UNITED STATES SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
| | | | | |
☒þ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 20222023
OR
| | | | | |
| ☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from ____ to ____
Commission file number: 001-37798
Selecta Biosciences,Cartesian Therapeutics, Inc.
(Exact name of registrant as specified in its charter)
| | | | | | | | | | | |
| Delaware | 26-1622110 | |
| (State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) | |
| 65 Grove Street, Watertown, MA704 Quince Orchard Road, Gaithersburg, MD | 0247220878 | |
| (Address of principal executive offices) | (Zip Code) | |
(617) 923-1400
Registrant’s telephone number, including area code
| | | | | | | | |
| Securities registered pursuant to Section 12(b) of the Act: |
| | |
| Title of each class | Trading Symbol(s) | Name of each exchange on which registered |
| Common Stock, $0.0001 par value per share | SELBRNAC | The Nasdaq Stock Market LLC |
| | |
| Securities registered pursuant to Section 12(g) of the Act: |
| | |
| Title of each class |
| Contingent Value Rights |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No ýþ
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No ýþ
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports); and (2) has been subject to such filing requirements for the past 90 days. Yes ☒þ No o
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☒þ No o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| | | | | | | | | | | | | | | | | | | | |
| Large accelerated filer | ☐ | | Accelerated filer | ☐þ | |
| Non-accelerated filer | ☒☐ | | Smaller reporting company | ☒þ | |
| | | | Emerging growth company | ☐ | |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attested to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. oþ
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. o
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). o
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒þ
The aggregate market value of common stock held by non-affiliates of the registrant based on the closing price of the registrant’s common stock as reported on the Nasdaq Stock Market on June 30, 2022,2023, the last business day of the registrant’s most recently completed second quarter, was $149,708,988.$128,805,952.
As of February 24, 2023March 1, 2024, the registrant had 153,318,915161,948,618 shares of common stock, par value $0.0001 per share, outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s definitive Proxy Statement relating to its 20222024 Annual Meeting of Stockholders to be filed with the Securities and Exchange Commission are incorporated by reference into Part III of this Annual Report on Form 10-K.
FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K, or the Annual Report, contains forward-looking statements. We intend such forward-looking statements to be covered by the safe harbor provisions for forward-looking statements contained in Section 27A of the Securities Act of 1933, as amended, or the Securities Act, and Section 21E of the Securities Exchange Act of 1934, as amended, or the Exchange Act. All statements other than statements of historical facts contained in this Annual Report, including statements regarding our future results of operations and financial position, business strategy, prospective products, product approvals, research and development costs, timing and likelihood of success, the plans and objectives of management for future operations and future results of anticipated products, the impact of the resurgence of the COVID-19 pandemic or emergence of another pandemic on our business and operations and our future financial results, and the period over which we estimate our existing cash and cash equivalents will be sufficient to fund our future operating expenses and capital expenditure requirements are forward-looking statements. These statements involve known and unknown risks, uncertainties and other important factors that may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by the forward-looking statements.
In some cases, you can identify forward-looking statements by terms such as “may,” “will,” “should,” “expect,” “plan,” “anticipate,” “could,” “intend,” “target,” “project,” “contemplate,” “believe,” “estimate,” “predict,” “potential”, or “continue” or the negative of these terms or other similar expressions. The forward-looking statements in this Annual Report are only predictions. We have based these forward-looking statements largely on our current expectations and projections about future events and financial trends that we believe may affect our business, financial condition and results of operations. These forward-looking statements speak only as of the date of this Annual Report and are subject to a number of important factors that could cause actual results to differ materially from those in the forward-looking statements, including the factors described under the sections in this Annual Report titled “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” as well as the following:
•our expectations regarding the conversion of the Series A Preferred Stock (as defined below) into our common stock;
•any future payouts under the contingent value right, or CVR, issued to our holders of record as of the close of business on December 4, 2023;
•our ability to achieve the expected benefits or opportunities and related timing with respect to the Merger (as defined below) or to monetize any of our legacy assets;
•our future results of operations and financial position, business strategy, and the length of time that we believe our existing cash resources will fund our operations;
•our market size and our potential growth opportunities;
•our preclinical and future clinical development activities;
•the efficacy and safety profile of our product candidates;
•the potential therapeutic benefits and economic value of our product candidates;
•the timing and results of preclinical studies and clinical trials;
•the expected impact of macroeconomic conditions, including inflation, increasing interest rates and volatile market conditions, current or potential bank failures;
•global events, including the ongoing conflicts between Russia and Ukraine and between Hamas and Israel and geopolitical tensions in China on our operations;
•the receipt and timing of potential regulatory designations, approvals and commercialization of product candidates;
•potential litigation related to the Merger (as defined below) instituted against us or our directors;
•our ability to prevent or minimize the effects of litigation and other contingencies;
•our ability to realize any benefits or opportunities from the Merger;
•our status as a preclinical and development-stage company and our expectation to incur losses in the future;future, and the possibility that we never achieve or maintain profitability;
•uncertainties with respect to our ability to access future capital needs and our need to raise additional funds;capital;
•our ability to build amaximize the value of our pipeline of product candidates and develop and commercialize such pipeline;candidates;
•our unproven approach to therapeutic intervention;
•our ability to enroll patients in clinical trials, timely and successfully complete those trials and receive necessary regulatory approvals;
•our ability to continue to grow our manufacturing capabilities and resources;
•our ability to manufacture our product candidates, which in some cases are manufactured on a patient-by-patient basis;
•our ability to access manufacturing facilities and to receive or manufacture sufficient quantities of our product candidates;
•our ability to maintain our existing or future collaborations or licenses;licenses and to seek new collaborations, licenses or partnerships;
•the continuing impact of resurgence of the COVID-19 pandemic on our operations, the continuity of our business, including our preclinical studies and clinical trials, and general economic conditions;
•our ability to protect and enforce our intellectual property rights;
•federal, state, and foreign regulatory requirements, including U.S. Food and Drug Administration, or FDA, regulation of our product candidates;
•our ability to obtain and retain key executives and attract and retain qualified personnel; and
•developments relating to our competitors and our industry, including the impact of government regulation; and
•our ability to successfully manage our growth.regulation.
Moreover, we operate in an evolving environment. New risks and uncertainties may emerge from time to time, and it is not possible for management to predict all risk and uncertainties.
You should read this Annual Report and the documents that we reference in this Annual Report completely and with the understanding that our actual future results may be materially different from what we expect. We qualify all of our forward-looking statements by these cautionary statements. Except as required by applicable law, we do not plan to publicly update or revise any forward-looking statements contained herein, whether as a result of any new information, future events, changed circumstances or otherwise.
PART I
Item 1. Business
Our Corporate History and Background
The Company (formerly known as Selecta Biosciences, Inc., or Selecta) was incorporated in Delaware on December 10, 2007, and is headquartered in Gaithersburg, Maryland. On November 13, 2023, the Company and the Delaware corporation which, immediately prior to the Merger (as defined below), was known as Cartesian Therapeutics, Inc., or Old Cartesian, entered into an Agreement and Plan of Merger, or the Merger Agreement, by and among the Company, Sakura Merger Sub I, Inc., a Delaware corporation and a wholly owned subsidiary of the Company, or First Merger Sub, Sakura Merger Sub II, LLC, a Delaware limited liability company and wholly owned subsidiary of the Company, or Second Merger Sub, and Old Cartesian. Pursuant to the Merger Agreement, and simultaneously with execution thereof, (i) First Merger Sub merged with and into Old Cartesian, pursuant to which Old Cartesian was the surviving corporation, or the First Step Surviving Corporation, and became a wholly owned subsidiary of the Company, or the First Merger, and (ii) immediately following the First Merger, Old Cartesian (as the First Step Surviving Corporation) merged with and into Second Merger Sub, pursuant to which Second Merger Sub was the surviving company, or the Surviving Company, and continued under the name “Cartesian Bio, LLC”, or the Second Merger and, together with the First Merger, the Merger. In connection with the Merger and pursuant to the Merger Agreement, the Company (which was known as Selecta Biosciences, Inc. until immediately prior to the Merger) changed its corporate name to Cartesian Therapeutics, Inc.
Overview
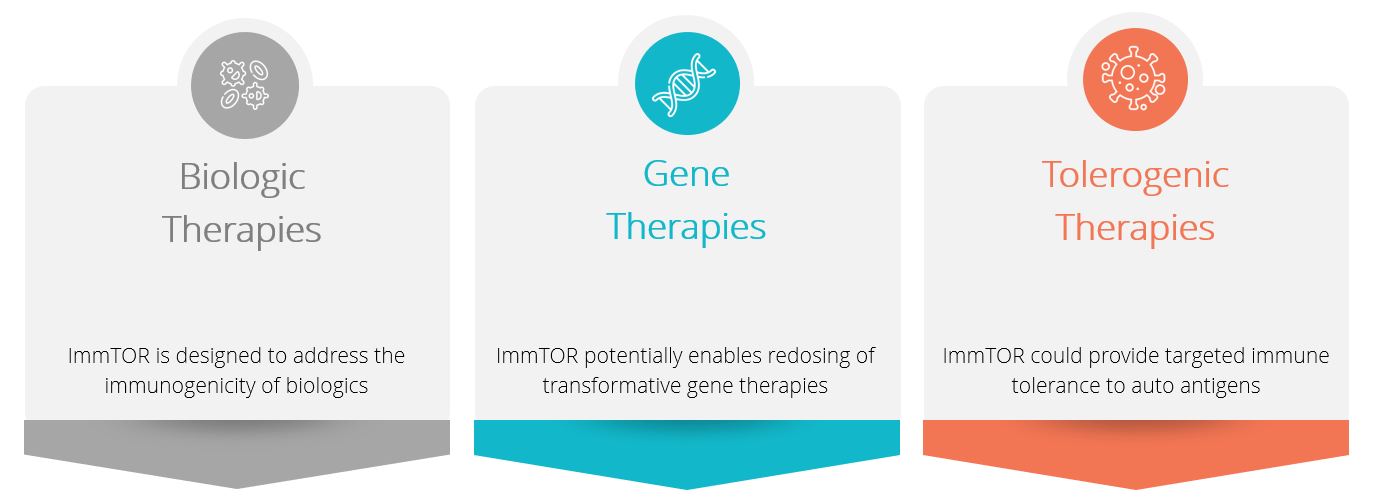
We are a clinical-stage biotechnology company leveraging our ImmTOR® platform to develop tolerogenicdeveloping mRNA cell therapies designed to selectively mitigate unwanted immune responses. With a proven ability to induce tolerance to highly immunogenic proteins, ImmTOR hasfor the potential to amplify the efficacytreatment of biologic therapies, including redosing of life-saving gene therapies, as well as restore the body's natural self-tolerance in autoimmune diseases. We have severalleverage our proprietary technology and partnered programsmanufacturing platform to introduce one or more mRNA molecules into cells to enhance their function. Unlike DNA, mRNA degrades naturally over time without integrating into the cell’s genetic material. Therefore, our mRNA cell therapies are distinguished by their capacity to be dosed repeatedly like conventional drugs, administered in our pipeline focused on enzyme therapies, gene therapies,an outpatient setting, and given without pre-treatment chemotherapy required with many conventional cell therapies. In an open-label Phase 2 clinical trial in patients with myasthenia gravis, or MG, a chronic autoimmune diseases.
In preclinical studies,disease that causes disabling muscle weakness and fatigue, we have observed that ImmTOR may have synergistic activityour lead product candidate, Descartes-08, generated a deep and durable clinical benefit.
Autoimmune diseases, where the immune system mistakenly attacks the body, are a family of more than 80 disorders. Autoimmune diseases are typically treated with interleukin-2,immunosuppressant medications, such as steroids. These treatments must be administered continually and carry risks, including infection, osteoporosis, and metabolic disease. Newer agents that block the complement pathway or IL-2, molecules that have been engineered toinhibit the neonatal Fc receptor, or FcRn, must also typically be selective for regulatory T cells, or Tregs. Treg-selective IL-2 molecules have been shown to transiently expand all pre-existing Tregs in preclinical and clinical studies conducted by others. We have observed in preclinical studies that the combination of ImmTOR, a Treg-selective IL-2 molecule and an antigen exhibited substantial synergistic activity in inducing and expanding antigen-specific Tregs beyond ImmTOR alone with evidence of enhanced durability of immune tolerance and the potential for ImmTOR dose sparing. This combination of ImmTOR with a Treg selective IL-2 molecule represents an evolution of the ImmTOR platform, which we call ImmTOR-IL™.administered continually. We believe this combination has the potential to bethere is a "first-in-class" antigen specific IL-2 therapysignificant unmet need for autoimmune disease.outpatient treatments, completed over a short period of time, that provide deep, durable clinical benefit.
We believe ImmTOR and ImmTOR-ILCell therapies have the potential to enable novel therapeutic modalitiesprovide this benefit, but conventional cell therapies that use DNA are associated with toxicities, including cytokine release syndrome, neurotoxicity, transformation to cancer, and death. Further, conventional cell therapies typically require pre-treatment with chemotherapy, which suppresses the immune system and increases the risk of infection, anemia, and neurotoxicity. As a result, conventional DNA cell therapies typically require close monitoring in autoimmune disease as well as enhance bothan inpatient setting, increasing the efficacytotal cost of care and safety of biologicgenerally limiting their reach to only the sickest patients.
We believe our mRNA cell therapies (including gene therapies) and improve product candidates under development. In clinical trials, ImmTOR has been observed to inhibit the formation of neutralizing antibodies to adeno-associated virus (AAV) capsids, potentially enabling re-dosing of gene therapies. Additionally, based on preclinical data in AAV gene therapies, we believe that ImmTOR hashave the potential to improve efficacy and safety by increasing transgene expression, reducing hepatic inflammation and inhibiting undesired immune responsesdeliver deep, durable clinical benefit to both the AAV capsid and the transgene product that can occur with the first dose of gene therapy.
In biologic therapies, clinical activity of ImmTOR in humans has been observed with pegadricase, a highly immunogenic pegylated uricase enzyme being developed for the treatmentbroad group of patients with chronic refractory gout. The combinationautoimmune diseases because they can be administered over a short period of ImmTORtime, in an outpatient setting, and pegadricase is currently being evaluated in a Phase 3 clinical trial that wewithout pre-treatment chemotherapy.
We are conducting on behalf ofleveraging our partner Swedish Orphan Biovitrum AB, or Sobi.
We intendproprietary technology and manufacturing platform, RNA Armory®, to pursue development ofdevelop mRNA cell therapies for autoimmune diseases where expansion of either all Tregsacross three modalities. Our mRNA CAR-T modality is a personalized approach that collects a patient’s T-cells and uses mRNA to introduce a chimeric antigen receptor, or antigen-specific Tregs has been shown to, or we believe is, likely to have a beneficial effect. We believe that ImmTOR and ImmTOR-IL have the potential to unlock antigen-specific therapies for autoimmune diseases and that ImmTOR-IL can further improve the efficacy and safety profile of biologic therapies beyond ImmTOR alone.
We have developed a portfolio of wholly owned and partnered candidates across gene therapies, biologic therapies and tolerogenic therapies for autoimmune diseases that leverage our ImmTOR platform. We plan to continue to develop proprietary compounds and pursue collaboration-driven development in certain disease areas, which could include strategic collaborations, out-licensing, and in-licensing transactions.
Our ImmTOR Platform
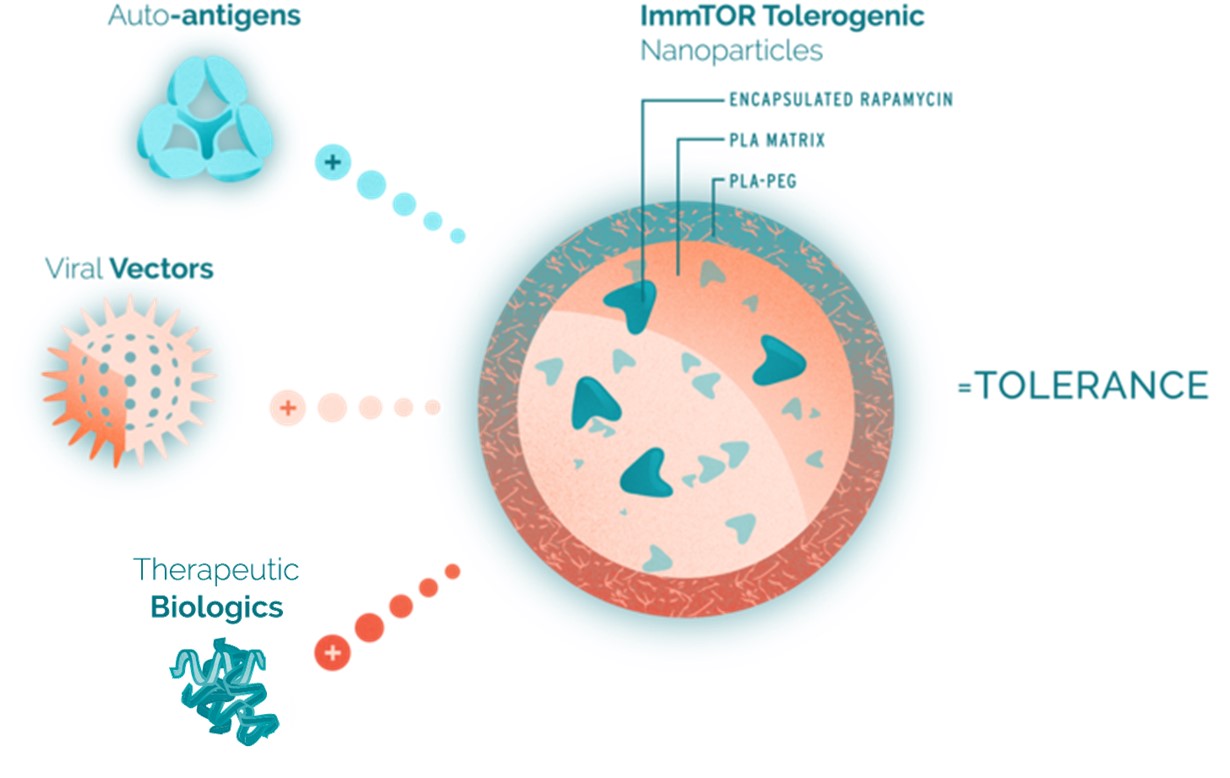
ImmTOR consists of biodegradable nanoparticles encapsulating the immunomodulator rapamycin. Rapamycin is the active ingredient of Rapamune, an immunosuppressant that has been used extensively in humans and is currently FDA-approved as a prophylaxis of organ rejection in kidney transplant patients aged 13 or older. The rapamycin component of ImmTOR is embedded in a matrix of synthetic polymers called poly(D,L-lactide), or PLA, and poly(D,L-lactide)-block-poly(ethylene-glycol), or PLA-PEG. PLA is part of the broader poly(lactic-co-glycolic acid), or PLGA, family of biodegradable polymers that have more than 30 years of commercial use and are formulation components in a number of approved products. Polyethylene glycol, or PEG, has been widely studied in clinical trials and is also a formulation component in many approved biologic products.
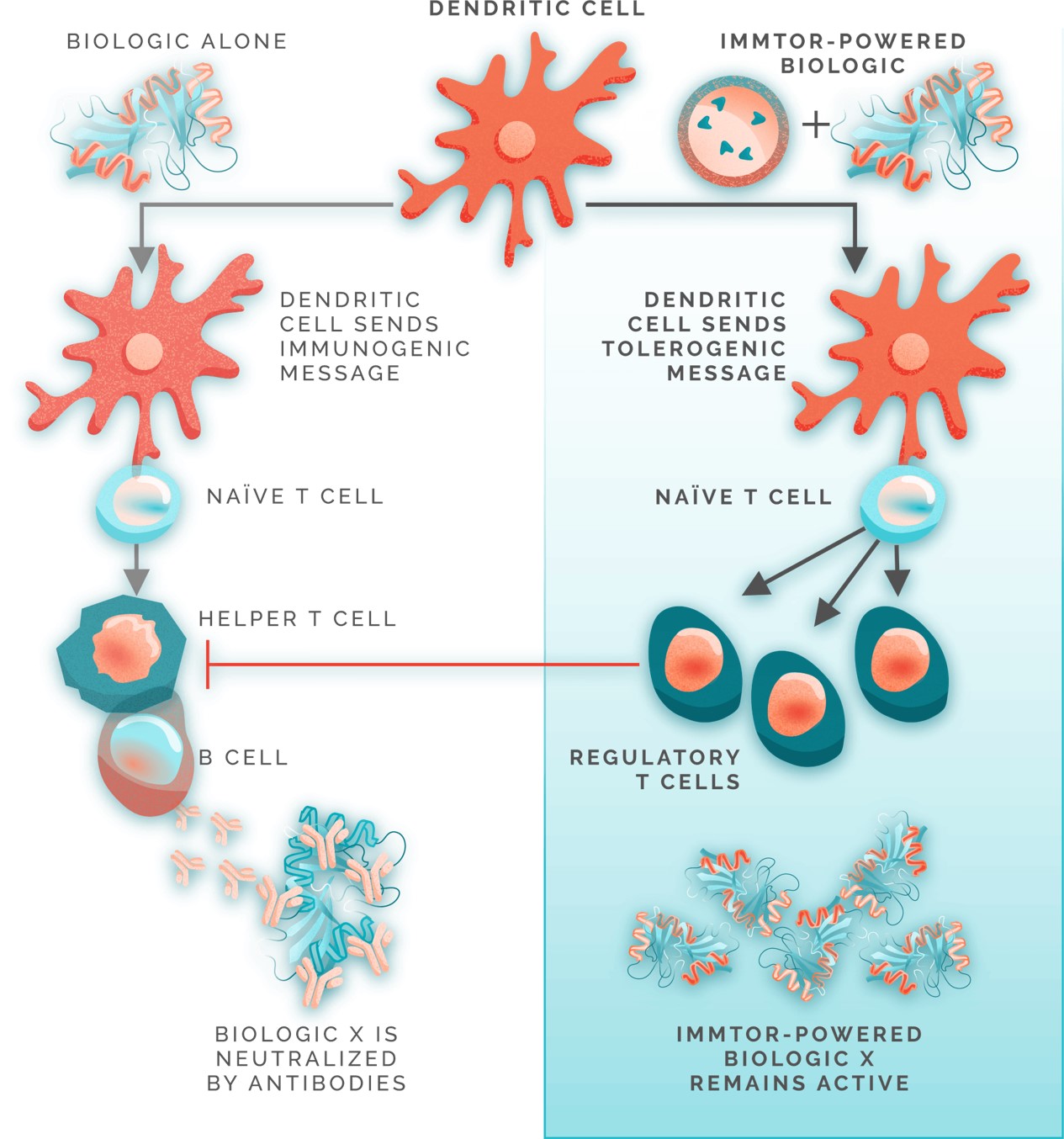
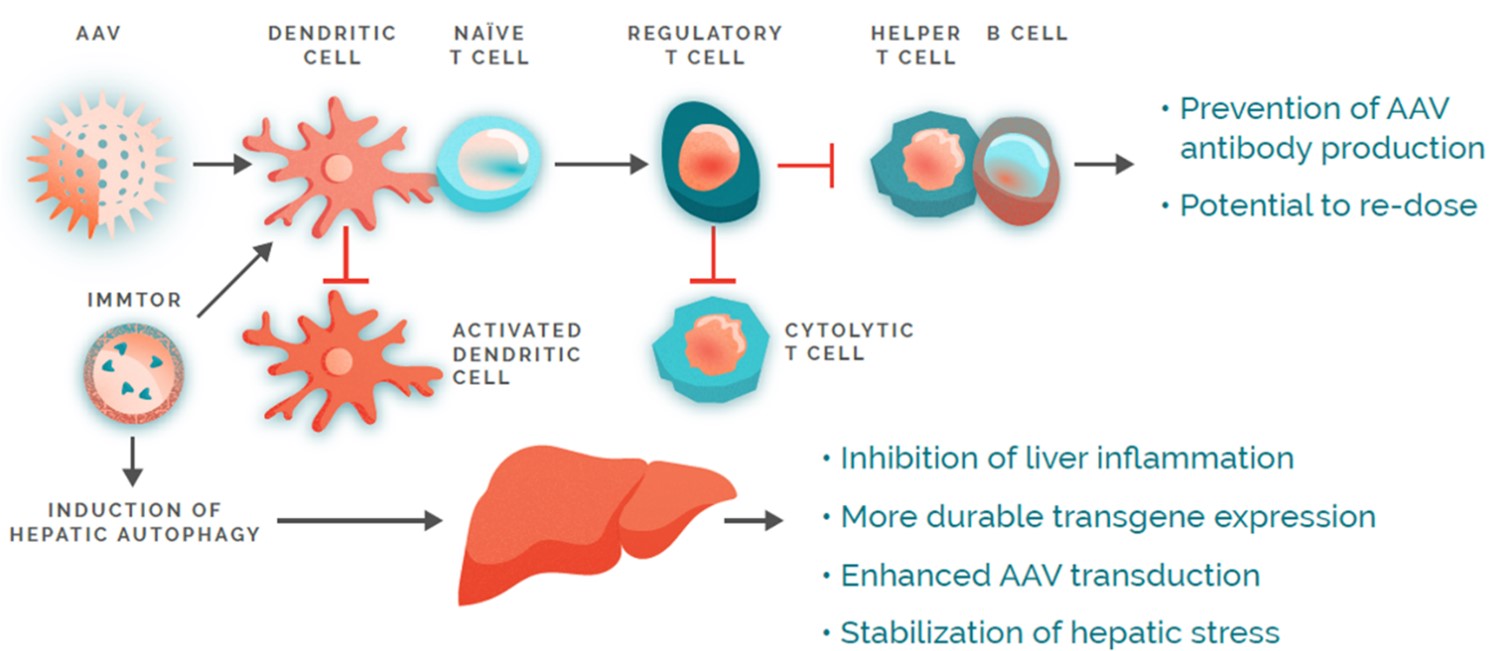
Our nanoparticles are designed to remain intact after injectionCAR, into the bodycell. The CAR redirects the T-cells to target and accumulate predominantlydestroy pathogenic self-reactive cells. Our mRNA MSC modality is an allogeneic approach that introduces one or more mRNAs into donor-sourced mesenchymal stem cells, or MSCs, enabling them to produce proteins that target key pathways involved in lymph nodes, the spleen,autoimmunity. These cells are banked and the liver, where immune responses are coordinated. The nanoparticles are designed to be processed by specialized immune cells, such as dendriticadministered off-the-shelf to any patient. Our mRNA in situ modality is designed to deliver mRNA into a patient’s lymph node to generate CAR-T cells and other antigen-presenting cells,proteins that initiate and regulate immune responses. ImmTOR is intended to induce a tolerogenic phenotype in these antigen-presenting cells, which then process and present co-administered antigens in a manner that results in the induction of antigen-specific regulatory T cells. To mitigate unwanted immune responses by inducing precision immune tolerance in the body, we administer our ImmTOR with the desired antigen, such as an auto-antigen in the case of an autoimmune disease, a viral vector in the case of our gene therapy program, or a therapeutic enzyme, as depicted in the figure below.
target autoimmunity
In the case of a biologic drug, ImmTOR is designed to be administered in conjunction with such biologic drugs to mitigate the formation of ADAs without requiring the alteration of the drug or its dose regimen. As a result, we believe ImmTOR may provide us with significant opportunities to co-administer ImmTOR with a variety of biologic drugs, including therapeutic enzymes and gene therapies. We believe each pairing of ImmTOR with a biologic drug also offers us the opportunity to pursue distinct proprietary product candidates, which we believe have the potential to be separately patented, approved and marketed. ImmTOR is manufactured in facilities subject to current good manufacturing practice, or cGMP, requirements using well-defined commercial operations, which, we believe, further enhances the scalability of our tolerance programs.
In preclinical studies, we observed that delivering an antigen together with ImmTOR provided the appropriate signals in vivo to induce regulatory T cells, which, in turn, inhibited effector immune responses, such as the formation of ADAs. In additional preclinical studies, we observed that ImmTOR labeled with a fluorescent dye selectively accumulated in the spleen and the liver following intravenous dosing, where it was processed by antigen-presenting cells, such as dendritic cells.. The figure below depicts a model of how we believe ImmTOR would be taken up by a dendritic cell in the spleen. We believe that both the biologic drug and ImmTOR are taken up and processed by dendritic cells and other antigen presenting cells in a manner that may result in the activation of antigen-specific regulatory T cells, which can potentially block the activation of helper T cells, thus mitigating the formation of ADAs.
ImmTOR-IL
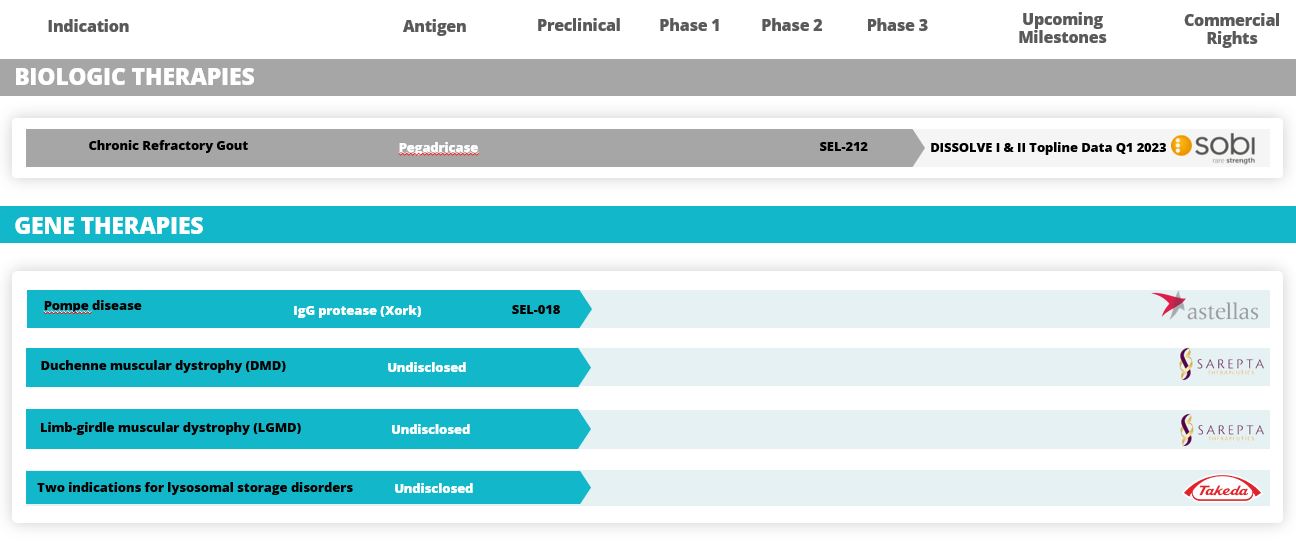
The table below summarizes key information about our development pipeline.

Our lead product candidate, Descartes-08, is an autologous mRNA CAR-T directed against the B cell maturation antigen, or BCMA, that we are developing for the treatment of autoimmune diseases. Descartes-08 has been granted Orphan Drug Designation by the FDA for the treatment of MG. Descartes-08 was observed to be safe and well-tolerated in a Phase 1b/2a trial of 14 patients with MG who received outpatient treatment without pre-treatment chemotherapy. All seven participants who received six once-weekly infusions at the highest dose continued to experience marked and long-lasting clinical improvement across validated MG disease scoring systems at month nine follow-up. At month 12, five of these seven participants maintained clinically meaningful improvement. One participant, who lost response after one year, experienced rapid improvement in clinical scores after re-treatment, which was ongoing at month six of follow-up. Clinical responses correlated with large reductions in autoantibody titers. We are currently enrolling in a Phase 2b randomized, double-blind, placebo-controlled trial in patients with MG, for which we expect to report topline results in mid-2024.
We lookare also developing Descartes-08 for waysthe treatment of other autoimmune diseases. We have received FDA allowance for our investigational new drug application, or IND, for a Phase 2 trial of Descartes-08 for the treatment of patients with systemic lupus erythematosus, or SLE, a chronic autoimmune disease that causes systemic inflammation affecting multiple organ systems. We expect this Phase 2 trial to enhanceinitiate in the first half of 2024.
Descartes-15 is our ImmTOR platform. Recentnext-generation autologous anti-BCMA mRNA CAR-T. In preclinical studies, we have observed Descartes-15 to be 10-fold more potent than Descartes-08. We intend to test the safety of Descartes-15 in an open label, single-arm Phase 1 trial in patients with relapsed/refractory multiple myeloma. This program has already received IND allowance from the FDA and is expected to enroll the first patient in the first half of 2024. We expect that these Phase 1 trial data generated bywill inform our scientific teams suggest that ImmTOR may have profound synergistic activity with engineered IL-2 molecules that are selectiveclinical development plan for Tregs. The IL-2 pathway influences critical aspects of both immune stimulation and immune regulation. Tregs express a high affinity form of the IL-2 receptor. Low doses of IL-2 have been shown by others to selectively activate Tregs resulting in expansion of pre-existing Tregs. Clinical trials of low dose IL-2 have generated evidence of efficacyDescartes-15 in autoimmune diseases. Other investigators have shown that IL-2 can be engineered to selectively bind to the high affinity IL-2 receptor and expand pre-existing Tregs.
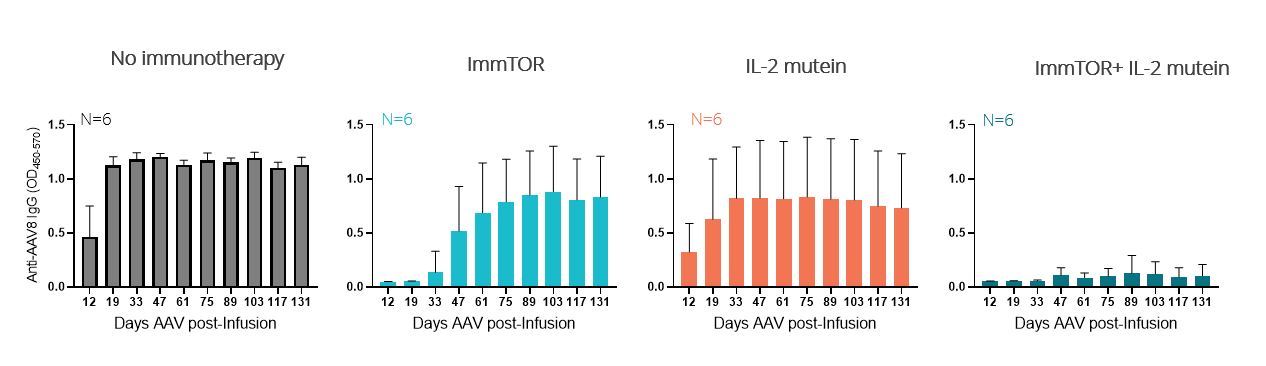
In our preclinical studies, we observed that ImmTOR combined with a Treg-selective IL-2 mutant protein, or IL-2 mutein, exhibited substantial synergistic activity in increasing the percentage and durability of total Treg expansion in the spleen. We believe that this combination has the potential to be a best-in-class therapy in diseases where expansion of total Treg may prove beneficial. The tables below show the synergistic expansion of total Treg we observed with treatment with ImmTOR and a Treg-selective IL-2 mutein. In the study, seven C57BL/6 mice per group were untreated, treated with ImmTOR alone, treated with IL-2 mutein alone or treated with a combination of ImmTOR and IL-2 mutein. Expansion of CD4+, CD25+ and FoxP3+ T regulatory cells in the spleen was assessed at four, seven and 14 days after treatment.
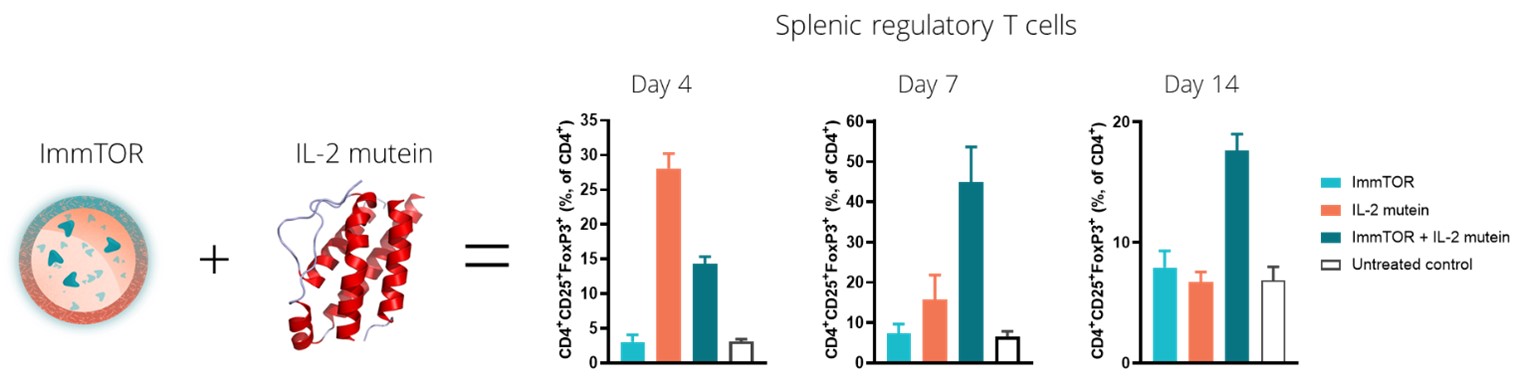
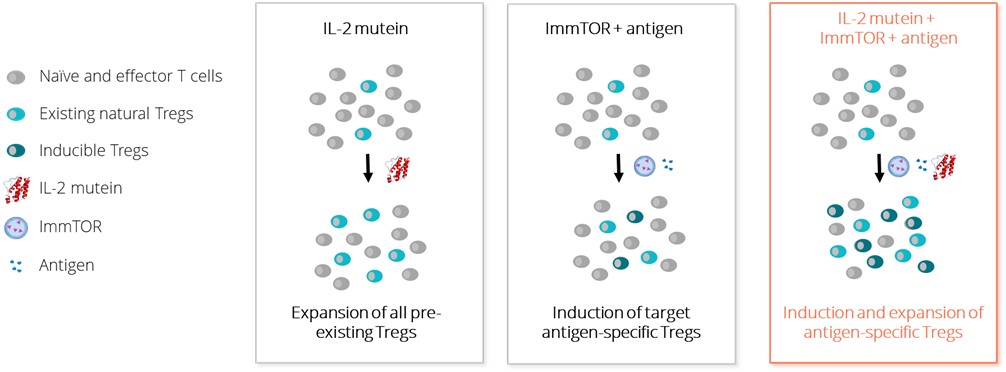
We believe that the power of ImmTOR is the ability to induce antigen-specific Treg to co-administered antigens. The ability to induce antigen-specific Treg may be beneficial in autoimmune diseases where the auto-antigens are well characterized. We believe the combination of a Treg-selective IL-2 with ImmTOR and an antigen has the potential to induce and/or expand antigen specific Treg. The image below illustrates the possibility that the combination of a Treg-selective IL-2 with ImmTOR plus an antigen could give rise to the induction and/or expansion of an antigen-specific Treg.
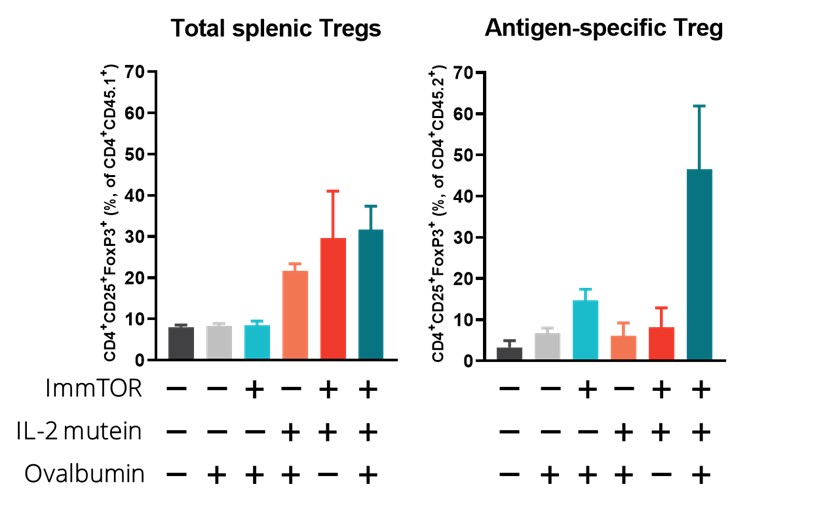
In a preclinical study, we evaluated the potential of a Treg-selective IL-2 to further expand antigen-specific Treg when combined with ImmTOR and an antigen. In this preclinical study, transgenic T cells expressing a T cell receptor specific for the antigen ovalbumin were adoptively transferred into wildtype mice. The next day, the mice were treated, left untreated or treated with ovalbumin or various combinations of ovalbumin with ImmTOR and/or IL-2 mutein. When total Treg were evaluated, we observed no increase an expansion of total Treg by IL-2 mutein + ovalbumin with a further increase in animals treated with ovalbumin + IL-2 mutein + ImmTOR. As we had expected, ImmTOR + ovalbumin alone did not increase total Treg. However, when ovalbumin-specific Treg were evaluated, we observed a significant increase in antigen-specific Treg in animals treated with the combination of ovalbumin + ImmTOR + IL-2 mutein. Animals treated with only ImmTOR + ovalbumin also were observed to have an increase in ovalbumin-specific Tregs but at levels that were approximately three-fold lower than those in the former group. The following graphs show expansion of antigen-specific Treg in mice treated with ImmTOR plus IL-2 mutein plus ovalbumin antigen.
Immune homeostasisLimitations of Current DNA-Based Cell Therapy Treatments in Autoimmune Disease
Conventional DNA cell therapies have been associated with cytokine release syndrome, neurological toxicities and Parkinsonism, infection, risk of secondary malignancy, and death. The acute toxicities are from exponential amplification of the modified cell, and the pre-treatment chemotherapy administered to enable cell amplification.
Conventional DNA-engineered CAR-T cells are in clinical development for several autoimmune diseases. DNA CAR-T cells are typically administered to patients in a subtherapeutic dose, which means that the cells must proliferate to reach therapeutic numbers in the body. However, this proliferation is not controlled in magnitude or duration, varies from patient to patient, and can be unpredictable. This proliferation occurs because the CAR gene is irreversibly integrated into the T-cell’s genome, causing a dynamic process balancing immune stimulatory and immune tolerizing signals. This balance is thought to be mediatedcascade in part bywhich every daughter cell carries the ratio of antigen-specific Treg to antigen-specific effector Tsame CAR as the parent cells. The expansionresulting unconstrained proliferation frequently exceeds the toxicity threshold, leading to serious adverse events. In November 2023, the FDA announced that it is investigating the risk of antigen-specific TregsT-cell malignancies in approved DNA CAR-T cell immunotherapies.
The proliferation of DNA CAR-T cells has typically required pre-treatment chemotherapy, usually fludarabine and cyclophosphamide administered for several days before CAR-T cell treatment. This chemotherapy is toxic, suppressing the immune system and increasing the risk of infection, anemia, and neurotoxicity.
Given these risks and requirements, conventional DNA cell therapies are administered under close monitoring in an inpatient setting, increasing their cost and limiting their reach to only the sickest patients.
Our Autoimmune Disease Solution
We believe that mRNA cell therapy has the potential to better withstandbe a potent immune stimulatory signalsyet safer alternative to DNA cell therapy for treating autoimmune diseases. We believe the mRNA cell therapies we are developing have the potential to deliver deep, durable clinical benefit to many patients with autoimmune diseases because they can be administered over a short period of time, in an outpatient setting, and provide better durabilitywithout pre-treatment chemotherapy. These attributes may extend the reach and potential of immune tolerance. In a preclinical study, we evaluated the ability of ImmTOR and IL-2 mutein to mitigate the immunogenicitymRNA cell therapy to a co-administered AAV vector. Mice were injectedbroader group of patients with a doseautoimmunity.
mRNA CAR-T cells locate their target, become activated, and proliferate like DNA CAR-T cells. However, because mRNA does not replicate and degrades naturally over time, the maximum number of 2.7E12 vg/kg AAV8 on Day 0mRNA molecules can be determined by the dose. The actual number of mRNA molecules declines to zero over time. The number of mRNA molecules determines the degree of CAR protein expression, and 5.0E12 vg/kg on Day 56 with or without IL-2 mutein and/or ImmTOR on the same days. Mice treated with AAV alone showed a robust antibody response to the AAV capsid, as we had expected. Co-treatment with IL-2 mutein on Days 0 and 56 showed a modest attenuationpersistence of the antibody response. Co-treatmentmRNA molecules determines the duration of mRNA CAR-T cell activity. Thus, unlike DNA CAR-T cells, our mRNA CAR-T cells provide pharmacokinetic control. In other words, a patient’s exposure to our cells is determined by the dose. The time, course and duration of that exposure are substantially determined by the nature of the mRNA we use. Therefore, while DNA CAR-T therapies are administered at subtherapeutic levels, we can administer a therapeutic number of mRNA CAR-T cells and re-dose these cells over time, much like a conventional drug. Because the mRNA cannot be replicated, we believe, and have thus far observed, that mRNA CAR-T cells do not cause the types of severe toxicity associated with ImmTOR on Days 0 and 56 showed a dose-responsive effect on the antibody response, withDNA CAR-T cells. Also, because mRNA CAR-T is dosed at a therapeutic dose and does not rely on cell proliferation to reach the therapeutic window, there is no need to administer pre-treatment chemotherapy. The graphs below contrast our mRNA cell therapy approach with that of 200 µg ImmTOR providing inhibition of antibodies against AAV through at least Day 75, or 19 days after the second dose. At Day 91, some animals showed breakthroughconventional DNA cell therapy.

As of the antibody response. ImmTOR doses2023 safety cutoff date, we have administered Descartes-08 to over 60 patients suffering from one of 50MG, multiple myeloma, and 100 µg alone were sub-optimal. However, when ImmTOR was combined with IL-2 mutein, weother diseases in open-label trials on an outpatient basis, many at community clinics. We have not observed inhibitionproduct-related cytokine release syndrome, neurotoxicity or infection of antibody formation through Day 117, 61 days after the second dose and the last time point measured. Importantly full inhibition of anti-AAV immunoglobulin G, or IgG, antibodies was observed even when IL-2 mutein was combined with 50 and 100 µg doses of ImmTOR. We believe these results suggest that the addition of a Treg-selective IL-2 to ImmTOR has the potential to increase the durability of tolerance allow for dose-sparing of ImmTOR and thus, potentially enabling chronic dosing of ImmTOR-IL in the treatment of autoimmune disease.any grade. The following graphs illustrate that the combination of ImmTOR and IL-2 mutein mitigated the formation of anti-AAV antibodies in response to a high vector dose of 5E13 vg/kg. These results suggest that ImmTOR-IL has the potential to enable repeat dosing of AAV vectors at 5E13 vg/kg which could be used as a strategy to avoid the toxicities that have been observed at doses of 1E14 vg/kg or higher in some clinical trials.
adverse events observed—headache, nausea and fever—were self-limited and resolved within 72 hours of onset. One participant with MG with a history of allergic reaction to biologics developed hives after the third infusion and was hospitalized for monitoring. The patient’s hives resolved completely after a brief course of steroids.
Our Product Candidates
Descartes-08
Our ImmTOR platform haslead product candidate is Descartes-08, a broad range of potential applications. Our product development strategy is builtfirst-in-class mRNA CAR-T. Descartes-08 targets BCMA, which exists on the following three distinct pillars.
Biologic therapies. Biologic therapiessurface of long-lived plasma cells, or LLPCs, and plasmacytoid dendritic cells, or pDCs. LLPCs, which can survive for decades, are the main producers of disease-causing autoantibodies. pDCs, which secrete type-I interferons, may also play a class of biologic drugs frequently usedcritical role in autoimmunity. While the lead indication for Descartes-08 is MG, we believe that Descartes-08 has potential to treat rare diseases. Through our analysis of biologic drugs, including in our preclinical studies, we have observed that enzymes foreign to the human body,other autoimmune diseases, such as enzymes derived from microbes or replacement enzymes inlupus.
Descartes-08 for the caseTreatment of patients that are deficient inMG
Overview
Descartes-08 has been granted Orphan Drug Designation by the specific enzyme, are especially prone to causing undesired immune responses. Our partnered product candidate, SEL-212, which is currently in Phase 3 clinical development, consists of ImmTOR co-administered with pegadricase, a pegylated uricase enzyme of fungal origin. This is an example of an immunogenic enzyme that we are combining with ImmTOR with the intention of improving the enzyme’s efficacy and safety. We believe that ImmTOR has the potential to enable and expand the use of enzymes derived from microbial sources, such as bacterial immunoglobulin A, or IgA, proteaseFDA for the treatment of IgA nephropathy.MG. We selectedchose MG as our lead indication because the pathogenesis for MG is common to many autoimmune diseases.
Background Information About MG
MG is a next generation IgA protease candidate from a commensal bacteriarare autoimmune disease that causes debilitating muscle weakness and fatigue. It is estimated to affect over 120,000 patients in the U.S. and Europe. MG patients develop antibodies that lead to an immunological attack on critical signaling proteins at the junction between nerve and muscle cells, thereby inhibiting the ability of nerves to communicate properly with a lower levelmuscles. This results in muscle weakness in tissues throughout the body, potentially manifesting in partial paralysis of baseline ADAs from IGAN Biosciences for the treatment of IgA nephropathy. By combining ImmTOR with this next generation IgA protease candidate, we believe our novel approach has the potential to mitigate the formation of new ADAseye movements, problems in chewing and address the underlying pathophysiologyswallowing, respiratory problems, speech difficulties and weakness in skeletal muscles. The symptoms of the disease.
Additionally, we believe advances in protein engineering may lead to innovative therapeutic enzymes with improved pharmaceutical properties or entirely novel specificity and/or activity, but which may be recognized as foreign by the immune system. We are partnering with Ginkgo Bioworks, or Ginkgo, to design novel enzymes and proteins with transformative therapeutic potential whichdisease can be paired with ImmTOR to advance treatments for orphantransient and rare diseases. We intend to seek, if appropriate, licenses to other enzymes to evaluate in combination with ImmTOR.
Gene therapies. We believe gene therapies have the potential to address key unmet medical needs for many rare genetic diseases, but undesired immune responses to the viral vectors used for gene replacement, augmentation and editing may be restricting their broader use. AAV immunogenicity and AAV toxicity represent two major challenges for the gene therapy field; in many cases these two issues are inextricably linked. Immunogenicity of AAV vectors is thought to cause or exacerbate manyearly stages of the adverse events associated with AAV gene therapy. Inductiondisease can remit spontaneously. However, as the disease progresses, symptom-free periods become less frequent and disease exacerbations can last for months. Disease symptoms reach their maximum levels within two to three years in approximately 80% of acute inflammationpatients. Up to 20% of MG patients experience a respiratory crisis at least once in their lives. During the crisis phase, decline in respiratory function can become life-threatening. Patients in crisis often require intubation and capsid-specific CD8 T cells by AAV gene therapy is thought to contribute to observationsmechanical ventilation.
There are no known cures for MG and the current standard of hepatotoxicity, which has been associated with losscare consists of transgene expression. The formationchronic use of neutralizing antibodies against AAV after initial treatment with AAV mediated gene therapies effectively preventssteroids and other immunosuppressants. These treatments must be administered continually and carry risks such as infection, osteoporosis, and metabolic diseases. Newer agents, such as those that block the possibilitycomplement pathway or inhibit FcRn, are typically administered continually.
Clinical Development
To date, we have completed the Phase 1b portion of re-dosingthe Phase 1/2 trial of Descartes-08 in patients who may benefit from additional doses due to either the failure to achieve therapeutic benefit or loss of transgene expression over time. Additionally, a significant number of patients who would benefit from treatment by gene therapies are ineligible due to pre-existing immunity to the AAV vectors from a natural infection. This pre-existing immunity could potentially be addressed through an IgG protease pre-treatment to open a dosing window for AAV gene therapies. We believe that the combination of ImmTOR and Xork could simultaneously address the two key issues facing the AAV gene therapy modality and make them more accessible while also making them safer and more durable.
We have observed that ImmTOR, when used in combination with AAV gene therapy vectors, inhibited the immune response to the viral vector and enabled successful re-dosing in both mice and non-human primates. Currently, the ability to re-administer systemic AAV gene therapy is limited by the development of neutralizing antibodies. The ability to safely re-dose AAV may help achieve therapeutic benefit in patients who are under-dosed; it may also help restore transgene expression in patients, particularly pediatric patients, who may lose expression over time as they grow. In a study conducted in non-human primates, or NHP, we observed that co-administration of AAV vector and ImmTOR resulted in higher and more durable transgene expression after the first dose of gene therapyMG, as well as robust inhibitionthe primary readout of anti-AAV8 immunoglobulin G,the Phase 2a portion of the trial. We continue to enroll patients to the Phase 2b portion of the trial.
The primary objective of the Phase 1b portion of the trial was to determine the maximum tolerated dose of Descartes-08 for patients with MG. To assess the safety and manufacturability of Descartes-08, the product candidate was administered in three ascending doses (3.5 x106 cells/kg; 17.5 x106 cells; 52.5 x106 cells/kg) to three patients with MG. After each infusion, patients were observed for at least one week, and a higher dose level was administered if there were no significant adverse effects observed at the initial dose. We observed Descartes-08 to be well-tolerated by the three patients who participated in this portion of the trial with no cytokine release syndrome or IgGother serious product-related adverse events.
The primary objective of the Phase 2a portion of the trial was to determine the optimal dosing schedule for patients with MG using the highest dose level tested in Phase 1b (52.5 x106 cells/kg). This portion of the trial was designed to assess the safety and neutralizing antibodies. We believepreliminary efficacy of Descartes-08 when administered across three different treatment schedules (six doses given twice-weekly, once-weekly, or once-monthly). This portion of the observationtrial evaluated 11 patients with particularly advanced disease as assessed by both patient and clinician-reported outcomes. 79% of the 14 patients included in the Phase 1b and Phase 2a portions of the trial were classified at screening to have Class III or IV disease, as defined by the Myasthenia Gravis Foundation of America, indicating they have moderate-to-severe weakness affecting their muscles.
The results of the Phase 2a portion of the trial, published in the Lancet Neurology in July 2023, indicated that co-administrationafter six weekly infusions of AAV vectorDescartes-08, the average improvement in all disease severity scores was three-to-five-fold greater than what is considered clinically meaningful by expert consensus. As shown in the figure below, clinical improvements persisted in all patients at month nine, and ImmTOR can lead to higher transgene expression illustratesin five of the potential forseven remaining patients at a final, 12-month follow-up. Of the two participants who lost response, one was retreated and experienced rapid improvement in clinical scores, which was ongoing at month six of
dosing lower levelsfollow-up. Descartes-08 was observed to be well-tolerated with no reports of AAV gene therapies when combined with ImmTOR. Integrating ImmTOR into a gene therapy protocol has the potential to provide a first dose benefit by enhancing liver-directed transgene expression and durability, as well as the potential to enable re-dosing to restoredose-limiting toxicities, cytokine release syndrome or augment transgene expression.neurotoxicity.
We have observed in preclinical studies that ImmTOR may also have hepatoprotective effects in mouse models of inflammation. As hepatotoxicity is associated with higher vector doses, one potential strategy that we are pursuing is to use ImmTOR to enable multiple lower doses of AAV vectors to mitigate toxicity risks associated with high vector doses. We are concurrently working with our partner Ginkgo to develop a proprietary AAV capsid with improved transduction for liver-directed gene therapy. The below illustration depicts the potential benefits of ImmTOR in systemic AAV gene therapy.

A–C: Mean change from Baseline (line) and standard error (bands) in Myasthenia Gravis Activities of Daily Living Score (MG-ADL, A), Quantitative Myasthenia Gravis Score (QMG, B), Myasthenia Gravis Composite Score (MGC, C) during 12 months of follow-up for MG-001 participants who received six once-weekly doses (n=7). MG-ADL is self-reported; MGC and QMG are neurologist-assessed. D: Change from Baseline in MGC Score after initial dosing and retreatment in a participant experiencing relapse at Month 12. E: Relative change in serum anti-acetylcholine receptor antibody levels in the three participants with detectable antibodies at baseline. Each line represents one patient.
All three participants with detectable anti-acetylcholine receptor antibody levels before treatment had an average 42% reduction in antibody levels by month six. These reductions deepened to 68% by month nine and persisted at month 12. In summary, we observed continued clinical improvement and autoantibody reductions after BCMA-directed mRNA CAR-T treatment for MG that persisted through the one-year follow-up period.
We conducted a human proof-of-conceptare currently enrolling patients in the Phase 2b randomized, double-blind, placebo-controlled portion of the Phase 1/2 trial. We expect to report topline results in mid-2024. The trial, (SEL-399) in healthy volunteers who were treated with an empty AAV8 capsid (EMC-101), which is an AAV capsid containing no transgene, aloneexpected to have at least 30 completers, is designed to assess the primary endpoint of the proportion of patients achieving a five-point or greater reduction in combination with ImmTOR. This clinical trial was conductedtheir MGC score at day 85. Patients will receive six weekly infusions at the dose established in Belgium in partnership with Asklepios BioPharmaceutical, Inc., or AskBio (a Bayer AG subsidiary)Phase 1b (52.5 x106 cells/kg). The goaltrial also involves a crossover component, in which any patient originally assigned to placebo will be given the opportunity to receive Descartes-08 after completing trial treatment.
Secondary endpoints are designed to assess a variety of additional clinical outcomes, including determining safety and tolerability, quantifying the SEL-399 clinical trial waseffect of Descartes-08 over one year, assessing changes through day 85 in QMG, MG QoL 15R, MG Composite and MG post-intervention status and comparing the effect of Descartes-08 versus placebo on MG scales through day 85 in patients who cross over from placebo to evaluateDescartes-08.
Descartes-08 for the appropriate doseTreatment of ImmTOR in humans to mitigateSystemic Lupus Erythematosus
Overview
We are also developing Descartes-08 for the formationtreatment of antibodies to AAV capsids used in gene therapies. Top-line results indicatedSLE, a chronic autoimmune disease that AAV8 empty capsids elicited peak median anti-AA8 neutralizing antibody, or NAb, titers of 1:6875. Median day 30 NAb titers were reduced to titers of 1:25 and 1:5 in the 0.15 mg/kg and 0.3 mg/kg ImmTOR cohorts, respectively, representingcauses systemic inflammation which affects multiple organ systems.
Background Information About Systemic Lupus Erythematosus
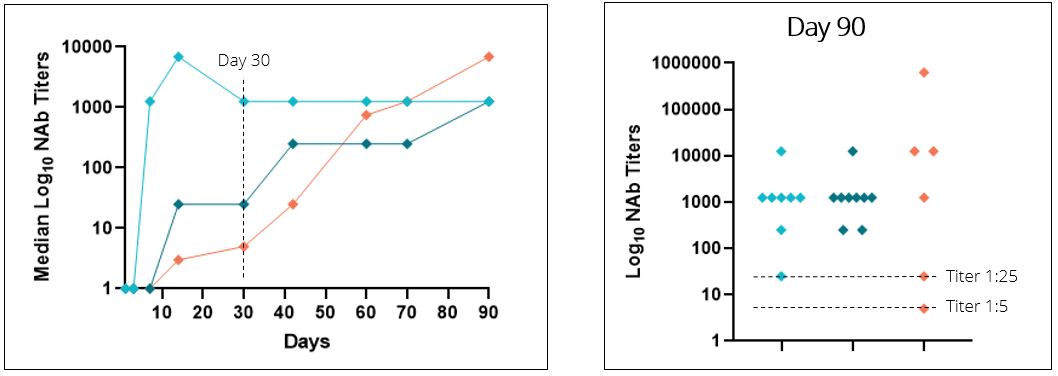
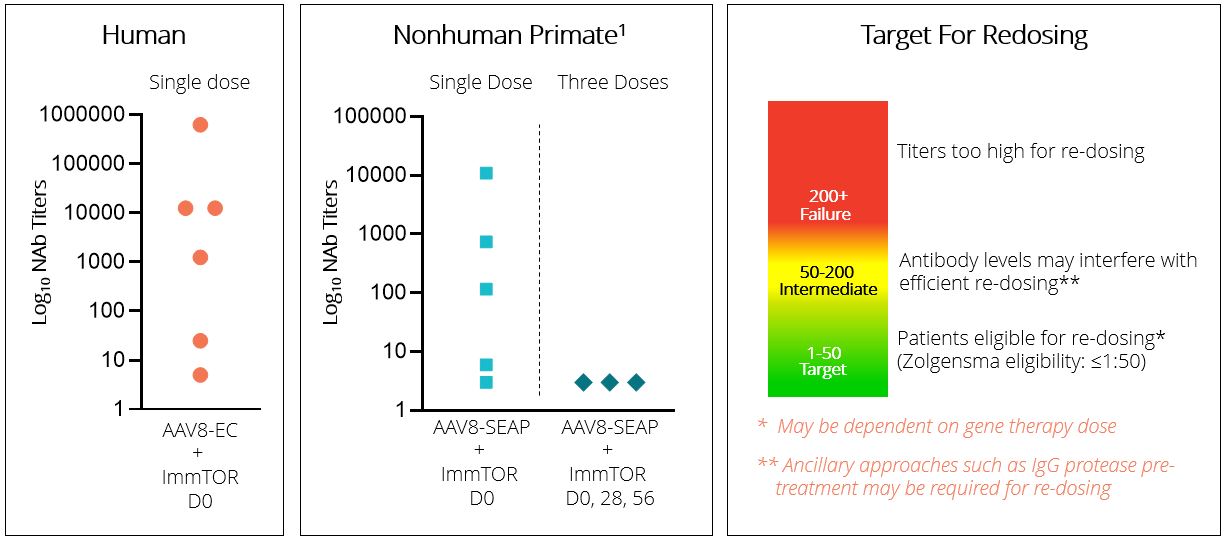
SLE is a 50-fold and 250-fold difference, respectively, compared to the median of control subjects dosed with AAV8 empty capsid alone, as depicted in the figure below. Further, we observedchronic, immune-mediated connective tissue disease that at Day 30, six of six, or 100%, of subjects that received 0.3 mg/kg exhibited NAb titers of 1:25 or less, and four of six, or 67%, of those subjects at this dose exhibited NAb titers of 1:5 or less. We observed at Day 30 that six of nine, or 67%, of subjects that received 0.15 mg/kg of ImmTOR exhibited NAb titers of 1:25 or less, and two of nine, or 22%, of subjects at this dose had a titer of 1:5 or less. At Day 90, two of six subjects in the 0.3 mg/kg cohort were observed to have sustained control of neutralizing antibodies with titers of 1:25 or less. Consistent with preclinical data, we observed that the single dose ImmTOR cohorts showed delayed formation of neutralizing antibodies which eventually reached similar median levels of neutralizing antibodies to the control group by Day 90. Similar data were observed in NHP receiving a single dose of ImmTOR with an AAV gene therapy vector in a preclinical study. Importantly, we observed that two additional doses of ImmTOR, or a total of three monthly doses, provided durable inhibition of neutralizing antibodies in NHP. Although we could not evaluate three-monthly doses of ImmTOR in healthy volunteers, we intend to employ a three-monthly dose regimen in clinical trials performed in patients. ImmTOR showed safety results consistent with prior human studies and was generally well tolerated. No serious adverse events were reported.can impact nearly all major organ systems. The most common treatment-related adverse events included mild-to-moderate stomatitismanifestations of SLE are cutaneous and rash. We believe this promising trial in healthy volunteers provides support for the potential usemusculoskeletal symptoms, although neurological, gastrointestinal, hematological, and renal symptoms are regularly observed as well. Patients with SLE are at a substantially increased risk of ImmTOR for the inhibition of neutralizing antibodies to AAV8 in gene therapy clinical trials. The following graphs depict the effect of ImmTOR on the formation of neutralizing antibodies to AAV8 capsid in humans and NHP.
Finally, pre-existing neutralizing antibodies, which develop asDescartes-15
Descartes-15 is a result of prior infection with wildtype AAV, are a major exclusion factor in clinical trials causing many potential patientsnext-generation, autologous anti-BCMA mRNA CAR-T. Using our proprietary technology and manufacturing platform, we designed Descartes-15 to be ineligible for gene therapy.more resistant than Descartes-08 to recycling of the CAR upon multiple antigen exposures. We have licensedbelieve this is a bacterial IgG protease, or Xork, from Genovis AB (publ.), or Genovis. IgG proteases have been shown by othersparticularly important feature to transiently cleave IgG and enable dosingincrease the durability of AAV vectorsCAR expression on the surface of these cells. We observed that Descartes-15 was 10-fold more potent than Descartes-08 in preclinical studies, as illustrated in the presence of pre-existing antibodies in NHP. However, IgG proteases, being of bacterial origin, are themselves immunogenic. The most commonly studied IgG protease, called IdeS or imlifidase, is derived from Streptococcus pyogenes, a common human pathogen. Most healthy individuals have been exposed to S. pyogenes and have pre-existing antibodies against IdeS. We believe that Xork is a differentiated product candidate, as it is derived from a Streptoccocal species that does not infect humans and so the enzyme shows very low cross reactivity to naturally existing antibodies in most human serum. We plan to develop Xork with the intention of enabling access to AAV gene therapy for those patients who are currently excluded due to pre-existing anti-AAV antibodies.below charts. In JanuaryNovember 2023, we announced an exclusive licensingreceived IND allowance from the FDA to initiate the Phase 1 trial to test the safety of Descartes-15 in patients with multiple myeloma.

Next Steps
We intend to leverage our preclinical and clinical observations from the Descartes-08 development agreementprogram and the Descartes-15 Phase 1 program to inform our clinical strategy for Xork to be developedDescartes-15 for use with AT845, Astellas Gene Therapies' investigational AAV-basedthe treatment of autoimmune diseases.
Allogeneic Product Candidate
Descartes-33 is our allogeneic mRNA MSC in preclinical development for Late-Onset Pompe disease in adults.treatment of autoimmune diseases. We are responsible fordeveloping Descartes-33 to deliver a combination of therapeutic proteins that target key drivers in the early development activities and manufacturingpathogenesis of Xork and will maintain the rights for the development of additional indications beyond Pompe disease.
Through our analysis of genetic diseases, we have identified applications and patient segments that we believe would benefit from our ImmTOR platform. Our initial area of focus is on genetic metabolic diseases but may also include lysosomal storage diseases and genetic muscular diseases. We believe we are the first company to systematically pursue the development of AAV gene therapy product candidates with the goal of enabling repeat administration. We have engaged third parties with experience in gene therapy and rare diseases to support the development of our proprietary products. We also have licensed our ImmTOR platform to AskBio, Sarepta Therapeutics, Inc., or Sarepta, and Takeda Pharmaceuticals USA, Inc., or Takeda, for certain pre-specified targets.autoimmunity.
Tolerogenic Therapies for Autoimmune Disease: Autoimmune diseases are caused by a breakdown in natural tolerance to our own self-antigens. With over 24 million Americans afflicted with autoimmune diseases, there is a large unmet medical need. As the ImmTOR platform is designed to induce or expand antigen specific T regulatory cells, we believe the ImmTOR platform has the potential to treat autoimmune diseases by restoring self-tolerance to auto-antigens.Manufacturing
Recent preclinical data generated by our scientific team suggest that ImmTOR mayWe have profound synergistic activity with engineered IL-2 molecules that are selective for Tregs. The IL-2 pathway influences critical aspects of both immune stimulationestablished wholly owned internal manufacturing and immune regulation. Tregs express a high affinity form of the IL-2 receptor. Low doses of IL-2 have been shown by others to selectively activate Tregs resulting in expansion of pre-existing Tregs.
In our preclinical studies, we observed that ImmTOR combined with a Treg-selective IL-2 molecule exhibited substantial synergistic activity in increasing the percentage and durability of total Treg expansion in the spleen. We believe that this combination has the potential to be a best-in-class therapy in diseases where expansion of total Treg may prove beneficial. This antigen specificity differentiates ImmTOR-IL from other IL-2 molecule approaches which do not show an antigen specific T-cell expansion. Thus, we believe that not only is ImmTOR-IL a potentially "best-in-class" IL-2 where generalized T cell expansion can be beneficial, but also a “first-in-class” antigen specific IL-2 therapy. Furthermore, in our preclinical studies, when we combined ImmTOR-IL with an antigen, we measured an approximately three-fold increase in antigen-specific T regulatory cells versus ImmTOR alone.
Our Product Candidates
Below is a summary of our ongoing discovery, research and development programs:capabilities, which allow us to optimize processes rapidly and in an iterative manner. Our main manufacturing facility is located in Gaithersburg, Maryland and operates under current good manufacturing practice, or cGMP. This facility enhances our control of product quality and production schedules and costs, allowing us to move assets from discovery to preclinical to clinical development quickly. We also entered into an agreement to lease additional manufacturing space located in Frederick, Maryland to transition and expand our clinical and commercial manufacturing capabilities for our maturing pipeline of innovative mRNA cell therapies for the treatment of autoimmune disease.
Our cGMP cell manufacturing facilities, with their dedicated quality management system, are also capable of mRNA production used in Descartes-08. We manufacture Descartes-08 in-house and are typically able to process and release lots for infusion within approximately three weeks. Our autologous cell therapy product candidates, including Descartes-08, are manufactured on a patient-by-patient basis. We have optimized our manufacturing processes through over 200 cGMP runs. We also maintain FDA-reviewed human umbilical cord MSC cell collection and banking operations.
Intellectual Property
In addition to patent protection, we also rely on trade secrets, know-how, trademarks, confidential information, other proprietary information and continuing technological innovation to develop, strengthen and maintain our competitive position. We seek to protect and maintain the confidentiality of proprietary information to protect aspects of our business that are not amenable to, or that we do not consider appropriate for, patent protection. Although we take steps to protect our proprietary information and trade secrets, including through contractual means with our employees, consultants, contractors and collaborators, third parties may independently develop substantially equivalent proprietary information and techniques or otherwise gain access to our trade secrets or disclose our technology. Thus, we may not be able to meaningfully protect our trade secrets. It is our policy to require our employees, consultants, outside scientific collaborators, sponsored researchers and other advisors to execute confidentiality and invention assignment agreements upon the commencement of employment or consulting relationships with us. However, such confidentiality agreements can be breached, and we may not have adequate remedies for any such breach. For more information regarding the risks related to our intellectual property, see the section titled “Risk factors—Risks related to intellectual property.”
Key Agreements
Biogen License Agreement
On September 8, 2023, we entered into a non-exclusive, sublicensable, worldwide, perpetual patent license agreement, or the Biogen Agreement, with Biogen MA, Inc, or Biogen, to research, develop, make, use, offer, sell and import products or processes containing or using an engineering T-cell modified with an mRNA comprising, or encoding a protein comprising, certain sequences licensed under the Biogen Agreement for the prevention, treatment, palliation and management of autoimmune diseases and disorders, excluding cancers, neoplastic disorders, and paraneoplastic disorders. We are not obligated to pay Biogen any expenses, fees, or royalties.
We may terminate the Biogen Agreement for any reason or no reason, and Biogen may terminate the agreement after a notice-and-cure period of 30 days if we fail to pay a fee owed to Biogen or for any other material breach of the agreement. The Biogen Agreement will otherwise expire when all claims of all issued patents within the patents and patent applications licensed
Biologic Therapies – Chronic Refractory Gout
SEL-212 consists of ImmTOR co-administered with pegadricase. Our pegadricase consiststo us under the Biogen Agreement have expired or been finally rendered revoked, invalid or unenforceable by a decision of a yeast-derived uricase modifiedcourt or government agency.
NCI License Agreement
Effective September 16, 2019, we entered into a nonexclusive, worldwide license agreement, or the NCI Agreement, with polyethylene glycol moieties. Uricase is an enzyme endogenous to all mammals, except for humansthe U.S. Department of Health and certain primates, which converts serum urate to the more soluble metabolite, allantoin. There is a natural limit to the amount of serum urate that can be excretedHuman Services, represented by the kidneys, which decreases with ageNational Cancer Institute of the National Institutes of Health, or NCI.
Under the NCI Agreement, we were granted a license under certain NCI patents and can be reduced by some medications. By converting serum uratepatent applications designated in the agreement, to allantoin, uricase provides an additional way formake, use, sell, offer and import products and processes within the body to reduce serum urate.
Pegadricase is designedscope of the patents and applications licensed under the NCI Agreement when developing and manufacturing anti-BCMA CAR-T cell products for the treatment of patientsMG, pemphigus vulgaris, and immune thrombocytopenic purpura according to methods designated in the NCI Agreement.
In connection with chronic gout, refractoryour entry into the NCI Agreement, we paid to standard serum urate lowering treatment, by breaking downNCI a one-time $100,000 license royalty payment. Under the excess serum urate, or SU,NCI Agreement, we are further required to pay NCI a low five-digit annual royalty. We must also pay earned royalties on net sales in a low single-digit percentage and pay up to $0.8 million in benchmark royalties upon our achievement of designated benchmarks that are based on the commercial development plan agreed between the parties.
Under the NCI Agreement, we must use reasonable commercial efforts to bring licensed products and licensed processes to the more soluble allantoin. However,point of Practical Application (as defined in the immune responseNCI Agreement). Upon our first commercial sale, we must use reasonable commercial efforts to pegadricase limits administrationmake licensed products and licensed processes reasonably accessible to a single dose which is effective for less than one month. The addition of ImmTOR to pegadricase (SEL-212) allows multiple monthly doses to be administered, thus reducing serum urate for a prolonged time.
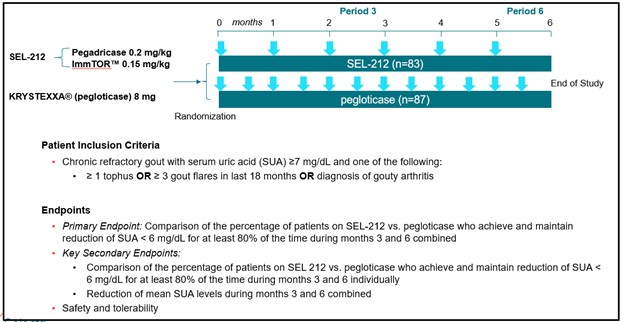
In preclinical studies and in our Phase 1b and Phase 2 clinical trials, we observed that ImmTOR, when co-administered with pegadricase, SEL-212 substantially reduced the formation of associated ADAs. We believe that SEL-212 serves as proof of concept for the ImmTOR platform in ameliorating the unwanted immune response to an immunogenic biologic. SEL-212 is in two pivotal Phase 3 studies versus placebo, which we refer to as DISSOLVE I and DISSOLVE II, with top-line data expected first quarter of 2023. SEL-212 has been licensed (except as to Greater China) to Sobi, pursuant to our license and development agreement dated June 11, 2020, with Sobi, or the Sobi License.
Enrollment into the DISSOLVE II trial is complete and the study is being conducted in the United States public. After our first commercial sale, we must make reasonable quantities of licensed products or materials produced via licensed processes available to patient assistance programs and four countries across eastern Europe. Asdevelop educational materials detailing the licensed products. Unless we obtain a result of the ongoingwaiver from NCI, we must have licensed products and rapidly evolving geopolitical situation in Ukraine and Russia, we have proactively undertaken mitigation steps to prioritize the safety of our patients and investigators, as well as address any potential disruptions. We have reserved existing clinical trial supplies in these countries for those already enrolled in the DISSOLVE II trial. In agreement with our study partner, Sobi, we increased enrollment in DISSOLVE II to 153 subjects for potential loss of subjects enrolled in Russia and Ukraine due to operational or other issues arising from instability in the region. As of the filing of this Annual Report, we expect to report DISSOLVE I and DISSOLVE II topline results in the first quarter of 2023.
The market for gout therapy
Gout is a painful and potentially disabling form of arthritis associated with elevation of SU levels caused by an overproduction of serum urate and/or an inability of the kidneys to excrete adequate amounts of serum urate from the body. High concentrations of SU lead to formation of serum urate crystals in joints and tissues, causing pain, inflammation and joint damage, and increase the risk for other conditions, including cardiovascular, cardiometabolic, joint and kidney disease.
Patients who are unable to reduce their SU levels below 6.0 mg/dL with oral drugs are defined as having refractory gout. Chronic refractory gout constitutes a subset of gout patients exhibiting chronic high SU levels and painful and damaging serum urate deposits. We estimate that there are approximately 160,000 chronic refractory gout patients in the U.S.
We believe SEL-212 may potentially address several key unmet needs in the treatment of chronic refractory gout: the durable control of SU levels, the elimination of painful and damaging serum urate deposits, reduction in incidence and severity of flares, based on our preclinical studies, clinical trials, and market research. SEL-212 is designed to address these unmet medical needs while improving the dosing regimen to a once-monthly treatment.
SEL-212 Clinical Development
We conducted Phase 1a, Phase 1b, and Phase 2 clinical trials for SEL-212 at multiple siteslicensed processes manufactured substantially in the United States. These trials were designedPrior to demonstrate the inductionfirst commercial sale, upon NCI’s request, we are obligated to provide NCI with commercially reasonable quantities of an immune responselicensed products made through licensed processes to pegadricase and the ability of ImmTORbe used for in vitro research.
Additionally, we must use reasonable commercial efforts to mitigate that immune response to both single and multiple doses. The table below summarizes the trial designs and objectives of these clinical trials.
| | | | | | | | |
| Design | Objective |
Phase 1a
SEL-212/101 | •Subjects with hyperuricemia
•Single dose of pegadricase
•n=22
| •Evaluate safety and tolerability
•Define dose of pegadricase that would support monthly dosing
•Demonstrate formation of ADAs
|
Phase 1b
SEL-212/101 | •Subjects with hyperuricemia
•Single dose of SEL-212
•n=53
| •Evaluate safety and tolerability
•Proof-of-biological activity:
•Mitigation of ADAs after single dose
•Select dose to take forward into Phase 2
|
Phase 2
SEL-212/201 | •Subjects with symptomatic gout and hyperuricemia
•Repeated monthly dosing of SEL ‑212
•n=152
| •Evaluate safety and tolerability
•Clinical proof-of-concept:
•Demonstrate sustained reduction of SU with repeat dosing
•Select dose to take intoinitiate a Phase 3
|
In the Phase 1a trial, we observed that a single dose of pegadricase rapidly reduced SU levels below 6.0 mg/dL for each dose level, although SU levels returned close to baseline by day 30. Consistent with our preclinical studies in animals, pegadricase induced uricase-specific ADAs in all patients with varying levels in this Phase 1a trial. The following table depicts the observations from the single ascending dose trial of pegadricase in patients with hyperuricemia. In this trial, patients with SU > 6 mg/dL at screening were assigned to one of five cohorts receiving a single IV infusion of pegadricase (0.1, 0.2, 0.4, 0.8 or 1.2 mg/kg). Each cohort consisted of five patients, except for cohort 5 (1.2 mg/kg) which enrolled two patients.
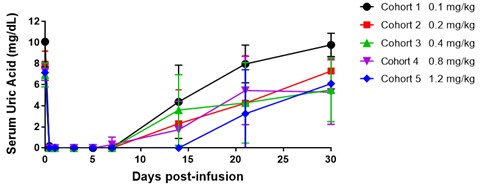
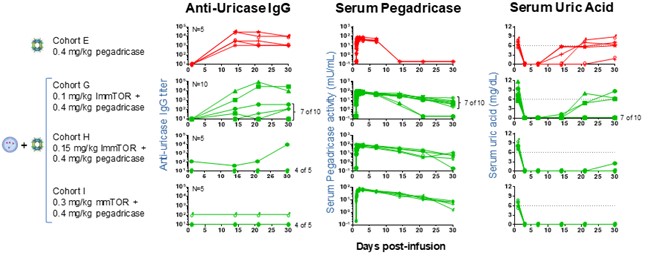
In the Phase 1b clinical trial, we observed the potential for increasing doses of ImmTOR to mitigate the formation of pegadricase ADAs and sustain the anti-uricase enzyme activity through 30 days. The cohort of subjects who received a single 0.4 mg/kg dose of pegadricase alone showed development of uricase ADAs, a short pegadricase half-life, and poor control of SU at 30 days. Three cohorts who received 0.4 mg/kg of pegadricase plus ascending doses of ImmTOR (0.1, 0.15 and 0.3 mg/kg) showed a dose related reduction in uricase ADAs, an increase in pegadricase half-life, and substantial reduction in SU. This graphs below depict the observed correlation of SU with anti-uricase IgG and serum pegadricase activity. In these graphs, anti-uricase IgG titers, serum pegadricase activity, and SU are plotted against time for individual patients in cohorts A, G, H, and I. Each line represents an individual patient.
The Phase 2 trial included patients with symptomatic gout and elevated SU levels in an open-label, multiple ascending-dose clinical trial of SEL-212a licensed product by the fourth quarter of 2024, submit a biologics license application, or BLA, with respect to demonstrate sustained reductiona licensed product by the fourth quarter of SU2026, and identifymake a first commercial sale of a licensed product by the best dose for Phase 3. Five monthly dosesfourth quarter of SEL-212 reduced SU2028.
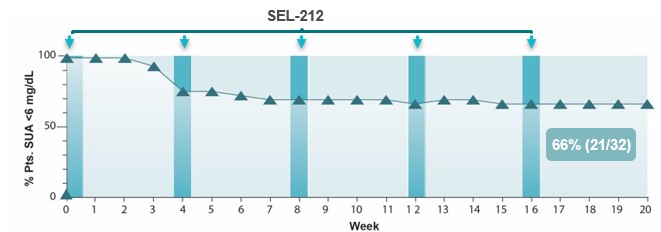
The NCI Agreement terminates upon the expiration of the last to less than 6.0 mg/dLexpire of the patent rights licensed thereunder, if not sooner terminated. NCI has the right to terminate this agreement, after giving written notice and providing a cure period in 21accordance with its terms, if we are in default of 32 evaluable patients, or 66%. In distinct contrast, pegadricase alone reduced SU to less than 6.0 mg/dL in only three of 19 patients one month after a single injection (data from pegadricase alone cohorts from the SEL-037/101, SEL-212/101, and SEL-212/201 trials). We believe these results support our view that the addition of ImmTOR to pegadricase is able dramatically enhance the efficacy of pegadricase in the long-term reduction of SU in gout patients. The chart below depicts SU after five monthly doses of SEL-212.
material obligation. We have observed that SEL‑212 and its components, ImmTOR and pegadricase, were generally well-toleratedthe unilateral right to terminate the agreement in the Phase 1a, 1b, and 2 clinical trials. In the Phase 1a trial we observed no serious adverse events,any country or SAEs, and that pegadricase was well tolerated at the five dose levels tested. Of the six SAEs in the Phase 1b trial, three were determinedterritory by giving NCI 60 days’ written notice. We agreed to not to be related to study drug by investigators. Of the remaining three SAEs that were determined to possiblyindemnify NCI against any liability arising out of our, sublicensees’ or likely be related to study drug, two were cases of stomatitis that occurred at the highest dose of ImmTOR tested (0.5 mg/kg), leading us to define 0.3 mg/kg as the maximum tolerated dose of ImmTOR in this patient population. The remaining SAE was a case of drug hypersensitivity that occurred at a dose of 0.1 mg/kg of ImmTOR in combination with 0.4 mg/kg of pegadricase. In the Phase 2 trial, 20 subjects reported a total of 23 SAEs, nine in the five dose combination cohorts, seven of which were reported to be not related or unlikely related to study drug, and two of which were infusion reactions. All SAEs were successfully treated without residual effects.
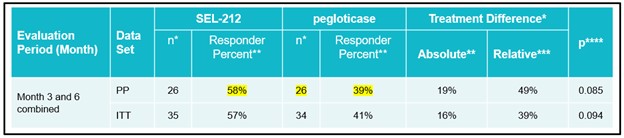
Phase 2 head-to-head clinical trial
In March 2019, we initiated a non-registrational Phase 2 head-to-head clinical trial of SEL-212, which we refer to as COMPARE, in which SEL-212 was compared against the current FDA-approved therapy for chronic refractory gout, pegloticase, in multiple clinical sites in the United States.
In September 2020, we announced top-line results from the COMPARE trial. We observed a numerically higher response rate to pegloticase on the primary endpoint during months three and six combined but did not meet the primary endpoint of statistical superiority. SEL-212 did generate a statistically significant higher response rate to pegloticase during the third month of treatment, as well as a statistically significant greater overall reduction in mean SU levels in SEL-212 versus pegloticase in months three and six combined. We observed a numerically higher response rate of SEL-212 during the sixth month of treatment. In patients with tophi at baseline, we observed substantially higher responder rates for SEL-212 compared to pegloticase on the primary endpoint, and a statistically significant reduction in mean SU levels when compared to pegloticase.
Approximately 41% of patients in the Phase 2 COMPARE trial had visible tophi at baseline, which is lower than we expected for the general refractory gout population. However, in these most severe patients with tophi, SEL-212 was superior to pegloticase with a 58% responder rate for SEL-212 versus a 39 % responder rate for pegloticase. Response rates observed in patients with tophi at baseline are depicted below.
SEL-212 showed a favorable safety profile and was well-tolerated. There were no deaths during the study. There were no notable differences in serious TEAEs, treatment-related serious TEAEs, or infusion reactions between the two groups. A full analysis of safety signals, including gout flare incidence and severity, requires evaluationparties’ use of the full data setlicensed patent rights and will be reported together with the full efficacy analysis in a manuscript in a medical journal.
Phase 3 DISSOLVE clinical program
In September 2020, we commenced the Phase 3 clinical program of SEL-212, which we refer to as DISSOLVE. We are responsible for the execution of the DISSOLVE program and are being reimbursed by Sobi on a quarterly basis for expenses incurredlicensed products or licensed processes developed in connection with these activities. The Phase 3 clinical program consists of two double-blinded, placebo-controlled trials of SEL-212. We refer to these trials as DISSOLVE I and DISSOLVE II. Each trial was expected to enroll up to 120 patients, with up to 40 patients receiving 0.1 mg/kg of ImmTOR and 0.2 mg/kg of pegadricase, up to 40 patients receiving 0.15 mg/kg of ImmTOR and 0.2 mg/kg of pegadricase, and up to 40 patients receiving placebo. An additional 15 patients were added to the study sample size to account for potential treatment discontinuations due to the ongoing COVID-19 pandemic as a result of the emergence of COVID-19 variants which were not accounted for in the sample size calculations. In agreement with our study partner, Sobi, we increased enrollment in DISSOLVE II to 153 subjects for potential loss of subjects enrolled in Russia and Ukraine due to operational or other issues arising from instability in the region.licensed patent rights.
We commenced enrollment of DISSOLVE I in September 2020 and DISSOLVE II in December 2020. In December 2021, we announced the completion of enrollment of the DISSOLVE I trial. DISSOLVE I has a six-month primary endpoint followed by a six-month safety extension. In June 2022, we announced the completion of enrollment of the DISSOLVE II trial. DISSOLVE II will have a six-month primary endpoint with no extension. The primary endpoint of the DISSOLVE program is
the maintenance of SU levels below 6 mg/dL at six months. As of the filing of this Annual Report, we expect to report DISSOLVE I and DISSOLVE II topline results in first quarter of 2023.
Biologic Therapies – IgA Nephropathy
The second development program in biologic therapies is in IgA nephropathy, an autoimmune kidney disease that occurs when immune complexes of a subclass of antibodies called immunoglobulin A1, or IgA1, accumulates in the kidneys. Previous studies in animal models conducted at independent laboratories demonstrated that bacterial IgA protease has the potential to remove injurious IgA immune complexes from kidneys and reduce inflammation, fibrosis, and hematuria. We believe these results suggest that IgA protease can potentially decrease the rate of disease progression and possibly even reverse the disease. The barrier to IgA protease commercialization has been the bacterial origin of the protease, which makes it highly immunogenic. Based on the learnings of SEL-212, we believe the combination of ImmTOR with an IgA protease could potentially enable repeated dosing to treat IgA nephropathy.
In clinical trials, we have observed ImmTOR mitigating the formation of ADAs to immunogenic enzymes. We intend to combine an IgA protease with our ImmTOR platform to develop a novel combination product candidate for the treatment of IgA nephropathy and IgA-mediated diseases. In December 2022, we announced the selection of a next generation IgA protease candidate from IGAN Biosciences for its IgA nephropathy program.
In October 2020, we entered into an Option andAstellas License Agreement or the IGAN Agreement, with IGAN Biosciences, Inc., or IGAN. Pursuant to the IGAN Agreement, IGAN granted us an exclusive license to research, evaluate, and conduct pre-clinical development activities on IGAN’s proprietary IgA proteases. We had an initial option term of 24 months, during which we could elect to obtain an exclusive license to further develop and commercialize the product candidate to treat all IgA-mediated diseases, including IgA nephropathy, Linear IgA bullous dermatitis, IgA pemphigus, and Henoch-Schonlein purpura (also known as IgA vasculitis).
In May 2022, we entered into Amendment No. 1 to the IGAN Agreement, or the First IGAN Amendment, with IGAN. The First IGAN Amendment provided an extension to the option term of 24 months from the date of the First IGAN Amendment.
In December 2022, we exercised our option to opt-in to an exclusive license agreement and selected a next generation IgA protease candidate from a commensal bacteria with a lower level of baseline ADAs from IGAN for the treatment of IgA nephropathy. By combining ImmTOR with this next generation IgA protease candidate, we believe our novel approach has the potential to mitigate the formation of new ADAs and address the underlying pathophysiology of the disease.
In October 2021, we entered into a Collaboration and License Agreement, or the First Ginkgo Agreement, with Ginkgo. Pursuant to the First Ginkgo Agreement, Ginkgo will leverage their high throughput enzyme discovery, design and screening capabilities to identify and further optimize a proprietary, next generation IgA protease. We have an exclusive license to further develop and commercialize the discovered IgA proteases to treat all IgA-mediated diseases, including IgA nephropathy, Linear IgA bullous dermatitis, IgA pemphigus, and Henoch-Schonlein purpura.
These collaborations build on extensive preclinical as well as strong clinical data from our Phase 2 COMPARE trial for the treatment of chronic refractory gout that we believe further supports ImmTOR’s potential for sustained therapeutic benefit when combined with immunogenic enzymatic therapies.
We are continuing to perform pre-clinical IND enabling studies.
Gene Therapies – Methylmalonic Acidemia
Our lead therapeutic gene therapy program, SEL-302, is intended to use ImmTOR to enhance the treatment of methylmalonic acidemia, or MMA, an inherited disorder in which the body is unable to process certain proteins and fats (lipids) properly. This program was previously being conducted under our collaboration with AskBio. In October and November 2020, we received rare pediatric disease designation and orphan drug designation, respectively, from the FDA for MMA-101, for the treatment of MMA due to methylmalonyl-CoA mutase, or MMUT gene mutations. See “⸺Licenses and Collaborations⸺AskBio” for more information. In April 2021, we were notified by AskBio that it intended to opt-out of development of the MMA indication. The feasibility study and license agreement with AskBio, or AskBio Collaboration Agreement, otherwise remains in effect. We filed an IND to conduct a Phase 1/2 clinical trial of our SEL-302 product candidate in pediatric patients with methylmalonic acidemia in the third quarter of 2021. ImmTOR manufacturing, controlled by us, continues to proceed in accordance with our expectations and we have not observed any impact to any of our ImmTOR programs. In October and November 2020, we received rare pediatric disease designation and orphan drug designation, respectively, from the FDA for MMA-101, for the treatment of MMA due to methylmalonyl-CoA mutase, or MMUT gene mutations. In December 2022, we initiated ReiMMAgine, the Phase 1/2 clinical trial of SEL-302. The ReiMMAgine trial is now enrolling patients and aims to evaluate the safety, tolerability and efficacy of SEL-302.
Gene Therapies – IgG Protease (Xork)
We have exclusively licensed Xork, an IgG-specific protease from Genovis, an enzyme technology company. We plan to develop Xork, either alone or in combination with our ImmTOR platform, with the goal of enabling the dosing of transformative gene therapies in patients with pre-existing AAV immunity due to natural exposures to AAV viruses. Currently, significant proportions of the potential patient populations for many gene therapy trials are ineligible for treatment by AAV mediated gene therapies due to pre-existing antibodies which limits transduction efficiency of the therapy and could trigger potentially dangerous immune responses. IgG proteases are derived from bacteria. Xork exhibits low cross-reactivity to antibodies in normal human serum and is differentiated from IgG proteases derived from Streptococcus pyogenes, a common human pathogen.
Because of this, the commercial potential of many gene therapies may be significantly limited due to the ineligibility of large segments of the target patient population. This is particularly important for gene therapies given the low incidence and small prevalent populations of rare monogenic diseases which many gene therapies target. Patients that are ineligible for gene therapy due to pre-existing immunity may have few or no alternative therapies available to them.
Xork, as a pre-treatment in advance of an AAV gene therapy, can potentially open a treatment window by cleaving the IgG antibodies and transiently reducing their concentration.
IgG proteases are bacterial proteins, which are themselves immunogenic. We believe that Xork is differentiated from other IgG proteases since it is not derived from a human pathogen and thus, exhibits low cross-reactivity with human sera. Additionally, to further reduce any potential immunogenicity of the Xork enzyme, we plan to explore the combination of Xork and ImmTOR. The following images depict how Xork cleaves human IgG specifically and efficiently, but shows low cross reactivity to human sera compared to IdeS, an IgG protease derived from the common human pathogen Streptococcus pyogenes.
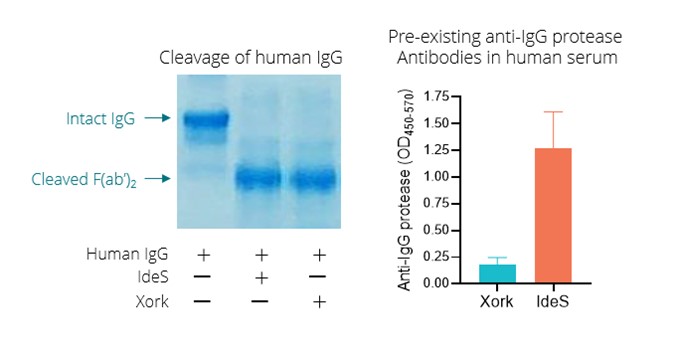
We believe that the combination of Xork and ImmTOR has the potential to address two of the major challenges to AAV gene therapy, pre-existing immunity that limits eligibility and de novo immunogenicity that can adversely affect safety and durability and which prevents the ability to re-dose AAV. We plan to explore the application of Xork in combination with our wholly owned gene therapies, as well as explore partnering opportunities for the enzyme with other gene therapy companies.
In January 2023, we announced an exclusive licensing and development agreement for Xork to be developed for use with AT845, Astellas Gene Therapies' investigational AAV-based treatment for Late-Onset Pompe disease in adults. We are responsible for the early development activities and manufacturing of Xork and will maintain the rights for the development of additional indications beyond Pompe disease.
Tolerogenic Therapies for Autoimmune Disease – ImmTOR & ImmTOR-IL
We intend to apply our ImmTOR platform to treat autoimmune diseases. In preclinical studies, we have observed ImmTOR’s ability to induce antigen-specific T regulatory cells. We believe that ImmTOR, in combination with an autoantigen of interest, could create self-tolerance to auto-antigens and thus be a novel approach to the treatment of autoimmune diseases.
Additionally, in preclinical studies we have observed ImmTOR, in combination with IL-2 molecules, expanding T-regulatory cells beyond IL-2 alone and we intend to pursue a combination of ImmTOR and IL-2 (ImmTOR-IL) in diseases
where general T cell expansion has shown a therapeutic benefit. Additionally, we have observed in preclinical studies that the combination of ImmTOR, a Treg-selective IL-2 molecule and an antigen, exhibited substantial synergistic activity in inducing and expanding antigen specific regulatory T cells when ImmTOR and IL-2 is combined with an antigen of interest. We intend to pursue and develop treatments for autoimmune diseases with well-defined antigens using either ImmTOR or ImmTOR-IL.
Cyrus Biotechnology, Inc., a collaboration partner, is engineering a proprietary IL-2 protein to combine with the ImmTOR platform to potentially mitigate unwanted immune responses by reducing the inherent immunogenicity of the protein while also promoting immune tolerance. The IL-2 pathway influences critical aspects of both immune stimulation and immune regulation, through the development and expansion of Treg cells. These Treg cells are a specialized subpopulation of T cells involved in suppressing certain immune responses and maintaining the body’s self-tolerance. In preclinical studies investigating the effects of ImmTOR in combination with a Treg-selective IL-2 molecule we have observed a substantial synergistic activity in increasing the percentage and durability of Treg expansion in the spleen.
Our lead autoimmune diseases indication is PBC, a T cell driven autoimmune disease that causes progressive destruction of the bile ducts. Patients with PBC are in need of a highly targeted, liver-directed approach to treating the root cause of the disorder. We believe PBC has a well-defined target antigen, significant unmet medical need, and is well suited to the application of our ImmTOR immune tolerance platform, as preclinical data suggest that ImmTOR has the potential to enhance the tolerogenic environment in the liver and provide a hepatoprotective benefit.
Licenses and Collaborations
We intend to both partner and strategically out-license ImmTOR, ImmTOR-IL and Xork for use with other products that are outside our focus to larger biopharmaceutical companies. We believe our ImmTOR platform may also be of interest to biopharmaceutical companies with novel biologic development concepts or product candidates in clinical development that have demonstrated initial efficacy but are experiencing issues with safety or sustained efficacy due to inhibitory ADAs. Additionally, we may strategically in-license products to combine with ImmTOR and develop novel therapeutic products independently. Our key partnerships are listed below.
In-licenses
Ginkgo Bioworks Holdings, Inc.
In October 2021, we entered into the First Ginkgo Agreement with Ginkgo. Under the First Ginkgo Agreement, Ginkgo will design next generation IgA proteases with potentially transformative therapeutic potential. In return, Ginkgo is eligible to earn both upfront research and development fees and milestone payments, including certain milestone payments in the form of our common stock, clinical and commercial milestone payments of up to $85 million in cash, as well as downstream value in the form of royalties on sales.
On January 3, 2022, we entered into a Collaboration and License Agreement, or the Second Ginkgo Agreement, with Ginkgo. Under the Second Ginkgo Agreement, we will engage with Ginkgo to design novel AAV capsids with potentially improved transduction, enhanced tissue tropism and reduced immunogenicity. In return, Ginkgo is eligible to earn both upfront research and development fees and milestone payments, including certain milestone payments in the form of our common stock, clinical and commercial milestone payments of up to $207 million in cash for each of a specified number of products which have the potential to total, in the aggregate, up to $1.1 billion. Ginkgo is also entitled to potential further downstream value in the form of royalties on sales.
Genovis AB (publ.)
In October 2021, we entered into a strategic licensing agreement with Genovis, or the Genovis Agreement. Under the Genovis Agreement, we paid to Genovis an upfront payment in exchange for an exclusive license to Genovis’ Xork enzyme technology for all therapeutic uses in humans, excluding research, preclinical, diagnostic, and other potential non-therapeutic applications of the enzyme. Genovis is eligible to earn development and sales-based milestones and sublicensing fees, as well as tiered royalties on worldwide sales in the low double digits.
Cyrus Biotechnology, Inc.
In September 2021, we entered into a Collaboration and License Agreement with Cyrus, or the Cyrus Agreement, pursuant to which Cyrus agreed to grant us an exclusive, worldwide license to certain intellectual property in order to form a protein engineering collaboration combining the ImmTOR platform with Cyrus’ engineered protein therapeutics. We expect that novel engineered protein therapeutic candidates from the partnership will be used to expand our proprietary pipeline and further bolster the ImmTOR platform. In return for the licensed intellectual property, we made an upfront payment and will pay certain discovery, development, and sales-based milestones which could potentially total up to approximately $1.5 billion across multiple programs.
In June 2022, we mutually agreed with Cyrus that the preclinical key in-vitro success milestone had been achieved.
IGAN Biosciences, Inc.
In October 2020, we entered into the IGAN Agreement with IGAN. Pursuant to the IGAN Agreement, IGAN granted us an exclusive license to research, evaluate, and conduct preclinical development activities on IGAN’s proprietary IgA proteases. We have an option term of 24 months, or the Option Term, during which we can elect to obtain an exclusive license to further develop and commercialize the product to treat all IgA-mediated diseases, including IgA nephropathy, Linear IgA bullous dermatitis, IgA pemphigus, and Henoch-Schonlein purpura.
Upon execution of the IGAN Agreement, we paid IGAN a one-time upfront payment of $0.5 million and we would owe additional payments to IGAN if we were to opt-in to an exclusive license agreement, as well as upon the achievement of certain development and sales milestones. During the option term, we may terminate the IGAN Agreement immediately for any reason upon written notice to IGAN. If we opt-in to an exclusive license agreement, we may terminate the IGAN Agreement upon 120 days’ written notice.
On May 25, 2022, we entered into Amendment No. 1 to the IGAN Agreement, or the First IGAN Amendment, with IGAN. The First IGAN Amendment provided an extension to the option term of 24 months from the date of the First IGAN Amendment.
On December 14, 2022, we exercised our option to opt-in to an exclusive license agreement and selected a next generation IgA protease candidate from a commensal bacteria with a lower level of baseline ADAs from IGAN for the treatment of IgA nephropathy. By combining ImmTOR with this next generation IgA protease candidate, we believe our novel approach has the potential to mitigate the formation of new ADAs and address the underlying pathophysiology of the disease.
Out-licenses
Astellas Therapeutics, Inc.
In January8, 2023, we entered into a License and Development Agreement, or the Astellas Agreement, with AstellasAudentes Therapeutics, Inc., or Astellas. Under this agreement,the Astellas obtainedAgreement, we granted Astellas an exclusive license to the soleCompany’s IdeXork technology arising from Xork, to develop and exclusive right to commercialize Xork for use in Pompe disease in combination with an Astellas gene therapy investigational or authorized product,product. Xork, a bacterial IgG protease licensed from Genovis AB (publ.), or Genovis, is licensed pursuant to an Exclusive License Agreement with Genovis, or the Genovis Agreement. Astellas paid to us a current focus on AT845. We received a $10$10.0 million upfront payment and are eligible to receive $340.0 million for certain additional development and commercial milestones plus royalties on any potential commercial sales where Xork is used as a pre-treatment for AT845. As a resultupon signing of the sublicense of Xork to Astellas we made a $4.0 million payment to Genovis in February 2023.
Takeda Pharmaceuticals USA, Inc.
In October 2021, we entered into a strategic licensing agreement with Takeda, or the Takeda Agreement. Under the Takeda Agreement, and we granted Takeda an exclusive license to our ImmTOR technology initially for two specified disease indications within the field of lysosomal storage disorders. Under the terms of the Takeda Agreement, we received an upfront payment and are entitled to receive up to $1.124 billion$340.0 million in future additional payments over the course of the partnership that are contingent on the achievement of various development and regulatory milestones and, if commercialized, sales thresholds for annual net sales where Xork is used as a pre-treatment for an Astellas investigational or commercial milestones or Takeda’s election to continue its activities at specified development stages.authorized product. We are also eligible for tiered royalty payments ranging from low to high single digits. Any proceeds received from milestone payments or royalties on future commercial salesrelating to Xork would be required to be distributed to holders of CVRs, net of certain deductions.
Pursuant to the Astellas Agreement, we will have the exclusive right and responsibility to complete research and development of Xork products and to conduct all preclinical studies and clinical trials for Xork for use in Pompe disease with an Astellas gene therapy investigational or authorized product, or the Xork Development Services. Astellas will reimburse us for 25% of all budgeted costs incurred to complete the development of Xork for use in Pompe disease with an Astellas gene therapy investigational or authorized product. We will have control and responsibility over regulatory filings, including any IND, BLA, and marketing authorization applications relating to the licensed products.product. Astellas will have the exclusive right and responsibility to research, develop, and commercialize Astellas products used in combination with Xork and will have control and responsibility over all regulatory filings, including any IND, BLA, and marketing authorization applications, relating to Astellas products and Astellas products used in combination with Xork.
Sobi License Agreement
On June 11, 2020, we entered into a License and Development Agreement with Swedish Orphan Biovitrum AB (publ.)
In June 2020, we announced that we had entered into(Publ), or Sobi, which was amended on October 31, 2023, or, as so amended, the Sobi License, pursuantLicense. Pursuant to whichthe Sobi License, we agreed to grant Sobi an exclusive, worldwide (except as to Greater China) license to develop, manufacture and commercialize the SEL-212 drug candidate, which is currently in development for the treatment of chronic refractory gout. In September 2020, pursuantThe SEL-212 drug candidate is a pharmaceutical composition containing a combination of SEL-037, or the Compound, and ImmTOR. Pursuant to the Sobi License, in consideration of the license, Sobi paidagreed to pay us a one-time, upfront payment of $75$75.0 million. Sobi has also agreed to make milestone payments totaling up to $630$630.0 million to us upon the achievement of various development and regulatory milestones and, if commercialized, sales thresholds for annual net sales of SEL-212, and tiered royalty payments ranging from the low double digits on the lowest sales tier to the high teens on the highest sales tier. In July 2022, weAny proceeds received $10.0 million for the completion of the enrollment of the DISSOLVE II trial.
Additionally, Sobi purchased an aggregate of 5,416,390 shares of our common stock at a purchase price of $4.6156 per share for aggregate gross proceeds of $25 million, which we referfrom milestone payments or royalties relating to as the Sobi Private Placement. The closingLicense would be required to be distributed to holders of the Sobi Private Placement occurred on July 31, 2020.CVRs, net of certain deductions.
UnderPursuant to the Sobi License, we will have operational oversightagreed to supply (at cost) quantities of the Compound and ImmTOR as necessary for completion of the two Phase 3 DISSOLVE clinical programtrials of SEL-212 (DISSOLVE I and DISSOLVE II) that commenced in September 2020, at Sobi’s expense.and a six-month placebo extension. We expectwere required to report DISSOLVE I and DISSOLVE II topline results in first quarter of 2023.
Sarepta Therapeutics, Inc.
In June 2020, we entered into a research license and option agreement with Sarepta, or the Sarepta Agreement. Pursuant to the agreement, we granted Sarepta a license to research and evaluate ImmTOR in combination with Sarepta’s AAV gene therapy or gene editing technology, using viral or non-viral delivery, or the Sarepta Product, to treat Duchenne Muscular Dystrophy and certain Limb-Girdle Muscular Dystrophy subtypes, or the Sarepta Indications. Sarepta will have an option term of 24 months during which it can opt-in to obtain an exclusive license to further develop and commercialize the Sarepta Product
to treat at least one Sarepta Indication, with a potential to extend the option term if Sarepta pays an additional fee to us. Sarepta made an upfront payment of $2.0 million to us upon signingsupply quantities of the agreement, and we are eligible to receive additional payments under the option term. If Sarepta opts-in to an exclusive license agreement, we could receive option exercise payments per indication and we would be entitled to significant development and commercial milestone payments and tiered royalties ranging from the mid-to-high single digits based on net sales. In June 2021, we received a payment of $3.0 million for the achievement of certain pre-clinical milestones. In August 2022, we received a payment of $2.0 for a nine-month extension to Sarepta’s options to both Duchenne muscular dystrophy and certain limb-girdle muscular dystrophies and a payment of $4.0 million for the achievement of certain pre-clinical milestones.
Asklepios Biopharmaceutical, Inc.
In August 2019, we entered into a feasibility study and license agreement with AskBio, or the AskBio Collaboration Agreement. The initial product candidate being developed under this collaboration is gene therapy for MMA which can cause severe developmental defects and premature death as a result of an accumulation of toxic metabolites. We previously conducted preclinical studies for this product candidate and will leverage that previous work within the collaboration. In April 2021, we were notified by AskBio that it intended to opt-out of development of the MMA indication. The feasibility study and license agreement with AskBio, or AskBio Collaboration Agreement, otherwise remains in effect.
Massachusetts Institute of Technology
In 2008, we entered into a license agreement with the Massachusetts Institute of Technology, or MIT, which we refer to in its amended form as the MIT License. We amended the MIT License in January 2010, August 2013, November 2016, December 2019 and June 2020. Under the MIT License, we acquired an exclusive worldwide license, with the right to grant sublicenses, to develop, make, sell, use and import certain licensed products that are therapeutic or prophylactic vaccines and use certain licensed processes in the exercise ofCompound until all rights to the licensed products,Compound and any materials needed to manufacture the manufacture, sale and practiceCompound were transferred to Sobi, which transfer occurred upon the execution of which are covered by patent rights owned or controlled by MIT, including patents jointly owned with Brigham and Women’s Hospital, or Brigham, the President and Fellows of Harvard College, the Immune Disease Institute and the Children’s Medical Center Corporation. Our exclusivity is subject to certain retained rights of these institutions and other third parties.
Upon our entry into the MIT License, we paid MIT a non-refundable license issue fee, reimbursed certain of MIT’s costs and issued shares of our common stock to MIT and the other institutional patent owners which were subject to certain anti-dilution, registration and other protective rights. We are obligated to pay MIT creditable annual maintenance fees, low-single-digit running royalty on annual net sales, developmental milestones up to an aggregate of $1.5 million, a mid-single digit percentage of certain payments we receive from corporate partners and a specified percentage of certain income received from sublicensees after 2009 between 10% and 30%. We may terminate the MIT License at any time upon six months’ written notice. MIT has the right to terminate the MIT License immediately upon written notice to us if we cease to carry on our business relatedAmendment No. 1 to the MIT License fail to maintain insurance as required under the MIT License, file for bankruptcy, fail to pay amounts due under the MIT License, challenge or assist others in bringing a challenge to MIT’s patents or fail to cure material breach within 60 days’ written notice thereof. Absent early termination, the MIT License will continue until the expiration or abandonment of the last to expire patent right subject to the MIT License.
In June 2020, we entered into a Fifth Amendment to the MIT License, or the MIT Amendment, which was effective as of May 15, 2020. Pursuant to the MIT Amendment, certain of our diligence obligations were extended to the second quarter of 2021, including a diligence obligation to commence a Phase 3 trial for a licensed product by a specific date in the second quarter of 2021 or to file an IND for a licensed product by a specific date in the third quarter ofand Development Agreement on October 31, 2023. Additionally, certain of our development and regulatory milestones and payments upon achievement of such milestones were adjusted.
Shenyang Sunshine Pharmaceutical Co., Ltd.
In 2014, we entered into a license agreement with 3SBio, as amended in 2017, which we refer to as the 3SBio License. Pursuant to the 3SBio License, we were granted an exclusive license to certain pegadricase-related patents and related know-how owned or in-licensed by 3SBio for the worldwide (except for Greater China and Japan) development and commercialization of products based thereupon for human therapeutic, diagnostic and prophylactic use. We are also granted a worldwide (except for Greater China) exclusive license to develop, commercialize and manufacture or have manufactured products combining our proprietary ImmTOR platform with pegadricase or related compounds supplied by 3SBio (or otherwise supplied if our rights to manufacture are in effect) for human therapeutic, diagnostic and prophylactic use. We were also granted a co-exclusive license to manufacture and have manufactured pegadricase and related compounds for our preclinical and clinical use or, if the 3SBio License is terminated for 3SBio’s material breach, for any use under the 3SBio License. In addition, the 3SBio License, as amended, permits us to utilize one or more third parties to provide up to 20% of our commercial supply of pegadricase. Otherwise, except in the case of a supply shortage on the part of 3SBio, we are obligated to obtain at least 80% of our supply of such compounds for Phase 3 clinical trials and commercial use from 3SBio under the terms of our separate Commercial Supply Agreement with 3SBio dated August 1, 2019.
Under the 3SBio License we have paid to 3SBio an aggregate of $7.0 million in upfront and milestone-based payments. We are required to make future payments to 3SBio contingent upon the occurrence of events related to the achievement of clinical and regulatory approval milestones of up to an aggregate of $15.0 million for products containing our ImmTOR platform. We are also required to pay 3SBio tiered royalties on annual worldwide net sales related to the pegadricase component of products at percentages ranging from the low-to-mid single digits for products containing our ImmTOR platform, subject to specified reductions. These royalties are payable, on a country-by-country and product-by-product basis until the later of (i) the date that all of the patent rights for that product have expired in that country or (ii) a specified number of years from the first commercial sale of such product in such country.
The 3SBio License expires on the date of expiration of all of our royalty payment obligations unless earlier terminated by either party for an uncured material default of the other party or for the other party’s bankruptcy. We may also terminate the 3SBio License on a country-by-country or product-by-product basis for any reason effective upon 60 days’ prior written notice to 3SBio or, with respect to a given product, immediately upon written notice to 3SBio if we identify a safety or efficacy concern related to such product.
Manufacturing
We manufacture ImmTOR using a scalable, self-assembly nanoemulsion process with well-defined, pharmaceutical unit operations. This proprietary, highly specialized and precisely controlled manufacturing process enables us to reproducibly manufacture ImmTOR across many production scales, from milligram-scale at the laboratory bench to hundreds of grams to multi-kilogram scale for commercial production. We have also developed and executed the required detailed analytic characterization of our products.
For our most advanced product candidate, SEL-212, we are producing ImmTOR at an approximately 400-gram scale process, which, at the doses being tested in the Phase 3 DISSOLVE program, we believe will be suitable for commercialization. The process is designed such that this same equipment is capable of potentially producing up to a one-kilogram batch size scale. As our nanoparticle manufacturing process is compact, and therefore also portable, our strategy is to transfer our custom designed process skids to a contract manufacturing organization, or CMO, and have the CMO produce the nanoparticles under our direction. This is the strategy we use for production of clinical supplies for clinical trials and would be the expected strategy for commercial production.
The pegadricase enzyme for SEL-212 is produced by fermentation in E. coli and is sourced from 3SBio in China. Through a licensing arrangement, we own exclusive worldwide rights to pegadricase outside of China, with co-ownership of rights in Japan and with 3SBio owning all rights in China. Under this arrangement, 3SBio hasSobi agreed to supply pegadricasereimburse us for the SEL-212 program. There is also a second supplier for pegadricase in the United States.
Our AAV vector product candidates are produced using the established triple transfection production process at CMOs. We work closely with our CMOs who have platform AAV production processes that we are ableall budgeted costs incurred to leverage for our specific AAV product candidates.
Intellectual Property
Our ImmTOR platform is designed to deliver precise instructions to the immune system as a result of the natural predisposition of the immune system to interrogate nanoparticles such as viruses. In connection with our founding, we licensed multiple patent families, including a patent family based in part on the pioneering research performed by our co-founders at Harvard University, MIT and Brigham. We have also conducted extensive research in-house to further research this area, leading to key discoveries regarding and uses for our ImmTOR technology. We have aggressively sought to extend and protect the proprietary intellectual property underlying the composition and use of ImmTOR, such as for antigen-specific immunotolerance.
We endeavor to protect our nanoparticle technology, which we consider fundamental to our business, by seeking, maintaining and defending patent and other intellectual property rights, whether developed internally or licensed from third parties, relating to our program, product candidates, their methods of use and the processes for their manufacture. Our practice is to strive to protect our intellectual property by, among other methods, pursuing and obtaining patent protection in the United States and in jurisdictions outside of the United States related to our proprietary technology, inventions, improvements, programs and product candidates that are commercially important to the operation and growth of our business. We also rely on trade secrets and know-how relating to our proprietary technology, programs and product candidates, and continue innovating and seeking in-licensing opportunities to maintain, advance and fortify our proprietary position in our nanoparticle-based immunotherapy program and product candidates as well as to develop new programs and other product candidates. Our commercial success will depend in part on our ability to obtain and maintain patent and other proprietary protection for our technology programs, inventions and improvements; to preserve the confidentiality of our trade secrets; to maintain our licenses to use intellectual property owned or controlled by third parties; to defend and enforce our proprietary rights, including our patents; and to operate without infringing the patents and proprietary rights of third parties.
We have developed and in-licensed numerous patents and patent applications and possess substantial know-how and trade secrets relating to our technology programs and product candidates. Our patent portfolio contains a number of issued patents in the United States and certain foreign jurisdictions. We also own a number of pending patent applications in the United States and certain foreign jurisdictions. These patents and patent applications include claims related to:
•tolerance immunotherapy programs;
•methods and compositions related to our proprietary nanoparticles in a variety of applications, including tolerance applications, such as:
◦mitigating anti-drug antibodies and/or their effects associated with protein drugs, such as for chronic refractory gout, including coverage for ImmTOR co-administered with pegadricase, which related patents are expected to expire between 2032 and 2041, and
◦genetic therapies (such as viral vector gene therapy), including coverage for ImmTOR co-administered with a viral vector, which related patents are expected to expire between 2032 and 2042; and
•complete development and commercialization of SEL-212, including both composition of matter and method of treatment claims (there are multiple patent families with claims that cover the SEL-212 product, one of which is a licensed, issued U.S. patent that expired in August 2021).
In addition, we have exclusively or non-exclusively licensed intellectual property, including U.S. issued patents, foreign issued patents, and pending applications in both the U.S. and foreign jurisdictions. The licensed patents and patent applications cover various aspects of the technology being developed by us, including claims directed to compositions of matter and methods of use, and have been filed in various countries worldwide including in North America, Europe and Asia, with material expiration dates varying to, if claims are issued, 2042. In addition to filing and prosecuting patent applications in the United States, we often file analogous patent applications in the European Union and in additional foreign countries where we believe such filing is likely to be beneficial, including but not limited to Australia, Brazil, Canada, China, India, Israel, Japan, Mexico and/or South Korea.costs incurred while conducting and completing the Phase 3 DISSOLVE trials, except for any costs of additional development activities required that are related to ImmTOR and that are unrelated to SEL-212. Sobi will have control and responsibility over all regulatory filings, including any IND, BLA, and marketing authorization applications relating to the licensed product.
Each patent’s term dependsThe transactions contemplated by the Sobi License were consummated on July 28, 2020. Sobi may terminate the Sobi License for any reason upon 180 days’ written notice, whereby all rights granted under the lawsSobi License would revert back to us. In addition, if Sobi were to terminate the Sobi License, we have the option to obtain a license to all patents and know-how necessary to exploit SEL-212 in existence as of the countriestermination date from Sobi in which they are obtained. The patent term in most countries in which we file is 20 years from the earliest date of filing of a non-provisional patent application. Notably, the term of U.S. patents may be extended duereturn for making an equitable royalty payment to delays incurred due to compliance with FDA or by delays encountered during prosecution that are caused by the USPTO. For example, the Hatch-Waxman Act permits a patent term extension for FDA-approved drugs of up to five years beyond the expiration of the patent, depending upon the length of time the drug is under regulatory review. There is a limit to the amount of time a patent may be extended in the United States; no patent extension can extend the remaining term of a patent beyond a total of 14 years from the date of product approval and only one patent applicable to an approved drug may be extended. Similar patent term extensions are available in Europe and other jurisdictions for patents that cover regulatory-approved drugs. Currently, we own or license patents and patent applications with expected material expiration dates ranging from 2032 to 2043. However, the actual patent protection period varies on a product-by-product basis, from country-to-country, and depends upon many factors, including the type of patent, the scope of its coverage, the availability of regulatory-related extensions, the availability of legal remedies in a particular country and the validity and enforceability of the patent.Sobi.
Competition
The biotechnology and pharmaceutical industries are characterized by rapidly advancing technologies, intense competition, and a strong emphasis on proprietary products. We face potential competition from many different sources, including pharmaceutical and biotechnology companies, academic institutions, and governmental agencies, and public and private research institutions. Product candidates that we successfully develop and commercialize may compete with existing therapies and new therapies that may become available in the future.
Our competitors may have significantly greater financial resources, established presence in the market, expertise in research and development, manufacturing, preclinical and clinical testing, obtaining regulatory approvals and reimbursement and marketing approved products than we do. These competitors also compete with us in recruiting and retaining qualified scientific, sales, marketing and management personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies.
The key competitive factors affecting the success of any other tolerance or immune modulationcell therapy product candidates that we develop, if approved, are likely to be their efficacy, safety, convenience, price, the level of generic competition and the availability of reimbursement from government and other third-party payors.
Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are more effective, have fewer or less severe side effects, are more convenient or are less expensive than any products that we may develop. Our competitors also may obtain FDA or other regulatory approval for their products more rapidly than we may
obtain approval for ours. In addition, our ability to compete may be affected in many cases by insurers or other third-party payors seeking to encourage the use of generic or biosimilar products.
SEL-212Descartes-08 may compete with othersproducts of other companies in the goutMG market, including pegloticase, which contains a pegylated uricase similar to the pegadricase component of SEL-212Argenx SE, UCB S.A., Johnson & Johnson, Alexion Pharmaceuticals, Inc. and is indicated for the treatment of refractory gout. HorizonCabaletta Bio, Inc.
Other companies developing CAR-T therapies include large, fully integrated pharmaceutical companies such as Novartis AG, Gilead Sciences, Inc., through its Kite Pharma, plc, whose affiliates own pegloticase, may find other approaches to eliminate undesired immunogenicity to pegloticase. Long-term treatment with global immunosuppressive products may increase the susceptibility to contract infections, tumorsInc. subsidiary, Bristol-Myers Squibb Company, AstraZeneca PLC and may lead to organ failure.
Large companies with active research to prevent the formation of ADAsJanssen Pharmaceuticals, Inc. and treat allergies or autoimmune diseases include Eli Lilly, Roche Holding AG, Sanofi S.A., Pfizer Inc., and Merck & Co., Inc. Small, early-stage biopharmaceutical companies active in the research for new technologies to induce antigen-specific immune tolerance include Abatasuch as Kyverna Therapeutics, Anokion SA, Cour Pharmaceutical Development Company, Inc., Cue Biopharma, Parvus Therapeutics, REGiMMUNE Corporation, Topas Therapeutics GmbH, Sangamo Therapeutics, and SQZ Biotechnologies. Biopharmaceutical companies active in the research for MMA include BridgeBio Pharma,Cabaletta Bio, Inc., HemoShear Therapeutics, Inc., LogicBio, Poseida, and Moderna. Biopharmaceutical companies active in the research for IgA nephropathy include Omeros, Travere, Novartis, Ionis, Visterra, Chinook Therapeutics, Calliditas Therapeutics AB, Vera Therapeutics, and Eledon Pharmaceuticals. Biopharmaceutical companies active in autoimmune disease research include Calliditas Therapeutics AB, CymaBay, Genfit, and Intercept Pharmaceuticals. Large biopharmaceutical companies active in the IL-2 research include Abbvie, Inc., Amgen, Bristol Myers Squibb, Eli Lilly, Sanofi S.A., and Merck & Co., Inc.
Government Regulation
Government authorities in the United States, at the federal, state and local level, and in other countries extensively regulate, among other things, the research, development, testing, manufacturing, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, post-approval monitoring and reporting, marketing and export and import of products such as those we are developing.
SEL-212 isWe believe our cell therapy product candidates are subject to regulation in the United States as a combination product. If marketed individually, each component would be subject to different regulatory pathways and would require approval of independent marketing applications by the FDA. A combination product, however, is assigned to a Center that will have primary jurisdiction over its regulation based on a determination of the combination product’s primary mode of action, which is the single mode of action that provides the most important therapeutic action. In the case of SEL-212, we believe that the primary mode of action is attributable to the biologic component of the product. In the case of SEL-212, which we believe will be regulated as a therapeutic biologic, the FDA’s Center for Drug Evaluation and Research,“biologics” or CDER, will have primary jurisdiction over premarket development.“biological products”. We expect to seek approval of SEL-212Descartes-08 through a single BLA reviewed by CDER,FDA’s Center for Biologics Evaluation and we do not expect that the FDA will require a separate marketing authorization for each constituent of SEL-212.Research, or CBER.
Biological products are subject to regulation under the Federal Food, Drug, and Cosmetic Act, or FD&C Act and the Public Health Service Act, or PHS Act, and other federal, state, local and foreign statutes and regulations. SEL-212Descartes-08 and any other product candidates that we develop must be approved by the FDA before they may be legally marketed in the United States and by the appropriate foreign regulatory agency before they may be legally marketed in foreign countries.
We regard our mRNA-modified products as cell therapy products and not as genetic engineering or gene therapy products, because mRNA modifications are not embodied in DNA or incorporated into a genome. However, it is possible that in some jurisdictions, regulations on genetic engineering or genetic therapy may intentionally or unintentionally apply to our technology. This could create additional regulatory burden.
U.S. biological products development processBiological Products Development Process
The process required by the FDA before a biologic, including a genecell therapy, may be marketed in the United States is summarized below.
Biological product candidates are preclinically tested before any testing is done in humans. These tests, or non-clinical studies, include laboratory evaluations of product chemistry, toxicity and formulation, as well as animal studies to assess the potential safety and activity of the product candidate. The conduct of the preclinical tests must comply with federal regulations and requirements including good laboratory practices, or GLPs.
The clinical study sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and a proposed clinical protocol, to the FDA as part of the IND which must become effective before human clinical trials may begin. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA places the clinical study on a clinical hold within that 30-day time period. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical study can begin. The FDA may also impose clinical holds on a biological product candidate at any time before or during clinical trials due to safety concerns, non-compliance with regulatory requirements, or non-compliance.other issues. If the FDA imposes a clinical hold, trials may not recommence without FDA authorization and then only under terms authorized by the FDA. In addition to these requirements, biological product candidates may also require evaluation and assessment by an institutional biosafety committee, or IBC, that reviews and oversees research utilizing recombinant or synthetic nucleic acid molecules at that institution.an institution participating in a clinical trial.
Clinical trials are conducted under protocols detailing among other things, the objectives of the clinical study, dosing procedures, patient selection and exclusion criteria, and the parameters to be used to monitor patient safety. Each protocol and
any amendments to the protocol must be submitted to the FDA as part of the IND. Clinical trials must be conducted and monitored in accordance with the FDA’s regulations, comprising theincluding with respect to good clinical practice, or GCP, requirements, including the requirement that all research subjects provide informed consent. Further, each clinical study must be reviewed and approved by an independent institutional review board, or IRB, at or servicing each institution at which the clinical study will be conducted. Human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
•Phase 1.The biological product candidate is initially introduced into healthy human patients and tested for safety.
•Phase 2.The biological product candidate is evaluated in a limited patient population of patients or healthy volunteers to identify the maximum tolerated dose, recommended Phase 2 dose, possible adverse effects and safety risks,risks. For the types of products and therapeutic areas we focus on, Phase 1 studies will generally be done in patients and not healthy volunteers.
Phase 2. The biological product candidate is evaluated in a broader population to evaluate safety further and preliminarily evaluate the efficacy of the product for specific targeted diseases, and to determine dosage tolerance,the optimal dosage and dosing schedule.
•Phase 3.Clinical trials are undertaken to further evaluate dosage, clinical efficacy, potency, and safety in an expanded patient population at geographically dispersed clinical study sites. These clinical trials are intended to establish the overall risk/benefit ratio of the product candidate and provide an adequate basis for product labeling.
Cell and gene therapy products may differ from the traditional clinical trial phases. For example, clinical trials for cell and gene therapy products are often structured as a hybrid Phase 1/2 study where a small group of participants with the disease are enrolled and both safety and efficacy tests are performed.
Post-approval clinical trials, sometimes referred to as Phase 4 clinical trials, may be conducted after initial marketing approval to gain additional experience from the treatment of patients in the intended therapeutic indication, particularly for long-term safety follow-up.
The FDA or the sponsor or itsa separate data safety monitoring board may suspend or terminate a clinical study at any time on various grounds. Similarly, an IRB can suspend or terminate approval of a clinical study at its institution if the clinical study is not being conducted in accordance with the IRB’s requirements or if the biological product candidate has been associated with unexpected serious harm to patients.patients or otherwise in the interest of patient welfare.
Sponsors of clinical trials of FDA-regulated products, including biologics, are also required to register and disclose certain clinical trial information, which is publicly available at www.clinicaltrials.gov. Concurrent with clinical trials, companies must also finalize a process for manufacturing the product in commercial quantities in accordance with GMP requirements.
After the completion of clinical trials of a biological product candidate, FDA approval of a BLA must be obtained before commercial marketing of the biological product. The BLA must include results of product development, laboratory and animal trials, human trials, information on the manufacture and composition of the product, proposed labeling and other relevant information. In addition, under the Pediatric Research Equity Act, or PREA, a BLA or supplement to a BLA must contain a pediatric assessment unless the applicant has obtained a waiver or deferral. Pediatric assessment contains data gathered from pediatric studies using appropriate formulations for each age group for which the assessment is required and other data adequate to assess the safety and effectiveness of the biological product candidate for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The Food and Drug Administration Safety and Innovation Act, or FDASIA, requires that sponsors planning to submit a BLASponsors with an application for a biological product that in certain circumstances nnew active ingredient, new indication, new dosage form, new dosing regimen, or new route of administration must submit an initial Pediatric Study Plan, or PSP, (or a deferral or waiver, as appropriate) within sixty60 days after a Type Cof an end-of-Phase 2 meeting or as may be agreed between the sponsor and FDA. Unless otherwise required by regulation, PREA does not apply to any biological product for an indication for which orphan designation has been granted.
Under the Prescription Drug Fee User Act, as amended, or PDUFA, as amended, each BLA must be accompanied by a substantial user fee. Fee waiver or reductions are available under certain circumstances, including for the first application filed by a small business. In addition, no user fees are assessed on BLAs on products designated as orphan drugs unless the product also includes a non-orphan indication.
Within 60 days following submission of the application, the FDA reviewsconducts a preliminary review of a BLA to determine whether they are sufficiently complete to permit substantive review. The FDA may request additional information before deciding whether to accept a BLA for completeness.filing. The FDA may refuse to file any BLA that it deems incomplete or otherwise not reviewable and may request additional information. OnceIf the submission is accepted for filing, the FDA substantively reviews the BLA to determine, among other things, whether the proposed product is safe, pure and potent, and manufactured in accordance with cGMP requirements.appropriate procedures and controls to ensure product quality. The FDA may refer applications for novel biological products or biological products that present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a non-binding recommendation on approval. The FDA may waive the review by an advisory committee and is not bound by the recommendation of an advisory committee, but it often follows such recommendations. During the biological product approval process, the FDA also will review proposed product labeling and will determine whether a Risk Evaluation and Mitigation Strategy, or REMS, is necessary to assure the safe use of the biological product candidate. If the FDA concludes a REMS is needed, the sponsor of the BLA must submit a proposed REMS; the FDA will not approve the BLA without a REMS, if required.
Before approving a BLA, the FDA will inspect the facilities in which the product is manufactured to determine whether the manufacturing processes and facilities are in compliance with GMPs.cGMPs. The FDA may also audit the clinical investigation sites to determine that they have complied with good clinical practices.
Notwithstanding the submission of relevant data and information, the FDA may ultimately deny approval.approval or seek additional information from the applicant. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently than the applicant interprets the same data. The FDA may also raise questions about product manufacturing and quality control. If the FDA denies approval of a BLA in its then-current form, the FDA will issue a complete response letter detailing deficiencies in the application. If a response letter is issued, the applicant may either resubmit the BLA, addressing all of the deficiencies identified in the letter, or withdraw the application.
One of the performance goals agreed to by the FDA under PDUFA is to review 90% of standard BLAs in 10 months from the filing date and 90% of priority BLAs in six months from the filing date, whereupon a review decision is to be made. Two additional months are added to these timelines for new molecular entities. The FDA does not always meet its PDUFA goal dates for standard and priority BLAs.
Orphan Designation
Prior to the submission of a BLA, the FDA may grant orphan designation to drugs or biologics intended to treat a rare disease or condition that affects fewer than 200,000 individuals in the United States, or if it affects more than 200,000
individuals in the United States, there is no reasonable expectation that the cost of developing and marketing the product for this type of disease or condition will be recovered from sales in the United StatesStates. After the FDA grants orphan designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Orphan designation does not convey any advantage in or shorten the duration of the regulatory review and approval process.
In the United States, orphan designation entitles a party to financial incentives such as opportunities for grant funding towards clinical trial costs, tax advantages and user-fee waivers. In addition, if a product receives the first FDA approval for the indication for which it has orphan designation, the product is entitled to orphan exclusivity, which means the FDA may not approve any other application to market the same product"same drug" for the same indication for a period of seven years, except in limited circumstances, such as a showing of clinical superiority over the product with orphan exclusivity or where the manufacturer with orphan exclusivity is unable to assure sufficient quantities of the approveapproved orphan product. Competitors, however, may receive approval of different products for the indication for which the orphan product has exclusivity or obtain approval for the same product but for a different indication for which the orphan product has exclusivity. Orphan product exclusivity also could block the approval of one of our products for seven years if a competitor obtains approval of the same biological product as defined by the FDA or if our product candidate is determined to be contained within the competitor’s product for the same indication or disease. If a drug or biological product designated as an orphan product receives marketing approval
Descartes-08 has been granted Orphan Drug Designation for an indication broader than what is designated, it may not be entitled to orphan product exclusivity.
Rare Pediatric Disease Designation
The Rare Pediatric Disease designation program allows for a sponsor who receives an approval for a product to potentially qualify for a voucher that can be redeemed to receive a priority reviewthe treatment of a subsequent marketing application for a different product.MG.
Expedited Development and Review Programs
The FDA has aoffers various programs, including the Fast Track program, Breakthrough Therapy designation, and the RMAT designation that isare intended to expedite or facilitate the process for reviewing new biological products that meet certain criteria. Specifically, new biological products are eligible for Fast Track designation if they are intended to treat a serious or life-threatening disease or condition and demonstrate the potential to address unmet medical needs for the disease or condition. Fast Track designation applies to the combination of the product and the specific indication for which it is being studied. The sponsor of a new biologic may request that the FDA designate the biologic as a Fast Track product at any time during the clinical development of the product.
Any product submitted to the FDA for marketing, including under a Fast Track program, may be eligible for other types of FDA programs intended to expedite development and review, such as priority review and accelerated approval. Any product is eligible for priority review if it treats a serious condition and, if approved, would provide a significant improvement in safety or effectiveness of treatment, diagnosis, or prevention compared to marketed products. available therapies.
Additionally, a product may be eligible for accelerated approval. Biological products studiedThe FDA may approve a product for their safety and effectiveness in treatinga serious or life-threatening illnesses and that provide meaningful therapeutic benefit over existing treatments may be eligible for accelerated approval, which means that they may be approveddisease or condition based on the basis of studies establishinga determination that the product has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. As a condition of approval, the FDA may require that a sponsor of a biological product subject to accelerated approval perform adequate and well-controlled post-marketing clinical studies to confirm such benefit. The Food and Drug Omnibus Reform Act of 2022, or FDORA, added the failure to conduct post-approval studies with due diligence or to submit timely progress reports on such studies to the list of prohibited acts under the FD&C Act, which means that any such failures, whether they result from a sponsor’s actions or the actions of third parties, could provide the basis for enforcement actions. In addition, the FDA currently requires as a condition for accelerated approval pre-approval ofthat promotional materials be submitted prior to use, which could adversely impact the timing of the commercial launch of the product. Fast Track designation, priority review and accelerated approval do not change the standards for approval but may expedite the development or approval process.
In addition, under the provisions of The Food and Drug Safety and Innovation Act, or FDASIA, the FDA established a Breakthrough Therapy Designation which is intended to expedite the development and review of products that treat serious or life-threatening diseases or conditions. A breakthrough therapy is defined as a drug that is intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition, and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. The designation includes all of the features of Fast Track designation, as well as more intensive FDA interaction and guidance. Fast Track, priority review, accelerated approval, and breakthrough therapy designations do not change the standards for approval and may not necessarily expedite the development or approval process.
In 2016, the 21st Century Cures Act established what the FDA describes as a regenerative medicine adventure therapy, or RMAT, designation. The RMAT designation program is intended to facilitate an efficient development program for, and expedite review of, any product that meets the following criteria: (i) the product qualifies as an RMAT, which is defined as a cell therapy, therapeutic tissue engineering product, human cell and tissue product, or any combination product using such therapies or products, with limited exceptions; (ii) the product is intended to treat, modify, reverse, or cure a serious or life-threatening disease or condition; and (iii) preliminary clinical evidence indicates that the product has the potential to address
unmet medical needs for such a disease or condition. RMAT designation provides all the benefits of Breakthrough Therapy Designation, including early interactions to discuss any potential surrogate or intermediate endpoints to be used to support accelerated approval, eligibility for rolling review and potential eligibility for priority review. Product candidates granted RMAT designation may also be eligible for accelerated approval on the basis of a surrogate or intermediate endpoint reasonably likely to predict long-term clinical benefit, or reliance upon data obtained from a meaningful number of clinical trial sites, including through expansion of trials to additional sites, as appropriate.
Post-approval Requirements
Rigorous and extensive FDA regulation of biological products continues after approval, particularly with respect to cGMP requirements. Manufacturers of our products are required to comply with applicable requirements in the cGMP regulations, including quality control and quality assurance and maintenance of records and documentation. Other post-approval requirements applicable to biological products include record-keeping requirements, reporting of adverse effects and reporting updated safety and efficacy information.
We also must comply with the FDA’s advertising and promotion requirements, such as those related to direct-to-consumer advertising, the prohibition on promoting products for uses or in patient populations that are not described in the product’s approved labeling, (knownknown as “off-label use”), industry-sponsored scientific and educational activities and promotional activities involving the internet.requirement to balance information provided about a product’s benefits with important safety information. Discovery of previously unknown problems or the failure to comply with the applicable regulatory requirements may result in restrictions on the marketing of a product or withdrawal of the product from the market as well as possible civil or criminal sanctions. Failure to comply may subject an applicant or manufacturer to administrative or judicial civil or criminal sanctions, expensive and onerous government investigations, and adverse publicity.
Conventional DNA-modified CAR-T cell products have been subject to extensive post-approval surveillance requirements. Because the mRNA of our products is temporary, we do not believe that our mRNA-modified products will be subject to requirements of this nature, although other post-approval requirements will apply.
Biosimilars and Exclusivity
The Patient Protection and Affordable Care Act, or ACA, includes a subtitle called the Biologics Price Competition and Innovation Act of 2009, or BPCIA, which created an abbreviated approval pathway for biological products that are biosimilar to or interchangeable with an FDA-licensed reference biological product.
Biosimilarity, which requires that there be no clinically meaningful differences between the biological product and the reference product in terms of safety, purity and potency, can be shown through analytical studies, animal studies and a clinical study or studies. However, complexities associatedThe FDA has approved a number of products under these provisions.
To the Company’s knowledge, the definition of “biosimilar” with the larger,regard to an mRNA-modified cell therapy has not been expressly stated in statute, regulation, or guidance, and often more complex, structureshas not been reviewed by a court. The regulatory pathway for a biosimilar to one of biologicalour products as well as the processes by which such products are manufactured, pose significant hurdles to implementation of the abbreviated approval pathway that are still being worked out by the FDA.thus remains somewhat uncertain.
Under the BPCIA, an application for a biosimilar product may not be submitted to the FDA until four years following the date that the reference product was first licensed by the FDA. In addition, the approval of a biosimilar product may not be made effective by the FDA until 12 years from the date on which the reference product was first licensed. During this 12-year period of exclusivity, another company may still market a competing version of the reference product if the FDA approves a full BLA for the competing product containing the sponsor’s own preclinical data and data from adequate and well-controlled clinical trials to demonstrate the safety, purity and potency of their product.
A biological product canmay also obtain pediatric market exclusivity in the United States. PediatricFor a biological product, pediatric exclusivity, if granted, adds six months to existing regulatory exclusivity periods and patent terms.periods. This six-month exclusivity, which runs from the end of other exclusivity protection, or patent term, may be granted based on the voluntary completion of a pediatric study in accordance with an FDA-issued “Written Request” for such a study.study or studies.
The BPCIA is complex and continues to be interpreted and implemented by the FDA. As a result, the ultimate impact, implementation and meaning of the BPCIA is subject to significant uncertainty.
Government Regulation Outside of the United States
Whether or not we obtain FDA approval for a product, we must obtain the requisite approvals from regulatory authorities in foreign countries outside the United States prior to the commencement of clinical studies or marketing of the product in those countries.
The requirements and process governing the conduct of clinical studies, product licensing, pricing and reimbursement vary from country to country. In all cases, the clinical studies are conducted in accordance with GCP and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki.
In the European Economic Area, or EEA, which is composed of the 27 member states of the European Union, or EU, plus Norway, Iceland and Liechtenstein, medicinal products can only be commercialized after obtaining a Marketing Authorization, or MA.
There are two types of MAs.
•The CommunityEU MA, which is issued by the European Commission through the Centralized Procedure, based on the opinion of the Committee for Medicinal Products for Human Use, or CHMP, of the European Medicines Agency, or EMA, and which is valid throughout the entire territory of the EEA. The Centralized Procedure is mandatory for certain types of products, such as biotechnology medicinal products, orphan medicinal products, advanced therapy medicinal products (such as(comprising gene therapy, somatic cell therapy and tissue engineered products), among others. The Centralized Procedure is optional for other products containing a new active substance not yet authorized in the EEA, or for other products that constitute a significant therapeutic, scientific or technical innovation or which are in the interest of public health in the EU. Under the Centralized Procedure the maximum timeframe for the evaluation of a marketing authorization application is 210 days (excluding clock stops, when additional written or oral information is to be provided by the applicant in response to questions asked by the CHMP). Accelerated evaluation might be granted by
the CHMP in exceptional cases. Under the accelerated procedure the standard 210 days review period is reduced to 150 days.
•National MAs, which are issued by the competent authorities of the Member States of the EEA and only cover their respective territory, are available for products not falling within the mandatory scope of the Centralized Procedure. Where a product has already been authorized for marketing in a Member State of the EEA, this National MA can be recognized in another Member States through the Mutual Recognition Procedure. If the product has not received a National MA in any Member State at the time of application, it can be approved simultaneously in various Member States through the Decentralized Procedure.
To obtain regulatory approval of an investigational biologicalmedical product under European UnionEU regulatory systems, we must submit a marketing authorization application, which is similar to the U.S. BLA. The European UnionEU also provides opportunities for market exclusivity. Upon receiving marketing authorization, new chemical entities“new active substances” generally receive eight years of data exclusivity, which prevents regulatory authorities in the European UnionEU from referencing the innovator’s data to assess a generic or biosimilar application, and an additional two years of market exclusivity, during which no generic or biosimilar product can be marketed. However, there is no guarantee that a product will be considered by the European Union’sEU’s regulatory authorities to be a new chemical entity,active substance, and products may not qualify for data exclusivity. Products receiving orphan designation in the European UnionEU can receive ten years of market exclusivity, during which time no marketing authorization application shall be accepted, and no marketing authorization shall be granted for a similar medicinal product for the same indication. An orphan product can also obtain an additional two years of market exclusivity in the European UnionEU for pediatric studies. No extension to any supplementary protection certificate can be granted on the basis of pediatric studies for orphan indications.
The criteria for designating an “orphan medicinal product” in the European UnionEU are similar in principle to those in the United States. Under Article 3 of Regulation (EC) 141/2000, a medicinal product may be designated as orphan if (1)(i) it is intended for the diagnosis, prevention or treatment of a life-threatening or chronically debilitating condition; (2)(ii) either (a) such condition affects no more than five in 10,000 persons in the European UnionEU when the application is made, or (b) the product, without the benefits derived from orphan status, would not generate sufficient return in the European UnionEU to justify investment; and (3)(iii) there exists no satisfactory method of diagnosis, prevention or treatment of such condition authorized for marketing in the European Union,EU, or if such a method exists, the product will be of significant benefit to those affected by the condition, as defined in Regulation (EC) 847/2000. Orphan medicinal products are eligible for certain financial and exclusivity incentives.
For other countries outside of the European Union,EU, such as countries in Eastern Europe, Latin America or Asia, the requirements governing the conduct of clinical studies, product licensing, pricing and reimbursement vary from country to country. In all cases, again, the clinical studies are conducted in accordance with GCP and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki.
When conducting clinical trials in the EU, we must adhere to the provisions of the European UnionEU Clinical Trials Directive (Directive 2001/20/EC) and the laws and regulations of the EU Member States implementing them. These provisions require, among other things, that the prior authorization of an Ethics Committee and the competent Member State authority is obtained before commencing the clinical trial. In 2014, the EU passed the Clinical Trials Regulation (Regulation 536/2014), which will replace the current Clinical Trials Directive, to ensure that the rules for clinical trials are identical throughout the European Union.EU.
We are also subject to data privacy and security laws in the jurisdictions outside of the U.S. in which we are established, run clinical trials or in which we sell or market our products once approved. For example, in Europe we are subject to Regulation (EU) 2016/679 (General Data Protection Regulation, or GDPR) in relation to our collection, control, processing and other use of personal data (i.e., data relating to an identifiable living individual). We process personal data in relation to participants in our clinical trials in the EEA.,EEA, including the health and medical information of these participants. The GDPR is directly applicable in each E.U.EU Member State, however, it provides that E.U.EU Member States may introduce further conditions, including limitations which could limit our ability to collect, use and share personal data (including health and medical information), or could cause our compliance costs to increase, ultimately having an adverse impact on our business. The GDPR
imposes accountability and transparency obligations regarding personal data. We are also subject to E.U.EU rules with respect to cross-border transfers of personal data out of the E.U.EU and EEA. We are subject to the supervision of local data protection authorities in those E.U.EU jurisdictions where we are established or otherwise subject to the GDPR. A breach of the GDPR could result in significant fines, regulatory investigations, reputational damage, orders to cease/ change our use of data, enforcement notices, as well potential civil claims including class action type litigation where individuals suffer harm. Moreover, the United Kingdom leaving the E.U.EU could also lead to further legislative and regulatory changes. It remains unclear how the United Kingdom data protection laws or regulations will develop in the medium to longer term and how data transfer to the United Kingdom from the E.U.EU will be regulated. However, the United Kingdom has transposed the GDPR into domestic law with the Data Protection Act 2018, which remains in force following the United Kingdom’s departure from the EU.
Other Healthcare Laws
The federal Anti-Kickback Statute prohibits, among other things, any person or entity from knowingly (regardless of knowledge of this specific statute) and willfully offering, paying, soliciting, receiving or providing any remuneration, directly or indirectly, overtly or covertly, to induce or in return for purchasing, leasing, ordering, or arranging for or recommending the purchase, lease, or order of any item or service reimbursable, in whole or in part, under Medicare, Medicaid or other federal healthcare programs. The term “remuneration” has been broadly interpreted to include anything of value. The Anti-Kickback Statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on the one hand and prescribers, purchasers, and formulary managers, and other third parties on the other. The majority of states also similar have anti-kickback laws, which in some cases are more restrictive.apply to items and services reimbursed by private insurance.
The federal false claims and civil monetary penalties laws, including the civil False Claims Act, prohibit any person or entity from, among other things, knowingly presenting, or causing to be presented, a false, fictitious or fraudulent claim for payment to, or approval by, the federal government or knowingly making, using, or causing to be made or used a false record or statement material to a false or fraudulent claim to the federal government, or from knowingly making a false statement to avoid, decrease or conceal an obligation. A claim includes “any request or demand” for money or property presented to the U.S. government. Manufacturers can be held liable under false claims laws, even if they do not submit claims to the government, where they are found to have caused submission of false claims by, among other things, providing incorrect coding or billing advice about their products to customers that file claims, or by engaging or off-label promotion to customers that file claims. Violation of the federal Anti-Kickback Statute may also constitute a false or fraudulent claim for purposes of the federal civil False Claims Act. Actions under the civil False Claims Act may be brought by the Department of Justice or as a qui tam action by a private individual in the name of the government. Violations of the civil False Claims Act can result in very significant monetary penalties and treble damages, and may be accompanied by additional civil monetary penalties against individuals. Many states also have similar fraud and abuse statutes or regulations that apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor.
The federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, prohibits, among other actions, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payors, knowingly and willfully embezzling or stealing from a healthcare benefit program, willfully obstructing a criminal investigation of a healthcare offense, and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
In addition, there has been a recent trend of increased federal and state regulation of payments made to physicians and certain other healthcare providers. The ACA imposed, among other things, new annual reporting requirements through the Physician Payments Sunshine Act for coveredrequires applicable manufacturers forto annually report certain payments and “transfers of value” provided to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors), certain other health care professionals beginning in 2022, and teaching hospitals,providers, as well as ownership and investment interests held by physicians and their immediate family members.
Sanctions under these federal and state fraud and abuse laws may include civil monetary penalties and criminal fines, exclusion from government healthcare programs, and imprisonment.
We may also be subject to data privacy and security regulation by both the federal government and the states in which we conduct our business. HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, or HITECH, and their respective implementing regulations, impose specified requirements relating to the privacy, security and transmission of individually identifiable health information held by covered entities and their business associates. Among other things, HITECH made HIPAA’s security standards directly applicable to, as well as imposed certain other privacy obligations on, “business associates,” defined as independent contractors or agents of covered entities that create, receive, maintain or transmit protected health information in connection with providing a service for or on behalf of a covered entity. HITECH also increased the civil and criminal penalties that may be imposed and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorney’s fees and costs associated with pursuing federal civil actions. Even when HIPAA does not apply, according to the Federal Trade Commission, or the FTC, failing to take appropriate steps to keep consumers’ personal information secure constitutes unfair acts or practices in or affecting commerce in violation of Section 5(a) of the Federal Trade Commission Act, or the FTCA, 15 U.S.C § 45(a).
Coverage and Reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of any pharmaceutical or biological products for which we obtain regulatory approval. In the United States and markets in other countries, patients who are prescribed treatments for their conditions and providers performing the prescribed services generally rely on third-party payors to reimburse all or part of the associated healthcare costs. Sales of any products for which we receive regulatory approval for commercial sale will therefore depend, in part, on the availability of coverage and adequate reimbursement from third-party payors. Third-party payors include government authorities, managed care plans, private health insurers and other organizations.
The process for determining whether a third-party payor will provide coverage for a pharmaceutical or biological product typically is separate from the process for setting the price of such product or for establishing the reimbursement rate that the payor will pay for the product once coverage is approved. Third-party payors may limit coverage to specific products on an approved list, also known as a formulary, which might not include all of the FDA-approved products for a particular indication.
A decision by a third-party payor not to cover our product candidates could reduce physician utilization of our products once approved and have a material adverse effect on our sales, results of operations and financial condition. Moreover, a third-party payor’s decision to provide coverage for a pharmaceutical or biological product does not imply that an adequate reimbursement rate will be approved. Adequate third-party reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development.
Healthcare Reform
A primary trend in the U.S. healthcare industry and elsewhere is cost containment. Government authorities and other third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medical products.
Federal, state and local governments in the U.S. have established and continue to consider policies to limit the growth of healthcare costs, including the cost of prescription drugs. Recently there has also been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products, which has resulted in several Congressional inquiries and proposed and enacted legislation designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for prescription drugs.
At the federal level, for example, the Inflation Reduction Act of 2022, or IRA, was signed into law. Key provisions of the IRA include the following, among others:
•The IRA requires manufacturers to pay rebates for Medicare Part B and Part D drugs whose price increases exceed inflation.
•The IRA eliminates the so-called “donut hole” under Medicare Part D beginning in 2025 by significantly lowering the beneficiary maximum out-of-pocket cost and requiring manufacturers to subsidize, through a newly established manufacturer discount program, 10% of Part D enrollees’ prescription costs for brand drugs below the out-of-pocket maximum and 20% once the out-of-pocket maximum has been reached.
•The IRA delays the rebate rule that would require pass through of pharmacy benefit manager rebates to beneficiaries.
•The IRA directs the Centers for Medicare and Medicaid Services, or CMS, to engage in price-capped negotiation for certain Medicare Part B and Part D products. Specifically, the IRA’s Price Negotiation Program applies to high-expenditure single-source drugs and biologics that have been approved for at least seven or 11 years, respectively, among other negotiation selection criteria, beginning with 10 high-cost drugs paid for by Medicare Part D starting in 2026, followed by 15 Part D drugs in 2027, 15 Part B or Part D drugs in 2028, and 20 Part B or Part D drugs in 2029 and beyond. The negotiated prices will be capped at a statutorily determined ceiling price. There are certain statutory exemptions from the IRA’s Price Negotiation Program, such as for a drug that has only a single orphan drug designation and is approved only for an indication or indications within the scope of such designation. The IRA’s Price Negotiation Program is currently the subject of legal challenges.
Manufacturers that fail to comply with the IRA may be subject to various penalties, including civil monetary penalties or a potential excise tax. The IRA permits the Secretary of Health and Human Services, or the HHS Secretary, to implement many of the IRA’s provisions through guidance, as opposed to regulation, for the initial years. The effect of the IRA is anticipated to have significant effects on the pharmaceutical industry and may reduce the prices pharmaceutical manufacturers can charge and reimbursement pharmaceutical manufacturers can receive for approved products, among other effects.
In addition, other legislative changes have been proposed and adopted in the United States since the ACA was enacted.States. This included aggregate reductions of Medicare payments to providers of 2% per fiscal year, which went into effect on April 1, 2013, and, due to subsequent legislative amendments, will stay in effect through 2027 unless additional Congressional action is taken. On January 2, 2013, the American Taxpayer Relief Act was signed into law, which, among other things, further reduced Medicare
payments to several providers, including hospitals and imaging centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. Recently there
The Biden administration has indicated that lowering prescription drug prices is a priority. On October 14, 2022, President Biden signed an executive order to lower prescription drug costs for Americans. In response to this directive, the HHS Secretary announced and the Center for Medicare and Medicaid Innovation is developing three new models intended to lower drug costs under Medicare and Medicaid, including establishing a new approach for administering outcomes-based agreements for cell and gene therapies. President Biden also been heightened governmental scrutiny oversigned an executive order on July 9, 2021 affirming the manner in which manufacturers set prices for their marketed products, which has resulted in several Congressional inquiries and proposed and enacted legislation designedadministration’s policy to, among other things, bring more transparencysupport legislative reforms that would lower the prices of prescription drugs, including by supporting the development and market entry of lower-cost generic drugs and biosimilars, and support the enactment of a public health insurance option. Among other things, the executive order directs the HHS Secretary to productprovide a report on actions to combat excessive pricing reviewof prescription drugs, continue to clarify and improve the relationship between pricingapproval framework for generic drugs and manufacturer patient programs,identify and reformaddress any efforts to impede generic drug competition, enhance the domestic drug supply chain, reduce the price that the federal government program reimbursement methodologies.pays for drugs, and address price gouging in the industry. The executive order also directs the FDA to work with states and Indian Tribes that propose to develop section 804 Importation Programs in accordance with the Medicare Prescription Drug, Improvement, and Modernization Act of 2003, and the FDA’s implementing regulations. The FDA released such implementing regulations on September 24, 2020, which went into effect on November 30, 2020, providing guidance for states to build and submit importation plans for drugs from Canada. In response, authorities in Canada have passed rules designed to safeguard the Canadian drug supply from shortages. On January 5, 2024, the FDA authorized Florida’s Agency for Health Care Administration’s drug importation proposal, the first step toward Florida facilitating importation of certain prescription drugs from Canada.
Employees and Human Capital Resources
At Selecta Biosciences,Cartesian Therapeutics, we consider human capital to be an essential driver of our business and successful strategy creation and execution. Our people, driven by our Collaborative, Pioneering,collaborative, pioneering, and Patient Focusedpatient-focused culture, propel our business forward, strengthening us for long-term success.
As of December 31, 2022,2023, we had 6438 employees, 4726 of whom are primarily engaged in research and development activities and 1712 in corporate functions. 61%37 of our employees are employed by us on a full-time basis. 73.6% of our employees have at least one of a Masters, PhD, or MD degree. Our 2022 annualized voluntary turnover rate was 9.82%. All employees reside and work in the United States and our employees are not represented by a labor union. We consider our employee relations to be strong and in good standing.
Our goal is to continually engage our talented and diverse workforce to drive value creation both for our business and ultimately our patient populations. We believe in a proactive approach to talent management focusing on retention of key talent, critical role successor identification, and impactful employment development. Additional priority areas intended to drive engagement include successful recruitment of diverse talent, continual promotion of professional development at all levels, introduction, and evolution of business-friendly HRhuman resources solutions, coupled with an intentional culture dialog aimed to drive a high engagement, high performance, patient centric culture.
To further drive attraction and retention of our high-quality, experienced, and diverse workforce, we invest in the physical, emotional, and financial well-being of our employees. These investments include a competitive mix of compensation and generous insurance benefits. To assist employees with the rising cost of healthcare, we pay 100% of an employee’s deductible and co-insurance payments. All employees are eligible to participate in our equity compensation programs. All employees are awarded new hire equity and annual equity as well as the opportunity to participate in our Employee Stock Purchase Plan.equity. Employees are also eligible to receive an annual cash bonus and to participate in a 401(k) retirement plan with an industry competitive company match.
Available Information
We file electronically with the SEC our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, proxy statements and other information. Our SEC filings are available to the public over the Internet at the SEC's website at http://www.sec.gov. We make available on our website at www.selectabio.com,www.cartesiantherapeutics.com, free of charge, copies of these reports as soon as reasonably practicable after filing or furnishing these reports with the SEC. The hyperlink to our website is included as an inactive textual reference only, and the information on our website is not incorporated by reference in this Annual Report on Form 10-K or in any other filings we make with the SEC.
RISK FACTORS SUMMARY
Investing in our common stock involves various risks. You should carefully read and consider the matters discussed in this Annual Report under the heading “Risk Factors,” which include the following risks:
•We are a clinical-stagedevelopment-stage company and have incurred significant losses since our inception. We expect to incur losses for the foreseeable future and may never achieve or maintain profitability.
•We will need substantial additional funding in order to complete development of our product candidates and commercialize our products, if approved. If we are unable to raise capital when needed and on terms favorable to us, we could be forced to delay, reduce or eliminate our product development programs or commercialization efforts.
•The terms ofWe develop our credit facility place restrictions on our operating and financial flexibility.
•OurmRNA-based product candidates are based onby leveraging our ImmTORproprietary technology and our manufacturing platform, RNA Armory®, which is an unproven approach designed to induce antigen-specific immune tolerancethe treatment of autoimmune disease. We are early in most of our clinical development efforts and may not be successful in our efforts to biologicbuild a pipeline of product candidates and develop marketable drugs.
•Regulatory authorities in the United States and European Union have limited experience in reviewing and approving gene therapy products.
•Clinical drug development is inherently risky and involves a lengthy and expensive process, with an uncertain outcome.
•The COVID-19 pandemicwhich is subject to a number of factors, many of which are outside of our control. We may continueincur additional costs or experience delays in completing, or ultimately be unable to adversely impactcomplete, the development and commercialization of our business, including sourcing raw materials and supplies to produce our product candidates, our preclinical studies and clinical trials.
•Geopolitical events, instability and wars can adversely affect both our clinical operations and supply chains that we rely on to advance our drug candidates.
•We rely on 3SBio in China as our primary supplier of pegadricase and on other third parties for the manufacture of our product candidates for preclinical and clinical testing, and expect to continue to do so for the foreseeable future.grow our manufacturing capabilities and resources and we must incur significant costs to develop this expertise and/or rely on third parties to manufacture our products.
•We rely, and expect to continue to rely, on third parties to conduct our clinical trials, and those third parties may not perform satisfactorily, including by failing to meet deadlines for the completion of such trials.
•If we or our licensors are unable to adequately protect our proprietary technology, or obtain and maintain issued patents that are sufficient to protect our product candidates, others could compete against us more directly, which would negatively impact our business.
•We may suffer from effects of macroeconomic instability and inflation.
•We would be reliant on Sobi to execute the marketing and sale of SEL-212, if SEL-212 were approved.
•We may not have the funds necessary to fulfill our obligation to repurchase certain warrants.
•We have been in the past and may in the future be subject to securities class action lawsuits.stockholder litigation.
•The failure to successfully integrate the businesses of Selecta and Old Cartesian in the expected timeframe would adversely affect the Company’s future results.
•We have identified a material weakness in our internal control over financial reporting and may identify additional material weaknesses in the future or otherwise fail to maintain an effective system of internal controls, which may result in material misstatements of our consolidated financial statements or cause us to fail to meet our periodic reporting obligations.
Item 1A. Risk Factors
Investing in our common stock involves a high degree of risk. You should consider carefully the risks described below, together with the other information included or incorporated by reference in this Annual Report. If any of the following risks occur, our business, financial condition, results of operations and future growth prospects could be materially and adversely affected. In these circumstances, the market price of our common stock could decline. Other events that we do not currently anticipate or that we currently deem immaterial may also affect our business, prospects, financial condition and results of operations.
Risks Related to the Development of our Product Candidates
OurWe develop our mRNA-based product candidates are based onby leveraging our ImmTORproprietary technology and our manufacturing platform, RNA Armory®, which is an unproven approach designed to induce antigen-specific immune tolerance to biologic drugs.the treatment of autoimmune disease. We are very early in most of our clinical development efforts and may not be successful in our efforts to use our ImmTOR platform to build a pipeline of product candidates and develop marketable drugs.
All of ourOur mRNA approach to develop product candidates are derived from our ImmTOR platform, whichfor the treatment of autoimmune diseases is an unproven approach to induce antigen-specific immune tolerance and to mitigate the immunogenicity of biologic therapies currently being implemented to treat patients. We are developing our ImmTOR platform to restore self-tolerance to autoantigens and potentially treat autoimmune diseases, to be co-administered with AAV gene therapies to potentially enable redosing of said gene therapies and improve and enable activityapproach. Our most advanced product candidate, Descartes-08 is in biologics (including therapeutic enzymes and other immunogenic drugs).
While we have completed our early development clinical trials and a Phase 2 clinical trial for SEL-212, wedevelopment. We have not completed a clinical trial for any other product candidate, nor have we demonstrated ourthe ability to successfully complete any Phase 3 or other pivotal clinical trials, obtain regulatory approvals, manufacture a commercial product, or arrange for a third party to do so on our behalf, or conduct other sales and marketing activities necessary for successful product commercialization. We may have problems identifying new product candidates and applying our technologies to these other areas. Even if we are successful in identifying new product candidates, they may not be suitable for
clinical development, including as a result of manufacturing difficulties, harmful side effects, limited efficacy or other characteristics that indicate that they are unlikely to be products that will receive marketing approval and achieve market acceptance. The success of our product candidates will depend on several factors, including the following:
•design, initiation and completion of preclinical studies and clinical trials with positive results;
•reliance on third parties, including but not limited to collaborators, licensees, clinical research organizations and contract manufacturing organizations;
•receipt of marketing approvals from applicable regulatory authorities;
•obtaining and maintaining patent and trade secret protection and regulatory exclusivity for our product candidates and not infringing or violating patents or other intellectual property of third parties;
•manufacturability, manufacturing, logistics, and stability of our cell therapies, including autologous cell therapies;
•growing our internal cGMP manufacturing capabilities to support commercial manufacturing or making arrangements with third-party manufacturers for, or establishing, commercial manufacturing capabilities, or establishing such capabilities ourselves;manufacturers;
•launching commercial sales of our products, if and when approved, whether alone or in collaboration with others;
•acceptance of our products, if and when approved, by patients and the medical community;
•effectively competing with other therapies;
•obtaining and maintaining coverage and adequate reimbursement by third-party payors, including government payors, for our products, if approved;
•maintaining an acceptable safety profile of our products following approval; and
•maintaining and growing an organization of scientists and business peoplebusinesspeople who can develop and commercialize our product candidates and technology.
The occurrenceOur failure to successfully execute on of any of the foregoing for any reason would effectively prevent or delay approval of our lead and other product candidates. The ongoing COVID-19 pandemic may continue to adversely impact our business, including our preclinical studies and clinical trials.
We are applying our ImmTOR platform to antigen-specific immune tolerance for gene therapy involving gene augmentation, replacement or editing. Regulatory authorities in the United States and European Union have limited experience in reviewing and approving gene therapy products, which could affect the time and data required to obtain marketing authorization of any of our product candidates.
Our future success depends in part on our successful development of viable gene therapy product candidates utilizing our ImmTOR platform.
The regulatory approval process for gene therapy product candidates can be more lengthy, rigorous and expensive than the process for other better known or more extensively studied product candidates and technologies. Since we are developing novel treatments for diseases in which there is little clinical experience with new endpoints and methodologies, there is heightened risk that the FDA, the European Medicines Agency, or the EMA, or comparable regulatory bodies may not consider the clinical trial endpoints to provide clinically meaningful results, and the resulting clinical data and results may be more difficult to analyze. This may be a particularly significant risk for many of the genetically defined diseases for which we may develop product candidates alone or with collaborators due to small patient populations for those diseases, and designing and executing a rigorous clinical trial with appropriate statistical power is more difficult than with diseases that have larger patient populations. Regulatory agencies administering existing or future regulations or legislation may not allow production and marketing of products utilizing genome editing technology in a timely manner or under technically or commercially feasible conditions. Even if our product candidates obtain required regulatory approvals, such approvals may later be withdrawn as a result of changes in regulations or the interpretation of regulations by applicable regulatory agencies.
Changes in applicable regulatory guidelines may lengthen the regulatory review process for our product candidates, require additional studies or trials, increase development costs, lead to changes in regulatory positions and interpretations, delay or prevent approval and commercialization of such product candidates, or lead to significant post-approval limitations or restrictions. Additionally, adverse developments in clinical trials conducted by others of gene therapy products or products created using genome editing technology, or adverse public perception of the field of genome editing, may cause the FDA, the EMA and other regulatory bodies to revise the requirements for approval of any product candidates we may develop or limit the
use of products utilizing genome editing technologies, either of which could materially harm our business. Furthermore, regulatory action or private litigation could result in expenses, delays or other impediments to our research programs or the development or commercialization of current or future product candidates.
Even if we comply with applicable laws, rules, and regulations, and even if we maintain close coordination with the applicable regulatory authorities with oversight over our product candidates, our development programs may fail to succeed. Regulatory authorities have substantial discretion in the approval process and may refuse to accept a marketing application as deficient or may decide that our data is insufficient for approval and require additional preclinical, clinical or other studies. In addition, varying interpretations of the data obtained from preclinical and clinical testing could delay, limit or prevent marketing approval of a product candidate. Additionally, adverse developments in clinical trials conducted by others of gene therapy products or products created using genome editing technology, or adverse public perception of the field of genome editing, may cause the FDA and other regulatory bodies to revise the requirements for approval of any product candidates we may develop or limit the use of products utilizing genome editing technologies, either of which could materially harm our business. Delay or failure to obtain, or unexpected costs in obtaining, the regulatory approval necessary to bring a potential product to market would materially and adversely affect our business, financial condition, results of operations and prospects.
Clinical drug development is inherently risky and involves a lengthy and expensive process with an uncertain outcome.which is subject to a number of factors, many of which are outside of our control. We may incur additional costs or experience delays in completing, or ultimately be unable to complete, the development and commercialization of our product candidates.
Except for SEL-212Clinical development is expensive, time consuming and SEL-302, our product candidates are in preclinical development.involves significant risk. It is impossible to predict when or if any of our product candidates will prove effective and safe in humans or will receive regulatory approval, and the risk of failure through the development process is high. Before obtaining marketing approval from regulatory authorities for the sale of any product candidate, we must complete manufacturing and preclinical development and then conduct extensive clinical trials to demonstrate the safety and efficacy of our product candidates in humans. Manufacturing cell therapies, particularly those modified with mRNA, is a new field.
Preclinical development is costly and inherently uncertain. Early preclinical results may not be predictive of future results, however, if our technology proves to be ineffective or unsafe as a result of, among other things, adverse side effects, pre-existing anti-drug antibodies that can neutralize the viral vector and block gene transfer, or cellular immune response to the transduced cells, we may incur additional costs or experience delays in completing, or ultimately be unable to complete, the clinical development and commercialization of our product candidates.
Before obtaining marketing approval from regulatory authorities for the sale of our product candidates, we must conduct extensive clinical trials to demonstrate the safety and efficacy of the product candidate for its intended indications. Clinical testing is expensive, difficult to design and implement, can take many years to complete and its outcome is inherently uncertain. A failed clinical trial can occur at any stage of testing. Moreover, the outcome of preclinical testing and early clinical trials may not be predictive of the success of later clinical trials, and interim results of a clinical trial do not necessarily predict final results. For example, the topline clinical trial results we reported from our Phase 2 head-to-head COMPARE study of SEL-212 and the top-line data from our SEL-399 program may not be predictive of future results. Moreover, we may not be able to complete, or may be required to deviate from the current clinical trial protocol for a variety of reasons.
Many companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in clinical trials after achieving positive results in preclinical development or early-stage clinical trials, and we cannot be certain that we will not face similar setbacks. Serious adverse events, or SAEs, caused by, or other unexpected properties of, any product candidates that we may choose to develop could cause us, an institutional review board or regulatory authority to interrupt, delay or halt clinical trials of one or more of such product candidates and could result in a more restrictive label or the delay or denial of marketing approval by the FDA or comparable non-U.S. regulatory authorities. If any product candidate that we may choose to develop is associated with SAEs or other unexpected properties, we may need to abandon development or limit development of that product candidate to certain uses or subpopulations in which those undesirable characteristics would be expected to be less
prevalent, less severe or more tolerable from a risk-benefit perspective. In the SEL-212 Phase 1/2 clinical program, we have observed multiple SAEs, and future SAEs may occur causing us to incur additional costs or experience delays in completing, or causing us to ultimately be unable to complete, the development and commercialization of our product candidates, and delay or prevent our ability to obtain FDA approval. Moreover, preclinical and clinical data is often susceptible to varying interpretations and analyses, and many companies that believed their product candidates performed satisfactorily in preclinical studies and clinical trials nonetheless failed to obtain FDA or other regulatory authority approval. If we fail to produce positive results in clinical trials of our product candidates, the development timeline and regulatory approval and commercialization prospects for our product candidates, and, correspondingly, our business and financial prospects, would be negatively impacted.
In addition, we cannot be certain as to what type and how many clinical trials the FDA will require us to conduct before we may gain regulatory approval to market any of our product candidates in the United States or other countries, if any. Prior to approving a new therapeutic product, the FDA generally requires that safety and efficacy be demonstrated in two adequate and well-controlled clinical trials.
We may experience numerous unforeseen events during, or as a result of, clinical trials that could delay or prevent our ability to receive marketing approval for, or commercialize, our product candidates, including:
•clinical trials of our product candidates may produce unfavorable, incomplete or inconclusive results;
•we may be unable to manufacture our product candidates, which in some cases such as mRNA CAR-T, are manufactured on a patient-by-patient basis;
•regulators or institutional review boards may not authorize us or our investigators to commence a clinical trial or conduct a clinical trial at a prospective trial site or may place a clinical hold on existing clinical trials;
•we may experience delays in reaching, or fail to reach, agreement on acceptable terms with contract research organizations, or CROs, or clinical trial sites;
•we may be unable to recruit suitable patients to participate in a clinical trial, the number of patients required for clinical trials of our product candidates may be larger than we expect, enrollment in these clinical trials may be slower than we expect or participants may drop out of these clinical trials at a higher rate than we expect, or enrollment could be affected by the ongoing COVID-19 pandemic orconflicts in Ukraine and the ongoing conflict in Ukraine;Middle East;
•the number of clinical trial sites required for clinical trials of our product candidates may be larger than we expect;
•our third-party contractors may fail to comply with regulatory requirements or meet their contractual obligations to us in a timely manner, or at all;
•we may have to suspend or terminate clinical trials of our product candidates for various reasons, including a finding that the participants are being exposed to unacceptable health risks;
•investigators, regulators, data safety monitoring boards or institutional review boards may require that we or our investigators suspend or terminate clinical research, or we may decide to do so ourselves;
•investigators may deviate from the trial protocol, fail to conduct the trial in accordance with regulatory requirements or misreport study data;
•the cost of clinical trials of our product candidates may be greater than we expect or we may have insufficient resources to pursue or complete certain aspects of our clinical trial programs or to do so within the timeframe we planned;
•the supply or quality of raw materials or manufactured product candidates (whether provided by us or third parties) or other materials necessary to conduct clinical trials of our product candidates may be insufficient, inadequate or not available at an acceptable cost, or in a timely manner, or we may experience interruptions in supply;
•laboratories that we rely upon to perform certain quality control tests may become unavailable, or their services could be delayed;
•regulators may revise the requirements for approving our product candidates, or such requirements may not be as we expect;
•the FDA or comparable foreign regulatory authorities may disagree with our clinical trial design or our interpretation of data from preclinical studies and clinical trials, or may change the requirements for approval even after it has reviewed and commented on the design of our clinical trials;
•regarding trials managed by our existing or any future collaborators, our collaborators may face any of the above issues, and may conduct clinical trials in ways they view as advantageous to them but potentially suboptimal for us; and
•geopolitical events may affect international and overseas trial sites in ways beyond our control.
If we are required to conduct additional clinical trials or other testing of our product candidates beyond those that we currently contemplate, if we are unable to successfully complete clinical trials of our product candidates or other testing, if the results of these trials or tests are not positive or are only modestly positive or if there are safety concerns, or if we are forced to delay or abandon certain clinical trials or other testing in order to conserve capital resources, we may:
•be delayed in obtaining marketing approval for our product candidates, if at all;
•lose the support of collaborators, requiring us to bear more of the burden of research and development;
•obtain marketing approval in some countries and not in others;
•obtain approval for indications or patient populations that are not as broad as intended or desired;
•obtain approval with labeling that includes significant use or distribution restrictions or safety warnings;
•be subject to additional post-marketing testing requirements; or
•have a product removed from the market after obtaining marketing approval.
We could also encounter delays if a clinical trial is suspended or terminated. Authorities may impose such a suspension or termination due to a number of factors, including failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols, inspection of the clinical trial operations or trial site by the FDA or comparable foreign regulatory authorities resulting in the imposition of a clinical hold, unforeseen safety issues or adverse side effects, failure to demonstrate a
benefit from using a drug, changes in governmental regulations or administrative actions or lack of adequate funding to continue the clinical trial. In addition, changes in regulatory requirements and policies may occur, and we may need to amend clinical trial protocols to comply with these changes. Amendments may require us to resubmit our clinical trial protocols to institutional review boards, or IRBs, for reexamination, which may impact the costs, timing or successful completion of a clinical trial.
We filed an IND to conduct a Phase 1/2 clinical trial of our SEL-302 product candidate in pediatric patients with methylmalonic acidemia in the third quarter of 2021. ImmTOR manufacturing continues to proceed in accordance with our expectations, and we have not observed any impact to any of our ImmTOR programs.
Our product development costs will increase if we experience delays in clinical testing or in obtaining marketing approvals. We do not know whether any of our preclinical studies or clinical trials will begin as planned, will need to be restructured or will be completed on schedule, or at all. Significant preclinical or clinical trial delays also could shorten any periods during which we may have the exclusive right to commercialize our product candidates or allow our competitors to bring products to market before we do, potentially impairing our ability to successfully commercialize our product candidates and harming our business and results of operations.
If we experience delays or difficulties in the enrollment of patients in clinical trials, our receipt of necessary regulatory approvals could be delayed or prevented.
We may not be able to initiate or continue clinical trials for our product candidates if we are unable to locate and enroll a sufficient number of eligible patients to participate in these trials as required by the FDA or similar regulatory authorities outside the United States. In addition, from time to time our competitors have ongoing clinical trials for product candidates that treat the same indications as our product candidates, and patients who would otherwise be eligible for our clinical trials may instead enroll in clinical trials of our competitors’ product candidates. The safety of our patients and investigators continues to be our utmost priority. Our inability to enroll a sufficient number of patients for our clinical trials would result in significant delays and could require us to abandon one or more clinical trials altogether. Enrollment delays in our clinical trials may result in increased development costs for our product candidates, which could cause the value of our common stock to decline and limit our ability to obtain additional financing.
We may conduct clinical trials for product candidates at sites outside the United States, and the FDA may not accept data from trials conducted in such locations or the complexity of regulatory burdens may otherwise adversely impact us.
Opening trial sites outside the United States may involve additional regulatory, administrative and financial burdens, including compliance with foreign and local requirements relating to regulatory submission and clinical trial practices. Although the FDA may accept data from clinical trials conducted outside the United States, acceptance of these data is subject to certain conditions imposed by the FDA. For example, the clinical trial must be well designed and conducted and performed by qualified investigators in accordance with good clinical practices, or GCPs, and the FDA must be able to validate the data from the trial through an onsite inspection, if necessary. Generally, the patient population for any clinical trials conducted outside the United States must be representative of the population for which we intend to seek approval in the United States. In addition, while these clinical trials are subject to the applicable local laws, FDA acceptance of the data will be dependent upon its determination that the trials also complied with all applicable U.S. laws and regulations. Nonetheless, there can be no assurance that the FDA will accept data from trials conducted outside the United States. If the FDA does not accept the data from any trial that we conduct outside the United States, it would likely result in the need for additional clinical trials, which would be costly and time-consuming and delay or permanently halt our development of any applicable product candidates.
Additional risks inherent in conducting international clinical trials include:
•foreign regulatory requirements that could burden or limit our ability to conduct our clinical trials;
•increased costs and heightened supply constraints associated with the acquisition of standard of care drugs and/or combination or comparator agents for which we may bear responsibility in certain jurisdictions;
•administrative burdens of conducting clinical trials under multiple foreign regulatory schema;
•foreign exchange fluctuations;
•more burdensome manufacturing, customs, shipment and storage requirements;
•cultural differences in medical practice and clinical research;
•lack of consistency in standard of care from country to country;
•diminished protection of intellectual property in some countries; and
•changes in country or regional regulatory requirements; and
•geopolitical instability or wars in regions outside of the United States where we conduct clinical trials may impact ongoing clinical trials.
We may not be able to qualify for or obtain various designations from regulators that would have the potential to expedite the review process of one or more of our product candidates and even if we do receive one or more such designations there is no guarantee that they will ultimately expedite the process, or aid in our obtaining marketing approval or provide market exclusivity.
There exist several designations that we can apply for from the FDA and other regulators that would provide us with various combinations of the potential for expedited regulatory review, certain financial incentives as well as the potential for post-approval exclusivity for a period of time. These designations include but are not limited to orphan drug designation, breakthrough therapy designation, accelerated approval, fast track status and priority review for our product candidates. For example, we and AskBio receivedDescartes-08 has been granted orphan drug designation by the FDA for SEL-302 in November 2020.the treatment of MG. We expect to seek one or more of these designations for our other current and future product candidates. There can be no assurance that any of our other product candidates will qualify for any of these designations. There can also be no assurance that any of our product candidates that do qualify for these designations will be granted such designations or that the FDA will not revoke a designation it grants at a later date. date, or that Congress will not change the law about a designation.
Further, there can be no assurance that any of our product candidates that are granted such designations, including Descartes-08, will ever benefit from such designations or that the FDA would not withdraw such designations once granted. Were we to receive a designation that promised a period of market exclusivity, such as orphan drug exclusivity, such exclusivity may not effectively protect the product from competition because different drugs can be approved for the same condition. In particular, the scope of exclusivity afforded for mRNA-modified cell therapy products may not be well defined. Further with respect to orphan drug status, even after an orphan drug is approved, the FDA can subsequently approve the same drug for the same condition if the FDA concludes that the later drug is clinically superior if it is shown to be safer, more effective or makes a major contribution to patient care.
Interim, top-line and preliminary data from our clinical trials that we announce or publish from time to time may change as more patient data become available and are subject to audit and verification procedures that could result in material changes in the final data.
From time to time, we may publish interim, top-line or preliminary data from our clinical studies, which is based on a preliminary analysis of then-available data, and the results and related findings and conclusions are subject to change following a full analyses of all data related to the particular trial. We also make assumptions, estimations, calculations and conclusions as part of our analyses of data, and we may not have received or had the opportunity to fully and carefully evaluate all data. As a result, the top-line results that we report may differ from future results of the same studies, or different conclusions or considerations may qualify such results, once additional data have been received and fully evaluated. Preliminary or top-line data also remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary data we previously published. Interim data from clinical trials are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment continues and more patient data become available.
Interim, top-line or preliminary data may not be representative of final data. If final data is not as positive as earlier interim, top-line or preliminary we have released, our business prospects would be significantly harmed.
In addition, the information we choose to publicly disclose regarding a particular study or clinical trial is based on what is typically extensive information, and you or others may not agree with what we determine is the material or otherwise appropriate information to include in our disclosure. Any information we determine not to disclose may ultimately be deemed significant by you or others with respect to future decisions, conclusions, views, activities or otherwise regarding a particular
product candidate or our business. As a result, preliminary and top-line data should not be relied upon in making an investment decision in our securities.
Negative public opinion and increased regulatory scrutiny of gene therapy and genetic research may damage public perception of our product candidates or compromise our ability to conduct our business or obtain regulatory approvals for our product candidates.
Gene therapy remains a novel technology. Public perception may be influenced by claims that gene therapy is unsafe, and gene therapy may not gain the acceptance of the public or the medical community. In particular, our success will depend upon physicians specializing in the treatment of those diseases that our product candidates target and prescribing treatments that involve the use of our product candidates in lieu of, or in addition to, existing treatments they are already familiar with and for which greater clinical data may be available. More restrictive government regulations or negative public opinion would have a negative effect on our business or financial condition and may delay or impair the development and commercialization of our product candidates or demand for any products we may develop.
Our product candidates may cause undesirable side effects or have other properties that could delay or prevent their regulatory approval, limit the commercial profile of an approved label, or result in significant negative consequences following marketing approval, if any.
Undesirable side effects caused by our product candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials, could result in a more restrictive label or the delay or denial of regulatory approval by the FDA or other comparable foreign authorities and could result in decreased market acceptance of any of our product candidates, if approved. Further, therapies such as those we are developing involve unique side effects that could be more significant than side effects
from other types of therapies with singular components. Results of our clinical trials could reveal a high and unacceptable severity and prevalence of side effects. In such an event, our trials could be suspended or terminated, and the FDA or comparable foreign regulatory authorities could order us to cease further development of or deny approval of our product candidates for any or all targeted indications.
In November 2023, the FDA issued a statement that it is investigating serious risk of T-cell malignancy following BCMA-directed or CD19-directed autologous CAR-T cell immunotherapies. While the FDA noted that it currently believes that the overall benefits of these products continue to outweigh their potential risks for their approved uses, the FDA stated that it is investigating the identified risk of T-cell malignancy with serious outcomes, including hospitalization and death, and is evaluating the need for regulatory action. Further, in January 2024, the clinical developmentFDA announced it would require a so-called “boxed warning” be added to the prescribing information for all six then-currently approved CAR-T therapies. A boxed warning is the strongest safety labeling the FDA may require. However, because all currently approved CAR T-cell immunotherapies are in oncology indications, there can be no assurance that FDA will reach the same risk-benefit analysis in other indications.
While we believe our mRNA-based CAR-T product candidates may have a differentiated toxicity profile than currently approved DNA-based CAR-T therapies, there can be no assurance that the FDA would not treat Descartes-08 or any of SEL-212 over many years has required multiple clinical trials and resultedour other product candidates similar to approved DNA-based CAR-T therapies. The FDA’s investigation may impact the FDA’s review of product candidates that we are developing, or that we may seek to develop in the use of different formulations of ImmTOR. While we do not believe that such differences in formulation will affect the safety or the efficacy of SEL-212, we cannot guarantee that any such formulation changes will not negatively impact the results of any clinical trials related to SEL-212, orfuture, which may, among other things, result in a significant difference inadditional regulatory scrutiny of our product candidates, delay the safety and efficacytiming for receiving any regulatory approvals or impose additional post-approval requirements on any of SEL-212.our product candidates that receive regulatory approval.
TheAny drug-related side effects observed in our clinical trials could also affect patient enrollment in our clinical trials or the ability of any enrolled patients to complete such trials or result in potential product liability claims. Any of these occurrences may harm our business, financial condition and prospects significantly.
Additionally, if one or more of our product candidates receives marketing approval, and we or others later identify undesirable side effects caused by such products, a number of potentially significant negative consequences could result, including:
•regulatory authorities may withdraw approvals of such product;
•regulatory authorities may require the addition of labeling statements, such as a “black box”boxed warning or a contraindication;
•regulatory authorities may impose additional restrictions on the marketing of, or the manufacturing processes for, the particular product;
•we may be required to create a medication guide outlining the risks of such side effects for distribution to patients;
•we could be sued and held liable for harm caused to patients, or become subject to fines, injunctions or the imposition of civil or criminal penalties;
•our reputation may suffer; and
•we could be required to develop a risk evaluation and mitigation strategies, or REMS, plan to prevent, monitor and/or manage a specific serious risk by informing, educating and/or reinforcing actions to reduce the frequency and/or severity of the event.
Any of these events could prevent us from achieving or maintaining market acceptance of a particular product candidate, if approved, and could significantly harm our business, results of operations and prospects.
Risks Related to our Dependence on Third Parties and Manufacturing
We rely on 3SBio in China as our primary supplier of pegadricase and on other third parties for the manufacture of our product candidates for preclinical and clinical testing, and expect to continue to do so for the foreseeable future. Our reliance on third parties increases the risk that we will not have sufficient quantities of our product candidates or that such quantities may not be available at an acceptable cost, or in compliance with regulatory requirements, which could delay, prevent or impair our development or commercialization efforts.
We obtain the biologic pegadricase, a component of SEL-212, primarily from 3SBio in China. Under the 3SBio License, we have limited rights to manufacture pegadricase and while we have entered into a contract with a back-up supplier located outside of China, we expect to continue to rely on 3SBio as the primary supplier of pegadricase for the foreseeable future.
Any disruption in production or inability of 3SBio in China to produce adequate quantities of pegadricase to meet our needs could impair our ability to operate our business on a day-to-day basis and to continue our research and development of our future product candidates. Furthermore, since 3SBio is located in China, we are exposed to the possibility of product supply disruption and increased costs in the event of changes in the policies, laws, rules and regulations of the United States or Chinese governments, political unrest or unstable economic conditions in China. For example, trade tensions between the United States and China have been escalating in recent years. Most notably, several rounds of U.S. tariffs have been placed on Chinese goods being exported to the United States. Each of these U.S. tariff impositions against Chinese exports were followed by a round of retaliatory Chinese tariffs on U.S. exports to China. Pegadricase is subject to, and any other components we purchase from China may be subject to, these tariffs, which could increase our manufacturing costs and could make our products, if successfully developed and approved, less competitive than those of our competitors whose inputs are not subject to these tariffs.
Moreover, as a result of the COVID-19 pandemic, certain of our suppliers and CMOs in the United States, China and other countries may be affected, which could disrupt their activities. We could face difficulty sourcing key components necessary to produce supply of SEL-212, which may negatively affect our clinical development activities and our agreement with Sobi. If COVID-19 continues to impact U.S. business operations, including those of our CMOs and suppliers, we could face additional disruptions to our supply chain that could affect the supply of drug product for our preclinical studies and clinical trials.
Any of these matters could materiallyRisks Related to Manufacturing and adversely affect our business and results of operations. Any issues related to the manufacturing lots or similar action regarding pegadricase used in preclinical studies or clinical trials could delay the studies or trials or detract from the integrity of the trial data and its potential use in future regulatory filings. In addition, manufacturing interruptions or failure to comply or maintain compliance with regulatory requirements by 3SBio could significantly delay our clinical development of potential products and reduce third-party or clinical researcher interest and support of our proposed trials. These interruptions or failures could also impede commercialization of our future product candidates and impair our competitive position. Further, we may be exposed to fluctuations in the value of the local currency in China. Future appreciation of the local currency could increase our costs. These factors and increasing wage rates due to increased demand for skilled laborers and the declining availability of skilled labor in China could cause our labor costs to rise.Dependence on Third Parties
We rely, and expect to continue to rely, in additiongrow our manufacturing capabilities and resources and we must incur significant costs to 3SBio, on other third parties for the manufacture of our product candidates for supply in preclinical studies and clinical trials, as well as for commercial manufacture if any of our product candidates receive marketing approval. Our reliance on such third parties increases the risk that we will not have sufficient quantities of our product candidates on a timely basis develop this expertise and/or at all, or that such quantities will be available at an acceptable cost or quality, which could delay, prevent or impair our development or commercialization efforts. For example, we rely on third parties to manufacture our products.
We have growing manufacturing capabilities, and in order to continue to develop our current product candidates, apply for regulatory approvals and, if approved, commercialize future products, we will need to continue to develop, contract for, or otherwise arrange for any necessary external manufacturing capabilities.
We manufacture our product candidates internally. There are risks inherent in biological manufacturing and we may not meet our delivery time requirements or provide adequate amounts of material to meet our needs, and we may make errors in manufacturing, any of which could delay our clinical trials and result in additional expense to us.
Our autologous cell therapy product candidates, including Descartes-08, are made on a patient-by-patient basis, rendering their manufacture less predictable and requiring more demanding logistics.
We rely on one or more third-party laboratories to perform certain quality control tests. These laboratories could become unavailable, or provision of their services could be delayed.
Additionally, as we scale up our manufacturing, we may encounter further challenges. Furthermore, competition for supply from our manufacturers from other companies, a breach or violation by such manufacturers of their contractual or regulatory obligations or a dispute with such manufacturers would cause delays in our discovery and development efforts, as well as additional expense to us.
In developing manufacturing capabilities by building our own manufacturing facilities, we have incurred substantial expenditures, and expect to incur significant additional expenditures in the future. Also, we have had to, and will likely need to continue to recruit, hire, and train qualified employees to staff our facilities. If we are unable to manufacture sufficient quantities of material or if we encounter problems with our gene therapy preclinical materials. Gene therapy is a relatively new area for commercial biopharmaceutical developmentfacilities in the future, we may also need to secure alternative suppliers, and there are a limited number of CMOs with adequate facilities and expertise in this area. As a result,such alternative suppliers may not be available, or we may be unable to successfully manufactureenter into agreements with them on reasonable terms and in a timely manner. In addition, to the extent we or our gene therapy preclinical materials throughpartners rely on contract manufacturing organizations, or CMOs, to supply our product candidates, any delays or disruptions in supply could have a third party or scale upmaterial adverse impact on the manufactureresearch and development activities and potential commercialization of our gene therapyor our partners’ product candidatescandidates.
The manufacturing process for clinical testing or commercialization, if at all.
We may be unable to establish any agreements with third-party manufacturers on acceptable terms or at all, and even if we do rely on third-party manufacturers entails additional risks, including the:
•inability, failure or unwillingness of third-party manufacturers to comply with regulatory requirements, maintain quality assurance, meet our needs, specifications or schedules or continue to supply products to us;
•reduced control we have over product development, including with respect to our lead product candidate, due to our reliance on such third-party manufacturers,
•breach of manufacturing agreements by the third-party manufacturers;
•misappropriation or disclosure of our proprietary information, including our trade secrets and know-how;
•relationships that the third-party manufacturer may have with others, some of which may be our competitors, and, if it does not successfully carry out its contractual duties, does not meet expectations, experiences work stoppages, or needs to be replaced, we may needdevelop is subject to enter into alternative arrangements, which may not be available, desirable or cost-effective; and
•termination or nonrenewal of agreements by third-party manufacturers at times that are costly or inconvenient for us.
Additionally, if our contract manufacturers cannot successfully manufacture material that conforms to our specifications and the strict regulatory requirements of the FDA or others, they will not be able to secure and/or maintain regulatory approval for their manufacturing facilities. If the FDA or a comparableand foreign regulatory authority does not approve these facilities for the manufacture of our product candidates or if it withdraws any such approval in the future,process and we maywill need to find alternative manufacturing facilities, which would significantly impact our abilitymeet, or will need to develop, obtaincontract with CMOs who can meet, all applicable FDA and foreign regulatory approval for or market our product candidates, if approved.authority requirements on an ongoing basis. Our failure or the failure of our third-party manufacturers,any CMO to comply with applicable regulationsmeet required regulatory authority requirements could result in sanctions being imposed on us, including clinical holds, fines, injunctions, civil penalties,the delayed submission of regulatory applications, or delays suspension or withdrawal of approvals, license revocations, seizures or recalls of product candidates or products, operating restrictions and criminal prosecutions,in receiving regulatory approval for any of which could significantly and adversely affect supplies of our products. In addition, there are a limited number of manufacturers that operate under cGMP regulations that might be capable of manufacturingor our products. Therefore, ourcurrent or future collaborators’ product candidates and any future productscandidates.
To the extent that we may develop may compete with other products for access to manufacturing facilities. Any failure to gain access to these limited manufacturing facilities could severely impact the clinical development, marketing approval and commercialization of our product candidates.
Any performance failure on the part of ourhave existing, or enter into future, manufacturers could delay clinical developmentmanufacturing arrangements with third parties, we depend, and will depend in the future, on these third parties to perform their obligations in a timely manner and consistent with contractual and regulatory requirements, including those related to quality control and quality assurance. The failure of any CMO to perform its obligations as expected, or, marketing approval. We do not currently have arrangements in place for redundant supplyto the extent we manufacture all or a second source for required raw materials used in the manufactureportion of our product candidates ourselves, our failure to execute on our manufacturing requirements, could adversely affect our business in a number of ways, including:
•we or our current or future collaborators may not be able to initiate or continue clinical trials of product candidates that are under development;
•we or our current or future collaborators may be delayed in submitting regulatory applications, or receiving regulatory approvals, for our product candidates;
•we may lose the manufacturecooperation of finished product. Moreover, we often rely on one CMO to produce multiple product components. For instance, oneour collaborators;
•our facilities and those of our CMOs, produces several polymers usedand our products could be the subject of inspections by regulatory authorities that could have a negative outcome and result in our ImmTOR platform. If our current CMOs cannot perform as agreed, delays in supply;
•we may be required to replace such manufacturerscease distribution or recall some or all batches of our products or take action to recover clinical trial material from clinical trial sites; and
•ultimately, we may not be unableable to replace them on a timely basis or at all. Our currentmeet the clinical and expected future dependence upon otherscommercial demands for the manufacture of our product candidates or products could delay, prevent or impair our development and commercialization efforts.products.
If we are unable to maintain any of our existing collaborations, or if these arrangements are not successful, or we are unable to enter into future licenses,collaborations and licensing arrangements, our business could be adversely affected.
We have entered into collaborations with other parties, including pharmaceutical and biotechnology companies and universities,intend to develop products based on our ImmTOR platform, and such collaborations and licensing arrangements currently represent a significant portion of our product pipeline and are expected to represent a larger portion of our pipeline in the future. Certain of our collaborations have provided us with important funding for some of our development programs and we expect to receive additional funding under collaborations in the future although not all of our collaborations may result in funding to us, and certain collaborations, licenses and agreements may result in increased expenditures by us. Our existing collaborations, and any future collaborations, may pose a number of risks, including the following:
•collaborators have significant discretion in determining the efforts and resources that they will apply to these collaborations;
•collaborators may not perform their obligations as expected;
•collaborators may not pursue development and commercialization of any product candidates that achieve regulatory approval or may elect not to continue or renew development or commercialization programs based on preclinical or clinical trial results, changes in the collaborators’ strategic focus or available funding, or external factors, such as an acquisition, that divert resources or create competing priorities;
•collaborators may delay clinical trials, provide insufficient funding for a clinical trial program, stop a clinical trial or abandon a product candidate, repeat or conduct new clinical trials or require a new formulation of a product candidate for clinical testing;
•collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with our product candidates, which may cause collaborators to cease to devote resources to the commercialization of our product candidates;
•a collaborator with marketing and distribution rights to one or more of our product candidates that achieve regulatory approval may not commit sufficient resources to the marketing and distribution of such product or products;
•disagreements with collaborators, including disagreements over proprietary rights, contract interpretation or the preferred course of development, might cause delays or termination of the research, development or commercialization of product candidates, might lead to additional responsibilities for us with respect to product candidates, or might result in litigation or arbitration, any of which would be time-consuming and expensive;
•collaborators may not properly maintain or defend our intellectual property rights or may use our proprietary information in such a way as to invite litigation that could jeopardize or invalidate our intellectual property or proprietary information or expose us to potential litigation;
•collaborators may infringe the intellectual property rights of third parties, which may expose us to litigation and potential liability;
•collaborations may be terminated and we would potentially lose the right to pursue further development or commercialization of the applicable product candidates as well as have difficulty entering into a similar collaboration where the potential collaborator is aware of the prior termination;
•collaborators may learn about our technology and use this knowledge to compete with us in the future;
•there may be conflicts between different collaborators that could negatively affect those collaborations and potentially others; and
•the number and type of our collaborations could adversely affect our attractiveness to future collaborators or acquirers.
We are actively exploringexplore licenses and other strategic collaborations with additional pharmaceutical and biotechnology companies for development and potential commercialization of therapeutic products. However, we face significant competition in seeking appropriate collaborators. If we are unable to reach agreements with suitable collaborators on a timely basis, on acceptable terms, or at all, we may not be able to access specific antigens that would be suitable to development with our technology, have to curtail the development of a product candidate, reduce or delay its development program or one or more of our other development programs, delay its potential commercialization or reduce the scope of any sales or marketing activities, or increase our expenditures and undertake development or commercialization activities at our own expense. If we elect to fund and undertake development or commercialization activities on our own, we may need to obtain additional expertise and additional capital, which may not be available to us on acceptable terms or at all. If we fail to enter into collaborations and do not have sufficient funds or expertise to undertake the
necessary development and commercialization activities, we may not be able to further develop our product candidates or bring them to market or continue to develop our programs, and our business may be materially and adversely affected.
We rely, and expect to continue to rely, on third parties to conduct our clinical trials, and those third parties may not perform satisfactorily, including by failing to meet deadlines for the completion of such trials.
We rely, and expect to continue to rely, on third parties, such as CROs, clinical data management organizations, medical institutions and clinical investigators, to conduct and manage our clinical trials, including our ongoing Phase 3 DISSOLVE1/2 clinical program for SEL-212, consistingtrial of the DISSOLVE I and DISSOLVE II trials, which we have agreed to continue to run on behalf of Sobi, and for our other product candidates.Descartes-08. We also expect to rely on other third parties to store and distribute drug supplies for our clinical trials.
While we rely on these third parties for research and development activities, we remain responsible for ensuring that each of our clinical trials is conducted in accordance with the general investigational plan and protocols for the trial. Moreover, the FDA requires us to comply with GCP regulations, for conducting, recording and reporting the results of clinical trials to assure that data and reported results are credible and accurate and that the rights, safety and welfare of trial participants are protected. Other countries’ regulatory agencies also have requirements for clinical trials. If we or any of our CROs or third-party contractors fail to comply with applicable GCPs, the data generated in our clinical trials may be deemed unreliable and the FDA or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. In addition, our clinical trials must be conducted with product produced under cGMP regulations. Our failure to comply with these regulations may require us to repeat clinical trials, which would delay the regulatory approval process. We also are required to register ongoing clinical trials and post the results of completed clinical trials on a government-sponsored database, www.ClinicalTrials.gov,, within specified timeframes. Failure to do so can result in fines, adverse publicity, and civil and criminal sanctions.
Furthermore, these third parties may also have relationships with other entities, some of which may be our competitors. If these third parties do not successfully carry out their contractual duties, do not comply with confidentiality obligations, do not meet expected deadlines, experience work stoppages, terminate their agreements with us or need to be replaced, or do not conduct our clinical trials in accordance with regulatory requirements or our stated protocols, we may need to enter into new arrangements with alternative third parties, which could be difficult, costly or impossible, and our clinical trials may be extended, delayed or terminated, or may need to be repeated. If any of the foregoing occur, we may not be able to obtain, or may be delayed in obtaining, marketing approvals for our product candidates or in commercializing our product candidates.
We have no experience manufacturing our product candidates for commercial use, and if we decide to establish our own commercial manufacturing facility, we cannot assure you that we can manufacture our product candidates in compliance with regulations at a cost or in quantities necessary to make them commercially viable.
We have a pilot manufacturing facility at our Watertown, Massachusetts location where we conduct process development, scale-up activities and the manufacture of ImmTOR product candidates for preclinical use. We do not currently have any of our own manufacturing facilities that meet the FDA’s cGMP requirements for the production of any product candidates used in humans, and rely on our CMOs for clinical production. We currently have no plans to establish our own commercial manufacturing facilities and we will continue to rely on our partnership with CMOs who are currently producing ImmTOR at the commercial scale for SEL-212 using our proprietary process and equipment.
Risks Related to Commercialization of our Product Candidates and Legal Compliance Matters
Even if any of our product candidates receives marketing approval, it may fail to achieve the degree of market acceptance by physicians, patients, third-party payors and others in the medical community necessary for commercial success.
If any of our product candidates receives marketing approval, it may nonetheless fail to gain sufficient market acceptance by physicians, patients, third-party payors and others in the medical community. If our product candidates do not achieve an adequate level of acceptance, we may not generate significant product revenues and we may not become profitable. The degree of market acceptance of our product candidates, if approved for commercial sale, will depend on several factors, including:
•the efficacy, safety and potential advantages compared to alternative treatments;
•our ability to manufacture and distribute cell therapies in a timely and secure manner;
•our ability to offer our products for sale at competitive prices;
•the convenience and ease of administration compared to alternative treatments;
•product labeling or product insert requirements of the FDA or foreign regulatory authorities, including any limitations or warnings contained in a product’s approved labeling, including any black box warning or REMS;
•the willingness of the target patient population to try new treatments and of physicians to prescribe these treatments;
•our ability to hire and retain a sales force in the United States;force;
•the strength of marketing and distribution support;
•the availability of third-party coverage and adequate reimbursement for our product candidates, once approved;
•the prevalence and severity of any side effects; and
•any restrictions on the use of our products together with other medications.
We currently have no sales organization and expect to rely on Sobi for the marketing and sale of SEL-212, if approved.organization. If we are unable to establish effective sales, marketing and distribution capabilities, or enter into agreements with third parties with such capabilities, we may not be successful in commercializing our product candidates if and when they are approved.
We do not have a sales or marketing infrastructure and have no experience in the sale, marketing or distribution of pharmaceutical products. To achieve commercial success for any product candidate for which we obtain marketing approval, we will need to establish a sales and marketing organization or make arrangements with third parties to perform sales and marketing functions and we may not be successful in doing so. We expect to rely on Sobi for the marketing and sale of SEL-212, if approved. For our other product candidates, we expect to build a focused sales and marketing infrastructure to market or co-promote our product candidates in the United States and potentially elsewhere, if and when they are approved. There are risks involved with establishing our own sales, marketing and distribution capabilities. For example, recruiting and training a sales force is expensive and time-consuming and could delay any product launch. This may be costly, and our investment would be lost if we cannot retain or reposition our sales and marketing personnel.
We face substantial competition, including from biosimilars, which may result in others discovering, developing or commercializing competing products before or more successfully than we do.
The development and commercialization of new drug and biologic products and technologies is highly competitive and is characterized by rapid and substantial technological development and product innovations. We are aware that pharmaceutical and biotechnology companies, offer or are pursuing the development of pharmaceutical products or technologies that may address one or more indications that our product candidates target, as well as smaller, early-stage companies, that offer or are pursuing the development of pharmaceutical products or technologies that may address one or more indications that our product candidates target. We face competition with respect to our current product candidates and will face competition with respect to any product candidates that we may seek to develop or commercialize in the future, from major pharmaceutical companies, specialty pharmaceutical companies and biotechnology companies worldwide.
Many of the companies against which we are competing or against which we may compete in the future have significantly greater financial resources, established presence in the market and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and reimbursement for product candidates and in marketing approved products than we do.
These third parties compete with us in recruiting and retaining qualified scientific, sales and marketing and management personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are more effective, have fewer or less severe side effects, are more convenient or are less expensive than any products that we may develop. Our competitors also may obtain FDA or other regulatory approval for their products more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market, especially for any competitor developing a competing immunomodulating therapeuticcell therapy product that will likely share our same regulatory approval requirements. In addition, our ability to compete may be affected in many cases by insurers or other third-party payors seeking to encourage the use of generic or biosimilar products.
We expect the product candidates we develop will be regulated as biological products, or biologics, and therefore they may be subject to competition sooner than anticipated.
The Biologics Price Competition and Innovation Act of 2009, or the BPCIA, was enacted as part of the Affordable Care Act to establish an abbreviated pathway for the approval of biosimilar and interchangeable biological products. The regulatory pathway establishes legal authority for the FDA to review and approve biosimilar biologics, including the possible designation of a biosimilar as “interchangeable” based on its similarity to an approved biologic. Under the BPCIA, an application for a biosimilar product cannot be approved by the FDA until 12 years after the reference product was approved under a biologics license application, or BLA. The law is still being interpreted and implemented by the FDA, and as a result, its ultimate impact, implementation, and meaning are subject to uncertainty. However, any such processes could have a material adverse effect on the future commercial prospects for our biological products.
We believe that any product candidate approved in the United States as a biological product under a BLA should qualify for the 12-year period of exclusivity. However, there is a risk that this exclusivity could be shortened due to Congressional action or otherwise, or that the FDA will not consider the subject product candidates to be reference products for competing products, potentially creating the opportunity for generic competition sooner than anticipated. Moreover, the extent to which a biosimilar, once approved, will be substituted for any one of the reference products in a way that is similar to traditional generic substitution for non-biological products is not yet clear, and will depend on a number of marketplace and regulatory factors that are still developing.
Even if we are able to commercialize any of our product candidates, the products may become subject to unfavorable pricing regulations or third-party coverage or reimbursement policies, any of which would have a material adverse effect on our business.
Significant uncertainty exists as to the coverage and reimbursement status of any product candidates for which we obtain regulatory approval, especially novel products like our genecell therapy product candidates, and may be particularly difficult because of the higher prices associated with gene therapysuch product candidates. Our ability to commercialize any product candidates successfully will depend, in part, on the extent to which coverage and adequate reimbursement for these products and related treatments will be available from government health administration authorities, private health insurers and other organizations. Government authorities and third-party payors, such as private health insurers and health maintenance organizations, decide which medications they will pay for and establish reimbursement levels.
Obtaining and maintaining adequate reimbursement for our products may be difficult. We cannot be certain if we will obtain an adequate level of reimbursement for our products by third-party payors. Even if we do obtain adequate levels of reimbursement, third-party payors, such as government or private healthcare insurers, carefully review and question the coverage of, and challenge the prices charged for, products. Government authorities and third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medications. Third-party payors often require that pharmaceutical companies provide them with predetermined discounts from list prices and are challenging the prices charged for products. We may also be required to conduct expensive pharmacoeconomic studies to justify coverage and reimbursement or the level of reimbursement relative to other therapies. Some third-party payors may require pre-approval of coverage for new and innovative therapies, such as our product candidates, before they will provide reimbursement. If coverage and reimbursement are not available or reimbursement is available only to limited levels, we may not be able to successfully commercialize any product candidate for which we obtain marketing approval.
There may be significant delays in obtaining reimbursement for newly approved products, and coverage may be more limited than the purposes for which the product is approved by the FDA or similar regulatory authorities outside of the United States. Moreover, eligibility for reimbursement does not imply that a product will be paid for in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution. Interim reimbursement levels for new products, if applicable, may also not be sufficient to cover our costs and may not be made permanent. Reimbursement rates may vary according to the use of the product and the clinical setting in which it is used, may be based on reimbursement levels already set for lower cost products and may be incorporated into existing payments for other services. Net prices for products may be reduced by mandatory discounts or rebates required by government healthcare programs or private payors and by any future relaxation of laws that presently restrict imports of products from countries where they may be sold at lower prices than in the United States. Our inability to promptly obtain coverage and adequate reimbursement rates from both government-funded and private payors for any approved products that we develop could have a material adverse effect on our operating results, our ability to raise capital needed to commercialize products and our overall financial condition.
The regulations that govern marketing approvals, pricing, coverage and reimbursement for new products vary widely from country to country. Current and future legislation may significantly change the approval requirements in ways that could involve additional costs and cause delays in obtaining approvals. Some countries require approval of the sale price of a product before it can be marketed. In many countries, the pricing review period begins after marketing or product licensing approval is granted. In some foreign markets, prescription pharmaceutical pricing remains subject to continuing governmental control, including possible price reductions, even after initial approval is granted. As a result, we might obtain marketing approval for a product in a particular country, but then be subject to price regulations that delay our commercial launch of the product, possibly for lengthy time periods, and negatively impact the revenues we are able to generate from the sale of the product in that country.Adverse pricing limitations may hinder our ability to recoup our investment in one or more product candidates, even if our product candidates obtain marketing approval. There can be no assurance that our product candidates, if they are approved for sale in the United States or in other countries, will be considered medically necessary for a specific indication or cost-effective, or that coverage or an adequate level of reimbursement will be available.
Moreover, there is heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products, which has resulted in several Congressional inquiries and proposed bills designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drug products. There can be no assurance that our product candidates, will not be subject to heightened governmental scrutiny, unfavorable regulatory inquiry or action, or Congressional inquiry.
Product liability lawsuits against us could cause us to incur substantial liabilities and limit commercialization of any products that we may develop.
We face an inherent risk of product liability exposure related to the testing of our product candidates in clinical trials and will face an even greater risk if we commercially sell any products that we may develop. If we cannot successfully defend ourselves against claims that our product candidates or products caused injuries, we will incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
•regulatory investigations, product recalls or withdrawals, or labeling, marketing or promotional restrictions;
•decreased demand for any product candidates or products that we may develop;
•injury to our reputation and significant negative media attention;
•loss of clinical trial participants or increased difficulty in enrolling future participants;
•significant costs to defend the related litigation or to reach a settlement;
•substantial payments to trial participants or patients;
•loss of revenue;
•reduced resources of our management to pursue our business strategy;
•the inability to commercialize any products that we may develop;
•distraction of management’s attention from our primary business; and
•substantial monetary awards to patients or other claimants;claimants.
We maintain general liability, product liability and umbrella liability insurance. Our existing insurance coverage may not fully cover potential liabilities that we may incur. We may need to increase our insurance coverage as we expand our clinical trials or if we commence commercialization of our product candidates. Insurance coverage is increasingly expensive. We may not be able to maintain insurance coverage at a reasonable cost or in an amount adequate to satisfy any liability that may arise. A product liability claim or series of claims brought against us could cause our share price to decline and, if judgments exceed our insurance coverage, could adversely affect our results of operations and business, including preventing or limiting the commercialization of any product candidates we develop.
Our relationships with healthcare providers, customers and third-party payors will be subject to applicable anti-kickback, fraud and abuse and other healthcare laws and regulations, which could expose us to criminal sanctions, civil penalties, exclusion from government healthcare programs, contractual damages, reputational harm and diminished profits and future earnings.
Arrangements with physicians, others who may be in a position to generate business for us, and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that constrain the business or financial arrangements and relationships through which we market, sell and distribute any products for which we obtain marketing approval. Restrictions under applicable federal and state healthcare laws and regulations include the following:
•the federal Anti-Kickback Statute, which prohibits, among other things, persons and entities from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward, or in return for, either the referral of an individual for, or the purchase, order or recommendation of, any good or service for which payment may be made under a federal healthcare program such as Medicare and Medicaid. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it to have committed a violation;
•the federal False Claims Act, which impose criminal and civil penalties against individuals or entities for knowingly presenting, or causing to be presented, to the federal government claims for payment that are false or fraudulent. Private individuals (e.g., whistleblowers) can bring these actions on behalf of the government; in addition, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act;
•the Health Insurance Portability and Accountability Act of 1996, or HIPAA, which imposes criminal and civil liability for, among other things, executing or attempting to execute a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it to have committed a violation;
•HIPAA, as amended by HITECH and itstheir respective implementing regulations, which also imposesimpose obligations, including mandatory contractual terms, on certain types of people and entities with respect to safeguarding the privacy, security and transmission of individually identifiable health information;
•the federal Physician Payments Sunshine Act, or the Sunshine Act, which requires applicable manufacturers of certain products for which payment is available under a federal healthcare program to report annually to the government information related to certain payments or other “transfers of value” made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors), certain other health care professionals beginning in 2022, and teaching hospitals, as well as ownership and investment interests held by the physicians and their immediate family members;
•analogous state laws and regulations, such as state anti-kickback and false claims laws, which may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by third-party payors, including private insurers; and requirements to comply with federal and pharmaceutical industry compliance guidelines;
•state data privacy and price transparency laws, many of which differ from each other in significant ways and often are broader than and not preempted by HIPAA or the Sunshine Act, thus complicating compliance efforts; by way of example, the California Consumer Privacy Act, or CCPA, which went into effect January 1, 2020, among other things, creates new data privacy obligations for covered companies and provides new privacy rights to California residents, including the right to opt out of certain disclosures of their information. The CCPA also creates a private right of action with statutory damages for certain data breaches, thereby potentially increasing risks associated with a data breach. Although the law includes limited exceptions, including for “protected health information” maintained by a covered entity or business associate, it may regulate or impact our processing of personal information depending on the context; and
•◦similar healthcare laws and regulations in the European UnionEU and other jurisdictions, including reporting requirements detailing interactions with and payments to healthcare providers and laws governing the privacy and security of certain protected information, such as the General Data Protection Regulation, or GDPR, which imposes obligations and restrictions on the collection and use of personal data relating to individuals located in the European UnionEU (including health data); in addition, the United Kingdom leaving the EU could also lead to further legislative and regulatory changes. It remains unclear how the United Kingdom data protection laws or regulations will develop in the medium to longer term and how data transfer to the United Kingdom from the E.U.EU will be regulated. However, the United Kingdom has transposed the GDPR into domestic law with the Data Protection Act 2018, which remains in force following the United Kingdom’s departure from the EUEU.
Efforts to ensure that our business arrangements with third parties comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices, including our relationships with physicians and other healthcare providers, some of whom may recommend, purchase and/or prescribe our product candidates, if approved, may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other governmental laws and regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, imprisonment, exclusion of products from government funded healthcare programs, such as Medicare and Medicaid, disgorgement, contractual damages, reputational harm, diminished profits and the curtailment or restructuring of our operations. If any of the physicians or other healthcare providers or entities with whom we expect to do business is found to be not in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs. The risk of our being found in violation of these laws is increased by the fact that many of them have not been fully interpreted by the regulatory authorities or the courts, and their provisions are open to a variety of interpretations.
Recently enacted and future legislation may increase the difficulty and cost for us to obtain marketing approval of and commercialize our product candidates and affect the prices we may obtain.
In the United States and some foreign jurisdictions, there have been a number of legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay marketing approval of our product candidates, restrict or regulate post-approval activities and affect our ability to profitably sell any product candidates for which we obtain marketing approval.
For example, the Patient Protection and Affordable Care Act of 2010, or the ACA, is a sweeping law intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for the healthcare and health insurance industries, impose new taxes and fees on the health industry and impose additional health policy reforms.
Since its enactment, there have been judicial and Congressional challenges to certain aspects of the ACA. We cannot predict the ultimate content, timing or effect of any healthcare reform legislation or the impact of potential legislation on us.
We expect that the ACA, as well as other healthcare reform measures that may be adopted in the future, may result in additional reductions in Medicare and other healthcare funding, more rigorous coverage criteria, new payment methodologies and additional downward pressure on the price that we receive for any approved product. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability or commercialize our products.
Disruptions at the FDA and other government agencies caused by funding shortages or global health concerns could hinder their ability to hire, retain or deploy key leadership and other personnel, or otherwise prevent new or modified products from being developed, approved or commercialized in a timely manner or at all, which could negatively impact our business.
The ability of the FDA to review and or approve new products can be affected by a variety of factors, including government budget and funding levels, statutory, regulatory, and policy changes, the FDA’s ability to hire and retain key personnel and accept the payment of user fees, and other events that may otherwise affect the FDA’s ability to perform routine functions. Average review times at the agency have fluctuated in recent years as a result. In addition, government funding of
other government agencies that fund research and development activities is subject to the political process, which is inherently fluid and unpredictable. Disruptions at the FDA and other agencies may also slow the time necessary for new drugs and biologics to be reviewed and/or approved by necessary government agencies, which would adversely affect our business. If a prolonged government shutdown occurs, or if global health concerns continuewere to again prevent the FDA or other regulatory authorities from conducting their regular inspections, reviews, or other regulatory activities, it could significantly impact the ability of the FDA or other regulatory authorities to timely review and process our regulatory submissions, which could have a material adverse effect on our business.
We are subject to U.S. and certain foreign export and import controls, sanctions, embargoes, anti-corruption laws, and anti-money laundering laws and regulations. Compliance with these legal standards could impair our ability to compete in domestic and international markets. We can face criminal liability and other serious consequences for violations, which can have a material adverse effect on our business.
We are subject to export control and import laws and regulations, including the U.S. Export Administration Regulations administered by the U.S. Commerce Department’s Bureau of Industry and Security, U.S. customs regulations, various economic and trade sanctions regulations including those administered or enforced by relevant government authorities, such as by the U.S. Treasury Department’s Office of Foreign Assets Control or the U.S. Department of State, the U.S. Foreign Corrupt Practices Act of 1977, as amended, or the FCPA, the U.S. domestic bribery statute contained in 18 U.S.C. § 201, the U.S. Travel Act, the USAUniting and Strengthening America by Providing Appropriate Tools Required to Intercept and Obstruct Terrorism, or PATRIOT Act, and other state and national anti-bribery and anti-money laundering laws in the countries in which we conduct activities. U.S. sanctions laws and regulations may govern or restrict our business and activities in certain countries and with certain persons. Anti-corruption laws are interpreted broadly and prohibit companies and their employees, agents, contractors and other partners from authorizing, promising, offering or providing, directly or indirectly, improper payments or anything else of value to recipients in the public or private sector. We may engage third parties for clinical trials outside of the United States, to sell our product candidates abroad once we enter a commercialization phase, and/or to obtain necessary permits, licenses, patent registrations, and other regulatory approvals. We have direct or indirect interactions with officials and employees of government agencies or government-affiliated hospitals, universities and other organizations. We can be held liable for the corrupt or other illegal activities of our employees, agents, contractors and other partners, even if we do not explicitly authorize or have actual knowledge of such activities. Our violations of the laws and regulations described above may result in substantial civil and criminal fines and penalties, imprisonment, the loss of export or import privileges, debarment, tax reassessments, breach of contract and fraud litigation, reputational harm and other consequences.
If we or our contract manufacturers or other third parties we rely upon fail to comply with environmental, health and safety laws and regulations, we could become subject to fines or penalties or incur costs that could have a material adverse effect on our business.
We and our contract manufacturers and other third parties with whom we do business are subject to numerous environmental, health and safety laws and regulations, including those governing laboratory procedures and the handling, use, storage, treatment and disposal of hazardous materials and wastes. Our operations involve the use of hazardous and flammable materials, including biological materials and chemicals. Our operations also produce hazardous waste products. We generally contract with third parties for the disposal of these materials and wastes. We cannot eliminate the risk of contamination or injury from these materials. In the event of contamination or injury resulting from our use of hazardous materials, we could be held liable for any resulting damages, and any liability could exceed our resources. We also could incur significant costs associated with civil or criminal fines and penalties for failure to comply with such laws and regulations.
Although we maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees resulting from the use of hazardous materials, this insurance may not provide adequate coverage against potential liabilities. We do not maintain insurance for environmental liability or toxic tort claims that may be asserted against us in connection with our storage or disposal of biological, hazardous or radioactive materials.
In addition, we may incur substantial costs in order to comply with current or future environmental, health and safety laws and regulations. These current or future laws and regulations may impair our research, development or production efforts. The failure to comply with these laws and regulations also may result in substantial fines, penalties or other sanctions.
Risks Related to our Financial Position and Need for Additional Capital
We are a development-stage company and have incurred significant losses since our inception. We expect to incur losses for the foreseeable future and may never achieve or maintain profitability.
Except for the year ended December 31, 2022, we have incurred significant operating losses since our inception. OurWe incurred a net loss of $219.7 million, had net income wasof $35.4 million, for the year ended December 31, 2022, and incurred a net losses wereloss of $25.7 million and $68.9 million for each of the years ended December 31, 20212023, 2022, and 2020,2021, respectively. As of December 31, 2022,2023, we had an accumulated deficit of $394.9$614.6 million. To date, we have financed our operations primarily through public offerings and private placements of our securities, funding received from collaboration and license arrangements and oura credit facility. We currently have no source of product revenue, and we do not expect to generate product revenue for the foreseeable future. We haveHistorically we devoted substantially all
of our financial resources and efforts to developing our ImmTOR platform and following the closing of the Merger, or the Closing, we expect to devote substantially all of our financial resources and efforts to developing our mRNA-based therapies for the treatment of autoimmune diseases, identifying potential product candidates and conducting preclinical studies and our clinical trials. We are in the early stages of clinical development of most of our product candidates, and we have not completed development of any ImmTOR-enabled therapies.candidates. We expect to continue to incur significant expenses and operating losses for the foreseeable future. We expect that our expenses will increase substantially as we:
•continue the research and development of our product candidates;
•seek to enhanceincrease and evolvedevelop our ImmTOR platformmanufacturing and distribution capacities;
•discover and develop additional product candidates;
•seek to maintain and enter into collaboration, licensing and other agreements, including, but not limited to research and development, and/or commercialization agreements;
•seek regulatory approvals for any product candidates that successfully complete clinical trials;
•potentially establish a sales, marketing and distribution infrastructure and scale up externalinternal manufacturing capabilities to commercialize any products for which we may obtain regulatory approval;
•maintain, expand and protect our intellectual property portfolio, including through licensing arrangements;
•add clinical, scientific, operational, financial and management information systems and personnel, including personnel to support our product development and potential future commercialization efforts;
•experience any delays or encounter any issues with any of the above, including, but not limited to, failed studies, complex results, safety issues or other regulatory, manufacturing or scale-up challenges; and
•are exposed to broad macroeconomic conditions including inflation and supply chain tightness which could result in us paying more, or being unable, to access goods and services.
To become and remain profitable, we must succeed in developing and eventually commercializing products that generate significant revenue. This will require us to be successful in a range of challenging activities, including completing preclinical testing and clinical trials of our product candidates, discovering additional product candidates, obtaining regulatory approval and securing reimbursement for these product candidates, manufacturing, marketing and selling any products for which we may obtain regulatory approval, and establishing and managing our collaborations at various stages of a product candidate’s development. We are only in the preliminary stages of most of these activities. We may never succeed in these activities and, even if we do, may never generate revenues that are significant enough to achieve profitability.
Because of the numerous risks and uncertainties associated with pharmaceutical and biological product development, we are unable to accurately predict the timing or amount of increased expenses or when, or if, we will be able to achieve profitability. If we are required by the FDA or other regulatory authorities to perform studies in addition to those currently expected, or if there are any delays in completing our clinical trials or the development of any of our product candidates, our expenses could increase and product revenue could be further delayed.
We may not be able to sustain or increase profitability on a quarterly or annual basis. Our failure to remain profitable would depress our value and could impair our ability to raise capital, expand our business, maintain our research and development efforts, diversify our product offerings or continue our operations.
We will need substantial additional funding in order to complete development of our product candidates and commercialize our products, if approved. If we are unable to raise capital when needed and on terms favorable to us, we could be forced to delay, reduce or eliminate our product development programs or commercialization efforts.efforts.
We expect our expenses to increase in connection with our ongoing activities, particularly as we continue to develop our gene therapy pipeline, including our collaboration with AskBio, research and develop our autoimmune programs and advance the evolution of our ImmTOR platform in combination with IL-2, and continue research and development for other product candidates. Additionally, if we obtain regulatory approval for any of our product candidates, we
expect to incur significant commercialization expenses related to product manufacturing, marketing, sales and distribution. Accordingly, we will need to obtain substantial additional funding to continue operations. If we are unable to raise capital when needed or on attractive terms, we could be forced to delay, reduce or eliminate our clinical trials, our other research and development programs or any future commercialization efforts.
We believe that our existing cash, cash equivalents and restricted cash as of December 31, 20222023, combined with net proceeds received subsequent to December 31, 2023 in connection with our November 2023 private placement, will enable us to fund our current planned operations into mid-2024, though we may realize additional cash resources uponoperating expenses and capital expenditure requirements for at least the achievement of certain contingent collaboration milestones and wenext 12 months. We may pursue additional cash resources through public or private equity or debt financings, by establishing collaborations with other companies or through the monetization of potential royalty and/or milestone payments pursuant to our existing collaboration and license arrangements. Management’s expectations with respect to our ability to fund current and long-term planned operations are based on estimates that are subject to risks and uncertainties. If actual results are different from management’s estimates, we may need to seek additional strategic or financing opportunities
sooner than would otherwise be expected. However, there is no guarantee that any collaboration milestones will be achieved or that any of these strategic or financing opportunities will be executed on favorable terms, and some could be dilutive to existing stockholders. If we are unable to obtain additional funding on a timely basis, we may be forced to significantly curtail, delay, or discontinue one or more of our planned research or development programs or be unable to expand our operations, meet long-term obligations or otherwise capitalize on our commercialization of our product candidates. We have based this estimate on assumptions that may prove to be wrong, and we could use our capital resources sooner than we currently expect.
Our future capital requirements will depend on many factors, including:
•the timing for stockholder approval of the conversion of our Series A Non-Voting Convertible Preferred Stock, par value $0.0001 per share, or Series A Preferred Stock, into shares of our common stock and any redemptions of Series A Preferred Stock for cash;
•the scope, progress, results and costs of our clinical trials, preclinical development, manufacturing, laboratory testing and laboratory testing;logistics;
•the number of product candidates that we pursue and the speed with which we pursue development;
•our collaboration agreements remaining in effect, our entering into additional collaboration agreements and our ability to achieve milestones under these agreements;
•our headcount growth and associated costs;
•the costs, timing and outcome of regulatory review of our product candidates;
•the costs and timing of future commercialization activities, including manufacturing, marketing, sales and distribution, for any of our product candidates for which we receive marketing approval;
•the revenue, if any, from commercial sales of our product candidates for which we receive marketing approval;
•the costs and timing of preparing, filing and prosecuting patent applications, maintaining and enforcing our intellectual property rights and defending any intellectual property-related claims;
•the effect of competing technological and market developments; and
•the extent to which we acquire or invest in businesses, products and technologies, including entering into licensing or collaboration arrangements for product candidates.
The Certificate of Designation of Preferences, Rights and Limitations of the Series A Non-Voting Convertible Preferred Stock, or the Certificate of Designation, contains a provision granting each holder of the Series A Preferred Stock the option to require us to redeem any or all of such holder’s shares of Series A Preferred Stock beginning on the date that is 18 months following Closing; provided, however, that no holder will have the right to seek redemption of any shares of Series A Preferred Stock to the extent that such holder would otherwise be unable to convert such shares of Series A Preferred Stock due to the common stock beneficial ownership limitation contained in the Certificate of Designation. The per-share redemption price is the average closing trading price of the common stock for the ten preceding trading days ending on, and including, the trading day immediately prior to the date a notice of conversion is delivered to us. We could be required to use a significant amount of our cash resources on hand to satisfy this redemption obligation, particularly if holders of Series A Preferred Stock exercise their redemption right with respect to a significant number of shares of Series A Preferred Stock or at a time when the trading price of our common stock is elevated. Further, in the event that we do not have sufficient cash on hand to satisfy our redemption obligations, we may need to raise additional capital to satisfy these potential obligations. Any redemption payments could materially limit the amount of cash we have available to fund our operations.
Any additional fundraising efforts may divert our management from their day-to-day activities, which may adversely affect our ability to develop and commercialize our product candidates. In addition, we cannot guarantee that future financing will be available in sufficient amounts or on terms acceptable to us, if at all. Market volatility resulting from the COVID-19 pandemic,ongoing conflicts in Ukraine and the ongoing conflict in UkraineMiddle East and current global macroeconomic conditions or other factors could also adversely impact our ability to access capital as and when needed. Moreover, the terms of any financing may adversely affect the holdings or the
rights of our stockholders, and the issuance of additional securities, whether equity or debt, by us, or the possibility of such issuance, may cause the market price of our shares to decline. The sale of additional equity or convertible securities would dilute all of our stockholders. The incurrence of indebtedness could result in increased fixed payment obligations and we may be required to agree to certain restrictive covenants, such as limitations on our ability to incur additional debt, limitations on our ability to acquire, sell or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business. We could also be required to seek funds through arrangements with collaborators or others at an earlier stage than otherwise would be desirable and we may be required to relinquish rights to some of our technologies or product candidates or otherwise agree to terms unfavorable to us, any of which may have a material adverse effect on our business, operating results and prospects.
If we are unable to obtain funding on a timely basis, we may be required to significantly curtail, delay or discontinue one or more of our research or development programs, including our clinical trial programs, or the commercialization of any product candidates, or be unable to sustain or expand our operations or otherwise capitalize on our business opportunities, as desired, which could materially affect our business, financial condition and results of operations.
The terms of our credit facility place restrictions on our operating and financial flexibility. If we raise additional capital through debt financing, the terms of any new debt could further restrict our ability to operate our business.
On August 31, 2020, we entered into a term loan, or the 2020 Term Loan, of up to $35.0 million, consisting of term loans in an aggregate amount of $25.0 million, or the Term A Loan, and term loans in an aggregate amount of $10.0 million, or the Term B Loan, governed by a loan and security agreement among us and Oxford Finance LLC, or Oxford, as collateral agent and a lender, and Silicon Valley Bank, or SVB, as a lender. The Term A Loan was funded in full on August 31, 2020, the proceeds of which were used to repay our previously existing 2017 term loan and for general corporate and working capital purposes, and the draw period relating to the Term B Loan expired on September 30, 2021.
The 2020 Term Loan is secured by a lien on substantially all of our assets, other than intellectual property, provided that such lien includes any rights to payments and proceeds from the sale, licensing or disposition of intellectual property. We also granted Oxford a negative pledge with respect to our intellectual property.
Failure to satisfy our current and future debt obligations, including covenants to take or avoid specific actions, under the 2020 Term Loan could result in an event of default, our lenders could accelerate all of the amounts due. If we raise any additional debt financing, the terms of such additional debt could further restrict our operating and financial flexibility.
Our ability to use our net operating loss and research and development tax credit carryforwards to offset future taxable income may be subject to certain limitations.
We have net operating loss carryforwards, or NOLs, for federal and state income tax purposes that may be available to offset our future taxable income, if any. In general, under Sections 382 and 383 of the U.S. Internal Revenue Code of 1986, as amended, or the Code, a corporation that undergoes an “ownership change” is subject to limitations on its ability to use its pre-change NOLs to offset future taxable income. If the U.S. Internal Revenue Service, or IRS, challenges our analysis that existing NOLs will not expire before utilization due to previous ownership changes, or if we undergo an ownership change, in connection with or after a public offering, our ability to use our NOLs could be limited by Section 382 of the Code. Future changes in our stock ownership, some of which are outside of our control, could result in an ownership change under Sections 382 and 383 of the Code.
Furthermore, our ability to use NOLs of companies that we may acquire in the future may be subject to limitations. As a result, we may not be able to use a material portion of the NOLs reflected on our balance sheet, even if we attain profitability. The reduction of the corporate tax rate under the TCJA may cause a reduction in the economic benefit of our NOLs and other deferred tax assets available to us. Under current law, NOLs that arose before January 1, 2018 will beginmay be carried forward up to expire in 2041.20 years. NOLs that arose after 2017 may be used to offset at most 80% of our taxable income to the extent not offset by pre-2018 NOLs.NOLs and such NOLs can be carried forward indefinitely. As a result, we may become required to pay federal income taxes in future years despite having generated losses for federal income tax purposes in prior years.
Risks Related to our Intellectual Property
If we or our licensors are unable to adequately protect our proprietary technology, or obtain and maintain issued patents that are sufficient to protect our product candidates, others could compete against us more directly, which would negatively impact our business.
Our success depends in large part on our ability to obtain and maintain patent and other intellectual property protection in the United States and other countries with respect to our proprietary technology and products. We seek to protect our proprietary position by filing patent applications in the United States and abroad related to our novel technologies and product candidates. We also rely on trade secrets to protect aspects of our business that are not amenable to, or that we do not consider appropriate for, patent protection.
The patent prosecution process is expensive and time-consuming, and we may not be able to file and prosecute all necessary or desirable patent applications at a reasonable cost, in a timely manner or in all jurisdictions. As we reach the statutory deadlines for deciding whether and where to initiate prosecution in specific foreign jurisdictions by filing national stage applications based on our Patent Cooperation Treaty, or PCT, applications, we will have to decide whether and where to pursue patent protection for the various inventions claimed in our patent portfolio, and we will only have the opportunity to obtain patents in those jurisdictions where we pursue protection. It is also possible that we will fail to identify patentable aspects of our research and development output before it is too late to obtain patent protection. It is possible that defects of form in the preparation or filing of our patents or patent applications may exist, or may arise in the future, such as, with respect to proper priority claims, inventorship, claim scope or patent term adjustments. If there are material defects in the form or preparation of our patents or patent applications, such patents or applications may be invalid and unenforceable. Moreover, our competitors may independently develop equivalent knowledge, methods and know-how. Any of these outcomes could impair our ability to prevent competition from third parties, which may have an adverse impact on our business. We also cannot guarantee that any of our patent searches or analyses, including but not limited to the identification of relevant patents, the scope of patent claims or the expiration of relevant patents, are complete and thorough, nor can we be certain that we have identified each and every patent and pending application in the United States and abroad that is relevant to or necessary for the commercialization of our product candidates in any jurisdiction.
In some circumstances, we may not have the right to control the preparation, filing and prosecution of patent applications, or to maintain the patents covering technology that we license from third parties. We may also require the cooperation of our licensors to enforce any licensed patent rights, and such cooperation may not be provided. Therefore, these patents and applications may not be prosecuted and enforced in a manner consistent with the best interests of our business. Moreover, we have obligations under our licenses, and any failure to satisfy those obligations could give our licensor the right to terminate the license. Termination of a necessary license could have a material adverse impact on our business.
Some of our patent licenses are non-exclusive. In those cases, a competitor could obtain a license to the same or similar technology from the licensor. We have at least one exclusive patent license that is restricted to a particular field of use. A competitor could obtain a license to a similar technology outside of that field of use.
We cannot provide any assurances that the issued patents we currently own, or any future patents, include claims with a scope sufficient to protect our product candidates or otherwise provide any competitive advantage.
Further, it is possible that a patent claim may provide coverage for some but not all parts of a product candidate or third-party product. These and other factors may provide opportunities for our competitors to design around our patents.
Moreover, other parties may have developed technologies that may be related or competitive to our approach, and may have filed or may file patent applications, and may have received or may receive patents that may overlap or conflict with our patent applications, either by claiming similar methods or by claiming subject matter that could dominate our patent position. In addition, it may be some time before we understand how the patent office reacts to our patent claims and whether they identify prior art of relevance that we have not already considered.
Publications of discoveries in the scientific literature often lag behind the actual discoveries, and patent applications in the United States and other jurisdictions are typically not published until 18 months after filing, or in some cases not at all. Therefore, we cannot know with certainty whether we were the first to make the inventions claimed in any owned patents or pending patent applications, or that we were the first to file for patent protection of such inventions, nor can we know whether those from whom we may license patents were the first to make the inventions claimed or were the first to file. For these and other reasons, the issuance, scope, validity, enforceability and commercial value of our patent rights are subject to a level of uncertainty. Our pending and future patent applications may not result in patents being issued that protect our technology or products, in whole or in part, or which effectively prevent others from commercializing competitive technologies and products. Changes in either the patent laws or interpretation of the patent laws in the United States and other countries may diminish the value of our patents or narrow the scope of our patent protection.
We may be subject to a third-party preissuance submission of prior art to the U.S. Patent and Trademark Office, or USPTO, or other patent office, or become involved in opposition, derivation, reexamination, inter partes review, post-grant review or interference proceedings challenging our patent rights or the patent rights of others. An adverse determination in any such submission, proceeding or litigation could reduce the scope of, or invalidate, our patent rights, allow third parties to commercialize our technology or products and compete directly with us, without payment to us, or result in our inability to manufacture or commercialize product candidates without infringing third-party patent rights. In addition, if the breadth or strength of protection provided by our patents and patent applications is threatened, it could dissuade companies from collaborating with us to license, develop or commercialize current or future product candidates. Furthermore, an adverse decision in an interference proceeding can result in a third party receiving the patent right sought by us, which in turn could affect our ability to develop, market or otherwise commercialize our product candidates. The issuance, scope, validity, enforceability and commercial value of our patents are subject to a level of uncertainty.
The patent position of biotechnology and pharmaceutical companies generally is highly uncertain, involves complex legal and factual questions and has in recent years been the subject of much litigation. Due to legal standards relating to patentability, validity, enforceability and claim scope of patents covering biotechnological and pharmaceutical inventions, our ability to obtain, maintain and enforce patents is uncertain and involves complex legal and factual questions. Even if issued, a patent’s validity, inventorship, ownership or enforceability is not conclusive. Accordingly, rights under any existing patent or any patents we might obtain or license may not cover our product candidates, or may not provide us with sufficient protection for our product candidates to afford a commercial advantage against competitive products or processes, including those from branded and generic pharmaceutical companies.
In addition to the protection afforded by patents, we rely on trade secret protection and confidentiality agreements to protect proprietary know-how, information, or technology that is not covered by our patents. Although our agreements require all of our employees to assign their inventions to us, and we require all of our employees, consultants, advisors and any other third parties who have access to our trade secrets, proprietary know-how and other confidential information and technology to enter into appropriate confidentiality agreements, we cannot be certain that our trade secrets, proprietary know-how, and other confidential information and technology will not be subject to unauthorized disclosure or that our competitors will not otherwise gain access to or independently develop substantially equivalent trade secrets, proprietary know-how, and other information and technology. Furthermore, the laws of some foreign countries do not protect proprietary rights to the same extent or in the same manner as the laws of the United States. As a result, we may encounter significant problems in protecting
and defending our intellectual property globally. If we are unable to prevent unauthorized disclosure of our intellectual property related to our product candidates and technology to third parties, we may not be able to establish or maintain a competitive advantage in our market, which could adversely affect our business and operations.
Any litigation to enforce or defend our patent rights, even if we were to prevail, could be costly and time-consuming and would divert the attention of our management and key personnel from our business operations. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded if we were to prevail may not be commercially meaningful. Even if we are successful, domestic or foreign litigation, or USPTO or foreign patent office proceedings, may result in substantial costs and distraction to our management. We may not be able, alone or with our licensors or potential collaborators, to prevent misappropriation of our proprietary rights, particularly in countries where the laws may not protect such rights as fully as in the United States. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation or other proceedings, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation or other proceedings. In addition, during the course of this kind of litigation or proceedings, there could be public announcements of the results of hearings, motions or other interim proceedings
or developments or public access to related documents. If investors perceive these results to be negative, the market price for our common stock could be adversely affected.
If we are unable to protect the confidentiality of our trade secrets and know-how, our business and competitive position would be harmed.
In addition to seeking patents for some of our technology and product candidates, we also rely on trade secrets, including unpatented know-how, technology and other proprietary information, to maintain our competitive position. We seek to protect these trade secrets, in part, by entering into non-disclosure and confidentiality agreements with parties who have access to them, such as our employees, corporate collaborators, outside scientific collaborators, contract manufacturers, consultants, advisors and other third parties. We also seek to enter into confidentiality and invention or patent assignment agreements with our employees, advisors and consultants. Despite these efforts, any of these parties may breach the agreements and disclose our proprietary information, including our trade secrets, and we may not be able to obtain adequate remedies for such breaches. Our trade secrets may also be obtained by third parties by other means, such as breaches of our physical or computer security systems. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive and time-consuming, and the outcome is unpredictable. In addition, some courts inside and outside the United States are less willing or unwilling to protect trade secrets. Moreover, if any of our trade secrets were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent them, or those to whom they communicate it, from using that technology or information to compete with us. If any of our trade secrets were to be disclosed to, or independently developed by, a competitor, our competitive position would be harmed.
Changes in U.S. patent law could diminish the value of patents in general, thereby impairing our ability to protect our product candidates.
As is the case with other biotechnology companies, our success is heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the biotechnology industry involves both technological and legal complexity, and is therefore costly, time-consuming and inherently uncertain. In addition, recent patent reform legislation could further increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents. The Leahy-Smith Act America Invents Act, or the Leahy-Smith Act, included provisions that affect the way patent applications are prosecuted and may also affect patent litigation, including first-to-file provisions. A third party that files a patent application in the USPTO before us could therefore be awarded a patent covering an invention of ours even if we had made the invention before it was made by the third party. This requires us to be cognizant of the time from invention to filing of a patent application. Thus, for our U.S. patent applications containing a priority claim after March 16, 2013, the date such provisions became effective, there is a greater level of uncertainty in the patent law. Moreover, some of the patent applications in our portfolio will be subject to examination under the pre-Leahy-Smith Act law and regulations, while other patents applications in our portfolio will be subject to examination under the law and regulations, as amended by the Leahy-Smith Act. This introduces additional complexities into the prosecution and management of our portfolio.
In addition, the Leahy-Smith Act limits where a patentee may file a patent infringement suit and provides opportunities for third parties to challenge any issued patent in the USPTO. These provisions apply to all of our U.S. patents, even those issued before March 16, 2013. Because of a lower evidentiary standard in USPTO proceedings compared to the evidentiary standard in U.S. federal court necessary to invalidate a patent claim, a third party could potentially provide evidence in a USPTO proceeding sufficient for the USPTO to hold a claim invalid even though the same evidence would be insufficient to invalidate the claim if first presented in a federal court action.
Accordingly, a third party may attempt to use the USPTO procedures to invalidate our patent claims because it may be easier for them to do so relative to challenging the patent in a federal court action. It is not clear what, if any, impact the Leahy-Smith Act will have on the operation of our business. However, the Leahy-Smith Act and its implementation could increase the
uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents, all of which could have a material adverse effect on our business and financial condition.
In addition, recent U.S. Supreme Court rulings have narrowed the scope of patent protection available in certain circumstances and weakened the rights of patent owners in certain situations. From time to time, the U.S. Supreme Court, other federal courts, the U.S. Congress or the USPTO may change the standards of patentability, and any such changes could have a negative impact on our business.
Depending on these and other decisions by the U.S. Congress, the federal courts and the USPTO, the laws and regulations governing patents could change or be interpreted in unpredictable ways that would weaken our ability to obtain new patents or to enforce any patents that may issue to us in the future. In addition, these events may adversely affect our ability to defend any patents that may issue in procedures in the USPTO or in courts.
Third parties may initiate legal proceedings alleging that we are infringing their intellectual property rights, the outcome of which would be uncertain and could have a material adverse effect on the success of our business.
Our commercial success depends upon our ability, and the ability of our collaborators, to develop, manufacture, market and sell our product candidates and use our proprietary technologies without infringing the proprietary rights of third parties. There is considerable intellectual property litigation in the biotechnology and pharmaceutical industries. While no such litigation has been brought against us and we have not been held by any court to have infringed a third party’s intellectual property rights, we cannot guarantee that our technology, product candidates or use of our product candidates do not infringe third-party patents.
We are aware of numerous patents and pending applications owned by third parties, and we monitor patents and patent applications in the fields in which we are developing product candidates, both in the United States and elsewhere. However, we may have failed to identify relevant third-party patents or applications. For example, applications filed before November 29, 2000 and certain applications filed after that date that will not be filed outside the United States remain confidential until patents issue. Moreover, it is difficult for industry participants, including us, to identify all third-party patent rights that may be relevant to our product candidates and technologies because patent searching is imperfect due to differences in terminology among patents, incomplete databases and the difficulty in assessing the meaning of patent claims. We may fail to identify relevant patents or patent applications or may identify pending patent applications of potential interest but incorrectly predict the likelihood that such patent applications may issue with claims of relevance to our technology. In addition, we may be unaware of one or more issued patents that would be infringed by the manufacture, sale or use of a current or future product candidate, or we may incorrectly conclude that a third-party patent is invalid, unenforceable or not infringed by our activities. Additionally, pending patent applications that have been published can, subject to certain limitations, be later amended in a manner that could cover our technologies, our product candidates or the use of our product candidates.
The biotechnology and pharmaceutical industries are characterized by extensive litigation regarding patents and other intellectual property rights. Other parties may allege that our product candidates or the use of our technologies infringes patent claims or other intellectual property rights held by them or that we are employing their proprietary technology without authorization. We may become party to, or threatened with, future adversarial proceedings or litigation regarding intellectual property rights with respect to our product candidates and technology, including interference or derivation proceedings before the USPTO and similar bodies in other countries. Third parties may assert infringement claims against us based on existing intellectual property rights and intellectual property rights that may be granted in the future.
Patent and other types of intellectual property litigation can involve complex factual and legal questions, and their outcome is uncertain. If we are found, or believe there is a risk we may be found, to infringe a third party’s intellectual property rights, we could be required or may choose to obtain a license from such third party to continue developing and marketing our product candidates and technology. However, we may not be able to obtain any such license on commercially reasonable terms or at all. Even if we were able to obtain a license, it could be non-exclusive, thereby giving our competitors access to the same technologies licensed to us. We could be forced, including by court order, to cease commercializing the infringing technology or product. In addition, we could be found liable for monetary damages, including treble damages and attorneys’ fees if we are found to have willfully infringed a patent. A finding of infringement could prevent us from commercializing our product candidates or force us to cease some of our business operations, which could materially harm our business.
Claims that we have misappropriated the confidential information or trade secrets of third parties could have a similar negative impact on our business.
Even if we are successful in such proceedings, we may incur substantial costs and divert management time and attention in pursuing these proceedings, which could have a material adverse effect on us. Patent litigation is costly and time-consuming. We may not have sufficient resources to bring these actions to a successful conclusion. There could be public announcements of the results of hearings, motions or other interim proceedings or developments and if securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our common stock.
Any of these risks coming to fruition could have a material adverse impact on our business.
We may become involved in lawsuits to protect or enforce our patents or other intellectual property, and our issued patents covering our product candidates could be found invalid or unenforceable or could be interpreted narrowly if challenged in court.
Competitors may infringe our intellectual property, including our patents or the patents of our licensors. As a result, we may be required to file infringement claims to stop third-party infringement or unauthorized use. This can be expensive, particularly for a company of our size, and time-consuming. If we initiated legal proceedings against a third party to enforce a patent, if and when issued, covering one of our product candidates, the defendant could counterclaim that the patent covering our product candidate is invalid and/or unenforceable. In patent litigation in the United States, defendant counterclaims alleging invalidity and/or unenforceability are commonplace. Grounds for a validity challenge include alleged failures to meet any of several statutory requirements, including lack of novelty, obviousness or non-enablement, or failure to claim patent-eligible subject matter. Grounds for unenforceability assertions include allegations that someone connected with the prosecution of the patent withheld relevant information from the USPTO, or made a misleading statement, during prosecution. Third parties may also raise similar claims before administrative bodies in the United States or abroad, even outside the context of litigation. Such mechanisms include re-examination, post-grant review, inter partes review, interference proceedings and equivalent
proceedings in foreign jurisdictions, such as opposition proceedings. Such proceedings could result in revocation or amendment of our patents in such a way that they no longer cover our product candidates or competitive products. The outcome following legal assertions of invalidity and unenforceability is unpredictable. With respect to validity, for example, we cannot be certain that there is no invalidating prior art, of which we and the patent examiner were unaware during prosecution. If a defendant were to prevail on a legal assertion of invalidity and/or unenforceability, we would lose at least part, and perhaps all, of the patent protection on our product candidates. Moreover, even if not found invalid or unenforceable, the claims of our patents could be construed narrowly or in a manner that does not cover the allegedly infringing technology in question. Such a loss of patent protection would have a material adverse impact on our business.
The lives of our patents may not be sufficient to effectively protect our products and business.
Patents have a limited lifespan. In the United States, the natural expiration of a patent is generally 20 years after its first effective non-provisional filing date. Although various extensions may be available, the life of a patent, and the protection it affords, is limited. Even if patents covering our product candidates, proprietary technologies and their uses are obtained, once the patent life has expired, we may be open to competition. In addition, although upon issuance in the United States a patent’s life can be increased based on certain delays caused by the USPTO, this increase can be reduced or eliminated based on certain delays caused by the patent applicant during patent prosecution. Given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. If we do not have sufficient patent life to protect our product candidates, proprietary technologies and their uses, our business and results of operations will be adversely affected.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for noncompliance with these requirements.
Periodic maintenance fees on any issued patent are due to be paid to the USPTO and foreign patent agencies in several stages over the lifetime of the patent and, in some jurisdictions, during the pendency of a patent application. The USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. While an inadvertent lapse can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction.
Noncompliance events that could result in abandonment or lapse of a patent or patent application include, but are not limited to, failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. In such an event, our competitors might be able to enter the market, which would have an adverse effect on our business.
If we fail to comply with our obligations in our intellectual property licenses and funding arrangements with third parties, we could lose rights that are important to our business.
We are party to multiple license agreements that impose, and we may enter into additional licensing and funding arrangements with third parties that may impose, diligence, development and commercialization timelines, milestone payment, royalty, insurance and other obligations on us. Under our existing licensing agreements, we are obligated to pay royalties on net product sales of product candidates or related technologies to the extent they are covered by the agreement. Our results of operations will be affected by the level of royalty payments that we are required to pay to third parties. We cannot precisely predict the amount, if any, of royalties that we will be required to pay to third parties in the future. Any disagreements with the counterparty over the amount of royalties owed could lead to litigation, which is costly. In addition, if we fail to comply with our obligations under current or future license agreements, our counterparties may have the right to terminate these agreements, in which event we might not be able to develop, manufacture or market any product candidate that is covered by these agreements, or may face other penalties under the agreements. Such an occurrence could materially adversely affect the value of product candidates being developed using rights licensed to us under any such agreement. Termination of these agreements or reduction or elimination of our rights under these agreements may result in our having to negotiate new or reinstated agreements with less favorable terms, or cause us to lose our rights under these agreements, including our rights to important intellectual property or technology. Furthermore, our counterparties may allege that we are operating outside the scope of the licenses granted and terminate our license or otherwise require us to alter development, manufacturing or marketing activities.
We may not be successful in obtaining or maintaining necessary rights to our product candidates through acquisitions and in-licenses.
We currently have rights to certain intellectual property, through licenses from third parties and under patents and patent applications that we own, to develop our product candidates. Because we may find that our programs require the use of proprietary rights held by third parties, the growth of our business may depend in part on our ability to acquire, in-license or use these proprietary rights. We may be unable to acquire or in-license compositions, methods of use, processes or other third-party intellectual property rights from third parties that we identify as necessary for our product candidates. The licensing and acquisition of third-party intellectual property rights is a competitive area, and a number of more established companies are also
pursuing strategies to license or acquire third-party intellectual property rights that we may consider attractive. These established companies may have a competitive advantage over us due to their size, financial resources and greater clinical development and commercialization capabilities. In addition, companies that perceive us to be a competitor may be unwilling to assign or license rights to us. We also may be unable to license or acquire third-party intellectual property rights on terms that would allow us to make an appropriate return on our investment.
If we are unable to successfully obtain rights to required third-party intellectual property rights or maintain the existing intellectual property rights we have, we may have to abandon development of that program and our business and financial condition could suffer.
We may be subject to claims by third parties asserting that our employees or we have misappropriated their intellectual property, or claiming ownership of what we regard as our own intellectual property.
Many of our employees were previously employed at universities or other biotechnology or pharmaceutical companies, including our competitors or potential competitors. We may also engage advisors and consultants who are concurrently employed at universities or other organizations or who perform services for other entities.
Although we try to ensure that our employees, advisors and consultants do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or our employees, advisors or consultants have used or disclosed intellectual property, including trade secrets or other proprietary information, of any such party’s former or current employer or in violation of an agreement with another party. Although we have no knowledge of any such claims being alleged to date, if such claims were to arise, litigation may be necessary to defend against any such claims.
In addition, while it is our policy to require our employees, consultants, advisors and contractors who may be involved in the development of intellectual property to execute agreements assigning such intellectual property to us, we may be unsuccessful in executing such an agreement with each party who in fact develops intellectual property that we regard as our own. Our and their assignment agreements may not be self-executing or may be breached, and we may be forced to bring claims against third parties, or defend claims they may bring against us, to determine the ownership of what we regard as our intellectual property. Similarly, we may be subject to claims that an employee, advisor or consultant performed work for us that conflicts with that person’s obligations to a third party, such as an employer, and thus, that the third party has an ownership interest in the intellectual property arising out of work performed for us. Litigation may be necessary to defend against these claims.
Although we have no knowledge of any such claims being alleged to date, if such claims were to arise, litigation may be necessary to defend against any such claims.
If we fail in prosecuting or defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. Even if we are successful in prosecuting or defending against such claims, litigation could result in substantial costs and be a distraction to management.
We will not seek to protect our intellectual property rights in all jurisdictions throughout the world and we may not be able to adequately enforce our intellectual property rights even in the jurisdictions where we seek protection.
Filing, prosecuting and defending patents on product candidates in all countries and jurisdictions throughout the world would be prohibitively expensive, and our intellectual property rights in some countries outside the United States could be less extensive than in the United States, assuming that rights are obtained in the United States and assuming that rights are pursued outside the United States. In this regard, in addition to the United States, we also seek to protect our intellectual property rights in other countries. The statutory deadlines for pursuing patent protection in individual foreign jurisdictions are based on the priority date of each of our patent applications. For all of the patent families in our portfolio, including the families that may provide coverage for our lead product candidate, the relevant statutory deadlines have not yet expired. Therefore, for each of the patent families that we believe provide coverage for our lead product candidate, we will need to decide whether and where to pursue additional protection outside the United States. In addition, the laws of some foreign countries, do not protect intellectual property rights to the same extent as federal and state laws in the United States. Consequently, for our existing patent rights outside the United States and any foreign patent rights we may decide to pursue in the future, we may not be able to obtain relevant claims and/or we may not be able to prevent third parties from practicing our inventions in all countries outside the United States, or from selling or importing products made using our inventions in and into the United States or other jurisdictions.
Competitors may use our technologies in jurisdictions where we do not pursue and obtain patent protection to develop their own products and further, may export otherwise infringing products to territories where we have patent protection, but enforcement is not as strong as in the United States. These products may compete with our product candidates and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing. Even if we pursue and obtain issued patents in particular jurisdictions, our patent claims or other intellectual property rights may not be effective or sufficient to prevent third parties from so competing.
If we do not obtain additional protection under the Hatch-Waxman Act and similar foreign legislation extending the terms of our patents for our product candidates, our business may be harmed.
Depending upon the timing, duration and specifics of FDA regulatory approval for our product candidates, one or more of our U.S. patents may be eligible for limited patent term restoration under the Drug Price Competition and Patent Term Restoration Act of 1984, or the Hatch-Waxman Act. The Hatch-Waxman Act permits a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. Patent term restorations, however, are limited to a maximum of five years and cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval by the FDA.
The application for patent term extension is subject to approval by the USPTO, in conjunction with the FDA. It takes at least six months to obtain approval of the application for patent term extension. We may not be granted an extension because of, for example, failing to apply within applicable deadlines, failing to apply prior to expiration of relevant patents or otherwise failing to satisfy applicable requirements. Moreover, the applicable time period or the scope of patent protection afforded could be less than we request. If we are unable to obtain patent term extension or restoration or the term of any such extension is less than we request, the period during which we will have the right to exclusively market our product will be shortened, our competitors may obtain earlier approval of competing products and our ability to generate revenues could be materially adversely affected.
Risks Related to our Operations
Our future success depends on our ability to retain key executives and to attract, retain and motivate qualified personnel.
We are highly dependent on Carsten Brunn, Ph.D., our President and Chief Executive Officer, as well as the other principal members of our management, scientific and clinical team.teams. Although we have entered into employment agreements or offer letters with Dr. Brunn and other executive officers, each of them may terminate their employment with us at any time. We do not maintain “key person” insurance for any of our executives or other employees.
Recruiting and retaining qualified scientific, clinical, manufacturing, technology and sales and marketing personnel will also be critical to our success. The loss of the services of our executive officers or other key employees could impede the achievement of our research, development and commercialization objectives and seriously harm our ability to successfully implement our business strategy. Furthermore, replacing executive officers and key employees may be difficult and may take an extended period of time because of the limited number of individuals in our industry with the breadth of skills and experience required to successfully develop, gain regulatory approval of and commercialize product candidates. Competition to hire from this limited pool is intense, and we may be unable to hire, train, retain or motivate these key personnel on acceptable terms given the competition among numerous pharmaceutical and biotechnology companies for similar personnel. We also experience competition for the hiring of scientific and clinical personnel from universities and research institutions. In addition, we rely on consultants and advisors, including scientific and clinical advisors, to assist us in formulating our research and development and commercialization strategy. Our consultants and advisors may be employed by employers other than us and may have commitments under consulting or advisory contracts with other entities that may limit their availability to us. If we are unable to continue to attract and retain high quality personnel, our ability to pursue our growth strategy will be limited.
We have incurred increased costs as a result of operating as a public company, and our management will be required to devote substantial time to compliance initiatives and corporate governance practices.
As a public company, we have incurred and expect to continue to incur significant legal, accounting and other expenses. If we are unable to maintain effective internal control over financial reporting, we may not have adequate, accurate or timely financial information, and we may be unable to meet our reporting obligations as a public company or comply with the requirements of the SEC or Section 404.404 of the Sarbanes-Oxley Act of 2002. This could result in a restatement of our financial statements, the imposition of sanctions, including the inability of registered broker dealers to make a market in our common stock, or investigation by regulatory authorities. Any such action or other negative results caused by our inability to meet our reporting requirements or comply with legal and regulatory requirements or by disclosure of an accounting, reporting or control issue could adversely affect the trading price of our securities and our business. Material weaknesses in our internal control over financial reporting could also reduce our ability to obtain financing or could increase the cost of any financing we obtain. This could result in an adverse reaction in the financial markets due to a loss of confidence in the reliability of our financial statements.
We have identified a material weakness in our internal control over financial reporting and may identify additional material weaknesses in the future or otherwise fail to maintain an effective system of internal controls, which may result in material misstatements of our consolidated financial statements or cause us to fail to meet our periodic reporting obligations.
In connection with the audit of our consolidated financial statements as of and for the year ended December 31, 2023, we identified a material weakness in our internal control over financial reporting and concluded that our internal control over financial reporting was not effective as of December 31, 2023. There are no material accounting errors or omissions within the consolidated financial statements as a result of this material weakness.We concluded that we did not design and implement effective internal controlsspecifically related to the documentation of the assumptions supporting the valuation of the in-process intangible assets in connection with the Old Cartesian material business combination and the initial and ongoing contingent value right obligation issued at the time to legacy Selecta stockholders. This includes a lack of sufficient documentation to provide evidence of the associated management review controls.
In response to the identified material weakness above, we, with the oversight of the Audit Committee of the Board of Directors, or the Audit Committee, intend to take comprehensive actions to remediate the material weakness in internal control over financial reporting. We expect to re-evaluate the scope and level of precision for conducting and documenting the reviews over significant acquisitions and contingent value rights including the review of prospective financial information used in valuation reports produced by third-party specialists supporting the accounting for business combinations and contingent value rights. The remediation efforts are intended both to address the identified material weakness and to enhance our overall financial control environment.
This material weakness and any other failure to maintain effective internal control over financial reporting could result in a loss of confidence in the reliability of our financial statements which could have a negative impact on the trading price of our common stock and harm our ability to raise additional capital on acceptable terms or at all.
A variety of risks associated with maintaining our subsidiary in Russia or expanding operations internationally could adversely affect our business.
In addition to our U.S. operations, we maintain a wholly owned subsidiary in Russia, Selecta (RUS). However, we are in the process of winding down these operations.all remaining operations of this subsidiary. We may face risks associated with winding down the operations of our subsidiary in Russia, or with any international operations, including possible unfavorable regulatory, pricing and reimbursement, legal, political, tax and labor conditions, and risks associated with our compliance with evolving international sanctions, which could harm our business. We may also rely on collaborators to commercialize any approved product candidates outside of the United States. Doing business internationally involves a number of risks, including but not limited to:
•multiple, conflicting and changing laws and regulations, such as privacy regulations, tax laws, export and import restrictions, employment laws, regulatory requirements and other governmental approvals, permits and licenses;
•failure by us to obtain and maintain regulatory approvals for the use of our product candidates in various countries;
•additional potentially relevant third-party patent rights;
•complexities and difficulties in obtaining protection of and enforcing our intellectual property rights;
•difficulties in staffing and managing foreign operations;
•complexities associated with managing multiple-payor reimbursement regimes, government payors or patient self-pay systems;
•limits on our ability to penetrate international markets;
•financial risks, such as longer payment cycles, difficulty collecting accounts receivable, the impact of local and regional financial crises on demand and payment for our product candidates and exposure to foreign currency exchange rate fluctuations, which could result in increased operating expenses and reduced revenues;
•natural disasters, political and economic instability, including wars, events of terrorism and political unrest, outbreak of disease, including the COVID-19 pandemic, boycotts, curtailment of trade and other business restrictions, economic sanctions, and economic weakness, including inflation;
•changes in diplomatic and trade relationships;
•challenges in enforcing our contractual and intellectual property rights, especially in those foreign countries that do not respect and protect intellectual property rights to the same extent as the United States;
•restriction on cross-border investment, including enhanced oversight by the Committee on Foreign Investment in the United States and substantial restrictions on investment from China;
•certain expenses including, among others, expenses for travel, translation and insurance;
•legal risks, including use of the legal system by the government to benefit itself or affiliated entities at our expense, including expropriation of property;
•regulatory and compliance risks that relate to maintaining accurate information and control over sales and activities that may fall within the purview of the FCPA its books and records provisions, or its anti-bribery provisions; and
•risks that we may suffer reputational harm as a result of our operations in Russia.
Any of these factors could significantly harm our future international expansion and operations and, consequently, our results of operations.
Our business and operations, including our development programs, could be materially disrupted in the event of system failures, security breaches, violations of data protection laws or data loss or damage by us or third parties on which we rely, including our CROs or other contractors or consultants.
Our internal computer systems and those of third parties on which we rely, including our CROs and other contractors and consultants, are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. If such an event were to occur and cause interruptions in our operations, it could have a material adverse effect on our business operations, including a material disruption of our development programs. Unauthorized disclosure of sensitive or confidential patient or employee data, including personally identifiable information, whether through breach of computer systems, systems failure, employee negligence, fraud or misappropriation, or otherwise, or unauthorized access to or through our information systems and networks, whether by our employees or third parties, could result in negative publicity, legal liability and damage to our reputation. Unauthorized disclosure of personally identifiable information could also expose us to sanctions for violations of data privacy laws and regulations around the world. To the extent that any disruption or security breach resulted in a loss of or damage to our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and the further development of our product candidates could be delayed. For example, the loss of or damage to clinical trial data, such as from completed or ongoing clinical trials, for any of our product candidates would likely result in delays in our marketing approval efforts and significantly increased costs in an effort to recover or reproduce the data.
We have previously been, and expect to remain, the target of cyber-attacks. As we become more dependent on information technologies to conduct our operations, cyber incidents, including deliberate attacks, such as ransomware attacks, and attempts to gain unauthorized access to computer systems and networks, may increase in frequency and sophistication. These incidents pose a risk to the security of our systems and networks, the confidentiality and the availability and integrity of our data and these risks apply both to us, and to third parties on whose systems we rely for the conduct of our business. While we do not believe the effect of these incidents has historically been material to our results of operations, financial condition or prospects, cyber threats are persistent and constantly evolving. Such threats have increased in frequency, scope and potential impact in recent years, which increases the difficulty of detecting and successfully defending against them. As cyber threats continue to
evolve, we may be required to incur additional expenses in order to enhance our protective measures or to remediate any information security vulnerability. There can be no assurance that we or our third-party providers will be successful in preventing cyber-attacks or mitigating their effects. Similarly, there can be no assurance that our collaborators, CROs, third-party logistics providers, distributors and other contractors and consultants will be successful in protecting our clinical and other data that is stored on their systems. Any cyber-attack or destruction or loss of data could have a material adverse effect on our business and prospects. In addition, we may suffer reputational harm or face litigation or adverse regulatory action as a result of cyber-attacks or destruction or loss of data and may incur significant additional expense to implement further data protection measures. It is also possible that unauthorized access to data may be obtained through inadequate use of security controls by our suppliers or other vendors.
Although we have general liability insurance coverage, our insurance may not cover all claims, continue to be available on reasonable terms or be sufficient in amount to cover one or more large claims. Additionally, the insurer may disclaim coverage as to any claim. The successful assertion of one or more large claims against us that exceed or are not covered by our insurance coverage or changes in our insurance policies, including premium increases or the imposition of large deductible or co-insurance requirements, could have a material adverse effect on our business, prospects, operating results and financial condition.
Acquisitions or joint ventures could disrupt our business, cause dilution to our stockholders and otherwise harm our business.
We may acquire other businesses, product candidates or technologies as well as pursue strategic alliances, joint ventures, technology licenses or investments in complementary businesses. We have not made any acquisitions to date, and our ability to do so successfully is unproven. Any of these transactions could be material to our financial condition and operating results and expose us to many risks, including:
•disruption in our relationships with future customers or with current or future distributors or suppliers as a result of such a transaction;
•unexpected liabilities related to acquired companies;
•difficulties integrating acquired personnel, technologies and operations into our existing business;
•diversion of management time and focus from operating our business to acquisition integration challenges;
•increases in our expenses and reductions in our cash available for operations and other uses;
•possible write-offs or impairment charges relating to acquired businesses; and
•inability to develop a sales force for any additional product candidates.
Foreign acquisitions involve unique risks in addition to those mentioned above, including those related to integration of operations across different cultures and languages, currency risks and the particular economic, political and regulatory risks associated with specific countries.
Also, the expected benefit of any acquisition may not materialize. Future acquisitions or dispositions could result in potentially dilutive issuances of our equity securities, the incurrence of debt, contingent liabilities or amortization expenses or write-offs of goodwill, any of which could harm our financial condition. We cannot predict the number, timing or size of future joint ventures or acquisitions, or the effect that any such transactions might have on our operating results.
Risks Related to our Common Stock
The market price of our common stock may be volatile and fluctuate substantially, which could result in substantial losses for purchasers of our common stock.
The trading price of our common stock is likely to be volatile and could be subject to wide fluctuations in response to various factors, some of which are beyond our control. The stock market in general and the market for smaller biopharmaceutical companies in particular have experienced extreme volatility that has often been unrelated to the operating performance of particular companies. As a result of this volatility, you may not be able to sell your common stock at or above the price at which you purchased. The market price for our common stock may be influenced by many factors, including:
•the success of competitive products or technologies;
•results or progress, or changes in approach or timelines, of clinical trials of our product candidates or those of our competitors;
•failure or discontinuation of any of our development programs;
•commencement of, termination of, or any development related to any collaboration or licensing arrangement;
•regulatory or legal developments in the United States and other countries;
•development of new product candidates that may address our markets and make our product candidates less attractive;
•changes in physician, hospital or healthcare provider practices that may make our product candidates less useful;
•announcements by us, our collaborators or our competitors of significant acquisitions, strategic partnerships, joint ventures, collaborations or capital commitments;
•announcement or market expectation of additional financing efforts;
•developments or disputes concerning patent applications, issued patents or other proprietary rights;
•the recruitment or departure of key personnel;
•the level of expenses related to any of our product candidates or clinical development programs;
•failure to meet or exceed financial estimates, projections or development timelines of the investment community or that we provide to the public;
•the results of our efforts to discover, develop, acquire or in-license additional product candidates or products;
•actual or expected changes in estimates as to financial results, development timelines or recommendations by securities analysts;
•variations in our financial results or those of companies that are perceived to be similar to us;
•changes in the structure of healthcare payment systems;
•sale of common stock by us or our stockholders in the future as well as the overall trading volume of our common stock;
•changes in the composition of our stockholder base;
•activity in the options market for shares of our common stock;
•market conditions in the pharmaceutical and biotechnology sectors;
•general economic, industry and market conditions; and
•the other factors described in this “Risk“Risk Factors” section.
Our executive officers, directors, and principal stockholders, if they choose to act together, will continue to have the ability to control or significantly influence all matters submitted to stockholders for approval.
Our executive officers, directors and stockholders who own more than 5% of our outstanding common stock and their respective affiliates, in the aggregate, hold shares representing approximately 38.4%60.1% of our outstanding voting stock as of December 31, 2022.2023, and assuming the conversion of all shares of Series A Preferred Stock into common stock and reflecting the completion of the November 2023 private placement, which occurred subsequent to December 31, 2023. As a result, if these stockholders choose to act together, they would be able to control or significantly influence all matters submitted to our stockholders for approval, as well as our management and affairs. For example, these persons, if they choose to act together, would control or significantly influence the election of directors, the composition of our management and approval of any merger, consolidation or sale of all or substantially all of our assets.
SalesFuture sales of a substantial number of shares of our common stock in the public market, or the perception in the market that the holders of a large number of shares intend to sell shares, could reduce the market price of our common stock.
SalesConcurrently and in connection with the execution of the Merger Agreement, certain Old Cartesian securityholders, as of immediately prior to the Merger, and certain of our directors and officers as of immediately prior to the Merger entered into lock-up agreements with us, pursuant to which each such stockholder is subject to a substantial numberlockup on the sale or transfer of shares of our common stock held by each such stockholder, including those shares received by Old Cartesian securityholders in the Merger, for a period of 180 days from the Closing. Upon expiration of this 180-day lockup period, these shares will become eligible for sale in the public market.
On November 13, 2023, we also entered into a Registration Rights Agreement, or the RRA, with holders of common stock and Series A Preferred Stock signatory thereto. Pursuant to the RRA, we are obligated to prepare and file a resale registration statement with the SEC by the Filing Deadline (as defined therein). We agreed to use our reasonable best efforts to cause this registration statement to be declared effective by the SEC within 45 calendar days of the Filing Deadline (or within 90 calendar days of the Filing Deadline if the SEC reviews the registration statement). Once such registration statement is declared effective, the shares to which the registration statement relates will no longer constitute restricted securities and may be sold freely in the public markets, subject to lapse on any related contractual restrictions related thereto of any holder party thereto, and subject to any restrictions that may be applicable to any control securities.
If our stockholders sell, indicate an intention to sell, or it is perceived that they will sell substantial amounts of our common stock in the public market could occur at any time. These sales, orafter legal restrictions on resale lapse, the perception in the market that the holders of a large number of shares intend to sell shares, could reduce the markettrading price of our common stock reduce. As of December 31, 2022, we had 153,042,435could decline. In addition, shares of common stock outstanding. Also, as of December 31, 2022, 15,844,651 and 1,705,558 shares ofour common stock that are subject to our outstanding options or restricted stock unit awards, respectively, under our outstanding equity plans arewill become eligible for sale in the public market to the extent permitted by the provisions of various vesting schedules,agreements and RuleRules 144 and 701 under the Securities Act. Additionally, as of December 31, 2022, up to 31,228,279 shares of common stock are issuable upon exercise of outstanding warrants. If these additional shares of common stock are sold, or it is perceived that they will be sold,
Anti-takeover provisions in the public market, the trading price of our common stock could decline.
We may not have the funds necessary to fulfill our obligation to repurchase certain warrants.
Under certain circumstances, holders of certain warrants may require us to repurchase the remaining unexercised portion of such warrants for an amount of cash equal to the value of the warrant as determined in accordance with the Black-Scholes option pricing modelcharter documents and under Delaware law and the terms of the warrants. Our ability to repurchase the warrants depends onsome of our ability to generate cash flow in the future. To some extent, this is subject to general economic, financial, competitive, legislative and regulatory factors and other factors that are beyond our control. We cannot be certain that we will maintain sufficient cash reserves or that our business will generate cash flow from operations at levels sufficient to permit us to repurchase the warrants.
Provisions in our restated certificate of incorporation and restated bylaws and under Delaware lawcontracts could make an acquisition of our company, which may be beneficial to our stockholders,us more difficult and may prevent attempts by our stockholders to replace or remove our current management.
Provisions in our restated certificate of incorporation, as amended, or the Charter, and ouramended and restated bylawsby-laws may discourage, delay or prevent a merger,an acquisition or othera change in controlmanagement. These provisions include a prohibition on actions by written consent of our company that stockholders may consider favorable, including transactions in which a stockholder might otherwise receive a premium for its shares. These provisions could also limitand the price that investors might be willing to pay in the future for sharesability of our common stock, thereby depressing the market price of our common stock. In addition, because our board of directors, is responsible for appointingor the membersBoard of our management team, these provisions may frustrate or prevent any attempts by our stockholdersDirectors, to replace or remove our current management by making it more difficult for stockholders to replace members of our board of directors.
Moreover,issue preferred stock without stockholder approval. In addition, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the General Corporation Law of the State of Delaware,DGCL, which prohibits a person who ownsstockholders owning in excess of 15% of our outstanding voting stock from merging or combining with usus.
Although we believe these provisions collectively will provide for a periodan opportunity to receive higher bids by requiring potential acquirers to negotiate with our Board of three years afterDirectors, they would apply even if the dateoffer may be considered beneficial by some stockholders. In addition, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove then current management by making it more difficult for stockholders to replace members of the transaction inBoard of Directors, which is responsible for appointing the person acquired in excessmembers of 15%management.
Furthermore, our restated certificate of incorporationCharter specifies that, unless we consent in writing to the selection of an alternative forum, the Court of Chancery of the State of Delaware will be the sole and exclusive forum for most legal actions involving claims brought against us by stockholders. We believe this provision benefits us by providing increased consistency in the application of Delaware law by chancellors particularly experienced in resolving corporate disputes, efficient administration of cases on a more expedited schedule relative to other forums and protection against the burdens of multi-forum litigation. However, the provision may have the effect of discouraging lawsuits against our directors, officers, employees and agents as it may limit any stockholder’s ability to bring a claim in a judicial forum that such stockholder finds favorable for disputes with us or our directors, officers, employees or agents.
In addition, the Certificate of Designation relating to our Series A Preferred Stock may delay or prevent a change in control of our Company. At any time while at least 30% of the originally issued Series A Preferred Stock remains issued and outstanding, we may not consummate a Fundamental Transaction (as defined in the Certificate of Designation) or any merger or consolidation of the Company with or into another entity or any stock sale to, or other business combination in which the stockholders of the Company immediately before such transaction do not hold at least a majority of the capital stock of the Company immediately after such transaction, without the affirmative vote of the holders of a majority of the then outstanding shares of the Series A Preferred Stock. This provision of the Certificate of Designation may make it more difficult for us to enter into any of the aforementioned transactions.
We have been in the past and may in the future be subject to securities class action lawsuits.stockholder litigation.
In the past, securities class action litigation has often been brought against a company following a decline in the market price of its securities. This risk is especially relevant for us because biopharmaceutical companies have experienced significant stock price volatility in recent years. Involvement in such litigation, could result in substantial costs and a diversion of management’s attention and resources, which could harm our business.
On February 21, 2024, Paul Wymer, a purported stockholder of our Company, filed an action against us and members of our Board of Directors in the U.S. District Court for the Southern District of New York, titled Wymer v. Cartesian Therapeutics, Inc., et al., No. 24-cv-01288. The complaint alleges that the defendants violated Sections 14(a) and 20(a) of the Exchange Act by failing to disclose purportedly material information to our stockholders in our Preliminary and Definitive Proxy Statements filed on January 31, 2024, and February 14, 2024, respectively, in connection with the solicitation of stockholder approval of a proposal to convert our Series A Preferred Stock into our common stock, subject to certain beneficial ownership limitations, or the Conversion Proposal. The complaint seeks injunctive relief enjoining or rescinding the Merger, issuance of an amended proxy statement, and attorneys' fees and costs. Additional similar lawsuits may be filed. We believe this lawsuit is without merit and intend to vigorously defend against this plaintiff’s claims.
On February 7, 2024, Justin Sloan, a purported stockholder of our Company, filed a putative class action on behalf of himself and similarly situated stockholders of the Company against our Company and members of our Board of Directors in the Court of Chancery of the State of Delaware, titled Sloan v. Barabe, et al., No. 2024-0105. The complaint alleges that the individual defendants breached their fiduciary duties by failing to disclose purportedly material information to our Company’s stockholders in our Preliminary Proxy Statement filed on January 31, 2024 in connection with the solicitation of stockholder approval of the Conversion Proposal. The complaint seeks a temporary injunction against the stockholder vote on the Conversion Proposal, compensatory damages, pre-and post-judgment interest, and attorneys’ fees and costs. At a telephonic hearing on February 28, 2024, the Court denied the Plaintiff’s motion to expedite the proceedings, rejecting Plaintiff's argument that the lawsuit raised colorable disclosure claims warranting expedited treatment. Additional similar lawsuits may be filed. We believe this lawsuit is without merit and intend to vigorously defend against this plaintiff’s claims.
On August 3, 2020, a stockholder of Selecta filed a stockholder derivative action, purportedly on behalf of Selecta and against certain current and former members of the Company’s Board of Directors, as well as one affiliated company owned by a current board member, in the Court of Chancery of the State of Delaware, namely Franchi v. Barabe, et al. The complaint alleges that the individual defendants breached their fiduciary duties and committed corporate waste when they authorized a private placement transaction, announced on December 19, 2019, at a price allegedly below fair value. The complaint further alleges that the four defendant directors who participated in the private placement were unjustly enriched in connection with the transaction. On September 25, 2020, the defendants filed a motion to dismiss the lawsuit. On November 6, 2020, the plaintiff filed an amended complaint, and the defendants filed a second motion to dismiss on January 8, 2021. On December 31, 2020, we received a litigation demand letter from two other putative stockholders relating to the same private placement transaction. On April 12, 2021, the Court of Chancery in the State of Delaware granted a motion to stay the litigation pending a review by a Special Committee appointed by the Company’s Board of Directors. While the litigation was stayed, the parties reached an agreement in principle to settle the matter, and on March 18, 2022, they submitted a Stipulation and Agreement of Settlement and other documentation to the Court for its approval of the settlement. On July 21, 2022, the Court held a settlement hearing, at which the settlement was approved. On August 1, 2022, the Court entered an Order and Final Judgment which dismissed the action, and all claims contained therein, with prejudice. We could receive other demands or be subject to other litigation. While weWe intend to vigorously defend against any demands which we believe to be without merit, theremerit.
There can be no assurance as to the outcome of any stockholder litigation. Unfavorable outcomes in securities class action litigation could require us to pay extensive damages, which could delay or prevent our ability to develop our product candidates and harm our operations.
Risks Related to the Merger
There is no guarantee that the Merger will increase stockholder value.
In November 2023 we merged with Old Cartesian. We cannot guarantee that implementing the Merger and related transactions will not impair stockholder value or otherwise adversely affect our business. The Merger poses significant integration challenges between our businesses and management teams which could result in management and business disruptions, any of which could harm our results of operation, business prospects, and impair the value of the Merger to our stockholders.
Pursuant to the terms of the Merger Agreement, we are required to recommend that our stockholders approve the conversion of shares of our Series A Preferred Stock into shares of our common stock. We cannot guarantee that our stockholders will approve this matter, and if they fail to do so we may be required to settle such shares in cash and our operations may be materially harmed.
Under the terms of the Merger Agreement, we agreed to call and hold a meeting of our stockholders to obtain the requisite approvals for the conversion of shares of Series A Preferred Stock into shares of our common stock, and, if such approval is not obtained at that meeting, to seek to obtain such approvals at an annual or special stockholders' meeting to be held at least every six months thereafter until such approval is obtained, which would be time-consuming and costly. Additionally, beginning on the date that is 18 months from the date of the Closing, the holders of our then-outstanding shares of Series A Preferred Stock will be entitled to elect to have such shares of Series A Preferred Stock redeemed for cash at a price per share equal to the ten-day trailing average closing trading price of the common stock at such time, as described in our Certificate of Designation relating to the Series A Preferred Stock. If we are forced to cash settle a significant amount of the Series A Preferred Stock, it could materially affect our results of operations.
The failure to successfully integrate the businesses of Selecta and Old Cartesian in the expected timeframe would adversely affect our future results.
Our ability to successfully integrate the operations of Selecta and Old Cartesian will depend, in part, on our ability to realize the anticipated benefits and cost savings from the Merger. If we are not able to achieve these objectives, the anticipated benefits and cost savings of the Merger may not be realized fully, or at all, or may take longer to realize than expected, and the value of our common stock may be adversely affected. In addition, the integration of Selecta's and Old Cartesian’s respective businesses will be a time-consuming and expensive process. Proper planning and effective and timely implementation will be critical to avoid any significant disruption to our operations. It is possible that the integration process could result in the loss of key employees, the disruption of our business or the identification of inconsistencies in standards, controls, procedures and policies that adversely affect our ability to maintain relationships with customers, suppliers, distributors, creditors, lessors, clinical trial investigators or managers or to achieve the anticipated benefits of the Merger. Delays encountered in the integration process could have a material adverse effect on our operating results and financial condition, including the value of its common stock.
We have incurred substantial expenses related to the integration of Old Cartesian.
We have incurred substantial expenses in connection with the Merger and the subsequent integration of Old Cartesian with Selecta. There are a large number of processes, policies, procedures, operations, technologies and systems that must be integrated, including purchasing, accounting and finance, sales, billing, payroll, research and development, marketing and benefits. Both we and Old Cartesian have incurred significant transaction expenses in connection with the drafting and negotiation of the Merger Agreement and significant severance expenses as a result of the Merger. While we and Old Cartesian have assumed that a certain level of expenses will be incurred, there are many factors beyond our control that could affect the total amount or the timing of the integration expenses. Moreover, many of the expenses that have been and will be incurred are, by their nature, difficult to estimate accurately. These integration expenses have resulted in our taking significant charges against earnings following the completion of the Merger, and the amount and timing of such charges are uncertain at present.
Item 1B. Unresolved Staff Comments
Not applicable.
Item 1C. Cybersecurity
One of the key responsibilities of our Board of Directors is informed oversight of our risk management process, including risks from cybersecurity threats. Our Board of Directors is responsible for monitoring and assessing strategic risk exposure, and our executive officers are responsible for the day-to-day management of the material risks we face. Our Board of Directors administers its cybersecurity risk oversight function directly and through the Audit Committee, which conducts regular risk assessments related to all matters affecting the enterprise, including cybersecurity, and receives periodic reports on the Company’s cybersecurity risks and activities.
Our Chief Financial Officer and our Senior Director, Head of IT and Informatics are the Company employeesprimarily responsible for assessing and managing material risks from cybersecurity threats with assistance from third-party service providers. Our Chief Financial Officer has served as a biotechnology executive for 15 years, whose responsibilities have included direct oversight of his companies' cybersecurity risks. Our Senior Director, Head of IT and Informatics has served as an information technology professional for over ten years and has held senior IT positions at multiple biotechnology companies, where his primary responsibilities included maintaining direct oversight over his companies' cybersecurity risks.
We have established policies and processes for assessing, identifying, and managing material risk from cybersecurity threats, and have integrated these processes into our overall risk management systems and processes. We routinely assess material risks from cybersecurity threats, including any potential unauthorized occurrence on or conducted through our information systems that may result in adverse effects on the confidentiality, integrity, or availability of our information systems or any information residing therein.
We conduct periodic risk assessments to identify cybersecurity threats, as well as assessments in the event of a material change in our business practices that may affect information systems that are vulnerable to such cybersecurity threats. These risk assessments include identification of reasonably foreseeable internal and external risks, the likelihood and potential damage that could result from such risks, and the sufficiency of existing policies, procedures, systems, and safeguards in place to manage such risks.
Following these risk assessments, we re-design, implement, and maintain reasonable safeguards to minimize identified risks; address any identified gaps in existing safeguards; and regularly monitor the effectiveness of our safeguards. Primary responsibility for assessing, monitoring and managing our cybersecurity risks is delegated to our Senior Director, Head of IT and Informatics, who reports on IT operations, risk mitigation and assessment efforts, and other general cybersecurity matters to our Chief Financial Officer, to manage the risk assessment and mitigation process.
The cybersecurity risk management program includes tools and activities to prevent, detect, and analyze current and emerging cybersecurity threats, and plans and strategies to address threats and incidents. As part of our overall risk management system, we monitor and test our safeguards and train our employees on these safeguards, in collaboration with our internal IT function and management.
We engage consultants or other third parties in connection with our risk assessment processes. These service providers assist us in designing and implementing our cybersecurity policies and procedures, and monitoring and testing our safeguards. We require each third-party service provider to certify that it has the ability to implement and maintain appropriate security measures, consistent with all applicable laws, to implement and maintain reasonable security measures in connection with their work with us, and to promptly report any suspected breach of its security measures that may affect our Company.Our Chief Financial Officer and Senior Director, Head of IT and Informatics provide periodic briefings to the Audit Committee regarding our Company’s cybersecurity risks and activities, including any recent cybersecurity incidents and related responses, cybersecurity systems testing, activities of third parties, and related matters. The Audit Committee provides regular updates to the full Board of Directors on such reports.
We have not encountered cybersecurity challenges that have materially impaired our operations or financial standing. For additional information regarding risks from cybersecurity threats, please refer to Item 1A, “Risk Factors,” in this Annual Report on Form 10-K.
Item 2. Properties
Our corporate headquarters are currently located at 65 Grove Street, Watertown, Massachusetts704 Quince Orchard Road, Gaithersburg, Maryland and consistconsists of 32,2947,909 total square feet of leased office, laboratory, and laboratorymanufacturing space under a lease that expires in January 2027. Additionally, we lease approximately 32,294 total square feet of office and laboratory space in Watertown, Massachusetts under a lease that expires in May 2028.
In February 2024, we entered into an agreement to lease 19,199 square feet consisting of integrated manufacturing and office space in Frederick, Maryland. The initial term is expected to commence no later than April 1, 2024 and terminate seven full lease years following, which is expected to be May 2031.
We also lease approximately 2,500 square feet of office and laboratory space in Moscow, Russia on a month-to-month basis.
Item 3. Legal Proceedings
On August 3, 2020,February 7, 2024, Justin Sloan, a purported stockholder of Selectaour Company, filed a stockholder derivativeputative class action purportedly on behalf of Selectahimself and similarly situated stockholders of our Company against certain currentus and former members of the Company’sour Board of Directors as well as one affiliated company owned by
a current board member, in the Court of Chancery of the State of Delaware, namely Franchititled Sloan v. Barabe, et al., No. 2024-0105. The complaint alleges that the individual defendants breached their fiduciary duties and committed corporate waste when they authorized a private placement transaction, announcedby failing to disclose purportedly material information to our stockholders in our Preliminary Proxy Statement filed on December 19, 2019, at a price allegedly below fair value. The complaint further alleges that the four defendant directors who participated in the private placement were unjustly enrichedJanuary 31, 2024 in connection with the transaction. On September 25, 2020, the defendants filed a motion to dismiss the lawsuit. On November 6, 2020, the plaintiff filed an amended complaint, and the defendants filed a second motion to dismiss on January 8, 2021. On December 31, 2020, we received a litigation demand letter from two other putative stockholders relating to the same private placement transaction. On April 12, 2021, the Courtsolicitation of Chancery in the State of Delaware granted a motion to stay the litigation pending a review by a Special Committee appointed by the Company’s Board of Directors. While the litigation was stayed, the parties reached an agreement in principle to settle the matter, and on March 18, 2022, they submitted a Stipulation and Agreement of Settlement and other documentation to the Court for itsstockholder approval of the settlement. On July 21, 2022,Conversion Proposal. The complaint seeks a temporary injunction against the stockholder vote on the Conversion Proposal, compensatory damages, pre- and post-judgment interest, and attorneys’ fees and costs. At a telephonic hearing on February 28, 2024, the Court helddenied the Plaintiff’s motion to expedite the proceedings, rejecting Plaintiff's argument that the lawsuit raised colorable disclosure claims warranting expedited treatment. Additional similar lawsuits may be filed. We believe this lawsuit is without merit and intend to vigorously defend against this plaintiff’s claims.
On February 21, 2024, Paul Wymer, a settlement hearing, at whichpurported stockholder of our Company, filed an action against us and members of our Board of Directors in the settlement was approved. On August 1, 2022,U.S. District Court for the Court enteredSouthern District of New York, titled Wymer v. Cartesian Therapeutics, Inc., et al., No. 24-cv-01288. The complaint alleges that the defendants violated Sections 14(a) and 20(a) of the Exchange Act by failing to disclose purportedly material information to our stockholders in our Preliminary and Definitive Proxy Statements filed on January 31, 2024, and February 14, 2024, respectively, in connection with the solicitation of stockholder approval of the Conversion Proposal. The complaint seeks injunctive relief enjoining or rescinding the Merger, issuance of an Orderamended proxy statement, and Final Judgment which dismissed the action,attorneys' fees and all claims contained therein, with prejudice.costs. Additional similar lawsuits may be filed. We believe this lawsuit is without merit and intend to vigorously defend against this plaintiff’s claims.
Item 4. Mine Safety Disclosures
Not applicable.
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Market Information
Our common stock is publicly traded on The Nasdaq Stock Market under the symbol “SELB.“RNAC.”
Holders
As of February 24, 2023,March 1, 2024, there were approximately 153,318,915161,948,618 shares of our common stock outstanding held by approximately 4789 holders of record. The actual number of stockholders is greater than this number of record holders, and includes stockholders who are beneficial owners, but whose shares are held in street name by brokers and other nominees.
Dividends
We have never declared or paid any cash dividends on our capital stock. We intend to retain future earnings, if any, to finance the operation and expansion of our business and do not expect to pay any cash dividends in the foreseeable future. Any future determination related to our dividend policy will be made at the discretion of our boardBoard of directorsDirectors after considering our financial condition, results of operations, capital requirements, business prospects and other factors the boardBoard of directorsDirectors deems relevant, and subject to the restrictions contained in any future financing instruments. In addition, our loan and security agreement with Oxford and SVB currently prohibits us from paying dividends on our equity securities, and any future debt agreements may likewise preclude us from paying dividends. See “Management’s Discussion and Analysis of Financial Condition and Results of Operations⸻Liquidity and Capital Resources.”
Stock Performance Graph
The graph set forth below compares the cumulative total stockholder return on our common stock between December 31, 20172018 and December 31, 2022,2023, with the cumulative total return of (a) the Nasdaq Composite Index and (b) the Nasdaq Biotechnology Index, over the same period. This graph assumes the investment of $100 at the market close on December 31, 20172018 in our common stock, the Nasdaq Composite Index and the Nasdaq Biotechnology Index and assumes the reinvestment of dividends, if any. The stock price performance of the following graph is not necessarily indicative of future stock price performance.
Comparison Of Cumulative Total Return Selecta Biosciences,Cartesian Therapeutics, Inc.,
NASDAQ COMPOSITE INDEX AND NASDAQ BIOTECHNOLOGY INDEXNasdaq Composite Index and Nasdaq Biotechnology Index
This performance graph shall not be deemed “soliciting material” or to be “filed” with the SEC for purposes of Section 18 of the Exchange Act, or otherwise subject to the liabilities under that Section, and shall not be deemed to be incorporated by reference into any of our filings under the Securities or the Exchange Act.
Securities Authorized for Issuance Under Equity Compensation Plans
Information about our equity compensation plans in Item 12 of Part III of this Annual Report is incorporated herein by reference. Any future determination to pay dividends will be made at the discretion of our Board of Directors and will depend
on various factors, including applicable laws, our results of operations, financial condition, future prospects, then applicable contractual restrictions and any other factors deemed relevant by our Board of Directors. Investors should not purchase our common stock with the expectation of receiving cash dividends.
Purchases of Equity Securities by the Issuer or Affiliated Purchasers
We did not repurchase any of our equity securities during the quarter ended December 31, 2022.2023.
Recent Sales of Unregistered Securities and Use of Proceeds from Registered Securities
None.
Item 6. [Reserved]
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
The following discussion and analysis of our financial condition and results of operations should be read in conjunction with our consolidated financial statements and related notes appearing elsewhere in this Annual Report on Form 10-K.Report. This discussion and other parts of this Annual Report on Form 10-K contain forward-looking statements that involve risks and uncertainties, such as statements regarding our plans, objectives, expectations, intentions and projections. Our actual results could differ materially from those discussed in these forward-looking statements. Important factors that could cause or contribute to such differences include, but are not limited to, those discussed in Item 1A. “Risk Factors.” A discussion of the year ended December 31, 20212022 compared to the year ended December 31, 20202021 has been reported previously in our Annual Report on Form 10-K for the year ended December 31, 2021,2022, filed with the SEC on March 10, 2022,2, 2023, under the heading “Management’s Discussion and Analysis of Financial Condition and Results of Operations.”
Overview
We are a clinical-stage biotechnology company leveraging our ImmTOR® platform to develop tolerogenicdeveloping mRNA cell therapies designed to selectively mitigate unwanted immune responses. With a proven ability to induce tolerance to highly immunogenic proteins, ImmTOR has the potential to amplify the efficacy of biologic therapies, including redosing of life-saving gene therapies, as well as restore the body's natural self-tolerance in autoimmune diseases. We have several proprietary and partnered programs in our pipeline focused on enzyme therapies, gene therapies, and autoimmune diseases.
In preclinical studies, we have observed that ImmTOR may have synergistic activity with interleukin-2, or IL-2, molecules that have been engineered to be selective for regulatory T cells, or Tregs. Treg-selective IL-2 molecules have been shown to transiently expand all pre-existing Tregs in preclinical and clinical studies conducted by others. We have observed in preclinical studies that the combination of ImmTOR, a Treg-selective IL-2 molecule and an antigen exhibited substantial synergistic activity in inducing and expanding antigen-specific Tregs beyond ImmTOR alone with evidence of enhanced durability of immune tolerance and the potential for ImmTOR dose sparing. This combination of ImmTOR with a Treg selective IL-2 molecule represents an evolution of the ImmTOR platform, which we call ImmTOR-IL™. We believe this combination has the potential to be a "first-in-class" antigen specific IL-2 therapy for autoimmune disease.
We believe ImmTOR and ImmTOR-IL have the potential to enable novel therapeutic modalities in autoimmune disease as well as enhance both the efficacy and safety of biologic therapies (including gene therapies) and improve product candidates under development. In clinical trials, ImmTOR has been observed to inhibit the formation of neutralizing antibodies to adeno-associated virus (AAV) capsids, potentially enabling re-dosing of gene therapies. Additionally, based on preclinical data in AAV gene therapies, we believe that ImmTOR has the potential to improve efficacy and safety by increasing transgene expression, reducing hepatic inflammation and inhibiting undesired immune responses to both the AAV capsid and the transgene product that can occur with the first dose of gene therapy.
In biologic therapies, clinical activity of ImmTOR in humans has been observed with pegadricase, a highly immunogenic pegylated uricase enzyme being developed for the treatment of autoimmune diseases. We leverage our proprietary technology and manufacturing platform to introduce one or more mRNA molecules into cells to enhance their function. Unlike DNA, mRNA degrades naturally over time without integrating into the cell’s genetic material. Therefore, our mRNA cell therapies are distinguished by their capacity to be dosed repeatedly like conventional drugs, administered in an outpatient setting, and given without pre-treatment chemotherapy required with many conventional cell therapies. In an open-label Phase 2 clinical trial in patients with MG, a chronic refractory gout. The combination of ImmTORautoimmune disease that causes disabling muscle weakness and pegadricase is currently being evaluated infatigue, we observed that our lead product candidate, Descartes-08, generated a Phase 3deep and durable clinical trial that webenefit.
We are conducting on behalf ofleveraging our partner Swedish Orphan Biovitrum AB, or Sobi.
We intendproprietary technology and manufacturing platform, RNA Armory®, to pursue development ofdevelop mRNA cell therapies for autoimmune diseases where expansion of either all Tregsacross three modalities. Our mRNA CAR-T modality is a personalized approach that collects a patient’s T-cells and uses mRNA to introduce a CAR into the cell. The CAR redirects the T-cells to target and destroy pathogenic self-reactive cells. Our mRNA MSC modality is an allogeneic approach that introduces one or antigen-specific Tregs has been shownmore mRNAs into donor-sourced MSCs, enabling them to or we believeproduce proteins that target key pathways involved in autoimmunity. These cells are banked and are designed to be administered off-the-shelf to any patient. Our mRNA in situ modality is likelydesigned to havedeliver mRNA into a beneficial effect. We believepatient’s lymph node to generate CAR-T cells and other proteins that ImmTORtarget autoimmunity.
Merger
On November 13, 2023, the Company and ImmTOR-IL haveOld Cartesian entered into the potentialMerger Agreement. In connection with the Merger and pursuant to unlock antigen-specific therapies for autoimmune diseases and that ImmTOR-IL can further improve the efficacy and safety profile of biologic therapies beyond ImmTOR alone.
Impact of Global Events
COVID-19
We continueMerger Agreement, the Company changed its corporate name to closely monitor how the COVID-19 pandemic is affecting our employees, business, preclinical studies and clinical trials. Disruptions caused by the COVID-19 pandemic may result in difficulties or delays in initiating, enrolling, conducting or completing our planned and ongoing clinical trials, and the incurrence of unforeseen costsCartesian Therapeutics, Inc., with Old Cartesian surviving as a result of supply chain, preclinical study, or clinical trial delays.
While the COVID-19 pandemic has not had a material impact on our clinical programs aswholly owned subsidiary of the dateCompany, as summarized in Note 3 of the accompanying notes to the consolidated financial statements appearing elsewhere in this Annual Report, it could have an impact on our ability to commence preclinical studies and clinical trials of our IgA nephropathy, gene therapy, and autoimmune disease programs, and our ability to obtain supply of both active drug substances and finished drug product as well as efficient execution of the overall supply chain for SEL-212 and our other programs.
At this time, any impact of COVID-19 on our business, revenues, results of operations and financial condition will largely depend on future developments, which are highly uncertain and cannot be predicted with confidence, such as the duration of the
pandemic, the emergence of new virus variants, travel restrictions and social distancing in the United States and other countries, business closures, disruptions, mandated stay at home orders or lockdowns, supply chain disruptions, the ultimate impact on financial markets and the global economy, and the effectiveness of actions taken in the United States and other countries to contain and treat the disease.
Report.
Financial Operations
To date, we have financed our operations primarily through public offerings and private placements of our securities, funding received from research grants, collaboration and license arrangements and oura credit facility. We do not have any products approved for sale and have not generated any product sales.
Except for the year ended December 31, 2022, we have incurred significant operating losses since our inception. We had net income of $35.4 million and incurred a net loss of $25.7$219.7 million and had net income of $35.4 million for the years ended December 31, 20222023 and 2021,2022, respectively. As of December 31, 2022,2023, we had an accumulated deficit of $394.9$614.6 million. We expect to continue to incur significant expenses and operating losses for at least the next several yearsforeseeable future as we:
•advance Descartes-08 for MG into Phase 3 development;
•continue the researchto develop our preclinical and development of our otherclinical-stage product candidates as well as product candidates that we may be developing jointly with collaboration partners;
•seek to enhance our ImmTOR platform and discover and develop additional product candidates;
•seek to enter into collaboration, licensing and other agreements, including, but not limited to research and development, and/or commercialization agreements;
•seek regulatory approvals for any product candidates that successfully complete clinical trials;
•potentially establish a sales, marketing and distribution infrastructure and scales-up external manufacturing capabilities to commercialize any products for which we may obtain regulatory approval;
•maintain, expand and protect our intellectual property portfolio, including through licensing arrangements; and
•add clinical, scientific, operational, financial and management information systems and personnel, including personnel to support our product development and potential future commercialization efforts and to support our operations as a public company.arrangements.
Until we can generate substantial product revenues, we expect to finance our cash needs through a combination of equity offerings, debt financings, and license and collaboration agreements. We may be unable to raise capital when needed or on reasonable terms, if at all, which would force us to delay, limit, reduce or terminate our product development or future commercialization efforts. We will need to generate significant revenues to achieve profitability, and we may never do so.
Concurrently with the closing of the Merger, we entered into a securities purchase agreement in which we agreed to issue 149,330.115 shares of Series A Preferred Stock, in exchange for aggregate gross proceeds of $60.25 million, or the November 2023 Private Placement. We granted customary registration rights to investors in connection with the November 2023 Private Placement.
We believe that our existing cash, cash equivalents, and restricted cash and marketable securities as of December 31, 20222023, combined with net proceeds from the November 2023 Private Placement received subsequent to December 31, 2023 will enable us to fund our operating expenses and capital expenditure requirements into mid-2024.mid-2026. We have based this estimate on assumptions that may prove to be wrong, and we could use our capital resources sooner than we currently expect.
We intend to seek collaboration partners for the assets in the development programs that we are no longer actively advancing.
The consolidated financial information presented below includes the accounts of Selecta Biosciences,Cartesian Therapeutics, Inc. and our wholly owned subsidiaries, Selecta (RUS) LLC, a Russian limited liability company, or Selecta (RUS), and Selecta Biosciences Security Corporation, a Massachusetts securities corporation.corporation, and Cartesian Bio, LLC, a Delaware limited liability company, which is a variable interest entity for which we are the primary beneficiary. All intercompany accounts and transactions have been eliminated.
Collaboration and license revenue
To date, we have not generated any revenue from product sales. Our revenue consists primarily of collaboration and license revenue, which includes amounts recognized related to upfront and milestone payments for research and development funding under collaboration and license agreements. We expect that any revenue we generate will fluctuate from quarter to quarter because of the timing and amounts of fees, research and development reimbursements and other payments from collaborators. We do not expect to generate revenue from product sales for at least the next several years. If we or our collaborators fail to complete the development of our product candidates in a timely manner or fail to obtain regulatory approval as needed, our ability to generate future revenue will be harmed, and will affect the results of our operations and financial position. For further description of the agreements underlying our collaboration and license revenue, see Notes 2 and 1214 to our consolidated financial statements included elsewhere in this Annual Report.
Research and development
Our research and development expenses consist of external research and development costs, which we track on a program-by-program basis and primarily include CMO-relatedcontract manufacturing organization related costs and fees paid to CROs,contract research organizations, and internal research and development costs, which are primarily compensation expenses for our research and development employees, lab supplies, analytical testing, allocated overhead costs and other related expenses. Our internal research and development costs are often devoted to expanding our programs and are not necessarily allocable to a specific target.
We have incurred a total of $436.4 million in research and development expenses from inception through December 31, 2022, with a majority of the expenses being spent on the development of SEL-212 and the remainder being spent on our various discovery and preclinical stage product candidate programs and the general expansion of our technology platform.
We expense research and development costs as incurred. Conducting a significant amount of research and development is central to our business model. Product candidates in clinical development generally have higher development costs than those in earlier stages of development, primarily due to the size, duration and cost of clinical trials. The successful development of our clinical and preclinical product candidates is highly uncertain. Clinical development timelines, the probability of success and development costs can differ materially from our expectations. For example, if the FDA or another regulatory authority were to require us to conduct clinical trials beyond those which we currently expect will be required for the completion of clinical development of a product candidate, or if we experience significant delays in enrollment in any of our clinical trials, we could be required to expend significant additional financial resources and time to complete any clinical development.
The following table sets forth the components of our research and development expenses during the periods indicated (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | Year Ended December 31, | | |
| | | | | | 2022 | | 2021 | | 2020 | | |
| SEL-212 | | | | | $ | 26,801 | | | $ | 31,446 | | | $ | 32,288 | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Other research and development expenses | | | | | 45,576 | | | 37,290 | | | 22,217 | | | | | |
| Total research and development expenses | | | | | $ | 72,377 | | | $ | 68,736 | | | $ | 54,505 | | | | | |
Note: Certain prior period expenses have been reclassified to conform to current year presentation.
In June 2020, we and Swedish Orphan Biovitrum AB, or Sobi, entered into a License and Development Agreement, which was amended in October 2023, or, as so amended, the Sobi License. Pursuant to the Sobi License, clinical trial costs incurred to complete development of the product candidate SEL-212, including but not limited to costs incurred while conducting and completing the Phase 3 DISSOLVE trials will befor SEL-212, were reimbursed by Sobi. These costs, when reimbursed, will be were
recognized as revenue consistent with the revenue recognition methodology disclosed in Note 1214 to our consolidated financial statements included elsewhere in this Annual Report. The reimbursable costs exclude any costs of additional development activities required that arewere related to the ImmTOR platform and that arewere unrelated to SEL-212.
In January 2023, we and Audentes Therapeutics, Inc., or Astellas, entered into a License and Development Agreement, or the Astellas Agreement. Pursuant to the Astellas Agreement, Astellas agreed to reimburse us for 25% of all budgeted costs incurred to complete the development of Xork, a bacterial IgG protease licensed from Genovis AB (publ.), or Genovis, for use in Pompe disease with an Astellas gene therapy investigational or authorized product. These costs, when reimbursed, will be recognized as revenue consistent with the revenue recognition methodology disclosed in Note 14 to our consolidated financial statements included elsewhere in this Annual Report.
General and administrative
General and administrative expenses consist primarily of salaries and related benefits, including stock-based compensation, related to our executive, finance, business development and support functions. Other general and administrative expenses include facility-related costs not otherwise allocated to research and development expenses, travel expenses for our general and administrative personnel and professional fees for auditing, tax and corporate legal services, including intellectual property-related legal services.
Investment income
Investment income consists primarily of interest income earned on our cash, cash equivalents and marketable securities.
Interest expense
Interest expense consists of interest expense on amounts borrowed under our credit facilities.facilities and loss on extinguishment of debt.
Other income, net
Other income, net consists primarily of sublease income during the year ended December 31, 2022 and was de minimis during the years ended December 31, 2021 and December 31, 2020.income.
Change in fair value of warrant liabilities
Common warrants classified as liabilities are remeasured quarterly at fair value, utilizing a Black-Scholes valuation methodology, with the change in fair value recognized as a component of earnings.
Change in fair value of contingent value right liability
The CVR liability is remeasured quarterly at fair value, utilizing a discounted cash flow valuation methodology, with the change in fair value recognized as a component of earnings.
Change in fair value of Series A Preferred Stock forward contract liabilities
The forward contract liability associated with the delayed issuance of the Series A Preferred Stock related to the Merger and November 2023 Private Placement is remeasured quarterly at fair value, utilizing the market price of our common stock, with the change in fair value recognized as a component of earnings.
Foreign currency transaction gain (loss)
The functional currency of our Russian subsidiarySelecta (RUS) is the Russian ruble. In addition to holding cash denominated in Russian rubles, our Russian bank accounts also hold cash balances denominated in U.S. dollars to facilitate payments to be settled in U.S. dollars or other currencies. As of each of December 31, 20222023 and 2021,2022, we maintained cash of $0.2 million and $0.3 million, respectively, in Russian bank accounts in denominations of both Russian rubles and U.S. dollars. The amounts denominated in U.S. dollars and used in transacting the day-to-day operations of our Russian subsidiary are subject to transaction gains and losses, which are reported as incurred.
Results of Operations
Comparison of the Years Ended December 31, 20222023 and 20212022
Collaboration and license revenue
The following is a comparisonDuring the year ended December 31, 2023, we recognized $26.0 million of collaboration and license revenue, compared to $110.8 million for the yearsyear ended December 31, 2022, and 2021 (in thousands, except percentages):a decrease of $84.8 million, or 77%. The decrease was primarily due to a
| | | | | | | | | | | | | | | | | | | | | | | |
| | Year Ended December 31, | | Increase |
| | 2022 | | 2021 | | (decrease) |
| | | | | |
| Collaboration and license revenue | $ | 110,777 | | | $ | 85,077 | | | $ | 25,700 | | | 30 | % |
| | | | | | | |
During the years ended December 31, 2022 and 2021, wedecrease of revenue recognized $82.6 million and $83.5 million, respectively, under the license agreement with Sobi License resulting from both the shipment of clinical supply and the reimbursement of costs incurred for the Phase 3 DISSOLVE clinical program. Additionally, during the year ended December 31, 2022, weprogram partially offset by an increase for revenue recognized $10.2 million under the Sarepta agreement, $9.2 million upon the mutual termination of the Spark License Agreement (as defined below), $7.0 million upon the mutual termination of the AskBio License Agreement (as defined below), and $1.8 million under the TakedaAstellas Agreement.
Research and development expenses
The following is a comparison of research and development expenses for the years ended December 31, 2022 and 2021 (in thousands, except percentages):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | Year ended December 31, | | | | Increase |
| | | | | | 2022 | | 2021 | | | | (decrease) |
| SEL-212 | | | | | $ | 26,801 | | | $ | 31,446 | | | | | $ | (4,645) | | | (15) | % |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Other research and development expenses | | | | | 45,576 | | | 37,290 | | | | | 8,286 | | | 22 | % |
| Total research and development expenses | | | | | $ | 72,377 | | | $ | 68,736 | | | | | $ | 3,641 | | | 5 | % |
Note: Certain prior period expenses have been reclassified to conform to current year presentation.
DuringFor the year ended December 31, 20222023, our research and development expenses increased by $3.6were $71.8 million, compared to $72.4 million for the year ended December 31, 2022, a decrease of $0.6 million, or 5%, as com1%. pared to 2021. The increasedecrease in cost was primarily the result of reductions in expenses incurred for preclinical and clinical programs due to the preclinical programs, increased personnelstrategic reprioritization partially offset by expenses andincurred for stock compensation expense.and personnel expenses.
General and administrative expenses
The following is a comparison of general and administrative expenses for the years ended December 31, 2022 and 2021 (in thousands, except percentages):
| | | | | | | | | | | | | | | | | | | | | | | |
| | Year Ended December 31, | | Increase |
| | 2022 | | 2021 | | (decrease) |
| | | | | |
| General and administrative | $ | 23,862 | | | $ | 20,938 | | | $ | 2,924 | | | 14 | % |
During the year ended December 31, 2022,2023, our general and administrative expenses increased by $2.9were $40.6 million, compared to $23.9 million for the year ended December 31, 2022, an increase of $16.7 million, or 14%, as compared to 2021.70%. The increase in costs was primarily the result of expenses incurred for stock compensation, personnel expenses, and personnel expenses.professional fees incurred in connection with the Merger.
Investment income
Investment income for the year ended December 31, 2023 was $5.0 million, compared to $2.1 million and de minimis for the yearsyear ended December 31, 2022, and 2021, respectively.an increase of $2.9 million, or 138%. The increase in investment income was due to increased investment balances and higher interest rates.
Foreign currency transaction gain (loss)
We recognized de minimis foreign currency translation adjustments during each of the years ended December 31, 20222023 and 2021.2022.
Interest expense
Interest expense for the year ended December 31, 2023 was $3.0$2.8 million and $2.8compared to $3.0 million for the yearsyear ended December 31, 2022, and 2021, respectively, representinga decrease of $0.2 million, or 7%. Interest expense for the year ended December 31, 2023 comprised interest expense and amortization of the carrying costs of our credit facilities.facilities and loss on extinguishment of debt.
Change in fair value of warrant liabilities
For the year ended December 31, 2022,2023, we recognized $20.9$12.7 million of income from the decrease in the fair value of warrant liabilities, compared to $20.9 million for the year ended December 31, 2022, a decrease of $8.2 million or 39.2%. Fair value of warrant liabilities was determined utilizing the Black-Scholes valuation methodology. The decrease in warrant value was primarily driven by a decrease in the Company’s share price. per-share price of our common stock.
Change in fair value of contingent value right liability
For the year ended December 31, 2021,2023 we recognized $2.3$18.3 million of loss fromexpense associated with the increase in the fair value of warrant liabilitiesCVR liability. The fair value of the CVR liability was determined utilizing a discounted cash flow valuation methodology. The increase in CVR value was primarily driven by a slight increase in the Company's share price, offset by the decrease in outstanding warrants dueinterest rates from the Merger to exercise.December 31, 2023 and the corresponding impact on the discount rate used in our discounted cash flow valuation. There was no CVR liability prior to the Merger and as such no CVR liability is reflected in our consolidated financial statements as of or for any period prior to the year ended December 31, 2023.
Change in fair value of Series A Preferred Stock forward contract liabilities
For the year ended December 31, 2023 we recognized $149.6 million of expense associated with the increase in the fair value of Series A Preferred Stock forward contract liabilities. The increase in Series A Preferred Stock value was primarily driven by an increase in the per-share price of our common stock since the date of the Merger and November 2023 Private Placement. A portion of the Series A Preferred Stock forward contract liability was settled during the year ended December 31, 2023 and there was no such forward contract liability prior to the Merger. There was no Series A Preferred Stock forward contract liability prior to the Merger and as such no Series A Preferred Stock forward contract liability is reflected in our consolidated financial statements as of or for any period prior to the year ended December 31, 2023.
Other income, net
OtherDuring the year ended December 31, 2023, we recognized other income, net wasof $0.7 million, compared to $0.3 million and de minimis for the yearsyear ended December 31, 2022, and 2021, respectively.an increase of $0.4 million, or 133%. The increase was primarily driven by sublease income.
Income taxes
During the year ended December 31, 2023, we recognized a current tax benefit of $19.0 million relating to the benefit of legacy Selecta tax attributes that reduced deferred tax liabilities during the year. For the year ended December 31, 2022, we recognized a $0.6 million benefit for penalty abatements received. For
Net (loss) income
Net loss for the year ended December 31, 2021, we recognized $16.02023 was $219.7 million as compared to net income of expense for the income taxes primarily related to the license agreement with Sobi upon the Company’s election to opt out of the installment sale method of taxation. For income tax purposes, the transfer of trademark and product rights is treated as a sale and the net proceeds from the sale are taxed under the default installment method as cash is received by the Company. During the year ended December 31, 2021, the Company completed an analysis of future tax obligations under the default installment sale method versus making a timely filed election on its 2020 tax return due October 15, 2021 to elect out of the installment sale method for income tax purposes. As a result, the Company elected out of the default installment sale treatment with the filing of its 2020 tax return. In the elect out method, the Company was initially taxed based upon the estimated fair value of all present and future proceeds from the sale and the Company utilized all of its available net operating losses and income tax credits, which served to reduce the federal and state tax liability. As the Company recognizes future cash proceeds under the Sobi license, the Company will exclude such amounts from taxable income up to the originally estimated fair value that was previously taxed.
Net income (loss)
Net income$35.4 million for the year ended December 31, 2022, increased to $35.4a decrease of $255.1 million, as compared to a net loss of $25.7 million in 2021or 721%. The change was primarily due to increaseddecreased collaboration and license revenue increase in the benefit fromand expenses associated with the change in fair value of warrant liabilities,the CVR liability and Series A Preferred Stock forward contract liability, and increased general and administrative expenses, partially offset by an increase in income tax benefit, partially offset by increased research and development expenses.
Liquidity and Capital Resources
Except for net income of $35.4 million for the year ended December 31, 2022, we have incurred recurring net losses since our inception. We expect that we will continue to incur losses and that such losses will increase for the foreseeable future. We expect that our research and development and general and administrative expenses will continue to increase and, as a result, we will need additional capital to fund our operations, which we may raise through a combination of equity offerings, debt financings, third-party funding, potential royalty and/or milestone monetization transactions and other collaborations and strategic alliances.
From our inception through December 31, 2022, we have raised an aggregateOn a pro forma basis, giving effect to the receipt of $723.3$40.0 million to fund our operations, which includes $118.5 million from the sale of preferred stock, $11.1 million in government grant funding, $36.7 million from borrowings under our credit facilities past and present, $250.2 million from our collaborations and license agreements, $64.5 million in combined net proceeds from our initial public offering, $185.2 million in combined net proceeds from private placements and follow-on offerings of our common stock, and, throughNovember 2023 Private Placement received subsequent to December 31, 2022, $57.1 million in aggregate net proceeds from “at-the-market” offerings of our common stock.
As of December 31, 2022,2023, our cash, cash equivalents, and restricted cash and marketable securities were $136.2$118.3 million as of December 31, 2023, of which $1.6$1.4 million was restricted cash related to lease commitments and $0.2 million was held by our Russian subsidiary designated solely for use in its operations.
In addition to our existing cash equivalents, we from time to time have received and may receive in the future research and development funding pursuant to our collaboration and license agreements. Currently, funding from payments under our collaboration agreements represent our only source of committed external funds.
Liability associated with the contingent value rights agreement, or CVR Agreement, entered into on December 6, 2023, will be settled solely through cash flow received under the Sobi License and any other Gross Proceeds (as such term is defined in the CVR Agreement) net of certain agreed deductions. Under the CVR Agreement, 100% of all milestone payments, royalties, and other amounts paid to us or our controlled entities under the Sobi License, and any other Gross Proceeds, in each case net of certain agreed deductions, will be distributed to holders of the CVRs. There is no contractual obligation for us to fund any amount related to the CVR liability.
Collaboration and License Agreements
In-licenses
In September 2023, we entered into the Biogen Agreement, a non-exclusive, sublicensable, worldwide, perpetual patent license agreement with Biogen, to research, develop, make, use, offer, sell and import products or processes containing or using an engineering T-cell modified with an mRNA comprising, or encoding a protein comprising, certain sequences licensed under the Biogen Agreement for the prevention, treatment, palliation and management of autoimmune diseases and disorders, excluding cancers, neoplastic disorders, and paraneoplastic disorders. We are not obligated to pay Biogen any expenses, fees, or royalties. For further description of the Biogen Agreement, see Note 16 to our consolidated financial statements included elsewhere in this Annual Report.
Effective September 2019, we entered into the NCI Agreement, a nonexclusive, worldwide license agreement with NCI. Under the NCI Agreement, we were granted a license under certain NCI patents and patent applications designated in the agreement, to make, use, sell, offer and import products and processes within the scope of the patents and applications licensed under the NCI Agreement when developing and manufacturing anti-BCMA CAR-T cell products for the treatment of MG, pemphigus vulgaris, and immune thrombocytopenic purpura according to methods designated in the NCI Agreement. In connection with our entry into the NCI Agreement, we paid to NCI a one-time $0.1 million license royalty payment. Under the NCI Agreement, we are further required to pay NCI a low five-digit annual royalty. We must also pay earned royalties on net sales in a low single-digit percentage and pay up to $0.8 million in benchmark royalties upon our achievement of designated benchmarks that are based on the commercial development plan agreed between the parties. For further description of the NCI Agreement, see Note 16 to our consolidated financial statements included elsewhere in this Annual Report.
In October 2021, we and Ginkgo Bioworks Holdings, Inc., or Ginkgo, entered into a Collaboration and License Agreement, or the First Ginkgo Agreement, and paid Ginkgo a $0.5 million one-time upfront payment. In June 2022, we paid $0.5 million and issued 892,857 shares of our common stock then-valued at $1.0 million to Ginkgo for the achievement of certain preclinical milestones under the First Ginkgo Agreement. In January 2022, we entered into a Collaboration and License Agreement, or the Second Ginkgo Agreement, and paid Ginkgo a $1.5 million one-time upfront payment. In July 2023, we paid $1.0 million and issued 1,339,285 shares of our common stock then-valued at $1.5 million to Ginkgo for the achievement of certain preclinical milestones under the Second Ginkgo Agreement. For further description of the First Ginkgo Agreement and the Second Ginkgo Agreement, see Note 16 to our consolidated financial statements included elsewhere in this Annual Report.
Additionally, in October 2021, we entered into an Exclusive License Agreement with Genovis, or the Genovis Agreement, and paid Genovis a $4.0 million one-time upfront payment. In February 2023, as a result of the sublicense of Xork to Astellas, we made a $4.0 million payment to Genovis.
the Genovis Agreement, see Note 16 to our consolidated financial statements included elsewhere in this Annual Report. On September 7, 2021, we entered into a Collaboration and License Agreement, or the Cyrus Agreement, with Cyrus Biotechnology, Inc., or Cyrus, and purchased 2,326,934 shares of Cyrus’ Series B Preferred Stock, par value $0.0001 per share at a purchase price of $0.8595 per share for an aggregate purchase price of $2.0 million. In October 2023, we notified Cyrus of our termination of the Cyrus Agreement, effective December 29, 2023. For further description of the Cyrus Agreement, see Note 16 to our consolidated financial statements included elsewhere in this Annual Report.
Out-licenses
In January 2023, we entered into the Astellas Agreement with Astellas. Under this agreement, Astellas obtained the sole and exclusive right to commercialize Xork for use in Pompe disease in combination with an Astellas gene therapy investigational or authorized product, with a current focus on AT845. In connection with entry into this agreement, we received a $10 million upfront payment and are eligible to receive $340.0 million for certain additional development and commercial milestones plus royalties on any potential commercial sales where Xork is used as a pre-treatment for AT845. As a result of the sublicense of Xork to Astellas, we made a $4.0 million payment to Genovis in February 2023. For further description of the Astellas Agreement, see Note 14 to our consolidated financial statements included elsewhere in this Annual Report.
On October 1, 2021, we entered into a License Agreement, or the Takeda Agreement.Agreement, with Takeda Pharmaceuticals USA, Inc. We received a $3.0 million upfront payment and arewere entitled to receive up to $1.124 billion in future additional payments over the course of the partnership that arewere contingent on the achievement of development or commercial milestones or Takeda’s election to continue its activities at specified development stages. The Takeda Agreement was terminated effective July 25, 2023. For further description of the Takeda Agreement, see Note 14 to our consolidated financial statements included elsewhere in this Annual Report.
In June 2020, we entered into the Sobi License. Sobi paid us a one-time, upfront payment of $75 million, and upon the closing of thea private placement of our common stock to Sobi Private Placement,at a price of $4.6156 per share, we received an additional $25 million from Sobi in consideration for Sobi’s purchase of our common stock at $4.6156 per share.Sobi. We are eligible to receive $630 million in milestone payments upon the achievement of various development and regulatory milestones and sales thresholds for annual net sales of SEL-212, and tiered royalty payments ranging from the low double digits on the lowest sales tier to the high teens on the highest sales tier. Sobi has agreed to fund the Phase 3 clinical program of SEL-212, which commenced in September 2020. In July 2022, we received $10.0 million for the completion of the enrollment of the DISSOLVE II trial. Proceeds from milestone payments and royalties on sales of SEL-212, if any, are required to be distributed, net of certain agreed deductions, to holders of the CVRs. For further description of the Sobi License, see Note 14 to our consolidated financial statements included elsewhere in this Annual Report.
Additionally, in June 2020, we and Sarepta Therapeutics, Inc., or Sarepta, entered into a Research License and Option Agreement, or the Sarepta Agreement. Sarepta paid us a $2.0 million upfront payment upon closing and a $3.0 million for the achievement of certain pre-clinical milestones in June 2021. In August 2022, we received a payment of $2.0 million in exchange for a nine-month extension to Sarepta's options to both Duchenne muscular dystrophy and certain limb-girdle muscular dystrophies and a payment of $4.0 million for the achievement of certain non-clinical milestones. In March 2023, we were notified by Sarepta that Sarepta would not be exercising its exclusive option under the Sarepta Agreement. The Sarepta Agreement terminated upon the expiration of the option in March 2023. For further description of the Sarepta Agreement, see Note 14 to our consolidated financial statements included elsewhere in this Annual Report.
In December 2019, we and Asklepios BioPharmaceutical, Inc., or AskBio, entered into a license agreement, with AskBio, or the AskBio License Agreement. Pursuant to the AskBio License Agreement, AskBio haspreviously exercised its option to exclusively license intellectual property rights covering ImmTOR to research, develop, and commercialize certain adeno-associated virus, or AAV, gene therapy products utilizing ImmTOR, and targeting the GAA gene, or derivatives thereof, to treat Pompe Disease. We received $7.0 million of upfront fees pursuant to the AskBio License Agreement. In November 2022, the AskBio License Agreement was mutually terminated. For further description of the AskBio License Agreement, see Note 14 to our consolidated financial statements included elsewhere in this Annual Report.
Financings
On August 6, 2020, we filed a universal shelf registration statement on Form S-3 (File No. 333-241692) with the Securities and Exchange Commission, or the SEC, to sell an aggregate amount of up to $200.0 million of certain of its securities. The shelf registration statement was declared effective by the SEC on August 14, 2020. Concurrent with the filing, we entered into a sales agreement, or the 2020 Sales Agreement, with Jefferies LLC, as sales agent, pursuant to which we were permitted, from time to time, to issue and sell common stock with an aggregate value of up to $50.0 million in an “at-the-market” offering. On October 8, 2021, we delivered notice to Jefferies LLC that we were terminating the 2020 Sales Agreement, with effect as of October 19, 2021.
On October 25, 2021, we entered into a Sales Agreement, or the 2021 Sales Agreement, with Leerink Partners LLC, or Leerink Partners (and then known as SVB Leerink LLCLLC), to sell shares of our common stock, from time to time, through an “at the market” equity offering program under which SVB Leerink Partners will act as sales agent. The shares of common stock sold pursuant to the 2021 Sales Agreement, willif any, would be issued and sold pursuant to such shelfa registration statement on Form S-3 and related prospectus supplement, filed on October 25, 2021 with the Securities and Exchange Commission, or SEC, for remaining aggregate gross sales proceeds of up to $75.0$51.0 million.
During the year ended December 31, 2023, we sold no shares of our common stock pursuant to the 2021 Sales Agreement. During the year ended December 31, 2022, we sold 774,544 shares of our common stock pursuant to the 2021 Sales Agreement for aggregate net proceeds of $2.1 million, after deducting commissions and other transaction costs. During the year ended December 31, 2021, we sold 13,767,511 shares of our common stock pursuant to the 2021 and 2020 Sales Agreements, as applicable, for aggregate net proceeds of $51.9 million, after deducting commissions and other transaction costs.
On April 11, 2022, we sold an aggregate of 27,428,572 shares of our common stock at a purchase price of $1.41 per share and warrants to purchase an aggregate of 20,571,429 shares of common stock at a purchase price of $1.55 per share underlying each common warrant for net proceeds of $36.9 million, after deducting commissions and other transaction costs.
Indebtedness
On August 31, 2020,November 13, 2023, we entered into a securities purchase agreement with (i) Dr. Timothy A. Springer, a member of our Board of Directors; (ii) TAS Partners LLC, an affiliate of Dr. Springer, and (iii) Seven One Eight Three Four Irrevocable Trust, a trust associated with Dr. Murat Kalayoglu, a co-founder and the former chief executive officer of Old Cartesian, who joined our Board of Directors effective immediately after the effective time of the Merger, providing for the November 2023 Private Placement. In the November 2023 Private Placement, we issued and sold an aggregate of 149,330.115 shares of Series A Preferred Stock for an aggregate purchase price of $60.25 million, of which 50,189.789 shares of Series A Preferred Stock were issued and sold in the year ended December 31, 2023 for gross proceeds of $20.25 million, and 99,140.326 shares of Series A Preferred Stock were issued and sold subsequent to December 31, 2023 for gross proceeds of $40.0 million.
Indebtedness
We previously maintained a term loan of up to $35.0 million, or the 2020 Term Loan, consisting of term loans in an aggregate amount ofwhich $25.0 million or the Term A Loan, and term loanswas funded in an aggregate amount of $10.0 million, or the Term B Loan, governed byAugust 2020. In September 2023, we entered into a loan and security agreement among us andpayoff letter with Oxford Finance LLC or Oxford, as collateral agent and a lender, and Silicon Valley Bank, or SVB,a division of First-Citizens Bank & Trust Company (successor by purchase to the Federal Deposit Insurance Corporation as Receiver for SVBB (as successor to Silicon Valley Bank)), the lenders under the term loan, pursuant to which we paid all outstanding amounts under such term loan, together with accrued interest and a lender.prepayment penalty, resulting in the full extinguishment of such term loan. The Term A Loantotal payoff amount was funded in full on August 31, 2020, or the Funding Date. The second draw period expired on September 30, 2021 and the Term B Loan is no longer available to be drawn by the Company.
On March 21, 2022, we entered into a Second Amendment to Loan and Security Agreement, or the Second Amendment, which amended the Loan and Security Agreement. The Second Amendment extends the date on which amortization payments in respect$22.3 million, consisting of the 2020 Term Loan will commence to April 1, 2023. Thereafter, amortization payments will be paid monthly in equal installmentsremaining principal amount due of principal and interest to fully amortize$19.8 million, the outstanding principal overfinal payment fee of $2.3 million, the remaining term of the 2020 Term
Loan, subject to recalculation upon a change in the prime rate. The Second Amendment was determined to be a loan modification, and the $0.1 million fee was recorded as an addition to the debt discount on the effective date.
On September 20, 2022, we entered into a Third Amendment to the Loan and Security Agreement, or the Third Amendment, which amended the Loan and Security Agreement. The Third Amendment was entered into in connection with the expansion of our corporate headquarters to provide for an increaseprepayment penalty of $0.2 million, and less than $0.1 million of accrued interest.
If in the letter of credit for a total of $1.6 million which renews automatically each year.
The 2020 Term Loan is secured by a lien on substantially all of our assets, other than intellectual property, provided that such lien on substantially all assets includes any rights to payments and proceeds from the sale, licensing or disposition of intellectual property. We also granted Oxford a negative pledge with respect to our intellectual property.
The 2020 Term Loan contains customary covenants and representations, including but not limited to financial reporting obligations and limitations on dividends, indebtedness, collateral, investments, distributions, transfers, mergers or acquisitions, taxes, corporate changes, deposit accounts, and subsidiaries. The 2020 Term Loan also contains other customary provisions, such as expense reimbursement, non-disclosure obligations as well as indemnification rights.
The events of default under the 2020 Term Loan include, but are not limited to, our failure to make any payments of principal or interest under the 2020 Term Loan or other transaction documents, our breach or default in the performance of any covenant under the 2020 Term Loan or other transaction documents, the occurrence of a material adverse event, making a false or misleading representation or warranty in any material respect under the 2020 Term Loan, our insolvency or bankruptcy, any attachment or judgment on our assets of at least approximately $0.5 million, or the occurrence of any default under any of our agreements or obligations involving indebtedness in excess of approximately $0.5 million. If an event of default occurs, Oxford is entitled to take enforcement action, including acceleration of amounts due under the 2020 Term Loan. Iffuture we raise any additionalseek debt financing, the terms of such additional debt could further restrict our operating and financial flexibility.
For a further descriptionflexibility by imposing liens on our assets and covenants on the operation of the 2020 Term Loan, see Note 9 to our consolidated financial statements included elsewhere in this Annual Report.business.
Future funding requirements
As of the date of this Annual Report, we have not generated any revenue from product sales. We do not know when, or if, we will generate revenue from product sales. We will not generate significant revenue from product sales unless and until we obtain regulatory approval and commercialize one of our current or future product candidates. Our primary uses of capital are, and we expect will continue to be, compensation and related expenses, third-party clinical research and development services, laboratory and related supplies, clinical costs, legal and other regulatory expenses, milestone and royalty payments for in-licenses, and general overhead costs. We expect that we will continue to generate losses for the foreseeable future, and we expect the losses to increase as we continue the development of, and seek regulatory approvals for, our product candidates, and begin to commercialize any approved products. We are subject to risks in the development of our products, and we may encounter unforeseen expenses, difficulties, complications, delays and other unknown factors that may adversely affect our business. We expect that we will need substantial additional funding to support our continuing operations.
The Certificate of Designation contains a provision granting each holder of the Series A Preferred Stock the option to require us to redeem any or all of such holder’s then-outstanding shares of Series A Preferred Stock beginning on the date that is 18 months following the Closing; provided, however, that no holder will have the right to seek redemption of any shares of Series A Preferred Stock to the extent that such holder would otherwise be unable to convert such shares of Series A Preferred Stock due to the common stock beneficial ownership limitation applicable to such holder. The per-share redemption price would be the average closing trading price of the common stock for the ten preceding trading days ending on, and including, the trading day immediately prior to the date a notice of conversion is delivered to us. We could be required to use a significant
amount of our cash resources on hand to satisfy this redemption obligation, particularly if our stockholders do not ever approve the Conversion Proposal and no shares of Series A Preferred Stock are automatically converted into common stock, or generally if holders of Series A Preferred Stock exercise their redemption right with respect to a significant number of shares of Series A Preferred Stock or at a time when the trading price of our common stock is elevated. Further, in the event that we do not have sufficient cash on hand to satisfy our redemption obligations, we may need to raise additional capital to satisfy these potential obligations. Any redemption payments could materially limit the amount of cash we have available to fund our operations and the potential need to redeem shares of Series A Preferred Stock may limit the flexibility with which we seek to operate our business.
As of December 31, 2022,2023, we had an accumulated deficit of $394.9$614.6 million. We anticipate operating losses to continue for the foreseeable future due to, among other things, costs related to research, development of our product candidates, conducting preclinical studies and clinical trials, and our administrative organization. We will require substantial additional financing to fund our operations and to continue to execute our strategy, and we will pursue a range of options to secure additional capital.
We are continually evaluatingregularly evaluate various potential sources of additional funding such as strategic collaborations, license agreements, debt issuance, potential royalty and/or milestone monetization transactions and the issuance of equity instruments to fund our operations. If we raise additional funds through strategic collaborations and alliances, which may include existing collaboration partners, we may have to relinquish valuable rights to our technologies or product candidates, or grant licenses on terms that are not favorable to us. To the extent that we raise additional capital through the sale of equity instruments, the ownership interest of our existing shareholdersstockholders will be diluted, and other preferences may be necessary that adversely affect the rights of existing shareholders.stockholders.
We believe that our existing cash, cash equivalents, and restricted cash and marketable securities as of December 31, 20222023 combined with net proceeds from the November 2023 Private Placement received subsequent to December 31, 2023 will enable us to fund our current planned operations into mid-2024, though we may realize additional cash resources uponoperating expenses and capital expenditure requirements for at least the achievement of certain contingent collaboration milestones and wenext 12 months. We may pursue additional cash resources through public or private equity or debt financings, by establishing collaborations with other companies or through the monetization of potential royalty and/or milestone payments pursuant to our existing collaboration and license arrangements. Management’s expectations with respect to our ability to fund current and long-term planned operations are based on estimates that are subject to risks and uncertainties. If actual results are different from management’s estimates, we may need to seek additional strategic or financing opportunities sooner than would otherwise be expected. However, there is no guarantee that any collaboration milestones will be achieved or that any of these strategic or financing opportunities will be executed on favorable terms, and some could be dilutive to existing stockholders. If we are unable to obtain additional funding on a timely basis, we may be forced to
significantly curtail, delay, or discontinue one or more of our planned research or development programs or be unable to expand our operations, meet long-term obligations or otherwise capitalize on our commercialization of our product candidates.
Our future capital requirements will depend on many factors, including:
•the numbertiming for stockholder approval of product candidates that we pursue;
•our collaboration agreements remaining in effect, our entering into additional collaboration agreements and our ability to achieve milestones under these agreements;
•the cost of manufacturing clinical suppliesconversion of our product candidates;
•Series A Preferred Stock into shares of our headcount growthcommon stock and associated costs;any redemptions of Series A Preferred Stock for cash;
•the scope, progress, results and costs of our clinical trials, preclinical development, manufacturing, laboratory testing and clinical trials for logistics;
•the number of product candidates that we pursue and the speed with which we pursue development;
•our other product candidates;headcount growth and associated costs;
•the costs, timing and outcome of regulatory review of our product candidates;
•the costs and timing of future commercialization activities, including manufacturing, marketing, sales and distribution, for any of our product candidates for which we receive marketing approval;
•the revenue, if any, received from commercial sales of our product candidates for which we receive marketing approval;
•the costs and timing of preparing, filing and prosecuting patent applications, maintaining and enforcing our intellectual property rights and defending any intellectual property-related claims;
•the effect of competing technological and market developments; and
•the extent to which we acquire or invest in businesses, products and technologies, including entering into licensing or collaboration arrangements for product candidates.
Summary of Cash Flows
| | | | | | | | | | | | | | | | | | | | |
| | | Year Ended December 31, |
| (In thousands) | | | | 2022 | | 2021 | | 2020 |
| Cash (used in) and provided by: | | | | | | | | |
| Operating activities | | | | $ | (31,631) | | | $ | (60,382) | | | $ | 34,881 | |
| Investing activities | | | | (15,002) | | | (17,140) | | | (741) | |
| Financing activities | | | | 39,215 | | | 52,897 | | | 14,431 | |
| Effect of exchange rate changes on cash | | | | 20 | | | (3) | | | (58) | |
| Net change in cash, cash equivalents, and restricted cash | | | | $ | (7,398) | | | $ | (24,628) | | | $ | 48,513 | |
| | | | | | | | | | | | | | | | | | | | |
| | | Year Ended December 31, |
| (In thousands) | | | | 2023 | | 2022 | | 2021 |
| Cash (used in) and provided by: | | | | | | | | |
| Operating activities | | | | $ | (51,161) | | | $ | (31,631) | | | $ | (60,382) | |
| Investing activities | | | | 34,609 | | | (15,002) | | | (17,140) | |
| Financing activities | | | | (13,145) | | | 39,215 | | | 52,897 | |
| Effect of exchange rate changes on cash | | | | (53) | | | 20 | | | (3) | |
| Net change in cash, cash equivalents, and restricted cash | | | | $ | (29,750) | | | $ | (7,398) | | | $ | (24,628) | |
Operating activities
Net cash used in operating activities for the year ended December 31, 20222023 was $31.6$51.2 million compared to $60.4$31.6 million in the same period in 2021.2022. The decreaseincrease in net cash used in operating activities of $19.6 million was primarily due to $28.8$56.2 million of net income,loss, adjusted for non-cash items, and uses$5.0 million of cash of approximately $60.4 million forprovided by changes in operating assets and liabilities.liabilities, in each case during the year ended December 31, 2023.
Investing activities
Net cash provided by investing activities for the year ended December 31, 2023 was $34.6 million compared to net cash used in investing activities of $15.0 million in the same period in 2022, an increase of $49.6 million. The net cash provided by investing activities for the year ended December 31, 2023 was primarily proceeds from the maturities of marketable securities and cash assumed in the Merger offset by purchases of property and equipment. The net cash used in investing activities for the year ended December 31, 2022 was $15.0 million compared to net cash used in investing activities of $17.1 million in the same period in 2021. The net cash used in investing activities in 2022 was to purchase marketable securities and property and equipment, offset by proceeds from the maturities of marketable securities.
Financing activities
Net cash used in financing activities for the year ended December 31, 2023 was $13.1 million compared to net cash provided by financing activities of $39.2 million in the same period in 2022, a decrease of $52.3 million. The net cash used in investingfinancing activities in 2021for the year ended December 31, 2023 was primarily to purchase marketable securitiesthe result of repayments of principal on outstanding debt and to investsettlement of equity awards in Cyrus Biotechnology,the Merger partially offset by proceeds from the maturities of marketable securities.
Financing activities
NetNovember 2023 Private Placement. The net cash provided by financing activities for the year ended December 31, 2022 was $39.2 million compared to net cash provided by financing activities of $52.9 million in the same period in 2021. The net cash provided by financing activities in 2022 was primarily the result of net proceeds from issuance of common stock and warrants to purchase common warrantsstock and the issuance of common stock in the "at-the-market"“at-the-market” offerings. The net cash providedoffering contemplated by financing activities inthe 2021 was primarily the result of net proceeds from Sales Agreement.“at-the-market” offerings and from the exercise of stock options.
Research and development contract obligations
Under our license agreement with MIT, milestone payments are due upon the occurrence of certain events and royalty payments commence upon our commercialization of a product. As of December 31, 2022, contractual obligations were $0.4 million. We have assumed license payments are fully offset by royalty payments in 2028.
Recent Accounting Pronouncements
For a discussion of recently adopted or issued accounting pronouncements please see Note 2 to our consolidated financial statements included elsewhere in this Annual Report.
Off-Balance Sheet Arrangements
As of December 31, 2022,2023, we did not have any off-balance sheet arrangements as defined in the rules and regulations of the Securities and Exchange Commission.
Critical Accounting Policies and Use of Estimates
Our management’s discussion and analysis of our financial condition and results of operations is based on our consolidated financial statements, which have been prepared in accordance with U.S. GAAP. The preparation of these consolidated financial statements requires us to make estimates and judgements that affect the reported amounts of assets, liabilities, revenues and expenses, and disclosure of contingent assets and liabilities in our consolidated financial statements. Actual results may differ from these estimates under different assumptions or conditions and could have a material impact on our reported results. While our significant accounting policies are more fully described in the Notesnotes to our consolidated financial statements included elsewhere in this Annual Report, on Form 10-K, we believe the following accounting policies to be the most critical in understanding the judgments and estimates we use in preparing our consolidated financial statements:
Revenue Recognition
Revenue is recognized when a customer obtains control of promised goods or services, in an amount that reflects the consideration which the entity expects to receive in exchange for those goods or services. Pursuant to ASC Topic 606, Revenue from Contracts with Customers (ASC 606), a customer is a party that has contracted with an entity to obtain goods or services that are an output of the entity’s ordinary activities in exchange for consideration. To determine revenue recognition for arrangements that an entity determines are within the scope of ASC 606, we perform the following five steps: (i) identify the
contract(s) with a customer; (ii) identify the performance obligations in the contract, including whether they are distinct in the context of the contract; (iii) determine the transaction price, including the constraint on variable consideration; (iv) allocate the transaction price to the performance obligations in the contract; and (v) recognize revenue when (or as) we satisfy each performance obligation. We only apply the five-step model to contracts when it is probable that the entity will collect the consideration it is entitled to in exchange for the goods or services it transfers to the customer. At contract inception, once the contract is determined to be within the scope of ASC 606, we assess the goods or services promised within each contract and determine those that are performance obligations, and assess whether each promised good or service is distinct. If a promised good or service is not distinct, it is combined with other promised goods or services into a performance obligation. We then recognize as revenue the amount of the transaction price that is allocated to the respective performance obligation when (or as) the performance obligation is satisfied. For example, certain performance obligations associated with Swedish Orphan Biovitrum, or Sobi, Sarepta Therapeutics, Inc., or Sarepta, and Takeda Pharmaceuticals USA, Inc., or Takeda,the Astellas Agreement, (see Note 12)14) will be satisfied over time, and revenue will be recognized usingutilizing the output method, based on the proportion of actual deliveries to the total expected deliveries over the initial term.input method.
Collaboration and License Revenue: We currently generate revenue through collaboration and license agreements with strategic collaborators for the development and commercialization of product candidates. Collaboration and license agreements with customers are generally accounted for in accordance with ASC 606. We analyze collaboration arrangements by first assessing whether they are within the scope of ASC Topic 808, Collaborative Arrangements (ASC 808), and evaluate whether such arrangements involve joint operating activities performed by parties that are both active participants in the activities and exposed to significant risks and rewards that are dependent on the commercial success of such activities. Collaboration agreements with customers that are not within the scope of ASC 808 are accounted for in accordance with ASC 606. To the extent the collaboration agreement is within the scope of ASC 808, we also assess whether any aspects of the agreement are within the scope of other accounting literature (specifically ASC 606). If we conclude that some or all aspects of the agreement are distinct and represent a transaction with a customer, we account for those aspects of the arrangement within the scope of ASC 606. We recognize the shared costs incurred that are not within the scope of other accounting literature as a component of the related expense in the period incurred by analogy to ASC Topic 730, Research and Development (ASC 730), and record reimbursements from counterparties as an offset to the related research and development costs. In determining the appropriate amount of revenue to be recognized as we fulfill our obligations under the agreements in accordance with ASC 606, we perform the five steps above. As part of the accounting for the arrangement, we must develop assumptions that require judgment to determine the stand-alone selling price for each performance obligation identified in the contract. We use key assumptions to determine the stand-alone selling price, which may include market conditions, reimbursement rates for
personnel costs, development timelines and probabilities of regulatory success. The assumptions used to determine the stand-alone selling price and our satisfaction of performance obligations have a material effect on our collaboration and license revenue and may prove to be wrong.
The terms of our arrangements typically include one or more of the following: (i) upfront fees; (ii) milestone payments related to the achievement of development, regulatory, or commercial goals; (iii) royalties on net sales of licensed products; (iv) reimbursements or cost-sharing of research and development (R&D) expenses; and (v) profit/loss sharing arising from co-promotion arrangements.
Licenses of Intellectual Property: If the license to our intellectual property is determined to be distinct from the other promised goods and services identified in the arrangement, we recognize revenues from non-refundable, upfront fees allocated to the license when the license is transferred to the customer and the customer is able to use and benefit from the license. If not distinct, the license is combined with other promised goodgoods and services in the contract. For licenses that are combined with other promised goodgoods and services, we assess the nature of the combined performance obligation to determine whether the combined performance obligation is satisfied over time or at a point in time and, if over time, the appropriate method of measuring progress for purposes of recognizing revenue. We evaluate the measure of progress each reporting period and, if necessary, adjust the measure of performance and related revenue recognition. Optional licenses are evaluated to determine if they are issued at a discount, and therefore, represent material rights and should be accounted for as separate performance obligations.
Milestone Payments: At the inception of each arrangement that includes developmental and regulatory milestone payments, we evaluate whether the achievement of each milestone specifically relates to our efforts to satisfy a performance obligation or transfer a distinct good or service within a performance obligation. If the achievement of a milestone is considered a direct result of our efforts to satisfy a performance obligation or transfer a distinct good or service and the receipt of the payment is based upon the achievement of the milestone, the associated milestone value is allocated to that distinct good or service. If the milestone payment is not specifically related to our efforts to satisfy a performance obligation or transfer a distinct good or service, the amount is allocated to all performance obligations using the relative standalone selling price method. We also evaluate the milestone to determine whether they are considered probable of being reached and estimates the amount to be included in the transaction price using the most likely amount method. If it is probable that a significant revenue reversal would not occur, the associated milestone value is included in the transaction price to be allocated, otherwise, such amounts are constrained and excluded from the transaction price. At the end of each subsequent reporting period, we re-evaluate the probability of achievement of such development milestones and any related constraint, and if necessary, adjust our estimate of the transaction price. Any such adjustments to the transaction price are allocated to the performance obligations on the same
basis as at contract inception. Amounts allocated to a satisfied performance obligation shall be recognized as revenue, or as a reduction of revenue, in the period in which the transaction price changes.
Manufacturing Supply Services: Arrangements that include a promise for future supply of drug substance or drug product for either clinical development or commercial supply at the customer’s discretion are evaluated to determine if they are distinct and optional. For optional services that are distinct, we assess if they are priced at a discount, and therefore, provide a material right to the licensee to be accounted for as separate performance obligations.
Royalties: For arrangements that include sales-based royalties, including milestone payments based on the level of sales, and the license is deemed to be the predominant item to which the royalties relate, we will recognize revenue at the later of (i) when the related sales occur, or (ii) when the performance obligation to which some or all of the royalty has been allocated has been satisfied (or partially satisfied) in accordance with the royalty recognition constraint.
Clinical Trial Costs
Clinical trial expenses are a significant component of research and development expenses, and we outsource a significant portion of these costs to third parties. Third party clinical trial expenses include patient costs, clinical research organization costs and costs for data management. The accrual for site and patient costs includes inputs such as estimates of patient enrollment, patient cycles incurred, clinical site activations, and other pass-through costs. Payments for these activities are based on the terms of the individual arrangements, which may differ from the pattern of costs incurred, and are reflected on the consolidated balance sheets as a prepaid asset or accrued clinical trial cost. These third party agreements are generally cancellable, and related costs are recorded as research and development expenses as incurred. Non-refundable advance clinical payments for goods or services that will be used or rendered for future R&Dresearch and development activities are recorded as a prepaid asset and recognized as expense as the related goods are delivered or the related services are performed. We also record accruals for estimated ongoing clinical research and development costs. When evaluating the adequacy of the accrued liabilities, we analyze progress of the studies, including the phase or completion of events, invoices received and contracted costs. Significant judgments and estimates may be made in determining the accrued balances at the end of any reporting period. Actual results could differ from the estimates made by us materially affecting our results of operations. The historical clinical accrual estimates made by us have not been materially different from the actual costs.
Warrant Liabilities
In December 2019, we issued common warrants in connection with a securities purchase agreement between us and a group of institutional investors and certain members of our boardBoard of directors,Directors, or the 2019 Warrants. Pursuant to the terms of these common warrants,the 2019 Warrants, we could be required to settle the common warrants in cash in the event of certain acquisitions of us and, as a result, the 2019 Warrants are required to be measured at fair value and reported as a liability on the balance sheet.
In April 2022, we issued warrants in connection with an underwritten offering of shares of common stock and warrants to purchase shares of common stock, or the 2022 Warrants. Pursuant to the terms of the 2022 Warrants, the Companywe could be required to settle the 2022 Warrants in cash in the event of an acquisition of the Companywe are acquired under certain circumstances and, as a result, the 2022 Warrants are required to be measured at fair value and reported as a liability on the balance sheet.
We recorded the fair value of the 2019 Warrants and 2022 Warrants upon issuance using the Black-Scholes valuation model, and are required to revalue the common warrants at each reporting date with any changes in fair value recorded on our statement of operations. In December 2022, we amended the terms of the outstanding 2019 Warrants held by certain members of our boardBoard of directorsDirectors to remove the cash settlement provision (the(as so amended, the Amended 2019 Warrants). As a result, the Amended 2019 Warrants were remeasured at fair value on December 20, 2022 and reclassified from a liability to equity on the balance sheet.
Inputs used to determine estimated fair value of the common warrant liabilities include the estimated fair value of the underlying stock at the valuation date, the estimated term of the warrants, risk-free interest rates, expected dividends and the expected volatility of the underlying stock. The estimates used to determine the fair value of these common warrants represent our best estimates, but may prove to be wrong. Therefore, the change in fair value of warrant liabilities could be materially different in the future.
Contingent Value Right Liability
The CVRs distributed pursuant to the terms of the CVR Agreement represent financial instruments that are accounted for under the fair value option election in ASC 825, Financial Instruments, or ASC 825. Under the fair value option election, the CVRs are initially measured at the aggregate estimated fair value of the CVRs and will be subsequently remeasured at estimated fair value on a recurring basis at each reporting period date. The estimated fair value of the CVR liability was determined using the discounted cash flow method to estimate future cash flows associated with the legacy assets, including the expected milestone and royalty payments under the Sobi License, net of deductions. Changes in fair value of the liability are presented within change in fair value of CVRs in the consolidated statements of operations and comprehensive income (loss). The liability value is based on significant inputs not observable in the market such as estimated cash flows, estimated
probabilities of success, and risk-adjustment discount rates, which represent a Level 3 measurement within the fair value hierarchy.
Stock-Based Compensation
We account for all stock-based compensation granted to employees and non-employees using a fair value method. Stock-based compensation is measured at the grant date fair value and is recognized over the requisite service period of the awards, usually the vesting period, on a straight-line basis, net of estimated forfeitures. To the extent that actual forfeitures differ from our estimates, the differences are recorded as a cumulative adjustment in the period the estimates were adjusted. Stock-based compensation expense recognized in the consolidated financial statements is based on awards that ultimately vest.
The assumptions used in determining the fair value of stock-based awards represent our best estimates, but the estimates involve inherent uncertainties and the application of our judgment. As a result, if factors change and we use significantly different assumptions or estimates, our stock-based compensation expense could be materially different in the future.
Smaller Reporting Company
We qualify as a “smaller reporting company” under the rules of the Securities Act and the Exchange Act. As a result, we may choose to take advantage of certain scaled disclosure requirements available specifically to smaller reporting companies. We will remain a smaller reporting company until the last day of the fiscal year in which the aggregate market value of our common stock held by non-affiliated persons and entities, or our public float, is more than $700 million as of the last business day of our most recently completed second fiscal quarter, or until the fiscal year following the year in which we have at least $100 million in revenue and at least $250 million in public float as of the last business day of our most recently completed second fiscal quarter.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
The market risk inherent in our financial instruments and in our financial position represents the potential loss arising from adverse changes in interest rates. As of December 31, 20222023 and 2021,2022, we had cash, cash equivalents, restricted cash and marketable securities of $136.2$78.3 million and $129.4$136.2 million, respectively, consisting of non-interest and interest-bearing money market accounts, U.S. government agency securities and treasuries, corporate bonds and commercial paper. Our primary exposure to market risk is interest rate sensitivity, which is affected by changes in the general level of U.S. interest rates. Due to the short-term and the low risk profile of our money market accounts and marketable securities, and our current plan to hold marketable securities to maturity,, an immediate 100 basis point change in interest rates would not have a material effect on the fair market value of our cash equivalents or short-term marketable securities.equivalents.
Item 8. Financial Statements and Supplementary Data
The consolidated financial statements together with the report of our independent registered public company accounting firm, required to be filed pursuant to this Item 8 are appended to this Annual Report. An index of those consolidated financial statements is found in Item 15.
Item 9. Changes in and Disagreements With Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
Evaluation of Disclosure Controls and Procedures
Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, evaluated, as of the end of the period covered by this Annual Report, on Form 10-K, the effectiveness of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act). Based on that evaluation, our Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures were not effective at the reasonable assurance level as of December 31, 2022.2023 because of the material weakness in internal control over financial reporting discussed below.
Management’s Annual Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over our financial reporting, as such term is defined in Rule 13a-15(f) and 15d-15(f) under the Exchange Act.
Our management conducted an assessment of the effectiveness of our internal control over financial reporting based on the criteria set forth in “Internal Control - Integrated Framework (2013)” issued by the Committee of Sponsoring Organizations of the Treadway Commission.
Management’s assessment of and conclusion on the effectiveness of internal control over financial reporting did not include the internal controls of Old Cartesian, which is included in our consolidated financial statements as of and for the year ended December 31, 2023 and constituted 2% and 1% of total assets and total liabilities, respectively, as of December 31, 2023 and 0% and 1% of revenues and operating expenses, respectively, for the year then ended.
Based on this assessment, our management concluded that, as of December 31, 2022,2023, our internal control over financial reporting was not effective.
As a result of its review, management identified a material weakness. A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that a reasonable possibility exists that a material misstatement of our annual or interim financial statements would not be prevented or detected on a timely basis. There are no material accounting errors or omissions within the consolidated financial statements as a result of this material weakness. Management concluded that it did not design and implement effective internal controls specifically related to the documentation of the assumptions supporting the valuation of the in-process intangible assets in connection with the Old Cartesian material business combination and the initial and ongoing contingent value right obligation issued at the time to legacy Selecta stockholders. This includes a lack of sufficient documentation to provide evidence of the associated management review controls.
Remediation Plans for Material Weakness in Internal Control over Financial Reporting
We are committed to maintaining a strong internal control environment. In response to the identified material weakness above, we, with the oversight of the Audit Committee, intend to take comprehensive actions to remediate the material weakness in internal control over financial reporting. We expect to re-evaluate the scope and level of precision for conducting and documenting the reviews over significant acquisitions and contingent value rights including the review of prospective financial information used in valuation reports produced by third-party specialists supporting the accounting for business combinations and contingent value rights. The remediation efforts are intended both to address the identified material weakness and to enhance our overall financial control environment.
Inherent Limitations of Internal Controls
While we believe we have a robust and efficient system of internal and disclosure controls and procedures, our management, including our Chief Executive Officer and Chief Financial Officer, recognize that it is impossible for our disclosure controls and procedures or our internal controls to prevent all errors and all fraud. A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within our company have been detected. These inherent limitations include the realities that judgments in decision-making can be faulty, and that breakdowns can occur because of a simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people, or by management override of the control. The design of any system of controls also is based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions. Over time, controls may become inadequate because of changes in conditions, or the degree of compliance with the policies or procedures may deteriorate. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate. Because of the inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and not be detected.
Changes in Internal Control over Financial Reporting
ThereExcept for our identification and assessment of the material weakness described above, there have been no changes in our internal control over financial reporting as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act during the three months ended December 31, 20222023 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Attestation Report of the Registered Public Accounting Firm
ThisErnst & Young LLP has independently assessed the effectiveness of our internal control over financial reporting as of December 31, 2023 and its report is included below.
Report of Independent Registered Public Accounting Firm
To the Stockholders and the Board of Directors of Cartesian Therapeutics, Inc.
Opinion on Internal Control over Financial Reporting
We have audited Cartesian Therapeutics, Inc.’s internal control over financial reporting as of December 31, 2023, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (the COSO criteria). In our opinion, because of the effect of the material weakness described below on the achievement of the objectives of the control criteria, Cartesian Therapeutics, Inc. (and subsidiaries) (the Company) has not maintained effective internal control over financial reporting as of December 31, 2023, based on the COSO criteria.
As indicated in the accompanying Management’s Annual Report on Form 10-K doesInternal Control Over Financial Reporting, management’s assessment of and conclusion on the effectiveness of internal control over financial reporting did not include the internal controls of Cartesian Bio, LLC, formerly known as Cartesian Therapeutics, Inc. (Old Cartesian), which is included in the 2023 consolidated financial statements of the Company and constituted 2% and 1% of total assets and total liabilities, respectively, as of December 31, 2023 and 0% and 1% of revenues and operating expenses, respectively, for the year then ended. Our audit of internal control over financial reporting of the Company also did not include an attestationevaluation of the internal control over financial reporting of Old Cartesian.
A material weakness is a deficiency, or combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of the company’s annual or interim financial statements will not be prevented or detected on a timely basis. The following material weakness has been identified and included in management’s assessment. Management has identified a material weakness in the design and operation of controls related to the valuation of the in-process intangible assets acquired as part of the Old Cartesian business combination and the Selecta Biosciences, Inc. contingent value right obligation.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the consolidated balance sheet of the Company as of December 31, 2023 and 2022, the related consolidated statements of operations and comprehensive income (loss), changes in convertible preferred stock and stockholders’ equity (deficit) and cash flows for each of the three years in the period ended December 31, 2023, and the related notes and our report dated March 7, 2024 expressed an unqualified opinion thereon.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our registeredaudit. We are a public accounting firm dueregistered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects.
Our audit included obtaining an exemption establishedunderstanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for “smallerour opinion.
Definition and Limitations of Internal Control Over Financial Reporting
A company’s internal control over financial reporting companies.”is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ Ernst & Young LLP
Boston, Massachusetts
March 7, 2024
Item 9B. Other Information
None.During the fiscal quarter ended December 31, 2023, none of our officers or directors, as defined in Rule 16a-1(f), informed us of the adoption, modification or termination of any "Rule 10b5-1 trading arrangement" or a "non-Rule 10b5-1 trading arrangement," as those terms are defined in Item 408 of Regulation S-K .
Item 9C. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections
Not applicable.
PART III
Item 10. Directors, Executive Officers and Corporate Governance
Incorporated by reference from the information in our Proxy Statement for our 20222024 Annual Meeting of Stockholders, which we will file with the SEC within 120 days of the end of the fiscal year to which this Annual Report on Form 10-K relates.
Item 11. Executive Compensation
Incorporated by reference from the information in our Proxy Statement for our 20222024 Annual Meeting of Stockholders, which we will file with the SEC within 120 days of the end of the fiscal year to which this Annual Report on Form 10-K relates.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
Incorporated by reference from the information in our Proxy Statement for our 20222024 Annual Meeting of Stockholders, which we will file with the SEC within 120 days of the end of the fiscal year to which this Annual Report on Form 10-K relates.
Item 13. Certain Relationships and Related Transactions, and Director Independence
Incorporated by reference from the information in our Proxy Statement for our 20222024 Annual Meeting of Stockholders, which we will file with the SEC within 120 days of the end of the fiscal year to which this Annual Report on Form 10-K relates.
Item 14. Principal Accountant Fees and Services
Incorporated by reference from the information in our Proxy Statement for our 20222024 Annual Meeting of Stockholders, which we will file with the SEC within 120 days of the end of the fiscal year to which this Annual Report on Form 10-K relates.
PART IV
Item 15. Exhibits, Financial Statement Schedules
(a)(1) Financial Statements
See the “Index to Consolidated Financial Statements” on page F-1 below for the list of financial statements filed as part of this report.
(a)(2) Financial Statement Schedules
All financial schedules have been omitted because the required information is either presented in the consolidated financial statements filed as part of this Annual Report on Form 10-K or the notes thereto or is not required.
(a)(3) Exhibits
The following is a list of exhibits filed as part of this Annual Report on Form 10-K.
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | Incorporated by Reference |
Exhibit
Number | | Exhibit Description | | Form | | File No. | | Exhibit | | Filing
Date |
| | | | 8-K | | 001-37798 | | 3.1 | | 6/29/2016 |
| | | | 8-K | | 001-37798 | | 3.1 | | 6/21/2022 |
| | | | 8-K | | 001-37798 | | 3.2 | | 8/19/2022 |
| | | | S-1 | | 333-211555 | | 4.2 | | 5/24/2016 |
| | | | S-1 | | 333-211555 | | 4.5 | | 5/24/2016 |
| | | | S-1 | | 333-211555 | | 4.6 | | 5/24/2016 |
| | | | 8-K | | 001-37798 | | 4.1 | | 6/28/2017 |
| | | | 8-K | | 001-37798 | | 10.2 | | 12/26/2019 |
| | | | 10-Q | | 001-37798 | | 4.1 | | 8/6/2020 |
| | | | 10-Q | | 001-37798 | | 4.2 | | 11/5/2020 |
| | | | 8-K | | 001-37798 | | 4.1 | | 12/26/2019 |
| | | | — | | — | | — | | Filed herewith |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | Incorporated by Reference |
Exhibit
Number | | Exhibit Description | | Form | | File No. | | Exhibit | | Filing
Date |
| | Agreement and Plan of Merger, dated November 13, 2023, by and among Selecta Biosciences, Inc., Sakura Merger Sub I, Inc., Sakura Merger Sub II, LLC, and Cartesian Therapeutics, Inc. | | 8-K | | 001-37798 | | 2.1 | | 11/13/2023 |
| | | | 8-K | | 001-37798 | | 3.1 | | 6/29/2016 |
| | | | 8-K | | 001-37798 | | 3.1 | | 6/21/2022 |
| | | | 8-K | | 001-37798 | | 3.3 | | 11/13/2023 |
| | | | 10-Q | | 001-37798 | | 3.2 | | 11/13/2023 |
| | | | S-1 | | 333-211555 | | 4.2 | | 5/24/2016 |
| | | | S-1 | | 333-211555 | | 4.5 | | 5/24/2016 |
| | | | S-1 | | 333-211555 | | 4.6 | | 5/24/2016 |
| | | | 8-K | | 001-37798 | | 4.1 | | 6/28/2017 |
| | | | 8-K | | 001-37798 | | 10.2 | | 12/26/2019 |
| | | | 10-Q | | 001-37798 | | 4.1 | | 8/6/2020 |
| | | | 10-Q | | 001-37798 | | 4.2 | | 11/5/2020 |
| | | | 8-K | | 001-37798 | | 4.1 | | 12/26/2019 |
| | | | 10-K | | 001-37798 | | 4.8(b) | | 3/2/2023 |
| | | | 8-K | | 001-37798 | | 4.1 | | 9/3/2020 |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | 8-K | | 001-37798 | | 4.1 | | 9/3/2020 |
| | | | 8-K | | 001-37798 | | 4.1 | | 4/6/2022 |
| | | | — | | — | | — | | Filed herewith |
| | | | S-1/A | | 333-211555 | | 10.2 | | 6/8/2016 |
| | | | S-1/A | | 333-211555 | | 10.3 | | 6/8/2016 |
| | | | S-8 | | 333-230501 | | 10.1 | | 3/25/2019 |
| | | | S-1/A | | 333-211555 | | 10.1 | | 6/20/2016 |
| | | | — | | — | | — | | Filed herewith |
| | | | S-1 | | 333-211555 | | 10.5 | | 5/24/2016 |
| | | | S-1 | | 333-211555 | | 10.7(a) | | 5/24/2016 |
| | | | S-1 | | 333-211555 | | 10.7(b) | | 5/24/2016 |
| | | | S-1 | | 333-211555 | | 10.7(c) | | 5/24/2016 |
| | | | S-1 | | 333-211555 | | 10.7(d) | | 5/24/2016 |
| | | | S-1 | | 333-211555 | | 10.7(e) | | 5/24/2016 |
| | | | 8-K/A | | 001-37798 | | 10.3(a) | | 12/14/2016 |
| | | | 8-K/A | | 001-37798 | | 10.3(b) | | 12/14/2016 |
| | | | 8-K/A | | 001-37798 | | 10.3(c) | | 12/14/2016 |
| | | | 10-K | | 001-37798 | | 10.7(i) | | 3/12/2020 |
| | | | 10-Q | | 001-37798 | | 10.1 | | 8/6/2020 |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | 8-K | | 001-37798 | | 4.1 | | 4/6/2022 |
| | | | 8-K | | 001-37798 | | 2.1 | | 11/13/2023 |
| | | | 8-K | | 001-37798 | | 10.2 | | 11/13/2023 |
| | | | 8-K | | 001-37798 | | 3.4 | | 11/13/2023 |
| | | | — | | — | | — | | Filed herewith |
| | | | S-1/A | | 333-211555 | | 10.2 | | 6/8/2016 |
| | | | S-1/A | | 333-211555 | | 10.3 | | 6/8/2016 |
| | | | S-8 | | 333-276486 | | 99.2 | | 1/12/2024 |
| | | | S-1/A | | 333-211555 | | 10.1 | | 6/20/2016 |
| | | | S-8 | | 333-276486 | | 99.1 | | 1/12/2024 |
| | | | — | | — | | — | | Filed herewith |
| | | | S-1 | | 333-211555 | | 10.5 | | 5/24/2016 |
| | | | | | | | | | |
| | | | | | | | | | |
| | | | | | | | | | |
| | | | | | | | | | |
| | | | | | | | | | |
| | | | | | | | | | |
| | | | | | | | | | |
| | | | | | | | | | |
| | | | | | | | | | |
| | | | | | | | | | |
| | | | | | | | | | |
| | | | 10-Q | | 001-37798 | | 10.6 | | 8/11/2017 |
| | | | S-1 | | 333-211555 | | 10.10 | | 5/24/2016 |
| | | | 10-Q | | 001-37798 | | 10.3 | | 11/8/2019 |
| | | | 10-Q | | 001-37798 | | 10.1 | | 11/3/2022 |
| | | | — | | — | | — | | Filed herewith |
| | | | — | | — | | — | | Filed herewith |
| | | | — | | — | | — | | Filed herewith |
| | | | — | | — | | — | | Filed herewith |
| | | | 8-K | | 001-37798 | | 10.2 | | 9/27/2018 |
| | | | 10-K | | 001-37798 | | 10.15 | | 3/2/2023 |
| | | | 10-Q | | 001-37798 | | 10.2 | | 8/6/2020 |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | 10-Q | | 001-37798 | | 10.6 | | 8/11/2017 |
| | | | S-1 | | 333-211555 | | 10.10 | | 5/24/2016 |
| | | | 10-Q | | 001-37798 | | 10.3 | | 11/8/2019 |
| | | | 10-Q | | 001-37798 | | 10.1 | | 11/3/2022 |
| | | | 8-K | | 001-37798 | | 10.2 | | 9/27/2018 |
| | | | S-1/A | | 333-211555 | | 10.18 | | 6/8/2016 |
| | | | S-1/A | | 333-211555 | | 10.21 | | 6/8/2016 |
| | | | 10-Q | | 001-37798 | | 10.1 | | 11/5/2020 |
| | | | — | | — | | — | | Filed herewith |
| | | | — | | — | | — | | Filed herewith |
| | | | 10-Q | | 001-37798 | | 10.2 | | 8/6/2020 |
| | | | 8-K | | 001-37798 | | 10.2 | | 6/28/2017 |
| | | | 8-K | | 001-37798 | | 10.1 | | 8/20/2019 |
| | | | 8-K | | 001-37798 | | 10.1.1 | | 9/3/2020 |
| | | | 10-Q | | 001-37798 | | 10.3 | | 11/9/2021 |
| | | | 8-K | | 001-37798 | | 10.1 | | 3/21/2022 |
| | | | 10-Q | | 001-37798 | | 10.2 | | 11/3/2022 |
| | | | S-1 | | 333-211555 | | 21.1 | | 5/24/2016 |
| | | | — | | — | | — | | Filed herewith |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | — | | — | | — | | Filed herewith |
| | | | — | | — | | — | | Filed herewith |
| | | | — | | — | | — | | Filed herewith |
| | | | 8-K | | 001-37798 | | 10.2 | | 6/28/2017 |
| | | | 8-K | | 001-37798 | | 10.1 | | 8/20/2019 |
| | | | 8-K | | 001-37798 | | 10.1.1 | | 9/3/2020 |
| | | | 10-Q | | 001-37798 | | 10.3 | | 11/9/2021 |
| | | | 8-K | | 001-37798 | | 10.1 | | 3/21/2022 |
| | | | 10-Q | | 001-37798 | | 10.2 | | 11/3/2022 |
| | Fourth Amendment to Loan and Security Agreement, dated March 31, 2023, between Selecta Biosciences, Inc., Oxford Finance LLC, as Collateral Agent and as a lender, and Silicon Valley Bank, a division of First-Citizens Bank & Trust Company (successor by purchase to the Federal Deposit Insurance Corporation as Receiver for Silicon Valley Bridge Bank, N.A. (as successor to Silicon Valley Bank)), as a lender | | 10-Q | | 001-37798 | | 10.1 | | 5/4/2023 |
| | | | — | | — | | — | | Filed herewith |
| | | | 8-K | | 001-37798 | | 10.3 | | 11/13/2023 |
| | | | 8-K | | 001-37798 | | 10.1 | | 11/13/2023 |
| | | | — | | — | | — | | Filed herewith |
| | | | — | | — | | — | | Filed herewith |
| | | | — | | — | | — | | Filed herewith |
| | | | — | | — | | — | | Filed herewith |
| | | | — | | — | | — | | Furnished herewith |
| | | | — | | — | | — | | Filed herewith |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | — | | — | | — | | Filed herewith |
| | | | — | | — | | — | | Filed herewith |
| | | | — | | — | | — | | Furnished herewith |
| 101.INS | | Inline XBRL Instance Document - the Instance Document does not appear in the interactive data file because its XBRL tags are embedded within the Inline XBRL Document | | — | | — | | — | | Filed herewith |
| 101.SCH | | Inline XBRL Taxonomy Extension Schema Document | | — | | — | | — | | Filed herewith |
| 101.CAL | | Inline XBRL Taxonomy Extension Calculation Linkbase Document | | — | | — | | — | | Filed herewith |
| 101.DEF | | Inline XBRL Taxonomy Extension Definition Linkbase Document | | — | | — | | — | | Filed herewith |
| 101.LAB | | Inline XBRL Taxonomy Extension Label Linkbase Document | | — | | — | | — | | Filed herewith |
| 101.PRE | | Inline XBRL Taxonomy Extension Presentation Linkbase Document | | — | | — | | — | | Filed herewith |
| 104 | | Cover Page Interactive Data File (formatted as Inline XBRL and contained in Exhibit 101) | | — | | — | | — | | Filed herewith |
| | # Indicates management contract or compensatory plan. | | | | | | | | |
| | † Portions of this exhibit (indicated by asterisks)* Certain annexes, schedules and exhibits have been omitted pursuant to Item 601(a)(5) of Regulation S-K. The Company agrees to furnish supplementally a request forcopy of any omitted attachment to the SEC on a confidential treatmentbasis upon request. |
| | † Certain confidential information contained in this exhibit, marked by brackets and asterisks, has been omitted pursuant to Rule 24h-2 underItem 601(b)(10)(iv) of Regulation S-K because the Securities Exchange Actinformation (i) is not material and (ii) is the type of 1934.information that the Company both customarily and actually treats as private and confidential. |
Item 16. Form 10-K Summary
None.
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| | | | | | | | | | | |
| | | SELECTA BIOSCIENCES,CARTESIAN THERAPEUTICS, INC. |
| | | | |
Date: March 2, 20237, 2024 | | By: | /s/ Carsten Brunn, Ph.D. |
| | | | Carsten Brunn, Ph.D. |
| | | | President and Chief Executive Officer |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed by the following persons on behalf of the registrant in the capacities and on the dates indicated.
| | | | | | | | | | | | | | |
| Signature | | Title | | Date |
| | | | |
| /s/ Carsten Brunn, Ph.D. | | President and Chief Executive Officer, and Director | | March 2, 20237, 2024 |
| Carsten Brunn, Ph.D. | | (Principal Executive Officer) | | |
| | | | |
| /s/ Blaine Davis | | Chief Financial Officer | | March 2, 20237, 2024 |
| Blaine Davis | | (Principal Financial and Accounting Officer) | | |
| | | | |
| /s/ Carrie S. Cox | | Director | | March 2, 20237, 2024 |
| Carrie S. Cox | | | | |
| | | | |
| | | | |
| | | | |
| | | | |
| /s/ Timothy C. Barabe | | Director | | March 2, 20237, 2024 |
| Timothy C. Barabe | | | | |
| | | | |
| /s/ Nishan de Silva, M.D. | | Director | | March 2, 20237, 2024 |
| Nishan de Silva, M.D. | | | | |
| | | | |
/s/ Scott D. MyersMurat Kalayoglu, M.D., Ph.D. | | Director | | March 2, 20237, 2024 |
Scott D. MyersMurat Kalayoglu, M.D., Ph.D. | | | | |
| | | | |
/s/ Aymeric SallinMichael Singer, M.D., Ph.D. | | Director | | March 2, 20237, 2024 |
Aymeric SallinMichael Singer, M.D., Ph.D. | | | | |
| | | | |
| /s/ Timothy Springer, Ph.D. | | Director | | March 2, 20237, 2024 |
| Timothy Springer, Ph.D. | | | | |
| | | | |
| /s/ Patrick Zenner | | Director | | March 2, 20237, 2024 |
| Patrick Zenner | | | | |
Selecta Biosciences,Cartesian Therapeutics, Inc. and Subsidiaries
| | | | | | | | | | | |
| Index to Consolidated Financial Statements | Pages | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
Report of Independent Registered Public Accounting Firm
To the Stockholders and the Board of Directors of Selecta Biosciences,Cartesian Therapeutics, Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Selecta Biosciences,Cartesian Therapeutics, Inc. and subsidiaries (the Company) as of December 31, 20222023 and 2021,2022, the related consolidated statements of operations and comprehensive income (loss), changes in convertible preferred stock and stockholders’ equity (deficit) and cash flows for each of the three years in the period ended December 31, 2022,2023, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 20222023 and 2021,2022, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2022,2023, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company's internal control over financial reporting as of December 31, 2023, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework), and our report dated March 7, 2024 expressed an adverse opinion thereon.
Basis for Opinion
These financial statements are the responsibility of the Company’sCompany's management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits.audit. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB)PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our auditsaudit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, anOur audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our auditsaudit also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provideaudit provides a reasonable basis for our opinion.
Critical Audit Matter
The critical audit matter communicated below is a matter arising from the current period audit of the financial statements that was communicated or required to be communicated to the audit committeeAudit Committee and that: (1) relates to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective or complex judgments. The communication of the critical audit matter does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the accounts or disclosures to which it relates.
| | | | | | | | |
| | Accrued and Prepaid Clinical Trial Expense |
Description of the Matter | | As discussed in Note 2 to the consolidated financial statements, the Company records research and development expenses, which include expenses related to clinical trials, as incurred. The Company’s determination of clinical trial costs incurred, as well as the related accrued and prepaid expenses at each reporting period, incorporates judgment and utilizes various assumptions. Such assumptions include progress of the studies, including the phase or completion of events, invoices received and contracted costs. Payments for these activities are based on the terms of the individual arrangements, which may differ from the pattern of costs incurred.
Auditing the Company’s accrued and prepaid research and development expenses was especially challenging due to the significant judgment required to estimate the expenses incurred but not yet invoiced. While the Company’s estimates of accrued and prepaid clinical trial expenses are primarily based on information received related to each study from its vendors, the Company makes an estimate for additional costs incurred based on management judgment. Additionally, due to the long duration of clinical trials and the timing of invoicing received from third parties, the actual amounts incurred are not always known at the time the financial statements are issued.
|
| | | | | | | | |
| | Valuation of in-process research and development acquired in a business combination |
| Description of the Matter | | As described in Note 3, on November 13, 2023, the Company acquired Cartesian Therapeutics, Inc. in a stock for stock transfer, which was accounted for as a business combination using the acquisition method of accounting. The acquired intangible assets consisted of in-process research and development which had estimated acquisition-date fair values of $150.6 million.
Auditing the acquisition date fair value of the in-process research and development was complex due to the significant judgment required in estimating the fair value. In particular, the fair value estimate required the use of valuation methodologies that were sensitive to significant assumptions (e.g., projected revenue growth rates, including forecasted selling prices and unit volumes, and discount rates applied to the in-process research and development), which are affected by expected future market or economic conditions. |
| | |
| How We Addressed the Matter in Our Audit | | To evaluatetest the accruedestimated fair value of the acquired in-process research and prepaid clinical trial expenses,development intangible assets, our audit procedures included, among others, assessing the appropriateness of the valuation methodology and testing the accuracysignificant assumptions discussed above and the completeness and accuracy of the underlying data used by the Company. For example, we evaluated the reasonableness of assumptions used to determine the projected revenue growth rates by comparing the forecasted assumptions to projected industry growth rates, and other factors considered by management in developing the estimates andmodel. We involved our valuation specialist to assist in evaluating the significant assumptions that arevaluation methodologies and discount rates used by management to estimate the recorded accruals and prepayments. We obtained third party confirmations from the clinical research organization to further test the underlying data used in management’s estimate. We corroborated the progress ofvalue in-process research and development activities associated with clinical trials through discussion withintangible assets. We also performed sensitivity analyses to evaluate the Company’schanges in the fair value of the acquired in-process research and development personnelintangible assets that overseewould result from changes in the research and development activities. We inspected the Company’s third-party contracts, amendments, and any pending change orders to assess the impact on amounts recorded. In addition, we performed analytics over fluctuations in accruals and prepaids by vendor throughout the period subject to audit and compared subsequent invoices received from third parties to amounts accrued.significant assumptions. |
/s/ Ernst & Young LLP
We have served as the Company’s auditor since 2009.
Boston, Massachusetts
March 2, 20237, 2024
Selecta Biosciences,Cartesian Therapeutics, Inc. and Subsidiaries
Consolidated Balance Sheets
(Amounts in thousands, except share data and par value)
| | | | | | | | | | | |
| | December 31, | | December 31, |
| | 2022 | | 2021 |
| | | |
| Assets | | | |
| Current assets: | | | |
| Cash and cash equivalents | $ | 106,438 | | | $ | 114,057 | |
| | | |
| Marketable securities | 28,164 | | | 13,998 | |
| Accounts receivable | 6,596 | | | 9,914 | |
| Unbilled receivables | 3,162 | | | — | |
| Prepaid expenses and other current assets | 3,778 | | | 6,474 | |
| Total current assets | 148,138 | | | 144,443 | |
| Non-current assets: | | | |
| Property and equipment, net | 2,794 | | | 2,142 | |
| Right-of-use asset, net | 11,617 | | | 9,829 | |
| Long-term restricted cash | 1,311 | | | 1,379 | |
| Investments | 2,000 | | | 2,000 | |
| Other assets | 26 | | | 90 | |
| Total assets | $ | 165,886 | | | $ | 159,883 | |
| Liabilities and stockholders’ equity | | | |
| Current liabilities: | | | |
| Accounts payable | $ | 316 | | | $ | 224 | |
| Accrued expenses | 14,084 | | | 10,533 | |
| Loan payable | 8,476 | | | 5,961 | |
| Lease liability | 1,608 | | | 1,049 | |
| Income taxes payable | — | | | 601 | |
| Deferred revenue | 593 | | | 53,883 | |
| | | |
| Total current liabilities | 25,077 | | | 72,251 | |
| Non-current liabilities: | | | |
| | | |
| Loan payable, net of current portion | 17,786 | | | 19,673 | |
| Lease liability, net of current portion | 10,055 | | | 8,598 | |
| Deferred revenue | — | | | 11,417 | |
| Warrant liabilities | 19,140 | | | 25,423 | |
| | | |
| Total liabilities | 72,058 | | | 137,362 | |
| Commitments and contingencies (Note 17) | | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| Stockholders’ equity: | | | |
| Preferred stock, $0.0001 par value; 10,000,000 shares authorized; no shares issued and outstanding as of December 31, 2022 and December 31, 2021 | — | | | — | |
| Common stock, $0.0001 par value; 350,000,000 and 200,000,000 shares authorized as of December 31, 2022 and December 31, 2021, respectively; 153,042,435 and 123,622,965 shares issued and outstanding as of December 31, 2022 and December 31, 2021, respectively | 15 | | | 12 | |
| Additional paid-in capital | 493,308 | | | 457,391 | |
| | | |
| Accumulated deficit | (394,937) | | | (430,316) | |
| Accumulated other comprehensive loss | (4,558) | | | (4,566) | |
| Total stockholders’ equity | 93,828 | | | 22,521 | |
| Total liabilities and stockholders’ equity | $ | 165,886 | | | $ | 159,883 | |
| | | | | | | | | | | |
| | December 31, | | December 31, |
| | 2023 | | 2022 |
| | | |
| Assets | | | |
| Current assets: | | | |
| Cash and cash equivalents | $ | 76,911 | | | $ | 106,438 | |
| | | |
| Marketable securities | — | | | 28,164 | |
| Accounts receivable | 5,870 | | | 6,596 | |
| Unbilled receivables | 2,981 | | | 3,162 | |
| Prepaid expenses and other current assets | 4,967 | | | 3,778 | |
| Total current assets | 90,729 | | | 148,138 | |
| Non-current assets: | | | |
| Property and equipment, net | 2,113 | | | 2,794 | |
| Right-of-use asset, net | 10,068 | | | 11,617 | |
| In-process research and development assets | 150,600 | | | — | |
| Goodwill | 48,163 | | | — | |
| Long-term restricted cash | 1,377 | | | 1,311 | |
| Investments | 2,000 | | | 2,000 | |
| Other assets | — | | | 26 | |
| Total assets | $ | 305,050 | | | $ | 165,886 | |
| Liabilities, convertible preferred stock, and stockholders’ (deficit) equity | | | |
| Current liabilities: | | | |
| Accounts payable | $ | 3,150 | | | $ | 316 | |
| Accrued expenses and other current liabilities | 15,572 | | | 14,084 | |
| Loan payable | — | | | 8,476 | |
| Lease liability | 2,166 | | | 1,608 | |
| | | |
| Deferred revenue | 2,311 | | | 593 | |
| Warrant liabilities | 720 | | | — | |
| Contingent value right liability | 15,983 | | | — | |
| Forward contract liabilities | 28,307 | | | — | |
| | | |
| Total current liabilities | 68,209 | | | 25,077 | |
| Non-current liabilities: | | | |
| | | |
| Loan payable, net of current portion | — | | | 17,786 | |
| Lease liability, net of current portion | 8,789 | | | 10,055 | |
| Deferred revenue, net of current portion | 3,538 | | | — | |
| Warrant liabilities, net of current portion | 5,674 | | | 19,140 | |
| Contingent value right liability, net of current portion | 342,617 | | | — | |
| Deferred tax liabilities, net | 15,853 | | | — | |
| | | |
| Total liabilities | 444,680 | | | 72,058 | |
| Commitments and contingencies (Note 19) | | | |
| | | |
| Series A Preferred Stock, $0.0001 par value; 548,375 and no shares authorized as of December 31, 2023 and December 31, 2022, respectively; 435,120.513 and no shares issued and outstanding as of December 31, 2023 and December 31, 2022, respectively | 296,851 | | | — | |
| Options for Series A Preferred Stock | 3,703 | | | — | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| Stockholders’ (deficit) equity: | | | |
| Preferred stock, $0.0001 par value; 9,451,625 and 10,000,000 shares authorized as of December 31, 2023 and December 31, 2022, respectively; no shares issued and outstanding as of December 31, 2023 and December 31, 2022 | — | | | — | |
| Common stock, $0.0001 par value; 350,000,000 shares authorized as of December 31, 2023 and December 31, 2022; 161,927,821 and 153,042,435 shares issued and outstanding as of December 31, 2023 and December 31, 2022, respectively | 16 | | | 15 | |
| Additional paid-in capital | 179,047 | | | 493,308 | |
| | | |
| Accumulated deficit | (614,647) | | | (394,937) | |
| Accumulated other comprehensive loss | (4,600) | | | (4,558) | |
| Total stockholders’ (deficit) equity | (440,184) | | | 93,828 | |
| Total liabilities, convertible preferred stock, and stockholders’ (deficit) equity | $ | 305,050 | | | $ | 165,886 | |
The accompanying notes are an integral part of these consolidated financial statements.
Selecta Biosciences,Cartesian Therapeutics, Inc. and Subsidiaries
Consolidated Statements of Operations and Comprehensive Income (Loss)
(Amounts in thousands, except share and per share data)
| | | | | Year Ended December 31, |
| | | | 2022 | | 2021 | | 2020 |
| | | | | Year Ended December 31, |
| | | | | | | | 2023 | | | | | | | | | | 2023 | | 2022 | | 2021 |
| | Collaboration and license revenue | |
| Collaboration and license revenue | |
| Collaboration and license revenue | Collaboration and license revenue | | $ | 110,777 | | | $ | 85,077 | | | $ | 16,597 | |
| Operating expenses: | Operating expenses: | | |
| Research and development | Research and development | | 72,377 | | | 68,736 | | | 54,505 | |
| Research and development | |
| Research and development | |
| General and administrative | General and administrative | | 23,862 | | | 20,938 | | | 18,913 | |
| Total operating expenses | Total operating expenses | | 96,239 | | | 89,674 | | | 73,418 | |
| Operating income (loss) | | 14,538 | | | (4,597) | | | (56,821) | |
| Operating (loss) income | |
| Investment income | Investment income | | 2,073 | | | 44 | | | 260 | |
| Loss on extinguishment of debt | | — | | | — | | | (461) | |
| | Foreign currency transaction gain (loss), net | |
| Foreign currency transaction gain (loss), net | |
| Foreign currency transaction gain (loss), net | Foreign currency transaction gain (loss), net | | (22) | | | — | | | 56 | |
| Interest expense | Interest expense | | (3,031) | | | (2,844) | | | (1,556) | |
| Change in fair value of warrant liabilities | Change in fair value of warrant liabilities | | 20,882 | | | (2,339) | | | (10,443) | |
| Change in fair value of contingent value right liability | |
| Change in fair value of forward contract liabilities | |
| Other income, net | Other income, net | | 330 | | | 15 | | | 89 | |
| Income (loss) before income taxes | | 34,770 | | | (9,721) | | | (68,876) | |
| Income tax (expense) benefit | | 609 | | | (15,966) | | | — | |
| Net income (loss) | | 35,379 | | | (25,687) | | | (68,876) | |
| (Loss) income before income taxes | |
| Income tax benefit (expense) | |
| Net (loss) income | |
| | Other comprehensive income (loss): | | |
| Other comprehensive (loss) income: | |
| Other comprehensive (loss) income: | |
| Other comprehensive (loss) income: | |
| Foreign currency translation adjustment | Foreign currency translation adjustment | | 18 | | | (2) | | | (40) | |
| Unrealized loss on marketable securities | | (10) | | | (1) | | | — | |
| Total comprehensive income (loss) | | $ | 35,387 | | | $ | (25,690) | | | $ | (68,916) | |
| Foreign currency translation adjustment | |
| Foreign currency translation adjustment | |
| Unrealized gain (loss) on marketable securities | |
| Total comprehensive (loss) income | |
| | | Net income (loss) per share: | | |
| Net (loss) income per share: | |
| | Net (loss) income per share: | |
| | Net (loss) income per share: | |
| Basic | |
| Basic | |
| Basic | Basic | | $ | 0.24 | | | $ | (0.22) | | | $ | (0.68) | |
| Diluted | Diluted | | $ | 0.10 | | | $ | (0.22) | | | $ | (0.68) | |
| Weighted average common shares outstanding: | | | | | | |
| Weighted-average common shares outstanding: | |
| Basic | |
| Basic | |
| Basic | Basic | | 144,758,555 | | | 114,328,798 | | | 101,202,176 | |
| Diluted | Diluted | | 145,874,889 | | | 114,328,798 | | | 101,202,176 | |
The accompanying notes are an integral part of these consolidated financial statements.
Selecta Biosciences,Cartesian Therapeutics, Inc. and Subsidiaries
Consolidated Statements of Changes in Convertible Preferred Stock and Stockholders’ Equity (Deficit)
(Amounts in thousands, except share data)
| | | | | | | | | | Accumulated | |
| | | | Additional | | | | | other | | Stockholders’ |
| | Common stock | | paid-in | | | Accumulated | | comprehensive | | Equity |
| | Shares | Amount | | capital | | | deficit | | loss | | (Deficit) |
| Balance at December 31, 2019 | 86,325,547 | | $ | 9 | | | $ | 348,664 | | | | $ | (335,753) | | | $ | (4,523) | | | $ | 8,397 | |
| Issuance of common stock under Employee Stock Purchase Plan | 110,212 | | — | | | 184 | | | | — | | | — | | | 184 | |
| Issuance of common stock upon exercise of options | 76,128 | | — | | | 193 | | | | — | | | — | | | 193 | |
| Issuance of vested restricted stock units | 93,750 | | — | | | — | | | | — | | | — | | | — | |
| | Issuance of common stock through at-the-market offering, net | 1,069,486 | | — | | | 2,108 | | | | — | | | — | | | 2,108 | |
| Issuance of common stock through private placement | 5,416,390 | | — | | | 10,268 | | | | — | | | — | | | 10,268 | |
| | Issuance of common stock upon exercise of pre-funded warrants | 8,342,128 | | 1 | | | — | | | | — | | | — | | | 1 | |
| Issuance of common stock upon exercise of warrants | 6,637,608 | | 1 | | | 24,262 | | | | — | | | — | | | 24,263 | |
| Other financing fees | — | | — | | | (370) | | | | — | | | — | | | (370) | |
| Issuance of common warrants with long-term debt, net | — | | — | | | 444 | | | | — | | | — | | | 444 | |
| Stock-based compensation expense | — | | — | | | 5,422 | | | | — | | | — | | | 5,422 | |
| Currency translation adjustment | — | | — | | | — | | | | — | | | (40) | | | (40) | |
| Net loss | — | | — | | | — | | | | (68,876) | | | — | | | (68,876) | |
| Balance at December 31, 2020 | 108,071,249 | | $ | 11 | | | $ | 391,175 | | | | $ | (404,629) | | | $ | (4,563) | | | $ | (18,006) | |
| Issuance of common stock under Employee Stock Purchase Plan | 58,794 | | — | | | 161 | | | | — | | | — | | | 161 | |
| Issuance of common stock upon exercise of options | 447,492 | | — | | | 778 | | | | — | | | — | | | 778 | |
| Issuance of vested restricted stock units | 201,250 | | — | | | — | | | | — | | | — | | | — | |
| Issuance of common stock through at-the-market offering, net | 13,767,511 | | 1 | | | 51,933 | | | | — | | | — | | | 51,934 | |
| | Issuance of common stock upon exercise of warrants | 1,076,669 | | — | | | 5,624 | | | | — | | | — | | | 5,624 | |
| | Stock-based compensation expense | — | | — | | | 7,720 | | | | — | | | — | | | 7,720 | |
| Currency translation adjustment | — | | — | | | — | | | | — | | | (2) | | | (2) | |
| Unrealized loss on marketable securities | — | | — | | | — | | | | — | | | (1) | | | (1) | |
| Net loss | — | | — | | | — | | | | (25,687) | | | — | | | (25,687) | |
| Balance at December 31, 2021 | 123,622,965 | | $ | 12 | | | $ | 457,391 | | | | $ | (430,316) | | | $ | (4,566) | | | $ | 22,521 | |
| | | Series A | | Series A | | | | | | Additional | | | | | | other | | Stockholders’ |
| | | Preferred stock | | Preferred Stock | | | Common stock | | paid-in | | | | Accumulated | | comprehensive | | (Deficit) |
| | | Shares | Amount | | Amount | | | Shares | Amount | | capital | | | | deficit | | loss | | Equity |
| Balance at December 31, 2020 | |
| Issuance of common stock under Employee Stock Purchase Plan | |
| Issuance of common stock upon exercise of options | |
| Issuance of vested restricted stock units | |
| | Issuance of common stock through at-the-market offering, net | |
| Issuance of common stock through at-the-market offering, net | |
| Issuance of common stock through at-the-market offering, net | |
| | Issuance of common stock upon exercise of warrants | |
| | Issuance of common stock upon exercise of warrants | |
| | Issuance of common stock upon exercise of warrants | |
| | Stock-based compensation expense | |
| | Stock-based compensation expense | |
| | Stock-based compensation expense | |
| Currency translation adjustment | |
| Unrealized loss on marketable securities | |
| Net loss | |
| Balance at December 31, 2021 | |
| Issuance of common stock under Employee Stock Purchase Plan | |
| Issuance of common stock upon exercise of options | |
| Issuance of vested restricted stock units | |
| Issuance of common stock through at-the-market offering, net | |
| Issuance of common stock and common warrants | |
| Issuance of common stock, license agreement | |
| | Reclassification of warrant liabilities | |
| | Issuance of common stock under Employee Stock Purchase Plan | 120,877 | | — | | | 189 | | | | — | | | — | | | 189 | |
| Issuance of common stock upon exercise of options | 71,190 | | — | | | 156 | | | | — | | | — | | | 156 | |
| Issuance of vested restricted stock units | 131,430 | | — | | | — | | | | — | | | — | | | — | |
| Issuance of common stock through at-the-market offering, net | 774,544 | | — | | | 2,121 | | | | — | | | — | | | 2,121 | |
| | Reclassification of warrant liabilities | |
| | Issuance of common stock and common warrants | 27,428,572 | | 3 | | | 21,477 | | | | — | | | — | | | 21,480 | |
| Issuance of common stock, license agreement | 892,857 | | — | | | 1,000 | | | | — | | | — | | | 1,000 | |
| | Reclassification of warrant liabilities | Reclassification of warrant liabilities | — | | — | | | 780 | | | | — | | | — | | | 780 | |
| Stock-based compensation expense | Stock-based compensation expense | — | | — | | | 10,194 | | | | — | | | — | | | 10,194 | |
| Currency translation adjustment | Currency translation adjustment | — | | — | | | — | | | | — | | | 18 | | | 18 | |
| Unrealized loss on marketable securities | Unrealized loss on marketable securities | — | | — | | | — | | | | — | | | (10) | | | (10) | |
| Net income | Net income | — | | — | | | — | | | | 35,379 | | | — | | | 35,379 | |
| Balance at December 31, 2022 | Balance at December 31, 2022 | 153,042,435 | | $ | 15 | | | $ | 493,308 | | | | $ | (394,937) | | | $ | (4,558) | | | $ | 93,828 | |
| Issuance of Series A Preferred Stock in private placement | |
| Issuance of Series A Preferred Stock in connection with the Merger and settlement of related forward contract | |
| Issuance of Series A Preferred Stock in connection with private placement and settlement of related forward contract | |
| Issuance of common stock under Employee Stock Purchase Plan | |
| | Issuance of vested restricted stock units | |
| Issuance of vested restricted stock units | |
| Issuance of vested restricted stock units | |
| Issuance of common stock forward in connection with the Merger | |
| Issuance of common stock in connection with the Merger and settlement of related forward contract | |
| Issuance of replacement options in Merger | |
| | Issuance of common stock, license agreement | |
| | Issuance of common stock, license agreement | |
| | Issuance of common stock, license agreement | |
| | Settlement of outstanding equity awards at Merger | |
| Settlement of outstanding equity awards at Merger | |
| Settlement of outstanding equity awards at Merger | |
| Distribution of contingent value rights | |
| Stock-based compensation expense | |
| Currency translation adjustment | |
| Unrealized gain on marketable securities | |
| Net loss | |
| Balance at December 31, 2023 | |
The accompanying notes are an integral part of these consolidated financial statements.
Selecta Biosciences,Cartesian Therapeutics, Inc. and Subsidiaries
Consolidated Statements of Cash Flows
| | | | | Year Ended December 31, |
| | | 2022 | | 2021 | | 2020 |
| | | Year Ended December 31, |
| | | | 2023 | | | | | | 2023 | | 2022 | | 2021 |
| | Cash flows from operating activities | Cash flows from operating activities | | (Amounts in thousands) |
| Net income (loss) | | $ | 35,379 | | | $ | (25,687) | | | $ | (68,876) | |
| Adjustments to reconcile net income (loss) to net cash used in operating activities: | | |
| Cash flows from operating activities | |
| Cash flows from operating activities | | | | | (Amounts in thousands) |
| Net (loss) income | |
| Adjustments to reconcile net (loss) income to net cash used in operating activities: | |
| Depreciation and amortization | |
| Depreciation and amortization | |
| Depreciation and amortization | Depreciation and amortization | | 1,287 | | | 1,252 | | | 734 | |
| Amortization of premiums and discounts on marketable securities | Amortization of premiums and discounts on marketable securities | | (375) | | | 57 | | | — | |
| Non-cash lease expense | Non-cash lease expense | | 1,337 | | | 1,119 | | | 1,127 | |
| (Gain) on disposal of property and equipment | | (147) | | | — | | | (52) | |
| Impairment of Right of use asset | |
| Loss (gain) on disposal of property and equipment | |
| Stock-based compensation expense | Stock-based compensation expense | | 11,194 | | | 7,720 | | | 5,422 | |
| Non-cash interest expense | Non-cash interest expense | | 953 | | | 1,012 | | | 620 | |
| Warrant liabilities revaluation | Warrant liabilities revaluation | | (20,882) | | | 2,339 | | | 10,443 | |
| Contingent value right liability revaluation | |
| Forward contract liabilities revaluation | |
| Loss on extinguishment of debt | Loss on extinguishment of debt | | — | | | — | | | 461 | |
| Provision (benefit) for deferred taxes | |
| | Changes in operating assets and liabilities: | Changes in operating assets and liabilities: | | | | | |
| Changes in operating assets and liabilities: | |
| Changes in operating assets and liabilities: | |
| Accounts receivable | |
| Accounts receivable | |
| Accounts receivable | Accounts receivable | | 3,318 | | | (2,690) | | | (2,224) | |
| Unbilled receivable | Unbilled receivable | | (3,162) | | | — | | | — | |
| Prepaid expenses, deposits and other assets | Prepaid expenses, deposits and other assets | | 2,471 | | | (1,451) | | | (4,418) | |
| Accounts payable | Accounts payable | | 92 | | | (219) | | | (57) | |
| Income taxes payable | Income taxes payable | | (601) | | | 601 | | | — | |
| Deferred revenue | Deferred revenue | | (64,707) | | | (45,496) | | | 94,462 | |
| | Accrued expenses and other liabilities | Accrued expenses and other liabilities | | 2,212 | | | 1,061 | | | (2,761) | |
| Net cash (used in) provided by operating activities | | (31,631) | | | (60,382) | | | 34,881 | |
| Accrued expenses and other liabilities | |
| Accrued expenses and other liabilities | |
| Net cash used in operating activities | |
| Cash flows from investing activities | Cash flows from investing activities | | | | | | |
| Cash assumed in acquisition of Old Cartesian | |
| Cash assumed in acquisition of Old Cartesian | |
| Cash assumed in acquisition of Old Cartesian | |
| Proceeds from maturities of marketable securities | Proceeds from maturities of marketable securities | | 19,700 | | | 16,400 | | | — | |
| Payment made for investments | Payment made for investments | | — | | | (2,000) | | | — | |
| Purchases of marketable securities | Purchases of marketable securities | | (33,501) | | | (30,455) | | | — | |
| | Purchases of property and equipment | Purchases of property and equipment | | (1,201) | | | (1,085) | | | (815) | |
| Proceeds from the sale of property and equipment | | — | | | — | | | 74 | |
| Purchases of property and equipment | |
| Purchases of property and equipment | |
| | Net cash used in investing activities | | (15,002) | | | (17,140) | | | (741) | |
| | Net cash provided by (used in) investing activities | |
| | Net cash provided by (used in) investing activities | |
| | Net cash provided by (used in) investing activities | |
| Cash flows from financing activities | Cash flows from financing activities | | | | | | |
| Proceeds from issuance of long-term debt, net of expenses | | — | | | — | | | 24,736 | |
| Repayments of principal on outstanding debt | | — | | | — | | | (19,313) | |
| | Proceeds from issuance of Series A Preferred Stock, gross in private placement | |
| | Proceeds from issuance of Series A Preferred Stock, gross in private placement | |
| | Proceeds from issuance of Series A Preferred Stock, gross in private placement | |
| Repayments of principal, final payment fee, and prepayment penalty on debt | |
| | Debt amendment fee included in debt discount | Debt amendment fee included in debt discount | | (110) | | | — | | | — | |
| | | Debt amendment fee included in debt discount | |
| | Debt amendment fee included in debt discount | |
| | Net proceeds from issuance of common stock- at-the-market offering | |
| Net proceeds from issuance of common stock- at-the-market offering | |
| Net proceeds from issuance of common stock- at-the-market offering | Net proceeds from issuance of common stock- at-the-market offering | | 2,121 | | | 51,958 | | | 2,108 | |
| Net proceeds from issuance of common stock and common warrants | Net proceeds from issuance of common stock and common warrants | | 36,859 | | | — | | | — | |
| Net proceeds from issuance of common stock- private placement | | — | | | — | | | 10,268 | |
| Issuance costs paid for December 2019 financing | | — | | | — | | | (4,381) | |
| Other financing fees | | — | | | — | | | (343) | |
| Proceeds from exercise of pre-funded and common warrants | | — | | | — | | | 979 | |
| | Settlement of outstanding equity awards at Merger | |
| | Settlement of outstanding equity awards at Merger | |
| | Settlement of outstanding equity awards at Merger | |
| Proceeds from exercise of stock options | Proceeds from exercise of stock options | | 156 | | | 778 | | | 193 | |
| Proceeds from issuance of common stock under Employee Stock Purchase Plan | Proceeds from issuance of common stock under Employee Stock Purchase Plan | | 189 | | | 161 | | | 184 | |
| Net cash provided by financing activities | | 39,215 | | | 52,897 | | | 14,431 | |
| Net cash (used in) provided by financing activities | |
| Effect of exchange rate changes on cash | Effect of exchange rate changes on cash | | 20 | | | (3) | | | (58) | |
| Net change in cash, cash equivalents, and restricted cash | Net change in cash, cash equivalents, and restricted cash | | (7,398) | | | (24,628) | | | 48,513 | |
| Cash, cash equivalents, and restricted cash at beginning of period | Cash, cash equivalents, and restricted cash at beginning of period | | 115,436 | | | 140,064 | | | 91,551 | |
| Cash, cash equivalents, and restricted cash at end of period | Cash, cash equivalents, and restricted cash at end of period | | $ | 108,038 | | | $ | 115,436 | | | $ | 140,064 | |
| Supplement cash flow information | Supplement cash flow information | | | | | | |
| Cash paid for interest | Cash paid for interest | | $ | 2,248 | | | $ | 2,002 | | | $ | 1,018 | |
| Noncash investing and financing activities | | | | | |
| Cash paid for interest | |
| Cash paid for interest | |
| Non-cash investing and financing activities | |
| Issuance of common stock, license agreement in stock-based compensation expense | |
| Issuance of common stock, license agreement in stock-based compensation expense | |
| Issuance of common stock, license agreement in stock-based compensation expense | Issuance of common stock, license agreement in stock-based compensation expense | | $ | 1,000 | | | $ | — | | | $ | — | |
| Cashless warrant exercise | Cashless warrant exercise | | $ | — | | | $ | 5,624 | | | $ | 21,790 | |
| Reclassification of warrant liability to equity | Reclassification of warrant liability to equity | | $ | 780 | | | $ | — | | | $ | 1,494 | |
| Fair value of warrants issued in connection with issuance of long-term debt | | $ | — | | | $ | — | | | $ | 444 | |
| Purchase of property and equipment not yet paid | Purchase of property and equipment not yet paid | | $ | 17 | | | $ | 224 | | | $ | 4 | |
|
The accompanying notes are an integral part of these consolidated financial statements.
Selecta Biosciences,Cartesian Therapeutics, Inc. and Subsidiaries
Notes to Consolidated Financial Statements
1. Description of the Business
Cartesian Therapeutics, Inc., or the Company, (formerly known as Selecta Biosciences, Inc., or the Company, Selecta) was incorporated in Delaware on December 10, 2007, and is basedheadquartered in Watertown, Massachusetts.Gaithersburg, Maryland. The Company is a clinical-stage biotechnology company developing mRNA cell therapies for the treatment of autoimmune diseases leveraging the Company's ImmTOR®its proprietary technology and manufacturing platform to develop toloreogenic therapies that selectively mitigate unwanted immune responses. With a proven abilityintroduce one or more mRNA molecules into cells to induce tolerance to highly immunogenic proteins, theenhance their function. The Company believes ImmTOR hasits mRNA cell therapies have the potential to amplify the efficacydeliver deep, durable clinical benefit to a broad group of biologic therapies, including redosingpatients with autoimmune diseases because they can be administered over a short period of life-saving gene therapies, as well as restore the body's natural self-tolerancetime, in autoimmune diseases. The Company has several proprietaryan outpatient setting, and partnered programs in its pipeline focused on enzyme therapies, gene therapies, and autoimmune diseases.without pre-treatment chemotherapy.
Since inception,On November 13, 2023, the Company has devoted its efforts principallyacquired, in accordance with the terms of the Agreement and Plan of Merger, or the Merger Agreement, the assets of the Delaware corporation which, immediately prior to researchthe Merger (as defined below), was known as Cartesian Therapeutics, Inc., or Old Cartesian, as disclosed in Note 3. The transaction was structured as a stock-for-stock transaction pursuant to which all of Old Cartesian’s outstanding shares of capital stock were exchanged based on a fixed exchange ratio for consideration of 6,723,639 shares of the common stock, $0.0001 per share, of the Company and development384,930.724 shares of its technologythe newly designated Series A Non-Voting Convertible Preferred Stock, $0.0001 per share, or the Series A Preferred Stock. The Series A Preferred Stock is intended to have economic rights similar to the common stock, but with only limited voting rights. Additionally, the Company assumed all outstanding stock options of Old Cartesian. The common stock and product candidates, recruiting managementSeries A Preferred Stock related to the Merger were issued on December 5, 2023. For additional information, see Note 3.
In connection with the Merger, the Company entered into a definitive agreement, or the Securities Purchase Agreement, for a private investment in public equity transaction, or the November 2023 Private Placement, with the Investors (as defined below). The Securities Purchase Agreement provides for the issuance to the Investors of an aggregate of 149,330.115 shares of Series A Preferred Stock for an aggregate purchase price of approximately $60.25 million. For additional information, see Note 11.
In connection with the Merger, a contractual contingent value right, or CVR, was distributed to the holders of record of the Company's common stock and technical staff, acquiring operating2022 Warrants as of the close of business on December 4, 2023, but was not distributed to holders of shares of common stock or Series A Preferred Stock issued to stockholders of Old Cartesian or the Investors in the transactions. Holders of the CVRs will be entitled to receive certain payments from proceeds received by the Company, if any, related to the disposition or monetization of the Company's legacy assets and raising capital. following the issuance of the CVRs. For additional information, see Note 6.
The Company is subject to risks common to companies in the biotechnology industry including, but not limited to, new technological innovations, protection of proprietary technology, dependence on key personnel, compliance with government regulations and the need to obtain additional financing. Product candidates currently under development will require significant additional research and development efforts, including extensive preclinical and clinical testing and regulatory approval, prior to commercialization. These efforts require significant amounts of additional capital, adequate personnel infrastructure and extensive compliance-reporting capabilities.
The Company’s product candidates are in pre-clinical and clinical development. There can be no assurance that the Company’s research and development will be successfully completed, that adequate protection for the Company’s intellectual property will be obtained, or maintained, that any products developed will obtain necessary government regulatory approval or that any approved products will be commercially viable. Even if the Company’s product development efforts are successful, it is uncertain when, if ever, the Company will generate significant revenue from product sales. The Company operates in an environment of rapid change in technology and substantial competition from pharmaceutical and biotechnology companies. In addition, the Company is dependent upon the services of its employees and consultants.
Liquidity and Management’s Plan
The future success of the Company is dependent on its ability to develop its product candidates and ultimately upon its ability to attain and sustain profitable operations. The Company is subject to a number of risks similar to other early-stage life science companies, including, but not limited to, successful development of its product candidates, raising additional capital with favorable terms, protection of proprietary technology and market acceptance of any approved future products. The successful development of product candidates requires substantial working capital, which may not be available to the Company on favorable terms or at all.
To date, the Company has financed its operations primarily through public offerings and private placements of its securities, funding received from research grants, collaboration and license arrangements and itsa credit facility. The Company currently has no source of product revenue, and it does not expect to generate product revenue for the foreseeable future. To
date, the Company’s revenue has primarily been from collaboration agreements. The Company has devoted substantially all of its financial resources and efforts to developing its ImmTOR platform,existing product candidates, identifying potential product candidates and conducting preclinical studies and clinical trials. The Company is in the early stages of development of its product candidates, and it has not completed development of any ImmTOR-enabled therapies.product candidates.
As of December 31, 2022,2023, the Company’s cash, cash equivalents, and restricted cash and marketable securities were $136.2$78.3 million, of which $1.6$1.4 million was restricted cash related to lease commitments and $0.2 million was held by its Russian subsidiary designated solely for use in its operations. The Company believes the cash, cash equivalents and restricted cash and marketable securities as of December 31, 20222023 combined with net proceeds of $40.0 million received subsequent to December 31, 2023 from the November 2023 Private Placement will enable it to fund its current planned operations for at least the next twelve months from the date of issuance of these financial statements, though it may realize additional cash resources upon the achievement of certain contingent collaboration milestones or it may pursue additional cash resources through public or private equity or debt financings or by establishing collaborations with other companies. Management’s expectations with respect to its ability to fund current and long term planned operations are based on estimates that are subject to risks and uncertainties. If actual results are different from management’s estimates, the Company may need to seek additional strategic or financing opportunities sooner than would otherwise be expected. However, there is no guarantee that any collaboration milestones will be achieved or that any of these strategic or financing opportunities will be executed on favorable terms, and some could be dilutive to existing stockholders. Further, the liability associated with the CVR Agreement (as defined below) will be settled solely through cash flow received under the Company's License and Development Agreement, or as so amended, the Sobi License, with Swedish Orphan Biovitrum AB (publ.), or Sobi, and any other Gross Proceeds (as defined in the CVR Agreement) net of certain agreed deductions. Under the CVR Agreement, 100% of all milestone payments, royalties and other amounts paid to the Company or controlled entities under the Sobi License, and any other Gross Proceeds will be distributed, net of specified deductions, to holders of the CVRs. There is no obligation to the Company to fund any amount related to the CVR liability. See Note 6.
The Certificate of Designation of Preferences, Rights, and Limitations of the Series A Non-Voting Convertible Preferred Stock, or the Certificate of Designation, contains a provision granting each holder of the Series A Preferred Stock the option to require the Company to redeem any or all of such holder’s then-outstanding shares of Series A Preferred Stock beginning on the date that is 18 months following the date of the closing of the Merger, November 13, 2023, at a price per share equal to the ten-day trailing average closing trading price of the common stock at such time; provided, however, that no holder will have the right to seek redemption of any shares of Series A Preferred Stock to the extent that such holder would otherwise be unable to convert such shares of Series A Preferred Stock due to the common stock beneficial ownership limitation applicable to such holder. The Company could be required to use a significant amount of its cash resources on hand to satisfy this redemption obligation, particularly if its stockholders do not ever approve a proposal to convert the Company’s Series A Preferred Stock into common stock, or generally if holders of Series A Preferred Stock exercise their redemption right with respect to a significant number of shares of Series A Preferred Stock or at a time when the trading price of the Company’s common stock is elevated. Further, in the event that the Company does not have sufficient cash on hand to satisfy its redemption obligations, the Company may need to raise additional capital to satisfy these potential obligations. Any redemption payments could materially limit the amount of cash the Company has available to fund our operations and the potential need to redeem shares of Series A Preferred Stock may limit the flexibility with which the Company seeks to operate its business.
If the Company is unable to obtain additional funding on a timely basis, it may be forced to significantly curtail, delay, or discontinue one or more of its planned research or development programs or be unable to expand its operations or otherwise capitalize on its commercialization of its product candidates. As of December 31, 2022,2023, the Company had an accumulated deficit of $394.9$614.6 million. The Company anticipates operating losses to continue for the
foreseeable future due to, among other things, costs related to research and development of its product candidates and its administrative organization.
At this time, any impact of COVID-19 on the Company’s business, revenues, results of operations and financial condition will largely depend on future developments, which are highly uncertain and cannot be predicted with confidence, such as the duration of the pandemic, the emergence of new virus variants, travel restrictions and social distancing in the United States and other countries, business closures, disruptions, mandated stay at home orders or lockdowns, supply chain disruptions, the ultimate impact on financial markets and the global economy, and the effectiveness of actions taken in the United States and other countries to contain and treat the disease.
Guarantees and Indemnifications
As permitted under Delaware law, the Company indemnifies its officers, directors, consultants and employees for certain events or occurrences that happen by reason of the relationship with, or position held at, the Company. Through December 31, 2022,2023, the Company had not experienced any losses related to these indemnification obligations, and no claims were outstanding. The Company does not expect significant claims related to these indemnification obligations and, consequently, concluded that the fair value of these obligations is negligible, and no related reserves were established.
2. Summary of Significant Accounting Policies
Principles of Consolidation
The consolidated financial statements include the accounts of the Company and its wholly owned subsidiaries, Selecta (RUS), LLC, or Selecta (RUS), a Russian limited liability corporation, and Selecta Biosciences Security Corporation, a Massachusetts securities corporation.corporation, and Cartesian Bio, LLC, a Delaware limited liability company, which is a variable interest entity for which the Company is the primary beneficiary. All significant intercompany accounts and transactions have been eliminated.
Use of Estimates
The preparation of consolidated financial statements in conformity with U.S. GAAP requires the Company’s management to make estimates and assumptions that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities as of the date of the consolidated financial statements and the reported amounts of revenues and expenses during the reporting period. The Company’s management considers many factors in selecting appropriate financial accounting policies and controls, and bases its estimates on historical experience and other market-specific or other relevant assumptions that it believes to be reasonable under the circumstances. In preparing these consolidated financial statements, management used significant estimates in the following areas, among others: estimated fair value of the intangible assets acquired in connection with the Merger, estimated fair value of the CVRs, deferred income taxes, revenue recognition and estimating accrued research and development expenses. The Company assesses the above estimates on an ongoing basis; however, actual results could materially differ from those estimates.
Segment Information
The Company views its operations and manages its business in one operating segment, which prior to the Merger related to the research and development of nanoparticle immunomodulatory drugs for the treatment and prevention of human diseases.diseases and subsequent to the Merger relates to the research and development of cell therapy product candidates.
Cash Equivalents, Restricted Cash, Marketable Securities and Investments
Cash equivalents include all highly liquid investments maturing within 90 days from the date of purchase. Marketable securities consist of securities with remaining maturities greater than 90 days when purchased. The Company classifies these marketable securities as available-for-sale and records them at fair value in the accompanying consolidated balance sheets. Marketable securities with less than one year until maturity are classified as short term, while marketable securities with maturities greater than one year are classified as long term. Unrealized gains or losses are included in accumulated other comprehensive income (loss). Premiums or discounts from par value are amortized to investment income over the life of the underlying investment. Although available to be sold to meet operating needs or otherwise, securities are generally held through maturity. The cost of securities sold is determined based on the specific identification method for purposes of recording realized gains and losses.
WeThe Company has also investin the past invested in equity securities of companiesa company whose securities are not publicly traded and where fair value is not readily available. These investments areThis investment is recorded using cost minus impairment adjusted for changes in observable prices, depending on our ownership percentage and other factors that suggest we have significant influence. We monitor these investmentsThe Company monitors this investment to evaluate whether any increase or decline in theirits value has occurred, based on the implied value of recent company financings, public market prices of comparable companies and general market conditions. These investments areThis investment is included in investments in ourthe consolidated balance sheets.
Concentrations of Credit Risk and Off-Balance Sheet Risk
Financial instruments that potentially subject the Company to concentration of credit risk consist primarily of cash, cash equivalents, short-term deposits and marketable securities, investments, and accounts receivable. Cash and cash equivalents are
deposited with federally insured financial institutions in the United States and may, at times, exceed federally insured limits. Management believes that the financial institutions that hold the Company’s deposits are financially creditworthy and, accordingly, minimal risk exists with respect to those balances. The Company also maintains cash in Russian bank accounts in denominations of both Russian rubles and U.S. dollars. As of December 31, 2022,2023, the Company maintained approximately $0.2 million in Russian bank accounts in denominations of both Russian rubles and U.S. dollars.
Fair Value of Financial Instruments
The Company’s financial instruments consist mainly of cash equivalents, restricted cash, accounts payable, loans payable, marketable securities, investments, warrants to purchase common stock, forward contract liabilities, and common warrants.contingent value rights. The carrying amounts of cash equivalents, restricted cash, accounts receivable, and accounts payable approximate their estimated fair value due to their short-term maturities.
Accounting standards define fair value as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. A three-level hierarchy is used to prioritize the inputs to valuation techniques used to measure fair value. The hierarchy gives the highest priority to unadjusted quoted prices in active markets for identical assets or liabilities (Level 1 measurements), and the lowest priority to unobservable inputs (Level 3 measurements). The three levels of the fair value hierarchy are described below:
Level 1—Level 1 inputs are quoted prices in active markets for identical assets or liabilities that the reporting entity has the ability to access at the measurement date.
Level 2—Level 2 inputs are inputs other than quoted prices included within Level 1 that are observable for the asset or liability, either directly or indirectly. If the asset or liability has a specified (contractual) term, a Level 2 input must be observable for substantially the full term of the asset or liability.
Level 3—Level 3 inputs are unobservable inputs for the asset or liability in which there is little, if any, market activity for the asset or liability at the measurement date.
To the extent that a valuation is based on models or inputs that are less observable or unobservable in the market, the determination of fair value requires more judgment. Accordingly, the degree of judgment exercised by the Company in determining fair value is greatest for instruments categorized in Level 3. A financial instrument’s level within the fair value hierarchy is based on the lowest level of any input that is significant to the fair value measurement. The fair value of warrant liabilities isand contingent value rights are determined using Level 3 inputs.
Fair value is a market-based measure considered from the perspective of a market participant rather than an entity-specific measure. Therefore, even when market assumptions are not readily available, the Company’s own assumptions are set to reflect those that market participants would use in pricing the asset or liability at the measurement date. The Company uses prices and inputs that are current as of the measurement date, including during periods of market dislocation. In periods of market dislocation, the observability of prices and inputs may change for many instruments. This condition could cause an instrument to be reclassified within levels in the fair value hierarchy.
The carrying amounts reflected in ourthe consolidated balance sheet for investments approximate fair value and are assessed for impairment quarterly.
Property and Equipment
Property and equipment are recorded at cost and depreciated using the straight-line method over the estimated useful lives of the respective assets, generally seven years for furniture and fixtures, five years for laboratory equipment, software and office equipment and three years for computer equipment. Leasehold improvements are amortized over their useful life or the life of the lease, whichever is shorter. Major additions and betterments are capitalized. Maintenance and repairs, which do not improve or extend the life of the respective assets, are charged to operations as incurred. Costs incurred for construction in progress are recorded as assets and are not amortized until the construction is substantially complete and the assets are ready for their intended use.
Impairment of Long-Lived Assets
The Company reviews long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying value of an asset may not be recoverable. In order to determine if assets have been impaired, assets are tested at the lowest level for which identifiable independent cash flows are available, which is at the entity level (“asset group”).available. An impairment loss is recognized when the sum of projected undiscounted cash flows is less than the carrying value of the asset group. The measurement of the impairment loss to be recognized is based on the difference between the fair value and the carrying value of the asset group. BasedThe Company recognized a $0.7 million impairment charge on management’s evaluation,a right-of-use asset during the fair value of the asset group, measured as the market capitalization of the Company exceeds its carrying value.year ended December 31, 2023.
Debt Issuance Costs
Debt issuance costs and fees paid to lenders are recorded as a direct deduction from the face amount of the related debt. Debt issuance costs are amortized over the term of the related debt using the effective interest method and recorded as interest expense.
Accumulated Other Comprehensive Income (Loss)
Comprehensive income (loss) is defined as the change in the equity of a business entity during a period from transactions and other events and circumstances from non-owner sources. It includes all changes in equity during a period except those resulting from investments by owners and distributions to owners. Comprehensive income (loss) consists of: (i) all components of net income (loss) and (ii) all components of comprehensive income (loss) other than net income (loss), referred to as other comprehensive income (loss). Other comprehensive income (loss) is comprised of unrealized gains and losses on debt securities and foreign currency translation adjustments.
Revenue Recognition
Revenue is recognized when a customer obtains control of promised goods or services, in an amount that reflects the consideration which the entity expects to receive in exchange for those goods or services. Pursuant to ASC Topic 606, Revenue from Contracts with Customers (ASC 606), a customer is a party that has contracted with an entity to obtain goods or services that are an output of the entity’s ordinary activities in exchange for consideration. To determine revenue recognition for arrangements that an entity determines are within the scope of ASC 606, the Company performs the following five steps: (i) identify the contract(s) with a customer; (ii) identify the performance obligations in the contract, including whether they are distinct in the context of the contract; (iii) determine the transaction price, including the constraint on variable consideration; (iv) allocate the transaction price to the performance obligations in the contract; and (v) recognize revenue when (or as) the
Company satisfies each performance obligation. The Company only applies the five-step model to contracts when it is probable that the entity will collect the consideration it is entitled to in exchange for the goods or services it transfers to the customer. At contract inception, once the contract is determined to be within the scope of ASC 606, the Company assesses the goods or services promised within each contract and determines those that are performance obligations, and assesses whether each promised good or service is distinct. If a promised good or service is not distinct, it is combined with other promised goods or services into a performance obligation. The Company then recognizes as revenue the amount of the transaction price that is allocated to the respective performance obligation when (or as) the performance obligation is satisfied. For example, certain performance obligations associated with Swedish Orphan Biovitrum,the License and Development Agreement, or Sobi, SareptaAstellas Agreement, entered into with Audentes Therapeutics, Inc., or Sarepta, and Takeda Pharmaceuticals USA, Inc., or Takeda,Astellas, (see Note 12)14) will be satisfied over time, and revenue will be recognized using the output method, based on the proportion of actual deliveries to the total expected deliveries over the initial term.input method.
Collaboration and License Revenue: The Company currently generates its revenue through collaboration and license agreements with strategic collaborators for the development and commercialization of product candidates. Collaboration and license agreements with customers are generally accounted for in accordance with ASC 606. The Company analyzes collaboration arrangements by first assessing whether they are within the scope of ASC Topic 808, Collaborative Arrangements (ASC 808), and evaluates whether such arrangements involve joint operating activities performed by parties that are both active participants in the activities and exposed to significant risks and rewards that are dependent on the commercial success of such activities. Collaboration agreements with customers that are not within the scope of ASC 808 are accounted for in accordance with ASC 606. To the extent the collaboration agreement is within the scope of ASC 808, the Company also assesses whether any aspects of the agreement are within the scope of other accounting literature (specifically ASC 606). If the Company concludes that some or all aspects of the agreement are distinct and represent a transaction with a customer, the Company accounts for those aspects of the arrangement within the scope of ASC 606. The Company recognizes the shared costs incurred that are not within the scope of other accounting literature as a component of the related expense in the period incurred by analogy to ASC Topic 730, Research and Development (ASC 730), and records reimbursements from counterparties as an offset to the related research and development costs. In determining the appropriate amount of revenue to be recognized as it fulfills its obligations under the agreements in accordance with ASC 606, the Company performs the five steps above. As part of the accounting for the arrangement, the Company must develop assumptions that require judgment to determine the stand-alone selling price for each performance obligation identified in the contract. The Company uses key assumptions to determine the stand-alone selling price, which may include market conditions, reimbursement rates for personnel costs, development timelines and probabilities of regulatory success.
The terms of the Company’s arrangements typically include one or more of the following: (i) upfront fees; (ii) milestone payments related to the achievement of development, regulatory, or commercial goals; (iii) royalties on net sales of licensed products; (iv) reimbursements or cost-sharing of research and development (R&D) expenses; and (v) profit/loss sharing arising from co-promotion arrangements.
Licenses of Intellectual Property: If the license to the Company’s intellectual property is determined to be distinct from the other promised goods and services identified in the arrangement, the Company recognizes revenues from non-refundable, upfront fees allocated to the license when the license is transferred to the customer and the customer is able to use and benefit from the license. If not distinct, the license is combined with other promised goods and services in the contract. For licenses that are combined with other promised goods and services, the Company assesses the nature of the combined performance
obligation to determine whether the combined performance obligation is satisfied over time or at a point in time and, if over time, the appropriate method of measuring progress for purposes of recognizing revenue. The Company evaluates the measure of progress each reporting period and, if necessary, adjusts the measure of performance and related revenue recognition. Optional licenses are evaluated to determine if they are issued at a discount, and therefore, represent material rights and accounted for as separate performance obligations.
Milestone Payments: At the inception of each arrangement that includes developmental and regulatory milestone payments, the Company evaluates whether the achievement of each milestone specifically relates to the Company’s efforts to satisfy a performance obligation or transfer a distinct good or service within a performance obligation. If the achievement of a milestone is considered a direct result of the Company’s efforts to satisfy a performance obligation or transfer a distinct good or service and the receipt of the payment is based upon the achievement of the milestone, the associated milestone value is allocated to that distinct good or service. If the milestone payment is not specifically related to the Company’s effort to satisfy a performance obligation or transfer a distinct good or service, the amount is allocated to all performance obligations using the relative standalone selling price method. The Company also evaluates the milestone to determine whether they are considered probable of being reached and estimates the amount to be included in the transaction price using the most likely amount method. If it is probable that a significant revenue reversal would not occur, the associated milestone value is included in the transaction price to be allocated, otherwise, such amounts are constrained and excluded from the transaction price. At the end of each subsequent reporting period, the Company re-evaluates the probability of achievement of such development milestones and any related constraint, and if necessary, adjusts its estimate of the transaction price. Any such adjustments to the transaction price are allocated to the performance obligations on the same basis as at contract inception. Amounts allocated to a satisfied
performance obligation shall be recognized as revenue, or as a reduction of revenue, in the period in which the transaction price changes.
Manufacturing Supply Services: Arrangements that include a promise for future supply of drug substance or drug product for either clinical development or commercial supply at the customer’s discretion are evaluated to determine if they are distinct and optional. For optional services that are distinct, the Company assesses if they are priced at a discount, and therefore, provide a material right to the licensee to be accounted for as separate performance obligations.
Royalties: For arrangements that include sales-based royalties, including milestone payments based on the level of sales, and the license is deemed to be the predominant item to which the royalties relate, the Company will recognize revenue at the later of (i) when the related sales occur, or (ii) when the performance obligation to which some or all of the royalty has been allocated has been satisfied (or partially satisfied) in accordance with the royalty recognition constraint.
Research and Development Costs
Costs incurred in the research and development of the Company’s products are expensed as incurred. Research and development expenses include costs incurred in performing research and development activities, including salaries and benefits, stock-based compensation expenses, facilities cost, overhead costs, contract services, supplies and other outside costs. Nonrefundable advance payments for goods and services that will be used in future research and development activities are expensed when the activity has been performed or when the goods have been received rather than when the payment is made.
Clinical Trial Costs
Clinical trial expenses are a significant component of research and development expenses, and the Company outsources a significant portion of these costs to third parties. Third party clinical trial expenses include patient costs, clinical research organization costs and costs for data management. The accrual for site and patient costs includes inputs such as estimates of patient enrollment, patient cycles incurred, clinical site activations, and other pass-through costs. Payments for these activities are based on the terms of the individual arrangements, which may differ from the pattern of costs incurred, and are reflected on the consolidated balance sheets as a prepaid asset or accrued clinical trial cost. These third party agreements are generally cancellable, and related costs are recorded as research and development expenses as incurred. Non-refundable advance clinical payments for goods or services that will be used or rendered for future R&Dresearch and development activities are recorded as a prepaid asset and recognized as expense as the related goods are delivered or the related services are performed. The Company also records accruals for estimated ongoing clinical research and development costs. When evaluating the adequacy of the accrued liabilities, the Company analyzes progress of the studies, including the phase or completion of events, invoices received and contracted costs. Significant judgments and estimates may be made in determining the accrued balances at the end of any reporting period. Actual results could differ from the estimates made by the Company. The historical clinical accrual estimates made by the Company have not been materially different from the actual costs.
In June 2020, the Company and Sobi entered into a licenseLicense and development agreement, or the Sobi License.Development Agreement, which was amended in October 2023. Pursuant to the Sobi License, clinical trial costs incurred to complete development of the SEL-212 product candidate, including but not limited to costs incurred while conducting and completing the Phase 3 DISSOLVE trials, will bewere reimbursed by Sobi. These costs, when reimbursed, were recognized as revenue consistent with the revenue recognition methodology disclosed in Note 14. The reimbursable costs exclude any costs of additional development activities required that are related to the ImmTOR platform and that are unrelated to SEL-212.
In January 2023, the Company and Astellas entered into the Astellas Agreement. Pursuant to the Astellas Agreement, Astellas will reimburse the Company for 25% of all budgeted costs incurred to complete the development of Xork for use in Pompe disease with an Astellas gene therapy investigational or authorized product. These costs, when reimbursed, will be recognized as revenue consistent with the revenue recognition methodology disclosed in Note 12. The reimbursable costs exclude any costs of additional development activities required that are related to ImmTOR and that are unrelated to SEL-212.
Income Taxes
The Company provides deferred tax assets and liabilities for the expected future tax consequences of temporary differences between the Company’s financial statement carrying amounts and the tax basis of assets and liabilities using enacted tax rates expected to be in effect in the years in which the differences are expected to reverse. A valuation allowance is provided to reduce the deferred tax assets to the amount that will more-likely-than-not be realized.
The Company determines whether it is more likely than not that a tax position will be sustained upon examination. If it is not more-likely-than-not that a position will be sustained, none of the benefit attributable to the position is recognized. The tax benefit to be recognized for any tax position that meets the more-likely-than-not recognition threshold is calculated as the largest amount that is more than 50% likely of being realized upon resolution of the contingency. The Company accounts for interest and penalties related to uncertain tax positions as part of its provision for income taxes. To date, the Company has not incurred interest and penalties related to uncertain tax positions.
Warrants
The Company determines the accounting classification of warrants that are issued, as either liability or equity, by first assessing whether the warrants meet liability classification in accordance with ASC 480-10, Accounting for Certain Financial Instruments with Characteristics of both Liabilities and Equity, and then in accordance with ASC 815-40, Accounting for Derivative Financial Instruments Indexed to, and Potentially Settled in, a Company’s Own Stock. Under ASC 480, warrants are considered liability classified if the warrants are mandatorily redeemable, obligate the issuer to settle the warrants or the underlying shares by paying cash or other assets, or must or may require settlement by issuing variable number of shares.
If warrants do not meet liability classification under ASC 480-10, the Company assesses the requirements under ASC 815-40, which states that contracts that require or may require the issuer to settle the contract for cash are liabilities recorded at fair value, irrespective of the likelihood of the transaction occurring that triggers the net cash settlement feature. If the warrants do not require liability classification under ASC 815-40, in order to conclude equity classification, the Company assesses whether the warrants are indexed to its common stock and whether the warrants are classified as equity under ASC 815-40 or other applicable GAAP. After all relevant assessments are made, the Company concludes whether the warrants are classified as liability or equity. Liability classified warrants are required to be accounted for at fair value both on the date of issuance and on subsequent accounting period ending dates, with all changes in fair value after the issuance date recorded in the statements of operations as a gain or loss. Equity classified warrants are accounted for at fair value on the issuance date with no changes in fair value recognized after the issuance date.
Stock-Based Compensation
The Company accounts for all stock-based compensation granted to employees and non-employees using a fair value method. Stock-based compensation is measured at the grant date fair value and is recognized over the requisite service period of the awards, usually the vesting period, on a straight-line basis, net of estimated forfeitures. To the extent that actual forfeitures differ from the Company’s estimates, the differences are recorded as a cumulative adjustment in the period the estimates were adjusted. Stock-based compensation expense recognized in the consolidated financial statements is based on awards that ultimately vest.
Net (Loss) Income (Loss) Per Share
The Company calculatesapplies the two-class method to compute basic and diluted net (loss) income (loss) per share attributable to common stockholders when it has issued shares that meet the definition of participating securities. The two-class method determines net (loss) income per share for each class of common and participating securities according to dividends declared or accumulated and participation rights in undistributed earnings. The two-class method requires (loss) income available to common stockholders for the period to be allocated between common and participating securities based upon their respective rights to share in the earnings as if all (loss) income for the period had been distributed. The Company's Series A Preferred Stock and 2022 Warrants participate in any dividends declared by the Company and are therefore considered to be participating securities. The participating securities are not required to participate in the losses of the Company, and therefore during periods of loss there is no allocation required under the two-class method.
Basic net (loss) income per share attributable to common stockholders is computed by dividing the net (loss) income (loss) attributable to common stockholders by the weighted average number of shares of common sharesstock outstanding duringfor the period. Diluted net (loss) income attributable to common stockholders is computed by adjusting net (loss) income per share attributable to common stockholders to reallocate undistributed earnings based on the potential impact of dilutive securities. Diluted net (loss) income per share attributable to common stockholders is calculatedcomputed by dividing the diluted net (loss) income attributable to common stockholders by the weighted-averageweighted average number of shares of common equivalent sharesstock outstanding for the period, including anypotential dilutive effect fromcommon shares. For purpose of this calculation, outstanding options to purchase common stock options,and Series A Preferred Stock, forward contracts to issue Series A Preferred Stock, restricted stock units, warrants to purchase common stock, and employee stock purchase plan stock, using the treasury stock method.contingently issuable shares, and Series A Preferred Stock are considered potential dilutive common shares.
Contingent Liabilities
The Company accounts for its contingent liabilities in accordance with ASC No. 450, Contingencies. A provision is recorded when it is both probable that a liability has been incurred and the amount of the loss can be reasonably estimated. With respect to legal matters, provisions are reviewed and adjusted to reflect the impact of negotiations, estimated settlements, legal rulings, advice of legal counsel and other information and events pertaining to a particular matter.
Leases
The Company accounts for its leases in accordance with ASC Topic 842, Leases (ASC 842), and determines whether the arrangement is or contains a lease based on the unique facts and circumstances present. Most leases with a term greater than one year are recognized on the balance sheet as right-of-use assets, lease liabilities and, if applicable, long-term lease liabilities. The Company elected not to recognize leases with an original term less than one year on its balance sheet. Operating lease right-of-use (ROU) assets and their corresponding lease liabilities are recorded based on the present value of lease payments over the expected remaining lease term. The interest rate implicit in lease contracts is typically not readily determinable. As a result, the Company
Company utilizes its incremental borrowing rates, which are the rates incurred to borrow, on a collateralized basis over a similar term, an amount equal to the lease payments in a similar economic environment.
In accordance with the guidance in ASC 842, the fixed and in-substance fixed contract consideration must be allocated to lease and non-lease components based on their relative fair values. Non-components of a contract (e.g., administrative tasks that do not transfer a good or service to the Company, reimbursement or payment of a lessor’s cost, etc.) do not receive an allocation of the consideration in the contract. Although allocation of consideration of lease and non-lease components is required, the Company elected the practical expedient to not separate lease components (e.g. land, building, etc.) and non-lease components (e.g., common area maintenance, consumables, etc.). The lease component results in an operating right-of-use asset being recorded on the balance sheet and amortized on a straight-line basis as lease expense. Right-of-use assets and operating lease liabilities are remeasured upon certain modifications to leases using the present value of remaining lease payments and the estimated incremental borrowing rate upon lease modification.
The Company enters into lease agreements with terms generally ranging from 2-8two to eight years. Some of the Company’s lease agreements include Company options to either extend and/or early terminate the lease, the costs of which are included in its operating lease liabilities to the extent that such options are reasonably certain of being exercised. Leases with renewal options allow the Company to extend the lease term typically between 1one and 5five years. When determining the lease term, renewal options reasonably certain of being exercised are included in the lease term. When determining if a renewal option is reasonably certain of being exercised, the Company considers several economic factors, including but not limited to, the significance of leasehold improvements incurred on the property, whether the asset is difficult to replace, underlying contractual obligations, or specific characteristics unique to that particular lease that would make it reasonably certain that the Company would exercise such option. Renewal and termination options were generally not included in the lease term for the Company’s existing operating leases. Leases with an initial term of 12 months or less are not recorded on the balance sheet; the Company recognizes lease expense for these leases on a straight-line basis over the lease term.
Acquisitions
The Company evaluates acquisitions of assets and other similar transactions to assess whether or not the transaction should be accounted for as a business combination or asset acquisition by first applying a screen to determine if substantially all of the fair value of the gross assets acquired is concentrated in a single identifiable asset or group of similar identifiable assets. If the screen is met, the transaction is accounted for as an asset acquisition. If the screen is not met, further determination is required as to whether or not the Company has acquired inputs and processes that have the ability to create outputs, which would meet the requirements of a business. If determined to be a business combination, the Company accounts for the transaction under the acquisition method of accounting which requires the acquiring entity in a business combination to recognize the fair value of all assets acquired, liabilities assumed, and any non-controlling interest in the acquiree and establishes the acquisition date as the fair value measurement point. Accordingly, the Company recognizes assets acquired and liabilities assumed in business combinations based on the fair value estimates as of the date of acquisition. In accordance with ASC 805, Business Combinations, or ASC 805, the Company recognizes and measures goodwill as of the acquisition date, as the excess of the fair value of the consideration paid over the fair value of the identified net assets acquired.
Goodwill
Goodwill represents the amount of consideration paid in excess of the fair value of the identified net assets acquired as a result of the Company’s business acquisitions accounted for using the acquisition method of accounting. Goodwill is not amortized and is subject to impairment testing at a reporting unit level on an annual basis or when a triggering event occurs that may indicate the carrying value of the goodwill is impaired. An entity is permitted to first assess qualitative factors to determine if a quantitative impairment test is necessary. Such qualitative factors include macroeconomic conditions, industry and market considerations, cost factors, overall financial performance and other relevant events. Further testing is only required if the entity determines, based on the qualitative assessment, that it is more likely than not that the fair value of the reporting unit is less than its carrying amount.
The Company evaluates goodwill for impairment at least annually on October 1 and whenever facts and circumstances indicate that their carrying amounts may not be recoverable. For the year ended December 31, 2023, the Company determined that there was no impairment to goodwill.
Indefinite-Lived Intangible Assets
Indefinite-lived intangible assets consist of in-process research and development, or IPR&D. The fair values of IPR&D assets acquired in business combinations are capitalized. These assets are treated as indefinite-lived intangible assets until completion or abandonment of the projects, at which time the assets are amortized over the remaining useful life or written off, as appropriate.
Intangible assets with indefinite lives, including IPR&D, are tested for impairment if impairment indicators arise and, at a minimum, annually. However, an entity is permitted to first assess qualitative factors to determine if a quantitative impairment test is necessary. Further testing is only required if the entity determines, based on the qualitative assessment, that it is more
likely than not that an indefinite-lived intangible asset’s fair value is less than its carrying amount. Otherwise, no further impairment testing is required. The indefinite-lived intangible asset impairment test consists of a one-step analysis that compares the fair value of the intangible asset with its carrying amount. If the carrying amount of an intangible asset exceeds its fair value, an impairment loss is recognized in an amount equal to that excess. The Company considers many factors in evaluating whether the value of its intangible assets with indefinite lives may not be recoverable, including, but not limited to, expected growth rates, the cost of equity and debt capital, general economic conditions, the Company’s outlook and market performance of the Company’s industry and recent and forecasted financial performance.
The Company evaluates indefinite-lived intangible assets for impairment at least annually on October 1 and whenever facts and circumstances indicate that their carrying amounts may not be recoverable. For the year ended December 31, 2023, the Company determined that there was no impairment to the IPR&D assets.
Series A Preferred Stock
The Company records the Series A Preferred Stock upon issuance at its fair value.The fair value includes the original issuance price, the settlement of any related forward contract, and is less issuance costs. The Company classifies its Series A Preferred Stock outside of stockholders’ equity as the redemption of such shares is outside the Company’s control. The Company does not adjust the carrying value of the Series A Preferred Stock to redemption value until it is probable of becoming redeemable, which the Company did not conclude was probable as of December 31, 2023.
Series A Preferred Stock Options
The Company classifies a portion of the fair value of the vested stock options for Series A Preferred Stock equal to the estimated redemption value on the measurement date outside of stockholders’ equity, as the redemption of the shares underlying the options are outside the Company’s control. Any fair value in excess of the estimated redemption value is recognized as additional paid-in capital. The estimated redemption value is based on the intrinsic value of the option. The Company does not adjust the carrying value of the stock options for Series A Preferred Stock until the underlying Series A Preferred Stock is probable of becoming redeemable. The Company concluded the redemption was not probable of occurring as of December 31, 2023. The Company records the stock options for Series A Preferred Stock based on the intrinsic value of the vested options.
Variable Interest Entities
The Company evaluates its variable interests in variable interest entities, or VIEs, and consolidates VIEs when the Company is the primary beneficiary. The Company determines whether it is the primary beneficiary of a VIE based on its assessment of whether the Company possesses both (i) the power to direct the activities that most significantly affect the VIE’s economic performance and (ii) the obligation to absorb losses that could be significant to the VIE or the right to receive benefits that could be significant to the VIE. The Company reevaluates the accounting for its VIEs upon the occurrence of events that could change the primary beneficiary conclusion.
Contingent Value Right Liability
The CVRs distributed by the Company pursuant to the terms of the CVR Agreement represent financial instruments that are accounted for under the fair value option election in ASC 825, Financial Instruments, or ASC 825. Under the fair value option election, the CVRs are initially measured at the aggregate estimated fair value of the CVRs and will be subsequently remeasured at estimated fair value on a recurring basis at each reporting period date. The estimated fair value of the CVR liability was determined using the discounted cash flow method to estimate future cash flows associated with the legacy assets, including the expected milestone and royalty payments under the Sobi License, net of deductions. Changes in fair value of the liability are presented within change in fair value of contingent value right liability in the consolidated statements of operations and comprehensive income (loss). The liability value is based on significant inputs not observable in the market such as estimated cash flows, estimated probabilities of success, and risk-adjustment discount rates, which represent a Level 3 measurement within the fair value hierarchy.
Forward Contract Liabilities
The Company accounts for contracts related to the future issuance of Series A Preferred Stock as a liability because the underlying shares of Series A Preferred Stock include a redemption feature that may require the Company to settle the instrument by transferring an asset. The forward contract liability is carried at fair value through the date the underlying Series A Preferred Stock are issued. The fair value of the forward contract liability was initially measured based on the fair value of the Series A Preferred Stock issued in the November 2023 Private Placement (see Note 11), less the purchase price, if any. Subsequent measurement of the fair value of the forward contract liability is based on the market price of the Company’s common stock, which represents the redemption and conversion value of the Series A Preferred Stock, less the purchase price, if any, on an as-converted basis. The remeasurement of the forward contract liability is based on Level 2 inputs within the fair value hierarchy as it’s based on observable market data. Changes in fair value of the liability are presented within change in fair value of forward contract liabilities in the consolidated statements of operations and comprehensive income (loss).
Recent Accounting Pronouncements
Recently Adopted
In May 2021, the FASB issued ASU 2021-04, Earnings Per Share (Topic 260), Debt – Modifications and Extinguishments (Subtopic 470-50), Compensation – Stock Compensation (Topic 718), and Derivatives and Hedging – Contracts in Entity’s Own Equity (Subtopic 815-40): Issuer's Accounting for Certain Modifications or Exchanges of Freestanding Equity-Classified Written Call Options. ASU 2021-04 provides guidance as to how entities should account for a modification of the terms or conditions or an exchange of a freestanding equity-classified written call option (i.e., a warrant) that remains equity-classified after modification or exchange as an exchange of the original instrument for a new instrument. An entity should apply the guidance provided in ASU 2021-04 prospectively to modifications or exchanges occurring on or after the effective date. The Company adopted the new standard effective January 1, 2022, and there was no impact on its consolidated financial statements.
In August 2020, the FASB issued ASU 2020-06, Debt – Debt with Conversion and Other Options (Subtopic 470-20) and Derivatives and Hedging – Contracts in Entity’s Own Equity (Subtopic 815-40). ASU 2020-06 simplifies the accounting for certain financial instruments with characteristics of liabilities and equity, including convertible instruments and contracts on an entity’s own equity. The Company has adopted ASU 2020-06 as of October 1, 2022, with an effective date of January 1, 2022, using the modified retrospective method. The adoption of ASU 2020-06 had no impact on the Company's consolidated financial statements and disclosures.
Not Yet Adopted
In June 2016, the FASB issued ASU 2016-13, Financial Instruments-Credit Losses (Topic 326), Measurement of Credit Losses on Financial Instruments. Subsequently, in November 2018, the FASB issued ASU 2018-19, Codification Improvements to Topic 326, Financial Instruments-Credit Losses. ASU 2016-13 requires entities to measure all expected credit losses for most financial assets held at the reporting date based on an expected loss model which includes historical experience, current conditions, and reasonable and supportable forecasts. ASU 2016-13 also requires enhanced disclosures to help financial statement users better understand significant estimates and judgments used in estimating credit losses. This ASU is effective for smaller reporting companies for fiscal years beginning after December 15, 2022, with early adoption permitted. The adoption of ASU 2016-13 is not expected to have anCompany adopted the new standard effective January 1, 2023, using a modified retrospective transition method, and there was no impact on the Company’sits consolidated financial positionstatements or results of operations upon adoption.
Not Yet Adopted
In November 2023, the FASB issued ASU 2023-07, Segment Reporting (Topic 280): Improvements to Reportable Segment Disclosures (ASU 2023-07), which requires an enhanced disclosure of significant segment expenses on an annual and interim basis. This guidance will be effective for the annual periods beginning the year ended December 31, 2024, and for interim periods beginning January 1, 2025. Early adoption is permitted. Upon adoption, the guidance should be applied retrospectively to all prior periods presented in the financial statements. The Company does not expect the adoption of this guidance to have a material impact on its consolidated financial statements.
In December 2023, the FASB issued ASU 2023-09, Income Taxes (Topic 740): Improvements to Income Tax Disclosures (ASU 2023-09), which improves the transparency of income tax disclosures by requiring consistent categories and greater disaggregation of information in the effective tax rate reconciliation and income taxes paid disaggregated by jurisdiction. It also includes certain other amendments to improve the effectiveness of income tax disclosures. This guidance will be effective for the annual periods beginning the year ended December 31, 2025. Early adoption is permitted. Upon adoption, the guidance can be applied prospectively or retrospectively. The Company does not expect the adoption of this guidance to have a material impact on its consolidated financial statements.
3. Merger
On November 13, 2023, the Company merged with Old Cartesian in accordance with the terms of the Merger Agreement, by and among Selecta, Sakura Merger Sub I, Inc., a wholly owned subsidiary of Selecta, or First Merger Sub, Sakura Merger Sub II, LLC, a wholly owned subsidiary of Selecta, or Second Merger Sub, and Old Cartesian. Pursuant to the Merger Agreement, First Merger Sub merged with and into Old Cartesian, pursuant to which Old Cartesian was the surviving corporation and became a wholly owned subsidiary of Selecta, or the First Merger. Immediately following the First Merger, Old Cartesian merged with and into Second Merger Sub, pursuant to which Second Merger Sub was the surviving entity, or the Second Merger and, together with the First Merger, the Merger. In connection with the Second Merger, Old Cartesian changed its name to Cartesian Bio, LLC.
The Merger was intended to qualify as a tax-free reorganization for U.S. federal income tax purposes. As a result of the Merger, Selecta changed its corporate name to Cartesian Therapeutics, Inc. and its common stock began trading on the Nasdaq Global Market under the new trading symbol “RNAC” beginning on November 14, 2023.
The Merger Agreement was unanimously approved by the board of directors, or the Board of Directors, of Selecta and the board of directors of Old Cartesian. The Merger was consummated substantially concurrently with the entry into the Merger Agreement and was not subject to approval of the Company's stockholders.
Under the terms of the Merger Agreement, following the consummation of the Merger on November 13, 2023, or the Closing Date, in exchange for 100% of the outstanding shares of capital stock of Old Cartesian immediately prior to the effective time of the First Merger, the Company agreed to issue to the stockholders of Old Cartesian (i) 6,723,639 shares of the Company’s common stock and (ii) 384,930.724 shares of Series A Preferred Stock. The issuance of the shares of common stock and Series A Preferred Stock occurred on December 5, 2023 which was after the December 4, 2023 record date for the distribution of the CVRs (see Note 6); as such, the Old Cartesian stockholders did not have rights as holders of common stock or holders of Series A Preferred Stock until such issuance on December 5, 2023. In addition, all outstanding stock options to
3.purchase Old Cartesian common stock were assumed by the Company and converted into stock options to purchase (i) shares of the Company’s common stock or (ii) shares of the Company’s Series A Preferred Stock on terms substantially identical to those in effect prior to Merger Agreement, except for adjustments to the underlying number of shares and the exercise price based on the Merger Agreement exchange ratio.
Pursuant to the Merger Agreement, the Company agreed to hold a stockholders’ meeting to submit the following proposals to a vote of its stockholders: (i) the approval of the conversion of shares of Series A Preferred Stock into shares of common stock, or the Conversion Proposal, and (ii) either or both of (A) the approval of an amendment to the Company’s restated certificate of incorporation, as amended, or the Charter, to increase the number of shares of common stock authorized under the Charter and (B) the approval of an amendment to the Charter to effect a reverse stock split of all outstanding shares of common stock, in either case (A) or (B) by a number of authorized shares or at a stock split ratio, as the case may be, sufficient to allow the conversion of all shares of Series A Preferred Stock issued in the Merger.
The Company concluded the acquisition resulted in the Company obtaining a controlling financial interest in a VIE in accordance with ASC 810, Consolidation. The Company determined that Old Cartesian was considered to be a VIE as it did not have sufficient equity to finance its activities without additional subordinated financial support. Prior to the Closing Date, the primary source of funding for Old Cartesian had been preferred stock financings. The Company acquired all of the outstanding shares of Old Cartesian and, therefore, is the sole equity holder and primary beneficiary. The Company has the obligation to the absorb losses and right to receive the benefits of Old Cartesian, and the power to direct the activities that most significantly affect the economic performance of Old Cartesian which the Company considers to be its development activities. Therefore, the Company is the primary beneficiary. Further, the Company concluded the VIE qualified as a business and accounted for the transaction as the acquisition of a business in accordance with ASC 805.As the primary beneficiary, the Company was the acquirer in the transaction.
The Company exchanged the right to receive shares of common stock and Series A Preferred Stock for all of the outstanding equity of Old Cartesian.The Company determined the rights to receive shares exchanged in the Merger represent a forward contract.The fair value of the forward contracts was determined based on the fair value of shares of common stock and Series A Preferred Stock underlying the forward contracts as of the acquisition date. The total purchase price consists of the fair value of the forward contracts in addition to a portion of the fair value of options exchanged in the transaction related to prior service. Under the acquisition method, the total purchase price of the acquisition was allocated to the net tangible and identifiable intangible assets acquired and liabilities assumed based on their estimated fair values as of the date of the acquisition.
The total fair value of the consideration of $168.5 million as of the Closing Date is summarized as follows (in thousands):
| | | | | |
| Forward contract to issue common stock | $ | 2,713 | |
| Forward contract to issue Series A Preferred Stock | 155,308 | |
| Stock options allocated to consideration paid | 10,444 | |
| Total consideration | $ | 168,465 | |
The Company recorded the assets acquired and liabilities assumed as of the Closing Date based on the information available at that date. The following table presents the allocation of the purchase price to the estimated fair values of the assets acquired and liabilities assumed as of the Closing Date (in thousands):
| | | | | |
| Assets acquired: | As of November 13, 2023 |
| Cash and cash equivalents | $ | 6,561 | |
| Prepaid expenses and other current assets | 309 | |
| Property and equipment, net | 215 | |
| Right-of-use asset, net | 915 | |
| In-process research and development assets | 150,600 | |
| Goodwill | 48,163 | |
| $ | 206,763 | |
| Liabilities assumed | |
| Accrued expenses and other current liabilities | $ | 2,530 | |
| Lease liability | $ | 292 | |
| Lease liability, net of current portion | $ | 623 | |
| Deferred tax liability | $ | 34,853 | |
| $ | 38,298 | |
| |
| Net assets acquired | $ | 168,465 | |
The fair value of IPR&D assets were capitalized as of the Closing Date and will be accounted for as indefinite-lived intangible assets until completion or disposition of the assets or abandonment of the associated research and development efforts. Upon successful completion of the development efforts, the carrying value of the respective IPR&D asset will be amortized over its estimated useful life. Until that time, the IPR&D assets will be subject to impairment testing and will not be amortized. The goodwill recorded related to the Merger is the excess of the fair value of the consideration transferred by the acquirer over the fair value of tangible assets, identifiable intangible assets and assumed liabilities as of the Closing Date and is not deductible for tax purposes. The goodwill balance is primarily attributable to the value of the assembled workforce and deferred tax liabilities associated with the transaction.
The following summarizes the Company’s intangible assets acquired in the Merger and their carrying value as of December 31, 2023 (in thousands):
| | | | | | | | | | | | | | | | | | | | |
| | Acquisition Date
Fair Value | | Impairment | | Carrying Value at
December 31, 2023 |
| Descartes-08 for MG | | $ | 93,900 | | | $ | — | | | $ | 93,900 | |
| Descartes-08 for SLE | | 56,700 | | | — | | | 56,700 | |
| Total in-process research and development assets | | $ | 150,600 | | | $ | — | | | $ | 150,600 | |
The fair value of the intangible assets was estimated using the income approach in which the after-tax cash flows were discounted to present value. The cash flows are based on estimates used to price the transaction, and the discount rates applied were benchmarked with reference to the implied rate of return from the transaction model as well as the weighted average cost of capital.
For the period from November 13, 2023 to December 31, 2023, Old Cartesian’s revenue and net loss within the consolidated statements of operations and comprehensive (loss) income were $0.0 million and $1.6 million, respectively.
The following unaudited pro forma financial information reflects the consolidated results of operations of the Company as if the Merger had taken place on January 1, 2022. The unaudited pro forma financial information is not necessarily indicative of the results of operations as they would have been had the transactions been effected on the assumed date (in thousands):
| | | | | | | | | | | | | | |
| | Year Ended December 31, |
| | 2023 | | 2022 |
| Revenue | | $ | 26,004 | | | $ | 112,226 | |
| Net (loss) income | | $ | (232,259) | | | $ | 29,607 | |
The Company’s transaction costs of $4.9 million were expensed as incurred and included in general and administrative expense in the consolidated statements of operations and comprehensive (loss) income.
The forward contract related to the common stock was recorded as additional paid-in capital as the instrument is indexed to the Company’s common stock.The forward contract related to the Series A Preferred Stock was recorded as a liability as the underlying Series A Preferred Stock has a redemption feature that may require the Company to settle the instrument by transferring an asset.The forward contract was measured at fair value through the date of settlement through the issuance of the shares of Series A Preferred Stock on December 5, 2023.
4. Marketable Securities and Investments
No marketable securities were held as of December 31, 2023. The following table summarizes the marketable securities held as of December 31, 2022 and 2021 (in thousands):
| | Amortized
cost | | | Amortized
cost | | Unrealized gains | | Unrealized losses | | Fair
value |
| | | | Amortized
cost | | Unrealized gains | | Unrealized losses | | Fair
value |
| December 31, 2022 | December 31, 2022 | | | | | | | |
| | December 31, 2022 | |
| | December 31, 2022 | |
| U.S. government agency securities and treasuries | |
| U.S. government agency securities and treasuries | |
| U.S. government agency securities and treasuries | U.S. government agency securities and treasuries | $ | 13,566 | | | $ | — | | | $ | (9) | | | $ | 13,557 | |
| Corporate bonds | Corporate bonds | 1,953 | | | — | | | (2) | | | 1,951 | |
| Commercial paper | Commercial paper | 12,656 | | | — | | | — | | | 12,656 | |
| | Total | Total | $ | 28,175 | | | $ | — | | | $ | (11) | | | $ | 28,164 | |
| | December 31, 2021 | |
| | Corporate bonds | $ | 2,007 | | | $ | — | | | $ | (1) | | | $ | 2,006 | |
| Commercial paper | 11,992 | | | — | | | — | | | 11,992 | |
| | Total | Total | $ | 13,999 | | | $ | — | | | $ | (1) | | | $ | 13,998 | |
| Total | |
All marketable securities held at December 31, 2022 and 2021 had maturities of less than 12 months when purchased and are classified as short-term marketable securities on the accompanying consolidated balance sheet. During the years ended December 31, 2022 and 2021, there were no marketable securities adjusted for other than temporary declines in fair value. The Company does not intend to sell its investments and it is not more likely than not that the Company will be required to sell the investments before recovery of their amortized cost bases, which may be maturity. Investments
As of December 31, 20222023 and 2021,2022, the Company has a $2.0 million investment in Cyrus Biotechnology, Inc., or Cyrus, pursuant to the Company's Collaboration and License Agreement with Cyrus, or the Cyrus Agreement. The Company’s maximum exposure to loss related to this variable interest entityVIE is limited to the carrying value of the investment. See Note 1416 for details.
4.5. Net Loss(Loss) Income Per Share
The Company has reported net income for the year ended December 31, 2022 and a net loss for the years ended December 31, 2023 and 2021, and 2020.net income for the year ended December 31, 2022. The Company used the treasury stock method to determine the number of dilutive shares. The following table sets forth the computation of basic and diluted net (loss) income (loss) per share (in thousands, except share and per-share data):
| | | |
| | | | | Year Ended December 31, | | | | Year Ended December 31, |
| | | | 2022 | | 2021 | | 2020 | | | | | | | | | 2023 | | 2022 | | 2021 |
| Numerator: | Numerator: | | | | | | |
| Net income (loss) | | $ | 35,379 | | | $ | (25,687) | | | $ | (68,876) | |
| Net (loss) income | |
| Net (loss) income | |
| Net (loss) income | |
| Less: CVR distribution to participating securities | |
| Net (loss) income allocable to shares of common stock - basic | |
| Less: Change in fair value of warrants | Less: Change in fair value of warrants | | (20,882) | | | — | | | — | |
| Adjusted net income (loss) | | $ | 14,497 | | | $ | (25,687) | | | $ | (68,876) | |
| Net (loss) income allocable to shares of common stock - diluted | |
| Denominator: | Denominator: | | | | | | | Denominator: | | | | | | | | | | |
| Weighted-average common shares outstanding - basic | Weighted-average common shares outstanding - basic | | 144,758,555 | | | 114,328,798 | | | 101,202,176 | |
| Dilutive effect of employee equity incentive plans and outstanding warrants | Dilutive effect of employee equity incentive plans and outstanding warrants | | 1,116,334 | | | — | | | — | |
| Weighted-average common shares used in per share calculations - diluted | Weighted-average common shares used in per share calculations - diluted | | 145,874,889 | | | 114,328,798 | | | 101,202,176 | |
| Net loss per share: | | | | | | |
| Net (loss) income per share: | |
| Basic | |
| Basic | |
| Basic | Basic | | $ | 0.24 | | | $ | (0.22) | | | $ | (0.68) | |
| Diluted | Diluted | | $ | 0.10 | | | $ | (0.22) | | | $ | (0.68) | |
The following table represents the potential dilutive shares of common sharesstock excluded from the computation of the diluted net (loss) income (loss) per share for all periods presented, as the effect would have been anti-dilutive:
| | | | | Year Ended December 31, |
| | | | 2022 | | 2021 | | 2020 |
| Options, RSUs and ESPP shares | | 17,800,034 | | | 11,492,002 | | | 7,909,583 | |
| | | | | | Year Ended December 31, |
| | | | | | | | | | | 2023 | | 2022 | | 2021 |
| Warrants to purchase common stock | Warrants to purchase common stock | | 213,339 | | | 10,735,980 | | | 12,378,016 | |
| Series A Preferred Stock | |
| Forward contract to issue Series A Preferred Stock | |
| Common stock options, RSUs and ESPP shares | |
| Series A Preferred Stock options | |
| Total | Total | | 18,013,373 | | | 22,227,982 | | | 20,287,599 | |
5.6. Fair Value Measurements
The following tables present the Company’s assets and liabilities that are measured at fair value on a recurring basis as of December 31, 20222023 and 20212022 (in thousands):
| | December 31, 2023 | | | December 31, 2023 |
| Total | | | Total | | Level 1 | | Level 2 | | Level 3 |
| Assets: | |
| Money market funds (included in cash equivalents) | |
| Money market funds (included in cash equivalents) | |
| Money market funds (included in cash equivalents) | |
| | | December 31, 2022 |
| | Total | | Level 1 | | Level 2 | | Level 3 |
| Assets: | | | | | | | |
| Money market funds (included in cash equivalents) | $ | 53,552 | | | $ | 53,552 | | | $ | — | | | $ | — | |
| Marketable securities: | |
| U.S. government agency securities and treasuries | 13,557 | | | — | | | 13,557 | | | — | |
| Corporate bonds | 1,951 | | | — | | | 1,951 | | | — | |
| Commercial paper | 12,656 | | | — | | | 12,656 | | | — | |
| | Total assets | |
| | Total assets | |
| | | Total assets | Total assets | $ | 81,716 | | | $ | 53,552 | | | $ | 28,164 | | | $ | — | |
| | Liabilities: | Liabilities: | |
| Liabilities: | |
| Liabilities: | |
| Warrant liabilities | Warrant liabilities | $ | 19,140 | | | $ | — | | | $ | — | | | $ | 19,140 | |
| Warrant liabilities | |
| Warrant liabilities | |
| Contingent value right liability | |
| Forward contract liabilities | |
| Total liabilities | Total liabilities | $ | 19,140 | | | $ | — | | | $ | — | | | $ | 19,140 | |
| | | | | | | | | | | | | | | | | | | | | | | |
| December 31, 2021 |
| Total | | Level 1 | | Level 2 | | Level 3 |
| Assets: | | | | | | | |
| Money market funds (included in cash equivalents) | $ | 66,563 | | | $ | 66,563 | | | $ | — | | | $ | — | |
| Marketable securities: | | | | | | | |
| Corporate bonds | 2,006 | | | — | | | 2,006 | | | — | |
| Commercial paper | 11,992 | | | — | | | 11,992 | | | — | |
| | | | | | | |
| Total assets | $ | 80,561 | | | $ | 66,563 | | | $ | 13,998 | | | $ | — | |
| | | | | | | |
| Liabilities: | | | | | | | |
| Warrant liabilities | $ | 25,423 | | | $ | — | | | $ | — | | | $ | 25,423 | |
| Total liabilities | $ | 25,423 | | | $ | — | | | $ | — | | | $ | 25,423 | |
| | | | | | | | | | | | | | | | | | | | | | | |
| December 31, 2022 |
| Total | | Level 1 | | Level 2 | | Level 3 |
| Assets: | | | | | | | |
| Money market funds (included in cash equivalents) | $ | 53,552 | | | $ | 53,552 | | | $ | — | | | $ | — | |
| | | | | | | |
| Marketable securities: | | | | | | | |
| U.S. government agency securities and treasuries | 13,557 | | | — | | | 13,557 | | | — | |
| Corporate bonds | 1,951 | | | — | | | 1,951 | | | — | |
| Commercial paper | 12,656 | | | — | | | 12,656 | | | — | |
| | | | | | | |
| Total assets | $ | 81,716 | | | $ | 53,552 | | | $ | 28,164 | | | $ | — | |
| | | | | | | |
| Liabilities: | | | | | | | |
| Warrant liabilities | $ | 19,140 | | | $ | — | | | $ | — | | | $ | 19,140 | |
| Total liabilities | $ | 19,140 | | | $ | — | | | $ | — | | | $ | 19,140 | |
There were no transfers within the fair value hierarchy during the years ended December 31, 20222023 or 2021.2022.
Cash, Cash Equivalents, and Restricted Cash
As of December 31, 20222023 and 2021, the2022, money market funds were classified as cash and cash equivalents on the accompanying consolidated balance sheets as they mature within 90 days from the date of purchase.
As of December 31, 2022,2023, the Company had restricted cash balances relating to a secured letter of credit in connection with its lease for the Company’s prior headquarters (see Note 89 included elsewhere in this Annual Report). Short-term restricted cash is included within prepaid expenses and other current assets in the consolidated balance sheets. The Company’s consolidated statement of cash flows includes the following as of December 31, 2023, 2022 2021 and 20202021 (in thousands):
| | | Year Ended December 31, |
| | 2022 | | 2021 | | 2020 |
| | Year Ended December 31, | |
| | Year Ended December 31, | |
| | Year Ended December 31, | |
| | | | 2023 | | | | | | 2023 | | 2022 | | 2021 |
| Cash and cash equivalents | Cash and cash equivalents | | $ | 106,438 | | | $ | 114,057 | | | $ | 138,685 | |
| Short-term restricted cash | Short-term restricted cash | | 289 | | | — | | | — | |
| Long-term restricted cash | Long-term restricted cash | | 1,311 | | | 1,379 | | | 1,379 | |
| Total cash, cash equivalents, and restricted cash | Total cash, cash equivalents, and restricted cash | | $ | 108,038 | | | $ | 115,436 | | | $ | 140,064 | |
Marketable Securities
AsNo marketable securities were held as of December 31, 2023. Marketable securities held as of December 31, 2022 marketable securitiesand classified as Level 2 within the valuation hierarchy consist of U.S. government agency securities and treasuries, corporate bonds and commercial paper. Marketable securities represent holdings of available-for-sale marketable debt securities in accordance with the Company’s investment policy. The Company estimates the fair value of these marketable securities by taking into consideration valuations that include market pricing based on real-time trade data for the same or similar securities, and other observable inputs. The amortized cost of available-for-sale debt
securities is adjusted for amortization of premiums and accretion of discounts to the earliest call date for premiums or to maturity for discounts.
Loans Payable
At December 31, 2022, in light of the recent issuance of the Term A Loan under the 2020 Term Loan, the Company believes the carrying value approximates the fair value of the loan.
Warrants to Purchase Common WarrantsStock
In December 2019, the Company issued warrants to purchase common warrantsstock in connection with a private placement, of common shares, or the 2019 Warrants. Pursuant to the terms of the 2019 Warrants, the Company could be required to settle the common warrants2019 Warrants in cash in the event of certain acquisitions of the Company and, as a result, the common warrants are required to be measured at fair value and reported as a liability on the balance sheet. Pursuant to the Stipulation and Agreement of Settlement discussed in Note 17, onOn December 20, 2022, the Company amended the terms of the outstanding 2019 Warrants held by certain members of its Board of Directors, or the Company's board of directorsAmended 2019 Warrants, to remove the cash settlement provision (the Amended 2019 Warrants).provision. As a result, the Amended 2019 Warrants were remeasured at fair value on December 20, 2022 and reclassified from a liability to equity on the balance sheet. Refer to Note 1012 for further discussion on the equity classifiedequity-classified Amended 2019 Warrants.
In April 2022, the Company issued warrants in connection with an underwritten offering, of shares of common stock and warrants to purchase shares of common stock, or the 2022 Warrants. Pursuant to the terms of the 2022 Warrants, the Company could be required to settle the 2022 Warrants in cash in the event of an acquisition of the Company under certain circumstances and, as a result, the 2022 Warrants are required to be measured at fair value and reported as a liability on the balance sheet.
The Company recorded the fair value of the 2019 Warrants and the 2022 Warrants upon issuance using the Black-Scholes valuation model and is required to revalue the 2019 Warrants and the 2022 Warrants at each reporting date, with any changes in
fair value recorded in the statement of operations and comprehensive income (loss). The valuations of the 2019 Warrants and the 2022 Warrants are classified as Level 3 of the fair value hierarchy due to the need to use assumptions in the valuations that are both significant to the fair value measurement and unobservable, including the stock price volatility and the expected life of the 2019 Warrants and the 2022 Warrants. Generally, increases (decreases) in the fair value of the underlying stock and estimated term would result in a directionally similar impact to the fair value measurement. The changes in the fair values of the warrants are reflected in the statement of operations and comprehensive income (loss) for the years ended December 31, 2023, 2022 2021 and 2020.2021.
The estimated fair values of the 2019 Warrants the Amended 2019 Warrants on the modification date, and the 2022 Warrants were determined using the following inputs to the Black-Scholes simulation valuation:
Estimated fair value of the underlying stock. The Company estimates the fair value of the common stock based on the closing stock price at the end of each reporting period.
Risk-free interest rate. The risk-free interest rate is based on the U.S. Treasury at the valuation date commensurate with the expected remaining life assumption.
Dividend rate. The dividend rate is based on the historical rate, which the Company anticipates will remain at zero.
Expected life. The expected life of the 2019 Warrants and the 2022 Warrants is assumed to be equivalent to their remaining contractual terms which expire on December 23, 2024 and April 11, 2027, respectively.
Volatility. The Company estimates stock price volatility based on the Company’s historical volatility and the historical volatility of peer companies for a period of time commensurate with the expected remaining life of the warrants.
A summary of the Black-Scholes pricing model assumptions used to record the fair value of the 2019 Warrants liability is as follows:
| | | | | | | | | | | |
| December 31, |
| | 2022 | | 2021 |
| Risk-free interest rate | 4.74 | % | | 0.97 | % |
| Dividend yield | — | | | — | |
| Expected life (in years) | 1.98 | | 2.98 |
| Expected volatility | 79.92 | % | | 96.10 | % |
A summary of the Black-Scholes pricing model assumptions used to record the fair value of the Amended 2019 Warrants liability on the modification date is as follows:
| | | | | | | |
| | | At Modification
December 20, |
| | | 2022 |
Risk-free interest rate | | | 4.64 | % |
Dividend yield | | | — | |
Expected life (in years) | | | 2.01 |
Expected volatility | | | 79.77 | % |
| | | | | | | | | | | |
| December 31, |
| | 2023 | | 2022 |
| Risk-free interest rate | 4.79 | % | | 4.74 | % |
| Dividend yield | — | | | — | |
| Expected life (in years) | 0.98 | | 1.98 |
| Expected volatility | 83.67 | % | | 79.92 | % |
A summary of the Black-Scholes valuation model assumptions used to record the fair value of the 2022 Warrants liability is as follows:
| | | | | | | | | | | |
| | | At Issuance |
| December 31, | | April 11, |
| | 2022 | | 2022 |
| Risk-free interest rate | 4.22 | % | | 2.79 | % |
| Dividend yield | — | | | — | |
| Expected life (in years) | 4.28 | | 5.00 |
| Expected volatility | 98.05 | % | | 96.00 | % |
Changes in Level 3 Liabilities Measured at Fair Value on a Recurring Basis | | | | | | | | | | | |
| December 31, |
| | 2023 | | 2022 |
| Risk-free interest rate | 4.01 | % | | 4.22 | % |
| Dividend yield | — | | | — | |
| Expected life (in years) | 3.28 | | 4.28 |
| Expected volatility | 84.09 | % | | 98.05 | % |
The following table reflects a roll-forward of fair value for the Company’s Level 3 warrant liabilities (see Note 10)12), for the year ended December 31, 20222023 (in thousands):
| | | | | |
| Warrant liabilities |
Fair value as of December 31, 2021 | $ | 25,423 | |
Issuances | 15,379 | |
Exercises | — | |
Reclassification of warrant liability to equity upon modification | (780) | |
Change in fair value | (20,882) | |
| Fair value as of December 31, 2022 | $ | 19,140 | |
| |
| |
| |
| Change in fair value | (12,746) | |
| Fair value as of December 31, 2023 | $ | 6,394 | |
| |
| |
| |
Contingent Value Right
6.On December 6, 2023, as contemplated by the Merger Agreement, the Company entered into the CVR Agreement, pursuant to which each holder of common stock as of December 4, 2023 or a 2022 Warrant was distributed a CVR, issued by the Company for each share of common stock held directly or underlying a 2022 Warrant held by such holder as of December 4, 2023. Holders of warrants other than the 2022 Warrants will be entitled to receive, upon exercise of such warrants and in accordance with the terms of the warrants, one CVR per each share of common stock underlying such warrants.
Each CVR entitles its holder to distributions of the following, pro-rated on a per-CVR basis, during the period ending on the date on which the Royalty Term (as defined in the Sobi License) ends, or the Termination Date:
•100% of all milestone payments, royalties and other amounts paid to the Company or its controlled affiliates, or the Company Entities, under the Sobi License or, following certain terminations of the Sobi License, any agreement a Company Entity enters into that provides for the development and commercialization of SEL-212; and
•100% of all cash consideration and the actual liquidation value of any and all non-cash consideration of any kind that is paid to or is actually received by any Company Entity prior to the Termination Date pursuant to an agreement relating to a sale, license, transfer or other disposition of any transferable asset of the Company existing as of immediately prior to the Merger, other than those exclusively licensed under the Sobi License or which the Company Entities are required to continue to own in order to comply with the Sobi License.
The distributions in respect of the CVRs will be made on a semi-annual basis, and will be subject to a number of deductions, subject to certain exceptions or limitations, including for (i) certain taxes payable on the proceeds subject to the CVR distribution, (ii) certain out of pocket costs incurred by the Company Entities, including audit and accounting fees incurred in connection with reporting obligations relating to the CVRs and other expenses incurred in the performance of their obligations and other actions under the CVR Agreement, (iii) a fixed semi-annual amount of $0.75 million for general and administrative overhead, (iv) payments made and remaining obligations on lease liabilities of Selecta immediately prior to the Merger and (v) amounts paid and remaining obligations with regard to the Xork product candidate. Each of the deductions described in (iv) and (v) will be made only if certain milestone payments under the Sobi License are made and are also subject to certain adjustments as contemplated in the CVR Agreement.
The CVRs represent financial instruments that are accounted for under the fair value option election in ASC 825, Financial Instruments. Under the fair value option election, the CVRs are initially measured at the aggregate estimated fair value of the CVRs and will be subsequently remeasured at estimated fair value on a recurring basis at each reporting period date. The liability was recorded at the date of approval, November 13, 2023, as a dividend. The estimated fair value of the CVR liability was determined using the discounted cash flow method to estimate future cash flows associated with the legacy assets, including the expected milestone and royalty payments under the Sobi License, net of deductions. Changes in fair value of the CVR liability are presented in the consolidated statements of operations and comprehensive income (loss). The liability value is based on significant inputs not observable in the market such as estimated cash flows, estimated probabilities of success, and risk-adjustment discount rates, which represent a Level 3 measurement within the fair value hierarchy. The significant inputs used to estimate the fair value of the CVR liability, which represented a financial instrument being accounted for under the fair value option, were as follows:
| | | | | | | | | | | |
| December 31, 2023 | | At Issuance
November 13, 2023 |
| Estimated cash flow dates | 2024 - 2038 | | 2024 - 2038 |
| Estimated probability of success | 95.0 | % | | 95.0 | % |
| Risk-adjusted discount rate | 13.7 | % | | 14.4 | % |
The following table reflects a roll-forward of fair value for the Company's Level 3 CVR liability for the year ended December 31, 2023 (in thousands):
| | | | | |
| CVR liability |
| Fair value as of December 31, 2022 | $ | — | |
| Issuances | 340,300 | |
| Change in fair value | 18,300 | |
| Fair value as of December 31, 2023 | $ | 358,600 | |
Forward Contract Liabilities
Merger Consideration
In connection with the Merger, the Company entered into a contract for the issuance of 384,930.724 shares of Series A Preferred Stock as part of the consideration transferred. The fair value of the forward contract at the Closing Date (defined below) was $155.3 million. The non-cash settlement of this liability occurred on December 5, 2023 with the issuance of the Series A Preferred Stock for $261.8 million.
November 2023 Private Placement
The Company entered into a contract for the issuance of 149,330.115 shares of Series A Preferred Stock as part of the November 2023 Private Placement which was settled in multiple tranches. The Company determined the obligation to issue
148,710.488 shares of Series A Preferred Stock to Dr. Timothy A. Springer, a member of the Company’s Board of Directors, and TAS Partners LLC, an affiliate of Dr. Springer, represented a forward contract. See Note 11. The fair value of the forward contract liability on November 13, 2023 was insignificant as the fair value of the underlying Series A Preferred Stock was equal to the purchase price of the Series A Preferred Stock as agreed upon in the November 2023 Private Placement. The non-cash settlement of a portion of the liability occurred on December 13, 2023 with the issuance of the first tranche of the Series A Preferred Stock for $14.8 million.
The following table presents changes in the forward contract liabilities for the periods presented (in thousands):
| | | | | |
| Forward contract liabilities |
| Fair value as of December 31, 2022 | $ | — | |
| Issuances | 155,308 | |
| Settlements | (276,601) | |
| Change in fair value | 149,600 | |
| Fair value as of December 31, 2023 | $ | 28,307 | |
7. Property and Equipment
Property and equipment consists of the following (in thousands):
| | | | December 31, | | December 31, |
| | | 2022 | | 2021 | | 2023 | | 2022 |
| Laboratory equipment | Laboratory equipment | $ | 6,001 | | | $ | 5,134 | |
| Computer equipment and software | Computer equipment and software | 697 | | | 731 | |
| Leasehold improvements | Leasehold improvements | 57 | | | 45 | |
| Furniture and fixtures | Furniture and fixtures | 453 | | | 332 | |
| Office equipment | Office equipment | 192 | | | 163 | |
| Construction in process | Construction in process | 599 | | | 534 | |
| Total property and equipment | Total property and equipment | 7,999 | | | 6,939 | |
| Less accumulated depreciation | Less accumulated depreciation | (5,205) | | | (4,797) | |
| Property and equipment, net | Property and equipment, net | $ | 2,794 | | | $ | 2,142 | |
Depreciation expense was $0.7 million, $0.6$0.7 million and $0.6 million for the years ended December 31, 2023, 2022 2021 and 2020,2021, respectively.
7.8. Accrued Expenses
Accrued expenses consist of the following (in thousands):
| | | | December 31, | | December 31, |
| | | 2022 | | 2021 | | 2023 | | 2022 |
| Payroll and employee related expenses | Payroll and employee related expenses | $ | 4,242 | | | $ | 3,179 | |
| | Accrued patent fees | Accrued patent fees | 696 | | | 309 | |
| Accrued patent fees | |
| Accrued patent fees | |
| Accrued external research and development costs | Accrued external research and development costs | 7,274 | | | 4,339 | |
| Accrued professional and consulting services | Accrued professional and consulting services | 985 | | | 815 | |
| Accrued interest | Accrued interest | 222 | | | 170 | |
| Other | Other | 665 | | | 1,721 | |
| Accrued expenses | Accrued expenses | $ | 14,084 | | | $ | 10,533 | |
8.9. Leases
65 Grove Street Lease
In July 2019, the Company entered into a lease with BRE-BMR Grove LLC for 25,078 square feet of laboratory and office space located at 65 Grove Street, Watertown, Massachusetts, or the HeadquartersWatertown Lease. As part of the HeadquartersWatertown Lease, the Company incurred $0.8 million in non-reimbursable construction costs. The lease began in March 2020, when the Company took control of the office space, and the lease term is 8 years. The discount rate of 8.9% was determined based on the Company’s incremental borrowing rate adjusted for the lease term, including any reasonably certain renewal periods. In connection with the HeadquartersWatertown Lease, the Company secured a letter of credit from Silicon Valley Bank, a division of First-Citizens Bank & Trust Company (successor by purchase to the Federal Deposit Insurance Corporation as Receiver for Silicon
Valley Bridge Bank, N.A. (as successor to Silicon Valley Bank)), or SVB, for $1.4$1.6 million, of which $0.3 million is recognized as short-term restricted cash and $1.3 million is recognized as long-term restricted cash, as of December 31, 2021.2022.
On September 1, 2022, the Company entered into an amendment, or the Lease Agreement Amendment, to its lease agreement with BRE-BMR Grove LLC, originally entered into on July 23, 2019, or the Lease Agreement, to expand the Company’s corporate headquarterslaboratory and office space located at 65 Grove Street, Watertown, Massachusetts by approximately 7,216 square feet. The lease term began on September 1, 2022, consistent with when the Company took control of the office space and the expected lease term is 5.7 years. The discount rate of 11.3% was determined based on the Company’s incremental borrowing rate adjusted for the lease term including any reasonably certain renewal periods. Rent payments began in November 2022, and the base rent for the first year is $0.1 million per month. In connection with the Lease Agreement Amendment, the Company entered into an amendment to its Loan and Security Agreement with Oxford, as Collateral Agent and a Lender, and SVB, as a Lender, on September 20, 2022, or the Third Amendment, to provide for an increase of $0.2 million in the letter of credit for a total of $1.6 million as of December 31, 2022, which renews automatically each year. The Company recorded the right-of-use asset and operating lease liabilities of $3.2 million during the year ended December 31, 2022 as control of the premises was transferred to the Company.
On October 6, 2022, the Company entered into a sublease agreement with Nexo Therapeutics, Inc., or the Tenant, to sublease approximately 7,216 square feet of space currently rented by the Company at the corporate headquarters located at 65 Grove Street, Watertown, Massachusetts. The sublease commenced on October 24, 2022, when the Company, the Tenantsublessee and BRE-BMR Grove LLC, executed thea Consent to Sublease. The term of the sublease expires on March 31, 2024 with no option to extend the sublease term. Sublease income is included within other income, net in the consolidated statements of operations and comprehensive income (loss).
As a result of the sublease agreement and Consent to Sublease, rent payments to BRE-BMR Grove LLC for the lease of the office space increased. The change of consideration in the contract was accounted for as a lease modification and the right-of-use asset and lease liability were remeasured at the modification date of October 24, 2022. The discount rate of 11.9% was determined based on the Company’s incremental borrowing rate adjusted for the lease term including any reasonably certain renewal periods as of October 24, 2022, resulting in a decrease of less than $0.1 million to both the right-of-use asset and lease liabilities.
In May 2023, the Company received notice from BRE-BMR Grove LLC that the requirements to reduce the amount of the letter of credit for the Watertown Lease had been met. In connection therewith, in June 2023, the Company secured a letter of credit from JPMorgan Chase Bank, N.A. for $1.4 million, which is recognized as long-term restricted cash as of December 31, 2023, and renews automatically each year. The $1.6 million letter of credit with SVB was released from restriction and returned to the Company on July 17, 2023, and therefore was reclassified into cash and cash equivalents in the consolidated balance sheets.
On October 31, 2023, in connection with entering into Amendment No. 1 to the License and Development Agreement with Sobi as described in Note 14, the Company entered into a sublease agreement with Sobi to sublease approximately 5,600 square feet of space currently rented by the Company at 65 Grove Street, Watertown, Massachusetts for which Sobi paid $1.0 million upfront rental payment. The sublease commenced on November 6, 2023, when the Company, Sobi, and BRE-BMR Grove LLC, executed a Consent to Sublease. The term of the sublease expires on November 5, 2024 with no option to extend the sublease term. As of December 31, 2023, deferred rent of $0.8 million is included within accrued expenses and other current liabilities in the consolidated balance sheets.
During the year ended December 31, 2023, the Company determined that the right-of-use asset related to the operating lease for approximately 7,216 square feet at 65 Grove Street was partially impaired as of November 30, 2023. As a result, the Company recognized a $0.7 million right-of-use asset impairment charge with $0.6 million and $0.1 million recognized in research and development and general and administrative operating expense categories, respectively, on its consolidated statements of operations and comprehensive income (loss) during the year ended December 31, 2023.
704 Quince Orchard Road Leases
In connection with the Merger, the Company acquired two operating leases for office and laboratory space in Gaithersburg, Maryland. The leases expire in January 2027 and do not contain any renewal rights. The discount rate of 11.5% was determined based on the Company’s incremental borrowing rate adjusted for the lease term.
Moscow, Russia Lease
The Company has a month-to-month facility agreement for itsSelecta (RUS)'s Moscow, Russia office. Rent expense is recognized as incurred.
Rent expense for the years ended December 31, 2023, 2022 and 2021 and 2020 was $3.8 million, $3.2 million, and $2.9 million, and $2.7 million, respectively.
For the years ended December 31, 2023, 2022 2021 and 2020,2021, the components of lease costs were as follows (in thousands):
| | | | | | | | | | | | | | | | | |
| Year Ended
December 31, |
| 2022 | | 2021 | | 2020 |
| Operating lease cost | $ | 2,276 | | | $ | 2,023 | | | $ | 2,096 | |
| Variable lease cost | 910 | | | 834 | | | 624 | |
| Short-term lease cost | 11 | | | 10 | | | 10 | |
| Less sublease income | (176) | | | — | | | — | |
| Total lease cost | $ | 3,021 | | | $ | 2,867 | | | $ | 2,730 | |
| | | | | | | | | | | | | | | | | |
| Year Ended
December 31, |
| 2023 | | 2022 | | 2021 |
| Operating lease cost | $ | 2,828 | | | $ | 2,276 | | | $ | 2,023 | |
| Variable lease cost | 965 | | | 910 | | | 834 | |
| Short-term lease cost | 8 | | | 11 | | | 10 | |
| Less sublease income | (1,172) | | | (176) | | | — | |
| Total lease cost | $ | 2,629 | | | $ | 3,021 | | | $ | 2,867 | |
The maturity of the Company’s operating lease liabilities as of December 31, 20222023 were as follows (in thousands):
| | December 31, | |
| 2022 | |
| December 31, | |
| December 31, | |
| December 31, | |
| 2023 | |
| 2023 | |
| 2023 | 2023 | $ | 2,668 | | |
| 2024 | 2024 | 2,740 | | |
| 2024 | |
| 2024 | |
| 2025 | |
| 2025 | |
| 2025 | 2025 | 2,818 | | |
| 2026 | 2026 | 2,902 | | |
| 2026 | |
| 2026 | |
| 2027 | 2027 | 2,990 | | |
| 2027 | |
| 2027 | |
| 2028 | |
| 2028 | |
| 2028 | |
| Thereafter | |
| Thereafter | |
| Thereafter | Thereafter | 946 | | |
| Total future minimum lease payments | Total future minimum lease payments | 15,064 | | |
| Less imputed interest | 3,401 | | |
| Total future minimum lease payments | |
| Total future minimum lease payments | |
| Less: Imputed interest | |
| Less: Imputed interest | |
| Less: Imputed interest | |
| Total operating lease liabilities | Total operating lease liabilities | $ | 11,663 | | |
| Total operating lease liabilities | |
| Total operating lease liabilities | |
The supplemental disclosure for the statement of cash flows related to operating leases were as follows (in thousands):
| | | | | | | | | | | |
| December 31, |
| 2022 | | 2021 |
| Cash paid for amounts included in the measurement of lease liabilities: | $ | 2,048 | | | $ | 1,812 | |
| | | |
| | | |
| | | |
| | | | | | | | | | | |
| December 31, |
| 2023 | | 2022 |
| Cash paid for amounts included in the measurement of lease liabilities: | $ | 2,696 | | | $ | 2,048 | |
| | | |
| | | |
| | | |
Other than the initial recording and modification of the right-of-use asset and lease liability for the HeadquartersWatertown Lease during the year ended December 31, 2022 and the impairment on the right-of-use asset for the Watertown Lease and the assumption of the right-of-use assets and lease liabilities in 2020 and 2022,connection with the Merger during the year ended December 31, 2023, which were non-cash, the changes in the Company’s right-of-use asset and lease liability for the years ended December 31, 20222023 and 20212022 are reflected in the non-cash lease expense and accrued expenses and other liabilities, respectively, in the consolidated statements of cash flows.
The following summarizes additional information related to operating leases:
| | December 31, |
| 2022 | | 2021 |
| December 31, | | | December 31, |
| 2023 | | | 2023 | | 2022 |
| Weighted-average remaining lease term | Weighted-average remaining lease term | 5.4 years | | 6.4 years | Weighted-average remaining lease term | 4.3 years | | 5.4 years |
| Weighted-average discount rate | Weighted-average discount rate | 9.7 | % | | 8.9 | % | Weighted-average discount rate | 9.9 | % | | 9.7 | % |
9.10. Debt
2020 Term Loan
On August 31, 2020, the Company entered into a term loan of up to $35.0 million, or the 2020 Term Loan, consisting of term loans in an aggregate amount of $25.0 million, or the Term A Loan and term loans in an aggregate amount of $10.0 million, or the Term B Loan, governed by a loan and security agreement, or the LoanSecurity Agreement between the Company andwith Oxford Finance LLC, or Oxford, as Collateral Agent and a Lender, and SVB, as a Lender. The Term A Loan was funded in full on August 31, 2020,Silicon Valley Bank, or the Funding Date. The second draw period expired on September 30, 2021Loan and Security Agreement, and such facility, the 2020 Term Loan. On March 10, 2023, Silicon Valley Bank was closed by the California Department of Financial Protection and Innovation, and the Federal Deposit Insurance Corporation, or the FDIC, was appointed as receiver. On March 13, 2023, the FDIC announced that all of Silicon Valley Bank’s deposits and substantially all of its assets had been transferred to a newly created, full-service, FDIC-operated bridge bank, Silicon Valley Bridge Bank, N.A., or SVBB. SVBB assumed all loans that were previously held by Silicon Valley Bank. On March 27, 2023, First-Citizens Bank & Trust Company assumed all of SVBB’s customer deposits and certain other liabilities and acquired substantially all of SVBB’s loans and certain other assets from the FDIC, including the 2020 Term B Loan is no longer available to be drawn by the Company.Loan.
On March 21, 2022,September 11, 2023, the Company entered into a Second Amendmentpayoff letter with Oxford and SVB, pursuant to itswhich the Company paid all outstanding amounts under the 2020 Term Loan, together with accrued interest and a prepayment penalty, resulting in the full extinguishment of the 2020 Term Loan. The Second Amendment extended the date on which amortization payments in respecttotal payoff amount was $22.3 million, consisting of the 2020 Term Loan will commence to April 1, 2023. Thereafter, amortization payments will be paid monthly in equal installments of principal and interest to fully amortize the outstanding principal over the remaining term of the 2020 Term Loan, subject to recalculation upon a change in the prime rate.
The Second Amendment was determined to be a loan modification, and the $0.1 million fee was recorded as an addition to the debt discount on the effective date.
The 2020 Term Loan will mature on August 1, 2025. Each advance under the Term Loan accrues interest at a floating per annum rate equal to the greater of (a) 7.90%, and (b) the lesser of (x) the sum of (i) the prime rate reported in The Wall Street Journal on the last business day of the month that immediately precedes the month in which the interest will accrue, and (ii) 4.65% and (y) 10.00%. The Term Loan provides for interest-only payments on a monthly basis until April 1, 2023. Thereafter, amortization payments will be payable monthly in equal installments of principal and interest to fully amortize the outstanding principal over the remaining term of the loan, subject to recalculation upon a change in the prime rate. The Company may prepay the Term Loan in full but not in part provided that the Company (i) provides ten days’ prior written notice to Collateral Agent, (ii) pays on the date of such prepayment (A) all outstanding principal plus accrued and unpaid interest, and (B) a prepayment fee of between 3.0% and 1.0% of the aggregate original principal amount advanced bydue of $19.8 million, the lender depending on the timing of the prepayment. Amounts outstanding during an event of default are payable upon SVB’s demand and shall accrue interest at an additional rate of 5.0% per annum of the past due amount outstanding. At the end of the loan term (whether at maturity, by prepayment in full or otherwise), the Company shall make a final payment to the lender in the amount of 9.0% of the aggregate original principal amount advanced by the lender. The final payment fee totalingof $2.3 million, is recorded as a loan discount.
The Term Loan is secured by a lien on substantially allthe prepayment penalty of the assets of the Company, other$0.2 million, and less than intellectual property, provided that such lien on substantially all assets includes any rights to payments and proceeds from the sale, licensing or disposition of intellectual property. The Company has also granted the Collateral Agent a negative pledge with respect to its intellectual property.
The Loan Agreement contains customary covenants and representations, including but not limited to financial reporting obligations and limitations on dividends, indebtedness, collateral, investments, distributions, transfers, mergers or acquisitions, taxes, corporate changes, deposit accounts, and subsidiaries. The Loan Agreement also contains other customary provisions, such as expense reimbursement, non-disclosure obligations as well as indemnification rights for the benefit of the Collateral Agent.
The events of default under the Loan Agreement include, but are not limited to, the Company’s failure to make any payments of principal or interest under the Loan Agreement or other transaction documents, the Company’s breach or default in the performance of any covenant under the Loan Agreement or other transaction documents, the occurrence of a material adverse change, the Company making a false or misleading representation or warranty in any material respect under the Loan Agreement, the Company’s insolvency or bankruptcy, any attachment or judgment on the Company’s assets of at least $0.5 million, or the occurrence of any default under any agreement or obligation of the Company involving indebtedness in excess of $0.5 million. If an event of default occurs, the Collateral Agent is entitled to take enforcement action, including acceleration of amounts due under the Loan Agreement.
The Company assessed all terms and features of the 2020 Term Loan to identify any potential embedded features that would require bifurcation. As part of this analysis, the Company assessed the economic characteristics and risks of the 2020 Term Loan, including any put, call, and contingent features. The Company determined that the interest rate collar and prepayment call option did not require bifurcation; whereas the contingent put option and default (contingent) interest rate feature met bifurcation criteria resulting in immaterial amounts.
Warrants
On August 31, 2020, in connection with the 2020 Term A Loan, the Company issued warrants to the Lenders to purchase an aggregate of 196,850 shares of its common stock at an exercise price equal to $2.54 per share. In accordance with ASC 815-40, these warrants are classified as permanent equity in the accompanying consolidated balance sheets and will expire ten years from the date of issuance. The initial grant date fair value of the warrants was $0.4 million as determined by the Black-Scholes valuation model and recorded to stockholders' equity, with the SVB portion allocated to the reacquisition price of the 2017 Term Loan and the Oxford fair value portion as a loan discount to the Term A Loan.
Payoff
On the Funding Date, the Company entered into a payoff letter with SVB, pursuant to which the Company utilized $13.7$0.1 million of the 2020 Term Loan to pay off all outstanding obligations under the previous term loan, consisting of the principal payment, final prepayment and accrued interest.
During the year ended December 31, 2020,2023, the Company recognizedrecorded a loss of $0.7 million on the extinguishment of debt in the amount2020 Term Loan, consisting of the prepayment penalty of $0.2 million and the write-off of $0.5 million determined asof unamortized debt issuance costs and venture debt termination fee, which was included within interest expense in the difference between the reacquisition priceconsolidated statements of operations and carrying value at August 31, 2020.comprehensive income (loss).
As of December 31, 20222023, the Company had no outstanding borrowings, and 2021,as of December 31, 2022, the outstanding principal balance under the 2020 Term Loan was $25.0 million.
Total 2020 Term Loan and unamortized debt discount balances as of December 31, 2022 are as follows (in thousands):
| | | | | |
Face value | $ | 25,000 | |
Venture debt termination fee | 2,250 | |
Less: Debt discount | (988) | |
Less: Current portion of loan payable | (8,476) | |
Loan payable, net of current portion | $ | 17,786 | |
Future minimum principal payments on the 2020 Term Loan as of December 31, 2022 are as follows (in thousands):
| | | | | |
| Year ended: | |
| |
| 2023 | 7,759 | |
| 2024 | 10,345 | |
| 2025 | 6,896 | |
| Total minimum principal payments | $ | 25,000 | |
During the years ended December 31, 2023, 2022 2021 and 2020,2021, the Company recognized $2.1 million, $3.0 million $2.8 million and $1.6$2.8 million respectively of interest expense related to the 2020 Term Loan and the previous term loan.Loan.
10.11. Series A Preferred Stock
The Certificate of Designation was filed on November 13, 2023, which provided for the designation of shares of the Series A Preferred Stock and authorized the issuance of 548,375 shares of Series A Preferred Stock.
Additionally on November 13, 2023, the Company entered into the Securities Purchase Agreement with (i) Dr. Timothy A. Springer, a member of the Company’s Board of Directors; (ii) TAS Partners LLC, an affiliate of Dr. Springer, and (iii) Seven One Eight Three Four Irrevocable Trust, a trust associated with Dr. Murat Kalayoglu, a co-founder and the former chief executive officer of Old Cartesian, who joined the Company’s Board of Directors effective immediately after the effective time of the Merger, or the Investors. Pursuant to the Securities Purchase Agreement, the Company agreed to issue and sell an aggregate of 149,330.115 shares of Series A Preferred Stock for an aggregate purchase price of $60.25 million in the November 2023 Private Placement.
In the November 2023 Private Placement Dr. Timothy A. Springer agreed to settle his purchases in three tranches of shares of Series A Preferred Stock, the first for a purchase price of $10.0 million and each thereafter for a purchase price of approximately $20.0 million, with the three tranches settling 30, 60, and 90 days, respectively, following the Closing Date. TAS Partners LLC agreed to settle its purchase for approximately $10.0 million within 30 days following the Closing Date. The first, second and third tranches were settled on December 13, 2023, January 12, 2024 and February 11, 2024, respectively, under which (i) 24,785.081 shares of Series A Preferred Stock were issued to each of TAS Partners LLC and Dr. Timothy A. Springer in the first tranche, (ii) 49,570.163 shares of Series A Preferred Stock were issued to Dr. Timothy A. Springer in the second tranche, and (iii) 49,570.163 shares of Series A Preferred Stock were issued to Dr. Timothy A. Springer in the third tranche. On November 15, 2023, the Company issued 619.627 shares of Series A Preferred Stock to Seven One Eight Three Four Irrevocable Trust for $0.25 million.
The Company determined the obligation to issue 148,710.488 shares of Series A Preferred Stock to Dr. Springer and TAS Partners LLC represented a forward contract and was accounted for as a liability with changes in fair value recorded in earnings. A portion of the liability was settled with the initial issuance of 49,570.162 shares of Series A Preferred Stock on December 13, 2023 (see Note 6).
On December 5, 2023, the Company issued 384,930.724 shares of Series A Preferred Stock as part of its consideration transferred in connection with the Merger which settled the related forward contract liability (see Note 6).
As of December 31, 2023, the Company had 435,120.513 shares of Series A Preferred Stock issued and outstanding.
In accordance with the guidance in ASC 480, Distinguishing Liabilities from Equity the Series A Preferred Stock is classified outside of stockholders’ equity because the shares of Series A Preferred Stock contain redemption features that are not solely within the control of the Company. The Series A Preferred Stock is not currently redeemable, nor is it probable that the instrument will become redeemable, as it is only redeemable upon the occurrence of a contingent event. Accordingly, no accretion has been recognized for the Series A Preferred Stock and it will not be accreted until it is probable that the shares of Series A Preferred Stock will become redeemable.
The Series A Preferred Stock had the following rights and preferences as of December 31, 2023:
Conversion
Prior to the stockholder approval of the Conversion Proposal, the Series A Preferred Shares are not convertible. Following the stockholder approval of the Conversion Proposal, each share of Series A Preferred Stock will automatically convert into 1,000 shares of common stock, subject to certain limitations, including that a holder of Series A Preferred Stock is prohibited from converting shares of Series A Preferred Stock into shares of common stock if, as a result of such conversion, such holder, together with its affiliates, would beneficially own more than a specified percentage (to be established by the holder between 0% and 19.9%) of the total number of shares of common stock issued and outstanding immediately after giving effect to such
conversion; provided, however, that such beneficial ownership limitation does not apply to Dr. Springer, TAS Partners LLC, or any of their respective affiliates.
Each share of Series A Preferred Stock outstanding that is not otherwise automatically converted into common stock as a result of the beneficial ownership limitation shall be convertible at any time at the option of the holder following stockholder approval of the Conversion Proposal, only to the extent the beneficial ownership limitation does not apply to the shares of Series A Preferred Stock to be converted.
Redemption
Each share of Series A Preferred Stock will be redeemable at the option of the holder at any time following the date that is 18 months after the initial issuance date of the Series A Preferred Stock, other than any shares of Series A Preferred Stock that would not be convertible into shares of common stock as a result of the beneficial ownership limitation referred to above. The amount payable upon redemption will be equal to the average closing sale price of the common stock listed over the ten consecutive trading days ending on, and including, the day immediately prior to the redemption date multiplied by the number of shares of common stock the Series A Preferred Stock would be convertible into.
Dividends
Holders of Series A Preferred Stock are entitled to receive dividends on shares of Series A Preferred Stock on an as-converted basis equal to the dividends paid on shares of the common stock; provided, however, that holders of Series A Preferred Stock (or any shares of common stock into which the Series A Preferred Stock are convertible) are not entitled to any CVRs or any amounts paid under the CVR Agreement.
Voting
Except as otherwise required by law, the Series A Preferred Stock does not have voting rights. However, as long as any shares of Series A Preferred Stock are outstanding, the Company will not, without the affirmative vote of the holders of a majority of the then-outstanding shares of the Series A Preferred Stock, (a) alter or change adversely the powers, preferences or rights given to the Series A Preferred Stock, (b) alter or amend the Certificate of Designation, (c) amend the Charter or other organizational documents in any manner that adversely affects any rights of the holders of Series A Preferred Stock, (d) issue further shares of Series A Preferred Stock (other than in connection with the exercise of the stock options to purchase Series A Preferred Stock) or increase or decrease (other than by conversion) the number of authorized shares of Series A Preferred Stock, (e) prior to the stockholder approval of the Conversion Proposal or at any time while at least 30% of the originally issued Series A Preferred Stock remains issued and outstanding, consummate either (A) a Fundamental Transaction (as defined in the Certificate of Designation) or (B) any merger or consolidation of the Company or other business combination in which the stockholders of the Company immediately before such transaction do not hold at least a majority of the capital stock of the Company immediately after such transaction, (f) amend or fail to comply with, in any manner that would be reasonably likely to prevent, impede or materially delay the conversion (or the stockholder approval thereof), or terminate, any of the stockholder support agreements entered into in connection with the Merger, or the Support Agreements, or agree to any transfer, sale or disposition of such shares subject to the Support Agreements (except for such transfers, sales or dispositions with respect to which the approval of the Company is not required pursuant to the applicable Support Agreement) or (g) enter into any agreement with respect to any of the foregoing.
Liquidation
The holders of Series A Preferred Stock shall rank on parity with the common stockholders as to distributions of assets upon liquidation, dissolution or winding up of the Company, whether voluntarily or involuntarily.
Upon any liquidation, dissolution or winding-up of the Company, whether voluntary or involuntary each holder of Series A Preferred Stock shall be entitled to receive out the assets of the Company equal to of the same amount that a holder of common stock would receive if the Series A Preferred Stock were fully converted, which shall be paid pari passu with holders of common stock, plus an amount equal to any dividends declared but unpaid. If the assets available for distribution are not sufficient to pay the holders of the Series A Preferred Stock pursuant to the preceding sentence, the assets will be distributed ratably to the holders of the Series A Preferred Stock and common stock.
Reserved Shares
As of December 31, 2023, the Company has authorized shares of Series A Preferred Stock for future issuance as follows:
| | | | | | | |
| As of | | |
| December 31, 2023 | | |
| Shares reserved for issuance in November 2023 Private Placement | 99,140.326 | | | |
| Outstanding Series A Preferred Stock options | 14,112.299 | | | |
| Total | 113,252.625 | | | |
12. Equity
Equity Financings
Merger
On December 5, 2023, the Company issued 6,723,639 shares of common stock as part of its consideration transferred in connection with the Merger which settled the related equity-classified forward contract (see Note 3).
Underwritten Offering
On April 6, 2022, the Company entered into an underwriting agreement with SVB Securities LLC (now known as Leerink Partners LLC), as representative of the several underwriters named therein, relating to an underwritten offering of 27,428,572 shares or the Shares, of the Company’s common stock and warrants2022 Warrants to purchase up to 20,571,429 shares of common stock, or the 2022 Warrants.stock. The offering of the Sharessuch shares and the 2022 Warrants is referred to as the 2022 Offering. Each Shareshare and accompanying 2022 Warrant to purchase 0.75 shares of common stock was sold at a combined offering price of $1.41. The exercise price for the 2022 Warrants is $1.55 per share. The Company received net proceeds from the 2022 Offering of approximately $36.9 million.
The 2022 Warrants are subject to adjustment in the event of certain stock dividends and distributions, stock splits, stock combinations, reclassifications or similar events affecting the Company’s common stock and also upon any distributions for no consideration of assets to the Company’s stockholders. Each 2022 Warrant is exercisable at any time and from time to time after issuance. In the event of certain corporate transactions, the holders of the 2022 Warrants will be entitled to receive the kind and amount of securities, cash or other property that the holders would have received had they exercised the Warrants immediately prior to such transaction. Therefore, the Company is required to account for the 2022 Warrants as liabilities and record the 2022 Warrants at fair value. The 2022 Warrants do not entitle the holders thereof to any voting rights or any of the other rights or privileges to which holders of Common Stock are entitled.
“At-the-Market” Offerings
2020 Sales Agreement
On August 6, 2020, the Company filed a universal shelf registration statement on Form S-3 (File No. 333-241692) with the Securities and Exchange Commission, or the SEC, to sell an aggregate amount of up to $200.0 million of certain of its securities. The shelf registration statement was declared effective by the SEC on August 14, 2020. Concurrent with the filing of the shelf registration statement, the Company entered into a sales agreement, or the 2020 Sales Agreement with Jefferies LLC, as sales agent, pursuant to which the Company was permitted, from time to time, to issue and sell common stock with an aggregate value of up to $50.0 million in an “at the market offering.” On October 8, 2021, the Company delivered notice to Jefferies LLC that the Company was terminating the 2020 Sales Agreement, with effect as of October 19, 2021.
2021 Sales Agreement
On October 25, 2021, the Company entered into a Sales Agreement, or the 2021 Sales Agreement, with Leerink Partners LLC (then known as SVB Leerink LLCLLC), or Leerink Partners, pursuant to which the Company may sell shares of the Company’s common stock, from time to time, through an “at the market” equity offering program under which SVB Leerink Partners will act as sales agent. The shares of common stock sold pursuant to the 2021 Sales Agreement, willif any, would be issued and sold pursuant to such shelfa registration statement on Form S-3 and related prospectus supplement,to be filed on October 25, 2021by the Company with the SEC, for aggregate remaining gross sales proceeds of up to $75.0$51.0 million.
During the year ended December 31, 2023, the Company sold no shares of its common stock pursuant to the 2021 Sales Agreement. During the year ended December 31, 2022, the Company sold 774,544 shares of its common stock pursuant to the 2021 Sales Agreement for aggregate net proceeds of $2.1 million, after deducting commissions and other transaction costs. During
the year ended December 31, 2021, the Company sold 13,767,511 shares of its common stock pursuant to the 2021 and 2020 Sales Agreements for aggregate net proceeds of $51.9 million, after deducting commissions and other transaction costs.
June 2020 Sobi Stock Purchase
On June 11, 2020, the Company entered into a stock purchase agreement with Sobi, pursuant to which the Company sold an aggregate of 5,416,390 shares of its common stock at a purchase price equal to $4.6156 per share, which represented 120% of the 10-day volume-weighted average price of the Company’s common stock prior to signing, for aggregate gross proceeds of $25.0 million, or the Sobi Private Placement. The closing of the Sobi Private Placement occurred on July 31, 2020.
In accordance with ASC 815, this forward sale treatment qualified as equity classification as the shares are not within the scope of ASC 480. The gross proceeds of $25.0 million were determined to include a premium to the fair value of the Company’s shares as of July 28, 2020 of approximately $14.5 million. As a result, such amount was included in the transaction price for revenue recognition of the Sobi License. See Note 1214 for details.
Also on June 11, 2020, the Company entered into a registration rights agreement, (asas amended by that certain letter agreement, dated as of November 4, 2020, or the “SobiSobi Registration Rights Agreement”)Agreement, with Sobi, pursuant to which the Company agreed to prepare and file a registration statement with respect to the resale of the shares of common stock acquired in the Sobi Private Placement. The Company will be required to file this resale registration statement within 30 days following receipt by the Company of a written request from Sobi to file such resale registration statement, and to have the registration
statement declared effective within ten (10) Business Daysbusiness days after the SEC informs the Company that no review of such resale registration statement will be made or that the SEC has no further comments on such resale registration statement.
December 2019 Financing
On December 18, 2019, the Company entered into a securities purchase agreement, or the 2019 Purchase Agreement, with a group of institutional investors and certain members of the Board of Directors. Pursuant to the 2019 Purchase Agreement, the Company sold an aggregate of 37,634,883 shares of its common stock at a purchase price of $1.46 per share, warrants to purchase an aggregate of 22,988,501 shares of common stock at a purchase price of $0.125 per share underlying each common warrant, and pre-funded warrants to purchase an aggregate of 8,342,128 shares of common stock at a purchase price of $1.46 per share, all with five year terms, or the 2019 PIPE. The closing of the 2019 PIPE occurred on December 23, 2019. The exercise price of the pre-funded warrants is $0.0001 per share and the exercise price for the common warrants is $1.46 per share. In the event of a certain sale of the Company, the terms of the common warrants require us to make a payment to such common warrant holders based on a Black-Scholes valuation (using variables as specified in the warrants). This provision does not apply to the pre-funded warrants. Therefore, the Company is required to account for the common warrants as liabilities and record them at fair value, while the pre-funded warrants met the criteria to be classified as permanent equity.
The Company recorded the fair value of the 2019 Warrants of $40.7 million upon issuance using the Black-Scholes valuation model. Issuance costs were allocated between the equity component with an offset to additional paid-in capital and the liability component recorded as expense on a relative fair value basis. Total net proceeds from the equity offering was $65.6 million, after deducting transaction costs and commissions of $4.4 million.
As discussed in Note 5,6, the Company remeasured the Amended 2019 Warrants at the fair value of $0.8 million on December 20, 2022 and reclassified this amount to additional paid-in capital.
The remaining 2019 Warrants liability and the 2022 Warrants liability were revalued as of December 31, 20222023 at $19.1$6.4 million. During the years ended December 31, 2023, 2022 2021 and 2020,2021, the Company recorded a decrease of $12.7 million and $20.9 million and an increase of $2.3 million and $10.4 million, respectively, in the fair value of the warrants in the consolidated statements of operations and comprehensive income (loss).
June 2017 Financing
In June 2017, the Company entered into a securities purchase agreement, or the Institutional Purchase Agreement, with a select group ofcertain institutional investors or the Institutional Investors and a securities purchase agreement with Timothy A. Springer, Ph.D., a member of the boardBoard of directors,Directors, or the Springer Purchase Agreement, for a private placement of the Company’s securities, or the 2017 PIPE. Pursuant to the Institutional Purchase Agreement, the Company sold an aggregate of 2,750,000 shares of its common stock at a purchase price equal to $16.00 per share. Pursuant to the Springer Purchase Agreement, the Company sold to Dr. Springer an aggregate of 338,791 shares of common stock at a purchase price equal to $17.71 per share, which was equal to the most recent consolidated closing bid price on the Nasdaq Stock Market on June 23, 2017, and warrants to purchase up to 79,130 shares of common stock, or the Warrant Shares, exercisable at $17.71 per Warrant Share, and with a term of five years. The equity classifiedequity-classified warrants expired induring the year ended December 31, 2022.
Warrants
The following is a summary of warrant activity for the years ended December 31, 20222023 and 2021:2022:
| | Number of Warrants | |
| Equity
classified | | Liability classified | | Total | | Weighted average
exercise price |
| Outstanding at December 31, 2020 | 292,469 | | | 12,085,547 | | | 12,378,016 | | | $ | 1.60 | |
| Exercises | — | | | (1,642,036) | | | (1,642,036) | | | 1.46 | |
| | Number of Warrants | |
| Equity
classified | |
| Equity
classified | |
| Equity
classified | | | Liability classified | | Total | | Weighted average
exercise price |
| Outstanding at December 31, 2021 | Outstanding at December 31, 2021 | 292,469 | | | 10,443,511 | | | 10,735,980 | | | $ | 1.62 | |
| | Issuance | |
| Issuance | |
| Issuance | Issuance | — | | | 20,571,429 | | | 20,571,429 | | | 1.55 | |
| Canceled | Canceled | (79,130) | | | — | | | (79,130) | | | 17.71 | |
| Reclassification of warrant liability to equity on modification | Reclassification of warrant liability to equity on modification | 2,022,987 | | | (2,022,987) | | | — | | | $ | 1.46 | |
| Outstanding at December 31, 2022 | Outstanding at December 31, 2022 | 2,236,326 | | | 28,991,953 | | | 31,228,279 | | | $ | 1.53 | |
| | Canceled | |
| | Canceled | |
| | Canceled | |
| | Outstanding at December 31, 2023 | |
| Outstanding at December 31, 2023 | |
| Outstanding at December 31, 2023 | |
Common Stock
On June 17, 2022, at the 2022 Annual Meeting of Stockholders, the Company’s stockholders approved an amendment to the Company’s Restated Certificate of Incorporation, or the Charter, to increase the number of authorized shares of common stock, par value $0.0001 per share, from 200,000,000 to 350,000,000 shares. On June 21, 2022, the Company filed a Certificate of Amendment to the Charter with the Secretary of State of the State of Delaware, which became effective upon filing.
As of December 31, 2022,2023, the Company had 350,000,000 shares of common stock authorized for issuance, $0.0001 par value per share, with 153,042,435161,927,821 shares issued and outstanding. The voting, dividend and liquidation rights of the common stockholders are subject to and qualified by the rights, powers and preferences of the preferred stock. The common stock has the following characteristics:
Voting
The commonCommon stockholders are entitled to one vote for each share of common stock held with respect to all matters voted on by the stockholders of the Company.
Dividends
The commonCommon stockholders are entitled to receive dividends, if and when declared by the Board of Directors. Through December 31, 2022,2023, no cash dividends have been declared or paid on common stock.
Liquidation
Upon liquidation of the Company, the common stockholders are entitled to receive all assets of the Company available for distribution to such stockholders.
Reserved Shares
The Company has authorized shares of common stock for future issuance as follows:
| | | | | | | | | | | |
| | As of |
| | December 31, 2022 | | December 31, 2021 |
| Exercise of warrants | 31,228,279 | | | 10,735,980 | |
| Shares available for future stock incentive awards | 5,782,591 | | | 6,039,564 | |
| | | |
| Unvested restricted stock units | 1,705,558 | | | 394,450 | |
| Outstanding common stock options | 15,844,651 | | | 11,039,873 | |
| Total | 54,561,079 | | | 28,209,867 | |
| | | | | | | |
| As of | | |
| December 31, 2023 | | |
| Exercise of warrants | 31,224,703 | | | |
| Shares available for future stock incentive awards | 35,836,268 | | | |
| | | |
| | | |
| Outstanding common stock options | 23,306,661 | | | |
| Total | 90,367,632 | | | |
As described in Note 11, prior to the stockholder approval of the Conversion Proposal, the Series A Preferred Shares are not convertible. Following the stockholder approval of the Conversion Proposal, each share of Series A Preferred Stock will automatically convert into 1,000 shares of common stock.
11.13. Stock Incentive Plans
The Company maintainsmaintained the 2008 Stock Incentive Plan, or the 2008 Plan, for employees, consultants, advisors, and directors. The 2008 Plan provided for the granting of incentive and non-qualified stock option and restricted stock awards as determined by the Board. In connection with the Merger, all outstanding awards issued under the 2008 Plan were cancelled, and the Board formally terminated the 2008 Plan.
In June 2016, the Company’s stockholders approved the 2016 Incentive Award Plan, or the 2016 Plan, which authorized 1,210,256 shares of common stock for future issuance under the 2016 Plan and the Company ceased granting awards under the 2008 Plan. Upon the effective date of the 2016 Plan, awards issued under the 2008 Plan remainremained subject to the terms of the 2008
Plan. Awards granted under the 2008 Plan that expire, lapseexpired, lapsed or terminate becometerminated became available under the 2016 Plan as shares available for future grants.
Additionally, pursuant to the terms of the 2016 Plan, the Board is authorized to grant awards with respect to common stock, and may delegate to a committee of one or more members of the Board or executive officers of the Company the authority to grant options and restricted stock units. On December 9, 2020, the Board established a Stock Option Committee authorized to grant awards to certain employees and consultants subject to conditions and limitations within the 2016 Plan. In January 20222023 and 2021,2022, the number of shares of common stock that may be issued under the 2016 Plan was increased by 4,944,9196,121,697 and 4,322,8504,944,919 shares, respectively. As of December 31, 2022, 1,719,0152023, 22,504,503 shares remain available for future issuance under the 2016 Plan.
In September 2018, the Company’s 2018 Employment Inducement Incentive Award Plan, or the 2018 Inducement Incentive Award Plan was adopted by the Board without stockholder approval pursuant to Rule 5635(c)(4) of the Nasdaq Stock Market LLC listing rules, which authorized 1,175,000 shares of its common stock for issuance. In March 2019, the Board approved thean amendment and restatement of the 2018 Inducement Incentive Award Plan to reserve an additional 2,000,000 shares of the Company’s common stock for issuance thereunder. In December 2023, the Board approved an amendment and restatement of the 2018 Inducement Incentive Award Plan to reserve an additional 1,825,000 shares of the Company’s common stock for issuance thereunder. As of December 31, 2022,2023, there are 425,8584,500,858 shares available for future grant under the 2018 Inducement Incentive Award Plan.
In accordance with the Merger Agreement, the Company assumed Old Cartesian’s 2016 Stock Incentive Plan, or the Old Cartesian Plan. The Old Cartesian Plan permits the granting of options or restricted stock to employees, officers, directors, consultants and advisors to the Company. The unvested common stock options and Series A Preferred Stock options assumed by the Company in connection with the Merger generally vest over a four-year period. Additionally, the stock options granted
have a contractual term of ten years and only full shares can be exercised as per the individual award agreements. As of December 31, 2023, there are 3,848,809 shares available for future grant under the Old Cartesian Plan.
In connection with the Merger, the outstanding stock options to purchase Old Cartesian common stock were converted into stock options to purchase 23,306,661 shares of common stock and 14,112.299 shares of Series A Preferred Stock of the Company. These replacement awards were revalued at their acquisition-date fair value and then attributed to pre and post-combination service. This resulted in $2.6 million attributed to post-combination service to be recognized as stock-based compensation expense over the remaining terms of the replacement awards, of which $0.2 million was recognized as research and development expense in the consolidated statements of operations and comprehensive (loss) income during the year ended December 31, 2023.
Settlement of Equity Compensation Awards
Upon consummation of the First Merger, the equity compensation awards of the Company outstanding as of the date of the Merger were settled as follows: (i) each unvested option to acquire shares of common stock and each unvested restricted stock unit award with respect to shares of common stock was accelerated and vested in full at the effective time of the First Merger; (ii) each option to acquire shares of common stock was canceled and in exchange therefore, former holders became entitled to receive an amount in cash equal to the product of (A) the total number of shares of common stock subject to the unexercised portion the stock option (determined after giving effect to the accelerated vesting) multiplied by (B) the excess, if any, of $2.06, or the Cash-out Amount, over the applicable exercise price per share of common stock under such stock option; and (iii) each restricted stock unit award with respect to shares of common stock was cancelled and the former holder of such canceled restricted stock unit became entitled, in exchange therefor, to receive an amount in cash equal to the product of (A) the total number of shares of common stock deliverable under such restricted stock unit (determined after giving effect to the accelerated vesting) multiplied by (B) the Cash-out Amount. Stock options with an exercise price in excess of the Cash-out Amount received no cash payment.
The modification to accelerate the vesting of all awards upon the Merger resulted in full recognition of unrecognized compensation of $13.1 million, of which $5.9 million and $7.2 million was classified as research and development expense and general and administrative expense, respectively, in the consolidated statements of operations and comprehensive (loss) income. In addition, with the exception of any options with an exercise price greater than $2.06 per share, all awards were settled in cash for an amount equal to $2.06 less any exercise price associated with the awards. The total cash payment made to the holders of stock options and restricted stock units was $9.4 million. The fair value of the awards prior to the settlement was recorded to additional paid-in capital in an amount of $6.2 million and the amount in excess of fair value was recognized as additional stock-based compensation expense in an amount of $3.2 million, of which $1.5 million and $1.7 million was classified as research and development expense and general and administrative expense, respectively, in the consolidated statements of operations and comprehensive (loss) income.
Stock-Based Compensation Expense
Stock-based compensation expense by classification included within the consolidated statements of operations and comprehensive income (loss), including $1.5 million recognized as stock-based compensation expense upon the achievement of a technical milestone by Ginkgo Bioworks Holdings, Inc., or Ginkgo, during the year ended December 31, 2023 and $1.0 million recognized as stock-based compensation expense upon the issuance of common stock to Ginkgo during the year ended December 31, 2022 as described in Note 16, was as follows (in thousands):
| | | |
| | | | | Year Ended December 31, | | | | Year Ended December 31, |
| | | | 2022 | | 2021 | | 2020 | | | | | | | | | 2023 | | 2022 | | 2021 |
| Research and development | Research and development | | $ | 5,061 | | | $ | 3,204 | | | $ | 2,271 | |
| General and administrative | General and administrative | | 6,133 | | | 4,516 | | | 3,151 | |
| Total stock-based compensation expense | Total stock-based compensation expense | | $ | 11,194 | | | $ | 7,720 | | | $ | 5,422 | |
Stock Options
The fair value of the stock options assumed in connection with the Merger was calculated using a Black-Scholes option pricing model based on the following weighted-average assumptions:
| | | | | | | | | | | |
| Common Stock | | Series A Preferred Stock |
| Risk-free interest rate | 4.83 | % | | 4.92 | % |
| Dividend yield | — | | | — | |
| Expected term | 3.59 | | 3.29 |
| Expected volatility | 83.77 | % | | 83.87 | % |
| Weighted-average fair value of common stock or Series A Preferred Stock, as applicable | $ | 0.40 | | | $ | 403.47 | |
The estimated grant date fair values of employee stock option awards granted under the 2016 Plan and the 2018 Inducement Incentive Award Plan were calculated using the Black-Scholes option pricing model, based on the following weighted-average assumptions:
| | | Year Ended December 31, |
| | | | | Year Ended December 31, | |
| | | | | Year Ended December 31, | |
| | | | | Year Ended December 31, | |
| | | | 2022 | | 2021 | | 2020 | | | | | | | | | | 2023 | | 2022 | | 2021 |
| Risk-free interest rate | Risk-free interest rate | | 2.24 | % | | 0.79 | % | | 1.11 | % | Risk-free interest rate | | | | | | | | | 3.95 | % | | 2.24 | % | | 0.79 | % |
| Dividend yield | Dividend yield | | — | | | — | | | — | |
| Expected term | Expected term | | 6.02 | | 6.03 | | 6.04 | Expected term | | | | | | | | | 5.94 | | 6.02 | | 6.03 |
| Expected volatility | Expected volatility | | 92.21 | % | | 95.04 | % | | 91.00 | % | Expected volatility | | | | | | | | | 94.64 | % | | 92.21 | % | | 95.04 | % |
| Weighted-average fair value of common stock | Weighted-average fair value of common stock | | $ | 2.63 | | | $ | 3.58 | | | $ | 2.49 | |
The expected term of the Company's stock options granted to employees has been determined utilizing the "simplified" method for awards that qualify as "plain-vanilla" options. Under the simplified method, the expected term is presumed to be the midpoint between the vesting date and the end of the contractual term. The Company utilizes this method due to lack of historical exercise data and the plain nature of its stock-based awards. Expected volatilities are based on the Company’s historical volatility.
The weighted average grant date fair value of stock options granted to employees during the years ended December 31, 2023, 2022 and 2021 was $0.90, $1.99, and 2020 was $1.99, $2.73 and $1.86 respectively.
As of December 31, 2022,2023, total unrecognized compensation expense related to unvested employeecommon stock options and Series A Preferred Stock options was $14.8$1.4 million and $1.0 million, respectively, which is expected to be recognized over a weighted average period of 2.6 years.
2.4 years and 2.5 years, respectively.
The following table summarizes the stock option activity under the 2008 Plan, the 2016 Plan, and the 2018 Inducement Incentive Award Plan:Plan, and Old Cartesian Plan for options for common stock:
| | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | Weighted-average | | |
| | | | | | remaining | | Aggregate |
| | Number of | | Weighted-average | | contractual term | | intrinsic value |
| | options | | exercise price ($) | | (in years) | | (in thousands) |
| Employees | | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| Outstanding at December 31, 2021 | 10,616,800 | | | $ | 3.99 | | | 8.19 | | $ | 4,982 | |
| Granted | 6,714,220 | | | $ | 2.63 | | | | | |
| Exercised | (69,928) | | | $ | 2.23 | | | | | |
| Forfeited | (1,682,680) | | | $ | 3.73 | | | | | |
| Outstanding at December 31, 2022 | 15,578,412 | | | $ | 3.44 | | | 7.57 | | $ | 4 | |
| | | | | | | |
| Vested at December 31, 2022 | 6,985,993 | | | $ | 4.18 | | | 6.50 | | $ | — | |
| Vested and expected to vest at December 31, 2022 | 14,749,359 | | | $ | 3.49 | | | 7.64 | | $ | 4 | |
| | | | | | | |
| Non-employee consultants | | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| Outstanding at December 31, 2021 | 423,073 | | | $ | 6.34 | | | 3.85 | | $ | 42 | |
| | | | | | | |
| | | | | | | |
| Forfeited | (156,834) | | | $ | 3.44 | | | | | |
| Outstanding at December 31, 2022 | 266,239 | | | $ | 8.05 | | | 5.08 | | $ | — | |
| | | | | | | |
| Vested at December 31, 2022 | 266,239 | | | $ | 8.05 | | | 5.08 | | $ | — | |
| Vested and expected to vest at December 31, 2022 | 266,239 | | | $ | 8.05 | | | 5.08 | | $ | — | |
| | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | Weighted-average | | |
| | Number of | | | | remaining | | Aggregate |
| | Common Stock | | Weighted-average | | contractual term | | intrinsic value |
| | options | | exercise price ($) | | (in years) | | (in thousands) |
| Employees | | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| Outstanding at December 31, 2022 | 15,578,412 | | | $ | 3.44 | | | 7.57 | | $ | 4 | |
| Granted | 5,477,200 | | | $ | 1.15 | | | | | |
| Assumed in connection with Merger | 23,306,661 | | | $ | 0.10 | | | | | |
| Exercised | — | | | $ | — | | | | | |
| Forfeited | (2,215,020) | | | $ | 2.68 | | | | | |
| Cancelled/settled in connection with the Merger | (18,840,592) | | | $ | 2.86 | | | | | |
| Outstanding at December 31, 2023 | 23,306,661 | | | $ | 0.10 | | | 6.50 | | $ | 13,760 | |
| | | | | | | |
| Vested at December 31, 2023 | 18,067,999 | | | $ | 0.10 | | | 6.13 | | $ | 10,725 | |
| Vested and expected to vest at December 31, 2023 | 23,306,661 | | | $ | 0.10 | | | 6.50 | | $ | 13,760 | |
| | | | | | | |
| Non-employee consultants | | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| Outstanding at December 31, 2022 | 266,239 | | | $ | 8.05 | | | 5.08 | | $ | — | |
| | | | | | | |
| | | | | | | |
| Forfeited | — | | | $ | — | | | | | |
| Cancelled/settled in connection with the Merger | (266,239) | | | $ | 8.05 | | | | | |
| Outstanding at December 31, 2023 | — | | | $ | — | | | — | | $ | — | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
The following table summarizes the stock option activity under the Old Cartesian Plan for options for Series A Preferred Stock:
| | | | | | | | | | | | | | | | | | | | | | | |
| | Number of | | | | Weighted-average | | |
| | Series A | | | | remaining | | Aggregate |
| | Preferred Stock | | Weighted-average | | contractual term | | intrinsic value |
| | options | | exercise price ($) | | (in years) | | (in thousands) |
| Employees | | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| Outstanding at December 31, 2022 | — | | | $ | — | | | — | | $ | — | |
| | | | | | | |
| Assumed in connection with Merger | 14,112.299 | | | $ | 79.94 | | | | | |
| | | | | | | |
| | | | | | | |
| Outstanding at December 31, 2023 | 14,112.299 | | | $ | 79.94 | | | 5.91 | | $ | 8,601 | |
| | | | | | | |
| Vested at December 31, 2023 | 10,860.441 | | | $ | 71.67 | | | 5.15 | | $ | 6,709 | |
| Vested and expected to vest at December 31, 2023 | 14,112.299 | | | $ | 79.94 | | | 5.91 | | $ | 8,601 | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
Restricted Stock Units
During the year ended December 31, 2022,2023, the Company granted 813,2001,054,600 restricted stock awards with a weighted average fair value of $3.31$1.13 per share based on the closing price of the Company’s common stock on the date of grant to employees under the 2016 Plan, which will vestvested over a four yearfour-year term and 699,500 restricted stock awards with a weighted average fair value of $1.70 per share based on the closing price of the Company’s common stock on the date of grant to employees under the 2016 Plan, which will vest over a two year term.. Forfeitures are estimated at the time of grant and are adjusted, if necessary, in subsequent periods if actual forfeitures differ from those estimates. The Company has estimated a forfeiture rate of 10% for restricted stock awards to employees based on historical experience.
UnrecognizedThere was no unrecognized compensation expense for alland no outstanding restricted stock units was $3.4 million as of December 31, 2022, which is expected to be recognized over a weighted average period of 2.4 years.2023.
The following table summarizes the Company’s restricted stock units under the 2016 Plan and 2018 Inducement Incentive Award Plan:
| | | | Number of shares | | Weighted average
grant date
fair value ($) | | Number of shares | | Weighted average
grant date
fair value ($) |
| Unvested at December 31, 2021 | 394,450 | | | $ | 3.45 | |
| Unvested at December 31, 2022 | |
| Granted | Granted | 1,512,700 | | | 2.57 | |
| Vested | Vested | (131,430) | | | 4.10 | |
| | Forfeited | Forfeited | (70,162) | | | 3.30 | |
| Unvested at December 31, 2022 | 1,705,558 | | | $ | 2.62 | |
| Forfeited | |
| Forfeited | |
| Cancelled/settled in connection with the Merger | |
| Unvested at December 31, 2023 | |
Employee Stock Purchase Plan
In June 2016, the Company approved the 2016 Employee Stock Purchase Plan, or the ESPP, which authorized 173,076 shares of common stock for future issuance under the ESPP to participating employees. In January 20222023 and 2021,2022, the number of shares of common stock authorized for issuance under the ESPP was increased by 1,530,424 shares and 1,236,229 shares, and 1,080,711 shares,
respectively. During the year ended December 31, 2022,2023, the Company issued 120,877186,044 shares of common stock under the ESPP. As of December 31, 2022, 3,637,7182023, 4,982,098 shares remain available for future issuance under the ESPP. In connection with the Merger, the Board suspended the current ESPP offering period.
For each of the years ended December 31, 20222023 and 2021,2022, the Company recognized $0.1 million of stock-based compensation expense under the ESPP, respectively.ESPP.
12.14. Revenue Arrangements
Astellas Gene Therapies
In January 2023, the Company entered into the Astellas Agreement, with Astellas. Under the Astellas Agreement, the Company granted Astellas an exclusive license to the Company’s IdeXork technology arising from Xork, to develop and commercialize Xork for use in Pompe disease in combination with an Astellas gene therapy investigational or authorized product. Xork, Genovis’ IgG Protease, is licensed pursuant to an Exclusive License Agreement with Genovis, or the Genovis Agreement, as described in Note 16 to these consolidated financial statements. Astellas paid a $10.0 million upfront payment to the Company upon signing of the Astellas Agreement, and the Company is entitled to receive up to $340.0 million in future additional payments over the course of the partnership that are contingent on the achievement of various development and regulatory milestones and, if commercialized, sales thresholds for annual net sales where Xork is used as a pre-treatment for an Astellas investigational or authorized product. The Company is also eligible for tiered royalty payments ranging from low to high single digits. Any proceeds received from milestone payments or royalties relating to Xork would be required to be distributed to holders of CVRs, net of certain deductions.
Pursuant to the Astellas Agreement, the Company will have the exclusive right and responsibility to complete research and development of Xork products and to conduct all preclinical studies and clinical trials for Xork for use in Pompe disease with an Astellas gene therapy investigational or authorized product, or the Xork Development Services. Astellas will reimburse the Company for 25% of all budgeted costs incurred to complete the development of Xork for use in Pompe disease with an Astellas gene therapy investigational or authorized product. The Company will have control and responsibility over regulatory filings, including any investigational drug applications, biologics license applications, and marketing authorization applications relating to the licensed product. Astellas will have the exclusive right and responsibility to research, develop, and commercialize Astellas products used in combination with Xork and will have control and responsibility over all regulatory filings, including any investigational drug applications, biologics license applications, and marketing authorization applications, relating to Astellas products and Astellas products used in combination with Xork.
The Company determined the Astellas Agreement represents a service arrangement under the scope of ASC 606. The Company determined that the sublicense of Xork to Astellas, the licensed know-how, and the Xork Development Services represent a single promise and performance obligation to be transferred to Astellas over time due to the nature of the promises in the contract. As such, the Company will recognize the transaction price as revenue utilizing the input method to measure the progress of satisfying the single performance obligation to Astellas.
In determining the transaction price, the Company concluded the upfront payment of $10.0 million and development cost reimbursements of $5.5 million will be included in the initial transaction price. All other development milestones will be fully constrained and will only be included in the transaction price when the applicable milestone is deemed probable of achievement. Each of these variable consideration items were evaluated under the most likely amount method to determine whether such amounts were probable of occurrence, or whether such amounts should be constrained until they become probable. As part of its evaluation of the constraint, the Company considered numerous factors, including that receipt and timing of such development milestones is outside the control of the Company and probability of success criteria is estimated. The Company will re-evaluate the transaction price in each reporting period, as uncertain events are resolved, or as other changes in circumstances occur. In accordance with ASC 606, the Company will only recognize revenue associated with sales-based milestones and royalties when the subsequent sales thresholds are reached and underlying sales occur, respectively. The Company determined that a significant financing component does not exist in its arrangement with Astellas. The Company also determined the options to negotiate additional fields, enter into a clinical supply agreement, and enter into a commercial supply agreement do not represent material rights under the Astellas Agreement. Astellas has the right to terminate the Astellas Agreement in its entirety or on a field-by-field basis, upon 90 days’ written notice to the Company.
As of December 31, 2023, the Company recorded $2.3 million as a short-term contract liability and $3.5 million as a long-term contract liability, representing deferred revenue associated with the Astellas Agreement. As of December 31, 2023, the Company recorded a receivable of $0.3 million, representing billings for the Xork Development Services that are subject to
reimbursement by Astellas. Revenue of $5.5 million related to the Astellas Agreement was recognized during the year ended December 31, 2023.
Takeda Pharmaceuticals USA, Inc.
License and Development Agreement
On October 1, 2021, the Company entered into a License Agreement, or the Takeda Agreement, with Takeda. Under the Takeda Agreement, the Company granted Takeda an exclusive license to the Company’s ImmTOR technology initially for two specified disease indications within the field of lysosomal storage disorders. Takeda paid a $3.0 million upfront payment to the Company upon signing of the Takeda Agreement, and the Company iswas entitled to receive up to $1.124 billion in future additional payments over the course of the partnership that arewere contingent on the achievement of development or commercial milestones or Takeda’s election to continue its activities at specified development stages. The Company iswas also eligible for tiered royalties on future commercial sales of any licensed products.
Pursuant to the Takeda Agreement, the Company determined the Takeda Agreement representsrepresented a service arrangement under the scope of ASC 606, and given the reversion of the rights under the Takeda Agreement representsrepresented a penalty in substance for a termination by Takeda, the contract term would remain the stated term of the Takeda Agreement. The Company determined that the research license, the licensed know-how, and the manufactured supply and delivery of materials representrepresented a single promise and performance obligation to be transferred to Takeda over time due to the nature of the promises in the contract. The delivery of the manufactured supply iswas the predominant promise within the arrangement, as it iswas essential to the utility of the licensed intellectual property. The material to be supplied by the Company to Takeda iswas unique to the Company and cannot be obtained by other vendors. As such, consideration in the initial transaction price will bewas allocated to the single performance obligation and the recognition period would not extend beyond the initial contractual period. The Company will recognizerecognized the revenue associated with the upfront payment and combined single performance obligation utilizing the output method over the term that manufactured supply iswas delivered to Takeda.
In determining the transaction price, the Company concluded the payment associated with all the performance milestones will bewas fully constrained and only be included in the transaction price when the respective milestone iswas deemed probable of achievement. Each of these variable consideration items were evaluated under the most likely amount method to determine whether such amounts were probable of occurrence, or whether such amounts should be constrained until they become probable. As part of its evaluation of the constraint, the Company considered numerous factors, including that receipt and timing of such study milestones is outside the control of the Company and probability of success criteria is estimated. The Company will re-evaluatere-evaluated the transaction price in each reporting period, as uncertain events arewere resolved, or as other changes in circumstances occur.occurred. Takeda hashad the right to exercise covenant release rights on a field-by-field basis. If Takeda exercisesexercised its covenant release rights, we could receivehave received exercise payments per indication and would behave been entitled to significant development and commercial milestone payments and tiered royalties on commercial sales. The Company determined that a significant financing component doesdid not exist in its arrangement with Takeda. The Company also determined the options to negotiate additional fields, pursue other products, enter into a supply agreement explore additional fields, and pursue additional development under the initial fields dodid not represent material rights under the agreement. Takeda hashad the right to terminate the Takeda Agreement in its entirety or on a field-by-field basis, upon 90 days’ written notice to the Company.
On March 9, 2023, the Company was notified by Takeda of the achievement of the milestone event related to the completion of a non-clinical milestone for one of the specified disease indications within the field of lysosomal storage disorders under the Takeda Agreement. Accordingly, the Company received a milestone payment of $0.5 million during the year ended December 31, 2023.
The Takeda Agreement was terminated effective July 25, 2023, following Takeda’s decision to discontinue discovery and pre-clinical activities in adeno-associated virus, or AAV, gene therapy.
As of December 31, 2023, the Company recorded no contract liability. As of December 31, 2022, the Company recorded $0.1 million as a short-term contract liability and no long-term contract liability representing deferred revenue associated with this agreement. As of December 31, 2021, the Company recorded $1.0 million as a short-term contract liability and $1.0 million as a long-term contract liability, respectively, representing deferred revenue associated with this agreement. Revenue of $1.8$0.6 million and $1.0$1.8 million related to the Takeda Agreement was recognized during the years ended December 31, 20222023 and 2021,2022, respectively.
Swedish Orphan Biovitrum
License and Development Agreement
On June 11, 2020, the Company and Sobi entered into the Sobi License.a License and Development Agreement. Pursuant to the Sobi License, the Company has agreed to grant Sobi an exclusive, worldwide (except as to Greater China) license to develop, manufacture and commercialize the Company’s SEL-212 drug candidate, which is currently in development for the treatment of chronic refractory gout. The SEL-212 drug candidate is a pharmaceutical composition containing a combination of SEL-037, or the Compound, and ImmTOR. Pursuant to the Sobi License, in consideration of the license, Sobi agreed to pay the Company a one-time, upfront payment of $75.0 million. Sobi has also agreed to make milestone payments totaling up to $630.0 million to the Company upon the achievement of various development and regulatory milestones and, if commercialized, sales thresholds for annual net sales
annual net sales of SEL-212, and tiered royalty payments ranging from the low double digits on the lowest sales tier to the high teens on the highest sales tier. Any proceeds received from milestone payments or royalties relating to the Sobi License would be required to be distributed to holders of CVRs, net of certain deductions.
Pursuant to the Sobi License, the Company has agreed to supply (at cost) quantities of the Compound and ImmTOR as necessary for completion of the two Phase 3 clinical trials of SEL-212 (DISSOLVE I and DISSOLVE II) and a 6-monthsix-month placebo extension. The Company iswas required to supply quantities of the Compound until all rights to the Compound and any materials needed to manufacture the Compound arewere transferred to Sobi.Sobi, which transfer occurred upon the execution of Amendment No. 1 to the License and Development Agreement on October 31, 2023. Sobi has agreed to reimburse the Company for all budgeted costs incurred to complete development of SEL-212, including but not limited to costs incurred while conducting and completing the Phase 3 DISSOLVE trials, except for any costs of additional development activities required that are related to ImmTOR and that are unrelated to SEL-212. Sobi will have control and responsibility over all regulatory filings, including any investigational drug applications (IND), biologics license applications (BLA), and marketing authorization applications (MAA) relating to the licensed product.
The transactions contemplated by the Sobi License were consummated on July 28, 2020. Sobi may terminate the Sobi License for any reason upon 180 days’ written notice to the Company, whereby all rights granted under the Sobi License would revert back to the Company. In addition, if Sobi were to terminate the Sobi License, the Company has the option to obtain a license to all patents and know-how necessary to exploit SEL-212 in existence as of the termination date from Sobi in return for making an equitable royalty payment to Sobi.
Additionally, on June 11, 2020, the Company entered into the Sobi Purchase Agreement in connection with the Sobi License. The closing of the Sobi Private Placement occurred on July 31, 2020, following the closing of the transactions contemplated under the Sobi License. See Note 1012 for details.
The Company determined that the Sobi License represents a service arrangement under the scope of ASC 606. In addition, given the Sobi License and Sobi Purchase Agreement were executed contemporaneously and negotiated as a package with a single commercial objective, the Company will account for the two agreements as a single contract. The term of the Sobi License commenced upon the effective date of July 28, 2020 and will continue on a product-by-product basis until the royalty terms for each country have expired. The royalty term for a given product begins upon the first commercial sale of the product in a country and ends at the later of ten years from the first commercial sale, expiration of the last valid patent claim covering the product and expiration of all regulatory exclusivity periods for the product in a country. Given the reversion of the rights under the Sobi License represents a penalty in substance for a termination by Sobi, the contract term would remain the stated term of the Sobi License.
The Company determined that the Sobi License contains three distinct performance obligations due to the nature of the promises in the contract, which includes conducting the Phase 3 DISSOLVE trials, Sobi’s option to set-up a second source supplier, and a combined obligation comprised of the delivery of the license to SEL-212, transfer of the know-how and the manufacturing and delivery of SEL-212 supply for development, or the Combined License Obligation. As the set-up of a second source supplier is optional for Sobi and the Company will be reimbursed at cost for its efforts in the subsequent set-up and technology transfer, the option for this future service was determined to be at a significant and incremental discount to its standalone selling price and treated as a material right in the arrangement, namely a distinct performance obligation.
In determining the transaction price, the Company concluded the upfront payment of $75.0 million and the $5.0 million development milestone associated with the dosing of the first patient in the Phase 3 DISSOLVE trials will bewere included in the transaction price. All other development milestones will be fully constrained and only be included in the transaction price when the respective milestone is deemed probable of achievement. Each of these variable consideration items was evaluated under the most likely amount method to determine whether such amounts were probable of occurrence, or whether such amounts should be constrained until they become probable. As part of the evaluation of the constraint, the Company considered numerous factors, including that receipt of such milestones is outside the control of the Company and probability of success criteria is estimated. The Company re-evaluates the transaction price in each reporting period, as uncertain events are resolved. In accordance with ASC 606, the Company will only recognize revenue associated with sales-based milestones and royalties when the subsequent sales thresholds are reached and underlying sales occur, respectively. In connection with the Sobi Purchase Agreement, the Company determined that the gross proceeds of $25.0 million from the Sobi Private Placement included a premium to the fair value of the Company’s shares as of July 28, 2020 equal to approximately $14.5 million. The premium amount is included in the transaction price for revenue recognition. The Company estimates and includes in the transaction price the total reimbursements to be received from Sobi for both the manufacturing and delivery of the Compound and ImmTOR as well as conducting the Phase 3 DISSOLVE trials. The Company determined that a significant financing component does not exist in its arrangement with Sobi.
The Company allocated the transaction price based on the relative standalone selling prices of the three distinct performance obligations. The Company estimated the standalone selling price of conducting the Phase 3 DISSOLVE trials by forecasting its anticipated costs and applying a margin reflective of the industry. The Company must determine the standalone
selling price of the second source supplier option by determining the discount given to Sobi multiplied by the likelihood that Sobi will exercise the option in the future. Similar to the Phase 3 program estimate, the Company estimated the discount of the
option by forecasting the set-up costs and applying a margin that is reflective of the industry. As the Company will be providing the set-up and technology transfer services and the future supply at cost, the discount of the option is equal to the margin amount. The Company considered discussions with Sobi as well as probability of regulatory success of SEL-212 in determining the likelihood of exercise. The Company estimated the standalone selling price of the Combined License Obligation by utilizing a discounted cash flow model.
The Company determined that the delivery of the supply to Sobi best represents the pattern of delivery of the Combined License Obligation as the supply is essential to the utility of the license and know-how. The Company will recognize the revenue allocated to the Combined License Obligation by utilizing the output method. The Company estimated the total supply of the Compound and ImmTOR to be required during the clinical trial period and will recognize revenue as this supply is shipped for use in the clinical trials. The Company will recognize the revenue allocated to the conducting of the Phase 3 DISSOLVE trials obligation by utilizing the input method. The Company estimated the total budgeted costs to be incurred over the Phase 3 DISSOLVE trials and will recognize revenue as these costs are incurred. The Company’s costs best represent the pattern of transfer as these will capture all performance of the trials completed to date and can be readily measured. The Company will recognize the revenue allocated to the second source supplier option when the future services and goods are transferred.
On June 29, 2022, the Company completed enrollment of the DISSOLVE II trial. The completion of enrollment of the DISSOLVE II trial resulted in the achievement of a development milestone and a $10.0 million payment obligation from Sobi to the Company. This amount was added to the overall transaction price and payment was received during the year ended December 31, 2022.
As of DecemberOn October 31, 2022,2023, the Company recorded no short-term or long-term contract liability. As of December 31, 2021,and Sobi entered into Amendment No. 1 to the License and Development Agreement, pursuant to which the Company recorded $37.5 million, asgranted Sobi an exclusive license to manufacture ImmTOR solely in connection with Sobi’s development of SEL-212 under the License and Development Agreement and transferred certain contracts and manufacturing equipment to Sobi. Additionally, in connection with entry into the amendment, Sobi agreed to make employment offers to certain of the Company’s employees engaged in ImmTOR manufacturing activities on or prior to a short-term contract liabilityspecified date, and $5.1 million, as a long-term contract liability, representing deferred revenue associated with this agreement. In addition, as of December 31, 2022 the Company has recorded $0.1 millionagreed not to terminate the employment of contract assets relatedsuch employees prior to incremental costs that would not have been incurred ifsuch specified date. The Company maintains no responsibilities to Sobi to manufacture, or supply Sobi with, ImmTOR under the Sobi License had not been obtained, of which $0.1 million is presented in prepaid expenses and other current assets and less than $0.1 million is presented in other assets in the accompanying consolidated balance sheets. Amortization of contract assets was $0.6 million for the year ended December 31, 2022.License.
As of December 31, 20222023 and 2021,2022, the Company recorded a total outstanding receivable of $5.0$4.6 million and $9.9$5.0 million, respectively, representing billings for the Phase 3 DISSOLVE program that are subject to reimbursement by Sobi. AsAdditionally, as of December 31, 2023 and 2022, the Company recorded a total unbilled receivable of $3.0 million and $3.2 million, respectively, representing revenue earned but not yet billed for the Phase 3 DISSOLVE program. Revenue of $82.6$19.4 million and $83.5$82.6 million related to the Sobi License was recognized during the years ended December 31, 20222023 and 2021,2022, respectively, inclusive of $13.4$1.1 million of revenue recognized from performance obligations related to prior periods as a result of the change in transaction price during the year ended December 31, 2022.2023.
Sarepta Therapeutics, Inc.
Research License and Option Agreement
In June 2020, the Company and Sarepta Therapeutics, Inc., or Sarepta, entered into a Research License and Option Agreement, or the Sarepta Agreement. Pursuant to the Sarepta Agreement, the Company agreed to grant Sarepta a license under the Company’s intellectual property rights covering the Company’s antigen-specific biodegradable nanoparticle encapsulating ImmTOR to research and evaluate ImmTOR in combination with Sarepta’s adeno-associated virus gene therapy technology, or gene editing technology, using viral or non-viral delivery, to treat Duchenne Muscular Dystrophy and certain Limb-Girdle Muscular Dystrophy subtypes, or the Indications. Sarepta initially had an option term of 24 months during which it could opt-in to obtain an exclusive license to further develop and commercialize the Product to treat at least one Indication, with a potential to extend the option term for an additional fee. The Company will supply ImmTOR to Sarepta for clinical supply on a cost-plus basis.
Sarepta paid a $2.0 million upfront payment to the Company upon signing of the Sarepta Agreement, and the Company is eligible to receive additional preclinical payments during the option term. If Sarepta opts-in to an exclusive license agreement, the Company could receive option exercise payments per Indication upon execution of the exclusive license, and the Company would be entitled to significant development and commercial milestone payments and tiered royalties ranging from the mid-to-high single digits based on net sales.
Pursuant to the Sarepta Agreement, the Company determined the Sarepta Agreement represents a service arrangement under the scope of ASC 606, with a 24 month24-month contract duration. Given the reversion of the rights under the Sarepta Agreement represents a penalty in substance for a termination by Sarepta, the contract term would remain the stated term of the Sarepta Agreement.
The Company determined that the Sarepta Agreement and supply obligation including the delivery of the research license, the licensed know-how, the manufactured supply and delivery of materials represent a single promise and performance obligation to be transferred to Sarepta over time due to the nature of the promises in the contract. The delivery of the
manufactured supply is the predominant promise within the arrangement, as it is essential to the utility of the licensed intellectual property. As such, consideration in the initial transaction price will be allocated to the single performance obligation based on the contractual price.
In determining the transaction price, the Company concluded the payment associated with all the performance milestones will be fully constrained and only be included in the transaction price when the respective milestone is deemed probable of achievement. Each of these variable consideration items was evaluated under the most likely amount method to determine whether such amounts were probable of occurrence, or whether such amounts should be constrained until they become probable. As part of its evaluation of the constraint, the Company considered numerous factors, including that receipt of such study milestones is outside the control of the Company and probability of success criteria is estimated.
The Company also determined the option to enter into a future commercial license agreement and extend the term of the option does not represent a material right since it was not priced at an incremental discount. Sarepta may terminate the Sarepta Agreement for any reason upon 30 days’ written notice to the Company. The Sarepta Agreement contains other customary terms and conditions, including representations and warranties, covenants, termination, and indemnification obligations in favor of each party. During the year ended December 31, 2020, the Company and Sarepta entered into two amendments relating to an additional feasibility study. During the year ended December 31, 2021, the Company and Sarepta entered into a third amendment relating to the additional feasibility study. During the year ended December 31, 2022, the Company and Sarepta entered into two amendments relating to the additional feasibility study.
On April 13, 2021, the Company was notified by Sarepta of the achievement of the milestone event related to the completion of a non-clinical study for Duchenne muscular dystrophy and certain limb-girdle muscular dystrophies under the Sarepta Agreement. Accordingly, the Company received a milestone payment of $3.0 million during the three months ended June 30, 2021.
On June 10, 2022, the Company was notified by Sarepta that Sarepta would be extending their options under the Sarepta Agreement. In exchange for a nine-monthnine-month extension to Sarepta’s options to both Duchenne muscular dystrophy and certain limb-girdle muscular dystrophies, the Company received a milestone payment of $2.0 million during the three monthsyear ended September 30,December 31, 2022.
On June 15, 2022, the Company was notified by Sarepta of the achievement of a milestone event related to certain preclinical study milestones under the Sarepta Agreement. Accordingly, the Company received a milestone payment of $4.0 million during the three monthsyear ended September 30,December 31, 2022.
On March 13, 2023, the Company was notified by Sarepta that Sarepta would not be exercising its exclusive option under the Sarepta Agreement. The Sarepta Agreement terminated upon the expiration of the option in March 2023.
As of December 31, 2022, one milestone remained constrained. The2023, the Company will re-evaluate the transaction price in each reporting period, as uncertain events are resolved. The Company is recognizing the revenue associated with the transaction price and combined single performance obligation utilizing the output method, over the 33-month term as the manufactured supply is delivered to Sarepta.
recorded no contract liability. As of December 31, 2022, and 2021, the Company recorded $0.5 million and $4.6 million, respectively, as a short-term contract liability representing deferred revenue associated with this agreement. Revenue of $10.2$0.5 million and $0.4$10.2 million related to the Sarepta Agreement was recognized during the years ended December 31, 2023 and 2022, and 2021, respectively, inclusive of $0.9 million of revenue recognized from performance obligations related to prior periods as a result of the change in transaction price during the year ended December 31, 2022.respectively.
Asklepios Biopharmaceutical, Inc.
License Agreement for Pompe Disease
In December 2019, the Company and Asklepios Biopharmaceutical, Inc., or AskBio, entered into a license agreement, or the AskBio License Agreement. Pursuant to the AskBio License Agreement, AskBio exercised its option to exclusively license the Company’s intellectual property rights covering the Company’s ImmTOR platform to research, develop, and commercialize certain AAV gene therapy products utilizing ImmTOR, and targeting the GAA gene, or derivatives thereof, to treat Pompe Disease.
On November 18, 2022, both parties agreed to mutually terminate the AskBio License Agreement. Therefore, the remaining contract liability of $7.0 million was recognized as revenue during the period ended December 31, 2022.
Spark Therapeutics, Inc.
In December 2016, the Company entered into a license and option agreement, or the Spark License Agreement, with Spark Therapeutics, Inc., or Spark, pursuant to which the Company and Spark agreed to collaborate on the development of gene therapies for certain targets utilizing the ImmTOR platform. The Spark License Agreement provided Spark with certain exclusive, worldwide, royalty bearing licenses to the Company’s intellectual property, allowing Spark to develop and commercialize gene therapies in combination with ImmTOR for Factor VIII, an essential blood clotting protein relevant to the treatment of hemophilia A, the initial target.
Additionally, in connection with the Spark License Agreement, the Company entered into a Stock Purchase Agreement with Spark, or the Spark Purchase Agreement. Pursuant to the Spark Purchase Agreement, Spark purchased a total of $15.0 million of the Company’s common stock including 197,238 shares for an aggregate purchase price of $5.0 million at the initial closing, 324,362 shares of common stock for an aggregate purchase price of $5.0 million on June 8, 2017, and 205,254 shares of common stock from the Company for an aggregate purchase price of $5.0 million on October 31, 2017.
On January 18, 2022, both parties agreed to mutually terminate the Spark License Agreement. Therefore, the remaining contract liability of $9.2 million was recognized as revenue during the year ended December 31, 2022.
During the year ended December 31, 2021, revenueTransaction Price Allocated to Future Performance Obligations
Remaining performance obligations represent the transaction price of contracts for which work has not been performed (or has been partially performed). As of December 31, 2022,2023, the aggregate amount of the transaction price allocated to remaining performance obligations was $0.6$5.8 million.
Contract Balances from Contracts with Customers (Astellas, Takeda, Sobi, Sarepta, AskBio, and Spark)
The following table presents changes in the Company’s contract liabilities during the year ended December 31, 20222023 (in thousands):
| | Balance at | | Balance at |
| beginning of period | | Additions | | Deductions | | end of period |
| Balance at | | | Balance at | | | | | | Balance at |
| beginning of period | | | beginning of period | | Additions | | Deductions | | end of period |
| Contract liabilities: | Contract liabilities: | | | | | | | |
| Deferred revenue | Deferred revenue | $ | 65,300 | | | $ | 16,000 | | | $ | (80,707) | | | $ | 593 | |
| Deferred revenue | |
| Deferred revenue | |
| Total contract liabilities | Total contract liabilities | $ | 65,300 | | | $ | 16,000 | | | $ | (80,707) | | | $ | 593 | |
13.15. Related-Party Transactions
November 2023 Securities Purchase Agreement
On November 13, 2023, the Company entered into the Securities Purchase Agreement with (i) Dr. Timothy A. Springer, (ii) TAS Partners LLC, an affiliate of Dr. Springer, and (iii) Seven One Eight Three Four Irrevocable Trust, a trust associated with Dr. Murat Kalayoglu, in which the Company agreed to issue and sell an aggregate of 149,330.115 shares of Series A Preferred Stock for an aggregate purchase price of $60.25 million (see Note 11). The November 2023 Private Placement includes a delayed settlement mechanism, and as a result, the below issuances and sales to related parties of the Company were made during the year ended December 31, 2023.
| | | | | | | | | | | | | | |
| Name | | Shares of Series A Preferred Stock purchased | | Total aggregate purchase price |
| Timothy A. Springer, Ph.D. | | 24,785.081 | | | $ | 10,000,000 | |
| TAS Partners LLC (affiliate of Timothy A. Springer, Ph.D.) | | 24,785.081 | | | $ | 10,000,000 | |
| Seven One Eight Three Four Irrevocable Trust (affiliate of Murat Kalayoglu, MD, Ph.D.) | | 619.627 | | | $ | 250,000 | |
April 2022 Offering
During the year ended December 31, 2022, the Company completed the 2022 Offering as described in Note 10.12. The following table sets forth the number of shares of Common Stock and 2022 Warrants purchased in the 2022 Offering by directors and executive officers, as of the time of the Offering, and related parties thereto:
| | Name | Name | | Shares of Common Stock purchased | | 2022 Warrants purchased | | Total aggregate purchase price | Name | | Shares of Common Stock purchased | | 2022 Warrants purchased | | Total aggregate purchase price |
| TAS Partners, LLC (affiliate of Timothy A. Springer, Ph.D.) | | 6,681,600 | | | 5,011,200 | | | $ | 9,421,056 | |
| TAS Partners LLC (affiliate of Timothy A. Springer, Ph.D.) | |
Warrant liability reclassification
During the year ended December 31, 2022, the Company amended the terms of certain of the outstanding 2019 Warrants held by members of the Company's boardBoard of directorsDirectors and remeasured the Amended 2019 Warrants as described in Notes 5 and 10.Note 6.
Consulting Services
The Company entered into consulting agreements with its founders to serve on its Scientific Advisory Board, effective January 1, 2020 to December 31, 2021, under which they were paid quarterly for their services. The Company incurred expenses for consulting services provided by its founders totaling $0.1 million during each of the yearsyear ended December 31, 2021 and 2020.2021. No expenses were incurred for the yearyears ended December 31, 2023 and 2022.
14.16. Collaboration and License Agreements
Biogen MA, Inc.
On September 8, 2023, the Company entered into a non-exclusive, sublicensable, worldwide, perpetual patent license agreement, or the Biogen Agreement with Biogen MA, Inc., or Biogen to research, develop, make, use, offer, sell and import products or processes containing or using an engineering T-cell modified with an mRNA comprising, or encoding a protein comprising, certain sequences licensed under the Biogen Agreement for the prevention, treatment, palliation and management
of autoimmune diseases and disorders, excluding cancers, neoplastic disorders, and paraneoplastic disorders. The Company is not obligated to pay Biogen any expenses, fees, or royalties.
The Company may terminate the Biogen Agreement for any reason or no reason, and Biogen may terminate the agreement after a notice-and-cure period of 30 days if the Company fails to pay a fee owed to Biogen or for any other material breach of the agreement. The Biogen Agreement will otherwise expire when all claims of all issued patents within the patents and patent applications licensed to the Company under the Biogen Agreement have expired or been finally rendered revoked, invalid or unenforceable by a decision of a court or government agency.
National Cancer Institute of the National Institutes of Health
Effective September 16, 2019, the Company entered into a nonexclusive, worldwide license agreement, or the NCI Agreement with the U.S. Department of Health and Human Services, represented by the National Cancer Institute of the National Institutes of Health, or NCI.
Under the NCI Agreement, the Company was granted a license under certain NCI patents and patent applications designated in the agreement, to make, use, sell, offer and import products and processes within the scope of the patents and applications licensed under the NCI Agreement when developing and manufacturing anti-BCMA CAR-T cell products for the treatment of myasthenia gravis, pemphigus vulgaris, and immune thrombocytopenic purpura according to methods designated in the NCI Agreement.
In connection with the Company's entry into the NCI Agreement, Old Cartesian paid to NCI a one-time $0.1 million license royalty payment. Under the NCI Agreement, the Company is further required to pay NCI a low five-digit annual royalty. The Company must also pay earned royalties on net sales in a low single-digit percentage and pay up to $0.8 million in benchmark royalties upon the Company's achievement of designated benchmarks that are based on the commercial development plan agreed between the parties.
Under the NCI Agreement, the Company must use reasonable commercial efforts to bring licensed products and licensed processes to the point of Practical Application (as defined in the NCI Agreement). Upon the Company's first commercial sale, the Company must use reasonable commercial efforts to make licensed products and licensed processes reasonably accessible to the United States public. After the Company's first commercial sale, the Company must make reasonable quantities of licensed products or materials produced via licensed processes available to patient assistance programs and develop educational materials detailing the licensed products. Unless the Company obtains a waiver from NCI, the Company must have licensed products and licensed processes manufactured substantially in the United States. Prior to the first commercial sale, upon NCI’s request, the Company is obligated to provide NCI with commercially reasonable quantities of licensed products made through licensed processes to be used for in vitro research.
Additionally, the Company must use reasonable commercial efforts to initiate a Phase 3 clinical trial of a licensed product by the fourth quarter of 2024, submit a BLA with respect to a licensed product by the fourth quarter of 2026, and make a first commercial sale of a licensed product by the fourth quarter of 2028.
The NCI Agreement terminates upon the expiration of the last to expire of the patent rights licensed thereunder, if not sooner terminated. NCI has the right to terminate this agreement, after giving written notice and providing a cure period in accordance with its terms, if the Company is in default of a material obligation. The Company has the unilateral right to terminate the agreement in any country or territory by giving NCI 60 days’ written notice. The Company agreed to indemnify NCI against any liability arising out of the Company's, sublicensees’ or third parties’ use of the licensed patent rights and licensed products or licensed processes developed in connection with the licensed patent rights.
Ginkgo Bioworks Holdings, Inc.
Collaboration and License Agreements
On October 25, 2021, the Company entered into a Collaboration and License Agreement, or the First Ginkgo Agreement, with Ginkgo Bioworks Holdings, Inc., or Ginkgo. Under the First Ginkgo Agreement, Ginkgo will design next generation IgA proteases with potentially transformative therapeutic potential. In return, Ginkgo is eligible to earn both upfront research and
development fees and milestone payments, including certain milestone payments for fixed fair values in the form of Selectathe Company's common stock, clinical and commercial milestone payments of up to $85.0 million in cash. The First Ginkgo Agreement was assessed for collaboration components and was determined not to be within the scope of ASC 808 as the risk and rewards are not shared by both parties. The Company will expense costs related to the First Ginkgo Agreement as incurred until regulatory approval is received in accordance with ASC 730. The Company is accounting for the contingently issuable shares to be issued in exchange for the license obtained from Ginkgo as a liability classified stock-based compensation arrangement with a non-employee which will be recognized when achievement of the milestones is probable. The Company will assess the capitalization of costs incurred after the receipt of regulatory approval and, if applicable, will amortize these payments based on the expected useful life of each asset, typically based on the expected commercial exclusivity period. The Company is also obligated to pay Ginkgo
tiered royalties ranging from low-single digit to high-single digit percentages of annual net sales of collaboration products which will be expensed as the commercial sales occur.
On January 3, 2022, the Company entered into a Collaboration and License Agreement, or the Second Ginkgo Agreement, with Ginkgo. Under this agreement, the Company will engage with Ginkgo to develop AAV capsids designed to enhance transduction efficiency and transgene expression. In return, Ginkgo is eligible to earn both upfront research and development fees and milestone payments, including certain milestone payments in the form of shares of the Company’s common stock, clinical and commercial milestone payments of up to $207 million in cash. The Second Ginkgo Agreement was assessed for collaboration components and was determined not to be within the scope of ASC 808 as the risk and rewards are not shared by both parties. The Company will expense costs related to the Second Ginkgo Agreement as incurred until regulatory approval is received in accordance with ASC 730. The Company is accounting for the contingently issuable shares of common stock to be issued in exchange for the license obtained from Ginkgo as a liability-classified, stock-based compensation arrangement with a non-employee which will be recognized when achievement of the milestones is probable. The Company will assess the capitalization of costs incurred after the receipt of regulatory approval and, if applicable, will amortize these payments based on the expected useful life of each asset, typically based on the expected commercial exclusivity period. The Company is also obligated to pay Ginkgo tiered royalties ranging from low-single digit to high-single digit percentages of annual net sales of collaboration products which will be expensed as the commercial sales occur.
On June 13, 2022, the Company was notified of the achievement of the midpoint of the technical development plan under the First Ginkgo Agreement by Ginkgo. This milestone resulted in the payment of $0.5 million and issuance of 892,857 shares of the Company’s common stock then-valued at $1.0 million to Ginkgo during the year ended December 31, 2022.
On July 19, 2023, the Company and Ginkgo mutually agreed that the completion of the technical development plan’s midpoint task under the Second Ginkgo Agreement had been achieved as of June 30, 2023. This milestone resulted in the payment of $1.0 million and issuance of 1,339,285 shares of the Company’s common stock then-valued at $1.5 million to Ginkgo during the year ended December 31, 2023.
Genovis AB (publ.)
License Agreement
On October 21, 2021, the Company entered into an Exclusive License Agreement, or the Genovis Agreement with Genovis. Under the Genovis Agreement, the Company paid to Genovis an upfront payment in exchange for an exclusive license to Genovis’ IgG Protease, or Xork, enzyme technology across all therapeutic uses in humans, excluding research, preclinical, diagnostic and other potential non-therapeutic applications of the enzyme. Genovis is eligible to earn from the Company development and sales-based milestones and sublicensing fees. The Genovis Agreement was assessed for collaboration components and was determined not to be within the scope of ASC 808 as the risk and rewards are not shared by both parties. The Company will expense costs related to the Genovis Agreement as incurred until regulatory approval is received in accordance with ASC 730. The Company will assess the capitalization of costs incurred after the receipt of regulatory approval and, if applicable, will amortize these payments based on the expected useful life of each asset, typically based on the expected commercial exclusivity period. The Company is also obligated to pay Genovis tiered royalties of low double digit percentages of worldwide annual net sales of collaboration products which will be expensed as the commercial sales occur.
In February 2023, the Company made a $4.0 million payment to Genovis as a result of the sublicense of Xork to Astellas. See Note 14 to these consolidated financial statements for further discussion on the Astellas Agreement.
Cyrus Biotechnology, Inc.
Collaboration and License Agreement
On September 7, 2021, the Company and Cyrus Biotechnology, Inc., or Cyrus, entered into a collaboration and license agreement, or the Cyrus Agreement. Pursuant to the Cyrus Agreement, Cyrus agreed to grant the Company an exclusive, worldwide license to certain intellectual property to form a protein engineering collaboration combining the Company’s ImmTOR platform with Cyrus’ ability to redesign protein therapeutics. The lead program iswas a proprietary interleukin-2, or IL-2, protein agonist designed to selectively promote expansion of regulatory T cells for treatment of patients with autoimmune diseases and other deleterious immune conditions. In return for the licensed intellectual property, the Company made an upfront payment and iswas obligated to pay certain discovery, development, and sales-based milestones which could have potentially totaltotaled up to approximately $1.5 billion across multiple programs. The Cyrus Agreement was assessed for collaboration components and was determined not to be within the scope of ASC 808 as the risk and rewards are not shared by both parties. The Company will expenseexpensed costs related to the Cyrus Agreement as incurred until regulatory approval is received in accordance with ASC 730. The Company will assessassessed the capitalization of costs incurred after the receipt of regulatory approval and, if applicable, will amortizewould have amortized these payments based on the expected useful life of each asset, typically based on the expected commercial exclusivity period. The Company was also obligated to pay Cyrus tiered
period. The Company is also obligated to pay Cyrus tiered royalties ranging from mid-single digit to low-double digit percentages of annual net sales of collaboration products which will bewould have been expensed as the commercial sales occur.
On June 13, 2022, the Company and Cyrus mutually agreed that the preclinical key in-vitro success milestone had been achieved.
In October 2023, the Company notified Cyrus of its termination of the Cyrus Agreement, effective December 29, 2023.
Stock Purchase Agreement
Additionally, on September 7, 2021, the Company entered into a stock purchase agreement, or the Series B Preferred Stock Purchase Agreement, in connection with the Cyrus Agreement. Pursuant to the Series B Preferred Stock Purchase Agreement, the Company purchased 2,326,934 shares of Cyrus’ Series B Preferred Stock, par value $0.0001 per share, at a purchase price of $0.8595 per share for $2.0 million.
In accordance with ASC 810, the Company has a variable interest in Cyrus resulting from its equity investment. The Company will share in Cyrus’ expected losses or receive a portion of its expected returns and absorb the variability associated with changes in the entity’s net assets. However, the Company is not the primary beneficiary as it does not have the power to direct the activities most significant to Cyrus, and therefore it is not required to consolidate Cyrus. The Company has recognized the $2.0 million investment of Cyrus’ Series B Preferred Stock at cost on the purchase date.
On June 13, 2022, the Company and Cyrus mutually agreed that the preclinical key in-vitro success milestone had been achieved.
As of December 31, 2022,2023, no impairment indicators are present and therefore the carrying value of the investment in Cyrus is $2.0 million on the accompanying consolidated balance sheet. The Company’s maximum exposure to loss related to this variable interest entityVIE is limited to the carrying value of the investment. The Company has not provided financing to Cyrus other than the amount contractually required by the Series B Preferred Stock Purchase Agreement.
Asklepios Biopharmaceutical, Inc.
Feasibility Study and License Agreement
In August 2019, the Company entered into a feasibility study and license agreement with AskBio, or the AskBio Collaboration Agreement. Pursuant to the AskBio Collaboration Agreement, the Company and AskBio agreed to license intellectual property rights to each other as part of a collaboration to research, develop, and commercialize certain AAV gene therapy products utilizing the Company’s ImmTOR platform to enable re-dosing of such AAV gene therapy products to treat serious rare and orphan genetic diseases for which there is a significant unmet medical need.
Pursuant to the AskBio Collaboration Agreement, the Company and AskBio agreed to conduct proof of concept studies to potentially validate the use of ImmTOR in conjunction with AskBio’s AAV gene therapy, or SEL-302, (previously disclosed as MMA-101, in combination with ImmTOR) for the treatment of methylmalonic acidemia, or MMA, to mitigate the formation of neutralizing anti-AAV capsid antibodies, or the POC Studies.antibodies. On April 29, 2021, the Company was notified by AskBio that it intended to opt-out of development of the MMA indication. The feasibility study and license agreement with AskBio, or AskBio Collaboration Agreement, otherwise remains in effect. Consequently, the Company has assumed all rights to the MMA program and intends to continue to progress the SEL-302 program through clinical development. The Company filed an IND to conduct a Phase 1/2 clinical trial of its SEL-302 product candidate in pediatric patients with methylmalonic acidemia in the third quarter of 2021. In December 2022, we initiated ReiMMAgine, the Phase 1/2 clinical trial of SEL-302. The ReiMMAgine trial is now enrolling patients and aims to evaluate the safety, tolerability and efficacy of SEL-302.
The SEL-399 program combined an empty AAV capsid (EMC-101), which is an AAV capsid containing no transgene, with ImmTOR and is being conducted in partnership with AskBio. Building on the preclinical data the Company has generated showing ImmTOR’s effect on mitigating or reducing the formation of neutralizing antibodies to AAV gene therapies, the Company completed a clinical trial of SEL-399 in healthy adult volunteers in Belgium. The goal of the SEL-399 clinical trial was to demonstrate the appropriate dose of ImmTOR in humans to mitigate the formation of antibodies to AAV capsids used in gene therapies. This promising study in healthy volunteers provides support for the potential use of ImmTOR for the inhibition of neutralizing antibodies to AAV8 in gene therapy clinical trials.
The Company and AskBio will shareshared responsibility for the research, development and commercialization of products developed under the SEL-399 program collaboration. The parties will also shareshared research, development, and commercialization costs equally for all collaboration products, but with a right of either party to opt out of certain products, and thereby no longernot be required to share costs for such products. Each party will receivewould have received a percentage of net profits under the collaboration equal to the percentage of shared costs borne by such party in the development of such product. Pursuant to the AskBio Collaboration Agreement, AskBio iswas responsible for manufacturing the AAV capsids and AAV vectors and the Company iswas responsible for manufacturing ImmTOR.
The Company and AskBio Collaboration Agreement is considered to be within the scope of ASC 808, as both parties are active participants and exposedmutually agreed to the risks and rewards of the collaborative activity. The Company evaluated the termstermination of the AskBio Collaboration Agreement, and have identified the following promises in the arrangement (1) conducting research and development activities to develop and commercialize products under the collaboration, or the R&D Services, (2) granting a non-exclusive, non-transferable, royalty-free, fully paid up, worldwide license to certain intellectual property of the Company, or the IP Rights, for the purpose of performing the POC Studies, or the Research License, (3) granting an exclusive,
nontransferable, worldwide license to the IP Rights for use in certain indications, or the Collaboration License, (4) providing manufactured supply of preclinical and clinical ImmTOR, or the Manufactured Supply, (5) participation on identified steering committees responsible for the oversight of the collaboration, or the JSC Participation, and (6) granting an exclusive option to obtain a license under the IP Rights to research, develop and commercialize Licensed Products. The Company determined that the R&D Services, Research License, Collaboration License, Manufactured Supply, and JSC Participation were not capable of being distinct, and therefore must be combined into a single performance obligation. Therefore, promises (1) through (5) identified above were combined into a single performance obligation. Furthermore, the Company evaluated the Option Agreement and determined that it does not provide AskBio with a material right under ASC 606 as the option was not priced at a discount (see discussion of the option exercise in Note 12). The Company noted that AskBio did not meet the definition of a customer within the scope of ASC 606 for any distinct performance obligations as the Company concluded that such items were not an output of the Company’s ordinary activities. As such, the Company determined that the entire arrangement would be accounted for within the scope of ASC 808. In accordance with ASC 808, collaboration expenses are recognized within R&D expense and selling, general and administrative expense on the Company’s condensed consolidated statements of operations.
Under certain collaborative arrangements, the Company is entitled to reimbursement of certain R&D expense. Activities under collaborative arrangements for which the Company is entitled to reimbursement are considered to be collaborative activities under the scope of ASC 808. For these units of account, the Company does not analogize to ASC 606 or recognize revenue. Rather, the Company analogizes to the guidance in ASC 730, which requires that reimbursements from counterparties be recognized as an offset to the related costs. In accordance with ASC 730, the Company records reimbursement payments received from collaborators as reductions to R&D expense.effective December 13, 2023.
For the years ended December 31, 20222023 and 2021,2022, the Company recognized $0.9$0.1 million and $2.7$0.9 million, respectively, of collaboration expense under the AskBio Collaboration Agreement in which actual costs incurred by both parties approximate a 50% cost share.
Massachusetts Institute of Technology
In November 2008, the Company entered into an exclusive patent license agreement, or the MIT License, with the Massachusetts Institute of Technology, or MIT, under which the Company received an exclusive royalty-bearing license to utilize patents held by MIT in exchange for upfront consideration and annual license maintenance fees. Such fees are expensed as incurred and have not been material to any period presented.
In June 2020, the Company entered into a Fifth Amendment, or the MIT Amendment, to the MIT License, which is effective as of May 15, 2020. Pursuant to the MIT Amendment, certain of the Company’s diligence obligations were extended. The extension included the obligation to commence a Phase 3 trial for a licensed product by the second quarter of 2021 or to file an IND (or equivalent) with the FDA or comparable European regulatory agency for a licensed product by the second quarter of 2023. Additionally, certain of the Company’s development and regulatory milestones and payments upon achievement of such milestones were adjusted.
As of December 31, 2022, and in connection with the execution of the Spark License Agreement, the Company has made contractual payments pursuant to the MIT License totaling $2.2 million. In connection with the Spark Purchase Agreement and the calculated premium paid by Spark for the equity investments made as described in Note 12, the Company has made additional contractual payments pursuant to the MIT license totaling $0.4 million as of December 31, 2022. The Company made no additional payments during the year ended December 31, 2022.
Shenyang Sunshine Pharmaceutical Co., Ltd
In May 2014, the Company entered into a license agreement, or the 3SBio License, with Shenyang Sunshine Pharmaceutical Co., Ltd., or 3SBio. The Company has paid to 3SBio an aggregate of $7.0 million in upfront and milestone-based payments under the 3SBio License as of December 31, 2022.2023. The Company is required to make future payments to 3SBio contingent upon the occurrence of events related to the achievement of clinical and regulatory approval milestones of up to an aggregate of $15.0 million for products containing the Company’s ImmTOR platform.
15.17. Income Taxes
The Company provides for income taxes under ASC 740. Under ASC 740, the Company provides deferred tax assets and liabilities for the expected future tax consequences of temporary differences between the Company’s financial statement carrying amounts and the tax bases of assets and liabilities using enacted tax rates expected to be in effect in the years in which the differences are expected to reverse.
On June 11, 2020,November 13, 2023, the Company entered intoacquired, in accordance with the Sobi License (see note 12).terms of the Merger Agreement, the assets of Old Cartesian. In September 2020, Sobi paidaccordance with ASC 805-740-25-3, recognition of deferred tax assets and liabilities is required for substantially all temporary differences and acquired tax carryforwards and credits. The Company has computed estimated temporary differences and acquired tax carryforwards and credits as of the transaction date. The Company will not have tax basis in IPR&D booked as part of the purchase accounting. For accounting purposes, the IPR&D will not be amortized and only subject to impairment review and testing. Though the tax effects may be delayed indefinitely, ASC 740-10-55-63 states that “deferred tax liabilities may not be eliminated or reduced because a reporting entity may be able to delay the settlement of those liabilities by delaying the events that would cause taxable temporary differences to reverse.” As such, the Company a one-time upfront payment of $75.0 million. Sobi has also agreedcan potentially only utilize indefinite-lived assets as it relates to make milestone payments totaling up to $630.0 million tothis indefinite lived intangible deferred tax liability reversal.As such, the Company upon achievementhas booked a deferred tax liability for the portion of various developmentthe liability that cannot be reduced based on scheduling. Additionally, a portion of this target deferred tax liability is offset with the Company's pre-Merger deferred tax assets on a combined basis, and regulatory milestones and sales thresholds for annual net salesas such the portion of
SEL-212, and tiered royalty payments ranging from low double digits on deferred tax liability reduced by the lowest sales tierCompany's pre-Merger deferred tax assets has been charged to high teens on the highest sales tier.income rather than to goodwill.
For income tax purposes, the transfer of trademark and product rights is treated as a sale and the net proceeds from the sale are taxed under the default installment method as cash is received by the Company. During the year endingended December 31, 2021,2023, the Company completed an analysisrecognized a current tax benefit, of future tax obligations under the default installment sale method versus making a timely filed election on its 2020 tax return due October 15, 2021 to elect out of the installment sale method for income tax purposes. As a result, the Company elected out of the default installment sale treatment with the filing of its 2020 tax return. In the elect out method, the Company was initially taxed based upon the estimated fair value of all present and future proceeds from the sale and the Company utilized all of its available net operating losses and income tax credits, which served to reduce the federal and state tax liability. As the Company recognizes future cash proceeds under the Sobi license, the Company will exclude such amounts from taxable income up to the originally estimated fair value that was previously taxed.
$19.0 million. For the year ended December 31, 2022, the Company recognized a current tax benefit for penalty abatements received of $0.6 million. For the year ended December 31, 2021, the Company recognizedhad recorded a tax expense of $16.0 million, inclusive of penalties and interest of $1.3 million assessed as of December 31, 2021. For the year ended December 31, 2020, the Company did not record a current or deferred income tax expense or benefit. The following table reconciles the federal statutory income tax rate to the Company’s effective income tax rate:
| | Year Ended December 31, |
| 2022 | | 2021 | | 2020 |
| Year Ended December 31, | | | Year Ended December 31, |
| 2023 | | | 2023 | | 2022 | | 2021 |
| Statutory U.S. federal rate | Statutory U.S. federal rate | 21.0 | % | | 21.0 | % | | 21.0 | % | Statutory U.S. federal rate | 21.0 | % | | 21.0 | % | | 21.0 | % |
| State income taxes - net of federal benefit | State income taxes - net of federal benefit | 1.6 | % | | (166.0 | %) | | 5.8 | % | State income taxes - net of federal benefit | 2.3 | % | | 1.6 | % | | (166.0) | % |
| Permanent items | Permanent items | (18.6 | %) | | 8.3 | % | | (2.9 | %) | Permanent items | (1.6) | % | | (18.6) | % | | 8.3 | % |
| Research tax credits | Research tax credits | (3.2 | %) | | 55.0 | % | | 1.0 | % | Research tax credits | 0.6 | % | | (3.2) | % | | 55.0 | % |
| Deferred revenue | Deferred revenue | — | % | | 156.5 | % | | — | % | Deferred revenue | — | % | | — | % | | 156.5 | % |
| Other | Other | — | % | | (3.7 | %) | | — | % | Other | — | % | | — | % | | (3.7) | % |
| Change in fair value of forward contract liabilities | | Change in fair value of forward contract liabilities | (13.2) | % | | — | % | | — | % |
| Valuation allowance, net | Valuation allowance, net | (4.4 | %) | | (230.1 | %) | | (23.6 | %) | Valuation allowance, net | 2.8 | % | | (4.4) | % | | (230.1) | % |
| Stock-based compensation | Stock-based compensation | 1.8 | % | | (5.2 | %) | | (1.3 | %) | Stock-based compensation | (3.9) | % | | 1.8 | % | | (5.2) | % |
| Effective income tax rate | Effective income tax rate | (1.8 | %) | | (164.2 | %) | | — | % | Effective income tax rate | 8.0 | % | | (1.8) | % | | (164.2) | % |
The tax effects of temporary differences that give rise to the Company’s net deferred tax assets are as follows (in thousands):
| | | | | | | | | | | |
| Year Ended December 31, |
| 2022 | | 2021 |
| Deferred Tax Assets | | | |
| Net operating loss carryforwards | $ | 17,015 | | | $ | 13,932 | |
| Research and development credits | 2,806 | | | 1,426 | |
| Stock-based compensation expense | 5,892 | | | 3,955 | |
| Other expenses | 1,697 | | | 745 | |
| Deferred revenue | 83,417 | | | 102,583 | |
| Operating lease liabilities | 3,186 | | | 2,636 | |
| R&E capitalization | 9,588 | | | — | |
| Patent costs | 7,472 | | | 6,843 | |
| | | |
| | | |
| Gross deferred tax assets | 131,073 | | | 132,120 | |
| | | |
| Deferred Tax Liabilities | | | |
| Depreciation | $ | (81) | | | $ | (90) | |
| Operating lease right-of-use assets | (3,174) | | | (2,685) | |
| | | |
| | | |
| Gross deferred tax liabilities | (3,255) | | | (2,775) | |
| Net deferred tax assets before valuation allowance | 127,818 | | | 129,345 | |
| Valuation allowance | (127,818) | | | (129,345) | |
| Net deferred tax assets | $ | — | | | $ | — | |
| | | | | | | | | | | |
| Year Ended December 31, |
| 2023 | | 2022 |
| Deferred Tax Assets | | | |
| Net operating loss carryforwards | $ | 29,841 | | | $ | 17,015 | |
| Research and development credits | 5,649 | | | 2,806 | |
| Stock-based compensation expense | 8 | | | 5,892 | |
| Other expenses | 705 | | | 1,697 | |
| Deferred revenue | 84,626 | | | 83,417 | |
| Operating lease liabilities | 2,718 | | | 3,186 | |
| R&E Capitalization | 19,778 | | | 9,588 | |
| Patent and license costs | 9,140 | | | 7,472 | |
| Gross deferred tax assets | 152,465 | | | 131,073 | |
| Deferred Tax Liabilities | | | |
| Intangible assets | $ | (41,144) | | | $ | — | |
| Depreciation | (128) | | | (81) | |
| Operating lease right-of-use assets | (2,751) | | | (3,174) | |
| Gross deferred tax liabilities | (44,023) | | | (3,255) | |
| Net deferred tax assets before valuation allowance | 108,442 | | | 127,818 | |
| Valuation allowance | (124,295) | | | (127,818) | |
| Net deferred tax assets/(liabilities) | $ | (15,853) | | | $ | — | |
The Company has provided a full valuation allowance against its net deferred tax assets, outside of the indefinite tax liability booked as part of the Merger. The Company believes that it is more likely than not that the net deferred tax assets will not be realized.
Realization of future tax benefits is dependent on many factors, including the Company’s ability to generate taxable income within the net operating loss carryforward period.income. The Company has evaluated the positive and negative evidence bearing upon the realizability of its deferred tax assets and concluded that it is more likely than not that the Company will not realize the
benefit of its net deferred tax assets. The valuation allowance decreased by $3.5 million for the year ended December 31, 2023, primarily as a result of tax benefit booked as part of the Merger. The valuation allowance decreased by $1.5 million for the year ended December 31, 2022, primarily as a result ofpre-tax income and credits. The valuation allowance increased by $22.4 million for the year ended December 31, 2021, primarily as a result of an increase in deferred revenue. As of December 31, 2022,2023, the Company is in the process of closingwinding down operations in Russia and does not expect any tax liability.liability relating to such operations.
At December 31, 2022,2023, the Company has federal net operating loss carryforward of $62.4$108.7 million, which can be carried forward indefinitely, but will be subject to an 80% limitation and state net operating loss carryforward of $61.8$110.3 million, which will expire at various times through 2042.2043. The Company has $2.3$4.9 million and $0.6$0.9 million, respectively, of federal and state research and development tax credit carryforwards, which will expire at various times through 2042.2043. Utilization of the NOL carryforwards and research and orphan drug credit carryforwards may be subject to a substantial annual limitation under Section 382 of the Internal Revenue Code of 1986, as amended, (the Code),or the Code, and similar state law due to ownership changes that could occur in the future.
These ownership changes may limit the amount of carryforwards that can be utilized annually to offset future taxable income. If the Company experiences a change of control, as defined by Section 382 of the Code and similar state law, utilization of the NOL carryforwards or research and orphan drug credit carryforwards may be subject to an annual limitation under Section 382 of the Code, which is determined by first multiplying the value of the Company’s stock at the time of the ownership change by the applicable long-term tax-exempt rate, and then could be subject to additional adjustments, as required. Any limitation may result in expiration of a portion of the NOL carryforwards or research and orphan drug credit carryforwards before utilization. The Company performed an analysis of ownership changes through December 31, 2022.2023. Based on this analysis, the Company does not believe that any of its tax attributes through December 31, 20222023 will expire unutilized due to Section 382 limitations. To the extent the Company enters into future equity transactions, there could be a limitation on the Company’s tax attributes.
The Company applies ASC 740, Income Taxes to uncertain tax positions. As of the adoption date on January 1, 2010 and through December 31, 2022,2023, the Company had no unrecognized tax benefits or related interest and penalties accrued.
During 2021,2023, the Company completed a detailed study of its research and development and orphan drug credits through December 31, 2020. The2022. As a result, the Company generated research credits for the years ended December 31, 2022 and 2021, but has not conducted a formal study to documentadjusted its qualified activities. This study may result in an adjustment to the Company’s research and development credit carryforwards; however, until a study is completed and any adjustment is known, no amounts are being presented as an uncertain tax position. A full valuation allowance has been provided against our research and development credits and, if an adjustment is required, this adjustment would be offset by an adjustment to the deferred tax asset established forbalances and the impacts are included in the research tax credits and development credit carryforwards andstate income taxes - net of federal benefit lines in the valuation allowance.effective rate reconciliation above.
The Tax Cuts and Jobs Act of 2017 requires taxpayers to capitalize and amortize, rather than deduct, research and experimental, or R&E, expenditures under section 174 for tax years beginning after December 31, 2021. This ruleThese rules became effective for the
Company during the year and resulted inended December 31, 2022. As a result, the capitalization ofCompany has capitalized R&E costs of $38.1 million.$44.7 million and $29.3 million for the years ended December 31, 2023 and December 31, 2022, respectively. The Company will amortize these costs for tax purposes over 5five years if the R&E was performed in the U.S. and over 15 years if the R&E was performed outside the U.S.
Interest and penalty charges, if any, related to unrecognized tax benefits would be classified as income tax expense in the accompanying statement of operations. As of December 31, 2022,2023, the Company had no accrued interest related to uncertain tax positions.
The statute of limitations for assessment by the Internal Revenue Service and Massachusetts tax authorities is open for tax years since inception as the Company claimed research tax credits on its 2020 tax return which remains open for examination for the 2020 year as well as for any year in which a credit has been claimed for. The Company files income tax returns in the United States and Massachusetts. There are currently no federal, state or foreign audits in progress.
16.18. Defined Contribution Plan
The Company maintains a defined contribution plan, or the 401(k) Plan, under Section 401(k) of the Internal Revenue Code. The 401(k) Plan covers all employees who meet defined minimum age and service requirements and allows participants to defer a portion of their annual compensation on a pretax basis. The 401(k) Plan provides for matching contributions on a portion of participant contributions pursuant to the 401(k) Plan’s matching formula. As of January 2022, all matching contributions vest ratably over two years and participant contributions vest immediately. Contributions by the Company totaled $0.3 million, $0.2$0.3 million, and $0.1$0.2 million during each of the years ended December 31, 2023, 2022 2021 and 2020,2021, respectively.
17.19. Commitments and Contingencies
As of December 31, 2022,2023, the Company was not a party to any litigation that could have a material adverse effect on the Company’s business, financial position, results of operations or cash flows.
On August 3, 2020, a stockholder of Selecta filed a stockholder derivative action, purportedly on behalf of Selecta and against certain current and former members of the Company’s Board of Directors, as well as one affiliated company owned by a current board member, in the Court of Chancery of the State of Delaware, namely Franchi v. Barabe, et al. The complaint alleges that the individual defendants breached their fiduciary duties and committed corporate waste when they authorized a private placement transaction, announced on December 19, 2019, at a price allegedly below fair value. The complaint further alleges that the four defendant directors who participated in the private placement were unjustly enriched in connection with the transaction. On September 25, 2020, the defendants filed a motion to dismiss the lawsuit. On November 6, 2020, the plaintiff filed an amended complaint, and the defendants filed a second motion to dismiss on January 8, 2021. On December 31, 2020, we received a litigation demand letter from two other putative stockholders relating to the same private placement transaction. On April 12, 2021, the Court of Chancery in the State of Delaware granted a motion to stay the litigation pending a review by a Special Committee appointed by the Company’s Board of Directors. While the litigation was stayed, the parties reached an agreement in principle to settle the matter, and on March 18, 2022, they submitted a Stipulation and Agreement of Settlement and other documentation to the Court for its approval of the settlement. On July 21, 2022, the Court held a settlement hearing, at which the settlement was approved. On August 1, 2022, the Court entered an Order and Final Judgment which dismissed the action, and all claims contained therein, with prejudice.
Other
As permitted under Delaware law, the Company indemnifies its directors for certain events or occurrences while the director is, or was, serving at the Company’s request in such capacity. The term of the indemnification is for the director’s lifetime. The maximum potential amount of future payments the Company could be required to make is unlimited; however, the Company has directors’ insurance coverage that limits its exposure and enables it to recover a portion of any future amounts paid. The Company also has indemnification arrangements under certain of its facility leases that require it to indemnify the landlord against certain costs, expenses, fines, suits, claims, demands, liabilities, and actions directly resulting from certain breaches, violations, or non-performance of any covenant or condition of the Company’s lease. The term of the indemnification is for the term of the related lease agreement. The maximum potential amount of future payments the Company could be required to make under these indemnification agreements is unlimited. To date, the Company had not experienced any material losses related to any of its indemnification obligations, and no material claims with respect thereto were outstanding.
The Company is a party in various other contractual disputes and potential claims arising in the ordinary course of business. The Company does not believe that the resolution of these matters will have a material adverse effect the Company’s business, financial position, results of operations or cash flows.
18.20. Restructuring
In April 2023, in light of current market conditions, the Board of Directors, took steps to extend the Company's cash runway by pausing further development of SEL-302 for the treatment of MMA, and conducting a targeted headcount reduction. On August 17, 2023, the Company announced additional steps to extend cash runway and maximize value for stockholders by continuing to prioritize development of SEL-212 and support of its collaboration with Astellas for Xork, and pausing further development of all of the Company’s other clinical and preclinical product candidates that it was no longer actively advancing.
As a result of these measures, the Company implemented a restructuring plan resulting in an approximate 79% reduction of the Company's existing headcount by December 31, 2023. The Company recognized restructuring expenses consisting of one-time cash severance payments and other employee-related costs of $6.4 million during the year ended December 31, 2023. Cash payments for employee related restructuring charges of $2.5 million were paid as of December 31, 2023. The Company recorded $5.6 million and $0.8 million based on each employee's role to research and development and general and administrative operating expense categories, respectively, on its consolidated statements of operations and comprehensive income (loss) for the year ended December 31, 2023.
The following table summarizes the change in the Company's accrued restructuring balance (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | |
| Beginning Balance | | | | | | Ending Balance |
| December 31, 2022 | | Charges | | Payments | | December 31, 2023 |
| Severance liability | $ | — | | | $ | 6,431 | | | $ | 2,535 | | | $ | 3,896 | |
21. Subsequent Events
Collaboration and License Agreements
On January 8, 2023,February 28, 2024, the Company entered into a License and Development Agreementlease agreement with Astellas Therapeutics, Inc.,7495 RP, LLC, or the AstellasLandlord, pursuant to which it agreed to lease from the Landlord the manufacturing space located at 7495 New Horizon Way, Frederick, Maryland, or the Frederick Lease Agreement. UnderThe space consists of 19,199 leasable square feet of integrated manufacturing and office space. The initial term of the AstellasFrederick Lease Agreement is expected to commence no later than April 1, 2024, once the Landlord has obtained legal possession of the solepremises free of the existing tenant and exclusive rightdelivered full possession of the premises to commercialize Xork for use in Pompe disease with an Astellas gene therapy investigationalthe Company, or authorized product, withthe Commencement Date. The Frederick Lease Agreement will terminate seven full lease years following the Commencement Date which, assuming a current focus on AT845.Commencement Date of April 1, 2024, will be May 31, 2031. The Company received a $10 million upfront payment in February 2023 and is eligiblewill have one option to receive $340.0 million for certain additional development and commercial milestones plus royalties on any potential commercial sales where Xork is used as a pre-treatment for AT845. As a resultextend the term of the sublicenseFrederick Lease Agreement for a period of Xork to Astellas,five years. The base rent for the Company made a $4.0initial term is $0.1 million payment to Genovis in February 2023.