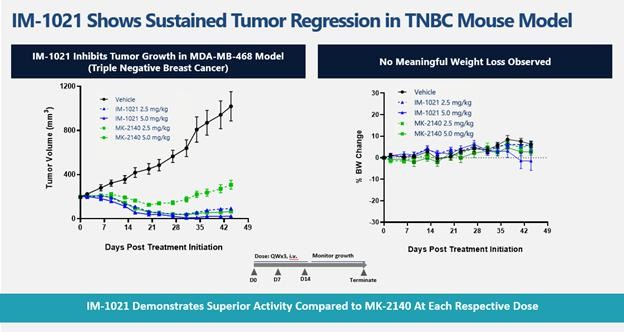
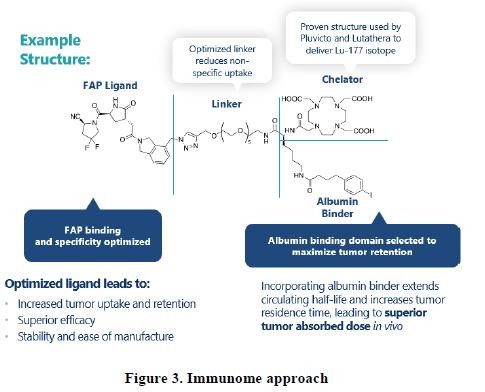
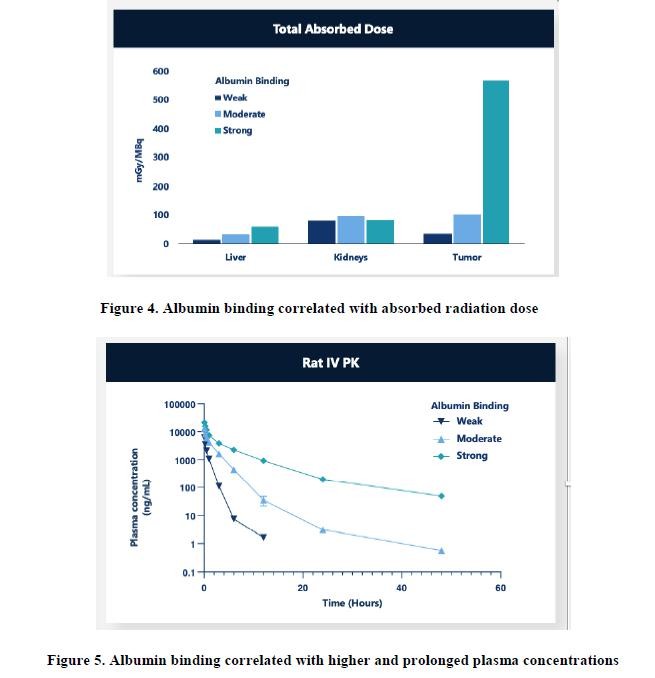
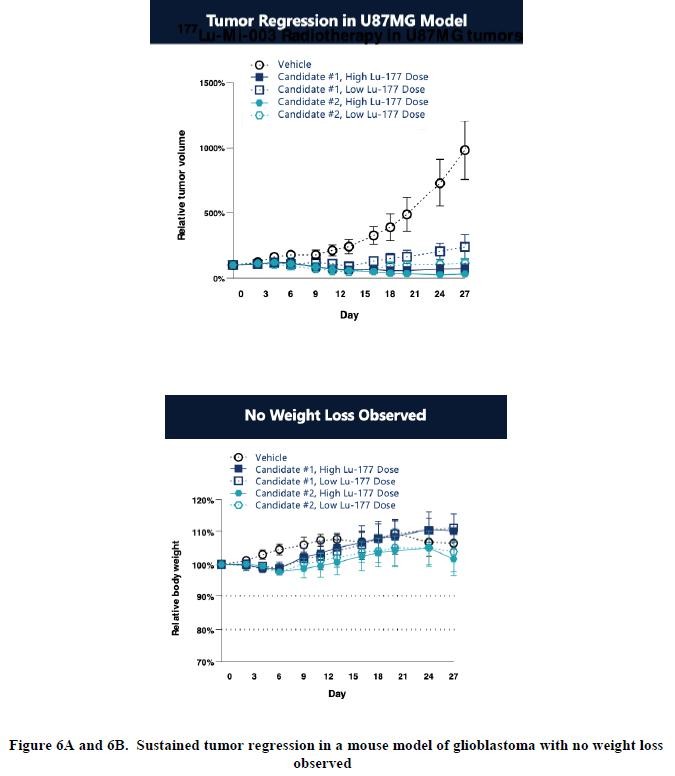
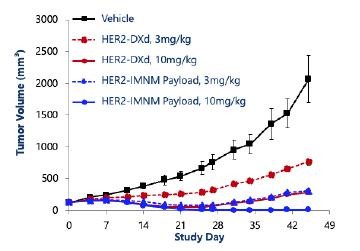
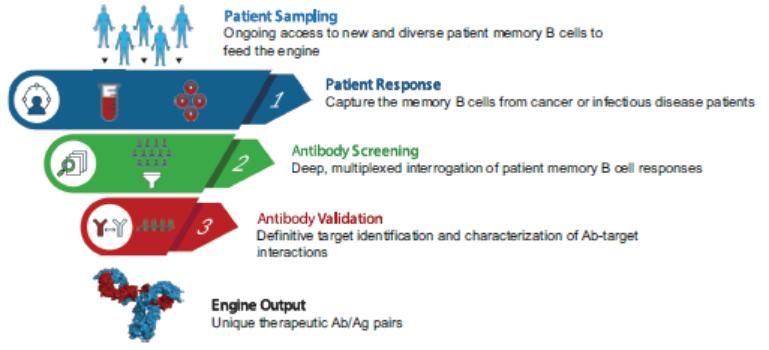
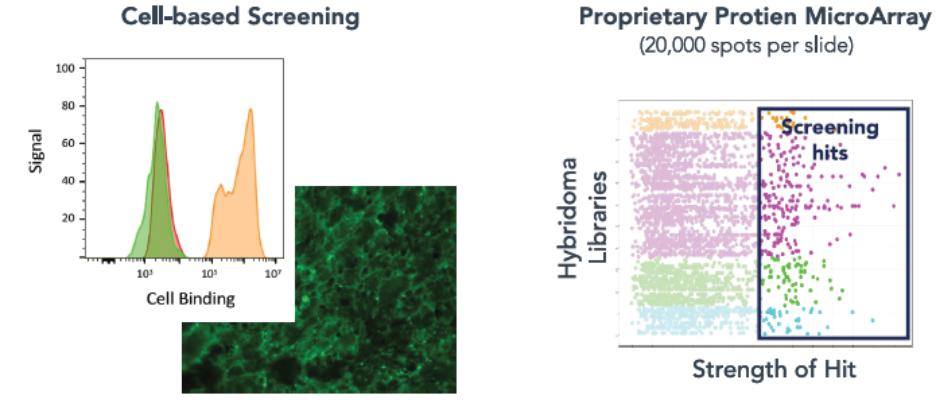
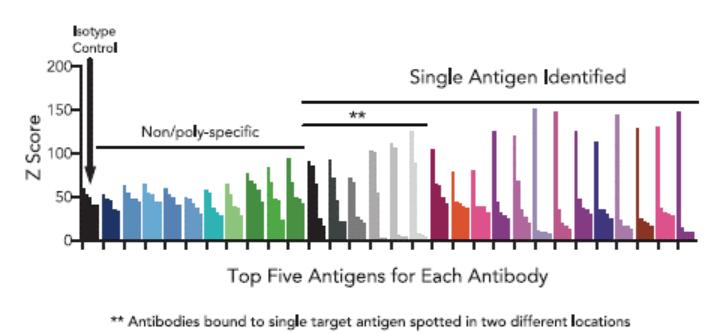
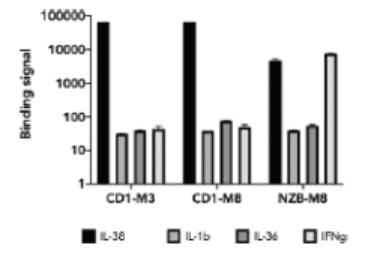
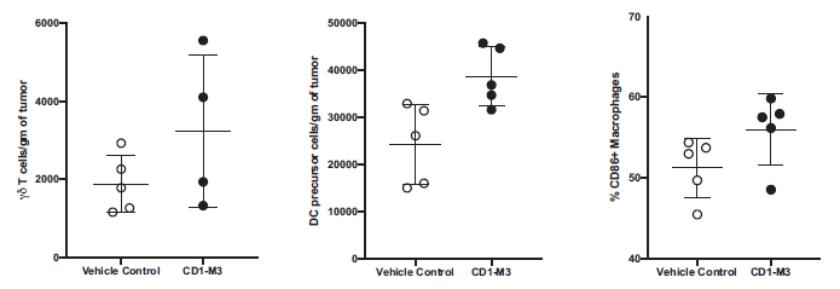
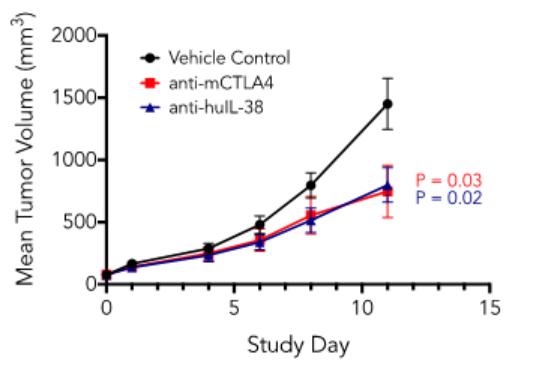
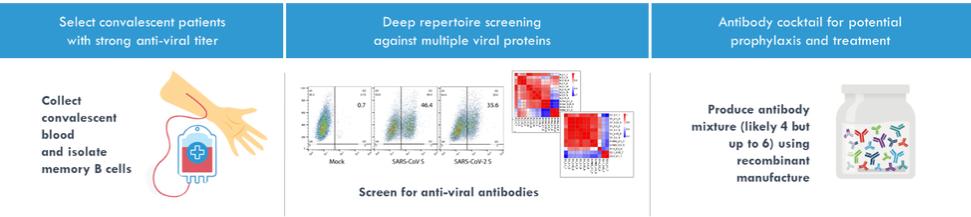
effect onthese attacks, including retaliatory cyber-attacks, that could materially disrupt our business, financial condition, results ofsystems and operations, and prospects. We do not currently maintain “key man” insurance on any of our executive officers.
In November 2020, our board of directors appointed Diane Marcou as our Interim Chief Financial Officer until a permanent successor can be named. We have an ongoing search in process to select the Company's next Chief Financial Officer. Leadership transitions can be inherently difficult to manage, and an inadequate transition to a permanent Chief Financial Officer may cause disruption within the Company. In addition, if we are unable to attract and retain a qualified candidate to become the permanent Chief Financial Officer in a timely manner, our financial performancesupply chain, and ability to meet operational goalsconduct our business as presently conducted.
We and strategic plansthe third parties upon which we rely are subject to a variety of evolving threats, including but not limited to social-engineering attacks (including through deep fakes, which may be increasingly more difficult to identify as fake, and phishing attacks), malicious code (such as viruses and worms), malware (including as a result of advanced persistent threat intrusions), denial-of-service attacks, credential stuffing, credential harvesting, personnel misconduct or error, ransomware attacks, supply-chain attacks, software bugs, server malfunctions, software or hardware failures, loss of data or other information technology assets, adware, telecommunications failures, earthquakes, fires, floods, attacks enhanced or facilitated by AI, and other similar threats.
In particular, severe ransomware attacks are becoming increasingly prevalent and can lead to significant interruptions in our operations, ability to provide our products or services, loss of sensitive data and income, reputational harm, and diversion of funds. Extortion payments may alleviate the negative impact of a ransomware attack, but we may be unwilling or unable to make such payments due to, for example, applicable laws or regulations prohibiting such payments.
Remote work has become more common and has increased risks to our information technology systems and data, as more of our employees utilize network connections, computers and devices outside our premises or network, including working at home, while in transit and in public locations.
Future or past business transactions (such as acquisitions or integrations) could expose us to additional cybersecurity risks and vulnerabilities, as our systems could be adversely impacted.negatively affected by vulnerabilities present in acquired or integrated entities’ systems and technologies. Furthermore, we may discover security issues that were not found during due diligence of such acquired or integrated entities, and it may be difficult to integrate companies into our information technology environment and security program.
The relationships that our key management team members have cultivated within our industry make us particularly dependent upon their continued employment with us. We are dependentrely on the continued service of our technical personnel because of the highly technical nature of our product candidatesthird parties and technologies to operate critical business systems to process sensitive data in a variety of contexts, including, without limitation, cloud-based infrastructure, data center facilities, encryption and the specialized nature of the regulatory approval process. Because our management team and key employees are not obligated to provide us with continued service, they could terminate their employment with us at any time without penalty. Our future success will depend in large part on our continued ability to attract and retain other highly qualified scientific, technical and management personnel, as well as personnel with expertise in clinical testing, manufacturing, governmental regulation and commercialization. We face competition for personnel from other companies, universities, public and private research institutions, government entitiesauthentication technology, employee email, and other organizations.
As of December 31, 2020, we had 23 full-time employees. Our focus on the development of our potential future product candidates will require adequate staffing. We may need to hire and retain new employees to execute our future clinical development and manufacturing plans. We cannot provide assurance that we will be able to hire or retain adequate staffing levels to advance our platform, develop our potential future product candidates or run our operations or to accomplish all of our objectives.
We may experience difficulties in managing our growth and expanding our operations.
We have limited experience in product development or clinical trials for any product candidate. As our potential future product candidates enter and advance through preclinical studies and any clinical trials, we will need to expand our development, regulatory and manufacturing capabilities or contract with other organizations to provide these capabilities for us. We may also experience difficulties in the discovery and development of new potential future product candidates using the Immunome discovery engine if we are unable to meet demand as we grow our operations. In the future, we also expect to have to manage additional relationships with collaborators, suppliers and other organizations.functions. Our ability to managemonitor these third parties’ information security practices is limited, and these third parties may not have adequate information security measures in place. If the third parties upon which we rely experience a security incident or other interruption, we could experience adverse consequences. While we may be entitled to damages if the third parties upon which we rely fail to satisfy their privacy or security-related obligations to us, any award may be insufficient to cover our operationsdamages, or we may be unable to recover such award. In addition, supply-chain attacks have increased in frequency and future growthseverity, and we cannot guarantee that third parties’ infrastructure in our supply chain or our third-party partners’ supply chains have not been compromised.
While we have implemented security measures designed to protect against security incidents, there can be no assurance that these measures will require usbe effective. We take steps designed to continue to improvedetect, mitigate and remediate vulnerabilities in our operational, financial and management controls, reportinginformation security systems and procedures and secure adequate facilities for(such as our operational needs. Wehardware and/or software, including that of third parties upon which we rely), but we may not be able to implement improvementsdetect, mitigate, and remediate all such vulnerabilities including on a timely basis. Further, we may experience delays in developing and deploying remedial measures and patches designed to our management information and control systems in an efficientaddress identified vulnerabilities.
Any of the previously identified or timely manner and may discover deficiencies in existing systems and controls.
Our employees, principal investigators, consultants and commercial partners may engage in misconductsimilar threats could cause a security incident or other improper activities, including noncompliance with regulatory standards and requirements.
We are exposed to the risk of fraud or other misconduct by our employees, principal investigators, consultants and commercial partners. Misconduct by employees could include intentional failures to comply with FDA regulations, provide accurate information to the FDA, comply with manufacturing standards we may establish, comply with federal and state healthcare fraud and abuse laws and regulations, report financial information or data accurately or disclose unauthorized activities to us. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Such misconduct could also involve the improper use of information obtained in the course of clinical trials, whichinterruption that could result in regulatory sanctions and serious harmunauthorized, unlawful, or accidental acquisition, modification, destruction, loss, alteration, encryption, disclosure of, or access to our reputation. For example, individuals conductingsensitive data or our information technology systems, or those of the non-interventionalthird parties upon whom we rely. A security incident or other interruption could disrupt our ability (and that of third parties upon whom we rely) to conduct our business as presently conducted. We may expend significant resources or modify our business activities (including our clinical studies that we sponsor through which we obtain antibodies for development into potential antibody-based therapeuticstrial activities) to try to protect against security incidents. Certain data privacy and security obligations may violate applicable lawsrequire us to implement and regulations regarding patients’ personalmaintain specific security measures or industry-standard or reasonable security measures to protect our information technology systems and sensitive data. It is not always possible to identify and deter