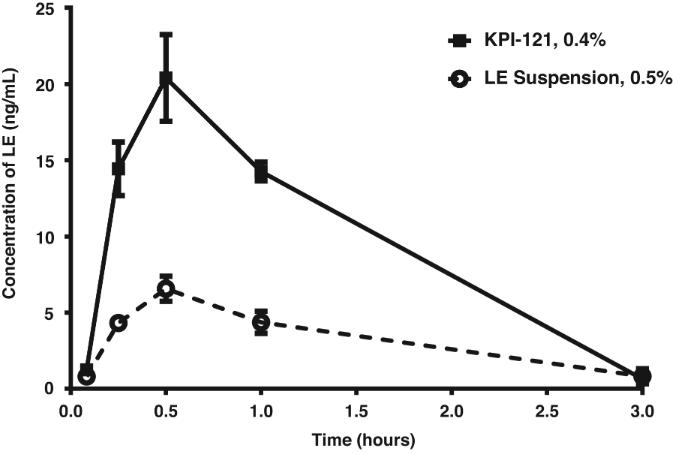
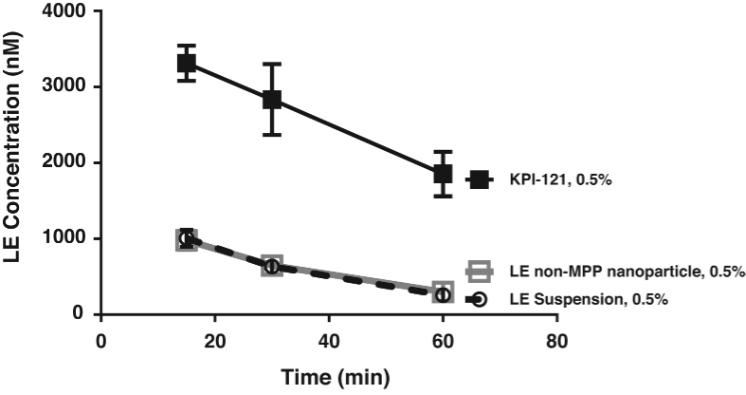
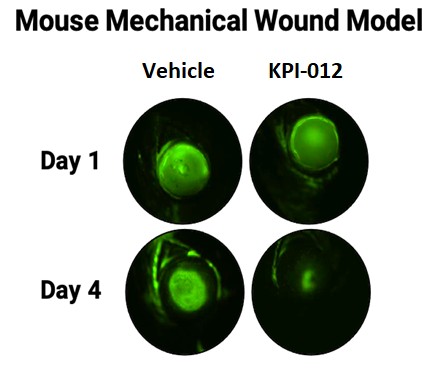
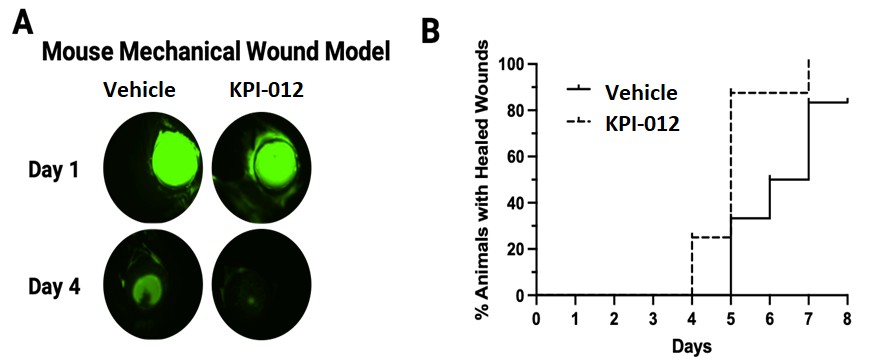
On August 9, 2018,only if the corresponding milestone is achieved again in a subsequent calendar year. To date, we filed our 2018 Shelf Registration under whichhave not received any such milestone payments. We now have no revenue-generating commercial products, and although we could initially offer and sellare eligible to receive up to $250.0$325.0 million of a variety of securities including common stock, preferred stock, warrants, depositary shares, debt securities, purchase contracts, purchase units or any combination of such securities during the three-year period that commenced upon the 2018 Shelf Registration becoming effective. Under the 2018 Shelf Registration,in milestone-based payments from Alcon, there can be no assurance as to when we may periodically offer onereceive such milestone payments or more types of securities in amounts, at prices and on terms announced, if and when the securities are ever offered.
On October 1, 2018, we entered into the Athyrium Credit Facility with Athyrium for up to $110.0 million. The Athyrium Credit Facility provided for a Term Loan A in the aggregate principal amount of $75.0 million, and a Term Loan B in the aggregate principal amount of $35.0 million. On October 1, 2018,milestone payments we borrowed the entire principal amount of the Term Loan A. We did not satisfy the conditions to draw downmay receive, if any of the Athyrium Term Loan B funds, and as a result, the Term Loan B funds are no longer available. The maturity date of the Athyrium Credit Facility is October 1, 2024, the six-year anniversary of the close. The Term Loan A bears interest at a rate of 9.875% per annum, with quarterly, interest-only payments until the fourth anniversary of the Term Loan A. The unpaid principal amount of the Term Loan A is due and payable in quarterly installments starting at the end of the fourth anniversary of the loan..
On October 5, 2018, we sold 7,500,000 shares of common stock in an underwritten offering pursuant to the 2018 ShelfOfferings under Registration at a public offering price of $8.25 per share, before underwriting discounts and commissions. Statements
In addition, the underwriters were granted an overallotment option to purchase an additional 1,125,000 shares of the common stock at the same public offering price, less underwriting discounts and commissions. On October 11, 2018, the underwriters exercised in full their option to purchase the overallotment shares. The total number of shares sold by us in the offering was 8,625,000 shares, resulting in net proceeds to us, after underwriting discounts and offering expenses, of $66.1 million. In connection with the filing of a registration statement on Form S-3 with the 2018SEC, or the 2020 Shelf Registration, we entered into aan amended and restated sales agreement with Jefferies, or the Amended and Restated Sales Agreement, pursuant to which we could issue and sell, from time to time, up to an aggregate of $50.0$75.0 million of our common stock in an ATM Offering, through Jefferies, as sales agent. As ofunder our at-the-market offering. During the year ended December 31, 2019,2022, we had issuedsold an aggregate of 2,592,934148,461 shares of our common stock under the ATM Offering,Amended and Restated Sales Agreement, resulting in net proceeds of $1.0 million. From January 1, 2023 to us of $13.1 million. During the first quarter of 2020,January 10, 2023, we issued an aggregate of 2,352,671sold 245,887 shares of our common stock under the ATM Offering,Amended and Restated Sales Agreement, resulting in net proceeds to us of $12.5$10.0 million. On MarchJanuary 10, 2020,2023, the Amended and Restated Sales Agreement terminated in accordance with its terms when we suspendedcompleted the sale of $75.0 million of our shares of common stock thereunder. As of the date of termination of the Amended and terminated the prospectus related to the ATM Offering.
On March 11, 2020,Restated Sales Agreement, we had sold 16,000,000an aggregate of 565,974 shares of our common stock inunder such agreement for aggregate gross proceeds of $75.0 million.
On January 19, 2023, we entered into a new sales agreement with Jefferies, or the Open Market Sale Agreement, pursuant to which we may issue and sell, from time to time, shares of our common stock through Jefferies under our at-the-market offering. We filed a prospectus supplement relating to the Open Market Sale Agreement under our 2020 Shelf Registration, or the 2020 Shelf ATM Prospectus Supplement, pursuant to which we could offer and sell shares of common stock having an underwrittenaggregate offering price of up to $40.0 million under the Open Market Sale Agreement. From January 19, 2023 to May 11, 2023, we sold 229,378 shares of our common stock under our at-the-market offering pursuant to the 2018Open Market Sale Agreement under the 2020 Shelf Registration, at a public offering price of $7.89 per share, resulting in net proceeds of $118.2 million, after underwriting discounts, commissions, and offering expenses. In addition,$4.9 million.
On March 3, 2023, we filed a shelf registration statement on Form S-3 with the underwriters ofSEC, or the offering were granted the option for a period of 30 days to purchase up to an additional 2,400,000 shares of common stock offered in the public offering at the public offering price, less underwriting discounts, commissions, and offering expenses. On April 3, 2020, the underwriters exercised their option and purchased an additional 979,371 shares of common stock at $7.89 per share, resulting in net proceeds to us of $7.2 million, after underwriting discounts, commissions, and offering expenses. The total number of shares sold by us in the offering2023 Shelf Registration, which was 16,979,371, resulting in total net proceeds to us, after underwriting discounts and offering expenses, of $125.4 million.
declared effective on May 11, 2023. Under the 20182023 Shelf Registration we have issued an aggregate of 30,549,976 shares of common stock, including under the ATM Offering, resulting in aggregate gross proceeds to us of $231.7 million. There was $18.3 million of securities available to be issued under the 2018 Shelf Registration as of December 31, 2020.
On May 7, 2020, we filed our 2020 Shelf Registration, under which we may offer and sell up to $350.0 million of a variety of securities including common stock, preferred stock, warrants, depositary shares, debt securities, subscription rights or units during the three-year period that commenced upon the 2020 Shelf Registration becoming effective.units. In connectionaccordance with the filingterms of the 2020 Shelf Registration, we entered into an amended and restated sales agreement with Jefferies, pursuant to whichOpen Market Sale Agreement, we may issue and sell, from time to time, up to an aggregate of $75.0$40.0 million of our common stock under our ATM Offering. Duringin an at-the-market equity offering through Jefferies. Upon effectiveness of the fourth quarter of 2020,2023 Shelf Registration, we issued an aggregate of 2,821,059 sharesceased any further offers or sales of our common stock underpursuant to the 2020 Shelf ATM Offering, resulting in net proceeds to us of $20.6 million. In January 2021,Prospectus Supplement and the 2020 Shelf Registration. During the year ended December 31, 2023, we issued and sold an additional 2,552,457256,256 shares of our common stock under our ATM Offering, resulting inat-the-market offering pursuant to the 2023 Shelf Registration for total net proceeds of $3.6 million.
During the year ended December 31, 2023, we sold an aggregate of 731,521 shares of our common stock pursuant to (1) our Amended and Restated Sales Agreement and our Open Market Sale Agreement under the 2020 Shelf Registration and (2) the Open Market Sale Agreement under the 2023 Shelf Registration, for total net proceeds of $18.5 million.
Loan Agreement
On May 4, 2021, we entered into the Loan Agreement with Oxford Finance, in its capacity as lender, or the Lender, and in its capacity as collateral agent, or Agent, pursuant to which a term loan of up to an aggregate principal amount of $125.0 million became available to us, consisting of $18.2 million. Asa tranche A term loan that was disbursed on the closing date of the dateLoan Agreement in the aggregate principal amount of this Annual Report on Form 10-K, there was $35.0$80.0 million and additional tranches that are no longer available to us. Through June 30, 2023, the term loan bore interest at a floating rate equal to the greater of shares30-day LIBOR and 0.11%, plus 7.89%. Effective July 1, 2023, the term loan bears interest at a floating rate equal to the greater of common stock remaining under(a) 8.00% and (b) the ATM Offering that we may issuesum of (i) the 1-Month CME Term Secured Overnight Financing Rate, or SOFR, (ii) 0.10% and (iii) 7.89%. Certain of the customary negative covenants limit our and certain of our subsidiaries’ ability, among other things, to incur future debt, grant liens, make investments, make acquisitions, distribute dividends, make certain restricted payments and sell assets, subject in each case to certain exceptions. In connection with our entry into the future and, excludingpurchase agreement for the funds designatedsale of our Commercial Business to beAlcon, on May 21, 2022, we entered into an amendment