According to the Crohn's & Colitis Foundation, IBD is diagnosed in nearly 1 in 100 Americans, resulting in a population of approximately 2.4 million patients in the United States. In 2022, global sales for UC therapies were approximately $7.7 billion, and the market is expected to grow to $10.6 billion by 2028. In 2022, global sales for CD therapies were estimated to be $16.2 billion, with anticipated growth to $18.9 billion by 2028.
For many years, tumor necrosis factor-alpha (“TNF-α”) antibody drugs were the primary treatment for moderate-to-severe IBD. Humira®and Remicade® are injectable and infused, respectively. Approximately one third of IBD patients do not respond to TNF-α antibody drugs and approximately another 30% to 40% become refractory within the first year of treatment. Additionally, TNF-α antibody drugs may predispose patients to an increased risk of serious infection and the development of anti-drug antibodies, which over time can cause loss of drug response. More recently, antibody products focused on potentially safer mechanisms of action have been gaining market share. One such product is Takeda’s Entyvio®, which targets the α4β7 integrin pathway. Takeda reported 2022 sales of Entyvio® of approximately $6.4 billion. Similarly, Johnson & Johnson’s Stelara®, which targets the Interleukin 12 (“IL-12”) and Interleukin 23 (“IL-23”) pathways, has gained significant traction. Johnson & Johnson global sales of Stelara® (approved for psoriasis, psoriatic arthritis, moderate-to-severe CD and UC) were $10.9 billion in 2023. Three anti-IL-23 mAbs are in Phase 3 trials or beyond in IBD: Tremfya®, Skyrizi® and Ely Lilly and Company’s mirikizumab. The development of oral medicine has been an unmet need and priority in IBD. The pan-JAK inhibitor Xeljanz® was approved in UC (but not CD) in 2018. The label contains black box warnings for “an increased risk of serious heart-related events such as heart attack or stroke, cancer, blood clots, and death.” The more selective JAK1/3 inhibitor Rinvoq® was approved in 2022 for UC and CD. The label carries the same black box warnings. The S1P1 modulator class of oral small molecules has also demonstrated efficacy in IBD, with Zeposia® approved in UC (but not CD) in 2021, and etrasimod approved in UC in 2023. The S1P1 class is associated with immunosuppression, cardiac, pulmonary and ocular toxicities.
The development of new, potent and targeted orally delivered therapies for IBD may offer safer and more effective treatment options, alone or in combination, for moderate-to-severe IBD patients. In addition, many clinicians continue to advocate for earlier introduction of targeted therapeutics in mild-to-moderate IBD to prevent disease progression and irreversible gastrointestinal damage. Given that the most effective agents in IBD induce remission in no more than 30% of patients, there has been much recent interest in combination therapies to break through this “therapeutic ceiling.” In 2022, JNJ reported results of the VEGA study, the first randomized double bind clinical trial to assess the combination of an anti-TNF (Simponi®) with an anti-IL-23 (Tremfya®) in moderate-to-severe UC. In the Phase 2a POC trial, investigators found 83.1% of patients in the treatment group achieved a clinical response and 36.6% of patients treated with the combination therapy achieved clinical remission. The high rates of clinical response and remission are both higher than the response and remission rates of patients treated with guselkumab alone (74.6%; 21.1%) and golimumab alone (61.1%; 22.2%). Hence, we believe the IL-23 inhibition mechanism is a potentially paradigm shifting combination strategy to improve remission rates in UC.
JNJ-2113: AN ORAL IL-23 RECEPTOR ANTAGONIST
JNJ License and Collaboration Agreement
We have a worldwide license and collaboration agreement with JNJ to research, develop and co-detail our IL-23 receptor (“IL-23R”) antagonist compounds for all indications, including IBD. The agreement with JNJ was amended in May 2019 to expand the collaboration by supporting efforts towards second-generation IL-23R antagonists; and in July 2021 to, among other things, enable JNJ to independently research and develop collaboration compounds for multiple indications in the IL-23 pathway and further align our financial interests. See “ItemPart II, Item 7. Management’s“Management’s Discussion and Analysis – Overview” and Note 3 to the Consolidated Financial Statements included elsewhere in this Annual Report on Form 10-K for additional information.
Our novel peptides have potential applicability JNJ is an experienced innovator in a wide range of therapeutic areas in addition to GI diseases. Our third product candidate, PTG-300,therapeutics targeting the IL-23 pathway. Stelara® is a mimic of the hormone hepcidin that we are developing for the treatment of anemiamonoclonal antibody targeting IL-12 and IL-23 through their common p40 subunit is approved in certain rare blood disorders, with an initial focus on beta-thalassemia. In the fourth quarter of 2017, wepsoriasis, psoriatic arthritis, CD and UC. Stelara® generated $10.9 billion in sales in 2023. Tremfya® is a specific IL-23 monoclonal antibody. It is approved in psoriasis and psoriatic arthritis and has completed a successful Phase 1 study of PTG-3003 trials in healthy volunteers which established pharmaceutical proof of concept. In 2018, we anticipate filing an IND in the United States and related clinical trial applications outside the United States and initiating a Phase 2 study of PTG-300 in patients with beta-thalassemia. PTG-300 has received an orphan drug designation from the FDA for the treatment of beta-thalassemia.
In addition, we continue to use our peptide technology platform to discover product candidates against targets in disease areas with significant unmet medical needs. In 2018, we anticipate initiating IND-enabling studies for a fourth product candidate, an oral peptide targeting a GI condition other than IBD.
Our Product Candidates
PTG‑100
PTG‑100 has the potential to be a first in class oral, α4β7 antagonist for the treatment of IBD. The α4β7 integrin is one of the most GI-specific biological targets for IBD. It is a cell surface protein present on T cells that plays an important role in the trafficking of T cells to the GI tissue compartment by binding to MAdCAM‑1, an extracellular protein that resides mostly in the GI vasculature.
We are leveraging several factors to inform and guide the clinical development of PTG‑100 for the treatment of IBD. First, PTG‑100 shares the same α4β7 integrin target as the injectable antibody drug vedolizumab, marketed as Entyvio®, for the treatment of moderate-to-severe UC and CD. Second, we utilized pharmacodynamic (“PD”) biomarker assays similar to those describedTremfya® generated $3.1 billion in scientific publications as used with Entyvio® and other antibodies as indicators of target engagement to establish proof-of-concept (“POC”)sales in our Phase 1 clinical trial with PTG‑100. These PD data include dose dependent increases in receptor occupancy and decreases in receptor expression.2023. We believe that we can utilize published information describing the development and regulatory path of Entyvio® and other approved antibody drugs for IBD to help inform the design of our clinical development studies.
We have completed extensive pre-clinical studies of PTG‑100 in which we established pharmacological POC, including effects on T cell trafficking and mucosal healing similar to the comparator α4β7 rodent antibody, DATK‑32. Following the submission and approval of a Clinical Trial Notification (“CTN”) in Australia in December 2015, we initiated a Phase 1 clinical trial, comprised of single ascending dose (“SAD”) and multiple ascending dose (“MAD”) components, each of which evaluated safety, pharmacokinetics (“PK”), and PD-based POC in healthy subjects. The Phase 1 clinical trial was completed in June 2016. Dose escalation proceeded up to 1,000 mg in both singlepsoriasis and multiple dosing. There were no serious adverse events reportedIBD, there is an urgent need for safe and effective oral therapies. It is notable that Stelara® lost patent exclusivity in the trial, and no dose-limiting toxicities were observed. All reported adverse events were of mild to moderate severity. There were no dose-dependent increases observed for any adverse events. The most frequent adverse events reported by subjects on PTG‑100 were headache and upper respiratory tract infection. These events were also observed in subjects who received placebo.
2023 with biosimilar competition expected.
We initiated a global Phase 2b randomized, double-blinded, placebo-controlled dose-finding clinical trial in the fourth quarter of 2016 to assess the safety and efficacy of PTG‑100 in moderate-to-severe UC patients. We anticipate that the trial will enroll approximately 240 subjects at approximately 100 sites in the United States, Canada, Europe (Western, Central, and Eastern)JNJ-2113 (formerly known as PN-235), Asia, Australia, and New Zealand. The primary objectives of our Phase 2b clinical trial are to evaluate the safety and tolerability of PTG‑100 and its efficacy in the induction of remission in subjects with moderate-to-severe UC. Secondary objectives are to select PTG‑100 induction doses for continued development, to characterize PTG‑100 plasma concentrations and pharmacodynamic responses, and to evaluate any immunogenicity over 12 weeks. The trial will include subjects who have had prior exposure to tumor necrosis factor-alpha (“TNF‑α”) inhibitors and subjects who have not been treated with biologics. Subjects will be randomized to one of four dose arms (150mg/300mg/900mg PTG‑100 or placebo) for 12 weeks of once-daily oral dosing, followed by four weeks of safety follow-up. An interim futility analysis is expected to be performed in the first quarter of 2018, and if futility criteria are not met, one or two PTG‑100 doses will be selected for continued randomization of the remaining subjects. We expect to complete the study and report top-line data in the fourth quarter of 2018. We expect that this trial will support end-of-Phase 2 meetings with global health authorities and enable the initiation of a Phase 3 pivotal program.
The primary endpoints are consistent with those used in the clinical development of previously approved drugs for UC. The trial is statistically powered to detect a clinically meaningful difference in induction of remission in subjects with moderate-to-severe UC who are treated with PTG‑100 compared to placebo. The evaluation of clinical remission is based on the Mayo Score, which is a well-established composite assessment that utilizes patient-reported outcomes and endoscopic improvement. Secondary efficacy endpoints will include endoscopic response, clinical response, endoscopic remission, change in endoscopic subscore, change in stool frequency and rectal bleeding subscores, change in fecal calprotectin, change in the IBD questionnaire, change in Mayo score and change in partial Mayo score, from baseline to multiple points during the induction period.
We plan to develop PTG‑100 initially for the treatment of moderate-to-severe UC and CD, as well as chronic pouchitis. Subsequent indications may include mild-to-moderate UC, CD, and pediatric IBD, the latter being an orphan indication.
PTG‑200
Our second oral, GI-restricted peptide product candidate is PTG‑200, a potential first-in-class IL‑23Rorally delivered IL-23R specific antagonist for the potential treatment of IBD. Interleukin‑23 (“IL‑23”),psoriasis, psoriatic arthritis and IBD indications, was discovered through our peptide technology platform. IL-23, a member of the IL‑12IL-12 family of pro-inflammatory cytokines, is a protein that regulates inflammatory and immune function and plays a key role in the development of IBD. By blocking the IL‑23 receptor with PTG‑200 in the GI tissue compartment,IL-23R, we expect to reduce inflammationbelieve JNJ-2113 may improve disease symptoms while potentially minimizing the risk of systemic side effects due to its GI-restricted nature. The IL‑23 pathway is targeted by the IL‑12 and IL‑23 antagonist infused antibody drug ustekinumab, marketed as Stelara®, for psoriasis, psoriatic arthritis, and moderate-to-severe CD.
We have completed pre-clinical POC studies and IND-enabling studies for PTG‑200, and we initiated a Phase 1 clinical trial ineffects. During the fourth quarter of 2017.2021, a decision was made by JNJ to advance development of our IL-23R antagonist JNJ-2113. For JNJ-2113, JNJ is primarily responsible for the conduct of all further development, and we were primarily responsible for the discovery, IND-enabling studies and the initial Phase 1 study.
Clinical Development of JNJ-2113
A Phase 1 study was initiated for JNJ-2113 in December 2020. The Phase 1 study which is beingfor JNJ-2113 was designed to determine the safety, tolerability and pharmacokinetics of JNJ-2113 in 107 healthy volunteers. The study was conducted in Australia, isthree parts: a SAD component, a MAD component, and a randomized, crossover solid dose comparison component. The primary endpoint was safety as measured by number and severity of adverse events. Secondary outcomes included pharmacokinetics measurements of peak concentration and area under the curve. This Phase 1 study was completed in September 2021. Results of the Phase 1 study demonstrated that administration of JNJ-2113 was well-tolerated. No serious adverse events or dose-limiting toxicities were observed. The pharmacokinetic and pharmacodynamic parameters of JNJ-2113 were consistent with those predicted by pre-clinical studies.
In February 2022, JNJ initiated FRONTIER 1, a 255-patient Phase 2b clinical trial of JNJ-2113 in moderate-to-severe plaque psoriasis, which was completed in December 2022. FRONTIER 1 was a randomized, multicenter, double-blind, placebo-controlled singletrial that evaluated three once-daily dosages and multiple dose-escalationtwo twice-daily dosages of JNJ-2113 taken orally. The primary endpoint of the trial was the proportion of patients achieving PASI-75 at 16 weeks. In July 2023, we announced updated positive topline results from the trial, which were presented by JNJ at the World Congress of Dermatology in approximately eighty healthy volunteers. Secondary endpointsSingapore. JNJ-2113 achieved the trial’s primary and secondary efficacy endpoints. A statistically significant greater proportion of patients who received JNJ-2113 achieving PASI-75 responses as well as PASI-90 and PASI-100 responses compared to placebo at week 16 in all five of the trial’s treatment groups. A clear dose response was observed across an eight-fold dose range. Treatment was well tolerated, with no meaningful difference in frequency of adverse events across treatment groups versus placebo.
JNJ has initiated five additional JNJ-2113 trials, including:
| ● | ICONIC-LEAD – A 600-patient randomized, controlled Phase 3 trial to evaluate the safety and efficacy of JNJ-2113 compared with placebo in participants with moderate-to-severe plaque psoriasis, with PASI-90 and IGA score of 0 (clear) or 1 (almost clear) as co-primary endpoints; |
| ● | ICONIC-TOTAL – A 300-patient randomized, controlled Phase 3 trial to evaluate the efficacy and safety of JNJ-2113 compared with placebo for the treatment of plaque psoriasis in participants with at least moderate severity affecting special areas (scalp, genital, and/or palms of the hands and soles of the feet) with overall IGA score of 0 or 1 as the primary endpoint; |
| ● | ICONIC ADVANCE 1 – A 750-patient randomized, controlled Phase 3 trial to evaluate the effectiveness of JNJ-2113 in participants with moderate-to-severe plaque psoriasis compared to placebo and Sotyktu (“deucravacitinib”). The trial’s primary co-endpoints are PASI-90 and IGA score of 0 or 1; |
| ● | ICONIC ADVANCE 2 – A 675-patient Phase 3 trial similarly designed to ICONIC ADVANCE 1, which is expected to start enrolling patients later in 2024; and |
| ● | ANTHEM-UC – A 240-patient Phase 2b randomized, controlled trial to evaluate the safety and effectiveness of JNJ-2113 compared with placebo in participants with moderate-to-severely active UC. |
All of the trials in the ICONIC program will use the once-daily, immediate release formulation from the previously completed FRONTIER 1 study. The estimated primary completion date for the ICONIC-LEAD and ICONIC-TOTAL trials is November 2024 (see NCT06095115 and NCT06095102, respectively, at clinicaltrials.gov). The estimated primary completion dates for the ICONIC-ADVANCE 1 and ICONIC-ADVANCE 2 trials are March 2025 and April 2025, respectively (see NCT06143878 and NCT06220604, respectively, at clinicaltrials.gov). The estimated primary completion date for the ANTHEM-UC trial is May 2025 (see NCT06049017 at clinicaltrials.gov). Other Phase 2 trials of JNJ-2113 include the identificationSUMMIT trial of the maximally tolerated dose and the evaluation of pharmacokinetic parameters.
We have a worldwide license and collaboration agreement with Janssen to co-develop and co-detail PTG-200 for all indications as described in Item 7. “Management’s Discussion and Analysis – Overview” and Note 3 to the Consolidated Financial Statements included elsewhere in this Annual Report on Form 10-K. We and Janssen currently plan to develop PTG‑200 initiallyJNJ-2113 for the treatment of moderate-to-severe CD, potentially followedplaque psoriasis, and FRONTIER 2, a long-term extension study, both of which were completed by UC.JNJ in 2023.
PTG‑300
PTG‑300 is an injectable peptide that mimics the effect of the hormone hepcidin. We are developing PTG-300 for the treatment the chronic anemia that arises from insufficient red blood cell production, known as ineffective erythropoiesis,At JNJ’s Innovative Medicines Enterprise Business Review in certain rare blood disorders, including beta-thalassemia. In these diseases, excessive quantities of iron
in the bone marrow inhibit the production of red blood cells causing anemia. In healthy individuals, hepcidin regulates iron levels by inhibiting iron absorption from the GI tract and by limiting macrophage release of iron in the bone marrow. Individuals with beta-thalassemia and MDS often have insufficient hepcidin to maintain appropriate iron levels. Because of stability issues, complexity of synthesis and solubility limitations, direct hepcidin replacement is not a practical therapeutic approach. We developed PTG‑300 as a stable, soluble, manufacturable hepcidin mimetic that could potentially prevent excessive iron accumulation and iron toxicity with weekly sub-cutaneous injections.
We completed IND-enabling studies during the first half of 2017 and completed a Phase 1 clinical trial during the fourth quarter of 2017. The Phase 1 study demonstrated that PTG-300 induces dose-related reductions in serum iron, which persist beyond 72 hours at higher dose levels. In the study PTG-300 produced dose-dependent increases in blood exposure and was well tolerated, with no serious adverse events or dose-limiting toxicities.
In 2018, we anticipate filing an IND in the United States and related clinical trial applications outside the United States and initiating a Phase 2 study of PTG-300 in patients with beta-thalassemia. PTG-300 has received orphan designation from the FDA for the treatment of beta-thalassemia.
Additional Product Candidates
We are currently researching potential oral and injectable peptide-based product candidates for a range of conditions including, but not restricted to, GI diseases. In 2018, we anticipate initiating IND-enabling studies for a fourth product candidate, an oral peptide targeting a GI condition other than IBD.
The Evolution of Antibody Drugs for Targeted Therapy and Their Limitations
Before FDA approval of antibody drugs, chemically synthesized oral small molecules were the standard-of-care for the treatment of many diseases. However, small molecules are often not capable of affecting the critical processes involved in diseases, for example by blocking protein-protein interactions (“PPIs”) or by mimicking naturally occurring molecules. It is estimated that small molecules cannot be developed as drugs for the treatment of up to 80% of all identified potential disease targets. With the availability of antibody drugs, targeted therapy for many PPI-driven diseases became feasible.
Despite their growing use, antibody drugs present several limitations for patients including, but not limited to, the following:
| ·
| | Antibody drugs may have significant safety issues. Antibody drugs are typically administered at high concentrations in order to attain appropriate therapeutic levels at distal sites of a disease. High systemic exposure of immunomodulatory agents can increase the risks of use for patients:
|
| ·
| | Elevated risk of serious or opportunistic infection, malignancy and severe hypersensitivity events. Many antibody drugs are immunosuppressive, which may lead to increased risk of serious or opportunistic infection, such as tuberculosis, histoplasmosis and hepatitis B, or malignancy. Further, injection or infusion may increase the risk of severe hypersensitivity reactions including anaphylaxis.
|
| ·
| | Long half-life resulting in delayed clearance from the bloodstream. Antibody drugs are large molecules engineered to have long half-lives and to circulate in the bloodstream for extended periods of time. This longevity can be potentially problematic for patients who experience adverse reactions and cannot readily eliminate the drug from their systems.
|
| ·
| | Immunogenicity reactions can lead to loss of response or possible safety risks. Antibody drugs may induce natural immunogenic responses from the body including the introduction of anti-drug antibodies (“ADAs”). These ADAs can neutralize the action of the therapeutic antibody either by enhancing its clearance or blocking its function, either of which can result in loss of therapeutic response. ADAs can cause immunogenic reactions in patients leading to possible adverse events, frequently necessitating drug withdrawal.
|
| ·
| | Antibody drugs are expensive. Compared to other classes of therapeutics, the complexity and size of antibody drugs can result in high manufacturing, storage and administration costs. To date, these costs have not been significantly reduced through the introduction of biosimilar drugs.
|
| ·
| | Injections or infusions are associated with significant patient burden. Antibody drugs are large proteins that are not stable when orally administered. As a consequence, antibody based therapies are administered primarily by injection or infusion into systemic circulation even when the target of the antibody is a specific site in the body. Injections or infusions as a mode of delivery can increase patient burden, including site reactions and systemic hypersensitivity, inconvenience, and needle anxiety and phobia, each of which may negatively affect patient compliance
|
Our Therapeutics Platform
Our novel peptide therapeutics platform may provide important benefits over existing non-targeted small molecule, injectable antibody, and conventional peptide therapeutics. In addition, our platform represents a major step forward in the evolution of peptides as therapeutics. Most of the more than 60 currently FDA approved peptides have unstructured shapes, leading to chemical and biological stability limitations, which confine their use to injectable drugs. In contrast, our peptide technology platform allows us to identify constrained peptides that can serve as a starting point for discovery and development of selective and potent peptides that may be orally delivered if desired. The well-folded conformation in our constrained peptides is typically derived by disulfide bonds, a structural feature inherent in many naturally occurring peptides.
Our IBD Solution: Oral, GI-Restricted Peptides as Targeted Therapies
For the IBD targets of interest, the size and nature of our peptides is carefully selected and modified so as to acquire the desired potency and specificity, and also to restrict their presence to the GI tissue compartment when administered orally. These features translate to oral, GI-restricted, selective and potent peptide drug candidates with specific advantages compared to antibody drugs:
| ·
| | Oral administration. We are developing our peptide therapeutics in a convenient capsule or tablet form intended for oral administration. We believe oral administration may reduce many of the problems and limitations associated with injections or infusions, including injection site pain and local reactions, inconvenience, anxiety, high rates of immunogenicity and potential safety risks.
|
| ·
| | Potential for improved safety and tolerability compared to antibody drugs.
|
| ·
| | Oral and GI-restricted delivery minimizes systemic exposure in the blood. Oral GI-restricted delivery results in lower drug levels in the blood that may provide the potential for an enhanced safety profile over antibody drugs.
|
| ·
| | Peptides can be cleared more quickly from systemic circulation. Small molecules and peptides below a size threshold can be rapidly cleared from blood circulation by kidney filtration and excretion. Rapid clearance may be beneficial especially if patients need to discontinue therapy. In contrast, antibody drugs, because of their long plasma half-life, may take months to clear from blood circulation leaving patients exposed to continued or increased safety risk.
|
| ·
| | The likelihood of much lower immunogenicity of small stable peptides compared to antibody drugs reduces the risk of loss of response. We believe that ADAs are less likely to be elicited against constrained peptides, due to their small size, lack of epitope density, resistance to proteolysis, oral tolerance, and minimal systemic absorption.
|
| ·
| | Potential for localized delivery to site of disease. We believe oral dosing of GI-restricted peptides results in substantially higher drug concentrations in the diseased GI tissue compartment compared to injectable
|
antibody drugs. This targeted delivery to the site of action may lead to more immediate and significant target engagement at the site of active disease in the GI tissue compartment.
|
| ·
| | Cost-effective and less complex manufacturing. Because of their size and stability, we believe that our oral, GI-restricted peptide product candidates can be produced, stored and shipped in a more cost-effective manner than many antibody drugs.
|
In chronic GI diseases such as IBD, we believe that our oral, GI-restricted peptide product candidates may offer improved delivery, the potential for improved safety and tolerability, and cost efficiencies that may provide an overall benefit to patients, payers, and physicians.
Overview of Inflammatory Bowel Disease
Inflammatory bowel disease is a group of chronic autoimmune and inflammatory conditions of the colon and small intestine, consisting primarily of UC and CD, and characterized by abdominal pain, diarrhea, weight loss, fatigue and anemia. In UC, inflammation starts in the rectum and generally extends proximally in a continuous manner through the entire colon. In CD, the disease most commonly affects the small intestine and the proximal large intestine. These chronic diseases tend to run in families and they affect males and females equally. Both UC and CD have periods of various intensity and severity, and when a patient is symptomatic, the disease is considered to be in an active or flare stage. Approximately 25% of UC cases occur in persons under the age of 20.
Market Overview
According to the Crohn’s & Colitis Foundation of America, there were an estimated 1.6 million IBD patients in the United States in 2013, an increase of approximately 200,000 patients since 2011. As many as 70,000 new cases of IBD are diagnosed in the United States each year. As of 2008, annual direct treatment costs for patients with IBD in the United States were estimated to exceed $6.3 billion, while indirect costs such as missed work days were estimated to cost an additional $5.5 billion. In 2015, GlobalData estimated that the UC market reached approximately $4.8 billion across seven major markets: the United States, France, Germany, Italy, Spain, the United Kingdom and Japan. By the end of 2025, the UC market is expected to reach $5.5 billion, reflecting a compound annual growth rate of 1.3%. In 2016, GlobalData estimated that the CD market reached approximately $9.2 billion across those same seven major markets. By the end of 2026, the CD market is expected to reach $13.4 billion, representing a compound annual growth rate of 3.8%.
History of IBD Treatments
Non-Targeted Therapies
Sulfasalazine was discovered as the first non-targeted therapy for treatment of UC. Non-targeted therapies continued to evolve, including the introduction of corticosteroids for treatment of moderate UC in the 1950s. Subsequently, the immunosuppressive drug mercaptopurine was identified for UC in the 1960s, azathiopurine was developed in the 1970s, followed by 5‑aminosalicylic acid. While these oral, non-targeted broad-spectrum anti-inflammatory agents and non-specific immunomodulators continue to be part of the IBD treatment paradigm, especially in mild-to-moderate IBD, these drugs are often ineffective, and corticosteroid and oral immunosuppressive drugs may have significant and disabling adverse effects that limit their use.
TNF- α and α4β7 Integrin Targeted Antibody Drugs
Recent advances in molecular biology and genomics ushered in the development of the potent and highly targeted biologic drugs. TNF‑α was identified as a cytokine, a protein involved in cell signaling, that plays an important role in the inflammatory processes associated with IBD. In developing therapies against TNF‑α, small molecule antagonists that directly bind TNF‑α and other PPI targets have yet to be discovered and approved as therapeutics for the treatment of IBD. Thus, monoclonal antibody drugs emerged as a new class of therapeutics that can inhibit TNF‑α activiy. There are currently five TNF‑α antibody drugs (Humira®, Remicade®, Cimzia®, Simponi® and Inflectra® (infliximab biosimilar))
approved for the treatment of UC and/or CD. In 2014, Entyvio®, an intravenously administered antibody that selectively targets the α4β7 integrin, was approved for the treatment of adult patients with moderate-to-severe UC or CD where one or more standard therapies have not resulted in an adequate response. Entyvio® sales were approximately $530 million in 2015 and are projected to peak at approximately $2 billion. In 2016, Stelara®, a subcutaneously administered antibody that targets the IL-23 and IL-12 cytokines, was approved for the treatment of adult patients with moderate to severe active CD wo had failed to respond to, or were intolerant of, standard therapies or a TNF-α antibody.
While antibody drugs have greatly improved the treatment of IBD, they generally serve as the last-line of treatment before surgery due to their potential for severe adverse effects, diminishing efficacy over time, inherent limitations as injectable-based therapies, and high costs of therapy.
The Evolving IBD Treatment Paradigm
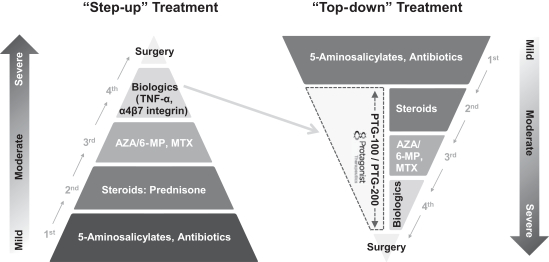
Inducing and maintaining clinical remission is the primary goal of treatment for IBD patients. The current treatment paradigm for IBD is considered a “step-up” approach. It involves a sequential “step-up” in treatment to more potent but higher risk therapies according to the level of severity of the patient’s disease. Thus, targeted biologic therapies are generally reserved for patients with moderate-to-severe disease who have failed to respond to non-targeted oral therapies including 5‑ASA agents, corticosteroids and non-specific immunomodulators. As a result, only a portion of IBD patients currently receive a targeted antibody therapy.
For moderate-to-severe IBD patients, physicians may prescribe TNF‑α antibody drugs, Entyvio®, or, in the case of CD, Stelara®, to induce and maintain clinical remission. Patients who are transitioned to these targeted antibody drugs may fail to respond to treatment or lose response to some or all of these agents over time and may ultimately require surgery. Approximately 50% to 73% of CD and 65% of UC patients fail to reach remission with TNF‑α antibodies. Furthermore, 30% to 40% of UC patients and approximately 40% of CD patients treated with TNF‑α antibody drugs stop responding to these agents over time (secondary non-responders) at a rate of approximately 10% to 13% per year. Of the CD patients who initially benefit from TNF‑α antibody drugs, 25% to 40% of these patients develop intolerable or serious adverse events or lose their response within the first year of therapy. Currently, a common approach for IBD patients with lack of efficacy or loss of response to TNF‑α antibody drugs is to switch such patients to other TNF‑α antibody drugs. Although this is initially successful in 40% to 60% of patients, there remains a lack of treatment options for patients who lose responses to multiple TNF‑α antibody drugs. Further, patient non-adherence with TNF‑α antibody drugs in IBD has been reported to be between approximately 30% to 45% resulting in a greater need for hospitalization.
We believe the development of new, potent and targeted therapies for IBD with oral delivery may offer more effective treatment options for moderate-to-severe IBD patients. In addition, many clinicians are already advocating for an earlier introduction of targeted therapeutics in IBD to reduce, replace or delay the use of corticosteroids and non-specific oral immunomodulators. This treatment approach is often referred to as a “top-down” approach as therapeutics that are currently at the top of the “step-up” pyramid are moved down to earlier in the treatment paradigm (see Figure 1). We believe we are well-positioned to be leaders in this shift from “step-up” to “top-down” therapy. Our oral, GI-restricted, and targeted peptide drugs work on the same specific targets as injectable antibody drugs and have the potential to offer improved patient safety, improved compliance and convenience and reduced immunogenicity as compared to antibody drugs. In addition, key opinion leaders are increasingly viewing the a4β7 integrin antagonist Entyvio® as a preferable alternative to TNF‑α blockers for the treatment of IBD due to its improved safety profile. Taken together, we believe that these trends may result in our product candidates, if approved, being used more broadly than antibody drugs in moderate-to-severe IBD patients and potentially being used for the treatment of mild-to-moderate disease.
Figure 1: Transforming the Existing IBD Treatment Paradigm with Oral Targeted Therapy Drugs
PTG‑100: AN ORAL α4β7 INTEGRIN ANTAGONIST
PTG‑100 was discovered through our peptide technology platform and is being developedDecember 2023, JNJ highlighted JNJ-2113 as a potential first-in-classfirst- and best-in class targeted oral GI-restricted α4β7 integrin-specific antagonist initially for patients with moderate-to-severe UC.
Mechanism of Action and Rationale
Integrins, such as α4β7, are transmembrane proteins that regulate cellular movement into extravascular tissue and play an important role in modulating the inflammatory reaction in the gut. The α4β7 integrin is expressed on the surface of T cells, immune cells that help defend against foreign and potentially harmful substances that enter the body. The development of UC is driven by the migration of α4β7 T cells into the GI tissue compartment and their subsequent activation within the GI tissue compartment. The entry of α4β7 T cells into the GI tissue compartment is facilitated by the PPI between the α4β7 integrin and its corresponding ligand, MAdCAM‑1, which is primarily expressed in the GI tissue compartment. Hence, the binding of α4β7 to MAdCAM‑1 can be categorized as a GI-specific interaction and has been identified as an IBD-specific targeted therapeutic approach. By blocking the binding of a4β7 integrin to MAdCAM‑1, PTG‑100 may prevent T cells from entering the GI tissue compartment, thereby reducing the inflammation that leads to the clinical manifestations of UC.
α4β7 for IBD is targeted by FDA-approved Entyvio® (vedolizumab), which has demonstrated safety and efficacy in patients with moderate-to-severe UC and CD. Since PTG‑100 targets the same biological pathway as Entyvio®, we utilized similar PD-based POC in our pre-clinical studies and Phase 1 clinical trial to inform and guide our Phase 2b development program. We sourced these PD biomarker assays from public scientific publications and do not maintain any contractual arrangement providing access to this information with the makers of these marketed products.
Translating PTG‑100’s Pre-Clinical POC to Clinical POC
We established a potentially efficacious dose range of PTG‑100 in mice by demonstrating similar pharmacologic activity between oral PTG‑100 and an injectable α4β7 antibody in mouse models of IBD. From this efficacious dose range in mice, approximately 6‑50 mg/kg per day, we were able to directly estimate a potentially efficacious dose range in humans through allometric scaling based on whole body surface areas, which we determined to be approximately 33‑300 mg per day.
Concurrently, we employed a complementary approach for establishing a potentially efficacious human dose range and early POC through specific blood PD response markers that reflect α4β7 integrin target engagement of PTG‑100 in the GI tissue compartment and correlated those PD measurements with efficacy responses in mouse colitis models. Target engagement is a critical feature for demonstrating that PTG‑100 can reach its intended target, thus inhibiting the trafficking of T cells into the GI tissue compartment. Our PD markers were monitored in mice and cynomolgus monkeys (“cyno”) and were similarly evaluated in normal healthy volunteers in our Phase 1 clinical trial. These blood PD responses demonstrated that PTG‑100 engaged its intended α4β7 target and helped guide human dosing for our Phase 2b clinical trial.
PTG‑100’s Pre-Clinical Proof-of-Concept Studies
Pre-clinical studies have demonstrated that PTG‑100 is a potent and highly selective α4β7IL-23 peptide antagonist with minimal systemic absorption. Mouse colitis models have further demonstrated that PTG‑100 can inhibit T cell trafficking in the gut similar to the actions of the mouse α4β7 antagonist antibody.
PTG‑100 potently inhibited binding of α4β7 to MAdCAM‑1 in several human biochemical enzyme-linked immunosorbent assays (“ELISA”) and cell adhesion (transformed and primary cells) assays in a low nanomolar concentration range sufficient to inhibit 50% of binding (“IC50”) comparable to vedolizumab. PTG‑100 exhibited greater than a 100,000‑fold selectivity against other structurally similar integrins, α4β1 and αLβ2, in cell adhesion assays which is comparable to the selectivity of vedolizumab. PTG‑100 was stable in in vitro assays simulating the GI tissue compartment, such as the small intestine and gastric stomach, with half-lives exceeding 12 hours and in human liver microsomes suggesting strong oral stability and the potential for once daily dosing in humans. PTG‑100 did not affect the growth of and was not metabolized by common members of the human intestinal microflora. In total, these drug properties provide evidence to characterize PTG‑100 as a potential first-in-class orally stable α4β7‑specific antagonist. Furthermore, these drug properties allowed us to demonstrate proof-of-concept in animal colitis studies.
Non-clinical metabolism and PK studies demonstrated that much greater amounts of PTG‑100 as measured by the maximum concentration (“Cmax”) as a percentage of total drug amount dosed orally, were present in the GI compartments, such as the small intestine, colon and feces compared to the systemic plasma and urine compartments of mice, rats, and cyno, thus confirming its GI-restricted properties. Further, PTG‑100 has an oral systemic bioavailability of less than 0.5%.
We designed mouse colitis studies similar to those used for antibody drugs targeting this pathway to specifically monitor T cell trafficking to and from the GI tissue compartment (Figure 2). PTG‑100 reduced α4β7 memory T cells migrating to the gut lymphoid tissues,across multiple indications, including the mesenteric lymph nodes (“MLN”) and Peyer’s patches (“PP”), under inflammatory conditions in the GI tissue compartment. Another example of the ability of PTG‑100 to inhibit T cell trafficking was demonstrated by the reduction in the number of α4β7 cells in colon lesions in colitis mice. Furthermore, treatment benefit was demonstrated through blinded video endoscopy analysis for mucosal damage, and assessment of the incidence of bloody feces, which represent symptoms and measurements of UC in humans. In all studies in mouse models of colitis, the effects of oral PTG‑100 were comparable to those of an injection of high doses of a positive control α4β7 antibody. This allows us to define the efficacious dose in mice with potential translation to the efficacious dose in humans.
Establishing Blood Pharmacodynamic Readouts of Target Engagement
We have used pre-clinical blood PD response markers that reflect target engagement in the GI tissue compartment and correlate with efficacy responses in mouse colitis studies to guide our dosing in human studies. Furthermore, we believe these pre-clinical blood PD responses, specifically receptor occupancy (“RO”) increases reflecting target engagement and receptor expression (“RE”) decreases reflecting subsequent pharmacologic activity, can be compared to the PD responses we observed in our Phase 1 clinical trial in healthy volunteers and ultimately can help to guide the dosing for evaluating clinical benefit in UC patients in the Phase 2b clinical trial. In the mouse colitis model, RO and RE were correlated with in vivo efficacy that can be extrapolated to the blood RO and RE observed in healthy mice and cyno. These PD markers from mice and cyno have specifically demonstrated increases in RO that peak at approximately 4 hours following a single dose and multiple doses and decreases in RE after multiple doses in healthy mice and colitis
mice. In translating the pre-clinical observations into a clinical setting, we are focused on evaluating dose- and time-dependent trends in RO and RE in our Phase 1 clinical trial that can be benchmarked to the animal data to give us greater confidence in progressing PTG‑100 in clinical trials. Emphasis is placed on trends and not on absolute numbers owing to differences in GI transit times in different species and absence of absolute scaling methods from animals to humans for GI-restricted drugs.
PTG‑100’s Non-GLP and GLP Safety Pharmacology and Toxicology Studies
To date, all toxicology and safety pharmacology studies have not identified any safety issues. Good Laboratory Practices (“GLP”) toxicology studies in rats and cyno over 42 days and 12 weeks of dosing showed that PTG‑100 was well-tolerated at all dose levels with no dose-limiting toxicities. GLP are those procedural and operational requirements specified by FDA regulation to ensure the validity and reliability of nonclinical studies. No adverse effects were seen in either rat or cyno studies at all doses tested. Standard safety pharmacology and genotoxicity studies were similarly negative. We are currently conducting chronic GLP toxicology studies to support our anticipated Phase 3 program.
PTG‑100’s Phase 1 Clinical Trial Overview
Following the submission and approval of a CTN, we initiated a Phase 1 randomized, double-blind, placebo-controlled clinical trial of PTG‑100 in 78 normal healthy male volunteers in Australia, which was completed in June 2016. The Phase 1 SAD and MAD components were conducted with a solution-based liquid formulation of PTG‑100. In the formulation bridging component of the trial, we compared the relative bioavailability of the liquid formulation to the capsule formulation that is being used in Phase 2b. In addition to determining the safety and tolerability and PK of PTG‑100, the SAD and MAD components of the trial evaluated PD-based POC through the assessment of α4β7 receptor occupancy that indicates target engagement and α4β7 target expression on peripheral blood memory T cells similar to what was done in the pre-clinical studies.
Safety and Tolerability
In both the SAD and MAD portions of the clinical trial, dose escalation proceeded from 100 mg up to the planned 1,000 mg dose level. There were no dose-limiting toxicities. There were no deaths or serious adverse events (“SAEs”) reported in the trial. All reported adverse events were of mild to moderate severity. There were no dose-dependent increases observed for any adverse events. The most frequent adverse events reported by subjects on PTG‑100 were headache and upper respiratory tract infection. These events were also observed in subjects who took placebo.
Pharmacokinetics
PTG‑100 plasma levels increased in a dose-dependent manner in both single and multiple dosing cohorts. Consistent with the pre-clinical data in mice, rats, and cyno, the blood levels of PTG‑100 were extremely low as determined by the Area Under the Curve (AUC, which is a pharmacokinetic measurement of drug exposure in blood plasma against time) and Cmax (maximum concentration), thus demonstrating the GI-restricted nature of the drug. There was no apparent evidence of drug accumulation at Day 14 in the MAD cohorts perhaps related to the relatively short half-life (“T1/2”) in the blood.
PTG‑100 fecal levels increased in a dose-dependent manner in the multiple dosing cohorts. Minimum drug levels of PTG‑100 were observed in urine samples, as expected, based on its characteristics as a GI-restricted drug with minimal systemic exposure.
Establishing Pharmacodynamic POC in Humans
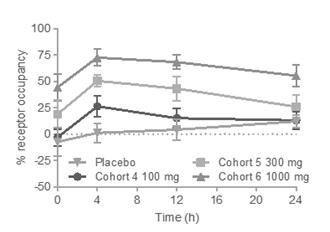
Data from our mouse colitis studies support our conclusion that blood RO is a correlate of target engagement in the GI tissue compartment in the dose ranges studied. In our Phase 1 clinical trial, blood RO on CD4+ memory α4β7+T cells increased in a dose-dependent and time-dependent manner. For RO in the SAD cohorts, treatment groups were significant compared to placebo at 100 mg (p<0.05), 300 mg (p<0.005) and 1,000 mg (p<0.0001). In the MAD cohorts,
treatment groups were significant compared to placebo at 100 mg (p<0.0005), 300 mg (p<0.0001) and 1,000 mg (p<0.0001) four hours post dose on Day 14 (Figure 2A).
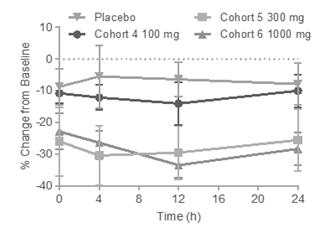
An additional parameter of pharmacologic activity that we measured was the change in α4β7 expression on the blood memory T cells. Based on in vitro studies comparing vedolizumab and PTG‑100, we expected that α4β7 expression would be reduced over time due to the internalization of the α4β7 receptor. Following single and multiple dose administration in the Phase 1 clinical trial, a dose-dependent and time-dependent reduction in α4β7 expression was observed, and it appears that the reduction in target expression may become saturated at 300 mg since a similar response was observed in the 1,000 mg cohort following both single and multiple dosing. For α4β7, downregulation of expression was significant in treatment groups, compared to placebo at 300 mg and 1,000 mg (p<0.01) (Figure 2B).
The single dose 300 mg cohort was evaluated under fasted and fed (standard high fat diet) conditions. Blood drug levels and blood RO of PTG‑100 were compared under both conditions. Based on data from this SAD component and previous pre-clinical studies, the MAD component of the clinical trial was conducted under fed conditions.
Thus, we observed dose-dependent and time-dependent target engagement and pharmacologic activity of PTG‑100 following single- and multiple-dose administration in healthy volunteers consistent with observations in the animal studies.
Figure 2: (A) Percent Receptor Occupancy and (B) Receptor Expression of α4β7 on CD4+ Memory T Cells in Blood of Healthy Humans Dosed for 14 Days
| |
A
| B
|
| 
|
Establishing Pharmacodynamic POC in Humans
Data from our mouse colitis studies support our conclusion that blood RO is a correlate of target engagement in the GI tissue compartment in the dose ranges studied. In our Phase 1 clinical trial, blood RO on CD4+ memory α4β7+T cells increased in a dose-dependent and time-dependent manner. For receptor occupancy in the SAD cohorts, treatment groups were significant compared to placebo at 100 mg (p<0.05), 300 mg (p<0.005) and 1,000 mg (p<0.0001). In the MAD cohorts, treatment groups were significant compared to placebo at 100 mg (p<0.0005), 300 mg (p<0.0001) and 1,000 mg (p<0.0001) four hours post dose on Day 14 (Figure 2A).
An additional parameter of pharmacologic activity that we measured was the change in α4β7 expression on the blood memory T cells. Based on in vitro studies comparing vedolizumab and PTG‑100, we expected that α4β7 expression would be reduced over time due to the internalization of the α4β7 receptor. Following single and multiple dose administration in the Phase 1 clinical trial, a dose-dependent and time-dependent reduction in α4β7 expression was observed, and it appears that the reduction in target expression may become saturated at 300 mg since a similar response
was observed in the 1,000 mg cohort following both single and multiple dosing. For α4β7, downregulation of expression was significant in treatment groups, compared to placebo at 300 mg and 1,000 mg (p<0.01) (Figure 2B).
The single dose 300 mg cohort was evaluated under fasted and fed (standard high fat diet) conditions. Blood drug levels and blood receptor occupancy of PTG‑100 were compared under both conditions. Based on data from this SAD component and previous pre-clinical studies, the MAD component of the clinical trial was conducted under fed conditions.
Thus, we observed dose-dependent and time-dependent target engagement and pharmacologic activity of PTG‑100 following single- and multiple-dose administration in healthy volunteers consistent with observations in the animal studies.
PTG‑200: AN ORAL IL‑23R ANTAGONIST
PTG‑200 was discovered through our peptide technology platform and is being developed as a potential first-in-class oral, GI-restricted antagonist that binds to the IL‑23R and specifically blocks its interaction with the IL‑23 cytokine. PTG‑200 will be initially studied in patients with moderate-to-severe CD potentially followed by UC and pediatric IBD.
Mechanism of Action and Rationale
IL‑23 is a member of the IL‑12 family of cytokines with pro-inflammatory and autoimmune properties. Cytokines are cell signaling proteins that are released by cells and affect the behavior of other cells. Binding of the IL‑23 ligand to the IL‑23R receptor leads to an expression of pro-inflammatory cytokines involved in the mucosal autocrine cascade that is an important pathway of many inflammatory diseases, including IBD. Furthermore, genetic analyses of IBD patients have implicated IL‑23R mutations as a risk factor associated with susceptibility to IBD. The antagonist infused antibody drug ustekinumab (marketed as Stelara® for psoriasis, psoriatic arthritis and IBD, with potential peak year sales projection of $5.0 billion plus. JNJ IL-23 mAb drugs Stelara and Tremfya generated $14.0 billion in revenues in 2023.
In February 2024, the JNJ-2113 Phase 2b FRONTIER 1 trial results in adults living with moderate-to-severe CD) is a p40 antagonist antibody that inhibits both the IL‑23 and IL‑12 pathways. Next-generation IBD antibody drugs, such as guselkumab, target the p19 subunit of the IL‑23 ligand and are specific to the IL‑23 pathway, which is believed to be an important driver of IBD pathology, while not blockading the IL‑12 pathway. IL‑12 is believed to be important in immune surveillance against the development of infections and malignancies.
We believe that the oral, GI-restricted nature of PTG‑200 will allow PTG‑200 to be a potent inhibitor of the IL‑23 pathway for the treatment of IBD. By targeting IL‑23R with our GI-restricted oral IL‑23R antagonist PTG‑200, we believe PTG‑200 will restore proper immune functionplaque psoriasis were published in the GI tissue compartment where there is active disease while minimizing the riskNew England Journal of systemic side effects. Several key cell types that reside in gut-associated lymphoid tissue (“GALT”), including T cells, innate lymphoid cells, and natural killer cells, increase their expression of IL‑23R during the progression of IBD. Therefore, the high concentrations of PTG‑200 in GALT will facilitate access and binding to IL‑23R expressed in the same tissue.
PTG‑200’s Pre-Clinical Proof-of-Concept Studies
PTG‑200 potently inhibited binding of IL‑23 to the IL‑23 receptor in several biochemical (ELISA) and cell (transformed and primary) signaling assays in a subnanomolar to low nanomolar concentration range sufficient to inhibit 50% of binding. PTG‑200 exhibited greater than a 50,000‑fold selectivity against other structurally similar receptors (IL‑12Rb1 and IL‑6R) thereby potentially reducing the risk of off target interactions. In total, these drug properties provide evidence to characterize PTG‑200 as a potential first-in-class orally stable IL‑23R-specific antagonist.
In PK studies in rats and cyno, PTG‑200 was GI-restricted with less than 0.5% oral systemic bioavailability in plasma or urine and principal exposure in the small intestine, colon, and feces. Similar results were observed in cyno.
We have also completed pre-clinical POC studies in rat 2, 4, 6‑trinitrobenzenesulfonic acid (TNBS) colitis models demonstrating that oral delivery of PTG‑200 and other prototype antagonists significantly improved disease outcomes, such as reducing body weight loss, reducing the increased colon weight/length ratio, and reducing the increased colon
macroscopic score which is comprised of assessments of colon adhesions, strictures, ulcers, and wall thickness in a dose dependent manner (Figure 3). Furthermore, PTG‑200 was found to reduce the increased histopathology summary score, which is comprised of assessments of mucosal and transmural inflammation, gland loss, and erosion parameters. Finally, PTG‑200 was able to reduce the expression of the pro-inflammatory IL‑23 induced cytokines in the colon and the IBD disease biomarker lipocalin (LCN2) in the serum and feces.
Figure 3: PTG‑200 Reduces Pathology in Rat TNBS-Induced Colitis
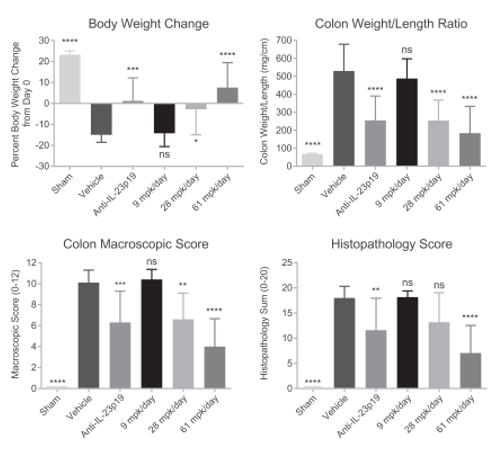
The efficacy of oral PTG‑200 seen in this IBD model was comparable to that of a positive control antibody against the rat IL‑23p19 subunit which was injected and therefore present in the systemic blood compartment. This allows us to define the efficacious dose range in rats (approximately 28‑61 mg/kg per day) with potential translation to the efficacious dose in humans.
PTG‑200’s Pre-Clinical Safety Studies
In pre-clinical safety and toxicity studies in rats, PTG‑200 was well-tolerated with no adverse events at the highest dose level tested.
Clinical Development Plans
We initiated a Phase 1 clinical trial of PTG‑200 in the fourth quarter of 2017 to evaluate safety, tolerability, and PK. Assuming successful completion of this study, we anticipate the next step in clinical development will be a randomized, double-blind, placebo-controlled Phase 2 POC clinical trial in patients with moderate-to-severe CD. We are developing PTG-200 in collaboration with Janssen. See Item 7. “Management’s Discussion and Analysis – Overview”
and Note 3 to the Consolidated Financial Statements included elsewhere in this Annual Report on Form 10-K for additional information.
PTG‑300: AN INJECTABLE HEPCIDIN MIMETIC
PTG‑300 was discovered through our peptide technology platform and is being developed as a novel mimetic of the hormone hepcidin to potentially treat anemia due to ineffective erythropoiesis in certain rare blood disorders with an initial focus on beta-thalassemia. In these diseases, excessive quantities of iron in the bone marrow inhibit the production of red blood cells, causing anemia. In healthy individuals, hepcidin regulates iron levels in the serum and the bone marrow. Because of stability issues complexity of synthesis and solubility limitations, direct hepcidin replacement is not a practical therapeutic approach. We designed PTG‑300 as a stable, soluble, manufacturable hepcidin mimetic that can treat anemia with weekly or less frequent subcutaneous delivery.
Mechanism of Action and Rationale
The molecular target of hepcidin is the cellular trans-membrane protein ferroportin, which functions as an export channel for intracellular iron in macrophages, liver hepatocytes, and duodenal enterocytes. By binding to the extracellular domain of ferroportin, hepcidin prevents the release of iron from cells. It can thus inhibit iron absorption from the GI tract by interacting with duodenal enterocytes and limit the release of iron in the bone marrow by interacting with macrophages. Excessive quantities of iron in the bone marrow induce ineffective erythropoiesis resulting in anemia. By mimicking hepcidin, PTG-300 may restore normal levels of iron in the bone marrow allowing for sufficient production of red blood cells. In addition, by limiting the release of iron into the blood, PTG-300 may inhibit the damage caused by excessive absorption of iron by vital organs.
Overview of Beta-Thalassemia and Current Therapies
We anticipate our initial clinical indication for PTG-300 will be the treatment of anemia in beta‑thalassemia. Beta-thalassemia patients frequently have elevated levels of iron in the blood serum and in the bone marrow. Elevated levels of iron in the serum can cause damage to vital organs in the form of cardiomyopathy or liver fibrosis and can make the patient more vulnerable to infections. In the bone marrow, elevated levels of iron can prevent red blood cells from fully developing, resulting in anemia. In addition, the resulting immature red blood cells can aggregate in the spleen and enlarge it to such an extent that it must be surgically removed. The elevated iron levels seen in beta-thalassemia patients are often caused by disease-related suppression of hepcidin production.
Existing treatment options for hepcidin related anemia and iron overload are limited. Erythropoietic-stimulating agents, such as erythropoietin, are commonly used. However, these agents are often insufficient to treat the patient’s anemia and do not address the issues related to excess serum levels of iron. Red blood cell transfusions can treat a patient’s anemia but exacerbate the patient’s iron overload and are burdensome. The iron overload caused by transfusions will often be treated with chelating agents. However, these agents work very slowly and have significant kidney and liver toxicity issues.
We believe that PTG-300 may be able to restore iron homeostasis to the bone marrow as well as reduce excess circulating iron. We anticipate that restoration of iron homeostasis in the bone marrow – and the resulting increase in red blood cell production – will result in the correction or amelioration anemia as measured by hemoglobin levels. In addition, may beta-thalassemia patients require regular transfusions to treat their anemia. For these patients, treatment with PTG-300 may reduce or eliminate the need for transfusions and related chelation treatments.
Beta-thalassemia is most prevalent in people of Mediterranean descent, such as Italians, Greeks and Turks, and is also found in people from the Arabian Peninsula, Iran, Africa, Southeast Asia and southern China. Globally, the prevalence of beta-thalassemia was estimated to be approximately 300,000 patients in 2008 according to the Centers for Disease Control. The disease is rarer in the United States where Decision Resources Group (“DRG”) estimates there are approximately 3,000 patients. In the major markets of the United States, Italy, Germany, UK, Spain, and France, DRG estimates there are approximately 16,000 diagnosed patients. Most patients with beta-thalassemia suffer from anemia
caused by hepcidin deficiency and a significant number are dependent on transfusions and chelating agents, which can cost between $50,000 to $70,000 per year in the United States.
PTG‑300’s Pre-clinical Proof-of-Concept Studies
In pre-clinical studies, we demonstrated that PTG‑300 can lower serum iron more effectively than hepcidin and maintain such lowered serum iron levels for at least 24 hours following a single subcutaneous injection (Figure 4). We have also demonstrated that PTG‑300 in a dose dependent manner can reduce serum iron in healthy mice, rats, and cyno.
Figure 4: Significant Difference Between PTG-300 and Synthetic Hepcidin in Lowering Serum Iron in Healthy Mice

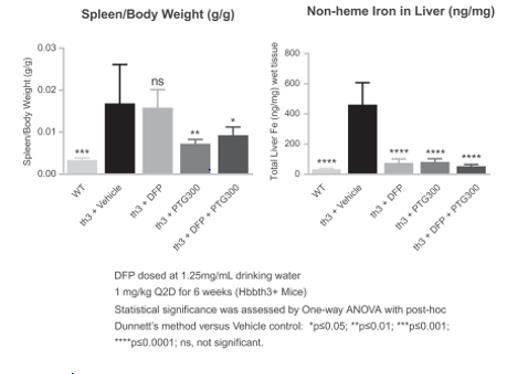
PTG‑300 was also able to address the underlying anemia present in a mouse genetic model of beta-thalassemia, as shown most prominently by the significant increase in red blood cell number (RBC) and hemoglobin (HGB) with the corresponding decrease in reticulocyte content (Figure 5). As a consequence we also observed a significant reduction in the pathological increases in spleen weight (splenomegaly) by addressing the underlying ineffective erythropoiesis. Furthermore, PTG‑300 was effective in reducing the increase in liver iron content. In contrast the oral iron chelator deferiprone (DFP) did not correct the anemia or the splenomegaly.
Figure 5: PTG‑300 Addresses Ineffective Erythropoiesis in Mouse Beta-thalassemia

PTG‑300’s Phase 1 Clinical Trial Overview
We completed IND-enabling studies during the first half of 2017 and completed a Phase 1 clinical trial during the fourth quarter of 2017. The Phase 1 randomized, placebo-controlled single ascending- and repeat-dose study was conducted to evaluate the safety, tolerability, pharmacokinetics and pharmacodynamics of PTG-300 in 62 normal healthy male volunteers.
The Phase 1 study demonstrated that PTG-300 induces dose-related reductions in serum iron, which persist beyond 72 hours at higher dose levels. These results were consistent with known activities of hepcidin and pre-clinical studies of PTG-300. In the study, PTG-300 produced dose-dependent increases in blood exposure, and was well tolerated, with no serious adverse events or dose-limiting toxicities. The most common adverse event was a transient and self-limited erythema (redness) at the injection site in some subjects which was largely dose-related. The study provided a pharmacodynamic proof of concept and established a range of doses that could be evaluated in the treatment of beta-thalassemia.
PTG‑300’s Development Program
In 2018, we anticipate filing an IND as well as ex-U.S. clinical trial applications and initiating a Phase 2 study of PTG-300 in patients with beta-thalassemia. In addition to the potential treatment of anemia in rare blood disorders, such as beta-thalassemia and myelodysplastic syndromes, PTG-300 therapy may also have potential benefit in other diseases such as hereditary hemochromatosis, polycythemia vera, siderophilic infections, and liver fibrosis and we are evaluating potential development pathways for those indications. PTG-300 has received orphan drug designation for the treatment of beta-thalassemia from the FDA.Medicine.
OUR PEPTIDE TECHNOLOGY PLATFORM
Our proprietary technology platform is purposefully built to exploit the advantages of constrained peptides, which are much smaller than antibody-based drugs and may be delivered orally but are big enough to bind and block the difficult targets that antibodies bind and modulate. The platform has been successfully applied to a diverse set of biological targets that has led to several pre-clinical and clinical-stageclinical stage peptide-based NCEs,new chemical entities, including our clinical-stageclinical stage product candidates, PTG‑100, PTG‑200 and PTG‑300, for a variety of clinical indications. Our platform is comprised of a series of tools and methods, including a combination of molecular design, phage display, oral stability assays, medicinal chemistry, surrogate biomarkers, formulations, in vitro biochemical, cell and tissue-based assays, and in vivo pharmacology and pharmacokinetic approaches. We apply this platform to the discovery and development of constrained peptides as new drug candidates.
The platform is used to develop potential drug candidates:candidates (agonists and antagonists): (i) using the structure of a target, when available, (ii)de novo when no target structure exists, or (iii) from publicly disclosed peptide starting points. In a structure-based approach, our proprietary molecular design software and structural database of several thousand constrained peptides, termed Vectrix™, are screened to identify suitable scaffolds. The scaffolds whichidentified form the basis of designing and constructing the first set of phage or chemical libraries. The initial hits are identified by either panning or screening such libraries, respectively. When structural information is unavailable for a target, hits are identified by panning a set of 34 proprietary cluster-based phage libraries consisting of millions of constrained peptides. Once the hits are identified, they are optimized using a set of peptide, peptide mimetic and medicinal chemistry techniques that include the incorporation of new or manipulation of existing cyclization-constraints, as well as natural or unnatural amino acids and chemical conjugation or acylation techniques. These techniques are applied to optimize potency, selectivity, stability, exposure and ultimately efficacy. For rusfertide, hit discovery and optimization relied exclusively on medicinal and computational chemistry, with no phage display, to develop potent and selective injectable candidates with enhanced stability and exposure in blood. For injectable products, stability in blood is determined using in vitro assay techniques to identify chemical and biological sites of degradation, which are then optimized while still maintaining potency and selectivity. Conjugation strategies are used to optimize the exposure of the injected peptide. For JNJ-2113, phage display is tightly coupled to medicinal chemistry, structural biology and oral stability techniques to develop potent, selective and orally delivered molecules. Oral stability is profiled in a series of in vitro and ex vivo oral-stability assays that portray the chemical and metabolic barriers a peptide will encounter as it transits the GI tract are used to identifyand systemic compartments as needed. These metabolically labile spots in the peptides. Such sites form the focus of medicinal-chemistry based optimizationpeptides are optimized using medicinal chemistry-based approaches to engineer oral stability. Finally, various stability while maintaining selectivity and potency. Various in vivo pharmacology tools are then used to quantify peptide exposure in relevant GI organs and tissues. Thesystemic compartments. This data can then be used to optimize required GI exposure and ultimately in vivo efficacy.
The key foundations of the platform include:
Molecular design tools and large database of constrained scaffolds
Through advances in genomics, molecular biology and structural genomic initiatives there has been an explosion in the number of known structures of potential new drug targets, including PPI targets. In particular, constrained peptides
have the required surface complexity to match or complement the large flat surfaces of PPI targets to provide potent and selective drug candidates. We believe existing commercial molecular design software is not suitable, as it has been developed to identify small molecules that plug cavities of enzymes and do not bind to PPI targets.
We have developed a database of all known structures of a sub-class of constrained peptides, known as disulfide-rich peptides (“DRPs”). We have collected approximately 4,500 DRP scaffolds that are found throughout nature, ranging from single cell organisms to humans. We have created a proprietary molecular design environment, called Vectrix™. A pattern matching algorithm within Vectrix™ allows the selection of an appropriately stable DRP scaffold using the structure of the target of interest. This molecular design process is used to identify constrained peptides as starting points for hit discovery, which are ultimately optimized into potent, selective peptides against targets which are not amenable to small molecule drug discovery.
Phage display techniques and cluster libraries
Phage display may be used to discover the original hit based on Vectrix™-derived scaffolds, optimize existing hits, or to identify hits against those targets in which no structural information exists. For the latter targets, a series of pre-existing phage libraries, termed cluster libraries, are used for hit discovery. This includes 20 proprietary libraries of structurally diverse DRPs that sample greater than 85% of their known structural diversity and 14 proprietary libraries that sample different protein loop geometries. Collectively these libraries provide immense potential for discovering hits at diverse targets as they are based on natural-DRP scaffolds with these characteristics.
Oral stability and in vitro and ex vivo assays
The GI tract provides a set of chemical and metabolic barriers that hinder the development of oral therapeutic agents. We have developed numerous in vitro and ex vivo systems that profile peptide candidates for their stability features needed for oral delivery, GI restriction, and transit through the entire GI tract. This includes profiling for chemical stability, specifically pH and redox stability, and metabolic stability against proteases and other enzymes that are either of human or microbial origin.
These in vitro assays identify metabolic weak spots of peptides, which can then be stabilized by peptidic and peptidomimetic modifications without losing potency.
Medicinal peptide chemistry
We have significant expertise in optimizing potency, selectivity, oral stability and exposure of constrained peptides using a combination of peptide-cyclization, natural and unnatural amino acids, and various conjugation and acylation techniques. With respect to PTG‑300, hit discovery and optimization relies exclusively on medicinal chemistry, with no phage display, to develop potent and selective injectable candidates with enhanced exposure in blood. For other targets, such as the discovery of PTG‑100 and PTG‑200, phage display is tightly coupled to medicinal chemistry and oral stability techniques to develop potent, selective and oral molecules that are GI-restricted.
In vivo pharmacology tools for GI restriction
When developing oral, GI-restricted constrained peptides, we correlate efficacy with potency and level of GI tissue compartment exposure. We have developed the required expertise and know-how to build PK and PD relationships to optimize physicochemical features of constrained peptides such that they are minimally absorbed and have the required degree of GI tissue compartment exposure over the required duration of time frame to achieve in vivoefficacy. This involves examining constrained peptide concentrations in variousis complemented by formulation technologies to enhance GI tissue compartments, blood, urine, and feces when delivered orally in rodents. In this fashion, we can understandsystemic exposure by exploiting the degree of tissue targeting, GI restriction and oralintrinsic stability that is required to achieve efficacy.
Future Applications of our Platform
oral peptides. Finally, various biomarkers are also developed to correlate exposure with efficacy to guide candidate selection, dose selection and provide preliminary POC of target engagement in clinical trials.
Discovery and Preclinical Activities
We believe we have built a versatile, well-validated and unique discovery platform. For example, this peptide technology platform has been used to develop product candidates for diverse target classes including G-protein-coupled receptors, (“GPCRs”), ion channels, transporters, cytokines and cytokinestheir receptors for a variety of therapeutic areas. In the future we may tackle other GI diseasesI&I, metabolic and blood disorders and expand our delivery techniquestechnology platform to include other organ/tissue systems, such as the lung and eye, which will provide potential opportunities to pursue a wider variety of diseases. In addition, the gutdiseases that may communicate with the immune, central nervous,include oral, topical and endocrine systems, providing the potential of our GI-restricted approach to treat metabolic, cancer and cardiovascular diseases. Lastly, wesystemic approaches. We also intend to progress our platform to achieve systemic bioavailability and activity with oral peptides, macrocycles and peptidomimetics, thereby enabling us to address systemic diseases.
Material Agreements
Janssen License and Collaboration Agreement
In May 2017, we and Janssen entered into an exclusive license and collaboration agreement (the “Janssen License and Collaboration Agreement”) for the development, manufacture and commercialization Examples of PTG-200 worldwide for the treatment of CD and UC. The Janssen License and Collaboration Agreement became effective on July 13, 2017. Upon the effectiveness of the agreement, we received a non-refundable, upfront cash payment of $50.0 million from Janssen. See “Item 7. Management’s Discussion and Analysis – Overview” and Note 3 to the Consolidated Financial Statements included elsewhere in this Annual Report on Form 10-K for additional information.
Research Collaboration and License Agreement with Zealand Pharma A/S
In June 2012, we entered into a Research Collaboration and License Agreement with Zealand Pharma A/S (“Zealand”)approach are our pre-clinical stage program to identify optimizean orally active hepcidin mimetic, as was reported at the American Society for Hematology’s virtual annual meeting in December 2020, the discovery and develop novel DRPs to discover a hepcidin mimetic. Under the termsdevelopment of the agreement, Zealand made an upfront paymentJNJ-2113, our IL-23R antagonist in collaboration with JNJ, and also funded the collaboration. See “Item 7. Management’s Discussion and Analysis – Contractual Obligations and Other Commitments” and Note 6 to the Consolidated Financial Statements included elsewhere in this Annual Report on Form 10-K for additional information.our recently announced IL-17 peptide antagonist program as described above.
Competition
The biotechnology and pharmaceutical industries are intensely competitive and subject to rapid and significant technological change. While we believe that our product candidates, technology, knowledge and experience provide us with certain competitive advantages, we face competition from established and emerging pharmaceutical and biotechnology companies, academic institutions, governmental agencies and public and private research institutions, among others. There are no
Rusfertide
Ruxolitinib, marketed as Jakafi®, was approved oral peptide-based α4β7 integrins and IL‑23 based product candidatesin 2014 for IBD.
We believe our principal competition in the treatment of IBDadults with PV who have inadequate response to or are intolerant to hydroxyurea. Approximately 5,300 PV patients are treated with Jakafi® each year. Besremi®, a ropeginterferon alfa-2b product indicated for the treatment of adults with PV, was approved with a black box warning in November 2021.
We are aware of other investigational compounds under clinical development for treatment of PV, including short interfering RNA approaches aimed at modulating or increasing endogenous hepcidin levels.
JNJ-2113
In psoriasis and psoriatic arthritis, competition will come from companies with approved injectable agents in the following therapeutic classes, among others:
| ·
| | Infused a4ß7 antibody: Takeda Pharmaceutical Company
|
| ·
| | Infused IL‑23 and IL‑12 antibody: Johnson & Johnson
|
| ·
| | Injectable or infused anti-TNFα therapy: AbbVie, Johnson & Johnson, Amgen, Pfizer, UCB S.A., Boehringer Ingelheim, Merck
|
We are also aware of several companies developing therapeutic product candidates for the treatment of IBD,IL-17 and IL-12/23 pathway, including but not limited to AbbVie, Allergan, Atlantic Healthcare Plc, Aprogen (biosimilar TNF-α antibody in Phase 3) Arena Pharmaceuticals, Inc.Cosentyx®, AstraZeneca, Biogen, Boehringer Ingelheim (adalimumab biosimilar in Pre-Registration)Taltz®, Bristol-Myers Squibb, Celgene (mongersen sodiumSiliq®, Tremfya®, and ozanimod hydrochloride inSkyrizi®. Bimekizumab (anti-IL-17A and F, UCB) has completed a positive Phase 3 clinical trials)program in psoriasis. Otezla® (Amgen) was the first oral agent approved in both psoriasis and psoriatic arthritis. The oral JAK inhibitors Xeljanz® (Pfizer) and Rinvoq® are approved in psoriatic arthritis. Several oral small molecules that inhibit the Janus kinase TYK2 are advancing in development. The Bristol Myers Squibb (“BMS”) TYK2 inhibitor, Sotyktu®, Eli Lillywas approved for psoriasis in 2022. Second generation allosteric TYK2 inhibitors from Nimbus Therapeutics (recently in-licensed by Takeda) are moving into Phase 3 development, and Company, Galapagos/Gilead (filgotinib ina molecule from Ventyx Biosciences has initiated Phase 3), Lycera Corp., Mitsubishi Tanabe Pharma Corporation, Pfizer (tofacitinib citrate in Pre-Registration), Roche/Genentech (etrolizumab in2 development. Several small molecules that inhibit IL-17 have completed Phase 3), Samsung Bioepis (adalimumab biosimilar in Pre-Registration), Sandoz (adalimumab biosimilar in Phase 3), Shire/Pfizer (PF-00547659), and UCB S.A.1 development.
We believe our principalIn IBD, competition in the treatment of chronic iron overload disorders, such as beta-thalassemia and MDS will come from products being developed by companies such as Acceleron (luspaterceptwith injectable agents in the anti-integrin class (Entyvio®, Takeda, approved) and the anti-IL-12/23 class that may be approved in the next several years, including JNJ’s Stelara® (approved in UC and CD), Abbvie’s risankizumab (Skyrizi®) (UC and CD Phase 3), JNJ’s guselkumab (Tremfya®) (UC and CD); and Eli Lilly’s mirikizumab (UC and CD).
In addition, orally delivered agents with novel mechanisms of action that are approved for or in development and may be approved for UC and/or CD prior to or shortly after the launch of our product candidates can have significant impact in the competitive environment, including,
| ● | JAK inhibitors: The pan-JAK tofacitinib (Xeljanz®) is approved in UC. The next-generation selective JAK1/3 inhibitors, including Abbvie’s upadacitinib (Rinvoq®) was approved in UC and |



