic Substances Control Act, may affect our business. These and other laws govern our use, handling and disposal of various biological, chemical, and radioactive substances used in, and wastes generated by, our operations. If our operations result in contamination of the environment or expose individuals to hazardous substances, we could be liable for damages and governmental fines. We believe that we are in material compliance with applicable environmental laws and that continued compliance therewith is unlikely to have a material adverse effect on our business. We cannot predict, however, how changes in these laws may affect our future operations.
U.S. Foreign Corrupt Practices Act and Similar International Laws
The U.S. Foreign Corrupt Practices Act, to which we are subject, prohibits corporations and individuals from engaging in certain activities to obtain or retain business or to influence a person working in an official capacity. It is illegal to pay, offer to pay or authorize the payment of anything of value to any foreign government official, government staff member, political party, or political candidate in an attempt to obtain or retain business or to influence otherwise a person working in an official capacity.
Other countries, including a number of EU member states, have laws of similar application, including anti-bribery or anti-corruption laws such as the UK Bribery Act. The UK Bribery Act prohibits giving, offering, or promising bribes to any person, as well as requesting, agreeing to receive, or accepting bribes from any person. Under the UK Bribery Act, a company that carries on a business or part of a business in the United Kingdom may be held liable for bribes given, offered or promised to any person in any country by employees or other persons associated with the company in order to obtain or retain business or a business advantage for the company. Liability under the UK Bribery Act is strict, but a defense of having in place adequate procedures designed to prevent bribery is available.
Government Regulation Outside of the United States
In addition to regulations in the United States, we will be subject to a variety of regulations in other jurisdictions governing, among other things, clinical trials and any commercial sales and distribution of our products. Because biologically sourced raw materials are subject to unique contamination risks, their use may be subjected to different types of restrictions in different countries.
Whether or not we obtain FDA approval for a product, we must obtain the required approvals from regulatory authorities in foreign countries prior to the commencement of clinical trials or marketing of the product in those countries. Certain countries outside of the United States have a similar process that requires the submission of a clinical trial application equivalent to an IND prior to the commencement of human clinical trials. In the European Union, for example, a clinical trial authorization, or CTA, must be submitted to each country’s national health authority and an independent ethics committee, much like the FDA and the IRB, respectively. Once the CTA is approved in accordance with a country’s requirements, clinical trials may start.
The requirements and process governing the conduct of clinical trials, product licensing, manufacturing, sales and marketing, pricing, and reimbursement vary from country to country. In all cases, the clinical trials are to be conducted in accordance with GCP, applicable regulatory requirements, and the ethical principles that have their origin in the Declaration of Helsinki.
If we fail to comply with applicable foreign regulatory requirements, we may be subject to, among other things, fines, suspension, or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution in those countries.
Employees and Human Capital
As of March 15, 2021,2024, we had 12 full-time employees.26 total employees, all of which were full-time. We are not a party to any collective bargaining agreements. We believe that we maintain good relations with our employees. Our employees are highly skilled, with many holding advanced degrees and having experience in drug development. We anticipate that the number of employees will remain approximately at this level throughout this year even as we progress our product pipeline.
We believe that our success depends in large part on our ability to attract and retain experienced and skilled employees. We endeavor to provide competitive compensation and benefits packages that reflect the highly competitive nature of our industry, as well as opportunities for professional development which are designed to attract, engage, retain and reward talented individuals who possess the skills necessary to support our business objectives, assist in the achievement of our strategic goals and increase stockholder value. We employ a pay for performance philosophy. Annual salary increases, promotional opportunities, incentive bonuses and stock option grants are available to all employees and are based on merit and include individual and corporate performance factors.
Much of our success is rooted in the diversity of our teams and our commitment to inclusion. We are committed to providing an environment of mutual respect and equal opportunity. We value diversity at all levels and continue to focus on extending our diversity and inclusion initiatives across our entire workforce. We believe that our business benefits from the different perspectives that a diverse workforce brings.
Corporate Information and Available Information.
We were incorporated in Delaware in April 2009. Our principal executive offices are located at 1601 Trinity Street, Bldg B, #3.312.09,3300 Bee Cave Road, #650-227, Austin, TX 78712,78746, and our telephone number is (877) 774-4679.
Our website address is www.genprex.com. The contents of, or information accessible through,On our website, are not partinvestors can obtain, free of this Annual Report on Form 10-K, and our website address is included in this document as an inactive textual reference only. We make our filings with the U.S. Securities and Exchange Commission (“SEC”), includingcharge, copies of our Annual Report on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, our Code of Business Conduct and allEthics, including disclosure related to any amendments or waivers thereto, other reports and any amendments thereto filed or furnished pursuant to those reports, available freeSection 13(a) or 15(d) of charge on our websitethe Exchange Act of 1934, as amended, as soon as reasonably practicable after we file such reportsmaterial electronically with, or furnish such reportsit to, the SEC. The public may readSecurities and copy the materials we file with the SEC at the SEC’s Public Reference Room at 100 F Street, NE, Washington, DC 20549. The public may obtain information on the operationExchange Commission (“SEC”). None of the Public Reference Roominformation posted on our website is incorporated by calling the SEC at 1-800-SEC-0330.reference into this Annual Report on Form 10-K. Additionally, the SEC maintains an internet site that contains reports, proxy and information statements and other information.information regarding us and other issuers that file electronically with the SEC. The address of the SEC’s website is www.sec.gov. The information contained in the SEC’s website is not intended to be a part of this filing.
We have proprietary rights to a number of trademarks, including GENPREX, ONCOPREX and REQORSA, that are used in this Annual Report on Form 10-K. Solely for convenience, the trademarks and trade names in this Annual Report on Form 10-K are generally referred to without the ® andor ™ symbols, but such references should not be construed as any indicator that their respective owners will not assert, to the fullest extent under applicable law, their rights thereto. All other trademarks, trade names and service marks appearing in this Annual Report on Form 10-K are the property of their respective owners.
We qualify as an “emerging growth company” as the term is used in The Jumpstart Our Business Startups Act of 2012 ("JOBS Act"), and therefore, we may take advantage of certain exemptions from various public company reporting requirements, including:
RISK FACTOR SUMMARY
|
Our business is subject to significant risks and uncertainties that make an investment in us speculative and risky. Below we summarize what we believe are the principal risk factors but these risks are not the only ones we face, and you should carefully review and consider the full discussion |
|
|
|
|
|
|
We will remain an “emerging growth company” until the earliest of (i) the last day of the fiscal year in which we have total annual gross revenues of $1.07 billion or more; (ii) the last day of our fiscal year followingrisk factors in the fifth anniversary ofsection titled “Risk Factors,” together with the date of our initial public offering; (iii) the date on which we have issued more than $1.0 billion in nonconvertible debt during the previous three years; or (iv) the date on which we are deemed to be a large accelerated filer under the rules of the SEC. We may choose to take advantage of some, but not all, of the available benefits of the JOBS Act. We have taken advantage of some of the reduced reporting requirementsother information in this Annual Report on Form 10-K. Accordingly,If any of the information contained herein mayfollowing risks actually occurs (or if any of those listed elsewhere in this Annual Report on Form 10-K occur), our business, reputation, financial condition, results of operations, revenue, and future prospects could be different than the information you receive from other public companies in which you hold stock. In addition, the JOBS Act providesseriously harmed. Additional risks and uncertainties that an emerging growth company can delay adopting newwe are unaware of, or revised accounting standards until such time as those standards apply to private companies. We have irrevocably elected not to avail ourselves of this exemption from new or revised accounting standards and, therefore,that we will be subject to the same new or revised accounting standards as other public companies thatcurrently believe are not emerging growth companies.material, may also become important factors that adversely affect our business.
Risks Related to Our Operations and Need for Additional Capital
| ● | We will require substantial additional funding, which may not be available to us on acceptable terms, or at all, and, if not so available, may require us to delay, limit, reduce or cease our operations. | |
| ● | Our recurring losses from operations have raised substantial doubt regarding our ability to continue as a going concern. | |
● | We have never been profitable, we have no products approved for commercial sale, and to date we have not generated any revenue from product sales. As a result, our ability to reduce our losses and reach profitability is unproven, and we may never achieve or sustain profitability. |
● | We have a limited operating history and we expect a number of factors to cause our operating results to fluctuate on an annual basis, which may make it difficult to predict our future performance. | |
| ● | Our ability to utilize our net operating loss carryforwards may be limited, resulting in income taxes sooner than currently anticipated. |
Risks Related to Development and Commercialization of Our Current and Future Product Candidates
● | Our success depends greatly on the success of our development of REQORSA for the treatment of NSCLC and SCLC, and our other product candidates, including GPX-002 for the treatment of diabetes. | |
| ● | If we are unable to secure contract manufacturers with capabilities to produce the products that we require, we could experience delays in conducting our planned clinical trials. | |
| ● | Negative public opinion and increased regulatory scrutiny of gene therapy and genetic research may damage public perception of our current and potential product candidates or adversely affect our ability to conduct our business or obtain regulatory approvals for our current and potential product candidates. | |
| ● | Conducting successful clinical studies may require the enrollment of large numbers of patients, and suitable patients may be difficult to identify and recruit. | |
| ● | Delays in the commencement, enrollment and completion of clinical trials could result in increased costs to us and delay or limit our ability to obtain regulatory approval for REQORSA and other current or future product candidates. | |
| ● | Fast track designation of our products by FDA and designation under any other FDA expedited development program may not actually lead to a faster development or regulatory review or approval process, nor will it assure FDA approval of our product candidates. | |
| ● | We may not be able to obtain or maintain orphan drug designation or exclusivity for our product candidates. |
| ● | A product candidate can fail at any stage of preclinical and clinical development. | |
● | REQORSA®, GPX-002, and any other product candidate we advance through clinical trials may not have favorable results in later clinical trials or receive regulatory approval. Even if we complete the necessary clinical trials, we cannot predict when, or if, we will obtain regulatory approval to commercialize our product candidates, and the approval may be for a narrower indication than we seek. | |
| ● | Even if we obtain regulatory approval of our current and future product candidates, the products may not gain market acceptance among physicians, patients, hospitals, treatment centers, third-party payors and others in the medical community. | |
| ● | REQORSA®, GPX-002, and other current or future product candidates may have undesirable side effects that may delay or prevent marketing approval, or, if approval is received, require them to be taken off the market, require them to include safety warnings or otherwise limit their sales. | |
| ● | If a product liability claim is successfully brought against us for uninsured liabilities, or such claim exceeds our insurance coverage, we could be forced to pay substantial damage awards that could materially harm our business. | |
| ● | Security breaches and other disruptions could compromise our information and expose us to liability, which would cause our business and reputation to suffer. | |
| ● | We face risks related to health epidemics and outbreaks, including COVID-19, which could significantly disrupt our preclinical studies and clinical trials. Our business has previously been adversely affected by the coronavirus pandemic which delayed our clinical trials and disrupted our supply chain. A potential resurgence of COVID-19, or any future disease outbreak, epidemic or pandemic, could disrupt our clinical trials and supply chain and materially adversely affect our business and operations. | |
| ● | We face competition from other biotechnology and pharmaceutical companies, particularly those that are gene therapy companies, and our operating results will suffer if we fail to compete effectively. |
Risks Related to Regulatory Approval and Marketing of Our Current and Future Product Candidates and Other Legal Compliance Matters
| ● | We cannot provide assurance that REQORSA, GPX-002, or any of our other current or future product candidates will receive regulatory approval, and without regulatory approval we will not be able to market them. | |
● | Even if we obtain regulatory approval for our product candidates, our products will remain subject to regulatory oversight. |
● | If the FDA does not find the manufacturing facilities of our current or future contract manufacturers acceptable for commercial production, we may not be able to commercialize REQORSA, GPX-002, or any of our other current or future product candidates. |
● | We may be subject, directly or indirectly, to federal and state healthcare fraud and abuse laws, false claims laws and other federal and state healthcare laws, and the failure to comply with such laws could result in substantial penalties. Our employees, independent contractors, consultants, principal investigators, CROs, commercial partners and vendors may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements. |
● | Coverage and reimbursement may be limited or unavailable in certain market segments for REQORSA, GPX-002, and our other current or future product candidates, if approved, which could make it difficult for us to sell REQORSA, GPX-002, and our other current or future product candidates profitably. | |
| ● | Concerns about gene therapy, genetic testing, and genetic research could result in new and/or additional government regulations and requirements that restrict or prohibit the processes we use or delay or prevent the regulatory approval of our current and potential product candidates. | |
| ● | Healthcare legislative reform measures may have a material adverse effect on our business and results of operations. | |
| ● | We are subject to a variety of risks associated with international operations which could materially adversely affect our business. | |
| ● | If we fail to comply with environmental, health and safety laws and regulations, we could become subject to fines or penalties or incur costs that could have a material adverse effect on the success of our business. |
Risks Related to Our Dependence on Third Parties
● | We may not be successful in establishing and maintaining development and commercialization collaborations, which could adversely affect our ability to develop our current and future product candidates and our financial condition and operating results could be adversely affected. |
● | We rely, in part, and expect to continue to rely, in part, on third parties to conduct, supervise and monitor our clinical trials, and if these third parties perform in an unsatisfactory manner, it may harm our business. |
| ● | We rely, and expect to continue to rely, on third parties to distribute, manufacture and perform release testing for our current and future product candidates and other key materials and if such third parties do not carry out their contractual duties or meet expected deadlines, we may not be able to obtain regulatory approvals for our product candidates. | |
| ● | We have completed and may in the future complete related party transactions that were not and may not be conducted on an arm’s length basis. | |
● | Disruptions in the global economy and supply chains may have a material adverse effect on our business, financial condition and results of operations. |
Risks Related to Our Intellectual Property
● | If we fail to comply with obligations pursuant to our license agreements, we could lose intellectual property and other rights that are important to our business; if we fail to obtain licenses to advance our research and development that may be required we may be unable to develop the affected product exclusively, on acceptable terms or at all. |
● | The intellectual property rights we have licensed from MD Anderson and the UP are subject to the rights of the U.S. government. |
● | If we are unable to protect our intellectual property rights or if our intellectual property rights are inadequate for our technology and product candidates, our competitive position could be harmed. |
● | Third-party claims of intellectual property infringement may prevent or delay our development and commercialization efforts. |
● | We may not be successful in obtaining or maintaining necessary rights to product components and processes for our development pipeline through acquisitions and in-licenses. |
● | Confidentiality agreements with employees and others may not adequately prevent disclosure of trade secrets and other proprietary information and may not adequately protect our intellectual property. |
● | Our reliance on third parties may require us to share our trade secrets, which increases the possibility that a competitor will discover them or that our trade secrets will be misappropriated or disclosed. |
● | We may be involved in lawsuits to protect or enforce our patents or the patents of our licensors, which could be expensive, time-consuming and unsuccessful. |
● | Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements. |
● | Issued patents covering our product candidates could be found invalid or unenforceable if challenged in court. |
● | We may be subject to claims that our employees, consultants or independent contractors have wrongfully used or disclosed confidential information of third parties or that our employees have wrongfully used or disclosed alleged trade secrets of their former employers. |
● | We may be subject to claims challenging the inventorship or ownership of our patents and other intellectual property. |
● | Changes in U.S. patent law could diminish the value of patents in general, thereby impairing our ability to protect our products. |
● | We have in the past and may again in the future have trademark applications in the United States and/or certain other countries, and failure to secure these registrations could adversely affect our business; additionally, we may need to enforce our trademark rights against third parties and expend significant resources to enforce such rights against infringement. |
● | We may not be able to protect our intellectual property rights throughout the world. |
Risks Related to Employee Matters and Managing Growth
● | We have no sales, marketing or distribution experience and we will have to invest significant resources to develop those capabilities or enter into acceptable third-party sales and marketing arrangements. |
● | We may not be able to manage our business effectively if we are unable to attract and retain key personnel and consultants. |
● | We may use our financial and human resources to pursue a particular research program or product candidate and fail to capitalize on programs or product candidates that may be more profitable or for which there is a greater likelihood of success. |
● | If we engage in future acquisitions or strategic partnerships, this may increase our capital requirements, dilute our stockholders, cause us to incur debt or assume contingent liabilities, and subject us to other risks. |
Risks Related to our Securities
● | The market price of our common stock may be highly volatile, and you may lose all or part of your investment. |
● | An active, liquid and orderly market for our common stock may not be sustained, and you may not be able to sell your common stock. |
● | We are currently listed on The Nasdaq Capital Market. If we are unable to maintain listing of our securities on Nasdaq or any stock exchange, our stock price could be adversely affected and the liquidity of our stock and our ability to obtain financing could be impaired and it may be more difficult for our stockholders to sell their securities. |
● | Unstable market and economic conditions may have serious adverse consequences on our business, financial condition and stock price. |
● | Failure to maintain effective internal control over our financial reporting in accordance with Section 404 of the Sarbanes-Oxley Act of 2002, as amended (“Sarbanes-Oxley Act”) could cause our financial reports to be inaccurate. |
● | We have no intention of declaring dividends in the foreseeable future. |
● | We will likely incur increased costs and devote additional management time to public company reporting and compliance obligations as a result of exiting “emerging growth company” status. |
● | Sales of a substantial number of shares of our common stock in the public market could cause our stock price to fall. |
● | Future sales and issuances of our securities could result in additional dilution of the percentage ownership of our stockholders and could cause our share price to fall. |
● | If securities or industry analysts do not publish research or reports about us, or if they adversely change their recommendations regarding our common stock, then our stock price and trading volume could decline. |
● | Certain provisions in our organizational documents could enable our board of directors to prevent or delay a change of control. |
● | Our Amended and Restated Certificate of Incorporation and Amended and Restated Bylaws contain an exclusive forum provision with respect to certain actions which may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable and discourage lawsuits against us or our current or former directors or officers and/or stockholders in such capacity. |
General Risk Factors
● | Obligations associated with being a public company in the United States are expensive and time-consuming, and our management will be required to devote substantial time to compliance matters. |
● | Business disruptions could seriously harm our future revenue and financial condition and increase our costs and expenses. |
● | We may be at risk of securities class action litigation. |
An investment in our common stock involves a high degree of risk. You should carefully consider the following risk factors and the other information in this Annual Report on Form 10-K before investing in our common stock. Our business and results of operations couldcould be seriously harmed by any of the following risks. The risks set out below are not the only risks we face. Additional risks and uncertainties not currently known to us or that we currently deem to be immaterial also may materially adversely affect our business, financial condition and/or operating results. If any of the following events occur, our business, financial condition and results of operations could be materially adversely affected. In such case, the value and trading price of our common stock could decline, and you may lose all or part of your investment.
Risks Related to Our Financial PositionOperations and Need for Additional Capital
We will require substantial additional funding, which may not be available to us on acceptable terms, or at all, and, if not so available, may require us to delay, limit, reduce or cease our operations.
We are using the proceeds from our recent sales of securities to advance REQORSA through clinical development, and to advance our other preclinical development programs as well as for other corporate purposes. Developing pharmaceutical products, including conducting preclinical studies and clinical trials, is expensive. We will require substantial additional future capital to complete clinical development and commercialize REQORSA and for preclinical and clinical development and commercialization of our gene therapy for diabetes, GPX-002.GPX-002, and our other product candidates. If the FDA requires that we perform additional preclinical studies or clinical trials beyond what we currently anticipate, our expenses will further increase beyond what we currently expect and the anticipated timing of any potential approval of REQORSA, GPX-002, and GPX-002our other current or future product candidates would likely be delayed. Furthermore, there can be no assurance that the costs to obtain regulatory approval of these product candidates will not increase.
We will continue to require substantial additional capital to continue our preclinical and clinical development and commercialization readiness activities. Because successful development of our current and potential product candidates is uncertain, we are unable to estimate the actual amount of funding we will require to complete research and development and commercialize our current and future product candidates.
The amount and timing of our future funding requirements will depend on many factors, including, but not limited to:
the progress, costs, results and timing of our preclinical development and clinical trials for REQORSA and GPX-002;
the outcome, costs and timing of seeking and obtaining FDA and any other regulatory approvals;
the ability of third parties to deliver materials and provide services for us;
the costs associated with securing and establishing commercialization and manufacturing capabilities;
market acceptance of our current and future product candidates;
the costs of acquiring, licensing or investing in businesses, products, product candidates and technologies;
our ability to maintain, expand and enforce the scope of our intellectual property portfolio, including the amount and timing of any payments we may be required to make, or that we may receive, in connection with the licensing, filing, prosecution, defense and enforcement of any patents or other intellectual property rights;
our need and ability to hire additional management and scientific and medical personnel;
the effect of competing drug candidates and new product approvals;
our need to implement additional internal systems and infrastructure, including financial and reporting systems; and
the economic and other terms, timing of and success of our existing licensing arrangements and any collaboration, licensing or other arrangements into which we may enter in the future.
● | the progress, costs, results and timing of our preclinical development and clinical trials for REQORSA, GPX-002, and other current or future product candidates; |
● | the outcome, costs and timing of seeking and obtaining FDA and any other regulatory approvals; |
● | the ability of third parties to deliver materials and provide services for us; |
● | the costs associated with securing and establishing commercialization and manufacturing capabilities; |
● | market acceptance of our current and future product candidates; |
● | the costs of acquiring, licensing or investing in businesses, products, product candidates and technologies; |
● | our ability to obtain, maintain, expand and enforce intellectual property rights for our products and product candidates, including the amount and timing of any payments we may be required to make, or that we may receive, in connection with the licensing, filing, prosecution, defense and enforcement of any patents or other intellectual property rights; |
● | our need and ability to hire additional management and scientific and medical personnel; |
● | the effect of competing drug candidates and new product approvals; |
● | our need to implement additional internal systems and infrastructure, including financial and reporting systems; and |
● | the economic and other terms, timing of and success of our existing licensing arrangements and any collaboration, licensing or other arrangements into which we may enter in the future. |
Some of these factors are outside of our control. Although we expect that our existing cash and marketable securities will be sufficient to fund our current operations and planned clinical trial activities into the third quarter of 2024, this period could be shortened if there are any significant increases in planned spending on current or additional development programs or more rapid progress of these development programs than anticipated. Furthermore, we believe that our existing capital will not be sufficient to enable us to complete the development and commercialization of REQORSA, GPX-002, and our other current or GPX-002.future product candidates. Accordingly, we expect that we will need to raise additional funds in the future.
We have raised and may continue to raise capital through a variety of sources that may be available to us. On March 1, 2023, we completed a registered direct offering, in which we sold to an accredited healthcare-focused institutional investor an aggregate of (i) 95,239 shares of our common stock and (ii) warrants to purchase up to 95,239 shares of our common stock, at a combined offering price of $42.00 per share of common stock and accompanying warrant, for net proceeds of approximately $3.6 million. On July 21, 2023, we completed a registered direct offering priced at the market under Nasdaq rules, in which we sold to accredited healthcare-focused institutional investors an aggregate of (i) 185,644 shares of our common stock, and (ii) warrants to purchase up to 185,644 shares of our common stock, at a combined offering price of $40.40 per share of common stock and accompanying warrant, for net proceeds of approximately $6.7 million. On December 13, 2023, we entered into an At The Market (“ATM”) Offering Agreement (the “Agreement”) with H.C. Wainwright & Co., LLC, serving as agent (“H.C. Wainwright” or the “Agent”) with respect to an at-the-market offering program under which we may offer and sell through the Agent, from time to time at our sole discretion, up to such number or dollar amount of shares of our common stock (the “Shares”) as registered on the prospectus supplement covering the ATM offering, as may be amended or supplemented from time to time. We have agreed to pay the Agent a commission equal to three percent (3%) of the gross sales proceeds of any Shares sold through the Agent under the Agreement, and also have provided the Agent with customary indemnification and contribution rights. As of December 31, 2023 we had not sold any Shares through the Agent under the Agreement. From January 1, 2024 through the date of filing of this Annual Report on Form 10-K, we have sold 158,474 Shares for net proceeds to us totaling $881,946 through the Agent under the Agreement. On March 21, 2024, we completed a registered direct offering, in which we sold to an institutional investor an aggregate of (i) 165,000 shares of our common stock, (ii) March 2024 Pre-Funded Warrants exercisable for up to an aggregate of 1,377,112 shares of our common stock, and (iii) March 2024 Common Warrants exercisable for up to an aggregate of 1,542,112 shares of our common stock, at a combined offering price of $4.215 per share of common stock (or $4.2149 per March 2024 Pre-Funded Warrant in lieu thereof) and accompanying March 2024 Common Warrant, for net proceeds of approximately $5.8 million. In connection with the March 2024 registered direct offering, we amended certain existing warrants to reduce the exercise price and extend the term thereof as discussed above in Part I, Item 1 of this Annual Report in the section captioned Recent Developments – March 2024 Registered Direct Offering”.
We may seek additional funding through a combination of equity offerings, drawdowns on our ATM pursuant to our At The Market Offering Agreement with H.C. Wainwright as Agent, debt financings, government or other third-party funding, commercialization, marketing and distribution arrangements and other collaborations, strategic alliances and licensing arrangements, some of which may require us to relinquish rights to some of our technologies or product candidates or otherwise agree to terms unfavorable to us. Additional funding may not be available to us on acceptable terms, if at all. In addition, the terms of any financing may adversely affect the holdings or the rights of our stockholders. Any new equity securities we issue could have rights, preferences, and privileges superior to those of holders of our existing capital stock. In addition, the issuance of additional shares by us may cause the market price of our shares to decline and result in dilution to our stockholders. Any debt financing secured by us in the future could involve restrictive covenants relating to our capital-raising activities and other financial and operational matters, which may make it more difficult for us to obtain additional capital and to pursue business opportunities.
If we are unable to obtain funding on a timely basis, we may be required to significantly curtail one or more of our researchpreclinical or clinical development programs, our ability to continue to support our business growth and to respond to business challenges could be significantly limited, and we may be required to curtail or cease operations. Accordingly, our business may fail, in which case you would lose the entire amount of your investment in our securities.
Our recurring losses from operations have raised substantial doubt regarding our ability to continue as a going concern.
We have recognized recurring losses, and as of December 31, 2023, had an accumulated deficit of approximately $133.9 million. We anticipate operating losses to continue for the foreseeable future due to, among other things expenses related to ongoing activities to research, develop and commercialize our product candidates. We expect the cash and cash equivalents of approximately $6.7 million at December 31, 2023 to be insufficient to meet our operating and capital requirements at least 12 months from the filing of this Annual Report on Form 10-K. Our forecast of the period of time through which our current financial resources will be adequate to support our operations and the costs to support our general and administrative and research and development activities are forward-looking statements and involve risks and uncertainties. The financial statements do not include any adjustments that might be necessary should we be unable to continue as a going concern.
As further described below, our ability to continue as a going concern is dependent on our ability to raise additional working capital through public or private equity or debt financings or other sources, which may include collaborations with third parties as well as disciplined cash spending. Should we be unable to raise sufficient additional capital, we may be required to undertake cost-cutting measures including delaying or discontinuing certain clinical activities. The sale of equity and convertible debt securities may result in dilution to our stockholders and certain of those securities may have rights senior to those of our common stock. If we raise additional funds through the issuance of preferred stock, convertible debt securities or other debt financing, these securities or other debt could contain covenants that would restrict our operations. Any other third-party funding arrangement could require us to relinquish valuable rights.
The source, timing and availability of any future financing will depend principally upon market conditions, and, more specifically, on the progress of our clinical development programs. Funding may not be available when needed, at all, or on terms acceptable to us. Lack of necessary funds may require us, among other things, to delay, scale back or eliminate some or all of our planned clinical trials. These factors among others create a substantial doubt about our ability to continue as a going concern.
We have never been profitable, we have no products approved for commercial sale, and to date we have not generated any revenue from product sales. As a result, our ability to reduce our losses and reach profitability is unproven, and we may never achieve or sustain profitability.
We have never been profitable and do not expect to be profitable in the foreseeable future. We have not yet submitted any drug candidates for approval by regulatory authorities in the United States or elsewhere. From our inception on April 1, 2009, to December 31, 2020,2023, we incurred an accumulated deficit of approximately $58.4$133.9 million. We incurred net losses of approximately $17.9$31.0 million and approximately $10.7$23.7 million for the years ended December 31, 20202023 and 2019,2022, respectively.
To date, we have devoted most of our financial resources to our corporate overhead and research and development, including our preclinical development activities, manufacturing processes and clinical trials. We have not generated any revenues from product sales. We expect to continue to incur losses for the foreseeable future, and we expect these losses to increase as we continue our development of, and seek regulatory approvals for, our current and potential product candidates, prepare for and begin the commercialization of any approved products, and add infrastructure and personnel to support our continuing product development efforts. We anticipate that any such losses could be significant for the next several years. If REQORSA, GPX-002, or any of our other potentialcurrent or future product candidates failsfail in clinical trials or does not gain regulatory approval, or if our drug candidates do not achieve market acceptance, we may never become profitable. These net losses and negative cash flows have had, and will continue to have, an adverse effect on our stockholders’ equity and working capital.
Because of the numerous risks and uncertainties associated with pharmaceutical product development, we are unable to accurately predict the timing or amount of increased expenses or if, or when, we will be able to achieve profitability. In addition, our expenses could increase if we are required by the FDA to perform studies or trials in addition to those currently expected, or if there are any delays in completing our clinical trials or the development of any of our drug candidates. The amount of future net losses will depend, in part, on the rate of future growth of our expenses and our ability to generate revenues.
We have a limited operating history and we expect a number of factors to cause our operating results to fluctuate on an annual basis, which may make it difficult to predict our future performance.
We are a clinical stage company with a limited operating history. Our operations to date have been limited to conducting clinical and preclinical research.research and development. We have not yet obtained any regulatory approvals for any of our drug candidates. Consequently, any predictions made about our future success or viability may not be accurate. Our operating results are expected to significantly fluctuate from quarter to quarter and year to year due to a variety of factors, many of which are beyond our control. Factors relating to our business that may contribute to these fluctuations include:
any delays in regulatory review and approval (assuming that our data support approval) of our current and potential product candidates in clinical development, including our ability to receive approval from the FDA for REQORSA or GPX-002;
delays in the commencement, enrollment and timing of clinical trials;
the success of our clinical trials through all phases of clinical development;
potential side effects of our current and potential product candidates that could delay or prevent approval or cause an approved drug to be taken off the market;
our ability to obtain additional funding to develop our current and future product candidates;
our identification and development of additional drug candidates beyond REQORSA and GPX-002;
competition from existing products or new products that continue to emerge;
the ability of patients or healthcare providers to obtain coverage or sufficient reimbursement for our products;
our ability to adhere to clinical trial requirements directly or with third parties such as contract research organizations ("CROs");
our dependency on third-party manufacturers to manufacture our products and key ingredients;
our ability to establish or maintain collaborations, licensing or other arrangements, particularly with MD Anderson and UP;
our ability to defend against any challenges to our intellectual property including claims of patent infringement;
our ability to enforce our intellectual property rights against potential competitors;
our ability to secure additional intellectual property protection for our product candidates and associated technologies;
our ability to attract and retain key personnel to manage our business effectively; and
potential product liability claims.
| ● | any delays in regulatory review and approval (assuming that our data support approval) of our current and future product candidates in clinical development, including our ability to receive approval from the FDA for REQORSA or GPX-002; |
| ● | delays in the commencement, enrollment and timing of clinical trials; |
● | the success of our current and future product candidates through all phases of preclinical and clinical development, including the ability of our third-party suppliers or manufacturers to supply or manufacture our products on a timely, consistent basis in a manner sufficient and appropriate as is commensurate to meet our clinical trial timing, courses of treatment, and other requisite fulfillment considerations necessary to adequately advance our development programs; |
● | potential side effects of our current and future product candidates that could delay or prevent approval or cause an approved drug to be taken off the market; |
| ● | our ability to obtain additional funding to develop our current and future product candidates; |
| ● | our identification and development of additional drug candidates beyond REQORSA, GPX-002, and our other current product candidates; |
| ● | competition from existing products or new products that continue to emerge; |
| ● | the ability of patients or healthcare providers to obtain coverage or sufficient reimbursement for our products; |
| ● | our ability to adhere to clinical trial requirements directly or with third parties such as contract research organizations ("CROs"); |
| ● | our dependency on third-party suppliers or manufacturers to manufacture our key ingredients and/or raw materials, products and/or product components and successfully carry out a sustainable, reproducible and scalable manufacturing process in accordance with specifications or applicable regulations; |
| ● | our ability to establish or maintain collaborations, licensing, sponsored research or other arrangements, particularly with MD Anderson and UP and otherwise relating to REQORSA, and GPX-002; |
| ● | our ability to defend against any challenges to our intellectual property including claims of patent infringement; |
| ● | our ability to enforce our intellectual property rights against potential competitors; |
| ● | our ability to secure additional intellectual property protection for our product candidates and associated technologies as may be required or desirable as the development of the product candidates progresses; |
| ● | our ability to attract and retain key personnel to manage our business effectively; and |
| ● | potential product liability claims. |
Our ability to utilize our net operating loss carryforwards may be limited, resulting in income taxes sooner than currently anticipated.
As of December 31, 2023, we had federal net operating loss carryforwards (“NOLs”) of approximately $80.5 million for federal income tax purposes of which approximately $1.3 million will begin to expire in 2030 and approximately $79.2 million can be carried forward indefinitely. These NOLs may be used to offset future taxable income, to the extent we generate any taxable income, and thereby reduce or eliminate our future federal income taxes otherwise payable. Section 382 of the Internal Revenue Code of 1986, as amended (the “Code”), imposes limitations on a corporation’s ability to utilize NOLs if it experiences an ownership change as defined in Section 382. In general terms, an ownership change may result from transactions increasing the ownership of certain stockholders in the stock of a corporation by more than 50% over a three-year period. In the event that an ownership change has occurred, or were to occur, utilization of our NOLs would be subject to an annual limitation under Section 382 determined by multiplying the value of our stock at the time of the ownership change by the applicable long-term tax-exempt rate as defined in the Code. Any unused annual limitation may be carried over to later years. We may be found to have experienced an ownership change under Section 382 as a result of events in the past or the issuance of shares of common stock in the future. If so, the use of our NOLs, or a portion thereof, against our future taxable income may be subject to an annual limitation under Section 382, which may result in expiration of a portion of our NOLs before utilization.
The utilization of our NOLs may also be limited under state laws. In addition, under the 2017 Tax Cuts and Jobs Act (the “TCJA”), tax losses generated in taxable years beginning after December 31, 2017 may be utilized to offset no more than 80% of taxable income annually. This change may require us to pay federal income taxes in future years despite generating a loss for federal income tax purposes. There is also a risk that due to regulatory changes, such as suspensions on the use of NOLs, or other unforeseen reasons, our existing NOLs could expire or otherwise be unavailable to offset future income tax liabilities. For these reasons, we may not be able to realize a tax benefit from the use of our NOLs, whether or not we attain profitability.
Risks Related to Development and Commercialization of Our Current and Future Product Candidates
Our success depends greatly on the success of our development of REQORSA for the treatment ofNSCLC and SCLC, and ourother product candidates, including GPX-002 for the treatment of diabetes.
At this time, we are actively pursuing the development of REQORSA for NSCLC through our Acclaim-1 and GPX-002, aAcclaim-2 clinical trials and for SCLC through our Acclaim-3 clinical trial, which are all currently enrolling patients. We are also pursuing the development of preclinical stage gene therapy GPX-002 for diabetes. WeType 1 and Type 2 diabetes, as well as earlier discovery programs. In particular, we are dependent on the success of REQORSA in the near term.and GPX-002. We cannot provide you any assurance that we will be able to successfully advance REQORSA, GPX-002 or GPX-002any of our other current or future product candidates through the development process, or that any development problems we experience in the future will not cause significant delays or unanticipated costs, or that such development problems can be solved. We may also experience delays in developingthe development of a sustainable, reproducible and scalable manufacturing process or transferring that process to commercial partners, or developing or validating product release assays in a timely manner, which may prevent us from completing our clinical trials or commercializing our products on a timely or profitable basis, if at all. Immunotherapy, gene therapy and biopharmaceutical product development are highly speculative undertakings and involve a substantial degree of uncertainty. Because REQORSA, GPX-002 and our other potentialcurrent product candidates are based upon novel technology, it is difficult to predict whether, either as stand-alone therapies or in combination with other drugs, they will show consistently favorable results and to predict the time and cost of their development and of subsequently obtaining regulatory approval. We believe only a few gene therapy products have been approved in the United States or Europe. We have found it difficult to enroll patients in our clinical studies in the past, have experienced certain difficulties in enrolling patients in our current trials and may continue to find it difficult in the future, find it difficult to enroll patients in our clinical studies, including our Acclaim-1 and Acclaim-2 clinical trials, which could delay or prevent clinical studies of REQORSA or other current and potentialor future product candidates. We may encounter other delays in our preclinical or clinical studies, or we may fail to demonstrate safety and efficacy to the satisfaction of FDA and other regulatory authorities. We may not be successful in our efforts to identify or discover additional product candidates, or to develop product candidates that we have identified.
In addition, the clinical trial requirements of the FDA, the European Commission, the European Medicines Agency (“EMA”), the competent authorities of the Member States of the European Union (“EU”) and other regulatory authorities and the criteria these regulators use to determine the safety and efficacy of a product candidate vary substantially according to the type, complexity, novelty and intended use and market of such product candidates. The regulatory approval process for novel product candidates such as ours can be more expensive and take longer than for other, better known or more extensively studied product candidates. Even if we are successful in developing product candidates, it is difficult to determine how long it will take or how much it will cost to obtain regulatory approvals for these product candidates in either the United States or the EU, or how long it will take to commercialize any other products for which we receive marketing approval. In addition, any future marketing authorization granted by the European Commission may not be indicative of what the FDA may require for approval and vice versa.
If we are unable to secure contract manufacturers with capabilities to produce the products that we require, we could experience delays in conducting our planned clinical trials.
Manufacturing REQORSA involves several manufacturing steps. Historically, part of our manufacturing process was conducted in manufacturing facilities at MD Anderson. We have transferred all of the steps of our manufacturing process to CDMOs and scaled-up clinical production in order to supply our Acclaim-1, Acclaim-2 and Acclaim-3 clinical trials. We also are preparing for commercial readiness for REQORSA through the development of an integrated supply chain network of manufacturing vendors and continue to work to identify cutting edge manufacturing technologies to optimize manufacturing processes and shelf life. With the advancement of the development of GPX-002, we also are working to optimize the manufacture of this product candidate and to source high quality and integrated vendors capable of producing it in accordance with GMP. Although we have contracted with CDMOs to produce our products, no assurance can be given that such CDMOs will be able to continue to produce the products that we require. In addition, the tremendous growth in the gene therapy sector has created increasing demand for the services of CDMOs with gene therapy capabilities which may impact our ability to schedule production runs of our products or product components to meet our needs on a timely basis. Furthermore, manufacturing gene-based therapies is complex and highly regulated and a CDMO with which we have contracted may fail to produce our products or product components timely or in accordance with our specifications or applicable regulations. We have experienced a variety of these challenges to varying degrees in connection with performance by our CDMOs, which have resulted in delays in our Acclaim-1, Acclaim-2 and Acclaim-3 clinical trials in the past. Changing our current or future contract manufacturers may be difficult and could be costly, which could result in our inability to manufacture our clinical product candidates and a delay in the development of our clinical product candidate. Further, in order to maintain our development timelines, any changes to, or the addition of a new, third-party contract manufacturers may result in our incurring higher costs to manufacture our clinical product candidates. Any delay in the availability of product supply or product component supply could result in a delay in our clinical trials, including our Acclaim-1, Acclaim-2 and Acclaim-3 clinical trials as well as the commencement of clinical trials for GPX-002.
Negative public opinion and increased regulatory scrutiny of gene therapy and genetic research may damage public perception of our current and potential product candidates or adversely affect our ability to conduct our business or obtain regulatory approvals for our current and potential product candidates.
Public perception may be influenced by claims that gene therapy is unsafe, and gene therapy may not gain the acceptance of the public or the medical community. In particular, our success will depend upon physicians prescribing treatments that involve the use of our product candidates in lieu of, or in addition to, existing treatments with which they are already familiar and for which greater clinical data may be available. More restrictive government regulations or negative public opinion would have a negative effect on our business or financial condition and may delay or impair the development and commercialization of our current and potential product candidates or demand for any products we may develop. Adverse events in our clinical trials, even if not ultimately attributable to our current and potential product candidates, and the resulting publicity could lead to increased governmental regulation, unfavorable public perception, potential regulatory delays in the testing or approval of our potential product candidates, stricter labeling requirements for those product candidates that are approved and a decrease in demand for any such product candidates. Concern about the environmental spread of our product, whether real or anticipated, could also hinder the commercialization of our products.
Prior to receiving REQORSA in our Acclaim-1 clinical trial, patients are required to undergo genetic screening to detect EGFR mutations and in the Acclaim-2 clinical trial genetic screening to detect PD-L1 as well as other mutations relevant to cancer.mutations. Genetic testing has raised concerns regarding the appropriate utilization and the confidentiality of information provided by genetic testing. Genetic tests for assessing a person’s likelihood of developing a chronic disease have focused public attention on the need to protect the privacy of genetic information. Genetic testing information is also subject to significant restrictions under both federal and state law. For example, concerns have been expressed that insurance carriers and employers may use these tests to discriminate on the basis of genetic information, resulting in barriers to the acceptance of genetic tests by consumers. This could lead to governmental authorities restricting genetic testing or calling for limits on or regulating the use of genetic testing, particularly for diseases for which there is no known cure. Any of the foregoing could decrease demand for REQORSA or our futureother product candidates.
Conducting successful clinical studies may require the enrollment of large numbers of patients, and suitable patients may be difficult to identify and recruit.
Patient enrollment in clinical trials and completion of patient participation and follow-up depends on many factors, including the size of the patient population; the nature of the trial protocol; the attractiveness of, or the discomforts and risks associated with, the treatments received by enrolled subjects; the availability of appropriate clinical trial investigators; support staff; and proximity of patients to clinical sites; ability to comply with the eligibility and exclusion criteria for participation in the clinical trial; and patient compliance. For example, patients may be discouraged from enrolling in our clinical trials if the trial protocol requires them to undergo extensive post-treatment procedures or follow-up to assess the safety and effectiveness of our product candidates or if they determine that the treatments received under the trial protocols are not attractive or involve unacceptable risks or discomforts. Patients may also not participate in our clinical trials if they choose to participate in contemporaneous clinical trials of competitive products.
In addition, our ability to successfully initiate, enroll and complete clinical trials in any foreign country is subject to numerous risks of conducting business in foreign countries, including:
| ● | difficulty in establishing or managing relationships with CROs and physicians;
|
| ● | different standards for the conduct of clinical trials; |
| ● | our inability to locate qualified local consultants, physicians and partners; and |
| ● | the potential burden of complying with a variety of foreign laws, medical standards and regulatory requirements, including the regulation of pharmaceutical and biotechnology products and treatments. |
If we have difficulty enrolling a sufficient number of patients to conduct our clinical trials as planned, we may need to delay, limit or terminate ongoing or planned clinical trials, any of which would have an adverse effect on our business.
Delays in the commencement, enrollment and completion of clinical trials could result in increased costs to us and delay or limit our ability to obtain regulatory approval for REQORSA and other current or futureproduct candidates.
Clinical development is very expensive and can take many years. Delays in the commencement, enrollment and/or completion of our Acclaim-1, Acclaim-2 and Acclaim-2Acclaim-3 clinical trials or any future clinical trials could increase our product development costs or delay or limit the regulatory approval of REQORSA or other product candidates. We have experienced delays in opening our clinical sites for our Acclaim-1 and Acclaim-2 trials in the past; for example, during the COVID-19 pandemic there was a delay due to a back-log in protocol review at the clinical trial sites. We do not know and cannot predict whether thefuture trials or studies of other current or future product candidates, including later stages of our Acclaim trials, and any for GPX-002, will begin as planned, if at all, and we do not know and cannot predict whether our Acclaim-1, or Acclaim-2 and Acclaim-3 clinical trials or any future trials or studies of other current or future product candidates will begin on time or will be completed on schedule, if at all. The start or end of a clinical study is oftenmay be delayed or halted due to regulatory requirements, changes in the proposed regulatory approval pathway for a drug candidate, manufacturing challenges, including delays or shortages in available raw materials required to manufacture the drug product, required clinical trial administrative actions, slower than anticipated patient enrollment, changing standards of care, availability or prevalence of use of a comparative drug or required prior therapy, clinical outcomes or financial constraints. For instance, delays or difficulties in patient enrollment or difficulties in retaining trial participants can result in increased costs, longer development times or termination of a clinical trial. Clinical trials of a new product candidate require the enrollment of a sufficient number of patients, including patients who are suffering from the disease the product candidate is intended to treat and who meet other eligibility criteria.criteria, such as mutation of the EGFR which is required for the Acclaim-1 trial and is present in a minority of NSCLC patients. Rates of patient enrollment are affected by many factors, including the size of the patient population, the eligibility criteria for the clinical trial, which include the age and condition of the patients and the stage and severity of disease, the nature of the protocol, the proximity of patients to clinical sites and the availability of effective treatments and/or availability of other investigational treatment options for the relevant disease. We are planning to initiatehave initiated our Acclaim-1, Acclaim-2 and Acclaim-2Acclaim-3 clinical trials pursuant to an existing IND. Prior to commencing either of these clinical trials, we intend to fileWe have previously filed with the FDA amendments to our IND consisting of an updated chemistry, manufacturing and controls section, and the protocol for the respective clinical trial. We cannot be sure that issues will not arise in the future in connection with the filing of thesepotential subsequent amendments or otherwise that willmight result in the FDA imposing a clinical hold which could result in the delay of either or bothany of these clinical trialstrials. For GPX-002 we have not yet filed an IND and cannot predict all of the challenges and issues that may arise in connection with the preparation and filing of an IND, or whether this IND will be filed at all.
Fast track designation of our products by FDA and designation under any other FDA expedited development program may not actually lead to a faster development or regulatory review or approval process, nor will it assure FDA approval of our product candidates.
REQORSA has received three fast track designations from the FDA. We may in the future seek additional fast track designations for our products and/or request breakthrough therapy designation, accelerated approval or priority review of applications for approval. FDA has broad discretion whether or not to grant these designations and requests, so even if we believe a particular product candidate is eligible for these designations, we cannot assure you that the FDA will grant them. Even with fast track designation and other FDA expedited development programs, we may not experience a faster development process, review or approval compared to conventional FDA procedures. The FDA may withdraw fast track designation and other expedited program designations if it believes that the requirements of the program are no longer met.
We may not be able to obtain or maintain orphan drug designation or exclusivity for our product candidates.
In August 2023, the FDA granted Orphan Drug Designation to REQORSA for the treatment of SCLC. Upon receipt of regulatory approval, orphan drug status will provide us with seven years of market exclusivity in the United States under the Orphan Drug Act. We may seek other orphan drug designations in the future in the United States and in the European Union for our product candidates. However, there is no guarantee that the FDA will grant orphan drug designation for any of our other drug candidates for any future indication, which would make us ineligible for the additional exclusivity and other benefits of orphan drug designation for those other drug candidates in the future. Moreover, there can be no assurance that another company also holding orphan drug designation for the same indication or which may receive orphan drug designation in the future will not receive approval prior to us, in which case our competitor would have the benefit of the seven years of market exclusivity, and we would be unable to commercialize our product for the same indication until the expiration of such seven-year period. Even if we are the first to obtain approval for the orphan drug indication, there are circumstances under which a competing product may be approved for the same indication during our seven-year period of exclusivity.
Under the Orphan Drug Act, the FDA may grant orphan drug designation to a drug intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making a drug available in the Unites States for this type of disease or condition will be recovered from sales of the product. Orphan drug designation must be requested before submitting an NDA or BLA. After the FDA grants orphan drug designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Orphan designation does not convey any advantage in or shorten the duration of regulatory review and approval process. In addition to the potential period of exclusivity, orphan designation makes a company eligible for grant funding of up to $400,000 per year for four years to defray costs of clinical trial expenses, tax credits for clinical research expenses and potential exemption from the FDA application user fee.
If a product that has orphan designation subsequently receives the first FDA approval for the disease or condition for which it has such designation, the product is entitled to orphan drug exclusivity, which means the FDA may not approve any other applications to market the same drug for the same indication for seven years, except in limited circumstances, such as (i) the drug’s orphan designation is revoked; (ii) its marketing approval is withdrawn; (iii) the orphan exclusivity holder consents to the approval of another applicant’s product; (iv) the orphan exclusivity holder is unable to assure the availability of a sufficient quantity of drug; or (v) a showing of clinical superiority to the product with orphan exclusivity by a competitor product. If a drug designated as an orphan product receives marketing approval for an indication broader than what is designated, it may not be entitled to orphan drug exclusivity. There can be no assurance that we will receive orphan drug designation for any of our drug candidates for any additional indications, if we elect to seek such designation. Even if orphan designation is granted it may be withdrawn by the FDA for non-compliance with regulations.
The accelerated approval pathway for our product candidates may not lead to a faster development or regulatory review or approval process and does not increase the likelihood that our product candidates will receive marketing approval.
Under the FDA’s accelerated approval program, the FDA may approve a drug for a serious or life-threatening illness that provides meaningful therapeutic benefit to patients over existing treatments based upon a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. We may seek accelerated approval for one or more of our product candidates on the basis of progression free survival, a surrogate endpoint that we believe is reasonably likely to predict clinical benefit.
For drugs granted accelerated approval, post-marketing confirmatory trials are required to describe the anticipated effect on irreversible morbidity or mortality or other clinical benefit. These confirmatory trials must be completed with due diligence and, in most cases, the FDA may require that the trial be designed, initiated, and/or fully enrolled prior to approval. If any of our competitors were to receive full regulatory approval for an indication for which we are seeking accelerated approval prior to our approval, the indication we are seeking may no longer qualify as a condition for which there is an unmet medical need and accelerated approval of our product candidate would be more difficult or may not occur. Moreover, the FDA may withdraw approval of our product candidate approved under the accelerated approval pathway if, for example:
the trial or trials required to verify the predicted clinical benefit of our product candidate fail to verify such benefit or do not demonstrate sufficient clinical benefit to justify the risks associated with the drug;
other evidence demonstrates that our product candidate is not shown to be safe or effective under the conditions of use;
If we are unable to secure contract manufacturers with capabilities to produce the products that we require, we could experience delays in conducting our planned clinical trials.
Manufacturing REQORSA involves several manufacturing steps. Historically, part of our manufacturing process was conducted in manufacturing facilities at MD Anderson. We recently transferred this portion of the process to CDMOs and also scaled-up clinical production in quantities we believe are sufficient for our Acclaim-1 and Acclaim-2 clinical trials. Although we have contracted with CDMOs to produce the products that we require, no assurance can be given that such CDMOs will be able to continue to produce the products that we require. In accordance with cGMP, changing manufacturers may require the re-validation of manufacturing processes and procedures, and may require further preclinical studies or clinical trials to show comparability between the materials produced by different manufacturers. Changing our current or future contract manufacturers may be difficult and could be costly, which could result in our inability to manufacture our clinical product candidates and a delay in the development of our clinical product candidate. Further, in order to maintain our development timelines in the event of a change in a third-party contract manufacturer, we may incur higher costs to manufacture our clinical product candidates.
A product candidate can fail at any stage of preclinical and clinical development.
The historical failure rate for product candidates is high due to scientific feasibility, safety, efficacy, changing standards of medical care and other variables. The results from preclinical testing or early clinical trials of a product candidate may not predict the results that will be obtained in later phase clinical trials of the product candidate. We, the FDA or other applicable regulatory authorities may suspend clinical trials of a product candidate at any time for various reasons, including, but not limited to, a belief that subjects participating in such trials are being exposed to unacceptable health risks or adverse side effects, or other adverse initial experiences or findings. We may not have the financial resources to continue development of, or to enter into collaborations for, a product candidate if we experience any problems or other unforeseen events that delay or prevent regulatory approval of, or our ability to commercialize, product candidates, including:
inability to obtain sufficient funds required for a clinical trial;
inability to reach agreements on acceptable terms with current or prospective CROs and trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites;
negative or inconclusive results from our clinical trials or the clinical trials of others for product candidates similar to ours, leading to a decision or requirement to conduct additional preclinical testing or clinical trials or abandon a program;
serious and unexpected side effects experienced by subjects in our clinical trials or by individuals using drugs similar to our current and future product candidates;
conditions imposed by the FDA or comparable foreign authorities regarding the scope or design of our clinical trials;
delays in enrolling research subjects in clinical trials;
high drop-out rates and high fail rates of research subjects;
inadequate supply or quality of product candidate components or materials or other supplies necessary for the conduct of our clinical trials;
greater than anticipated clinical trial costs;
poor effectiveness of our current and potential product candidates during clinical trials; or
unfavorable FDA or other regulatory agency inspection and review of a clinical trial site or vendor.
| ● | inability to obtain sufficient funds required for clinical development; |
| ● | inability to reach agreements on acceptable terms with current or prospective vendors, CROs and trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different vendors, CROs and trial sites; |
| ● | negative or inconclusive results from our clinical studies or the clinical studies of others for product candidates similar to ours, leading to a decision or requirement to conduct additional preclinical testing or clinical trials or abandon a program; |
| ● | serious and unexpected side effects experienced by subjects in our clinical trials or by individuals using drugs similar to our current and future product candidates; |
| ● | conditions imposed by the FDA or comparable foreign authorities regarding the scope or design of our clinical trials; |
| ● | unexpected results from preclinical testing and development; | |
| ● | inability or delays in enrolling research subjects in clinical trials; |
| ● | high drop-out rates and high fail rates of research subjects; |
| ● | inadequate supply or quality of product candidate components or materials or other supplies necessary for the conduct of preclinical or clinical testing; |
| ● | greater than anticipated clinical trial costs; |
| ● | poor effectiveness of our current and potential product candidates during clinical trials; or |
| ● | unfavorable FDA or other regulatory agency inspection and review of a clinical trial site or vendor. |
REQORSA®™ , GPX-002, and any other product candidate we advance through clinical trials may not have favorable results in later clinical trials or receive regulatory approval. Even if we complete the necessary clinical trials, we cannot predict when, or if, we will obtain regulatory approval to commercialize ourproduct candidates, and the approval may be for a narrower indication than we seek.
Clinical failure can occur at any stage of our clinical development. Clinical trials may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional clinical trials or nonclinical studies. In addition, data obtained from trials and studies are susceptible to varying interpretations, and regulators may not interpret our data as favorably as we do, which may delay, limit or prevent regulatory approval. Additionally, our partners, clients, other vendors, and/or other stakeholders may not agree with our interpretation(s) of data obtained from our clinical trials, which could potentially cause a variety of issues, including, but not limited to, delays, the necessity for additional studies and analyses, dependence on third-party validation, and/or other unforeseen challenges. Success in preclinical studies and early clinical trials does not ensure that subsequent clinical trials will generate the same or similar results or otherwise provide adequate data to demonstrate the efficacy and safety of a product candidate. Later-stage clinical trials could differ in significant ways from early-stage clinical trials, including changes to inclusion and exclusion criteria, efficacy endpoints, dosing regimen and statistical design. For example, the small number of patients in our completed Phase 1 Monotherapy clinical trial of REQORSA and the Phase 1/2 Combination Tarceva trial may make the results of these trials less predictive of the outcome of later clinical trials. In addition, although we have observed encouraging clinical activity in the Phase 1 Monotherapy and Phase 1/2 Combination Tarceva trial, the primary objectives of the Phase 1 Monotherapy Trial and the Phase 1 portion of the Phase 1/2 Combination Tarceva trial were safety and MTD and not to demonstrate efficacy. The assessments of clinical activity from these clinical trials, some of which were not pre-specified, may not be predictive of the results of later clinical trials of REQORSA. Furthermore, safety events may be observed in later trials that alter the anticipated risk-benefit profiles of REQORSA or other product candidates.
In addition, the design of a clinical trial can determine whether its results will support approval of a product and flaws in the design of a clinical trial may not become apparent until the clinical trial is well advanced. We may be unable to design and execute a clinical trial to support regulatory approval. Further, clinical trials of potential products often reveal that it is not practical or feasible to continue development efforts.
We do not know whether any clinical trials we or any of our potential future collaborators may conduct will demonstrate the consistent or adequate efficacy and safety that would be required to obtain regulatory approval and market any products. If REQORSA, GPX-002 or GPX-002 isother current or future product candidates are found to be unsafe or lack efficacy, we will not be able to obtain regulatory approval for it and our business would be harmed. If we are unable to bring REQORSA, GPX-002 or other product candidates to market, or to acquire other products that are on the market or can be developed, our ability to create stockholder value will be limited.
Regulatory authorities also may approve a product candidate for more limited indications than requested, or they may impose significant limitations in the form of narrow indications. These regulatory authorities may require warnings or precautions with respect to conditions of use or they may grant approval subject to the performance of costly post-marketing clinical trials. In addition, regulatory authorities may not approve the labeling claims or allow the promotional claims that are necessary or desirable for the successful commercialization of our product candidates. Any of the foregoing scenarios could materially harm the commercial prospects for our product candidates and materially and adversely affect our business, financial condition, results of operations and prospects.
Even if we obtain regulatory approval of our current and potentialfuture product candidates, the products may not gain market acceptance among physicians, patients, hospitals, treatment centers, third-party payors and others in the medical community.
Ethical, social and legal concerns about gene therapy and genetic research could result in additional regulations restricting or prohibiting the products and processes we may use. Even with the requisite approvals, the commercial success of REQORSA, GPX-002 and any of our other current or future product candidates will depend in part on the medical community, patients, and third-party payors accepting gene therapy products in general, and our current and future product candidates in particular, as medically useful, cost-effective and safe. Any product that we bring to the market may not gain market acceptance by physicians, patients, hospitals, treatment centers, third-party payors and others in the medical community. If these products do not achieve an adequate level of acceptance, we may not generate significant product revenue and may not become profitable. The degree of market acceptance of these product candidates, if approved for commercial sale, will depend on a number of factors, including:
the clinical indications for which our current and potential product candidates are approved;
physicians, hospitals, treatment centers and patients considering our product candidates as a safe and effective treatment;
the potential and perceived advantages of our product candidates over alternative treatments;
the prevalence and severity of any side effects;
product labeling or product insert requirements of the FDA or other regulatory authorities;
limitations or warnings contained in the labeling approved by the FDA;
the timing of market introduction of our product candidates as well as competitive products;
the cost of treatment – both in absolute terms and in relation to alternative treatments;
the availability of coverage, reimbursement and pricing by third-party payors and government authorities and the adequacy thereof;
the willingness of patients to pay out-of-pocket in the absence of coverage by third-party payors, including government authorities;
the willingness, ability and availability of healthcare providers that can comply with the transportation, handling, and temperature-controlled storage requirements associated with our product candidates;
relative convenience and ease of administration, including as compared to alternative treatments and competitive therapies; and
the effectiveness of our sales and marketing efforts, which are subject to various limitations under applicable law.
| ● | the clinical indications for which our current and potential product candidates are approved; |
| ● | physicians, hospitals, treatment centers and patients considering our product candidates as a safe and effective treatment; |
| ● | the potential and perceived advantages of our product candidates over alternative treatments; |
| ● | the prevalence and severity of any side effects; |
| ● | product labeling or product insert requirements of the FDA or other regulatory authorities; |
| ● | limitations or warnings contained in the labeling approved by the FDA; |
| ● | the timing of market introduction of our product candidates as well as competitive products; |
| ● | the cost of treatment – both in absolute terms and in relation to alternative treatments; |
| ● | the availability of coverage, reimbursement and pricing by third-party payors and government authorities and the adequacy thereof; |
| ● | the willingness of patients to pay out-of-pocket in the absence of coverage by third-party payors, including government authorities; |
| ● | the willingness, ability and availability of healthcare providers that can comply with the transportation, handling, and temperature-controlled storage requirements associated with our product candidates; |
| ● | relative convenience and ease of administration, including as compared to alternative treatments and competitive therapies; and |
| ● | the effectiveness of our sales and marketing efforts, which are subject to various limitations under applicable law. |
Our efforts to educate the medical community and third-party payors about the benefits of our product candidates may require significant resources and may never be successful.
REQORSA®™, GPX-002, and other current or futureproduct candidates may have undesirable side effects that may delay or prevent marketing approval, or, if approval is received, require them to be taken off the market, require them to include safety warnings or otherwise limit their sales.
In our Phase 1 clinical trial of REQORSA as a monotherapy, the only serious adverse events, defined as grade 3, 4 or 5 events under the Common Terminology Criteria for Adverse Events published by the U.S. Department of Health and Human Services, were grade 3 fever and grade 3 hypotension, and the only dose-limiting toxicities were two episodes of transient grade 3 hypophosphatemia (abnormally low levels of phosphate in the blood).
The Phase 1 portion of our Phase 1/2 Combination Tarceva Trial was a dose escalation study with the primary purpose of determining the MTD. Dose Limiting Toxicities were defined as grade 3, 4 or 5 events during the first cycle of treatment that were considered to be treatment related. At dose level 1, one subject had grade 3 adverse events of fatigue, muscle weakness and hyponatremia (low sodium level) and considered to be related to the study treatment (Tarceva). Therefore, three additional subjects were treated at this dose level (six subjects total), none of whom suffered a DLT. At dose level 2, there were no DLTs. At dose level 3, one subject had a grade 3 rash considered to be related to the study treatment (Tarceva); therefore, an additional three subjects were treated at this dose level (six subjects total). No additional subjects suffered a DLT at dose level 3. At dose level 4, there were no DLTs; thus, dose level 4 was determined to be the MTD.
Additional or unforeseenUndesirable side effects fromfor REQORSA, GPX-002, or any of our other current or future product candidates could arise either during clinical development or, if approved, after the approved product has been marketed. A showing that REQORSA, GPX-002, or any of our other current or future product candidates cause undesirable or unacceptable side effects could interrupt, delay or halt clinical trials and result in the failure to obtain or suspension or termination of marketing approval from the FDA and other regulatory authorities, or result in marketing approval from the FDA and other regulatory authorities only with restrictive label warnings.
If REQORSA, GPX-002, or any of our other current or future product candidates receives marketing approval and we or others later identify undesirable or unacceptable side effects caused by such products:
regulatory authorities may require the addition of labeling statements, specific warnings, a contraindication or field alerts to physicians and pharmacies;
we may be required to change instructions regarding the way the product is administered, conduct additional clinical trials or change the labeling of the product;
we may be subject to limitations on how we may promote the product;
sales of the product may decrease significantly;
regulatory authorities may require us to take our approved product off the market;
we may be subject to litigation or product liability claims; and
our reputation may suffer.
| ● | regulatory authorities may require the addition of labeling statements, specific warnings, a contraindication or field alerts to physicians and pharmacies; |
| ● | we may be required to change instructions regarding the way the product is administered, conduct additional clinical trials or change the labeling of the product; |
| ● | we may be subject to limitations on how we may promote the product; |
| ● | sales of the product may decrease significantly; |
| ● | regulatory authorities may require us to take our approved product off the market; |
| ● | we may be subject to litigation or product liability claims; and |
| ● | our reputation may suffer. |
Any of these events could prevent us or our potential future collaborators from achieving or maintaining market acceptance of our products or could substantially increase commercialization costs and expenses, which in turn could delay or prevent us from generating revenues from the sale of our products.
If a product liability claim is successfully brought against us for uninsured liabilities, or such claim exceeds our insurance coverage, we could be forced to pay substantial damage awards that could materially harm our business.
The use of any of our existing or future product candidates in clinical trials and the sale of any approved pharmaceutical products may expose us to significant product liability claims. We currently carry product liability insurance relating to our clinical trials only. Such insurance coverage may not protect us against any or all of the product liability claims that may be brought against us in the future. We may not be able to acquire or maintain adequate product liability insurance coverage at a commercially reasonable cost or in sufficient amounts or scope to protect us against potential losses. In the event a product liability claim is brought against us, we may be required to pay legal and other expenses to defend the claim, as well as uncovered damage awards resulting from a claim brought successfully against us. In the event our product candidate is approved for sale by the FDA or other regulatory agency and commercialized, we may need to substantially increase the amount of our product liability coverage. Defending any product liability claim or claims could require us to expend significant financial and managerial resources, which could have an adverse effect on our business.
Security breaches and other disruptions could compromise our information and expose us to liability, which would cause our business and reputation to suffer.
In the ordinary course of our business, we collect and store sensitive data, including intellectual property, our proprietary business information and that of our suppliers and business partners, as well as personally identifiable information of clinical trial participants and employees. Similarly, our CROs, contractors and consultants possess certain of our sensitive data. The secure maintenance of this information is critical to our operations and business strategy.
Despite ourthe implementation of security measures, our information technologyinternal computer systems and infrastructure may be vulnerable to attacks by hackers or breached due to employee error, malfeasance or other disruptions. Any such breach could compromise our networks and the information stored there could be accessed, publicly disclosed, lost or stolen. Any such access, disclosure or other lossthose of information, including our data being breached at our CROs, CMOs, and other contractors and consultants are vulnerable to damage from cyberattacks, malicious intrusion, computer viruses, unauthorized access, loss of data privacy, natural disasters, terrorism, war and telecommunication, electrical failures or other significant disruption. If such an event were to occur and cause interruptions in our operations, it could result in legal claims or proceedings, liability under laws that protect the privacya material disruption of personal information, disrupt our operations,drug development programs and damage our reputation which could adversely affect our business. Furthermore,commercialization efforts. For example, the loss of clinical trialstudy data from completed or ongoing clinical studies for any of our drug candidates could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach were to result in a loss of or damage to our data whichor applications or inappropriate disclosure of confidential or proprietary information, we could causeincur liability and the further development andor commercialization of our currentdrug candidates could be delayed.
While we have not experienced any such event to date, if such an event were to occur and potentialcause interruptions in our operations, it could result in a material disruption of our independent drug development programs. For example, the loss of clinical trial data from ongoing or future clinical trials for any of our product candidates could result in delays in regulatory approval efforts and significantly increase costs to recover or reproduce the data. Our information security systems are also subject to laws and regulations requiring that we take measures to protect the privacy and security of certain information we gather and use in our business. For example, HIPAA and its implementing regulations impose, among other requirements, certain regulatory and contractual requirements regarding the privacy and security of personal health information. In addition to HIPAA, numerous other federal, state, and international laws, including, without limitation, state security breach notification laws, state health information privacy laws and federal and state consumer protection laws, govern the collection, use, disclosure and storage of personal information. To the extent that any disruption or security breach were to result in a loss of or damage to data or applications, or inappropriate disclosure of confidential or proprietary information or personal health information, we could incur substantial liability, our reputation would be damaged, and the further development of our product candidates could be delayed.
Business disruptionsWe face risks related to health epidemics and outbreaks, including COVID-19, which could seriously harmsignificantly disrupt our preclinical studies and clinical trials. Our business has previously been adversely affected by the coronavirus pandemic which delayed our clinical trials and disrupted our supply chain. A potential resurgence of COVID-19, or any future revenuedisease outbreak, epidemic or pandemic, could disrupt our clinical trials and financial conditionsupply chain and increasematerially adversely affect our costsbusiness and expenses.operations.
Our operations, and those of our CROs, contractors and consultants, could be subject to power shortages, telecommunications failures, wildfires, water shortages, floods, earthquakes, hurricanes, typhoons, fires, extreme weather conditions, medicalDisease outbreaks, epidemics and other natural or man-made disasters or business interruptions. The occurrencepandemics, including COVID-19, in regions where we have concentrations of any of these business disruptions could seriously harm our operationsclinical trial sites and financial condition and increase our costs and expenses. Our ability to obtain clinical supplies of our current and potential product candidates could be disrupted if the operations of our contract manufacturers are affected by a man-made or natural disaster or other business interruption. Unfavorable global economic conditionsoperations, could adversely affect our business, financial condition, including by causing significant disruptions in our operations and/or resultsin the operations of operations.
We do not carry insurance for all categoriesmanufacturers and CROs upon whom we rely. Disease outbreaks, epidemics and pandemics may have negative impacts on our ability to initiate new clinical trial sites, enroll new patients and maintain existing patients who are participating in clinical trials, which may result in increased clinical trial costs, longer timelines and delays in our ability to obtain regulatory approvals of risk that our business may encounter. There canproduct candidates, if at all. For example, patient enrollment and recruitment could be no assurance that we will secure adequate insurance coverage or that any such insurance coverage will be sufficientdelayed due to local clinical trial site protocols designed to protect usstaff and patients from potential liability incertain outbreaks, which could delay the future. Any significant uninsured liability may require us to pay substantial amounts, which may adversely affectexpected timelines for data readouts of our financial positionpreclinical studies and results of operations.
Our businessclinical trials. Additionally, general supply chain issues may be adversely affected byexacerbated during disease outbreaks, epidemics or pandemics and may also impact the ongoing coronavirus pandemic.
The outbreakability of the novel coronavirus (COVID-19) evolved intoour clinical trial sites to obtain basic medical supplies used in our trials in a global pandemic. The coronavirus has spread to many regions oftimely fashion, if at all. Moreover, the world. The extent to which the coronavirus impactsdisease outbreaks, epidemics and pandemics may impact our business, results of operations and operating resultsfinancial position will depend on future developments, thatwhich are highly uncertain and cannot be accurately predicted including new information thatwith confidence. New health epidemics or pandemics may emerge concerning the coronavirusthat result in similar or more severe disruptions to our business. A future disease outbreak, epidemic or pandemic adversely affects our business, financial condition, results of operations and the actions to contain the coronavirus or treat its impact, among others.growth prospects.
As a result of the continuing spread of the coronavirus, ourOur business operations could be delayed or interrupted. For example, our clinical trials may bewere previously interrupted and delayed as a result of the COVID-19 pandemic. ManufacturingSpecifically, we experienced delays in engaging clinical sites as a result of a backlog of clinical trial protocols requiring site review created by an accumulation of protocols while clinical trials and testing,the clinical trial review process had been widely disrupted during the pandemic. Additionally, site initiation, participant recruitment and enrollment, participant dosing, distribution of clinical trial materials, study monitoring, data analysis, and laboratory research activities may behad been paused or delayed due to changes in hospital or university policies, federal, state or local regulations, prioritization of hospital resources toward pandemic efforts, or other reasons related to the COVID-19 pandemic. IfDuring the coronavirus continues to spread, some participantsCOVID-19 pandemic, we had also experienced disruptions in our supply chain regarding our manufacturing and clinical investigators may not be able to comply with clinical trial protocols. For example, quarantinestesting operations.
The future progression of the COVID-19 epidemic and its effects on our business and operations are uncertain. The impacts of a potential resurgence of COVID-19 could pose the risk that we or other travel limitations (whether voluntary or required) may impede participant movement, affect sponsor access to study sites, or interrupt healthcare services,our employees, suppliers, customers and weothers may be unable to conduct our clinical trials. Further, if the spreadrestricted or prevented from conducting business activities for indefinite or intermittent periods of the coronavirus pandemic continuestime, including as a result of employee health and our operations are adversely impacted, we risk a delay, default and/safety concerns, shutdowns, shelter in place orders, travel restrictions and other actions and restrictions that may be prudent or nonperformance under existing agreements which may increase our costs. These cost increases may not be fully recoverable or adequately coveredrequired by insurance. Infections and deaths related to the pandemic maygovernmental authorities. This could disrupt the United States’ healthcare and healthcare regulatory systems. Such disruptions could divert healthcare resources away from, or materially delay FDA review and/or approval with respect to, our clinical trials. It is unknown how long these disruptions could continue, were they to occur. Any elongation or de-prioritization of our clinical trials or delay in regulatory review resulting from such disruptions could materially affect the development and study of our product candidates.
We currently utilize third parties to, among other things, manufacture raw materials and to manufacture clinical product. If any third-party parties in the supply chain for materials used in the production of our product candidates or the third-party manufacturers of our product candidates themselves are adversely impacted by restrictions resulting from the coronavirus outbreak, our ability to manufactureoperate our business, including producing drug product candidates forand administering our preclinical and clinical trialsstudies. In addition, fluctuations in demand and researchother implications associated with the COVID-19 pandemic have resulted in, and development activities related thereto may be disrupted. In particular, we utilize lipidscould continue resulting in, certain supply chain constraints and challenges in the manufacture of REQORSA. Lipids are also used in the production of various COVID-19 vaccinesbroader markets and this has resulted in increased demand for this material and may limit the available supply.
The spread of the coronavirus,economy generally, which has caused a broadcould impact globally, including restrictions on travel and quarantine policies put into place by businesses and governments, may have a material economic effect on our business. While the potential economic impact brought by and the duration of the pandemic may be difficult to assess or predict, it has already caused, and is likely to result in further, significant disruption of global financial markets, which may reduce our ability to access capital either at all or on favorable terms. In addition, a recession, depression or other sustained adverse market event resulting from the spread of the coronavirus could materially and adversely affect our business and the value ofsupply sources, including our common stock.
The ultimate impact of the current pandemic, or any other health epidemic, is highly uncertain and subject to change. We do not yet know the full extent of potential delays or impacts on our business, our clinical trials, our research programs, healthcare systems or the global economy as a whole. However, these effects could have a material impact on our operations, and we will continue to monitor the situation closely.CDMOs.
We face competition from other biotechnology and pharmaceutical companies, particularly those that are gene therapy companies, and our operating results will suffer if we fail to compete effectively.
The biotechnology and pharmaceutical industries are intensely competitive and subject to rapid and significant technological change. This is particularly so in the fast-growing gene therapy space. We face competition from domestic and international competitors including major multinational pharmaceutical and biotechnology companies, specialty pharmaceutical and generic drug companies, academic institutions, government agencies and other public and private research institutions. Many of our competitors have greater financial and other resources, such as larger research and development staff and more experienced marketing and manufacturing organizations than we do. Large pharmaceutical companies, in particular, have extensive experience in clinical testing, obtaining regulatory approvals, recruiting patients and manufacturing pharmaceutical products. These companies also have significantly greater research, sales and marketing capabilities and collaborative arrangements in our target markets with leading companies and research institutions. Established pharmaceutical companies may also invest heavily to accelerate discovery and development of novel compounds or to in-license novel compounds that could make the product candidates that we develop obsolete. As a result of these factors, our competitors may succeed in obtaining patent protection and/or FDA or other regulatory approval or in discovering, developing and commercializing drugs for the indications that we are targeting before we do or may develop drugs that are deemed to be more effective or gain greater market acceptance than ours. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large, established companies.
If our competitors market products that are more effective, safer or less expensive or reach the market sooner than our future products, if any, we may not achieve commercial success. In addition, because of our limited resources, it may be difficult for us to stay abreast of the rapid changes in all technologies that are or may become competitive with ours. If we fail to stay at the forefront of technological change, we may be unable to compete effectively. Technological advances or products developed by our competitors may render our technologies or product candidates obsolete, less competitive or not economical.
Risks Related to Regulatory Approval and Marketing of Our Current and Future Product Candidates and Other Legal Compliance Matters
We cannot provide assurance that REQORSA,GPX-002, or any of our other current or future product candidates will receive regulatory approval, and without regulatory approval we will not be able to market them.
Our business currently depends largely on the successful development and commercialization of REQORSA, GPX-002, and our other current product candidates, REQORSA and/or GPX-002.candidates. Our ability to generate revenue related to product sales will depend on the successful development and regulatory approval of REQORSA for the treatment of cancer and/or GPX-002 for diabetes. Even if we complete the necessary clinical studies, we cannot predict when or if we will obtain regulatory approval to commercialize a product candidate. Further, even if we obtain regulatory approval, it may only apply to a narrower indication than we expect and our products will remain subject to regulatory scrutiny.
We currently have no products approved for sale and we cannot guarantee that we will ever have marketable products. The development of a product candidate and issues relating to its approval and marketing are subject to extensive regulation by the FDA in the United States and regulatory authorities in other countries, with regulations differing from country to country. We are not permitted to market our product candidates in the United States until we receive approval of a BLA from the FDA. We have not submitted any marketing applications for any of our current and potential product candidates.
BLAs must include extensive preclinical and clinical data and supporting information to establish the product candidate’s safety and effectiveness for each desired indication. BLAs must also include significant information regarding the chemistry, manufacturing and controls for the product. Obtaining approval of a BLA is a lengthy, expensive and uncertain process, and we may not be successful in obtaining approval. The FDA review processes can take years to complete, and approval is never guaranteed. If we submit a BLA to the FDA, the FDA must decide whether to file the BLA or refuse to file it. We cannot be certain that any submissions will be filed and reviewed by the FDA. In addition, regulators in other jurisdictions have their own procedures for approval of product candidates.
The FDA or regulators in other jurisdictions could delay, limit or deny approval of a product candidate for many reasons, including because they:
may not deem our product candidate to be safe and effective;
determine that the product candidate does not have an acceptable benefit-risk profile;
determine in the case of a BLA seeking accelerated approval that the BLA does not provide evidence that the product candidate represents a meaningful advantage over available therapies;
determine that the results on our primary endpoints are not clinically meaningful;
may not agree that the data collected from preclinical studies and clinical trials are acceptable or sufficient to support the approval of a BLA or other submission or to obtain regulatory approval, and may impose requirements for additional preclinical studies or clinical trials;
may determine that adverse events experienced by participants in our clinical trials represent an unacceptable level of risk;
may determine that population studied in the clinical trial may not be sufficiently broad or representative to assure safety in the full population for which we seek approval;
may disagree regarding the formulation or the specifications of our product candidates;
may not approve the manufacturing processes associated with our product candidate or may determine that a manufacturing facility does not have an acceptable compliance status; or
may change approval policies or adopt new regulations.
| ● | may not deem our product candidate to be safe and effective; |
| ● | determine that the product candidate does not have an acceptable benefit-risk profile; |
| ● | determine in the case of a BLA seeking accelerated approval that the BLA does not provide evidence that the product candidate represents a meaningful advantage over available therapies; |
| ● | determine that the results on our primary endpoints are not clinically meaningful; |
| ● | may not agree that the data collected from preclinical studies and clinical trials are acceptable or sufficient to support the approval of a BLA or other submission or to obtain regulatory approval, and may impose requirements for additional preclinical studies or clinical trials; |
| ● | may determine that adverse events experienced by participants in our clinical trials represent an unacceptable level of risk; |
| ● | may determine that population studied in the clinical trial may not be sufficiently broad or representative to assure safety in the full population for which we seek approval; |
| ● | may disagree regarding the formulation or the specifications of our product candidates; |
| ● | may not approve the manufacturing processes associated with our product candidate or may determine that a manufacturing facility does not have an acceptable compliance status; or |
| ● | may change approval policies or adopt new regulations. |
Even if a product is approved, the FDA may limit the indications for which the product may be marketed, require extensive warnings on the product labeling or require expensive and time-consuming clinical trials or reporting as conditions of approval. Regulatory authorities in countries outside of the United States also have requirements for approval of drug candidates with which we must comply prior to marketing in those countries. Obtaining regulatory approval for marketing of a product candidate in one country does not ensure that we will be able to obtain regulatory approval in any other country. Furthermore, regulatory approval for any of our future product candidates may be withdrawn after approval.
If we are unable to obtain approval from the FDA or other regulatory agencies for REQORSA, GPX-002, or GPX-002,our other current or future product candidates, or if, subsequent to approval, we are unable to successfully commercialize REQORSA, or GPX-002, or our other potentialcurrent or future product candidates, we will not be able to generate sufficient revenue to become profitable or to continue our operations.
In addition, the clinical trial requirements of the FDA, the EMA, and other regulatory agencies and the criteria these regulators use to determine the safety and efficacy of a product candidate vary substantially according to the type, complexity, novelty and intended use and market of the potential products. The regulatory approval process for novel product candidates such as ours can be more expensive and take longer than for other, better known or extensively studied pharmaceutical or other product candidates.
Regulatory requirements governing gene therapy products have changed frequently and may continue to change in the future. For example, in January 2017, the FDA Oncology Center of Excellence, or the Center of Excellence, was created to leverage the combined skills of regulatory scientists and reviewers with expertise in drugs, biologics, and devices (including diagnostics). While the Center of Excellence is designed to help expedite the development of oncology and malignant hematology-related medical products and support an integrated approach in the clinical evaluation of drugs, biologics and devices for the treatment of cancer, the Center of Excellence may, at times, create confusion within the FDA and especially in the Center of Biologics and Research, which is the primary review division for REQORSA. Gene therapy clinical trials conducted at institutions that receive funding for recombinant DNA research from the NIH, are also subject to review by NExTRAC. In August 2018, the NIH Director issued a statement describing a proposal intended to streamline the federal framework for oversight of gene therapy. The proposal, which the NIH developed in conjunction with the FDA, included amending the NIH Guidelines for Research Involving Recombinant or Synthetic Nucleic Acid Molecules, or NIH Guidelines, to eliminate duplicative review and reporting requirements for human gene transfer protocols. The statement also described NIH’s effort to refocus the role of the RAC to be closer to its original mandate – a transparent forum for science, safety, and ethics of emerging biotechnologies. Accordingly, the NIH Guidelines have been updated to reflect these changes. Additionally, before a clinical trial can begin at an NIH-funded institution, that institution’s IRB, and its Institutional Biosafety Committee will have to review the proposed clinical trial to assess the safety of the study. In addition, adverse developments in clinical trials of gene therapy products conducted by others may cause the FDA or other regulatory bodies to change the requirements for performing studies or for obtaining approval of any of our current and potential product candidates. The regulatory changes discussed herein as well as other existing and future regulatory developments may cause unexpected delays and challenges for companies seeking approval of gene therapy products, like REQORSA, GPX-002, and our other current or GPX-002.future product candidates.
These regulatory review committees and advisory groups, and the new guidelines they promulgate, may lengthen the regulatory review process, require us to perform additional studies, increase our development costs, lead to changes in regulatory positions and interpretations, delay or prevent approval and commercialization of our current and potential product candidates or lead to significant post-approval limitations or restrictions. As we advance our current and potential product candidates, we will be required to consult with these regulatory and advisory groups and comply with applicable guidelines. Any delay or failure to obtain, or unexpected costs in obtaining, the regulatory approval necessary to bring a potential product to market could decrease our ability to generate sufficient revenue to maintain our business.
Even if we obtain regulatory approval for our product candidates, our products will remain subject to regulatory oversight.
Our product candidates for which we obtain regulatory approval will be subject to ongoing regulatory requirements for manufacturing, labeling, packaging, storage, advertising, promotion, record-keeping and submission of safety and other post-market information. Any regulatory approvals that we receive for our product candidates also may be subject to the specific obligations imposed as a condition for marketing authorization by equivalent authorities in a foreign jurisdiction, particularly by the European Commission, limitations on the approved indicated uses for which the product may be marketed or to the conditions of approval, or contain requirements for potentially costly post-marketing testing, including Phase 4 clinical trials, and surveillance to monitor the quality, safety and efficacy of the product.
For example, in the United States, the holder of an approved BLA is obligated to monitor and report adverse events and any failure of a product to meet the specifications in the BLA. The holder of an approved BLA also must submit new or supplemental applications and obtain FDA approval for certain changes to the approved product, product labeling or manufacturing process. Advertising and promotional materials must comply with the Federal Food, Drug, and Cosmetic Act ("FDCA"(“FDCA”) and implementing regulations and are subject to FDA oversight and post-marketing reporting obligations, in addition to other potentially applicable federal and state laws.
In the EU, any future advertising and promotion of our products will be subject to EU laws governing promotion of medicinal products, interactions with physicians, misleading and comparative advertising and unfair commercial practices. In addition, other legislation adopted by individual EU Member States may apply to the advertising and promotion of medicinal products. These laws may limit or restrict the advertising and promotion of our products to the general public and may impose limitations on our promotional activities with health care professionals. These laws require that promotional materials and advertising for medicinal products are consistent with the product’s Summary of Product Characteristics ("SmPC"(“SmPC”) as approved by the competent authorities. The SmPC is the document that provides information to physicians concerning the safe and effective use of the medicinal product. It forms an intrinsic and integral part of the marketing authorization granted for the medicinal product. Promotion of a medicinal product that does not comport with the SmPC is considered to constitute off-label promotion, which is prohibited in the EU. The applicable laws at EU level and in the individual EU Member States also prohibit the direct-to-consumer advertising of prescription-only medicinal products. Violations of the rules governing the promotion of medicinal products in the EU could be penalized by administrative measures, fines and imprisonment.
In addition, product manufacturers and their facilities may be subject to payment of application and program fees and are subject to continual review and periodic inspections by FDA and other regulatory authorities for compliance with cGMP, requirements and adherence to commitments made in the BLA or foreign marketing application. If we, or a regulatory authority, discover previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured or disagree with the promotion, marketing or labeling of that product, a regulatory authority may impose restrictions relative to that product, the manufacturing facility or us, including requiring recall or withdrawal of the product from the market or suspension of manufacturing.
If we fail to comply with applicable regulatory requirements for any product following approval, a regulatory authority may:
| ● | issue a warning or untitled letter asserting that we are in violation of the law; |
| ● | seek an injunction or impose administrative, civil or criminal penalties or monetary fines; |
| ● | suspend or withdraw regulatory approval; |
| ● | suspend any ongoing clinical trials; |
| ● | refuse to approve a pending BLA or comparable foreign marketing application (or any supplements thereto) submitted by us or our strategic partners; |
| ● | restrict the marketing or manufacturing of the product; |
| ● | seize or detain the product or otherwise demand or require the withdrawal or recall of the product from the market; |
| ● | refuse to permit the import or export of products; |
| ● | request and publicize a voluntary recall of the product; or |
| ● | refuse to allow us to enter into supply contracts, including government contracts. |
issue a warning or untitled letter asserting that we are in violation of the law;
seek an injunction or impose administrative, civil or criminal penalties or monetary fines;
suspend or withdraw regulatory approval;
suspend any ongoing clinical trials;
refuse to approve a pending BLA or comparable foreign marketing application (or any supplements thereto) submitted by us or our strategic partners;
restrict the marketing or manufacturing of the product;
seize or detain the product or otherwise demand or require the withdrawal or recall of the product from the market;
refuse to permit the import or export of products;
request and publicize a voluntary recall of the product; or
refuse to allow us to enter into supply contracts, including government contracts.
Any government investigation of alleged violations of law could require us to expend significant time and resources in response and could generate negative publicity. The occurrence of any event or penalty described above may inhibit our ability to commercialize our product candidates and adversely affect our business, financial condition, results of operations and prospects.
If the FDA does not find the manufacturing facilities of ourcurrent orfuture contract manufacturers acceptable for commercial production, we may not be able to commercialize REQORSA, GPX-002, or any of our other current or future product candidates.
We do not have the internal infrastructure or facilities to manufacture ourselves REQORSA, or earlier stage GPX-002 which is in preclinical development, or any other current or future product candidate, and intend to rely on CDMOs for use in the conduct of our clinical trials or fortrial needs and commercial supply. However, our strategy could change in the future and we could choose to develop such infrastructure. Currently, we intend to continue to use CDMOs for the production of the active pharmaceutical ingredients and the formulation of drug product for the trials of REQORSA to be conducted or that will need to be conducted prior to seeking regulatory approval. Although we have produced a sufficient amount of REQORSA to initiate our Acclaim-1 and Acclaim-2 clinical trials, weWe do not have agreements for all of the steps relating to the ongoing supply of REQORSA or any of our other potential product candidates, and we may not be able to reach agreements with these or other contract manufacturers for sufficient supplies to complete clinical trialsdevelopment and commercialize REQORSA or any of our other product candidates, if itany of them are approved. The manufacture of gene therapy products is approved.complex, and for CDMOs with whom we have agreements, there is no guarantee that they will be able to perform as required on a timely, consistent basis under the applicable governing agreement. Additionally, the facilities used by our contract manufacturers to manufacture product candidates must be the subject of a satisfactory inspection before the FDA approves the product candidate manufactured at that facility. We are completely dependent on our third-party manufacturers for compliance with the requirements of U.S. and non-U.S. regulators for the manufacture of our finished products. If our manufacturers cannot successfully manufacture materials that conform to our specifications and the FDA’s cGMP standards and other requirements of any governmental agency to whose jurisdiction, we are subject, our product candidates will not be approved or, if already approved, may be subject to recalls. Reliance on third-party manufacturers entails risks including:
the possibility that we are unable to enter into manufacturing agreements with third parties to manufacture our product candidates;
the possibility that our contract manufacturers may breach the terms of their manufacturing agreements with us; and
the possibility of termination or nonrenewal of any manufacturing agreement we may enter into.
| ● | the possibility that we are unable to enter into manufacturing agreements with third parties to manufacture our product candidates on acceptable terms; |
| ● | the possibility that our contract manufacturers may breach the terms of their manufacturing agreements with us; |
| ● | the possibility that our contract manufacturers may experience failures in product production; and | |
| ● | the possibility of termination or nonrenewal of any manufacturing agreement we may enter into. |
Any of these factors could cause the delay of approval or commercialization of our product candidates, cause us to incur higher costs or prevent us from commercializing our product candidates successfully. Furthermore, if our product candidates are approved for commercialization and our contract manufacturers fail to deliver the required commercial quantities of finished product on a timely basis at commercially reasonable prices, we would likely be unable to meet demand for our products and could lose potential revenue. It may take several years to establish an alternative source of supply for our product candidates and to have any such new source approved by the government agencies that regulate our products.
We may be subject, directly or indirectly, to federal and state healthcare fraud and abuse laws, false claims laws and other federal and state healthcare laws, and the failure to comply with such laws could result in substantial penalties. Our employees, independent contractors, consultants, principal investigators, CROs, commercial partners and vendors may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements.
State and federal regulatory and enforcement agencies continue actively to investigate violations of health care laws and regulations, and the United States Congress continues to strengthen the arsenal of enforcement tools. The Bipartisan Budget Act of 2018 increased the criminal and civil penalties that can be imposed for violating certain federal health care laws, including the Anti-Kickback Statute. Enforcement agencies also continue to pursue novel theories of liability under these laws. Government agencies have increased regulatory scrutiny and enforcement activity with respect to programs supported or sponsored by pharmaceutical companies, including reimbursement and co-pay support, funding of independent charitable foundations and other programs that offer benefits for patients. Several investigations into these types of programs have resulted in significant civil and criminal settlements.
We are exposed to the risk of fraud, misconduct or other illegal activity by our employees, independent contractors, consultants, principal investigators, CROs, commercial partners and vendors. Misconduct by these parties could include intentional, reckless and/or negligent conduct that fails to: comply with the laws of the FDA and similar foreign regulatory bodies; provide true, complete and accurate information to the FDA and similar foreign regulatory bodies; comply with manufacturing standards we have established; comply with federal and state data privacy, security, fraud and abuse and other healthcare laws and regulations in the United States and similar foreign fraudulent misconduct laws; or report financial information or data accurately or to disclose unauthorized activities to us. If we obtain FDA approval for any of our product candidates and begin commercializing those products in the United States, our potential exposure under such laws would increase significantly, and our costs associated with compliance with such laws would likely also increase. These laws may impact, among other things, our current activities with principal investigators and research patients, as well as proposed and future sales, marketing and education programs and interactions with physicians and other health care providers. In particular, the promotion, sales and marketing of healthcare items and services, as well as certain business arrangements in the healthcare industry, are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, including off-label uses of our products, structuring and commission(s), certain customer incentive programs and other business arrangements generally. Activities subject to these laws also involve the improper use or misrepresentation of information obtained in the course of patient recruitment for clinical trials, creating fraudulent data in our preclinical studies or clinical trials or illegal misappropriation of drug product, which could result in regulatory sanctions and cause serious harm to our reputation. Additionally, we are subject to the risk that a person or government could allege such fraud or other misconduct, even if none occurred. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of fines or other sanctions. The laws that may affect our ability to operate include, but are not limited to:
the Federal Anti-Kickback Statute, which prohibits, among other things, individuals and entities from knowingly and willfully soliciting, receiving, offering or paying any remuneration (including any kickback, bribe, or rebate), directly or indirectly, overtly or covertly, in cash or in kind, to induce, or in return for, either the referral of an individual for, or the purchase, lease, order or recommendation of, any good, facility, item or service for which payment may be made, in whole or in part, under a federal healthcare program, such as the Medicare and Medicaid programs. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation;
federal civil and criminal false claims laws and civil monetary penalty laws, including the civil False Claims Act, which impose criminal and civil penalties, through government, civil whistleblower or qui tam actions, on individuals and entities for, among other things, knowingly presenting, or causing to be presented, claims for payment or approval from Medicare, Medicaid, or other third-party payors that are false, fictitious or fraudulent, or knowingly making, using or causing to be made or used, a false statement to avoid, decrease or conceal an obligation to pay money to the federal government. In addition, the government may assert that a claim including items and services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act;
HIPAA imposes criminal and civil liability for, among other things, knowingly and willfully executing, or attempting to execute, a scheme to defraud or to obtain any healthcare benefit program or obtain, by means of false or fraudulent pretenses, representations, or promises, any of the money or property owned by, or under the custody or control of, any healthcare benefit program, regardless of the payor (e.g., public or private), willfully obstructing a criminal investigation of a healthcare offense, and knowingly and willfully falsifying, concealing or covering up by any trick or device a material fact or making any materially false, fictitious or fraudulent statements in connection with the delivery of, or payment for, healthcare benefits, items or services relating to healthcare matters;
HIPAA, as amended by HITECH and their respective implementing regulations, which impose requirements on certain covered healthcare providers, health plans, and healthcare clearinghouses, as well as their respective business associates that perform services for them that involve the creation, use, maintenance or disclosure of, individually identifiable health information, relating to the privacy, security and transmission of individually identifiable health information without appropriate authorization;
the federal physician payment transparency requirements, sometimes referred to as the “Physician Payments Sunshine Act,” created under the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act (the Affordable Care Act) and its implementing regulations, which require certain manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) to report annually to the United States Department of Health and Human Services information related to payments or other transfers of value made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members;
the U.S. FDCA, which prohibits, among other things, the adulteration or misbranding of drugs;
the Foreign Corrupt Practices Act and similar worldwide anti-bribery laws, which generally prohibit companies and their intermediaries from making improper payments to non-U.S. officials for the purpose of obtaining or retaining business;
federal consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers; and
state and foreign equivalents of each of the healthcare laws described above, among others, some of which may be broader in scope and may apply regardless of the payor.
| ● | the Federal Anti-Kickback Statute, which prohibits, among other things, individuals and entities from knowingly and willfully soliciting, receiving, offering or paying any remuneration (including any kickback, bribe, or rebate), directly or indirectly, overtly or covertly, in cash or in kind, to induce, or in return for, either the referral of an individual for, or the purchase, lease, order or recommendation of, any good, facility, item or service for which payment may be made, in whole or in part, under a federal healthcare program, such as the Medicare and Medicaid programs. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation; |
| ● | federal civil and criminal false claims laws and civil monetary penalty laws, including the civil False Claims Act, which impose criminal and civil penalties, through government, civil whistleblower or qui tam actions, on individuals and entities for, among other things, knowingly presenting, or causing to be presented, claims for payment or approval from Medicare, Medicaid, or other third-party payors that are false, fictitious or fraudulent, or knowingly making, using or causing to be made or used, a false statement to avoid, decrease or conceal an obligation to pay money to the federal government. In addition, the government may assert that a claim including items and services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act; |
| ● | HIPAA imposes criminal and civil liability for, among other things, knowingly and willfully executing, or attempting to execute, a scheme to defraud or to obtain any healthcare benefit program or obtain, by means of false or fraudulent pretenses, representations, or promises, any of the money or property owned by, or under the custody or control of, any healthcare benefit program, regardless of the payor (e.g., public or private), willfully obstructing a criminal investigation of a healthcare offense, and knowingly and willfully falsifying, concealing or covering up by any trick or device a material fact or making any materially false, fictitious or fraudulent statements in connection with the delivery of, or payment for, healthcare benefits, items or services relating to healthcare matters; |
| ● | HIPAA, as amended by HITECH and their respective implementing regulations, which impose requirements on certain covered healthcare providers, health plans, and healthcare clearinghouses, as well as their respective business associates that perform services for them that involve the creation, use, maintenance or disclosure of, individually identifiable health information, relating to the privacy, security and transmission of individually identifiable health information without appropriate authorization; |
| ● | the federal physician payment transparency requirements, sometimes referred to as the “Physician Payments Sunshine Act,” created under the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act (the Affordable Care Act) and its implementing regulations, which require certain manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) to report annually to the United States Department of Health and Human Services information related to payments or other transfers of value made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members; |
| ● | the U.S. FDCA, which prohibits, among other things, the adulteration or misbranding of drugs; |
| ● | the Foreign Corrupt Practices Act and similar worldwide anti-bribery laws, which generally prohibit companies and their intermediaries from making improper payments to non-U.S. officials for the purpose of obtaining or retaining business; |
| ● | federal consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers; and |
| ● | state and foreign equivalents of each of the healthcare laws described above, among others, some of which may be broader in scope and may apply regardless of the payor. |
It is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent inappropriate conduct may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. Efforts to ensure that our business arrangements will comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental and enforcement authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law interpreting applicable fraud and abuse or other healthcare laws and regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of civil, criminal and administrative penalties, damages, disgorgement, monetary fines, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, contractual damages, reputational harm, diminished profits and future earnings, and curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations. In addition, the approval and commercialization of any of our current and potential product candidates outside the United States will also likely subject us to foreign equivalents of the healthcare laws mentioned above, among other foreign laws.
Coverage and reimbursement may be limited or unavailable in certain market segments for REQORSA, GPX-002, and our other current or future product candidates, if approved, which could make it difficult for us to sell REQORSA, GPX-002, and our other current or future product candidates profitably.
The commercial success of any current or future product candidate will depend upon the degree of market acceptance by physicians, patients, insurance companies and other third-party payors, and others in the medical community. Even if we obtain approval to commercialize our current and potential product candidates outside of the United States, a variety of risks associated with international operations could materially affect our business. Due to the novel nature of our technology, we face uncertainty related to pricing and reimbursement for our current and potential product candidates. The insurance coverage and reimbursement status of newly approved products is uncertain. Failure to obtain or maintain adequate coverage and reimbursement for new or current products could limit our ability to market those products and decrease our ability to generate revenue. If market opportunities for our current and potential product candidates are smaller than we believe they are, our revenues may be adversely affected and our business may suffer.
Successful sales of our products, if our current and potential product candidates are approved, depend on the availability of coverage and adequate reimbursement from third-party payors. In addition, because our current and potential product candidates represent new approaches to the treatment of cancer and diabetes, we cannot be sure that coverage and reimbursement will be available for, or accurately estimate the potential revenue from, our current and potential product candidates. Patients who are provided medical treatment for their conditions generally rely on third-party payors to reimburse all or part of the costs associated with their treatment. Coverage and adequate reimbursement from governmental healthcare programs, such as Medicare and Medicaid, and commercial payors are critical to new product acceptance.
Government authorities and other third-party payors, such as private health insurers and health maintenance organizations, decide which drugs and treatments they will cover and the amount of reimbursement. Coverage and reimbursement by a third-party payor may depend upon a number of factors, including, but not limited to, the third-party payor’s determination that use of a product is:
a covered benefit under its health plan;
safe, effective and medically necessary;
appropriate for the specific patient;
cost-effective; and
neither experimental nor investigational.
| ● | a covered benefit under its health plan; | |
| ● | safe, effective and medically necessary; | |
| ● | appropriate for the specific patient; | |
| ● | cost-effective; and | |
| ● | neither experimental nor investigational. |
In the United States, no uniform policy of coverage and reimbursement for products exists among third-party payors, and coverage and reimbursement for products can differ significantly from payor to payor. As a result, obtaining coverage and reimbursement approval of a product from a government or other third-party payor is a time-consuming and costly process that could require us to provide to each payor supporting scientific, clinical and cost-effectiveness data for the use of our products and to justify the level of coverage and reimbursement relative to other therapies, with no assurance that coverage and adequate reimbursement will be obtained. Third party payors may also have difficulty in determining the appropriate coverage of REQORSA and our other potentialcurrent and future product candidates that are combination products, if approved, due to the fact that they are combination products that include another drug. To the extent there are any delays in determining such coverage or inadequate coverage and reimbursement for all aspects of our combination therapies, it would adversely affect the market acceptance, demand and use of our current and potential product candidates. Any denial in coverage or reduction in reimbursement from Medicare or other government programs may result in a similar denial or reduction in payments from private payors, which may adversely affect our future profitability.
We intend to seek approval to market our current and potential product candidates in both the United States and in selected foreign jurisdictions. If we obtain approval in one or more foreign jurisdictions for our current and potential product candidates, we will be subject to rules and regulations in those jurisdictions. In some foreign countries, particularly those in the European Union, the pricing of biologics is subject to governmental control and other market regulations which could put pressure on the pricing and usage of our current and potential product candidates. In these countries, pricing negotiations with governmental authorities can take considerable time after obtaining marketing approval of a product candidate. In addition, market acceptance and sales of our current and potential product candidates will depend significantly on the availability of coverage and adequate reimbursement from third-party payors for our current and potential product candidates and may be affected by existing and future health care reform measures.
Concerns about gene therapy, genetic testing, and genetic research could result in new and/or additional government regulations and requirements that restrict or prohibit the processes we use or delay or prevent the regulatory approval of our current and potential product candidates.
Ethical, social, and legal concerns about gene therapy, genetic testing, and genetic research could result in additional regulations restricting or prohibiting the processes we may use. Federal and state agencies, congressional committees and foreign governments have expressed interest in further regulating biotechnology. More restrictive regulations or claims that our products are unsafe or pose a hazard could prevent us from commercializing any products. New government requirements may be established that could delay or prevent regulatory approval of our current and potential product candidates. It is impossible to predict whether legislative changes will be enacted, regulations, policies, or guidance changed, or interpretations by agencies or courts changed, or what the impact of such changes, if any, may be.
Healthcare legislative reform measures may have a material adverse effect on our business and results of operations.
Third-party payors, whether domestic or foreign, or governmental or commercial, are developing increasingly sophisticated methods of controlling healthcare costs. In both the United States and certain foreign jurisdictions, there have been a number of legislative and regulatory changes to the health care system that could affect our ability to sell our products profitably. In particular the Affordable Care Act and its implementing regulations, among other things, subjected biological products to potential competition by lower-cost biosimilars, revised the methodology by which rebates owed by manufacturers to the state and federal government for covered outpatient drugs and certain biologics, including our current and potential product candidates, under the Medicaid Drug Rebate Program are calculated, increased the minimum Medicaid rebates owed by most manufacturers under the Medicaid Drug Rebate Program, extended the Medicaid Drug Rebate program to utilization of prescriptions of individuals enrolled in Medicaid managed care organizations, subjected manufacturers to new annual fees and taxes for certain branded prescription drugs, and provided incentives to programs that increase the federal government’s comparative effectiveness research. There
More recently, other legislative changes that affect the pharmaceutical industry have been judicialproposed and Congressional challenges to certain aspects of the Affordable Care Act. In November 2020,adopted in the United States Supreme Court held oral arguments on the U.S. Court of Appeals for the Fifth Circuit’s decision that held that the individual mandate is unconstitutional. It is uncertain how the United States Supreme Court will rule on this case or how healthcare measures of the Biden administration will impactsince the Affordable Care Act and our business.
Since January 2017, former President Trump signed two Executive Orders designed to delaywas enacted. For example, the implementation of certain provisions of the Affordable Care Act or otherwise circumvent some of the requirements for health insurance mandated by the Affordable Care Act. Concurrently, Congress has considered legislation that would repeal or repeal and replace all or part of the Affordable Care Act. While Congress has not passed comprehensive repeal legislation, two bills affecting the implementation of certain taxes under the Affordable Care Act have been signed into law. The Tax Cuts and JobsInflation Reduction Act of 2017 includes a provision repealing, effective January 1, 2019, the tax-based shared responsibility payment imposed by the Affordable Care Act on certain individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate.” Additionally, on January 22, 2018, former President Trump signed a continuing resolution on appropriations for fiscal year 2018 that delayed the implementation of certain Affordable Care Act-mandated fees, including the so-called “Cadillac” tax on certain high cost employer-sponsored insurance plans, the annual fee imposed on certain health insurance providers based on market share, and the medical device excise tax on non-exempt medical devices. Further, the Bipartisan Budget Act of 2018,2022 included, among other things, amended the Affordable Care Act, effective January 1, 2019,a provision that authorizes CMS to close the coverage gap in mostnegotiate a “maximum fair price” for a limited number of high-cost, single-source drugs every year, and another provision that requires drug companies to pay rebates to Medicare drug plans, commonly referredif prices rise faster than inflation. In addition, various states have adopted or are considering adopting laws that require pharmaceutical companies to as the “donut hole.” Congress also could consider subsequent legislationprovide notice prior to repeal or repealraising prices and replace other elements of the Affordable Care Act.to justify price increases. We continue to evaluate the possible impact of the Affordable Care Act, as amended, and the possible repeal and/or replacement of the Affordable Care Act on our business.
In late 2018, the Centers for Medicare & Medicaid Services, or CMS, issued an advance notice of proposed rulemaking describing a potential mandatory model to test Medicare reimbursement based on an “International Pricing Index,” or IPI. More recently, CMS published an interim final ruleexpect that establishes a Most Favored Nation, or MFN, Model for Medicare Part B drug payment. This regulation would substantially change the drug reimbursement landscape as it bases Medicare Part B payment for 50 selected drugs on prices in foreign countries instead of average sales price, or ASP, and establishes a fixed add-on payment in place of the current 6 percent (4.3 percent after sequestration) of ASP. The MFN drug payment amount is expected toadditional healthcare reform measures will be lower than the current ASP-based limit because U.S. drug prices are generally the highestadopted in the world. On December 28, 2020,future, any of which could limit the United States District Courtamounts that federal and state governments will pay for healthcare products and services, and in Northern California issued a nationwide preliminary injunction against implementationturn could significantly reduce the projected value of the interim final rule,certain development projects and it faces uncertain prospects for implementation.reduce our profitability.
There have been, and likely will continue to be, legislative and regulatory proposals at the foreign, federal and state levels directed at broadening the availability of healthcare and containing or lowering the cost of healthcare. We cannot predict the initiatives that may be adopted in the future. The continuing efforts of the government, insurance companies, managed care organizations and other payors of healthcare services to contain or reduce costs of healthcare and/or impose price controls may adversely affect:
the demand for our current and potential product candidates, if we obtain regulatory approval;
our ability to set a price that we believe is fair for our products;
our ability to generate revenue and achieve or maintain profitability;
the level of taxes that we are required to pay; and
the availability of capital.
| ● | the demand for our current and potential product candidates, if we obtain regulatory approval; | |
| ● | our ability to set a price that we believe is fair for our products; | |
| ● | our ability to generate revenue and achieve or maintain profitability; | |
| ● | the level of taxes that we are required to pay; and | |
| ● | the availability of capital. |
We are subject to a variety of risks associated with international operations which could materially adversely affect our business.
We anticipate that we will be subject to additional risks in commercializing our product candidates outside the United States, including the following, any one or combination of which could have a material adverse effect on our business:
different regulatory requirements for approval of product candidates in foreign countries;
reduced protection for intellectual property rights;
unexpected changes in tariffs, trade barriers and regulatory requirements;
economic weakness, including inflation, or political instability in particular foreign economies and markets;
compliance with tax, employment, immigration and labor laws for employees living or traveling abroad;
foreign currency fluctuations, which could result in increased operating expenses and reduced revenues, and other obligations incident to doing business in another country;
workforce uncertainty in countries where labor unrest is more common than in the United States;
production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; and
business interruptions resulting from geopolitical actions, including war and terrorism or natural disasters including earthquakes, typhoons, floods, fires and medical epidemics.
| ● | different regulatory requirements for approval of product candidates in foreign countries; | |
| ● | reduced protection for intellectual property rights; | |
| ● | unexpected changes in tariffs, trade barriers and regulatory requirements; | |
| ● | economic weakness, including inflation, or political instability in particular foreign economies and markets; | |
● ● | compliance with tax, employment, immigration and labor laws for employees living or traveling abroad; compliance with international data privacy laws, including GDPR; | |
| ● | foreign currency fluctuations, which could result in increased operating expenses and reduced revenues, and other obligations incident to doing business in another country; | |
| ● | workforce uncertainty in countries where labor unrest is more common than in the United States; | |
| ● | production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; and | |
| ● | business interruptions resulting from geopolitical actions, including war and terrorism or natural disasters including earthquakes, typhoons, floods, fires and medical epidemics. |
If we fail to comply with environmental, health and safety laws and regulations, we could become subject to fines or penalties or incur costs that could have a material adverse effect on the success of our business.
We may become subject to federal, state, local, and foreign laws and regulations governing the use, manufacture, storage, handling and disposal of hazardous materials and certain waste products, including numerous environmental, health and safety laws and regulations, such as those governing laboratory procedures and the handling, use, storage, treatment and disposal of hazardous materials and wastes. Our operations may in the future involve the use of hazardous materials, including chemicals and biological materials. Our operations may also produce hazardous waste products. We generally contract with third parties for the disposal of these materials and wastes. We cannot eliminate the risk of contamination or injury from these materials. In the event of contamination or injury resulting from our use of hazardous materials, we could be held liable for any resulting damages, and any liability could exceed our resources. We also could incur significant costs associated with civil or criminal fines and penalties. In addition, we may incur substantial costs in order to comply with current or future environmental, health and safety laws and regulations. These current or future laws and regulations may impair our research, development or production efforts. Failure to comply with these laws and regulations may also result in substantial fines, penalties or other sanctions.
Risks Related to Our Dependence on Third Parties
We may not be successful in establishing and maintaining development and commercialization collaborations, which could adversely affect our ability to develop certain of our current and future product candidates and our financial condition and operating results.results could be adversely affected.
Because developing pharmaceutical products, conducting clinical trials, obtaining regulatory approval, establishing manufacturing capabilities and marketing approved products are expensive, we have and may continue to enter into collaborations with companies that have the required expertise. Additionally, if any of our product candidates receive marketing approval, we may enter into sales and marketing arrangements with third parties. If we are unable to enter into arrangements on acceptable terms, or at all, we may be unable to effectively market and sell our products in our target markets. We expect to face competition in engaging collaborators. We may not be successful in our efforts to establish and implement collaborations or other alternative arrangements for the development of our current and potential product candidates.
When we collaborate with a third party for development and commercialization of a product candidate, we can expect to relinquish some or all of the control over the timing and future success of that product candidate to such third party.
One or more of our collaboration partners may not devote sufficient resources to the development and commercialization of our product candidates or may otherwise fail in their commercializationthese efforts. The terms of any collaboration or other arrangement that we establish may contain provisions that are not favorable to us. In addition, any collaboration that we enter into may not be successful in the development and commercialization of our product candidates. In some cases, we may be responsible for continuing preclinical and initial clinical development of a product candidate or research program under a collaboration arrangement, and the payment we receive from our collaboration partner may be insufficient to cover the cost of such development. If we are unable to reach agreements with suitable collaborators for our product candidates, we may face increased costs, we may be forced to limit the number of our product candidates we can commercially develop or the territories in which we commercialize them. As a result, we may be unable to commercialize products or programs if we are unable to engage a suitable collaborator, which may have a material adverse effect on our operating results and financial condition.
We rely, in part, and expect to continue to rely, in part, on third parties to conduct, supervise and monitor our clinical trials, and if these third parties perform in an unsatisfactory manner, it may harm our business.
We rely in part on CROs and clinical trial sites to ensure our clinical trials are conducted properly and on time. While we have or will have agreements governing their activities, we may have limited influence over their actual performance because we control only certain aspects of our CROs’ activities. Nevertheless, we are responsible for ensuring that each of our clinical trials is conducted in accordance with the applicable protocol and legal, regulatory and scientific standards, and our reliance on the CROs does not relieve us of our regulatory responsibilities. These CROs may also have relationships with other commercial entities, including our competitors, for whom they may also be conducting clinical trials or other drug development activities that could harm our competitive position.
We and our CROs are required to comply with good clinical practices (“GCPs”) for conducting, recording and reporting the results of clinical trials to assure that the data and reported results are credible and accurate and that the rights, integrity and confidentiality of clinical trial participants are protected. The FDA enforces these GCPs through periodic inspections of study sponsors, principal investigators and clinical trial sites. If we or our CROs fail to comply with applicable GCPs, the clinical data generated in our clinical trials may be deemed unreliable, and the FDA may require us to perform additional clinical trials before approving any marketing applications. In addition, our ongoing and future clinical trials will require a sufficient number of test subjects to evaluate the safety and effectiveness of our current and potential product candidates. Accordingly, if our CROs fail to comply with applicable regulations or fail to recruit a sufficient number of patients, we may be required to repeat such clinical trials, which would delay the regulatory approval process.
If our CROs do not successfully carry out their contractual duties or obligations, fail to meet expected deadlines, or if the quality or accuracy of the clinical data they obtain is compromised due to their failure to adhere to our clinical protocols or regulatory requirements, or for any other reasons, our clinical trials may be extended, delayed or terminated, and we may not be able to obtain regulatory approval for or successfully commercialize our product candidates. As a result, our financial results and the commercial prospects may be harmed, our costs could increase and our ability to generate revenues could be limited or delayed.
We rely, and expect to continue to rely, on third parties to distribute, manufacture and perform release testing for our current and future product candidates and other key materials and if such third parties do not carry out their contractual duties or meet expected deadlines, we may not be able to obtain regulatory approvals for our product candidates.
We rely, and expect to continue to rely on third-party CDMOs to produce REQORSA and expect to do so with GPX-002 and other current and future product candidates and other key materials and on third-party contract testing organizations, or CTOs, for the establishment and performance of validated product release assays, but we have not entered into binding agreements with any such CMOs or CTOs to support commercialization.assays. Additionally, any CDMO may not have specific experience producing our product candidates at commercial levels and may not achieve the necessary regulatory approvals or produce our products at the quality, quantities, locations and timing needed to support commercialization. We do not have full control of these CDMOs, and they may prioritize other customers or be unable to provide us with enough manufacturing capacity to meet our objectives. We may change our manufacturing process, and there can be no guarantee that the regulatory authorities will approve any new process in a timely manner, or ever. Also, as a consequence of the manufacturing change, there may be a requirement to conduct additional preclinical safety or efficacy studies, develop new manufacturing and release assays and/or repeat all or part of the ascending dose safety study in animals or humans. Regulatory requirements ultimately imposed could adversely affect our ability to test, manufacture or market products.
Historically, part of our manufacturing process was conducted in manufacturing facilities at MD Anderson. We have completed the technology transfer from MD Anderson to experienced commercial contract development and manufacturing organizations and have scaled-up clinical production of REQORSA in quantities we believe are sufficientappropriate for our Acclaim-1, Acclaim-2 and Acclaim-2Acclaim-3 clinical trials. With the advancement of the development of GPX-002, we also are working to optimize the manufacture of this product candidate and to source high quality and integrated vendors capable of producing it in accordance with GMP. No assurance can be given that such contract manufacturers will be able to, and will receive all approvals to, produce product sufficient for all of our clinical trial needs moving forward or for commercialization. In accordance with cGMPs, changing manufacturers may require the re-validation of manufacturing processes and procedures, and may require further preclinical studies or clinical trials to show comparability between the materials produced by different manufacturers. Changing our contract manufacturers may be difficult and could be costly if we do make such a change, which could result in our inability to manufacture our product candidates and a delay in the development of our product candidates and their commercial sale, should they be approved. Further, in order to maintain our development timelines in the event of a change in a third-party contract manufacturer, we may incur higher costs to manufacture our product candidates. There can be no guarantee that the regulatory authorities will approve any new process in a timely manner or ever. Regulatory requirements ultimately imposed could adversely affect our ability to test, manufacture or market products.
In connection with our manufacturing activities, we may encounter technical or scientific issues related to manufacturing or development that we may be unable to resolve in a timely manner or with available funds. Further, we have not fully completed the characterization and validation activities necessary for commercial and regulatory approvals. If our manufacturing and testing partners do not enable such regulatory approvals, our commercialization efforts may be harmed. If such third-party manufacturers are unable to produce REQORSA, GPX-002, or futureother product candidates in the necessary quantities, or in compliance with cGMP or in compliance with pertinent regulatory requirements, and within our planned time frame and cost parameters, the development and sales of our products, if approved, would be materially harmed. The manufacturing processes used by our contract manufacturers to manufacture product candidates must be approved by the FDA as part of our BLA package and the facilities used by our contract manufacturers must maintain a compliance status acceptable to the FDA pursuant to inspections that will be conducted after we submit our BLA to the FDA. Although we provide specifications, we do not control the manufacturing process of, and are completely dependent on, our contract manufacturing partners for compliance with cGMPs for the manufacture of our product candidates. If our CDMOs cannot successfully manufacture material that conforms to our specifications and the strict regulatory requirements of the FDA or other regulatory agencies, we will not be able to secure and/or maintain regulatory approval covering their manufacturing facilities. In addition, we have limited control over the ability of our contract manufacturers to maintain adequate quality control, quality assurance and qualified personnel. If the FDA or a comparable foreign regulatory authority does not approve these facilities for the manufacture of our product candidates or if it withdraws any such approval in the future, we may need to find alternative manufacturing facilities, which would significantly affect our ability to develop, obtain regulatory approval for or market our future product candidates, if approved. In addition, any failure to achieve and maintain compliance with these laws, regulations and standards could subject us to the risk that we may have to suspend the manufacturing of our current and future product candidates or that obtained approvals could be revoked, which would adversely affect our business and reputation.
In addition, any significant disruption in our supplier relationships could harm our business. We source key materials, devices and equipment from third parties, either directly through agreements with suppliers or indirectly through our manufacturers who have agreements with suppliers. There is a small number of suppliers for certain key materials and components that are used to manufacture our current and future product candidates. Such suppliers may not sell these key materials to our manufacturers at the times or quantities we need them or on commercially reasonable terms. We may not have any control over the process or timing of the acquisition of these key materials by our manufacturers.
We also expect to rely on other third parties to store and distribute our products for our clinical trials. Any performance failure on the part of these third parties could delay clinical development or marketing approval of our current and future product candidates or commercialization of our products, if approved, producing additional losses and depriving us of potential product revenue.
We have completed and may in the future complete related party transactions that were not and may not be conducted on an arm’sarm’s length basis.
Our leadingIf we are successful in commercializing REQORSA, our lead drug candidate, we will owe IRI a 1% royalty of our product sales. REQORSA is based upon patents and related technology covered by the 1994 MD Anderson License Agreement, under which we have rights to patents covering use of various genes, including the TUSC2 gene, for treatment of cancer, as well as know-how and related intellectual property. In 2007, the 1994 MD Anderson License Agreement was sublicensed by Introgen to IRI and in 2009 this sublicense was assigned by IRI to us, and we granted back to IRI a nonexclusive, royalty-free license to use and practice the licensed technology for non-commercial research purposes. As consideration for this assignment, we agreed to assume all of IRI’s obligations to MD Anderson under the 1994 MD Anderson License Agreement, including ongoing patent related expenses and royalty obligations. IRI also agreed in 2011, pursuant to the 2011 IRI Collaboration Agreement, to provide additional technology licensing opportunities and services to us in return for monthly payments and our obligation to pay to IRI a royalty of 1% on sales of products licensed to us under the 1994 MD Anderson License Agreement. We also granted a non-exclusive, royalty-free sublicense to IRI in 2011 for non-commercial research purposes. IRI’s obligations to provide additional technology licensing opportunities and services to us, and our obligation to make monthly payments to IRI, were terminated in 2012; however, our obligation to pay the 1% royalty to IRI upon sales of products licensed to us under the 1994 MD Anderson License Agreement is ongoing. This royalty obligation continues for 21 years after the later of the termination of the 1994 MD Anderson License Agreement and the termination of the sublicense assigned by IRI to us.
IRI is controlled by Rodney Varner and his immediate family members.IRI is owned by trusts for the benefit of Mr. Varner’s descendants. Mr. Varner is currently Chairman of our board of directors, having joined our board of directors on August 15, 2012, and has been our Chief Executive Officer since August 29, 2012 and President since August 10, 2020; accordingly, in 2009 and 2011, when the above referenced agreements between IRI and Genprex were entered into, Mr. Varner was neither a member of our board of directors nor an executive officer of Genprex. When the 2011 IRI Collaboration Agreement was entered into, Mr. Varner was deemed to be an “affiliate” of the Company due to his beneficial ownership of approximately 39% of our issued and outstanding shares at that time. Although we believe that these transactions were conducted on an arm’s length basis, it is possible that the terms were less favorable to us than they might have been in a transaction with an unrelated party.
Disruptions in the global economy and supply chains may have a material adverse effect on our business, financial condition and results of operations.
The disruptions to the global economy since the COVID-19 global pandemic commenced in 2020 has impeded global supply chains, resulting in longer lead times and also increased critical component costs and freight expenses. We have experienced this impact most notably in our manufacturing operations due to the delay in our ability to acquire raw materials for our drug product. This delay has the potential to impact the timing of the conduct of our clinical trials. We have taken steps to minimize the impact of these increased costs by working closely with our suppliers and locating redundant or comparable sources. Despite the actions we have undertaken to minimize the impacts from disruptions to the global economy, there can be no assurances that unforeseen future events in the global supply chain, and inflationary pressures, will not have a material adverse effect on our business, financial condition and results of operations.
Risks Related to Our Intellectual Property
If we fail to comply with obligations pursuant to our license agreements, we could lose intellectual property and other rights that are important to our business.business; if we fail to obtain licenses to advance our research and development that may be required we may be unable to develop the affected product exclusively, on acceptable terms or at all.
Pursuant to the 1994 MD Anderson License Agreement and subsequent Amendments thereto, as well as the 2020 MD Anderson License Agreement, we hold worldwide, exclusive license rights to certain inventions covering the therapeutic use of TUSC2 and other genes and polypeptides that have been shown to have cancer fighting properties, as well as a number of related technologies. In addition, pursuant to the UP License Agreement,Agreements, UP granted us a worldwide, exclusive license to certain licensed technology, and a worldwide, non-exclusive license to use certain related know-how, all related to diabetes gene therapy. In addition, we expect to enter into additional license agreements in the future. Our existing and future license agreements may impose various payment and other obligations on us. If we fail to comply with our obligations under these agreements, our licensors may have the right to terminate our licenses, in which event we would not be able to market products covered by such licenses.
Moreover, in the event we need to obtain licenses from third parties to advance our research and development or allow commercialization of our product candidates, including additional technology that may be required or advisable to advance REQORSA or GPX-002, we may fail to obtain any of such licenses at a reasonable cost or on reasonable terms, if at all. In that event, we may be required to expend significant time and resources to develop or license replacement technology. If we are unable to develop or license replacement technology, we may be unable to develop or commercialize the affected product candidates, which could harm our business significantly. We cannot provide any assurances that third-party patents do not exist which might be enforced against our current product candidates or future products, resulting in either an injunction prohibiting our sales, or, with respect to our sales, an obligation on our part to pay royalties and/or other forms of compensation to third parties.parties or that we may be estopped from acquiring such rights due to prior art. Specifically, mice and NHP studies for which we have disclosed data relating to our diabetes program includes a technology to which we do not have exclusive rights, though we expect, but are not guaranteed, that we will have exclusive rights to the final product(s) developed under this program.
In certain cases, patent prosecution of our licensed technology may be controlled solely by the licensor. If our licensors fail to obtain and maintain patent or other protection for the proprietary intellectual property we license from them, we could lose our rights to the intellectual property or our exclusivity with respect to those rights, and our competitors could market competing products using the intellectual property. In addition, in certain cases, we control the prosecution of patents resulting from licensed technology. In the event we breach any of our obligations related to such prosecution, we may incur significant liability to our licensing partners. Licensing of intellectual property is of critical importance to our business and involves complex legal, business and scientific issues and is complicated by the rapid pace of scientific discovery in our industry. Disputes may arise regarding intellectual property subject to a licensing agreement, including, but not limited to:
the scope of rights granted under the license agreement and other interpretation-related issues;
the extent to which our technology and processes infringe intellectual property of the licensor that is not subject to the licensing agreement;
the sublicensing of patent and other rights under our collaborative development relationships;
our diligence obligations under the license agreement and what activities satisfy those diligence obligations;
the ownership of inventions and know-how resulting from the joint creation or use of intellectual property by our licensors and us and our partners; and
the priority of invention of patented technology.
| ● | the scope of rights granted under the license agreement and other interpretation-related issues; |
| ● | the extent to which our technology and processes infringe intellectual property of the licensor that is not subject to the licensing agreement; |
| ● | the sublicensing of patent and other rights under our collaborative development relationships; |
| ● | our diligence obligations under the license agreement and what activities satisfy those diligence obligations; |
| ● | the ownership of inventions and know-how resulting from the joint creation or use of intellectual property by our licensors and us and our partners; and |
| ● | the priority of invention of patented technology. |
If disputes over intellectual property that we have licensed prevent or impair our ability to maintain our current licensing arrangements on acceptable terms, we may be unable to successfully develop and commercialize the affected product candidates.
The intellectual property rights we have licensed from MD Anderson and the UPare subject to the rights of the U.S. government.
The rights we have obtained pursuant to our license agreements with MD Anderson and the UP are made subject to the rights of the U.S. government to the extent that the technology covered by the licensed intellectual property was developed under a funding agreement between such institution and the U.S. government. Additionally, to the extent there is any conflict between our license agreement and applicable laws or regulations, applicable laws and regulations will prevail. Similarly, to the extent there is any conflict between our license agreement with one of these institutions and the institution’s funding agreement with the US government, the terms of the funding agreement will prevail. Some, and possibly all, of our licensed intellectual property rights have been developed in the course of research funded by the U.S. government. As a result, the U.S. government may have certain rights to intellectual property embodied in our current or future products pursuant to the Bayh-Dole Act of 1980. Government rights in certain inventions developed under a government-funded program include a non-exclusive, non-transferable, irrevocable worldwide license to use inventions for any governmental purpose. In addition, the U.S. government has the right to require us, or an assignee or exclusive licenseesublicensee to such inventions, to grant licenses to any of these inventions to a third party if the U.S. government determines that adequate steps have not been taken to commercialize the invention, that government action is necessary to meet public health or safety needs, that government action is necessary to meet requirements for public use under federal regulations, or that the right to use or sell such inventions is exclusively licensed to an entity within the U.S. and substantially manufactured outside the U.S. without the U.S. government’s prior approval. Additionally, we may be restricted from granting exclusive licenses for the right to use or sell our inventions created pursuant to such agreements unless the licensee agrees to additional restrictions (e.g., manufacturing substantially all of the invention in the U.S.). The U.S. government also has the right to take title to these inventions if we failour licensors failed to disclose the invention to the government and failin a timely manner and/or failed to file an application to register the intellectual property within specified time limits. In addition, the U.S. government may acquire title in any country in which a patent application is not filed within specified time limits. Furthermore, certain inventions are subject to transfer restrictions during the term of these agreements and for a period thereafter, including sales of products or components, transfers to foreign subsidiaries for the purpose of the relevant agreements, and transfers to certain foreign third parties. If any of our intellectual property becomes subject to any of the rights or remedies available to the U.S. government or third parties pursuant to the Bayh-Dole Act of 1980, this could impair the value of our intellectual property and could adversely affect our business.
If we are unable to protect our intellectual property rights or if our intellectual property rights are inadequate for our technology and product candidates, our competitive position could be harmed.
Our commercial success will depend in part on our ability and the ability of our licensors to obtain and maintain patent and other intellectual property protection in the United States and other countries with respect to our proprietary technology and products. We rely on trade secret, patent, copyright and trademark laws, and confidentiality, licensing and other agreements with employees and third parties, all of which offer only limited protection. We seek to protect our proprietary position by filing and prosecuting patent applications in the United States and abroad related to our novel technologies and products that are important to our business.
The patent positions of biotechnology and pharmaceutical companies generally are highly uncertain, involve complex legal and factual questions and have in recent years been the subject of much litigation. As a result, the issuance, scope, validity, enforceability and commercial value of our patents, including those patent rights licensed to us by third parties, are highly uncertain. The steps we or our licensors have taken to protect our proprietary rights may not be adequate to preclude misappropriation of our proprietary information or infringement of our intellectual property rights, both inside and outside of the United States. Further, the examination process may require us or our licensors to narrow the claims for our pending patent applications, which may limit the scope of patent protection that may be obtained if these applications issue. The rights already granted under any of our currently issued patents or those licensed to us and those that may be granted under future issued patents may not provide us with the proprietary protection or competitive advantages we are seeking. If we or our licensors are unable to obtain and maintain patent protection for our technology and products, or if the scope of the patent protection obtained is not sufficient, our competitors could develop and commercialize technology and products similar or superior to ours, and our ability to successfully commercialize our technology and products may be adversely affected. It is also possible that we or our licensors will fail to identify patentable aspects of inventions made in the course of our development and commercialization. It is also possible that as research and development progresses, the direction of our intellectual property strategy and patent portfolio will change, resulting in strategic business decisions to allow certain patents or patent applications to be abandoned or lapse.
With respect to patent rights, we do not know whether any of the pending patent applications relating to any of our current and future product candidates will result in the issuance of patents that effectively protect our technology or products, or if any of our licensors’ issued patents will effectively prevent others from commercializing competitive technologies and products. Publications of discoveries in the scientific literature often lag behind the actual discoveries, and patent applications in the United States and other jurisdictions are typically not published until 18 months after filing or in some cases not at all, until they are issued as a patent. Therefore, we cannot be certain that we or our licensors were the first to make the inventions claimed in our owned or licensed patents or pending patent applications, or that we or our licensors were the first to file for patent protection of such inventions.
Our pending applications cannot be enforced against third parties practicing the technology claimed in such applications unless and until a patent issues from such applications. Because the issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability, issued patents that we have licensed from third parties may be challenged in the courts or patent offices in the United States and abroad. Such challenges may result in the loss of patent protection, the narrowing of claims in such patents or the invalidity or unenforceability of such patents, which could limit our ability to stop others from using or commercializing similar or identical technology and products, or limit the duration of the patent protection for our technology and products. Protecting against the unauthorized use of our or our licensor’s patented technology, trademarks and other intellectual property rights is expensive, difficult and may in some cases not be possible. In some cases, it may be difficult or impossible to detect third-party infringement or misappropriation of our intellectual property rights, even in relation to issued patent claims, and proving any such infringement may be even more difficult.
Third-party claims of intellectual property infringement may prevent or delay our development and commercialization efforts.
Our commercial success depends in part on our avoiding infringement of the patents and proprietary rights of third parties. There is a substantial amount of litigation, both within and outside the United States, involving patent and other intellectual property rights in the biotechnology and pharmaceutical industries, including patent infringement lawsuits, interferences, oppositions and inter partes reexaminationother post grant proceedings before the US Patent and Trademark Office ("USPTO"(“USPTO”) and corresponding foreign patent offices. Numerous US and foreign issued patents and pending patent applications, which are owned by third parties, exist in the fields in which we are pursuing development candidates. As the biotechnology and pharmaceutical industries expand and more patents are issued, the risk increases that our current and future product candidates may be subject to claims of infringement of the patent rights of third parties.
Third parties may in the future assert that we are employing their proprietary technology without authorization. There may be third-party patents or patent applications with claims to materials, formulations, methods of manufacture or methods for treatment related to the use or manufacture of our product candidates. Because patent applications can take many years to issue, there may be currently pending patent applications which may later result in issued patents that our product candidates may infringe. In addition, third parties may obtain patents in the future and claim that use of our technologies infringes upon these patents. If any third-party patents were held by a court of competent jurisdiction to cover the manufacturing process of any of our product candidates, or any final product itself, the holders of any such patents may be able to block our ability to develop and commercialize such product candidate until such patents expire unless we obtained a license under the applicable patents, which license may not be available on acceptable terms, if at all, or until such patents expire.all.
Parties making claims against us may obtain injunctive or other equitable relief, which may hinder our ability to further develop and commercialize one or more of our product candidates. Defense of these claims, regardless of their merit, may involve substantial litigation expense and diversion of our management’s attention. In the event of a successful claim of infringement against us, we may have to pay substantial damages, including treble damages and attorneys’ fees for willful infringement, pay royalties, redesign our infringing products or obtain one or more licenses from third parties, which licenses may not be on acceptable terms or require substantial time and monetary expenditure.
We may not be successful in obtaining or maintaining necessary rights to product components and processes for our development pipeline through acquisitions and in-licenses.
Presently we believe that we have the necessary rights to the intellectual property, through licenses or other rights from third parties to develop our product candidates. Because our programs may involve additional product candidates that may require the use of proprietary rights held by third parties, the growth of our business may depend in part on our ability to acquire, in-license or use these proprietary rights. In addition, our product candidates may require specific formulations to work effectively and efficiently and these rights may be held by others. We may be unable to acquire or in-license any compositions, methods of use, processes or other third-party intellectual property rights from third parties on reasonable terms, if at all. The licensing and acquisition of third-party intellectual property rights is a competitive area, and a number of more established companies are also pursuing strategies to license or acquire third-party intellectual property rights that we may consider attractive. These established companies may have a competitive advantage over us due to their size, cash resources and greater clinical development and commercialization capabilities.
If we are unable to successfully obtain rights to required third-party intellectual property, rights, our business, financial condition and prospects for growth could suffer.
Confidentiality agreements with employees and others may not adequately prevent disclosure of trade secrets and other proprietary information and may not adequately protect our intellectual property.property.
We rely on trade secrets to protect our proprietary technologies, especially where we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. We rely in part on confidentiality agreements with our employees, consultants, outside scientific collaborators, sponsored researchers and other advisors to protect our trade secrets and other proprietary information. These agreements may not effectively prevent disclosure of confidential information and may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. In addition, others may independently discover our trade secrets and proprietary information. Costly and time-consuming litigation could be necessary to enforce and determine the scope of our proprietary rights, and failure to obtain or maintain trade secret protection could adversely affect our competitive business position.
Our reliance on third parties may require us to share our trade secrets, which increases the possibility that a competitor will discover them or that our trade secrets will be misappropriated or disclosed.
Because we rely on and intend to continue to rely on third parties to manufacture our current and future product candidates, and because we collaborate with various organizations and academic institutions on the advancement of our product candidates, we must, at times, share trade secrets with them. We seek to protect our proprietary technology in part by entering into confidentiality agreements and, if applicable, material transfer agreements, collaborative research agreements, consulting agreements or other similar agreements with our manufacturers, collaborators, advisors, employees and consultants prior to beginning research or disclosing proprietary information. These agreements typically limit the rights of the third parties to use or disclose our confidential information, such as trade secrets. Despite the contractual provisions of such agreements, the need to share trade secrets and other confidential information increases the risk that such trade secrets may become known by our competitors, may be inadvertently incorporated into the technology of others, may be used inappropriately to create new inventions or may be disclosed or used in violation of such agreements.
Our academic collaborators typically have rights to publish data, provided that we are notified in advance and may delay publication for a specified time in order to secure our intellectual property rights arising from the collaboration. In other cases, publication rights are controlled exclusively by us, although in some cases we may share these rights with other parties. We may also conduct joint research and development programs that may require us to share trade secrets under the terms of research and development partnerships or similar agreements. Despite our efforts to protect our trade secrets, our competitors may discover our trade secrets through breach of these agreements, independent development or publication of information including our trade secrets in cases where we do not have proprietary or otherwise protected rights at the time of publication. Given that our proprietary position is based, in part, on our know-how and trade secrets, a competitor’s discovery of our trade secrets or other unauthorized use or disclosure may impair our competitive position and have a material adverse effect on our business.
We may be involved in lawsuits to protect or enforce our patents or the patents of our licensors, which could be expensive, time-consuming and unsuccessful.
Competitors may infringe our patents or the patents of our licensors. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time-consuming. In addition, in an infringement proceeding, a court may decide that a patent is not valid, is unenforceable and/or is not infringed, or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation or defense proceedings could put one or more of our patents at risk of being invalidated or interpreted narrowly and could put our patent applications at risk of not issuing.
Interference proceedings provoked by third parties or brought by us may be necessary to determine the priority of inventions with respect to our patents or patent applications or those of our licensors. An unfavorable outcome could require us to cease using the related technology or to attempt to license rights to it from the prevailing party. Our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms. Our defense of litigation or interference proceedings may fail and, even if successful, may result in substantial costs and distract our management and other employees. We may not be able to prevent, alone or with our licensors, misappropriation of our intellectual property rights, particularly in countries where the laws may not protect those rights as fully as in the United States.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. There could also be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a material adverse effect on the price of our common stock.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees, renewal fees, annuity fees and various other governmental fees on patents and/or applications will be due to be paid to the USPTO and various governmental patent agencies outside of the United States in several stages over the lifetime of the patents and/or applications. We employ an outside firm and rely on our outside counsel to pay these fees due to non-US patent agencies. The USPTO and various non-US governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. Although an inadvertent lapse can be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which non-compliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, our competitors might be able to enter the market which would have a material adverse effect on our business.
Issued patents covering ourproduct candidates could be found invalid or unenforceable if challenged in court.
If we or one of our licensing partners, including MD Anderson or UP, initiate legal proceedings against a third party to enforce a patent covering one of our product candidates, the defendant could counterclaim that the patent covering our product candidate is invalid and/or unenforceable. In patent litigation in the United States, defendant counterclaims alleging invalidity and/or unenforceability are commonplace. Grounds for a validity challenge could be an alleged failure to meet any of several statutory requirements, including lack of novelty, obviousness or non-enablement. Grounds for an unenforceability assertion could be an allegation that someone connected with prosecution of the patent withheld relevant information from the USPTO, or made a misleading statement, during prosecution. Third parties may also raise similar claims before administrative bodies in the United States or abroad, even outside the context of litigation. Such mechanisms include re-examination, post grant review and equivalent proceedings in foreign jurisdictions (e.g., opposition proceedings). Such proceedings could result in revocation or amendment to our patents in such a way that they no longer cover our current and potential product candidates. The outcome following legal assertions of invalidity and unenforceability is unpredictable. With respect to the validity question, for example, we cannot be certain that there is no invalidating prior art, of which we and the patent examiner were unaware during prosecution. If a defendant were to prevail on a legal assertion of invalidity and/or unenforceability, we would lose at least part, and perhaps all, of the patent protection on our current and potential product candidates. Such a loss of patent protection would have a material adverse impact on our business.
We may be subject to claims that our employees, consultants or independent contractors have wrongfully used or disclosed confidential information of third parties or that our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
We currently and in the future may employ individuals who were previously employed at universities or other biotechnology or pharmaceutical companies, including our competitors. Although we try to ensure that our employees, consultants and independent contractors do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or our employees, consultants or independent contractors have inadvertently or otherwise used or disclosed intellectual property, including trade secrets or other proprietary information, of any of our employee’s former employer or other third parties. Litigation may be necessary to defend against these claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel, which could adversely impact our business. Even if we are successful in defending against such claims, litigation could result in substantial costs and result in a diversion of management’s attention.
We may be subject to claims challenging the inventorship or ownership of our patents and other intellectual property.
We may be subject to claims that former employees, collaborators or other third parties have an ownership interest in our patents or other intellectual property. We may have potential ownership disputes arising, for example, from conflicting obligations of consultants, collaborators or others who are involved in developing our current and potential product candidates. Litigation may be necessary to defend against these and other claims challenging inventorship or ownership. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights, such as exclusive ownership of, or right to use, valuable intellectual property. Such an outcome could have a material adverse effect on our business. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.
Changes in U.S. patent law could diminish the value of patents in general, thereby impairing our ability to protect our products.
The United States has enacted and is currently implementing wide-ranging patent reform legislation. Recent U.S. Supreme Court rulings have narrowed the scope of patent protection available in certain circumstances and weakened the rights of patent owners in certain situations. In addition to increasing uncertainty with regard to our ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on decisions by the U.S. Congress, the federal courts and the USPTO, the laws and regulations governing patents could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce our existing patents and patents that we might obtain in the future.
We have not yet registeredin the past and may again in the future have trademark for REQORSA™,applications in the United States and/or certain other countries, and failure to secure such registrationthese registrations could adversely affect our business.business; additionally, we may need to enforce our trademark rights against third parties and expend significant resources to enforce such rights against infringement.
WhileWe have obtained trademark registrations in the United States for GENPREX, ONCOPREX and REQORSA and we may in the future have submitted apending trademark application forapplications in the mark "REQORSA," this mark has not yet been approved by the USPTO.United States and/or certain other countries. During trademark registration proceedings, our application may be rejected. Although we would be given an opportunity to respond to the rejection of a trademark application, we may be unable to overcome such rejection. In addition, with respect to the USPTO and comparable agencies in many foreign jurisdictions, third parties are given an opportunity to oppose pending trademark applications and to seek to cancel registered trademarks. Opposition or cancellation proceedings may be filed against our trademark, and our trademark may not survive such proceedings. Additionally, we may need to enforce our trademark rights against third parties and expend significant additional resources to enforce such rights against infringement. Moreover, any name we propose to use with our current and potential product candidates in the United States must be approved by the FDA, regardless of whether we have registered it, or applied to register it, as a trademark. The FDA typically conducts a review of proposed product names, including an evaluation of the potential for confusion with other product names. If the FDA objects to any of our proposed proprietary product names, we may be required to expend significant additional resources in an effort to identify a suitable substitute name that would qualify under applicable trademark laws, not infringe the existing rights of third parties and be acceptable to the FDA.
We may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting and defending patents with respect to product candidates in all countries throughout the world would be prohibitively expensive, and our intellectual property rights in some countries outside the United States can be less extensive than those in the United States. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as federal and state laws in the United States. Consequently, we may not be able to prevent third parties from practicing our inventions in all countries outside the United States, or from selling or importing products made using our inventions in and into the United States or other jurisdictions. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and further, may export otherwise infringing products to territories where we have patent protection, but enforcement is not as strong as that in the United States. These products may compete with our products and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents, trade secrets and other intellectual property protection, particularly those relating to biotechnology products, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly and our patent applications at risk of not issuing and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate, and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
Risks Related to Employee Matters and Managing Growth
We have no sales, marketing or distribution experience and we will have to invest significant resources to develop those capabilities or enter into acceptable third-party sales and marketing arrangements.
We have no sales, marketing or distribution experience. To develop sales, distribution and marketing capabilities, we will have to invest significant amounts of financial and management resources, some of which will need to be committed prior to any confirmation that our product candidates will be approved by the FDA. For product candidates for which we decide to perform sales, marketing and distribution functions ourselves or through third parties, we could face a number of additional risks, including that we or our third-party sales collaborators may not be able to build and maintain an effective marketing or sales force, and we may experience difficulty in managing the growth of our organization. If we use third parties to market and sell our products, we may have limited or no control over their sales, marketing and distribution activities on which our future revenues may depend.
We may not be able to manage our business effectively if we are unable to attract and retain key personnel and consultants.
As of March 15, 2021,2024, we had 12 full-time employees.26 total employees, all of which were full-time. As we advance our product candidates through preclinical studies and clinical trials, we will need to increase our clinical trial management, product development, manufacturing, regulatory, and administrative headcount to manage these programs. In addition, to meet our obligations as a public company, we may need to increase our general and administrative capabilities. We may not be able to attract or retain qualified personnel and consultants due to the intense competition for qualified personnel and consultants among biotechnology, pharmaceutical and other businesses. If we are not able to attract and retain necessary personnel and consultants to accomplish our business objectives, we may experience constraints that will significantly impede the achievement of our development objectives, our ability to raise additional capital and our ability to implement our business strategy.
We are highly dependent on the development, manufacturing, regulatory, commercialization and business development expertise of our management team, key employees and consultants. Any of our executive officers or key employees or consultants may terminate their employment or engagement with us at any time. If we lose one or more of our executive officers or key employees or consultants, our ability to implement our business strategy successfully could be seriously harmed. Replacing executive officers, key employees and consultants may be difficult and may take an extended period of time because of the limited number of individuals in our industry with the breadth of skills and experience required to develop, gain regulatory approval of and commercialize products successfully. Competition to hire and retain employees and consultants from this limited pool is intense, and we may be unable to hire, train, retain or motivate these additional key personnel and consultants. Our failure to retain key personnel or consultants could materially harm our business.
As previously reported in 2022, our Chairman, President and Chief Executive Officer, Rodney Varner, had been diagnosed with a cutaneous lymphoma and has been undergoing chemotherapy and other treatments since then. Mr. Varner has been continuing in his roles at the Company during this period and expects to continue to do so, although he has limited some business activities and travel. Our board of directors had taken steps in 2022 to ensure smooth continuity of business priorities and operations during periods when Mr. Varner was receiving treatments or otherwise unavailable due to his condition. For business continuity, our board of directors has determined that Ryan Confer, our Chief Financial Officer, can and shall act in covering when Mr. Varner is unavailable.
In addition to our management team and key employees, we have scientific and clinical advisors and consultants who assist us in formulating our research, development and clinical strategies. These advisors are not our employees and may have commitments to, or consulting or advisory contracts with, other entities that may limit their availability to us and typically they will not enter into non-compete agreements with us. If a conflict of interest arises between their work for us and their work for another entity, we may lose their services. In addition, our advisors may have arrangements with other companies to assist those companies in developing products or technologies that may compete with ours.
We may use our financial and human resources to pursue a particular research program or product candidate and fail to capitalize on programs or product candidates that may be more profitable or for which there is a greater likelihood of success.
Because we have limited resources, we may forego or delay pursuit of opportunities with certain programs or product candidates or for indications that later prove to have greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities. Our spending on research and development programs for product candidates may not yield any commercially viable products. If we do not accurately evaluate the commercial potential or target market for a particular product candidate, we may relinquish valuable rights to that product candidate through strategic collaboration, licensing or other royalty arrangements in cases in which it would have been more advantageous for us to retain sole development and commercialization rights to such product candidate, or we may allocate internal resources to a product candidate in a therapeutic area in which it would have been more advantageous to enter into a partnering arrangement.
If we engage in future acquisitions or strategic partnerships, this may increase our capital requirements, dilute our stockholders, cause us to incur debt or assume contingent liabilities, and subject us to other risks.
We may evaluate and enter into various acquisitions and strategic partnerships, including licensing or acquiring additional products, intellectual property rights, technologies, or businesses. Any potential acquisition or strategic partnership may entail numerous risks, including:
increased operating expenses and cash requirements;
the assumption of additional indebtedness or contingent liabilities;
the issuance of our equity securities;
assimilation of operations, intellectual property and products of an acquired company, including difficulties associated with integrating new personnel;
difficulties in achieving anticipated cost savings, synergies, business opportunities, and growth prospects from any business combination;
the diversion of our management’s attention from our existing product programs and initiatives in pursuing such a strategic merger or acquisition;
retention of key employees, the loss of key personnel, and uncertainties in our ability to maintain key business relationships;
risks and uncertainties associated with the other party to such a transaction, including the prospects of that party and their existing products or product candidates and marketing approvals; and
our inability to generate revenue from acquired technology and/or products sufficient to meet our objectives in undertaking the acquisition or even to offset the associated acquisition and maintenance costs.
| ● | increased operating expenses and cash requirements; | |
| ● | the assumption of additional indebtedness or contingent liabilities; | |
| ● | the issuance of our equity securities; | |
| ● | assimilation of operations, intellectual property and products of an acquired company, including difficulties associated with integrating new personnel; | |
| ● | difficulties in achieving anticipated cost savings, synergies, business opportunities, and growth prospects from any business combination; | |
| ● | the diversion of our management’s attention from our existing product programs and initiatives in pursuing such a strategic merger or acquisition; | |
| ● | retention of key employees, the loss of key personnel, and uncertainties in our ability to maintain key business relationships; | |
| ● | risks and uncertainties associated with the other party to such a transaction, including the prospects of that party and their existing products or product candidates and marketing approvals; and | |
| ● | our inability to generate revenue from acquired technology and/or products sufficient to meet our objectives in undertaking the acquisition or even to offset the associated acquisition and maintenance costs. |
Moreover, we may not be able to locate suitable acquisition opportunities and such inability could impair our ability to grow or obtain access to technology or products that may be important to the development of our business.
Risks Related to our Securities
The market price of our common stock may be highly volatile, and you may lose all or part of your investment.
The market price of our common stock has been volatile in the past and is likely to be volatile.volatile in the future. Our stock price could be subject to wide fluctuations in response to a variety of factors, including the following:
inability to obtain additional funding;
adverse results or delays in preclinical or clinical trials;
reports of adverse events in other gene therapy products or clinical trials of such products;
| ● | inability to obtain additional funding; | |
| ● | adverse results or delays in preclinical or clinical trials; | |
● ● | reports of adverse events in other gene therapy products or clinical trials of such products; manufacturing and supply issues related to our existing or future products; | |
| ● | any delay in filing an IND or BLA for our product candidates and any adverse development or perceived adverse development with respect to the FDA’s review of that IND or BLA; | |
| ● | failure to develop successfully and commercialize our product candidates; | |
| ● | failure to maintain our existing strategic collaborations or enter into new collaborations; | |
| ● | failure by us or our licensors and strategic collaboration partners to prosecute, maintain or enforce our intellectual property rights; | |
| ● | changes in laws or regulations applicable to our products and product candidates; | |
| ● | inability to obtain adequate product supply for our product candidates or inability to do so at acceptable prices; | |
| ● | adverse regulatory decisions; | |
| ● | introduction of new products, services or technologies by our competitors; | |
| ● | failure to meet or exceed financial projections we may provide to the public; | |
| ● | failure to meet or exceed the financial projections of the investment community; | |
| ● | the perception of the pharmaceutical and biotechnology industries by the public, legislatures, regulators and the investment community; | |
| ● | announcements of significant acquisitions, strategic partnerships, joint ventures, capital commitments or other material corporate transactions or events by us, our strategic collaboration partners or our competitors; | |
● | disputes or other developments relating to proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our technologies; | |
● | additions or departures of key scientific or management personnel; | |
● | significant lawsuits, including patent or stockholder litigation; | |
● | changes in the market valuations of similar companies; | |
● | sales of our common stock by us or our stockholders; | |
● | trading volume of our common stock; | |
| ● | material announcements or changes impacting our common stock or our capitalization, or the perception that such changes could occur; and | |
● | General economic conditions in the United States and abroad; and
other events or factors, many of which may be out of our control, including, but not limited to, pandemics, |
In addition, companies trading in the stock market in general, and The Nasdaq Capital Market in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies. Broad market and industry factors may negatively affect the market price of our common stock, regardless of our actual operating performance.
An active, liquid and orderly market for our common stock may not be sustained, and you may not be able to sell your common stock.
Our common stock trades on the Nasdaq Capital Market. We cannot assure you that an active trading market for our common stock will be sustained. The lack of an active market may impair your ability to sell the common stock at the time you wish to sell them or at a price that you consider reasonable. An inactive market may also impair our ability to raise capital by selling common stock and may impair our ability to acquire other businesses, applications or technologies using our common shares as consideration, which, in turn, could materially adversely affect our business.
We are currently listed on The Nasdaq Capital Market. If we are unable to maintain listing of our securities on Nasdaq or any stock exchange, our stock price could be adversely affected and the liquidity of our stock and our ability to obtain financing could be impaired and it may be more difficult for our stockholders to sell their securities.
Although our common stock is currently listed on The Nasdaq Capital Market, we may not be able to continue to meet the exchange’s minimum listing requirements, including the minimum bid price requirement and other applicable corporate governance requirements, or those of any other national exchange. If we are unable to maintain listing on Nasdaq, or if a liquid market for our common stock does not develop or is sustained, our common stock may remain thinly traded. The Listing Rules of Nasdaq require listing issuers to comply with certain standards in order to remain listed on its exchange. If, for any reason, we should fail to maintain compliance with these listing standards and Nasdaq should delist our securities from trading on its exchange and we are unable to obtain listing on another national securities exchange, a reduction in some or all of the following may occur, each of which could have a material adverse effect on our stockholders:
the liquidity of our common stock;
the market price of our common stock;
our ability to obtain financing for the continuation of our operations;
the number of investors that will consider investing in our common stock;
the number of market makers in our common stock;
the availability of information concerning the trading prices and volume of our common stock; and
the number of broker-dealers willing to execute trades in shares of our common stock;
● | the liquidity of our common stock; |
● | the market price of our common stock; |
● | our ability to obtain financing for the continuation of our operations; |
● | the number of investors that will consider investing in our common stock; |
● | the number of market makers in our common stock; |
● | the availability of information concerning the trading prices and volume of our common stock; and |
● | the number of broker-dealers willing to execute trades in shares of our common stock; |
Unstable market and economic conditions may have serious adverse consequences on our business, financial condition and stock price.
Global credit and financial markets have experienced volatility and disruptions in past years, including severely diminished liquidity and credit availability, declines in consumer confidence, declines in economic growth, increases in unemployment rates, concerns about medical epidemics, and uncertainty about economic stability. We cannot assure you that further deterioration in credit and financial markets and confidence in economic conditions will not occur, particularly as a result of the ongoing COVID-19 pandemic.occur. Our general business strategy may be adversely affected by any such economic downturn, volatile business environment or continued unpredictable and unstable market conditions. If the current equity and credit markets deteriorate, or do not improve, it may make any necessary debt or equity financing more difficult, more costly and more dilutive. Failure to secure any necessary financing in a timely manner and on favorable terms could have a material adverse effect on our growth strategy, financial performance and stock price and could require us to delay or abandon clinical development plans. In addition, there is a risk that one or more of our current service providers, manufacturers and other partners may not survive difficult economic times, which could directly affect our ability to attain our operating goals on schedule and on budget.
We may be at risk of securities class action litigation.
We may be at risk of securities class action litigation. In the past, biotechnology and pharmaceutical companies have experienced significant stock price volatility, particularly when associated with binary events such as clinical trials and product approvals. If we face such litigation, it could result in substantial costs and a diversion of management’s attention and resources, which could harm our business and results in a decline in the market price of our common stock.
Failure to maintain effective internal control over our financial reporting in accordance with Section 404 of the Sarbanes-Oxley Act of 2002, as amended (“Sarbanes-Oxley Act”) could cause our financial reports to be inaccurate.
We are required pursuant to Section 404 of the Sarbanes-Oxley Act, or Section 404, to maintain internal control over financial reporting and to assess and report on the effectiveness of those controls. This assessment includes disclosure of any material weaknesses identified by our management in our internal control over financial reporting. Although we prepare our financial statements in accordance with accounting principles generally accepted in the United States, our internal accounting controls may not meet all standards applicable to companies with publicly traded securities. If we fail to implement any required improvements to our disclosure controls and procedures, we may be obligated to report control deficiencies, in which case we could become subject to regulatory sanction or investigation. Further, such an outcome could damage investor confidence in the accuracy and reliability of our financial statements.
Effective internal controls over financial reporting are necessary for us to provide reliable financial reports and, together with adequate disclosure controls and procedures, are designed to prevent fraud. Any failure to implement required or improved controls, or difficulties encountered in their implementation, could cause us to fail to meet our reporting obligations. We have designed, implemented and tested the internal control over financial reporting required to comply with this obligation, which was and is time consuming, costly, and complicated. A control system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the control system’s objectives will be met.
Our management has concluded that our internal controls over financial reporting were, and continue to be, ineffective, and as of the year ended December 31, 20202023 as a result of a material weaknessweaknesses in our internal controls due to the lack of segregation of duties.duties between accounting and other functions and the absence of sufficient depth of in-house accounting personnel with the ability to properly account for complex transactions. While management is working to remediate thethese material weakness,weaknesses, there is no assurance that such changes, when economically feasible and sustainable, will remediate the identified material weaknesses or that the controls will prevent or detect future material weaknesses. If we are not able to remediate the material weaknesses or maintain effective internal control over financial reporting, this could result in a material misstatement in our consolidated financial statements including related disclosures, may be inaccurate,and a failure to meet our reporting and financial obligations, which could have a material adverse effect on our business.
We have no intention of declaring dividends in the foreseeable future.
We have never declared or paid cash dividends on our capital stock, and we do not currently anticipate declaring any dividends in the foreseeable future. We anticipate that we will retain all future earnings for the development, operation, and expansion of our business. Any future determination to declare dividends will be made at the discretion of our board of directors and will depend on, among other factors, our financial condition, operating results, capital requirements, contractual restrictions, general business conditions and other factors that our board of directors may deem relevant. Investors in our common stock should not expect to receive dividend income on their investment, and investors will be dependent on the appreciation of our common stock, if any, to earn a return on their investment.
We arewill likely incur increased costs and devote additional management time to public company reporting and compliance obligations as a result of exiting “emerging growth company” status.
We no longer qualify as an emerging growth company and smaller reporting company, and we cannot be certain ifunder the reduced reporting requirements applicableJOBS Act (our eligibility to emerging growth companies and smaller reporting companies will make our common stock less attractive to investors.
We arequalify as an emerging growth company as defined inended on December 31, 2023, the JOBS Act. For as long aslast day of our fiscal year following the fifth anniversary of our initial public offering). While we continue to bewere an emerging growth company, we maywere able to take advantage of exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies, including not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements and exemptions from the requirements of holding nonbinding advisory votes on executive compensation and stockholder approval of any golden parachute payments not previously approved. We will remain an “emerging growth company” until the earliest of (i) the last day of the fiscal year in which
Now that we have total annual gross revenues of $1.07 billion or more; (ii) the last day of our fiscal year following the fifth anniversary of the date of our initial public offering; (iii) the date on which we have issued more than $1 billion in nonconvertible debt during the previous three years; or (iv) the date on which we are deemed to be a large accelerated filer under the rules of the SEC.
Even after we no longer qualify as anexited emerging growth company status and we may still qualify as a “smaller reporting company” which would allow usare no longer eligible to take advantage of many of the samecorresponding exemptions, from disclosure requirements including not being requiredwe expect our management and other personnel to devote more time and the Company to incur additional costs to comply with the auditor attestationmore stringent reporting requirements of Section 404 of the Sarbanes-Oxley Act and reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements. We cannot predict if investors will find our common stock less attractive because we may rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile.
Under the JOBS Act, emerging growth companies can also delay adopting new or revised accounting standards until such time as those standards applyapplicable to private companies; however, we have irrevocably elected not to avail ourselves of this exemption from new or revised accounting standards and, therefore, will be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies. As a result, changes in rules of U.S. generally accepted accounting principles or their interpretation, the adoption of new guidance or the application of existing guidance to changes inWe expect that compliance with these requirements will increase our business could significantly affect our financial position and results of operations.
Financial reporting obligations of being a public company in the United States are expensive and time-consuming, and our management will be required to devote substantial time to compliance matters.
As a publicly traded company we incur significant legal accounting and other expenses. The obligations of being a public company in the United States require significant expenditures and places significant demands on our management and other personnel, including costs resulting from public company reporting obligations under the Securities Exchange Act of 1934, as amended (“Exchange Act”) and the rules and regulations regarding corporate governance practices, including those under the Sarbanes-Oxley Act, the Dodd-Frank Wall Street Reform and Consumer Protection Act, and the listing requirements of The Nasdaq Capital Market. These rules require the establishment and maintenance of effective disclosure and financial controlscompliance costs and procedures, internal control over financial reporting and changes in corporate governance practices, among many other complex rules that are often difficult to implement, monitor and maintain compliance with. Moreover, despite recent reforms made possible by the JOBS Act, the reporting requirements, rules, and regulations will make some activities more time-consumingtime consuming and costly, particularly after we are no longer an “emerging growth company”costly. We cannot predict or “smaller reporting company.” In addition, we expect these rules and regulations to make it more difficult and more expensive for us to obtain director and officer liability insurance. Our management and other personnel will need to devote a substantialestimate the amount of time to ensure that we comply with all of these requirements and to keep pace with new regulations, otherwiseadditional costs we may fall outincur as a result of compliance and risk becoming subject to litigationour exit from emerging growth company status, or being delisted, among other potential consequences.the timing of the incurrence of such costs.
Sales of a substantial number of shares of our common stock in the public market could cause our stock price to fall.
If our existing stockholders sell, or indicate an intention to sell, substantial amounts of our common stock in the public market, the market price of our common stock could decline. As of December 31, 2023, we had outstanding options to purchase an aggregate of 285,883 shares of our common stock at a weighted average exercise price of $121.11 per share and warrants to purchase an aggregate of 346,440 shares of our common stock at a weighted average exercise price of $57.79 per share. The exercise of such outstanding options and warrants will result in further dilution of your investment, even if there is no relationship between such sales and the performance of our business.
We are unable to predict the effect that sales may have on the market price of our common stock. If any shares of our common stock are sold, or if it is perceived that they will be sold, in the public market, the market price of our common stock could decline.
Future salesand issuances of our securities could result in additional dilution of the percentage ownership of our shareholdersstockholders and could cause our share price to fallfall..
We expect that significant additional capital will be needed in the future to continue our planned operations, including research and development, increased marketing, hiring new personnel, commercializing our products, and continuing activities as an operating public company. To the extent we raise additional capital by issuing equity securities, our shareholdersstockholders may experience substantial dilution. We may sell common stock, convertible securities or other equity securities in one or more transactions at prices and in a manner we determine from time to time. If we sell common stock, convertible securities or other equity securities in more than one transaction, investors may be materially diluted by subsequent sales. SuchWe may sell a substantial number of shares of our common stock pursuant to our existing At The Market Offering Agreement with H.C. Wainwright, pursuant to which we have the discretion to deliver placement notices to H.C. Wainwright at any time throughout the term of the At The Market Offering Agreement covering up to such number or dollar amount of shares of our common stock as registered on the prospectus supplement covering the ATM offering, as may be amended or supplemented from time to time; pursuant to such agreement, we have the discretion, subject to market demand, to vary the timing, prices, and quantity of shares sold, and there is no minimum or maximum sales price. Any sales of equity securities, whether pursuant to our At The Market Offering Agreement or otherwise, may also result in material dilution to our existing shareholders,stockholders, and new investors could gain rights superior to our existing shareholders.stockholders.
If securities or industry analysts do not publish research or reports about us, or if they adversely change their recommendations regarding our common stock, then our stock price and trading volume could decline.
The trading market for our common stock will be influenced by the research and reports that industry or securities analysts publish about us, our industry and our market. If no analyst elects to cover us and publish research or reports about us, the market for our common stock could be severely limited and our stock price could be adversely affected. In addition, if one or more analysts ceases coverage of us or fails to regularly publish reports on us, we could lose visibility in the financial markets, which in turn could cause our stock price or trading volume to decline. If one or more analysts who elect to cover us issue negative reports or adversely change their recommendations regarding our common stock, our stock price could decline.
Certain provisions in our organizational documents could enable our board of directors to prevent or delay a change of control.
We are authorized to issue up to 10,000,000 shares of preferred stock, none of which are outstanding as of March 15, 2021.2024. This preferred stock may be issued in one or more series, the terms of which may be determined at the time of issuance by our board of directors without further action by stockholders. The terms of any series of preferred stock may include voting rights (including the right to vote as a series on particular matters), preferences as to dividend, liquidation, conversion and redemption rights and sinking fund provisions. The issuance of any preferred stock could materially adversely affect the rights of the holders of our common stock, and therefore, reduce the value of our common stock. The ability of our board of directors to issue preferred stock also could have the effect of discouraging unsolicited acquisition proposals, thus adversely affecting the market price of our common stock.
In addition, our organizational documents contain provisions that may have the effect of discouraging, delaying or preventing a change of control of, or unsolicited acquisition proposals, that a stockholder might consider favorable. These include provisions:
requiring at least 66-2/3% of the voting power of all of our then-outstanding shares of capital stock entitled to vote generally in the election of directors, voting together as a single class, to amend the Amended and Restated Bylaws;
providing that the authorized number of directors may be changed only by resolution of the board of directors;
providing that the directors may only be removed with cause and the affirmative vote of the holders of at least 66-2/3% of the voting power of all of our then outstanding shares of capital stock entitled to vote generally at the election of directors;
providing that all vacancies, including newly created directorships, may, except as otherwise required by law, be filled by the affirmative vote of a majority of directors then in office, even if less than a quorum;
dividing our board of directors into three classes;
requiring that any action to be taken by our stockholders must be effected at a duly called annual or special meeting of stockholders and not be taken by written consent;
providing that stockholders seeking to present proposals before a meeting of stockholders or to nominate candidates for election as directors at a meeting of stockholders must provide notice in writing in a timely manner and also specify requirements as to the form and content of a stockholder’s notice;
that do not provide for cumulative voting rights (therefore allowing the holders of a majority of the shares of common stock entitled to vote in any election of directors to elect all of the directors standing for election, if they should so choose); and
providing that special meetings of our stockholders may be called only by the Chairman of the board, our Chief Executive Officer or by the board of directors pursuant to a resolution adopted by a majority of the total number of authorized directors.
● | requiring at least 66-2/3% of the voting power of all of our then-outstanding shares of capital stock entitled to vote generally in the election of directors, voting together as a single class, to amend the Amended and Restated Bylaws; |
● | providing that the authorized number of directors may be changed only by resolution of the board of directors; |
● | providing that the directors may only be removed with cause and the affirmative vote of the holders of at least 66-2/3% of the voting power of all of our then outstanding shares of capital stock entitled to vote generally at the election of directors; |
● | providing that all vacancies, including newly created directorships, may, except as otherwise required by law, be filled by the affirmative vote of a majority of directors then in office, even if less than a quorum; |
● | dividing our board of directors into three classes; |
● | requiring that any action to be taken by our stockholders must be effected at a duly called annual or special meeting of stockholders and not be taken by written consent; |
● | providing that stockholders seeking to present proposals before a meeting of stockholders or to nominate candidates for election as directors at a meeting of stockholders must provide notice in writing in a timely manner and also specify requirements as to the form and content of a stockholder’s notice; |
● | that do not provide for cumulative voting rights (therefore allowing the holders of a majority of the shares of common stock entitled to vote in any election of directors to elect all of the directors standing for election, if they should so choose); and |
● | providing that special meetings of our stockholders may be called only by the Chairman of the board, our Chief Executive Officer or by the board of directors pursuant to a resolution adopted by a majority of the total number of authorized directors. |
In addition, Delaware law makes it difficult for stockholders that recently have acquired a large interest in a corporation to cause the merger or acquisition of the corporation against the directors’ wishes. Under Section 203 of the Delaware General Corporation Law, a Delaware corporation may not engage in any merger or other business combination with an interested stockholder for a period of three years following the date that the stockholder became an interested stockholder except in limited circumstances, including by approval of the corporation’s board of directors.
Market and economic conditions may negatively impact our business, financial condition and share price.
Concerns over inflation, energy costs, geopolitical issues, the U.S. mortgage market and a declining real estate market, unstable global credit markets and financial conditions, and volatile oil prices have led to periods of significant economic instability, diminished liquidity and credit availability, declines in consumer confidence and discretionary spending, diminished expectations for the global economy and expectations of slower global economic growth going forward, increased unemployment rates, and increased credit defaults in recent years. Our general business strategy may be adversely affected by any such economic downturns, volatile business environments and continued unstable or unpredictable economic and market conditions. If these conditions continue to deteriorate or do not improve, it may make any necessary debt or equity financing more difficult to complete, more costly, and more dilutive. Failure to secure any necessary financing in a timely manner and on favorable terms could have a material adverse effect on our growth strategy, financial performance, and share price and could require us to delay or abandon development or commercialization plans.
Our Amended and Restated Certificate of Incorporation andAmended and Restated Bylaws containscontain an exclusive forum provision with respect to certain actions which may limit a stockholder’sstockholder’s ability to bring a claim in a judicial forum that it finds favorable and discourage lawsuits against us or our current or former directors or officers and/or stockholders in such capacity.
Our Amended and Restated Certificate of Incorporation, as may be further amended and restated from time to time, and Amended and Restated Bylaws, as may be further amended from time to time, provide that, unless we consent in writing to the selection of an alternative forum, the following actions must be brought solely and exclusively in the Court of Chancery of the State of Delaware (i) any derivative action or proceeding brought on behalf of us; (ii) any action asserting a claim of breach of a fiduciary duty owed by any director, officer or other employee of the Company to us or our stockholders; (iii) any action asserting a claim against us or any director or officer or other employee of the Company arising pursuant to any provision of the DGCL, our certificate of incorporation or our bylaws; or (iv) any action asserting a claim against us or any director or officer or other employee of the Company governed by the internal affairs doctrine. We believe that the exclusive forum provision may not apply to suits brought to enforce any liability or duty created by the Securities Act or the Exchange Act or any other claim for which the federal courts have exclusive jurisdiction. We believe that to the extent that any such claims may be based upon federal law claims, Section 27 of the Exchange Act creates exclusive federal jurisdiction over all suits brought to enforce any duty or liability created by the Exchange Act or the rules and regulations thereunder. Furthermore, we believe that Section 22 of the Securities Act creates concurrent jurisdiction for federal and state courts over all suits brought to enforce any duty or liability created by the Securities Act or the rules and regulations thereunder. This choice of forum provision may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, officers or other employees, which may discourage such lawsuits against us and our directors, officers or other employees. Alternatively, if a court were to find the choice of forum provision contained in our Amended and Restated Certificate of Incorporation or Amended and Restated Bylaws to be inapplicable or unenforceable in an action, we may incur additional costs associated with resolving such action in other jurisdictions, which could have a material adverse effect on our business, results of operations, and financial condition.
General Risk Factors
Obligations associated with being a public company in the United States are expensive and time-consuming, and our management will be required to devote substantial time tocompliance matters.
As a publicly traded company we incur significant legal, accounting and other expenses. The obligations of being a public company in the United States require significant expenditures and places significant demands on our management and other personnel, including costs resulting from public company reporting obligations under the Securities Exchange Act of 1934, as amended (“Exchange Act”) and the rules and regulations regarding corporate governance practices, including those under the Sarbanes-Oxley Act, the Dodd-Frank Wall Street Reform and Consumer Protection Act, and the listing requirements of The Nasdaq Capital Market. These rules require the establishment and maintenance of effective disclosure and financial controls and procedures, internal control over financial reporting and changes in corporate governance practices, among many other complex rules that are often difficult to implement, monitor and maintain compliance with. Moreover, the reporting requirements, rules, and regulations will make some activities more time-consuming and costly, particularly now that we are no longer an “emerging growth company” (our eligibility to qualify as an emerging growth company ended on December 31, 2023, the last day of our fiscal year following the fifth anniversary of our initial public offering). Further, in the event that in the future we were to no longer be eligible to qualify as a “smaller reporting company,” and/or if we become subject to the requirements applicable to accelerated filers or large accelerated filers, including complying with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act, our compliance burdens and expenses will further increase. In addition, and as a general matter, we expect the foregoing public company rules and regulations to make it more difficult and more expensive for us to obtain director and officer liability insurance. Our management and other personnel will need to devote a substantial amount of time to ensure that we comply with all of these requirements and to keep pace with new regulations, otherwise we may fall out of compliance and risk becoming subject to litigation or being delisted, among other potential consequences.
Business disruptions could seriously harm our future revenue and financial condition and increase our costs and expenses.
Our operations, and those of our CROs, contract manufacturers and other contractors, vendors and consultants, could be subject to power shortages, telecommunications failures, wildfires, water shortages, floods, earthquakes, hurricanes, typhoons, fires, extreme weather conditions, medical epidemics and pandemics and other natural or man-made disasters or business interruptions. The occurrence of any of these business disruptions could seriously harm our operations and financial condition and increase our costs and expenses. Our ability to obtain clinical supplies of our current and potential product candidates could be disrupted if the operations of our contract manufacturers are affected by a man-made or natural disaster or other business interruption.
In addition, the global macroeconomic environment could be negatively affected by, among other things, new variants of the COVID-19 pandemic or other epidemics, instability in global economic markets, increased U.S. trade tariffs and trade disputes with other countries, supply chain weaknesses, instability in the global credit markets, severely diminished liquidity and credit availability, rising interest and inflation rates, declines in consumer confidence, declines in economic growth, increases in unemployment rates, uncertainty about economic stability, instability in the geopolitical environment, the Russian invasion of the Ukraine and other political tensions, including in the Middle East, and foreign governmental debt concerns. In 2023, the closures of Silicon Valley Bank, or SVB, and Signature Bank and their placement into receivership with the Federal Deposit Insurance Corporation, or the FDIC, created bank-specific and broader financial institution liquidity risk and concerns. Although the Department of the Treasury, the Federal Reserve, and the FDIC jointly released a statement that depositors at SVB and Signature Bank would have access to their funds, even those in excess of the standard FDIC insurance limits, under a systemic risk exception, future adverse developments with respect to specific financial institutions or the broader financial services industry may lead to market-wide liquidity shortages, impair the ability of market participants to access near-term working capital needs, and create additional market and economic uncertainty. There can be no assurance that any such continuing or future credit and financial market instability, liquidity shortages and a deterioration in confidence in economic conditions will not occur. Such challenges have caused, and may continue to cause, uncertainty and instability in local economies and in global financial markets, which could adversely affect our business, financial condition or results of operations. If these conditions continue to deteriorate or do not improve, it may make any necessary debt or equity financing more difficult to complete, more costly, and more dilutive. Failure to secure any necessary financing in a timely manner and on favorable terms could have a material adverse effect on our growth strategy, financial performance, and share price and could require us to delay or abandon development or commercialization plans.
We do not carry insurance for all categories of risk that our business may encounter. There can be no assurance that we will secure adequate insurance coverage or that any such insurance coverage will be sufficient to protect us from potential liability in the future. Any significant uninsured liability may require us to pay substantial amounts, which may adversely affect our financial position and results of operations.
We may be at risk of securities class action litigation.
We may be at risk of securities class action litigation. In the past, biotechnology and pharmaceutical companies have experienced significant stock price volatility, particularly when associated with binary events such as clinical trials and product approvals. If we face such litigation, it could result in substantial costs and a diversion of management’s attention and resources, which could harm our business and results in a decline in the market price of our common stock.
Item 1B. Unresolved Staff Comments.
None.
Cybersecurity Risk Management
We, like other companies in our industry, face several cybersecurity risks in connection with our business. Our business strategy, results of operations, and financial condition have not, to date, been affected by risks from cybersecurity threats. During the reporting period, we have not experienced any material cyber incidents, nor have we experienced a series of immaterial incidents, which would require disclosure.
In the ordinary course of our business, we use, store and process data including data of our employees, partners, collaborators, and vendors. To effectively prevent, detect, and respond to cybersecurity threats, we maintain a cyber risk management program, which is comprised of a wide array of policies, standards, architecture, and processes. The cyber risk management program falls under the responsibility of our Director of IT & GxP Quality Systems, who has more than 15 years of experience, including cross-functional expertise in IT compliance and GXP systems. Under the guidance of our Director of IT & GxP Quality Systems, reporting up to our Chief Financial Officer, we develop, maintain, and evidence the policies, standards, and processes in a manner consistent with applicable legal requirements. We also utilize a variety of cybersecurity software from reputable vendors in cybersecurity.
We have implemented a cybersecurity risk management program that is designed to identify, assess, and mitigate risks from cybersecurity threats to this data and our systems and ensure the effectiveness of our security controls. Our cybersecurity risk management program is Systems and Organization Controls (SOC)-certified and incorporates a number of components, including information security program assessments, continuous monitoring of critical risks from cybersecurity threats using automated tools, internal audits, and employee training. We deploy a wide range of security tools across the environment, require multifactor authentication, hold client data on a separate virtual private cloud (VPC), and implement access control policies that further limit access to data within the systems.
We periodically conduct internal audits and are consistently evaluating our cyber readiness. As a result, we have not identified any material cybersecurity risks and are continuously hardening our environment, systems and infrastructure to reduce our vulnerability to attack. Additionally, our program includes regular cybersecurity training for all employees.
For further information regarding cybersecurity risks, please refer to “Risk Factors – Risks Related to Development and Commercialization of Our Current and Future Product Candidates” and other risks described in the “Risk Factors” section of this Annual Report on Form 10-K.
Governance
Our board of directors is responsible for the oversight of cybersecurity risk management. The board of directors delegates oversight of the cybersecurity risk management program to the audit committee of the board of directors (the “Audit Committee”). Our Chief Financial Officer reports to the Audit Committee. The Audit Committee updates the board of directors on our cybersecurity risk management program, including any critical cybersecurity risks, ongoing cybersecurity initiatives and strategies, and applicable regulatory requirements and industry standards on an annual and as-needed basis.
Our corporate and executiveprincipal offices are located at 1601 Trinity Street, Bldg. B, #3.312.09,3300 Bee Cave Road, #650-227, Austin, Texas 78712. Our lease for such office expires on April 30, 2021.78746. We operate primarily as a virtual company, and we believe our current facilities and those that we believe are available to us are sufficient to meet our current needs and that suitable space will be available as and when needed. We do not own any real property.
From time to time, we may becomebe involved in various lawsuits and legal proceedings. Litigationproceedings that arise during the ordinary course of business. Although the results of legal proceedings cannot be predicted with certainty, we do not currently have any pending litigation to which we are a party or to which our property is subject that we believe to inherent uncertainties,be material. Regardless of the outcome, litigation can be costly and an adverse result in these or othertime consuming, and it can divert management’s attention from important business matters may arise from time to time that may harmand initiatives, negatively impacting our business. We are currently not aware of any such legal proceedings or claims that will have, individually or in the aggregate, a material adverse effect on our business, financial condition or operating results.overall operations.
Item 4. Mine Safety Disclosures.
Not applicable.
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
Market Information
Our common stock began trading on The Nasdaq Capital Market under the symbol “GNPX” on March 29, 2018. Prior to that date, there was no public trading market for our common stock.
Holders of Record
As of March 15, 2021,25, 2024, there were approximately 155158 stockholders of record of our common stock. The actual number of stockholders is greater than this number of record holders and includes stockholders who are beneficial owners but whose shares are held in street name by brokers and other nominees. This number of holders of record also does not include stockholders whose shares may be held in trust or by other entities.
Dividend Policy
We have never declared or paid cash dividends on our capital stock. We currently intend to retain all available funds and any future earnings for use in the operation of our business and do not anticipate paying any dividends on our common stock in the foreseeable future. Any future determination to declare dividends will be made at the discretion of our board of directors and will depend on, among other factors, our financial condition, operating results, capital requirements, contractual restrictions, general business conditions and other factors that our board of directors may deem relevant.
Recent Sales of Unregistered Securities
For the yearquarter ended December 31, 2020,2023, we issued and sold the following unregistered securities:
1) | On October | |
|
|
The foregoing issuance of securities was not registered under the Securities Act or the securities laws of any state, and the securities were offered and issued in reliance on the exemption from registration under the Securities Act afforded by Section 4(a)(2).
During the year ended December 31, 2023, there were no other unregistered sales of our securities except as previously reported in a Current Report on Form 8-K or a Quarterly Report on Form 10-Q.
Item 6. Selected Financial Data.[Reserved]
As a smaller reporting company, we are not required to provide the information required by this item.
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
This Management’s Discussion and Analysis of Financial Condition and Results of Operations (“MD&A”) contains certain forward-looking statements. Historical results may not indicate future performance. Our forward-looking statements reflect our current views about future events, are based on assumptions and are subject to known and unknown risks and uncertainties that could cause actual results to differ materially from those contemplated by these statements. Factors that may cause differences between actual results and those contemplated by forward- looking statements include, but are not limited to, those discussed in “Risk Factors.” We undertake no obligation to publicly update or revise any forward-looking statements, including any changes that might result from any facts, events, or circumstances after the date hereof that may bear upon forward-looking statements. Furthermore, we cannot guarantee future results, events, levels of activity, performance, or achievements. All amounts in this report are in United States (“U.S.”) dollars, unless otherwise noted.
Overview
We are a clinical stage gene therapy company focused on developing life-changing treatmentspioneering the development of gene-based therapies for cancer and diabetes.large patient populations with unmet medical needs. Our lead cancer drug candidate, REQORSA™ Immunogene therapy drug (sometimes referred to as GPX-001), is being developed to treat NSCLC. The active agent in REQORSA is a TUSC2 gene expressing plasmid that is encapsulatedoncology platform utilizes our systemic, non-viral ONCOPREX® Delivery System which uses lipid-based nanoparticles in a DOTAP cholesterol nanoparticle. TUSC2 is alipoplex form to deliver tumor suppressor gene which has both tumor killing (via apoptosis) and immunomodulatory effects. We utilize our novel proprietary ONCOPREX® Nanoparticle Delivery System to deliver the TUSC2 gene expressing plasmidgene-expressing plasmids to cancer cells. The TUSC2 geneproduct is one of a series of genes whose therapeutic useadministered intravenously, where it is coveredtaken up by our exclusive worldwide licenses from MD Anderson.
We are planning to initiate our Acclaim-1 and Acclaim-2 clinical trialstumor cells that then express tumor suppressor proteins that were deficient in 2021. Acclaim-1 is a Phase 1/2 clinical trial using a combination of REQORSA with AstraZeneca PLC’s Tagrisso® in patients with late-stage NSCLC with mutated epidermal growth factor receptors ("EGFRs") whose disease progressed after treatment with Tagrisso. In January 2020, we received FDA Fast Track Designation for the Acclaim-1 patient population. Acclaim-2 is a Phase 1/2 clinical trial using a combination of REQORSA with Merck & Co.’s Keytruda® in late-stage NSCLC patients who are low expressors (1% to 49%) of the PD-L1.
Intumor. Our diabetes we are developing a gene therapy that is exclusively licensed from the University of Pittsburgh of the Commonwealth System of Higher Education ("University of Pittsburgh") for the treatment of Type 1 and Type 2 diabetes. This potential treatmenttechnology is designed to work in Type 1 diabetes by transforming alpha cells in the pancreas into functional beta-like cells, which can produce insulin but aremay be distinct enough from beta cells to evade the body’s immune system. OurIn Type 2 diabetes, product candidateour technology is currently being evaluated in preclinical studies.believed to work by replenishing and rejuvenating exhausted beta cells that make insulin.
Oncology Platform
Utilizing our non-viral ONCOPREX Nanoparticle Delivery System, we are developing cancer treatments that are designed to administer cancer fighting genes. We encapsulate the gene-expressing plasmids using ONCOPREX lipid nanoparticles, and administer them intravenously, where they are then taken up by tumor cells and express proteins that are missing or found in low quantities in the tumor cells. With ourOur lead oncology drug candidate, REQORSA® Immunogene Therapy (generic name: quaratusugene ozeplasmid), previously referred to as GPX-001, is initially being developed in combination with prominent, approved cancer drugs to treat Non-Small Cell Lung Cancer (“NSCLC”) and Small Cell Lung Cancer (“SCLC”). REQORSA there ishas a multimodal mechanism of action whereby REQORSAit interrupts cell signaling pathways that cause replication and proliferation of cancer cells, re-establishes pathways for apoptosis, or programmed cell death, in cancer cells, and modulates the immune response against cancer cells. In early studies, REQORSA has also been shown to block mechanismsbe complementary with targeted drugs and immunotherapies. Our strategy is to develop REQORSA in combination with current approved therapies and we believe REQORSA’s unique attributes position it to provide treatments that create drug resistance.improve on these current therapies for patients with NSCLC, SCLC, and possibly other cancers.
Acclaim – 1: We currently are enrolling and treating patients in the Phase 2a expansion portion of our Phase 1/2 Acclaim-1 clinical trial. The Acclaim-1 trial uses a combination of REQORSA and AstraZeneca’s Tagrisso® in patients with late-stage NSCLC that has activating epidermal growth factor receptor (“EGFR”) mutations and progression after treatment with Tagrisso. Following the May 2023 completion of the Phase 1 dose escalation portion of the study, the Acclaim-1 Safety Review Committee (“Acclaim-1 SRC”) approved advancement from the Phase 1 dose escalation portion to the Phase 2a expansion portion of the study. Based on a review of safety data which showed no dose limiting toxicities (“DLTs”), the Acclaim-1 SRC determined that the recommended Phase 2 dose (“RP2D”) of REQORSA will be 0.12 mg/kg. This was the highest dose level delivered in the Phase 1 portion of the study and is twice the highest dose level delivered in the Company’s prior clinical trial combining REQORSA with Tarceva® for the treatment of late-stage lung cancer. We opened the Phase 2a expansion portion of the study and enrolled and dosed the first patient in January 2024. The Phase 2a expansion portion of the trial is expected to enroll approximately 66 patients; half will be patients who received only prior Tagrisso treatment and the other half will be patients who received prior Tagrisso treatment and chemotherapy. The aim is to determine toxicity and efficacy profiles of patients with different eligibility criteria. There will be an interim analysis following the treatment of 19 patients in each cohort. We expect to complete the enrollment of 19 patients in each cohort of the Phase 2a expansion portion of the study by the end of 2024, and thus we expect the interim analyses in early 2025. The Food and Drug Administration (“FDA”) has granted Fast Track Designation for the Acclaim-1 treatment combination of REQORSA and Tagrisso in NSCLC patients who have progressed after Tagrisso treatment.
Acclaim – 2: We currently are enrolling and treating patients in the Phase 1 dose escalation portion of our Phase 1/2 Acclaim-2 clinical trial. The Acclaim-2 trial uses a combination of REQORSA and Merck & Co.’s Keytruda® in patients with late-stage NSCLC whose disease has progressed after treatment with Keytruda. Patients are currently being treated at the 0.06 mg/kg dose level in the first cohort of patients and, subject to Acclaim-2 Safety Review Committee (“Acclaim-2 SRC”) approval, will be treated at successive dose levels of 0.09 mg/kg and 0.12 mg/kg. In March 2023, we amended the Acclaim-2 protocol to include additional treatments in the control group with the goal of accelerating enrollment in the study by making the trial more attractive to a wider variety of investigators. We expect enrollment in the dose escalation portion of the study to be completed in the second half of 2024. We will then initiate and evaluate patients in the Phase 2a expansion portion of the study at the maximum tolerated dose (the “MTD”) or RP2D. The FDA has granted Fast Track Designation for the Acclaim-2 treatment combination of REQORSA and Keytruda in NSCLC patients who have progressed after Keytruda treatment.
The expansion portion of both the Acclaim-1 and Acclaim-2 trials are Phase 2 studies. The expansion portion of these studies provides us the advantage of early insight into drug effectiveness in defined and distinct patient populations at the MTD or RP2D in order to better evaluate efficacy and increase the likelihood of a successful randomized Phase 2 trial which will follow the expansion portion of each study.
Diabetes Gene Therapy
Diabetes Gene Therapy
In diabetes, we have exclusively licensed from the University of Pittsburgh of the Commonwealth System of Higher Education (“University of Pittsburgh”) multiple technologies relating to the development of a gene therapy product for each of Type 1 and Type 2 diabetes. The same general novel approach is used in each of Type 1 and Type 2 diabetes whereby an adeno-associated virus (“AAV”) vector containing the Pdx1 and MafA genes is administered directly into the pancreatic duct. In humans, this can be done with a routine endoscopy procedure. Our diabetes gene therapy, also referred to as GPX-002, was developed by lead researcher Dr. George Gittes,product candidates are currently being evaluated and optimized in preclinical studies at the Rangos Research Center atUniversity of Pittsburgh. GPX-002 is being developed using the UPMC Children’s Hospital. This potentialsame construct for the treatment of both Type 1 diabetes and Type 2 diabetes. GPX-002 for Type 1 diabetes is designed to work by transforming alpha cells in the pancreas into functional beta-like cells, which can produce insulin but aremay be distinct enough from beta cells to evade the body’s immune system. In a similar approach, GPX-002 for Type 2 diabetes (formerly known as GPX-003), where autoimmunity is not at play, is believed to work by replenishing and rejuvenating exhausted beta cells that make insulin. We finalized the components of the diabetes construct to take forward for nonclinical studies and in December 2023, we submitted a request to meet with the FDA to obtain their guidance on the nonclinical studies needed to file an Investigational New Drug (“IND”) application and initiate first-in-human studies. As a result of the FDA’s response, the Company will continue with its planned additional nonclinical studies before requesting regulatory guidance in 2024 for the IND-enabling studies. In October 2023, we entered into a one-year extension to our August 2022 sponsored research agreement with the University of Pittsburgh for the use of GPX-002 in a non-human primate (“NHP”) model in Type 2 diabetes. The therapy utilizesextension includes a procedurerevised research plan to encompass our most recent technologies to which we acquired exclusive rights from the University of Pittsburgh in which an adeno-associated virus vector deliversJuly 2023. These include a MafB promoter to drive expression of the Pdx1 and MafA genestranscription factors that can potentially be used for both Type 1 and Type 2 diabetes. See also “Note 7 – Commitments and Contingencies” to the pancreas.
JOBS Act
On April 5, 2012, the JOBS Act was enacted. Section 107 of the JOBS Act provides that an “emerging growth company” can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act for complying with new or revised accounting standards. In other words, an “emerging growth company” can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. Although we are an emerging growth company, we have irrevocably elected not to avail ourselves of this extended transition period and, as a result, we will adopt new or revised accounting standards on the relevant dates on which adoption of such standards is required for other public companies. We have implemented all new accounting pronouncements that are in effect and may affect our financial statements included in this Annual Report on Form 10-K. In February 2023, the Company’s research collaborators at the University of Pittsburgh presented preclinical data in a NHP model of Type 1 diabetes highlighting the therapeutic potential of GPX-002 at the 16th International Conference on Advanced Technologies & Treatments for Diabetes (ATTD 2023) in Berlin, Germany. The statistically significant study results showed the treated animals had decreased insulin requirements, increased c-peptide levels, and we do not believe that there are any other new accounting pronouncements that have been issued that would haveimproved glucose tolerance compared to baseline. In April 2023, the Company hosted a material impact on our financial position or results of operations.
Notwithstanding the foregoing, subject to certain conditions set forth in the JOBS Act, as an “emerging growth company,” we intend to rely on certain exemptions, including, without limitation, the exemption from the requirements (i) to provide an auditor’s attestation report on our system of internal controls over financial reporting pursuant to Section 404(b) of the Sarbanes-Oxley Act, and (ii) to comply with any requirement that may be adopted by the Public Company Accounting Oversight Board regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements, known as the auditor discussion and analysis. We will remain an “emerging growth company” until the earliest of (i) the last day of the fiscal year in which we have total annual gross revenues of $1.07 billion or more; (ii) the last day of our fiscal year following the fifth anniversary of the date of our initial public offering; (iii) the date on which we have issued more than $1.0 billion in nonconvertible debt during the previous three years; or (iv) the date on which we are deemed to be a large accelerated filer under the rules of the SEC.
Recently Issued Accounting Pronouncements
A description of recently issued accounting pronouncements that may potentially impact our financial position and results of operations is disclosed in Note 2 to our financial statements appearing in this Annual Report on Form 10-K.
Critical Accounting Policies and Significant Judgments and Estimates
Our financial statements have been prepared in accordance with generally accepted accounting principles in the United States ("GAAP"(“GAAP”). The preparation of these financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements, as well as the reported expenses incurred during the reporting periods. Our estimates are based on our historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
We believe that the following accounting policies are the most critical to aid in fully understanding and evaluating our reported financial results, and they require our most difficult, subjective or complex judgments, resulting from the need to make estimates about the effect of matters that are inherently uncertain.
Research and Development Costs
We record accrued expenses for costs invoiced from research and development activities conducted on our behalf by third-party service providers, which include the conduct of preclinical studies and clinical trials and use of contract research, manufacturing, and manufacturingtesting activities. We record the costs of research and development activities based upon the amount of services provided, and we include these costs in accrued liabilities in the condensed balance sheets and within research and development expense in the condensed statements of operations. These costs are a significant component of our research and development expenses. Purchased materials to be used in future research are valued at cost and capitalized and included in research and development supplies.
We estimate the amount of work completed through discussions with internal personnel and external service providers as to the progress or stage of completion of the services and the agreed-upon fee to be paid for such services. We make significant judgments and estimates in determining the accrued balance in each reporting period. As actual costs become known, we adjust our accrued estimates. Although we do not expect our estimates to be materially different from amounts actually incurred, our understanding of the status and timing of services performed, the number of patients enrolled and the rate of patient enrollment in any of our clinical trials may vary from our estimates and could result in our reporting amounts that are too high or too low in any particular period. Our accrued expenses are dependent, in part, upon the receipt of timely and accurate reporting from CROscontract research organizations (“CROs”) and other third-party service providers. To date, there have been no material differences from our accrued expenses to actual expenses.
Income Taxes
Deferred tax assets or liabilities are recorded for temporary differences between financial statement and tax basis of assets and liabilities, using applicable rates in effect for the year in which the differences are expected to reverse. A valuation allowance is recorded if it is more likely than not that a deferred tax asset will not be realized. We have provided a full valuation allowance on our deferred tax assets, which primarily consist of cumulative net operating losses from April 1, 2009 (inception) to December 31, 2020.2023. Due to our history of operating losses since inception and losses expected to be incurred in the foreseeable future, a full valuation allowance was considered necessary.
Impairment of Long-Lived Assets
Management reviews long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount may not be realizable or at a minimum annually during the fourth quarter of the year. If an evaluation is required, the estimated future undiscounted cash flows associated with the asset are compared to the asset’s carrying value to determine if an impairment of such asset is necessary. The effect of any impairment would be to expense the difference between the fair value of such asset and its carrying value.
Off-Balance Sheet Arrangements
As of December 31, 2020 and 2019, we did not have any off-balance sheet arrangements as defined in Item 303(a)(4)(ii) of Regulation S-K or any commitments or contractual obligations.
Components of our Results of Operations and Financial Condition
Operating expenses
We classify our operating expenses into three categories: research and development, general and administrative, and depreciation.
Research and development. Research and development expenses consist primarily of:
costs incurred to conduct research, such as the discovery and development of our current and potential product candidates;
| ● | costs incurred to conduct research, such as the discovery and development of our current and potential product candidates; |
costs related to production and storage of clinical supplies, including fees paid to contract manufacturers, manufacturing consultants, and cold-storage facilities;
| ● | costs related to the production and storage of supplies for engineering purposes and storage and usage of clinical supplies, including waste created in the process of producing clinical materials, spoilage, and testing of clinical materials; |
fees paid to clinical consultants, clinical trial sites and vendors, including CROs in conjunction with implementing and monitoring our clinical trials and acquiring and evaluating clinical trial data, including all related fees, such as patient screening fees, laboratory work, and statistical compilation and analysis;
| ● | costs related to the use of contract manufacturers, manufacturing consultants, testing organizations, cold-storage facilities, and logistics service providers; |
| ● | fees paid to clinical consultants, clinical trial sites and vendors, including CROs in conjunction with implementing and monitoring our clinical trials and acquiring and evaluating clinical trial data, including all related fees, such as patient screening fees, laboratory work, and statistical compilation and analysis; |
costs related to compliance with drug development regulatory requirements;
| ● | costs related to compliance with drug development regulatory requirements; and |
| ● | costs related to staffing and personnel associated with research and development activities, including wages, taxes, benefits, leases, overheads, supplies, and share-based compensation. |
We recognize all research and development costs as they are incurred. Clinical trial costs, contract manufacturing and other development costs incurred by third parties are expensed as the contracted work is performed.
We expect our research and development expenses to increase in the future as we (i) continue to advance our current and potentialfuture product candidates into and through clinical trials, (ii) transition some of our manufacturing activities to new vendors for a variety of reasons, such as weto incorporate more advanced processes and scale production, including any additional work that has been or may be required to successfully adapt our process to these new processes, (iii) pursue regulatory approval of our current and potential product candidates in the United States and Europe, and as we(iv) expand our research programs to include new therapies and new therapy combinations. The process of conducting the necessary preclinical and clinical research to obtain regulatory approval is costly and time-consuming. The actual probability of success for our current and potential product candidates may be affected by a variety of factors including the quality of our current and potential product candidates, early clinical data, investment in our clinical program, competition, manufacturing capability and commercial viability.viability, and limited contracted partners. We may never succeed in achieving regulatory approval for any of our current or future product candidates. As a result of the uncertainties discussed above, we are unable to determine the duration and completion costs of our research and development projects or when and to what extent we will generate revenue from the commercialization and sale of our product candidates, if at all.
General and administrative. General and administrative expense consists of personnel related costs, which include salaries, as well as the costs of professional services, such as accounting and legal, travel, facilities, information technology and other administrative expenses. We expect our general and administrative expense to increase in future periods due to the anticipated growth of our business and related infrastructure as well as accounting, insurance, investor relations, legal, information technology, and other costs associated with being a public company.
Depreciation. Depreciation expense consists of depreciation from our fixed assets consisting of our property, equipment, and furniture. We depreciate our assets over their estimated useful life. We estimate furniture and computer and office equipment to have a 5-yearfive-year life.
Results of Operations
Comparison of the Years Ended December 31, 20202023 and 20192022
The following summarizes our results of operations for the years ended December 31, 20202023 and 2019.2022.
Research and Development Expense. Research and development ("(“R&D"&D”) expense was $7,302,923$17,616,605 for the year ended December 31, 20202023 as compared to $1,967,007$11,510,074 for the year ended December 31, 2019.2022. This increase of $5,335,916,$6,106,531, or 271%53%, is primarily due to the hiring of new employees and consultants to develop strategy for and execute on the launch of our Acclaim-1 and Acclaim-2 clinical trials, major advancements(i) changes in our manufacturing programs providingdue to our transition to more capable contract development and manufacturing organizations (“CDMOs”), (ii) increased manufacturing and testing of our drug product for our Acclaim-1 and Acclaim-2 clinical trials, and research of novel therapeutic approaches for the treatment of cancer using REQORSA and immunotherapies. These R&D activities will continue throughout 2021 and thereafter and will continue to include costs related to the launch and conduct of the Acclaim-1 and Acclaim-2 clinical trials, the development and execution on related manufacturing strategies and processes required(iii) increase in third-party vendors to support these,our manufacturing and potentially other,preclinical and clinical programs, and additional preclinical research.programs.
General and Administrative Expense. General and administrative ("(“G&A"&A”) expense for the year ended December 31, 20202023 was $10,635,881$13,443,961 as compared to $8,702,596$12,295,070 for the year ended December 31, 2019.2022. The increase of $1,933,285,$1,148,891, or 22%9% is mostlyprimarily due to (i) an increase in financing costsprofessional services and legal feesthird-party vendors to support corporate programs, and (ii) greater share-based compensation associated with our registered direct offerings in 2020 as well as greater than normal share-based compensation expense associated with the accelerated vesting of options for a former executive pursuant to his separation agreement. Notwithstanding these expenses, some G&A expenses decreased for the year ended December 31, 2020 due to reclassification of expenses associated with R&D personnel to more accurately reflect expenses associated with their job function as well as reduced travelemployees and office expenses as the result of travel restrictions and social distancing guidelines put in place as a result of the COVID-19 pandemic.service providers.
Interest Income. Interest income was $18,811$215,109 and $27,905$90,098 for the years ended December 31, 20202023 and 2019,2022, respectively. This decreaseincrease of $9,094, or 33%%,$125,011 was primarily due to changes in balances and a significant dropincrease in interest rates of our money market instruments for the year ended December 31, 20202023 as opposedcompared to the prior year.
Interest Expense. There was no interest expense for the years ended December 31, 20202023 and 20192022 because we satisfied allhad no debt obligations and repaid all short-term loans prior to 2019.obligations. As of December 31, 2020,2023, we had no outstanding debt.
Depreciation Expense. Depreciation expense was $22,777$15,004 and $13,070$25,575 for the years ended December 31, 20202023 and 2019,2022, respectively. The increasedecrease of $9,707,$10,571, or 74%41%, in depreciation was driven by increased purchaseprimarily due to the timing of purchases of computer equipment for use by employees and manufacturing partnersnew employees.
Net Loss. We had a net loss of $30,860,461, for research activities in the fiscal year ended in December 31, 2020.2023 compared to a net loss of $23,740,621 for the fiscal year ended December 31, 2022. The increase of $7,119,840, or 30%, in net loss was primarily due to the expansion of our manufacturing to more qualified CDMOs as well as increased manufacturing and testing of our drug product to support our Acclaim-1 and Acclaim-2 clinical trials.
Liquidity and Capital Resources
From inception through December 31, 2020,2023, we have never generated revenue from product sales and have incurred net losses in each year. As of December 31, 2020,2023, we had an accumulated deficit of $58,422,229.$133,688,280. We have funded our operations primarily through the sale and issuance of capital stock. During 2019,For the year ended December 31, 2022, we sold 3,167,986 sharesan aggregate of common stock and warrants to purchase 3,167,98698 shares of common stock for total net proceeds of $1,178,491$4,532 pursuant to a registered direct offering.our 2022 ATM facility as governed by the Equity Distribution Agreement (as further described below) and issued 2,925 shares of common stock upon the exercise of options for gross proceeds of $1,755. For the year ended December 31, 2020,2023, we sold an aggregate of 16,697,8841,342 shares of common stock for total net proceeds of $34,493,423$78,355 pursuant to our 2022 ATM facility as governed by the Equity Distribution Agreement (as further described below).
On November 18, 2022, we entered into an Equity Distribution Agreement with JMP Securities, with respect to an at-the-market offering program (our “2022 ATM Facility”) under which we could offer and sell, from time to time at our sole discretion, shares of our common stock, having an aggregate offering price of up to $50.0 million. We agreed to pay JMP Securities a commission equal to three percent (3%) of the gross sales proceeds of any shares sold under the Equity Distribution Agreement, and also provided JMP Securities with customary indemnification and contribution rights. For the year ended December 31, 2022, we sold 98 shares of our common stock for net proceeds to us totaling $4,532. For the year ended December 31, 2022, we sold 1,342 shares of our common stock for net proceeds to us totaling $78,355. On December 12, 2023, we provided notice to JMP Securities of our termination of the 2022 ATM Facility. The termination of the Equity Distribution Agreement with JMP Securities was effective as of December 13, 2023.
On March 1, 2023, we completed a registered direct offeringsoffering in which we sold 95,239 shares of our common stock and issued 7,104,524warrants to purchase 95,239 shares of our common stock to an accredited healthcare-focused institutional investor for aggregate net proceeds of approximately $3.6 million. On July 21, 2023, we raised approximately $6.7 million in net proceeds in a registered direct offering pursuant to which we sold 185,644 shares of common stock and warrants to purchase up to 185,644 shares of common stock.
On December 13, 2023, we entered into an At The Market (“ATM”) Offering Agreement (the “Agreement”) with H.C. Wainwright & Co., LLC, serving as agent (the “Agent”) with respect to an at-the-market offering program under which we may offer and sell through the Agent, from time to time at our sole discretion, up to such number or dollar amount of shares of our common stock (the “Shares”) as registered on the prospectus supplement covering the ATM offering, as may be amended or supplemented from time to time. We have agreed to pay the Agent a commission equal to three percent (3%) of the gross sales proceeds of $3,857,886 from warrantany Shares sold through the Agent under the Agreement, and option exercises.also have provided the Agent with customary indemnification and contribution rights. As of December 31, 2023 we had not sold any Shares through the Agent under the Agreement. From January 1, 2024 through the date of filing of this Annual Report on Form 10-K, subsequent to the December 31, 2023 balance sheet date, we have sold 158,474 Shares for net proceeds to us totaling $881,946 through the Agent under the Agreement.
As of December 31, 2020,2023, we had $27,319,685$6,737,629 in cash. Subsequent to year-end 2023, on March 21, 2024, we raised approximately $5.8 million in net proceeds in a registered direct offering pursuant to which we sold (i) 165,000 shares of our common stock, (ii) pre-funded warrants exercisable for up to an aggregate of 1,377,112 shares of our common stock, and (iii) warrants exercisable for up to an aggregate of 1,542,112 shares of our common stock. In connection with the March 2024 registered direct offering, we amended certain existing warrants to reduce the exercise price and extend the term thereof. See “Note 9 – Subsequent Events – March 2024 Registered Direct Offering” to the Financial Statements included in this Annual Report on Form 10-K. Additionally, as noted above, we received net proceeds totaling $881,946 through the Agent under the ATM Agreement subsequent to year-end 2023.
We do not expect to generate revenue from product sales unless and until we successfully complete development of, obtain regulatory approval for and begin to commercialize one or more of our current andor potential product candidates, or other product candidates to which we may acquire rights, which we expect will take a number of years and which is subject to significant uncertainty. Accordingly, we anticipate that we will need to raise additional capital to fund our future operations, which include conducting our Acclaim-1, Acclaim-2, and Acclaim-2Acclaim-3 clinical trials (of which Acclaim-1, Acclaim-2 and Acclaim-3 are currently enrolling) and completing preclinical work for potential other oncology candidates and completing preclinical work and conducting clinical trials for our diabetes program. We expect enrollment in each of the cohorts of the Phase 2a expansion portion of the Acclaim-1 trial to be completed by the end of 2024. We expect enrollment in the Phase 1 dose escalation portion of the Acclaim-2 trial to be completed by the second half of 2024. Enrollment in the Acclaim-3 clinical trial is expected to be initiatedcompleted during the second half of 2024 after which we expect to commence the Phase 2 portion of the Acclaim-3 trial in 2021.the second half of 2024. Until such time as we can generate substantial revenue from product sales, if ever, we expect to finance our operating activities through a combination of equity offerings, drawdowns on our ATM pursuant to our Agreement with the Agent, and debt financings and we may seek to raise additional capital through strategic collaborations.collaborations or transactions. However, we may be unable to raise additional funds or enter into such arrangements when needed on favorable terms, or at all, which would have a negative impact on our financial condition and could force us to delay, limit, reduce or terminate our research and development programs or commercialization efforts or grant rights to others rights to develop or market product candidates that we would otherwise prefer to develop and market ourselves. Failure to receive additional funding could cause us to curtail or cease our operations. Furthermore, even if we believe we have sufficient funds for our current or future operating plans, we may seek additional capital due to favorable market conditions or strategic considerations.
Based on our current cash, we estimate that we will be able to fund our expenditure requirements for our current operations and planned clinical trial activities into the third quarter of 2024. We have based this estimatethese estimates on assumptions that may prove to be wrong, and we could utilize our available capital resources sooner than we currently plan due to incorrect assumptions or due to a decision to expand our activities beyond those currently planned. We previously have experienced delays in engaging clinical sites as a result of disruptions at these clinical sites caused by the COVID-19 pandemic. We also have experienced delays in clinical trial enrollment as a result of competition for patients and additional time required in connection with our transition to the new third party CDMO and the manufacture of final drug product. Delays in the conduct of our trials could result in utilizing our capital resources sooner without advancing our clinical trials as anticipated.
The following table sets forth the primary sources and uses of cash for the years ended December 31, 20202023 and 2019:2022:
Years Ended December 31, | Years Ended December 31, | |||||||||||||||
2020 | 2019 | 2023 | 2022 | |||||||||||||
Net cash used in operating activities | $ | (13,935,086 | ) | $ | (6,918,721 | ) | $ | (24,738,603 | ) | $ | (17,621,498 | ) | ||||
Net cash used in investing activities | (2,337,250 | ) | (946,899 | ) | (71,214 | ) | (59,735 | ) | ||||||||
Net cash provided by financing activities | 41,589,529 | 1,267,194 | 10,593,377 | 6,426 | ||||||||||||
Net increase (decrease) in cash | 25,317,193 | (6,598,426 | ) | |||||||||||||
Net decrease in cash | (14,216,440 | ) | (17,674,807 | ) | ||||||||||||
Short Term Cash Requirements
We believe that our existing cash is sufficient to fund our expected short-term needs into the third quarter of 2024, but will need additional fundraising activities and cash on hand by such time. We currently have certain fixed cash obligations with respect to development of materials used in our clinical studies and payment obligations associated with our ongoing conduct and monitoring of our Acclaim clinical trials, and we expect that we will have insufficient cash to cover these requirements through fiscal year 2024 without raising additional working capital. The foregoing conditions raise substantial doubt regarding our ability to continue as a going concern for a period of one year after the date the financial statements included in this Annual Report are issued. Lack of necessary funds may require us, among other things, to delay, scale back or eliminate some or all of our ongoing and/or planned clinical trials.
Long Term Cash Requirements
We regularly evaluate our business plans and strategy. These evaluations often result in changes to our business plans and strategy, some of which may be material and significantly change our cash requirements. Ongoing business development activity may require us to use some of our liquidity for an acquisition, or additional capital to fund newly acquired operations. If we raise additional funds by issuing equity securities, our existing security holders will likely experience dilution; and the incurring of indebtedness would result in increased debt service obligations and could require us to agree to operating and financial covenants that could restrict operations.
Our future capital requirements depend on many factors, including, but not limited to:
| ● | the costs and timing of our development activities and preclinical and clinical trials; |
| ● | the cost of manufacturing our existing and future products; |
| ● | the expenses needed to attract and retain skilled personnel; |
| ● | the costs associated with being a public company; |
| ● | the costs associated with additional business development or mergers and acquisitions activity, including acquisition-related costs, earn-outs or other contingent payments and costs of developing and commercializing any technologies to which we obtain rights; |
| ● | third-party costs associated with the development and commercialization of our existing and future products and the ability of our development partners to satisfy our requirements on a timely basis; |
| ● | the scope and terms of our business plans from time to time, and our ability to realize upon our business plans; and |
| ● | the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing possible patent claims, including litigation costs and the outcome of any such litigation. |
Cash used in operating activities
Net cash used in operating activities was $13,935,086$24,738,603 and $6,918,721$17,621,498 for the years ended December 31, 20202023 and 2019,2022, respectively. The increase of $7,016,365,$7,117,105, or 101%%40%, in net cash used in operating activities in 20202023 was primarily due to an increaseus advancing our manufacturing programs to support enrollment of patients in financing costs and legal fees associated with our fundraising activities in early and late 2020 and significant increases to our headcount and service providers in preparation for the launch of our Acclaim-1 and Acclaim-2 clinical trials planned for 2021.in the year ended December 31, 2023 compared to December 31, 2022 when our Acclaim-1 and Acclaim-2 clinical trials were initiated but enrolling patients at the slower, safety-oriented, Phase 1 portion per their respective protocols.
Cash used in investing activities
Net cash used in investing activities was $2,337,250$71,214 and $946,899$59,735 for the years ended December 31, 20202023 and 2019,2022, respectively. The increase in net cash used inprovided by investing activities of $1,390,351,$11,479, or 147%19%, was primarily due to a major investmentgreater use of R&D materials associated with testing and clinical use in manufacturing materials forour Acclaim clinical trials in the year ended December 31, 2020 that are currently being used to manufacture REQORSA for our planned clinical trials. Investments in property and equipment and intellectual property increased marginally for the year ended December 31, 20202023 compared to the year ended December 31, 2019.2022. Changes in intellectual property and research and development supplies during this period were negligible.
Cash provided by financing activities
Net cash provided by financing activities was $41,589,529$10,593,377 and $1,267,194$6,426 for the years ended December 31, 20202023 and 2019,2022, respectively. The increase of $40,322,335,$10,586,951, or 3,182%100%, in net cash provided by financing activities was primarily due to our selling common stockthe registered direct offerings in February 2023 and July 2023 and there were no comparable capital raising activities throughout the year ended December 31, 2020.2022.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk.
As a smaller reporting company, we are not required to provide the information requiredcalled for by this item.
Item 8. Financial Statements and Supplementary Data.
The financial statements and supplementary data required by this item are included after Part IV of this Annual Report on Form 10-K beginning on page F-1.
Item 9. Changes in and Disagreements With Accountants on Accounting and Financial Disclosure.
None.Not applicable.
Item 9A. Controls and Procedures.
Evaluation of Disclosure Controls and Procedures
As required by Rules 13a-15(b) and 15d-15(b) of the Exchange Act, our management with the participation of our Chief Executive Officer and our Chief Financial Officer, evaluated the effectiveness of our disclosure controls and procedures as of December 31, 2020.2023. The term “disclosure controls and procedures” as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act, means controls and other procedures of a company that are designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is accumulated and communicated to a company’s management, including its principal executive and principal financial officer, as appropriate to allow timely decisions regarding required disclosure. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives, and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on the evaluation of our disclosure controls and procedures as of December 31, 2020,2023, our Chief Executive Officer and our Chief Financial Officer concluded that, as of such date, our disclosure controls and procedures were not effective due to material weaknessweaknesses in our internal control over financial reporting duerelated to a lack of segregation of duties.duties between accounting and other functions and the absence of sufficient depth of in-house accounting personnel with the ability to properly account for complex transactions.
Management’s Report on Internal Control over Financial Reporting
Our principal executive officer and our principal accounting and financial officer are responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rules 13a-15(f) and 15d-15(f). Management conducted an assessment of the effectiveness of our internal control over financial reporting as of December 31, 2020.2023. In making this assessment, management used the criteria described in Internal Control-Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Our management concluded that our internal controls over financial reporting were, and continue to be, ineffective as of December 31, 20202023 due to a material weaknessweaknesses in our internal controls due to the lack of segregation of duties.duties and insufficient depth of in-house accounting personnel.
It should be noted that any system of controls, however well designed and operated, can provide only reasonable and not absolute assurance that the objectives of the system are met. In addition, the design of any control system is based in part upon certain assumptions about the likelihood of certain events. Because of these and other inherent limitations of control systems, there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions, regardless of how remote.
In light of the material weakness described below, we performed additional analysis and other post-closing procedures to ensure our financial statements were prepared in accordance with generally accepted accounting principles. Accordingly, we believe that the financial statements included in this report fairly present, in all material respects, our financial condition, results of operations and cash flows for the periods presented.
During the last quarter of fiscal 2020year ended December 31, 2023 and as our operational activities increased, management determined and continues to determine that it does not have sufficient segregation of duties within its accounting functions nor does it have sufficient depth of in-house accounting personnel with the ability to properly account for complex transactions, which is aare basic internal control.controls. Due to our size, and nature, segregation of all conflicting duties may not always be possible and may not be economically feasible. However, to the extent possible, the initiation of transactions, the custody of assets and the recording of transactions should be performed by separate individuals. Our size and nature also do not allow for our accounting staff to have depth of expertise in all areas that might be desirable, such as expertise in accounting for a variety of complex transactions. Management evaluated the impact of our failure to maintain effective segregation of duties and sufficient depth of personnel on our assessment of our internal control over financial reporting and has concluded that thethese control deficiency represents adeficiencies represent material weakness.weaknesses.
Remediation Plans
Management has begun implementing ais actively engaged in remediation planefforts to address the control deficiency that ledmaterial weakness identified in the management’s evaluation of internal controls and procedures. The remediation efforts, which have been or are in the process of being implemented, are intended to address the identified material weakness and include:
| ● | new accounting software, processes, and workflows to further segregate duties among limited accounting staff; |
| ● | specific review procedures, including the added involvement of our General Counsel to review all accounting transactions following a given period in an effort to enhance accuracy of reporting; |
| ● | specific review procedures, including the added involvement of our manufacturing staff, to enhance controls associated with the tracking and reporting of inventory values in our supply chain; |
| ● | a formal Disclosure Committee that has oversight responsibility for the accuracy and timeliness of disclosures made by the Company through controls and procedures and the monitoring of their integrity and effectiveness; |
| ● | additional hiring of staff and development of accounting processes and policies to further segregate accounting responsibilities and increase the depth of our expertise in accounting for a variety of complex transactions; and |
| ● | additional training, testing, and certification of key accounting, finance, IT, and legal team members. |
During the year ended December 31, 2023, we took actions to remediate the material weakness. Theweakness relating to our internal controls over financial reporting including:
| ● | continued evaluation and documentation of policies, processes, and controls, both manual and automated; |
| ● | identification and implementation of improvements to information technology and security controls and supporting control documentation; |
| ● | improvements to software workflows to further segregate duties, enhance accuracy of vendor billing, and ensure transparency and oversight from vendor or project managers, department leaders, legal team members, and finance team members; |
| ● | initiation and completion of training programs, testing, and certification for key accounting and finance personnel related to internal controls and implementation of COSO Framework; |
| ● | evaluation of existing capabilities of accounting and finance staff related to complex technical accounting items associated with U.S. GAAP and engagement with external advisory firms to provide technical support as necessary; |
| ● | the hiring of a director-level IT employee specialized in the evaluation and implementation of IT and secure policies, processes, and control documentation, and evaluation of inventory management and quality management software systems and associated implementation to ensure effective workflow processes and effective control environment of these systems; and |
| ● | the hiring and integration of an associate general counsel with SEC reporting experience into our internal controls over financial reporting process to assist in the review and oversight of financial reporting and disclosure controls. |
As management continues to evaluate and work to improve its internal control over financial reporting, we may take additional measures to address control deficiencies, or we may modify certain of the remediation plan includes implementingmeasures described above. While remediation efforts are active, management requires additional time to demonstrate the following:
new accounting software, processes, and workflows to further segregate duties among limited accounting staff;
specific review procedures, including the added involvementoperating effectiveness of our General Counsel, hired in 2020, to review all accounting transactions followingremediation efforts. The material weaknesses cannot be considered remediated until the applicable remedial controls operate for a givensufficient period in an effort to enhance accuracy of reporting;
specific review procedures, including the added involvement of our VP of Manufacturing, to enhance controls associated with the reporting of inventory values;
We currently plan to have our enhanced review procedures and documentation standards in place and operating by the end of 2021. Our goal is to remediate this material weakness by the end of 2022, subject to there being sufficient opportunities to conclude,concluded, through testing, that the enhancedthese controls are operating effectively.
There were no changes in our internal control over financial reporting that occurred during the year ended December 31, 20202023 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Inherent Limitations of Disclosure Controls and Internal Control over Financial Reporting
Because of their inherent limitations, our disclosure controls and procedures and our internal control over financial reporting may not prevent material errors or fraud. A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. The effectiveness of our disclosure controls and procedures and our internal control over financial reporting is subject to risks, including that the controls may become inadequate because of changes in conditions or that the degree of compliance with our policies or procedures may deteriorate.
None.Rule 10b5-1 Trading Arrangements and Non-Rule 10b5-1 Trading Arrangements
During the fiscal quarter ended December 31, 2023, none of our officers or directors, as those terms are defined in Rule 16a-1(f), adopted or terminated a “Rule 10b5-1 trading arrangement” or a “non-Rule 10b5-1 trading arrangement,” as those terms are defined in Item 408 of Regulation S-K.
Item 9C. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections.
Not applicable.
Item 10. Directors, Executive Officers and Corporate Governance.
The information required by this Item 10 is incorporated herein by reference to the information that will be contained in our definitive proxy statement (the “Proxy Statement”) for the 20212024 annual meeting of stockholders (the “Annual Meeting”), which is expected to be filed not later than 120 days after the end of our fiscal year ended December 31, 2020.2023.
We have adopted a written Code of Business Conduct and Ethics, or Ethics Code, that applies to all of our officers, directors and employees. The Ethics Code is available on our website at www.genprex.com/investors/corporate-governance. We intend to satisfy the disclosure requirement under Item 5.05 of Form 8-K regarding an amendment to, or a waiver from, a provision of our Ethics Code that applies to our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions and that relates to any element of the code of ethics definition enumerated in paragraph (b) of Item 406 of the SEC’s Regulation S-K, by posting such information on our website, www.genprex.com.
Item 11. Executive Compensation.
The information required by this Item 11 is incorporated herein by reference to the information that will be contained in our Proxy Statement for the Annual Meeting.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
The information required by this Item 12 is incorporated herein by reference to the information that will be contained in our Proxy Statement for the Annual Meeting.
Item 13. Certain Relationships and Related Transactions, and Director Independence.
The information required by this Item 13 is incorporated herein by reference to the information that will be contained in our Proxy Statement for the Annual Meeting.
Item 14. Principal Accountant Fees and Services.
The information required by this Item 14 is incorporated herein by reference to the information that will be contained in our Proxy Statement for the Annual Meeting.
Item 15. ExhibitsExhibit and Financial Statement Schedules.
(a)(1) | Financial statements. |
The financial statements and supplementary data required by this item begin on page F-1.
(a)(2) | Financial Statement Schedules. |
All schedules are omitted because the required information is inapplicable or the information is presented in the financial statements and the related notes.
(a)(3) | Exhibits. |
* | Filed herewith. |
| ** | Furnished herewith. |
+ | Indicates management contract or compensatory plan. |
| ++ | Pursuant to Item 601(b)(10) of Regulation S-K, certain confidential portions of this exhibit were omitted by means of marking such portions with an asterisk because the identified confidential portions (i) are not material and (ii) would be competitively harmful if publicly disclosed. |
None.
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this annual report on Form 10-K to be signed on its behalf by the undersigned, thereunto duly authorized.
| GENPREX, INC. | |
Date: | By: | /s/ J. Rodney Varner |
|
| J. Rodney Varner |
|
| Chief Executive Officer |
|
| (Principal Executive Officer) |
Pursuant to the requirements of the Securities Exchange Act of 1934, as amended, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Signature |
| Title |
| Date |
/s/ J. Rodney Varner |
| Chief Executive Officer and
|
|
|
J. Rodney Varner |
| (Principal Executive Officer) |
|
|
|
|
|
|
|
/s/ Ryan M. Confer |
| Chief Financial Officer |
| |
Ryan M. Confer |
| (Principal Financial and Accounting Officer) |
|
|
|
|
|
|
|
/s/ Brent Longnecker | Member of the Board of Directors | |||
Brent Longnecker | ||||
/s/ Jose Antonio Moreno Toscano | Member of the Board of Directors | |||
Jose Antonio Moreno Toscano | ||||
/s/ Will R. Wilson, Jr. | Member of the Board of Directors | |||
Will R. Wilson, Jr. |
INDEX TO FINANCIAL STATEMENTS
Report of Independent Registered Public Accounting Firm (Auditor ID # 100) |
| |
| Report of Independent Registered Public Accounting Firm (Auditor ID # 0229) | F-3 | |
|
| |
Statements of |
| |
Statements of Changes in Stockholders’ Equity - December 31, 2023 and 2022 | ||
Statements of Cash Flows - December 31, 2023 and 2022 | ||
|
Report of Independent Registered Public Accounting Firm
To the Board of Directors and
Stockholders of
Genprex, Inc.
Austin, Texas:
Opinion on the Financial Statements
We have audited the accompanying balance sheetssheet of Genprex, Inc. (the “Company”) atas of December 31, 2020 and 2019,2023, and the related statements of operations, changes in stockholders’ equity, and cash flows for eachthe year then ended, and the related notes (collectively referred to as the “financial statements”). In our opinion, the 2023 financial statements present fairly, in all material respects, the financial position of the yearsCompany as of December 31, 2023, and the results of its operations and its cash flows for the year then ended in conformity with accounting principles generally accepted in the two-year periodUnited States of America.
Substantial Doubt Regarding Going Concern
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 2 to the financial statements, the Company has an accumulated deficit at December 31, 2023 and, since inception, has suffered significant operating losses and negative cash flows from operations that raise substantial doubt about its ability to continue as a going concern. Management's plans in regard to these matters are also described in Note 2. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Revisions to 2022 Financial Statements
The financial statements of the Company as of and for the year ended December 31, 2020,2022, before the effects of the adjustments to retrospectively apply the change in accounting related to the reverse stock split described in Notes 2 and 4 to the financial statements, were audited by other auditors whose report, dated March 31, 2023, expressed an unqualified opinion on those statements. We audited the adjustments to the 2022 financial statements to retrospectively apply the February 2, 2024, reverse stock split, as described in Notes 2 and 4 to the financial statements. In our opinion, such retrospective adjustments are appropriate and have been properly applied. However, we were not engaged to audit, review, or apply any other procedures to the 2022 financial statements of the Company other than with respect to the retrospective adjustments and procedures related to certain state and federal tax information as described in the following paragraph and, accordingly, we do not express an opinion or any other form of assurance on the 2022 financial statements taken as a whole.
The financial statements of the Company as of and for the year ended December 31, 2022, excluding the tabular federal and state income tax information for the 2022 financial statements disclosed in Note 8, were audited by other auditors whose report, dated March 31, 2023, expressed an unqualified opinion on those statements. We audited the tabular federal and state income tax information for the 2022 financial statements disclosed in Note 8. In our opinion, such information included in the table for 2022 is fairly stated, in all material respects, as of and for the year ended December 31, 2022. However, we were not engaged to audit, review, or apply any other procedures to the 2022 financial statements of the Company other than with respect to the tabular federal and state income tax information for the 2022 financial statements disclosed in Note 8 and procedures related to retroactive adjustments as a result of the reverse stock split as described in the preceding paragraph, and, accordingly, we do not express an opinion or any other form of assurance on the 2022 financial statements taken as a whole.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audit. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) ("PCAOB") and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audit we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the entity’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audit included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audit also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audit provides a reasonable basis for our opinion.
Critical Audit Matters
The critical audit matters communicated below are matters arising from the current period audit of the financial statements that were communicated or required to be communicated to the audit committee and that: (1) relates to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective or complex judgments. The communication of the critical audit matters does not alter in any way our opinion on the financial statements, taken as a whole, and we are not, by communicating the critical audit matters below, providing a separate opinion on the critical audit matters or on the accounts or disclosures to which they relate.
Accounting for Accrued Research and Development Costs
Critical Audit Matter Description
The Company records accrued expenses for estimated costs of research and development activities conducted by third-party service providers, which include the conduct of preclinical studies and clinical trials, and contract manufacturing activities. The Company records the estimated costs of research and development activities based upon the estimated amount of services provided and include the costs incurred but not yet invoiced within other accrued liabilities in the balance sheet and within research and development expense in the statement of operations. These costs can be a significant component of the Company’s research and development expenses.
The Company estimates the amount of work completed through discussions with internal personnel and external service providers as to the progress of the services and the agreed-upon fee to be paid for such services. The Company makes estimates in determining the accrued balance in each reporting period. Clinical trial related contracts vary in duration, and may be for a fixed amount, based on the achievement of certain contingent events or deliverables, a variable amount based on actual costs incurred, capped at a certain limit or contain a combination of these elements. Estimates include costs associated with services provided by contract organizations for preclinical and clinical development, and manufacturing of the Company’s product candidates. In the case of clinical trials, the Company relies on estimates of the progress of the clinical trials and related expenses incurred. As actual costs become known, the Company adjusts its accrued estimates.
We identified accrued research and development costs as a critical audit matter given the estimation involved in accounting for accrued research and development costs, as well as the material weakness identified by the Company in its internal controls over financial reporting. This required extensive audit effort related to the estimation of accrued research and development costs.
How the Critical Audit Matter Was Addressed in the Audit
Our audit procedures related to accrued research and development costs included the following, among others: we obtained an understanding and evaluated the design and implementation of controls over the estimation of accrued preclinical studies, clinical trials and manufacturing expenses. We also read a sample of research, collaboration, and manufacturing agreements and contracts, as well as amendments thereto.
We also evaluated publicly available information (such as press releases and investor presentations) and board of directors’ materials regarding the status of preclinical studies, clinical trial and manufacturing activities. For a selection of agreements and contracts, we compared the amount of accrual at the end of the prior period to current year activity and evaluated the accuracy of the Company’s estimation methodology. For the selection of agreements, we obtained a written confirmation of the status of clinical trials and manufacturing from the Company’s third-party service providers. We also made selections of specific amounts recognized as research and development expense as well as those recognized as accrued expenses to evaluate management’s estimate of the vendor’s progress and performed additional procedures.
Accounting for Equity Financing and Warrant Issuances
Critical Audit Matter Description
The Company evaluates its equity issuances to determine if those contracts or embedded components of those contracts qualify as derivative instruments requiring separate recognition in the Company’s financial statements. As described in Note 4, on March 1, 2023, the Company completed a registered direct offering (“RDOs”), in which the Company sold to an accredited healthcare-focused institutional investor an aggregate of 95,239 shares of its common stock and warrants to purchase up to 95,239 shares of its common stock, at a combined offering price of $42.00 per share of common stock and accompanying warrant. On July 21, 2023, the Company completed another registered direct offering priced at the market under Nasdaq rules, in which the Company sold to accredited healthcare-focused institutional investors an aggregate of (i) 185,644 shares of its common stock, and (ii) warrants to purchase up to 185,644 shares of its common stock, at a combined offering price of $40.40 per share of common stock and accompanying warrant. Also, the Company agreed to issue to the Placement Agent warrants to purchase up to an aggregate of 11,140 shares of the Company’s common stock.
We identified the accounting for equity financing and warrant issuances as a critical audit matter because of the complexity in applying the accounting framework, as well as the material weakness identified by the Company in its internal controls over financial reporting. This required extensive audit effort related to application of accounting framework.
How the Critical Audit Matter Was Addressed in the Audit
Addressing the matter involved performing procedures and evaluating audit evidence in connection with forming our overall opinion on the financial statements. These procedures included, among others, obtaining an understanding of, evaluating the design of the operating effectiveness of controls over management’s process for the accounting treatment, reading the agreements, and evaluating the accounting for the RDO warrant issuance. Testing management’s process included (i) evaluating the internal controls and application of the accounting framework used by management; (ii) testing the mathematical accuracy of management’s calculations; and (iii) testing the completeness and accuracy of the underlying data used. Professionals with specialized skill and knowledge were used to assist in (i) evaluating management’s application of the accounting framework and (ii) testing the mathematical accuracy of management’s calculation and conclusion related to the accounting classification of the 2023 RDO warrant issuances.
/s/ WithumSmith+Brown, PC
We have served as the Company’s auditor since 2023.
East Brunswick, New Jersey
April 1, 2024
PCAOB ID Number 100
Report of Independent Registered Public Accounting Firm
To the Board of Directors and
Stockholders of Genprex, Inc.
Austin, Texas
Opinion on the Financial Statements
We have audited, before (1) the effects of the adjustments to retrospectively apply the effects of the reverse stock split described in Note 2 – Summary of Significant Accounting Policies – Reverse Stock Split and Note 4 – Equity – Reverse Stock Split (Reverse Stock Split) and (2) the inclusion of tabular federal and state income tax information for the 2022 financial statements disclosed in Note 8 – Income Taxes, the accompanying balance sheet of Genprex, Inc. (the “Company”) at December 31, 2022, and the related statements of operations, changes in stockholders’ equity, and cash flows for the year ended December 31, 2022, and the related notes (collectively referred to as the financial statements). In our opinion, before (1) the effects of the adjustments to retrospectively apply the reverse stock split described in Note 2 – Summary of Significant Accounting Policies – Reverse Stock Split and Note 4 – Equity – Reverse Stock Split (Reverse Stock Split) and (2) the inclusion of tabular federal and state income tax information for the 2022 financial statements disclosed in Note 8 – Income Taxes, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2020 and 2019,2022, and the results of its operations and its cash flows for each of the years in the two-year periodyear ended December 31, 2020,2022, in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits.audit. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our auditsaudit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our auditsaudit included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provideaudit provides a reasonable basis for our opinion.
Critical Audit Matters
The critical audit matters communicated below are matters arising from the current period audit of the financial statements that were communicated or required to be communicated to the audit committee and that: (1) relate to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the financial statements, taken as whole, and we are not, by communicating the critical matters below, providing separate opinions on the critical audit matters or on the accounts or disclosures to which they relate.
Valuation of equity transactions
As described in the Equity Note to the financial statements, the Company has complex equity transactions including the use of stock options and warrants. The estimates that management used in calculating the price value depend on assumptions specific to the nature of the management service activities with regard to the amount of the price model.
The principal consideration for our determination surrounding equity transactions as a critical audit matter is the significant judgment by management when developing the valuation of options and warrants. This, in turn, led to a high degree of auditor judgment, subjectivity, and effort in performing procedures and evaluating management’s significant assumptions related to the price model used to calculate equity transactions.
Addressing the matter involved performing procedures and evaluating audit evidence in connection with forming our overall opinion on the financial statements. These procedures included evaluating the use of the fair value-based method of accounting for stock-based compensation for options granted to employees, independent consultants and contractors. The Company measures options granted at fair value determined as of the grant date, and recognizes the expense over the periods in which the related services are rendered based on the terms and conditions of the awards. Evaluation of management’s assumptions related to the price model and evaluating whether assumptions used by management were reasonable considering the current and past performance of equity, the consistency, and whether these assumptions were consistent with evidence obtained in other areas of the audit.
/s/ Daszkal Bolton LLP | |
| |
| |
| |
We served as the Company’s auditor from 2014 through March 2023.
Boca Raton, FL
March 31, 2023
Balance Sheets
| Year Ended | ||||||||
| December 31, | ||||||||
2023 | 2022 | |||||||
Assets | ||||||||
Current assets: | ||||||||
Cash and cash equivalents | $ | 6,737,629 | $ | 20,954,069 | ||||
Accounts receivable | — | 34,852 | ||||||
Prepaid expenses and other | 794,138 | 484,224 | ||||||
Total current assets | 7,531,767 | 21,473,145 | ||||||
Property and equipment, net | 7,859 | 23,032 | ||||||
Other assets: | ||||||||
Security deposits | 10,000 | 21,818 | ||||||
Research and development supplies | 2,347,488 | 2,864,937 | ||||||
Intellectual property, net | 773,478 | 702,095 | ||||||
Total other assets | 3,130,966 | 3,588,850 | ||||||
Total assets | $ | 10,670,592 | $ | 25,085,027 | ||||
Liabilities and Stockholders' Equity | ||||||||
Current liabilities: | ||||||||
Accounts payable | $ | 1,397,610 | $ | 442,925 | ||||
Other current liabilities | 1,856,598 | 2,367,362 | ||||||
Total current liabilities | 3,254,208 | 2,810,287 | ||||||
Stockholders' equity: | ||||||||
Common stock $0.001 par value: 200,000,000 shares authorized; 1,485,902 and 1,202,677 shares issued and outstanding, respectively | 1,486 | 1,203 | ||||||
Additional paid-in capital | 141,103,178 | 125,101,356 | ||||||
Accumulated deficit | (133,688,280 | ) | (102,827,819 | ) | ||||
Total stockholders' equity | 7,416,384 | 22,274,740 | ||||||
Total liabilities and stockholders' equity | $ | 10,670,592 | $ | 25,085,027 | ||||
See accompanying notes to the financial statements.
Balance SheetsStatements of Operations
2020 | 2019 | |||||||
Assets | ||||||||
Current assets: | ||||||||
Cash | $ | 27,319,685 | $ | 2,002,492 | ||||
Accounts receivable | 127 | 655 | ||||||
Prepaid expenses and other | 384,553 | 171,716 | ||||||
| Supplies | 3,011,042 | 801,780 | ||||||
Total current assets | 30,715,407 | 2,976,643 | ||||||
Property and equipment, net | 39,441 | 44,654 | ||||||
Other assets: | ||||||||
Security deposits | 10,741 | 21,732 | ||||||
Intellectual property, net | 601,625 | 491,200 | ||||||
Total other assets | 612,366 | 512,932 | ||||||
Total assets | $ | 31,367,214 | $ | 3,534,229 | ||||
Liabilities and Stockholders' Equity | ||||||||
Current liabilities: | ||||||||
Accounts payable and accrued expenses | $ | 192,968 | $ | 436,258 | ||||
Other current liabilities | 257,756 | 74,426 | ||||||
Total current liabilities | 450,724 | 510,684 | ||||||
Investment unit | — | — | ||||||
Commitments and contingencies | ||||||||
Stockholders' equity: | ||||||||
Common stock $0.001 par value: 200,000,000 shares authorized; 43,117,681 and 19,263,841 shares issued and outstanding, respectively | 43,118 | 19,264 | ||||||
Additional paid-in capital | 89,295,601 | 43,483,740 | ||||||
Accumulated deficit | (58,422,229 | ) | (40,479,459 | ) | ||||
Total stockholders' equity | 30,916,490 | 3,023,545 | ||||||
Total liabilities and stockholders' equity | $ | 31,367,214 | $ | 3,534,229 | ||||
Year Ended | ||||||||
December 31, | ||||||||
2023 | 2022 | |||||||
Revenues | $ | — | $ | — | ||||
Cost and expenses: | ||||||||
Depreciation | 15,004 | 25,575 | ||||||
Research and development | 17,616,605 | 11,510,074 | ||||||
General and administrative | 13,443,961 | 12,295,070 | ||||||
Total costs and expenses | 31,075,570 | 23,830,719 | ||||||
Operating loss | (31,075,570 | ) | (23,830,719 | ) | ||||
Interest income | 215,109 | 90,098 | ||||||
Net loss | $ | (30,860,461 | ) | $ | (23,740,621 | ) | ||
Net loss per share — basic and diluted | $ | (22.56 | ) | $ | (19.80 | ) | ||
Weighted average number of common shares — basic and diluted | 1,367,747 | 1,198,837 | ||||||
See accompanying notes to the financial statements
Statements of Operations
Year Ended | ||||||||
December 31, | ||||||||
2020 | 2019 | |||||||
Revenues | $ | — | $ | — | ||||
Cost and expenses: | ||||||||
Depreciation | 22,777 | 13,070 | ||||||
Research and development | 7,302,923 | 1,967,007 | ||||||
General and administrative | 10,635,881 | 8,702,596 | ||||||
Total costs and expenses | 17,961,581 | 10,682,673 | ||||||
Operating loss | (17,961,581 | ) | (10,682,673 | ) | ||||
Interest income | 18,811 | 27,905 | ||||||
Net loss | $ | (17,942,770 | ) | $ | (10,654,768 | ) | ||
Net loss per share — basic and diluted | $ | (0.51 | ) | $ | (0.66 | ) | ||
Weighted average number of common shares — basic and diluted | 35,522,875 | 16,026,980 | ||||||
See accompanying notes to the financial statementsstatements.
Statements of Changes in Stockholders’ Equity
Common Stock | ||||||||||||||||||||
Shares | Amount | Additional Paid-In Capital | Accumulated Deficit | Total | ||||||||||||||||
Balance at December 31, 2018 | 15,239,148 | $ | 15,240 | $ | 38,690,586 | $ | (29,824,691 | ) | $ | 8,881,135 | ||||||||||
Issuance of stock for cash | 3,517,986 | 3,518 | 1,263,676 | — | 1,267,194 | |||||||||||||||
Issuance of stock for services | 506,707 | 506 | 469,082 | — | 469,588 | |||||||||||||||
Share based compensation | — | — | 3,060,396 | — | 3,060,396 | |||||||||||||||
Net loss | — | — | — | (10,654,768 | ) | (10,654,768 | ) | |||||||||||||
Balance at December 31, 2019 | 19,263,841 | $ | 19,264 | $ | 43,483,740 | $ | (40,479,459 | ) | $ | 3,023,545 | ||||||||||
Issuance of stock for cash | 23,802,408 | 23,802 | 41,565,727 | — | 41,589,529 | |||||||||||||||
Issuance of stock for services | 51,432 | 52 | 154,596 | — | 154,648 | |||||||||||||||
Share based compensation | — | — | 4,091,538 | — | 4,091,538 | |||||||||||||||
Net loss | — | — | — | (17,942,770 | ) | (17,942,770 | ) | |||||||||||||
Balance at December 31, 2020 | 43,117,681 | $ | 43,118 | $ | 89,295,601 | $ | (58,422,229 | ) | $ | 30,916,490 | ||||||||||
Common Stock | ||||||||||||||||||||
Shares | Amount | Additional Paid-In Capital | Accumulated Deficit | Total | ||||||||||||||||
Balance at December 31, 2021 | 1,196,893 | 1,197 | 120,362,992 | (79,087,198 | ) | 41,276,991 | ||||||||||||||
Issuance of common stock for cash | 3,023 | 3 | 6,423 | — | 6,426 | |||||||||||||||
Issuance of common stock for services | 2,761 | 3 | 163,229 | — | 163,232 | |||||||||||||||
Share-based compensation | — | — | 4,568,712 | — | 4,568,712 | |||||||||||||||
Net loss | — | — | — | (23,740,621 | ) | (23,740,621 | ) | |||||||||||||
Balance at December 31, 2022 | 1,202,677 | 1,203 | 125,101,356 | (102,827,819 | ) | $ | 22,274,740 | |||||||||||||
Issuance of common stock and warrants for cash net of issuance costs | 282,725 | 282 | 10,593,095 | — | 10,593,377 | |||||||||||||||
Issuance of common stock for services | 500 | 1 | 19,009 | — | 19,010 | |||||||||||||||
Share-based compensation | — | — | 5,389,718 | — | 5,389,718 | |||||||||||||||
Net loss | — | — | — | (30,860,461 | ) | (30,860,461 | ) | |||||||||||||
Balance at December 31, 2023 | 1,485,902 | 1,486 | 141,103,178 | (133,688,280 | ) | $ | 7,416,384 | |||||||||||||
See accompanying notes to the financial statementsstatements.
Statements of Cash Flows
| Year Ended | ||||||||||||||||
| December 31, | ||||||||||||||||
2020 | 2019 | 2023 | 2022 | |||||||||||||
Cash flows from operating activities: | ||||||||||||||||
Net loss | $ | (17,942,770 | ) | $ | (10,654,768 | ) | $ | (30,860,461 | ) | $ | (23,740,621 | ) | ||||
Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||||||||||
Depreciation | 22,777 | 13,070 | 15,004 | 25,575 | ||||||||||||
Share based compensation | 4,246,186 | 3,529,984 | ||||||||||||||
Share-based compensation | 5,408,728 | 4,731,944 | ||||||||||||||
Changes in operating assets and liabilities: | ||||||||||||||||
Accounts receivable | 528 | 8,642 | 34,852 | (34,852 | ) | |||||||||||
Prepaid expenses and other | (212,837 | ) | 65,135 | (309,914 | ) | 27,125 | ||||||||||
Research and development supplies | 517,449 | 157,465 | ||||||||||||||
Deposits | 10,991 | (3,647 | ) | 11,818 | (13,127 | ) | ||||||||||
Accounts payable and accrued expenses | (247,290 | ) | 141,189 | |||||||||||||
Accounts payable | 954,686 | (530,269 | ) | |||||||||||||
| Other current liabilities | 187,329 | (18,326 | ) | (510,765 | ) | 1,755,262 | ||||||||||
Net cash used in operating activities | (13,935,086 | ) | (6,918,721 | ) | (24,738,603 | ) | (17,621,498 | ) | ||||||||
Cash flows from investing activities: | ||||||||||||||||
Additions to property and equipment | (17,564 | ) | (33,370 | ) | ||||||||||||
Reductions to property and equipment | 169 | — | ||||||||||||||
Additions to intellectual property | (110,424 | ) | (111,749 | ) | (71,383 | ) | (59,735 | ) | ||||||||
| Additions to research and development supplies | (2,209,262 | ) | (801,780 | ) | ||||||||||||
Net cash used in investing activities | (2,337,250 | ) | (946,899 | ) | (71,214 | ) | (59,735 | ) | ||||||||
Cash flows from financing activities: | ||||||||||||||||
Proceeds from issuances of common stock | 41,589,529 | 1,267,194 | ||||||||||||||
Net proceeds from issuances of common stock | 10,593,377 | 6,426 | ||||||||||||||
Net cash provided by financing activities | 41,589,529 | 1,267,194 | 10,593,377 | 6,426 | ||||||||||||
Net increase (decrease) in cash | 25,317,193 | (6,598,426 | ) | |||||||||||||
Cash, beginning of year | 2,002,492 | 8,600,918 | ||||||||||||||
Cash, end of year | $ | 27,319,685 | $ | 2,002,492 | ||||||||||||
Net decrease in cash | (14,216,440 | ) | (17,674,807 | ) | ||||||||||||
Cash and cash equivalents, beginning of year | 20,954,069 | 38,628,876 | ||||||||||||||
Cash and cash equivalents, end of year | $ | 6,737,629 | $ | 20,954,069 | ||||||||||||
See accompanying notes to the financial statementsstatements.
Notes to Financial Statements
Note 1 – Description of Business and Basis of Presentation
We are
We are planning to initiate our Acclaim-1 and Acclaim-2 clinical trials in 2021. Acclaim-1 is a Phase 1/2 clinical trial using a combination of REQORSA with AstraZeneca PLC’s Tagrisso® in patients with late-stage NSCLC with mutated epidermal growth factor receptors ("EGFRs") whose disease progressed after treatment with Tagrisso. In January 2020, we received Food and Drug Administration ("FDA") Fast Track Designation for the Acclaim-1 patient population. Acclaim-2 is a Phase 1/2 clinical trial using a combination of REQORSA with Merck & Co.’s Keytruda® in late-stage NSCLC patients who are low expressors (1% to 49%) of the protein programmed death-ligand 1 ("PD-L1").
InCompany’s diabetes we are developing a gene therapy that is exclusively licensed from the University of Pittsburgh of the Commonwealth System of Higher Education ("University of Pittsburgh") for the treatment of Type 1 and Type 2 diabetes. This potential treatmenttechnology is designed to work in Type 1 diabetes by transforming alpha cells in the pancreas into functional beta-like cells, which can produce insulin but aremay be distinct enough from beta cells to evade the body’s immune system. OurIn Type 2 diabetes, product candidatethe Company’s technology is currently being evaluated in preclinical studies.believed to work by replenishing and rejuvenating exhausted beta cells that make insulin.
Oncology Platform
Oncology Platform
Utilizing our non-viral ONCOPREX Nanoparticle Delivery System, we are developing cancer treatments that are designed to administer cancer fighting genes. We encapsulate the gene-expressing plasmids using ONCOPREX lipid nanoparticles, and administer them intravenously, where they are then taken up by tumor cells and express proteins that are missing or found in low quantities in the tumor cells. With ourGenprex’s lead oncology drug candidate, REQORSA® Immunogene Therapy (generic name: quaratusugene ozeplasmid), previously referred to as GPX-001, is initially being developed in combination with prominent, approved cancer drugs to treat Non-Small Cell Lung Cancer (“NSCLC”) and Small Cell Lung Cancer (“SCLC”). REQORSA there ishas a multimodal mechanism of action whereby REQORSAit interrupts cell signaling pathways that cause replication and proliferation of cancer cells, re-establishes pathways for apoptosis, or programmed cell death, in cancer cells, and modulates the immune response against cancer cells. In early studies, REQORSA has also been shown to block mechanismsbe complementary with targeted drugs and immunotherapies. The Company’s strategy is to develop REQORSA in combination with current approved therapies and the Company believes REQORSA’s unique attributes position it to provide treatments that create drug resistance.improve on these current therapies for patients with NSCLC, SCLC, and possibly other cancers.
We believeAcclaim – 1: The Company is currently enrolling and treating patients in the Phase 2a expansion portion of its Phase 1/2 Acclaim-1 clinical trial. The Acclaim-1 trial uses a combination of REQORSA and AstraZeneca’s Tagrisso® in patients with late-stage NSCLC that ourhas activating epidermal growth factor receptor (“EGFR”) mutations and progression after treatment with Tagrisso. Following the May 2023 completion of the Phase 1 dose escalation portion of the study, the Acclaim-1 Safety Review Committee (“Acclaim-1 SRC”) approved advancement from the Phase 1 dose escalation portion to the Phase 2a expansion portion of the study. Based on a review of safety data which showed no dose limiting toxicities (“DLTs”), the Acclaim-1 SRC determined that the recommended Phase 2 dose (“RP2D”) of REQORSA will be 0.12 mg/kg. This was the highest dose level delivered in the Phase 1 portion of the study and is twice the highest dose level delivered in the Company’s prior clinical trial combining REQORSA with Tarceva® for the treatment of late-stage lung cancer. Genprex opened the Phase 2a expansion portion of the study and enrolled and dosed the first patient in January 2024. The Phase 2a expansion portion of the trial is expected to enroll approximately 66 patients; half will be patients who received only prior Tagrisso treatment and the other half will be patients who received prior Tagrisso treatment and chemotherapy. The aim is to determine toxicity and efficacy profiles of patients with different eligibility criteria. There will be an interim analysis following the treatment of 19 patients in each cohort. The Company expects to complete the enrollment of 19 patients in each cohort of the Phase 2a expansion portion of the study by the end of 2024, and thus the Company expects the interim analyses in early 2025. The Food and Drug Administration (“FDA”) has granted Fast Track Designation for the Acclaim-1 treatment combination of REQORSA and Tagrisso in NSCLC patients who have progressed after Tagrisso treatment.
Acclaim – 2: The Company is currently enrolling and treating patients in the Phase 1 dose escalation portion of its Phase 1/2 Acclaim-2 clinical trial. The Acclaim-2 trial uses a combination of REQORSA and Merck & Co.’s Keytruda® in patients with late-stage NSCLC whose disease has progressed after treatment with Keytruda. Patients are currently being treated at the 0.06 mg/kg dose level in the first cohort of patients and, subject to Acclaim-2 Safety Review Committee (“Acclaim-2 SRC”) approval, will be treated at successive dose levels of 0.09 mg/kg and 0.12 mg/kg. In March 2023, Genprex amended the Acclaim-2 protocol to include additional treatments in the control group with the goal of accelerating enrollment in the study by making the trial more attractive to a wider variety of investigators. The Company expects enrollment in the dose escalation portion of the study to be completed in the second half of 2024. The Company will then initiate and evaluate patients in the Phase 2a expansion portion of the study at the maximum tolerated dose (the “MTD”) or RP2D. The FDA has granted Fast Track Designation for the Acclaim-2 treatment combination of REQORSA and Keytruda in NSCLC patients who have progressed after Keytruda treatment.
The expansion portion of both the Acclaim-1 and Acclaim-2 trials are Phase 2 studies. The expansion portion of these studies provides the Company with the advantage of early insight into drug effectiveness in defined and distinct patient populations at the MTD or RP2D in order to better evaluate efficacy and increase the likelihood of a successful randomized Phase 2 trial which will follow the expansion portion of each study.
Diabetes Gene Therapy
Our
Capital Requirements, Liquidity and Going Concern Considerations
OurThe Company’s financial statements are prepared in accordance with U.S. generally accepted accounting principles ("GAAP"(“US GAAP”) applicable to a going concern, which contemplates the realization of assets and liquidation of liabilities in the normal course of business. However, as shown in the accompanying financial statements, we havethe Company has sustained substantial losses from operations since inception and have has no current source of revenue. In addition, we havethe Company has used, rather than provided, cash in ourits operations. We expectGenprex expects to continue to incur significant expenditures to further clinical trials for the commercial development of ourits patents.
ManagementThe Company recognizes that weit must obtain additional capital resources to successfully commercialize our intellectual property.its product candidates. To date, we haveGenprex has received funding in the form of equity and debt, and we planthe Company plans to seek additional funding in the future. However, no assurances can be given that weit will be successful in raising additional capital. If we are the Company is not able to timely and successfully raise additional capital, the timing of ourits clinical trials, financial condition and results of operations will continue to may be materially and adversely affected. These financial statements do not include any adjustments relating to the recoverability and classification of recorded asset amounts and classification of liabilities.
Genprex believes that its current cash and cash equivalents will be sufficient to fund expenditure requirements for its necessary operations and expected clinical trial activities into the third quarter of 2024. Genprex has based these estimates, however, on assumptions that may prove to be wrong, and could spend available financial resources much faster than it currently expects. The Company will need to raise additional funds to continue funding its development and operations. The Company plans to secure such additional funding, although there are no guarantees or commitments for additional funding.
As a result of its recurring losses from operations and the need for additional financing to fund its operating and capital requirements, there is uncertainty regarding the Company’s ability to maintain liquidity sufficient to operate its business effectively, which raises substantial doubt as to the Company’s ability to continue as a going concern. These financial statements do not include any adjustments that might be necessary if Genprex is unable to continue as a going concern.
Note 2 – Summary of Significant Accounting Policies
The Company’sGenprex’s financial statements have been prepared in accordance with US GAAP. Accordingly, they do not include all the information and footnotes required by generally accepted accounting principles for complete financial statements. In ourthe Company’s opinion the financial statements include all adjustments (consisting of normal recurring accruals) necessary to make the financial statements not misleading. The results of operations for any interim periods are not necessarily indicative of results to be expected for the full year. A summary of ourthe Company’s significant accounting policies consistently applied in the preparation of the accompanying financial statements follows.
CapitalReverse Stock Split
In connection with the Company’s initial public offering ("IPO"On February 2, 2024, Genprex completed a 1-for-40 reverse stock split (“Reverse Split”) in April 2018, all of the Company’s preferred stockits issued and non-voting common stock were converted intooutstanding shares of the Company’s common stock. The Company’s common stock was then forward-split at a ratioReverse Split did not change the number of 6.6841954-to-1. Furthermore, prior to the closing of the IPO, the Company’s Certificate of Incorporation was amended and restated to provide the Company with the authority to issue up to 210,000,000 shares of stock consisting of 200,000,000authorized shares of common stock at aor par value of $0.001value. All references in these financial statements to shares, share prices, exercise prices, and other per share and 10,000,000 shares of preferred stock atinformation in all periods have been adjusted, on a par value of $0.001 per share.retroactive basis, to reflect the Reverse Split (See Note 4 – Equity – Reverse Stock Split).
Reclassification of Prior Year Presentation
Certain prior year amounts have been reclassified for consistency with the current year presentation. These reclassifications had no effect on the reported results of operations. An adjustment has been made to the Statement of Cash Flows for the year ended fiscal year ended December 31, 2022 to reclassify research and development supplies from an investing activity to an operating activity.
Use of Estimates
The preparation of ourthe Company’s financial statements in conformity with US GAAP requires usthe Company to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of expenses during the reporting period. Actual results could differ from those estimates.
Cash and Cash Equivalents
We considerGenprex considers all highly liquid short-term investments with an initial maturity of three months or less to be cash equivalents. Any amounts of cash in financial institutions which exceed FDICFederal Deposit Insurance Corporation (“FDIC”) insured limits expose usthe Company to cash concentration risk. We have noGenprex has cash equivalents,in a money market account and had $27,091,596$6,490,117 and $1,761,278$20,679,538 in excess of FDIC insured limits of $250,000$250,000 at December 31, 20202023 and December 31, 20192022, respectively. Any loss incurred or a lack of access to such funds could have a significant adverse impact on the Company’s financial condition, results of operations, and cash flows.
Net Loss Per Share
Basic net loss per share is calculated by dividing the net loss by the weighted-average number of shares of common stock outstanding for the period, without consideration for potential dilutive shares of common stock, which includes common stock equivalents consisting of (i) 632,323 unexercised options granted by the Company’s board of directors and warrants to purchase shares of common stock, and (ii) 51,862 unvested restricted stock units to purchase shares of common stock granted by the Company’s board of directors as of December 31, 2023.
Fair Value of Financial Instruments
The carrying amounts reported in the balance sheet for cash, money-market savings account, accounts receivable, and accounts payable and accrued expensespayables approximate fair value because of the immediate or short-term maturity of these financial instruments.
Accounting Standards Codification ("ASC") 820, Fair Value Measurements and Disclosures, defines fair value, provides a consistent framework for measuring fair value under GAAP and expands fair value financial statement disclosure requirements. ASC 820’s valuation techniques are based on observable and unobservable inputs. Observable inputs reflect readily obtainable data from independent sources, while unobservable inputs reflect our market assumptions. ASC 820 classifies these inputs into the following hierarchy:
|
| |
|
| |
|
|
Property and Equipment
Furniture and equipment are stated at cost.cost less accumulated depreciation. Depreciation is calculated using the straight-line method over the estimated useful lives of the assets, which range from three to five years. Routine maintenance and repairs are charged to expense as incurred and major renovations or improvements are capitalized.
Research and Development Materials Costs
Research and development expenditures consist of costs incurred to conduct research, develop engineering materials for further study, and development activities.develop clinical strategies for current and future programs associated with the Company's preclinical and Phase 1/2 clinical trials. These expenditures are expensed in the period incurred and include payments to collaborative research partners, manufacturing partners and consultants, and clinical strategy partners, wages and associated employee benefits, facilities, and overhead costs. These expenditures relate to our preclinical, Phase 1, and Phase 2 clinical trials and are expensed as incurred. Purchased materials
Materials acquired to be used in clinical research, that have an alternative future researchuse, are capitalized when the materials are acquired, and included in research and development supplies. These supplies are recognized as expense as they are consumed through use for testing or clinical activities, or have spoiled. The costs of materials that were acquired for a particular research and development activity and have no alternative future use are expensed in the period acquired.
Research and development supplies purchased and capitalized for future use was $3,011,042were $2,347,488 and $801,780$2,864,937 at December 31, 20202023 and December 31, 20192022, respectively.
Awards
In 2010, we were awarded $4.5 million from the State of Texas Emerging Technology Fund ("TETF"). The award was received in two tranches of $2.25 million during 2010 and 2011. The award proceeds were used for the development and future commercialization of our nanomolecular therapy product for the treatment of cancer. In consideration for the award, we provided the TETF with an "Investment Unit", consisting of (i) a Promissory Note ("Note") and (ii) a right to purchase our equity shares ("Warrant"). The funds received for this award were assigned to the Investment Unit, and classified separately from equity as "mezzanine" in the balance sheet.
In 2010, we also were awarded approximately $244,500 from the U.S. Treasury Department for our QTDP Program Nanoparticle Therapy for Lung Cancer. The award was received during 2011 for our historical activities, and required no prospective expenditures. We accounted for these funds received as revenue at that time.
Intellectual Property
Intellectual property consists of legal and related costs associated with patents, trademarks, and other proprietary technology and rights developed, acquired, or licensed by or maintained by usGenprex that we believeit believes contribute to a probable economic benefit toward such patents and activities. These costs incurred in connection with obtaining and maintaining intellectual property protection, such as patent applications and filing fees associated with and patent maintenance,protection, are capitalized. Intellectual property is stated at cost, to be amortized on a straight-line basis over the estimated useful lives of the assets.
Accounting for Stock-Based Compensation
We useGenprex uses the fair value-based method of accounting for stock-based compensation for options granted to employees, independent consultants and contractors. We measureThe Company measures options granted at fair value determined as of the grant date and recognize the expense over the periods in which the options vest or are expected to vest and related services are rendered based on the terms and conditions of the award. Generally, where the award only has a service condition, the requisite service period is the same as the vesting period.
Financial InstrumentsLong-Lived Assets
We have elected the Fair Value Option to account for the Investment Unit at fair value as a combined hybrid financial instrument containing a Warrant and a Note (see Note 4 - Investment Unit Note). Prior to its exercise, the Warrant component was not classified within equity, as the exercise price of the warrants was affected by the market price of our stock in a future qualifying financing transaction and was not considered to be indexed to our own stock. The Note is not classified within liabilities, as our management can determine the timing of the repayment obligation, if any. As a result, the Warrant and Note that comprised the Investment Unit were aggregated and classified within the mezzanine section of the balance sheet.
Due to the contingent terms of the financial instruments, changes in the fair value of the Investment Unit were calculated and realized in earnings. There were no changes in the fair value of the Investment Unit at December 31, 2020.
In August 2019, the remaining articles of the Investment Unit were terminated.
Long-Lived Assets
We reviewGenprex reviews long-lived assets and certain identifiable intangibles held and used for possible impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. In evaluating the fair value and future benefits of its intangible assets, managementthe Company performs an analysis of the anticipated undiscounted future net cash flow of the individual assets over the remaining amortization period. We recognizeThe Company recognizes an impairment loss if the carrying value of the asset exceeds the discounted expected future cash flows. During the years ended December 31, 20202023 and December 31, 20192022, there were no deemed impairments of ourthe Company’s long-lived assets.
Recent Accounting Developments
Accounting pronouncements issued but not effective until after December 31, 20202023 are not expected to have a significant effect on ourthe Company’s financial condition, results of operations, or cash flows.
Note 3 – Intellectual Property
On February 11, 2020, we entered into an exclusive license agreement with the UniversityAs of Pittsburgh for patented gene therapy technologies relating to the potential treatment of type 1 and type 2 diabetes.
On May 4, 2020, the Company entered into an exclusive worldwide license agreement with The Board of Regents of the University of Texas System on behalf of MD Anderson relating to a portfolio of 16 patent applications and related technology for the treatment of cancer using the Company’s lead drug candidate and immunotherapies.
We haveDecember 31, 2023, Genprex owned or had exclusive license agreements on 28 issued21 granted patents and 1723 pending patent applications worldwide for technologies developed in-house or by researchers at the National Cancer Institute, MD Anderson, the University of Texas Southwestern Medical Center, and the University of Pittsburgh. These patents comprise various therapeutic, diagnostic, technical and processing claims. These license rights will be amortized on a straight-line basis over the estimated period of useful lives of the underlying patents or the license agreements.
Note 4 – Investment Unit
The TETF was created as an incentive for economic development to the Texas economy by providing financial support that leverages private investment for the creationUniversity of high-quality technology jobs in Texas. The award received required us to comply with certain performance conditions to ensure the monies the Company received were used for development activities in the state of Texas, and that we maintained our corporate nexus in Texas. Further, in connection with the award, the Company issued an Investment Unit to the TETF. On September 25, 2017 and again on August 16, 2019, the Company entered into Termination Agreements with the Texas Treasury Safekeeping Trust Company, the entity managing and controlling TETF interests, that terminated Article II and the all remaining Articles of the Investment Unit, respectively, so that the entirety of the Investment Unit was effectively terminated. As further described below, the Investment Unit consisted of a Promissory Note and a Right to Purchase.
Promissory NotePittsburgh
The Promissory Note was an obligation to repay the $4.5 million principal amount, with interest accrued at 8% per annum, but only if an event of default occurred prior to August 13, 2020. If no event of default occurred prior to August 13, 2020, the Promissory Note and all related interest would be cancelled. The Note was cancelled on August 16, 2019.
Consistent with the stated objectives of the TETF, an event of default that would trigger the repayment obligation under the Promissory Note was a failure to maintain our principal place of business or our principal executive offices headquartered in the State of Texas (referred to as the “Residency Requirement”) until August 13, 2020.
Warrant
The Warrant was an obligation to issue (a Right to purchase by the TETF) shares of the same class of stock as issued in the Company's next financing transition (“First Qualifying Financing Transaction”), at 80% of the per share transaction value (effectively a 20% discount). Alternatively, the TETF could exercise its right to purchase at any time prior to the occurrence of a First Qualifying Financial Transaction for $0.001 per share.
The Warrant included a provision that required changes in the strike price, driven by the pricing of the First Qualifying Financing Transaction. As a result, the Warrant embedded in the Investment Unit was accounted for as a derivative financial instrument and classified outside of equity under ASC 815-40-15 as the settlement adjustment from the future transaction did not permit for the strike price to be considered fixed.
On March 12, 2014,February 11, 2020, Genprex entered into an exclusive license agreement with the TETF exercised its Right to PurchaseUniversity of Pittsburgh for $0.001 per share, and we issuedpatented gene therapy technologies relating to the TETFpotential treatment of Type 1 and Type 2 diabetes. This license was first amended on August 17, 2022, to extend the milestone related to the filing of a new investigational drug (“IND”) application. This license was amended again on November 3, 2022, to include a new licensed glucagon promoter technology related to Type 1 diabetes and set FDA and clinical milestones related to the glucagon technology.
On November 22, 2022, Genprex entered into an aggregateexclusive license agreement with the University of 184,797Pittsburgh relating to the transformation of macrophages enabling them to reduce autoimmunity activity in Type 1 diabetes.
On December 29, 2022, Genprex entered into an exclusive license agreement with the University of Pittsburgh relating to the use of an insulin promoter in combination with the Company’s existing gene therapy, including the Pdx1 and MafA transcription factors, as a potential treatment for Type 2 diabetes.
On July 14, 2023, Genprex entered into an exclusive license agreement with the University of Pittsburgh related to a gene therapy for both Type 1 and Type 2 diabetes using a MafB promoter to drive expression of the Pdx1 and MafA transcription factors.
The University of Texas MD Anderson Cancer Center
On May 4, 2020, Genprex entered into an exclusive worldwide license agreement with The Board of Regents of the University of Texas System on behalf of MD Anderson relating to a portfolio of patent applications and related technology for the treatment of cancer using the Company’s lead drug candidate and immunotherapies. See “Note 7 - Commitments and Contingencies - Commitments - MD Anderson” for information on additional agreements involving MD Anderson licensed rights and technologies.
Note 4 – Equity
Reverse Stock Split
On February 2, 2024, Genprex completed a 1-for-40 reverse stock split of its issued and outstanding shares of our Series B preferredcommon stock. These shares were subsequently forward-split and converted to 1,235,219The Reverse Split did not change the number of authorized shares of common stock or par value. All references in connection with our IPO. these consolidated financial statements to shares, share prices, exercise prices, and other per share information in all periods have been adjusted, on a retroactive basis, to reflect the split.
Accounting for the Investment Unit
We accounted for the Investment Unit as a hybrid financial instrument under Financial Accounting Standards Board ("FASB") Statement 155, and measured the Investment Unit at the amount of proceeds received from the TETF award. The First Qualifying Financial Transaction occurred during December 2013, resulting in an adjustment to the fair value of the Investment Unit in the amount of approximately $2.5 million. The TETF exercised the Warrant for $0.001 per share. We received notice of purchase from the TETF during March 2014, and issued 184,797 shares of Series B preferred stock, which has since been converted to 1,235,219 shares of common stock upon completion of the Company’s IPO. Upon exercise by the TETF of the Warrant, the remaining component within the Investment Unit was the Promissory Note. The Investment Unit was valued at zero, because our obligation to repay the Promissory Note arose from an event of default (a failure to maintain the Texas Residency Requirement), which was an event which rested entirely within our control.
Note 5 – Equity
Registered Direct Offerings
On November 22, 2019, the Company completed a registered direct offering (“2019 RDO”), whereby the Company sold to investors an aggregate of 3,167,986 shares of the Company’s common stock at $0.40 per share and warrants to purchase up to 3,167,986 shares of the Company’s common stock at an exercise price of $0.46 per share. The warrants were first exercisable on May 22, 2020. The Company received net proceeds of approximately $1,093,000 after commissions and expenses. Additionally, the placement agent was issued warrants to purchase common stock equal to 7% of the aggregate number of shares of common stock issued and issuable pursuant to the 2019 RDO (including shares underlying any warrants), or 443,518 shares of common stock at an exercise price of 125% of the 2019 RDO price per share, or $0.50 per share.
In connection with the closing of the 2019 RDO, the Company further adjusted the warrants to purchase up to 2,283,740 shares of the Company's common stock, which had been issued as part of the Company's May 9, 2018 private placement and adjusted in August 2018 to (i) reduce the exercise price for each share from $4.25 per share to $0.46 per share, (ii) extend the date upon which such warrants could be exercised to May 22, 2020, and (iii) extend the termination date of such warrants by six months and one day.
On January 21, 2020, the CompanyMarch 1, 2023, Genprex completed a registered direct offering, in which the Company sold to an accredited healthcare-focused institutional investor 961,000an aggregate of 95,239 shares of the Company’sits common stock and warrants to purchase up to 95,239 shares of its common stock, at $0.24a combined offering price of $42.00 per share of common stock and accompanying warrant. The warrants are exercisable immediately upon issuance, expire 5 years from the date of issuance and have an exercise price of $44.00 per share. The Company received net proceeds of approximately $200,000$3.6 million after $400,000 of commissions and expenses.expenses, excluding any proceeds that may be received in the future from any exercise of the warrants.
On January 23, 2020, the CompanyJuly 21, 2023, Genprex completed a registered direct offering priced at the market under Nasdaq rules, in which the Company sold to accredited healthcare-focused institutional investors an aggregate of 7,620,000(i) 185,644 shares of its common stock, and (ii) warrants to purchase up to 185,644 shares of its common stock, at a combined offering price of $40.40 per share of common stock and accompanying warrant. The warrants are exercisable immediately upon issuance, expire 5 years from the date of issuance and have an exercise price of $35.40 per share. Also, the Company agreed to issue to H.C. Wainwright & Co., LLC or its designees (the “Placement Agent”) warrants to purchase up to an aggregate of 11,140 shares of the Company’s common stock at $1.05stock. The warrants issued to the Placement Agent have substantially the same terms as the warrants issued to the investors except that the Placement Agent warrants have an exercise price of $50.50 per share. The Companyshare and expire on July 18, 2028. Genprex received net proceeds of approximately $7.2$6.7 million after approximately $800,000 of commissions and expenses.expenses, excluding any proceeds that may be received in the future from any exercise of the warrants.
See “Note 9 - Subsequent Events - March 2024 Registered Direct Offering” for a description of the Company's registered direct offering of shares, pre-funded warrants, and warrants in March 2024 resulting in net proceeds of approximately $5.8 million.
At-The Market Offering
On February 19, 2020,November 18, 2022, Genprex entered into an Equity Distribution Agreement with JMP Securities LLC (“JMP Securities”) pursuant to which the Company amendedmay sell from time to time, at its option, shares of its common stock through JMP Securities, as sales agent (the “2022 ATM Facility”), up to an aggregate offering price of $50 million. Sales of the shares were made under the Company’s previously filed Registration Statement on Form S-3S-3 (Reg. No.333-239134), by means of ordinary brokers’ transactions on the NASDAQ Global Market or otherwise. Additionally, under the terms of the Sales Agreement, the shares could be sold at market prices, at negotiated prices or at prices related to increase the maximum offering size by approximately $3,000,000. On February 21, 2020,prevailing market price. Genprex agreed to pay JMP Securities a commission of 3.0% of the gross proceeds from the sale of the shares. During the year ended December 31, 2023, the Company completed a registered direct offeringsold 1,342 shares of common stock for aggregate net proceeds of $78,355 under the amended S-3 Registration Statement, in2022 ATM Facility. On December 12, 2023, the Company provided notice to JMP Securities of its termination of the 2022 ATM Facility. The termination of the Equity Distribution Agreement with JMP Securities was effective as of December 13, 2023.
On December 13, 2023, Genprex entered into an At The Market (“ATM”) Offering Agreement (the “Agreement”) with H.C. Wainwright & Co., LLC, serving as agent (the “Agent”) with respect to an at-the-market offering program (the “2023 ATM Facility”) under which the Company may offer and sell through the Agent, from time to time at its sole discretion, up to such number or dollar amount of shares of its common stock (the “Shares”) as registered on the prospectus supplement covering the ATM offering, as may be amended or supplemented from time to time. Any Shares offered and sold pursuant to investors an aggregatethis Agreement will be issued pursuant to the Company’s currently effective shelf Registration Statement on Form S-3 (File No.333-271386) filed with the SEC on April 21, 2023, which was declared effective on June 9, 2023. The Company has agreed to pay the Agent a commission equal to three percent (3%) of 5,000,000 sharesthe gross sales proceeds of any Shares sold through the Agent under the Agreement, and also have provided the Agent with customary indemnification and contribution rights. As of December 31, 2023, the Company had not sold any Shares under the 2023 ATM Facility.
See “Note 9 – Subsequent Events – 2023 ATM Facility” for a description of the Company’s common stock at $3.50 per share. The Company receivedusage of the 2023 ATM Facility after December 31, 2023 resulting in net proceeds of approximately $16 million after commissions and expenses.
On December 24, 2020, the Company completed a registered direct offering, in which the Company sold to an accredited investor 3,116,884 shares of the Company’s common stock at $3.85 per share. The Company received net proceeds of approximately $11.2 million after commissions and expenses.$881,946.
Stock Issuances
During the year ended December 31, 20202023, weGenprex issued (i) 16,697,884 shares of common stock from registered direct offerings for cash proceeds of $37,731,643, (ii) 1,277,743 shares of common stock from the exercise of options for cash proceeds of $1,320,155, (iii) 5,511,599 shares of common stock from the exercise of warrants for cash proceeds of $2,537,731, (iv) 199,630 shares of common stock from the exercise of cashless warrants, and (v) 51,432500 shares of common stock for serviceservices provided to us,the Company valued at $154,648.$19,200 to the Chairman of its Scientific Advisory Board, and (ii) 500 shares of common stock upon the exercise of options by a former board member of the Company.
During the year ended December 31, 20192022, weGenprex issued (i) 3,167,986 shares of common stock from the Company’s 2019 RDO for cash proceeds of $1,267,194, (ii) 506,707500 shares of common stock for serviceservices provided to us,the Company valued at $469,588, and (iii) we issued 350,000$42,400 to the Chairman of its Scientific Advisory Board, (ii) 342 shares of common stock held in abeyanceupon the exercise of warrants on a cashless basis, (iii) 2,925 shares of common stock upon the exercise of options by an executive of the Company, and (iv) 1,919 shares of common stock for an investor from our May 2018 private placement.services provided to the Company, valued at $99,010, to consultants.
Preferred Stock
In connection with the Company’s IPO, all preferred stock included in Series A through Series G, totaling 1,394,953 shares were converted into 9,324,177 shares of common stock in association with the forward-split (See Note 2 - Capital Stock). Upon the completion of the IPO, the CompanyGenprex is authorized to issue 10,000,000 shares of preferred stock at a par value of $0.001 per share, none of which are outstanding as of December 31, 20202023.
Common Stock
Upon the completion of the IPO, all of the Company’s non-voting common stock automatically converted into voting common stock on a one-to-one basis. Immediately following the completion of the IPO, the CompanyGenprex is authorized to issue 200,000,000 shares of common stock at a par value of $0.001 per share, all of which is voting common stock. There were 43,117,6811,485,902 shares of common stock outstanding at December 31, 20202023.
Common Stock Purchase Warrants
Common stock purchase warrant activity for the years ended December 31, 2020 2023 and 20192022 respectively are as follows:
| Number of Warrants | Weighted Avg. Exercise Price | |||||||
Outstanding at January 1, 2019 | 3,864,552 | $ | 2.36 | |||||
Issued | 3,611,504 | 0.47 | ||||||
Cancelled or expired | — | — | ||||||
Exercised | — | — | ||||||
Outstanding at December 31, 2019 | 7,476,056 | $ | 1.45 | |||||
Issued | 550,000 | 2.41 | ||||||
Cancelled or expired | (44,528 | ) | 0.50 | |||||
Exercised | (5,826,781 | ) | 0.47 | |||||
Outstanding at December 31, 2020 | 2,154,747 | $ | 4.37 | |||||
| Exercisable at December 31, 2020 | 1,904,747 | $ | 4.61 | |||||
2023 | 2022 | ||||||||
Number of | Weighted Average | Number of | Weighted Average | ||||||
Warrants | Exercise Price | Warrants | Exercise Price | ||||||
Outstanding at January 1, | 53,695 | $ 172.81 | 55,121 | $ 175.65 | |||||
Warrants issued | 296,273 | 38.75 | 2,575 | 61.55 | |||||
Warrants exercised | — | — | 342 | 20.00 | |||||
Warrants cancelled or expired | 3,528 | 209.77 | 3,659 | 151.66 | |||||
Outstanding at December 31, | 346,440 | $ 57.79 | 53,695 | $ 172.81 | |||||
Exercisable at December 31, | 340,585 | $ 57.36 | 47,548 | $ 183.42 | |||||
InDuring the year ending ended December 31, 20202023, (i) investors and placement agents of the Company's May 2018 private placement and 2019 RDO exercised warrants to purchase 5,511,599 shares of common stock for cash proceeds of $2,537,731, (ii) the CompanyGenprex issued 315,182 shares of common stock and cancelled 44,528 shares of common stock to placement agents of the 2019 RDO for the exercise of warrants via cashless exercise, and (iii) the Company issued(i) warrants to purchase up to 550,000an aggregate of 4,250 shares of common stock to service providers including 500,000at exercise prices ranging from $26.00 to $66.00 per share, the fair market value of a share of common stock on the date of issuance, (ii) warrants to purchase up to 95,239 shares of common stock to Cancer Revolution, LLC accredited healthcare-focused institutional investors in connection with the registered direct offering completed on March 1, 2023, at an exercise price of $2.27$44.00 per share, and 50,000(iii) warrants to purchase up to 185,644 shares of common stock to Capital City Technical Consulting, Inc. accredited healthcare-focused institutional investors in connection with the registered direct offering completed on July 21, 2023, at an exercise price of $3.81$35.40 per share, and (iv) warrants to purchase up to 11,140 shares of common stock to the Placement Agent in connection with the registered direct offering completed on July 21, 2023, at an exercise price of $50.50 per share. InDuring the year ending ended December 31, 20202023, wethe Company was deemed to cancel (i) warrants to purchase up to an aggregate of 960 shares of common stock upon termination of warrants previously issued to placement agents associated with the Company’s Initial Public Offering (“IPO”) in March 2018, and (ii) warrants to purchase up to an aggregate of 2,568 shares of common stock upon termination of warrants to a service provider. During the year ended December 31, 2023, the Company recorded share-based compensation of $450,000$133,558, respectively, associated with Company milestone-basedthe vesting and issuance of the Cancer Revolution, LLC warrants. We expect to record $124,000 of share-based compensation for time-based vesting over the next three years and another $300,000 of share-based compensation based on performance-based vesting.
InDuring the year ending ended December 31, 20192022, weGenprex issued (i) issueda warrant to purchase up to 1,250 shares of common stock to a service provider at an exercise price of $55.20 per share, the fair market value of a share of common stock on the date of issuance, (ii) a warrant to purchase up to 1,250 shares of common stock to a service provider at an exercise price of $59.60 per share, the fair market value of a share of common stock on the date of issuance, (iii) a warrant, previously accounted as a warrant issuable to a consultant in consideration of services provided at the Company’s IPO, to purchase up to 75 shares of common stock at an exercise price of $200.00 per share, the fair market value of a share of common stock at the Company’s IPO, and (iv) 342 shares of common stock to a placement agent associated with a registered direct offering in November 2019 upon the exercise of warrants on a cashless basis. During the year ended December 31, 2022, the Company was deemed to cancel (i) warrants to purchase 3,167,986up to an aggregate of 257 shares on common stock upon forfeiture of ourwarrant shares associated with a cashless exercise, and (ii) warrants to purchase up to an aggregate of 3,402 shares on common stock upon termination associated with prior service providers.
As of December 31, 2023, Genprex had outstanding warrants to purchase 346,440 shares of common stock at $0.46 per share to the investors in the Company’s 2019 RDO, (ii) issued warrants to purchase 443,518 shares of our common stock at $0.58 per share to the underwriter of the Company’s 2019 RDO, and (iii) reduced thea weighted average exercise price of the warrants,$57.79 that have been issued to various consultants, investors, inand placement agents of the Company’s May 2018 private placement,Company. These warrants vest immediately or over periods ranging up to 12 months, are exercisable for a period of up to five years, enable the holders to purchase 2,283,740 shares of the Company’s common stock at exercise prices ranging from $4.25$26.00 to $288.80 per share and have per-share fair values ranging from $13.85 to $0.46 per share. $185.00, based on Black-Scholes-Merton pricing models. The following assumptions were used in calculation of fair market value of options via Black-Scholes-Merton pricing models for the year ended December 31, 2023. Assumptions for the year ended December 31, 2022 are similar to those listed for the year ended December 31, 2023.
Twelve Months Ended December31, 2023 | ||
Expected term (in years): | 2.5 - 3.0 | |
Risk-free rate: | 5.33% - 5.52% | |
Volatility: | 83.42% | |
Dividend yield: | 0% |
On January 29, 2018, the Company entered into an agreement with a consultant whereby the Company agreed to grant warrants to purchase 6,000 shares of our common stock at $5.00 per share in consideration of services valued at $30,000 provided to the Company. As of December 31, 2020, the Company has not issued these warrant shares.
2018 Equity Incentive Plan
The Company’sGenprex’s board of directors and stockholders have approved and adopted the Company’s 2018 Equity Incentive Plan (“(“2018 Plan”), which became effective on the completion of the IPO on April 3, 2018. The 2018 Plan provides for the grant of incentive stock options that are intended to qualify under Section 422 of the Internal Revenue Code of 1986, as amended (“ISOs”), nonstatutory stock options, stock appreciation rights, restricted stock awards, restricted stock unit awards, performance-based stock awards other forms of equity compensation and performanceperformance-based cash awards. ISOs may be granted only to employees.employees of the Company. All other awards may be granted to employees, including officers, and to the Company’s non-employee directors and consultants and affiliates.of the Company.
A total of 4,160,000 shares of common stock are available under the 2018 Plan, which includes 554,963 shares of common stock reserved for issuance under our 2009 Equity Incentive Plan that were added to the 2018 Plan. No further grants will be made under the 2009 Plan and any shares subject to outstanding stock options under the 2009 Plan that would otherwise be returned to the 2009 Plan will instead be added to the shares initially reserved under the 2018 Plan.
In addition, theThe number of shares of common stock reserved for issuance under the 2018 Plan will automatically increase on January 1 of each year (the “evergreen provision”), beginning on January 1, 2019 by 5% of the total number of shares of the Company’s common stock outstanding on December 31 of the preceding calendar year, or a lesser number of shares determined by the Company’s board of directors or a committee appointed to administer the 2018 Plan. On January 1, 2023 and 2022, the number of shares of common stock reserved for issuance under the 2018 Plan was increased by an aggregate of 60,132 and 59,843 shares, respectively. As of December 31, 2023, a total of 11,686 shares of common stock remain available for issuance for future awards under the 2018 Plan. Subsequent to the year ended December 31, 2023, an additional 74,294 shares of common stock became available for issuance for future awards under the 2018 Plan pursuant to the evergreen provision thereof.
2018 Employee Stock Purchase Plan
Genprex’s board of directors and stockholders approved and adopted the Company’s 2018 Employee Stock Purchase Plan (“ESPP”), which became effective on April 3, 2018. The ESPP has not yet been utilized as a benefit available to the Company’s employees. The ESPP authorizes the issuance of 5,202 shares of the Company’s common stock pursuant to purchase rights that may be granted to its eligible employees. The number of shares of common stock reserved for issuance under the ESPP is automatically increased on January 1 of each calendar year, beginning on January 1, 2019, by 2% of the total number of shares of the Company’s common stock outstanding on December 31 of the preceding calendar year, or a lesser number of shares determined by the administrator of the 2018 Plan. On January 1, 2019 and 2020, the number of shares of common stock reserved for issuance under the 2018 Plan was increased by an aggregate of 761,957 and 963,192 shares, respectively.
2018 Employee Stock Purchase Plan
The Company’s board of directors and stockholders have approved and adopted the Company’s 2018 Employee Stock Purchase Plan (“ESPP”), which became effective on the completion of the IPO on April 3, 2018. The ESPP authorizes the issuance of 208,500 shares of the Company’s common stock pursuant to purchase rights granted to our eligible employees. The number of shares of common stock reserved for issuance will automatically increase on January 1 of each calendar year, from January 1, 2019 by the lesser of 2% of the total number of shares of our common stock outstanding on December 31 of the preceding calendar year or a number determined by the administrator of the ESPP. The administrator of the ESPP which is our Board of Directors, determined not to increase the number of shares reserved for issuance under the ESPP on January 1, 2019 or January 1, 2020 due to the ESPP not currently being utilized.2023.
Stock Options
As of December 31, 20202023, the Company hasGenprex had outstanding stock options to purchase 6,844,069285,883 shares of common stock that have been granted to various officers,executives, employees, directors, employees, vendors and independent contractors.contractors of the Company, including outstanding stock options to purchase 25,417 shares of common stock issued as inducement grants, outside of the 2018 Plan, associated with the hiring of new executives in 2021 and 2023. These options can vest immediately or over periods ranging from twelve (12)12 to forty-eight (48)48 months, are exercisable for a period of up to ten years, and enable the holders to purchase shares of ourthe Company’s common stock at exercise prices ranging from $0.001 - $9.80.$18.00 to $140.00 per share. The per-share fair values of these options range from $0.001$12.62 to $7.93,$98.84, based on Black-Scholes-Merton pricing models with the following assumptions:
| ||||
| ||||
| ||||
|
| Twelve Months Ended December 31, 2023 | Twelve Months Ended December 31, 2022 | |||
Expected term (in years): | 6.0 | 10 | ||
Risk-free rate: | 4.60% – 5.37% | 0.07% – 4.77% | ||
Volatility: | 83.14% - 83.42% | 75.98% - 88.38% | ||
Dividend yield: | 0% | 0% |
In
During the year ending ended December 31, 20202023, the CompanyGenprex (i) granted stock options to purchase an aggregate of 2,466,5298,251 shares of the Company'sCompany’s common stock with exercise prices ranging from $1.28$18.00 to $4.42$60.40 per share to employees, board members, and consultants, (ii) cancelled options to purchase 327,6406,245 shares of common stock at an exercise prices ranging from $50.80 to $142.00 per share in connection with the termination of certain employees, and (iii) issued 500 shares of the Company’s common stock upon the exercise of options held by a former board member with an exercise price of $11.92 per share.
During the year ended December 31, 2022, Genprex (i) granted stock options to purchase an aggregate of 74,600 of the Company’s common stock with exercise prices ranging from $50.80 to $140.00 per share to executives and employees, (ii) cancelled options to purchase 1,668 shares of common stock at exercise prices ranging from $5.29$80.00 to $9.80 due to expiration$146.40 per share in connection with the termination of options and separation of a former executive,certain employees, and (iii) issued 1,277,7432,925 shares of the Company'sCompany’s common stock upon the exercise of options held by former board members and a formeran executive with an exercise prices ranging from $0.015 to $2.15price of $0.60 per share.
In the year ending December 31, 2019, Company granted stock options to employees and consultants to purchase 1,744,300 shares of common stock with exercise prices ranging from $0.30 to $1.62 per share and cancelled options to purchase 297,058 shares of common stock due to the inactivity of service providers.
The weighted average remaining contractual term for the outstanding options at December 31, 2020 2023 and 20192022 is 7.066.13 and 7.457.08 years, respectively.
Stock option activity for the years ended December 31, 2020 2023 and 20192022, respectively, is as follows:
| Number of Shares | Weighted Avg. Exercise Price | |||||||
Outstanding at January 1, 2019 | 4,535,681 | $ | 3.31 | |||||
Options granted | 1,744,300 | 1.48 | ||||||
Options exercised | — | — | ||||||
Options expired | (297,058 | ) | — | |||||
Outstanding at December 31, 2019 | 5,982,923 | $ | 2.66 | |||||
Options granted | 2,466,529 | 2.87 | ||||||
Options exercised | (1,277,743 | ) | 1.03 | |||||
Options expired | (327,640 | ) | 8.31 | |||||
Outstanding at December 31, 2020 | 6,844,069 | $ | 2.81 | |||||
| Exercisable at December 31, 2020 | 4,491,545 | $ | 2.75 | |||||
2023 | 2022 | |||||||
Number of | Weighted Average | Number of | Weighted Average | |||||
Options | Exercise Price | Options | Exercise Price | |||||
Outstanding at January 1, | 284,377 | $ 123.19 | 214,370 | $ 134.14 | ||||
Options granted | 8,251 | 30.14 | 74,600 | 86.45 | ||||
Options exercised | 500 | 11.92 | 2,925 | 0.60 | ||||
Options expired or cancelled | 6,245 | 104.42 | 1,668 | 101.88 | ||||
Outstanding at December 31, | 285,883 | $ 121.11 | 284,377 | $ 123.19 | ||||
Exercisable at December 31, | 237,859 | $ 124.73 | 167,957 | $ 129.19 | ||||
Restricted Stock Units
A summary of the RSU activity under the 2018 Plan during the year ended December 31, 2023, is presented below. These amounts include RSUs granted to executives, other employees, and board members. There was no RSU activity for the year ended December 31, 2022.
2023 | ||||
Number of | Weighted Average | |||
Units | Grant Date Fair Value | |||
Outstanding at January 1, | — | $ — | ||
Restricted stock units granted | 57,119 | 60.08 | ||
Restricted stock units forfeited or cancelled | 5,257 | 66.00 | ||
Outstanding at December 31, | 51,862 | $ 59.48 | ||
Share-Based Compensation
In the year ending ended December 31, 20202023, the Company'sGenprex’s total share-based compensation was approximately $4.3$5.4 million, consisting of $4.4 million and $1.0 million associated with G&A expense and R&D expense, respectively, which approximately $3.6 million represents the vesting of options and warrants issued to service providers, executives, employees, and board members. Themembers of the Company. As of December 31, 2023, the Company’s total compensation cost related to non-vested time-based stock option awards, RSUs, and warrants granted to executives, employees, and board members, and service providers and not yet recognized was approximately $4.7 million, the year ending December 31, 2020. The Companyconsisting of $3.8 million and $0.9 million associated with G&A expense and R&D expense, respectively. Genprex expects to record this stock-based compensation expense over the next three years using a graded vesting method. As of December 31, 20202023, the weighted average term over which these expenses are expected to be recognized are 2.15is 1.12 years.
As of December 31, 20202023, there are no performance-based stock option awards outstanding. outstanding and one performance-based warrant outstanding issued to a service provider of the Company. Genprex’s total compensation cost related to the non-vested performance-based warrant not yet recognized was approximately $300,000. The entirety of this warrant may be recognized and recorded upon the achievement of certain milestones.
In the year ended December 31, 2019,2022, the Company'sCompany’s total share-based compensation was approximately $3.5$4.7 million with approximately $3.0$4.6 million representing the vesting of options issued to service providers, employees, and board members.members of the Company.
Note 5 - 401(k) Savings Transactions
In 2022, Genprex established a defined-contribution savings plan under Section 401(k) of the Internal Revenue Code (“401(k) Plan”) and established an employer matching program for participants in the 401(k) Plan. The 401(k) Plan covers all employees who meet defined minimum age and service requirements, and allows participants to defer a portion of their annual compensation on a pre-tax basis. Genprex incurred $141,564 and $74,281 of expense for matching contributions to the 401(k) Plan for the years ended December 31, 2023 and 2022, respectively.
Note 6 - Related Party Transactions
Introgen Research Institute
Introgen Research Institute (“IRI”) is a Texas-based technology company currently affiliated withformed by Rodney Varner, ourGenprex’s President, Chief Executive Officer and director.Chairman of the Board and IRI’s sole officer. IRI is owned by trusts of which Mr. Varner’s descendants are the sole beneficiaries. In April 2009, prior to Mr. Varner becoming an officer and director of ourthe Company in August 2012, weGenprex entered into an Assignment and Collaboration Agreement with IRI, providing usthe Company with the exclusive right to commercialize a portfolio of intellectual property. This agreement was amended in 2011 to include additional sublicensing of additional intellectual property made available to IRI from MD Anderson.
Viet Ly
The Company entered into a consulting agreement with Viet Ly on April 19, 2018. The Company agreed to pay Mr. Ly $175,000 initially, with compensation variable from time-to-time as determined by the Company, for strategic consulting services. The Company paid Mr. Ly an aggregate of $28,500 during the quarter ended September 30, 2020 for strategic services. In addition, in April 2020, the Company issuedAnderson (See Note 7 – Commitments and Contingences – Commitments – MD Anderson Cancer Revolution LLC, an entity owned by Mr. Ly, a warrant to purchase up to 500,000 shares of common stock at the fair market value of the common stock on the date of issuance that vests based on the achievement of Company milestones. Center).
Note 7 - Commitments and Contingencies
LeasesCommitments
On April 16, 2018, the Company executed a service agreement with CIC Innovation Communities, LLC to establish and lease offices at the Cambridge Innovation Center in Cambridge, Massachusetts. On April 1, 2020, the Company provided notice of cancellation of our lease in the Cambridge Innovation Center in Cambridge, Massachusetts, effective April 30, 2020.
On April 16, 2018, the Company executed a space utilization agreement with the Board of Regents of the University of Texas System to establish and lease offices at the Dell Medical School in Austin, Texas. The lease runs through April 30, 2021 and the Company pays $462 per month to occupy this location.
Commitments
MD Anderson
We have entered into a clinical study agreement with the MD Anderson, to administer the Company’s phase 1/2 clinical trial, combining REQORSA-nanoparticles and Tarceva in Stage 4 lung cancer patients. The trial was expected to run through the end of 2018 with a projected total cost of approximately $2 million. Payments are due and payable when invoiced throughout the clinical trial period. The agreement may be terminated at any time. In 2020, the Company received from the Food and Drug Administration Fast Track Designation ("FTD") for its Acclaim-1 trial which combines REQORSA plus Tagrisso in patients who have previously failed Tagrisso treatment. Given the FTD and with Tagrisso now considered a new standard of care in the US for NSCLC with an EGFR mutation, we are no longer enrolling ONC-002 and plan to initiate Acclaim-1 and Acclaim-2 in 2021.
In July 2018, the CompanyGenprex entered into a two-yeartwo-year sponsored research agreement with MD Anderson to sponsor preclinical studies focused on the combination of REQORSA with an immunotherapy with a projected total cost of approximately $2 million. Payments are due and payable when invoiced throughout the clinical trial period. The agreement may be terminated at any time. This agreement has beenwas extended through beyond the original expiration date. This agreement expired in May 2022.In August 2022, Genprex entered into a three-year sponsored research agreement with MD Anderson to sponsor preclinical studies focused on REQORSA and other potential product candidates in oncology to resensitize NSCLC and SCLC to targeted therapies and immunotherapies with a projected total cost of approximately $2.9 million. As of December 31, 2023, Genprex has paid approximately $1.2 million toward this commitment.
In 2009, we2011, Genprex agreed to assume certain contractual and other obligations of IRI in consideration for the sublicense rights, expertise, and assistance associated with the assignment of certain technologies and intellectual property. Weproperty originally licensed to another party under the 1994 License Agreement with MD Anderson (“Original MD Anderson License Agreement”). These technologies and intellectual property were later sublicensed to IRI (the “IRI Sublicense”). Genprex also agreed to pay royalties of one percent (1%)1% on sales of resulting certain licensed products for a period of 21 years following the termination of the lastlater of the Technology SublicenseOriginal MD Anderson License Agreement dated March 7, 2007 by and between Introgen Therapeutics, Inc., andthe IRI and weSublicense. The Company assumed patent prosecution costs and an annual minimum royalty of $20,000 payable to the National Institutes of Health.
On March 3, 2021, Genprex entered into an amendment (the “MD License Amendment”) to the Patent and Technology License Agreement dated May 4, 2020, with MD Anderson. The MD License Amendment grants the Company with a worldwide, exclusive, sublicensable license to an additional portfolio of six patents and one patent application and related technology for methods for treating cancer by administration of a TUSC2 therapy in conjunction with EGFR inhibitors or other anti-cancer therapies in patients predicted to be responsive to TUSC2 therapy. Pursuant to the MD License Amendment, Genprex agreed to (i) pay annual maintenance fees ranging from the mid five figures to the low six figures, (ii) total milestone payments of $6,150,000, (iii) a one-time fee in the mid five figures and (iv) certain patent related expenses. As of December 31, 2023, Genprex has paid approximately $320,000 toward this commitment.
National Institutes of Health
Our $191,393 paymentGenprex has a royalty obligation to the National Institutes of Health (“NIH”) represented a current obligation, of which $15,393 of 2016 patent prosecution costs were paid in the fourth quarter of 2016 and $176,000 was included in Accounts Payable at December 31, 2016 (consisting of accrued annual royalties of $140,000 and patent costs of $36,000). During the first quarter of 2017, we modified the terms of our accrued royalty obligation to NIH. Under the modified agreement, NIH agreed to extinguish $120,000 of the accrued royalties payable to them in consideration for payment by us of (i) accrued patent costs of $36,000, (ii) a royalty payment of $20,000, and (iii) a contingent payment of $240,000, increasing at $20,000 per year starting in 2018, to be paid upon ourthe Company’s receipt of FDA approval. The payments for the patent costs of $36,000 and royalties of $20,000 were paid during the second quarter of 2017.
As a result of our modified agreement with theapproval using NIH we have recognized the exchange of the $120,000 fixed obligation for the $240,000 contingent obligation as a $120,000 reduction to intellectual property expense (classified within General and Administrative Expense) during the first quarter of 2017.technology. The $240,000 contingent obligation which increases annually by $20,000 and is $300,000 as of $360,000 and $340,000 for the years ended December 31, 20202023 and 2022, respectively, will be recognized when we obtainthe Company obtains regulatory approval (the event that triggers the payment obligation).
University of Pittsburgh
As part of our License Agreement withPursuant to an exclusive license agreement dated February 11, 2020 by and between Genprex and the University of Pittsburgh, in February 2020, weamended on August 17, 2022, and amended again on November 3, 2022, Genprex agreed to pay (i) an initial licensing fee of $25,000, (ii) annual maintenance fees of $25,000 for the firstthree years and $40,000 for each subsequent year following the first anniversary of the agreement, (iii) royalties betweenranging from 1.5% to 3% of net sales of licensed technologies, (iv) an annual minimalminimum royalty payment of $250,000 per year beginning in the year of the first commercial sale of licensed technology, (v) a share of non-royalty sublicense income of 20%, and (vi) milestone payments of an aggregate of $3,975,000. The$3,975,000 in milestone payments related to the usage of a glucagon promoter and gene therapy technologies to potentially treat Type 1 diabetes. Unless earlier terminated pursuant to its terms, the agreement expires upon the later of (i) 20 years after the first commercial sale of the licensed technology thereunder and (ii) expiration of the last valid claim under the patent rights, subjectrights. As of December 31, 2023, Genprex has paid approximately $110,000 toward this commitment.
Pursuant to an exclusive license agreement dated November 22, 2022 by and between Genprex and the University of Pittsburgh, Genprex agreed to pay (i) an initial licensing fee of $25,000, (ii) annual maintenance fees of $25,000 for the firstthree years and $40,000 for each subsequent year following the first anniversary of the agreement, (iii) royalties ranging from 1.5% to 3% of net sales of licensed technologies, (iv) an annual minimum royalty payment of $250,000 per year beginning in the year of the first commercial sale of licensed technology, (v) a share of non-royalty sublicense income of 20%, and (vi) an aggregate of $3,975,000 in milestone payments related to the usage of a macrophage technology and gene therapy technologies to potentially treat Type 1 diabetes. Unless earlier terminationterminated pursuant to its terms, the termsagreement expires upon the later of (i) 20 years after the first commercial sale of the agreement.licensed technology thereunder and (ii) expiration of the last valid claim under the patent rights. As of December 31, 2023, Genprex has paid approximately $50,000 toward this commitment.
Pursuant to an exclusive license agreement dated December 29, 2022 by and between Genprex and the University of Pittsburgh, Genprex agreed to pay (i) an initial licensing fee of $25,000, (ii) annual maintenance fees of $25,000 for the firstthree years and $40,000 for each subsequent year following the first anniversary of the agreement, (iii) royalties ranging from 1.5% to 3% of net sales of licensed technologies, (iv) an annual minimum royalty payment of $250,000 per year beginning in the year of the first commercial sale of licensed technology, (v) a share of non-royalty sublicense income of 20%, and (vi) an aggregate of $3,975,000 in milestone payments related to the usage of an insulin promoter and gene therapy technologies to potentially treat Type 2 diabetes. Unless earlier terminated pursuant to its terms, the agreement expires upon the later of (i) 20 years after the first commercial sale of the licensed technology thereunder and (ii) expiration of the last valid claim under the patent rights. As of December 31, 2023, Genprex has paid approximately $25,000 toward this commitment.
Pursuant to an exclusive license agreement dated July 14, 2023, by and between Genprex and the University of Pittsburgh, Genprex agreed to pay (i) an initial licensing fee of $25,000, (ii) annual maintenance fees of $25,000 for the first year, $50,000 for the second and third years, and $100,000 for the fourth year and each subsequent year following the fourth anniversary of the agreement thereafter until the anniversary prior to the year of the first commercial sale, (iii) royalties ranging from 1.5% to 3% of net sales of licensed technologies, (iv) an annual minimum royalty payment of $250,000 per year beginning in the year of the first commercial sale of licensed technology, (v) a share of non-royalty sublicense income of 20%, and (vi) an aggregate of $4,225,000 in milestone payments related to the usage of an MafB promoter and gene therapy technologies to potentially treat Type 1 and Type 2 diabetes. Unless earlier terminated pursuant to its terms, the agreement expires upon the later of (i) 20 years after the first commercial sale of the licensed technology thereunder and (ii) expiration of the last valid claim under the patent rights. As of December 31, 2023, Genprex has paid approximately $25,000 toward this commitment.
Contract Development and Manufacturing Organization
Genprex entered into a three-year development services agreement in July 2022, amended in each of January 2023 and March 2023, with a contract development and manufacturing organization to manufacture good manufacturing practices (“GMP”) grade materials for use in the Company’s clinical trials with a projected total cost of approximately $4.5 million. As of December 31, 2023, Genprex has paid approximately $2.6 million toward this commitment.
Contingencies
From time to time, we the Company may become subject to threatened and/or asserted claims arising in the ordinary course of ourits business. ManagementThe Company is not aware of any matters, either individually or in the aggregate, that are reasonably likely to have a material impact on our Company’sits financial condition, results of operations or liquidity.
Note 8 - Significant Events – Income Taxes
In March 2020, the outbreakComponents of COVID-19 caused by a novel strain of the coronavirus was recognized as a pandemic by the World Health Organization. The pandemic has become increasingly widespread in the United States, including markets in which the Company operates or may operate in the future. The COVID-19 pandemic has had a notable impact on general economic conditions, including, but not limited to, the temporary closures of many businesses, “shelter in place” orders and other governmental regulations, reduced consumer spending due to both job losses and other effects attributable to the COVID-19, in addition to many other unknowns. To date, the Company has not experienced any material impact on its financial results or operations as a result of the COVID-19 pandemic. The extent to which the COVID-19 pandemic could impact the Company's operations or financial results is uncertain. The Company continues to monitor the impact of the COVID-19 pandemic closely.
Note 9 – Income Taxes
The provision for income taxes differs from the amount computed by applying the statutory federal income tax rate to income before provisionbenefit for income taxes. The sources the year ended December 31, 2023 and tax effects of the differences2022, respectively, are as follows:
| December 31, 2022 | |||||||
| ||||||||
Federal | — | — | ||||||
State | — | — | ||||||
Total | — | — | ||||||
Deferred Tax Expense (Benefit): | ||||||||
Federal | — | — | ||||||
State | — | — | ||||||
Total | — | — | ||||||
Total Provision for Income Taxes | — | — | ||||||
Temporary differences between financial statement carrying amount and tax basis of assets and liabilities that give rise to significant portions of the deferred tax assets and liabilities at December 31, 2023 and 2022, respectively, are as follows:
December 31, 2023 | December 31, 2022 | |||||||
Deferred tax assets: | ||||||||
Allowance for Doubtful Accounts | — | — | ||||||
Interest Expense | — | — | ||||||
Intangible Assets - R&D Expenses | 4,710,079 | 1,986,102 | ||||||
Accrued Expenses | 684,699 | 561,496 | ||||||
Tax Credits | 1,434,851 | 728,701 | ||||||
Stock Compensation Expense | 306,906 | 94,237 | ||||||
Net Operating Losses | 16,915,002 | 14,464,170 | ||||||
Total Deferred Income Tax Assets | 24,051,538 | 17,834,706 | ||||||
Deferred Income Tax Liabilities: | ||||||||
Fixed Assets | (1,650 | ) | (4,354 | ) | ||||
Intangible Assets | — | — | ||||||
Prepaid Expenses | (629,136 | ) | (710,643 | ) | ||||
Lease - Right of Use | — | — | ||||||
Unrealized Gain/Loss | — | — | ||||||
Total Deferred Income Tax Liabilities | (630,786 | ) | (714,997 | ) | ||||
Less Valuation Allowance | (23,420,752 | ) | (17,119,710 | ) | ||||
Net Deferred Income Tax Asset | — | — | ||||||
At December 31, 20202023 and 2022, the Company has ahad federal net operating loss carryforwardlosses of approximately $44.6$80.5 million for Federal and state purposes. This loss$68.9 million, respectively. Net deferred tax assets are mainly comprised of temporary differences between financial statement carrying amount and tax basis of assets and liabilities.
In assessing the ability to realize the deferred tax assets, management considers whether it is more likely than not that some portion or all of the deferred tax assets will notbe available to offsetrealized. The ultimate realization of deferred tax assets is dependent on the generation of future taxable income. If not used,income during the period in which these temporary differences become deductible. Management considers the projected future taxable income and prudent and feasible tax planning strategies in making this carryforwardassessment. As of December 31, 2023 and 2022, valuation allowances of $23.4 million and $17.1 million have been recorded, respectively.
The Company has federal research and development (“R&D”) credit carryforwards of $1.4 million which will begin to expire in 2029.2037.
Effective January 1, 2009 the Company adopted ASC 740-10, the provision formerly FASB Interpretation No.48, Accounting for Uncertainty in Income Taxes, (“FIN48”) and began evaluating tax positions utilizing a two-step process. The deferredfirst step is to determine whether it is more-likely-than-not that a tax asset relatingposition will be sustained upon examination based on the technical merits of the position. The second step is to measure the operating loss carryforward has been fully reserved at December 31, 2020benefit to be recorded from tax positions that meet the more-likely-than-not recognition threshold by determining the largest amount of tax benefit that is greater than 50 percent likely of being realized upon ultimate settlement and December 31, 2019. The principal differences betweenrecognizing that amount in the operating loss for income tax purposes and reporting purposes are shares issued for services and share-based compensation and a temporary difference in depreciation expense.financial statements.
Note 109 – Subsequent Events
Reserves of 2018 Equity Incentive Plan and 2018 Employee Stock Purchase Plan
On January 1, 2021, 2024, the total shares of common stock reserved under the 2018 Equity Incentive Plan increased 2,155,884by 74,294 shares. On March 17, 2021, 14, 2024, the Company's BoardCompany’s board of Directorsdirectors determined that no additional shares would be reserved during the 20212024 fiscal year for the 2018 Employee Stock Purchase PlanESPP given that no shares have yet been issued under the plan.ESPP.
Share Issuances
On January 1, 2021,2, 2024, Genprex issued 125 shares of common stock to the CompanyChairman of the Company’s Scientific Advisory Board in consideration for services. On January 3, 2024, Genprex issued 5,0006,250 shares of common stock to a service provider in consideration of services.the Company. On March 18, 2021, the Company14, 2024, Genprex issued 38,056 shares of common stock for $61,651 in cash to a former executive upon the exercise of a stock option granted under the 2018 Equity Incentive Plan.
Option & Warrant Issuances
On February 10, 2021, the Company’s Board of Directors approved the (i) grant of stock options under the 2018 Equity Incentive Plan to purchase a total of 210,000 shares of common stock to employees and consultants, and (ii) an issuance of a warrant to purchase up to 17,50030,000 shares of common stock to a consultant, at the fair market valueservice provider of the common stock onCompany.
2023 ATM Facility
From January 1, 2024 through the date of issuance.filing of this Annual Report on Form 10-K, Genprex has sold 158,474 shares of its common stock for aggregate net proceeds to the Company totaling $881,946 under the 2023 ATM Facility.
Reverse Stock Split
At Genprex’s special meeting of stockholders held on December 14, 2023, the Company’s stockholders granted the Company’s board of directors the discretion to effect a reverse stock split of the Company’s issued and outstanding common stock through an amendment (the “Certificate of Amendment”) to the Company’s Amended and Restated Certificate of Incorporation, as amended and restated to date, at a ratio of not less than 1-for-10 and not more than 1-for-50, such ratio to be determined by the Company’s board of directors. On January 19, 2024, the Company’s board of directors approved a 1-for-40 reverse stock split and authorized the filing of the Certificate of Amendment for the Reverse Split with the Secretary of State of the State of Delaware. The Reverse Split became effective in accordance with the terms of the Certificate of Amendment on February 2, 2024. The Certificate of Amendment did not change the number of authorized shares of common stock or the par value. All share and per share amounts in this Annual Report on Form 10-K have been adjusted to reflect the Reverse Stock Split.
Departure of a Named Executive Officer
Effective as of February 4, 2024, Catherine Vaczy’s employment with the Company as Senior Vice President, General Counsel, and Chief Strategy Officer was terminated.
March 2024 Registered Direct Offering
On February 10, 2021, March 21, 2024, the Company completed a registered direct offering, in which the Company sold to investorsan institutional investor an aggregate of 4,000,000(i) 165,000 shares of the Company's common stock, (ii) pre-funded warrants (the “March 2024 Pre-Funded Warrants”) exercisable for up to an aggregate of 1,377,112 shares of common stock, and (iii) warrants (the “March 2024 Common Warrants”) exercisable for up to an aggregate of 1,542,112 shares of common stock. The offering price for each share of common stock and accompanying March 2024 Common Warrant was $4.215, and the offering price for each March 2024 Pre-Funded Warrant and accompanying March 2024 Common Warrant was $4.2149. The March 2024 Pre-Funded Warrants are exercisable immediately upon issuance at $6.25an exercise price of $0.0001 per share.share and will expire when exercised in full. The March 2024 Common Warrants are exercisable immediately upon issuance at an exercise price of $4.09 per share and will expire in 5 years from the date of issuance. The Company received net proceeds of approximately $23.2$5.8 million after commissions and expenses.
License Agreement
On expenses, excluding any proceeds that may be received in the future from any exercise of the March 3, 2021,2024 Common Warrants. In connection with the offering, the Company entered into an amendment (the “MD License Amendment”)also amended certain existing warrants to the Patent and Technology License Agreement dated May 4, 2020 with MD Anderson. The MD License Amendment grants the Company a worldwide, exclusive, sublicensable licensepurchase up to an additional portfolio of six patents and one patent application and related technology for methods for treating cancer by administration of a TUSC2 therapy in conjunction with EGFR inhibitors or other anti-cancer therapies in patients predicted to be responsive to TUSC2 therapy. Pursuant to the MD License Amendment, the Company agreed to (i) pay annual maintenance fees ranging from the mid five figures to the low six figures, (ii) total milestone payments of $6,150,000, (iii) a one-time fee in the mid five figures and (iv) certain patent related expenses.
Executive Compensation
On March 19, 2021, the Company's Compensation Committee approved (i) annual incentive awards of an aggregate of $599,667 and (ii) the grant of stock options under the 2018 Equity Incentive Plan to purchase an aggregate of 1,095,000194,248 shares of the Company's common stock that were previously issued to Company executives.investors in March 2023 and July 2023, with exercise prices of $44.00 and $35.40 per share and expiration dates of March 1, 2028 and July 21, 2028 for $0.125 per amended warrant, such that the amended warrants have a reduced exercise price of $4.09 per share and an expiration date of five years from the closing of this offering.