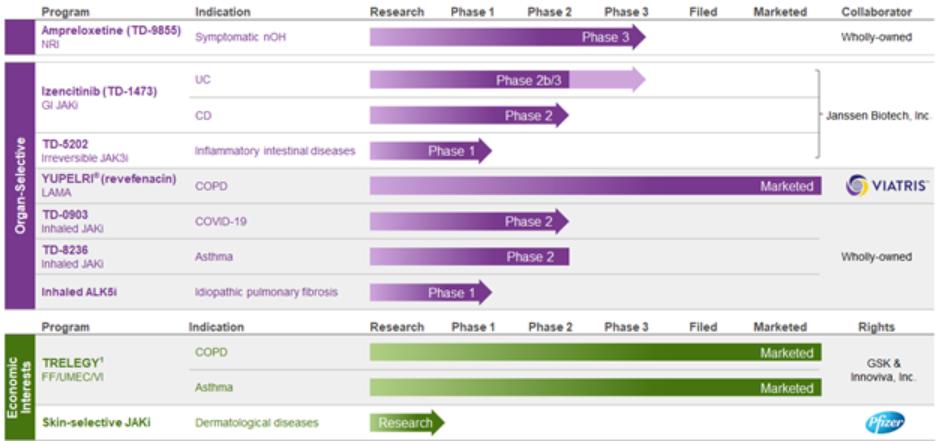
Gut-selective Pan-JAK Inhibitor Program (Izencitinib)and OHSA #1. Throughout the study, there was no indication of worsening of supine hypertension among any of the patient sub-groups. Data suggest that ampreloxetine was well-tolerated and no new safety signals were identified among any of the patient sub-groups.
JAK inhibitors function by inhibitingIn June 2022, we held a Type C meeting with the activityFDA. From this meeting, we aligned on a path to a New Drug Application (“NDA”) filing with one additional Phase 3 clinical study (CYPRESS) in MSA patients with symptomatic nOH, using the OHSA composite score as the primary endpoint. This Phase 3 study was initiated in the first quarter of one or more2023, and the study is currently open to recruitment with the expectation of enrolling the final patient into the open label period of the Janus kinase familystudy in the second half of enzymes (JAK1, JAK2, JAK3, TYK2)2024. In May 2023, we announced that play a key role in cytokine signaling. Inhibiting these JAK enzymes interferes with the JAK/STAT signaling pathway and, in turn, modulates the activity of a wide range of pro-inflammatory cytokines. JAK inhibitors are currently approvedFDA granted Orphan Drug Designation status to ampreloxetine for the treatment of rheumatoid arthritis, myelofibrosis, and ulcerative colitis and have demonstrated therapeutic benefit forsymptomatic nOH in patients with Crohn’s disease. However, these products are known to have side effects based on their systemic exposure. In izencitinib, our program goal is to develop an orally administered, gut-selective pan-JAK inhibitor specifically designed to distribute adequately and predominantly to the tissues of the intestinal tract, treating inflammation in those tissues while minimizing systemic exposure. We believe izencitinib could be a potential treatment for a range of inflammatory intestinal diseases, and it is in development for the treatment of ulcerative colitis and Crohn’s disease.MSA.
BasedIn July 2022, Royalty Pharma Investments 2019 ICAV (“Royalty Pharma”) agreed to invest up to $40.0 million to advance the development of ampreloxetine in MSA in exchange for unsecured low single-digit royalties. Royalty Pharma’s $40.0 million investment in ampreloxetine included a $25.0 million upfront payment received in July 2022 and an additional $15.0 million payment upon the first regulatory approval of ampreloxetine. In exchange, Royalty Pharma will receive future unsecured royalties of 2.5% on positive results from a Phase 1b exploratory study in ulcerative colitisannual ampreloxetine global net sales up to $500.0 million and following dialogues with4.5% on annual global net sales over $500.0 million. If ampreloxetine regulatory approval is not achieved or if ampreloxetine sales are never recognized, the FDA and European Medicines Agency (“EMA”) regarding study design, we advanced izencitinib into two clinical studies in inflammatory intestinal diseases. The Phase 2 (DIONE) study is a twelve-week randomized, double-blind, placebo-controlled study designed to evaluate the efficacy and safety of patients with Crohn’s disease, which began dosing patients in late 2018. The Phase 2b/3 (RHEA) study is a randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of eight weeks induction and 44 weeks maintenance therapy in patients with ulcerative colitis, which began dosing patients in early 2019. As described above and in Item 1A. Risk Factors, the COVID-19 pandemic has impacted the timeline for our clinical trials. Data from the Phase 2b portion of the ulcerative colitis study and the Phase 2 Crohn’s disease studies is expected in the third quarter of 2021.
amounts invested by Royalty Pharma would not be repaid by us.
Irreversible JAK3 Inhibitor (TD-5202)
TD-5202 is an investigational, orally administered, gut-selective, irreversible JAK3Skin-selective Pan-JAK inhibitor that has demonstrated a high affinity for the JAK3 enzyme. Through the selective inhibition of JAK3, TD-5202 interferes with the JAK/STAT signaling pathway and, in turn, modulates the activity of select pro-inflammatory cytokines, including IL-2, IL-15, and IL-21 which play a central role in the pathogenesis of T-cell mediated disease, including inflammatory intestinal disease, such as celiac disease. Importantly, TD-5202 is specifically designed to act locally within the intestinal wall thereby limiting systemic exposure.Program
In September 2019, we announced the initiation of a Phase 1 single ascending dose and multiple ascending dose trial designed to evaluate the safety and tolerability of TD-5202 in healthy participants, plus assess plasma pharmacokinetics of TD-5202 to confirm circulating levels are low, consistent with a gut-selective approach. In February 2020, we announced that data from the Phase 1 study indicated that TD-5202 was generally well tolerated as a single oral dose up to 2000 milligrams and as a twice-daily oral dose up 2000 milligrams total per day given for ten consecutive days in healthy participants.
We are developing izencitinib and TD-5202 in collaboration with Janssen as part of the companies’ global co-development and commercialization agreement for novel, gut-selective JAK inhibitors.
Janssen Biotech Collaboration
In February 2018,December 2019, we announcedentered into a global co-development and commercializationlicense agreement with JanssenPfizer Inc. (“Pfizer”) for izencitinibour preclinical skin-selective, locally acting pan-JAK inhibitor program (the “Pfizer Agreement”). The compounds in this program are designed to target validated pro-inflammatory pathways and related back-up compounds for inflammatory intestinal diseases, including ulcerative colitis and Crohn's disease.are specifically designed to possess skin-selective activity with minimal systemic exposure. Under the terms of the agreement, we received an upfront payment of $100.0 million and will be eligible to receive up to an additional $900.0 million in potential payments, inclusive of a potential opt-in payment following completion of the Phase 2 Crohn’s study and the Phase 2b induction portion of the ulcerative colitis study. At that time, Janssen can elect to obtainPfizer Agreement, Pfizer had an exclusive license to develop, manufacture and commercialize izencitinibcertain compounds for all uses other than gastrointestinal, ophthalmic, and certain related compounds by paying usrespiratory applications. We received an upfront cash payment of $10.0 million in 2019, and in March 2022, we received a fee$2.5 million development milestone payment from Pfizer for the first patient dosed in a Phase 1 clinical trial of $200.0 million. Upon such election,the skin-selective pan-JAK inhibitor program. In June 2023, we and Janssen will jointly develop and commercialize izencitinib in inflammatory intestinal diseases, and we and Janssen will share profits and losses inreceived notice from Pfizer terminating the US and expenses relatedPfizer Agreement, effective as of October 7, 2023, at which time the skin-selective pan-JAK inhibitor program was returned to a potential Phase 3 program (67% to Janssen; 33% to Theravance Biopharma). In addition, we would receive royalties on ex-US sales at double-digit tiered percentage royalty rates.us.
Economic Interests and Other Assets
Mid- and Long-Term Economic Interest in TRELEGY®
In July 2022, we completed the sale of all of our equity interests in Theravance Respiratory Company, LLC (“TRC”) representing our 85% economic interest in the sales-based royalty rights on worldwide net sales of GSK plc's (“GSK”) TRELEGY ELLIPTA (“TRELEGY”) to Royalty Pharma for approximately $1.1 billion in upfront cash while retaining future value through the right to receive contingent milestone payments and certain outer year-royalties (the “TRELEGY Royalty Transaction”).
From and after January 1, 2023, for any calendar year starting with the year ended December 31, 2023 and ending with the year December 31, 2026, upon certain milestone minimum royalty amounts for TRELEGY being met, Royalty Pharma is obligated to make certain cash payments to us (the “Milestone Payment(s)”). As of January 1, 2024, a total of $200.0 million in potential Milestone Payments remain available to us. For the next potential Milestone Payment, we are eligible to receive either (i) $25.0 million if Royalty Pharma receives $240.0 million or more in royalty payments from GSK with respect to 2024 TRELEGY global net sales, which we would expect to occur in the event TRELEGY global net sales are approximately $2.86 billion; or (ii) $50.0 million if Royalty Pharma receives $275.0 million or more in royalty payments from GSK with respect to 2024 TRELEGY global net sales, which we would expect to occur in the event TRELEGY global net sales exceed approximately $3.21 billion. Fourth quarter of 2023 global net sales were $737.0 million which represented an increase of 35% year-over-year, and total 2023 global net sales were $2.7 billion which represented an increase of 28% year-over-year.
In addition to potential Milestone Payments, we will receive from Royalty Pharma 85% of the royalty payments on TRELEGY payable (a) for sales or other activities occurring on and after January 1, 2031 related to TRELEGY in the