on our Uncovering Rare Obesity™ (URO) sponsored genetic testing program, fuel MC4R pathway research, disease education and awareness and patient finding.
With approximately 140 employees in the United States and Europe as of January 31, 2022, a rapidly expanding network of key opinion leaders, and an increasing number of treated patients, we are focused on changing the paradigm for the treatment of rare genetic disordersdiseases of obesity. We believe setmelanotide, for which we have exclusive worldwide rights, has the potential to serve as replacement therapy for the treatment of melanocortin‑4, or MC4, pathway deficiencies. MC4 pathway deficiencies result in the disruption of satiety signalsOur focused disease awareness and energy homeostasis in the body, which, in turn, leads to intense feelings of hunger and to obesity. Our developmentpatient finding efforts are initially focused on obesity related to six single gene‑related, or monogenic, MC4 pathway deficiencies—pro‑opiomelanocortin, or POMC, leptin receptor, or LEPR, Bardet‑Biedl syndrome, Alström syndrome, POMC heterozygous, and POMC epigenetic disorders—for which there are currently no effective or approved treatments. We believe that the MC4 pathway is a compelling target for treating these genetic disorders because of its critical role in regulating appetite and weight by promoting satiety and weight control, and that peptide therapeutics are uniquely suited for activating this target.
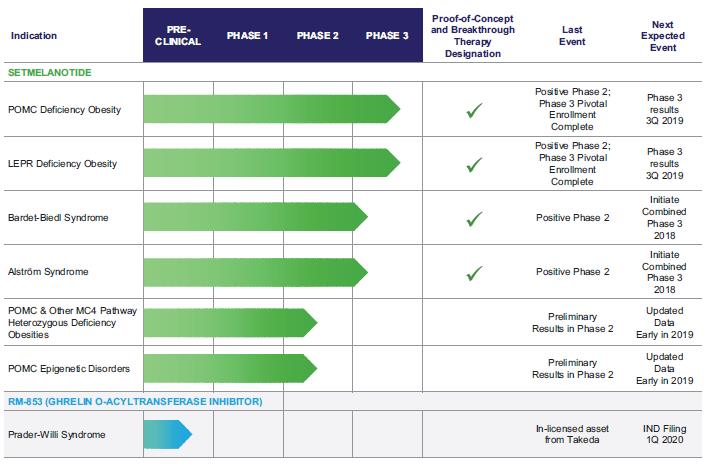
We have demonstrated proof of concept in Phase 2 clinical trials in POMC deficiency obesity, LEPR deficiency obesity, Bardet‑Biedl syndrome and Alström syndrome, four genetic disorders of extreme and unrelenting appetite and obesity, in which setmelanotide dramatically reduced both weight and hunger. The U.S. Food and Drug Administration, or the FDA, has acknowledged the importance of these results by giving setmelanotide Breakthrough Therapy designation for the treatment of obesity associated with genetic defects upstream of the MC4 receptor in the leptin‑melanocortin pathway. This designation currently covers four Breakthrough Therapy designation indications: POMC deficiency obesity, LEPR deficiency obesity, Bardet-Biedl syndrome and Alström syndrome. Setmelanotide is currently in Phase 3 development for POMC deficiency obesity and LEPR deficiency obesity, and we have initiated a combined Phase 3 trial for Bardet-Biedl syndrome and Alström syndrome. In addition, in late June 2018 setmelanotide was designated as Priority Medicne, or PRIME, by the European Medicines Agency, or EMA, Committee for Medicinal Products for Human Use, or CHMP. We have completed enrollment in the pivotal cohorts for both our POMC deficiency obesity Phase 3 clinical
3
trial and our LEPR deficiency obesity Phase 3 clinical trial. We expect to report initial Phase 3 data from these trials in the third quarter of 2019, and subsequently plan to file a new drug application, or NDA, for FDA approval for these two indications concurrently. We believe that we have demonstrated proof of concept in our Phase 2 clinical trial in Bardet‑Biedl syndrome and in Alström syndrome, and met with the FDA in May 2018 to discuss a combined pivotal Phase 3 clinical trial in these indications. Based on these discussions with the FDA, we have initiated and started to enroll patients in December 2018. We have an ongoing Phase 2 clinical trial in POMC heterozygous deficiency obesity and POMC epigenetic disorders. We reported initial, preliminary results in these additional Phase 2 indications in June 2018, and plan to provide a further update for these indications early in 2019. In total, more than 330 obese subjects and patients have been treated with setmelanotide in previous and ongoing clinical trials in which setmelanotide demonstrated statistically significant weight loss with good tolerability.
Obesity is epidemic in the United States and current treatment approaches have demonstrated limited long‑term success for most obese patients. We are taking a different approach to obesity drug development by leveraging new understanding of the genetic causes of severe obesity to develop innovative therapies that we believe have the potential for compelling efficacy. Setmelanotide’s unique mechanism of action at MC4R enables a targeted approach to treating very severe obesity in patients with specific, monogenic defects in the MC4 signaling pathway. By restoring impaired function in this pathway, setmelanotide can serve as replacement therapy for genetic deficiencies, with the potential for dramatic improvements in weight and appetite. We believe we are at the forefront of developing a therapeutic option to improve treatment outcomes in subtypes of severe obesity caused by genetically‑defined defects in the MC4 pathway.
Setmelanotide activates MC4R, which is part offuel the key pathway that can independently regulate energy homeostasis, which refers to the body’s energy balance, and appetite. The critical role of the MC4 pathway in weight regulation was validated with the discovery that single genetic defects along this pathway result in early onset and severe obesity. An expanding set of severe obesity genetic defects are now identified that involve genes in the pathway which are either upstream of MC4R—for example POMC deficiency obesity and LEPR deficiency obesity—or genes that are downstream of MC4R or affect MC4R itself. We are focusing setmelanotide clinical development on patients with monogenic upstream genetic defects in which obesity is life‑threatening but the downstream MC4 pathway is fully functional. We believe setmelanotide has the potential to restore lost activity in the MC4 pathway by bypassing the defects upstream of MC4R, and activating the MC4 pathway below such defects. In this way, setmelanotide may serve as replacement therapy to reestablish weight and appetite control in patients with these genetic disorders.
The first generation of MC4R agonists were predominantly small molecules that failed in clinical trials due to safety issues, particularly increases in blood pressure, in addition to having limited efficacy. In contrast, setmelanotide, a novel eight amino acid peptide, retains the specificity and functionality of the naturally occurring hormone that activates MC4R, and has exhibited preliminary evidence of efficacy without adversely affecting blood pressure in our Phase 1 and ongoing Phase 2 clinical trials. We are currently evaluating setmelanotide, which is administered by subcutaneous, or SC, injection, for the treatment of six genetic disorders of obesity: POMC deficiency obesity, LEPR deficiency obesity, Bardet‑Biedl syndrome, Alström syndrome, POMC heterozygous deficiency obesity, and POMC epigenetic disorders. We have positive Phase 2, proof of concept results for four of these indications thus far—POMC deficiency obesity, LEPR deficiency obesity, Bardet-Biedl syndrome and Alström syndrome, for which we are in Phase 3 development.
POMC deficiency obesity is a life‑threatening, ultra‑rare orphan disease, with approximately 50 patients reported to date. Ultra‑rare orphan diseases are generally categorized as those that affect fewer than 20 patients per million. We estimate that our addressable patient population for this disorder is approximately 100 to 500 patients in the United States. Patients with POMC deficiency have unrelenting hunger, or hyperphagia, that begins in infancy and they develop severe, early onset obesity. POMC deficiency obesity results from two different homozygous genetic defects, both upstream of MC4R, that result in loss of function in the MC4 pathway. Currently, there is no approved treatment for the obesity and hyperphagia associated with this genetic disorder. We have completed enrollment of the pivotal cohort in our Phase 3 open label, single arm, multinational trial to evaluate the safety and efficacy of setmelanotide for POMC deficiency obesity, with setmelanotide administered once daily by SC, injection for 12 months and we plan to report initial Phase 3 data in the third quarter of 2019. Previously, we completed a positive Phase 2 clinical trial in which two patients were enrolled and received treatment. These results were published in the New England Journal of Medicine in July 2016. The first patient in this trial lost 146.6 lbs. over 118 weeks, from a baseline weight of 341.7 lbs. and the second patient lost 89.3 lbs. over 64 weeks, from a baseline weight of 336.9 lbs. Both patients experienced substantial reductions in hunger, with
4
hunger scores falling to one to two from baseline scores of nine to 10. Hunger scores were measured using a Likert score of zero to 10, where zero represents no hunger and 10 represents extreme hunger. Setmelanotide was generally well tolerated in this Phase 2 trial.
LEPR deficiency obesity is an ultra‑rare orphan disease that results in hyperphagia and severe early‑onset obesity, with an estimated prevalence of 1% of subjects with severe, early‑onset obesity. We estimate that our addressable patient population for this disorder is approximately 500 to 2,000 patients in the United States. Like other deficiencies upstream in the MC4 pathway, LEPR deficiency results in loss of function in the MC4 pathway. Therefore, patients with this indication also manifest hyperphagia and severe obesity from early childhood. Currently, there is no approved treatment for the obesity and hyperphagia associated with LEPR deficiency obesity. We have completed enrollment of the pivotal cohort in our Phase 3 open label, single arm, multinational trial to evaluate the safety and efficacy of setmelanotide for LEPR deficiency obesity, with setmelanotide administered once daily by SC injection for 12 months, and we plan to report initial Phase 3 data in the third quarter of 2019. Previously, we completed a positive Phase 2 clinical trial in which three patients were enrolled and received treatment in this trial each experiencing significant weight loss and substantial reductions in hunger. These results were published in Nature Medicine in May 2018. Setmelanotide was generally well tolerated in this Phase 2 trial.
Based on our POMC deficiency obesity and LEPR deficiency obesity Phase 2 results, the FDA granted setmelanotide Breakthrough Therapy designation for the treatment of obesity associated with genetic defects upstream of the MC4 receptor in the leptin‑melanocortin pathway, which includes both POMC deficiency obesity and LEPR deficiency obesity, enabling an expedited path to approval of setmelanotide for these two indications. The FDA has granted orphan drug designation for setmelanotide for the treatment of POMC deficiency obesity and LEPR deficiency obesity.
Bardet‑Biedl syndrome is a life‑threatening, ultra‑rare orphan disease with a prevalence of approximately one in 100,000 in North America. We estimate that our addressable patient population for Bardet‑Biedl syndrome obesity is approximately 1,500 to 2,500 patients in the United States. Bardet‑Biedl syndrome is a monogenic disorder that causes severe obesity and hyperphagia as well as vision loss, polydactyly, kidney abnormalities, and other signs and symptoms. Currently there are no approved or effective therapies for Bardet‑Biedl syndrome. We have demonstrated proof of concept based on data from nine patients in our Phase 2 clinical trial in Bardet‑Biedl syndrome, indicating that this is also a setmelanotide‑responsive, upstream MC4 pathway disorder. Six of these nine patients showed significant weight loss and seven patients showed clear improvements in every hunger assessment. We reported preliminary Phase 2 results in the fourth quarter of 2017, and provided an update on these patients in the second quarter of 2018. Based on these results, the FDA recently included Bardet‑Biedl syndrome under our existing Breakthrough Therapy designation for setmelanotide. We provided an additional update on these patients in January 2019. Setmelanotide has been generally well tolerated in this trial.
Alström syndrome is a life‑threatening, ultra‑rare orphan disease with a prevalence of approximately one in one million in North America. We estimate that our addressable patient population for Alström syndrome is approximately 500 to 1,000 patients worldwide. Alström syndrome is a monogenic disorder, closely related to Bardet‑Biedl syndrome, that causes childhood obesity and hyperphagia as well as progressive vision loss, deafness, cardiomegaly, insulin resistance and other signs and symptoms. Currently there are no approved or effective therapies for Alström syndrome. We believe we have recently demonstrated proof of concept in our Phase 2 clinical trial in Alström syndrome, indicating that this is also a setmelanotide responsive, upstream MC4 pathway disorder. Based on these results, the FDA recently included Alström syndrome in our existing Breakthrough Therapy designation for setmelanotide. Setmelanotide has so far been generally well tolerated in this trial.
We met with the FDA in May 2018 to discuss a combined pivotal Phase 3 clinical trial in both Bardet‑Biedl syndrome and Alström syndrome. Based on these discussions with the FDA, we initiated this trial and started enrolling patients in December 2018.
We are also focusing on additional monogenic, upstream MC4 pathway deficiencies for which setmelanotide can function as replacement therapy and provide activation of the pathway downstream of the defect, promoting satiety and weight control. We have enrolled patients in Phase 2 proof of concept trials for POMC epigenetic disorders, and for POMC heterozygous deficiency obesity, for which we estimate our addressable population is approximately 4,000 patients in the
5
United States. For all of these patients, hyperphagia and obesity can have significant health consequences for which there is currently no approved treatment. We reported initial, preliminary results from these trials in June 2018, and plan to provide a further update for these indications early in 2019.
In addition to our development of setmelanotide, in April 2018, we announced that we had acquired exclusive, worldwide rights from Takeda Pharmaceutical Company Limited, or Takeda, to develop and commercialize T‑3525770, now called RM‑853. RM‑853 is a potent, orally available ghrelin o‑acyltransferase, or GOAT, inhibitor currently in preclinical development for Prader‑Willi Syndrome, or PWS. PWS is a rare genetic disorder that results in hyperphagia and early‑onset, life‑threatening obesity, for which there are no approved therapeutic options. RM‑853 is currently in pre‑clinical development. We anticipate filing an Investigational New Drug application, or IND, for RM‑853 with the FDA in 2020.
Our company was founded in November 2008 by former biopharmaceutical executives who have successfully developed, commercialized and in‑licensed innovative pharmaceutical products, and we have subsequently expanded our senior management team to further broaden our team’s experience in developing, registering and commercializing new drugs. In addition, our scientific advisory board, or SAB, members have extensive clinical expertise in obesity, endocrinology and metabolic diseases. We intend to leverage the experience of our senior management team and SAB to develop and commercialize setmelanotide. Through our senior management team’s network of industry contacts, we will continue to evaluate additional product candidate licensing and acquisition opportunities.
Our setmelanotide patent portfolio includes composition of matter patents for setmelanotide that expire in the United States in 2027, with possible patent term extension to 2032 under the Hatch‑Waxman Act. Additional patent coverage may be provided due to pending formulation patent applications, if such pending formulation applications were issued.
Our Strategy
Our goal is to be a leader in developing and commercializing targeted therapies for genetic deficiencies that result in life‑threatening metabolic disorders. The key componentselements of our strategy, are:including:
| Rapidly |
|
|
|
|
6
|
|
|
| ● | Ensure global access to IMCIVREE: We are |
| ● | Leverage genetic testing programs to support clinical trial enrollment and commercial launch activities: We are committed to expanding our obesity DNA database by expanding access and availability to genetic testing for individuals with early-onset, severe obesity and hyperphagia. Approximately 50% of |
| ● | Lifecyle Management: As we make IMCIVREE available in our initial indications |
|
|
Ultimately, we intend to establish our own commercial sales and marketing organization in the United States and other core strategic markets. We expect that this sales organization will target physicians treating these rare genetic disorders of obesity, including pediatric and adult endocrinologists. We believe that building our own commercial operations will deliver a greater return on our product investment than if we license the rights to commercialize these products to third parties. We may also selectively establish partnerships in markets outside the United States for sales, marketing and distribution.
|
|
7
Our Product Pipeline
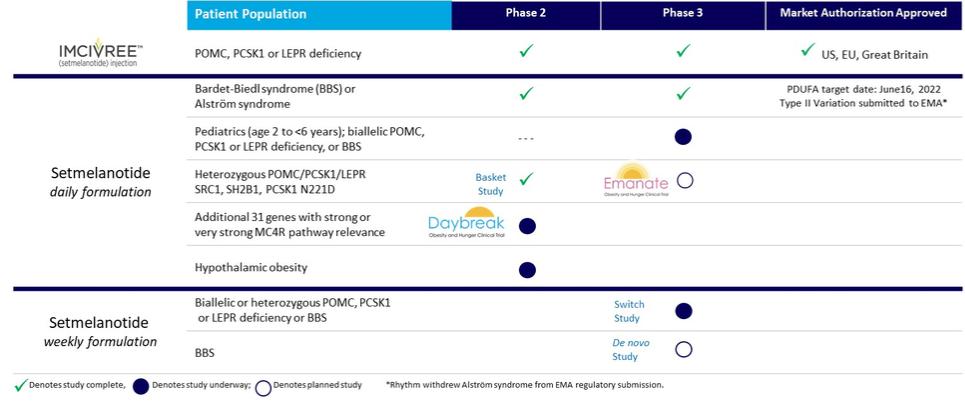
The following chart depicts key information regarding the development of setmelanotide, including the indications we are pursuing within MC4MC4R pathway deficiencies and the current state of developmentdevelopment:
Market Overview
Early-onset, Severe Obesity, Hyperphagia, and our expected upcoming milestones:the MC4R Pathway
Market Overview
Recent Advances in the Understanding of Obesity
Diet and lifestyle modifications remain the cornerstones of weight loss therapy, but they are limited by a lack of long‑term success for most obese patients. The long‑term efficacy of these interventions and for existing drug therapies is often limited by the counter‑regulatory mechanisms of the human body. For example, with diet induced weight loss, typically there is a large decrease in energy expenditure that offsets that weight loss. Accordingly, the discovery that the MC4MC4R pathway can regulateregulates both appetiteenergy intake (hunger) and energy homeostasis separately—helping maintain theexpenditure to balance between food intake and energy burn—caloric expenditure has definedmade it an important target for therapeutics. In addition to obesity due to POMC, deficiency obesity andPCSK1 or LEPR deficiency obesity,deficiencies, recent advances in genetic studies have identified several diseases characterized at least in part with early-onset, severe obesity and hyperphagia that are the result of genetic defectsvariants affecting the MC4MC4R pathway, including Bardet‑Biedl syndrome,BBS, Alström syndrome, certain variants of the POMC, heterozygousPCSK1, LEPR, SRC1 and SH2B1 genes, as well as MC4R deficiency obesity and POMC epigenetic disorders.deficiencies in upwards of 31 additional genes with strong or very strong relevance to the MC4R pathway. With a deeper understanding of this critical signaling pathway, we are taking a different approach to drug development by focusing on specific genetic deficienciesvariants affecting the MC4MC4R pathway. We believe that this approach has the potential to provide dramatic improvements in weightobesity and appetitehyperphagia by restoring lost function in the MC4MC4R pathway.
8
Obesity Caused by Rare Genetic DeficienciesVariants Affecting the MC4MC4R Pathway
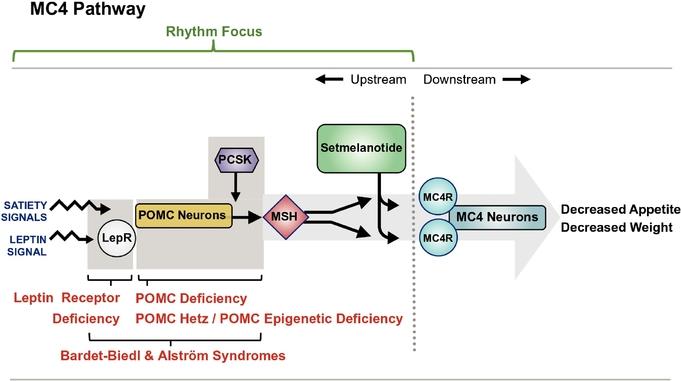
The MC4MC4R pathway, serves a critical role in the control of food intake and energy balance. Its activity decreases appetite and caloric intake, and increases energy expenditure, with MC4R acting as the final step in the signaling pathway. This important hypothalamic, or lower brainstem, pathwaywhich has been the focus of extensive scientific investigation for many years, regulates hunger, caloric intake, and we have a deep understanding of this mechanism,energy expenditure, which is unlike the targets of most other anti‑obesity therapies. As a result, we believe we can better predict the efficacy and safety profile expected from modulating this target.consequently affect body weight. The critical role of the MC4MC4R pathway in weight regulation was also validated withis supported by the discoveryobservation that single genetic defectsgene variants at many points in this pathway result in early onset,early-onset, severe obesity.
The MC4MC4R pathway is illustrated in the figure below, from the activation of the pathway to the resulting decrease in appetite and weight.below. Under normal conditions, POMC neurons are activated by brainadiposity and satiety signals, including those resulting from the hormone leptin acting through the LEPR. POMC neurons
8
produce a protein, which is specifically processed by the proprotein convertase subtilisin/kexin 1, or PCSK,PCSK1 enzyme, into melanocyte stimulating hormone, or MSH, the natural ligand, or activator forof the MC4R. When upstream genetic mutationsvariants disrupt this pathway, it can lead to insufficient MC4R activation and downstream signaling; the result of which is hyperphagia and severe obesity.
The figure below also illustrates some of the genes that are upstream of the MC4R and the potential effect variants in those genes may have on the activation of the MC4R, which regulates food intake and energy expenditure.
Setmelanotide Development Targets: Upstream Deficiencies Affecting the MC4R Pathway
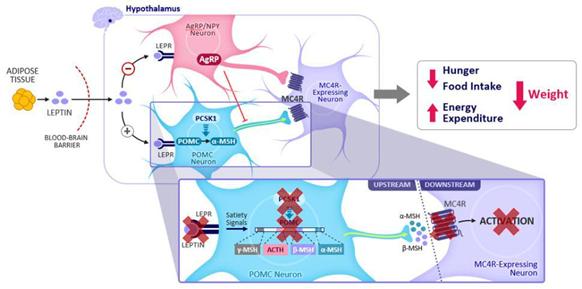
AgRP, agouti-related protein; LEPR, leptin receptor; MC4R, melanocortin-4 receptor; MSH, melanocyte-stimulating hormone; ACTH, adrenocorticotropic hormone; PCSK1, proprotein convertase subtilisin/kexin-type 1; POMC, proopiomelanocortin; . Reference: Yazdi FT et al. PeerJ. 2015;3:e856.
We are focused on developing setmelanotide for genetic disorders that result in defectsarise due to variants in this pathway that are upstream of the MC4R. With our expanding clinical development program, we are evaluating setmelanotide in Phase 2 and 3 trials for the treatment of obesity due to variants in one of 36 genes associated with the MC4R pathway. Setmelanotide has the potential to restore lost function in this pathway by activating the intact MC4 pathway belowMC4R-expressing neuron downstream of the genetic defect.impairment. In this way, we believe setmelanotide acts as replacement therapy.
The figure below also illustrates somerestorative therapy, to restore lost signaling of the upstream MC4 pathway deficienciesMC4R pathway.
Epidemiology Estimates of Rare Genetic Diseases of the MC4R Pathway
While obesity is epidemic in the United States and elsewhere, we are focused on rare genetic diseases of obesity, most often characterized by early-onset, severe obesity and hyperphagia. Of the tens of millions of individuals with obesity in the United States, the U.S. Center for Disease Control (CDC) estimates that there are approximately 5 million individuals whose severe obesity had early onset between the targetsages of our development activities.
Setmelanotide Development Targets: Upstream Deficiencies Affecting2 and 5 years old. The table below summarizes, using the MC4 Pathwayestimations described below, the indications currently approved or under active clinical investigation.
Approved by the U.S. FDA and authorized by the EC and Great Britain’s MHRA1 | |
Obesity due to biallelic POMC or PCSK1 deficiency | ~100 – 500 U.S. patients |
Obesity due to biallelic LEPR deficiency | ~500 – 2,000 U.S. patients |
Under review by U.S. FDA with PDUFA target date of June 16, 2022, and under review by EU EMA | |
Bardet-Biedl syndrome | ~1,500 – 2,500 U.S. patients |
Alström syndrome | ~500 U.S. patients |
9
Setmelanotide currently being evaluated in Phase 2 or Phase 3 trials | |
Heterozygous POMC or PCSK1 variants | ~45,000 U.S. patients |
Heterozygous LEPR variants | ~25,000 U.S. patients |
SRC1 variants | ~20,000 U.S. patients |
SH2B1 variants | ~23,000 U.S. patients |
PCSK1 N221D variant | ~100,000 U.S. patients |
MC4R variants | ~10,000 U.S. patients* |
Hypothalamic obesity | >4,500 U.S. patients** |
Epidemiology1. Approved for use in patients six years of Rare Genetic Disordersage and older.
These calculations rely on internal and proprietary sequencing data and current estimated responder rates to setmelanotide therapy, and they assume a U.S. population of 327 million, of which 1.7% have early-onset, severe obesity (Hales et al in JAMA – April 2018: Trends in Obesity and Severe Obesity Prevalence in US Youth and Adults by Sex and Age, 2007-2008 to 2015-2016); * Estimated prevalence of U.S. patients with rescuable variants of the MC4 Pathway
Clinical Epidemiology
The figure below summarizes the indications on which we are focusing for the development of setmelanotide, including our clinical epidemiology estimates for the addressable patient populations within these indications.
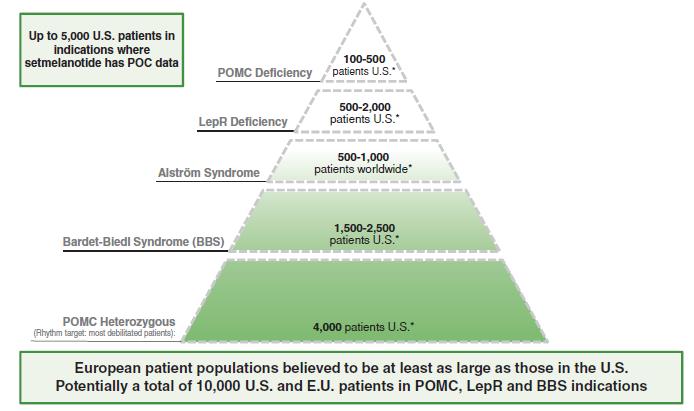
*The patient numbers above areMC4R; ** Internal Company estimate is based on Company estimates from clinical publications.reported incidence of hypothalamic obesity following craniophryngioma and long-term survival rates, Zacharia, et al., Neuro-Oncology 14(8):1070–1078, 2012. doi:10.1093/neuonc/nos142; and Muller, et al., Neuro-Oncology 17(7), 1029–1038, 2015 doi:10.1093/neuonc/nov044.
†Epidemiological estimates are not yet available for POMC epigenetic disorders.
We believe that the patient populations in the European UnionEurope are at least as large as those in the United States. However,While our sequencing data include patients from the United States and Europe, we do not have comparable epidemiological data from the European Unioncountries and these estimates are therefore based solely on applying relative population percentages to the Company‑derivedRhythm-derived estimates described above.
Genetic Epidemiology Studies
We have estimatedPrevalence estimates for Bardet-Biedl syndrome vary markedly between populations, from 1 in 160,000 in northern European populations with higher prevalence rates in some additional regions throughout the patient populations for our rare genetic disorders of obesity primarily by identifying patients or by estimating from clinical epidemiology information. Another method toworld. Our estimate the size of these ultra‑rare populations is by genetic epidemiology using newly available large genomic databases of either full genome or exome sequences. We have begun some substantial efforts, both directly and with collaborators, with a series of such databases. Much of our preliminary work has utilized a database of approximately 140,000 genomes, representative of the U.S. population. The results of this analysis were published in May 2018 in the Journal of Clinical Endocrinology and Metabolism, a leading journal in this field, which we believe validates the scientific and epidemiological importance of our findings.
The results from this effort have been supportive of our clinical epidemiological estimates, even when using conservative genetic epidemiology assumptions. The results support estimates ofthat the number of patients with BBS in the United
10
States who are homozygous deficient in the POMC gene, which is one of the two genetic defects causing POMC deficiency obesity, and who are homozygous deficient in the LEPR gene, are at least as large, or larger than, our clinical epidemiology assumptions, as provided above. In addition, the estimates of patients who are homozygous deficient in the PCSK1 gene, which is the other genetic defect causing POMC deficiency obesity, may be substantially larger than our clinical epidemiology assumptions. Overall, our clinical epidemiology based estimates for POMC deficiency obesity (up to 500 U.S. patients including both POMC and PCSK1 gene disorders) and LEPR deficiency obesity (up to 2,000 U.S. patients) support upbetween 1,500 to 2,500 U.S. patients combined. Our preliminary genetic epidemiology estimates suggestpeople and that it is a total of approximately 13,000 U.S. patients with these disorders, representing a 5‑fold increaseslightly higher number in our clinical epidemiology based estimates.
We have already begun to expand our genetic epidemiology analysis into other genomic databases that contain patients with different demographics, and are actively working with a series of academic and industry collaborators. Our published data support that while individual mutations may cluster in specific demographic groups, these rare genetic disorders are seen widely across the demographic spectrum. The ongoing expansion of genomic data available in other forums also has the effect of supporting this effort. An important improvement in this effort will be working with data linked to phenotypic information to better characterize the genetic information we are analyzing.
Europe. We believe the separate analyses that we have completed using clinical epidemiologyBBS community in EU member states and genetic epidemiology provide a robust range of patient population estimates for these rare disorders. However, to be conservative, we reference the clinical epidemiology figures in our descriptions of our target indications.
Obesity Caused by Upstream Genetic Deficiencies Affecting the MC4 Pathway
We have completed four positive Phase 2 trials of setmelanotide that provide proof of concept for four upstream MC4 pathway genetic defects in which obesityGreat Britain is life‑threatening but the downstream MC4 pathway is fully functional: For POMC deficiency obesity, LEPR deficiency obesity,particularly well established and Bardet‑Biedl syndrome, we estimate an aggregate addressable population of up to 5,000 people in the United States. In addition, we estimate that Alström syndrome has an addressable patient population of up to 1,000 people worldwide.
POMC Deficiency Obesity
POMC deficiency obesity is an ultra‑rare genetic disorder, with severe, early onset obesity, defined here as a body mass index, or BMI, of greatermore advanced than 40 kg/m2, and hyperphagia as hallmark clinical features. Patients with POMC deficiency obesity are extremely rare. There are approximately 50 patients with POMC deficiency obesity noted in a series of published case reports, each mostly reporting a single or small number of patients. However, we estimate that our addressable patient population for this disorder is approximately 100 to 500 patients in the United States, as mostwe believe there are approximately 1,500 patients diagnosed and being cared for at academic centers in Europe. Additionally, our genetic sequencing data support our belief that these prevalence estimates may be lower than the actual disease prevalence.
For patients with genetic variants of the reported cases are from a smallMC4R pathway, the rarity and the genetic pathophysiology of our target indications means that there is no comprehensive patient registry or other method of establishing with precision the actual number of academic research centers,patients. As a result, we have had to rely on other available sources to derive clinical prevalence estimates for our target indications. We recently updated our prevalence estimates in 2021 based on sequencing data from approximately 45,000 individuals with obesity, and becauserates of response to setmelanotide in our exploratory Phase 2 Basket study. Because the published epidemiology studies for these genetic deficiencies are based on relatively small population samples, and are not amenable to robust statistical analyses, it is possible that these projections may significantly under- or overestimate the addressable population. While our projected estimates of the aggregate total addressable population continues to expand with the addition of new genes, the addressable population faces the challenges of a rare disease population. The disease must be suspected by the physician, confirmed by genetic testing and then setmelanotide responsiveness confirmed by a 12-16 week trial with the product candidate.
Limitations of Current Therapies
Although drugs approved for general obesity potentially can be used in patients with obesity and MC4R pathway variants, all currently available products have limited efficacy and treat symptoms without addressing the underlying biology of MC4R impairment. For example, drugs which delay gastric emptying may cause a patient to feel full and eat less, but are also often associated with nausea and vomiting as a consequence of the delayed emptying. In the case of individuals with MC4R pathway variants, these therapies also do not specifically address the impaired signaling in this central energy regulating pathway. Similarly, bariatric surgery which has been shown to be quite effective in the general population with obesity, may be unsuccessful in patients with MC4R variants for the same reason.
10
IMCIVREE™ (setmelanotide)
IMCIVREE was approved in November 2020 by the U.S. FDA for chronic weight management in adult and pediatric patients 6 years of age and older with obesity due to POMC, PCSK1 or LEPR deficiency is often unavailableconfirmed by genetic testing demonstrating variants in POMC, PCSK1, or LEPR genes that are interpreted as pathogenic, likely pathogenic, or of uncertain significance, and rarely performed. However, our genetic epidemiological estimates are several times higher. Most patients are not currently diagnosedin July and September 2021, respectively, by the EC and Great Britain’s MHRA for the treatment of obesity and the control of hunger associated with genetically confirmed loss-of-function biallelic POMC, including PCSK1, deficiency or biallelic LEPR deficiency in adults and children 6 years of age and above. These approvals were based on discussionsPhase 3 data demonstrating a statistically significant and clinically meaningful reductions in weight and hunger in patients 12 years old or older with experts in rare diseases, we believesevere obesity due to POMC, PCSK1 or LEPR deficiency. As an MC4 receptor agonist, IMCIVREE is designed to restore impaired MC4 receptor pathway activity arising due to genetic impairments upstream of the numberMC4 receptor.
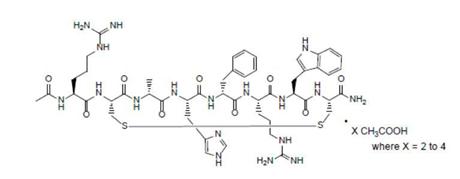
IMCIVREE contains setmelanotide acetate, a melanocortin 4 receptor (MC4R) agonist. Setmelanotide is an 8 amino acid cyclic peptide analog of diagnosed cases will increase several‑foldendogenous melanocortin peptide α-MSH. The chemical name for setmelanotide acetate is acetyl-L-arginyl-L-cysteinyl-D-alanyl-L-histidinyl-D-phenylalanyl-L-arginyl-L-tryptophanyl-L-cysteinamide cyclic (2→8)-disulfide acetate. Its molecular formula is C49H68N18O9S2 (anhydrous, free-base), and molecular mass is 1117.3 Daltons (anhydrous, free-base).
The chemical structure of setmelanotide is:
IMCIVREE injection is a sterile, clear to slightly opalescent, colorless to slightly yellow solution. Each 1 mL of IMCIVREE contains 10 mg of setmelanotide provided as setmelanotide acetate, which is a salt with increased awareness2 to 4 molar equivalents of this disorderacetate, and the availabilityfollowing inactive ingredients: 100 mg N-(carbonyl-methoxypolyethylene glycol 2000)-1,2-distearoyl- glycero-3- phosphoethanolamine sodium salt, 8 mg carboxymethylcellulose sodium (average MWt 90,500), 11 mg mannitol, 5 mg phenol, 10 mg benzyl alcohol, 1 mg edetate disodium dihydrate, and Water for Injection. The pH of new treatments.IMCIVREE is 5 to 6.
Obesity due to POMC, PCSK1 or LEPR deficiency are ultra-rare diseases caused by variants in POMC, PCSK1 or LEPR genes that impair the MC4 receptor pathway. People living with obesity due to POMC, PCSK1 or LEPR deficiency struggle with hyperphagia, an extreme, insatiable hunger, beginning at a young age and resulting in early-onset, severe obesity.
Obesity due to POMC or PCSK1 deficiency is caused by the loss of both genetic copies of either the gene for POMC or the gene for PCSK.PCSK1. This results either in loss of POMC neuropeptide synthesis, in the case of homozygousbiallelic (compound heterozygous and homozygous) deficiency in the POMC gene, or in disruption of the required processing of the POMC neuropeptide product to MSH by the PCSKPCSK1 enzyme, in the case of homozygousbiallelic deficiency in the PCSKPCSK1 gene. The end result of both of these two homozygousbiallelic genetic defectsvariants is lackdeficiency of MSH to bind and activate MC4R, ultimately leading to the lack of stimulation of downstream MC4MC4R neurons and causing severe, early onsetearly-onset obesity and hyperphagia. POMC homozygousor PSCK1 biallelic deficiency may also be associated with other hormonal deficiencies, such as hypoadrenalism, as well as other characteristics of MSH deficiency such as red hair and fair skin.
POMC11
POMC/PCSK1 deficiency is characterized by voracious infant feeding, rapid weight gain and severe obesity, often in early infancy, with patients demonstrating remarkable weight increases many standard deviations fromabove the normal weight growth curves. These patients and their caregivers have attemptedoften attempt to stabilize body weight with the help of psychologists, nutritionists and pediatric endocrinologists, all without significant success. We have completed enrollment insuccess, since none of these interventions addresses the pivotal
11
cohorts for our POMCthe impact of the POMC/PSCK1 deficiency obesity Phase 3 clinical trial, and expecton the MC4R pathway.
Obesity due to report initial Phase 3 data in the third quarter of 2019. Currently there are no approved or effective therapies for POMC deficiency obesity.
Leptin Receptor Deficiency Obesity
LEPR deficiency obesity is an ultra‑rareultra-rare genetic disorderdisease that causes hyperphagia and severe, early onsetearly-onset obesity. LEPR deficiency accounts for an estimated 1% of cases of severe, early onset obesity. Based on clinical epidemiology studies in small cohorts of patients with severe, early onset obesity, we estimate that our addressable patient population for this disorder is approximately 500 to 2,000 patients in the United States, through our genetic epidemiological estimates suggest the number may be moderately higher.
Leptin’s role in obesity has been elucidated by characterization of severely obese people with homozygoussevere obesity and biallelic mutations that impair the activity of leptin, including disruption of signaling at the LEPR, known as LEPR deficiency obesity. Under normal conditions, leptin is released from adipose (fat) tissue as a signal of the body’s energy reserves, and can activate POMC neurons and the downstream MC4, butMC4R to signal for decreased energy intake and increased energy expenditure. However, like other deficiencies upstream in the MC4MC4R pathway, lack of signaling at LEPR results in loss of function in the MC4 pathway.MC4R pathway and loss of signaling of downstream MC4R expressing neurons, resulting in hyperphagia and early-onset severe obesity.
LikePivotal Phase 3 Clinical Trials Evaluating Setmelanotide in POMC deficiency obesity,and LEPR Deficiency Obesities
We assessed the safety and efficacy of IMCIVREE in two pivotal trials that were identically designed: one-year, open-label studies, each with an eight-week, double-blind withdrawal period. The studies enrolled patients with homozygous or presumed compound heterozygous pathogenic, likely pathogenic variants, or VUS, for either the POMC, PCSK1 or LEPR gene. In both studies, adult patients had a body mass index (BMI) of ≥30 kg/m2. Weight in pediatric patients was ≥95th percentile using growth chart assessments.
Efficacy analyses were conducted in 21 patients who had completed at least one year of treatment at the time of a pre-specified data cutoff. Of the 21 patients included in the efficacy analysis in both pivotal studies, 62% were adults and 38% were aged 16 years or younger. In Study 1, 50% of patients were female, 70% were White, and the median baseline BMI was 40.0 kg/m2 (range: 26.6-53.3). In Study 2, 73% of patients were female, 91% were White, and the median baseline BMI was 46.6 kg/ m2 (range: 35.8-64.6).
In the POMC/PCSK1 study, 80% of patients with obesity due to POMC or PCSK1 deficiency met the primary endpoint, achieving a ≥10% weight loss after one year of treatment with IMCIVREE. In the LEPR study, 46% of patients with obesity due to LEPR deficiency obesity exhibitmet the primary endpoint by achieving a ≥10% weight loss after 1 year of treatment with IMCIVREE.
Development of Setmelanotide for Additional Indications
Bardet-Biedl and Alström syndromes
Bardet-Biedl syndrome (BBS) is a life-threatening, ultra-rare orphan disease. BBS is a disease that causes hyperphagia and severe obesity frombeginning in early childhood. LEPR deficiency is also associated with hypogonadism and reduced immune function. We have completed enrollment of the pivotal cohort for our LEPR deficiency obesity Phase 3 clinical trial, and expect to report initial Phase 3 data in the third quarter of 2019. It is our intention to file our NDA for LEPR deficiency obesity concurrently with our NDA for POMC deficiency obesity, as enrollment of our pivotal cohort completed almost simultaneously. Currently there are no approved or effective therapies for LEPR deficiency obesity.
Bardet‑Biedl Syndrome
Bardet‑Biedl syndrome is a life‑threatening, ultra‑rare orphan disease with a prevalence of approximately one in 100,000 in North America. We estimate that our addressable patient population for Bardet‑Biedl syndrome obesity is approximately 1,500 to 2,500 patients in the United States. Bardet‑Biedl syndrome is a monogenic disorder that causes severe obesity and hyperphagiachildhood, as well as vision loss, polydactyly, kidney abnormalities, and other signs and symptoms. For Bardet‑Biedl syndrome patients with BBS, hyperphagia and obesity can have significant health consequences.
Bardet‑Biedl syndrome BBS is part of a class of disorders called ciliopathies, or disorders associated with the impairment of cilia function in cells. Cilia are hair‑likehair-like cellular projections that play a fundamental role in the regulation of several biological processes, including satiety signaling. Cilia dysfunction in the hypothalamus, including in the MC4R pathway, is thought to contribute to hyperphagia and obesity in Bardet‑Biedl syndrome. Bardet‑Biedl syndromeBBS. BBS is a genetically heterogeneous disease that is caused by as many as 21 separate Bardet‑BiedlBardet-Biedl loci defectsvariants that result in a similar syndrome though each Bardet‑Biedl syndrome patient only has one of these defects.
clinical manifestations. Recent scientific studies identify deficiencies affecting the MC4MC4R pathway as a potential cause of the obesityhyperphagia and hyperphagiaobesity associated with Bardet‑Biedl syndromeBBS, and demonstrate that an MC4R agonist can directly impact these symptoms. Studies in mouse models of Bardet‑Biedl syndrome show that deficiencies in the MC4 pathway contribute to the obesity and hyperphagia in Bardet‑Biedl syndrome, with animals developing hyperphagic tendencies as early as 10 weeks of age. Notably, these mice have decreased leptin receptor signaling, with the essential hallmarks of failure to activate POMC neurons. The potential utility of MC4 agonists is also supported by studies in Bardet‑Biedl syndrome rodent models, where mice have responded to an MC4 agonist resulting in reduced food intake and body weight. We have demonstrated proof of concept in Bardet‑Biedl syndrome indicating that this is also a setmelanotide‑responsive, upstream MC4 pathway disorder. We reported preliminary Phase 2 results for Bardet‑Biedl syndrome in the fourth quarter of 2017, and updated the clinical status of these patients in the second quarter of 2018. We have had discussions with the FDA on a Phase 3 design and submitted a draft Phase 3 protocol for FDA review. The FDA has indicated that this Phase 3 trial will include both Bardet‑Biedl syndrome and Alström syndrome patients. We started to enroll patients in this Phase 3 trial in December 2018. Currently there are no approved or effective therapies for Bardet‑Biedl syndrome.
12
Alström SyndromeBBS.
Alström syndrome is a life‑threatening, ultra‑rarelife-threatening, ultra-rare orphan disease with a prevalence of approximately one in 1,000,000 in North America. We estimate that our addressable patient population for Alström syndrome is approximately 500 to 1,000 patients worldwide. Alström syndromedisease. It is a monogenic disorder that causes childhoodhyperphagia and obesity and hyperphagiabeginning in early childhood, as well as progressive vision loss, deafness, cardiomegaly, insulin resistance and other signs and symptoms. Variable features include short stature, cardiomyopathy, and progressive lung, liver, and kidney dysfunction. Symptoms of Alström syndrome first appear in infancy, and progressive development of multi‑organ
12
multi-organ pathology leads to a reduced life expectancy, with survival rare beyond the age of 50.
Alström syndrome is a ciliopathy caused by mutations in the ALMS1 gene, which has been shown to be important for cilia function. Like Bardet‑Biedl syndrome, recent scientific studies identify genetic deficiencies affecting the MC4 signaling pathway as a potential cause of the obesity and hyperphagia associated with Alström syndrome. Studies in a mouse model of Alström syndrome show a reduction in the number of cilia in specific neurons in the hypothalamus that are critical for MC4 pathway signaling. While Alström syndrome is less well studied than Bardet‑Biedl syndrome, the similar pathophysiology of cilial dysfunction and clinical presentation support that deficiencies in the MC4 pathway are implicated in the obesity and hyperphagia observed in Alström syndrome. We believe we have demonstrated proof of concept in our Phase 2 clinical trial in Alström syndrome and met with the FDA in May 2018 to discuss a combined pivotal Phase 3 clinical trial in both Bardet‑Biedl syndrome and Alström syndrome. Based on these discussions with the FDA, we initiated this trial and started enrolling patients in December 2018. Currently there are no approved or effective therapies for Alström syndrome.
Other Upstream Genetic Defects in the MC4 Pathway
In addition to POMC deficiency obesity, LEPR deficiency obesity, Bardet‑Biedl syndrome and Alström syndrome, there are other upstream, MC4 pathway deficiencies for which we believe setmelanotide may function as replacement therapy, including defects that partially modulate POMC activity, such as POMC heterozygous deficiency obesity and POMC epigenetic disorders.
POMC Heterozygous Deficiency Obesity
POMC heterozygous deficiency results in a strong predisposition to obesity, though the epidemiology and clinical characterization of these patients is less well known. POMC heterozygous deficiency obesity is caused by the loss of one of the two genetic copies of either the gene for POMC or the gene for PCSK. An estimated 2% of severe, early onset obesity patients have POMC heterozygous deficiency obesity, which is much more common than the ultra‑rare POMC deficiency obesity in which both copies of either the POMC or PCSK genes are impaired. We believe that the most severe POMC heterozygous deficiency obesity patients may be suitable for treatment with setmelanotide. We estimate that our addressable patient population within severe POMC heterozygous deficiency obesity is approximately 4,000 patients in the United States, based on epidemiology studies in small cohorts of patients with severe early onset obesity and adult obesity. Animal models support that such heterozygous deficiency in the critical MC4 pathway can result in a strong predisposition to severe obesity. The effect of heterozygous deficiency was first demonstrated in MC4R heterozygous deficiency obesity.
We are also studying patients with other MC4 pathway gene heterozygous mutations, including patients with heterozygous mutations in genes such as LEPR and Bardet‑Biedl, as well as patients with heterozygous mutations in more than one gene in the MC4 pathway. For simplicity, in this prospectus we refer to the group of such heterozygous patients as patients with POMC heterozygous deficiency, but we will be transitioning to more general language, such as patients with Heterozygous Mutations in the MC4 Pathway, as more of these patients enter our Phase 2 trials.
It is thought that the obesity of patients with POMC heterozygous deficiency may have a broader spectrum of severity than POMC deficiency obesity. Therefore, our focus will be on the most severe of the POMC heterozygous deficiency obesity patients, with our estimate that only a small percentage of these patients will benefit from targeted therapy with substantial efficacy. As a result, we have initiated a Phase 2 proof of concept trial to confirm our hypothesis that the subset of patients with very severe POMC heterozygous deficiency obesity may be highly responsive to
13
setmelanotide therapy. We have an ongoing Phase 2 clinical trial in POMC heterozygous deficiency obesity and reported initial, preliminary results in June 2018. There are currently no approved or effective therapies for POMC heterozygous deficiency obesity.
POMC Epigenetic Disorders
Recent scientific studies have identified patients with obesity due to a partial lack of MSH that is caused by epigenetic POMC variant. Given the recent discovery of these epigenetic disorders, there is currently no epidemiology data that defines the prevalence of POMC epigenetic disorders. However, we believe that these are rare disorders. Epigenetics implies DNA modifications, which can change gene expression without altering the DNA sequence itself. The most stable epigenetic modification is called DNA methylation. Recently, our academic collaborators in Berlin have described a POMC hypermethylation variant, which correlates with increased body weight in children and adults. Therefore, the presence of the POMC epigenetic variant leads to an increased risk of obesity based on reduced POMC gene activity. We expect that these patients under‑express the POMC gene product and as a result have a partial MSH deficiency. We have initiated a Phase 2 proof of concept trial to confirm our hypothesis that the subset of patients with very severe POMC epigenetic disorders may be highly responsive to setmelanotide therapy. We have an ongoing Phase 2 clinical trial in POMC epigenetic disorders and reported initial, preliminary results in June 2018. There are currently no approved or effective therapies for these disorders.
Other MC4 Disorders
Based on setmelanotide’s biochemical structure and mechanism of action, we believe setmelanotide has the potential to serve as replacement therapy for other rare genetic disorders of obesity which have pathophysiology upstream of the MC4 receptor. We are conducting research activities to study which potential disorders tied to the pathway may benefit from setmelanotide therapy. Our basket study protocols, which enable enrollment of new populations with disorders tied to the pathway, allow us to study new potential indications without the administrative and regulatory burden of initiating a separate clinical study de novo for each new indication.
Obesity Caused by Downstream Genetic Deficiencies Affecting the MC4 Pathway
MC4 Receptor Heterozygous Deficiency Obesity
MC4 Receptor, or MC4R, heterozygous deficiency is caused by the absence or loss of function of one genetic copy of the gene for MC4R. Consistent with POMC heterozygous deficiency, MC4R heterozygous deficiency results in a strong predisposition to early onset and severe obesity. MC4R heterozygous deficiency is the most common genetic cause of obesity. An epidemiological study performed in Europe in 2006 reported a prevalence of 2.6% of genetic defects in the MC4R gene in the obese population with a BMI of greater than 30 kg/m2, and studies performed in both Europe and the United States in 2000 and 2003, respectively, reported a prevalence of up to 4% of these genetic defects in more severely obese populations with a BMI of greater than 35 kg/m2. These prevalence rates suggest that there are approximately one million people in the United States with obesity caused by a mutation of the MC4R gene.
These patients have a higher risk than the general population for early onset obesity and complications such as diabetes. Furthermore, MC4R deficiency may offset the beneficial effects of diet and exercise for sustained weight loss, limiting treatment options for these individuals. There are currently no approved or effective therapies for MC4 heterozygous deficiency obesity.
While setmelanotide appears to show strong efficacy in a Phase 1b trial for the treatment of MC4R heterozygous deficiency obesity patients, it is downstream of where setmelanotide interacts with the MC4R, and we are currently focusing instead on genetic defects that are upstream of the MC4R. This is because we believe that many of these upstream genetic disorders cause even more severe, often life‑threatening obesity, and because setmelanotide has the potential to restore lost function in these upstream disorders, delivering more compelling efficacy. We have conducted additional research that was published in Nature Medicine in May 2018, which suggests that a sizable number of individuals with obesity who carry MC4R mutations, and were previously assessed functionally normal, may respond to setmelanotide treatment. This may open the possibility of new indications for setmelanotide in the future.
14
Expanding Attention to the Diagnosis of Genetic Obesity
The Endocrine Society issued new Pediatric Obesity Guidelines in January 2017 that, for the first time, recommend genotyping patients with severe pediatric obesity and hyperphagia. These guidelines estimate that up to 7% of patients with extreme pediatric obesity have a genetic mutation, including genetic MC4 pathway deficiencies, that drives their obesity. The guidelines also suggest that this percentage of severe pediatric obesity patients will increase, with newer methods and wider awareness of the need for genetic testing.
We are supporting several initiatives to expand the diagnosis of genetic obesity, including a genotyping study, called GO‑ID, and a patient registry, called TEMPO. The objective of GO-ID is to develop a screening algorithm for selecting patients to be genotyped and identified with POMC deficiency obesity and LEPR deficiency obesity, and to guide further genotyping efforts. We have expanded our panel of obesity‑related genes used in our GO‑ID study in the second half of 2018. TEMPO is a commitment to understanding the ongoing impact and burden of disease on patients and their caregivers. This registry facilitates enhanced understanding of these conditions in the medical community and builds upon ongoing collaborations with existing patient registries in syndromic conditions such as Bardet-Biedl syndrome.
We are also conducting genetic obesity epidemiology analysis of MC4 pathway genetic defects in a large representative sample of the U.S. population, and we expect to expand the genetic databases in our research efforts on an ongoing basis. The first results of this research were published in May 2018 in the Journal of Clinical Endocrinology and Metabolism, a leading journal in this field. An important improvement in this effort will be working with data linked to phenotypic information to better characterize the genetic information we are analyzing. In addition, we tested associations between BMI and loss of function mutation burden in various populations to further define the MC4 pathway and its potential impact on obesity, showing that the cumulative allele burden, or number of mutations along the MC4 pathway in a single individual, predisposes to more obesity.
Limitations of Current Therapies
Although drugs approved for general obesity can potentially be used in obese patients with MC4 pathway deficiencies, all have limited efficacy and aim to treat symptoms rather than addressing the underlying biology. There are currently no treatments approved specifically for obesity and hyperphagia in POMC deficiency obesity, LEPR deficiency obesity, Bardet‑Biedl syndrome, Alström syndrome, POMC heterozygous deficiency obesity, or POMC epigenetic disorders. Bariatric surgery is not an option in patients with upstream defects in the MC4 pathway who have severe obesity and hyperphagia.
Setmelanotide: A First‑in‑Class MC4R Agonist in Three Phase 3 Programs
Setmelanotide is a potent, first‑in‑class, MC4R agonist peptide administered by daily SC injection. Setmelanotide is in Phase 3 for the treatment of two rare genetic disorders of obesity caused by MC4 pathway deficiencies, and we have initiated a combined Phase 3 clinical trial in two additional rare genetic disorders. We also have an ongoing Phase 2 clinical trial in several other MC4 pathway disorders. MC4R modulates a key pathway in humans that regulates energy homeostasis and food intake.
The critical role of the MC4 pathway in weight regulation was validated with the discovery that single genetic defects in this pathway result in severe, early onset obesity. The first generation MC4R agonists were small molecules that failed in clinical trials primarily due to safety issues, particularly increases in blood pressure, as well as limited efficacy. Setmelanotide is a peptide that retains the specificity and functionality of the naturally occurring hormone that activates MC4R, with demonstrated efficacy and little, if any, signal of increases in blood pressure. In total, more than 330 obese subjects and patients have been treated with setmelanotide in previous and ongoing clinical trials in which setmelanotide demonstrated significant weight loss with good tolerability.
Clinical Development in Rare Genetic Disorders of Obesity Caused by MC4 Pathway Deficiencies
Setmelanotide is currently in Phase 3 development for POMC deficiency obesity and LEPR deficiency obesity, and we are initiating a combined Phase 3 trial for Bardet‑Biedl syndrome and Alström syndrome. We have completed
15
enrollment in the pivotal cohorts for both our POMC deficiency obesity Phase 3 clinical trial and our LEPR deficiency obesity Phase 3 clinical trial. We expect to report initial Phase 3 data from these trials in the third quarter of 2019, and subsequently plan to file for regulatory approval for these two indications concurrently. We believe that we have demonstrated proof of concept in our Phase 2 clinical trial in Bardet‑Biedl syndrome and in Alström syndrome, and met with the FDA in May 2018 to discuss a combined pivotal Phase 3 clinical trial in both Bardet‑Biedl syndrome and Alström syndrome. Based on these discussions with the FDA, we initiated this trial and started enrolling patients in December 2018. We continue to study setmelanotide in these indications in Phase 2 development in long‑term extensions. We are also enrolling patients for the treatment of other rare monogenic disorders of obesity, including POMC and other heterozygous deficiency obesity, and POMC epigenetic disorders. We hypothesize that all of these disorders are genetically defined deficiencies upstream in the MC4 pathway.
We are enrolling patients in two very similar Phase 2 clinical trials, each of which is designed to capture a broad range of indications under one investigational protocol, or a basket study. We believe that we have demonstrated proof of concept both in Bardet‑Biedl syndrome and in Alström syndrome, indicating that these closely related syndromes are also setmelanotide‑responsive, upstream MC4 pathway disorders. We reported initial, preliminary results for POMC heterozygous deficiency obesity and POMC epigenetic patients in June 2018. We plan to provide a further update for these indications early in 2019. We have also completed a Phase 2 trial in PWS.
Based on FDA consultations to date, and the FDA granting Breakthrough Therapy designation for POMC deficiency obesity and LEPR deficiency obesity, and including Bardet‑Biedl syndrome and Alström syndrome under the existing Breakthrough Therapy designation for setmelanotide, we believe we can seek indications for obesity caused by upstream defects in the MC4 pathway with rapid paths to approval, as compared to typical obesity drug candidates, because of the high unmet need and rare prevalence of these disorders. We expect to use the results of our Phase 3 clinical trials of setmelanotide in POMC deficiency obesity and LEPR deficiency obesity, which will run concurrently, as the foundation for proceeding directly to approval for these indications.
We believe our data in POMC deficiency obesity, LEPR deficiency obesity, Bardet‑Biedl syndrome and Alström syndrome provide strong proof of concept that setmelanotide, when targeted for deficiencies affecting the upstream portion of the MC4 pathway, can provide compelling efficacy for weight loss and decrease in hunger. There are still several genes to explore that are involved in signaling upstream in the MC4 pathway, which although not a current focus, may provide opportunities for further research in the future. Proof of concept for substantial weight loss in patients with downstream, heterozygous mutations of the MC4R gene itself has also been achieved in a small, four‑week, Phase 1b clinical trial. While these downstream defects are not our current area of focus, based on new scientific research, we believe there is evidence supporting the potential for substantial weight loss efficacy in a setting of a partially defective, downstream defect in the MC4 pathway, which impacts a significantly larger population.
Initial setmelanotide clinical trials were in patients with general obesity, which provided preliminary evidence of the safety and efficacy of the drug, and were the foundation for the Phase 2 trials in rare genetic disorders of obesity. In these trials, setmelanotide has generally achieved weight loss without adversely increasing blood pressure. These trials in the general obese population are described below.
The following table outlines our ongoing and planned setmelanotide trials in rare monogenic disorders of obesity.
16
Setmelanotide: Key Clinical Programs in Monogenic MC4 Pathway Disorders of Defined Obesity
|
|
|
| |||||
|
|
|
|
| ||||
|
|
|
|
| ||||
|
|
|
|
| ||||
|
|
|
|
| ||||
|
|
|
|
| ||||
|
|
|
|
| ||||
|
|
|
|
|
|
|
|
| |||||
|
|
|
|
| ||||
|
|
|
|
| ||||
|
|
|
|
| ||||
|
|
|
|
| ||||
|
|
|
|
| ||||
|
|
|
|
| ||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
17
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Setmelanotide: Clinical Development Program in Genetically Defined Obesity
Phase 2 Clinical Development in POMC Deficiency Obesity
We have completed a Phase 2 proof of concept, open label clinical trial, Study RM‑493‑011, in two patients with POMC deficiency obesity in which these patients have been treated with setmelanotide for more than two years, resulting in profound reductions of hyperphagia and body weight, with good tolerability. The patients in this trial have provided proof of concept for the compelling effect of setmelanotide in this disorder. We are close to completing enrollment in the pivotal cohort of patients in our Phase 3 trial for this indication. The results of the Phase 2 trial were published in July 2016 in the New England Journal of Medicine, which we believe validates the scientific and clinical importance of our Phase 2 findings, and the accompanying editorial described the trial as demonstrating impressive hunger reduction and weight loss as well as improved insulin sensitivity. A brief update on these patients was also included in a publication in Nature Medicine in May 2018.
The first setmelanotide‑treated patient was a 20‑year‑old woman, who at three months of age experienced the onset of obesity and hyperphagia. In spite of enormous efforts, the patient was never able to stabilize her body weight, except for brief periods, and she has remained hyperphagic. Ahead of our trial, the patient’s self‑reported trial hunger score was eight to nine out of 10 points, representing extreme hunger. She was entered into the trial at adulthood because of her
18
severe obesity, with a baseline weight of 155 kg, or 341.7 lbs., and a BMI of 49.8 kg/m2, and significant risk of comorbidities and a reduced life expectancy.
The trial, initially a 13‑week, open label, ascending dose Phase 2 trial, was approved by the German Federal Institute for Drugs and Medical Devices, with open‑label one‑year extensions, and was planned to include approximately four to six patients with genetically confirmed POMC deficiency obesity. After efficacy‑gated dose escalation, aiming for weekly weight loss of approximately two kg, or 4.4 lbs., the primary endpoint was weight loss, with other key endpoints including hunger score, body composition, insulin and glucose parameters, metabolic and cardiovascular risk factors, energy expenditure and general safety and tolerability.
After 13 weeks of therapy, with approximately the first four weeks at sub‑therapeutic doses, our initial patient demonstrated weight loss of 25.8 kg, or 56.9 lbs., representing 16.7% of her initial body weight, with approximately two to three kilograms per week of weight loss demonstrated at the highest 1.5 mg/day dose. Hunger scores, measured using a Likert score of zero to 10, where zero represents no hunger and 10 represents extreme hunger, mirrored the rate of weight loss, moving from scores of eight to nine prior to our trial to zero to one, as the patient was treated with increasing doses of setmelanotide. After termination of the 13‑week main trial, the patient underwent a three‑week withdrawal period off drug and regained 4.8 kg, or 10.6 lbs., with a return to moderate to severe hunger. Following approval to restart setmelanotide treatment, there was an immediate reduction of hunger and subsequently a continuation of body weight loss. This patient was on continuous treatment for 106 weeks, with a total weight loss of 65.6 kg, or 144.6 lbs., representing 42.3% of her initial body weight. There was no apparent difference in the rate of weight loss during the initial extension phase versus the main trial, however over time, the rate of weight loss has slowed, though this patient has continued to lose weight. The patient’s need for continued therapy was supported by a short period of withdrawal after the patient had been treated for over one year. Reducing her daily dose from 1.5 mg/day to 1.0 mg/day resulted in an increase in her hunger scores from one to two points to four to five points, resulting in the patient requesting to be returned to her 1.5 mg/day dose, after which her hunger scores returned to one to two points. This data supports the physiological prediction that pharmacological treatment for this condition to suppress hunger will be required chronically. After approximately 106 weeks of treatment, her dose was reduced to 1 mg/day, and while her weight remained stable from week 106 to approximately week 130, her hunger scores increased to three to four points on the lower dose before returning to two points.
The results for this patient are shown in the figure below.
19
The Clinical Course of the Initial Patient in the Setmelanotide POMC Deficiency Obesity Phase 2 Trial through Week 118(1)
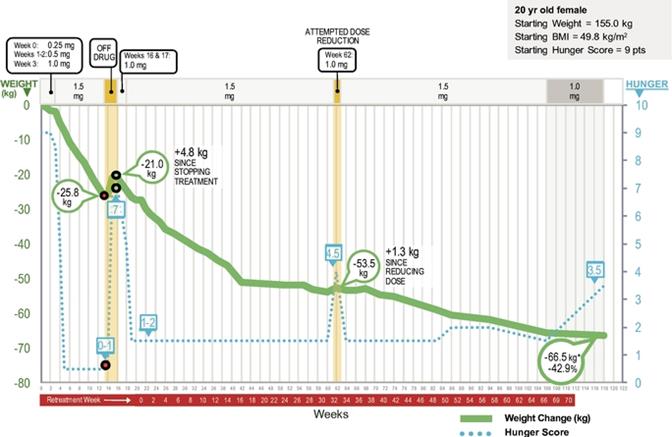
*Figures represent cumulative weight lost in kgs through approximately 118 weeks of treatment. See text for additional details.
(1)Daily setmelanotide dose adjustments over time are indicated at the top of the panel.
In general, diet induced weight loss in patients with general obesity is accompanied by significant counter‑regulatory effects, including reductions in energy expenditure and increases in hunger. These lead to weight regain in the majority of patients. In contrast, the initial patient in our trial did not manifest these counter‑regulatory responses, even after six months of therapy and a tremendous reduction of body weight. This data supports an effect of setmelanotide on energy expenditure independent from the profound effects on hyperphagia, corroborating results from previous trials of setmelanotide in patients with general obesity. Also of note, the reduction in body weight was mainly due to a loss of body fat mass, and lean body mass was not greatly altered. In this initial patient, setmelanotide was also associated with excellent tolerability, additional favorable changes in cardiovascular risk parameters, or lipids, and improvements in blood pressure and heart rate.
MC4R activation also causes improvements in glucose and insulin parameters in animal models, independent of weight loss. As shown in the figure below, for the initial patient in our POMC deficiency proof of concept trial, setmelanotide demonstrated a marked improvement in insulin resistance during treatment. While weight loss likely played an important role in this improvement, we believe the independent effect of MC4R agonism may also have contributed.
20
Setmelanotide Treatment Effects on Insulin Resistance (Insulin Response in Oral Glucose Tolerance
Test) at Baseline, After 13 Weeks of Treatment (Phase 1), and at Approximately 26 Weeks During the Long‑term Extension for our POMC Initial Patient
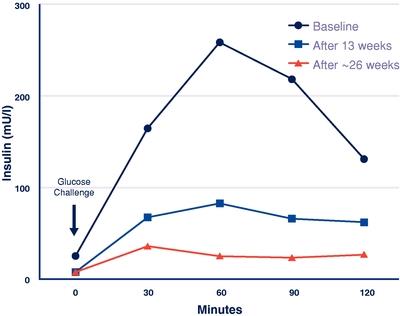
Results are also available for treatment with setmelanotide of a second patient with POMC deficiency obesity. The second patient is a 26‑year old woman who also experienced early onset of obesity and hyperphagia. Like the first patient, in spite of significant efforts, she was never able to stabilize her body weight, and she has remained hyperphagic. Ahead of our trial, the patient’s self‑reported trial hunger score was nine out of 10 points, representing extreme hunger, and her weight and BMI at trial entry were 152.8 kg, or 336.9 lbs., and 54.1 kg/m2, respectively.
After 42 weeks of therapy at the 1.5 mg/day dose, our second patient demonstrated weight loss of 40.6 kg, or 89.5 lbs., representing 26.6% of her initial body weight, with approximately two to three kilograms per week of weight loss demonstrated initially. Hunger scores, measured using a Likert score of zero to 10, where zero represents no hunger and 10 represents extreme hunger, mirrored the rate of weight loss, with scores moving from nine prior to the trial to one on most weeks during the trial, as the patient was treated with increasing doses of setmelanotide. Similar to the initial patient, setmelanotide demonstrated an improvement in insulin resistance during treatment in our second POMC deficiency obesity patient. This patient continued on active treatment for 64 weeks on therapy and her weight stabilized at a weight loss of 40.5 kg, or 89.3 lbs. However, a non‑serious febrile infection made an increase of the hydrocortisone treatment necessary. Due to a misunderstanding, this planned temporary elevated dosage of hydrocortisone was not returned to her basal dose of hydrocortisone after a few days as planned; as a result, her hunger feelings started to increase, with an associated increase in hunger score to 7 points, followed by weight gain. After adaptation of the hydrocortisone dosage during her next visit, the hunger feeling and weight decreased again and she has continued on treatment for 100 weeks in total.
21
The Clinical Course of our Second Patient in the Setmelanotide POMC Deficiency Obesity Phase 2 Trial through Week 64(1)
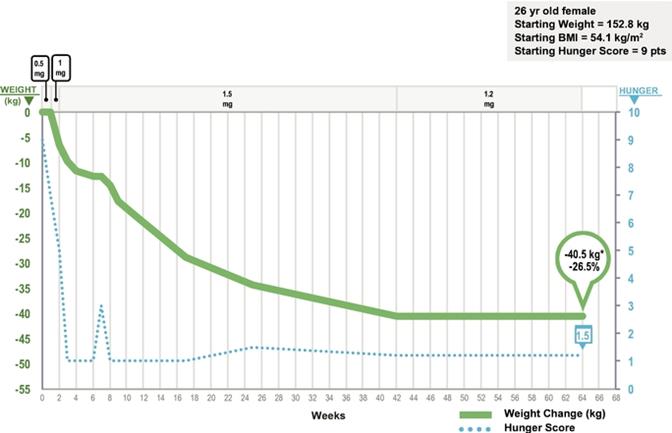
*Figures represent cumulative weight lost in kgs through approximately 64 weeks of treatment. See text for additional details.
(1)Daily setmelanotide dose adjustments over time are indicated at the top of the panel.
Setmelanotide was generally well tolerated in the POMC deficiency obesity Phase 2 trial, with few adverse events, all mild and infrequent, and all previously reported in other clinical trials. These included reduced appetite and tanning of skin and nevi, or moles, intermittent and mild injection site reactions, and in rare instances tiredness, dry mouth, and gastrointestinal symptoms. The single serious adverse event was an influenza immunization reaction, which resulted in an overnight hospitalization and was considered unrelated to trial drug. A similar immunization reaction had occurred in this patient in a previous influenza immunization prior to treatment, and the patient has continued on setmelanotide since that event.
The results from the initial patients in our POMC deficiency obesity proof of concept trial are compelling, but these data have limitations due to open label treatment. However, the strong treatment effect is supported by these patients’ long histories of weight gain and hyperphagia prior to treatment, and a strong dose response in the dose escalation phase. More importantly, the biology of this disorder has been well studied, and the clinical responses in these patients were strongly predicted by the deep understanding of the role of the MC4 pathway in appetite and weight regulation. The interruption of treatment effectively allowed the first patient to serve as her own control, demonstrating an immediate and rapid increase in hunger and weight after a short‑term treatment withdrawal, and a rapid response to re‑treatment, thereby further demonstrating the strong effect of setmelanotide. The greater than two years of treatment of our first patient, and the greater than one year of treatment for our second patient also support the ability of setmelanotide to be effective for longer treatment periods. Finally, our data supports that this indication will require chronic treatment.
22
Most importantly, this initial proof of concept data provides support for the belief that setmelanotide will restore activity in patients with upstream defects in the MC4 pathway, by helping patients lose weight and reduce hyperphagia. This was further supported by our other proof of concept results in MC4 pathway rare genetic obesities, such as LEPR deficiency obesity, Bardet-Biedl syndrome and Alström syndrome. Similarly, we would expect efficacy in other upstream MC4 pathway genetic disorders, others of which are under study in Phase 2 proof of concept trials.
Phase 3 Clinical Development in POMC Deficiency Obesity
After discussions with the FDA as part of our Breakthrough Therapy designation, we initiated our Phase 3 trial in POMC deficiency obesity in January 2017, Study RM‑493‑012. This is an open label, one‑year trial, including a double‑blind placebo‑controlled withdrawal period, of setmelanotide in POMC deficiency obesity. This pivotal trial is assessing long‑term efficacy of setmelanotide given once daily by SC injection. The trial begins with an initial period of dose titration lasting between two and 12 weeks where the individual patient’s therapeutic dose will be established by upwards dose titration in two week intervals. Thereafter, patients will continue on active treatment at their individually titrated optimal therapeutic dose for an additional 10 weeks, for a total combined dosing duration of 12 weeks at the individual patient’s therapeutic dose. Patients who demonstrate at least five kilograms weight loss at the end of the open label treatment period will continue onto the double‑blind, variably‑timed, placebo‑controlled, withdrawal period lasting eight weeks inclusive of a four‑week period of placebo treatment. Following the withdrawal period, all patients will complete an additional period of setmelanotide treatment to bring the total therapeutic dosing period to approximately one year.
Based on discussions with the FDA, we plan to file an NDA with the FDA based on approximately one-year data from a pivotal cohort of 10 patients in this trial. We also are enrolling supplemental patients who may not complete one year of treatment at the time of NDA filing, including patients between six and eleven years of age under the implementation of a pediatric amendment, to provide additional important data regarding the use of setmelanotide in people living with POMC deficiency obesity. The primary endpoint of the trial will be a categorical analysis of responders for weight, defined as patients achieving a 10% change from baseline, with mean percentage change in weight from baseline as the key secondary endpoint. Hunger-related endpoints are also key secondary endpoints in the study. Other secondary endpoints are safety and tolerability, change in body fat mass and glucose parameters, and the effect of withdrawal of setmelanotide in the double‑blind, placebo controlled period. We have also obtained Scientific Advice from the European Medicines Agency, or EMA, in relation to the protocol for this trial, which is currently enrolling in the United States, United Kingdom, Germany, France and Belgium.
We have completed enrollment of the pivotal cohort in June 2018 and we expect to report initial Phase 3 data from this pivotal trial in the third quarter of 2019.
Phase 2 Clinical Development in LEPR Deficiency Obesity
Leptin’s role in obesity has been elucidated by characterization of severely obese people with homozygous mutations that impair the activity of leptin, including disruption of signaling at the LEPR, known as LEPR deficiency obesity. To study setmelanotide in this indication we initially amended our Phase 2 clinical trial in POMC deficiency obesity, Study RM‑493‑011, to also include this new and related genetically defined population of severely obese patients. We then completed this part of the Phase 2 proof of concept, open label clinical trial in patients with LEPR deficiency obesity by treating three patients in this trial, who demonstrated weight loss and hunger reduction as outlined below. The results of this trial were published in May 2018 in Nature Medicine, which we believe validates the scientific and clinical importance of our Phase 2 findings.
Results from treatment with setmelanotide in these three LEPR deficiency obesity patients are available. The first LEPR deficiency obesity patient was a 23‑year old male, who experienced early onset of obesity and hyperphagia. After little success in controlling his weight, he underwent, and failed, a gastric banding procedure, and had regained over 20 kg in the last year since his procedure. Ahead of our trial, the patient’s self‑reported trial hunger score was nine out of 10 points, representing extreme hunger, and his weight and BMI at trial entry were 130.6 kg, or 287.9 lbs., and 39.9 kg/m2, respectively. After initiation and upwards dose titration over 13 weeks of setmelanotide treatment, the patient demonstrated prompt and striking reductions in appetite and body weight with a total loss of 17.5 kg body weight, representing 13.4%
23
of his initial body weight. Hunger scores decreased from nine points at baseline to one to two points at 13 weeks. With continued treatment for 26 weeks, the patient lost 28.2 kg, or 62.2 lbs., representing 21.6% of his initial body weight, and during that interval, he had a hunger score of one to two. Subsequently, on this dose, his weight has remained stable over an additional 35 weeks, with total weight loss of 25.1 kg or 55.3 lbs., representing 19.2% of his initial body weight over the 61 weeks of treatment. The weight loss was predominantly caused by a reduction in body fat and resting energy expenditure stayed stable during this period. This patient also had pre‑trial insulin levels that were elevated as examined by an oral glucose tolerance test, as were glucose values, demonstrating insulin resistance. These values improved with setmelanotide treatment. Notably, there was also an improvement in the patient’s lipid profile over 13 weeks of setmelanotide treatment. Setmelanotide was generally well tolerated in this LEPR deficiency obesity trial.
The Clinical Course of our Initial Patient in the Setmelanotide LEPR Deficiency Obesity Phase 2 Trial through Week 49(1)
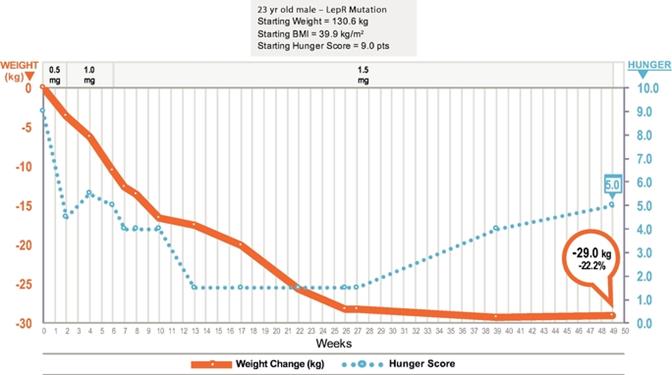
*Figures represent cumulative weight loss in kgs through approximately 49 weeks of treatment. See text for additional details.
(1)Daily setmelanotide dose adjustments over time are indicated at the top of the panel.
The second LEPR deficiency obesity patient is a 22‑year old male who also experienced early onset of obesity and hyperphagia. His growth curve since infancy demonstrated early onset, severe obesity that had continued through his whole life, with little ability to control his weight gain. Ahead of our trial, the patient’s self‑reported trial hunger score was nine to ten out of ten points, and his weight and BMI at trial entry were 122.1 kg, or 269.2 lbs., and 40.7 kg/m2, respectively. He was treated with setmelanotide and his dose was escalated up to 1.5 mg once daily with only modest effects on weight and hunger scores. However, when he was advanced to 2 mg once daily, he demonstrated prompt and striking reductions in appetite and body weight. After 17 weeks, including 11 weeks on his therapeutic dose of 2 mg/day, he had lost 9.6 kg, or 21.2 lbs., representing 7.9% of his body weight, and his hunger score dropped to two to three points. By 36 weeks of total treatment, he had lost 13.9 kg, or 30.6 lbs., representing 11.4% of his body weight. Despite good tolerance of treatment and this marked weight loss, the patient still felt dissatisfaction at the rate of weight loss and independently discontinued his setmelanotide administration for approximately 2 weeks. During this time he regained 5.2 kg or 11.5 lbs., and his hunger score increased to nine out of ten points, or severe hunger. The patient reported that he felt hungry every hour, and
24
was struggling to control his appetite. As a result, treatment was re‑initiated at the 2 mg dose, and was then increased to 2.5 mg per day with the goal of accelerating his weight loss. As a result, he demonstrated a significant reduction in hunger, with his hunger score decreasing from nine to two out of ten points, and a reduction in body weight. The patient remains on treatment with good tolerability. This patient had few metabolic abnormalities at baseline, but he was hyperinsulinemic as examined by an oral glucose tolerance test. After 13 weeks of treatment, the hyperinsulinemia started to improve and the blood glucose levels during the oral glucose tolerance test normalized.
Our Second and Third Patients in the Setmelanotide LEPR Deficiency Obesity Phase 2 Trial(1)
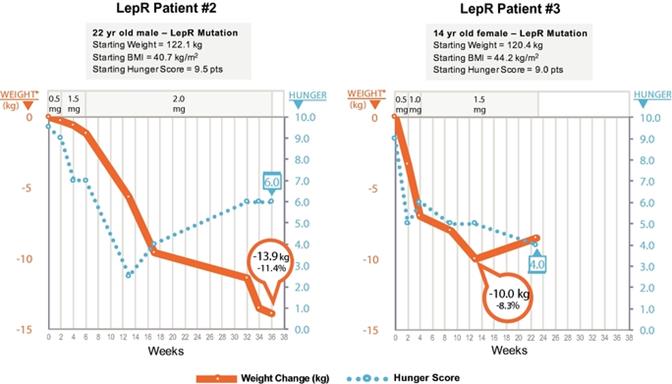
*Figures represent cumulative weight loss in kgs; further details are in the text.
*Figures show patient data only during weeks in which patients were in compliance with the trial protocol.
(1)Daily setmelanotide dose adjustments over time are indicated at the top of the panel.
The third LEPR deficiency obesity patient is a 14‑year‑old female adolescent, and the first adolescent patient treated with setmelanotide. Her growth curve since infancy demonstrated early onset, severe obesity her whole life, with little ability to control her weight gain. Ahead of our trial, the patient’s self‑reported trial hunger score was nine out of 10 points, and her weight and BMI at trial entry were 120.4 kg, or 265.4 lbs., and 44.2 kg/m2, respectively. The patient lost 10 kg, or 22 lbs., representing 8.3% of her body weight, and her hunger score reduced to five out of 10 points in the first 13 weeks on a dose of 1.5 mg per day. Before and during treatment, the patient exhibited behavior that compromised compliance with the treatment regimen. This manifested as a misunderstanding that led her to perform the injections late in the afternoon and at an incorrect, lower dosage of 1.0 mg setmelanotide. Considering that we have noted a dose threshold for therapeutic benefit and SC setmelanotide injections have a 10‑ to 12‑hour half‑life, we concluded that the combination of a lower dose and the occurrence of the highest drug concentration at night most likely precipitated an interval of weight regain. In support of this explanation, her feeling of hunger was reduced during the night but started to increase during the day. Eventually, these errors were reported and corrected. Following such correction, careful monitoring was instituted to ensure that the patient performed the injections in the morning as instructed, leading again to reduced hunger.
25
This trial provides the second proof of concept for the effectiveness of setmelanotide in patients with upstream defects in the MC4 pathway, showing marked weight reduction and decreases in hunger in patients with LEPR deficiency obesity. In addition, the efficacy of the drug correlates well with periods of setmelanotide treatment and withdrawal, as in POMC deficiency obesity. Based on this proof of concept for the compelling efficacy of setmelanotide in this disorder, and the FDA granting of Breakthrough Therapy designation, we transitioned the LEPR development program to Phase 3.
Phase 3 Clinical Development in LEPR Deficiency Obesity
Our LEPR deficiency obesity development program is now in Phase 3. We have completed enrollment of the 10 patients in the pivotal cohort in the second quarter of 2018 and we expect to report initial Phase 3 data from this pivotal trial in the third quarter of 2019. This is an open label, one‑year trial, including a double‑blind placebo‑controlled withdrawal period, of setmelanotide in LEPR deficiency obesity. This pivotal trial will assess long‑term efficacy of setmelanotide given once daily by SC injection in LEPR deficiency obesity, and is planned to be of very similar design to the Phase 3 ongoing trial in POMC deficiency obesity. The trial begins with an initial period of dose titration lasting between two and 12 weeks where the individual patient’s therapeutic dose will be established by upwards dose titration in two‑week intervals. Thereafter, patients continue on active treatment at their individually titrated optimal therapeutic dose for an additional 10 weeks, for a total combined dosing duration of 12 weeks at the individual patient’s therapeutic dose. Patients who demonstrate at least five kilograms weight loss at the end of the open label treatment period will continue onto the double‑blind, variably‑timed, placebo‑controlled, withdrawal period. Following the withdrawal period, all patients will complete an additional period of setmelanotide treatment to bring the total therapeutic dosing period to approximately one year.
We have completed enrollment of the pivotal cohort of 10 patients in this trial, aged 12 years and older. We also are enrolling supplemental patients, who may not complete one year of treatment at the time of NDA filing, including patients between six and eleven years of age under the implementation of a pediatric amendment, to provide additional important data regarding the use of setmelanotide in people living with LEPR deficiency obesity.
Completion of enrollment for the pivotal cohort in this Phase 3 trial was contemporaneous with completion of enrollment for our POMC deficiency obesity pivotal trial, and so we are planning to file an NDA for each of these indications concurrently.
The primary and key secondary endpoints will be identical to those for our POMC deficiency obesity pivotal trial. This trial is currently being conducted in the United States, the United Kingdom, France, Netherlands, and Germany. Based on the FDA granting Breakthrough Therapy designation for the treatment of obesity associated with genetic defects upstream of the MC4 receptor in the leptin‑melanocortin pathway, which includes the LEPR deficiency obesity indication, we believe we can seek indications for this type of obesity with a faster path to approval, as compared to typical obesity drug candidates, because of the high unmet need and rare prevalence of this disorder.
Phase 2 Clinical Development in Bardet‑Biedl Syndrome
Bardet‑Biedl syndrome is a life‑threatening, orphan disease with prevalence of approximately one in 100,000 in North America. We estimate that the addressable patient population for Bardet‑Biedl syndrome is approximately 1,500 to 2,500 patients in the United States. It is a rare monogenic disorder that causes severe obesity and hyperphagia as well as vision loss, polydactyly, kidney abnormalities, and other signs and symptoms. Bardet‑Biedl syndrome is part of a class of disorders called ciliopathies, or disorders associated with the impairment of cilia function in cells. Cilia are hair‑like cellular projections that play a fundamental role in the regulation of several biological processes, including satiety signaling. Cilia dysfunction is thought to contribute to hyperphagia and obesity in Bardet‑Biedl syndrome. Bardet‑Biedl syndrome is a genetically heterogeneous disease that is caused by as many as 21 separate Bardet‑Biedl loci defects resulting in a similar syndrome, though each Bardet‑Biedl syndrome patient only has one of these defects.
The role of abnormal cilia development and function in obesity has been elucidated in animal models, most strongly for Bardet‑Biedl syndrome. Studies in mouse models of Bardet‑Biedl syndrome show that deficiencies in the MC4 pathway contribute to the obesity and hyperphagia in Bardet‑Biedl syndrome, with animals developing hyperphagic tendencies early in life. Notably, these mice have decreased leptin receptor signaling, with the essential hallmarks of failure
26
to activate POMC neurons. This is supported in Bardet‑Biedl syndrome rodent models, where the mice respond to an MC4 agonist resulting in reduced food intake and body weight. The relation of Bardet‑Biedl syndrome gene mutations to the MC4 pathway is supported by clinical data. Patients with Bardet‑Biedl syndrome have higher leptin than expected for their degree of adiposity, or leptin resistance, which is consistent with the notion that ciliopathy‑induced leptin signaling dysfunction is associated with leptin resistance.
Overall, these data support that the phenotypes of these ciliopathies, while complex with additional clinically important features along with obesity and hyperphagia, may be responsive to setmelanotide treatment, and will be investigated in our proof of concept Phase 2 trial.
We are studying Bardet‑Biedl syndrome patients who are severely obese, and have provided proof of concept that Bardet‑Biedl syndrome patients will also demonstrate decreased hunger and significant weight loss, similar to that seen in patients with POMC deficiency obesity, or LEPR deficiency obesity. We have enrolled the first five patients in this trial, have reported preliminary results for these patients in a presentation at the Obesity Week 2017 meeting, and updated the clinical status of these patients in June 2018. In the second quarter of 2018, the FDA agreed to include Bardet‑Biedl syndrome under our existing Breakthrough Therapy designation for setmelanotide. We have had ongoing discussions with the FDA regarding a Phase 3 trial and initiated this trial and began to enroll patients in December 2018. The FDA has indicated that this Phase 3 trial will include both Bardet‑Biedl syndrome and Alström syndrome patients.
For the Phase 2 trial, additional assessments of hunger using daily hunger scores and questionnaires were also obtained. We are using these new assessments in all our ongoing Phase 2 and Phase 3 trials and for future trials. These new assessments are as follows:
|
|
|
|
|
|
|
|
We believe that proof of concept in Bardet‑Biedl syndrome has been demonstrated by improvements in hunger and weight reduction, supporting that this is a setmelanotide‑responsive, MC4 pathway disorder. The age of the patients ranged from 12 to 61 years. The starting weights of the patients ranged from 51.3 to 162.7 kg and BMI ranged from 37 to 51. The starting hunger scores for the adult patients ranged from 6 to 9 points on the 10‑point scale, with higher scores indicating more hunger and the SEQ scores for the two adolescent patients were 1, and 2 for a third adolescent patient.
27
Description of the Nine Bardet‑Biedl patients in the Phase 2 Proof of Concept study
|
|
|
|
|
|
|
|
|
|
|
Patient Number |
| Age |
| Bardet‑Biedl |
| Starting |
| Starting |
| Starting Hunger Score |
1..................................................... |
| 25 |
| 1 |
| 147.5 |
| 44 |
| Most hungry score = 9 |
2..................................................... |
| 61 |
| 2 |
| 99.4 |
| 44 |
| Most hungry score = 7 |
3..................................................... |
| 16 |
| 10 |
| 121.6 |
| 44 |
| FPD = 6/ SEQ = 1 |
4..................................................... |
| 17 |
| 12 |
| 98.3 |
| 42 |
| Most hungry score = 6 |
5..................................................... |
| 12 |
| 1 |
| 119.3 |
| 49 |
| FDP = 15/SEQ = 1 |
6..................................................... |
| 16 |
| 5 |
| 122.3 |
| 43 |
| Most hungry score = 9 |
7..................................................... |
| 14 |
| 4 |
| 88.6 |
| 37 |
| Most hungry score = 7 |
8..................................................... |
| 13 |
| * |
| 51.3 |
| 51 |
| FPD = 9 / SEQ = 2 |
9..................................................... |
| 31 |
| 1 |
| 162.7 |
| 48 |
| Most hungry score = 6 |
Three patients with Bardet‑Biedl syndrome type 1 mutations and one each with Bardet‑Biedl syndrome types 2, 4, 5, 10, 12 mutations and one patient with a BBS mutation that was not established were enrolled. Total treatment durations lasted between 41 and 65 weeks. Six of the nine patients showed clinically important, marked weight loss of 27.0 kg, 14.1 kg, 24.2 kg, 13.7 kg, 13.7 kg and 21.2 kg, or 59.5 lbs., 31.1 lbs., 53.4 lbs., 30.2 lbs., 30.2 lbs. and 46.7 lbs. respectively, and a decrease of 18.3%, 14.2%, 19.9%, 11.2%, 15.5% and 21.6%, respectively. Hunger scores were reduced by 44.4%, 85.7%, 100.0%, 66.0%, 21.0% and 66.7%, respectively, in these six patients. Summary of the data is shown below and with six out of nine patients showing efficacy of > 10% weight loss supported moving into phase 3.
Summary of the Phase 2 data for Our Six Patients in the Setmelanotide Bardet‑Biedl Syndrome Phase 2 Trial who showed improvements in both weight and hunger
|
|
|
|
|
|
|
|
|
Bardet‑Biedl Patient Number |
| Weight loss |
| Weight loss |
| Weight loss |
| Decrease in |
1........................................................... |
| 27.0 |
| 59.5 |
| 18.3% |
| 44.4% |
2........................................................... |
| 14.1 |
| 31.1 |
| 14.2% |
| 85.7% |
3........................................................... |
| 24.2 |
| 53.4 |
| 19.9% |
| 100.0% |
4........................................................... |
| 21.2 |
| 46.7 |
| 21.6% |
| 66.7% |
6........................................................... |
| 13.7 |
| 30.2 |
| 11.2% |
| 66.0% |
7........................................................... |
| 13.7 |
| 30.2 |
| 15.5% |
| 21.0% |
Mean..................................................... |
| 19.0 kg |
| 41.9 lbs. |
| 16.8% |
| 64.0% |
For six of the nine Bardet‑Biedl syndrome patients who responded to treatment with setmelanotide, mean weight loss reached 16.8% of body weight, and mean hunger decreased by 64.0%. The time course of individual patient weight loss and hunger scores for these four patients who were treated for longer-term duration (50-65 weeks) are shown below.
28
Patients in the Setmelanotide Bardet‑Biedl syndrome Phase 2 Trial who showed improvements in both weight and hunger(1)(2)(3)
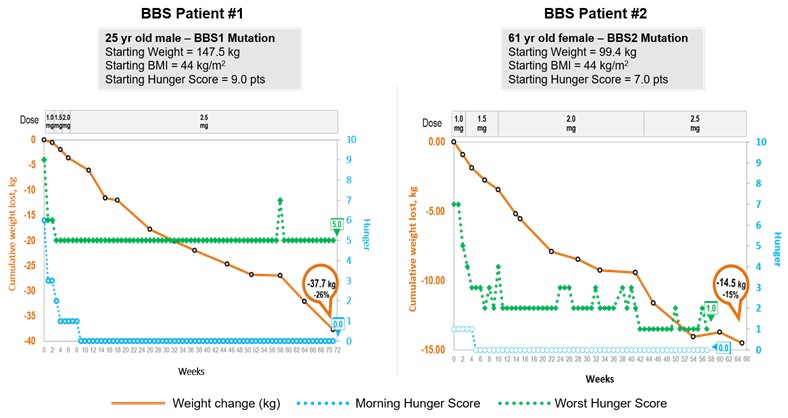
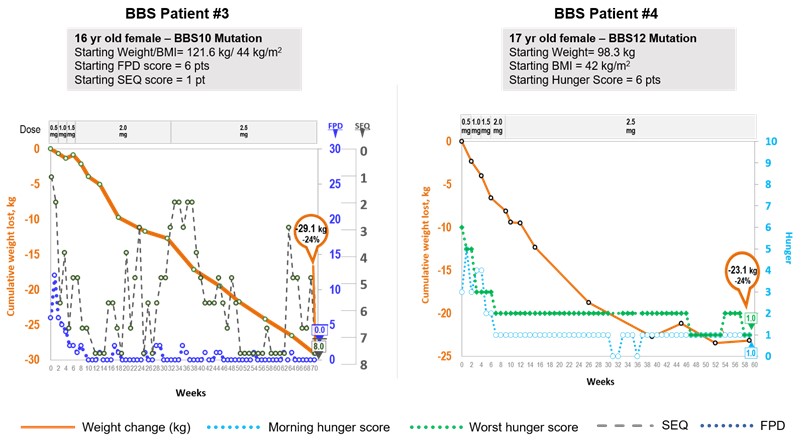
*Figures represent cumulative weight lost in kgs
FPD: Food Problem Diary; Score Range 0 to 30, higher score means worse result
29
SEQ: Significant Event Questionnaire, which counts significant food behavior events rarely seen in this population (Y/N for 8 behaviors), so maximum score of 8 points means greatest improvement. Shown in reverse scale so downward movement equals improvement for clarity.
(1)Daily setmelanotide dose adjustments over time are indicated at the top of the panel.
(2)In some cases, dates for entry of weight and hunger assessment data may differ within a single patient.
(3)Data as of August 2018.
The fifth patient, a patient with a Bardet‑Biedl syndrome type 1 mutation, showed marked improvement in hunger scores with a 53.3% decrease, but did not demonstrate any body weight change after 33 weeks on treatment, including a final 12‑week test period on 3.0 mg daily. However, the weight curve for this patient indicated a slowing of prior childhood weight gain upon treatment with setmelanotide, as indicated with an arrow in her pediatric growth chart, below. This patient was a 12‑year‑old with Type 1 diabetes who entered the trial with extremely poor glucose control, with an average blood sugar level, or HbA1c, of 10.1%. We have been investigating the reason for the inconsistency between her improvement in hunger and lack of weight loss. During her treatment, her HbA1c showed an improvement to 7.6%. Following treatment discontinuation, the patient gained 5.9 kg, appetite and hunger returned to baseline levels and HbA1c increased to 11.7%.
Overall setmelanotide was generally well tolerated in all patients in this Bardet‑Biedl syndrome Phase 2 proof of concept study. Adverse events associated with setmelanotide treatment included increased pigmentation of the skin/nvi and mild injection site reactions. No discontinuations were due to adverse events. No serious adverse events were reported in BBS patients. No clinically significant detrimental changes in blood pressure or heart rate have been reported.
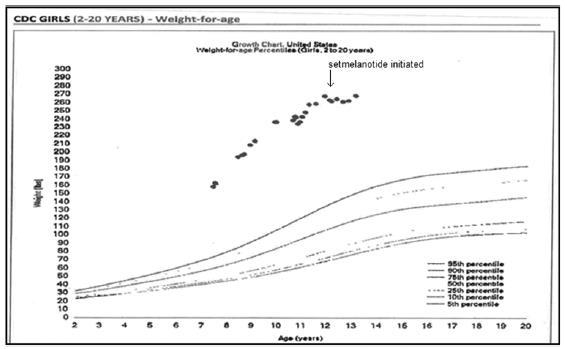
Phase 2 Proof of Concept Study of Patients with Alström Syndrome
We are conducting a Phase 2, proof of concept trial in Alström syndrome, which is a life‑threatening, ultra‑rare orphan disease with a prevalence of approximately one in 1,000,000 in North America and we estimate that the addressable patient population for Alström syndrome obesity is approximately 500 to 1,000 patients worldwide. Alström syndrome shares many clinical features with Bardet‑Biedl syndrome,BBS, including obesityhyperphagia and hyperphagia,obesity, and is also characterized by progressive vision loss, deafness, congestive heart failure, hyperinsulinemia and type 2 diabetes mellitus. Similarly,
30
Alström syndrome is a ciliopathy caused by mutations in the ALMS1 gene, which has also been shown to be important for cilia function. Like Bardet‑Biedl syndrome,BBS, recent scientific studies identify genetic deficiencies affecting the MC4MC4R signaling pathway as a potential cause of the obesityhyperphagia and hyperphagiaobesity associated with Alström syndrome. Studies in a mouse model of Alström syndrome show a reduction in the number of cilia in specific neurons in the hypothalamus that are critical for MC4 pathway signaling. While Alström syndrome is less well studied than Bardet‑Biedl syndrome,BBS, the similar pathophysiology of cilialcilia dysfunction and clinical presentation support our belief that deficiencies in the MC4MC4R pathway are implicated in the obesityhyperphagia and hyperphagiaobesity observed in Alström syndrome. Therefore, we hypothesize that setmelanotide treatment can be applied to treat Alström syndrome.
We are studyingIn September 2021, we submitted an sNDA to the FDA for setmelanotide for the treatment of obesity and control of hunger in adult and pediatric patients 6 years of age and older with BBS or Alström syndrome, patients who are severely obese. We believe,and in October 2021, we submitted a Type II variation application to the EMA for the same indications and patient populations. These submissions were based on based on data from our initial proof of concept data, shown below, demonstrates that Alström syndrome patients may also experience decreased hunger and significant weight loss similar to that seen in patients with POMC deficiency obesity, LEPR deficiency obesity, or Bardet‑Biedl syndrome. We have enrolled patients in this trial and have updated the clinical status of these patients in June 2018. The FDA has also recently included Alström syndrome under our existing Breakthrough Therapy designation. As mentioned for Bardet‑Biedl syndrome, we have had ongoing discussions with the FDA regarding apivotal Phase 3 trial and initiated this trial and started to enroll patients in December 2018. The FDA has indicated that this Phase 3 trial will include both Bardet‑Biedl syndrome and Alström syndrome patients.
As of October 2018, four Alström syndrome patients have been treated with setmelanotide in the Phase 2 study. The age of the patients range from 12 to 21 years. The starting weights of the patients range from 70.7 to 108.1 kg and BMI ranged from 28 to 47. The starting hunger scores for the adult patients ranged from 2 to 7 points on the 10-point scale.
The initial Alström syndrome patient in our Phase 2 clinical trial was a 12‑year‑old male, starting weight of 78.6 kg, or 173.2 lbs., with a BMI of 28.2 kg/m2 (above the 98th percentile for his age). At baseline, his initial “worst hunger” score was 5.5 points, and his morning hunger score was 5 points. As of October 2018, this patient has been treated for 56 weeks including titration, most of that time on a dose of 2 mg/day. During the course of 56 weeks of treatment, he experienced weight loss of 16.4 kg, or 36.2 lbs., which represented a 20.9% weight loss and a 45.5% decrease in hunger. Clinical assessment by the Dykens hyperphagia questionnaire, another instrument also collected in this patient only, demonstrated a baseline of 31 out of 55 possible points, which improved to 11 points, or a 64.5% improvement in
31
hyperphagia. Because his body weight was approaching ideal body weight, and he is a growing child, his dose was reduced to 1.5 mg/day, at 32 weeks and 0.5 mg six days/week at 50 weeks, with stabilization of his weight.
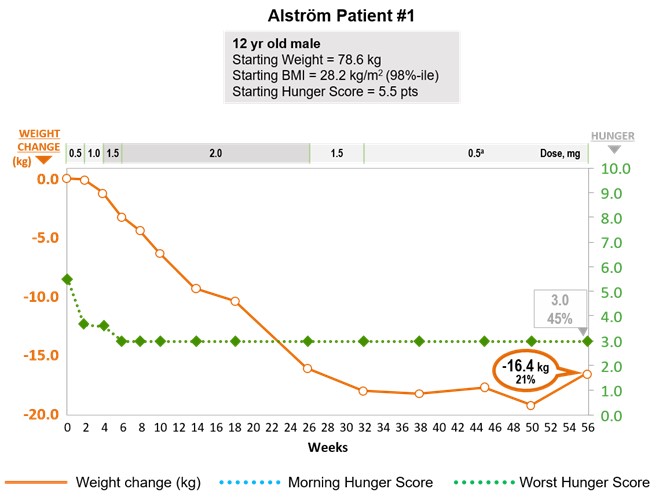
Three additional Alström patients (2 adolescents, 1 adult) have been enrolled in this Phase 2 study and the data is summarized below. One of these patients is showing promising responses in weight loss and hunger, one is too early to determine, but one patient, the adult, did not show improvements in either weight or hunger and was discontinued.
|
|
|
|
|
|
|
|
|
Alström Syndrome Patient Number |
| Weight loss |
| Weight loss |
| Weight loss |
| Change in |
1........................................................... |
| 16.4 |
| 36.2 |
| 21.0% |
| 6/3 |
2........................................................... |
| 3.8 |
| 8.4 |
| 5.4% |
| 7/2 |
3........................................................... |
| 1.1 |
| 4.6 |
| 1.1% |
| NA |
4........................................................... |
| 5.0 |
| 11.2 |
| 5.5% |
| 2/5 |
Overall, setmelanotide was generally well tolerated in all patients in this Alström syndrome Phase 2 proof of concept study. Adverse events associated with setmelanotide treatment included increased pigmentation of the skin/nevi and injection site reactions. No serious adverse events or discontinuations due to adverse events were reported. No clinically significant increases in blood pressure were observed that resulted in the development of hypertension.
Phase 3 Clinical Development in Bardet‑Biedl and Alström Syndrome
Since the FDA agreed to include Bardet‑Biedl syndrome and Alström syndrome under our Breakthrough Therapy designation, we began in May 2018 an ongoing discussion about Phase 3 trial design with the FDA, and have received preliminary guidance on many aspects of the Phase 3 development programs in Bardet‑Biedl syndrome and Alström syndrome. Based on these discussions, we are started a combined Phase 3 study to include both Bardet‑Biedl and Alström patients in a single Phase 3 trial, including at least 20 Bardet‑Biedl syndrome patients and at least six Alström syndrome
32
patients, aged six years and older. The combined trial is likely to lead to a more rapid path for approval of these two indications than two separate trials. Like previous setmelanotide Phase 3 trials in POMC and LEPR deficiency obesities, we are planning an open label, approximately one‑year trial, assessing long term efficacy of setmelanotide given once daily by SC injection. We may also plan to enroll supplemental patients who may not complete one year of treatment at the time of NDA filing. We expect to discuss with the FDA the primary endpoint of the trial. Potential endpoints will likely include a categorical analysis of responders for weight, with mean percentage change in weight from baseline as a key secondary endpoint. Hunger‑related endpoints are also likely to be key secondary endpoints in the study.
We are planning the trial to begin with an initial period of dose titration, using a simplified titration scheme over two weeks for adults, and possibly slightly longer for pediatric patients. This trial may include a short, 12‑week, randomized, placebo‑controlled period. Following the 12‑week, double‑blind, placebo controlled period, all patients will complete an additional period of active setmelanotide treatment to complete the approximately one‑year trial. We are also discussing with the FDA a potential alternative primary endpoint for growing pediatric patients that will capture the therapeutic benefits of setmelanotide in the pediatric population.
We have continued ongoing discussion with the FDA, completed the Phase 3 protocol, initiated the Phase 3 study and enrolled our first patients in December of 2018. This study will be conducted in both the United States and Europe.
Phase 2 Proof of Concept Studies Focused on Patients with Monogenic Disorders of the MC4 Pathway: Heterozygous Mutations in the MC4 Pathway and Epigenetic Disorders of the MC4 Pathway
We are conducting Phase 2, proof of concept trials in a variety of monogenic, upstream disorders of the MC4 pathway, including POMC and other MC4 pathway gene heterozygous mutations, and epigenetic disorders of the MC4 pathway. These trials are Phase 2 open label, single arm, proof of concept trials assessing the effect of setmelanotide on the rare genetic disorders of obesity described below. We hypothesize that these disorders may be genetically‑defined deficiencies upstream in the MC4 pathway. Each trial includes a three‑month proof of concept phase at which weight loss, hunger and other metabolic parameters will be evaluated. If patients demonstrate significant weight loss and acceptable safety and tolerability, they will continue treatment for evaluation of setmelanotide’s effects for a total of one year. Similar to our previous trials, this trial begins with an initial period of dose titration where the individual patient’s therapeutic dose is established by upwards dose titration in two week intervals. We plan to enroll from 20 up to 100 patients between these two rare genetic populations. We are conducting these trials, as we did for the ongoing Bardet‑Biedl syndrome and Alström syndrome Phase 2 trial described above, under basket protocols, which are designed to capture a broad range of patient populations to be treated under one investigational protocol. We believe this approach is efficient for studying many potential indications, and we intend to add additional populations to these basket protocols over the next one to two years.
The genetic disorders we are studying in our additional Phase 2 proof of concept trials are outlined below, and initial, preliminary Phase 2 data for each indication is summarized.
a.Clinical Development in MC4 Pathway Heterozygous Deficiency Obesity
MC4 pathway heterozygous deficiency obesity is caused by the loss of one of the two genetic copies of either the genes for POMC, PCSK, LEPR, or other genes in the MC4 pathway. Animal models support that such heterozygous deficiency in the critical leptin‑melanocortin pathway can result in a strong predisposition to severe obesity. The effect of genetic heterozygous deficiency obesity was first demonstrated for another gene in the MC4 pathway: MC4R heterozygous deficiency obesity. Later data also supported that POMC heterozygous deficiency obesity also results in a strong predisposition to obesity, though the epidemiology and clinical characterization of these patients is less well known. An estimated 2% of severe, early onset obesity patients have POMC heterozygous deficiency obesity, which is much more common than the ultra‑rare POMC deficiency obesity in which both copies of either the POMC or PCSK genes are impaired. Our initial clinical focus is on the most severely obese MC4 pathway heterozygous patients to test the hypothesis that severely obese heterozygous POMC patients might also respond substantially to setmelanotide treatment.
We are studying patients who are severely obese, or whose BMI is equal to or greater than 40kg/m2, and who are heterozygous deficient for POMC, LEPR, or other genes in the MC4 pathway. These patients have a
33
heterozygous genetic mutation resulting in full or partial loss of MC4 pathway signaling to the downstream MC4R. The purpose of studying these patients in this trial is to provide proof of concept that severely impaired MC4 pathway heterozygous deficiency obesity patients will also demonstrate significant weight loss, similar to though possibly of less magnitude, as that seen in patients with POMC deficiency obesity or LEPR deficiency obesity. We are enrolling this trial at sites in the United States and Europe, and reported initial, preliminary results in June 2018.
Five heterozygous patients have been enrolled, mostly for short durations, in this Phase 2 trial. The longest treated patient, on treatment for 34 weeks including titration, is showing promising responses in weight loss of 23.1 kg, or 50.9 lbs. which represented a 15.4% decrease in body weight, and also showed an 80% decrease in hunger score. A second patient has shown 9.5 kg, or 21.0 lbs. of weight loss, which represented a 6.5% decrease in body weight, and also showed a moderate 20% decrease in hunger score. One patient did not show improvements in either weight or hunger and was discontinued due to lack of efficacy, and two patients discontinued early due to tolerability issues related to muscle cramps or tanning, respectively. Four of the five patients treated to date were enrolled at a single site. We are currently opening a number of additional sites with relevant pre‑identified heterozygous patients in their databases.
POMC heterozygous deficiency obesity is complex in many ways, in that there appears to be variable penetrance, or manifestation of heterozygous mutations. As a result, we intend to study a larger number of heterozygous patients in Phase 2, to more carefully delineate those patients most debilitated and those who will have the best response to setmelanotide, before we discuss the design for a Phase 3 study with the FDA. We intend to provide an update on a larger number of heterozygous patients in the first quarter of 2019.
Mutations that are of particular interest include mutations to the section of the POMC gene that are translated into the protein, beta‑melanocyte stimulating hormone, or β‑MSH. These mutations have been implicated in human and canine obesity. In 2002, researchers identified one specific heterozygote mutation of β‑MSH, called the R236G mutation, in two children with extreme childhood obesity. This R236G mutation results in an abnormal β‑MSH protein with a markedly reduced ability to activate the MC4R itself, and may also prevent other natural MC4R ligands from activating the MC4R. These combined effects may result in more significant obesity than other heterozygous mutations. The overall prevalence of this mutation is rare, 0.7% of the obese population is estimated to carry this mutation, but our genotyping study of 560 patients with early onset, childhood obesity has identified five heterozygote patients with the R236G mutation, all with severe obesity. This mutation may be a focus when enrolling our Phase 2 trial in POMC heterozygous obesity. We are studying patients who are heterozygous deficient for other genes in the MC4 pathway, though less is known about the epidemiology and clinical impact of these other heterozygous deficiency obesities. We have also hypothesized that patients who carry high allelic burden in this pathway, or who have genetic disorders in two or more of the genes of the MC4 pathway, and who therefore might have some impairment at more than one location in the MC4 pathway, might also be responsive to setmelanotide.
b.Clinical Development in Patients with Epigenetic Changes at the POMC receptor
In our proof of concept Phase 2 trials, we are studying patients suffering from obesity due to a partial lack of MSH due to an epigenetic POMC variant. Epigenetics changes are DNA modifications that can change gene expression without altering the DNA sequence itself. The most stable epigenetic modification is called DNA methylation. Recently, our academic collaborators in Berlin have described a POMC hypermethylation variant, which correlates with increased body weight in children and adults. Therefore, the presence of the POMC genetic/epigenetic variant leads to an increased risk for obesity based on reduced POMC gene activity. We expect that these patients under express the POMC gene product and as a result have a partial MSH deficiency.
There is convincing evidence that such epigenetic variants are potentially major factors for an increased individual risk to develop obesity later in life, and we hypothesize that the most obese patients in their populations may benefit from treatment with setmelanotide. However, epigenetic variation is likely not the only reason for the development of obesity in this patient group, because these variants are also observed in normal weight
34
individuals, although to a lesser extent. At this point, no epidemiology data is available to estimate the size of the POMC epigenetic deficiency obese population.
We are studying patients who are severely obese, or whose BMI is equal to or greater than 40kg/m2, and who have hypermethylation at the POMC gene. The purpose of studying these patients in this trial is to provide proof of concept that severely impaired, epigenetic POMC variant obesity patients will also demonstrate significant weight loss similar to, though possibly of less magnitude, as that seen in patients with POMC deficiency obesity or LEPR deficiency obesity. We are enrolling this trial at sites in the United States and Europe, and reported initial, preliminary results in June 2018.
Two epigenetic patients have been enrolled, one of whom is just starting titration. The longer treated patient, on treatment for 27 weeks including titration, is a 20‑year‑old female, with a starting weight of 182.2 kg, or 401.7 lbs., and a BMI of 55.0 kg/m2. Her starting hunger score was 7.5 points. As she was titrated, her hunger score decreased 60% to three points and she began to lose weight. Due to a vacation, she withdrew from treatment for approximately four weeks and her hunger score returned to baseline but she did not regain weight over that period. With resumption of treatment, her hunger score decreased to 3.5 points, and at week 27, she had lost 16.8 kg, or 37.0 lbs., which represented a loss of 9.2% of body weight.
Phase 1b Clinical Development in Patients with Heterozygous MC4R Gene Mutations
Early studies in downstream MC4 pathway defects demonstrated good efficacy and tolerability, and served as a foundation for potentially greater efficacy in upstream MC4 pathway deficiencies. We established proof of concept for efficacy of setmelanotide in patients with an MC4R heterozygous genetic mutationBBS or Alström syndrome. As we first reported in one cohortDecember 2020, the trial met its primary endpoint and all key secondary endpoints, with statistically significant and clinically meaningful reductions in weight and hunger at 52 weeks on therapy. All patients who met the primary endpoint, defined as at least 10 % weight loss, had BBS and none had Alström syndrome. However, data from this Phase 3 trial are supported by results from the Phase 2 trial, which suggested that treatment with setmelanotide may result in decreased weight and hunger in patients with Alström syndrome. In addition, data from a predefined exploratory endpoint showed that in patients with BBS or Alström syndrome who were younger than 18 years old and likely still growing in height, setmelanotide treatment was associated with clinically meaningful reductions in BMI Z scores. The BMI Z score, or BMI standard deviation score, represents the number of patientsstandard deviations from median BMI by child’s age and sex, and represents a unit of measure that accounts for height as well as weight in growing children and adolescents. Based on our discussion with the FDA and the EMA, we believe the decision or decisions to approve and/or authorize setmelanotide for BBS and Alström would be made independently, and recently we have decided to withdraw the Alström syndrome indication from the pending Type II variation application based on feedback from the EMA. In November 2021, we announced that the FDA accepted for filing our sNDA with Priority Review and assigned a PDUFA goal date of June 16, 2022 for our sNDA.
Based on the data included in our regulatory applications, we believe setmelanotide has the potential to address the early-onset, severe obesity and hyperphagia that characterize these diseases. In addition to the topline data reported from our Phase 1b clinical trial. This clinical3 trial of setmelanotide in patients with BBS or Alström syndrome, we have also presented several poster and oral presentations featuring the topline data from the pivotal cohort as well as additional updated data also including the supplemental cohort at medical meetings during 2021. Our pivotal, Phase 3 trial that combined both BBS and Alström syndrome was a double‑blind, placebo‑controlled, randomized Phase 1b clinical trial designed to evaluate the effectmultinational, open-label, single-arm study consisting of setmelanotide on weight loss and safety in obese patients with a heterozygous mutation of the MC4R gene. The initial cohort of eight patients was treated for four weeks with setmelanotide or placebo. The setmelanotide group showed weight loss of 3.48 kg, or 7.67 lbs., approximately 2.62 kg, or 5.78 lbs., more weight loss than the placebo group, which showed weight loss of 0.85 kg, or 1.87 lbs. Other parameters supporting weight loss were also positively impacted by setmelanotide. We believe that these results support the hypothesis that setmelanotide can be effective in weight loss in MC4R deficient patients, and provide evidence of the minimum expected treatment effect of setmelanotide, approximately 0.9 kg/week, or 1.98 lbs./week, of weight loss over four weeks, even in a situation where setmelanotide’s action is on a downstream MC4 pathway that is no longer fully functional due to heterozygous MC4R mutations. However, our focus is on upstream disorders of the MC4 pathway where we hypothesize that setmelanotide can serve as replacement therapy and provide more compelling efficacy.
35
The following figure depicts preliminary data relating to our setmelanotide Phase 1b clinical trial in MC4 heterozygous deficiency obesity patients:
Setmelanotide Phase 1b Trial MC4 Heterozygous Patients: Placebo Subtracted Differences(1)(2)
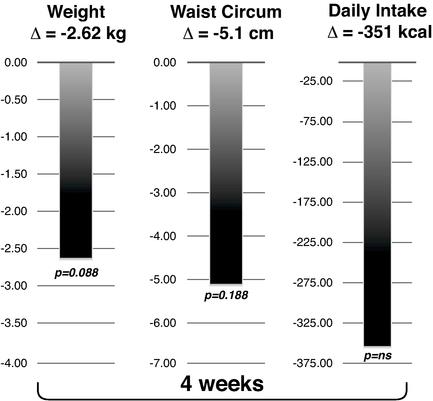
(1)Over four52 weeks of treatment with setmelanotide. Participants were blinded and randomized for the first 14 weeks of the trial to receive either placebo or setmelanotide 0.01 mg/kg/day by continuous SC infusion.
(2)Preliminary data.
In general, we consider a p‑value of 0.05 to be significant. P‑values are an indication of statistical significance reflectingtherapy. Those participants who began the probability of an observation occurring due to chance alone. However, it is not possible to determine a p‑valuetrial on setmelanotide continued therapy for very small sample sizes, such as one‑ or two‑patient trials.
Other Clinical and Scientific Initiatives in Genetic Obesity
Genotyping Study
Leveraging new understanding of severe obesity caused by specific genetic defects has the potential to improve both diagnosis and treatment for specific types of life‑threatening obesity. We have expanded our genotyping study—the Genetic Obesity ID | Genotyping Study—in which eligible patients are genotyped for rare genetic disorders of obesity. The goal is to develop a screening algorithm for selecting patients to be genotyped and identified with POMC deficiency obesity and LEPR deficiency obesity, and to guide further genotyping efforts; in addition, it is our expectation that patients who can participate in our clinical trials will be genetically identified. We are currently including approximately 100 genes which, in medical and scientific literature, have been associated with obesity, including other genes associated with the MC4 pathway. Individuals may enter the study through one of three arms including: a history of severe, early onset obesity, along with hyperphagia, high BMI, and individuals within three months of bariatric surgery. The study is currently
36
enrolling in the United States and Europe and is expected to expand to a total of 140 sites worldwide. We plan52 weeks, while those on placebo went on to work with these investigators to publish the resultsreceive 52 weeks of this study and guidance on the usesetmelanotide therapy after completion of the algorithm for screening, to enable more systematic diagnoses of these rare genetic disorders of obesity.
Unique Mechanism of Action of Setmelanotide at the MC4R
We recently published new molecular evidence of setmelanotide action on MC4R in Nature Medicine in May 2018, which demonstrates a unique mechanism of action compared to the endogenous activator, MSH, and first generation MC4R agonists. With agonism of the MC4R, setmelanotide appears to use different signaling pathways inside the MC4R cell, and to better compete away the natural antagonist at MC4R (AgRP). This may explain the remarkable efficacy of setmelanotide for appetite control in individuals with severe hyperphagia, and may also suggest why setmelanotide does not clinically increase blood pressure or heart rate, compared to former MC4R agonists. Further research in this area is planned as well.
Setmelanotide: Clinical Development Program in Prader‑Willi Syndrome
PWS is a life threatening, orphan multigenic disease with prevalence estimates ranging from approximately one in 8,000 to one in 52,000, with at least 8,000 diagnosed14-week placebo period. All patients in the United States. A hallmark of PWS is hyperphagia, leading to severehad obesity, and other complications. For PWS patients, hyperphagia and obesity are the greatest threats to their health, and these patients are likely to die prematurely as a result of choking, stomach rupture, or from complications caused by morbid obesity.
The genetics of PWS are complex, involving many genes on chromosome 15 that are not properly expressed. Recent discoveries highlight that a defect in one of these, the melanoma antigen family L2, or MAGEL2, gene, in rodent models impairs the function of POMC neurons, which are key components of the MC4 pathway. Studies have suggested a link between defects in MAGEL2 in some humans with obesity, hyperphagia, autism spectrum disorders, reduced intellectual ability and most other aspects of behavior and metabolism associated with PWS. However, the connection of PWS with the MC4 pathway is complex.
We have completed a Phase 2 proof of concept, double‑blind, placebo‑controlled, randomized clinical trial in PWS, Study RM‑493‑010, which enrolled 40 patients for four weeks of active setmelanotide treatment, administered once daily by SC injection. This trial was intended to assess the effects of setmelanotide on weight reduction, and PWS‑specific hyperphagia‑related behaviors, as PWS patients do not respond to hunger questionnaires, as well as determine its safety profile. Based on the data from this Phase 2 clinical trial, we do not believe we will be positioned to proceed directly into a Phase 3 clinical trial.
The trial included a two‑week run‑in period, a four‑week double blind, randomized, placebo‑controlled parallel group main trial, a two‑week double‑blind, randomized, placebo‑controlled withdrawal period during which half of the trial patients were randomized to either continue to receive their therapy or be switched to the alternative therapy, from active to placebo, or vice versa, and a two‑week active‑treatment extension. There were four treatment arms in the trial: placebo (N=14); 0.5 mg of setmelanotide SC injection daily (N=4), 1.5 mg of setmelanotide SC injection daily (N=12), and 2.5 mg of setmelanotide SC injection daily (N=10). Patients were 17 to 54 years of age, with a mean BMI of 39.4 kg/m2, and with a genetically confirmed diagnosis of PWS. Primary endpoints for the trial included safety and tolerability, weight loss and hyperphagia, with hyperphagia to be measured by a PWS hyperphagia observer reported outcome, or ORO, questionnaire. Secondary endpoints included dual‑energy x‑ray absorptiometry measurements, pharmacokinetics, effects during the randomized withdrawal stage, and effects on quality of life and food‑related and other behaviors. Primary evaluations were assessed at the end of the four‑week double blind parallel group stage, as well as after the withdrawal stage and open label extension.
The results of the trial showed modest effects on hyperphagia, which did not approach statistical significance, and no effect on weight, though there may have been some small evidence of clinically‑important weight loss in the very small group of patients who were randomized to the highest dose of setmelanotide over the longest interval of treatment (N=4 patients, post‑hoc evaluation, non‑significant). There was good safety and tolerability, providing support for the 2.5 mg daily dose, with only injection site reactions common in both active and placebo groups. There were no serious adverse
37
events, no significant safety issues or changes in labs or other safety parameters, and the one discontinuation was due to injection site reactions.
PWS is a complex multigenic disease, and the hypothesis that PWS is an upstream MC4 pathway disorder is supported primarily on the role of only one of those genes, MAGEL2, in animal models of obesity. Our results may support that PWS is not an upstream MC4 pathway disorder. Alternatively, other design factors may have influenced the outcome of this trial, and we have started initial planning for future Phase 2 trials in PWS that address these potential factors: longer duration of treatment, younger patient population, improved setmelanotide pharmacokinetics, consideration of higher doses, and operational limitations of the completed Phase 2 trial.
In addition to our plans to further evaluate setmelanotide in PWS, we also plan to pursue the development of RM‑853, our preclinical candidate GOAT inhibitor for PWS. Additionally, given setmelanotide and RM‑853’s distinct mechanisms of action, we plan to explore opportunities to evaluate the two compounds in combination, as we believe there may be complementary effects.
Setmelanotide Clinical Development in General Obesity Patients
Initial studies in general obesity provided preliminary evidence of efficacy and of good tolerability, and served as a foundation for the clinical development of setmelanotide. The general obese population is defined as having a BMI of equal to or greater than 30 kg/m2 for patients aged 16 years or older or weight greater than 97th percentile for age and sex on growth chart assessment for patients 6 to 15 years of age. The primary endpoint was the proportion of participants (aged 12 years or older) who achieved at least 10 % reduction in body weight from baseline after 52 weeks of setmelanotide treatment. Key secondary endpoints further assessed changes in body weight and hunger, and daily maximal hunger score was based on participant responses to scoring their “most” hunger during the day using a numerical rating scale ranging from 0 to 10, where 0 is not hungry at all and 10 is the hungriest possible. Recognizing that adolescents normally gain height and weight, an analysis was performed to assess changes in BMI Z scores in participants with BBS who were younger than 18. This trial enrolled 38 participants, of which 32 were confirmed to have BBS and 6 were confirmed to have Alström syndrome. Five study participants who were under 12 years old and two participants who were equal to or more than 12 years old and who discontinued before receiving active therapy were not included in the primary analysis.
In this Phase 3 trial in participants with BBS or Alström syndrome, setmelanotide was associated with significant and clinically meaningful reductions in body weight and hunger, with outcomes driven by responses in individuals with
13
BBS. Overall, this study met its primary and key secondary efficacy endpoints demonstrating that approximately 52 weeks of treatment with setmelanotide produced:
| ● | A statistically significant proportion of patients who achieved at least 10% reduction in body weight over approximately 52 weeks of treatment; |
| ● | Statistically significant and clinically meaningful reductions in mean body weight over approximately 52 weeks of treatment; |
| ● | Statistically significant and clinically meaningful reductions in the mean weekly average of the Daily Hunger Questionnaire over approximately 52 weeks of treatment, whether analyzed by most/worst score over 24 hours, average score over 24 hours, or morning hunger score; and |
| ● | A statistically significant proportion of patients who achieved an equal to or greater than 25% improvement in the weekly average of the Daily Hunger score over approximately 52 weeks, whether analyzed by most/worst score over 24 hours, average score over 24 hours, or morning hunger score. |
In the primary analysis, a total of 28 patients who had BBS were older than 12 years, 15 were adults and 13 were adolescents between the ages of 12 and 18. As presented at The Endocrine Society Annual Meeting in March 2021, data after approximately 52 weeks of treatment with setmelanotide demonstrated:
| ● | Of the 15 adult patients with BBS: |
| o | there was a -9.1% mean percentage change from baseline in BMI; |
| o | 46.7%, or seven adults, achieved at least 10% weight reduction from baseline; and |
| o | 60%, or nine adults, achieved at least 5% weight reduction from baseline; |
| ● | Of the 14 patients who were younger than 18 years old: |
| o | there was a -9.5% mean percentage change from baseline in BMI; |
| o | 85.7%, or 12 adolescents, had a reduction from baseline in BMI Z of greater than 0.2; and |
| o | there was a -0.75 point mean change from baseline in BMI Z score. |
We also presented the 14-week, placebo-controlled data from this trial at the 59th Annual European Society for Paediatric Endocrinology (ESPE) Meeting in September 2021. Patients with BBS treated with setmelanotide achieved an average BMI reduction of −1.5 kg/m2 (or a decrease of 3.8%) at Week 14 compared with patients on placebo who had negligible change from baseline (P<0.05). Treatment with setmelanotide during that 14-week period also significantly reduced the Daily Hunger Questionnaire’s Most/Worst Hunger score compared to placebo in patients in the pivotal cohort aged 12 years or older, with directionally consistent results observed when the analysis was repeated using all patients (from the pivotal and supplemental cohorts).
In November 2021, we presented the first-ever data on the health-related quality of life (HRQOL) and experience of patients with BBS at The Obesity Society’s ObesityWeek®. This research was conducted as a post-hoc analysis of data from our Phase 3 study, using the self-reported Pediatric Quality of Life Inventory (PedsQL) or the Impact of Weight on Quality of Life Questionnaire-Lite (IWQOL-Lite), both of which are 100-point scales, with zero being the worst and 100 being the best. Key findings include:
| ● | 85% of patients reported clinically meaningful improvements in their HRQOL status after one year of setmelanotide therapy, or preserved their non-impaired HRQOL status; |
| ● | In adult patients, changes in their IWQOL score were clinically meaningful with a mean increase of 12 points after one year on setmelanotide therapy from 74.9 at baseline; |
14
| ● | In pediatric patients, changes in their PedsQL score were clinically meaningful with a mean increase of 11.2 after one year on setmelanotide therapy from 67.2 at baseline; |
| ● | For the subset of patients without cognitive impairment, clinically meaningful improvements in outcomes such as body mass index and hunger mirrored their improvements in HRQOL; |
| ● | HRQOL improvements were sustained over the 52-week trial period. |
On February 16, 2022, we announced positive interim data from our long-term extension study evaluating setmelanotide in patients with BBS. Of the patients enrolled in our long-term extension trial, 19 individuals with BBS had reached 24 months on therapy. As of a data cutoff date of October 29, 2021:
•The mean percent reduction in BMI from pivotal trial baseline was -14.3% (n=19);
•The mean percent reduction in body weight from pivotal trial baseline among patients 18 years of age or older was -14.9% (n=6);
•The mean reduction in BMI Z score from pivotal trial baseline among patients younger than 18 was -0.72 (n=12). For one patient who was 17 years old when enrolled and at baseline in the pivotal trial, BMI Z score could not be calculated as this patient was 20 years old at 24 months on therapy.
Overall, 34 patients with BBS transitioned from our phase 3 pivotal trial into our long-term extension study. We anticipate reporting updated data from this trial at a medical meeting in the spring of 2022. Consistent with prior clinical observations, setmelanotide was generally well-tolerated in the long-term extension study and no new safety signals were observed.
We studied patients with Alström syndrome and severe obesity in two clinical trials, our exploratory Phase 2 Basket study and our pivotal Phase 3 study in conjunction with BBS. As stated above, our Phase 3 study included three patients with Alström syndrome in the primary analysis and none of them met the primary endpoint of equal to or greater than 10 % weight loss. However, there were signals of potential efficacy in some patients, and based on those signals and data from the Phase 2 Basket study, we believe setmelanotide has the potential to address the unmet medical need in patients with Alström syndrome, hyperphagia and severe obesity.
A total of 12 patients with Alström syndrome entered these trials, with eight of them having been exposed to setmelanotide for at least 26 weeks, and overall patients had setmelanotide exposure up to 79 weeks. As the majority (67%) of patients were less than 18 years of age and still growing in height, we believe changes in BMI Z scores are a more appropriate measure of setmelanotide’s potential effects than change in body weight. Reductions in BMI Z scores were observed in 5 of the 6 patients (or 83%) under the age of 18 years who had at least 26 weeks of exposure to setmelanotide, with robust clinically meaningful reductions greater than 0.3 points observed in 3 of these patients. Clinically meaningful reductions in body weight (equal to or greater than 5%) were observed in 2 of the 8 (or 25%) patients with Alström syndrome with at least 26 weeks on setmelanotide, with one 12-year-old patient losing more than 20% of initial body weight at baseline. With respect to hunger, 4 of the 6 (or 66.7%) patients with evaluable data demonstrated clinically meaningful reductions of 1 to 2 points in hunger, as measured by the most/worst hunger question of the Daily Hunger Questionnaire. Five of the eight patients with Alström syndrome treated for at least 26 weeks decided to continue treatment with setmelanotide in our ongoing extension study.
In addition to positive results from our pivotal Phase 3 clinical trial, we included in our regulatory submissions a series of comprehensive individual patient narratives supporting our belief that setmelanotide, has the potential to address the hyperphagia and early-onset, severe obesity that characterize these syndromes.
Additional MC4R Pathway Genetic Variants
With significant advances in gene sequencing and analysis, we have deepened our understanding of the relationship between genetics and severe obesity. We also are advancing a broad clinical development program evaluating
15
setmelanotide in several ongoing and planned clinical trials, and we are leveraging the largest known DNA database focused on obesity - with approximately 45,000 sequencing samples as of December 2021 - to improve the understanding, diagnosis and care of people living with severe obesity due to certain variants in genes associated with the MC4R pathway. There remains a significant unmet need with no effective therapeutic options for patients with these rare genetic diseases of obesity, and we believe setmelanotide has the potential to address the hyperphagia and severe obesity associated with these rare genetic diseases.
Having achieved proof-of-concept in our ongoing exploratory Phase 2 Basket Study evaluating setmelanotide in patients with severe obesity driven by variants in several different MC4R pathway associated genes, we expect to initiate in the first half of 2022, the pivotal Phase 3 EMANATE clinical trial to evaluate setmelanotide in five independent sub-studies in patients with variants in one of five specific genes within the MC4R pathway. In addition to EMANATE, we also initiated the Phase 2 DAYBREAK clinical trial to evaluate setmelanotide in patients who carry a confirmed variant in one or more of 31 genes with strong or very strong relevance to the MC4R pathway. Additionally, in our ongoing exploratory Phase 2 Basket Study, we are evaluating setmelanotide in patients with obesity arising due to heterozygote loss of function mutations in the MC4R gene itself. We also have a separate trial evaluating setmelanotide in patients with hypothalamic obesity, or HO, a severe obesity that arises after injury to the hypothalamus (such as from a tumor or the treatment of a tumor in this location). We anticipate reporting data from both of these Phase 2 studies in the first half of 2022.
Phase 3 EMANATE Trial
In the first half of 2022, we expect to initiate our pivotal Phase 3 EMANATE clinical trial, a randomized, double-blind, placebo-controlled trial, designed to evaluate setmelanotide in five independent sub-studies in patients with obesity due to: a heterozygous variant of the POMC/PCSK1 genes or LEPR gene, certain variants of the SRC1 gene or the SH2B1 gene, or PCSK1 N221D deletions within the MC4R pathway. The epidemiology estimates for the indications being studied in our Phase 3 EMANATE trial suggest that between 100,000 to 200,000 U.S. patients with one of these genetic deficiencies have the potential to respond to setmelanotide.
POMC, PCSK1 and LEPR are core genes of the MC4R pathway. Heterozygous variants in POMC, PCSK1 and LEPR have been associated with clinical obesity that may be due to a MC4R pathway dysfunction. Obesity due to rare variants in the SRC1 gene is an autosomal dominant disorder that is characterized by early-onset severe obesity and hyperphagia, as SRC1 variants found in individuals with severe obesity significantly impaired leptin-induced POMC expression (Yang et al 2019, Nat Comm. 10, Article 1718). Specifically, SRC1 is a transcriptional coactivator that has links to both the leptin receptor and to POMC. When the leptin receptor is activated, SRC1 is activated through a cascade of events that then drives the expression of POMC. Individuals who have heterozygous loss-of-function variants in their SRC1 genes can have insufficient leptin receptor activation of the MC4R pathway as a result of decreased POMC expression. This decreases the amount of available MSH to activate the MC4R, consequently resulting in hyperphagia and obesity in these individuals. Obesity due to variants in the SHWB1 gene is a rare genetic disease that is characterized by early-onset severe obesity, hyperphagia, hyperinsulinemia, and reduced final height. SH2B1 variants can arise through either DNA variants in the SH2B1 gene or through chromosomal deletions (chromosome 16) that encompass the SH2B1 gene. In both cases, dysfunction/loss of only one copy of the SH2B1 gene is sufficient to give rise to obesity and hyperphagia. The SH2B1 protein has been shown to have direct links to the MC4R-pathway. Specifically, SH2B1 is an adapter protein that amplifies the signal coming through the leptin receptor. In individuals who carry heterozygote loss of function mutations in SH2B1 or a chromosomal deletion that remove the SH2B1 from the chromosome, individuals may have insufficient leptin receptor activity activation of their MC4R pathway. This gives rise to a well-documented form of severe early-onset obesity and hyperphagia.
Proof of concept achieved in exploratory Phase 2 Basket Study
In January 2021, we announced proof-of-concept data from our exploratory Phase 2 Basket Study in multiple patient cohorts of patients with severe obesity due to a variant in one of the two alleles in the POMC, PCSK1, or LEPR genes (PPL HET obesity), as well as the SRC1 and SH2B1 genes. We provided subsequently furnished updated data in multiple presentations at medical meetings throughout 2021. The ongoing Phase 2 Basket Study is an open label study designed to evaluate setmelanotide in patients with obesity defined as Body Mass Index (BMI) ≥ 30 kg/m2 for patients 16
16
years of age or older or BMI≥ 95th percentile for age and gender for patients between 6 and 16 years old. Patients were stratified by cohort according to their genetic variant. The primary endpoint of the study is the percent of patients in each subgroup showing at least a 5% loss of body weight over three months (“clinical responders”).
PPL HET Obesity (POMC, LEPR, PCSK1) highlights included:
| ● | Overall, 12 of 35 patients (34.3%) achieved the primary endpoint. This full analysis includes six patients who withdrew early; |
| ● | Mean reduction from baseline in body weight over three months across all 35 patients was -3.7%, which includes both clinical responders and non-responders; and |
| ● | Among the 12 patients who achieved the primary endpoint (responder group), the mean reduction from baseline in body weight over three months was -10.1%. |
In our initial clinical trials,analyses, we delivered setmelanotideare applying variant classification guidelines from the American College of Medical Genetics, or ACMG (as described in Richards, et al., 2015), to patient cohort stratification. Specific variants of the POMC, LEPR, PCSK1, SRC1 or SH2B1 gene may be classified based on published data as being pathogenic, likely pathogenic, likely benign or benign, or classified as a variant of unknown significance or VOUS. As genetics of obesity remains an emerging field, the vast majority of variants in genes associated with continuous SC infusion using an insulin pump. More recently, our administration has been convertedthe MC4R pathway are classified as VOUS. Our hypothesis was that patients with genetic variants that indicate a higher degree of pathogenicity would be more likely to a once daily SC injectable formulation.have impaired pathway signaling and therefore more likely to respond to setmelanotide. In addition, we have an ongoing trialdecided to assess the pharmacokineticsstudy a cohort of a new, long‑acting formulation of setmelanotide.
The table below summarizes the setmelanotide studies that we conducted in general obese patients under IND # 112595 submitted to the Division of Metabolism and Endocrinology Products, Center for Drug Evaluation and Research, FDA.
38
Completed and Ongoing Setmelanotide Clinical Trials in the General Obese Population
|
|
|
|
| ||||
|
|
|
|
| ||||
|
|
| ||||||
| ||||||||
|
|
|
|
| ||||
|
|
| ||||||
|
| |||||||
|
|
|
|
| ||||
|
|
| ||||||
| ||||||||
| ||||||||
|
|
|
|
| ||||
|
| |||||||
| ||||||||
|
|
|
|
| ||||
|
|
|
| |||||
|
| |||||||
| ||||||||
|
|
|
|
| ||||
|
| |||||||
| ||||||||
| ||||||||
|
|
|
|
| ||||
|
| |||||||
| ||||||||
| ||||||||
| ||||||||
|
|
|
|
| ||||
|
|
|
| |||||
|
|
|
SC=subcutaneous.
Phase 2 Clinical Development in the General Obese Population
a.Phase 2 Clinical Trial Results with Continuous Infusion
We conducted our first Phase 2 clinical trial of setmelanotide using continuous SC infusion. This was a 12‑week, Phase 2 proof of concept clinical trial in general obese patients using the SC continuous infusion formulation of setmelanotide delivered by an insulin pump. We treated approximately 74 obese patients with either placebo or setmelanotide at a dose of 1.0 mg over 24 hours, with no serious adverse events or other safety indications from laboratory tests, electrocardiograms or vital signs noted in the setmelanotide treatment group. Evaluationan N221D variant of the pharmacokinetics, or blood levels, of setmelanotidePCSK1 gene. This is a common variant which has been associated with obesity in scientific and medical literature.
| ● | Patients with PPL HET obesity were stratified into three pre-specified cohorts by classification of their genetic variants according to ACMG guidelines; |
| ● | Four of eight patients (50.0%) with a pathogenic or likely pathogenic variant achieved greater than 5% weight loss over three months; |
| ● | Four of eight patients (50.0%) with the N221D variant of the PCSK1 gene achieved greater than 5% weight loss over three months; and |
| ● | Four of 19 patients (21.1%) with a variant of unknown significance (VUS) achieved greater than 5% weight loss over three months. |
In September 2021, we presented updated interim data from this clinical trial demonstrated that the SC continuous infusion method of drug administration was not optimal. A large numberSRC1 and SH2B1 cohorts at the at the 59th Annual European Society for Paediatric Endocrinology (ESPE) Meeting. The data presented were based on an interim analysis of patients did not meet the target pharmacokinetic exposureswho completed 12 weeks of therapy. These presentations included analyses that showed setmelanotide that our Phase 1 clinical trials suggested would have to be achieved in order for setmelanotide to show efficacy. This clinical trial did not demonstrate statistically significant weight loss compared to the placebo. We believe patients in this clinical trial lacked adequate exposure to setmelanotide, and concluded that all future efficacy clinical trials in obese patients should be conducted using
39
the SC injection method. This belief is based on a prior Phase 1 pharmacokinetic trial, which used the SC injection formulation and demonstrated higher pharmacokinetic exposures in obese patients.
b.Phase 2 Clinical Trial with Once Daily SC Injection
We conducted a three‑stage, randomized, placebo‑controlled, Phase 2 12‑week general obesity trial, with approximately 100 obese patients, using our SC injection formulation, primarily with once daily dosing. We designed this Phase 2 clinical trial to bridge between the earlier clinical trials that used continuous infusion and all future clinical trials that use the formulation for once daily SC injection. Therefore, the primary purpose of the staged approach in this trial was to assess if appropriate pharmacokinetic targets could be reached with the new SC injection, first in an in‑patient setting similar to the setting where robust weight loss was demonstrated in the Phase 1 general obesity trial, and then in an outpatient setting.
Overall, setmelanotide demonstrated significant weight loss over 12‑weeks in all stages, with placebo subtractedclinically meaningful weight loss or BMI Z reduction in 30% (9 of 30) of study participants with obesity due to variants of the differenceSRC1 gene and clinically meaningful weight loss or BMI Z reduction in 43% (15 of 35) of study participants with obesity due to variants of the SH2B1 gene, including 16p11.2 chromosomal deletions.
Specifically in the amountSRC1 cohort, a total of weight gained or lost30 patients with obesity and deficiency in the active treatment group as compared to the placebo treatment group, from baseline of −2.78% to −4.69% and p‑values ranging from 0.005 to <0.001. However, weight loss was more pronounced and consistentSRC1 gene were enrolled in the full analysis set of this study. These patients had a mean BMI of 45.4 kg/m2 or BMI Z of 3.0 at baseline. Highlights of these data, as of a cut-off date of March 16, 2021, include:
| ● | Nine of 30 (or 30%) of patients achieved a clinically meaningful response to setmelanotide at three months, as defined by weight loss of 5% or greater from baseline, or for patients under 18 years old, a reduction of at least 0.15 in BMI Z score: |
| o | In adult patients 18 years or older, six of 20 (or 30%) achieved 5% or greater weight loss at three months; |
17
| o | In patients younger than 18 years, three of 10 (or 30%) achieved a BMI Z reduction of 0.15% or more at three months. |
| ● | Across all enrolled patients, the mean overall weight loss from baseline to three months among patients 18 years and older (a sample of 20) was -4.0% (a standard deviation of 3.3%), and the mean overall BMI Z score reduction from baseline to three months among patients younger than 18 years (n=10) was -0.21 (a standard deviation of 0.23). |
In addition, these interim data showed a clear separation between patients who responded to setmelanotide treatment at three months and those who did not:
| ● | The mean body weight reduction for adult patients who responded (n= 6) was 7.9% (90% confidence interval (CI), −9.7 to −6.0), as compared to 2.3% (90% CI, −3.2 to −1.4) for adult patients who did not respond (a sample of 14); |
| ● | The mean BMI Z reduction for patients younger than 18 years who responded (n= 3) was 0.48 (90% CI, −0.95 to −0.01), as compared to 0.09 (90% CI, −0.11 to −0.07) for those who did not respond (n= 7). |
In the SH2B1 cohort, treateda total of 35 patients with an initial four‑week, observed dosing, inpatient period, for whichobesity and 16p11.2 deletions that include the SH2B1 gene or deficiency in the SH2B1 gene were enrolled in the full analysis set of this study. These patients had a mean BMI of 47.2 kg/m2 or BMI Z of 3.6 at baseline. Highlights of these interim data, as of a cut-off date of March 16, 2021, include:
| ● | Fifteen of 35 (or 42.9%) of patients achieved a clinically meaningful response to setmelanotide at three months, as defined by weight loss of 5% or greater from baseline, or for patients under 18 years old, a reduction of at least 0.15 in BMI Z score: |
| o | In patients 18 or older, eight of 22 (or 36.4%) achieved 5% or greater weight loss at three months; |
| o | In patients younger than 18 years, seven of 13 (or 53.8%) achieved a BMI Z reduction of 0.15% or more at three months. |
Across all enrolled patients, the mean overall placebo subtracted weight loss from baseline to three months among patients 18 years and older (n= 22) was -3.1% (a standard deviation of 3.9%), and the mean overall BMI Z score reduction from baseline to three months among patients younger than 18 years (n= 13) was -0.15 (a standard deviation of 0.13). In addition, the interim data showed a clear separation between patients who responded to setmelanotide treatment at week 12 rangedthree months and those who did not:
| ● | The mean body weight reduction for adult patients who responded (n= 8) was 7.2% (90% CI, −8.6 to −5.8), as compared to 0.8% (90% CI, −1.9 to 0.3) for adult patients who did not respond (n= 14); |
| ● | The mean BMI Z reduction for patients younger than 18 years who responded (n= 7) was 0.25 (90% CI, −0.29 to −0.21), as compared to 0.03 (90% CI, −0.08 to 0.02) patients younger than 18 years who did not respond (n= 7). |
Consistent with prior clinical experience, setmelanotide was generally well tolerated in each of these rare genetic diseases of obesity as of the cutoff date. The most common treatment-emergent adverse events, or TEAEs, included mild injection site reactions, hyperpigmentation, and nausea and vomiting, which occurred early in the treatment course. There were no SAEs related to treatment with setmelanotide.
MC4 Receptor Deficiency Obesity
In the first half of 2022, we anticipate reporting interim data from −3.87%ongoing exploratory Phase 2 Basket Study from a cohort of patients with MC4R deficiency obesity arising due to −4.69%, all with p‑valuesheterozygote loss of less than 0.005, withfunction mutations in the MC4 receptor gene itself. This is one of the most pronouncedwell-known and prevalent forms of monogenic early-onset severe obesity.
18
Based on a comprehensive ongoing biochemical screening study, we believe setmelanotide may have the potential to address MC4R loss of function in a defined subset of this broader population, specifically individuals who carry MC4R loss of function variants that we believe can be rescued by setmelanotide (e.g. may not responsive to the endogenous ligand MSH, but would otherwise respond normally to setmelanotide).
Hypothalamic Obesity
Hypothalamic obesity, or HO, is a severe obesity that arises from mechanical hypothalamic injury. Lesions of the hypothalamus can derive from various types of tumors (e.g., craniopharyngiomas, gliomas, pituitary adenomas, hamartomas) or may be caused by surgeries and/or radiotherapies for the treatment of these same tumor types. These hypothalamic lesions, whether caused by the tumor itself and/or the treatment of the tumor, can disrupt the MC4R pathway. Moreover, patients with HO display high degree of hyperleptinemia and hyperinsulinemia. Alpha-melanocortin stimulating hormone (MSH) can be detectable in blood, and its levels can change depending on different energy states; however, in patients with craniopharyngioma or post-surgical treatment for it, α-MSH levels are significantly reduced. Reduced serum α-MSH levels may suggest melanocortin pathway deficiency, which might explain obesity in these patients.
As of February 1, 2022, we have completed enrollment in our Phase 2, multi-center, open-label, proof-of-concept study designed to assess the effect of setmelanotide over 16 weeks on weight-related assessments in patients aged 6 to 28 years who are affected by HO. We enrolled more than 15 subjects across 3-5 clinical sites in the United States. The primary endpoint is the percentage of subjects aged >12 years with ≥5 % body weight loss duringfrom baseline to week 16, compared to a historic control of <5 % in this subject population. All enrolled patients will have obesity, defined as a BMI ≥35 kg/m2 for subjects ≥16 years of age or BMI ≥99th percentile for age and gender for subjects 6 to <16 years of age based on the in‑patient period. The once daily SC injection formulation also showed consistentU.S. Centers for Disease Control and predictable pharmacokinetic measurements during the four‑week inpatient intervalPrevention criteria.
We anticipate announcing results from a preliminary data analysis from this trial in the first stage, validating the characteristicshalf of the SC injection formulation. However, this trial demonstrated challenges in drug administration and compliance when administered in an outpatient setting in the general obese population.2022.
Setmelanotide Phase 2 SC Injection Trial 4‑week In‑Patient Dosing Period: Percent Weight Loss for
Setmelanotide 1.5 mg/day SC injection vs Placebo over 26 daysWeekly Formulation of Observed Dosing
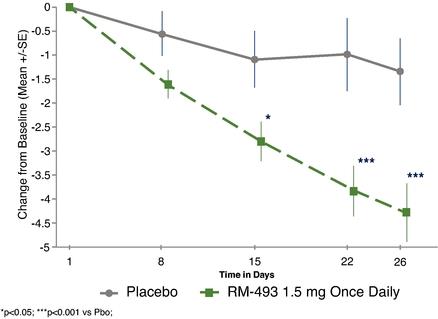
Phase 1 Clinical Development in the General Obese Population
We have completed a Phase 1 single‑ascending dose, or SAD, clinical trial of setmelanotide, as well as five cohorts in a Phase 1 multiple‑ascending dose, or MAD, clinical trial of setmelanotide. Both clinical trials were in healthy obese subjects, and included a double‑blind, placebo‑controlled randomized escalating dose design. Subjects received
40
treatment in these Phase 1 clinical trials for one day at doses up to 0.1 mg/kg/day, which is a total daily dose of approximately 10 mg/day, and for up to 28 days at doses up to 0.015 mg/kg/day, which is a total daily dose of approximately 1.5 mg/day.Setmelanotide
In the SAD clinical trial, our extensive monitoring of heart rate and blood pressure did not demonstrate any clinically meaningful changes with setmelanotide treatment compared with placebo. Similarly, in the MAD clinical trial, there was no evidence of any notable changes in cardiovascular parameters compared to placebo when assessed by 24‑hour ambulatory blood pressure monitoring, or ABPM. We determined that the terminal half‑life of setmelanotide is approximately nine to ten hours, making it suitable for once daily dosing.
Four cohorts of the Phase 1 MAD clinical trial that included doses of greater than 0.01 mg/kg/day, which is approximately 1 mg/day, for two to four weeks, demonstrated placebo subtracted weight loss differences. Most panels showed statistically significant, placebo subtracted weight reduction that ranged from 0.6 to 1.4 kg/week, with a mean of approximately 0.9 kg/week over the two to four weeks of treatment in Phase 1.
Setmelanotide: Phase 1b General Obesity Patients: Placebo Subtracted Differences(1)(2)
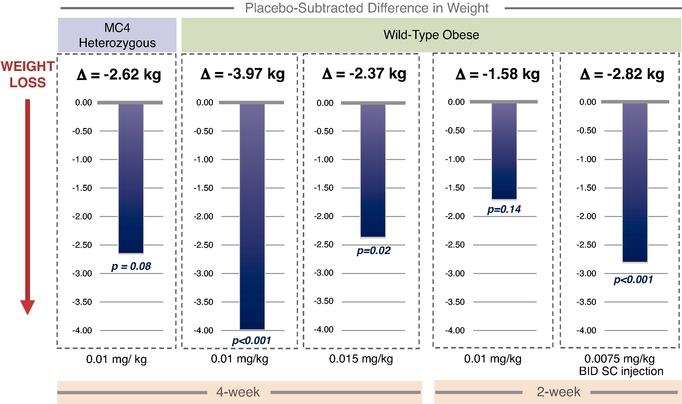
|
|
|
|
Δ = Placebo subtracted weight loss from baseline.
BID = Two times per day.
41
Phase 1 Energy Expenditure Clinical Trial
In collaboration with the National Institute of Diabetes, Digestive and Kidney Diseases, we investigated setmelanotide in a Phase 1 clinical trial to determine the effects of setmelanotide on energy expenditure, a mechanism for weight loss, in addition to the well‑known effects of MC4R agonists on appetite and food intake. Twelve obese adults were randomized to receive setmelanotide or placebo by continuous SC infusion over 72 hours, followed immediately by crossover to the other treatment. Setmelanotide showed statistically significant 6.85% increases in resting energy expenditure, supporting a role for setmelanotide in weight regulation. This trial provided the first clinical demonstration that MC4R activation with setmelanotide increases resting energy expenditure in obese humans.
Long‑Acting Setmelanotide Pharmacokinetic Trial
In addition to developing the once daily SC injectable formulation of setmelanotide that we are using in our ongoing clinical trials, in collaboration with Camurus AB, or Camurus, we have developed a once weekly, long‑actingonce-weekly, long-acting formulation of setmelanotide using FluidCrystal® technology. When injected subcutaneously, aqueous body fluid ismay be absorbed by the excipient lipid phase, which formsmay then form a gel‑likegel-like depot consisting of liquid crystals formed in situ leading to slow diffusion of setmelanotide from the depot. We believe that this formulation may be more convenient and less burdensome for patients and their families.
We have compelling preclinicalIn October 2021, we presented full data from our trial that evaluated the weekly formulation of setmelanotide in patients with obesity at the Obesity Medicine Association’s (OMA) Overcoming Obesity 2021 Conference. The data showed that otherwise healthy people with obesity treated with the long‑acting formulation:weekly formulation of setmelanotide (QW) achieved comparable weight loss to those treated with the daily formulation, and that both weekly and daily formulations of setmelanotide (QD) were observed to be generally well tolerated. A total of 82 individuals aged 18 to 55 years with body mass index (BMI) ≥35 kg/m2 were included in monkeys, the terminal half‑lifedata analysis. Based on the data from the study, we concluded the safety profile of QW setmelanotide was similar to that of the long‑actingQD formulation; in healthy volunteers, peak and trough QW setmelanotide concentrations were generally consistent with the concentrations of the QD formulation; and weight and hunger change at Week 12 was comparable between QW and QD setmelanotide. Notably, the weight and hunger score changes in otherwise healthy individuals with obesity receiving both formulations were lower than those reported separately in patients with rare genetic diseases of obesity associated with an impaired MC4R pathway who received setmelanotide. Additionally, pharmacokinetic, or PK, analyses showed similar trough drug concentrations for the daily and weekly formulations over the duration of therapy. The weekly formulation is approximately 105 hours,of setmelanotide demonstrated a consistent 24-hour PK range and in rats, approximately 92 hours. Two‑was detected steadily over one week, toxicology studies in rats have been completed,with a trough concentration consistent with the trough concentration of the daily formulation.
Weekly setmelanotide administration was generally well tolerated, with no serious TEAEs, and the long‑acting formulation was well tolerated. Duringsafety results were similar to the two‑week dosing period, animals given setmelanotide had dose‑related, statistically significant lower body weights, from −9.8% to −11.7%, compared to those given placebo controls. Food consumption for animals given setmelanotide was also lower compared to controls, which decreased by approximately −20.5%.
Two parts of this clinical pharmacokinetic trial are completed, defining the single‑dosedaily administration and multiple‑dose pharmacokinetics of this formulation. The first part, Part A, is an ascending‑dose, placebo‑controlled, up to three sequential panel PK trial, and PK and safety/tolerability will be collected for approximately 14 days. Dose for the three panels will range from 2.5 mg up to 30 mg given as a single SC injection. The second part, Part B, is a placebo‑controlled, single panel of 12 normal healthy obese patients who received four once‑weekly injections of 10 mg setmelanotide long‑acting formulation.
The results from Part A demonstrate that a 10 mg single subcutaneous dose showed a profile that was consistent with onceprior clinical experience. The most commonly reported TEAEs, rates of which were generally similar between individuals treated with the weekly dosing withand daily formulations, included
19
injection site reaction, hyperpigmentation, nausea, headache and vomiting. Of the 249 reported injection site reactions, 247 (≥99%) were classified as mild.
In December 2021, we initiated a mean pharmacokinetic half‑life of 123 hours. Following the completion of the single‑dose part, we completed Part B, with multiple dosing in orderPhase 3 trial to evaluate the extended‑release, once‑weekly formulation of setmelanotide in patients with rare genetic diseases of obesity. The weekly switch trial is a randomized, double-blind switch trial in patients with obesity due to biallelic or heterozygous POMC, PCSK1 or LEPR deficiency or a clinical diagnosis of BBS with genetic confirmation, who were previously enrolled in our long-term, open-label extension trial. We expect to enroll 30 patients, randomized 1:1 to receive once-weekly setmelanotide and once-daily placebo, or once-daily setmelanotide and once-weekly placebo for 13 weeks. Following the 13-week randomized treatment period, patients will crossover to an open-label, 13-week study in which all patients will receive once-weekly setmelanotide. Multiple dosingThe primary efficacy endpoint is a responder analysis, based on the proportion of patients with no weight gain defined as a change of 5 % or less from baseline to week 13.
In addition, we plan to initiate a second trial to evaluate the weekly formulation demonstrated tolerability and pharmacokinetics that support further clinical development. While this data is preliminary, and this formulation is anticipated to be only ready for submission in 2021, or later, this simpler dosing regimen may provide improvements in patient convenience, and may provide additional advantagesof setmelanotide in the pediatric population.second half of 2022. This de novo trial will be a randomized, double-blind Phase 3 clinical trial in patients with BBS who live outside the United States. We expect to enroll 40 patients, randomized 1:1 to receive 30 mg of setmelanotide or placebo once weekly for 18 weeks. Following the 18-week treatment period, patients will continue on treatment, or crossover from placebo to active therapy, for an additional 14 weeks. The primary efficacy endpoint is the mean change from baseline in body weight after approximately 18 weeks of once weekly dosing.
42
Mean setmelanotide concentrations (ng/mL) in plasma during weeks 1 and 4 following 10 mg subcutaneous weekly injections of a Camurus formulation
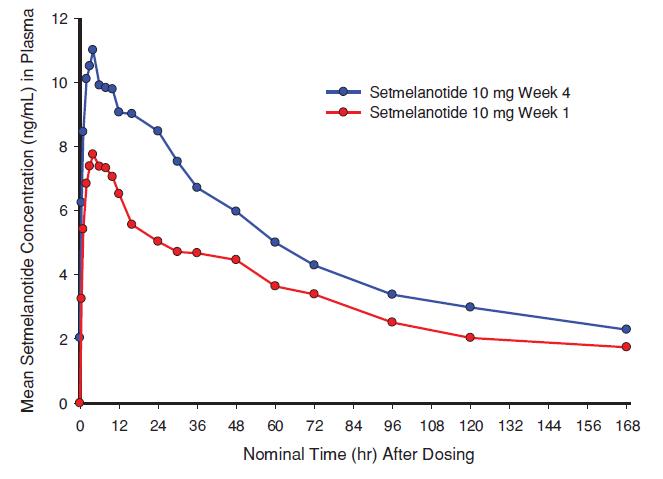
Safety and Tolerability Results
Historically, clinical data with other MC4R therapies suggested that MC4R‑mediatedMC4R-mediated side effects may include changes in blood pressure and heart rate, increased erections in males, changes in libido and sexual function in females, and nausea and vomiting. As a result, primarily due to concerns about blood pressure and heart rate changes, nonewe are not aware of these therapies have proceeded to commercialization and noany other MC4R agonists are currently in the clinic for the treatment of obesity and/or hyperphagia. It is noteworthy that the pattern of effects differed among each of the other MC4R therapies, underscoring the complex physiology of MC4R. With setmelanotide, there has been little, if any, evidence of blood pressure or heart rate changes, preliminarily supporting an important differentiation of setmelanotide from previous MC4R therapies. Careful monitoringMonitoring for blood pressure and heart rate changes, as well as other potential adverse events, or AEs, is included in all setmelanotide clinical trials.
Because of these first generation MC4MC4R therapy failures, the setmelanotide program employed an intensive preclinical screening program to assess clinical candidates for blood pressure and heart rate effects, along with efficacy. The cornerstone of this preclinical screening program was a significant investment in obese primate studies which validated setmelanotide as a promising compound for clinical development. More recently, new research supporting a unique mechanism of action of setmelanotide, compared to earlier MC4R agonists and the endogenous ligand MSH, was published in May 2018 in Nature Medicine.
43
Medicine.
Setmelanotide was generally well tolerated in our Phase 1, Phase 2 and Phase 23 clinical trials and in very preliminary data from our ongoing Phase 3 trials.to date. Overall, except as outlined below, the number and patterns of adverse events wasAEs were generally low, and the intensity of the adverse eventsAEs was generally mild, and infrequently led to clinical trial discontinuation.
There has been only a single serious adverse event possibly attributed to setmelanotide in our clinical trials. In our Phase 2 clinical trial with once daily SC injection, one patient was hospitalized for unusual chest pain, but no evidence of any serious respiratory or cardiac cause was found after careful evaluation, and the event was attributed to musculoskeletal pain. There were no treatment‑related changes in physical examination, except as noted below, and few, if any, clinically relevant changes in electrocardiograms, laboratory data and/or anti‑drug antibodies. Overall, there have been ten other serious adverse events in the full development program, in addition to the serious adverse event described above: seven others on setmelanotide, mostly from open label trials, including left arm numbness, influenza immunization reaction, pancreatitis secondary to pre‑existing gallstones, pneumonia, hypoglycemia due to acute adrenal insufficiency, due to non‑compliance with hydrocortisone regimen, a depressive episode with elective admission to a psychiatry ward in a patient with history of depression and who continued on treatment, and rotavirus‑related gastroenteritis. There were also three serious adverse events during treatment with placebo, consisting of biliary dyskinesia, severe groin strain, and pelvic inflammatory disease. None of these serious adverse events was considered related to setmelanotide treatment.
To demonstrate that setmelanotide has the potential to provide a safe cardiovascular profile, we extensively validated setmelanotide in obese primate preclinical studies, with special attention to cardiovascular effects. The results of these studies supported testing in clinical trials. In the clinical trials, we monitored blood pressure and heart rate extensively, primarily by 24‑hour ABPM. In most clinical trials, there were multiple 24‑hour ABPM periods, both on a pre‑treatment and post‑treatment basis. Trial‑by‑trial review of the 24‑hour ABPM data shows little, if any, evidence of changes in heart rate and/or blood pressure even at the highest doses tested in Phase 1 and Phase 2 clinical trials. We have also conducted an analysis of 24‑hour ABPMs that were obtained pre‑dose and post‑dose across completed studies, which was presented at the Obesity Society in 2015. This included 128 patients, of which 79 were active and 49 were on a placebo. Overall, there was little, if any, evidence of blood pressure or heart rate changes evident from baseline versus placebo in any trial, preliminarily supporting an important differentiation of setmelanotide from previous MC4 therapies. While the preliminary data are encouraging, there will be continued focus on potential cardiovascular risk until addressed in larger and longer clinical trials.
44
Setmelanotide Phase 2 SC Injection Trial: 24‑hr ABPM (All Studied Patients),
Showing No Adverse Effect of Setmelanotide on Blood Pressure or Heart Rate
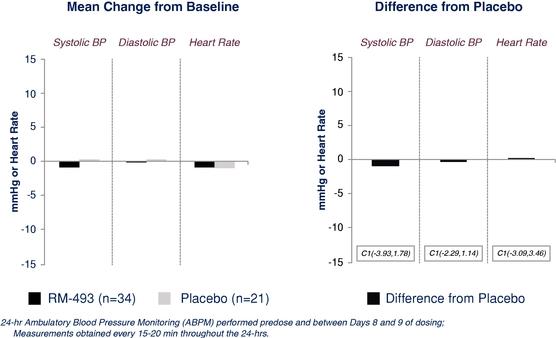
In the majority of our trials, there waswe observed a small increase in frequency of penile erections in male patients, as well as signs of sexual arousal in a small number of female patients. These symptoms were infrequent, generally mild, not painful, and short‑lived.short-lived. Most often these symptoms were reported in the first week of treatment. There was a small incidence of nausea and vomiting, as well as injection site reactions, both of which usually were reported as mild, early in treatment, and short‑lived.short-lived. A small number of patients had dose reductions and/or discontinued treatment due to nausea and vomiting.
We also noted darkening of skin and skin lesions, such as moles and freckles, in mostapproximately half of the patients who received setmelanotide. This was likely caused by activation of the closely related MC1 receptor, the receptor that mediates skin darkening in response to sun exposure. This was observed generally after one to two weeks of treatment,
20
most often plateaued by two to four weeks of treatment, and like sun‑relatedsun-related tanning, generally returned to baseline after cessation of exposure.
Additional adverse events noted at somewhat greater rates numerically forOverall, the most common AEs reported among setmelanotide comparedtreated patients have been skin hyperpigmentation, injection site reactions, nausea, headache, vomiting, decreased appetite, and diarrhea.
Life-Cycle Management and Preclinical Development
We continue to placebo acrossadvance the clinical program include fatigue and related symptoms, diarrhea, arthralgia, back pain and headache, but most investigators reported these effects to be unrelated to setmelanotide.
While general obese patients are not currently the focus of setmelanotide studies, the FDA and EMA consider the risk and benefit information observed to date with setmelanotide in general obese patients to be supportive of the continued development of this therapy. These data from general obese patients do not raise any new safety concerns and suggest that substantial benefit, as evidenced by weight loss, is possible.
Preclinical Development
Preclinical studies demonstrated the efficacy of setmelanotide in suppressing food intake and body weight gain in diet‑induced obese mice, rats, dogs, and rhesus macaques, as well as in genetic models of obesity, including leptin‑deficient ob/ob mice and obese Zucker, or fa/fa (leptin‑receptor deficient), rats. Furthermore, setmelanotide is associated with restoring insulin sensitivity in nonclinical models of obesity in rodents and lowering of plasma triglycerides, cholesterol, and free fatty acids.
45
In particular, we demonstrated activity in obese non‑human primates, where approximately 13% weight loss was demonstrated with eight weeks of treatment, without evidence of cardiovascular toxicity. We also studied obese primates in crossover studies to confirm the lack of cardiovascular toxicity by setmelanotide in obese primates. These preclinical studies also confirmed the cardiovascular effects of previous MC4 therapies that had produced cardiovascular toxicity in humans. In contrast, setmelanotide was without cardiovascular effects in head‑to‑head studies.
Lastly, the toxicology program to support the NDA filingour once-weekly formulation of setmelanotide for POMC deficiency obesityall indications in which setmelanotide is completed. We completed three‑month toxicology studiesapproved or in rats and monkeys, with doses and exposures that are more than 300‑fold greater than those at the anticipated clinical doses without evidence of clinically relevant toxicological findings. Similarly, we have also completed chronic toxicity studies (6‑month rat, 9‑month monkey), which in rats provided 219‑ (maximum concentration) and 106‑times (area under the curve), respectively, and in monkeys 282‑ and 82‑times, respectively, the exposures at the anticipated clinical doses compared to the No‑Observed‑Adverse‑Effect‑Level(s) in animals. We have evaluated the potential reproductive and development effects of setmelanotide in rats and rabbits with administration by SC injection, to support the administration of setmelanotide in women of child‑bearing potential. In addition, a juvenile toxicology study has been completed that will support dosing in pediatric patients less than 12 years of age.development. In addition, we are planning carcinogenicity studies, the longest of which is expected to be two years. The FDA has allowed us to defer carcinogenicity studies until after approvalhave initiated development of an NDAauto-injector device designed to make administration of our once-weekly product candidate easier and more convenient for setmelanotide. We believe this also to be true for the EMA, however, the EMA has not yet provided firm guidance on the need for submission of results of one or more carcinogenicity studies at the time of filing of an application for marketing authorizationour patients.
IMCIVREE is approved in the EU.
RM‑853, a Preclinical Ghrelin O‑Acyltransferase Inhibitor
In addition to our development of setmelanotide, in April 2018, we announced that we had acquired exclusive, worldwide rights from Takeda to developUnited States and commercialize RM‑853. RM‑853 is a potent, orally available GOAT inhibitor currently in preclinical development for PWS. PWS is a rare genetic disorder that results in hyperphagia and early‑onset, life‑threatening obesity, for which there are no approved therapeutic options. RM‑853 is currently in pre‑clinical development. We anticipate filing an IND for RM‑853 with the FDA in 2020.
Ghrelin is an orexigenic peptide, secreted by the stomach and proximal small intestine in response to a negative energy balance. Ghrelin can play a key physiological role in stimulating appetite and promoting food intake, thereby maintaining overall energy balance. In people living with PWS, levels of active ghrelin are elevated, contributing to hyperphagia, which leads to severe obesity. RM‑853 is designed to block GOAT, the key enzyme involvedauthorized in the productionEU and Great Britain for patients 6 years of the active formage and older. Rare genetic diseases of ghrelin,obesity often present early in life, and we have identified children with the expected effectgenetic obesity under 6 years of lowering active ghrelin levels. This blockage also increases the levels of des‑acyl‑ghrelin, or DAG, a ghrelin precursor; high levels of DAG are believed to have independent beneficial effects on the control of appetite and tissue homeostasis, which might add to the potential efficacy of RM‑853 in PWS.age who we believe may potentially benefit from treatment with setmelanotide. In preclinical research, RM‑853 prevented body weight gain and reduced fat mass in high fat‑fed mice, with a favorable pharmacokinetic, pharmacodynamic, and safety profile. We plan to complete preclinical studies of RM‑853 and file an IND with the FDA in approximately the first quarter of 2020. Under the terms2022, we announced initiation of the agreement,Phase 3 trial evaluating daily setmelanotide in pediatric patients who are between the ages of 2 and 6 years old. The trial is a multi-center, one-year, open-label Phase 3 trial in pediatric patients with obesity due to biallelic POMC, PCSK1 or LEPR deficiency or a clinical diagnosis of BBS with genetic confirmation. The primary efficacy endpoint is a responder analysis, based on the proportion of patients who experience a decrease from baseline over one year in BMI Z of ≥0.2. In addition, we will assume sole responsibilityhave initiated discussions with the EMA to modify our EMA-approved Pediatric Investigation Plan (PIP) to be consistent with our plans for the treatment of these younger patients. We expect to meet all our PIP requirements by 2024.
We have initiated a program to identify next generation MC4R agonists based on the setmelanotide chemical space that we believe will have the potential to avoid cardiovascular AEs and MC1R activation, the latter of which results in hyperpigmentation. This program is expected to result in a clinical development candidate that will be an MC4R agonist matching setmelanotide’s cardiovascular safety but without the potential to cause hyperpigmentation. We expect to identify a lead compound from this program for preclinical development in 2022.
Genetic Sequencing and Patient Finding
We continue to expand our sequencing efforts in individuals living with early-onset, severe obesity to support research, patient finding and community building efforts in order to better understand rare genetic diseases of obesity. Our obesity DNA database contains sequencing samples from approximately 45,000 individuals, and we are using these data to support research, patient finding and community building while forging a better understanding of rare genetic diseases of obesity. Our sequencing data has come from four distinct sources in recent years: the Genetic Obesity ID | Genotyping Study, a global product developmentnetwork of collaborations with obesity researchers with individual sample collections, institutional biobanks and Uncovering Rare Obesity or URO. By bringing additional awareness to these rare genetic diseases of obesity, our sequencing efforts have the potential to help foster patient communities and drive medical action in these populations.
Uncovering Rare Obesity
Moving forward, we expect that Uncovering Rare Obesity, or URO, our free genetic testing program designed to help determine if individuals have an underlying genetic cause of their severe obesity, will become the primary driver of how we collect sequencing samples and identify patients. As severe obesity is epidemic in the United States, we are focused on identifying people with early-onset obesity that may be caused by certain rare genetic variants. As part of these efforts, we have launched Uncovering Rare Obesity in order to increase access to genetic testing. As of December 31, 2021, 2,758 United States health care providers have requested 28,171 Uncovering Rare Obesity kits, and 13,029 sequencing tests have been ordered and patient samples collected.
This program complements several initiatives designed to advance the understanding of genetic causes of severe obesity, and Uncovering Rare Obesity broadens these efforts and brings access to genetic testing into the community setting. Currently available physician-ordered genetic testing panels are often cost prohibitive, while many consumer genetic tests are incomplete when it comes to genetic disorders of obesity. This makes it difficult to confirm an underlying
21
genetic cause of severe obesity. We believe the program marks an important step in the understanding of these disorders that might help patients and their families find new diagnosis and treatment strategies in the years ahead.
Our partner Prevention Genetics, a Clinical Laboratory Improvement Amendments-College of American Pathologists of CLIA/CAP-certified independent laboratory, conducts the genetic testing for Uncovering Rare Obesity. This program covers the cost of the test and excludes office visit, copay, sample collection, and any other related costs to a participant. In addition, as part of the program, licensed genetic counselors from PWN Health, a leading provider of professional guidance for diagnostic and genetic testing, are available to advise participating individuals.
Commercial Efforts for IMCIVREE
We are focused on making IMCIVREE available globally. In the United States, we have executed on our strategy of making IMCIVREE commercially available to patients with obesity due to POMC, PCSK1 or LEPR deficiency, and we are building out the infrastructure and teams in preparation for a commercial launch for patients with BBS and potentially Alstrom syndrome, if our sNDA is approved by FDA. We expect IMCIVREE will be commercially available in the key European markets, UK and Israel in 2022. Although the total number of patients potentially addressable by setmelanotide may not be so rare, individually populations with each of these MC4R pathway-related genetic variants are rare and affected patients face many of the same challenges as any classically rare patient population. There is little or no awareness about these rare genetic diseases of obesity, and the patients suffering from them are lost in the health care system, with limited educational resources and no effective treatments for their condition. All our efforts and services described above are designed to address the challenges of rare diseases and lay the groundwork for potential future launches, with a focus on scalability.
We are working with the broader community of physicians, patients and families to improve the path to an accurate diagnosis as we support disease education through our medical and commercial team efforts. Additionally, we have patient support programs in place to provide support to patients, including genetic counseling, reimbursement support inclusive of co-pay and patient assistance programs, and IMCIVREE injection training.
The ongoing efforts outlined to support the POMC, PCSK1 and LEPR launch lay the foundation for future commercialization efforts, including potential for BBS and Alström if setmelanotide is ultimately approved in these indications. All disease education efforts supporting awareness of RM‑853.rare genetic diseases of obesity and testing will also uncover BBS and Alström patients. In addition, compared to POMC, PCSK1 and LEPR, BBS and Alström syndrome are syndromic diseases where patients suffer from multiple symptoms beyond early-onset obesity and hyperphagia. This allows for a tailored approach to disease education efforts to differentiate individuals with BBS and Alström syndrome from the broader population with obesity.
Following marketing authorization by both the European Commission and Great Britain’s MHRA for biallelic POMC, PCSK1 and LEPR deficiency, we are focused on achieving market access and reimbursement for IMCIVREE with country level authorities. As of March 1, 2022:
| ● | The German Federal Joint Committee (G-BA) has unanimously voted to exclude IMCIVREE from its lifestyle exemption list for POMC, PCSK1and LEPR deficiency obesities. In Germany, drugs classified as lifestyle drugs which including those designed to effect weight loss, smoking cessation or hair loss are not eligible for reimbursement. This first-ever exclusion marks an important recognition that IMCIVREE is designed to treat rare genetic diseases that manifest as obesity and that this group of diseases is distinct from general obesity. With this exemption status, IMCIVREE would be eligible for national coverage and reimbursement. We expect first commercial sales in Germany in the second quarter of 2022. |
| ● | The French Haute Autorité de Santé (HAS) has granted paid early access for IMCIVREE for patients with POMC, PCSK1 or LEPR deficiency obesity which we expect to commence in the second quarter of 2022. |
| ● | We have submitted reimbursement dossiers in seven countries, including France, Germany, Italy, Spain, the Netherlands, Israel, and the United Kingdom, with positive feedback from EU payers and favorable HTA ratings from authorities in several countries. |
22
We have established initial partnerships in Asia and the Middle East, and we are actively exploring opportunities to provide setmelanotide to patients in South America.
Competition
The biotechnology and pharmaceutical industries are intensely competitive and subject to rapid and significant technological change. We have competitors in a number of jurisdictions, many of which have substantially greater name recognition, commercial infrastructures and financial, technical and personnel resources than we have. Established competitors may invest heavily to quickly discover and develop compounds that could make setmelanotide obsolete or uneconomical. Any new product that competes with an approved product may need to demonstrate compelling advantages in efficacy, convenience, tolerability and safety to be commercially successful. Other competitive factors, including generic competition, could force us to lower prices or could result in reduced sales. In addition, new products developed by others could emerge as competitors to setmelanotide. If we are not able to compete effectively against our current and future competitors, our business will not grow, and our financial condition and operations will suffer.
46
ThereCurrently, IMCIVREE is the only approved treatment for weight management in patients with obesity due to POMC, PCSK1 or LEPR deficiencies, and there are no current pharmacologicalapproved treatments for regulating hunger and hyperphagia‑hyperphagia related behaviors of patients with PWS. In contrast withearly-onset, severe obesity and hyperphagia that are the absenceresult of companies who have disclosed efforts to study upstream disordersgenetic variants affecting the MC4R pathway, including BBS, Alström syndrome, certain variants of the MC4 pathway, we are awarePOMC, PCSK1, LEPR, SRC1 and SH2B1 genes, as well as MC4R deficiency obesity. Bariatric surgery is not an appropriate treatment option for these genetic diseases of several companies investigating or developing therapies intended to treat hungerobesity because the severe obesity and hyperphagia associated with PWS. The different companiesthese diseases are considered to be risk factors for bariatric surgery. Also, existing therapies indicated for general obesity, including glucagon-like peptide-1 (GLP-1) receptor agonists, such as Wegovy®, and compoundsglucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) agonists, such as tirzepatide which is being investigated as a treatment for obesity, do not specifically restore function impaired by genetic deficiencies in development, ofthe MC4R pathway, which we are aware, involve multiple mechanismsbelieve is the root cause of action. The companieshyperphagia and their compounds include; Millendo Therapeutics Inc. (AZP‑531), Soleno Therapeutics (Diazoxide Choline Controlled Release), Zafgen (ZGN‑1258), GLWL Research Inc. (GLWL‑01), Insys Therapeutics Inc. (Oral Cannabidiol Solution) and Calm Therapeutics.obesity in patients with MC4R genetic variants.
Licensing Agreements
Ipsen Pharma S.A.S.
In February 2010, the Predecessor Company entered intoPursuant to a license agreement with Ipsen Pharma S.A.S., or Ipsen, pursuant to which Ipsen granted to itwe have an exclusive, sublicensable, worldwide license to certain patents and other intellectual property rights to research, develop, and commercialize compounds that were discovered or researched by Ipsen in the course of conducting its MC4MC4R program or that otherwise were covered by the licensed patents. Rights under the license included the right to research, develop and commercialize setmelanotide. Pursuant to the license, Ipsen also granted to the Predecessor Companywe have a non‑exclusive,non-exclusive, sublicensable, worldwide license to certain patents and other intellectual property rights that were licensed by Ipsen from a third party or that Ipsen may develop in the future to research, develop, and commercialize any of the compounds exclusively licensed by Ipsen pursuant to the license. See “Item 7. Management's Discussion and Analysis of Financial Condition and Results of Operations—Corporate Background and Distribution.”
On March 21, 2013, the LLC entity completed the Corporate Reorganization pursuant to which, among other things, the existing license with Ipsen with respect to the MC4 program is now held separately by us. As a result we hold the rights to the MC4 program, including the rights to develop and commercialize setmelanotide. See “Item 7. Management's Discussion and Analysis of Financial Condition and Results of Operations—Corporate Background and Distribution.”
Under the terms of the Ipsen license agreement, Ipsen willis eligible to receive payments of up to $40.0 million upon the achievement of certain development and commercial milestones in connection with the development, regulatory approval and commercialization of applicable licensed products, and royalties on future sales of the licensed products. Substantially all of the aggregate payments under the Ipsen license agreement are for milestones that may be achieved no earlier than first commercial sale of the applicable licensed product.product, and as of December 31, 2021, we have paid $4.0 million in clinical and regulatory milestones and $5.0 million in commercial milestones. Royalties in the mid‑singlemid-single digits on future sales of the applicable licensed products will be due under the Ipsen license agreement on a licensed product‑by‑licensedproduct-by-licensed product and country‑by‑countrycountry-by-country basis until the later of the date when sales of a licensed product in a particular country are no longer covered by patent rights licensed pursuant to the Ipsen license agreement and the tenth anniversary of the date of the first commercial sale of the applicable licensed product in the applicable country. The term of the Ipsen license agreement continues until the expiration of the applicable royalty term on a country‑by‑countrycountry-by-country and product‑by‑product-by- product basis. Upon expiration of the term of the agreement, the licensed rights granted to us under the agreement, to the extent they remain in effect at the time of expiration, will thereafter become irrevocable, perpetual and fully paid‑uppaid-up licenses that survive the expiration of the term. We have a right to terminate the license agreement at any time during the term for any reason on 180 days’ written notice to Ipsen. Ipsen has a right to terminate the agreement prior to expiration
23
of its term for our material breach of the agreement, our failure to initiate or complete development of a licensed product or our bringing an action seeking to have an Ipsen license patent right declared invalid. Upon any early termination of the license agreement not due to Ipsen’s material breach, all licensed rights granted under the license agreement will terminate.
Camurus
In January 2016, we entered into a license agreement for the use of Camurus’ drug delivery technology, FluidCrystal, to formulate setmelanotide with Camurus. Under the terms of the agreement, Camurus granted us a worldwide license to the FluidCrystal technology to formulate setmelanotide and to develop, manufacture, and commercialize this new formulation for once‑weeklyonce-weekly dosing, administered as a SC injection. The license granted to us is specific to the FluidCrystal technology incorporating setmelanotide. Under the terms of the license agreement, we are responsible for manufacturing, development, and commercialization of the setmelanotide FluidCrystal formulation
47
worldwide. Camurus received a non‑refundablenon-refundable and non‑creditablenon-creditable upfront payment of $0.5 million in January 2016, and is eligible to receive progressive payments of approximately $65.0 million, of which the majority are sales milestones. As of December 31, 2021, we have made $1.3 million of milestone payments to Camrus. In addition, Camurus is eligible to receive tiered, mid to mid‑high, single digitmid-high, single-digit royalties on future sales of the product.
The term of the agreement continues until the expiration of the applicable royalty term on a country‑by‑countrycountry-by-country and product‑by‑productproduct-by-product basis. Upon expiration of the term of the agreement, the licensed rights granted to us under the agreement, to the extent they remain in effect at the time of expiration, will thereafter become irrevocable, perpetual and fully paid‑uppaid-up licenses that survive the expiration of the term. We have a right to terminate the license agreement at any time during the term for any reason upon 90 days’ written notice to Camurus. Camurus has a right to terminate the agreement prior to expiration of its term for our material breach of the agreement, if we voluntarily or involuntarily file for bankruptcy, or for our bringing an action seeking to have a Camurus license patent right declared invalid. Upon any early termination of the license agreement not due to Camurus’ material breach, all licensed rights granted under the license agreement will terminate.
Takeda
In March 2018, we acquired exclusive, worldwide rights from Takeda to develop and commercialize RM‑853. RM‑853RM-853. RM-853 is a potent, orally available GOAT inhibitor currently in preclinical development for Prader-Willi Syndrome, or PWS. PWS is a rare genetic disorder that results in hyperphagia and early‑onset, life‑threateningearly-onset, life-threatening obesity, for which there are no approved therapeutic options. We will assume sole responsibility for the global product development and commercialization of RM‑853.RM-853. Takeda received an upfront fee of $4.4 million which we settled in April 2018 with shares of our common stock, and is eligible to receive milestone payments of approximately $140.0 million, most of which are payable upon regulatory approval or are sales milestones. In addition, Takeda is eligible to receive back‑endback-end development milestones, and single‑digitsingle-digit royalties on future RM‑853RM-853 sales.
Among other obligations under our agreement with Takeda, Takeda has a right of first negotiation under certain circumstances to sublicense the assets we acquired from Takeda in the territory of Japan. This right of first negotiation remains in effect until the earlier of five years from the date of the agreement, consummation of a change in control, or sublicense to a third party. This may delay or limit our ability to enter into certain transactions with respect to this product candidate.
The term of the agreement continues until the expiration of the applicable royalty term on a country‑by‑countrycountry-by-country and product‑by‑productproduct-by-product basis. Upon expiration of the term of the agreement, the licensed rights granted to us under the agreement, to the extent they remain in effect at the time of expiration, will thereafter become irrevocable, perpetual and fully paid‑uppaid-up licenses that survive the expiration of the term. We have a right to terminate the license agreement at any time during the term for any reason upon 90 days’ written notice to Takeda. Takeda has a right to terminate the agreement prior to expiration of its term for our material breach of the agreement, if we voluntarily or involuntarily file for bankruptcy, or for our bringing an action seeking to have a Takeda license patent right declared invalid. Upon any early termination of the license agreement not due to Takeda’s material breach, all licensed rights granted under the license agreement will terminate.
Commercial Operations24
Our commercial strategies center around creatingRareStone Group Ltd.
In December 2021, we entered into an Exclusive License Agreement with RareStone, or the RareStone License. Pursuant to the RareStone License, we granted to RareStone an exclusive, sublicensable, royalty-bearing license under certain patent rights and know-how to develop, manufacture, commercialize and otherwise exploit any pharmaceutical product that contains setmelanotide in the diagnosis, treatment or prevention of conditions and diseases in humans in China, including mainland China, Hong Kong and Macao. RareStone has a well‑informed, supportive geneticright of first negotiation in the event that Rhythm chooses to grant a license to develop or commercialize the licensed product in Taiwan.
According to the terms of the RareStone License, RareStone has agreed to seek local approvals to commercialize IMCIVREE for the treatment of obesity community of institutions, healthcare providers, patients, caregivers, and payershyperphagia due to support our ongoing researchbiallelic proopiomelanocortin (POMC), proprotein convertase subtilisin/kexin type 1 (PCSK1) or leptin receptor (LEPR) deficiency, as well as Bardet-Biedl and developmentAlström syndromes. Additionally, RareStone agreed to fund efforts to transform the care ofidentify and enroll patients from China in Rhythm’s global EMANATE trial, a Phase 3, randomized, double-blind, placebo-controlled trial to evaluate setmelanotide in five independent sub-studies in patients with MC4 pathway deficiencies.obesity due to a heterozygous variant of POMC/PCSK1 or LEPR; certain variants of the SRC1 gene, certain variants of the SH2B1 gene, or PCSK1 N221D deletions within the MC4R pathway. According to the terms of the RareStone License, RareStone made an upfront payment to Rhythm of $7.0 million and issued $5.0 million in equity to Rhythm. Rhythm will be eligible to receive development and commercialization milestones of up to $62.5 million, as well as tiered royalty payments on annual net sales of IMCIVREE.
Our commercial priorities for the launch of setmelanotide include:
|
|
48
|
|
|
|
|
|
Our management team understands the complexity of rare diseases and we believe has the necessary expertise to be a true partner to patients, caregivers, advocacy, and healthcare teams leading to shared success. We intend to establish a specialty sales force and develop an organizational infrastructure that will support an extensive network of endocrinologists and other physicians treating severe childhood obesity and rare genetic disorders of obesity which in turn we believe will help establish genetic obesity centers of excellence. Our goal is for our field personnel to work directly with patients, caregivers and healthcare providers to facilitate therapy initiation and adherence. We also expect to partner with existing and new advocacy organizations to further educate our patient population on genetic obesity and support coverage for setmelanotide. In addition, we intend to establish our own commercial sales and marketing organization in the United States and core strategic markets and to selectively establish partnerships in markets outside the United States for sales, marketing and distribution.
Patents and Proprietary Rights
We have in‑licensedin-licensed a large patent portfolio from Ipsen for our melanocortin programs. The portfolio includes multiple patent families, and all of these in‑licensedin-licensed patent families are being prosecuted or maintained by Ipsen in consultation with us. We have also filed patent applications in fivesix families which are exclusively owned and maintained by us that relate to the melanocortin program.
Our MC4MC4R portfolio of licensed and exclusively owned patent families, which includes setmelanotide, consists of 1213 patent families currently being prosecuted or maintained, which include applications and patents directed to compositions of matter, formulations and methods of treatment using setmelanotide. As of June 8, 2018,December 31, 2020, the portfolio for the MC‑4MC4 program consists of 14 issued United States patents and 54228 issued non‑Unitednon-United States patents across eight8 of the 1213 families. We are actively pursuing eightThere also 13 pending United States patent applications and 43 non‑United76 pending non-United States applications in 1224 jurisdictions.
In the patent family directed to selected MC4MC4R receptor agonists, including the composition of matter for setmelanotide, we have three3 issued United States patents and 26108 issued non‑Unitednon-United States patents, including Australia, Canada, China, Europe, Hong Kong, India, Israel, Japan, Korea, New Zealand, Russia and Singapore. The standard 20‑year20-year term for patents in this family would expire in 2026, but two of the United States patents are expected to expire in 2027 due to patent term adjustments. Patent term extensions for delays in marketing approval may also extend the terms of patents in this family.family, and we have filed for patent term extension in the United States that, if granted, would extend the composition of matter patent protection to 2032.
In addition to the patents and patent applications discussed above, we co‑ownco-own one patent family with Charité‑Universitä-Universitätsmedizin Berlin, which has been filed in 21 jurisdictions. We have also filedco-own one application in the United States co‑ownedpatent family with the University of Strasbourg and the French National Institute of Health and Medical Research. These applicationsResearch, which has been filed in 4 jurisdictions. Both of these patent families relate to the melanocortin program and, have not yet entered active prosecution.program.
We have also in‑licensedin-licensed a patent family from Takeda directed to the composition of matter and methods of use of ghrelin O‑acetyltransferaseO-acetyltransferase inhibitors, including RM‑853.RM-853. This patent family includes one1 issued United States patent, nine issued non‑Unitednon-United States patents including China, Europe, and Japan, and one pendingallowed application in Canada. The standard 20‑year20-year term for the patents in this family will expire in 2033, though patent term extensions for delays in marketing approval may also extend the terms of patents in this family.
4925
Intellectual Property Protection Strategy
We currently seek, and intend to continue seeking, patent protection whenever commercially reasonable for any patentable aspects of setmelanotide and related technology or any new products or product candidates we acquire in the future. Where our intellectual property is not protected by patents, we may seek to protect it through other means, including maintenance of trade secrets and careful protection of our proprietary information. Our license from Ipsen for the melanocortin program require Ipsen, subject to certain exceptions and upon consultation with us, to prosecute and maintain its patent rights as they relate to the licensed compounds and methods. If Ipsen decides to cease prosecution or maintenance of any of the licensed patent rights, we have the option to take over prosecution and maintenance of those patents and Ipsen will assign to us all of its rights in such patents. For those patent rights that we own exclusively, we control all prosecution and maintenance activities.
The patent positions of biopharmaceutical companies are generally uncertain and involve complex legal, scientific and factual questions. In addition, the coverage claimed in a patent application can be significantly reduced before the patent is issued, and its scope can be reinterpreted after issuance. Consequently, we do not know whether the product candidate we in‑licensein-license will be protectable or remain protected by enforceable patents. We cannot predict whether the patent applications we are currently pursuing will issue as patents in any particular jurisdiction, and furthermore, we cannot determine whether the claims of any issued patents will provide sufficient proprietary protection to protect us from competitors, or will be challenged, circumvented or invalidated by third parties. Because patent applications in the United States and certain other jurisdictions are maintained in secrecy for 18 months, and since publication of discoveries in the scientific or patent literature often lags behind actual discoveries, we cannot be certain of the priority of inventions covered by pending patent applications. This potential issue is exacerbated by the fact that, prior to March 16, 2013, in the United States, the first to make the claimed invention may be entitled to the patent. On March 16, 2013, the United States transitioned to a “first to file” system in which the first inventor to file a patent application may be entitled to the patent. Therefore,For applications filed prior to the institution of the “first to file” system, we may have to participate in interference proceedings declared by the United States Patent and Trademark Office, or PTO, or a foreign patent office to determine priority of invention. Moreover, we may have to participate in other proceedings declared by the United States PTO or a foreign patent office, such as post‑grantpost-grant proceedings and oppositions, that challenge the validity of a granted patent. Such proceedings could result in substantial cost, even if the eventual outcome is favorable to us.
Although we currently have issued patents directed to a number of different attributes of our products, and pending applications on others, there can be no assurance that any issued patents would be held valid by a court of competent jurisdiction. An adverse outcome could subject us to significant liabilities to third parties, require disputed rights to be licensed from third parties or require us to cease using specific compounds or technology. To the extent prudent, we intend to bring litigation against third parties that we believe are infringing our patents.
The term of individual patents depends upon the legal term of the patents in the countries in which they are obtained. In most countries in which we file, the patent term is 20 years from the earliest date of filing a non‑provisionalnon-provisional patent application. In the United States, a patent’s term may be lengthened by patent term adjustment, which compensates a patentee for administrative delays by the United States PTO in granting a patent, or may be shortened if a patent is terminally disclaimed over another patent with an earlier expiration date.
As mentioned above, in the United States, the patent term of a patent that covers an FDA‑approvedFDA-approved drug may also be eligible for patent term extension, which permits patent term restoration as compensation for the patent term lost during the FDA regulatory review process. Setmelanotide has received FDA approval and we have filed for patent term extension on that product. In the future, if and when our other pharmaceutical products receive FDA approval, we expect to apply for patent term extensions on patents covering those products. We intend to seek patent term adjustments and extensions to any of our issued patents in any jurisdiction where these are available, however there is no guarantee that the applicable authorities, including the FDA in the United States, will agree with our assessment of whether such extensions should be granted, and even if granted, the length of such adjustments or extensions.
To protect our rights to any of our issued patents and proprietary information, we may need to litigate against infringing third parties, or avail ourselves of the courts or participate in hearings to determine the scope and validity of those patents or other proprietary rights. These types of proceedings are often costly and could be very time‑consumingtime-consuming to
26
us, and we cannot be certain that the deciding authorities will rule in our favor. An unfavorable decision could result in
50
the invalidation or a limitation in the scope of our patents or forfeiture of the rights associated with our patents or pending patent applications. Any such decision could result in our key technologies not being protectable, allowing third parties to use our technology without being required to pay us licensing fees or may compel us to license needed technologies from third parties to avoid infringing third‑partythird-party patent and proprietary rights. Such a decision could even result in the invalidation or a limitation in the scope of our patents or could cause us to lose our rights under existing issued patents or not to have rights granted under our pending patent applications.
In addition, we intend to seek orphan drug exclusivity in jurisdictions in which it is available. A prerequisite to orphan drug exclusivity in the United States and in the European Union is orphan drug designation. An orphan drug designation may be granted, subject to fulfillment of specific criteria, where a drug is developed specifically to treat a rare or uncommon medical treatment. If a product which has an orphan drug designation subsequently receives the first regulatory approval for the indication for which it has such designation, the product is entitled to orphan exclusivity, meaning that the applicable regulatory authority may not approve any other applications to market the same drug for the same indication, except in certain very limited circumstances, for a period of seven years in the United States and 10 years in the European Union. Orphan drug exclusivity does not prevent competitors from developing or marketing different drugs for an indication. We have received orphan drug designation in the United States for the use of setmelanotide for five indications and approval for two of those indications.
We also rely on trade secret protection for our confidential and proprietary information. Although we take steps to protect our proprietary information and trade secrets, including through contractual means with our employees and consultants, no assurance can be given that others will not independently develop substantially equivalent proprietary information and techniques or otherwise gain access to our trade secrets or disclose such technology, or that we can meaningfully protect our trade secrets. It is our policy to require our employees, consultants, outside scientific collaborators, sponsored researchers and other advisors to execute confidentiality agreements upon the commencement of employment or consulting relationships with us. These agreements provide that all confidential information developed or made known to the individual during the course of the individual’s relationship with us is to be kept confidential and not disclosed to third parties except in specific circumstances. In the case of employees, the agreements provide that all inventions conceived by the individual will be our exclusive property. There can be no assurance, however, that these agreements will provide meaningful protection or adequate remedies for our trade secrets in the event of unauthorized use or disclosure of such information.
Manufacturing
We currently contract with avarious third partyparties for the manufacture of setmelanotide and intend to continue to do so in the future. We have entered into a process development and manufacturing services agreementservice agreements with CordenPharma International, formerlyour CMOs, Corden Pharma Brussels S.A, or Corden (formerly Peptisyntha SA prior to its acquisition by CordenPharma International,Corden), PolyPeptide Group, Braine L’Alleud, or Peptisyntha, under which Peptisyntha will providePolypeptide, Neuland Laboratories, and Recipharm Monts S.A.S for certain process development and manufacturing services for regulatory starting materials and/or drug substance, or API, and drug product in connection with the manufacture of setmelanotide. We have also entered into commercial supply agreements with both Polypeptide and Recipharm. Under the agreement,our agreements, we pay Peptisynthathese third parties for services and/or manufacture of setmelanotide in accordance with the terms of mutually agreed upon work orders, which we and Peptisyntha may enter into from time to time. The agreement also providesWe may need to engage additional third-party suppliers to manufacture our clinical and commercial drug supplies in the future. In connection with our commercialization of setmelanotide or any future product candidate, we have engaged and could need to engage other third parties to assist in manufacturing and/or supply chain related aspects. While there are a limited number of companies that subjectcan produce raw materials and API in the quantities and with the quality and purity that we require for our product, based on our diligence to certain conditions, for a period following each product launch date, we believe our current network of manufacturing partners are able to fulfill these requirements, and are capable of continuing to expand capacity as needed. Additionally, we have, and will source from Peptisynthacontinue to evaluate further relationships with additional suppliers to increase overall capacity as well as further reduce risks associated with reliance on a portionlimited number of our requirementssuppliers for that product being sourced from non‑affiliate third parties.manufacturing. Under the agreement,current agreements, each party is subject to customary indemnification provisions.
The Peptisyntha agreement will continue, unless earlier terminated pursuant to its terms, until the later of six years from the July 17, 2013 effective date or the completion of all services under all work plans executed in accordance with the terms of the agreement prior to the sixth anniversary of its effective date. The agreement may be extended by us continuously for additional two‑year periods upon written notice to Peptisyntha. We also may terminate the agreement or any work order thereunder upon at least 30 days’ prior written notice to Peptisyntha.
We have also entered into a process development and manufacturing services agreement with Recipharm Monts S.A.S., or Recipharm, under which Recipharm will provide certain process development and manufacturing services in connection with the manufacture of setmelanotide. Under the agreement, we pay Recipharm for services in accordance with the terms of mutually agreed upon work orders, which we and Recipharm may enter into from time to time. Under the agreement, each party is subject to customary indemnification provisions. The Recipharm agreement will continue, unless earlier terminated pursuant to its terms, until the later of three years from the December 21, 2016 effective date or
5127
the completion of all services under all work plans executed in accordance with the terms of the agreement prior to the third anniversary of its effective date. The agreement may be extended by us continuously for additional two‑year periods upon written notice to Recipharm. We also may terminate the agreement or any work order thereunder upon at least 60 days’ prior written notice to Recipharm.
Our contract manufacturing agreements give us visibility into the expected future cost of producing setmelanotide at commercial scale. Based upon a range of prices of currently‑marketedcurrently-marketed therapies indicated for orphan diseases, we believe that our cost of goods for setmelanotide will be highly competitive.
We currently have no plans to build our own clinical or commercial scale manufacturing capabilities. To meet our projected needs for clinical supplies to support our activities through regulatory approval and commercial manufacturing, the contract manufacturing organizations, or CMOs with whom we currently work willmay need to increase scale of production or we expect that we willmay need to secure additional capacity or seek alternate suppliers. We have not currently identified alternatebelieve that our current suppliers in the event the currentand CMOs we utilize are unableable to scale production.production to meet our clinical and commercial demands. Because we rely on these CMOs, we have personnel with pharmaceutical development and manufacturing experience who are responsible for maintaining our CMO relationships.
Setmelanotide is distributed in the U.S. through our specialty pharmacy and in the EU/UK through third-party service providers that deliver the medication to patients. We plan to continue building out our network for commercial distribution in jurisdictions in which setmelanotide is approved.
Regulatory Matters
Government Regulation and Product Approvals
Government authorities in the United States, at the federal, state and local level, and in other countries and jurisdictions, including the European Union, extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, recordkeeping, labeling,record-keeping, promotion, advertising, promotion, distribution, marketing post‑approval monitoring and reporting, including pharmacovigilance,export and import and export of pharmaceuticaldrug products. The processes for obtaining marketing approvalsA new drug must be approved by the FDA through NDA process or by comparable foreign regulatory authorities through similar applications before it may be legally marketed in the United States and in foreign countries and jurisdictions,jurisdictions. We, along with any third-party contractors, will be required to navigate the various preclinical, clinical and commercial approval requirements of the governing regulatory agencies of the countries in which we wish to conduct studies or seek approval of our products and product candidates. The process of obtaining regulatory approvals and the subsequent compliance with applicable federal, state, local and foreign statutes and regulations and other competent authorities, require the expenditure of substantial time and financial resources.
Review and Approval of Drugs in the United StatesU.S. Drug Development Process
In the United States, the FDA approves drug productsregulates drugs under the Federalfederal Food, Drug, and Cosmetic Act or FDCA,(FDCA) and associatedits implementing regulations. Biological products, onThe process of obtaining regulatory approvals and the other hand, are licensedsubsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources. The process required by the FDA under the Public Health Service Act, or PHSA. With passage of the Biologics Price Competition and Innovation Act of 2009, Congress amended the definition of “biological product” in the PHSA so as to excludebefore a chemically synthesized polypeptide from licensure under the PHSA. Rather, the Act provided that such products woulddrug may be treated as drugs under the FDCA. Subsequently, through final guidance issued in April 2015, the FDA indicated that a “chemically synthesized polypeptide” is any alpha amino acid polymer that is made entirely by chemical synthesis and is less than 100 amino acids in size. Accordingly, based on this FDA guidance, we believe that our products will not be treated as biologics subject to approval of a biologics license application, or BLA, by the FDA, and rather will be treated as drug products subject to approval of a new drug application, or NDA, by the FDA pursuant to the FDCA.
An applicant seeking approval to market and distribute a new drug productmarketed in the United States must typically undertakegenerally involves the following:
| ●completion |
|
|
|
|
52
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The failure to comply with applicable requirements under the FDCA and other applicable lawsregulations;
28