Merger of Celladon Corporation and Eiger BioPharmaceuticals, Inc.
On March 22, 2016, Celladon Corporation, or Celladon, and privately-held Eiger BioPharmaceuticals, Inc., or Private Eiger, completed a business combination in accordance with the terms of the Agreement and Plan of Merger and Reorganization, or the Merger Agreement, dated as of November 18, 2015, by and among Celladon, Celladon Merger Sub, Inc., a wholly-owned subsidiary of Celladon, or Merger Sub, and Private Eiger, pursuant to which Merger Sub merged with and into Private Eiger, with Private Eiger surviving as a wholly-owned subsidiary of Celladon. This transaction is referred to herein as “the Merger.” Immediately following the Merger, Celladon changed its name to “Eiger BioPharmaceuticals, Inc.” In connection with the closing of the Merger, our common stock began trading on The NASDAQ Global Market under the ticker symbol “EIGR” on March 23, 2016.
Business
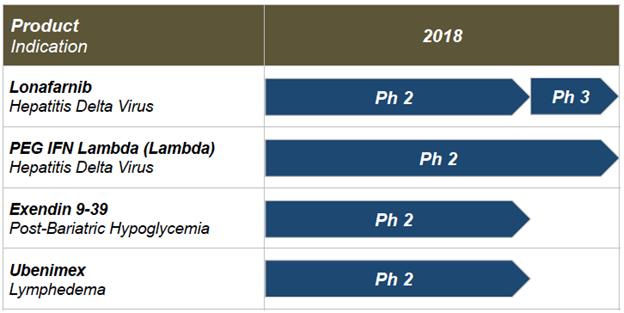
Our current project timelines, planned development and regulatory pathways are illustrated below. As discussed above, prior clinical experience by our licensors with the product candidates has supported and guided our understanding of safety in advancing these products in our clinical development programs. Specifically, we in-licensed lonafarnib from Merck Sharp & Dohme Corp, or Merck, in 2010; licensed ubenimex from Nippon Kayaku Co., Ltd., or Nippon Kayaku, in 2015; and licensed Lambda from Bristol-Myers Squibb, or BMS, in April 2016. We have relied upon Merck’s, Nippon Kayaku’s and BMS’s prior Phase 1/2/3 clinical data, manufacturing and experience with these three molecules to proceed directly into Phase 2 clinical trials following authorization by the U.S. Food and Drug Administration.
Note: All dates represent our current expectations. Actual timing may vary.
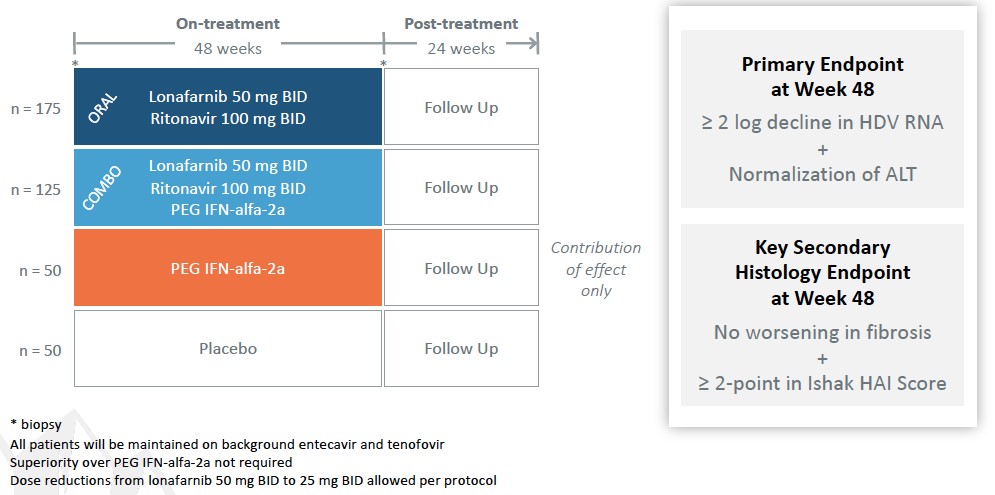
Our product candidate pipeline includes four Phase 2 programs:
|
|
Lonafarnib, or LNF, is an orally bioavailable, small molecule in Phase 2 clinical trials for HDV infection and is our most advancedlead program. HDV is the most severe form of viral hepatitis for which there is currently no approvedFDA-approved therapy. Chronic HDV infection can lead to a rapid progression to liver cirrhosis, a greater likelihood of developing liver cancer, and has the highest fatality rate of all the chronic hepatitis infections.
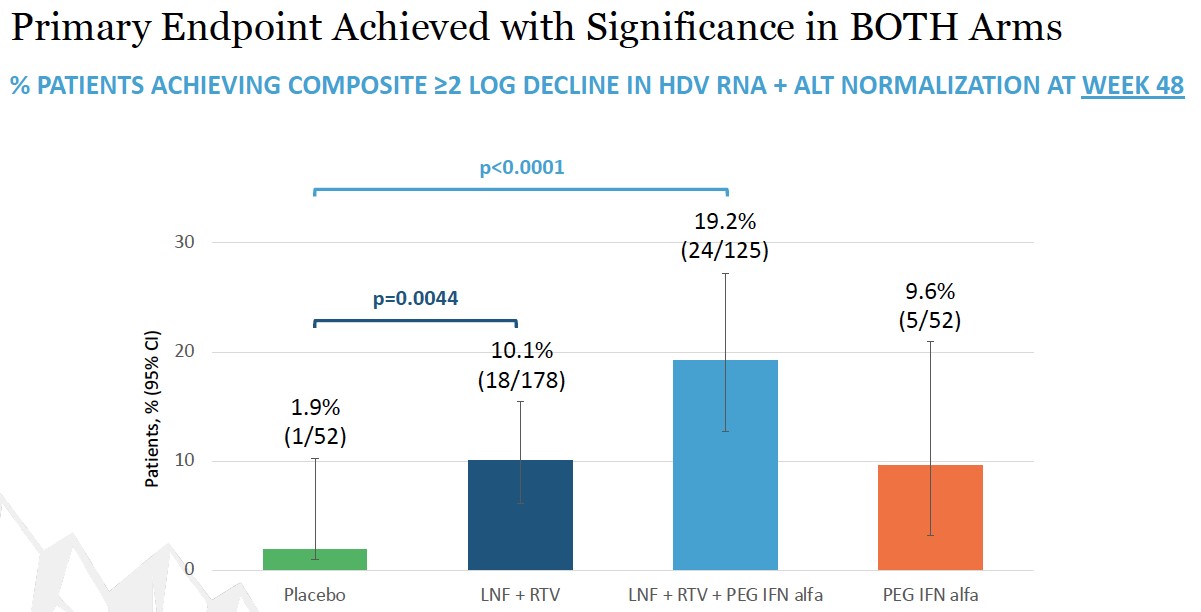
placebo. Topline Week 48 data announced in December 2022 demonstrated that both lonafarnib-based regimens showed statistical significance versus placebo on the primary endpoint. Week 72 data is expected in mid-2023
| Lambda |
Pegylated interferon lambda (Lambda) is our second program treating HDV.in clinical development for HDV and is currently in Phase 3. Lambda is a well-characterized, late-stage, first in class,first-in-class, type III, well-tolerated interferon or IFN,(IFN), that stimulates immune responses that are critical for the development of host protection during viral infections. Lambda targets type III IFN receptors which are distinct from the type I IFN, receptors targeted by IFN-alfa. These type III receptors are highly expressed on hepatocytes with limited
expression on hematopoietic and central nervous system cells, which has been demonstrated to reduce the off-target effects associated with other IFNs and improve the tolerability of Lambda. Although Lambda does not use the IFN-alfa receptor, signaling through either the IFN-lambda or IFN-alfa receptor complexes results in the activation of the same Jak-STAT signal transduction cascade.
We licensed worldwide rights to Lambdalambda from BMSBristol Myers Squibb in April 2016. Lambda has been administered in clinical trials involving over 3,0004,000 patients infected with the Hepatitis B Virus or HBV, or(HBV), Hepatitis C Virus or HCV.(HCV), Hepatitis D Virus (HDV), and SARS-CoV-2. Lambda has not been approved for any indication.
|
|
Exendin 9-39 is the third Phase 2 program andwell-characterized peptide that we are developing this candidate as a treatment for PBH. PBHcongenital hyperinsulinism (HI), an ultra-rare, pediatric metabolic disorder. HI is the most frequent cause of persistent hypoglycemia in neonates and children and is characterized by fasting and protein-induced hypoglycemia and results in permanent brain damage with neurodevelopmental deficits in up to 50% of patients. Near-total pancreatectomy is often indicated and leads to life-long insulin-dependent diabetes (IDDM). Safe and effective therapies are urgently needed to prevent brain damage, IDDM and death.
We
In December 2016, Eiger filed an Investigational New Drug application for exendin 9-39 in the United States. Exendin 9-39reduced postprandial insulin peak during MMTT was also statistically significant.
|
|
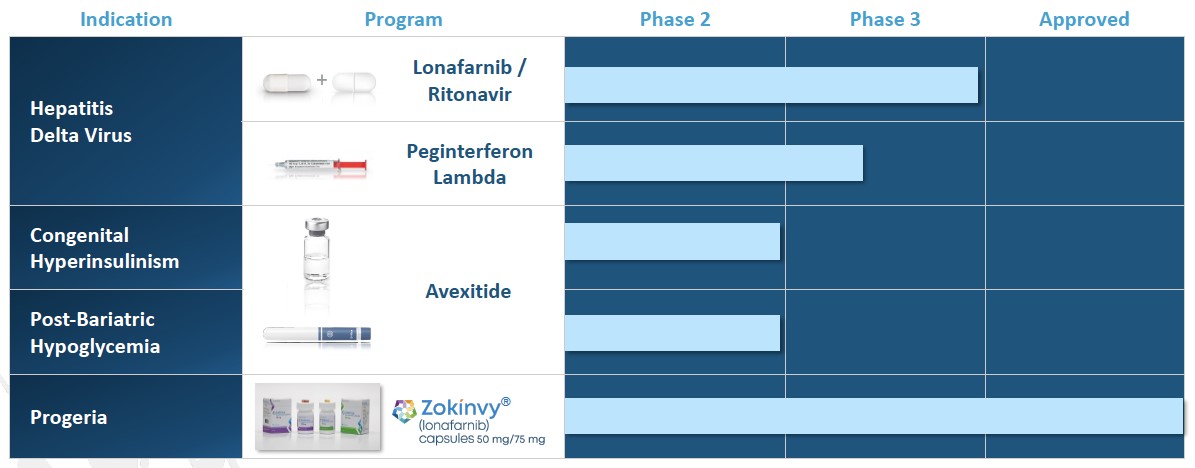
Our fourth NIPHS describes a spectrum of acquired metabolic disorders characterized by inappropriately high insulin levels (hyperinsulinemia) and low blood glucose levels (hypoglycemia), which includes PBH. Avexitide for the treatment of PBH has also been granted Breakthrough Therapy designation by the FDA. Following End of Phase 2 program involves clinical developmentand Scientific Advice meetings with regulatory agencies, we have agreement on a single pivotal Phase 3 study.
Ubenimex is a well-characterized, oral, small-molecule inhibitor of leukotriene A4 hydrolase, or LTA4H, the enzyme responsible for converting the inflammatory mediator leukotriene A4, or LTA4, to leukotriene B4, or LTB4.
Researchers at Stanford have demonstrated for the first time that LTB4 is elevated in both animal models of lymphedema as well as human lymphedema and that elevated LTB4 is associated with tissue inflammation and impaired lymphatic function. In that research, applying inhibitors of LTB4 promoted physiologic lymphatic repair and reversed lymphedema in treated animals. Eiger is developing ubenimexapproved therapy for lymphedema based on its distinct mechanism of action impacting lymphangiogenesis as published in Science Translation Medicine (Tian et al, May 2017). Wethese indications. There are currently conducting a Phase 2 clinical trial, or the ULTRA Study, treating subjects with both primary lymphedema and secondary lymphedema with ubenimex. We completed enrollment of 54approximately 20 identified patients in the ULTRA StudyU.S. who are eligible for treatment with Zokinvy.
Ubenimex was exclusively licensed from Nippon Kayaku,mortality by 60% (p=0.0064) and increased average survival time by at least 2.5 years. The most commonly reported adverse reactions were gastrointestinal (vomiting, diarrhea, nausea), and most were mild or moderate (Grade 1 or 2) in severity. Many patients with HGPS have received continuous Zokinvy therapy for usemore than 10 years.
In January 2018, Phase 2 LIBERTY study results in PAH demonstrated no improvement overall or in key subgroups for bothlicense agreement with Merck to include not only all uses of LNF related to the primary efficacy endpointtreatment of pulmonary vascular resistance (PVR) and the secondary endpoint of 6-minute walk distance (6MWD). No safety signals attributed to ubenimex were identified in the preliminary analysis. Further analysis of data, including biomarkers is ongoing, although we will discontinue development of ubenimex in PAH based on these results. We plan to continue to study ubenimex in lymphedema.
HGPS, but also progeroid laminopathies.
We
Amgen.
•Advance our existing product candidates through late-stage clinical trials, generating meaningful clinical results;
•Work with U.S. and international regulatory authorities for expeditious, efficient development pathways toward registration;
•Prepare for commercialization of each program;
•Use our industry relationships and experience to source, evaluate and in-license well-characterized product candidates to continue pipeline development; and
|
|
•Identify potential commercial or distribution partners for our products in relevant territories.
Our Product Candidates
Lonafarnib in HDV
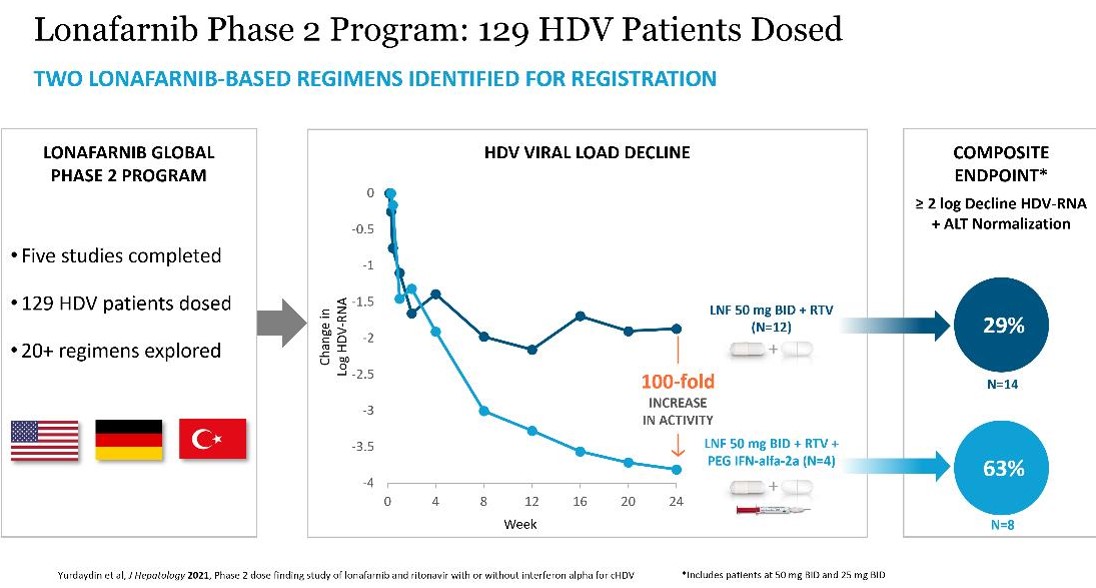
Lonafarnib, or LNF, is a small molecule that we in-licensed from Merck in 2010 and that we are advancing for the treatment of HDV infection. LNF is a well-characterized, orally active inhibitor of farnesyl transferase, an enzyme involved in modification of proteins through a process called prenylation. HDV uses this prenylation process inside host liver cells to complete a key step in its life cycle. LNF inhibits the prenylation step of HDV replication inside liver cells and blocks the virus life cycle at the stage of assembly. Since prenylation is carried out by a host enzyme, there is a higher barrier to develop viral resistance mutations to LNF therapy. We have generated clinical results in over 129 HDV-infected patients in Phase 2 trials, across international study sites, demonstrating rapid decreases in HDV viral loads and no measurable levels of resistance. We have completed five Phase 2 clinical trials including Proof of Concept (NIH), LOWR HDV – 1 (Ankara, Turkey), LOWR HDV – 2 study (Ankara, Turkey), LOWR HDV – 3 (NIH) and LOWR HDV – 4 (Hannover, Germany) in over 129 HDV-infected patients, across international study sites, demonstrating rapid decreases in HDV viral loads and no resistance. In February 2018, we met with the FDA and, subject to agreement on a proposed Phase 3 clinical trial design, have an opportunity for a potentially pivotal single Phase 3 trial as the basis for an NDA filing.
Lambda in HDV
Lambda is a well-characterized, late-stage, first in class, type III interferon, or IFN, that we in-licensed from BMS in April 2016 for the treatment of HDV. Lambda stimulates immune responses that are critical for the development of host protection during viral infections. Lambda targets type III IFN receptors which are distinct from the type I IFN receptors targeted by IFN-alfa. These type III receptors are highly expressed on hepatocytes with limited expression on hematopoietic and central nervous system cells, which in BMS’s clinical trials has demonstrated to reduce the off-target effects associated with other IFNs and improve the tolerability of Lambda. Although Lambda does not use the IFN-alfa receptor, signaling through either the lambda or IFN-alfa receptor complexes results in the activation of the same Jak-STAT signal transduction cascade. Lambda has not been approved for any indication. We are developing Lambda as both a monotherapy and a combination therapy with lonafarnib. Currently, we are conducting a Phase 2 monotherapy study using Lambda to treat HDV and have completed recruitment of 33 patients and are currently dosing at four international sites.
As part of the FDA meeting in February 2018, Eiger discussed the potential regulatory pathways for a Lonafarnib / Ritonavir / Lambda combination regimen including possible study designs and clinical endpoints. The current status of discussion with the FDA is as follows:
|
|
Agency has agreed that the next Eiger study can be a single, registration trial in HDV.
Eiger expects written minutes from the agency (by the end of March 2018) and plans to announce additional details on its clinical development efforts in HDV during the second quarter of this year.
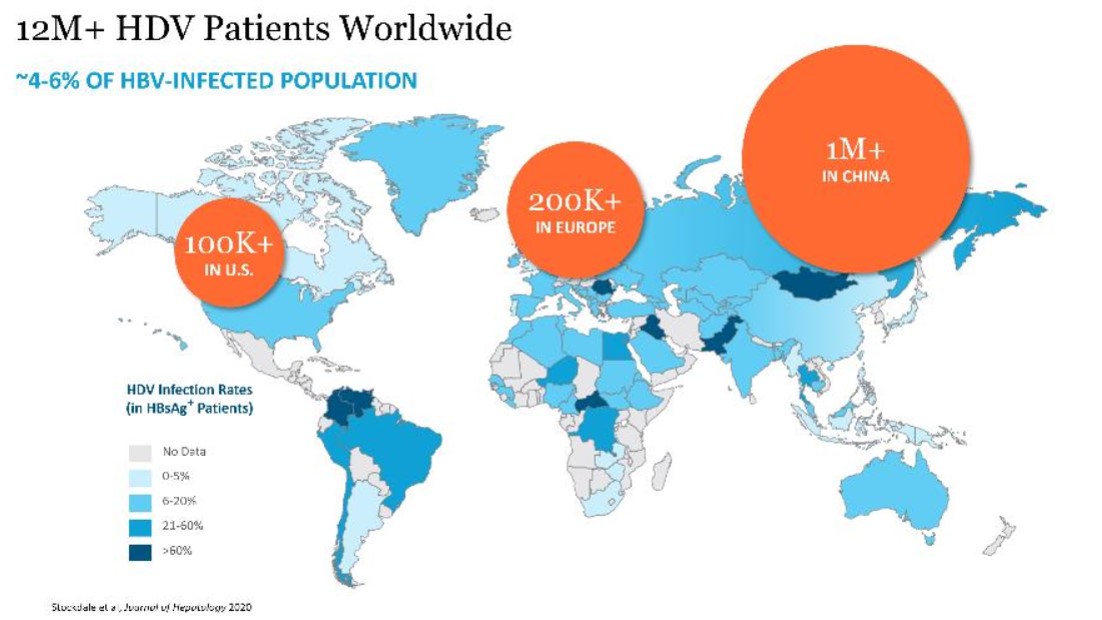
Hepatitis Delta Virus Overview
Hepatitis delta infection
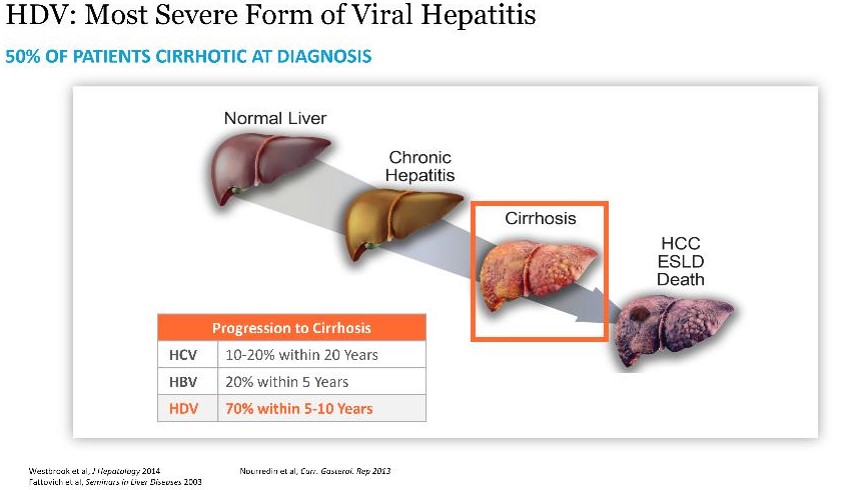
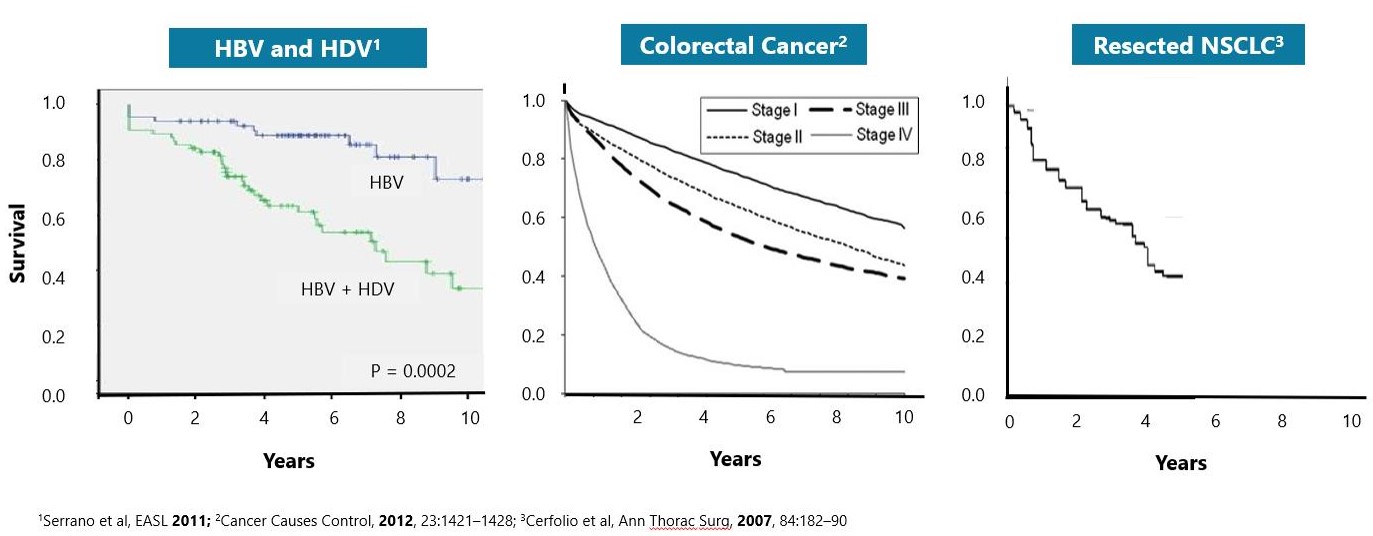
HDV is the most severe form of viral hepatitis. HDV can be acquired either by co-infection (a simultaneous co-infection with HDV and HBV) or by super-infection (HDV infection of someone already harboring a chronic HBV infection). Both co-infection and super-infection with HDV result in more severe complications compared to infection with HBV alone. These complications include a greater likelihood of experiencing liver failure in acute infections and a rapid progression to liver cirrhosis, with an increased chance of developing liver cancer in chronic infections. HDV has the highest fatality rate of all the hepatitis infections at up to 20%. Although HDV/HBV simultaneous co-infection in adults usually resolves completely, in some cases it can become fulminant hepatitis, which carries a very high mortality rate. In the case of super-infections, the predominant form of HDV, HDV super-infection leads to a more severe form of disease than chronic HBV mono-infection. In a study published in 1987 in the Journal of Infectious Diseases (Fattovich, G. et al. “Influence of Hepatitis Delta Virus Infection on Progression to Cirrhosis in Chronic Hepatitis Type B,” J Infect Dis, 1987; 155:931), histological liver deterioration was observed in 77% of HBV patients co-infected with HDV over a 15-year follow-up period, versus 30% of patients infected with HBV alone (p<0.01). In a 2013 study of chronic HBV patients published in the Journal of Gastroenterology and Hepatology, (Gish, R. et al. “Coinfection with hepatitis B and D: epidemiology, prevalence and disease in patients in Northern California,” J Gastroenterol Hepatol, 2013; 28(9):1521), cirrhosis was present in 73% of HBV patients co-infected with HDV, compared to only 22% of those infected with HBV alone. Patients co-infected with HDV are more than twice-as-likely to develop liver-related complications, cirrhosis, or require liver transplants than matched patients infected with HBV alone.
There are diagnostic tests in use today in clinical laboratories to detect anti-HDV antibodies in serum. These tests are currently able to detect acute HDV infections after four weeks, but they are poor tests for active HDV infections.
LNF and lambda.
HDV Replication and Farsenylation
Farnesylation
LNFLonafarnib (LNF) for HDV
LNF Clinical Data
LNF has been tested in
•POC Study (Placebo-controlled LNF monotherapy) (N=14)
The National Institutes of Health, or the2 NIH conducted a 14 patient, double blind, placebo-controlled, proof of conceptproof-of-concept study which was the first ever to evaluate LNF in patients infected with HDV. Patients either received LNF 100 mg (group 1) or LNF 200 mg (group 2) twice daily, or BID, for 28 days with six months of follow-up. Both groups enrolled six treatment participants and two placebo participants. The two placebo patients from group 1 later received open-label LNF as group 2 participants. Doses of 100 mg and 200 mg of LNF administered BID demonstrated a dose dependent decrease in viral loads of 0.73 and 1.54 log decline, respectively, in 28 days. The results were published in The Lancet Infectious Diseases Journal in 2015.
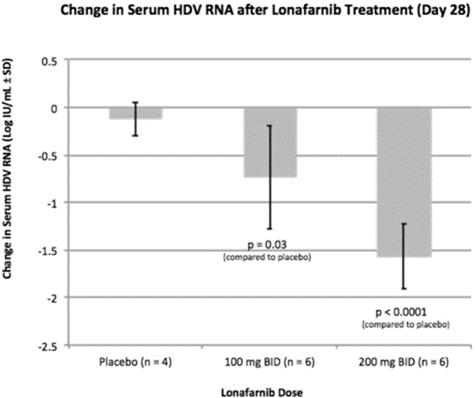
As shown in the table above, statistically significant decreases in HDV RNA viral load were demonstrated by both the 100 mg ofwith two LNF BID (p=0.03) and 200 mg of LNF BID (p<0.0001) active groups versus the placebo.placebo for 28-days. A statistically significant correlation between increasing LNF serum levels and decreasing HDV RNA viral loads was also demonstrated. observed, demonstrating that higher serum levels resulted in greater decline in HDV RNA.
A p-value is a statistical measure of the probability that the difference in two values could have occurred by chance. The smaller the p-value, the greater the statistical significance and confidence in the result. Typically, results are considered statistically significant if they have a p-value less than 0.05, meaning that there is less than a one-in-20 likelihood that the observed results occurred by chance. The FDA requires that sponsors demonstrate the effectiveness and safety of their product candidates through the conduct of adequate and well-controlled studies in order to obtain marketing approval. Typically, the FDA requires a p-value of less than 0.05 to establish the statistical significance of a clinical trial, although there are no laws or regulations requiring that clinical data be statistically significant, or that require a specific p-value, in order for the FDA to grant approval.
In 2014, we initiated the LOWR HDV (Lonafarnib With Ritonavir in HDV) Phase 2 Program. The objective of this program is to identify dose(s) and regimen(s) for registration. To date, 129 HDV subjects have been dosed with LNF in multiple studies including:
LOWR HDV – 1 Study (Combination: LNF with RTV or PEG IFN-α)
LOWR HDV – 2 Study (Dose Finding: LNF + RTV ± PEG IFN-α)
LOWR HDV – 3 Study (QD Dosing: LNF + RTV)
LOWR HDV – 4 Study (Dose-Escalation: LNF + RTV)
LOWR HDV—1 (LOnafarnib With and without Ritonavir in HDV - 1) Phase 2 Study
The LOWR HDV—1 trial studied LNF in 21 subjects who were enrolled into one of seven groups for durations of 4-12 weeks (three patients in each group): LNF 200 mg BID (12 weeks), LNF 300 mg BID (12 weeks), LNF 100 mg TID (5 weeks), LNF 100 mg BID + RTV 100 mg QD (8 weeks), LNF 100 mg BID + PEG-IFN-alfa 180 mcg QW (8 weeks), LNF 200 mg BID + PEG-IFN-alfa 180 mcg QW (8 weeks) and LNF 300 mg BID + PEG-IFN-alfa 180 mcg QW (8 weeks).
In LNF monotherapy treatment groups, increasing the dosageRTV-boosting of LNF from 100 mg three times a dayfor up to 200 mg twice a day to 300 mg twice a day led to greater reductions in viral loads at Week 4 (1.2 logs versus 1.6 logs versus 2.0 logs). However, increasing the dosage24 weeks of LNF also led to increasing gastrointestinal, or GI, intolerability and was not considered for longer term dosing.
In the LNF-RTV combination arm of LOWR HDV—1, 100 mg of LNF BID was combined with 100 mg of RTV once daily. RTV is a pharmacokinetic or PK,(PK) enhancer known to inhibit the metabolism of LNF, allowing lower doses of LNF to be administered, while resulting in higher systemic concentrations of LNF.
The additionPhase 2 LOWR HDV studies identified two LNF-based regimens that can achieve clinically meaningful composite endpoints of 100 mg of RTV once daily to 100 mg LNF BID led to a four- to five-fold increase in the serum concentration of LNF in treated patients compared to LNF 100 mg BID alone. This dose combination led to a mean viral load decrease of 2.4 logs after 28 days of treatment, which is a greater than three-fold reduction in viral load compared to the NIH data of a mean viral load decrease of 0.74 logs after 28 days of monotherapy treatment of 100 mg LNF BID. Extending dosing to Week 8 resulted in a 3.2 viral load decline. Importantly, when therapy was discontinued the viral loads rebounded, which we believe indicates that LNF treatment was eliciting an antiviral effect. The addition of 180 mcg of PEG-IFN-alfa once weekly to 100 mg LNF BID was also more active in reducing HDV RNA versus studies with either agent alone. This dose combination led to a greater reduction in viral load, compared to the NIH results on monotherapy treatment with 100 mg LNF BID, with a mean decrease of 0.74 logs versus 1.8 logs after four weeks. Extending dosing to eight weeks resulted in a 3.0 logs viral load decline. Importantly, when therapy was discontinued the viral loads rebounded. The mean change in HDV RNA for the patients receiving eight weeks of treatment of 100 mg LNF BID in combination with RTV and 100 mg LNF BID in combination with PEG-IFN-alfa is shown below. Viral loads for LNF 200 mg and 300 mg BID in combination with PEG-IFN-alfa was not shown since these dosages were intolerable (all patients discontinued) for future development. LOWR HDV-1 did not include a placebo arm and, as such, statistical significance could not be determined.

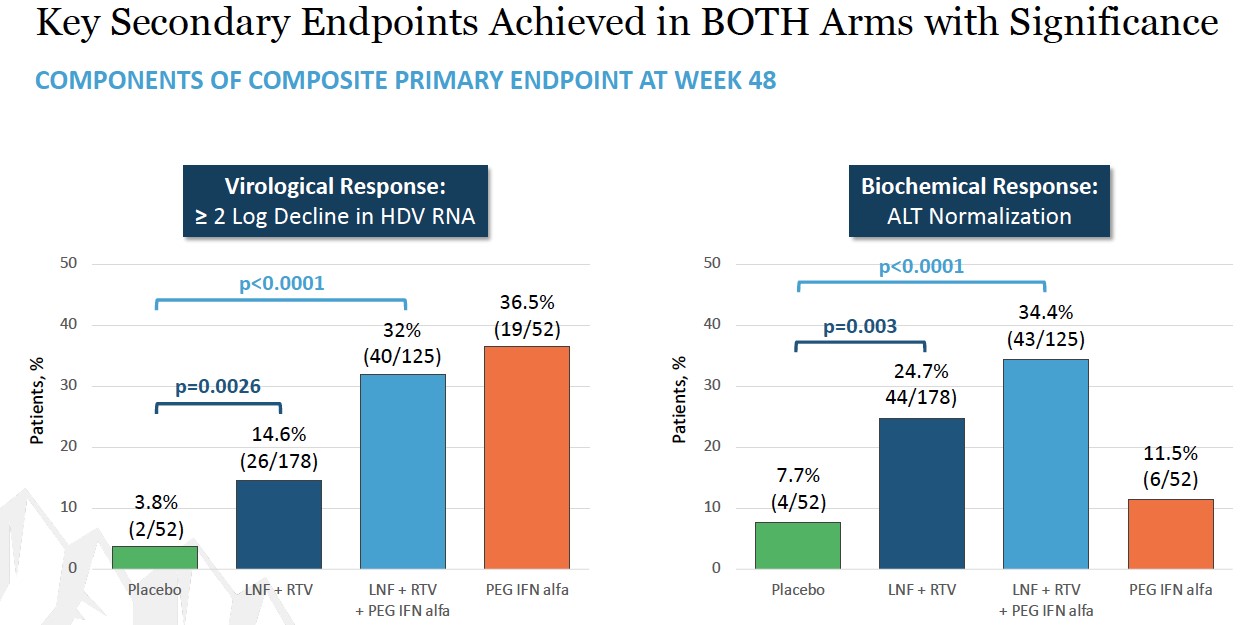
Liver enzymes are often elevated during infections with viral hepatitis, a sign of damage being done to liver cells. In both LNF combination cohorts, all HDV patients enrolled had elevated alanine aminotransferase, or ALT, a liver enzyme that is a surrogate marker of inflammation, prior to receiving any treatment. By the end of eight weeks of combination therapy with LNF and RTV or LNF and PEG-IFN-alfa, all patients’ ALT liver enzymes normalized or trended toward normal while on therapy.
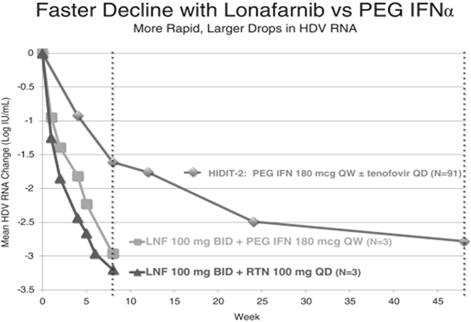
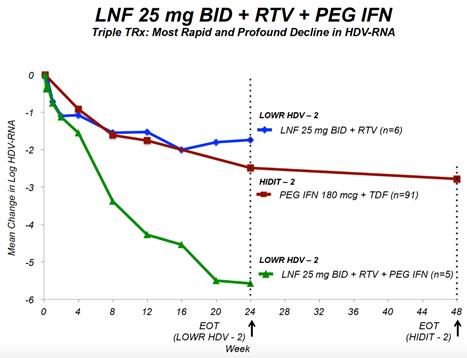
In the three patients receiving LNF in combination with RTV and the three patients receiving LNF in combination with PEG-IFN-alfa, we observed decreases in HDV RNA viral load of approximately 3.2 logs and 3.0 logs after eight weeks of treatment, respectively. For comparison, and as shown in the figure below, published data from the HIDIT-2 trial of PEG-IFN-alfa in 91 HDV infected patients demonstrated a mean decline in HDV RNA of approximately 1.6 logs and 2.7 logs after 8 weeks and 48 weeks, respectively. The HIDIT-2 (Hep-Net International Delta Hepatitis International Trial-II) was a multicenter randomized trial studying effects of PEG-IFN-alfa plus tenofovir in chronic HDV patients, and is the largest clinical study to date in HDV. The HIDIT-2 trial was conducted on 91 patients, whereas the LOWR HDV—1 study was conducted on an aggregate of 21 patients, with three patients per treatment arm. If the LOWR HDV—1 trial was conducted on a larger group of patients, the mean HDV RNA decline may differ≥ 2 logs from the 3.2 logbaseline and 3.0 log declines after eight weeksnormalized ALT at Week 24: all-oral regimen of treatment observed in the three patient arms receiving LNF combination treatment in the LOWR HDV—1 trial. However, based on clinical results to date, we expect all patients who are treated with LNF to show a viral load response.
LOWR HDV—2 (LOnafarnib With Ritonavir in HDV - 2) Phase 2 Study
LOWR HDV – 2 is a dose-finding Phase 2 study of multiple doses of LNF boosted by RTV with and without PEG-IFN-alfa in 58 subjects for 24-48 weeks of treatment with 24 weeks of follow-up, with the aim to identify regimen(s) with improved tolerability for the longer-term registration studies. LOWR HDV – 2 (conducted as an extension of LOWR HDV – 1, collectively EIG-300) was conducted at Ankara University in Turkey and we have identified and certain good clinical practice violations at this site that may impact certain data and information that we plan to submit to the FDA.
Fifty-eight subjects were enrolled into one of ten groups of different LNF with RTV and/or PEG-IFN-alfa combinations for 12 or 24 or 48 weeks as follows: Group 1: LNF 100 mg BID + RTV 50 mg BID; Group 2: LNF 100 mg BID + RTV 100 mg QD; Group 3: LNF 150 mg QD + RTV 100 mg QD; Group 4: LNF 100 mg QD + RTV 100 mg QD; Group 5: LNF 75 mg BID + RTV 100 mg BID with PEG-IFN-alfa 180 mcg QW added at week 12; Group 6: LNF 50 mg BID +boosted with RTV 100 mg BID; Group 7:twice daily and combination regimen of LNF 50 mg BID + RTV 100 mg BID with PEG-IFN-alfa 180 mcg QW added at week 12; Group 8: LNF 50 mg BID + RTV 100 mg BID + PEG-IFN-alfa 180 mcg QW; Group 9: LNFor 25 mg BID +boosted with RTV 100 mg BID; and Group 10: LNF 25 mg BID + RTV 100 mg BID + PEG-IFN-alfa 180 mcg QW.
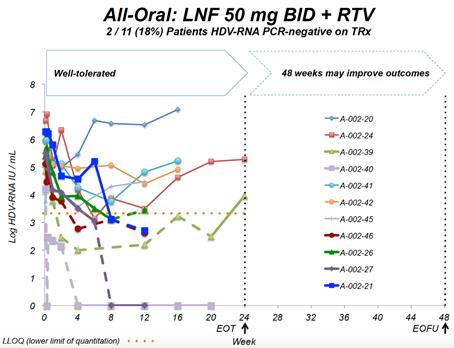
End of study results were presented at EASL 2017 in Amsterdam, Netherlands. Key findings were that in the all-oral LNF 50 mg BID + RTV 100 mg BID regimen, 7 of 14 (50%) patients demonstrated ≥ 2 log decline or PCR-negative at Week 24. Combination regimens of LNF 25 mg BID + RTV 100 mg BID + PEG IFN-α 180 mcg QW resulted in the highest response rates of 5 of 7 (71%) patients achieving ≥ 2 log decline or PCR-negative at Week 24 and the majority of patients normalized ALT at Week 24. In addition, GI AEs predominantly were predominantly mild and moderate. Reported data used a research use only assay at Ankara University.
LOWR HDV—3 (LOnafarnib With Ritonavir in HDV - 3) Phase 2 Study
LOWR HDV – 3 was a double-blind, randomized, placebo-controlled study designed to evaluate the efficacy and tolerability of once-daily doses of LNF – 50 mg, 75 mg and 100 mg – each combined with RTV 100 mg once daily for 12 (N=9) or 24 (N=12) weeks. Twenty-one patients with chronic hepatitis delta were randomized into one of six treatment groups. LOWR HDV – 3 was conducted at the National Institutes of Health (NIH) Bethesda, MD. This study has completed.
End of study results were presented at EASL 2017 in Amsterdam, Netherlands. After 12 weeks of therapy, the median log HDV RNA decline from baseline was 1.60 log IU/mL (LNF 50 mg), 1.33 (LNF 75 mg) and 0.83 (LNF 100 mg) (p=0.001)PEG IFN-alfa-2a (see figures below). In subjects treated for 24 weeks, HDV RNA levels significantly differed from placebo (p=0.04). During the study, 6 patients achieved ≥ 2 log decline in HDV RNA; HDV RNA levels became undetectable in one subject and < LLOQ in three subjects. ALT normalization was achieved in 47% of patients. Adverse events were mild to moderate and included nausea, vomiting, dyspepsia, anorexia, diarrhea, and weight loss. There were no treatment discontinuations for adverse events.
The LOWR HDV-3 study demonstrated that the all-oral combination of once-daily ritonavir boosted lonafarnib was safe and tolerable in patients for up to 6 months of therapy and demonstrated antiviral activity. Reported data used Robogene® HDV RNA Quantification Kit 2.0.
LOWR HDV—4 (LOnafarnib With Ritonavir in HDV - 4) Phase 2 Study
LOWR HDV – 4 was an open-label study to evaluate the efficacy and tolerability of dose-escalation of LNF combined with RTV administered twice daily for dosing durations of 24 weeks. Fifteen patients were initiated at LNF 50 mg and RTV 100 mg twice daily, and dose-escalated up to LNF 100 mg twice daily at the discretion of the investigator and patient tolerability. LOWR HDV – 4 was conducted at Hannover Medical School in Hannover, Germany.
End of study results were presented at EASL 2017 in Amsterdam, Netherlands. At end of treatment, 5 of 15 (33%) patients reached and maintained LNF 100 mg BID + RTV through EOT; 1 of 5 (20%) patients achieved undetectable HDV-RNA, and 1 of 5 (20%) patients achieved HDV-RNA < LLOQ. ALT normalization was demonstrated in 53% patients.
In follow-up visits, 1 of 15 (7%) patients remained HDV-RNA < LLOQ and 3 of 15 (20%) patients dropped > 2 logs from baseline. Gastrointestinal AEs were mostly grade 1-2; 8 of 15 (53%) patients required dose reduction and 2 of 15 (13%) patients were discontinued. Reported data used Robogene® HDV RNA Quantification Kit 2.0.
Key findings from the LOWR HDV Program demonstrate that LNF (all-oral) can achieve HDV-RNA negativity on-treatment, and that the most robust HDV-RNA on-treatment on-anti-viral activity is observed in LNF triple therapy with PEG-IFN-alfa. Findings demonstrate that LNF-based regimens can normalize ALTs in 60% of patients. WithThese dosing regimens of LNF 25 and 50 mg BID identifiedwere associated with predominantly grade 1 GI AEs amongst per-protocol treated patient.
Our Second HDV Therapeutic Approach:Program: Peginterferon Lambda (lambda) for HDV
demonstrated to reduce the off-target effects associated with other IFNs and improve the tolerability of Lambdalambda (Chan 2016). Although Lambdalambda does not use the IFN-alfa receptor, signaling through either the IFN-lambda or IFN-alfa receptor complexes results in the activation of the same Jak-STAT signal transduction cascade.
a combination with LNF + RTV.
A head-to-head study comparing the safety and efficacy of lambda versus PEG-IFN-alfa was reported in 2016 by Chan et al. In this study, HBeAg(+) patients were treated with either Lambda (n=80) or PEG-IFN-alfa (n=83) for 48 weeks. A subset of on-treatment safety data is summarized in the table below. Lambda is generally better-tolerated when compared to PEG-IFN-alfa. Lower rates of flu-like symptoms and musculoskeletal symptoms were observed with lambda versus PEG-IFN-alfa.

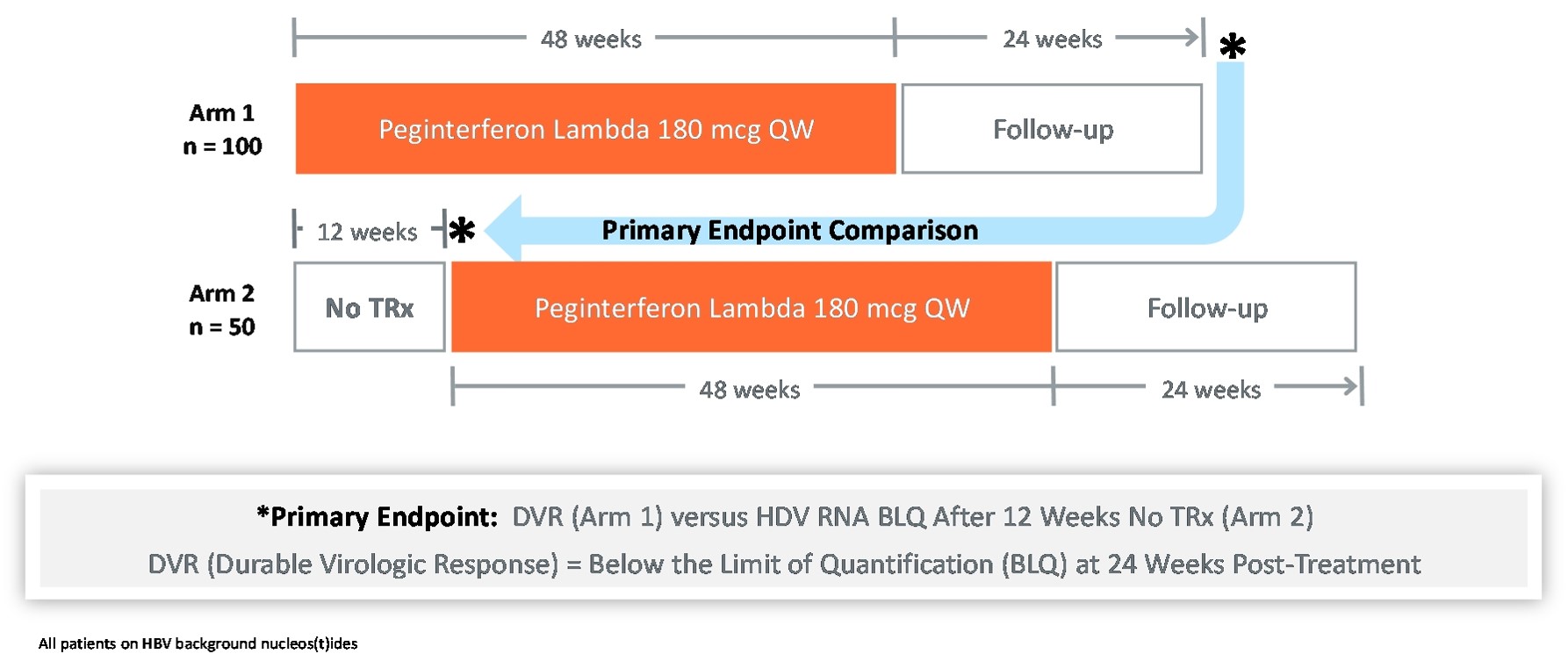
LIMT HDV
The LIMT HDV Phase 2 Clinical Trial is
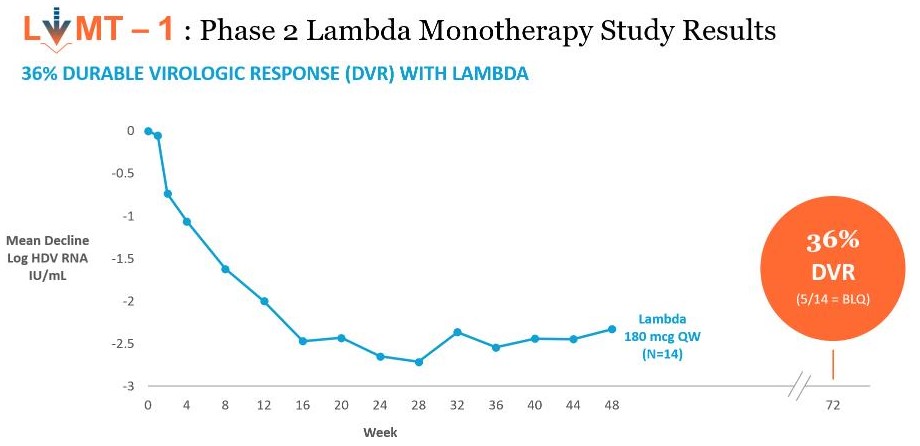
Interim
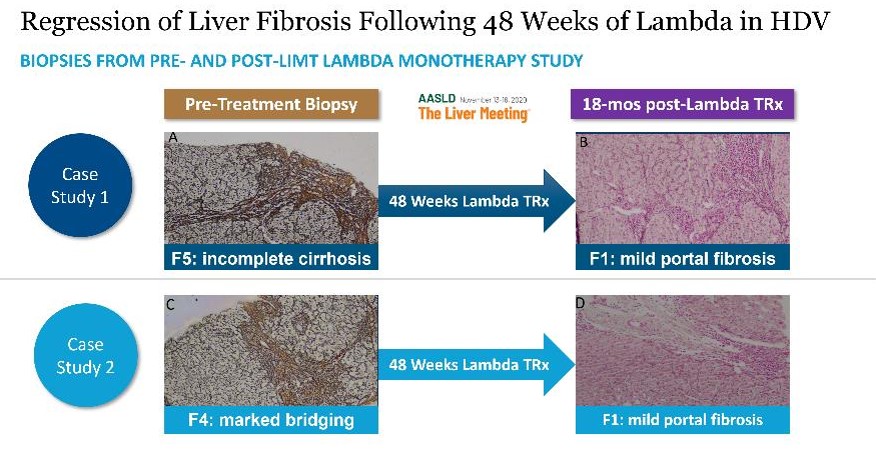
Interim data shows that Lambda demonstrates comparable anti-HDV activity to historical PEG-alfa and that Lambda is well toleratedpatients were HDV-RNA negative at end of treatment. At Week 48, patients in the majority120 μg lambda treated group experienced a 1.5 log
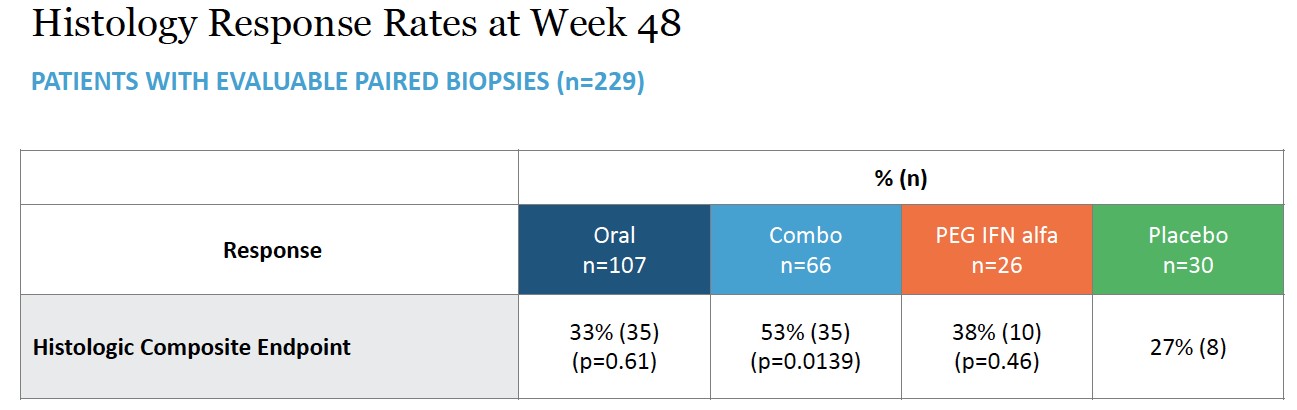
condition. Severe cases have historically been surgically managed with near-total to total pancreatectomy, which results in insulin dependent diabetes and is associated with a greater than 6% surgical mortality risk. called PREVENT in October 2018. The PBH. To date, our third-party manufacturers have met the manufacturing requirements for the product candidates. We expect third-party manufacturers to be capable of providing sufficient quantities of our product candidates to meet anticipated All of our third-party manufacturers are subject to periodic audits to confirm compliance with applicable regulations and must pass inspection before we can manufacture our products for commercial sales. GMP products from all CMOs. third-party CMOs. We also rely on trade secrets, know-how, and continuing innovation to develop and maintain our competitive position. We cannot be certain that patents will be granted with respect to any of our pending patent applications or with respect to any patent applications filed by us in the future, nor can we be sure that any of our existing patents or any patents granted to us in the future will be commercially useful in protecting our technology. intellectual property owned by others; (iv) preserve the confidentiality of our trade secrets, and (v) operate without infringing the valid and enforceable patents and other proprietary rights of others. In addition to maintaining our existing proprietary assets, we seek to strengthen our proprietary positions when economically reasonable to do so. Our ability to augment our proprietary position relies on its: (i) know-how; (ii) ability to access technological innovations, and (iii) ability to in-license technology when appropriate. (PCT). Other Proprietary Rights and Processes There are other therapies that are used or may be used for our targeted indications, and these other products in clinical development or marketed for other indications may be used in competition with our product candidates if we are able to identify potential market opportunities of interest. For example, HDV has not been generally identified as a target for development compared to hepatitis B or hepatitis C, and products on the market or in development for those indications may potentially be tested in HDV as the understanding of the potential medical need for therapies in this indication become more widely understood. the study is expected to complete by the end of 2023. The term of the EGI APA extends until expiration of all payment obligations, and we may terminate the agreement upon notice to EGI. EGI may terminate the EGI APA if we fail to use commercially reasonable efforts to develop and commercialize licensed products. In addition, each party may terminate the EGI APA for the other party’s uncured material breach or bankruptcy. In the event of any termination, other than termination by us for EGI’s breach, we will assign the purchased assets back to EGI. Asset Purchase Agreement with Tracey McLaughlin and Colleen Craig avexitide. License Agreement with Bristol-Myers Squibb Company Under the BMS License Agreement, BMS granted us an exclusive, worldwide, license to research, develop, manufacture, and sell products within the specified periods, subject to our extension rights. Preclinical tests include laboratory evaluation of the product’s chemistry, formulation, and toxicity, as well as animal studies to characterize and assess the potential safety, •Phase 1. The •Phase 2. The trials are conducted using a limited patient population for the purposes of preliminarily determining the effectiveness of the •Phase 3. If a compound demonstrates evidence of efficacy and has an acceptable safety profile in the Phase 2 clinical trials, then Phase 3 clinical trials are undertaken to obtain additional information from an expanded and diverse patient population, at multiple, geographically dispersed clinical trial sites, in randomized controlled studies often with a double-blind design to maximize the reproducibility of the study results. Typically, a minimum of two positive Phase 3 clinical trials are submitted to support the product’s marketing application. These Phase 3 clinical trials are intended to provide sufficient data demonstrating evidence of the safety, efficacy, purity and Sponsors of clinical trials for investigational After the FDA evaluates the application and conducts its inspections, it issues either an approval letter or a complete response letter. A complete response letter generally outlines the deficiencies contained in the submission and may require substantial additional testing or information in order for the FDA to reconsider the application, including potentially significant, expensive and time-consuming requirements related to clinical trials, nonclinical studies or manufacturing. Even if such data are submitted, the FDA may ultimately decide that the NDA or BLA does not satisfy the criteria for approval. Data from clinical trials are not always conclusive, and the FDA may interpret data differently than we do. If and when those deficiencies have been addressed to the FDA’s satisfaction in a resubmission of the application, the FDA will typically issue an approval letter. The FDA has committed to reviewing such resubmissions in two or six months depending on the type of additional information requested. FDA approval is never guaranteed. The FDA may refuse to approve an application if applicable regulatory criteria are not satisfied. industry-sponsored scientific and educational activities and promotional activities involving the Internet. Failure to comply with these requirements can result in adverse publicity as well as significant penalties, including the issuance of warning letters directing a company to correct any deviations from the FDA’s standards. The FDA may also impose a requirement that future advertising and promotional materials be pre-cleared by the FDA, and the product. intended to induce prescribing, purchases or recommendations may be subject to scrutiny if they do not qualify for an exception or safe harbor. Failure to meet all of the requirements of a particular applicable statutory exception or regulatory safe harbor does not make the conduct per se illegal under the Anti-Kickback Statute. Instead, the legality of the arrangement will be evaluated on a case-by-case basis based on a cumulative review of all of its facts and circumstances. Several courts have interpreted the statute’s intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals of federal healthcare covered business, the Anti-Kickback Statute has been violated. Violations of the federal Anti-Kickback Statute are punishable by imprisonment for up to ten years and statutory fines of up to $100,000. Additional criminal fines can be imposed under federal U.S. criminal procedure laws. If our operations are found to be in violation of any of the Additionally, coverage policies and third‑party reimbursement rates may change at any time. Therefore, even if favorable coverage and reimbursement status is attained for one or more of our products, less favorable coverage policies and reimbursement rates may be implemented in the future. and Human Capital We have also agreed with The Progeria Research Foundation to make Zokinvy available to progeria (HGPS and processing-deficient progeroid laminopathies) patients under an expanded access program that may not result in payment to us. Future clinical trials of new therapies for progeria conducted by third parties may also result in patients converting from commercially reimbursed Zokinvy to product provided through clinical trials and result in lower revenues received by us. •continue the clinical development of our product candidates; •in-license or acquire additional product candidates; •undertake the manufacturing or have manufactured our product candidates; •advance our programs into larger, more expensive clinical studies; •identify •seek regulatory and marketing approvals and reimbursement for our product candidates; •establish a sales, marketing, and distribution infrastructure to commercialize any products for which we may obtain marketing approval and market ourselves; •seek to identify, assess, acquire, and/or develop other product candidates; •make milestone, royalty or other payments under third-party license agreements; •seek to maintain, protect, and expand our intellectual property portfolio; •seek to attract and retain skilled personnel; •create additional infrastructure to support our operations as a public company and our product development and planned future commercialization efforts; and experience any delays or encounter issues with the development and potential for regulatory approval of our clinical candidates such as safety issues, clinical trial accrual delays, longer follow-up for planned studies, additional major studies, or supportive studies necessary to support marketing approval. •completing research and development of our product candidates; •obtaining additional and maintaining current regulatory and marketing approvals for our product candidates; •manufacturing product candidates and establishing and maintaining supply and manufacturing relationships with third parties that meet regulatory requirements and our supply needs in sufficient quantities to meet market demand for our product candidates, if approved; •marketing, launching and commercializing product candidates for which we obtain regulatory and marketing approval, either directly or with a collaborator or distributor; •gaining market acceptance of our product candidates as treatment options; •addressing any competing products; •protecting and enforcing our intellectual property rights, including patents, trade secrets, and know-how; •negotiating favorable terms in and maintaining any collaboration, licensing, or other arrangements into which we may enter; •obtaining reimbursement or pricing for our product candidates that supports profitability; and •attracting, hiring, and retaining qualified personnel. Even if regulatory approval or authorization for use in these jurisdictions. product candidates significantly less competitive and potentially not viable investments for further development. Although we •the FDA or comparable foreign regulatory authorities may disagree with the design, size or implementation of our clinical studies; •the population studied in the clinical program may not be sufficiently broad or representative to assure safety in the full population for which we seek approval; •the FDA or comparable foreign regulatory authorities may disagree with our interpretation of data from our development efforts; •the data collected from clinical studies of our product candidates may not be sufficient or complete or meet the regulatory requirements to support the submission of •the FDA or comparable foreign regulatory authorities may find failures in our manufacturing processes, validation procedures and specifications, or facilities of our third-party manufacturers with which we contract for clinical and commercial supplies that may delay or limit our ability to obtain regulatory approval for our product candidates; and •the approval policies or regulations of the FDA or comparable foreign regulatory authorities may significantly change in a manner rendering our NDA, BLA or other submission insufficient for approval. conduct larger, well-controlled studies in our proposed indications to verify the results obtained to date and to support any regulatory submissions for further clinical development. A number of companies in the biopharmaceutical industry have suffered significant setbacks in advanced clinical studies due to lack of efficacy or adverse safety profiles despite promising results in earlier, smaller clinical studies. Moreover, clinical data are often susceptible to varying interpretations and analyses. We do not know whether any Phase 2, Phase 3, or other clinical studies we have conducted or may conduct will demonstrate consistent or adequate safety, efficacy, For example, in 2018 we announced negative results from two of our Phase 2 clinical trials of ubenimex in two different indications, and as a result we have terminated further development of ubenimex. •delays in reaching agreement on acceptable terms with •delays in obtaining required Institutional Review Board •failure to permit the conduct of a study by regulatory authorities, after review of an investigational new drug •delays in recruiting qualified patients, or patients dropping out of, in our clinical •failure to perform the clinical studies in accordance with the FDA’s •occurrence of adverse events associated with our product candidates; •changes in regulatory requirements and guidance that require amending or submitting new clinical protocols; •the cost of clinical studies of our product candidates; •negative or inconclusive results from our clinical trials which may result in our deciding, or regulators requiring us, to conduct additional clinical studies or abandon development programs in other ongoing or planned indications for a product candidate; and •delays in reaching agreement on acceptable terms with third-party manufacturers and the time for manufacture of sufficient quantities of our product candidates for use in clinical studies. For example, the ECG abnormalities seen with lonafarnib in HGPS and PL patients has the potential to impact the labeling for lonafarnib-based regimens for HDV. Our avexitide product candidate has been studied in 39 HI patients and over 70 PBH patients, and the most common adverse events are injection site bruising/reaction, nausea, and headache. There is no guarantee that additional or more severe side effects will not be identified through ongoing clinical studies for other uses of avexitide in clinical trials. •regulatory authorities may withdraw approvals of such products; •regulatory authorities may require additional warnings on the label; •we could be sued and held liable for harm caused to patients; and •our reputation may suffer. •issue warning letters; •impose civil or criminal penalties; •suspend or withdraw regulatory approval; •suspend any of our ongoing clinical studies; •refuse to approve pending applications or supplements to approved applications submitted by us; •impose restrictions on our operations, including closing our contract manufacturers’ facilities; or •require a product recall. candidate products. •We may be unable to identify manufacturers on acceptable terms or at all; •Our third-party manufacturers might be unable to timely formulate and manufacture our product or produce the quantity and quality required to meet our clinical and commercial needs, if any; •Contract manufacturers may not be able to execute our manufacturing procedures appropriately; •Our future contract manufacturers may not perform as agreed or may not remain in the contract manufacturing business for the time required to supply our clinical trials or to successfully produce, store and distribute our products; •Manufacturers are subject to ongoing periodic unannounced inspection by the FDA and corresponding state agencies to ensure strict compliance with cGMP and other government regulations and corresponding foreign standards. We do not have control over third-party manufacturers’ compliance with these regulations and standards; •We may not own, or may have to share, the intellectual property rights to any improvements made by our third-party manufacturers in the manufacturing process for our product candidates; and •Our third-party manufacturers could breach or terminate their agreement with us. Each of these risks could delay our clinical trials, the approval Moreover, we expect that the sales of Zokinvy to patients with progeria will have limited profits given the ultra-rare nature of these diseases. substantially greater financial, technical, and other resources, such as larger research and development staff and experienced marketing and manufacturing organizations. Additional mergers and acquisitions in the biotechnology and pharmaceutical industries may result in even more resources being concentrated in our competitors. As a result, these companies may obtain regulatory approval more rapidly than we are able to and may be more effective in selling and marketing their products as well. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large, established companies. Competition may increase further as a result of advances in the commercial applicability of technologies and greater availability of capital for investment in these industries. Our competitors may succeed in developing, acquiring, or licensing on an exclusive basis, products that are more effective or less costly than any product candidate that we may develop, or achieve earlier patent protection, regulatory approval, product commercialization, and market penetration than we do. Additionally, technologies developed In addition, we have established an expanded access program in order to make Zokinvy available for patients with progeria, which requires additional resources and costs to support. •the timing of our receipt of any marketing and commercialization licensures; •the prevalence and severity of the disease and any side effects; •the clinical indications for which approval is granted, including any limitations or warnings contained in a product’s approved labeling; •the convenience and ease of administration; •the cost of treatment; •the willingness of the patients and physicians to accept these •the marketing, sales and distribution support for the product; •the pricing and availability of third-party insurance coverage and reimbursement. For example, Zokinvy for patients with HGPS and processing-deficient progeroid laminopathies provided under an expanded access program may not result in reimbursement. In the United States, parties may challenge their validity, enforceability, or scope, which may result in such patents being narrowed, found unenforceable or invalidated. Furthermore, even if they are unchallenged, our patents and patent applications may not adequately protect our intellectual property, provide exclusivity for our product candidates, or prevent others from designing around our claims. Any of these outcomes could impair our ability to prevent competition from third parties, which may have an adverse impact on our business. Although we have licensed a number of patents covering methods of use and certain compositions of matter, we do not have complete patent protection for our product candidates. For example, in connection with the are typically not published until 18 months after filing, or in some cases not at all. We therefore cannot be certain that it or our licensors were the first to make the invention claimed in our owned and licensed patents or pending applications, or that we or our licensor were the first to file for patent protection of such inventions. Assuming the other requirements for patentability are met, in the United States prior to March 15, 2013, the first to make the claimed invention is entitled to the patent, while outside the United States, the first to file a patent application is entitled to the patent. After March 15, 2013, under the Leahy-Smith America Invents Act through development. Accordingly, there may be third-party patents that would impair our ability to commercialize product candidates and we cannot assure you that we could obtain a license, or even if available, whether such license might be obtained on commercially reasonable terms. Even in those situations where we conduct a freedom to operate analysis, there can be no assurance that we would identify all relevant or necessary patents and patent applications that may apply to the manufacture and commercialization of our product candidates. Because patent applications can take many years to issue, there may be currently pending patent applications that may later result in issued patents that our product candidates may If we are unable to successfully obtain and maintain rights to required third-party intellectual property, we may have to abandon development of that product candidate or pay additional amounts to the third party, and our business and financial condition could suffer. If we fail to comply with obligations in the agreements under which we license intellectual property and other rights from third parties or otherwise experience disruptions to our business relationships with our licensors, we could lose license rights that are important to our business. We may not be able to protect our intellectual property rights throughout the world. on January 3, 2023, Sriram Ryali notified the Company of his resignation as the Company’s Chief Financial Officer, effective January 20, 2023, and on February 17, 2023, Erik Atkisson resigned as our General Counsel, Corporate Secretary and Chief Compliance Officer. business and result in increased costs or loss of revenue, other financial and reputational harm to us, theft of •our research or business development methodology or search criteria and process may be unsuccessful in identifying potential product candidates; •we may not be able or willing to assemble sufficient resources to acquire or discover additional product candidates; •our product candidates may not succeed in preclinical or clinical testing; •our potential product candidates may be shown to have harmful side effects or may have other characteristics that may make the products unmarketable or unlikely to receive marketing approval; •competitors may develop alternatives that render our product candidates obsolete or less attractive; •product candidates we develop may be covered by third parties’ patents or other exclusive rights; •the market for a product candidate may change during our program so that such a product may become unreasonable to continue to develop; •a product candidate may not be capable of being produced in commercial quantities at an acceptable cost, or at all; and •a product candidate may not be accepted as safe and effective by patients, the medical community, or third-party payors. Further, it is possible that additional governmental action is taken in response to the COVID-19 pandemic. •the federal Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, receiving, offering or paying anything of value as remuneration, directly or indirectly, to induce, or in return for, the purchase or recommendation of an item or service reimbursable under a federal healthcare program, such as the Medicare and Medicaid programs; •federal civil and criminal false claims laws and civil monetary penalty laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other third-party payors that are false or fraudulent; •The Physician Payments Sunshine Act, which requires manufacturers of drugs, devices, biologics, and medical supplies to report annually to •state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws that may apply to items or services reimbursed by any third-party payors, including commercial insurers, state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government, or otherwise restrict payments that may be made to healthcare providers and other potential referral sources; state laws that require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing We face potential product liability, and, if successful claims are brought against us, we may incur substantial liability and costs. If the use or misuse of our product candidates harm patients or is perceived to harm patients even when such harm is unrelated to our product candidates, our regulatory approvals could be revoked or otherwise negatively impacted, and we could be subject to costly and damaging product liability claims. •impairment of our business reputation; •initiation of investigations by regulators; •withdrawal of clinical trial participants; •costs due to related litigation; •distraction of management’s attention from our primary business; •substantial monetary awards to patients or other claimants; •the inability to commercialize our product candidates; •product recalls, withdrawals or labeling, marketing or promotional restrictions; and •decreased demand for our product candidates, if approved for commercial sale. commercialization efforts, research and development efforts and business operations, environmental damage resulting in costly clean-up and liabilities under applicable laws and regulations governing the use, storage, handling, and disposal of these materials and specified waste products. Although we believe that the safety procedures utilized by our licensors and our third-party manufacturers for handling and disposing of these materials generally comply with the standards prescribed by these laws and regulations, we cannot guarantee that this is the case or eliminate the risk of accidental contamination or injury from these materials. In such an event, we may be held liable for any resulting damages and such liability could exceed our resources and state or federal or other applicable authorities may curtail our use of certain materials and/or interrupt our business operations. Furthermore, environmental laws and regulations are complex, change frequently, and have tended to become more stringent. We cannot predict the impact of such changes and cannot be certain of our future compliance. We do not currently carry biological or hazardous waste insurance coverage. operations and prospects. region may disrupt our supply chain or limit our ability to obtain sufficient materials for our drug products. treat COVID-19. In addition, to the extent the ongoing COVID-19 pandemic adversely affects our business and results of this ‘‘Risk Factors’’ section. •results or delays in preclinical studies or clinical trials; •our decision to initiate a clinical trial, not to initiate a clinical trial or to terminate an existing clinical trial; •unanticipated serious safety concerns related to the use of any of our product candidates; •reports of adverse events in other gene therapy products or clinical trials of such products; •inability to obtain additional funding; •any delay in filing an IND, NDA, BLA, or •our ability to obtain regulatory approvals for our product candidates, and delays or failures to obtain such approvals; •failure of any of our product candidates, if approved, to achieve commercial success; •failure to obtain •failure to maintain our existing third-party license and supply agreements; •failure by our licensors to prosecute, maintain, or enforce our intellectual property rights; •changes in laws or regulations applicable to our product candidates; •adverse regulatory authority decisions; •introduction of new products, services, or technologies by our competitors; •failure to meet or exceed financial and development projections we may provide to the public; •failure to meet or exceed the financial and development projections of the investment community; •the perception of the pharmaceutical industry by the public, legislatures, regulators, and the investment community; •announcements of significant acquisitions, strategic partnerships, joint ventures, or capital commitments by us or our competitors; •disputes or other developments relating to proprietary rights, including patents, litigation matters, and our ability to obtain patent protection for our technologies; •additions or departures of key personnel; •significant lawsuits, including patent or stockholder litigation; •if securities or industry analysts do not publish research or reports about our business, or if they issue adverse or misleading opinions regarding our business and stock; •changes in the market valuations of similar companies; •general market or macroeconomic conditions; •sales of our common stock by us or our stockholders in the future; •trading volume of our common stock; •announcements by commercial partners or competitors of new commercial products, clinical progress or the lack thereof, significant contracts, commercial relationships or capital commitments; •adverse publicity relating to the hepatitis market generally, including with respect to other products and potential products in such markets; •the introduction of technological innovations or new therapies that compete with potential products of ours; •changes in the structure of health care payment systems; and •period-to-period fluctuations in our financial results. who have not previously managed and operated a public company. These executive officers and other personnel will need to devote substantial time to gaining expertise regarding operations as a public company and compliance with applicable laws and regulations. In addition, it may be more difficult for us to attract and retain qualified individuals to serve on our board of directors or as executive officers, which may adversely affect investor confidence and could cause our business or stock price to suffer. Price Range High Low Year Ended December 31, 2017 First Quarter $ 12.65 $ 10.15 Second Quarter $ 11.60 $ 6.10 Third Quarter $ 12.05 $ 7.13 Fourth Quarter $ 14.50 $ 10.26 Year Ended December 31, 2016 First Quarter $ 25.80 $ 12.90 Second Quarter $ 23.10 $ 17.06 Third Quarter $ 20.63 $ 13.15 Fourth Quarter $ 14.75 $ 10.71 Holders of Record 2022. a focus on enhancing long-term shareholder value while fulfilling the promise of advancing our high-potential product candidates for patients with serious diseases. other period costs. •expenses incurred under agreements with consultants, contract research organizations and clinical trial sites that conduct research and development activities on our behalf; •laboratory and vendor expenses related to the execution of clinical trials; •contract manufacturing expenses, primarily for the production of clinical trial supplies; •license fees associated with our license agreements; and •internal costs that are associated with activities performed by our research and development organization and generally benefit multiple programs. These costs are not separately allocated by product candidate. Unallocated internal research and development costs consist primarily of: ◦personnel costs, which include salaries, benefits and stock-based compensation expense; ◦allocated facilities and other expenses, which include expenses for rent and maintenance of facilities and depreciation expense; and ◦regulatory expenses and technology license fees related to development activities. The largest component of our operating expenses has historically been the investment in clinical trials, including contract manufacturing Year Ended December 31, 2017 2016 2015 Product candidates: Lonafarnib HDV $ 4,284 $ 5,237 $ 2,052 Lambda HDV 3,892 7,244 — Exendin 9-39 PBH 3,173 2,984 115 Ubenimex PAH (terminated in January 2018) 9,789 10,393 648 Ubenimex Lymphedema 2,132 2,271 198 Internal research and development costs 6,249 4,885 5,104 Total research and development expense $ 29,519 $ 33,014 $ 8,117 The COVID-19 pandemic presents additional risks and uncertainties associated with developing drugs, We expect our selling, general and administrative expenses to increase in the future due to sales and marketing activities from the commercialization of our product candidates. (expense) income, net 2022. Costs liabilities. We record forfeitures when they occur. following assumptions: prior to exercise. 2021 Years Ended December 31, Increase / (Decrease) % Change 2017 2016 Operating expenses: Research and development $ 29,519 33,014 $ (3,495 ) (11 )% General and administrative 12,001 13,106 $ (1,105 ) (8 )% Total operating expenses 41,520 46,120 (4,600 ) Loss from operations (41,520 ) (46,120 ) 4,600 Interest expense (1,524 ) (690 ) (834 ) 121 % Interest income 410 97 313 323 % Other income (expense), net 186 (374 ) 560 (150 )% Net loss $ (42,448 ) $ (47,087 ) $ 4,639 Years Ended December 31, Increase / (Decrease) % Change 2016 2015 Operating expenses: Research and development $ 33,014 $ 8,117 $ 24,897 307 % General and administrative 13,106 4,855 $ 8,251 170 % Total operating expenses 46,120 12,972 33,148 Loss from operations (46,120 ) (12,972 ) (33,148 ) Interest expense (690 ) (350 ) $ (340 ) 97 % Interest income 97 — $ 97 100 % Other expense, net (374 ) — $ (374 ) 100 % Net loss $ (47,087 ) $ (13,322 ) $ (33,765 ) Innovatus loan. income in offerings that are seemed “at the market” offerings as defined in Rule 415 under the Securities Act (2020 ATM Facility). In COVID-19 pandemic, among other events. Our primary uses of capital are, and we expect will continue to be, funding Years Ended December 31, 2017 2016 2015 Net cash provided by (used in): Operating activities $ (38,372 ) $ (37,970 ) $ (9,134 ) Investing activities 22,657 (4,194 ) (45 ) Financing activities 19,994 65,142 13,180 Net increase in cash and cash equivalents $ 4,279 $ 22,978 $ 4,001 Contractual Obligations and Other Commitments Payments due by period Total Less than 1 year 1 – 3 Years 3 – 5 Years More than 5 years Operating lease obligations (1) $ 3,222 $ 634 $ 1,203 $ 1,276 $ 109 Term loan debt (2) $ 15,000 $ 2,083 $ 10,000 $ 2,917 $ — Interest on term loan debt (3) $ 3,641 $ 1,156 $ 1,284 $ 1,201 $ — Total $ 21,863 $ 3,873 $ 12,487 $ 5,394 $ 109 We are obligated to make future payments to third parties under asset purchase and license agreements, including royalties and payments that become due and payable on the achievement of certain development and commercialization milestones. December 31, 2022. the other. : December 31, 2017 2016 Assets Current assets: Cash and cash equivalents $ 32,035 $ 27,756 Short-term marketable securities 9,744 32,180 Prepaid expenses and other current assets 712 581 Total current assets 42,491 60,517 Property and equipment, net 79 76 Other assets 312 143 Total assets $ 42,882 $ 60,736 Liabilities and Stockholders’ Equity Current liabilities: Accounts payable $ 3,183 $ 2,639 Accrued liabilities 2,084 2,649 Current portion of long term debt 2,002 — Total current liabilities 7,269 5,288 Long term debt, net 13,091 14,727 Total liabilities $ 20,360 $ 20,015 Commitments and contingencies (Note 15) Stockholders’ equity: Common stock, $0.001 par value, 200,000,000 shares authorized as of December 31, 2017 and 2016; 10,526,599 and 8,356,659 shares issued and outstanding as of December 31, 2017 and 2016, respectively 11 8 Additional paid-in capital 141,320 117,086 Accumulated other comprehensive loss (3 ) (15 ) Accumulated deficit (118,806 ) (76,358 ) Total stockholders’ equity 22,522 40,721 Total liabilities and stockholders’ equity $ 42,882 $ 60,736 Year Ended December 31, 2017 2016 2015 Operating expenses: Research and development $ 29,519 $ 33,014 $ 8,117 General and administrative 12,001 13,106 4,855 Total operating expenses 41,520 46,120 12,972 Loss from operations (41,520 ) (46,120 ) (12,972 ) Interest expense (1,524 ) (690 ) (350 ) Interest income 410 97 — Other income (expense), net 186 (374 ) — Net loss $ (42,448 ) $ (47,087 ) $ (13,322 ) Net loss per share, basic and diluted $ (4.86 ) $ (7.84 ) $ (62.19 ) Weighted-average common shares outstanding, basic and diluted 8,727,935 6,007,027 214,228 Consolidated Statements of Comprehensive Loss Year Ended December 31, 2017 2016 2015 Net loss $ (42,448 ) $ (47,087 ) $ (13,322 ) Other comprehensive loss: Unrealized gain (loss) on marketable securities, net 12 (15 ) — Comprehensive loss $ (42,436 ) $ (47,102 ) $ (13,322 ) Convertible Preferred Stock Common Stock Additional Paid-In Accumulated Other Comprehensive Accumulated Total Stockholders’ Shares Amount Shares Amount Capital Loss Deficit Equity (Deficit) Balance at December 31, 2014 1,519,274 $ 15,366 193,850 $ - $ 1,114 $ - $ (15,949 ) $ 531 Issuance of Series A-1 convertible preferred stock, net of $22 of issuance costs 1,089,828 7,201 — — — — — 7,201 Issuance of common stock in connection with a license and asset purchase agreement — — 15,378 — 211 — — 211 Issuance of common stock upon stock option exercises — — 64,765 — — — — — Stock-based compensation expense — — — — 227 — — 227 Net loss — — — — — — (13,322 ) (13,322 ) Balance at December 31, 2015 2,609,102 22,567 273,993 — 1,552 — (29,271 ) (5,152 ) Issuance of common stock upon private placement, net of $1,300 of issuance cost — — 1,954,390 2 32,106 — — 32,108 Issuance of common stock upon conversion of convertible promissory note — — 350,040 — 6,129 — — 6,129 Issuance of common stock upon exercise of warrants — — 61,254 — 1,057 — — 1,057 Issuance of common stock to Eiccose upon private placement — — 96,300 — 1,661 — — 1,661 Conversion of preferred stock into common stock (2,609,102 ) (22,567 ) 2,609,102 3 22,564 — — — Issuance of common stock upon reverse merger — — 1,596,959 2 27,388 — — 27,390 Issuance of common stock upon execution of license agreement — — 157,587 — 3,172 — — 3,172 Issuance of common stock upon public offering, net of $1,800 of issuance costs — — 1,250,000 1 18,228 — — 18,229 Issuance of common stock upon exercise of stock option — — 7,034 — 39 — — 39 Stock-based compensation expense — — — — 3,190 — — 3,190 Unrealized loss on marketable securities, net — — — — — (15 ) — (15 ) Net loss — — — — — — (47,087 ) (47,087 ) Balance at December 31, 2016 — — 8,356,659 8 117,086 (15 ) (76,358 ) 40,721 Issuance of common stock upon public offering, net of $376 issuance costs — — 2,143,525 3 19,797 — — 19,800 Issuance of common stock upon ESPP purchase — — 16,186 — 142 — — 142 Issuance of common stock upon stock option exercise — — 10,229 — 52 — — 52 Stock-based compensation expense — — — — 4,243 — — 4,243 Unrealized gain on marketable securities, net — — — — — 12 — 12 Net loss — — — — — — (42,448 ) (42,448 ) Balance at December 31, 2017 — $ — 10,526,599 $ 11 $ 141,320 $ (3 ) $ (118,806 ) $ 22,522 Year Ended December 31, 2017 2016 2015 Operating Activities Net loss $ (42,448 ) $ (47,087 ) $ (13,322 ) Adjustments to reconcile net loss to net cash used in operating activities: Depreciation 41 23 11 Amortization of premiums on marketable securities (53 ) (41 ) — Stock-based compensation 4,243 3,190 227 Non-cash interest expense 366 685 350 Gain on intellectual property sale (200 ) — — Issuance of common stock in connection with a license and asset purchase agreement — 3,172 211 Change in fair value of obligation to issue shares to Eiccose — 204 1,457 Change in fair value of warrants liability — 165 — Changes in operating assets and liabilities: Prepaid expenses and other current assets (131 ) 326 (685 ) Other non-current assets (169 ) (89 ) (46 ) Accounts payable 544 699 1,881 Accrued liabilities (565 ) 783 782 Net cash used in operating activities (38,372 ) (37,970 ) (9,134 ) Investing Activities Purchase of marketable securities (24,524 ) (34,154 ) — Proceeds from maturities of marketable securities 47,025 2,000 — Proceeds from intellectual property sale 200 — — Cash received from merger transaction — 28,018 — Purchase of property and equipment (44 ) (58 ) (45 ) Net cash provided by (used in) investing activities 22,657 (4,194 ) (45 ) Financing Activities Proceeds from issuance of common stock upon public offering, net of issuance cost 19,800 18,229 — Proceeds from borrowings in connection with term loan, net of issuance cost — 14,759 — Proceeds from issuance of common stock upon private placement, net of issuance cost — 32,108 — Proceeds from issuance of common stock upon ESPP purchase 142 — — Proceeds from issuance of common stock upon options exercises 52 39 — Proceeds from issuance of common stock upon warrants exercises — 7 — Proceeds from issuance of convertible promissory note, net of issuance costs — — 5,979 Proceeds from issuance of preferred stock, net of issuance costs — — 7,201 Net cash provided by financing activities 19,994 65,142 13,180 Net increase in cash and cash equivalents 4,279 22,978 4,001 Cash and cash equivalents at beginning of period 27,756 4,778 777 Cash and cash equivalents at end of period $ 32,035 $ 27,756 $ 4,778 Supplemental disclosure of cash flow information Non-cash investing and financing activities: Conversion of warrant liability to common stock upon private placement $ — $ 1,050 $ — Issuance of common stock in connection with a license agreement — 3,172 211 Issuance of common stock to Eiccose upon private placement — 1,661 — Non-cash net liabilities assumed in reverse merger — 671 — Conversion of convertible promissory note to common stock upon private placement — 6,129 — Conversion of preferred stock to common stock upon reverse merger — 22,567 — Issuance of warrants in connection with convertible promissory note — — 885 Management believes that the currently available resources will be sufficient to fund its planned operations Lab equipment 3 years Costs expenses. Manufacturing costs related to products undergoing regulatory approval are expensed as research and development costs until receipt of such approval. Advance payments for research and development activities are deferred as prepaid expenses. The prepaid amounts are expensed as the related services are performed. model inputs. The Company accounts for forfeitures of stock-based awards when they occur. December 31, 2017 2016 2015 Options to purchase common stock 1,467,051 1,212,044 254,058 Warrants to purchase common stock 10,180 10,180 — Convertible preferred stock — — 2,609,102 Total 1,477,231 1,222,224 2,863,160 In June 2016, the In There were no assets or liabilities classified as Level 3 as of December 31, 2022 and 2021. December 31, 2017 Level 1 Level 2 Level 3 Total Financial Assets: Money market funds $ 19,612 $ — $ — $ 19,612 Corporate debt securities — 6,501 — 6,501 Commercial paper — 3,243 — 3,243 Total $ 19,612 $ 9,744 $ — $ 29,356 December 31, 2016 Level 1 Level 2 Level 3 Total Financial Assets: Money market funds $ 9,657 $ — $ — $ 9,657 Corporate debt securities — 11,469 — 11,469 Commercial paper — 22,891 — 22,891 Total $ 9,657 $ 34,360 $ — $ 44,017 Year Ended December 31, 2016 Balance at December 31, 2015 $ 2,342 Change in fair value of common stock warrants and obligation to issue common stock to Eiccose (1) 369 Settlement of warrant liability upon exercise of common stock warrants (1,050 ) Settlement of Eiccose obligation upon issuance of common stock (1,661 ) Balance at December 31, 2016 $ — December 31, 2017 Amortized cost Unrealized gain Unrealized loss Estimated Fair Value Cash equivalents: Money market funds $ 19,612 $ — $ — $ 19,612 Total cash equivalents $ 19,612 $ — $ — $ 19,612 Marketable securities: Corporate debt securities $ 6,503 $ — $ (2 ) $ 6,501 Commercial paper 3,244 — (1 ) 3,243 Total marketable securities $ 9,747 $ — $ (3 ) $ 9,744 December 31, 2016 Amortized cost Unrealized gain Unrealized loss Estimated Fair Value Cash equivalents: Money market funds $ 9,657 $ — $ — $ 9,657 Corporate debt securities 2,180 — — 2,180 Total cash equivalents $ 11,837 $ — $ — $ 11,837 Marketable securities: Corporate debt securities $ 9,294 $ — $ (5 ) $ 9,289 Commercial paper 22,901 3 (13 ) 22,891 Total marketable securities $ 32,195 $ 3 $ (18 ) $ 32,180 December 31, 2017 2016 Lab equipment $ 36 $ 35 Computer equipment and software 150 107 Total property and equipment 186 142 Less: accumulated depreciation (107 ) (66 ) Property and equipment, net $ 79 $ 76 December 31, 2017 2016 Compensation and related benefits $ 1,262 $ 1,299 Contract research costs 634 834 Consulting costs 87 106 Franchise tax 56 97 Contract manufacturing costs 4 122 Other 41 191 Total accrued liabilities $ 2,084 $ 2,649 Cash and cash equivalents $ 28,018 Prepaid and other assets 198 Current liabilities (857 ) Non-current liabilities (12 ) Net acquired tangible assets 27,347 Estimated total purchase consideration $ 27,347 Bristol-Meyers Squibb License Agreement EGI Asset Purchase Agreement Of the 46,134 options to purchase common stock, 15,378 shares vest monthly over four years as services are provided by the Sellers and 30,756 vest upon the earlier of the first commercial sale of the product or the approval of new drug application by the U.S. 2020. December 31, 2017 2016 Face value of long term debt $ 15,000 $ 15,000 Exit fee 1,125 1,125 Unamortized debt discount associated with exit fee, debt issuance costs and loan origination fees (1,032 ) (1,398 ) Total long term debt 15,093 14,727 Less: current portion of long term debt (2,002 ) — Long term debt, net $ 13,091 $ 14,727 Year ending December 31, 2018 $ 3,239 2019 5,839 2020 5,445 2021 4,118 Total future payments 18,641 Less: unamortized interest (2,516 ) Less: exit fee (1,125 ) Face value of term loan $ 15,000 In May 2017, the Company and Theragene Pharmaceuticals, Inc. To-date, no consideration has been earned or is expected to be earned. December 31, 2017 2016 Options issued and outstanding 1,467,051 1,212,044 Options available for future grants 799,375 646,778 Warrants to purchase common stock issued and outstanding 10,180 10,180 Total 2,276,606 1,869,002 Shares Available for Grant Number of Options Weighted- Average Exercise Price Per Option Weighted- Average Remaining Contractual Life (in Years) Aggregate Intrinsic Value Outstanding as of December 31, 2016 646,778 1,212,044 $ 14.06 9.00 $ 2,414 Additional options authorized 417,833 — — Granted (652,200 ) 652,200 $ 10.91 Exercised — (10,229 ) $ 5.04 Forfeited or cancelled 386,964 (386,964 ) $ 14.13 Outstanding as of December 31, 2017 799,375 1,467,051 $ 12.70 8.41 $ 4,511 Vested and exercisable as of December 31, 2017 630,727 $ 13.82 8.01 $ 2,117 2022. Year Ended December 31, 2017 2016 2015 Expected term (in years) 5.27 - 6.08 5.27 - 6.08 5.00 - 6.08 Volatility 79.00% - 80.00% 73.91% - 78.00% 77.58% - 97.62% Risk free interest rate 1.63% - 2.23% 1.21% - 2.27% 1.44% - 1.75% Dividend yield — — — Expected Term: The prior to exercise. price volatility. qualify as an employee stock purchase plan under Section 423 of the U.S. Internal Revenue Code and is administered by the Company’s board of directors. Year Ended December 31, 2017 2016 2015 Remaining contractual term (in years) 6.00 – 10.00 6.75 – 10.00 7.75 – 10.00 Volatility 83.62% – 106.13% 84.42% – 98.13% 85.83% – 90.50% Risk-free interest rate 1.90% – 2.43% 1.37% – 2.50% 1.73% – 2.24% Dividend yield — — — Year Ended December 31, 2017 2016 2015 Research and development $ 1,214 $ 737 $ 64 General and administrative 3,029 2,453 163 Total stock-based compensation expense $ 4,243 $ 3,190 $ 227 administrative expenses. As of December 31, 11. Income Taxes state taxes. Year Ended December 31, 2017 2016 2015 Federal statutory income tax rate 34.00 % 34.00 % 34.00 % State income taxes, net of federal benefit 0.36 (0.21 ) 6.11 Federal and state tax credits 5.10 4.68 2.54 Change in valuation allowance (8.04 ) (36.67 ) (42.14 ) Stock-based compensation (1.06 ) (1.26 ) (0.49 ) Other, net — (0.54 ) (0.02 ) Change in tax law (30.36 ) — — Provision (benefit) for income taxes — % — % — % December 31, 2017 2016 Deferred tax assets: Net operating loss carryforwards $ 20,230 $ 19,882 Tax credits 9,514 5,344 Depreciation and amortization 1,700 3,025 Accruals and reserves 1,158 936 Gross deferred tax assets 32,602 29,187 Valuation allowance (32,602 ) (29,187 ) Net deferred tax assets $ — $ — The Company had $10.8 million of federal Orphan Drug credit carryforwards available to reduce future taxable income that will begin to expire in 12/31/2041. Additional ownership changes in the future could result in additional limitations on the Company’s NOL and tax credit carryforwards. Year Ended December 31, 2017 2016 2015 Balance at beginning of year $ 1,831 $ 404 $ 99 Additions based on tax positions related to prior year — 19 46 Additions based on tax positions related to current year 1,387 1,408 259 Balance at end of year $ 3,218 $ 1,831 $ 404 Contingencies Year ending December 31, Amounts $ 634 2019 593 2020 610 2021 629 2022 647 Thereafter 109 Total minimum payments $ 3,222 2022. Exhibit Description of Document 2.1 3.1 3.2 3.3 3.4 4.1 4.2 10.2+ 10.7+ 10.8 24.1 Inline XBRL Instance Inline XBRL Taxonomy Extension Schema Document Inline XBRL Taxonomy Extension Calculation Linkbase Document Inline XBRL Taxonomy Extension Definition Linkbase Document Inline XBRL Taxonomy Extension Label Linkbase Document Inline XBRL Taxonomy Extension Presentation Linkbase Document Date: /s/ David David Director, Interim President and Chief Executive Officer (Principal Executive Officer) Eiger BioPharmaceuticals, Inc. Date: /s/ (Acting Principal Financial and Accounting Officer) Chairman of the Board of Directors Member of the Board of Directors Member of the Board of Directors Member of the Board of Directors Member of the Board of Directors LNFLNF/RTV and Lambdain developing LNF and Lambda is to reduce viral load in such a manner as to achieve durable suppressionapproval of the virus to below the level of quantification (<LLOQ), the pointthese treatments for HDV, where upon withdrawal of the therapy, the infection does not return to quantifiable levels. Evidence that academic investigatorsthere is an urgent, unmet medical need. Key opinion leaders have gathered suggestssuggested that combinations of LNFLNF/RTV and Lambdalambda with other antiviral agents may hold promisedemonstrate an even greater antiviral effect.longer duration24 weeks and underwent off-treatment follow-up for 24 weeks. The primary endpoint was >2 log decline in HDV RNA at end of 24-weeks of treatment. The secondary endpoint was an improvement in histology (>2-point improvement in histological activity index and no progression in fibrosis) at end of follow-up. LIFT-1 was conducted within the National Institutes of Health (NIH) at the NIDDK. The final end-of-treatment and end-of-study data were reported in November 2020 at the AASLD conference and are summarized in the figure below.sustained, long-term reduction6 of viral load.We20 patients (30%) achieved the secondary endpoint of > 2 point improvement in HAI.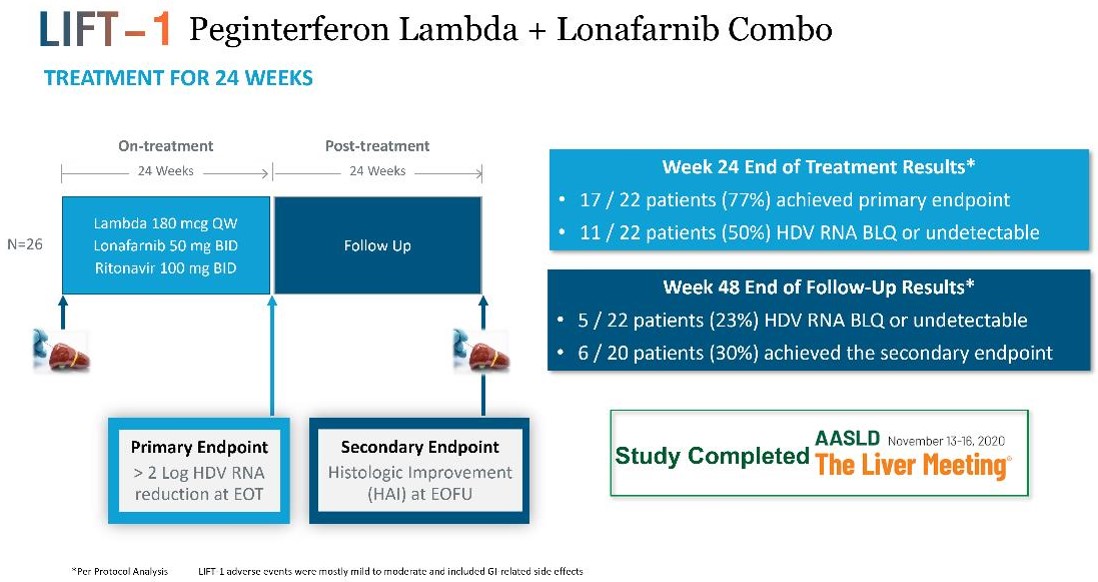
believebe conducted within the (NIH) at the NIDDK. If positive, data from LIFT-2 will provide additional support for this combination therapy.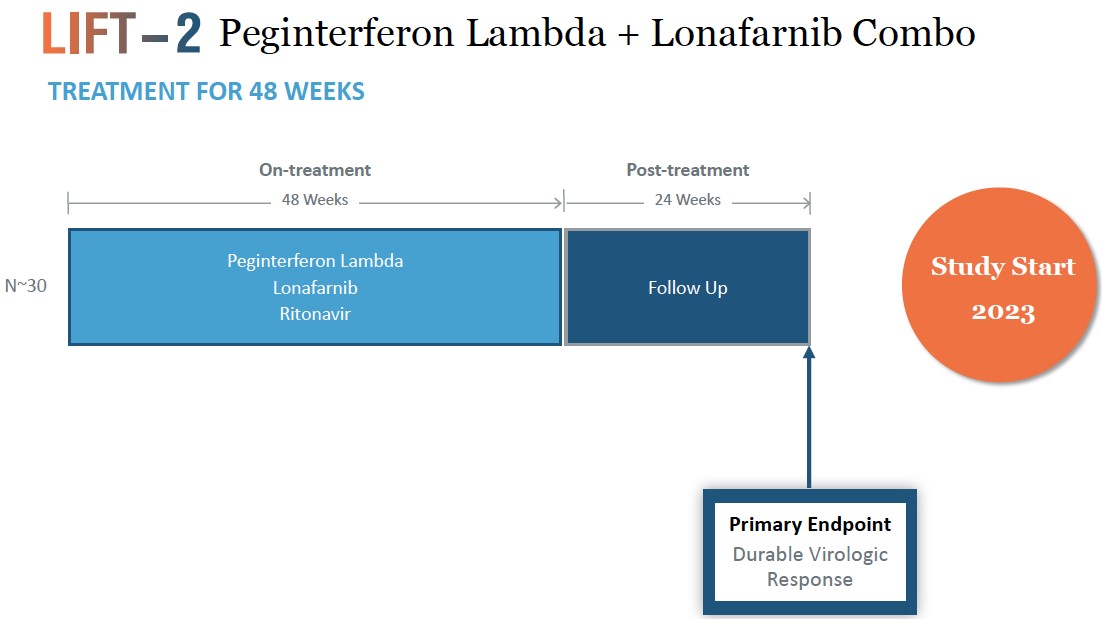
LNFavexitide may effectively reduce fasting and Lambdapostprandial hypoglycemia in combinationpatients with other antiviral agents may contribute to long-term benefit for patients, which may represent an alternative path to regulatory approval. In a study published in Plos One in 2014 (Romeo, R. et al. “High Serum LevelsHI. Avexitide treatment was well tolerated, with no serious drug-related adverse events (AEs) reported. Data from each of HDV RNA Are Predictors of Cirrhosis and Liver Cancer in Patients with Chronic Hepatitis Delta,” Plos One, 2014; 9:1), high serum levels of HDV were found to be a predictor of cirrhosis and liver cancer development. In a study published in Gastroenterology in 2004 (Farci, P. et al. “Long-Term Benefit of Interferon Therapy of Chronic Hepatitis D: Regression of Advanced Hepatic Fibrosis,” Gastroenterol, 2004; 126:1740), researchers demonstrated that lower frequencies of clinical events, leading to improvements in overall liver health and reductionsthe three studies are presented in the ratesfigures below. Avexitide for treatment of developing hepatic complications, could be achievedHI has been granted Rare Pediatric Disease designation by the FDA.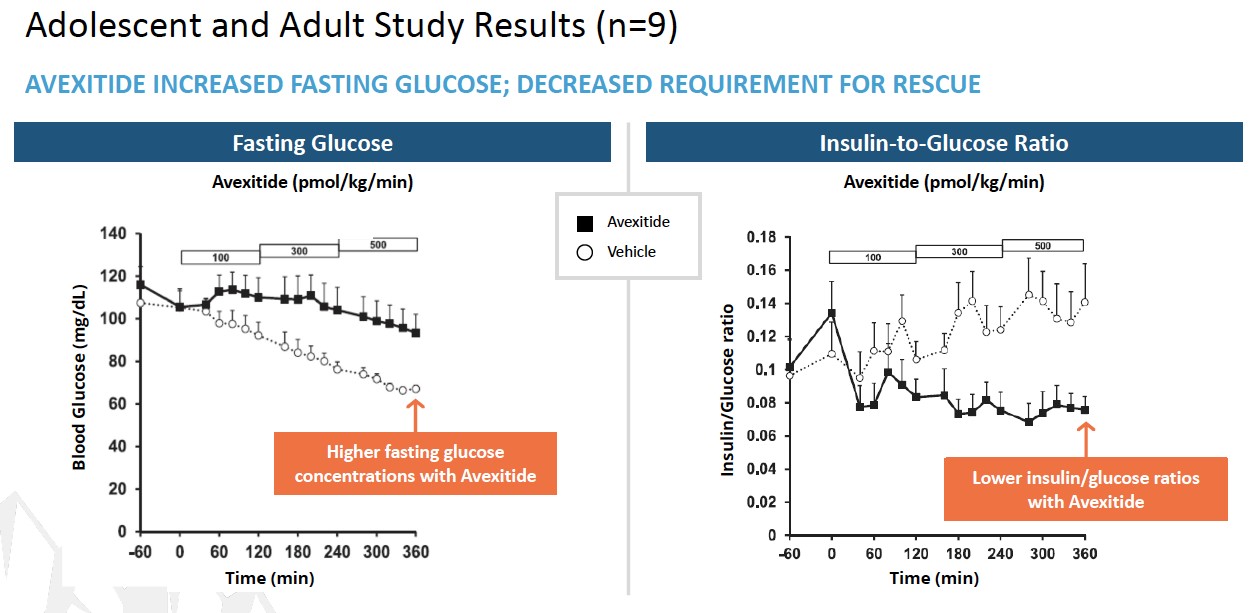
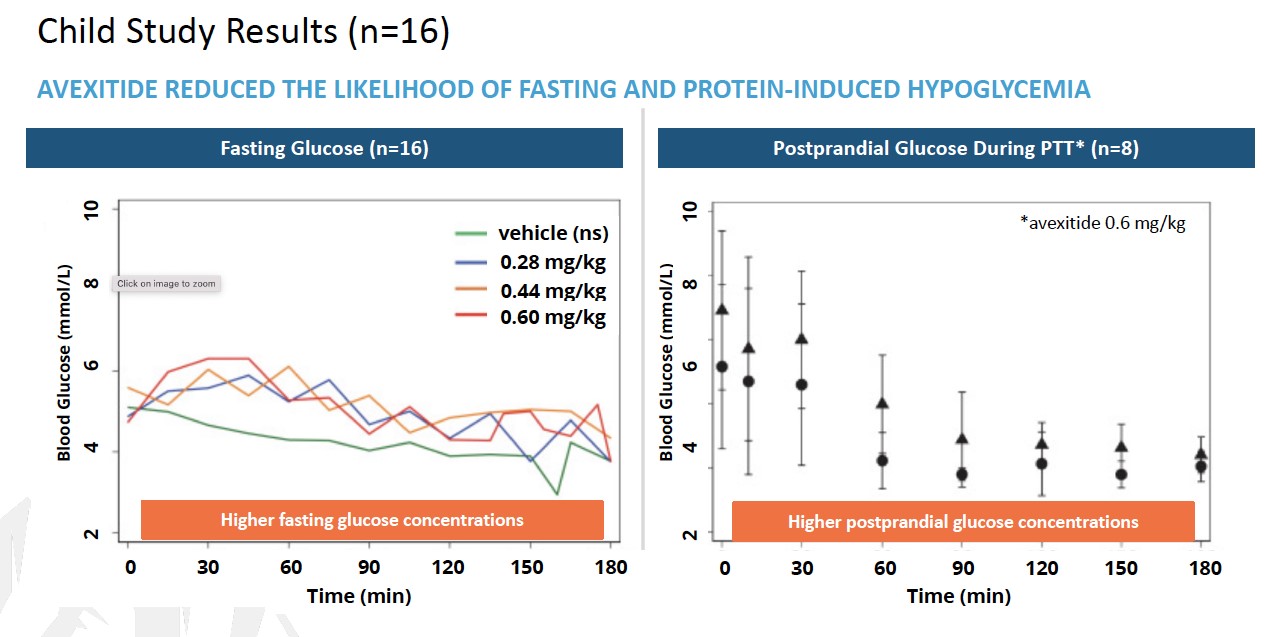
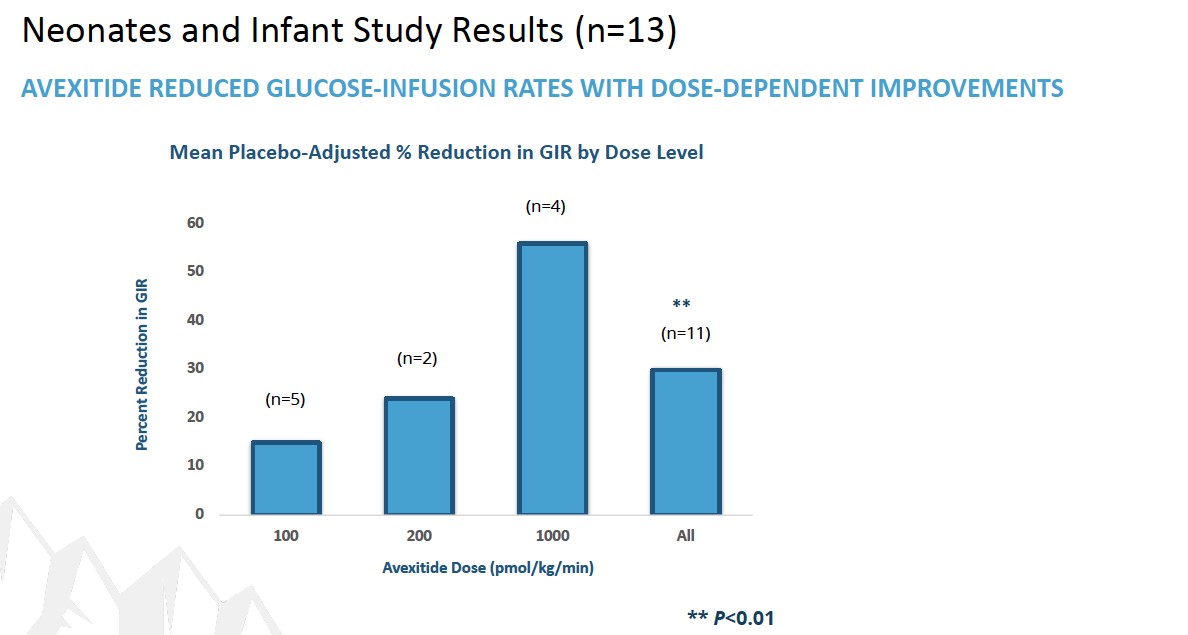
HDV infected patients who were treatedcongenital hyperinsulinism (HI) by the end of the year. In connection with high dose IFN-alfa and who experienced biochemical response and sometimes as little as 2 log declines in viral load. A 2014 Hepatology study by Heidrich suggests that transient suppression of HDV replication in patients treated with PEG-IFN-alfa improves the clinical long-term outcome, as not a single patient in their study with a post-treatment Week 24 HDV RNA response experienced a clinical event, including those patients who experienced viral rebound. We believe that these studies suggest that eradication of HDV RNA may not be necessary in patients treated with IFNs to achieve a substantial clinical benefit and improve long-term outcomes.Exendin 9-39 for Post-Bariatric HypoglycemiaExendin 9-39 is the second most advanced product candidate in our pipeline. Exendin 9-39 is a glucagon-like peptide-1, or GLP-1, receptor antagonist. GLP-1 is a gut-derived incretin hormone released by intestinal “L” cells after meals. Incretin hormones, such as GLP-1, enhance the secretion of insulin from pancreatic beta cells in a glucose-dependent manner, thereby lowering blood glucose levels after meals. Exendin 9-39 blocks GLP-1 from binding to the GLP-1 receptor, inhibiting the GLP-1-mediated incretin effect. We are developing exendin 9-39 as a treatment for PBH, which is characterized by an exaggerated incretin response, with patients exhibitingPhase 3, startup activities, we observed low levels of glucose and excessively high levels of insulinproduct-related impurities in the blood after meals. This formfinished drug product. Although not unusual for this class of compounds, we are working with our CMO's to control and qualify these materials and plan to initiate dosing when an adequate supply of materials with a sufficient shelf life has been released. Avexitide has been granted Orphan Drug designation in the U.S. by the FDA for the treatment of hyperinsulinemic hypoglycemia (which includes HI) and has also been granted Rare Pediatric Disease designation making it potentially eligible for a priority review voucher upon regulatory approval. Avexitide is a debilitating and potentially life-threatening condition. Gastric bypass procedures are widely performed and are increasing in frequency for medically complicated obesity. There is no approvedthe only investigational therapy for PBH and the unmet medical need is high.We have demonstrated clinical proof of concept in 36 patients suffering from PBHHI that exendin 9-39 can prevent an exaggerated fall in blood sugar following a meal, or post-prandial hypoglycemia, in affected patients. Data has been generated using both intravenous delivery and SC delivery. Pharmacokinetics indicate thatgranted Breakthrough Therapy designation by the SC delivery could enable once or twice a day pre-prandial dosing. We have developed a novel liquid formulation for SC injection and have completed a Phase 1 PK study with this new formulation of exendin 9-39 in healthy volunteers in mid-2017. We completed a Phase 2 multiple ascending dosing trial (up to 3 days) in affected patients with our exendin 9-39 SC novel liquid formulation in 2017, and have initiated a Phase 2, 28 day study (PREVENT) in affected patients using of the new SC formulation in Q1 2018. We continue to expect data from the PREVENT study in the second half of 2018.Post-BariatricFDA.Surgically-alteredSurgically altered nutrient transit, such as a Roux-en-Y procedure, causesresulting from bariatric surgeries can cause early nutrient sensing by the intestinal “L” cells, resulting inleading to enhanced secretion of GLP-1 leading tocausing elevated insulin secretion. This effect may play a primary role in the early resolution of Type 2 diabetes after surgery. A number of synthetic analogs of GLP-1, or agonists, have been approved for the treatment of Type 2 diabetes including Byetta™ (exenatide), Victoza™ (liraglutide), and Trulicity™ (dulaglutide). These drugs, all agonists, bind to the GLP-1 receptor and enhance the release of insulin in a glucose-dependent manner. In patients with PBH, excessive secretion of GLP-1 and/or exaggerated sensitivity to GLP-1 results in dysfunctional insulin release, leading to severe, debilitating hypoglycemia. GLP-1 receptor antagonists compete with endogenous GLP-1 and hashave the potential to prevent dysfunctional insulin release and resultant symptomatic hypoglycemia.Approximately 200,000 bariatric surgical procedures are performed each year in the United States, and another 100,000 are performed each year in Europe. Approximately 30% of these bariatric surgeries are Roux-en-Y gastric bypass procedures.Our Next Product Candidate: Exendin 9-39 to Treat Post-Bariatric HypoglycemiaExendin 9-39 is a well-characterized, competitive antagonist of GLP-1 at its receptor. Exendin 9-39 is a 31 amino acid fragment of exenatide, a commercially available GLP-1 agonist, brand named Byetta™ used in the treatment of type 2 diabetes. Exendin 9-39 blocks the GLP-1 receptor and leads to reduced post-prandial levels of insulin secreted by the pancreas. While exenatide has been approved for the treatment of type 2 diabetes, exendin 9-39, is a new molecular entity and has never been approved nor commercialized for any indication.We in three clinical studies with exendin 9-39 that pharmacologic blockade of the GLP-1 receptor can preventreduce hypoglycemia in affected patients and mitigate symptoms of hypoglycemia.associated symptoms. We believe that exendin 9-39avexitide may represent the first targeted medical treatment for patients with PBH. In the threestudies completed studies,to date, there werehave been no significant adverse drug reactions attributed to exendin 9-39. These single-dose and multiple-doseavexitide.1/2 studies were conducted under two investigator INDs for the study of exendin 9-39 for PBH.first exendin 9-39PREVENT study was a Phase 1, double-blinded crossover2, multicenter, randomized, single-blind, placebo-controlled cross-over study wherein eight patients with PBH were randomly assigned to receive IV infusion of exendin 9-39 or placebo during an oral glucose tolerance test, or OGTT (Craig et al, Diabetologia 2016). The trial assessed patient blood glucose and insulin levels andassess the presence and severity of symptoms of hypoglycemia. Hypoglycemia was defined as glucose levels falling to or below 50 mg/dL.In this trial, IV infusion of exendin 9-39 raised the postprandial glucose nadir by over 70% and lowered the area under the curve insulin by 57%, normalizing both parameters relative to healthy nonsurgical controls, and preventing hypoglycemia in all eight participants. In contrast, during placebo infusion every patient became hypoglycemic, requiring investigator intervention with administration of IV dextrose when patient plasma glucose fell to a level of 50 mg/dL or less.To assess for the presence and severity of symptoms of hypoglycemia during IV infusion of exendin 9-39 versus placebo, patients completed severity-grade questionnaires every 30 minutes during each 180-minute OGTT period. The severity-grade questionnaires showed that, on average patients experienced fewer and less severe hypoglycemic symptoms during IV infusion of exendin 9-39 as compared to during IV infusion of placebo (p<0.001). While symptoms reported by subjects during the glucose rise (from T=0 to peak glucose) were unchanged by exendin 9-39 infusion, both autonomic (p=0.002) and neuroglycopenic (p=0.001) symptoms reported during the glucose fall period (from peak to nadir glucose) were reduced.The second clinical proof of concept study, a Phase 2 clinical trial, was a single ascending dose, or SAD, and exendin 9‑39 was administered subcutaneously in eight patients with PBH. This was the first investigation involving the SC administration of exendin 9‑39 in human subjects and was designed to examine the PK, PD, and local tolerability of SC exendin 9‑39 in patients with PBH. After metabolic and symptomatic responses to a baseline 75 g OGTT were evaluated, patients returned for a repeat OGTT with administration of a single exendin 9‑39 dose, ranging from approximately 10–30 mg (0.13–0.38 mg/kg).In all eight patients undergoing the OGTT, exendin 9-39 administration prevented hypoglycemia and reduced symptoms of hypoglycemia. The baseline OGTT resulted in a high peak in plasma glucose concentration for all eight patients, followed by a rapid, steep decline, with all patients requiring rescue with IV dextrose at a plasma glucose concentration of 50 mg/dL. In contrast, prevention of hypoglycemia occurred at all dose levels of SC exendin 9-39 tested, with all patients completing the 180-minute OGTT without requiring intervention with IV dextrose. While early glycemic responses (fasting plasma glucose, peak postprandial glucose, time to peak glucose, and AUC glucose from 0–60 minutes postmeal) were unchanged by administration of SC exendin 9-39, late glycemic responses (nadir glucose, time to nadir glucose, AUC glucose from 0–180 minutes) were significantly improved. The average nadir glucose was increased by 61%, as shown in the figure below.Exendin 9-39 SC Injection SAD Study Results
*p < 0.01, **p < 0.001, and ***p < 0.0001for PBH patients with SC exendin 9-39 injection vs no injection.Source: Craig et al, ADA 2016.Symptoms of PBH were assessed using the Edinburgh Hypoglycemia Symptom Scale, which was completed by patients every 30 minutes during each 180-minute OGTT. Patients used the scale to report the presence and severity of autonomic or neuroglycopenic symptoms or symptoms of malaise. SC exendin 9-39 reduced symptoms of PBH overall and during the glucose fall period without altering symptoms during the glucose rise period. While symptoms associated with PBH were observed during this study, no adverse reactions attributed to exendin 9-39 were identified, and no injection site reactions were reported in any patients in this study.
P-value by paired two-tailed Student's t-test.Source: Craig et al, ADA, 2016b.A third study with exendin 9-39 completed in June 2017. This was a Phase 2 trial evaluating the safety, efficacy and PK profilesafety of multiple ascending doses28-day dosing of subcutaneously administered exendin 9-39avexitide in patients with PBH. A liquidtotal of 18 patients were enrolled and a lyophilized formulationtreated with two dosing regimens (once daily and twice daily) of avexitide for 28 days. All patients participated in three 14-day treatment periods, involving placebo SCexendin 9-39 were evaluatedinjections, and twice-daily avexitide SC injections. Patients self-administered injections in the MAD study.Key findings from thisoutpatient setting throughout the study demonstrated that both SC exendin 9-39 liquidduration and lyophilized formulations reduced postprandial hyperinsulinemic hypoglycemia, reduced hypoglycemic symptomsadditionally underwent in-clinic MMTT provocations with concomitant blood draws and were well tolerated with no related adverse events. In addition, SC exendin 9-39 liquid formulation improved postprandialsymptom assessments following each 14-day treatment period. Participants' metabolic and clinical parameters with comparable or greater activity versus the lyophilized formulation. The liquid formulation produced a pharmacokinetic profile which may confer a longer duration of action versus the lyophilized formulation.Exendin 9-39 SC Injection MAD Study Glycemic Results
Source: Craig et al ADA Poster, June 2017.The mean postprandial insulin peak was reduced by 51%, while fasting insulin was not raised, in patients who received doses of ≥ 0.2 mg/kg.Exendin 9-39 SC Injection MAD Study Results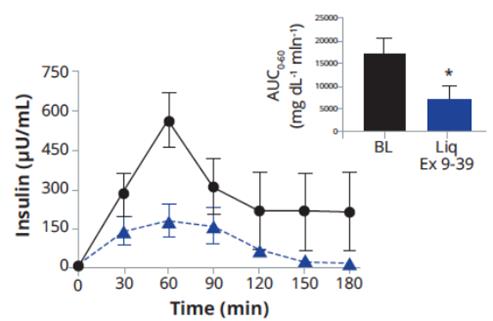
Source: Craig et al ADA Poster, June 2017.Lymphedema Disease OverviewAbout LymphedemaLymphedema is the build-up of fluid in soft body tissues when the lymph system has been damaged or blocked. It is characterized by swelling due to abnormal transport of lymphatic fluid and thickening or hardening of the skin in affected areas. As fluid builds up, swelling occurs, usually in an arm or a leg, but can also affect other parts of the body. Lymphedema often causes long-term physical, psychological and social problems for patients and significantly impacts quality of life. There are currently no approved pharmacological treatments for lymphedema and the unmet medical need is high.Lymphedema can be either primary, meaning it is congenital or occurs on its own, or secondary, meaning it is caused by another disease or condition. Primary lymphedema is caused by the absence of certain lymph vessels at birth or by abnormalitiesoutcomes were assessed in the lymphatic vessels. It can be divided into three forms, depending on ageoutpatient setting by electronic diary, self -monitoring of onset. The prevalence of primary lymphedema is less than 200,000blood glucose (SMBG), and continuous glucose monitoring (CGM) and postprandial glucose, insulin, and symptom responses were assessed during MMTT provocation in the United States and less than 5 in 10,000 in the European Union, and expected to be eligible for orphan drug designation by regulatory authorities. Secondary lymphedema usually develops as a result of a blockage or interruption that alters the flow of lymph through the lymphatic system and can develop from an infection, malignancy, surgery, scar tissue formation, trauma, radiation, or other cancer treatment.Primary lymphedema and secondary lymphedema can both be debilitating disorders with negative impact on quality of life and a large unmet medical need exists for an effective therapy. There is no approved pharmacologic treatment for lymphedema. Available treatments include compression garments, massage and exercise. Several agents such as coumarin have been tested in investigator-initiated clinical trials but have shown no clinical efficacy.Ubenimex for LymphedemaA study conducted at Stanford demonstrated that LTB4 is elevated in both animal models of lymphedema and human lymphedema. Elevated LTB4 is associated with tissue inflammation and impaired lymphatic function. Targeted pharmacologic inhibition of LTB4 promotes physiologic lymphatic repair and reverses lymphedema disease in treated animals.Researchers at Stanford demonstrated a novel function of LTB4 in the pathogenesis of lymphedema suggesting that blocking the effects of LTB4 may be a promising and potentially safe new therapeutic strategy for this disease. We initiated a clinical study to explore if blocking the effects of LTB4 may be useful as a new treatment for lymphedema. We in-licensed ubenimex from Nippon Kayaku in 2015 and have relied on Nippon Kayaku’s prior Phase 1 clinical data and experience with ubenimex to understand safety. Nippon Kayaku conducted four Phase 1 studies in healthy subjects and cancer patients to study metabolite determination, metabolism and excretion, drug absorption, and a pharmacokinetic study in lymphoma patients.In the metabolite determination study, ubenimex was rapidly absorbed following oral administration of single doses ranging from 10 mg to 200 mg, reaching a maximum serum level between 30 minutes and three hours after dosing. Mean peak concentrations after 30 mg, 100 mg and 200 mg were 2.2 µg/mL at one hour, 2.5 µg/mL at three hours, and 7.4 µg/mL at two hours, respectively.In the metabolism and excretion study, 84% to 94% of the administered doses of ubenimex was recovered in urine within 24 hours of dosing.In the absorption study, prolonged administration of ubenimex to cancer patients showed rapid absorption of the drug and maximum peak levels which ranged from 30 minutes to three hours in most patients. In a small study of eight patients receiving 30 mg of ubenimex daily, delayed α-phase decrease, an initial phase of rapid decrease of concentration of the drug in the plasma, was observed in patients with renal cancer compared to patients with bladder cancer, suggesting that clearance of ubenimex may be slower in patients with impaired renal function. The pharmacokinetics did not appear to change over time with repeated administration of ubenimex.In a Phase 1b study performed in bone marrow transplant lymphoma patients in the United States, PK evaluation was performed in groups of ten patients receiving 10 mg of ubenimex QD, 30 mg of ubenimex QD, 30 mg of ubenimex three times a day, or TID, or 60 mg of ubenimex TID, in each case for up to 60 days. The mean AUC and Cmax increased with increasing doses of 10 mg, 30 mg, 90 mg or 180 mg ubenimex daily. At all doses, no accumulation was apparent over the six days.Preclinical LTB4 Data in LymphedemaAn animal model of lymphedema was used to mimic the physiological changes seen in lymphedema patients. In this model, acquired lymphedema was surgically induced in the tails of mice through the ablation of lymphatic trunks. As the tail volume increases, there is an accumulation of fibroblasts, fat cells and skin cells in the tail, and poor clearance of immune cells from the tail. As lymphedema is established in this model, the levels of LTB4 in serum rise significantly. For surgical controls (sham animals), skin incision alone was performed without lymphatic cautery. Normal controls did not go under any surgical manipulation. When serum from human lymphedema patients was examined, the LTB4 levels were also significantly (p<0.0001) elevated compared to normal controls (control n=18, lymphedema patients n=8).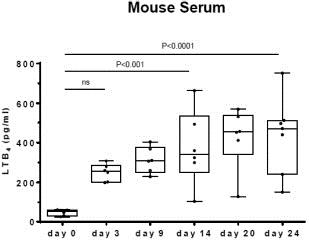

In animal models, ubenimex significantly reduced tail volume (p<0.0001, sham n=13, saline n=14, ubenimex n=14). Sham surgery (placebo surgery) is a faked surgical intervention that omits the step thought to be therapeutically necessary. In clinical trials of surgical interventions, sham surgery is an important scientific control. This is because it isolates the specific effects of the treatment as opposed to the incidental effects caused by anesthesia, the incisional trauma, pre- and postoperative care, and the patient’s perception of having had a regular operation. Thus, sham surgery serves an analogous purpose to placebo drugs, neutralizing biases such as the placebo effect.
Ubenimex reversed lymphedema-induced tissue remodeling in animal models. Thickness of both the epidermis and dermis were reduced.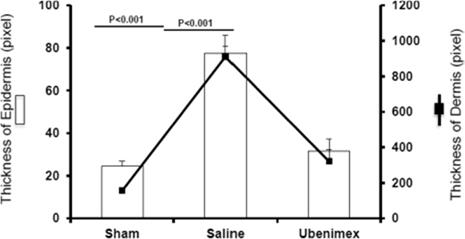
Our Planned Solution: Ubenimex for LymphedemaClinical PlanWe in-licensed ubenimex from Nippon Kayaku in 2015 and have relied on Nippon Kayaku’s prior Phase 1 clinical data and experience with ubenimex to understand safety. We filed an. IND for Ubenimex for the treatment of lymphedema with FDA in December 2015. Our Phase 2 clinical proof of concept trial for ubenimex in lymphedema is called ULTRA (Ubenimex Lymphedema Trial to Restore Activity), and is designed to assess efficacy as well as safety. As of December 31, 2017, the trial was fully enrolled with a total of 54 patients enrolled across 4 international sites. The primary endpoint is a measure of change in skin fold thickness from baseline. Secondary endpoints include change in limb volume from baseline and patient reported outcomes, including quality of life. Based on the proposed mechanism of action of ubenimex, as a potential anti-proliferative and a potential disease modifying agent, dosing in the ULTRA trial is six months, which we believe represents sufficient time to demonstrate activity.Ubenimex for Pulmonary Arterial HypertensionIn January 2018, topline results for the Phase 2 LIBERTY study results in PAH were announced. Ubenimex demonstrated no improvement overall or in key subgroups for both the primary efficacy endpoint of PVRimproved postprandial glucose nadir during MMTT was achieved with statistical significance with avexitide 30 mg BID and 60 mg QD, with fewer participants requiring glycemic rescue during each of the active dosing regimens than during placebo dosing. The secondary endpoint of 6-minute walk. No safety signals attributed to ubenimex were identifiedreduced postprandial insulin peak during MMTT was also statistically significant with avexitide 30 mg BID and 60 mg QD. The primary and secondary endpoints are shown in the preliminary analysis. Further analysisfigure below.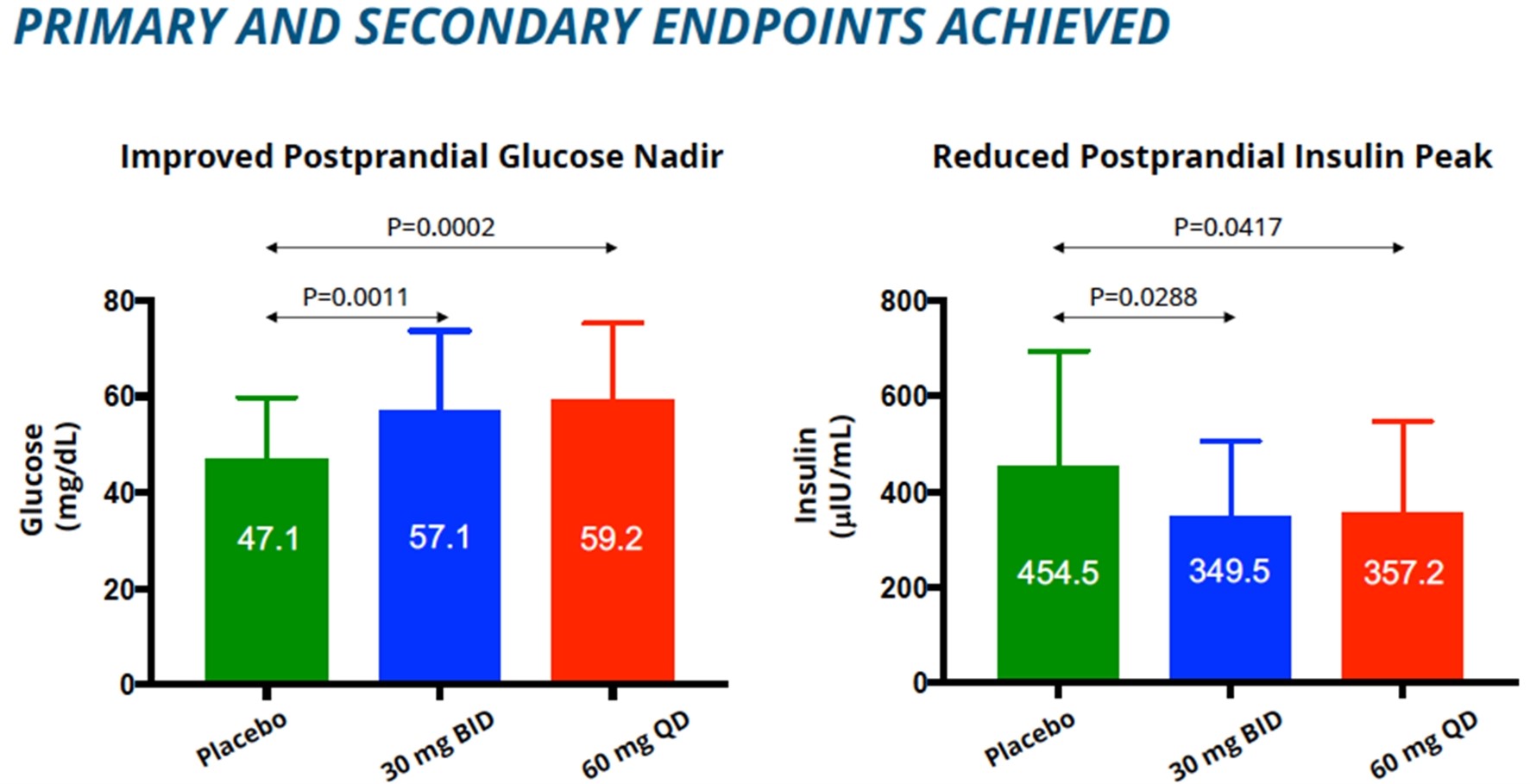
Number of Episodes in 14 Day Period Placebo 4.03 2.81 1.56 Change from Placebo NA -1.24
(p=0.072)-2.51
(p=0.001)2.01 1.21 0.81 Change from Placebo NA -0.77
(p=0.040)1.96 1.50 0.86 Change from Placebo N/A -0.49
(p=0.224) -1.09
(p=0.014)ongoing, althoughdefined as number of episodes in a 14 day periodwill discontinue developmentreceived concurrence with the FDA and EMA on a single Phase 3 trial for avexitide for treatment of ubenimex in PAH based on these results. for preclinical and clinical studies and intend to do so in the future. We do not own or operate manufacturing facilities for the production of clinical trial quantitiesmaterials of our product candidates and have no plans to build our own clinical or commercial scale manufacturing capabilities. We believe that the use of contractedcontract manufacturing organizations or CMOs,(CMOs) eliminates the need for us to directly invest in manufacturing facilities, and equipment and additional staff. Although we rely on contract manufacturers, our personnel and consultants have extensive manufacturing experience overseeing our CMOs.full scalefull-scale commercial demands but have not assessed these capabilities beyond the supply of clinical material.material for products in clinical development. We have identified or plan to identify commercial contract manufacturers as we move our product candidates to Phase 3 clinical trials. We believe there are alternate sources of manufacturing that could be identified and enabled to satisfy our clinical and commercial requirements, however, we cannot be certain that identifying and establishing alternative relationships with such sources can be successful, cost effective, or completed on a timely basis without significant delay in the development or commercialization of our product candidates. (LNF)LNFlonafarnib (LNF) Phase 2 clinical studies for the treatment of HDV and HGPS was manufactured by Merck. We have successfully completed the technology transfer for manufacture of the LNF drug substance and the LNF drug product to our third-party manufacturers. All future HDV clinical trials will be conducted with product manufactured by these CMOs.PEGylated Interferon These manufacturers produce our commercial supply for HGPS and processing-deficient progeroid laminopathies. (Lambda)PEGylated Interferon Lambda product.lambda program. As part of the license agreement, sufficient inventory of drug substance and drug product was obtained to complete our Phase 2 and initiate our Phase 3 clinical trials. We have completed the first FMP drug product manufacturing campaign in 2017 at a new manufacturing facility. The drug substance CMO remains the same CMO contracted by BMSmanufactured additional batches of lambda products.no changeslambda are anticipated for the drug substance manufacturing process.Exendin 9-39 drug product for exendin 9-39 for the treatment of PBH for Phase 2 clinical studies is manufactured by a third-party CMO.UbenimexNippon Kayaku manufactures the drug substance and drug product used for ubenimex Phase 2the production of avexitide clinical studiestrial material for the treatment of PAHHI and lymphedema. In 2017, we completed the process of transferring the drug substance process from Nippon Kayaku toPBH is manufactured by our CMO. Since then, we have successfully manufactured a new formulation for the drug product which is intended to improve dosing compliance and reduce capsule burden.of matter, method(s) ofdosage, formulation, use, andmanufacturing process, patents covering manufacture and/or formulation.among others. We have also licensed patents and patent applications that cover certain of our product candidates and/or their manufacture, use, or formulation.orphan drugOrphan Drug designation and New Chemical Entity or NCE,(NCE) and Biologic License Application or BLA,(BLA) exclusivities, as well as trade secrets and carefully monitor our proprietary information to protect all aspects of our business. on new dosage forms, methods of treatment, and compositions of matter for our product candidates. We file and prosecute patent applications in the United States and Europe and, when appropriate, additional countries, including Japan, Korea, Canada and China.or the USPTO,(the USPTO) and certain other patent offices are maintained in secrecy for a minimum of 18 months, and publications of discoveries in the scientific or patent literature often lag far behind the actual discoveries themselves. For these reasons, we cannot be certain that inventions claimed in pending patent applications were not invented by another party prior to our invention or disclosed or claimed in a patent application filed before the effective filing date of our applications, in either of which case the claims may not be patentable to us. For certain applications with an effective filing date prior to March 13, 2013, we may have to participate in interference proceedings declared by the USPTO to determine priority of invention. Also, while we are not currently participating in any interferences or post-grant challenge proceedings, such as patent oppositions, post-grant reexamination proceedings, inter parties review proceedings and patent litigation, that seek to invalidate claims of pending patent applications or issued patents, we may have to participate in such proceedings in the future. Such proceedings could result in substantial cost, even if the eventual outcome is favorable to us.or the PCT.145154 PCT contracting states, providing a cost-effective means for seeking patent protection in numerous regions or countries. Conversion of a PCT application into an application in any of the contracting states typically occurs about 30 months after a priority application is filed, or about 18 months after the PCT application filing date. An applicant must undertake prosecution within the allotted time in the patent offices of any, or a combination, of the contracting states or in a regional patent office it determines to undertake patent issuance in protection in such country or territory.Ourand/or their uses in one or more indicationsas of interest to usDecember 31, 2022 are covered by in-licensed patents and patent applications and by our own patent applications.Lonafarnib (LNF)summarized below.in-licensedlicensed from Merck a portfolio of patents and know how covering the compound, formulations of the compound, and synthesis, but these expire before the anticipated launch date of the LNF product candidate. Wefiled one US application and two PCT applications that claimobtained patent protection for the use of LNF in combination with RTV and/or optionally other drugs for the treatment of HDV infection.One PCT Eiger’s U.S. Patent Nos. 10,076,512; 10,828,283; and 11,311,519 entitled, Treatment of Hepatitis Delta Virus Infection, include claims covering a broad range of RTV-boosted LNF doses and durations with and without interferons. The patents have terms that extend at least until 2035. Additional claims are being pursued in a continuation application. The European Patent Office, the Chinese Patent office and the Japanese Patent Office have also granted patents with claims covering a broad range of LNF boosted with RTV dosing regimens for the treatment of HDV infection. These patents will have a term that extends to 2035. In Europe, China and Japan, additional claims are being pursued in divisional applications. We have now obtained patent protection with claims covering treatment of HDV with LNF boosted with RTV the U.S., Europe, China and Japan, which are key major pharmaceutical markets. A corresponding patent application claiming the use of LNF in combinationlonafarnib boosted with RTVritonavir is pending in Korea. Additionally, US Patent 10,835,496 and one has matured into patent applications in the aOffice (EPO)issued claiming particular dosage forms for ritonavir-boosted lonafarnib for the treatment of HDV infections. A Notice of Allowance was also received for an application in South Korea. These patents extend protection until at least 2036.Japan, Korea and China. Anyprogeroid laminopathies. The patents that issue from this these applications will expire in 2035, but aprovide protection until at least 2024 and an application for patent term extension (as described below)(PTE) that could extend the protection for one of up to five years is availablethe patents until 2029 has been filed. We have also filed a patent application in the United States,U.S. directed to methods of treating HGPS and we expect LNF to be eligible for this additional protection. In addition, provided it is the first indication in which LNF is approved, we expect LNF to be eligible for NCE status, whichprogeroid laminopathies that if granted provides five years of regulatory exclusivity.issued would provide further protection until at least 2039. In addition, LNF has been granted orphan drugOrphan Drug designation by the FDA and the EMA in this indication, which respectively may provide up to seven and ten years of regulatory exclusivity.We have filed an additional PCT application for LNF/RTV combination drug products useful for treating HDV and this application is pending. Any patents that issue from this application will expire in 2036.We have not yet determined the countries in which we will pursue potential patent protection from our currently pending PCT applications, but even if we determine to make such filings, our efforts may not result in the issuance of patents as a result.Pegylated interferon-lambda (Lambda). Lambdalambda modified by polyethylene glycol derivatization or Lambda.(lambda). The key United States composition of matter patent in this portfolio expires in 2025, but we expect to be eligible for the full five years of patent term extension for that patent. In addition, we expect Lambdaregulatory approval for lambda to be filed under a BLA, and so Lambdawhich if granted would be eligible forprovide 12 years reference product exclusivity (4 years in filing exclusivity; 12 years for data), as well as orphan drugOrphan Drug exclusivity for treatment of HDV infection.this indication. continuation/divisional applications in the U.S. and Europe. Applications are also pending in China, Japan and Korea. Any patents that issue from these applications will expire in 2037.Lambdalambda in HDV.Exendin 9-39HDV that has matured into patent applications in the United States, Europe, Australia, Brazil, Canada, China, Eurasia, Israel, India, Japan, South Korea, Mexico, and South Africa. Any patents that issue from these applications should offer protection until at least 2039.exendin 9-39avexitide and other agents in the treatment of hypoglycemia associated with bariatric surgery, including in PBH. The PCT applications have matured into patent applications in the United States, the European Patent Office (EPO), Australia, Brazil, Canada,USPTO issued US 10,639,354, US 10,660,937, US 10,993,992, and Chile. Any patents that issue from these applications will expire in 2036 without extension and upUS 10,993,991, which provide protection at least until 2036. Up to five years of patent term extension will be available in the United States. WeThe European Patent Office has also expect exendin 9-39granted two patents with claims covering use of avexitide to be eligible for orphan drugtreat hypoglycemia, which will expire in 2036. Two Australian patents and a Chilean patent have also issued with similar claims, which will also expire in 2036 Additional claims are being pursued in continuation/divisional applications in the U.S. and Europe. Applications are also pending in Canada, and Chile. Any patents that issue from these applications will expire in 2036. Additionally, avexitide has been granted Orphan Drug designation in this indication,the treatment of hyperinsulinemic hypoglycemia by the FDA and the EMA, which provides seven years and ten years of regulatory exclusivity in the United States and Europe, respectively.Ubenimex.Lymphedema. Wealso in-licensed from Stanford applications pending in the U.Sobtained patent protection for formulations of avexitide and EPO that claim the use of ubenimex and other agentsthese formulations in the treatment of lymphedema.hypoglycemia associated with bariatric surgery. The USPTO issued US 11,020,484, which will provide protection at least until 2037. Additional claims are being pursued in continuation application. Applications are also pending in Europe, Australia, Brazil, Canada, Chile, China, Hong Kong, Israel, India, and Japan. Any patents that issue from these applications will expire in 2037.EPO will expire in 2036. Any US patent may be eligible for patent term extension of up to five years in the United States.Regulatory Exclusivity and Patent Term Extension. If ubenimex is approved, it would be entitled to NCE exclusivity,Europe, provide protection until 2028. There are continuation applications pending from which would provide five years of regulatory exclusivity for the approved product. In addition, the FDA has granted orphan drug designation to ubenimex for the treatment of PAH, and we are seeking orphan drug designation for ubenimex for the treatment of lymphedema. Orphan drug designation, if obtained, may provide seven years of regulatory exclusivity for each indication upon NDA approval. However, patent term extension, as described below, will be available only for the first of the two indications to be approved.Patent TermIn the United States, the patent term for an FDA-approved drug may be eligible for a patent term extension, or a PTE. The Hatch-Waxman Act of 1984 permits restoration of a portion of the patent term of a U.S. patent as compensation for the patent term lost during product development and the FDA regulatory review process if approval of the application for the product is the first permitted commercial marketing of a drug or biological product containing the active ingredient. The length of the PTE is based on the length of time it takes for the drug to complete the pre-market regulatory approval requirements. The time required for approval of a NDA or BLA and 50% of the time spent in testing phase, reduced by any periods of lack of diligence, are credited up to a maximum five-year extension. The PTE cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval. Only one patent per approved drug may be extended and a patent can only be extended once; thus, even if a single patent is applicable to multiple products, it can only be extended based on one product.Similar provisions to extend the term of a patent that covers an approved drug may be available in certain other foreign jurisdictions. For example, in Europe, a supplementary protection certificate (commonly referred to as a SPC), if granted, may extend certain patent rights for up to five years. In addition, in Europe, marketing approval obtained through the EMA may provide a period of ten years of regulatory data exclusivity from the time of approval. When possible, depending upon the length of clinical trials and other factors involved in the filing of NDAs and BLAs for our products, we expect to apply for patent term extension for patents covering our product candidates and their methods of use both in the United States and any foreign jurisdiction where available. There is no guarantee, however, that the applicable authorities will agree to grant extensions, and if granted, what the length of those extensions will be.surgery-induced hypoglycemia, associated with bariatric surgery and lymphedema,COVID-19, these conditions are where various treatments from many companies are used and where many public and private universities and research organizations are actively engaged in the discovery, research and development of product candidates. As a result, there are and will likely continue to be extensive resources invested in the discovery and development of new products to treat these unmet medical needs. We anticipate facing intense and increasing competition as new products enter the market and advanced technologies become available.Hepatitis delta virus:•HDV: Gilead (conditional approval in Europe, BLA submitted in the U.S.), Vir (Phase 2), Janssen Research & Development, LLC (Phase 2), Replicor, Inc. (Phase 2), Hepatera LtdPharmaEssentia (Phase 2)Lymphedema: Novartis (Phase 2). Merck & Co., Inc., or Merck, which provides us with the exclusive right to develop and commercialize lonafarnib. As consideration for such exclusive right, we issued Private Eiger convertible preferred stock with a fair value of $0.5 million when the agreement was executed in September 2010. This preferred stock was converted to 27,350 shares of common stock upon the Merger. In addition, we are obligated to pay Merck up to an aggregate of $27.0 million in development milestones and will be required to pay tiered royalties based on aggregate annual net sales of all licensed products ranging from mid-single to low double-digit royalties on net sales. Our obligation to pay royalties to Merck expires on a country-by-country and product-by-product basis on the later of the expiration of the last to expire patent assigned to us under the agreement which is estimated to be in the first half of 2018; or on the tenth anniversary of the first commercial sale of the product. In May 2015, the first regulatory milestone was achieved, and we paid the related milestone payment of $1.0 million to Merck. No additional milestone payments were incurred during the yearsyear ended December 31, 20172021. Upon completion of D-LIVR, we will owe Merck a $1.0 million milestone. We announced primary topline data of D-LIVR in December 2022, and 2016., or EGI, (EGI) dated December 8, 2010 or the(the EGI APA.APA). Dr. Jeffrey Glenn, a member of our board of directors, is the sole owner of EGI.farnesyl transferasefarnesyltransferase inhibitors as anti-viral agents and methods to treat viral infection with those inhibitors. We also purchased all assets including intellectual property rights related to the use of inhibitors of prenylation, prenyl cysteine methyltransferase, and a specified protease as anti-viral agents and methods to treat viral infection with those inhibitors. We are obligated to use commercially reasonable efforts to develop and commercialize the licensed products in major markets.License Agreement with Janssen Pharmaceutica NVIn December 2014, we, through our wholly-owned subsidiary EB Pharma, LLC, or EBP, entered a License Agreement with Janssen Pharmaceutica NV, or Janssen, dated December 19, 2014, or the Janssen License Agreement.Under the Janssen License Agreement, Janssen granted us an exclusive, worldwide, license to develop, manufacture, and sell products containing the compound tipifarnib for all therapeutic and diagnostic uses in humans, including any such uses for human virology diseases, but excluding oncology diseases.We are responsible for the development of at least one product in a major market country and for commercialization of products in all countries where necessary authorization is obtained, both at our cost and expense. We may manufacture, develop, and commercialize the products itself or we may grant one or more sublicenses for such purposes. However, for a period of time following completion of the proof of concept trial, Janssen has a first right of negotiation for an exclusive license back from us to develop and commercialize tipifarnib in any country in the world.Under the Janssen License Agreement, we are obligated to make development milestone payments in aggregate of up to $38.0 million, sales milestone payments in aggregate of up to $65.8 million, and pay a tiered royalty, ranging from the mid-single to low double digits, based on aggregate annual net sales of all licensed products. If we grant a sublicense, we are obligated to pay Janssen a portion of the sublicensing income received. As of December 31, 2017,2022, the product has not reached commercialization and no milestones have been paid.The Janssen License Agreement will continue for so long as we owe royalty payments to Janssen under the agreement or for so long as there is a valid patent claim under the agreement, whichever is longer. Both parties have the right to terminate the agreement for the other party’s uncured material breach of the agreement or for the other party’s bankruptcy. Janssen also has the right to terminate the agreement if we fail to meet certain specified diligence obligations. In addition, we have the right to terminate the agreement without cause at any time.License Agreement with Nippon Kayaku Co., Ltd.In May 2015, Eiccose, LLC, or Eiccose, and Nippon Kayaku Co., Ltd, or NK, entered into a License Agreement, or the NK License, dated May 1, 2015 pursuant to which NK granted Eiccose an exclusive license to develop, manufacture, and sell ubenimex outside certain identified Asia countries, including Japan, for the treatment of PAH and other inflammatory disease involving leukotriene B4. Eiccose assigned the NK License to us as part of the Eiccose asset purchase described below.Under the NK License, we are responsible for the development and commercialization of ubenimex in our territory at our cost and expense. We will purchase ubenimex for development and commercial use from NK at agreed transfer prices under a separate supply agreement, but we have the option to manufacture and supply the product for Phase 3 studies and/or commercial use. If we exercise the manufacturing option, NK will transfer the manufacturing of the product to us or our contract manufacturer, at our cost and expense, and we will pay NK a running, mid-single-digit royalty on the net sales of ubenimex sold in our territory or, if the parties agree, a lump-sum payment, for the use of NK’s manufacturing know-how.Under the NK License, we also granted back to NK an exclusive license to develop, manufacture, and sell ubenimex for the treatment of PAH and other inflammatory disease involving leukotriene B4 in the Asia countries comprising the NK territory. NK is responsible for the development and commercialization of ubenimex in the licensed indications in its territory at its own cost and expense. NK will pay us a running, mid-single-digit royalty on net sales of ubenimex in the specified indications in NK’s territory.The NK License Agreement will continue for so long as the parties and their sublicensees continue to develop and commercialize ubenimex for the treatment of PAH and other inflammatory disease involving leukotriene B4. Both parties have the right to terminate the agreement for the other party’s uncured material breach, and NK also has the right to terminate the agreement if we fail to meet certain specified diligence obligations. In addition, the parties may terminate the agreement if further development of the product is commercially, financially, or otherwise not advisable.In September 2015, weor the Sellers,(the Sellers) dated September 25, 2015 or the(the Exendin APA.APA). We also entered into a consulting agreement with the Sellers as part of the agreement.exendin 9-39avexitide from the Sellers, including an assignment of a license agreement with Stanford which covered exclusive rights with respect to the compound exendin 9-39.avexitide. Under the assigned Stanford exclusive license agreement, we are obligated to pay Stanford a low, single-digit royalty on net sales after the first commercial sale of any product developed based on exendin 9-39.exendin 9-39avexitide subject to certain reductions and exceptions. Our obligation to pay royalties expires on the expiration of the last to expire patent assigned to us under the agreement. We also agreed to retain each of the Sellers as consultants pursuant to consulting agreements, each with a term of one year, subject to annual renewal. The consulting agreement with Dr. Tracey McLaughlin was extendedagreements related to go through December 31, 2017. The consulting agreement with Dr. Colleen Craig expired in the year ended December 31, 2016.Exendin APA have expired. During the year ended December 31, 2017, upon the successful completion of the Phase 2 trials, the development milestone was achieved, and we paid the related milestone payment of $0.1 million to each of the Sellers.Exclusive Agreement with the Board of Trustees of the Leland Stanford Junior University—LymphedemaIn October 2015, as part of the assets we purchased from Eiccose, we acquired and were assigned an Exclusive Agreement between Eiccose and the Board of Trustees of Stanford dated October 27, 2015, or the Stanford Lymphedema Agreement.Under the Stanford Lymphedema Agreement, Stanford granted us an exclusive, worldwide license under specified patent rights related to the treatment of lymphedema, to manufacture, use, and sell products covered by the licensed patents for all uses.We are responsible for the development and commercialization of any products under the license at our cost and expense, and are obligated to use commercially reasonable efforts to achieve certain specified milestones. In consideration of the license, we paid to Stanford a low, single-digit equity interest and are obligated to make development and commercial milestone payments in aggregate of up to $0.5 million as well as a low, single-digit royalty on net sales of any products. As of December 31, 2017, the product has not reached commercialization and no milestones have been paid.Stanford may terminate the agreement for our uncured material breach or bankruptcy. Stanford also has the right to terminate the agreement if we fail to develop and commercialize products in accordance with certain specified diligence obligations. We have the right to terminate the agreement without cause at any time., together BMS, the BMS Purchase Agreement and the BMS License Agreement.Product,Product) for all therapeutic and diagnostic uses in humans and animals.million. million. The BMS Purchase Agreement grants BMS certain registration rights with respect to the shares of common stock delivered, and BMS has agreed to certain trading and other restrictions with respect to the shares purchased.2017,2022.product has not reached commercializationUPenn/CHOP Agreement in its entirety for any reason by providing prior written notice to UPenn and noCHOP. UPenn or CHOP may terminate the UPenn/CHOP Agreement, upon a written notice, for our failure to achieve the specified diligence milestones have been paid.or FDC Act,(FDC Act) and it’sits implementing regulations. Our Lambdalambda product candidate is additionally subject to regulation as a biologic under the Public Health Service Act. The FDA subjects drugs and biologics to extensive pre and post market regulation. Failure to comply with the FDC Act and other federal and state statutes and regulations may subject a company to a variety of administrative or judicial sanctions, such as FDA refusal to approve pending NDAs,new drug applications (NDAs), BLAs, withdrawal of approvals, clinical holds, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties or criminal penalties.drug,product, can be marketed in the United States. The process required by the FDA before a new drugproduct may be marketed in the United States is long, expensive, and inherently uncertain. DrugProduct development in the United States typically involves completion of preclinical laboratory and animal tests, submission to the FDA of an Investigational New DrugIND application, or IND, which must become effective before clinical testing may commence, approval by an independent institutional review board or IRB,(IRB) at each clinical site before each trial may be initiated, performance of adequate and well controlled clinical trials to establish the safety, and effectivenessefficacy, purity and/or potency of the drugproduct for each indication for which FDA approval is sought, submission to the FDA of an NDA or BLA, satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product is produced, and FDA review and approval of the NDA or BLA. Developing the data to satisfy FDA pre-market approval requirements typically takes many years and the actual time required may vary substantially based upon the type, complexity, and novelty of the product, disease or indication.and efficacy, purity and/or potency of the product. The conduct of the preclinical tests must comply with federal regulations and requirements, including good laboratory practice or GLP,(GLP) regulations. These preclinical results are submitted to the FDA as part of an IND along with other information, including information about the product’s chemistry, manufacturing and controls, and a proposed clinical trial protocol. Long term preclinical studies including reproductive toxicity and carcinogenicity may be initiated or continue after the IND is submitted.does raiseraises any concerns or questions and places the clinical trial on a clinical hold, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. As a result, a submission of an IND may not result in FDA authorization to commence a clinical trial. A separate submission to an existing IND must also be made for each successive clinical trial conducted during product development.or GCP,(GCP) requirements for conducting, monitoring, recording and reporting the results of clinical trials, in order to ensure that the data and results are scientifically credible and accurate and that the trial subjects are adequately informed of the potential risks of participating in clinical trials; and (ii) with protocols that detail, among other things, the objectives of the trial, the parameters to be used in monitoring safety, and the effectivenessefficacy, purity and/or potency criteria to be evaluated. Each protocol involving testing on U.S. patients and subsequent protocol amendments must be submitted to the FDA as part of the IND.drugproduct candidate has been associated with unexpected serious harm to patients, or the IRB may impose other conditions. The study sponsor or the FDA may also suspend or discontinue a clinical trial at any time on various grounds, including a determination that the subjects are being exposed to an unacceptable health risk.drugproduct candidate is initially introduced into healthy human subjects or patients with the target disease or condition, and is tested to assess safety, dosage tolerance, pharmacokinetics and pharmacological activity, and, when possible, to ascertain evidence of efficacy. The drugproduct candidate may also be tested in patients with severe or life-threatening diseases to gain an early indication of its effectiveness.drugproduct in that particular indication, ascertaining dosage tolerance, discerning the optimal dosage, and identifying possible adverse effects and safety risks.safetypotency of the drugproduct such that the FDA can evaluate the overall benefit-risk of the drugproduct and provide adequate information for the labeling and package insert for the drug.product. Trials conducted outside of the United States under similar, GCP-compliant conditions in accordance with local applicable laws may also be acceptable to the FDA in support of product approval.drugsproducts must publicly disclose certain clinical trial information, including detailed trial design. These requirements are subject to specific timelines and apply to most Phase 3 clinical trials of FDA-regulated products.drug.product. Such post approval trials are typically referred to as Phase 4 clinical trials.drugproduct in commercial quantities in accordance with current good manufacturing practice or cGMP,(cGMP) requirements. The manufacturing process must be capable of consistently producing quality batches of the drugproduct candidate and, among other things, the manufacturer must develop methods for testing the identity, strength, quality and purity of the final drug product. Additionally, appropriate packaging must be selected and tested, and stability studies must be conducted to demonstrate that the drugproduct candidate does not undergo unacceptable deterioration over its shelf life.product and establishmentprogram user fees. These fees are typically increased annually.or PDUFA,(PDUFA) guidelines that are currently in effect, the FDA has agreed to certain performance goals in the review of applications. Standard applications are generally reviewed within ten months of filing, or twelve months from submission. Although the FDA often meets its user fee performance goals, the FDAit can extend these timelines if necessary, and FDA review may not occur on a timely basis. The FDA usually refers applications for novel drugs,products, or drugsproducts that present difficult questions of safety, efficacy, purity and/or efficacy,potency, to an advisory committee—a panel of independent experts, typically including clinicians and other scientific experts—for review, evaluation, and a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound bydrugproduct with specific prescribing information for specific indications. The approval for a drugproduct may be significantly more limited than requested in the application, including limitations on the specific diseases and dosages or the indications for use, which could restrict the commercial value of the product. The FDA may also require that certain contraindications, warnings, or precautions be included in the product’s package insert, or labeling.or REMS,(REMS) to help ensure that the benefits of the drugproduct outweigh the potential risks. REMS can include medication guidelines, communication plans for healthcare professionals, and elements to assure safe use or ETASU.(ETASU). ETASU can include, but are not limited to, special training or certification for prescribing or dispensing-includingdispensing, including dispensing only under certain circumstances, special monitoring, and the use of patient registries. The requirement for a REMS or use of a companion diagnostic with a drugproduct can materially affect the potential market and profitability of the drug.product. Moreover, product approval may require, as a condition of approval, substantial post-approval testing and surveillance to monitor the drug’sproduct’s safety, efficacy, purity and/or efficacy.potency. The FDA may also condition approval on, among other things, changes to proposed labeling or development of adequate controls and specifications.fee requirementsfees for any marketed products, and the establishments at which such products are manufactured, as well as new application fees for supplemental applications with clinical data.orphan drugOrphan Drug designation to products intended to treat a rare disease or condition—generally one that affects fewer than 200,000 individuals in the United States. Orphan drugDrug designation must be requested before submitting the NDA or BLA. After the FDA grants orphan drugOrphan Drug designation, the FDA publicly discloses the drug’sproduct’s identity and its intended orphan use. Orphan drugDrug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process. The first active moiety to be approved to treat a disease with FDA’s orphan drugOrphan Drug designation is entitled to a seven-year period of marketing exclusivity in the United States fordrugproduct for the same orphan indication, regardless of patent status, except in limited circumstances, such as a showing of clinical superiority to the product with orphan exclusivity or if the FDA finds that the holder of the orphan exclusivity has not shown that it can assure the availability of sufficient quantities of the orphan drugOrphan Drug to meet the needs of patients with the disease or condition for which the drugproduct was designated. Orphan drugDrug exclusivity does not prevent the FDA from approving a different chemical/biological entity for the same disease or condition. An orphan drugOrphan Drug designation also does not preclude the same drugproduct from being developed for a different disease or condition. Among the other benefits of orphan drugOrphan Drug designation are tax credits for certain research expenses and a waiver of the application user fee.drugs,products, including standards and regulations for direct-to-consumer advertising, off-label promotion,companyCompany may face federal and/or state civil and criminal investigations and prosecutions.Drugsdrug.drug.product. In addition, the FDA may place conditions on an approval that could restrict the distribution or use of the product. Furthermore, manufacture, packaging, labeling, storage and distribution procedures must continue to conform to cGMPs after approval. Manufacturers and certain of their subcontractors are required to register their establishments with the FDA and certain state agencies and are subject to periodic unannounced inspections by the FDA and certain state agencies to assess compliance with ongoing regulatory requirements, including cGMPs. Changes to the manufacturing process are strictly regulated and often require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP requirements and impose reporting and documentation requirements upon the sponsor and any third-party manufacturers that the sponsor may decide to use. Accordingly, manufacturers must continue to expend time, money, and effort in the areas of production and quality control to maintain compliance with cGMPs. Failure to comply with the statutory and regulatory requirements can subject a manufacturer to possible legal or regulatory action, such as warning letters, suspension of manufacturing, seizure of product, injunctive action or possible civil penalties. We cannot be certain that we or our present or future third-party manufacturers or suppliers will be able to comply with the cGMP regulations and other ongoing FDA regulatory requirements. If we or our present or future third-party manufacturers or suppliers are not able to comply with these requirements, the FDA may halt our clinical trials, require us to recall a drugproduct from distribution or withdraw approval of the NDA for that drug.product. Regulatory authorities may also withdraw product approvals, request product recalls, or impose marketing restrictions through labeling changes or product removals upon discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes.“anyany request or demand”demand for money or property presented to the U.S. government. Several pharmaceuticalPharmaceutical and other healthcare companies have been prosecuted under these laws for, among other things, allegedly providing free product or other items or services of value to customers with the expectation that the customers wouldrecommend the use of certain products or bill federal programs for the product. Other companiessuch products. Companies have been prosecuted for causing false claims to be submitted because of, for example, the companies’alleged marketing of products for unapproved, and thus non-reimbursable, uses.uses, for the alleged submission of incorrect government price reporting data to certain Federal health care programs, for alleged violations of the federal Anti-Kickback Statute, and for other sales and marketing practices. Violations of the False Claims Act can result in civil penalties of up more than $25,000 per false claim or statement (an amount adjusted annually for inflation) plus three times the amount of damages sustained by the government. In addition, the civil monetary penalties statute imposes penalties against any person or entity that, among other things, is determined to have presented or caused to be presented a claim to a federal health program that the person knows or should know is for an item or service that was not provided as claimed or is false or fraudulent.or HIPAA,(HIPAA) created new federal criminal statutes that prohibit knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payors and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services.regulation bylaws and regulations, including both foreign and domestic, in the federal government and the stateslocations in which we conduct our business. HIPAA as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH, and theirits respective implementing regulations including the final Omnibus Rule published on January 25, 2013, imposes specifiedspecific requirements relating to the privacy, security and transmission of individually identifiable health information. Among other things, HITECH makes HIPAA’s security standards directly applicableinformation and requires the adoption of administrative, physical and technical safeguards to business associates, defined as service providers of covered entities that create, receive, maintain or transmit protected health information in connection with providing a service for or on behalf of a covered entity. HITECH also created four new tiers of civil monetary penalties and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorneys’ fees and costs associated with pursuing federal civil actions.protect such information. In addition, manycertain state laws govern the privacy and security of health information in certain circumstances, some of which are more stringent than HIPAA and many of which differ from HIPAA and each other in significant ways and may not have the same effect, thus complicating compliance efforts.Failure to submit timely, accurately and completely the required information for all payments, transfers of value and ownership or investment interests may result in civil monetary penalties of up to an aggregate of $0.2 million per year and up to an aggregate of $1.0 million per year for “knowing failures.” Covered manufacturers must submit reports by the 90th day of each calendar year.healthhealthcare regulatory laws described above or any other laws that apply to it, we may be subject to penalties, including potentially significant criminal, civil and/or administrative penalties, damages, fines, disgorgement, imprisonment, exclusion from participation in government healthcare programs, contractual damages, reputational harm, administrative burdens, diminished profits and future earnings, and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations.drugs.products. Whether or not we obtain FDA approval for a drug,product, we or our collaborators must obtain approval of the drug by the comparable regulatory authorities of foreign countries before commencing clinical trials or marketing of the drugproduct in those countries. The approval process varies from country to country and the time to approve may be longer or shorter than that required for FDA approval. Further, to the extent that any of our products are sold in a foreign country, we may be subject to additional foreign laws and regulations, which may include, for instance, applicable post-marketing requirements, including safety surveillance, anti-fraud and abuse laws and implementation of corporate compliance programs and reporting of payments or transfers of value to healthcare professionals.oursuch products once approved and have a material adverse effect on our sales, results of operations and financial condition. Moreover, a third-party payor’s decision to provide coverage for a drug product does not imply that an adequate reimbursement rate will be approved. Adequate third-party reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development. Additionally, coverage and reimbursement for drug products can differ significantly from payor to payor. One third-party payor’s decision to cover a particular medical product or service does not ensure that other payors will also provide coverage for the medical product or service or will provide coverage at an adequate reimbursement rate. As a result, the coverage determination process will require us to provide scientific and clinical support for the use of our products to each payor separately and will be a time-consuming process.By wayWe expect that additional state and federal healthcare reform measures will be adopted in the future, any of example,which could limit the amounts that third-party payors will pay for healthcare products and services, which could result in reduced demand for our products once approved or additional pricing pressures.oras amended by the ACA containswas passed, which substantially changed the way health care is financed by both governmental and private insurers, and significantly impacts the U.S. pharmaceutical industry. This law was designed to expand access to health insurance coverage for uninsured and underinsured individuals while containing overall healthcare costs. The framework of the ACA and other healthcare reforms continues to evolve as a result of executive, legislative, regulatory, and administrative developments.For example, Congress has enacted several laws that modify certain provisions that may reduceof the profitability of drug products. InACA, such as removing penalties, starting January 2017, Congress voted1, 2019, for not complying with the ACA’s individual mandate to adopt a budget resolution for fiscal year 2017, or the Budget Resolution, that authorizescarry health insurance and delaying the implementation of legislationthat would repeal portionscertain ACA-mandated fees. In addition, the 2020 federal spending package permanently eliminated, effective January 1, 2020, the ACA-mandated “Cadillac” tax on high-cost employer-sponsored health coverage and medical device tax and, effective January 1, 2021, the health insurer tax.the ACA. Further, on January 20, 2017, President Trump signed an Executive Order directing federal agencies with authorities and responsibilities2021 temporarily increased premium tax credit assistance established under the ACA to waive, defer, grant exemptionshelp eligible individuals cover premiums for health insurance purchased through the health insurance marketplace and removed the 400% federal poverty level limit that otherwise applies for purposes of eligibility to receive premium tax credits. The Inflation Reduction Act of 2022 extended this increased tax credit assistance and removal of the 400% federal poverty limit through 2025.On January 28, 2021, President Biden issued an executive order instructing certain governmental agencies to review and reconsider their existing policies and rules that limit access to healthcare, including among others, reexamining Medicaid demonstration projects and waiver programs that include work requirements, and policies that create unnecessary barriers to obtaining access to health insurance coverage through Medicaid or the ACA. The ACA has also faced various judicial challenges, and on June 17, 2021, the U.S. Supreme Courtdelayrepeal of all or parts of the ACA or the implementation of any provision ofnew health care legislation, could result in significant changes to the ACA that would impose a fiscalhealth care system, which may prevent us from being able to generate revenue, attain profitability or regulatory burden on states, individuals, healthcare providers, health insurers, or manufacturers of pharmaceuticals or medical devices. Congress also could consider subsequent legislation to replace elements of the ACA that are repealed.commercialize our drugs. We expect that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand or lower pricing for our products once approvedproduct candidates, or additional pricing pressures.$29.5$75.3 million, $33.0$64.4 million and $8.1$41.6 million for the years ended December 31, 2017, 20162022, 2021 and 2015,2020, respectively.2017,2022, we had a total of 1656 full-time employees in the United States eightand Europe, 39 of whom were primarily engaged in manufacturing and research and development activities, and eight17 of whom were engaged in general management and administration. Five11 of our employees have either an M.D. or a Ph.D. None of our employees are represented by a labor union or subject to a collective bargaining agreement. We have never experienced any work stoppage and consider our relations with our employees to be good.Corporation.Corporation (Celladon). In April 2012, Celladon reincorporated in Delaware and had its initial public offering in February of 2014.On March 22, 2016, Private Eiger BioPharmaceuticals, Inc. (Private Eiger) completed its merger (Merger) with Celladon in accordance with the terms of the Merger Agreement. Pursuant to the Merger Agreement, Celladon Merger Sub, Inc., a wholly-owned subsidiary of Celladon (Merger Sub) merged with and into Private Eiger, with Private Eiger becoming a wholly-owned subsidiary of Celladon and the surviving corporation of the Merger. Immediately following the Merger, Celladon changed its name to “EigerEiger BioPharmaceuticals, Inc.” In connection with the Merger, our common stock began trading on The NASDAQNasdaq Global Market with the ticker symbol “EIGR”EIGR on March 23, 2016. Our principal executive offices are located at 350 Cambridge Avenue, Suite 350,2155 Park Blvd in Palo Alto, California 94306, and our telephone number is 650-272-6138. Our corporate website address is www.eigerbio.com. The contents of our website are not incorporated into this Annual Report on Form 10-K and our reference to the URL for our website is intended to be an inactive textual reference only.We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012. We will remain an emerging growth company until the earlier of (1) the last day of the fiscal year (a) following the fifth anniversary of the completion of our initial public offering in February 2014, (b) in which we have total annual gross revenue of at least $1.1 billion, or (c) in which we are deemed to be a large accelerated filer, which means the market value of our common stock that is held by non-affiliates exceeded $700.0 million as of the prior June 30th, and (2) the date on which we have issued more than $1.0 billion in non-convertible debt during the prior three-year period. We refer to the Jumpstart Our Business Startups Act of 2012 in this Annual Report on Form 10-K as the “JOBS Act,” and references to “emerging growth company” have the meaning associated with it in the JOBS Act.ItemRiskRisk Factorsclinical development-stagecommercial-stage biopharmaceutical company with a limited operating history. We have incurred net losses in each year since our inception. For the years ended December 31, 2017, 20162022, 2021 and 2015,2020 we reported a net loss of $42.4$96.8 million, $47.1$33.9 million and $13.3$65.1 million, respectively. As of December 31, 2017,2022, we had an accumulated deficit of $118.8$437.2 million. Our prior losses, combined with expected future losses, have had and may continue to have an adverse effect on our stockholders’ equity (deficit) and working capital.theour currently available resources will be sufficient to fund our planned operations for at least the next 12 months following the issuance date of these consolidated financial statements. WeOur assumption does not reflect the possibility that we may not be able to access a portion of our existing cash, cash equivalents and short-term securities due to market and other conditions. For example, on March 10, 2023, the Federal Deposit Insurance Corporation (FDIC) was appointed receiver of Silicon Valley Bank (SVB), where we maintain an operating account with a balance that exceeded the FDIC insurance limit. Although, in this case the cash in our accounts at Silicon Valley Bank did not impact our operations or our conclusion that we have sufficient capital to fund our operations for at least the next 12 months, we cannot be certain that future market corrections or other conditions will not impact our ability to access our cash, cash equivalents and short-term securities. On March 12, 2023, the U.S. Treasury Department, the Federal Reserve and theamountamounts and timing of our future funding requirements will depend on many factors, including our ability to achieve regulatory approval and the pace and results of our clinical development efforts. Failure to raise capital as and when needed, on favorable terms or at all, would have a negative impact on our financial condition and our ability to develop our product candidates.four Phase 2our clinical development programs for potentially three indications. In addition, our recent meeting within various clinical studies, particularly the FDA in February 2018 may allow usD-LIVR pivotal study, to initiate a single potentially pivotal Phase 3 study assupport the basis for the filingsubmission of an NDA for lonafarnib-based regimens for use in which case we wouldan HDV indication. We may need significant additional resources in order to fundaggressively move lonafarnib-based regimens forward successfully based on the conduct of that potentially pivotal study. While we have not yet commenced pivotal clinical studies for any product candidate and itdiscussions with the FDA. It may be several years, if ever, before we complete pivotal clinical studies and have aadditional product candidatecandidates approved for commercialization, wecommercialization. We expect to invest significant funds into theseour clinical candidates to advance these compounds to potential regulatory approval.aone or more additional product candidate,candidates, our future revenue will depend upon the size of any markets in which our product candidates may receive approval, and our ability to achieve sufficient market acceptance, pricing, reimbursement from third-party payors, and adequate market share for our product candidates in those markets. Even if we obtain adequate market share for our product candidates, because the potential markets in which our product candidates may ultimately receive regulatory approval could be very small, we may never become profitable despite obtaining such market share and acceptance of our products.
•initiate additional nonclinical, clinical, or other studies for our product candidates;
•commercialize and provide expanded access to Zokinvy for the treatment of HGPS and processing-deficient PL•initiate additional nonclinical, clinical, or other studies for our product candidates; educate and develop potential commercial opportunities, such as lonafarnib for HDV,lonafarnib-based regimens, peginterferon lambda for HDV, exendin 9-39and avexitide for PBHHI and ubenimex forlymphedema;We haverevenue from product salesrevenue and may never be profitable.no productsone product approved for commercialization in the U.S. and have never generated any revenue.EU for two ultra-rare diseases. The first is Zokinvy, which works to (i) reduce the risk of mortality in HGPS, and (ii) treatprocessing-deficient progeroid laminopathies with either heterozygous LMNA mutation with progerin-like protein accumulation, or homozygous or compound heterozygous ZMPSTE24 mutations. Zokinvy was approved by the FDA in November 2020 and launched commercially in the U.S. in January 2021, under the exceptional circumstances procedure. In July 2022, our MAA for Zokinvy was approved by the EC, based on a recommendation by the EMA. The Marketing Authorization (MA) is subject to the EMA’s continued review on an annual basis of new efficacy and safety information which may become available. Our ability to generate substantial revenue and achieve profitability depends on our ability to (i)obtain the regulatory and marketing approvals necessary to commercialize Zokinvy in foreign jurisdictions, alone or with strategic collaboration partners, and (ii) to successfully complete the development of, and obtain the regulatory and marketing approvals necessary to commercialize, one or more of our product candidates.candidates in the U.S. or foreign jurisdictions. We do not anticipate generating revenue fromsignificant product salesrevenue for the foreseeable future. Our ability to generate future revenue from product salesrevenue depends heavily on our success in many areas, including, but not limited to:one or more of thewe obtain additional product candidates that we develop is approvedapprovals for commercial sale, we anticipate incurring significant costs associated with commercializing any approved product candidate. Our current pipeline of product candidates has been in-licensed from third parties and we will have to develop or acquire manufacturing capabilities in order to continue development and potential commercialization of our product candidates. Additionally, if we are not able to generate sufficient revenue from the sale of any approved products, we may never become profitable.loanLoan and security agreementSecurity Agreement we entered into with Oxford Finance LLC, or Oxford Finance,Innovatus Life Sciences Lending Fund I, LP (Innovatus) in December 2016, or the Oxford Loan. ThisJune 2022 (the Innovatus Loan). The Innovatus Loan was a $25.0$75.0 million debt financing arrangement with Oxford FinanceInnovatus wherein we borrowed the first tranche of $15.0$40.0 million upon closing of the debt financing in December 2016. Our ability to access the final $10.0 million under the Oxford Loan is subject to our ability to achieve certain clinical development milestones, which we may not be able to meet and which and could adversely affect our liquidity.June 2022. The OxfordInnovatus Loan is secured by the perfected first priority liens on the Company's assets, including a commitment by the Company to not allow any liens to be placed upon the Company’s intellectual property.our assets. The OxfordInnovatus Loan includes customary events of default, including failure to pay amounts due, breaches of covenants and warranties, material adverse effect events, certain cross defaults and judgments, and insolvency. we will we will be able to obtain additional funding if and when necessary to fund our entire portfolio of product candidates to meet our projected plans. If we are unable to obtain funding on a timely basis, we may be required to delay or discontinue one or more of our development programs or the commercialization of any product candidates or be unable to expand our operations or otherwise capitalize on potential business opportunities, which could materially affect our business, financial condition, and results of operations.our loan and security agreementthe Innovatus Loan restrict our business and operations in many ways and if we do not effectively managecomply with our covenants, our financial conditions and results of operations could be adversely affected. In addition, we may not meet the milestones required to access the final loan available under the agreement and may also not provide sufficient cash to meet the repayment obligations of our debt incurred under the loan and security agreement.OxfordInnovatus Loan provides for up to $25.0$75.0 million in term loans due on July 1, 2021,August 31, 2027, of which $15.0$40.5 million in term loans have been borrowed to date.principal is outstanding as of December 31, 2022. All of our current and future assets, except for intellectual property, are secured forsecure our borrowings under the loan and security agreement.Innovatus Loan. The loan and security agreementInnovatus Loan requires that we comply with certain covenants applicable to us, including among other things, covenants restricting dispositions, changes in business, management, ownership or business locations, mergers or acquisitions, indebtedness, encumbrances, distributions, investments, transactions with affiliates and subordinated debt, any of which could restrict our business and operations, particularly our ability to respond to changes in our business or to take specified actions to take advantage of certain business opportunities that may be presented to us. Our failure to comply with any of the covenants could result in a default under the loan and security agreement,Innovatus Loan, which could permit the lenders to declare all or part of any outstanding borrowings to be immediately due and payable, or to refuse to permit additional borrowings under the loan and security agreement.payable. If we are unable to repay those amounts, the lenders under the loan and security agreementInnovatus Loan could proceed against the collateral granted to them to secure that debt, whichand our inability to use or dispose of those assets would seriously harm our business. In addition, should we be unable to comply with these covenants or if we default on any portion of our outstanding borrowings, the lenders can also impose a 5.0% penalty, and restrict access to additional borrowings under the loan and security agreement. Moreover, our ability to accessagreement, and accelerate the final $10.0 millionmaturity of the debt. Any default under the loan and security agreement is subject to our ability to achieve certain clinical development milestones, which we may not be able to meet and which and could adverselyInnovatus Loan would materially affect our liquidity. In addition, although we expect to borrow additional funds under the loan and security agreement, before we do so, we must first satisfyourselves that we will have access to future alternate sources of capital, including cash flow from our own operations, equity capital markets or debt capital markets in order to repay any principal borrowed, which we may be unable to do, in which case, our liquidity and ability to fund our operations mayand complete our planned clinical trials and regulatory filings would be substantially impaired. heavily dependent on the success of our product candidates, which are in the earlyvarious stages of clinical development. Certain of our product candidates have produced results in academic settings to date or for other indications than those that we contemplate, and we cannot give any assurance that we will generate data for any of our product candidates sufficient to receive regulatory approval in our planned indications, which will be required before they can be commercialized.successfully further develop, obtain regulatory approvalapprovals for, and commercialize one or more of these product candidates. We currently generate noOur NDA for Zokinvy to reduce the risk of mortality in HGPS, and for treatment of processing-deficient progeroid laminopathies (PL) with either heterozygous LMNA mutation with progerin-like protein accumulation, or homozygous or compound heterozygous ZMPSTE24 mutations, was approved in November 2020. Prior to the U.S. Zokinvy commercial launch in 2021, we had not generated revenue from sales of any drugs,products and we may never be able to develop or commercialize additional product candidates. In addition, we have a product candidate.Wecommitment to provide access to Zokinvy for patients with HGPS and PL with either heterozygous LMNA mutation with progerin-like protein accumulation, or homozygous or compound heterozygous ZMPSTE24 mutations, for no or minimal cost to those patients.fourthree product candidates that are in Phase 3 clinical development, lonafarnib-based regimens for HDV, peginterferon lambda for HDV, and avexitide for HI development program. Avexitide for PBH has completed Phase 2 development programs focused on three separate indications. For oneclinical trials. It may be years before our studies are completed, and new studies initiated, if at all.In addition, none of our product candidates have advanced into a pivotal study for our proposed indications and it may be years before such study is initiated and completed, if at all. Although our February 2018 discussions with the FDA with respect to the entry of lonafarnib into a single potentially pivotal clinical trial have been positive, Likewise, there can be no assurance that we would be able to design a singlethe data obtained from foreign clinical trial that would enablesites in studies not conducted under an NDA filing if successful andinvestigational new drug application, or IND, will be accepted in any eventsupport of an application for regulatory approval or authorization for usetiming of our discussions with the FDA are uncertain andU.S. Similarly, data obtained from foreign clinical trial sites may not meet our timelinesbe accepted by other foreign regulatory authorities in support of an application for the planned initiation of a Phase 3 study of lonafarnib by the end of 2018.orphan drugand maintaining Orphan Drug designation for our product candidates, which is an uncertain process. The regulatory approval processes of the FDA and comparable foreign authorities are lengthy, time consuming, and inherently unpredictable. If we are unable to obtain orphan drugor maintain Orphan Drug designation or regulatory approval for our product candidates, our business willwould be substantially harmed.orphan drugand maintain Orphan Drug designation from regulatory authorities in major markets. Without the potential protection of this regulatory exclusivity upon approval, many of our product candidates would otherwise not justify investment. While we assess the potential for obtaining orphan drugOrphan Drug designation at the time that we contemplate the acquisition of product candidates and we intend to timely file for such designation, there can be no assurance that we will obtain orphan drugOrphan Drug designation or beorphan drugand maintain Orphan Drug designation would make ouralready obtained orphan drugcurrently have Orphan Drug designation for foursome of our product candidates in threemultiple targeted indications, failure to demonstrate significant benefit over existing approved drugsproducts in pivotal clinical trials may lead to marketing approval but without qualifying for orphan drugOrphan Drug protection in some regions, such as in Europe.not obtained U.S. and EMA regulatory approval for anyone product, candidate,Zokinvy, and it is possible that none of our existingcurrent product candidates or any future product candidates we may seek to develop in the future will ever obtain regulatory approval.Applicationsa new drug application, oran NDA, BLA, or other submission or to obtain regulatory approval in the United States or foreign jurisdictions. For example, we identified significant good clinical practice, or GCP, violations in the LOWR HDV – 1 and LOWR HDV – 2 studies. We alerted the FDA to the discovery of GCP violations. These findings may bring into question the validity of the data set and may require repeat clinical studies to confirm the observations in question. Further studies may be needed to confirm the rigor, robustness and validity of any problematic safety and efficacy data identified;orphan drugOrphan Drug designation for fourfive of our product candidates in our planned indicationsdevelopment programs to date, there can be no assurance that the FDA or comparable foreign regulatory authorities will grant our similar status for our other proposed development indications or other product candidates in the future.and safetypurity and/or potency with respect to the proposed indication for use sufficient to obtain regulatory approval to receive regulatory approval or market our drugproduct candidates.and efficacy, purity and/or potency to the satisfaction of applicable regulatory authorities.CROscontract research organizations (CROs) and clinical study sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and clinical study sites;or IRB,(IRB) approval at each clinical study site;or IND,(IND) or equivalent foreign application or amendment;studies;
•feasibility of continuous trial execution in countries impacted by war, geopolitical conflict and other humanitarian crises;GCPgood clinical practice (GCP) requirements, or applicable foreign regulatory guidelines;patients dropping out of our clinical studies;orphan drugor maintain Orphan Drug designation exclusivity and to successfully commercialize our product candidates and may harm our business and results of operations.Undesirable side effects, or unforeseen side effects due to incomplete or inadequate safety data collection from LOWR HDV – 1 and LOWR HDV – 2 clinical studies, caused by our product candidates could cause us or regulatory authorities to interrupt, delay, or terminate or conduct additional or larger clinical studies or even if approved, result in a restrictive label or delay regulatory approval by the FDA or comparable foreign authorities.For example, ourAdditionally, while we have a licenseOur peginterferon lambda product candidate is well-characterized and has been studied in thousands of HBV and HCV patients, and the most common adverse events seen are moderate headache, pyrexia, fatigue, and myalgia. ALT flares that were seen result from vigorous antiviral immunological response to another farnesyltransferase inhibitor compound, tipifarnib, from Janssen Pharmaceutica, N.V.,treatment, not due to direct hepatotoxicity. There is no guarantee that additional or Janssen, Janssen has granted rights to tipifarnib to Kura Oncology, Inc., or Kura, in oncology and negative results or undesirablemore severe side effects from Kura’swill not be identified through ongoing clinical trialsstudies for a compound with a similar mechanismother uses of action may negatively impact the perception of lonafarnib for anti-viral indications. Merck may also grant rights to other anti-viral or potentially other indications to other third parties.peginterferon lambda. Undesirable side effects, other properties, and negative results for other indications may negatively impact the development and potential for approval of our product candidates for our proposed indications.
•we may be required to create a Risk Evaluation and Mitigation Strategy (REMS) plan, which could include a medication guide outlining the risks of such side effects for distribution to patients, a communication plan for healthcare providers, and/or other elements to assure safe use;Evenapprovalapprovals for a product candidate, we will remainbe subject to additional ongoing regulatory requirements.or cGMP,(cGMP) regulations and corresponding foreign regulatory manufacturing requirements. As such, we and our contract manufacturers will be subject to continual review and inspections to assess compliance with cGMP and adherence to commitments made in any NDA, or marketing authorization application,BLA, or MAA.IV4 clinical trials, and surveillance to monitor the safety, and efficacy, purity and/or potency of the product candidate. We will be required to report certain adverse reactions and production problems, if any, to the FDA and comparable foreign regulatory authorities.drugproduct safety issues could result in delays in product development or commercialization, or increased costs to assure compliance. If our original marketing approval for a product candidate was obtained through an accelerated approval pathway, we could be required to conduct a successful post-marketing clinical study in order to confirm the clinical benefit for our products. An unsuccessful post-marketing study or failure to complete such a study could result in the withdrawal of marketing approval.
However, physicians may, in their independent medical judgment, prescribe legally available products for off-label uses. The FDA does not regulate the behavior of physicians in their choice of treatments, but the FDA does restrict manufacturer’s communications on the subject of off-label use of their products.For example, we identified significant GCP violations in the LOWR HDV – 1 and LOWR HDV – 2 studies. We alerted the FDA to the discovery or GCP violations. These findings may call into question the validity of the data set and may require repeat clinical studies to confirm the observations in question. Further studies may be needed to confirm the rigor, robustness and validity of any problematic safety and efficacy data identified. We cannot assure you that our CROs and other vendors will meet these requirements, or that upon inspection by any regulatory authority, such regulatory authority will determine that efforts, including any of our clinical studies, comply with applicable requirements. Our failure to comply with these laws, regulations and guidelines may require us to repeat clinical studies or conduct larger additional studies, which would be costly and delay the regulatory approval process.future clinical studies. With respectstudies, however, our trials could be delayed if quality, stability, or other issues occur in relation to our lambda program, as partthe manufacture of the license agreement, we obtained a substantial inventoryany unique batch. If these CMOs are not able to provide usfrom BMS sufficient to initiatefor our clinical trials. During 2017 we transferredtrials or in support of our commercialization of potential products on a timely basis, or at all, whether due to production shortages or other supply interruptions resulting from any delay, our clinical trials or regulatory approval may be delayed, or could impair our ability to generate revenue from the manufacturing technology to our third-partyvendors. With respect to our ubenimex programs, we have relied on Nippon Kayaku to provide us withsale of such product to conduct our trials in 2016 and 2017 and have now completed the process of transferring the manufacturing of ubenimex to our third-party vendors in the United States and such drug product will be adequate to complete ongoing and future clinical studies.drug product, or fail to do so at acceptable quality levels or prices our product candidates could be stopped, delayed, or made less profitable.mustwill be approvedsubject to pre-approval inspection by the FDA pursuant to inspections that will be conducted after we submit our marketing applications to the FDA.FDA or comparable foreign regulatory authorities. We do not control the manufacturing process of, and are completely dependent on, our contract manufacturing partners for compliance with the regulatory requirements, known as cGMPs, for manufacture of our product candidates. If our contract manufacturers cannot successfully manufacture material that conforms to our specifications and the strict regulatory requirements of the FDA, EMA or others, they willour future applications may not be able to secure and/or maintainapproved by regulatory approval for their manufacturing facilities. In addition, weauthorities, which would significantly delay our commercialization plans and increase our costs. We have no control over the ability of our contract manufacturers to maintain adequate quality control, quality assurance and qualified personnel.personnel, and in the past we have experienced quality control issues with product manufactured by our contract manufacturers. If the FDA or a comparable foreign regulatory authority does not approve these facilities for the manufacture of our product candidates or if it withdraws any such approval in the future, we may need to find alternative manufacturing facilities, which would significantly impact our ability to develop, obtain regulatory approval for or market our product candidates, if approved.nevernot be able to develop aadditional commercially viable product.ifof any of our product candidates by the FDA or comparable foreign regulatory authorities or the commercialization of our product candidates or result in higher costs or deprive us of potential product revenue. In addition, we rely on third parties to perform release testing on our product candidates prior to delivery to patients. If these tests are not conducted appropriately and test data is not reliable, patients could be put at risk of serious harm and could result in product liability suits.for lonafarnib and lambda, HDV is associated with hepatitis B virus infection, which is a pre-requisite for the replication of HDV. Althoughalthough we believe that theour lonafarnib-based regimens and peginterferon lambda data are supportive of the increased severity of hepatitis in the presence of hepatitis D and hepatitis B virus co-infection compared to hepatitis B alone,antiviral activity against HDV, there can be no assurance that our clinical trials will successfully address this condition. Likewise, the potentially addressable patient population for each of our product candidates may be limited or may not be amenable to treatment with our product candidates, and new patients may become increasingly difficult to identify or gain access to, which would adversely affect our results of operations and our business.Inc., Johnson & Johnson, Replicor, Inc., Hepatera, Arrowhead Pharmaceuticals, Novartis International, AG,Zealand Pharmaceuticals, Xeris Pharmaceuticals, Rezolute, Hanmi Pharmaceutical, and XerisCrinetics Pharmaceuticals as well as other smaller companies or biotechnology startups and large multinational pharmaceutical companies. Many of our competitors haveWe currentlynolimited recent experience selling and marketing our product candidates and we currently have noa small sales and marketing or sales organization. To successfully commercialize anyZokinvy and additional products that may result from our development programs, we will need to invest in and developexpand these capabilities, either on our own or with others, which would be expensive, difficult and time consuming. Any failure or delay in the timely development of our internal commercialization capabilities could adversely impact the potential for success of our products.lack of priorrecent, limited experience in marketing and selling biopharmaceutical products, we may rely on future collaborators to commercialize our products. If collaborators do not commit sufficient resources to commercialize our future products and we are unable to develop the necessary marketing and sales capabilities on our own, we will be unable to generate sufficient product revenue to sustain or grow our business. We may be competing with companies that currently have extensive and well-funded marketing and sales operations, in particular in the markets our product candidates are intended to address. Without appropriate capabilities, whether directly or through third-party collaborators, we may be unable to compete successfully against these more established companies.candidatecandidates will depend upon the degree of market acceptance by physicians, patients, third-party payors, and others in the medical community.therapies;
•the publicity concerning our products or competing products and treatments;
•the success of our physician education programs;•the publicity concerning our products or competing products and treatments; andand safetypurity and/or potency profile upon approval, market acceptance of the product remains uncertain. Efforts to educate the medical community and third-party payors on the benefits of the products may require significant investment and resources and may never be successful. If our products fail to achieve anorphan drugOrphan Drug designated indications where the eligible patient population is small. Sales of our product candidatesproducts will depend substantially, both domestically and abroad, on the extent to which the costs of our product candidates will be paid for or reimbursed by health maintenance, managed care, pharmacy benefit and similar healthcare management organizations, or government authorities, private health insurers, and other third-party payors. If coverage and reimbursement are not available, or are available only in limited amounts, we may have to subsidize or provide products for free or we may not be able to successfully commercialize our products.or CMS,(CMS), an agency within the U.S. Department of Health and Human Services (HHS), as CMS decides whether and to what extent a new drug will be covered and reimbursed under Medicare. Private payors tend tooften follow the coverage reimbursement policies established by CMS to a substantial degree. It is difficult to predict what CMS and other payors will decide with respect to reimbursementcoverage for products such as ours and what reimbursement codes our products may receive.they may not coverinadequate coverage or provide adequate payment for our products.the increasing trend toward managedincreased and continued efforts to limit or reduce healthcare including the increasing influence of health maintenance organizations and additional legislative changes.spending. The downward pressure on healthcare costs in general, particularly prescription drugs, has and is expected to continue to increase in the future. As a result, profitability of our products may be more difficult to achieve even if they receive regulatory approval.orphan drugOrphan Drug designation as well as patent rights for our product candidates and any future product candidates. If we are unable to obtain or maintain exclusivity from the combination of these approaches, we may not be able to compete effectively in our markets.orphan drugOrphan Drug designation may be obtained in the major markets of the world. In addition, we rely or will rely upon a combination of patents, trade secret protection, and confidentiality agreements to protect the intellectual property related to our technologies and product candidates. For example, the portfolio of patents licensed from Merck expires before the anticipated launch date of thelonafarnib product candidate. Our success depends in large part on our and our licensors’ ability to obtain regulatory exclusivity and maintain patent and other intellectual property protection in the United States and in other countries with respect to our proprietary technology and products.orphan drugOrphan Drug if it is intended to treat a rare disease or condition, defined as a patient population of fewer than 200,000 in the United States, or a patient population greater than 200,000 in the United States where there is no reasonable expectation that the cost of developing the drugproduct will be recovered from sales in the United States. In the European Union (the EU), the EMA’s Committee for Orphan Medicinal Products or COMP,(COMP) grants orphan drugOrphan Drug designation to promote the development of products that are intended for the diagnosis, prevention, or treatment of a life-threatening or chronically debilitating condition affecting not more than five in 10,000 persons in the European Union.EU. Additionally, designation is granted for products intended for the diagnosis, prevention, or treatment of a life-threatening, seriously debilitating or serious and chronic condition when, without incentives, it is unlikely that sales of the drug in the European UnionEU would be sufficient to justify the necessary investment in developing the drug or biological product or where there is no satisfactory method of diagnosis, prevention, or treatment, or, if such a method exists, the medicine must be of significant benefit to those affected by the condition.orphan drugOrphan Drug designation entitles a party to financial incentives such as opportunities for grant funding towards clinical trial costs, tax advantages, and user-fee waivers. In addition, if a product receives the first FDA approval for the indication for which it has orphan drugOrphan Drug designation, the product is entitled to orphan drugOrphan Drug exclusivity, which means the FDA may not approve any other application to market the same drugproduct for the same indication for a period of seven years, except in limitedEuropean Union, orphan drugEU, Orphan Drug designation entitles a party to financial incentives such as reduction of fees or fee waivers and ten years of market exclusivity following drug or biological product approval. This period may be reduced to six years if the orphan drugOrphan Drug designation criteria are no longer met, including where it is shown that the product is sufficiently profitable not to justify maintenance of market exclusivity.orphan drugOrphan Drug designation is especially important for our products for which orphan drugOrphan Drug designation may be available. For eligible drugs,products, we plan to rely on the exclusivity period under the Orphan Drug Act to maintain a competitive position. If we do not obtain orphan drugor maintain Orphan Drug exclusivity for our drug products and biologicbiological products that do not have broad patent protection, our competitors may then sell the same drug to treat the same condition sooner than if we had obtained orphan drugOrphan Drug exclusivity and our revenue will be reduced.orphan drug designationOrphan Drug designations for lonafarnibeach of our development programs in the United States and Europe, we may not be the first to obtain marketing approval for any particular orphan indication due to the uncertainties associated with developing pharmaceutical products. Further, even if we obtain orphan drugOrphan Drug exclusivity for a product, that exclusivity may not effectively protect the product from competition because different drugsproducts with different active moieties can be approved for the same condition. Even after an orphan drugOrphan Drug is approved, the FDA or EMA can subsequently approve the same drugproduct with the same active moiety for the same condition if the FDA or EMA concludes that the later drugproduct is safer, more effective, or makes a major contribution to patient care. Orphan drugDrug designation neither shortens the development time or regulatory review time of a product candidate nor gives the product candidate any advantage in the regulatory review or approval process.in-licensesin-licensed may fail to result in issued patents with claims that cover our product candidates in the United States or in other foreign countries. There is no assurance that all potentially relevant prior art relating to our patents and patent applications has been found, which can invalidate a patent or prevent a patent from issuing from a pending patent application. Even if patents do successfully issue, and even if such patents cover our product candidates, thirdpatent coverage for lonafarnib expires beforearbitration with PRF described in Part I, Item 3 Legal Proceedings below, PRF is indirectly challenging the anticipated launch date.validity of our in-license of certain patents covering the methods of treating HGPS and progeroid laminopathies. Likewise,For example, Kura has an exclusive license to tipifarnib for use in cancer indications while we have a license for anti-viral indications. As a result of Kura’s right to use the same compound in a different indication, it is possible that development and sales may impact our product development and commercialization efforts. If we cannot obtain and maintain effective protection of exclusivity from our regulatory efforts and intellectual property rights, including patent protection, for our product candidates, we may not be able to compete effectively, and our business and results of operations would be harmed.or USPTO.(USPTO). For example, a patent term can be reduced based on certain delays caused by the patent applicant during patent prosecution.ubenimex, lonafarnib,lonafarnib-based regimens, peginterferon lambda and exendin 9-39,avexitide, a substantial portion of the potential commercial opportunity will likely rely on patent term extensions, and we cannot provide any assurances that any such patent term extensions will be obtained and, if so, for how long. As a result, we may not be able to maintain exclusivity for our products for an extended period after regulatory approval, which would negatively impact our business and results of operations. If we do not have sufficient patent terms or regulatory exclusivity to protect our products, our business and results of operations will be adversely affected.or the(the Leahy-Smith Act,Act) enacted on September 16, 2011, the United States has moved to a first to file system. The Leahy-Smith Act also includes a number of significant changes that affect the way patent applications will be prosecuted and may also affect patent litigation. The effects of these changes are currently unclear as the USPTO must still implement various regulations, the courts have yet to address any of these provisions and the applicability of the act and new regulations on specific patents discussed herein have not been determined and would need to be reviewed. In general, the Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents, all of which could have a material adverse effect on our business and financial condition., and if patents issue with respect to any such application and we become aware of such issuance, we would have to determine its impact on our efforts to develop and commercialize our product candidates and the strategy for obtaining a license or contesting any such issued patent.Stanford license agreementsagreement for our ubenimex and lonafarnib product candidates.lonafarnib. We may not be able to achieve the required diligence milestones in a timely manner, which may result in a right of termination by Stanford,the license agreement being terminated, and we may be unable to successfully negotiate an extension or waiver of those termination rights. Any termination of license agreements with third parties with respect to our product candidates would be expected to negatively impact our business prospects.or ANDA,(ANDA) seeking approval of a generic copy of an approved innovator product marketed under an NDA. Generally, in place of clinical studies intended to demonstrate safety or effectiveness, an ANDA applicant usually needs only to submit data demonstrating that its product has the same active ingredient(s), strength, dosage form, route of administration and that it is bioequivalent to the branded product. Under the Hatch-Waxman Act, a manufacturer may also submit an NDA underexclusivity (e.g.,exclusivity. The FDCA, provides a period of five years of non-patent exclusivity for a new drug containing a new chemical entities,entity (NCE). During the exclusivity period, the FDA may not accept for review an ANDA or a 505(b)(2) NDA submitted by another company for another version of such product candidate where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement. The FDCA also provides three years of marketing exclusivity for changesan NDA, 505(b)(2) NDA or supplement to an approved drug requiring aNDA if new clinical study, seven yearsinvestigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for orphan drugs), which precludeexample, for new indications, dosages or strengths of an existing product candidate. This three-year exclusivity covers only the conditions associated with the new clinical investigations and does not prohibit the FDA from approving ANDAs for product candidates containing the original active agent for other conditions of use. Five-year and three-year exclusivity will not delay the submission or approval (or in some circumstances, FDA filingof a full NDA. However, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the nonclinical studies and review of) an ANDA or 505(b)(2) NDA relyingadequate and well-controlled clinical trials necessary to demonstrate safety and effectiveness.FDA’s findinginvestments we have made in those drug candidates. Our future revenues, profitability and cash flows could also be materially and adversely affected and our ability to obtain a return on the investments we have made in those drug candidates may be substantially limited if our drug candidates, if and when approved, are not afforded the appropriate periods of safety and effectiveness for the innovative drug. non-patent exclusivity.product candidatesproducts in the Orange Book after approval by the FDA, ANDAs and 505(b)(2) NDAs with respect to those product candidatesproducts would be required to include a certification as to each listed patent indicating whether the ANDA applicant does or does not intend to challenge the patent. We cannot predict whether any patents issuing from our pending patent applications will be eligible for listing in the Orange Book, how any generic competitor would address such patents, whether we would sue on any such patents, or the outcome of any such suit. Likewise, certain of our license agreements, for example for ubenimex, do not include patents or patent applications outside of the United States as our licensor elected not to file in foreign jurisdictions. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as federal and state laws in the United States. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and may also export infringing products to territories where we have patent protection, but enforcement is not as strong as that in the United States. These products may compete with our products and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.to attract, retain,a member of our Board, and motivate other qualified personnel.
The Board has appointed Dr. David Apelian as the Company’s Interim President and Chief Executive Officer, and Dr. Apelian will remain a member of the Board. We are highly dependent on David Cory, ourDr. Apelian until we hire a full-time non-interim President and Chief Executive Officer, the loss of whose services may adversely impact the achievement of our objectives. Mr. Cory could leave our employment at any time, as he is an “at will” employee. and will need to attract and retain appropriate finance and other personnel.orincluding appropriate finance and legal personnel, the loss of the services of Mr. CoryDr. Apelian, or the failure to hire a fulltime President and Chief Executive Officer and other qualified management personnel, may impede the progress of our research, development, and commercialization objectives and would negatively impact our ability to succeed in our in-licensing strategy.2017,2022, we had 1656 full-time employees. As our development and commercialization plans and strategies develop, we expect to need additional managerial, operational, manufacturing, sales, marketing, financial, legal, and other resources. Our management may need to divert a disproportionate amount of their attention away from our day-to-day activities and devote a substantial amount of time to managing these growth activities. We may not be able to effectively manage the expansion of our operations, which may result in weaknesses in our infrastructure, operational mistakes, loss of business opportunities, loss of employees, and reduced productivity among remaining employees. Our expected growth could require significant capital expenditures and may divert financial resources from other projects, such as the development of additional product candidates. If our management is unable to effectively manage our growth, our expenses may increase more than expected, our ability to generate and/or grow revenue could be reduced and we may not be able to implement our business strategy. Our future financial performance and our ability to commercialize product candidates and compete effectively will depend, in part, on our ability to effectively manage any future growth.
Failure to comply with existing or future laws and regulations related to privacy or data security could lead to government enforcement actions (which could include civil or criminal fines or penalties), private litigation, other liabilities, and/or adverse publicity. Compliance or the failure to comply with such laws could increase the costs of our products and services, could limit their use or adoption, and could otherwise negatively affect our operating results and business.or(IT) systems, and the availability of data related to our products, services and operations. IT systems. IT systems and data are vulnerable to risks and damages from a variety of sources, including catastrophe or natural disaster, telecommunications or network failures, malicious human acts, andbreaches of security, cyber-attacks, loss of power or other natural disasters.or man-made events. Moreover, despite network security and back-up measures, some ofwe and our vendors frequently defend against and ourrespond to data security attacks and incidents, and vendors’ servers are potentially vulnerable to physical or electronic break-ins, computer viruses, software vulnerabilities, ransomware attacks and similar disruptive problems. If our business continuity and disaster recovery plans and procedures were disrupted, inadequate or unsuccessful in the event of a problem, we could experience a material adverse interruption of our operations.business.containreform the delivery and payment for healthcare costs.items and services. For example, in March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or the ACA was passed, which substantially changeschanged the way health care is financed by both governmental and private insurers, and significantly impacts the U.S. pharmaceutical industry. Some ofThis law was designed to expand access to health insurance coverage for uninsured and underinsured individuals while at the provisions of the Affordable Care Act have yet to be fully implemented, and since its enactment, there have been judicial and Congressional challenges to numerous provisionssame time containing overall healthcare costs. The framework of the ACA and other healthcare reforms continues to evolve as well as recent efforts by theTrump administration to repeal or replace certain aspectsa result of the ACA. Since January 2017, President Trumpexecutive, legislative, regulatory, and administrative developments. For example, Congress has signed two Executive Orders designed to delay the implementation of anyenacted several laws that modify certain provisions of the ACA or otherwise circumvent somesuch as removing penalties, starting January 1, 2019, for not complying with the ACA’s individual mandate to carry health insurance and delaying the implementation of certain ACA-mandated fees. In addition, the requirements2020 federal spending package permanently eliminated, effective January 1, 2020, the ACA-mandated “Cadillac” tax on high-cost employer-sponsored health coverage and medical device tax and, effective January 1, 2021, the health insurer tax. More recently, for calendar years 2021 and 2022, the American Rescue Plan Act of 2021 temporarily increased premium tax credit assistance established under the ACA to help eligible individuals cover premiums for health insurance mandated bypurchased through the health insurance marketplace and removed the 400% federal poverty level limit that otherwise applies forThe Trump administration hasThere have also announced that it will discontinue the payment of cost-sharing reduction, or CSR, paymentsbeen numerous historical challenges and amendments to insurance companies until Congress approves the appropriation of funds for the CSR payments. The losscertain aspects of the CSR payments is expectedACA, and on June 17, 2021, the U.S. Supreme Court dismissed the most recent judicial challenge to increase premiumsthe ACA brought by several states without specifically ruling on certain policies issued by qualified health plans under the ACA. A bipartisan bill to appropriate funds for CSR payments has been introduced in the Senate, but the future of that bill is uncertain. Further, each chamber of Congress has put forth multiple bills designed to repeal or repeal and replace portionsconstitutionality of the ACA. Although noneIt is unclear how the healthcare reform measures of these measuresthe Biden administration or other efforts, if any, to challenge, repeal, amend or replace the ACA will impact the ACA and our business.enacted by Congress to date, Congress may consider other legislation to repeal and replace elementsheightened governmental scrutiny recently over pharmaceutical pricing practices in light of the ACA.rising cost of prescription drugs and biologics. Such scrutiny has resulted in Congressional and federal agency inquiries regarding pricing and related practices, as well as proposed and enacted federal and state legislation and regulation designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for products. For example, the American Rescue Plan Act of 2021 included among its provisions a sunset of the ACA’s cap on pharmaceutical manufacturers’ rebate liability under the Medicaid Drug Rebate Program. Previously, under the ACA, manufacturers’ rebate liability was capped at 100% of the average manufacturer price for a covered outpatient drug. Effective January 1, 2024, manufacturers’ Medicaid Drug Rebate Program rebate liability will no longer be capped, potentially resulting in a manufacturer paying more in Medicaid Drug Rebate Program rebates than it receives on the sale of certain covered outpatient drugs. Additionally, on July 9, 2021, President Biden issued an Executive Order to promote competition in the U.S. economy that included several initiatives addressing prescription drugs. Among other provisions, the Executive Order stated that the Biden administration will “support aggressive legislative reforms that would lower prescription drug prices, including by allowing Medicare to negotiate drug prices, by imposing inflation caps, and through other related reforms.” In response to the Executive Order, on September 9, 2021, HHS issued a Comprehensive Plan for Addressing High Drug Prices that identified potential legislative policies and administrative tools that Congress and the agency can pursue in order to make drug prices more affordable and equitable, improve and promote competition throughout the prescription drug industry, and foster scientific innovation. In August 2022, President Biden signed into law the Inflation Reduction Act of 2022, which implements substantial changes to the Medicare program, including drug pricing reforms and changes to the Medicare Part D benefit design.Among other reforms, the Inflation Reduction Act of 2022 imposes inflation rebates on drug manufacturers for products reimbursed under Medicare Parts B and D if the prices of those products increase faster than inflation; implements changes to the Medicare Part D benefit that, beginning in 2025, will cap benefit annual out-of-pocket spending at $2,000, while imposing new discount obligations for pharmaceutical manufacturers; and, beginning in 2026, establishes a “maximum fair price” for a fixed number of pharmaceutical and biological products covered under Medicare Parts B and D following a price negotiation process with the Centers for Medicare and Medicaid Services. The Biden administration continues to direct the Department of Health and Human Services to consider new healthcare payment and delivery models that would lower drug costs and promote access to innovative therapies for beneficiaries enrolled in the Medicare and Medicaid programs.We cannot predict how, or to what extent, the Biden administration’s drug pricing policies will affect our products.
At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. Any repeal and replace legislation aimed at further healthcare reform may have the effect of limiting the amounts that government agencies will pay for healthcare products and services. Policy changes, including potential modification or repeal of all or parts of the ACA or the implementation of new health care legislation, could result in significant changes to the health care system, which may prevent us from being able to generate revenue, attain profitability or commercialize our drugs. We expect that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand or lower pricing for our product candidates, or additional pricing pressures.European UnionEU and other potentially significant markets for our product candidates, government authorities and third-party payors are increasingly attempting to limit or regulate the price of medical products and services, particularly for new and innovative products and therapies, which has resulted in lower average selling prices. For example, in the United States, there have been several recent Congressional inquiries and proposed bills designed to, among other things, bring more transparency to drug pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drugs. Furthermore, theThe increased emphasis on managed healthcare in the United States and on country and regional pricing and reimbursement controls in the European UnionEU will put additional pressure on product pricing, reimbursement and usage, which may adversely affecthashave not fully complied, with such laws, itwe could face substantial penalties.If we obtain FDA approval for any of our product candidates and begin commercializing those products in the United States, ourpenalties, sanctions or other liability.and physician sunshine laws, the EU GDPR and/or the UK GDPR and other regulations. These laws may impact, among other things, our proposedresearch, sales, marketing, education and educationpatient assistance programs. In addition, we may be subject to patient privacy regulation by both theforeign, federal, government and the statesstate governments in which we conduct our business. The laws that may affect our ability to operate include:the federal Health Insurance Portability and Accountability Act of 1996, or •HIPAA, which created new federal criminal statutes that prohibit executing a scheme to defraud any healthcare benefit program and making false statements relating to healthcare matters;
•HIPAA and its implementing regulations impose certain requirements on certain covered entity healthcare providers, health plans, and healthcare clearinghouse and their business associates that perform certain services involving the use or disclosure of individually identifiable health information as well as their covered subcontractors, relating to the privacy, security, and transmission of individually identifiable health information;the U.S. Department of Health and Human ServicesHHS information related to payments and other transfers of value to physicians other healthcare providers,(defined to include doctors, dentists, optometrists, podiatrists, and chiropractors), teaching hospitals, physician assistants, nurse practitioners, clinical nurse specialists, anesthesiologist assistants, certified registered nurse anesthetists, and certified nurse midwives, as well as ownership and investment interests held by physicians and other healthcare providers and their immediate family members and applicable group purchasing organizations;expenditures,expenditures; state and local laws requiring the registration of pharmaceutical sales representatives; and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts.criminaladministrative penalties, damages, disgorgement, fines, sanctions, exclusion from participation in government health care programs, such as Medicare and Medicaid, imprisonment, corporate integrity oversight and reporting obligations, contractual damages, reputational harm, diminished profits and future earnings, and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations.States; Canada; Sydney, Australia; Ankara, Turkey; Hannover, Germany; Karachi, Pakistan; Auckland,States, Canada, Australia, Turkey, Germany, Pakistan, New Zealand, Mongolia, Spain, France, Bulgaria, Romania, Taiwan, Sweden, Italy, Belgium, Switzerland, United Kingdom, Greece, Moldova, Ukraine, Russia, and Jerusalem and Beersheba, Israel, and accordingly, we are subject to risks associated with doing business globally, including commercial, political, and financial risks. Emerging regions, such as Eastern Europe, Latin America, Asia, and Africa, as well as more developed markets, such as the United Kingdom, France, Germany, and Australia, provide clinical study opportunities for us. In addition, we are subject to potential disruption caused by military conflicts; potentially unstable governments or legal systems; civil or political upheaval or unrest; local labor policies and conditions; possible expropriation, nationalization, or confiscation of assets; problems with repatriation of foreign earnings; economic or trade sanctions; closure of markets to imports; anti-American sentiment; terrorism or other types of violence in or outside the United States; health pandemics; and a significant reduction in global travel. For example, both Turkey and Pakistan are key regions for clinical activity relating to Hepatitis Delta Virus, and furtherfinancial results.Ourareis located in the San Francisco Bay Area, which has in the past experienced severe earthquakes, and other natural disasters.disasters, and an outbreak of COVID-19. We do not carry earthquake insurance. Earthquakes or other natural disasters could severely disrupt our operations or those of our collaborators, and have a material adverse effect on our business, results of operations, financial condition, and prospects. If a natural disaster, terrorist attack, power outage, or other event occurred that prevented us from using or damaged critical elements of our business and operations (such as the manufacturing facilities of our third-party contract manufacturers) our business may be disrupted for a substantial period of time. We have limited or no disaster recovery and business continuity plans in place currently and our business would be impaired in the event of a serious disaster or similar event. We may incur substantial expenses to develop and implement any disaster recovery and business continuity plans, which could have a material adverse effect on our business.Risks RelatedCelladon’s Historical Business OperationsWeadversely affect our business and could materially and adversely affect our operations and those of our manufacturers and other third parties with whom we conduct business.subjectUnited States. While many of securities class action lawsuits that were filed against Celladon in 2015, and additional securities litigationthese materials may be brought against usobtained by more than one supplier, including suppliers outside of China, Japan, Canada, Italy and the United States, port closures and other restrictions resulting from the coronavirus outbreak in the future.July 2015, following Celladon’s announcementsaddition, our clinical trials have been and may continue to be affected by the COVID-19 pandemic. Site initiation and patient enrollment has been delayed, due to prioritization of hospital resources toward the COVID-19 pandemic, travel restrictions imposed by governments, and the inability to access sites for initiation and monitoring.negative CUPID 2COVID-19 pandemic, the extent and length of which is uncertain, we may be required to develop and implement additional clinical study policies and procedures designed to help protect study participants from the COVID-19 virus, which may include using telemedicine visits, remote monitoring of patients and clinical sites, and measures to ensure that data from clinical studies that may be disrupted as result of the pandemic are collected pursuant to the study protocol and consistent with GCPs, with any material protocol deviation reviewed and approved by the site IRB.suspensionvalue of further research and development activitiesmarket for our common stock will depend on future developments that are highly uncertain and cannot be predicted with confidence at this time, such as the subsequent declinesultimate duration of the price of its common stock, three putative class actions were filedpandemic, travel restrictions, quarantines, social distancing and business closure requirements in the U.S. District Court forand in other countries, and the Southern Districteffectiveness of California against Celladonactions taken globally to contain and certainits current and former officers. The complaints generally alleged thatoperations, it may also have the defendants violated Sections 10(b) and 20(a)effect of heightening many of the Securities Exchange Act of 1934, as amended, or the Exchange Act, by making materially falseother risks and misleading statements regarding the clinical trial program for MYDICAR, thereby artificially inflating the price of Celladon’s common stock. The complaints sought unspecified monetary damages and other relief, including attorneys’ fees. On December 9, 2015, the district court consolidated the threeputative securities class actions and appointed a lead plaintiff to represent the putative class. The lead plaintiff filed a consolidated amended complaint on February 29, 2016.On October 7, 2016, the district court granted defendants’ motion to dismiss the consolidated amended complaint and granted leave to amend within 60 days from the date of the district court’s order. The lead plaintiff subsequently filed a notice of intent not to amend the consolidated amended complaint and instead indicated that it intended to appeal the district court’s decision. On December 9, 2016, the district court closed the case.On December 28, 2016, the lead plaintiff filed a notice to the United States Court of Appeals for the Ninth Circuit appealing the district court’s order dismissing the consolidated amended complaint. On May 5, 2017, the lead plaintiff filed his opening brief. On July 5, 2017, the defendants filed their appellate brief response. On August 18, 2017, the lead plaintiff filed his reply appellate brief.It is possible that additional suits will be filed, or allegations made by stockholders, with respect to these same or other matters and also naming us and/or our former officers and directors as defendants. We believe that we have meritorious defenses and intend to defend these lawsuits vigorously. We are not able to predict or reasonably estimate the ultimate outcome or possible losses relating to these claims. While we and Celladon’s former directors and officers have a separate liability insurance policy dedicated to any claims that may arise from premerger events, there is no assurance that the coverage will be sufficient. In addition, any such litigation could resultuncertainties described elsewhere in substantial costs and a diversion of our management’s attention and resources, which could harm our business.NDAMAA for any of our product candidates and any adverse development or perceived adverse development with respect to the FDA’s review of that IND, NDA, or NDA;orphan drugor maintain Orphan Drug designation;
•any inability to obtain adequate supply of our product candidates or the inability to do so at acceptable prices;•any inability to obtain adequate supply of our product candidates or the inability to do so at acceptable prices;companies.companies, including very recently in connection with the ongoing COVID-19 pandemic, which has resulted in decreased stock prices for many companies notwithstanding the lack of a fundamental change in their underlying business models or prospects. These broad market fluctuationsand industry factors, including potentially worsening economic conditions and other adverse effects or developments relating to the ongoing COVID-19 pandemic, may also adversely affect the trading price of our common stock.NASDAQNasdaq Stock Market LLC. These rules and regulations impose significant legal and financial compliance costs and make some activities more time-consuming and costly. For example, our management team consists of certain executive officersVivo Ventures Fund VI, L.P.Columbia Threadneedle Investments, 683 Capital Management, BlackRock Institutional Trust, and Interwest Partners X, L.P.,The Vanguard Group, and their respective affiliated entities, own substantial ownership interest in our common stock and any decision to sell a significant number of shares may negatively impact the price of our common stock.The recently passed comprehensive tax reform bill could adversely affect our business and financial condition.On December 22, 2017, President Trump signed into law new tax legislation, or the Tax Act, which significantly changes the Internal Revenue Code of 1986, as amended. The Tax Act, among other things, contains significant changes to corporate taxation, including reduction of the corporate tax rate from a top marginal rate of 35% to a flat rate of 21%; limitation of the tax deduction for interest expense to 30% of adjusted earnings (except for certain small businesses); for net operating losses generated after 2017, limitation of the deduction to 80% of current year taxable income, indefinite carryforward of net operating losses and elimination of net operating loss carrybacks; changes in the treatment of offshore earnings regardless of whether they are repatriated; mandatory capitalization of research and development expenses beginning in 2022; immediate deductions for certain new investments instead ofdeductions for depreciation expense over time; further deduction limits on executive compensation; and modifying, repealing and creating many other business deductions and credits, including the reduction in the orphan drug credit from 50% to 25% of qualifying expenditures. We continue to examine the impact this tax reform legislation may have on our business. Notwithstanding the reduction in the corporate income tax rate, the overall impact of the Tax Act is uncertain and our business and financial condition could be adversely affected. The impact of this tax reform on holders of our common stock is also uncertain and could be adverse. This annual report does not discuss any such tax legislation or the manner in which it might affect us or our stockholders in the future. We urge our stockholders to consult with their legal and tax advisors with respect to such legislation.Because the Merger resulted in an ownership change under Section 382 of the Internal Revenue Code ourOur net operating loss carryforwards and certain other tax attributes are now subject to limitations.If“ownership change”ownership change within the meaning of Section 382 of the Internal Revenue Code of 1986, as amended, or Section 382,(Section 382) the corporation’s net operating lossNOL carryforwards and certain other tax attributes arising from before the ownership change are subject to limitations on use after the ownership“ownership change.” In general, an ownership change occurs if there is a cumulative change in the corporation’s equity ownership by certain stockholders that exceeds fifty percentage points over a rolling three-year period. Similar rules may apply under state tax laws. The MergerOur merger with Celladon resulted in such an ownership change and, accordingly, Celladon’s and Eiger’s net operating lossNOL carryforwards and certain other tax attributes will be subject to further limitations on their use. WeIn addition, we assessed whether Eiger had an ownership change, as defined by Section 382 of the Code, as a result of the Merger and other stock issuances that occurred from our formation through December 31, 2016.2020. Based upon this assessment, no reduction waswe have experienced ownership changes on April 20, 2016, October 18, 2018 and December 31, 2020. Due to these ownership changes, reductions were made to the federalour NOL and state NOL carryforwards or federal and state tax credit carryforwards under these rules. Additional ownership changes in the future could result in additional limitations on the combined organization’sour net operating loss and tax credit carryforwards. Consequently, even if we achieve profitability, we may not be able to utilize a material portion of our net operating lossNOL carryforwards and other tax attributes, which could have a material adverse effect on cash flow and results of operations. A full valuation allowance has been provided for the entire amount of our remaining net operating losses.350 Cambridge Avenue, Suite 350,2155 Park Blvd in Palo Alto, California 94306 in a facility we lease encompassing 3,8778,029 square feet of office space. The lease for this office space expires in March 2018, has one two-year renewal option prior to expiration and includes rent escalation clauses through the lease term.In March 2017, we entered into a non-cancelable facility lease agreement for an office facility at 366 Cambridge Avenue in Palo Alto, California 94306. The lease commenced on April 1, 2017 and expires 12 months after the commencement date. The lease has one twelve-month renewal option prior to expiration and includes rent escalation clauses through the lease term.In October 2017, we entered into a non-cancelable facility lease agreement for 8,029 square feet of office space located at 2171 Park Blvd in Palo Alto, California 94306. The lease commences on March 1, 2018 and expireswas to expire five years after the commencement date. The lease hashad one three-year renewal option prior to expiration and includesincluded rent escalation clauses through the lease term.July 2015, following Celladon’s announcementsdetermining whether to discuss a pending matter, we consider both quantitative and qualitative factors to assess materiality, such as, among others, the amount of damages alleged and the nature of other relief sought, if specified; our view of the negative CUPID 2 data and the suspension of further research and development activities and the subsequent declinesmerits of the priceclaims and of its common stock, threethe strength of our defenses; and whether the action purports to be, or is, a class action the jurisdiction in which the proceeding is pending.actions wereaction complaint was filed in the U.S.United States District Court for the SouthernNorthern District of California against Celladon and certain of its current and former officers. The complaints generally allegedalleging that the defendantscompany and two former executives violated Sections 10(b) and 20(a) of the Securities Exchange Act of 1934, as amended, or the Exchange Act, by making materially false and misleading statementsSEC Rule 10b-5. The complaint alleges generally that between March 2021 and October 2022 material misstatements and omissions were made to shareholders regarding the clinical trial programTOGETHER study of peginterferon lambda for MYDICAR, thereby artificially inflating the pricetreatment of Celladon’s common stock.COVID-19 as well as the likelihood of FDA approval of an Emergency Use Authorization for peginterferon lambda. The complaints sought unspecified monetary damages and other relief, including attorneys’ fees. On December 9, 2015, the district court consolidated the three putative securities class actions andCourt appointed a lead plaintiff on March 2, 2023. The litigation is currently ongoing.representsupply quantities of a drug as requested by PRF. PRF also has a claim for declaratory relief regarding the putative class. The lead plaintiffgrant of licenses under the PRF Collaboration Agreement. On January 18, 2023, the Company filed a consolidated amended complaint on February 29, 2016.On October 7, 2016, the district court granted defendants’ motion to dismiss the consolidated amended complaint and granted leave to amend within 60 days from the date of the district court’s order. The lead plaintiff subsequently filed a notice of intent not to amend the consolidated amended complaint and instead indicated that it intended to appeal the district court’s decision. On December 9, 2016, the district court closed the case.On December 28, 2016, the lead plaintiff filed a notice to the United States Court of Appeals for the Ninth Circuit appealing the district court’s order dismissing the consolidated amended complaint. On May 5, 2017, the lead plaintiff filed his opening brief. On July 5, 2017, the defendants filed their appellate brief response. The Plaintiff subsequently filed their response to the Company’s July 5, 2017 filingDemand denying PRF’s claims, contesting the arbitrability of PRF’s claim for declaratory relief, and asserting a counterclaim for declaratory relief related to the contractual provision underlying PRF’s original drug supply claim. To give the parties an opportunity to discuss a potential negotiated resolution of their dispute, the arbitration has been suspended through the end of 2023. As aAugust 19, 2017. Upon these filings,hold, and the next stepfinal hearing, originally scheduled for May 9 – 12, 2023, in the process would be to wait for oral arguments to be scheduled by the 9th Circuit Court of Appeals.It is possible that additional suits will be filed, or allegations made by stockholders, with respect to these same or other matters and also naming us and/or our former officers and directors as defendants. We believe that we have meritorious defenses and intend to defend these lawsuits vigorously. We are not able to predict or reasonably estimate the ultimate outcome or possible losses relating to these claims. While we and Celladon’s former directors and officers have a separate liability insurance policy dedicated to any claims that may arise from premerger events, there is no assurance that the coverage will be sufficient. In addition, any such litigation could result in substantial costs and a diversion of our management’s attention and resources, which could harm our business.NASDAQNasdaq Global Market on January 30, 2014. Prior to January 30, 2014, there was no public market for our common stock. The following table sets forth the high and low sales prices per share of our common stock as reported on The NASDAQ Global Market for the period indicated, adjusted for the reverse stock split.2, 2018,13, 2023, there were approximately 2820 stockholders of record of our common stock. Certain shares are held in “street” name and accordingly, the number of beneficial owners of such shares is not known or included in the foregoing number. Plan Category (1) (2) 6,784,590 $ 9.00 2,764,517 — — — Total 6,784,590 $ 9.00 2,764,517 Selected Financial DataAs a “smaller reporting company” as defined by Rule 12b-2Reservedindicates,indicated, references to the terms the “combined company,” “Eiger,” the “Company,” “we,” “our” and “us” refer to Eiger BioPharmaceuticals, Inc. (formerly known as Celladon Corporation) and its subsidiaries after the merger described herein. The term “Private Eiger” refers to privately-held Eiger BioPharmaceuticals, Inc. prior to its merger with Celladon Merger Sub, Inc. a wholly-owned subsidiary of Celladon Corporation. The term “Celladon” refers to Celladon Corporation and its subsidiaries prior to the Merger.Introductionclinical stagecommercial-stage biopharmaceutical company focused on bringingthe development of innovative therapies for hepatitis delta virus (HDV) and other serious diseases. All five of our rare disease programs have FDA Breakthrough Therapy designation.market novelcontrol and qualify these materials and plan to initiate dosing in Phase 3 when an adequate supply of materials with a sufficient shelf-life has been released.candidatesZokinvy (lonafarnib), to reduce the risk of mortality of Hutchinson-Gilford progeria syndrome (HGPS) and for treatment of processing-deficient progeroid laminopathies (PL) with either heterozygous LMNA mutation with progerin-like protein accumulation, or homozygous or compound heterozygous ZMPSTE24 mutations, on November 20, 2020. Collectively known as progeria, these are ultra-rare and rapidly fatal genetic conditions of accelerated aging in children. On July 20, 2022, we announced that the European Commission (EC) granted Eiger a centralized marketing authorization (MA) under the exceptional circumstances procedure for Zokinvy for the treatment of rare diseases. Since our founding in 2008, we have worked with investigators at Stanford UniversityHGPS and evaluated a number of potential development candidates from pharmaceutical companies to comprise a pipeline of novel product candidates. Our resulting pipeline includes four Phase 2 candidates addressing three distinct rare diseases. The programs have several aspects in common: the disease targets representPL, ultra-rare and rapidly fatal genetic conditions of high medical needaccelerated aging in children. The MA is subject to the EMA’s continued review on an annual basis of new efficacy and safety information which are inadequately treatedmay become available. The EC's centralized MA is valid in all 27 EU member states plus Iceland, Liechtenstein, and Norway. Regulatory review is ongoing by current standard of care; the therapeutic approaches are supported by an understanding of disease biologyUK's Medicine and mechanismHealthcare products Regulatory Agency (MHRA) as elucidated by our academic research relationships; prior clinical experience with the product candidates guides an understanding of safety; and the development paths leverage the experience and capabilities of our experienced, commercially focused management team. The pipeline includes lonafarnib for Hepatitis Delta Virus, or HDV, lambda for HDV, exendin 9-39 for PBH and ubenimex for lymphedema. We plan to deliver data from three ongoing Phase 2 clinical trials over the coursepart of the next twelve months.InUK's European Commission Decision Reliance Procedure, which has been extended to apply across Great Britain until December 31, 2023.2018, Phase 2 LIBERTY study results2021 and in PAH demonstrated no improvement overall orEurope in key subgroups for bothNovember 2022 and started to record product revenue in the primary efficacy endpointsfirst quarter of PVR and 6-minute walk distance. Further analysis2021. Our first European sales were recognized in the fourth quarter of data, including biomarkers is ongoing, although we plan to discontinue development of ubenimex in PAH based on the LIBERTY study resultsno products approved for commercial sale and have not generated any revenue from product sales. We have never been profitable and havehistorically incurred operating losses in each year since inception and we do not anticipate that we will achieve profitability inexpect to incur losses for the near term. Ourforeseeable future. We had a net losses were $42.4loss of $96.8 million, $47.1$33.9 million and $13.3$65.1 million for the years ended December 31, 2017, 20162022, 2021 and 2015,2020, respectively. As of December 31, 2017,2022, we had an accumulated deficit of $118.8$437.2 million. Substantially all of our operating losses have resulted from expenses incurred in connection with our research and development programs and from selling, general and administrative costs associated with our operations.add personnel necessaryupgrades to operate as a public company with an advanced clinical candidate pipelineour information technology systems. Weapproval.CelladonMarch 22, 2016,December 8, 2022, we completedannounced topline Week 48 data from the merger between Private Phase 3 D-LIVR study (N=407) evaluating lonafarnib in two regimens in patients with chronic HDV: lonafarnib boosted with ritonavir alone (all-oral) and in combination with peginterferon alfa (combination). The composite primary endpoint was a ≥2 log decline in HDV RNA and normalization of alanine aminotransferase (ALT) at the end of 48 weeks of treatment compared to placebo.and Celladonplans to engage with regulatory agencies, beginning with a request for a pre-NDA meeting with FDA in accordanceQ1 2023 with a meeting anticipated in Q2 2023, to discuss pathways for regulatory submissions. The full D-LIVR dataset, including analyses of the 24-week post-treatment period, would be included in potential regulatory submissions.termsFDA, we submitted a pre-EUA meeting request to the FDA as well as additional morbidity and mortality outcomes data and analyses from the investigator-sponsored TOGETHER study. This included further statistical modeling and efficacy analyses of the Agreementstudy's primary and Plansecondary endpoints and long-term follow-up data that we believe continues to support the topline outcomes reported in March. In response, the FDA denied the request for a pre-EUA meeting. Citing its concerns with the conduct of Merger, datedthe TOGETHER study, the FDA concluded that any authorization request based on these data is unlikely to meet the statutory criteria for issuance of an EUA in the current context of the pandemic.18, 2015, by and among Private Eiger, Celladon and Celladon Merger Sub,2022, sales of Zokinvy launched commercially in Europe through our wholly owned subsidiary in Ireland.or the Merger. Also on March 22, 2016,which was executed in connection with,May 2022.priorindirect costs related to the completionmanufacturing of the Merger, we effected a fifteenZokinvy for one reverse stock splitcommercial sale, including third-party manufacturing costs, third party logistics costs, write down of our common stock, or the Reverse Stock Split,inventories, and changed our name to “Eiger BioPharmaceuticals, Inc.”On November 18, 2015, in connection with the Merger, we entered into a subscription agreement, or the Subscription Agreement, with investors for the sale of shares of our common stock, or the Private Placement, which closed on March 22, 2016.Immediately prior to and in connection with the Merger, each share of Private Eiger’s preferred stock outstanding was converted into shares of Private Eiger’s common stock at an exchange ratio of one share of common stock for each share of preferred stock.Under the terms of the Merger Agreement, at the effective time of the Merger, Celladon issued shares of common stock to Private Eiger stockholders, at an exchange ratio of approximately 0.09 shares of common stock, after taking into account the Reverse Stock Split, in exchange for each share of Private Eiger’s common stock outstanding immediately prior to the Merger. The exchange ratio was calculated by a formula that was determined through arms-length negotiations between Celladon and Private Eiger. Immediately after the Merger, the former Private Eiger equity holders beneficially owned approximately 78% of post-merger Eiger’s common stock. The Merger was accounted for as a reverse asset acquisition.Financial Operations Overviewcapabilitiesarrangements, clinical trial material related costs and other research and development activities. However, we do not allocate internal research and development costs, such as salaries, benefits, stock-based compensation expense and indirect costs to product candidates on a program-specific basis. The following table shows our research and development expenses for the years ended December 31, 2017, 20162022, 2021 and 20152020 (in thousands):Year Ended December 31, 2022 2021 2020 Product candidates: Lonafarnib $ 25,643 $ 32,732 $ 25,051 Peginterferon Lambda 21,563 17,322 6,128 Avexitide 9,596 3,082 1,891 Internal research and development costs 18,480 11,300 8,520 Total research and development expense $ 75,282 $ 64,436 $ 41,590 continue tomay evaluate opportunities to acquire or in-license other product candidates and technologies, which may result in higher research and development expenses due to license feefees and/or milestone payments.General and expenses for outside professional services, including legal, audit, accounting services, insurance costs and costs associated with being a public company.company, and commercial related expenses. Personnel costs consist of salaries, benefits and stock-based compensation. Allocated expenses consist of facilities and other expenses, which include direct and allocated expenses for rent and maintenance of facilities, depreciation expense and other supplies. As a result of becoming a public company following completion of the Merger,Our expenses include costs related to compliance with the rules and regulations of the SEC and NASDAQ, additionalNasdaq, insurance, investor relations, bankbanking fees and other administrative expenses and professional services. for the year ended December 31, 2017, consists of interest and amortization of the debt discount related toon our borrowing under the Oxford Loan executed in December 2016. Interest expense for the year ended December 31, 2016, consists of interest and amortization of the debt discount related to the outstanding convertible promissory notes issued in November 2015, which were converted into common stock in March 2016, or the Notes.long-term borrowings.marketabledebt securities and cash equivalents.Income (Expense), Net(expense), net in 2017 primarily consists of the $0.2 million payment received for MYDICAR sale.Other income (expense), net in 2016 consistsloss on extinguishment of the change in fair value of the obligation to issue common stock to Eiccose and the change in fair value of warrant liability.The change in fair value of the obligation to issue common stock to Eiccose was related to our obligation to issue shares to Eiccose upon the closing of the next round of financing that resulted in at least $25.0 million in gross proceeds to us. Upon the closing of the Private Placement on March 22, 2016, we issued 96,300 fully vested shares of our common stock to Eiccose in settlement of this obligation. In connection with this transaction we remeasured the fair value of the obligation to issue common stock at the settlement date and the change in fair value of $0.2 million was recognized within other income (expense), netdebt recorded during the year ended December 31, 2016. Upon the settlement of the obligation with the issuance of shares on March 22, 2016, the liability was reclassified to common stock and additional paid-in capital within stockholders’ equity (deficit).In connection with our issuance of the Notes, we issued warrants to the noteholders to purchase shares of our common stock at an exercise price of $0.11 per share, on a post-Merger and post-Reverse Stock Split basis, or the Warrants. The number of shares into which the Warrants could be exercised was equal to the warrant coverage amount divided by the per share price of the equity securities sold in a qualified financing and thus was accounted for as a liability. Upon the closing of the Private Placement on March 22, 2016, the number of shares of common stock issuable upon exercise of the Warrants was fixed and the fair value remeasured at that date, and the warrants were automatically exercised. During the year ended December 31, 2016, we recognized a loss related to the change in fair value of the warrant liability of $0.2 million. The warrant liability was reclassified to common stock and additional paid-in capital within stockholders’ equity (deficit), upon the exercise of the Warrants and issuance of shares on March 22, 2016.Expensesrecord accrued expensesaccrue for estimated costs of research and development activities conducted by externalthird-party service providers, which include the conduct of preclinical and clinical researchstudies, and contract formulation and manufacturing activities. We record the estimated costs of research and development activities based upon the estimated amount of services provided but not yet invoiced and includeincludes these costs in accrued liabilities in the consolidated balance sheetsheets and within research and development expenseexpenses in the consolidated statementstatements of operations. We record accrued expensesaccrue for these costs based on factors such as estimates of the estimated amount of work completed and in accordance with agreements established with these externalour third-party service providers.We estimate the amount of work completed through discussions with internal personnel and external service providers as to the progress or stage of completion of the services and the agreed-upon fee to be paid for such services. We make judgments and estimates in determining the accrued liabilities balance in each reporting period. As actual costs become known, we adjust our accrued estimates.granted to employeesand restricted stock units based on the estimated fair value of the awards on the date of grant.grant date. We estimate the grant date fair value of stock options, and the resulting stock-based compensation expense, using the Black-Scholes option-pricing model. The grant date fair value of the stock-based awards is generally recognized on a straight-line basis over the requisite service period, which is generally the vesting period of the respective awards.requiresincludes the use of subjective assumptions, which determine the fair value of stock-based awards. These assumptions include:OurThe expected term represents the periodnumber of years that our stock-based awards are expected towe estimate, based primarily on historical experience, that the options will be outstanding and is determined using the simplified method (based on the mid-point between the vesting date and the end of the contractual term).Since we have only been publicly traded for a short period and do not have adequate trading history for our common stock, theThe expected volatility was estimatedfor stock options is based on the average volatility for comparable publicly traded biopharmaceutical companies over a period equal to the expected termCompany's historical stock price volatility.Black-Scholes assumptions and prior to adoption of ASU 2016-09 in the first quarter of 2017, we estimated our forfeiture rate based on actual forfeiture experience, analysis of employee turnover behavior, and other factors. The impact from any forfeiture rate adjustment was recognized in full in the period of adjustment and if the actual number of future forfeitures differed from our estimates, we were required to record adjustments to stock-based compensation in future periods. Following the adoption of ASU 2016-09 we made an accounting policy election to account for forfeitures as they occur. This change did not have a material impact to the consolidated financial statements.Prior to the completion of the Merger in March 2016, the fair value of the shares of common stock underlying our share-based awards were estimated on each grant date by our Board of Directors. In ordercontractual term to determine the non-employee awards’ fair value at the grant date. The contractual term of our common stock underlying option grants, our Board of Directors considered, among other things, timely valuations of our common stock prepared by an unrelated third-party valuation firm in accordance withoptions granted under the guidance provide by the American Institute of Certified Public Accountants Practice Guide, Valuation of Privately-Held-Company Equity Securities Issued as Compensation. Given the absence of a public trading market for our common stock, our Board of Directors exercised reasonable judgment and considered a number of objective and subjective factors to determine the best estimate of the fair value of our common stock, including our stage of development; progress of our research and development efforts; the rights, preferences and privileges of our preferred stock relative to those of our common stock; equity market conditions affecting comparable public companies and the lack of marketability of our common stock. After the completion of the Merger, ourPlan is 10 years. Our Board of Directors determined the fair value of each share of underlying common stock based on the closing price of our common stock as reported by the NASDAQNasdaq Global Market on the date of grant.
We estimate the fair value of restricted stock units based on the closing market price of our common stock on the date of grant and the resulting stock-based compensation expense is recognized on a straight-line basis over the requisite service period, which is generally the vesting period of the awards.20172022 and 201620172022 and 20162021 (in thousands):Year Ended December 31, $
Change%
Change2022 2021 $
Change%
ChangeProduct revenue, net Product revenue, net $ 12,734 $ 12,142 Other revenue Other revenue 750 — 750 * Total revenue Total revenue 13,484 12,142 1,342 11 % Costs and operating expenses: Costs and operating expenses: Cost of sales Cost of sales 1,837 745 1,092 147 % Research and development 75,282 64,436 10,846 17 % Selling, general and administrative Selling, general and administrative 29,105 23,900 5,205 22 % Total costs and operating expenses Total costs and operating expenses 106,224 89,081 17,143 19 % Loss from operations (92,740) (76,939) (15,801) 21 % Interest expense (4,132) (3,559) (573) 16 % Interest income 1,082 158 924 585 % Other (expense) income, net Other (expense) income, net (963) 46,487 (47,450) (102) % Loss before provision for income taxes Loss before provision for income taxes (96,753) (33,853) (62,900) 186 % Provision for income taxes Provision for income taxes 23 64 (41) (64) % Net loss $ (96,776) $ (33,917) $ (62,859) 185 % Research and development expensesResearch and development expenses decreased by $3.5 million to $29.5 million2017,2022, from $33.0the same period of 2021.same period in 2016. The decrease was primarily due to a $5.2 millionyear ended December 31, 2022 reflects the upfront payment received from AnGes, Inc. under our License Agreement with Bristol Myers Squibb Companythe terms of the MDA, which was executed in 2016. There were no similar payments in 2017. The decrease was partially offset by a $0.7 million increase in compensation and personnel related expenses due to an increase in headcount, a $0.5 million increase in stock-based compensation expense due to an increase in headcount, $0.3 million increase in facility and insurance expenses and $0.2 million in milestone payments to two Stanford University inventors for the asset purchase agreement.General and administrative expensesGeneral and administrative expenses decreasedMay 2022.$12.0$1.8 million for the year ended December 31, 2017, from $13.1 million for the same period in 2016. The decrease was due to a $2.5 million decrease in legal, consulting, advisory and accounting services due to the incremental expenses incurred as a result of the Merger in the first quarter 2016. The decrease was partially offset by a $0.6 million increase in stock-based compensation expense, a $0.5 million increase in compensation and personnel related expenses due to an increase in headcount and a $0.3 million increase in facility and insurance expenses.Interest expenseInterest expense increased by $0.8 million to $1.5 million for the year ended December 31, 2017,20222016. Interest expense in 2017 consisted2021. The increase was primarily due to a non-conforming batch of interestinventory that was written off during the year ended December 31, 2022.amortization of the debt discount related to the Oxford Loan borrowings in December 2016. Interest expense in 2016 consisted of interestdevelopment expensesamortization of the debt discount related to the Notes outstanding prior to their conversion into common stock in March 2016.Interest incomeInterest incomedevelopment expenses increased by $0.3$10.8 million to $0.4$75.2 million for the year ended December 31, 2017,2022, from $0.12016. The increase was primarily due to the interest earned on our investments in marketable securities and cash equivalents being for the entire year of 2017, compared to 2016 only being for one quarter as the funds were not invested until the fourth quarter of 2016.Other income (expense), net changed by $0.62022, from to $0.2 million of other income for the year ended December 31, 2017, from $0.4 million of other expense for same period in 2016. Other income in 2017 consisted of the $0.2 million payment received from Theragene for MYDICAR sale. Other expense in 2016 primarily consisted of the change in fair value of the obligation to issue common stock to Eiccose and the change in fair value of warrant liability, that were settled upon the Merger.Comparison of the Years Ended December 31, 2016 and 2015The following table summarizes results of operations for the years ended December 31, 2016 and 2015 (in thousands):Research and developmentResearch and development expenses increased by $24.9 million to $33.0 million for the year ended December 31, 2016, from $8.1 million2015.2021. The increase was primarily due to a $15.0$2.9 million increase in clinical expenditures due to increased program activity,outside services, including consulting, advisory and accounting services, a $5.2 million expense related to upfront payments under our License Agreement with Bristol-Meyers Squibb Company (the BMS License Agreement), a $2.2$2.1 million increase in compensation and personnelheadcount related expenses and a $0.7$0.3 million increase in stock-based compensationother operating expenses, all to support company growth. The increases were partially offset by a $0.1 million decrease in sales and marketing expense. due to an increase in headcount, a $1.6 million increase in consulting fees related to increased program activity and a $0.2 million increase in facility related and insurance expenses.General and administrativeGeneral and administrative expenses$8.3$0.6 million compared to $13.1 million for the year ended December 31, 2016, from $4.9 million for the same period in 2015.2021. The increase was primarily due to a $3.4 million increase in consulting, advisory, legal and accounting services incurred in connection with the Merger with Celladon and being a public company, a $2.3 million increase in stock-based compensation expense and a $1.4 million increase in compensation and personnel related expenses due to an increase in headcount, a $0.6 million increase in litigation expensesthe outstanding principal balance of our long-term borrowings related to the Celladon shareholder law suit and a $0.5 million increase in facility related and insurance expenses.expenseexpenseincome increased by $0.3$0.9 million compared to $0.7 million for the year ended December 31, 2016, from $0.4 million for same period in 2015. Interest expense consisted of interest and amortization of the debt discount related to the Notes outstanding prior to their conversion into common stock in March 2016.2021. The increase was primarily due to a longer outstandingan increase in the average balance of money market funds and available-for-sale securities during 2022 along with an increase in interest rates.20162021. The change was primarily due to $46.5 million net proceeds received from the sale of our rare pediatric disease priority review voucher to AbbVie Inc in 2021 and a $1.1 million loss on early extinguishment of the Oxford loan in 2022.2015.the same period in 2021. The change was primarily due to the tax expense related to state taxes.expense,(expense) income, netOther expense, net in the consolidated statement of $0.3operations and comprehensive loss. As part of the agreement with Innovatus, we are required to maintain a cash balance of not less than 5% of the aggregate principal amount of funded and outstanding term loans at all times. We are currently eligible to draw the $17.5 million under Tranche B, but have not done so as of December 31, 2022.year ended December 31, 2016, primarily consistssale of the change in faircommon stock with an aggregate value of the obligation to issue$5.0 million. On June 1, 2022, we issued 749,053 shares of common stock to Eiccose andInnovatus at a per share purchase price of $6.6751.changemarket” offerings as defined in fair valueRule 415 under the Securities Act, under Eiger’s effective shelf registration statement (the 2022 ATM Facility). In April 2022, we completed offerings from the 2022 ATM facility for a total of warrant liability. We did not have any such items outstanding during the year ended 2,686,288 shares of our common stock resulting in net proceeds of $20.8 million, after deducting commissions costs. No additional offerings were completed since April 2022. As of December 31, 2015.Sources of LiquidityIn June 2016,2022333-212114)333-251497) with the Securities and Exchange Commission (SEC), which was declared effective by the SEC on December 31, 2020 and permits the offering,offer, issuance and sale by us of up to a maximum aggregate offering price of $125.0$200.0 million of our common stock, preferred stock, debt securities and warrants. UpIn connection with the filing of the registration statement, we entered into an Open Market Sale AgreementSM with Jefferies LLC (Jefferies), pursuant to which we could sell up to a maximum of $25.0 million of the maximum aggregate offering price of $125.0 million may be issued and sold pursuant to an At-The-Market, or ATM, financing facility under a sales agreement with Cantor Fitzgerald & Co. On August 23, 2016, we completed an underwritten public offering of 1,250,000 shares of common stock at an offering price of $16.00 for gross cash proceeds of $20.0 million under our shelf registration statement. As a result of the sale, our aggregate offering price was reduced to $105.0 million.In February and March 2017, pursuant to ATM, we completed our underwritten public offering of 4,537 shares of common stock for gross cash proceeds of $53,000 under the shelf registration statement on Form S-3 (File No. 333-212114).On October 31, 2017, we issued 2,132,961 shares of common stock pursuant to an underwriting agreement with BTIG, LLC at a public offering price of $10.00 per share, for gross proceeds of $21.3 million, which resulted in approximately $19.8 million of net proceeds to the Company after deducting underwriting fees and offering expenses.In December 2017, we filed a second shelf registration statement on Form S-3 (File No. 333-221972) with the Securities and Exchange Commission which permits the offering, issuance and sale by us of up to a maximum aggregate offering price of $125.0$50.0 million of our common stock preferred stock, debt securities and warrants.December 2017, pursuant to ATM,March 2022, we completed our underwritten public offeringthe sale of 6,027all shares of common stock for gross cash proceeds of $73,000available under the shelf registration statement on Form S-3 (File No. 333-212114).As of December 31, 2017, we had $32.0 million of cash2020 ATM Facility, and cash equivalents, $9.7 million of short-term marketable securities and an accumulated deficit of $118.8 million. the 2020 ATM Facility was terminated.these consolidatedthis Annual Report on Form 10-K. However, if our anticipated operating results are not achieved in future periods, we believe that planned expenditures may need to be reduced or we would be required to raise funding in order to fund our operations. Additionally, our ability to raise additional capital may be adversely impacted by potential worsening global economic conditions and the recent disruptions to, and volatility in, the credit and financial statements.In December 2016, we entered into a secured loan agreement with Oxford Finance LLC, pursuant to which we borrowed $15.0 millionmarkets in the United States and will be permitted to borrow up to an additional $10.0 million upon achievement of positive top line dataworldwide resulting from the lonafarnib Phase 2 trials in HDV, which was achieved in the fourth quarter of 2016, plus positive top line Phase 2 data from at least one of the following programs, including: (i) Lambda in HDV, (ii) exendin 9-39 in PBH based on the Company’s own IND, (iii) ubenimex in PAH, or (iv) ubenimex in Lymphedema.We haverevenue from product sales.revenue. We do not know when, or if, we will generate any revenue fromlaunched our first commercial product, sales.Zokinvy, in January 2021. We do not expect to generate any revenue from product sales unless and until we obtain regulatory approval for and commercialize any of our product candidates. At the same time, we expect our expenses to increase in connection with our ongoing development and manufacturing activities, particularly as we continue the research, development, manufacture and clinical trials of, and seek regulatory approval for our product candidates.research efforts and the development of our product candidates, sales and marketing costs for commercialization of Zokinvy and other product candidates, compensation and related expenses, hiring additional staff, including clinical, scientific, operational, manufacturing, financial, and managementCash FlowsThe following table summarizes our cash flows for the periods indicated (in thousands):Cash flows from operating activitiesCash used in operating activities for the year ended December 31, 2017 was $38.4 million and primarily consisted of a net loss of $42.4 million and gain on intellectual property sale of $0.2 million from MYDICAR sale, which was partially offset by $4.2 million of stock-based compensation expense and $0.3 million of non-cash interest related to amortization of debt discount. Additionally, cash used in operating activities reflected changes in net operating assets due to an increase of $0.2 million in other non-current assets primarily associated with the deposit paid for the new facility lease entered into in October 2017 and an increase of $0.1 million in prepaid expenses and other current assets primarily associated with the timing of payments.Cash used in operating activities for the year ended December 31, 2016 was $38.0 million, and primarily consisted of a net loss of $47.1 million, offset by $3.2 million expense related to a non-cash issuance of common stock to Bristol-Meyers Squibb in connection with the BMS License Agreement, $3.2 million of stock-based compensation expense, $0.7 million of non-cash interest expense related to the Notes outstanding prior to their conversion into common stock in March 2016 and $0.4 million change in fair value of warrant liability and obligation to issue shares to Eiccose. Additionally, cash used in operating activities reflected changes in net operating assets due to an increase of $1.5 million in accounts payable and accrued expenses and other liabilities primarily associated with increase in business activity, and decrease of $0.3 million in prepaid expenses and other current assets.Cash used in operating activities for the year ended December 31, 2015 was $9.1 million and primarily consisted of a net loss of $13.3 million, offset by $1.5 million change in fair value of obligation to issue shares to Eiccose, $0.4 million of non-cash interest expense related to the Notes, $0.2 million expense related to a non-cash issuance of common stock to Dr. Tracey McLaughlin and Dr. Colleen Craig in connection with the acquisition of assets related to the compound exendin 9-39 and $0.2 million of stock-based compensation expense. Additionally, cash used in operating activities reflected changes in net operating assets primarily due to an increase of $2.7 million in accounts payable and accrued expenses and other liabilities primarily associated with increase in business activity, offset by a $0.7 million increase in prepaid expenses and other current assets primarily associated with the prepayment of a license agreement.Cash flows from investing activitiesNet cash provided by investing activities for the year ended December 31, 2017 was $22.7 million. The net cash increase was primarily due to $47.0 million of proceeds from maturities of marketable securities and a $0.2 million payment received from Theragene for the sale of MYDICAR related assets, partially offset by $24.5 million purchase of marketable securities.Net cash used in investing activities for the year ended December 31, 2016 was $4.2 million. The net cash increase was primarily due to $34.2 million for the purchase of marketable securities, partially offset by $28.0 million of proceeds received upon the consummation of the Merger and $2.0 million of proceeds from maturities of marketable securities.Cash used in investing activities for the year ended December 31, 2015 was related to the purchase of property and equipment.Cash flows from financing activitiesCash provided by financing activities for the year ended December 31, 2017 was $20.0 million and consisted of $19.8 million of proceeds from the issuance of common stock upon public offering, net of issuance costs, and $0.2 million of proceeds received from the issuance of common stock upon ESPP purchase and options exercises.Cash provided by financing activities for the year ended December 31, 2016 was $65.1 million and consisted of $32.1 million of proceeds from the issuance of common stock in the Private Placement on March 22, 2016, net of issuance costs, $18.2 million of net proceeds from the issuance of common stock in the underwritten public offering, after deducting underwriting discounts and commissions and expenses payable by us, and $14.8 million of proceeds from borrowings in connection with Oxford loan, net of issuance costs.Cash provided by financing activities for the year ended December 31, 2015 consisted of $7.2 million of proceeds from the issuance of convertible preferred stock, net of issuance costs and $6.0 million of proceeds from issuance of the Notes, net of issuance costs.following table summarizesextended lease will commence on March 1, 2023 and expires on February 28, 2024. We also have additional operating leases that are not considered significant for disclosure. Refer to Note 13 of our consolidated financial statements included within Item 8 of this Annual Report on Form 10-K for a description of our contractual obligations atas of December 31, 2017 (in thousands):
2022.(1)Represents future rent payments under facility lease contracts.
Asset and License Agreements(2)Represents the Oxford first tranche Loan of $15.0 million.(3)Includes an exit fee on the Oxford Loan of $1.125 million due at maturity.We have notRefer to Note 5, Note 6 and Note 7 in our consolidated financial statements included these potential paymentwithin Item 8 of this Annual Report on Form 10-K for a description of our contractual obligations in the table above as the amount and timing of such payments are not known.Oxford Finance Term LoanDecember 30, 2016,August 11, 2018, we entered into a Product Development Agreement and a First Project Agreement (the Product Agreements), pursuant to which we will receive development program support services for the OxfordHDV program. The services are to be provided from July 1, 2018 through the new drug application (NDA) filing. The Product Agreements can be terminated by either party due to material breach or by us without cause with 90 days prior written notice. Refer to Note 5 of our consolidated financial statements included within Item 8 of this Annual Report on Form 10-K for additional details.$25.0 million.up to $75.0 million funded in three tranches with a maturity date of August 31, 2027. The Oxford Loan bears interest at a floating rate per annum interest rate of the Innovatus Loan is equal to the sum of (a) the greater of either(i) the 30-day U.S. Dollar LIBOR reportedPrime Rate published in the Money Rates section of the Wall Street Journal (or any successor thereto) and (ii) 3.5%, plus 6.41%(b) 3.75%; provided that, at the election of the Borrower, up to 2.25% of such rate shall be payable in-kind until the third anniversary of the closing date. We received $40.0 million in June 2022 on the Closing Date under Tranche A. The remaining $35.0 million is divided into two tranches (Tranche B and Tranche C). The $17.5 million under each of Tranche B and Tranche C will be available for a period commencing on the later of (a) the first date that we achieve certain development and regulatory milestones applicable to each Tranche and (b) November 1, 2022. Both Tranche B and Tranche C draw periods end on the earlier of (a) June 30, 2024 or 6.95%,(b) an event of default.only payments through July 1, 2018 followed by 36 equal monthly paymentsand exit fees in connection with our loans outstanding owed to Oxford Finance LLC. Refer to Note 8 of principalour consolidated financial statements included within Item 8 of this Annual Report on Form 10-K for additional details.interest until maturity at July 1, 2021. AtProduct Agreement with Patheon, Inc. (Patheon) for the timemanufacturing of final payment,lonafarnib capsules and packaging of bottles for commercial sale. Under the terms of the agreements, we are required to provide Patheon with annual volume forecasts of capsules and Patheon will manufacture 80 percent of actual manufacturing volume. If we order more than 20 percent of manufacturing volume from other manufacturers, we are required to pay an exit fee70 percent of 7.5%purchase price to Patheon for the shortfall. The initial terms of the original principal balance of the Oxford Loan, which was $1.125 million atMMSA and Product Agreement end on December 31, 2017. The loan is secured by2025 with automatic renewal for successive two-year terms, unless earlier terminated pursuant to the perfected first priority liens on our assets, including our commitmentterms of each agreement, or upon either party’s notice of termination to not allow any liens to be placed upon our intellectual property. The Oxford Loan includes customary events of default, including failure to pay amounts due, breaches of covenants and warranties, material adverse effect events, certain cross defaults and judgments, and insolvency. As of December 31, 2017, we were in compliance with all loan terms.Year Ended December 31, 2022 2021 Net cash used in operating activities $ (83,010) $ (71,342) Net cash provided by investing activities 9,632 61,532 Net cash provided by financing activities 76,955 3,167 Net increase (decrease) in cash and cash equivalents $ 3,577 $ (6,643) In January 2016, the FASB issued ASU No. 2016-01, RecognitionMeasurement of Financial Assets and Financial Liabilities. ASU No. 2016-01 supersedes and amends the guidance to classify equity securities with readily determinable fair values into different categories (that is, trading or available-for-sale) and require equity securities to be measured at fair value with changes in the fair value recognized through net income. The amendments allow equity investments that do not have readily determinable fair values to be remeasured at fair value either upon the occurrence of an observable price change or upon identification of an impairment. The amendments also require enhanced disclosures about those investments. ASU No. 2016-01 is effectiveyet adopted for annual reporting beginning after December 15, 2017, including interim periods within the year of adoption, and calls for prospective application. The Company is currently in the process of evaluating the impact that the standard will have on its consolidated financial statements.In February 2016, the FASB issued ASU 2016-02, Leases (Topic 842), which requires lessees to recognize most leases on their balance sheet. The standard requires use of the modified retrospective transition method, with elective relief, which requires application of the guidance for all periods presented. The new standard will be effective for fiscal years beginning afterended December 15, 2018. Early adoption is permitted. The Company is currently in the process of evaluating the impact the standard will have on its consolidated financial statements.In March 2016, the FASB issued ASU 2016-09, Compensation-Stock Compensation (Topic 718): Improvements to Employee Share-Based Payment Accounting, which is intended to simplify several aspects of the accounting for employee share-based payment transactions, including the accounting and reporting of income taxes, the determination of forfeiture rates, classification of awards as either equity or liabilities, and classification on the statement of cash flows. ASU 2016-09 is effective for fiscal years and interim periods within those years beginning after December 15, 2016 and early adoption is permitted. The Company adopted this ASU in the first quarter of 2017. As a result of adopting this standard, the Company made an accounting policy election to account for forfeitures as they occur. This change did not have a material impact to the consolidated financial statements.In June 2016, the FASB issued ASU No. 2016-13, Financial Instruments—Credit Losses (Topic 326). The standard changes how entities will measure credit losses for most financial assets and certain other instruments that are not measured at fair value through net income. Financial assets measured at amortized cost will be presented at the net amount expected to be collected by using an allowance for credit losses. The standard is effective for fiscal years and interim periods beginning after December 15, 2019. Early adoption is permitted for all periods beginning afterDecember 15, 2018. The Company is currently in the process of evaluating the impact that the standard will have on its consolidated financial statements. In August 2016, the FASB issued ASU No. 2016-15, "Statement of Cash Flows (Topic 230): Classification of Certain Cash Receipts and Cash Payments. ASU 2016-15 identifies how certain cash receipts and cash payments are presented and classified in the Statement of Cash Flows. The standard is effective for fiscal years and interim periods beginning after December 15, 2017. The standard should be applied retrospectively and early adoption is permitted, including adoption in an interim period. The Company does not expect the standard to have a material impact on its consolidated financial statements.Aboutabout Market RiskStatementsStatements and Supplementary Data20172022 and 2016,2021, the related consolidated statements of operations, comprehensive loss, stockholders’ equity, (deficit), and cash flows for each of the years in the three‑yearthree-year period ended December 31, 2017,2022, and the related notes (collectively, the consolidated financial statements). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 20172022 and 2016,2021, and the results of its operations and its cash flows for each of the years in the three‑yearthree-year period ended December 31, 2017,2022, in conformity with U.S. generally accepted accounting principles./s/ KPMG LLP
9, 201816, 2023Year Ended December 31, 2022 2021 Assets Current assets: Cash and cash equivalents $ 25,798 $ 22,221 Short-term debt securities 73,150 66,594 Accounts receivable, net 1,749 2,576 Inventories 2,853 2,612 Prepaid expenses and other current assets 13,985 9,361 Total current assets 117,535 103,364 Long-term debt securities — 17,262 Property and equipment, net 696 613 Operating lease right-of-use assets 561 653 Other assets 1,347 4,510 Total assets $ 120,139 $ 126,402 Liabilities and Stockholders’ Equity Current liabilities: Accounts payable $ 8,975 $ 7,765 Accrued liabilities 15,655 13,699 Current portion of operating lease liabilities 491 628 Debt, current portion — 7,809 Total current liabilities 25,121 29,901 Debt, net of current portion 39,625 23,986 Operating lease liabilities 83 116 Total liabilities 64,829 54,003 Stockholders’ equity: 44 35 Additional paid-in capital 492,759 412,930 Accumulated other comprehensive loss (300) (149) Accumulated deficit (437,193) (340,417) Total stockholders’ equity 55,310 72,399 Total liabilities and stockholders’ equity $ 120,139 $ 126,402 Year Ended December 31, 2022 2021 2020 Product revenue, net $ 12,734 $ 12,142 $ — Other revenue 750 — — Total revenue 13,484 12,142 — Costs and operating expenses: Cost of sales 1,837 745 — Research and development 75,282 64,436 41,590 Selling, general and administrative 29,105 23,900 20,559 Total costs and operating expenses 106,224 89,081 62,149 Loss from operations (92,740) (76,939) (62,149) Interest expense (4,132) (3,559) (3,594) Interest income 1,082 158 704 Other (expense) income, net (963) 46,487 (12) Loss before provision for income taxes (96,753) (33,853) (65,051) Provision for income taxes 23 64 — Net loss $ (96,776) $ (33,917) $ (65,051) Net loss per common share, basic and diluted $ (2.32) $ (1.00) $ (2.31) Weighted-average common shares outstanding, basic and diluted 41,628,207 33,944,342 28,143,391 Seeto theare an integral part of these consolidated financial statements.Consolidated Statements of Operations(In thousands, except share and per share amounts)See accompanying notes to the consolidated financial statements.Eiger BioPharmaceuticals, Inc.Year Ended December 31, 2022 2021 2020 Net loss $ (96,776) $ (33,917) $ (65,051) Other comprehensive loss: Unrealized loss on available-for-sale debt securities, net Unrealized loss on available-for-sale debt securities, net (51) (141) (50) Foreign currency translation adjustment Foreign currency translation adjustment (100) — — Comprehensive loss $ (96,927) $ (34,058) $ (65,101) Seeto theare an integral part of these consolidated financial statements. (Deficit) Common Stock Additional Paid-In Capital Accumulated Other Comprehensive Income (Loss) Accumulated Deficit Total Stockholders’ Equity Shares Amount Balance at December 31, 2019 24,523,381 $ 24 $ 297,863 $ 42 $ (241,449) $ 56,480 Issuance of common stock upon offering at-the-market, net of $3,231 of issuance costs 9,267,760 10 96,750 — — 96,760 Issuance of common stock upon ESPP purchase 25,645 — 186 — — 186 Issuance of common stock upon stock option exercise 61,700 — 520 — — 520 Vesting of common stock under Product Development Agreement — — 217 — — 217 Stock-based compensation expense — — 5,973 — — 5,973 Unrealized loss on available-for-sale debt securities, net — — — (50) — (50) Net loss — — — — (65,051) (65,051) Balance at December 31, 2020 33,878,486 34 401,509 (8) (306,500) 95,035 Issuance of common stock upon offering at-the-market, net of $417 of issuance costs 565,938 1 2,684 — — 2,685 Issuance of common stock upon ESPP purchase 37,619 — 257 — — 257 Issuance of common stock upon stock option exercise 53,028 — 361 — — 361 Issuance of common stock upon release of restricted stock units 33,750 — — — — — Vesting of common stock under Product Development Agreement — — 218 — — 218 Stock-based compensation expense — — 7,901 — — 7,901 Unrealized loss on available-for-sale debt securities, net — — — (141) — (141) Net loss — — — — (33,917) (33,917) Balance at December 31, 2021 34,568,821 35 412,930 (149) (340,417) 72,399 Issuance of common stock upon offering at-the-market, net of $2,068 of commissions and issuance costs 8,528,074 8 66,102 — — 66,110 Issuance of common stock upon exercise of stock options 47,367 — 234 — — 234 Issuance of common stock to lender 749,053 1 4,999 — — 5,000 Vesting of common stock issued under Product Development Agreement — — 19 — — 19 Issuance of common stock upon ESPP purchase 48,115 — 158 — — 158 Issuance of common stock upon release of restricted stock units 132,854 — — — — — Stock-based compensation expense — — 8,317 — — 8,317 Unrealized loss on available-for-sale debt securities, net — — — (51) — (51) Cumulative translation adjustment — — — (100) — (100) Net loss — — — — (96,776) (96,776) Balance at December 31, 2022 44,074,284 $ 44 $ 492,759 $ (300) $ (437,193) $ 55,310 Seeto theare an integral part of these consolidated financial statements.statementsYear Ended December 31, 2022 2021 2020 Operating activities Net loss $ (96,776) $ (33,917) $ (65,051) Adjustments to reconcile net loss to net cash used in operating activities: Gain from sale of priority review voucher — (46,493) — Income related to asset purchase agreement — (281) — Depreciation and amortization 292 276 167 Inventory write down 1,043 — — Amortization of debt securities premiums and discounts 682 996 124 Loss on extinguishment of debt 1,144 — — Non-cash interest expense 1,238 776 804 Reduction in the carrying amount of right-of-use assets 550 523 478 Common stock issued under Product Development Agreement 19 218 217 Stock-based compensation 8,317 7,901 5,973 Change in operating assets and liabilities: Accounts receivable 828 (2,576) — Inventories (1,367) (2,424) — Prepaid expenses and other current assets (196) (701) (3,563) Other assets (1,265) (607) (1,392) Accounts payable 1,152 3,234 (1,899) Accrued liabilities 1,957 2,309 1,491 Operating lease liabilities (628) (576) (534) Net cash used in operating activities (83,010) (71,342) (63,185) Investing activities Purchase of debt securities available-for-sale (75,101) (84,647) (128,295) Proceeds from maturities of debt securities available-for-sale 85,073 99,630 83,766 Proceeds related to asset purchase agreement — 281 — Proceeds from sale of priority review voucher — 95,000 — Payments related to priority review voucher — (48,507) — Purchase of property and equipment (340) (225) (258) Net cash provided by (used in) investing activities 9,632 61,532 (44,787) Financing activities Issuance of common stock upon offering at-the-market, net of commissions 66,402 3,040 96,779 Proceeds from issuance of common stock to lender 5,000 — — Proceeds from debt 39,841 — — Repayment of debt (33,277) — — Proceeds from issuance of common stock upon stock option exercises 234 361 520 Proceeds from issuance of common stock upon ESPP purchase 157 257 186 Payment of debt issuance costs (1,116) (175) — Common stock offering costs (286) (316) (22) Net cash provided by financing activities 76,955 3,167 97,463 Net increase (decrease) in cash and cash equivalents 3,577 (6,643) (10,509) Cash and cash equivalents at beginning of the year 22,221 28,864 39,373 Cash and cash equivalents at end of the year $ 25,798 $ 22,221 $ 28,864 Supplemental disclosure of cash flow information: Interest paid $ 2,885 $ 2,783 $ 2,791 Income taxes paid $ 64 $ — $ — Property and equipment purchases included in accounts payable and accrual $ 36 $ — $ — Common stock offering costs included in accounts payable and accrual $ 6 $ — $ — Right-of-use assets obtained in exchange for lease liabilities $ 458 $ — $ — Seeto theare an integral part of these consolidated financial statements.1.Description of Business“Company”Company or “Eiger”)Eiger) was incorporated in the State of Delaware on November 6, 2008. Eiger is a commercial-stage biopharmaceutical company focused on the development of innovative therapies for hepatitis delta virus (HDV), the most severe form of viral hepatitis, and other serious diseases. All five of the Company’s rare disease programs have been granted Breakthrough Therapy designation by the U.S. Food and Drug Administration (FDA).clinical-stage biopharmaceutical company committed to bringing to market novel productswell-characterized peptide, as a treatment for the treatment of rare diseases. The Company has builtcongenital hyperinsulinism (HI), an ultra-rare pediatric metabolic disorder, and post-bariatric hypoglycemia (PBH), a diverse portfolio of well-characterized product candidates with the potential to address diseasesdebilitating and potentially life-threatening condition. There are currently no approved therapies for which the unmet medical need is high, the biology for treatment is clear, and for which an effective therapy is urgently needed. these disorders.itWales. The Company operates in one segment.Reverse MergerOn March 22, 2016, Eiger completed its merger with Celladon Corporation (“Celladon”) in accordance with the terms of the Agreement and Plan of Merger and Reorganization, dated November 18, 2015 (the “Merger Agreement”), by and among Celladon, Celladon Merger Sub, Inc. (“Merger Sub”) and Eiger (the “Merger”). Pursuant to the Merger Agreement, Merger Sub merged with and into Eiger, with Eiger becoming a wholly-owned subsidiary of Celladon and the surviving corporation of the Merger. Pursuant to the terms and subject to the conditions set forth in the Merger Agreement, Eiger stockholders became the majority stockholders of the surviving company. In connection with, and immediately prior to, the closing of the Merger, on March 22, 2016, Celladon filed an amendment to its amended and restated certificate of incorporation with the Secretary of State of the State of Delaware to affect a fifteen-for-one reverse stock split of its common stock (the “Reverse Stock Split”). In connection with and immediately following the consummation of the Merger, on March 22, 2016, Celladon filed an amendment to its amended and restated certificate of incorporation with the Secretary of State of the State of Delaware to change its name to Eiger BioPharmaceuticals, Inc. The Company’s shares of common stock listed on the NASDAQ Global Market, previously trading through the close of business on Tuesday, March 22, 2016 under the ticker symbol “CLDN,” commenced trading on the NASDAQ Global Market, on a post-reverse stock split adjusted basis, under the ticker symbol “EIGR” on March 23, 2016. On March 22, 2016, a Certificate of Merger was filed with the Secretary of State of the State of Delaware to affect the Merger of Merger Sub with and into Eiger. See Note 5 for further details.The Company, or Eiger, as used in the accompanying notes to the consolidated financial statements, refers to Private Eiger prior to the completion of the Merger and Public Eiger subsequent to the completion of the Merger.Reverse Stock Split and Exchange RatioOn March 22, 2016, and prior to the closing of the Merger, Celladon completed a fifteen-for-one reverse stock split. As a result of the reverse stock split, every fifteen shares of Celladon common stock outstanding immediately prior to the Merger were combined and reclassified into one share of Celladon common stock. No fractional shares were issued in connection with the reverse stock split.The holders of shares of Eiger common stock outstanding immediately prior to the Merger received approximately 0.09 shares of Celladon common stock in exchange for each share of Eiger common stock in the Merger. Following the reverse stock split and the Merger on March 22, 2016, the combined company had 6,945,401 shares of common stock outstanding.The accompanying consolidated financial statements and notes to the consolidated financial statements give retroactive effect to the reverse stock split for all periods presented.2017,2022, the Company had $32.0$98.9 million of cash, cash equivalents and short-term securities, comprised of $25.8 million of cash and cash equivalents and $9.7$73.1 million of short-term marketable securities. In addition, thedebt securities available-for-sale. The Company had an accumulated deficit of $118.8$437.2 million and negative cash flows from operating activities.activities as of December 31, 2022. As the Company continues to incur losses, its transition to profitability will depend on the successful development, approval, and commercialization of product candidates and on the achievement of sufficient revenues to support its cost structure. The Company expectsmay never achieve profitability, and until it does, the Company will need to continue to incur losses for the next several years.raise additional capital. focused on completing its three ongoing Phase 2 development programs and initial preparations for advancing its lead program into Phase 3, for at least the next 12 months following the issuance date of these consolidated financial statements. However, if the Company’s anticipated operating results are not achieved in future periods, managementthe Company believes that planned expenditures willmay need to be reduced or it would be required to raise funding in order to extend the time period over which the then-available resources would be able to fund the operations. Additionally, the Company’s operations.2.Summary of Significant Accounting Policies and Consolidationsubsidiarysubsidiaries, EBPI Merger Inc., EB Pharma LLC, Eiger BioPharmaceuticals Europe Limited, and EigerBio Europe Limited, have been prepared in conformityaccordance with accounting principles generally accepted in the United States of America, (“U.S. GAAP”).(U.S. GAAP) and follow the requirements of the Securities and Exchange Commission (the SEC) for annual reporting. All intercompany balances and transactions have been eliminated in consolidation.estimates, including those related to accrued research and development expenses, stock-based compensation and income taxes.estimates. The Company bases its estimates on historical experience and on various other market-specific and relevant assumptions that the Company believes to be reasonable under the circumstances. Actual results could differ from those estimates.For each product candidate, thethe fourits product candidates. If any of the single source suppliers in any of the supply chains fail to satisfy the Company’s requirements on a timely basis, it could suffer delays in its clinical development programs and activities, which could adversely affect its operating results.Short-Term MarketableAll short-term marketableLong-term securities consist of debt securities classified as available-for-carried at fairclassified as Level 2 because their value is based upon quotedon valuations using significant inputs derived from, or corroborated by, observable market prices.data. Unrealized gains and losses on available-for-sale debt securities are excluded from earnings and are reported as a component of accumulated other comprehensive loss. The cost of available-for-sale securities sold is based on the specific-identification method. Realized gains and losses on the sale of marketabledebt securities are determined using the specific-identification method and recorded in other (expense) income, (expense), net on the accompanying consolidated statements of operations.Lab equipment5 years5 years Furniture 5 years Leasehold improvements Shorter of remaining lease term or 5 years Computer equipment and software 2017,2022, the Company has not impaired any long-lived assets.Expensesexpenseexpenses in the accompanying consolidated statements of operations. The Company accrues for these costs based on factors such as estimates of the work completed and in accordance with agreements established with its third-party service providers. The Company makes judgments and estimates in determining the accrued liabilities balance in each reporting period. As actual costs become known, the Company adjusts its accrued liabilities.expense.regarding subjective variables.Stock-based awards and stock options issued to non-employee consultants are recorded at fair value and remeasured at the end of each period as they vest using thefor Black-Scholes option pricing model. Expense is recognized over the vesting period which is generally the same as the service period.hadexperiences a change of ownership, utilization of the net operating loss and tax credit carryforwards may be restricted. (deficit) except those resulting from and distributions to stockholders. The Company’s unrealized gains and losses on available-for-saledebt securities and foreign currency translation adjustment represent the only componentcomponents of other comprehensive loss that are excluded from the reported net loss and that are presented in the consolidated statements of comprehensive loss.December 31, 2022 2021 2020 Options to purchase common stock 6,143,183 5,262,185 3,697,075 Restricted stock units (unvested) Restricted stock units (unvested) 641,407 623,000 37,500 ESPP ESPP 334,751 48,159 81,169 Total 7,119,341 5,933,344 3,815,744 In January 2016, the FASB issued ASU No. 2016-01, Recognition and Measurement of Financial Assets and Financial Liabilities. ASU No. 2016-01 supersedes and amends the guidance to classify equity securities with readily determinable fair values into different categories (that is, trading or available-for-sale) and require equitysecurities to be measured at fair value with changes in the fair value recognized through net income. The amendments allow equity investments that do not have readily determinable fair values to be remeasured at fair value either upon the occurrence of an observable price change or upon identification of an impairment. The amendments also require enhanced disclosures about those investments. ASU No. 2016-01 is effective for annual reporting beginning after December 15, 2017, including interim periods within the year of adoption, and calls for prospective application. The Company is currently in the process of evaluating the impact that the standard will have on its consolidated financial statements.In February 2016, the FASB issued ASU 2016-02, Leases (Topic 842), which requires lessees to recognize most leases on their balance sheet. The standard requires use of the modified retrospective transition method, with elective relief, which requires application of the guidance for all periods presented. The new standard will be effective for fiscal years beginning after December 15, 2018. Early adoption is permitted. The Company is currently in the process of evaluating the impact that the standard will have on its consolidated financial statements.In March 2016, the FASB issued ASU 2016-09, Compensation-Stock Compensation (Topic 718): Improvements to Employee Share-Based Payment Accounting, which is intended to simplify several aspects of the accounting for employee share-based payment transactions, including the accounting and reporting of income taxes, the determination of forfeiture rates, classification of awards as either equity or liabilities, and classification on the statement of cash flows. ASU 2016-09 is effective for fiscal years and interim periods within those years beginning after December 15, 2016 and early adoption is permitted. The Company adopted this ASU in the first quarter of 2017. As a result of adopting this standard, the Company made an accounting policy election to account for forfeitures as they occur. This change did not have a material impact on the Company’s consolidated financial statements.FASBFinancial Accounting Standards Board (FASB) issued ASUAccounting Standards Updates (ASU) No. 2016-13, Financial Instruments—Credit Losses (Topic 326)The standard is effective for fiscal years and interim periods beginning after December 15, 2019. Early adoption is permitted for all periods beginning after December 15, 2018. The Company is currently in the process of evaluating the impact the standard will have on its consolidated financial statements.August 2016,April 2019, the FASB issued ASU No. 2016-15, "Statement2019-04, Codification Improvements to Topic 326, Financial Instruments – Credit Losses, Topic 815, Derivatives and Hedging, and Topic 825, Financial Instruments, which clarifies and corrects certain unintended applications of Cash Flowsthe guidance contained in each of the amended Topics. Additionally, in May 2019, the FASB issued ASU No. 2019-05, Financial Instruments – Credit Losses (Topic 230): Classification326), which provides an option to irrevocably elect to measure certain individual financial assets at fair value instead of Certain Cash Receiptsamortized cost. In November 2019, the FASB issued ASU No. 2019-10, Financial Instruments – Credit Losses (Topic 326), Derivatives and Cash Payments.Hedging (Topic 815), and Leases (Topic 842), whichdefers the effective date for ASU 2016-15 identifies how certain cash receipts and cash payments are presented and classified in the Statement of Cash Flows. The standard is effectiveNo. 2016-13 for smaller reporting companies to fiscal years and interim periods beginning after December 15, 2017. The standard should be applied retrospectively and early adoption is permitted,2022, including adoption in an interim period.periods within those fiscal years. The Company does not expect the impact of adopting this standard to be material.3.Fair Value MeasurementsAtAs of December 31, 20172022 and 2016,2021, the carrying amount of cash equivalents, accounts receivable, prepaid expenses and other current assets, accounts payable and accrued liabilities approximated their estimateestimated fair value due to their relatively short maturities. Management believes the terms of long termits long-term debt reflectsreflect current market conditions for an instrument with similar terms and maturity, therefore the carrying value of the Company’s debt approximated its fair value.marketabledebt securities and commercial paper consist of available-for-sale securities and are classified as Level 2 because their value is based on valuations using significant inputs derived from or corroborated by observable market data.between Level 1, Level 2into or out of Level 3 of the fair value hierarchy during the periods presented.December 31, 2022 Level 1 Level 2 Level 3 Total Financial assets: Financial assets: Money market funds $ 11,546 $ — $ — $ 11,546 Commercial paper Commercial paper — 8,913 — 8,913 Corporate debt securities — 28,642 — 28,642 U.S. government bonds U.S. government bonds — 39,563 — 39,563 Total $ 11,546 $ 77,118 $ — $ 88,664 December 31, 2021 Level 1 Level 2 Level 3 Total Financial assets: Financial assets: Money market funds $ 13,520 $ — $ — $ 13,520 Corporate debt securities — 41,511 — 41,511 U.S. government bonds U.S. government bonds — 42,345 — 42,345 Total $ 13,520 $ 83,856 $ — $ 97,376 2017.The following table provides a reconciliation of liabilities measured at fair value using Level 3 significant unobservable inputs as of December 31, 2016 (in thousands):
2022 and 2021.(1)Changes in fair value of the obligation to issue common stock and the common stock warrant liability are recorded in other income (expense), net on the accompanying consolidated statements of operations.marketabledebt securities and the gross unrealized holding gains and losses (in thousands):December 31, 2022 Amortized cost Unrealized gain Unrealized loss Estimated Fair
ValueCash equivalents: Money market funds $ 11,546 $ — $ — $ 11,546 Commercial paper Commercial paper 3,968 — — 3,968 Total cash equivalents $ 15,514 $ — $ — $ 15,514 Debt securities: Debt securities: U.S. government bonds U.S. government bonds $ 39,646 $ 3 $ (86) $ 39,563 Corporate debt securities 28,759 — (117) 28,642 Commercial paper 4,945 — — 4,945 Total debt securities Total debt securities $ 73,350 $ 3 $ (203) $ 73,150 Classified as: Classified as: Cash equivalents Cash equivalents $ 15,514 Short-term debt securities Short-term debt securities 73,150 $ 88,664 December 31, 2021 Amortized cost Unrealized gain Unrealized loss Estimated Fair
ValueCash equivalents: Money market funds $ 13,520 $ — $ — $ 13,520 Total cash equivalents Total cash equivalents $ 13,520 $ — $ — $ 13,520 Debt securities: Debt securities: Corporate debt securities $ 41,576 $ — $ (65) $ 41,511 U.S. government bonds U.S. government bonds 42,429 — (84) 42,345 Total debt securities Total debt securities $ 84,005 $ — $ (149) $ 83,856 Classified as: Classified as: Cash equivalents Cash equivalents $ 13,520 Short-term debt securities Short-term debt securities 66,594 Long-term debt securities Long-term debt securities 17,262 $ 97,376 As of December 31, 2017, the contractual maturity of the available-for-sale marketable securities is less than one year. investmentssecurities for other-than-temporary impairment loss. The Company considers factors such as the duration, severity and the reason for the decline in value, the potential recovery period and its intent to sell. For debt securities, it also considers whether (i) it is more likely than not that the Company will be required to sell the debt securities before recovery of their amortized cost basis, and (ii) the amortized cost basis cannot be recovered as a result of credit losses. As ofDuring the year ended December 31, 2017,2022, the Company did not recognize any other-than-temporary impairment losses. All marketableThe unrealized losses of $0.2 million as of December 31, 2022 include debt securities with unrealized losses of $21,000 that have been in athe loss position for lessmore than twelve 12 months. However, the Company is planning to hold these securities until maturity and expects to recover the amortized cost basis.4.Balance Sheet ComponentsDecember 31, 2022 2021 Raw materials $ 1,703 $ 1,056 Work-in-progress 884 1,468 Finished goods 266 88 Total inventories $ 2,853 $ 2,612 December 31, 2022 2021 Computer equipment and software Computer equipment and software $ 736 $ 669 Furniture Furniture 116 116 Leasehold improvements Leasehold improvements 101 101 Lab equipment 271 271 Construction in progress Construction in progress 367 59 Total property and equipment 1,591 1,216 Less: accumulated depreciation (895) (603) Property and equipment, net $ 696 $ 613 2017, 20162022, 2021 and 20152020 was $41,000, $23,000approximately $0.3 million, $0.3 million and $11,000,$0.2 million, respectively.
Prepaid Expenses and Other Current AssetsDecember 31, 2022 2021 Prepaid contract manufacturing costs $ 3,542 $ 3,695 Short term deposits 4,542 54 Prepaid research costs 2,822 3,253 Prepaid insurance 586 631 Prepaid marketing 753 469 Other 1,740 1,259 Total prepaid expenses and other current assets $ 13,985 $ 9,361 December 31, 2022 2021 Compensation and related benefits $ 6,167 $ 3,131 Contract research costs 4,188 4,760 Contract manufacturing costs 2,101 3,288 Product revenue reserves Product revenue reserves 1,373 1,846 Other 1,826 674 Total accrued liabilities $ 15,655 $ 13,699 5.Reverse MergerOn March 22, 2016, Eiger completedMergerTrustees of the University of Pennsylvania and the Children’s Hospital of PhiladelphiaCelladon as discussedthe Trustees of the University of Pennsylvania (UPenn) and the Children’s Hospital of Philadelphia (CHOP), under which the Company obtained an exclusive, royalty-bearing, worldwide license to develop, manufacture and sell certain Glucagon Like Peptide-1 (GLP-1) receptor antagonist(s) products to treat all human and animal conditions. The Company also obtained an exclusive, royalty-bearing, sublicensable, worldwide license to certain data developed by CHOP. The Company is responsible for the development and commercialization of the licensed products at its sole cost and expense.Note 1. For accounting purposes, Eigerresearch and development expenses for the year ended December 31, 2019. In addition, the Company is consideredobligated to have acquired Celladonpay UPenn a specified annual license maintenance fee, up to $2.5 million upon marketing authorization in one or more countries, and up to $18.0 million in commercial milestones. The Company will also be required to pay UPenn a flat royalty in the Merger. Eiger was determinedlow-single digits on net sales of all licensed products by the Company, subject to specified reductions and offsets, and specified minimum annual royalty payments. The Company’s obligation to pay royalties expires on a product-by-product and country-by-country basis, on the later of: (i) the expiration of the last valid claim in the licensed patents in any country, or (ii) the tenth anniversary of the first commercial sale of the product in such country. No milestones have been achieved as of December 31, 2022.accounting acquirer based uponnew drug application (NDA) filing. As consideration, the termsCompany has committed to pay fees of approximately $10.0 million in cash and stock over four years, including approximately $0.8 million for expert consultant fees and pass through costs to vendors, plus certain incentive-based regulatory milestone fees up to $1.0 million. As part of the Merger and other factors including; (i) Eiger security holders owned approximately 78% ofaggregate payment, the combined company immediately following the closing of the Merger, (ii) Eiger directors held all of the board seats in the combined company, and (iii) Eiger management held all key positions in the management of the combined company. The Merger was accounted for as an asset acquisition rather than business combination because the assets acquired and liabilities assumed by Eiger did not meet the definition of a business as defined by U.S. GAAP. The net assets acquired in connection with this transaction were recorded at their estimated acquisition date fair values as of March 22, 2016, the date the Merger with Celladon was completed.Immediately prior to the effective date of the Merger and in connection with the Private Placement, the Notes converted intoCompany issued 115,526 shares of common stock of Eiger. Further, allsubject to quarterly vesting requirements related to successful performance of the Warrants were exercised for common stock (see Note 9)services and all sharesachievement of preferred stockbudget timeline set forth in the Product Agreements. Eiger converted into shares of common stock of Eiger.At the effective date of the Merger, Celladon issued shares of its common stock to Eiger stockholders, at an exchange rate of approximately 0.09 shares of common stock, after taking into account the Reverse Stock Split, in exchange for each share of Eiger common stock outstanding immediately prior$19,000, $0.2 million and $0.2 million, respectively, related to the Merger.shares issued under the exchange rate was calculatedCompany is obligated to: (i) exclusively supply lonafarnib to PRF for use in clinical trials and non-clinical research in progeria at the Company’s expense, (ii) prepare and be the sponsor of any NDA submission for lonafarnib for the treatment of progeria to the FDA, (iii) use commercially reasonable efforts to file a NDA for progeria by a formula that was determined through arms-length negotiations between Celladon and Eiger. The combined Company assumed all of the outstanding options, whether or not vested, under the Eiger 2009 Equity Incentive Plan (the “Eiger Plan”) with such options henceforth representing the right to purchasespecified date, (iv) submit a number of shares of Celladon common stock equal to approximately 0.09 multiplied by the number of shares of Eiger common stock previously represented by such options.Immediately after the Reverse Stock Split and the Merger on March 22, 2016, there were 6,945,401 shares of the combined Company’s common stock outstanding. In addition, immediately after the Merger, pre-Merger Eiger stockholders, warrant holders and option holders owned approximately 78% of the aggregate number of shares of the combined Company’s common stock, and the stockholders of Celladon immediately prior to the Merger owned approximately 22% of the aggregate number of shares of the combined Company’s common stock (on a fully diluted basis).On March 22, 2016, Celladon had 1,596,959 shares of common stock outstandingRare Pediatric Disease designation and a market capitalizationrequest for expedited approval in connection with a NDA filing, (v) establish a patient support program in progeria, and (vi) use commercially reasonable efforts to develop a pediatric formulation of $27.5lonafarnib for use in progeria.estimated fair valuePRF Collaboration Agreement continues for an initial term of the net assetsten years and automatically renews for subsequent renewal terms of Celladon on March 22, 2016 was $27.3 million. The fair value of Celladon’s common stock on the Merger closing date was above the fair value of Celladon’s net assets. As Celladon’s net assets were predominantly comprised of cash offset by current liabilities, the fair value of Celladon’s net assets as of March 22, 2016 was considered to be the best indicator of the fair value and, therefore, the estimated purchase consideration.two years each unless either party terminates earlier.The following table summarizes the net assets acquired based on their estimated fair values as of March 22, 2016 (in thousands):6.License Agreements(“BMS”)(BMS) entered into a License Agreement (the “BMSBMS License Agreement”)Agreement) and a Common Stock Purchase Agreement (the “BMSBMS Purchase Agreement”)Agreement).the proprietary BMS molecule known as PEG-interferon Lambda-1a (the “Licensed Product”)(peginterferon lambda or the Licensed Product) for all therapeutic and diagnostic uses in humans and animals. The Company is responsible for the development and commercialization of the Licensed Product at its sole cost and expense. The Company paid BMS $2.0 million and issued 157,587 shares of its common stock at an aggregate fair value of $3.2 million in April 2016. The BMS License Agreement required the Company to make an upfront payment of $2.0 million in cash and issue $3.0 million in Company common stock (see below) andalso includes development and regulatory milestone payments totaling $61.0 million and commercial sales milestones of up to $128.0 million. The Company is obligated to pay BMS annual net sales royalties in the range of mid-single to mid-teens, depending on net sales levels. In addition, ifthe fourth quarter of 2020, the Company grants a sublicense, the Company is obligated to pay BMS a portion of the sublicensing income received.The Company paid BMS an upfront payment of $2.0 millionrecorded in cash in April 2016, which was charged to research and development expense in the consolidated statement of operations as there is no future alternative use for the intellectual property licensed.The Company issued BMSa $3.0 million milestone, triggered on successful demonstration of proof of concept, as defined by the BMS License Agreement, in common stock as an element of the upfront payment. The BMS Purchase Agreement provided for the issuance of 157,587 shares of common stock ofa Phase 2 clinical trial. In March 2022, the Company to BMSrecorded a $5.0 million milestone expense in consideration of the license grantedresearch and development, which was related to the Companyinitiation of a Phase 3 clinical trial, as defined under the BMS License Agreement. The BMS Purchase Agreement grants BMS certain registration rights with respect to the shares of common stock delivered, and BMS has agreed to certain trading and other restrictions with respect to the shares issued. In April 2016, the Company issued 157,587 common shares to BMS for an aggregate fair value of $3.2 million, which was charged to research and development expense in the consolidated statement of operations as there is no future alternative use for the intellectual property licensed.(“Merck”)(Merck), which provides the Company with the exclusive right to develop, manufacture, and sell products containing the compounds lonafarnib for the treatment of all human viruses except certain specified viruses such as hepatitis B and hepatitis C alone. As part of consideration, for such exclusive right, the Company issued Private Eiger convertible preferred27,350 shares of common stock with a fair value of $0.5 million when the agreement was executed in September 2010. This preferred stock was converted to 27,350 shares of common stock upon the Merger. In addition, themillion. The Company is obligated to pay Merck up to an aggregate of $27.0 million in development milestones and will be required to pay tiered royalties based on aggregate annual net sales of all licensed products ranging from mid-single to low double-digit royalties on net sales. The Company’s obligation to pay royalties to Merck expires on a country-by-country and product-by-product basis on the later of the expiration of the last to expire patent assigned to the Company under the agreement, which is estimated to be in the first half of 2018; or on the tenth anniversary of the first commercial sale of the product. In May 2015, the first regulatory milestone was achieved and the Company paid the related milestone payment of $1.0 million to Merck. The amount has been recorded as a charge to research and development expense during the year ended December 31, 2015. No additional milestones have been achieved during the years endedas of December 31, 2017 and 2016.2022. The next milestone of $1.0 million is due upon successful completion of the Phase 3 study, which is expected by end of 2023.In December 2014,On May 15, 2018, the Company entered into aan amendment to the exclusive license agreement with Janssen Pharmaceutica NV, (“Janssen”),Merck, which provides for expansion of the existing exclusively licensed field of use under the license agreement with Merck to include all uses of lonafarnib related to the treatment of Hutchinson-Gilford progeria syndrome (HGPS) in humans at no cost to the Company. On November 3, 2020, the Company withentered into an amendment to the exclusive worldwide license agreement with Merck which expanded the indication to develop,also include progeroid laminopathies, collectively called progeria. The Company has the sole responsibility and the continuing obligation for the manufacture and sell products containingsupply of lonafarnib to the compound tipifarnib for all therapeutic and diagnostic uses in humans, including any such uses for human virology diseases, but excluding oncology diseases. The Company is responsible for the development of at least one product in a major market country and for commercialization of products in all countries where necessary authorization is obtained, at its sole cost and expense. The Company may manufacture, develop, and commercialize the products itself or grant one or more sublicenses for such purposes. However, for a period of time following completion of the proof of concept trial, Janssen has a first right of negotiation for an exclusive license back from the Company to develop and commercialize tipifarnib in any country in the world. The agreementPRF. Merck will continue for so long as the Company owe royalty payments to Janssen under the agreement or for so long as there is a valid patent claim under the agreement, whichever is longer.In connection with this license agreement, the Company is obligated to make developmentnot receive milestone payments in aggregaterelation to lonafarnib for the treatment of up to $38.0 million, sales milestoneprogeria or any royalty payments in aggregate up to $65.8 million and will be required to pay tiered royalties based on aggregate annual netfor sales of all licensed products ranging from mid-singlea specified quantity of lonafarnib to low double-digit royalties of net sales. As of December 31, 2017,treat the product has not reached commercializationcurrently estimated progeria patient population worldwide.no milestones have been paid.Related License Agreements7.Asset Purchase Agreements and Related License Agreements(“EGI”)(EGI). Dr. Jeffrey Glenn, a founder and director of the Company, is the sole owner of EGI. Pursuant to the agreement, the Company purchased all of the assets including the intellectual property rights related to the use of farnesyl transferasefarnesyltransferase inhibitors as anti-viral agents and methods to treat viral infections with those inhibitors and inhibitors of prenylation, prenyl cysteine methyltranferasemethyltransferase and a protease as anti-viral agents and methods to treat viral infection with those inhibitors. The Company paid EGI an upfront payment of $0.4 million when the agreement was executed in December 2010. Additionally, the Company will pay EGI a low single-digit royalty based on aggregate annual net sales of products developed using the intellectual property. Within the first ten years after commercialization, the Company may make a one-time payment of $0.5 million for each contract for the three types of product related to such intellectual property that would reduce the payment term for the three products to the tenth anniversary of the first commercial sale. The obligation to pay royalties expires on a country-by-country and product-by-product basis on the later of either when the product is no longer sold in any country or the earliest of the tenth anniversary of the first commercial sale of the product. As of December 31, 2017, theThe product hasis currently not achieved regulatory approval.In November 2012, the Company entered into an agreement with EGI whereby the Company sold all of the assets related to the compound clemizole, including any related intellectual property. EGI will pay to the Company a high single-digit royalty on future aggregate annual net sales, subject to certain reductions and exceptions. EGI’s obligation to pay royalties expires on a country-by-country and product-by-product basis on the later of either expiration of the last to expire patent sold to EGI under the agreement or the earliest of the tenth anniversary of the first commercial sale of the product. As of December 31, 2017, the product has not achieved regulatory approval.Exendin 9-39development.“Sellers”)Sellers), whereby the Company purchased all of the assets related to the compound avexitide (formerly known as exendin 9-399-39) including any related intellectual property from the Sellers (the “Exendin APA”)Exendin APA). The Company also entered into a consulting agreement with the Sellers as part of the agreement. The Company issued 15,378 shares of common stock that were valued at $0.2 million and optionswith the option to purchase 46,134 shares of common stock with an exercise price of $2.06 per share when the agreement was executed in September 2015.Food and Drug AdministrationFDA (the milestone-vested options).Merger,merger, the Company issued additional “top-up”top-up options to Dr. Tracey McLaughlin and Dr. Colleen Craigthe Sellers to purchase an aggregate of 48,544 shares of common stock, pursuant to the terms of the Exendin APA, with an exercise price of $17.25 per share. The top-up options consist of both time-vested and milestone-vested options.non-employee share-basedstock-based compensation expense as the awards vest over time, with the unvested portion revalued each period.time. The fair value of the milestone-vested options will be recognized as research and development expense when it is probable that the earliest milestone will be achieved at their then fair value.value as of the ASU 2018-07 adoption date. During the years ended December 31, 2017, 20162022, 2021 and 2015,2020, the Company recognized $44,000, $0.3$0 million, $0.1 million and $15,000$0.1 million of non-employee compensation expense related to the time-vested options, respectively. No expense was recognized for the milestone vested options during the years ended December 31, 2017, 20162022, 2021, and 2015.exendin 9-39,avexitide, subject to certain reductions and exceptions. The Company’s obligation to pay royalties expires on the expiration of the last to expire patent assigned to the Company under the agreement. Additionally, the Company has assumed the license agreement the Sellers had previously entered into with the Board of Trustees of the Leland Stanford Junior University (“Stanford”)(Stanford). The Company is obligated to pay a royalty to Stanford in the low single-digits on annual net sales after the first commercial sale of any products developed based on exendin 9-39.avexitide. As of December 31, 2017, upon2022, the Company has paid a total of $0.1 million in milestone payments to each of the Sellers related to the successful completion of the Phase 2 trials,trials.development milestoneAbbVie Agreement with AbbVie to sell its rare pediatric disease priority review voucher (the PRV), which was achievedawarded on November 20, 2020 upon FDA approval of Zokinvy. The AbbVie Agreement contains customary representations, warranties, covenants, and indemnification provisions subject to certain limitations.paid$95.0 million. The transaction closed in January 2021. Such consideration was shared with the related milestone payment of $0.1 million to eachPRF in accordance with the terms of the Sellers.Eiccose PurchasePRF Collaboration and Supply Agreement, and Related Stanford and Nippon License AgreementsIn October 2015, the Company entered into an asset purchase agreement with Eiccose whereby Eiccose sold all of the assets related to the treatment of pulmonary arterial hypertension (“PAH”), treatment of lymphedema and products containing ubenimex for the treatment of disorders involving LTB4, and any related intellectual property to the Company (the “Eiccose APA”). David Cory, the President, Chief Executive Officer and a director of the Company, is the sole managing member and significant equity interest holder of Eiccose. The Company made a payment to Eiccose of $0.1 million representing reimbursement of certain previously incurred expenses, including payments and accrued amounts owed to Stanford in connection with the license agreement for the treatment of Lymphedema (the “Lymphedema License Agreement”) and the license agreement for the treatment of PAH (the “PAH License Agreement”). The Eiccose APA also provided that, upon a next round of financing pursuant to which the Company sold sharesand PRF will equally share any net proceeds from the sale of capital stock resulting in gross proceeds toa priority review voucher that the Company may receive as the sponsor of at least $25.0a rare pediatric disease product application. The Company retained $46.5 million of proceeds from the Company would issue to Eiccose fully vested sharessale of the Company’s common stock equal to 1.75%PRV, net of related payments, and recorded the total number of the Company’s outstanding capital stock, before Merger. In October 2015, the Company recorded $1.5 millionamount in research and development expenses and a corresponding liability representing the fair value of the Company’s obligation to issue common stock to Eiccose.On March 22, 2016, the Company issued to Eiccose 96,300 fully vested shares of the Company’s common stock pursuant to the terms of the Eiccose APA. In connection with this transaction the Company remeasured the fair value of the obligation to issue common stock at the settlement date and the change in fair value of $0.2 million was recognized within other (expense) income, (expense), net in the consolidated statement of operations duringfor the year ended December 31, 2016. Upon the settlement2021.the obligation with the issuance of shares on March 22, 2016, the liability was reclassified to common stock and additional paid-in capital within stockholders’ equity (deficit).The Company is also obligated to pay to Eiccose an aggregate of up to a maximum of $10.0 million of commercial milestones in connection with future sales of the product and royalties in the low single-digits based on aggregate annual net sales following the first commercial sale of any product. As of December 31, 2017, the product has not reached commercialization and no milestones have been paid. In June 2017, Eiccose was disbanded. TheCompany’s commercial milestone obligations and sales royalties remain, however have been transferred to the previous shareholders of Eiccose.In addition, as a result of this agreement, the Company has assumed the license agreements Eiccose had previously entered into. These include the PAH License Agreement, the Lymphedema License Agreement for the treatment of lymphedema and the license agreement with Nippon Kayaku Co., Ltd, (“Nippon”). As part of the agreement, Nippon is obligated to make a payment for royalties in the low single-digits of sales to the Company. In connection with the PAH License Agreement and the Lymphedema License Agreement, the Company is obligated to make development and commercial milestone payments of up to $0.5 million in the aggregate under each contract, increasing annual license maintenance fees ranging from $10,000 to $75,000 over the term of each license agreement and royalty payments in low single-digits on annual net sales after the first commercial sale of a product under each license. For the year ended December 31, 2015, the Company recorded $0.2 million to research and development expense including $0.1 million for license fees and $0.1 million for the reimbursement of incurred expenses in connection with the Eiccose APA. For the years ended December 31, 2017 and 2016, no amounts have been recorded in connection with the Eiccose APA.8.DebtIn December 2016, the Company entered into an aggregate $25.0 million loan with Oxford Finance LLC (the “Oxford Loan”). The loan matures on July 1, 2021. The Company borrowed $15.0 million in December 2016 (“Tranche A”). The remaining $10.0 million (“Tranche B”) will be available to the Company upon achievement of positive top line data from the lonafarnib Phase 2 trial in HDV, which was achieved in the fourth quarter of 2016, plus positive top line Phase 2 data from at least one of the following programs: (i) Lambda in HDV, (ii) Exendin 9-39 in PBH based on the Company’s own IND, (iii) ubenimex in PAH, or (iv) ubenimex in Lymphedema.The Oxford Loan bears interest at a floating rate per annum equal to the greater of either the 30-day U.S. Dollar LIBOR reported in the Wall Street Journal plus 6.41% or 6.95%. The Company is required to repay the Tranche A in 18 monthly interest only payments followed by 36 equal monthly payments of principal and interest commencing on the first day of the month following the funding of Tranche A. If the Company receives the Tranche B funds, then the interest only period is extended by six months followed by 30 equal monthly payments of principal plus accrued interest. At the time of final payment, the Company is required to pay an exit fee of 7.5% of the original principal balance of each tranche, which will be $1.1 million for Tranche A. The Company recorded as a liability and debt discount the exit fee at the origination of the term loan. In addition, the Company incurred loan origination fees and debt issuance costs of $0.3 million which were recorded as a direct deduction from the carrying amount of the related debt liability as a debt discount. The Company is also required to pay a 5.0% success fee within 30 days following the FDA’s approval of the Company’s first product. This fee is enforceable within 10 years from the funding of Tranche A. In connection with the execution of the Loan Agreement, the Company agreed to certain customary representations and warranties.The loan is secured by the perfected first priority liens on the Company's assets, including a commitment by the Company to not allow any liens to be placed upon the Company’s intellectual property. The Oxford Loan includes customary events of default, including failure to pay amounts due, breaches of covenants and warranties, material adverse effect events, certain cross defaults and judgments, and insolvency. If the Company is unable to comply with these covenants or if the Company defaults on any portion of the outstanding borrowings, the lenders can also impose a 5.0% penalty and restrict access to additional borrowings under the loan and security agreement. The Company was in compliance with the terms under the Oxford Loan as of December 31, 2017 and 2016.The Company is permitted to make voluntary prepayments of the Oxford Loan with a prepayment fee, calculated as of the loan origination date, equal to (i) 3.0% of the loan prepaid during the first 12 months, (ii) 2.0% of the loan prepaid in months 13-24 and (iii) 1.0% of the loan prepaid thereafter. The Company is required to make mandatory prepayments of the outstanding loan upon the acceleration by lender following the occurrence of an event of default, along with a payment of the final payment, the prepayment fee and any other obligations that are due and payable at the time of prepayment.The Company accounts for the amortization of the debt discount utilizing the effective interest method. The Company recorded interest expense of $1.1 million and $5,000 for the years ended December 31, 2017 and 2016, respectively. Long-term debt and unamortized discount balances are as follows (in thousands):As of December 31, 2017, future minimum payments of principal, exit fee and interest expense under the Oxford Loan were as follows (in thousands):9.Convertible Promissory Note and Warrant Purchase AgreementOn November 12, 2015, the Company entered into a convertible note and warrant purchase agreement (the “Note and Warrant Purchase Agreement”) with three lenders under which the Company issued the Notes for an aggregate principal amount of $6.0 million and the Warrants exercisable for shares of the Company’s equity securities at a purchase price of $0.11 per share, on a post-Merger and post-Reverse Stock Split basis. The terms of the Notes included a provision whereby the Notes would be automatically converted into equity securities from a qualified financing with proceeds of at least $25.0 million. The terms of the Warrants included a provision whereby the Warrants would be automatically exercised if the Company consummated a public offering including a reverse merger (“PO”). If the PO did not occur on or prior to February 28, 2016, the warrant coverage amount was equal to 17.5% of the outstanding principal balance of the Notes. The number of warrant shares into which the Warrants could be exercised was equal to the warrant coverage amount divided by the per share price of the equity securities sold in a qualified financing for an exercise price of $0.11 per share, on a post-Merger and post-Reverse Stock Split basis. The Warrants also include a provision whereby in the event of a PO which would result in the automatic exercise of the Warrants and the automatic conversion of the Notes, the exercise price of the warrants would be paid by cancelling any unpaid interest on the Notes.On November 18, 2015, the Company entered into the Subscription Agreement with the holders of the Notes and new investors for the sale of 2,304,430 shares of its common stock at purchase price of $17.14 per share for total gross proceeds of $39.5 million. The proceeds were comprised of approximately $6.0 million from the conversion of the Notes and approximately $33.5 million of cash.The Company allocated the aggregate proceeds from the issuance of the Notes first to the Warrants based on the warrants’ fair value and then the residual proceeds were allocated to the debt obligation. As of December 31, 2015, the fair value of warrants of $0.9 million was recorded as a debt discount to be amortized as interest expense over the term of the Note using the effective interest rate method. The fair value of the Warrants was also recorded as a corresponding warrant liability.In addition, the Company incurred debt issuance costs of $21,000 in connection with the Note and Warrant Purchase Agreement. The debt issuance costs were being amortized to interest expense over the term of the Note using the effective interest rate method.Upon the closing of the Private Placement on March 22, 2016, immediately prior to the closing of the Merger, the outstanding balance of the Notes totaling approximately $6.0 million was converted into 350,040 shares of the Company’s common stock. The Warrants were exercised for 61,254 shares of the Company’s common stock. During the year ended December 31, 2016, the Company recognized a loss related to the change in fair value of the Warrants of $0.2 million. The warrant liability was reclassified to common stock and additional paid-in capital within stockholders’ equity (deficit), upon the exercise of the Warrants and issuance of shares on March 22, 2016.For the year ended December 31, 2016, the Company recognized $0.7 million related to the accrued interest and amortization of the debt discount within interest expense on the Company’s consolidated statements of operations. The discount was fully amortized upon the conversion of the Notes.10.Sale of Assets(“Theragene”)(Theragene) entered into an asset purchase agreement (“Theragene APA”)(Theragene APA), whereby the Company sold all of the assets related to MYDICAR including any related intellectual property for a cash payment of $0.2 million and 475,000 shares of common stock of Theragene. At any time after the Theragene APA execution date and until Theragene has received cumulative gross proceeds of $4.0 million (“Proceeds Date”)(Proceeds Date) from all equity financing transactions occurring after the Theragene APA execution date, if Theragene issues additional shares of its common stock without consideration or for a consideration per share less than $6.00 per share then Theragene will issue additional shares of its common stock to the Company concurrently with such issue, in an amount such that the per share consideration multiplied by the aggregate number of common stock shares issued to the Company will equal $2.85$2.9 million. Additionally, the Company may exercise a put option at any time after the Proceeds Date, where upon written notice from the Company, Theragene will repurchase the 225,000 shares of its common stock held by the Company (“Option Shares”)(Option Shares) at an aggregate purchase price equal to the greater of $1.35$1.4 million or the aggregate fair market value of the Option Shares as of the date of the receipt of such notice. The Company is also eligible to receive contingent consideration up to a maximum $45.0 million in cash, based upon Theragene achieving certain specified future milestones. In addition, the Company is also eligible to receive up to 8% royalties on net sales of any future Theragene products covered by or involving the related patents or know-how until the 20thtwentieth anniversary of the Theragene APA.During the year ended December 31, 2017, the Company received a cash payment of $0.2 million, which was recognized as other income (expense), net in the consolidated statement of operations. anthe owner of 475,000 shares of common stock of Theragene and held a put option for 225,000 shares of common stock of Theragene, which were recognized as a cost method investment with carrying value of zero.December 31, 2022 2021 Face value of debt $ 40,531 $ 30,000 Exit fee 2,600 3,277 Unamortized debt discount associated with exit fee, debt issuance costs and loan origination fees (3,506) (1,482) Total debt, net 39,625 31,795 Less: current portion of debt — (7,809) Debt, net $ 39,625 $ 23,986 2017, there was no change in2022, future minimum payments of principal, exit fee and interest expense under the fair value of our equity investment in Theragene.Innovatus Loan were as follows (in thousands):11.Stockholders’ EquityYear ending December 31, 2023 $ 3,686 2024 3,780 2025 4,407 2026 4,814 2027 48,185 Total future payments 64,872 Less: unamortized interest (21,741) Less: exit fee (3,506) Face value of term loan $ 39,625 2017,2022, no dividends had been declared by the Board of Directors.
In December 2020, the Company filed a shelf registration statement on Form S-3 (File No. 333-251497) with the Securities and Exchange Commission, which permits the offering, issuance and sale by the Company up to a maximum aggregate offering price of $200.0 million of its common stock, preferred stock, debt securities and warrants. Up to a maximum of $50.0 million of the maximum aggregate offering price of $200.0 million may be issued and sold pursuant to an ATM financing facility (the 2020 ATM Facility) under a sales agreement with Jefferies. In December 2021, the Company completed ATM offerings for a total of 565,938 shares of common stock under the 2020 ATM Facility. The offerings were made under the Company’s effective shelf registration statement and resulted in net proceeds to the Company of $3.0 million, after deducting commissions. In January and March 2022, the Company completed ATM offerings for a total of 5,841,786 shares of common stock under the 2020 ATM Facility. The offerings were made under the Company’sDecember 31, 2022 2021 Options issued and outstanding 6,143,183 5,262,185 Options available for future grants 1,976,460 1,327,645 Restricted and performance stock units outstanding Restricted and performance stock units outstanding 641,407 623,000 Shares available for issuance under ESPP Shares available for issuance under ESPP 788,057 671,172 Total 9,549,107 7,884,002 WarrantsThe Company issued the Warrants in connection with the issuance of the Notes (see Note 9). As of December 31, 2015, the Company accounted for the Warrants as a liability at fair value as the number of shares were not fixed and determinable at the issuance date. The Company adjusted the liability for changes in fair value until the exercise of the Warrants in March 2016, when the number of shares to be exercised became fixed, and the Warrants were automatically exercised into common stock. The warrant liability was immediately reclassified to common stock and additional paid in capital within stockholders’ equity (deficit). The change in fair value of the warrant liability was recognized as a component of other income (expense), net in the consolidated statements of operations.The Company assumed from Celladon fully exercisable warrants outstanding for the purchase of 10,180 shares of common stock. The warrants have an exercise price of $84.15 per share and expire in October 2018.12.Stock Option PlanIn 2009, the Company adopted the 2009 or the Plan. Under the Plan, shares ofcommon stock have been reserved for the issuance of stock options to employees, directors, and consultants under terms and provisions established by the Board of Directors.Directors adopted and in August 2016 the Company’s stockholders approved the Amended and Restated 2013 Equity Incentive Plan (the Restated 2013 Plan). Under the terms of the Restated 2013 Plan, the Company may grant stock options, stock appreciation rights, restricted stock, restricted stock units and other awards to individuals who are then employees, officers, non-employee directors or consultants of the Company. Under the terms of the Restated 2013 Plan, options may be granted at an exercise price not less than fair market value. For employees holding more than 10% of the voting rights of all classes of stock, the exercise prices for incentive and non-statutory stock options may not be less than 110% of fair market value, as determined by the Board of Directors. The terms of options granted under the Restated 2013 Plan may not exceed ten years. The vesting schedule of newly issued option grants is generally four years.As discussed in Note 5, the Company assumed all of the outstanding options, whether or not vested, under the Eiger Plan, with such options henceforth representing the right to purchase a number of shares of the Company’s common stock equal to approximately 0.09 multiplied by the number of shares of Eiger common stock previously represented by such options. For accounting purposes, however,December 31, 2022, the Company is deemedauthorized to have assumedissue up to 8,127,807 shares under the CelladonRestated 2013 Equity Incentive Plan.Becauseis consideredapproved the 2021 Inducement Plan to reserve 850,000 shares of its common stock to be the acquirerused exclusively for accounting purposes, the pre-Merger vested stock options granted by Celladon are deemedgrants of awards to have been exchanged for equity awardsindividuals that were not previously employees or directors of the Company and, as a material inducement to such individuals’ entry into employment with the portionCompany within the meaning of Rule 5635(c)(4) of the acquisition date fair valueNasdaq Listing Rules. As of these equity awards attributableDecember 31, 2022, there were 380,000 shares remaining available to pre-Merger service to Celladon were accounted for as a component ofbe issued under the consideration transferred, which was inconsequential to the consolidated financial statements.The exchange of options to purchase shares of Eiger common stock for options to purchase shares of the Combined Company, was accounted for as a modification of the awards because the legal exchange of the awards is considered a modification of Eiger stock options. The modification of the stock options did not result in any incremental compensation expense as the modification did not increase the fair value of the stock options.In June 2016, the Company’s board of directors adopted and in August 2016 the Company’s stockholders approved the amended and restated 2013 Equity Incentive Plan (the “Restated 2013 Plan”), which increased the number of shares reserved for grant by 1,296,683 shares. Under the terms of the Restated 2013 Plan, the Company may grant stock options, stock appreciation rights, restricted stock, restricted stock units and other awards to individuals who are then employees, officers, non-employee directors or consultants of the Company. All awards granted prior to the approval of the Restated 2013 Plan remain subject to the terms of the previous plans and the applicable award agreements. planplans during the yearsyear ended December 31, 2017 and 20162022 (in thousands, except shareoption and per share data):Shares
Available for
GrantsNumber of
OptionsWeighted-
Average
Exercise
PriceWeighted-
Average
Remaining
Contractual
Life
(in Years)Aggregate
Intrinsic
ValueOutstanding as of December 31, 2021 1,327,645 5,262,185 $ 10.24 7.25 $ 530 Additional options authorized 1,728,441 Granted (2,061,100) 2,061,100 $ 5.74 Restricted stock units granted (298,150) — Performance stock units granted (30,000) — Exercised (47,367) $ 4.93 Forfeited 1,132,735 (1,132,735) $ 9.03 Restricted stock units forfeited 126,889 — Performance stock units cancelled 50,000 — Outstanding as of December 31, 2022 1,976,460 6,143,183 $ 9.00 6.00 $ — Vested and exercisable as of December 31, 2022 3,726,809 $ 10.30 4.50 $ — $13.95$1.18 as of December 31, 2017.2017, 20162022, 2021 and 20152020 was $0.1 million, $0.1 million and $0.9$0.3 million, respectively.Stock Options Granted to Employees2017, 20162022, 2021 and 2015,2020, the Company granted employees stock options for the purchase of 579,700, 902,028 and 166,793 shares, respectively, with a weighted-average grant date fair value of $7.66, $10.40options granted were $3.65, $5.58 and $1.09$4.78 per share, respectively. The total grant date fair value of employee options that vested during the years ended December 31, 2017, 20162022, 2021 and 20152010 was $5.0$7.3 million, $2.0$6.9 million and $6,000,$5.6 million, respectively. to employees by estimating the fair value of stock-based awards using the Black-Scholes option pricing model and amortizingamortizes the fair value of the stock-based awards granted over the applicable vesting period of the awards on a straight-line basis. The fair value of employee stock options was estimated using the following weighted-average assumptions:Year Ended December 31, 2022 2021 2020 Expected term (in years) 5.27-6.08 5.27-6.08 5.00-6.08 Contractual term (in years) Contractual term (in years) — — 10.00 Volatility 68.71%-88.51% 68.96%-71.40% 73.00%-77.00% Risk free interest rate 1.76%-4.23% 0.62%-1.35% 0.39%-1.37% Dividend yield — — — Each of these inputs is subjective and generally requires judgment to determine.Fair Value of Common Stock: Prior to the Merger, the fair value of the shares of common stock underlying stock options was determined by the Company’s Board of Directors. In order to determine the fair value of the common stock at the time of grant of the option, the Board of Directors considered, among other things, valuations performed by an independent third-party. Because there was no public market for the Company’scommon stock, the Board of Directors exercised reasonable judgment and considered a number of objective and subjective factors to determine the best estimate of the fair value of the Company’s common stock, including important developments in the Company’s operations, sales of convertible preferred stock, actual operating results and financial performance, the conditions in the life sciences industry and the economy in general, the stock price performance and volatility of comparable public companies, and the lack of liquidity of the Company’s common stock, among other factors. Following the Merger, the Company’s Board of Directors determined the fair value of each share of underlying common stock based on the closing price of the Company’s common stock as reported on the date of grant. Company’s expected term represents the periodnumber of years the Company estimates, based primarily on historical experience, that the Company’s stock-based awards are expected tooptions will be outstanding and is determined using the simplified method (based on the mid-point between the vesting date and the end of the contractual term for employee options).Since the Company does not have a long trading history for its common stock, theThe expected volatility was estimatedfor stock options is based on the average volatility for comparable publicly traded biotechnology companies over a period equal to the expected term of theCompany's historical stock option grants. The comparable companies were chosen based on their similar size, or stage in the life cycle.Options GrantedPurchase PlanNon-EmployeesCompany grantsnumber of shares of common stock optionsinitially reserved for issuance under the 2013 ESPP was 100,000 shares with an automatic annual increase to non-employees in exchangethe shares issuable under the 2013 ESPP to the lower of (i) 1% of the total number of shares of common stock outstanding on December 31 of the preceding calendar year, and (ii) 165,000 shares of common stock, unless a lower number determined by the board of directors. As of December 31, 2022, a total of 948,831 shares are reserved for services rendered.issuance and 788,057 shares available for future issuance under the 2013 ESPP. During the years ended December 31, 2017, 20162022, 2021 and 2015,2020, employees purchased 48,115 shares for $0.2 million, 37,619 shares for $0.3 million and 25,645 shares for $0.2 million, respectively, under the 2013 ESPP.to non-employees stock options for 72,500, 48,54430,000 and 46,134 shares,270,000 PSUs, with a weighted-average grant date fair value of $6.87 and $7.92 per share, respectively. Stock-basedThe performance-based metrics include the achievement of certain revenue targets and clinical and regulatory milestones. During the year ended December 31, 2022 and 2021, the Company recorded $0.5 million and $0.2 million of stock-based compensation expense related to stock options granted to non-employeesthe PSUs as it is recognized asexpected that all milestones will be achieved by the stock options are earnedtarget dates, respectively. This expense is included in selling, general and will fluctuate as the estimated fair value of the common stock fluctuates until the awards vest. The Company believes that the estimated fair value of the stock options is more readily measurable than the fair value of the services rendered.The fair value of the stock options granted to non-employees is estimated at each reporting date using the Black-Scholes option-pricing model using similar assumptions as for employees except that the expected term is based on the options’ remaining contractual term instead of the simplified method:Stock-Based Compensation ExpenseTotal stock-based compensation expense recognized for options granted to employees and non-employees was as follows (in thousands):2017,2022, no PSUs have vested as the performance-based metrics of the PSUs have not yet been achieved. During the year ended December 31, 2022 and 2021, the Company granted 298,150 and 371,500 RSUs, respectively, with a weighted-average grant date fair value of $5.22 and $9.16 per share, respectively.employeeRSUs and PSUs was $3.0 million, which the Company expects to recognize over an estimated weighted-average period of 2.0 years.Shares Weighted-
Average
Grant Date
Fair ValueUnvested shares as of December 31, 2021 623,000 $ 8.63 Granted 328,150 $ 5.37 Vested (132,854) $ 9.40 Forfeited (176,889) $ 7.63 Unvested shares as of December 31, 2022 641,407 $ 7.08 Year Ended December 31, 2022 2021 2020 Research and development $ 3,159 $ 2,252 $ 1,494 Selling, general and administrative 5,158 5,649 4,479 Total $ 8,317 $ 7,901 $ 5,973 $6.8$10.1 million, which the Company expects to recognize over an estimated weighted average period of 2.72.70 years.13.2017, 2016 and 2015.2020. The Company has incurred net operating losses for alleffective tax rate in each period differs from the periods presented. The Company has not reflected any benefit of such net operating loss carryforwards inU.S. statutory tax rate primarily due to the accompanying consolidated financial statements. The Company has established a full valuation allowance against itsallowances on the Company’s deferred tax assets dueas it is more likely than not that some or all of the Company’s deferred tax assets will not be realized. The tax expense recorded for the year ended December 31, 2022 relates to the uncertainty surrounding the realization of such assets.Year Ended December 31, 2022 2021 2020 Federal statutory income tax rate 21.00 % 21.00 % 21.00 % Change in valuation allowance Change in valuation allowance (25.99) (31.07) (26.75) Tax credits Tax credits 4.15 12.14 6.43 State income taxes, net of federal benefit 2.39 0.73 0.33 Stock-based compensation (0.98) (3.02) (1.00) Other, net (0.59) 0.03 (0.01) Provision for income taxes Provision for income taxes (0.02) % (0.19) % — % December 31, 2022 2021 Deferred tax assets: Net operating loss carryforwards $ 63,069 $ 60,273 Tax credits 9,985 5,486 Depreciation and amortization 2,920 1,863 Stock-based compensation Stock-based compensation 3,429 3,004 Section 174 Capitalization Section 174 Capitalization 15,581 — Accruals and reserves 1,431 646 Lease liabilities Lease liabilities 131 156 Gross deferred tax assets 96,546 71,428 Valuation allowance (96,418) (71,291) Total deferred tax assets Total deferred tax assets 128 137 Deferred tax liabilities: Deferred tax liabilities: Right-of-use assets Right-of-use assets (128) (137) Total deferred tax liabilities Total deferred tax liabilities (128) (137) Net deferred tax assets $ — $ — 20172022 and 2016.2021. The net change in the valuation allowance increased by $3.4 million and $17.3 million duringfor the years ended December 31, 20172022 and 2016,2021 increased by $25.1 million and $10.5 million, respectively.2017,2022, the Company had approximately $88.4$292.3 million and $25.5$25.4 million, respectively, of federal and state operating loss carryforwards available to reduce future taxable income that will begin to expire in 2030 and 2028, respectively, for federal and state tax purposes.2017,2022, the Company also had research and development tax credit carryforwards of approximately $0.2$0 million and $0.9$3.1 million for federal and state purposes available to offset future taxable income tax, respectively. If not utilized, the federal carryforwards will expire in various amounts beginning in 2028, and the state credits can be carried forward indefinitely.As of December 31, 2017,had orphan drug tax credit carryforwards of approximately $11.8 million for federal purposes available to offset future taxable income tax. If not utilized, the federal carryforwards will begin to expire in 2033 .Utilization of the NOL and tax credit carryforwards may be subject to a substantial annual limitation due tohas experienced an ownership change limitations that have occurred or that could occur in future, as required byunder Section 382 of the Internal Revenue Code (the Code) of 1986, as well as similar stateamended. The Company’s merger with Celladon resulted in such an ownership change and, foreign provisions. These ownership changes may limit the amount ofaccordingly, Celladon’s NOL carryforwards and certain other tax credit carryforwards that canattributes will be utilized annuallysubject to offset future taxable income and tax, respectively.further limitations on their use. The Company assessed whether it had an ownership change, as defined by Section 382 of the Code occurred from its formation through December 31, 2017.2021. Based upon this assessment, no reduction wasthe Company experienced ownership changes on April 20, 2016, October 18, 2018 and December 31, 2020. Due to the April 20, 2016 and October 18, 2018 ownership changes, reductions were made to the federalCompany’s NOL and state NOL carryforwards or federal and state tax credit carryforwards under these rules.2017, 20162022, 2021 and 20152020 is as follows (in thousands):Year Ended December 31, 2022 2021 2020 Balance at beginning of year $ 1,950 $ 451 $ 3,277 Additions based on tax positions related to prior year — — — Additions based on tax positions related to current year 1,586 1,499 81 Reductions based on tax positions related to prior year Reductions based on tax positions related to prior year (44) — (2,907) Reductions based on tax positions related to current year Reductions based on tax positions related to current year — — — Balance at end of year $ 3,492 $ 1,950 $ 451 $3.2$3.5 million of unrecognized tax benefits is recognized, there would not be an effect on the effective tax rate. The Company does not expect the unrecognized tax benefits to change significantly over the next 12 months. AtAs of December 31, 2017,2022, the unrecognized tax benefits for uncertain tax positions were offset against deferred tax assets and would not affect the income tax rate if recognized due to the Company being in a full valuation allowance position.statementconsolidated statements of operations. The Company files income tax returns in the U.S. federal and state jurisdictions. All periods since inception are subject to examination by U.S. federal and state jurisdictions. There were no such interest or penalties during the years ended December 31, 2017, 20162022, 2021 and 2015.2020.14.Legal MattersIn July 2015, following Celladon’s announcementsnegative CUPID 2 dataCompany's patent or other intellectual property rights and contractual rights. The Company is defending all current litigation matters, including the litigation matters discussed below. Although there can be no assurances and the suspensionoutcome of further research and development activitiesthese matters is currently not determinable, the Company currently believes that none of these claims or proceedings are likely to have a material adverse effect on the Company's financial position.subsequent declinesamount of loss can be reasonably estimated. The Company evaluated developments in legal proceedings, claims or investigations that could affect the priceamount of its common stock, threeany accrual, as well as any developments that would result in a loss contingency to become both probable and reasonably estimable. The Company has not recorded any accrual for loss contingencies associated with such legal claims or litigation discussed herein as they are in the early stages and an estimate of loss, if any, cannot be made.actions wereaction complaint was filed in the U.S.United States District Court for the SouthernNorthern District of California against Celladon and certain of its current and former officers. The complaints generally allegedalleging that the defendantscompany and two former executives violated Sections 10(b) and 20(a) of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), by making materially false and misleading statementsSEC Rule 10b-5. The complaint alleges generally that between March 2021 and October 2022 material misstatements and omissions were made to shareholders regarding the clinical trial programTOGETHER study of peginterferon lambda for MYDICAR, thereby artificially inflating the pricetreatment of Celladon’s common stock.COVID-19 as well as the likelihood of FDA approval of an Emergency Use Authorization for peginterferon lambda. The complaints sought unspecified monetary damages and other relief, including attorneys’ fees. On December 9, 2015, the district court consolidated the three putative securities class actions andCourt appointed a lead plaintiff on March 2, 2023. The litigation is currently ongoing.representsupply quantities of a drug as requested by PRF. PRF also has a claim for declaratory relief regarding the putative class. The lead plaintiffgrant of licenses under the PRF Collaboration Agreement. On January 18, 2023, the Company filed a consolidated amended complaint on February 29, 2016.On October 7, 2016, the district court granted defendants’ motion to dismiss the consolidated amended complaint and granted leave to amend within 60 days from the date of the district court’s order. The lead plaintiff subsequently filed a notice of intent not to amend the consolidated amended complaint and instead indicated that it intended to appeal the district court’s decision. On December 9, 2016, the district court closed the case.On December 28, 2016, the lead plaintiff filed a notice to the United States Court of Appeals for the Ninth Circuit appealing the district court’s order dismissing the consolidated amended complaint. On May 5, 2017, the lead plaintiff filed his opening brief. On July 5, 2017, the defendants filed their appellate brief response. The Plaintiff subsequently filed their response to the Company’s July 5, 2017 filing on August 19, 2017. Upon these filings,Demand denying PRF’s claims, contesting the next step inarbitrability of PRF’s claim for declaratory relief, and asserting a counterclaim for declaratory relief related to the process would becontractual provision underlying PRF’s original drug supply claim. To give the parties an opportunity to wait for oral arguments to be scheduled bydiscuss a potential negotiated resolution of their dispute, the 9th Circuit Court of Appeals.It is possible that additional suits will be filed, or allegations made by stockholders, with respect to these same or other matters and also naming the Company and/or Celladon’s former officers and directors as defendants. The Company believes that itarbitration has meritorious defenses and intends to defend these lawsuits vigorously. The Company is not able to predict or reasonably estimate the ultimate outcome or possible losses relating to these claims.15.Commitments and ContingenciesLease AgreementIn March 2015, the Company entered into a non-cancelable facility lease agreement for an office facility in Palo Alto, California. The lease commenced on April 1, 2015 and expires 36 months after the commencement date. The lease has one two-year renewal option prior to expiration and includes rent escalation clausesbeen suspended through the lease term. Scheduled rent increasesend of 2023. As a result, all arbitration activities are recognized as deferred rentnow on hold, and are amortized on a straight-line basis over the term of the lease. The Companyfinal hearing, originally scheduled for May 9 – 12, 2023, in Boston, Massachusetts, has provided a security deposit of $21,000 as collateral for the lease.In October 2016, the Palo Alto lease was modified to include two additional suites, bringing the total leased space to 3,877 square feet. The lease commenced on January 4, 2017been taken off calendar.expires in March 2018. The lease has one two-year renewal option prior to expiration and includes rent escalation clauses through the lease term. The security deposit was increased to $49,000, which is included in other assets in the Company’s consolidated balance sheet as of December 31, 2017 and 2016.In March 2017, the Company entered into a non-cancelable facility lease agreement for an office facility in Palo Alto, California. The lease commenced on April 1, 2017 and expires 12 months after the commencement date. The lease has one twelve-month renewal option prior to expiration and includes rent escalation clauses through the lease term. In April 2017, the Company provided a security deposit of $27,000 as collateral for the lease, which is included in other assets in the Company’s consolidated balance sheet as of December 31, 2017.21712155 Park BlvdBlvd. in Palo Alto, California 94306.California. The lease commencescommenced on March 1, 2018 and expires five years after the commencement date.was to expire in February 2023. The lease has onehad a three-year renewal option prior to expiration and includesexpiration. The lease included rent escalation clauses throughthroughout the lease term. In October 2017, the Company provided a security deposit of $0.3 million, which ismillion. In February 2023, the Company amended the lease to extend the lease by one year with a one year renewal option. The extended lease will commence on March 1, 2023 and expire on February 28, 2024. The Company accounted for the amendment as a lease modification in accordance with ASC Topic 842. The Company also has additional operating leases that are included in other assets inits lease accounting but are not considered significant for disclosure.consolidated balance sheetoperating lease liabilities as of December 31, 2017.Future aggregate minimum lease payments under the non-cancelable operating leases are2022 were as follows (in thousands):Undiscounted lease payments December 31, 2022 2023 528 2024 84 2025 1 Total undiscounted payments 613 Less: imputed interest 39 Present value of future lease payments 574 Less: current portion of operating lease liabilities 491 Operating lease liabilities $ 83 $0.3 millionfor the years ended December 31, 2022, 2021 and 2020, respectively. Under the terms of the lease agreements, the Company is also responsible for certain variable lease payments that are not included in the measurement of the lease liability. Variable lease payments for the operating leases were $0.1 million for the years ended December 31, 2017, 20162022, 2021 and 2015,2020, respectively.
The operating cash outflows for the operating lease liabilities were $0.7 million, $0.6 million and $0.7 million for the years ended December 31, 2022, 2021 and 2020, respectively. As of December 31, 2022, the weighted-average remaining lease term and weighted-average discount rate were 1.2 years and 12.82%, respectively.The Company has not included these potential payment obligations in the table above asHowever, the amount and timing of suchthese payments are not known.
Manufacturing Service Agreement16.Related Party Transactions7,6, the Company entered into license agreements with EGI, which is owned by the founder of the Company.As disclosed in Note 7, in 2015an asset purchasea separation agreement and general release with Eiccose, for whichDavid Cory, the Company’s chief executive officer was the sole managing memberCompany's former President and significant equity interest holder. In June 2017, Eiccose was disbanded. The Company’s commercial milestone obligations and sales royalties remain, however have been transferredChief Executive Officer. Pursuant to the previous shareholdersseparation agreement, Mr. Cory will be entitled to receive, (i) $1.5 million continuation of Eiccose.his base salary and target bonus for a period of 18 months; (ii) $0.4 million cash payment equal to Mr. Cory's target bonus for 2022; (iii) reimbursement for COBRA continuation coverage for Mr. Cory and his eligible dependents for a period of 18 months; (iv) accelerated vesting of 50% of Mr. Cory's unvested stock options, performance-based restricted stock units, and time-based RSUs outstanding as of the separation date; (v) reimbursement of Mr. Cory's legal fees associated with the negotiation of the Separation Agreement up to $10,000. The Company accrued $2.0 million related to David Cory's separation for salary and benefits as of December 31, 2022. Expense related to stock options acceleration will be recorded in Q1 2023.
Silicon Valley Bank ClosureAccountantsAccountants on Accounting and Financial DisclosureNone2017,2022, the end of the period covered by this report.2017,2022, our management assessed the effectiveness of our internal control over financial reporting using the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission in Internal Control-Integrated Framework (2013 Framework). Based on this assessment, our management concluded that, as of December 31, 2017,2022, our internal control over financial reporting was effective based on those criteria.Pursuant to Regulation S-K 308(b), this Annual Report on Form 10-K does not include an attestation report of our company’s registered public accounting firm regarding internal control over financial reporting.Except as otherwise described above under “Management’s Report on Internal Control over Financial Reporting”, there material changes in our internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act) that occurred during the fiscal quarteryear ended December 31, 2017,2022, that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting. III III. OTHER INFORMATION20182022 Annual Meeting of Stockholders within 120 days after the end of the fiscal year ended December 31, 2017.NASDAQNasdaq Global Market, by filing a Current Report on Form 8-K with the SEC, disclosing such information.20182023 Annual Meeting of Stockholders within 120 days after the end of the fiscal year ended December 31, 2017.2022.20182023 Annual Meeting of Stockholders within 120 days after the end of the fiscal year ended December 31, 2017.2022.20182023 Annual Meeting of Stockholders within 120 days after the end of the fiscal year ended December 31, 2017.2022.AccountingAccountant Fees and Services20182023 Annual Meeting of Stockholders within 120 days after the end of the fiscal year ended December 31, 2017.2022.(a)Financial Statements and Financial Statement Schedules
(a)Financial Statements and Financial Statement Schedules1.Financial Statements
2.Financial Statement Schedules2.Financial Statement Schedules3.Exhibits
Number10.1+10.1+ Exhibit
NumberDescription of Document10.4+ 10.510.6+ 10.6+ 10.9+ 10.10+ 10.11+ 10.12+ 10.13+ 10.14† 10.15†10.10* 10.16†Exhibit
NumberDescription of Document 10.18† 10.19† 10.20† 10.21† 10.22 10.23† 10.24 10.25 10.26 10.27 10.2810.17* 10.18* 10.19 10.20 10.21 21.110.23+ 10.24+ 10.25+ 10.26* 10.27* 10.28+ 10.29+ 10.30* 10.31 10.32 10.33 10.34+ 10.35+ 10.36+ 10.37 10.38** 10.39* 10.40 10.41**+ 10.42**+ 10.43** 21.1** 23.1 31.1Exhibit
NumberDescription of Document31.2** 32.1101.INSDocument101.SCH101.CAL101.DEF101.LAB101.PRE104 The cover page from the Company’s Annual Report on Form 10-K for the year ended December 31, 2022, has been formatted in Inline XBRL and is contained in Exhibit 101. †Confidential treatment has been granted with respect to certain portionsexhibit. Omitted portionsexhibit have been filed separately with the SEC.omitted as being immaterial and would be competitively harmful if publicly disclosed.
** Filed herewith.Eiger BioPharmaceuticals, Inc. 9 March 2018By: By:A. CoryA. Cory9 March 2018By: By:James WelchJames WelchChief Financial OfficerCoryApelian and James Welch,Michelle Maynard, and each of them, as his or her attorneys-in-fact, with the power of substitution, for him or her in any and all capacities, to sign any amendments to this report, and to file the same, with exhibits thereto and other documents in connection therewith with the Securities and Exchange Commission, hereby ratifying and confirming all that said attorneys-in-fact, and each of them, or his or her substitute or substitutes may do or cause to be done by virtue hereof.SignatureTitleDateSignature Title Date /s/ David A. CoryDavid Cory9 March 2018David Apelian /s/ James WelchJames WelchChief Financial Officer9 March 2018Michelle Maynard /s/ Thomas J. Dietz Thomas J. Dietz9 March 2018Thomas J. Dietz /s/ David ApelianDavid Apelian9 March 2018Evan Loh /s/ Evan LohEvan Loh9 March 2018Jeffrey Glenn /s/ Jeffrey GlennJeffrey Glenn9 March 2018Christine Murray /s/ Eldon MayerAmit K. Sachdev9 March 2018Eldon Mayer/s/ Kim Sablich Member of the Board of Directors March 16, 2023 Kim Sablich /s/ Lisa Kelly-Croswell Member of the Board of Directors March 16, 2023 Lisa Kelly-Croswell 116