UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| | | | | | | | |
| ☑ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the Fiscal Year Ended December 31, 20212023
| | | | | | | | |
| ☐ | Transition Report Pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 |
Commission File Number: 001-36257
TRAVERE THERAPEUTICS, INC.
(Exact Name of Registrant as specified in its Charter)
| | | | | |
| Delaware | 27-4842691 |
| (State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) |
| | |
3611 Valley Centre Drive, Suite 300
San Diego, CA 92130
(Address of Principal Executive Offices)
888-969-7879
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| | | | | | | | | | | | | | |
| Title of each class | Trading Symbol(s) | Name of each exchange on which registered | |
| Common Stock, par value $0.0001 per share | TVTX | The Nasdaq Global Market | |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. ☑ Yes ☐ No
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. ☐ Yes ☑ No
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. ☑ Yes ☐ No
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). ☑ Yes ☐ No
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company or an emerging growth company. See definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and "emerging growth company" in Rule 12b-2 of the Exchange Act).
| | | | | | | | | | | |
| Large Accelerated Filer | ☑ | Accelerated Filer | ☐ |
| Non-Accelerated Filer | ☐ | Smaller Reporting Company | ☐ |
| | Emerging growth Company | ☐ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 7(a)(2)(B) of the Securities Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☑
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). ☐ Yes ☑ No
State the aggregate market value of the voting and non-voting common equity held by non-affiliates computed by reference to the price at which the common equity was last sold, or the average bid and asked price of such common equity, as of the last business day of the registrant's most recently completed second fiscal quarter. $879,383,041.$1,145,413,509.
The number of shares of outstanding common stock, par value $0.0001 per share, of the registrant as of February 22, 202215, 2024 was 63,211,565.76,098,485.
DOCUMENTS INCORPORATED BY REFERENCE: Portions of the Proxy Statement for the registrant’s 20222024 Annual Meeting of Stockholders, to be filed within 120 days after the conclusion of the registrant's fiscal year ended December 31, 2021,2023, are incorporated by reference into Part III of this Annual Report on Form 10-K.
FORM 10-K REPORT INDEX
CAUTIONARY STATEMENT REGARDING FORWARD-LOOKING STATEMENTS
Certain information contained in this Annual Report on Form 10-K of Travere Therapeutics, Inc., a Delaware corporation (the “Company”) include forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. The statements herein which are not historical reflect our current expectations and projections about the Company’s future results, performance, liquidity, financial condition, prospects and opportunities and are based upon information currently available to the Company's management and is subject to its interpretation of what are believed to be significant factors affecting the Company's business, including many assumptions regarding future events. Such forward-looking statements include statements regarding, among other things:
•the potential full regulatory approval of FILSPARI for Immunoglobulin A nephropathy (IgAN);
•the estimated addressable U.S. patient population for FILSPARI under the indication approved via accelerated approval; and the estimated addressable patient populations if full approval is granted;
•estimated patient populations related to our other products and products in development;
•the anticipated opinion by the EMA’s Committee for Human Medicinal Products (“CHMP”) on the conditional marketing authorization ("CMA") application for sparsentan for the treatment of IgAN, and the timing thereof;
•expectations regarding our pivotal Phase 3 HARMONY Study to support the potential approval of pegtibatinase for the treatment of classical homocystinuria (HCU);
•statements regarding our studies of sparsentan for focal segmental glomerulosclerosis (FSGS), plans for conducting additional analyses of FSGS data, and intentions to engage with regulators to evaluate potential regulatory pathways for a sparsentan FSGS indication;
•our ability to produce, sustain and expand sales of our products;
•our ability to develop, acquire and/or introduce new products;products including expectations with regard to clinical trials and preclinical studies;
•our projected future sales, profitability, savings and other financial metrics;
•our future financing plans;
•our anticipated needs for working capital;
•the anticipated trends in our industry;
•acquisitions of other companies or assets that we might undertake in the future;
•our operations in the United States and abroad, and the domestic and foreign regulatory, economic and political conditions; and
•competition existing today or that will likely arise in the future.
Forward-looking statements, which involve assumptions and describe our future plans, strategies and expectations, are generally identifiable by use of the words “may,” “should,” “expect,” “anticipate,” “estimate,” “believe,” “intend,” “seek,” or “project” or the negative of these words or other variations on these words or comparable terminology. Actual results, performance, liquidity, financial condition and results of operations, prospects and opportunities could differ materially from those expressed in, or implied by, these forward-looking statements as a result of various risks, uncertainties and other factors, including the ability to raise sufficient capital to continue the Company’s operations. Actual events or results may differ materially from those discussed in forward-looking statements as a result of various factors, including, without limitation, the risks outlined under “Risk Factors” and matters described in this Annual Report generally. In addition, statements that "we believe" and similar statements reflect our beliefs and opinions on the relevant subject. These statements are based upon information available to us as of the date of this Annual Report on Form 10-K, and while we believe such information formsprovides a reasonable basis for suchthese statements, such information may be limited or incomplete, and our statements should not be read to indicate that we have conducted an exhaustive inquiry into, or review of, all potentially available relevant information. These statements are inherently uncertain and youinvestors are cautioned not to not unduly rely upon these statements.
In light of these risks and uncertainties, there can be no assurance that the forward-looking statements contained in this Annual Report will in fact occur. Potential investors should not place undue reliance on any forward-looking statements. Except as expressly required by the federal securities
laws, there is no undertaking to publicly update or revise any forward-looking statements, whether as a result of new information, future events, changed circumstances or any other reason.
The specific discussions in this Annual Report about the Company include financial projections and future estimates and expectations about the Company’s business. The projections, estimates and expectations are presented in this Annual Report only as a guide about future possibilities and do not represent actual amounts or assured events. All the projections and estimates are based exclusively on the Company management’s own assessment of the business, the industry in which it works and the economy at large and other operational factors, including capital resources and liquidity, financial condition, fulfillment of contracts and opportunities. The actual results may differ significantly from the projections.
Potential investors should not make an investment decision based solely on the Company’s projections, estimates or expectations.
Risk Factor Summary
Below is a summary of material factors that make an investment in our common stock speculative or risky. This summary does not address all of the risks that we face. Additional discussion of the risks summarized in this risk factor summary, and other risks that we face, can be found under the heading “Risk Factors” in Item 1A of Part I of this Annual Report on Form 10-K and should be carefully considered, together with other information in this Annual Report on Form 10-K and our other filings with the SEC before making investment decisions regarding our common stock.
•Our future prospects are highly dependent upon our ability to successfully develop and execute commercialization strategies for our products, including FILSPARI (sparsentan) to reduce proteinuria in adults with primary IgAN, and to attain market acceptance among physicians, patients and healthcare payers.
•Our clinical trials are expensive and time-consuming and may fail to demonstrate the safety and efficacy of our product candidates, including sparsentan and pegtibatinase (TVT-058), which could prevent or significantly delay their regulatory approval.
•The planned eGFR data cut from the DUPLEX Study may not support accelerated approval submissions in the U.S. and/or Europe.candidates.
•Success in preclinicalnonclinical testing and early clinical trials does not ensure that later clinical trials will be successful.
•An extended delay in the rateCommunications and/or feedback from regulatory authorities related to our clinical trials does not guarantee any particular outcome from or timeline for regulatory review, and expedited regulatory review pathways may not actually lead to faster development or approval.
•In order to operate our business and increase adoption and sales of enrollment in our ongoing Phase 1/2 Study of pegtibatinase (TVT-058), asproducts, we need to continue to develop our commercial organization, including maintaining a result of the COVID-19 pandemic or otherwise, may delay our timelines for analyzing future data from the study.highly experienced and skilled workforce with qualified sales representatives.
•Interim, topline and preliminary data from our clinical trials that we announce or publish from time to time may change materially as more patient data become available and are subject to audit and verification procedures that could result in material changes in the final data.
•The commercial success of Chenodal, Cholbam and Thiola depends on them being considered to be effective drugs with advantages over other therapies.are completed.
•We are subject toface substantial generic and other competition, and recent developments relatingour operating results will suffer if we fail to generic competition for pharmaceutical products could cause our product sales and business to be negatively impacted.compete effectively.
•ChangesHealthcare reform initiatives, unfavorable pricing regulations, and changes in reimbursement practices of third-party payers or patients' access to insurance coverage could affect the pricing of and demand for our products and/or the prices at which they are sold.products.
•We are dependent on third parties to manufacture and distribute our pharmaceutical products who may not fulfill their obligations.products.
•If we are unable to maintain an effective and specialized sales force, we will not be able to commercialize our products in the United States successfully.
•Our products may not achieve or maintain expected levels of market acceptance or commercial success.
•If theThe market opportunities for our products and product candidates aremay be smaller than we believe they are, our revenues may be adversely affected and our business may suffer.are.
•Our product candidates may cause undesirable side effects or have other properties that could delay or prevent their regulatory approval or commercialization.
•We do not currently have patent protection for certain of our commercial products. If we are unable to obtain and maintain protection for the intellectual property relating to our technology and products, thetheir value of our technology and products willmay be adversely affected.
•We expect to rely on orphan drug status to develop and commercialize certain of our product candidates, but our orphan drug designations may not confer marketing exclusivity or other expected commercial benefits.
•Any drugs we develop may become subject to unfavorable pricing regulations, third-party reimbursement practices or healthcare reform initiatives, thereby harming our business.
•We face potential product liability exposure far in excess of our limited insurance coverage.
•We face substantial competition, which may result in others discovering, developing or commercializing products before or more successfully than we do. Our operating results will suffer if we fail to compete effectively.
•The COVID-19 pandemic could materially adversely affect our business, results of operations and financial condition.
•Our limited operating history makes it difficult to evaluate our future prospects, and our profitability in the future is uncertain.
•We depend on a highly experienced and skilled workforce to grow and operate our business. If we are unable to attract, retain and engage our employees, we may not be able to grow effectively.
•We will likely experience fluctuations in operating results and could incur substantial losses.losses, and the market price for shares of our common stock may be volatile.
•Negative publicity regarding any of our products could impair our ability to market any such product and may require us to spend time and money to address these issues.
•We may need substantial funding and may be unable to raise capital when needed, which would force us to delay, reduce or eliminateneeded. Our indebtedness could adversely affect our product development programs or commercialization efforts.financial condition.
•The market price for sharesWe might not receive some or all of the potential milestone payments from the sale of our common stock may be volatile and purchasersbile acid product portfolio for the treatment of our common stock could incur substantial losses.rare liver diseases.
•We may be unable to successfully integrate new products or businesses we may acquire.
•We may become involved in certain litigation matters, any of which could result in substantial costs, divert management's attention and otherwise have a material adverse effect on our business, operating results or financial condition.
•We are subject to significant ongoing regulatory obligations and oversight, which may result in significant additional expense and may limit our commercial success.
•We are subject to stringent and changing obligations related to data privacy and security. Our actual or perceived failure to comply with such obligations may result in adverse business consequences.
•Our ability to use net operating loss carryforwards and certain other tax attributes to offset future taxable income and taxes may be subject to limitations.
•We are dependent on information technology systems, infrastructure and data, which exposes us to data security risks.
•Business disruptions could seriously harm our future revenue and financial condition and increase our costs and expenses.
•Our indebtedness could adversely affect our financial condition.
•We may be unable to raise the funds necessary to repurchase the $276.0 million aggregate principal amount of 2.50% Convertible Senior Notes due 2025 ("2025 Notes") for cash following a fundamental change, or to pay any cash amounts due upon conversion, and our future indebtedness may limit our ability to repurchase the 2025 Notes or pay cash upon their conversion.
PART I
In this Annual Report on Form 10-K, unless the context requires otherwise, the terms “we”, “our”, “us”, “Travere” and the “Company” refer to Travere Therapeutics, Inc., a Delaware corporation, as well as our direct and indirect subsidiaries.
We own or have rights to various trademarks used in our business, including those referenced in the subsection of Item 1 below titled “Trademarks”. Our logos and trademarks are the property of Travere Therapeutics, Inc. All other brand names or trademarks appearing in this report are the property of their respective holders.
ITEM 1. BUSINESS
Those statements in the following discussion that are not historical in nature should be considered forward-looking statements that are inherently uncertain. Actual results and the timing of the events may differ materially from those contained in these forward-looking statements due to a number of factors, including those discussed in the “Cautionary Statement Regarding Forward-Looking Statements” and “Risk Factors” set forth elsewhere in this Annual Report.
Overview
We are a biopharmaceutical company headquartered in San Diego, California, focused on identifying, developing and delivering life-changing therapies to people living with rare kidney liver, and metabolic diseases. Our approach centers on advancing our innovative pipeline with multiple late-stage clinical programs targeting rare diseases with significant unmet medical needs. Our lead pipeline candidate, sparsentan,In February 2023, the U.S. Food and Drug Administration (FDA) granted accelerated approval to our first development program, FILSPARI® (sparsentan), which is an investigational product candidateindicated to reduce proteinuria in adults with primary IgAN at risk of rapid disease progression. Sparsentan is also in late-stage development for focal segmental glomerulosclerosis (FSGS). IgAN and IgA nephropathy (IgAN) –FSGS are rare kidney disorders that often lead to end-stage kidney disease.failure. We are also developingadvancing pegtibatinase, (TVT-058)a novel investigational enzyme replacement therapy for the treatment of classical homocystinuria (HCU), a genetic disorder caused by a deficiency in a pivotal enzyme essential to the body.In December 2023, we initiated the pivotal Phase 3 HARMONY Study to support the potential approval of pegtibatinase as the first disease modifying therapy for HCU. Our early research efforts include partnering with patient advocacy groups and government researchers to identify potential therapeutics for NGLY1 deficiency and Alagille syndrome conditions(ALGS), a condition with no approved treatment options. In addition, we continue to evaluate potential opportunities to expand our pipeline and approved products through licenses and acquisitions of products in areas that will serve rare disease patients with serious unmet medical need and that we believe offer attractive growth characteristics. Our research and development efforts are at the forefront of our mission to address the unmet needs of patients and we support this innovation by reinvesting revenues from our commercialized products, Chenodal®, Cholbam®, Thiola® and Thiola EC®.products. We are committed to ensuring broad access and educational and diagnostic support for patients.
Our Strategy
Our vision is to become a leading biopharmaceutical company dedicated to the delivery of innovation and hope to patients in the global rare disease community. In order to achieve our vision, we intend to:
•Focus on developing products to treat rare diseases characterized by severe unmet medical needs. We believe that our research, development, and commercialization capabilities in rare disease represent distinct competitive advantages. We leverage our development capabilities in rare disease to focus on advancing therapeutic candidates with life-changing potential. Given these capabilities, the well-established regulatory model and the ability to demonstrate clinical effects in small clinical studies, we believe that we can successfully bring new therapies to patients living with severe unmet medical needs.
•Leverage our commercialization expertise to effectively deliver the therapies we develop. Our strategy is to maximize the benefit and value of our commercial products through commercial execution and expertise needed to successfully support rare disease patients. Our approach may vary depending on the product and will be based on a number of factors including capital necessary to execute on each option, size of the market and terms of potential collaboration and/or licensing offers from other pharmaceutical and biotechnology companies with respect to jurisdictions outside the United States.
•Develop a sustainable pipeline by employing disciplined decision criteria in the evaluation of potential in-licensing candidates. We seek to build a sustainable product pipeline by employing multiple therapeutic approaches and by developing or acquiring orphan drug candidates. We seek to augment our internally developed pipeline projects by selectively and strategically acquiring pipeline assets that will add value to the portfolio. We continue to evaluate potential in-licensing, out-licensing and other potential relationships with other pharmaceutical or biotechnology companies. We intend to mitigate risk by employing rigorous decision criteria, favoring therapeutic candidates that have undergone at least some clinical study. Our decision to acquire rights to a therapeutic candidate also depends on the scientific merits of the available clinical data; the identifiable orphan patient population; the economic terms of any proposed acquisition of rights; the projected amount of capital required to develop the therapeutic candidate; and the economic potential of the therapeutic
candidate, should it be commercialized. We believe this strategy minimizes our clinical development risk and allows us to accelerate the development and potential commercialization of current and future therapeutic candidates.
•Listen to patients. Leadership in rare disease demands attention beyond innovative medicines. By listening to patients and leaders in the rare disease community, including those who have traditionally been underserved, we are focusing on barriers that prevent some patients from accessing the incredible innovation that our industry is delivering, including access to clinical trials and rare disease specialists.
•Support earlier diagnosis. We support efforts in furtherance of enabling earlier diagnosis. The growth of our commercial business reflects the strong capabilities of our commercial and medical teams to work across multiple medical specialties to help patients find a diagnosis.
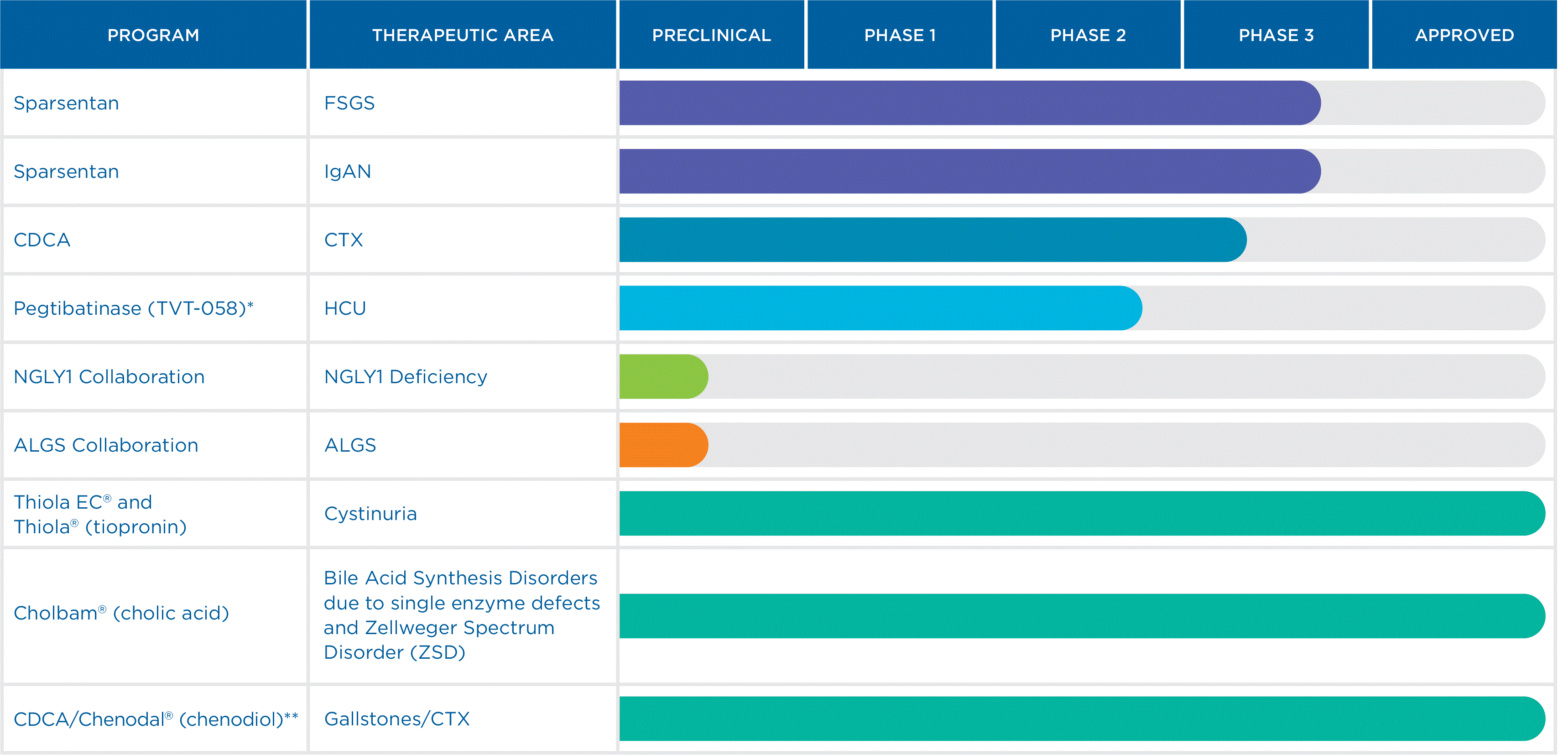
Our Pipeline and Approved Products
We have a diversified pipeline designed to address areas of high unmet need in rare kidney liver, and metabolic diseases. We invest revenues from our commercial portfolio into our pipeline with the goal of delivering new treatments for diseases with limited or no approved or limited therapies.
The following table summarizes the status of our clinical programs, preclinical programs and approved products, each of which is described in further detail below.
| | | | | |
| 1 | | | | | On February 17, 2023, the FDA granted accelerated approval of FILSPARI® (sparsentan) to reduce proteinuria in adults with primary IgAN at risk of rapid disease progression, generally a urine protein-to-creatinine ratio (UPCR) ≥1.5 gram/gram. On September 21, 2023, we announced topline two-year confirmatory secondary endpoint results from the pivotal Phase 3 PROTECT Study, as described below. |
*2 | Pegtibatinase (TVT-058) is currently in a Phase 1/2 clinical study. |
** | CDCA is not indicated for CTX but has received a medical necessity determination inOn May 1, 2023, we announced topline primary efficacy results from the US by the FDA for CTX. Travere Therapeutics is conducting apivotal Phase 3 clinical trial to examine the safety and efficacy of CDCA (Chenodal®) for the treatment of CTX.DUPLEX Study, as described below. |
Clinical Programs:FILSPARI® (sparsentan)
SparsentanOn February 17, 2023, the FDA granted accelerated approval of FILSPARI® (sparsentan) to reduce proteinuria in adults with primary IgAN at risk of rapid disease progression, generally a UPCR ≥1.5 gram/gram. FILSPARI became commercially available in the U.S. beginning the week of February 27, 2023, and we are providing a comprehensive patient support program throughout the patient’s treatment journey.
Sparsentan,This indication was granted under accelerated approval based on reduction in proteinuria. The continued approval of FILSPARI may be contingent upon confirmation of a Dual Endothelin Angiotensin Receptor Antagonist (DEARA),clinical benefit in the Phase 3 PROTECT Study. As described in more detail below, in September 2023, we announced topline two-year confirmatory secondary endpoint results from the PROTECT Study, and in December 2023, we announced the completion of a successful pre-NDA meeting with the FDA for FILSPARI in IgAN. Following our regulatory engagement, we plan to submit a supplemental New Drug Application (sNDA) in the first quarter 2024 for conversion of the existing U.S. accelerated approval of FILSPARI to full approval.
FILSPARI, a once-daily, oral medication is designed to selectively target two critical pathways in the disease progression of IgAN (endothelin 1 and angiotensin-II) and is the first and only non-immunosuppressive therapy approved for the treatment of this condition.
FILSPARI is a novel investigational product candidate.dual endothelin angiotensin receptor antagonist (DEARA). Pre-clinical data have shown that blockade of both endothelin type A and angiotensin II type 1 pathways in forms of rare chronic kidney disease, reduces proteinuria, protects podocytes and prevents glomerulosclerosis and
mesangial cell proliferation. Sparsentan has been granted Orphan Drug Designation for the treatment of FSGS and IgAN in the U.S. and Europe. Sparsentanthe European Economic Area countries (the “EEA”) and FILSPARI has been granted seven years of Orphan Drug Exclusivity in the U.S. for the reduction of proteinuria in adults with primary IgAN at risk of rapid disease progression.
IgAN is characterized by hematuria, proteinuria, and variable rates of progressive renal failure. With an estimated prevalence of up to 150,000 people in the United States and greater numbers in Europe and Asia, IgAN is the most common primary glomerular disease. Most patients are diagnosed between the ages of 16 and 35, with up to 40% progressing to kidney failure within 15 years. FILSPARI is the first and only non-immunosuppressive therapy approved for this condition. We estimate approximately 30,000 to 50,000 patients in the United States to be addressable under FILSPARI's accelerated approval indication statement.
Data to support the accelerated approval of FILSPARI was generated from the Phase 3 PROTECT Study, the largest head-to-head interventional study to date in IgAN. It is a global, randomized, multicenter, double-blind, parallel-arm, active-controlled clinical trial that evaluated the safety and efficacy of 400mg of sparsentan, compared to 300mg of irbesartan, in 404 patients ages 18 years and up with IgAN and persistent proteinuria despite available ACE or ARB therapy, and is currently being evaluatedongoing in two pivotalthe open label extension phase of the study.
The PROTECT Study protocol provided for an unblinded analysis of at least 280 patients to be performed after 36 weeks of treatment to evaluate the primary efficacy endpoint - the change in proteinuria (UPCR) at week 36 from baseline. Secondary efficacy endpoints include the rate of change in estimated glomerular filtration rate (eGFR) following the initiation of randomized treatment over 58-week and 110-week periods, as well as the rate of change in eGFR over 52-week and 104-week periods following the first six weeks of randomized treatment in approximately 380 patients. In August 2021, we announced positive topline interim results from the ongoing Phase 3 PROTECT Study. The PROTECT Study met its pre-specified interim primary efficacy endpoint with statistical significance. After 36 weeks of treatment, patients receiving FILSPARI achieved a mean reduction in proteinuria from baseline of 49.8 percent, compared to a mean reduction in proteinuria from baseline of 15.1 percent for irbesartan-treated patients (p<0.0001). Results from the interim assessment in the PROTECT Study showed that FILSPARI was well tolerated with a clearly defined safety profile that has been consistent across all clinical studiestrials conducted to date. In PROTECT, at the interim assessment, the most common adverse reactions (≥ 5%) were peripheral edema, hypotension (including orthostatic hypotension), dizziness, hyperkalemia, and anemia. FILSPARI is available only through a risk evaluation and mitigation strategy (REMS) approved by the FDA.
Per request from the FDA, the efficacy data contained in rarethe FDA-approved label under accelerated approval is a post-hoc sensitivity analysis that evaluates the first 281 randomized patients, a subset of the full trial population. The mean reduction in proteinuria from baseline in the post-hoc sensitivity analysis is 45% for FILSPARI versus 15% for the active control, irbesartan. Both the pre-specified and post-hoc sensitivity analyses have demonstrated that FILSPARI achieves a rapid and sustained reduction in proteinuria, with statistically significant and clinically meaningful improvement compared to the active comparator irbesartan.
In September 2023, we announced topline two-year confirmatory secondary endpoint results from the PROTECT Study; eGFR total and chronic slope are the secondary confirmatory endpoints for the U.S. and the EU, respectively. FILSPARI demonstrated long-term kidney diseases, including:function preservation and achieved a clinically meaningful difference in eGFR total and chronic slope versus irbesartan, narrowly missing statistical significance in eGFR total slope while achieving statistical significance in eGFR chronic slope for purposes of regulatory review in the EU. All topline efficacy endpoints favored FILSPARI as compared to irbesartan. A preliminary review of the safety results through 110 weeks of treatment indicates FILSPARI was generally well-tolerated and the overall safety profile in the study has been consistent between treatment groups. In December 2023, we announced the completion of a successful pre-NDA meeting with the FDA for FILSPARI in IgAN. Following our regulatory engagement, we plan to submit a supplemental New Drug Application (sNDA) in the first quarter of 2024 for conversion of the existing U.S. accelerated approval of FILSPARI to full approval.
In August 2022, we and Vifor (International) Ltd. ("CSL Vifor"), with whom we entered into a license and collaboration agreement ("License Agreement") in September 2021, announced that the EMA had accepted for review the conditional marketing authorization ("CMA") application of sparsentan for the treatment of IgAN in the EU. We anticipate an opinion by the Committee for Medicinal Products for Human Use (the "CHMP") in the first quarter of 2024.
In January 2024, we announced our entry into an exclusive licensing agreement with Renalys Pharma, Inc. ("Renalys"), to bring sparsentan to patients in Japan and other countries in Asia, for the treatment of IgAN. Renalys will hold regional rights to sparsentan for Japan, South Korea, Taiwan, Brunei, Cambodia, Indonesia, Laos, Malaysia, Myanmar, the Philippines, Singapore, Thailand, and Vietnam. Following successful meetings with the Pharmaceuticals and Medical Devices Agency (PMDA) in 2023, Renalys plans to initiate an open label registrational study of sparsentan in Japan in the second quarter of 2024 to support potential approval of sparsentan in Japan. Results from the urine protein/creatinine ratio (UP/C) endpoint in the study are expected in the second half of 2025 to support a submission for approval to PMDA. Under the terms of the licensing agreement, Renalys will be responsible for development, regulatory matters, and commercialization in the licensed territories.
Clinical-Stage Programs:
Sparsentan for the treatment of FSGS
Sparsentan has been granted Orphan Drug Designation for the treatment of FSGS in the U.S. and the EEA.
•Focal segmental glomerulosclerosis ("FSGS")FSGS is a leading cause of end-stage kidney disease (ESKD)failure and nephrotic syndrome. There are currently no United States Food and Drug Administration ("FDA") approvedFDA-approved pharmacologic treatments for FSGS and there remains a high unmet need for patients living with FSGS as off-label treatments such as ACE/ARBs, steroids, and immunosuppressant agents are effective in only a subset of patients and use of some of these off-label treatments may be further inhibited by their safety profiles. Every year approximately 5,400 patients are diagnosed with FSGS and we estimate that there are more than 40,000 FSGS patients in the United States and a similar number in Europe with approximately half of them being candidates for sparsentan.
In 2016, we generated positive data from our Phase 2 DUET study in FSGS. In 2018, we announced the initiation of the Phase 3 DUPLEX studyclinical trial designed to serve as the basis for an NDA and MAA filing for sparsentan for the treatment of sparsentan in FSGS.FSGS (the "DUPLEX Study"). The DUPLEX Study is a global, randomized, multicenter, double-blind, parallel-arm, active-controlled clinical trial evaluating the safety and efficacy of sparsentan in 371 patients. The DUPLEX Study protocol provided for an unblinded analysis of at least 190 patients to be performed after 36 weeks of treatment to evaluate the interim efficacy endpoint - the proportion of patients achieving a FSGS partial remission of proteinuria endpoint (FPRE), which is defined as urine protein-to-creatinine ratio (Up/C)(UPCR) ≤1.5 g/g and a >40% reduction in Up/CUPCR from baseline, at week 36. In February 2021, we announced that the ongoing Phase 3 DUPLEX Study achieved its pre-specified interim FSGS partial remission of proteinuria endpoint following the 36-week interim period. After 36 weeks of treatment, 42.0 percent of patients receiving sparsentan achieved FPRE, compared to 26.0 percent of irbesartan-treated patients (p=0.0094). A preliminary reviewFollowing engagement with the FDA on the interim proteinuria analysis and a subsequent eGFR data-cut, we elected to forego the previously planned submission for accelerated approval and pursue a potential traditional approval upon completion of the DUPLEX Study.
In May 2023, we announced topline primary efficacy results from the interim analysis suggest that to datepivotal Phase 3 DUPLEX Study of sparsentan in the study, sparsentan has been generally well-tolerated and the overall safety results in the study to date have been generally comparable between treatment groups.FSGS. The confirmatory primary endpoint of the DUPLEX Study designed to support fulltraditional regulatory approval iswas the rate of change in eGFR over 108 weeks of treatment. AsAt the end of the time108-week double-blind period, sparsentan was observed to have a 0.3 mL/min/1.73m2 per year (95% CI: -1.74, 2.41) favorable difference on eGFR total slope and a 0.9 mL/min/1.73m2 per year (95% CI: -1.27, 3.04) favorable difference on eGFR chronic slope compared to the active control irbesartan, which was not statistically significant. After 108 weeks of the interim analyses, available long-term eGFR datatreatment, sparsentan achieved a mean reduction in proteinuria from baseline of 50%, compared to 32% for the confirmatory endpoint were limited. Consistent withirbesartan. Although the DUPLEX Study protocol, patients will continue indid not achieve its two-year primary endpoint with statistical significance over the active control irbesartan, we are encouraged by the results, including the secondary endpoints on proteinuria and topline exploratory endpoints, including renal outcomes, which trended favorably for sparsentan. In addition, a blinded manner to assessreview of the treatment effect on eGFR slope oversafety results through 108 weeks of treatment indicate sparsentan was generally well-tolerated and the overall safety profile in the confirmatory endpoint analysis. The DUPLEX Study is fully enrolled and toplinestudy to date was generally consistent between treatment groups.
In December 2023, we announced that we completed our planned Type C meeting with the FDA to discuss previously reported results from the confirmatory endpoint are expectedPhase 3 DUPLEX Study of sparsentan in FSGS. The FDA acknowledged the first half of 2023.
In May 2021, we provided a regulatory update regardinghigh unmet need for approved therapies as well as the sparsentanchallenges in studying FSGS program, including feedbackbut indicated that the two-year results from the Phase 3 DUPLEX Study alone are not sufficient to support an sNDA submission. The FDA thatacknowledged the work being done by the larger nephrology community to better understand proteinuria and eGFR as endpoints in clinical trials of FSGS and indicated a willingness to continue to engage with us on a potential path forward for sparsentan in FSGS following our consideration of additional data would be neededevidence. Together with CSL Vifor, we also plan to potentially support a submissionengage with the EMA to determine the potential for accelerated approval under subpart H. At a subsequent Type A meeting, we andvariation to the FDA reached alignment on a pathway for us to proceed with a submission for accelerated approval, pending additional supportive eGFR data. We intend to provide the FDA with additional eGFR data from the ongoing DUPLEX Study in the first half of 2022, and if such data are supportive, submit an application for accelerated approval in the United States in mid-2022.
In mid-2022, we and Vifor (International) Ltd. ("Vifor Pharma"), with whom we entered into a license and collaboration agreement ("License Agreement") in September 2021, plan to submit an application for conditional marketing authorization ("CMA")Conditional Marketing Authorization (CMA) of sparsentan for the treatment of FSGS, if the MAA of sparsentan in IgA nephropathy is approved. Given the high unmet need of FSGS patients, with no medicines currently approved for the condition, and IgAN in Europe. the challenges associated with studying FSGS due to its heterogeneity and other attributes, we are conducting additional analyses of FSGS data and intend to engage with regulators to evaluate potential regulatory pathways for a sparsentan FSGS indication.
If sparsentan receives marketing authorization in any of the licensed territories covered by the exclusive license to CSL Vifor, PharmaCSL Vifor will be responsible for all commercialization activities in such licensed territories. If sparsentan receives marketing authorization in any of the territories covered by the exclusive license to Renalys, Renalys will be responsible for all development, regulatory matters, and commercialization activities in such licensed territories. We remain responsible for the clinical development of sparsentan in the applicable territories and we will retain all rights to sparsentan in the United States and rest of world outside of the territories licensed territories,to CSL Vifor and Renalys, provided that CSL Vifor Pharma has a right of negotiation to expand the licensed territories into Canada, China, Brazil and/or Mexico.
•Immunoglobulin A nephropathy ("IgAN") is characterized by hematuria, proteinuria, and variable rates of progressive renal failure. With an estimated prevalence of more than 100,000 people in the United States and greater numbers in Europe and Asia, IgAN is the most common primary glomerular disease. Most patients are diagnosed between the ages of 16 and 35, with up to 40% progressing to end stage kidney disease within 15 years. There are currently no non-immunosuppressive treatments for IgAN approved by the FDA. The current standard of care is renin-angiotensin-aldosterone system ("RAAS") blockade with immunosuppression also being commonly used for patients with significant proteinuria or rapidly progressive glomerulonephritis. In 2018, we announced that the first patient had been dosed in the PROTECT Study, a global, randomized, multicenter, double-blind, parallel-arm, active-controlled pivotal Phase 3 clinical trial evaluating the safety and efficacy of sparsentan in 404 patients with IgAN.
The PROTECT Study protocol provided for an unblinded analysis of at least 280 patients to be performed after 36 weeks of treatment to evaluate the primary efficacy endpoint - the change in proteinuria (urine protein-to-creatinine ratio) at week 36 from baseline. Secondary efficacy endpoints include the rate of change in eGFR following the initiation of randomized treatment over 58-week and 110-week periods, as well as the rate of change in eGFR over 52-week and 104-week periods following the first six weeks of randomized treatment in approximately 380 patients. In August 2021, we announced positive topline interim results from the ongoing Phase 3 PROTECT Study. The PROTECT Study met its pre-specified interim primary efficacy endpoint with statistical significance. After 36 weeks of treatment, patients receiving sparsentan achieved a mean reduction in proteinuria from baseline of 49.8 percent, compared to a mean reduction in proteinuria from baseline of 15.1 percent for irbesartan-treated patients (p<0.0001). We believe that preliminary eGFR data available at the time of the interim analysis are indicative of a potential clinically meaningful treatment effect after two years of treatment. Preliminary results at the time of the interim assessment suggested that sparsentan had been generally well-tolerated to date in the study and consistent with its overall observed safety profile. The PROTECT Study is fully enrolled and is scheduled to continue as planned on a blinded basis to assess the treatment effect on eGFR slope over 110 weeks in the confirmatory endpoint analysis. Topline results from the confirmatory endpoint analysis are expected in the second half of 2023.
Subsequent to the announcement of the interim results from the PROTECT Study, we conducted pre-NDA interactions with the FDA, during which we confirmed alignment with our plan to submit in the first quarter of 2022 an NDA for accelerated approval of sparsentan for IgAN.
In mid-2022, we and Vifor Pharma plan to submit a combined application for conditional marketing authorization ("CMA") of sparsentan for the treatment of FSGS and IgAN in Europe.
Pegtibatinase (TVT-058)
Pegtibatinase (TVT-058) is a novel investigational human enzyme replacement candidate being evaluated for the treatment of classical homocystinuria (HCU). Classical HCU is a rare metabolic disorder characterized by elevated levels of plasma homocysteine that can lead to vision, skeletal, circulatory and central nervous system complications. It is estimated that there are at least 3,500approximately 7,000 to 10,000 people living with HCU in the United States with similar numbers in Europe.globally. Pegtibatinase has been granted Rare Pediatric Disease, and Fast Track and Breakthrough Therapy designations by the FDA, as well as orphan drug designation in the United States and European Union. Pegtibatinase is currently being evaluated in
In December 2021, we announced positive topline results from the Phase 1/2 COMPOSE Study, a double blind, randomized, placebo-controlled dose escalation study to assess its safety, tolerability, pharmacokinetics, pharmacodynamics and clinical effects in patients with classical HCU.
In December 2021, we announced positive topline results from the Phase 1/2 COMPOSE Study. Pegtibatinase demonstrated dose-dependent reductions in total homocysteine (tHcy) during the 12 weeks of treatment, and in the highest dose cohort to date evaluating 1.5mg/kg of pegtibatinase twice weekly (BIW), treatment with pegtibatinase resulted in rapid and sustained reductions in total homocysteine (tHcy) through 12 weeks of treatment, including a 55.1% mean relative reduction in tHcy from baseline as well as maintenance of tHcy below a clinically meaningful threshold of 100 μmol. Additionally, in a dose-dependent manner in the study to date, methionine levels were
substantially reduced and cystathionine levels were substantially elevated following treatment with pegtibatinase, suggesting that pegtibatinase acts in a manner similar to the native CBS enzyme. To date in
In May 2023, we announced positive topline results from the study, pegtibatinase has been generally well-tolerated, with no discontinuations due to treatment-related adverse events.
Based on these results,sixth cohort of the Company is in the process of engaging with regulators to establish next steps for a pivotal development program to ultimately support potential approvals of pegtibatinase for the treatment of HCU. In parallel, the Company has initiated one additional cohort in thePhase 1/2 COMPOSE Study, which was initiated to inform and refine formulation work for future development and commercial purposes and to further evaluate the dose response curve for pegtibatinase.pegtibatinase, and to further inform our pivotal development program to ultimately support potential approval of pegtibatinase for the treatment of HCU.In this cohort, five patients were randomized in a blinded fashion to receive 2.5 mg/kg of lyophilized pegtibatinase or placebo twice weekly (BIW), with four patients assigned to the treatment group. In this highest dose cohort to date, treatment with pegtibatinase resulted in rapid and sustained reductions in total homocysteine (tHcy), with a 67.1% mean relative reduction in tHcy from baseline, as well as maintenance of mean tHcy below the clinically meaningful threshold of 100 μmol, over weeks 6 to 12.In the double-blind period, pegtibatinase was generally well-tolerated, with no discontinuations due to treatment-related adverse events.
In December 2023, we initiated the pivotal Phase 3 HARMONY Study to support the potential approval of pegtibatinase for the treatment of classical HCU. The HARMONY Study is a global, randomized, multi-center, double-blind, placebo-controlled Phase 3 clinical trial designed to evaluate the efficacy and safety of pegtibatinase as a novel treatment to reduce total homocysteine (tHcy) levels. Topline results from the HARMONY Study are expected in 2026.
We acquired pegtibatinase as part of the November 2020 acquisition of Orphan Technologies Limited ("Orphan").
Chenodal
Chenodal (chenodeoxycholic acid or CDCA) is a naturally occurring bile acid that is approved for the treatment of people with radiolucent stones in the gallbladder. While indicated for radiolucent stones in the gallbladder, Chenodal has been recognized as the standard of care for cerebrotendinous xanthomatosis (CTX) for more than three decades, although it is not currently labeled for this indication. CTX is a rare, progressive and underdiagnosed bile acid synthesis disorder affecting many parts of the body. In January 2020, we randomized the first patients in our Phase 3 RESTORE Study to evaluate the effects of Chenodal in adult and pediatric patients with CTX, and the study enrollment remains open. The pivotal study is intended to support an NDA submission for marketing authorization of Chenodal for CTX in the United States.Limited.
Preclinical Programs:
We are a participant in twoa Cooperative Research and Development Agreements ("CRADAs")Agreement (CRADA), which formforms a multi-stakeholder approach to pool resources with leading experts, and incorporate the patient perspective early in the therapeutic identification and development process. We have partneredare partnering with the National Institutes of Health’s National Center for Advancing Translational Sciences ("NCATS")(NCATS) and a leading patient advocacy organizations, CDG Care andorganization, Alagille Syndrome Alliance, aimed at the identification of potential small molecule therapeutics for NGLY1 deficiency and Alagille syndrome ("ALGS"), respectively.(ALGS). There are no treatment options currently approved for these diseases.ALGS.
ApprovedWe are party to a collaboration agreement with PharmaKrysto Limited and their early-stage cystinuria discovery program, whereby we are responsible for funding all research and development expenses for the pre-clinical activities associated with the cystinuria program.
Other Commercial Products:
Thiola and Thiola EC (tiopronin)
Thiola and Thiola EC are approved by the FDA for the treatment of cystinuria, a rare genetic cystine transport disorder that causes high cystine levels in the urine and the formation of recurring kidney stones. Due to the larger stone size, cystine stones may be more difficult to pass, often requiring surgical procedures to remove. More than 80 percent of people with cystinuria develop their first stone by the age of 20. More than 25 percent will develop cystine stones by the age of 10. Recurring stone formation can cause loss of kidney function in addition to substantial pain and loss of productivity associated with renal colic and stone passage. While a portion of people living with the disease are able to manage symptoms through diet and fluid intake, the prevalence of cystinuria in the US is estimated to be 10,000 to 12,000, indicating that there may be as many as 4,000 to 5,000 affected individuals with cystinuria in the US that would be candidates for Thiola or Thiola EC.
In June 2019 we announced that the FDA approved 100 mg and 300 mg tablets of Thiola EC, an enteric-coated formulation of Thiola, to be used for the treatment of cystinuria. Thiola EC offers the potential for administration with or without food, and the ability to reduce the number of tablets necessary to manage cystinuria. Thiola EC became available to patients in July 2019.
In May 2021, a generic option for the 100 mg version of the original formulation of Thiola (tiopronin tablets) became available. Whileavailable and in June 2022, a second option for the impact to100 mg version of the original formulation of Thiola (tiopronin tablets) was approved. These generic versions of the original formulation of Thiola have impacted our business has been minimal to date, we are not able to estimate any future impact, including the impact ofsales, and these or additional generic entrants, if any.
Cholbam (cholic acid)
Theversions of either formulation could have a material adverse impact on sales. During 2023, two generic versions of Thiola EC (100 mg and 300 mg) were approved by the FDA. Following receipt of paragraph IV certification notices from these manufacturers, we and our licensor Mission Pharmacal entered into agreements with these manufacturers in order to settle disputes around patent validity and infringement, providing for a license entry date of April 1, 2026 of these generic versions of Thiola EC (100mg and 300mg), or earlier under certain circumstances. On January 30, 2024, the FDA approved Cholbam (cholic acid capsules) in March 2015, the first FDA-approved treatment for pediatrican additional generic version of Thiola EC (100mg and adult patients with bile acid synthesis disorders due300mg). Accordingly, Thiola EC is subject to single enzyme defects, and for adjunctive treatmentimmediate generic competition, which could impact our sales of patients with peroxisome biogenesis disorder-Zellweger spectrum disorder. The effectiveness of Cholbam has been demonstrated in clinical trials for bile acid synthesis disorders and the adjunctive treatment of peroxisomal disorders. An estimated 200 to 300 patients are current candidates for therapy.
Chenodal (chenodiol)
Chenodal is a synthetic oral form of chenodeoxycholic acid ("CDCA"), a naturally occurring primary bile acid synthesized from cholesterol in the liver. The FDA approved Chenodal for the treatment of people with radiolucent stones in the gallbladder. In 2010, Chenodal was granted orphan drug designation for the treatment of cerebrotendinous xanthomatosis ("CTX"), a rare autosomal recessive lipid storage disease. We acquired Chenodal in March 2014.
While Chenodal is not labeled for CTX, it received a medical necessity determination in the US by the FDA and has been used as the standard of care for more than three decades. We are working to obtain FDA approval of Chenodal for the treatment of CTX and initiated a Phase 3 clinical trial for this indication in January 2020. The prevalence of CTX is estimated in the literature to be as high as 1 in 70,000 in the overall population. Pathogenesis of CTX involves deficiency of the enzyme 27-hydroxylase (encoded by the gene CYP27A1), a rate-limiting enzyme in the synthesis of primary bile acids, including CDCA, from cholesterol. The disruption of primary bile acid synthesis in CTX leads to toxic accumulation of cholesterol and cholestanol in most tissues. Patients may present with intractable diarrhea, premature cataracts, tendon xanthomas, atherosclerosis, and cardiovascular disease in childhood and adolescence. Neurological manifestations of the disease, including dementia and cognitive and cerebellar deficiencies, emerge during late adolescence and adulthood. The types, combinations and severity of symptoms can be different from person to person, and making diagnosis challenging and often delayed. Oral administration of CDCA has been shown to normalize primary bile acid synthesis in patients with CTX.
Our Strategy
Our vision is to become a leading biopharmaceutical company dedicated to the delivery of innovation and hope to patients in the global rare disease community. In order to achieve our vision, we intend to:
•Focus on developing products to treat rare diseases characterized by severe unmet medical needs. We believe that our research, development, and commercialization capabilities in rare disease represent distinct competitive advantages. We leverage our development capabilities in rare disease to focus on advancing therapeutic candidates with life-changing potential. Given these capabilities, the well-established regulatory model and the ability to demonstrate clinical effects in small clinical studies, we believe that we can successfully bring new therapies to patients living with severe unmet medical needs.Thiola EC.
•Develop a sustainable pipeline by employing disciplined decision criteria in the evaluation of potential in-licensing candidates. We seek to build a sustainable product pipeline by employing multiple therapeutic approaches and by developing or acquiring orphan drug candidates. We seek to augment our internally developed pipeline projects by selectively and strategically acquiring pipeline assets that will add value to the portfolio. We continue to evaluate potential in-licensing, out-licensing and other potential relationships with other pharmaceutical or biotechnology companies. We intend to mitigate risk by employing rigorous decision criteria, favoring drug candidates that have undergone at least some clinical study. Our decision to acquire rights to a drug candidate also depends on the scientific merits of the available clinical data; the identifiable orphan patient population; the economic terms of any proposed acquisition of rights; the projected amount of capital required to develop the drug candidate; and the economic potential of the drug candidate, should it be commercialized. We believe this strategy minimizes our clinical development risk and allows us to accelerate the development and potential commercialization of current and future drug candidates.
•Leverage our commercialization expertise to effectively deliver the therapies we develop. As we move our drug candidates through development toward regulatory approval, we intend to build upon and strengthen our existing commercialization infrastructure and expertise needed to successfully support rare disease patients. Our approach may vary depending on the product and will be based on a number of factors including capital necessary to execute on each option, size of the market and terms of potential collaboration and/or licensing offers from other pharmaceutical and biotechnology companies with respect to jurisdictions outside the United States.
•Listen to patients. Leadership in rare disease demands attention beyond innovative medicines. By listening to patients and leaders in the rare disease community, including those who have traditionally been underserved, we are focusing on barriers that prevent some patients from accessing the incredible innovation that our industry is delivering, including access to clinical trials and rare disease specialists.
•Support earlier diagnosis. We support efforts in furtheranceSale of enabling earlier diagnosis. For example,Bile Acid Product Portfolio
On July 16, 2023, we provide a cholestasis gene panelentered into an Asset Purchase Agreement (the “Purchase Agreement”) with Mirum Pharmaceuticals, Inc. ("Mirum Pharmaceuticals" or “Mirum”), pursuant to help families gain understandingwhich Mirum agreed to purchase substantially all of their infant's condition. The growththe assets primarily related to our business of our commercial business reflectsdevelopment, manufacture (including synthesis, formulation, finishing or packaging) and commercialization of Chenodal and Cholbam (also known as Kolbam, and together with Chenodal, the strong capabilities of our commercial and medical teams to work across multiple medical specialties to help patients find a diagnosis.“Products”), collectively, the "bile acid business". On August 31, 2023, we consummated the transactions contemplated by the Purchase Agreement (the "Closing").
Competition
The pharmaceutical and biotechnology industries are intensely competitive and subject to rapid and significant technological change. Many of our competitors are larger than our company and have substantially greater financial, marketing and technical resources than we have.
The development and commercialization of new products to treat rare diseases is highly competitive, and we expect considerable competition from major pharmaceutical, biotechnology and specialty pharmaceutical companies. As a result, there are, and will likely continue to be, extensive research and substantial financial resources invested in the discovery and development of new orphan drug products.
Our competition will be determined in part by the potential indications for which drugstherapies are developed and ultimately approved by regulatory authorities. The speed with which we can develop products, complete preclinical testing, clinical trials, approval processes, and supply commercial quantities to market are expected to be important competitive factors. We expect that competition among products approved for sale will be based on various factors, including product efficacy, safety, reliability, availability, price, reimbursement, patent position, and regulatory exclusivity.
Chenodal
There are other approved chenodeoxycholic acid products available outside of the United States. Furthermore, Chenodal is subject to immediate competition from compounded and generic entrants, as the Abbreviated New Drug Application ("ANDA") for this drug has no remaining patent or non-patent exclusivity.
Cholbam
There are currently no FDA-approved treatments in the United States that compete with Cholbam. In mid-March 2022, Cholbam will have no remaining regulatory exclusivity and could be subject to generic competition.
Pegtibatinase
Current treatment options for homocystinuria (HCU) are limited to protein-restricted diet and supplemental use of vitamin B6 and betaine.
According to public sources, Aeglea BioTherapeutics, Inc. (AGLE-177) and Codexis, Inc. (CDX-6512) are developing enzyme replacement therapies for the treatment of HCU. Additionally, Synlogic Inc. / Gingkoand Ginkgo Bioworks Holdings, Inc. are developing a microbiome therapy, SYNB1353, for the management of HCU.HCU and have completed a Phase 1 study in healthy volunteers.
There are other pre-clinical development programs in HCU that may enter the clinic.
Thiola
In May 2021, Teva Pharmaceuticals,October 2022, Mission was granted a U.S. affiliatepatent covering the treatment of Teva Pharmaceutical Industries Ltd. announced its launchcystinuria by administering Thiola EC with food (US Patent No. 11,458,104, “the ‘104 patent”) and has listed this patent in the FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations (the Orange Book). Following Mission’s listing of the first available'104 patent in the Orange Book, and as of December 31, 2023, Mission has received three paragraph IV notice letters from three generic manufacturers notifying Mission that each has filed an ANDA seeking approval of a proposed generic version of Thiola EC (tiopronin) 100 mg and 300 mg oral tablets before expiration of the '104 patent and asserting that the '104 patent is not infringed and/or is invalid, with each such ANDA having been filed prior to the granting and listing of the ‘104 patent. The ANDAs filed by Par Pharmaceutical Inc. (Par) and Amneal EU, Limited (Amneal) for a generic version of Thiola EC (100 mg and 300 mg) were approved by the FDA in February and August 2023, respectively. Under our agreement with Mission, we have the right to enforce the '104 patent. We and Mission have entered into agreements with each of Par and Amneal in order to settle patent invalidity and infringement disputes related to the '104 patent and providing for a license entry date of April 1, 2026 for their generic versions of Thiola EC (100mg and 300mg), or earlier under certain circumstances. On January 30, 2024, an ANDA filed by Torrent Pharmaceuticals Limited (Torrent) for a generic version of Thiola EC (100mg and 300mg) was approved by the FDA. Accordingly, Thiola EC is subject to immediate generic competition.
As of January 31, 2024, two generic versions of the original formulation of THIOLA® (tiopronin) tablets have been approved in the United StatesStates. Availability of THIOLA® (tiopronin) tablets.either product is subject to the discretion of either ANDA holder (Teva Pharmaceuticals USA Inc. and Par Pharmaceutical Inc.).Furthermore, Thiola and Thiola EC areis subject to further competition from compounded and generic entrants, if any.
Penicillamine agents, including Cuprimine and Depen are FDA approved for the treatment of cystinuria, as well as Wilson’s disease. Additional generic versions of penicillamine have been approved by the FDA and some have entered the market.
Captopril is not FDA approved for the treatment of cystinuria but has been prescribed for patients with cystinuria.
Advicenne Pharma is developing ADV-7103, a microtablet formulation containing potassium citrate monohydrate and potassium bicarbonate, for the potential oral treatment of cystinuria.
Based on public sources, there are other preclinical assets in development that may enter the clinic for the treatment of cystinuria.
FILSPARI/Sparsentan
FILSPARI is the first and only non-immunosuppressive therapy approved to reduce proteinuria in adults with IgAN at risk of rapid disease progression. There are currently no non-immunosuppressive pharmacological treatments approved by the European Commission for IgAN or by the FDA or the EMA for FSGS or IgAN in the US or Europe.
For FSGS, the current standard of care includes steroids, ACE/ARBs, calcineurin inhibitors, dialysis, and renal transplant.
Based on public sources, Chinook Therapeutics, Dimerix Bioscience, Goldfinch Bio, Horizon Therapeutics, Pfizer Inc., Reata Pharmaceuticals and Vertex Pharmaceuticals may have programs in clinical developmentEuropean Commission for the treatment of FSGS or related conditions.
Additionally, there are several Phase 1 assets in development and other pre-clinical programs that may enter the clinic for the treatment of FSGS or related conditions.FSGS.
For IgAN, the currenttraditional standard of care ishas been renin-angiotensin-aldosterone system (RAAS) blockade with immunosuppression. RAAS blockageblockade is also commonly used in patients with significant proteinuria or rapidly progressive glomerulonephritis.
In December 2021,2023, the FDA approvedgranted full approval for Calliditas’ budesonide delayed release capsules (Tarpeyo) to reduce the loss of kidney function in adults with primary IgAN at risk for disease progression. The same product was previously granted accelerated approval in December 2021 to reduce proteinuria in adults with primary IgAN at risk of rapid disease progression, generally at urine protein-to-creatinine ratio (UPCR)a UPCR ≥1.5 g/g. Although there are currently no pharmacological treatments approved bygram/gram. Calliditas' partner, Everest Medicines, has additionally filed for approval for the EMA for IgANsame product in Europe (orother jurisdictions including South Korea, Taiwan, and Singapore. In July 2022, the UK), public sources indicate thatEuropean Commission issued a conditional marketing authorization to Calliditas (along with EU/UK licensee, Stada Arzneimittel) submittedfor the same product (branded as Kinpeygo) for the treatment of primary IgAN in adults at risk of rapid disease progression with a UPCR ≥1.5 gram/gram. In September 2023, Calliditas and Stada filed for full marketing authorization of Kinpeygo in the EU.
In the fourth quarter of 2023, Novartis announced Phase 3 data from their MAA dossier for budesonide delayed release capsulesIgAN studies with iptacopan (Factor B complement inhibitor) and atrasentan (endothelin receptor antagonist), and we anticipate potential approvals could begin in 2021.2024. Additionally, Novartis has also announced the commencement of a Phase 3 IgAN study with zigakibart (an anti-APRIL monoclonal antibody (mAb)). Both atrasentan and zigakibart were obtained by Novartis via the acquisition of Chinook Therapeutics in 2023.
We are aware of three additional Phase 3 IgAN programs that are ongoing and/or enrolling in the United States and Europe. Roche initiated their Phase 3 study with (licensed via Ionis Pharmaceuticals) sefaxersen (Factor B complement inhibitor) and Vera Therapeutics with atacicept (BAFF / APRIL inhibitor). Otsuka Corporation started their Phase 3 study in IgAN with sibeprenlimab (anti-APRIL mAb) in 2022. Based on public sources, Alpine Immune Sciences Inc., Arrowhead Pharmaceuticals, AstraZeneca (Alexion(with its subsidiary Alexion Pharmaceuticals), AlnylamBioCity Pharmaceutics Corporation, BioCryst Pharmaceuticals, Arrowhead Pharmaceuticals, BioCryst Pharmaceuticals,, Calliditas Therapeutics / Stada / Everest, Chinook Therapeutics / SanReno Therapeutics, DiaMedica Therapeutics, Eledon Pharmaceuticals, Everest Medicines, Human Immunology Biosciences Inc., Ionis Pharmaceuticals, / Roche,Jiangsu HengRui Medicine Corporation, MorphoSys, Novartis (and subsidiaries), Omeros Corporation, Otsuka (Visterra), Reata Pharmaceuticals, RemeGen, Roche, Stada Arzneimittel, Takeda Pharmaceuticals, and Vera Therapeutics, mayand Viatris Pharmaceuticals Japan Inc. have programs in clinical development for the treatment of IgAN. Some of the listed companies have several assets that are in development for IgAN and some of these programs may no longer be active or may be deprioritized in favor of other indications.
Additionally, there are several pre-clinical, Phase 1, and later-stage programs/assets in development and other pre-clinical programs(including those that are academia-sponsored) that may enter the clinic for the treatment of IgAN or related conditions.conditions (such as diabetic and non-diabetic chronic disease).
In 2021,2021-2023, there were global (including United States, European Union, United Kingdom and Japan) regulatory label expansions of two SGLT2 inhibitors, AstraZeneca's Farxiga/Forxiga and Boehringer Ingelheim and Eli Lilly’s Jardiance, in chronic kidney disease, which could potentially be competitive with or complementary to sparsentan for the treatment of IgAN and/or FSGS. ThereNotably, Travere has and continues to generate combination data with this class of therapies. Additionally, although patients diagnosed with IgAN are additional marketed SGLT2 inhibitors which may also expand their current labels to include non-diabeticnot part of the label (nor were they included in the initial pivotal studies), Bayer’s non-steroidal mineralocorticoid receptor antagonist, Kerendia, could be used in patients with diabetic kidney disease and theoretically, those with concurrent IgAN and is currently studied in patients with chronic kidney disease.disease without diabetes. Finally, there are ERAs (endothelin receptor antagonists) that are developed for CKD and CKD-related conditions which could potentially be used (if approved) in CKD patients without a specific IgAN diagnosis. In December 2022 and January 2023, Idorsia Pharmaceuticals submitted an application for approval of aprocitentan with FDA and EMA, with potential approvals expected in 2024. The pivotal study was in patients with resistant hypertension, including those with Stage 3 & 4 CKD. In November 2023, AstraZeneca initiated a registrational fixed-dose combination (FDC) study with zibotentan and Farxiga in CKD patients with high proteinuria. Patients with IgAN are not excluded from the study.
For FSGS, the current standard of care includes steroids, ACE/ARBs, calcineurin inhibitors, dialysis, and renal transplant. Moreover, as an adjuvant apheresis device which works to remove lipoproteins from blood, the Liposorber® LA-15 System (Kaneka Pharma America LLC) is FDA-approved for the treatment of adult and pediatric patients with Nephrotic Syndrome associated with primary FSGS when standard options are unsuccessful or post-transplant with FSGS recurrence. A post-approval study with this device is ongoing. Based on public sources, Amgen Inc., AstraZeneca / Ionis Pharmaceuticals, Boehringer Ingelheim, Dimerix Bioscience, Maze Therapeutics, Novartis, River 3 Renal Corp., Vertex Pharmaceuticals, and ZyVersa Therapeutics may have programs in clinical development for the treatment of FSGS or related conditions. Some of these programs may no longer be active or may be deprioritized in favor of other indications.
Additionally, there are several pre-clinical, Phase 1, and later-stage programs/assets in development (including those that are academia-sponsored) that may enter the clinic for the treatment of FSGS or related conditions.
Licenses and Royalties
Ligand License Agreement
In 2012, we entered into a license agreement with Ligand Pharmaceuticals, Inc. ("Ligand"), granting us a worldwide licensesublicense for the development, manufacture and commercialization of sparsentan, which we are developing for the treatment of FSGS and IgAN.FILSPARI (sparsentan). Under the license agreement, Ligand granted us a sublicense under certain of its patents and other intellectual property in connection with the development and commercialization of sparsentan. Under the license agreement, Ligand is obligated to transfer to us certain information, records, regulatory filings, materials and inventory controlled by Ligand and relating to or useful for developing sparsentan. We must use commercially reasonable efforts to develop and commercialize sparsentan in specified major market countries and other countries in which we believe it is commercially reasonable to develop and commercialize such products.
As consideration for the license, we are required to make substantial payments upon the achievement of certain milestones, totaling up to $114.1 million. ShouldIn March 2023, we commercialize sparsentan or any products containing anycapitalized a $23.0 million milestone payment to Ligand (and Bristol-Myers Squibb Company ("BMS")) that was triggered upon the accelerated approval of FILSPARI in February 2023. Pursuant to the licensed compounds,Ligand License Agreement, we will beare obligated to pay to Ligand (and BMS) an escalating annual royalty between 15% and 17% of net sales of allsparsentan, with payments due quarterly. We began incurring costs associated with such products. Through 2021, we have made aggregate milestone paymentsroyalties following the February 2023 approval of FILSPARI (sparsentan). For the year ended December 31, 2023, the Company capitalized $4.4 million to Ligandintangible assets for royalties owed on net sales of $7.2 million under the terms of the license agreement.FILSPARI.
Under the terms of the license agreement, Bristol-Myers Squibb Company ("BMS")BMS has a right of first negotiation and Ligand has a right of second negotiation with respect to any license arrangement for a licensed compound, except to the extent such rights may be waived.
Absent early termination, theThe license agreement will continue until neither party has any further payment obligations under the agreement and is expected to continue for approximately 10 to 20 years from the effective date. Ligand may terminate the license agreement due to (i) our insolvency, (ii) our material uncured breach of the agreement, (iii) our failure to use commercially reasonable efforts to develop and commercialize sparsentan as described above or (iv) certain other conditions. We may terminate the license agreement due to a material uncured breach of the agreement by Ligand.
Thiola License Agreement
In 2014, we entered into a license agreement with Mission Pharmacal, Company ("Mission"), pursuant to which we obtained an exclusive, royalty-bearing license to market, sell and commercialize Thiola (tiopronin) in the United States and Canada, and a non-exclusive license to use know-how relating to Thiola to the extent necessary to market Thiola. We paid Mission an up-front license fee of $3.0 million and through May 28, 2024over the term of the agreement will pay guaranteed minimum royalties during each calendar year the greater of $2.0 million, representing the guaranteed minimum royalty, or 20% of our net sales of Thiola in the United States and Canada.
Canada during each calendar year. In October 2015, the license agreement was amended to allow for us to secure enough active pharmaceutical ingredient ("API")API to ensure an adequate level of safety stock to prevent an interruption in the supply of Thiola and to prepare for a reformulation development project.
In March 2016, the license agreement was amended to, among other things, include a new formulation development project for tiopronin tablets.
In November 2017, we amended the license agreement was amended to extend the license term at a minimum, through May 2029.
In November 2018, the license agreement was amended to remove all territorial restrictions on our license rightsrights. As consideration for the expanded territory, we paid an up-front fee of $0.3 million and provide that we will pay guaranteed minimum royalties equaling the greater of $0.1 million, representing the guaranteed minimum royalty, or 20% of our Thiola net sales generated outside of the United States during each calendar year.
Intellectual Property
The proprietary nature of, and protection for, our product candidates and our discovery programs, processes and know-how are important to our business. We have sought patent protection in the United States and certain other jurisdictions for sparsentan, where available and when appropriate. Our policy is to pursue, maintain and defend patent rights, whether developed internally or licensed from third parties, and to protect the technology, inventions and improvements that are commercially important to the development of our business.
Our commercial success will depend in part on obtaining and maintaining patent protection and for our current and future product candidates, their use in treating particular diseases, and the methods used to develop and manufacture them, as well as successfully defending these patents against third-party challenges. Our ability to stop third parties from making, using, selling, offering to sell, or importing our products depends on the extent to which we have rights under valid and enforceable patents that cover these activities. We cannot be sure that patents will be granted with respect to any of our pending patent applications or with respect to any patent applications filed by us in the future, nor can we be sure that any of our existing patents or any patents that may be granted to us in the future will be commercially useful in protecting our product candidates, discovery programs and processes.
FILSPARI
As of December 31, 2021,2023, our patent portfolio for sparsentanFILSPARI (sparsentan) was comprised of fivesix distinct patent families, one of which is exclusively licensed from Ligand (the “Ligand patent family”). The other fourfive patent families are owned by Travere (the "Travere patent families"). We previously had one additional licensed patent family related to sparsentan that was owned by BMS, exclusively licensed to Ligand and sub-licensed to us by Ligand. However, as of December 31, 2019, the BMS patent family is expired in 2019 and therefore is no longer in force.
The Ligand patent family is directed to methods of using sparsentan in the treatment of various diseases, including glomerulosclerosis and IgAN. As of December 31, 2021,2023, this patent family included two US patents (US Patent No. 9,662,312, which we refer to herein as the ‘312 patent, and US Patent No. 9,993,461, which we refer to herein as the ‘461 patent), a pending US application (Application Serial No. 17/368,239,951,830, filed July 6, 2021)September 23, 2022), two European patents (European Patent No. EP2732818, which we refer to herein as the European ‘818 patent, and European Patent No. EP3222277, which we refer to herein as the European ‘277 patent), a pending European application, and two granted Hong Kong patents. The ‘312 patent and the European ‘818 patent claim the use of sparsentan for treating glomerulosclerosis. The ‘461 patent and the European '277 patent each claim both the use of sparsentan for treating glomerulosclerosis and the use of sparsentan for treating IgAN. We expect the US and foreign patents in this patent family to expire in March 2030. In November 2020, a third party filed an opposition to the European ‘277 patent and we are vigorously defending the European '277 against the opposition. If we were to be unsuccessful in doing so, we would expect to rely on the data and/or marketing exclusivity that we expect tomay be available in Europe,the EU, as described below under “Regulatory Exclusivity.”
The first Travere patent family is directed to methods of using sparsentan in the treatment of various kidney diseases, including focal segmental glomerulosclerosis (FSGS) and IgAN, e.g., by achieving a specified urine protein to creatinineprotein-to-creatinine ratio. As of December 31, 2021,2023, this patent family was comprised of a granted U.S. patent (U.S. Patent No. 10,864,197, which we refer to herein as the ‘197 patent) and pending patent applications in the US, Australia, Brazil, Canada, China, the European Patent Office, Hong Kong, Japan, Korea, New Zealand and South Africa. The ‘197 patent claims use of sparsentan for treating Alport syndrome.
The second Travere patent family is directed to methods of using sparsentan for treating hearing loss associated with Alport syndrome. As of December 31, 2021,2023, this patent family was comprised of a granted U.S. patent (U.S. Patent No. 11,207,299) and pending patent applications in the US, Australia Brazil, Canada, China, the European Patent Office, Hong Kong, Israel, Japan, Korea, Mexico, New Zealand and South Africa.
The third Travere patent family is directed to an amorphous form of sparsentan. As of December 31, 2021,2023, this patent family was comprised of pending patent applications in the United States, Australia, Brazil, Canada, China, the European Patent Office, Hong Kong, India, Israel, Japan, Korea, Mexico, New Zealand, Russia and South Africa.
The fourth Travere patent family is comprised of a seriespending international patent application related to methods of treating kidney diseases or disorders.
The fifth Travere patent family is comprised of a U.S. provisional patent applications.application, related to methods of treating a kidney disease or disorder comprising sparsentan, and a SGLT2 inhibitor.
It is possible, assuming that sparsentan achieves regulatory approval and depending upon the dateWe have filed an application for patent term extension of any such approval, that the term of either the ‘312 patent or the ‘461 patent, which may be extended under the provisions of the Hatch-Waxman Act. Patent term extension also may be available in certain foreign jurisdictions upon regulatory approval. Likewise, it is possible, assuming thatif sparsentan achieves regulatory approval in Europethe EU and depending upon the date of any such approval, that the term of either the European ‘818 patent or the European '277 patent may be extended in various European countries under the provisions governing Supplementary Protection Certificates (SPCs).
Pegtibatinase
As of December 31, 2021,2023, our patent portfolio for pegtibatinase was comprised of six distinct patent families, which we obtained upon our acquisition of Orphan Technologies Limited (now Travere Therapeutics Switzerland GmbH). Orphan Technologies obtained the rights to the first five patent families under an exclusive license agreement with the University of Colorado, while the sixth patent family was owned by Orphan Technologies. The first three patent families are owned by the University of Colorado and exclusively licensed to Travere Therapeutics Switzerland GmbH (the “CU patent families”). The first CU patent family is directed to human cystathionine β-synthase variants and methods for their production, the second CU patent family is directed to methods of purifying human cystathionine β-synthase variants, and the third CU patent family is directed to compositions comprising human cystathionine β-synthase variants and methods of treating homocystinuria. The next two families are co-owned by the University of Colorado and Travere Therapeutics Switzerland GmbH, with the University of Colorado’s interest exclusively licensed to Travere Therapeutics Switzerland GmbH (the “co-owned patent families”). The first co-owned patent family is directed to methods of pegylating human cystathionine β-synthase variants, while the second co-owned patent family is directed to pharmaceutical formulations comprising pegylated human cystathionine β-synthase variants and their use in treating homocystinuria. Lastly, the sixth patent family is owned by Travere Therapeutics Switzerland GmbH (the “Travere patent family”). The Travere patent family is directed to methods for treating human cystathionine β-synthase deficiency in patients with elevated homocysteine levels.
Thiola
Our patent portfolio for Thiola is comprised of a patent family which is exclusively licensed from Mission Pharmacal (the “Mission patent family”). The Mission patent family is directed to a new formulation of Thiola, known as Thiola EC. As of December 31, 2021,2023, this patent family included a granted US patent application filed in 2018.(US Patent No. 11,458,104, which we refer to herein as the ‘104 patent) and a pending US patent application. The '104 patent claims a method for treating cystinuria by administering a formulation of tiopronin with food.
Regulatory Exclusivity
If we obtain marketing approval for sparsentan for the treatment of FSGS in the United States or in certain jurisdictions outside of the United States, or for sparsentan for the treatment of IgAN in certain jurisdictions outside of the United States, we may be eligible for regulatory exclusivity for the approved drug.therapy. For example, in the United States, an FDA-approved drug producttherapy may be eligible to receive five years of new chemical entity ("NCE") exclusivity or, for drugs granted an orphan designation by the FDA, seven years of orphan drug exclusivity ("ODE"Orphan Drug Exclusivity" or "ODE"). In addition, both the five-year NCE period and the seven-year ODE period may be extended by six months upon submission of satisfactory written reports to the FDA of clinical studies conducted in pediatric populations.
Likewise, if we obtain marketing approval for pegtibatinase in the United States or in certain jurisdictions outside of the United States, we may be eligible for regulatory exclusivity for the approved product. Pegtibatinase is a biologic product and therefore its application for approval would be via a biologic license application (“BLA”) (vs. an NDA). In the United States, an FDA-approved biologic product may be eligible to receive twelve years of regulatory exclusivity. In addition, the twelve-year BLA exclusivity period may be extended by six months upon submission of satisfactory written reports to the FDA of clinical studies conducted in pediatric populations.
In the European Union and European Free Trade Association ("EFTA") member states, a new drug productcountries, innovative medicinal products approved by the EMAEuropean Commission may receive eight years of data exclusivity and 10 years of marketingmarket exclusivity. The 10-year period of marketingmarket exclusivity may extend to 11 years if, during the eight-year period of data exclusivity, the product receives marketing authorization for a second therapeutic that provides a significant clinical benefit in comparison to existing therapies. Additionally, upon approval by the EMA,European Commission, orphan drugs may receive 10 years of market exclusivity or, in the case of orphan drugs for which a pediatric investigational plan (PIP) has been completed, 12 years of market exclusivity. This period of market exclusivity may, however, be reduced to six years if, at the end of the fifth year, it is established that the product no longer meets the criteria on the basis of which it received orphan medicinal product destination, including where it can be demonstrated on the basis of available evidence that the original orphan medicinal product is sufficiently profitable not to justify maintenance of market exclusivity or where the prevalence of the condition has increased above the threshold. The new drugregulatory exclusivity and the orphan drug exclusivityperiods run concurrently from the date of EMAEuropean Commission approval. There can be no assurance that we will qualify for any such regulatory exclusivity, or that any such exclusivity will prevent competitors from seeking approval solely on the basis of their own studies. See “Government Regulation” below.
ChenodalFILSPARI
ChenodalFILSPARI (sparsentan) received orphan drug designation in the United States and in the European Union for the treatment of CTXIgAN in 2010. Consequently, if Chenodal is2021 and FSGS in 2015. In the first chenodeoxycholic acid product to gain FDA approval for the treatment of CTX, we expect it will beUnited States, FILSPARI has been granted seven years of marketing exclusivity in the US for that indication. Currently Chenodal does not have regulatory exclusivity in the US.
Cholbam
Upon approval in March 2015, Cholbam received seven years of orphan drug exclusivity in the United StatesOrphan Drug Exclusivity for the treatment for pediatric and adult patientsreduction of proteinuria in adults with bile acid synthesis disorders due to single enzyme defects and for patients with peroxisomal disorders. This marketing exclusivity in the US expires in March 2022.primary IgAN at risk of rapid disease progression, generally a UPCR ≥1.5 gram/gram.
Thiola
Thiola does not have regulatory exclusivity in the United States.
Trademarks
Our trademark portfolio includes both Travere-owned and Travere-licensed trademarks and is comprised of various US and foreign registered trademarks and pending trademark applications relating to our company name, our commercial products (Thiola,(FILSPARI, Thiola, EC, Chenodal and Cholbam)Thiola EC), and our product candidates (i.e. sparsentan).sparsentan.
More specifically, as of December 31, 2021,2023, our trademark portfolio included an allowed US trademark application and foreign registered trademark and trademark applications for the mark “Travere Therapeutics”, US and foreign trademark applications and foreign registered trademarks relating to sparsentan, one registered US trademark and one registered Canadian trademark for the mark “CHENODAL”, one allowed US trademark application directed to the Chenodal logo, onea registered US trademark for the mark “MANCHESTER PHARMACEUTICALS”, one registered US trademark for the mark “KEEP IT BELOW THE LINE”, registered US and foreign trademarks for the mark “CHOLBAM”, a registered European Community trademark for the mark “KOLBAM”“TOTALCARE”, a registered US trademark for the mark “TOTAL CARE HUB”, a registered US trademark for the Total Care HubTotalCare logo, and a registered US trademark for a leaves logo. In addition, under our license agreement with Mission we have an exclusive license to use Mission’s trademarks related to Thiola and Thiola EC, including three registered US trademarks and one registered Canadian trademark for the mark “THIOLA”, and one registered US trademark for the mark "Thiola EC"., in the United States and Canada.
Trade Secrets
In addition to patents, we rely on trade secrets and know-how to develop and maintain our competitive position. We seek to protect our proprietary data and processes, in part, by confidentiality agreements and invention assignment agreements with our employees, consultants, scientific
advisors, contractors, and partners. These agreements are designed to protect our proprietary information. We also seek to preserve the integrity and confidentiality of our data, trade secrets and know-how by maintaining physical security of our premises and physical and electronic security of our information technology systems. Trade secrets and know-how can be difficult to protect. Consequently, we anticipate that trade secrets and know-how will, over time, be disseminated within the industry through independent development, the publication of journal articles, and the movement of personnel skilled in the art from academic to industry scientific positions.
Manufacturing
Nexgen Pharma, which was acquired by LGM Pharma in 2020, manufactures Chenodal, New Zealand PharmaSTA Pharmaceutical Hong Kong Limited manufactures the active pharmaceutical ingredient for Cholbam, Patheon Inc. formulatesFILSPARI. Catalent Pharma Solutions manufactures FILSPARI and packages Cholbam,performs primary packaging. PCI Pharma Services performs secondary packaging and serialization for FILSPARI. Mission Pharmacal manufactures Thiola and Thiola EC.
We intend to continue to use our financial resources to accelerate development of our drugtherapeutic candidates rather than establishing our own manufacturing facilities. We intend to meet our preclinical and clinical trial manufacturing requirements by establishing relationships with third-party manufacturers and other service providers to perform these services for us. Because we rely on these third parties, we have personnel with pharmaceutical development and manufacturing experience who are responsible for maintaining our third-party manufacturing relationships.
Should any of our drugtherapeutic candidates obtain marketing approval, we anticipate establishing relationships with third-party manufacturers and other service providers in connection with the commercial production of our products. We have some flexibility in securing other manufacturers to produce our drugtherapeutic candidates; however, our alternatives may be limited due to proprietary technologies or methods used in the manufacture of some of our drugtherapeutic candidates.
Sales, Marketing and Distribution
During fiscal 2021,In 2023, we continued to utilize our specialty sales force to market our FDA-approved products in the US. In order to commercializeThrough our clinical drug candidates if and when they are approved for sale in the US or elsewhere, we will need to increase our marketing, sales and distribution capabilities. In February 2021, we entered into a limited co-promotion arrangement with Albireo Pharma, Inc. (“Albireo”), whereby our Cholbam dedicated sales representatives will dedicate a portion of their efforts to promoting Albireo’s product, Bylvay (odevixibat), in the Unites States. The initial term of the arrangement is two years from the July 2021 launch of Bylvay, terminable at will by either party after one year following launch.
Commercialization
Through deep understanding of patient and healthcare provider needs, we believe we are able to:
•serve patients living with rare disease that have limited treatment options;
•drive optimum performance of our marketed products;
•educate and train healthcare providers about our products and the diseases for which they are approved to treat;
•support access to and reimbursement coverage for our products without significant restrictions; and
•minimize the number of patients who discontinue treatment or have low compliance with our products by providing patients with support services and disease education, to the extent and in the manner permitted under applicable laws, to help them utilize our products in a manner consistent with the label and maximize the benefits of treatment.treatment; and
•successfully launch new treatment options once approved.
Our US commercial initiatives are designed to support patients living with rare diseases and clinicians treating these patients. We believe that it is possible to commercialize our products in the United States with a relatively small specialty sales force. Nephrologists are the primary call point for FILSPARI. The primary call points for Thiola and Thiola EC include urologists and nephrologists. The primary call points for Cholbam are gastroenterologists, hepatologists, and metabolic specialists. We do not promote Chenodal.
Our sales force is differentiated by its high level of experience, averaging more than 1520 years in pharmaceutical sales including over five years of experience in rare disease. Our commercial management and operations team has an average of more than 15 years of pharmaceutical experience focused on specialty and rare disease.
Our small marketing team,and patient access teams, supported by third-party agencies with rare disease experience, drivesdrive our commercialization and disease awareness efforts in the United States. Specifically, we implement a variety of industry accepted programs to educate physicians, including direct-to-physician contact by sales representatives, peer-to-peer educational programs, and participation in targeted medical convention programs.
We distribute FILSPARI through three direct to patient pharmacies, and operate Travere TotalCare, pursuant to which we provide our comprehensive patient support services. This patient support program for FILSPARI in the United States provides services, assistance and resources that help patients understand IgAN, manage the insurance process, fill their prescriptions and initiate treatment.
We distribute our other products through one direct to patient pharmacy, Eversana, (formerly Dohmen Life Science Services), who also provides our comprehensive patient support services in the US for Thiola and Thiola EC (i.e., the Total Care Hub)Travere TotalCare). This patient support program (for all US commercial products) includes a case-managed approach to patient education, insurance verification and reimbursement support, co-pay and other financial assistance for eligible patients, monitoring and support of adherence, and 24/7 access to pharmacist counseling.
We are also in the expansion planning phases to coordinate similar marketing efforts in other countries where our products may be approved or available through named patient sales. Outside the US, including Europe, we plan to continue to partner with local distributors and certain field-based personnel as necessary to conduct permitted commercial activities.
In September 2021,August 2022, we and CSL Vifor, with whom we entered into a license and collaboration agreement ("License Agreement") with Vifor (International) Ltd. ("Vifor Pharma"). In mid-2022, we and Vifor Pharma plan to submit anin September 2021, announced that the EMA had accepted for review the CMA application for conditional marketing authorization ("CMA") of sparsentan for the treatment of FSGS and IgAN in Europe.the EU. We anticipate an opinion by the Committee for Medicinal Products for Human Use (the "CHMP") in the first quarter of 2024. If sparsentan receives marketing authorization in any of the licensed territories, CSL Vifor Pharma will be responsible for all commercialization activities in such licensed territories. We remain responsible for the clinical development of sparsentan and will retain all rights to sparsentan in the United States and rest of world outside of the licensed territories, provided that CSL Vifor Pharma has a right of negotiation to expand the licensed territories into Canada, China, Brazil and/or Mexico.
In January 2024, we announced our entered into an exclusive licensing agreement with Renalys, to bring sparsentan for the treatment of IgAN to patients in Japan and other countries in Asia. Renalys will hold regional rights to sparsentan for Japan, South Korea, Taiwan, Brunei, Cambodia, Indonesia, Laos, Malaysia, Myanmar, the Philippines, Singapore, Thailand, and Vietnam. Following successful meetings with the Pharmaceuticals and Medical Devices Agency (PMDA) in 2023, Renalys plans to initiate an open label registrational study of sparsentan in Japan in the second quarter of 2024 to support potential approval of sparsentan in Japan. Results from the urine protein/creatinine ratio (UP/C) endpoint in the study are expected in the second half of 2025 to support a submission for approval to PMDA. Under the terms of the agreement, Renalys will be responsible for development, regulatory matters, and commercialization in the licensed territories.
Medical Affairs
We have a global medical affairs team located in the United States and Europe which supports data dissemination, education, external stakeholder engagement and data generation in therapeutic areas relevant to our pipeline and commercial assets. The responsibilities of our medical affairs personnel include execution of real-world evidence studies, scientific exchange with external stakeholders, medical communication through scientific publications and presentations at medical congresses, providing medical information support to HCPs and patients related to our pipeline and our post-approval clinical commitments, organizeorganizing medical advisory boards to obtain input from experts and practitioners, and supportsupporting grants for investigator sponsored research and independent medical education programs via grants on medical topics relevant to our products and diseases.
Government Regulation
Regulation by government authorities in the United States and foreign countries is a significant factor in the development, manufacture and marketing of our proposed products and in our ongoing research and product development activities. All of our pipeline products will require regulatory approval by government agenciescompetent regulatory authorities prior to commercialization. In particular, human therapeutic products are subject to rigorous preclinical studies and clinical trials and other approval procedures of the FDA and similar regulatory authorities in foreign countries. Various federal and state statutes and regulations also govern or influence analytical testing, manufacturing, safety evaluation, labeling, storage, inspection and record-keeping related to such products and their marketing. The process of obtaining these approvals and the subsequent compliance with appropriate federal and state statutes and regulations require the expenditure of substantial time and financial and human resources.
FDA Drug Approval Process
In the United States, pharmaceutical products (drugs and biologics) are subject to extensive regulation by the FDA. The Federal Food, Drug, and Cosmetic Act, and other federal and state statutes and regulations, govern, among other things, the research, development, testing, manufacture, storage, recordkeeping, approval, labeling, promotion and marketing, distribution, post-approval monitoring and reporting, sampling and import and export of pharmaceutical products. Failure to comply with applicable US requirements may subject a company to a variety of administrative or judicial sanctions, such as FDA refusal to approve pending new drug or new biologic license applications, or NDAs and BLAs, respectively, warning or untitled letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties and criminal prosecution.
Firms are also subject to potential inspection by FDA. We cannot market a drug product candidate in the United States until the drug has received FDA approval. The key steps required before a drug may be marketed in the United States generally include the following:
•completion of extensive preclinical laboratory tests, animal studies, toxicology, pharmacology, and formulation studies in accordance with the FDA’s GLP regulations;
•submission to the FDA of an IND, and oversight by an Institutional Review Board, for human clinical testing, which must become effective before human clinical trials may begin;
•performance of adequate and well-controlled human clinical trials (phase 1-3) in accordance with Good Clinical Practices ("GCP") requirements to establish the safety and efficacy of the drug for each proposed indication;
•submission to the FDA of an NDA or BLA after completion of allthe required pivotal clinical trials;
•satisfactory completion of anany FDA pre-approval inspection of the manufacturing facility or facilities at which the active pharmaceutical ingredient, or API, and finished drug product are produced and tested to assess compliance with current Good Manufacturing Practices ("cGMPs"); and
•FDA review and approval of the NDA prior to any commercial marketing or sale of the drug in the United States.BLA.
Satisfaction of FDA pre-market approval requirements typically takes many years and the actual time required may vary substantially based upon the type, complexity and novelty of the product or disease.
Preclinical tests include laboratory evaluation of product chemistry, formulation and toxicity, as well as animal trials to assess the characteristics and potential safety and efficacy of the product. The conduct of the preclinical tests must comply with federal regulations and requirements, including good laboratory practices. The results of preclinical testing are submitted to the FDA as part of an IND along with other information, including information about product chemistry, manufacturing and controls and a proposed clinical trial protocol. Long-term preclinical tests, such as animal tests of reproductive toxicity and carcinogenicity, may continue after the IND is submitted.
A 30-day waiting period after the submission of each IND is required prior to the commencement of clinical testing in humans. If the FDA has neither commented on nor questioned the IND within this 30-day period, the clinical trial proposed in the IND may begin. If the FDA raises concerns or questions about the conduct of the trial, such as whether human research subjects will be exposed to an unreasonable health risk, the IND sponsor must resolve any outstanding FDA concerns or questions before clinical trials can proceed.
Clinical trials involve the administration of the investigational new drug or biologic to healthy volunteers or patients under the supervision of a qualified investigator. Clinical trials must be conducted in compliance with federal regulations, including GCP requirements, as well as under protocols detailing the objectives of the trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. Each protocol and subsequent protocol amendments must be submitted to the FDA as part of the IND. Certain studies must also be posted on clinicaltrials.gov.
The FDA may order the temporary, or permanent, discontinuation of a clinical trial at any time, or impose other sanctions, if it believes that the clinical trial either is not being conducted in accordance with FDA requirements or presents an unacceptable risk to the clinical trial patients. The study protocol and informed consent information for patients in clinical trials also must be submitted for approval to an institutional review board, or IRB, for approval at each site at which the clinical trial will be conducted. An IRB also may require the clinical trial at its site to be halted, either temporarily or permanently, for failure to comply with the IRB’s requirements, or may impose other conditions.
Clinical trials to support NDAs for marketing approval are typically conducted in three sequential phases, but the phases may overlap. In Phase 1, the initial administration of the drug to healthy human subjects or patients, the drug is tested to assess pharmacological actions, side effects associated with increasing doses and, if possible, early evidence on effectiveness. Phase 2 usually involves trials in a limited patient population to determine metabolism, pharmacokinetics, the effectiveness of the drug for a particular indication, dosage tolerance and optimum dosage, and to identify common adverse effects and safety risks. If a drug demonstrates evidence of effectiveness and an acceptable safety profile in Phase 2 evaluations, Phase 3 clinical trials, also called pivotal trials, are undertaken to obtain the additional information about clinical efficacy and safety in a larger number of patients, typically at geographically dispersed clinical trial sites, to permit the FDA to evaluate the overall benefit-risk relationship of the drug and to provide adequate information for the labeling of the drug. In most cases the FDA requires two adequate and well controlled Phase 3 clinical trials to demonstrate the efficacy of the drug. A single Phase 3 clinical trial with other confirmatory evidence may be sufficient in rare instances where the study is a large multicenter trial demonstrating internal consistency and a statistically very persuasive finding of a clinically meaningful effect on mortality, irreversible morbidity or prevention of a disease with a potentially serious outcome and where confirmation of the result in a second Phase 3 trial would be impractical or unethical. A sponsor may choose to pursue a Special Protocol Assessment, or SPA, agreement with FDA for the design of a Phase 3 trial. A SPA agreement is intended to provide assurance that if the agreed upon clinical trial protocols are followed and the clinical trial endpoints are achieved, the data may serve as the primary basis for an efficacy claim in support of an NDA. A SPA agreement is not binding on the FDA if previously unrecognized public health concerns arise during the performance of the clinical trial, if other new scientific concerns regarding product candidate safety or efficacy arise or if the sponsoring company fails to comply with the agreed upon clinical trial protocols.
After completion of the required clinical testing, an NDA or BLA submission is prepared and submitted to the FDA. FDA approval of the NDAsubmission is required before marketing of the product in the United States may begin. The NDAsubmission must include the results of all preclinical, clinical and other testing and a compilation of data relating to the product’s pharmacology, chemistry, manufacture and controls. The cost of preparing and submitting an NDAa submission is substantial. The submission of most NDAs and BLAs is additionally subject to a substantial application user fee, and the sponsor of an approved NDA or BLA is also subject to an annual program user fee. These fees typically are increased annually.
The FDA has 60 days from its receipt of an NDA or BLA to determine whether the application will be accepted for filing based on the agency’s threshold determination that it is sufficiently complete to permit substantive review. Once the submission is accepted for filing, the FDA begins an in-depth review. The FDA has agreed to certain performance goals in the review of NDAs.NDAs and BLAs. Most such applications for standard review drug products are reviewed within 10 months of filing; most applications for priority review drugs are reviewed within six months of filing. Priority review can be applied to drugs to treat serious conditions that the FDA determines offer significant improvement in safety or effectiveness. The review process for both standard and priority review may be extended by the FDA for three additional months to consider certain late-submitted information, or information intended to clarify information already provided in the submission.
The FDA also may refer applications for novel drug products, or drug products that present difficult questions of safety or efficacy, to an advisory committee—typically a panel that includes clinicians and other experts—for review, evaluation and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendations.
Before approving an NDA or BLA, the FDA typically will inspect one or more clinical sites to assure compliance with GCPs. Additionally, the FDA willmay inspect the facility or the facilities at which the drug is manufactured. The FDA will not approve the product unless compliance with cGMPs is satisfactory and the NDAsubmission contains data that provide substantial evidence that the drug is safe and effective in the indication studied.
After the FDA evaluates the NDA or BLA and the manufacturing facilities, it issues either an approval letter or a complete response letter. A complete response letter generally outlines the deficiencies in the submission and may require substantial additional testing, or information, in order for the FDA to reconsider the application. If, or when, those deficiencies have been addressed to the FDA’s satisfaction in a resubmission of the NDA or BLA, the FDA will issue an approval letter. The FDA has committed to reviewing such resubmissions in two or six months depending on the type of information included.
An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications. As a condition of NDA approval, the FDA may require risk evaluation and mitigation strategies ("REMS")a REMS to ensure that the benefits of the drug outweigh the potential risks. REMS can include a medication guide, a communication plan for healthcare professionals and elements to assure safe use, such as special training and certification requirements for individuals who prescribe or dispense the drug, requirements that patients enroll in a registry and other measures that the FDA deems necessary to assure the safe use of the drug. The requirement for REMS can materially affect the potential market and profitability of the drug. Moreover, product approval may require substantial post-approval testing and surveillance to monitor the drug’s safety or efficacy. Once granted, product approvals may be withdrawn if compliance with regulatory standards is not maintained or problems are identified following initial marketing.
Changes to some of the conditions established in an approved application, including changes in indications, labeling, or manufacturing processes or facilities, require submission and FDA approval of a new NDA or NDA supplement before the change can be implemented. An NDA supplement for a new indication typically requires clinical data similar to that in the original application, and the FDA uses the same procedures and actions in reviewing NDA supplements as it does in reviewing NDAs. Such supplements typically are reviewed within 10 months of receipt.
Orphan Drugs
Under the Orphan Drug Act, the FDA may grant orphan drug designation to drugs intended to treat a rare disease or condition—generally a disease or condition that affects fewer than 200,000 individuals in the US orphanUS. Orphan drug designation must be requested before submitting an NDA. After the FDA confers orphan drug status, the generic identity of the drug and its potential orphan indication are disclosed publicly by the FDA. Orphan drug designation in and of itself does not convey any advantage in, or shorten the duration of, the regulatory review and approval process. The first NDA applicant to receive FDA approval for a particular active ingredient to treat a particular indication with FDA orphan drug designation is entitled to a seven-year exclusive marketing period in the US for that product, for that indication. During the seven-year exclusivity period, the FDA may not approve any other applications to market the same drug for the same disease, except in limited circumstances, such as a showing of clinical superiority to the product with orphan drug exclusivity. Orphan drug exclusivity does not prevent FDA from approving a different drug for the same disease or condition, or the same drug for a different disease or condition. Prior to FDA approval, orphan designation provides incentives for sponsors including tax credits for clinical research expenses, the opportunity to obtain government grant funding to support clinical research, and an exemption from FDA user fees.
Fast Track DesignationExpedited Development and Review Programs
A sponsor may seek to develop and obtain approval of its products under programs designed to accelerate the development, FDA review and approval of new products that meet certain criteria.
Fast track is a process designed by the FDA to facilitate the development of drugs to treat serious conditions through expediting their review. The purpose is to get important new drugs to patients earlier. Fast Track addresses a broad range of serious conditions. Determining whether a condition is serious is a matter of judgment, but generally is based on whether the drug will have an impact on such factors as survival, day-to-day functioning, or the likelihood that the condition, if left untreated, will progress from a less severe condition to a more serious one.
A drug that receives Fast Track designation is eligible for some or all of the following:
•more frequent meetings with FDA to discuss the drug's development plan and ensure collection of appropriate data needed to support drug approval;
•more frequent written communication from FDA about such things as the design of the proposed clinical trials and use of biomarkers;
•eligibility for Accelerated Approval and Priority Review, if relevant criteria are met; and
•rollingRolling Review, which means that a drug company can submit completed sections of its Biologic License Application (BLA)NDA or NDABLA for review by FDA, rather than waiting until every section is completed before the entire application can be reviewed. BLANDA or NDABLA review usually does not begin until the drug company has submitted the entire application to the FDA.
Once a drug receives Fast Track designation, early and frequent communication between the FDA and a drug company is encouraged throughout the entire drug development and review process. The frequency of communication assures that questions and issues are resolved quickly, often leading to earlier drug approval and access by patients.
Breakthrough therapy designation is available if the product is intended, alone or in combination with one or more other drugs or biologics, to treat a serious or life-threatening condition and preliminary clinical evidence indicates that the product may demonstrate substantial improvement over currently approved therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. If the FDA designates a breakthrough therapy, it may take actions appropriate to expedite the development and review of the application, which may include holding meetings with the sponsor and the review team throughout the development of the therapy; providing timely advice to, and interactive communication with, the sponsor regarding the development of the product candidate to ensure that the development program to gather the nonclinical and clinical data necessary for approval is as efficient as practicable; involving senior managers and experienced review staff, as appropriate, in a collaborative, cross- disciplinary review; assigning a cross-disciplinary project lead for the FDA review team to facilitate an efficient review of the development program and to serve as a scientific liaison between the review team and the sponsor; and considering alternative clinical trial designs when scientifically appropriate, which may result in smaller trials or more efficient trials that require less time to complete and may minimize the number of patients exposed to a potentially less efficacious treatment. Breakthrough therapy designation comes with the benefits of Fast Track designation, which means that the sponsor may submit sections of the NDA or BLA for review on a rolling basis.
Accelerated Approval
Under the FDA’s Subpart H accelerated approval regulations, FDA may approve a drug or biologic for a serious or life-threatening illness that provides meaningful therapeutic benefit to patients over existing treatments based upon a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments.
In clinical trials, a surrogate endpoint is a measurement of laboratory or clinical signs of a disease or condition that substitutes for a direct measurement of how a patient feels, functions, or survives. Surrogate endpoints can often be measured more easily or more rapidly than clinical endpoints. A drug candidate approved on this basis is subject to rigorous post-marketing compliance requirements, including the completion of Phase 4 or post-approval clinical trials to confirm the effect on the clinical endpoint.endpoint with regular reporting to FDA on the status of such trials. Failure to conduct required post-approval studies, or confirm a clinical benefit during post-marketing studies, will allow FDA to withdraw the drug from the market on an expedited basis. All promotional materials for drug candidates approved under accelerated regulations are subject to prior review by FDA.
The Hatch-Waxman Amendments: Orange Book Listing
In seeking approval for a drug through an NDA, applicants are required to list with the FDA each patent whose claims cover the applicant’s product or its use for the relevant application. Upon approval of a drug, each of the patents listed in the application for the drug is then published in the FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the Orange Book. Drugs listed in the Orange Book can, in turn, be cited by potential generic competitors in support of approval of an ANDA. An ANDA provides for marketing of a drug product that has the same active ingredients in the same strengths and dosage form as the listed drug and has been shown through bioequivalence testing to be therapeutically equivalent to the listed drug. Other than the requirement for bioequivalence testing, ANDA applicants are not required to conduct, or submit results of, preclinical or clinical tests to prove the safety or effectiveness of their drug product. Drugs approved in this way are commonly referred to as “generic equivalents” to the listed drug, and can often be substituted by pharmacists under prescriptions written for the original listed drug.
The ANDA applicant is required to certify to the FDA concerning all patents listed for the approved product in the FDA’s Orange Book. Specifically, the applicant must certify that: (i) the required patent information has not been filed; (ii) the listed patent has expired; (iii) the listed patent has not expired, but will expire on a particular date and approval is sought after patent expiration; or (iv) the listed patent is invalid or will not be infringed by the new product. The ANDA applicant may also elect to submit a section viii statement certifying that its proposed ANDA label does not contain (or carves out) any language regarding the patented method-of-use rather than certify to a listed method-of-use patent. If the applicant does not challenge the listed patents, the ANDA application will not be approved until all the listed patents claiming the referenced product have expired.
A certification that the new product will not infringe the already approved product’s listed patents, or that such patents are invalid, is called a Paragraph IV certification. If the ANDA applicant has provided a Paragraph IV certification to the FDA, the applicant must also send notice of the Paragraph IV certification to the NDA and patent holders once the ANDA has been accepted for filing by the FDA. The NDA and patent holders may then initiate a patent infringement lawsuit in response to the notice of the Paragraph IV certification. TheUnless the submission of the ANDA pre-dates the listing of the patent in the Orange Book, the filing of a patent infringement lawsuit within 45 days of the receipt of a Paragraph IV certification automatically prevents the FDA from approving the ANDA until the earlier of 30 months, expiration of the patent, settlement of the lawsuit, or a decision in the infringement case that is favorable to the ANDA applicant.
The ANDA application also will not be approved until any applicable non-patent exclusivity listed in the Orange Book for the referenced product has expired.
Post-Approval Requirements
Once an NDA is approved, a product will be subject to certain post-approval requirements. For instance, the FDA closely regulates the post-approval marketing and promotion of drugs, including standards and regulations for direct-to-consumer advertising, off-label promotion, industry-sponsored scientific and educational activities and promotional activities involving the internet and social media. Drugs may be marketed only for the approved indications and in accordance with the provisions of the approved labeling.
Adverse event reporting and submission of periodic reports is required following FDA approval of an NDA. The FDA also may require post-marketing testing, known as Phase 4 testing, REMS, surveillance to monitor the effects of an approved product, or restrictions on the distribution or use of the product. In addition, quality-control, drug manufacturing, packaging and labeling procedures must continue to conform to cGMPs after approval. Drug manufacturers and certain of their subcontractors are required to register their establishments with the FDA and certain state agencies. Registration with the FDA subjects entities to periodic unannounced inspections by the FDA, during which the agency inspects manufacturing facilities to assess compliance with cGMPs. Accordingly, manufacturers must continue to expend time, money and effort in the areas of production and quality-control to maintain compliance with cGMPs. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or failure to comply with regulatory requirements, may result in, among other things:
•restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market and/or product recalls;
•fines, warning letters or holds on post-approval clinical trials;
•refusal of the FDA to approve pending applications or supplements to approved applications, or suspension or revocation of product approvals;
•product seizure or detention, or refusal to permit the import or export of products; or
•injunctions or the imposition of civil or criminal penalties.
Pricing and Reimbursement
A portion of our end-userproduct demand for our approved drugstherapies comes from patients covered under Medicaid, Medicare and other federal and state government-related programs such as TRICARE and the Department of Veterans Affairs, or the VA. As required by Federal regulations, we provide rebates and discounts in connection with these programs.
Our commercial success depends in significant part on the extent to which coverage and adequate reimbursement for these products will be available from third-party payers, including government health administration authorities, private health insurers and other organizations. Third-party payers determine which medications they will cover and establish reimbursement levels. Even if a third-party payer covers a particular product, the resulting reimbursement payment rates may not be adequate or may require co-payments that patients find unacceptably high. Patients who are prescribed medications for the treatment of their conditions, and their prescribing physicians, generally rely on third-party payers to reimburse all or part of the costs associated with prescription drugs.therapies. Patients are unlikely to use our products unless coverage is provided and reimbursement is adequate to cover all or a significant portion of the cost of our products. Therefore, coverage and adequate reimbursement is critical to product acceptance. Further, coverage policies and third-party payer reimbursement rates may change at any time. Even if favorable coverage and reimbursement status is attained, less favorable coverage policies and reimbursement rates may be implemented in the future.
Government authorities and other third-party payers have and are developing increasingly sophisticated methods of controlling healthcare costs, such as by limiting coverage and the amount of reimbursement for particular medications. Increasingly, third-party payers are requiring that drugtherapeutic companies provide them with predetermined discountsdiscounts/rebates from list prices as a condition of coverage, are using restrictive formularies and preferred drugtherapy lists to leverage greater discounts in competitive classes, and are challenging the prices charged for medical products. Third party payers also are carefully evaluating the medical necessity and cost-effectiveness of medical products and services, in addition to a product's safety and efficacy, which may require us to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and cost-effectiveness of our products. Further, no uniform policy requirement for coverage and reimbursement for drug productstherapies exists among third-party payers in the United States. Therefore, coverage and reimbursement for drug productstherapies can differ significantly from payer to payer. As a result, the coverage determination process is often a time-consuming and costly process that will require us to provide scientific and clinical support for the use of our products to each payer separately, with no assurance that coverage and adequate reimbursement will be applied consistently or obtained in the first instance.
In addition, it is possible that future legislation in the US and other jurisdictions could be enacted which could potentially influence the coverage and reimbursement rates for our products and also could further impact the levels of discounts and rebates paid to federal and state government entities.entities, as well as commercial payers. Any legislation that influences these areas could impact, in a significant way, our ability to generate revenues from sales of products that, if successfully developed, we bring to market.
There have been a number of enacted or proposed legislative and regulatory changes affecting the healthcare system and pharmaceutical industry that could affect our commercial success. For example, in March 2010, President Obama signed into law the Patient Protection and Affordable Care
Act, as amended by the Health Care and Education Reconciliation Act of 2010, (collectively, the “PPACA”) a law intended to, among other things, broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against healthcare fraud and abuse, add new transparency requirements for healthcare and health insurance industries, impose new taxes and fees on pharmaceutical and medical device manufacturers and impose additional health policy reforms. Specifically, by way of example, the PPACA revised the definition of “average manufacturer price” for reporting purposes, which could increase the amount of Medicaid drug rebates to states. PPACA also increased the mandated Medicaid rebate from 15.1% to 23.1% of the average manufacturer price, expanded the rebate to Medicaid managed care utilization and increased the types of entities eligible for the federal 340B drug discount program. There have been executive, judicial and Congressional challenges to certain aspects of the PPACA. For example, President Trump signed several Executive Orders and other directives designed to delay the implementation of certain provisions of the PPACA or otherwise circumvent some of the requirements for health insurance mandated by the PPACA. Concurrently, Congress considered legislation to repeal or repeal and replace all or part of the PPACA. While Congress has not passed comprehensive repeal legislation, several bills affecting the implementation of certain taxes under the PPACA have been signed into law. Legislation enacted in 2017, informally titled the Tax Cuts and Jobs Act of 2017 (“Tax Act”), includes a provision which repealed, effective January 1, 2019, the tax-based shared responsibility payment imposed by the PPACA on certain individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate”. In addition, the 2020 federal spending package permanently eliminates, effective January 1, 2020, the PPACA-mandated “Cadillac” tax on high-cost employer-sponsored health coverage and medical device tax and, effective January 1, 2021, also eliminates the health insurer tax. The Bipartisan Budget Act of 2018, or the BBA, among other things, amended the PPACA, effective January 1,2019, to increase the discount that is owed by pharmaceutical manufacturers who participate in Medicare Part D from 50% to 70%, and close the coverage gap in most Medicare drug plans, commonly referred to as the “donut hole.” On June 17, 2021, the U.S. Supreme Court dismissed a challenge on procedural grounds that argued the PPACA is unconstitutional in its entirety because the "individual mandate" was repealed by Congress. Thus, the PPACA will remain in effect in its current form. Prior to the U.S. Supreme Court ruling,Further, on January 28, 2021,August 16, 2022, President Biden issued an executive order that initiated a special enrollment periodsigned the Inflation Reduction Act of 2022 (“IRA”) into law, which among other things, extends enhanced subsidies for purposes of obtainingindividuals purchasing health insurance coverage in PPACA marketplaces through plan year 2025. The IRA also eliminates the PPACA marketplace."donut hole" under the Medicare Part D program beginning in 2025 by significantly lowering the beneficiary maximum out-of-pocket cost and creating a new manufacturer discount program. The executive orderIRA, discussed in more detail below, also instructedestablished new pricing pressures, including government price setting for certain governmental agencies to reviewtherapies and reconsider their existing policies and rules that limit access to healthcare, including among others, reexamining Medicaid demonstration projects and waiver programs that include work requirements, and policies that create unnecessary barriers to obtaining access to health insurance coverage through Medicaid or the PPACA. It is possible that the PPACA will be subject to judicial or Congressional challenges in the future. It is unclear how such challenges and the healthcare reform measures of the Biden administration will impact the PPACA and our business.mandatory rebates on price increases over inflation.
In addition, other legislative changes have been proposed and adopted since the PPACA was enacted. For example, in August 2011, the President Obama signed into law the Budget Control Act of 2011, which, among other things, created the Joint Select Committee on Deficit Reduction to recommend to Congress proposals in spending reductions. The Joint Select Committee on Deficit Reduction did not achieve a targeted deficit reduction of at least $1.2 trillion for fiscal years 2012 through 2021, triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions to Medicare payments to providers of up to 2% per fiscal year, which went into effect beginning on April 1, 2013 and, due to subsequent legislative amendments, including the BBA, and the Infrastructure Investment and Jobs Act, will stay in effect throughuntil 2031, except for a temporary suspension from May 1, 2020 through March 31, 2022 due to the COVID-19 pandemic, unless additional Congressional action is taken. Under current legislation the actual reduction in Medicare payments will vary from 1% in 2022 to up to 3% in the final fiscal year of this sequester. Additionally, in January 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, reduced Medicare payments to several providers, including hospitals and imaging centers.
Moreover, the Drug Supply Chain Security Act imposes additional obligations on manufacturers of pharmaceutical products related to product tracking and tracing. Legislative and regulatory proposals have been made to expand post-approval requirements and restrictalter sales and promotional activities for pharmaceutical products.
There has been increasing legislative and enforcement interest in the United States with respect to drugtherapy pricing practices. Specifically, there have been several recent US Congressional inquiries and numerous proposed and enacted legislation at both the state and federal levels designed to, among other things, bring more transparency to drugtherapy pricing, reduce the cost of prescription drugstherapies under Medicare, expedite generic competition, review the relationship between pricing and manufacturer patient programs, institute drugtherapy re-importation, and reform government program reimbursement methodologies for drugs.therapies. At the federal level, the Trump administration used several means to propose or implement drug pricing reform, including through federal budget proposals, executive orders and policy initiatives. For example, on July 24, 2020 and September 13, 2020, the Trump administration announced several executive orders related to prescription drug pricing that attempt to implement several of the administration’s proposals. The FDA concurrently released a final rule and guidance in September, 2020, implementing a portion of the importation executive order providing pathways for states to build and submit importation plans for drugs from Canada. Further, on November 20, 2020, the U.S. Department of Health and Human Services, or HHS, finalized a regulation removing safe harbor protection for price reductions from pharmaceutical manufacturers to plan sponsors under Part D, either directly or through pharmacy benefit managers, unless the price reduction is required by law. The implementation of the rule has been delayed by the Biden administration from January 1, 2022 to January 1, 2023 in response to ongoing litigation. The rule also creates a new safe harbor for price reductions reflected at the point-of-sale, as well as a new safe harbor for certain fixed fee arrangements between pharmacy benefit managers and manufacturers, the implementation of which have also been delayed until January 1, 2023. On November 20, 2020, the Centers for Medicare & Medicaid Services, or CMS, issued an interim final rule implementing President Trump’s Most Favored Nation executive order, which would tie Medicare Part B payments for certain physician-administered drugs to the lowest price paid in other economically advanced countries, effective January 1, 2021. As a result of litigation challenging the Most Favored Nation model, on December 27, 2021, CMS published a final rule that rescinded the Most Favored Nation model interim final rule. In July 2021, the Biden administration released an executive order, “Promoting Competition in the American Economy,” with multiple provisions aimed at prescription drugs. In response to Biden’s
executive order, on September 9, 2021, HHSthe U.S. Department of Health and Human Services (HHS) released a Comprehensive Plan for Addressing High Drug Prices that outlines principles for drug pricing reform and sets out a variety of potential legislative policies that Congress could pursue as well as potential administrative actions HHS can take to advance these principles. No legislation or administrative actions have been finalizedFurther, the IRA, among other things (i) directs HHS to implement these principles.negotiate the price of certain high-expenditure, single-source drugs and biologics covered under Medicare and (ii) imposes rebates under Medicare Part B and Medicare Part D to penalize price increases that outpace inflation. These provisions take effect progressively starting in fiscal year 2023. On August 29, 2023, HHS announced the list of the first ten drugs that will be subject to price negotiations, although the Medicare drug price negotiation program is currently subject to legal challenges. In response to the Biden administration’s October 2022 executive order, on February 14, 2023, HHS released a report outlining three new models for testing by the CMS Innovation Center which will be evaluated on their ability to lower the cost of drugs, promote accessibility, and improve quality of care. It is unclear whether these or similar policy initiativesthe models will be implementedutilized in any health reform measures in the future. Further, on December 7, 2023, the Biden administration announced an initiative to control the price of prescription drugs through the use of march-in rights under the Bayh-Dole Act. On December 8, 2023, the National Institute of Standards and Technology published for comment a Draft Interagency Guidance Framework for Considering the Exercise of March-In Rights which for the first time includes the price of a product as one factor an agency can use when deciding to exercise march-in rights. While march-in rights have not previously been exercised, it is uncertain if that will continue under the new framework. At the state level, legislatures are increasingly passing legislation and implementing regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. For example, on January 5, 2024, the FDA approved Florida’s Section 804 Importation Program (SIP) proposal to import certain drugs from Canada for specific state healthcare programs. It is unclear how this program will be implemented, including which drugs will be chosen, and whether it will be subject to legal challenges in the United States or Canada. Other states have also submitted SIP proposals that are pending review by the FDA. Any such approved importation plans, when implemented, may result in lower drug prices for products covered by those programs.
We expect that the PPACA, the IRA, as well as other federal and state healthcare reform measures that have been and may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on the price that we receive for any of our products, and could seriously harmaffect our future revenues. In addition, it is possible that future legislation in the United States and other jurisdictions could be enacted which could potentially impact the reimbursement rates for the products we are developing and may develop in the future and also could further
impact the levels of discounts and rebates paid to federal and state government entities. Any legislation that impacts these areas could impact, in a significant way, our ability to generate revenues from sales of products that, if successfully developed, we bring to market. For example, the U.S. government is considering targeted price controls and reference pricing based on foreign single-payer country access policies which may adversely affect our revenues. Further, it is possible that additional governmental action is taken in response to the COVID-19 pandemic.
Health Care Regulatory Laws
In addition to FDA marketing restrictions and regulation of pharmaceutical products, several other types of state and federal laws have been applied to restrict and regulate certain business practices in the pharmaceutical industry in recent years. These laws include, without limitation, anti-kickback statutes and false claims laws, data privacy and security laws, and transparency laws regarding payments or other items of value provided to healthcare providers.
The federal healthcare program anti-kickback statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving remuneration to induce; or in return for; purchasing, leasing, ordering or arranging for the purchase, lease or order of any healthcare item or service reimbursable under Medicare, Medicaid, or other federally financed healthcare programs. This statute has been interpreted broadly to apply to arrangements between pharmaceutical manufacturers on the one hand and prescribers, purchasers, and formulary managers, among others, on the other. Although there are a number of statutory exceptions and regulatory safe harbors protecting certain common activities from prosecution or other regulatory sanctions, the exceptions and safe harbors are drawn narrowly, and practices that involve remuneration that may induce prescribing, purchases, or recommendations may be subject to scrutiny if they do not qualify for an exemption or safe harbor. Failure to meet all of the requirements of a particular applicable statutory exception or regulatory safe harbor does not make the conduct per se illegal under the anti-kickback statute. Instead, the legality of the arrangement will be evaluated on a case-by-case basis based on a cumulative review of all its facts and circumstances. Several courts have interpreted the statute’s intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals of federal healthcare covered business, the federal anti-kickback statute has been violated. Additionally, the PPACA amended the federal anti-kickback statute to a stricter standard such that a person or entity no longer needs to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. In addition, the PPACA codified case law that a claim including items or services resulting from a violation of the federal anti-kickback statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act.
Federal false claims laws, including the civil False Claims Act, prohibit any person or entity from knowingly presenting, or causing to be presented, a false claim for payment to the federal government, or knowingly making, or causing to be made, a false statement to have a false claim paid. This includes claims made to programs where the federal government reimburses, such as Medicaid, as well as programs where the federal government is a direct purchaser, such as when it purchases off the Federal Supply Schedule. The False Claims Act contains qui tam provisions, which allow a private individual, or relator, to bring a civil action on behalf of the federal government alleging that the defendant submitted a false claim to the federal government, and to share in any monetary recovery. In recent years, the number of suits brought by private individuals has increased dramatically. For example, pharmaceutical and other healthcare companies have been prosecuted under these laws for allegedly inflating drug prices they report to pricing services, which in turn were used by the government to set Medicare and Medicaid reimbursement rates, and for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. In addition, certain marketing practices, including off-label promotion, may also violate federal false claims laws.
Also, many states have similar fraud and abuse statutes or regulations, including state anti-kickback and false claims laws, thatwhich apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payer.
The US Foreign Corrupt Practices Act, and similar worldwide anti-bribery laws generally prohibit companies and their intermediaries from making improper payments to government officials for the purpose of obtaining or retaining business. Our policies mandate compliance with these anti-bribery laws. We operate in parts of the world that have experienced governmental corruption to some degree and in certain circumstances, strict compliance with anti-bribery laws may conflict with local customs and practices or may require us to interact with doctors and hospitals, some of which may be state controlled, in a manner that is different than in the United States. We cannot assure you that our internal control policies and procedures will protect us from reckless or criminal acts committed by our employees or agents. Violations of these laws, or allegations of such
violations, could disrupt our business and result in criminal or civil penalties or remedial measures, any of which could have a material adverse effect on our business, financial condition and results of operations and could cause the market value of our common stock to decline.
The federal Health Insurance Portability and Accountability Act of 1996 ("HIPAA"), created new federal criminal statutes that prohibit, among other actions, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payers, and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Like the federal anti-kickback statute, the PPACA amended the intent standard for certain healthcare fraud provisions under HIPAA such that a person or entity no longer needs to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
In addition, we may be subject to data privacy and security regulation by both the federal government and the states in which we conduct our business. HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act or HITECH,("HITECH"), and their respective implementing regulations, imposes specified requirements relating to the privacy, security and transmission of individually identifiable health information. Among other things, HITECH, through its implementing regulations, makes certain of HIPAA’s privacy and security standards directly applicable to business associates, defined as a person or organization, other than a member of a covered entity’s workforce, that creates, receives,
maintains or transmits protected health information for or on behalf of a covered entity for a function or activity regulated by HIPAA and their covered subcontractors.
Additionally, the federal Physician Payments Sunshine Act within the PPACA, and its implementing regulations, require that certain manufacturers of drugs, devices, biologicals and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) to annually report information related to certain payments or other transfers of value made or distributed to physicians and teaching hospitals, or to entities or individuals at the request of, or designated on behalf of, physicians (defined to include to include doctors, dentists, optometrists, podiatrists and chiropractors), other healthcare professionals (such as physician assistants and nurse practitioners), and teaching hospitals and to report annually certain ownership and investment interests held by physicians and their immediate family members.
Further, certain states require implementation of commercial compliance programs and marketing codes, compliance with the pharmaceutical industry’s voluntary compliance guidelines, and compliance with the applicable compliance guidance promulgated by the federal government. Other various state level requirements include restricting payments or the provision of other items of value that may be made to healthcare providers and other potential referral sources; restricting various marketing practices; requiring prescription drug companies to report expenses relating to the marketing and promotion of drug products; requiring the posting of information relating to clinical studies and their outcomes; requiring the registration of sales representatives; requiring the reporting of certain information related to drug pricing; and requiring drug manufacturers to track and report information related to payments, gifts, compensation, and other items of value to physicians and other healthcare providers. Additionally, states that have not implemented these types of regulations are considering similar proposals. Compliance with these laws is difficult and time consuming, and companies that do not comply with these state laws face civil penalties.
If our operations are found to be in violation of any of the health regulatory laws described above or any other laws that apply to us, we may be subject to significant penalties, including imprisonment, criminal fines, civil monetary penalties, administrative penalties, disgorgement, exclusion from participation in federal healthcare programs, contractual damages, injunctions, recall or seizure of products, total or partial suspension of production, reputational harm, administrative burdens, additional oversight and reporting obligations if we become subject to a corporate integrity agreement or similar agreement to resolve allegation of non-compliance with these laws, diminished profits and future earnings, and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations.
Foreign Regulation
In addition to regulations in the United States, we are subject to a variety of foreign regulations governing clinical trials and commercial sales and distribution of our products. Whether or not we obtain FDA approval for a product, we must obtain approval by the comparable regulatory authorities of foreign countries before we can commence clinical trials and approval of foreign countries or economic areas, such as the European Union, before we may market products in those countries or areas. The approval process and requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from place to place, and the time may be longer or shorter than that required for FDA approval.
Clinical Trials and Marketing Authorization in the European Union
In Europe, athe EU, clinical trials are governed by the Clinical Trials Regulation (EU) No 536/2014, or CTR, which entered into application on January 31, 2022 repealing and replacing the former Clinical Trials Directive 2001/20, or CTD. The CTR is intended to harmonize and streamline clinical trial authorizations, simplify adverse-event reporting procedures, improve the supervision of clinical trials and increase transparency. Specifically, the Regulation, which is directly applicable in all EU Member States, introduces a streamlined application (“CTA”) must currentlyprocedure through a single-entry point, the "EU portal", the Clinical Trials Information System, or CTIS; a single set of documents to be prepared and submitted for the application; as well as simplified reporting procedures for clinical trial sponsors. A harmonized procedure for the assessment of applications for clinical trials has been introduced and is divided into two parts. Part I assessment is led by the competent authorities of a reference Member State selected by the trial sponsor and relates to clinical trial aspects that are considered to be scientifically harmonized across EU Member States. This assessment is then submitted to the competent national regulatory authorityauthorities of all concerned Member States in which the trial is to be conducted for their review. Part II is assessed separately by the competent authorities and to an independent ethics committeeEthics Committees in each countryconcerned EU Member State. Individual EU Member States retain the power to authorize the conduct of clinical trials on their territory.
The extent to which on-going clinical trials will be governed by the CTR will depend on the duration of the individual clinical trial. For clinical trials in relation to which we intendan application for approval was made on the basis of the CTD before January 31, 2023, the CTD will continue to conductapply on a transitional basis until January 31, 2025. By that date, all ongoing trials will become subject to the provisions of the CTR. The CTR will apply to clinical trials. Oncetrials from an earlier date if the CTA is approved in accordance with that country’s requirements,related clinical trial development may proceed in that country. Underapplication was made on the new Regulation on Clinical Trials, which took effect in January 2022, there will be a centralized application procedure where one national authority takesbasis of the lead in reviewing the application and the other national authorities have more limited involvement. Any substantial changes to the trial protocolCTR or other information submitted withif the clinical trial applications must be notifiedhas already transitioned to or approved by the relevant competent authorities and ethics committees. CTR framework before January 31, 2025.
In all cases, the clinical trials must be conducted in accordance with good clinical practicesGCP and otherthe applicable regulatory requirements and medicinesthe ethical principles that have their origin in the Declaration of Helsinki. Medicines used in clinical trials must be manufactured in accordance with good manufacturing practices. A clinical trial may onlythe guidelines on cGMP and in a GMP licensed facility, which can be undertaken subject to certain conditions. The relevant ethics committee must give its opinion, before a clinical trial commences, on any issue requested. Clinical trials information must be entered into a European database. There are strict requirements in relation to the labeling and packaging of our product candidates, the verification of compliance with the provisions on good clinical and manufacturing practice and the notification of adverse events and serious adverse reactions.
Under European Union regulatory systems, a company may not market a medicinal product without a marketing authorization.
There are four proceduresIn the EU, medicinal products can only be commercialized after a related marketing authorization (“MA”), has been granted. To obtain an MA for submittinga product in the EU, an applicant must submit a Marketing Authorization Application (“MAA”) in Europe:, either under a centralized procedure administered by the EMA or one of the procedures administered by the competent national authorities of EU Member States: (i) the national procedure, (ii) the mutual recognition procedure (“MRP”);procedure; or (iii) the decentralized procedure (“DCP”) and (iv) the centralized procedure (“CP”).procedure. The submission strategy for a given product will depend on the nature of the product, the target indication(s), the history of the product and the marketing plan. The centralized procedure provides for the grant of a single MA by the European Commission that is valid throughout the European Economic Area (which is comprised of the 27 EU Member States plus Norway, Iceland and Liechtenstein). The centralized procedure is compulsory for certain medicinal products which are produced by biotechnology processes, advanced therapy medicinal products, and for thoseproducts which are designated as orphan medicinal products. Besidesproducts and products with a new active substance indicated for the treatment of HIV/AIDS, cancer, neurodegenerative diseases, diabetes, auto-immune and other immune dysfunctions and viral diseases. For products falling underwith a new active substance indicated for the mandatory scope, the centralized procedure is also optional for medicinaltreatment of other diseases and products that constitute a significant therapeutic, scientific or technical innovation i.e. new active substances or other medicinal products that constitute a significant therapeutic, scientific or technical innovation, that contain an active substance not authorized in the European Union before May 20, 2004are highly innovative or for which a centralized procedure would beprocess is in the interest of patients.
The centralized procedure leads to approval of the product in all 27 EU member states and in Norway, Iceland and Liechtenstein, collectively referred to herein as the EEA. Submission of one MAA thus leads to one assessment process and onepatients, authorization that allows access to the market of the entire EEA. The process ofthrough the centralized procedure is triggered whenoptional on related approval.
Under the applicant submits an MAA tocentralized procedure, the EMA. The letter of intent also initiates the assignment of the Rapporteur and Co-Rapporteur, who are the appointed members of theEMA’s Committee for Human Medicinal Products (“CHMP”) representing two EU member states.conducts the initial assessment of a product. Under the centralized procedure, the maximum timeframe for the evaluation of an MAA by the EMA is 210 days. This excludes so-called clock stops, during which additional written or oral information is to be provided by the applicant in response to questions asked by the CHMP. At the end of the review period, the CHMP provides an opinion to the European Commission. If this opinion is opinion favorable, the Commission may then adopt a decision to grant a marketing authorization. In exceptional cases, the CHMP might perform an accelerated review of an MAA in no more than 150 days.days, excluding clock stops. This is usually when the product targets an unmet medical need and is expected to be of major interest from the point of view of public health and, in particular, from the viewpoint of therapeutic innovation. The CHMP can, however, revert to the standard time limit for the centralized procedure if it considers that it is no longer appropriate to conduct an accelerated assessment.
When usingUnlike the MRPcentralized authorization procedure, the decentralized MA procedure requires a separate application to, and leads to separate approval by, the competent authorities of each EU Member State in which the product is to be marketed. This application is identical to the application that would be submitted to the EMA for authorization through the centralized procedure. The reference EU Member State prepares a draft assessment and drafts of the related materials within 120 days after receipt of a valid application. The resulting assessment report is submitted to the concerned EU Member States who, within 90 days of receipt, must decide whether to approve the assessment report and related materials. If a concerned EU Member State cannot approve the assessment report and related materials due to concerns relating to a potential serious risk to public health, disputed elements may be referred to the Heads of Medicines Agencies’ Coordination Group for Mutual Recognition and Decentralised Procedures – Human, or DCP,CMDh, for review. The subsequent decision of the European Commission is binding on all EU Member States.
The mutual recognition procedure allows companies that have a medicinal product already authorized in one EU Member State to apply for this authorization to be recognized by the competent authorities in other EU Member States. Like the decentralized procedure, the mutual recognition procedure is based on the acceptance by the competent authorities of the EU Member States of the MA of a medicinal product by the competent authorities of other EU Member States. The holder of a national MA may submit an application to the competent authority of an EU Member State requesting that this authority recognize the MA delivered by the competent authority of another EU Member State.
An MA has, in principle, an initial validity of five years. The MA may be renewed after five years on the basis of a re-evaluation of the risk-benefit balance by the EMA or by the competent authority of the EU Member State in which the original MA was granted. To support the application, the MA holder must provide the EMA or the competent authority with a consolidated version of the Common Technical Document providing up-to-date data concerning the quality, safety and efficacy of the product, including all variations introduced since the MA was granted, at least nine months before the MA ceases to be valid. The European Commission or the competent authorities of the EU Member States may decide on justified grounds relating to pharmacovigilance, to proceed with one further five-year renewal period for the MA. Once subsequently definitively renewed, the MA shall be valid for an unlimited period. Any authorization which is not followed by the actual placing of the medicinal product on the EU market (for a centralized MA) or on the market of the authorizing EU Member State within three years after authorization ceases to be valid (the so-called sunset clause).
In the EU, a “conditional” MA may be granted in cases where all the required safety and efficacy data are not yet available. The European Commission may grant a conditional MA for a medicinal product if it is demonstrated that all of the following criteria are met: (i) the benefit-risk balance of the medicinal product is positive; (ii) it is likely that the applicant will be able to provide comprehensive data post-authorization; (iii) the medicinal product fulfils an unmet medical need; and (iv) the benefit of the immediate availability to patients of the medicinal product is greater than the risk inherent in the fact that additional data are still required. The conditional MA is subject to conditions to be fulfilled for generating the missing data or ensuring increased safety measures. It is valid for one year and must selectbe renewed annually until all related conditions have been fulfilled. Once any pending studies are provided, the conditional MA can be converted into a traditional MA. However, if the conditions are not fulfilled within the timeframe set by the EMA and approved by the European Commission, the MA will cease to be renewed.
An MA may also be granted “under exceptional circumstances” where the applicant can show that it is unable to provide comprehensive data on efficacy and safety under normal conditions of use even after the product has been authorized and subject to specific procedures being introduced. These circumstances may arise in particular when the intended indications are very rare and, in the state of scientific knowledge at that time, it is not
possible to provide comprehensive information, or when generating data may be contrary to generally accepted ethical principles. Like a conditional MA, an MA granted in exceptional circumstances is reserved to medicinal products intended to be authorized for treatment of rare diseases or unmet medical needs for which the applicant does not hold a complete data set that is required for the grant of a standard MA. However, unlike the conditional MA, an applicant for authorization in exceptional circumstances is not subsequently required to provide the missing data. Although the MA “under exceptional circumstances” is granted definitively, the risk-benefit balance of the medicinal product is reviewed annually, and the MA will be withdrawn if the risk-benefit ratio is no longer favorable.
Pediatric Development in the European Union
In the EU, member statesRegulation (EC) No 1901/2006 provides that all MAAs for new medicinal products have to include the results of trials conducted in the pediatric population, in compliance with a pediatric investigation plan, or PIP, agreed with the EMA’s Pediatric Committee, or PDCO. The PIP sets out the timing and measures proposed to generate data to support a pediatric indication of the medicinal product for which MA is being sought. The PDCO can grant a deferral of the obligation to seek approval. Inimplement some or all of the measures provided in the PIP until there are sufficient data to demonstrate the efficacy and safety of the product in adults. Further, the obligation to provide pediatric clinical trial data can be waived by the PDCO when these data are not needed or appropriate because the product is likely to be ineffective or unsafe in children, the disease or condition for which the product is intended occurs only in adult populations, or when the product does not represent a significant therapeutic benefit over existing treatments for pediatric patients. Once the MA is obtained in all EU Member States and study results are included in the product information, even when negative, the product is eligible for a six-month extension to the Supplementary Protection Certificate, or SPC, if any is in effect at the time of authorization or, in the case of an MRP, the applicant must initially receive national approval in one EU member state. This will be the so-called reference member state (“RMS”) for the MRP. Then, the applicant seeks approval for the product in other EU member states, the so-called concerned member states, inorphan medicinal products, a second step: the mutual recognition process. For the DCP, the applicant will approach all chosen member states at the same time. To do so, the applicant will identify the RMS that will assess the submitted MAA and provide the other selected member states with the conclusions and resultstwo-year extension of the assessment.orphan market exclusivity.
An innovator company enjoys a period of “data exclusivity” during which their pre-clinical and clinical trials data may not be referenced in the regulatory filings of another company (typically a generic company) for the same drug substance.
Upon receiving an MA, innovative medicinal products are generally entitled to receive eight years of data exclusivity and 10 years of market exclusivity. Data exclusivity, if granted, prevents regulatory authorities in Europe isthe EU from referencing the innovator’s data to assess a generic application or biosimilar application for eight years from the date of first authorization in Europe with an additional period of two years of “market exclusivity.” This is the period of time duringinnovative product, after which a generic companyor biosimilar MAA can be submitted, and the innovator’s data may be referenced. The market exclusivity period prevents a successful generic or biosimilar applicant from commercializing its product in the EU until 10 years have elapsed from the initial MA of the reference product in the EU. The overall ten-year period may, occasionally, be extended for a further year to a maximum of 11 years if, during the first eight years of those ten years, the MA holder obtains an authorization for one or more new therapeutic indications which, during the scientific evaluation prior to their authorization, are held to bring a significant clinical benefit in comparison with existing therapies. However, there is no guarantee that a product will be considered by the EU’s regulatory authorities to be a new chemical/biological entity, and products may not marketqualify for data exclusivity.
Orphan Designation in the European Union
In the EU, Regulation (EC) No. 141/2000, as implemented by Regulation (EC) No. 847/2000 provides that a medicinal product can be designated as an equivalent generic versionorphan medicinal product by the European Commission if its sponsor can establish that: (i) the product is intended for the diagnosis, prevention or treatment of life-threatening or chronically debilitating conditions; (ii) either (a) such conditions affect not more than 5 in 10,000 persons in the EU when the application is made, or (b) the product without the benefits derived from orphan status, would not generate sufficient return in the EU to justify the necessary investment in developing the medicinal product; and (iii) there exists no satisfactory authorized method of diagnosis, prevention, or treatment of the originator’s pharmaceuticalcondition that has been authorized in the EU, or even if such method exists, the product will be of significant benefit to those affected by that condition.
Regulation (EC) No 847/2000 sets out further provisions for implementation of the criteria for designation of a medicinal product as an orphan medicinal product. An additional one yearapplication for the designation of a medicinal product as an orphan medicinal product must be submitted at any stage of development of the medicinal product but before filing of an MAA. An MA for an orphan medicinal product may only include indications designated as orphan. For non-orphan indications treated with the same active pharmaceutical ingredient, a separate marketing authorization has to be sought.
Orphan medicinal product designation entitles an applicant to incentives such fee reductions or fee waivers, protocol assistance, and access to the centralized marketing authorization procedure. Upon grant of a marketing authorization, orphan medicinal products are entitled to a ten-year period of market exclusivity may be obtained wherefor the innovator company is granted aapproved therapeutic indication, which means that the EMA cannot accept another marketing authorization within the above eight-year period for a significant new indication for the relevant medicinal product.
Additionally, medicines that meet the criteria for orphan designation benefit from ten years of market exclusivity once they are approved for marketing in the EU. This protects them from market competition by similar medicines for the same therapeutic indication. Specifically, the competent regulatory authorities will not for a period of ten years following approval of an orphan designated product accept another application for marketing authorization, grant a marketing authorization, or accept an application to extend an existingfor a similar product and the European Commission cannot grant a marketing authorization for the same therapeutic indication in respect of a similar medicinal product. Each orphan designation carries the potential for a market exclusivity for the particular indication. Thus, a medicine that has several separate orphan designations for different indications can have a separate orphan marketing exclusivity for each orphan indication.
The Paediatric Regulation provides that an application for a new marketing authorization must include the results of all trials performed and details of all information collected in compliance with an agreed pediatric investigation plan (PIP), unless a deferral or waiver applies on the basis that pediatric use is not relevant. This requirement can also be deferred by agreement. Further, each ten-year period of marketingten years. The period of market exclusivity based upon an orphan designation is extended by two years for orphan medicinal products that have also complied with an agreed PIPPIP. No extension to any supplementary protection certificate can be granted on the basis of pediatric studies for orphan indications. Orphan medicinal product designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process.
The period of market exclusivity may, however, be reduced to six years if, at the end of the fifth year, it is established that the product no longer meets the criteria on the basis of which it received orphan designation.
Whenmedicinal product destination, including where it can be demonstrated on the application for marketing authorizationbasis of available evidence that the original orphan medicinal product is made,sufficiently profitable not to justify maintenance of market exclusivity or where the competent authority responsible for granting a marketing authorization must verify whetherprevalence of the application complies withcondition has increased above the relevant requirements, including compliance with the agreed PIP. Assuming it does, the marketing authorizationthreshold. Additionally, an MA may be granted and the relevant results are included in the summary ofto a similar medicinal product characteristics (“SmPC”) for the product, along with a statement indicating compliance with the agreed PIP. In the case of non-orphan products, the applicant then receives the six-month extension to the SPC (the “pediatric extension”). However, the six-month pediatric extension to SPC is not available for orphan designated products, or where the product has received an additional one year of marketing exclusivity by obtaining approval for a second marketing authorization for a significant new indication within the first eight years following the first marketing authorization. Where the six-month pediatric extension is available, it is not necessary for the product to actually be indicated for use in the pediatric population (for example, if the results show that that would not be appropriate).same
orphan indication during the 10 year period if: (i) if the applicant consents to a second original orphan medicinal product application, (ii) if the manufacturer of the original orphan medicinal product is unable to supply sufficient quantities; or (iii) if the second applicant can establish that its product, although similar, is safer, more effective or otherwise clinically superior to the original orphan medicinal product. A company may voluntarily remove a product from the register of orphan products.
Post-authorization Requirements
Where an MA is granted in relation to a medicinal product in the EU, the holder of the MA is required to comply with a range of regulatory requirements applicable to the manufacturing, marketing, promotion and sale of medicinal products. Similar to the United States, both MA holders and manufacturers of medicinal products are subject to comprehensive regulatory oversight by the EMA, the European Commission and/or the competent regulatory authorities of the individual EU Member States. The holder of an MA must establish and maintain a pharmacovigilance system and appoint an individual qualified person for pharmacovigilance who is responsible for oversight of that system. Key obligations include expedited reporting of suspected serious adverse reactions and submission of periodic safety update reports, or PSURs.
All new MAAs must include a risk management plan, or RMP, describing the risk management system that the company will put in place and documenting measures to prevent or minimize the risks associated with the product. The regulatory authorities may also impose specific obligations as a condition of the MA. Such risk- minimization measures or post-authorization obligations may include additional safety monitoring, more frequent submission of PSURs or the conduct of additional clinical trials or post-authorization safety studies.
In the EU, the advertising and promotion of medicinal products are subject to both EU and EU Member States’ laws governing promotion of medicinal products, interactions with physicians and other healthcare professionals, misleading and comparative advertising and unfair commercial practices. General requirements for advertising and promotion of medicinal products, such as direct-to-consumer advertising of prescription medicinal products are established in EU law. However, the details are governed by regulations in individual EU Member States and can differ from one country to another. For example, applicable laws require that promotional materials and advertising in relation to medicinal products comply with the product’s Summary of Product Characteristics, or SmPC, which may require approval by the competent national authorities in connection with an MA. The SmPC is the document that provides information to physicians concerning the safe and effective use of the product. Promotional activity that does not comply with the SmPC is considered off-label and is prohibited in the EU.
Other Laws and Regulatory Processes
We are subject to a variety of financial disclosure and securities trading regulations as a public company in the United States, including laws relating to the oversight activities of the Securities and Exchange Commission (“SEC”), and Nasdaq rules under which our stock is listed. In addition, the Financial Accounting Standards Board (“FASB”), the SEC, and other bodies that have jurisdiction over the form and content of our accounts, our financial statements and other public disclosures are constantly considering and interpreting proposals and existing pronouncements designed to ensure that companies best display relevant and transparent information relating to their respective businesses.
Our present and future business has been and will continue to be subject to various other laws and regulations. Various laws, regulations and recommendations relating to safe working conditions, laboratory practices, the experimental use of animals, and the purchase, storage, movement, import and export and use and disposal of hazardous or potentially hazardous substances used in connection with our research work are or may be applicable to our activities. Certain agreements entered into by us involving exclusive license rights or acquisitions may be subject to national antitrust regulatory control, the effect of which cannot be predicted. The extent of government regulation which might result from future legislation or administrative action also cannot be predicted with accuracy.
Employees and Human Capital Management
As of January 31, 2022,2024, we had 310380 full-time employees, with most of those based in the United States and a small number outside of the United States. We consider the intellectual capital and well-being of our employees to be an important driver of our business and key to our future success. The biopharmaceutical industry is very competitive and we believe that our future success largely depends upon our continued ability to attract, develop and retain highly skilled employees.employees as our operations expand, as well as our continued focus on our culture and patient centricity. Our workforce primarily consists of college-educated workers with experience in the biopharmaceutical industry, many of whom have advanced degrees. Our employees primarily focus on our drug development and commercialization efforts, including sales and general and administrative operational support of those functions. Currently, we rely on third-party contract manufacturers and conduct our discovery research efforts via collaborations and/or contracted third-party engagements. None of our employees in the United States are represented by a labor union or covered by collective bargaining agreements. We consider our current employee relations to be good.
We are committed to creating a multigenerational, multiracial, and multiculturalmultibackground workforce by welcoming each member’s unique experiences, backgrounds and talents and empowering them to reach their highest potential. We desire to have our organization be representative of the communities we aim to serve with our treatments, and we are placing an increased focus on ways in which we can gain greater ethnic and racial diversity within our workforce, as well as increase genderother underrepresented groups, both throughout the organization and ethnic/racial diversity in leadership roles. We have an internal diversity council that is open to all employees of our organization, and has a focus onsupport a number of initiatives that directly impact our human capital, including efforts to further diversity, equity, inclusion and equitybelonging ("DEIB") initiatives
within our recruitment, retention and developmental programs, and increase cultural awareness and engagement. We disclose information about our DEIB efforts on our website. Information on our website is not incorporated in this annual report on Form 10-K.
We strive to provide compensation, benefits and support services that help meet the varying needs of our employees. In the United States, our total compensation package includes competitive pay, including opportunities for performance-based bonuses; comprehensive healthcare benefits; paid time off and paid holidays, and the opportunity for equity ownership through our equity incentive plan and our employee stock purchase plan.plan, as well as a suite of wellness focused offerings introduced during the pandemic. We also sponsor a 401(k) plan that includes a discretionary matching contribution. A similar package of benefits areis provided to our employees outside of the United States, subject to regional differences. In 2021, we further enhanced and expanded our employee benefits in response to the continued impact of the ongoing COVID-19 pandemic as we identified additional ways in which we could further support our workforce, as well as attract new talent as we seek to grow our organization in the current tight labor market, and high competition for skilled employees within our industry. For example, we continued to provide resources to enable employees to work more efficiently and effectively from home and expanded our focus on well-being.
By focusing on employee retention, engagement and development opportunities, we believe we also improve our ability to support our clinical trials, our pipeline, our business and our operations. We value the growth and professional development of our employees. We do this through clear organizational, team, and individual goal setting, performance measurement, customized professional development, and employee training and development sessions on various topics. Our success also depends on our ability to respond to the needs of employees and weemployees. We do this by listening to our employees through many avenues, for example, throughincluding formal engagement initiatives, such as surveys, as well as informal listening sessions hosted by senior leaders, including our CEO and our human resources department. Also, during 2021The response rate for our latest employee engagement survey, which was conducted in November 2023, was 85%. During 2023 we convened a team of mid and senior level managers focusedcontinued to focus on culture who meet regularly to discuss the specifics of howways to maximize a healthy,people-centered, inclusive, and recognition-based company culture.culture, building on initiatives that have been implemented over the last several years. Also during 2023, we continued to enhance our hybrid workforce program that provides a variety of virtual and in-person collaboration opportunities. As we continue to navigate through these challenging times during a global pandemic,new ways of working, we take pride in the role we play in helping to ensure our employees are as productive and engaged as possible and their physical and emotional health, well-being and well-beingsafety remains a key priority. We are committed to providing a safe and healthy working environment for our employees and to avoiding adverse impact to the environment and the communities in which we do business.
Available Information
We were incorporated in the state of Delaware in February 2011. Our website address is travere.com. We post links on our website to the following filings as soon as reasonably practicable after they are electronically filed with or furnished to the SEC: annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, proxy statements, and any amendments to those reports filed or furnished pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934, as amended. All such filings are available through our website free of charge. The SEC also maintains an internet site at www.sec.gov that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC.
ITEM 1A. RISK FACTORS
Our business, as well as an investment in our common stock, is highly speculative in nature and involves a high degree of risk. Our securities should be purchased only by persons who can afford to lose their entire investment. Carefully consider the risks and uncertainties described below together with all of the other information included herein, including the financial statements and related notes, before deciding to invest in our common stock. If any of the following risks actually occur, they could adversely affect our business, prospects, financial condition and results of operations. In such event(s), the market price of our common stock could decline and result in a loss of part or all of your investment. Accordingly, prospective investors should carefully consider, along with other matters referred to herein, the following risk factors in evaluating our business before purchasing any shares of our common stock.
Risks Related to the DevelopmentCommercialization of our Product CandidatesOur Products
Our clinical trials may fail to demonstrate the safety and efficacy of our product candidates, including sparsentan and pegtibatinase (TVT-058), which could prevent or significantly delay their regulatory approval.
Before obtaining regulatory approval for the sale of any of our current or future product candidates, including sparsentan and pegtibatinase (TVT-058), we must subject these product candidates to extensive preclinical and clinical testing to demonstrate their safety and efficacy for humans. Clinical trialsprospects are expensive, time-consuming and may take years to complete.
We may experience numerous unforeseen events during, or as a result of, preclinical or nonclinical testing and the clinical trial process that could delay or preventhighly dependent upon our ability to obtain regulatory approval orsuccessfully develop and execute commercialization strategies for our products, including FILSPARI, and to attain market acceptance among physicians, patients and healthcare payers.
Our ability to generate significant product revenues and to achieve commercial success in the near-term will depend almost entirely on our ability to successfully commercialize our product candidates, including:
•our preclinical or nonclinical tests or clinical trials may produce negative or inconclusive results, and we may decide, or regulators may require us,products in the United States, including FILSPARI (sparsentan) to conduct additional preclinical testing or clinical trials or we may abandon projects that we expect to be promising;
•regulators may require us to conduct studiesreduce proteinuria in adults with primary IgAN at risk of the long-term effects associated with the use of our product candidates;
•regulators or institutional review boards may not authorize us to commence a clinical trial or conduct a clinical trial at a prospective trial site;
•rapid disease progression, which was approved by the FDA or any non-United States regulatory authority may impose conditions on us regardingin February 2023 under the scope or designFDA’s accelerated approval regulations.
As a newly-approved product for a rare disease that had no previously-approved non-immunosuppressive treatment, the successful launch and commercialization of our clinical trials or may require usFILSPARI is subject to resubmit our clinical trial protocolsmany risks. There are numerous examples of unsuccessful product launches and failures to institutional review boards for re-inspection due to changes in the regulatory environment;
•the numbermeet high expectations of patients required for our clinical trials may be largermarket potential, including by pharmaceutical companies with more experience and resources than we anticipate or participants may drop out ofhave. While we have established our clinical trials at a higher rate thancommercial team and U.S. sales force, we anticipate;
•our third-party contractors or clinical investigators may fail to comply with regulatory requirements or fail to meet their contractual obligations to us in a timely manner;
•we might have to suspend or terminate one or more of our clinical trials if we, regulators or institutional review boards determine that the participants are being exposed to unacceptable health risks;
•regulators or institutional review boards may require that we hold, suspend or terminate clinical research for various reasons, including noncompliance with regulatory requirements;
•the cost of our clinical trials may be greater than we anticipate;
•the supply or quality of our product candidates or other materials necessary to conduct our clinical trials may be insufficient or inadequate or we may not be able to reach agreements on acceptable terms with prospective clinical research organizations; and
•the effects of our product candidates may not be the desired effects or may include undesirable side effects or the product candidates may have other unexpected characteristics.
These risks and uncertainties impact all of our clinical programs that we pursue and have been amplified by the ongoing COVID-19 pandemic, as described below. We will only obtain regulatory approval to commercialize a product candidate if we can demonstrate to the satisfaction of the FDA, and in the case of foreign commercialization, to the applicable foreign regulatory authorities, in well-designed and conducted clinical trials, that our product candidates are safe and effective and otherwise meet the appropriate standards required for approval for a particular indication.
Our product development costs will also increase if we experience delays in testing or approvals. We do not know whether any preclinical tests or clinical trials will be initiated as planned, will need to be restructured or will be completed on schedule, if at all. Significant preclinical or clinical trial delays also could shortencontinue to train and further develop the patent protection period during which we may have the exclusive right to commercialize our product candidates. Such delays could allow our competitors to bring products to market before we do and impair our ability to commercialize our products or product candidates.
If we are required to conduct additional clinical trials or other testing of our product candidates beyond those that we currently contemplate, if we are unableteam in order to successfully complete our clinical trials or other testing, ifcoordinate the resultsongoing launch and commercialization of these trials or tests are not positive or are only modestly positive or if there are safety concerns, we may:
•be delayed in obtaining, or may not be able to obtain, marketing approval for one or more of our product candidates;
•FILSPARIobtain approval for indications that are not as broad as intended or entirely different than those indications for which we sought approval; and
•have the product removed from the market after obtaining marketing approval.
In February 2021, we announced that our ongoing pivotal Phase 3 DUPLEX Study of sparsentan in focal segmental glomerulosclerosis (“FSGS”) achieved its pre-specified interim FSGS partial remission of proteinuria endpoint (“FPRE”) after 36 weeks of treatment and in August 2021, we announced that our ongoing pivotal Phase 3 PROTECT Study of sparsentan in IGA Nephropathy (“IgAN”) achieved its pre-specified primary efficacy endpoint after 36 weeks of treatment. Pursuant to the DUPLEX and PROTECT Study protocols, patients are to continue in a blinded manner to assess the treatment effect on eGFR slope over two years in the confirmatory endpoint analyses of the studies. Given that interim results from the studies have been publicly announced, it is possible that we may see a higher than anticipated attrition rate in one or both of these studies. To the extent that an insufficient number of patients choose to remain in either study for the full two years, it could jeopardize our ability to complete the studies and submit for full regulatory approval for sparsentan in FSGS and/or IgAN.
We may not be able to initiate or continue clinical trials in the rare diseases in which we are focused if we are unable to locate a sufficient number of eligible patients willing and able to participate in the clinical trials required by the FDA or foreign regulatory agencies. In addition, as other companies and researchers may be concurrently developing therapies for the same or similar indications that we are focused on, we could face competition for a limited number of patients, investigators and clinical trial sites willing to participate in clinical trials. Our inability to enroll and maintain a sufficient number of patients for any of our current or future clinical trials would result in significant delays or may require us to abandon one or more clinical trials altogether.
In January 2020, we randomized the first patients in a Phase 3 clinical trial to evaluate the effects of Chenodal in adult and pediatric patients with CTX. The pivotal study, known as the RESTORE study, is intended to support an NDA submission for marketing authorization of Chenodal for CTX in the United States. While ChenodalThere are many factors that could cause the launch and commercialization of FILSPARI to be unsuccessful, including a number of factors that are outside our control. Because no non-immunosuppressive product has previously been used as the standard of care for CTX for over three decades, it is not labeled for CTX and as such we cannot market this drug candidate for the treatment of CTX unless and until it receives FDA approval for this indication. If we experience delays in obtaining approval or if we fail to obtain approval of Chenodal for the treatment of CTX, our business, financial condition and results of operations could be adversely affected.
The planned eGFR data cut from the DUPLEX Study may not support accelerated approval submissions in the U.S. and/or Europe.
In February 2021, we announced that our ongoing pivotal Phase 3 DUPLEX Study of sparsentan in FSGS achieved its pre-specified interim FSGS partial remission of proteinuria endpoint (“FPRE”) after 36 weeks of treatment. The confirmatory primary endpoint of the DUPLEX Study to support full regulatory approval is the rate of change in eGFR over 108 weeks of treatment. As of the time of the interim analyses, available long-term eGFR data for the confirmatory endpoint were limited. In May 2021, we provided a regulatory update regarding the sparsentan FSGS program, including feedback fromapproved by the FDA that the available data from the previously announced interim assessment of the DUPLEX Study would not be adequate to support an accelerated approval in the U.S. at this time. At a subsequent Type A meeting, we and the FDA reached alignment on a pathway for us to proceed with a submission for accelerated approval, pending additional supportive eGFR data. While we intend to provide the FDA with additional eGFR data from the ongoing DUPLEX Study in the first half of 2022, and if such data are supportive, submit an application for accelerated approval in the U.S. in mid-2022, there is no guarantee that such eGFR data will be supportive, or that the FDA will agree with our assessment as to whether it, together with the data from the previously announced interim assessment of the DUPLEX Study will be adequate to support an accelerated approval in the U.S. Similarly, we intend to provide such data to the EMA via a submission of sparsentan for FSGS, and there is no guarantee that the data will support such a submission.
Success in preclinical testing and early clinical trials does not ensure that later clinical trials will be successful.
Success in preclinical testing and early clinical trials does not ensure that later clinical trials will be successful.For example, although we observed favorable responses with the physician-initiated treatment of fosmetpantotenate in PKAN patients outside the United States, the Phase 3 FORT Study evaluating the safety and efficacy of fosmetpantotenate compared to placebo in patients with PKAN did not meet its primary endpoint, did not demonstrate a difference between treatment groups, and did not meet its secondary endpoint. In addition, there can be no assurance that the
positive eGFR results from the open-label portion of the DUET study of sparsentan in FSGS will be repeated in the Phase 3 clinical trial. Similarly, the positive pre-clinical data we have seen from pegtibatinase (TVT-058) being tested in a mouse model of homocystinuria and the positive topline results we reported in December 2021 from the ongoing Phase 1/2 clinical trial of pegtibatinase (TVT-058) may not be replicated in future studies. We cannot assure that any current or future clinical trials of sparsentan or pegtibatinase (TVT-058) will ultimately be successful.
Before obtaining regulatory approval to conduct clinical trials of our product candidates, we must conduct extensive preclinical tests to demonstrate the safety of our product candidates in animals. Preclinical testing is expensive, difficult to design and implement, and can take many years to complete. In addition, during the clinical development process, additional nonclinical toxicology studies are routinely conducted concurrently with the clinical development of a product candidate. If any of our product candidates show unexpected findings in concurrent toxicology studies, we could experience potentially significant delays in, or be required to abandon, development of that product candidate. A failure of one or more of our nonclinical studies can occur at any stage of testing.
Communications and/or feedback from the FDA or EMA related to our current or planned future clinical trials does not guarantee any particular outcome from regulatory review for such clinical trials.
Communications and/or feedback from the FDA or EMA related to our current or future clinical trials does not guarantee any particular outcome from regulatory review for such clinical trials.
In 2018 we initiated the following Phase 3 clinical trials of sparsentan: 1) a single Phase 3 clinical trial designed to serve as the basis for an NDA and MAA filing for sparsentan for the treatment of FSGS (the “DUPLEX Study”), and 2) a single Phase 3 clinical trial designed to serve as the basis for an NDA and MAA filing for sparsentan for the treatment of IgAN, (the “PROTECT Study”). We are conductingit is difficult to estimate FILSPARI’s market potential or the DUPLEX Studytime it will take to increase patient and the PROTECT Study under the Subpart H pathway for potential accelerated approval in the United States,physician awareness of FILSPARI and in Europe we plan to pursue potential Conditional Marketing Authorization, in both jurisdictions based on change in proteinuria. Recognition of change in proteinuria as a surrogate endpoint in kidney disease is a relatively new regulatory development, and, as the field continues to evolve, new learnings may impact regulatory viewpoints. For example, in the DUPLEX Study in conjunction with ongoing FDA dialogue to enable submission for potential approval on the subpart H pathway, in November 2020 we adopted an eGFR measurement over a 108-week period following randomizedcurrent treatment to support full approval. However, in May 2021, we received feedback from the FDA that the available data from the previously announced interim assessment of the DUPLEX Study would not be adequate to support an accelerated approval in the U.S. at this time. While we believe that we and the FDA have reached alignment on a pathway for us to proceed with a submission for accelerated approval, pending additional supportive eGFR data, additional data from the DUPLEX Study may not be sufficient to support an NDA under Subpart H for accelerated approval. For both indications, there is no guarantee that the FDA will accept the NDA for filing, as the FDA has the authority to refuse to file NDAs for a variety of reasons. Further, even if the FDA accepts the NDAs for filing, there is no guarantee that either NDA will be granted priority review during the application review process. If the application is not granted priority review, the review and potential approval timeline will extend beyond our current stated timing assumptions.
If the FDA or EMA agree to review our regulatory submissions for accelerated approval/conditional marketing authorization, we expect that the FDA’s and EMA’s determination as to whether the sufficiency of the data from the DUPLEX and PROTECT Studies supports an accelerated approval/conditional marketing authorization in either jurisdiction will be made during the application review process based on the totality of the data, including eGFR data available for review from the respective studies. There can be no assurance that the FDA or EMA will deem our achievement of any interim endpoint or measurement in the DUPLEX or PROTECT Studies to be sufficient to grant accelerated approval or Conditional Marketing Authorization for sparsentan for the treatment of FSGS or IgAN, respectively.
There can be no guarantee that the data generated from the DUPLEX Study will be sufficient to serve as the basis for an NDA filing, or that additional data from the DUPLEX Study will be sufficient to support an NDA under Subpart H for accelerated approval. In addition, our statistical modeling that supports proceeding with the DUPLEX Study on the Subpart H pathway is based on data from other FSGS studies. To the extent that the model population is not representative of the DUPLEX Study population, the FDA may not agree that the new results continue to support a Subpart H pathway. Furthermore, even if sparsentan is granted accelerated approval for FSGS, there can be no assurance that the post-marketing confirmatory data will support full approval of sparsentan as a treatment for FSGS.
Although we have announced that our ongoing pivotal Phase 3 PROTECT Study of sparsentan in IgAN achieved its pre-specified primary efficacy endpoint after 36 weeks of treatment and we have reached alignment with the FDA on proceeding with a submission for accelerated approval of sparsentan for IgAN, there can be no assurance that the study will proceed as planned and there can be no guarantee that the data generated from the study will be sufficient to serve as the basis for an NDA filing, including an NDA under Subpart H for accelerated approval or support Conditional Marketing Authorization in the EU. Furthermore, even if sparsentan is granted accelerated approval for IgAN, there can be no assurance that the post-marketing confirmatory data will support full approval of sparsentan as a treatment for IgAN.
In addition, because both the DUPLEX Study and PROTECT Study are evaluating the same compound for the treatment of chronic kidney diseases and utilizing similar endpoints, the risk of success or failure for the two studies may, depending on the outcomes of the studies, end up being correlated.
An extended delay in the rate of enrollment or data collection in our ongoing Phase 1/2 Study of pegtibatinase (TVT-058), as a result of the COVID-19 pandemic or otherwise, may delay our timelines for analyzing future data from the study.
While we have recently completed enrollment of the initially planned dose cohorts, we are currently enrolling an additional patient cohort in the clinical trial of pegtibatinase (TVT-058) for homocystinuria, a rare disease. Given that this development candidate is still undergoing required testing, we may not be able to initiate or continue clinical trials if we are unable to locate a sufficient number of eligible patients willing and able to participate in the clinical trial required by the FDA or foreign regulatory agencies. In addition, as other companies and researchers may be concurrently developing therapies for the same or similar indications that we are focused on, we could face competition for a limited number of patients, investigators and clinical trial sites willing to participate in clinical trials. Our inability to enroll and maintain a sufficient number of patients for any of our current or future clinical trials would result in significant delays or may require us to abandon one or more clinical trials altogether.
If the rate of enrollment in the ongoing Phase 1/2 Study of pegtibatinase (TVT-058) is slower than we anticipate, due to the COVID-19 pandemic or otherwise, or if there are barriers to data collection or monitoring activities due to the COVID-19 pandemic, our timelines for analyzing results from the Phase 1/2 Study of pegtibatinase (TVT-058) could be delayed.
Interim, topline and preliminary data from our clinical trials that we announce or publish from time to time may change as more patient data become available and are subject to audit and verification procedures that could result in material changes in the final data.
From time to time, we may publicly disclose preliminary or topline or interim data from our clinical studies, which is based on a preliminary analysis of then-available data, and the results and related findings and conclusions are subject to change following a more comprehensive review of the data related to the particular study. We also make assumptions, estimations, calculations and conclusions as part of our analyses of data, and we may not have received or had the opportunity to fully and carefully evaluate all data. As a result, the topline results that we report may differ from future results of the same studies, or different conclusions or considerations may qualify such results, once additional data have been received and fully evaluated. Topline data also remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary data we previously published. As a result, topline data should be viewed with caution. From time to time, we may also disclose interim data from our clinical trials. Interim data from clinical trials that we may complete are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment and dosing continues and more patient data become available. Adverse differences between preliminary or interim data and final or confirmatory data could significantly harm our business prospects.
Further, others, including regulatory agencies, may not accept or agree with our assumptions, estimates, calculations, conclusions or analyses or may interpret or weigh the importance of data differently, which could impact the value of the particular program, the approvability or commercialization of the particular product candidate or product and our company in general. In addition, the information we choose to publicly disclose regarding a particular study or clinical trial is based on what is typically extensive information, and you or others may not agree with what we determine is the material or otherwise appropriate information to include in our disclosure, and any information we determine not to disclose may ultimately be deemed significant with respect to future decisions, conclusions, views, activities or otherwise regarding a particular drug, drug candidate or our business. If the topline data that we report differ from actual results, or if others, including regulatory authorities, disagree with the conclusions reached, our ability to obtain approval for, and commercialize, our product candidates may be harmed, which could harm our business, operating results, prospects or financial condition.
Even if any of our product candidates receives regulatory approval, we and/or a collaborative partner will be subject to ongoing regulatory obligations and continued regulatory review, which may result in significant additional expense.
Any regulatory approvals that any of our product candidates receives may be subject to significant limitations on the approved indicated uses for which the product may be marketed or to the conditions of approval, or contain requirements for potentially costly post-marketing testing, including Phase 4 clinical trials, and surveillance to monitor the safety and efficacy of the product candidate.
In addition, if the FDA or a comparable foreign regulatory authority approves any product candidates, those products will be subject to extensive and ongoing regulatory requirements, including for the manufacturing, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion, import, export, recordkeeping, conduct of potential post-marketing studies and post-market submission requirements. These requirements include submissions of safety and other post-marketing information and reports, registration, as well as continued compliance with current good manufacturing practices and good clinical practices, for any clinical trials that we conduct post-approval. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, undesirable side effects caused by the product, problems encountered by our third-party manufacturers or manufacturing processes, or failure to comply with regulatory requirements, either before or after product approval, may result in, among other things:
•restrictions on the marketing, manufacturing, or distribution of the product;
•requirements to include additional warnings on the label;
•requirements to create or enhance a medication guide outlining the risks to patients;paradigms.
•withdrawal of the productIn September 2023, we announced topline two-year confirmatory secondary endpoint results from the market;
•voluntary or mandatory product recalls;
•requirements to change the way the product is administered or for us to conduct additional clinical trials;
•fines, warning or untitled letters or holds on clinical trials;
•refusal by the FDA to approve pending applications or supplements to approved applications filed by us or our strategic partners, or suspension or revocation of product license approvals;
•product seizure or detention, or refusal to permit the import or export of products;
•injunctions or the imposition of civil or criminal penalties;PROTECT Study. While FILSPARI demonstrated long-term kidney function preservation and
•harm to our reputation.
For example, we have certain post-marketing requirements achieved a clinically meaningful difference in estimated glomerular filtration rate (eGFR) total and commitments associated with Cholbam. Further, we face risks relating to those post-marketing obligations,chronic slope versus irbesartan as well as the commercial acceptancestatistical significance in eGFR chronic slope for purposes of Cholbam. If the regulatory approval for Chenodal, Cholbam and/or Thiola are withdrawn for any reason, it would have a material adverse impact on our sales and profitability.
The independent clinical investigators and contract research organizations that we rely upon to conduct our clinical trials may not be diligent, careful or timely, and may make mistakes,review in the conduct of our trials.
We depend on independent clinical investigators and contract research organizations (“CROs”) to conduct our clinical trials under agreements with us. The CROs play a significant roleEU., the PROTECT Study narrowly missed statistical significance in eGFR total slope, which was the pre-specified confirmatory endpoint in the conductU.S. In December 2023, we announced the completion of our clinical trials. Failurea successful pre-NDA meeting with the FDA for FILSPARI in IgAN. We plan to submit a supplemental New Drug Application (sNDA) in the first quarter of 2024 for conversion of the CROs to meet their obligations could adversely affect clinical development of our product candidates. The independent clinical investigators are not our employees and we cannot control the timing or amount of resources they devote to our studies. If their performance is substandard, it could delay or preventexisting U.S. accelerated approval of our FDA applications. Moreover, these independent investigators and CROs may also have relationships with other commercial entities, some of which may compete with us. If independent investigators and CROs allocate their resourcesFILSPARI to assist our competitors at our expense, it could harm our competitive position. In response to the COVID-19 pandemic, we have engaged or intend to engage providers of home health and remote monitoring services to assist with the ongoing conduct of our clinical trials in an effort to mitigate disruption caused by COVID-19 related issues. The introduction of new third parties into our ongoing clinical trials increases the risks associated with our dependence on third parties, including the risk that substandard performance by, or competing interests of, such third parties could have a negative impact on our clinical trials. Furthermore,full approval. However, there is no guarantee that the utilizationFDA will accept our sNDA submission for filing, that the FDA's accelerated approval of such home health providersFILSPARI will continue, or remote monitoring servicesthat FILSPARI will be successful in mitigating disruptions to our clinical trials caused byreceive full approval for IgAN. Further, if the COVID-19 pandemic.
Risks Related toFDA grants full approval of FILSPARI for IgAN, there is no guarantee that the Commercialization of Our Products
FDA will approve an expanded label. The commercial success of Chenodal, CholbamFILSPARI depends on the extent to which patients and physicians accept and adopt FILSPARI for IgAN patients. For example, if the addressable patient population suffering from primary IgAN is smaller than we estimate, if it proves difficult to educate physicians as to the availability and potential benefits of FILSPARI, or if physicians are unwilling to prescribe or patients are unwilling to take FILSPARI, the commercial potential of FILSPARI will be limited. We also do not know how physicians, patients and payers will respond to the pricing of FILSPARI, the confirmatory endpoint data from the Phase 3 PROTECT Study, the results of our ongoing interactions with regulators, and any future publications. Physicians may not prescribe FILSPARI and patients may be unwilling to use FILSPARI if coverage is not provided or reimbursement is inadequate to cover a significant portion of the cost. Thus, significant uncertainty remains regarding the commercial potential of FILSPARI. If the launch or commercialization of FILSPARI is unsuccessful or perceived as disappointing, the price of our common stock could decline significantly and long-term success of the product and our company could be harmed.
In order to operate our business and increase adoption and sales of our products, we need to continue to develop our commercial organization, including maintaining a highly experienced and skilled workforce with qualified sales representatives.
In order to successfully commercialize our products in the United States, we have built a specialized sales force. In order to successfully commercialize any approved products, we must continue to build our sales, marketing, distribution, managerial and other non-technical capabilities. Factors that may hinder our ability to successfully market and commercially distribute our products include:
•inability of sales personnel to obtain access to or educate adequate numbers of physicians on the benefits and safety of prescribing our products;
•inability to recruit, retain and Thiolaeffectively manage adequate numbers of effective sales personnel;
•lack of complementary products to be offered by sales personnel, which may put us at a competitive disadvantage relative to companies that have more extensive product lines; and
•unforeseen delays, costs and expenses associated with maintaining our sales organization.
If we are unable to maintain an effective sales force for our products, including the recently expanded sales force for FILSPARI or any other potential future approved products, we may not be able to generate sufficient product revenue in the United States. In addition, until the commencement of our commercial launch in February 2023, no one in our sales force had promoted FILSPARI or any other medicine for the treatment of IgAN patients. We are required to expend significant time and resources to train our sales force to be credible in educating physicians and pharmacists on the benefits of our products. In addition, we must continually train our sales force to ensure that a consistent and appropriate message about our products is being delivered to our potential customers. We currently have limited resources compared to some of our competitors, and the continued development of our own commercial organization to market our products and any additional products we may develop or acquire will be expensive and time-consuming. We also cannot be certain that we will be able to continue to successfully develop this capability.
We have granted exclusive licenses to third parties for the commercialization of sparsentan in certain territories outside of the United States, including Europe, Australia, New Zealand, Japan, South Korea, Taiwan and the ASEAN member states. If these third parties do not effectively engage or maintain their sales force for sparsentan if approved in the applicable territories, our ability to recognize milestone payments and royalties from the sales in such territories will be adversely affected.
We will need to continue to expend significant time and resources to train our sales forces to be credible in discussing our products with the specialists treating the patients indicated under the product’s label. In addition, if we are unable to effectively train our sales force and equip them with effective marketing materials our ability to successfully commercialize our products could be diminished, which would have a material adverse effect on our business, results of operations and financial condition.
We are dependent on third parties for the successful commercialization of sparsentan in certain key territories outside of the United States, if approved, and such third parties' commercialization efforts may fail to meet our expectations. We may not be able to establish additional collaborations or other arrangements for sparsentan in other territories, which may adversely impact our ability to generate product revenue in additional jurisdictions.
We have granted exclusive licenses to third parties for the commercialization of sparsentan in certain territories outside of the United States, including Europe, Australia, New Zealand, Japan, South Korea, Taiwan and the ASEAN member states. Consequently, the commercial success of sparsentan in these territories will depend in significant part on the efforts of such third parties, over which we will have limited control. In August 2022, Vifor Pharma Group was acquired by CSL Limited, parent company to CSL Behring and is now operating under the brand CSL Vifor. We do not currently know what effect, if any, this acquisition will ultimately have on our relationship with CSL Vifor. While our agreement with CSL Vifor remains in place following the acquisition, there is no guarantee that our collaboration with CSL Vifor will not be affected, adversely or otherwise, by the change in ownership. Moreover, in connection with the acquisition of CSL Vifor and related restructuring, substantially less resources could be devoted to the commercialization of sparsentan in the territories licensed to CSL Vifor, or such efforts could be discontinued entirely. If we are unable to establish sales and marketing capabilities or enter into agreements with third parties to market and sell sparsentan in territories outside of the United States, if approved, our ability to generate product revenue outside of the United States may be limited.
The commercial success of our products depends on them being considered to be effective drugs with advantages over other therapies.
The commercial success of our products Chenodal, CholbamFILSPARI and Thiola, and, if approved, sparsentan for the treatment of FSGS, depends on them being considered to be effective drugs with advantages over other therapies. A number of factors, as discussed in greater detail below, may adversely impact the degree of acceptance of these products, including their efficacy, safety, price and benefits over competing therapies, as well as the coverage and reimbursement policies of third-party payers, such as government and private insurance plans.
If unexpected adverse events are reported in connection with the use of any of these products, physician and patient acceptance of the product could deteriorate and the commercial success of such product could be adversely affected. We are required to report to the FDA events associated with our products relating to death or injury. Adverse events could result in additional regulatory controls, such as a requirement for costly post-approval clinical studies or revisions to our approved labeling which could limit the indications or patient population for a product or could even lead to the withdrawal of a product from the market.
We are subject toface substantial generic and other competition, and recent developments relatingour operating results will suffer if we fail to generic competition for pharmaceutical products could cause our product sales and business to be negatively impacted.compete effectively.
Under the Hatch-Waxman Amendments of the Federal Food, Drug, and Cosmetic Act, a pharmaceutical manufacturer may file an ANDA seeking approval of a generic copy of an approved innovator product or an NDA under Section 505(b)(2) that relies on the FDA’s prior findings of safety and effectiveness in approving the innovator product. A Section 505(b)(2) NDA may be for a new or improved version of the original innovator product. Certain of ourOur product Thiola, and products from which we may receive milestone payments including Thiola,Cholbam and Chenodal, are subject to immediate competition from compounded and generic entrants, as the ANDA and
and/or NDA for these drug products have no remaining or current patent or non-patent exclusivity. In MayApril 2021, a generic option for the 100 mg version of the original formulation of Thiola (tiopronin tablets) was approved by the FDA and an additional generic alternativesoption of the original formulation of Thiola (tiopronin tablets) was approved in June 2022 and during the year ended December 31, 2022, we experienced a decrease in total net product revenues compared to the year ended December 31, 2021, which was due in part to competition from generic tiopronin tablets (100 mg version of the original formulation). Additional generic versions of Thiola may be approved in the future. In February 2023, August 2023 and January 2024, generic versions of Thiola EC (100 mg and 300 mg) were approved by the FDA. Our future net product revenues from Thiola and/or Thiola EC may be materially impacted by competition from existing or additional generic versions of Thiola or Thiola EC.
In addition, there have been a number of recent regulatory and legislative initiatives designed to encourage generic competition for pharmaceutical products, including expedited review procedures for generic manufacturers and incentives designed to spur generic competition of branded drugs. In particular, the FDA and the U.S. Federal Trade Commission (“FTC”) have been focused on brand companies’ denial of drug supply to potential generic competitors for testing. In December 2019, the CREATES Act was enacted, which provides a legislatively defined private right of action under which generic companies can bring suit against companies who refuse access to product for the bioequivalence testing needed to support approval of a generic product.
We have completed our response to a civil investigative demand from the FTC related to the marketing, sale, distribution and pricing of our products, including Thiola. While the investigation remains open, at this time the FTC has not indicated that it has additional questions for us and has not initiated any claim or proceeding against us relating to these matters.
We cannot currently predict the specific outcome or impact on our business of such regulatory and legislative initiatives, litigation or investigation. However, it is our policy, which is in compliance with the CREATES Act, to evaluate requests for samples of our branded products, and to provide samples in response to bona fide requests from qualified third parties, including generic manufacturers, subject to specified conditions. We have provided and are in the process of providing samples to certain generic manufacturers.
If additional generic versions of Thiola or Thiola EC, any generic versions of Cholbam,FILSPARI following the expiration of patent or regulatory exclusivity for the product, or generic versions of Chenodal or any of our other current or future products are approved, sales of that product likely would be negatively impacted, which could have a material adverse impact on our salesrevenue and profitability. BothIf generic versions of Cholbam or Chenodal are approved, our potential to receive milestone payments from the original formulationsale of Thiola and Thiola EC are subject to generic competition, and a generic version of either formulation could have a material adverse impact on sales of Thiola EC.our bile acid product portfolio may be negatively impacted. In addition, the defense of litigation and response to investigation requests could result in substantial costs, reputational impact, and the diversion of management attention and resources.
ChangesThe Drug Price Competition and Patent Term Restoration Act (commonly referred to as the "Hatch-Waxman Act") requires an ANDA applicant seeking FDA approval of its proposed generic product prior to the expiration of an Orange Book-listed patent (as defined below) to certify that the applicant believes that the patent is invalid, unenforceable and/or will not be infringed by the manufacture, use or sale of the drug for which the application has been submitted (a paragraph IV certification) and notify the NDA and patent holder of such certification (a paragraph IV notice). Upon receipt of a paragraph IV notice, the Hatch-Waxman Act allows the patent holder, with proper basis, to bring an action for patent infringement against the ANDA filer, asking that the proposed generic product not be approved until after the patent expires. For ANDAs that are filed (“received”) after the listing of the patent in the Orange Book, if the patent holder commences a lawsuit within 45 days from receipt of the paragraph IV notice, the Hatch-Waxman Act provides a 30-month stay during which time the FDA cannot finally approve the generic’s application. If the litigation is resolved in favor of the ANDA applicant during the 30-month stay period, the stay is lifted and the FDA may finally approve the ANDA if it is otherwise ready for approval. For ANDAs that are filed (“received”) before the listing of the patent in the Orange Book, the 30-month stay provision of the Hatch-Waxman Act does not apply. It also may be possible, depending on the approved label, for an ANDA applicant to elect to submit a section viii statement certifying that its proposed ANDA label does not contain (or carves out) any language regarding the patented method-of-use rather than certify to a listed method-of-use patent.
In October 2022, our licensor, Mission Pharmacal Company, was granted a patent covering the treatment of cystinuria by administering Thiola EC with food (US Patent No. 11,458,104, “the ‘104 patent”) and listed this patent in the FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations (the "Orange Book"). Following Mission’s listing of the '104 patent in the Orange Book, and as of December 31, 2023, Mission has received three paragraph IV notice letters from three generic manufacturers notifying Mission that each has filed an ANDA seeking approval of a proposed generic version of Thiola EC (tiopronin) 100 mg and 300 mg oral tablets before expiration of the '104 patent and asserting that the '104 patent is not infringed and/or is invalid, with each such ANDA having been filed prior to the granting and listing of the ‘104 patent. The ANDAs filed by Par Pharmaceutical Inc. (Par) and Amneal EU, Limited (Amneal) for generic versions of Thiola EC (100 mg and 300 mg) were approved by the FDA in February and August 2023, respectively. Under our agreement with Mission, we have the right to enforce the '104 patent. We and Mission have entered into agreements with each of Par and Amneal in order to settle patent invalidity and infringement disputes related to the '104 patent and providing for a license entry date of April 1, 2026 for their generic versions of Thiola EC (100mg and 300mg), or earlier under certain circumstances.
There is no guarantee that the ‘104 patent will withstand any challenge at the Patent and Trademark Office or in litigation, if initiated. Patent litigation is expensive and time consuming, requires significant resources, may absorb significant time of our management and has an unpredictable outcome. If we determine not to pursue patent litigation or the patent is not upheld in litigation or administrative review or if a generic competitor is found not to infringe this patent, the resulting generic competition will likely negatively affect our business, financial condition and results of operations. Following the FDA’s approval in January 2024 of an ANDA for a generic version of Thiola EC (100 mg and 300 mg), Thiola EC is subject to immediate generic competition.
Healthcare reform initiatives, unfavorable pricing regulations, and changes in reimbursement practices of third-party payers or patients' access to insurance coverage could affect the pricing of and demand for our products and/or the prices at which they are sold.products.
The business and financial condition of healthcare-related businesses will continue to be affected by efforts of governments and third-party payers to contain or reduce the cost of healthcare through various means. In the United States and some foreign jurisdictions, there have been a number of legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay marketing approval for sparsentan, pegtibatinase (TVT-058),our current product candidates or any otherfuture product candidate that we develop, restrict or regulate post-approval activities and affect our ability to profitably sell sparsentan, pegtibatinase, (TVT-058), or any other product candidate for which we obtain marketing approval.
Our products are sold to patients whose healthcare costs are met by third-party payers, such as government programs, private insurance plans and managed-care programs. These third-party payers are increasingly attempting to contain healthcare costs by limiting both coverage and the level of reimbursement for medical products and services. Levels of reimbursement, if any, may be decreased in the future, and future healthcare reform legislation, regulations or changes to reimbursement policies of third-party payers may otherwise adversely affect the demand for and price levels of our products, which could have a material adverse effect on our sales and profitability.
Economic, social, and congressional pressure may result in individuals and government entities increasingly seeking to achieve cost savings through mechanisms that limit coverage or payment for our products. For example, state Medicaid programs are increasingly requesting manufacturers to pay supplemental rebates and are requiring prior authorization for use of drugs. Managed care organizations continue to seek price discounts and, in some cases, to impose restrictions on the coverage of particular drugs. Government efforts to reduce Medicaid expenses may lead to increased use of managed care organizations by Medicaid programs. This may result in managed care organizations influencing prescription decisions for a larger segment of the population and a corresponding constraint on prices and reimbursement for our products.
In addition, patients’ access to employer sponsored insurance coverage may be negatively impacted by the COVID-19 pandemic or other economic factors that result in increased rates of unemployment. To the extent patients taking our approved therapies become unemployed and experience a reduction to, or increased costs associated with, their insurance coverage, demand for our products could decline, which could have a material adverse effect on our sales and profitability, either as a result of decreased sales of our products and/or increased provision by us of free product to uninsured or commercially insured patients. The extent and duration of this potential impact on our business is currently unknown.
We are dependent on third parties to manufacture and distribute our pharmaceutical products who may not fulfill their obligations.products.
We have no manufacturing capabilities and rely on third partythird-party manufacturers who are sole source suppliers for manufacturing of Chenodal, CholbamFILSPARI and Thiola. The facilities used by our third-party manufacturers must be approved by the FDA.FDA and comparable foreign regulatory authorities. Our dependence on third parties for the manufacture of our products may harm our profit margin on the sale of products and our ability to deliver products on a timely and competitive basis. Because we are ultimately responsible for ensuring that our API and finished products are manufactured in accordance with cGMP regulations and similar regulatory requirements outside the United States, it is critical that we maintain effective management practices and oversight with respect to our third-party manufacturers, including routine auditing. If our third-party manufacturers are unable to manufacture to specifications or in compliance with applicable regulatory requirements, our ability to commercialize our products will be adversely impacted and could affect our ability to gain market acceptance for our products and negatively impact our revenues.
We currently have no in-house distribution channels for Chenodal, CholbamFILSPARI or Thiola and we are dependent on a third-party distributor, Eversana,distributors to distribute such products. We rely on this distributor for all of our proceeds from sales of Chenodal, Cholbam and Thiola in the United States. The outsourcing of our distribution function is complex, and we may experience difficulties that could reduce, delay or stop shipments of such products. If we encounter such distribution problems, and we are unable to quickly enter into a similar agreement with another distributor on substantially similar terms, distribution of Chenodal, CholbamFILSPARI and/or Thiola could become disrupted, resulting in lost revenues, provider dissatisfaction, and/or patient dissatisfaction.
Governments outside the United States tend to impose strict price controls and reimbursement approval policies, which may adversely affect our prospects for generating revenue.
Outside the United States, reimbursement and healthcare payment systems vary significantly by country, and many countries have instituted price ceilings on specific products and therapies. In some countries, particularly EU countriesMember States and EFTA member states,countries, the pricing of prescription pharmaceuticals is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take considerable time (6 to 12 months or longer) after the receipt of marketing approval for a product. ToThe EU provides options for EU Member States to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. An EU Member State may approve a specific price for the medicinal product, it may refuse to reimburse a product at the price set by the manufacturer or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. Many EU Member States also periodically review their reimbursement procedures for medicinal products, which could have an adverse impact on reimbursement status.
Moreover, to obtain reimbursement or pricing approval in some countries, we may be required to conduct a clinical trial that compares the cost effectiveness of our product candidate to other available therapies. This Health Technology Assessment (“HTA”) of medicinal products is becoming an increasingly common part of the pricing and reimbursement procedures in some EU Member States, including those representing the larger markets. The HTA process is the procedure to assess therapeutic, economic and societal impact of a given medicinal product in the national healthcare systems of the individual country. The outcome of an HTA will often influence the pricing and reimbursement status granted to these medicinal products by the competent authorities of individual EU Member States. The extent to which pricing and reimbursement decisions are influenced by the HTA of the specific medicinal product currently varies between EU Member States. In December 2021, Regulation No 2021/2282 on HTA amending Directive 2011/24/EU, was adopted in the EU. This Regulation, which entered into force in January 2022 and will apply as of January 2025, is intended to boost cooperation among EU Member States in assessing health technologies, including new medicinal products, and providing the basis for cooperation at EU level for joint clinical assessments in these areas. The Regulation foresees a three-year transitional period and will permit EU Member States to use common HTA tools, methodologies, and procedures across the EU, working together in four main areas, including joint clinical assessment of the innovative health technologies with the most potential impact for patients, joint scientific consultations whereby developers can seek advice from HTA authorities, identification of emerging health technologies to identify promising technologies early, and continuing voluntary cooperation in other areas. Individual EU Member States will continue to be responsible for assessing non-clinical (e.g., economic, social, ethical) aspects of health technologies, and making decisions on pricing and reimbursement. If we or our partners are unable to maintain favorable pricing and reimbursement status in EU Member States for product candidates that we or our partners may successfully develop and for which we or our partners may obtain regulatory approval, any anticipated revenue from and growth prospects for those products in the EU could be negatively affected.
In addition, certain governmental authorities may conduct reviews of reimbursement previously provided and assert for various reasons that amounts need to be repaid. For example, in October 2021 our distributor/exploitant in France for our previously marketed product Kolbam (which has since been divested) informed us that they had received a notice that the price previously paid for Kolbam during its period on the market in France had been recalculated by the agency responsible for pharmaceutical pricing in France, with such notice asserting amounts owed for repayment. Based on the ongoing review process, we expect that we will need to repay the amounts being asserted prior to pursuing a formal appeal, which we currently estimate to be approximately $5 million. While we cannot currently estimatepredict the likelihoodamount that any of such asserted amount willwe may ultimately need to be repaidrepay, following the currently ongoing review process
and any applicablefuture potential appeal procedures, we may ultimately determine the need to repay all or a portion of the amounts being asserted. Fromproceedings, from 2015 through 2020, the period during which we had sales of Kolbam in France, our aggregate revenues from sales of Kolbam in France attributable to all purchasers/payers were approximately $8 million. If reimbursement of our products is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels or subject to re-assessment and recoupment procedures, our prospects for generating revenue outside of the United States, if any, could be adversely affected and our business may suffer.
We are dependent on Vifor Pharma for the successful commercialization of sparsentan in certain key territories outside of the United States, if approved. We may not be able to establish additional collaborations or other arrangements for sparsentan in other territories, which may adversely impact our ability to generate product revenue in additional jurisdictions.
Pursuant to the terms of the License Agreement, we granted an exclusive license to Vifor Pharma for the commercialization of sparsentan in the Licensed Territories, which consist of Europe, Australia and New Zealand. Consequently, the commercial success of sparsentan in the Licensed Territories will depend in significant part on the efforts of Vifor Pharma, over which we will have limited control. In December 2021, it was announced that CSL Limited, parent company to CSL Behring, intends to initiate a tender offer to acquire Vifor Pharma. We do not currently know what effect, if any, such acquisition, if consummated, will have on our relationship with Vifor Pharma. While our agreement with Vifor Pharma provides that it will remain in place following any acquisition, there is no guarantee that our collaboration with Vifor Pharma will not be affected, adversely or otherwise, by a change in ownership of Vifor Pharma. Moreover, in connection with CSL Limited's acquisition of Vifor Pharma, if consummated, and any related restructuring, substantially less resources could be devoted to the commercialization of sparsentan in the Licensed Territories, or such efforts could be discontinued entirely. If we are unable to establish sales and marketing capabilities or enter into agreements with third parties to market and sell sparsentan, if approved, in additional territories, our ability to generate product revenue outside of the United States and the Licensed Territories may be limited.
We may not be able to rely on orphan drug exclusivity for our products.
Regulatory authorities in some jurisdictions, including the United States and Europe,the EU, may designate drugs for relatively small patient populations as orphan drugs, providing eligibility for orphan drug exclusivity upon regulatory approval if certain jurisdictional-specific conditions are met. For example, Cholbam wasFILSPARI has been granted orphan drug designation infor the United Statestreatment of IgAN and upon FDA approval of the marketing application in March 2015 washas been awarded seven years of orphan drug exclusivity which expires in March 2022.the United States to reduce proteinuria in adults with primary IgAN at risk of rapid disease progression, generally a urinary protein-to-creatinine ratio ("UPCR") ≥1.5 gram/gram. Generally, if a product with an orphan drug designation subsequently receives the first marketing approval for the indication for which it has such designation, that product is entitled to a period of marketing exclusivity, which precludes the applicable regulatory authority from approving another marketing application for the same drug for the same indication for that time period. The applicable period is seven years in the United States and ten years in Europethe EU or, in the case of orphan drugs for which a pediatric investigation plan has been completed, 12 years. Even though we have been awarded orphan drug designation in the United States and Europethe EU for sparsentan for the treatment of IgAN and FSGS and IgAN and for the pegtibatinase for the treatment of HCU, we may not be able to maintain it in Europethe EU and the orphan drug designation may not result in orphan drug exclusivity in the United States for FSGS or Europe upon approval.the EU if approved. For example, if a competitive product that contains the same active moiety and treats the same disease as our product is shown to be clinically superior to our product, any orphan drug exclusivity we have obtained will not block the approval of such competitive product and we may effectively lose orphan drug exclusivity. Similarly, if a competitive product that contains the same active moiety and treats the same disease as our product candidate is approved for orphan drug exclusivity before our product candidate, we may not be able to obtain approval for our product candidate until the expiration of the competitive product’s orphan drug exclusivity unless our product candidate is shown to be clinically superior to the competitive product.
Risks Related to the Development of our Product Candidates
Our clinical trials are expensive and time-consuming and may fail to demonstrate the safety and efficacy of our product candidates.
Before obtaining regulatory approval for the sale of any of our current or future product candidates, we must subject these product candidates to extensive nonclinical and clinical testing to demonstrate their safety and efficacy for humans. Clinical trials are expensive, time-consuming and may take years to complete.
We may experience numerous unforeseen events during, or as a result of, preclinical or nonclinical testing and the clinical trial process that could delay or prevent our ability to obtain, or impact our willingness to pursue, regulatory approval or commercialize our product candidates, including:
•regulators may require us to conduct studies of the long-term effects associated with the use of our product candidates;
•regulators, institutional review boards or ethics committees may not authorize us to commence a clinical trial or conduct a clinical trial at a prospective trial site;
•the FDA or any non-United States regulatory authority may impose conditions on us regarding the scope or design of our clinical trials or may require us to resubmit our clinical trial protocols to institutional review boards or ethics committees for re-inspection due to changes in the regulatory environment;
•the number of patients required for our clinical trials may be larger than we anticipate or participants may drop out of our clinical trials at a higher rate than we anticipate;
•our third-party contractors or clinical investigators may fail to comply with regulatory requirements or fail to meet their contractual obligations to us in a timely manner;
•we might have to suspend, vary or terminate one or more of our clinical trials if we, regulators or institutional review boards or ethics committees determine that the participants are being exposed to unacceptable health risks;
•regulators, institutional review boards or ethics committees may require that we hold, suspend, vary or terminate clinical research for various reasons, including noncompliance with regulatory requirements;
If we are unable to maintain an effective and specialized sales force, we will not be able to commercialize our products in the United States successfully.
In order to successfully commercialize our products in the United States, we have built a specialized sales force. Factors that may hinder our ability to successfully market and commercially distribute our products include:
•inabilitythe cost of sales personnel to obtain access toour clinical trials or educate adequate numbers of physicians on the benefits and safety of prescribing our products;anticipated commercialization costs may be greater than we anticipate;
•inabilitythe supply or quality of our product candidates or other materials necessary to recruit, retain and effectively manage adequate numbers of effective sales personnel;
•lack of complementary products toconduct our clinical trials may be offered by sales personnel, which may put us at a competitive disadvantage relative to companies that haveinsufficient or inadequate, or more extensive product lines; and
•unforeseen delays, costs and expenses associated with maintaining our sales organization.
Ifexpensive than we are unable to maintain our sales force for our products,originally anticipated, or we may not be able to generate sufficientreach agreements on acceptable terms with prospective suppliers or clinical research organizations; and
•the effects of our product revenuecandidates may not be the desired effects or may include undesirable side effects or the product candidates may have other unexpected characteristics.
We will only obtain regulatory approval to commercialize a product candidate if we can demonstrate to the satisfaction of the FDA, and in the United States. Similarly,case of foreign commercialization, to the applicable foreign regulatory authorities, in well-designed and conducted clinical trials, that our product candidates are safe and effective and otherwise meet the appropriate standards required for approval for a particular indication.
Conducting clinical trials effectively in pursuit of regulatory approval requires significant resources, and the costs of conducting clinical trials varies depending on a number of factors, including the dosage of the study therapy, trial size and duration. These costs may prove greater than we originally anticipated, which may result in us choosing to abandon or forgo clinical trials that we deem clinically promising as we actively strategize over time with respect to the allocation of our resources.
Our product development costs will also increase if Vifor doeswe experience delays in testing or approvals. We do not effectively engageknow whether any nonclinical tests or maintain its sales force for sparsentan,clinical trials will be initiated as planned, will need to be restructured or will be completed on schedule, if at all. Significant nonclinical or clinical trial delays also could shorten the patent protection period during which we may have the exclusive right to commercialize our product candidates. In addition, such delays could allow our competitors to bring products to market before we do and impair our ability to recognize milestones payments and royalties from the Licensed Territories will be adversely affected.
We will need to continue to expend significant time and resources to train our sales forces to be credible, persuasive and compliant in discussingcommercialize our products with the specialists treating the patients indicated under the product’s label. In addition,or product candidates.
If we are required to conduct additional clinical trials or other testing of our product candidates beyond those that we currently contemplate, if we are unable to effectively trainsuccessfully complete our sales forceclinical trials or other testing, if the results of these trials or tests are not positive or are only modestly positive or if there are safety concerns, we may:
•be delayed in obtaining, or may not be able to obtain, marketing approval for one or more of our product candidates;
•obtain approval for indications that are not as broad as intended or entirely different than those indications for which we sought approval; and equip them
•have the product removed from the market after obtaining marketing approval.
For example, in our pivotal Phase 3 DUPLEX Study of sparsentan in FSGS, although we achieved the pre-specified interim FSGS partial remission of proteinuria endpoint after 36 weeks of treatment, the study did not achieve the primary efficacy eGFR slope endpoint over 108 weeks of treatment. While we intend to continue to engage with effectivethe FDA to explore a potential path forward for a supplemental New Drug Application (sNDA) in the U.S. and work with our collaborator CSL Vifor to engage with the European Medicines Agency ("EMA") to also explore a potential regulatory path forward in FSGS in the EU based on the DUPLEX data, there is no guarantee that we will be able to establish a pathway to a potential submission of sparsentan for FSGS based on the results from the DUPLEX Study, that the FDA and/or EMA will support an application for sparsentan in FSGS, or that sparsentan will be approved for FSGS.
Also, in our pivotal Phase 3 PROTECT Study of sparsentan in IgAN, although we achieved the pre-specified primary efficacy proteinuria endpoint after 36 weeks of treatment, and after 110 weeks of treatment, FILSPARI demonstrated long-term kidney function preservation and achieved a clinically meaningful difference in estimated glomerular filtration rate (eGFR) total and chronic slope versus irbesartan, the study narrowly missed statistical significance in eGFR total slope while achieving statistical significance in eGFR chronic slope for purposes of regulatory review in the EU. In December 2023, we announced the completion of a successful pre-NDA meeting with the FDA for FILSPARI in IgAN. We plan to submit a supplemental New Drug Application (sNDA) in the first quarter of 2024 for conversion of the existing U.S. accelerated approval of FILSPARI to full approval. However, there is no guarantee that the FDA will accept our sNDA submission for filing, that the FDA's accelerated approval of FILSPARI will continue, or that FILSPARI will receive full approval for IgAN. Furthermore, if the FDA grants full approval for FILSPARI for IgAN, there is no guarantee that the FDA will approve an expanded label.
We may not be able to initiate or continue clinical trials in the rare diseases on which we are focused if we are unable to locate a sufficient number of eligible patients willing and able to participate in the clinical trials required by the FDA or foreign regulatory authorities. In addition, as other companies and researchers may be concurrently developing therapies for the same or similar indications that we are focused on, we could face competition for a limited number of patients, investigators and clinical trial sites willing to participate in clinical trials. Our inability to enroll and maintain a sufficient number of patients for any of our current or future clinical trials would result in significant delays or may require us to abandon one or more clinical trials altogether.
Success in nonclinical testing and early clinical trials does not ensure that later clinical trials will be successful.
Success in nonclinical testing and early clinical trials does not ensure that later clinical trials will be successful.For example, while we saw trends in favor of sparsentan in the two-year confirmatory endpoint analysis in the DUPLEX Study in FSGS, the positive eGFR results from the open-label portion of the DUET study of sparsentan in FSGS were not replicated in the Phase 3 clinical trial with statistical significance. Similarly, while the Phase 3 PROTECT Study of FILSPARI in IgAN demonstrated long-term kidney function preservation in IgAN and met the endpoint for eGFR chronic slope for the purposes of regulatory review in the EU, and all topline efficacy endpoints favored FILSPARI as compared to the active control (irbesartan), the study narrowly missed statistical significance with respect to the eGFR total slope endpoint. Similarly, the positive nonclinical data we have seen from pegtibatinase being tested in a mouse model of homocystinuria and the positive topline results we reported in December 2021 and May 2023 from the ongoing Phase 1/2 clinical trial of pegtibatinase may not be replicated in future studies. We cannot assure that any current or future clinical trials of sparsentan or pegtibatinase will ultimately be successful. Before obtaining regulatory approval to conduct clinical trials of our product candidates, we must conduct extensive nonclinical tests to demonstrate the safety of our product candidates in animals. Nonclinical testing is expensive, difficult to design and implement, and can take many years to complete. In addition, during the clinical development process, additional nonclinical toxicology studies are routinely conducted concurrently with the clinical development of a product candidate. If any of our product candidates show unexpected findings in concurrent toxicology studies, we could experience potentially significant delays in, or be required to abandon, development of that product candidate. A failure of one or more of our nonclinical studies can occur at any stage of testing.
Communications and/or feedback from regulatory authorities related to our current or planned future clinical trials does not guarantee any particular outcome from or timeline for regulatory review, and expedited regulatory review pathways may not actually lead to faster development or approval.
Communications and/or feedback from regulatory authorities, including the FDA or EMA, related to our current or future clinical trials does not guarantee any particular outcome from or timeline for regulatory review for such clinical trials, and expedited regulatory review pathways may not actually lead to faster development or approval.
In 2018 we initiated the Phase 3 DUPLEX Study and the Phase 3 PROTECT Study. We initiated the DUPLEX Study and the PROTECT Study under the Subpart H pathway for potential accelerated approval in the United States, and potential conditional marketing materialsauthorization in the EU, in both jurisdictions based on change in proteinuria. Recognition of change in proteinuria as a surrogate endpoint in kidney disease is a relatively new regulatory development, and, as the field continues to evolve, new learnings may impact regulatory viewpoints.
In February 2023, the FDA granted accelerated approval to FILSPARI (sparsentan) to reduce proteinuria in adults with primary IgAN at risk of rapid disease progression, generally a UPCR ≥1.5 gram/gram. In September 2023, we announced topline two-year confirmatory secondary endpoint results from the PROTECT Study. While FILSPARI demonstrated long-term kidney function preservation and achieved a clinically meaningful difference in estimated glomerular filtration rate (eGFR) total and chronic slope versus irbesartan as well as statistical significance in eGFR chronic slope for purposes of regulatory review in the EU, the PROTECT Study narrowly missed statistical significance in eGFR total slope, which was the pre-specified confirmatory endpoint in the U.S. In December 2023, we announced the completion of a successful pre-NDA meeting with the FDA for FILSPARI in IgAN. We plan to submit a supplemental New Drug Application (sNDA) in the first quarter of 2024 for conversion of the existing U.S. accelerated approval of FILSPARI to full approval. However, there is no guarantee that the FDA will accept our sNDA submission for filing, that the FDA's accelerated approval of FILSPARI will continue, or that FILSPARI will receive full approval for IgAN. In January 2024, we announced that, following submission of the two-year results from the PROTECT Study of FILSPARI in IgAN and a corresponding procedural review clock-stop, we and our collaborator CSL Vifor anticipate an opinion by the Committee for Medicinal Products for Human Use (CHMP) on our conditional marketing authorization (CMA) application for sparsentan for the treatment of IgAN in the EU in the first quarter of 2024. There is no guarantee that sparsentan will be approved for the treatment of IgAN in the EU, or that our timelines will not be delayed notwithstanding the availability of an expedited regulatory review pathway.
In May 2023, we announced that the DUPLEX Study did not achieve its two-year primary endpoint with statistical significance over the active control irbesartan. Although we are encouraged by the topline results for the secondary endpoints on proteinuria and topline exploratory endpoints, including renal outcomes, which trended favorably for sparsentan, and we are continuing to analyze the data to further evaluate the potential for sparsentan as a treatment for FSGS and are engaging with the regulators to explore a potential path to a submission for sparsentan in FSGS, there is no guarantee that we or our collaborator CSL Vifor will be able to establish a pathway to a potential submission of sparsentan for FSGS based on the results from the DUPLEX Study, that the FDA and/or EMA will support an application for sparsentan in FSGS, or that sparsentan will be approved for FSGS.
In December 2023, we initiated the pivotal Phase 3 HARMONY Study to support the potential approval of pegtibatinase for the treatment of classical HCU. The HARMONY Study is a global, randomized, multi-center, double-blind, placebo-controlled Phase 3 clinical trial designed to evaluate the efficacy and safety of pegtibatinase as a novel treatment to reduce total homocysteine (tHcy) levels. Topline results from the HARMONY Study are expected in 2026. Although the FDA has granted Fast Track and Breakthrough Therapy designations to pegtibatinase for the treatment of HCU, there is no guarantee that our pivotal Phase 3 Harmony Study will be successful or that pegtibatinase will be approved for HCU in the future, on an expedited timeline or at all.
Obtaining access to an expedited program (such as Fast Track and Breakthrough Therapy designations) may not in fact lead to faster development timelines or achieve faster review or approval than conventional FDA procedures. We may experience delays in approval timelines attributable to,
among other things, acquiring sufficient supply of our product to conduct clinical trials, identifying and resolving issues relating to chemistry, manufacturing and controls, or conducting additional nonclinical or clinical studies. In addition, the FDA may withdraw access to an expedited program if it believes the access or designation is no longer supported by the data from our program.
Interim, topline and preliminary data from our clinical trials that we announce or publish may change materially as more patient data become available and audit and verification procedures are complete.
From time to time, we may publicly disclose preliminary or topline or interim data from our clinical studies, which is based on a preliminary analysis of then-available data, and the results and related findings and conclusions are subject to change following a more comprehensive review of the data related to the particular study. We also make assumptions, estimations, calculations and conclusions as part of our analyses of data, and we may not have received or had the opportunity to fully and carefully evaluate all data. As a result, the topline results that we report may differ from future results of the same studies, or different conclusions or considerations may qualify such results, once additional data have been received and fully evaluated. Topline data also remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary data we previously published. As a result, topline data should be viewed with caution. From time to time, we may also disclose interim data from our clinical trials. Interim data from clinical trials that we may complete are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment and dosing continues and more patient data become available. Adverse differences between preliminary or interim data and final or confirmatory data could significantly harm our business prospects.
Further, others, including regulatory authorities, may not accept or agree with our assumptions, estimates, calculations, conclusions or analyses or may interpret or weigh the importance of data differently, which could impact the value of the particular program, the approvability or commercialization of the particular product candidate or product and our company in general. In addition, the information we choose to publicly disclose regarding a particular study or clinical trial is based on what is typically extensive information, and you or others may not agree with what we determine is the material or otherwise appropriate information to include in our disclosure, and any information we determine not to disclose may ultimately be deemed significant with respect to future decisions, conclusions, views, activities or otherwise regarding a particular therapy, therapeutic candidate or our business. If the topline data that we report differ from actual results, or if others, including regulatory authorities, disagree with the conclusions reached, our ability to obtain approval for, and commercialize, our product candidates may be harmed, which could harm our business, operating results, prospects or financial condition.
We and/or a collaborative partner are or will be subject to ongoing regulatory obligations and continued regulatory review for our approved products and any product candidates that receive regulatory approval.
The FDA’s accelerated approval of FILSPARI is limited to adults with primary IgAN who are at risk of rapid disease progression, generally a UPCR ≥1.5 gram/gram. The continued approval of FILSPARI may be contingent upon confirmation of a clinical benefit in the Phase 3 PROTECT Study. In September 2023, we announced data from the Phase 3 PROTECT Study as further described herein, including in the risk factor titled “Our future prospects are highly dependent upon our ability to successfully commercializedevelop and execute commercialization strategies for our products, couldincluding FILSPARI, and to attain market acceptance among physicians, patients and healthcare payers.” Any future regulatory approvals that sparsentan or any of our other product candidates receives may be diminished,subject to significant limitations on the approved indicated uses for which the product may be marketed or to the conditions of approval, or contain requirements for potentially costly post-marketing testing, including Phase 4 clinical trials, and surveillance to monitor the safety and efficacy of the product candidate.
In addition, our products, including FILSPARI, and any of our product candidates that are approved by the FDA or a comparable foreign regulatory authority, are or will be subject to extensive and ongoing regulatory requirements, including for the manufacturing, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion, import, export, recordkeeping, conduct of potential post-marketing studies and post-market submission requirements. These requirements include submissions of safety and other post-marketing information and reports, registration, as well as continued compliance with current good manufacturing practices and good clinical practices, for any clinical trials that we conduct post-approval. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, undesirable side effects caused by the product, problems encountered by our third-party manufacturers or manufacturing processes, or failure to comply with regulatory requirements, either before or after product approval, may result in, among other things:
•restrictions on the marketing, manufacturing, or distribution of the product;
•requirements to include additional warnings on the label;
•requirements to create or enhance a medication guide outlining the risks to patients;
•withdrawal of the product from the market;
•voluntary or mandatory product recalls;
•requirements to change the way the product is administered or for us to conduct additional clinical trials;
•fines, warning or untitled letters or holds on clinical trials;
•refusal by the FDA or comparable foreign regulatory authorities to approve pending applications or supplements to approved applications filed by us or our strategic partners, or suspension, variation or revocation of product license approvals;
•product seizure or detention, or refusal to permit the import or export of products;
•injunctions or the imposition of civil or criminal penalties; and
•harm to our reputation.
For example, we have certain post-marketing requirements and commitments associated with FILSPARI. Further, we face risks relating to those post-marketing obligations, as well as the commercial acceptance of FILSPARI. If the regulatory approval for FILSPARI and/or Thiola are withdrawn for any reason, it would have a material adverse effectimpact on our business, resultssales and profitability. Furthermore, if the regulatory approval for Chenodal and/or Cholbam are withdrawn for any reason, it would reduce the chance that we will receive any or all of operationsthe milestone payments from the sale of our bile acid product portfolio in August 2023.
The third-party clinical investigators and financial conditioncontract research organizations that we rely upon to conduct our clinical trials may not be diligent, careful or timely, and may make mistakes, in the conduct of our trials.
We depend on third-party clinical investigators and contract research organizations (“CROs”) to conduct our clinical trials under agreements with us. The CROs play a significant role in the conduct of our clinical trials. Failure of the CROs to meet their obligations could adversely affect clinical development of our product candidates. The third-party clinical investigators are not our employees and we cannot control the timing or amount of resources they devote to our studies. If their performance is substandard, it could delay or prevent approval of our FDA applications. Moreover, these third-party investigators and CROs may also have relationships with other commercial entities, some of which may compete with us. If third-party investigators and CROs allocate their resources to assist our competitors at our expense, it could harm our competitive position. The introduction of new third parties into our ongoing clinical trials increases the risks associated with our dependence on third parties, including the risk that substandard performance by, or competing interests of, such third parties could have a negative impact on our clinical trials.
Risks Related to our Products and Product Candidates
Our products may not achieve or maintain expected levels of market acceptance or commercial success.
The success of our products is dependent upon achieving and maintaining market acceptance. Commercializing products is time consuming, expensive and unpredictable. There can be no assurance that we will be able to, either by ourselves or in collaboration with our partners or through our licensees, successfully commercialize new products or current products or gain market acceptance for such products. New product candidates that appear promising in development may fail to reach the market or may have only limited or no commercial success.
Further, the discovery of significant problems with a product similar to one of our products that implicate (or are perceived to implicate) an entire class of products could have an adverse effect on sales of the affected products. Accordingly, new data about our products, or products similar to our products, could negatively impact demand for our products due to real or perceived side effects or uncertainty regarding efficacy and, in some cases, could result in product withdrawal.
Our current products, including FILSPARI, and any productsproduct candidates that receive marketing approval, that we or a collaboration partner bring to the market including sparsentan and pegtibatinase (TVT-058), if they receive marketing approval, may not gain market acceptance by physicians, patients, third-party payers, and others in the medical community. If these products do not achieve an adequate level of acceptance, we may not generate significant product revenue and we may not become profitable. The degree of market acceptance of our current products and product candidates, if approved for commercial sale, will depend on a number of factors, including:
•the prevalence and severity of any side effects, including any limitations or warnings contained in a product’s approved labeling;
•the efficacy and potential advantages over alternative treatments;
•the pricing of our product candidates;
•the relative convenience and ease of administration;
•the willingness of the target patient population to try new therapies and of physicians to prescribe these therapies;
•the strength of marketing and distribution support and timing of market introduction of competitive products;
•publicity concerning our products or competing products and treatments; and
•sufficient third-party insurance coverage and reimbursement.
the NDA review process for sparsentan for IgAN, the FDA required us to include a REMS and a boxed warning on the label regarding mandatory birth control for patients of child-bearing potential regarding risk of embryo-fetal toxicity, as has been required for other approved endothelin antagonists, and a REMS and boxed warning on the label for liver monitoring regarding potential risk of hepatotoxicity, as has been required for certain other approved endothelin antagonists. As part of the liver monitoring REMS, monthly monitoring of each patient is required for the first year the patient is on treatment, and quarterly thereafter. While we have taken efforts to streamline the REMS with the cadence of typical patient monitoring and have implemented convenience-focused features within the REMS program, the existence of monthly liver monitoring has the potential to be viewed as an impediment to prescribing FILSPARI. Also, while we intend to utilize our continued clinical trial experience with FILSPARI and post-marketing data gathering commitment to potentially support modifying or lifting of the liver monitoring REMS in the future following sufficient experience with FILSPARI and if supported by the data, there is no guarantee that the data will support this endeavor, or even if we believe it does, that the FDA will agree with it. Even if a potential or current product displays a favorable efficacy and safety profile in preclinicalnonclinical and clinical trials, market acceptance of the product will not be known until after it is launched. The efforts by us or any applicable collaboration partner to educate patients, the medical community, and third-party payers on the benefits of our products may require significant resources and may never be successful. Such efforts to educate the marketplace may require more resources than are required by the conventional marketing technologies employed by our competitors.
If theThe market opportunities for our products and product candidates aremay be smaller than we believe they are, our revenues may be adversely affected and our business may suffer.are.
Certain of the diseases that our current and future product candidates are being developed to address, such as IgAN, FSGS and IgAN,HCU, are relatively rare. Our projections of both the number of people who have these diseases, as well as the subset of people with these diseases who have the potential to benefit from treatment with our product candidates, may not be accurate.
Currently, most reported estimates of the prevalence of IgAN, FSGS and HCU are based on studies of small subsets of the population of specific geographic areas, which are then extrapolated to estimate the prevalence of the diseases in the broader world population. As new studies are performed the estimated prevalence of these diseases may change. There can be no assurance that the prevalence of IgAN, FSGS or HCU in the study populations accurately reflect the prevalence of these diseases in the broader world population.
The FDA-approved label of FILSPARI is currently limited to adult patients with IgAN at risk of rapid disease progression, generally a UPCR ≥1.5 gram/gram. Based on our interactions with the FDA, we believe that the FDA has imposed the rapid disease progression limitation on the FILSPARI label because of the accelerated approval pathway under which the product has been approved, and that there may be an opportunity to further expand the label to cover a broader population of IgAN patients based on the confirmatory data from the PROTECT Study, pending favorable regulatory review. However, there can be no guarantee that this will be the case. For additional information, see the risk factor titled “Our future prospects are highly dependent upon our ability to successfully develop and execute commercialization strategies for our products, including FILSPARI, and to attain market acceptance among physicians, patients and healthcare payers.”
If our estimates of the prevalence of IgAN, FSGS or IgANHCU or of the number of patients who may benefit from treatment with sparsentan or pegtibatinase prove to be incorrect or if regulatory approval is conditioned on label restrictions that limit the approved patient population, the market opportunities for our product candidates may be smaller than we believe they are, our prospects for generating revenue may be adversely affected and our business may suffer.
Our product candidates may cause undesirable side effects or have other properties that could delay or prevent their regulatory approval or commercialization.
Undesirable side effects caused by our product candidates could interrupt, delay or halt clinical trials and could result in the denial of regulatory approval by the FDA or other regulatory authorities for any or all targeted indications, and in turn prevent us from commercializing our product candidates and generating revenues from their sale.
Additionally, if sparsentan receives marketing approval, we expect the FDA will require us to include a REMS and mandatory birth control for women of child-bearing age regarding risk of embryo-fetal toxicity, as has been required for other approved endothelin antagonists.
In addition, if any of our product candidates receive marketing approval and we or others later identify undesirable side effects caused by the product:
•regulatory authorities may require the addition of restrictive labeling statements;
•regulatory authorities may withdraw, suspend or vary their approval of the product; and
•we may be required to change the way the product is administered or conduct additional clinical trials.
Any of these events could prevent us from achieving or maintaining market acceptance of the affected product or could substantially increase the costs and expenses of commercializing the product candidate, which in turn could delay or prevent us from generating significant revenues from its sale or adversely affect our reputation.
We do not currently have patent protection for certain of our commercial products. If we are unable to obtain and maintain protection for the intellectual property relating to our technology and products, thetheir value of our technology and products will be adversely affected.
Our success will depend in large part on our ability to obtain and maintain protection in the United States and other countries for the intellectual property covering, or incorporated into, our technology and products. The patent situation in the field of biotechnology and pharmaceuticals generally is highly uncertain and involves complex legal, technical, scientific and factual questions. We do not have, and do not expect to obtain, patent protection for Thiola, Chenodal or Cholbam.the original formulation of Thiola. Additionally, although we have a license to a granted U.S. patent covering the treatment of cystinuria by administering Thiola EC with food (U.S. Patent No. 11,458,104, "the '104 patent”), as well as a pending U.S. patent application directed to Thiola EC, and/or its use for treating cystinuria, we do not know whether thisthe pending U.S. patent application or any future patent applicationsapplication will result in a granted patent covering Thiola EC or its use for treating cystinuria.EC. More generally, we may not be able to obtain additional issued patents relating to our technology or products. Even if issued, patents issued to us or our licensors may be challenged, narrowed, invalidated, held to be unenforceable or circumvented, which could limit our ability to stop competitors from marketing similar products or reduce the term of patent protection we may have for our products. In addition, in certain circumstances with respect to method of use patents, an ANDA applicant may certify that its proposed ANDA label does not contain (or carves out) any language regarding the patented method-of-use rather than certify to a listed method-of-use patent. On January 30, 2024, the FDA approved Torrent Pharmaceuticals Limited’s (Torrent) ANDA for Thiola EC (100mg and 300mg), and accordingly, Thiola EC is now subject to immediate generic competition. Changes in either patent laws or in interpretations of patent laws in the United States and other countries may diminish the value of our intellectual property or narrow the scope of our patent protection.
Patent laws vary by country. Some countries have compulsory licensing laws under which a patent owner may be required to grant licenses to third parties. Some countries do not grant or enforce patents related to medical treatments, or limit enforceability in the case of a public emergency. In addition, many countries limit the enforceability of patents against government agencies or government contractors. If we are unable to obtain or enforce patents related to medical treatments in certain countries, or we or any of our licensors is forced to grant a license to third parties with respect to any patents relevant to our business, our business may be adversely affected.
The intellectual property systems in other countries can be destabilized as a result of political events, during which the ability to obtain, maintain and enforce intellectual property protection in the affected country may be uncertain and evolving. For example, as a result of the ongoing war between Ukraine and Russia, Russian officials have suggested that they may treat patents or patent applications owned by parties from certain countries, including the United States, as unenforceable and/or provide for zero compensation compulsory licenses to such patents or patent applications. Recent court decisions in Russia have raised questions about the strength of trademark protections in Russia. The U.S. government’s response to political events may also negatively affect our ability to obtain, maintain and enforce intellectual property protection in the affected country. For example, the U.S. government has issued sanctions against Russia related to the ongoing war in Ukraine, and as a result of these sanctions, it may not be possible to pay fees necessary for prosecution and maintenance of Russian patent applications and patents in the absence of licenses or exclusions set forth by the U.S. government authorizing transactions in connection with intellectual property. Payments for trademark protection may be similarly impacted. The U.S. Department of the Treasury has issued General License No. 31, authorizing such transactions to allow filing, prosecution and maintenance of Russian patents and trademarks. Uncertainties regarding political events, including the ongoing war between Ukraine and Russia, as well as any resulting losses of intellectual property protection, could harm our business.
Our product candidate sparsentanFILSPARI is covered by U.S. Patent No. 6,638,937, which expired in 2019 and to which we have an exclusive license. In addition, U.S. Patent No. 9,662,312, to which we also have an exclusive license and which was granted on May 30, 2017 and expires in 2030, covers the use of sparsentan for treating glomerulosclerosis, including FSGS. And U.S. Patent No. 9,993,461, to which we also have an exclusive
license and which was granted on June 12, 2018 and expires in 2030, covers the use of sparsentan for treating IgA nephropathyIgAN as well as glomerulosclerosis, including FSGS.
For products we develop based on a new chemical entity not previously approved by the FDA, we expect that in addition to the protection afforded by our patent filings that we will be able to obtain five years regulatory exclusivity via the provisions of the Food, Drug, and Cosmetic Act ("FDC"FDC Act") Act and possibly seven years regulatory exclusivity via the orphan drug provisions of the FDC Act. In the case of sparsentan, the periods of regulatory exclusivity may, if certain conditions are satisfied, be extended by six months on the basis of pediatric exclusivity, thereby resulting in exclusivity periods of 5.5 years and 7.5 years, respectively. In addition, we may be able to obtain up to five years patent term extension (to compensate for regulatory approval delay) for one patent covering such a product for its FDA-approved use. Such a patent, like the periods of regulatory exclusivity, also may be extended by a further six months on the basis of pediatric exclusivity if certain conditions are satisfied.
The degree of future protection for our proprietary rights is uncertain, and we cannot ensure that:
•we or our licensors were the first to make the inventions covered by each of our pending patent applications;
•we or our licensors were the first to file patent applications for these inventions;
•others will not independently develop similar or alternative technologies or duplicate any of our technologies;
•any patents issued to us or our licensors that provide a basis for commercially viable products will provide us with any competitive advantages or will not be challenged by third parties;
•we will develop additional proprietary technologies that are patentable;
•we will file patent applications for new proprietary technologies promptly or at all;
•the claims we make in our patents will be upheld by patent offices in the United States and elsewhere;
•our patents will not expire prior to or shortly after commencing commercialization of a product; and
•the patents of others will not have a negative effect on our ability to do business.
We have negotiated a license agreement with Ligand Pharmaceuticals for the rights to sparsentan which we are initially developing for the treatment of FSGSIgAN and IgAN.FSGS. This license subjects us to various commercialization, reporting and other obligations. If we were to default on our obligations, we and CSL Vifor Pharma could lose our rights to sparsentan. We have obtained a U.S. patent and European patent each covering the use of sparsentan for treating glomerulosclerosis, including FSGS, as well as a second U.S. patent and a second European patent each covering both the use of sparsentan for treating IgAN and the use of sparsentan for treating glomerulosclerosis, including FSGS. In addition, in 2020 we obtained a U.S. patent covering the use of sparsentan for the treatment of Alport syndrome. However, we cannot be certain that we will be able to obtain patent protection for various other potential indications for sparsentan, or whether, if granted, we would be able to enforce such patents. Additionally, in November 2020, a third party filed an opposition to our second European patent (European Patent No. EP3222277, “the ‘277 EP Patent”), in the European Patent Office ("EPO"). While we are vigorously defending the ‘277 EP Patent against the opposition, there is no guarantee that we will be successful in doing so.
Our patents also may not afford us protection against competitors with similar technology. Because patent applications in the United States and many other jurisdictions are typically not published until 18 months after filing, or in some cases not at all, and because publications of discoveries in the scientific literature often lag behind the actual discoveries, neither we nor our licensors can be certain that we or they were the first to make the inventions claimed in our or their issued patents or pending patent applications, or that we or they were the first to file for protection of the inventions set forth in these patent applications. If a third party has also filed a United States patent application prior to the effective date of the relevant provisions of the America Invents Act (i.e. before March 16, 2013) covering our product candidates or a similar invention, we may have to participate in an adversarial proceeding, known as an interference, declared by the USPTO to determine priority of invention in the United States. The costs of these proceedings could be substantial, and it is possible that our efforts could be unsuccessful, resulting in a loss of our United States patent position.
We cannot assure you that third parties will not assert patent or other intellectual property infringement claims against us with respect to technologies used in our products. If patent infringement suits were brought against us, we may be unable to commercialize some of our products which could severely harm our business. Litigation proceedings, even if not successful, could result in substantial costs and harm our business.
We expect to rely on orphan drug status to develop and commercialize certain of our products and product candidates, but our orphan drug designations may not confer marketing exclusivity or other expected commercial benefits.
We expect to rely on orphan drug exclusivity for sparsentan and potential future product candidates that we may develop. Orphan drug status currently confers seven years of marketing exclusivity in the United States under the FDC Act, and up to ten years of marketing exclusivity in Europethe EU for a particular product in a specified indication or, in the case of orphan drugs for which a pediatric investigation plan has been completed, 12 years. The FDA and EMAEuropean Commission have granted orphan designation for Chenodal, sparsentan and pegtibatinase (TVT-058) for the treatment of CTX, FSGS, IgAN and homocystinuria, respectively.FSGS, and pegtibatinase for the treatment of homocystinuria. While we have been granted these orphan designations, we will not be able to rely on these designations to exclude other companies from manufacturing or selling these molecules for the same indication beyond these time frames. Furthermore, in Europe,the EU, orphan drug status is re-evaluated in connection with the marketing authorization review process and a product candidate must re-qualify as of such time in order to maintain orphan drug status. In addition, anystatus and benefit from the potential regulatory exclusivity periods related to marketing authorizations granted to orphan products. The period of market exclusivity in Europe canthe EU may, however, be reduced from ten years to six years if, at the initial designation criteria have significantly changed since the market authorizationend of the fifth year, it is established that the product no longer meets the criteria on the basis of which it received orphan medicinal product destination, including where it can be demonstrated on the basis of available evidence that the original orphan medicinal product is sufficiently profitable not to justify maintenance of market exclusivity or where the prevalence of the condition has increased above the threshold. Additionally, a marketing authorization may be granted to a similar medicinal product with the same orphan indication during the 10 year period if: (i) the applicant consents to a second original orphan medicinal product application, (ii) the manufacturer of the original orphan medicinal product is unable to supply sufficient quantities; or (iii) the second applicant can establish that its product, although similar, is safer, more effective or otherwise clinically superior to the original orphan medicinal product. A company may voluntarily remove a product from the register of orphan products.
For any product candidate for which we have been granted orphan drug designation in a particular indication, it is possible that another company also holding orphan drug designation for the same product candidate will receive marketing approval for the same indication before we do. If that were to happen, our applications for that indication may not be approved until the competing company's period of exclusivity expires. Even if we are the first to obtain marketing authorization for an orphan drug indication in the United States, there are circumstances under which a competing product may be approved for the same indication during the seven-year period of marketing exclusivity, such as if the later product is shown to be clinically superior to our orphan product, or if the later product is deemed a different product than ours. Further, the seven-year marketing exclusivity
would not prevent competitors from obtaining approval of the same product candidate as ours for indications other than those in which we have been granted orphan drug designation, or for the use of other types of products in the same indications as our orphan product.
Any drugstherapies we develop may become subject to unfavorable pricing regulations, third-party reimbursement practices or healthcare reform initiatives, thereby harming our business.
In March 2010, President Obama signed the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010 (collectively, the "PPACA"), a sweeping law intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for healthcare and health insurance industries, impose new taxes and fees on the health industry and impose additional health policy reforms. The PPACA revised the definition of “average manufacturer price” for reporting purposes, which could increase the amount of Medicaid drug rebates to states. The PPACA also increased the mandated Medicaid rebate from 15.1% to 23.1% of the average manufacturer price, expanded the rebate to Medicaid managed care utilization and increased the types of entities eligible for the federal 340B drug discount program. Further, the law imposed a significant annual fee on companies that manufacture or import certain branded prescription drug products. There have been executive, judicial, Congressional, and political challenges to certain aspects of the PPACA. For example, President Trump signed several Executive Orders and other directives designed to delay the implementation of certain provisions of the PPACA or otherwise circumvent some of the requirements for health insurance mandated by the PPACA. Concurrently, Congress considered legislation to repeal or repeal and replace all or part of the PPACA. While Congress has not passed comprehensive repeal legislation, several bills affecting the implementation of certain taxes under the PPACA have been signed into law. For example, legislation enacted in 2017 informally titled the Tax Cuts and Jobs Act ("Tax Act") includes a provision which repealed, effective January 1, 2019, the tax-based shared responsibility payment imposed by the PPACA on certain individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate”. In addition, the 2020 federal spending package eliminated, effective January 1, 2020, the PPACA-mandated “Cadillac” tax on high-cost employer-sponsored health coverage and medical device tax and, effective January 1, 2021, also eliminated the health insurer tax. The Bipartisan Budget Act of 2018 ("BBA"), among other things, amended the PPACA, effective January 1, 2019, to increase the discount that is owed by pharmaceutical manufacturers who participate in Medicare Part D from 50 percent to 70 percent, and closed the coverage gap in most Medicare drug plans, commonly referred to as the “donut hole”. On June 17, 2021 the U.S. Supreme Court dismissed a challenge on procedural grounds that argued the PPACA is unconstitutional in its entirety because the “individual mandate” was repealed by Congress. Thus, the PPACA will remain in effect in its current form. Further, prior to the U.S. Supreme Court ruling, on January 28, 2021,August 16, 2022, President Biden issued an executive order that initiated a special enrollment periodsigned the Inflation Reduction Act of 2022 ("IRA") into law, which among other things, extends enhanced subsidies for purposes of obtainingindividuals purchasing health insurance coverage in PPACA marketplaces through plan year 2025. The IRA also eliminates the PPACA marketplace. The executive order also instructed certain governmental agencies to review"donut hole" under the Medicare Part D program beginning in 2025 by significantly lowering the beneficiary maximum out-of-pocket cost and reconsider their existing policies and rules that limit access to healthcare, including among others, reexamining Medicaid demonstration projects and waiver programs that include work requirements, and policies that create unnecessary barriers to obtaining access to health insurance coverage through Medicaid or the PPACA.a newly established manufacturer discount program. It is possible that the PPACA will be subject to judicial or Congressional challenges in the future. It is unclear how any such challenges and theadditional healthcare reform measures of the Biden administration will impact the PPACA and our business.
In addition, other legislative changes have been proposed and adopted since the PPACA was enacted. For example, in August 2011, the President Obama signed into law the Budget Control Act of 2011, which, among other things, created the Joint Select Committee on Deficit Reduction to recommend to Congress proposals in spending reductions. The Joint Select Committee on Deficit Reduction did not achieve a targeted deficit reduction of at least $1.2 trillion for fiscal years 2012 through 2021, triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions to Medicare payments to providers of up to 2% per fiscal year, which went into effect beginning on April 1, 2013 and, due to subsequent legislative amendments, including the BBA and the Infrastructure Investment and Jobs Act, will stay in effect through 2031until 2032 unless additional Congressional action is taken. The COVID-19 relief support legislation suspended the 2% Medicare sequester from May 1, 2020 through March 31, 2022. Under current legislation, the actual reduction in Medicare payments will vary from 1% in 2022 to up to 3% in the final fiscal year of
this sequester. Additionally, in January 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, reduced Medicare payments to several providers, including hospitals and imaging centers.
If we are unable to obtain and maintain coverage and adequate reimbursement from governments or third-party payers for any products that we may develop or if we are unable to obtain acceptable prices for those products, our prospects for generating revenue and achieving profitability will suffer.
Our prospects for generating revenue and achieving profitability will depend heavily upon the availability of coverage and adequate reimbursement for the use of our approved product candidates from governmental and other third-party payers, both in the United States and in other markets. Reimbursement by a third-party payer may depend upon a number of factors, including the third-party payer’s determination that use of a product is:
•a covered benefit under its health plan;
•safe, effective and medically necessary;
•appropriate for the specific patient;
•cost-effective; and
•neither experimental nor investigational.
Obtaining reimbursement approval for a product from each government or other third-party payer is a time consuming and costly process that could require us to provide supporting scientific, clinical and cost effectiveness data for the use of our products to each payer. We may not be able to provide data sufficient to gain acceptance with respect to reimbursement. Additionally, we might need to conduct post-marketing studies in order to demonstrate the cost-effectiveness of any future products to such payers’ satisfaction. Such studies might require us to commit a significant amount of management time and financial and other resources. Even when a payer determines that a product is eligible for reimbursement, the payer may impose coverage limitations that preclude payment for some uses that are approved by the FDA or non-United States regulatory authorities. Also, prior authorization for a product may be required. In addition, there is a risk that full reimbursement may not be available for high-priced products. Moreover, eligibility for coverage does not imply that any product will be reimbursed in all cases or at a rate that allows us to make a profit or even cover our costs. Interim payments for new products, if applicable, may also not be sufficient to cover our costs and may not be made permanent. Further, coverage policies and third-party payer reimbursement rates may change at any time. Even if favorable coverage and reimbursement status is attained, less favorable coverage policies and reimbursement rates may be implemented in the future.
A primary trend in the United States healthcare industry and elsewhere is toward cost containment. We expect the changes made by PPACA, other legislation impacting the Medicare program and the 340B program, and the increasing emphasis on managed care to continue to put pressure on
pharmaceutical product pricing. As these concerns continue to grow over the need for tighter oversight, there remains the possibility that the Heath Resources and Services Administration or another agency under the U.S. Department of Health and Human Services ("HHS") will propose regulations or that Congress will explore changes to the 340B program through legislation. For example, on November 30, 2018, the U.S. Health Resources & Services Administration published its final rule regarding the calculation of 340B ceiling price and imposition of civil monetary penalties on manufacturers for knowingly and intentionally overcharging covered entities, which became effective on January 1, 2019. Pursuant to the final rule, after January 1, 2019, manufacturers must calculate 340B program ceiling prices on a quarterly basis. Moreover, manufacturers could be subject to a $5,000 penalty for each instance where they knowingly and intentionally overcharge a covered entity under the 340B program.
There have also been a number of initiatives pending at the state and federal level that could negatively impact the reimbursement for products approved under the accelerated approval pathway in the United States.States by restricting patient access or establishing differential payment models. Certain states are also in the process of establishing Patient Drug Affordability Boards with the authority in some cases to set upper payment limits.
Further, there has been increasing legislative and enforcement interest in the United States with respect to drug pricing practices, including several recent U.S. congressional inquiries and federal and state legislation designed to, among other things, increase drug pricing transparency, expedite generic competition, review relationships between pricing and manufacturer patient assistance programs, and reform government program drug reimbursement methodologies. At the federal level, the Trump administration used several means to propose or implement drug pricing reform, including through federal budget proposals, executive orders and policy initiatives. For example, on July 24, 2020 and September 13, 2020, President Trump announced several executive orders related to prescription drug pricing that attempt to implement several of the Trump administration proposals. The FDA concurrently released a final rule and guidance in September 2020, implementing a portion of the importation executive order providing pathways for states to build and submit importation plans for drugs from Canada. Further, on November 20, 2020, HHS finalized a regulation removing safe harbor protection for price reductions from pharmaceutical manufacturers to plan sponsors under Part D, either directly or through pharmacy benefit managers, unless the price reduction is required by law. The implementation of the rule has been delayed by the Biden administration from January 1, 2022 to January 1, 2023 in response to ongoing litigation. The rule also creates a new safe harbor for price reductions reflected at the point-of-sale, as well as a new safe harbor for certain fixed fee arrangements between pharmacy benefit managers and manufacturers, the implementation of which have also been delayed until January 1, 2023. On November 20, 2020, the Centers for Medicare & Medicaid Services ("CMS") issued an interim final rule implementing President Trump’s Most Favored Nation ("MFN") executive order, which would tie Medicare Part B payments for certain physician-administered drugs to the lowest price paid in other economically advanced countries, effective January 1, 2021. As a result of litigation challenging the MFN model, on December 27, 2021, CMS published a final rule that rescinded the MFN
model interim final rule. Additionally, on March 11, 2021, President Biden signed the American Rescue Plan Act of 2021 into law, which eliminates the statutory Medicaid drug rebate cap, currently set at 100% of a drug’s average manufacturer price, for single source and innovator multiple source drugs, beginning January 1, 2024. Further, in July 2021, the Biden administration released an executive order that included multiple provisions aimed at prescription drugs. In response to Biden’s executive order, on September 9, 2021, HHS released a Comprehensive Plan for Addressing High Drug Prices that outlines principles for drug pricing reform. The plan sets out a variety of potential legislative policies that Congress could pursue as well as potential administrative actions HHS can take to advance these principles. No legislation or administrative actions have been finalized to implement these principles. In addition, Congressthe IRA, among other things (i) directs HHS to negotiate the price of certain high-expenditure, single-source drugs and biologics covered under Medicare and (ii) imposes rebates under Medicare Part B and Medicare Part D to penalize price increases that outpace inflation. These provisions take effect progressively starting in fiscal year 2023. On August 29, 2023, HHS announced the list of the first ten drugs that will be subject to price negotiations, although the Medicare drug price negotiation program is considering drug pricingcurrently subject to legal challenges. HHS has and will continue to issue and update guidance as partthese programs are implemented. It is currently unclear how the IRA will be implemented but it is likely to have a significant impact on the pharmaceutical industry. In addition, in response to the Biden administration’s October 2022 executive order, on February 14, 2023, HHS released a report outlining three new models for testing by the Center for Medicare and Medicaid Innovation which will be evaluated on their ability to lower the cost of other reform measures.drugs, promote accessibility, and improve quality of care. It is unclear whether these or similar policy initiativesthe models will be implementedutilized in any health reform measures in the future. Further, on December 7, 2023, the Biden administration announced an initiative to control the price of prescription drugs through the use of march-in rights under the Bayh-Dole Act. On December 8, 2023, the National Institute of Standards and Technology published for comment a Draft Interagency Guidance Framework for Considering the Exercise of March-In Rights which for the first time includes the price of a product as one factor an agency can use when deciding to exercise march-in rights. While march-in rights have not previously been exercised, it is uncertain if that will continue under the new framework. At the state level, legislatures are increasingly passing legislation and implementing regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. Also, thereFor example, on January 5, 2024, the FDA approved Florida’s Section 804 Importation Program (SIP) proposal to import certain drugs from Canada for specific state healthcare programs. It is unclear how this program will be implemented, including which drugs will be chosen, and whether it will be subject to legal challenges in the United States or Canada. Other states have been reportsalso submitted SIP proposals that are pending review by the U.S. government is considering targeted price controls and reference pricing based on foreign single-payer country access policies, which, ifFDA. Any such approved importation plans, when implemented, could adversely affect our revenues.may result in lower drug prices for products covered by those programs.
Any reduction in reimbursement from Medicare, Medicaid or other government-funded programs may result in a similar reduction in payments from private payers. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability or commercialize our drugs.therapies. Additionally, we are currently unable to predict what additional legislation or regulation, if any, relating to the healthcare industry may be enacted in the future or what effect recently enacted federal legislation or any such additional legislation or regulation would have on our business.
It is also possible that additional governmental action is taken in response to the COVID-19 pandemic.
We face potential product liability exposure far in excess of our limited insurance coverage.
The use of any of our potential products in clinical trials, and the sale of any approved products, may expose us to liability claims. These claims might be made directly by consumers, health care providers, pharmaceutical companies or others selling our products. We have obtained limited product liability insurance coverage for our clinical trials in the amount of $10 million per occurrence and $25$30 million in the aggregate. However, our insurance may not reimburse us or may not be sufficient to reimburse us for any expenses or losses we may suffer. Moreover, insurance coverage is becoming increasingly expensive, and we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses due to liability. We intend to expand our insurance coverage to include the sale of commercial products ifas we obtain marketing approval for additional product candidates in development, but we may be unable to obtain commercially reasonable product liability insurance for any products approved for marketing.insurance. On occasion, juries have awarded large judgments in class action lawsuits based on drugs that had unanticipated side effects. A successful product liability claim or series of claims brought against us would decrease our cash reserves and could cause our stock price to fall.
We face substantial competition, which may result in others discovering, developing or commercializing products before or more successfully than we do. Our operating results will suffer if we fail to compete effectively.
Several of our competitors have substantially greater financial, research and development, distribution, manufacturing and marketing experience and resources than we do and represent substantial long-term competition for us. Other companies may succeed in developing and marketing products that are more effective and/or less costly than any products that may be developed and marketed by us, or that are commercially accepted before any of our products. Factors affecting competition in the pharmaceutical and drugtherapeutic industries vary, depending on the extent to which a competitor is able to achieve a competitive advantage based on its proprietary technology and ability to market and sell drugs.therapeutics. The industry in which we compete is characterized by extensive research and development efforts and rapid technological progress. Although we believe that our
orphan drug status and proprietary position with respect to sparsentan may give us a competitive advantage, new developments are expected to continue and there can be no assurance that discoveries by others will not render such potentialour products and product candidates noncompetitive. Furthermore, competitors could enter the market with generic versions of our products. For example, a generic option for the 100 mg version of the original formulation of TholaThiola (tiopronin tablets) was approved by the FDA in May 2021.2021 and a second 100 mg version of the original formulation of Thiola (tiopronin tablets) was approved by the FDA in June 2022. Also, as of February 1, 2024, three generic options for the 100 mg and 300 mg versions of Thiola EC have been approved by the FDA.
Our competitive position also depends on our ability to enter into strategic alliances with one or more large pharmaceutical and contract manufacturing companies, attract and retain qualified personnel, develop effective proprietary products, implement development and marketing plans, obtain patent protection, secure adequate capital resources and successfully sell and market our approved products. There can be no assurance that we will be able to successfully achieve all of the foregoing objectives.
Use of third parties to manufacture our products and product candidates may increase the risk that we will not have sufficient quantities of our product and product candidates or such quantities at an acceptable cost, and clinical development and commercialization of our product and product candidates could be delayed, prevented or impaired.
We do not own or operate manufacturing facilities for clinical or commercial production of our products or product candidates. We have limited personnel with experience in drug manufacturing and we lack the resources and the capabilities to manufacture any of our product candidates on a clinical or commercial scale. We outsource all manufacturing and packaging of our preclinical,nonclinical, clinical, and commercial products to third parties. The manufacture of pharmaceutical products requires significant expertise and capital investment, including the development of advanced manufacturing
techniques and process controls. Manufacturers of pharmaceutical products often encounter difficulties in production, particularly in scaling up initial production and in maintaining required quality control. These problems include difficulties with production costs and yields and quality control, including stability of the product candidate.
We intend to rely on third-party manufacturers for the long-term commercial supply of FILSPARI and for our development stage product candidates, including sparsentan and pegtibatinase (TVT-058).candidates. We expect the manufacturers of each product or product candidate to, at least initially and potentially for a significant period of time, be single source suppliers to us. Reliance on third-party manufacturers entails risks to which we may not be subject if we manufactured our product candidates or products ourselves, including:
•reliance on the third party for regulatory compliance and quality assurance;
•limitations on supply availability resulting from capacity and scheduling constraints of the third parties;
•less control over cost increases resulting from inflationary pressures affecting raw materials and other supply chain components;
•impact on our reputation in the marketplace if manufacturers of our products fail to meet the demands of our customers;
•the possible breach of the manufacturing agreement by the third party because of factors beyond our control; and
•the possible termination or nonrenewal of the agreement by the third party, based on its own business priorities, at a time that is costly or inconvenient for us.
The failure of any of our contract manufacturers to maintain high manufacturing standards could result in injury or death of clinical trial participants or patients using our products. Such failure could also result in product liability claims, product recalls, product seizures or withdrawals, delays or failures in testing or delivery, cost overruns or other problems that could seriously harm our business or profitability.
Our contract manufacturers will beare required to adhere to FDA regulations setting forth cGMP.cGMP and comparable foreign regulatory authority requirements. These regulations cover all aspects of the manufacturing, testing, quality control and recordkeeping relating to our product candidates and any products that we may commercialize. Our manufacturers may not be able to comply with cGMP regulations or similar regulatory requirements outside the United States. Our manufacturers are subject to unannounced inspections by the FDA, state regulators and similar regulators outside the United States.States to monitor and ensure compliance with cGMP. We are ultimately responsible for ensuring that our API and finished products are manufactured in accordance with cGMP regulations and similar regulatory requirements outside the United States, and it is therefore critical that we maintain effective management practices and oversight with respect to our third-party manufacturers, including routine auditing. Our failure, or the failure of our third-party manufacturers, to comply with applicable regulations could result in sanctions being imposed on us, including shutdown of the third-party vendor or invalidation of drug product lots or processes, fines, injunctions, civil penalties, failure of regulatory authorities to grant marketing approval of our product candidates, delays, suspension, variation or withdrawal of approvals, license revocation, seizures or recalls of product candidates or products, operating restrictions and criminal prosecutions, any of which could significantly and adversely affect regulatory approval and supplies of our product candidates.
Our product and any products that we may develop may compete with other product candidates and products for access to manufacturing facilities. There are a limited number of manufacturers that operate under cGMP regulations and that are both capable of manufacturing for us and willing to do so. The ongoing COVID-19A health epidemic or pandemic and associated vaccine or treatment development and manufacturing efforts have increasedmay increase demand for the services supplied by many third partythird-party manufacturers, including some of those that we utilize for our products and product candidates, and there has recently been, andwhich may continue to be,result in decreased availability of manufacturing slots at many such facilities. If the third parties that we engage to manufacture products for our developmental or commercial products should halt or cease to continue to do so for any reason, we likely would experience interruptions in cash flows and/or delays in advancing our clinical trials while we identify and qualify replacement suppliers, and we may be unable to obtain replacement supplies on terms that are favorable to us. Later relocation to another manufacturer will also require notification, review and other regulatory approvals from the FDA and other regulators and will subject our production to further cost and instability in the availability of our product candidates. In addition, if we are not able to obtain adequate supplies of our products and product candidates, or the drug substances used to manufacture them, it will be more difficult for us to sell our products and to develop our product candidates. This could greatly reduce our competitiveness. On March 27, 2020, President Trump signed into law the Coronavirus Aid, Relief,competitiveness and Economic Security Act (the "CARES Act") in response to the COVID-19 pandemic. Throughout the COVID-19 pandemic, there has been public concern over the availability and accessibility of critical medical products, and the Act enhances FDA’s existing authority with respect to drug shortage measures. Under the Act, manufacturers must have in place a risk management plan that identifies and evaluates the risks to the supply of approved drugs for certain serious diseases or conditions for each establishment where the drug or active pharmaceutical ingredient is manufactured. The risk management plan will be subject to FDA review during an inspection. If we experience shortages in the supply of our marketed products,negatively affect our results could be materially impacted.of operations.
Our current and anticipated future dependence upon others for the manufacture of our products and product candidates may adversely affect our future profit margins and our ability to develop product candidates and commercialize our marketed products and any other products that may obtain regulatory approval on a timely and competitive basis.
Materials necessary to manufacture our products and product candidates may not be available on commercially reasonable terms, or at all, which may delay the development and commercialization of our products and product candidates.
We rely on the manufacturers of our products and product candidates to purchase from third-party suppliers the materials necessary to produce the compounds for our preclinicalnonclinical and clinical studies and rely on these other manufacturers for commercial distribution if we obtain marketing approval for any of our product candidates. Suppliers may not sell these materials to our manufacturers at the time we need them or on commercially reasonable terms and all such prices are susceptible to fluctuations in price and availability due to transportation costs, government regulations, price
controls, and changes in economic climate or other foreseen circumstances. We do not have any control over the process or timing of the acquisition of these materials by our manufacturers. In addition, inflation and/orand global supply chain disruptions, as well as past disruptions related to COVID-19 and potential future disruptions related to a future health epidemic or pandemic, wars, armed conflicts, and global geopolitical tension, including between the U.S. and China, have had and may continue to have a negative impact on our manufacturers’ ability to acquire the materials necessary for our business. Moreover, we currently do not have any agreements for the commercial production of these materials. If our manufacturers are unable to obtain these materials for our preclinicalnonclinical and clinical studies, product testing and potential regulatory approval of our product candidates would be delayed, significantly impacting our ability to develop our product candidates. If our manufacturers or we are unable to purchase these materials after regulatory approval has been obtained for our product candidates, the commercial launch of our product candidates would be delayed or there would be a shortage in supply, which would materially affect our ability to generate revenues from the sale of our product candidates. For example, in 2021 a membrane used in pegtibatinase (TVT-058) drug substance manufacturing became more difficult to acquire due to the same or similar membranes being used in certain of the COVID-19 vaccine manufacturing and we continue to see challenges with securing materials used in the pegtibatinase manufacturing process that are in short supply as a result of the pandemic.processes. While we believe our contingency plans will enableenabled us to continueinitiate the ongoing clinicalPhase 3 study of pegtibatinase (TVT-058) with the currently available clinical supplies,on our desired timeline, there is no guarantee that we will not continue to face additionalsupply challenges or shortages of this membrane, or other materials necessary to manufacture pegtibatinase (TVT-058) or our other products and product candidates. If our risk mitigation plans are not successful in overcoming these challenges, our pegtibatinase program or other products and product candidates, could be delayed.
Risks Related to Our Business
The COVID-19 pandemic could materially adversely affect our business, results of operations and financial condition.
The ongoing COVID-19 pandemic continues to impact domestic and worldwide economic activity, including global supply and financial markets. The COVID-19 pandemic also poses the risk that we or our clinical trial subjects, employees, contractors, collaborators, suppliers and vendors may be prevented from conducting certain clinical trials or other business activities for an indefinite period of time, including due to travel restrictions, quarantines, “stay-at-home” and "shelter-in-place" orders or shutdowns that have been or may be requested or mandated by governmental authorities, or that our or their ability to conduct operations will be negatively impacted by staffing shortages while employees quarantine as a result of exposure to or transmission of the virus. In addition, the COVID-19 pandemic could impact personnel at third-party manufacturing facilities in the United States and other countries, including China, or the availability or cost of materials, which could potentially disrupt the supply chain for our commercial products, our product candidates or the comparator products in our ongoing clinical trials.
The timelines and conduct of our ongoing clinical trials may be affected by the COVID-19 pandemic. For example, in 2020 we experienced a reduction in the rates of patient enrollment in our ongoing clinical trials as a result of the pandemic. Clinical site initiation and patient enrollment may be further delayed due to prioritization of hospital resources toward the COVID-19 pandemic and patients’ ability or willingness to participate in clinical trials. For those patients who are enrolled and desire to continue in the clinical trials, some patients may not be able or willing to comply with clinical trial protocols if quarantines or governmental orders impede patient movement or interrupt healthcare services. Similarly, we may face increased challenges with the ability to recruit and retain patients and principal investigators and site staff who, as healthcare providers, may have heightened exposure to COVID-19, which could adversely impact our clinical trial operations, timelines and outcomes. In addition, we rely on independent clinical investigators, contract research organizations (CROs) and other third-party service providers to assist us in managing, monitoring and otherwise carrying out our clinical trials, and the pandemic may affect their ability to devote sufficient time and resources to our programs or to travel to sites to perform work for us. While we remain in close contact with our CROs, clinical sites and suppliers to attempt to assess the impacts that COVID-19 may have on our clinical trials and projected timelines and we continue to implement appropriate mitigating measures in accordance with recent FDA guidance in an effort to ensure the ongoing safety of the patients in our clinical trials and the continued collection of high quality data, there is no guarantee that such efforts will be successful. As challenging as conducting clinical trials is during normal times, the risks, operational challenges and costs of conducting clinical trials has increased substantially during the pandemic. In addition, restrictions caused by the ongoing COVID-19 pandemic may result in impediments to obtaining biopsies, which could impact the ability to timely obtain diagnoses of IgAN or FSGS.
Beginning in March 2020, substantially all of our workforce began working remotely either all or substantially all of the time as a result of applicable stay-at-home and shelter-in-place orders. The effects of these orders and our related remote-work policies may negatively impact productivity, disrupt our business and delay our development programs, regulatory and commercialization timelines, the magnitude of which will depend, in part, on the length and severity of the restrictions and other limitations on our ability to conduct our business in the ordinary course. In addition, as the applicable orders have recently begun to be lifted and certain of our employees begin to return to the office, we cannot guarantee that our workforce will not face an outbreak that could adversely impact our operations.
While the long-term economic impact and the duration of the COVID-19 pandemic may be difficult to predict, the widespread pandemic has resulted in, and may continue to result in, significant disruption of global financial markets, which could reduce our ability to access capital and could negatively affect our liquidity and the liquidity and stability of markets for our common stock and convertible notes. In addition, a further market correction, recession or depression resulting from the spread of COVID-19 could materially adversely affect our business and the value of our common stock and convertible notes.
Moreover, the COVID-19 pandemic continues to evolve, and the extent to which the COVID-19 pandemic may impact our business, results of operations and financial position will depend on future developments, which are highly uncertain and cannot be predicted with confidence, such as the ultimate geographic spread of the disease, the duration of the pandemic, travel restrictions and social distancing in the United States and other
countries, business closures or business disruptions, global supply challenges, and the effectiveness of actions taken in the United States and other countries to contain and treat the disease.
Our limited operating history makes it difficult to evaluate our future prospects, and our profitability in the future is uncertain.
We face the problems, expenses, difficulties, complications and delays, many of which are beyond our control, associated with any business in its early stages and have a limited operating history on which an evaluation of our prospects can be made. Such prospects should be considered in light of the risks, expenses and difficulties frequently encountered in the establishment of a business in a new industry, characterized by a number of market entrants and intense competition, and in the shift from development to commercialization of new products based on innovative technologies.
We have experienced significant growth over the past five years in the number of our employees and the scope of our operations. We have expanded our sales and marketing, compliance and legal functions in addition to expansion of all functions to support a commercial organization. We have also expanded our operations in connection with the commercial launch of FILSPARI in the United States, including by adding additional members to our sales force. To appropriately manage for our anticipated future, growth, we must continue to implement and improve our managerial, operational and financial systems, expand our facilities and continue to recruit, train and train additionalretain qualified personnel.personnel as needed, and successfully integrate any changes into our existing business. To succeed, we must recruit, retain, manage and motivate qualified clinical, scientific, technical, commercial and management personnel, and we face significant competition for experienced personnel.
Due to our limited resources, we may not be able to effectively manage the expansion of our operations or recruit, train and train additionalretain qualified personnel.personnel, including in connection with the ongoing commercial launch of FILSPARI in the United States. The physical expansionmanagement of changes to our operations may lead to significant costs and may divert our management and business development resources. Any inability on the part of our management to manage growth or other changes in our organization could delay the execution of our business plans or disrupt our operations.
Factors that may inhibit our efforts to commercialize our products without strategic partners or licensees include:
•our inability to recruit and retain adequate numbers of effective sales and marketing personnel;
•the inability of sales personnel to obtain access to or educate adequate numbers of physicians to prescribe our products;
•the lack of complementary products to be offered by our sales personnel, which may put us at a competitive disadvantage against companies with broader product lines;
•unforeseen costs associated with expanding our own sales and marketing team for new products or with entering into a partnering agreement with an independent sales and marketing organization; and
•efforts by our competitors to commercialize competitive products.
Moreover, though we generate revenues from product sales arrangements, we may incur significant operating losses over the next several years. Our ability to achieve profitable operations in the future will depend in large part upon successful in-licensing of products approved by the FDA, selling and manufacturing these products, completing development of our products, obtaining regulatory approvals for these products, and bringing these products to market. The likelihood of the long-term success of our company must be considered in light of the expenses, difficulties and delays frequently encountered in the development and commercialization of new drug products,therapeutics, competitive factors in the marketplace, as well as the regulatory environment in which we operate.
In addition, we may encounter unforeseen expenses, difficulties, complications, delays and other known and unknown factors.
We depend on a highly experienced and skilled workforce to grow and operate our business. If we are unable to attract, retain and engage our employees, we may not be able to grow effectively.
The execution of our strategic objectives and future success will depend upon our continued ability to identify, hire, develop, motivate and retain a highly qualified workforce. We depend on contributions from our employees, and, in particular, our senior management team, to execute efficiently and effectively. Our success further depends on our ability to attract, retain and motivate highly skilled mid-level and senior managers as well as team members at various levels in the scientific, development, medical and commercial areas of the business.business, particularly in connection with our ongoing commercial launch of FILSPARI in the United States.
Our headquarters are based in San Diego, California. This region is home to many other biopharmaceutical companies and many academic and research institutions. Competition for qualified key talent in our market is intense and may limit our ability to hire and retain employees on acceptable terms, or at all. As a result, we may not be able to retain our existing employees or hire new employees quickly enough to meet our needs.
To induce valuable employees to remain at our company, in addition to salary, cash incentives and other employee benefits, we have provided stock options and restricted stock unit ("RSU") awards that vest over time. The value to employees of stock options and RSU awards that vest over time may be significantly affected by movements in our stock price that are beyond our control and may at any time be insufficient to counteract more lucrative offers from other companies. Current market conditions and the potential for extreme stock price volatility exacerbates this risk. Despite our efforts to retain valuable employees, members of our management, scientific, development and commercial teams may terminate their employment with us on short notice. All of our employees have at-will employment, which means that they
could leave our employment at any time, with or without notice. We do not maintain “key person” insurance policies on the lives of any of our employees.
If we fail to effectively manage our hiring and retention needs, our ability to meet our strategic objectives and our business and operating results may be adversely impacted.
Health epidemics or pandemics could materially adversely affect our business, results of operations and financial condition.
A health epidemic or pandemic poses the risk that we or our clinical trial subjects, employees, contractors, collaborators, suppliers and vendors may be prevented from conducting certain clinical trials or other business activities for an indefinite period of time, including due to travel restrictions, quarantines, “stay-at-home” and "shelter-in-place" orders or shutdowns that have been or may be requested or mandated by governmental authorities, or that our or their ability to conduct operations will be negatively impacted by staffing shortages while employees quarantine as a result of exposure to or transmission of the virus. In addition, a health epidemic or pandemic could impact personnel at third-party manufacturing facilities in the United States and other countries, including China, or the availability or cost of materials, which could potentially disrupt the supply chain for our commercial products, our product candidates or the comparator products in our ongoing clinical trials.
The timelines and conduct of our ongoing clinical trials previously have been affected by COVID-19 and we may experience similar delays or interruptions due to other health epidemics or pandemics in the future. For example, in 2020 we experienced a reduction in the rates of patient
enrollment in our ongoing clinical trials as a result of the COVID-19 pandemic. New health epidemics or pandemics may emerge that result in similar or more severe disruptions to our business, which could adversely impact our business and operating results.
Our strategic reorganization and the associated workforce reduction may not result in the level of savings that we currently anticipate, could result in costs and expenses that are greater than expected, and could disrupt our business.
In December 2023, we announced a strategic reorganization including an approximate 20% workforce reduction focused on non-field-based employees in an effort to align our resources on the ongoing FILSPARI launch and the pivotal Phase 3 HARMONY Study to support the potential approval of pegtibatinase as the first potential disease-modifying treatment for HCU. There is a chance that we will not realize the level of savings and improvements in our cost structure that we currently anticipate, and there may be unforeseen difficulties, delays or unexpected costs. If we are unable to realize the expected operational efficiencies and cost savings, our operating results and financial condition would be adversely affected. Furthermore, our strategic reorganization may be disruptive to our operations. For example, our workforce reductions could yield unanticipated consequences, such as increased difficulties in implementing our business strategy, including retention of our remaining employees. Employee litigation related to the headcount reduction could be costly and prevent management from fully concentrating on the business.
We will likely experience fluctuations in operating results and could incur substantial losses.
We expect that our operating results will vary significantly from quarter-to-quarter and year-to-year as a result of investments in research and development, specifically our clinical and preclinicalnonclinical development activities. We have not completed development of any drugs and we anticipate that certain of our expenses will continue to increase, substantially as we:
•continuedepending on factors including but not limited to: the open label portioncontinuation and cost of DUETour clinical trials and conduct the Phase 3 trials of sparsentan;
•continue the research and development of additional product candidates,candidates; the costs involved in seeking and obtaining marketing approvals for our products, and in maintaining quality systems standards for our products; the timing of, and costs involved in, commercial activities, including pegtibatinase (TVT-058);
•expand ourproduct marketing, sales and marketing infrastructuredistribution, costs related to commercialize our current products and any new products for which we may obtain regulatory approval; and
•expand operational, financial, and management information systems and personnel, including personnel to support product development efforts and our obligations as a public company.
Furthermore, the extent of the ultimate impact of the COVID- 19 pandemic on our operational and financial performance will depend on various developments, including the duration and spread of the pandemic, and its impact on potential customers, employees, and vendors, all of which cannot be reasonably predicted at this time.
To attain and sustain profitability, we must succeed in developing and commercializing drugstherapies with significant market potential. This will require us to be successful in a range of challenging activities, including the discovery of product candidates, successful completion of preclinicalnonclinical testing and clinical trials of our product candidates, obtaining regulatory approval for these product candidates and manufacturing, marketing and selling those products for which we may obtain regulatory approval. We are only in the preliminary stages of these activities. We may not be successful enough in these activities to generate revenues that are substantial enough to recoup the expenses we have expended in conducting these activities to achieve profitability. Pursuant to the Ligand License Agreement, we are obligated to pay to Ligand an escalating annual royalty between 15% and 17% of net sales of FILSPARI and any other products containing sparsentan or related compounds, which will impact our potential future profit from the commercialization of FILSPARI in the United States and sparsentan for the treatment of IgAN in the EU, if approved, as well as sparsentan for the treatment of FSGS, if approved. Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our failure to become or remain profitable could depress the market price of our common stock and could impair our ability to raise capital, expand our business, diversify our product offerings or continue our operations. A decline in the market price of our common stock may also cause a loss of a part or all of your investment.
Negative publicity regarding any of our products could impair our ability to market any such product and may require us to spend time and money to address these issues.
If any of our products or any similar products distributed by other companies prove to be, or are asserted to be, harmful to consumers and/or subject to FDA or comparable foreign regulatory authority enforcement action, our ability to successfully market and sell our products could be impaired. Because of our dependence on patient and physician perceptions, any adverse publicity associated with illness or other adverse effects resulting from the use or misuse of our products or any similar products distributed by other companies could limit the commercial potential of our products and expose us to potential liabilities.
We may not have sufficient insurance to cover our liability in any current or future litigation claims either due to coverage limits or as a result of insurance carriers seeking to deny coverage of such claims.
We face a variety of litigation-related liability risks. Our certificate of incorporation, bylaws, other applicable agreements, and/or Delaware law require us to indemnify (and advance expenses to) our current and past directors and officers and employees from reasonable expenses related to the defense of any action arising from their service to us, including circumstances under which indemnification is otherwise discretionary. While our directors and officers are included in a director and officer liability insurance policy, which covers all our directors and officers in some circumstances, our insurance coverage does not cover all of our indemnification obligations and may not be adequate to cover any indemnification or other claims against us. In addition, the underwriters of our present coverage may seek to avoid coverage in certain circumstances based upon the terms of the respective policies. If we incur liabilities that exceed our coverage under our directors and officers insurance policy or incur liabilities not covered by our insurance, we would have to self-fund any indemnification amounts owed to our directors and officers and employees in which case our results of operations and financial condition could be materially adversely affected. Further, if D&O insurance becomes prohibitively expensive to maintain in the future, we may be unable to renew such insurance on economic terms or unable to renew such insurance at all. The potential lack of D&O
insurance may make it difficult for us to retain and attract talented and skilled directors and officers to serve our company, which could adversely affect our business.
We may need substantial funding and may be unable to raise capital when needed, which would force us to delay, reduce or eliminate our product development programs or commercialization efforts.needed.
We expect our general and research and development expenses to increase in connection with our ongoing and planned activities, particularly as we conduct Phase 3 clinical trials of sparsentan, and conduct any other later-stage clinical trials of our product candidates. In addition, subject to obtaining regulatory approvalin connection with the commercial launch of any of our product candidates,FILSPARI in the United States, we expecthave begun to incur significant commercialization expenses and expect to continue to incur significant commercialization expenses for FILSPARI and any other future approved products, including for product sales and marketing, securing commercial quantities of product from our manufacturers, and product distribution. Our expenses have and may continue to increase as a result of increasing inflation in the United States and abroad. We currently have no additional commitments or arrangements for any additional financing to fund the research and development and commercial launch of our product candidates. General market conditions, resulting from theincluding high interest rates and stock price volatility, actual or anticipated bank failures, and ongoing issues arising fromglobal geopolitical tensions, including the COVID-19 pandemic,wars and other armed conflicts, as well as market conditions affecting companies in the life sciences industry in general, may make it difficult for us to seek financing from the capital markets on attractive terms, or at all.
Management believes our ability to continue our operations depends on our ability to sustain and grow revenue, results of operations and our ability to access capital markets when necessary to accomplish our strategic objectives. Management believes that we may incur losses in the immediate future. We expect that our operating results will vary significantly from quarter-to-quarter and year-to-year as a result of investments in research and development, specifically our clinical and preclinicalnonclinical development activities. We expect to finance our cash needs from cash on hand and results of operations, and depending on results of operations we may either need additional equity or debt financing, or need to enter into strategic alliances on products in development to continue our operations until we can achieve sustained profitability and positive cash flows from operating activities. Additional funds may not be available to us when we need them on terms that are acceptable to us, or at all. If adequate funds are not available to us on a timely basis, we may be required to reduce or eliminate research development programs or commercial efforts.
Our future capital requirements will depend on many factors, including:
•the timing, progress, cost and results of our pre-clinicalclinical trials, preclinical studies and clinical studiesother discovery and research and development activities;
•the timing of, sparsentanand costs involved in, seeking and obtaining marketing approvals for FSGSour products, and in maintaining quality systems standards for our products;
•the timing of, and costs involved in, commercial activities, including product marketing, sales and distribution;
•our ability to successfully commercialize FILSPARI in adult patients with IgAN, pegtibatinase (TVT-058)and to obtain regulatory approval for HCU, Chenodal for CTX, and anysuccessfully commercialize our other drugor future product candidates;
•the costs, timing and outcome of regulatory review ofincreases or decreases in revenue from our product candidates;marketed products, including decreases resulting from generic entrants or health epidemics or pandemics;
•debt service obligations on the 2025 Notes and 2029 Notes;
•the number and development requirements of other product candidates that we pursue;
•the costsour ability to manufacture sufficient quantities of commercialization activities, including product marketing, sales and distribution;
•the emergence of competing technologies and other adverse market developments;our products to meet expected demand;
•the costs of preparing, filing and prosecuting patent applications and maintaining, enforcing and defending intellectual property related claims;
•our ability to enter into collaboration, licensing or distribution arrangements and the terms and timing of these arrangements;
•the potential need to expand our business, resulting in additional payroll and other overhead expenses;
•the potential in-licensing of other products or technologies;
•the emergence of competing products and technologies and other adverse market developments;
•the extent to which we acquire or invest in businesses, products and technologies; and
•our ability to establish collaborationsthe potential impacts of inflation and obtain milestone, royalty or other payments from any such collaborators.resulting cost increases.
The market price for shares of our common stock may be volatile and purchasers of our common stock could incur substantial losses.
The price of our stock is likely to be volatile. The stock market in general, and the market for biotechnology companies in particular, have experienced extreme volatility that has often been unrelated to the operating performance of particular companies. The market price for our common stock has been in the past, and may be in the future, influenced by many factors, including:
•results of clinical trials of our product candidates or those of our competitors;
•our entry into or the loss of a significant collaboration;
•regulatory or legal developments in the United States and other countries, including changes in the health care payment systems;
•our ability to obtain and maintain marketing approvals from the FDA or similar regulatory authorities outside the United States;
•variations in our financial results or those of companies that are perceived to be similar to us;
•changes in the structure of healthcare payment systems;
•market conditions in the pharmaceutical and biotechnology sectors and issuance of new or changed securities analysts’ reports or recommendations;
•general economic, industry and market conditions;conditions, including the impacts thereon of inflation and high interest rates, actual or anticipated bank failures, wars, armed conflicts and global geopolitical tensions;
•results of clinical trials conducted by others on drugstherapies that would compete with our product candidates;
•developments or disputes concerning patents or other proprietary rights;
•public concern over our product candidates or any products approved in the future;
•litigation;
•communications from government officials regarding health care costs or pharmaceutical pricing;
•future sales or anticipated sales of our common stock by us or our stockholders; and
•the other factors described in this “Risk Factors” section.
In addition, the stock markets, and in particular, the Nasdaq GlobalStock Market, have experienced extreme price and volume fluctuations that have affected and continue to affect the market prices of equity securities of many pharmaceutical companies. The realization of any of the above risks or any of a broad range of other risks, including those described in these “Risk Factors” could have a dramatic and material adverse impact on the market price of our common stock.
We might not receive some or all of the potential milestone payments from the sale of our bile acid product portfolio for the treatment of rare liver diseases.
On July 16, 2023, we entered into a definitive asset purchase agreement (the “Purchase Agreement”) with Mirum Pharmaceuticals, Inc. (“Mirum”), pursuant to which we agreed to sell to Mirum, subject to the terms of the Purchase Agreement, our bile acid product portfolio including Chenodal and Cholbam (also known as Kolbam) (the “Products”). The closing of the transaction occurred on August 31, 2023. A portion of the consideration for the sale is in the form of milestone payments that will only be payable upon the achievement of certain milestones based on specified amounts of annual net sales of the Products (the “Milestone Events”). There is a risk that any or all of the Milestone Events might not be achieved and that any or all of the consideration tied to the achievement of the Milestone Events might not be received.
We may be unable to successfully integrate new products or businesses we may acquire.
We intend tomay in the future expand our product pipeline by pursuing acquisition of pharmaceutical products. If an acquisition is consummated, the integration of the acquired business, product or other assets into our company may also be complex and time- consumingtime-consuming and, if such businesses, products and assets are not successfully integrated, we may not achieve the anticipated benefits, cost-savings or growth opportunities. Potential difficulties that may be encountered in the integration process include the following:
•integrating personnel, operations and systems, while maintaining focus on producing and delivering consistent, high quality products;
•coordinating geographically dispersed organizations;
•distracting employees from operations;
•retaining existing customers and attracting new customers; and
•managing inefficiencies associated with integrating the operations of the Company.acquired company or product into our own operations.
Furthermore, these acquisitions and other arrangements, even if successfully integrated, may fail to further our business strategy as anticipated, expose us to increased competition or challenges with respect to our products or geographic markets, and expose us to additional liabilities associated with an acquired business, product, technology or other asset or arrangement. Any one of these challenges or risks could impair our ability to realize any benefit from our acquisitions or arrangements after we have expended resources on them.
Product liability lawsuits against us could cause us to incur substantial liabilities and to limit commercialization of any products that we may develop.
Our business exposes us to potential liability risks inherent in the research, development, manufacturing and marketing of pharmaceutical products. If any of our product candidates in clinical trials or commercialized products harm people, we may be subject to costly and damaging product liability claims. We have clinical trial insurance and commercial product liability coverage. However, this insurance may not be adequate to cover all claims. We may be exposed to product liability claims and product recalls, including those which may arise from misuse or malfunction of, or design flaws in, such products, whether or not such problems directly relate to the products and services we have provided. If we cannot successfully defend ourselves against claims that our product candidates or products caused injuries, we will incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
•decreased demand for any product candidates or products that we may develop;
•damage to our reputation;
•regulatory investigations that could require costly recalls or product modifications;
•withdrawal of clinical trial participants;
•costs to defend the related litigation;
•substantial monetary awards to trial participants or patients, including awards that substantially exceed our product liability insurance, which we would then be required to pay from other sources, if available, and would damage our ability to obtain liability insurance at reasonable costs, or at all, in the future;
•loss of revenue;
•the diversion of management’s attention from managing our business; and
•the inability to commercialize any products that we may develop.
A successful product liability claim or a series of claims brought against us could cause our stock price to fall and, if judgments exceed our insurance coverage, could decrease our available cash and adversely affect our business.
We may become involved in certain litigation matters, any of which could result in substantial costs, divert management's attention and otherwise have a material adverse effect on our business, operating results or financial condition.
From time to time we may become involved in certain litigation matters, including those described in Note 11 of the Consolidated Financial Statements included in this report. Although we intend to vigorously defend our interests in each matter, there is no guarantee that we will be successful and we may have to pay damages awards or otherwise may enter into settlement arrangements in connection with such matters. Any such payments or settlement arrangements could have material adverse effects on our business, operating results or financial condition. Even if we are successful in defending our interests in each matter, litigation with respect to such matters could result in substantial costs and significant adverse impact on our reputation and divert management's attention and resources, which could have a material adverse effect on our business, operating results or financial condition.
We are subject to significant ongoing regulatory obligations and oversight, which may result in significant additional expense and may limit our commercial success.
We are subject to significant ongoing regulatory obligations, such as safety reporting requirements and additional post-marketing obligations, including regulatory oversight of the promotion and marketing of our products. In addition, the manufacture, quality control, labeling, packaging, safety surveillance, adverse event reporting, storage and recordkeeping for our products are subject to extensive and ongoing regulatory requirements. If we become aware of previously unknown problems with any of our products, a regulatory agencyauthority may impose restrictions on our products, our contract manufacturers or us. If we, our products and product candidates, or the manufacturing facilities for our products and product candidates fail to comply with applicable regulatory requirements, a regulatory agency,authority, including the FDA, may send enforcement letters, mandate labeling changes, suspend, vary or withdraw regulatory approval, suspend, vary or terminate any ongoing clinical trials, refuse to approve pending applications or supplements filed by us, suspend or impose restrictions on manufacturing operations, request a recall of, seize or detain a product, seek criminal prosecution or an injunction, or impose civil or criminal penalties or monetary fines. In such instances, we could experience a significant drop in the sales of the affected products, our product revenues and reputation in the marketplace may suffer, and we could become the target of lawsuits.
We are also subject to regulation by supranational, national, regional, state and local agencies and regulatory authorities, including but not limited to the FDA, CMS,the Centers for Medicare & Medicaid Services ("CMS"), Department of Justice, the Federal Trade Commission, the HHS Office of Inspector General and other regulatory bodies. The FDC Act, Social Security Act, Public Health Service Act and other federal and state statutes and regulations, and comparable foreign regulatory acts, govern to varying degrees the research, development, manufacturing and commercial activities relating to prescription pharmaceutical products, including preclinicalnonclinical testing, clinical research, approval, production, labeling, sale, distribution, post-market surveillance, advertising, dissemination of information, promotion, marketing, and pricing to government purchasers and government health care programs. Our manufacturing partners are subject to many of the same requirements.
Companies may not promote drugs for “off-label” uses-thatuses—that is, uses that are not described in the product’s labeling and that differ from those approved by the FDA or other applicable regulatory agencies.authorities. However, a company may share truthful and not misleading information that is otherwise consistent with the product’s labeling. A company that is found to have improperly promoted off-label uses may be subject to significant liability, including civil and administrative remedies as well as criminal sanctions. In addition, management’s attention could be diverted from our business operations and our reputation could be damaged.
The federal health care program Anti-Kickback statuteStatute prohibits, among other things, knowingly and willfully offering, paying, soliciting, or receiving remuneration to induce or in return for purchasing, leasing, ordering or arranging for the purchase, lease or order of any health care item or service reimbursable under Medicare, Medicaid or other federally financed healthcare programs. This statute has been interpreted broadly to apply to
arrangements that pharmaceutical companies have with prescribers, purchasers and formulary managers, among others. Further, the PPACA, among other things, amends the intent requirement of the federal anti-kickback statuteAnti-Kickback Statute so that a person or entity no longer needs to have actual knowledge of this statute or specific intent to violate it. In addition, the PPACA provides that the government may assert that a claim including items or services resulting from a violation of the federal anti-kickback statuteAnti-Kickback Statute constitutes a false or fraudulent claim for purposes of the civil False Claims Act. Although there are a number of statutory exceptions and regulatory safe harbors under the federal anti-kickback statuteAnti-Kickback Statute protecting certain common manufacturer business arrangements and activities from prosecution, the exceptions and safe harbors are drawn narrowly and an arrangement must meet all of the conditions specified in order to be fully protected from scrutiny under the federal anti-kickback statute.Anti-Kickback Statute. We seek to comply with the exceptions and safe harbors whenever possible, but our practices, such as our patient assistance programs and prompt pay discounts with certain customers, may not in all cases meet all of the criteria for protection from anti-kickbackAnti-Kickback liability and may be subject to scrutiny.
The federal false claims laws, including the federal False Claims Act, prohibit any person or entity from knowingly presenting, or causing to be presented, a false claim for payment to the federal government, or knowingly making, or causing to be made, a false statement to get a false claim paid. Additionally, the civil monetary penalties statute imposes penalties against any person or entity that, among other things, is determined to have presented or caused to be presented a claim to a federal health program that the person knows or should know is for an item or service that was not provided as claimed or is false or fraudulent. Many pharmaceutical and other health care companies have been investigated and have reached substantial financial settlements with the federal government under the federal False Claims Act for a variety of alleged marketing activities, including providing free product to customers with the expectation that the customers would bill federal programs for the product; providing consulting fees, grants, free travel, and other benefits to physicians to induce them to prescribe the company’s products; and inflating prices reported to private price publication services, which may be used by states to set drug payment rates under government health care programs. Companies have been prosecuted for causing false claims to be submitted because of the marketing of their products for unapproved uses. Pharmaceutical and other health care companies have also been prosecuted on other legal theories of Medicare and Medicaid fraud.
Legislative and regulatory proposals have been made to expand post-approval requirements and restrict sales and promotional activities for pharmaceutical products. It is not clear whether additional legislative changes will be enacted, or whether the FDA regulations, guidance or interpretations will be changed, or what the impact of such changes on the marketing approvals of any Travere products, if any, may be. In addition, increased scrutiny by the U.S. Congress of the FDA’s approval process may significantly delay or prevent marketing approval, as well as subject us to more stringent product labeling and post-marketing testing and other requirements.
We also could become subject to government investigations and related subpoenas. Such subpoenas are often associated with previously filed qui tam actions, or lawsuits filed under seal under the federal False Claims Act. Qui tam actions are brought by private plaintiffs suing on behalf of the federal government for alleged violations of the federal False Claims Act. The time and expense associated with responding to such subpoenas, and any related qui tam or other actions, may be extensive, and we cannot predict the results of our review of the responsive documents and underlying facts or the results of such actions. Responding to government investigations, defending any claims raised, and any resulting fines, restitution, damages and penalties, settlement payments or administrative actions, as well as any related actions brought by stockholders or other third parties, could have a material impact on our reputation, business and financial condition and divert the attention of our management from operating our business.
The number and complexity of both federal and state laws continues to increase, and additional governmental resources are being added to enforce these laws and to prosecute companies and individuals who are believed to be violating them. In particular, the PPACA includes a number of provisions aimed at strengthening the government’s ability to pursue anti-kickbackAnti-Kickback and false claimsFalse Claims Act cases against pharmaceutical manufacturers and other healthcare entities, including substantially increased funding for healthcare fraud enforcement activities, enhanced investigative powers, amendments to the federal False Claims Act that make it easier for the government and whistleblowers to pursue cases for alleged kickback and false claim violations and public reporting of certain payments and transfers of value by certain pharmaceutical manufacturers to physicians and teaching hospitals nationwide. We anticipate that government scrutiny of pharmaceutical sales and marketing practices will continue for the foreseeable future and subject us to the risk of further government investigations and enforcement actions. Responding to a government investigation or enforcement action would be expensive and time-consuming, and could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
In February 2021, we entered into a limited co-promotion arrangement with Albireo Pharma, Inc. ("Albireo"), providing for our Cholbam dedicated sales representatives to dedicate a portion of their efforts to promoting Albireo's product, Bylvay (odevixibat), in the United States following approval. In July 2021, Albireo announced that the U.S. Food & Drug Administration ("FDA") has approved Bylvay (odevixibat) for the treatment of pruritis in patients with Progressive Familial Intrahepatic Cholestasis ("PFIC"). In addition to our activities in connection with promoting our own products, if our or Albireo's sales representatives violate or are perceived to have violated any applicable regulatory requirement in promoting Bylvay (odevixibat), we could become subject to investigations, litigation, and/or penalties as described above, reputational harm, as well as contractual liabilities associated with the Albireo co-promotion agreement, any of which could have a material adverse effect on our business.
The U.S. Foreign Corrupt Practices Act, and similar worldwide anti-bribery laws generally prohibit companies and their intermediaries from making improper payments to government officials for the purpose of obtaining or retaining business. Our policies mandate compliance with these anti-bribery laws. We operate in parts of the world that have experienced governmental corruption to some degree and in certain circumstances, strict compliance with anti-bribery laws may conflict with local customs and practices or may require us to interact with doctors and hospitals, some of which may be state controlled, in a manner that is different than in the United States. We cannot assure that our internal control policies and
procedures will protect us from reckless or criminal acts committed by our employees or agents. Violations of these laws, or allegations of such violations, could disrupt our business and result in criminal or civil penalties or remedial measures, any of which could have a material adverse effect on our business, financial condition and results of operations and could cause the market value of our common stock to decline.
The federal Health Insurance Portability and Accountability Act of 1996 ("HIPAA"), created new federal criminal statutes that prohibit, among other actions, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payers, and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Like the federal anti-kickback statute,Anti-Kickback Statute, the PPACA amended the intent standard for certain healthcare fraud provisions under HIPAA such that a person or entity no longer needs to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
Additionally, the federal Physician Payments Sunshine Act within the PPACA, and its implementing regulations, require that certain manufacturers of drugs, devices, biologicals and medical supplies to report annually information related to certain payments or other transfers of value made or distributed to physicians (defined to include doctors, dentists, optometrists, podiatrists, and chiropractors), other healthcare professionals (such as physician assistants and nurse practitioners), and teaching hospitals, or to entities or individuals at the request of, or designated on behalf of, physicians and teaching hospitals and to report annually certain ownership and investment interests held by physicians and their immediate family members.
Moreover, the Drug Supply Chain Security Act imposes obligations on manufacturers of pharmaceutical products related to product tracking and tracing. We are not sure whether additional legislative changes will be enacted, or whether the current regulations, guidance or interpretations will be changed, or what the impact of such changes on our business, if any, may be.
Also, many states have similar fraud and abuse statutes or regulations, including state anti-kickback and false claims laws, that apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payer. Further, certain states require implementation of commercial compliance programs and marketing codes, compliance with the pharmaceutical industry’s voluntary compliance guidelines, and compliance with the applicable compliance guidance promulgated by the federal government. Other various state level requirements include restricting payments or the provision of other items of value that may be made to healthcare providers and other potential referral sources; restricting various marketing practices; requiring prescription drug companies to report expenses relating to the marketing and promotion of drug products; requiring the posting of information relating to clinical studies and their outcomes; requiring the registration of sales representatives; requiring the reporting of certain information related to drug pricing; and requiring drug manufacturers to track and report information related to payments, gifts, compensation, and other items of value to physicians and other healthcare providers.
If our operations are found to be in violation of any of the health regulatory laws described above or any other laws that apply to us, we may be subject to significant penalties, including imprisonment, criminal fines, civil monetary penalties, administrative penalties, disgorgement, exclusion from participation in federal healthcare programs, contractual damages, injunctions, recall or seizure of products, total or partial suspension of production, reputational harm, administrative burdens, additional oversight and reporting obligations if we become subject to a corporate integrity
agreement or similar agreement to resolve allegation of non-compliance with these laws, diminished profits and future earnings, and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations.
We are also subject to foreign requirements comparable to those established above. Outside the United States, interactions between pharmaceutical companies and health care professionals are also governed by strict laws, such as national anti-bribery laws of European countries, national sunshine rules, regulations, industry self-regulation codes of conduct and physicians’ codes of professional conduct. Failure to comply with these requirements could result in reputational risk, public reprimands, administrative penalties, fines or imprisonment.
If we are not able to obtain and maintain required regulatory approvals, we will not be able to commercialize our products, and our ability to generate revenue will be materially impaired.
Our product candidates, once approved, and the activities associated with their manufacture, marketing, distribution, and sales are subject to extensive regulation by the FDA and other regulatory agencies in the United States and by comparable authorities in other countries. Failure to adhere to regulations set out by these bodies for one or more of our commercial products could prevent us from commercializing the product candidate in the jurisdiction of the regulatory authority. We have only limited experience in meeting the regulatory requirements incumbent on the sale of drugs in the United States and elsewhere, and expect to rely on third-partiesthird parties to assist us in these processes. If these third parties fail to adequately adhere to the regulations governing drug distribution and promotion, we may be unable to sell our products, which could have a material effect on our ability to generate revenue.
Our product candidates and the activities associated with their development and commercialization, including testing, manufacture, safety, efficacy, recordkeeping, labeling, storage, approval, advertising, promotion, sale and distribution, are subject to comprehensive regulation by the FDA and other regulatory agencies in the United States and by comparable authorities in other countries. Failure to obtain regulatory approval for a product candidate will prevent us from commercializing the product candidate in the jurisdiction of the regulatory authority. We have only limited experience in filing and prosecuting the applications necessary to obtain regulatory approvals and expect to rely on third-party contract research organizations to assist us in this process.
Securing FDA approval requires the submission of extensive preclinicalnonclinical and clinical data and supporting information to the FDA for each therapeutic indication to establish the product candidate’s safety and efficacy. Securing FDA approval also requires the submission of information about the product manufacturing process to, and successful inspection of manufacturing facilities by, the FDA. Our future products may not be effective, may be only moderately effective or may prove to have undesirable or unintended side effects, toxicities or other characteristics that may preclude our obtaining regulatory approval or prevent or limit commercial use. Comparable requirements are applicable outside the United States.
Our product candidates may fail to obtain regulatory approval for many reasons, including:
•our failure to demonstrate to the satisfaction of the FDA or comparable regulatory authorities that a product candidate is safe and effective for a particular indication;
•the results of clinical trials may not meet the level of statistical significance required by the FDA or comparable regulatory authorities for approval;
•our inability to demonstrate that a product candidate’s benefits outweigh its risks;
•our inability to demonstrate that the product candidate presents an advantage over existing therapies;
•the FDA’s or comparable regulatory authorities’ disagreement with the manner in which we interpret the data from preclinicalnonclinical studies or clinical trials;
•failure of the third-party manufacturers with which we contract for clinical or commercial supplies to satisfactorily complete an FDA or comparable foreign regulatory authority pre-approval inspection of the facility or facilities at which the product is manufactured to assess compliance with the FDA’s cGMP regulations or comparable foreign regulatory authority requirements to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity; and
•a change in the approval policies or regulations of the FDA or comparable regulatory authorities or a change in the laws governing the approval process.
The process of obtaining regulatory approvals is expensive, often takes many years, if approval is obtained at all, and can vary substantially based upon a variety of factors, including the type, complexity and novelty of the product candidates involved. Changes in regulatory approval policies during the development period, changes in or the enactment of additional statutes or regulations, or changes in regulatory review for each submitted product application may cause delays in the approval or rejection of an application. The FDA and non-United States regulatory authorities have substantial discretion in the approval process and may refuse to accept any application or may decide that our data is insufficient for approval and
require additional preclinical,nonclinical, clinical or other studies. In addition, varying interpretations of the data obtained from preclinicalnonclinical and clinical testing could delay, limit or prevent regulatory approval of a product candidate. Any regulatory approval we ultimately obtain may be limited or subject to restrictions or post approval commitments that render the approved product not commercially viable. Any FDA or other regulatory approval of our product candidates, once obtained, may be suspended, varied or withdrawn, including for failure to comply with regulatory requirements or if clinical or manufacturing problems follow initial marketing.
We are subject to stringent and changing U.S. and foreign laws, regulations, and rules, contractual obligations, industry standards, policies and other obligations related to data privacy and security. Our actual or perceived failure to comply with such obligations could lead to regulatory investigations or actions; litigation; fines and penalties; disruptions of our business operations; reputational harm; loss of revenue or profits; loss of customers or sales; and other adverse business consequences.
In the ordinary course of business, we collect, receive, store, process, generate, use, transfer, disclose, make accessible, protect, secure, dispose of, transmit, and share (collectively, processing)"process") personal data and other sensitive information, including proprietary and confidential business data, trade secrets, intellectual property, data we collect about trial participants in connection with clinical trials, and sensitive third-party data. Our data processing activities may subject us to numerous data privacy and security obligations, such as various laws, regulations, guidance, industry standards, external and internal privacy and security policies, contracts, and other obligations that govern the processing of personal data by us and on our behalf.
In the United States, federal, state, and local governments have enacted numerous data privacy and security laws, including data breach notification laws, personal data privacy laws, and consumer protection laws.laws (e.g., Section 5 of the Federal Trade Commission Act) and other similar laws (e.g., wiretapping laws). For example, HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act ("HITECH"), and their respective implementing regulations, imposes specifiedspecific requirements relating to the privacy, security and transmission of individually identifiable health information. Among other things, HITECH, through its implementing regulations, makes certain of HIPAA’s privacy and security standards directly applicable to business associates, defined as a person or organization, other than a member of a covered entity’s workforce, that creates, receives, maintains or transmits protected health information for or on behalf of a covered entity for a function or activity regulated by HIPAA as well as their covered subcontractors. In addition,
Additionally, in the past few years, numerous U.S. states—including California, Virginia, Colorado, Connecticut, and Utah—have enacted comprehensive privacy laws that impose certain obligations on covered businesses, including providing specific disclosures in privacy notices and affording residents with certain rights concerning their personal data. As applicable, such rights may include the right to access, correct, or delete certain personal data, and to opt-out of certain data processing activities, such as targeted advertising, profiling, and automated decision-making. The exercise of these rights may impact our business and ability to provide our products and services. Certain states also impose stricter requirements for processing certain personal data, including sensitive information, such as conducting data privacy impact assessments. These state laws allow for statutory fines for noncompliance. For example, the California Consumer Privacy Act of 2018, (“CCPA”) imposes obligations on businesses to which it applies. These obligations include, without limitation, providing specific disclosures in privacy notices, affording California residents certain rights related to their personal data, and requiring businesses subject to the CCPA to implement certain measures to effectuate California residents’ personal data rights. The CCPA allows for statutory fines for noncompliance (up to $7,500 per violation). In addition, it is anticipated thatas amended by the California Privacy Rights Act of 2020 (“CPRA”), effective January 1, 2023, will expand the CCPA. For example, the CPRA establishes a new(collectively “CCPA”), applies to personal data of consumers, business representatives, and employees who are California Privacy Protection Agencyresidents, and requires businesses to implementprovide specific disclosures in privacy notices, and enforceafford California residents certain privacy rights related to their personal data, such as those noted below. The CCPA allows for fines for noncompliance (up to $7,500 per intentional violation) and allows private litigants affected by certain data breaches to recover significant statutory damages. Although the CCPA (as amended), which could increase(like other U.S. comprehensive privacy laws) exempts some data processed in the riskcontext of an enforcement action. Otherclinical trials, the CCPA increases compliance costs and potential liability with respect to other personal data we maintain about California residents. Similar laws are being considered in several other states, have enacted data privacy laws. For example, Virginia passed its Consumer Data Protection Act, and Colorado passed the Colorado Privacy Act, both of which differ from the CPRA and become effective in 2023. Additional data privacy and security legislation has been proposedas well as at the federal, state and local levels, and we expect more jurisdictions to pass similar laws in recent years, which, alongthe future.
We may also be subject to new laws governing the privacy of consumer health data, including reproductive, sexual orientation, and gender identity privacy rights. For example, Washington’s My Health My Data Act (“MHMD”) broadly defines consumer health data, places restrictions on processing consumer health data (including imposing stringent requirements for consents), provides consumers certain rights with existingrespect to their health data, and creates a private right of action to allow individuals to sue for violations of the law. Other states are considering and may adopt similar laws. California also recently passed a law protecting privacy of abortion-related records and other reproductive healthcare services.
Additionally, under various privacy laws and other obligations, we may be required to obtain certain consents to process personal data. For example, some of our data processing practices may be challenged under wiretapping laws, since we obtain consumer information from third parties through various methods. These practices may be subject to increased challenges by class action plaintiffs. Our inability or failure to obtain consent for these practices could increase our potential liability, increase compliance costs, or adversely affect our business.result in adverse consequences, including class action litigation and mass arbitration demands.
Outside the United States, an increasing number of laws, regulations, and industry standards apply to data privacy and security. For example, the European Union’s General Data Protection Regulation (“EU GDPR”), the United Kingdom’s GDPR (“UK GDPR”) (EU GDPR and UK GDPR, collectively "GDPR"), Brazil’s General Data Protection Law (Lei Geral de Proteção de Dados Pessoais, or “LGPD”) (Law No. 13,709/2018), and China’s Personal Information Protection Law (“PIPL”) impose strict requirements for processing personal data. For example, the EU GDPR imposes significant and complex burdens on processing personal data, particularly for processing “special category personal data” (such as personal data related to health and genetic information), which
could be relevant to our operations in the context of our conduct of clinical trials and is of interest to relevant regulators. Under the EU GDPR, government regulators may impose temporary or definitive bans on data processing, as well as fines of up to 20 million euros under the EU GDPR, 17.5 million pounds sterling under the UK GDPR or, in each case, or 4% of annual global revenue, whichever
is greater. Further, under the GDPR, individuals may initiate litigation related to processing of their personal data.data, as well as consumer protection organizations authorized at law to represent data subjects' interests.
In addition, privacy advocates and industry groups around the world have proposed, and may propose, standards with which we are legally or contractually bound to comply.comply, or may become subject to in the future. We are also bound by contractual obligations related to data privacy and security, and our efforts to comply with such obligations may not be successful. Additionally, we publish privacy policies, marketing materials and other statements, such as compliance with certain certifications, regarding data privacy and security. If these policies, materials or statements are found to be deficient, lacking in transparency, deceptive, unfair, or misrepresentative of our practices, we may be subject to investigation, enforcement actions by regulators or other adverse consequences.
CertainIn the ordinary course of business, we may transfer personal data from Europe and other jurisdictions to the United States or other countries. Europe and other jurisdictions have enacted laws requiring data to be localized or limiting the transfer of personal data to other countries. In particular, the European Economic Area ("EEA") and the United Kingdom ("UK") have significantly restricted the transfer of personal data to the United States and other countries whose privacy laws it generally believes are inadequate. Other jurisdictions may adopt similarly stringent interpretations of their data localization laws and cross-border personal data transfer laws, which could make it more difficult to transfer information across jurisdictions (such as transferring or receiving personal data that originatesprevent us from conducting business in the EU, the UK, Switzerland or in other foreign jurisdictions). Existingcertain countries. Although there are currently various mechanisms that facilitate cross-border personal data transfers may change or be invalidated. For example, absent appropriate safeguards or other circumstances, the EU GDPR generally restricts the transfer of personal data to countries outside of the European Economic Area (“EEA”) that the European Commission does not consider to provide an adequate level of data privacy and security, such as the United States. The European Commission released a set of “Standard Contractual Clauses” (“SCCs”) that are designed to be a valid mechanism to facilitate personal data transfers out of the EEA to these jurisdictions. Currently, these Standard Contractual Clauses are a valid mechanismused to transfer personal data outside offrom the EEA but there exists some uncertainty regarding whetherand UK to the SCCs will remain a valid mechanism. Additionally, the SCCs impose additionalUnited States in compliance burdens,with law, such as conducting transfer impact assessmentsthe EU Standard Contractual Clauses ("EU SCCs"), the UK’s International Data Transfer Agreement / International Data Transfer Addendum to determine whether additional security measures are necessary to protect the at-issue personal data. As we incorporateEU SCCs, and the new SCCs into our contractual arrangements, we may be required to expend significant resources to update our contractual arrangements and to comply with such obligations. In addition, SwitzerlandEU-U.S. Data Privacy Framework and the UK similarly restrictextension thereto (which allows for transfers to relevant U.S.-based organizations who self-certify compliance and participate in the applicable frameworks), these mechanisms are subject to legal challenges, and there is no assurance that we can satisfy or rely on the Data Privacy Framework to lawfully transfer personal data transfers outside of those jurisdictions to countries such as the United States that do not provide an adequate level of personal data protection, and have also passed or are considering laws requiring local data residency or otherwise impeding the transfer of personal data across borders, any of which could increase the cost and complexity of doing business.States.
If we are unable to implement a valid compliance mechanism for cross-border personal data transfers, or if the requirements for a legally-compliant transfer are too onerous, we may face significant adverse consequences, including increased exposure to regulatory actions, substantial fines and injunctions against processing or transferring personal data from Europe. Inability to import personal data from Europe to the United States may significantly and negatively impact our business operations, including by limiting our ability to conduct clinical trialstrial activities in Europe and elsewhere; limiting our ability to collaborate with CROs, service providers, contractors and other companies that are subject to such cross-border data transfer or localization laws; the need to relocate part of or all of our business or data processing activities to other jurisdictions (such as Europe) at significant expense; or requiring us to increase our personal data processing capabilities and infrastructure in foreign jurisdictions at significant expense. Additionally, companies that transfer personal data out of the EEA and UK to other jurisdictions, particularly to the United States, are subject to increased scrutiny from regulators, individual litigants, and activities groups. Some European regulators have ordered certain companies to suspend or permanently cease certain transfers out of Europe for allegedly violating the GDPR’s cross-border data transfer limitations.
Our obligations related to data privacy and security (and consumers’ data privacy expectations) are quickly changing in an increasingly stringent fashion, creating some uncertainty as to the effective future legal framework.uncertainty. Additionally, these obligations may be subject to differing applications and interpretations, which may be inconsistent or conflict among jurisdictions. Preparing for and complying with these obligations requires significant resources and may necessitate changes to our information technologies, systems, and practices and to those of any third parties that process personal data on our behalf. Although we endeavor to comply with all applicable data privacy and security obligations, we may at times fail (or be perceived to have failed) to do so. Moreover, despite our efforts, our personnel or third parties upon whom we rely may fail to comply with such obligations, which could negatively impact our business operations and compliance posture. For example, any failure by a third-party service provider to comply with applicable law, regulations, or contractual obligations could result in adverse effects, including proceedings against us by governmental entities or others. If we or any of our partners fail to comply or are perceived to have failed to comply with applicable regulatory requirements,obligations, we or they could be subject to a range of regulatory actions, litigation (including class actions), or litigationmass arbitration demands that could affect our or our partners' ability to commercialize our products and conduct necessary research and development, and could harm or prevent sales of the affected products, or could substantially increase the costs and expenses of commercializing and marketing our products. In particular, plaintiffs have become increasingly more active in bringing privacy-related claims against companies, including class claims and mass arbitration demands. Some of these claims allow for the recovery of statutory damages on a per violation basis, and, if viable, carry the potential for monumental statutory damages, depending on the volume of data and the number of violations. Any threatened or actual government enforcement action or litigation could also generate adverse publicity and require that we devote substantial resources that could otherwise be used in other aspects of our business. Compliance with applicable federal, state, and foreign laws is difficult and time consuming, and companies that violate them may face substantial penalties. The potential sanctions include significant criminal fines, civil monetary penalties, administrative penalties, disgorgement, exclusion from participation in federal health care programs, individual imprisonment, injunctions, recall or seizure of products, total or partial suspension of production, reputational harm, administrative burdens, interruption or cessation of clinical trials, additional oversight and reporting obligations if we become subject to a corporate integrity agreement or similar agreement to resolve allegations of non-compliance with these laws, diminished profits and future earnings, and the curtailment or restructuring of our operations, and other sanctions. Because of the breadth of these laws, it is possible that some of our business activities could be subject to challenge under one or more of these laws. Such a challenge, irrespective of the underlying merits of the challenge or the ultimate outcome of the matter, could have a material adverse effect on our business, financial condition, results of operations and growth prospects. Moreover, clinical trial subjects and other individuals about whom we or our potential collaborators obtain personal information, as well as the providers who share this information with us, may limit our ability to collect, use and disclose the information. Claims that we have violated individuals’ privacy rights, failed to comply with data protection laws, or breached our contractual obligations, even if we are not found liable, could be expensive and time-consuming to defend and could result in adverse publicity that could harm our business.
If our information technology systems or data, or those of third parties upon which we rely, are or were compromised, we could experience adverse impacts resulting from such compromise, including, but not limited to, regulatory investigations or actions; litigation; fines and penalties; interruptions to our commercial operations, such as clinical trials;trials or other operations; harm to our reputation; loss of revenue or profits; and a loss of sales and other adverse consequences.
In the ordinary course of our business, we and our third-party service providers may collect, receive, store, process generate, use, transfer, disclose, make accessible, protect, secure, dispose of, transmit, and share (collectively, processing) proprietary, confidential, and sensitive data, including personal data (such as health-related data)data and data related to our clinical trials), intellectual property, and trade secrets (collectively, sensitive information). We may rely upon third-party service providers and technologies to operate critical business systems to process sensitive information in a variety of contexts, including, without limitation, third-party providers of cloud-
based infrastructure, encryption and authentication technology, employee email, and other functions. Our ability to monitor these third parties’ information security practices is limited, and these third parties may not have adequate information security measures in place. We may share or receive sensitive information with or from third parties.
Cyberattacks, malicious internet-based activity, and online and offline fraud are prevalent and continue to increase. These threats are becoming increasingly difficult to detect. These threats come from a variety of sources, including traditional computer “hackers,” threat actors, personnel (such as through theft or misuse), "hacktivists", organized criminal threat actors, sophisticated nation-states, and nation-state-supported actors. Some actors now engage and are expected to continue to engage in cyberattacks, including without limitation nation-state actors for geopolitical reasons and in conjunction with military conflicts and defense activities. During times of war and other major conflicts, we and the third parties upon which we rely may be vulnerable to a heightened risk of these attacks, including retaliatory cyberattacks that could materially disrupt our systems and operations, supply chain, and ability to produce, sell and distribute our products. For example, we have third parties upon which we rely to support our business located in unstable regions and regions experiencing (or expected to experience) geopolitical or other conflicts, including in Israel, where businesses have experienced an increase in cyberattacks in relation to the Israel/Hamas conflict. We and the third parties upon which we rely may be subject to a variety of other evolving threats, including, but not limited to, social-engineering attacks (including through deep fakes, which may be increasingly more difficult to identify as fake, and phishing attacks), malicious code (such as viruses and worms), malware (including as a result of advanced persistent threat intrusions), denial-of-service attacks, (such as credential stuffing),stuffing, credential harvesting, personnel misconduct or error, ransomware attacks, supply-chain attacks, software bugs, server malfunctions, software or hardware failures, loss of data or other information technology assets, adware, telecommunications failures, attacks enhanced or facilitated by artificial intelligence, and other similar threats. RansomwareIn particular, ransomware attacks, including those from organized criminal threat actors, nation-states and nation-state supported actors, are becoming increasingly prevalent and severe and can lead to significant interruptions, delays, or outages in our operations, ability to provide our products, disruption of clinical trials, loss of data (including data related to clinical trials), loss of income, significant extra expenses to restore data or systems, reputational loss and the diversion of funds. To alleviate the financial, operational and reputational impact of a ransomware attack, it may be preferable to make extortion payments, but we may be unwilling or unable to do so (including, for example, if applicable laws prohibit such payments). Similarly, supply chain attacks have increased in frequency. The Company’s dataAdditionally, hybrid and information systems may also fail for reasons other than a security incident or breach, such as server malfunctions, software or hardware failures,remote work has become more common and telecommunications failures. Additionally, the COVID-19 pandemic and our remote workforce poseshas increased risks to our information technology systems and data, as more of our employees work from home, utilizingutilize network connections, computers, and devices outside our premises.premises or network, including working at home, while in transit, and in public locations. Future or past business transactions (such as acquisitions or integrations) could also expose us to additional cybersecurity risks and vulnerabilities, as our systems could be negatively affected by vulnerabilities present in acquired or integrated entities’ systems and technologies. Furthermore, we may discover security issues that were not found during due diligence of such acquired or integrated entities, and it may be difficult to integrate companies into our information technology environment and security program.
We rely upon third parties and technologies to operate critical business systems to process sensitive information in a variety of contexts, including, without limitation, third-party providers of cloud-based infrastructure, encryption and authentication technology, employee email, and other functions. We also rely on third parties to provide certain products, including active pharmaceutical ingredients, to operate our business, including in China. Our ability to monitor these third parties’ information security practices is limited, and these third parties may not have adequate information security measures in place. While we may be entitled to damages if the third parties upon which we rely fail to satisfy their privacy or security-related obligations to us, any award may be insufficient to cover our damages, or we may be unable to recover such award. In addition, supply-chain attacks have increased in frequency and severity, and we cannot guarantee that third parties’ infrastructure in our supply chain or our third-party partners’ supply chains have not been compromised. We may share or receive sensitive information with or from third parties.
While we have implemented security measures designed to protect against security incidents, there can be no assurance that these measures will be effective. We take steps designed to detect, mitigate and remediate vulnerabilities in our information security systems (such as our hardware and/or software, including that of third parties upon which we rely), but we may not be able to detect, mitigate, and remediate all such vulnerabilities including on a timely basis. Further, we may experience delays in developing and deploying remedial measures and patches designed to address identified vulnerabilities. Vulnerabilities could be exploited and result in a security incident.
Any of the previously identified or similar threats could cause a security incident or other interruption. A security incident or other interruption that could result in unauthorized, unlawful, or accidental acquisition, modification, destruction, loss, alteration, encryption, disclosure of, or access to our sensitive information.information or our information technology systems, or those of the third parties upon whom we rely. A security incident or other interruption could disrupt our ability (and that of third parties upon whom we rely) to provide our products and services.products. We may expend significant resources or modify our business activities (including our clinical trial activities) to try to protect against security incidents. Certain data privacy and security obligations may require us to implement and maintain specific security measures, industry-standard or reasonable security measures to protect our information technology systems and sensitive information.
While we have implemented security measures designed to protect against security incidents, there can be no assurance that these measures will be effective. We may be unable in the future to detect vulnerabilities in our information technology systems (including our products) because such threats and techniques change frequently, are often sophisticated in nature, and may not be detected until after a security incident has occurred. Despite our efforts to identify and remediate vulnerabilities, if any, in our information technology systems, our efforts may not be successful and could result in a material disruption of our programs and operations. For example, the loss of clinical trial data from completed or ongoing clinical trials for any of our product candidates could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. Further, we may experience delays in developing and deploying remedial measures designed to address any such identified vulnerabilities
Applicable data security laws, privacy policies and data securitypublic company disclosure obligations may require us to notify relevant stakeholders of anycertain security incidents, including affected individuals, customers, regulators and regulators.investors. Such disclosures are costly, and the disclosures or the failure to comply with such requirements, could lead to adverse consequences. If we (or a third party upon whom we rely) experience a security incident or are perceived to have experienced a security incident, we may experience adverse consequences. These consequences may include: government enforcement
actions (for example, investigations, fines, penalties, audits, and inspections); additional reporting requirements and/or oversight; restrictions on processing sensitive information (including personal data); litigation (including class claims); indemnification obligations; negative publicity; reputational harm; monetary fund diversions; diversion of management attention; interruptions in our operations (including availability of data); financial loss;loss and other similar harms. Security incidentsFor example, the loss of clinical trial data from completed or ongoing clinical trials for any of our product candidates could result in delays in our regulatory approval efforts and attendant consequencessignificantly increase our costs to recover or reproduce the data.
Whether a cybersecurity incident is reportable to our investors may cause customersnot be straightforward, may take considerable time to stop usingdetermine, and may be subject to change as the investigation of the incident progresses, including changes that may significantly alter any initial disclosure that we provide. Moreover, experiencing a material cybersecurity incident and any mandatory disclosures could lead to negative publicity, loss of customer, investor or partner confidence in the effectiveness of our products or services, deter new customers from using our products or services,cybersecurity measures, diversion of management’s attention, governmental investigations, lawsuits, and negatively impact our ability to growthe expenditure of significant capital and operate our business.other resources.
Our contracts may not contain limitations of liability, and even where they do, there can be no assurance that limitations of liability in our contracts are sufficient to protect us from liabilities, damages, or claims related to our data privacy and security obligations. In addition, our insurance coverage may not be adequate or sufficient to protect us from or to mitigate liabilities arising out of our privacy and security practices or that such coverage will continue to be available on commercially reasonable terms or at all, or that such coverage will pay future claims.
ChangesIn addition to experiencing a security incident, third parties may gather, collect, or infer sensitive information about us from public sources, data brokers, or other means that reveals competitively sensitive details about our organization and could be used to undermine our competitive advantage or market position. Sensitive information of us or our customers could also be leaked, disclosed, or revealed as a result of or in connection with our employee’s, personnel’s, or vendor’s use of generative AI technologies.
Uncertainties in the interpretation and application of existing, new and proposed tax laws and regulations could materially affect our tax obligations and effective tax rate.
The tax regimes to which we are subject or under which we operate are unsettled and may be subject to significant change. The issuance of additional guidance related to existing or future tax laws, or changes to tax laws or regulations proposed or implemented by the current or a future U.S. presidential administration, Congress, or taxing authorities in other jurisdictions, including jurisdictions outside of the United States, could materially affect our tax obligations and effective tax rate. To the extent that are applied adversely to us or our customers maysuch changes have a material adverse effectnegative impact on us, including as a result of related uncertainty, these changes may adversely impact our business, cash flow, financial condition, or results of operations.operations, and cash flows.
New income, sales, use or otherThe amount of taxes we pay in different jurisdictions depends on the application of the tax laws statutes, rules, regulationsof various jurisdictions, including the United States, to our international business activities, tax rates, new or ordinancesrevised tax laws, or interpretations of tax laws and policies, and our ability to operate our business in a manner consistent with our corporate structure and intercompany arrangements. The taxing authorities of the jurisdictions in which we operate may challenge our methodologies for pricing intercompany transactions pursuant to our intercompany arrangements or disagree with our determinations as to the income and expenses attributable to specific jurisdictions. If such a challenge or disagreement were to occur, and our position was not sustained, we could be enacted at any time,required to pay additional taxes, interest, and penalties, which could affect theresult in one-time tax treatmentcharges, higher effective tax rates, reduced cash flows, and lower overall profitability of our domesticoperations. Our financial statements could fail to reflect adequate reserves to cover such a contingency. Similarly, a taxing authority could assert that we are subject to tax in a jurisdiction where we believe we have not established a taxable connection, often referred to as a “permanent establishment” under international tax treaties, and foreign earnings. Any new taxessuch an assertion, if successful, could adversely affectincrease our domesticexpected tax liability in one or more jurisdictions.
Effective January 1, 2022, the Tax Cuts and international businessJobs Act of 2017 eliminated the option to deduct research and development expenses for tax purposes in the year incurred and requires taxpayers to capitalize and subsequently amortize such expenses over five years for research activities conducted in the United States and over 15 years of research activities conducted outside the United States. Unless the United States Department of the Treasury issues regulations that narrow the application of this provision to a smaller subset of our research and development expenses or the provision is deferred, modified, or repealed by Congress, in future years we may experience a material decrease in our cash flows from operations and our business and financial performance. Further, existing tax laws, statutes, rules, regulations or ordinances could be interpreted, changed, modified or applied adversely to us. For example, the Biden administration and Congress have proposed various U.S. federal tax law changes, which if enacted could have a material impact on our business, cash flow, financial condition or results of operations. In addition, it is uncertain if and to what extent various states will conform to the Tax Act, the CARES Act, or any newly enacted federal tax legislation. Changesan offsetting similarly sized increase in corporate tax rates, the
realization ofour net deferred tax assets relating to our operations, the taxationover these amortization periods. The actual impact of foreign earnings, and the deductibility of expenses under the Tax Act or future reform legislation could have a material impactthis provision will depend on the value of our deferred tax assets, could result in significant one-time charges, and could increase our future U.S. tax expense.
Our effective tax rate may fluctuate, and we may incur obligations in tax jurisdictions in excess of accrued amounts.
Our effective tax rate is derived from a combination of applicable tax rates in the various places that we operate. In preparing our financial statements, we estimatemultiple factors, including the amount of tax thatresearch and development expenses we will become payable in each of such places. Nevertheless,incur and whether we conduct our effective tax rate may be different than experienced inresearch and development activities inside or outside the past due to numerous factors, including changes in the mix ofUnited States and our profitability from state to state, the results of examinations and audits of our tax filings, our inability to secure or sustain acceptable agreements with tax authorities, changes in accounting for income taxes and changes in tax laws. Any of these factors could cause us to experience an effective tax rate significantly different from previous periods or our current expectations and may result in tax obligations in excess of amounts accrued in our financial statements.overall net operating loss position.
Our ability to use net operating loss carryforwards and certain other tax attributes to offset future taxable income and taxes may be subject to limitations.
Under current law, our federal net operating losses ("NOLs") generated in tax years beginning after December 31, 2017, may be carried forward indefinitely, but the deductibility of such federal NOLs in tax years beginning after December 31, 2020, is limited to 80% of taxable income. As of December 31, 2021,2023, we had federal net operating loss ("NOL")NOLs of $85.8$154.3 million. It is uncertain if and to what extent various states will conform to federal tax laws. In addition, under Sections 382 and 383 of the Internal Revenue Code of 1986, as amended, and corresponding provisions of state law, if a corporation undergoes an “ownership change,” which is generally defined as a greater than 50% change, by value, in its equity ownership over a three-year period, the corporation’s ability to use its pre-change NOL carryforwards and other pre-change U.S. tax attributes (such as research tax credits) to offset its post-change income or taxes may be limited. We completed a study to analyze whether any ownership changes occurred through March 31, 2023, and determined no ownership changes
occurred. We are in the process of updating our analysis of owner shifts to determine whether an ownership change occurred since March 31, 2023. It is possible that we have experienced an ownership change in the past. In addition, we may experience ownership changes in the future as a result of subsequent shifts in our stock ownership, some of which may be outside of our control.
As a result, our federal NOL carryforwards may be subject to a percentage limitation if used to offset income in tax years following an ownership change. In addition, it is possible that we have in the past undergone, and in the future may undergo, additional ownership changes that could limit our ability to use all of our pre-change NOL carryforwards and other pre-change tax attributes (such as research tax credits) to offset our post-change income or taxes. Similar provisions of state tax law may also apply to limit our use of accumulated state tax attributes. In addition, at the state level, there may be periods during which the use of NOL carryforwards is suspended or otherwise limited, which could accelerate or permanently increase state taxes owed. As a result, we may be unable to use all or a material portion of our NOL carryforwards and other tax attributes, which would harm our future operating results by effectively increasing our future tax obligations.
Changes in funding for the FDA, the SEC and other government agencies or regulatory authorities could hinder their ability to hire and retain key leadership and other personnel, prevent new products and services from being developed or commercialized in a timely manner, or otherwise prevent those agencies from performing normal functions on which the operation of our business may rely, which could negatively impact our business.
The ability of the FDA to review and approve new products can be affected by a variety of factors, including government budget and funding levels, ability to hire and retain key personnel and accept payment of user fees, and statutory, regulatory, and policy changes. Average review times at the agency have fluctuated in recent years as a result. In addition, government funding of the SEC and other government agencies on which our operations may rely, including those that fund research and development activities is subject to the political process, which is inherently fluid and unpredictable.
Disruptions at the FDA and other agencies may also slow the time necessary for new drugstherapies to be reviewed and/or approved by necessary government agencies, which would adversely affect our business. For example, over the last several years, the U.S. government has shut down several times and certain regulatory agencies, such as the FDA and the SEC, have had to furlough critical FDA, SEC and other government employees and stop critical activities. If a prolonged government shutdown occurs, or if the FDA or EDA experience resource constraints, continue to arise from the COVID-19 pandemic, it could significantly impact the ability of the FDAapplicable regulatory agency to timely review and process our regulatory submissions, which could have a material adverse effect on our business. Further, future government shutdowns could impact our ability to access the public markets and obtain necessary capital in order to properly capitalize and continue our operations. Comparable considerations may be applicable in relation to foreign regulatory authorities.
Our business could be negatively impacted by environmental, social and corporate governance (ESG) matters or our reporting of such matters.
There is an increasing focus from certain investors, employees, partners, and other stakeholders concerning ESG matters. While we have internal efforts directed at ESG matters and preparations for increased future disclosures, we may be perceived by certain stakeholders as not acting responsibly in connection with these matters, which could negatively impact us. Moreover, the SEC has recently proposed, and may continue to propose, certain mandated ESG reporting requirements, such as the SEC's proposed rules designed to enhance and standardize climate-related disclosures, which, if finally approved, would significantly increase our compliance and reporting costs and may also result in disclosures that certain investors or other stakeholders deem to negatively impact our reputation and/or that harm our stock price. In addition, given our business model, we currently do not report our environmental emissions and absent a legal requirement to do so we currently do not plan to report our environmental emissions, and lack of reporting could result in certain investors declining to invest in our common stock.
The withdrawal of the United Kingdom from the European Union, commonly referred to as “Brexit,” may adversely impact our ability to obtain regulatory approvals of our product candidates in the European Union,United Kingdom, result in restrictions or imposition of taxes and duties for importing our product candidates into the European Union,United Kingdom, and may require us to incur additional expenses in order to develop, manufacture and commercialize our product candidates in the European Union.United Kingdom.
FollowingThe United Kingdom’s (“UK”) withdrawal from the result of a referendum in 2016, the United Kingdom left the European UnionEU on January 31, 2020, commonly referred to as “Brexit.” PursuantBrexit, has changed the regulatory relationship between the UK and the EU. The Medicines and Healthcare products Regulatory Agency, or MHRA, is now the UK’s standalone regulator for medicinal products and medical devices. Great Britain (England, Scotland and Wales) is now a third country to the formal withdrawal arrangements agreed betweenEU. Northern Ireland will, with regard to EU regulations, continue to follow the United KingdomEU regulatory rules for now.
The UK regulatory framework in relation to clinical trials is governed by the Medicines for Human Use (Clinical Trials) Regulations 2004, as amended, which is derived from the CTD, as implemented into UK national law through secondary legislation. On January 17, 2022, the MHRA launched an eight-week consultation on reframing the UK legislation for clinical trials, and which aimed to streamline clinical trials approvals, enable innovation, enhance clinical trials transparency, enable greater risk proportionality, and promote patient and public involvement in clinical trials. The UK Government published its response to the European Union,consultation on March 21, 2023 confirming that it would bring forward changes to the United Kingdom was subject to alegislation. These resulting legislative amendments will determine how closely the UK regulations will align with the CTR. In October 2023, the MHRA
transition periodannounced a new Notification Scheme for clinical trials which enables a more streamlined and risk-proportionate approach to initial clinical trial applications for Phase 4 and low-risk Phase 3 clinical trial applications.
Marketing authorizations in the UK are governed by the Human Medicines Regulations (SI 2012/1916), as amended. Since January 1, 2021, an applicant for the EU centralized procedure marketing authorization can no longer be established in the UK. As a result, since this date, companies established in the UK cannot use the EU centralized procedure and instead must follow one of the UK national authorization procedures or one of the remaining post-Brexit international cooperation procedures to obtain a marketing authorization to market products in the UK. All existing EU marketing authorizations for centrally authorized products were automatically converted or grandfathered into UK marketing authorization, effective in Great Britain only, free of charge on January 1, 2021, unless the marketing authorization holder opted-out of this possibility. Northern Ireland currently remains within the scope of EU authorizations in relation to centrally authorized medicinal products. Accordingly, until the Windsor Framework is implemented in Northern Ireland on January 1, 2025, products falling within the scope of the EU centralized procedure can only be authorized through UK national authorization procedures in Great Britain.
The MHRA has also introduced changes to national marketing authorization procedures. This includes introduction of procedures to prioritize access to new medicines that ended December 31, 2020, or the Transition Period, during which EU rules continued to apply. A trade and cooperation agreement, or the Trade and Cooperation Agreement, that outlines the future trading relationship between the United Kingdomwill benefit patients, including a 150-day assessment route, a rolling review procedure and the European Union was agreed in December 2020.
International Recognition Procedures which entered into application on January 1, 2024. Since a significant proportion ofJanuary 1, 2024, the regulatory framework in the United Kingdom applicable to our business and our product candidates is derived from EU directives and regulations, Brexit has had, andMHRA may continue to have, a material impactrely on the regulatory regime with respect toInternational Recognition Procedure, or IRP, when reviewing certain types of marketing authorization applications. This procedure is available for applicants for marketing authorization who have already received an authorization for the development, manufacture, importation, approvalsame product from a reference regulator. These include the FDA, the EMA, and commercializationnational competent authorities of our product candidates in the United Kingdom or the European Union. For example, Great Britain is no longer covered by the centralized procedures for obtaining EU-wide marketing authorizationindividual EEA countries. A positive opinion from the EMA and andCHMP, or a separatepositive end of procedure outcome from the mutual recognition or decentralized procedures are considered to be authorizations for the purposes of the IRP.
There is no pre-marketing authorization orphan designation for medicinal products in the UK. Instead, the MHRA reviews applications for orphan designation in parallel to the corresponding marketing authorization application. The criteria are essentially the same as those in the EU, but have been tailored for the market. This includes the criterion that prevalence of the condition in Great Britain, rather than the EU, must not be more than five in 10,000. Upon the grant of a marketing authorization with orphan status, the medicinal product will benefit from up to 10 years of market exclusivity from similar products in the approved orphan indication. The start of this market exclusivity period will be required to marker ourset from the date of first approval of the product candidates in Great Britain. It is currently unclear whether the Medicines & Healthcare products Regulatory Agency ("MHRA") in the U.K. is sufficiently prepared to handle the increased volume of marketing authorization applications that it is likely to receive.
While the Trade and Cooperation Agreement provides for the tariff-free trade of medicinal products between the UK and the EU there may be additional non-tariff costs to such trade which did not exist prior to the end of the Transition Period. Further, should the UK diverge from the EU from a regulatory perspective in relation to medicinal products, tariffs could be put into place in the future. We could therefore, both now and in the future, face significant additional expenses (when compared to the position prior to the end of the Transition Period) to operate our business, which could significantly and materially harm or delay our ability to generate revenues or achieve profitability of our business. Any further changes in international trade, tariff and import/export regulations as a result of Brexit or otherwise may impose unexpected duty costs or other non-tariff barriers on us. These developments, or the perception that any of them could occur, may significantly reduce global trade and, in particular, trade between the impacted nations and the UK It is also possible that Brexit may negatively affect our ability to attract and retain employees, particularly those from the EU. These developments, or the perception that any of them could occur, may significantly reduce global trade and, in particular, trade between the impacted nations and the United Kingdom.
Business disruptions could seriously harm our future revenue and financial condition and increase our costs and expenses.
Our operations, and those of our third-party manufacturers, CROs and other contractors and consultants, could be subject to disruptions resulting from earthquakes, power shortages, telecommunications failures, water shortages, floods, hurricanes, typhoons, fires, extreme weather conditions, medicalhealth epidemics or pandemics, wars and other geopolitical conflicts, and other natural or man-made disasters or business interruptions, for which we are predominantly self-insured. The occurrence of any of these business disruptions could seriously harm our operations and financial condition and increase our costs and expenses.
Our corporate headquarters are located in San Diego, California, an area prone to wildfires and earthquakes. These and other natural disasters could severely disrupt our operations, and have a material adverse effect on our business, results of operations, financial condition and prospects. If a natural disaster, power outage or other event occurred that prevented us from using all or a significant portion of our headquarters, that damaged critical infrastructure, such as the manufacturing facilities of our third-party contract manufacturers, or that otherwise disrupted operations, it may be difficult or, in certain cases, impossible for us to continue our business for a substantial period of time. Any disaster recovery and business continuity plans we have in place may prove inadequate in the event of a serious disaster or similar event. We may incur substantial expenses as a result of the limited nature of our disaster recovery and business continuity plans, which, could have a material adverse effect on our business.
In addition, we rely on third-party manufacturers, some of whom are located in China, to manufacture API for FILSPARI and certain of our product candidates, including sparsentan.candidates. Any disruption in production or inability of our manufacturers in China to produce or ship adequate quantities to meet our needs, whether as a result of a natural disaster or other causes (such as the COVID-19staffing shortages, or a health epidemic or pandemic), could impair our ability to meet commercial demand for FILSPARI, to operate our business on a day-to-day basis and to continue our research and development of our product candidates. In addition, we are exposed to the possibility of product supply disruption and increased costs in the event of changes in the policies of the United States or Chinese governments (such as tariffs on chemical intermediates we use that are manufactured in China), political unrest or unstable economic conditions in China. Any recall of the manufacturing lots or similar action regarding our API used in clinical trials could delay the trials or detract from the integrity of the trial data and its potential use in future regulatory filings. In addition, manufacturing interruptions or failure to comply with regulatory requirements by any of these manufacturers could significantly delay clinical development of potential products and reduce third-party or clinical researcher interest and support of proposed trials. These interruptions or failures could also impede commercialization of our product candidates and impair our competitive position.
We have previously identified a material weakness in our internal control over financial reporting. If additional material weaknesses in our internal control over financial reporting are discovered or occur in the future, our consolidated financial statements may contain material misstatements and we could be required to restate our financial results, which could adversely affect our stock price and result in an inability to maintain compliance with applicable stock exchange listing requirements.
We previously concluded that there was a matter that constituted a material weakness in our internal control over financial reporting that has since been remediated. A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting such that there is a reasonable possibility that a material misstatement of our annual or interim consolidated financial statements will not be prevented or detected on a timely basis. As disclosed under Item 9A of our Annual Report on Form 10-K for the fiscal year ended December 31, 2022, there was a material weakness in our internal control over financial reporting as of December 31, 2022 because we did not design effective controls and procedures to evaluate the accounting for a certain pre-launch inventory contract affecting the timing of recognition of research and development expense. As a result of the material weakness, we added controls for the timely accounting evaluation of research and development contracts that were intended to ensure appropriate expense recognition of certain pre-launch inventory. As of December 31, 2023, the material weakness had been remediated.
If additional material weaknesses in our internal control over financial reporting are discovered or occur in the future, or if we are unable to maintain effective internal control over financial reporting or disclosure controls and procedures for any reason, our ability to record, process and report financial information accurately, and to prepare financial statements within required time periods, could be adversely affected, which could subject us to litigation or investigations requiring management resources and payment of legal and other expenses and negatively impact the price of our common stock. In addition, we could be subject to sanctions or investigations by the SEC or other regulatory authorities, which would require additional financial and management resources.
Furthermore, investor perceptions of our company may suffer as a result of the previous material weakness or any future material weakness in our internal controls, and this could cause a decline in the market price of our stock. Any failure of our internal control over financial reporting could have a material adverse effect on our stated operating results, result in an adverse opinion on our internal control over financial reporting from our independent registered public accounting firm, and harm our reputation.
Adverse developments affecting the financial services industry could adversely affect our current and projected business operations and our financial condition and results of operations.
Adverse developments that affect financial institutions, such as events involving liquidity that are rumored or actual, have in the past and may in the future lead to bank failures and market-wide liquidity problems. For example, on March 10, 2023, Silicon Valley Bank (SVB) was closed by the California Department of Financial Protection and Innovation, which appointed the Federal Deposit Insurance Corporation (FDIC) as receiver. Similarly, on March 12, 2023, Signature Bank and Silvergate Capital Corp. were each swept into receivership. In addition, on May 1, 2023, the FDIC seized First Republic Bank and sold its assets to JPMorgan Chase & Co. It is uncertain whether the U.S. Department of Treasury, FDIC and Federal Reserve Board will provide access to uninsured funds in the future in the event of the closure of other banks or financial institutions, or that they would do so in a timely fashion.
Although we assess our banking relationships as we believe necessary or appropriate, our access to cash in amounts adequate to finance or capitalize our current and projected future business operations could be significantly impaired by factors that affect the financial institutions with which we have banking relationships. These factors could include, among others, events such as liquidity constraints or failures, the ability to perform obligations under various types of financial, credit or liquidity agreements or arrangements, disruptions or instability in the financial services industry or financial markets, or concerns or negative expectations about the prospects for companies in the financial services industry. These factors could also include factors involving financial markets or the financial services industry generally. The results of events or concerns that involve one or more of these factors could include a variety of material and adverse impacts on our current and projected business operations and our financial condition and results of operations. These could include, but may not be limited to, delayed access to deposits or other financial assets or the uninsured loss of deposits or other financial assets; or termination of cash management arrangements and/or delays in accessing or actual loss of funds subject to cash management arrangements.
In addition, widespread investor concerns regarding the U.S. or international financial systems could result in less favorable commercial financing terms, including higher interest rates or costs and tighter financial and operating covenants, or systemic limitations on access to credit and liquidity sources, thereby making it more difficult for us to acquire financing on acceptable terms or at all. Any decline in available funding or access to our cash and liquidity resources could, among other risks, adversely impact our ability to meet our operating expenses, financial obligations or fulfill our other obligations, result in breaches of our financial and/or contractual obligations or result in violations of federal or state wage and hour laws. Any of these impacts, or any other impacts resulting from the factors described above or other related or similar factors not described above, could have material adverse impacts on our liquidity and our current and/or projected business operations and financial condition and results of operations.
We maintain our cash at financial institutions, often in balances that exceed federally insured limits.
We maintain the majority of our cash and cash equivalents in accounts at banking institutions in the United States that we believe are of high quality. Cash held in these accounts often exceed the FDIC insurance limits. If such banking institutions were to fail, we could lose all or a portion of amounts
held in excess of such insurance limitations. In the event of failure of any of the financial institutions where we maintain our cash and cash equivalents, there can be no assurance that we would be able to access uninsured funds in a timely manner or at all. Any inability to access or delay in accessing these funds could adversely affect our business and financial position.
Risks Related to our Indebtedness and Investments
Our indebtedness could adversely affect our financial condition.
As of December 31, 2021,2023, we had approximately $276$385 million of total debt outstanding, classified as long term. As a result of our indebtedness, a portion of our cash flow will be required to pay interest and principal on the 2025 Notes and 2029 Notes if the notes are not converted to shares of common stock prior to maturity. We may not generate sufficient cash flow from operations or have future borrowings available to enable us to repay our indebtedness or to fund other liquidity needs.
Our indebtedness pursuant to the 2025 Notes and 2029 Notes could have important consequences. For example, it could:
•make it more difficult for us to satisfy our obligations with respect to any other debt we may incur in the future;
•increase our vulnerability to general adverse economic and industry conditions;
•require us to dedicate a substantial portion of our cash flow from operations to payments on our indebtedness and related interest, thereby reducing the availability of our cash flow to fund working capital, capital expenditures and other general corporate purposes;
•limit our flexibility in planning for, or reacting to, changes in our business and the industry in which we operate;
•increase our cost of borrowing;
•place us at a competitive disadvantage compared to our competitors that may have less debt; and
•limit our ability to obtain additional financing for working capital, capital expenditures, acquisitions, debt service requirements or general corporate purposes.
We expect to use cash flow from operations and outside financings to meet our current and future financial obligations, including funding our operations, debt service and capital expenditures. Our ability to make these payments depends on our future performance, which will be affected by financial, business, economic and other factors, many of which we cannot control. Our business may not generate sufficient cash flow from operations in the future, which could result in our being unable to repay indebtedness, or to fund other liquidity needs. If we do not generate sufficient cash from operations, we may be forced to reduce or delay our business activities and capital expenditures, sell assets, obtain additional debt or equity capital or restructure or refinance all or a portion of our debt, including the 2025 Notes and 2029 Notes, on or before maturity. We cannot make any assurances that we will be able to accomplish any of these alternatives on terms acceptable to us, or at all. In addition, the terms of existing or future indebtedness may limit our ability to pursue any of these alternatives. In addition, we may from time to time seek to retire or purchase our outstanding debt, including the 2025 Notes or 2029 Notes, through cash purchases and/or exchanges for equity securities, in open market purchases, privately negotiated transactions or otherwise. Such repurchases or exchanges, if any, will depend on prevailing market conditions, our liquidity requirements, contractual restrictions, and other factors. The amounts involved in any such transactions, individually or in the aggregate, may be material. Further, any such purchases or exchanges may result in us acquiring and retiring a substantial amount of such indebtedness, which could impact the trading liquidity of such indebtedness.
We may be unable to raise the funds necessary to repurchase the 2025 Notes and 2029 Notes for cash following a fundamental change, or to pay any cash amounts due upon conversion, and our future indebtedness may limit our ability to repurchase the 2025 Notes and 2029 Notes or pay cash upon their conversion.
Noteholders may require us to repurchase their 2025 Notes and 2029 Notes following a fundamental change at a cash repurchase price generally equal to the principal amount of the 2025 Notes and 2029 Notes to be repurchased, plus accrued and unpaid interest to, but excluding, the fundamental change repurchase date. In addition, upon conversion, we willwould satisfy part or all of our conversion obligation in cash unless we electelected to settle conversions solely in shares of our common stock.
We may not have enough available cash or be able to obtain financing at the time we are required to repurchase the 2025 Notes and 2029 Notes or pay the cash amounts due upon conversion of the 2025 Notes and 2029 Notes. In addition, applicable law, regulatory authorities and the agreements governing our future indebtedness may restrict our ability to repurchase the 2025 Notes and 2029 Notes or pay the cash amounts due upon conversion of the 2025 Notes and 2029 Notes. Our failure to repurchase the 2025 Notes and 2029 Notes or to pay the cash amounts due upon conversion of the 2025 Notes and 2029 Notes when required will constitute a default under the base and supplemental indentures that will govern the
2025 Notes and 2029 Notes, which we refer to collectively as the “indenture.” We may not have sufficient funds to satisfy all amounts due under the other indebtedness and the 2025 Notes and 2029 Notes.
A default under the 2025 Notes or 2029 Notes may have a material adverse effect on our financial condition.
If an event of default under the 2025 Notes or 2029 Notes occurs, the principal amount of the 2025 Notes or the 2029 Notes, as applicable, plus accrued and unpaid interest (including additional interest, if any) may be declared immediately due and payable, subject to certain conditions set forth in the indenture governing such notes. Events of default include, but are not limited to:
•failure to pay (for more than 30 days) interest when due;
•failure to pay principal when due;
•failure to deliver shares of common stock upon conversion of a 2025 Notes;Note or 2029 Note;
•failure to provide notice of a fundamental change;
•acceleration on our other indebtedness in excess of $10 million (other than indebtedness that is non-recourse to us); or
•certain types of bankruptcy or insolvency involving us.
Accordingly, the occurrence of a default under the 2025 Notes or 2029 Notes, unless cured or waived, may have a material adverse effect on our results of operations.
Provisions of the 2025 Notes and 2029 Notes could discourage an acquisition of us by a third party.
Certain provisions of the 2025 Notes and 2029 Notes could make it more difficult or more expensive for or prevent a third party to acquire us. Upon the occurrence of certain transactions constituting a fundamental change, holders of the 2025 Notes and 2029 Notes will have the right, at their option, to require us to repurchase all of their 2025 Notes and 2029 Notes or any portion of the principal amount of such Notes in integral multiples of $1,000. We may also be required to increase the conversion rate for conversions in connection with certain fundamental changes.
Conversion of the Notes may dilute the ownership interest of existing stockholders, including holders who had previously converted their 2025 Notes or 2029 Notes.
To the extent we issue shares of common stock upon conversion of the 2025 Notes or 2029 Notes, the conversion of some or all of the 2025 Notes or 2029 Notes will dilute the ownership interests of existing stockholders. Any sales in the public market of shares of the common stock issuable upon such conversion could adversely affect prevailing market prices of shares of our common stock. In addition, the existence of the 2025 Notes and 2029 Notes may encourage short selling by market participants because the conversion of the 2025 Notes and 2029 Notes could depress the price of shares of our common stock.
General Risk Factors
Unstable market, economic and geopolitical conditions may have serious adverse consequences on our business, financial condition and stock price.
The global credit and financial markets have experienced extreme volatility and disruptions, including as a result of inflation and high interest rates, bank failures, wars, armed conflicts and global geopolitical tension, and may experience disruptions in the future. These disruptions can result in severely diminished liquidity and credit availability, increase in inflation, declines in consumer confidence, declines in economic growth, increases in unemployment rates and uncertainty about economic stability. There can be no assurance that further deterioration in credit and financial markets and confidence in economic conditions will not occur. Our general business strategy may be adversely affected by any such economic downturn, volatile business environment, higher inflation, or unpredictable and unstable market conditions. If the current equity and credit markets deteriorate, it may make any necessary debt or equity financing more difficult, more costly and more dilutive. Failure to secure any necessary financing in a timely manner and on favorable terms could have a material adverse effect on our operations, growth strategy, financial performance and stock price and could require us to delay or abandon clinical development plans.
Other international and geopolitical events could also have a serious adverse impact on our business. For instance, in February 2022, Russia initiated military action against Ukraine. In response, the United States and certain other countries imposed significant sanctions and trade actions against Russia and could impose further sanctions, trade restrictions, and other retaliatory actions. While we cannot predict the broader consequences, the conflict and retaliatory and counter-retaliatory actions could materially adversely affect global trade, currency exchange rates, inflation, regional economies, and the global economy, which in turn may increase our costs, disrupt our supply chain, impair our ability to raise or
access additional capital when needed on acceptable terms, if at all, or otherwise adversely affect our business, financial condition, and results of operations.
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
ITEM 1C. CYBERSECURITY
Risk Management and Strategy
We have implemented and maintain various information security processes designed to identify, assess and manage material risks from cybersecurity threats to our critical computer networks, third party hosted services, communications systems, hardware and software, and our critical data, including intellectual property, confidential information that is proprietary, strategic or competitive in nature, and data related to patients and clinical trials (“Information Systems and Data”).
Various members of our management team, IT department and other employees, including but not limited to the individuals on our cybersecurity incident management team, help identify, assess and manage our cybersecurity threats and risks. We manage, identify and assess risks from cybersecurity threats by monitoring and evaluating our threat environment and risk profile using various methods including, for example: through the use of automated tools, including but not limited to tools for monitoring, geolocation, remote wiping, threat detection, intrusion detection and prevention (including through the use of machine learning, a form of artificial intelligence), patch management, distributed denial of service (DDoS) protection and forensics; conducting (directly or through third parties) regular audits and threat assessments for internal and external threats; subscribing to reports and services that identify cybersecurity threats; analyzing reports of threats and actors; conducting vulnerability assessments to identify vulnerabilities; evaluating our and our industry’s risk profile; conducting tabletop incident response exercises; and evaluating threats reported to us.
Depending on the environment, we implement and maintain various technical, physical, and organizational measures, processes, standards and policies designed to manage and mitigate material risks from cybersecurity threats to our Information Systems and Data, including, for example: incident response plans and procedures, disaster recovery/business continuity plans, risk assessments, implementation of security standards and certifications, encryption of data, network security controls, data segregation, access controls, physical security, asset management, tracking and disposal, systems monitoring, vendor risk management program, employee training and penetration testing.
Our assessment and management of material risks from cybersecurity threats are integrated into our overall risk management processes. For example, cybersecurity risk is addressed as a component of our enterprise risk management program, and members of our management team, IT department and other relevant team members work together to prioritize our risk management processes, mitigate cybersecurity threats that are more likely to lead to a material impact to our business, and report regularly to our board of directors on cybersecurity matters.
We use third-party service providers to assist us from time to time to identify, assess, and manage material risks from cybersecurity threats, including for example managed cybersecurity service providers, threat intelligence service providers, dark web monitoring services, and other cybersecurity software providers.
We use third-party service providers to perform a variety of functions throughout our business, including but not limited to application providers, hosting companies, contract manufacturing organizations and contract research organizations. We have a vendor management program to oversee, identify and manage cybersecurity risks associated with our use of these providers. The program includes a risk assessment for vendors that may include, depending on the vendor and nature of services being performed, security questionnaires, review of the vendor's written security program, review of security assessments, audits and reports, vulnerability scans related to the vendor, security assessment calls with the vendor's security personnel, and the imposition of certain contractual obligations on the vendor, among other elements, in accordance with the processes outlined in our internal vendor selection, management, and oversight process policy and other internal guidelines. More specifically, the level of assessment may depend on the following: the nature of the services provided and the data the vendors may collect, retain, and utilize, the sensitivity of the Information Systems and Data at issue, and the identity of the provider.
For a description of the risks from cybersecurity threats that may materially affect us and how they may do so, see our risk factors under Part 1. Item 1A. Risk Factors in this Annual Report on Form 10-K, including the risk factor captioned “If our information technology systems or data, or those of third parties upon which we rely, are or were compromised, we could experience adverse impacts resulting from such compromise, including, but not limited to, regulatory investigations or actions; litigation; fines and penalties; interruptions to our commercial operations, clinical trials or other operations; harm to our reputation; loss of revenue or profits; loss of sales and other adverse consequences.”
Governance
Our board of directors addresses our cybersecurity risk management as part of its general oversight function.
Our cybersecurity risk assessment and management processes are implemented and maintained by various members of our management team, IT department and other employees, including but not limited to the individuals on our cybersecurity incident management team, which includes individuals who have a diverse combination of relevant expertise, experience, education and training, with representation from our IT, legal, human resources, compliance, risk and privacy functions, among others. Our team includes individuals with relevant experience in enterprise risk management and disclosure controls and procedures. Additionally, certain members of our IT department have experience managing cybersecurity programs and are specifically assigned cybersecurity oversight.
Certain members of our management team and IT department are responsible for hiring appropriate personnel, helping to integrate cybersecurity risk considerations into our overall risk management strategy, communicating key priorities to relevant personnel, approving budgets, helping prepare for cybersecurity incidents, approving cybersecurity processes, and reviewing security assessments and other security-related reports.
Our cybersecurity incident response processes are designed to escalate certain cybersecurity incidents to members of management depending on the circumstances, including in some cases to our executive team. Our cybersecurity incident management team, and other individuals as needed, work to help us mitigate and remediate cybersecurity incidents of which we are notified. In addition, our incident response processes include a procedure for reporting certain cybersecurity incidents to the board of directors.
The board of directors receives regular reports from management concerning our cybersecurity risk management program. The board also receives various summaries and/or presentations related to cybersecurity threats, risks and mitigation.
Commencing in 2024, our Nominating / Corporate Governance Committee is taking the lead on behalf of the board of directors on oversight of our cybersecurity risk management program.
ITEM 2. PROPERTIES
We lease the following locations to conduct our business: | | | | | | | | | | | |
| Location | Address | Lease Expiration | Square Feet |
| San Diego, California | 3611 Valley Centre Drive, Suite 300 | August 31, 2028 | 149,123103,677 |
| Dublin, Ireland | 3 Mount Street Crescent, 2nd Floor | September 30, 2027 | 1,960 |
We believe these facilities are adequate to conduct our business.
For additional information regarding our lease agreements, see Note 18 of the Consolidated Financial Statements included in this report.
ITEM 3. LEGAL PROCEEDINGS
The information required by this Item is incorporated herein by reference to the Legal Proceedings paragraph of Note 11 ofNotes to the Consolidated Financial Statements includedStatements--Note 11 Commitments and Contingencies: Legal Proceedings in Item 15 of this report.Annual Report on Form 10-K.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
PART II
ITEM 5. MARKET FOR REGISTRANT'S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information
Our common stock is listed for quotation on the Nasdaq Global Market under the trading symbol “TVTX” and is part of the Nasdaq Biotechnology Index (Nasdaq: NBI).
As of February 22, 2022, the last reported sale price of our Common Stock as reported by the Nasdaq was $27.94.
As of February 22, 2022,15, 2024, we had approximately 177175 holders of record of our common stock.
Performance Graph
The following is not deemed “filed” with the Securities and Exchange Commission and is not to be incorporated by reference into any filing we make under the Securities Act of 1933, as amended, whether made before or after the date hereof and irrespective of any general incorporation by reference language in such filing.
Our common stock is traded on the Nasdaq Global Market and is a component of both the Nasdaq Composite Index and the Nasdaq Biotechnology Index. The total return for our common stock and for each index assumes the reinvestment of dividends, although dividends have never been declared on our common stock, and is based on the returns of the component companies weighted according to their capitalizations as of the end of each monthly period. The Nasdaq-Composite tracks the aggregate price performance of equity securities of companies traded on the Nasdaq National Market. The Nasdaq Biotechnology Index contains securities and tracks the aggregate price performance of equity securities of Nasdaq-listed companies classified according to the Industry Classification Benchmark as either Biotechnology or Pharmaceuticals which also meet other eligibility criteria. The comparisons shown in the graph are based upon historical data and we caution that the stock price performance shown in the graph is not indicative of, nor intended to forecast, the potential future performance of our stock.

Dividends
Since inception we have not paid any dividends on our common stock. We currently do not anticipate paying any cash dividends in the foreseeable future on our common stock. Although we intend to retain our earnings, if any, to finance the exploration and growth of our business, our Board of Directors will have the discretion to declare and pay dividends in the future. Payment of dividends in the future will depend upon our earnings, capital requirements and other factors which our Board of Directors may deem relevant.
ITEM 6. RESERVED[RESERVED]
ITEM 7. MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
Our discussion and analysis of our financial condition and results of operations for 20212023 as compared to 20202022 are discussed below and should be read in conjunction with our audited Consolidated Financial Statements, including the notes thereto. For a discussion of our financial condition and results of operations for 20202022 as compared to 2019,2021, except as set forth below, please refer to Item 7, “Management’s Discussion and Analysis of Financial Condition and Results of Operations” in our 20202022 Annual Report on Form 10-K, except as set forth below.which discussion is incorporated by reference herein. Overview
We are a biopharmaceutical company headquartered in San Diego, California, focused on identifying, developing and delivering life-changing therapies to people living with rare kidney liver, and metabolic diseases. Our approach centers on advancing our innovative pipeline with multiple late-stage clinical programs targeting rare diseases with significant unmet medical needs. Upon approval of any of our late-stage programs, we intend to leverage the skills of our talented commercial organization which has successfully identified, supported and treated patients prescribed our approved products over the last ten years.
Uncertainty Related
FILSPARI® (sparsentan)
On February 17, 2023, the FDA granted accelerated approval of FILSPARI® (sparsentan) to reduce proteinuria in adults with primary IgAN at risk of rapid disease progression, generally a UPCR ≥1.5 gram/gram. FILSPARI became commercially available in the COVID-19 PandemicU.S. beginning the week of February 27, 2023, and we are providing a comprehensive patient support program throughout the patient’s treatment journey.
WhileThis indication was granted under accelerated approval based on reduction in proteinuria. The continued approval of FILSPARI may be contingent upon confirmation of a clinical benefit in the impactPhase 3 PROTECT Study. As described in more detail below, in September 2023, we announced topline two-year confirmatory secondary endpoint results from the PROTECT Study, and in December 2023, we announced the completion of a successful pre-NDA meeting with the FDA for FILSPARI in IgAN. Following our regulatory engagement, we plan to submit a supplemental New Drug Application (sNDA) in the first quarter 2024 for conversion of the ongoing COVID-19 pandemic did not haveexisting U.S. accelerated approval of FILSPARI to full approval.
FILSPARI, a material adverse effect on our financial position or resultsonce-daily, oral medication is designed to selectively target two critical pathways in the disease progression of operationsIgAN (endothelin 1 and angiotensin-II) and is the first and only non-immunosuppressive therapy approved for the year ended December 31, 2021, we have been monitoring the developments and assessing areas where there is potential for our business to be impacted. Astreatment of December 31, 2021, the majority of our labor force is remote, which could, among other things, negatively impact our ability to conduct research and development activities, engage in sales-related initiatives, or efficiently conduct day-to-day operations. Remote work operations also heighten the risk of cyber-attacks and make it more difficult for companies to protect their confidential information. Circumstances arising from the pandemic have slowed and could continue to slow the pace of enrollment in our clinical trials or otherwise hinder patients' abilities to comply with the clinical trial protocols and could ultimately delay the availability of results and analysis of outcomes. Disruptions in the supply chain could negatively impact our ability to source materials or manufacture and distribute product. While to date we have not experienced a material reduction in demand for our commercialized products as a result of the pandemic, we could experience a decrease in new patient identification and increased requests for patient assistance due to increased levels of unemployment, either of which would negatively impact our revenues and hinder our cash flows. Similarly, we could face challenges with regard to healthcare programs, including access and changes in coverage. Growth in revenue could also be impeded by these factors. The financial markets have been subject to significant volatility that could impact our ability to enter into, modify, and negotiate favorable terms and conditions relative to equity and debt financing activities. We had $552.9 million in cash and cash equivalents and available-for-sale securities as of December 31, 2021, which we believe provides sufficient capital to fund our operations for at least the next twelve months. While we have not yet experienced a material impact to date, the full magnitude of the pandemic cannot be measured at this time, and therefore any of the aforementioned circumstances, as well as other factors, may cause our results of operations to vary substantially from year to year and quarter to quarter.condition.
License and Collaboration Agreement with Vifor Pharma
On September 15, 2021, we entered into a License Agreement with Vifor Pharma, pursuant to which we granted an exclusive license to Vifor Pharma for the commercialization of sparsentan in the Licensed Territories. Under the terms of the License Agreement, we received an upfront payment of $55.0 million in September 2021, and will be eligible for up to $135.0 million in aggregate regulatory and market access related milestone payments and up to $655.0 million in aggregate sales-based milestone payments for a total potential value of up to $845.0 million. We are also entitled to receive tiered double-digit royalties of up to 40 percent of annual net sales of sparsentan in the Licensed Territories.
The Agreement includes a sublicense to Vifor Pharma under our license agreement with Ligand Pharmaceuticals, Inc. (“Ligand”). We remain obligated to make payments to Ligand upon achievement of certain regulatory and sales milestones, as well as an escalating annual royalty between 15 percent and 17 percent of global net sales of licensed products.
Acquisition of Orphan Technologies Limited
In November 2020, we completed the acquisition of Orphan Technologies Limited (“Orphan”), including Orphan’s rare metabolic disorder drug pegtibatinase (TVT-058). We acquired Orphan by purchasing all of the outstanding shares. In exchange for the shares, we made an upfront cash payment at closing of $90.0 million plus closing adjustments, net liabilities assumed, and transaction expenses of $1.2 million, $1.8 million, and $4.2 million, respectively. Under the Agreement, we have also agreed to make contingent cash payments up to an aggregate of $427.0 million based on the achievement of certain development, regulatory and commercialization events as set forth in the Agreement, as well as additional tiered mid-single digit royalty payments based upon future net sales of any pegtibatinase products in the US and Europe, subject to certain reductions as set forth in the Agreement, and a contingent payment in the event a pediatric rare disease voucher for any pegtibatinase product is granted. In December 2021, Orphan was re-domiciled and renamed Travere Therapeutics Switzerland Gmbh ("TTSG").
Research and Development
Sparsentan
Sparsentan, a Dual Endothelin Angiotensin Receptor Antagonist (DEARA),FILSPARI is a novel investigational product candidate.dual endothelin angiotensin receptor antagonist (DEARA). Pre-clinical data have shown that blockade of both endothelin type A and angiotensin II type 1 pathways in forms of rare chronic kidney disease, reduces proteinuria, protects podocytes and prevents glomerulosclerosis and mesangial cell proliferation. Sparsentan has been granted Orphan Drug Designation for the treatment of FSGS and IgAN in the U.S. and Europe. Sparsentanthe EEA and FILSPARI has been granted seven years of Orphan Drug Exclusivity in the U.S. for the reduction of proteinuria in adults with primary IgAN at risk of rapid disease progression.
IgAN is characterized by hematuria, proteinuria, and variable rates of progressive renal failure. With an estimated prevalence of up to 150,000 people in the United States and greater numbers in Europe and Asia, IgAN is the most common primary glomerular disease. Most patients are diagnosed between the ages of 16 and 35, with up to 40% progressing to kidney failure within 15 years. FILSPARI is the first and only non-immunosuppressive therapy approved for this condition. We estimate approximately 30,000 to 50,000 patients in the United States to be addressable under FILSPARI's accelerated approval indication statement.
Data to support the accelerated approval of FILSPARI was generated from the Phase 3 PROTECT Study, the largest head-to-head interventional study to date in IgAN. It is a global, randomized, multicenter, double-blind, parallel-arm, active-controlled clinical trial that evaluated the safety and efficacy of 400mg of sparsentan, compared to 300mg of irbesartan, in 404 patients ages 18 years and up with IgAN and persistent proteinuria despite available ACE or ARB therapy, and is currently being evaluatedongoing in two pivotalthe open label extension phase of the study.
The PROTECT Study protocol provided for an unblinded analysis of at least 280 patients to be performed after 36 weeks of treatment to evaluate the primary efficacy endpoint - the change in proteinuria (UPCR) at week 36 from baseline. Secondary efficacy endpoints include the rate of change in estimated glomerular filtration rate (eGFR) following the initiation of randomized treatment over 58-week and 110-week periods, as well as the rate of change in eGFR over 52-week and 104-week periods following the first six weeks of randomized treatment in approximately 380 patients. In August 2021, we announced positive topline interim results from the ongoing Phase 3 PROTECT Study. The PROTECT Study met its pre-specified interim primary efficacy endpoint with statistical significance. After 36 weeks of treatment, patients receiving FILSPARI achieved a mean reduction in proteinuria from baseline of 49.8 percent, compared to a mean reduction in proteinuria from baseline of 15.1 percent for irbesartan-treated patients (p<0.0001). Results from the interim assessment in the PROTECT Study showed that FILSPARI was well tolerated with a clearly defined safety profile that has been consistent across all clinical studiestrials conducted to date. In PROTECT, at the interim assessment, the most common adverse reactions (≥ 5%) were peripheral edema, hypotension (including orthostatic hypotension), dizziness, hyperkalemia, and anemia. FILSPARI is available only through a risk evaluation and mitigation strategy (REMS) approved by the FDA.
Per request from the FDA, the efficacy data contained in rarethe FDA-approved label under accelerated approval is a post-hoc sensitivity analysis that evaluates the first 281 randomized patients, a subset of the full trial population. The mean reduction in proteinuria from baseline in the post-hoc sensitivity analysis is 45% for FILSPARI versus 15% for the active control, irbesartan. Both the pre-specified and post-hoc sensitivity analyses have demonstrated that FILSPARI achieves a rapid and sustained reduction in proteinuria, with statistically significant and clinically meaningful improvement compared to the active comparator irbesartan.
In September 2023, we announced topline two-year confirmatory secondary endpoint results from the PROTECT Study; eGFR total and chronic slope are the secondary confirmatory endpoints for the U.S. and the EU, respectively. FILSPARI demonstrated long-term kidney diseases, including:function preservation and achieved a clinically meaningful difference in eGFR total and chronic slope versus irbesartan, narrowly missing statistical significance in eGFR total slope while achieving statistical significance in eGFR chronic slope for purposes of regulatory review in the EU. All topline efficacy endpoints favored FILSPARI as compared to irbesartan. A preliminary review of the safety results through 110 weeks of treatment indicates FILSPARI was generally well-tolerated and the overall safety profile in the study has been consistent between treatment groups. In December 2023, we announced the completion of a successful pre-NDA meeting with the FDA for FILSPARI in IgAN. Following our regulatory engagement, we plan to submit a supplemental New Drug Application (sNDA) in the first quarter of 2024 for conversion of the existing U.S. accelerated approval of FILSPARI to full approval.
•In August 2022, we and ViforFocal segmental glomerulosclerosis (International) Ltd. ("FSGS"CSL Vifor"), with whom we entered into a license and collaboration agreement ("License Agreement") in September 2021, announced that the EMA had accepted for review the conditional marketing authorization ("CMA") application of sparsentan for the treatment of IgAN in the EU. We anticipate an opinion by the Committee for Medicinal Products for Human Use (the "CHMP") in the first quarter of 2024.
In January 2024, we announced our entry into an exclusive licensing agreement with Renalys Pharma, Inc. ("Renalys"), to bring sparsentan to patients in Japan and other countries in Asia, for the treatment of IgAN. Renalys will hold regional rights to sparsentan for Japan, South Korea, Taiwan, Brunei, Cambodia, Indonesia, Laos, Malaysia, Myanmar, the Philippines, Singapore, Thailand, and Vietnam. Following successful meetings with the Pharmaceuticals and Medical Devices Agency (PMDA) in 2023, Renalys plans to initiate an open label registrational study of sparsentan in Japan in the second quarter of 2024 to support potential approval of sparsentan in Japan. Results from the urine protein/creatinine ratio (UP/C) endpoint in the study are expected in the second half of 2025 to support a submission for approval to PMDA. Under the terms of the licensing agreement, Renalys will be responsible for development, regulatory matters, and commercialization in the licensed territories.
Clinical-Stage Programs:
Sparsentan for the treatment of FSGS
Sparsentan has been granted Orphan Drug Designation for the treatment of FSGS in the U.S. and the EEA.
FSGS is a leading cause of end-stage kidney disease (ESKD)failure and nephrotic syndrome. There are currently no United States Food and Drug Administration ("FDA") approvedFDA-approved pharmacologic treatments for FSGS and there remains a high unmet need for patients living with FSGS as off-label treatments such as ACE/ARBs, steroids, and immunosuppressant agents are effective in only a subset of patients and use of some of these off-label treatments may be further inhibited by their safety profiles. Every year approximately 5,400 patients are diagnosed with FSGS and we estimate that there are more than 40,000 FSGS patients in the United States and a similar number in Europe with approximately half of them being candidates for sparsentan.
In 2016, we generated positive data from our Phase 2 DUET study in FSGS. In 2018, we announced the initiation of the Phase 3 DUPLEX studyclinical trial designed to serve as the basis for an NDA and MAA filing for sparsentan for the treatment of sparsentan in FSGS.FSGS (the "DUPLEX Study"). The DUPLEX Study is a global, randomized, multicenter, double-blind, parallel-arm, active-controlled clinical trial evaluating the safety and efficacy of sparsentan in 371 patients. The DUPLEX Study protocol provided for an unblinded analysis of at least 190 patients to be performed after 36 weeks of treatment to evaluate the interim efficacy endpoint - the proportion of patients achieving a FSGS partial remission of proteinuria endpoint (FPRE), which is defined as urine protein-to-creatinine ratio (Up/C)(UPCR) ≤1.5 g/g and a >40% reduction in Up/CUPCR from baseline, at week 36. In February 2021, we announced that the ongoing Phase 3 DUPLEX Study achieved its pre-specified interim FSGS partial remission of proteinuria endpoint following the 36-week interim period. After 36 weeks of treatment, 42.0 percent of patients receiving sparsentan achieved FPRE, compared to 26.0 percent of irbesartan-treated
patients (p=0.0094). A preliminary reviewFollowing engagement with the FDA on the interim proteinuria analysis and a subsequent eGFR data-cut, we elected to forego the previously planned submission for accelerated approval and pursue a potential traditional approval upon completion of the DUPLEX Study.
In May 2023, we announced topline primary efficacy results from the interim analysis suggest that to datepivotal Phase 3 DUPLEX Study of sparsentan in the study, sparsentan has been generally well-tolerated and the overall safety results in the study to date have been generally comparable between treatment groups.FSGS. The confirmatory primary endpoint of the DUPLEX Study designed to support fulltraditional regulatory approval iswas the rate of change in eGFR over 108 weeks of treatment. AsAt the end of the time108-week double-blind period, sparsentan was observed to have a 0.3 mL/min/1.73m2 per year (95% CI: -1.74, 2.41) favorable difference on eGFR total slope and a 0.9 mL/min/1.73m2 per year (95% CI: -1.27, 3.04) favorable difference on eGFR chronic slope compared to the active control irbesartan, which was not statistically significant. After 108 weeks of the interim analyses, available long-term eGFR datatreatment, sparsentan achieved a mean reduction in proteinuria from baseline of 50%, compared to 32% for the confirmatory endpoint were limited. Consistent withirbesartan. Although the DUPLEX Study protocol, patients will continue indid not achieve its two-year primary endpoint with statistical significance over the active control irbesartan, we are encouraged by the results, including the secondary endpoints on proteinuria and topline exploratory endpoints, including renal outcomes, which trended favorably for sparsentan. In addition, a blinded manner to assessreview of the treatment effect on eGFR slope oversafety results through 108 weeks of treatment indicate sparsentan was generally well-tolerated and the overall safety profile in the confirmatory endpoint analysis. The DUPLEX Study is fully enrolled and toplinestudy to date was generally consistent between treatment groups.
In December 2023, we announced that we completed our planned Type C meeting with the FDA to discuss previously reported results from the confirmatory endpoint are expectedPhase 3 DUPLEX Study of sparsentan in FSGS. The FDA acknowledged the first half of 2023.
In May 2021, we provided a regulatory update regardinghigh unmet need for approved therapies as well as the sparsentanchallenges in studying FSGS program, including feedbackbut indicated that the two-year results from the Phase 3 DUPLEX Study alone are not sufficient to support an sNDA submission. The FDA thatacknowledged the work being done by the larger nephrology community to better understand proteinuria and eGFR as endpoints in clinical trials of FSGS and indicated a willingness to continue to engage with us on a potential path forward for sparsentan in FSGS following our consideration of additional data would be neededevidence. Together with CSL Vifor, we also plan to potentially support a submissionengage with the EMA to determine the potential for accelerated approval under subpart H. At a subsequent Type A meeting, we andvariation to the FDA reached alignment on a pathway for us to proceed with a submission for accelerated approval, pending additional supportive eGFR data. We intend to provide the FDA with additional eGFR data from the ongoing DUPLEX Study in the first half of 2022, and if such data are supportive, submit an application for accelerated approval in the U.S. in mid-2022.
In mid-2022, we and Vifor (International) Ltd. ("Vifor Pharma"), with whom we entered into a license and collaboration agreement ("License Agreement") in September 2021, plan to submit an application for conditional marketing authorization ("CMA")Conditional Marketing Authorization (CMA) of sparsentan for the treatment of FSGS, if the MAA of sparsentan in IgA nephropathy is approved. Given the high unmet need of FSGS patients, with no medicines currently approved for the condition, and IgAN in Europe. the challenges associated with studying FSGS due to its heterogeneity and other attributes, we are conducting additional analyses of FSGS data and intend to engage with regulators to evaluate potential regulatory pathways for a sparsentan FSGS indication.
If sparsentan receives marketing authorization in any of the licensed territories covered by the exclusive license to CSL Vifor, PharmaCSL Vifor will be responsible for all commercialization activities in such licensed territories. If sparsentan receives marketing authorization in any of the territories covered by the exclusive license to Renalys, Renalys will be responsible for all development, regulatory matters, and commercialization activities in such licensed territories. We remain responsible for the clinical development of sparsentan in the applicable territories and we will retain all rights to sparsentan in the United States and rest of world outside of the territories licensed territories,to CSL Vifor and Renalys, provided that CSL Vifor Pharma has a right of negotiation to expand the licensed territories into Canada, China, Brazil and/or Mexico.
•Immunoglobulin A nephropathy ("IgAN") is characterized by hematuria, proteinuria, and variable rates of progressive renal failure. With an estimated prevalence of more than 100,000 people in the United States and greater numbers in Europe and Asia, IgAN is the most common primary glomerular disease. Most patients are diagnosed between the ages of 16 and 35, with up to 40% progressing to end stage kidney disease within 15 years. There are currently no non-immunosuppressive treatments for IgAN approved by the FDA. The current standard of care is renin-angiotensin-aldosterone system ("RAAS") blockade with immunosuppression also being commonly used
for patients with significant proteinuria or rapidly progressive glomerulonephritis. In 2018, we announced that the first patient had been dosed in the PROTECT Study, a global, randomized, multicenter, double-blind, parallel-arm, active-controlled pivotal Phase 3 clinical trial evaluating the safety and efficacy of sparsentan in 404 patients with IgAN.
The PROTECT Study protocol provided for an unblinded analysis of at least 280 patients to be performed after 36 weeks of treatment to evaluate the primary efficacy endpoint - the change in proteinuria (urine protein-to-creatinine ratio) at week 36 from baseline. Secondary efficacy endpoints include the rate of change in eGFR following the initiation of randomized treatment over 58-week and 110-week periods, as well as the rate of change in eGFR over 52-week and 104-week periods following the first six weeks of randomized treatment in approximately 380 patients. In August 2021, we announced positive topline interim results from the ongoing Phase 3 PROTECT Study. The PROTECT Study met its pre-specified interim primary efficacy endpoint with statistical significance. After 36 weeks of treatment, patients receiving sparsentan achieved a mean reduction in proteinuria from baseline of 49.8 percent, compared to a mean reduction in proteinuria from baseline of 15.1 percent for irbesartan-treated patients (p<0.0001). We believe that preliminary eGFR data available at the time of the interim analysis are indicative of a potential clinically meaningful treatment effect after two years of treatment. Preliminary results at the time of the interim assessment suggested that sparsentan had been generally well-tolerated to date in the study and consistent with its overall observed safety profile. The PROTECT Study is fully enrolled and is scheduled to continue as planned on a blinded basis to assess the treatment effect on eGFR slope over 110 weeks in the confirmatory endpoint analysis. Topline results from the confirmatory endpoint analysis are expected in the second half of 2023.
Subsequent to the announcement of the interim results from the PROTECT Study, we conducted pre-NDA interactions with the FDA, during which we confirmed alignment with our plan to submit in the first quarter of 2022 an NDA for accelerated approval of sparsentan for IgAN.
In mid-2022, we and Vifor Pharma plan to submit a combined application for conditional marketing authorization ("CMA") of sparsentan for the treatment of FSGS and IgAN in Europe.
Pegtibatinase (TVT-058)
Pegtibatinase (TVT-058) is a novel investigational human enzyme replacement candidate being evaluated for the treatment of classical homocystinuria (HCU). Classical HCU is a rare metabolic disorder characterized by elevated levels of plasma homocysteine that can lead to vision, skeletal, circulatory and central nervous system issues.complications. It is estimated that there are at least 3,500approximately 7,000 to 10,000 people living with HCU in the United States with similar numbers in Europe.globally. Pegtibatinase has been granted Rare Pediatric Disease, and Fast Track and Breakthrough Therapy designations by the FDA, as well as orphan drug designation in the United States and European Union. Pegtibatinase is currently being evaluated in
In December 2021, we announced positive topline results from the Phase 1/2 COMPOSE Study, a double blind, randomized, placebo-controlled dose escalation study to assess its safety, tolerability, pharmacokinetics, pharmacodynamics and clinical effects in patients with classical HCU.
In December 2021, we announced positive topline results from the Phase 1/2 COMPOSE Study. Pegtibatinase demonstrated dose-dependent reductions in total homocysteine (tHcy) during the 12 weeks of treatment, and in the highest dose cohort to date evaluating 1.5mg/kg of pegtibatinase twice weekly (BIW), treatment with pegtibatinase resulted in rapid and sustained reductions in total homocysteine (tHcy) through 12 weeks of treatment, including a 55.1% mean relative reduction in tHcy from baseline as well as maintenance of tHcy below a clinically meaningful threshold of 100 μmol. Additionally, in a dose-dependent manner in the study to date, methionine levels were substantially reduced and cystathionine levels were substantially elevated following treatment with pegtibatinase, suggesting that pegtibatinase acts in a manner similar to the native CBS enzyme. To date in
In May 2023, we announced positive topline results from the study, pegtibatinase has been generally well-tolerated, with no discontinuations due to treatment-related adverse events.
Based on these results,sixth cohort of the Company is in the process of engaging with regulators to establish next steps for a pivotal development program to ultimately support potential approvals of pegtibatinase for the treatment of HCU. In parallel, the Company has initiated one additional cohort in thePhase 1/2 COMPOSE Study, which was initiated to inform and refine formulation work for future development and commercial purposes and to further evaluate the dose response curve for pegtibatinase.pegtibatinase, and to further inform our pivotal development program to ultimately support potential approval of pegtibatinase for the treatment of HCU.In this cohort, five patients were randomized in a blinded fashion to receive 2.5 mg/kg of lyophilized pegtibatinase or placebo twice weekly (BIW), with four patients assigned to the treatment group. In this highest dose cohort to date, treatment with pegtibatinase resulted in rapid and sustained reductions in total homocysteine (tHcy), with a 67.1% mean relative reduction in tHcy from baseline, as well as maintenance of mean tHcy below the clinically meaningful threshold of 100 μmol, over weeks 6 to 12.In the double-blind period, pegtibatinase was generally well-tolerated, with no discontinuations due to treatment-related adverse events.
In December 2023, we initiated the pivotal Phase 3 HARMONY Study to support the potential approval of pegtibatinase for the treatment of classical HCU. The HARMONY Study is a global, randomized, multi-center, double-blind, placebo-controlled Phase 3 clinical trial designed to evaluate the
efficacy and safety of pegtibatinase as a novel treatment to reduce total homocysteine (tHcy) levels. Topline results from the HARMONY Study are expected in 2026.
We acquired pegtibatinase as part of the November 2020 acquisition of Orphan Technologies Limited.
Chenodal
Chenodal (chenodeoxycholic acid or CDCA) is a naturally occurring bile acid that is approved for the treatment of people with radiolucent stones in the gallbladder. While indicated for radiolucent stones in the gallbladder, Chenodal has been recognized as the standard of care for cerebrotendinous xanthomatosis (CTX) for more than three decades, although it is not currently labeled for this indication. CTX is a rare, progressive and underdiagnosed bile acid synthesis disorder affecting many parts of the body. In January 2020, we randomized the first patients in our Phase 3 RESTORE Study to evaluate the effects of Chenodal in adult and pediatric patients with CTX, and the study enrollment remains open. The pivotal study is intended to support an NDA submission for marketing authorization of Chenodal for CTX in the United States.
Preclinical ProgramsPrograms:
We are a participant in twoa Cooperative Research and Development Agreements ("CRADAs")Agreement (CRADA), which formforms a multi-stakeholder approach to pool resources with leading experts and incorporate the patient perspective early in the therapeutic identification and development process. We have partneredare partnering with the National Institutes of Health’s National Center for Advancing Translational Sciences ("NCATS")(NCATS) and a leading patient advocacy organizations, CDG Care andorganization, Alagille Syndrome Alliance, aimed at the identification of potential small molecule therapeutics for NGLY1 deficiency and Alagille syndrome ("ALGS"), respectively.(ALGS). There are no treatment options currently approved for these diseases.ALGS.
Approved ProductsWe are party to a collaboration agreement with PharmaKrysto Limited and their early-stage cystinuria discovery program, whereby we are responsible for funding all research and development expenses for the pre-clinical activities associated with the cystinuria program.
Other Commercial Products:
Thiola and Thiola EC (tiopronin)
Thiola and Thiola EC are approved by the FDA for the treatment of cystinuria, a rare genetic cystine transport disorder that causes high cystine levels in the urine and the formation of recurring kidney stones. Due to the larger stone size, cystine stones may be more difficult to pass, often requiring surgical procedures to remove. More than 80 percent of people with cystinuria develop their first stone by the age of 20. More than 25 percent will develop cystine stones by the age of 10. Recurring stone formation can cause loss of kidney function in addition to substantial pain and loss of productivity associated with renal colic and stone passage. While a portion of people living with the disease are able to manage symptoms through diet and fluid intake, the prevalence of cystinuria in the US is estimated to be 10,000 to 12,000, indicating that there may be as many as 4,000 to 5,000 affected individuals with cystinuria in the US that would be candidates for Thiola or Thiola EC.
In June 2019 we announced that the FDA approved 100 mg and 300 mg tablets of Thiola EC, an enteric-coated formulation of Thiola, to be used for the treatment of cystinuria. Thiola EC offers the potential for administration with or without food, and the ability to reduce the number of tablets necessary to manage cystinuria. Thiola EC became available to patients in July 2019.
In May 2021, a generic option for the 100 mg version of the original formulation of Thiola (tiopronin tablets) became available. Whileavailable and in June 2022, a second option for the 100 mg version of the original formulation of Thiola (tiopronin tablets) was approved. These generic versions of the original formulation of Thiola have impacted our sales, and these or additional generic versions of either formulation could have a material adverse impact on sales. During 2023, two generic versions of Thiola EC (100 mg and 300 mg) were approved by the FDA. Following receipt of paragraph IV certification notices from these manufacturers, we and our licensor Mission Pharmacal entered into agreements with these manufacturers in order to settle disputes around patent validity and infringement, providing for a license entry date of April 1, 2026 of these generic versions of Thiola EC (100mg and 300mg), or earlier under certain circumstances. On January 30, 2024, the FDA approved an additional generic version of Thiola EC (100mg and 300mg). Accordingly, Thiola EC is subject to immediate generic competition, which could impact our sales of Thiola EC.
Sale of Bile Acid Product Portfolio
On July 16, 2023, we entered into an Asset Purchase Agreement (the “Purchase Agreement”) with Mirum Pharmaceuticals, Inc. ("Mirum Pharmaceuticals" or “Mirum”), pursuant to which Mirum agreed to purchase substantially all of the assets primarily related to our business of development, manufacture (including synthesis, formulation, finishing or packaging) and commercialization of Chenodal and Cholbam (also known as Kolbam, and together with Chenodal, the “Products”), collectively, the "bile acid business". On August 31, 2023, we consummated the transactions contemplated by the Purchase Agreement (the "Closing"). In connection with the Closing, we received an upfront cash payment of $210.0 million. Pursuant to the Purchase Agreement, after the Closing, we are eligible to receive up to $235.0 million upon the achievement of certain milestones based on specified amounts of annual net sales (tiered from $125.0 million to $500.0 million) of the Products.
A $226.0 million gain, net of tax, was recognized on the transaction as a component of net income from discontinued operations in the Consolidated Statements of Operations. The bile acid business has been minimal to date, we are not able to estimate any future impact, includingclassified as a discontinued operation for all periods presented and is excluded from the impactfollowing discussion of additional generic entrants, if any.
Cholbam (cholic acid)
The FDA approved Cholbam (cholic acid capsules) in March 2015, the first FDA-approved treatment for pediatric and adult patients with bile acid synthesis disorders due to single enzyme defects, and for adjunctive treatmentresults of patients with peroxisome biogenesis disorder-Zellweger spectrum disorder. The effectiveness of Cholbam has been demonstrated in clinical trials for bile acid synthesis disorders and the adjunctive treatment of peroxisomal disorders. An estimated 200 to 300 patients are current candidates for therapy.
Chenodal (chenodiol)
Chenodal is a synthetic oral form of chenodeoxycholic acid ("CDCA"), a naturally occurring primary bile acid synthesized from cholesterolour continuing operations in the liver. The FDA approved Chenodalresults of operations. Refer to Note 19 of our Consolidation Financial Statements for the treatment of people with radiolucent stones in the gallbladder. In 2010, Chenodal was granted orphan drug designation for the treatment of cerebrotendinous xanthomatosis ("CTX"), a rare autosomal recessive lipid storage disease. We acquired Chenodal in March 2014.additional information.
While Chenodal is not labeled for CTX, it received a medical necessity determination in the US by the FDA and has been used as the standard of care for more than three decades. We are working to obtain FDA approval of Chenodal for the treatment of CTX and initiated a Phase 3 clinical trial for this indication in January 2020. The prevalence of CTX is estimated in the literature to be as high as 1 in 70,000 in the overall population. Pathogenesis of CTX involves deficiency of the enzyme 27-hydroxylase (encoded by the gene CYP27A1), a rate-limiting enzyme in the synthesis of primary bile acids, including CDCA, from cholesterol. The disruption of primary bile acid synthesis in CTX leads to toxic accumulation of cholesterol and cholestanol in most tissues. Patients may present with intractable diarrhea, premature cataracts, tendon xanthomas, atherosclerosis, and cardiovascular disease in childhood and adolescence. Neurological manifestations of the disease, including dementia and cognitive and cerebellar deficiencies, emerge during late adolescence and adulthood. The types, combinations and severity of symptoms can be different from person to person, and making diagnosis challenging and often delayed. Oral administration of CDCA has been shown to normalize primary bile acid synthesis in patients with CTX.
Financial Overview
Acquired IPR&D
We assess whether IPR&D assets acquired from others in an asset acquisition have alternative future use (in research and development projects or otherwise) at the acquisition date. If such assets have alternative future use, they are capitalized and recognized as an intangible asset. If such assets do not have alternative use, nor is there an alternative indication that the Company plans to pursue, the upfront consideration transferred, including any contingent consideration that is probable and reasonably estimable, is charged to expense at the acquisition date. We expensed the
acquired IPR&D associated withStrategic Reorganization
In December 2023, we implemented an approximate 20% workforce reduction focused on non-field-based employees in an effort to align our resources on the acquisition of Orphan Technologies, which includedongoing FILSPARI launch and the developmental product candidate for the treatment of classical homocystinuria (HCU), pegtibatinase (TVT-058).
Research and Development Costs
Research and development costs include expenses related to sparsentan, discontinued programs fosmetpantotenate and CNSA-001, and our other pipeline programs, including the recently acquired pegitbatinase (TVT-058) program. We expense all research and development costs as they are incurred. Our research and development costs are comprised of salaries and bonuses, benefits, non-cash share-based compensation, license fees, milestones under license agreements, costs paid to third-party contractors to perform research, conduct clinical trials, and develop drug materials and delivery methods, and associated overhead expenses and facilities costs. We charge direct internal and external program costs to the respective development programs. We also incur indirect costs that are not allocated to specific programs because such costs benefit multiple development programs and allow us to increase our pharmaceutical development capabilities. These consist of internal shared resources related to the development and maintenance of systems and processes applicable to all of our programs.
We record expenses in connection with our clinical trials under contracts with contract research organizations ("CROs") that support conducting and managing clinical trials. The financial terms and activities of these agreements vary from contract to contract and may result in uneven expense levels. Generally, these agreements set forth activities that drive the recording of expenses such as start-up, initiation activities, enrollment, treatment of patients, or the completion of other clinical trial activities.
Expenses related to clinical trials are accrued based on our estimates and/or representations from service providers regarding work performed, including actual level of patient enrollment, completion of patient studies and progress of the clinical trials. Other incidental costs related to patient enrollment or treatment are accrued when reasonably certain. If the amounts we are obligated to pay under our clinical trial agreements are modified (for instance, as a result of changes in the clinical trial protocol or scope of work to be performed), we adjust our accruals accordingly on a prospective basis. Revisions to our contractual payment obligations are charged to expense in the period in which the facts that give rise to the revision become reasonably certain.
We currently have threepivotal Phase 3 clinical trials in process that are in varying stages of activity, with ongoing non-clinical support studies. As such, clinical trial expenses will vary depending on the all the factors set forth above and may fluctuate significantly from quarter to quarter.
At any point in time, we typically have various early stage research and drug discovery projects. Our internal resources, employees and infrastructure are not directly tied to any one research or drug discovery project and are typically deployed across multiple projects. As such, we do not maintain information regarding costs incurred for these early stage research and drug discovery programs on a project-specific basis.
We routinely engage vendors and service providers for scientific research, clinical trial, regulatory compliance, manufacturing and other consulting services. We also make grants to research and non-profit organizations to conduct research which may lead to new intellectual properties that we may subsequently license under separately negotiated license agreements. Such grants may be funded in lump sums or installments.
The following table summarizes our research and development expenses during the years ended December 31, 2021, 2020 and 2019. The internal costs include personnel, facility costs, and discovery and research related activities associated with our pipeline. The external program costs reflect external costs attributable to our clinical development candidates and preclinical candidates selected for further development. Such expenses primarily include third-party contract costs relating to clinical trial activities, nonclinical studies and manufacturing.
| | | | | | | | | | | | | | | | | |
| For the Year Ended December 31, |
| | | (in thousands) | | |
| 2021 | | 2020 | | 2019 |
| External service provider costs: | |
| Sparsentan | $ | 103,350 | | | $ | 70,486 | | | $ | 48,533 | |
| Fosmetpantotenate (discontinued) | (124) | | 1 | (2,295) | | 1 | 31,193 | |
| Censa (discontinued) | — | | | — | | | 3,166 | |
| Pegtibatinase | 27,823 | | | 1,962 | | | — | |
| Other product candidates | 6 | | | — | | | 496 | |
| General | 18,780 | | | 15,433 | | | 19,638 | |
| Total external service provider costs | 149,835 | | | 85,586 | | | 103,026 | |
| Internal personnel costs | 60,493 | | | 46,187 | | | 37,937 | |
| Total research and development | $ | 210,328 | | | $ | 131,773 | | | $ | 140,963 | |
1Credit received in 2021 and 2020.
Most of our product development programs are in clinical trials which are highly uncertain and may not result in approved products. Completion dates and completion costs can vary significantly for each product candidate and are difficult to project. Given the uncertainty associated with clinical trial enrollments and the risks inherent in the development process, we are unable to determine the duration and completion costs of current or future clinical trials of our product candidates, or if and to what extent we will generate revenues, if any, from the commercialization and sale of any of our product candidates.
Selling, General and Administrative
Selling, general and administrative expenses consist of salaries and bonuses, benefits, non-cash share-based compensation, legal and other professional fees, rent, depreciation and amortization, travel, insurance, business development, sales and marketing programs, and other operating expenses.
Other Income/Expenses
Other income/expenses consists of interest income and expense, finance expense and miscellaneous other income/expenses.
License Agreements
Ligand License Agreement
In 2012, we entered into a license agreement with Ligand, granting us a worldwide license for the development, manufacture and commercialization of sparsentan, which we are developing in connection with the treatment of FSGS. Under the license agreement, Ligand granted us a sublicense under certain of its patents and other intellectual property in connection with the development and commercialization of sparsentan. Under the license agreement, Ligand is obligated to transfer to us certain information, records, regulatory filings, materials and inventory controlled by Ligand and relating to or useful for developing sparsentan. We must use commercially reasonable efforts to develop and commercialize sparsentan in specified major market countries and other countries in which we believe it is commercially reasonable to develop and commercialize such products.
As consideration for the license, we are required to make payments upon the achievement of certain milestones, totaling up to $114.1 million. Should we commercialize sparsentan or any products containing any of the licensed compounds, we will be obligated to pay Ligand an escalating annual royalty between 15% and 17% of net sales of all such products. Through 2021, we made milestone payments to Ligand of $7.2 million under the license agreement.
Under the terms of the license agreement, Bristol-Myers Squibb Company (“BMS”) has a right of first negotiation and Ligand has a right of second negotiation with respect to any license arrangement for a licensed compound, except to the extent such rights may be waived.
The license agreement will continue until neither party has any further payment obligations under the agreement and is expected to continue for approximately 10 to 20 years from the effective date. Ligand may terminate the license agreement due to (i) our insolvency, (ii) our material uncured breach of the agreement, (iii) our failure to use commercially reasonable efforts to develop and commercialize sparsentan as described above or (iv) certain other conditions. We may terminate the license agreement due to a material uncured breach of the agreement by Ligand.
Thiola License Agreement
In 2014, we entered into a license agreement with Mission Pharmacal ("Mission"), pursuant to which we obtained an exclusive, royalty-bearing license to market, sell and commercialize Thiola (tiopronin) in the United States and Canada, and a non-exclusive license to use know-how relating to Thiola to the extent necessary to market Thiola. We paid Mission an up-front license fee of $3.0 million and will pay guaranteed minimum royalties during each calendar year the greater of $2.0 million or 20% of our net sales of Thiola in the United States and Canada.
In October 2015, the license agreement was amended to allow for us to secure enough API to ensure an adequate level of safety stock to prevent an interruption in the supply of Thiola and to prepare for a reformulation development project.
In March 2016, the license agreement was amended to, among other things, include a new formulation development project for tiopronin tablets.
In November 2017, we amended the license agreement to extend the term through May of 2029.
In November 2018, the license agreement was amended to remove all territorial restrictions on our license rights. As consideration for the expanded territory we paid an up-front fee of $0.3 million and will pay guaranteed minimum royalties equaling the greater of $0.1 million or 20% of our Thiola net sales generated outside of the United States during each calendar year.
See Note 9 to Consolidated Financial Statements for further discussion.
License and Collaboration Agreement with Vifor Pharma
On September 15, 2021, we entered into a License Agreement with Vifor Pharma, pursuant to which we granted an exclusive license to Vifor Pharma for the commercialization of sparsentan in the Licensed Territories. Under the terms of the License Agreement, we received an upfront payment of $55.0 million in September 2021, and will be eligible for up to $135.0 million in aggregate regulatory and market access related milestone payments and up to $655.0 million in aggregate sales-based milestone payments for a total potential value of up to $845.0 million. We are also entitled to receive tiered double-digit royalties of up to 40 percent of annual net sales of sparsentan in the Licensed Territories.
The License Agreement includes a sublicense to Vifor Pharma under our license agreement with Ligand Pharmaceuticals, Inc. (“Ligand”). We remain obligated to make payments to Ligand upon achievement of certain regulatory and sales milestones, as well as an escalating annual royalty between 15 percent and 17 percent of global net sales of licensed products.
See Note 4 to Consolidated Financial Statements for further discussion.
Results of Operations
Revenue
The following table provides information regarding revenue, including net product sales and license and collaboration revenue (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Year Ended December 31, | | Year Ended December 31, |
| | 2021 | | 2020 | | Change | | 2020 | | 2019 | | Change |
| Tiopronin products | $ | 115,122 | | | $ | 108,883 | | | $ | 6,239 | | | $ | 108,883 | | | $ | 95,638 | | | $ | 13,245 | |
| Bile acid products | 95,654 | | | 89,438 | | | 6,216 | | | 89,438 | | | 79,700 | | | 9,738 | |
| Total net product sales | 210,776 | | | 198,321 | | | 12,455 | | | 198,321 | | | 175,338 | | | 22,983 | |
| License and collaboration revenue | 16,714 | | | — | | | 16,714 | | | — | | | — | | | — | |
| Total revenue | $ | 227,490 | | | $ | 198,321 | | | $ | 29,169 | | | $ | 198,321 | | | $ | 175,338 | | | $ | 22,983 | |
Net product sales for the years ended December 31, 2021, 2020 and 2019 were $210.8 million, $198.3 million and $175.3 million, respectively, and consisted of sales of Thiola ("Tiopronin products"), Chenodal and Cholbam ("Bile acid products").
The increase in net product sales for the year ended December 31, 2021 as compared to the same period in 2020, is due to increased patient counts for all products.
We use a direct-to-patient distributor. Under this distribution model, we record revenues when customers take control of the product (at delivery).
License and collaboration revenue for the year ended December 31, 2021 was $16.7 million. We had no license or collaboration revenue for the years ended December 31, 2020 and 2019. License and collaboration revenue recognized in 2021 was attributable to the license and collaboration agreement entered into with Vifor Pharma in September 2021. We recognized $12.0 million from the license based upon the allocation of the stand-alone selling price, and $4.7 million from the clinical development activities based upon the ratio of costs incurred to total estimated costs.
Operating Expenses
The following table provides information regarding operating expenses (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Year Ended December 31, | | Year Ended December 31, |
| | 2021 | | 2020 | | Change | | 2020 | | 2019 | | Change |
| Cost of goods sold | $ | 6,784 | | | $ | 6,126 | | | $ | 658 | | | $ | 6,126 | | | $ | 5,234 | | | $ | 892 | |
| Research and development | 210,328 | | | 131,773 | | | 78,555 | | | 131,773 | | | 140,963 | | | (9,190) | |
| Selling, general and administrative | 149,883 | | | 135,799 | | | 14,084 | | | 135,799 | | | 128,951 | | | 6,848 | |
| Change in fair value of contingent consideration | 22,260 | | | 3,655 | | | 18,605 | | | 3,655 | | | 15,051 | | | (11,396) | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| Impairment of L-UDCA IPR&D intangible asset | — | | | — | | | — | | | — | | | 25,500 | | | (25,500) | |
| Write off of L-UDCA contingent consideration | — | | | — | | | — | | | — | | | (18,000) | | | 18,000 | |
| Acquired IPR&D expense | — | | | 97,131 | | | (97,131) | | | 97,131 | | | — | | | 97,131 | |
| Impairment of long-term investment | — | | | — | | | — | | | — | | | 15,000 | | | (15,000) | |
| | $ | 389,255 | | | $ | 374,484 | | | $ | 14,771 | | | $ | 374,484 | | | $ | 312,699 | | | $ | 61,785 | |
2021 versus 2020 results
Operating expenses for the year ended December 31, 2021, were $389.3 million compared to $374.5 million for the year ended December 31, 2020, an increase of $14.8 million.
Cost of goods sold increased by $0.7 million due to increased sales.
Research and development costs increased by $78.6 million due to increases in clinical trial expenses, chemistry manufacturing and control costs, preclinical study expenses, and related expenses associated with the advancement of our programs in development, particularly sparsentan and pegtibatinase, which saw increases in external service provider costs of $32.9 million and $25.9 million, respectively. Internal personnel costsHARMONY Study to support these programs also increased by $14.3 million.
Selling, general and administrative expenses increased by $14.1 million due to increased personnel expenses, including increased headcountthe potential approval of pegtibatinase as we preparethe first potential disease-modifying treatment for commercialization of sparsentan and enhance support for other programs in development.
Fair value of contingent consideration increased by $18.6 million due to changes in discount factors, along with changes in revenue forecasts and timing of payments.
Acquired IPR&D of $97.1 million in 2020 was due to the November 2020 acquisition of Orphan Technologies Limited, which included the developmental rare metabolic disorder drug pegtibatinase.
Other Income/Expenses
The following table provides information regarding other income (expenses) (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Year Ended December 31, | | Year Ended December 31, |
| | 2021 | | 2020 | | Change | | 2020 | | 2019 | | Change |
| | | | | | | | | | | |
| Interest income | $ | 1,993 | | | $ | 5,003 | | | $ | (3,010) | | | $ | 5,003 | | | $ | 10,055 | | | $ | (5,052) | |
| Interest expense | (20,141) | | | (19,050) | | | (1,091) | | | (19,050) | | | (18,828) | | | (222) | |
| Other income (expense), net | 231 | | | 1,420 | | | (1,189) | | | 1,420 | | | (314) | | | 1,734 | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | $ | (17,917) | | | $ | (12,627) | | | $ | (5,290) | | | $ | (12,627) | | | $ | (9,087) | | | $ | (3,540) | |
Other expense for the year ended December 31, 2021 was $17.9 million compared to other expense of $12.6 million for the year ended December 31, 2020, which represents a change of $5.3 million. The change was primarily attributable to a decrease in interest income earned on our available-for-sale marketable debt securities. The decrease in interest income earned was due to a decline in short-term interest rates.
Income Tax Benefit (Provision)
We follow ASC 740, Income Taxes, which requires recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have been included in the financial statements or tax returns. Under this method, deferred tax assets and liabilities are based on the differences between the financial statement and tax basis of assets and liabilities using enacted tax rates in effect for the year in which the differencesHCU. These restructuring adjustments are expected to reverse. Deferred tax assets are reduced by a valuation allowanceresult in an estimated annualized savings of approximately $25.0 million beginning in 2024, and an estimated non-recurring charge of approximately $12.0 million to the extent management concludes it is more likely than not that the asset will not be realized.
The standard addresses the determination$14.0 million, of whether tax benefits claimed or expected to be claimed on a tax return should be recorded in the financial statements. Under ASC 740, we may recognize the tax benefit from an uncertain tax position only if it is more likely than not that the tax position will be sustained on examination by the tax authorities, based on the technical merits of the position. The tax benefitswhich $11.4 million was recognized in the financial statements from such a position should be measured based on the largest benefit that has a greater than fifty percent likelihood of being realized upon ultimate settlement. ASC 740 also provides guidance on de-recognition, classification, interest and penalties on income taxes, accounting in interim periods and requires increased disclosures. Our policy is to record estimated interest and penalty related to the underpayment of income taxes or unrecognized tax benefits as a component of its income tax provision.
The change in tax provision of $0.4 million for fiscal 2021 as compared to a tax benefit of $19.4 million for fiscal 2020 was due to the income tax benefit related to the CARES Act NOL carryback that was recognized in 2020.
Inflation
Inflation generally affects us by increasing our salaries and fees paid to third-party contract service providers.
Liquidity and Capital Resources
fourth quarter 2023. We have financed our operations through a combination of borrowings, sales of our equity securities, and revenues generated from our commercialized products and license and collaboration agreements. Research and development activities have required significant capital investment and are expected to continue to require significant cash expenditure in the future, particularly as our pipeline of drug candidates and our employee headcount have expanded. In addition, we continue to evaluate potential opportunities to expand our pipeline and approved products through licenses and acquisitions of products in areasexpect that we believe offer attractive growth characteristics, which may require considerable upfront capital to pursue as well as possible contingent payments upon the future achievement of certain milestones.
We believe that our available cash and short-term investments as of the date of this filing will be sufficient to fund our anticipated level of operations beyond the next 12 months. Management believes that our operating results will vary from quarter to quarter and year to year depending upon various factors including revenues, general and administrative expenses, and research and development expenses.
For the years ended December 31, 2021 and 2020, we had the following balances and financial performance (in thousands):
| | | | | | | | | | | |
| December 31, 2021 | | December 31, 2020 |
| Revenue | $ | 227,490 | | | $ | 198,321 | |
| Net loss | (180,091) | | | (169,431) | |
| Cash & cash equivalents | 165,753 | | | 84,772 | |
| Marketable debt securities | 387,129 | | | 276,817 | |
| Accumulated deficit | (765,966) | | | (585,875) | |
| Stockholders' equity | 302,112 | | | 211,213 | |
| Working capital | $ | 458,739 | | | $ | 317,747 | |
| Working capital ratio | 4.70 | | | 4.43 | |
Collaboration and License Proceeds
License and Collaboration Agreement with Vifor Pharma
On September 15, 2021, we entered into a License Agreement with Vifor Pharma, pursuant to which we granted an exclusive license to Vifor Pharma for the commercialization of sparsentan in the Licensed Territories. Under the terms of the License Agreement, we received an upfront payment of $55.0 million in September 2021, and will be eligible for up to $135.0 million in aggregate regulatory and market access related milestone payments and up to $655.0 million in aggregate sales-based milestone payments for a total potential value of up to $845.0 million. We are also entitled to receive tiered double-digit royalties of up to 40 percent of annual net sales of sparsentan in the Licensed Territories.
The Agreement includes a sublicense to Vifor Pharma under our license agreement with Ligand Pharmaceuticals, Inc. (“Ligand”). We remain obligated to make payments to Ligand upon achievement of certain regulatory and sales milestones, as well as an escalating annual royalty between 15 percent and 17 percent of global net sales of licensed products.
Equity Offerings
2021 Underwritten Public Offering of Common Stock
In February 2021, we sold an aggregate of 7.5 million shares of our common stock in an underwritten public offering, at a price to the public of $26.75 per share. The net proceeds to us from the offering, after deducting the underwriting discounts and offering expenses, were $189.3 million.
2020 Underwritten Public Offering of Common Stock
In June 2020, we sold an aggregate of 7.5 million shares of our common stock in an underwritten public offering, at a price to the public of $15.50 per share. The net proceeds to us from the offering, after deducting the underwriting discounts and offering expenses, were $108.7 million.
At-the-Market Equity Offering
In February 2020, we entered into an Open Market Sale Agreement ("ATM Agreement") with Jefferies LLC, as agent (“Jefferies”), pursuant to which we may offer and sell, from time to time through Jefferies, shares of our common stock having an aggregate offering price of up to $100.0 million. Of the $100.0 million originally authorized for sale under the ATM Agreement, approximately $28.6 million were sold under our prior registration statement on Form S-3 (Registration Statement No. 333-227182). An additional $31.7 million were sold under our effective registration statement on Form S-3 (Registration Statement No. 333-259311).
In 2020, the Company sold 867,806 shares under the ATM Agreement, resulting in gross proceeds of $23.5 million. In 2021, the Company sold a combined 1,240,640 shares under the ATM Agreement, resulting in gross proceeds of $36.8 million. Through December 31, 2021, we have sold a
total of 2,108,446 shares under the ATM Agreement, resulting in gross proceeds of $60.3 million. As of December 31, 2021, an aggregate amount of $39.7 million remained eligible for sale under the facility.
Authorized Shares of Common Stock
On May 14, 2021, in connection with the Company’s 2021 Annual Meeting of Stockholders, the Company’s stockholders approved, among other matters, a Certificate of Amendment (“Certificate of Amendment”) to the Company’s Certificate of Incorporation to increase the number of shares of common stock authorized for issuance thereunder from 100,000,000 to 200,000,000. Effective May 18, 2021, the Certificate of Amendment was filed with the Secretary of State of the State of Delaware.
Operating Leases
Future Minimum Rental Commitments
We have future minimum rental commitments totaling $43.7 million arising from our operating leases. These commitments represent the aggregate base rent through August 2028.
Contingent Cash Payments
Acquisition Agreement with Orphan Technologies Limited
In November 2020, we completed the acquisition of Orphan Technologies Limited (“Orphan”), including Orphan’s rare metabolic disorder drug pegtibatinase. We acquired Orphan by purchasing all of its outstanding shares. In exchange for the shares, we made an upfront cash payment at closing of $90.0 million plus closing adjustments, net liabilities assumed, and transaction expenses of $1.2 million, $1.8 million, and $4.2 million, respectively. Under the Agreement, we also agreed to make contingent cash payments up to an aggregate of $427.0 million based on the achievement of certain development, regulatory and commercialization events as set forth in the Agreement, as well as additional tiered mid-single digit royalty payments based upon future net sales of any pegtibatinase products in the US and Europe, subject to certain reductions as set forth in the Agreement, and a contingent payment in the event a pediatric rare disease voucher for any pegtibatinase product is granted.
License Agreement Obligations
See discussion above under the heading "License Agreements".
Borrowings
Convertible Senior Notes Due 2025
On September 10, 2018, we completed a registered underwritten public offering of $276 million aggregate principal amount of 2.50% Convertible Senior Notes due 2025 ("2025 Notes"), and entered into a base indenture and supplemental indenture agreement ("2025 Indenture") with respect to the 2025 Notes. The 2025 Notes will mature on September 15, 2025 ("Maturity Date”), unless earlier repurchased, redeemed, or converted. The 2025 Notes are senior unsecured obligations of ours and bear interest at an annual rate of 2.50%, payable semi-annually in arrears on March 15 and September 15 of each year.
The net proceeds from the issuance of the 2025 Notes were approximately $267.2 million, after deducting commissions and the offering expenses payable by us. A portion of the net proceeds from the 2025 Notes were used by us to repurchase $23.4 million aggregate principal amount of its then-outstanding 4.5% senior convertible notes due in 2019 in privately-negotiated transactions.
The initial conversion rate for the 2025 Notes is 25.7739 shares of our common stock per $1,000 principal amount of 2025 Notes, which represents an initial conversion price of approximately $38.80 per share. If a “make-whole fundamental change” (as defined in the 2025 Indenture) occurs, then we will, in certain circumstances, increase the conversion rate for a specified period of time.
The 2025 Notes will be redeemable, in whole or in part, at our option at any time, and from time to time, on or after September 15, 2022 and, in the case of any partial redemption, on or before the 40th scheduled trading day before the Maturity Date, at a cash redemption price equal to the principal amount of the 2025 Notes to be redeemed, plus accrued and unpaid interest, if any, to, but excluding, the redemption date, but only if the last reported sale price per share of our common stock exceeds 130% of the conversion price on each of at least 20 trading days during the 30 consecutive trading days ending on, and including, the trading day immediately before the date we send the related redemption notice. If a fundamental change (as defined in the 2025 Indenture) occurs, then, subject to certain exceptions, holders may require us to repurchase their 2025 Notes at a cash repurchase price equal to the principal amount of the 2025 Notes to be repurchased, plus accrued and unpaid interest, if any, to, but excluding, the fundamental change repurchase date.
The effective interest rate on the liability components of the 2025 Notes for the period from the date of issuance was 7.7%.
Convertible Senior Notes Due 2019
On May 29, 2014, the Company entered into a Note Purchase Agreement relating to a private placement by the Company of $46.0 million aggregate principal amount of 2019 Notes which were convertible into shares of the Company’s common stock at an initial conversion price of $17.41 per share. The conversion price was subject to customary anti-dilution protection. The 2019 Notes bore interest at a rate of 4.5% per annum, payable semiannually in arrears on May 15 and November 15 of each year. The maturity date of the 2019 Notes was on May 30, 2019, and there were no contractual payments due prior to that date.
In September 2018, the Company used part of the net proceeds from the issuance of the 2025 Notes to repurchase $23.4 million aggregate principal amount of the 2019 Notes in privately-negotiated transactions for approximately $40.2 million in cash. The partial repurchase of the 2019 Notes resulted in a $17.0 million loss on early extinguishment of debt in September 2018.
In May 2019,incur the remaining $22.6 million outstanding principal amount of 2019 Notes was converted by the holders thereof into approximately 1.3 million shares of common stock. At December 31, 2019, there was no outstanding balance on the 2019 Notes.
Interest Expense
Total interest expense recognized for the years ended December 31, 2021, 2020 and 2019 was $20.1 million, $19.1 million, and $18.8 million, respectively.
Funding Requirements
We believe that our available cash and short-term investments as of the date of this filing will be sufficient to fund our anticipated level of operations beyond the next 12 months. This belief is based on many factors, some of which are beyond our control. Factors that may affect financing requirements include, but are not limited to:
•the rate of progress and cost of our clinical trials, preclinical studies and other discovery and research and development activities, including any delays resulting from the COVID-19 pandemic;
•the timing of, andestimated restructuring costs involved in, seeking and obtaining marketing approvals for our products, and in maintaining quality systems standards for our products;
•increases or decreases in revenue from our marketed products, including decreases in revenue resulting from the COVID-19 pandemic, if any;
•debt service obligations on the 2025 Notes;
•our ability to manufacture sufficient quantities of our products to meet expected demand;
•the costs of preparing, filing, prosecuting, maintaining and enforcing any patent claims and other intellectual property rights, litigation costs and the results of litigation;
•our ability to enter into collaboration, licensing or distribution arrangements and the terms and timing of these arrangements;
•the potential need to expand our business, resulting in additional payroll and other overhead expenses;
•the potential in-licensing of other products or technologies; and
•the emergence of competing technologies or other adverse market or technological developments.
Future cash requirements will also depend on the extent to which we acquire or invest in additional complementary businesses, products and technologies.
Cash Flows
The following table summarizes our cash flows for the periods set forth below (in thousands):
| | | | | | | | | | | | | | | | | |
| Twelve Months Ended December 31, |
| | 2021 | | 2020 | | 2019 |
| Net cash used in operating activities | $ | (14,792) | | | $ | (42,743) | | | $ | (58,214) | |
| Net cash (used in) provided by investing activities | (137,623) | | | (61,327) | | | 19,863 | |
| Net cash provided by (used in) financing activities | 231,683 | | | 127,713 | | | (2,077) | |
| Effect of exchange rate changes on cash | 1,713 | | | (1,307) | | | (9) | |
| Net increase (decrease) in cash & cash equivalents | 80,981 | | | 22,336 | | | (40,437) | |
| | | | | |
| Cash & cash equivalents, beginning of period | 84,772 | | | 62,436 | | | 102,873 | |
| Cash & cash equivalents, end of period | $ | 165,753 | | | $ | 84,772 | | | $ | 62,436 | |
Management considers marketable debt securities to be available to fund current operations, and they are classified as available for sale and included within current assets in our Consolidated Balance Sheets. Therefore, cash and short-term investments available to fund operations is $552.9 million as of December 31, 2021.
Cash Flows from Operating Activities
Operating activities used $14.8 million of cash during the year ended December 31, 2021 compared to $42.7 million of cash used for the year ended December 31, 2020. The decrease in cash used was primarily attributable to the $55.0 million upfront payment received in connection with the license and collaboration agreement entered into with Vifor Pharma in September 2021, along with increased net product sales, offset by increased research and development expenses.
Cash Flows from Investing Activities
Cash used in investing activities for the year ended December 31, 2021 was $137.6 million compared to cash used of $61.3 million for the year ended December 31, 2020. The increase in cash used was due to the increase in net purchases of marketable debt securities, offset by expenses arising from the acquisition of Orphan Technologies in 2020.
Cash Flows from Financing Activities
For the year ended December 31, 2021, cash provided by financing activities was $231.7 million compared to cash provided of $127.7 million for the year ended December 31, 2020. The increase in cash provided was due to the February 2021 issuance of stock through an underwritten public offering that provided $189.3 million and an At-The-Market public offering that provided $36.8 million.2024.
Critical Accounting Policies and Estimates
Management makes certain judgments and uses certain estimates and assumptions when applying accounting principles generally accepted in the United States (“GAAP”) in the preparation of our Consolidated Financial Statements. We evaluate our estimates and judgments on an ongoing basis and base our estimates on historical experience and on assumptions that we believe to be reasonable under the circumstances. Our experience and assumptions form the basis for our judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may vary from what we anticipate and different assumptions or estimates about the future could change our reported results. We believe the following accounting policies are the most critical to us, in that they require our most difficult, subjective or complex judgments in the preparation of our Consolidated financial statements. For further information, see Note 2, Summary of Significant Accounting Policies, to our Consolidated Financial Statements, which outlines our application of significant accounting policies.
Revenue Recognition
We recognize revenue when the customer obtains control of promised goods or services, in an amount that reflects the consideration which the entity expects to receive in exchange for those goods or services. To determine revenue recognition for arrangements that an entity determines are within the scope of Accounting Standards Codification ("ASC") 606, Revenue from Contracts with Customers ("ASC 606"), the entity performs the following five steps: (i) identify the contract(s) with a customer; (ii) identify the performance obligations in the contract; (iii) determine the transaction price; (iv) allocate the transaction price to the performance obligations in the contract; and (v) recognize revenue when (or as) the entity satisfies a performance obligation. We only apply the five-step model to contracts when it is probable that the entity will collect substantially all the consideration it is entitled to in exchange for the goods or services it transfers to the customer.
We recognize revenues from product sales are recognized when the customer obtains control of the product, which occurs upon delivery to our customer. We receive payments from our product sales based on terms that generally are within 30 days of delivery of product to the patient.
Revenues from product sales are recorded at the net sales price, which includes provisions resulting from discounts, rebates and co-pay assistance that are offered to its customers, health care providers, payorspayers and other indirect customers relating to the sale of our products. In order to determine the transaction price, we estimate, utilizing the expected value method, the amount of variable consideration to which we will be entitled. These provisions are based on the amounts earned or to be claimed on the related sales and are classified as a reduction of accounts receivable (if the amount is payable to the customer) or as a current liability (if the amount is payable to a party other than a customer). Calculating these provisions involves estimates and judgements. Where appropriate, these reserves take into consideration our historical experience, current contractual and statutory requirements and specific known market events and trends. Overall, these reserves reflect our best estimates of the amount of consideration to which it is entitled based on the terms of the contract. If actual results in the future vary from the provisions, we will adjust the provision, which would affect net product revenue and earnings in the period such variances become known. Our historical experienceFor the years ended December 31, 2023, 2022 and 2021, the Company recorded a net product revenue increase of $0.4 million, $0.2 million, and $0.2 million, respectively, related to performance obligations satisfied in previous periods.
Government Rebates: We calculate the rebates that we will be obligated to provide to government programs and deduct these estimated amounts from our gross product sales at the time the revenues are recognized. Allowances for government rebates and discounts are established based on an estimated allocation of payers and the government-mandated discounts applicable to government-funded programs. Rebate discounts are included in accrued expenses in the accompanying consolidated balance sheets.
Commercial Rebates: We calculate the rebates we incur according to any contracts with certain commercial payers and deduct these amounts from our gross product sales at the time the revenues are recognized. Allowances for commercial rebates are established based on actual payer information, which is that such adjustmentsreasonably estimated at the time of delivery for applicable products. Rebate discounts are included in accrued expenses in the accompanying consolidated balance sheets.
Prompt Pay Discounts: We offer discounts to certain customers for prompt payments. We accrue for the calculated prompt pay discount based on the gross amount of each invoice for those customers at the time of sale.
Product Returns: Consistent with industry practice, we offer our customers a limited right to return product purchased directly from us, which is principally based upon the product’s expiration date. Historically, returns have been immaterial.
Co-pay Assistance: We offer a co-pay assistance program, which is intended to provide financial assistance to qualified commercially insured patients with prescription drug co-payments required by payers. The calculation of the accrual for co-pay assistance is based on an estimate of claims and the estimated cost per claim associated with product that has been recognized as revenue.
Payments received under collaboration and licensing agreements may include non-refundable fees at the inception of the arrangements, milestone payments for specific achievements and royalties on the sale of products. At the inception of arrangements that include milestone payments, we use judgement to evaluate whether the milestones are probable of being achieved and estimates the amount to include in the transaction price utilizing the most likely amount method. If it is probable that a significant revenue reversal will not occur, the estimated amount is included in the transaction price. Milestone payments that are not within our or the licensee’s control, such as regulatory approvals, are considered to be constrained due to a high degree of uncertainty and are not included in the transaction price until those approvals are received.such uncertainty is resolved. At the end of each reporting period, we re-evaluate the probability of achievement of development milestones and any related constraint and adjustsadjust the estimate of the overall transaction price, if necessary. As of December 31, 2023, our evaluation concluded that all such milestones associated with our collaboration and licensing agreements remained constrained and therefore no adjustment to the respective transaction price was necessary. We recognize aggregate sales-based milestones and royalty payments from product sales of which the license is deemed to be the predominant item to which the royalties relate, at the later of when the related sales occur or when the performance obligation to which the sales-based milestone or royalty has been allocated has been satisfied. If it is probable that a significant revenue reversal will not occur, we estimate the sales-based milestone and royalty payments using the most likely amount method.
We utilize significant judgement to develop estimates of the stand-alone selling price for each distinct performance obligation based upon the relative stand-alone selling price. Variable consideration that relates specifically to our efforts to satisfy specific performance obligations is allocated entirely to those performance obligations. The stand-alone selling price for license-related performance obligations requires judgement in developing assumptions to project probability-weighted cash flows based upon estimates of forecasted revenues, clinical and regulatory timelines and discount rates. The stand-alone selling price for clinical development performance obligations is based on forecasted expected costs of satisfying a performance obligation plus an appropriate margin. In 2023, there were no new collaboration and licensing agreements with customers which required an estimate of the stand-alone selling price for an underlying performance obligation.
If the licenses to intellectual property are determined to be distinct from the other performance obligations identified in the arrangement, we recognize revenues from non-refundable, upfront fees allocated to the license when the license is transferred to the licensee and the licensee is able to benefit from the license. For licenses that are not distinct from other promises, we apply judgment to assess the nature of the combined performance obligation to determine whether the combined performance obligation is satisfied over time or a point in time and, if over time, the appropriate method of measuring progress for purposes of recognizing revenue from non-refundable, upfront fees. We evaluate the measure of progress each reporting period and, if necessary, adjust the related revenue recognition accordingly.
The selection of the method to measure progress towards completion requires judgment and is based on the nature of the products or services to be provided. Revenue is recorded proportionally as costs are incurred. We generally utilize the cost-to-cost method of progress because it best measures the transfer of control to the customer which occurs as we incur costs. Under the cost-to-cost measure of progress, the extent of progress towards completion is measured based on the ratio of costs incurred to date to the total estimated costs at completion of the performance obligation. We use judgment to estimate the total costs expected to complete the clinical development performance obligations, which include subcontractor costs, labor, materials, other direct costs and an allocation of indirect costs. We evaluate these cost estimates and the progress each reporting period and adjustsadjust the measure of progress, if necessary.
Changes in assumptions where management utilizes significant judgement could have a material impact on the revenue we recognize.
Clinical Trial Expenses
We record expenses in connection with our clinical trials under contracts with contract research organizations (CROs)("CROs") that support conducting and managing clinical trials.trials, as well as contract manufacturing organizations ("CMOs") for the manufacture of drug product supplies to support clinical development. The financial terms and activities of these agreements vary from contract to contract and may result in uneven expense levels. Generally, these agreements set forth activities that drive the recording of expenses such as start-up, and initiation activities, enrollment, and treatment of patients, or the completion of other clinical trial activities.activities, and in the case of CMOs, costs associated with the production of drug product supplied and the procurement of raw materials to be consumed in the manufacturing process.
Expenses related to clinical trials are accrued based on our estimates and/or representations from service providers regarding workof the progress of services performed, including actual level of patient enrollment, completion of patient studies and progress of the clinical trials.trials or the delivery of goods. We currently have one Phase 1/2 clinical trial and three Phase 3 clinical trials in process that are in varying stages of activity, with ongoing non-clinical support trials that are significant and changes in estimates and representations from service providers could have a material impact on expenses we recognize.
Stock-Based Compensation
We account for share-based compensation arrangements in accordance with ASC 718, Compensation – Stock Compensation, which requires the measurement and recognition of compensation expense for all share-based payment awards to be based on estimated fair values. We use the Black-Scholes option valuation model to estimate the fair value of itsour stock options at the date of grant. The Black-Scholes option valuation model
requires the input of subjective assumptions to calculate the value of stock options. For restricted stock units, the value of the award is based on our stock price at the grant date. For performance-based restricted stock unit awards, the value of the award is based on our stock price at the grant date, with consideration given to the probability of the performance condition being achieved. We use historical data and other information to estimate the expected price volatility for stock option awards and the expected exercise activity for all awards. Expense is recognized over the vesting period for all awards and commences at the grant date for time-based awards and upon our determination that the achievement of such performance conditions is probable for performance-based awards. The valuation, including the assumptions, require significant judgment by management.
Contingent Consideration
We record contingent consideration resulting from a business combination at its fair value onThe following weighted average assumptions were used in the acquisition date. On a quarterly basis, we revalue these obligations and record increases or decreases from their fair value as an adjustmentBlack-Scholes options pricing model to the Consolidated Statements of Operations and Comprehensive Loss. In estimatingestimate the fair value of stock options for the specified reporting periods:
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| | 2023 | | 2022 | | 2021 |
| Risk free rate | 3.9 | % | | 1.7 | % | | 0.6 | % |
| Expected volatility | 50 | % | | 50 | % | | 59 | % |
| Expected life (in years) | 6.4 | | 6.4 | | 6.4 |
| Expected dividend yield | — | | | — | | | — | |
For the years ended December 31, 2023, 2022 and 2021, non-cash stock-based compensation expense totaled $44.2 million, $38.1 million and $29.6 million, respectively.
Inventory
We capitalize inventory costs associated with our contingent consideration, we useproducts after regulatory approval, when, based on management’s judgment, future commercialization is considered probable and the future economic benefit is expected to be realized. Until the date at which regulatory approval has been received, costs related to the production of inventory are recorded as research and development expenses as incurred.
We periodically analyze our inventory levels to identify inventory that may expire prior to expected sale or has a Monte Carlo Simulation. Changescost basis in excess of its estimated realizable value, and write down such inventory as appropriate. In addition, our products are subject to contingent consideration obligations can result from changes to discount rates, accretionstrict quality control and monitoring which our suppliers and manufacturers perform throughout their manufacturing process. If certain batches or units of the liabilityproduct no longer meet quality specifications or become obsolete due to the passage of time, changes in revenue forecasts and changes in our estimates of the likelihood or timing of achieving commercial revenue milestones. The determination of the contingent consideration liabilities requires significant judgments including the appropriateness of the valuation model and reasonableness of estimates and assumptions included in the forecasts of future net sales and the discount rates appliedexpiration, we record a charge to write down such forecasts. Changes in these estimates and assumptions could have a significant impact on the fair value of the contingent consideration liabilities.inventory to its estimated realizable value.
Impairment of Intangible Assets subject to amortization
Intangible assets subject to amortization include certain license agreements and purchased technologies. Intangible assets subject to amortization are reviewed for impairment whenever events or circumstances indicate that the carrying amount of these assets may not be recoverable and are also reviewed annually to determine whether any impairment is necessary.
Income Taxes
We follow ASC 740, Income Taxes, which requires recognitionare subject to generic competition, and if additional generic versions of deferred tax assetsThiola and liabilitiesThiola EC, any generic versions of FILSPARI following the expiration of patent or regulatory exclusivity for the expectedproduct, or any of our current or future tax consequencesproducts, are approved, sales of events that product likely would be negatively impacted, which could have a material adverse impact on the recoverability of certain related intangible assets. Generic versions of Thiola and Thiola EC have been included inapproved and have impacted sales, and these or additional generic versions of either formulation could have a material adverse impact on sales and the financial statements or tax returns. Under this method, deferred taxrecoverability of the intangible assets and liabilities are baseddepending on the differences between the financial statement and tax basis of assets and liabilities using enacted tax rates in effect for the year in which the differences are expected to reverse. Deferred tax assets are reduced by a valuation allowance to the extent management concludes it is more likely than not that the asset will not be realized.
The standard addresses the determination of whether tax benefits claimed or expected to be claimed on a tax return should be recorded in the financial statements. Under ASC 740, we may recognize the tax benefit from an uncertain tax position only if it is more likely than not that the tax position will be sustained on examination by the tax authorities, based on the technical meritstiming of the position. The tax benefits recognized inmarket entry and the financial statements from such a position should be measured basedrelated impact on the largest benefit that has a greater than fifty percent likelihood of being realized upon ultimate settlement.net sales.
Recently Issued Accounting Pronouncements
See Note 2 to the Consolidated Financial Statements for discussion.
Results of Operations
Unless noted otherwise, the discussion below, and the revenue and expense amounts discussed below, are based on and relate to our continuing operations.
Revenue
For further background on our net product sales and license and collaboration revenue, see Revenue Recognition under our Critical Accounting Estimates.
The following table provides information regarding revenue, including net product sales and license and collaboration revenue (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Year Ended December 31, | | Year Ended December 31, |
| | 2023 | | 2022 | | Change | | 2022 | | 2021 | | Change |
| Tiopronin products | $ | 98,329 | | | $ | 97,970 | | | $ | 359 | | | $ | 97,970 | | | $ | 115,122 | | | $ | (17,152) | |
| FILSPARI | 29,208 | | | — | | | 29,208 | | | — | | | — | | | — | |
| Total net product sales | 127,537 | | | 97,970 | | | 29,567 | | | 97,970 | | | 115,122 | | | (17,152) | |
| License and collaboration revenue | 17,701 | | | 11,490 | | | 6,211 | | | 11,490 | | | 16,714 | | | (5,224) | |
| Total revenue | $ | 145,238 | | | $ | 109,460 | | | $ | 35,778 | | | $ | 109,460 | | | $ | 131,836 | | | $ | (22,376) | |
Net product sales
The $29.6 million increase in total net product revenues for the year ended December 31, 2023 compared to the year ended December 31, 2022 was primarily due the launch of FILSPARI in February 2023 and subsequent sales throughout the year.
The $17.2 million decrease in total net product revenues for the year ended December 31, 2022 as compared to the year ended December 31, 2021, which consisted of Thiola net sales, was a result of generic competition.
License and collaboration revenue
The increase in license and collaboration revenue for the year ended December 31, 2023 compared to the year ended December 31, 2022 was due to a $3.3 million sale of active pharmaceutical ingredients to CSL Vifor in March 2023 in preparation for a potential European Union approval and subsequent commercial launch, along with a $2.9 million increase in amortization of deferred revenue under the cost-to-cost model, which saw total costs for clinical development activities decrease as a result of the restructuring announced in December 2023. We recognize costs for clinical development activities in research and development; costs related to sale of active pharmaceutical ingredients are recognized in cost of goods sold.
The decrease in license and collaboration revenue for the year ended December 31, 2022 as compared to the year ended December 31, 2021 was primarily due to the recognition of $12.0 million in license revenue upon entering into the CSL Vifor License Agreement in September 2021, offset by an increase in collaboration revenue associated with the CSL Vifor License Agreement. Collaboration revenue, derived from the performance of clinical development activities and based upon the ratio of costs incurred to total estimated costs, increased by $6.8 million from $4.7 million in 2021 to $11.5 million in 2022, primarily due to a full year of performance in 2022 as compared to a partial year of performance in 2021.
Operating Expenses
The following table provides information regarding operating expenses (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Year Ended December 31, | | Year Ended December 31, |
| | 2023 | | 2022 | | Change | | 2022 | | 2021 | | Change |
| Cost of goods sold - product sales | $ | 8,406 | | | $ | 4,420 | | | $ | 3,986 | | | $ | 4,420 | | | $ | 3,818 | | | $ | 602 | |
| Cost of goods sold - license and collaboration | 3,044 | | | — | | | 3,044 | | | — | | | — | | | — | |
| Total cost of goods sold | 11,450 | | | 4,420 | | | 7,030 | | | 4,420 | | | 3,818 | | | 602 | |
| Research and development | 244,990 | | | 227,333 | | | 17,657 | | | 227,333 | | | 201,159 | | | 26,174 | |
| Selling, general and administrative | 265,542 | | | 197,520 | | | 68,022 | | | 197,520 | | | 126,284 | | | 71,236 | |
| Restructuring | 11,394 | | | — | | | 11,394 | | | — | | | — | | | — | |
| Total operating expenses | $ | 533,376 | | | $ | 429,273 | | | $ | 104,103 | | | $ | 429,273 | | | $ | 331,261 | | | $ | 98,012 | |
Cost of goods sold
Cost of goods sold includes the cost of inventory sold, third party manufacturing and supply chain costs, product shipping, tracking and handling costs, and provisions for excess and obsolete inventory.
Prior to the February 2023 FDA accelerated approval of FILSPARI (sparsentan), we recognized approximately $7.5 million in research and development expenses related to the production of active pharmaceutical ingredients to support the commercial launch of FILSPARI. For the year ended December 31, 2023, sales of FILSPARI primarily consisted of zero-cost inventories. As of December 31, 2023, we had $7.2 million of zero-cost inventory remaining. We expect to continue to benefit from the sale of previously expensed inventories through at least 2025.
We began capitalizing inventory costs associated with FILSPARI (sparsentan) following the February 2023 approval for treatment in IgAN. At December 31, 2023, our evaluation of excess inventory and obsolescence considered certain minimum purchase obligations, which in combination with lower forecasted sales of FILSPARI resulted in a $3.2 million charge to cost of goods sold. The charge to cost of goods sold included a $2.1 million write-down of inventory balances and $1.1 million accrued for firm purchase commitments.
For the year ended December 31, 2023 compared to the year ended December 31, 2022, our cost of goods sold - license and collaboration increased by $3.0 million due to the sale of active pharmaceutical ingredients to CSL Vifor in March 2023.
Research and development expenses
Research and development costs include expenses related to sparsentan, pegtibatinase and our other pipeline programs. We expense all research and development costs as they are incurred. Our research and development costs are comprised of salaries and bonuses, benefits, non-cash share-based compensation, license fees, milestones under license agreements, costs paid to third-party contractors to perform research, conduct clinical trials, and develop drug materials and delivery methods, manufacture drug product supplies to support clinical development, and associated overhead expenses and facilities costs. We charge direct internal and external program costs to the respective development programs. We also incur indirect costs that are not allocated to specific programs because such costs benefit multiple development programs and allow us to increase our pharmaceutical development capabilities. These consist of internal shared resources related to the development and maintenance of systems and processes applicable to all of our programs.
We currently have one Phase 1/2 clinical trial and three Phase 3 clinical trials in process that are in various stages of activity, with ongoing non-clinical support trials. As such, clinical trial expenses will vary depending on the all the factors set forth above and may fluctuate significantly from quarter to quarter and year to year.
We routinely engage vendors and service providers for scientific research, clinical trial, regulatory compliance, manufacturing and other consulting services. We also make grants to research and non-profit organizations to conduct research which may lead to new intellectual properties that we may subsequently license under separately negotiated license agreements. Such grants may be funded in lump sums or installments.
The following table provides information regarding research and development expenses (in thousands):
| | | | | | | | | | | | | | | | | |
| For the Year Ended December 31, |
| 2023 | | 2022 | | 2021 |
| External service provider costs: | |
| Sparsentan | $ | 91,702 | | | $ | 87,598 | | | $ | 103,350 | |
| Pegtibatinase | 50,780 | | | 42,100 | | | 27,823 | |
| General and other product candidates | 17,850 | | | 21,971 | | | 9,493 | |
| Total external service provider costs | 160,332 | | | 151,669 | | | 140,666 | |
| Internal personnel costs | 84,658 | | | 75,664 | | | 60,493 | |
| Total research and development | $ | 244,990 | | | $ | 227,333 | | | $ | 201,159 | |
For the year ended December 31, 2023 compared to the year ended December 31, 2022, our research and development expenses increased by $17.7 million. Internal personnel costs to support all programs increased by $9.0 million, reflecting increased headcount along with rising labor costs driven in part by inflation. External service provider costs increased by $8.7 million, which was largely driven by increases in investment toward the December 2023 initiation of the Phase 3 HARMONY Study for pegtibatinase. While costs associated with the development of sparsentan have been decreasing throughout the year as our Phase 3 programs advance towards completion, we observed a year over year increase as a result of increased investment in Medical Affairs initiatives supporting the IgAN program.
For the year ended December 31, 2022 compared to the year ended December 31, 2021, our research and development costs increased by $26.2 million due to increases in clinical trial expenses, chemistry manufacturing and control costs, and related expenses associated with the advancement of our programs in development. Internal personnel costs to support all programs increased by $15.2 million, reflecting increased headcount along with rising labor costs. External service provider costs increased by $11.0 million, which included a $14.3 million increase in such costs for the development of pegtibatinase along with a $12.5 million increase in other general research and development costs, offset by a $15.8 million decrease in external service provider costs associated with the development of sparsentan as the program progresses through the development cycle.
Selling, general and administrative expenses
Selling, general and administrative expenses consist of salaries and bonuses, benefits, non-cash share-based compensation, legal and other professional fees, rent, depreciation and amortization, travel, insurance, business development, sales and marketing programs, and other operating expenses.
For the year ended December 31, 2023 compared to the year ended December 31, 2022, our selling, general and administrative expenses increased by $68.0 million due to the onboarding of the dedicated sales force and other related commercial preparations and activity to support the U.S. launch of FILSPARI. Increases include an increase in combined employee compensation and stock compensation costs of $22.0 million, an increase in commercial support expenses of $19.1 million, an increase in various professional services expenses of $2.0 million, and an increase in various legal expenses of $5.0 million, along with incremental increases in information technology expense, legal fees, travel expense, and depreciation and amortization. A change in the estimated useful life of the Thiola intangible asset and the accelerated amortization recognized in connection with the change also contributed to the increase in expense.
For the year ended December 31, 2022 compared to the year ended December 31, 2021 our selling, general and administrative expenses increased by $71.2 million due to increased headcount as a result of operational growth, including rising labor costs, and commercial launch preparations in anticipation of the U.S. launch of FILSPARI. Increases include combined employee compensation and stock compensation costs of $31.9 million, increases in commercial support expenses of $15.6 million and increases in various professional services expenses of $12.0 million. Incremental increases in information technology expense, legal fees, travel expense, and depreciation and amortization also contributed to the increase in selling, general and administrative expenses.
Restructuring expenses
In December 2023, we initiated a restructuring plan that resulted in a reduction of our workforce by approximately 20%, primarily impacting non-field-based employees. Restructuring adjustments are expected to result in an estimated annualized savings of approximately $25.0 million beginning in 2024, and a total estimated non-recurring charge of approximately $12.0 million to $14.0 million. For the year ended December 31, 2023, we recognized restructuring costs of $11.4 million, comprised of one-time termination benefits, including severance, continuation of health insurance coverage, and other benefits for a specified period of time.
Other Income/Expenses
Other income/expenses consists of interest income and expense, finance expense and miscellaneous other income/expenses.
The following table provides information regarding other income (expenses) (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Year Ended December 31, | | Year Ended December 31, |
| | 2023 | | 2022 | | Change | | 2022 | | 2021 | | Change |
| Interest income | $ | 21,768 | | | $ | 6,276 | | | $ | 15,492 | | | $ | 6,276 | | | $ | 1,993 | | | $ | 4,283 | |
| Interest expense | (11,334) | | | (11,014) | | | (320) | | | (11,014) | | | (19,682) | | | 8,668 | |
| Other income, net | 1,594 | | | 974 | | | 620 | | | 974 | | | 231 | | | 743 | |
| Loss on extinguishment of debt | — | | | (7,578) | | | 7,578 | | | (7,578) | | | — | | | (7,578) | |
| Total other income (expense), net | $ | 12,028 | | | $ | (11,342) | | | $ | 23,370 | | | $ | (11,342) | | | $ | (17,458) | | | $ | 6,116 | |
The $23.4 million change in our total other income (expense), net for the year ended December 31, 2023 compared to the year ended December 31, 2022, is primarily attributable to a $15.5 million increase in interest income in 2023, which was due to increases in short-term interest rates and higher average balances on our interest-bearing security investments, and the $7.6 million loss on extinguishment of debt that was recognized in 2022 in connection with the partial repurchase of the Convertible Senior Notes due 2025. For the year ended December 31, 2023, we also recognized other income of $1.0 million in connection with the transition services agreement entered into with Mirum as part of our sale of the bile acid business.
The $6.1 million change in our total other income (expense), net for the year ended December 31, 2022 compared to the year ended December 31, 2021 was primarily due to loss on extinguishment of debt in connection with the partial repurchase in 2022 of the Convertible Senior Notes due 2025, offset by reduced interest expense due to adoption of ASU 2020-06, Accounting for Convertible Instruments and Contracts in an Entity's Own Equity ("ASU 2020-06") as debt discount is no longer amortized. In 2021, we recognized $10.3 million in interest expense for the amortization of the debt discount in accordance with the previous accounting guidance. The increase in interest income was driven by the impact of increases in short-term interest rates on our interest-bearing security investments.
Income Tax Benefit (Provision)
The tax provision was $0.2 million for fiscal 2023 compared to $0.3 million for fiscal 2022, a decrease of $0.1 million. The tax provision was $0.4 million for fiscal 2021.
Discontinued Operations
Results of discontinued operations are as follows (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Year Ended December 31, | | Year Ended December 31, |
| | 2023 | | 2022 | | Change | | 2022 | | 2021 | | Change |
| Income from discontinued operations, net of tax | $ | 264,934 | | | $ | 52,986 | | | $ | 211,948 | | | $ | 52,986 | | | $ | 37,201 | | | $ | 15,785 | |
The $211.9 million increase in income from discontinued operations, net of tax for the year ended December 31, 2023 compared to the year ended December 31, 2022 is primarily due to the August 31, 2023 sale of our bile acid business, which resulted in a gain, net of tax, of $226.0 million. The gain consists of net consideration, including the upfront payment and the deduction of investment banker fees owed upon the Closing, plus the derecognition of the carrying value of the net liabilities included in the transaction and the immaterial tax due on the sale. The increase arising from the gain was offset by four months less of bile acid business operations as a result of the divestiture on August 31, 2023.
The $15.8 million increase in income from discontinued operations, net of tax for the year ended December 31, 2022 compared to the year ended December 31, 2021 was due in part to a $6.9 million increase in net sales of our bile acid products. The remainder of the increase in income from discontinued operations, net of tax, was primarily due to changes in the fair value of contingent consideration arising from updated revenue projections and changes in market driven discount rates.
See Note 19 to our Consolidated Financial Statements for further discussion.
Liquidity and Capital Resources
We have financed our operations through a combination of borrowings, sales of our equity securities, and revenues generated from our commercialized products, along with proceeds from license and collaboration agreements and the divestiture of our bile acid business. We experienced significant growth in recent years in the number of our employees and the scope of our operations. We also expanded our sales and marketing, compliance and legal functions in addition to expansion of all functions to support a commercial organization, including by adding additional members to our sales force in connection with the recent commercial launch of FILSPARI in the United States for IgAN. In December 2023, we implemented an approximate 20% workforce reduction focused on non-field-based employees in an effort to align our resources on the ongoing FILSPARI launch and the pivotal Phase 3 HARMONY Study to support the potential approval of pegtibatinase as the first potential disease-modifying treatment for HCU. These restructuring adjustments are expected to result in an estimated annualized savings of approximately $25.0 million beginning in 2024.
We believe that our available cash and short-term investments as of the date of this filing, together with anticipated cash generated from operations, will be sufficient to fund our anticipated level of operations beyond the next 12 months. We expect that our operating results will vary from quarter-to-quarter and year-to-year depending upon various factors including revenues, selling, general and administrative expenses, and research and development expenses, particularly with respect to our clinical and preclinical development activities. Our ability to fund our operations in subsequent years will depend upon certain factors which are beyond our control and may require us to obtain additional debt or equity capital or refinance all or a portion of our debt, including the 2025 Notes and 2029 Notes, on or before maturity. Though we generate revenues from product sales arrangements, we may incur significant operating losses over the next several years. Our ability to achieve profitable operations in the future will depend in large part upon completing development of products in our pipeline, obtaining regulatory approvals for these products and bringing these products to market, along with potential in-licensing of additional products approved by the FDA and selling and manufacturing these products.
For the years ended December 31, 2023 and 2022, we had the following balances and financial performance (in thousands):
| | | | | | | | | | | |
| December 31, 2023 | | December 31, 2022 |
| | | |
| | | |
| | | |
| | | |
| Cash and cash equivalents | $ | 58,176 | | | $ | 61,688 | |
| Marketable debt securities, at fair value | $ | 508,675 | | | $ | 388,557 | |
| Convertible debt | $ | 377,263 | | | $ | 375,545 | |
| Accumulated deficit | $ | (1,125,622) | | | $ | (1,014,223) | |
| Stockholders' equity | $ | 200,810 | | | $ | 42,851 | |
| Net working capital* | $ | 438,867 | | | $ | 344,274 | |
| Net working capital ratio** | 3.47 | | | 3.42 | |
* Current assets less current liabilities
**Current assets divided by current liabilities | | | |
As of December 31, 2023, we had cash and cash equivalents of $58.2 million and available-for-sale marketable debt securities of $508.7 million. Substantial sources of funds since the start of 2023, as summarized further below, include an upfront cash payment of $210.0 million in connection with the sale of our bile acid product portfolio, and net proceeds of $215.8 million from an underwritten public offering of our common stock and pre-funded warrants to purchase shares of our common stock.
Over the next 12 months, our expected financial obligations include, but are not limited to, funding our operations, operating lease payments, interest payments on our outstanding debt, anticipated milestone payments, royalties on sales of our existing commercialized products, research and development expenses pertaining to clinical and preclinical development activities across our pipeline, expenses associated with the launch of FILSPARI. Sources of cash over this period include net revenues from sales of our products, the sale or maturity of investments in our portfolio of marketable debt securities, and certain earned and potential milestone payments. We anticipate achieving milestones with both FILSPARI and pegtibatinase that will result in us making net payments of approximately $50.0 million during the next 12 months.
Beyond the next 12 months and over the foreseeable future, our known commitments and potential financial obligations will likely include ongoing operations funding, operating lease payments, interest payments on our outstanding debt, royalties on sales of our existing commercialized products, research and development expenses pertaining to clinical and preclinical development activities across our pipeline, milestone and royalty payments associated with FILSPARI, pegtibatinase, and other developmental programs based upon the achievement of certain agreement-specific criteria, along with sales-based royalties and the repayment of principal on the outstanding 2025 Notes and 2029 Notes upon their respective maturities. Potential sources of cash over this time horizon may include net revenues from sales of our existing products and, if commercialized, our pipeline products, licensing revenue, the sale or maturity of marketable debt securities in our investment portfolio, the refinancing of all or a portion of our debt, including the 2025 Notes and 2029 Notes, on or before maturity, or the issuance of additional debt or equity. In addition, depending on prevailing market conditions, our liquidity requirements, contractual restrictions, and other factors, we may also from time to time seek to retire or purchase our outstanding debt, including the 2025 Notes or 2029 Notes, through cash purchases and/or exchanges for equity securities, in open market purchases, privately negotiated transactions or otherwise. In light of global and macroeconomic conditions, including rising interest rates, liquidity concerns at, and failures of, banks and other financial institutions, and volatility in the capital markets, we may not be able to successfully conduct financing or refinancing activity on favorable terms or at all.
Purchase Agreement Proceeds
Sale of Bile Acid Product Portfolio
On July 16, 2023, we entered into the Purchase Agreement with Mirum, pursuant to which Mirum agreed to purchase substantially all of the assets primarily related to our business of development, manufacture and commercialization of the Products, which comprised our bile acid business. Upon the Closing of the transaction on August 31, 2023, we received an upfront cash payment of $210.0 million. Pursuant to the Purchase Agreement, we are eligible to receive up to $235.0 million upon the achievement of certain milestones based on specified amounts of annual net sales (tiered from $125.0 million to $500.0 million) of the Products.
Collaboration and License Proceeds
License and Collaboration Agreement with CSL Vifor
In September, 2021, we entered into a License Agreement with CSL Vifor, pursuant to which we granted an exclusive license to CSL Vifor for the commercialization of sparsentan in the Licensed Territories. Under the terms of the License Agreement, we received an upfront payment of $55.0 million in September 2021, and will be eligible for up to $135.0 million in aggregate regulatory and market access related milestone payments and up to $655.0 million in aggregate sales-based milestone payments for a total potential value of up to $845.0 million. We are also entitled to receive tiered double-digit royalties of up to 40 percent of annual net sales of sparsentan in the Licensed Territories.
See Note 4 to Consolidated Financial Statements for further discussion.
Equity Offerings
2023 Underwritten Public Offering of Common Stock
In February 2023, we sold an aggregate of approximately 9.7 million shares of our common stock and pre-funded warrants to purchase 1.25 million shares of our common stock in an underwritten public offering, at a price to the public of $21.00 per share of common stock and $20.9999 per pre-funded warrant. The pre-funded warrants are exercisable immediately, subject to certain beneficial ownership limitations which can be modified by the respective holders with at least 61 days' notice, and are exercisable for one share of our common stock. The exercise price of each pre-funded warrant is $0.0001 per share of common stock. The net proceeds to us from the offering, after deducting the underwriting discounts and offering expenses, were approximately $215.8 million.
At-the-Market Equity Offering
In February 2020, we entered into an Open Market Sale Agreement ("ATM Agreement") with Jefferies LLC, as agent (“Jefferies”), pursuant to which we may offer and sell, from time to time through Jefferies, shares of our common stock having an aggregate offering price of up to $100.0 million. Of the $100.0 million originally authorized for sale under the ATM Agreement, approximately $28.6 million were sold under our prior registration statement on Form S-3 (Registration Statement No. 333-227182). An additional $51.9 million were sold under our effective registration statement on Form S-3 (Registration Statement No. 333-259311), which included $20.1 million in the year ended December 31, 2022. We did not sell any shares under the ATM Agreement during the year ended December 31, 2023. As of December 31, 2023, an aggregate amount of $19.5 million remained eligible for sale under the ATM Agreement.
Operating Leases
Future Minimum Rental Commitments
As of December 31, 2023, we have future minimum rental commitments totaling $31.9 million arising from our operating leases. These commitments represent the aggregate base rent through August 2028.
See Note 18 to Consolidated Financial Statements for further discussion.
Purchase Commitments
Manufactured Product
Certain of our contractual arrangements with contract manufacturing organizations ("CMOs") require binding forecasts or commitments to purchase minimum amounts for the manufacture of drug product supply, which may be material to our financial statements.
Royalties and Contingent Cash Payments
Ligand License Agreement
In 2012, we entered into an agreement with Ligand Pharmaceuticals, Inc. ("Ligand") for a worldwide sublicense to develop, manufacture and commercialize sparsentan (the “Ligand License Agreement”). As consideration for the license, the Company is required to make substantial payments upon the achievement of certain milestones, totaling up to $114.1 million. Through December 31, 2023, we have paid $41.4 million for contractual milestone payments under the Ligand License Agreement, which includes a $23.0 million milestone payment to Ligand (and Bristol-Myers Squibb Company ("BMS")) in March 2023 that was triggered upon the accelerated approval of FILSPARI in February 2023. Following commercialization of sparsentan or any products containing related compounds, we are obligated to pay to Ligand an escalating royalty between 15% and 17% of net sales of all such products, with payments due quarterly. We began incurring costs associated with such royalties following the February 2023 approval of FILSPARI (sparsentan). For the year ended December 31, 2023, we capitalized $4.4 million to intangible assets for royalties owed on net sales of FILSPARI.
The license agreement will continue until neither party has any further payment obligations under the agreement and is expected to continue for up to 20 years from the effective date. Ligand may terminate the license agreement due to (i) our insolvency, (ii) our material uncured breach of the agreement, (iii) our failure to use commercially reasonable efforts to develop and commercialize sparsentan as described above or (iv) certain other conditions. We may terminate the license agreement due to a material uncured breach of the agreement by Ligand.
See Note 9 to our unaudited Consolidated Financial Statements for further discussion.
Thiola License Agreement
In 2014, we entered into a license agreement with Mission Pharmacal ("Mission"), pursuant to which we obtained an exclusive, royalty-bearing license to market, sell and commercialize Thiola (tiopronin) in the United States and Canada, and a non-exclusive license to use know-how relating to Thiola to the extent necessary to market Thiola. Under the terms of the license agreement, as subsequently amended, which runs through May 2029, we are obligated to pay to Mission the greater of $2.1 million, representing the guaranteed minimum royalty, or 20% of our Thiola net sales generated globally during each calendar year.
See Note 9 to Consolidated Financial Statements for further discussion.
Acquisition of Orphan Technologies Limited
In November 2020, we completed the acquisition of Orphan Technologies Limited (“Orphan”), including Orphan’s rare metabolic disorder drug pegtibatinase. We acquired Orphan by purchasing all of its outstanding shares. Under the Stock Purchase Agreement ("the Agreement"), we agreed to make contingent cash payments up to an aggregate of $427.0 million based on the achievement of certain development, regulatory and commercialization events as set forth in the Agreement, as well as additional tiered mid-single digit royalty payments based upon future net sales of any pegtibatinase products in the US and Europe, subject to certain reductions as set forth in the Agreement, and a contingent payment in the event a pediatric rare disease voucher for any pegtibatinase product is granted.
Stock Purchase and Collaboration Agreement with PharmaKrysto
On March 8, 2022, we entered into a Collaboration Agreement with PharmaKrysto Limited (“PharmaKrysto”), a privately held pre-clinical stage company related to PharmaKrysto's early-stage cystinuria discovery program, and concurrently therewith entered into a Stock Purchase Agreement with PharmaKrysto (together, the "Agreements"). Pursuant to the terms of the Agreements, we acquired 5% of the outstanding common shares and are required to purchase an additional 5% of the outstanding common shares for $1.0 million upon the occurrence of a specified pre-clinical milestone. The Agreements also require us to fund all research and development expenses for the pre-clinical activities associated with the cystinuria program, which are expected to be approximately $5.0 million. In addition, the Agreements grant us an option to purchase the remaining outstanding shares of PharmaKrysto for $5.0 million upon the occurrence of a subsequent pre-clinical milestone prior to expiration of the option on March 8, 2025. If we elect to exercise the option, we would be required to perform commercially reasonable clinical diligence obligations. In addition, we would be required to make cash milestone payments totaling up to an aggregate $16.0 million upon the achievement of certain development and regulatory milestones, plus tiered royalty payments of less than 4% on future net sales of a product, if approved. We have the right to terminate the Agreements and return the shares for a nominal price at any time upon 60 days' notice, subject to survival of contingent obligations, if any.
See Note 5 to Consolidated Financial Statements for further discussion.
Borrowings
Convertible Senior Notes Due 2029
On March 11, 2022, we completed a registered underwritten public offering of $316.3 million aggregate principal amount of 2.25% Convertible Senior Notes due 2029 (“2029 Notes”). We issued the 2029 Notes under an indenture, dated as of September 10, 2018, as supplemented by the second supplemental indenture, dated as of March 11, 2022 (collectively, the “2029 Indenture”). The 2029 Notes will mature on March 1, 2029, unless earlier repurchased, redeemed, or converted. The 2029 Notes are senior unsecured obligations of ours and bear interest at an annual rate of 2.25%, payable semi-annually in arrears on March 1 and September 1 of each year, beginning on September 1, 2022. The 2029 Notes do not contain any financial or operating covenants or any restrictions on the payment of dividends, the issuance of other indebtedness or the issuance or repurchase of securities by us.
Convertible Senior Notes Due 2025
On September 10, 2018, we completed a registered underwritten public offering of $276.0 million aggregate principal amount of 2.50% Convertible Senior Notes due 2025 ("2025 Notes"), and entered into a base indenture and supplemental indenture agreement ("2025 Indenture") with respect to the 2025 Notes. The 2025 Notes will mature on September 15, 2025, unless earlier repurchased, redeemed, or converted. The 2025 Notes are senior unsecured obligations of the Company and bear interest at an annual rate of 2.50%, payable semi-annually in arrears on March 15 and September 15 of each year, beginning on March 15, 2019. On March 11, 2022, coinciding with the issuance of the 2029 Notes, we completed our repurchase of $207.1 million aggregate principal amount of 2025 Notes for cash. After giving effect to the repurchase, the total remaining principal amount outstanding under the 2025 Notes as of December 31, 2023 was $68.9 million. The 2025 Notes do not contain any financial or operating covenants or any restrictions on the payment of dividends, the issuance of other indebtedness or the issuance or repurchase of securities by us.
See Note 7 to Consolidated Financial Statements for further discussion.
Funding Requirements
We believe that our available cash and short-term investments as of the date of this filing will be sufficient to fund our anticipated level of operations beyond the next 12 months. We expect to use cash flows from operations and, when necessary, outside financings, to meet our current and future financial obligations, including funding our operations, debt service and capital expenditures. Our ability to make these payments depends on our future performance, which will be affected by financial, business, economic, regulatory and other factors, many of which we cannot control. Factors that may affect financing requirements include, but are not limited to:
•the timing, progress, cost and results of our clinical trials, preclinical studies and other discovery and research and development activities;
•the timing and outcome of, and costs involved in, seeking and obtaining marketing approvals for our products, and in maintaining quality systems standards for our products;
•the timing of, and costs involved in, commercial activities, including product marketing, sales and distribution;
•our ability to successfully commercialize FILSPARI for IgAN, to obtain full regulatory approval for, and successfully commercialize, FILSPARI for the treatment of IgAN, and to obtain regulatory approval for, and successfully commercialize, sparsentan for FSGS and our other or future product candidates;
•increases or decreases in revenue from our marketed products, including decreases in revenue resulting from generic entrants or health epidemics or pandemics;
•debt service obligations on the 2025 Notes and 2029 Notes;
•the number and development requirements of other product candidates that we pursue;
•our ability to manufacture sufficient quantities of our products to meet expected demand;
•the costs of preparing, filing, prosecuting, maintaining and enforcing any patent claims and other intellectual property rights, litigation costs and the results of litigation;
•our ability to enter into collaboration, licensing or distribution arrangements and the terms and timing of these arrangements;
•the potential need to expand our business, resulting in additional payroll and other overhead expenses;
•the potential in-licensing of other products or technologies;
•the emergence of competing technologies or other adverse market or technological developments; and
•the impacts of inflation and resulting cost increases.
Future capital requirements will also depend on the extent to which we acquire or invest in additional complementary businesses, products and technologies.
Cash Flows
The following table summarizes our cash flows for the periods set forth below (in thousands):
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| | 2023 | | 2022 | | 2021 |
| Net cash used in operating activities | $ | (280,021) | | | $ | (186,291) | | | $ | (14,792) | |
| Net cash provided by (used in) investing activities | 55,776 | | | (32,553) | | | (137,623) | |
| Net cash provided by financing activities | 218,752 | | | 117,573 | | | 231,683 | |
| Effect of exchange rate changes on cash | 1,981 | | | (2,794) | | | 1,713 | |
| Net (decrease) increase in cash and cash equivalents | (3,512) | | | (104,065) | | | 80,981 | |
| | | | | |
| Cash and cash equivalents, beginning of year | 61,688 | | | 165,753 | | | 84,772 | |
| Cash and cash equivalents, end of year | 58,176 | | | 61,688 | | | 165,753 | |
| Marketable debt securities, at fair value | 508,675 | | | 388,557 | | | 387,129 | |
| Total cash and cash equivalents and marketable debt securities | $ | 566,851 | | | $ | 450,245 | | | $ | 552,882 | |
Management considers marketable debt securities to be available to fund current operations, and they are classified as available for sale and included within current assets in our Consolidated Balance Sheets. Therefore, cash and short-term investments available to fund operations is $566.9 million as of December 31, 2023.
Cash Flows from Operating Activities
Operating activities used $280.0 million of cash during the year ended December 31, 2023 compared to $186.3 million of cash used for the year ended December 31, 2022. The increase in cash used was primarily attributable to increased research and development and sales, general and administrative expenses.
Cash Flows from Investing Activities
Cash provided by investing activities for the year ended December 31, 2023 was $55.8 million compared to cash used of $32.6 million for the year ended December 31, 2022. The change was due to $207.4 million in net proceeds from the sale of our bile acid business in the third quarter 2023, offset by an increase in net purchases of marketable debt securities and the $23.0 million milestone payment to Ligand (and BMS) in March 2023 that was triggered upon the accelerated approval of FILSPARI in February 2023.
Cash Flows from Financing Activities
For the year ended December 31, 2023, cash provided by financing activities was $218.8 million compared to cash provided of $117.6 million for the year ended December 31, 2022. The increase in cash provided was due to the March 2023 issuance of common stock and pre-funded warrants through an underwritten public offering that provided $215.8 million in net proceeds, compared with net proceeds of $95.0 million from the March 2022 issuance of the 2029 Notes and repurchase of the 2025 Notes and $19.5 million in net proceeds from sales under the ATM Agreement in 2022.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Our primary exposure to market risk is related to changes in interest rates. As of December 31, 2021,2023, we had cash equivalents and marketable debt securities of approximately $552.9$566.9 million, consisting of money market funds, U.S. government agency debt, corporate debt and commercial paper. This exposure to market risk is interest rate sensitivity, which is affected by changes in the general level of U.S. interest rates, particularly because our investments are in short-term debt securities. Our marketable debt securities are subject to interest rate risk and will fall in value if market interest rates continue to increase. Due to the short-term duration of our investment portfolio and the low risk profile of our investments, a change in interest rates of 100 basis points would have approximately a $1.7$4.2 million impact on our investments.
The marketable debt securities held in our investment portfolio may subject us to credit risk, though our investment policy limits interest-bearing security investments to certain types of instruments issued by institutions with primarily investment grade credit ratings and places restrictions on maturities and concentration by asset class and issuer. Given these policy restrictions and our emphasis on preserving capital and liquidity while enhancing overall returns, we have not experienced material credit-related losses with our securities holdings.
We are also exposed to market risk related to changes in foreign currency exchange rates. From time to time, we enter into contracts with vendors that are located outside of the United States, which contracts are denominated in foreign currencies. We are subject to fluctuations in foreign currency rated in connection with these agreements. We do not currently hedge our foreign currency exchange rate risk.
Inflation generally affects us by increasing our salaries and fees paid to third-party contract service providers. Inflationary pressures have primarily impacted our operations through increased labor costs. While we continue to monitor the effects of macroeconomic factors, inflationary pressures have not affected our current outlook or business objectives.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
The Consolidated Financial Statements and supplementary data of Travere Therapeutics, Inc. required by this Item are described in Item 15 of this Annual Report on Form 10-K and are presented beginning on page F-1.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
We maintain disclosure controls and procedures that are designed to ensure that information required to be disclosed in our Exchange Act reports is recorded, processed, summarized and reported within the timelines specified in the SEC’s rules and forms, and that such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure. In designing and evaluating the disclosure controls and procedures, management recognized that any controls and procedures, no matter how well designed and operated, can only provide reasonable assurance of achieving the desired control objectives, and in reaching a reasonable level of assurance, management necessarily was required to apply its judgment in evaluating the cost-benefit relationship of possible controls and procedures.
As required by Rule 13a-15(b) of the Exchange Act, we carried out an evaluation, under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, of the effectiveness of the design and operation of our disclosure controls and procedures (as defined in Exchange Act Rule 13a–15(e) and 15d-15(e)), as of the end of the year covered by this report. Based on the foregoing, our Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures were effective at the reasonable assurance level.
Management’s Annual Report on Internal Control Over Financial Reporting
Internal control over financial reporting refers to the process designed by, or under the supervision of, our Chief Executive Officer and Chief Financial Officer, and effected by our board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles, and includes those policies and procedures that:
(1) Pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets;
(2) Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorization of our management and directors; and
(3) Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements.
Internal control over financial reporting cannot provide absolute assurance of achieving financial reporting objectives because of its inherent limitations. Internal control over financial reporting is a process that involves human diligence and compliance and is subject to lapses in judgment and breakdowns resulting from human failures. Internal control over financial reporting also can be circumvented by collusion or improper management override. Because of such limitations, there is a risk that material misstatements may not be prevented or detected on a timely basis by internal control over financial reporting. However, these inherent limitations are known features of the financial reporting process. Therefore, it is possible to design into the process safeguards to reduce, though not eliminate, this risk. Management is responsible for establishing and maintaining adequate internal control over financial reporting for the company.
Management has used the framework set forth in the report entitled Internal Control-Integrated Framework (2013 framework) published by the Committee of Sponsoring Organizations of the Treadway Commission, known as COSO, to evaluate the effectiveness of our internal control over financial reporting. Based on this assessment, our Chief Executive Officer and Chief Financial Officer concluded that our internal control over financial reporting was effective as of December 31, 2021. BDO USA,2023. Ernst & Young LLP ("EY"), our independent registered public accounting firm, has issued an attestation report on our internal control over financial reporting as of December 31, 2021,2023, which is included herein.
Changes in Internal Control Over Financial Reporting
ThereAn evaluation was also performed under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, of any change to our internal control over financial reporting that occurred during the fourth quarter of 2023 and that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
As disclosed in Part II, Item 9A of the Company’s Annual Report on Form 10-K for the year ended December 31, 2022, our Chief Executive Officer and Chief Financial Officer concluded that our internal control over financial reporting was not effective as of December 31, 2022 due to the following material weakness: We did not design effective controls and procedures to evaluate the accounting for a certain pre-launch inventory contract affecting the timing of recognition of research and development expense. Such material weakness did not result in a restatement of previously issued annual consolidated financial statements or interim consolidated financial statements. The material weakness has been fully remediated as of December 31, 2023 and as of the date of this report.
Other than the aforementioned material weakness remediation, there have not been any changes in our internal control over financial reporting during the quarter ended December 31, 20212023 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Report of Independent Registered Public Accounting Firm
Shareholders
To the Stockholders and the Board of Directors
of Travere Therapeutics, Inc.
San Diego, California
Opinion on Internal Control over Financial Reporting
We have audited Travere Therapeutics, Inc. and its subsidiaries’ (the “Company’s”) internal control over financial reporting as of December 31, 2021,2023, based on criteria established in Internal Control – Control—Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (the “COSO criteria”)COSO criteria). In our opinion, the CompanyTravere Therapeutics, Inc. and subsidiaries (the Company) maintained, in all material respects, effective internal control over financial reporting as of December 31, 2021,2023, based on the COSO criteria.criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (“PCAOB”)(PCAOB), the consolidated balance sheetssheet of the Company and subsidiaries as of December 31, 2021 and 2020,2023, the related consolidated statements of operations and comprehensive loss, stockholders’ equity and cash flows for each of the three years in the periodyear ended December 31, 2021,2023, and the related notes and our report dated February 24, 2022,20, 2024 expressed an unqualified opinion thereon.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Item 9A, Management’s Annual Report on Internal Control overOver Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit of internal control over financial reporting in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects.
Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audit also includedrisk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ BDO USA,Ernst & Young LLP
San Diego, California
February 24, 202220, 2024
ITEM 9B. OTHER INFORMATION
None.Trading Arrangements
None of the Company’s directors or officers adopted or terminated a Rule 10b5-1 trading arrangement or a non-Rule 10b5-1 trading arrangement during the quarter ended December 31, 2023.
There are no other items required to be disclosed.
ITEM 9C. DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS
Not applicable.
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
Information required by this item and not included below will be contained under the captions "Election of Directors,"Directors" and "Information Regarding the Board of Directors and Corporate Governance,"Governance", in our Definitive Proxy Statement for our 20222024 Annual Meeting of Stockholders, to be filed pursuant to Regulation 14A with the Securities and Exchange Commission within 120 days of December 31, 20212023 (the "Proxy Statement"). Such information is incorporated herein by reference.
Our Board of Directors consists of the following members:
Roy D. Baynes, M.D., Ph.D. has served as a director of the Company since June 2016. Since July 2022, Dr. Baynes has served as Executive Vice President and Chief Medical Officer of Eikon Therapeutics, Inc., a privately-held biotechnology company. Prior to Eikon Therapeutics, Inc., until April 2022 he served as Senior Vice President and Head of Global Clinical Development at Merck Research Laboratories, the research division of Merck and Co., Inc., commencing in December 2013 and as Chief Medical Officer of Merck and Co, Inc., a global healthcare company, commencing in July 2016. Prior to his roles at Merck, Dr. Baynes served as Senior Vice President of Oncology, Inflammation and Respiratory Therapeutics at Gilead Sciences, Inc., a biopharmaceutical company, from January 2012 to December 2013. Prior to Gilead, Dr. Baynes held positions of increasing responsibility at Amgen Inc., a biotechnology company, including Vice President of Global Clinical Development and Therapeutic Area Head for Hematology/Oncology. Before joining Amgen, Dr. Baynes was the Charles Martin Professor of Cancer Research at the Barbara Ann Karmanos Cancer Institute, a National Cancer Institute-designated Comprehensive Cancer Center, at Wayne State University. Dr. Baynes has authored more than 150 publications and is a member or fellow of several international medical societies. Dr. Baynes currently serves on the Board of Directors of Natera, Inc., a genetic testing and diagnostics company, and on the Board of Directors of CatalYm GmbH, a privately-held Germany based biotechnology company. Previously he served on the Board of Directors of Atara Biotherapeutics, Inc., a T-cell immunotherapy company. Dr. Baynes received his medical degree and doctorate in philosophy from the University of the Witwatersrand in South Africa, and completed his medical training in the Department of Hematology and Oncology at Johannesburg Hospital.
Suzanne Bruhn, Ph.D.has served as a director of the Company since April 2020. Since November 2023, Dr. Bruhn has served as Chief Executive Officer of The Charcot-Marie-Tooth Association (CMTA), a nonprofit organization dedicated to finding a cure for CMT, a rare, debilitating peripheral neuropathy. Previously, from May 2019 to December 2023, Dr. Bruhn served as the President and Chief Executive Officer of Tiaki Therapeutics Inc., a biotechnology company. Dr. Bruhn served as President and Chief Executive Officer of Proclara Biosciences, Inc., a biotechnology company, from April 2017 to September 2018, and as President and Chief Executive Officer of Promedior, Inc., a biotechnology company, from May 2012 to November 2015. Currently, Dr. Bruhn serves on the Boards of Directors of Pliant Therapeutics, Inc., Vigil Neuroscience, Inc., and Mind Medicine Inc. (MindMed). Previously, Dr. Bruhn served as a member of the Board of Directors of Raptor Pharmaceuticals Corp., a pharmaceutical company, from April 2011 until it was acquired by Horizon Pharma plc in October 2016, as a member of the Board of Directors of Novelion Therapeutics, Inc., a biopharmaceutical company, from October 2017 to January 2020, as a member of the Board of Directors of Aeglea BioTherapeutics, Inc., a clinical stage biotechnology company, from February 2017 to August 2020, and on the Board of Directors of Avalo Therapeutics, Inc. (formerly Cerecor, Inc.), a biopharmaceutical company from April 2020 to November 2021. Earlier in her career Dr. Bruhn served in roles of increasing responsibility at Shire Human Genetic Therapies (formerly Transkaryotic Therapies), including Senior Vice President, Strategic Planning and Program Management. Dr. Bruhn received her B.S. degree in Chemistry from Iowa State University and her Ph.D. in Chemistry from Massachusetts Institute of Technology.
Timothy Coughlin has served as a director of the Company since March 2015. Mr. Coughlin is the former Chief Financial Officer of Neurocrine Biosciences, Inc., a biopharmaceutical company that received FDA approval for INGREZZA® (valbenazine) and ORILISSA® (elagolix), both of which were discovered and developed during his tenure at Neurocrine from 2002 to 2018. Mr. Coughlin currently serves on the board of directors of Fate Therapeutics, Inc., and aTyr Pharma, Inc., both biotechnology companies, and previously served on the Board of Directors of Peloton Therapeutics, Inc. prior to its sale to Merck in 2019. Prior to joining Neurocrine, he was with Catholic Health Initiatives, a nationwide integrated healthcare delivery system, where he served as Vice President, Financial Services. Earlier in his career Mr. Coughlin served as a Senior Manager in the Health Sciences practice of Ernst & Young LLP and its predecessors. Mr. Coughlin holds a master's degree in international business from San Diego State University and a bachelor’s degree in accounting from Temple University. Mr. Coughlin is a certified public accountant in both California and Pennsylvania.
Eric Dube, Ph.D. has served as President and Chief Executive Officer of the Company and as a member of our board of directors since January 2019. Previously, Dr. Dube served as the Head of North America of ViiV Healthcare Limited, a pharmaceuticals company, since January 2018. From June 2015 to December 2017, Dr. Dube served as Sr. Vice President and Head, Global Respiratory Franchise, of GlaxoSmithKline Pharmaceuticals plc (“GSK”), a pharmaceutical company. From February 2013 to May 2015, Dr. Dube served as Senior Vice President and Business Unit Head, Respiratory Japan of GSK. Earlier in his career, Dr. Dube held positions of increasing responsibility at GSK including senior leadership roles in Strategy, Planning & Operations, Oncology, Managed Markets and Marketing. Dr. Dube currently serves on the Board of Directors for the Biotechnology Innovation Organization (BIO), and for Reneo Pharmaceuticals, Inc., a clinical stage pharmaceutical company
focused on the development and commercialization of therapies for patients with rare genetic mitochondrial diseases. Previously, Dr. Dube served on the Board of Trustees for AIDS United, and on the Board of Directors for Biocom California. Dr. Dube holds a B.S. from Santa Clara University and a M.A. and Ph.D. from Cornell University.
Gary Lyons has served as a director of the Company since October 2014 and Chair of the Company since May 2016. Previously, Mr. Lyons was the founding President and Chief Executive Officer of Neurocrine Biosciences, Inc. and he remains a member of its Board of Directors. Prior to joining Neurocrine, Mr. Lyons held a number of senior management positions at Genentech, Inc., including Vice President of Business Development and Vice President of Sales. Mr. Lyons currently serves on the Board of Directors of Rigel Pharmaceuticals, Inc., a biotechnology company focused on developing drugs for the treatment of inflammatory/autoimmune and metabolic diseases. Previously, Mr. Lyons served on the Board of Directors of Eledon Pharmaceuticals, Inc. (formerly Novus Therapeutics, Inc.), from May 2017 to June 2023, and on the Board of Directors of Fresh Tracks Therapeutics, Inc. (formerly known as Brickell Biotech, Inc.) from August 2019 to September 2023. Mr. Lyons is also a Senior Advisor for HealthCare Royalty Partners. Mr. Lyons holds a B.A. in Marine Biology from the University of New Hampshire and an M.B.A. from Northwestern University’s J.L. Kellogg Graduate School of Management.
Jeffrey Meckler has served as a director of the Company since October 2014. Since August 2021, Mr. Meckler has served as Chief Executive Officer of Indaptus Therapeutics, Inc. (formerly Intec Inc.), a biopharmaceutical company. From April 2017 to August 2021, Mr. Meckler served as Chief Executive Officer and Vice Chair of the Board of Intec Pharma, Ltd. He served as Chief Executive Officer and a director of Cocrystal Pharma, Inc., a pharmaceutical company, from April 2015 to July 2016, as a director of QLT, Inc., an ultra-orphan ophthalmic biotechnology company based in Canada, from June 2012 to November 2016, as well as the Managing Director of The Andra Group, a life sciences consulting firm. Previously, Mr. Meckler acted as a director of several biopharmaceutical companies and medical device companies. Earlier in his career, Mr. Meckler held a series of positions at Pfizer Inc. in Manufacturing Systems, Market Research, Business Development, Strategic Planning and Corporate Finance, which included playing a significant role in acquisitions and divestitures. Mr. Meckler is the past President and continues to serve on the board of Children of Bellevue, a non-profit organization focused on advocating and developing pediatric programs at Bellevue Hospital Center. Mr. Meckler holds a B.S. in Industrial Management and M.S. in Industrial Administration from Carnegie Mellon University. In addition, Mr. Meckler received a J.D. from Fordham University School of Law.
John A. Orwin has served as a director of the Company since March 2017. Since April 2018, Mr. Orwin has served as the President and Chief Executive Officer of Atreca, Inc., a biopharmaceutical company. From June 2013 through June 2017, Mr. Orwin served as Chief Executive Officer of Relypsa, Inc. and from June 2013 through March 2017 also served as President of Relypsa and served on its board of directors from June 2013 until Relypsa’s acquisition by the Galenica Group in September 2016. Prior to Relypsa, Mr. Orwin served as President and Chief Operating Officer of Affymax, Inc., a biotechnology company, from April 2010 to January 2011, and as Affymax’s Chief Executive Officer and a member of the board of directors from February 2011 to May 2013. Previously, Mr. Orwin served as Vice President and then Senior Vice President of the BioOncology Business Unit at Genentech, Inc. (now a member of the Roche Group), a biotechnology company, and served in various executive-level positions at Johnson & Johnson, a life sciences company. Prior to such roles, Mr. Orwin held senior marketing and sales positions at various life sciences and pharmaceutical companies, including Alza Corporation (acquired by Johnson & Johnson), SangStat Medical Corporation (acquired by Genzyme), Rhone-Poulenc Rorer Pharmaceuticals, Inc. (merged with Sanofi-Aventis) and Schering-Plough Corporation (merged with Merck). Mr. Orwin currently serves as the Chair of the Board of Directors of CARGO Therapeutics (formerly Syncopation Life Sciences), a biotechnology company, and as the Chair of the Board of Directors for AnaptysBio, Inc., a clinical-stage biotechnology company, and as the Chair of the Board of Directors of the privately held company Nested Therapeutics which is focused on precision oncology medicine. Previously, Mr. Orwin served as a member of the Board of Directors of Seagen Inc. from January 2014 to December 2023, the board of directors of NeurogesX, Inc., and on the board of directors of Array BioPharma Inc. from November 2012 until its acquisition by Pfizer in July of 2019. Mr. Orwin received a B.A. in Economics from Rutgers University and an M.B.A. from New York University.
Sandra Poole has served as a director of the Company since May 2019. Since July 2020, Ms. Poole has served as the Chief Operating Officer of Mythic Therapeutics, a clinical stage biotechnology company focused on the development of novel antibody-drug conjugates (ADCs) for cancer therapy. Prior to Mythic, Ms. Poole served as the Chief Operating Officer of LogicBio Therapeutics, Inc., a genome editing company from April 2018 to March 2019, and of Candel Therapeutics, a biotechnology company, from January 2020 to March 2020. Prior to LogicBio, Ms. Poole held executive roles of increasing responsibility at ImmunoGen, Inc., a company developing antibody-drug conjugates (ADCs) to treat cancer, including serving as Senior Vice President of Technical Operations from September 2014 to July 2015, Executive Vice President, Technical Operations from July 2015 to October 2016 and Executive Vice President, Technical Operations and Commercial Development from October 2016 to January 2017. From February 2017 to March 2018 Ms. Poole served as managing director of S.E.P. Life Sciences Consulting LLC (formerly S. Poole Consulting LLC), a biopharmaceutical consulting company. Before joining ImmunoGen, Ms. Poole spent more than 15 years in CMC product development and manufacturing leadership positions at Genzyme (now Sanofi), ultimately becoming SVP of Biologics Manufacturing, overseeing global manufacturing of six commercial products across five manufacturing sites in the US and EU. Previously, Ms. Poole served on the Supervisory Board for Valneva, SE a France based vaccine company, and on the Board of Directors of ViaCyte, a privately held biotechnology until its acquisition by Vertex Pharmaceuticals Incorporated in July of 2022. Ms. Poole holds an M.A.Sc. and a B.A.Sc. in chemical engineering from the University of Waterloo (Canada).
Ron Squarer has served as a director of the Company since April 2017. Since June 2023, Mr. Squarer has served as Chair of the Board of Directors of Deciphera Pharmaceuticals, Inc., a commercial-stage biopharmaceutical company, where he has served as a Director since December 2019. Mr. Squarer has also served as Chair of the Board of Directors of ADC Therapeutics SA, a commercial-stage biopharmaceutical
company, since March 2020. Mr. Squarer has extensive commercial, development and executive leadership expertise from a greater than 25-year career in the pharmaceutical industry. Previously, Mr. Squarer served as the Chief Executive Officer and a member of the Board of Directors of Array BioPharma, Inc., an oncology focused biopharmaceutical company from April 2012 to July 2019, when Array BioPharma, Inc. was acquired by Pfizer, Inc. at an enterprise value of approximately $11.4 billion. Prior to this, Mr. Squarer held positions of increasing responsibility with Hospira Inc., a global pharmaceutical and medical device company, including serving as Senior Vice President, Chief Commercial Officer, where he was responsible for delivering $4 billion in annual revenue and leading more than 2,000 employees worldwide. Mr. Squarer joined Hospira from Mayne Pharma, an oncology-focused, global pharmaceutical company, where he served as Senior Vice President, Global Corporate and Business Development when Mayne was sold to Hospira for $2 billion in 2007. Prior to Mayne Pharma, Mr. Squarer held senior management roles at both Pfizer, Inc., focused on global oncology commercial development, and at SmithKline Beecham Pharmaceuticals (now GlaxoSmithKline) in the U.S. and Europe. Mr. Squarer holds an MBA from the Kellogg School of Management, Northwestern University and a bachelor’s degree in biochemistry from the University of California, Berkeley.
Ruth Williams-Brinkley has served as a director of the Company since September 2021. In January 2024, Ms. Williams-Brinkley retired as President of the Kaiser Foundation Health Plan of the Mid-Atlantic States, where she led all of Kaiser Permanente’s care delivery and health plan operations in Washington, D.C., suburban Maryland, Baltimore, and Northern Virginia since June 2020. She joined Kaiser Permanente in 2017 as President of Kaiser Foundation Health Plan and Hospitals of the Northwest, in Portland, Oregon. Prior to joining Kaiser, Ms. Williams-Brinkley served as CEO of KentuckyOne Health, an affiliate of CommonSpirit Health, from 2011 to 2017, as President and CEO of Carondelet Health Network in Tucson, Arizona, an affiliate of Ascension Health, from 2008 to 2011, and as President and CEO of Memorial Health Care System of Chattanooga, Tennessee, an affiliate of CommonSpirit Health, from 2002 to 2008. Currently Ms. Williams-Brinkley serves on the Board of Directors of Natera, Inc., on the Board of Trustees of the University of Phoenix, and on the not for profit Boards of DePaul University in Chicago, Illinois, and Allina Health. Previously, she served as a member of the Board of Directors of Results Physiotherapy, a private care delivery company until it was acquired by Upstream Rehabilitation in 2021, and Chattem, Inc. until it was acquired by Sanofi in 2009. Earlier in her career, Ms. Williams-Brinkley held various nursing staff and management roles of increasing responsibility. Ms. Williams-Brinkley received her B.S. and Master of Science in Nursing from DePaul University and is a Life Fellow of the American College of Healthcare Executives.
In addition to Dr. Dube, our executive officers are as follows:
Christopher Cline has served as the Chief Financial Officer of the Company since August 2022. Mr. Cline brings more than 15 years of industry experience in investor relations, corporate communications, and financial strategy, planning and analysis to the chief financial officer position. Previously, Mr. Cline served as senior vice president, investor relations and corporate communications at the Company. Since joining the Company in 2014, Mr. Cline has been responsible for leading engagement with the investment community, as well as building a developed corporate communications infrastructure and strategy. Prior to the Company, Mr. Cline was a member of the global investor relations group at Elan Corporation, plc, and the financial planning and analysis group at Phase Forward. Mr. Cline is a CFA charter holder and holds a degree in finance from the Williams College of Business at Xavier University.
Peter Heerma has served as Chief Commercial Officer of the Company since October 2019. Previously, Mr. Heerma served as Global Product General Manager for oncology and cardiovascular products at Amgen Inc., a biotechnology company, from December 2015 to September 2019. From December 2003 until November 2015, Mr. Heerma held roles of increasing responsibility at Abbott Laboratories (“Abbott”), and following Abbott’s spin-off of AbbVie, Inc., a biopharmaceutical company, at AbbVie. These roles included Senior Director of Portfolio Strategy for hepatology and nephrology, Senior Director and Asset Team Lead for HCV, diabetic nephropathy, and neuroscience development projects (all AbbVie), Director of Commercial Strategy Renal Care, International Marketing Director, Business Unit Manager of hospital products, and Product Manager for obesity and cardiovascular products (all Abbott). Mr. Heerma holds a Master of Science in European business administration and business law from the Lund University in Sweden and a Bachelor of Science in retail management and marketing from Stenden University in the Netherlands.
Jula Inrig has served as Chief Medical Officer of the Company since January 2022. Previously, Dr. Inrig served as Global Head of the Renal Center of Excellence at IQVIA, a global provider of analytics, technology solutions, and clinical research services to the life sciences industry, from September 2017 to December 2021, where she helped develop the design, execution and strategy of clinical trials leading to FDA and European Commission approvals in autosomal dominant polycystic kidney disease (ADPKD), and diabetic kidney disease. From August 2012 to March 2015 Dr. Inrig served as Medical Director, and then as Senior Medical Director from April 2015 to December 2021 and was responsible for the execution of numerous global clinical trials, including pivotal phase 3 trials in FSGS, IgAN and lupus nephritis. From March 2013 to January 2018 Dr. Inrig served on the board of directors for the Kidney Health Initiative, a public-private partnership with the FDA, working to improve the development of therapies for patients with kidney disease. Dr. Inrig has authored or co-authored over 50 peer-reviewed publications and editorials and is currently a member of several professional medical societies. Dr. Inrig is board certified in nephrology and internal medicine and has served on the faculty at the University of California, Irvine, and as an adjunct in the Department of Medicine at the Duke University School of Medicine. Dr. Inrig holds a B.A. from California State University, Sacramento, and received her M.D. from Loma Linda University and completed her internal medicine residency, her nephrology fellowship and Masters of Health Science at Duke University.
Elizabeth E. Reed has served as Senior Vice President, General Counsel and Corporate Secretary of the Company since January 2017. Previously, Ms. Reed served as Vice President, General Counsel and Secretary of Celladon Corporation, a publicly traded biotechnology company, from June 2014 to March 2016 and served as a legal consultant for companies in the life sciences industry from 2013 to June 2014 and again during 2016. From 2001 to 2012, Ms. Reed led the legal function at Anadys Pharmaceuticals, Inc., a publicly traded biopharmaceutical company, including
serving as Senior Vice President, Legal Affairs, General Counsel and Corporate Secretary until Anadys’ acquisition by Roche. Prior to Anadys, Ms. Reed was an attorney with the law firms Cooley LLP and Brobeck, Phleger & Harrison LLP. Ms. Reed is a member of the State Bar of California and received her B.S. in Business Administration from the Haas School of Business at the University of California, Berkeley and holds a J.D., cum laude, from Harvard Law School.
William E. Rote has served as Senior Vice President of Research & Development of the Company since February 2017. Previously, Dr. Rote led clinical development at Ardea Biosciences, a wholly owned subsidiary of AstraZeneca, serving as Vice President, Clinical Development from September 2014 to July 2016. From 2003 to 2014, Dr. Rote held numerous positions of increasing responsibility at Amylin Pharmaceuticals, Inc., a biopharmaceutical company, including Vice President, Site Head for Research & Development from September 2012 to July 2014, Vice President, Research & Product Development from January 2010 to September 2012, and Vice President, Corporate Development, New Ventures, from 2007 to 2010, among others. Prior to Amylin, Dr. Rote served as Executive Director, Development of Corvas International, a biopharmaceutical company. He earned both his Ph.D. in Pharmacology and B.S. in Pre-Medicine from Pennsylvania State University and received postdoctoral training from the University of Michigan.
We have adopted a Code of Business Conduct that applies to our directors and employees (including our principal executive officer, principal financial officer, principal accounting officer and controller), and have posted the text of the policy on our website (https://ir.travere.com/governance-documents). In addition, we intend to promptly disclose on our website in the future (i) the date and nature of any amendment (other than technical, administrative or other non-substantive amendments) to the policy that applies to our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions and relates to any element of the code of ethics definition enumerated in Item 406(b) of Regulation S-K, and (ii) the nature of any waiver, including an implicit waiver, from a provision of the policy that is granted to one of these specified individuals that relates to one or more of the elements of the code of ethics definition enumerated in Item 406(b) of Regulation S-K, the name of such person who is granted the waiver and the date of the waiver.
ITEM 11. EXECUTIVE COMPENSATION
Information required by this item will be contained under the captions “Compensation Discussion and Analysis,” “Executive Compensation,” “Director Compensation Summary” and “Compensation Committee Interlocks and Insider Participation” in the Proxy Statement. Such information is incorporated herein by reference.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
Information required by this item will be contained under the captions “Security Ownership of Certain Beneficial Owners and Management” and “Securities Authorized for Issuance Under Equity Compensation Plans” in the Proxy Statement. Such information is incorporated herein by reference.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
Information required by this item will be contained under the caption “Transaction With Related Persons” and “Information Regarding the Board of Directors and Corporate Governance” in the Proxy Statement. Such information is incorporated herein by reference.
ITEM 14. PRINCIPAL ACCOUNTING FEES AND SERVICES
Information required by this item will be contained under the caption “Principal Accountant Fees and Services” in the Proxy Statement. Such information is incorporated herein by reference.
PART IV
ITEM 15. EXHIBITS, FINANCIAL STATEMENT SCHEDULES
(a) The financial statements at page F-1 are incorporated by reference to a part of this Annual Report on Form 10-K.
Financial statement schedules are omitted because they are not applicable or the required information is shown in the financial statements or notes thereto.
(b) Exhibits: The exhibits to this report are listed in the exhibit index below.
| | | | | | | | |
| Exhibit No. | | Description |
| | | |
| 1.1 | | |
2.1+2.1∞ | | |
2.2+ | | |
2.3*2.2* | | |
| 2.3↡∞ | | |
| 3.1 | | |
| 3.2 | | |
| 3.3 | | |
| 3.4 | | |
| 3.5 | | |
| 3.6 | | |
| 4.1 | | |
| 4.2 | | |
| 4.3 | | |
| 4.4 | | |
| 4.5 | | |
| 4.6 | | |
| 10.1 | | |
| 10.2† | | |
| 10.3† | | |
| 10.4† | | |
10.5† | | |
10.6†10.5† | | |
1
| | | | | | | | |
10.7†10.6† | | |
10.8†10.7† | | |
| 10.8† | | |
| 10.9† | | |
| | | | | | | | |
10.9† | | |
| 10.10† | | |
10.11† | | |
10.12† | | |
10.13† | | |
10.14† | | |
10.15†10.11† | | |
10.16†10.12† | | |
10.17†10.13† | | |
10.18†10.14† | | |
10.19†10.15† | | |
10.20†10.16† | | |
10.21†10.17† | | |
10.22†10.18† | | |
10.23†10.19† | | |
10.24†10.20† | | |
10.25+¥10.21+↡ | | Sublicense Agreement, dated February 16, 2012, by and among Ligand Pharmaceuticals Incorporated, a Delaware corporation, Pharmacopeia, Inc., a Delaware limited liability company, and the Company, a Delaware limited liability company (incorporated by reference to Exhibit 10.25 to the Company’s Annual Report on Form 10-K for the year ended December 31, 2021, filed with the SEC on February 24, 2022). |
10.26*10.22* | | |
10.27*10.23* | | |
10.2810.24 | | |
10.2910.25∞ | | |
10.30 | | |
10.3110.26 | | |
10.3210.27 | | |
10.33+10.28∞ | | |
10.34+10.29∞ | | |
10.35+10.30∞ | | |
10.3610.31 | | |
| | | | | | | | |
10.37+10.32∞ | | |
10.38¥10.33↡* | | |
| | | | | | | | |
10.39¥10.34↡* | | |
| 10.35↡* | | |
10.40¥10.36↡* | | |
| 10.37↡* | | |
10.4110.38 | | |
10.42*10.39* | | |
10.4310.40 | | |
| 21.1 | | |
| 23.1 | | |
| 23.2 | | |
| 24.1 | | |
| 31.1 | | |
| 31.2 | | |
| 32.1 | | |
| 32.2 | | |
| 97 | | |
| 101.INS | | Inline XBRL Instance Document. |
| 101.SCH | | Inline XBRL Taxonomy Extension Schema Document. |
| 101.CAL | | Inline XBRL Taxonomy Extension Calculation Linkbase Document. |
| 101.DEF | | Inline XBRL Taxonomy Extension Definition Linkbase Document. |
| 101.LAB | | Inline XBRL Taxonomy Extension Label Linkbase Document. |
| 101.PRE | | Taxonomy Extension Presentation Linkbase Document. |
| 104 | | The cover page to this Annual Report on Form 10-K has been formatted in Inline XBRL. |
| | | | | | | | |
+ | | We have received confidential treatment of certain portions of this agreement, which have been omitted and filed separately with the SEC pursuant to Rule 406 under the Securities Act of 1933, as amended, or Rule 24b-2 of the Securities Exchange Act of 1934, as amended. |
| † | | Indicates management contract or compensatory plan. |
¥↡ | | Schedules have been omitted pursuant to Item 601(a)(5) of Regulation S-K. The Company undertakes to furnish supplemental copies of any of the omitted schedules upon request by the SEC. |
| * | | Certain portions of this exhibit are omitted pursuant to Item 601(b)(10)(iv). |
| ∞ | | Certain confidential information contained in this Exhibit, marked in brackets, has been omitted, because it is both not material and of the type of information that the registrant treats as private or confidential. |
ITEM 16. FORM 10-K SUMMARY
None.
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| | | | | | | | |
Date: February 24, 202220, 2024 | Travere Therapeutics, Inc. |
| | |
| By: | /s/ Eric M. Dube |
| | Name: Eric M. Dube |
| | Title: Chief Executive Officer |
POWER OF ATTORNEY
Know all persons by these presents, that each person whose signature appears below constitutes and appoints Eric Dube and Laura Clague,Christopher Cline, and each of them, as his attorneys-in-fact and agents, each with power of substitution in any and all capacities, to sign any amendments to this annual report on Form 10-K, and to file the same with exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, hereby ratifying and confirming all that the attorney-in-fact or his substitute or substitutes may do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated:
| | | | | | | | | | | | | | | | | | | | |
| Signature | | Title | | Date | | |
| | | | | | |
/s/ Eric M. Dube | | Chief Executive Officer and Director (Principal Executive Officer) | | | | |
Eric M. Dube | | | | February 24, 202220, 2024 | | |
| | | | | | | |
/s/ Laura ClagueChristopher Cline | | Chief Financial Officer (Principal Financial Officer) | | | | |
Laura ClagueChristopher Cline | | | | February 24, 202220, 2024 | | |
| | | | | | |
| /s/ Sandra Calvin | | Senior Vice President Corporate Controller and Chief Accounting Officer (Principal Accounting Officer) | | | | |
| Sandra Calvin | | | | February 24, 202220, 2024 | | |
| | | | | | | |
/s/ Stephen Aselage | | Director | | | | |
Stephen Aselage | | | | February 24, 2022 | | |
| | | | | | |
| /s/ Roy D. Baynes | | Director | | | | |
| Roy D. Baynes | | | | February 24, 202220, 2024 | | |
| | | | | | | |
| /s/ Suzanne Bruhn | | Director | | | | |
| Suzanne Bruhn | | | | February 24, 202220, 2024 | | |
| | | | | | |
| /s/ Timothy Coughlin | | Director | | | | |
| Timothy Coughlin | | | | February 24, 202220, 2024 | | |
| | | | | | | |
| /s/ Gary Lyons | | Director | | | | |
| Gary Lyons | | | | February 24, 202220, 2024 | | |
| | | | | | |
| /s/ Jeffrey A. Meckler | | Director | | | | |
| Jeffrey A. Meckler | | | | February 24, 202220, 2024 | | |
| | | | | | |
| /s/ John A. Orwin | | Director | | | | |
| John A. Orwin | | | | February 24, 202220, 2024 | | |
| | | | | | |
| /s/ Sandra E. Poole | | Director | | | | |
| Sandra E. Poole | | | | February 24, 202220, 2024 | | |
| | | | | | |
| /s/ Ron Squarer | | Director | | | | |
| Ron Squarer | | | | February 24, 202220, 2024 | | |
| | | | | | |
| /s/ Ruth Williams-Brinkley | | Director | | | | |
| Ruth Williams-Brinkley | | | | February 24, 202220, 2024 | | |
TRAVERE THERAPEUTICS, INC. AND SUBSIDIARIES
INDEX TO FINANCIAL STATEMENTS
| | | | | |
| | Page |
| ID: 42) | |
| Report of Independent Registered Public Accounting Firm (BDO USA, P.C., San Diego, California, PCAOB ID: 243) | |
| Financial Statements | |
| |
| |
| |
| |
| |
Report of Independent Registered Public Accounting Firm
ShareholdersTo the Stockholders and the Board of Directors
of Travere Therapeutics, Inc.
San Diego, California
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheetssheet of Travere Therapeutics, Inc. and subsidiaries (the “Company”)Company) as of December 31, 2021 and 2020,2023, the related consolidated statements of operations and comprehensive loss, stockholders’ equity and cash flows for each of the three years in the periodyear ended December 31, 2021,2023 and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2021 and 2020,2023, and the results of its operations and its cash flows for each of the three years in the periodyear ended December 31, 2021,2023, in conformity with accounting principlesU.S. generally accepted in the United States of America.accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (“PCAOB”)(PCAOB), the Company's internal control over financial reporting as of December 31, 2021,2023, based on criteria established in Internal Control – IntegratedControl-Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”)(2013 framework), and our report dated February 24, 202220, 2024 expressed an unqualified opinion thereon.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audit included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audit also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audit provides a reasonable basis for our opinion.
Critical Audit Matter
The critical audit matter communicated below is a matter arising from the current period audit of the financial statements that was communicated or required to be communicated to the audit committee and that: (1) relates to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective or complex judgments. The communication of the critical audit matter does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the accounts or disclosuresto which it relates.
Government rebate deductions from revenue
| | | | | | | | |
| Description of the Matter | As described in Note 3 to the consolidated financial statements under the caption “Deductions from Revenue”, the Company calculates the rebates that it will be obligated to provide to government programs and deducts these estimated amounts from its gross product sales at the time the revenues are recognized. Allowances for government rebates and discounts are established based on an estimated allocation of payers and the government-mandated discounts applicable to government-funded programs. Where appropriate, these allowances take into consideration the Company’s historical experience, current statutory requirements and specific known market events and trends. Estimated government rebates are included in accrued expenses on the consolidated balance sheet.
Auditing the government rebate deductions from revenue was complex and required significant auditor judgment because the related accruals were dependent on certain subjective assumptions. Such subjective assumptions included estimated allocations of payers and estimated government-mandated discounts applicable to government-funded programs, each of which are adjusted for the Company’s historical experience, current statutory requirements and specific known market events and trends, where appropriate. |
| How We Addressed the Matter in Our Audit | We obtained an understanding, evaluated the design, and tested the operating effectiveness of controls over the Company’s government rebate deductions from revenue process. This included testing controls over management’s review of the significant assumptions described above and inputs into the rebate calculations. For example, we tested controls over actual product sales and the accuracy of forecasting estimated allocations of payers. The testing was inclusive of management’s controls to evaluate the accuracy of its judgments when compared to actual rebates paid, rebate validation and processing, and controls to ensure that the data used to evaluate and support the significant assumptions was complete, accurate and, where applicable, verified to external data sources.
To test the government rebate deductions from revenue, our audit procedures included, among others, understanding and evaluating the significant assumptions and underlying data used in management’s calculations. Our testing of significant assumptions included a lookback analysis to evaluate the historical accuracy of management’s estimates by comparing actual activity to previous estimates and performed sensitivity analyses over the subjective assumptions to evaluate the completeness of the reserves. As a part of our procedures, we evaluated the reasonableness of the Company’s assumptions considering recent sales trends and regulatory factors related to government rebates. |
/s/ Ernst & Young LLP
We have served as the Company’s auditor since 2023.
San Diego, California
February 20, 2024
Report of Independent Registered Public Accounting Firm
Shareholders and Board of Directors
Travere Therapeutics, Inc.
San Diego, California
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheet of Travere Therapeutics, Inc. (the “Company”) as of December 31, 2022, and the related consolidated statements of operations and comprehensive loss, stockholders’ equity, and cash flows for each of the two years in the period ended December 31, 2022, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2022, and the results of its operations and its cash flows for each of the two years in the periods ended December 31, 2022, in conformity with accounting principles generally accepted in the United States of America.
Change in Accounting Principle
As discussed in Note 2 to the consolidated financial statements, effective January 1, 2022, the Company adopted Accounting Standards Update (“ASU”) No. 2020-06, Debt — Debt with Conversion and Other Options (“Subtopic 470-20”) and Derivatives and Hedging — Contracts in Entity’s Own Equity (“Subtopic 815-40”): Accounting for Convertible Instruments and Contracts in an Entity’s Own Equity(“ASU 2020-06”).
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the auditaudits to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matters
The critical audit matters communicated below are matters arising from the current period audit of the consolidated financial statements that were communicated or required to be communicated to the audit committee and that: (1) relate to accounts or disclosures that are material to the consolidated financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matters below, providing separate opinions on the critical audit matters or on the accounts or disclosures to which they relate.
Business Combination-Related Contingent Consideration
As described in Note 8 to the consolidated financial statements, the Company acquired two businesses, related to the Cholbam and Chenodal products, whose purchase price included potential future payments that are contingent on the achievement of certain milestones and percentages of future net sales derived from the products acquired. The Company recorded business combination-related contingent consideration liabilities at their fair value on the acquisition date and revalues them at the end of each reporting period.
We identified the determination of the fair value of business combination-related contingent consideration as a critical audit matter. The determination of the fair value of business combination-related contingent consideration requires management to make significant judgments including the appropriateness of the valuation model selected, the reasonableness of estimates and assumptions utilized in forecasts of future net sales and the discount rates applied to such forecasts. Changes in these judgments could have a significant impact on the fair value of the business combination-
related contingent consideration. Auditing these elements involved especially challenging auditor judgment due to the subjectivity, nature and extent of auditor effort required to address the matter, including the extent of specialized skills and knowledge needed.
The primary procedures we performed to address this critical audit matter included:
•Testing the design and operating effectiveness of certain controls over the development of the significant assumptions used in the valuation model selected, including controls over assumptions related to: (i) forecasts of future net sales and (ii) discount rates applied to the forecasts.
•Assessing the reasonableness of certain significant assumptions used in the valuation model, by: (i) reviewing the products’ historical performance, (ii) evaluating the reasonableness of significant assumptions (including net sales projections) against budgets and the current performance of the Company, and (iii) performing sensitivity analyses to test the potential effect of changes in certain assumptions on the valuation.
•Utilizing professionals with specialized skills and knowledge in valuation to assist in evaluating the appropriateness of the valuation models utilized by management and to assess the reasonableness of certain estimates and assumptions used by management to develop the discount rates applied to the net sales forecasts.
License and Collaboration Agreement with Vifor
As discussed in Note 4 to the consolidated financial statements, in September 2021, the Company entered into a license and collaboration agreement with Vifor (International) Ltd. (“Vifor Pharma”). Under the terms of the agreement, the Company granted Vifor Pharma an exclusive license to commercialize sparsentan in Europe, Australia, and New Zealand. In exchange for the license, the Company received an upfront payment of $55.0 million during 2021 and is eligible to receive additional regulatory and market access related milestones and sales-based milestones and royalties.
We have identified the identification of performance obligations under the Vifor license and collaboration agreement as a critical audit matter. Determining the distinct performance obligations included in the agreement required management to make judgements with respect to the appropriate application of relevant authoritative accounting guidance. Auditing management’s judgments and the resulting impacts on the accounting and revenue recognition for the Vifor license and collaboration agreement required increased auditor effort.
The primary procedures we performed to address this critical audit matter included:
a.Assessing the key terms in the license and collaboration agreement under the relevant authoritative accounting guidance to evaluate management’s conclusion that the license and collaboration agreement contains two distinct performance obligations.
a.Utilizing professionals with increased experience and expertise in technical accounting for license and collaboration agreements to assist in evaluation of management’s conclusions with respect to the performance obligations identified.
/s/ BDO USA, LLPP.C.
We have served as the Company’s auditor since 2014.from 2014 to 2023.
San Diego, California
February 24, 202223, 2023, except for the effects of discontinued operations discussed in Notes 1 and 19, as to which the date is February 20, 2024.
TRAVERE THERAPEUTICS, INC. AND SUBSIDIARIES
CONSOLIDATED BALANCE SHEETS
(In thousands, except share and per share amounts)
| | December 31, 2021 | | December 31, 2020 |
| December 31, 2023 | | | December 31, 2023 | | December 31, 2022 |
| Assets | Assets | | | | Assets | | | |
| Current assets: | Current assets: | | | | Current assets: | | | |
| Cash and cash equivalents | Cash and cash equivalents | $ | 165,753 | | | $ | 84,772 | |
| Marketable debt securities, at fair value | Marketable debt securities, at fair value | 387,129 | | | 276,817 | |
| Accounts receivable, net | Accounts receivable, net | 15,914 | | | 15,925 | |
| Inventory, net | Inventory, net | 7,313 | | | 7,608 | |
| Tax receivable | 247 | | | 17,142 | |
| Prepaid expenses and other current assets | Prepaid expenses and other current assets | 6,471 | | | 8,143 | |
| | Current assets of discontinued operations | |
| Total current assets | Total current assets | 582,827 | | | 410,407 | |
| Long-term inventory, net | |
| Property and equipment, net | Property and equipment, net | 11,106 | | | 9,418 | |
| Operating lease right of use assets | Operating lease right of use assets | 23,196 | | | 25,675 | |
| | Intangible assets, net | Intangible assets, net | 148,435 | | | 154,125 | |
| Other assets | Other assets | 11,069 | | | 7,814 | |
| Non-current assets of discontinued operations | |
| Total assets | Total assets | $ | 776,633 | | | $ | 607,439 | |
| Liabilities and Stockholders' Equity | Liabilities and Stockholders' Equity | | | | Liabilities and Stockholders' Equity | | | |
| Current liabilities: | Current liabilities: | | | | Current liabilities: | | | |
| Accounts payable | Accounts payable | $ | 15,144 | | | $ | 12,133 | |
| Accrued expenses | Accrued expenses | 75,180 | | | 56,793 | |
| | Deferred revenue, current portion | Deferred revenue, current portion | 16,268 | | | — | |
| Business combination-related contingent consideration | 7,400 | | | 17,400 | |
| Operating lease liabilities, current portion | Operating lease liabilities, current portion | 3,908 | | | 357 | |
| Other current liabilities | Other current liabilities | 6,188 | | | 5,977 | |
| | Current liabilities of discontinued operations | |
| Total current liabilities | Total current liabilities | 124,088 | | | 92,660 | |
| Convertible debt | Convertible debt | 226,581 | | | 215,339 | |
| Deferred revenue, less current portion | Deferred revenue, less current portion | 20,379 | | | — | |
| Business combination-related contingent consideration, less current portion | 59,700 | | | 47,700 | |
| Operating lease liabilities, less current portion | Operating lease liabilities, less current portion | 31,497 | | | 28,336 | |
| Other non-current liabilities | Other non-current liabilities | 12,276 | | | 12,191 | |
| | Non-current liabilities of discontinued operations | |
| Total liabilities | Total liabilities | 474,521 | | | 396,226 | |
| Commitments and Contingencies (See Note 11) | 0 | | 0 |
| Commitments and contingencies (See Note 11) | | Commitments and contingencies (See Note 11) | | | |
| Stockholders' Equity: | Stockholders' Equity: | | | | Stockholders' Equity: | | | |
| Preferred stock $0.0001 par value; 20,000,000 shares authorized; 0 issued and outstanding as of December 31, 2021 and 2020 | — | | | — | |
| Common stock $0.0001 par value; 200,000,000 and 100,000,000 shares authorized; 62,491,498 and 52,248,431 issued and outstanding as of December 31, 2021 and 2020, respectively | 6 | | | 5 | |
| Preferred stock $0.0001 par value; 20,000,000 shares authorized; no shares issued and outstanding as of December 31, 2023 and 2022 | |
| Common stock $0.0001 par value; 200,000,000 shares authorized; 75,367,117 and 64,290,570 issued and outstanding as of December 31, 2023 and 2022, respectively | |
| Additional paid-in capital | Additional paid-in capital | 1,068,634 | | | 797,985 | |
| Accumulated deficit | Accumulated deficit | (765,966) | | | (585,875) | |
| Accumulated other comprehensive loss | Accumulated other comprehensive loss | (562) | | | (902) | |
| Total stockholders' equity | Total stockholders' equity | 302,112 | | | 211,213 | |
| Total liabilities and stockholders' equity | Total liabilities and stockholders' equity | $ | 776,633 | | | $ | 607,439 | |
TRAVERE THERAPEUTICS, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
(In thousands, except share and per share amounts)
| | | | Years Ended December 31, | | Year Ended December 31, |
| | | 2021 | | 2020 | | 2019 | | 2023 | | 2022 | | 2021 |
| Net product sales | Net product sales | $ | 210,776 | | | $ | 198,321 | | | $ | 175,338 | |
| License and collaboration revenue | License and collaboration revenue | 16,714 | | | — | | | — | |
| Total revenue | Total revenue | 227,490 | | | 198,321 | | | 175,338 | |
| Operating expenses: | Operating expenses: | | | | | Operating expenses: | | | | | |
| Cost of goods sold | Cost of goods sold | 6,784 | | | 6,126 | | | 5,234 | |
| Research and development | Research and development | 210,328 | | | 131,773 | | | 140,963 | |
| Selling, general and administrative | Selling, general and administrative | 149,883 | | | 135,799 | | | 128,951 | |
| Change in fair value of contingent consideration | 22,260 | | | 3,655 | | | 15,051 | |
| | Impairment of L-UDCA IPR&D intangible asset | — | | | — | | | 25,500 | |
| Write off of L-UDCA contingent consideration | — | | | — | | | (18,000) | |
| Acquired IPR&D expense | — | | | 97,131 | | | — | |
| Impairment of long-term investment | — | | | — | | | 15,000 | |
| Restructuring | |
| Total operating expenses | Total operating expenses | 389,255 | | | 374,484 | | | 312,699 | |
| Operating loss | Operating loss | (161,765) | | | (176,163) | | | (137,361) | |
| Other Income (expense), net: | | | | | |
| Other income (expenses), net: | | Other income (expenses), net: | | | |
| Interest income | Interest income | 1,993 | | | 5,003 | | | 10,055 | |
| Interest expense | Interest expense | (20,141) | | | (19,050) | | | (18,828) | |
| Other income (expense), net | 231 | | | 1,420 | | | (314) | |
| | Total other expense, net | (17,917) | | | (12,627) | | | (9,087) | |
| Loss before (provision) benefit for income taxes | (179,682) | | | (188,790) | | | (146,448) | |
| | Income tax (provision) benefit | (409) | | | 19,359 | | | 21 | |
| | Other income, net | |
| Loss on extinguishment of debt | |
| Total other income (expense), net | |
| Loss from continuing operations before income tax provision | |
| Income tax provision on continuing operations | |
| Loss from continuing operations, net of tax | |
| Income from discontinued operations, net of tax | |
| Net loss | Net loss | $ | (180,091) | | | $ | (169,431) | | | $ | (146,427) | |
| | Basic and diluted net loss per common share | $ | (3.01) | | | $ | (3.56) | | | $ | (3.46) | |
| Per share data | |
| Per share data | |
| Per share data | |
| Basic and diluted: | |
| Basic and diluted: | |
| Basic and diluted: | |
| Net loss from continuing operations | |
| Net loss from continuing operations | |
| Net loss from continuing operations | |
| Net income from discontinued operations | |
| Net loss per common share | |
| | Basic and diluted weighted average common shares outstanding | 59,832,287 | | | 47,539,631 | | | 42,339,961 | |
| Weighted average common shares outstanding | |
| Weighted average common shares outstanding | |
| Weighted average common shares outstanding | |
| | | Comprehensive loss: | Comprehensive loss: | | | | | |
| | Comprehensive loss: | |
| | Comprehensive loss: | | | | |
| Net loss | Net loss | $ | (180,091) | | | $ | (169,431) | | | $ | (146,427) | |
| Foreign currency translation gain (loss) | 1,515 | | | (1,452) | | | 92 | |
| Unrealized (loss) gain on debt securities | (1,175) | | | (176) | | | 2,163 | |
| Unrealized loss on defined benefit pension plan | |
| Foreign currency translation (loss) gain | |
| Unrealized gain (loss) on marketable debt securities | |
| Comprehensive loss | Comprehensive loss | $ | (179,751) | | | $ | (171,059) | | | $ | (144,172) | |
The accompanying notes are an integral part of these consolidated financial statements.
TRAVERE THERAPEUTICS, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF STOCKHOLDERS' EQUITY
(In thousands, except share amounts)
| | | Common Stock | | | Additional Paid in Capital | | Accumulated Other Comprehensive Income (Loss) | | Accumulated
Deficit | | Total
Stockholders'
Equity |
| | Shares | | Amount | | | |
| | BALANCE - DECEMBER 31, 2018 | 41,389,524 | | | $ | 4 | | | | $ | 589,795 | | | $ | (1,529) | | | $ | (270,017) | | | $ | 318,253 | |
| Share based compensation | — | | | — | | | | 20,463 | | | — | | | — | | | 20,463 | |
| | Unrealized gain/(loss) on marketable debt securities | — | | | — | | | | — | | | 2,163 | | | — | | | 2,163 | |
| Foreign currency translation adjustments | — | | | — | | | | — | | | 92 | | | — | | | 92 | |
| Issuance of common shares under the equity incentive plan and proceeds from exercise. | 272,730 | | | — | | | | 1,618 | | | — | | | — | | | 1,618 | |
| Employee stock purchase program stock purchase and expense | 129,324 | | | — | | | | 2,444 | | | — | | | — | | | 2,444 | |
| Convertible debt issue | 1,297,343 | | | — | | | | 22,590 | | | — | | | — | | | 22,590 | |
| Net loss | — | | | — | | | | — | | | — | | | (146,427) | | | (146,427) | |
| BALANCE - DECEMBER 31, 2019 | 43,088,921 | | | $ | 4 | | | | $ | 636,910 | | | $ | 726 | | | $ | (416,444) | | | $ | 221,196 | |
| Share based compensation | — | | | — | | | | 22,733 | | | — | | | — | | | 22,733 | |
| Issuance of common stock under the equity incentive plan and proceeds from exercise | 649,075 | | | — | | | | 3,895 | | | — | | | — | | | 3,895 | |
| Equity offering, net of issuance costs of $7.2 million | 7,475,000 | | | 1 | | | | 108,691 | | | — | | | — | | | 108,692 | |
| Issuance of common stock under At-The-Market offering, net of offering costs | 867,806 | | | — | | | | 22,828 | | | 22,828 | |
| Unrealized gain/(loss) on marketable debt securities | — | | | — | | | | — | | | (176) | | | — | | | (176) | |
| Foreign currency translation adjustments | — | | | — | | | | — | | | (1,452) | | | — | | | (1,452) | |
| | Employee stock purchase program stock purchase and expense | 167,629 | | | — | | | | 2,928 | | | — | | | — | | | 2,928 | |
| Net loss | — | | | — | | | | — | | | — | | | (169,431) | | | (169,431) | |
| Common Stock | |
| Common Stock | |
| Common Stock | | | | | Additional Paid-in Capital | | Accumulated Other Comprehensive Loss | | Accumulated
Deficit | | Total
Stockholders'
Equity |
| Shares | |
| BALANCE - DECEMBER 31, 2020 | |
| BALANCE - DECEMBER 31, 2020 | |
| BALANCE - DECEMBER 31, 2020 | BALANCE - DECEMBER 31, 2020 | 52,248,431 | | | $ | 5 | | | | $ | 797,985 | | | $ | (902) | | | $ | (585,875) | | | $ | 211,213 | |
| Share based compensation | Share based compensation | — | | | — | | | | 29,862 | | | — | | | — | | | 29,862 | |
| Issuance of common stock under the equity incentive plan and proceeds from exercise | Issuance of common stock under the equity incentive plan and proceeds from exercise | 1,302,966 | | | — | | | | 12,841 | | | — | | | — | | | 12,841 | |
| Employee stock purchase program stock purchase and expense | |
| Equity offering, net of issuance costs of $12.2 million | Equity offering, net of issuance costs of $12.2 million | 7,532,500 | | | 1 | | | | 189,278 | | | — | | | — | | | 189,279 | |
| Issuance of common stock under At-The-Market offering, net of offering costs | 1,240,640 | | | — | | | | 35,656 | | | — | | | — | | | 35,656 | |
| Unrealized gain/(loss) on marketable debt securities | — | | | — | | | | — | | | (1,175) | | | — | | | (1,175) | |
| Issuance of common stock under At-The-Market offering, net of issuance costs of $1.1 million. | |
| Foreign currency translation adjustments | Foreign currency translation adjustments | — | | | — | | | | — | | | 1,515 | | | — | | | 1,515 | |
| Employee stock purchase program stock purchase and expense | 166,961 | | | — | | | | 3,012 | | | — | | | — | | | 3,012 | |
| Unrealized loss on marketable debt securities | |
| Net loss | Net loss | — | | | — | | | | — | | | — | | | (180,091) | | | (180,091) | |
| BALANCE - DECEMBER 31, 2021 | BALANCE - DECEMBER 31, 2021 | 62,491,498 | | | $ | 6 | | | | $ | 1,068,634 | | | $ | (562) | | | $ | (765,966) | | | $ | 302,112 | |
| Cumulative-effect adjustment from adoption of ASU 2020-06 | |
| Share based compensation | |
| Issuance of common stock under the equity incentive plan and proceeds from exercise | |
| Employee stock purchase program purchase and expense | |
| Issuance of common stock under At-The-Market offering, net of issuance costs of $0.6 million. | |
| Unrealized loss on defined benefit pension plan | |
| Foreign currency translation adjustments | |
| Unrealized loss on marketable debt securities | |
| Net loss | |
| BALANCE - DECEMBER 31, 2022 | |
| Share based compensation | |
| Issuance of common stock under the equity incentive plan and proceeds from exercise | |
| Employee stock purchase program purchase and expense | |
| Equity offering, net of issuance costs of $12.6 million | |
| Issuance of pre-funded common stock warrants, net of issuance costs of $1.6 million | |
| Unrealized loss on defined benefit pension plan | |
| Foreign currency translation adjustments | |
| Unrealized gain on marketable debt securities | |
| Net loss | |
| BALANCE - DECEMBER 31, 2023 | |
TRAVERE THERAPEUTICS, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands) | | | | | | | | | | | | | | | | | |
| | For the year ended December 31, |
| | 2021 | | 2020 | | 2019 |
| Cash Flows from Operating Activities: | | | | | |
| Net loss | $ | (180,091) | | | $ | (169,431) | | | $ | (146,427) | |
| Adjustments to reconcile net loss to net cash used in operating activities: | | | | | |
| Share based compensation | 30,766 | | | 23,614 | | | 21,105 | |
| Depreciation and amortization | 26,617 | | | 24,579 | | | 20,408 | |
| Change in estimated fair value of contingent consideration | 22,260 | | | 3,655 | | | (2,948) | |
| Payments from change in fair value of contingent consideration | (14,375) | | | (12,407) | | | (5,661) | |
| Amortization of debt discount and deferred financing costs | 11,242 | | | 10,478 | | | 9,903 | |
| Loss on allowance for inventory | 2,110 | | | 2,225 | | | 1,468 | |
| Amortization of premiums (discounts) on investments | 2,008 | | | 703 | | | (788) | |
| Acquired IPR&D expense | — | | | 97,131 | | | — | |
| Impairment of intangible assets | — | | | — | | | 25,500 | |
| Loss on impairment of long-term investment | — | | | — | | | 15,000 | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| Other | 4,692 | | | 2,752 | | | 1,602 | |
| Changes in operating assets and liabilities, net of asset acquisition: | | | | | |
| Accounts receivable | (59) | | | 2,301 | | | (5,184) | |
| Inventory | (1,815) | | | (5,067) | | | (1,958) | |
| Prepaid expenses and other current assets | (1,438) | | | (4,314) | | | 208 | |
| Tax receivable | 17,099 | | | (15,746) | | | 555 | |
| Change in lease assets and liabilities, net | 5,577 | | | (1,203) | | | (4,854) | |
| Accounts payable | 2,770 | | | (6,036) | | | 9,255 | |
| Accrued expenses and other current liabilities | 19,545 | | | 4,023 | | | 4,602 | |
| Deferred revenue, current and non-current | 38,300 | | | — | | | — | |
| Net cash used in operating activities | (14,792) | | | (42,743) | | | (58,214) | |
| Cash Flows from Investing Activities: | | | | | |
| Purchase of fixed assets | (5,101) | | | (6,767) | | | (195) | |
| Purchase of intangible assets | (19,050) | | | (17,799) | | | (15,370) | |
| | | | | |
| Proceeds from the sale/maturity of marketable debt securities | 433,604 | | | 273,482 | | | 259,140 | |
| Purchase of marketable debt securities | (547,076) | | | (214,964) | | | (223,712) | |
| Purchased IPR&D, net of cash acquired | — | | | (95,279) | | | — | |
| Net cash (used in) provided by investing activities | (137,623) | | | (61,327) | | | 19,863 | |
| Cash Flows from Financing Activities: | | | | | |
| Payment of acquisition-related contingent consideration | (6,104) | | | (7,648) | | | (3,388) | |
| | | | | |
| Payment of guaranteed minimum royalty | (2,100) | | | (2,100) | | | (2,084) | |
| | | | | |
| Proceeds from exercise of stock options | 12,841 | | | 3,895 | | | 1,618 | |
| Proceeds from the issuance of common stock in At-the-Market equity offering | 35,656 | | | 22,828 | | | — | |
| Proceeds from the issuance of common stock, net of issuance costs | 189,278 | | | 108,692 | | | — | |
| | | | | |
| | | | | |
| | | | | |
| Proceeds from the issuance of common stock under the employee stock purchase program | 2,112 | | | 2,046 | | | 1,777 | |
| Net cash provided by (used in) financing activities | 231,683 | | | 127,713 | | | (2,077) | |
| Effect of exchange rate changes on cash | 1,713 | | | (1,307) | | | (9) | |
| Net increase (decrease) in cash and cash equivalents | 80,981 | | | 22,336 | | | (40,437) | |
| Cash and cash equivalents, beginning of year | 84,772 | | | 62,436 | | | 102,873 | |
| Cash and cash equivalents, end of year | $ | 165,753 | | | $ | 84,772 | | | $ | 62,436 | |
| Supplemental Disclosure of Cash Flow Information: | | | | | |
| Cash paid for interest | $ | 7,327 | | | $ | 6,901 | | | $ | 7,669 | |
| Cash refunded (paid) for income taxes | $ | 16,420 | | | $ | 3,373 | | | $ | (656) | |
| Non-cash Investing and financing activities: | | | | | |
| | | | | |
| Accrued royalty in excess of minimum payable to the sellers of Thiola | $ | 19,362 | | | $ | 18,168 | | | $ | 15,815 | |
| | | | | |
| | | | | |
| | | | | | | | | | | | | | | | | |
| | For the year ended December 31, |
| | 2023 | | 2022 | | 2021 |
| Cash Flows From Operating Activities: | | | | | |
| Net loss | $ | (111,399) | | | $ | (278,482) | | | $ | (180,091) | |
| Net income from discontinued operations | 264,934 | | | 52,986 | | | 37,201 | |
| Net loss from continuing operations | (376,333) | | | (331,468) | | | (217,292) | |
| Adjustments to reconcile net loss to net cash used in operating activities: | | | | | |
| Depreciation and amortization | 38,530 | | | 20,719 | | | 12,746 | |
| Share based compensation | 44,246 | | | 38,143 | | | 29,566 | |
| | | | | |
| | | | | |
| Loss on extinguishment of debt | — | | | 7,578 | | | — | |
| Loss on allowance for inventory | 3,039 | | | 1,039 | | | 498 | |
| Amortization of debt discount and issuance costs | 1,718 | | | 1,622 | | | 11,242 | |
| Amortization of (discounts) premiums on investments | (7,456) | | | (700) | | | 2,008 | |
| Other | (3,626) | | | 1,346 | | | 4,219 | |
| Changes in operating assets and liabilities: | | | | | |
| Accounts receivable | (11,264) | | | (1,060) | | | 853 | |
| Inventory | (39,420) | | | (1,209) | | | 117 | |
| Tax receivable | (35) | | | (374) | | | 17,099 | |
| Prepaid expenses and other current and non-current assets | (4,657) | | | (5,196) | | | (1,439) | |
| Change in lease assets and liabilities, net | (986) | | | (984) | | | 5,577 | |
| Accounts payable | 24,763 | | | 2,055 | | | 2,467 | |
| Accrued expenses | 22,321 | | | 22,181 | | | 16,678 | |
| Deferred revenue, current and non-current | (15,779) | | | (12,929) | | | 38,300 | |
| Other current and non-current liabilities | (418) | | | (1,609) | | | 1,123 | |
| Net cash used in operating activities - continuing operations | (325,357) | | | (260,846) | | | (76,238) | |
| Net cash provided by operating activities - discontinued operations | 45,336 | | | 74,555 | | | 61,446 | |
| Net cash used in operating activities | (280,021) | | | (186,291) | | | (14,792) | |
| Cash Flows From Investing Activities: | | | | | |
| Proceeds from the sale/maturity of marketable debt securities | 334,575 | | | 381,993 | | | 433,604 | |
| Purchase of marketable debt securities | (443,942) | | | (385,389) | | | (547,076) | |
| Purchase of fixed assets | (668) | | | (191) | | | (5,101) | |
| Purchase of intangible assets | (41,591) | | | (28,366) | | | (19,050) | |
| | | | | |
| Other | — | | | (600) | | | — | |
| Net cash used in investing activities - continuing operations | (151,626) | | | (32,553) | | | (137,623) | |
| Net cash provided by investing activities - discontinued operations | 207,402 | | | — | | | — | |
| Net cash provided by (used in) investing activities | 55,776 | | | (32,553) | | | (137,623) | |
| Cash Flows From Financing Activities: | | | | | |
| Payment of guaranteed minimum royalty | (2,100) | | | (2,100) | | | (2,100) | |
| | | | | |
| Proceeds from issuance of 2029 convertible senior notes | — | | | 316,250 | | | — | |
| Payment of debt issuance costs | — | | | (9,882) | | | — | |
| Repurchase of 2025 convertible senior notes including premium | — | | | (211,324) | | | — | |
| Proceeds from the issuance of common stock, net of issuance costs | 191,198 | | | — | | | 189,278 | |
| Proceeds from the issuance of pre-funded warrants, net of issuance costs | 24,630 | | | — | | | — | |
| | | | | | | | | | | | | | | | | |
| Proceeds from exercise of stock options | 3,240 | | | 4,585 | | | 12,841 | |
| Proceeds from the issuances under the employee stock purchase program | 3,166 | | | 2,978 | | | 2,112 | |
| Proceeds from the issuance of common stock in At-the-Market equity offering | — | | | 19,545 | | | 35,656 | |
| Net cash provided by financing activities - continuing operations | 220,134 | | | 120,052 | | | 237,787 | |
| Net cash used in financing activities - discontinued operations | (1,382) | | | (2,479) | | | (6,104) | |
| Net cash provided by financing activities | 218,752 | | | 117,573 | | | 231,683 | |
| Effect of exchange rate changes on cash | 1,981 | | | (2,794) | | | 1,713 | |
| Net (decrease) increase in cash and cash equivalents | (3,512) | | | (104,065) | | | 80,981 | |
| Cash and cash equivalents, beginning of year | 61,688 | | | 165,753 | | | 84,772 | |
| Cash and cash equivalents, end of year | $ | 58,176 | | | $ | 61,688 | | | $ | 165,753 | |
| Supplemental Disclosure of Cash Flow Information: | | | | | |
| Operating cash flows used for operating leases | $ | 6,315 | | | $ | 6,020 | | | $ | 2,205 | |
| Cash paid for interest | $ | 8,838 | | | $ | 10,159 | | | $ | 7,327 | |
| Cash (paid) refunded for income taxes | $ | (578) | | | $ | (996) | | | $ | 16,420 | |
| Non-cash investing and financing activities: | | | | | |
| | | | | |
| Accrued royalty in excess of minimum payable to the sellers of Thiola | $ | 16,206 | | | $ | 16,067 | | | $ | 19,362 | |
The accompanying notes are an integral part of these consolidated financial statements.
TRAVERE THERAPEUTICS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 1. DESCRIPTION OF BUSINESS
Organization and Description of Business
Travere Therapeutics, Inc. (“we”, “our”, “us”, “Travere” and the “Company”) refers to Travere Therapeutics, Inc., a Delaware corporation, as well as our direct and indirectits subsidiaries. Travere is a fully integrated biopharmaceutical company headquartered in San Diego, California focused on identifying, developing and delivering life-changing therapies to people living with rare kidney and metabolic diseases. WeThe Company regularly evaluateevaluates and, where appropriate, actacts on opportunities to expand ourits product pipeline through licenses and acquisitions of products in areas that will serve patients with serious or rare diseasesunmet medical need and that we believethe Company believes offer attractive growth characteristics.
The ongoing novel coronavirus (COVID-19) pandemic has resulted in travel restrictions, quarantines, “stay-at-home”Discontinued Operations - Sale of Bile Acid Product Portfolio
On July 16, 2023, Travere entered into an Asset Purchase Agreement (the “Purchase Agreement”) with Mirum Pharmaceuticals, Inc. ("Mirum Pharmaceuticals" or “Mirum”), pursuant to which Mirum agreed to purchase from Travere substantially all of the assets primarily related to Travere’s business of development, manufacture (including synthesis, formulation, finishing or packaging) and “shelter-in-place” orderscommercialization of Chenodal and extended shutdownCholbam (also known as Kolbam, and together with Chenodal, the “Products”), collectively, the "bile acid business". On August 31, 2023, the Company and Mirum consummated the transactions contemplated by the Purchase Agreement (the “Closing”). In connection with the Closing, Mirum paid Travere an upfront cash payment of $210.0 million. Pursuant to the Purchase Agreement, Travere is eligible to receive up to $235.0 million upon the achievement of certain businesses around the world. While the impactmilestones based on specified amounts of annual net sales (tiered from $125.0 million to $500.0 million) of the COVID-19 pandemic did not haveProducts. The Company has reflected the bile acid business as a material adverse effectdiscontinued operation in the Consolidated Financial Statements for all periods presented. See Note 19 for further discussion.
Unless otherwise noted, amounts and disclosures throughout the Notes to the Consolidated Financial Statements relate to the Company's continuing operations.
Approved Products:
FILSPARI® (sparsentan)
On February 17, 2023, the U.S. Food and Drug Administration (the "FDA") granted accelerated approval of FILSPARI® (sparsentan) to reduce proteinuria in adults with primary IgAN at risk of rapid disease progression, generally a UPCR ≥1.5 gram/gram. FILSPARI, a once-daily, oral medication is designed to selectively target two critical pathways (endothelin 1 and angiotensin-II) in the disease progression of IgAN.
This indication was granted under accelerated approval based on our financial position or resultsreduction in proteinuria. The continued approval of operationsFILSPARI may be contingent upon confirmation of a clinical benefit in the Company's Phase 3 clinical trial of sparsentan for the twelve months endedtreatment of IgAN (the "PROTECT Study"). In September 2023, the Company announced topline two-year confirmatory secondary endpoint results from the PROTECT Study and in December 31,2023, the Company announced the completion of a successful pre-NDA meeting with the FDA for FILSPARI in IgAN. Following its regulatory engagement, the Company plans to submit a supplemental New Drug Application (sNDA) for conversion of the existing U.S. accelerated approval of FILSPARI to full approval.
In August 2022, the Company and Vifor (International) Ltd. ("CSL Vifor"), with whom the Company entered into a license and collaboration agreement ("License Agreement") in September 2021, these governmental actions and similar actionsannounced that may be enactedthe European Medicines Agency (the "EMA") had accepted for review the MAA of sparsentan for the treatment of IgAN in the future, and the widespread economic disruption arising from the pandemic, have the potential to materially impact our business and influence our business decisions. The extent and duration of the pandemic is unknown, and the future effects on our business are uncertain and difficult to predict.EU. The Company is continuing to monitorawaiting an opinion by the eventsCommittee for Medicinal Products for Human Use (the "CHMP").
Thiola® and circumstances surrounding the COVID-19 pandemic, which may require adjustments to the Company’s estimatesThiola EC® (tiopronin tablets)
Thiola® and assumptionsThiola EC® (tiopronin tablets) are approved in the future.United States for the prevention of cystine (kidney) stone formation in patients with severe homozygous cystinuria.
Clinical-Stage Programs:
Sparsentan isfor the treatment of FSGS
Sparsentan remains a novel investigational product candidate andwhich has been granted Orphan Drug Designation for the treatment of focal segmental glomeruloscleroisglomerulosclerosis (FSGS) and immunoglobulin A nephropathy (IgAN) in the U.S. and Europe. Sparsentan is currently being evaluated in two pivotalthe EEA. In December 2023, the Company announced that it had completed its planned Type C meeting with the FDA to discuss previously reported results from the Phase 3 clinical studiesDUPLEX Study of sparsentan in rare kidney diseases.FSGS. The FDA acknowledged the high unmet need for approved therapies as well as the challenges in studying FSGS but indicated that the two-year results from the Phase 3 DUPLEX Study alone were not sufficient to support an sNDA submission. Together with CSL Vifor, the Company also plans to engage with the EMA to determine the potential for a subsequent variation to the Conditional Marketing Authorization (CMA) of sparsentan for the treatment of FSGS, subject to a review decision on the pending application for CMA of sparsentan in IgAN. The Company is conducting additional analyses of FSGS data and intends to engage with regulators to evaluate potential regulatory pathways for a sparsentan FSGS indication.
Pegtibatinase
Pegtibatinase (PegtibatinasTVT-058)e is a novel investigational human enzyme replacement candidate being evaluated for the treatment of classical homocystinuria (HCU). Pegtibatinase has been granted Rare Pediatric Disease, and Fast Track and Breakthrough Therapy designations by the FDA, as well as orphan drug designation in the United States and European Union. Pegtibatinase is currently being evaluatedIn May 2023, the Company announced positive topline results from cohort 6 in the Phase 1/2 COMPOSE Study. In December 2023, the Company initiated the pivotal Phase 3 HARMONY Study to assess its safety, tolerability, pharmacokinetics, pharmacodynamics and clinical effects in patients withsupport the potential approval of pegtibatinase for the treatment of classical HCU. The HARMONY Study is a global, randomized, multi-center, double-blind, placebo-controlled Phase 3 clinical trial designed to evaluate the efficacy and safety of pegtibatinase as a novel treatment to reduce total homocysteine (tHcy) levels. Topline results from the HARMONY Study are expected in 2026. The Company acquired pegtibatinase as part of the November 2020 acquisition of Orphan Technologies Limited.
Chenodal (chenodeoxycholic acid or CDCA) is a naturally occurring bile acid that is approved for the treatment of people with radiolucent stones in the gallbladder. In January 2020, we randomized the first patients in our Phase 3 RESTORE Study to evaluate the effects of Chenodal in adult and pediatric patients with cerebrotendinous xanthomatosis (CTX), and the study enrollment remains open. The pivotal study is intended to support an NDA submission for marketing authorization of Chenodal for CTX in the United States.
Preclinical Programs:
We areThe Company is a participant in twoa Cooperative Research and Development AgreementsAgreement ("CRADAs"CRADA"), which formforms a multi-stakeholder approach to pool resources with leading experts and incorporate the patient perspective early in the therapeutic identification and development process. We have partneredThe Company is partnering with the National Institutes of Health’s National Center for Advancing Translational Sciences ("NCATS") and a leading patient advocacy organizations, CDG Care andorganization, Alagille Syndrome Alliance, aimed at the identification of potential small molecule therapeutics for NGLY1 deficiency and Alagille syndrome ("ALGS"), respectively.. There are no treatment options currently approved for these diseases.ALGS.
Approved products:
•Chenodal (chenodiol tablets)The Company is approved inparty to a collaboration agreement with PharmaKrysto Limited and their early-stage cystinuria discovery program, whereby the United StatesCompany is responsible for funding all research and development expenses for the treatment of patients suffering from gallstones in whom surgery poses an unacceptable health risk due to disease or advanced age.
•Cholbam® (cholic acid capsules) is approved inpre-clinical activities associated with the United States for the treatment of bile acid synthesis disorders due to single enzyme defects and is further indicated for adjunctive treatment of patients with peroxisomal disorders.
•Thiola® and Thiola EC® (tiopronin tablets) are approved in the United States for the prevention of cystine (kidney) stone formation in patients with severe homozygous cystinuria.
NOTE 2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
A summary of the significant accounting policies applied in the preparation of the accompanying consolidated financial statements follows:
Principles of Consolidation
The consolidated financial statements represent the consolidation of the accounts of the Company, and its subsidiaries and variable interest entities for which the Company has been determined to be the primary beneficiary, in conformity with accounting principles generally accepted in the United States ("U.S. GAAP"). All intercompany accounts and transactions have been eliminated in consolidation. See Note 5 for further discussion of variable interest entities (“VIE”) that the Company consolidates.
Use of Estimates
In preparing financial statements in conformity with U.S. GAAP, management is required to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements, as well as the reported amounts of expenses during the reporting period. Due to inherent uncertainty involved in making estimates, actual results reported in future periods may be affected by changes in these estimates. On an ongoing basis, the Company evaluates its estimates and assumptions. These estimates and assumptions include revenue recognition, forecasting probability-weighted cash flows based upon estimates of forecasted revenues, clinical and regulatory timelines and discount rates, valuing equity securities in share-based payments, estimating expenses of contracted research organizations, estimating reserves for inventory, estimating the useful lives of depreciable and amortizable assets, goodwill impairment, estimating the fair value of contingent consideration, estimating of valuation allowances and uncertain tax positions, and estimates associated with the assessment of impairment for long-lived assets.
Revenue Recognition
The Company recognizes revenue when its customer obtains control of promised goods or services, in an amount that reflects the consideration which the entity expects to receive in exchange for those goods or services. To determine revenue recognition for arrangements that an entity determines are within the scope of Accounting Standards Codification ("ASC") 606, Revenue from Contracts with Customers ("ASC 606"), the entity performs the following five steps: (i) identify the contract(s) with a customer; (ii) identify the performance obligations in the contract; (iii) determine the transaction price; (iv) allocate the transaction price to the performance obligations in the contract; and (v) recognize revenue when (or as) the entity satisfies a performance obligation. The Company only applies the five-step model torecognizes revenue from contracts when it is probable that the entity will collect substantially all the consideration it is entitled to in exchange for the goods or services it transfers to the customer. See Note 3 and Note 4 for further discussion.
Payments received under collaboration and licensing agreements may include non-refundable fees at the inception of the arrangements, milestone payments for specific achievements and royalties on the sale of products. At the inception of arrangements that include milestone payments, the Company uses judgement to evaluate whether the milestones are probable of being achieved and estimates the amount to include in the transaction price utilizing the most likely amount method. If it is probable that a significant revenue reversal will not occur, the estimated amount is included in the transaction price. Milestone payments that are not within the Company or the licensee’s control, such as regulatory approvals, are considered to be constrained due to a high degree of uncertainty and are not included in the transaction price until those approvals are received.such uncertainty is resolved. At the end of each reporting period, the Company re-evaluates the probability of achievement of development milestones and any related constraint and adjusts the estimate of the overall transaction price, if necessary. The Company recognizes aggregate sales-based milestones and royalty payments from product sales of which the license is deemed to be the predominant item to which the royalties relate, at the later of when the related sales occur or when the performance obligation to which the sales-based milestone or royalty has been allocated has been satisfied. If itRevenue from collaboration and licensing agreements may also include sales of inventory, at cost plus a margin, which is probable that a significant revenue reversal will not occur, the Company estimates the sales-based milestonerecorded in license and royalty payments using the most likely amount method.collaboration revenue.
The Company utilizes significant judgement to develop estimates of the stand-alone selling price for each distinct performance obligation based upon the relative stand-alone selling price. Variable consideration that relates specifically to the Company’s efforts to satisfy specific performance obligations is allocated entirely to those performance obligations. The stand-alone selling price for license-related performance obligations requires judgement in developing assumptions to project probability-weighted cash flows based upon estimates of forecasted revenues, clinical and regulatory timelines and discount rates. The stand-alone selling price for clinical development performance obligations is based on forecasted expected costs of satisfying a performance obligation plus an appropriate margin.
If the licenses to intellectual property are determined to be distinct from the other performance obligations identified in the arrangement and have stand-alone functionality, the Company recognizes revenues from non-refundable, upfront fees allocated to the license when the license is transferred to the licensee and the licensee is able to benefit from the license. For licenses that are not distinct from other promises, the Company applies judgment to assess the nature of the combined performance obligation to determine whether the combined performance obligation is satisfied over time or a point in time and, if over time, the appropriate method of measuring progress for purposes of recognizing revenue from non-refundable, upfront fees. The Company evaluates the measure of progress each reporting period and, if necessary, adjusts the related revenue recognition accordingly.
The selection of the method to measure progress towards completion requires judgment and is based on the nature of the products or services to be provided. Revenue is recorded proportionally as costs are incurred. The Company generally utilizes the cost-to-cost method of progress because it
best measures the transfer of control to the customer which occurs as the Company incurs costs. Under the cost-to-cost measure of progress, the extent of progress towards completion is measured based on the ratio of costs incurred to date to the total estimated costs at completion of the performance obligation. The Company uses judgment to estimate the total costs expected to complete the clinical development performance obligations, which include subcontractor costs, labor, materials, other direct costs and an allocation of indirect costs. The Company evaluates these cost estimates and the progress each reporting period and adjusts the measure of progress, if necessary.
Inventory, Related Reserves and Cost of Goods Sold
Inventory, which is recorded at the lower of cost or net realizable value, includes materials and other direct and indirect costs and is valued using the first-in, first-out method. The Company periodically analyzes its inventory levels to identify inventory that may expire prior to expected sale or has a cost basis in excess of its estimated realizable value, and writes down such inventory as appropriate. In addition, the Company's products are subject to strict quality control and monitoring which the Company’s manufacturers perform throughout their manufacturing process. The Company does not directly manufacture any product. The Company has a single supplier for its product Thiola, and utilizes contract service providers for the manufacture of the active pharmaceutical ingredient for FILSPARI and the manufacture of primary packaging, secondary packaging and serialization for its product FILSPARI. The inventory reserve was $2.4 million and zero at December 31, 2023 and 2022, respectively.
Inventory, net of reserves, consisted of the following at December 31, 2023 and 2022 (in thousands):
| | | | | | | | | | | |
| December 31, 2023 | | December 31, 2022 |
| Raw materials | $ | 33,790 | | | $ | 2,397 | |
| Work in process | 4,727 | | | — | |
| Finished goods | 2,387 | | | 2,126 | |
| Total inventory | $ | 40,904 | | | $ | 4,523 | |
| Classified as: | | | |
| Inventory, net | $ | 9,410 | | | $ | 4,523 | |
| Long-term inventory, net | 31,494 | | | — | |
| Total inventory | $ | 40,904 | | | $ | 4,523 | |
The balance classified as long-term inventory consists of raw materials and work in process for FILSPARI as of December 31, 2023. The Company maintains levels of these inventories beyond a one-year production plan to limit exposure to potential supply disruption. Such inventories are classified as long-term.
Cost of goods sold includes the cost of inventory sold, third party manufacturing and supply chain costs, product shipping, tracking and handling costs, and provisions for excess and obsolete inventory. Cost of goods sold also includes the cost of goods sold under the Company's license and collaboration agreements, which currently consists of the sale of active pharmaceutical ingredients to the Company's collaboration partner, at cost plus a margin.
The following table summarizes cost of goods sold for the years ended December 31, 2023, 2022 and 2021 (in thousands):
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2023 | | 2022 | | 2021 |
| Cost of goods sold - product sales | $ | 8,406 | | | $ | 4,420 | | | $ | 3,818 | |
| Cost of goods sold - license and collaboration | 3,044 | | | — | | | — | |
| Total cost of goods sold | $ | 11,450 | | | $ | 4,420 | | | $ | 3,818 | |
Capitalization of Inventory Costs
Prior to the regulatory approval of the Company's therapeutic candidates, the Company incurs expenses for the manufacture of drug product supplies to support clinical development that could potentially be available to support the commercial launch of those therapies. The Company capitalizes inventory costs associated with its products after regulatory approval, when, based on management’s judgment, future commercialization is considered probable and the future economic benefit is expected to be realized. Until the date at which regulatory approval has been received, costs related to the production of inventory are recorded as research and development expenses as incurred. Any eventual sale of previously expensed ("zero-cost") inventories may impact future margins, for any periods in which those inventories are sold.
Prior to the February 2023 FDA accelerated approval of FILSPARI (sparsentan), the Company recognized approximately $7.5 million in research and development expenses related to the production of active pharmaceutical ingredients to support the commercial launch of FILSPARI. For the year ended December 31, 2023, sales of FILSPARI primarily consisted of zero-cost inventories. As of December 31, 2023, the Company had $7.2 million of zero-cost inventory remaining. The Company expects to continue to benefit from the sale of previously expensed inventories through at least 2025. The Company began capitalizing inventory costs associated with FILSPARI (sparsentan) following the February 2023 approval for treatment in IgAN.
At December 31, 2023, the Company's evaluation of excess inventory and obsolescence considered certain minimum purchase obligations, which in combination with lower forecasted sales of FILSPARI resulted in a $3.2 million charge to cost of goods sold. The charge to cost of goods of sold included a $2.1 million write-down of inventory balances and $1.1 million accrued for firm purchase commitments.
Research and Development CostsExpenses
Research and development includes expenses related to sparsentan, pegtibatinase, and the Company's other pipeline programs. The Company expenses all research and development costs as they are incurred. The Company's research and development costs are comprisedcomposed of salaries and bonuses, benefits, share-based compensation, license fees, milestones under license agreements, costs paid to third-party contractors to perform research, conduct clinical trials, andcosts to develop drug materials and delivery devices, costs to manufacture drug product supplies to support clinical development, and associated overhead expenses and facilities costs. The Company charges direct internal and external program costs to the
respective development programs. The Company also incurs indirect costs that are not allocated to specific programs because such costs benefit multiple development programs and allow us to increase our pharmaceutical development capabilities. These consist of internal shared resources related to the development and maintenance of systems and processes applicable to all of our programs.
Nonrefundable advance payments for goods and services to be received in the future for use in research and development activities are recorded as prepaid expenses. The prepaid amounts are expensed as the related goods are delivered or the services are performed, or when it is no longer expected that the goods will be delivered or the services rendered.
During the three months ended December 31, 2022, the Company recorded an out of period adjustment that decreased research and development expenses and related accrued expenses by $7.7 million, $1.5 million of which was previously recorded in fiscal year 2022 and $6.2 million that was originally recorded in fiscal year 2021. The adjustment was the result of a certain pre-launch inventory contract that was not properly evaluated for accounting implications at inception. The Company evaluated the impact of the adjustment and concluded it is not material, individually and in the aggregate, to 2022 or any prior period financial statements.
Clinical Trial Expenses
The Company records expenses in connection with its clinical trials under contracts with contract research organizations (CROs)("CROs") that support conducting and managing clinical trials.trials, as well as contract manufacturing organizations ("CMOs") for the manufacture of drug product supplies to support clinical development. The financial terms and activities of these agreements vary from contract to contract and may result in uneven expense levels. Generally, these agreements set forth activities that drive the recording of expenses such as start-up, and initiation activities, enrollment, and treatment of patients, or the completion of other clinical trial activities.activities, and in the case of CMOs, costs associated with the production of drug product supplied and the procurement of raw materials to be consumed in the manufacturing process.
Expenses related to clinical trials are accrued based on our estimates and/or representations from service providers regarding workof the progress of services performed, including actual level of patient enrollment, completion of patient studies and progress of the clinical trials.trials or the delivery of goods. Other incidental costs related to patient enrollment or treatment are accrued when reasonably certain. If the amounts we are obligated to pay under our clinical trial agreements are modified (for instance, as a result of changes in the clinical trial protocol or scope of work to be performed), the Company adjusts its accrualsestimates accordingly on a prospective basis. Revisions to the Company's contractual payment obligations are charged to expense in the period in which the facts that give rise to the revision become reasonably certain.
The Company currently has 3one Phase 1/2 clinical trial and three Phase 3 clinical trials in process that are in varying stages of activity, with ongoing non-clinical support trials. As such, clinical trial expenses will vary depending on the all the factors set forth above and may fluctuate significantly from quarter to quarter.
Share-Based Compensation
The Company recognizes all employee share-based compensation as a cost in the financial statements.within research and development expenses and selling, general, and administrative expenses. Equity-classified awards principally related to stock options, restricted stock units (“RSUs”) and performance stock units ("PSUs"), are measured at the grant date fair value of the award. The Company determines grant date fair value of stock option awards using the Black-Scholes option-pricing model. The fair value of RSUs and PSUs are determined using the closing price of the Company’s common stock on the grant date. For service based vesting grants, expense is recognized over the requisite service period based on the number of options or shares expected to ultimately vest.period. For PSUs, expense is recognized over the implicit service period, assuming vesting is probable. No expense is recognized for PSUs if it is not probable the vesting criteria will be satisfied. Forfeitures are accounted for as they occur.
| | | | | | | | | | | |
| Expiration Term | | Vesting Term |
| Stock Options | 10 years | | 3 to 4 years |
| Restricted Stock Units | ---- | | 1 to 4 years |
Earnings (Loss) Per Share
The Company calculates basic earnings per share by dividing net income/(loss) by the weighted average number of shares outstanding during the period. Pre-funded warrants issued and sold by the Company to purchase shares of its common stock are included in the calculation of basic net loss per common share if the exercise price of the pre-funded warrant represents little consideration and is non-substantive in relation to the price paid for the warrant, and if the warrants are immediately exercisable with no further vesting conditions or contingencies associated with them.
The Company's diluted earnings/(loss) per share computation includes the effect, if any, of shares that would be issuable upon the exercise of outstanding stock options, derivative warrant liability, convertible debt and RSUs, reduced by the number of shares which are assumed to be purchased by the Company from the resulting proceeds at the average market price during the year, when such amounts are dilutive to the earnings per share calculation.
In accordance with ASC 260,
Cash and Cash Equivalents
The Company considers all highly liquid marketable securities with an original maturity of three months or less to be cash equivalents. Due to the short-term maturity of such investments, the carrying amounts are a reasonable estimate of fair value.
Concentration of Credit Risk
The Company maintains its cash and cash equivalents at insured financial institutions, the balances of which may, at times, exceed federally insured limits. Generally, these deposits may be redeemed upon demand, and the Company believes there is minimal risk of losses on such balances.
The Company monitors its investments with counterparties with the objective of minimizing concentrations of credit risk. The Company's investment policy is to invest only in institutions that meet high credit quality standards and established limits on the amount and time to maturity of investments with any individual counterparty. The policy also requires that investments are only entered into with corporate and financial institutions that meet high quality standards.
Marketable Debt Securities
The Company accounts forclassified marketable debt securities held as “available-for-sale” in accordance with ASC 320, “Investments - Debt Securities” (“ASC 320”).and carries them at fair value. The Company classifies these investments as current assets, even if the maturity when acquired by the Company is greater than one year due to the ability to liquidate within the next 12 months. The amortized cost of marketable debt securities is adjusted for amortization of premiums and carries them at fair value.accretion of discounts to maturity. Such amortization and accretion, as well as interest and dividends, is included in interest income. Unrealized gains and losses on marketable debt securities are recorded as a separate component of stockholders’ equity as accumulated other comprehensive loss, unless an impairment is determined to be the result of credit-related factors or the Company intends to sell the security or it is more likely than not that the Company will be required to sell the security before recovery. Unrealized losses that are determined to be credit-related are recorded as an allowance against the amortized cost basis. Realized gains or losses on debt security transactions and declines in value that are determined to be the result of credit losses, if any, are reported in other income or expense in the Consolidated Statements of Operations and Comprehensive Loss. The cost of securities sold is based on the specific identification method. Marketable debt securities are maintained at one financial institution and are governed by the Company’s investment policy. See Note 6 for further discussion.
TradeAccounts Receivables, Net
Trade accounts receivable are recorded net of reserves for prompt pay discounts and expected credit losses. Estimates for allowances for credit losses are determined based on existing contractual obligations, historical payment patterns and individual customer circumstances. The allowance for credit losses was zero at both December 31, 20212023 and 2020,2022, respectively. For the years ended December 31, 2021, 20202023, 2022 and 2019,2021, bad debt expense recorded in the Consolidated Statements of Operations and Comprehensive Loss was immaterial. The Company's evaluation and application of ASU No. 2016-13, Financial Instruments - Credit Lossescredit losses for the current period included an assessment of our aged trade receivables balances and their underlying credit risk characteristics. Our evaluation of past events, current conditions, and reasonable and supportable forecasts about the future resulted in an expectation of immaterial credit losses.
Inventory, Related Reserves and Cost of Goods SoldSupplier Concentration Risk
Inventory, which is recorded at the lower of cost or net realizable value, includes materials, labor, and other direct and indirect costs and is valued using the first-in, first-out method. The Company periodically analyzes its inventory levels to identify inventory that may expire prior to expected sale or has a cost basis in excess of its estimated realizable value, and writes down such inventory as appropriate. In addition, the Company's products are subject to strict quality control and monitoring which the Company’s manufacturers perform throughout their manufacturing process. The Company does not directly manufacture any product. The Company has singleno manufacturing capabilities and relies on third party manufacturers who are sole source suppliers for products Chenodalmanufacturing of its products. The Company intends to rely on third-party manufacturers for the long-term commercial supply of FILSPARI and Thiola, and prospectively arranges for manufacture from contract service providers for its development stage product Cholbam.candidates, including sparsentan for the treatment of FSGS and pegtibatinase. The inventory reserve was $4.1 millionCompany expects the manufacturers of each product or product candidate to, at least initially and $3.6 million at December 31, 2021 and 2020, respectively.
Inventory, netpotentially for a significant period of reserve, consisted oftime, be single source suppliers to the following at December 31, 2021 and 2020 (in thousands):
| | | | | | | | | | | |
| December 31, 2021 | | December 31, 2020 |
| Raw material | $ | 5,205 | | | $ | 3,219 | |
| | | |
| Finished goods | 2,108 | | | 4,389 | |
| Total inventory | $ | 7,313 | | | $ | 7,608 | |
Cost of goods sold includes the cost of inventory sold, third party manufacturing and supply chain costs, product shipping and handling costs, and provisions for excess and obsolete inventory.Company.
Segment Information
The Company currently operates in 1one operating segment focused on the development and commercialization of innovative therapies for people with serious and life-threatening rare diseases and medical conditions. The Company is not organized by market and is managed and operated as one business. A single management team reports to the chief operating decision maker who comprehensively manages the entire business. The
Company does not operate any separate lines of business or separate business entities with respect to its products. Accordingly, the Company does not accumulate discrete financial information with respect to separate products, other than revenues, and does not have separately reportable segments.
Property and Equipment, net
Property and equipment are stated at cost, net of accumulated depreciation. Depreciation is computed using the straight-line method over the related estimated useful lives as presented in the table below. Significant additions and improvements are capitalized, while repairs and maintenance are
charged to expense as incurred. Property and equipment purchased for specific research and development projects with no alternative use is expensed as incurred.
The major classifications of property and equipment, including their respective expected useful lives, consist of the following:
| | | | | |
| Computers and equipment | 3 years |
| Furniture and fixtures | 7 years |
| Leasehold improvements | Shorter of length of lease or life of the asset |
Leases
The Company determines whether a contract is, or contains, a lease at inception. The Company classifies each of its leases as operating or financing considering factors such as the length of the lease term, the present value of the lease payments, the nature of the asset being leased, and the potential for ownership of the asset to transfer during the lease term. Leases with terms greater than one year are recognized on the Consolidated Balance Sheets as Right-of-use assets and Lease liabilities and are measured at the present value of the fixed payments due over the expected lease term minus the present value of any incentives, rebates or abatementsabatement expected to be received from the lessor. Options to extend a lease are typically excluded from the expected lease term as the exercise of the option is typically not reasonably certain. The interest rate implicit in lease contracts is typically not readily determinable. As such, the Company utilizes the appropriate incremental borrowing rate, which is the rate incurred to borrow on a collateralized basis an amount equal to the lease payments over a similar term and in a similar economic environment.
In addition to rent, the leases may require the Company to pay additional amounts for taxes, insurance, maintenance, and other expenses, which are generally referred to as non-lease components. The Company has elected to not separate lease and non-lease components. Only the fixed costs for lease components and their associated non-lease components are accounted for as a single lease component and recognized as part of the Right-of-use assets and Lease liabilities. The Company records expense to recognize fixed lease payments, including payment escalation, on a straight-line basis over the expected lease term. Costs determined to be variable and not based on an index or rate are not included in the measurement of the lease liability and are expensed as incurred.
The Company has made an accounting policy election to not recognize short-term leases, or leases that have a lease term of 12 months or less at commencement date, within its Consolidated Balance Sheets and to recognize those lease payments in the Consolidated Statements of Operations and Comprehensive Loss on a straight-line basis over the lease term.
Intangible Assets, Net
The Company's intangible assets consist of licenses and purchased technology. Intangible assets with definite lives are amortized on a straight-line basis over their estimated useful lives and are reviewed periodically for impairment.
Intangible assets related to in-process research and development (IPR&D) projects are considered to be indefinite-lived until the completion or abandonment of the associated research and development efforts. During the period the assets are considered indefinite-lived, they will not be amortized but will be tested for impairment. If and when development is complete, which generally occurs when regulatory approval to market a product is obtained, the associated assets are deemed finite-lived and are amortized over a period that best reflects the economic benefits provided by these assets.
Intangible Assets with Cost Accumulation Model
In 2014, the Company entered into a license agreement with Mission Pharmacal in which the Company obtained the exclusive right to license the trademark of Thiola. The acquisition of the Thiola license qualified as an asset acquisition under the principles of ASC 805, Business Combinations ("ASC 805") in effect at the time of acquisition. The license agreement requires the Company to make royalty payments based on net sales of Thiola. The liability for royalties in excess of the annual contractual minimum is recognized in the period in which the royalties become probable and estimable, which is typically in the period corresponding with the respective sales. The Company records an offsetting increase to the cost basis of the asset under the cost accumulation model.model ("Thiola Intangible"). The additional cost basis is subsequently amortized over the remaining useful life.
In the second quarter of 2023, the Company reduced the estimated useful life of the license agreement.Thiola Intangible to better reflect the pattern of projected future cash flows, resulting in incremental expense of $3.7 million recorded in selling, general, and administrative. The change in estimated useful life has been accounted for as a change in accounting estimate and the remaining carrying amounts of the Thiola Intangible will be amortized prospectively over the new useful life.
Consistent with all prior periods since Thiola was acquired, the Company has not accrued any liability for future royalties in excess of the annual contractual minimum at December 31, 20212023 as such royalties are not yet probable and estimable.
Variable Interest Entity
The Company reviews each investment and collaboration agreement to determine if it has a variable interest in the entity. In assessing whether the Company has a variable interest in the entity as a whole, the Company considers and makes judgements regarding the purpose and design of the entity, the value of the licensed assets to the entity, the value of the entity’s total assets and the significant activities of the entity. If the Company has a variable interest in the entity as a whole, the Company assesses whether or not the Company is a primary beneficiary of that VIE, based on a number of factors, including: (i) which party has the power to direct the activities that most significantly affect the VIE’s economic performance, (ii) the
parties’ contractual rights and responsibilities pursuant to the collaboration agreement, and (iii) which party has the obligation to absorb losses of or the right to receive benefits from the VIE that could be significant to the VIE. If the Company determines that it is the primary beneficiary of a VIE at the onset of the collaboration, the collaboration is treated as a business combination and the Company consolidates the financial statements of the VIE into the Company’s consolidated financial statements. On a quarterly basis, the Company evaluates whether it continues to be the primary beneficiary of the consolidated VIE. If the Company determines that it is no longer the primary beneficiary of a consolidated VIE, it deconsolidates the VIE in the period in which the determination is made.
Assets and liabilities recorded as a result of consolidating the financial results of the VIE into the Company’s consolidated balance sheet do not represent additional assets that could be used to satisfy claims against the Company’s general assets or liabilities for which creditors have recourse to the Company’s general assets.
Goodwill
Goodwill represents the excess of purchase price over fair value of net assets acquired in a business combination and is not amortized. Goodwill is subject to impairment testing at least annually or when a triggering event occurs that could indicate a potential impairment. The Company has 1one segment and 1one reporting unit and as such reviews goodwill for impairment at the consolidated level.
For the years ended December 31, 2021, 20202023, 2022 and 20192021 there were no impairments to goodwill.
Impairment of Long-Lived Assets
The Company's long-lived assets are primarily comprised of intangible assets, right-of-use assets, and property and equipment. The Company evaluates its finite-lived intangible assets, right-of-use assets, and property and equipment for impairment whenever events or changes in circumstances indicate the carrying value of an asset or group of assets may not be recoverable. If these circumstances exist, recoverability of assets to be held and used is measured by a comparison of the carrying amount of an asset group to future undiscounted net cash flows expected to be generated by the use and eventual disposition of the asset group. If such assets are considered to be impaired, the impairment to be recognized is measured by the amount by which the carrying amount of the assets exceeds the fair value of the assets.
In addition, indefinite-lived intangible assets comprised of IPR&D, are reviewed for impairment annually and whenever events or changes in circumstances indicate that it is more likely than not that the asset is impaired by comparing the fair value to the carrying value of the asset. To determine the fair value of the asset, the Company used the multi-period excess earnings method of the income approach. The more significant assumptions inherent in the application of this method include: the amount and timing of projected future cash flows (including revenue, cost of sales, research and development costs, and sales and marketing expenses), and the discount rate selected to measure the risks inherent in the future cash flows.
There were no impairments related to finite-lived intangible assets in the years ended December 31, 2021, 20202023, 2022 and 2019.
Contingent Consideration
The Company records contingent consideration resulting from a business combination at its fair value on the acquisition date. On a quarterly basis, the Company revalues these obligations and record increases or decreases from their fair value as an adjustment to the consolidated statement of operations. Changes to contingent consideration obligations can result from changes to discount rates, accretion of the liability due to the passage of time, changes in revenue forecasts and changes in our estimates of the likelihood or timing of achieving commercial revenue milestones.2021.
Income Taxes
The Company follows ASC 740, Income Taxes ("ASC 740"), which requires recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have been included in the financial statements or tax returns. Under this method, deferred tax assets and liabilities are based on the differences between the financial statement and tax basis of assets and liabilities using enacted tax rates in effect for the year in which the differences are expected to reverse. Deferred tax assets are reduced by a valuation allowance to the extent management concludes it is more likely than not that the asset will not be realized.
The standard addresses the determination of whether tax benefits claimed or expected to be claimed on a tax return should be recorded in the financial statements. Under ASC 740, the Company may recognize the tax benefit from an uncertain tax position only if it is more likely than not that the tax position will be sustained on examination by the tax authorities, based on the technical merits of the position. The tax benefits recognized in the financial statements from such a position should be measured based on the largest benefit that has a greater than fifty percent likelihood of being realized upon ultimate settlement. ASC 740 also provides guidance on de-recognition, classification, interest and penalties on income taxes, accounting in interim periods and requires increased disclosures. The Company’s policy is to record estimated interest and penalty related to the underpayment of income taxes or unrecognized tax benefits as a component of its income tax provision.
Foreign Currency Translation
Functional and presentation currency
Items included in the financial statements of each entity comprising the Company are measured using the currency of the primary economic environment in which the entity operates (the functional currency).
Transactions and balances
Foreign currency transactions in each entity comprising the Company are remeasured into the functional currency of the entity using the exchange rates prevailing at the respective transaction dates. Foreign exchange gains and losses resulting from the settlement of such transactions and from the remeasurement at year-end exchange rates of monetary assets and liabilities denominated in foreign currencies are recognized within Other operating expense, net except for changes related to the liability related to the sale of future royalties which are recorded in Other non-operating (income) expense,income (expense), net in the Consolidated Statements of Operations and Comprehensive Loss.
The aggregate gain arising from foreign exchange transactions included in other income (expense), net for the years ended December 31, 2023, 2022 and 2021 was $0.5 million, $1.4 million, and $0.3 million, respectively.
The results and financial position of the Company that have a functional currency different from the US dollar are translated as follows:
a.assets and liabilities presented are translated at the closing exchange rate as of December 31, 20212023 and 2020;2022;
b.income and expenses for the statements of operations and comprehensive loss are translated at average exchange rates that are relevant for the respective periods for which the income and expenses occurred; and
c.significant transactions use the exchange rate on the date of the transaction;transaction.
All resulting exchange differences arising from such translations are recognized directly in comprehensive income and presented as a separate component of equity.
The following table summarizes the foreign currency translation adjustment included in accumulated other comprehensive loss for the year ended December 31, 2023, 2022 and 2021 (in thousands):
| | | | | | | | | | | | | | | | | |
| Foreign Currency Translation Adjustments included in Accumulated Other Comprehensive Loss |
| 2023 | | 2022 | | 2021 |
| Balance at January 1, | $ | 415 | | | $ | (92) | | | $ | (1,607) | |
| Foreign currency translation adjustments | (1,871) | | | 507 | | | 1,515 | |
| Balance at December 31, | $ | (1,456) | | | $ | 415 | | | $ | (92) | |
Reclassifications
Certain reclassifications have been made to the prior year financial statements in order to conform to the current year’s presentation, including reclassifying operating lease rightthe prior period carrying values of usethe assets and operating lease liabilities from otherincluded in the sale of the bile acid business as assets and other liabilities respectively onof discontinued operations within the Consolidated Balance Sheets.Sheets, and revenues and expenses attributable to the bile acid business as net income from discontinued operations. These reclassifications did not have an impact on total assets or total liabilities ofin the Consolidated Balance Sheets.Sheets or net loss in the Consolidated Statements of Operations.
Patents
The Company expenses external costs, such as filing fees and associated attorney fees, incurred to obtain issued patents and patent applications pending. The Company also expenses costs associated with maintaining and defending patents subsequent to their issuance in the period incurred.
Legal Contingencies
The Company may, from time to time, be involved in various claims and legal actions that arise in the ordinary course of business. The Company accrues for legal contingencies when it is determined probable that a liability has been incurred and the amount of the loss can be reasonably estimated. See Note 11 for further discussion.
Discontinued Operations
Discontinued operations is presented when there is a disposal of a component or a group of components that in the Company's judgement represents a strategic shift that will have a major effect on the Company's operations and financial results. Results of operations directly related to discontinued operations are aggregated into a single line item in the Consolidated Statements of Operations for all periods presented. See Note 19 for further discussion.
Restructuring
Restructuring charges consist primarily of employee severance, one-time termination benefits related to the reduction of its workforce, and other costs. Liabilities for costs associated with a restructuring activity are recognized when the liability is incurred and are measured at fair value. One-time termination benefits are expensed at the date the entity notifies the employee, unless the employee must provide future service, in which case the benefits are expensed ratably over the service period. Termination benefits are calculated based on a regional benefit practices and local statutory requirements.
In December 2023, the Company initiated a restructuring plan that resulted in a reduction of its workforce, primarily impacting non-field-based employees. One-time termination benefits include severance, continuation of health insurance coverage, and other benefits for a specified period of time. The Company estimates that it will incur a total of $12.0 million to $14.0 million in non-recurring charges in connection with the restructuring, of which the Company has recognized $11.4 million for the year ended December 31, 2023. As of December 31, 2023, the Company recorded accrued restructuring costs of $11.4 million, which is included in accrued expenses of the Consolidated Balance Sheets. No cash payments were made related to the restructuring during 2023. The Company expects that it will incur the remaining estimated restructuring costs during 2024. The Company intends to pay all restructuring plan amounts during 2024.
| | | | | | | | |
| | 2023 |
| Liability balance at January 1, | | $ | — | |
| Restructuring expenses | | 11,394 | |
| | |
| Foreign currency impact | | 27 | |
| Liability balance at December 31, | | $ | 11,421 | |
Recently Adopted Accounting Pronouncements
In August 2020, the FASB issued ASU No. 2020-06, Accounting for Convertible Instruments and Contracts in an Entity's Own Equity. The ASU includes amendments to the guidance on convertible instruments and the derivative scope exception for contracts in an entity's own equity in Subtopic 815-40 and simplifies the accounting for convertible instruments which include beneficial conversion features or cash conversion features by removing certain separation models in Subtopic 470-20. Additionally, the ASU requires entities to use the "if-converted" method when calculating diluted earnings per share for convertible instruments. The adoption of the new standard impacted the Company's accounting for its Convertible Senior Notes Due 2025 (2025 Notes), discussed in Note 7, which were previously accounted for using the cash conversion model applied under ASC 470-20, Debt with Conversion and Other Options ("ASC 470-20"). The Company adopted ASU 2020-06 on January 1, 2022 using the modified retrospective method. The cumulative effect of the accounting change as of January 1, 2022 increased the carrying amount of the 2025 Notes by $44.7 million, reduced additional paid-in capital by $74.9 million, and reduced accumulated deficit by $30.2 million.
Recently Issued Accounting Pronouncements
From time to time, new accounting pronouncements are issued by the Financial Accounting Standards Board ("FASB") or other standard setting bodies. Unless otherwise discussed, the Company believes that the impact of recently issued standards that are not yet effective will not have a material impact on its consolidated financial position or results of operations upon adoption.
In August 2020,December 2023, the FASB issued ASU No. 2020-06, Accounting for Convertible Instruments and Contracts in an Entity's Own Equity. 2023-09, Improvements to Income Tax Disclosures.The ASU includes amendments to the guidance on convertible instruments and the derivative scope exception for contracts in an entity's own equity and simplifies the This new standard does not change accounting for convertible instruments which include beneficial conversion features or cash conversion features by removing certain separation models in Subtopic 470-20. Additionally,income taxes but requires new disclosures focusing on two areas, the ASU will require entities to use the "if-converted" method when calculating diluted earnings per share for convertible instruments. The ASUeffective rate reconciliation and taxes paid. This new standard is effective for fiscal yearspublic business entities for annual periods beginning after December 15, 2021, including2024. Early adoption is permitted. The Company expects to adopt this new standard beginning in fiscal 2025 when it becomes effective.
In November 2023, the FASB issued ASU No. 2023-07, Improvements to Reportable Segment Disclosure. The FASB amended the guidance in ASC 280, Segment Reporting ("ASC 280"), to require a public entity to disclose significant segment expenses and other segment items on an annual and interim basis and to provide in interim periods within those fiscal years. The new standard will impact the Company's accounting for its Convertible Senior Notes Due 2025 (2025 Notes), discussed in Note 7, whichall disclosures about a reportable segment's profit or loss and assets that are currently accounted for usingrequired annually. Public entities with a single reportable segment are required to provide the cash conversion model appliednew disclosures and all the disclosures required under ASC 470-20, Debt with Conversion and Other Options ("ASC 470-20"). Under the280. The guidance is applied retrospectively to all periods presented in financial statements, unless it is impracticable. This new guidance the Company intends to apply the modified retrospective method as the transition approach atis effective for public business entities for annual periods beginning after December 15, 2023. Early adoption which will result in adjustments to the January 1, 2022 opening balances of convertible debt, additional paid-in capital, and accumulated deficit. Additionally, from January 1, 2022, theis permitted. The Company will no longer incur non-cash interest expense for the amortizationadopt this new guidance beginning in fiscal 2024 when it becomes effective.
NOTE 3. REVENUE RECOGNITION
Product Sales, Net
Product sales consist of Bile Acid products (ChenodalFILSPARI and Cholbam) and Tiopronintiopronin products (Thiola and Thiola EC). The Company sells its products to specialty pharmacies and through direct-to-patient distributors worldwide, with the United States and Canada representing 98% and 2%99% of net product sales, respectively, based on the product shipment destination.sales.
The Company sells FILSPARI to three direct-to-patient specialty pharmacies. The Company sells its other products to patients and pharmacies, with distribution facilitated through a single direct-to-patient distributor. Revenues from product sales are recognized in satisfaction of a single performance obligation when the customer obtains control of the Company’s product, which occursproduct. For FILSPARI, sales are recognized upon delivery of the product to the customer.specialty pharmacies. The Company receives payments from its FILSPARI sales based on terms that are generally 30 days from shipment of the product to the specialty pharmacy. For the Company's other products, product sales are recognized upon delivery to the patient. The Company receives payments from sales of its other products, primarily through third party payers, based on terms that generally are within 30 days of delivery of product to the patient. Contracts do not contain significant financing components based on the typical period of time between performance of services and collection of consideration.
Deductions from Revenue
Revenues from product sales are recorded at the net sales price, which includes provisions resulting from discounts, rebates and co-pay assistance that are offered to its customers, health care providers, payers and other indirect customers relating to the Company’s sales of its products. These
provisions are based on the estimates of the amounts earned or to be claimed on the related salessales. These amounts are treated as variable consideration, estimated and recognized as a reduction of the transaction price at the time of the sale, using the most likely amount method, and are classified as a reduction of accounts receivable (if the amount is payable to thea customer) or as a current liability (if the amount is payable to a party other than a customer). The Company includes these estimated amounts in the transaction price to the extent it is probable that a significant reversal of cumulative revenue recognized for such transactions will not occur. Where appropriate, these reserves take into consideration the Company’s historical experience, current contractual and statutory requirements and specific known market events and trends. Overall, these reserves reflect the Company’s best estimates of the amount of consideration to which it is entitled based on the terms of the contract. If actual results in the future vary from the Company’s provisions, the Company will adjust the provision,estimate, which would affect net product revenue and earnings in the period such variances become known. Our historical experience is that such adjustments have been immaterial.For the years ended December 31, 2023, 2022 and 2021, the Company recorded a net product revenue increase of $0.4 million, $0.2 million, and $0.2 million, respectively, related to performance obligations satisfied in previous periods.
Government Rebates:We calculate The Company calculates the rebates that weit will be obligated to provide to government programs and deductdeducts these estimated amounts from ourits gross product sales at the time the revenues are recognized. Allowances for government rebates and discounts are established based on actual payer information, which is reasonablyan estimated at the timeallocation of delivery,payers and the government-mandated discounts applicable to government-funded programs. Rebate discounts are included in accrued expenses in the accompanying Consolidated Balance Sheets.consolidated balance sheets.
Commercial Rebates: WeThe Company calculatecalculates the rebates that we incur dueit incurs according to any contracts with certain commercial payers and deductdeducts these amounts from ourits gross product sales at the time the revenues are recognized. Allowances for commercial rebates are established based on actual payer information, which is reasonably estimated at the time of delivery.delivery for applicable products. Rebate discounts are included in accrued expenses in the accompanying Consolidated Balance Sheets.consolidated balance sheets.
Prompt Pay Discounts: We offerThe Company offers discounts to certain customers for prompt payments. We accrueThe Company accrues for the calculated prompt pay discount based on the gross amount of each invoice for those customers at the time of sale.
Product Returns:Consistent with industry practice, we offer ourthe Company offers its customers a limited right to return product purchased directly from the Company, which is principally based upon the product’s expiration date. Generally, shipments are only made upon a patient prescription thusHistorically, returns are minimal.have been immaterial.
Co-pay Assistance: We offerThe Company offers a co-pay assistance program, which is intended to provide financial assistance to qualified commercially insured patients with prescription drug co-payments required by payers. The calculation of the accrual for co-pay assistance is based on an identificationestimate of claims and the estimated cost per claim associated with product that has been recognized as revenue.
The following table summarizes net product sales for the twelve monthsyear ended December 31, 2021, 20202023, 2022 and 20192021 (in thousands):
| | | | | | | | | | | | | | | | | |
| Twelve Months Ended December 31, |
| 2021 | | 2020 | | 2019 |
| Tiopronin products | $ | 115,122 | | | $ | 108,883 | | | $ | 95,638 | |
| Bile acid products | 95,654 | | | 89,438 | | | 79,700 | |
| Total net product revenue | $ | 210,776 | | | $ | 198,321 | | | $ | 175,338 | |
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2023 | | 2022 | | 2021 |
| Tiopronin products | $ | 98,329 | | | $ | 97,970 | | | $ | 115,122 | |
| FILSPARI | 29,208 | | | — | | | — | |
| Total net product sales | $ | 127,537 | | | $ | 97,970 | | | $ | 115,122 | |
NOTE 4. COLLABORATION AND LICENSE AGREEMENTS
OnIn September 15, 2021, the Company entered into a license and collaboration agreement (“License Agreement”) with Vifor (International) Ltd. (“Vifor Pharma”CSL Vifor”), pursuant to which the Company granted an exclusive license to CSL Vifor Pharma for the commercialization of sparsentan in Europe, Australia and New Zealand ("Licensed Territories"). CSL Vifor Pharma also has first right of negotiation to expand the licensed territories into Canada, China, Brazil and/ or Mexico. Under the terms of the License Agreement, the Company received an upfront payment of $55.0 million and will be eligible for up to $135.0 million in aggregate regulatory and market access related milestone payments and up to $655.0 million in aggregate sales-based milestone payments for a total potential value of up to $845.0 million. The Company is also entitled to receive tiered double-digit royalties of up to 40 percent of annual net sales of sparsentan in the Licensed Territories.
Under the License Agreement, CSL Vifor Pharma will be responsible for all commercialization activities in the Licensed Territories. The Company remains responsible for the worldwide clinical development of sparsentan through regulatory approval as defined and will retain all rights to sparsentan in the United States and rest of world outside of the Licensed Territories. Development costs for any post regulatory approval development activities, subject to approval by both parties, will be borne by the Company and CSL Vifor Pharma as defined, respectively. The License Agreement will remain in effect, unless terminated earlier, until the expiration of all royalty terms for sparsentan in the licensed territories. Each party has the right to terminate the License Agreement for the other party’s uncured material breach, insolvency or if the time required for performance under the License Agreement by the other party is extended due to a force majeure event that continues for six months or more.
The Company assessed the License Agreement and determined that it meets both criteria to be considered a collaborative agreement within the Scope of ASC 808, Collaborative Arrangements ("ASC 808") of active participation by both parties and exposures to significant risks and rewards dependent on the commercial success of the activities. Both parties participate on joint steering and other committees overseeing the collaboration
activities. Also, both parties are exposed to significant risks and rewards based on the economic outcomes of regulatory approvals and commercialization of sparsentan.
The Company determined the transaction price under the License Agreement totaled $55.0 million, consisting of the fixed non-refundable upfront payment. The variable regulatory and access related milestones were excluded from the transaction price given the substantial uncertainty related to their achievement. Sales-based milestone payments and royalties on net sales were excluded from the transaction price and will be recognized at the later of when the related sales occur or when the performance obligation to which the sales-based milestone or royalty has been allocated have been satisfied.
The Company concluded that CSL Vifor Pharma represented a customer and applied relevant guidance from ASC 606 to evaluate the accounting under the License Agreement. In accordance with this guidance, the Company concluded that the promise to grant the license is distinct from the promise to provide clinical development services resulting in 2two performance obligations as the license has stand-alone functionality due to sparsentan being a later stage asset and the clinical development services are primarily related to achieving regulatory approval.obligations. As a result, the Company allocated $12.0 million of the transaction price, based on the performance obligations' relative standalone selling prices, to the license.license, which was recognized in full in 2021. The remaining $43.0 million of the transaction price was allocated to the clinical development activities and recorded as deferred revenue, which will be recognized over the development period based upon the ratio of costs incurred to date to the total estimated costs.
The Company recognized a total of $16.7$17.7 million in license and collaboration revenue for the twelve monthsyear ended December 31, 2021,2023, which consisted of $12.0$3.3 million from the sale of active pharmaceutical ingredients to CSL Vifor, and $14.4 million for clinical development activities, based upon the ratio of costs incurred to total estimated costs. A decrease in total costs for clinical development activities due to the restructuring announced in December 2023 resulted in a $2.9 million increase in amortization of deferred revenue under the cost-to-cost model for the year ended December 31, 2023. For the year ended December 31, 2022, the Company recognized a total of $11.5 million in license and $4.7 million from thecollaboration revenue for clinical development activities, based upon the ratio of costs incurred to total estimated costs.
Deferred revenue related to the clinical development activities as of December 31, 20212023 was $36.6$8.9 million. Of this amount, $16.3$7.1 million was classified as current as of December 31, 2021,2023, based upon amount expected to be realized within the next year.
In February 2021, the Company entered into a limited co-promotion agreement with Albireo Pharma, Inc. ("Albireo"), whereby the Company's Cholbam dedicated sales representatives will dedicate a portion of their efforts to promoting Albireo's product, Bylvay (odevixibat), in the United States. The initial term of the arrangement is two years from the July 2021 launch of Bylvay, terminable at will by either party after one year following launch. For the year ended December 31, 2021, the Company recognized $1.8 million, offset against selling, general, and administrative expenses.
NOTE 5. ACQUISITIONS
Acquisition of Orphan Technologies Limited
In November 2020, the Company completed the acquisition of Orphan Technologies Limited (“Orphan”), including Orphan’s rare metabolic disorder drug pegtibatinase (TVT-058). The Company acquired Orphan by purchasing all of the outstanding shares. In exchange for the shares, the Company made an upfront cash payment at closing of $90.0 million plus closing adjustments, net liabilities assumed, and transaction expenses of $1.2 million, $1.8 million, and $4.2 million, respectively. Under the Agreement, the Company has also agreed to make contingent cash payments up to an aggregate of $427.0 million based on the achievement of certain development, regulatory and commercialization events as set forth in the Agreement, as well as additional tiered mid-single digit royalty payments based upon future net sales of any pegtibatinase products in the US and Europe, subject to certain reductions as set forth in the Agreement, and a contingent payment in the event a pediatric rare disease voucher for any pegtibatinase product is granted.
The Company applied the principles of ASC 805 in determining the proper accounting treatment for the acquisition. Substantially all of the value of the assets acquired was concentrated within pegtibatinase, and as of the acquisition date, the Company did not anticipate any economic benefit to be derived from pegtibatinase other than the primary indication. Accordingly, the transaction was treated as an asset acquisition with amounts charged to expense for the acquired in-process research and development on the date of acquisition. The Company incurred $97.1 million in expenses related to the Orphan acquisition, which was recognized in 2020.
In accordance with ASC 450, contingent cash payments will be accrued for when it is probable that a liability has been incurred and the amount can be reasonably estimated. As of December 31, 2022, deferred revenue related to the clinical development activities was $22.9 million, of which $12.0 million was classified as current. The Company estimates that the remainder of the deferred revenue balance will be fully realized by mid-2025.
The following table sets forth a summary of changes in deferred revenue for the years ended December 31, 2023, 2022 and 2021 no contingent cash payments have been accrued.(in thousands):
| | | | | | | | | | | | | | | | | |
| Deferred Revenue |
| 2023 | | 2022 | | 2021 |
| Balance at January 1, | $ | 22,907 | | | $ | 36,647 | | | $ | — | |
| New consideration resulting in deferred revenue | — | | | — | | | 55,000 | |
| License and collaboration revenue | (14,363) | | | (11,490) | | | (16,714) | |
| Foreign currency impact | 387 | | | (2,250) | | | (1,639) | |
| Balance at December 31, | $ | 8,931 | | | $ | 22,907 | | | $ | 36,647 | |
NOTE 5. VARIABLE INTEREST ENTITIES
Stock Purchase and Collaboration Agreement with PharmaKrysto
On March 8, 2022, the Company entered into a Collaboration Agreement with PharmaKrysto Limited (“PharmaKrysto”), a privately held pre-clinical stage company related to PharmaKrysto's early-stage cystinuria discovery program, and concurrently therewith entered into a Stock Purchase Agreement with PharmaKrysto (together, the "Agreements"). Pursuant to the terms of the Agreements, the Company paid PharmaKrysto's shareholders $0.6 million in cash to purchase 5% of the outstanding common shares of PharmaKrysto and $0.4 million to PharmaKrysto as a one-time signing fee. Under the Collaboration Agreement, the Company will fund all research and development expenses for the pre-clinical activities associated with the cystinuria program, which are estimated to be approximately $5.0 million. The Agreements require the Company to purchase an additional 5% of the outstanding common shares for $1.0 million upon the occurrence of a specified pre-clinical milestone, and granted an option to the Company to purchase the remaining outstanding shares of PharmaKrysto for $5.0 million upon the occurrence of a subsequent pre-clinical milestone prior to expiration of the option on March 8, 2025. If the Company elects to exercise the option, it would be required to perform commercially reasonable clinical diligence obligations. In addition, it would be required to make cash milestone payments totaling up to an aggregate $16.0 million upon the achievement of certain development and regulatory milestones, plus tiered royalty payments of less than 4% on future net sales of a product, if approved. The Company has the right to terminate the Agreements and return the shares for a nominal price at any time upon 60 days’ notice, subject to survival of contingent obligations, if any.
The Company determined that PharmaKrysto is a VIE because it lacks the resources to conduct the cystinuria clinical program and the limitation on the residual returns through the Company's option to purchase the remaining outstanding shares. The Company further concluded that it is the primary beneficiary of the VIE due to the Company's ultimate control over the research and development program, and its obligation, subject to continuation of the collaboration, to fund 100% of research and development costs of the program pursuant to the terms of the Collaboration Agreement.
The upfront payments were expensed to research and development and other income (expense), net upon initial consolidation. The consolidated assets and liabilities as of December 31, 2023 and 2022 were immaterial. The results of operations were not significant for the years ended December 31, 2023 and 2022. The Company is not required to provide additional funding other than the contractually required amounts disclosed above. The creditors and beneficial holders of PharmaKrysto have no recourse to the general credit or assets of the Company.
NOTE 6. MARKETABLE DEBT SECURITIES
The Company's marketable debt securities as of December 31, 20212023 and 20202022 were comprisedcomposed of available-for-sale commercial paper and corporate and government debt securities. These securities are carried at fair value, with the unrealized gains and losses reported in accumulated other comprehensive income (loss), unless an impairment is determined to be the result of credit-related factors or the Company intends to sell the security or it is more likely than not that the Company will be required to sell the security before recovery. The amortized cost of marketable debt securities is adjusted for amortization of premiums and accretion of discounts to maturity. Such amortization and accretion is included in interest income. Realized gains and losses and declines in value that are determined to be the result of credit losses, if any, on available-for-sale securities are included in other income or expense. Unrealized losses that are determined to be credit-related are also recorded as an allowance against the amortized cost basis. The cost of securities sold is based on the specific identification method. Interest and dividends on securities classified as available-for-sale are included in interest income. All available-for-sale securities are classified as current assets, even if the maturity when acquired by the Company is greater than one year due to the ability to liquidate within the next 12 months.
Marketable debt securities consist of the following (in thousands):
| | | | | | | | | | | |
| As of December 31, |
| 2021 | | 2020 |
| Marketable debt securities: | | | |
| Commercial paper | $ | 127,379 | | | $ | 135,145 | |
| Corporate debt securities | 233,319 | | | 98,646 | |
| Securities of government sponsored entities | 26,431 | | | 43,026 | |
| Total marketable debt securities | $ | 387,129 | | | $ | 276,817 | |
The following is a summary of short-term marketable debt securities classified as available-for-sale as of December 31, 2021 (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Contractual Maturity (in years) | | Amortized Cost | | Unrealized Gains | | Unrealized Losses | | Aggregate Estimated Fair Value |
| Marketable debt securities: | | | | | | | | | |
| Commercial paper | Less than 1 | | $ | 127,435 | | | $ | — | | | $ | (57) | | | $ | 127,378 | |
| Corporate debt securities | Less than 1 | | 113,001 | | | — | | | (97) | | | 112,904 | |
| Securities of government-sponsored entities | Less than 1 | | 21,909 | | | — | | | (5) | | | 21,904 | |
| Total maturity less than 1 year | | | 262,345 | | | — | | | (159) | | | 262,186 | |
| Corporate debt securities | 1 to 2 | | 120,705 | | | — | | | (289) | | | 120,416 | |
| Securities of government-sponsored entities | 1 to 2 | | 4,549 | | | — | | | (22) | | | 4,527 | |
| Total maturity 1 to 2 years | | | 125,254 | | | — | | | (311) | | | 124,943 | |
| Total available-for-sale marketable debt securities | | | $ | 387,599 | | | $ | — | | | $ | (470) | | | $ | 387,129 | |
The following is a summary of short-term marketable debt securities classified as available-for-sale as of December 31, 2020 (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Contractual Maturity (in years) | | Amortized Cost | | Unrealized Gains | | Unrealized Losses | | Aggregate Estimated Fair Value |
| Marketable debt securities: | | | | | | | | | |
| Commercial paper | Less than 1 | | $ | 135,161 | | | $ | 1 | | | $ | (17) | | | $ | 135,145 | |
| Corporate debt securities | Less than 1 | | 92,906 | | | 723 | | | — | | | 93,629 | |
| Securities of government-sponsored entities | Less than 1 | | 43,031 | | | — | | | (5) | | | 43,026 | |
| Total maturity less than 1 year | | | 271,098 | | | 724 | | | (22) | | | 271,800 | |
| Corporate debt securities | 1 to 2 | | 5,013 | | | 4 | | | — | | | 5,017 | |
| | | | | | | | | |
| Total maturity 1 to 2 years | | | 5,013 | | | 4 | | | — | | | 5,017 | |
| Total available-for-sale marketable debt securities | | | $ | 276,111 | | | $ | 728 | | | $ | (22) | | | $ | 276,817 | |
During 2021, the Company had less than $0.1 million in realized gains on marketable debt securities. During 2020, the Company had $0.1 million in realized gains on marketable debt securities. The Company received proceeds from the sale or maturity of marketable debt securities of $433.6 million, $273.5 million and $259.1 million for 2021, 2020 and 2019, respectively.
The primary objective of the Company’s investment portfolio is to preserve capital and liquidity while enhancing overall returns. The Company’s investment policy limits interest-bearing security investments to certain types of instruments issued by institutions with primarily investment grade credit ratings and places restrictions on maturities and concentration by asset class and issuer.
Marketable debt securities consisted of the following (in thousands):
| | | | | | | | | | | |
| As of December 31, |
| 2023 | | 2022 |
| Marketable debt securities: | | | |
| Commercial paper | $ | 34,458 | | | $ | 123,647 | |
| Corporate debt securities | 368,323 | | | 224,055 | |
| Securities of government-sponsored entities | 105,894 | | | 40,855 | |
| Total available-for-sale marketable debt securities | $ | 508,675 | | | $ | 388,557 | |
The following is a summary of short-term marketable debt securities classified as available-for-sale as of December 31, 2023 (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Remaining Contractual Maturity
(in years) | | Amortized Cost | | Unrealized Gains | | Unrealized Losses | | Aggregate Estimated Fair Value |
| Marketable debt securities: | | | | | | | | | |
| Commercial paper | Less than 1 | | $ | 34,450 | | | $ | 25 | | | $ | (17) | | | $ | 34,458 | |
| Corporate debt securities | Less than 1 | | 133,463 | | | 29 | | | (408) | | | 133,084 | |
| Securities of government-sponsored entities | Less than 1 | | 81,334 | | | 36 | | | (274) | | | 81,096 | |
| Total maturity less than 1 year | | | 249,247 | | | 90 | | | (699) | | | 248,638 | |
| Corporate debt securities | 1 to 2 | | 233,969 | | | 1,444 | | | (174) | | | 235,239 | |
| Securities of government-sponsored entities | 1 to 2 | | 24,718 | | | 106 | | | (26) | | | 24,798 | |
| Total maturity 1 to 2 years | | | 258,687 | | | 1,550 | | | (200) | | | 260,037 | |
| Total available-for-sale marketable debt securities | | | $ | 507,934 | | | $ | 1,640 | | | $ | (899) | | | $ | 508,675 | |
The following is a summary of short-term marketable debt securities classified as available-for-sale as of December 31, 2022 (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Remaining Contractual Maturity
(in years) | | Amortized Cost | | Unrealized Gains | | Unrealized Losses | | Aggregate Estimated Fair Value |
| Marketable debt securities: | | | | | | | | | |
| Commercial paper | Less than 1 | | $ | 124,301 | | | $ | 2 | | | $ | (656) | | | $ | 123,647 | |
| Corporate debt securities | Less than 1 | | 155,841 | | | — | | | (1,355) | | | 154,486 | |
| Securities of government-sponsored entities | Less than 1 | | 7,473 | | | — | | | (80) | | | 7,393 | |
| Total maturity less than 1 year | | | 287,615 | | | 2 | | | (2,091) | | | 285,526 | |
| Corporate debt securities | 1 to 2 | | 70,195 | | | 33 | | | (659) | | | 69,569 | |
| Securities of government-sponsored entities | 1 to 2 | | 33,702 | | | 6 | | | (246) | | | 33,462 | |
| Total maturity 1 to 2 years | | | 103,897 | | | 39 | | | (905) | | | 103,031 | |
| Total available-for-sale marketable debt securities | | | $ | 391,512 | | | $ | 41 | | | $ | (2,996) | | | $ | 388,557 | |
During 2023 and 2022, realized gains on marketable debt securities were immaterial. As of December 31, 2023 and December 31, 2022, the accrued interest receivable related to the Company's marketable debt securities was $4.6 million and $1.9 million, respectively, and was recorded in prepaid expenses and other current assets on the Consolidated Balance Sheets.
The Company reviews the available-for-sale marketable debt securities for declines in fair value below the cost basis each quarter. For any security whose fair value is below its amortized cost basis, the Company first evaluates whether it intends to sell the impaired security, or will otherwise be more likely than not required to sell the security before recovery. If either are true, the amortized cost basis of the security is written down to its fair value at the reporting date. If neither circumstance holds true, the Company assesses whether any portion of the unrealized loss is a result of a credit loss. Any amount deemed to be attributable to credit loss is recognized in the income statement, with the amount of the loss limited to the difference between fair value and amortized cost and recorded as an allowance for credit losses. The portion of the unrealized loss related to factors other than credit losses is recognized in other comprehensive income (loss).
The following is a summary
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Less Than 12 Months | | 12 Months or Greater | | Total |
| Description of Securities | | Fair Value | | Unrealized Losses | | Fair Value | | Unrealized Losses | | Fair Value | | Unrealized Losses |
| Commercial paper | | $ | 122,380 | | | $ | 57 | | | $ | — | | | $ | — | | | $ | 122,380 | | | $ | 57 | |
| Corporate debt securities | | 231,879 | | | 386 | | | — | | | — | | | 231,879 | | | 386 | |
| Securities of government-sponsored entities | | 26,431 | | | 27 | | | — | | | — | | | 26,431 | | | 27 | |
| Total | | $ | 380,690 | | | $ | 470 | | | $ | — | | | $ | — | | | $ | 380,690 | | | $ | 470 | |
The following is a summary of available-for-sale marketable debt securities in an unrealized loss position with no credit losses reported as of December 31, 20202023 (in thousands):
| | Less Than 12 Months | | 12 Months or Greater | | Total |
| | Less Than 12 Months | | | | Less Than 12 Months | | 12 Months or Greater | | Total |
| Description of Securities | Description of Securities | | Fair Value | | Unrealized Losses | | Fair Value | | Unrealized Losses | | Fair Value | | Unrealized Losses | Description of Securities | | Fair Value | | Unrealized Losses | | Fair Value | | Unrealized Losses | | Fair Value | | Unrealized Losses |
| Commercial paper | Commercial paper | | $ | 112,148 | | | $ | 17 | | | $ | — | | | $ | — | | | $ | 112,148 | | | $ | 17 | |
| | Corporate debt securities | |
| Securities of government-sponsored entities | Securities of government-sponsored entities | | 43,026 | | | 5 | | | — | | | — | | | 43,026 | | | 5 | |
| Total | Total | | $ | 155,174 | | | $ | 22 | | | $ | — | | | $ | — | | | $ | 155,174 | | | $ | 22 | |
The following is a summary of available-for-sale marketable debt securities in an unrealized loss position with no credit losses reported as of December 31, 2022 (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Less Than 12 Months | | 12 Months or Greater | | Total |
| Description of Securities | | Fair Value | | Unrealized Losses | | Fair Value | | Unrealized Losses | | Fair Value | | Unrealized Losses |
| Commercial paper | | $ | 117,853 | | | $ | 656 | | | $ | — | | | $ | — | | | $ | 117,853 | | | $ | 656 | |
| Corporate debt securities | | 99,066 | | | 1,041 | | | 107,964 | | | 973 | | | 207,030 | | | 2,014 | |
| Securities of government-sponsored entities | | 31,402 | | | 263 | | | 4,456 | | | 63 | | | 35,858 | | | 326 | |
| Total | | $ | 248,321 | | | $ | 1,960 | | | $ | 112,420 | | | $ | 1,036 | | | $ | 360,741 | | | $ | 2,996 | |
As of December 31, 20212023 and December 31, 2020,2022, the amortized cost of the available-for-sale marketable debt securities in an unrealized loss position was $381.2$269.7 million and $155.2$363.7 million, respectively.
As of December 31, 20212023 and December 31, 2020,2022, the Company does not intend to sell these investments and it is not more likely than not that the Company will be required to sell the investments before recovery of their amortized cost basis. The decrease in unrealized losses for the year ended December 31, 2023 was primarily due to fluctuations in short-term interest rates. The Company does not believe the unrealized losses incurred during the period are due to credit-related factors. Liquidity issues that arose from economic circumstances surrounding the COVID-19 pandemic have eased and theThe credit ratings of the securities held remain of the highest quality. While certain securities in the portfolio may be downgraded momentarily, the Federal Reserve has allowed institutions to continue to issue debt where there is need, with the government itself purchasing such securities. Moreover, the Company continues to receive payments of interest and principal as they become due, and our expectation is that those payments will continue to be received timely. Uncertainty surrounding the COVID-19 pandemic, as well as other factorsFactors unknown to us at this time may cause actual results to differ and require adjustments to the Company’s estimates and assumptions in the future.
NOTE 7. CONVERTIBLE SENIOR NOTES PAYABLE
The composition of the Company’s convertible senior notes are as follows (in thousands):
| | | | | | | | | | | |
| | December 31, 2023 | | December 31, 2022 |
| 2.25% convertible senior notes due 2029 | $ | 316,250 | | | $ | 316,250 | |
| 2.50% convertible senior notes due 2025 | 68,904 | | | 68,904 | |
| Unamortized debt issuance costs - 2.25% convertible senior notes due 2029 | (7,348) | | | (8,750) | |
| Unamortized debt issuance costs - 2.50% convertible senior notes due 2025 | (543) | | | (859) | |
| Total convertible senior notes, net of unamortized debt discount and debt issuance costs | $ | 377,263 | | | $ | 375,545 | |
Convertible Senior Notes Due 2029
On March 11, 2022, the Company completed a registered underwritten public offering of $316.3 million aggregate principal amount of 2.25% Convertible Senior Notes due 2029 (“2029 Notes”), which includes $41.3 million aggregate principal amount of 2029 Notes sold pursuant to the full exercise of the underwriters’ option to purchase additional 2029 Notes. The Company issued the 2029 Notes under an indenture, dated as of September 10, 2018, as supplemented by the second supplemental indenture, dated as of March 11, 2022 (collectively, the “2029 Indenture”). The 2029 Notes will mature on March 1, 2029, unless earlier repurchased, redeemed, or converted. The 2029 Notes are senior unsecured obligations of the Company and bear interest at an annual rate of 2.25%, payable semi-annually in arrears on March 1 and September 1 of each year, beginning on September 1, 2022.
The Company received net proceeds from the issuance of the 2029 Notes of $306.4 million, after deducting commissions and offering expenses of $9.9 million. At December 31, 2023, accrued interest on the 2029 Notes of $2.4 million is included in accrued expenses in the accompanying Consolidated Balance Sheets. The 2029 Notes comprise the Company’s senior, unsecured obligations and are (i) equal in right of payment with the Company’s existing and future senior, unsecured indebtedness; (ii) senior in right of payment to the Company’s existing and future indebtedness that is expressly subordinated to the 2029 Notes; (iii) effectively subordinated to the Company’s existing and future secured indebtedness, to the extent of the value of the collateral securing that indebtedness; and (iv) structurally subordinated to all existing and future indebtedness and other liabilities, including trade payables.
Holders may convert their 2029 Notes at their option only in the following circumstances: (1) during any calendar quarter commencing after the calendar quarter ending on June 30, 2022 (and only during such calendar quarter), if the last reported sale price per share of the Company’s common stock for each of at least 20 trading days, whether or not consecutive, during the period of 30 consecutive trading days ending on, and including, the last trading day of the immediately preceding calendar quarter exceeds 130% of the conversion price on the applicable trading day; (2) during the five consecutive business days immediately after any 10 consecutive trading day period (such 10 consecutive trading day period, the “measurement period”) if the trading price per $1,000 principal amount of 2029 Notes for each trading day of the measurement period was less than 98% of the product of the last reported sale price per share of the Company’s common stock on such trading day and the conversion rate on such trading day; (3) upon the occurrence of certain corporate events or distributions of the Company’s common stock; (4) if the Company calls the 2029 Notes for redemption; and (5) at any time from, and including, December 1, 2028 until the close of business on the scheduled trading day immediately before the maturity date. The Company will settle conversions by paying or delivering, as applicable, cash, shares of the Company’s common stock, or a combination of cash and shares of the Company’s common stock, at the Company’s election, based on the applicable conversion rate. The initial conversion rate for the 2029 Notes is 31.3740 shares of the Company’s common stock per $1,000 principal amount of 2029 Notes, which represents an initial conversion price of approximately $31.87 per share. If a “make-whole fundamental change” (as defined in the 2029 Indenture) occurs, then the Company will, in certain circumstances, increase the conversion rate for a specified period of time.
The 2029 Notes will be redeemable, in whole or in part at the Company’s option at any time, and from time to time, on or after March 2, 2026 and, in the case of any partial redemption, on or before the 40th scheduled trading day before the maturity date, at a cash redemption price equal to the principal amount of the 2029 Notes to be redeemed, plus accrued and unpaid interest, if any, to, but excluding, the redemption date but only if the last reported sale price per share of the Company’s common stock exceeds 130% of the conversion price on (1) each of at least 20 trading days, whether or not consecutive, during the 30 consecutive trading days ending on, and including, the trading day immediately before the date the Company sends the related redemption notice; and (2) the trading day immediately before the date the Company sends such notice. However, the Company may not redeem less than all of the outstanding 2029 Notes unless at least $100.0 million aggregate principal amount of 2029 Notes are outstanding and not called for redemption as of the time the Company sends the related redemption notice. In addition, calling any 2029 Note for redemption will constitute a make-whole fundamental change with respect to that 2029 Note, in which case the conversion rate applicable to the conversion of that 2029 Note will be increased in certain circumstances if it is converted after it is called for redemption. If a fundamental change (as defined in the 2029 Indenture) occurs, then, except as described in the 2029 Indenture, holders may require the Company to repurchase their 2029 Notes at a cash repurchase price equal to the principal amount of the 2029 Notes to be repurchased, plus accrued and unpaid interest, if any, to, but excluding, the fundamental change repurchase date. In the event of conversion, holders would forgo all future interest payments, any unpaid accrued interest and the possibility of further stock price appreciation. Upon the receipt of conversion requests, the settlement of the 2029 Notes will be paid pursuant to the terms of the 2029 Indenture. In the event that all of the 2029 Notes are converted, the Company would be required to repay the principal amount and any conversion premium in any combination of cash and shares of its common stock at the Company’s option. In addition, calling the 2029 Notes for redemption will constitute a “make-whole fundamental change."
The Company incurred approximately $9.9 million of debt issuance costs relating to the issuance of the 2029 Notes, which were recorded as a reduction to the 2029 Notes on the Consolidated Balance Sheets. The debt issuance costs are being amortized and recognized as additional interest expense over the expected life of the 2029 Notes using the effective interest method. We determined the expected life of the debt is equal to the seven-year term of the 2029 Notes. The effective interest rate on the 2029 Notes is 2.74%.
Convertible Senior Notes Due 2025
On September 10, 2018, the Company completed itsa registered underwritten public offering of the "2025$276.0 million aggregate principal amount of 2.50% Convertible Senior Notes due 2025 ("2025 Notes"), and entered into a base indenture and supplemental indenture agreement ("2025 Indenture") with respect to the 2025 Notes. The 2025 Notes will mature on September 15, 2025, ("Maturity Date”), unless earlier repurchased, redeemed, or converted. The 2025 Notes are senior unsecured obligations of the Company and bear interest at an annual rate of 2.50%, payable semi-annually in arrears on March 15 and September 15 of each year, beginning on March 15, 2019.
The composition of the Company’s 2025 Notes are as follows (in thousands):
| | | | | | | | | | | |
| | December 31, 2021 | | December 31, 2020 |
2.50% convertible senior notes due 2025 | $ | 276,000 | | | $ | 276,000 | |
| Unamortized debt discount | (46,045) | | | (56,384) | |
| Unamortized debt issuance costs | (3,374) | | | (4,277) | |
| Total 2025 Notes, net of unamortized debt discount and debt issuance costs | $ | 226,581 | | | $ | 215,339 | |
The net proceeds from the issuance of the 2025 Notes were approximately $267.2 million, after deducting commissions and the offering expenses of $8.8 million payable by the Company. A portionAt December 31, 2023, accrued interest of $0.5 million is included in accrued expenses in the accompanying Consolidated Balance Sheets. The 2025 Notes comprise the Company’s senior, unsecured obligations and are (i) equal in right of payment with the Company’s existing and future senior, unsecured indebtedness; (ii) senior in right of payment to the Company’s existing and future indebtedness that is expressly subordinated to the 2025 Notes; (iii) effectively subordinated to the Company’s existing and future secured indebtedness, to the extent of the net proceeds fromvalue of the 2025 Notes were used by the Companycollateral securing that indebtedness; and (iv) structurally subordinated to repurchase $23.4 million aggregate principal amountall existing and future indebtedness and other liabilities, including trade payables.
Holders may convert their 2025 Notes at their option only in the following circumstances: (1) during any calendar quarter commencing after the calendar quarter ending on December 31, 2018 (and only during such calendar quarter), if the last reported sale price per share of the Company’s common stock for each of at least 20 trading days, whether or not consecutive, during the period of 30 consecutive trading days ending on, and including, the last trading day of the immediately preceding calendar quarter exceeds 130% of the conversion price on the applicable trading day; (2) during the five consecutive business days immediately after any 10 consecutive trading day period (“measurement period”) if the trading price per $1,000 principal amount of 2025 Notes for each trading day of the measurement period was less than 98% of the product of the last reported sale price per share of the Company’s common stock on such trading day and the conversion rate on such trading day; (3) upon the occurrence of certain corporate events or distributions on the Company’s common stock; (4) if the Company calls the 2025 Notes for redemption; and (5) at any time from, and including, May 15, 2025 until the close of business on the scheduled trading day immediately before the Maturity Date.maturity date. The Company will settle conversions by paying or delivering, as applicable, cash, shares of the Company’s common stock, or a combination of cash and shares of the Company’s common stock, at the Company’s election, based on the applicable conversion rate.
The initial conversion rate for the 2025 Notes is 25.7739 shares of the Company’s common stock per $1,000 principal amount of 2025 Notes, which represents an initial conversion price of approximately $38.80 per share. If a “make-whole fundamental change” (as defined in the 2025 Indenture) occurs, then the companyCompany will, in certain circumstances, increase the conversion rate for a specified period of time.
The 2025 Notes will be redeemable, in whole or in part, at the Company’s option at any time, and from time to time, on or after September 15, 2022 and, in the case of any partial redemption, on or before the 40th scheduled trading day before the Maturity Date,maturity date, at a cash redemption price equal to the principal amount of the 2025 Notes to be redeemed, plus accrued and unpaid interest, if any, to, but excluding, the redemption date, but only if the last reported sale price per share of the Company’s common stock exceeds 130% of the conversion price on each of at least 20 trading days during the 30 consecutive trading days ending on, and including, the trading day immediately before the date the Company sends the related redemption notice. If a fundamental change (as defined in the 2025 Indenture) occurs, then, subject to certain exceptions, holders may require the Company to repurchase their 2025 Notes at a cash repurchase price equal to the principal amount of the 2025 Notes to be repurchased, plus accrued and unpaid interest, if any, to, but excluding, the fundamental change repurchase date.
As of December 31, 2021, the 2025 Notes had a market price of $1,089 per $1,000 principal amount, or $300.6 million. In the event of conversion, holders would forgo all future interest payments, any unpaid accrued interest and the possibility of further stock price appreciation. Upon the receipt of conversion requests, the settlement of the 2025 Notes will be paid pursuant to the terms of the 2025 Indenture. In the event that all of the 2025
Notes are converted, the Company would be required to repay the $276.0 million in principal valueamount and any conversion premium in any combination of cash and shares of its common stock at the Company’s option. In addition, calling the 2025 Notes for redemption will constitute a “make whole“make-whole fundamental change.”"
The 2025 Notes are the Company’s general unsecured obligations that rank senior in rightCompany incurred approximately $8.8 million of payment to all of its indebtedness that is expressly subordinated in right of paymentdebt issuance costs relating to the 2025 Notes, and equal in right of payment to the Company’s unsecured indebtedness.
The 2025 Notes are currently classified on the Company’s consolidated balance sheet at December 31, 2021 as long-term convertible debt.
Under ASC 470-20, an entity must separately account for the liability and equity components of convertible debt instruments (such as the 2025 Notes) that may be settled entirely or partially in cash upon conversion, in a manner that reflects the issuer’s economic interest cost. The liability component of the instrument is valued in a manner that reflects the market interest rate for a similar nonconvertible instrument at the date of issuance. The initial carrying value of the liability component was $198.6 million. The equity component of $77.4 million, representing the conversion option, was determined by deducting the fair value of the liability component from the par value of the 2025 Notes and was recorded in additional paid-in capital on the Consolidated Balance Sheets at the issuance date. That equity component is treated as a discount on the liability component of the 2025 Notes, which iswere recorded as a reduction to the 2025 Notes on the Consolidated Balance Sheets. The debt issuance costs are being amortized and recognized as additional interest expense over the seven-year termexpected life of the 2025 Notes using the effective interest rate method. The equity component is not re-measured as long as it continues to meetCompany determined the conditions for equity classification. The Company allocated the total transaction costs of approximately $8.8 million related to the issuanceexpected life of the 2025 Notesdebt is equal to the liability and equity components of the 2025 Notes based on their relative values. Transaction costs attributable to the liability component are amortized to interest expense over the seven-year term of the 2025 Notes, and transaction costs attributable to the equity component are netted with the equity component in stockholders’ equity.
Notes. The effective interest rate on the liability components2025 Notes is 2.98%.
On March 11, 2022, the Company completed its repurchase of $207.1 million aggregate principal amount of 2025 Notes for cash, including accrued and unpaid interest, for a total of $213.8 million. This transaction involved a contemporaneous exchange of cash between the Company and holders of the 2025 Notes participating in the issuance of the 2029 Notes. Accordingly, we evaluated the transaction for modification or extinguishment accounting in accordance with ASC 470-50, Debt – Modifications and Extinguishments on a creditor-by creditor basis depending on whether the exchange was determined to have substantially different terms. The repurchase of the 2025 Notes and issuance of the 2029 Notes were deemed to have substantially different terms based on the present value of the cash flows or significant difference between the value of the conversion option immediately prior to and after the exchange. Therefore, the repurchase of the 2025 Notes was accounted for as a debt extinguishment. The Company recorded a $7.6 million loss on extinguishment of debt on its Consolidated Statements of Operations for the yearsyear ended December 31, 2021, 20202022, which included the write-off of related deferred financing costs of $3.4 million. After giving effect to the repurchase, and 2019 was 7.7%. The following table sets forthas of December 31, 2023, the total interest expense recognized related toremaining principal amount outstanding under the 2025 Notes (in thousands):was $68.9 million.
| | | | | | | | | | | | | | | | | |
| Twelve Months Ended December 31, |
| 2021 | | 2020 | | 2019 |
| Contractual interest expense | $ | 6,900 | | | $ | 6,900 | | | $ | 6,900 | |
| Amortization of debt discount | 10,339 | | | 9,578 | | | 8,874 | |
| Amortization of debt issuance costs | 903 | | | 900 | | | 896 | |
| Total interest expense for the 2025 Notes | $ | 18,142 | | | $ | 17,378 | | | $ | 16,670 | |
The 2025 and 2029 Notes do not contain any financial or operating covenants or any restrictions on the payment of dividends, the issuance of other indebtedness or the issuance or repurchase of securities by the Company. The 2025 Indenture contains customaryThere were no events of default with respectfor the 2025 Notes or 2029 Notes at December 31, 2023.
The 2025 and 2029 Notes are classified on the Company's Consolidated Balance Sheets at December 31, 2023 as long-tern convertible debt.
Interest Expense
The following table sets forth total interest expense recognized related to the 2025 and 2029 Notes including that upon certain events of default, 100% of the principal and accrued and unpaid interest on the 2025 Notes will automatically become due and payable.(in thousands):
Interest Expense | | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2023 | | 2022 | | 2021 |
| Contractual interest expense | $ | 8,838 | | | $ | 8,433 | | | $ | 6,900 | |
| Amortization of debt discount | — | | | — | | | 10,339 | |
| Amortization of debt issuance costs | 1,718 | | | 1,622 | | | 903 | |
| Total interest expense for the 2025 and 2029 Notes | $ | 10,556 | | | $ | 10,055 | | | $ | 18,142 | |
Total interest expense recognized for the years ended December 31, 2023, 2022 and 2021 2020 and 2019 was $20.1$11.3 million, $19.1$11.0 million and $18.8$19.7 million, respectively.
NOTE 8. FAIR VALUE MEASUREMENTS
The Company accounts for financial instruments in accordance with ASC 820, “Fair Value Measurements and Disclosures” (“ASC 820”). ASC 820 establishesutilizes a fair value hierarchy that prioritizes the inputs to valuation techniques used to measure fair value. The hierarchy gives the highest priority to unadjusted quoted prices in active markets for identical assets or liabilities (Level 1 measurements) and the lowest priority to unobservable inputs (Level 3 measurements). The three levels of the fair value hierarchy under ASC 820 are described below:
Level 1 – Unadjusted quoted prices in active markets that are accessible at the measurement date for identical, unrestricted assets or liabilities;
Level 2 – Quoted prices in markets that are not active or financial instruments for which all significant inputs are observable, either directly or indirectly; and
Level 3 – Prices or valuations that require inputs that are both significant to the fair value measurement and unobservable.
The valuation techniques used to measure the fair value of the Company’s debt securities and all other financial instruments, all of which have counter-partiescounterparties with high credit ratings, were valued based on quoted market prices or model driven valuations using significant inputs derived from or corroborated by observable market data. Based on the fair value hierarchy, the Company classified marketable debt securities within Level 2.
The Company acquired 2 businesses, related to the Cholbam and Chenodal products, whose purchase price included potential future payments that are contingent on the achievement of certain milestones and percentages of future net sales derived from the products acquired. The Company recorded contingent consideration liabilities at their fair value on the acquisition date and revalues them at the end of each reporting period. In estimating the fair value of the Company’s contingent consideration, the Company uses a Monte Carlo Simulation. The determination of the contingent consideration liabilities requires significant judgments including the appropriateness of the valuation model and reasonableness of estimates and assumptions included in the forecasts of future net sales and the discount rates applied to such forecasts. Changes in these estimates and assumptions could have a significant impact on the fair value of the contingent consideration liabilities.
Discount rates used to determine the fair value at December 31, 2021 and 2020 are as follows:
| | | | | | | | | | | | | | | | | | | | |
| | Revenue Discount | | Payment Discount |
| | Cholbam | | Chenodal | | |
| December 31, 2021 | | 6.25% | | 7.25% | | 6.48% |
| December 31, 2020 | | 6.50% | | 8.50% | | 7.45% |
Based on the fair value hierarchy, the Company classified the fair value of contingent consideration within Level 3 because valuation inputs are based on projected revenues discounted to a present value.
Financial instruments with carrying values approximating fair value include cash and cash equivalents, accounts receivable, and accounts payable, due to their short-term nature. As of December 31, 2021,2023 and 2022, the fair value of the Company's 2.5%2.50% Convertible Senior Notes due 2025 was $300.6$58.3 million and $62.9 million, respectively. As of December 31, 2023 and 2022, the fair value of the Company's 2.25% Convertible Senior Notes due 2029, which were issued in 2022, was $212.1 million and $283.0 million, respectively. The fair values were estimated utilizing market quotations and are considered Level 2.
The following table presents the Company’s asset and liabilities,assets, measured and recognized at fair value on a recurring basis, classified under the appropriate level of the fair value hierarchy as of December 31, 20212023 (in thousands):
| | | | | As of December 31, 2023 | | Fair Value Hierarchy at December 31, 2023 |
| Total carrying and estimated fair value | | | Total carrying and estimated fair value | | Quoted prices in active markets (Level 1) | | Significant other observable inputs (Level 2) | | Significant unobservable inputs (Level 3) |
| Assets: | |
| Cash and Cash Equivalents | |
| Cash and Cash Equivalents | |
| Cash and Cash Equivalents | |
| Marketable debt securities, available-for-sale | |
| Total | |
| | | | As of December 31, 2021 | | Fair Value Hierarchy at December 31, 2021 |
| | | Total carrying and estimated fair value | | Quoted prices in active markets (Level 1) | | Significant other observable inputs (Level 2) | | Significant unobservable inputs (Level 3) |
| Asset: | | | | | | | |
| Cash and Cash Equivalents | $ | 165,753 | | | $ | 165,753 | | | $ | — | | | $ | — | |
| Marketable debt securities, available-for-sale | 387,129 | | | — | | | 387,129 | | | — | |
| Total | $ | 552,882 | | | $ | 165,753 | | | $ | 387,129 | | | $ | — | |
| Liabilities: | | | | | | | |
| Business combination-related contingent consideration | $ | 67,100 | | | $ | — | | | $ | — | | | $ | 67,100 | |
| Total | $ | 67,100 | | | $ | — | | | $ | — | | | $ | 67,100 | |
The following table presents the Company’s assets, and liabilities that are measured and recognized at fair value on a recurring basis, classified under the appropriate level of the fair value hierarchy as of December 31, 20202022 (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | |
| | As of December 31, 2020 | | Fair Value Hierarchy at December 31, 2020 |
| | Total carrying and
estimated fair value | | Quoted prices in
active markets
(Level 1) | | Significant other
observable inputs
(Level 2) | | Significant
unobservable
inputs (Level 3) |
| Asset: | | | | | | | |
| Cash and Cash Equivalents | $ | 84,772 | | | $ | 84,772 | | | $ | — | | | $ | — | |
| Marketable debt securities, available-for-sale | 276,817 | | | — | | | 276,817 | | | — | |
| Total | $ | 361,589 | | | $ | 84,772 | | | $ | 276,817 | | | $ | — | |
| Liabilities: | | | | | | | |
| | | | | | | |
| Business combination-related contingent consideration | $ | 65,100 | | | $ | — | | | $ | — | | | $ | 65,100 | |
| Total | $ | 65,100 | | | $ | — | | | $ | — | | | $ | 65,100 | |
The following table sets forth a summary of changes in the estimated fair value of the Company's Level 3 business combination-related contingent consideration for the years ended December 31, 2021 and 2020 (in thousands):
| | | | | | | | | | | |
| Fair Value Measurements of Acquisition-Related Contingent Consideration (Level 3) |
| 2021 | | 2020 |
| Balance at January 1, | $ | 65,100 | | | $ | 70,900 | |
| | | |
| Changes in the fair value of contingent consideration | 22,260 | | | 3,655 | |
| Contractual payments | (17,444) | | | (7,003) | |
| Contractual payments included in accrued liabilities at December 31 | (2,700) | | | (2,488) | |
| Foreign currency impact | (116) | | | 36 | |
| Balance at December 31, | $ | 67,100 | | | $ | 65,100 | |
| | | | | | | | | | | | | | | | | | | | | | | |
| | As of December 31, 2022 | | Fair Value Hierarchy at December 31, 2022 |
| Total carrying and estimated fair value | | Quoted prices in active markets (Level 1) | | Significant other observable inputs (Level 2) | | Significant unobservable inputs (Level 3) |
| Assets: | | | | | | | |
| Cash and Cash Equivalents | $ | 61,688 | | | $ | 61,688 | | | $ | — | | | $ | — | |
| Marketable debt securities, available-for-sale | 388,557 | | | — | | | 388,557 | | | — | |
| Total | $ | 450,245 | | | $ | 61,688 | | | $ | 388,557 | | | $ | — | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
NOTE 9. INTANGIBLE ASSETS
Amortizable Intangible Assets
Ligand License Agreement
In 2013,2012, the Company entered into a $2.5 millionan agreement with Ligand Pharmaceuticals, Inc. ("Ligand") for a worldwide sublicense to develop, manufacture and commercialize a drug technology compound including sparsentan (the “Ligand License Agreement”). The cost of the Ligand License Agreement, which is presented net of amortization in the accompanying Consolidated Balance Sheets in intangible assets, net, is being amortized to research and development on a straight-line basis through September 30, 2023. As consideration for the license, we arethe Company is required to make substantial payments upon the achievement of certain milestones, totaling up to $114.1 million. Through 2021, we have madeIn March 2023, the Company capitalized a $23.0 million milestone paymentspayment to Ligand (and Bristol-Myers Squibb Company ("BMS")) that was triggered upon the accelerated approval of $7.2 million under the terms ofFILSPARI in February 2023. Pursuant to the Ligand License Agreement. Should we commercialize sparsentan or any products containing related compounds, we will beAgreement, the Company is obligated to pay to Ligand (and BMS) an escalating annual royalty between 15% and 17% of net sales of allsparsentan, with payments due quarterly. The Company began incurring costs associated with such products.
In September 2015,royalties following the Ligand License Agreement was amended to facilitate sub-licensing in Asia-Pacific. As consideration forFebruary 2023 approval of FILSPARI (sparsentan). For the amendmentyear ended December 31, 2023, the Company paid $1.0 million.
In March 2018, the Ligand License Agreement was amendedcapitalized $4.4 million to update certain development milestones set forth in the Sublicense Agreement to comport with the current development timelineintangible assets for sparsentan. As consideration, the Company paid Ligand $4.6 million, which replaced the amount that would have been due upon initiationroyalties owed on net sales of FILSPARI. The cost of the first Phase 3 trial for sparsentan.
Manchester Pharmaceuticals LLC
In 2014, the Company acquired intangible assets with finite lives related to the Chenodal product rights, trade names,$23.0 million milestone payment and customer relationships with the values of $67.8 million, $0.2 million, and $0.4 million, respectively. The useful lives related to the acquired product rights, trade names, and customer relationships are expected to be approximately 16, 1 and, 10 years, respectively. Amortization of product rights, trade names and customer relationshipsroyalty payments are being recorded inamortized to selling, general and administrative expense over their respective lives.administration on a straight-line basis through April 30, 2033
Thiola License Agreement
The Company entered into a license agreement with Mission Pharmacal in 2014, in which the Company obtained an exclusive, royalty-bearing license to market, sell and commercialize Thiola (tiopronin) in the United States and Canada, and a non-exclusive license to use know-how relating to Thiola to the extent necessary to market Thiola. The initial term of the license is 10 years and will automatically renew thereafter for periods of one year.
The acquisition of the Thiola license qualified as an asset acquisition under the principles of ASC 805 in effect at the time of acquisition. The license agreement requires the Company to make royalty payments based on net sales of Thiola. The liability for royalties in excess of the annual contractual minimum is recognized in the period in which the royalties become probable and estimable, which is typically in the period corresponding with the respective sales. The Company records an offsetting increase to the cost basis of the asset under the cost accumulation model. The additional cost basis is subsequently amortized over the remaining life of the license agreement.
The Company paid Mission an up-front license fee of $3$3.0 million and during the term of the agreement will pay guaranteed minimum royalties during each calendar year the greater of $2$2.0 million, representing the guaranteed minimum royalty, or twenty percent (20%)20% of the Company’s net sales of Thiola through May 28, 2024.during each calendar year.
In November 2017, the Company amended its agreement with Mission to extend the term of the current exclusive U.S. and Canada licensing agreement by an additional five years, to 2029. The royalty rate and guaranteed minimum payment were also extended through the new agreement term. Upon execution of the amendment, the Company capitalized an additional $5.9 million in intangible assets and recorded a guaranteed minimum liability for the same amount.
In November 2018, the Company amended its agreement with Mission to remove all territorial restrictions on our license. As consideration for the expanded territory, the Company paid an up-front fee of $0.3 million and will pay guaranteed minimum royalties equaling the greater of $0.1 million, representing the guaranteed minimum, or 20% of our Thiolathe Company's net sales of Thiola generated outside of the United States during each calendar year. Upon execution of the amendment, the Company capitalized an additional $1.0 million in intangible assets and recorded a guaranteed minimum liability of $0.7 million related to this amendment.
The present value of guaranteed minimum royalties payable using a discount rate of ranging from approximately 7% to 11% based on the Company’s then borrowing rate is $12.3$9.7 million and $13.4$11.1 million as of December 31, 20212023 and 2020,2022, respectively. As of December 31, 2021,2023, the guaranteed
minimum royalty current and long-term liability was approximately $2.1 million and $7.6 million, respectively, and is recorded as Other Liability in the Consolidated Balance Sheets. As of December 31, 2022, the guaranteed minimum royalty current and long-term liability was approximately $2.1 million and $10.2$9.0 million, respectively, and is recorded as Other Liability in the Consolidated Balance Sheet. As of December 31, 2020, the guaranteed minimum royalty current and long-term liability was approximately $2.1 million and $11.3 million, respectively, and is recorded as Other Liability in the Consolidated Balance Sheet.Sheets. The Company has capitalized $123.3$155.6 million related to the Thiola intangible asset which consists of the up-front license fee, professional fees, present value of the guaranteed minimum royalties and any additional payments through 2021December 31, 2023 in excess of minimum royalties. In 20212023 the Company added $19.4$16.2 million to the intangible asset related to the royalties in excess of the minimum. Consistent with all prior periods since Thiola was acquired, the Company has not accrued any liability for royalties in excess of the annual contractual minimum at December 31, 2021,2023, as such royalties are not yet probable and estimable.
There are 7.4 years remaining Amortizable intangible assets as of December 31, 2023 (in the term of the license agreement.thousands):
Cholbam Asset Purchase
On March 31, 2015, the Company completed its acquisition from Asklepion of all worldwide rights, titles and ownership of Cholbam, including all related contracts, data assets, intellectual property, regulatory assets and the PRV. The Company capitalized $75.9 million and $7.3 million for the U.S. and international economic interest, respectively. | | | | | | | | | | | | | | | | | | | | | | | |
| | Useful Life | | Gross Carrying Amount | | Accumulated Amortization | | Net Book Value |
| Thiola license | 12 | | $ | 155,589 | | | $ | (77,311) | | | $ | 78,278 | |
| Ligand license | 11 | | 27,448 | | | (2,036) | | | 25,412 | |
| Total amortizable intangible assets | | | $ | 183,037 | | | $ | (79,347) | | | $ | 103,690 | |
Amortizable intangible assets as of December 31, 20212022 (in thousands):
| | | | | | | | | | | | | | | | | | | | |
| | Useful Life | Gross Carrying
Amount | | Accumulated
Amortization | | Net Book Value |
| Thiola License | 15 | $ | 123,316 | | | $ | (37,992) | | | $ | 85,324 | |
| Chenodal Product Rights | 16 | 67,849 | | | (32,938) | | | 34,911 | |
| Economic Interest - U.S. revenue Cholbam | 10 | 75,900 | | | (51,258) | | | 24,642 | |
| Ligand License | 11 | 7,900 | | | (5,875) | | | 2,025 | |
| Economic Interest - International revenue Cholbam | 10 | 7,982 | | | (7,483) | | | 499 | |
| Manchester Customer Relationships | 10 | 403 | | | (313) | | | 90 | |
| Internal use software | 5 | 207 | | | (199) | | | 8 | |
| Total | | $ | 283,557 | | | $ | (136,058) | | | $ | 147,499 | |
Amortizable intangible assets as of December 31, 2020 (in thousands):
| | | | | | | | | | | | | | | | | | | | |
| | Useful Life | Gross Carrying
Amount | | Accumulated
Amortization | | Net Book Value |
| Thiola License | 15 | $ | 103,992 | | | $ | (28,117) | | | $ | 75,875 | |
| Chenodal Product Rights | 16 | 67,849 | | | (28,700) | | | 39,149 | |
| Economic Interest - U.S. revenue Cholbam | 10 | 75,900 | | | (43,675) | | | 32,225 | |
| Ligand License | 11 | 7,900 | | | (4,717) | | | 3,183 | |
| Economic Interest - International revenue Cholbam | 10 | 8,250 | | | (5,673) | | | 2,577 | |
| Manchester Customer Relationships | 10 | 403 | | | (273) | | | 130 | |
| Internal use software | 5 | 207 | | | (157) | | | 50 | |
| Manchester trade name | 1 | 175 | | | (175) | | | — | |
| Total | | $ | 264,676 | | | $ | (111,487) | | | $ | 153,189 | |
| | | | | | | | | | | | | | | | | | | | | | | |
| Useful Life | | Gross Carrying Amount | | Accumulated Amortization | | Net Book Value |
| Thiola license | 12 | | $ | 139,383 | | | $ | (50,324) | | | $ | 89,059 | |
| Ligand license | 11 | | 19,400 | | | (12,139) | | | 7,261 | |
| Total amortizable intangible assets | | | $ | 158,783 | | | $ | (62,463) | | | $ | 96,320 | |
The following table summarizes amortization expense for the twelve monthsyear ended December 31, 2021, 20202023, 2022 and 20192021 (in thousands):
| | | | | | | | | | | | | | | | | |
| 2021 | | 2020 | | 2019 |
| Research and development | $ | 1,158 | | | $ | 1,162 | | | $ | 1,158 | |
| Selling, general and administrative | 23,788 | | | 21,275 | | | 18,549 | |
| Total amortization expense | $ | 24,946 | | | $ | 22,437 | | | $ | 19,707 | |
| | | | | | | | | | | | | | | | | |
| 2023 | | 2022 | | 2021 |
| Selling, general and administrative | $ | 29,021 | | | $ | 12,339 | | | $ | 9,888 | |
| Research and development | 7,261 | | | 6,264 | | | 1,158 | |
| Total amortization expense | $ | 36,282 | | | $ | 18,603 | | | $ | 11,046 | |
As of December 31, 2021,2023, amortization expense for the next five years and thereafter is expected to be as follows (in thousands):
| | 2022 | $ | 25,040 | |
| 2023 | 24,241 | |
| 2024 | 2024 | 23,408 | |
| 2025 | 2025 | 17,621 | |
| 2026 | 2026 | 15,751 | |
| 2027 | |
| 2028 | |
| Thereafter | Thereafter | 41,438 | |
| Total | Total | $ | 147,499 | |
Goodwill
As of December 31, 20212023 and 2020,2022, the Company had goodwill of $0.9$0.8 million.
NOTE 10. ACCRUED EXPENSES
Accrued expenses consist of the following at December 31, 20212023 and 20202022 (in thousands):
| | 2021 | | 2020 |
| 2023 | | | 2023 | | 2022 |
| Compensation related costs | |
| Research and development | Research and development | $ | 26,841 | | | $ | 10,166 | |
| Compensation related costs | 25,998 | | | 17,912 | |
| Accrued royalties and contingent consideration | 8,402 | | | 7,857 | |
| Government rebates payable | 7,493 | | | 10,707 | |
| Sales discounts, rebates, and allowances | |
| Transition services accrual | |
| Accrued restructuring costs | |
| Selling, general and administrative | |
| Accrued royalties | |
| Miscellaneous accrued | Miscellaneous accrued | 3,302 | | | 6,207 | |
| Selling, general and administrative | 3,144 | | | 3,944 | |
| Total accrued expenses | Total accrued expenses | $ | 75,180 | | | $ | 56,793 | |
NOTE 11. COMMITMENTS AND CONTINGENCIES
Commitments
Certain of the Company's contractual arrangements with contract manufacturing organizations ("CMOs") require binding forecasts or commitments to purchase minimum amounts for the manufacture of drug product supply, which may be material to the Company's financial statements.
Contingencies
In October 2021, our Kolbam distributor in France notified us that the French authorities are asking for reimbursement for a portion of Kolbam sales in France during the periods from 2015-2020. During this period,November 2020, the Company hadcompleted the acquisition of Orphan Technologies Limited (“Orphan”), including Orphan’s rare metabolic disorder drug pegtibatinase. The Company acquired Orphan by purchasing all of the outstanding shares. Under the Agreement, the Company has also agreed to make contingent cash payments up to an aggregate revenues fromof $427.0 million based on the achievement of certain development, regulatory and commercialization events as set forth in the Agreement, as well as additional tiered mid-single digit royalty payments based upon future net sales of Kolbamany pegtibatinase products in Francethe US and Europe, subject to certain reductions as set forth in the Agreement, and a contingent payment in the event a pediatric rare disease voucher for any pegtibatinase product is granted. In accordance with ASC 450, contingent cash payments will be accrued for when it is probable that a liability has been incurred and the amount can be reasonably estimated. As of December 31, 2023, no contingent cash payments have been accrued.
The Company anticipates that it may achieve certain milestones with both FILSPARI and pegtibatinase that will result in the Company making net payments of approximately $8.0 million. At this time,$50.0 million in the Company is not able to estimate the potential liability that may be incurred, if any.next 12 months.
Legal Proceedings
From time to time in the normal course of business, the Company is subject to various legal matters such as threatened or pending claims or litigation. Although the results of claims and litigation cannot be predicted with certainty, the Company does not believe it is a party to any claim or litigation in which the outcome, of which, if determined adversely to it, would individually or in the aggregate be reasonably expected to have a material adverse effect on its results of operations or financial condition.
NOTE 12. STOCKHOLDERS’ EQUITY
Common Stock
The Company is currently authorized to issue up to 200,000,000 shares of $0.0001 par value common stock. All issued shares of common stock are entitled to vote on a 1 share/1 vote basis.
Preferred Stock
The Company is currently authorized to issue up to 20,000,000 shares of $0.0001 par value preferred stock, of which 1,000 shares are designated Class "A" Preferred shares. Class A Preferred Shares are not entitled to interest, have certain liquidation preferences, special voting rights and other provisions. No preferred stock has been issued to date.
20152018 Equity Incentive Plan
On June 8, 2015,The Company's 2018 Equity Incentive Plan (the "2018 Plan") is the successor to and continuation of the Company's stockholders approved the 2015 Equity Incentive Plan (the "2015 Plan"). The 2015 and the Company's 2014 Equity Incentive Plan is intended as the successor to(the "2014 Plan", and continuation of the Company’s 2014 Incentive Compensation Plan. Stockholders approved 1.4 million new shares to be issued undertogether with the 2015 Plan, in addition to 0.6 million unallocatedthe "Prior Plans"). Unallocated shares remainingunder the Prior Plans are no longer available for issuance under the 2014 Incentive Compensation Plan that werePrior Plans, and have instead been added to the 2015 Plan. Options issued under the 2015 Plan will generally expire ten years from the date of grant and vest over a three-year or four-year period.
On May 18, 2016, the Company's stockholders approved an amendment to the 2015 Plan (the "Amended 2015 Plan"). The amendment provides for an additional 1.6 million new shares to be issued under the Amended 2015 Plan, in addition to 0.7 million unallocated shares remaining available for issuance. The amendment also includes a provision that on or after March 21, 2016, the number of shares available for issuance under the Amended 2015 Plan will be reduced by 1 share for each share subject to a stock option or stock appreciation right and by 2.0 shares for each share subject to any other type of stock award issued pursuant to the Amended 2015 Plan, and any such shares will return to the share reserve at the same rates upon cancellation or other forfeiture of such awards or shares.
On May 17, 2017, the Company's stockholders approved an amendment to the Amended 20152018 Plan. The amendment2018 Plan, as amended, and including the unallocated shares of the Prior Plans, provides for an additional 1.8a total of 15.7 million new shares to be issued, underplus the Amended 2015 Plan.
2018 Equity Incentive Plan
On May 9, 2018, the Company's stockholders approved the 2018 Equity Incentive Plan (the "2018 Plan"). The 2018 Plan is intended as the successor to and continuation of the Amended 2015 Plan. Stockholders approved 1.8 million newPrior Plans' returning shares, to be issuedif any, which become available for grant under the 2018 Plan in additionfrom time to 1.6 million unallocated shares remaining available for issuance under the Amended 2015 Plan that were added to the 2018 Plan.time. Options issued under the 2018 Plan will generally expire ten years from the date of grant and vest over a four-year period.
On May 9, 2019, the Company’s stockholders approved an amendment to the 2018 Plan that increased the number As of authorizedDecember 31, 2023, there were approximately 4.5 million shares issuablereserved for future issuance under the 2018 Plan by 2.0 million shares.
On May 15, 2020, the Company's stockholders approved an amendment to the 2018 Plan that increased the number of authorized shares issuable under the 2018 Plan by 2.4 million shares.
On May 14, 2021, the Company’s stockholders approved an amendment to the 2018 Plan that increased the number of authorized shares issuable under the 2018 Plan by 3.2 million shares.Plan.
2017 Employee Stock Purchase Plan
The 2017 Employee Stock Purchase Plan ("2017 ESPP") originated with 380,000 shares of common stock available for issuance. Beginning on January 1, 2018, and ending on (and including) January 1, 2026, the number of shares of common stock available for issuance under the 2017 ESPP shallmay increase by an amount equal to the lesser of (i) one percent (1%) of the total number of shares of common stock outstanding on December 31st of the preceding calendar year andor (ii) 300,000 shares of common stock.
Substantially all employees are eligible to participate in the 2017 ESPP and, through payroll deductions, can purchase shares on established dates semi-annually. The purchase price per share sold pursuant to the 2017 ESPP will be the lower of (i) 85% of the fair market value of common stock on the first day of the offering period or (ii) 85% of the fair market value on the purchase date. Each offering period will span up to six months. Purchases may be up to 15% of qualified compensation, with an annual limit of $25,000. The 2017 ESPP is intended to qualify as an “employee stock purchase plan” under Section 423 of the Internal Revenue Code.
As of December 31, 2021,2023, there were approximately 1,580,0002,180,000 shares authorized and 989,0521,100,657 shares reserved for future issuance under the 2017 ESPP.
Stock Options
The fair values of stock option grants during the years ended December 31, 2021, 20202023, 2022 and 20192021 were calculated on the date of grant using the Black-Scholes option pricing model. Compensation expense is recognized over the period of service, generally the vesting period. During the year ended December 31, 2021, 1,850,407 stock options were granted by the Company. The following weighted average assumptions were used in the Black-Scholes options pricing model to estimate the fair value of stock options for the specified reporting periods:
| | Twelve Months Ended December 31, |
| Year Ended December 31, | | | Year Ended December 31, |
| | | 2021 | | 2020 | | 2019 | | 2023 | | 2022 | | 2021 |
| Risk free rate | Risk free rate | 0.6 | % | | 1.5 | % | | 2.3 | % | Risk free rate | 3.9 | % | | 1.7 | % | | 0.6 | % |
| Expected volatility | Expected volatility | 59 | % | | 64 | % | | 68 | % | Expected volatility | 50 | % | | 50 | % | | 59 | % |
| Expected life (in years) | Expected life (in years) | 6.4 | | 6.3 | | 6.2 | Expected life (in years) | 6.4 | | 6.4 |
| Expected dividend yield | Expected dividend yield | — | | | — | | | — | |
The risk-free interest rate was based on rates established by the Federal Reserve. The Company’s expected volatility was based on analysis of the Company’s historical volatility. In years prior to 2020, the assessment also took into account the volatilities of guideline companies due to limited history of the Company's own activity. The expected life of the Company’s options was determined using the Company's historical exercise activity. In years prior to 2020, the Company applied the simplified method as a result of limited historical data regarding the Company’s exercise activity. The dividend yield is based upon the fact that the Company has not historically paid dividends and does not expect to pay dividends in the foreseeable future.
The following table summarizes our stock option activity and related information for the year ended December 31, 2021:2023:
| | | | | Weighted Average | | |
| | Shares Underlying Options | | Exercise Price | | Remaining Contractual Term (in years) | | Aggregate Intrinsic Value (in thousands) |
| Outstanding at December 31, 2020 | 8,242,996 | | | $ | 18.97 | | | 6.43 | | $ | 71,641 | |
| | | Weighted Average | |
| Shares Underlying
Options | |
| Shares Underlying
Options | |
| Shares Underlying
Options | | | Exercise
Price | | Remaining
Contractual
Term (in years) | | Aggregate
Intrinsic Value
(in thousands) |
| Outstanding at December 31, 2022 | |
| Granted | Granted | 1,850,407 | | | 25.32 | | | — | | | — | |
| Forfeited and expired | Forfeited and expired | (427,289) | | | 24.37 | | | — | | | — | |
| Exercised | Exercised | (779,830) | | | 16.47 | | | — | | | 7,538 | |
| Outstanding at December 31, 2021 | 8,886,284 | | | $ | 20.26 | | | 6.27 | | $ | 96,577 | |
| Outstanding at December 31, 2023 | |
| Vested and expected to vest at December 31, 2023 | |
The aggregate intrinsic value of stock options exercised in the years ended December 31, 2023, 2022 and 2021 was $1.4 million, $5.1 million, and $7.5 million, respectively.
The following table summarizes our stock options exercisable at December 31, 2021, 20202023, 2022 and 2019:2021:
| | | | | | | | | | | | | | | | | | | | | | | |
| | | | Weighted Average | | |
| Shares Underlying Options | | Exercise Price | | Remaining Contractual Term (in years) | | Aggregate Intrinsic Value (in thousands) |
| 2019 | 4,936,995 | | | $ | 18.93 | | | 5.62 | | $ | 4,490 | |
| 2020 | 5,488,207 | | | $ | 19.37 | | | 5.36 | | $ | 46,626 | |
| 2021 | 6,011,349 | | | $ | 19.34 | | | 5.16 | | $ | 71,063 | |
| | | | | | | | | | | | | | | | | | | | | | | |
| | | Weighted Average | | |
| Shares Underlying
Options | | Exercise
Price | | Remaining
Contractual
Term (in years) | | Aggregate
Intrinsic Value
(in thousands) |
| Exercisable at December 31, 2021 | 6,011,349 | | | $ | 19.34 | | | 5.16 | | $ | 71,063 | |
| Exercisable at December 31, 2022 | 6,999,669 | | | $ | 20.22 | | | 4.62 | | $ | 21,806 | |
| Exercisable at December 31, 2023 | 7,942,964 | | | $ | 20.85 | | | 3.95 | | $ | — | |
The weighted average grant date fair value of options granted was $14.00, $9.54,$11.66, $13.45, and $12.11$14.00 during the years ended December 31, 2021, 20202023, 2022 and 2019,2021, respectively. The aggregate intrinsic value for outstanding options is calculated as the difference between the exercise price of the underlying awards and the closing price of the Company’s common stock of $31.04, $27.25$8.99, $21.03 and $14.20$31.04 as of December 31, 2021, 20202023, 2022 and 2019,2021, respectively. Unrecognized compensation cost associated with unvested stock options amounts to $30.0$24.1 million as of December 31, 2021,2023, which will be expensed over a weighted average remaining vesting period of 2.52.3 years.
In connection with the retirement of the Company's former Chief Financial Officer, the Board of Directors approved a modification to extend the deadline to exercise each stock option held to the earlier of three months following the last vesting date or the original expiration date of the option, and to continue vesting on the original schedule of any underlying unvested stock options and restricted stock units. The modification resulted in incremental compensation cost of $2.6 million for the year ended December 31, 2023.
Restricted Stock Units
As of December 31, 2021,2023, there was approximately $23.2$46.8 million of unrecognized compensation cost related to restricted stock units ("RSUs") granted. This amount is expected to be recognized over a weighted average period of 2.42.5 years.
The following table summarizes our restricted stock unit activity for the year ended December 31, 2021:2023:
| | | Number of RSUs | | Weighted Average Grant Date Fair Value |
| Unvested December 31, 2020 | 1,109,942 | | | $ | 17.84 | |
| Number of
RSUs | | | Number of
RSUs | | Weighted Average
Grant Date Fair Value |
| Unvested December 31, 2022 | |
| Granted | Granted | 911,523 | | | 25.06 | |
| Vested | Vested | (408,136) | | | 18.08 | |
| Forfeited/cancelled | Forfeited/cancelled | (139,380) | | | 21.62 | |
| Unvested December 31, 2021 | 1,473,949 | | | $ | 21.88 | |
| Unvested December 31, 2023 | |
The fair value of restricted stock units vested for the years ended December 31, 2023, 2022 and 2021 was $18.7 million, $11.5 million, and $7.4 million, respectively. The weighted average grant date fair value for stock awards granted during the years ended December 31, 2023, 2022 and 2021 was $23.30, $21.21, and $18.08, respectively.
Performance-based Stock Units
PSUsPerformance-based stock units ("PSUs") are subject to vest only if certain specified criteria are achieved. As of December 31, 2021,2023, there was less than $0.1approximately $2.0 million of unrecognized compensation cost related to performance-based stock units ("PSUs") granted.PSUs granted and deemed probable of vesting. This amount is expected to be recognized over a weighted average period of 0.50.9 years.
The following table summarizes our performance-based stock unit activity for the year ended December 31, 2021:2023:
| | | | Number of PSUs | | Weighted Average Grant Date Fair Value | | Number of
PSUs | | Weighted Average
Grant Date Fair Value |
| Unvested December 31, 2020 | 167,500 | | | $ | 16.48 | |
| Unvested December 31, 2022 | |
| Granted | Granted | — | | | — | |
| Vested | Vested | (115,000) | | | 15.46 | |
| Forfeited/cancelled | Forfeited/cancelled | — | | | — | |
| Unvested December 31, 2021 | 52,500 | | | $ | 18.73 | |
| Unvested December 31, 2023 | |
The fair value of PSUs vested for the years ended December 31, 2023, 2022 and 2021 was $0.3 million, $0.4 million, and $1.8 million, respectively. The weighted average grant date fair value for performance-based stock awards granted during the years ended December 31, 2023, 2022 and 2021 was $15.46, $25.26, and $15.46, respectively.
Share Based Compensation
Total non-cash stock-based compensation expense consisted of the following for the years ended December 31, 2021, 20202023, 2022 and 20192021 (in thousands):
| | | | Twelve Months Ended December 31, | | Year Ended December 31, |
| | | 2021 | | 2020 | | 2019 | | 2023 | | 2022 | | 2021 |
| Selling, general and administrative expenses | Selling, general and administrative expenses | $ | 18,134 | | | $ | 14,247 | | | $ | 14,195 | |
| Research and development expenses | Research and development expenses | 12,632 | | | 9,367 | | | 6,910 | |
| Total | $ | 30,766 | | | $ | 23,614 | | | $ | 21,105 | |
| Total stock-based compensation expense | |
NOTE 13. NET LOSS PER COMMON SHARE
Basic and diluted net lossincome (loss) per common share is calculated by dividing net loss applicable to common stockholders by the weighted-average number of common shares outstanding during the period. In accordance with ASC 260, Earnings per Share, if a company had a discontinued operation, the company uses income from continuing operations, adjusted for preferred dividend and similar adjustments, as its control number to determine whether potential common shares are dilutive.
As discussed in Note 15, as part of its February 2023 underwritten public offering, the Company issued and sold pre-funded warrants to purchase 1.25 million shares of its common stock at a price to the public of $20.9999 per pre-funded warrant. The pre-funded warrants are exercisable immediately and are exercisable for one share of the Company's common stock. The exercise price of each pre-funded warrant is $0.0001 per share of common stock. Since the $0.0001 price per share represents little consideration and is non-substantive in relation to the $20.9999 price per pre-funded warrant and the $21.00 price per share of the common stock offered to the public, and as the warrants are immediately exercisable with no further vesting conditions or contingencies associated with them, the shares underlying the warrants are therefore included in the calculation of basic net loss per common share.
The Company’s potentially dilutive shares, which include outstanding stock options, restricted stock units, performance-based stock units, and shares issuable upon conversion of the 2025 Notes and 2029 Notes, are considered to be common stock equivalents and are not included in the calculation of diluted net loss per share because their effect is anti-dilutive.
Basic and diluted net loss per share is calculated as follows (net loss amounts are stated in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| For the year ended December 31, |
| 2021 | | 2020 | | 2019 |
| Shares | | Net loss | | EPS | | Shares | | Net loss | | EPS | | Shares | | Net loss | | EPS |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| Basic and diluted loss per share | 59,832,287 | | | $ | (180,091) | | | $ | (3.01) | | | 47,539,631 | | | $ | (169,431) | | | $ | (3.56) | | | 42,339,961 | | | $ | (146,427) | | | $ | (3.46) | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| For the year ended December 31, |
| 2023 | | 2022 | | 2021 |
| Shares | | Net (loss) income | | EPS | | Shares | | Net (loss) income | | EPS | | Shares | | Net (loss) income | | EPS |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| Continuing operations | 74,267,418 | | | $ | (376,333) | | | $ | (5.07) | | | 63,758,515 | | | $ | (331,468) | | | $ | (5.20) | | | 59,832,287 | | | $ | (217,292) | | | $ | (3.63) | |
| Discontinued operations | 74,267,418 | | | $ | 264,934 | | | $ | 3.57 | | | 63,758,515 | | | $ | 52,986 | | | $ | 0.83 | | | 59,832,287 | | | $ | 37,201 | | | $ | 0.62 | |
| Basic and diluted loss per share | 74,267,418 | | | $ | (111,399) | | | $ | (1.50) | | | 63,758,515 | | | $ | (278,482) | | | $ | (4.37) | | | 59,832,287 | | | $ | (180,091) | | | $ | (3.01) | |
For the years ended December 31, 2021, 20202023, 2022 and 2019,2021, the following sharesweighted-average number of common stock equivalents were excluded because they were anti-dilutive:
| | For the year ended December 31, |
| 2021 | | 2020 | | 2019 |
| For the year ended December 31, | | | For the year ended December 31, |
| 2023 | | | 2023 | | 2022 | | 2021 |
| Convertible debt | Convertible debt | 7,113,402 | | | 7,113,402 | | | 7,632,414 | |
| Restricted stock | 1,569,470 | | | 1,368,096 | | | 599,298 | |
| Options | Options | 9,214,188 | | | 8,349,310 | | | 7,644,251 | |
| | Restricted stock units and performance-based stock units | |
| Total anti-dilutive shares | Total anti-dilutive shares | 17,897,060 | | | 16,830,808 | | | 15,875,963 | |
NOTE 14. INCOME TAXES
For financial reporting purposes, net loss from continuing operations before income taxes includes the following components (in thousands):
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2021 | | 2020 | | 2019 |
| United States | $ | (124,132) | | | $ | (65,881) | | | $ | (129,452) | |
| Foreign* | (55,550) | | | (122,909) | | | (16,996) | |
| Total | $ | (179,682) | | | $ | (188,790) | | | $ | (146,448) | |
*Foreign losses in 2020 include charges relate to IPR&D. See Note 5 for further discussion. | | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2023 | | 2022 | | 2021 |
| United States | $ | (293,283) | | | $ | (268,887) | | | $ | (161,333) | |
| Foreign | (82,827) | | | (62,268) | | | (55,550) | |
| Total | $ | (376,110) | | | $ | (331,155) | | | $ | (216,883) | |
The components of the provision (benefit) for income taxes, in the Consolidated Statements of Operations are as follows (in thousands):
| | | | 2021 | | 2020 | | 2019 | | 2023 | | 2022 | | 2021 |
| Current | Current | | | | | |
| Federal | Federal | $ | — | | | $ | (19,436) | | | $ | (319) | |
| Federal | |
| Federal | |
| State | State | 412 | | | 74 | | | 298 | |
| Foreign | Foreign | (3) | | | 3 | | | — | |
| | 409 | | | (19,359) | | | (21) | |
| 223 | |
| Deferred | Deferred | | |
| Federal | Federal | — | | | — | | | — | |
| Federal | |
| Federal | |
| State | State | — | | | — | | | — | |
| — | | | — | | | — | |
| Total tax provision (benefit) | $ | 409 | | | $ | (19,359) | | | $ | (21) | |
| — | |
| Total tax provision | |
The following is a reconciliation of the statutory federal income tax rate to the Company’s effective tax rate expressed as a percentage of loss before income taxes:
| | | | 2021 | | 2020 | | 2019 | | 2023 | | 2022 | | 2021 |
| Statutory rate - federal | Statutory rate - federal | (21.00) | % | | (21.00) | % | | (21.00) | % | Statutory rate - federal | (21.00) | % | | (21.00) | % | | (21.00) | % |
| State taxes, net of federal benefit | State taxes, net of federal benefit | (2.71) | % | | (1.57) | % | | (4.35) | % | State taxes, net of federal benefit | (3.19) | % | | (3.62) | % | | (2.92) | % |
| Foreign rate differential | Foreign rate differential | 3.28 | % | | 6.65 | % | | — | % | Foreign rate differential | 1.29 | % | | 0.81 | % | | 2.72 | % |
| IPR&D | — | % | | 6.50 | % | | — | % |
| Federal IRS Audit settlement | — | % | | 1.62 | % | | — | % |
| | Nondeductible executive compensation | |
| | Nondeductible executive compensation | |
| | Nondeductible executive compensation | Nondeductible executive compensation | 0.48 | % | | — | % | | — | % | 1.50 | % | | 0.41 | % | | 0.40 | % |
| Excess tax benefits associated with share-based awards | Excess tax benefits associated with share-based awards | 0.19 | % | | — | % | | — | % | Excess tax benefits associated with share-based awards | 0.68 | % | | (0.17) | % | | 0.15 | % |
| Other permanent differences | Other permanent differences | 0.15 | % | | 0.09 | % | | 0.24 | % | Other permanent differences | 0.37 | % | | 0.17 | % | | 0.12 | % |
| Tax credits | Tax credits | (10.87) | % | | (4.49) | % | | (15.17) | % | Tax credits | (1.13) | % | | (5.29) | % | | (8.65) | % |
| Return to provision adjustments and other true-ups | Return to provision adjustments and other true-ups | 2.42 | % | | 4.88 | % | | 0.44 | % | Return to provision adjustments and other true-ups | 4.19 | % | | (0.17) | % | | 2.00 | % |
| CARES Act-Net operating loss carryback | — | % | | (9.58) | % | | — | % |
| | Adoption of ASU 2020-06 | |
| Adoption of ASU 2020-06 | |
| Adoption of ASU 2020-06 | | — | % | | (3.54) | % | | — | % |
| Other | Other | 0.20 | % | | (0.10) | % | | 2.32 | % | Other | 0.57 | % | | — | % | | 0.18 | % |
| Change in valuation allowance | Change in valuation allowance | 28.09 | % | | 6.75 | % | | 37.50 | % | Change in valuation allowance | 16.78 | % | | 32.49 | % | | 27.19 | % |
| Income tax provision (benefit) | Income tax provision (benefit) | 0.23 | % | | (10.25) | % | | (0.02) | % | Income tax provision (benefit) | 0.06 | % | | 0.09 | % | | 0.19 | % |
The significant components of the Company’s deferred tax assets and liabilities as of December 31, 20212023 and 20202022 are as follows (in thousands):
| | | | 2021 | | 2020 | | 2023 | | 2022 |
| Deferred Tax Assets: | Deferred Tax Assets: | | | |
| Net operating loss | Net operating loss | $ | 38,618 | | | $ | 19,114 | |
| Net operating loss | |
| Net operating loss | |
| Research and development and other tax credits | Research and development and other tax credits | 69,213 | | | 46,294 | |
| Capitalized research and development | |
| Contingent consideration | Contingent consideration | 16,726 | | | 16,311 | |
| Other accrued expenses | Other accrued expenses | 8,678 | | | 7,489 | |
| Stock based compensation | Stock based compensation | 18,003 | | | 21,886 | |
| Charitable contributions | Charitable contributions | 3,427 | | | 2,575 | |
| Intangible assets greater than book | Intangible assets greater than book | 48,801 | | | 45,520 | |
| Interest expense limitation | Interest expense limitation | 2,802 | | | 1,659 | |
| Operating lease liabilities | Operating lease liabilities | 8,825 | | | 7,314 | |
| Tax basis depreciation greater than book depreciation | Tax basis depreciation greater than book depreciation | 13 | | | 76 | |
| Difference in basis of loan costs | |
| 296,473 | |
| Deferred Tax Liabilities: | |
| Operating lease right of use assets | |
| Operating lease right of use assets | |
| Operating lease right of use assets | |
| | Prepaid assets | |
| Prepaid assets | |
| Prepaid assets | |
| (5,793) | |
| | 215,106 | | | 168,238 | |
| Deferred Tax Liabilities: | | | |
| | Operating lease right of use assets | (5,782) | | | (6,544) | |
| Convertible debt | (11,020) | | | (13,868) | |
| | (16,802) | | | (20,412) | |
| | Net deferred tax assets before valuation allowance | |
| Net deferred tax assets before valuation allowance | |
| Net deferred tax assets before valuation allowance | Net deferred tax assets before valuation allowance | 198,304 | | | 147,826 | |
| Valuation allowance | Valuation allowance | (198,304) | | | (147,826) | |
| Total deferred tax assets | Total deferred tax assets | $ | — | | | $ | — | |
The Company has established a full valuation allowance against its U.S. federal, state, and foreign deferred tax assets due to the uncertainty surrounding the realization of such assets in future periods. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during the periods in which temporary differences become deductible. Management considers the scheduled reversal of deferred liabilities and tax planning strategies in making this assessment and evaluates the recoverability of the deferred tax assets as of each reporting date. At such time as it is determined that it is more likely than not that deferred assets are realizable, the valuation allowance will be reduced accordingly and recorded as a tax benefit.
The Company has recorded a valuation allowance of $198.3$290.7 million as of December 31, 20212023 to reflect the estimated amount of deferred tax assets that may not be realized. The Company increaseddecreased its valuation allowance by $50.5 million and $63.1$2.7 million for the yearsyear ended December 31, 2021 and 2020, respectively.
At2023, compared to a $95.1 million increase for the year ended December 31, 2021,2022.
As of December 31, 2023, the Company had available unused U.S. federal and state net operating loss (“NOL”) carryforwards of $85.8$154.3 million and $151.2$208.4 million, respectively, all of which are fully offset by a valuation allowance. Additionally, the Company had an interest expense limitation carryforward of $12.6$8.0 million that is fully offset by a valuation allowance. The federal NOL and interest expense limitation carryforward have an indefinite life. The state NOL carryforwards will begin to expire in 2022.2024 unless previously utilized, except for $39.8 million of the state net operating losses that have an indefinite carryforward period. In addition, at December 31, 2021,2023, the Company had federal orphan drug tax credit carryforwards of $61.7$86.8 million that begin to expire in 2035 unless utilized, federal research and development tax credit carryforwards of $5.4$4.8 million that begin to expire in 2033 unless utilized, state research and development tax credit carryforwards of $10.1$8.0 million that begin to expire in 2030 unless utilized and $1.3 million that have an indefinite carryforward period, and California Competes tax credit carryforwards of $2.0 million that begin to expire in 2022.2024. Pursuant to Internal Revenue Code Sections 382 and 383, use of the Company’s federal net operating loss and credit carryforwards may be limited upon a cumulative change in ownership of more than 50% within a three-year period.
At The Company completed a study to analyze whether any ownership changes had occurred through March 31, 2023, and determined no ownership changes occurred. The Company is in the process of updating its analysis of owner shifts to determine whether an ownership change occurred since March 31, 2023. Any change in ownership should not impact the utilization of tax attributes for the year ended December 31, 2021,2023, and due to the Company’s full valuation allowance any subsequent ownership changes will not impact the Company’s effective tax rate.
As of December 31, 2023, the Company had Irish NOL carryforwards of $15.9$16.1 million which are fully offset by a valuation allowance and have an indefinite life. The Company also had Swiss NOL carryforwards of $77.3$251.1 million which are fully offset by a valuation allowance and begin to expire in 20232024, as well as Federal Act on Tax Reform and AHV Financing (“TRAF”) cantonal tax benefits of $526.2 million which expire in 2029.
The Company accounts for uncertain tax benefits in accordance with the provisions of ASC 740-10 of the Accounting for Uncertainty in Income Taxes. As of December 31, 2021,2023, the Company had $7.8$22.9 million in unrecognized tax benefits, all of which $7.5 millionare related to orphan drug and research and development tax credits which wasand were recorded as a reduction to the deferred tax assets with a corresponding reduction in the Company’s valuation allowance of $7.5$22.9 million. To the extent unrecognized tax benefits are recognized at a time when a valuation allowance does not exist, the recognition of the $7.5$22.9 million tax benefit would reduce the effective tax rate.
The Company does not anticipate that the amount of unrecognized tax benefits as of December 31, 20212023 will change materially within the following 12 months.
A reconciliation of the Company's unrecognized tax benefits for the years 20212023 and 20202022 is provided in the following table (in thousands):
| | 2021 | | 2020 |
| 2023 | | | 2023 | | 2022 |
| Balance as of January 1: | Balance as of January 1: | $ | 5,193 | | | $ | 235 | |
| Increase in current period positions | Increase in current period positions | 2,255 | | | 1,187 | |
| Increase in prior period positions | |
| Decrease in prior period positions | Decrease in prior period positions | — | | | (235) | |
| Increase in prior period positions | 377 | | | 4,006 | |
| Decrease due to settlements with tax authorities | |
| Balance as of December 31: | Balance as of December 31: | $ | 7,825 | | | $ | 5,193 | |
The Company files income tax returns in the U.S. federal jurisdiction, various state and local, and foreign jurisdictions. With few exceptions related to the carryforward of unutilized tax attributes, the Company’s income tax returns are open to examination by federal authorities for the years ended December 31, 2018,2019, and forward, and for state and foreign tax authorities, for the years ended December 31, 2017,2014, and forward. The Company's Swiss income tax returns are open to examination for the years ended December 31, 2018, and forward and the Company's Irish tax returns are open to examination for the years ended December 31, 2019, and forward.
The Company recognizes interest and penalties as a component of income tax expense. Interest and penalties for the year ended December 31, 2021 were less than $0.1 million. The Company did not recognize any interest or penalties for the year ended December 31, 2023. Interest and penalties for the years ended December 31, 20202022 and 2019.2021 were less than $0.1 million.
NOTE 15. EQUITY OFFERINGS
Underwritten Public OfferingsOffering of Common Stock
In February 2021, we2023, the Company sold an aggregate of 7.5approximately 9.7 million shares of ourits common stock and pre-funded warrants to purchase 1.25 million shares of its common stock in an underwritten public offering, at a price to the public of $26.75$21.00 per share.share of common stock and $20.9999 per pre-funded warrant. The pre-funded warrants are exercisable immediately, subject to certain beneficial ownership limitations which can be modified by the respective holders with at least 61 days' notice, and are exercisable for one share of the Company's common stock. The exercise
price of each pre-funded warrant is $0.0001 per share of common stock. The net proceeds to usthe Company from the offering, after deducting the underwriting discounts and offering expenses, were $189.3approximately $215.8 million.
The pre-funded warrants were classified as a component of permanent stockholders' equity within additional paid-in capital and were recorded at the issuance date using a relative fair value allocation method. The pre-funded warrants are equity classified because they (i) are freestanding financial instruments that are legally detachable and separately exercisable from the equity instruments, (ii) are immediately exercisable, (iii) do not embody an obligation for the Company to repurchase its shares, (iv) permit the holders to receive a fixed number of shares of common stock upon exercise, (v) are indexed to the Company's common stock and (vi) meet the equity classification criteria. In June 2020,addition, such pre-funded warrants do not provide any guarantee of value or return. The Company valued the pre-funded warrants at issuance, concluding that their sale price approximated their fair value, and allocated the aggregate net proceeds from the sale proportionately to the common stock and prefunded warrants, including approximately $24.6 million allocated to the pre-funded warrants and recorded as a component of additional paid-in capital.
In February 2021, the Company sold an aggregate of 7.5 million shares of its common stock in an underwritten public offering, at a price to the public of $15.50$26.75 per share. The net proceeds to the Company from the offering, after deducting the underwriting discounts and offering expenses, were $108.7$189.3 million.
At-the-Market Equity Offering
In February 2020, the Company entered into an Open Market Sale Agreement ("ATM Agreement") with Jefferies LLC, as agent (“Jefferies”), pursuant to which the Company may offer and sell, from time to time through Jefferies, shares of its common stock having an aggregate offering price of up to $100.0 million. Of the $100.0 million originally authorized for sale under the ATM Agreement, approximately $28.6 million were sold under ourthe Company's prior registration statement on Form S-3 (Registration Statement No. 333-227182). An additional $31.7$51.9 million were sold under ourthe Company's effective registration statement on Form S-3 (Registration Statement No. 333-259311).
In 2020,, which included $20.1 million in the year ended December 31, 2022. The Company sold 867,806did not sell any shares under the ATM Agreement resulting in gross proceeds of $23.5 million. In 2021,during the Company sold a combined 1,240,640 shares under the ATM Agreement, resulting in gross proceeds of $36.8 million. Throughyear ended December 31, 2021, the Company has sold a total of 2,108,446 shares under the ATM Agreement, resulting in gross proceeds of $60.3 million.2023. As of December 31, 2021,2023, an aggregate amount of $39.7$19.5 million remained eligible for sale under the facility.
Authorized Shares of Common Stock
On May 14, 2021, in connection with the Company’s 2021 Annual Meeting of Stockholders, the Company’s stockholders approved, among other matters, a Certificate of Amendment (“Certificate of Amendment”) to the Company’s Certificate of Incorporation to increase the number of shares of common stock authorized for issuance thereunder from 100,000,000 to 200,000,000. Effective May 18, 2021, the Certificate of Amendment was filed with the Secretary of State of the State of Delaware.ATM Agreement.
NOTE 16. RETIREMENT PLAN
401(k) Savings Plan
The Company has a 401(k) defined contribution savings plan for the benefit of all eligible employees. Employer matching contributions were $1.3$2.7 million, $1.2$2.1 million, and $1.0$1.3 million for the years ended December 31, 2021, 20202023, 2022 and 2019,2021, respectively.
Switzerland Defined Benefit Plan
The Company maintains a defined benefit pension plan covering employees of its Swiss subsidiary, Travere Therapeutics Switzerland GmbH (the "Swiss Plan"). The Swiss Plan is a government-mandated retirement fund that provides employees with a minimum benefit. Employer and employee contributions are made to the Swiss Plan based on various percentages of participants' salaries and wages that vary according to the participants' age and other factors. As of December 31, 2023, the projected benefit obligations under the Swiss Plan were approximately $3.3 million, and plan assets were approximately $2.5 million. The funded status of the Swiss Plan is included in other long-term liabilities on the Company's Consolidated Balance Sheets.
NOTE 17. PROPERTY AND EQUIPMENT
Property, plant and equipment, net consisted of the following (in thousands):
| | December 31, |
| 2021 | | 2020 |
| December 31, | | | December 31, |
| 2023 | | | 2023 | | 2022 |
| Computers and equipment | Computers and equipment | $ | 1,950 | | | $ | 595 | |
| Furniture and fixtures | Furniture and fixtures | 2,783 | | | 1,791 | |
| Leasehold improvements | Leasehold improvements | 8,790 | | | 4,630 | |
| Construction-in-progress | Construction-in-progress | 631 | | | 6,339 | |
| 14,154 | | | 13,355 | |
| 14,922 | |
| Less: Accumulated depreciation | Less: Accumulated depreciation | (3,048) | | | (3,937) | |
| Total property and equipment, net | Total property and equipment, net | $ | 11,106 | | | $ | 9,418 | |
The construction-in-process balance consists of costs related to the Company’s leasehold improvements at its facilities in San Diego, California.
Non-cash investing activities for the years ending December 31, 2021 and 2020 included accrued capital expenditures of $0.1 million and $1.9 million, respectively. There were no accrued capital expenditures for the year ending December 31, 2019.
Depreciation expense for the years ended December 31, 2023, 2022 and 2021 2020 and 2019 was $1.7$2.2 million, $2.1 million and $0.7$1.7 million, respectively.
NOTE 18. LEASES
As of December 31, 2021,2023, the Company had 1two operating leases, including one operating lease with Kilroy Realty, L.P. (the "Landlord") for office space located in San Diego, California, which was entered into in April 2019 and subsequently amended in May 2020. Coinciding with ourthe Company's ability to direct the use of the office space which occurred in phases over 2020, and utilizing a discount rate equal to ourthe Company's estimated incremental borrowing rate, the Company established ROU assets totaling $34.6 million and lease liabilities totaling $34.5 million. The total ROU asset and lease liability at measurement were each offset by lease incentives associated with tenant improvement allowances totaling $7.9 million.
The initial term of the office lease ends in August 2028, and the Landlord has granted the Company an option to extend the term of the lease by a period of 5 years. At this time,lease inception, it iswas not reasonably certain that wethe Company will extend the term of the lease and therefore the renewal period has been excluded from the aforementioned ROU asset and lease liability measurements. The measurement of the lease term occurs from the February 2021 occupancy date of the office space.
The Company also has one operating lease with Esprit Investments Limited for office space deliveredlocated in September 2020.Dublin, Ireland, which was entered into in October 2022. The aggregate base rent due over the initial term of the office lease is approximately $49.5ends in September 2027. The lease provides the option to extend the term of the lease by a period of 5 years, although at lease inception, it was not reasonably certain that the Company will elect this option and therefore the renewal period has been excluded from the initial lease measurement. Utilizing a discount rate equal to the Company's borrowing rate, the Company established an ROU asset and corresponding lease liability of $0.4 million.
FollowingThe following is a schedule of the future minimum rental commitments for our operating leases reconciled to the lease liability and ROU asset as of December 31, 20212023 (in thousands):
| | December 31, 2021 |
| 2022 | $ | 6,020 | |
| 2023 | 6,200 | |
| December 31, 2023 | | | December 31, 2023 |
| 2024 | 2024 | 6,386 | |
| 2025 | 2025 | 6,578 | |
| 2026 | 2026 | 6,775 | |
| 2027 | |
| 2028 | |
| Thereafter | Thereafter | 11,760 | |
| Total undiscounted future minimum payments | Total undiscounted future minimum payments | 43,719 | |
| | Present value discount | Present value discount | (8,314) | |
| Total operating lease liability | 35,405 | |
| Total lease liability | |
| Unamortized lease incentives | Unamortized lease incentives | (6,559) | |
| Cash payments in excess of straight-line lease expense | Cash payments in excess of straight-line lease expense | (5,650) | |
| Total ROU asset | Total ROU asset | $ | 23,196 | |
The weighted-average remaining lease term and weighted-average discount rate of the Company's operating leases are as follows:
| | | | | | | | | | | |
| December 31, |
| 2023 | | 2022 |
| Weighted-average remaining lease term in years | 4.7 | | 5.7 |
| Weighted-average discount rate | 6.48 | % | | 6.48 | % |
For the twelve monthsyear ended December 31, 2021, 20202023, 2022 and 20192021 the Company recorded $4.9 million, $2.0$5.0 million, and $2.4$4.9 million, respectively, in expense related to operating leases, including amortized tenant improvement allowances.
NOTE 19. DIVESTITURES
Discontinued Operations
Sale of Bile Acid Product Portfolio
On August 31, 2023, the Company closed the sale of its bile acid business to Mirum Pharmaceuticals pursuant to the terms of the Purchase Agreement dated July 16, 2023 between the Company and Mirum. The assets sold consisted of substantially all of the assets primarily related to the Company’s business of development, manufacture (including synthesis, formulation, finishing or packaging) and commercialization of the products, Chenodal and Cholbam (also known as Kolbam). In connection with the Closing, the Company received an upfront cash payment of $210.0 million.
Pursuant to the Purchase Agreement, after the Closing, the Company is eligible to receive up to $235.0 million upon the achievement of certain milestones based on specified amounts of annual net sales (tiered from $125.0 million to $500.0 million) of the Products. The Company will recognize the contingent consideration receivable in earnings when the target annual sales for the milestones are met and the contingency is resolved.
The Company's sale of the bile acid business resulted in a gain, net of tax, of $226.0 million. The net gain consists of net consideration, including the upfront payment and the deduction of investment banker fees owed upon the Closing, plus the derecognition of the carrying value of the net liabilities included in the transaction and the immaterial tax due on the sale.
The Company and Mirum have also entered into a transition services agreement ("TSA") pursuant to which the Company has agreed to perform certain services for a period of time following the Closing, with respect to Mirum’s use and operation of the assets purchased in the Purchase Agreement. The TSA is designed to ensure and facilitate an orderly transfer of business operations, and the consideration to be received by the Company primarily consists of cost reimbursement. For the year ended December 31, 2023, the Company recognized $1.0 million, included in continuing operations within other income (expense), net, of which the Company collected $0.6 million as of December 31, 2023. As part of the TSA, the Company is collecting certain receivables related to purchased assets for a period of time and remitting them to Mirum. The transition services accrual as of December 31, 2023 was $12.3 million, and is included in accrued expenses in the accompanying consolidated balance sheets. TSA services provided by the Company are not anticipated to last beyond 12 months post-close.
The Company determined that the divestiture represents a strategic shift that will have a major effect on the Company's operations and financial results, and has therefore reflected the bile acid business as a discontinued operation for all periods presented.
Results of discontinued operations are as follows (in thousands):
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2023 | | 2022 | | 2021 |
| Net product sales | $ | 66,164 | | | $ | 102,558 | | | $ | 95,654 | |
| Total revenue | 66,164 | | | 102,558 | | | 95,654 | |
| Operating expenses: | | | | | |
| Cost of goods sold | 1,899 | | | 3,172 | | | 2,966 | |
| Research and development | 6,118 | | | 8,447 | | | 9,169 | |
| Selling, general and administrative | 19,500 | | | 22,686 | | | 23,599 | |
| Change in fair value of contingent consideration | (473) | | | 15,006 | | | 22,260 | |
| Total operating expenses | 27,044 | | | 49,311 | | | 57,994 | |
| Operating income | 39,120 | | | 53,247 | | | 37,660 | |
| Other income (expenses), net: | | | | | |
| Interest expense | (191) | | | (261) | | | (459) | |
| Gain on disposal of discontinued operations, net of tax | 226,005 | | | — | | | — | |
| Total other income (expense), net | 225,814 | | | (261) | | | (459) | |
| Income from discontinued operations before income tax | 264,934 | | | 52,986 | | | 37,201 | |
| Income tax on discontinued operations | — | | | — | | | — | |
| Net income from discontinued operations | $ | 264,934 | | | $ | 52,986 | | | $ | 37,201 | |
Supplemental cash flow information related to leases isAssets and liabilities of discontinued operations presented in the Consolidated Balance Sheets as followsof December 31, 2022 include the following (in thousands):
| | | | | | | | | | | | | | | | | |
| 2021 | | 2020 | | 2019 |
| Operating cash flows used for operating leases | $ | 2,205 | | | $ | 2,537 | | | $ | 2,639 | |
| | | | | | | | |
| | December 31, 2022 |
| Assets | | |
| Current assets: | | |
| Inventory | | $ | 2,399 | |
| Prepaid expenses and other current assets | | 591 | |
| Total current assets of discontinued operations | | 2,990 | |
| Intangible assets, net | | 47,965 | |
| Other assets | | 377 | |
| Total assets of discontinued operations | | $ | 51,332 | |
| | |
| Liabilities | | |
| Current liabilities: | | |
| Business combination-related contingent consideration, current portion | | $ | 7,000 | |
| Total current liabilities of discontinued operations | | 7,000 | |
| Business combination-related contingent consideration, less current portion | | 64,200 | |
| Total liabilities of discontinued operations | | $ | 71,200 | |
NOTE 20. QUARTERLY FINANCIAL INFORMATION (UNAUDITED)
The following table presents selected consolidated statements of operations data for each quarter for the fiscal years ended December 31, 2023 and 2022 (unaudited, in thousands, except for per share data):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Quarter | |
| | First | | | Second | | | Third | | | Fourth | |
| For the year ended December 31, 2023: | | | | | | | | | | | | |
| Total revenue | | $ | 30,888 | | | | $ | 32,196 | | | | $ | 37,095 | | | | $ | 45,059 | | |
| Total operating expenses | | 128,257 | | | | 136,147 | | | | 129,680 | | | | 139,292 | | |
| Operating loss | | (97,369) | | | | (103,951) | | | | (92,585) | | | | (94,233) | | |
| Loss from continuing operations before income tax provision | | (96,486) | | | | (101,867) | | | | (89,229) | | | | (88,528) | | |
| Loss from continuing operations, net of tax | | (96,564) | | | | (101,932) | | | | (89,241) | | | | (88,596) | | |
| Income (loss) from discontinued operations, net of tax | 1 | | 10,233 | | | | 16,302 | | | | 239,976 | | | | (1,577) | | |
| Net (loss) income | | (86,331) | | | | (85,630) | | | | 150,735 | | | | (90,173) | | |
| (Loss) income per common share - basic and diluted: | | | | | | | | | | | | |
| Continuing operations | | $ | (1.42) | | | | $ | (1.34) | | | | $ | (1.17) | | | | $ | (1.16) | | |
| Discontinued operations | | 0.15 | | | | 0.21 | | | | 3.14 | | | | (0.02) | | |
| Net (loss) income per common share | | $ | (1.27) | | | | $ | (1.13) | | | | $ | 1.97 | | | | $ | (1.18) | | |
| For the year ended December 31, 2022: | | | | | | | | | | | | |
| Total revenue | | $ | 23,412 | | | | $ | 28,634 | | | | $ | 28,075 | | | | $ | 29,339 | | |
| Total operating expenses | | 95,393 | | | | 107,160 | | | | 110,679 | | | | 116,041 | | |
| Operating loss | | (71,981) | | | | (78,526) | | | | (82,604) | | | | (86,702) | | |
| Loss from continuing operations before income tax provision | | (81,694) | | | | (79,970) | | | | (83,918) | | | | (85,573) | | |
| Loss from continuing operations, net of tax | | (81,746) | | | | (80,023) | | | | (84,063) | | | | (85,636) | | |
| Income from discontinued operations, net of tax | 1 | | 5,775 | | | | 12,991 | | | | 14,407 | | | | 19,813 | | |
| Net loss | | (75,971) | | | | (67,032) | | | | (69,656) | | | | (65,823) | | |
| (Loss) income per common share - basic and diluted: | | | | | | | | | | | | |
| Continuing operations | | $ | (1.29) | | | | $ | (1.25) | | | | $ | (1.31) | | | | $ | (1.33) | | |
| Discontinued operations | | 0.09 | | | | 0.20 | | | | 0.22 | | | | 0.30 | | |
| Net loss per common share | | $ | (1.20) | | | | $ | (1.05) | | | | $ | (1.09) | | | | $ | (1.03) | | |
| | | | | |
| 1 | On August 31, 2023, the Company sold its bile acid business, resulting in a gain, net of tax, of $226.0 million. The Company has reflected the bile acid business as a discontinued operation in the Consolidated Financial Statements for all periods presented. See Note 19 for results of discontinued operations and further discussion. |
NOTE 19.21. SUBSEQUENT EVENTS
At-the-Market Equity Offering
FromOn January 1, 2022 through February 22, 2022,25, 2024, the Company sold 701,600 sharesannounced that it has entered into an exclusive licensing agreement with Renalys Pharma, Inc. ("Renalys"), to bring sparsentan, a dual endothelin angiotensin receptor antagonist for the treatment of its common stock underIgAN, to patients in Japan and other countries in Asia. Renalys will hold regional rights to sparsentan for Japan, South Korea, Taiwan, Brunei, Cambodia, Indonesia, Laos, Malaysia, Myanmar, the ATM Agreement, resultingPhilippines, Singapore, Thailand, and Vietnam.
Under the terms of the agreement, Travere will receive an upfront payment and be eligible to receive up to $120 million in gross proceedsregulatory, development and sales-based milestone payments. As part of $20.1 million. After giving effectthe collaboration, Travere has obtained a minority equity stake in Renalys and will be eligible to suchreceive tiered royalties on net sales as of February 22, 2022, an aggregate amount of $19.6 million remained eligiblesparsentan in the licensed territories. Renalys will be responsible for sale underdevelopment, regulatory matters, and commercialization in the facility.licensed territories.