UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| (Mark One) | |||||
| ☒ | |||
| ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |||
For the fiscal year ended December 31, 20192023
OR
| ☐ | |||||
| TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |||||
For the transition period from _________ to _________
Commission File Number: 001-38984
CASTLE BIOSCIENCES, INC.
(Exact name of registrant as specified in its charter)
| Delaware | 77-0701774 | |||||||
| (State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) | |||||||
| 77546 | ||||||||
| (Address of principal executive offices) | (Zip Code) | |||||||
(866) 788-9007
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Trading Symbol(s) | Name of each exchange on which registered | ||||||
| Common Stock, $0.001 par value per share | CSTL | The Nasdaq Global Market | ||||||
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨☐ No x☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨☐ No x☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x☒ No ¨☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes x☒ No ¨☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of ‘‘large accelerated filer,’’ ‘‘accelerated filer,’’ ‘‘smaller reporting company,’’ and ‘‘emerging growth company’’ in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | Accelerated filer | ||||||||||
| Non-accelerated filer | Smaller reporting company | ||||||||||
| Emerging growth company | |||||||||||
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☒
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant's executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨☐ No x☒
The registrant’saggregate market value of voting and non-voting common stock was not publicly tradedequity held by non-affiliates of the registrant as of theJune 30, 2023 (the last business day of the registrant’s most recently completed second fiscal quarter.quarter) was $311 million based on the closing price of the registrant’s common stock on June 30, 2023, as reported by the Nasdaq Global Market.
As of February 28, 2020,21, 2024, there were 17,192,35127,449,983 shares of common stock, $0.001 par value $0.001 per share, issued and outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s definitive proxy statement to be filed with the Securities and Exchange Commission, or SEC, subsequent to the date hereof pursuant to Regulation 14A in connection with the registrant’s 20202024 Annual Meeting of Stockholders, are incorporated by reference into Part III of this Annual Report on Form 10-K. We intendThe registrant intends to file such proxy statement with the SEC not later than 120 days after the conclusion of the registrant’sits fiscal year ended December 31, 2019.
2023.
Table of Contents
| Page | |||||||||
| Item 1. | |||||||||
| Item 1A. | |||||||||
| Item 1B. | |||||||||
| Item | |||||||||
| Item 2. | |||||||||
| Item 3. | |||||||||
| Item 4. | |||||||||
| Item 5. | |||||||||
| Item 6. | |||||||||
| Item 7. | |||||||||
| Item 7A. | |||||||||
| Item 8. | |||||||||
| Item 9. | |||||||||
| Item | |||||||||
| Item 9B. | |||||||||
| Item 9C. | |||||||||
| Item 10. | |||||||||
| Item 11. | |||||||||
| Item 12. | |||||||||
| Item 13. | |||||||||
| Item 14. | |||||||||
| Item 15. | |||||||||
| Item 16. | |||||||||
F- | |||||||||
1
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains forward-looking statements.statements within the meaning of the Private Securities Litigation Reform Act of 1995 that are subject to risks and uncertainties. The forward-looking statements are contained principally in the sections entitled “Risk Factors,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and “Business.” These statements relate to future events or to our future financial performance and involve known and unknown risks, uncertainties and other factors which may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by the forward-looking statements. Forward-looking statements include, but are not limited to, statements about:
•estimates of our total addressable market (“TAM”), future revenue and addressable patient populations, expenses, capital requirements and our needs for additional financing;
•expectations with respect to reimbursement for our products, including third-party payor reimbursement and coverage decisions;
•anticipated cost, timing and success of our products in development,product candidates, and our plans to research, develop and commercialize new tests;
•the impact of geopolitical and macroeconomic developments, such as the Israel-Hamas war, and the ongoing conflict between Ukraine and Russia and related sanctions on our business;
•our ability to obtain funding for our operations, including funding necessary to complete the expansion of our operations and development of our product candidates;pipeline products;
•the implementation of our business model and strategic plans for our products, technologies and businesses;business;
•expectations with respect to acquisitions of businesses, assets, products or technologies;
•our ability to manage and grow our business by expanding our sales to existing customers, or introducing our products to new customers;customers, addressing areas of high clinical need or reducing healthcare costs;
•our ability to develop and maintain sales and marketing capabilities;
•regulatory developments in the United States and foreign countries;
•the performance of our third-party suppliers;
•the success of competing diagnostic products that are or become available;
•our ability to attract and retain key personnel; and
•our expectations regarding our ability to obtain and maintain intellectual property protection for our products and our ability to operate our business without infringing on the intellectual property rights of others.
In some cases, you can identify these statements by terms such as “anticipate,” “believe,” “could,” “estimate,” “expects,” “intend,” “may,” “plan,” “potential,” “project,” “should,” “will,” “would” or the negative of those terms, and similar expressions that convey uncertainty of future events or outcomes. These forward-looking statements reflect our management’s beliefs and views with respect to future events and are based on estimates and assumptions as of the date of this Annual Report on Form 10-K and are subject to risks and uncertainties. In addition, statements that “we believe” and similar statements reflect our beliefs and opinions on the relevant subject. These statements are based upon information available to us as of the date of this Annual Report on Form 10-K, and while we believe such information forms a reasonable basis for such statements, such information may be limited or incomplete, and our statements should not be read to indicate that we have conducted an exhaustive inquiry into, or review of, all potentially available relevant information. These statements are inherently uncertain and investors are cautioned not to unduly rely upon these statements. We discuss many of the risks associated with the forward-looking statements in this Annual Report on Form 10-K in greater detail underin the headingsection entitled “Risk Factors.” Moreover, we operate in a very competitive and rapidly changing environment. New risks emerge from time to time. It is not possible for our management to predict all risks, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements we may make. Given these uncertainties, you should not place undue reliance on these forward-looking statements.
2
RISK FACTORS SUMMARY
We face many risks and uncertainties, as more fully described in this Annual Report on Form 10-K under the heading “Risk Factors.” Some of these risks and uncertainties are summarized below. The summary below does not contain all of the information that may be important to you, and you should read this summary together with the more detailed discussion of these risks and uncertainties contained in “Risk Factors.”
Risks Related to our Financial Condition
•A significant portion of our revenue comes from a small number of third-party payors.
•Due to how we recognize revenue, our quarterly and annual revenues may not reflect our underlying business.
•We have incurred significant losses since inception, and we may never achieve profitability.
•We are an early, commercial-stage company and have a limited operating history, which may make it difficult to evaluate our current business and predict our future performance.
•Our quarterly and annual operating results and cash flows may fluctuate in the future, which could cause the market price of our stock to decline substantially.
•If our internal control over financial reporting is not effective, we may not be able to accurately report our financial results or file our periodic reports in a timely manner, which may cause adverse effects on our business and may cause investors to lose confidence in our reported financial information and may lead to a decline in our stock price.
•We may need to raise additional capital to fund our existing operations, commercialize new products, or expand our operations.
Risks Related to our Business
•Our revenue currently depends primarily on sales of DecisionDx®-Melanoma and our other dermatologic tests, and we will need to generate sufficient revenue from this and other products to grow our business.
•Unfavorable U.S. and global economic conditions could adversely affect our business, financial condition, results of operations or cash flows.
•Public health crises, such as pandemics or similar outbreaks, could adversely impact our business.
•Billing for our products is complex and requires substantial time and resources to collect payment.
•We rely on third parties for sample collection, preparation and delivery.
•We rely on our database of samples for some of the development and improvement of our products. Depletion or loss of our samples could significantly harm our business.
•If our primary clinical laboratory facilities become damaged or inoperable, or we are required to vacate our existing facilities, our ability to perform our tests and pursue our research and development efforts may be jeopardized.
•New product development involves a lengthy and complex process, and we may be unable to develop and commercialize, or receive reimbursement for, on a timely basis, or at all, new products.
•We rely on limited or sole suppliers for some of the reagents, equipment, chips and other materials used by our products, and we may not be able to find replacements or transition to alternative suppliers.
•The sizes of the TAM for our current and future products have not been established with precision and may be smaller than we estimate.
•The diagnostic testing industry is subject to rapid change, which could make our current or future products obsolete.
Risks Related to Reimbursement and Government Regulation
•We generally have limited reimbursement coverage for our products, and if third-party payors, including government and commercial payors, do not provide sufficient coverage of, or adequate reimbursement for, our products, our commercial success, including revenue, will be negatively affected.
3
•We conduct business in a heavily regulated industry and failure to comply with federal, state and foreign laboratory licensing requirements including those established by the Centers for Medicare and Medicaid (“CMS”) and the applicable requirements of the U.S. Food and Drug Administration (“FDA”) or any other regulatory authority, could cause us to lose the ability to perform our tests, experience disruptions to our business, or become subject to administrative or judicial sanctions.
•Interim, topline and preliminary data from our clinical studies that we announce or publish from time to time may change as more data become available and are subject to audit and verification procedures that could result in material changes in the final data.
•Changes in healthcare policy, statutes or regulations, or our ability to comply with applicable healthcare requirements, could have a material adverse effect on our business and operations.
Risks Related to Intellectual Property
•If we are unable to obtain and maintain sufficient intellectual property protection for our technology, our ability to successfully commercialize our products may be impaired.
•Our commercial success depends significantly on our ability to operate without infringing upon the intellectual property rights of third parties.
•We depend on information technology systems that we license from third parties. Any failure of such systems or loss of licenses to the software that comprises an essential element of such systems could significantly harm our business.
Risks Related to Employee Matters and Managing Growth and Other Risks Related to Our Business
•We are highly dependent on the services of our key personnel, including our President and Chief Executive Officer.
•Our employees, clinical investigators, consultants, speakers, and vendors and any current or potential commercial partners may engage in misconduct or other improper activities, including non-compliance with regulatory standards and requirements and insider trading.
•We have engaged in, and may continue to engage in, strategic transactions, such as the acquisition of businesses, assets, products or technologies, which could be disruptive to our existing operations, divert the attention of our management team and adversely impact our liquidity, cash flows, financial condition and results of operations.
•Product or professional liability lawsuits against us could cause us to incur substantial liabilities and could limit our commercialization of our products.
•Our business could be adversely affected by natural disasters, public health crises and other events beyond our control.
Risks Related to Ownership of Our Common Stock
•The price of our common stock may be volatile or may decline regardless of our operating performance, and you may lose all or part of your investment.
•We have broad discretion in the use of working capital and may not use it effectively or in ways that increase our share price.
•We have and may continue to enter into related party transactions that create conflicts of interest, or the appearance of conflicts of interest, which may harm our business and cause our stock price to decline.
•The concentration of our stock ownership will likely limit your ability to influence corporate matters, including the ability to influence the outcome of director elections and other matters requiring stockholder approval.
•Delaware law and provisions in our amended and restated certificate of incorporation and amended and restated bylaws could make a merger, tender offer or proxy contest difficult, thereby depressing the trading price of our common stock.
•Our amended and restated certificate of incorporation provides that the Court of Chancery of the State of Delaware is the exclusive forum for certain disputes between us and our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers or employees.
4
PART I
Item 1. Business.
As used in this Annual Report on Form 10-K, unless the context indicates or otherwise requires, “Castle Biosciences,” “the Company,” “we,” “us,”Biosciences”, the “Company”, “we”, “us”, and “our” refer to Castle Biosciences, Inc., a Delaware Corporation.
Overview
Our Testing Solutions
Our tests are a commercial-stage dermatological cancer company focused on providing physicians and their patients withdesigned to deliver personalized clinically actionable genomic information to make more accurate treatmenthelp better inform care decisions. We believe that the traditional approachFor our tissue based tests, we use multi-analyte assays with algorithmic analysis (“MAAA”) to developing a treatment plan for certain cancers using clinical and pathology factors alone can be improved by incorporating personalized genomic information. Our non-invasive genomic products utilize proprietary algorithmscharacterize an individual patient’s biology to provide an assessment of a patient’sinform specific risk of metastasis or recurrenceprogression.
Test Portfolio and Market Overview
The foundation of theirour business is our dermatologic cancer allowing physiciansfranchise. We currently offer five commercially available proprietary MAAA tests for use in the dermatologic, gastroenterology and ocular fields and a proprietary pharmacogenomic (“PGx”) test to identifyguide optimal drug treatment for patients who are likelydiagnosed with depression, anxiety and other mental health conditions.
Maintaining commercial success for our existing test portfolio requires generating ongoing evidence, such as clinical use documentation, to benefit from an escalationsupport appropriate clinician adoption, reimbursement success and guideline inclusion. The clinical validity and utility of care as well as those who may avoid unnecessary medicalour test portfolio is supported by peer-reviewed publications and surgical interventions. Our lead product, ongoing clinical studies. Collectively, approximately 140 peer-reviewed articles have been published demonstrating the analytical validity, clinical validity and clinical utility of the tests in our portfolio.
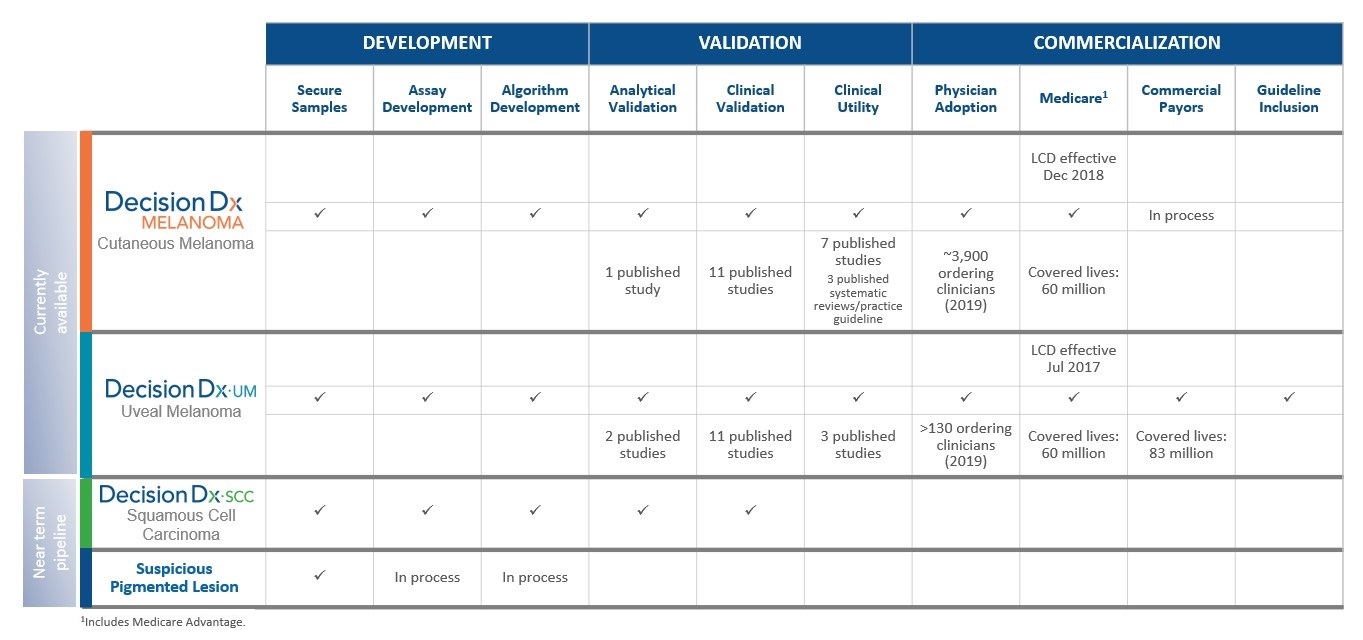
DecisionDx-Melanoma
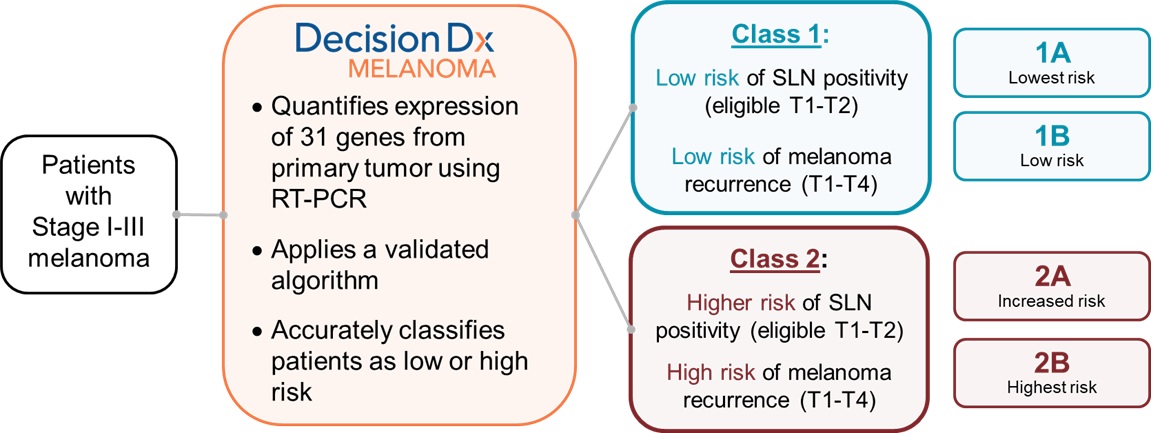
DecisionDx-Melanoma is aour proprietary multi-generisk stratification gene expression profile or GEP,(“GEP”) test that predicts the risk of metastasis, or recurrence, for patients diagnosed with invasive cutaneous melanoma (“CM”), a deadly skin cancer. We also market DecisionDx-UM,
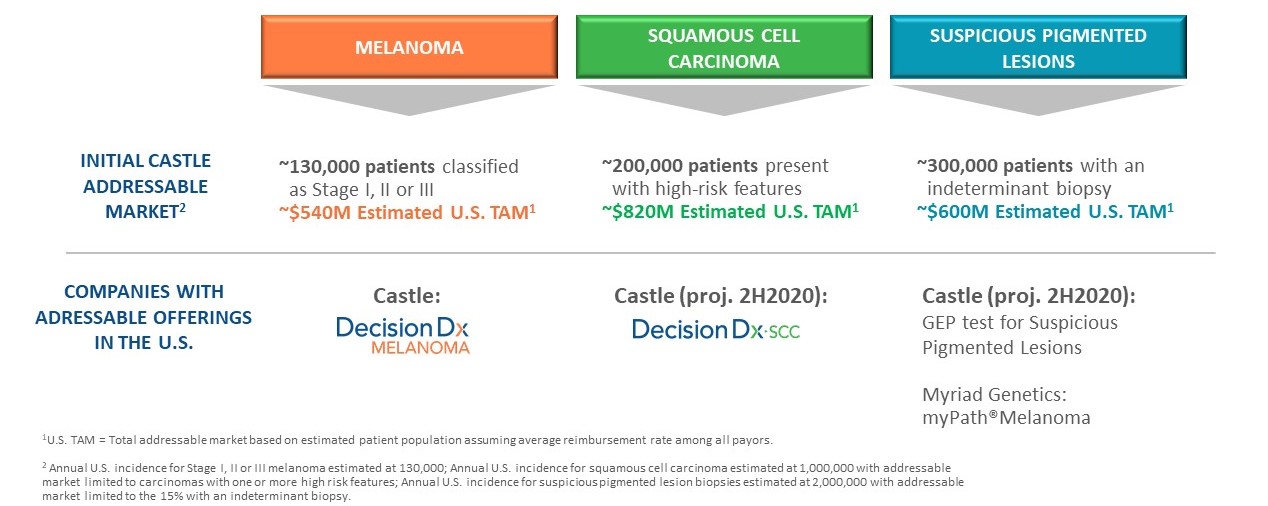
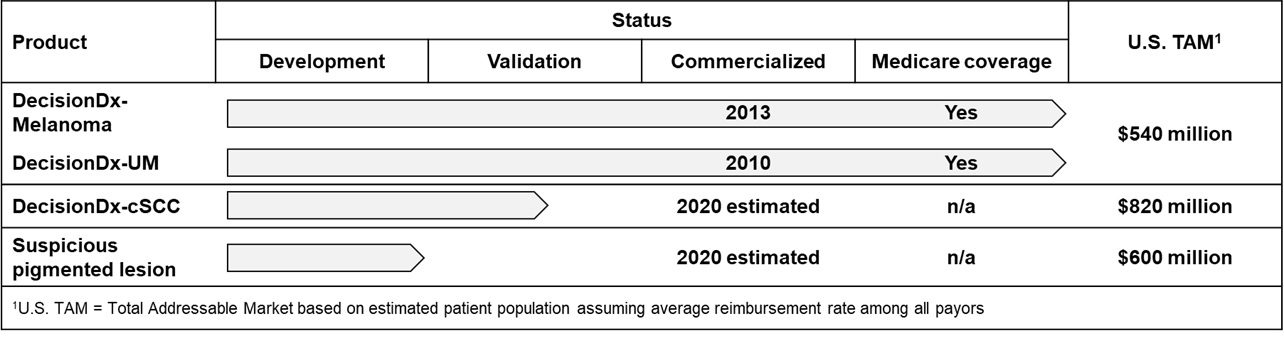
In a typical year, we estimate approximately 130,000 patients are diagnosed with invasive CM in the United States, representing an estimated U.S. TAM of approximately $540 million. This estimated annual incidence number is based upon a calculation using data from the U.S. Surveillance, Epidemiology, and End Results registries and subsequently adjusted for the documented underreporting of melanoma diagnoses which range from 30%-72%. Based on currently available data, we estimate the targetable clinician base treating melanoma is a proprietary GEP test that predicts the risk of metastasis for patients with uveal melanoma, a rare eye cancer.between 11,000 and 15,000. Based on the substantial clinical evidence that we have developed, we have received Medicare coverage for both of our products, which representsDecisionDx-Melanoma. We estimate that approximately 50% of our addressable patient population. We also have two proprietary products in late-stage development that address cutaneous squamous cell carcinoma, or SCC, and suspicious pigmented lesions, which are indications with high clinical need in dermatological cancer.
As of December 31, 2023, 49 peer-reviewed articles, six of which were published in 2023, support the clinical validity, clinical utility and impact on outcomes of our DecisionDx-Melanoma test. Based on our published data, we believe is underreported. In addition, our two late-stage proprietary products in development target approximately 200,000 patients diagnosed with SCC with high-risk features and approximately 300,000 patients with suspicious pigmented lesions without a definitive diagnosis of skin cancer. We estimatehave shown that the total addressable U.S. market for these three indications is approximately $2.0 billion.
5
DecisionDx-SCC
DecisionDx®‑SCC is our proprietary non-invasive genomic DecisionDx-Melanoma product to address this diagnostic discordance in patients with Stage I-III cutaneous melanoma. The product interrogates the biology of a patient’s tumor by analyzing the gene expression profile of 31 genes, a process made possible by our proprietary algorithm, developed using machine learning techniques. DecisionDx-Melanoma reports the risk of metastasis or recurrence for a patient’s melanoma into two classes and two subclasses, ranging from Class 1A, the lowest risk group, through Class 2B, the highest risk group, based on the genomics of the patient’s tumor. Physicians and patients use this additional tumor-specific genomic information, along with traditional staging criteria, to make better-informed decisions about how to manage the disease.
MyPath Melanoma
MyPath® Melanoma is our proprietary GEP test offering for patients with difficult-to-diagnose melanocytic lesions. Of the two million suspicious pigmented lesions are unusual-looking lesions that may be melanoma. There are approximately two million skin biopsies performed specifically for the diagnosis of melanomabiopsied annually in the United States. Approximately 15% of these biopsies are classified as indeterminate, in which case a pathologist cannot make a definitive diagnosis as to whether the biopsy is benign or malignant. Using a lower target reimbursement rate,States, we estimate the U.S. TAM at $600 million.
As of December 31, 2023, MyPath Melanoma has been clinically validated through three tissues comprising the uveal tractstudies and varyis supported by location17 peer-reviewed publications.
TissueCypher
TissueCypher® is our proprietary risk stratification spatial omics test designed to predict future development of high-grade dysplasia (“HGD”) and/or esophageal cancer in patients with non-dysplastic (“ND”), indefinite dysplasia (“IND”) or low-grade dysplasia (“LGD”) Barrett’s esophagus (“BE”).
There are approximately 90% occurring in choroid, 5%four million patients in the ciliary bodyU.S. currently diagnosed with BE and 5% inapproximately 415,000 patients annually undergo an endoscopic biopsy with a subsequent diagnosis of ND, IND or LGD BE, representing an estimated U.S. TAM of approximately $1 billion.
As of December 31, 2023, our TissueCypher test is supported by 14 peer-reviewed clinical validation and utility studies.
DecisionDx-UM
DecisionDx®-UM is our proprietary risk stratification GEP test that helps healthcare providers predict the iris. Uvealrisk of metastasis for patients with uveal melanoma may(“UM”), also be referred to as ocular melanoma.
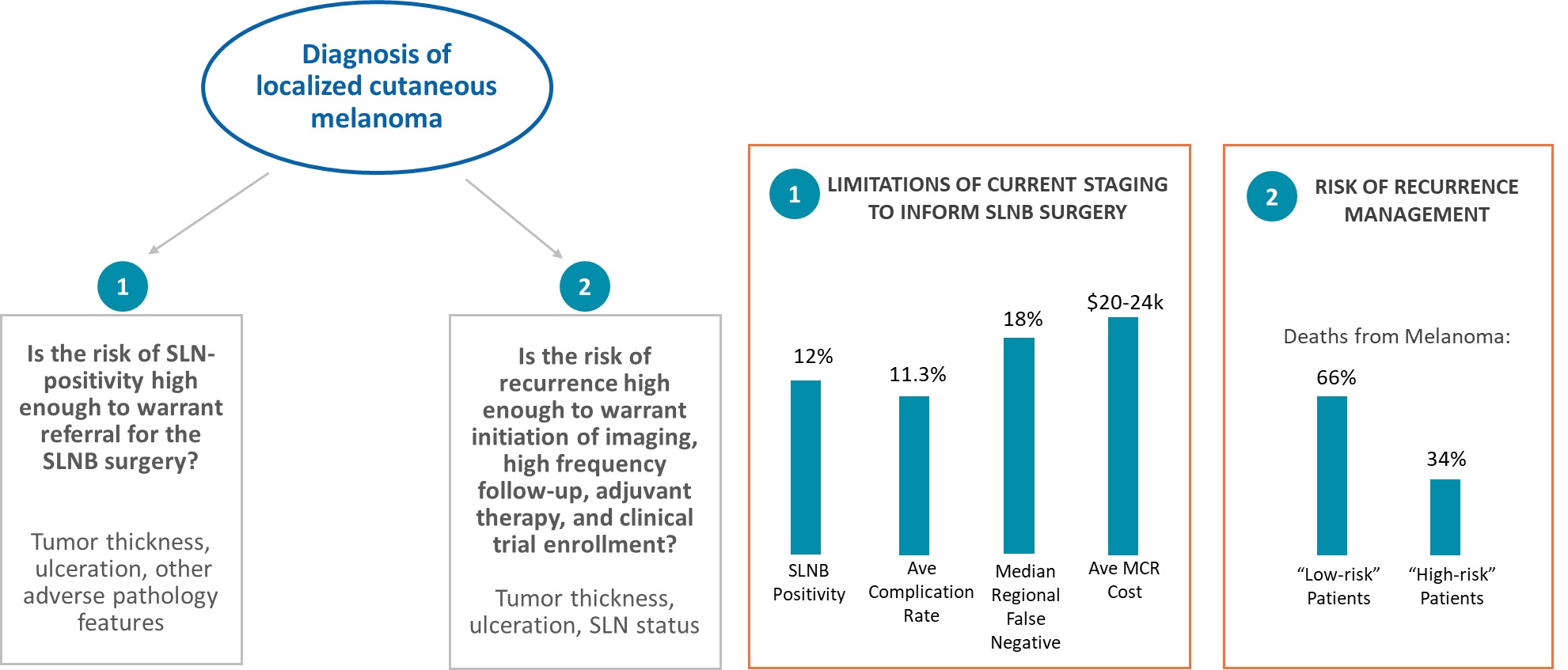
The dermatologic skin cancer market has significant unmet clinical needs, as clinical and pathology staging systems have traditionally applied a population-wide approachMedicare eligible population represents close to estimate an individual patient’s risk of metastasis and have not incorporated the genomics of a patient’s tumor biology. This is unlike the diagnostic process applied to other solid tumors, such as breast and prostate cancer, where the broader use of genomics to understand tumor biology has led to individualized patient treatment plans. Not incorporating tumor biology leads to a discordance between the estimated and actual risk of metastasis, which results in over- and under-treatment as well as increased healthcare costs.
DecisionDx-UM has been clinically validated by an independent prospective, multi-center study, by multiple retrospective and prospective single-center studies. As of December 31, 2023, our DecisionDx-UM test is supported by 25 peer-reviewed publications.
IDgenetix
IDgenetix® is our proprietary PGx test that helps guide optimal drug treatment for high-patients diagnosed with major depressive disorder, schizophrenia, bipolar disorder, anxiety disorders, social phobia, obsessive-compulsive personality disorder, post-traumatic stress disorder, and low-risk patients. For example, a 2014 study compared the AJCC version 7 and National Comprehensive Cancer Network, or NCCN, systemsattention deficit hyperactivity disorder. IDgenetix is designed to assess concordance between the AJCC and NCCN systems. The AJCC system classified 82% as low risk while the NCCN system classified 13% as low risk. As such, this level of discordance results in the risk assessment staging systems minimally impactingprovide important genetic information to clinicians to help guide personalized treatment plans with patients frequently being over- and under-treated.
We estimate a U.S. TAM of approximately $5 billion associated with this study, NCCNtest. We began offering the IDgenetix test following our acquisition of AltheaDx, Inc. (“AltheaDx”), in April 2022.
IDgenetix is supported by a published, peer-reviewed randomized controlled trial that demonstrated clinical utility over the standard of care when physicians used IDgenetix prior to prescribing a sensitivity of 96% while PPVmedication. The trial was 7% and NPV was 90.5%. The low PPV means that 93 out of 100 NCCN high risk SCC’s did not actually metastasize. AJCC and BWH demonstrated a sensitivity of 38.5% and 25%, respectively, PPV of 33% and 35%, respectively and NPV of 88% and 86%, respectively. If one relies just upon NCCN, the low PPV means that developing an adjuvant treatment plan that includes radiation, or chemotherapy or complete lymph surgical dissection, or a combination of these, for a high-risk patient may be appropriate for the one out of fourteen high-risk patients who will metastasize but not for the remaining thirteen patients who would not have metastasized. For AJCC and BWH, the PPV does improve but it also means that two out of three patients would be recommended for an adjuvant treatment plan who will not benefit. These accuracy metrics have created significant discordance in the approach to managing patients with high-risk features, from one of the spectrum being intervention for all high-risk patients to “watch and wait” for all high-risk patients. The end result is an unacceptableconducted across 20 independent clinical discordance in the approach to treatment plans and significant over- and under-treatment for a diagnosis that leads to the most skin cancer deaths insites within the United States.States specializing in psychiatry, internal medicine, obstetrics & gynecology, and family medicine. As of December 31, 2023, our IDgenetix test is supported by 19 peer-reviewed publications.
6
We usehave significant expertise in developing proprietary algorithms, conducting clinical studies and using the gene expression profile of an individual patient’s tumor biology to inform specific prognosis of metastasis or recurrence and aid the decision-making process of the treating physician and their patient to help optimize health outcomes and reduce healthcare costs.necessary instrumentation required for efficiently developing our pipeline products. Due to the biological complexity of skin cancers,diseases, developing accurate products takes scientific diligence, stringent clinical protocols, machine learning expertise, proprietary algorithms and significant investments of time and capital. In addition, the underlying tissue samples and associated clinical outcomes data required to develop and validate these products are difficult to obtain. Once successfully developed
In 2021, we announced the launch of our innovative pipeline initiative to develop a genomic test, or series of tests, aimed at predicting response to systemic therapy in patients with moderate to severe atopic dermatitis, psoriasis and validated, commercial success requiresrelated inflammatory skin conditions. In the generation of ongoing evidence such as clinical use documentation to support appropriate physician adoption, reimbursement success and guideline inclusion.
In addition to the U.S. TAM for both products have been published since completioneach of the initial clinical validation studies and have confirmedtests in our portfolio disclosed above, we estimate that the accuracy oftests in our products. Also, multiple clinical impact studies have demonstrated a significant impact on physician decisionspipeline add an additional estimated $5.7 billion to alter their treatment plan when the results of our test are considered in concert with the traditional clinical and pathology factors. Both of our currently marketed proprietary products are reimbursed by Medicare under positive coverage policies. In addition, we have received widespread positive private payor coverage and positive guideline inclusion for DecisionDx-UM, our first melanoma test. Since commercial launch, we have processed more than 60,000 clinical patient samples.
We expect to continue to pursue pipeline initiatives for tests that will branch out upstream, downstream and parallel to our test reports, physicians changed a patient’s treatmentexisting tests, within or adjacent to our established dermatological call points.
Acquisitions
From time to time, we may consider engaging in more than 50%transactions such as acquisitions of cases, indicating physician confidence in the evidence underlying our reports.
In April 2022, we acquired AltheaDx, a commercial-stage molecular diagnostics company specializing in the United States. We have received positive local coverage determinations, or LCDs, providing Medicare coveragefield of PGx testing services, for bothtotal consideration of $47.6 million, consisting of $30.5 million in cash and $17.1 million in shares of our common stock, adding the IDgenetix test to our portfolio.
In December 2021, we extended our commercial products. These LCDs facilitate reimbursementportfolio of proprietary tests into the gastroenterology market through our acquisition of Cernostics, Inc. (“Cernostics”) and the TissueCypher platform for total consideration of $49.0 million.
In May 2021, we acquired Myriad myPath, LLC and MyPath Melanoma test from Medicare, which represents approximately 50% of the addressable patient population. We also have third-party payor coverage for over 100 million lives for DecisionDx-UM and over 14 million lives for DecisionDx-Melanoma.

7
Test Report Volume and Revenue
The number of test reports we discovered, developed and completed validation for DecisionDx-Melanoma. This productgenerate is designeda key indicator that we use to help physicians identify high-risk patients with Stage I and II melanomas based on biological information, or expression, from 31 genes within their tumor tissue. DecisionDx-Melanoma does not change a physician’s standard diagnostic workflow for suspicious pigmented lesions, which includes performing the initial biopsy procedure and placing the biopsied tissue in formalin. The dermatopathologist then embeds the specimen in a paraffin block, cuts sections that are stained for viewing under a microscope and makes a diagnosis of invasive melanoma. We then extract and purify RNA from sections of the remaining specimen to runassess our test. We report test results in two classes and two subclasses. Class 1A represents the lowest risk group, Class 1B represents a low risk group, Class 2A represents an increased risk group and Class 2B represents the highest risk group.
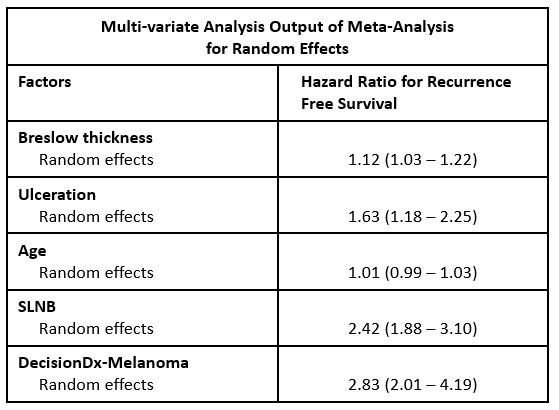
| Study | Design | # of Patients | % Change in Management |
| Berger et al. CMRO 2016 | Prospectively tested cohort, multi-center. Retrospective pre test / post test management. | 156 | 53% |
| Dillon et al. SKIN J Cutan Med 2018 | Prospective, multi-center: pre test / post test management. | 247 | 49% |
| Farberg et al. J Drugs Derm 2017 | 169 physician impact study: patient vignettes with pre test / post test management. | n/a | 47-50% |
| Schuitevoerder et al. J Drugs Derm 2018 | Prospectively tested cohort, single center. Retrospective pre test / post test management; and modeling of prospective cohort. | 91 | 52% |
below:
| Years Ended December 31, | |||||||||||||||||||||||||||||
| 2023 | 2022 | 2021 | 2020 | 2019 | |||||||||||||||||||||||||
| DecisionDx-Melanoma | 33,330 | 27,803 | 20,328 | 16,232 | 15,529 | ||||||||||||||||||||||||
DecisionDx‑SCC(1) | 11,442 | 5,967 | 3,510 | 485 | — | ||||||||||||||||||||||||
Diagnostic GEP offering(2) | 3,962 | 3,561 | 2,662 | 73 | — | ||||||||||||||||||||||||
| Dermatologic Total | 48,734 | 37,331 | 26,500 | 16,790 | 15,529 | ||||||||||||||||||||||||
| DecisionDx-UM | 1,674 | 1,711 | 1,618 | 1,395 | 1,526 | ||||||||||||||||||||||||
TissueCypher(3) | 9,100 | 2,128 | 27 | — | — | ||||||||||||||||||||||||
IDgenetix(4) | 10,921 | 3,249 | — | — | — | ||||||||||||||||||||||||
| Grand Total | 70,429 | 44,419 | 28,145 | 18,185 | 17,055 | ||||||||||||||||||||||||
| Net Revenues (in thousands) | $ | 219,788 | $ | 137,039 | $ | 94,085 | $ | 62,649 | $ | 51,865 | |||||||||||||||||||


(2)Includes MyPath Melanoma and provideDiffDx®-Melanoma. On November 2, 2020, we commercially launched our DiffDx-Melanoma test. We began offering MyPath Melanoma following our acquisition of the Myriad MyPath Laboratory on May 28, 2021. We offered both MyPath Melanoma and DiffDx-Melanoma under our Diagnostic GEP offering until February 2023 when we suspended the clinical offering of DiffDx-Melanoma.
(3)We began offering the TissueCypher on December 3, 2021, following our acquisition of Cernostics. Our TissueCypher test report volumes primarily derived from processed backlog orders. We temporarily paused accepting additional orders in July 2023 and resumed accepting new orders in a better PPV while maintaining similar NPV.phased approach in September 2023. We believe this product candidate will enable more informed clinical decisions regarding adjuvant intervention and other management decisions. We have ongoing multi-center studies involving more than 75 U.S. centers. Our development analysis has identified a proprietary gene expression profile algorithm that exhibits significant differential expression between non-recurrent and recurrent cases. Data fromcompleted processing of our initial validation study was presented at the American Society of Dermatologic Surgerypre-existing backlog orders in October 2019. 2023 and continue to accept new orders as of December 31, 2023.
(4)We have ongoing studiesbegan offering the IDgenetix test on April 26, 2022, following our acquisition of AltheaDx. Includes both single-gene and based upon our current progress we intend to commercially launch DecisionDx-SCC in the second half of 2020.multi-gene tests.
Our Commercial Channel
Sales and Marketing
Our sales and marketing efforts are currentlyprimarily focused on the United StatesU.S. skin cancer, market.gastroenterology and mental health markets. We employ a direct sales and marketing strategy to educate dermatologists, surgeonsclinicians and other physiciansassociated personnel on the clinical and economic benefits of our products. Our sales approach is highly technical, and our team is trained to articulate the scientific and clinical evidence behind our products and how they influence the clinical care pathway and ultimately improve patient outcomes. Our sales force is focused on educating and informing the entire patient care team, which consists of treating clinicians, nurses, laboratory and pathology personnel, and finance administrators, on the appropriate use and value of our test.
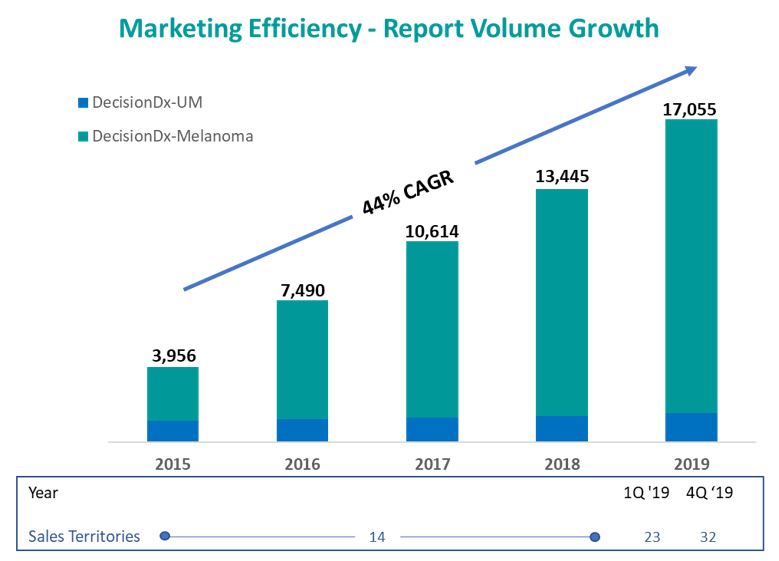
We increased our outside sales territories from 14 to 23 in the first quarter of 2019 and also added supporting inside sales associates, medical affairs and marketing staff. We believed that this increase in customer-facing personnel would increase the adoption of our products as we would be able educate more physicians on the clinical benefits of our products. We did see the promotion responsiveness that we anticipated and conducted a second expansion in December 2019 such that our outside sales territories now total 32. Based upon current knowledge, we believe that an optimally efficient dermatology salesforce to support our DecisionDx-Melanoma test ranges between 35 and 45 individuals. However, we will also continue to evaluate our mix of outside sales territories, inside sales support, marketingassess market response in determining further commercial expansions and medical affairs in the context of our DecisionDx-Melanoma test and our near-term pipeline product and adjust our investments based upon these evaluations.
Medical Affairs
We also deploy an experienced medical affairs group to assist education of treating physiciansclinicians and key opinion leaders, to identify and engage sites for our sponsored clinical studies and to evaluate collaborative study opportunities. Our medical affairs strategy complements our sales, and marketing and clinical research operations efforts.
We design our test reports with input from physicianswill continue to provide an easy to read risk classification and present specific actionable information to enable improved treatment decisions between patients and their physicians. We update our test reports as new data become available that may impact physicians’ treatment decisions based onassess the usemarket needs in determining further expansions of our tests. For example, the most recent update to our DecisionDx-Melanoma report incorporates data from our prospective multicenter 1,421 patient study showing how our tests can predict the likelihood of a patient experiencing an SLN-positive result from the SLNB surgery.medical affairs team.

Reimbursement
The primary source of revenue for our products is reimbursement from third-party payors, which includes government payors, such as Medicare, and commercial payors, such as insurance companies. Achieving broad coverage and reimbursement of our current products by third-party payors and continued Medicare coverage are key components of our financial success. De novo coverage by government and
We bill third-party payors for our pipeline tests will be important over time.
8
which meet certain criteria for Medicare and Medicare Advantage beneficiaries. A “covered life” means a public comment period that opened on October 7, 2019 and closed on November 21, 2019. Based upon our analysis from the close of the public comment period to issuance of the final LCD for other MolDX LCDs, we estimate that this expanded LCD may be final in the second half of 2020. However, there is no assurance that the timing of our LCD will match the recent experience of other MolDX LCDs.
The Medicare rates discussed below are prior to giving effect to applicable sequestration in effect from time to time as described in further detail under "Government Regulation and to guide surveillance and referral to medical oncology for those patients. Similar to cutaneous melanoma the median age at diagnosis for uveal melanoma is estimated at 58-62 years old, therefore the Medicare eligible population represents close to 45% of the addressable market.Product Approval—Healthcare Reform" included in Part 1, Item 1, “Business”, in this Annual Report on Form 10-K.
Commercial Third-Party Payors
We are actively engaged in efforts to achieve broad coverage and reimbursement for our current and future products, followed by contracting with commercial payors. Achieving positive coverage reduces the need for appeals and reduces failures to collect from the patient’s commercial insurance payor. Even with positive coverage decisions, we still experience delays in time to payment. Achieving in-network contracts with third-party payors can shorten the time required to receive payments. Implementing our strategy includes our managed care and medical affairs teams educating third-party payors regarding our strong clinical utility and outcomes data, which we believe validates the value of our products and will persuade more third-party payors to provide value-based reimbursement.
We have broad positive policy coverage for our DecisionDx-UM test weand have executed contracts with certain commercial payors. For our other tests, we engage third-party payors for positive coverage and anticipate increases in contracting in 2020 and 2021. We also have received positive policy recommendations from many third-party technical assessment review groups.
Dependence on Third-Party Payors
We receive a substantial portion of our revenue from a small number of third-party payors, primarily Medicare, BlueCross BlueShield affiliates and Medicare Advantage plans.payors. Our revenue from patients covered by Medicare Medicare Advantage plans, United Healthcare and BlueCross BlueShield plans, as a percentage of total revenue, was 49%, 29%, 6% and 6%, respectively, for the year ended December 31, 2023. Additionally, there was a commercial payor from which 14% of our revenue from patients was derived for the year ended December 31, 2023.
Government Payors
Medicare coverage is limited to items and services that are within the scope of a Medicare benefit category and that are reasonable and necessary for the diagnosis or treatment of an illness or injury. The controlling Medicare regulation for guiding the assessment of reasonable and necessary of diagnostic laboratory tests is 42 CFR. Section 410.32(a). Medicare Administrative Contractors (“MACs”) can provide coverage through evidentiary based reviews as well as more formal processes such as development of local coverage determinations (“LCD”).
Our laboratories are located in Phoenix, Arizona and Pittsburgh, Pennsylvania. The MAC responsible for administering claims for laboratory services located in Arizona is Noridian Healthcare Solutions, LLC (“Noridian”). Noridian has a joint operating agreement with Palmetto GBA MolDX (“Palmetto”) where Palmetto is responsible for reviewing genomic based tests. The MAC responsible for administering claims for laboratory services located in Pennsylvania is Novitas Solutions (“Novitas”).
Medicare
DecisionDx-Melanoma
Palmetto issued a final expanded test-specific LCD for DecisionDx-Melanoma, effective November 22, 2020. With this expanded LCD and the accompanying billing and coding articles, we estimate that a significant majority of the DecisionDx-Melanoma tests performed for Medicare patients will meet the coverage criteria. Noridian adopted the same coverage policy as Palmetto and also issued an expanded final LCD for DecisionDx-Melanoma, effective December 6, 2020. On May 19, 2022, Palmetto finalized an LCD that converted the DecisionDx-Melanoma test-specific LCD to a “foundational” LCD with Noridian issuing the same on June 16, 2022. The final LCDs did not result in any changes in coverage.
DecisionDx-UM
Palmetto issued a final test-specific LCD for DecisionDx-UM, which became effective in July 2017, and Noridian issued a similar LCD that became effective in September 2017. We estimate that a significant majority of the DecisionDx-UM tests performed for Medicare patients will meet the coverage criteria.
9
MyPath Melanoma and DiffDx-Melanoma
MyPath Melanoma was covered under a test-specific LCD policy through Noridian that became effective in June 2019. BlueCross BlueShield plansEffective August 6, 2023, Palmetto and Noridian issued LCDs that converted the test-specific MyPath Melanoma LCD to a “foundational” LCD and provided coverage for both MyPath Melanoma and DiffDx-Melanoma.
DecisionDx‑SCC
We issue our DecisionDx-SCC tests from our Pittsburgh and Phoenix labs, with a majority of tests being issued from our Pittsburgh lab. As previously discussed, Novitas is the MAC responsible for administering claims for test reports issued by our Pittsburgh laboratory.
We requested and received an evidentiary review of our DecisionDx-SCC by Novitas during the first quarter of 2022. Based upon this review, DecisionDx-SCC test began receiving coverage in April 2022.
On June 9, 2022, Novitas posted a draft oncology biomarker LCD that proposes to rely upon evidentiary reviews sourced from three databases for all oncology biomarker tests: ClinGen, OncoKB and National Comprehensive Cancer Network (“NCCN”). We believe the purpose of the proposals in this draft LCD are to streamline future reviews. Two of the databases do not review GEP tests and NCCN has not yet, to our knowledge, reviewed DecisionDx-SCC. If finalized as proposed, then DecisionDx-SCC would not have been included as a covered test in the associated billing and coding article. The comment period for the draft LCD ended on September 6, 2022.
On June 2, 2023, Novitas posted a finalized oncology biomarker LCD pursuant to which the DecisionDx-SCC test would no longer be covered by Medicare Advantage plans representeffective July 17, 2023. However, on July 6, 2023, Novitas suspended the final version of the LCD and announced its intent to post a new proposed LCD for comment and presentation at an aggregationopen meeting. On July 27, 2023, Novitas posted a nearly identical proposed oncology biomarker LCD that continues to intend to rely upon evidentiary reviews sourced from three databases: ClinGen, OncoKB and NCCN. The proposed LCD also recommends non-coverage for our DecisionDx-SCC test. The comment period for the proposed LCD ended on September 9, 2023. We cannot predict whether this LCD will be finalized as proposed or what the timing of any final LCD might be.
As previously discussed, the Palmetto MolDX program oversees MAAA tests that are reported from our Phoenix laboratory and Noridian is the MAC responsible for administering claims for test reports issued by our Phoenix laboratory. In the second quarter of 2020, we submitted our technical assessment dossier for DecisionDx-SCC to Palmetto and Noridian. The dossier was accepted as complete in the third quarter of 2020. On June 8, 2023, both Palmetto and Noridian posted a preliminary draft LCD recommending no coverage for DecisionDx-SCC. The comment period for the draft LCDs ended on July 22, 2023.
Advanced Diagnostic Laboratory Tests
Advanced Diagnostic Laboratory Test (“ADLT”) status is a designation granted by CMS for clinical diagnostic laboratory tests offered and furnished by a single laboratory and covered under Medicare Part B that meet one of the following criteria:
Criterion A: The test:
•Is an analysis of multiple payors making independentbiomarkers of DNA, RNA or proteins;
•When combined with an empirically derived algorithm, yields a result that predicts the probability a specific individual patient will develop a certain condition or conditions, or respond to a particular therapy or therapies;
•Provides new clinical diagnostic information that cannot be obtained from any other test or combination of tests; and
•May include other assays.
Criterion B: The test is cleared or approved by the FDA. Laboratories requesting ADLT status under this criterion are required to submit documentation of premarket approval (“PMA”) or premarket notification from the FDA.
All five of our commercially available proprietary MAAA tests have been reviewed by the CMS and have been granted ADLT status. ADLT status is not an indication of future coverage.
10
Medicare Reimbursement Rates
DecisionDx-Melanoma
On May 17, 2019, CMS determined that DecisionDx-Melanoma meets the criteria for “new ADLT” status. Our rate is set annually based upon the median private payor rate for the first half of the second preceding calendar year. For example, the rate for 2023 was set using median private payor rate data from January 1, 2021 to June 30, 2021. Our rate for 2022 and 2023 was $7,193 per test and is $7,193 for 2024.
DecisionDx-UM
On May 17, 2019, CMS determined that DecisionDx-UM meets the criteria for “existing advanced diagnostic laboratory test” status, also referred to as “existing ADLT” status. Our rate is set annually based upon the median private payor rate for the first half of the second preceding calendar year. For example, the rate for 2023 was set using median private payor rate data from January 1, 2021 to June 30, 2021. Our rate for 2022 and 2023 was $7,776 per test and is $7,776 for 2024.
MyPath Melanoma
On September 6, 2019, MyPath Melanoma was approved as a “new ADLT”. Our rate is set annually based upon the median private payor rate for the first half of the second preceding calendar year. Our rate for 2022 was $1,950 per test. Our 2023 rate was set at $1,755 per test, based on data submitted by the predecessor owner of the Myriad MyPath Laboratory relating to the first half of 2021. Our 2024 rate is set at $1,950 per test.
DiffDx-Melanoma
In the second quarter of 2022, we obtained a Proprietary Laboratory Analyses (“PLA”) code for DiffDx-Melanoma. In 2023, DiffDx-Melanoma went through the CMS gapfill process which concluded in September 2023 with CMS posting a final MAC-specific gapfill rate of $1,950 per test. Our rate for 2024 is $1,950 per test.
Diagnostic GEP Offering
Our Diagnostic GEP offering included MyPath Melanoma and DiffDx-Melanoma. We began offering MyPath Melanoma following our acquisition of the Myriad MyPath Laboratory on May 28, 2021. Our internal data indicates that we have improved the technical performance of MyPath Melanoma such that it is now comparable to the technical performance of DiffDx-Melanoma. As such, following an internal assessment of the clinical value of offering both tests, we made the decision to suspend the clinical offering of DiffDx-Melanoma in February 2023.
DecisionDx‑SCC
In the first quarter of 2022, we requested that Novitas conduct a medical review of our DecisionDx-SCC test. That review was completed towards the end of that quarter. In the second quarter of 2022, following the completion of a requested medical review and pricing of our DecisionDx-SCC test by Novitas, we obtained a PLA code and began receiving reimbursement decisions; however, these plans often basefrom Novitas for DecisionDx-SCC at a rate of $3,873 per test.
On June 30, 2023, CMS determined DecisionDx-SCC meets the criteria for “new ADLT” status. Effective July 1, 2023 and through March 31, 2024, CMS set the initial period rate equal to the list price of $8,500 per test. Effective April 1, 2024 and through December 31, 2025, the published CLFS rate for DecisionDx-SCC will be based on the median private payor rates received between July 1, 2023 and November 30, 2023. We submitted the median private payor data to CMS during the data reporting period in December 2023. Future rates will be set annually based upon the median private payor rate for the first half of the second preceding calendar year. ADLT status determines the process by which the rate is set and is not an indication of Medicare coverage.
11
TissueCypher
TissueCypher is processed in our Pittsburgh, Pennsylvania laboratory and falls under the Medicare jurisdiction managed by Novitas. From January 1, 2022 through March 31, 2022, we received payments for claims according to the published CLFS rate at $2,513 per test. On March 24, 2022, CMS determined TissueCypher meets the criteria for “new ADLT” status. ADLT status exempts TissueCypher from what is called the “14-day rule,” which simplifies the billing process for Medicare patients. From April 1, 2022 through December 31, 2022, CMS set the initial period rate equal to the original list price of $2,350 per test. Effective January 1, 2023, the published CLFS rate for TissueCypher was set at $4,950 per test, which will remain effective through December 31, 2024. This rate is based on the median private payor rates received between April 1, 2022 and August 31, 2022. Thereafter, the rate will be set annually based upon the median private payor rate for the first half of the second preceding calendar year.
IDgenetix
IDgenetix is currently covered under a Noridian LCD policy and accompanying billing and coding article developed by MolDX. The Medicare coverage includes depression and the following seven additional mental health conditions beyond major depressive disorder: schizophrenia, bipolar disorder, anxiety disorders, social phobia, obsessive-compulsive personality disorder, post-traumatic stress disorder and attention deficit hyperactivity disorder. IDgenetix has historically been billed to Medicare using an unspecified CPT code along with the IDgenetix test-specific MolDX Z-code (the “IDgenetix Z-Code”). We acquired AltheaDx and the IDgenetix test in April 2022 and received Medicare reimbursement decisions on common guidelinesat a rate of approximately $1,500 per test. In February 2023, MolDX notified us that as part of its annual CPT code updates, IDgenetix should shift billing to a different multi-test generic gene sequencing CPT code (the “New CPT Code”) and continue using the IDgenetix Z-Code beginning in March 2023. The New CPT Code was set at $917 per test while the test went through CMS’s Gapfill pricing process. We believed the new CPT Code, in conjunction with the IDgenetix Z-Code, did not describe all of the components of the IDgenetix test and thus, was not appropriate for IDgenetix. We subsequently obtained a test-specific PLA CPT code which can influence multiple plans simultaneously.became effective October 1, 2023. In November 2023, CMS posted its final CLFS determination which crosswalks our PLA CPT code to an existing PLA code at a rate of $1,336 per test effective January 1, 2024.
Competition
We are focused on providing high value diagnostic and prognostic solutions for dermatological cancers. We believe, today,improving health through innovative tests that there is limited existing competition for our products that provide evidence-based genomic solutions to physicians and their patients.guide patient care.
We believe the principal competitive factors in our target markets include:
•Proprietary, disciplined approach to genomic and proteomic analysis including the use of proprietary deep learning, machine learning, artificial intelligence and other techniques to identify and optimize genebiomarker selection and algorithmic approaches to answer the clinically important questions with accuracyaccurate tests. This involves the ability to design and efficiently conduct the right clinical studies at the right time;
•Research and development investments to document the quality, quantity, consistency and strength of the clinical validity data, the impact our products have on clinical use, and demonstration of net health outcome improvement that reduce health system costs;
•Maintaining a strong reputation with the treating physicianclinician by providing consistent, transparent, and clinically relevant information that will improve the appropriate management of their patients;
•Ease of use in accessing our products, reimbursement support for the physician and their patientour patients and laboratory reports that clearly communicate the clinically relevant data points;
•Demonstrated ability to work with, and secure coverage and reimbursement from, governmental and commercial payors;
•Ability to efficiently commercialize pipeline products to the same customer base asboth our current products.and our pipeline products
We believe we compete favorably on the factors described above.
Today, our principal competition for DecisionDx-Melanoma is existing traditional clinical and pathology staging criteria. While some clinical and pathology criteria have changed over time, this approach has been the standard of care in the United States for many years, and physiciansclinicians may be unwilling to accept the validity of the published data and adopt our test until it has become incorporated into national guidelines. In December 2019, we did see positive improvements in the NCCN guidelines as it related to our DecisionDx-Melanoma test. However, it is too early to understand the impact on physician adoption and commercial payer coverage. In addition, we currently face, or may in the near future,
12
face, competition from a limited number of companies who are working in this disease space, such as NeracareSkylineDx/Tempus/Quest, AMLo/Avero and SkylineDx.Neracare. In the future, we may face additional competitors.
We are unaware of late-stage work being performed to develop and validate a product that would compete with DecisionDx-SCC. We believe that the current primary competitor for DecisionDx-SCC is existing traditional clinical and pathology staging criteria. In the future, we may face additional competitors.
DecisionDx-UM competes with a subsidiary of LabCorp and several academic laboratories, all of which have had tests available for several years. To date, our data has demonstrated that DecisionDx-UM is clinically and statistically superior to these products. In the future, we may face additional competitors.
With respect to IDgenetix, our pipeline product for suspicious pigmented lesions suchcompetition arises from other parties using the same or similar methods as well as alternative methods of PGx testing. IDgenetix competes with Myriad Genetics. If we are successful in validating a clinically useful product, then we expect to compete favorably with them.Genetics’s GeneSight test, Genomind’s PGx test, and tests from numerous other commercial and academic laboratories.
Laboratory Operations
Raw Materials and Suppliers
We procure certain reagents, equipment, chips/cards and other materials used to perform our tests from sole suppliers such as ThermoFisher Scientific, Inc., and Qiagen, Inc. Some of these items are unique to these suppliers and vendors. While we have developed alternate sourcing strategies for these materials and vendors and while we have experienced no business interruption due to an inability to source these materials, we cannot be certain whether these strategies will be effective or whether alternative sources will be available when we need them. If these suppliers can no longer provide us with the materials we need to perform our test services, they do not meet our quality specifications, or we cannot obtain acceptable substitute materials, our business would likely be negatively affected.
License Agreement with The Washington University
In November 2009, we entered into a license agreement or the License Agreement,(the “License Agreement”) with WUSTLThe Washington University in St. Louis, Missouri (“WUSTL”) to license certain patent rights and technical information from WUSTL for the development of melanoma products or the Products,(the “Products”), and services or the Services.(the “Services”). The rights licensed under this agreement are used in DecisionDx-UM only.
Under the License Agreement, we obtain an exclusive, worldwide, royalty-bearing license to certain patent rights owned by WUSTL or the Patent Rights,(the “Patent Rights”) and a non-exclusive, worldwide license to certain technical information and research property owned by WUSTL, with the right to grant sublicenses under certain conditions, in order to develop the Products and the Services. WUSTL retains the right to use the Patent Rights for research purposes.
The Patent Rights that we license pursuant to the License Agreement have been generated through the use of U.S. government funding and are therefore subject to certain federal regulations. See “Risk Factors—Risks Related to Intellectual Property—Our in-licensed intellectual property has been discovered through government funded programs and thus may be subject to federal regulations such as “march-in” rights, certain reporting requirements
13
and a preference for U.S.-based companies, and compliance with such regulations may limit our exclusive rights, and limit our ability to contract with non-U.S. manufacturers.”
Under the License Agreement, we are required to use best efforts to carry out the activities under an agreed-upon development plan or the Development Plan,(the “Development Plan”) and meet any and all milestones set forth in the Development Plan. We are required to make milestone payments to WUSTL upon successful completion of development and commercialization milestones as set forth in the Development Plan. For each Product or Service that receives FDA approval, premarket approvalPMA or premarket notification, we are obligated to make a milestone payment to WUSTL in the mid-four digits. For the issuance of the first U.S. patent and the first foreign patent, we are obligated to make aggregate milestone payments to WUSTL in the low-five digits.
Under the License Agreement, we were obligated to pay WUSTL an initial license issue fee in the low-five digits. We are also obligated to make royalty payments to WUSTL equal to (i) a percentage in the mid-single digits of our and any of our affiliates’ or sub-licensees’ net sales of the Products and (ii) a percentage in the low-single digits of our and any of our affiliates’ or sub-licensees’ revenue from the Services. We are also obligated to make royalty payments to WUSTL in the low-to-mid single digit percentage of net sales, with minimum royalty payments to WUSTL every six-month period following the first commercial sale.
The term of the License Agreement will continue for ten years following the last-to-expire valid claim relating to the Patent Rights, unless terminated earlier. WUSTL may terminate the License Agreement upon written notice in the event of (i) our material breach if such breach remains uncured for 90 days, (ii) the exercise of certain rights by us with respect to the Patent Rights and/or the licensed technical information outside the scope of the License Agreement, or (iii) for certain insolvency-related events. We may terminate the License Agreement without cause upon written notice to WUSTL and payment of any amount due to WUSTL under the License Agreement.
Intellectual Property
Our core technology for our products is related to methods and devices for analysis of genetic expression. Using this technology, we are able to provide a more accurate prediction of a patient’s metastatic risk as compared to other methods. We have secured and continue to pursue intellectual property rights globally, including through patent protection covering analysis of metastasis in cutaneous melanoma, as well asthe treatment of cutaneous squamous cell carcinoma.SCC, BE and gastroenterology, and PGx for mental illness. We also rely on trademarks, trade secrets, know-how, continuing technological innovation and potential in-licensing opportunities to develop and maintain our proprietary position. For more information, please see “Risk Factors—Risks Related to Intellectual Property.”
14
Patents and Patent Applications
We have developed a global patent portfolio that as of the date of this Annual Report on Form 10-K,December 31, 2023, is comprised ofas follows:
| Number of Applications and Patents | ||||||||||||||||||||
| Commercial Focus | United States | International | Total | |||||||||||||||||
| Owned Patent Families | ||||||||||||||||||||
| Methods for predicting risk of metastasis in cutaneous melanoma | 3 | 19 | 22 | |||||||||||||||||
| Methods of diagnosing and treating patients with pigmented skin lesions | 1 | — | 1 | |||||||||||||||||
| Methods of diagnosing and treating patients with cutaneous squamous cell carcinoma | 2 | 19 | 21 | |||||||||||||||||
| Determining Prognosis and Treatment based on Clinical-Pathologic Factors and Continuous Multigene-Expression Profile Scores | 1 | — | 1 | |||||||||||||||||
| Diagnosing and Treating Atopic Dermatitis and/or Psoriasis | 2 | — | 2 | |||||||||||||||||
| Diagnosing and Treating Uveal Melanoma | 1 | — | 1 | |||||||||||||||||
| Genes and gene signatures for diagnosis and treatment of melanoma | 7 | 33 | 40 | |||||||||||||||||
| Method for automated tissue analysis | 2 | 3 | 5 | |||||||||||||||||
| Systems and compositions for diagnosing BE and methods of using same | 3 | 13 | 16 | |||||||||||||||||
| Methods of predicting progression of BE | 2 | 22 | 24 | |||||||||||||||||
| Expression profiling using microarrays | 1 | — | 1 | |||||||||||||||||
| Licensed Portfolio from WUSTL | ||||||||||||||||||||
| Method for predicting risk of metastasis | 2 | — | 2 | |||||||||||||||||
| Compositions and methods for detecting cancer metastasis | 2 | 2 | 4 | |||||||||||||||||
| Total | 29 | 111 | 140 | |||||||||||||||||
Included in the table above are 16 issued patents, including five issued U.S. patents and nine pending patent applications, including three U.S. applications. Our patent portfolio consists of three owned patent families and an exclusively in-licensed portfolio from WUSTL, which includes six pending and77 issued patents or patent applications across two families.international patents. This global patent portfolio has filing dates ranging from 20092007 to 2018,2023, and therefore are projected to expire between 20292027 and 2038,2043, subject to any patent term extension or patent term adjustment that might be available in a particular jurisdiction. The owned and licensed families contain issued patents and pending applications that relate to devices, systems, and methods for macromolecular analysis, and reflect our active and ongoing research programs. The commercial foci of these patent families are discussed below.
Individual patents extend for varying periods depending on the date of filing of the patent application or the date of patent issuance and the legal term of patents in the countries in which they are obtained. Generally, patents issued for regularly filed applications in the United States are granted a term of 20 years from the earliest effective non-provisional filing date. In addition, in certain instances, a patent term can be extended to recapture a period due to delay by the United States Patent and Trademark Office or USPTO(the “USPTO”) in issuing the patent as well as a portion of the term effectively lost as a result of the FDA regulatory review period. However, as to the FDA component, the restoration period cannot be longer than five years and the total patent term including the restoration period must not exceed 14 years following FDA approval. The duration of foreign patents varies in accordance with provisions of applicable local law, but typically is also 20 years from the earliest effective non-provisional filing date. However, the actual protection afforded by a patent varies on a product-by-product basis, from country to country, and depends upon many factors, including the type of patent, the scope of its coverage, the availability of regulatory-related extensions, the availability of legal remedies in a particular country and the validity and enforceability of the patent.
Trademarks and Trade Secrets
As of the date of this Annual Report on Form 10-K, our U.S. trademark portfolio contained seven16 trademark registrations.
We rely upon trade secrets, know-how, continuing technological innovation and potential in-licensing opportunities to develop and maintain our competitive position. We seek to protect our intellectual property and proprietary technology, in part, by entering into confidentiality agreements and intellectual property assignment agreements with our employees, consultants, corporate partners and, as applicable, our advisors. These agreements are designed to protect our proprietary information and, in the case of the invention assignment agreements, to grant us ownership of technologies that are developed through a relationship with an employee or a third party. These agreements may be breached, and we may not have adequate remedies for any breach. We additionally seek to preserve the
15
integrity and confidentiality of our data and trade secrets, such as our proprietary algorithms, by maintaining the physical security of our premises and physical and electronic security of our information technology systems. While we have confidence in these individuals, organizations and systems, agreements or security measures may be breached, and we may not have adequate remedies for any breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors. To the extent that our commercial partners, collaborators, employees and consultants use intellectual property owned by others in their work for us, disputes may arise as to the rights in related or resulting know-how and inventions.
Government Regulation and Product Approval
Regulations
Clinical Laboratory Improvement Amendments of 1988
As a clinical reference laboratory, we are required to hold certain federal, state and local licenses, certifications and permits to conduct our business. Under CLIA, we are required to hold a certificate applicable to the type of laboratory tests we perform and to comply with standards applicable to our operations, including test processes, personnel, facilities administration, equipment maintenance, recordkeeping, quality systems and proficiency testing. We must maintain CLIA compliance and certification to be eligible to bill for diagnostic services provided to Medicare beneficiaries.
To renew our CLIA certificate, we are subject to survey and inspection every two years to assess compliance with program standards. Because we areour Phoenix, Arizona laboratory is a College of American Pathologists, or CAP accredited laboratory, CMS does not perform thismay defer the survey and inspection and relies on our CAP survey and inspection.to those conducted by CAP. We may also be subject to additional unannounced inspections. The regulatory and compliance standards applicable to the testing we perform may change over time,periodically, and any such changes couldare published by CAP. Our SOPs, documents & records are updated accordingly and as needed. Any such changes may have a material effect on our business.
Penalties for non-compliance with CLIA requirements include suspension, limitation or revocation of the laboratory’s CLIA certificate, as well as directed plan of correction, state on-site monitoring, civil money penalties, civil injunctive suit or criminal penalties.
State Laboratory Licensing
In addition to federal certification requirements of laboratories under CLIA, CLIA provides that states may adopt laboratory regulations and licensure requirements that are more stringent than those under federal law. Such laws, among other things, establish standards for the day-to-day operation of a clinical reference laboratory, includingwhich includes ensuring personnel have the adequate knowledge and training and skills required of personnel andto maintain quality control. We currently provide laboratory services in all 50 states andstates. Additionally, we maintain out-of-state laboratory licenses in New York, California, Maryland, Pennsylvania and Rhode Island.
Because we receive specimens from the state of New York, our clinical reference laboratory is required to be licensed by New York, underhave a lab director with a specific certificate of qualification and is subject to biennial New York laws and regulations.state inspections to ensure the lab is compliant with New York lawlicensing standards. New York regulations also mandatesmandate proficiency testing for laboratories licensed under New York state law, regardless of whether such laboratories are located in New York. If a laboratory is out of compliance with New York statutory or regulatory standards, the New York State Department of Health or NYSDOH,(the “NYSDOH”) may suspend, limit, revoke or annul the laboratory’s New York license, censure the holder of the license, or assess civil money penalties. We have received writtenformal approval from the NYSDOH to offer the following of our proprietary DecisionDx-Melanoma DecisionDx-UM and DecisionDx-PRAME products in New York. If we wereassays to be found out of compliance with New York laboratory requirements,patients: DecisionDx-Melanoma, DecisionDx-CMSeq, DecisionDx-UM, DecisionDx-PRAME, DecisionDx-UMSeq, DecisionDx-SCC, MyPath Melanoma, and DiffDx-Melanoma. We have been given conditional approval for IDgenetix from the NYSDOH while we could be subjectwork through the formal review process. In July 2022, we submitted TissueCypher for review by the NYSDOH. We had been given clearance to such sanctions, which could harmtest New York state patients by the New York’s Clinical Laboratory Evaluation Program while our business.applications were under review. On September 12, 2023, we received our Clinical Laboratory Permit from NYSDOH for our Pennsylvania laboratory.
Federal Oversight of Laboratory Developed Tests
The laws and regulations governing the marketing of diagnostic products are evolving, extremely complex, and in many instances, there are no significant regulatory or judicial interpretations of these laws and regulations. Clinical laboratory tests are regulated under CLIA, as administered by CMS, as well as by applicable state laws. In addition, the Federal Food, Drug and Cosmetic Act or FDCA,(the “FDCA”) defines a medical device to include any instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article, including a
16
component part, or accessory, intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease, in man or other animals. Our in vitro testing products are considered by the FDA to be subject to regulation as medical devices. Among other things, pursuant to the FDCA and its implementing regulations, the FDA regulates the research, testing, manufacturing, safety, labeling, storage, recordkeeping, pre-market clearance or approval, marketing and promotion, and sales and distribution of medical devices in the United States to ensure that medical products distributed domestically are safe and effective for their intended uses. In addition, the FDA regulates the export of medical devices manufactured in the United States to international markets.
Although the FDA has statutory authority to assure that medical devices are safe and effective for their intended uses, the FDA has generally exercised its enforcement discretion and not enforced applicable regulations with respect to in vitro diagnostics that are designed, manufactured, and used within a single laboratory for use only in that laboratory. These tests are referred to as LDTs.Laboratory Developed Tests (“LDTs”). As a result, we believe our diagnostic services are currently subject to the FDA’s enforcement discretion and are not currently subject to the FDA’s oversight. However, reagents, instruments, software or components provided by third parties and used to perform LDTs may be subject to regulation.
In recent years, the FDA has stated its intention to modify its enforcement discretion policy with respect to LDTs. For example,Most recently, on July 31, 2014, the FDA notified Congress of its intent to modify, in a risk-based manner, its policy of enforcement discretion with respect to LDTs. On October 3, 2014,2023, the FDA issued two draft guidance documents entitled “Framework for Regulatory Oversight of Laboratory Developed Tests (LDTs),” or the Framework Guidance, and “FDA Notification and Medical Device Reporting for Laboratory Developed Tests (LDTs),” or the Reporting Guidance. The Framework Guidance states that FDA intends to modifyproposed regulations under which it would phase out its policy of enforcement discretion with respect to LDTs in a risk-based manner consistent with the classification of medical devices generally in Classes I through III. The Reporting Guidance would further enable FDA to collect information regarding the LDTs currently being offered for clinical use through a notification process, as well as to enforce its regulations for reporting safety issues and collecting information on any known or suspected adverse events related to the use of an LDT.
Medical Device Regulatory Framework
Although we currently market our proprietary testing products as LDTs, which are currently subject to enforcement discretion, we could be subject to more onerous FDA compliance obligations in the future. Specifically, if the FDA begins to actively regulate LDTs, then, unless an exemption applies, each new or significantly modified medical device we seek to commercially distribute in the United States will require either a premarket notification to the FDA requesting permission for commercial distribution under Section 510(k) of the FDCA, also referred to as a 510(k) clearance, or approval from the FDA of a premarket approval, or PMA, application. Both the 510(k) clearance and PMA processes can be resource intensive, expensive, and lengthy, and require payment of significant user fees.
Device Classification
Under the FDCA, medical devices are classified into one of three classes-Class I, Class II or Class III depending on the degree of risk associated with each medical device and the extent of control needed to provide reasonable assurances with respect to safety and effectiveness.
Class I includes devices are those with the lowest risk to the patient and are those for which safety and effectiveness can be reasonably assured by adherence to a set of FDA regulations, referred to as the General Controls for Medical Devices, which require compliance with the applicable portions of the FDA’s Quality System Regulation, facility registration and product listing, reporting of adverse events and malfunctions, and appropriate, truthful and non-misleading labeling and promotional materials. Some Class I devices also require premarket clearance by the FDA through the 510(k) premarket notification process described below. Most Class I products are exempt from the premarket notification requirements.
Class II devices are those that are subject to the General Controls, and Special Controls as deemed necessary by the FDA to ensure the safety and effectiveness of the device. These Special Controls can include performance standards, patient registries, FDA guidance documents and post-market surveillance. Most Class II devices are subject to premarket review and clearance by the FDA. Premarket review and clearance by the FDA for Class II devices is accomplished through the 510(k) premarket notification process.
Class III devices include devices deemed by the FDA to pose the greatest risk such as life-supporting or life-sustaining devices, or implantable devices, in addition to those deemed novel and not substantially equivalent following the 510(k) process. The safety and effectiveness of Class III devices cannot be reasonably assured solely by the General Controls and Special Controls described above. Therefore, these devices are subject to the PMA application process, which is generally more costly and time-consuming than the 510(k) process. Through the PMA application process, the applicant must submit data and information demonstrating reasonable assurance of the safety and effectiveness of the device for its intended use to the FDA’s satisfaction. Accordingly, a PMA typically includes, but is not limited to, extensive technical information regarding device design and development, pre-clinical
17
and clinical trial data, manufacturing information, labeling and financial disclosure information for the clinical investigators in device studies. The PMA application must provide valid scientific evidence that demonstrates to the FDA’s satisfaction a reasonable assurance of the safety and effectiveness of the device for its intended use.
The Investigational Device Process
In the United States, absent certain limited exceptions, human clinical trials intended to support medical device clearance or approval require an investigational device exemption or IDE,(“IDE”), application. Some types of studies deemed to present “non-significant risk” are deemed to have an approved IDE once certain requirements are addressed and Institutional Review Board, or IRB approval is obtained. If the device presents a “significant risk” to human health, as defined by the FDA, the sponsor must submit an IDE application to the FDA and obtain IDE approval prior to commencing the human clinical trials. The IDE application must be supported by appropriate data, such as animal and laboratory testing results, showing that it is safe to test the device in humans and that the testing protocol is scientifically sound. Generally, clinical trials for a significant risk device may begin once the IDE application is approved by the FDA and the study protocol and informed consent are approved by appropriate IRBs at the clinical trial sites. Submission of an IDE will notenot necessarily result in the ability to commence clinical trials, and although the FDA’s approval of an IDE allows clinical testing to go forward for a specified number of subjects, it does not bind the FDA to accept the results of the trial as sufficient to prove the product’s safety and efficacy, even if the trial meets its intended success criteria.
All clinical trials must be conducted in accordance with the FDA’s IDE regulations that govern investigational device labeling, prohibit promotion and specify an array of recordkeeping, reporting and monitoring responsibilities of study sponsors and study investigators. Clinical trials must further comply with the FDA’s good clinical practice regulations for IRB approval and for informed consent and other human subject protections. Required records and reports are subject to inspection by the FDA. The results of clinical testing may be unfavorable, or, even if the intended safety and efficacy success criteria are achieved, may not
be considered sufficient for the FDA to grant marketing approval or clearance of a product. The commencement or completion of any clinical trial may be delayed or halted, or be inadequate to support approval of a PMA application, for numerous reasons.
The 510(k) Clearance Process
Under the 510(k) clearance process, the manufacturer must submit to the FDA a premarket notification, demonstrating that the device is “substantially equivalent” to a legally marketed predicate device. A predicate device is a legally marketed device that is not subject to a PMA, i.e., a device that was legally marketed prior to May 28, 1976 (pre-amendments device) and for which a PMA is not required, a device that has been reclassified from Class III to Class II or I, or a device that was previously found substantially equivalent through the 510(k) process. To be “substantially equivalent,” the proposed device must have the same intended use as the predicate device, and either have the same technological characteristics as the predicate device or have different technological characteristics and not raise different questions of safety or effectiveness than the predicate device. Clinical data is sometimes required to support substantial equivalence.
After a 510(k) premarket notification is submitted, the FDA determines whether to accept it for substantive review. If it lacks necessary information for substantive review, the FDA will refuse to accept the 510(k) notification. If it is accepted for filing, the FDA begins a substantive review. By statute, the FDA is required to complete its review of a 510(k) notification within 90 days of receiving the 510(k) notification. As a practical matter, clearance often takes longer, and clearance is never assured. Although many 510(k) premarket notifications are cleared without clinical data, the FDA may require further information, including clinical data, to make a determination regarding substantial equivalence, which may significantly prolong the review process. If the FDA agrees that the device is substantially equivalent, it will grant clearance to commercially market the device.
If the FDA determines that the device is not “substantially equivalent” to a predicate device, or if the device is automatically classified into Class III, the device sponsor must then fulfill the much more rigorous premarketing requirements of the PMA approval process, or seek reclassification of the device through the de novo process. The de novo classification process is an alternate pathway to classify medical devices that are automatically classified into Class III, but which are low to moderate risk. A manufacturer can submit a petition for direct de novo review if the manufacturer is unable to identify an appropriate predicate device and the new device or new use of the device presents a moderate or low risk. De novo classification may also be available after receipt of a “not substantially equivalent” letter following submission of a 510(k) to FDA.
After a device receives 510(k) clearance, any modification that could significantly affect its safety or effectiveness, or that would constitute a new or major change in its intended use, will require a new 510(k) clearance or, depending on the modification, could require a PMA application. The FDA requires each manufacturer to determine whether
18
the proposed change requires a new submission in the first instance, but the FDA can review any such decision and disagree with a manufacturer’s determination. Many minor modifications are accomplished by a letter-to-file in which the manufacturemanufacturer documents the change in an internal letter-to-file. The letter-to-file is in lieu of submitting a new 510(k) to obtain clearance for such change. The FDA can always review these letters to file in an inspection. If the FDA disagrees with a manufacturer’s determination regarding whether a new premarket submission is required for the modification of an existing 510(k)-cleared device, the FDA can require the manufacturer to cease marketing and/or recall the modified device until 510(k) clearance or approval of a PMA application is obtained. In addition, in these circumstances, the FDA can impose significant regulatory fines or penalties for failure to submit the requisite application(s).
The PMA Approval Process
Following receipt of a PMA application, the FDA conducts an administrative review to determine whether the application is sufficiently complete to permit a substantive review. If it is not, the agency will refuse to file the PMA. If it is, the FDA will accept the application for filing and begin the review. The FDA has 180 days to review a filed PMA application, althoughhowever, in practice the application review of an application moreprocess often occurs over a significantly longer period of time.exceeds this deadline. During this review period, the FDA may request additional information or clarification of information already provided, and the FDA may issue a major deficiency letter to the applicant, requesting the applicant’s response to deficiencies communicated by the FDA.
Before approving or denying a PMA, an FDA advisory committee may review the PMA at a public meeting and provide the FDA with the committee’s recommendation on whether the FDA should approve the submission, approve it with specific conditions, or not approve it. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
Prior to approval of a PMA, the FDA may conduct inspections of the clinical trial data and clinical trial sites, as well as inspections of the manufacturing facility and processes. Overall, the FDA review of a PMA application generally takes between one and three years but may take significantly longer. The FDA can delay, limit or deny approval of a PMA application for many reasons, including:
•the device may not be shown safe or effective to the FDA’s satisfaction;
•the data from pre-clinical studies and/or clinical trials may be found unreliable or insufficient to support approval;
•the manufacturing process or facilities may not meet applicable requirements; and
•changes in FDA approval policies or adoption of new regulations may require additional data.
If the FDA evaluation of a PMA is favorable, the FDA will issue either an approval letter, or an approvable letter, the latter of which usually contains a number of conditions that must be met in order to secure final approval of the PMA. When and if those conditions have been fulfilled to the satisfaction of the FDA, the agency will issue a PMA approval letter authorizing commercial marketing of the device, subject to the conditions of approval and the limitations established in the approval letter. If the FDA’s evaluation of a PMA application or manufacturing facilities is not favorable, the FDA will deny approval of the PMA or issue a not approvable letter. The FDA also may determine that additional tests or clinical trials are necessary, in which case the PMA approval may be delayed for several months or years while the trials are conducted and data is submitted in an amendment to the PMA, or the PMA is withdrawn and resubmitted when the data are available. The PMA process can be expensive, uncertain and lengthy and a number of devices for which the FDA approval has been sought by other companies have never been approved by the FDA for marketing.
New PMA applications or PMA supplements are required for modification to the manufacturing process, equipment or facility, quality control procedures, sterilization, packaging, expiration date, labeling, device specifications, ingredients, materials or design of a device that has been approved through the PMA process. PMA supplements often require submission of the same type of information as an initial PMA application, except that the supplement is limited to information needed to support any changes from the device covered by the approved PMA application and may or may not require as extensive technical or clinical data or the convening of an advisory panel, depending on the nature of the proposed change.
In approving a PMA application, as a condition of approval, the FDA may also require some form of post-approval study or post-market surveillance, whereby the applicant conducts a follow-up study or follows certain patient groups for a number of years and makes periodic reports to the FDA on the clinical status of those patients when necessary to protect the public health or to provide additional or longer termlonger-term safety and effectiveness data for the
19
device. The FDA may also approve a PMA application with other post-approval conditions intended to ensure the safety and effectiveness of the device, such as, among other things, restrictions on labeling, promotion, sale, distribution and use. New PMA applications or PMA supplements may also be required for modifications to any approved diagnostic tests, including modifications to our manufacturing processes, device labeling and device design, based on the findings of post-approval studies.
Federal and State Physician Self-Referral Prohibitions
We are subject to the federal physician self-referral prohibitions, commonly known as the Stark Law, and to comparable state laws. Together these restrictions generally prohibit us from billing a patient or any governmental or private payerpayor for certain designated health services, including clinical laboratory services, when the physician ordering the service, or any member of such physician’s immediate family, has a financial interest, such as an ownership or investment interest in or compensation arrangement with us, unless the arrangement meets an exception to the prohibition.
Sanctions for a Stark Law violation include the following:
•denial of payment for the services provided in violation of the prohibition;
•refunds of amounts collected by an entity in violation of the Stark Law;
•a civil penalty for each bill or claim for a service arising out of the prohibited referral;
•the imposition of up to three times the amounts for each item or service wrongfully claimed;
•possible exclusion from federal healthcare programs, including Medicare and Medicaid; and
•a civil penalty for each arrangement or scheme that the parties know (or should know) has the principal purpose of circumventing the Stark Law’s prohibition.
These prohibitions apply regardless of any intent by the parties to induce or reward referrals or the reasons for the financial relationship and the referral. In addition, knowing violations of the Stark Law may also serve as the basis for liability under the federal False Claims Act or the FCA,(the “FCA”), which can result in additional civil and criminal penalties.
Federal and State Anti-Kickback Laws
The federal Anti-Kickback Statute or the AKS,(the “AKS”) makes it a felony for a person or entity, including a clinical laboratory, to knowingly and willfully offer, pay, solicit or receive any remuneration, directly or indirectly, overtly or covertly, in cash or in kind, in order to induce business that is reimbursable under any federal health carehealthcare program. A violation of the AKS may result in imprisonment for up to ten years and fines for each violation and administrative civil money penalties, including an additional amount of up to three times the amount of the remuneration paid. Convictions under the AKS result in mandatory exclusion from federal health carehealthcare programs for a minimum of five years. In addition, The U.S. Department of Health and
Human Services or HHS,(“HHS”) has the authority to impose civil assessments and fines and to exclude health carehealthcare providers and others engaged in prohibited activities from Medicare, Medicaid and other federal health carehealthcare programs. In addition, the government may assert that a claim that includes items or services resulting from a violation of the AKS constitutes a false or fraudulent claim under the FCA, which is discussed in greater detail below. Additionally, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
Although the AKS applies only to items and services reimbursable under any federal health carehealthcare program, a number of states have passed statutes substantially similar to the AKS that apply to all payors. Penalties of such state laws include imprisonment and significant monetary fines.
Federal and state law enforcement authorities scrutinize arrangements between health carehealthcare providers and potential referral sources to ensure that the arrangements are not designed as a mechanism to induce patient care referrals or induce the purchase or prescribing of particular products or services. Generally, courts have taken a broad interpretation of the scope of the AKS, holding that the statute may be violated if merely one purpose of a payment arrangement is to induce referrals or purchases.
In addition to statutory exceptions to the AKS, regulations provide for a number of safe harbors. If an arrangement meets the provisions of a safe harbor, it is deemed not to violate the AKS. An arrangement must fully comply with each element of an applicable safe harbor in order to qualify for protection.
20
Failure to meet the requirements of the safe harbor, however, does not render an arrangement illegal. Rather, the government may evaluate such arrangements on a case-by-case basis, taking into account all facts and circumstances.
The Eliminating Kickbacks in Recovery Act
The Eliminating Kickbacks in Recovery Act of 2018 (“EKRA”) prohibits knowingly and willfully soliciting or receiving any remuneration (including any kickback, bribe or rebate) directly or indirectly, overtly or covertly, in cash or in kind, in return for referring a patient or patronage to a laboratory; or paying or offering any remuneration (including any kickback, bribe or rebate) directly or indirectly, overtly or covertly, in cash or in kind, to induce a referral of an individual to a laboratory or in exchange for an individual using the services of that laboratory. EKRA was enacted to help reduce opioid-related fraud and abuse. However, EKRA defines the term “laboratory” broadly and without reference to any connection to substance use disorder treatment. EKRA applies to all payors including commercial payors and government payors. The law includes a limited number of exceptions, some of which closely align with corresponding AKS exceptions and safe harbors, and others that materially differ. Currently, there is no regulation interpreting or implementing EKRA, nor any guidance released by a federal agency regarding the scope of EKRA.
Other Federal and State Health CareHealthcare Laws
In addition to the requirements discussed above, several other health carehealthcare fraud and abuse laws could have an effect on our business. For example, provisions of the Social Security Act permit Medicare and Medicaid to exclude an entity that charges the federal health carehealthcare programs substantially in excess of its usual charges for its services. The terms “usual charge” and “substantially in excess” are subject to varying interpretations.
The FCA prohibits, among other things, a person from knowingly presenting, or causing to be presented, a false or fraudulent claim for payment or approval and from, making, using, or causing to be made or used, a false record or statement material to a false or fraudulent claim in order to secure payment or retaining an overpayment by the federal government. In addition to actions initiated by the government itself, the statute authorizes actions to be brought on behalf of the federal government by a private party having knowledge of the alleged fraud.fraud, through the FCA’s “qui tam” or whistleblower provision. Because the complaint is initially filed under seal, the action may be pending for some time before the defendant is even aware of the action. If the government intervenes and is ultimately successful in obtaining redress in the matter or if the plaintiff succeeds in obtaining redress without the government’s involvement, then the plaintiff will receive a percentage of the recovery. Finally, the Social Security Act includes its own provisions that prohibit the filing of false claims or submitting false statements in order to obtain payment. Several states have enacted comparable false claims laws which may be broader in scope and apply regardless of payor.
The civil monetary penalties statute imposes penalties against any person or entity that, among other things, is determined to have presented, or caused to be presented, a claim to a federal health program that the person knows or should know is for an item or service that was not provided as claimed or is false or fraudulent. A person who offers or provides to a Medicare or Medicaid beneficiary any remuneration, including waivers of co-payments and deductible amounts (or any part thereof), that the person knows or should know is likely to influence the beneficiary’s selection of a particular provider, practitioner or supplier of Medicare or Medicaid payable items or services may be liable under the civil monetary penalties statute. Moreover, in certain cases, providers who routinely waive copaymentsco-payments and deductibles for Medicare and Medicaid beneficiaries, for example, in connection with patient assistance programs, can also be held liable under the AKS and False Claims Act.the FCA. One of the statutory exceptions to the prohibition is non-routine, unadvertised waivers of copaymentsco-payments or deductible amounts based on individualized determinations of financial need or exhaustion of reasonable collection efforts. The U.S. Department of Health and Human Services Office of Inspector General of HHS, or OIG,(the “OIG”) emphasizes, however, that this exception should only be used occasionally to address special financial needs of a particular patient. Although this prohibition applies only to federal healthcare program beneficiaries, applicable state laws related to, among other things, unlawful schemes to defraud, excessive fees for services, tortious interference with patient contracts and statutory or common law fraud, may also be implicated for similar practices offered to patients covered by commercial payor.payors.
The federal Health Insurance Portability and Accountability Act of 1996 or HIPAA,(“HIPAA”) created newadditional federal criminal statutes that prohibit, among other actions, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payors, and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of, or payment for, healthcare benefits, items or services. Like the AKS, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
21
The Physician Payments Sunshine Act, enacted as part of the ACA,Patient Protection and Affordable Care Act of 2010, as amended among other things,by the Health Care and Education Reconciliation Act of 2010 (collectively, the “ACA”) also imposed annual reporting requirements on manufacturers of certain devices, drugs and biologics for certain payments and transfers of value by them and in some cases their distributors to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors), other healthcare professionals (such as physician assistants and nurse practitioners) and teaching hospitals, and certain other healthcare professionals as of January 1, 2022; as well as information regarding ownership and investment interests held by physicians and their immediate family members. Any failure to comply with these reporting requirements could result in significant fines and penalties. Because we manufacture our ownand other companies with LDTs solely for use by or within our own laboratory,are considered healthcare providers rather than device manufacturers, we believe that we are exempt from these reporting requirements. We cannot assure you, however, that the government will agree with our determination,determination. Despite maintaining it has clear regulatory authority over LDTs, the FDA generally has not regulated them and a determination that wehas traditionally exercised enforcement discretion, choosing not to enforce applicable statutory and regulatory requirements. Therefore, most of these tests have violated these laws and regulations,neither undergone premarket review nor received FDA clearance, authorization or a public announcement that we are being investigatedapproval for possible violations,marketing. We will continue to monitor the FDA’s position as changes in this respect could adverselymaterially affect our business, prospects, results of operations or financial condition.
State equivalents of each of the above federal laws, such as anti-kickback and false claims laws, that may impose similar or more prohibitive restrictions, and may apply to items or services reimbursed by non-governmental third-party payors, including private insurers.
If our operations are found to be in violation of any of the fraud and abuse laws described above or any other laws that apply to us, we may be subject to penalties, including potentially significant criminal, civil and administrative penalties, damages, fines, disgorgement, imprisonment, exclusion from participation in government healthcare programs, contractual damages, reputational harm, integrity oversight and reporting obligations, diminished profits and future earnings, and the curtailment or restructuring of our operations.
International Regulations
Many countries in which we may offer any of our testing products in the future have anti-kickback regulations prohibiting providers from offering, paying, soliciting or receiving remuneration, directly or indirectly, in order to induce business that is reimbursable under any national health carehealthcare program. In situations involving physicians employed by state-funded institutions or national health carehealthcare agencies, violation of the local anti-kickback law may also constitute a violation of the U.S. Foreign Corrupt Practices Act or FCPA.(“FCPA”).
The FCPA prohibits any U.S. individual, business entity or employee of a U.S. business entity to offer or provide, directly or through a third party, including any potential distributors we may rely on in certain markets, anything of value to a foreign government official with corrupt intent to influence an award or continuation of business or to gain an unfair advantage, whether or not such conduct violates local laws. In addition, it is illegal for a company that reports to the SEC to have false or inaccurate books or records or to fail to maintain a system of internal accounting controls. We will also be required to maintain accurate information and control over sales and distributors’ activities that may fall within the purview of the FCPA, its books and records provisions and its anti-bribery provisions.
The standard of intent and knowledge in the Anti-Bribery cases is minimal-intent and knowledge are usually inferred from thatthe fact that bribery took place. The accounting provisions do not require intent. Violations of the FCPA’s anti-bribery provisions for corporations and other business entities are subject to a fine of up to $2 million and officers, directors, stockholders, employees, and agents are subject to a fine of up to $100,000 and imprisonment for up to five years. Other countries, including the United Kingdom (“UK”) and other OECD Anti-Bribery Convention members, have similar anti-corruption regulations, such as the United Kingdom Anti-Bribery Act.
When marketing our testing products outside of the United States, we may be subject to foreign regulatory requirements governing human clinical testing, prohibitions on the import of tissue necessary for us to perform our testing products or restrictions on the export of tissue imposed by countries outside of the United States or the import of tissue into the United States, and marketing approval. These requirements vary by jurisdiction, differ from those in the United States and may in some cases require us to perform additional pre-clinical or clinical testing. In many countries outside of the United States, coverage, pricing and reimbursement approvals are also required.
Privacy and Security Laws
Health Insurance Portability and Accountability Act
Under HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act or HITECH,(“HITECH”) HHS has issued regulations to protect the privacy and provide for the security of protected health information or PHI,(“PHI”) used or disclosed by certain entities including health carehealthcare providers, such as us. HIPAA also regulates
22
standardization of data content, codes and formats used in certain health carehealthcare transactions and standardization of identifiers for health plans and providers. Penalties for violations of HIPAA and HITECH laws and regulations include significant civil and criminal penalties.
Three standards have been promulgated under HIPAA’s and HITECH’s regulations: the Standards for Privacy of Individually Identifiable Health Information, which restrict the use and disclosure of certain individually identifiable health information, the Standards for Electronic Transactions, which establish standards for common healthcare transactions, such as claims information, plan eligibility, payment information and the use of electronic signatures, and the Security Standards for the
Protection of Electronic Protected Health Information, which require covered entities and business associates to implement and maintain certain security measures to safeguard certain electronic health information, including the adoption of administrative, physical and technical safeguards to protect such information.
The HIPAA privacy regulations cover the use and disclosure of PHI by covered entities as well asand business associates, which are defined to include subcontractors that create, receive, maintain, or transmit PHI on behalf of a business associate.covered entity, as well as their covered subcontractors. They also set forth certain rights that an individual has with respect to his or her PHI maintained by a covered entity, including the right to access or amend certain records containing PHI, or to request restrictions on the use or disclosure of PHI. The HIPAA security regulations establish requirements for safeguarding the confidentiality, integrity, and availability of PHI that is electronically transmitted or electronically stored. HITECH, among other things, established certain health information security breach notification requirements. A covered entity must notify any individual whose PHI is breached according to the specifications set forth in the breach notification rule. The HIPAA privacy and security regulations establish a uniform federal “floor” and do not preempt state laws that are more stringent or provide individuals with greater rights with respect to the privacy or security of, and access to, their records containing PHI or insofar as such state laws apply to personal information that is broader in scope than PHI.
Individuals (or their personal representatives, as applicable) have the right to access test reports directly from laboratories and to direct thatthe copies of those reports to be transmitted to persons or entities designated by the individual.
HIPAA authorizes state attorneys general to file suit on behalf of their residents for violations. Courts are able to award damages, costs and attorneys’ fees related to violations of HIPAA in such cases. While HIPAA does not create a private right of action allowing individuals to file suit against us in civil court for violations of HIPAA, its standards have been used as the basis for duty of care cases in state civil suits such as those for negligence or recklessness in the misuse or breach of PHI. In addition, HIPAA mandates that the Secretary of HHS conduct periodic compliance audits of HIPAA covered entities, such as us, and their business associates for compliance with the HIPAA privacy and security standards. It also tasks HHS with establishing a methodology whereby harmed individuals who were the victims of breaches of unsecured PHI may receive a percentage of the civil monetary penalty paid by the violator.
As a covered entity with downstream vendors and subcontractors and, in certain instances, as a business associate of other covered entities with whom we have entered into a business associate agreement, we have certain obligations under HIPAA regarding the use and disclosure of any PHI that may be provided to us. HIPAA and HITECH impose civil and criminal penalties against covered entities and business associates for noncompliance with privacy and security requirements. Further, various states, such as California and Massachusetts, have implemented similar privacy laws and regulations that impose restrictive requirements regulating the use and disclosure of health information and other personally identifiable information.information (“PII”).
Numerous other federal, state and foreign laws, including consumer protection laws and regulations, govern the collection, dissemination, use, access to, confidentiality and security of patient health information. We intend to continue to comprehensively protect all personal information and to comply with all applicable laws regarding the protection of such information.
Reimbursement for Clinical Laboratory Services
We generate revenue on our products from several sources, including third-party payors, laboratory services intermediaries, and self-paying individuals. Depending on the billing arrangement and applicable law, we must bill various third-party payors, such as insurance companies, Medicare, Medicaid, and patients, all of which have different billing requirements. Compliance with applicable laws and regulations as well as internal compliance policies and procedures adds further complexity to the billing process. CMS establishes new procedures and continuously evaluates and implements changes to the reimbursement process for billing the Medicare program.
23
To receive reimbursement from third-party payors, we bill our tests using a variety of CPT codes, as defined by the AMA.AMA CPT Editorial Panel. For many of thethose genetic tests we conduct there maythat do not be an appropriatehave a dedicated CPT code, for some of the genes in a panel, in which case the testtests may be billed under a miscellaneous code for an unlisted molecular pathology procedure. Because these miscellaneous codes do not describe a specific service, the third-party payor claim may need to be examined to determine the service that was provided, whether the service was appropriate and medically necessary and whether payment should be rendered. This process can require a letter of medical necessity and other types of medical documentation from the ordering physician and it can result in a delay in processing the claim, a lower reimbursement amount, or denial of the claim.
With the evolution of genetic testing, we have seen individual third-party payors’ medical coverage policies around the CPT codes we bill and their associated payment rates change over time, resulting in changes to our reimbursement revenues.reimbursement. We believe all of our products provide significant clinical value and reduction in downstream healthcare spend, as evidenced in research studies and clinical publications, which we believe will continue to support and drive third-party payor reimbursement.
Under Medicare, payment for our laboratory tests areproducts like ours is generally made under the Clinical Laboratory Fee Schedule, or CLFS with payment amounts assigned to specific procedure billing codes. In April 2014, Congress passed the Protecting Access to Medicare Act (PAMA)(“PAMA”), which included substantial changes to the way in which clinical laboratory services will be paid under Medicare. Under PAMA, certain laboratories that receive the majority of their Medicare revenue from payments made under the CLFS or the Physician Fee Schedule arewere required to report to CMS beginning in 2017 and every three years thereafter (or annually for ADLTs), private payor payment rates and volumes for their tests. CMS uses this data to calculate a weighted median payment rate for each test, which will be used to establish revised Medicare CLFS reimbursement rates for the test. Laboratories that fail to report the required payment information may be subject to substantial civil monetary penalties. As required under PAMA, CMS will use the ratesWe bill Medicare for our products, and volumes reported by laboratoriestherefore we are subject to develop Medicare payment rates for laboratory tests equal to the volume-weighted median of the private payor payment rates for the tests. On June 23, 2016, CMS published the final rule implementing the reporting and rate-setting requirements under PAMA. See “Reimbursement—Government Payors” above for additional information.
PAMA also authorizes the adoption of new, temporary billing codes and/or unique test identifiers for FDA-cleared or approved tests as well as ADLTs. The AMA’s CPT Editorial Panel has approved a proposal to create a new section of billingnow issues PLA codes to facilitate implementationin support of this section of PAMA. These proprietary laboratory analysesPLA codes may be requested by a clinical laboratory or manufacturer to specifically identify their test. If approved, the codes are issued by the AMA on a quarterly basis. Our DecisionDx-UM test was granted a Category I MAAA CPT code and was effective January 1, 2020. Our DecisionDx-Melanoma test was granted a Category I MAAA CPT code that will beand was effective January 1, 2021. Our MyPath Melanoma test was granted a PLA CPT code prior to our May 2021 acquisition of the Myriad MyPath Laboratory. Our TissueCypher test was granted a PLA CPT code prior to our December 2021 acquisition of Cernostics. Our DecisionDx-SCC and DiffDx-Melanoma tests were granted PLA CPT codes effective April 1, 2022. In 2023, IDgenetix test was granted a new PLA CPT code which became effective October 1, 2023.
Healthcare Reform
In March 2010, the Patient Protection and Affordable Care Act of 2010, as amended by the Health Care and Education Reconciliation Act of 2010, collectively the ACA was enacted in the U.S. The ACA made a number of substantial changes tobecame law. This law substantially changed the way healthcare is financed by both by governmentalgovernment and private insurers. There remain judicialcommercial third-party payors, and Congressional challenges to certain aspects of the ACA, as well as efforts by the current administration to repeal or replace certain aspects of the ACA. For example,significantly impacted our industry. Among other things, the ACA required each medical device manufacturermanufacturers to pay a sales tax equal to 2.3% of the price for which such manufacturer sells its medical devices. However,devices, and began to apply to sales of taxable medical devices after December 31, 2012, but was suspended in 2016. Further, the 2020 federal spending package permanently eliminated, effective January 1, 2020, the ACA-mandated medical device tax and “Cadillac” tax on high-cost employer-sponsored health coverage and, effective January 1, 2021, also eliminateseliminated the health insurer tax. The ACA also contains a number of other provisions, including provisions governing enrollment in federal and state healthcare programs, reimbursement matters and fraud and abuse, which we expect will impact our industry and our operations in ways that we cannot currently predict.
On August 2, 2011, the Budget Control Act of 2011 was signed into law, which, among other things, reduced Medicare payments to providers by 2% per fiscal year, effective on April 1, 2013 and, due to subsequent legislative amendments to the statute including the Infrastructure Investment and Jobs Act, will remain in effect through 2032, unless additional Congressional action is taken.
24
We anticipate there will continue to be proposals by legislators at both the federal and state levels, regulators and commercial third-party payors to reduce costs while expanding individual healthcare benefits. Certain of these changes could impose additional limitations on the prices we will be able to charge for our products, the coverage of or the amounts of reimbursement available for our products from third-party payors, including government and commercial payors.
Human Capital Resources
Overview
Our vision is to transform disease management by keeping people first: patients, clinicians, employees and investors. We understand the importance of maintaining a strong corporate culture with our employees at the center, based on the cornerstones we laid in 2008 at our inception: trust, excellence, collaboration, integrity, innovation and excitement. We strive to find members of our team who embody the values of our company. As of December 31, 2019,2023, we had 135 employees, of whom 133 were610 full-time employees. During the year ended December 31, 2023, we added 67 employees to our team, a 12% increase from 2022. We face competition for experienced, qualified personnel in our industry, particularly for highly skilled scientists, laboratory technicians and salespeople versed in diagnostic testing services.
The tables below provide information on the distribution of our employees by functional area and by location as of December 31, 2023:
| Number of Employees | |||||
| Laboratory Testing Operations | 164 | ||||
| Research & Development | 103 | ||||
| Sales & Marketing | 197 | ||||
| Administrative & General | 146 | ||||
| Total as of December 31, 2023 | 610 | ||||
| Number of Employees | |||||
| Friendswood, Texas | 105 | ||||
| Phoenix, Arizona | 181 | ||||
| Pittsburgh, Pennsylvania | 62 | ||||
| Home-based | 262 | ||||
| Total as of December 31, 2023 | 610 | ||||
Our employees are not represented by labor unions or covered by collective bargaining agreements. We consider our relationship with our employees to be good.
Diversity, Equity and Inclusion
We are committed to fostering, cultivating and preserving a culture of diversity, equity and inclusion (“DEI”). We are a company whose mission is improving health through innovative tests that guide patient care. Keeping people first—patients, clinicians, employees and investors—highlights a critical part of the patient-centric work we do.
Our DEI initiatives are applicable—but not limited—to our practices and policies on recruitment and selection; compensation and benefits; professional development and training; promotions; transfers; social and recreational programs; layoffs; terminations; and the ongoing cultivation of a work environment built on the premise of equity and belonging for employees of all backgrounds. We are committed to maintaining:
•Respectful communication and cooperation between all employees
•Teamwork and employee participation that enables the representation of all groups and employee perspectives
•Employer and employee contributions to the communities we serve to promote a greater understanding and respect for diversity
25
•Equitable policies, processes and practices
All of our employees have a responsibility to treat others with dignity and respect at all times. All employees are expected to exhibit conduct that promotes inclusion and belonging in the workplace, at work functions on or off the work site, and at all other company-sponsored events. Our DEI strategy and programs hinge upon three core pillars:
1.Recruiting a diverse workforce
2.Building a culture of inclusion
3.Promoting transparency
To ensure we are cultivating an authentic company culture, we will take the following actions:
•Conduct annual diversity awareness/unconscious bias training
•Monitor diversity data, including compensation data
•Offer mentorship programs or networking groups
•Support employee resource groups
As of December 31, 2023, our employees were 64% female and 36% male. Our overall employee population as of December 31, 2023 was 65% White, 21% Hispanic or Latino, 6% Asian, 3% Black or African-American and 5% two or more races (not Hispanic or Latino) and other. In executive positions, which we define as Executive Director or Regional Business Director level and above, our employee population as of December 31, 2023 was 72% White, 16% Hispanic or Latino and 12% other (not Hispanic or Latino). Females represented 39% of employees in executive positions.
Affirmative Action
Our DEI practices reaffirm our belief in and commitment to equal employment opportunity (“EEO”) for all employees and applicants in all aspects of employment.
We have developed and maintained a written Affirmative Action Program (“AAP”). Our President and Chief Executive Officer supports the AAP and urges each employee to commit to carrying out the intent of the AAP and this statement. We maintain an audit and reporting system to determine overall compliance with its EEO mandates. The EEO Administrator oversees the AAP development, modification, implementation, effectiveness and reporting requirements, and conducts management updates.
We strive to ensure all aspects of employment, including recruitment, selection, job assignment, training, compensation, benefits, discipline, promotion, transfer, layoff and termination processes remain free of illegal or unethical discrimination based upon race, color, religion, sex (including pregnancy, sexual orientation, gender identity or transgender status), age, national origin, genetic information, marital status, political affiliation, disability, status as a parent, protected veteran status, or a person’s relationship or association with a protected veteran. Regular review helps ensure compliance with this policy.
Employee Engagement
We value the unique perspective our employees bring to the organization and encourage open channels of communication. In June 2023, we conducted our third annual employee engagement survey to understand what was working well at Castle and what opportunities we had for improvement. We received feedback from over 95% of our employees and achieved an engagement score of 86%, meaning that 86% of our employees are engaged or enthusiastically engaged in the culture at Castle. Our engagement score was higher than the healthcare benchmark average of 75% for other healthcare companies who conducted the same employee engagement survey in 2023.
Compensation, Benefits and Professional Development
We are committed to offering competitive benefits and compensation packages to our employees. In addition to competitive base pay, we offer the following benefits, among others, to our full-time employees:
•a defined contribution 401(k) plan with employer matching contributions;
•an annual bonus opportunity;
•equity compensation, including stock options and/or restricted stock units (“RSUs”) and an employee stock purchase plan;
•medical, dental and vision plans;
26
•paid maternity, paternity and adoption leave policies;
•paid holidays and paid time off; and
•an employee assistance program.
We survey all new hires 90 days after the start of their employment to solicit feedback on employee engagement. We provide performance reviews at least once per year, with pay raises commensurate with market and performance indicators. Our turnover remains low for the year ended December 31, 2023.
We prioritize and encourage internal growth and professional development of our employees. To encourage employee development, we offer a professional development reimbursement program to eligible employees who attend job-related professional development activities.
Corporate and Other Information
We were incorporated in Delaware in September 2007. Our principal executive offices are located at 820505 S. Friendswood Drive, Suite 201,401, Friendswood, Texas 77456 and our telephone number is (866) 788-9007. Our corporate website address is www.CastleBiosciences.com. Information contained on, or accessible through, our website is not considered part of this Annual Report on Form 10-K or our other filings with the SEC. Our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and any amendments to such reports filed or furnished pursuant to Section 13(a) and 15(d) of the Securities Exchange Act of 1934, as amended or the Exchange Act,(the “Exchange Act”) are available free of charge on our website as soon as reasonably practicable after we electronically file such material with, or furnish it to, the SEC.
This Annual Report on Form 10-K contains references to our trademarks and to trademarks belonging to other entities. Solely for convenience, trademarks and trade names referred to in this Annual Report on Form 10-K, including logos, artwork and other visual displays, may appear without the ® or TM symbols, but such references are not intended to indicate, in any way, that their respective owners will not assert, to the fullest extent under applicable law, their rights thereto. We do not intend our use or display of other companies’ trade names or trademarks to imply a relationship with, or endorsement of or sponsorship of us by, any other companies.
27
Item 1A. Risk Factors.
Risk Factors
You should consider carefully the risks described below, as well as the other information in this Annual Report on Form 10‑K, before deciding whether to purchase, hold or sell shares of our common stock. The occurrence of any of the following risks could harm our business, financial condition, results of operations and/or growth prospects or cause our actual results to differ materially from those contained in forward-looking statements we have made in this report and those we may make from time to time. You should consider all of the factors described as well as the other information in thethis Annual Report on Form 10-K, including our consolidated financial statements and the related notes and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” when evaluating our business. If any of the following risks actually occur, our business, financial condition, results of operations and future growth prospects could be materially and adversely affected. In these circumstances, the market price of our common stock could decline and you may lose all or part of your investments.investment. Additional risks and uncertainties not presently known to us or that we currently deem immaterial also may impair our business operations.
Risks Related to Our Financial Condition
Our revenue for our test reports provided for patients covered by Medicare and United Healthcare as a percentage of total revenue, was 49% and 6%, respectively,53% for the years ended December 31, 2023 and 2022, respectively. Additionally, there was a commercial payor from which 14% of our revenue from patients were derived for the year ended December 31, 2019, and 36% and 12%, respectively, for the year ended December 31, 2018. In addition, our current accounts receivable balances for Medicare and United Healthcare, as a percentage of our total current accounts receivable, were 7% and 9%, respectively, as of December 31, 2019, and 54% and 7%, respectively, as of December 31, 2018. There were no long-term accounts receivable balances for Medicare and United Healthcare as of December 31, 2019, but long-term accounts receivable for Medicare and United Healthcare as a percentage of total long-term accounts receivable were and 0% and 15%, respectively, as of December 31, 2018.2023. If our largest current payors were to significantly reduce, or cease to pay, the amount they reimburse for our products, or if they do not reach favorable coverage and reimbursement decisions for our products, or attempt to recover amounts they had already paid, it could have a material adverse effect on our business, financial condition and results of operations and cause significant fluctuations in our results of operations.
Due to how we recognize revenue, our quarterly and annual revenues may not reflect our underlying business.
We have concluded that our contracts include variable consideration because the amounts paid by Medicare or commercial health insurance carriers may be paid at less than our standard rates or not paid at all, with such differences considered implicit price concessions. Variable consideration attributable to these price concessions is measured at the expected value using the ‘‘most likely amount’’ method under Accounting Standards Codification (‘‘ASC’’) Topic 606, Revenue from Contracts with Customers, or (“ASC 606.606”). The amounts are determined byestimated using historical average collection rates by test type and payor category taking into consideration the range of possible outcomes, the predictive value of our past experiences, the time period of when uncertainties expect to be resolved and the amount of consideration that is susceptible to factors outside of our influence, such as the judgment and actions of third parties. Determining variable consideration through a consideration of these factors involves a significant level of estimation uncertainty, and our estimations may turn out to be incorrect. Such variable consideration is included in the transaction price only to the extent it is probable that a significant reversal in the amount of cumulative revenue recognized will not occur when the uncertainties with respect to the amount are resolved. Variable consideration may be constrained and excluded from the transaction price in situations where there is no contractually agreed upon reimbursement coverage or in the absence of a predictable pattern and history of collectability with a payor. Variable consideration for Medicare claims for which there are no existing positive coverage decisions, including those claims undergoing appeal, is deemed to be fully constrained when the payment of such claims is subject to approval by an Administrative Law Judge, or ALJ, at an appeal hearing, due to factors outside our influence (i.e.(e.g., judgment or actions of third parties) and the uncertainty of the amount to be received is not expected to be resolved for a long period of time. Variable consideration is evaluated each reporting period and adjustments are recorded as increases or decreases in revenues. As a result of the timing and amount of adjustments for variable consideration, our operating results and comparisons of such results on a period-to-period basis may be difficult to understand and may not be meaningful. In addition, these fluctuations in revenue may make it difficult for us, for research analysts and for investors to accurately forecast our revenue and operating results. If our revenue or operating results fall below expectations, the price of our common stock would likely decline.
We have incurred significant losses since inception, and we may never achieve or sustain profitability.
Since our inception, we have had a history of net losses. AsFor the year ended December 31, 2023, we had a net loss of $57.5 million, and as of December 31, 2019,2023, we had a cash balance of approximately $98.8 million and an accumulated deficit of approximately $52.2$218.4 million. We cannot predict if we will achieve sustained profitability in the near future or at all. We expect that ourto incur losses will continue forin the foreseeable future as we plan to invest significant additional funds toward the expansion of our commercial organization, the conduct of clinical utility and
28
validity studies to support adoption of our products and the development or acquisition of additional products. AsWe also expect increases in our stock-based compensation expense in future periods due to additional awards outstanding, attributable to increased headcount. Additionally, our performance could be affected by the impacts of geopolitical and macroeconomic developments, such as the invasion of Ukraine by Russia and related sanctions or the Israel-Hamas war, economic slowdowns, labor shortages, recessions or market corrections, supply chain disruptions, inflation and monetary policy shifts, bank failures or other disruptions in the banking system or financing markets, rising interest rates and tightening of credit markets resulting from the conflict or other evolving macroeconomic developments. Due to the requirements associated with being a public company, including those associated with no longer qualifying as a smaller reporting company and becoming an accelerated filer, we
We are an early, commercial-stage company and have a limited operating history, which may make it difficult to evaluate our current business and predict our future performance.
We are an early commercial-stage company and have a limited operating history. Our limited operating history may make it difficult to evaluate our current business and this makes predictions about our future success or viability subject to significant uncertainty. In particular, we intend to use a portion of our working capital to increase our headcount, including through the expansion of our laboratory testing operations, sales and marketing and research and development teams, which will increase our operating costs in a manner not historically reflected in our consolidated financial statements. In combination with our other anticipated increased operating expenses in connection with becoming a public company, theseThese anticipated changes in our operating expenses may make it difficult to evaluate our current business, assess our future performance relative to prior performance and accurately predict our future performance.
We will continue to encounter risks and difficulties frequently experienced by early commercial-stage companies, including those associated with increasing the size of our organization and the prioritization of our commercial, research and business development activities. If we do not address these risks successfully, our business could suffer.
Changes in financial accounting standards or practices may cause adverse, unexpected financial reporting fluctuations and affect our reported operating results.
Accounting principles generally accepted in the United States of America are(“U.S. GAAP”) is subject to interpretation by the Financial Accounting Standards Board (“FASB”), the SEC, and various bodies formed to promulgate and interpret appropriate accounting principles. A change in accounting standards or practices can have a significant effect on our reported results and may even affect our reporting of transactions completed before the change is effective. New accounting pronouncements and varying interpretations of accounting pronouncements have occurred and may occur in the future. Changes to existing rules or the questioning of current practices may adversely affect our reported financial results or the way we conduct our business.
Our quarterly and annual operating results and cash flows may fluctuate in the future, which could cause the market price of our stock to decline substantially.
Numerous factors, many of which are outside our control may cause or contribute to significant fluctuations in our quarterly and annual operating results. These fluctuations may make financial planning and forecasting uncertain. In addition, these fluctuations may result in unanticipated decreases in our available cash, which could negatively affect our business and prospects. In addition, one or more of such factors may cause our revenue or operating expenses in one period to be disproportionately higher or lower relative to the others. As a result, comparing our operating results on a period-to-period basis may be difficult to understand and may not be meaningful. You should not rely on our past results as indicative of our future performance.
In addition, a significant portion of our operating expense is relatively fixed in nature, and planned expenditures are based in part on expectations regarding future revenue. Accordingly, unexpected revenue shortfalls could decrease our gross margins and cause significant changes in our operating results from quarter to quarter. If this occurs, the trading price of our stock could fall substantially.
29
This variability and unpredictability caused by factors such as those described above could also result in our failing to meet the expectations of industry or financial analysts or investors for any period. If our revenue or operating results fall below the expectations of analysts or investors or below any guidance we may provide, or if the guidance we provide is below the expectations of analysts or investors, the price of our common stock could decline substantially. Such a stock price decline could occur even when we have met any previously publicly stated guidance we may provide.
Effective internal control over financial reporting is necessary for us to provide reliable financial reports in a timely manner. In connection with the audits of our financial statements for the years ended December 31, 2018 and 2017, we concluded that
A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of the annual or interim consolidated financial statements will not be prevented or detected on a timely basis.
We may need to raise additional capital to fund our existing operations, commercialize new products or expand our operations.
We believe our existing cash and cash equivalents, marketable investment securities and anticipated cash generated from sales of our products will be sufficient to fund our operating expensesoperations for at least the foreseeable future.next 12 months. If our available cash balancesand cash equivalents, marketable investment securities and anticipated cash generated from sales of our products are insufficient to satisfy our liquidity requirements including because of lower demand for our products, lower than currently expected rates of reimbursement from third-party payors or other risks described in this Annual Report on Form 10-K, we may finance our cash needs through a combination of equity offerings, debt financings, collaborations, strategic alliances and marketing, distribution or licensing arrangements. We do not currently have any committed external source of funds. In addition, we may seek additional capital due to favorable market conditions or strategic considerations, even if we believe we have sufficient funds for our current or future operating plans.
We may consider raising additional capital in the future to expand our business, to pursue strategic investments, to take advantage of financing opportunities or for other reasons, including to:
•increase our sales and marketing efforts for the DecisionDx-Melanoma, DecisionDx-SCC, MyPath Melanoma, DecisionDx-UM, TissueCypher and IDgenetix tests and address competitive developments;developments among these or future commercial products;
•fund ongoing evidence development offor our existing products as well as additional pipeline products, including for SCC and suspicious pigmented lesions, in addition to other programs in development;programs;
•expand our laboratory testing facility and related testing capacity;
•expand our technologies into other types of skin cancer management and detection products;dermatological, ocular, gastrointestinal or mental health disorders;
•acquire, license or invest in technologies;
•acquire or invest in complementary businesses or assets; and
•finance capital expenditures and general and administrative expenses.
30
Our present and future funding requirements will depend on many factors, including:
•our ability to achieve revenue growth;
•our rate of progress in establishing payor coverage and reimbursement arrangements with third-party payors;
•our rate of progress in, and cost of the sales, marketing, coverage and reimbursement activities associated with, establishing adoption of our lead product, DecisionDx-Melanoma, among our other products;
•the cost of expanding our laboratory operations and offerings, including our sales, marketing, coverage and reimbursement efforts;
•our rate of progress in, and cost of research and development activities associated with, diagnostic products in research and early development;
•the potential cost of, and delays in, the development of new products as a result of changes in regulatory oversight applicable to our products;
•acquisitions of businesses, assets, products or technologies;
•the duration and effects of elevated inflation;
•the effects on our operations of general political and economic conditions and evolving macroeconomic developments, including geopolitical and macroeconomic developments, such as the ongoing conflict between Ukraine by Russia and related sanctions or the Israel-Hamas war, public health crises, economic slowdowns, labor shortages, recessions or market corrections, supply chain disruptions, inflation and monetary policy shifts, bank failures or other disruptions in the banking system or financing markets, rising interest rates and tightening of credit markets resulting from the conflict or other evolving macroeconomic developments; and
•the effect of competing technological and market developments.
To the extent that we raise additional capital through the sale of equity or convertible debt securities, your ownership interest will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect your rights as a common stockholder. Debt financing and preferred equity financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making acquisitions or capital expenditures or declaring dividends.
If we raise additional funds through collaborations, strategic alliances or marketing, distribution or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or products, or grant licenses on terms that may not be favorable to us.
Any disruptions to, or volatility in, the credit and financial markets or any deterioration in overall economic conditions may make any necessary debt or equity financing more difficult to obtain, more costly and/or more dilutive. If we are unable to raise additional funds through equity or debt financings or other arrangements when needed, we may be required to delay, limit, reduce or terminate our commercialization, research and development efforts or grant rights to third parties to market and/or develop products that we would otherwise prefer to market and develop ourselves.
Risks Related to Our Business
Our revenue currently depends primarily on sales of DecisionDx-Melanoma, and we will need to generate sufficient revenue from this and other products to grow our business.
We believe that our long-term commercial success, and ability to generate revenue, will depend on our ability to develop and market additional products, such ason our pipeline products for SCC and suspicious pigmented lesions. Our ability to derive revenue from DecisionDx-Melanoma, DecisionDx-UMincrease market penetration for our existing and anypotential future products that we commercialize is uncertain and depends on our ability to obtain favorable coverage and reimbursement policies from government payors, likesuch as Medicare, and from private payors, likesuch as insurance companies.
Without positive coverage policies, our products may not be reimbursed and we may not be able to recognize revenue. If we are unable to increase sales and expand coverage and reimbursement for
DecisionDx-Melanoma and our other tests, develop and commercialize other products, and successfully obtain coverage and adequate
31
reimbursement for such products, our revenue and our ability to achieve and sustain profitability would be impaired, and the market price of our stock could decline substantially.
Unfavorable U.S. and global economic conditions could adversely affect our business, financial condition, results of operations or cash flows.
Our results of operations could be adversely affected by general conditions in the U.S. and global economies, the U.S. and global financial markets and adverse macroeconomic developments. U.S. and global market and economic conditions have been, and continue to be, disrupted and volatile due to many factors, including public health crises such as the COVID-19 pandemic, geopolitical and macroeconomic developments, such as the Israel-Hamas war and the ongoing conflict between Ukraine and Russia and related sanctions, economic slowdowns, labor shortages, recessions or market corrections, supply chain disruptions, inflation and monetary policy shifts, liquidity concerns, at, and failures of, banks and other financial institutions or other disruptions in the banking system or financial markets, rising interest rates and tightening of credit markets resulting from the conflict or other evolving macroeconomic developments, among others. General business and economic conditions that could affect our business, financial condition or results of operations include fluctuations in economic growth, debt and equity capital markets, liquidity of the global financial markets, the availability and cost of credit, investor and consumer confidence, and the strength of the economies in which we, our collaborators, our manufacturers and our suppliers operate.
A severe or prolonged global economic downturn could result in a variety of risks to our business. For example, inflation rates, particularly in the United States, have increased recently to levels not seen in years, and increased inflation may result in increases in our operating costs (including our labor costs), reduced liquidity and limits on our ability to access credit or otherwise raise capital on acceptable terms, if at all. In addition, the U.S. Federal Reserve has raised, and may again raise, interest rates in response to concerns about inflation, which coupled with reduced government spending and volatility in financial markets may have the effect of further increasing economic uncertainty and heightening these risks. Furthermore, the recent closures of Silicon Valley Bank, Signature Bank and First Republic Bank have resulted in broader financial institution liquidity risk and concerns. Future adverse developments with respect to specific financial institutions or the broader financial services industry may lead to market-wide liquidity shortages that could materially harm our business and financial condition. In this regard, we continue to maintain our cash deposits with banking institutions, often in balances that exceed the current Federal Deposit Insurance Corporation insurance limits, and the failure of any bank in which we deposit our funds could reduce the amount of cash we have available for our operations, limit our access to additional capital on favorable terms, or at all, or delay our ability to access such funds or collect receivables, which could negatively affect our financial condition and our ability to pursue our business strategy.
Risks of a prolonged global economic downturn are particularly true in Europe, which is undergoing a continued severe economic crisis. A weak or declining economy could also strain our suppliers and manufacturers, possibly resulting in supply disruption. Any of the foregoing could harm our business and we cannot anticipate all of the ways in which the current economic climate and financial market conditions could adversely impact our business.
Additionally, financial markets around the world experienced volatility following the invasion of Ukraine by Russia in February 2022. In response to the invasion, the United States, UK and European Union (“EU”), along with others, imposed significant new sanctions and export controls against Russia, Russian banks and certain Russian individuals and may implement additional sanctions or take further punitive actions in the future. The full economic and social impact of the sanctions imposed on Russia (as well as possible future punitive measures that may be implemented), as well as the counter measures imposed by Russia, in addition to the ongoing military conflict between Ukraine and Russia, which could conceivably expand into the surrounding region, remains uncertain; however, both the conflict and related sanctions have resulted and could continue to result in disruptions to trade, commerce, pricing stability, credit availability and/or supply chain continuity in both Europe and globally, and has introduced significant uncertainty into global markets. In particular, the Russia-Ukraine conflict has contributed to rapidly rising costs of living (driven largely by higher energy prices) in Europe and other advanced economies. Further, a weak or declining economy could strain our suppliers, manufacturers and collaborators, possibly resulting in additional supply disruption for our product candidates. As a result, our business and results of operations may be adversely affected by the ongoing conflict between Ukraine and Russia, particularly to the extent it escalates to involve additional countries, further economic sanctions or wider military conflict. If economic conditions in Europe and other key markets for our business and the business of our suppliers, manufacturers and collaborators remain uncertain or deteriorate further, we could experience adverse effects on our business, financial condition, results of operations or cash flows.
32
Billing for our products is complex and requires substantial time and resources to collect payment.
Billing for clinical laboratory testing services is complex, time-consuming and expensive. Depending on the billing arrangement and applicable law, we bill various payors, including Medicare, Medicaid, private insurance companies, private healthcare institutions, and patients, all of which have different billing requirements. We generally bill third-party payors for products and pursue reimbursement on a case-by-case basis where pricing contracts are not in place. To the extent laws or contracts require us to bill patient co-payments or co-insurance, we must also comply with these requirements. We may also face increased risk in our collection efforts, including potential write-offs of accounts receivable and long collection cycles, which could adversely affect our business, results of operations and financial condition.
Several factors make the billing process complex, including:
•differences between the billing rates and reimbursement rates for our products;
•compliance with complex federal and state regulations related to billing government healthcare programs, including Medicare, Medicaid, Veterans Health Administration and TRICARE;
•risk of government audits related to billing;
•disputes among payors as to which party is responsible for payment;
•differences in coverage and information and billing requirements among payors, including the need for prior authorization and/or advanced notification;
•the effect of patient co-payments or co-insurance and our ability to collect such payments from patients;
•changes to billing codes used for our products;
•changes to requirements related to our current or future clinical studies, including our registry studies, which can affect eligibility for payment;
•ongoing monitoring provisions of LCDs for our products, which can affect the circumstances under which a claim would be considered medically necessary;
•incorrect or missing billing information; and
•the resources required to manage the billing and claims appeals process.
We use standard industry billing codes, known as CPT codes to bill for our products. If these codes were to change, there is a risk of an error being made in the claim adjudication process. Such errors can occur with claims submission, third-party transmission or in the processing of the claim by the payor. Claim adjudication errors may result in a delay in payment processing or a reduction in the amount of the payment we receive.
As we introduce new products, we may need to add new codes to our billing process as well as our financial reporting systems. Failure or delays in effecting these changes in external billing and internal systems and processes could negatively affect our collection rates, revenue and cost of collecting.
Additionally, our billing activities require us to implement compliance procedures and oversight, train and monitor our employees, and undertake internal audits to evaluate compliance with applicable laws and regulations as well as internal compliance policies and procedures. When payors deny our claims, we may challenge the reason, low payment amount or payment denials. Payors also conduct external audits to evaluate payments, which add further complexity to the billing process. If the payor makes an overpayment determination, there is a risk that we may be required to return all or some portion of prior payments we have received.
Additionally, the Patient Protection and Affordable Care Act of 2010, as amended by the Health Care and Education Reconciliation Act of 2010, collectively the ACA requires providers and suppliers to report and return any overpayments received from government payors under the Medicare and Medicaid programs within 60 days of identification. Failure to identify and return such overpayments exposes the provider or supplier to liability under federal false claims laws. These billing complexities, and the related uncertainty in obtaining payment for our products, could negatively affect our revenue and cash flow, our ability to achieve profitability, and the consistency and comparability of our results of operations.
In addition to the complexities noted above, we rely upon a third-party software application in the administration of our billing and collection process. Any significant disruption in our billing operations or the discovery of a deficiency in the design of our billing process could adversely impact our ability to generate and send invoices, calculate revenues, track payments and collect our accounts receivable. Although to date we have not experienced any disruptions or identified any deficiencies with our billing process or billing system, there can be no assurances that
33
any disruptions or deficiencies will not occur in the future. Additionally, any failure in the design or operation of our internal controls related to our billing and collection processes could adversely impact our ability to conclude on the effectiveness of our internal control over financial reporting and could cause our auditor to issue an adverse opinion on our internal control over financial reporting.
We rely on third parties for tumor sample collection, preparation and delivery. Any defects in sample collection or preparation by such third parties and any delays in delivery of such samples could cause errors in our test reports and delayaffect our ability to deliver test reports in a timely manner or at all, which could significantly harm our business.
The tumor tissue samples that we test are biopsied (if applicable), preserved, prepared and delivered to us by third parties, including dermatopathologists and laboratory facilities. As such, we rely on these third parties to prepare, label and deliver the tissue samples that we test in compliance with applicable laws and guidelines, and in a timely manner. Therefore, the accuracy and correctness of the test reports that we deliver are dependent on proper chain of custody and appropriate methods of sample collection or preparation utilized by these third parties, and our ability to timely deliver reports is dependent upon the ability of these third parties to provide these samples to us in a timely manner. The ability of these third parties to provide these samples to us in a timely manner could be delayed by events beyond our control, including but not limited to operational problems, natural disasters and public health crises. Any errors in any part of the sample collection or preparation process could render us unable to process tests, or deliver test reports, or cause us to deliver incorrect test reports, potentially resulting in harm to patients whose physiciansclinicians implement a change in treatment decisions based upon our test report. If we are unable to timely deliver test reports, physiciansclinicians may be less likely to recommend and order our products.products and our revenues could be adversely affected. The occurrence of any of the foregoing could significantly harm our reputation and our results of operations, causing significant harm to our business.
We rely on our database of tumor samples for some of the development and improvement of our products. Depletion or loss of our tumor samples could significantly harm our business.
The development and validation of accurate products is a complex process that requires access to tumor tissue specimens and long-term outcomes data. Our research and development efforts to improve our existing commercial products and develop new pipeline products may require the depletion of our existing database of tumor samples. If our tumor samples are lost or destroyed, or substantially depleted before we are able to generate meaningful data, we may be unable to improve our existing products, continue the development of pipeline products or validate product candidates. While we have historically been able to create and maintain a large sample bank to expand the clinical use of our products and develop new products, we may be unable to do so in the
future. If we were unable to maintain or replenish our sample bank, we may be unable to improve our products or develop new products.
If one or more of our soleprimary clinical laboratory facility becomesfacilities become damaged or inoperable or we are required to vacate our existing facility, our ability to conduct our laboratory analysis and pursue our research and development efforts may be jeopardized.
We currently perform all of our testing and store our database of tumor samples at a singleboth our Phoenix, Arizona and Pittsburgh, Pennsylvania clinical laboratory facility in Phoenix, Arizona.facilities. Our facilityfacilities and equipment could be harmed or rendered inoperable by natural or man-made disasters, including war, fire, earthquake, power loss, communications failure, terrorism, burglary, public health crises (including restrictions that may be imposed on businesses by state and local governments under stay-at-home or similar orders and mandates such as those imposed during the COVID-19 pandemic) or other events, which may make it difficult or impossible for us to perform our testing services for some period of time or to receive and store samples. The inability to perform tests or to reduce the backlog of sample analysis that could develop if our facility becomesfacilities become inoperable, for even a short period of time, may result in the loss of revenue, loss of customers or harm to our reputation, and we may be unable to regain that revenue, those customers or repair our reputation in the future. Furthermore, integral parties in our supply chain are operating from single sites, increasing their vulnerability to natural disasters and man-made disasters or other sudden, unforeseen and severe adverse events.
In addition, the loss of our tumortissue samples due to such events could limit or prevent our ability to conduct research and development analysis on existing tests as well as tests in active pipeline development.
While we have a business continuity plan in place, and intentionally built out two clinical laboratories in adjacent buildings in Phoenix, Arizona to not only support our facilitygrowth but to provide certain operational redundancy, our facilities and the equipment we use to perform our testing and research and development could be unavailable or costly and time-consuming to repair or replace. It would be difficult, time-consuming and expensive to rebuild our facility,facilities, to locate and qualify a new facility, replace certain pieces of equipment or license or transfer our
34
proprietary technology to a third-party, particularly in light of licensure and accreditation requirements. Even in the unlikely event that we are able to find a third party with such qualifications to enableenabling us to resume our operations, we may be unable to negotiate commercially reasonable terms.
We carry insurance for damage to our property and the disruption of our business, but this insurance may not cover all of the risks associated with damage or disruption to our business, may not provide coverage in amounts sufficient to cover our potential losses and may not continue to be available to us on acceptable terms, if at all.
Our current or future products may not achieve or maintain significant commercial market acceptance.
We believe our commercial success is dependent upon our ability to continue to successfully market and sellcommercialize our products, to continue to expand our current relationships and develop new relationships with healthcare providers, to expand and maintain coverage for our products, and to develop and commercialize new products. Our ability to achieve and maintain commercial market acceptance of our existing and future products will depend on a number of factors, including:
•our ability to increase awareness of our products through successful clinical utility and validity studies;
•the rate of adoption of our products by physicians and other healthcare providers;
•our ability to achieve guideline inclusion for our products;
•the timeliness with which we can provide our clinical reports to the ordering physician;clinician;
•the timing and scope of any regulatory approval for our products, if such approvals become required, and maintaining ongoing compliance with regulatory requirements;
•our ability to obtain and maintain positive coverage decisions for our products from government and commercial payors;
•our ability to obtain and maintain adequate reimbursement from third-party payors, includingsuch as Medicare, Medicare Advantage plans, United Healthcare and BlueCross BlueShield plans, which accounted for an aggregate of approximately 90%49% and 83%53% of our total revenue from test reports for the years ended December 31, 20192023 and 2018, respectively;2022, respectively, with an additional third-party payor accounting for 14% of our revenue from test reports for the year ended December 31, 2023;
•the impact of our investments in research and development and commercial growth;
•negative publicity regarding our or our competitors’ products resulting from scientific publications, or defects or errors in the products; and
•our ability to further validate our products through clinical research and accompanying publications.
We cannot assure you that we will be successful in addressing each of these factors or other factors that might affect the market acceptance of our products. If we are unsuccessful in achieving and maintaining market acceptance of our products, our business and results of operations will suffer.
New product development involves a lengthy and complex process, and we may be unable to develop and commercialize, or receive reimbursement for, on a timely basis, or at all, new products.
We continually seek to develop new product offerings, which requires us to devote considerable resources to research and development. For example, beforeBefore we can commercialize oura new pipeline products for SCC and suspicious pigmented lesions,product, we will need to expend significant resources in order to conduct substantial research and development, including clinical utility and validity studies, and further develop and scale our laboratory processes and infrastructure to accommodate additional products. For example, in 2021, we launched our innovative pipeline to develop a genomic test, or series of genomic tests, aimed at predicting response to systemic therapy in patients with moderate to severe psoriasis, atopic dermatitis and related inflammatory skin conditions. With this launch, we initiated a large prospective, multi-center clinical study to develop and validate this inflammatory skin disease pipeline program. We announced early discovery data from this study in October 2023 and are targeting launch of this pipeline program by the end of 2025.
Our product development process takes time and involves a high degree of risk, and such development efforts may fail for many reasons, including failure of the product to perform as expected, failure to successfully complete analytic and clinical validation, or failure to demonstrate the clinical utility of the product.
As we develop new products, we will have to make significant investments in research and development, marketing, selling, coverage and reimbursement activities. Typically, few research and development projects result in a commercialized product, and there can be no assurance that we will be able to successfully develop new products that can be commercialized. At any point, we may abandon development of a product or we may be required to
35
expend considerable resources conducting research, which would adversely affect the timing for generating potential revenue from a new product and our ability to invest in other products in our pipeline. If a clinical validation study fails to demonstrate the prospectively defined endpoints of the study or if we fail to sufficiently demonstrate analytical validity or clinical utility, we might choose to abandon the development of the product, which could harm our business. In addition, competitors may develop and commercialize competing products or technologies faster than us or at a lower cost.
We may experience limits on our revenue if we are unable to increase and support adoption of our products by physicians and other healthcare providers.
Physicians and other healthcare providers may be unwilling to adopt our products due to their reliance on existing traditional clinical and pathology staging criteria and our ability to generate revenue from our products would be significantly impaired if we were unable to educate physicians, healthcare providers, patients and third-party payors about the benefits and advantages of our products. We will need to continue to educate physicians and pathologists about the benefits and cost-effectiveness of our products through published papers, presentations at scientific conferences, one-on-one marketing efforts by our sales force and one-on-one education by our medical affairs team. However, physicians and other healthcare providers may be reluctant to adopt our products in circumstances where our products are not incorporated into the current standard of care or practice guidelines. For example, while clinical utility of DecisionDx-Melanoma has been demonstrated in peer reviewedpeer-reviewed publications, the SLNB surgery is the most widely used pathology staging tool by clinicians for determining a cutaneous melanoma patient’s metastatic risk. Whether healthcare providers adopt DecisionDx-Melanoma as a complementary or triage diagnostic method relative to the SLNB surgery will depend on our ability to increase awareness of DecisionDx-Melanoma and its clinical validation.
In addition, all of our testing services are performed by our certified laboratorylaboratories located in Phoenix, Arizona and Pittsburgh, Pennsylvania, under the Clinical Laboratory Improvement Amendments of 1988, or CLIA rather than by local laboratory or pathology practices. Accordingly, it may be difficult for us to collect samples from pathologists, and pathologists may be reluctant to support our testing services.
We rely on limited or sole suppliers for some of the reagents, equipment, chips and other materials used by our products, and we may not be able to find replacements or transition to alternative suppliers.
We rely on limited or sole suppliers for certain reagents and other materials and components that we use for our products. Some of these items are unique to these suppliers and vendors. While we have developed alternate sourcing strategies for these materials and vendors, we cannot be certain whether these strategies will be effective or the alternative sources will be available when we need them. If these suppliers can no longer provide us with the materials we need, if the materials do not meet our quality specifications or are otherwise unusable, if we cannot obtain acceptable substitute materials, or if we elect to change suppliers, an interruption in laboratory operations could occur, we may not be able to deliver patient reports on a timely basis, or at all, and we may incur higher one-time switching costs. Any such interruption may significantly affect our future revenue, cause us to incur higher costs, and harm our customer relationships and reputation. In addition, in order to mitigate these risks, we maintain inventories of these supplies at higher levels than would be the case if multiple sources of supply were available. If our testing volume decreases or we switch suppliers, we may hold excess supplies with expiration dates that occur before use which would adversely affect our losses and cash flow position. As we introduce any new products, we may experience supply issues as we ramp up test volume.volume, or encounter additional disruptions to trade, commerce, pricing stability, credit availability and global supply chain continuity as a result of the invasion of Ukraine by Russia, particularly if we contract with suppliers with operations or commercial relationships in Eastern Europe or to the extent the conflict escalates to involve additional countries, further economic sanctions or wider military conflict. If we should encounter delays or difficulties in securing, reconfiguring or revalidating the equipment, reagents or other materials we require for our products, our business, financial condition, results of operations and reputation could be adversely affected.
If our products do not meet the expectations of physiciansclinicians and patients, our operating results, reputation and business could suffer.
Our success depends on physicianclinician and patient confidence that we can provide reliable, high-quality information that will improve treatment outcomes, lower healthcare costs and enable better patient care. We believe that patients, physicians and other healthcare providers are likely to be particularly sensitive to defects and errors in our products, including if our products fail to accurately predict risk of metastasis with high accuracy from samples, and there can be no guarantee that our products will meet their expectations. As a result, the failure of our products to perform as expected could significantly impair our operating results and our reputation, including if we become subject to legal claims arising from any defects or errors in our products or reports.
36
If we are unable to compete successfully, our business will suffer and we may be unable to increase or sustain our revenue or achieve profitability.
We face competition from companies and academic institutions that have either developed or may seek to develop products intended to compete with our products. Potential competitors within the broader genomics profiling space based on tissue sample collection include laboratory companies such as Laboratory Corporation of America and Myriad Genetics, and other companies which have strong infrastructures capable of supporting the commercialization of diagnostic services.
In addition, competitors may develop their own versions of our solutions in countries where we do not have patents or where our intellectual property rights are not recognized and compete with us in those countries, including encouraging the use of their solutions by physiciansclinicians in other countries.
Some potential competitors may have longer operating histories, larger customer bases, greater brand recognition and market penetration, substantially greater financial, technological and research and development resources and selling and marketing capabilities, and more experience dealing with third-party payors. As a result, they may be able to respond more quickly to changes in customer requirements, devote greater resources to the development, promotion and sale of their products than we do or sell their products at prices designed to win significant levels of market share. We may not be able to compete effectively against these organizations. Increased competition and cost-saving initiatives on the part of governmental entities and other third-party payors are likely to result in pricing pressures, which could harm our sales, profitability or ability to gain market share. In addition, competitors may be acquired by, receive investments from or enter into other commercial relationships with larger, well-established and well-financed companies. Certain potential competitors may be able to secure key inputs from vendors on more favorable terms, devote greater resources to marketing and promotional campaigns, adopt more aggressive pricing policies and devote substantially more resources to test development than we can. In addition, companies or governments that control access to testing through umbrella contracts or regional preferences could promote our competitors or prevent us from performing certain services. If we are unable to compete successfully against current and future competitors, our business will suffer and we may be unable to increase market acceptance and sales of our products, which could prevent us from increasing our revenue or achieving profitability and could cause our stock price to decline. As we add new tests and services, we will face many of these same competitive risks for these new tests.
Impairment of our goodwill or other intangible assets could have a material adverse effect on our operating results and financial condition.
Goodwill represents the excess of amounts paid for acquiring businesses over the fair value of the net assets acquired, and intangible assets are measured at fair value upon the acquisition of a business for purposes of such calculations. As of December 31, 2023, our goodwill and other intangible assets balances were $10.7 million and $106.6 million, respectively. Goodwill is evaluated for impairment annually, or more frequently if conditions warrant, by comparing the carrying value of a reporting unit to its estimated fair value. Intangible assets with finite lives are reviewed for impairment when events or circumstances indicate that their carrying value may not be recoverable. Declines in operating results, divestitures, sustained market declines and other factors could result in an impairment of goodwill or other intangible assets and, in turn, a charge to net income or loss. Any future charges could have a material adverse effect on our results of operations or financial condition.
On June 2, 2023, a MAC finalized an LCD pursuant to which the DecisionDx-SCC test would no longer be covered by Medicare effective July 17, 2023. On June 5, 2023, our stock price decreased significantly and did not recover before June 30, 2023. In response to this trigger, we tested goodwill for impairment at June 30, 2023. We elected to bypass the optional qualitative assessment and proceeded directly to the quantitative assessment. In conducting our interim test, we concluded that our business consists of a single reporting unit. To measure the fair value of our reporting unit, we used a market approach whereby we calculated our total market capitalization on the impairment test date, based on the closing price of our common stock as reported on the Nasdaq Global Market, and applied a reasonable control premium. The control premium was based on an analysis of control premiums paid in recent acquisitions of companies in the same or similar industry as us. Our impairment test indicated that the fair value of our reporting unit exceeded its carrying value by 13% and therefore no impairment was indicated. In July 2023, the MAC suspended the LCD and then posted a new draft LCD for comment that is substantially the same as the LCD that was to become effective.
During the third quarter of 2023, we continued to monitor our market capitalization against the carrying value of our reporting unit and did not observe any significant changes since our impairment test at June 30, 2023. We have performed our annual impairment test, as of October 1, 2023, and we have not identified any additional indicators of impairment to date.
37
Factors that could result in a future impairment of goodwill include declines in the price of our common stock, increased competition, changes in macroeconomic developments, unfavorable government or regulatory developments and changes in coverage or reimbursement conditions.
The sizes of the marketsTAM for our current and future products have not been established with precision and may be smaller than we estimate.
Our estimates of the total addressable marketsTAM for the DecisionDx-Melanoma, DecisionDx-UM, DecisionDx-SCC, MyPath Melanoma, TissueCypher and our products in developmentIDgenetix tests are based on a number of internal and third-party estimates, including, without limitation, the annual rate of patients with the applicable form of skin cancer,indications, the list price of our products relative to the reimbursement we expect to receive from third-party payors and the assumed prices at which we can sell our products in markets that have not been established. For example, we estimate that the total addressable market for DecisionDx-Melanoma is approximately $540 million, which is based, in part, on our review of multiple recent publications which show that diagnosis of melanoma is underreported by 30% to 40%. While we believe our assumptions and the data underlying our estimates are reasonable, these assumptions and estimates may not be correct and the conditions supporting our assumptions or estimates may change at any time, thereby reducing the predictive accuracy of these underlying factors. As a result, our estimates of the annual total addressable marketTAM for our current or future products may prove to be incorrect. If the actual number of patients who would benefit from our products, the price at which we can sell future products, or the annual total addressable marketTAM for our products is smaller than we have estimated, it may impair our sales growth and have an adverse impact on our business.
The diagnostic testing industry is subject to rapid change, which could make our current or future products obsolete.
Our industry is characterized by rapid changes, including technological and scientific breakthroughs, frequent new product introductions and enhancements and evolving industry standards, all of which could make our current products and the other products we are developing obsolete. Our future success will depend on our ability to keep pace with the evolving needs of
Our business operations may subject us to disputes, claims, government investigations and lawsuits, which may be costly and time-consuming and could materially and adversely impact our financial position and results of operations.
From time to time, we may become involved in disputes, claims, government investigations and lawsuits relating to our business operations. In particular, we may face claims related to the safety of our products, intellectual property matters, financial arrangements with health care providers, regulatory compliance, product promotional practices, and documentation, coding and billing practices, employment matters, tax matters, commercial disputes, competition, sales and marketing practices, environmental matters, personal injury, insurance coverage, and acquisition or divestiture-related matters. Any dispute, claim, government investigation or lawsuit may divert management’s attention away from our business, we may incur significant expenses in addressing or defending any dispute, claim or lawsuit, and we may be required to pay damage awards or settlements or become subject to equitable remedies that could adversely affect our operations and financial results. For example, as described further in “Item 3. Legal Proceedings,” on February 1, 2024 we received a subpoena from United States Department of Health and Human Services Office of Inspector General. This inquiry, and any potential resulting claim asserted against us, with or without merit, could be time-consuming, expensive to address and divert management’s attention and other resources. These claims also could subject us to significant liability for damages and harm our reputation. Our insurance and indemnities may not cover all claims that may be asserted against us.
Additionally, the associated uncertainty could lead to increased volatility in our stock price and governmental enforcement action may result in substantial fines, penalties or administrative remedies, including exclusion from government reimbursement programs and entry into corporate integrity agreements with governmental agencies, which would entail significant obligations and costs.
38
Risks Related to Reimbursement and Government Regulation
We currentlygenerally have limited reimbursement coverage for our lead product, DecisionDx-Melanoma,products, and if third-party payors, including government and commercial payors, do not provide sufficient coverage of, or adequate reimbursement for, our products, our commercial success, including revenue, will be negatively affected.
Our revenue depends on achieving broad coverage and adequate reimbursement for our products from third-party payors, including both government and commercial third-party payors. If third-party payors do not provide coverage of, or do not provide adequate reimbursement for, a substantial portion of the list price of our products, we may need to seek additional payment from the patient beyond any co-payments and deductibles, which may adversely affect demand for our products. Coverage determinations by a third-party payor may depend on a number of factors, including, but not limited to, a third-party payor’s determination of whether our products are appropriate, medically necessary or cost-effective. If we are unable to provide third-party payors with sufficient evidence of the clinical utility and validity of our products, they may not provide coverage, or may provide limited coverage, which will adversely affect our revenues and our ability to succeed. To the extent that more competitors enter our markets, the availability of coverage and the reimbursement rate for our products may decrease as we encounter pricing pressure from these competitors.
Since each third-party payor makes its own decision as to whether to establish a policy to cover our products, enter into a contract with us and set the amount it will reimburse for a product, these negotiations are a time-consuming and costly process, and they do not guarantee that the third-party payor will provide coverage or adequate reimbursement for our products. In addition, the determinations by a third-party payor whether to cover our products and the amount it will reimburse for them are often made on an indication-by-indication basis.
In cases where there is no coverage policy or we do not have a contracted rate for reimbursement as a participating provider, the patient is typically responsible for a greater share of the cost of the product, which may result in further delay of our revenue, increase our collection costs or decrease the likelihood of collection.
Our claims for reimbursement from third-party payors may be denied upon submission, and we may need to take additional steps to receive payment, such as appealing the denials. Such appeals and other processes are time-consuming and expensive and may not result in payment. Third-party payors may perform audits of historically paid claims and attempt to recoup funds years after the funds were initially distributed if the third-party payors believe the funds were paid in error or determine that our products were medically unnecessary. If a third-party payor audits our claims and issues a negative audit finding, and we are not able to overturn the audit findings through appeal, the recoupment may result in a material adverse effect on our revenue. Additionally, in some cases commercial third-party payors for whom we are not a participating provider may elect at any time to review claims previously paid and determine the amount they paid was too much. In these situations, the third-party payor will typically notify us of their decision and then offset whatever amount they determine they overpaid against amounts they owe us on current claims. We cannot predict when, or how often, a third-party payor might engage in these reviews and we may not be able to dispute these retroactive adjustments.
Under ASC 606, we recognize revenue at the amount we expect to be entitled, subject to a constraint for variable consideration, in the period in which our tests are delivered to the treating physician.clinician. We have determined that our contracts contain variable consideration under ASC 606 because the amounts paid by third-party payors may be paid at less than our standard rates or not paid at all, with such differences considered implicit price concessions. Variable consideration is recognized only to the extent it is probable that a significant reversal of revenue will not occur in future periods when the uncertainties are resolved. We consider variable consideration to be fully constrained (and therefore not recognized) for Medicare claims when the payment of such claims is subject to approval by an ALJ at an appeal hearing, due to the level of uncertainty and timing of the outcome.
Variable consideration is evaluated each reporting period and adjustments are recorded as increases or decreases in revenues. Variable consideration for Medicare claims that are not covered by Medicare, including those claims undergoing appeal, is deemed to be fully constrained due to factors outside our influence (e.g., judgment or actions of third parties) and the uncertainty of the amount to be received is not expected to be resolved for a long period of time. For these fully constrained claims, we generally recognize revenue in the period the uncertainties are resolved, if favorable. Due to the outcome of ALJ hearings, potential future changes in Medicare coverage policies and appeal cycles, insurance coverage policies, contractual rates and other trends in the reimbursement of our tests, our revenues may fluctuate significantly from period to period.
Although we are an in-network participating provider with some commercial third-party payors, including several Blue Cross Blue Shield plans, and certain large, national commercial third-party payors, including Aetna, other commercial third-party payors have issued non-coverage policies that currently categorize DecisionDx-UM and DecisionDx-Melanomaour tests as experimental or investigational. If we are not successful in obtaining coverage from third-party payors, in reversing existing non-coveragenon-
39
coverage policies, or if other third-party payors issue similar non-coverage policies, this could have a material adverse effect on our business and operations.
On June 2, 2023, Novitas posted a finalized oncology biomarker LCD pursuant to which the DecisionDx-SCC test would no longer be covered by Medicare effective July 17, 2023. However, on July 6, 2023, Novitas suspended the final version of the LCD and announced its intent to post a new proposed LCD for comment and presentation at an open meeting. On July 27, 2023, Novitas posted a nearly identical proposed oncology biomarker LCD that continues to intend to rely upon evidentiary reviews sourced from three databases: ClinGen, OncoKB and NCCN. The proposed LCD also recommends non-coverage for our DecisionDx-SCC test. The comment period for the proposed LCD ended on September 9, 2023. We cannot predict whether this LCD will be finalized as proposed or what the timing of any final LCD might be.
Under Medicare, payment for products like ours is generally made under the CLFS with payment amounts assigned to specific procedure billing codes. Medicare reimbursement rates for our tests are subject to change and may decrease from those currently in effect. For example, in February 2023, MolDX notified us that IDgenetix should shift billing to a different multi-test generic gene sequencing CPT code and continue using the IDgenetix Z-Code beginning in March 2023. As a result of this change, the Medicare reimbursement rate for the IDgenetix multi-gene panel decreased from approximately $1,500 to $917 per test. We subsequently obtained a test-specific PLA CPT code which became effective October 1, 2023. In November 2023, CMS posted its final CLFS determination which crosswalks our PLA CPT code to an existing PLA code at a rate of $1,336 per test effective January 1, 2024.
In April 2014, Congress passed the Protecting Access to Medicare Act of 2014, or PAMA which included substantial changes to the way in which clinical laboratory services are paid under Medicare. Under PAMA, certain laboratories wereare required to report to CMS beginning in 2017 and every three years thereafter (or annually for advanced diagnostic laboratory tests, or ADLTs), commercial third-party payor payment rates and volumes for each test they perform. CMS uses this data to calculate a weighted median payment rate for each test, which will be used to establish revised Medicare CLFS reimbursement rates for the test. Laboratories that fail to report the required payment information may be subject to substantial civil monetary penalties. We bill Medicare for our products, and therefore we are subject to reporting requirements under PAMA.
If we are unable to obtain and maintain adequate reimbursement rates from commercial third-party payors, this may adversely affect our Medicare rate. It is unclear what impact new pricing structures, such as those adopted under PAMA, may have on our business, financial condition, results of operations or cash flows.
The U.S. federal government continues to show significant interest in pursuing health carehealthcare reform and reducing health carehealthcare costs. Similarly, commercial third-party payors may seek to reduce costs by limiting coverage or reducing reimbursement for our products. Any government-adopted reform measures or changes to commercial third-party payor coverage and
reimbursement policies could cause significant pressure on the pricing of, and reimbursement for, health carehealthcare products and services, including our products, which could decrease demand for our products, and adversely affect our sales and revenue.
In addition, some third-party payors have implemented, or are in the process of implementing, laboratory benefit management programs, often using third-party benefit managers to manage these programs. The stated goals of these programs are to help improve the quality of outpatient laboratory services, support evidence-based guidelines for patient care and lower costs. The impact on laboratories, such as ours, of active laboratory benefit management by third parties is unclear, and we expect that it could have a negative impact on our revenue in the short term. It is possible that third-party payors will resist reimbursement for the products that we offer, in favor of less expensive
40
products, may require pre-approval for our products or may impose additional pricing pressure on and substantial administrative burden for reimbursement for our products.
We expect to continue to focus substantial resources on increasing coverage and reimbursement for our current products and any future products we may develop. We believe it may take several years to achieve broad coverage and adequate contracted reimbursement with a majority of third-party payors for our products.
However, we cannot predict whether, under what circumstances, or at what payment levels third-party payors will cover and reimburse our products. If we fail to establish and maintain broad adoption of, and coverage and reimbursement for, our products, our ability to generate revenue could be harmed and our future prospects and our business could suffer.
Our products are currently marketed as laboratory developed tests, and any changes in regulations or the U.S. Food and Drug Administration’sFDA’s enforcement discretion for laboratory developed tests, or violations of regulations by us, could adversely affect our business, prospects, results of operations or financial condition.
The diagnostics industry is highly regulated, and we cannot assure you that the regulatory environment in which we operate will not change significantly and adversely in the future. In many instances, there are no significant regulatory or judicial interpretations of these laws and regulations. Although the FDA has statutory authority to assure that medical devices are safe and effective for their intended uses, the FDA has generally exercised its enforcement discretion and not enforced applicable regulations with respect to in vitro diagnostics that are designed, manufactured and used within a single laboratory. These tests are referred to as laboratory developed tests, or LDTs. We currently market our products as LDTs.
The FDA has adopted a policy of enforcement discretion with respect to LDTs whereby the FDA does not actively require premarket review of LDTs or otherwise impose its requirements applicable to other medical devices on LDTs. However,On October 3, 2023, the FDA has stated its intention to modifyissued proposed regulations under which it would phase out its enforcement discretion policy with respect to LDTs. The FDA could ultimately modify its current approach to LDTs inover a way thatperiod of four years. Although the proposed regulation is subject to a period of notice and comment, if finalized as proposed, we would subjectbe required to obtain 510(k) or PMA for certain of our products marketed as LDTs to the enforcement of additional regulatory requirements. Moreover, legislative measures have recently been proposed in Congress that, if ultimately enacted, could provide the FDA with additional authority to require premarket review of and regulate LDTs. If and when such changes to the regulatory framework occur, we could for the first timetests by October 1, 2027. We would also be subject to enforcement of regulatory requirements as a device manufacturer such as registration and listing requirements, medical device reporting requirements and the requirements of the FDA’s Quality System Regulation. We may be required to conduct clinical trials prior to continuing to sell our existing products or launching any other products we may develop. This may increase the cost of conducting, or otherwise harm, our business.
If the FDA begins to actively regulate our diagnostic products, we may be required to obtain premarket clearance under Section 510(k) of the U.S. Federal Food, Drug and Cosmetic Act, or FDCA or a premarket approval, or PMA. The process for submitting a 510(k) premarket notification and receiving FDA clearance usually takes from three to 12 months, but it can take significantly longer and clearance is never guaranteed. The process for submitting and obtaining FDA approval of a PMA is much more costly, lengthy and uncertain. It generally takes from one to three years or even longer, and approval is not guaranteed. PMA approval typically requires extensive clinical data and can be significantly longer, more expensive and more uncertain than the 510(k) clearance process. Despite the time, effort and expense expended, there can be no assurance that a particular device ultimately will be cleared or approved by the FDA through either the 510(k) clearance process or the PMA process on a timely basis, or at all. Moreover, there can be no assurance that any cleared or approved labeling claims will be consistent with our current claims or adequate to support continued adoption of and reimbursement for our products. If premarket review is required for some or all of our products, the FDA may require that we stop selling our products pending
clearance or approval, which would negatively impact our business. Even if our products are allowed to remain on the market prior to clearance or approval, demand or reimbursement for our products may decline if there is uncertainty about our products, if we are required to label our products as investigational by the FDA, or if the FDA limits the labeling claims we are permitted to make for our products. As a result, we could experience significantly increased development costs and a delay in generating additional revenue from our products, or from other products now in development.pipeline products.
If the FDA imposes significant changes to the regulation of LDTs it could reduce our revenues or increase our costs and adversely affect our business, prospects, results of operations or financial condition.
41
We conduct business in a heavily regulated industry, and failure to comply with federal, state and foreign laboratory licensing requirements including those established by CMS and the applicable requirements of the FDA or any other regulatory authority, could cause us to lose the ability to perform our tests, experience disruptions to our business, or become subject to administrative or judicial sanctions.
The diagnostics industry is highly regulated, and the laws and regulations governing the marketing of diagnostic tests are extremely complex. Areas of the regulatory environment that may affect our ability to conduct business include, without limitation:
•federal and state laws applicable to test ordering, documentation of tests ordered, billing practices and claims payment and/or regulatory agencies enforcing those laws and regulations;
•federal and state fraud and abuse laws;
•federal and state laboratory anti-mark-up laws;
•coverage and reimbursement levels by Medicare, Medicaid, other governmental payors and private insurers;
•restrictions on coverage of and reimbursement for tests;
•federal and state laws governing laboratory testing, including CLIA, and state licensing laws;laws and accreditation requirements;
•federal and state laws and enforcement policies governing the development, use and distribution of diagnostic medical devices, including LDTs;
•federal, state and local laws governing the handling and disposal of medical and hazardous waste;
•federal and state Occupational Safety and Health Administration rules and regulations; and
In particular, the FDCA defines a medical device to include any instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article, including a component, part, or accessory, intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease, in man or other animals. Our products are considered by the FDA to be subject to regulation as medical devices, and marketed under FDA’s policy of enforcement discretion for LDTs. Among other things, pursuant to the FDCA and its implementing regulations, the FDA regulates the research, testing, manufacturing, safety, labeling, storage, recordkeeping, premarket clearance or approval, marketing and promotion, and sales and distribution of medical devices in the United States to ensure that medical products distributed domestically are safe and effective for their intended uses. In addition, the FDA regulates the import and export of medical devices manufactured between the United States and international markets.
CLIA Certifications
We are also subject to CLIA, a federal law that regulates clinical laboratories that perform testing on specimens derived from humans for the purpose of providing information for the diagnosis, prevention or treatment of disease. CLIA regulations establish specific standards with respect to personnel qualifications, facility administration, proficiency testing, quality control, quality assurance and inspections. Any testing subject to CLIA regulation must be performed in a CLIA certifiedCLIA-certified or accredited lab. CLIA certification or accreditation is also required in order for us to be eligible to bill state and federal healthcare programs, as well as commercial third-party payors, for our products. We
CAP maintains a clinical laboratory accreditation program. While not required for the operation of a CLIA-certified laboratory, many private insurers require CAP accreditation as a condition to contracting with clinical laboratories to cover their tests. In addition, some countries outside the United States require CAP accreditation as a condition to permitting clinical laboratories to test samples taken from their citizens. CAP accredited laboratories are surveyed for compliance with CAP standards every two years in order to maintain accreditation. Failure to maintain CAP accreditation could have a current CLIA accreditation undermaterial adverse effect on the Collegesales of American Pathologists, or CAP, programour products and the results of our operations. Therefore, to conduct our tests at our clinical reference laboratory in Phoenix, Arizona.
We have a current CLIA accreditation under the CAP program to conduct our tests at our clinical reference laboratories in Phoenix, Arizona. The most recent CAP inspection of our Phoenix, Arizona laboratories occurred in October 2022.
42
We currently have a CLIA certificate of registration for our Pittsburgh, Pennsylvania laboratory which expires in February 2024. In November 2022, our Pittsburgh, Pennsylvania passed CAP inspection and received CAP accreditation.
In addition, certain states require our laboratorylaboratories to be licensed in suchspecific states in order to test specimens from those states. Accordingly, our laboratory is alsolaboratories are licensed by California, Maryland, Pennsylvania, Rhode Island and New York, Pennsylvania and Rhode Island.York. Other states may have similar requirements ordo not currently require additional licensure but they may adopt similar requirements in the future.
Although we have obtained licenses from states where we believe we are required to be licensed, we may become aware of other states that require out-of-state laboratories to obtain licensure in order to accept specimens from the state, and it is possible that other states currently have such requirements or will have such requirements in the future.
In order to test specimens from New York, LDTs must be approved by the New York State Department of Health, or NYSDOH on a test-by-test basis before they are offered. Our laboratory director and laboratory operations must also be separately qualified to be a laboratory director inand approved through the state of New York. DecisionDx-Melanoma, DecisionDx-CMSeq, DecisionDx-UM, DecisionDx-PRAME, DecisionDx-UMSeq, DecisionDx-SCC, MyPath Melanoma and DecisionDx-MelanomaDiffDx-Melanoma have each been approvedapproved. We have been given conditional approval for IDgenetix from the NYSDOH while we work our way through the formal approval. In July 2022, we submitted TissueCypher for review by the NYSDOH and had been given clearance to test New York state patients by the New York’s Clinical Laboratory Evaluation Program while our applications were under review. On September 12, 2023, we received our Clinical Laboratory Permit from NYSDOH for our Pennsylvania laboratory. Our laboratory director has been qualified by the NYSDOH. We are subject to periodic inspection by the NYSDOH and are required to demonstrate ongoing compliance with the NYSDOH regulations and standards. Our most recent inspection was in October 2022 and we were deemed to be compliant with the NYSDOH regulations and standards. To the extent the NYSDOH had identified any instances of non-compliance, and we arewere unable to remedy such non-compliance, the State of New York could withdraw approval for our products.products to test samples from New York state. We will need to seek the NYSDOH approval of any future LDTs we develop and want to offer for clinical testing to New York residents, and there can be no assurance that we will be able to obtain such approval.
We may also be subject to regulation in foreign jurisdictions as we seek to expand international utilization of our products or such jurisdictions adopt new licensure requirements, which may require review of our products in order to offer them or may have other limitations such as restrictions on the transport of human tissue samples necessary for us to perform our tests that may limit our ability to make our products available outside of the United States. Complying with licensure requirements in new jurisdictions may be expensive, time-consuming and subject us to significant and unanticipated delays.
Failure to comply with applicable clinical laboratory licensure requirements may result in a range of enforcement actions, including suspension, limitation or revocation of our CLIA accreditation and/or state licenses, imposition of a directed plan of action, onsite monitoring, civil monetary penalties, criminal sanctions and revocation of the laboratory’s approval to receive Medicare and Medicaid payment for its services, as well as significant adverse publicity. Any sanction imposed under CLIA, its implementing regulations, or state or foreign laws or regulations governing clinical laboratory licensure or our failure to renew our CLIA accreditation, or a state or foreign license, could have a material adverse effect on our business, financial condition and results of operations. Even if we were able to bring our laboratory back into compliance, we could incur significant expenses and potentially lose revenue in doing so.
Doing business with the public sector, including the U.S. government, subjects us to risk of audits, investigations, sanctions and penalties.
We have entered into, and may enter into in the future, contracts with the U.S. government or other governmental entities, and this subjects us to statutes and regulations applicable to companies doing business with the government. For example, we have a U.S. Federal Supply Schedule contract with the Veterans Health Administration covering our covering all of our tests with the exception of DecisionDx-UM. Government contracts normally contain additional requirements that may increase our costs of doing business, reduce our profits (or increase our losses) and expose us to liability for failure to comply with these terms and conditions. Such requirements may include mandatory socioeconomic compliance requirements, including labor requirements, non-discrimination and affirmative action programs and environmental compliance requirements. Being a government contractor also subjects us to reviews, audits and investigations regarding our compliance. If we fail to comply with our obligations associated with being a government contractor, our contracts may be subject to termination, and we
43
may be subject to financial and/or other liability under our contracts, which could adversely affect our results of operations.
The FDA may modify its enforcement discretion policy with respect to LDTs in a risk-based manner, and we may become subject to extensive regulatory requirements and may be required to conduct additional clinical trials prior to continuing to sell our existing tests or launching any other tests we may develop, which may increase the cost of conducting, or otherwise harm, our business.
If the FDA changes or ends its policy of enforcement discretion with respect to LDTs, whether by finalization of regulations initial proposed on October 3, 2023, or otherwise, and our products become subject to the FDA’s requirements for premarket review of medical devices, we may be required to cease commercial sales of our products and conduct clinical trials prior to making submissions to the FDA to obtain premarket clearance or approval. If we are required to conduct such clinical trials, delays in the commencement or completion of clinical trials could significantly increase our product development costs and delay commercialization of any currently marketed testing that we may be required to cease selling or the commercialization of any future tests that we may develop. Many of the factors that may cause or lead to a delay in the commencement or completion of clinical trials may also ultimately lead to delay or denial of regulatory clearance or approval. The commencement of clinical trials may be delayed due to insufficient patient enrollment, which is a function of many factors, including the size of the patient population, the nature of the protocol, the proximity of patients to clinical sites and the eligibility criteria for the clinical trial.
The FDA requires medical device manufacturers to comply with, among other things, current good manufacturing practices for medical devices, known as the Quality System Regulation, which requires manufacturers to follow elaborate design, testing, control, documentation and other quality assurance procedures during the manufacturing process; the medical device reporting regulation, which requires that manufacturers report to the FDA if their device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if it were to recur; labeling regulations, including the FDA’s general prohibition against promoting products for unapproved or ‘‘off-label’’ uses; and the reports of corrections and removals regulation, which requires manufacturers to report to the FDA if a device correction
or removal was initiated to reduce a risk to health posed by the device or to remedy a violation of the FDCA caused by the device which may present a risk to health.
Even if we were able to obtain FDA clearance or approval for one or more of our products, if required, a diagnostic test may be subject to limitations on the indications for which it may be marketed or to other regulatory conditions. In addition, such clearance or approval may contain requirements for costly post-market testing and surveillance to monitor the safety or efficacy of the test.
In addition, the FDA’s and other regulatory authorities’ policies may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approvals. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing authorization that we may have obtained and we may not achieve or sustain profitability, which would adversely affect our business, prospects, financial condition and results of operations.
Furthermore, government funding of the FDA other government agencies on which our operations may rely is subject to the political process, which is inherently fluid and unpredictable. Disruptions at the FDA and other government agencies may impact the ability of such agencies to timely review and process our regulatory and other submissions, which could have a material adverse effect on our business.
Interim, topline and preliminary data from our clinical studies that we announce or publish from time to time may change as more data become available and are subject to audit and verification procedures that could result in material changes in the final data.
From time to time, we may publicly disclose preliminary or topline or data from our clinical studies, which is based on a preliminary analysis of then-available data, and the results and related findings and conclusions are subject to change following a more comprehensive review of the data related to the particular study. We also make assumptions, estimations, calculations and conclusions as part of our analyses of data, and we may not have received or had the opportunity to fully and carefully evaluate all data. As a result, the topline results that we report may differ from future results of the same studies, or different conclusions or considerations may qualify such results once additional data have been received and fully evaluated. Topline data also remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary data we previously published. As a result, topline data should be viewed with caution until the final data are available. From
44
time to time, we may also disclose interim data from our clinical studies. Interim data from clinical studies that we may complete are subject to the risk that one or more of the clinical outcomes may materially change as more patient data becomebecomes available. Adverse differences between preliminary or interim data and final data could significantly harm our reputation and marketing efforts.
Further, others, including healthcare providers or payors, may not accept or agree with our assumptions, estimates, calculations, conclusions or analyses or may interpret or weigh the importance of data differently, which could impact the value of the particular program, the approvability or commercialization of the particular product candidate or product and our company in general. In addition, the information we choose to publicly disclose regarding a particular study is based on what is typically extensive information, and you or others may not agree with what we determine is the material or otherwise appropriate information to include in our disclosure, and any information we determine not to disclose may ultimately be deemed significant with respect to future decisions, conclusions, views, activities or otherwise regarding our business. If the topline or interim data that we report differ from actual results, or if others, including healthcare providers or payors, disagree with the conclusions reached, our ability to commercialize, our product candidates may be harmed, which could harm our business, operating results, prospects or financial condition.
Changes in health carehealthcare policy could increase our costs, decrease our revenues and impact sales of and reimbursement for our products.
In March 2010, the ACA became law. This law substantially changed the way health carehealthcare is financed by both government and commercial third-party payors, and significantly impacted our industry. The ACA contains a number of provisions that are expected to impact our business and operations, some of which in ways we cannot currently predict, including those governing enrollment in state and federal health care programs, reimbursement changes and fraud and abuse, which impact existing state and federal health care programs and will result in the development of new programs. Among other things, the ACA required medical device manufacturers to pay a sales tax equal to 2.3% of the price for which such manufacturer sells its medical devices, and began to apply to sales of taxable medical devices after December 31, 2012, but was suspended in 2016. Further, the 2020 federal spending package permanently eliminated, effective January 1, 2020, the medical device tax and “Cadillac” tax on high-cost employer-sponsored health coverage and, effective January 1, 2021, also eliminateseliminated the health insurer tax.
Since 2016, there have been efforts to repeal all or part of the ACA, and the currentprevious administration and the U.S. Congress have taken action to roll back certain provisions of the ACA. The current administration andFor example, on June 17, 2021, the U.S. Congress may take further action regarding the ACA, including, but not limited to, repeal or replacement.
On August 2, 2011, the Budget Control Act of 2011 was signed into law, which, among other things, reduced Medicare payments to providers by 2% per fiscal year, effective on April 1, 2013 and, due to subsequent legislative amendments to the statute, including the Infrastructure Investment and Jobs Act, will remain in effect through 2029,until 2032, unless additional Congressional action is taken.
We anticipate there will continue to be proposals by legislators at both the federal and state levels, regulators and commercial third-party payors to reduce costs while expanding individual healthcare benefits. Certain of these changes could impose additional limitations on the prices we will be able to charge for our products, the coverage of or the amounts of reimbursement available for our products from third-party payors, including government and commercial payors.
We are subject to numerous federal and state healthcare statutes and regulations, and complying with laws pertaining to our business is an expensive and time-consuming process. If we are unable to comply, or have not fully complied, with such laws, we could face substantial penalties and a material adverse effect to our business and operations.
Physicians, other healthcare providers and third-party payors play a primary role in the recommendation of our products. Our arrangements with healthcare providers, third-party payors and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that affect the business and financial arrangements and relationships through which we market and sell our products. The laws that affect our ability to operate include, but are not limited to:
45
or service reimbursable, in whole or in part, under a federal healthcare program, such as the Medicare and Medicaid programs. The term ‘‘remuneration’’ has been broadly interpreted to include anything of value, such as specimen collection materials or test kits. There are a number of statutory exceptions and regulatory safe harbors protecting some common activities from prosecution, however these are drawn narrowly. Additionally, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. Violations are subject to civil and criminal fines and monetary penalties of up to $100,000 for each violation, plus up to three times the remuneration involved, imprisonment of up to ten years and exclusion from government healthcare programs. In addition, the ACA codified case law that a claim including items or services resulting from a violation of the AKS constitutes a false or fraudulent claim for purposes of the federal False Claims Act, or the FCA;
•the Stark Law, which prohibits a physician from making a referral for certain designated health services covered by the Medicare or Medicaid program, including laboratory and pathology services, if the physician or an immediate family member of the physician has a financial relationship with the entity providing the designated health services and prohibits that entity from billing, presenting or causing to be presented a claim for the designated health services furnished pursuant to the prohibited referral, unless an exception applies. Sanctions for violating the Stark Law include denial of payment, civil monetary penalties and exclusion from the federal health carehealthcare programs. Failure to refund amounts received as a result of a prohibited referral on a timely basis may constitute a false or fraudulent claim and may result in civil penalties and additional penalties under the FCA;
•federal civil and criminal false claims laws and civil monetary penalty laws, such as the FCA, which can be enforced by private citizens through civil qui tam actions, prohibitsaction, and civil monetary penalty laws prohibit individuals or entities from, among other things, knowingly presenting, or causing to be presented through distribution of template medical necessity language or other coverage and reimbursement information, false, fictitious or fraudulent claims for payment or approval by the federal government, including federal health carehealthcare programs, such as Medicare and Medicaid, and knowingly making, using or causing to be made or used a false record or statement material to a false or fraudulent claim, or knowingly making a false statement to improperly avoid, decrease or conceal an obligation to pay money to the federal government. In addition, a claim including items or services resulting from a violation of the AKS constitutes a false or fraudulent
claim for purposes of the FCA. Private individuals can bring False Claims ActFCA ‘‘qui tam’’ actions, on behalf of the government and such individuals, commonly known as ‘‘whistleblowers,’’ may share in amounts paid by the entity to the government in fines or settlement. When an entity is determined to have violated the federal civil False Claims Act,FCA, the government may impose civil fines and penalties, plus treble damages, and exclude the entity from participation in Medicare, Medicaid and other federal healthcare programs;
•the EKRA prohibits payments for referrals to recovery homes, clinical treatment facilities, and laboratories. EKRA’s reach extends beyond federal healthcare programs to include private insurance (i.e., it is an “all payor” statute). For purposes of EKRA, the term “laboratory” is defined broadly and without reference to any connection to substance use disorder treatment. The law includes a limited number of exceptions, some of which closely align with corresponding federal AKS exceptions and safe harbors, and others that materially differ;
•HIPAA, which, among other things, imposes criminal liability for executing or attempting to execute a scheme to defraud any healthcare benefit program, including private third-party payors, knowingly and willfully embezzling or stealing from a healthcare benefit program, willfully obstructing a criminal investigation of a healthcare offense, and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement or representation, in connection with the delivery of or payment for healthcare benefits, items or services. Like the AKS, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation;
•HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH, and their implementing regulations, which imposes privacy, security and breach reporting obligations with respect to individually identifiable health information upon entities subject to the law, such as health plans, healthcare clearinghouses and certain healthcare providers, known as covered entities, and their respective business associates, individuals or entities that perform services for them that involve individually identifiable health information.information as well as their covered subcontractors. Failure to comply with the HIPAA privacy and security standardsHIPAA’s obligations can result in civil monetary penalties, and, in certain circumstances, criminal penalties. HITECH also created new tiers of civil monetary penalties, amended HIPAA to make civil and criminal penalties directly applicable to business associates, and gave state attorneys general new
46
authority to file civil actions for damages or injunctions in U.S. federal courts to enforce HIPAA and seek attorneys’ fees and costs associated with pursuing federal civil actions;
•state laws that prohibit other specified practices, such as billing physicians for tests that they order or providing tests at no or discounted cost to induce physician or patient adoption; insurance fraud laws; waiving coinsurance, copayments,co-payments, deductibles, and other amounts owed by patients; billing a state Medicaid program at a price that is higher than what is charged to one or more other third-party payors employing, exercising control over or splitting professional fees with licensed professionals in violation of state laws prohibiting fee splitting or the corporate practice of medicine and other professions; and
•federal and state consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers;
•the federal transparency requirements under the Physician Payments Sunshine Act, created under the ACA, which requires, among other things, certain manufacturers of drugs, devices, biologics and medical supplies reimbursed under Medicare, Medicaid, or the Children’s Health Insurance Program to annually report to CMS information related to payments and other transfers of value provided to physicians certain(defined to include doctors, dentists, optometrists, podiatrists and chiropractors), other healthcare professionals (such as physician assistants and nurse practitioners) and teaching hospitals and information regarding physician ownership and investment interests, including such ownership and investment interests held by a physician’s immediate family members. Failure to submit required information may result in civil monetary penalties for all payments, transfers of value or ownership or investment interests that are not timely, accurately, and completely reported in an annual submission, and may result in liability under other federal laws or regulations. We believe that we are exempt from these reporting requirements. We cannot assure you, however, that our regulators, principally the federal government, will agree with our determination, and a determination that we have violated these laws and regulations, or a public announcement that we are being investigated for possible violations, could adversely affect our business;
•the prohibition on reassignment of Medicare claims, which, subject to certain exceptions, precludes the reassignment of Medicare claims to any other part;
•state and foreign law equivalents of each of the above federal laws, such as anti-kickback and false claims laws, thatwhich may impose similar or more prohibitive restrictions, and may apply to items or services reimbursed by any non-governmental third-party payors, including private insurers; and
•federal, state, local and foreign laws that govern the privacy and security of health information or personally identifiable information in certain circumstances, including state health information privacy and data breach notification laws which govern the collection, use, disclosure, and protection of health-related and other personal information,data, many of which differ from each other in significant ways and often aremay not be pre-empted by HIPAA, thus complicating compliance efforts.
As a clinical laboratory, our business practices may face additional scrutiny from government regulatory agencies such as the Department of Justice, the U.S. Department of Health and Human Services Office of Inspector General, or OIG and CMS. Certain arrangements between clinical laboratories and referring physicians have been identified in fraud alerts issued by the OIG as implicating the AKS. The OIG has stated that it is particularly concerned about these types of arrangements because the choice of laboratory, as well as the decision to order laboratory tests, typically are made or strongly influenced by the physician, with little or no input from patients. Moreover, the provision of payments or other items of value by a clinical laboratory to a referral source could be prohibited under the Stark Law unless the arrangement meets all criteria of an applicable exception. The government has been active in enforcement of these laws as they apply to clinical laboratories.
We have entered into consulting and scientific advisory board arrangements, speaking arrangements and clinical research agreements with physicians and other healthcare providers, including some who could influence the use of our products. Because of the complex and far-reaching nature of these laws, regulatory agencies may view these transactions as prohibited arrangements that must be restructured, or discontinued, or for which we could be subject to other significant penalties. We could be adversely affected if regulatory agencies interpret our financial relationships with providers who may influence the ordering of and use of our products to be in violation of applicable laws.
The scope and enforcement of each of these laws is uncertain and subject to rapid change in the current environment of healthcare reform. Federal and state enforcement bodies have recently increased their scrutiny of interactions between healthcare companies, healthcare providers and other third parties, including charitable foundations, which has led to a number of investigations, prosecutions, convictions and settlements in the healthcare industry. It is possible that governmental authorities may conclude that our business practices, including
47
our consulting arrangements with physicians, as well as our financial assistance programs, do not comply with current or future statutes, regulations, agency guidance or case law involving applicable healthcare laws. Responding to investigations can be time and resource-consuming and can divert management’s attention from the business. Any such investigation or settlement could increase our costs or otherwise have an adverse effect on our business.
Ensuring that our business arrangements with third parties comply with applicable healthcare laws and regulations is costly. If our operations are found to be in violation of any of these laws or any other current or future governmental laws and regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, disgorgement, individual imprisonment, exclusion from government funded healthcare programs, such as Medicare and Medicaid, contractual damages, reputational harm, diminished profits and future earnings, additional reporting obligations and oversight if we become subject to a corporate integrity agreement or other agreement to resolve allegations of non-compliance with these laws, and the curtailment or restructuring of our operations, any of which could substantially disrupt our operations. If any of the physicians or other healthcare providers or entities with whom we do business is found to be not in compliance with applicable laws, they may be subject to significant criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs.
We are subject to certain U.S. anti-corruption, anti-money laundering, export control, sanctions, and other trade laws and regulations and may become subject to their similar foreign equivalents. We can face serious consequences for violations.
U.S. and foreign anti-corruption, anti-money laundering, export control, sanctions, and other trade laws and regulations or collectively, Trade Laws, prohibit, among other things, companies and their employees, agents, legal counsel, accountants, consultants, contractors, and other partners from authorizing, promising, offering, providing, soliciting, or receiving, directly or indirectly, corrupt or improper payments or anything else of value to or from recipients in the public or private sector. Violations of Trade Lawsthese trade laws can result in substantial criminal fines and civil penalties, imprisonment, the loss of trade privileges, debarment, tax reassessments, breach of contract and fraud litigation, reputational harm, and other consequences. We have direct or indirect interactions with officials and employees of government agencies or government-affiliated hospitals, universities, and other organizations. We also expect that we may engage in non-U.S. activities over time. We expect to rely on third-party suppliers and/or third parties to obtain necessary permits, licenses, and patent registrations. We can be held liable for the corrupt or other illegal activities of our personnel, agents, or partners, even if we do not explicitly authorize or have prior knowledge of such activities. Any violations of the laws and regulations described above may result in substantial civil and criminal fines and penalties, imprisonment, the loss of export or import privileges, debarment, tax reassessments, breach of contract and fraud litigation, reputational harm and other consequences.
In the ordinary course of our business, we collect, store, use, transmit, disclose, or otherwise process (“Process”) confidential, proprietary, and store sensitive data, including protected health information, or PHI, personally identifiable information, or PII,personal data, credit card and other financial information, intellectual property and proprietary business information owned or controlled by ourselves or our customers, payors and other parties. We manageOur data processing activities may subject us to numerous data privacy and maintain our applicationssecurity obligations, such as laws, regulations, guidance, industry standards, external and data utilizing a combination of on-site systems, managed data centers,internal privacy and cloud-based data centers. We utilize external security policies, contracts and infrastructure vendors to manage parts of our data centers.
In the United States, numerous federal, state, and local governments have enacted data privacy and security laws, including federal health information such asprivacy laws, state data breach notification laws, state health information privacy laws, federal and state consumer protection laws (e.g., Section 5 of the Federal Trade Commission Act), and other similar laws (e.g., wiretapping laws). For example, HIPAA, as amended by HITECH, imposes specific requirements relating to the privacy, security, and transmission of individually identifiable health information. In the past few years, numerous U.S. states—including California, Virginia, Colorado, Connecticut, and Utah—have enacted comprehensive privacy laws that impose certain obligations on covered businesses, including providing specific disclosures in privacy notices and affording residents with certain rights concerning their personal data. As applicable, such rights may include the right to access, correct, or delete certain personal data, and to opt-out of certain data processing activities, such as targeted advertising, profiling, and automated decision-making. The
48
exercise of these rights may impact our business and ability to provide our products and services. Certain states also impose stricter requirements for processing certain personal data, including sensitive information, such as conducting data privacy impact assessments. These state laws allow for statutory fines for noncompliance. For example, the California Consumer Privacy Act of 2018, as amended by the California Rights Privacy Act of 2020 (“CPRA”) (collectively, the “CCPA”), applies to personal data of consumers, business representatives and employees who are California residents and requires businesses to provide specific disclosures in privacy notices and honor requests of such individuals to exercise certain privacy rights. The CCPA provides for fines of up to $7,500 per intentional violation and allows private litigants affected by certain data breaches to recover significant statutory damages. Similar laws are being considered in several other states, as well as at the federal and local levels, and we expect more states to pass similar laws in the future. Although these states, like the CCPA, exempt some personal data processed in the context of clinical trials, these developments, to the extent applicable to our business and operations, may complicate our compliance efforts and costs and increase legal risk for us and the third parties upon whom we rely.
Outside the United States, there are also an increasing number of laws, regulations, industry standards and other obligations concerning privacy and data security. For example, we may be subject to the European Union’s General Data Protection Regulation (“EU”) 2016/679 (“EU GDPR”) and the United Kingdom’s GDPR (“UK GDPR”) (collectively, “GDPR”). Under the GDPR, companies may face temporary or definitive bans on data processing and other corrective actions; fines of up to 20 million Euros under the EU GDPR, 17.5 million pounds sterling under the UK GDPR or, in each case, 4% of annual global revenue, whichever is greater; or private litigation related to processing of personal data brought by classes of data subjects or consumer protection organizations authorized at law to represent their interests.
Our employees and personnel use, or may use, generative artificial intelligence (“AI”) technologies to perform their work, and the disclosure and use of personal data in generative AI technologies is subject to various privacy laws and other privacy obligations. Governments have passed and are likely to pass additional laws regulating generative AI. Our use of this technology could result in additional compliance costs, regulatory penalties. Noticeinvestigations and actions, and consumer lawsuits. If we are unable to use generative AI, it could make our business less efficient and result in competitive disadvantages.
In the ordinary course of breaches must be madebusiness, we may transfer personal data from Europe and other jurisdictions to affected individuals, the Secretary of the Department of HealthUnited States or other countries. Europe and Human Services, and for extensive breaches, notice may needother jurisdictions have enacted laws requiring data to be madelocalized or limiting the transfer of personal data to other countries. In particular, the European Economic Area (“EEA”) and the UK have significantly restricted the transfer of personal data to the media or State Attorneys General. Such a notice could harm our reputationUnited States and our abilityother countries whose privacy laws it generally believes are inadequate. Other jurisdictions may adopt similarly stringent interpretations of their data localization and cross-border data transfer laws. Although there are currently various mechanisms that may be used to compete. Although we have implemented security measurestransfer personal data from the EEA and a formal, dedicated enterprise security programthe UK to prevent unauthorized accessthe United States in compliance with law, such as the EEA standard contractual clauses, the UK’s International Data Transfer Agreement / Addendum, and the EU-U.S. Data Privacy Framework and the UK extension thereto (which allows for transfers to patient data, such data is currently accessible through multiple channels,relevant U.S.-based organizations who self-certify compliance and participate in the Framework), these mechanisms are subject to legal challenges, and there is no guaranteeassurance that we can protect oursatisfy or rely on these measures to lawfully transfer personal data to the United States. If there is no lawful manner for us to transfer personal data from breach. Unauthorized access, lossthe EEA, the UK, or disseminationother jurisdictions to the United States, or if the requirements for a legally-compliant transfer are too onerous, we could also disruptface significant adverse consequences, including the interruption or degradation of our operations, (includingthe need to relocate part of or all of our abilitybusiness or data processing activities to conduct our analyses, provide test results, bill payers or patients, process claimsother jurisdictions (such as Europe) at significant expense, increased exposure to regulatory actions, substantial fines and appeals, provide customer assistance, conduct researchpenalties, the inability to transfer data and development activities, collect, process, and prepare company financial information, provide information about our productswork with partners, vendors and other patientthird parties, and physician education and outreach efforts throughinjunctions against our website, and manage the administrative aspectsprocessing or transferring of our business) and damage our reputation, any of which could adversely affectpersonal data necessary to operate our business. Additionally, companies that transfer personal data out of the EEA and UK to other jurisdictions, particularly to the United States, are subject to increased scrutiny from regulators, individual litigants, and activist groups. Some European regulators have ordered certain companies to suspend or permanently cease certain transfers of personal data out of Europe for allegedly violating the GDPR’s cross-border data transfer limitations.
In addition, privacy advocates and industry groups have proposed, and may in the future propose, standards with which we may obtain health information from third parties that are also subjectlegally or contractually bound to comply. In addition to data privacy and security requirements under HIPAA,laws, we are contractually subject to industry standards adopted by industry groups and may become subject to such obligations in the future. For example, we are subject to the Payment Card Industry Data Security Standard (“PCI DSS”). The PCI DSS requires companies to adopt certain measures to ensure the security of cardholder information, including using and maintaining firewalls, adopting proper password protections for certain devices and software, and restricting data access. Noncompliance with PCI-DSS can result in penalties ranging from $5,000 to $100,000 per
49
month by credit card companies, litigation, damage to our reputation, and revenue losses. We rely on vendors to process payment card data, and those vendors may be subject to PCI DSS, and our business may be negatively affected if our vendors are fined or suffer other consequences as amendeda result of PCI DSS noncompliance.
More generally, we are also bound by HITECH.
Obligations related to data privacy and security (and consumers’ data privacy expectations) are quickly changing in an increasingly stringent fashion, creating uncertainty as to the effective future legal framework. These laws and regulations are not necessarily preempted by HIPAA, particularly if a state affords greater protection to individuals than HIPAA. Where state laws are more protective, we have to comply with the stricter provisions. In addition to fines and penalties imposed upon violators, some of these state laws also afford private rights of action to individuals who believe their personal information has been misused. California’s patient privacy laws, for example, provide for penalties of up to $250,000 and permit injured parties to sue for damages. The interplay of federal and state lawsobligations may be subject to varying applications and interpretations, by courts and government agencies,which may be inconsistent or conflicting among jurisdictions, creating complex compliance issues for us and our clientsclients. Preparing for and potentially exposingcomplying with these obligations requires us to additional expense, adverse publicitydevote significant resources (including, without limitation, financial and liability. Further, as regulatory focus on privacy issues continues to increase and laws and regulations concerning the protection of personal information expand and become more complex, these potential riskstime-related resources).
These obligations may necessitate changes to our information technologies, systems, and practices and to those of any third parties that process personal data on our behalf. In addition, these obligations may require us to change our business model or to take on more onerous obligations in our contracts. Although we endeavor to comply with all applicable obligations, we may, at times, fail or be perceived to have failed to do so. Moreover, despite our efforts, our personnel or third parties upon whom we rely on may fail to comply with such obligations, which could intensify. Changesnegatively impact our business operations and compliance posture. Failure or perceived failure to comply with these obligations could result in laws significant consequences, including but not limited to government enforcement actions (e.g., investigations, fines, penalties, audits, inspections, and similar), litigation (including class-action claims) and mass arbitration demands, additional reporting requirements and/or regulations associated withoversight, bans on processing personal data, and orders to destroy or not use personal data. In particular, plaintiffs have become increasingly more active in bringing privacy-related claims against companies, including class claims and mass arbitration demands. Some of these claims allow for the enhanced protectionrecovery of statutory damages on a per violation basis, and, if viable, carry the potential for monumental statutory damages, depending on the volume of data and the number of violations. Any of these events could have a material adverse effect on our reputation, business, or financial condition, including but not limited to: inability to process personal data or to operate in certain types of sensitive data, such PHI or PII along with increased customer demands for enhanced data security infrastructure, could greatlyjurisdictions, increase our cost of providing our services, decrease demand for our services, reduce our revenue, and/or subject us to additional liabilities.
Ethical, legal and social concerns related to the use of genetic information could reduce demand for our products.
Genetic testing has raised ethical, legal, and social issues regarding privacy and the appropriate uses of the resulting information. Governmental authorities have, through the Genetic Information Nondisclosure Act of 2008, and could further, for social or other purposes, limit or regulate the use of genetic information or genetic testing or prohibit testing for genetic predisposition to certain conditions, particularly for those that have no known cure. Ethical and social concerns may also influence governmental authorities to deny or delay the issuance of patents for technology relevant to our business. While we do not currently perform genetic tests for genetic predisposition to certain conditions, these concerns may lead patients to refuse to use, or clinicians to be reluctant to order, our genomic tests or genetic tests for somatic mutations even if permissible. These and other ethical, legal and social concerns may limit market acceptance of our products or reduce the potential markets for our products, either of which could have an adverse effect on our business, financial condition, or results of operations.
Evolving expectations around corporate responsibility practices, specifically related to environmental, social and governance (“ESG”) matters, may expose us to reputational and other risks.
Investors, stockholders, customers, suppliers and other third parties are increasingly focusing on ESG and corporate social responsibility endeavors and reporting. Companies that do not adapt to or comply with the evolving investor or stakeholder expectations and standards, or that are perceived to have not responded appropriately, may suffer from reputational damage, which could result in the business, financial condition and/or stock price of a company being materially and adversely affected. Further, this increased focus on ESG issues may result in new regulations and/or third-party requirements that could adversely impact our business, or certain shareholders reducing or eliminating their holdings of our stock. Additionally, an allegation or perception that we have not taken sufficient action in these areas could negatively harm our reputation.
50
Risks Related to Intellectual Property
If we are unable to obtain and maintain sufficient intellectual property protection for our technology, or if the scope of the intellectual property protection obtained is not sufficiently broad, our competitors could develop and commercialize diagnostic tests similar or identical to ours, and our ability to successfully commercialize our products may be impaired.
We rely on patent protection as well as trademark, copyright, trade secret and other intellectual property rights protection as well as nondisclosure, confidentiality and other contractual restrictions to protect our brands and proprietary tests and technologies, all of which provide limited protection and may not adequately protect our rights or permit us to gain or keep any competitive advantage. If we fail to protect our intellectual property, third parties may be able to compete more effectively against us. In addition, we may incur substantial litigation costs in our attempts to recover or restrict use of our intellectual property.
As is the case with other life science companies, our success depends in large part on our ability to obtain and maintain protection of the intellectual property we may own solely or jointly with others or in-license from others, particularly patents, in the United States and other countries with respect to our products and technologies. We apply for patents covering our products and technologies and uses thereof, as we deem appropriate. However, obtaining and enforcing life sciences patents is costly, time-consuming and complex, and we may fail to apply for patents on important tests, services and technologies in a timely fashion or at all, or we may fail to apply for patents in potentially relevant jurisdictions. We may not be able to file and prosecute all necessary or desirable patent applications, or maintain, enforce and license any patents that may issue from such patent applications, at a reasonable cost or in a timely manner. It is also possible that we will fail to identify patentable aspects of our research and development output before it is too late to obtain patent protection.
We may not have the right to control the preparation, filing and prosecution of patent applications, or to maintain the rights to patents licensed from or to third parties. Therefore, these patents and applications may not be prosecuted and enforced in a manner consistent with the best interests of our business.
Our patent portfolio as of December 31, 2023 includes five16 issued U.S. patents and three13 pending U.S. patent applications, with foreign counterparts. It is possible that none of our pending patent applications will result in issued patents in a timely fashion or at all, and even if patents are granted, they may not provide a basis for intellectual property protection of commercially viable tests or services, may not provide us with any competitive advantages, or may be challenged and invalidated by third parties. It is possible that others will design around our future patented technologies. We may not be successful in defending any such challenges made against our patents or patent applications. Any successful third-party challenge to our patents could result in the unenforceability or invalidity of such patents and increased competition to our business. Even if our patents are held valid and enforceable, they may still be found insufficient to provide protection against competing products and services sufficient to achieve our business objectives. We may have to challenge the patents or patent applications of third parties, such as to counter infringement or unauthorized use. In addition, in an infringement proceeding, a court may decide that a patent of ours is invalid or unenforceable, or may refuse to enjoin the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. Even if we prevail against an infringer in a U.S. district court or foreign trial-level court, there is always the risk that the infringer will file an appeal and the initial court judgment will be overturned at the appeals court and/or that an adverse decision will be issued by the appeals court relating to the validity or enforceability of our patents. The outcome of patent litigation or other proceeding can be uncertain, and any attempt by us to enforce our patent rights against others or to challenge the patent rights of others may not be successful, or, if successful, may take substantial time and result in substantial cost, and may divert our efforts and attention from other aspects of our business.
The patent positions of life sciences companies can be highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. No consistent policy regarding the breadth of claims allowed in such companies’ patents has emerged to date in the United States or elsewhere. Courts frequently render opinions in the life sciences field that may affect the patentability of certain inventions or discoveries, including opinions that may affect the patentability of methods for analyzing or comparing DNA sequences.
In particular, the patent positions of companies engaged in the development and commercialization of genomic diagnostic tests are particularly uncertain. Various courts, including the U.S. Supreme Court, have rendered decisions that affect the scope of patentability of certain inventions or discoveries relating to certain diagnostic tests and related methods. These decisions state, among other things, that a patent claim that recites an abstract idea, natural phenomenon or law of nature (for example, the relationship between particular genetic variants and cancer) are not themselves patentable. Precisely what constitutes a law of nature is uncertain, and it is possible that certain
51
aspects of genetic diagnostics tests would be considered natural laws. Accordingly, the evolving case law in the United States may adversely affect our ability to obtain patents and may facilitate third-party challenges to any owned or licensed patents. The laws of some foreign countries do not protect intellectual property rights to the same extent as the laws of the United States, and we may encounter difficulties in protecting and defending such rights in foreign jurisdictions. The legal systems of many other countries do not favor the enforcement of patents and other intellectual property protection, particularly those relating to life science technologies, which could make it difficult for us to stop the infringement of our patents in such countries. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial cost and divert our efforts and attention from other aspects of our business.
To the extent our intellectual property offers inadequate protection, or is found to be invalid or unenforceable, we would be exposed to a greater risk of direct competition, and our competitive position could be adversely affected, as could our business. Both the patent application process and the process of managing patent disputes can be time-consuming and expensive. Moreover, if a third party has intellectual property rights that cover the practice of our technology, we may not be able to fully exercise or extract value from our intellectual property rights. The following examples are illustrative:
•others may be able to develop and/or practice technology that is similar to our technology or aspects of our technology, but that are not covered by the claims of the patents that we own or control, assuming such patents have issued or do issue;
•we or our licensors or any future strategic partners might not have been the first to conceive or reduce to practice the inventions covered by the issued patents or pending patent applications that we own or have exclusively licensed;
•we or our licensors or any future strategic partners might not have been the first to file patent applications covering certain of our inventions;
•others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our intellectual property rights;
•it is possible that our pending patent applications will not lead to issued patents;
•issued patents that we own or have exclusively licensed may not provide us with any competitive advantage, or may be held invalid or unenforceable, as a result of legal challenges by our competitors;
•our competitors might conduct research and development activities in countries where we do not have patent rights and then use the information learned from such activities to develop competitive tests for sale in our major commercial markets;
•third parties performing manufacturing or testing for us using our products or technologies could use the intellectual property of others without obtaining a proper license;
•parties may assert an ownership interest in our intellectual property and, if successful, such disputes may preclude us from exercising exclusive rights over that intellectual property;
•we may not develop or in-license additional proprietary technologies that are patentable;
•we may not be able to obtain and maintain necessary licenses on commercially reasonable terms, or at all; and
•the patents of others may have an adverse effect on our business.
Should any of these events occur, they could significantly harm our business and results of operations.
Changes in patent law in the United States and other jurisdictions could diminish the value of patents in general, thereby impairing our ability to protect our products.
As is the case with other life sciences companies, our success is heavily dependent on intellectual property, particularly patents relating to our research programs and products. Obtaining and enforcing patents in the life sciences industry involves both technological and legal complexity and is therefore costly, time consuming and inherently uncertain. Changes in either the patent laws or interpretation of the patent laws in the United States or the USPTO rules and regulations could increase these uncertainties and costs. Recent patentPatent reform legislation in the United States and other countries, including the Leahy-Smith America Invents Act or the AIA,(“AIA”), signed into law on September 16, 2011, could increase those uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents. The AIA includes a number of significant changes to U.S. patent law. These include provisions that affect the way patent applications are prosecuted,
52
redefine prior art and provide more efficient and cost-effective avenues for competitors to challenge the validity of patents. These include allowing third-party submission of prior art to the USPTO during patent prosecution and additional procedures to attack the validity of a patent in USPTO administeredUSPTO-administered post-grant proceedings, including post-grant review, inter partes review, and derivation proceedings. For applications filed after March 15, 2013 that do not claim the benefit of applications filed before that date, the AIA transitioned the United States from a first to invent system to a first-inventor-to-file system in which, assuming that the other statutory requirements are met, the first inventor to file a patent application will be entitled to the patent on an invention regardless of whether a third party was the first to invent the claimed invention. The AIA and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications, our ability to obtain future patents, and the enforcement or defense of our issued patents, all of which could have a material adverse effect on our business, financial condition, results of operations and prospects.
The U.S. Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations.
Depending on future actions by the U.S. Congress, the U.S. courts, the USPTO and the relevant law-making bodies in other countries, the laws and regulations governing patents could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce our existing patents and patents that we might obtain in the future.
Our in-licensed intellectual property has been discovered through government funded programs and thus may be subject to federal regulations such as ‘‘march-in’’ rights, certain reporting requirements and a preference for U.S.-based companies, and compliance with such regulations may limit our exclusive rights, and limit our ability to contract with non-U.S. manufacturers.
Intellectual property rights that have been in-licensed pursuant to our license agreement, or the License Agreement with WUSTL have been generated through the use of U.S. government funding, and are therefore subject to certain federal regulations. As a result, the United States federal government may retain certain rights to intellectual property embodied in our current or future product candidates under the Bayh-Dole Act. These federal government rights include a ‘‘nonexclusive, nontransferable, irrevocable, paid-up license’’ to use inventions for any governmental purpose. The Bayh-Dole Act also provides federal agencies with ‘‘march-in rights.’’ March-in rights allow the government, in specified circumstances, to require the contractor or successors in title to the patent to grant a ‘‘nonexclusive, partially exclusive, or exclusive license’’ to a ‘‘responsible applicant or applicants’’ if it determines that (1) adequate steps have not been taken to commercialize the invention, (2) government action is necessary to meet public health or safety needs or (3) government action is necessary to meet requirements for public use under federal regulations. If the patent owner refuses to do so, the government may grant the license itself.
The U.S. government also has the right to take title to these inventions if the licensor fails to disclose the invention to the government or fails to file an application to register the intellectual property within specified time limits. Intellectual property generated under a government funded program is also subject to certain reporting requirements, compliance with which may require us to expend substantial resources. In addition, the U.S. government requires that any products embodying any of these inventions or produced through the use of any of these inventions be manufactured substantially in the United States, and the License Agreement requires that we comply with this requirement. This preference for U.S. industry may be waived by the federal agency that provided the funding if the owner or assignee of the intellectual property can show that reasonable but unsuccessful efforts have been made to grant licenses on similar terms to potential licensees that would be likely to manufacture substantially in the United States or that under the circumstances domestic manufacture is not commercially feasible. This preference for U.S. industry may limit our ability to contract with non-U.S. product manufacturers for products covered by such intellectual property. To the extent any of our owned or future in-licensed intellectual property is also generated through the use of U.S. government funding, the provisions of the Bayh-Dole Act may similarly apply.
Issued patents covering our products and related technologies could be found invalid or unenforceable if challenged.
The issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability. Some of our patents or patent applications (including licensed patents) have been, are being or may be challenged at a future point in time in an opposition, nullification, derivation, reexamination, inter partes review, post-grant review or interference action in court or before patent offices or similar proceedings for a given period after allowance or grant, during which time third parties can raise objections against such grant. In the course of such proceedings, which may continue for a protracted period of time, the patent owner may be compelled to limit the scope of the allowed or granted claims thus attacked, or may lose the allowed or granted claims altogether. Any successful third-partythird-
53
party challenge to our patents in this or any other proceeding could result in the unenforceability or invalidity of such patents, which may lead to increased competition to our business, which could harm our business. In addition, if the breadth or strength of protection provided by our patents and patent applications is threatened, regardless of the outcome, it could dissuade companies from collaborating with us to license, develop or commercialize current or future diagnostic tests.
We may not be aware of all third-party intellectual property rights potentially relating to our products. Publications of discoveries in the scientific literature often lag behind the actual discoveries, and patent applications in the United States and other jurisdictions are typically not published until approximately 18 months after filing or, in some cases (e.g.(e.g., U.S. applications for which a request not to publish has been filed), not until such patent applications issue as patents. We might not have been the first to make the inventions covered by each of our pending patent applications and we might not have been the first to file patent applications for these inventions. To determine the priority of these inventions, we have and may have to participate in interference proceedings, derivation proceedings or other post-grant proceedings declared by the USPTO that could result in substantial cost to us. The outcome of such proceedings is uncertain. We can give no assurance that all of the potentially relevant art relating to our patents and patent applications has been found; overlooked prior art could be used by a third party to challenge the validity, enforceability and scope of our patents or prevent a patent from issuing from a pending patent application. As a result, we may not be able to obtain or maintain protection for certain inventions. No assurance can be given that other patent applications will not have priority over our patent applications. In addition, changes to the patent laws of the United States allow for various post-grant opposition proceedings that have not been extensively tested, and their outcome is therefore uncertain. Therefore, the validity, enforceability and scope of our patents in the United States and other countries cannot be predicted with certainty and, as a result, any patents that we own or license may not provide sufficient protection against our competitors. Furthermore, if third parties bring these proceedings against our patents, we could experience significant costs and management distraction.
Our commercial success depends significantly on our ability to operate without infringing upon the intellectual property rights of third parties.
The life sciences industry is subject to rapid technological change and substantial litigation regarding patent and other intellectual property rights. Our potential competitors in both the United States and abroad, may have substantially greater resources and are likely to make substantial investments in patent portfolios and competing technologies, and may apply for or obtain patents that could prevent, limit or otherwise interfere with our ability to make, use and sell our products. Numerous third-party patents exist in fields relating to our products and technologies, and it is difficult for industry participants, including us, to identify all third-party patent rights relevant to our products and technologies. Moreover, because some patent applications are maintained as confidential for a certain period of time, we cannot be certain that third parties have not filed patent applications that cover our products and technologies.
Patents could be issued to third parties that we may ultimately be found to infringe. Third parties may have or obtain valid and enforceable patents or proprietary rights that could block us from using our technology. Our failure to obtain or maintain a license to any technology that we require may materially harm our business, financial condition and results of operations. Furthermore, we would be exposed to a threat of litigation.
From time to time, we may be party to, or threatened with, litigation or other proceedings with third parties, including non-practicing entities, who allege that our products, components of our products, and/or proprietary technologies infringe, misappropriate or otherwise violate their intellectual property rights. The types of situations in which we may become a party to such litigation or proceedings include:
•we may initiate litigation or other proceedings against third parties seeking to invalidate the patents held by those third parties or to obtain a judgment that our products or technologies do not infringe those third parties’ patents;
•we may participate at substantial cost in International Trade Commission proceedings to abate importation of products that would compete unfairly with our products or technologies;
•if a competitor files patent applications that claim technology also claimed by us or our licensors, we or our licensors may be required to participate in interference, derivation or opposition proceedings to determine the priority of invention, which could jeopardize our patent rights and potentially provide a third party with a dominant patent position;
•if third parties initiate litigation claiming that our products or technologies infringe their patent or other intellectual property rights, we will need to defend against such proceedings;
54
•if third parties initiate litigation or other proceedings seeking to invalidate patents owned by or licensed to us or to obtain a declaratory judgment that their products, services, or technologies do not infringe our patents or patents licensed to us, we will need to defend against such proceedings;
•we may be subject to ownership disputes relating to intellectual property, including disputes arising from conflicting obligations of consultants or others who are involved in developing our products and technologies; and
•if a license to necessary technology is terminated, the licensor may initiate litigation claiming that our products or technologies infringe or misappropriate its patent or other intellectual property rights and/or that we breached our obligations under the license agreement, and we would need to defend against such proceedings.
These lawsuits and proceedings, regardless of merit, are time-consuming and expensive to initiate, maintain, defend or settle, and could divert the time and attention of managerial and technical personnel, which could materially adversely affect our business. Any such claim could also force us to do one or more of the following:
•incur substantial monetary liability for infringement or other violations of intellectual property rights, which we may have to pay if a court decides that the diagnostic test or technology at issue infringes or violates the third party’s rights, and if the court finds that the infringement was willful, we could be ordered to pay treble damages and the third party’s attorneys’ fees;
•stop manufacturing, offering for sale, selling, using, importing, exporting or licensing the diagnostic test or technology incorporating the allegedly infringing technology or stop incorporating the allegedly infringing technology into such test or technology;
•obtain from the owner of the infringed intellectual property right a license, which may require us to pay substantial upfront fees or royalties to sell or use the relevant technology and which may not be available on commercially reasonable terms, or at all;
•redesign our products and technologies so they do not infringe or violate the third party’s intellectual property rights, which may not be possible or may require substantial monetary expenditures and time;
•enter into cross-licenses with applicable third party, which could weaken our overall intellectual property position;
•lose the opportunity to license our technology to others or to collect royalty payments based upon successful protection and assertion of our intellectual property against others;
•find alternative suppliers for non-infringing technologies, which could be costly and create significant delay; or
•relinquish rights associated with one or more of our patent claims if our claims are held invalid or otherwise unenforceable.
Third parties may be able to sustain the costs of complex intellectual property litigation more effectively than we can because they have substantially greater resources. In addition, intellectual property litigation, regardless of its outcome, may cause negative publicity, adversely impact our business, cause delays, or prohibit us from marketing or otherwise commercializing our products and technologies. Any uncertainties resulting from the initiation and continuation of any litigation could have a material adverse effect on our ability to raise additional funds or otherwise have a material adverse effect on our business, results of operation, financial condition or cash flows.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. There could also be public announcements of the results of hearings, motions or other interim proceedings or developments, which could have a material adverse effect on the price of our common stock. If securities analysts or investors perceive these results to be negative, it could have a material adverse effect on the price of our common stock. The occurrence of any of these events may have a material adverse effect on our business, results of operation, financial condition or cash flows.
We depend on information technology systems that we license from third parties. Any failure of such systems or loss of licenses to the software that comprises an essential element of such systems could significantly harm our business.
We depend on information technology systems for significant elements of our operations, such as our laboratory information management systems,Laboratory Information Management System, including test validation, specimen tracking and quality control, our bioinformatics
55
analytical software systems, and our test report generating systems and billing systems. Essential elements of these systems depend on software that we license from third parties. If we are unable to maintain the licenses to this software or our software providers discontinue or alter the programs on which we rely, it could render our test reports unreliable or hinder our ability to generate accurate test reports, among other things. Any of the foregoing could have a material adverse effect on our competitive position, business, financial conditions, results of operations and prospects.
We rely on licenses from third parties, and if we lose these licenses or are not able to obtain licenses to third-party technology on reasonable grounds or at all, then we may not be able to continue to commercialize existing diagnostic tests, be subjected to future litigation and may not be able to commercialize new diagnostic tests in the future.
We are party to certain royalty-bearing license agreements that grant us rights to use certain intellectual property, including patents and patent applications, in certain specified fields of use. Although we intend to develop products and technologies through our own internal research, we may need to obtain additional licenses from others to advance our research, development and commercialization activities. Our license agreements impose, and we expect that future license agreements will impose, various development, diligence, commercialization and other obligations on us.
In the future, we may identify third-party technology we may need, including to develop or commercialize new diagnostic tests or services. In return for the use of a third party’s technology, we may agree to pay the licensor royalties based on sales of our solutions. Royalties are a component of the cost of our products or services and affect our margins. We may also need to negotiate licenses to patents or patent applications before or after introducing a commercialized test. The in-licensing and acquisition of third-party intellectual property rights is a competitive area, and a number of more established companies are also pursuing strategies to in-license or acquire third-party intellectual property rights for technologies that we may consider attractive or necessary.
These established companies may have a competitive advantage over us due to their size, cash resources and greater clinical development and commercialization capabilities. Furthermore, companies that perceive us to be a competitor may be unwilling to assign or license rights to us. In addition, we expect that competition for the in-licensing or acquisition of third-party intellectual property rights for technologies that are attractive to us may increase in the future, which may mean fewer suitable opportunities for us as well as higher acquisition or licensing costs. We may not be able to obtain necessary or strategic licenses to patents or patent applications, and our business may suffer if we are unable to enter into these licenses on acceptable terms or at all, if any necessary licenses are subsequently terminated, if the licensors fail to abide by the terms of the licenses or fail to prevent infringement by third parties, or if the licensed patents or other rights are found to be invalid or unenforceable.
In spite of our efforts, our licensors might conclude that we have materially breached our obligations under such license agreements and might therefore terminate the license agreements, thereby removing or limiting our ability to develop and commercialize tests and technology covered by these license agreements. If these in-licenses are terminated, or if the underlying patents fail to provide the intended exclusivity, competitors or other third parties might have the freedom to seek regulatory approval of, and to market, tests identical to ours and we may be required to cease our development and commercialization activities. For example, we license certain intellectual property from WUSTL that is incorporated into DecisionDx-UM. In 2019,2023, we provided more than 1,500over 1,600 test reports for DecisionDx-UM. If this license agreementthe License Agreement were terminated, we would be unable to continue to issue test reports and thus sales of DecisionDx-UM. Any of the foregoing could have a material adverse effect on our competitive position, business, financial conditions, results of operations and prospects.
Moreover, disputes may arise with respect to any one of our licensing agreements, including:
•the scope of rights granted under the license agreement and other interpretation-related issues;
•the extent to which our products, technology and processes infringe on intellectual property of the licensor that is not subject to the licensing agreement;
•the sublicensing of patent and other rights under our collaborative development relationships;
•our diligence obligations under the license agreement and what activities satisfy those diligence obligations;
•the inventorship and ownership of inventions and know-how resulting from the joint creation or use of intellectual property by our licensors and us and our partners; and
•the priority of invention of patented technology.
If we do not prevail in such disputes, we may lose any of such license agreements.
56
In addition, the agreements under which we currently license intellectual property or technology from third parties are complex, and certain provisions in such agreements may be susceptible to multiple interpretations.
The resolution of any contract interpretation disagreement that may arise could narrow what we believe to be the scope of our rights to the relevant intellectual property or technology, or increase what we believe to be our financial or other obligations under the relevant agreement, either of which could have a material adverse effect on our business, financial condition, results of operations and prospects. Moreover, if disputes over intellectual property that we have licensed prevent or impair our ability to maintain our current licensing arrangements on commercially acceptable terms, we may be unable to successfully develop and commercialize the affected diagnostic tests, which could have a material adverse effect on our business, financial conditions, results of operations and prospects.
Our failure to maintain such licenses could have a material adverse effect on our business, financial condition and results of operations. Any of these licenses could be terminated, such as if either party fails to abide by the terms of the license, or if the licensor fails to prevent infringement by third parties or if the licensed patents or other rights are found to be invalid or unenforceable. Absent the license agreements, we may infringe patents subject to those agreements, and if the license agreements are terminated, we may be subject to litigation by the licensor. Litigation could result in substantial costs and be a distraction to management. If we do not prevail, we may be required to pay damages, including treble damages, attorneys’ fees, costs and expenses, royalties or, be enjoined from selling our products or services, including DecisionDx-UM and DecisionDx-Melanoma, which could adversely affect our ability to offer our products or services, our ability to continue operations and our financial condition.
We may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting and defending patents on our products in all countries throughout the world would be prohibitively expensive. The requirements for patentability may differ in certain countries, particularly developing countries, and the breadth of patent claims allowed can be inconsistent. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as the laws of the United States, and we may encounter difficulties in protecting and defending such rights in foreign jurisdictions. Consequently, we may not be able to prevent third parties from practicing our inventions in all countries outside the United States, or from selling or importing products made using our inventions in and into the United States or other jurisdictions. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own tests or products and may also export infringing tests or products to territories where we have patent protection, but enforcement is not as strong as in the United States. These products may compete with our products. Our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of many other countries do not favor the enforcement of patents and other intellectual property protection, particularly those relating to life science technologies, which could make it difficult for us to stop the infringement of our patents in such countries. We do not have patent rights in certain foreign countries in which a market may exist. Moreover, in foreign jurisdictions where we do have patent rights, proceedings to enforce our patent rights could result in substantial cost and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly and our patent applications at risk of not issuing, and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license. We may not be able to stop a competitor from marketing and selling in foreign countries tests, products and services that are the same as or similar to our products and technologies, in which case our competitive position in the international market would be harmed.
If we are unable to protect the confidentiality of our trade secrets, the value of our technology could be materially adversely affected and our business could be harmed.
In addition to pursuing patents on our technology, we also rely on trade secrets, including unpatented know-how, technology and other proprietary information, to maintain our competitive position. We take steps to protect our trade secrets, in part, by entering into agreements, including confidentiality agreements, non-disclosure agreements and intellectual property assignment agreements, with our employees, consultants, academic institutions, corporate partners and, when needed, our advisers. However, we cannot be certain that such agreements have been entered into with all relevant parties, and we cannot be certain that our trade secrets and other confidential proprietary information will not be disclosed or that competitors will not otherwise gain access to our trade secrets or independently develop substantially equivalent information and techniques. For example, any of these parties may breach the agreements and disclose our proprietary information, including our trade secrets, and once disclosed,
57
Monitoring unauthorized disclosure is difficult, and we do not know whether the steps we have taken to prevent such disclosure are, or will be, adequate. If we were to enforce a claim that a third party had illegally obtained and was using our trade secrets, it would be expensive and time-consuming, and the outcome would be unpredictable. In addition, courts outside the United States may be less willing to protect trade secrets.
We also seek to preserve the integrity and confidentiality of our confidential proprietary information by maintaining physical security of our premises and physical and electronic security of our information technology systems, but it is possible that these security measures could be breached. If any of our confidential proprietary information were to be lawfully obtained or independently developed by a competitor, absent patent protection, we would have no right to prevent such competitor from using that technology or information to compete with us, which could harm our competitive position.
We may be subject to claims that our employees, consultants or independent contractors have wrongfully used or disclosed confidential information of third parties.
We do and may employ individuals who previously worked with universities or other companies, including potential competitors. We could in the future be subject to claims that we or our employees, consultants, or independent contractors have inadvertently or otherwise used or disclosed alleged trade secrets or other confidential information of current or former employers or competitors. Although we try to ensure that our employees, consultants and independent contractors do not use the intellectual property, proprietary information, know-how or trade secrets of others in their work for us, we may become subject to claims that we caused an individual to breach the terms of his or her non-competition or non-solicitation agreement, or that we or these individuals have, inadvertently or otherwise, used or disclosed the alleged trade secrets or other proprietary information of a current or former employer or competitor. Although, we are currently not subject to any such claims.
While we may litigate to defend ourselves against these claims, even if we are successful, litigation could result in substantial costs and could be a distraction to management and other employees. If our defenses to these claims fail, in addition to requiring us to pay monetary damages, a court could prohibit us from using technologies or features that are essential to our products, if such technologies or features are found to incorporate or be derived from the trade secrets or other proprietary information of the current or former employers. Therefore, we could be required to obtain a license from such third-party employer to commercialize our products or technology. Such a license may not be available on commercially reasonable terms or at all.
Moreover, any such litigation or the threat thereof may adversely affect our reputation, our ability to form strategic alliances or sublicense our rights to collaborators, engage with scientific advisors or hire employees or consultants, each of which would have an adverse effect on our business, results of operations and financial condition.
If our trademarks and trade names are not adequately protected, then we may not be able to build name recognition in our markets of interest and our business may be adversely affected.
We have not yet registered certain of our trademarks in all of our potential markets, although we have registered,registrations for, among others, DecisionDx, DiffDx-Melanoma, DecisionDx-UM, DecisionDx-Melanoma, DecisionDx-SCC, MyPath Melanoma, TissueCypher and DecisionDx-MelanomaIDgenetix in the United States. Our current or future registered or unregistered trademarks or trade names may be challenged, infringed, circumvented or declared generic or descriptive determined to be infringing on other marks. We may not be able to protect our rights to these trademarks and trade names or may be forced to stop using these names, which we need for name recognition by potential partners or customers in our markets of interest. During trademark registration proceedings, we may receive rejections. Although we would be given an opportunity to respond to those rejections, we may be unable to overcome such rejections. In addition, in the USPTO and in comparable agencies in many foreign jurisdictions, third parties are given an opportunity to oppose pending trademark applications and to seek to cancel registered trademarks. Opposition or cancellation proceedings may be filed against our trademarks, and our trademarks may not survive such proceedings. In addition, third parties have used trademarks similar and identical to our trademarks in foreign jurisdictions and have filed or may in the future file for registration of such trademarks. If they succeed in registering or developing common law rights in such trademarks, and if we are not successful in challenging such third-party rights, we may not be able to use these trademarks to market our products in those countries. If we are unable to establish name recognition based on our trademarks and trade names, we may not be able to compete
58
effectively and our business may be adversely affected. We may license our trademarks and trade names to third parties, such as distributors. Although these license agreements may provide guidelines for how our trademarks and trade names may be used, a breach of these agreements or
misuse of our trademarks and tradenames by our licensees may jeopardize our rights in or diminish the goodwill associated with our trademarks and trade names.
We may be subject to claims challenging the inventorship of our patents and other intellectual property.
We or our licensors may be subject to claims that former employees, collaborators or other third parties have an interest in our owned or in-licensed patents, trade secrets or other intellectual property as an inventor or co-inventor. For example, we or our licensors may have inventorship disputes arise from conflicting obligations of employees, consultants or others who are involved in developing our products. Litigation may be necessary to defend against these and other claims challenging inventorship or our or our licensors’ ownership of our owned or in-licensed patents, trade secrets or other intellectual property. If we or our licensors fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights, such as exclusive ownership of, right to use, or right to exclude others from using, intellectual property that is important to our products. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees. Any of the foregoing could have a material adverse effect on our business, financial condition, results of operations and prospects.
In addition, while it is our policy to require our employees and contractors who may be involved in the development of intellectual property to execute agreements assigning such intellectual property to us, we may be unsuccessful in executing such an agreement with each party who in fact develops intellectual property that we regard as our own. Our assignment agreements may not be self-executing or may be breached, and we may be forced to bring claims against third parties, or defend claims they may bring against us, to determine the ownership of what we regard as our intellectual property.
Obtaining and maintaining our patent protection depends on compliance with various required procedures, document submissions, fee payments and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees, renewal fees, annuity fees and various other governmental fees on patents and/or applications must be paid to the USPTO and various governmental patent agencies outside of the United States at several stages over the lifetime of the patents and/or applications. We have systems in place to remind us to pay these fees, and we employ an outside firm and rely on our outside counsel to pay these fees due to non-U.S. patent agencies. The USPTO and various non-U.S. governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. We employ reputable law firms and other professionals to help us comply, and in many cases, an inadvertent lapse can be cured by payment of a late fee or by other means in accordance with the applicable rules. However, there are situations in which non-compliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction, such as failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. If we, or our licensors, fail to maintain the patents and patent applications covering our products and technologies, potential competitors may be able to enter the market without infringing our patents and this circumstance would have a material adverse effect on our business.
Patent terms may be inadequate to protect our competitive position on our products for an adequate amount of time.
Patents have a limited lifespan, and the protection patents afford is limited. In the United States, if all maintenance fees are timely paid, the natural expiration of a patent is generally 20 years from its earliest U.S. non-provisional filing date. Various extensions may be available, but the term of a patent, and the protection it affords, is limited. Even if patents covering our products are obtained, once the patent term has expired, we may be open to competition from competitive tests or products. Given the amount of time required for the development, testing and regulatory review of potential new tests or products, patents protecting such tests or products might expire before or shortly after such tests or products are commercialized. As a result, our owned and licensed patent portfolio may not provide us with sufficient rights to exclude others from commercializing tests or other products similar or identical to ours.
59
Risks Related to Employee Matters and Managing Growth and Other Risks Related to Our Business
We are highly dependent on the services of our key personnel.
We are highly dependent on the services of our key personnel, including Derek J. Maetzold, our President and Chief Executive Officer. Although we have entered into agreements with themour key personnel regarding their employment, they are not for a specific term and each of may terminate their employment with us at any time, though we are not aware of any present intention of any of these individuals to leave us, except as follows. As previously disclosed, in November 2019, Federico A. Monzon, M.D., our Chief Medical Officer, notified us of his intention to resign effective on or about April 1, 2020. Dr. Monzon has delayed his resignation and we now expect that he will remain as our Chief Medical Officer in a reduced capacity during the next few
Our research and development programs and laboratory operations depend on our ability to attract and retain highly skilled scientists and technicians. We may not be able to attract or retain qualified scientists and technicians in the future due to the competition for qualified personnel among life science businesses, particularly near our sole laboratory facilityfacilities and office spaces located in Phoenix, Arizona.Arizona; Pittsburgh, Pennsylvania; and our corporate headquarters in Friendswood, Texas. We also face competition from universities and public and private research institutions in recruiting and retaining highly qualified scientific personnel. We may have difficulties locating, recruiting or retaining qualified salespeople. Recruiting and retention difficulties can limit our ability to support our research and development and sales programs. We may also experience employee turnover as a result of the ongoing “great resignation.” In response to competition, rising inflation rates and labor shortages, we may need to adjust employee cash compensation, which would affect our operating costs and our margins, or equity compensation, which would affect our outstanding share count and cause dilution to existing stockholders. All of our employees are at-will, which means that either we or the employee may terminate their employment at any time.
The 2019 Equity Incentive Plan (the “2019 Plan”) provides for automatic increases in the number of shares authorized for issuance annually through January 1, 2029, however, there can be no assurances that these increases will be adequate to support our requirements for future equity awards or that we will be able to obtain approval from our stockholders in the future should we require authorization for the issuance of additional shares. For example, for the year ended December 31, 2022, we had granted awards in excess of the number of shares authorized for issuance under our 2019 Plan. As of December 31, 2023, there were 366,432 shares available for grant under the 2019 Plan. In December 2022, our board of directors adopted a separate equity plan, the 2022 Inducement Plan (the “Inducement Plan”), to be used exclusively for grants of awards as an inducement material to new employees entering into employment with us, which was subsequently amended in November 2023 to increase the shares reserved under the plan. However, the Inducement Plan cannot be used to grant ongoing equity awards to existing employees. If we are unable to provide adequate or competitive equity compensation, we may have to adjust other elements of our compensation packages and may encounter difficulties attracting and retaining personnel.
Our employees, clinical investigators, consultants, speakers, vendors and any current or potential commercial partners may engage in misconduct or other improper activities, including non-compliance with regulatory standards and requirements and insider trading.
We are exposed to the risk of fraud or other misconduct by our employees, clinical study investigators, consultants, speakers, vendors and any potential commercial partners. Misconduct by these parties could include intentional, reckless and/or negligent conduct or disclosure of unauthorized activities to us that violates: federal laws and regulations or those of comparable foreign regulatory authorities, including those laws that require the reporting of true, complete and accurate information; manufacturing standards; federal and state health and data privacy, security, fraud and abuse, government price reporting, transparency reporting requirements, and other healthcare laws and regulations in the United States and abroad; sexual harassment and other workplace misconduct; or laws that require the true, complete and accurate reporting of financial information or data. Such misconduct could also involve the improper use of information obtained in the course of clinical studies, which could result in regulatory sanctions and cause serious harm to our reputation. We have adopted a code of conduct applicable to all of our employees, as well as a disclosure program and other applicable policies and procedures, but it is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant civil, criminal and administrative penalties, damages, fines, disgorgement, individual imprisonment, exclusion from government funded healthcare programs, such as Medicare, Medicaid and other federal healthcare programs, contractual damages, reputational harm, diminished profits and future earnings, additional integrity reporting and oversight obligations, and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations.
60
We may be unable to manage our future growth effectively, which could make it difficult to execute our business strategy.
We have experienced significant revenue growth in a short period of time. We may not achieve similar growth rates in future periods. You should not rely on our operating results for any prior periods as an indication of our future operating performance. To effectively manage our anticipated future growth, we must continue to maintain and enhance our financial, accounting, human resources, laboratory operations, customer support and sales administration systems, processes and controls. Failure to effectively manage our anticipated growth could lead us to over-invest or under-invest in development, operational and administrative infrastructure, result in weaknesses in our infrastructure, systems, or internal controls, give rise to operational mistakes, losses, loss of customers, productivity or business opportunities, and result in loss of employees and reduced productivity of remaining employees.
We also anticipate further growth in our business operations. ThisFor example, since May 2021, we have completed the acquisitions of Myriad MyPath Laboratory, Cernostics and AltheaDx, each of which we expect will contribute to our future growth. These acquisitions and other future growth could create strain on our organizational, administrative and operational infrastructure, including laboratory operations, quality control, customer service and sales organization management. We expect to increasecontinue increasing our headcount and to hire more specialized personnel in the future as we grow our business.business and expand our product offerings. We will need to continue to hire, train and manage additional qualified scientists, laboratory personnel, client and account services personnel, and sales and marketing staff and improve and maintain our technology to properlyeffectively manage our growth. If our new hires perform poorly, if we are unsuccessful in hiring, training, managing and integrating these new employees or if we are not successful in retaining our existing employees, our business may be harmed.
In addition, our anticipated growth could require significant capital expenditures and might divert financial resources from other projects such as the development of new diagnostic tests and services. As we commercialize additional diagnostic tests, we may need to incorporate new equipment, implement new technology systems, automate or otherwise improve the efficiency of our operational processes or hire new personnel with different qualifications. Failure to manage this growth or transition could result in turnaround time delays, higher costs, declining quality, deteriorating customer service, and slower responses to competitive challenges. A failure in any one of these areas could make
it difficult for us to meet market expectations for our products and could damage our reputation and the prospects for our business.
In July 2023, we elected to temporarily pause accepting additional TissueCypher orders to focus on scaling efforts and to work through a significant backlog of orders. In September 2023, we resumed accepting new orders for testing in a phased approach consistent with continued scaling activity aimed at accommodating current demand and future growth. As of mid-October 2023, we completed the pre-existing backlog orders. However, there can be no assurance that our efforts will be successful, which could damage our reputation and the prospects for our business.
We may not be able to maintain the quality or expected turnaround times of our products, or satisfy customer demand as it grows. Our ability to manage our growth properly will require us to continue to improve our operational, financial and management controls, as well as our reporting systems and procedures. The time and resources required to implement these new systems and procedures is uncertain, and failure to complete this in a timely and efficient manner could adversely affect our operations. If our management is unable to effectively manage our anticipated growth, our expenses may increase more than expected, our revenue could decline or grow more slowly than expected and we may be unable to implement our business strategy. The quality of our products and services may suffer, which could negatively affect our reputation and harm our ability to retain and attract customers.
61
Myriad MyPath Laboratory, Cernostics and AltheaDx, respectively. These and any other strategic acquisition transactions may entail numerous operational and financial risks, including:
•delays, difficulties and higher than expected costs associated with integration activities, such as those involving operational processes, regulatory and licensure compliance, personnel and information technology systems;
•difficulties in scaling and growing the Internal Revenue Codeoperations of 1986, as amended,acquired businesses in a cost-efficient manner;
•disruption of our existing business operations and diversion of management’s time, focus and attention;
•decreases in our liquidity and operating cash flows, increases in our overall operating costs, substantial amounts of amortization expense, increased capital expenditure requirements and non-recurring charges, including possible impairments of acquired assets and losses on the remeasurement of contingent consideration;
•incurrence of substantial debt or dilutive issuances of equity securities, the Code. The TCJA, among other things, contains significant changesassumption of additional liabilities, exposure to corporate taxation, including reductionunknown liabilities and being subject to disputes with former owners of acquired businesses;
•inability to retain key personnel of any acquired businesses; and
•failure to realize any of the corporate tax rate fromanticipated revenues, synergies, efficiencies or other benefits of a top marginal rate of 35%transaction within our estimated time frame or at all.
With regard to a flat rate of 21%, limitationour acquisitions of the tax deduction for interest expenseMyriad MyPath Laboratory, Cernostics and AltheaDx, actual results may differ materially from our plans and expectations. For example, there can be no assurances regarding our ability to 30% of adjusted earnings (except for certain small businesses), limitationsuccessfully scale and integrate the MyPath Melanoma, TissueCypher and IDgenetix tests into our commercial offerings and the ability of the deductioncombined strengths of Castle, the Myriad MyPath Laboratory, Cernostics or AltheaDx to position us for net operating losses generated after December 31, 2017continued growth and success as a leader in the diagnostics space. Further, there are inherent execution and business risks associated with managing the integration and growth objectives of more than one acquisition at the same time and such circumstances may have the effect of heightening the operational and financial risks related to 80%acquisitions noted above and the other risks described in this “Risk Factors” section.
In July 2023, we elected to temporarily pause accepting additional TissueCypher orders to focus on scaling efforts and to work through a significant backlog of orders. In September 2023, we resumed accepting new orders for testing in a phased approach consistent with continued scaling activity aimed at accommodating current year taxable incomedemand and eliminationfuture growth. As of net operating loss carrybacks, one time taxationmid-October 2023, we completed the pre-existing backlog orders. However, there can be no assurance that we will be successful in our efforts.
We are unable to predict the timing, size or nature of offshore earnings at reduced rates regardless ofany future transactions, whether they are repatriated, elimination of U.S. taxwill be completed or financed on foreign earnings (subject to certain important exceptions), immediate deductions for certain new investments instead of deductions for depreciation expense over time, and modifyingfavorable terms, if at all, or repealing many business deductions and credits. We do not expect the TCJA to have a material impact to our current projection of minimal cash taxes for the near future.
Our ability to use net operating lossesloss carryforwards and certain other tax attributes to offset future taxable income and taxes may be subject to limitations.
Under the TCJA,legislation known as the Tax Cuts and Jobs Act of 2017 (“TCJA”), as modified by the Coronavirus Aid, Relief, and Economic Security Act (“CARES Act”), federal net operating losses incurredNOLs generated in 2018 and in futuretaxable years beginning after December 31, 2017 may be carried forward indefinitely, but the deductibility of such federal net operating lossesNOL carryforwards is limited. It is uncertain if andlimited to what extent various states will conform to the TCJA. 80% of taxable income.
In addition, under Sections 382 and 383 of the Internal Revenue Code, and corresponding provisions of state law, if a corporation undergoes an ‘‘ownership change’’ (which is generally defined as a greater than 50% change (by value) in its equity ownership over a three-year period), the corporation’s ability to use its pre-change net operating lossNOL carryforwards and other pre-change tax attributes (such as research tax credits) to offset its post-change income or
62
taxes may be limited. For example, with respect to the NOLs we obtained in our acquisitions of Cernostics and AltheaDx, $36,347,000 of NOLs are expected to expire unused as a result of Section 382 limitations. We have experienced an ownership changechanges in the past and we may also experience additional ownership changes in the future as a result of subsequent shifts in our stock ownership, some of which may be outside of our control. If an ownership change occurs and our ability to use our net operating lossNOL carryforwards is materially limited, it would harm our future operating results by effectively increasing our future tax obligations. In addition, at the state level, there may be periods during which the use of NOL carryforwards is suspended or otherwise limited, which could accelerate or permanently increase state taxes owed.
New income, sales, use or other tax laws, statutes, rules, regulations or ordinances could be negatively impactedenacted at any time, which could adversely affect our business operations and financial performance. Further, existing tax laws, statutes, rules, regulations or ordinances could be interpreted, changed, modified or applied adversely to us. For example, the TCJA, the CARES Act and the IRA enacted many significant changes to the U.S. tax laws. Further guidance from the Internal Revenue Service and other tax authorities with respect to such legislation may affect us, and certain aspects of such legislation could be repealed or modified in future legislation. In addition, it is uncertain if and to what extent various states will conform to federal tax laws. Future tax reform legislation could have a material impact on the value of our deferred tax assets and could increase our future U.S. tax expense.
Effective January 1, 2022, the TCJA eliminated the option to deduct research and development expenses for tax purposes in the year incurred and requires taxpayers to capitalize and subsequently amortize such expenses over five years for research activities conducted in the United States and over 15 years for research activities conducted outside the United States. Unless the United States Department of the Treasury issues regulations that narrow the application of this provision to a smaller subset of our research and development expenses or the provision is deferred, modified, or repealed by cyber security threats.Congress, it could harm our future operating results by effectively increasing our future tax obligations. The actual impact of this provision will depend on multiple factors, including the amount of research and development expenses we will incur, whether we achieve sufficient income to fully utilize such deductions and whether we conduct our research and development activities inside or outside the United States.
In the ordinary course of business, we collect, store and transmitthe third parties upon which we rely (such as contractors and consultants) process proprietary, confidential, and sensitive information (including but not limited to intellectual property, proprietary business information and personal information)data).
Cyber-attacks, malicious internet-based activity, online and offline fraud and other similar activities threaten the confidentiality, integrity and availability of our proprietary, confidential, and sensitive information and information technology systems, and those of the third parties upon which we rely. Such threats are prevalent, continue to increase, and are becoming increasingly difficult to detect. These threats come from a variety of sources, including threat actors, traditional computer “hackers,” organized criminal threat actors, personnel (such as through theft or misuse), hacktivists, sophisticated nation-states, and nation-state-supported actors. Some actors now engage and are expected to continue to engage in cyber-attacks, including without limitation nation-state actors for geopolitical reasons and in conjunction with military conflicts and defense activities. During times of war and other major conflicts, we and the third parties upon which we rely may be vulnerable to a heightened risk of these attacks, including retaliatory cyber-attacks, that could materially disrupt our systems and operations, supply chain, and ability to produce, sell and distribute our goods and services. We and the third parties upon which we rely (such as our contractors and consultants) are vulnerable to a variety of evolving threats including but not limited to service interruptions, system malfunction, natural disasters, terrorism, war, public health crises, telecommunication and electrical failures, malware (including as a result of advanced persistent threat intrusions), malicious code (such as viruses and worms), ransomware, supply chain attacks, credential harvesting, denial-of-service attacks, credential stuffing, personnel misconduct or error, social engineering attacks (including through deep fakes, which may be increasingly more difficult to identify as fake, and phishing attacks), attacks enhanced or facilitated by AI, and other similar threats. In particular, severe ransomware attacks have become increasingly prevalent and severe and can lead to significant interruptions in our operations, loss of data and income, reputational harm, and diversion of funds. Extortion payments may alleviate the negative impact of a ransomware attack, but we may be unwilling or unable to
63
make such payments due to, for example, applicable laws or regulations prohibiting such payments. Future or past business transactions (such as acquisitions or integrations) could expose us to additional cybersecurity risks and vulnerabilities, as our systems could be negatively affected by vulnerabilities present in acquired or integrated entities’ systems and technologies. Furthermore, we may discover security issues that were not found during due diligence of such acquired or integrated entities, and it may be difficult to integrate companies into our information technology environment and security program.
Remote work has become more common and has increased risks to our information technology systems and data, as more of our employees utilize network connections, computers and devices outside our premises or network, including working at home, while in transit and in public locations.
We manage and maintain our applications and information utilizing a combination of on-site systems, managed data centers, and cloud-based data centers. It is critical that we do so in a secure manner to maintain the confidentiality, availability and integrity of such confidential information. We also have outsourced elements of our operations to third parties, including third-party service providers and technologies to help operate critical business systems to Process proprietary, confidential and sensitive information, and as a result we also manage a number of third-party contractors who have access to our proprietary, confidential information.and sensitive information including information related to our clinical trials. Our ability to monitor these third parties’ cybersecurity information security practices is limited, and these third parties may not have adequate information security measures in place. While we may be entitled to damages if our third-party service providers fail to satisfy their privacy or security-related obligations to us, any award may be insufficient to cover our damages, or we may be unable to recover such award. Additionally, supply-chain attacks have also increased in frequency and severity, and we cannot guarantee that third parties and infrastructure in our supply chain or our third-party partners’ supply chains have not been compromised or that they do not contain exploitable defects or bugs that could result in a breach of or disruption to our information technology systems (including our services) or the third-party information technology systems that support us and our services.
Any of confidentialthe previously identified or similar threats could cause a disruption or security incident, which could result in unauthorized, unlawful, or accidental loss of, damage to, modification of, destruction of, alteration of, encryption of, disclosure of, access to, or acquisition of our information or our information technology systems, or those of the third parties upon whom we rely. A security incident could disrupt our ability (and that theyof the third parties upon whom we rely) to provide our services.
We may expend significant resources or modify our business activities (including our clinical research activities) to try to protect against security incidents. Certain data privacy and security obligations may require us to implement and maintain specific security measures or industry-standard or reasonable security measures to protect our internal information technology systems and thosesensitive information.
If we or a third party upon whom we rely experience a security incident or are perceived to have experienced a security incident, we may experience government enforcement actions (for example, investigations, fines, penalties, audits, and inspections), additional reporting requirements and/or oversight, restrictions on Processing data (including personal data), litigation (including class action claims), indemnification obligations, negative publicity, reputational harm, monetary fund diversions, diversion of management attention, interruptions in our operations, and other harms.Such consequences may disrupt our operations (including our ability to conduct our analyses, provide test results, bill payors or patients, process claims and appeals, provide customer assistance, conduct research and development activities, collect, process, and prepare company financial information, provide information about our products and other patient and physician education and outreach efforts through our website, and manage the administrative aspects of our contractors and consultants are potentially vulnerable to breakdown or otherbusiness), damage or interruption from service interruptions, system malfunction, natural disasters, terrorism, war, public health crises and telecommunication and electrical failures, as well as security breaches from inadvertent or intentional actions by our employees, contractors, consultants, business partners, and/or other third parties, or from cyber-attacks by malicious third parties (including the deployment of harmful malware, ransomware, denial-of-service attacks, social engineering and other means to affect service reliability and threaten the confidentiality, integrity and availability
64
database, our ability to further validate, improve and therefore maintain and grow sales of DecisionDx-Melanoma could be significantsignificantly impaired.
In addition to experiencing a security incident, third parties may gather, collect, or infer sensitive information whichabout us from public sources, data brokers, or other means that reveals competitively sensitive details about our organization and could be used to undermine our competitive advantage or market position.
Additionally, sensitive information of the Company could be leaked, disclosed, or revealed as a result of or in significant legal and financial exposure and reputational damages that could potentially have an adverse effect onconnection with our business.employees’, personnel’s, or vendors’ use of generative AI technologies.
Product or professional liability lawsuits against us could cause us to incur substantial liabilities and could limit our commercialization of our products.
We face an inherent risk of product and professional liability exposure related to our products. The marketing, sale and use of our products could lead to the filing of product liability claims were someone to allege that our products identified or reported inaccurate or incomplete information, or otherwise failed to perform as designed. We may also be subject to liability for errors in, a misunderstanding of or inappropriate reliance upon, the information we provide in the ordinary course of our business activities.
If we cannot successfully defend ourselves against claims that our products caused injury or otherwise failed to function properly, we could incur substantial liabilities. Regardless of merit or eventual outcome, product liability claims may result in:
•decreased demand for our current tests any tests that we may develop, and the inability to commercialize such tests;
•injury to our reputation and significant negative media attention;
•reluctance of experts willing to conduct our clinical studies;
•initiation of investigations by regulators;
•significant costs to defend the related litigation and diversion of management’s time and our resources;
•substantial monetary awards to study subjects or patients;
•product recalls, withdrawals or labeling, or marketing or promotional restrictions; and
•loss of revenue.
We currently carry product liability insurance. However, the amount of this insurance may not be adequate to cover all liabilities that we may incur. Insurance coverage is increasingly expensive. We may not be able to maintain insurance coverage at a reasonable cost or in an amount adequate to satisfy any liability that may arise.
International expansion of our business exposes us to business, regulatory, political, operational, financial, and economic risks associated with doing business outside of the United States.
•multiple, conflicting and changing laws and regulations such as privacy regulations, tax laws, export and import restrictions, economic sanctions and embargoes, employment laws, regulatory requirements and other governmental approvals, permits and licenses;
•limits in our ability to penetrate international markets if we are not able to perform tests locally;
65
•logistics and regulations associated with shipping tissue samples, including infrastructure conditions and transportation delays;
•difficulties in staffing and managing foreign operations;
•failure to obtain regulatory approvals for the commercialization of our products in various countries;
•complexities and difficulties in obtaining intellectual property protection and enforcing our intellectual property;
•complexities associated with managing multiple payor reimbursement regimes, government payors, or patient self-pay systems;
•financial risks, such as longer payment cycles, difficulty collecting accounts receivable, the impact of local and regional financial crises on demand and payment for our products and exposure to foreign currency exchange rate fluctuations;
•natural disasters, political and economic instability, including wars, terrorism, and political unrest, outbreak of disease, boycotts, curtailment of trade and other business restrictions; and
•regulatory and compliance risks that relate to maintaining accurate information and control over sales and distributors’ activities that may fall within the purview of the U.S. Foreign Corrupt Practices Act, or FCPA, its books and records provisions, or its anti-bribery provisions.
Additionally, financial markets around the world experienced volatility following the invasion of Ukraine by Russia in February 2022. In response to the invasion, the United States, UK and EU, along with others, imposed significant new sanctions and export controls against Russia, Russian banks and certain Russian individuals and may implement additional sanctions or take further punitive actions in the future. The full economic and social impact of the sanctions imposed on Russia (as well as possible future punitive measures that may be implemented), as well as the counter measures imposed by Russia, in addition to the ongoing military conflict between Ukraine and Russia, which could conceivably expand into the surrounding region, remains uncertain; however, both the conflict and related sanctions have resulted and could continue to result in disruptions to trade, commerce, pricing stability, credit availability, and/or supply chain continuity, in both Europe and globally, and has introduced significant uncertainty into global markets. While we do not operate in Russia or Ukraine, as the adverse effects of this conflict continue to develop and potentially spread, both in Europe and throughout the rest of the world, our business and results of operations may be adversely affected, particularly to the extent this conflict escalates to involve additional countries, further economic sanctions or wider military conflict. Any of these factors could significantly harm our future international expansion and operations and, consequently, our revenue and results of operations.
Requirements associated with being a public company, including those associated with no longer qualifying as a smaller reporting company and becoming an accelerated filer, will continue to increase our costs significantly, as well as divert significant company resources and management attention.
We are subject to the reporting requirements of the Exchange Act or the other rules and regulations of the SEC and any securities exchange relating to public companies. Sarbanes-Oxley, as well as rules subsequently adopted by the SEC and The Nasdaq Stock Market LLC, or Nasdaq to implement provisions of Sarbanes-Oxley, impose significant requirements on public companies, including requiring establishment and maintenance of effective disclosure and financial controls and changes in corporate governance practices. Further, pursuant to the Dodd-Frank Wall Street Reform and Consumer Protection Act of 2010, the SEC has adopted additional rules and regulations in these areas, such as mandatory ‘‘say on pay’’ voting requirements that will apply to us when we cease to be an emerging growth company.requirements. Stockholder activism, the current political environment and the current high level of government intervention and regulatory reform may lead to substantial new regulations and disclosure obligations. For example, during 2022, the SEC adopted new rules covering pay versus performance disclosures, "clawback" policies and insider trading plans. Future changes in regulations and disclosure obligations which may lead to additional compliance costs and impact the manner in which we operate our business in ways we cannot currently anticipate. Compliance with the various reporting and other requirements applicable to public companies requires considerable time and attention of management. We cannot assure you that we will satisfy our obligations as a public company on a timely basis.
We will be subject to the reporting deadlines of an accelerated filer effective December 31, 2023. Beginning with our Quarterly Report on Form 10-Q for the three months ended March 31, 2024, we will no longer qualify as a smaller reporting company and we will be unable to take advantage of scaled disclosure requirements. We expect the rules and regulations applicable to public companies will continue to substantially increase our legal and financial compliance costs and to make some activities more time-consuming and costly. If we are unable to comply with these requirements diverton a timely basis or if the attention of our management and personnel is diverted from other business concerns, they it
66
could have a material adverse effect on our business, financial condition and results of operations. The increased costs will increase our net loss or decrease our net income, or increase our net loss, and may require us to reduce costs in other areas of our business or increase the prices of our products. In addition, as a public company,we expand, it may be more difficult or more costly for us to obtain certain types of insurance, including directors’ and officers’ liability insurance, and we may be forced to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. The impact of these events could also make it more difficult for us to attract and retain qualified personnel to serve on our board of directors, our board committees or as executive officers.
If we fail to comply with environmental, health and safety laws and regulations, we could become subject to fines or penalties or incur costs that could have a material adverse effect on the success of our business.
We, and the third parties with whom we share our facilities, are subject to numerous environmental, health and safety laws and regulations, including those governing laboratory procedures and the handling, use, storage, treatment and disposal of hazardous materials and wastes. Each of our operations involve the use of hazardous and flammable materials, including chemicals and biological and radioactive materials. Our operations also produce hazardous waste. We generally contract with third parties for the disposal of these materials and wastes. We cannot eliminate the risk of contamination or injury from these materials. We could be held liable for any resulting damages in the event of contamination or injury resulting from the use of hazardous materials by us or the third parties with whom we share our facilities, and any liability could exceed our resources. We also could incur significant costs associated with civil or criminal fines and penalties.
Although we maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees resulting from the use of hazardous materials, this insurance may not provide adequate coverage against potential liabilities. We do not maintain insurance for environmental liability or toxic tort claims that may be asserted against us in connection with our storage or disposal of biological, hazardous or radioactive materials.
In addition, we may incur substantial costs in order to comply with current or future environmental, health and safety laws and regulations. These current or future laws and regulations may impair our research and development. Failure to comply with these laws and regulations also may result in substantial fines, penalties or other sanctions.
Our business could be adversely impacted by inflation.
In 2021, the rate of inflation in the United States began to increase and then rose to levels not experienced in over 40 years, but began subsiding in the second half of 2022. We are experiencing inflationary pressures, primarily in increased personnel costs and price increases for certain lab supplies. We anticipate possible inflationary impacts on other cost areas in the future. The extent of any future impacts from inflation on our business and our results of operations will be dependent upon how long the elevated inflation levels persist and the extent to which the rate of inflation were to further increase, if at all, neither of which we are able to predict. If elevated levels of inflation were to persist or if the rate of inflation were to accelerate, the purchasing power of our cash and cash equivalents and marketable investment securities may be further diminished, our expenses could increase faster than anticipated and we may utilize our capital resources sooner than expected. Further, given the complexities of the reimbursement landscape in which we operate, our payors may be unwilling or unable to increase reimbursement rates to compensate for inflationary impacts. As such, the effects of inflation may adversely impact our results of operations, financial condition and cash flows.
Our business could be adversely affected by natural disasters, public health epidemicscrises and other events beyond our control.
Although we maintain crisis management plans, our business operations are subject to interruption by natural disasters and catastrophicother events and catastrophes beyond our control, including, but not limited to, earthquakes, floods, fires, tornadoes, hurricanes, power or other utility outages, telecommunications failures and public health issuescrises. Further, the ongoing conflict between Ukraine and epidemics. Further, outbreaks of epidemic diseases, such as coronavirus,Russia, or the fear of suchsimilar events, could provoke responses, including government-imposed travel restrictions that could impede the mobility and effectiveness of our sales force. In particular, as the coronavirus outbreak has recently spread to the United States, it mayforce, disrupt our operations or those of our suppliers and service providers. The ultimate impact of the coronavirus outbreakany of these or any other health epidemicsimilar events is highly uncertain and subject to change. We do not yet know the full extent of any potential delays or impacts on our business that may be attributable to the coronavirus outbreak. However, these effects could have a material adverse impact on our operations.
67
Risks Related to Ownership of Our Common Stock
The stock price of our common stock may be volatile or may decline regardless of our operating performance, and you may lose all or part of your investment.
The market price of our common stock may fluctuate significantly in response to numerous factors, many of which are beyond our control, including:
•our operating performance and the performance of other similar companies;
•our success in marketing and selling our products;
•our ability to achieve guideline inclusion for our products;
•reimbursement determinations by third-party payors, including MACs, and reimbursement rates for our products;
•changes in our projected operating results that we provide to the public, our failure to meet these projections or changes in recommendations by securities analysts that elect to follow our common stock;
•regulatory or legal developments in the United States and other countries;
•the level of expenses related to product development and clinical studies for our products;
•our ability to achieve product development goals in the timeframetimeframes we announce;
•announcements of clinical study results, regulatory developments, acquisitions, strategic alliances or significant agreements by us or by our competitors;
•the success or failure of our efforts to acquire, license or develop additional tests;
•recruitment or departure of key personnel;
•interest rates and the rate of inflation;
•the extent and duration of the impacts on our operations of general political and economic conditions, including the Israel-Hamas war, the ongoing conflict between Ukraine and Russia, economic slowdowns, recessions or market corrections, the duration and effects of elevated inflation, rising interest rates and tightening of credit markets resulting from the conflict or other evolving macroeconomic developments;
•trading activity by a limited number of stockholders who together beneficially own a significant percentage of our outstanding common stock;
•the size of our market float; and
•any other factors discussed in this Annual Report on Form 10-K.
For example, on June 5, 2023 our stock price decreased 49% after Novitas published a final LCD that would have impacted Medicare coverage for our DecisionDx-SCC test. In addition, the stock market in general, and diagnostic and life sciences companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies. Broad market and industry factors may negatively affect the market price of our securities, regardless of our actual operating performance. In the past, stockholders of other companies have filed securities class action litigation following periods of market volatility. If we were to become involved in securities litigation, it could subject us to substantial costs, divert resources and the attention of management from our business and adversely affect our business.
If there are substantial sales of shares of our common stock, the price of our common stock could decline.
The price of our common stock could decline if there are substantial sales of our common stock, particularly sales by our directors, executive officers and significant stockholders, or if there is a large number of shares of our common stock available for sale and the market perceives that sales will occur. As of February 28, 2020, we had 17,192,351 shares of common stock outstanding. Shares held by directors, executive officers and other affiliates are subject to volume limitations under Rule 144 under the Securities Act of 1933, as amended, or the Securities Act.
Certain of our stockholders have rights, subject to some conditions, to require us to file registration statements covering their shares or to include their shares in registration statements that we may file for ourselves or our stockholders. We have registered shares of common stock that we have issued and may issue under our employee equity incentive plans. As a result, these shares will be able to be sold freely in the public market upon issuance.
68
The market price of the shares of our common stock could decline as a result of the sale of a substantial number of our shares of common stock in the public market or the perception in the market that the holders of a large number of shares intend to sell their shares.
We have broad discretion in the use of working capital and may not use it effectively or in ways that increase our share price.
We cannot specify with any certainty the particular uses of working capital, but we currently expect such uses will include: funding selling and marketing activities, including expansion of our sales force to support the ongoing commercialization of current and future products; research and development related to the continued support of our current products, as well as the development of our product pipeline; and other general corporate purposes, including acquisitions and the additional costs associated with being a public company. The failure by our management to apply our working capital effectively could adversely affect our business and financial condition. Pending its use, we may invest working capital in a manner that does not produce income or that loses value. These investments may not yield a favorable return to our investors.
If securities or industry analysts do not publish research or publish inaccurate or unfavorable research about our business, our stock price and trading volume could decline.
The trading market for our common stock depends in part on the research and reports that securities or industry analysts publish about us or our business. If one or more of the analysts who cover us downgrade our common stock or publish inaccurate or unfavorable research about our business, our common stock price would likely decline. If one or more of these analysts cease coverage of us or fail to publish reports on us regularly, demand for our common stock could decrease, which might cause our common stock price and trading volume to decline.
Our disclosure controls and procedures may not prevent or detect all errors or acts of fraud.
We are subject to the periodic reporting requirements of the Exchange Act. We designed our disclosure controls and procedures to reasonably assure that information we must disclose in reports we file or submit under the Exchange Act is accumulated and communicated to management, and recorded, processed, summarized and reported within the time periods specified in the rules and forms of the SEC. We believe that any disclosure controls and procedures or internal controls and procedures, no matter how well-conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met.
These inherent limitations include the realities that judgments in decision-making can be faulty, and that breakdowns can occur because of simple error or mistake. For example, our directors or executive officers could inadvertently fail to disclose a new relationship or arrangement causing us to fail to make any related party transaction disclosures. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people or by an unauthorized override of the controls. Accordingly, because of the inherent limitations in our control system, misstatements due to error or fraud may occur and not be detected. In addition, we do not have a risk management program or processes or procedures for identifying and addressing risks to our business in other areas.
We have and may continue to enter into related party transactions that create conflicts of interest, or the appearance of conflicts of interest, which may harm our business and cause our stock price to decline.
We have entered into related party transactions that create conflicts of interest between our interests and the interests of our directors and executive officers. For example, we employ three children and a brother-in-law of Derek J. Maetzold, our President and Chief Executive Officer, three children and a son-in-law of Kristen M. Oelschlager, our Chief Operating Officer, and the son of Tobin W. Juvenal, our Chief Commercial Officer, in each case in non-officer positions. Additionally, Derek J. Maetzold and Daniel M. Bradbury, the chair of our board of directors, each served on the board of directors of AltheaDx, a commercial-stage molecular diagnostics company that we acquired in April 2022. Further, each of the following individuals was a direct or indirect beneficial owner of AltheaDx securities and received consideration in the transaction: Mr. Bradbury; Mr. Maetzold; Thomas Sullivan, John Maetzold and Peter Maetzold, immediate family members of Mr. Maetzold; Frank Stokes, our Chief Financial Officer; Tobin Juvenal, our Chief Commercial Officer; Kristen Oelschlager, our Chief Operating Officer; and Joshua Albers and Allysa Topel, immediate family members of Ms. Oelschlager.
These types of related party arrangements are an emerging growth companyrequired to be disclosed in our public filings based on certain criteria. We may engage in other transactions in the future involving our executive officers, directors and their family members and/or entities which they control or are affiliated, which could cause individuals in our management to seek to advance their economic interests or the economic interests of certain related parties above ours. Although we have a written policy on related party transactions that involves independent review and oversight by the audit
69
committee of our board of directors, there can be no assurances that conflicts of interest will not exist, or that we will be able to adequately address or mitigate any actual or perceived conflicts of interest, and stockholders, analysts, proxy advisory firms, the news media and other parties may view these transactions as representing conflicts of interest or as otherwise inappropriate, which may result in negative public perception and reputational harm, and could impair our ability to enter into new customer relationships or attract and retain employees. Potential, perceived and actual conflicts of interest could cause investors to question the independence of our management, the adequacy and effectiveness of our disclosure controls and procedures or the integrity of our corporate governance procedures and compensation practices, which could have a material adverse effect on the trading price of our common stock and our business, financial condition and results of operations.
We are a smaller reporting company and we cannot be certain if the reduced reportingscaled disclosure requirements applicable to emerging growthsmaller reporting companies will make our common stock less attractive to investors.
Revenue exceeded $100.0 million for the year ended December 31, 2022 and will be able to take advantagethe market value of these scaled disclosures for so long as our voting and non-voting common stock held by non-affiliates is less thanexceeded $250.0 million measuredas of June 30, 2023. Therefore, effective with our Quarterly Report on Form 10-Q for the last business daythree months ended March 31, 2024, we will no longer qualify as a smaller reporting company and will not be permitted to take advantage of our second fiscal quarter, or our annual revenue is less than $100.0 million during the most recently completed fiscal year and our voting and non-voting common stock held by non-affiliates is less than $700.0 million measured on the last business day of our second fiscal quarter.scaled disclosures.
We do not intend to pay dividends for the foreseeable future.
We have never declared nor paid cash dividends on our capital stock. We currently intend to retain any future earnings to finance the operation and expansion of our business, and we do not expect to declare or pay any dividends in the foreseeable future. In addition, the terms of the 2018 LSA precludes us from paying dividends without prior consent. Consequently, stockholders must rely on sales of their common stock after price appreciation, which may never occur, as the only way to realize any future gains on their investment.
The concentration of our stock ownership will likely limit your ability to influence corporate matters, including the ability to influence the outcome of director elections and other matters requiring stockholder approval.
Based upon shares outstanding as of February 28, 2020,December 31, 2023, our executive officers, directors and the known holders of more than 5% of our outstanding common stock, in the aggregate, beneficially owned approximately 48%43% of our common stock. As a result, these stockholders, acting together, will have significant influence over all matters that require approval by our stockholders, including the election of directors and approval of significant corporate transactions. Corporate actions might be taken even if other stockholders oppose them. This concentration of ownership might also have the effect of delaying or preventing a change of control of our company that other stockholders may view as beneficial.
Delaware law and provisions in our amended and restated certificate of incorporation and amended and restated bylaws could make a merger, tender offer or proxy contest difficult, thereby depressing the trading price of our common stock.
Provisions of our amended and restated certificate of incorporation and amended and restated bylaws may delay or discourage transactions involving an actual or potential change in our control or change in our management, including transactions in which stockholders might otherwise receive a premium for their shares or transactions that our stockholders might otherwise deem to be in their best interests. Therefore, these provisions could adversely affect the price of our common stock. Among other things, our amended and restated certificate of incorporation and amended and restated bylaws:
•permit our board of directors to issue up to 10,000,000 shares of preferred stock, with any rights, preferences and privileges as they may designate (including the right to approve an acquisition or other change in our control);
•provide that the authorized number of directors may be changed only by resolution of the board of directors;
•provide that the board of directors or any individual director may only be removed with cause and the affirmative vote of the holders of at least 66-2/3% of the voting power of all of our then outstanding common stock;
70
•provide that all vacancies, including newly created directorships, may, except as otherwise required by law, be filled by the affirmative vote of a majority of directors then in office, even if less than a quorum;
•divide our board of directors into three classes;
•require that any action to be taken by our stockholders must be effected at a duly called annual or special meetingmeetings of stockholders and not be taken by written consent;
•provide that stockholders seeking to present proposals before a meeting of stockholders or to nominate candidates for election as directors at a meeting of stockholders must provide notice in writing in a timely manner and also specify requirements as to the form and content of a stockholder’s notice;
•do not provide for cumulative voting rights (therefore allowing the holders of a majority of the shares of common stock entitled to vote in any election of directors to elect all of the directors standing for election, if they should so choose);
•provide that special meetings of our stockholders may be called only by the chairmanchairperson of the board, our Chief Executive Officer or by the board of directors pursuant to a resolution adopted by a majority of the total number of authorized directors;
•provide that, unless we consent in writing to the selection of an alternative forum, the Court of Chancery of the State of Delaware (or, if and only if the Court of Chancery of the State of Delaware lacks subject matter jurisdiction, any state court located within the State of Delaware or, if and only if all such state courts lack subject matter jurisdiction, the federal district court for the District of Delaware) will be the sole and exclusive forum for the following types of actions or proceedings under Delaware statutory or common law: (i) any derivative action or proceeding brought on our behalf; (ii) any action or proceeding asserting a claim of breach of a fiduciary duty owed by any of our current or former directors, officers or other employees to us or our stockholders; (iii) any action or proceeding asserting a claim against us or any of our current or former directors, officers or other employees, arising out of or pursuant to any provision of the Delaware General Corporation Law, our certificate of incorporation or our bylaws; (iv) any action or proceeding to interpret, apply, enforce or determine the validity of our certificate of incorporation or our bylaws; (v) any action or proceeding as to which the Delaware General Corporation Law confers jurisdiction to the Court of Chancery of the State of Delaware; and (vi) any action asserting a claim against us or any of our directors, officers or other employees governed by the internal affairs doctrine, in all cases to the fullest extent permitted by law and subject to the court’s having personal jurisdiction over the indispensable parties named as defendants; provided these provisions of our amended and restated certificate of incorporation and amended and restated bylaws will not apply to suits brought to enforce a duty or liability created by the Exchange Act or any other claim for which the federal courts have exclusive jurisdiction; and
•provide that unless we consent in writing to the selection of an alternative forum, the federal district courts of the United States of America shall be the exclusive forum for the resolution of any complaint asserting a cause of action arising under the Securities Act, subject to and contingent upon a final adjudication in the State of Delaware of the enforceability of such exclusive forum provision.
The amendment of any of these provisions, with the exception of the ability of our board of directors to issue shares of preferred stock and designate any rights, preferences and privileges thereto, would require approval by the holders of at least 66-2/3% of our then-outstanding common stock.
In addition, as a Delaware corporation, we are subject to Section 203 of the Delaware General Corporation Law. These provisions may prohibit large stockholders, in particular those owning 15% or more of our outstanding voting stock, from merging or combining with us for a certain period of time. A Delaware corporation may opt out of this provision by express provision in its original certificate of incorporation or by amendment to its certificate of incorporation or bylaws approved by its stockholders. However, we have not opted out of this provision.
These and other provisions in our amended and restated certificate of incorporation, amended and restated bylaws and Delaware law could make it more difficult for stockholders or potential acquirorsacquirers to obtain control of our board of directors or initiate actions that are opposed by our then-current board of directors, including delay or impede a merger, tender offer or
proxy contest involving our company. The existence of these provisions could negatively affect the price of our common stock and limit opportunities for you to realize value in a corporate transaction.
71
Our amended and restated certificate of incorporation provides that the Court of Chancery of the State of Delaware is the exclusive forum for certain disputes between us and our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers or employees.
Our amended and restated certificate of incorporation provides that, unless we consent in writing to the selection of an alternative forum, the Court of Chancery of the State of Delaware (or, if and only if the Court of Chancery of the State of Delaware lacks subject matter jurisdiction, any state court located within the State of Delaware or, if and only if all such state courts lack subject matter jurisdiction, the federal district court for the District of Delaware) will be the sole and exclusive forum for the following types of actions or proceedings under Delaware statutory or common law: (i) any derivative action or proceeding brought on our behalf; (ii) any action or proceeding asserting a claim of breach of a fiduciary duty owed by any of our current or former directors, officers or other employees to us or our stockholders; (iii) any action or proceeding asserting a claim against us or any of our current or former directors, officers or other employees arising out of or pursuant to any provision of the Delaware General Corporation Law, our amended and restated certificate of incorporation or amended and restated bylaws; (iv) any action or proceeding to interpret, apply, enforce or determine the validity of our amended and restated certificate of incorporation or our amended and restated bylaws; (v) any action or proceeding as to which the Delaware General Corporation Law confers jurisdiction to the Court of Chancery of the State of Delaware; and (vi) any action asserting a claim against us or any of our directors, officers or other employees that is governed by the internal affairs doctrine, in all cases to the fullest extent permitted by law and subject to the court’s having personal jurisdiction over the indispensable parties named as defendants; provided these provisions would not apply to suits brought to enforce a duty or liability created by the Exchange Act, or any other claim for which the federal courts have exclusive jurisdiction. In addition,This provision would not apply to suits brought to enforce a duty or liability created by the Exchange Act. Furthermore, Section 22 of the Securities Act creates concurrent jurisdiction for federal and state courts over all such Securities Act actions. Accordingly, both state and federal courts have jurisdiction to entertain such claims. To prevent having to litigate claims in multiple jurisdictions and the threat of inconsistent or contrary rulings by different courts, among other considerations, our amended and restated certificate of incorporation and amended and restated bylaws provide that unless we consent in writing to the selection of an alternative forum, the federal district courts of the United States of America shall be the exclusive forum for the resolution of any complaint asserting a cause of action arising under the Securities Act, subject to and contingent upon a final adjudication in the State of Delaware of the enforceability of such exclusive forum provision. While the Delaware courts have determined that such choice of forum provisions are facially valid, a stockholder may nevertheless seek to bring a claim in a venue other than those designated in the exclusive forum provisions. In such instance, we would expect to vigorously assert the validity and enforceability of the exclusive forum provisions of our amended and restated certificate of incorporation. This may require significant additional costs associated with resolving such action in other jurisdictions and there can be no assurance that the provisions will be enforced by a court in those other jurisdictions.
These choice of forum provisions may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, officers or other employees and may discourage these types of lawsuits. Furthermore, the enforceability of similar choice of forum provisions in other companies’ certificates of incorporation has been challenged in legal proceedings, and it is possible that a court could find these types of provisions to be inapplicable or unenforceable. If a court were to find the choice of forum provisions contained in our amended and restated certificate of incorporation to be inapplicable or unenforceable in an action, we may incur additional costs associated with resolving such action in other jurisdictions.
Item 1B. Unresolved Staff Comments.
None.
Item 1C. Cybersecurity.
Risk management and strategy
We have implemented and maintain various information security processes designed to identify, assess and manage material risks from cybersecurity threats to our critical computer networks, third party hosted services, communications systems, hardware and software, and our critical data, including intellectual property, confidential information that is proprietary, strategic or competitive in nature, and data related to our clinical trials and commercial and pipeline tests (“Information Systems and Data”).
Our Director of Cyber Security & Infrastructure (“DCSI”) oversees our information security function, which in conjunction with our security and engineering operations, Security Operations Center, a third-party managed
72
security provider, helps identify, assess and manage the Company’s cybersecurity threats and risks. This group identifies and assesses risks from cybersecurity threats by monitoring and evaluating our threat environment and the Company’s risk profile using various methods including, for example: manual and automated tools; subscribing to and analyzing reports of cybersecurity threats and threat actors; conducing scans of our environment; evaluating our and our industry’s risk profile and threats reported to us; coordinating with law enforcement concerning certain threats; conducting internal and external audits and threat assessments; conducting vulnerability assessments; using external intelligence feeds; and working with a third-party test our incident response processes.
Depending on the environment, we implement and maintain various technical, physical, and organizational measures, processes, standards and policies designed to manage and mitigate material risks from cybersecurity threats to our Information Systems and Data, including, for example: an incident response and business continuity plan; vulnerability management policy; risk assessments; implementing certain certifications and security standards; encrypting certain of our data; network security controls; segregating certain data; access and physical security controls; asset management, tracking, and disposal; monitoring our systems; employee training; penetration tests; maintaining cybersecurity insurance; and dedicated cybersecurity staff.
Our assessment and management of material risks from cybersecurity threats are integrated into the Company’s overall risk management processes. For example, cybersecurity risk is addressed as a component of the Company’s enterprise risk management program; our information security function works with management to prioritize our risk management processes and mitigate cybersecurity threats that are more likely to lead to a material impact to our business; our senior management evaluates material risks from cybersecurity threats against our overall business objectives and reports to the audit committee of the board of directors, which is primarily responsible for overseeing our risk management processes on behalf of our board of directors.
We use third-party service providers to assist us from time to time to identify, assess, and manage material risks from cybersecurity threats, including for example professional services firms (including legal counsel), threat intelligence service providers, cybersecurity consultants, cybersecurity software and managed service providers, penetration testing firms, and dark web monitoring services.
We use third-party service providers to perform a variety of functions throughout our business, such as application providers and hosting companies. We have vendor management processes to manage cybersecurity risks associated with our use of certain providers. These processes may include reviewing vendors’ written security programs and security assessments, conducting security assessment calls with vendor personnel, and imposing contractual obligations related to information security on certain of our vendors. Depending on the nature of the services provided, the sensitivity of the Information Systems and Data at issue, and the identity of the provider, our vendor management process may involve different levels of assessment designed to help identify cybersecurity risks associated with a provider and impose contractual obligations related to cybersecurity on the provider.
For a description of the risks from cybersecurity threats that may materially affect the Company and how they may do so, see our risk factors under Part 1. Item 1A. Risk Factors in this Annual Report on Form 10-K, including “If our information technology systems, or those of third parties upon which we rely, or our data are or were compromised, we could experience adverse consequences resulting from such compromise, including but not limited to regulatory investigations or actions; litigation; fines and penalties; disruptions of our business operations; reputational harm; loss of revenue or profits; loss of customers or sales; and other adverse consequences.”
Governance
Our board of directors addresses the Company’s cybersecurity risk management as part of its general oversight function. The board of directors’ audit committee oversees the risk assessment, risk management and internal controls over cyber security matters, as outlined in its committee charter.
Our cybersecurity risk assessment and management processes are implemented and maintained by certain Company management, including our DCSI. Our DCSI has over 20 years of experience as a security professional, has a CISSP certification, and has completed the Carnegie Melon Insider Risk Management Program.
The DCSI is responsible for hiring appropriate personnel, leading enterprise-wide cybersecurity strategy, helping to integrate cybersecurity risk considerations into the Company’s overall risk management strategy, and communicating key priorities to relevant personnel.
Our cybersecurity incident response plan is designed to escalate certain cybersecurity incidents to members of management depending on the circumstances, including Security Management. Security Management work with the Company’s incident response team to help the Company mitigate and remediate cybersecurity incidents of
73
which they are notified. In addition, the Company’s incident response plan includes reporting to the audit committee of the board of directors for certain cybersecurity incidents.
The DCSI provides periodic reports to our board of directors, as well as our Chief Operating Officer, Chief Executive Officer and other members of our senior management as appropriate, concerning the Company’s significant cybersecurity threats and risk and the processes the Company has implemented to address them. The audit committee also receives various reports, summaries or presentations related to cybersecurity threats, risk and mitigation.
Item 2. Properties.
We currentlyhave a lease agreement for approximately 4,19526,700 square feet of office space in Friendswood, Texas under an agreement that expires June 30, 2020, which we have the option to renew for an additional one-year period commencing on July 1, 2020. We have entered into an agreement to lease approximately 21,760 square feet of new office space in Friendswood, Texas to replace the existing Friendswood office space.is used as our corporate headquarters. This lease is expected commence on or prior to August 1,commenced in late 2020 and has a 60-month term, expiring in November 2025, with an option to renew for one additional five-year period. We also lease approximately 23,40046,000 square feet of laboratory and office space in Phoenix, Arizona under two agreements, expiring in July 2033 and October 2033. For both leases, there are options to renew for two additional five-year terms.
In April 2022, we entered into a lease agreement for 20,856 square feet of laboratory and office space in Pittsburgh, Pennsylvania, with a 10.5-year term and an agreement that expires July 31, 2027, with two optional renewal periodsoption to renew for one additional five-year period. We commenced operations in the Pittsburgh facility in the second quarter of five years each. We believe our existing facilities, together with the new2023.
In February 2024, we purchased approximately 23 acres of land in Friendswood, Texas office space, will be sufficient for our needs for the foreseeable future.purpose of developing a commercial office building which may be used as our future corporate headquarters. We have initiated planning for such facilities and do not expect substantial construction of such facilities would be ready for operational use before the end of 2025.
Item 3. Legal Proceedings.
From time to time, we may be involved in legal proceedings arising in the ordinary course of business. Legal proceedings, including litigation, government investigations and enforcement actions could result in material costs, occupy significant management resources and entail civil and criminal penalties, even if we ultimately prevail. On February 1, 2024, we received a subpoena from United States Department of Health and Human Services Office of Inspector General and are fully cooperating with the government’s inquiry. This inquiry, and any potential resulting claim asserted against us, with or without merit, could be time-consuming, expensive to address and divert management’s attention and other resources. These claims also could subject us to significant liability for damages and harm our reputation. Our insurance and indemnities may not cover all claims that may be asserted against us. We believe there is no threatened litigationare unable to predict the outcome and are unable to make a meaningful estimate of the amount or litigation pendingrange of loss, if any, that could have, individually or in the aggregate, a material adverse effect on our financial position, results of operations or cash flows.result from any unfavorable outcome.
Item 4. Mine Safety Disclosures.
Not applicable.
74
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
Market Information
Our Common Stock, $0.001 par value $0.001 per share, began tradingtrades on the NASDAQNasdaq Global Market under the symbol “CSTL” on July 25, 2019. Prior to that date, there was no public trading market for our common stock..
Holders of Record
As of February 28, 2020,21, 2024, there were approximately 161132 stockholders of record of our common stock, which does not include stockholders who hold shares held in street name.
Dividend Policy
We have never declared or paid cash dividends on our capital stock. We currently intend to retain all available funds and any future earnings for use in the operation of our business and do not anticipate paying any dividends on our common stock in the foreseeable future. Any future determination to declare dividends will be made at the discretion of our board of directors and will depend on, among other factors, our financial condition, operating results, capital requirements, contractual restrictions, general business conditions and other factors that our board of directors may deem relevant. In addition, the terms of the 2018 LSA, with Silicon Valley Bank, or SVB, and Oxford Finance LLC, or Oxford, prohibit us from paying dividends on our common stock without prior consent.
Recent Sales of Unregistered Equity Securities
Use of Proceeds from the IPO of Common Stock
On July 29, 2019, we completed the initial public offering of our common stock or the IPO,(our “IPO”), pursuant to which we issued and sold 4,600,000 shares of our common stock, including 600,000 shares associated with the full exercise of the underwriters’ option to purchase additional shares, at a price to the public of $16.00 per share.
The offer and sale of all of the shares of our common stock in the IPO were registered under the Securities Act pursuant to our Registration Statements on Form S-1, as amended (File Nos. 333-232369 and 333-232796), which were declared or became effective on July 24, 2019. SVB Leerink LLC and Robert W. Baird & Co. Incorporated acted as joint book-running managers for the IPO and as representatives of the underwriters. Canaccord Genuity LLC and BTIG, LLC acted as co-managers for the IPO.
There has been no material change in our planned use of the net proceeds from the initial public offering or IPO as described in the final prospectus filed with the SEC on July 26, 2019 relating to our Registration Statements on Form S-1 (File Nos. 333-232369 and 333-232796).
Since the effective date of our registration statement through December 31, 2019,2023, we have not used any of the net proceeds from the IPO. Pending such uses, we have invested, and plan to continue to invest, the balance of the net proceeds from the IPO in cash and cash equivalent securities or highly liquid investment securities.
Purchases of Equity Securities by the Issuer and Affiliated Purchasers
None.
Item 6. Selected Financial Data.[Reserved].
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
You should read the following discussion and analysis of financial condition and results of operations together with our consolidated financial statements and related notes included elsewhere in this Annual Report on Form 10-K. This discussion and other parts of this Annual Report on Form 10-K contain forward-looking statements that involve risk and uncertainties, such as statements of our plans, objectives, expectations and intentions. Our actual results, performance or achievements could differ materially from thoseany future results, performance or achievements discussed in these forward-looking statements. Factors that could cause or contribute to such differences include, but are not limited to, those discussed inunder the section titledheading “Special Note Regarding Forward-Looking Statements” and “Risk Factors.”
Overview
Castle Biosciences is a molecular diagnostics company offering innovative test solutions to aid clinicians in the diagnosis and treatment of dermatologic cancers, BE, UM, and in the treatment of mental health conditions.
75
Our Test Portfolio
We arecurrently offer five commercially available proprietary MAAA tests for use in the dermatologic, gastroenterology and ocular fields. We also offer a commercial-stage dermatological cancer company focused on providing physiciansproprietary PGx test to guide optimal drug treatment for patients diagnosed with depression, anxiety and their patientsother mental health conditions following our acquisition of AltheaDx in April 2022, as discussed below.
Currently, our revenue is primarily generated by our DecisionDx-Melanoma risk stratification test for cutaneous melanoma, which is supplemented by revenue generated from our DecisionDx-SCC risk stratification test for SCC, our TissueCypher risk stratification test for BE and our DecisionDx-UM risk stratification test for UM.
All five of our MAAA tests have been granted ADLT test status by CMS which means each test has demonstrated that (i) when combined with personalized, clinically actionable genomican empirically derived algorithm, it yields a result that predicts the probability a specific individual patient will develop a certain condition or conditions, or will respond to a particular therapy or therapies; and (ii) it provides new clinical diagnostic information to make more accurate treatment decisions.that cannot be obtained from any other test or combination of tests. We believe thatthis designation not only demonstrates our focus on developing and validating innovative tests but also enables our Medicare reimbursement rate to be set, over the traditional approachlong term, by the median private payor rate, which we believe provides a fair exchange of value. Further information about Medicare coverage and ADLT status with respect to developing a treatment plan for certain cancers using clinical and pathology factors aloneeach of our tests is inadequate and can be improved by incorporating personalized genomic information. set forth below.
Test Overview
Our non-invasive products utilize proprietary algorithms to provide an assessment of a patient’s specific risk of metastasis or recurrence of their cancer, allowing physicians to identify patients who are likely to benefit from an escalation of care as well as those who may avoid unnecessary medical and surgical interventions. Our lead product, Dermatologic Tests
DecisionDx-Melanoma is aour proprietary risk stratification GEP test that predicts the risk of metastasis or recurrence for patients diagnosed with invasive cutaneous melanoma,melanoma. In a deadly skin cancer. Wetypical year, we estimate more thanapproximately 130,000 patients are diagnosed with invasive cutaneous melanoma each year in the United States.States, representing an estimated U.S. TAM of approximately $540 million. We launched DecisionDx-Melanomaestimate that approximately 50% of patients diagnosed with CM are 65 years of age or older.
DecisionDx‑SCC is our proprietary GEP test for use in May 2013.patients with SCC, with one or more risk factors (also referred to as “high-risk” SCC). We also market estimate 20% of SCC, or 200,000 annually in the United States, are classified as high risk, representing an estimated U.S. TAM of approximately $820 million.
MyPath Melanoma is our proprietary GEP test for use in patients with a melanocytic lesion and uncertainty related to the malignancy of the lesion. We estimate approximately 300,000 patients each year present with a diagnostically ambiguous lesion, representing an estimated U.S. TAM of approximately $600 million.
Initially, we offered both our MyPath Melanoma test and our DiffDx-Melanoma test under an offering that we referred to as our Diagnostic GEP offering. However, following an internal assessment of the clinical value of offering both tests, we made the decision to suspend the clinical offering of DiffDx-Melanoma in February 2023 and now the focus of this offering is MyPath Melanoma.
Our Gastroenterology Test
TissueCypher is our proprietary risk stratification spatial omics test designed to predict future development of HGD and/or esophageal cancer in patients with ND, IND or LGD BE. We estimate a U.S. TAM of approximately $1 billion.
Our Uveal Melanoma Test
DecisionDx-UM which is a proprietary, risk stratification GEP test that predictshelps healthcare providers predict the risk of metastasis for patients with uveal melanoma,UM. We believe DecisionDx-UM is the standard of care in the management of newly diagnosed UM in the majority of ocular oncology practices in the United States. The Medicare eligible population represents close to 45% of the addressable market. We estimate a rare eye cancer.U.S. TAM of approximately $10 million.
Our Mental Health Test
IDgenetix is a PGx test that guides personalized mental health medication selection and management for patients with depression, anxiety and other mental health conditions. We launched DecisionDx-UMestimate a U.S. TAM of approximately $5 billion associated with this test. We began offering the IDgenetix test following our acquisition of AltheaDx in January 2010. BasedApril 2022.
Commercial Expansion Efforts
During the year ended December 31, 2022, we expanded our outside territories for our TissueCypher test to 16 sales territories. In late April 2022, we acquired AltheaDx and established a dedicated IDgenetix commercial team covering approximately 20 outside territories. In September 2022, we established a new commercial sales team
76
dedicated to our Diagnostic GEP offering and added additional outside territories for our TissueCypher test, which were fully integrated into our commercial operations by the end of the second quarter of 2023.
During the year ended December 31, 2023, we continued to expand our dermatologic and gastrointestinal commercial sales forces through territory and headcount expansions with focus being on our DecisionDx-Melanoma, DecisionDx-SCC, and TissueCypher tests.
We will continue to assess market response in determining further commercial expansions and commercial team structure.
Reimbursement
The primary source of revenue for our products is reimbursement from third-party payors, which includes government payors, such as Medicare, and commercial payors, such as insurance companies. Achieving broad coverage and reimbursement of our current products by third-party payors and continued Medicare coverage are key components of our financial success.
We bill third-party payors and patients for the substantial clinical evidence thattests we have developed, weperform. We have received Medicare coverage for our DecisionDx-Melanoma, DecisionDx-SCC, MyPath Melanoma, DecisionDx-UM, TissueCypher and IDgenetix tests which meet certain criteria for Medicare and Medicare Advantage beneficiaries, representing. A ‘‘covered life’’ means a subscriber, or a dependent of a subscriber, who is insured under an insurance carrier’s policy.
The Medicare rates discussed below are prior to giving effect to applicable sequestration in effect from time to time as described in further detail under "Government Regulation and Product Approval—Healthcare Reform" included in Item 1, Business, of this Annual Report on Form 10-K.
DecisionDx-Melanoma
DecisionDx-Melanoma tests are processed from our Phoenix laboratory and are covered under “foundational” LCDs finalized by MACs Palmetto and Noridian in the second quarter of 2022. Previously, DecisionDx-Melanoma was covered under an expanded LCD form the same MACs from the fourth quarter of 2020 until the effective date of the current LCD.
DecisionDx-Melanoma has met ADLT status, as determined by the CMS, since 2019. Since 2022, the rate for DecisionDx-Melanoma is set annually based upon the median private payor rate for the first half of the second preceding calendar year. For example, the rate for 2023 was set using median private payor rate data from January 1, 2021 to June 30, 2021. Our rate for 2022 and 2023 was $7,193 per test and is $7,193 for 2024.
DecisionDx-UM
DecisionDx-UM tests are processed from our Phoenix laboratory and are covered under LCDs finalized by MAC administrators Palmetto and Noridian in July 2017.
DecisionDx-UM has met the criteria of “existing advanced diagnostic laboratory test” status, also referred to as “existing ADLT” status, as determined by the CMS, since May 2019. Our rate is set annually based upon the median private payor rate for the first half of the second preceding calendar year. For example, the rate for 2023 was set using median private payor rate data from January 1, 2021 to June 30, 2021. Our rate for 2022 and 2023 was $7,776 per test and is $7,776 for 2024.
MyPath Melanoma and DiffDx-Melanoma
MyPath Melanoma was covered under a test-specific LCD policy through Noridian that became effective in June 2019. Effective August 6, 2023, Palmetto and Noridian issued LCDs that converted the test-specific MyPath Melanoma LCD to a “foundational” LCD and provided coverage for both MyPath Melanoma and DiffDx-Melanoma.
MyPath Melanoma was approved as a “new ADLT” in September 2019. Rates for our MyPath Melanoma test are set annually based upon the median private payor rate for the first half of the second preceding calendar year. Our rate for 2022 was $1,950 per test. Our 2023 rate was set at $1,755 per test, based on data submitted by the predecessor owner of the Myriad MyPath Laboratory relating to the first half of 2021. Our 2024 rate is set at $1,950 per test.
In the second quarter of 2022, we obtained a PLA code for DiffDx-Melanoma. In 2023, DiffDx-Melanoma went through the CMS gapfill process which concluded in September 2023 with CMS posting a final MAC-specific gapfill rate of $1,950 per test. Our rate for 2024 is $1,950 per test.
77
Diagnostic GEP Offering
Our Diagnostic GEP offering included MyPath Melanoma and DiffDx-Melanoma. We began offering MyPath Melanoma following our acquisition of the Myriad MyPath Laboratory on May 28, 2021. Our internal data indicates that we have improved the technical performance of MyPath Melanoma such that it is now comparable to the technical performance of DiffDx-Melanoma. As such, following an internal assessment of the clinical value of offering both tests, we made the decision to suspend the clinical offering of DiffDx-Melanoma in February 2023.
DecisionDx‑SCC
We issue our DecisionDx-SCC tests from our Pittsburgh and Phoenix labs, with a majority of tests being issued from our Pittsburgh lab. Novitas is the MAC responsible for administering claims for test reports issued by our Pittsburgh laboratory.
We requested and received an evidentiary review of our products, which represents approximately 50%DecisionDx-SCC by Novitas during the first quarter of 2022. Based upon this review, DecisionDx-SCC test began receiving coverage in April 2022.
On June 9, 2022, Novitas posted a draft oncology biomarker LCD that proposes to rely upon evidentiary reviews sourced from three databases for all oncology biomarker tests: ClinGen, OncoKB and NCCN. We believe the purpose of the proposals in this draft LCD were to streamline future reviews. Two of the databases do not review GEP tests and NCCN did not, to our addressable patient population.
On June 2, 2023, Novitas posted a finalized oncology biomarker LCD pursuant to which the DecisionDx-SCC test would no longer be covered by Medicare effective July 17, 2023. However, on July 6, 2023, Novitas suspended the final version of the LCD and announced its intent to post a new proposed LCD for comment and presentation at an open meeting. On July 27, 2023, Novitas posted a nearly identical proposed oncology biomarker LCD that continues to intend to rely upon evidentiary reviews sourced from three databases: ClinGen, OncoKB and NCCN. The proposed LCD also recommends non-coverage for our DecisionDx-SCC test. The comment period for the proposed LCD ended on September 9, 2023. We have processed over 60,000 clinical samples since commercial launchcannot predict whether this LCD will be finalized as proposed or what the timing of any final LCD might be.
Palmetto MolDX program oversees MAAA tests that are reported from our Phoenix laboratory and Noridian is the MAC responsible for administering claims for test reports issued by our annual revenue increasedPhoenix laboratory. In the second quarter of 2020, we submitted our technical assessment dossier for DecisionDx-SCC to Palmetto and Noridian. The dossier was accepted as complete in the third quarter of 2020. On June 8, 2023, both Palmetto and Noridian posted a preliminary draft LCD recommending no coverage for DecisionDx-SCC. The comment period for the draft LCDs ended on July 22, 2023.
Decision-SCC was reimbursed at a rate of $3,873 per test under a PLA code from $22.8 million in 2018second quarter of 2022 through June 30, 2023 when CMS determined DecisionDx-SCC meet the criteria for “new ADLT” status. Effective July 1, 2023 and through March 31, 2024, CMS set the initial period rate equal to $51.9 million in 2019. For the year endedlist price of $8,500 per test. Effective April 1, 2024 and through December 31, 2019, year-over-year growth in new ordering clinicians2025, the published CLFS rate for our DecisionDx-Melanoma test was 24.3%. Additionally, total ordering clinicians in 2019 for DecisionDx-Melanoma increased 32%DecisionDx-SCC will be based on the median private payor rates received between July 1, 2023 and November 30, 2023. We submitted the median private payor data to 3,927, year-over-year.
TissueCypher
TissueCypher is processed in our Pittsburgh, Pennsylvania laboratory and falls under the Medicare jurisdiction managed by Novitas. From January 1, 2022 through March 31, 2022, we received payments for claims according to the published CLFS rate at $2,513 per test. On March 24, 2022, CMS determined that TissueCypher meets the criteria for “new ADLT” status. ADLT status exempts TissueCypher from what is called the “14-day rule,” which simplifies the billing process for Medicare patients. From April 1, 2022 through December 31, 2019 and 2018 are presented in2022, CMS set the table below:
DecisionDx- Melanoma | DecisionDx-UM | Total | ||||||
| Q1 2019 | 3,232 | 360 | 3,592 | |||||
| Q2 2019 | 3,691 | 376 | 4,067 | |||||
| Q3 2019 | 4,126 | 356 | 4,482 | |||||
| Q4 2019 | 4,480 | 434 | 4,914 | |||||
| For the year ended December 31, 2019 | 15,529 | 1,526 | 17,055 | |||||
| Q1 2018 | 2,727 | 322 | 3,049 | |||||
| Q2 2018 | 2,899 | 382 | 3,281 | |||||
| Q3 2018 | 3,136 | 324 | 3,460 | |||||
| Q4 2018 | 3,270 | 385 | 3,655 | |||||
| For the year ended December 31, 2018 | 12,032 | 1,413 | 13,445 | |||||
78
IDgenetix
IDgenetix is currently covered under a Noridian LCD policy and accompanying billing and coding article developed by MolDX.
Our IDgenetix multi-gene panel was reimbursed by Medicare at approximately $1,500 per test from the April 2022 acquisition date through February 2023, when MolDX notified us that as part of its annual CPT code updates, IDgenetix should shift billing to “Information about certain metrics” below.
Delivered Test Reports
The number of test reports we generate is a key indicator that we use to assess our business. A test report is generated when we receive a sample in our laboratory, and then the relevant test information relating to that test is entered into our Laboratory Information Management System, the genomic profilelaboratory portion of the sampletest is performed, including proprietary algorithmic analysis of the combined biomarkers and a report providing the results of that profileis then generated which is sent to the physicianclinician who ordered the test.
The number of test reports delivered by us during the years ended December 31, 2023 and 2022 are presented in the table below:
| Proprietary Dermatologic GEP Tests | |||||||||||||||||||||||||||||||||||||||||||||||
| DecisionDx- Melanoma | DecisionDx-SCC | Diagnostic GEP offering (1) | Dermatologic Total | DecisionDx-UM | TissueCypher(3) | IDgenetix(2) | Grand Total | ||||||||||||||||||||||||||||||||||||||||
| Q1 2023 | 7,583 | 2,411 | 980 | 10,974 | 409 | 1,383 | 2,150 | 14,916 | |||||||||||||||||||||||||||||||||||||||
| Q2 2023 | 8,597 | 2,681 | 953 | 12,231 | 461 | 1,447 | 2,681 | 16,820 | |||||||||||||||||||||||||||||||||||||||
| Q3 2023 | 8,559 | 2,820 | 1,011 | 12,390 | 399 | 2,829 | 2,791 | 18,409 | |||||||||||||||||||||||||||||||||||||||
| Q4 2023 | 8,591 | 3,530 | 1,018 | 13,139 | 405 | 3,441 | 3,299 | 20,284 | |||||||||||||||||||||||||||||||||||||||
| For the year ended December 31, 2023 | 33,330 | 11,442 | 3,962 | 48,734 | 1,674 | 9,100 | 10,921 | 70,429 | |||||||||||||||||||||||||||||||||||||||
| Q1 2022 | 6,023 | 1,142 | 950 | 8,115 | 456 | 56 | — | 8,627 | |||||||||||||||||||||||||||||||||||||||
| Q2 2022 | 7,125 | 1,344 | 955 | 9,424 | 431 | 352 | 827 | 11,034 | |||||||||||||||||||||||||||||||||||||||
| Q3 2022 | 7,354 | 1,636 | 834 | 9,824 | 392 | 690 | 1,208 | 12,114 | |||||||||||||||||||||||||||||||||||||||
| Q4 2022 | 7,301 | 1,845 | 822 | 9,968 | 432 | 1,030 | 1,214 | 12,644 | |||||||||||||||||||||||||||||||||||||||
| For the year ended December 31, 2022 | 27,803 | 5,967 | 3,561 | 37,331 | 1,711 | 2,128 | 3,249 | 44,419 | |||||||||||||||||||||||||||||||||||||||
(1)Includes MyPath Melanoma and DiffDx-Melanoma. We bill third-party payorsoffered both MyPath Melanoma and patients forDiffDx-Melanoma under our Diagnostic GEP offering until February 2023 when we suspended the tests we perform. The majorityoffering of DiffDx-Melanoma, as discussed above.
(2)We began offering the IDgenetix test on April 26, 2022, following our acquisition of AltheaDx. Includes both single-gene and multi-gene tests.
(3)We temporarily paused accepting additional orders in July 2023 and resumed accepting new orders in a phased approach in September 2023. We completed processing of our pre-existing backlog orders in October 2023 and continue to accept new orders as of December 31, 2023.
For the years ended December 31, 2023 and 2022, our dermatologic test report volume increased by 30.5% and 40.9%, respectively, largely driven by continued growth from our DecisionDx-Melanoma and DecisionDx-SCC tests. Increases from our other tests (non-dermatologic), primarily IDgenetix and TissueCypher, also contributed to the overall volume increase. For a discussion of how we recognize revenue collections is paid by third-party insurers, including Medicare. We have received LCDs, which provide coverage forderived from our tests, refer to “Net Revenues” under “Components of Results of Operations” below.
79
In developing our DecisionDx-SCC test, we believed that meet certain criteriain addition to addressing significant unmet clinical needs, we would see opportunities for Medicare and Medicare Advantage beneficiaries, representing approximately 60 million covered lives. As it relates to DecisionDx-UM, we have contracts or have received positive medical policy decisions from additional payors representing approximately 83 million covered lives. A ‘‘covered life’’ means a subscriber, or a dependent of a subscriber, who is insured under an insurance policy for such an insurance carrier.
Information about certain metricsAbout Certain Metrics
The following provides additional information about certain metrics we have disclosed in this Management’s Discussion and Analysis of Financial Condition and Results of Operation.Operations.
Test Reports Delivered
Test reports delivered for DecisionDx-Melanoma and DecisionDx-UM represents the number of completed test reports delivered by us during the reporting period indicated. The period in which a test report is delivered does not necessarily correspond with the period in which the related revenue, if any, is recognized, due to the timing and amount of adjustments for variable consideration under ASC 606. We use this metric to evaluate the growth in adoption of our tests and to measure against our internal performance objectives. We believe this metric is useful to investors in evaluating the volume of our business activity from period to periodperiod-to-period that may not be discernible from our reported revenues under ASC 606.
Other Events
Impact of Macroeconomic Conditions
Macroeconomic conditions, including uncertainties associated with the numberIsrael-Hamas war, the ongoing conflict between Ukraine and Russia, economic slowdowns, public health crises, labor shortages, recessions or market corrections, supply chain disruptions, inflation and monetary policy shifts, liquidity concerns at, and failures of, clinicians who orderedbanks and other financial institutions or other disruptions in the DecisionDx-Melanoma test forbanking system or financing markets, rising interest rates and financial and credit market fluctuations, volatility in the first time duringcapital markets or other evolving macroeconomic developments, continued to have direct and indirect impacts on our business and could in the reportingfuture materially impact our results of operations and financial condition. We continue to actively monitor the impact of these macroeconomic factors on our results of operations, financial condition and cash flows. The extent of the impact of these factors on our operational performance and financial condition, including our ability to execute our business strategies and initiatives in the expected timeframe, will depend on future developments, which are uncertain and cannot be predicted; however, any continued or renewed disruption resulting from these factors could negatively impact our business.
Our Financial Results
Our net loss may fluctuate significantly from period specified. We believe this metric is useful in evaluatingto period, depending on the effectivenesstiming of our planned development activities, the growth of our sales and marketing effortsactivities and the timing of revenue recognition under ASC 606. We expect our expenses will increase substantially over time as we:
•execute clinical studies to generate evidence supporting our current and future product candidates;
•execute our commercialization strategy for our current and future commercial products;
•continue our ongoing and planned development of new products in establishing new relationships withour pipeline;
•seek to discover and develop additional product candidates;
•hire additional scientific and research and development staff; and
•add additional operational, financial and management information systems and personnel.
Factors Affecting Our Performance
We believe there are several important factors that have impacted, and that we expect will continue to impact, our operating performance and results of operations, including:
•Report volume. We believe that the number of reports we deliver to clinicians is an important indicator of the growth of adoption among the healthcare provider community. Our revenue and increasingcosts are affected by the adoptionvolume of our DecisionDx-Melanoma test. We also believe this metric provides useful information to investors in assessingtesting and mix of customers. Our performance depends on our ability to expand the use of the DecisionDx-Melanoma test. Since this metric is based upon the reporting period in which an order is placed, it does not necessarily correspond to the reporting period in which a test report was delivered or in which any revenue was recognized.
80
•Reimbursement. We believe that this metric provides useful informationexpanding reimbursement is an important indicator of the value of our products. Payors require extensive evidence of clinical utility, clinical validity, patient outcomes and health economic benefits in order to investors in assessingprovide reimbursement for diagnostic products. Our revenue depends on our ability to both expanddemonstrate the usevalue of our products to these payors.
•Gross margin. We believe that our gross margin is an important indicator of the DecisionDx-Melanoma test with newoperating performance of our business. Higher gross margins reflect the average selling price of our tests, as well as the operating efficiency of our laboratory operations.
•Expansion of our sales force and marketing programs. We believe the expansion of our direct sales force and marketing organization to educate clinicians and maintainpathologists on the value of our molecular testing products will significantly impact our performance.
•Integrating acquisitions. Revenue growth, operational results and advances to our business strategy depends on our ability to integrate any acquisitions into our existing clinician relationships. Since this metricbusiness and effectively scale their operations. The integration of acquired assets may impact our revenue growth, increase the cost of operations or may require management resources that otherwise would be available for ongoing development of our existing business.
•New product development. A significant aspect of our business is based upon the reporting periodour investment in which an order is placed, it does not necessarily correspondresearch and development activities, including activities related to the reporting period in which a test report was delivered or in which any revenue was recognized.development of new products. In addition to the development of new product candidates, we believe these studies are critical to gaining clinician adoption of new products and driving favorable coverage decisions by payors for such products.
Components of the Results of Operations
Net Revenues
We generate revenues from the sale of our products,products. Currently, our revenues are primarily derived from the sale of DecisionDx-Melanoma, DecisionDx-SCC, TissueCypher and DecisionDx-UM. We bill third-party payors and patients for the tests we perform.
Under ASC 606, we recognize revenue at the amount we expect to be entitled, subject to a constraint for variable consideration, in the period in which our tests are delivered to the treating physicians.clinicians. We have determined that our contracts contain variable consideration under ASC 606 because the amounts paid by third-party payors may be paid at less than our standard rates or not paid at all, with such differences considered implicit price concessions. Variable consideration is recognized only to the extent it is probable that a significant reversal of revenue will not occur in future periods when the uncertainties are resolved. We consider variable consideration to be fully constrained (and therefore not recognized) for Medicare claims when the payment of such claims is subject to approval by an ALJ at an appeal hearing, due to the level of uncertainty and timing of the outcome. Variable consideration is evaluated each reporting period and adjustments are recorded as increases or decreases in revenues. Importantly,Variable consideration for Medicare claims that are not covered by Medicare, including those claims undergoing appeal, is deemed to be fully constrained due to factors outside our influence (e.g., judgment or actions of third parties) and the uncertainty of the amount to be received is not expected to be resolved for a long period of time. For these fully constrained claims, we expect togenerally recognize any revenue adjudicated by the ALJ in the reporting period in which we are notified of the ALJ hearing outcome and ituncertainty is determined that such decision will not be appealed.favorably resolved, if at all. Due to ALJ hearings, potential future changes in Medicare coverage policies and appeal cycles, insurance coverage policies, contractual rates and other trends in the reimbursement of our tests, our revenues may fluctuate significantly from period to period. Our ability to recognize revenue for a test is dependent on the development of reimbursement experience and obtaining coverage decisions. For tests with limited reimbursement experience or no coverage, we recognize revenues on the basis of actual cash collections.
Our ability to increase our revenues will depend on our ability to further penetrate our target market,markets, and, in particular, generate sales through our direct sales force, develop and commercialize additional tests, including through acquisitions, obtain reimbursement from additional third-party payors and increase our reimbursement rate for tests performed. In the near term, our financial performance will be highly dependent on reimbursement for DecisionDx-Melanoma. The use of DecisionDx-Melanoma is not yet broadly covered under positive coverage policies, although many third-party payors have begun to reimburse for this test.
Cost of Sales (exclusive of amortization of acquired intangible assets)
The components of our cost of sales are material and service costs personnel costs, which includes stock-based compensation expense, equipment and infrastructure expenses associated with testing samples, personnel costs (including salaries, bonuses, benefits and stock-based compensation expense), electronic medical records,record set up costs, order and delivery systems, shipping charges to transport samples, third-party test fees, and allocated overhead including rent, information technology costs, equipment and facilities depreciation and utilities. Costs associated with performing teststesting samples are recorded when the test is
processed regardless of whether and when revenues are recognized with respect to that test. As a result, our cost of sales as a percentage of revenues may vary significantly from period to period because we do not recognize all revenues in the period in which the associated costs are incurred. We expect cost of sales in absolute dollars to increase as the number of tests we perform
81
increases. Additionally, we expect cost of sales to increase with the expansion of laboratory capacity and staffing in advance of the anticipated growth of our more recently launched tests and tests acquired through acquisitions. For example, we commenced operations in a new expanded laboratory facility in Pittsburgh, Pennsylvania in the second quarter of 2023.
Gross margin calculated as net revenue minus cost of sales, is aand gross margin percentage are key indicatorindicators we use to assess our business. For example,See the table in 2016, we transitioned the performance“Results of our genomic tests to our own dedicated clinical lab from a third-party contract lab. This transition increased the gross margin from our genomic tests while also increasing our level of controlOperations—Comparison of the performance of our testsyears ended December 31, 2023 and laboratory quality metrics.2022” for details.
Research and Development
Research and development expenses include costs incurred to develop our genomic tests, collect clinical samples and conduct clinical studies to develop and support our products. These costs consist of personnel costs including(including salaries, bonuses, benefits and stock-based compensation expense,expense), prototype materials, laboratory supplies, consulting costs, regulatory costs, electronic medical records set up costs, costs associated with setting up and conducting clinical studies and allocated overhead, including rent, information technology, equipment depreciation and utilities. We expense all research and development costs in the periods in which they are incurred. We expect our research and development expenses to increase in absolute dollars as we continue to invest in research and development activities related to developing enhanced and new products.
We expect to use a portion of our cash and cash equivalents and marketable investment securities to further support and accelerate our research and development activities, including important studies that are underway to support our DecisionDx-Melanoma test. For instance, in February 2023, we announced the publication of data from the DECIDE study presenting DecisionDx-Melanoma test results influenced 85% of clinicians’ decisions regarding the SLNB surgical procedure. Additionally, use of the tests’ results within current guideline recommendations led to a significant reduction in SLNB procedures performed, demonstrating the clinical value of the test to guide risk-aligned patient care. Also, in 2021, we initiated our large prospective, multi-center clinical study to develop, validate and bring to market a pipeline genomic test, or tests, aimed at predicting response to systemic therapy in patients with moderate to severe psoriasis, atopic dermatitis and related inflammatory skin conditions. As of December 31, 2023, there were more than 45 active clinical study sites and over 1,000 patients enrolled in this study. Assuming we are successful in validating a genomic tests, or tests, for one or more of these uses, then we expect to launch this pipeline test by the end of 2025.
Selling, General and Administrative
Selling, general and administrative or (“SG&A,&A”) expenses include executive, selling and marketing, legal, finance and accounting, human resources and billing and client services.functions. These expenses consist of personnel costs including(including salaries, bonuses, benefits and stock-based compensation expense,expense), direct marketing expenses, audit and legal expenses, consulting costs, training and medical education activities, payor outreach programs and allocated overhead, including rent, information technology, equipment depreciation, and utilities. In the near term, we expect increases in SG&A expenses related to compliance with the rules and regulations of the SEC and Nasdaq, investor relations activities, and additional insurance expenses. Other administrative and professional services expenses within SG&A are expected to increase with the scale of our business, but selling and marketing-related expenses are expected to increase significantly, consistent with our growth strategy.
Amortization of Acquired Intangible Assets
Amortization of acquired intangible assets is primarily associated with developed technology obtained through acquisitions, such as our acquisitions of Cernostics in December 2021 and AltheaDx in April 2022.
Change in Fair Value of Contingent Consideration
Change in fair value of contingent consideration is associated with our acquisitions of Cernostics and AltheaDx and the related contingent consideration of up to $50.0 million and $75.0 million, respectively, payable based on the achievement of certain commercial milestones relating to the year ended December 31, 2022 in the case of Cernostics, and the years ending December 31, 2022, 2023 and 2024, in the case of AltheaDx (the “Earnout Payments”). No Earnout Payments were paid relating to the year ended December 31, 2022 in connection with our acquisitions of Cernostics and AltheaDx since the applicable commercial milestones were not achieved. As of December 31, 2023 and December 31, 2022, our contingent consideration liability for AltheaDx was zero.
Interest and Other Non-Operating Income
Interest income consists primarily of interest earnedearnings on cash and cash equivalents, in interest-bearing accounts.primarily money market funds, and marketable investment securities, primarily short-term U.S. government obligations.
Interest Expense
Interest expense is primarily attributable to borrowings under our term debt, revolving linefinance leases.
82
Income Tax Expense (Benefit)
In connection with our acquisition of AltheaDx in April 2022, and taking into consideration the additional deferred tax liabilities resulting from such acquisition, we determined that a portion of our valuation allowance should be reduced, which was reflected in our income tax benefit for the year ended December 31, 2022. Our consolidated financial statements do not reflect any federal or state income tax benefits attributable to the netpre-tax losses we have incurred, due to the uncertainty of realizing a benefit from those items. As of December 31, 2019,2023, we had federal net operating lossNOL carryforwards of $57.4$197.1 million, of which $43.5$92.0 million will begin to expire in 20282029 if not utilized to offset federal taxable income, and $13.9$105.1 million may be carried forward indefinitely. Also, as of December 31, 2019,2023, we had state net operating lossNOL carryforwards of $42.2$114.3 million, which begin to expire in 2028 if not utilized to offset state taxable income.
83
Results of Operations
Comparison of the years endedYears Ended December 31, 20192023 and 20182022
The following table summarizes our results of operations for the periods indicated (in thousands, except percentages):
| Years Ended December 31, | Change | ||||||||||||||||||||||
| 2023 | 2022 | ||||||||||||||||||||||
| Net revenues | $ | 219,788 | $ | 137,039 | $ | 82,749 | 60.4 | % | |||||||||||||||
| Operating expenses and other operating income: | |||||||||||||||||||||||
| Cost of sales (exclusive of amortization of acquired intangible assets) | 44,982 | 32,009 | 12,973 | 40.5 | % | ||||||||||||||||||
| Research and development | 53,618 | 44,903 | 8,715 | 19.4 | % | ||||||||||||||||||
| Selling, general and administrative | 180,152 | 143,003 | 37,149 | 26.0 | % | ||||||||||||||||||
| Amortization of acquired intangible assets | 9,013 | 8,266 | 747 | 9.0 | % | ||||||||||||||||||
| Change in fair value of contingent consideration | — | (18,287) | 18,287 | 100.0 | % | ||||||||||||||||||
| Total operating expenses, net | 287,765 | 209,894 | 77,871 | 37.1 | % | ||||||||||||||||||
| Operating loss | (67,977) | (72,855) | 4,878 | 6.7 | % | ||||||||||||||||||
| Interest and other non-operating income | 10,623 | 3,968 | 6,655 | 167.7 | % | ||||||||||||||||||
| Interest expense | (11) | (17) | 6 | NM | |||||||||||||||||||
| Loss before income taxes | (57,365) | (68,904) | 11,539 | 16.7 | % | ||||||||||||||||||
| Income tax expense (benefit) | 101 | (1,766) | 1,867 | 105.7 | % | ||||||||||||||||||
| Net loss | $ | (57,466) | $ | (67,138) | $ | 9,672 | 14.4 | % | |||||||||||||||
(1) NA = Not applicable
(2) NM = Not meaningful
The following table indicates the amount of stock-based compensation expense (non-cash) reflected in the line items above (in thousands):
| Years Ended December 31, | |||||||||||||||||
| 2023 | 2022 | Change | |||||||||||||||
| Cost of sales (exclusive of amortization of acquired intangible assets) | $ | 4,938 | $ | 3,755 | $ | 1,183 | |||||||||||
| Research and development | 10,119 | 7,635 | 2,484 | ||||||||||||||
| Selling, general and administrative | 36,162 | 24,931 | 11,231 | ||||||||||||||
| Total stock-based compensation expense | $ | 51,219 | $ | 36,321 | $ | 14,898 | |||||||||||
The following table provides a disaggregation of net revenues by type (in thousands):
| Years Ended December 31, | |||||||||||||||||
| 2023 | 2022 | Change | |||||||||||||||
Dermatologic(1) | $ | 183,375 | $ | 124,809 | $ | 58,566 | |||||||||||
Non-Dermatologic(2) | 36,413 | 12,230 | 24,183 | ||||||||||||||
| Total net revenues | $ | 219,788 | $ | 137,039 | $ | 82,749 | |||||||||||
(1)Consists of DecisionDx-Melanoma, DecisionDx-SCC and our Diagnostic GEP offering.
(2)Consists of TissueCypher, DecisionDx-UM and IDgenetix.
84
| Years Ended December 31, | Increase (Decrease) | |||||||||||||
| 2019 | 2018 | |||||||||||||
| Net revenues | $ | 51,865 | $ | 22,786 | $ | 29,079 | 127.6 | % | ||||||
| Cost of sales | 7,310 | 5,297 | 2,013 | 38.0 | % | |||||||||
| Gross margin | 44,555 | 17,489 | 27,066 | 154.8 | % | |||||||||
| Operating expenses: | ||||||||||||||
| Research and development | 7,385 | 4,854 | 2,531 | 52.1 | % | |||||||||
| Selling, general and administrative | 29,842 | 16,471 | 13,371 | 81.2 | % | |||||||||
| Total operating expenses | 37,227 | 21,325 | 15,902 | 74.6 | % | |||||||||
| Operating income (loss) | 7,328 | (3,836 | ) | 11,164 | 291.0 | % | ||||||||
| Interest income | 312 | 24 | 288 | 1,200.0 | % | |||||||||
| Interest expense | (4,571 | ) | (2,274 | ) | (2,297 | ) | (101.0 | )% | ||||||
| Gain on extinguishment of debt | 5,213 | — | 5,213 | N/A | ||||||||||
| Other expense, net | (2,933 | ) | (272 | ) | (2,661 | ) | (978.3 | )% | ||||||
| Income (loss) before income taxes | 5,349 | (6,358 | ) | 11,707 | 184.1 | % | ||||||||
| Income tax expense | 72 | 9 | 63 | 700.0 | % | |||||||||
| Net income (loss) | $ | 5,277 | $ | (6,367 | ) | $ | 11,644 | 182.9 | % | |||||
| Years Ended December 31, | |||||||||||||||||
| 2023 | 2022 | Change | |||||||||||||||
| Net revenues | $ | 219,788 | $ | 137,039 | $ | 82,749 | |||||||||||
| Less: Cost of sales (exclusive of amortization of acquired intangible assets) | 44,982 | 32,009 | 12,973 | ||||||||||||||
| Less: Amortization of acquired intangible assets | 9,013 | 8,266 | 747 | ||||||||||||||
| Gross margin | $ | 165,793 | $ | 96,764 | $ | 69,029 | |||||||||||
| Gross margin percentage | 75.4 | % | 70.6 | % | 4.8 | % | |||||||||||
Net Revenues
Net revenues increased by $29.1 million, or 127.6%, to $51.9 million due to a combination of increased test volume and higher per-unit revenues. Approximately 94% of the increase is attributable to DecisionDx-Melanoma test revenues with the remainder primarily attributable to DecisionDx-UM. Forfor the year ended December 31, 2019, we experienced an increase in DecisionDx-Melanoma and DecisionDx-UM test volume of 26.9%2023 increased by $82.7 million, or 60.4%, to $219.8 million compared to the year ended December 31, 2018. Our 2019 per-unit revenues also benefited2022 due to a $58.6 million increase in revenue from the attainment of “new ADLT” statusour dermatologic tests and a $24.1 million increase in revenue from our non-dermatologic tests.
The increase from our dermatologic tests was primarily due to higher average selling price per unit for our DecisionDx-Melanoma test, effective July 1, 2019, which resulted inDecisionDx-SCC tests, where we began receiving Medicare reimbursement at a higher Medicarerate beginning in July 2023, increases in test report volume of 19.9%, for DecisionDx-Melanoma and 91.8%, for DecisionDx-SCC.
The increase in net revenue from our non-dermatologic tests of $24.1 million was primarily attributable to TissueCypher, due to a higher average selling price and higher test report volume, reflecting a higher 2023 reimbursement rate forthan our 2022 rates. Our IDgenetix test, which we acquired in connection with our acquisition of AltheaDx in April 2022, also contributed to the test,increase in non-dermatologic revenues during the period. Net revenue from our non-dermatologic tests as described above. Revenuesa percentage of total net revenue increased from 8.9% for the year ended December 31, 2019 were also positively impacted by2022 to 16.6% for the full-year effect of our final LCD for DecisionDx-Melanoma. The LCD was issued by Palmetto, effective December 3, 2018, and provided for payment of covered claims submitted for reimbursement beginning in February 2018. For the yearsyear ended December 31, 2019 and 2018, we recorded2023.
The increases in total net positiverevenues were partially offset by the effect of variations in revenue adjustments of $2.5 million and $0.3 million, respectively, related to tests delivered in previous periods, associated with changes in estimated variable consideration. The additional positiveconsideration, which were $4.5 million of net negative revenue adjustments in the current year primarily relate to recognition of revenue for certain tests delivered in prior periods for which no revenue was recognizable originally, but was recognized upon cash collection of payments for the tests in the current-year period.
Cost of Sales (exclusive of amortization of acquired intangible assets)
Cost of sales (exclusive of amortization of acquired intangible assets) for the year ended December 31, 2023 increased by $2.0$13.0 million, or 38.0%40.5%, compared to the year ended December 30, 2018,31, 2022, primarily due to increased expenditures on supplies, higher personnel costs, of suppliesthird-party services and services,rent. Supply and service expenses have increased due to higher laboratory activity, which is attributable to the higher activity levels,test report volumes. The increase in personnel costs primarily consist of higher salaries and wages, stock-based compensation expense and benefits expense, reflecting headcount additions made to support business growth as well as higher personnel costs due to additionalmerit and annual inflationary wage adjustment for existing employees. Increases in headcount, in addition to higher lab service expense and rent expense, have largely been attributable to commencement operations at our new Pittsburgh laboratory, testing operations. Our gross margin percentage was 85.9% forwhich opened in the year ended December 31, 2019, compared to 76.8% for the same period in 2018, with the improvement principally a resultsecond quarter of the increased operating leverage on the higher revenues. 2023.
Due to the nature of our business, a significant portion of our cost of sales expenses representrepresents fixed costs associated with our testing operations. Accordingly, our cost of sales expense will not necessarily increase or decrease commensurately with the change in net revenues from period to period. We expect our cost of sales expenses (exclusive of amortization of acquired intangible assets) to continue to increase in future periods as we hire additional laboratory personnel and related resources to support our expected growth in volume for our dermatologic, gastrointestinal, mental health and pipeline tests.
Gross Margin
Our gross margin percentage was 75.4% for the year ended December 31, 2023, compared to 70.6% for the same period in 2022. The increase was primarily due to higher net revenues, partially offset by higher supplies expenditures and higher laboratory personnel costs, higher laboratory overhead, to include lab service fees and rent, and variations in revenue adjustments related to tests delivered in previous periods.
85
Research and Development
Research and development expenses increased by $2.5$8.7 million, or 52.1%19.4%, for the year ended December 31, 2019,2023, compared to the year ended December 31, 2018,2022, primarily associated with increasesconsisting of higher personnel costs and higher clinical studies expense partially offset by lower expense from advisory boards and consulting services.
Higher personnel costs are primarily a result of higher salaries and wages, stock-based compensation expense, bonuses and benefits expense. Increases in personnel costs.salaries and wages, stock-based compensation expense, bonuses and benefit expenses were due to headcount expansions as well as merit and annual inflationary wage adjustment for existing employees. Higher research and development expense is attributable to higher costs for clinical studies, most of which was attributable to our other inflammatory skin disease pipeline studies. Advisory board and consulting costs decreased due to fewer conference and speaker events being held in 2023 than in 2022.
We expect research and development expense to increase as we continue to invest in ongoing pipeline initiatives as well as seek opportunities to branch out upstream, downstream and parallel to our existing commercial tests, within or adjacent to our established dermatology commercial call points.
Selling, General and Administrative
The following table provides a breakdown of SG&A expenseexpenses (in thousands):
| Years Ended December 31, | |||||||||||||||||
| 2023 | 2022 | Change | |||||||||||||||
| Sales and marketing | $ | 113,657 | $ | 86,607 | $ | 27,050 | |||||||||||
| General and administrative | 66,495 | 56,396 | 10,099 | ||||||||||||||
| Total selling, general and administrative expense | $ | 180,152 | $ | 143,003 | $ | 37,149 | |||||||||||
Sales and marketing expenses increased by $13.4$27.1 million, or 81.2%31.2%, for the year ended December 31, 20192023 compared to the year ended December 31, 2018. Approximately 52% of the increase is2022, primarily attributable to higher personnel costs, particularly due to increased headcount, which includesconsisting of salaries, stock-based compensation, bonuses benefits and stock-based compensation. In early 2019, we expanded our sales organization from 14 territories to 23 territories and implemented an additional expansion to 32 territories beginning in December 2019. The higher personnel costs also reflect expanded headcount in our administrative support functions.employee benefits. The remainder of the increase in sales and marketing expenses was primarily associated with higher professionaltraining events, conference fees, (principallytravel and other marketing costs incurred through our ongoing sales and marketing operations.
In 2023, we continued to focus on the expansion and development of both our dermatology-facing and non-dermatology-facing commercial outside sales forces, and ancillary teams, as we seek to expand awareness of our test portfolio for patients new clinicians. Salaries and stock-based compensation for our sales and marketing teams have increased through headcount expansions as well as through merit and annual inflationary wage adjustment for existing employees. Bonuses have increased through expanded headcount and through individual and company performance. Higher expense for travel, training events and conference fees were primarily attributable to reimbursementour sales and accounting), increased travel costs, increased spending for eventsmarketing operations. Stock-based compensation expense included in sales and conferences, higher insurancemarketing expense and other general increases.
General and administrative expenses increased by $10.1 million, or 17.9%, for the year ended December 31, 2023, compared to the year ended December 31, 2018, as a result of higher interest-bearing balances of cash and cash equivalents.2022. The higher cash and cash equivalents wereincrease is primarily attributable to proceeds from the IPOhigher personnel costs and the issuancehigher information technology and software-related costs. Increases in personnel costs reflect higher stock-based compensation and salaries. Personnel costs have increased through headcount expansions in our administrative support functions and through merit and annual inflationary wage adjustment for existing employees. Increases in personnel costs and information technology and software-related costs were partially offset by a decrease in professional service fees, most of convertible promissory noteswhich related to costs incurred through our acquisition of AltheaDx during 2019.
86
Amortization of Acquired Intangible Assets
Amortization of acquired intangible assets increased by $0.7 million for the year ended December 31, 2023, compared to the year ended December 31, 2018.2022. The effectincrease is primarily associated with amortization of developed technology attributable to the acquisition of AltheaDx in April 2022. Amortization of acquired intangible assets is projected to be approximately $9.0 million for the year ending December 31, 2024.
Change in Fair Value of Contingent Consideration
The change in fair value of contingent consideration for the year ended December 31, 2022 of $18.3 million, a net gain, was primarily related to the remeasurement of the issuanceEarnout Payments associated with our acquisition of Cernostics. There was no such activity for the Q1 2019 Notesyear ended December 31, 2023.
Interest and Other Non-Operating Income
Interest income increased by $6.7 million for the year ended December 31, 2023, compared to the year ended December 31, 2022, primarily as a result of higher interest rates and our purchases of marketable investment securities beginning in January and February 2019 added $1.7 millionthe third quarter of 2022.
Income Tax Expense (Benefit)
We recorded a minimal amount in interestincome tax expense for the year ended December 31, 2019 and consisted of the accrual of the contractual 8% interest plus the amortization of issuance costs and debt discount. The remainder of the increase is due to a combination of higher average outstanding balances and higher interest rates on our variable-rate debt under our banking credit facility. Average outstanding bank debt balances were approximately $4.62023. Our income tax benefit was $1.8 million higher during the year ended December 31, 2019 compared to the year ended December 31, 2018 and average interest rates increased by approximately 0.6% from the same period in 2018.
Stock-Based Compensation Expense
Stock-based compensation expense, which is more likely thanallocated among cost of sales, research and development expense and SG&A expense, totaled $51.2 million for the year ended December 31, 2023 compared to $36.3 million for the year ended December 31, 2022. The increase is primarily due to our annual grant of equity awards in December 2022, as well as new hire and merit based awards during 2023, partially offset by the impact of an annual grant of equity awards not that these benefits will notbeing issued in December 2023.
We expect material increases in stock-based compensation expense in future periods, attributable to both existing awards outstanding and anticipated additional grants to our current and future employees. We expect to complete an annual grant of equity awards to our employees in March 2024.
As of December 31, 2023, we had 610 employees compared to 543 as of December 31, 2022. As of December 31, 2023, the total unrecognized stock-based compensation cost related to outstanding awards was $85.5 million, which is expected to be realized.recognized over a weighted-average period of 2.3 years.
Liquidity and Capital Resources
Our principal sources of liquidity are our inception, wecash and cash equivalents, marketable investment securities and cash generated from the sale of our products. All of our marketable investment securities are considered investment grade, are readily available for use in current operations and have incurred significant operating losses and negative cash flows. For thecontractual maturities of one year ended December 31, 2018, our net loss was $6.4 million, our net cash used in operating activities was $12.3 million and we had an accumulated deficit of $57.5 million as of December 31, 2018. For the year ended December 31, 2019, we reported net income of $5.3 million (which includes the benefit of a $5.2 million non-cash debt extinguishment gain), our net cash provided by operating activities was $7.0 million and we had an accumulated deficit of $52.2 million as of December 31, 2019. We also have substantial indebtedness, the terms of which require us to meet a three-month trailing revenue covenant, which is currently tested quarterly.or less. As of December 31, 20192023 and 2018,December 31, 2022, we are in compliance with this covenant.had marketable investment securities of $144.3 million and $135.7 million, respectively. As of December 31, 2023 and 2022, we had cash and cash equivalents of $98.8 million and $122.9 million, respectively.
87
As mentioned above, we expect to use a portion of our convertible preferredcash and cash equivalents and marketable investment securities to further support and accelerate our research and development activities, including the clinical studies noted above in “Components of the Results of Operations—Research and Development.”
Public Offerings of Common Stock
On June 29, 2020 and July 2, 2020, we issued and sold 2,000,000 and 300,000 shares of our common stock, respectively, of our common stock in various private placements beginninga follow-on public offering at a price of $37.00 per share. We received $79.5 million in 2008, which we have used to fund our operations. In addition, we have obtained financing through term debt, a revolving line of creditaggregate net proceeds, after deducting underwriting discounts and convertible promissory notes, which are discussed further below.
Our primary uses of capital are, and we expect will continue to be, compensation and related expenses, clinical research and development services, laboratory operations, equipment and related supplies, legal and other regulatory expenses, and general administrative costs.costs and, from time to time, expansion of our laboratory and office facilities in support of our growth. We anticipate that a substantial portion of our capital resources and effortscash requirements in the foreseeable future will be focused onrelate to the further commercialization of our existingcurrently marketed products, the development of our future product candidates in our pipeline and the potential commercialization of our product candidates,these pipeline products, should their development be successful.
In April 2022, we acquired AltheaDx, for $30.5 million in cash and $17.1 million in shares of our common stock. We agreed to pay contingent consideration of up $75.0 million, 50% in cash and 50% in common stock, based on the achievement of certain commercial milestones relating to the years ending December 31, 2022, 2023 and 2024. The portions associated with the commercial milestones for the years ended December 31, 2023 and 2022 were $37.5 million and $35.0 million, respectively, and were not paid since the applicable commercial milestones were not met. The 2022 portion was exclusive of a catch-up payment in 2023 of $17.5 million if all 2023 commercial milestones had been fully met. Therefore, a potential payment obligation of up to $20.0 million with respect to the remaining commercial 2024 milestone remains as of December 31, 2023. In each case, the number of shares of our common stock that may be issued in connection with the commercial milestone payments is subject to limitations. Our actual liability with respect to these commercial milestone payments from our acquisition will depend, in part, on our ability to successfully grow IDgenetix (acquired from AltheaDx) revenue and timing thereof. See Note 6 of the consolidated financial statements for additional information on the acquisition of AltheaDx.
On July 10, 2023, following approval by our board of directors, we entered into a definitive agreement to purchase a plot of land located in Friendswood, Texas for a purchase price of $7.6 million, subject to certain adjustments, for the purpose of developing a commercial office building which may be used as our future corporate headquarters. Under the agreement, we had the option to terminate the contract within 90 days, for any reason. On October 4, 2023, we amended the definitive agreement to extend the option period by an additional 30 days. On January 9, 2024, we amended the definitive agreement to finalize easement right and related matters. On February 9, 2024, we closed on the purchase of the land for cash consideration of $7.2 million.
Since our inception, we have generally incurred significant losses and negative operating cash flows. For the year ended December 31, 2023 we had a net loss of $57.5 million and an accumulated deficit of $218.4 million as of December 31, 2023. Our ability to generate revenue sufficient to achieve profitability will depend heavily on the successful commercialization of our currently marketed products and general administrative costs.
the development and validation of genomic classifiers; and
88
•successful commencement and completion of clinical study protocols;
•successful identification and acquisition of tissue samples;
•the development and validation of genomic classifiers; and
•acceptance of new genomic tests by clinicians, patients and third-party payors including competitor actions.
Because of the numerous risks and uncertainties associated with research, development and commercialization of product candidates, we are unable to estimate theour exact amount of our working capital requirements. Our future funding requirements will depend on and could increase significantly as a result of, many factors, including those listed above.above as well as those listed in Part 1, Item 1A., “Risk Factors” in this Annual Report on Form 10-K.
| Years Ended December 31, | |||||||
| 2019 | 2018 | ||||||
| Term debt | $ | 26,688 | $ | 21,350 | |||
| Revolving line of credit | — | 5,000 | |||||
| Total principal amount | 26,688 | 26,350 | |||||
| Unamortized discount and issuance costs | (1,566 | ) | (1,850 | ) | |||
| Total long-term debt | 25,122 | 24,500 | |||||
| Less: Current portion of long-term debt | (5,833 | ) | — | ||||
| Long-term debt, less current portion | $ | 19,289 | $ | 24,500 | |||
We have entered into various operating and finance leases, which are primarily associated with our laboratory facilities and office space.
Total undiscounted future minimum payment obligations under our operating leases and finance leases as of December 31, 20192023 totaled approximately $6.4$24.4 million,. of which $2.4 million is payable in 2024 and $22.0 million is payable through the end of 2033. The leases expire on various dates through 20272033 and provide certain options to renew for additional periods.
We expect our lease obligations may increase in the future as we expand our facilities, operations and headcount in support of the anticipated growth in our portfolio of commercial products and pipeline tests. Refer to Note 9 to10 of the consolidated financial statements for additional information on our leasing arrangements.
Cash Flows
| Years Ended December 31, | ||||||||
| 2019 | 2018 | |||||||
| Net cash provided by (used in) operating activities | $ | 7,015 | $ | (12,295 | ) | |||
| Net cash used in investing activities | (937 | ) | (277 | ) | ||||
| Net cash provided by financing activities | 88,288 | 15,839 | ||||||
| Net increase in cash and cash equivalents | $ | 94,366 | $ | 3,267 | ||||
| Years Ended December 31, | |||||||||||||||||||||||||||||||||||
| 2023 | 2022 | ||||||||||||||||||||||||||||||||||
| Net cash used in operating activities | $ | (5,626) | $ | (41,655) | |||||||||||||||||||||||||||||||
| Net cash used in investing activities | (16,183) | (166,545) | |||||||||||||||||||||||||||||||||
| Net cash (used in) provided by financing activities | (2,298) | 1,515 | |||||||||||||||||||||||||||||||||
| Net change in cash and cash equivalents | (24,107) | (206,685) | |||||||||||||||||||||||||||||||||
| Cash and cash equivalents, beginning of year | 122,948 | 329,633 | |||||||||||||||||||||||||||||||||
| Cash and cash equivalents, end of year | $ | 98,841 | $ | 122,948 | |||||||||||||||||||||||||||||||
Operating Activities
Net cash provided byused in operating activities was $7.0$5.6 million for the year ended December 31, 2019 and was primarily attributable to net income of $5.3 million and net non-cash charges of $1.7 million (consisting of $2.1 million of fair value option adjustments, $1.9 million in amortization of debt discount and issuance costs, stock compensation expense of $1.2 million,
89
Net cash used in operating activities was $41.7 million for the year ended December 31, 2022, and was primarily attributable to the net loss of $67.1 million, the change in fair value of contingent consideration of $18.3 million, increases in accounts receivable of $6.2 million, deferred income taxes of $1.9 million, increases in inventory of $1.7 million and increases in accretion of discounts on marketable investment securities of $1.4 million, partially offset by stock compensation expense of $36.3 million, depreciation and amortization of $10.5 million and increases in accrued compensation of $8.5 million.
The $36.0 million decrease in net cash used in operating activities for the year ended December 31, 2023 compared to the year ended December 31, 2022 is primarily due to increases in collections from customers attributable to higher net revenues partially offset by increases in accrued compensationoperating expenditures. In part, the cash used during the year ended December 31, 2023 reflects the payment of $1.3annual cash bonuses to our employees as well as certain health care benefit payments totaling $17.7 million. In comparison, we paid $11.6 million during the same period in 2022 towards annual cash bonuses and non-cash charges of $0.9 million.certain health care benefits.
Investing Activities
Net cash used in investing activities for the years ended December 31, 2019 and 2018 consisted entirely of purchases of property and equipment.
Net cash used in investing activities was $166.5 million for the year ended December 31, 2022 and consisted primarily of purchases of marketable investment securities of $134.7 million, the cash portion of the AltheaDx purchase consideration of $27.0 million (net of underwriting discounts, commissionscash and issuance costs), $11.7cash equivalents acquired) and purchases of property and equipment of $5.6 million.
The $8.0 million of net proceeds from the issuance of the Q1 2019 Notes, $9.2 million of net proceeds from the issuance of the July 2019 Note, $1.8 million of net proceeds associated with an increase in cash used for the 2018 Term Loan in connection with an amendmentpurchase of property and equipment for the year ended December 31, 2023 compared to the 2018 LSAyear ended December 31, 2022 was primarily due to the build out and $1.2completion of our new laboratory and office facilities located in Pittsburgh, Pennsylvania. Fixed asset purchases for this build out included those made for laboratory equipment, leasehold improvements, furniture and fixtures, and information technology infrastructure.
Financing Activities
Net cash used in financing activities was $2.3 million for the year ended December 31, 2023, and consisted primarily of payment of employees’ taxes on vested RSUs of $5.1 million, partially offset by $2.7 million of proceeds from the exercise of stock options, partially offset by principal repayments of $1.8 million oncontributions to our line of credit.2019 Employee Stock Purchase Plan (the “ESPP”).
Net cash provided by financing activities was $15.8$1.5 million for the year ended December 31, 20182022, and consisted primarily of $10.4$2.5 million attributable to proceeds received from the issuance of preferred stock and preferred stock warrants, $4.4 million from the issuance of term debt and preferred stock warrants in connection with the 2018 LSA and $1.0 million in proceeds from our linecontributions to the ESPP and $0.8 million of credit.proceeds from exercise of common stock options, partially offset by payment of employees’ taxes on vested RSUs of $1.7 million.
In 2021, the rate of inflation in the United States increased, began subsiding in the second half of 2022 and Commitments
has remained stable in 2023. We do not currently have, nor did we have any off-balance sheet arrangements, as defined in the rules and regulations of the SEC,believe that inflation has had a material impact on our financial results during the periods presented.year ended December 31, 2023. We are unable to predict if the rate of inflation will increase in future periods.
Critical Accounting Policies and Significant Judgments and Estimates
Our consolidated financial statements are prepared in accordance with U.S. GAAP. The preparation of our consolidated financial statements and related disclosures requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, costs and expenses, and the disclosure of contingent assets and liabilities in our consolidated financial statements. We base our estimates on historical experience, known trends and events and various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. We evaluate our estimates and assumptions on an ongoing basis. Our actual results may differ from these estimates under different assumptions or conditions.
While our significant accounting policies are described in more detail in Note 2 to our audited consolidated financial statements, we believe that the following accounting policies are those most critical to the judgments and estimates used in the preparation of our consolidated financial statements.
Revenue Recognition
We recognize revenue in accordance with ASC 606.In accordance with ASC 606, we follow a five-step process to recognize revenues: (1) identify the contract with the customer;customer, (2) identify the performance obligations;obligations, (3)
90
determine the transaction price;price, (4) allocate the transaction price to the performance obligation;obligations and (5) recognize revenues when the performance obligations are satisfied.
We have determined that we have a contract with the patient when the treating physicianclinician orders the test. Our contracts generally contain a single performance obligation, which is the delivery of the test report, and we satisfy our performance obligation at a point-in-time upon the point in time when we deliverdelivery of the test report to the treating physician,clinician, at which point we can bill for the report. The amount
of revenue recognized reflects the amount of consideration to which we expect to receive,be entitled, or the transaction price, and considers the effects of variable consideration, which is discussed further below.consideration.
All of our revenues from contracts with customers are associated with the provision of diagnostic and prognostic testing services. Our revenues are primarily attributable to DecisionDx-Melanoma for cutaneous melanoma. We also provide a test for UM, DecisionDx-UM. We launched a test for patients with cutaneous SCC, DecisionDx-SCC in August 2020 and launched a test for use in patients with suspicious pigmented lesions, DiffDx-Melanoma in November 2020. We began offering a test for difficult-to-diagnose melanocytic lesions, MyPath Melanoma, following an asset acquisition completed in May 2021. We also provide a test for UM, DecisionDx-UM. We began offering the TissueCypher test for patients with BE following an asset acquisition completed in December 2021. We began offering a proprietary PGx test service focused on mental health IDgenetix, following a business combination completed in April 2022.
Once we satisfy our performance obligations and bill for the test report,service, the timing of the collection of payments may vary based on the payment practices of the third-party payor and the existence of contractually established reimbursement rates. Most of theThe payments for our test reportsservices are primarily made by third-party payors, including Medicare and commercial health insurance carriers. Certain contracts contain a contractual commitment of a reimbursement rate that differs from our list prices. However, absent a contractually committed reimbursement rate with a commercial carrier or governmental program, our diagnostic tests may or may not be covered by these entities’ existing reimbursement policies. In addition, patients do not enter into direct agreements with us that commit them to pay any portion of the cost of the tests in the event that their insurance provider declines to reimburse us. We may pursue, on a case-by-case basis, reimbursement from such patients in the form of co-payments and co-insurance, in accordance with the contractual obligations that we have with the insurance carrier or health plan. These situations may result in a delay in the collection of payments.
The Medicare claims that are covered by policy under an LCD are generally paid at the established rate by our Medicare contractor within 30 days from receipt. As the LCD currently only covers a portion of the DecisionDx-Melanoma test reports, we provide for patients covered by Medicare, we intend to attempt to expand this LCD coverage policy in the future. Medicare claims that were either submitted to Medicare prior to thea medical review and coverage, or an LCD’s effective date, or that are not otherwise covered, by the terms of the LCD, but that may meet the definition of being medically reasonable and necessary pursuant to the controlling Section 1862(a)(1)(A) of the Social Security Act may be paid upon initial claim submission or if denied for payment are generally appealed by us. If our appeal is successfuland may ultimately be paid at the first level (termed “redetermination”), second level (termed “reconsideration”) or third level of appeal (de(de novo hearing with an ALJ), payment may be made for all. A successful appeal at any of these levels results in payment.
In the absence of Medicare coverage or a portion of the claim.
91
related recognition of revenue in ASC 606 to not disclose information about our remaining performance obligations. Any incremental costs to obtain contracts are recorded as SG&A expense as incurredcurrent period for tests delivered in prior periods due to the short duration of our contracts. Contract balances consisted solely of accounts receivable (both current and noncurrent) as of December 31, 2019 and 2018.
constraint on variable consideration.
Stock-Based Compensation
Stock-based compensation expense for equity instruments issued to employees and non-employees, including thestock options, RSUs, performance-based restricted stock units (“PSUs”) and purchase rights issued under our 2019 Employee Stock Purchase Plan, orthe ESPP is measured based on the grant-dategrant date fair value of the awards. TheFor stock options and purchase rights granted under the ESPP, we estimate the grant date fair value using the Black-Scholes option-pricing valuation model. For RSUs and PSUs, we use the closing price of each employeeour common stock option is estimated on the date of grant usingto determine the Black-Scholes option-pricing valuation model and on the offering date for the ESPP.fair value. We recognize compensation costs on a straight-line basis for all employee stock-based compensation awards over the requisitewith only service period of the awards,conditions, which is generally the awards’ vesting period, which is typically four years for options and RSUs and the two-year offering period for the ESPP. PSUs vest upon the achievement of certain performance conditions and the provision of service with us through a specified period. Accruals of compensation cost for PSUs are based on the probable outcome of the performance conditions and are reassessed each reporting period. We recognize compensation cost for PSUs separately for each vesting tranche on a ratable basis over the requisite service period. The requisite service period for PSUs is based on an analysis of vesting requirements and performance conditions for the particular award. Under specific circumstances, certain employees may be eligible for accelerated vesting of a portion of their awards upon retirement, contingent upon meeting predefined criteria related to age, tenure, and notice. The determination of the requisite service period considers the employee's retirement eligibility and is reassessed at each reporting date. Forfeitures are accounted for as they occur.
Set forth below is a description of the significant assumptions used in the option pricing model:
•Expected term. The expected term is the period of time that granted options are expected to be outstanding. For stock options, we have set the expected term using the simplified method based on the weighted average of both the period to vesting and the period to maturity for each option, as we have concluded that our stock option exercise history does not provide a reasonable basis upon which to estimate the expected term. For the ESPP, the expected term is the period of time from the offering date to the purchase date.
•Expected volatility. BecausePreviously, because of the limited period of time our stock hashad been traded in an active market, we calculatecalculated expected volatility by using the historical stock prices of a group of similar companies looking back over the estimated life of the option or the ESPP purchase rightrights under the ESPP and averaging the volatilities of these companies.
In the third quarter of 2021, we adjusted this calculation to include our own stock price on a relative basis to the peer group in the calculation of expected volatility, as our common stock has now been traded in an active market for more than two years.•Risk-free interest rate. We base the risk-free interest rate used in the Black-Scholes valuation model on the market yield in effect at the time of option grant and at the offering date for the ESPP provided from the Federal Reserve Board’s Statistical Releases and historical publications from the Treasury constant maturities rates for the equivalent remaining terms.
•Dividend yield. We have not paid, and do not have plans to pay, cash dividends. Therefore, we use an expected dividend yield of zero in the Black-Scholes option valuation model.
The fair value of our common stock is also an assumption used to determine the fair value of stock options. Prior to our IPO, the estimated fair value of our common stock had been determined by our board of directors as of the date of each award, grant, with input from management, considering our most recently available third-party valuations of common stock and our board of directors’ assessment of additional objective and subjective factors that it believed were relevant and which may have changed from the date of the most recent valuation through the date of the grant, which intended all options granted to be exercisable at price per share not less than the per share fair value of our common stock underlying those options on the grant date. Subsequent to our IPO, the fair value of our common stock is the closing selling price per share of our common stock as reported on the Nasdaq Global Market on the date of grant or other relevant determination date.
92
The following table sets forth the assumptions used to determine the fair value of stock options:
| Years Ended December 31, | ||||||||||||||||||
| 2019 | 2018 | |||||||||||||||||
| Average expected term | 6 years | 6 years | ||||||||||||||||
| Years Ended December 31, | ||||||||||||||||||
| Years Ended December 31, | ||||||||||||||||||
| Years Ended December 31, | ||||||||||||||||||
| 2023 | ||||||||||||||||||
| 2023 | ||||||||||||||||||
| 2023 | ||||||||||||||||||
| Average expected term (years) | ||||||||||||||||||
| Average expected term (years) | ||||||||||||||||||
| Average expected term (years) | ||||||||||||||||||
| Expected stock price volatility | ||||||||||||||||||
| Expected stock price volatility | ||||||||||||||||||
| Expected stock price volatility | 56.74% - 60.17% | 57.77% - 58.29% | ||||||||||||||||
| Risk-free interest rate | 1.63% - 2.47% | 2.77% - 3.13% | ||||||||||||||||
| Risk-free interest rate | ||||||||||||||||||
| Risk-free interest rate | ||||||||||||||||||
| Dividend yield | —% | —% | ||||||||||||||||
| Dividend yield | ||||||||||||||||||
| Dividend yield | ||||||||||||||||||
The following table sets forth assumptions used to determine the fair value of the purchase rights issued under the ESPP:
| Years Ended December 31, | ||||||||||||||
| 2023 | 2022 | |||||||||||||
| Average expected term (years) | 1.3 | 1.3 | ||||||||||||
| Expected stock price volatility | 72.80% - 130.95% | 62.98% - 91.78% | ||||||||||||
| Risk-free interest rate | 4.74% - 5.33% | 0.60% - 3.45% | ||||||||||||
| Dividend yield | —% | —% | ||||||||||||
Intangible Assets and Goodwill
Intangible assets
Our intangible assets, which are comprised primarily of acquired developed technology, are considered to be finite-lived and are amortized on a straight-line basis over their estimated useful lives. Estimating the useful lives of our intangible assets requires considerable judgment. In determining the estimated useful lives, management considers factors such as historical experience, industry and regulatory factors, competition, patent expirations and commercial plans. If new information becomes available in future periods, we may be required to revise our estimated useful lives. If the revised useful lives are shorter than originally estimated, our future amortization expense will increase.
Goodwill
Our goodwill is not amortized but is tested for impairment on an annual basis or whenever events or changes in circumstances indicate that it may be impaired. We perform annual impairment reviews of our goodwill balance during the fourth quarter of each year, or more frequently if management believes indicators of impairment exist. We may perform a qualitative assessment to determine if it is necessary to perform a quantitative impairment test. If we determine that a quantitative impairment test is necessary, we apply the guidance in Accounting Standards Update (“ASU”) No. 2017-04, Intangibles—Goodwill and Other (Topic 350): Simplifying the Test for Goodwill Impairment, by comparing the fair value of the reporting unit to its carrying value, including the goodwill. If the carrying value exceeds the fair value, we recognize an impairment loss for the amount by which the carrying value exceeds fair value, up to the total amount of goodwill allocated to the reporting unit. We did not incur any goodwill impairment losses in any of the periods presented. We have concluded that our business is comprised of a single reporting unit. For our annual impairment test for the year ended December 31, 2023, we elected to bypass the qualitative assessment and proceeded directly to the quantitative assessment by comparing our reporting unit’s fair value to its carrying value. Since we have a single reporting unit, fair value of the reporting unit was measured at our total market capitalization on the impairment test date based on the closing price of our common stock. Factors that could result in an impairment of goodwill in the future include declines in the price of our common stock, increased competition, changes in macroeconomic developments and unfavorable government or regulatory developments.
On June 2, 2023, a MAC finalized an LCD pursuant to which the DecisionDx-SCC test would no longer be covered by Medicare effective July 17, 2023. On June 5, 2023, our stock price decreased significantly and did not recover before June 30, 2023. In response to this trigger, we tested goodwill for impairment at June 30, 2023. We elected to bypass the optional qualitative assessment and proceeded directly to the quantitative assessment. In conducting our interim test, we concluded that our business consists of a single reporting unit. To measure the fair value of our reporting unit, we used a market approach whereby we calculated our total market capitalization on the impairment test date, based on the closing price of our common stock as reported on the Nasdaq Global Market, and applied a reasonable control premium. The control premium was based on an analysis of control premiums paid in recent acquisitions of companies in the same or similar industry as us. Our impairment test indicated that the fair value of
93
| Years Ended December 31, | ||||
| 2019 | 2018 | |||
| Average expected term | 1.2 years | Not applicable | ||
| Expected stock price volatility | 61.12% - 68.89% | Not applicable | ||
| Risk-free interest rate | 1.56% - 1.80% | Not applicable | ||
| Dividend yield | —% | Not applicable | ||
On October 1, 2023, we performed our annual impairment test which indicated that the fair value of our reporting unit exceeded its carrying value by 34% and therefore no impairment was indicated. We continued to monitor for additional indicators of impairment through December 31, 2023 and did not observe any factors that could result in an impairment of goodwill.
Recent Accounting Pronouncements
In November 2023, the FASB issued ASU No. 2023-07, Segment Reporting (Topic 280)—Improvements to Reportable Segment Disclosures (“ASU 2023-07”), which require public companies disclose significant segment expenses and other segment items on an annual and interim basis and to provide in interim periods all disclosures about a reportable segment’s profit or loss and assets that are currently required annually. The guidance is effective for public entities for fiscal years beginning after December 15, 2023, and interim periods within fiscal years beginning after December 15, 2024. Early adoption is permitted. The guidance is applied retrospectively to all periods presented in the accompanying notes to our financial statements, included in this Annual Report on Form 10-K for a discussion of recent accounting pronouncements.
unless it is impracticable. We are an emerging growth company withincurrently evaluating the meaning of the JOBS Act. Section 107(b) of the JOBS Act provides that an emerging growth company can leverage the extended transition period, provided in Section 102(b) of the JOBS Act, for complying with new or revised accounting standards. Thus, an emerging growth company can delay the adoption of new or revised accounting standards thatimpact this update will have different effective dates for public and private companies until those standards apply to private companies. We have elected to use this extended transition period and, as a result,on ourconsolidated financial statements mayand disclosures.
We have evaluated all other recently issued, but not be comparable to companiesyet effective, accounting pronouncements and do not believe that comply with public company effective dates. We also intend to relythese accounting pronouncements will have any material impact on other exemptions provided by the JOBS Act, including without limitation, not being required to comply with the auditor attestation requirements of Section 404(b) of Sarbanes-Oxley.our consolidated financial statements or disclosures upon adoption.
Item 7A. QualitativeQuantitative and QuantitativeQualitative Disclosures About Market Risk.
As a smaller reporting company, we are not required to provide the information required by this Item.
Item 8. Financial Statements and Supplementary Data.
The financial statements and supplementary data required by this item are included after the Signature page of this Annual Report on Form 10-K beginning on page F-1.
Item 9. Changes in and Disagreements With Accountants on Accounting and Financial Disclosure.
None.
Item 9A. Controls and Procedures.
Evaluation of Disclosure Controls and Procedures
We maintain “disclosure controls and procedures,” as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act, that are designed to ensure that information required to be disclosed in the reports that we file or submit under the Exchange Act is (1) recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms and (2) accumulated and communicated to our management, including our principal executive officer and principal financial officer, to allow timely decisions regarding required disclosure. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures.
Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, evaluated the effectiveness of our disclosure controls and procedures as of December 31, 2019.2023. Based upon the evaluation, our Chief Executive Officer and Chief Financial Officer concluded that, as of such date,December 31, 2023, our disclosure controls and procedures were effective at the reasonable assurance level of December 31, 2019.
Management’s Report on Internal Control over Financial Reporting
94
officer and principal financial officer, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of consolidated financial statements for external purposes in accordance with U.S. GAAP.
As of December 31, 2023, our management assessed the effectiveness of our internal control over financial reporting using the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission in Internal Control-Integrated Framework (2013 Framework). Based on this assessment, our management concluded that, as of December 31, 2023, our internal control over financial reporting was effective based on those criteria.
Our independent registered public accounting firm, due to a transition period established by rulesKPMG LLP, has audited the effectiveness of the SEC for newly public companies.our internal control over financial reporting as of December 31, 2023, as stated in its report contained in Item 15 of this Annual Report on Form 10-K.
Changes in Internal Control over Financial Reporting
Item 9B. Other Information.
On November 5, 2023, Frank Stokes, our Chief Financial Officer, adopted a Rule 10b5-1 trading arrangement providing for the sale from time to time of an aggregate of up to 43,309 shares of our common stock. The trading arrangement is intended to satisfy the affirmative defense in Rule 10b5-1(c). The duration of the trading arrangement will be from February 5, 2024 until the earlier of the completion of all transactions under the trading arrangement or the termination of the plan. On November 6, 2023, Frank Stokes terminated a trading arrangement for the sale of the Company's common stock. Such trading arrangement was not intended to satisfy the affirmative defense conditions of Securities Exchange Act Rule 10b5-1(c), but complied with the then-applicable requirements of Rule 10b5-1(c) when adopted in May 11, 2023. Such trading arrangement provided for the sale of up to 4,000 shares between August 15, 2023 and the completion of all the transactions under the trading arrangement.
On December 5, 2023, Derek Maetzold, our Chief Executive Officer, adopted a Rule 10b5-1 trading arrangement providing for the sale from time to time of an aggregate of up to 228,103 shares of our common stock. The trading arrangement is intended to satisfy the affirmative defense in Rule 10b5-1(c). The duration of the trading arrangement will be from March 7, 2024 through September 6, 2024.
No other officers or directors, as defined in Rule 16a-1(f), adopted and/or terminated a Rule 10b5-1 trading arrangement or a non-Rule 10b5-1 trading arrangement, as defined in Regulation S-K Item 408, during the last fiscal quarter.
Item 9C. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections.
Not applicable.
95
PART III
Certain information required by Part III is omitted from this Annual Report on Form 10-K since we intend to file our definitive proxy statement for our 2023 Annual Meeting of Stockholders (the “Proxy Statement”) pursuant to Regulation 14A of the Exchange Act, not later than 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K, and certain information to be included in the Proxy Statement is incorporated herein by reference.
Item 10. Directors, Executive Officers and Corporate Governance.
The information required by this item and not set forth below will be contained in our definitive proxy statementis to be filed with the Securities and Exchange Commissionincluded in connection with our 2020 Annual Meeting of Stockholders, or the Proxy Statement whichas follows:
•The information relating to our executive officers is expected to be filed not later than 120 days afterincluded in the endsection entitled “Executive Officers,”
•The information relating to our directors and nominees for director is to be included in the section entitled “Proposal 1: Election of Directors,”
•The information relating to our fiscal year ended December 31, 2019,audit committee and audit committee financial expert is to be included in the section entitled “Information Regarding the Board of Directors and Corporate Governance,” and
•If required, the information regarding delinquent reports under Section 16(a) of the Exchange Act is to be included in the section entitled “Delinquent Section 16(a) Reports,”
•The information relating to our insider trading policies is to be included in the section entitled “Clawback Policy.”
Such information is to be included in the Proxy Statement and is incorporated herein by reference.
We have adopted a written Code of Business Conduct and Ethics or (“Ethics Code,Code”) that applies to all officers, directors and employees, including our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions. The Ethics Code is available on our website at www.CastleBiosciences.com. If we makeever were to amend or waive any substantive amendmentsprovision that applies to the Ethics Codeour principal executive officer, principal financial officer, principal accounting officer or grant any waiver from a provision of the Ethics Codeperson performing similar functions, we intend to satisfy our disclosure obligations, if any, with respect to any executive officersuch waiver or director, we will promptly disclose the nature of the amendment or waiverby posting such information on our website, or inrather than by filing a Current Report on Form 8-K.
Item 11. Executive Compensation.
The information required by this item willis to be set forthincluded in the Proxy Statement under the section entitled “Executive Compensation” and is incorporated herein by reference.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
The information required by this item willwith respect to equity compensation plans is to be set forthincluded in the Proxy Statement under the section entitled “Equity Compensation Plan Information” and the information required by this item with respect to security ownership of certain beneficial owners and management is to be included in the Proxy Statement under the section entitled “Security Ownership of Certain Beneficial Owners and Management” and in each case is incorporated herein by reference.
Item 13. Certain Relationships and Related Transactions, and Director Independence.
The information required by this item willis to be set forthincluded in the Proxy Statement under the sections entitled “Transactions with Related Persons and Indemnifications—Related Person Transactions Policy and Procedures” and is incorporated herein by reference.
Item 14. Principal AccountingAccountant Fees and Services.
Our independent registered public accounting firm is KPMG LLP, Houston, TX, Audit Firm ID: 185.
The information required by this item willis to be set forthincluded in the Proxy Statement under the section entitled “Proposal 2: Ratification of Selection of Independent Registered Public Accounting Firm” and is incorporated herein by reference.
96
PART IV
Item 15. Exhibits,Exhibit and Financial Statement Schedules.
(a)(1) Financial Statements.
The consolidated financial statements and supplementary data required by this item are included after the Signature page of this Annual Report on Form 10-K beginning on page F-1.
(a)(2) Financial Statement Schedules.
All schedules have been omitted because they are not required or because the required information is given in the Consolidated Financial Statements or Notes thereto.
(a)(3) Exhibits.
The exhibits listed in the Exhibit Index below are filed or incorporated by reference as part of this Annual Report.Report on Form 10-K.
Exhibit Index
| Exhibit Number | Description of document | |||||||
| 2.2^† | ||||||||
| 3.1 | ||||||||
| 3.2 | ||||||||
| 4.2 | ||||||||
| 4.3 | ||||||||
| 10.2+ | ||||||||
| 10.3+ | ||||||||
| 10.4+ | ||||||||
| 10.5+ | ||||||||
| 10.6+ | ||||||||
97
| Exhibit Number | Description of document | |||||||
| 10.7+ | ||||||||
| 10.8+ | ||||||||
| 10.9+ | ||||||||
| 10.12+ | ||||||||
| 10.13+ | ||||||||
| 10.14+ | ||||||||
| 10.15+ | ||||||||
| 10.18+ | ||||||||
| 10.19+ | ||||||||
| 10.20*+† | ||||||||
| 10.21 | ||||||||
| 10.25† | ||||||||
98
| 10.35† | ||||||||||
| 10.36 | ||||||||||
| 10.38*† | ||||||||||
| 10.39# | ||||||||||
| 23.1* | ||||||||||
| 31.1* | ||||||||||
| 31.2* | ||||||||||
| 32.1** | ||||||||||
| 101.INS* | Inline XBRL Instance | |||||||||
| 101.SCH* | Inline XBRL Taxonomy Extension | |||||||||
| 101.CAL* | Inline XBRL Taxonomy Extension Calculation | |||||||||
99
| Exhibit Number | Description of document | |||||||
| 101.DEF* | Inline XBRL Taxonomy Extension Definition | |||||||
| 101.LAB* | Inline XBRL Taxonomy Extension Label | |||||||
| 101.PRE* | Inline XBRL Taxonomy Extension Presentation | |||||||
| 104* | Cover Page Interactive Data File (embedded within the Inline XBRL and contained in Exhibit 101). | |||||||
** Furnished herewith.
+ Indicates management contract or compensatory plan.
# Pursuant to Item 601(b)(10) of Regulation S-K, certain portions of this exhibit have been omitted (indicated by “[***]”) because the Company has determined that the information is not material and is the type that the Company treats as private or confidential.
† Certain schedules or exhibits have been omitted pursuant to Item 601(a)(5) of Regulation S-K. A copy of any omitted schedule or exhibit will be furnished to the SEC upon request; provided, however, that we may request confidential treatment pursuant to Rule 24b-2 of the Exchange Act for any schedule or exhibit so furnished.
^ Pursuant to Item 601(b)(2) of Regulation S-K, certain portions of this exhibit have been omitted (indicated by “[***]”) because the Company has determined that the information is not material and is the type that the Company treats as private or confidential.
Item 16. Form 10-K Summary.
None.
100
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized, on March 10, 2020.
authorized.
| CASTLE BIOSCIENCES, INC. | |||||||||||||||||
| Date: | February 28, 2024 | By: | /s/ Derek J. Maetzold | ||||||||||||||
Derek J. Maetzold President and Chief Executive Officer (Principal Executive Officer) | |||||||||||||||||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the Registrant and in the capacity and on the dates indicated.
| SIGNATURE | TITLE | DATE | ||||||||||||
| /s/ Derek J. Maetzold | President, Chief Executive Officer and Director | |||||||||||||
| (Derek J. Maetzold) | (Principal Executive Officer) | |||||||||||||
| /s/ Frank Stokes | Chief Financial Officer | |||||||||||||
| (Frank Stokes) | (Principal Financial and Accounting Officer) | |||||||||||||
| /s/ Daniel M. Bradbury | ||||||||||||||
| (Daniel M. Bradbury) | ||||||||||||||
| /s/ | Member of the Board of Directors | |||||||||||||
| ( | ||||||||||||||
| /s/ Kimberlee S. Caple | Member of the Board of Directors | |||||||||||||
| ( | ||||||||||||||
| /s/ G. Bradley Cole | Member of the Board of Directors | |||||||||||||
| (G. Bradley Cole) | ||||||||||||||
| /s/ | Member of the Board of Directors | |||||||||||||
| ( | ||||||||||||||
| /s/ | Member of the Board of Directors | |||||||||||||
| ( | ||||||||||||||
| /s/ Tiffany P. Olson | Member of the Board of Directors | February 28, 2024 | ||||||||||||
| (Tiffany P. Olson) | ||||||||||||||
101
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
F-1
Report of Independent Registered Public Accounting Firm
To the Stockholders and Board of Directors
Castle Biosciences, Inc.:
We have audited the accompanying consolidated balance sheets of Castle Biosciences, Inc. and subsidiaries (the Company) as of December 31, 20192023 and 2018,2022, the related consolidated statements of operations, and comprehensive income (loss), convertible preferred stock andloss, stockholders’ equity, (deficit), and cash flows for each of the years in the two-year period ended December 31, 2019,2023, and the related notes (collectively, the consolidated financial statements). We also have audited the Company’s internal control over financial reporting as of December 31, 2023, based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of the Company as of December 31, 20192023 and 2018,2022, and the results of its operations and its cash flows for each of the years in the two-year period ended December 31, 2019,2023, in conformity with U.S. generally accepted accounting principles. Also in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2023 based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission.
Basis for OpinionOpinions
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the auditaudits to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. fraud, and whether effective internal control over financial reporting was maintained in all material respects.
Our audits of the consolidated financial statements included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinion.opinions.
Definition and Limitations of Internal Control Over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
F-2
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Critical Audit Matter
The critical audit matter communicated below is a matter arising from the current period audit of the consolidated financial statements that was communicated or required to be communicated to the audit committee and that: (1) relates to accounts or disclosures that are material to the consolidated financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of a critical audit matter does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the accounts or disclosures to which it relates.
Measurement of test revenue
As discussed in Notes 2 and 3 of the consolidated financial statements, test revenue is recognized when the performance obligation is satisfied, upon delivery of the test report. The amount of revenue recognized reflects the amount of consideration to which the Company expects to be entitled and considers the effects of variable consideration. The measurement of test revenue is determined by contractually established rates as well as historical average collection rates by test type and payor category taking into consideration the range of possible outcomes, the predictive value of the Company’s past experiences, the time period of when uncertainties expect to be resolved and the amount of consideration that is susceptible to factors outside of the Company’s influence, such as the judgment and actions of third parties. The Company reported net revenues of $219.8 million for the year ended December 31, 2023, a portion of which related to test revenue.
We identified the evaluation of the measurement of test revenue as a critical audit matter. Evaluating the measurement of test revenue, specifically the estimation of the amount of test revenue expected to be collectible related to commercial payors and certain others, required especially complex and subjective auditor judgment.
The following are the primary procedures we performed to address this critical audit matter. We evaluated the design and tested the operating effectiveness of certain internal controls related to the estimation of the expected collectible test revenue related to commercial payors and certain others. This included controls related to management’s review of the assumptions and inputs used in the determination of test revenue expected to be collectible from commercial payors and certain others. For a selection of tests, we evaluated certain historical data inputs that are used in management’s model to estimate test revenue expected to be collectible by comparing the data from the model to physician test requisition forms, proof of delivery, and cash collection activity. These data inputs included collection activity, amounts of historical claims, insurance payor categories, test dates, test types, claim aging categories, and revenue recognition dates. In addition, we inquired of the individuals who are responsible for monitoring and tracking the status of anticipated collections to understand the progress of these activities and any impact to management’s estimates.
/s/ KPMG LLP
We have served as the Company’s auditor since 2018.
F-3
CASTLE BIOSCIENCES, INC. BALANCE SHEETS (in thousands, except share and per share data) | |||||||
| December 31, | |||||||
| 2019 | 2018 | ||||||
| ASSETS | |||||||
| Current Assets | |||||||
| Cash and cash equivalents | $ | 98,845 | $ | 4,479 | |||
| Accounts receivable, net | 14,648 | 12,090 | |||||
| Inventory | 1,237 | 882 | |||||
| Prepaid expenses and other current assets | 1,951 | 675 | |||||
| Total current assets | 116,681 | 18,126 | |||||
| Long-term accounts receivable, net | 870 | 2,532 | |||||
| Property and equipment, net | 2,060 | 1,529 | |||||
| Intangible assets, net | — | 4 | |||||
| Other assets – long-term | 135 | 214 | |||||
| Total assets | $ | 119,746 | $ | 22,405 | |||
LIABILITIES, CONVERTIBLE PREFERRED STOCK AND STOCKHOLDERS’ EQUITY (DEFICIT) | |||||||
| Current Liabilities | |||||||
| Accounts payable | $ | 1,865 | $ | 1,451 | |||
| Accrued compensation | 5,779 | 4,571 | |||||
| Other accrued liabilities | 1,812 | 715 | |||||
| Current portion of long-term debt | 5,833 | — | |||||
| Total current liabilities | 15,289 | 6,737 | |||||
| Long-term debt | 19,289 | 24,500 | |||||
| Preferred stock warrant liability | — | 1,194 | |||||
| Deferred rent liability | 55 | 44 | |||||
| Total liabilities | 34,633 | 32,475 | |||||
| Commitments and Contingencies (Note 11) | |||||||
| Convertible Preferred Stock | |||||||
| Convertible preferred stock Series C, $0.001 par value, zero and 503,056 shares authorized as of December 31, 2019 and 2018, respectively; zero and 503,056 shares issued and outstanding as of December 31, 2019 and 2018, respectively; $0 and $2,417 aggregate liquidation preference as of December 31, 2019 and 2018, respectively. | — | 1,501 | |||||
| Redeemable convertible preferred stock Series A, B, D, E-1, E-2, E-2A, E-3 and F, $0.001 par value, zero and 9,640,493 shares authorized as of December 31, 2019 and 2018, respectively; zero and 9,456,775 shares issued and outstanding as of December 31, 2019 and 2018, respectively; $0 and $57,570 aggregate liquidation preference as of December 31, 2019 and 2018, respectively. | — | 44,995 | |||||
| Stockholders’ Equity (Deficit) | |||||||
| Common stock, $0.001 par value; 200,000,000 and 15,102,000 shares authorized as of December 31, 2019 and 2018, respectively; 17,130,907 and 1,916,224 shares issued and outstanding as of December 31, 2019 and 2018, respectively. | 17 | 2 | |||||
| Preferred stock, $0.001 par value; 10,000,000 and zero shares authorized as of December 31, 2019 and 2018, respectively; no shares issued and outstanding as of December 31, 2019 and 2018. | — | — | |||||
| Additional paid-in capital | 137,308 | 921 | |||||
| Accumulated deficit | (52,212 | ) | (57,489 | ) | |||
| Total stockholders’ equity (deficit) | 85,113 | (56,566 | ) | ||||
| Total liabilities, convertible preferred stock and stockholders’ equity (deficit) | $ | 119,746 | $ | 22,405 | |||
CASTLE BIOSCIENCES, INC.
CONSOLIDATED BALANCE SHEETS
(in thousands, except share and per share data)
| December 31, | |||||||||||
| 2023 | 2022 | ||||||||||
| ASSETS | |||||||||||
| Current Assets | |||||||||||
| Cash and cash equivalents | $ | 98,841 | $ | 122,948 | |||||||
| Marketable investment securities | 144,258 | 135,677 | |||||||||
| Accounts receivable, net | 38,302 | 23,476 | |||||||||
| Inventory | 7,942 | 3,980 | |||||||||
| Prepaid expenses and other current assets | 6,292 | 6,207 | |||||||||
| Total current assets | 295,635 | 292,288 | |||||||||
| Long-term accounts receivable, net | 1,191 | 1,087 | |||||||||
| Property and equipment, net | 25,433 | 14,315 | |||||||||
| Operating lease assets | 12,306 | 12,181 | |||||||||
| Goodwill and other intangible assets, net | 117,335 | 126,348 | |||||||||
| Other assets – long-term | 1,440 | 1,110 | |||||||||
| Total assets | $ | 453,340 | $ | 447,329 | |||||||
LIABILITIES AND STOCKHOLDERS’ EQUITY | |||||||||||
| Current Liabilities | |||||||||||
| Accounts payable | $ | 10,268 | $ | 4,731 | |||||||
| Accrued compensation | 28,945 | 24,358 | |||||||||
| Operating lease liabilities | 1,137 | 1,777 | |||||||||
| Other accrued and current liabilities | 7,317 | 5,262 | |||||||||
| Total current liabilities | 47,667 | 36,128 | |||||||||
| Noncurrent operating lease liabilities | 14,173 | 11,533 | |||||||||
| Deferred tax liability | 206 | 428 | |||||||||
| Other liabilities | 25 | 90 | |||||||||
| Total liabilities | 62,071 | 48,179 | |||||||||
| Commitments and Contingencies (Note 12) | |||||||||||
| Stockholders’ Equity | |||||||||||
| Preferred stock, $0.001 par value; 10,000,000 shares authorized as of December 31, 2023 and 2022; no shares issued and outstanding as of December 31, 2023 and 2022. | — | — | |||||||||
| Common stock, $0.001 par value; 200,000,000 authorized as of December 31, 2023 and 2022; 27,410,532 and 26,553,681 shares issued and outstanding as of December 31, 2023 and 2022, respectively. | 27 | 27 | |||||||||
| Additional paid-in capital | 609,477 | 560,409 | |||||||||
| Accumulated deficit | (218,371) | (160,905) | |||||||||
| Accumulated other comprehensive income (loss) | 136 | (381) | |||||||||
| Total stockholders’ equity | 391,269 | 399,150 | |||||||||
| Total liabilities and stockholders’ equity | $ | 453,340 | $ | 447,329 | |||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-4
CASTLE BIOSCIENCES, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
(in thousands, except per share data)
| Years Ended December 31, | ||||||||||||||
| 2023 | 2022 | |||||||||||||
| NET REVENUES | $ | 219,788 | $ | 137,039 | ||||||||||
| OPERATING EXPENSES AND OTHER OPERATING INCOME | ||||||||||||||
| Cost of sales (exclusive of amortization of acquired intangible assets) | 44,982 | 32,009 | ||||||||||||
| Research and development | 53,618 | 44,903 | ||||||||||||
| Selling, general and administrative | 180,152 | 143,003 | ||||||||||||
| Amortization of acquired intangible assets | 9,013 | 8,266 | ||||||||||||
| Change in fair value of contingent consideration | — | (18,287) | ||||||||||||
| Total operating expenses, net | 287,765 | 209,894 | ||||||||||||
| Operating loss | (67,977) | (72,855) | ||||||||||||
| Interest and other non-operating income | 10,623 | 3,968 | ||||||||||||
| Interest expense | (11) | (17) | ||||||||||||
| Loss before income taxes | (57,365) | (68,904) | ||||||||||||
| Income tax expense (benefit) | 101 | (1,766) | ||||||||||||
| Net loss | $ | (57,466) | $ | (67,138) | ||||||||||
| Loss per share, basic and diluted | $ | (2.14) | $ | (2.58) | ||||||||||
| Weighted-average shares outstanding, basic and diluted | 26,802 | 26,054 | ||||||||||||
CASTLE BIOSCIENCES, INC. STATEMENTS OF OPERATIONS AND COMPREHENSIVE INCOME (LOSS) (in thousands, except per share data) | ||||||||
| Years Ended December 31, | ||||||||
| 2019 | 2018 | |||||||
| NET REVENUES | $ | 51,865 | $ | 22,786 | ||||
| COST OF SALES | 7,310 | 5,297 | ||||||
| Gross margin | 44,555 | 17,489 | ||||||
| OPERATING EXPENSES | ||||||||
| Research and development | 7,385 | 4,854 | ||||||
| Selling, general and administrative | 29,842 | 16,471 | ||||||
| Total operating expenses | 37,227 | 21,325 | ||||||
| Operating income (loss) | 7,328 | (3,836 | ) | |||||
| Interest income | 312 | 24 | ||||||
| Interest expense | (4,571 | ) | (2,274 | ) | ||||
| Gain on extinguishment of debt (Note 7) | 5,213 | — | ||||||
| Other expense, net | (2,933 | ) | (272 | ) | ||||
| Income (loss) before income taxes | 5,349 | (6,358 | ) | |||||
| Income tax expense | 72 | 9 | ||||||
| Net income (loss) and comprehensive income (loss) | 5,277 | (6,367 | ) | |||||
| Convertible preferred stock cumulative dividends | 2,156 | 3,577 | ||||||
| Accretion of redeemable convertible preferred stock to redemption value | 130 | 219 | ||||||
| Net income (loss) and comprehensive income (loss) attributable to common stockholders | $ | 2,991 | $ | (10,163 | ) | |||
| Earnings (loss) per share attributable to common stockholders: | ||||||||
| Basic | $ | 0.35 | $ | (5.33 | ) | |||
| Diluted | $ | (0.21 | ) | $ | (5.33 | ) | ||
| Weighted-average shares outstanding: | ||||||||
| Basic | 8,584 | 1,906 | ||||||
| Diluted | 8,658 | 1,906 | ||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-5