| | | Large accelerated filer | ☐ | | Accelerated filer | ☐ | Non-accelerated filer | ☒ | | Smaller reporting company | ☒ | | | | Emerging growth company | ☐ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐ Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. Yes ☐ No ☒ Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ☐ No ☒ The aggregate market value of voting and non-voting common equity held by non-affiliates, based upon the closing price of the registrant’s common stock on the NASDAQ Global Market on June 30, 2020,2022, was approximately $280.6$537.9 million. As of February 23, 2021,24, 2023, there were 37,150,37649,278,861 shares of the registrant’s common stock, $0.0001 par value per share, outstanding.
ALTIMMUNE, INC. ANNUAL REPORT ON FORM 10-K TABLE OF CONTENTS
Forward-looking statements This Annual Report on Form 10-K for the year ended December 31, 20202022 (this “Annual Report”) contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended (the “Securities Act”), Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), and the Private Securities Litigation Reform Act of 1995. Written or oral statements that constitute forward-looking statements may be made by us or on our behalf. Words such as “expect,” “anticipate,” “intend,” “plan,” “believe,” “estimate,” “may,” “will,” “should,” “could,” “target,” “strategy,” “intend,” “project,” “guidance,” “likely,” “usually,” “potential,” or the negative of these words or variations of such words, similar expressions, or comparable terminology are intended to identify such forward-looking statements, although not all forward-looking statements contain these identifying words. There are a number of important risks and uncertainties that could cause our actual results to differ materially from those indicated by forward-looking statements. We may not actually achieve the plans, intentions or expectations disclosed in our forward-looking statements, and you should not place undue reliance on our forward-looking statements. Actual results or events could differ materially from the plans, intentions and expectations disclosed in the forward-looking statements we make. These forward-looking statements are based on current expectations, estimates, forecasts, and projections about the industry and markets in which we operate, and management’s beliefs and assumptions. These statements are not guarantees of future performance and involve certain risks, uncertainties, and assumptions that are difficult to predict and may cause our actual results, performance or achievements to be materially different from future results, performance or achievements expressed or implied by any forward-looking statements. These risks, uncertainties and other factors include, but are not limited to, risks associated with the following: | ● | our ability to develop and commercialize our current and future product candidates; |
| ● | our ability to expand our pipeline of product candidates and the success of future product candidate advancements, including the success of future preclinical and clinical trials, and our ability to commercialize our products; |
| ● | the reliability of the results of the studies relating to human safety and possible adverse effects resulting from the administration of our product candidates; |
| ● | our ability to obtain potential regulatory approvals on the timelines anticipated, or at all; |
| ● | our ability to obtain additional patents or extend existing patents on the timelines anticipated, or at all; |
| ● | our ability to identify and consummate potential future strategic partnerships or business combinations; |
| ● | our expectations regarding the potential market size and the size of the patient populations for our product candidates, if approved for commercial use; |
| ● | our anticipated financial or operational results; |
| ● | our ability to obtain additional capital resources; |
| ● | risks related to the direct or indirect impact of the COVID-19 pandemic and the conflict in Ukraineon the global economy, including causing or contributing to global supply chain disruption, price fluctuations, including increased costs for raw materials, and other significant economic effects; |
| ● | breaches of data privacy, or disruptions in our information technology systems; |
| ● | our ability to continue to satisfy the listing requirements of the NASDAQ Global Market; and |
| ● | risks detailed under the caption “Risk Factors” in this Annual Report and in our other reports filed with the U.S. Securities and Exchange Commission (“SEC”), from time to time hereafter. |
our ability to expand our pipeline of product candidates and the success of future product candidate advancements, including the success of future preclinical and clinical trials, and our ability to commercialize our products;
funding delays, reductions in or elimination of U.S. government funding and/or non-renewal of expiring funding under our agreement with the Biomedical Advanced Research and Development Authority (“BARDA”);
our ability to satisfy certain technical milestones under our contracts with BARDA that would entitle us to receive additional funding over the period of the agreement;
delays caused by third parties challenging government contracts awarded to us;
the receipt of future potential payments under government contracts or grants;
our ability to identify potential future government contracts or grant awards;
our ability to obtain potential regulatory approvals on the timelines anticipated, or at all;
our ability to obtain additional patents or extend existing patents on the timelines anticipated, or at all;
our ability to identify and consummate potential future strategic partnerships or business combinations;
our anticipated financial or operational results;
our ability to obtain additional capital resources;
breaches of data privacy, or disruptions in our information technology systems;
our ability to continue to satisfy the listing requirements of the NASDAQ Global Market; and
risks detailed under the caption “Risk Factors” in this Annual Report and in our other reports filed with the U.S. Securities and Exchange Commission (“SEC”), from time to time hereafter.
We have based the forward-looking statements included in this Annual Report on information available to us on the date of this annual report. Except as required by law we undertake no obligation to revise or update any forward-looking statements, whether as a result of new information, future events or otherwise. You are advised to consult any additional disclosures that we may make in reports that we, in the future, may file with the SEC, including annual reports on Form 10-K, quarterly reports on Form 10-Q and current reports on Form 8-K. All forward-looking statements included herein are expressly qualified in their entirety by the foregoing cautionary statements. Unless otherwise indicated, the information in this Annual Report is as of December 31, 2020.2022.
Note regarding trademarks “Altimmune,” our logo and other trademarks, trade names or service marks of the Company appearing in this Annual Report, including, AdCOVID, NasoShield, NasoVAX, T-COVID, HepTcell, Densigen, and RespirVec., are the property of the Company. The other trademarks, trade names and service marks appearing in this Annual Report are the property of their respective owners. We do not intend our use or display of other companies’ trademarks, trade names or service marks to imply an endorsement or sponsorship of us by such companies, or any relationship with such companies. Solely for convenience, trademarks and trade names referred to in this Annual Report may appear without the ® or TM symbol. Summary of Risk Factors The risk factors detailed in Item 1A entitled “Risk Factors” in this Annual Report on Form 10-K are the risks that we believe are material to our investors and a reader should carefully consider them. Those risks are not all of the risks we face and other factors not presently known to us or that we currently believe are immaterial may also affect our business if they occur. The following is a summary of the risk factors detailed in Item 1A: Risks Related to Our Business, Financing Requirements, Product Development and Clinical Trials | ● | our ability to raise capital |
| ● | our history of operating losses since our founding and the anticipation that we will continue to incur significant losses for the foreseeable future and may never achieve or maintain profitability |
| ● | our ability to develop and commercialize our current and future product candidates |
| ● | delays in our clinical trials or the failure of our trials to demonstrate the safety and efficacy of our product candidates to the satisfaction of applicable regulatory authorities |
| ● | our ability to enroll patients in our clinical trials |
| ● | our ability to predict the time and cost of product development for our product candidates |
| ● | our reliance on third parties to conduct preclinical studies and clinical trials for our product candidates |
| ● | the ability and timeline to recruit patients for our clinical trials and for our third party contractors to conduct our clinical trials |
| ● | supply chain and labor shortage impacts on our contract manufacturers ability to manufacture our clinical materials and supplies according to the specified time line |
| ● | the ability of additional third party contractors to perform various services required in support of our clinical trials and nonclinical studies |
| ● | credit and financial market impacts |
| ● | global economic conditions and uncertainties |
our ability to raise capital
our history of operating losses since our founding and the anticipation that we will continue to incur significant losses for the foreseeable future and may never achieve or maintain profitability
our ability to develop and commercialize our current and future product candidates
our ability to predict the time and cost of product development for our product candidates
our reliance on third parties to conduct preclinical studies and clinical trials for our product candidates
delays in our clinical trials or the failure of our trials to demonstrate the safety and efficacy of our product candidates to the satisfaction of applicable regulatory authorities
our ability to enroll patients in our clinical trials
Risks Related to the Development of Our COVID-19 Vaccine and Therapeutic
our ability to receive emergency use authorization or approval for any of our COVID-19 product candidates a timely manner, if at all, and the potential that our COVID-19 product candidates may never be authorized for emergency use or approved
unforeseen or unexpected challenges in the regulatory pathway for T-COVID and AdCOVID
Risks Related to the Regulatory Approval Process | ● | our ability to obtain required regulatory approvals, including in non-U.S. jurisdictions |
| ● | the potential that our product candidates have undesirable side effects or have other properties that delay or prevent their regulatory approval or limit their commercial potential |
| ● | the expense of the marketing approval process and ongoing regulatory review if our product candidates ever received regulatory approval |
our ability to obtain required regulatory approvals, including in non-U.S. jurisdictions
the potential that our product candidates have undesirable side effects or have other properties that delay or prevent their regulatory approval or limit their commercial potential
the expense and restrictions of the marketing approval process and ongoing regulatory review if our product candidates ever received regulatory approval
Risks Related to Our Intellectual Property | ● | the cost and difficulty of protecting our proprietary rights and the potential that our intellectual property rights do not adequately protect our product candidates |
| ● | our ability to protect our intellectual property rights throughout the world |
our ability to protect our intellectual property rights throughout the world
| ● | the adequacy of our patent terms to protect our competitive position on our products for an adequate amount of time |
| ● | third-party claims of intellectual property infringement or misappropriation, including circumstances involving our employees, independent contractors or consultants |
third-party claims of intellectual property infringement or misappropriation, including circumstances involving our employees, independent contractors or consultants
Risks Related to Commercialization of the Company’s Product Candidates | ● | our ability to attain significant market acceptance of our product candidates, if approved, among physicians, patients, third-party payers and others in the medical community |
| ● | our reliance on third parties to manufacture our products in sufficient quantities, or at sufficient yields, or obtain regulatory approvals for a manufacturing facility for our products and if approved, in sufficient quantities to meet commercial demand |
| ● | our reliance on third parties to manufacture our product candidates and related materials for our clinical trials and preclinical studies |
| ● | the ability of our contract manufacturers to manufacture any such product to the specifications and the quantities that are needed along the timelines that are specified |
our ability to attain significant market acceptance of our product candidates, if approved, among physicians, patients, third-party payers and others in the medical community
our reliance on third parties to manufacture our product candidates and related materials for our clinical trials and preclinical studies
our ability to manufacture our products in sufficient quantities, or at sufficient yields, or obtain regulatory approvals for a manufacturing facility for our products
Risks Related to our BARDA Contract and Other Government Programs
our ability to move forward with the NasoShield program without the BARDA anthrax contract award
the activities for which we received reimbursement under our BARDA contracts, which are cost-plus-fixed-fee contracts
our reliance for further revenues from grants, contracts and loans from the U.S. and other governments, non-profit entities and academic institutions
Risks Related to Reimbursement and Government Regulation | ● | our ability to obtain coverage and reimbursement in certain market segments for our product candidates, if they are approved |
| ● | the imposition of price controls |
| ● | our ability to comply with multiple substantial federal and state health care and other laws, and the complexity of our regulatory compliance obligations |
| ● | the unknown impact of recent health care reform legislation and other changes in the health care industry and in health care spending |
our ability to obtain coverage and reimbursement in certain market segments for our product candidates, if they are approved
the imposition of price controls
our ability to comply with multiple substantial federal and state health care and other laws, and the complexity of our regulatory compliance obligations
the unknown impact of recent health care reform legislation and other changes in the health care industry and in health care spending
Risks Related to our Securities | ● | the volatility of the trading price of our common stock and substantial price fluctuations on heavy volume |
PART I PART IItem 1. Business Overview Altimmune, Inc. is a clinical stage biopharmaceutical company focused on developing intranasal vaccines, immune modulating therapies and treatments for obesity and liver disease.diseases. Our diverse pipeline includes proprietary intranasal vaccineslead product candidate, pemvidutide (formerly known as ALT-801), is a GLP-1/glucagon dual receptor agonist that is being developed for COVID-19 (AdCOVID), anthrax (NasoShield)the treatment of obesity and influenza (NasoVAX); an intranasal immune modulating therapeutic for COVID-19 (T-COVID); and next generation peptide therapeutics for non-alcoholic steatohepatitis (“NASH”) (ALT-801) and. In addition, we are developing HepTcell, an immunotherapeutic agent designed to achieve a functional cure for chronic hepatitis B (HepTcell). Our business is the result of a merger between PharmAthene, Inc. (“PharmAthene”) and the business previously known as Altimmune, Inc. (“Private Altimmune”). On May 4, 2017, Private Altimmune merged with PharmAthene in a series of mergers and reorganizations (collectively, the “Mergers”) pursuant to an Agreement and Plan of Merger and Reorganization (the “PharmAthene Merger Agreement”) dated January 18, 2017, among Private Altimmune, PharmAthene, its wholly owned acquisition subsidiaries Mustang Merger Sub Corp I Inc. and Mustang Merger Sub II LLC. Upon closing of the Mergers, all equity instruments of Private Altimmune were exchanged for corresponding equity instruments of PharmAthene. Prior to the Mergers, PharmAthene was a publicly traded biodefense company engaged in Phase 2 clinical trials.B. Except where the context indicates otherwise, references to “we,” “us,” “our,” “Altimmune” or the “Company” refer for periods prior to the completion of the Mergers, to Private Altimmune and its subsidiaries, and for periods following the completion of the Mergers to the combined company and its subsidiaries.
AdCOVID
AdCOVID is an intranasal COVID-19 vaccine candidate designed to guard the respiratory tract from viral invasion and to provide downstream protection against viral spread through stimulation of both mucosal and systemic antibodies (IgA and IgG) as well as cell-mediated immunity. By stimulating mucosal immunity in the nasal cavity, a key point of entry and replication for the SARS-CoV-2 virus, AdCOVID has the potential to defend against both the infection and spread of the virus to others. AdCOVID’s intranasal delivery method provides an easier route of administration than an injection and may eliminate the need for administration by trained medical personnel. We believe nasal mucosal immunity has the potential to provide an effective protection at the site of viral entry and early replication, and may block transmission by shed virus. We believe AdCOVID has the potential to meet most of the World Health Organization’s published preferred attributes for a COVID-19 vaccine, including single dose, rapid onset of protection, immunity lasting one year, non-injected and temperature stability. In particular, data from our NasoVAX clinical trials demonstrated a strong serological response at two weeks that remained unchanged at 400 days, after a single dose. In addition, since it is expected to have extended stability at room temperature, AdCOVID may avoid the need for costly cold chain logistics. During 2020, we completed initial preclinical mouse studies in collaboration with the University of Alabama at Birmingham (“UAB”), and began manufacturing AdCOVID during the third quarter of 2020. We submitted an Investigation New Drug application (“IND”) with the U.S. Food and Drug Administration (“FDA”) in the fourth quarter of 2020 and received clearance from the FDA in February 2021 to initiate a Phase 1 clinical trial of AdCOVID. The Phase 1 clinical trial is currently enrolling and will evaluate the safety and immunogenicity of AdCOVID in up to 180 healthy adult volunteers between the ages of 18 and 55, with data expected in the second quarter of 2021.
T-COVID
T-COVID is an intranasal immune modulating therapeutic candidate based on the same replication-deficient adenovirus 5 (“RD-Ad5”) vector technology behind our other intranasal vaccine candidates, but it acts through a different mechanism. In preclinical studies sponsored by the National Institute of Allergy and Infectious Diseases, intranasal administration of RD-Ad5 vectors modulated the innate immune response to lethal challenge with a respiratory virus in mice and protected them from death. The immunomodulatory effects resulted in significantly decreased cellular inflammation and lower concentrations of IL-6 and other inflammatory cytokines in the lungs of treated animals compared to controls. Excessive production of inflammatory cytokines like IL-6 has been associated with the lung pathology and death in COVID-19. The protective effects were independent of any specific immunity or vaccine effects against the challenge virus. These protective effects were only observed with intranasal administration of RD-Ad5, and intramuscular administration provided no survival benefit.
With the support of the U.S. Army Medical Research & Development Command in collaboration with the Medical Technology Enterprise Consortium, we initiated a placebo-controlled Phase 1/2 double-blind clinical trial to evaluate the potential of T-COVID to prevent clinical worsening in patients with early COVID-19 during 2020. The trial is expected to enroll 96 community-based patients who are 18 years and older that present with fever, cough, or shortness of breath, with onset of symptoms within 48 hours, and a diagnosis of COVID-19 within 24 hours. The trial consists of three cohorts of increasing age and risk for complications of COVID-19, and patients are randomized 1:1 to NasoVAX or placebo administered as a single 0.5ml nasal spray within 24 hours of diagnosis. The primary endpoint of the trial is the proportion of patients with clinical worsening, defined as a 4% decrease in pulse oxygen saturation, or the need for hospitalization. Secondary endpoints will measure the average decrease in resting pulse oxygen saturation, average
increase in resting pulse rate and proportion of patients requiring oxygen supplementation and mechanical ventilation. In order to expedite the study, we have been permitted by the FDA to use existing lots of NasoVAX, which is an identical vector to T-COVID, in lieu of newly manufactured T-COVID. The protocol was recently amended such that 40% of patients in the final cohort, in which efficacy will primarily be assessed, to be either 65 years and older or have risk for complications of COVID-19. Based on these protocol changes, we expect to receive results from the Phase 1/2 trial in the second quarter of 2021.
NasoShield
NasoShield is an anthrax vaccine product candidate designed to provide rapid and stable protection after a single intranasal administration. It is being developed with the support of the U.S. Biomedical Advanced Research and Development Authority (“BARDA”) for post-exposure prophylaxis against anthrax following exposure to aerosolized B. anthracis spores. After an individual has been exposed to the spores that cause anthrax, B. anthracis bacteria multiply and release toxins within the host. Although antibiotic therapy is effective at eliminating the actively growing bacteria, vaccination is necessary to protect against the germination of dormant spores after the cessation of antibiotic therapy. Because NasoShield is intended to protect against anthrax after a single intranasal dose, we believe it may be a convenient and simple alternative to the only approved vaccine, which must be given as a series of three injections over 1 month. We believe the simplified immunization route and schedule, together with the reliable stability at ambient temperature may allow NasoShield to be deployed in an anthrax event more easily and faster than the currently approved vaccine. We commenced a Phase 1b trial of NasoShield in adults in 2020 which builds on the Phase 1a trial completed in 2018 and evaluates the effect of modified methods of intranasal dosing on NasoShield safety and immunogenicity. Results are expected in the first quarter of 2021.
NasoVAX
NasoVAX is a recombinant intranasal vaccine product candidate that is being developed for both seasonal and pandemic use. NasoVAX is believed to simultaneously activate the humoral, mucosal and cellular immune arms which may enable a more comprehensive immune response. The data from our Phase 2a trial with a monovalent NasoVAX vaccine indicated that NasoVAX was generally well-tolerated and achieved 100% seroprotection with serum antibody responses, which was comparable to published results of a licensed injected influenza vaccine. Statistically significant increases in mucosal antibody were noted as well as a robust T cell response directed against influenza. Approximately half of the subjects from the highest dose were evaluated between 12 and 14 months after initial dosing for additional immunogenicity assessment. The durability data show that the immune response elicited by NasoVAX was stable with no overall change in the antibody titer or level of seroprotection over an average of 13 months. The combination of serum antibody, mucosal antibody and T-cell response in combination with the durability data provides the potential for improved protection against influenza and suggests that NasoVAX could have a great impact on flu symptoms and shedding of the influenza virus. We are currently evaluating the development path of NasoVAX.
ALT-801Pemvidutide
We completed an acquisition in July 2019 of all of the equity interests of Spitfire Pharma, Inc. (“Spitfire”). Spitfire was a privately held, preclinical pharmaceutical company with the primary asset being pemvidutide, a novel peptide-based dual GLP-1/glucagon dual receptor agonist for the treatment of NASH. We refer to this product candidate as ALT-801, and it is designed to treat the obesity and the metabolic dysfunction that causes NASH. NASH, Obesity is a significant burden to the most severe formglobal healthcare systems and is implicated in two-thirds of non-alcoholic fatty liverthe leading causes of death from non-communicable diseases worldwide. Some of the leading co-morbidities of obesity include high blood pressure, high cholesterol, type 2 diabetes, heart disease, (“NAFLD”),stroke, gallbladder disease, osteoarthritis, sleep apnea and breathing problems, certain cancers and NASH. According to the Center for Disease Control and Prevention, the estimated annual medical cost of obesity in the U.S. was nearly $173.0 billion in 2019 dollars. Globally, the market size for weight loss alone was $2.4 billion in 2022 and is estimated that it will reach $54.0 billion by 2030. Previous approaches to the treatment of obesity have been associated with safety concerns, limiting the success of those approaches. NASH involves multiple metabolic pathways leading to the abnormal accumulation of liver fat, toxic lipid metabolites, and inflammation, leading to fibrosis or eventuallyand increased risk of death due to liver cancer. NAFLDfailure and cardiovascular disease. Non-alcoholic fatty liver disease (“NAFLD”), the fatty liver precursor to NASH, is present in up to 90%50% of obese patients, and approximatelyup to 20% of NAFLD patients progress to NASH. In addition, up to 40% of NASH patients develop NAFLD recurrence one year after liver transplant, which we believe indicates that the underlying metabolic disease is still present after transplant. We believe the treatment of obesity is thea cornerstone of treating NASH and the principal morbidities of NASH. In addition, clinical evidence from recent trials of potential NASH products indicates that reduction in liver fat may play an important role in the resolution of inflammation and fibrosis. We believe that combining a reduction in liver fat content with weight loss could be the optimal approach for NASH resolution. ALT-801’sPemvidutide’s dual agonist mechanism of action is designed to combine the activity of GLP-1 for the reduction of appetite and inflammation, with the direct activity of glucagon, on the liver, including increased energy expenditure, adipose browning lipolysis and mobilization of the liver fat. ALT-801fat through lipolysis and reduction of lipid synthesis. Pemvidutide incorporates a proprietary side chain, referred to as the EuPort domain, which is designed to enhance pharmacokinetics for tolerability in the gastrointestinal tract and permit weekly dosing. As observed in a well-established preclinical model of the disease, ALT-801pemvidutide is capable of inducing significant weight loss with concomitant decreases in liver fat content, inflammation and fibrosis, with superior results compared to elafibranorfibrosis. In a first-in-human, randomized, placebo-controlled, single-ascending and semaglutide. multiple-ascending dose study of pemvidutide in overweight and obese volunteers, at 12 weeks, we observed significant reductions in body weight, and in the absence of caloric restriction or lifestyle modification, as well as reduction in low density lipoprotein cholesterol (“LDL-C”).
In addition, ALT-801pemvidutide demonstrated improved metabolic function and exhibited pleiotropic effects in our preclinical testing across multiple metabolic pathways that are involved in NASH. We also observed in preclinicalthese studies that ALT-801pemvidutide resulted in more profound suppression of genes associated with steatosis, inflammation and stellate cell fibrosis by RNA sequencing comparedsequencing. Additionally, we have now observed profound decreases and normalization in liver fat content and significant reductions in body weight at 12 weeks in a Phase 1 trial of pemvidutide in healthy volunteers and at both 12 weeks and 24 weeks in a Phase 1b trial of pemvidutide in subjects with NAFLD as well as reductions and normalization in serum alanine aminotransferase (“ALT”) at both 12 weeks and 24 weeks in the Phase 1b trial. We believe that pemvidutide is the only NASH candidate in development with observed rapid effects in liver fat reduction and liver inflammation together with significant weight loss. 
Regulatory Basis for Clinical Trials of Pemvidutide On November 9, 2020, we announced that we received clearance from the Human Research and Ethics Committee and filed a Clinical Trial Notification with the Australian regulatory authority prior to elafibranorcommencing our first-in-human trial of pemvidutide. On September 28, 2021, we announced that we received clearance of our Investigational New Drug (“IND”) Application in NASH from the U.S. Food and semaglutide. We commencedDrug Administration (“FDA”) prior to commencing our Phase 1b trial of pemvidutide in NAFLD. On January 23, 2022, we further announced that we received clearance of our IND Application from the FDA prior to commencing our 48-week Phase 2 trial of pemvidutide in obesity. Phase 1 Clinical Trial Results – Obesity In September 2021, we announced the completion of a 12-week Phase 1 clinical trial of pemvidutide in Australia in overweight and obese adult volunteers in the fourth quarter of 2020.under a clinical trial application. The trial is expected to have bothwas a first-in-human, randomized, placebo-controlled, single-ascending and multiple-ascending dose arms over a 6-week treatment duration, with data readouts planned for each arm(“MAD”) study in the second quarter of 2021.non-diabetic overweight and obese volunteers. The endpoints of the Phase 1a1 trial are expectedwere to beassess the safety, tolerability and pharmacokinetics of pemvidutide, with a preliminary readoutprimary readouts on safety, pharmacokinetics and weight loss, resting energy expenditure, liver fat by MRI-PDFFloss. Additional readouts included metabolic and lipid profiles, cardiovascular measures and glucose homeostasis. Pending interim resultsAt 12 weeks, subjects receiving pemvidutide showed mean weight losses of 4.9%, 10.3% and 9.0% at the 1.2 mg, 1.8 mg and 2.4 mg doses, respectively, while the placebo group experienced a mean weight loss of 1.6%, and in the absence of caloric restriction or lifestyle modification. Weight loss occurred rapidly and consistently over 12 weeks. | | | | | | Summary of 12-week MAD weight loss findings | Characteristic | Treatment | | 1.2mg (n=7) | 1.8mg (n=9) | 2.4mg (n=11) | Pooled Placebo (n=7) | Baseline demographics | Age, years | Mean (SD) | 27.7 (10.5) | 32.0 (10.7) | 31.4 (11.7) | 35.3 (12.4) | Body weight (kg) | Mean (SD) | 90.5 (15.4) | 86.4 (12.9) | 91.9 (15.1) | 87.6 (14.3) | BMI (kg/m2) | Mean (SD) | 30.0 (3.9) | 30.1 (3.9) | 31.8 (2.9) | 31.0 (4.3) | Results | Weight loss (kg) | Mean (SD) | -4.7 (3.0) | -8.8 (3.0) | -8.4 (2.8) | -1.5 (3.0) | Weight loss (%) | Mean (SD) | -4.9 (2.9) % | -10.3 (3.4) %** | -9.0 (3.3) %* | -1.6 (3.0) % | *p < .01, **p < .005, compared to placebo |
The 1.8 mg dose cohort experienced the highest weight loss, with 100% of the single-subjects losing at least 5% of body weight and 55% of subjects losing at least 10% of body weight. The amounts of weight loss at the 1.8 mg and 2.4 mg doses were similar given the sample size and overlapping confidence intervals. No correlation was found between the magnitude of weight loss and either age or baseline body mass index (“BMI”). Favorable or statistically significant trends were observed in secondary measures, including reductions in systolic and diastolic blood pressure, serum lipids and HOMA-IR (a measure of insulin resistance). As seen in the table below, the effect on serum lipids was particularly striking and in the 1.8mg dose, included a greater than 25% decrease in LDL-C, which is known to increase the risk of cardiovascular disease, as well as significant decreases in total cholesterol and triglycerides. In addition, a rise in serum ketone bodies and a fall in serum tripalmitin was observed, consistent with the stimulatory effects of glucagon on hepatic beta-oxidation of lipids and suppressive effects of pemvidutide on triglyceride synthesis, respectively. 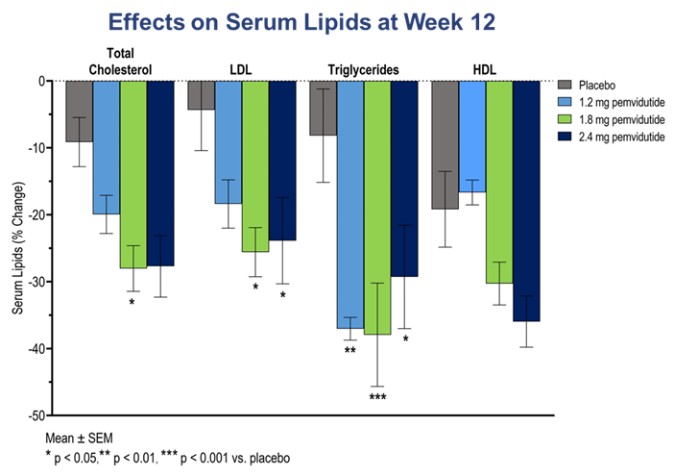
Side effects in this trial were mild to moderate, with no serious or severe treatment-emergent adverse events reported. No discontinuations due to adverse events were reported. Summary of 12-week MAD safety findings | Characteristic | Treatment | | 1.2mg (n=7) | 1.8mg (n=9) | 2.4mg (n=11) | Pooled Placebo (n=7) | Discontinuations due to adverse events (n) | 0 | 0 | 0 | 0 | Early withdrawal (n) | 1 | 0 | 2 | 2 | Gastrointestinal adverse events | Nausea | Mild | 14.3% | 55.6% | 45.5% | 14.3% | Moderate | 14.3% | 11.1% | 45.5% | 0% | Vomiting | Mild | 14.3% | 11.1% | 45.5% | 14.3% | Moderate | 0% | 11.1% | 27.3% | 0% | Diarrhea | Mild | 0% | 0% | 18.2% | 0% | Moderate | 0% | 0% | 0% | 0% | Constipation | Mild | 0% | 11.1% | 18.2% | 0% | Moderate | 0% | 11.1% | 9.1% | 0% | Other adverse events (n) | 0 | 2 | 1 | 0 |
Unlike the majority of other clinical trials with agents within the GLP-1 class, including dual agonists and tri-agonists, dose arms, we intendtitration, a gradual increasing of dose within a subject over a period of weeks to extendmonths to improve tolerability, was not used in the multiple-ascendingpemvidutide trial. Even without that dose armstitration, the symptoms experienced by subjects who received pemvidutide 1.2 mg and 1.8 mg were predominantly mild, did not require treatment and were consistent with known effects of GLP-1-based therapies. Tolerability was observed to decrease at the highest dose level. One subject receiving placebo and one subject receiving pemvidutide 1.8 mg had a 3 to 5-fold elevation in ALT levels over baseline that resolved rapidly after a pause in dosing. In this trial, no perturbations of glucose control, as assessed by fasting glucose and hemoglobin A1c, were observed in subjects with obesity/overweight with pre-diabetes; in fact, a reduction of insulin resistance was observed, as expected when significant weight loss is experienced. We are currently conducting a Phase 1 trial to establish the effects of pemvidutide on glucose control and confirm the reduction in insulin resistance in subjects with Type 2 diabetes. Phase 1 Clinical Trial Results – Liver Fat Content While the trial inclusion criteria for the MAD study did not pre-specify a minimum liver fat content (“LFC”), the trial did enroll a number of subjects with measurable LFC as determined by magnetic resonance imaging – proton density fat fraction (“MRI-PDFF”). A post-hoc analysis of the trial data through 6 weeks forshowed that 8 subjects had hepatic steatosis, defined as liver fat content greater than or equal to 5% at baseline (LFC range from 5.5% to 19.5% in these 8 subjects). LFC fell below the limit of detection (“LOD”), or less than 1.5%, within 6 weeks of treatment in all subjects with steatosis receiving the 1.8 mg or 2.4 mg dose of pemvidutide, representing a twelve-week, parallel-dosinggreater than 90% reduction in the liver fat content (see chart below). These findings reinforce the results from preclinical studies of pemvidutide, in which we observed statistically greater reductions in liver fat than an equivalent dose of semaglutide. We believe these findings support the potential combined beneficial effects of weight loss and glucagon agonism on liver fat content. Images of Representative MRI-PDFF Images at Baseline and Week 6 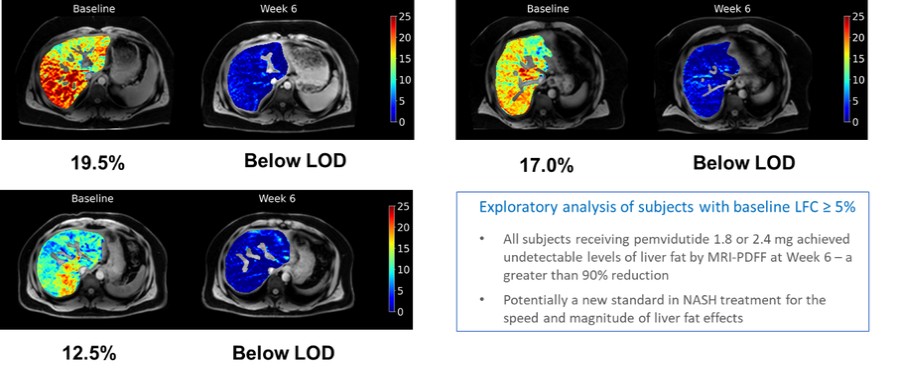
The table below displays the changes in liver fat content at Week 6 compared to baseline in the 8 subjects with steatosis at baseline: Treatment Group | Weight Loss (%) at Week 6 | MRI-PDFF | | | Baseline | Week 6 | Absolute ∆ at Week 6 (%) | Relative ∆ at Week 6 (%) | | | | | Individual | Mean | Individual | Mean | Placebo | 0.5 | 5.2 | 3.7 | 1.5 | 1.5 | 28.8 | 28.8 | pemvidutide 1.2 mg | 1.0 | 19.1 | 14.0 | 5.10 | 6.50 | 26.7 | 48.2 | | 5.1 | 11.2 | 3.4 | 7.80 | | 69.6 | | pemvidutide 1.8 mg | 4.4 | 12.4 | < LOD | 11.65 | 11.65 | 94.0 | 94.0 | pemvidutide 2.4 mg | 3.7 | 17.0 | < LOD | 16.25 | 11.50 | 95.6 | 91.9 | | 4.9 | 5.5 | < LOD | 4.75 | | 86.4 | | | 3.1 | 7.0 | < LOD | 6.25 | | 89.3 | | | 4.7 | 19.5 | < LOD | 18.75 | | 96.2 | | LOD (limit of detection) = 1.5%; for absolute and relative ∆, values < LOD are set at 0.75% |
Clinical Trial Results – 12-week Phase 1b (NAFLD) In September 2022, we announced the topline results from our 12-week Phase 1b clinical trial.trial of pemvidutide in subjects with NAFLD. The endpoints oftrial was a double-blind, placebo-controlled study. Subjects were randomized 1:1:1:1 to 1.2 mg, 1.8 mg, 2.4 mg pemvidutide or placebo administered weekly for 12 weeks. No dose titration was used with the Phase 1b trial are expected to be safety, tolerability, pharmacokinetics,1.2 mg or 1.8 mg dose, while a short 4-week dose titration was employed at the 2.4 mg dose. The primary efficacy endpoint was the percent (%) reduction in LFC from baseline, and the key secondary efficacy endpoint was the % weight loss decreasefrom baseline, both at 12 weeks of treatment. The trial was conducted without adjunctive diet and exercise interventions that are the standard for obesity trials. Ninety-four (94) subjects were randomized and treated at 13 sites across the U.S. Mean BMI at baseline was approximately 36 kg/m2 and mean LFC, as measured by MRI-PDFF, was approximately 22%. Twenty-seven (29%) subjects had type 2 diabetes at baseline, and approximately 75% of study subjects were of Hispanic ethnicity. The chart below details the baseline study demographics. Characteristic | Treatment | | Placebo (n = 24) | 1.2 mg (n=23) | 1.8 mg (n=23) | 2.4 mg (n=24) | Age, years | Mean (SD) | 47.9 (14) | 48.6 (11) | 50.3 (9) | 48.8 (8) | Sex | Female, n (%) | 14 (58.3%) | 9 (39.1%) | 12 (52.2%) | 15 (62.5%) | Race | White, n (%) | 21 (87.5%) | 21 (91.3%) | 20 (87.0%) | 24 (100%) | | Other, n (%) | 3 (12.5%) | 2 (8.7%) | 3 (13.0%) | 0 (0.0%) | Ethnicity | Hispanic, n (%) | 14 (58.3%) | 20 (87.0%) | 19 (82.6%) | 18 (75.0%) | | Non-Hispanic, n (%) | 10 (41.7%) | 3 (13.0%) | 4 (17.4%) | 6 (25.0%) | BMI, kg/m2 | Mean (SD) | 36.9 (4.7) | 36.3 (5.6) | 35.4 (3.9) | 35.3 (5.0) | Body weight, kg | Mean (SD) | 105.1 (20.8) | 102.4 (14.6) | 98.9 (19.7) | 98.2 (18.9) | Diabetes status | T2D, n (%) | 6 (25.0%) | 7 (30.4%) | 7 (30.4%) | 7 (33.3%) | LFC, % | Mean (SD) | 23.8 (9.2) | 21.6 (7.3) | 21.8 (8.0) | 20.2 (7.0) |
The trial met its primary endpoint in all pemvidutide treatment groups. As seen in the table below showing reduction in LFC as measured by MRI-PDFF in all subjects, at the 1.8 mg dose (with and without diabetes), pemvidutide achieved a mean reduction of liver fat content of 68.5%, with 94.4% of subjects achieving a 30% reduction in liver fat, (as72.2% achieving a 50% reduction in liver fat, and 55.6% of subjects achieving normalization of liver fat, defined as liver fat fraction of 5% or less. Endpoint | Treatment | | Placebo (n = 24) | 1.2 mg (n=20) | 1.8 mg (n=18) | 2.4 mg (n=20) | Absolute reduction, % | Mean (SE) | 0.2 (1.7) | 8.9 (1.8)** | 14.7 (1.7)** | 11.3 (2.0)** | Relative reduction, % | Mean (SE) | 4.4 (8.7) | 46.6 (8.1)** | 68.5 (9.7)** | 57.1 (8.0)** | 30% reduction | n (%) | 1 (4.2%) | 13 (65.0%)** | 17 (94.4%)** | 17 (85.0%)** | 50% reduction | n (%) | 0 (0.0%) | 8 (40.0%)** | 13 (72.2%)** | 14 (70.0%)** | Normalization (≤ 5% LFC) | n (%) | 0 (0.0%) | 4 (20.0%)* | 10 (55.6%)** | 10 (50.0%)** |
*p < .05, **p<.001 compared to placebo In addition, as shown in the below table, mean serum ALT levels declined in all subjects, and in subjects with baseline serum ALT above 30 IU/L, levels declined more than 17 IU/L at all dose levels and 27.0 IU/L in the 2.4 mg dose cohort. Endpoint | Treatment | | Placebo | 1.2 mg | 1.8 mg | 2.4 mg | | ALT, change from baseline, IU/L, LSM (SE) | n = 24 | n = 23 | n = 23 | n = 24 | | -6.2 (2.8) | -11.2 (3.1) | -13.8 (3.0)* | -13.6 (3.2)* | ALT, change from baseline, IU/L, LSM (SE), baseline ≥ 30 IU/L | n = 15 | n = 10 | n = 15 | n = 12 | | -12.6 (4.1) | -17.8 (4.8) | -20.8 (4.2) | -27.0 (4.8)* |
*p < .05 The trial also met its key secondary endpoint in all pemvidutide treatment groups. As portrayed in the following table, employing an efficacy estimand, mean weight losses of 4.9% (placebo-adjusted 4.7%) in subjects without diabetes and 4.4% in subjects with diabetes (placebo-adjusted 3.9%) were achieved at the 1.8 and 2.4 mg doses, respectively. Population | Treatment | | Placebo (n = 24) | 1.2 mg (n=23) | 1.8 mg (n=23) | 2.4 mg (n=24) | No diabetes, (% change) | LSM (SE)
| -0.2
(0.7) | -3.4**
(0.8) | -4.9**
(0.8) | -3.5**
(0.8) | Diabetes, (% change) | LSM (SE)
| -0.5
(1.3) | -3.3*
(1.1) | -3.8*
(1.2) | -4.4*
(1.3) | All subjects (% change) | LSM (SE)
| -0.2
(0.7) | -3.4**
(0.7) | -4.3**
(0.7) | -3.7**
(0.7) |
LSM least square mean; *p < .05, **p<.001 compared to placebo Pemvidutide was reported to be generally well tolerated. Gastrointestinal events comprised the majority of the adverse events (“AEs”). Even without dose titration, the symptoms experienced by subjects were predominantly mild and transient in nature, consistent with known GLP-1 class effects. No serious or severe AEs were reported. Two subjects treated with pemvidutide discontinued treatment due to AEs [1 (4.3%) at 1.8 mg and 1 (4.2%) at 2.4 mg], both secondary to gastrointestinal intolerability. No clinically significant ALT elevations (defined as an increase to 3-fold or greater the upper limit of normal) were observed. Glycemic control was unaffected, with no clinically meaningful changes in HbA1c or fasting glucose. Clinically meaningful reductions in systolic blood pressure were observed, along with the two to three beats per minute increase in heart rate typical for GLP-1 class of drugs. The table below summarizes the safety findings. Characteristic | Treatment | | Placebo (n = 24) | 1.2 mg (n=23) | 1.8 mg (n=23) | 2.4 mg (n=24) | Severe AEs | n (%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | SAEs | n (%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | AEs leading to treatment discontinuation | n (%) | 0 (0.0%) | 0 (0.0%) | 1 (4.3%) | 1 (4.2%) | Nausea | Mild, n (%) | 3 (12.5%) | 3 (13.0%) | 6 (26.1%) | 6 (25.0%) | | Mod, n (%) | 0 (0.0%) | 1 (4.3%) | 6 (26.1%) | 3 (12.5%) | Vomiting | Mild, n (%) | 0 (0.0%) | 3 (13.0%) | 2 (8.7%) | 2 (8.3%) | | Mod, n (%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | Diarrhea | Mild, n (%) | 4 (16.7%) | 3 (13.0%) | 5 (21.7%) | 1 (4.2%) | | Mod, n (%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | Constipation | Mild, n (%) | 0 (0.0%) | 3 (13.0%) | 4 (17.4%) | 1 (4.2%) | | Mod, n (%) | 0 (0.0%) | 1 (4.3%) | 0 (0.0%) | 0 (0.0%) |
Clinical Trial Results – 24-week Phase 1b (NAFLD) In December 2022, we announced the topline results from our 24-week (12-week extension) Phase 1b clinical trial of pemvidutide in subjects with NAFLD. Sixty-six (66) of the 83 subjects who completed the initial 12-week Phase 1b NAFLD trial consented to participate in this 12-week extension trial to receive a total of 24 weeks of treatment, and 64 subjects were enrolled. The trial was conducted without adjunctive diet and exercise interventions and the double-blinding of the trial was maintained during the extension study. The same endpoints as the 12-week parent NAFLD trial were employed, with a primary efficacy endpoint of percent (%) reduction in liver fat content; key secondary endpoints were reduction in liver inflammation, as measured by serum ALT levels and corrected T1 (“cT1”), and percent weight loss. The population of the 12-week extension trial had similar baseline characteristics as the population of the parent, 12-week Phase 1b NAFLD trial. At baseline, across all treatment groups, mean BMI was 36.7 kg/m2 and mean LFC, as measured by MRI-PDFF, standard)was 22.2%. Type 2 diabetes was present in 26.6% of subjects and lean body mass,73.4% of study subjects were of Hispanic ethnicity. The trial met its primary endpoint in all pemvidutide treatment groups. At the 1.8 mg and 2.4 mg doses, subjects receiving pemvidutide achieved mean relative reductions of liver fat content of 75.2% and 76.4%, respectively; 92.3% and 100% of subjects, respectively, achieved a 30% reduction in liver fat, 84.6% and 72.7% of subjects, respectively, achieved a 50% reduction in liver fat, and 53.8% and 45.5% of subjects, respectively, achieved normalization of liver fat. As in the 12-week Phase 1b NAFLD trial, statistically significant declines in mean serum ALT levels were observed in all pemvidutide-treated subjects, and in subjects with baseline serum ALT ≥30 IU/L, ALT levels declined at least 17 IU/L at all pemvidutide dose levels. In a subset of subjects evaluated for cT1 response, 75.0% and 100% of subjects receiving 1.8 mg or 2.4 mg pemvidutide, respectively, achieved an 80 millisecond (ms) decrease in cT1. cT1 value is an MRI-based quantitative metric for assessing a composite of liver inflammation and fibrosis. Elevated cT1 levels have been associated with increased risk of major adverse cardiac events (MACE) and major adverse liver outcomes (MALO), and an 80 ms reduction has been associated with a 2-point reduction of NAFLD Activity Score (NAS). The trial also met its key secondary weight loss endpoint in all pemvidutide treatment groups. Employing an efficacy estimand, mean weight losses of 7.2% (placebo-adjusted 6.0%) in subjects without diabetes and 6.2% (placebo-adjusted 4.8%) in all subjects were achieved at the 1.8 mg dose. A summary of the key primary and secondary efficacy findings is below: Endpoint | Treatment | | Placebo | 1.2 mg | 1.8 mg | 2.4 mg | Primary Endpoint—Liver Fat Content | n = 18 | n = 14 | n = 13 | n = 11 | Liver fat reduction, absolute, % change, LSM (SE) | 1.6 (0.8) | 11.2 (2.3) *** | 17.0 (2.4) *** | 15.6 (2.1) *** | Liver fat reduction, relative, % change, LSM (SE) | 14.0 (3.8) | 56.3 (11.6) *** | 75.2 (8.1) *** | 76.4 (5.9) *** | Proportion of subjects with 30% reduction, (%) | 5.6 | 76.9 **** | 92.3 **** | 100.0 **** | Proportion of subjects with 50% reduction, (%) | 0.0 | 61.5 *** | 84.6 **** | 72.7 **** | Proportion of subjects with normalization, (%) | 0.0 | 30.8 * | 53.8 *** | 45.5 ** | Secondary Endpoint—Markers of Inflammation | ALT, change from baseline, IU/L, LSM (SE) | n = 19 | n = 16 | n = 15 | n = 14 | | -2.2 (2.5) | -13.3 (3.7) ** | -13.7 (5.1) ** | -15.2 (5.8) ** | ALT, change from baseline, IU/L, LSM (SE), baseline ≥ 30 IU/ | n = 13 | n = 7 | n = 10 | n = 9 | | -3.1 (3.5) | -17.0 (7.6) * | -17.7 (7.2) * | -20.6 (9.8) * | Proportion of subjects with cT1 response, (%) | n = 6 | n = 7 | n = 4 | n = 2 | | 0.0 | 85.7 ** | 75.0 * | 100.0 * | Secondary Endpoint—Weight Loss | Weight loss, no diabetes, (% change), LSM (SE) | n = 14 | n = 13 | n = 9 | n = 11 | | 1.2 (0.7) | 5.2 (1.7) ** | 7.2 (1.1) *** | 5.8 (1.6) ** | Weight loss, diabetes, (% change), LSM (SE) † | n = 5 | n = 3 | n = 6 | n = 3 | | 3.4 (2.1) | 4.3 (1.9) | 5.3 (2.7) | 3.5 (2.5) | Weight loss, all subjects, (% change), LSM (SE | n = 19 | n = 16 | n = 15 | n = 14 | | 1.4 (0.7) | 5.1 (1.4) ** | 6.2 (1.3) *** | 5.2 (1.4) ** |
Normalization of liver fat defined as ≤ 5%; cT1 response define as an 80 ms change from baseline; LSM, least square mean † High variability due to the small numbers of diabetic subjects (n = 5, 3, 6, 3 in respective treatment groups) * p < .05; ** p < 0.01, *** p < 0.001, ****p < 0.0001 compared with placebo Pemvidutide was generally well astolerated. A total of three serious or severe AEs were reported, each unrelated to study drug administration (chest pain post-elective cardiac stent placement; Salmonella infection; and hypertension greater than three weeks after the completion of treatment). Three AEs led to treatment discontinuation, one being the Salmonella infection, and two gastrointestinal AEs, one (6.3%) at the 1.2 mg dose and one (6.7%) at the 1.8 mg dose. As expected, gastrointestinal events comprised the majority of AEs and were predominantly mild in nature. No clinically significant ALT elevations were observed. Meaningful reductions in systolic blood pressure were observed, and increases in heart rate, typical of the incretin class of agents, were minimal at zero to four beats per minute and independent of dose. Below is a summarization of the safety findings: Characteristic | Treatment | | Placebo (n = 19) | 1.2 mg (n=16) | 1.8 mg (n=15) | 2.4 mg (n=14) | Serious or severe AEs | n (%) | 1 (5.3%) | 1 (6.3%) | 1 (6.7%) | 0 (0.0 %) | AEs leading to treatment discontinuation | n (%) | 0 (0.0%) | 2 (12.5%) | 1 (6.7%) | 0 (0.0 %) | Nausea | Mild, n (%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 1 (7.1%) | | Moderate, n (%) | 0 (0.0%) | 0 (0.0%) | 3 (20.0%) | 0 (0.0%) | Vomiting | Mild, n (%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | | Moderate, n (%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | Diarrhea | Mild, n (%) | 1 (5.3%) | 0 (0.0%) | 1 (6.7%) | 0 (0.0%) | | Moderate, n (%) | 0 (0.0%) | 1 (6.3%) | 0 (0.0%) | 0 (0.0%) | Constipation | Mild, n (%) | 0 (0.0%) | 0 (0.0%) | 1 (6.7%) | 0 (0.0%) | | Moderate, n (%) | 1 (5.3%) | 1 (6.3%) | 0 (0.0%) | 0 (0.0%) | Systolic Blood Pressure, mm Hg, LSM (SE) | -2.3 (2.8) | -10.1 (4.2) * | -5.5 (3.7) | -12.0 (3.5) * | Diastolic Blood Pressure, mm Hg, LSM (SE) | -2.5 (1.5) | -2.9 (2.6) | -4.0 (3.7) | -3.8 (2.8) | Heart Rate, mmHg, LSM (SE) | -1.0 (1.7) | 3.7 (1.8) | 0.5 (2.8) | -0.1 (1.8) |
A total of 3 serious or severe adverse events (AEs) were reported, each unrelated to study drug administration (chest pain post-elective cardiac stent placement; Salmonella infection; and hypertension greater than 3 weeks after the completion of treatment), with only the Salmonella infection leading to treatment discontinuation. The other AEs leading to treatment discontinuation were mild (Grade 1) abdominal pain in 2 subjects. No significant ALT elevations were reported. *p < .05 compared with placebo. As detailed in the table below, glycemic control was maintained in subjects with diabetes, all pemvidutide groups demonstrating trends toward improvements in fasting glucose and either maintaining or demonstrating trends toward improvement in HbA1c over the 24 weeks of treatment: Characteristic | Treatment | | Placebo | 1.2 mg | 1.8 mg | 2.4 mg | Non-diabetes | n = 14 | n = 13 | n = 9 | n = 11 | Fasting glucose | Baseline, mg/dL, mean (SD) | 96.2 (12.4) | 99.4 (11.9) | 96.0 (12.4) | 99.3 (13.6) | Week 24, mg/dL, mean (SD) | 93.3 (12.1) | 99.1 (13.1) | 96.9 (12.5) | 98.4 (24.5) | HbA1c | Baseline, %, mean (SD) | 5.8 (0.2) | 5.7 (0.3) | 5.7 (0.2) | 5.5 (0.4) | Week 24, %, mean (SD) | 5.7 (0.3) | 5.8 (0.3) | 5.8 (0.3) | 5.6 (0.3) | Diabetes | n = 5 | n = 3 | n = 6 | n = 3 | Fasting glucose | Baseline, mg/dL, mean (SD) | 111.5 (19.2) | 132.1 (28.2) | 120.2 (37.1) | 147.4 (40.4) | Week 24, mg/dL, mean (SD) | 109.4 (14.8) | 123.4 (50.8) | 109.0 (13.1) | 75.5 (29.0) | HbA1c | Baseline, %, mean (SD) | 6.1 (0.6) | 7.8 (1.4 | 6.4 (0.5) | 6.8 (1.3) | Week 24, %, mean (SD) | 6.4 (1.1) | 7.4 (2.3 | 6.4 (0.3) | 6.3 (1.3) |
Clinical Development Plan We initiated a 48-week Phase 2 MOMENTUM obesity trial in the first half of 2022. The readouts of this trial will include safety, weight loss and other metabolic biomarkers. If successful,measures. The trial will include a 24-week interim analysis of approximately 160 subjects for safety, weight loss and other measures that we expect data fromwill read out in the first quarter of 2023. The trial is being conducted at approximately 30 sites in the U.S. The randomized, placebo-controlled trial enrolled approximately 320 non-diabetic subjects randomized 1:1:1:1 to receive either 1.2 mg, 1.8 mg, 2.4 mg pemvidutide or placebo weekly for 48 weeks. No dose titration is being used with the 1.2 mg or 1.8 mg dose, while a short 4-week dose titration is being employed at the 2.4 mg dose. The primary endpoint of the trial is the relative (percent) change in body weight at 48 weeks compared to baseline, with additional readouts including metabolic and lipid profiles, cardiovascular measures and glucose homeostasis. The trial is being conducted with adjunctive diet and exercise interventions that is typical in weight loss studies. On August 11, 2022, we announced that 167 subjects had been randomized and we further announced on September 28, 2022 the first dosing of all subjects in the MOMENTUM trial. We intend to initiate a biopsy-driven Phase 1b study Q3 2021.2 NASH trial in 2023. We expect this trial to enroll approximately 200 subjects across 4 treatment arms (placebo, pemvidutide 1.2, 1.8 and 2.4 mg) administered for a period of 48 weeks. The primary efficacy readout will be based on a biopsy readout at the end of either 24 or 48 weeks. These details may change by the time of protocol finalization. HepTcell HepTcell is an immunotherapeutic product candidate for patients chronically infected with the hepatitis B virus (“HBV”). Approximately 300 million people worldwide live with chronic HBV infection, including approximately 2.2 million in the United States. Chronic HBV infection can lead to serious complications, including cirrhosis and liver cancer. Approximately 780,000 people die per year worldwide due to cirrhosis and liver cancer. Current antivirals prevent disease progression but rarely clear chronic infection. HepTcell is designed to drive CD4+ and CD8+ T-cell responses against all HBV genotypes in patients of all ethnic backgrounds. Stimulating T-cell responses in chronically infected HBV patients has been challenging because chronic infection with HBV and elevated hepatitis B surface antigen (HBsAg) levels strongly diminishessuppresses T-cell immunity directed against the virus. HepTcell focuses the immune systemT cell response on discrete, highly conserved regions of the HBV proteome. We believe our approach allows HepTcell to break immune tolerance by activating T-cells against critical viral sequences with decreased probability of immune escape due to viral mutation. HepTcell is based on our synthetic peptide technology platform and is given by intramuscular injection. In 2018, we completed a Phase 1 trial in the United Kingdom and South Korea in adult patients with chronic HBV. The HepTcell Phase 1 trial was a double-blinded, placebo-controlled, randomized, dose-escalation study that enrolled 61 subjects with chronic HBV who were HBeAg-negative and well-controlled on licensed antivirals. A total of 41 patients received one of two dose levels of HepTcell, with and without IC31TM®, a depot-forming TLR9 adjuvant developed by Valneva SE, while 20 control patients received either placeboIC31® alone or IC31 alone.placebo. Patients received three injections each 28 days apart and were followed for six months after the final dose. All dose combinations showed excellent tolerabilitywere generally well-tolerated and met the primary endpoint of safety. In the two adjuvanted HepTcell arms, T-cell responses against HBV markedly increased over baseline compared to placebo. The chart below presents the immunogenicity against hepatitis B epitopes that was demonstrated in our Phase 1 clinical trial: 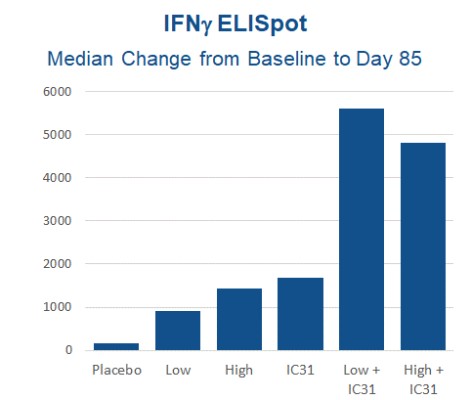
We initiated a Phase 2 studytrial during Q4the fourth quarter of 2020 in the United States, Canada, Europe and EuropeAsia that is a double-blind, randomized, placebo-controlled study of 80 adult patients with HBeAg-negative inactive CHB and HBsAg ≤ 100200 IU/mL.mL Patients with approximately low HBsAg are more likely to mount effective T cell responses against HBV than those with higher levels. The rationale for the study design is based in our understanding that HepTcell willcould be used in combination with newer direct acting agents that may be more effective than the current nucleosides analogs in reducing HBsAg to this level. Accordingly, selection of patients with HBsAg levels ≤ 200 IU/mL may mimic the eventual combination of HepTcell with the newer antiviral drugs in development. HepTcell is being administered in 6 doses at the low dose level of HepTcell plus IC31® at 4-week intervals for 24 weeks, and patients will be followed for one year to evaluate safety and durability of response. The primary efficacy endpoint is virological response, defined as a 1-log reduction in HBsAg levels from baseline or HBsAg clearance at 24 weeks. Secondary efficacy endpoints include reactivation of anti-HBV T cell responses HBsAg clearance, and other assessments of virologic response. We expect data from this studytrial in the first quarterhalf of 2022.2024. A follow-up phase will assess the safety and durability of response one year after completion of treatment. Our Strategy Key elements of our strategy include the following: | ● | Strategically partner or out-license certain product candidates at later stages of development to focus our efforts on early to mid-stage product development; |
Apply our RespirVec platform technologies to design and develop vaccines and therapeutics tailored to address a wide range of infectious diseases, including COVID-19, anthrax, and influenza;16
Apply our EuPort platform technologies to design and develop treatments for NASH, obesity, and other metabolic diseases;
Apply our Densigen platform technologies to design and develop immunotherapeutic products tailored to address acute and chronic infections;Table of Contents
| ● | In-license or acquire complementary metabolic or immunotherapeutic technologies and product candidates that are either synergistic or complementary to our capabilities to expand our pipeline; and |
| ● | Apply our EuPort platform technologies to design and develop treatments for obesity, NASH and other metabolic diseases. |
Strategically partner or out-license certain product candidates at later stages of development to focus our efforts on early to mid-stage product development; and
In-license or acquire complementary immunotherapeutic technologies and product candidates that are either synergistic or complementary to our capabilities to expand our pipeline.
Our Technology Platforms Certain product candidates are based on our proprietary platform technologies as described below.below: Intranasal Vaccines - RespirVec
NasoShield, NasoVAX, AdCOVID and T-COVID, our respiratory anti-infective product candidates, are derived from our RespirVec platform, which is designed to elicit rapid and long-term immune protection by intranasal delivery of adenovectored pathogen sequences. We believe that our RespirVec technology may be particularly well-suited for pandemic response to respiratory pathogens as a result of its ability to stimulate mucosal immunity in the nasal cavity, a site of viral attack. RespirVec is designed to stimulate serum neutralizing antibody and cellular immune response for a broad immune response and is stable at room temperature
for several months. We believe that the favorable stability profile of RespirVec vaccines, when combined with the simple intranasal route of administration, has the potential for efficient and inexpensive distribution of the vaccine in a pandemic.
RespirVec technology is comprised of intranasal delivery of replication-deficient adenoviruses and is protected by patents that we own or license.
Key aspects of our RespirVec technology, supported by findings in our preclinical studies and clinical trials, include its potential to:
enable intracellular expression of the vaccine antigen for authentic immune presentation;
mobilize the innate, cellular and mucosal immune systems, not just the antibody-based response triggered by conventional injectable vaccines;
elicit a more durable antibody response than typical licensed injectable vaccines;
provide a self-adjuvanting adenovector delivery system with the potential to improve immunogenicity; and
allow a rapid production cycle at anticipated lower costs.
Incretin-basedEuPort-based Peptide Technology – EuPort
EuPort is a platform technology exclusively licensed by us andthat comprises a hydrophobic domain (e.g., substituted or unsubstituted alkyl chain) and a hydrophilic group (e.g., saccharide) conjugated to a non-terminal amino acid of the peptide. PursuantThe technology, on which pemvidutide is based, allows the peptide to abind extensively to albumin, an abundant protein in the blood, slowing the elimination of the peptide and increasing its serum half-life, allowing for weekly instead of daily dosing, for example. EuPort technology may also slow the entry of the peptide into the circulation following subcutaneous injection which may lead to improvements in tolerability, cardiovascular risk and other characteristics of the peptide as have been observed with pemvidutide. We have license agreement between the Company and Mederis Diabetes, LLC (“Mederis”) (the “Mederis IP License Agreement”), we are the exclusive licensee of patent rights owned by Mederis to develop and commercialize surfactant functionalized (“EuPort domain”) incretin-basedoxynomodulin (GLP1/glucagon dual receptor agonist)-based peptide therapeutics including GLP-1, Glucagon, Oxyntomodulin, and variants thereof,based on EuPort technology for any indication, and Mederis has certain patent rights granted back to it for the use of the EuPort technology outside of the Company’s exclusive field of incretin-based peptide therapeutics. ALT-801, our GLP-1/ Glucagon dual agonist, was developed using the EuPort technology and is based on peptides at least four amino acids in length that bind receptors for Glucagon and/or GLP-1, conjugated to an alkyl saccharide surfactant, including an alkyl glycoside surfactant.indication.
Key aspects of our EuPort technology, supported by findings in our preclinical studies and clinical trial, include its potential to: | ● | increase the serum half-life of the peptide allowing for extended dosing intervals; and |
| ● | slow the entry of the peptide into the circulation, increasing the Tmax of the peptide, and potentially improving the tolerability of the peptide. |
Induce significant weight loss;
Enhance pharmacokinetics for gastrointestinal tract tolerability; and
Improve metabolic function and exhibit pleiotropic effects, including suppression of genes associated with steatosis, inflammation and stellate cell fibrosis by RNA sequencing.
Synthetic Peptide Technology - Densigen Densigen is our synthetic fluorocarbon peptide technology platform. HepTcell, an immunotherapeutic developed using our Densigen platform, is designed to activate T-cells to generate a cytotoxic immune response against intracellular pathogens. This synthetic peptide technology is based on peptides of 30 – 40 amino acids that comprise a high density of CD4 and CD8 T-cell epitopes selected to focus the T-cell response on highly conserved targets and allow diverse populations to respond to the product candidate. Densigen technology is protected by patents owned by us. Key aspects of our Densigen technology, supported by findings in our preclinical studies and clinical trials, include its potential to: | ● | elicit responses across multiple targets for the disease; |
| ● | direct an immune response precisely to specific antigen sites, thereby avoiding more reactive but less effective sites present in the full-length protein; and |
| ● | prompt a stronger immune response than naked peptides due to depot effect caused by attaching a biologically inert fluorocarbon chain to each peptide. |
elicit responses across multiple targets for the disease;
direct an immune response precisely to specific antigen sites, thereby avoiding more reactive but less effective sites present in the full-length protein; and
prompt a stronger immune response than naked peptides due to depot effect caused by attaching a biologically inert fluorocarbon chain to each peptide.
Competition The biopharmaceutical industry and the vaccine market areis intensely competitive and areis characterized by rapid technological progress. In general, competition among pharmaceutical products is based in part on product efficacy, safety, reliability, availability, price and patent position. An important factor is the relative timing of the market introduction of our products and our competitors’ products. Accordingly, the speed with which we can develop products, complete the clinical trials and approval processes and supply commercial quantities of the products to the market is an important competitive factor. Our competitive position also depends upon our ability to show differentiation with a product that is either more efficacious, particularly in the relevant target populations, and/offers a better safety or betolerability profile, is less expensive andor quicker to manufacture.manufacture, or represents a combination of these advantages. We also depend upon our ability to attract and retain qualified personnel, obtain patent protection or otherwise develop proprietary products or processes and secure sufficient capital resources for the often substantial period between technological conception and commercial sale. Large and established companies such as Eli Lilly, Roche, Novartis,Novo Nordisk and Pfizer, and Sanofi Pasteur, among others, compete in the same market as our product candidates. These companies compete with us with their greater experience and resources to support their research and development efforts, conduct testing and clinical trials, obtain regulatory approvals to market products, manufacture such products on a broad scale and market approved products. These companies also compete with us by having significantly greater research and marketing capabilities than we do and may also have products that have been approved or are in late stages of development and have collaborative arrangements in our target markets with leading companies and research institutions. Established pharmaceutical companies may also invest heavily to accelerate discovery and development of novel compounds or to in-license novel compounds that could make the products that we develop obsolete. We also face competition from smaller companies who, like us, rely on investors to fund research and development and compete for co-development and licensing opportunities from large and established pharmaceutical companies. We face competition from multiple biotechnology and bio-pharmaceutical companies, such as Pfizer, Moderna, AstraZeneca, Regeneron and Johnson & Johnson, that are in the process of developing vaccines and therapeutics against COVID-19. Certain of these vaccine and therapeutic technologies are being developed at a faster rate than AdCOVID and T-COVID or have superior immunogenicity or manufacturability attributes. We face competition for NasoShield, our single dose intranasal anthrax vaccine product candidate, from Emergent Biosolutions which manufactures the existing anthrax vaccine. Additionally, we generally face substantial competition for government funding from companies that develop products with government contracts and grants. We face competition for NasoVAX, our intranasal influenza candidate from Novavax, which is developing an influenza vaccine; and a number of companies of varying sizes are also pursuing the development of a “universal” flu vaccine. We face competition for ALT-801,pemvidutide, our dual GLP-1/glucagon dual agonist for the treatment of obesity and NASH. For obesity, we face competition from companies such as Novo Nordisk, whose GLP-1 agonist, brand named Wegovy, or compound name semaglutide, was approved for weight loss in June 2020. Other companies with potentially competitive candidates in development, include Eli Lilly with GLP-1/glucose-dependent insulinotropic polypeptide receptor (“GIP”) dual agonists, including Mounjaro, or compound name tirzepatide, which was approved in 2022 for diabetes but has also shown weight loss effect; Boehringer Ingelheim, Merck/Hanmi Pharmaceutical, AstraZeneca, Innovent Biologics/Eli Lilly, Carmot and D&D Pharma, with GLP-1/glucagon receptor dual agonists; Hanmi Pharmaceutical and Eli Lilly with GLP-1/glucagon/GIP triple agonists; Amgen with its GLP-1 agonist/GIP antagonist antibody; and Novo Nordisk with Amylin and Amylin-GLP-1 combination candidates. We face competition in NASH from companies such as Intercept Pharmaceuticals, which is developing a farnesoid X receptor (“FXR”) agonist; Madrigal Pharmaceuticals, Terns, Aligos and Viking Therapeutics, which isare developing an orally administered, small-molecule, liver-directed, thyroid hormone receptor (THR)(“THR”) β-selective agonist; and, Akero Therapeutics, 89Bio, Novo Nordisk, Boston Pharmaceuticals and Roche, which are developing fibroblast growth factor 21 analogs; Novo Nordisk, which is developing a fibroblast growth factor 21 analog.GLP-1 agonist; and, Inventiva, which is developing a pan-peroxisome proliferator-activated receptor (“PPAR”) agonist and orally based GLP-1 agents that either deliver GLP-1 monoagonist activity either as a peptide (Novo Nordisk) or small molecule (Pfizer, Eli Lilly). In addition, many other small companies are developing other new technologies directed towards obesity or NASH. Finally, we face competition for HepTcell, our immunotherapeutic HBV product candidate, from companies such as TransgeneGSK, Janssen, Vaccitech VBI Vaccines, all of which is developing an adenovirus-based vaccine; Arrowhead Pharmaceuticals, which is developing an HBV therapeutic vaccine; and Inovio, which isare developing a DNAtherapeutic vaccine delivered by in vivo electroporation. Any of theseagainst chronic HBV infection. In addition, many other companies may develop competing products more rapidly than we do.are developing direct acting antivirals against HBV. Intellectual Property We generally seek patent protection for our technology and product candidates in the United States and abroad. The patent coverage available to biotechnology companies is generally uncertain because it involves complex legal and factual considerations. Our success will depend, in part, on whether we can: | ● | obtain patents to protect our own technologies and product candidates; |
| ● | obtain licenses to use the technologies of third parties, which may be protected by patents; |
| ● | protect our trade secrets and know-how; and |
| ● | operate without infringing the intellectual property and proprietary rights of others. |
obtain patents to protect our own technologies and product candidates;
obtain licenses to use the technologies of third parties, which may be protected by patents;
protect our trade secrets and know-how; and
operate without infringing the intellectual property and proprietary rights of others.
We have relied upon certain proprietary trade secrets, know-how and continuing technological advances to develop a competitive position. In efforts to maintain confidentiality and ownership of trade secrets, proprietary information and developments, all of our employees are required to execute agreements regarding confidentiality and assign to us all rights to any inventions and processes they develop while they are employed by us. We may in the future use license agreements to access external products and technologies as well as to convey our own intellectual property to others. We will be able to protect our proprietary rights from unauthorized use by third parties only to the extent that our proprietary rights are covered by valid and enforceable patents or are effectively maintained as trade secrets.
Patent Rights Related to our RespirVec Platform Technology
Immunotherapy for Respiratory Pathogens — Intranasal Application of Adenoviral Vector Vaccines
We are developing a rapid and prolonged immunologic-therapeutic technology, for which we have a patent issued or allowed in the United States, Europe, South Korea and Japan for respiratory pathogens including influenza, coronavirus and anthrax; and pending applications in the United States, Europe, Japan, and other commercially relevant jurisdictions. The claims are directed to methods for inducing an immune response against respiratory pathogens including influenza, coronavirus and Bacillus anthracis, the causative agent of anthrax, comprising intranasal administration of an effective amount of E1 and/or E3 deleted adenovirus. The patent and, if issued, the patent(s) resulting from the pending applications have an expiration date no earlier than March 2032, not giving effect to any potential extensions and assuming payment of all associated fees.
We are also developing methods of administering and dosing the intranasal adenoviral vector vaccine compositions, for which we have an international (PCT) patent application filed, with expectation that in due course in jurisdictions of commercial consideration (e.g., United States, Europe, Japan) there will be national or regional patent applications based on the PCT. The claims are directed to transmucosal administration of our respiratory anti-infective product candidates based on our RespirVec platform technology. The patent(s), if issued, resulting from the pending application have an expiration date of no earlier than August 2040, not giving effect to any potential extensions and assuming payment of all associated fees.
PER.C6 Cell Line — In-Licensed from Janssen Vaccines & Prevention B.V. (Formerly Crucell Holland, B.V.) We are the non-exclusive licensee of patent rights held by Janssen Vaccines & Prevention B.V. (Formerly(formerly Crucell Holland, B.V.) (“Janssen”), covering a method of producing an adenoviral vector stock using cell lines including the PER.C6 cell line, which may be used for the development and manufacture of vaccine products. We entered into an amended license agreement with Janssen, effective as of October 4, 2005, which amended and restated our prior license agreements with Janssen. Under the amended license agreement, we obtained a non-exclusive, worldwide license (with the right to sublicense) under certain patent rights and know-how to use Janssen’s proprietary cell line to develop, manufacture and commercialize vaccines to prevent and/or treat influenza virus and anthrax infection in humans. In consideration for the license, we paid an up-front license fee, issued equity shares, and agreed to pay certain development-based milestone payments through approval of licensed products by the Food and Drug Administration (“FDA”),FDA, up to an aggregate amount of approximately $2.5 million. We also agreed to pay royalty payments as a percentage of net sales of products in any country where the manufacture of such product is covered by a valid claim of any licensed patent or uses licensed know-how, subject to a royalty stacking reduction and minimum annual royalty payments, until the expiration of the term of the amended agreement. We further amended our license agreement with Janssen, effective September 25, 2015, primarily to streamline our manufacturing license arrangements. Prior to the 2015 amendment, we entered into three-party manufacturing license agreements with each manufacturer and Janssen. The 2015 amendment enables us to directly grant sublicenses of certain of our rights under Janssen’s patent rights and know-how to manufacturers, subject to Janssen’s consent which may not be withheld if the manufacturer meets certain criteria. We may terminate the amended license agreement without cause, and the agreement contains customary provisions for either party to terminate prior to the expiration of the agreement. The amended license agreement expires on a product-by-product and country-by-country basis on the later of the date upon which the last of the licensed patents applicable to the relevant product expires or 15 years from the date of first commercial sale of the relevant product. The Janssen patent rights include patents issued in the United States with an expected expiration date no earlier than April 2020, in each case not giving effect to any potential extensions and assuming payment of all associated fees. Upon expiration of the amended license agreement, or if we terminate the amended license agreement for Janssen’s material breach, we retain the right to exploit the rights granted. The agreement’s current maintenance fee term is set to expire on April 2, 2023, and we have provided notice to Janssen that we do not intend to renew the agreement. On April 2, 2020, we entered into Amendment No. 3 to the Second Restated License Agreement and additionally entered into Amendment No. 4, 5 and 6 throughout 2020 (collectively, the “Amendments”), by and between us and Janssen (as amended by Amendment No. 1 to Second Restated License Agreement and Amendment No. 2 to Second Restated License Agreement, together with the Amendments, the “License Agreement”). Pursuant to the Amendment, the field of licenses granted to us for the use of the PER.C6 cell line under the License Agreement is expanded to cover COVID-19 caused by SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2), in addition to the existing licenses related to Bacillus anthracis and influenza virus. Pursuant to the Amendment, we agreed to pay certain additional development-based milestone payments through approval of licensed products by the FDA for the treatment or prevention of COVID-19, up to an aggregate amount of $1.2 million. We also agreed to pay royalty payments as a percentage of net sales of products to a royalty stacking reduction and minimum annual royalty payments, until the expiration of the term of the License Agreement, as amended. We additionally entered into Amendment No. 4, 5 and 6 throughout
2020 to add additional manufacturing partners related to manufacturing AdCOVID. As of December 31, 2020,2022, we have paid Janssen $2.7$3.0 million in cash and equity under the amended agreement.License Agreement. Patent Rights Related to our EuPort Platform Technology EuPort Technology — In-Licensed from Mederis Diabetes, LLC Pursuant to a license agreement between the Company and Mederis Diabetes, LLC (“Mederis”) (the “Mederis IP License Agreement”), we are the exclusive licensee of patent rights owned by Mederis to develop and commercialize surfactant functionalized (“EuPort domain”) incretin-based peptide therapeutics, including GLP-1, Glucagon, Oxyntomodulin,(GLP-1-glucagon)/oxyntomodulin, and variants thereof, including pemvidutide, for any indication, and Mederis has certain patent rights granted back to it for the use of the EuPort technology outside of the Company’s exclusive field of incretin-based peptide therapeutics. The EuPort domain comprises a hydrophobic domain (e.g., substituted or unsubstituted alkyl chain) and a hydrophilic group (e.g., saccharide) conjugated to a non-terminal amino acid of the peptide. Patents under Mederis IP License Agreement have been granted in the United States, Japan and Korea, and applications are pending in the United States, Japan as well as other commercially relevant jurisdictions. The claims are directed to peptides (at least four amino acids in length), including peptides that bind receptors for Glucagonglucagon and/or GLP-1, conjugated to an alkyl saccharide surfactant, including an alkyl glycoside surfactant. The patents and, if issued, the patent(s) resulting from the pending application(s) have an expiration date of no earlier than May 2032, not giving effect to any potential extensions and assuming payment of all associated fees. Patents subject to the Mederis IP License Agreement have also been granted in the United States, Australia, Israel and Japan, and applications are pending in the United States, Europe, Japan, China and other commercially relevant jurisdictions, wherein the claims are directed to specific GLP-1 and/or Glucagonglucagon peptides conjugated to the EuPort domain. The patents and, if issued, the patent(s) resulting from the pending application(s) have an expiration date of no earlier than May 2035. Patent Rights Related to Ourour Densigen Platform Technology Fluorocarbon Antigen Delivery Vectors We are developing a fluorocarbon antigen construct platform technology. Our patents covering this technology are issued in the United States, China, Japan and certain European countries, including the United Kingdom, Germany and France. Additional patents are issued in other commercially relevant jurisdictions and an application is pending in the United States. The claims are directed to the fluorocarbon linked antigen construct, compositions comprising the construct and methods of using the construct to stimulate an immune response. The patents and, if issued, the patent(s) resulting from the pending patent applications, are expected to have an expiration date no earlier than April 2025, not giving effect to any potential extensions and assuming payment of all associated fees. Formulation of Antigen Delivery Vectors — Manufacturing Process for the Final Formulation of the Antigen Delivery Vectors We are developing a manufacturing process for solubilizing certain fluorocarbon peptides and final lyophilized compositions thereof that are soluble in an aqueous solution, for which we have a patentpatents issued in the United States, Europe, and Japan and patent applications pending in the United StatesKorea and Japan as well as other commercially relevant jurisdictions.jurisdictions, and a patent application pending in the United States. The claims are directed to methods of solubilizing certain fluorocarbon antigen peptides using acetic acid formulations and manufactured lyophilized compositions thereof that are soluble in an aqueous solution. The patentpatents and, if issued, the patent(s) resulting from the pending patent applications, are expected to have an expiration date no earlier than December 2031, not giving effect to any potential extensions and assuming payment of all associated fees. Patent Rights Related to Ourour Product Candidates AdCOVID, COVID-19 Vaccines
We are developing an intranasal COVID-19 vaccine candidate based on our replication-deficient adenoviral vectored RespirVec platform technology, for which we have a patent application filed in the United States, Taiwan, Argentina and an international (PCT) patent application filed, with expectation that in due course in jurisdictions of commercial consideration (e.g., Europe, Japan) there will be national or regional patent applications based on the PCT. The claims are directed to the AdCOVID vaccine composition and its method of use. If issued, patent(s) resulting from the pending application(s) have an expiration date of no earlier than February 2041, not giving effect to any potential extensions and assuming payment of all associated fees.
AdCOVID is further covered by our patent rights related to our RespirVec platform technology, for which we have patent applications pending in the US, Europe and Japan. The claims are directed to a method of inducing an innate immune response against coronavirus following intranasal administration of an effective amount of E1 and/or E3 deleted adenovirus expressing a coronavirus antigen. The patent application(s), if issued, would have an expiration date no earlier than March 2032, not giving effect to any potential extensions and assuming payment of all associated fees. Use and dosing of AdCOVID is further covered by an international (PCT) patent application, from which we expect to file national and regional applications in the U.S., Europe and other commercially
relevant jurisdictions. The claims are directed to transmucosal administration of our respiratory anti-infective product candidates, including AdCOVID. If issued, the patents resulting from the pending applications have an expiration date of no earlier than August 2040, not giving effect to any potential extensions and assuming payment of all associated fees.
T-COVID, COVID-19 Therapeutic
We are developing an intranasal COVID-19 therapeutic candidate based on our replication-deficient adenoviral vectored RespirVec platform technology, for which we have a patent application filed in the United States, Taiwan, Argentina and an international (PCT) patent application filed, with expectation that in due course in jurisdictions of commercial consideration (e.g., Europe, Japan) there will be national or regional patent applications based on the PCT. The claims are directed to the T-COVID therapeutic composition and its method of use. If issued, patent(s) resulting from the pending application(s) have an expiration date of no earlier than February 2041, not giving effect to any potential extensions and assuming payment of all associated fees.
T-COVID is further covered by our patent rights related to our RespirVec platform technology, for which we have a granted patent in Europe and have recently received a Notice of Allowance from the United States Patent and Trademark Office (“USPTO”) for claims directed to a method of inducing an innate immune response against coronavirus. That patent, once issued, will have an expiration date of no earlier than March 2032. Use and dosing of T-COVID is further covered by an international (PCT) patent application, from which we expect to file national and regional applications in the U.S., Europe and other commercially relevant jurisdictions. The claims are directed to transmucosal administration of our respiratory anti-infective product candidates, including T-COVID. If issued, patent(s) resulting from the pending application(s) have an expiration date of no earlier than August 2040, not giving effect to any potential extensions and assuming payment of all associated fees.
NasoShield, Anthrax Vaccine
We are developing a rapid and prolonged immunologic-therapeutic technology for anthrax (NasoShield), for which we have a patent granted in the United States, Canada, Europe and Japan. Additional patent applications are pending in the United States, Europe, Japan and other commercially relevant jurisdictions. The issued and pending claims are directed to methods for inducing a rapid protective response against anthrax, comprising intranasal administration of an effective amount of E1 and/or E3-deleted adenovirus expressing a Bacillus anthracis antigen. The patent, if issued, resulting from the pending applications are expected to have an expiration date no earlier than March 2032, not giving effect to any potential extensions and assuming payment of all associated fees.
NasoShield is further covered by an issued and pending U.S. patent. The issued claims are directed to methods of inducing a protective immune response against inhalation anthrax, comprising intranasal administration of an adenoviral vector expressing anthrax protective antigen. The patent has an expiration date no earlier than July 2024, not giving effect to any potential extensions and assuming payment of all associated fees.
Use and dosing of NasoShield is further covered by an international (PCT) patent application, from which we expect to file national and regional applications in the US, Europe and other commercially relevant jurisdictions. The claims are directed to transmucosal administration of our respiratory anti-infective product candidates, including NasoShield. If issued, the patent(s) resulting from the pending application(s) have an expiration date of no earlier than August 2040, not giving effect to any potential extensions and assuming payment of all associated fees.
NasoVAX, an Influenza Vaccine
We are developing a rapid and prolonged immunologic-therapeutic technology for influenza, for which we have a patent issued in the United States, Europe and Japan for influenza and patent applications pending in the United States, Canada, Europe and Japan, as well as other commercially relevant jurisdictions. The issued and pending claims are directed to methods for inducing a rapid protective response against influenza, comprising intranasal administration of an effective amount of E1 and/or E3 deleted adenovirus. The patent and, if issued, the patent(s) resulting from the pending applications are expected to have an expiration date no earlier than March 2032, not giving effect to any potential extensions and assuming payment of all associated fees.
Use and dosing of NasoVAX is further covered by an international (PCT) patent application, from which we expect to file national and regional applications in the U.S., Europe and other commercially relevant jurisdictions. The claims are directed to transmucosal administration of our respiratory anti-infective product candidates, including NasoVAX. The patents, if issued, resulting from the pending application have an expiration date of no earlier than August 2040, not giving effect to any potential extensions and assuming payment of all associated fees.
Single dose NasoVAX formulations and their use for inducing a combined mucosal, humoral and T cell immune response against influenza virus is further covered by an international (PCT) patent application, from which we expect to file national and
regional applications in commercially relevant jurisdictions, and a pending U.S. patent application. The claims are directed to a single dose NasoVAX intranasal formulation and its use for inducing a combined immune response in humans providing durable immunity against influenza. If issued, patent(s) resulting from the pending application(s) have an expiration date of no earlier than April 2040, not giving effect to any potential extensions and assuming payment of all associated fees.
ALT-801,Pemvidutide, Dual GLP-1/Glucagon Dual Agonist for Obesity and NASH
We are the exclusive licensee of patent rights owned by Mederis Diabetes, LLC (“Mederis”) to develop and commercialize surfactant functionalized GLP-1/Glucagon/Oxyntomodulin-based(GLP-1-glucagon)/oxyntomodulin-based peptide therapeutics, and variants thereof, including pemvidutide, for any use including the treatment of obesity, metabolic syndrome, insulin resistance, diabetes and cardiovascular disease. Patents under this license agreement (“the Mederis IP License Agreement”)Agreement have been granted in the United States, Europe, Japan, Australia and Mexico with pending applications in the United States, Europe, Japan and Korea, as well as other commercially relevant jurisdictions. The claims are directed to GLP-1/glucagon dual agonist peptides conjugated to a surfactant and their use to treat metabolic syndrome, obesity and other related diseases. The patents and, if issued, the patent(s) resulting from the pending application(s) have an expiration date of no earlier than May 2032 and extending to May 2035, not giving effect to any potential extensions and assuming payment of all associated fees. Use of ALT-801pemvidutide for treating NASH is further covered by, and subject to the Mederis IP License Agreement, with a granted patent in the United States, and pending applications in the U.S.,United States, Europe, Japan and other commercially relevant jurisdictions. IfThe patents and, if issued, the patent(s) resulting from the pending application(s)patent applications, are expected to have an expiration date of no earlier than January 2039, not giving effect to any potential extensions and assuming payment of all associated fees. Use of ALT-801pemvidutide in methods with improved tolerability, dosing and therapeutic regimens is further covered in two pending U.S. provisional patent applications in the United States, Europe, Japan and Korea, as well as other commercially relevant jurisdictions, which are owned by us and not subject to the Mederis IP License Agreement, and from which we expect to file U.S. and international patent applications.Agreement. The claims are directed to liquid formulations and the use of ALT-801pemvidutide in a therapeutic dosing regimen with improved tolerability. If issued, the patent(s) resulting from the pending application(s) have an expiration date of no earlier than February 2041 not giving effect to any potential extensions and assuming payment of all associated fees. Use of pemvidutide in methods for inducing weight loss is further covered in a United States patent application and corresponding international (PCT) patent application owned by us and not subject to the Mederis IP License Agreement, and from which we expect to file National Phase patent applications in commercially relevant jurisdictions. The claims are directed to the use of pemvidutide in a therapeutic dosing regimen for chronic weight management. If issued, the patent(s) resulting from the pending application(s) have an expiration date of no earlier than December 2041, respectively,not giving effect to any potential extensions and assuming payment of all associated fees. Use of pemvidutide in methods for reducing body weight in a human with fatty liver disease is further covered in provisional United States patent applications owned by us and not subject to the Mederis IP License Agreement, and from which we expect to file a United States non-provisional patent application and an international (PCT) patent application. The claims are directed to the use of pemvidutide in methods for reducing body weight in a human with NASH or NAFLD, with or without also having type II diabetes. If issued, the patent(s) resulting from the pending application(s) have an expiration date of no earlier than September 2043, not giving effect to any potential extensions and assuming payment of all associated fees. Use of pemvidutide in methods for reducing the risk of cardiovascular (CV) disease is further covered in a provisional United States patent application owned by us and not subject to the Mederis IP License Agreement, and from which we expect to file a United States non-provisional patent application and an international (PCT) patent application. The claims are directed to the use of pemvidutide in methods for reducing the risk of cardiovascular (CV) disease in a human with or without also having type II diabetes. If issued, the patent(s) resulting from the pending application(s) have an expiration date of no earlier than November 2043, not giving effect to any potential extensions and assuming payment of all associated fees. HepTcell, Chronic Hepatitis B Immunotherapy We are developing an HBV immunotherapy technology directed to compositions comprising fluorocarbon constructs with specific peptide HBV antigen sequences. We have an issued patentpatents for this technology in the United States, Europe, Japan and Korea and pending applications in the United States, Europe, Japan China and Korea,China, as well as other commercially relevant jurisdictions. The claims are directed to HBV antigen peptide sequences comprising T-cell epitopes linked to fluorocarbon chains and compositions comprising a combination of HBV antigen peptide sequences. IfThe patents, and if issued, the patent(s) resulting from the pending patent applications, are expected to have an expiration date no earlier than December 2033, not giving effect to any potential extensions and assuming payment of all associated fees. HepTcell is also covered by the patents and patent applications relating to our Densigen platform technology. Use of HepTcell for treating patients with chronic hepatitis B viral (HBV)HBV infection is further covered by a pending U.S.United States provisional patent application, from which we expect to file U.S.United States and international (PCT) patent applications. The claims are directed to treating patients with chronic hepatitis B virus (HBV)HBV infection characterized with low hepatitis B surface antigen (HBsAg)(“HBsAg”). If issued, the patent(s) resulting from the pending patent application(s) are expected to have an expiration date no earlier than December 2041,2043, not giving effect to any potential extensions and assuming payment of all associated fees. Patent Rights Related to our RespirVec Platform Technology Immunotherapy for Respiratory Pathogens — Intranasal Application of Adenoviral Vector Vaccines While we are focusing primarily in areas other than intranasal application of adenoviral vector vaccines, we currently own patents and applications in multiple patent families related to intranasal vaccines for respiratory pathogens, including influenza, anthrax and COVID-19, using our RespirVec platform technology, with patents that have issued expiring starting not earlier than July 2024 and extending up to February 2040. We are evaluating whether we will continue to prosecute and maintain the patents and applications in these families given our current product focus. Government Contracts Substantially all of our revenues to date have been derived from grants and United States government contracts. We are currently closing out one U.S. government contract, and have no currently active U.S. government funded research programs. There can be no assurances that our remaining U.S. government contract will be continued, renewed beyond the base period, or that we can enter into new contracts or receive new grants to supply the U.S. or other governments with our products. The process of obtaining government contracts is lengthy and uncertain. U.S. government contracts typically are subject to audit by the government and contain termination provisions for the government allowing it to terminate at its discretion, which subjects us to additional risks. These risks include the ability of the U.S. government unilaterally to:
preclude us, either temporarily or for a set period of time, from receiving new contracts or extending our remaining contracts based on violations or suspected violations of laws or regulations;
terminate our remaining contracts either for the convenience of the government (at the government’s sole discretion, for example, if funds become unavailable or the government no longer wants the work) or for default (for failing to perform in accordance with the contract schedule and terms);
| •
| revise the scope and value of our contracts and/or the timing for work to be performed;
|
audit and object to our contract-related costs and fees, including allocated indirect costs;
control and potentially prohibit the export of our products;
claim rights to intellectual property, including our products, developed under the contract;
add or remove the terms and conditions in our contracts; and
cancel or amend planned procurements, including outstanding request for proposal solicitations.
Termination-for-convenience provisions generally enable us to recover only our costs incurred or committed, settlement expenses, and profit on the work completed prior to termination. Termination-for-default provisions do not permit these recoveries and would make us liable for excess costs incurred by the U.S. government in procuring undelivered items from another source.
MTEC COVID-19 Contract We are fundingfunded our Phase 1/2 clinical trial of T-COVID with a $4.7 million grant from the U.S. Army Medical Research & Development Command (“USAMRDC”). The competitive award was granted by USAMRDC in collaboration with the Medical Technology Enterprise Consortium (“MTEC”), a 501(c)(3) biomedical technology consortium working in partnership with the Department of Defense (“DoD”). Under the contract, MTEC payspaid us a firm fixed fee based on achieving certain milestones for conduct and completion of a Phase 1/2 study. As ofThrough December 31, 2020,2021, we have collected an aggregate of approximately $1.1$4.7 million in receivablescash under the contract.contract and received no further funding from this contract in 2022. We own the intellectual property rights to inventions made by us in the performance of work under the MTEC contracts provided that we disclose such inventions to the U.S. government and notifiesnotify the U.S. government of our election to retain title. The U.S. government will have a non-exclusive, non-transferable, irrevocable, paid-up license to practice, or have practiced for or on our behalf, such inventions throughout the world, in addition to other rights customarily reserved by the U.S. government for intellectual property generated using government funds. BARDA Anthrax Contract We arewere developing our NasoShield anthrax vaccine pursuant to a contract with BARDABiomedical Advanced Research and Development Authority (“BARDA”) that commenced in July 2016. Under this contract, BARDA payspaid us a fixed fee and reimburses certain of our costs for the research and development of an Ad5-vectored, protective antigen-based intranasal anthrax vaccine through current good manufacturing practice (“cGMP”) manufacture and conduct of a Phase 1 clinical trial dose ranging assessment of safety and immunogenicity. The contract consistsconsisted of an initial base performance period providing approximately $27.8$30.9 million in funding for the period July 2016 through JuneDecember 2021. BARDA hashad seven options to extend the contract to fund certain continued development and manufacturing activities for the anthrax vaccine, including Phase 2 clinical studiestrials for a three-year period. Each option, if exercised by BARDA, would providehave provided additional funding ranging from approximately $1.1 million to $34.4 million, providing a total contract potential of $133.7$136.8 million. As ofThrough December 31, 2020,2022, we have collected an aggregate of approximately $24.7$29.2 million in receivablescash under the current BARDA contract. BARDA did not extend the contract beyond the end of December 2021. We are currently expecting to collect final payments for indirect rate adjustments for the final two years of the contract. We have been audited by BARDA through 20162019 and have agreed on final indirect rates with the Defense Contract Audit Agency (“DCAA”)BARDA through 2016.2019. We own the intellectual property rights to inventions made by us in the performance of work under the BARDA contracts, provided that we disclose such inventions to the U.S. government and notifiesnotify the U.S. government of our election to retain title. The U.S. government will have a non-exclusive, non-transferable, irrevocable, paid-up license to practice, or have practiced for or on our behalf, such inventions throughout the world, in addition to other rights customarily reserved by the U.S. government for intellectual property generated using government funds. BARDA is a division of the U.S. Department of Health and Human Services (“HHS”) in the Office of the Assistant Secretary for Preparedness and Response that supports the advanced research and development, manufacturing, acquisition and stockpiling of medical countermeasures. Our contracts with BARDA, like those awarded by other U.S. government agencies, contain provisions not typically found in commercial contracts. Most notably, BARDA, or the U.S. government acting through BARDA, may terminate, modify or amend our contract, in whole or in part, for nearly any reason or no reason. NIAID Anthrax Contract
Through the third quarter of 2019, we were developing our SparVax-L anthrax vaccine pursuant to a contract with the National Institute of Allergy and Infectious Diseases (“NIAID”). SparVax-L, a recombinant protein-based anthrax vaccine, was designed to
require fewer doses and have a longer shelf life than the only currently licensed anthrax vaccine. We demonstrated a significant improvement in shelf life (two years at room temperature and six years at refrigerated temperatures) with a lyophilized formulation. Preclinical experiments showed it to be 100% protective with a two-dose regimen (administered on study Days 0 and 14 days) with higher protective (toxin neutralizing) antibodies than the currently licensed vaccine administered under the same schedule. Activities under this contract were completed during the quarter ended September 30, 2019 and no further funding is expected for this program. As a result of the contract completion and the U.S. government’s funding prioritization of only single dose anthrax vaccine candidates, we abandoned the project and impaired the $1.0 million net book value of the SparVax-L IPR&D asset in 2019.
United States Government Regulation BiologicalThe FDA regulates drugs and biological products such as our product candidates, are subject to regulation under the Federal Food, Drug, and Cosmetic Act (“FD&C Act”), the Public Health Service Act (“PHS Act”), the FDA regulations under TitleTitles 21 and 42 of the Code of Federal Regulations (21 CFR and 42 CFR), as well as other federal, state and local statutes and regulations. Both theThe FD&C Act and the PHS Act and their corresponding regulations govern, among other things, the testing, research, manufacturing, approval, safety, efficacy, labeling, packaging, storage, record keeping, distribution, import, export, reporting, sale, advertising and other promotional practices involving drugs and biological products. An investigational new drug (“IND”)IND application must be in effect before clinical testing of drugs and biological products can begin. FDA approval must be obtained before drugs and biological products can be marketed. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources, and each process may take several years to complete, although certain expedited programs potentially applicable to our product candidates, such as FDA fast track designation for certain new drugs with the potential to address unmet medical needs for certain serious or life-threatening conditions, may potentially expedite development and/or approval processes. Certain federal incentive programs are also potentially applicable to our product candidates, such as for “orphan drugs” that treat rare conditions, and programs supporting the development of bioterrorism medical countermeasures.conditions. Data obtained from clinical activities is not always conclusive and may be susceptible to varying interpretations, which could delay, limit or prevent regulatory approval. The FDA may not grant approval on a timely basis, or at all, and we may encounter difficulties or unanticipated costs in our efforts to secure necessary governmental approvals, which could delay or preclude us from marketing our product candidates. In addition, the FDA may limit the indications for use or place other conditions on any approvals that could restrict the commercial application of the products. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further testing requirements and FDA review and approval. In addition, our failure, or the failure of our third-party manufacturers, to comply with applicable regulations could result in sanctions being imposed on us, including clinical holds, fines, injunctions, civil penalties, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of product candidates or products, operating restrictions and criminal prosecutions, any of which could adversely affect our ability to commercialize our product candidates.
Drug andBiological Products Development Process The process required by the FDA before a drug or biological product may be marketed in the United States generally involves the following: | ● | completion of preclinical laboratory tests and animal studies according to applicable good laboratory practices (“GLP”), applicable requirements for the humane use of laboratory animals, such as the Animal Welfare Act or other applicable regulations; |
| ● | submission to the FDA of an application for an IND which must become effective before human clinical trials may begin; |
| ● | obtaining approval by an independent Institutional Review Board (“IRB”) at each clinical site before a clinical trial may be initiated at that site; |
| ● | performance of adequate and well-controlled human clinical trials according to the FDA’s regulations commonly referred to as good clinical practices (“GCP”) and any additional requirements for the protection of human research subjects and their health information, to establish the safety and efficacy of the proposed product for its intended use; |
submission to the FDA of an application for an IND which must become effective before human clinical trials may begin;
obtaining approval by an independent Institutional Review Board (“IRB”) at each clinical site before a clinical trial may be initiated at that site;
submission to the FDA of a biologics license application (“BLA”) for marketing approval that includes substantial evidence of safety, purity and potency from results of clinical trials, as well as the results of preclinical testing, detailed information about the chemistry, manufacturing and controls, and proposed labeling and packaging for the product candidate;
review of the product candidate by an FDA advisory committee, if applicable;
| •
● | submission to the FDA of a new drug application (“NDA”) or biologics license application (“BLA”) for marketing approval that includes substantial evidence of safety, purity and potency from results of clinical trials, as well as the results of preclinical testing, detailed information about the chemistry, manufacturing and controls, and proposed labeling and packaging for the product candidate; |
| ● | review of the product candidate by an FDA advisory committee, if applicable; |
| ● | satisfactory completion of an FDA inspection of the manufacturing facility or facilities where the biological product candidate is produced to assess compliance with cGMP and to confirm that the facilities, methods and controls are adequate to assure the biological product candidate’s identity, strength, quality and purity; |
| ● | satisfactory completion of potential FDA audits of the preclinical study and clinical trial sites that generated the data in support of the NDA or BLA; and |
| ● | FDA review and approval, or licensure, of the NDA or BLA, including agreement on post-marketing commitments, if applicable. |
satisfactory completion of potential FDA audits of the preclinical study and clinical trial sites that generated the data in support of the BLA; and
FDA review and approval, or licensure, of the BLA, including agreement on post-marketing commitments, if applicable.
Before testing any drug or biological product candidate in humans, the product candidate enters the preclinical study stage. Preclinical studies include laboratory evaluations of product chemistry, toxicity and formulation, as well as animal studies to assess the potential safety and activity of the product candidate. The conduct of certain preclinical studies must comply with federal regulations and requirements including GLP and the Animal Welfare Act. The clinical trial sponsor must submit the results of the preclinical studies, together with manufacturing information, analytical data, any available clinical data or literature and a proposed clinical trial protocol, to the FDA as part of the IND. Some preclinical studies may continue even after the IND is submitted. The FDA requires a 30-day waiting period after the filing of each IND before clinical trials may begin. The FDA may also place the clinical trial on a clinical hold within that 30-day time period. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. Even after the IND has gone into effect and clinical testing has begun, the FDA may also impose a partial or complete clinical holdshold on clinical trials due to safety concerns or non-compliance. IfA partial clinical hold would limit a trial, for example, to certain doses or for a certain length of time or to a certain number of subjects. A complete clinical hold order issued by the FDA imposeswould delay a proposed clinical hold, studiesstudy or cause suspension of an ongoing study until all outstanding concerns have been adequately addressed and the FDA has notified the company that investigations may not recommence without FDA authorization and then only under terms authorized by the FDA.proceed or resume. Accordingly, we cannot be sure that submission of an IND will result in the FDA allowing clinical trials to begin, or that, once the trials have begun, issues will not arise that suspend or terminate such studies. Clinical trials involve the administration of the biological product candidate to healthy volunteers or patients under the supervision of qualified investigators. Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria, and the parameters to be used to monitor subject safety, including stopping rules that assure a clinical trial will be stopped if certain adverse events (“AEs”), should occur. Each protocol and any amendments to the protocol must be submitted to the FDA as part of the IND during applicable phases of development. Clinical trials must be conducted and monitored in accordance with the FDA’s regulations and GCP requirements, which establish standards for conducting, recording data from, and reporting the results of clinical trials, with the goals of assuring that the data and results are credible and accurate and that study participants’ rights, safety, and well-being are protected. GCP requirements include the requirement that all research subjects provide informed consent. Further, each clinical trial must be reviewed and approved by an IRB at or servicing each institution at which the clinical trial will be conducted. An IRB is charged with protecting the welfare and rights of trial participants and considers such items as whether the risks to individuals participating in the clinical trials, not only from the investigational product itself but also from any required procedures or study visits to be conducted during the trial, are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the form and content of the informed consent that must be signed by each clinical trial subject or his or her legal representative and must monitor the clinical trial until completed. Human clinical trials are typically conducted in three sequential phases that may overlap or be combined: | ● | Phase 1. The drug or biological product is initially introduced into a small group of healthy human subjects (e.g., 10 to 20 volunteers) and tested for safety and to develop detailed profiles of its pharmacological and pharmacokinetic actions, determine side effects associated with increasing doses, and, if possible, gain early evidence of effectiveness. In the case of some products for severe or life-threatening diseases, especially when the product may be too inherently toxic to ethically administer to healthy volunteers, the initial human testing is often conducted in patients. |
| ● | Phase 2. The drug or biological product is evaluated in a larger but limited patient population (e.g., a few hundred patients with the disease or conduction under study) to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance, optimal dosage and dosing schedule. |
| ● | Phase 3. These clinical trials are undertaken to further evaluate dosage, clinical efficacy, potency and safety in an expanded patient population (e.g., several hundred to several thousand patients) at geographically dispersed clinical trial sites. These clinical trials are intended to establish the overall risk/benefit profile of the product and provide an adequate basis for product labeling. |
Phase 1. The biological product is initially introduced into a small group of healthy human subjects (e.g., 10 to 20 volunteers) and tested for safety and to develop detailed profiles of its pharmacological and pharmacokinetic actions, determine side effects associated with increasing doses, and, if possible, gain early evidence of effectiveness. In the case of some products for severe or life-threatening diseases, especially when the product may be too inherently toxic to ethically administer to healthy volunteers, the initial human testing is often conducted in patients.
Phase 2. The biological product is evaluated in a larger but limited patient population (e.g., a few hundred patients) to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance, optimal dosage and dosing schedule.
Phase 3. These clinical trials are undertaken to further evaluate dosage, clinical efficacy, potency and safety in an expanded patient population (e.g., several hundred to several thousand patients) at geographically dispersed clinical trial sites. These clinical trials are intended to establish the overall risk/benefit profile of the product and provide an adequate basis for product labeling.
Post-approval clinical trials, sometimes referred to as Phase 4 clinical trials, may be conducted after initial marketing approval. These clinical trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication, particularly for long-term safety follow-up.
During all phases of clinical development, regulatory agencies require extensive monitoring and auditing of all clinical activities, clinical data and clinical trial investigators. Annual progress reports detailing the results of the clinical trials must be submitted to the FDA. Written IND safety reports must be promptly submitted to the FDA and the investigators for serious and unexpected AEs, any findings from other studies, tests in laboratory animals or in vitro testing and other sources that suggest a significant risk for human subjects, or any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigator brochure. The sponsor must submit an IND safety report within 15 calendar days after the sponsor determines that the information qualifies for reporting. The sponsor also must notify the FDA of any unexpected fatal or life-threatening suspected adverse reaction within seven7 calendar days after the sponsor’s initial receipt of the information. Phase 1, Phase 2 and Phase 3 clinical trials may not be completed successfully within any specified period, if at all. The FDA or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug or biological product has been associated with unexpected serious harm to patients. In limited circumstances, FDA also permits the administration of investigational small molecule drug or biological products to patients under its expanded access regulatory authorities. Under the FDA’s expanded access authority, patients who are not able to participate in a clinical trial may be eligible for accessing investigational products, including through individual compassionate or emergency use in concert with their requesting physician. For medical countermeasure (“MCM”) focused products, in particular, FDA has recently indicated that its expanded access authorities may be important in enabling quicker access to investigational products intended for MCM uses in the COVID-19 pandemic. Concurrent with clinical trials, companies usually complete additional animal studies, develop additional information about the physical characteristics of the drug or biological product candidate and finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. To help reduce the risk of the introduction of adventitious agents with use of biological products, the PHS Act emphasizes the importance of manufacturing controls for products whose attributes cannot be precisely defined. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, the sponsor must develop methods for testing the identity, strength, quality, potency and purity of the final biological product. Additionally, appropriate packaging must be selected and tested, and stability studies must be conducted to demonstrate that the biological product candidate does not undergo unacceptable deterioration over its shelf life. Certain FDA programs are available to facilitate and expedite the development and review of new drugs intended to address unmet needs in the treatment of serious or life-threatening conditions. These expedited programs include fast track designation, breakthrough therapy designation, priority review and accelerated approval. Each of these programs has its own features and qualifying criteria. A sponsor must submit a request for fast track designation, breakthrough therapy designation, or priority review, which may or may not be granted by the FDA. For fast track and breakthrough therapy designations, FDA may later decide the product no longer meets the conditions for designation and may rescind the designation. For accelerated approval, a sponsor generally discusses the possibility of accelerated approval with the FDA during development, and the FDA may or may not agree that accelerated approval is an appropriate pathway for a particular drug. Some of these expedited programs could potentially apply to our product candidates, although this cannot be assured, and we do not currently have any products with expedited program designations. The sponsor of a clinical trial or the sponsor’s designated responsible party may be required to register certain information about the trial and disclose certain results on government or independent registry websites, such as ClinicalTrials.gov. Additionally, a manufacturer of an investigational drug for a serious disease or condition is required to make available, such as by posting on its website, its policy on evaluating and responding to requests for individual patient access to such investigational drug. This requirement applies on the earlier of the first initiation of a Phase 2 or Phase 3 trial of the investigational drug or, as applicable, 15 days after the drug receives a designation as a breakthrough therapy, fast track product, or regenerative medicine advanced therapy. Review and Approval Processes After the completion of clinical trials of a drug or biological product candidate, the FDA’s approval of aan NDA or BLA must be obtained before commercial marketing of the biological product may begin. The NDA or BLA must include results of product development, laboratory and animal studies, human studies, information on the manufacture and composition of the product, proposed labeling and other relevant information. In addition, under the Pediatric Research Equity Act, as amended, aan NDA or BLA or supplement to aan NDA or BLA generally must contain data to assess the safety and effectiveness of the biological product for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may grant deferrals for submission of data or full or partial waivers depending on the designated pathway for submission. The testing and approval processes require substantial time and effort and there can be no assurance that the FDA will accept the NDA or BLA for filing and, even if filed, that any approval will be granted on a timely basis, if at all.
Under the Prescription Drug User Fee Act (“PDUFA”), as amended, each NDA or BLA must be accompanied by a significant user fee. PDUFA also imposes an annual prescription drug product program fee for biologics. Fee waivers or reductions are available in certain circumstances, including a waiver of the application fee for the first application filed by a small business. Following submission of the application, the FDA reviews the NDA or BLA to determine if it is substantially complete before the agency accepts it for filing. The FDA may refuse to file any NDA or BLA that it deems incomplete or not properly reviewable at the time of submission and may request additional information. In this event, the NDA or BLA must be resubmitted with the additional information. The resubmitted application also is subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review of the NDA or BLA. FDA performance goals generally provide for action on aan NDA or BLA within 10 months of the 60-day filing date, orwhich would be within 12 months of its submission. That deadline can be extended under certain circumstances, including by the FDA’s requests for additional information. The targeted action date can also be shortened to 6 months of the 60-day filing date, or 8 months after submission, for products that are granted priority review designation because they are intended to treat serious or life-threatening conditions and demonstrate the potential to address unmet medical needs. The FDA reviews the NDA or BLA to determine, among other things, whether the proposed product is safe and potent, or effective for its intended use, has an acceptable purity profile, and whether the product is being manufactured in accordance with cGMP to assure the product’s identity, safety, quality, potency and purity. The FDA may refer applications for noveldrugs or biological products that are novel or biological products that present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions. During the NDA or BLA review process, the FDA also will determine whether a Risk Evaluation and Mitigation Strategy (“REMS”) is necessary to assure the safe use of the drug or biological product. If the FDA concludes a REMS is needed, the sponsor of the NDA or BLA must submit a proposed REMS; the FDA will not approve the NDA or BLA without a REMS, if required. A REMS could include medication guides, physician communication plans or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. Any of these limitations on approval or marketing could restrict the commercial promotion, distribution, prescription or dispensing of products. Before approving aan NDA or BLA, the FDA will inspect the facilities at which the product is manufactured. The FDA will not approve the NDA or BLA unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving aan NDA or BLA, the FDA will typically inspect one or more clinical sites to assure that the clinical trials were conducted in compliance with IND study requirements and GCP requirements. To assure cGMP and GCP compliance, an applicant must incur significant expenditure of time, money and effort in the areas of training, record keeping, production and quality control. Notwithstanding the submission of relevant data and information, the FDA may ultimately decide that the NDA or BLA does not satisfy its regulatory criteria for approval. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently than how we interpret the same data. If the agency decides not to approve the NDA or BLA in its present form, the FDA will issue a complete response letter that usually describes all of the specific deficiencies in the NDA or BLA identified by the FDA. The deficiencies identified may be minor, for example, requiring labeling changes; or major, for example, requiring additional clinical trials. Additionally, the complete response letter may include recommended actions that the applicant may take for the FDA to reconsider the application. If a complete response letter is issued, the applicant may either resubmit the NDA or BLA, addressing all of the deficiencies identified in the complete response letter, or withdraw the application. If a product receives regulatory approval, the approval may be significantly limited to specific diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings or precautions be included in the product labeling. The FDA may impose restrictions and conditions on product distribution, prescribing, or dispensing in the form of a REMS, or otherwise limit the scope of any approval. In addition, the FDA may require post-marketing clinical trials, sometimes referred to as Phase 4 clinical trials, designed to further assess a biological product’s safety and effectiveness, and testing and surveillance programs to monitor the safety of approved products that have been commercialized. Post-approval modifications to a drug product, such as changes in indications, labeling or manufacturing processes or facilities, may require development and submission of additional information or data in a new or supplemental NDA or BLA, which would also require prior FDA approval. Post-Approval Requirements After regulatory approval of a product is obtained, products are subject to extensive continuing extensive regulation and post-approval requirements. For example, as a condition of approval of aan NDA or BLA, the FDA may require post-marketing testing and surveillance to monitor the product’s safety or efficacy. In addition, holders of an approved NDA or BLA are required to submit annual reports, keep extensive records, tosubmit annual reports, report certain adverse reactions and production problems to the FDA, to provide updated safety and efficacy information, and to comply with requirements concerning advertising and promotional labeling for their products. Also, quality control and manufacturing procedures must continue to conform to cGMP regulations and practices, as well as the manufacturing conditions of approval set forth in the NDA or BLA. TheDrug manufacturers and their subcontractors and those supplying products, ingredients, and components are required to register their establishments with the FDA and certain state agencies, and the FDA periodically inspects manufacturing facilities to assess compliance with cGMP, which imposes certain procedural, substantive and recordkeeping requirements. Manufacturers and other parties involved in the drug supply chain for prescription drug products also must comply with product tracking and tracing requirements and notify the FDA of counterfeit, diverted, stolen and intentionally adulterated products or products that are otherwise unfit for distribution in the United States. Accordingly, manufacturers must continue to expend time,
money and effort in the area of production and quality control to maintain compliance with cGMP and other aspects of regulatory compliance. If, after receiving approval, a company makes a material change in manufacturing equipment, location, or process (all of which are, to some degree, incorporated in the NDA or BLA), additional regulatory review and approval may be required. Future FDA inspections may identify cGMP compliance issues at manufacturer facilities or at the facilities of third-party suppliers that may disrupt production or distribution or require substantial resources to correct and prevent recurrence of any deficiencies, and could result in fines or penalties by regulatory authorities. In addition, discovery of problems with a product or the failure to comply with applicable requirements may result in restrictions on a product, manufacturer or holder of an approved NDA or BLA, including withdrawal or recall of the product from the market or other voluntary, FDA-initiated or judicial action, including warning letters, fines, injunctions, civil penalties, license revocations, seizure, total or partial suspension of production or criminal penalties, any of which could delay or prohibit further marketing. Newly discovered or developed safety or efficacy data may require changes to a product’s approved labeling, including the addition of new warnings and contraindications. Certain U.S. Regulatory Incentives and Other Programs Priority Rule Voucher (PRV)Marketing Exclusivity and Patent Term Restoration
The 21st Century CuresDrug Price Competition and Patent Term Restoration Act (“Cures Act”), which was signed into law on December 13, 2016,of 1984, also referred to as the Hatch-Waxman Amendments, established acertain periods of marketing exclusivity for new priority review voucher (“PRV”) program for material threat MCMs. Upon approval of a material threat MCM application, the FDA will award a PRV provided certain criteria are met. A PRV voucher can be used to obtain priority review for a subsequent marketing application that would not otherwise have met the criteria for priority review. When a marketing application receives priority review, the FDA’s goal is to take action on that application within 6 months of the filing date. To be considered a material threat MCM application, the application must be: (i) intended for use to prevent or treat harm from a chemical, biological, radiological, or nuclear (“CBRN") agent (or intended to mitigate, prevent or treat harm caused by an MCM used against such agent) determined by the Department of Homeland Security to be a material threat; (ii) eligible for priority review; (iii) approved after the date of enactment of the Cures Act; and (iv) for a drug for which an active ingredient has not been previouslydrugs approved by the FDA. AlthoughFDA, including a five-year period of non-patent marketing exclusivity within the material threat MCM PRV programUnited States to the first applicant to gain approval of an NDA for a new chemical entity. A drug is separate and distinct froma new chemical entity if the FDA’sFDA has not previously approved any other PRV programs, FDA intends to implementnew drug containing the law consistently with implementationsame active moiety, which is the molecule or ion responsible for the action of the other PRV programs.
Animal Rule and Project BioShield Emergency Use Authorization
In 2002,drug substance. During the exclusivity period, the FDA amended its requirements applicablemay not accept for review an abbreviated new drug application (“ANDA”) or a 505(b)(2) NDA submitted by another applicant for such drug where the applicant does not own or have a right of reference to BLAsthe data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement. The Hatch-Waxman Amendment also established a three-year period of marketing exclusivity for an NDA, 505(b)(2) NDA, or supplement to permitan NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the approval of certain biologics thatapplicant are intended to treat or prevent serious or life-threatening conditions causeddeemed by exposure to lethal or permanently disabling toxic CBRN substances based on evidence of safety from trial in healthy human subjects and animal studies, and effectiveness from appropriate animal studies when human efficacy studies are not ethical or feasible. These regulations, also known as the “Animal Rule,” and published in the Code of Federal Regulations (21 CFR 601 Subpart H), authorize the FDA to rely on evidence from animal studiesbe essential to provide evidence of a product’s effectiveness under circumstances where there is a reasonably well-understood mechanism for the toxicity of the agent and its prevention or substantial reduction by the biological product, the effect is demonstrated in more than one animal species expected to react with a response predictive for humans, the animal study endpoint is clearly related to the desired benefit in humans, and the data or information on the kinetics and pharmacodynamics of the product or other relevant data or information, in animals and humans, allows selection of an effective dose in humans. Under these requirements, and with FDA’s prior agreement, biologics used to reduce or prevent the toxicity of CBRN substances may be approved for use in humans based on evidence of effectiveness derived from appropriate animal studies and any additional supporting data. Products evaluated for effectiveness under this rule are evaluated for safety under preexisting requirements for establishing the safety of new drug and biological products, including Phase 1 through Phase 2 clinical trials. Under certain circumstances a single animal species may be acceptable if that animal model is sufficiently well-characterized for predicting a response in humans. The Animal Rule also requires post-marketing studies, such as field studies, to verify and describe the product’s clinical benefit and assess its safety should an exigency exist that leads to the product being used in humans; the nature of these studies will be discussed with the FDA as part of the BLA process. Products approved under the Animal Rule are subject to additional requirements, such as restrictions imposed on distribution or labeling requirements to inform patients that the product’s approval was based on efficacy studies conducted in animals alone. The Animal Rule drug development pathway typically involves costs and time for nonclinical studies and animal models in excess of what would be expended in conducting human vaccine clinical trials not requiring compliance with the Animal Rule.
Under the Project BioShield Act of 2004 and subsequent amendments by the Pandemic and All-Hazards Preparedness Reauthorization Act of 2013 (“Project Bioshield”), the Secretary of HHS may, with the concurrence of the Secretary of the Department of Homeland Security (“DHS”), and upon the approval of the President, contract to purchase unapproved MCMs for the Strategic National Stockpile (“SNS”)application (e.g., in specified circumstances. The U.S. Congress is notified of a recommendation for a stockpile purchase after Presidential approval. Project BioShield specifies that a company supplying the countermeasure to the SNS is paid on delivery of a substantial portion of the countermeasure. To be eligible for purchase under these provisions, the Secretary of HHS must
determine that there are sufficient and satisfactory clinical resultsnew indications, dosages or research data, including data, if available, from preclinical studies and clinical trials, to support a reasonable conclusion that the countermeasure will qualify for FDA approval or licensing within eight years. The legislation also allows unlicensed products to be procured for the SNS so that they are available at the time an emergency is declared.
Project BioShield also allows the Secretary of HHS to authorize the emergency use of unapproved medical products or an unapproved usestrengths of an approved medical product under certain emergency circumstances (“Emergency Use Authorization”)drug). To exercise this authority,This three-year exclusivity covers only the conditions of use associated with the new clinical investigations and does not prohibit the FDA must conclude that:
from approving ANDAs for drugs containing the CBRN agentactive ingredient for which the countermeasure is designed to treat can cause serious or life-threatening disease; based on the totalityother conditions of scientific evidence available to the FDA, including data from adequate and well-controlled clinical trials, if available, it is reasonable to believe that the product may be effective in diagnosing, treating or preventing the disease;
the known and potential benefits of the product outweigh its known and potential risks; and
there is no adequate, approved, and available alternative to the product for diagnosing, preventing, or treating such disease or condition.
Some product candidates may be eligible both for consideration for procurement into the SNS and for Emergency Use Authorization, although there is no guarantee that our product candidates will meet the criteria set forth by HHS or the FDA for procurement and Emergency Use Authorization, respectively.use.
Marketing Exclusivity for Reference Biological Products
As part of the ongoing efforts of governmental authorities to lower health care costs by facilitating generic competition to pharmaceuticalFor biological products, the Biologics Price Competition and Innovation Act of 2009 (“BPCIA”), enacted as part of the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010 (collectively, the “Health Care Reform Law”), created a newan abbreviated regulatory approval pathway in the United States for biological products that are found to be “biosimilar” orto (and in some instances “interchangeable” withwith) a biological “reference product” previously licensed under aan NDA or BLA. This abbreviated approval pathway is intended to permit a biosimilar to come to market more quickly and less expensively by relying to some extent on the data generated by the reference product’s sponsor and the FDA’s previous review and approval of the reference product. Under the BPCIA, a biosimilar sponsor’s ability to seek or obtain approval through the abbreviated pathway is limited by periods of exclusivity granted by the FDA to the holder of the reference product’s BLA, andNDA or BLA. In general, no biosimilar application may be accepted by the FDA for review until 4 years after the date the reference product was first licensed by the FDA, and no biosimilar application, once accepted, may receive final approval until 12 years after the reference product was first licensed by the FDA.
While we would expect to be granted this 12-year period of exclusivity for our applicable product candidates, if approved, notably, this period of reference product market exclusivity applies only to the biosimilar pathway and will not, for example, provide protection against any biological product for a similar indication that achieves FDA approval under a traditional NDA or BLA based on the sponsor’s own research data. There is also risk that the 12-year period of biological reference product exclusivity could be shortened due to congressional action, or that the FDA will not consider our product candidates, if they are approved, to be reference products for competing products, potentially creating the opportunity for competition sooner than anticipated. Once approved, biosimilars likely would compete with, and in some circumstances may be deemed under the law to be “interchangeableinterchangeable with,” the previously approved reference product. The extent to which a biosimilar, once approved, willwould be substituted for any one of our product candidates, if approved, in a way that is similar to traditional generic substitution for non-biological products is not yet clear, and will depend on a number of marketplace and regulatory factors that are still developing. Although there isGiven this uncertainty, regarding the impact of this program, there is risk that, the approval ofif approved, a biosimilar competitor to one of our products if approved, could have an adverse impact on our business. In particular, a biosimilar could be significantly less costly to bring to market and priced significantly lower than our product, if approved by the FDA. Additionally, products approved under an NDA or BLA may qualify for the restoration of a portion of the patent term lost during product development and FDA review of the application, if approval of the application is the first permitted commercial marketing of a drug containing the active ingredient. The patent term restoration period is generally one-half the time between the effective date of the IND or the date of patent grant (whichever is later) and the date of submission of the NDA or BLA, plus the time between the date of submission of the NDA or BLA and the date of FDA approval of the product. The maximum period of restoration is five years, and the patent cannot be extended to more than 14 years from the date of FDA approval of the product. Only one patent claiming each approved product is eligible for restoration and the patent holder must apply for restoration within 60 days of approval. The USPTO, in consultation with the FDA, reviews and approves the application for patent term restoration. Pediatric Exclusivity Biologics,Drugs and biological products, such as our product candidates, may be eligible for pediatric exclusivity, an incentive intended to encourage medical product research for children. Pediatric exclusivity, if granted, addsmay add six months to existingcertain patents or regulatory exclusivity periods applicable to an approved drug and six months to regulatory exclusivity periods applicable to an approved biological products — namely, the four-year period during which the FDA will not consider an application for a biosimilar product, and the 12-year period during which the FDA will not approve a biosimilar application.product. This six-monthadditional six months of exclusivity which runs from the end of these exclusivity protection periods, may be granted based on the completion of one or more pediatric trials in response to a Written Request from the FDA. It is possible, but not assured, that certain of our current or future product candidates may be targeted to pediatric populations, such as our influenza vaccine candidate.populations.
Orphan Drug Designation The FDA may grant orphan drug designation to drugs intended to treat a “rare disease or condition” that affects fewer than 200,000 individuals in the United States, or that affects more than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making available in the United States a drug for such a disease or condition will be recovered from sales in the United States for that drug. Orphan drug designation must be requested before submitting an application for marketing approval. Orphan drug designation can provide opportunities for grant funding towardstoward clinical trial costs, tax advantages and FDA user fee exemptions. In addition, if a product that has an orphan drug designation subsequently receives FDA approval for the indication for which it has such designation, the product may be entitled to orphan drug exclusivity, which means the FDA would not approve any other application to market the same drug for the same indication for a period of seven years, except in limited circumstances, such as a showing of clinical superiority to the product with orphan exclusivity. It is possible, but not assured, that certain of our current or future product candidates may be targeted totarget rare diseases or conditions. U.S. Regulations Affecting Health Care Companies Pharmaceutical manufacturers with products that are reimbursed by U.S. federally funded health care programs such as Medicare and Medicaid are subject to so-called fraud and abuse laws including false claims and anti-kickback laws. The federal Anti-Kickback Law prohibits anyone from, among other things, knowingly and willingly, directly or indirectly, soliciting, receiving, offering, or paying any remuneration with the intent of (or in return for) generating referrals of individuals or purchases, leases, orders, or recommendationsarranging for servicesthe purchase, lease, or itemsorder of any healthcare item or service reimbursable by federal health care programs like Medicare and Medicaid. Courts have interpreted this law very broadly, including by holding that a violation has occurred if one purpose of the remuneration is to generate referrals even if there are other lawful purposes. Moreover, liability under the Anti-Kickback Law may be established without proving actual knowledge of the law or specific intent to violate. There are statutory exceptions and regulatory safe harbors that protect certain arrangements from prosecution or administrative sanctions, but the exceptions and safe harbors are drawn narrowly. The fact that an arrangement does not fall within a safe harbor does not necessarily render the conduct illegal under the Anti-Kickback Law, but the arrangement may be subject to scrutiny based on the facts and circumstances. Practices that involve remuneration to those who prescribe, purchase, or recommend pharmaceutical products, including certain discounts, or engaging such individuals as consultants, advisors and speakers, may be subject to scrutiny if they do not fit squarely within an exception or safe harbor. Moreover, there are no safe harbors for many common practices, such as educational and research grants, charitable donations, product support and patient assistance programs. Violations of the Anti-Kickback Law may be punished by civil and criminal penalties, damages, fines, or exclusion from participation in federal health care programs like Medicare and Medicaid. Many states have enacted similar laws, some of which apply regardless of payer. The Federal civil False Claims Act (“FCA”) prohibits any person from, among other things, knowingly presenting or causing to be presented a false or fraudulent claim for payment of government funds, or knowingly making, using, or causing to be made or used, a false record or statement material to a false or fraudulent claim to the federal government, or knowingly concealing or knowingly and improperly avoiding, decreasing, or decreasingconcealing an obligation to pay or transmit money or property to the government. The FCA is commonly used to sueenforced against those who submit allegedly false Medicare or Medicaid claims, as well as those who induce or assist others to submit a false claim. “False claims” can result not only from non-compliance with the express requirements of applicable governmental reimbursement programs, such as Medicaid or Medicare, but also from non-compliance with other laws, such as the Anti-Kickback Law or laws that require quality care in service delivery. Actions under the FCA may be brought by the government or by whistleblowers referred to as “relators,” who may initiate an action in the name of the government and the individual and may share in any monetary recovery. Violations of the FCA can result in treble damages, mandatory per claim penalties, and exclusion from participation in federal health care programs. Most states have adopted similar state false claims laws, some of which are broader than the FCA, and these state laws have their own penalties which may be in addition to FCA penalties. The Health Care Reform Law significantly strengthened the FCA and federal Anti-Kickback Law provisions, which could lead to the possibility of increased whistleblower or relator suits, and among other things, made clear that a federal Anti-Kickback Law violation can be a basis for federal FCA liability. The bringing of any FCA or other enforcement investigation or action, even if unsuccessful, could require us to devote resources to investigate and defend the action, as well as result in reputational harm. Failure to comply with fraud and abuse laws could result in significant civil and criminal penalties and costs, including the loss of licenses and the ability to participate in federal and state health care programs, and could have a material adverse effect on our business. In addition, many of these laws are vague, subject to modification, and are subject to evolving interpretation by prosecutorial and regulatory authorities, increasing the risk of noncompliance. We cannot predict whether changes in applicable law, or interpretation of laws, or changes in our services or marketing practices in response to changes in applicable law or interpretation of laws, could have a material adverse effect on our business. In addition to the above, several other laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, structuring and commission(s), certain customer incentive programs and other business arrangements
generally. Activities subject to these laws also involve the improper use of information obtained in the course of patient recruitment for clinical trials. Some state laws restrict whether and when pharmaceutical companies may provide meals to health care professionals or engage in other marketing-related activities; some states require certain compliance program elements and disclosures; and certain states and cities require identification or licensing of sales representatives. For example, the federal Health Insurance Portability and Accountability Act of 1996, and its implementing regulations (collectively, “HIPAA”), created additional federal criminal statutes that prohibitprohibits knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program or obtain, by means of false or fraudulent pretenses, representations, or promises, any of the money or property owned by, or under the custody or control of, any healthcare benefit program, regardless of the payorpayer (e.g., public or private) and knowingly and willfully falsifying, concealing or covering up by any trick or device a material fact or making any materially false statements in connection with the delivery of, or payment for, healthcare benefits, items or services relating to healthcare matters. Similar to the Anti-Kickback Law, a person or entity can be found guilty of violating the HIPAA fraud statute without actual knowledge of the statute or specific intent to violate it. Violations of HIPAA fraud provisions may result in criminal, civil and administrative penalties, fines and damages, including exclusion from participation in federal healthcare programs. Privacy Laws In the U.S., we may be subject to data privacy and security laws and regulations by both the federal government and the states in which we conduct our business. The legislative and regulatory landscape for privacy and data protection continues to evolve, and there has been an increasing focus on privacy and data protection issues which may affect our business. Numerous federal and state laws and regulations, including state data breach notification laws, state health information and/or genetic privacy laws and federal and state consumer protection laws (e.g., Section 5 of the FTC Act, HIPPA and the California Consumer Privacy Act (“CCPA”)), govern the collection, use, disclosure, and protection of health-related and other personal information. Many of these laws differ from each other in significant ways and may not have the same effect,be interpreted and applied in a manner that is inconsistent from one jurisdiction to another, thus complicating compliance efforts. Compliance with these laws is difficult, constantly evolving, and time consuming. Federal regulators, state attorneys general, and plaintiffs’ attorneys, including class action attorneys, have been and will likely continue to be active in this space. Any failure or perceived failure by us to comply with such laws may result in governmental enforcement actions, litigation, fines and penalties and/or adverse publicity, and could have an adverse effect on our reputation and business. The CCPA, for example establishes certain requirements for data use and sharing transparency, and provides California residents certain rights concerning their personal data. In November 2020, California voters approved the California Privacy Rights Act (“CPRA”) which introduced significant amendments to the CCPA and established and funded a dedicated California privacy regulator, the California Privacy Protection Agency. The amendments introduced by the CPRA became effective on January 1, 2023, and it is not yet fully clear how the CCPA and CPRA will be enforced and interpreted. The effects of the CCPA and CPRA are potentially significant and may require us to modify our data collection or processing practices and policies and to incur substantial costs and expenses in an effort to comply. Certain other states have passed legislation similar to the CCPA, which will provide individuals with new privacy rights and increase the privacy and security obligations of entities handling certain personal data of such individuals. For example, in March 2021, Virginia enacted the Consumer Data Protection Act which became effective on January 1, 2023. In July 2021, Colorado passed the Colorado Privacy Act, which will become effective on July 1, 2023. Additionally, in March 2022, Utah enacted the Utah Consumer Privacy Act, which will become effective on December 31, 2023. Also, in May 2022, Connecticut signed the Connecticut Data Privacy Act into law, which will become effective on July 1, 2023. A number of additional states have proposed bills for comprehensive privacy legislation, and it is possible that certain of these bills will pass. The existence of new comprehensive privacy laws in different states in the country, if enacted, could add additional complexity, variation in requirements, restrictions and potential legal risk. Such new laws could also require additional investment of resources in compliance programs, impact strategies regarding and the availability of personal data, and would result in increased compliance costs and/or changes in business practices and policies. On the federal level, HIPAA imposes requirements relating to the privacy, security and transmission of individually identifiable health information. We may obtain health information from third parties, such as research institutions, that are subject to privacy and security requirements under HIPAA. Although we are not directly subject to HIPAA other than with respect to providing certain employee benefits, we could potentially be subject to criminal penalties if we, our affiliates, or our agents knowingly obtain, use, or disclose individually identifiable health information maintained by a HIPAA-covered entity in a manner that is not authorized or permitted by HIPAA. In addition,Outside of the CCPA became effective on January 1, 2020U.S., the legislative and establishes certain requirementsregulatory landscape for privacy and data usesecurity continues to become more comprehensive. There has been increased attention to privacy and sharing transparency, and provides California residents certain rights concerningdata security issues that could potentially affect our business, including as a result of the use, disclosure, and retention of their personal data. The CCPA and its implementing regulations have already been amended multiple times since their enactment. Similarly, there are a number of legislative proposalsGeneral Data Protection Regulation in the United States, at bothEU and the federalU.K. and state level, that could impose new obligationsdata protection laws in the U.K. The EU GDPR (and the regulation as incorporated in U.K. law) imposes fines of up to EUR 20 million (£17.5 million) or limitations in areas affecting our business. These4% of the annual global revenue of a noncompliant company, for non-compliance. In addition, laws and regulations are evolvingenacted in the U.S., Europe, Asia and subject to interpretation,Latin America increases potential enforcement and may impose limitations on our activities or otherwise adversely affect our business. The CCPA and evolving legislation may require us, among other things, to update our notices and develop new processes internally and with our partners.litigation activity.
If we, our agents, or our third-party partners fail to comply or are alleged to have failed to comply with these or other applicable data protection and privacy laws and regulations, or if we were to experience a data breach involving personal information, we could be subject to government enforcement actions or private lawsuits. Any associated claims, inquiries, or investigations or other government actions could lead to unfavorable outcomes that have a material impact on our business including through significant penalties or fines, monetary judgments or settlements including criminal and civil liability for us and our officers and directors, increased compliance costs, delays or impediments in the development of new products, negative publicity, increased operating costs, diversion of management time and attention, or other remedies that harm our business, including orders that we modify or cease existing business practices. Outside the U.S., the legislative and regulatory landscape for privacy and data security continues to evolve. There has been increased attention to privacy and data security issues that could potentially affect our business, including the EU General Data Protection Regulation, which entered into effect on May 25, 2018 and imposes penalties up to 4% of annual global turnover. In addition, laws and regulations enacted in the United States, Europe, Asia, and Latin America increases potential enforcement and litigation activity.
U.S. Health Care Reform Law Our financial prospects could be affected by changes in health care spending and policy in the United States and abroad. We operate in a highly regulated industry and new laws, regulations, or judicial decisions, or new interpretations of existing laws, regulations, or decisions, related to, among other things, health care availability, or the method of delivery of or payment for health care products and services could negatively impact our business, operations and financial condition.
For example, in the United States there is significant interest in promoting health care reform, as evidenced by the enactment in the United States of the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010 (collectively, the “Health Care Reform Law.Law”). The Health Care Reform Law substantially changed the way health care is financed by both governmental and commercial payers and significantly impacts the pharmaceutical industry. The Health Care Reform Law contains provisions that may reduce the profitability of drug products, including, for example, by increasing the minimum rebates owed by manufacturers under the Medicaid Drug Rebate Program, extending the rebate program to individuals enrolled in Medicaid managed care plans, addressing a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected, and imposing certain annual fees based on pharmaceutical companies’ share of sales to federal health care programs. The Health Care Reform Law established and provided significant funding for a new Patient-Centered Outcomes Research Institute to coordinate and fund comparative effectiveness research. While the stated intent of comparative effectiveness research is to develop information to guide providers to the most efficacious therapies, outcomes of comparative effectiveness research could influence the reimbursement or coverage for therapies that are determined to be less cost effective than others. Should any of our products be approved for sale, but then determined to be less cost effective than alternative therapies, the levels of reimbursement for these products, or the willingness to reimburse at all, could be impacted, which could materially impact our financial results. Certain provisions of the Health Care Reform Law have been subject to judicial challenges, as well as efforts to repeal, replace or otherwise modify them or to alter their interpretation or implementation. For example, the Tax Cuts and Jobs Act enacted on December 22, 2017, eliminated the tax-based payment for individuals who fail to maintain minimum essential coverage under section 5000A of the Internal Revenue Code of 1986, as amended, commonly referred to as the “individual mandate,” effective January 1, 2019. Further, the Bipartisan Budget Act of 2018 among other things, amended the Medicare statute, effective January 1, 2019, to reduce the coverage gap in most Medicare prescription drug plans, commonly known as the “donut hole,” by raising the manufacturer discount under the Medicare Part D coverage gap discount program to 70%. Additional legislative changes, regulatory changes and judicial challenges related to the Health Care Reform Law remain possible.possible, but the nature and extent of such potential changes or challenges are uncertain at this time. It is unclear how the Health Care Reform Law, and its implementation, as well as efforts to repeal, replace or otherwise modify, or invalidate, the Health Care Reform Law, or portions thereof, and other health care reform measures that may be adopted in the future will affect our business. Another provision of the Health Care Reform Law, generally referred to as the Physician PaymentPayments Sunshine Act or Open Payments Program, requires manufacturers of drugs, devices, biologics, and medical supplies for which payment is available under Medicare, Medicaid, or the Children’s Health Insurance Program (with certain exceptions) to report annually to CMS information related to direct or indirect payments and other transfers of value made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals, as well as ownership and investment interests held in the company by physicians and their immediate family members. Effective January 1, 2022, these reporting obligations will extendhave been extended to include transfers of value made (starting in 2021) to certain non-physician providers such as physician assistants, nurse practitioners, clinical nurse specialists, certified nurse anesthetists and certified nurse-midwives. CMS publishes information from these reports on a publicly available website. Our compliance with these rules may also impose additional costs and may impact our relationships with physicians, teaching hospitals and the other non-physician health care providers. In addition, other legislative changes have been proposed and adopted since the Health Care Reform Law was enacted. For example, the Budget Control Act of 2011, as amended by the American Taxpayer Relief Act of 2012, among other things led to aggregate reductions in Medicare payments for all items and services, including prescription drugs and biologics, to service providers of, on average, 2% per fiscal year beginning April 1, 2013, and, due to subsequent legislation, will continue until 2030 (with the exception of a temporary suspension that lasted from May 1, 2020, through March 31, 2021) unless Congress takes additional action.2022 due to the COVID-19 pandemic). Following the suspension, a 1% payment reduction began April 1, 2022 and remained through June 30, 2022, The 2% payment reduction resumed on July 1, 2022. To offset the temporary suspension during the COVID-19 pandemic, in 2030, the sequestration will be 2.25% for the first half of the year, and 3% in the second half of the year. As another example, for calendar quarters beginning January 1, 2022, manufacturers will be required to report the average sales price for certain drugs under the Medicare program regardless of whether we participate in the Medicaid Drug Rebate Program. Previously, such reporting was only required for manufacturers that participated in that program. Starting in 2023, manufacturers must pay refunds to Medicare for single source drugs or biologicals, or biosimilar biological products, reimbursed under Medicare Part B and packaged in single-dose containers or single-use packages, for units of discarded drug reimbursed by Medicare Part B in excess of 10% of total allowed charges under Medicare Part B for that drug. Manufacturers that fail to pay refunds could be subject to civil monetary penalties of 125% of the refund amount. The Inflation Reduction Act (“IRA”) was signed into law in August 2022. The IRA includes several provisions that will impact our business to varying degrees, including provisions that create a $2,000 out-of-pocket cap for Medicare Part D beneficiaries, impose new mandatory discounts from manufacturers under Medicare Part D, allow the U.S. government to negotiate Medicare Part B and Part D pricing for certain high-cost single-source drugs and biologics without generic or biosimilar competition, and require companies to pay rebates to Medicare for drug prices that increase faster than inflation. The IRA provides a five-year temporary increase in Medicare Part B payment for certain qualifying biosimilars, and it also delays the rebate rule that would require pass through of pharmacy benefit manager rebates to beneficiaries. The effect of IRA on our business and the healthcare industry in general is not yet known. Additional legislative changes, regulatory changes or guidance could be adopted, which may impact the marketing approvals and reimbursement for our product candidates. Further, there has been increasing legislative, regulatory and enforcement interest in the United States with respect to drug pricing practices. There have been several Congressional inquiries and proposed and enacted federal and state legislation and regulatory initiatives designed to, among other things, bring more transparency to product pricing, evaluate the relationship between pricing and manufacturer patient programs, and reform government healthcare program reimbursement methodologies for drug products. For example, in May 2019, CMS issued a final rule to allow Medicare Advantage Plans the option of using step therapy, a type of prior authorization, for Part B drugs beginning January 1, 2020. And, on November 20, 2020, CMS issued an interim final rule to implement a “Most Favored Nation” demonstration project to test Medicare Part B reimbursement of certain separately payable drugs and biologicals based on international reference prices. The rule has become subject to judicial challenges, and federal courts have enjoined the rule at this time. If the rule survives judicial scrutiny, the Most Favored Nation model will subject certain drugs or biologicals identified by CMS as having the highest annual Medicare Part B spending to an alternative payment methodology based on international reference prices, with the list of products to be updated annually to add more products and products not to be removed absent limited circumstances. There has also been legislation that would establish an international reference price-based Medicare Part B drug and biological payment methodology.
It is possible that the Health Care Reform Law, as currently enacted or may be amended or otherwise modified in the future, as well as other health care reform measures that may be adopted in the future, may result in additional reductions in Medicare payment and other health care financing, more rigorous coverage criteria, and new payment methodologies and in additional downward pressure on coverage and payment and the price that we receive for any approved product. For example, under the IRA, Congress has enacted a program that allows Medicare to negotiate pricing for certain single-source drugs and biologics under Medicare Parts B and D. The IRA also imposes Medicare Part B and Part D inflation-based rebates, under which manufacturers owe additional rebates if the average sales price of a drug increases faster than the pace of inflation, based on a statutory reference period. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors.payers. We cannot predict the reform initiatives that may be adopted in the future or whether initiatives that have been adopted will be repealed or modified. These continuing health care reform initiatives may adversely affect: | ● | the demand for any product candidates for which we may obtain regulatory approval; |
| ● | our ability to set a price that we believe is fair for our products; |
| ● | our ability to obtain coverage and reimbursement approval for a product; |
| ● | our ability to generate revenues and achieve or maintain profitability; and |
| ● | the level of taxes that we are required to pay. |
Congress also could enact additional changes that affect our overall rebate liability and the information we report to the government as part of price reporting calculations. In addition, Congress could enact a drug price negotiation program under which the prices for certain high Medicare spend single source drugs would be capped by reference to the non-federal average manufacturer price. This or any other legislative change could impact the market conditions for our products. We further expect continued scrutiny on government price reporting and pricing more generally from Congress, agencies, and other bodies. our ability to set a price that we believe is fair for our products;
our ability to obtain coverage and reimbursement approval for a product;Table of Contents
our ability to generate revenues and achieve or maintain profitability; and
the level of taxes that we are required to pay.
Coverage and Reimbursement In the United States and markets in other countries, patients generally rely on third-party payorspayers to reimburse all or part of the costs associated with their treatment. Adequate coverage and reimbursement from governmental healthcare programs, such as Medicare and Medicaid, and commercial payorspayers is critical to new product acceptance. Our ability to successfully commercialize our product candidates will depend in part on the extent to which coverage and adequate reimbursement for these products and related treatments will be available from government health administration authorities, private health insurers and other organizations. Government authorities and third-party payors,payers, such as private health insurers and health maintenance organizations, decide which medications they will pay for and establish reimbursement levels. The availability of coverage and extent of reimbursement by governmental and private payorspayers is essential for most patients to be able to afford treatments such as gene therapy products. Sales of these or other product candidates that we may identify will depend substantially, both domestically and abroad, on the extent to which the costs of our product candidates will be paid by health maintenance, managed care, pharmacy benefit and similar healthcare management organizations, or reimbursed by government health administration authorities, private health coverage insurers and other third-party payors.payers. If coverage and adequate reimbursement is not available, or is available only to limited levels, we may not be able to successfully commercialize our product candidates. Even if coverage is provided, the approved reimbursement amount may not be high enough to allow us to establish or maintain pricing sufficient to realize a sufficient return on our investment. There is also significant uncertainty related to the insurance coverage and reimbursement of newly approved products and coverage may be more limited than the purposes for which the medicine is approved by the FDA or comparable foreign regulatory authorities. In the United States, the principal decisions about reimbursement for new medicines are typicallymay be made by CMS. CMS decides whether and to what extent acertain new medicinemedicines will be covered and reimbursed under Medicare and private payorspayers tend to follow CMS to a substantial degree. Environmental Regulations We are also subject to regulation under the Occupational Safety and Health Act, the Environmental Protection Act, the Toxic Substances Control Act, the Resource Conservation and Recovery Act and other present and potential federal, state or local regulations. These and other laws govern our use, handling and disposal of various biological and chemical substances used in, and waste generated by our operations. Our research and development involve the controlled use of hazardous materials, chemicals and viruses. Although we believe that our safety procedures for handling and disposing of such materials comply with the standards prescribed by state and federal regulations, the risk of accidental contamination or injury from these materials cannot be completely eliminated. In the event of such an accident, we could be held liable for any damages that result and any such liability could exceed our resources. Additionally, for formulations containing controlled substances, we are subject to Drug Enforcement Act regulations. Pricing Regulations There have been a number of federal and state legislative changes made over the last few years regarding the pricing of pharmaceutical and biological products, government control and other changes to the health care system of the United States. Concerns about drug pricing have beencontinue to be expressed by members of Congress and prior presidential administrations. It is uncertain how such legislative changes will be adopted or what actions federal, state or private payers for medical goods and services may take in response to such legislation. We cannot predict the effect such health care changes will have on our business, and no assurance can be given that any such reforms will not have a material adverse effect.
Non-U.S. Government Regulations European Drug Development Our products will also be subject to extensive regulatory requirements in the European Union.Union (“EU”). As in the United States, medicinal products can only be marketed if a marketing authorization from the competent regulatory agencies has been obtained. See “European Marketing Authorization” below. In the United States,EU, the various phases of preclinicalnew Clinical Trials Regulation 536/2014 has been applicable since January 31. 2022. The Clinical Trials Regulation repealed and clinical research in European Union are subject to significant regulatory controls. The EUreplaced the Clinical Trials Directive, (2001/20/EC) (Clinical Trials Directive) providesand introduced a complete overhaul of the existing regulation of clinical trials regulatory frameworkfor medicinal products in the European Union, but the European Union member states have transposedEU, including a new coordinated procedure for authorization of clinical trials that will be conducted in multiple EU Member States, and applied theincreased obligations on sponsors to publish clinical trial results. The transitory provisions of the new Clinical Trials Regulation provide that ongoing clinical trials previously authorized under the Clinical Trials Directive differently. This has ledcan remain under the Directive, or they can transition to significant variationsthe Regulation. By January 31, 2025, when all ongoing clinical trials must have transitioned to the new Regulation. The new Regulation is directly applicable in all Member States (and so does not require national implementing legislation in each Member State) and aims at simplifying and streamlining the approval of clinical studies in the regimesEU. The main characteristics of the different member states. Undernew Regulation include: a streamlined application procedure via a single-entry point through the current regime, beforeClinical Trials Information System, or CTIS; a single set of documents to be prepared and submitted for the application as well as simplified reporting procedures for clinical trial sponsors; and a harmonized procedure for the assessment of applications for clinical trials, which is divided in two parts (Part I contains scientific and medicinal product documentation and Part II contains the national and patient-level documentation). Part I is assessed by a coordinated review by the competent authorities of all EU Member States in which an application for authorization of a clinical trial can be initiated it must be approved inhas been submitted (Concerned Member States) of a draft report prepared by a Reference Member State. Part II is assessed separately by each Concerned Member State. Strict deadlines have also been established for the assessment of the European Union countries where theclinical trial is to be conducted by the relevant National Competent Authority (“NCA”), and one or more Ethics Committees (“ECs”), and a Clinical Trial Authorization must be obtained.applications. Similar to the FDA, Europe’sthe European Medicines Agency’s Committee for Medicinal Products for Human useUse (“CHMP”) has adopted ICH S6 as a guideline governing preclinical testing of biologics. Sponsors usually must conduct pharmacodynamic (“PD”) studies, such as in vitro binding assays and in vivo studies that assess the product’s pharmacologic activity and define its mechanism of action. Biologics typically undergo single- and repeat-dose toxicity studies using relevant species. Safety pharmacology studies, which evaluate the product’s functional effects on major body systems and specific organs, and local tolerance testing can be done separately or subsumed in toxicity testing. Sponsors also usually conduct single- and multiple-dose pharmacokinetic (“PK”) and/or toxicokinetic studies to assess absorption, disposition, exposure and clearance (in particular, antibody-mediated clearance), and explore dose-response relationships. This information is used to predict margins of safety for human studies. Immunogenicity testing might include screening and mechanistic studies. Clinical Trial Authorization in the European Union
The Clinical Trials Directive and European Commission guidance describe the steps that a sponsor must take before commencing a clinical trial in the European Union. According to these documents, a clinical trial may commence only if: (i) the anticipated therapeutic and public health benefits outweigh any foreseeable risks and inconveniences to the subjects; (ii) the trial subjects understand the objectives and risks of the trial and give informed, written consent to participate; (iii) the trial safeguards the physical and mental integrity of the subjects; and (iv) insurance covers the liability of the sponsor and investigator. To comply with these requirements, the trial sponsor must take certain steps. In general, the sponsor must take responsibility for trial conduct, appointment of an appropriate investigator, selection of the institution that will conduct the trial, quality control, data collection standards, protocol drafting, and creation of the investigator’s brochure. The sponsor then must apply for approval from both the ethics committee and the relevant NCA in the member state. Written authorization may be required for all biologics trials and is required for trials involving medicines containing genetically modified organisms, medicines for gene therapy, and medicines for somatic cell therapy (including xenogeneic cell therapy). The opinion of the ethics committee should be issued within 60 days. A review period of 30 days can be added for medicines requiring written authorization noted earlier, and for xenogeneic cell therapy, there are no time limits for authorization. This timeframe can be extended by an additional 90 days (in addition to the original 90 days) if the ethics committee consults a national group or committee. The trial may begin only if (i) the ethics committee has issued a favorable opinion and (ii) no competent authority has informed the applicant of grounds for non-acceptance.
Good Clinical Practices and Other Considerations for Clinical Trials Clinical trials of biologicsmedical products (including biologics) must comply with GCP, as described in Directive 2005/28/EC on Good Clinical Practice and the ICH E6 guideline, which the CHMP has adopted. The directiveDirective and guideline describe general governing principles for clinical trials. The rights, safety and well-being of trial subjects must prevail over the interests of science and society. Investigators must obtain freely given informed consent from every trial subject before each subject is enrolled. Clinical trial information must be handled, recorded and stored with respect for relevant confidentiality and privacy rules. Trials must comply with the ethical principles of the World Medical Association’s Declaration of Helsinki. Specific GCP guidelines apply to trials of advanced therapy medicinal products.products (i.e. gene therapy, somatic-cell therapy and tissue-engineered medicines). These guidelines regulate issues such as the donation, procurement and testing of human tissues and cells; the implementation of a traceability system; and specific rules on safety reporting and long-term follow-up. Under the Clinical Trials Directive,Regulation, special requirements apply to clinical trials conducted on minors and other persons not able to give informed legal consent. These requirements are intended to preserve the dignity of the trial subjects, confirm that the benefits of the trial outweigh the risks and ensure that subjects’ representatives give consent with as much involvement of the subject as possible. Competent authorities must record information regarding trials in the European database of clinical trials which is accessible only to other competent authorities, the European Medicines Agency (“EMA”), and the European Commission. CHMP has also issued a guideline on quality requirements during the clinical trial period for investigational medicinal products containing biological or biotechnology-derived substances. The guideline describes quality documentation that should be submitted to the competent authority as part of the sponsor’s investigational
medicinal product dossier (“IMPD”). The IMPD should include, among other things, (i) an adequate description of the process and process controls, including a flow chart of all successive steps and details of in-process testing and (ii) a description and justification of “any reprocessing during manufacture of the drug substance.” The guideline also recognizes that sponsors will improve and optimize their manufacturing processes during clinical development and describes the steps sponsors should take following these changes. Specifically, the sponsor must compare the quality attributes of the pre- and post-change biological active substances and relevant intermediates and conduct a comparability exercise where necessary. For first-in-human clinical trials, sponsors should use product representative of the material used during the non-clinical testing phase. Finally, with regard to characterization, the guideline requires details on the biological activity to be provided, recognizing that the extent of characterization data will further increase in later phases. Study Design Considerations General regulatory guidance on study design applies to biologics as well as small molecule medicines. According to the guidance, there is a “close, but variable correlation” between phase of development and type of study, but one type of trial can occur in several different phases. The guidance therefore identifies the most typical kind of study for each phase. Phase 1 usually involves the initial introduction of the investigational product into human subjects, and studies in this phase usually have non-therapeutic objectives. Specifically, Phase 1 studies typically investigate initial safety and tolerability, PK, PD and/or drug activity, to preliminarily determine the potential therapeutic benefit of a medicine. Phase 1 studies may be conducted in healthy volunteers or certain types of patients. If the medicine has significant potential toxicity (e.g., cytotoxic products), the trial will usually be conducted in patients. The most typical Phase 2 study is a therapeutic exploratory study that explores efficacy in narrowly defined, relatively homogenous groups of patients. Initially, studies may use a variety of designs (e.g., concurrent controls and comparisons with baseline status). Subsequent Phase 2 trials usually are randomized and concurrently controlled, allowing for evaluation of the medicine’s safety and efficacy for a particular indication. A major goal of this phase is to determine the dose(s) for Phase 3 trials. Phase 3 typically involves therapeutic confirmatory studies that are designed to verify the preliminary evidence obtained in Phase 2 and to provide a sufficient basis for marketing authorization. Phase 3 studies may also further explore the dose response relationship, or explore the drug’s use in wider populations, in different stages of disease, or in combination with another drug. With regard to medicines administered for long periods, extended exposure trials ordinarily occur during Phase 3, although the sponsor may start them in Phase 2. To ensure that clinical trials in all three phases of development will be adequate to support a Marketing Authorization Applicationmarketing authorization application (“MAA”), sponsors should design these trials with the MAA requirements in mind. Biologics in generalCertain biologics products need to comply with the requirements set out in Part III of the Annex I to Directive 2003/63/EC (which amends the core EU medicines legislation, Directive 2001/83/EC), and advanced therapy medicinal products need to comply with the requirements described in Part IV. Consultation with the European Medicines Agency A sponsor may obtain, from the EMA, scientific advice regarding clinical trial protocols.the development of a medicinal product. Although this advice does not bind the ethics committees or NCAsEMA and is not binding for purposes of a future MAA, it can be useful to guide developers generally in performing the appropriate preclinical and clinical tests for the product, or on more specific aspects such as guiding revisions to thea clinical trial protocol. EMA’s remarks will only address scientific issues and will generally focus on matters such as the selection of endpoints and comparator, the duration of treatment or follow-up and the design of pivotal studies. Advice also might address a sponsor’s proposal to deviate from a CHMP guideline. If the applicant decides not to follow the EMA’s advice, it should justify this decision in its MAA. EMA guidance details the procedures for requesting scientific advice. The fact that an applicant requests advice from EMA does not preclude it from also seeking advice from national competent authorities or from foreign regulators, such as the FDA. The process of obtaining advice from the national competent authorities is often less formal than requesting advice from the EMA, and such advice can prove helpful. Consequently, seeking such advice is a common choice among applicants. Generally, the parallel scientific procedure (a program shred by the EMA and FDA) is available for “important breakthrough drugs,” that is, products that the EMA and FDA have identified as falling within therapeutic areas of overlapping interest (e.g., oncology products, vaccines and blood products). The goal of these meetings is to provide clarity regarding the regulatory requirements of each region and the reasons for any differences between them. A sponsor requesting parallel scientific advice should authorize the agencies to exchange all information about the product, including trade secrets. After the parallel scientific advice procedure, each agency will provide its own independent advice on the questions at issue. There is no guarantee of harmonized advice or identical regulatory decisions on the approvability of the product. European Marketing Authorization In the European Economic Area (“EEA”), which includes the 27-member27 member states of the European UnionEU plus Norway, Iceland and Liechtenstein, medicinal products can only be placed on the market after approvalthe grant of a Marketing Authorization Application, or
MA.marketing authorization. The MAMAA is based on the results of pharmaceutical tests, preclinical tests and clinical trials conducted on the medicinal product in question. There are two types of MAs:
The Centralized MA, which is issued by the European Commission through the Centralized Procedure, based on the opinion of the CHMP and which is valid throughout the entire territory of the EEA. The Centralized Procedure is mandatory for certain types of drugs, such as biotechnology medicinal products, orphan medicinal products, and medicinal products containing new active substances indicated for certain diseases. The Centralized Procedure is optional for other drugs provided eligibility criteria are met. In all cases, to find out whether a product can be evaluated via the Centralized Procedure, applicants must always submit an eligibility request.
National MAs, which are issued by the competent authorities of the member states of the EEA and only cover their respective territory, are available for drugs not falling within the mandatory scope of the Centralized Procedure. Where a drug has already been authorized for marketing in a member state of the EEA, this National MA can be recognized in other member states through the Mutual Recognition Procedure. If the drug has not received a National MA in any member state at the time of application, it can be approved by multiple member states in parallel through the Decentralized Procedure.authorizations:
| ● | The centralized marketing authorization, which is issued by the European Commission through the centralized procedure, based on the opinion of the CHMP and which is valid throughout the entire territory of the EEA. The centralized procedure is mandatory for certain types of drugs, such as medicinal products derived from biotechnology processes (such as genetic engineering), orphan medicinal products, advanced-therapy medicinal products and medicinal products containing new active substances indicated for the treatment of HIV, AIDS, cancer, neurodegenerative disorders, diabetes, auto-immune and other immune dysfunctions and viral diseases. . The centralized procedure is optional for products containing a new active substance not yet authorized in the EU, or for products that constitute a significant therapeutic, scientific or technical innovation or which are in the interest of public health in the EU. . To find out whether a product can be evaluated via the centralized procedure, applicants should always submit an eligibility request to the EMA, including by a justification of eligibility for evaluation under the centralized procedure. |
| ● | National marketing authorizations, which are issued by the competent authorities of the member states of the EEA and only cover their respective territory, are available for drugs not falling within the mandatory scope of the centralized procedure. Where a drug has already been authorized for marketing in a member state of the EEA, this National marketing authorization can be recognized in other member states through the Mutual Recognition Procedure. If the drug has not received a National marketing authorization in any member state at the time of application, it can be approved by multiple member states in parallel through the Decentralized Procedure. |
Under the above described procedures, before granting the MA,marketing authorization, the EMA or the competent authorities of the Member Statesmember states of the EEA make an assessment of the risk-benefit balance of the drug on the basis of scientific criteria concerning its quality, safety and efficacy. The Marketing Authorization Application: Contents and Approval Standard Many biologics fall under the scope of the Centralized Procedure,centralized procedure, which, as mentioned above, is mandatory for medicines developed through biotechnological methods, such as recombinant DNA technology; controlled expression of genes coding for biologically active proteins in prokaryotes and eukaryotes, including transformed mammalian cells; and hybridoma and mAb methods. For example,Gene therapy and cell therapy gene therapy, vaccines from strains developed through recombinant DNA technology (including gene deletion), and “any medicinal product for which a monoclonal antibody is used at any stage in the manufacturing process”products are allalso subject to the Centralized Procedure.centralized procedure as advanced therapy medicinal products. Nonetheless, some biologics are still approved at the member state level. For example, manycertain types of vaccines do not fall within the mandatory scope of the Centralized Procedure.centralized procedure (although they may be eligible for the centralized procedure in the interest of public health). The EMA has published a guideline intended to harmonize the quality aspects to be included in summaries of product characteristics and patient information leaflets for human vaccines. With respect to the Centralized Procedure,centralized procedure, the approval standards for biotechnology products are the same as for chemically synthesized medicines. Both types of products must be safe and effective and have appropriate quality. Because of their special characteristics, however, biotechnology products must comply with several additional dossier requirements. The MAA for a biotechnology product must meetFor example, the standard dossier submission requirements, as described in Article 8 of the Medicines Directive (2001/83/EC). Consequently, the MAA must generally comply with the Common Technical Document format, including with respect to Module I (administrative information, including labeling and mock-ups), Module 2 (various summaries), Module 3 (chemical, pharmaceutical and biological information), Module 4 (non-clinical reports) and Module 5 (clinical study reports). MAAs for biologics also must meet special requirements. The applicant must thoroughly describe the manufacturing process and must: (i) provide information on the origin and history of the starting materials; (ii) demonstrate that the active substance complies with specific measures for preventing the transmission of animal and human spongiform encephalopathies; (iii) if cell banks are used, demonstrate that cell characteristics remain unchanged at the passage level for production (and beyond); (iv) provide information as to whether there are adventitious agents in seed materials, cell banks, pools of serum or plasma, and all other materials of biological origin, and, if it is not possible to avoid the presence of potentially pathogenic adventitious agents, show that further processing ensures elimination or inactivation of the agents; (v) if possible, base vaccine production on a seed lot system and established cell banks; (vi) in case of medicines derived from human blood or plasma, describe the origin, criteria and procedures for the collection, transportation and storage of the starting material; and (vii) describe the manufacturing facilities and equipment. Other special rules apply certain types of biological medicines. For example, for plasma-derived medicinal products, the applicant must provide an information dossier, the Plasma Master File. MAAs for vaccines other than for influenza need to contain a Vaccine Antigen Master File. Special rules also apply to advanced therapy medicinal products, including gene therapies, somatic cell therapies and tissue-engineered products. Data and Market Exclusivity in the European Union In the European Union,EU, new chemical entities (including both small molecules and biological medicinal productsproducts), sometimes referred to as new active substances, qualify for eight years of data exclusivity upon marketing authorization and an additional two years of market exclusivity. This data exclusivity if granted, prevents regulatory authorities in the European Uniongeneric or biosimilar applicants from referencing the innovator’s pre-clinical and clinical trial data to assesscontained in the dossier of the reference product when applying for a generic or biosimilar applicationmarketing authorization, for a period of eight years afterfrom the date on which the reference product was first authorized in the EU. After such eight year period, a generic or biosimilar marketing authorization can be submitted, and the innovator’s data may be referenced, but not approvedno generic or biosimilar product can be marketed for two years. The overall ten-year period may be extended to a maximum of 11 years if, during the period of data exclusivity, the marketing authorization holder obtains an authorization for one or more new therapeutic indications which, during the scientific evaluation prior to their authorization, are held to bring a significant clinical benefit in comparison with existing therapies. Even if an innovative medicinal product gains the prescribed period of data exclusivity, another company may market another version of the product if such company obtained a marketing authorization based on an application with a complete and independent data package of pharmaceutical tests, preclinical tests and clinical trials.
Orphan Designation in the European Union The European Commission is also able to grant orphan designation in respect of medicinal products. To qualify the medicinal product must be intended for the diagnosis, prevention or treatment of (i) a life-threatening or chronically debilitating condition affecting not more than 5 in 10,000 persons in the European UnionEU or (ii) a life-threatening, seriously debilitating or serious and chronic condition in the European UnionEU where without incentives it is unlikely that the marketing of the medicinal product in the European UnionEU would generate sufficient return to justify the necessary investment.investment in its development. Further, no satisfactory method of diagnosis, prevention or treatment of the condition in question must exist in the European UnionEU or, if such method exists, the medicinal product must be of significant benefit to those affected by that condition. Orphan medicinal products still remain subject to the same regulatory approval process, albeit that they are always assessed through the Centralized Procedure.centralized procedure. Effective September 19, 2018, sponsors applying for Orphan Designationorphan designation must use EMA’s secure online IRIS platform. However, sponsors of orphan medicinal products are eligible to benefit from a number of incentives offered, including certain assistance with development of the medicinal product, reduced fees for MA applicationsMAAs and protection from market competition once the medicinal product is authorized, as described below. Where an MAa marketing authorization in respect of an orphan medicinal product is granted, the European Commission, EMA and the competent authorities of the EU member states shall not, for a period of ten years, accept another application for an MA,a marketing authorization or grant an MAa marketing authorization or accept an application to extend an existing MA,authorization , for the same therapeutic indication, in respect of a similar medicinal product, unless: (i) the holder of the MAmarketing authorization for the original orphan medicinal product has given its consent to the second applicant; (ii) the holder of the MAmarketing authorization for the original orphan medicinal product is unable to supply sufficient quantities of the medicinal product; or (iii) the second applicant can establish the second medicinal is safer, more effective or otherwise clinically superior.superior than the authorized orphan product. A “similar medicinal product” is defined as a medicinal product containing a similar active substance or substances as contained in an authorized orphan medicinal product, and which is intended for the same therapeutic indication. Other government regulation in the European Union and United Kingdom The European UnionEU and the European UnionEU member states and the U.K. have extensive laws and regulations relating to a variety of other topics that would be of relevance for us if we are active in the European Union,EU and U.K., including but not limited to laws and regulations regarding data privacy, drug pricing and reimbursement, advertising and interactions with healthcare professionals. Other Jurisdictions In addition to regulations in the United States and the European Union,EU, we may be subject to a variety of regulations in other jurisdictions governing, among other things, clinical trials and any commercial sales and distribution of our product. As the United Kingdom is no longer a member state of the European Union,EU, this may also apply to the United Kingdom. Whether or not we obtain FDA approval for a product, we must obtain approval from comparable regulatory authorities in foreign countries before we can commence clinical trials in such countries and the approval of the regulators of foreign countries before we may market products in such countries. The approval process and requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from place to place, and the time may be longer or shorter than that required for FDA approval. Acceptance of Foreign Clinical Trials in the United States and Europethe European Union The FDA has issued regulations governing its acceptance of foreign clinical data not conducted under an IND to support IND applications or marketing authorizations, such as BLAs. FDA may accept a well-designed, well-conducted, non-IND foreign study as support for an IND or marketing application if the study was conducted in accordance with GCP and if FDA is able to validate the data from the study through an onsite inspection, if necessary. Where a marketing application is based solely on foreign data, additional requirements apply, including a demonstration that the foreign data are applicable to the U.S. population and U.S. medical practice. EU Directive 2001/83/EC allows for clinical trials conducted outside the European UnionEU to be taken into consideration during the review of an MAAa marketing authorization in the European UnionEU if such trials have been designed, implemented and reported based on principles equivalent to those of the Clinical Trials DirectiveRegulation, including with regard to good clinical practice and ethical principles. Moreover, they should comply with the ethical principles outlined in the Declaration of Helsinki. The applicant must submit a statement declaring such compliance as part of the MAA.marketing authorization. In December 2008 and April 2012, the EMA published a strategy paper on the acceptance of data from foreign clinical trials conducted in “third countries,” particularly those outside the “‘traditional’ Western European and North American research areas.” According to the 2008 strategy paper, there is a “growing concern both among regulators and in public debate about how well these trials are conducted from an ethical and scientific/organizational standpoint.” The EMA has called for increased cooperation between international regulatory authorities involved in the supervision of clinical trials and has put forth other proposals to address these issues.
Manufacturing and Source of Supply We do not have any manufacturing facilities. We currently rely, and expect to continue to rely, on third parties for the manufacture of our product candidates for preclinical studies and clinical trials, as well as for commercial manufacture if our product candidates receive marketing approval. To date, we have obtained materials for clinical trials and non-clinical studies from third-party manufacturers who are suppliers to us. For our product candidates, we intend to identify and qualify additional contract manufacturers to provide commercial scale manufacturing prior to submission of aan NDA or BLA to the FDA. Employees and Human Capital Management As of December 31, 2020,2022, we had 4352 full-time employees, 1118 of whom hold M.D. or Ph.D. degrees and 534 of whom hold other advanced degrees. In light of the COVID-19 pandemic, our headcount has grown by 72% in the past year in order to support our pipeline of research programs and product candidates. Of our total workforce, 2728 are engaged primarily in research and development activities and 1619 are engaged primarily in executive, finance and accounting, and administrative functions. As of December 31, 2020, 422022, 50 employees are located in the United States and one employee2 employees in the United Kingdom. Of our employees, 58% are female and 42% are male. None are represented by labor unions or covered by collective bargaining agreements. We consider our relations with our employees to be good. Corporate Culture Our values – compliance, collaboration, integrity and high performance – are built on the foundation that the employees we hire and the way we treat one another promote creativity, innovation and productivity, which spur our success. This culture depends in large part on our ability to attract, retain and develop a diverse population of talents and high-performing employees at all levels of our organization. Providing market competitive pay and benefit programs, opportunities to participate in the success they help create, while engaging employees in important dialogue regarding organization performance, we create a culture of inclusion in which all colleagues have the opportunity to thrive. The success of our business is fundamentally connected to the well-being of our employees. Compensation and Benefits Program Our compensation program is designed to attract and reward talented individuals who possess the skills necessary to support our business objectives, assist in the achievement of our strategic goals and create long-term value for our stockholders. We provide all of our employees with what we consider to be a very competitive mix of compensation and insurance benefits, for all our employees, as well as participation in our equity programs. Diversity and Inclusion
We believe that an equitable and inclusive environment with diverse teams produces more creative solutions, results in better, more innovative products and services and is crucial to our efforts to attract and retain key talent.
Hybrid Culture and COVID-19 Like other companies, we have learned to adapt during the ongoing COVID-19 pandemic. We have prioritized employee safety and transparency during the pandemic and continue to do so, ensuring all employees are set up to work remotely and providing clarity on office closures and evolving guidelines issued by the CDC, the WHO and state and local authorities, where possible. In the second quarter of 2020, we made the decision to move to a hybrid workplace model, which means that certain of our employees will have the option to be 100% remote, work full-time in our office, or have the flexibility to work between office and remotely until the pandemic is resolved. This move provides our employees with continued flexibility, through the resolution of the pandemic, to work in person, remotely or in a hybrid model. This will enable us to grow better as we develop our programs. Available Information Our stock is traded on the Nasdaq Global Market (“NASDAQ”) under the symbol “ALT”. Our principal executive offices located at 910 Clopper Road, Suite 201S, Gaithersburg, Maryland 20878. Our telephone number is (240) 654-1450, and our Internet website is www.altimmune.com and our investor relations website is located under the “Investors” tab. The information on, or that can be accessed through, our website is not part of this Annual Report and is not incorporated by reference herein. We make available our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, and Current Reports on Form 8-K, and amendments to these reports, free of charge through our website (www.altimmune.com) as soon as reasonably practicable after we electronically file such material with, or furnish such material to, the SEC. We also make available on our website reports filed by our executive officers and Directors on Forms 3, 4, and 5 regarding their ownership of our securities. Our Code of Business Conduct and Ethics, and any amendments to our Code of Business Conduct and Ethics, are also available on our website under the “Investors” tab. The SEC maintains a website that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC at www.sec.gov. Item 1A. Risk Factors In addition to the other information included in this Annual Report, the following risk factors should be carefully considered when evaluating an investment in us. These risk factors and other uncertainties may cause our actual future results or performance to differ materially from any future results or performance expressed or implied in the forward-looking statements contained in this report and in other public statements we make. In addition, because of these risks and uncertainties, as well as other variables
affecting our operating results, our past financial performance is not necessarily indicative of future performance. See “Forward-Looking statements” in Item 1 of this Annual Report. Risks Related to Our Business, Financing Requirements, Product Development and Clinical Trials We have incurred significant losses since our founding and anticipate that we will continue to incur significant losses for the foreseeable future and may never achieve or maintain profitability. We are a clinical-stage biotechnology company and have not yet generated revenues from product sales. To date, substantially all of our revenues have been derived from grants and contracts with governmental agencies, consisting primarily of our BARDA contract for our anthrax vaccine product candidate. We have incurred net losses in most periods since our inception, including a net loss of $84.7 million and $97.1 million for the years ended December 31, 2022 and 2021, respectively. As of December 31, 2022, we have an accumulated deficit of $377.9 million. To date, we have not received regulatory approvals for any products or generated any revenues from the sale of products, and we do not expect to generate any product revenues in the foreseeable future. We do not know whether or when we will generate product revenues or become profitable. We have devoted most of our financial resources to research and development, including preclinical and clinical development of our product candidates. We have not completed pivotal clinical trials for any product candidate. Our leading product candidates remain in early stage clinical development, and it may be several years, if ever, before we have a product candidate ready for commercialization. Even if we obtain regulatory approval to market a product candidate, our future revenues will depend upon the size of any markets in which our product candidates have received approval, our ability to achieve sufficient market acceptance, reimbursement from third-party payers and other factors. The net losses we incur may fluctuate significantly from quarter to quarter and year to year, such that a period-to-period comparison of our results of operations may not be a good indication of our future performance. In any particular quarter or quarters, our operating results could be below the expectations of securities analysts or investors, which could cause our stock price to decline. Our profitability depends on our ability to develop and commercialize our current and future product candidates. To become and remain profitable, we must succeed in developing and eventually commercializing products that generate significant revenue. This will require us to be successful in a range of challenging activities, including completing preclinical studies and clinical trials of our product candidates, discovering additional product candidates, obtaining regulatory approval for these product candidates, forming strategic partnerships and alliances with third parties and manufacturing, marketing and selling any products for which we may obtain regulatory approval. We are only in the preliminary stages of most of these activities. We may never succeed in these activities and, even if we do, we may never generate revenues that are significant enough to achieve profitability. If some or all of our product candidates do not prove to be safe, pure and efficacious, then we may have to abandon those product candidates altogether and we will be unable to generate revenues from sales of such products. We expect to continue to incur significant expenses and increasing operating losses for the foreseeable future. We anticipate that our expenses will increase significantly if and as we: | ● | continue our clinical trials for our product candidates; |
| ● | initiate additional preclinical studies, clinical trials or other studies or trials for our other product candidates; |
| ● | manufacture material for clinical trials and, if any product candidate is approved for marketing, for commercial sale; |
| ● | seek regulatory approvals for our product candidates that successfully complete clinical trials; |
| ● | establish a sales, marketing and distribution infrastructure to commercialize any products for which we may obtain marketing approval; |
| ● | seek to discover and develop additional product candidates; |
| ● | acquire or in-license other product candidates and technologies; |
| ● | make royalty, milestone or other payments under any in-license agreements; |
| ● | form strategic partnerships and/or make additional acquisitions; |
| ● | maintain, protect and expand our intellectual property portfolio; |
| ● | attract and retain skilled personnel; and |
| ● | create additional infrastructure to support our operations as a public company and our product development and planned future commercialization efforts. |
Because of the numerous risks and uncertainties associated with pharmaceutical product development, we are unable to accurately predict the timing or amount of increased expenses or when, or if, we will be able to achieve profitability. If we are required by the FDA or other regulatory authorities to perform studies in addition to those currently expected, or if there are any delays in completing our clinical trials or the development of any of our product candidates, our expenses could increase. Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our failure to become and remain profitable would depress the value of our company and could impair our ability to raise capital, expand our business, maintain our research and development efforts, diversify our product offerings or even continue our operations. Future conditions might require us to make substantial write-downs in our assets, which would adversely affect our balance sheet and results of operations. We review our long-lived tangible and intangible assets for impairment whenever events or changes in circumstances indicate that the carrying value of an asset may not be recoverable. We also test our indefinite-lived intangible assets for impairment at least annually in the fourth quarter, or when events or changes in the business environment indicate that the carrying value of the reporting unit may exceed its fair value. As of December 31, 2021, we recorded non-cash impairment charges of $11.4 million for construction-in-progress assets that were previously capitalized in connection with the discontinuation of AdCOVID. At December 31, 2022, we continued to carry $12.4 million of indefinite lived intangible assets. Any such significant write-downs of our long-lived assets in the future could adversely affect our balance sheet and results of operations. We will require substantial additional financing to achieve our goals, and a failure to obtain this necessary capital when needed would force us to delay, limit, reduce or terminate our product development or commercialization efforts. We do not expect to generate revenue from product sales, licensing fees, royalties, milestones, contract research or other sources in an amount sufficient to fully fund our operations for the foreseeable future, if ever. Therefore, we will use our existing cash resources, and will require additional funds to maintain our operations, continue our research and development programs, commence future preclinical studies and clinical trials, seek regulatory approvals and manufacture and market our products. As of December 31, 2022, our cash, cash equivalents, restricted cash and short-term investments were $184.9 million. Based on our current operating plan, we believe that our existing cash will be sufficient to fund our projected operating expenses and capital expenditure requirements for at least a twelve-month period from the issuance date of our December 31, 2022 financial statements. However, we do not expect that these funds will be sufficient to enable us to complete the clinical trials needed to seek marketing approval or commercialize any of our product candidates. Furthermore, our operating plan may change as a result of many factors currently unknown to us, and we may need additional funds sooner than planned. We believe that we will continue to expend substantial resources for the foreseeable future developing our product candidates. These expenditures will include costs associated with research and development, maintaining our intellectual property estate, potentially acquiring new technologies, obtaining regulatory approvals and manufacturing products, forming partnerships and strategic alliances, as well as marketing and selling products approved for sale, if any. In addition, other unanticipated costs may arise. Because the outcome of our planned and anticipated clinical trials is highly uncertain, we cannot reasonably estimate the actual amounts necessary to successfully complete the development and commercialization of our product candidates. Our future capital requirements depend on many factors, including: | ● | the progress, results and costs of our clinical trials for our leading product candidates; |
| ● | the scope, progress, results and costs of preclinical development, laboratory testing and clinical trials for our other product candidates; |
| ● | the amount of funding that we receive from other non-dilutive funding sources; |
| ● | the number and development requirements of other product candidates that we pursue; |
| ● | the timing of, and the costs involved in, obtaining regulatory approvals for our product candidates if clinical trials are successful and the outcome of regulatory review of our product candidates; |
| ● | our ability to contract with third-party manufacturing facilities for adequate supply and to establish processes that meet regulatory requirements for commercialization; |
| ● | the cost and timing of future commercialization activities for our products, if any of our product candidates are approved for marketing, including product manufacturing, marketing, sales and distribution costs; |
| ● | the revenue, if any, received from commercial sales of our product candidates for which we receive marketing approval; |
| ● | our ability to establish and maintain strategic partnerships, licensing or other arrangements and the financial terms of such agreements; and |
| ● | the costs involved in preparing, filing and prosecuting patent applications, and maintaining, defending and enforcing our intellectual property rights, including litigation costs and the outcome of such litigation. |
We may also seek additional capital due to favorable market conditions or strategic considerations even if we believe we have sufficient funds for our current or future operating plans. Additional funds may not be available when we need them on terms that are acceptable to us, or at all. If adequate funds are not available to us when needed, we may be required to delay, limit, reduce or terminate preclinical studies, clinical trials or other development activities for one or more of our product candidates or delay, limit, reduce or terminate our establishment of sales and marketing capabilities or other activities that may be necessary to commercialize our product candidates. Our ability to raise capital may be limited by applicable laws and regulations. Using a shelf registration statement on Form S-3 to raise additional capital generally takes less time and is less expensive than other means, such as conducting an offering under a Form S-1 registration statement. However, our ability to raise capital using a shelf registration statement may be limited by, among other things, SEC rules and regulations. Under SEC rules and regulations, if our public float (the market value of our common stock held by non-affiliates) is less than $75.0 million, then the aggregate market value of securities sold by us or on our behalf under our Form S-3 in any 12-month period is limited to an aggregate of one-third of our public float. While our public float is currently more than $75.0 million, we have been subject to this limitation in the past and we may be subject to it again in the future. If our ability to utilize a Form S-3 registration statement for a primary offering of our securities is limited to one-third of our public float, we may conduct such an offering pursuant to an exemption from registration under the Securities Act or under a Form S-1 registration statement, and we would expect either of those alternatives to increase the cost of raising additional capital relative to utilizing a Form S-3 registration statement. In addition, under current SEC rules and regulations, our common stock must be listed and registered on a national securities exchange in order to utilize a Form S-3 registration statement (i) for a primary offering, if our public float is not at least $75.0 million as of a date within 60 days prior to the date of filing the Form S-3 or a re-evaluation date, whichever is later, and (ii) to register the resale of our securities by persons other than us (i.e., a resale offering). While currently our common stock is listed on the NASDAQ Global Market, there can be no assurance that we will be able to maintain such listing. Our ability to timely raise sufficient additional capital also may be limited by NASDAQ’s stockholder approval requirements for transactions involving the issuance of our common stock or securities convertible into our common stock. For instance, NASDAQ requires that we obtain stockholder approval of any transaction involving the sale, issuance or potential issuance by us of our common stock (or securities convertible into our common stock) at a price less than the greater of book or market value, which (together with sales by our officers, directors and principal stockholders) equals 20% or more of our then outstanding common stock, unless the transaction is considered a “public offering” by NASDAQ. In addition, certain prior sales by us may be aggregated with any offering we may propose in the future, further limiting the amount we could raise in any future offering without stockholder approval. NASDAQ also requires that we obtain stockholder approval if the issuance or potential issuance of additional shares will be considered by NASDAQ to result in a change of control of our company. Obtaining stockholder approval is a costly and time-consuming process. If we are required to obtain stockholder approval for a potential transaction, we would expect to spend substantial additional money and resources. In addition, seeking stockholder approval would delay our receipt of otherwise available capital or alter the terms of the transaction, which may materially and adversely affect our ability to execute our business strategy, and there is no guarantee our stockholders ultimately would approve a proposed transaction. We have incurred significant losses since our founding and anticipate that we will continue to incur significant losses for the foreseeable future and may never achieve or maintain profitability.
We are a clinical-stage biotechnology company and have not yet generated revenues from product sales. To date, substantially all of our revenues have been derived from grants and contracts with governmental agencies, consisting primarily our BARDA contract for our anthrax vaccine product candidate. We have incurred net losses in most periods since our inception, including a net loss of $49.0 million and $20.5 million for the years ended December 31, 2020 and 2019, respectively. As of December 31, 2020, we have an accumulated deficit of $186.4 million. To date, we have not received regulatory approvals for any products or generated any revenues from the sale of products, and we do not expect to generate any product revenues in the foreseeable future. We do not know whether or when we will generate product revenues or become profitable.
We have devoted most of our financial resources to research and development, including preclinical and clinical development of our product candidates. We have not completed pivotal clinical trials for any product candidate. Our leading product candidates remain in early stage clinical development, and it may be several years, if ever, before we have a product candidate ready for commercialization. Even if we obtain regulatory approval to market a product candidate, our future revenues will depend upon the size of any markets in which our product candidates have received approval, our ability to achieve sufficient market acceptance, reimbursement from third-party payers and other factors.
The net losses we incur may fluctuate significantly from quarter to quarter and year to year, such that a period-to-period comparison of our results of operations may not be a good indication of our future performance. In any particular quarter or quarters, our operating results could be below the expectations of securities analysts or investors, which could cause our stock price to decline.
Our profitability depends on our ability to develop and commercialize our current and future product candidates.
To become and remain profitable, we must succeed in developing and eventually commercializing products that generate significant revenue. This will require us to be successful in a range of challenging activities, including completing preclinical studies and clinical trials of our product candidates, discovering additional product candidates, obtaining regulatory approval for these product candidates, forming strategic partnerships and alliances with third parties and manufacturing, marketing and selling any products for which we may obtain regulatory approval. We are only in the preliminary stages of most of these activities. We may never succeed in these activities and, even if we do, we may never generate revenues that are significant enough to achieve profitability. If some or all of our product candidates do not prove to be safe, pure and efficacious, then we may have to abandon those product candidates altogether and we will be unable to generate revenues from sales of such products.
We expect to continue to incur significant expenses and increasing operating losses for the foreseeable future. We anticipate that our expenses will increase significantly if and as we:
continue our clinical trials for our product candidates;
initiate additional preclinical studies, clinical trials or other studies or trials for our other product candidates;
manufacture material for clinical trials and, if any product candidate is approved for marketing, for commercial sale;
seek regulatory approvals for our product candidates that successfully complete clinical trials;
establish a sales, marketing and distribution infrastructure to commercialize any products for which we may obtain marketing approval;
seek to discover and develop additional product candidates;
acquire or in-license other product candidates and technologies;
make royalty, milestone or other payments under any in-license agreements;
form strategic partnerships and/or makes additional acquisitions;
maintain, protect and expand our intellectual property portfolio;
attract and retain skilled personnel; and
create additional infrastructure to support our operations as a public company and our product development and planned future commercialization efforts.
Because of the numerous risks and uncertainties associated with pharmaceutical product development, we are unable to accurately predict the timing or amount of increased expenses or when, or if, we will be able to achieve profitability. If we are required by the FDA or other regulatory authorities to perform studies in addition to those currently expected, or if there are any delays in completing our clinical trials or the development of any of our product candidates, our expenses could increase.
Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our failure to become and remain profitable would depress the value of our company and could impair our ability to raise capital, expand our business, maintain our research and development efforts, diversify our product offerings or even continue our operations.
Future conditions might require us to make substantial write-downs in our assets, which would adversely affect our balance sheet and results of operations.
We review our long-lived tangible and intangible assets for impairment whenever events or changes in circumstances indicate that the carrying value of an asset may not be recoverable. We also test our indefinite-lived intangible assets for impairment at least annually in the fourth quarter, or when events or changes in the business environment indicate that the carrying value of the reporting unit may exceed its fair value. During 2019, the development activities under our SparVax-L governmental contract were completed with no future funding identified. As a result of the contract completion and the US government’s funding prioritization of only single dose anthrax vaccine candidates, we impaired $1.0 million for the remaining net book value of the SparVax-L IPR&D asset during 2019. At December 31, 2020, we continued to carry $12.4 million of indefinite lived intangible assets. Any such significant write-downs of our long-lived assets in the future could adversely affect our balance sheet and results of operations.
We will require substantial additional financing to achieve our goals, and a failure to obtain this necessary capital when needed would force us to delay, limit, reduce or terminate our product development or commercialization efforts.
We do not expect to generate revenue from product sales, licensing fees, royalties, milestones, contract research or other sources in an amount sufficient to fully fund our operations for the foreseeable future, if ever. Therefore, we will use our existing cash resources, together with funding received from BARDA and MTEC, and will require additional funds to maintain our operations, continue our research and development programs, commence future preclinical studies and clinical trials, seek regulatory approvals and manufacture and market our products. As of December 31, 2020, our cash and short-term investment balance was $216.0 million. Based on our current operating plan, we believe that our existing cash will be sufficient to fund our projected operating expenses and capital expenditure requirements for at least a twelve-month period from the issuance date of our December 31, 2020 financial statements. However, we do not expect that these funds will be sufficient to enable us to complete the clinical trials needed to seek marketing approval or commercialize any of our product candidates. Furthermore, our operating plan may change as a result of many factors currently unknown to us, and we may need additional funds sooner than planned.
We believe that we will continue to expend substantial resources for the foreseeable future developing our product candidates. These expenditures will include costs associated with research and development, maintaining our intellectual property estate, potentially acquiring new technologies, obtaining regulatory approvals and manufacturing products, forming partnerships and strategic alliances, as well as marketing and selling products approved for sale, if any. In addition, other unanticipated costs may arise. Because the outcome of our planned and anticipated clinical trials is highly uncertain, we cannot reasonably estimate the actual amounts necessary to successfully complete the development and commercialization of our product candidates.
Our future capital requirements depend on many factors, including:
the progress, results and costs of our clinical trials for our leading product candidates;
the scope, progress, results and costs of preclinical development, laboratory testing and clinical trials for our other product candidates;
the amount of funding that we receive from BARDA, MTEC, other government agencies and other non-dilutive funding sources;
the number and development requirements of other product candidates that we pursue;
the timing of, and the costs involved in, obtaining regulatory approvals for our product candidates if clinical trials are successful and the outcome of regulatory review of our product candidates;
our ability to contract with third-party manufacturing facilities for adequate supply and to establish processes that meet regulatory requirements for commercialization;
the cost and timing of future commercialization activities for our products, if any of our product candidates are approved for marketing, including product manufacturing, marketing, sales and distribution costs;
the revenue, if any, received from commercial sales of our product candidates for which we receive marketing approval;
our ability to establish and maintain strategic partnerships, licensing or other arrangements and the financial terms of such agreements; and
the costs involved in preparing, filing and prosecuting patent applications, and maintaining, defending and enforcing our intellectual property rights, including litigation costs and the outcome of such litigation.
We may also seek additional capital due to favorable market conditions or strategic considerations even if we believe we have sufficient funds for our current or future operating plans. Additional funds may not be available when we need them on terms that are acceptable to us, or at all. If adequate funds are not available to us when needed, we may be required to delay, limit, reduce or terminate preclinical studies, clinical trials or other development activities for one or more of our product candidates or delay, limit, reduce or terminate our establishment of sales and marketing capabilities or other activities that may be necessary to commercialize our product candidates.
Raising additional capital may cause dilution to stockholders, restrict our operations or require us to relinquish rights to our technologies or product candidates on unfavorable terms. Until such time, if ever, as we can generate substantial product revenues, we expect to finance our cash needs through a combination of public or private equity offerings, debt financings, BARDA funding, MTEC funding and license and development agreements through strategic partnerships with third parties. To the extent that we raise additional capital through the sale of equity or convertible debt securities, your ownership interest will be diluted, and the terms may include liquidation or other preferences that
adversely affect your rights as a stockholder. Debt or preferred stock financing, if available, may involve agreements that include covenants limiting or restricting our ability to take certain actions, such as incurring additional debt, issuing additional equity, making capital expenditures or declaring dividends. If we raise additional funds through strategic partnerships with third parties, we may have to relinquish valuable rights to our technologies or product candidates, future revenue streams, research programs or product candidates, or otherwise grant licenses on terms that are not favorable. If we are unable to raise additional capital when needed, we may be required to delay, limit, reduce or terminate our product development or commercialization efforts for our leading product candidates or our preclinical product candidates, or grant rights to develop and market product candidates that we would otherwise prefer to develop and market ourselves. Our product candidates are in an early stage of development, there is a high risk of failure, and we may never succeed in developing marketable products or generating product revenue. Our preclinical and clinical results are not necessarily predictive of the final results of our ongoing or future clinical trials. We have completed a Phase 2 clinical trial of NasoVAX; have initiated a Phase 2 clinical trial of HepTcell; are conducting a Phase 1/2 clinical trial of T-COVID;HepTcell and are currently in Phase 2 and Phase 1 clinical development with NasoShield, ALT-801, and AdCOVID;.pemvidutide for multiple indications. Success in preclinical studies may not be predictive of similar results in humans during clinical trials, and successful results from early or small clinical trials of a product candidate may not be replicated in later and larger clinical trials. Interim results from clinical trials may not be predictive Clinical trials are expensive, time consuming and uncertain as to outcome, and we cannot guarantee that any of these activities will be successful. If the results of our ongoing or future clinical trials are inconclusive with respect to the efficacy of our product candidates, if we do not meet our clinical endpoints with statistical significance or if there are safety concerns or adverse events associated with our product candidates, we may be prevented or delayed in obtaining marketing approval for our product candidates, or we may determine to suspend development of or abandon specific product candidates. For example, we suspended the development of a Densigen platform-based product candidate, Flunisyn, which was being developed as a T-cell vaccine for the treatment of influenza, in favor of NasoVAX. Clinical trials with this product candidate showed that it was generally well-tolerated and able to induce robust T-cell responses against the viral sequences represented, but a comparison of the entire study population in later-stage clinical trials showed no statistical differences between the vaccinated and placebo groups for several measures of protection. In addition, we can offer no assurances that we have correctly estimated the resources or personnel necessary to seek partners, co-developers or acquirers for our programs. If a larger workforce or one with a different skillset is ultimately required to maintain these operations, we may be unable to maximize our existing programs. Interim and preliminary results from our clinical trials that we announce or publish from time to time may change as more patient data become available and are subject to additional audit, validation and verification procedures that could result in material changes in the final data. From time to time, we may publish interim data, including interim top-line results or preliminary results from our clinical trials. Any interim data and other results from our clinical trials may materially change as more patient data become available. Preliminary or top-line results also remain subject to audit, validation and verification procedures that may result in the final data being materially different from the interim and preliminary data we previously published. As a result, interim and preliminary data may not be predictive of final results and should be viewed with caution until the final data are available. We may also arrive at different conclusions, or considerations may qualify such results, once we have received and fully evaluated additional data. Differences between preliminary or interim data and final data could adversely affect our business. The manufacture of our product candidates is complex and we may encounter difficulties in production, particularly with respect to acquisition of materials, process development or scaling-up of our manufacturing capabilities. The manufacture of our product candidates is complex, highly-regulated and subject to multiple risks. The complex processes associated with the manufacture of our product candidates expose us to various manufacturing challenges and risks, which may include delays in manufacturing our product candidates, limits on our ability to increase manufacturing capacity, and the potential for product failure and product variation that may interfere with our clinical development plans and add additional costs. It is possible that we will make changes to our manufacturing process at various points during product development or commercialization for various reasons, such as controlling costs, achieving scale, decreasing processing time, increasing manufacturing success rate, or other reasons. Such changes can be costly and carry the risk that they will not achieve their intended objectives, or these changes could cause our product candidates to perform differently and affect the results of current or future clinical trials, or the performance of a commercialized product. In some circumstances, changes in the manufacturing process may require us to perform analytical or clinical comparability studies and to collect additional data prior to undertaking more advanced clinical trials, and such studies may introduce additional costs or delays to the program. We may be required to collect additional clinical data from any modified process prior to obtaining marketing approval for the product candidate produced with such modified process. If clinical data are not ultimately comparable to that seen in the earlier trials in terms of safety or efficacy, we may be required to make further changes to our process
and/or undertake additional clinical testing, either of which could significantly delay the clinical development or commercialization of the associated product candidate. In 2020, we initiated the manufacture of clinical lots of AdCOVID and in parallel began technology transfer to multiple commercial scale CMOs in preparation for process scale-up. Although we have added, and are continuing to add, employees with experience in manufacturing products similar to our product candidates, we have limited experience as a company manufacturing product candidates for use in clinical trials and no experience as a company manufacturing product candidates for commercial supply. We may never be successful in manufacturing product candidates in sufficient quantities or with sufficient quality for some or all of our clinical or commercial uses. Even if we are successful in developing our manufacturing capabilities sufficient for our clinical and commercial uses, our manufacturing capabilities could be affected by cost-overruns, unexpected delays, equipment failures, labor shortages, operator error, natural disasters, availability of qualified personnel, difficulties with logistics and shipping, problems regarding yields or stability of product, product contamination or other quality control issues, power failures, and numerous other factors that could prevent us from realizing the intended benefits of our manufacturing strategy and have a material adverse effect on our business.
Compliance with cGMP requirements and other quality or regulatory issues may arise during our internal efforts to manufacturing our product candidates, and with our current or any future CMOs.contract manufacturing organizations (“CMOs”). Furthermore, ongoing stability studies subsequent to manufacture must be periodically conducted to demonstrate that each of our product candidates do not undergo unacceptable deterioration over its shelf life. If issues affecting the quality of our product candidates, our manufacturing facilities, or those of our CMOs are discovered, such manufacturing facilities may need to be closed for an extended period of time to investigate and remedy the issue. To the extent any adversely affected material is being used in an ongoing clinical trial, the FDA could impose a clinical hold on our studytrial to investigate and remedy the quality issue. We cannot assure that any manufactured product or product candidate will not suffer a loss isin stability or that other issues relating to the manufacture of our product candidates will not occur in the future. Additionally, we and our CMOs may experience manufacturing difficulties due to resource constraints, material constraints, or as a result of labor disputes or unstable political environments. If we or our CMOs were to encounter any of these difficulties, our ability to provide our product candidate to patients in clinical trials, or to provide product for treatment of patients once approved, would be jeopardized. We may encounter substantial delays in our clinical trials, or our clinical trials may fail to demonstrate the safety and efficacy of our product candidates to the satisfaction of applicable regulatory authorities. Before obtaining marketing approval from regulatory authorities for the sale of our product candidates, we must conduct extensive clinical trials to demonstrate the safety and efficacy of the product candidates in humans. Clinical testing is expensive, time consuming and uncertain as to outcome. We cannot guarantee that clinical trials will be conducted as planned or completed on schedule, if at all. A failure of one or more clinical trials can occur at any stage of testing. Events that may prevent successful or timely completion of clinical development include: delays or failure in reaching a consensus with regulatory agencies on trial design;
delays or failure in reaching agreement on acceptable terms with prospective contract research organizations (“CROs”) and clinical trial sites;
delays or failure in obtaining required approvals from the Institutional Review Board (“IRB”) or other similar committees or bodies at each clinical trial site;
imposition of a clinical hold by regulatory agencies for any reason, including safety concerns raised by other clinical trials of similar product candidates that may reflect an unacceptable risk with the patient population, technology platform, product stability or after an inspection of clinical operations or trial sites;
failure to perform clinical trials in accordance with the FDA’s Good Clinical Practices (“GCP”) or applicable regulatory guidelines in other countries, including the United Kingdom;
delays or failure in the testing, validation, manufacturing and delivery of the product candidates to the clinical sites;
any shelter-in-place orders from local, state or federal governments or clinical trial site policies resulting from the COVID-19 pandemic that determine essential and non-essential functions and staff, which may impact the ability of site staff to conduct assessments, or result in delays to the conduct of the assessments, as part of our clinical trial protocols, or the ability to enter assessment results into clinical trial databases in a timeline manner;
| •
● | delays or failure in reaching a consensus with regulatory agencies on trial design; |
| ● | delays or failure in reaching agreement on acceptable terms with prospective contract research organizations (“CROs”) and clinical trial sites; |
| ● | delays or failure in obtaining required approvals from the IRB or other similar committees or bodies at each clinical trial site; |
| ● | imposition of a clinical hold by regulatory agencies for any reason, including safety concerns raised by other clinical trials of similar product candidates that may reflect an unacceptable risk with the patient population, technology platform, product stability or after an inspection of clinical operations or trial sites; |
| ● | failure to perform clinical trials in accordance with the FDA’s GCP or applicable regulatory guidelines in other countries, including the United Kingdom, Canada, Germany, Italy, Spain, Thailand and Australia; |
| ● | delays or failure in the testing, validation, manufacturing and delivery of the product candidates to the clinical sites, including as a result of supply chain delays in obtaining materials for the manufacture of our clinical trial materials; |
| ● | delays in testing, analysis and other activities by our third-party contractors required in conducting and analyzing our clinical trials as a result of the ongoing COVID-19 pandemic; |
| ● | any shelter-in-place orders from local, state or federal governments or clinical trial site policies resulting from the ongoing COVID-19 pandemic that determine essential and non-essential functions and staff, which may impact the ability of site staff to conduct assessments, or result in delays to the conduct of the assessments, as part of our clinical trial protocols, or the ability to enter assessment results into clinical trial databases in a timely manner; |
| ● | the number of patients required for our clinical trials may be larger than we anticipate, enrollment in our clinical trials may be slower than we anticipate or participants may, including as a result of the ongoing COVID-19 pandemic, withdraw from our clinical trials, fail to complete dosing or fail to return for post-treatment follow-up at higher rates than we anticipate, any of which could result in significant delay; |
| ● | withdrawal of clinical trial sites from our clinical trials, including as a result of changing standards of care or the ineligibility of a site to participate; |
| ● | occurrence of serious adverse events in clinical trials that are associated with the product candidate that are viewed to outweigh its potential benefits; |
| ● | our preclinical tests or clinical trials may produce negative or inconclusive results, and we may decide, or regulators or funders may require us, to conduct additional preclinical testing or clinical trials or to abandon projects that we expected to be promising; |
| ● | our third-party contractors (such as CROs, product manufacturers, or investigators) may fail to comply with regulatory requirements or meet their contractual obligations to us in a timely manner; |
| ● | fraudulent activity by a clinical researcher, if discovered, could preclude the submission of clinical data prepared by that researcher, lead to the suspension or substantive scientific review or one or more of our marketing applications by regulatory agencies; |
| ● | the cost of our clinical trials may be greater than we anticipate; |
| ● | additional trials may be necessary, including trials to analyze different dose strengths or dosing schemes; |
| ● | the regulatory requirements for product approval may not be explicit, may evolve over time and may diverge by jurisdiction; |
| ● | evolution in the standard of care that require amendments to ongoing clinical trials and/or the conduct of additional preclinical studies or clinical trials; or |
| ● | changes in regulatory requirements and guidance that require amending or submitting new clinical protocols. |
occurrence of serious adverse events in clinical trials that are associated with the product candidate that are viewed to outweigh its potential benefits;
our preclinical tests or clinical trials may produce negative or inconclusive results, and we may decide, or regulators or funders may require us, to conduct additional preclinical testing or clinical trials or to abandon projects that we expected to be promising;
our third-party contractors (such as CROs, product manufacturers, or investigators) may fail to comply with regulatory requirements or meet their contractual obligations to us in a timely manner;
fraudulent activity by a clinical researcher, if discovered, could preclude the submission of clinical data prepared by that researcher, lead to the suspension or substantive scientific review or one or more of our marketing applications by regulatory agencies;
the cost of our clinical trials may be greater than we anticipate;
the regulatory requirements for product approval may not be explicit, may evolve over time and may diverge by jurisdiction;
evolution in the standard of care that require amendments to ongoing clinical trials and/or the conduct of additional preclinical studies or clinical trials; or
changes in regulatory requirements and guidance that require amending or submitting new clinical protocols.
We, the FDA, other regulatory authorities outside the United States, or an IRB may suspend a clinical trial at any time for various reasons, including if it appears that the clinical trial is exposing participants to unacceptable health risks or if the FDA or one or more other regulatory authorities outside the United States find deficiencies in our IND or similar application outside the United States or the conduct of the trial. Delays, including delays caused by the above factors, can be costly and could negatively affect our ability to complete a clinical trial. For example, we have had delays in previous clinical trials, including those conducted for NasoVAX, as a result of clinical holds imposed by the FDA or other regulatory authorities and requests for additional or new information on vaccine product testing in connection with an Investigational New Drug (“IND”)IND submitted to the FDA. We have previously experienced multiple failures during the manufacturing of clinical materials for use in a future NasoVAX Phase 2 clinical trial.trial. We cannot give any assurance that we will be able to resolve any future clinical holds imposed by the FDA or other regulatory authorities outside of the United States, or any delay caused by manufacturing failures or other factors described above or any other factors, on a timely basis or at all. If we are not able to successfully initiate and complete clinical trials, we will not be able to obtain regulatory approval and will not be able to commercialize our product candidates. Even if our clinical trials are successfully completed as planned, the results may not support approval of our product candidates under the laws and regulations of the FDA or other regulatory authorities outside the United States. The clinical trial process may fail to demonstrate that our product candidates are both safe and effective for their intended uses. Pre-clinical and clinical data and analyses are often able to be interpreted in different ways. Even if we view our results favorably, if a regulatory authority has a different view, we may still fail to obtain regulatory approval of our product candidates. This, in turn, would significantly adversely affect our business prospects. We may find it difficult to enroll patients in our clinical trials, which could delay or prevent clinical trials of our product candidates. Identifying and qualifying patientssubjects to participate in clinical trials of our product candidates is critical to our success. The timing of our clinical trials depends on the speed at which we can recruit patientssubjects to participate in testing our product candidates. If patientssubjects are unwilling to participate in our trials because of the ongoing COVID-19 pandemic and restrictions on travel or healthcare institution policies, negative publicity from adverse events in the biotechnology industries, public perception of vaccine safety issues or for other reasons, including competitive clinical trials for similar patient populations, the timeline for recruiting patients,subjects, conducting studies and
obtaining regulatory approval of potential products may be delayed. These delays could result in increased costs, delays in advancing our product development, delays in testing the effectiveness of our technology or termination of the clinical trials altogether. We may not be able to identify, recruit and enroll a sufficient number of patients,subjects, or those with required or desired characteristics to achieve diversity in a trial, to complete our clinical trials in a timely manner. PatientSubject enrollment is affected by several factors, including: | ● | severity of the disease under investigation; |
| ● | design of the trial protocol; |
| ● | size of the patient population; |
| ● | eligibility criteria for the trial in question; |
| ● | perceived risks and benefits of the product candidate being tested; |
| ● | willingness or availability of subjects to participate in our clinical trials (including due to the COVID-19 pandemic); |
| ● | proximity and availability of clinical trial sites for prospective subjects; |
| ● | our ability to recruit clinical trial investigators with appropriate competencies and experience; |
| ● | availability of competing vaccines and/or therapies and related clinical trials; |
| ● | efforts to facilitate timely enrollment in clinical trials; |
| ● | our ability to obtain and maintain subject consents; |
| ● | patient referral practices of physicians; and |
| ● | ability to monitor subjects adequately during and after treatment. |
severity of the disease under investigation;
design of the trial protocol;
size of the patient population;
eligibility criteria for the trial in question;
perceived risks and benefits of the product candidate being tested;
willingness or availability of patients to participate in our clinical trials (including due to the COVID-19 pandemic);
proximity and availability of clinical trial sites for prospective patients;
our ability to recruit clinical trial investigators with appropriate competencies and experience;
availability of competing vaccines and/or therapies and related clinical trials;
efforts to facilitate timely enrollment in clinical trials;
our ability to obtain and maintain patient consents;
patient referral practices of physicians; and
ability to monitor patients adequately during and after treatment.
We may not be able to initiate or continue clinical trials if we cannot enroll a sufficient number of eligible patientssubjects to participate in the clinical trials required by regulatory agencies. Even if we enroll a sufficient number of eligible patientssubjects to initiate our clinical trials, we may be unable to maintain participation of these patientssubjects throughout the course of the clinical trial as required by the clinical trial protocol, in which event we may be unable to use the research results from those patients.subjects. For example, we may face difficulties in identifying patient populations with active disease to enroll in our HBV productPhase 2 clinical trial for HepTcell.of HepTcell in patients with chronic HBV. Other clinical trials involving patients with active HBV have sometimes faced difficulties in working with these patient populations, which may include significant numbers of individuals with difficulties with treatment compliance, such as active drug users. While we are developing strategies to address this issue, there is no guarantee that these strategies will prove successful. If we have difficulty enrolling and maintaining the enrollment of a sufficient number of patientssubjects to conduct our clinical trials as planned, we may need to delay, limit or terminate ongoing or planned clinical trials, any of which would have an adverse effect on our business. It may be difficult to predict the time and cost of product development. Unforeseen problems may prevent further development or approval of our product candidates. OurBecause our product candidates including vaccines and immunotherapies, involve novel therapeutic approaches, to activate the immune system. Consequently, it may be difficult to predict the time and cost of product development. For example, the RespirVec platform involves intranasally administered adenovectored vaccines, the Densigen platform involves synthetic peptide T-cell vaccines and the EuPort platform involves a novel peptide-based dual GLP-1/glucagon receptor agonist. Unforeseen problems with our
approaches to vaccines and therapies may prevent further development or approval of our product candidates. Because of the novelty of our approaches, there may be unknown safety risks associated with the vaccines and therapies that we develop or the clinical endpoints that we establish in trials may not be generally accepted by regulatory agencies, which may therefore require us to perform large field studies to demonstrate efficacy. There can be no assurance that any development problems we may experience in the future will not cause significant delays or unanticipated costs, or that such development problems can be solved. In addition, novel vaccine adjuvants, which are included in HepTcell, our product candidate based on the Densigen technology, may pose an increased safety risk to patients. Adjuvants are compounds that are added to vaccine antigens to enhance the activation and improve immune response and efficacy of vaccines. Development of vaccines with novel adjuvants requires evaluation in larger numbers of patients prior to approval than would be typical for therapeutic drugs. Guidelines for evaluation of vaccines with novel
adjuvants have been established by the FDA and other regulatory bodies and expert committees. Any vaccine, because of the presence of an adjuvant, may have side effects considered to pose too great a risk to patients to warrant approval of the vaccine. Traditionally, regulatory authorities have required extensive study of novel adjuvants because vaccines typically get administered to healthy populations, in particular infants, children and the elderly, rather than in people with disease. As a result, although it is anticipated that HepTcell is intended for the treatment of patients suffering from a disease, regulatory agencies such as the FDA may nevertheless require us to conduct extensive safety testing prior to approval to demonstrate a low risk of rare and severe adverse events caused by our product candidates that include novel vaccine adjuvants. If approved, the novel mechanism of action of the vaccines may adversely affect physician and patient perception and acceptance of our products. Public perception of vaccine safety issues, including adoption of novel vaccine mechanisms of action, may adversely influence willingness of subjects to participate in clinical trials, or if approved, to prescribe and receive novel vaccines. For example, GSK pulled from the market an approved vaccine to prevent Lyme disease (Lymerix) in February 2002 after anecdotal evidence of joint pain resulted in subjects’ unwillingness to receive the vaccine. The FDA found no evidence that the vaccine caused a safety risk; however, GSK pulled the vaccine due to low sales resulting from the negative public perception associated with the reports on joint pain. In addition, parental aversion to new vaccines or vaccines in general may adversely influence later stage clinical trials of our COVID-19 product candidates or, if approved, its commercial success. We rely, and expect to continue to rely, on third parties to conduct preclinical studies and clinical trials for our product candidates, and if they do not properly and successfully perform their obligations to us, we may not be able to obtain regulatory approvals for our product candidates. We rely, and expect to continue to rely, on third parties, such as CROs, clinical data management organizations, medical institutions and clinical investigators to assist in managing, monitoring and otherwise carrying out our clinical trials. We compete with many other companies for the resources of these third parties. The third parties on whom we rely generally may terminate their engagements at any time and causing us to enter into alternative arrangements would delay development and commercialization of our product candidates. Our reliance on these third parties for research and development activities will reduce our control over these activities but will not relieve us of our responsibilities. For example, the FDA and foreign regulatory authorities require compliance with applicable law, regulations and standards, including GCP, for designing, conducting, monitoring, recording, analyzing and reporting the results of clinical trials to assure that the data and results are credible and accurate and that the rights, integrity and confidentiality of trial participants are protected. Regulatory authorities enforce these GCP requirements through periodic inspections of trial sponsors, clinical investigators and trial sites. We cannot be certain that, upon inspection, such regulatory authorities will determine that any of our clinical trials complies with GCP requirements. Although we rely on third parties to conduct our clinical trials, we are responsible for ensuring that each of these clinical trials is conducted in accordance with applicable law, regulations and standards, including our general investigational plan and protocol. Furthermore, if these third parties do not successfully carry out their duties under their agreements, if the quality or accuracy of the data they obtain is compromised due to their failure to adhere to clinical trial protocols or to regulatory requirements, or if they otherwise fail to comply with clinical trial protocols or meet expected deadlines, then the clinical trials of our product candidates may not meet regulatory requirements. If clinical trials do not meet regulatory requirements or if these third parties need to be replaced, then preclinical development activities or clinical trials may be extended, delayed, suspended or terminated. If any of these events occur, we may not be able to obtain regulatory approval of our product candidates on a timely basis or at all. Any third parties conducting aspects of our preclinical studies or clinical trials will not be our employees and, except for remedies that may be available to us under our agreements with such third parties, we cannot control whether they devote sufficient time and resources to our preclinical studies and clinical trials. These third parties may also have relationships with other commercial entities, including our competitors, for whom they may also be conducting clinical trials or other product development activities, which could affect their performance on our behalf. If these third parties do not successfully carry out their contractual duties or obligations or meet expected deadlines, if they cannot perform their contractual duties or obligations due to the impacts of the ongoing COVID-19 pandemic on their operations or at the sites they are overseeing, if they need to be replaced or if the quality or accuracy of the preclinical or clinical data they obtain is compromised due to the failure to adhere to our protocols or regulatory requirements or for other reasons, our development timelines, including clinical development timelines, may be extended, delayed or terminated and we may not be able to complete development of, receive regulatory approval of or successfully commercialize our product candidates. As a result, our financial results and the commercial prospects for our product candidates would be harmed, our costs could increase and our ability to generate revenue could be delayed. If any of our relationships with these third parties terminates, we may not be able to enter into arrangements with alternative third parties or to do so on commercially reasonable terms. Switching or adding additional third partythird-party service providers involves
additional cost and requires management time and focus. In addition, there is a natural transition period when a new third partythird-party service provider begins work. As a result, delays may occur, which can materially impact our ability to meet our desired development timelines. We face substantial competition from other pharmaceutical and biotechnology companies, which may result in others discovering, developing or commercializing products before, or more successfully, than we do. The development and commercialization of new drug products is highly competitive. Our future success depends on our ability to demonstrate and maintain a competitive advantage with respect to the design, development and commercialization of our product candidates. Our objective is to design, develop and commercialize new products with superior efficacy, convenience, tolerability and safety. In many cases, the products that we intend to commercialize, if successfully commercialized, will compete with existing market-leading products. Many of our potential competitors have significantly greater financial, manufacturing, marketing, drug development, technical and human resources than we do. Large pharmaceutical companies, in particular, have extensive experience in clinical testing, obtaining regulatory approvals, recruiting patients and manufacturing pharmaceutical products. Large and established companies such as Eli Lilly, Roche, Novartis,Novo Nordisk and Pfizer, and Sanofi Pasteur, among others, compete in the same market as our product candidates. These companies compete with us with their greater experience and resources to support their research and development efforts, conduct testing and clinical trials, obtain regulatory approvals to market products, manufacture such products on a broad scale and market approved products. These companies also compete with us by having significantly greater research and marketing capabilities than we do and may also have products that have been approved or are in late stages of development and have collaborative arrangements in our target markets with leading companies and research institutions. Established pharmaceutical companies may also invest heavily to accelerate discovery and development of novel compounds or to in-license novel compounds that could make the products that we develop obsolete. We also face competition from smaller companies who, like us, rely on investors to fund research and development and compete for co-development and licensing opportunities from large and established pharmaceutical companies. We face competition from multiple biotechnologyfor pemvidutide, our dual GLP 1/glucagon dual agonist for the treatment of obesity and bio-pharmaceuticalNASH. For obesity, we face competition from companies such as Pfizer, Moderna,Novo Nordisk, whose GLP-1 agonist, brand named Wegovy, or compound name semaglutide, was approved in June 2021. Other companies with potentially competitive candidates in development, include Eli Lilly with GLP-1/glucose-dependent insulinotropic polypeptide receptor (“GIP”) dual agonists, including Mounjaro, or compound name tirzepatide, currently approved for type 2 diabetes; Boehringer Ingelheim, Merck/Hanmi Pharmaceutical, AstraZeneca, RegeneronInnovent Biologics/Eli Lilly, Carmot and Johnson & Johnson, that are in the processD&D Pharma, with GLP-1/glucagon receptor dual agonists; Hanmi Pharmaceutical and therapeutics against COVID-19. Certain of these vaccineEli Lilly with GLP-1/glucagon/GIP triple agonists; Amgen with its GLP-1 agonist/GIP antagonist antibody; and therapeutic technologies are being developed at a faster rate than AdCOVIDNovo Nordisk with Amylin and T-COVID or have superior immunogenicity or manufacturability attributes.Amylin-GLP-1 combination candidates. We face competition for NasoShield, our single dose intranasal anthrax vaccine product candidate, from Emergent Biosolutions which manufactures the existing anthrax vaccine. Additionally, we generally face substantial competition for government funding from companies that develop products with government contracts and grants. We face competition for NasoVAX, our intranasal influenza candidate from Novavax, which is developing an influenza vaccine; and a number of companies of varying sizes are also pursuing the development of a “universal” flu vaccine. We face competition for ALT-801, our dual GLP-1/glucagon dual agonist product candidate for the treatment ofin NASH from companies such as Intercept Pharmaceuticals, which is developing a farnesoid X receptor (“FXR”) agonist; Madrigal Pharmaceuticals, Terns, Aligos and Viking Therapeutics, which isare developing an orally administered, small-molecule, liver-directed, thyroid hormone receptor (THR)(“THR”) β-selective agonist; and, Akero Therapeutics, 89Bio, Novo Nordisk, Boston Pharmaceuticals and Roche, which are developing fibroblast growth factor 21 analogs; Novo Nordisk, which is developing a fibroblast growth factor 21 analog.GLP-1 agonist; and, Inventiva, which is developing a pan-peroxisome proliferator-activated receptor (“PPAR”) agonist and orally based GLP-1 agents that either deliver GLP-1 monoagonist activity either as a peptide (Novo Nordisk) or small molecule (Pfizer, Eli Lilly). In addition, many other small companies are developing other new technologies directed towards obesity or NASH. Finally, we face competition for HepTcell, our immunotherapeutic HBV product candidate, from companies such as TransgeneGSK, Janssen, Vaccitech VBI Vaccines, all of which is developing an adenovirus-based vaccine; Arrowhead Pharmaceuticals, which is developing an HBV therapeutic vaccine; and Inovio, which isare developing a DNAtherapeutic vaccine delivered by in vivo electroporation. Any of theseagainst chronic HBV infection. In addition, many other companies may develop competing products more rapidly than we do.are developing direct acting antivirals against HBV. As a result of all of these factors, our competitors may succeed in obtaining patent protection and/or FDA approval or discovering, developing and commercializing products before we do. In addition, any new product that we develop that competes with an approved product must demonstrate compelling advantages in efficacy, convenience, tolerability and safety in order to overcome price competition and to be commercially successful. If we are not able to compete effectively against potential competitors, our business will not grow, and our financial condition and operations will suffer. We are heavily dependent on the success of our leading product candidates, AdCOVID and ALT-801.candidate, pemvidutide. If we ultimately are unable to develop, obtain regulatory approval for or commercialize AdCOVID, ALT-801,pemvidutide, or any other product candidate, our business will be substantially harmed. We currently have no products approved for commercial distribution. Our business strategy is to build a pipeline of product candidates using our proprietary platforms, including our leading product candidates AdCOVID and ALT-801,candidate, pemvidutide, and to progress thosethe product candidatescandidate through clinical development for the treatment of different types of diseases. We may not be able to develop products that are safe and effective for all or any of the indications that we target. Even if we are successful in building a product pipeline, the potential product candidates that we identify may not be suitable for clinical development for a number of reasons, including causing harmful side effects or demonstrating other characteristics that indicate a low likelihood of receiving marketing
approval or achieving market acceptance. If our methods of identifying potential product candidates fail to produce a pipeline of potentially viable product candidates, then our success as a business will be dependent on the success of fewer potential product candidates, which introduces risks to our business model and potential limitations to any success we may achieve. Because we have limited financial and managerial resources, we must focus on a limited number of research programs and product candidates and on specific indications. As a result, we may forego or delay pursuit of opportunities with other product candidates or for other indications that later prove to have greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities. Our spending on current and future discovery and preclinical development programs and product candidates for specific indications may not yield any commercially viable products. Furthermore, until such time as we are able to build a broader product candidate pipeline, if ever, any adverse developments with respect to our leading product candidates, AdCOVID and ALT-801,candidate, pemvidutide, would have a more significant adverse effect on our overall business than if we maintained a broader portfolio of product candidates. Even if a current or future product candidate receives marketing approval, it may fail to achieve the degree of market acceptance by physicians, patients, third-party payorspayers and others in the medical community necessary for commercial success. If any current or future product candidate we develop receives marketing approval, whether as a single agent or in combination with other therapies, it may nonetheless fail to gain sufficient market acceptance by physicians, patients, third-party payors,payers, and others in the medical community. The degree of market acceptance of any product candidate, if approved for commercial sale, will depend on a number of factors, including: | ● | efficacy and potential advantages compared to alternative treatments; |
the ability to offer our products, if approved, for sale at competitive prices;
| ● | the ability to offer our products, if approved, for sale at competitive prices; |
| ● | convenience and ease of administration compared to alternative treatments; |
| ● | the willingness of the target patient population to try new therapies and of physicians to prescribe these therapies; |
| ● | the strength of marketing and distribution support; |
| ● | the ability to obtain sufficient third-party coverage and adequate reimbursement, including with respect to the use of the approved product as a combination therapy; |
| ● | the cost of treatment in relation to alternative treatments, including generic products; |
| ● | the extent and strength of our third-party manufacturer and supplier support; |
| ● | the extent and strength of marketing and distribution support; |
| ● | the limitations or warnings contained in a product’s approved labeling; |
| ● | distribution and use restrictions imposed by the FDA or other regulatory authorities outside the United States or that are part of a REMS or voluntary risk management plan; and |
| ● | the prevalence and severity of any side effects, including the tolerability and effect on comorbidities relative to alternative treatments. |
the willingness of the target patient population to try new therapies and of physicians to prescribe these therapies;
the strength of marketing and distribution support;
the ability to obtain sufficient third-party coverage and adequate reimbursement, including with respect to the use of the approved product as a combination therapy;
the cost of treatment in relation to alternative treatments, including generic products;
the extent and strength of our third party manufacturer and supplier support;
the extent and strength of marketing and distribution support;
the limitations or warnings contained in a product’s approved labeling;
distribution and use restrictions imposed by the FDA or other regulatory authorities outside the United States or that are part of a REMS or voluntary risk management plan; and
the prevalence and severity of any side effects.
For example, even if our products have been approved by the FDA, physicians and patients may not immediately be receptive to them and may be slow to adopt them. If our products do not achieve an adequate level of acceptance among physicians, patients and third party payors,third-party payers, we may not generate meaningful revenues and we may not become profitable. We may not be able to comply with the requirements of foreign jurisdictions in conducting trials within the United Kingdom, European Union Australia or any other foreign country. We are conducting our Phase 2 clinical trial of HepTcell in the U.S., United Kingdom, Canada, Germany Italy, Spain and Spain; conducting our ALT-801 Phase 1 clinical trial in Australia;Thailand; and future clinical trials may be conducted in other foreign jurisdictions. Our ability to successfully initiate, enroll and complete a clinical trial in any other foreign country is subject to numerous risks unique to conducting business in foreign countries, including: difficulty in establishing or managing relationships with CROs and physicians;
different standards for the approval and conduct of clinical trials;
our inability to locate qualified local consultants, physicians and partners;
| •
● | difficulty in establishing or managing relationships with CROs and physicians; |
| ● | different standards for the approval and conduct of clinical trials; |
| ● | our inability to locate qualified local consultants, physicians and partners; |
| ● | the potential burden of complying with a variety of foreign laws, medical standards and regulatory requirements, including the regulation of the conduct of clinical trials, pharmaceutical and biotechnology products and treatment; and |
| ● | the acceptability of data obtained from studies conducted outside the United States to the FDA in support of U.S. marketing authorizations, such as an NDA or BLA. |
the acceptability of data obtained from studies conducted outside the United States to the FDA in support of U.S. marketing authorizations, such as a BLA.
If we fail to successfully meet requirements for the conduct of clinical trials outside of the United States, we may be delayed in obtaining, or be unable to obtain, regulatory approval for our product candidates in the United States or in countries outside of the United States. If we fail to attract and keep senior management and key scientific personnel, we may be unable to successfully develop our products, conduct our clinical trials and commercialize our product candidates. We are highly dependent on members of our senior management, including Dr. Vipin Garg, our President and Chief Executive Officer, Will Brown,Richard Eisenstadt, our Chief Financial Officer, Dr. Scott Harris, our Chief Medical Officer, and Dr. M. Scot Roberts, our Chief Scientific Officer and Raymond Jordt, our Chief Business Officer. Although we have entered into employment agreements with each of these members of senior management and key employees, the loss of the services of any of these persons could impede the achievement of our research, development and commercialization objectives. Recruiting and retaining qualified scientific, clinical, manufacturing, sales and marketing personnel will also be critical to our success. The loss of the services of our executive officers or other key employees could impede the achievement of our research, development, commercialization and commercializationbusiness development objectives and seriously harm our ability to successfully implement our business strategy. Furthermore, replacing executive officers and key employees may be difficult and may take an extended period of time because of the limited number of individuals in our industry with the breadth of skills and experience required to successfully develop, gain regulatory approval of and commercialize products. Competition to hire from this limited pool is intense, and we may be unable to hire, train, retain or motivate these key personnel on acceptable terms given the competition among numerous pharmaceutical and biotechnology companies for similar personnel. We also experience competition for the hiring of scientific and clinical personnel from universities and research institutions. In addition, we rely on consultants and advisors, including scientific and clinical advisors, to assist us in formulating our research and development and commercialization strategy. Our consultants and advisors may be employed by employers other than the Company and may have commitments under consulting or advisory contracts with other entities that may limit their availability to the Company. If we are unable to continue to attract and retain high quality personnel, our ability to pursue our growth strategy will be limited. A pandemic, epidemic or outbreak of an infectious disease in the United States such as the COVID-19 pandemic may adversely affect our business. If a pandemic, epidemic or outbreak of an infectious disease occurs in the United States or worldwide, our business may be adversely affected. In December 2019, a novel strain of coronavirus, COVID-19, was identified in Wuhan, China. This virus continuesOur global operations expose us to spread globallyrisks associated with public health crises and has been declared a pandemic by the World Health Organization.epidemics/pandemics, such as COVID-19. The global spread of COVID-19 has impacted the global economycreated significant volatility, uncertainty and worldwide economic disruption, resulting in an economic slowdown of potentially extended duration and may impact our operations, including the potential interruption of our clinical trial activities, regulatory reviews and our supply chain. For example, the ongoing COVID-19 outbreakpandemic (including the Delta and Omicron variants and any future variants that may emerge) may delay preclinical testing and enrollment in our clinical trials due to prioritization of laboratory and hospital resources toward the outbreak or other factors, and some patients may be unwilling to enroll in our trials or be unable to comply with clinical trial protocols if quarantines impede patient movement or interrupt healthcare services, which would delay our ability to conduct clinical trials or release clinical trial results and could delay our ability to obtain regulatory approval and commercialize our product candidates. In addition, since the beginning of the COVID-19 pandemic, several vaccines for COVID-19 have received Emergency Use Authorization by the FDA and a number of those later received marketing approval. Additional vaccines may be authorized or approved in the future. The resultant demand for vaccines and potential for manufacturing facilities and materials to be commandeered under the Defense Production Act of 1950, or equivalent foreign legislation, may make it more difficult to obtain materials or manufacturing slots for the products needed for our clinical trials, which could lead to delays in these trials.
Furthermore, the spread of the virus may affect the operations of key governmental agencies, such as the FDA, which may delay the development of our product candidates. The spread of an infectious disease, including COVID-19, its subvariants and other SARS-CoV-2 viruses, may also result in the inability of our suppliers to deliver components or raw materials on a timely basis or at all. In addition, hospitals may reduce staffing and reduce or postpone certain treatments in response to the spread of an infectious disease. Third parties and CROs on which we rely may also reduce staffing which could impact our ability to continue preclinical testing and clinical trials on expected timeframes. Such events may result in a period of business disruption, and in reduced operations, or doctors and medical providers may be unwilling to participate in our clinical trials, any of which could materially affect our business, financial condition and results of operations. In response to COVID-19-related government and public health directives and orders, we have implemented work-from-home and hybrid policies for certain employees. The effects of these orders and our work-from-home policesand hybrid policies may negatively impact productivity, disrupt our business and delay our clinical programs and timelines, the magnitude of which will depend, in part, on the length and severity of the restrictions and other limitations on our ability to conduct our business in the ordinary course. These and similar, and perhaps more severe, disruptions in our operations could negatively impact our business, operating results and financial condition.
In addition, the trading prices for our common stock and other biopharmaceutical companies have been highly volatile as a result of the COVID-19 pandemic. As a result, we may face difficulties raising capital through sales of our common stock or such sales may be on unfavorable terms. The COVID-19 outbreak continues to rapidly evolve. The extent to which the coronavirusCOVID-19 pandemic may impact our business will depend on future developments, which are highly uncertain and cannot be predicted, including new information which may emerge concerning the severity of the coronavirus, the geographic spread of the disease, the duration of the outbreak, new strains of the virus, and any future variants that may emerge, and the actions to contain the coronavirus or treat its impact, including vaccination campaigns, travel restrictions and other social distancing restrictions in the United States and other countries, among others. A significant outbreak of coronavirus and other infectious diseases could result in a widespread health crisis that could adversely affect the economies and financial markets worldwide, resulting in an economic downturn that could impact our business, financial condition and results of operations.
Labor shortages and constraints in the supply chain could adversely affect our results of operations. In 2022, many companies experienced labor shortages and other labor-related issues, which were pronounced as a result of the ongoing COVID-19 pandemic. A number of factors may adversely affect the labor force available to us or increase labor costs, including high employment levels, federal unemployment subsidies, increased wages offered by other employers, vaccine mandates and other government regulations and our responses thereto. As more employers offer remote work, we may have more difficulty recruiting for jobs that require on-site attendance. We have recently observed an overall tightening and increasingly competitive labor market. If we are unable to hire and retain employees capable of performing at a high-level, our business could be adversely affected. A sustained labor shortage, lack of skilled labor, or increased turnover within our employee base, caused by COVID-19 or as a result of general macroeconomic factors, could have a material adverse impact on our business and operating results. In addition, recent developments in the national and worldwide supply chain slowdown, including as a result of the conflict in Ukraine, have resulted in increased cost and reduced supply for supplies and materials. It is impossible to predict how long this supply chain slowdown will last or how much it will impact our business operations, but it is likely that our costs will increase for supplies. Legal, political and economic uncertainty surrounding the exit of the United Kingdom (“U.K.”), from the European Union (“EU”) may be a source of instability in international markets, create significant currency fluctuations, adversely affect our operations in the U.K. and pose additional risks to our business, revenue, financial condition and results of operations. On June 23, 2016, the U.K. held a referendum in which a majority of the eligible members of the electorate voted to leave the EU. The U.K’sU.K.’s withdrawal from the EU is commonly referred to as Brexit. Pursuant to Article 50 of the Treaty on European Union, the U.K. ceased being a Member State of the EU on January 31, 2020. The U.K.’s withdrawal from the EU has created significant uncertainty concerning the future relationship between the U.K. and the EU. On December 24, 2020, the EU and U.K. reached an agreement in principle on the framework for their future relationship, the EU-UK Trade and Cooperation Agreement. The Agreement primarily focuses on ensuring free trade between the EU and the U.K. in relation to goods, including medicinal products. Among the changes that will now occur are that Great Britain (England, Scotland and Wales) will be treated as a third country. Northern Ireland will, with regard to EU regulations, continue to follow the EU regulatory rules. As part of the Agreement, the EU and the U.K. will recognize Good Manufacturing Practice (GMP)GMP inspections carried out by the other Party and the acceptance of official GMP documents issued by the other Party. The Agreement also encourages, although it does not oblige, the parties to consult one another on proposals to introduce significant changes to technical regulations or inspection procedures. Among the areas of absence of mutual recognition are batch testing and batch release. The U.K. has unilaterally agreed to accept EU batch testing and batch release for a period of at least 2two years until January 1, January 2023. However, the EU continues to apply EU laws that require batch testing and batch release to take place in the EU territory. This means that medicinal products that are tested and released in the U.K. must be retested and re-released when entering the EU market for commercial use. As regards marketing authorizations, Great Britain will have a separate regulatory submission process, approval process and a separate national marketing authorization. Northern Ireland will, however, continue to be covered by the marketing authorizations granted by the European Commission. This lack of clarity on future UKU.K. laws and regulations and their interaction with EU laws and regulations may negatively impact foreign direct investment in the UK,U.K., increase costs, depress economic activity, and restrict access to capital. The uncertainty concerning the UK’sU.K.’s legal, political and economic relationship with the EU after Brexit may be a source of instability in the international markets, create significant currency fluctuations, and/or otherwise adversely affect trading agreements or similar cross-border co-operation arrangements (whether economic, tax, fiscal, legal, regulatory or otherwise) beyond the date of Brexit. These developments, or the perception that any of them could occur, may have a significant adverse effect on global economic conditions and the stability of global financial markets, and could significantly reduce global market liquidity and limit the ability of key market participants to operate in certain financial markets. In particular, it could also lead to a period of considerable uncertainty in relation to the U.K. financial and banking markets, as well as on the regulatory process in Europe. Asset valuations, currency exchange rates and credit ratings may also be subject to increased market volatility. We may also face new regulatory costs and challenges that could have an adverse effect on our operations. Our acquisitions may expose us to unknown liabilities. Because we have acquired all the outstanding shares of most of our acquired companies, our investment in those companies are or will be subject to all of their liabilities other than their respective debts which we paid or will pay at the time of the acquisitions. If there are unknown liabilities or other obligations, our business could be materially affected. We may also experience issues relating to internal controls over financial reporting, issues that could affect our ability to comply with the Sarbanes-Oxley Act, tax examinations by the IRS or state tax authorities, or issues that could affect our ability to comply with other applicable laws. Tax laws could change.
The rules dealing with U.S. federal, state, and local income taxation are constantly under review by persons involved in the legislative process and by the Internal Revenue Service and the U.S. Treasury Department. Changes to tax laws (which changes may have retroactive application) could adversely affect us or holders of our common stock. In recent years, many such changes have been made and changes are likely to continue to occur in the future. Future changes in tax laws resulting from legislative, administrative or judicial decisions may have adverse tax consequences on our business, cash flow, financial condition or results of operations or to a holder of our common stock. Shareholders should consult with their legal and tax advisers regarding the implications of potential changes in tax laws on an investment in our common stock. We may not be able to utilize a significant portion of our net operating loss carryforwards, which could harm our results of operations. We had U.S. federal and state net operating loss carryforwards of approximately $45.7$134.3 million and $103.7 million, respectively, as of December 31, 2020. Of this2022, of which a portion of the federal and state amount $6.6of $7.1 million and $103.7 million, respectively, has a 20-year carry forward period that will expire at various dates beginning in 2021.2024. Under current law, the remaining federal amount of $39.1$127.2 million has an unlimitedindefinite life and generally may not be carried back to prior taxable years except that net operating losses generated in 2018, 2019 and 2020 may be carried back five taxable years. For net operating losses arising in taxable years beginning after December 31, 2017, we are permitted a net operating loss deduction that is limited to 80% of our taxable income in such year. The net operating loss carryforwards are reflective ofsubject to a 382-limitation related to ownership changes. Under Section 382 of the Code, a corporation that undergoes an “ownership change” is subject to limitations on its ability to utilize its pre-change net operating losses or NOLs,(“NOLs”), to offset futureU.S. federal and state taxable income. For these purposes, an ownership change generally occurs wherein the aggregate stockevent of a cumulative change in ownership of one or more stockholders or groupsthe Company of stockholders who owns at least 5% of a corporation’s stock increases its ownership by more than 50 percentage points over its lowest50% within any three-year period. We have not determined if we have experienced Section 382 ownership percentage withinchanges in the past and if a specified testing period.portion of our NOL and tax credit carryforwards are subject to an annual limitation under Section 382. Our existing NOLs are subject to limitations arising from previous ownership changes that impact the timing and amount. In addition, future changes in our stock ownership, many of which are outside of our control, could result in an ownership change. Accordingly, we may not be able to utilize a material portion of our NOLs and this could harm our future operating results by effectively increasing our future tax obligations. As of December 31, 2020,2022, we have recorded a valuation allowance of $18.7$57.2 million against our net deferred tax asset. Risks Related to the Development of COVID-19 Therapeutics
Our pursuit of potential therapeutic and prophylactic treatments for COVID-19 is at an early stage and subject to many risks. We may be unable to receive emergency use authorization or approval for any of our COVID-19 product candidates in a timely manner, if at all, and our COVID-19 product candidates may never be authorized for emergency use or approved.
The FDA cleared our IND application in 2020 to proceed with a Phase 1/2 clinical trial of T-COVID, our investigational agent for the treatment of early COVID-19. The FDA recently cleared our IND for AdCOVID, our intranasal vaccine candidate for COVID-19. We have initiated the Phase 1 clinical trial for AdCOVID and are enrolling in the T-COVID trial. We may experience difficulties or delays in enrolling patients in clinical trials due to the impact of the global COVID-19 pandemic or other reasons. Many of the risks related to the development of these product candidates are beyond our control, including risks related to clinical development, the regulatory submission process, potential threats to our intellectual property rights and manufacturing delays or difficulties. We may be unable to produce an efficacious and/or approved product for the treatment of patients with early COVID-19 in a timely manner, if at all.
The results of preclinical studies from our COVID-19 product candidates may not be predictive of the results of clinical trials, and the results of any early-stage clinical trials we commence may not be predictive of the results of the later-stage clinical trials. There can be no assurance that any of our clinical trials for our COVID-19 product candidates, or any other of our product candidates, will ultimately be successful or support further clinical development. In addition, the interpretation of the data from our clinical trials of T-COVID or AdCOVID by FDA and other regulatory agencies may differ from our interpretation of such data and the FDA or other regulatory agencies may require that we conduct additional studies or analyses. Any of these factors could delay or prevent us from receiving regulatory approval of T-COVID or AdCOVID and there can be no assurance that either product candidate will be approved in a timely manner, if at all.
If the COVID-19 outbreak is effectively contained or the risk of coronavirus infection is diminished or eliminated before we can successfully develop and manufacture our product candidate, the commercial viability of such product candidate may be diminished or eliminated. We are also committing financial resources and personnel to the development of this product candidate which may cause delays in or otherwise negatively impact our other development programs, despite uncertainties surrounding the longevity and extent of coronavirus as a global health concern. Our business could be negatively impacted by our allocation of significant resources to a global health threat that is unpredictable and could rapidly dissipate or against which our treatment, if successfully developed, may not be effective. Two vaccines for COVID-19 were granted Emergency Use Authorization by the FDA in late 2020, and more are likely to
be authorized in the coming months. In addition, other parties are currently developing therapeutic and vaccine candidates for COVID-19, which may be more efficacious than or may be approved prior to T-COVID or AdCOVID.
The regulatory pathway for T-COVID and AdCOVID is continually evolving, and may result in unexpected or unforeseen challenges.
The speed at which parties are acting to create and test many therapeutics and vaccines for COVID-19 is unusual, and evolving or changing plans or priorities within the FDA, including those based on new knowledge of COVID-19 and how the disease affects the human body, may significantly affect the regulatory timeline for our product candidates. Results from ongoing clinical trials and discussions with regulatory authorities may raise new questions and require us to redesign proposed clinical trials, including revising proposed endpoints or adding new clinical trial sites or cohorts of subjects. Any such developments could delay the development timeline for our product candidates and materially increase the cost of the development for such candidates.
Risks Related to the Regulatory Approval Process We cannot guarantee how long it will take regulatory agencies to review our applications for product candidates, and we may fail to obtain the necessary regulatory approvals to market our product candidates. If we are not able to obtain required regulatory approvals, we will not be able to commercialize our product candidates and our ability to generate revenue will be materially impaired. Our product candidates and the activities associated with their development and commercialization, including their design, research, testing, manufacture, safety, efficacy, recordkeeping, labeling, packaging, storage, approval, advertising, promotion, sale and distribution, are subject to comprehensive regulation by the FDA and other regulatory agencies in the United States and foreign jurisdictions. Failure to obtain marketing approval for our product candidates will prevent us from commercializing them in those markets. We have not received approval from regulatory authorities to market any product candidate in any jurisdiction, and it is possible that neither our current product candidates nor any product candidates that we may seek to develop in the future will ever obtain the appropriate regulatory approvals necessary for us to commence product sales. We expect to rely on third-party CROs and consultants to assist in filing and supporting the applications necessary to gain marketing approvals. Securing marketing approval requires the submission of extensive preclinical and clinical data and supporting information to regulatory authorities for each therapeutic indication of each of our product candidates to establish the product candidates’ safety and efficacy for such indications. Securing marketing approval also requires the submission of information about the product manufacturing process to, and inspection of manufacturing facilities by, regulatory authorities. The pathway to regulatory approvals is time consuming and unpredictable, involves substantial costs and consumes management time and attention. It is not possible to predict the timing or success of obtaining regulatory approvals with any degree of certainty, and as a result, it is difficult to forecast our future financial results or prospects. Any unexpected development in the regulatory approval process, including delays or denials of regulatory approvals or significant modifications to our product candidates required by our regulators, could materially and adversely affect our business, results of operations and financial condition, and could substantially harm our stock price. To obtain marketing approval, United States laws require: | ● | controlled research and human clinical testing; |
| ● | establishment of the safety and efficacy of the product for each use sought; |
| ● | government review and approval of a submission containing, among other things, manufacturing, preclinical and clinical data; and |
| ● | compliance with cGMP regulations. |
controlled research and human clinical testing;
establishment of the safety and efficacy of the product for each use sought;
government review and approval of a submission containing, among other things, manufacturing, pre-clinical and clinical data; and
compliance with cGMP regulations.
The process of reviewing and approving a drug is time-consuming, unpredictable, and dependent on a variety of factors outside of our control. The FDA and corresponding regulatory authorities in other jurisdictions have a significant amount of discretion in deciding whether or not to approve a marketing application. Our product candidates could fail to receive regulatory approval from the FDA or comparable regulatory authorities outside the United States for several reasons, including: disagreement with the design or implementation of our clinical trials;
failure to demonstrate that our candidate is safe and effective for the proposed indication;
| •
● | disagreement with the design or implementation of our clinical trials; |
| ● | failure to demonstrate that our candidate is safe and effective for the proposed indication; |
| ● | failure of clinical trial results to meet the level of statistical significance required for approval; |
| ● | failure to demonstrate that the product candidate’s benefits outweigh its risks; |
| ● | disagreement with our interpretation of preclinical or clinical data; and |
| ● | inadequacies in the manufacturing facilities or processes of third-party manufacturers. |
failure to demonstrate that the product candidate’s benefits outweigh its risks;
disagreement with our interpretation of pre-clinical or clinical data; and
inadequacies in the manufacturing facilities or processes of third-party manufacturers.
The FDA or a comparable regulatory authority outside the United States may require us to conduct additional pre-clinicalpreclinical and clinical testing, which may delay or prevent approval and our commercialization plans or cause us to abandon the development program. Further, any approval we receive may be for fewer or more limited indications than we request, may not include labeling claims necessary for successful commercialization of the product candidate, or may be contingent upon our conducting costly post-marketing clinical trials. Any of these scenarios could materially harm the commercial prospects of a product candidate, and our operations will be adversely effected.affected. Our product candidates may cause undesirable side effects or have other properties that delay or prevent their regulatory approval or limit their commercial potential. Significant adverse events caused by our product candidates or even competing products in development that utilize a common mechanism of action could cause us, an IRB or ethics committee, and/or regulatory authorities to interrupt, delay or halt clinical trials and could result in clinical trial challenges such as difficulties in patient recruitment, retention, and adherence, the denial of regulatory approval by the FDA or other regulatory authorities and potential product liability claims. Serious adverse events deemed to be caused by our product candidates could have a material adverse effect on the development of our product candidates and our business as a whole. The most common adverse events in the clinical trials evaluating the safety and tolerability of the product candidates developed using the Respirvec platform have been headaches, runny noses and sore throats. The most common adverse events observed in clinical trials for product candidates developed using the Densigen platform include injection site reactions, headache, malaise and fatigue. We have not yet completed a clinical trial using our EuPort platform. Our understanding of the relationship between our product candidates and these events, as well as our understanding of adverse events reported in future clinical trials of other product candidates, may change as we gather more information, and additional unexpected adverse events may be observed. In addition, the side effect profile of pharmaceutical drugs cannot be fully established based on preapproval clinical trials involving a limited number of patients. Routine review and analysis of post-marketing safety surveillance and clinical trials will provide additional information, for example, potential evidence of rare, population-specific or long-term adverse reactions, and may adversely affect the commercialization of the product, and even lead to the suspension or revocation of product marketing authorization. If we or others identify undesirable side effects caused by our product candidates either before or after receipt of marketing approval, a number of potentially significant negative consequences could result, including: | ● | our clinical trials may be put on hold; |
| ● | we may be unable to obtain regulatory approval for our product candidates; |
| ● | regulatory authorities may withdraw approvals of our products; |
| ● | regulatory authorities may require additional warnings on the label; |
| ● | a medication guide outlining the risks of such side effects for distribution to patients may be required; |
we may be unable to obtain regulatory approval for our product candidates;
| ● | we could be sued and held liable for harm caused to patients; and |
| ● | our reputation may suffer. |
regulatory authorities may require additional warnings on the label;
a medication guide outlining the risks of such side effects for distribution to patients may be required;
we could be sued and held liable for harm caused to patients; and
our reputation may suffer.
Any of these events could prevent us from achieving or maintaining marketing approvals for and market acceptance of our product candidates and could have a material adverse effect on our business and financial results. If we fail to obtain regulatory approval in non-U.S. jurisdictions, we will not be able to market our products in those jurisdictions. Receiving and maintaining regulatory approval of our product candidates in one jurisdiction does not mean that we will be successful in receiving or maintaining regulatory approval of our product candidates in other jurisdictions. We intend to market certain of our product candidates, if approved, in the United Kingdom and other international markets, in addition to the United States. Such marketing will require separate regulatory approvals in each market and compliance with numerous and varying regulatory requirements. The approval procedures vary among countries and may involve requirements for additional testing, and the time required to obtain approval may differ from that required to obtain FDA approval. In addition, in many
countries outside the United States, such as certain countries of the European Union, a vaccine or therapeutic must be approved for reimbursement, including the price that can be charged, before it can be approved for sale in that country. In these countries, pricing discussions with governmental authorities can take considerable time after the receipt of marketing approval for a product, and additional clinical research may be required to enable comparison of the cost effectiveness of our product candidate to other available alternatives. Approval by the FDA does not ensure approval by regulatory authorities in other countries or jurisdictions, and approval by one foreign regulatory authority does not ensure approval by regulatory authorities in other foreign countries or by the FDA. However, the failure to obtain approval in one jurisdiction may compromise our ability to obtain approval elsewhere. The foreign regulatory approval process may include all of the risks associated with obtaining FDA approval. We may not obtain foreign regulatory approvals on a timely basis, if at all. Even if we receive regulatory approval for our product candidates, such products will be subject to ongoing regulatory obligations and continued regulatory review, which may result in significant additional expense and other restrictions, and we may be subject to penalties if we fail to comply with regulatory requirements or experience unanticipated problems with our product candidates. Any regulatory approvals that we receive for our product candidates may be subject to limitations on the approved indicated uses for which the product may be marketed or to conditions of approval. We may also be required to conduct post-marketing testing, including Phase 4 clinical trials, and surveillance to monitor the safety and efficacy of the product potentially over many years. If the FDA or other regulatory authority approves any of our product candidates, the manufacturing processes, labeling, packaging, distribution, import, export, adverse event reporting, storage, advertising, promotion and recordkeeping for the product will be subject to extensive and ongoing regulatory requirements. These requirements include submissions of safety and other post-marketing information and reports, registration, as well as continued compliance with cGMP, and compliance with cGMP and GCP for any clinical trials that we conduct post-approval. Manufacturers and manufacturers’ facilities are also required to comply with extensive FDA and comparable foreign regulatory authority requirements, including ensuring that quality control and manufacturing procedures conform to cGMP regulations and applicable product tracking and tracing requirements. Additionally, under the Food and Drug Omnibus Reform Act of 2022 (“FDORA”), sponsors of approved drugs and biologics must provide six months’ notice to the FDA of any changes in marketing status, such as the withdrawal of a drug, and failure to do so could result in the FDA placing the product on a list of discontinued products, which would revoke the product’s ability to be marketed. Any such restrictions may result in significant additional expense or could limit sales of the approved product. If we, or any of the third parties on which we rely, fail to meet those requirements, the FDA or comparable regulatory authorities outside the United States could initiate enforcement action. Other consequences include the issuance of fines, warning letters, untitled letters or holds on clinical trials, product seizure or detention or refusal to permit the import or export of our product candidates, and permanent injunctions and consent decrees, including the imposition of civil or criminal penalties, among other consequences, that could significantly impair our ability to successfully commercialize a given product. Later discovery of previously unknown problems with an approved product, including adverse events of unanticipated severity or frequency, or with manufacturing operations or processes, may result in, among other things: | ● | restrictions on the marketing or manufacturing of the product, withdrawal of the product from the market or voluntary or mandatory product recalls; |
| ● | fines or warning letters, or clinical holds on clinical trials involving related product candidates; |
| ● | refusal by the FDA or other regulatory authorities to approve pending applications or supplements to approved applications submitted by us or suspension or revocation of product license approvals; |
| ● | product seizure or detention or refusal to permit the import or export of products; and |
| ● | injunctions or the imposition of civil, criminal and/or administrative penalties, damages, monetary fines, disgorgement, exclusion from participation in governmental reimbursement programs, such as Medicare, Medicaid and other federal health care programs and curtailment or restructuring of our operations. |
restrictions on the marketing or manufacturing of the product, withdrawal of the product from the market or voluntary or mandatory product recalls;
fines or warning letters, or clinical holds on clinical trials involving related product candidates;
refusal by the FDA or other regulatory authorities to approve pending applications or supplements to approved applications submitted by us or suspension or revocation of product license approvals;
product seizure or detention or refusal to permit the import or export of products; and
injunctions or the imposition of civil, criminal and/or administrative penalties, damages, monetary fines, disgorgement, exclusion from participation in governmental reimbursement programs, such as Medicare, Medicaid and other federal health care programs and curtailment or restructuring of our operations.
In addition, applicable regulatory policies of governmental authorities, such as the FDA, may change and additional government regulations may be enacted that could affect any regulatory approval that we may receive for our product candidates. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or not able to maintain regulatory compliance, we may lose any marketing approval that may have been obtained and we may not achieve or sustain profitability, which would adversely affect our business. Even if we complete the necessary preclinical studies and clinical trials, the marketing approval process is expensive, time-consuming and uncertain and may prevent us or any of our existing or future collaboration partners from obtaining approvals for the commercialization of our current product candidates and any other product candidate we develop. Any current or future product candidate we may develop and the activities associated with their development and commercialization, including their design, testing, manufacture, safety, efficacy, recordkeeping, labeling, storage, approval, advertising, promotion, sale, import, export and distribution, are subject to comprehensive regulation by the FDA and other regulatory authorities in the United States and by comparable authorities in other countries. Failure to obtain marketing approval for a product candidate will prevent us from commercializing the product candidate in a given jurisdiction. We have not received approval to market any product candidates from regulatory authorities in any jurisdiction and it is possible that none of the product candidates we
may seek to develop in the future will ever obtain regulatory approval. We have no experience in filing and supporting the applications necessary to gain marketing approvals and expect to rely on third-party CROs or regulatory consultants to assist us in this process. Securing regulatory approval requires the submission of extensive preclinical and clinical data and supporting information to the various regulatory authorities for each therapeutic indication to establish the biologic product candidate’s safety purity, efficacy and potency.effectiveness. Securing regulatory approval also requires the submission of information about the product manufacturing process to, and inspection of manufacturing facilities by, the relevant regulatory authority. Any product candidates we develop may not be effective, may be only moderately effective, or may prove to have undesirable or unintended side effects, toxicities or other characteristics that may preclude our obtaining marketing approval or prevent or limit commercial use. The process of obtaining marketing approvals, both in the United States and abroad, is expensive, may take many years if additional clinical trials are required, if approval is obtained at all, and can vary substantially based upon a variety of factors, including the type, complexity, and novelty of the product candidates involved. Changes in marketing approval policies during the development period, changes in or the enactment of additional statutes or regulations, or changes in regulatory review for each submitted product application, may cause delays in the approval or rejection of an application. The FDA and comparable authorities in other countries have substantial discretion in the approval process and may refuse to accept any application or may decide that our data are insufficient for approval and require additional preclinical, clinical or other studies. In addition, varying interpretations of the data obtained from preclinical and clinical testing could delay, limit, or prevent marketing approval of a product candidate. Any marketing approval we ultimately obtain may be limited or subject to restrictions or post-approval commitments that render the approved product not commercially viable. Additionally, product approvals may be delayed as a result of the COVID-19 pandemic. As of June 23, 2020, the FDA noted it is continuing to ensure timely reviews of applications for medical products during the COVID-19 pandemic in line with its user fee performance goals; however, the FDA may not be able to continue its current pace and approval timelines could be extended, including where a pre-approval inspection or an inspection of clinical sites is required and due to the COVID-19 pandemic and travel restrictions, the FDA is unable to complete such required inspections during the review period. In 2020, several companies announced receipt of complete response letters due to the FDA's inability to complete required inspections for their applications.
If we experience delays in obtaining approval or if we fail to obtain approval of any current or future product candidates we may develop, the commercial prospects for those product candidates may be harmed, and our ability to generate revenues will be materially impaired. If the FDA or comparable foreign regulatory authorities approve generic or biosimilar versions of any of our products that receive marketing approval, or if any product approvals we obtain do not provide us withobtain the exclusivity periods for our approved products that we hope to achieve, the sales of our products could be adversely affected. As part of the ongoing efforts of governmental authorities to lower health care costs by facilitating genericIf and when approved, our product candidates may face competition to pharmaceutical products, the BPCIA enacted as part of the Health Care Reform Law, created a new abbreviated regulatory approval pathway in the United States forfrom ANDA or 505(b)(2) product candidates referencing our drug product and from biosimilars product candidates referencing our biological product. Certain ANDAs, and certain biosimilar products that are found to be “biosimilar” to or “interchangeable” with a biological “reference product” previously licensed under a BLA. This abbreviated approval pathway is intended to permit a biosimilar to come to market more quickly and less expensively by relying to some extent on the data generated by the reference product’s sponsor and the FDA’s previous review and approval of the reference product. Under the BPCIA, a biosimilar sponsor’s ability to seek or obtain approval through the abbreviated pathway is limited by periods of exclusivity granted by the FDA to the holder of the reference product’s BLA, and no biosimilar application may be accepted by the FDA for review until four years after the date the reference product was first licensed by the FDA, and no biosimilar application, once accepted, may receive final approval until 12 years after the reference product was first licensed by the FDA.
Once approved, biosimilars likely would compete with, and in some circumstances may be deemed under applicable laws to be “interchangeable with,” the previously approved reference product. The extent to which a biosimilar,with” our biological product, once approved, willmay be substituted for any one of our product candidates, if approved, in a way that is similarsubject to traditional generic substitution for non-biological products is not yet clear, and will depend on a number of marketplace and regulatory factors that are still developing. Although there is uncertainty regarding the impact of this new program, it seems likely that if any of our product candidates are approved by the FDA, there is risk that the approval of a biosimilar competitor to one of our products could have an adverse impact on our business. In particular, a biosimilar could be significantly less costly to bring to market and priced significantly lower than our product, if approved by the FDA.applicable state laws.
We may also be subject to competition from biosimilar products in Europe. To date, many biosimilar products have been authorized by the European Commission, after application at EMA for a centralized marketing authorization. As in the United States the regulatory approval pathway for biosimilar products in Europe is abbreviated. A biosimilar sponsor must however still provide all of the preclinical and clinical data required to demonstrate the similarity of their product with the reference product. The level of data
required is assessed on a case by case basis, but it will be less than that required for an original biological product. The pathway is more complex than the abridged procedure that may be followed to obtain authorization of a generic version of a non-biological product, but it would still allow the biosimilar product to be brought to market more quickly and less expensively than our original product. That said, in the EU, applications for marketing authorizations in relation to biosimilar products are subject to the same data and market exclusivity rules that apply to generic non-biologic products so no biosimilar product can be approved or placed on the market during the period such exclusivity applies to our product. Marketing authorization of a biosimilar product in the EU does not guarantee that the biosimilar product may be substituted for the reference product. Interchangeability of a biosimilar product with the reference product is not assessed by the EMA but this determination is left to each of the member states. We cannot know at this stage the extent to which any biosimilar product would be interchangeable with our reference product, and this may vary between member states. We may pursue pediatric exclusivity for one or more of our product candidates but may not succeed in obtaining it. There is also a risk that a competitor may achieve pediatric exclusivity that would delay any potential approvals of our product candidates. In the United States, the FDA has the authority to award additional exclusivity for approved products where the sponsor conducts specified testing on pediatric or adolescent populations upon the written request of the FDA. If granted, pediatric exclusivity addsmay add six months to existingcertain patents or regulatory exclusivity periods applicablefor an approved drug, and to regulatory exclusivity periods for an approved biological products under the BPCIA — namely, the four-year period during which the FDA will not consider an application for a biosimilar product, and the twelve-year period during which the FDA will not approve a biosimilar application. This six-month exclusivity runs from the end of these exclusivity protection periods.product. In the EU, as well, pediatric studies are incentivized by the reward of additional exclusivity. Pediatric Investigation Plans (“PIPs”), are determined by the Pediatric Committee of the EMA. Where an application for a marketing authorization is submitted in respect of a medicinal product designated as an orphan medicinal product and that application contains the results of the PIP studies, market exclusivity for that orphan medicinal product is extended by two years. Where an application for a marketing authorization is submitted in respect of a medicinal product that is not designated as an orphan medicinal product and that application contains the results of the PIP studies, it may be possible to obtain a 6-month extension of a supplementary protection certificate extending patent protection for a medicinal product. Orphan drug designation presents yet another regulatory incentive that may be available to us and our competitors. The FDA may grant orphan drug designation to products intended to treat a “rare disease or condition” that affects fewer than 200,000 individuals in the United States, or affects more than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making available in the United States a drug for such disease or condition will be recovered from sales in the United States of such drug. Orphan drug designation must be requested before submitting an application for marketing approval. Orphan drug designation can provide opportunities for grant funding towards clinical trial costs, tax advantages and FDA user fee exemptions. In addition, if a product that has an orphan drug designation subsequently receives FDA approval for the indication for which it has such designation, the product may be entitled to orphan drug exclusivity, which means the FDA would not approve any other application to market the same drug for a period of seven years, except in limited circumstances, such as a showing of clinical superiority to the product with orphan exclusivity. In the European Union, orphan drug status offers similar but not identical benefits as those in the United States. We may pursue orphan drug designation for one or more of our product candidates but obtaining such designation cannot be assured. Even if we obtain orphan drug exclusivity, competitors may receive approval of either a different product for the same indication or the same product for a different indication but that could be used off-label in the orphan indication. Additionally, should a competitor receive orphan drug designation for a product to treat the same disease and same indication as one of our product candidates, there is a risk that the FDA or a comparable European regulatory body could delay approving our product candidate. Developing a drug product, such as NasoShield, to address biological warfare involves special considerations, including compliance with the “Animal Rule,” that may increase drug development delays and costs, and result in a longer and more uncertain regulatory approval process.
Under a special FDA procedure available for studying certain biological warfare products, such as NasoShield, the FDA makes available a research pathway known as the “Animal Rule,” which permits the conduct of clinical trials without exposing human subjects to deadly substances, such as anthrax. These regulations authorize the FDA to rely on evidence from animal studies to provide evidence of a product’s effectiveness under circumstances where there is a reasonably well-understood mechanism for the toxicity of the agent. Under these requirements, and with the FDA’s prior agreement, biologics used to reduce or prevent the toxicity of chemical, biological, radiological or nuclear substances may be approved for use in humans based on evidence of effectiveness derived from appropriate animal studies and any additional supporting data. Products evaluated for effectiveness under this rule are evaluated for safety under preexisting requirements for establishing the safety of new drug and biological products, including Phase 1 through Phase 2 clinical trials. Under certain circumstances a single animal species may be acceptable if that animal model is sufficiently well-characterized for predicting a response in humans. The animal study endpoint must be clearly related to the desired benefit in humans and the information obtained from animal studies must allow for selection of an effective dose in humans. The Animal Rule also requires post-marketing studies, such as field studies, to verify and describe the product’s clinical benefit and assess its safety should an exigency exist that leads to the product being used in humans; the nature of these studies will be discussed with
FDA as part of the BLA process. Products approved under the Animal Rule are subject to additional requirements, such as restrictions imposed on marketing or distribution or requirements to provide information to patients.
Compliance with the Animal Rule would generally require us to utilize animal model studies for efficacy and provide certain animal and human safety data in order to obtain FDA approval for our anthrax vaccine product candidate. The Animal Rule drug development pathway typically involves costs and delays in excess of what would be expended in conducting human vaccine clinical trials not requiring compliance with the Animal Rule. Although there is an alternative regulatory pathway available for biological warfare drug candidates, called Emergency Use Authorization, which avoids the Animal Rule’s reliance on animal models focused on efficacy, there can be no assurance that this alternative model will apply to our anthrax vaccine product candidate.
Developing appropriate animal models in compliance with the Animal Rule is a time-consuming and expensive research effort. Further, we may not be able to sufficiently demonstrate the animal correlation to the satisfaction of the FDA, as these corollaries are difficult to establish and are often unclear. The FDA may decide that our data are insufficient for approval and require additional non-clinical, clinical or other studies, refuse to approve our product candidates, or place restrictions on our ability to commercialize those approved products. As a general matter, complying with the Animal Rule involves a more uncertain pathway to regulatory approval, as relatively few products have been approved in this manner. This means that it may be particularly difficult for us to predict the timing or ultimate success of receiving FDA approval for NasoShield. Further, other countries have not, at this time, established criteria for review and approval of these types of products outside their normal review process; i.e., there is no Animal Rule equivalent, and consequently there can be no assurance that we will be able to make a submission for marketing approval in foreign countries based on such animal data.
Additionally, few facilities in the United States and internationally have the capability to perform animal testing with anthrax or otherwise assist us in qualifying the requisite animal models. We compete with other biodefense companies for access to this limited pool of highly specialized resources. We therefore may not be able to secure contracts to conduct testing of our anthrax vaccine product candidate in a predictable timeframe or at all.
Our NasoShield anthrax vaccine product candidate and our NasoVAX pandemic influenza vaccine product candidate may potentially be eligible for the Strategic National Stockpile (“SNS”) under Project BioShield, but there is no guarantee that our product candidates will meet the criteria set forth by HHS or the FDA for procurement and Emergency Use Authorization.
Under the Project BioShield Act of 2004 (“Project BioShield”), the Secretary of HHS may, with the concurrence of the Secretary of DHS and upon the approval of the President, contract to purchase unapproved medical countermeasures for the SNS, in specified circumstances. The U.S. Congress is notified of a recommendation for a stockpile purchase after Presidential approval. Project BioShield specifies that a company supplying the countermeasure to the SNS is paid on delivery of a substantial portion of the countermeasure. To be eligible for purchase under these provisions, the Secretary of HHS must determine that there are sufficient and satisfactory clinical results or research data, including data, if available, from preclinical studies and clinical trials, to support a reasonable conclusion that the countermeasure will qualify for approval or licensing within eight years. The legislation also allows unlicensed products to be procured for the SNS so that they are available at the time an emergency is declared.
Project BioShield also allows the Secretary of HHS to authorize the emergency use of medical products that have not yet been approved by the FDA. To exercise this authority, the Secretary of HHS must conclude that:
the agent for which the countermeasure is designed can cause serious or life-threatening disease;
based on the totality of scientific evidence available to the Secretary, including data from adequate and well-controlled clinical trials, if available, it is reasonable to believe that the product may be effective in diagnosing, treating or preventing the disease;
the known and potential benefits of the product outweigh its known and potential risks; and
there is no adequate, approved, and available alternative to the product for diagnosing, preventing, or treating such disease or condition.
Although this provision permits the Secretary of HHS to circumvent the FDA approval process, its use would be limited to rare circumstances. Our product candidates will be eligible both for consideration for procurement into the SNS and for use in the event of an emergency, although there is no guarantee that our product candidates will meet the criteria set forth by HHS or the FDA for procurement and Emergency-use Authorization, respectively. Both our NasoShield anthrax vaccine product candidate and our NasoVAX pandemic influenza vaccine product candidate may potentially be eligible for the SNS under Project BioShield.
Risks Related to Our Intellectual Property It is difficult and costly to protect our proprietary rights, and we may not be able to ensure their protection. If our patent position and other intellectual property rights do not adequately protect our product candidates, others could compete against us (including directly), which could materially harm our business, results of operations and financial condition. We rely upon a combination of patents, patent applications, trade secret protection and confidentiality agreements to protect the intellectual property related to our product candidates, platform technology and know-how. The patent position of biotechnology companies is generally uncertain, because it involves complex legal and factual considerations. The standards applied by the USPTO and foreign patent offices in granting patents are not always applied uniformly or predictably. For example, there is no uniform worldwide policy regarding patentable subject matter or the scope of claims allowable in biotechnology patents. In addition, some countries do not grant patent claims directed to methods of treating humans, and in these countries patent protection may not be available at all to protect our product candidates. The patent applications that we own or in-license may fail to result in issued patents with claims that cover our product candidates in the United States or in other countries. The patent prosecution process is expensive and time consuming, and our current or future licensors, licensees or collaborators may not be able to prepare, file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner. Moreover, in some circumstances, we may not have the right to control the preparation, filing and prosecution of patent applications, or to maintain the patents, covering technology that we license from or license to third parties, making us reliant on our licensors, licensees or collaborators. Therefore, these patents and patent applications may not be prosecuted and enforced in a manner consistent with the best interests of the Company’s business. If our current or future licensors, licensees or collaborators fail to establish, maintain or protect such patents and other intellectual property rights, such rights may be lost or impaired. If our licensors, licensees or collaborators are not fully cooperative or disagree with the Company as to the prosecution, maintenance or enforcement of any patent rights, such patent rights could be compromised. If patent applications we hold or have in-licensed with respect to our product candidates fail to issue, if their breadth or strength of protection is threatened, or if they fail to provide meaningful exclusivity for our product candidates, it could dissuade companies from collaborating with us. We and our licensors have filed several patent applications covering aspects of our product candidates. We cannot offer any assurance about which, if any, patents will issue, the breadth of any such patents or whether any issued patents will be found invalid or unenforceable or will be successfully challenged by third parties. Patent applications in the United States and most other countries are confidential for a period of time after filing, and some remain so until issued. We cannot be certain that our licensors were the first to satisfy the requirements necessary to secure patent rights relating to any particular invention. Furthermore, if third parties have filed such patent applications, an interference proceeding in the United States can be initiated by such third party, or by the USPTO itself, to determine who was the first to invent any of the subject matter covered by the patent claims of our patent applications. Even if patents do successfully issue and even if such patents cover our product candidates, third parties may challenge their validity, enforceability or scope, which may result in such patents being narrowed or invalidated. Any successful challenge to our patents or patent applications, or to any other patents or patent applications owned by or licensed to us, could deprive us of the rights necessary to prevent competition from third parties, which may impair the commercial success of any product candidate that we may develop. There is no assurance that all potentially relevant prior art relating to our patents and patent applications or those of our licensors has been found, and prior art that we have not identified could be used by a third party to invalidate a patent or prevent a patent from issuing from a pending patent application. Furthermore, even if they are unchallenged, our patents and patent applications, or those of our licensors, may not adequately protect our technology, provide exclusivity for our product candidates, prevent others from designing around our patents with similar products, or prevent others from operating in jurisdictions in which we did not pursue patent protection. Any of these outcomes could impair our ability to prevent competition from third parties, which may have an adverse impact on our business. Any loss of, or failure to obtain, patent protection could have a material adverse impact on our business. We may be unable to prevent competitors from entering the market with a product that is similar to or the same as our products. We may not be able to protect our intellectual property rights throughout the world. Filing, prosecuting and defending patents on product candidates in all countries throughout the world would be prohibitively expensive, and our intellectual property rights in some countries outside the United States may be less extensive than those in the United States. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as federal and state laws in the United States. Consequently, we may not be able to prevent third parties from practicing our inventions in all countries outside the United States, or from selling or importing products made using our inventions in and into the United States or other jurisdictions. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop
their own products and, further, may export otherwise infringing products to territories where we have patent protection, but enforcement rights are not as strong as those in the United States. These products may compete with our product candidates and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing. Many companies have encountered significant problems in protecting and defending intellectual property rights in some foreign jurisdictions. The legal systems of certain countries do not favor the enforcement of patents and other intellectual property protection, which could make it difficult for us to stop the infringement of our patents generally. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly and our patent applications at risk of not issuing, and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license. Patent terms may be inadequate to protect our competitive position on our products for an adequate amount of time. Patents have a limited lifespan. In most countries, including the United States, the natural expiration of a patent is 20 years from the date that the application for the patent is filed. In some cases, the term of a U.S. patent is shortened by a terminal disclaimer that reduces its term to that of an earlier-expiring patent. Various extensions of patent term may be available in particular countries; however, in all circumstances the life of a patent, and the protection it affords, has a limited term. If we encounter delays in obtaining regulatory approvals, the period of time during which we could market a product under patent protection could be reduced. We expect to seek extensions of patent terms where these are available in any countries where we are prosecuting patents. Such possible extensions include those permitted under the Drug Price Competition and Patent Term Restoration Act of 1984 in the United States, which permits a patent term extension of up to five years to cover an FDA-approved product. The actual length of the extension will depend on the amount of patent term lost while the product was in clinical trials. If any of our biologic products qualify as being a “first licensure” under the Biologics Price Competition and Innovation Act (“BPCIA”) provisions of the Affordable Care Act (“ACA”), we also expect to seek regulatory exclusivity for those products from the FDA, which can grant twelve (12) years of exclusivity under the BPCIA provisions of the ACA. However, the applicable authorities, including the USPTO and FDA in the United States, and any equivalent regulatory authority in other countries, may not agree with our assessment of whether such extensions are available or whether any of our products qualify as a first licensure, and may refuse to grant extensions to our patents, or may grant more limited extensions than we request, or may not grant regulatory exclusivity. If this occurs, our competitors may be able to take advantage of our investment in development and clinical trials by referencing our clinical and preclinical data, and then may be able to launch their product earlier than might otherwise be the case. We may become involved in lawsuits to protect or enforce our intellectual property, which could be expensive, time consuming and unsuccessful. Competitors may infringe our patents or misappropriate or otherwise violate our intellectual property rights. To counter infringement or unauthorized use, litigation may be necessary to enforce or defend our intellectual property rights, to protect our trade secrets and/or to determine the validity and scope of our own intellectual property rights or the proprietary rights of others. Such litigation can be expensive and time consuming, which could divert management resources and harm our business and financial results. Many of our current and potential competitors have the ability to dedicate substantially greater resources to litigate intellectual property rights than we can. Accordingly, despite our efforts, we may not be able to prevent third parties from infringing upon or misappropriating our intellectual property. Patent assertion, including initiating litigation, increases the likelihood that the accused third party will seek to narrow or invalidate our asserted patent. The scope and validity of our asserted patent may be challenged in a variety of post-grant proceedings before the USPTO and foreign patent offices. In addition, in an infringement proceeding, a court may decide that our asserted patent is invalid or unenforceable or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation proceeding or other legal proceeding could therefore put one or more of our patents at risk of being invalidated, held unenforceable or interpreted narrowly. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. Third-party claims of intellectual property infringement or misappropriation may prevent or delay our development and commercialization efforts. Our commercial success depends in part on our ability to develop, manufacture, market and sell our product candidates, and to use our or our licensors’ proprietary technologies without infringing the patents and proprietary rights of third parties. Numerous U.S. and foreign issued patents and pending patent applications owned by third parties exist in the fields in which we are developing and may develop our product candidates. As the biotechnology and pharmaceutical industries expand and more patents are issued, the risk
increases that our product candidates may be subject to claims of infringement of the patent rights of third parties. We may not have identified all U.S. and foreign patents or published patent applications that affect our business either by blocking our ability to commercialize our product candidates or by covering similar technologies that affect our market. Third parties may assert that we are employing their proprietary technology without authorization. There may be third-party patents or patent applications with claims, for example, to materials, formulations, methods of manufacture, methods of analysis and/or methods for treatment related to the use or manufacture of our product candidates. For instance, we and certain of our executive officers have been named in a lawsuit brought by a former employee, De-Chu Christopher Tang. The lawsuit asserts a number of claims, including claims that Dr. Tang owns certain portions of our intellectual property and that we wrongfully retained Dr. Tang’s lab notebooks after the conclusion of his employment in 2012. We have filed a motion to dismiss and believe the claims are without merit. In some cases, we may have failed to identify such relevant third-party patents or patent applications. For example, patent applications filed before November 29, 2000 and certain patent applications filed after that date that will not be filed outside the United States remain confidential until issued as patents. Except for the preceding exceptions, patent applications in the United States and elsewhere are generally published only after a waiting period of approximately 18 months after the earliest filing. Therefore, patent applications covering our platform technology or our product candidates could have been filed by others without our knowledge. Additionally, pending patent applications which have been published can, subject to certain limitations, be later amended in a manner that could cover our platform technologies or product candidates and/or the use, analysis and/or manufacture of our product candidates. If any third-party patents are held by a court of competent jurisdiction to cover aspects of our materials, formulations, methods of manufacture, methods of analysis and/or methods for treatment, the holders of any such patents may be awarded monetary damages, obtain injunctive or other equitable relief, or both. An award of monetary damages may be substantial and may include treble damages and attorneys’ fees for willful infringement. An award of injunctive relief could block our ability to develop and commercialize the applicable product candidate until such patent expired or unless we obtain a license. Such licenses may not be available on acceptable terms, if at all. Even if we were able to obtain a license, the rights may be non-exclusive, which could result in our competitors gaining access to the same intellectual property. Ultimately, we could be forced to redesign an infringing product, prevented from commercializing a product, or forced to cease some aspect of our business operations, if, as a result of actual or threatened patent infringement claims, we are unable to enter into licenses on acceptable terms. Defending against claims of patent infringement or misappropriation of trade secrets could be costly and time consuming, regardless of the outcome. Thus, even if we were to ultimately prevail, or to settle at an early stage, such litigation could burden us with substantial unanticipated costs. In addition, litigation or threatened litigation could result in significant demands on the time and attention of our management team, distracting them from the pursuit of other company business. We may face a claim of misappropriation if a third party believes that we inappropriately obtained and used trade secrets of such third party. If we are found to have misappropriated a third party’s trade secrets, we may be prevented from further using such trade secrets, limiting our ability to develop our product candidates, and we may be required to pay damages. During the course of any patent or other intellectual property litigation, there could be public announcements of the results of hearings, rulings on motions, and other interim proceedings in the litigation. If securities analysts or investors regard these announcements as negative, the perceived value of our product candidates, platform technology or intellectual property could be diminished. Accordingly, the market price of our common stock may decline. In addition, the uncertainties associated with litigation could have an adverse effect on our ability to raise the funds necessary to continue our clinical trials, continue our research programs, license necessary technology from third parties or enter into development partnerships that would help us bring our product candidates to market. We may be subject to claims that our employees, independent contractors or consultants have wrongfully used or disclosed alleged trade secrets of their former employers, or our employees may challenge the inventorship of our patents. As is common in the biotechnology and pharmaceutical industry, we employ individuals who were previously employed at other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Some of these individuals, including members of our senior management, executed proprietary rights, non-disclosure and non-competition agreements, or similar agreements, in connection with such previous employment. Although we use reasonable efforts to ensure that our employees, independent contractors and consultants do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or these individuals have used or disclosed intellectual property, including trade secrets or other proprietary information, of any such third party.
We may have inventorship disputes arise from conflicting obligations of employees, consultants or others who are involved in developing our product candidates. In addition, we may be subject to claims that former employees, collaborators or other third parties have an interest in our patents or other intellectual property as an inventor or co-inventor. Litigation may be necessary to defend against these claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. Such intellectual property rights could be awarded to a third party, and we could be required to obtain a license from such third party to commercialize our technology or products. Such a license may not be available on commercially reasonable terms or at all. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management. We have in-licensed a portion of our intellectual property, and, if we fail to comply with our obligations under these arrangements, we could lose such intellectual property rights or owe damages to the licensor of such intellectual property. We are a party to a number of license agreements that are important to our business, and we may enter into additional license agreements in the future. Certain of our in-licensed intellectual property covers, or may cover, RespirVec technology, EuPort technology, and certain of our product candidates including ALT-801.pemvidutide. Our existing license agreements impose, and we expect that future license agreements will impose, various diligence, milestone payment, royalty and other of our obligations. If there is any conflict, dispute, disagreement or issue of non-performance between the Company and our licensing partners regarding our rights or obligations under the license agreements, including any such conflict, dispute or disagreement arising from our failure to satisfy payment obligations under any such agreement, we may owe damages, our licensor may have a right to terminate the affected license, and our ability to utilize the affected intellectual property in our product discovery and development efforts and our ability to enter into collaboration or marketing agreements for an affected product candidate may be adversely affected. We may need to license certain intellectual property from third parties, and such licenses may not be available on commercially reasonable terms or at all. A third party may hold intellectual property, including patent rights, that is important or necessary to the development or commercialization of our product candidates. If the patented or proprietary technology of third parties is necessary for us to commercialize our product candidates, we would be required to obtain a license from these third parties. Such a license may not be available on commercially reasonable terms or at all, which could materially harm our business. Confidentiality agreements with employees and third parties may not prevent unauthorized disclosure of proprietary information. In addition to the protection afforded by patents, we rely on confidentiality agreements to protect trade secrets and proprietary know-how that may not be patentable or that we may elect not to patent, processes for which patents are difficult to enforce and any other elements of our technology and development processes that involve proprietary know-how, information or technology that is not covered by patents. In particular, we seek to protect our proprietary technology and processes, in part, by entering into confidentiality agreements with our employees, consultants, outside scientific advisors, contractors and collaborators. These agreements require that all confidential information developed by the individual or made known to the individual by the Company during the course of the individual’s relationship with us be kept confidential and not disclosed to third parties. We also enter into agreements with our employees that provide that any inventions conceived by the individual in the course of rendering services to the Company shall be our exclusive property. However, we may not obtain these agreements in all circumstances, and individuals with whom we have these agreements may not comply with their terms. Although we use reasonable efforts to protect our know-how, our employees, consultants, contractors or outside scientific advisors might intentionally or inadvertently disclose our know-how or other proprietary information to competitors. In addition, competitors may otherwise gain access to our know-how or independently develop substantially equivalent information and techniques. Enforcing a claim that a third party illegally obtained and is using any of our know-how is expensive and time consuming, and the outcome is unpredictable. In addition, courts outside the United States sometimes are less willing than U.S. courts to protect know-how. Misappropriation or unauthorized disclosure of our know-how could impair our competitive position and may have a material adverse effect on our business. If our trademarks and trade names are not adequately protected, then we may not be able to build name recognition in our markets of interest and our business may be adversely affected. Our registered or unregistered trademarks or trade names may be challenged, infringed, circumvented, declared generic or determined to be infringing on other marks. We may not be able to protect our rights to these trademarks and trade names, which we need to build name recognition among potential partners or customers in our markets of interest. At times, competitors may adopt trade names or trademarks similar to those of the Company’s, thereby impeding our ability to build brand identity and possibly leading to market confusion. In addition, there could be potential trade name or trademark infringement claims brought by owners of other registered trademarks or trademarks that incorporate variations of our registered or unregistered trademarks or trade names. For example, we have experienced threatened or actual opposition for two trademarks that we were pursuing. We decided to discontinue
our use of one of those trademarks, and the other matter was resolved on favorable terms. Although these matters have been resolved on terms that did not materially harm the Company, we may become subject to other trademark challenges in the future. If we are unable to establish long-term name recognition based on our trademarks and trade names, then we may not be able to compete effectively, and our business may be adversely affected. Risks Related to Commercialization of the Company’s Product Candidates Our future commercial success depends upon attaining significant market acceptance of our product candidates, if approved, among physicians, patients, third-party payers and others in the medical community. Even if we obtain marketing approval for our product candidates, or any other product candidates that we may develop or acquire in the future, the product may not gain market acceptance among physicians, third-party payers, patients and others in the medical community. Market acceptance of any approved products depends on a number of other factors, including: | ● | the efficacy and safety of the product, as demonstrated in clinical trials; |
| ● | the clinical indications for which the product is approved and the label approved by regulatory authorities for use with the product, including any warnings that may be required on the label; |
| ● | acceptance by physicians and patients of the product as a safe and effective treatment and the willingness of the target patient population to try new vaccines and/or therapies and of physicians to prescribe new vaccines and/or therapies; |
| ● | the cost, safety and efficacy of treatment in relation to alternative treatments; |
| ● | the availability of adequate course and reimbursement by third-party payers and government authorities; |
| ● | relative convenience and ease of administration; |
| ● | the prevalence and severity of adverse side effects; |
| ● | the effectiveness of our sales and marketing efforts; and |
| ● | the restrictions on the use of our products together with other medications, if any. |
the efficacy and safety of the product, as demonstrated in clinical trials;
the clinical indications for which the product is approved and the label approved by regulatory authorities for use with the product, including any warnings that may be required on the label;
acceptance by physicians and patients of the product as a safe and effective treatment and the willingness of the target patient population to try new vaccines and/or therapies and of physicians to prescribe new vaccines and/or therapies;
the cost, safety and efficacy of treatment in relation to alternative treatments;
the availability of adequate course and reimbursement by third-party payers and government authorities;
relative convenience and ease of administration;
the prevalence and severity of adverse side effects;
the effectiveness of our sales and marketing efforts; and
the restrictions on the use of our products together with other medications, if any.
Market acceptance is critical to our ability to generate significant revenue. Any product candidate, if approved and commercialized, may be accepted in only limited capacities or not at all. If any approved products are not accepted by the market to the extent that we expect, we may not be able to generate significant revenue and our business would suffer. We rely on, and expect to continue to rely on, third parties to manufacture our product candidates and related materials for our clinical trials and preclinical studies, and these third parties may not perform satisfactorily. We do not have any manufacturing facilities or personnel, and we rely on, and expect to continue to rely on, third-party manufacturers and suppliers to manufacture and supply vaccines, drug substance and drug product for our preclinical studies and clinical trials, and on related materials, such as anthrax, influenzaHBV products and HBV products.pemvidutide. We rely on a small number of third-party manufacturers and suppliers to manufacture and supply bulk drug substance and drug product and fill finished vaccines for our initial clinical trials. This reliance on a small number of third parties increases the risk that we will not have sufficient quantities of our product candidates or other products needed for our preclinical studies and clinical trials, or that such quantities will be manufactured for us at an acceptable cost or quality, which could delay, prevent or impair our development or commercialization efforts. Any of these third parties that we rely upon may terminate their engagement with us at any time. If we need to enter into alternative arrangements, it could delay our product development activities. In addition, our reliance on these third parties for manufacturing activities will reduce our control over these activities but will not relieve us of our responsibility to ensure compliance with all required regulations regarding manufacturing. Reliance on third-party manufacturers entails risks to which we would not be subject if we manufactured the product candidates itself, including: | ● | the inability to negotiate manufacturing agreements with third parties under commercially reasonable terms; |
reduced control as a result of using third-party manufacturers for all aspects of manufacturing activities, including regulatory compliance and quality assurance. We do not have control over third party manufacturers’ compliance with these regulations and standards, but we may ultimately be responsible for any of their failures;
| •
● | reduced control as a result of using third-party manufacturers for all aspects of manufacturing activities, including regulatory compliance and quality assurance. We do not have control over third party manufacturers’ compliance with these regulations and standards, but we may ultimately be responsible for any of their failures; |
| ● | delays as a result of manufacturing problems or re-prioritization of projects at a third-party manufacturer; |
| ● | our third-party manufacturers might be unable to formulate and manufacture our product candidates in the volume and of the quality required to meet our clinical and commercial needs, if any; |
| ● | termination or non-renewal of manufacturing agreements with third parties in a manner or at a time that is costly or damaging to us; |
| ● | the possible misappropriation of our proprietary information, including our trade secrets and know-how or infringement of third-party intellectual property rights by our contract manufacturers; and |
| ● | disruptions to the operations of our third-party manufacturers or suppliers caused by conditions unrelated to our business or operations, including the bankruptcy of the manufacturer or supplier. |
termination or non-renewal of manufacturing agreements with third parties in a manner or at a time that is costly or damaging to us;
the possible misappropriation of our proprietary information, including our trade secrets and know-how or infringement of third-party intellectual property rights by our contract manufacturers; and
disruptions to the operations of our third-party manufacturers or suppliers caused by conditions unrelated to our business or operations, including the bankruptcy of the manufacturer or supplier.
Any of these events could lead to preclinical study and clinical trial delays or failure to obtain regulatory approval or affect our ability to successfully commercialize future products. Some of these events could be the basis for FDA or other regulatory authority action, including clinical holds, fines, injunctions, civil penalties, license revocations, recall, seizure, total or partial suspension of production, or criminal penalties. In addition, our product candidates involve technically complex manufacturing processes, and even slight deviations at any point in the production process may lead to production failures and may cause the production of our product candidates to be disrupted, potentially for extended periods of time. For example, one of our third-party manufacturers failed on multiple occasions to successfully manufacture sufficient quantities of our NasoVAX product candidate. This failure required us to delay our planned multi-valent clinical trials. In some cases, the technical skills required to manufacture our products or product candidates may be unique or proprietary to the original contract manufacturer and we may have difficulty, or there may be contractual restrictions prohibiting us from, transferring such skills to a back-up or alternate supplier, or we may be unable to transfer such skills at all. Furthermore, a contract manufacturer may possess technology related to the manufacture of our product candidate that such contract manufacturer owns independently. This would increase our reliance on such contract manufacturer or require us to obtain a license from such contract manufacturer in order to have another contract manufacturer manufacture our product candidates. Third-party manufacturers may not be able to comply with applicable cGMP, regulations or similar regulatory requirements outside the United States. Our failure, or the failure of our third-party manufacturers, to comply with applicable regulations could result in sanctions being imposed, including clinical holds, fines, injunctions, civil penalties, delays, license revocation, seizures or recalls of product candidates or products, operating restrictions and criminal prosecutions, any of which could significantly and adversely affect supplies of our product candidates. In addition, if we are required to change contract manufacturers for any reason, we will be required to verify that the new contract manufacturer maintains facilities and procedures that comply with quality standards and with all applicable regulations. We will also need to verify, such as through a manufacturing comparability study, that any new manufacturing process will produce our product candidate according to the specifications previously submitted to the FDA or another regulatory authority. The delays associated with the verification of a new contract manufacturer could negatively affect our ability to develop product candidates or commercialize our products in a timely manner or within budget. In addition, changes in manufacturers often involve changes in manufacturing procedures and processes, which could require that we conduct bridging studies between our prior clinical supply used in our clinical trials and that of any new manufacturer. We may be unsuccessful in demonstrating the comparability of clinical supplies which could require the conduct of additional clinical trials. Our third-party manufacturers may be subject to damage or interruption from, among other things, fire, natural or man-made disaster, power loss, telecommunications failure, unauthorized entry, computer viruses, denial-of-service attacks, acts of terrorism, human error, vandalism or sabotage, financial insolvency, bankruptcy and similar events. For example, in December 2019, a novel strain Since March 2020, when foreign and domestic inspections byof facilities were largely placed on hold due to the ongoing COVID-19 pandemic, the FDA have largelyhas been on hold with FDA announcing plans in July 2020working to resume prioritized domesticpre-pandemic levels of inspection activities, including routine surveillance, bioresearch monitoring and pre-approval inspections. FDA has stated that foreign inspections not deemed mission-critical remain temporarily postponed, while those that are deemed “mission-critical” will be considered for inspection on a case-by-case basis.\ Should FDA determine that an inspection is necessary for approval of a marketing application and an inspection cannot be completed during the review cycle due to restrictions on travel, FDA has stated that it generally intends to issue a complete response letter. Further, if there is inadequate information to make a determination on the acceptability of a facility, FDA may defer action on the application until an inspection can be completed. In 2020,Throughout the duration of the public health emergency, several companies announced receipt of complete response letters due to the
FDA's FDA’s inability to complete required inspections for their applications. Regulatory authorities outside the U.S. may adopt similar restrictions or other policy measures in response to the COVID-19COVID 19 pandemic and may experience delays in their regulatory activities.
The extent to which the novel coronavirusongoing COVID-19 pandemic may impact our results will depend on future developments, which are highly uncertain and cannot be predicted, including new information which may emerge concerning the severity of COVID-19, the emergence of new variants, and the actions to contain COVID-19 or treat its impact, among others. Our product candidates and any products that we may develop may compete with other product candidates and products for access to manufacturing facilities. There are a limited number of manufacturers that operate under cGMP regulations and that might be capable of manufacturing for us. Any performance failure on the part of our existing or future manufacturers could delay clinical development or marketing approval. We have limited arrangements in place for redundant supply or a second source for bulk drug substance. If our current contract manufacturers cannot perform as agreed, we may be required to replace such manufacturers, and it may prove very difficult and time consuming to identify potential alternative manufacturers who could manufacture our product candidates. Accordingly, we may incur added costs and delays in identifying and qualifying any such replacement. Our current and anticipated future dependence upon others for the manufacture of our product candidates or products may adversely affect our future profit margins and our ability to commercialize any products that receive marketing approval on a timely and competitive basis. If we are unable to manufacture our products in sufficient quantities, or at sufficient yields, or are unable to obtain regulatory approvals for a manufacturing facility for our products, we may experience delays in product development, clinical trials, regulatory approval and commercial distribution. Completion of our clinical trials and commercialization of our product candidates require access to, or development of, facilities to manufacture our product candidates at sufficient yields and at commercial scale, and this manufacturing involves a complicated process with which we have limited experience. Even if clinical trials are successful, we still may be unable to commercialize a product due to difficulties in obtaining regulatory approval for our engineering processes or problems in scaling that process to commercial production. We have no experience manufacturing, or managing third parties in manufacturing, any of our product candidates in the volumes that will be necessary to support large-scale clinical trials or commercial sales. Efforts to establish these capabilities may not meet initial expectations as to scheduling, scale-up, reproducibility, yield, purity, cost, potency or quality. We expect to rely on third parties for the manufacture of clinical and, if approved for marketing, commercial quantities of our product candidates. These third-party manufacturers must also receive FDA or other applicable governmental authority approval before they can produce clinical material or commercial products. Our products may be in competition with other products for access to these facilities and may be subject to delays in manufacture if third parties give other products greater priority. We may not be able to enter into any necessary third-party manufacturing arrangements on acceptable terms, or on a timely basis. In addition, we may have to enter into technical transfer agreements and share our know-how with the third-party manufacturers, which can be time consuming and may result in delays. No known manufacturer has received FDA clearance to manufacture large scale quantities of commercial products with the modified version of adenovirus used in the production of product candidates based on our proprietary RespirVec technology. We or our contract manufacturers therefore will need to develop a scalable manufacturing process for any product candidates that we may develop and commercialize that use our RespirVec technology. Our contract manufacturing organizations may encounter technical or scientific issues related to development or manufacturing that we may be unable to resolve in a timely manner or with available funds. If we or our manufacturing partners are unable to scale the manufacturing process to produce commercial quantities of our product candidates, or our manufacturing partners do not pass required regulatory pre-approval inspections, our commercialization efforts may be adversely affected.
Our reliance on contract manufacturers may adversely affect our operations or result in unforeseen delays or other problems beyond our control. Because of contractual restraints and the limited number of third-party manufacturers with the expertise, required regulatory approvals and facilities to manufacture our products on a commercial scale, replacement of a manufacturer may be expensive and time consuming and may cause interruptions in the production of our product candidates. A third-party manufacturer may also encounter difficulties in production. These problems may include: difficulties with production costs, scale-up and yields;
unavailability of raw materials and supplies;
insufficient quality control and assurance;
shortages of qualified personnel;
| •
● | difficulties with production costs, scale-up and yields; |
| ● | unavailability of raw materials and supplies; |
| ● | insufficient quality control and assurance; |
| ● | shortages of qualified personnel; |
| ● | failure to comply with strictly enforced federal, state and foreign regulations that vary in each country where product might be sold; and |
| ● | lack of capital funding. |
Any delay or interruption in the manufacture of our products could have a material adverse effect on our business, financial condition, results of operations and cash flows. If we are unable to establish sales, marketing and distribution capabilities, we may not be successful in commercializing our product candidates if they are approved. We do not have a sales or marketing infrastructure and have no experience in the sale, marketing or distribution of pharmaceutical products. To achieve commercial success for any product for which we obtain marketing approval, and for which we decide to independently commercialize, we will need to establish a sales and marketing organization. In the future, we may build a focused sales and marketing infrastructure to market or co-promote some of our product candidates in the United States and in Europe, if and when they are approved. There are risks involved with our establishing our own sales, marketing and distribution capabilities. For example, recruiting and training a sales force is expensive and time consuming and could delay any product launch. If the commercial launch of a product candidate for which we recruit a sales force and establish marketing capabilities is delayed or does not occur for any reason, we would have prematurely or unnecessarily incurred these commercialization expenses. This may be costly, and our investment would be lost if we cannot retain or reposition our sales and marketing personnel. Factors that may inhibit our efforts to commercialize our products on our own include: | ● | our inability to recruit, train and retain adequate numbers of effective sales and marketing personnel; |
| ● | the inability of sales personnel to obtain access to physicians; |
| ● | the lack of adequate numbers of physicians to prescribe any future products; |
| ● | the lack of complementary products to be offered by sales personnel, which may put us at a competitive disadvantage relative to companies with more extensive product lines; and |
| ● | unforeseen costs and expenses associated with creating an independent sales and marketing organization. |
our inability to recruit, train and retain adequate numbers of effective sales and marketing personnel;
the inability of sales personnel to obtain access to physicians;
the lack of adequate numbers of physicians to prescribe any future products;
the lack of complementary products to be offered by sales personnel, which may put us at a competitive disadvantage relative to companies with more extensive product lines; and
unforeseen costs and expenses associated with creating an independent sales and marketing organization.
If we do not establish our own sales, marketing and distribution capabilities and instead enter into arrangements with third parties to perform these services, our product revenues and our profitability, if any, could be lower than if we were to market, sell and distribute any products that we develop ourselves. In addition, we may not be successful in entering into arrangements with third parties to sell, market and distribute our product candidates or may be unable to do so on terms that are favorable to us. We likely will have little control over such third parties, and any of them may fail to devote the necessary resources and attention to sell and market our products effectively. If we do not establish sales, marketing and distribution capabilities successfully, either on our own or in collaboration with third parties, we will not be successful in commercializing our product candidates. We may encounter difficulties in managing our growth and expanding our operations successfully. As we seek to advance our product candidates through clinical trials and commercialization, we will need to expand our development, regulatory, manufacturing, marketing and sales capabilities or contract with third parties to provide these capabilities for the Company. As our operations expand, we expect that we will need to manage additional relationships with various strategic partners, suppliers and other third parties. Future growth will impose significant added responsibilities on members of management. Our future financial performance and our ability to commercialize our product candidates and to compete effectively will depend, in part, on our ability to manage any future growth effectively. To that end, we must be able to manage our development efforts and clinical trials effectively and hire, train and integrate additional management, administrative and, if necessary, sales and marketing personnel. We may not be able to accomplish these tasks, and our failure to accomplish any of them could prevent us from successfully growing our business. We may not be successful in establishing and maintaining strategic partnerships, which could adversely affect our ability to develop and commercialize products. A key part of our strategy is to seek strategic partnerships in the future, including potentially with major biotechnology or pharmaceutical companies for late-stage development and commercialization of our product candidates. We face significant competition in seeking appropriate partners for our product candidates, and the negotiation process is time consuming and complex. In order for the Company to successfully partner our product candidates, potential partners must view these product candidates as
economically valuable in markets they determine to be attractive in light of the terms that we are seeking and other products available for licensing from other companies. Even if we are successful in our efforts to establish strategic partnerships, the terms that we agree upon may not be favorable to the Company, and we may not be able to maintain such strategic partnerships if, for example, development or approval of a product is delayed or sales of an approved product are disappointing. Any delay in entering into strategic partnership agreements related to our product candidates could delay the development and commercialization of our product candidates and reduce their competitiveness even if they reach the market. In addition, any future partnerships we may enter into pose a number of risks, including that our partners may breach their agreements with the Company, and we may not be able to adequately protect our rights under these agreements. Furthermore, prospective partners will likely negotiate for certain rights to control decisions regarding the development and commercialization of our product candidates, if approved, and may not conduct those activities in the same manner as we would. If we fail to establish and maintain strategic partnerships related to our product candidates, we will bear all of the risk and costs related to the development of any such product candidate, and we may need to seek additional financing, hire additional employees and otherwise develop expertise which we do not have and for which we have not budgeted. This could negatively affect the development of any unpartnered product candidate. We may acquire other businesses, form joint ventures or make investments in other companies or technologies that could negatively affect our operating results, dilute our stockholders’ ownership, increase our debt or cause us to incur significant expense. As part of our business strategy, we may pursue acquisitions of assets or licenses of assets, including preclinical, clinical or commercial stage products or product candidates, businesses, strategic alliances, joint ventures and collaborations, to expand our existing technologies and operations. In the future, we may not be able to find suitable partners or acquisition candidates, and we may not be able to complete such transactions on favorable terms, if at all. If we make any acquisitions, we may not be able to integrate these acquisitions successfully into our existing business, and we could assume unknown or contingent liabilities. Any future acquisitions also could result in the incurrence of debt, contingent liabilities or future write-offs of intangible assets or goodwill, any of which could have a negative impact on our cash flows, financial condition and results of operations. Integration of an acquired company also may disrupt ongoing operations and require management resources that we would otherwise focus on developing our existing business. We may experience losses related to investments in other companies, which could harm our financial condition and results of operations. We may not identify or complete these transactions in a timely manner, on a cost-effective basis, or at all, and we may not realize the anticipated benefits of any acquisition, license, strategic alliance or joint venture. To finance such a transaction, we may choose to issue shares of our common stock as consideration, which would dilute the ownership of our stockholders. If the price of our common stock is low or volatile, we may not be able to acquire other companies or fund a joint venture project using our stock as consideration. Alternatively, it may be necessary for us to raise additional funds for these activities through public or private financings or through the issuance of debt. Additional funds may not be available on terms that are favorable to the Company, or at all, and any debt financing may involve covenants limiting or restricting our ability to take certain actions. If product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to limit commercialization of our product candidates. We face an inherent risk of product liability as a result of the clinical testing of our product candidates and will face an even greater risk if we commercialize any products. For example, we may be sued if any product we develop allegedly causes injury or is found to be otherwise unsuitable during product testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability and a breach of warranties. Claims could also be asserted under state consumer protection acts. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our product candidates. We believe our anthrax countermeasures are covered under the general immunity provisions of the U.S. Public Readiness and Emergency Preparedness Act, or Public Readiness Act, but this cannot be assured. Also, there can be no assurance that the Secretary of the HHS will make other declarations in the future that cover any of our other product candidates or that the U.S. Congress will not act in the future to reduce coverage under the Public Readiness Act or to repeal it altogether. Additionally, we are considering applying for liability protection under the U.S. Support Anti-terrorism by Fostering Effective Technologies (SAFETY) Act of 2002 (the “SAFETY Act”) which may limit the claims and damages potentially faced by companies who provide certain “qualified” anti-terrorism products. However, we cannot be certain that we will be able to obtain or maintain coverage under the SAFETY Act.
Even a successful defense would require significant financial and management resources. Regardless of the merits or eventual outcome, liability claims may result in: | ● | decreased demand for any product candidates or products that we may develop; |
| ● | injury to our reputation and significant negative media attention; |
| ● | withdrawal of clinical trial participants; |
| ● | significant costs to defend the related litigations; |
| ● | a diversion of management’s time and the Company’s resources; |
| ● | substantial monetary awards to trial participants or patients; |
| ● | product recalls, withdrawals or labeling, marketing or promotional restrictions; |
| ● | the inability to commercialize any product candidates that we may develop; and |
| ● | a decline in our stock price. |
decreased demand for any product candidates or products that we may develop;
injury to our reputation and significant negative media attention;
withdrawal of clinical trial participants;
significant costs to defend the related litigations;
a diversion of management’s time and the Company’s resources;
substantial monetary awards to trial participants or patients;
product recalls, withdrawals or labeling, marketing or promotional restrictions;
the inability to commercialize any product candidates that we may develop; and
a decline in our stock price.
Failure to obtain and retain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the commercialization of products we develop. We currently carry liability insurance covering residual liability related to previously completed clinical trials in the amount of $5.0 million in the U.S.,worldwide product liability insurance covering our clinical trials in the United Kingdom in the amount of £5.0$10.0 million in the aggregate, and clinical trial liability insurance covering our clinical trials in various countries.aggregate. Although we maintain product liability insurance, any claim that may be brought against us could result in a court judgment or settlement in an amount that is not covered, in whole or in part, by our insurance or that is in excess of the limits of our insurance coverage. Our insurance policies also have various exclusions, and we may be subject to a product liability claim for which we have no coverage. We will have to pay any amounts awarded by a court or negotiated in a settlement that exceed our coverage limitations or that are not covered by our insurance, and we may not have, or be able to obtain, sufficient capital to pay such amounts. A breakdown in our information technology systems could result in a significant disruption to our business. Our operations and those of our business partners, such as CROs and others that manage sensitive data, are highly dependent on information technology systems, including Internet-based systems, which may be vulnerable to breakdown, wrongful intrusions, data breaches and malicious attack. Information security risks have generally increased in recent years. Our systems, and those of our third-party providers, are potentially vulnerable to data security breaches or cyberattack, whether by employees or others, which may expose sensitive data to unauthorized persons. A data security breach could lead to the loss of trade secrets or other intellectual property, the value of which may be contingent upon maintaining our confidentiality, or could lead to the public exposure of personal information (including sensitive personal medical information) of clinical trial participants, our employees and others, or adversely impact the conduct of scientific research and clinical trials, including the submission of research results to support marketing authorizations. This could require us to expend significant efforts and resources or incur significant expense to eliminate these problems and address related security concerns. In addition, procedures and safeguards must continually evolve to meet new data security challenges, and enhancing protections, and conducting investigations and remediation, may impose additional costs on the Company. If we were to suffer a breakdown in our systems, storage, distribution or tracing, we could experience significant disruptions affecting our business, reputational harm or claims against us by private parties and/or governmental agencies. In addition, the European Parliament and the Council of the European Union have adopted a new pan-European General Data Protection Regulation (“GDPR”), effective May 25, 2018, which increases privacy rights for individuals in Europe, extends the scope of responsibilities for data controllers and data processors and imposes increased requirements and potential penalties on companies, offering goods or services to individuals who are located in Europe or monitoring the behavior of such individuals (including by companies based outside of Europe). Noncompliance can result in penalties of up to the greater of EUR 20 million, or 4% of global company revenues. While we expect to have substantially compliant programs and controls in place to comply with the GDPR requirements, our compliance with the new regulation is likely to impose additional costs on us and we cannot predict whether the interpretations of the requirements, or changes in our practices in response to new requirements or interpretations of the requirements could have a material adverse effect on our business.
Risks Related to our BARDA Contract and Other Government Programs
Without the BARDA anthrax contract award, we would only be able to move forward with the NasoShield program at our own risk and without BARDA reimbursement and may therefore suspend or terminate it.
In recent financial periods, a significant portion of our revenues have been derived from our BARDA contract and other government contracts. For the years ended December 31, 2020 and 2019, BARDA funding for the development of NasoShield accounted for approximately 46% and 92% of our total consolidated revenue and grants and contracts, respectively. There are significant uncertainties and risks associated with our BARDA contract for our NasoShield anthrax vaccine program. Although our current BARDA contract that may fund our NasoShield anthrax vaccine program until 2021, including a $3.7 million award during the third quarter of 2019, the majority of the funds will be received during the three year period beginning in 2021 and are dependent on achieving positive clinical results during the initial two-year period to demonstrate interim safety and immune response to the vaccine in the Phase 1 clinical study. The results of the Phase 1 study obtained during 2018 met the endpoints for safety, however an appreciable immune response was not observed. We completed an investigation into the results, and the data from this study demonstrated that a simple modification to the method of intranasal dose administration had a dramatic impact on the resulting immunogenicity. These results suggest that the 2018 Phase 1 study of NasoShield in healthy adults might have shown a more robust immunogenic effect had a modified administration method been employed. The $3.7 million award is to perform a Phase 1b trial employing the modified administration method. BARDA will decide in its sole discretion whether to pursue any of the options under the contract and there can be no assurance that BARDA will elect to pursue any of the designated options. If BARDA does not pursue any of the options, BARDA could terminate the program, and we would not receive any further funds thereunder.
Our BARDA contracts are cost-plus-fixed-fee contracts that only reimburse certain specified activities.
Our BARDA contracts are cost-plus-fixed-fee contracts that only reimburse certain specified activities related to our anthrax vaccine program that have been previously authorized by BARDA. There is no guarantee that additional activities will not be needed and, if so, that BARDA will reimburse the Company for these activities. There are also significant requirements associated with operating as a federal government contractor, which include having appropriate accounting, and project tracking systems implemented and operational, and we may not be able to consistently meet these requirements. Performance under the BARDA contracts requires that we comply with appropriate regulations and operational mandates, which require us to engage internal and external expertise for compliance. Our ability to be regularly and fully reimbursed for our activities depends and will depend on our ability to comply and demonstrate compliance with such requirements. In the past, we have experienced delays in reimbursements under a BARDA contract on account of compliance issues, which we have had to dedicate substantial time and resources to remedy, including through modifications to our statement of work related to the program. In addition, under certain circumstances, BARDA may advise us to delay certain activities and invest additional time and resources before proceeding. If we follow such BARDA advice, overall program delays and costs associated with additional resources for which we have not planned may result. The costs associated with following such advice may or may not be reimbursed by BARDA under the contract. We may decide not to follow the advice provided by BARDA and instead pursue activities that we believe are in the best interest of our anthrax vaccine program and our business as a whole, even if BARDA would not reimburse us under our contract.
Most of our immediately foreseeable future revenues are contingent upon grants, contracts and loans from the U.S. and other governments, non-profit entities and academic institutions, and we may not achieve sufficient revenues from these sources either to maintain operations or eventually attain profitability.
Substantially all of our revenues to date have been derived from U.S. and European government grants, contracts and loans (such as our current BARDA contract), and from time to time, we may apply for additional contracts, grants or loans from government agencies, non-profit entities and academic institutions. Such contracts, grants or loans can be highly attractive, because they provide additional capital to fund the ongoing development of our technologies and product candidates without diluting our stockholders. However, there is often significant competition for these contracts, grants and loans, and the process of obtaining government and other contracts, grants and loans is lengthy and uncertain. Entities offering contracts, grants or loans may have requirements to apply for or to otherwise be eligible to receive certain contracts, grants or loans that our competitors may be able to satisfy that we cannot. In addition, such entities may make arbitrary decisions as to whether to offer contracts or make grants or loans, to whom the contracts, grants or loans will be awarded and the size of the contracts, grants or loans to each awardee. Even if we are able to satisfy the award requirements, there is no guarantee that we will be a successful awardee. Therefore, we may not be able to win any contracts, grants or loans in a timely manner, if at all, and there can be no assurance that existing government or other contracts, grants or loans will be renewed or that we can enter into new contracts or receive new grants or loans.
With respect to the BARDA funding we receive for our anthrax vaccine product candidate, if the U.S. government makes significant contract awards to our competitors, rather than to us, our business will be harmed and it is unlikely that we would ultimately be able to supply that particular treatment or product either in the United States or to foreign governments or other third parties. Further, changes in government budgets and agendas, funding strategies, cost overruns in our programs, or advances by our competitors, may result in changes in the timing of funding for, a decreased and de-prioritized emphasis on, or termination of, government contracts that support the development and/or procurement of the biodefense product we are developing. For example, the outbreak of Ebola in 2014 changed the near-term focus and priorities of BARDA to help ensure sufficient progress was being made on
a solution for that disease. This resulted in a delay of funding to some non-Ebola programs until Congress appropriated additional funds to BARDA specific for this purpose.
U.S. government funding is also subject to Congressional appropriations generally made on an annual basis even for multi-year contracts. More generally, due to the ongoing economic and political uncertainty, the U.S. government may reduce or delay spending in the biodefense field or eliminate funding of certain programs altogether, which could decrease the likelihood of future government contract awards or that the government would procure products from the Company. Future funding levels for BARDA for the advanced development and procurement of medical countermeasures are uncertain and may be subject to budget cuts and/or government shutdowns as the U.S. Congress and the President look to reduce the U.S. budget deficit. Potential reductions in funding could severely limit our ability to maintain, renew or enter into new contracts and therefore materially and adversely impact our business. A government shutdown could result in a suspension or delayed funding, which may materially and adversely affect our ability to continue our anthrax program.
Further, the 21st Century Cures Act (“Cures Act”), was signed into law on December 13, 2016 and, among other things, includes a provision requiring timely and accurate recommended utilization guidelines for MCMs, including for products in the Strategic National Stockpile. The Cures Act requires HHS to report to the appropriate committees of Congress when funding in the Special Reserve Fund (“SRF”), available for the procurement of MCMs falls below $1.5 billion and how the amount of funding will impact identified MCM priorities. The Cures Act ensures coordinated and efficient processes for executing MCM development and procurement programs by clarifying that the Director of BARDA carry out the programs funded by the SRF, as well as the procurement contracts, grants, and cooperative agreements under BARDA.
U.S. government agencies have special contracting requirements that give them the ability to unilaterally control contracts such as our BARDA contract.
U.S. government contracts, such as our BARDA contract, typically contain unilateral termination provisions for the government and are subject to audit and modification by the government at its sole discretion, which will subject the Company to additional risks during the term of such contracts. These risks include the ability of the U.S. government unilaterally to:
suspend or prevent the Company for a set period of time from receiving new U.S. government contracts or extending existing contracts based on violations or suspected violations of laws or regulations;
terminate our existing U.S. government contracts, including for poor performance or if funds become unavailable or are not provided to the applicable governmental agency;
reduce the scope and value of our U.S. government contracts and/or revise the timing for work to be performed;
audit and object to our contract-related costs and fees, including allocated indirect costs;
control and potentially prohibit the export of our products developed under the contract;
claim rights to products, including intellectual property, developed under the contract;
change certain terms and conditions in our U.S. government contracts; and
cancel outstanding Request for Proposal solicitations or Broad Agency Announcements.
The U.S. government will be able to terminate any of its contracts with the Company, including our BARDA contract, either for its convenience or if we default by failing to perform in accordance with the contract schedule and terms. Termination-for-convenience provisions generally enable us to recover only our costs incurred or committed, settlement expenses, and profit on the work completed prior to termination. Termination-for-default provisions do not permit these recoveries and would make us liable for excess costs incurred by the U.S. government in procuring undelivered items from another source.
The U.S. government’s determination to award any contracts may be challenged by an interested party, such as another bidder, at the U.S. Government Accountability Office (the “GAO”) or in federal court. If such a challenge is successful, a contract award may be re-evaluated and terminated.
The laws and regulations governing the procurement of goods and services by the U.S. government provide procedures by which other bidders and other interested parties may challenge the award of a government contract. Such challenges or protests could be filed with respect to any U.S. government contract awarded to the Company, including our BARDA contract, even if there are not any valid legal grounds on which to base the protest. If any such protests are filed, the government agency may decide, and in certain circumstances will be statutorily required, to suspend our performance under the contract while such protests are being considered by the GAO or the applicable federal court, thus potentially delaying delivery of goods and services and payment. In addition, we could be forced to expend considerable funds to defend any potential award. If a protest is successful, the government may be ordered to
terminate our contract and re-evaluate bids. The government could even be directed to award a potential contract to one of the other bidders.
Our business is subject to audit by the U.S. government, and may be subject to audit by foreign governments. A negative audit could adversely affect our business.
Our business is subject to audit by the U.S. government in part because of the funding we receive for our anthrax vaccine program under our BARDA contract. U.S. government agencies such as the Defense Contract Audit Agency (the “DCAA”) routinely audit and investigate government contractors. These agencies review a contractor’s performance under its contracts, cost structure and compliance with applicable laws, regulations and standards.
The DCAA also reviews the adequacy of, and a contractor’s compliance with, its internal control systems and policies, including the contractor’s purchasing, property, estimating, compensation and management information systems. Any costs found to be improperly allocated to a specific contract will not be reimbursed, while such costs already reimbursed must be refunded. If an audit uncovers improper or illegal activities, it may be subject to civil and criminal penalties and administrative sanctions, including termination of contracts, forfeiture of profits, suspension of payments, fines and suspension or prohibition from conducting business with the U.S. government. In addition, a contractor could suffer serious reputational harm if allegations of impropriety were made against it.
In the future, we may also be subject to audits by foreign governments, as we from time to time receive funding from non-U.S. government sources.
Laws and regulations affecting government contracts make it more costly and difficult for us to successfully conduct our business.
Our business plan includes the continued development of our anthrax vaccine candidate, NasoShield, pursuant to our BARDA contract in addition to applying for additional contracts, grants or loans from government agencies, non-profit entities and academic institutions. We must comply with numerous laws and regulations relating to the formation, administration and performance of government contracts, which can make it more difficult for us to retain our rights under these contracts. These laws and regulations affect how we conduct business with government agencies. Among the most significant government contracting regulations that affect our business are:
the Federal Acquisition Regulation (“FAR”) and agency-specific regulations supplemental to the FAR, which comprehensively regulate the procurement, formation, administration and performance of government contracts;
the business ethics and public integrity obligations, which govern conflicts of interest and the hiring of former government employees, restrict the granting of gratuities and funding of lobbying activities and incorporate other requirements such as the Anti-Kickback Act, the Procurement Integrity Act, the FCA and Foreign Corrupt Practices Act (“FCPA”);
export and import control laws and regulations; and
laws, regulations and executive orders restricting the use and dissemination of information classified for national security purposes and the exportation of certain products and technical data.
Foreign governments typically also have laws and regulations governing contracts with their respective agencies. These foreign laws and regulations affect how we and our customers conduct business and, in some instances, impose added costs on our business. Any changes in applicable laws and regulations could restrict our ability to maintain our existing contracts and obtain new contracts, which could limit our ability to conduct our business and materially and adversely affect our revenues and results of operations.
Risks Related to Reimbursement and Government Regulation Coverage and reimbursement may be limited or unavailable in certain market segments for our product candidates, if they are approved, which could make it difficult for us to sell our products profitably. Market acceptance and sales of any approved products will depend significantly on the availability of adequate coverage and reimbursement from third-party payers and may be affected by existing and future health care reform measures. Third-party payers, such as government health care programs, and private health insurers and health plans, decide which drugs they will provide coverage for and establish reimbursement levels. Coverage and reimbursement decisions by a third-party payer may depend upon a number of factors, including the third-party payer’s determination that use of a product is: a covered benefit under its health plan;
safe, effective and medically necessary;
| •
● | a covered benefit under its health plan; |
| ● | safe, effective and medically necessary; |
| ● | appropriate for the specific patient; |
| ● | neither experimental nor investigational. |
neither experimental nor investigational.
Third-party payers, whether foreign or domestic, or governmental or commercial, are developing increasingly sophisticated methods of controlling health care costs. Coverage and reimbursement can vary significantly from payer to payer. As a result, obtaining coverage and reimbursement approval for any approved product from each government and other third-party payer may require us to provide supporting scientific, clinical and cost-effectiveness data for the use of such products to each payer separately, with no assurance that we will be able to provide data sufficient to gain acceptance with respect to coverage and reimbursement. We cannot be sure that coverage or adequate reimbursement will be available for any of our product candidates, and we cannot be sure that coverage determinations or reimbursement amounts will not reduce the demand for, or the price of, our products. If reimbursement is not available or is available only to limited levels, we may not be able to commercialize certain of our products, even if they are approved by the FDA or other regulatory authorities. In addition, in the United States third-party payers are increasingly attempting to contain health care costs by limiting both coverage and the level of reimbursement of new drugs. As a result, significant uncertainty exists as to whether and how much third-party payers will reimburse patients for their use of newly approved drugs, which in turn will put pressure on the pricing of drugs. Price controls may be imposed, which may adversely affect our future profitability. In international markets, reimbursement and health care payment systems vary significantly by country, and many countries have instituted price ceilings on specific products and therapies. In some countries, particularly member states of the European Union, the pricing of prescription pharmaceuticals is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take considerable time after receipt of marketing approval for a product. In addition, there can be considerable pressure by governments and other stakeholders on coverage, prices and reimbursement levels, including as part of cost containment measures. Political, economic and regulatory developments may further complicate pricing negotiations, and pricing negotiations may continue after coverage and reimbursement has been obtained. Reference pricing used by various European Union member states and parallel distribution, or arbitrage between low-priced and high-priced member states, can further reduce revenues. In some countries, additional clinical research may be required to enable comparison of the cost-effectiveness of our product candidates, if they are approved, to other available vaccinesproducts in order to obtain or maintain coverage, reimbursement or pricing approval. Publication of discounts by third-party payers or authorities may lead to further pressure on the prices or reimbursement levels within the country of publication and other countries. In the United States, concerns about drug pricing have been expressed by members of Congress and prior presidential administrations. There can be no assurance that our product candidates, if approved, will be considered cost-effective by third-party payers, that an adequate level of reimbursement will be available or that the third-party payers’ reimbursement policies will not adversely affect our ability to sell our products profitably. If reimbursement of our products is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, our business could be adversely affected. We are subject to multiple and substantial federal and state health care and other laws, and the complexity of our regulatory compliance obligations is likely to increase in the event our product candidates are commercialized. Our business operations and activities may be directly or indirectly subject to various federal and state fraud and abuse laws, including, without limitation, the federal Anti-Kickback Statute and the federal FCA, and their implementing regulations, such as recent rules finalized by OIG which added safe harbor protections under the Anti-Kickback Statute for certain coordinated care and value-based arrangements among clinicians, providers, and others and that (with exceptions) became effective January 19, 2021. If we obtain FDA approval for any of our product candidates and begin commercializing those products in the United States, our potential exposure under such laws will increase significantly, and our costs associated with compliance with such laws are also likely to increase. These laws may impact, among other things, our current activities with principal investigators and research subjects, as well as proposed and future sales, marketing and education programs. In addition, we may be subject to patient privacy regulation by the federal government and state governments in which we conduct our business. In addition to the Anti-Kickback Statute, FCA and Physician Payments Sunshine Act and their implementing regulations, the laws that may affect our ability to operate include, but are not limited to: HIPAA, which created additional federal criminal statutes that prohibit knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program or obtain, by means of false or fraudulent pretenses, representations, or promises, any of the money or property owned by, or under the custody or control of, any healthcare benefit program, regardless of the payor
| ● | HIPAA, which created additional federal criminal statutes that prohibit knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program or obtain, by means of false or fraudulent pretenses, representations, or promises, any of the money or property owned by, or under the custody or control of, any healthcare benefit program, regardless of the payer (e.g., public or private) and knowingly and willfully falsifying, concealing or covering up by any trick or device a material fact or making any materially false statements in connection with the delivery of, or payment for, healthcare benefits, items or services relating to healthcare matters. Similar to the Anti-Kickback Statute, a person or entity can be found guilty of violating HIPAA without actual knowledge of the statute or specific intent to violate it. |
| •
● | federal consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers; |
| ● | federal government price reporting laws that require the calculation and reporting of complex pricing metrics to government programs, where such reported prices may be used in the calculation of reimbursement and/or discounts, on any of our product candidates that may be approved for marketing (participation in these programs and compliance with the applicable requirements may also subject us to potentially significant discounts on our products and increased infrastructure costs, and potentially limit our ability to offer certain marketplace discounts); |
| ● | the FCPA, which regulates certain financial relationships with foreign government officials (which could include, for example, certain medical professionals), and anti-bribery laws and related laws, and laws pertaining to the accuracy of our internal books and records, which have been the focus of increasing enforcement activity in recent years; and |
| ● | state law equivalents of each of the above federal laws, such as anti-kickback, false claims, consumer protection and unfair competition laws, which may apply to our business practices, including but not limited to, research, distribution, sales-and-marketing arrangements as well as submitting claims involving health care items or services reimbursed by any third-party payer, including commercial insurers; state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government that otherwise restricts payments that may be made to health care providers; state laws that require drug manufacturers to file reports with states regarding marketing information, such as the tracking and reporting of gifts, compensations and other remuneration and items of value provided to health care professionals and entities (compliance with such requirements may require investment in infrastructure to ensure that tracking is performed properly, and some of these laws result in the public disclosure of various types of payments and relationships, which could potentially have a negative effect on our business and/or increase enforcement scrutiny of the Company’s activities); and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways, with differing effects. |
federal government price reporting laws that require the calculation and reporting of complex pricing metrics to government programs, where such reported prices may be used in the calculation of reimbursement and/or discounts, on any of our product candidates that may be approved for marketing (participation in these programs and compliance with the applicable requirements may also subject us to potentially significant discounts on our products and increased infrastructure costs, and potentially limit our ability to offer certain marketplace discounts);
the FCPA, which regulates certain financial relationships with foreign government officials (which could include, for example, certain medical professionals), and anti-bribery laws and related laws, and laws pertaining to the accuracy of our internal books and records, which have been the focus of increasing enforcement activity in recent years; and
state law equivalents of each of the above federal laws, such as anti-kickback, false claims, consumer protection and unfair competition laws, which may apply to our business practices, including but not limited to, research, distribution, sales-and-marketing arrangements as well as submitting claims involving health care items or services reimbursed by any third-party payer, including commercial insurers; state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government that otherwise restricts payments that may be made to health care providers; state laws that require drug manufacturers to file reports with states regarding marketing information, such as the tracking and reporting of gifts, compensations and other remuneration and items of value provided to health care professionals and entities (compliance with such requirements may require investment in infrastructure to ensure that tracking is performed properly, and some of these laws result in the public disclosure of various types of payments and relationships, which could potentially have a negative effect on our business and/or increase enforcement scrutiny of the Company’s activities); and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways, with differing effects.
In addition, the regulatory approval and commercialization of any of our product candidates outside the United States will also subject us to foreign equivalents of the health care laws mentioned above, among other foreign laws, as well as compliance with the codes of practice of certain associations within such countries (for example, the Association of the British Pharmaceutical Industry (ABPI) in the United Kingdom). Efforts to help ensure that our business arrangements will comply with applicable health care laws and codes of practice may involve substantial costs. We have adopted policies and practices that are designed to help ensure that the Company, our employees, officers, agents, intermediaries and other third parties comply with applicable laws, but it is not always possible to assure compliance with applicable requirements, and the precautions we take to achieve compliance may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. It is possible that governmental and enforcement authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law interpreting applicable fraud and abuse or other health care laws and regulations. If our operations are found to be in violation of any of the laws described above or any other governmental regulations that apply to the Company, we may be subject to penalties, including, without limitation, civil, criminal and administrative penalties, damages, monetary fines, disgorgement, possible exclusion from participation in Medicare, Medicaid and other federal and state health care programs, individual imprisonment, contractual damages, reputational harm, diminished profits and future earnings, and curtailment or restructuring of our operations as well as additional reporting obligations and oversight if we become subject to a corporate integrity agreement or other agreement to resolve allegations of non-compliance with these laws. The impact of recent health care reform legislation and other changes in the health care industry and in health care spending on the Company is currently unknown and may adversely affect our business model. Our financial prospects could be affected by changes in health care spending and policy in the United States and abroad. We operate in a highly regulated industry and new laws, regulations, or judicial decisions, or new interpretations of existing laws, regulations, or decisions, related to, among other things, health care availability, or the method of delivery of or payment for health care products and services could negatively impact our business, operations and financial condition. For example, in the United States there is significant interest in promoting health care reform, as evidenced by the enactment in the United States of the Health Care Reform Law. The Health Care Reform Law substantially changed the way health care is financed by both governmental and commercial payers and significantly impacts the pharmaceutical industry. The Health Care Reform Law contains provisions that may reduce the profitability of drug products, including, for example, by increasing the minimum rebates owed by manufacturers under the Medicaid Drug Rebate Program, extending the rebate program to individuals enrolled in Medicaid managed care plans, addressing a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected, and imposing certain annual fees based on pharmaceutical companies’ share of sales to federal health care programs.
The Health Care Reform Law established and provided significant funding for a new Patient-Centered Outcomes Research Institute to coordinate and fund comparative effectiveness research. While the stated intent of comparative effectiveness research is to develop information to guide providers to the most efficacious therapies, outcomes of comparative effectiveness research could influence the reimbursement or coverage for therapies that are determined to be less cost effective than others. Should any of our products be approved for sale, but then determined to be less cost effective than alternative therapies, the levels of reimbursement for these products, or the willingness to reimburse at all, could be impacted, which could materially impact our financial results. Certain provisions of the Health Care Reform Law have been subject to judicial challenges, as well as efforts to repeal, replace or otherwise modify them or to alter their interpretation or implementation. For example, the Tax Cuts and Jobs Act enacted on December 22, 2017, eliminated the tax-based payment for individuals who fail to maintain minimum essential coverage under section 5000A of the Internal Revenue Code of 1986, as amended, commonly referred to as the “individual mandate,” effective January 1, 2019. Further, the Bipartisan Budget Act of 2018 among other things, amended the Medicare statute, effective January 1, 2019, to reduce the coverage gap in most Medicare prescription drug plans, commonly known as the “donut hole,” by raising the manufacturer discount under the Medicare Part D coverage gap discount program to 70%. Additional legislative changes, regulatory changes and judicial challenges related to the Health Care Reform Law remain possible. It is unclear how the Health Care Reform Law and its implementation, as well as efforts to repeal, replace or otherwise modify, or invalidate, the Health Care Reform Law, or portions thereof, will affect our business. We cannot predict what affect further changes to the Health Care Reform Law would have on our business, especially including under the Biden administration. Another provision of the Health Care Reform Law, generally referred to as the Physician Payment Sunshine Act or Open Payments Program, requires manufacturers of drugs, devices, biologics, and medical supplies for which payment is available under Medicare, Medicaid, or the Children’s Health Insurance Program (with certain exceptions) to report annually to CMS information related to direct or indirect payments and other transfers of value made to physicians (defined to include doctors, dentists, optometrists, podiatrists, and chiropractors) and teaching hospitals, as well as ownership and investment interests held in the company by physicians and their immediate family members. Effective January 1, 2022, these reporting obligations will extendhave been extended to include transfers of value made (starting in 2021) to certain non-physician providers such as physician assistants, nurse practitioners, clinical nurse specialists, certified nurse anesthetists, and certified nurse-midwives. CMS publishes information from these reports on a publicly available website. Our compliance with these rules may also impose additional costs and may impact our relationships with physicians, teaching hospitals, and other non-physician health care providers. In addition, other legislative changes have been proposed and adopted since the Health Care Reform Law was enacted. For example, the Budget Control Act of 2011, as amended by the American Taxpayer Relief Act of 2012, among other things led to aggregate reductions in Medicare payments for all items and services, including prescription drugs and biologics, to service providers of, on average, 2% per fiscal year beginning April 1, 2013, and, due to subsequent legislation, will continue until 2030 (with the exception of a temporary suspension from May 1, 2020, through March 31, 2021) unless Congress takes additional action. Additional legislative changes, regulatory changes or guidance could be adopted, which may impact the marketing approvals and reimbursement for our product candidates. For example, on July 9, 2021, President Biden issued an executive order directing the FDA to, among other things, continue to clarify and improve the approval framework for biosimilars, including the standards for interchangeability of biological products, facilitate the development and approval of biosimilar and interchangeable products, clarify existing requirements and procedures related to the review and submission of BLAs, and identify and address any efforts to impede biosimilar competition. Further, there has been increasing legislative, regulatory and enforcement interest in the United States with respect to drug pricing practices. There have been several Congressional inquiries and proposed and enacted federal and state legislation and regulatory initiatives designed to, among other things, bring more transparency to product pricing, evaluate the relationship between pricing and manufacturer patient programs, and reform government healthcare program reimbursement methodologies for drug products. For example, on November 20, 2020, CMS issued an interim final rule to implement a “Most Favored Nation” demonstration project to test Medicare Part B reimbursement of certain separately payable drugs and biologicals based on international reference prices. The rule has become subject to judicial challenges, and federal courts have enjoined the rule at this time. If the rule survives judicial scrutiny, the Most Favored Nation model will subject certain drugs or biologicals identified by CMS as having the highest annual Medicare Part B spending to an alternative payment methodology based on international reference prices, with the list of products to be updated annually to add more products and products not to be removed absent limited circumstances. There has also been legislation that would establish an international reference price-based Medicare Part B drug and biological payment methodology. It is possible that the Health Care Reform Law, as currently enacted or may be amended or otherwise modified in the future, as well as other health care reform measures that may be adopted in the future, may result in additional reductions in Medicare payment and other health care financing, more rigorous coverage criteria, and new payment methodologies and in additional downward pressure on coverage and payment and the price that we receive for any approved product. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from commercial payors.payers. We cannot predict the
reform initiatives that may be adopted in the future or whether initiatives that have been adopted will be repealed or modified. These continuing health care reform initiatives may adversely affect: | ● | the demand for any product candidates for which we may obtain regulatory approval; |
| ● | our ability to set a price that we believe is fair for our products; |
| ● | our ability to obtain coverage and reimbursement approval for a product; |
| ● | our ability to generate revenues and achieve or maintain profitability; and |
| ● | the level of taxes that we are required to pay. |
the demand for any product candidates for which we may obtain regulatory approval;
our ability to set a price that we believe is fair for our products;
our ability to obtain coverage and reimbursement approval for a product;
our ability to generate revenues and achieve or maintain profitability; and
the level of taxes that we are required to pay.
Certain business practices associated with the commercialization of pharmaceutical products are subject to scrutiny by regulatory authorities, as well as to lawsuits brought by private citizens under federal and state laws. Failure to comply with applicable law or an adverse decision in lawsuits may result in adverse consequences to the Company. The laws that would govern our conduct in the United States upon the commercialization of our product candidates are enforceable by criminal, civil and administrative penalties. Violations of laws such as anti-kickbackthe Anti-Kickback Statute and false claims laws,FCA, the FD&C Act, or any regulations promulgated under their authority, may result in jail sentences, fines or exclusion from federal and state programs, as may be determined by Medicare, Medicaid, the Department of Defense, other regulatory authorities and the courts. There can be no assurance that our activities will not come under the scrutiny of regulators and other government authorities or that our practices will not be found to violate applicable laws, rules and regulations or prompt lawsuits by private citizen “relators” under federal or state false claims laws. In the United States, among the laws that may affect our ability to operate and market our products include, but are not limited to: The federal Anti-Kickback Law prohibits, among other activities, any person from knowingly and willfully, directly or indirectly soliciting, receiving, offering or paying any remuneration with the intent of (or in return for) generating referrals of individuals or purchases, orders or recommendations for services or items reimbursable by federal health care programs like Medicare and Medicaid. Courts have interpreted this law very broadly, including by holding that a violation has occurred if even one purpose of the remuneration is to generate referrals, even if there are other lawful purposes. Moreover, liability under the Anti-Kickback Law may be established without proving actual knowledge of the law or specific intent to violate. There are statutory exceptions and regulatory safe harbors that protect certain arrangements from prosecution or administrative sanctions, but the exceptions and safe harbors are drawn narrowly. The fact that an arrangement does not fall within a safe harbor does not necessarily render the conduct illegal under the Anti-Kickback Law, but the arrangement may be subject to scrutiny based on the facts and circumstances. Practices that involve remuneration to those who prescribe, purchase, or recommend pharmaceutical products, including certain discounts, or engaging such individuals as consultants, advisors and speakers, may be subject to scrutiny if they do not fit squarely within an exception or safe harbor. Moreover, there are no safe harbors for many common practices, such as educational and research grants, charitable donations, product support and patient assistance programs. Violations of the Anti-Kickback Law may be punished by civil and criminal penalties, damages, fines, or exclusion from participation in federal health care programs like Medicare and Medicaid. In addition, the government may assert that a claim including items or services resulting from a violation of the Anti-Kickback law constitutes a false or fraudulent claim for purposes of the FCA.
The FCA prohibits any person from, among other things, knowingly presenting or causing to be presented a false or fraudulent claim for payment of government funds, or making, or causing to be made, a false statement material to a false or fraudulent claim, or knowingly concealing or knowingly and improperly avoiding, or decreasing, an obligation to pay or transmit money or property to the government. The FCA is commonly used to sue those who submit allegedly false Medicare or Medicaid claims, as well as those who induce or assist others to submit a false claim. “False claims” can result not only from non-compliance with the express requirements of applicable governmental reimbursement programs, such as Medicare or Medicaid, but also from non-compliance with other laws, such as the Anti-Kickback Law, FDA laws on off-label promotion, or laws that require quality care in service delivery. Actions under the FCA may be brought by the government or by whistleblowers referred to as relators, who may initiate an action in the name of the government and may share in any monetary recovery. Violations of the FCA can result in treble damages, mandatory per claim penalties, and exclusion from participation in federal health care programs.
| •
● | The federal Anti-Kickback Statute prohibits, among other activities, any person from knowingly and willfully, directly or indirectly soliciting, receiving, offering or paying any remuneration with the intent of (or in return for) generating referrals of individuals or purchases, orders or recommendations for services or items reimbursable by federal health care programs like Medicare and Medicaid. Courts have interpreted this law very broadly, including by holding that a violation has occurred if even one purpose of the remuneration is to generate referrals, even if there are other lawful purposes. Moreover, liability under the Anti-Kickback Statute may be established without proving actual knowledge of the law or specific intent to violate. There are statutory exceptions and regulatory safe harbors that protect certain arrangements from prosecution or administrative sanctions, but the exceptions and safe harbors are drawn narrowly. The fact that an arrangement does not fall within a safe harbor does not necessarily render the conduct illegal under the Anti-Kickback Statute, but the arrangement may be subject to scrutiny based on the facts and circumstances. Practices that involve remuneration to those who prescribe, purchase, or recommend pharmaceutical products, including certain discounts, or engaging such individuals as consultants, advisors and speakers, may be subject to scrutiny if they do not fit squarely within an exception or safe harbor. Moreover, there are no safe harbors for many common practices, such as educational and research grants, charitable donations, product support and patient assistance programs. Violations of the Anti-Kickback Statute may be punished by civil and criminal penalties, damages, fines, or exclusion from participation in federal health care programs like Medicare and Medicaid. In addition, the government may assert that a claim including items or services resulting from a violation of the Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the FCA. |
| ● | The FCA prohibits any person from, among other things, knowingly presenting or causing to be presented a false or fraudulent claim for payment of government funds, or making, or causing to be made, a false statement material to a false or fraudulent claim, or knowingly concealing or knowingly and improperly avoiding, or decreasing, an obligation to pay or transmit money or property to the government. The FCA is commonly used to sue those who submit allegedly false Medicare or Medicaid claims, as well as those who induce or assist others to submit a false claim. “False claims” can result not only from non-compliance with the express requirements of applicable governmental reimbursement programs, such as Medicare or Medicaid, but also from non-compliance with other laws, such as the Anti-Kickback Law, FDA laws on off-label promotion, or laws that require quality care in service delivery. Actions under the FCA may be brought by the government or by whistleblowers referred to as relators, who may initiate an action in the name of the government and may share in any monetary recovery. Violations of the FCA can result in treble damages, mandatory per claim penalties, and exclusion from participation in federal health care programs. |
| ● | HIPAA created additional federal criminal statutes that prohibit knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program or obtain, by means of false or fraudulent pretenses, representations, or promises, any of the money or property owned by, or under the custody or control of, any healthcare benefit program, regardless of the payorpayer (e.g., public or private) and knowingly and willfully falsifying, concealing or covering up by any trick or device a material fact or making any materially false statements in connection with the delivery of, or payment for, healthcare benefits, items or services relating to healthcare matters. Similar to the Anti-Kickback Law, a |
| | person or entity can be found guilty of violating the HIPAA fraud statute without actual knowledge of the statute or specific intent to violate it. |
| ● | The Physician Payments Sunshine Act, implemented as the Open Payments program, imposes reporting requirements for pharmaceutical, biologic, and device manufacturers regarding payments or other transfers of value made to physicians, teaching hospitals, and other healthcare providers, including investment interests in such manufacturers held by physicians and their immediate family members during the preceding calendar year. Annual reporting of such transfers of value by manufacturers has increased scrutiny of the financial relationships between industry and the physicians, teaching hospitals and other healthcare providers. Failure to submit required annual information may result in civil monetary penalties, which may increase significantly for “knowing failures.” |
The Physician Payments Sunshine Act, implemented as the Open Payments program, imposes reporting requirements for pharmaceutical, biologic, and device manufacturers regarding payments or other transfers of value made to physicians, teaching hospitals, and other healthcare providers, including investment interests in such manufacturers held by physicians and their immediate family members during the preceding calendar year. Annual reporting of such transfers of value by manufacturers has increased scrutiny of the financial relationships between industry and the physicians, teaching hospitals and other healthcare providers. Failure to submit required annual information may result in civil monetary penalties, which may increase significantly for “knowing failures.”77
Analogous state laws and regulations, including anti-kickback and false claims laws, which apply to items and services reimbursed under Medicaid or, in several states, regardless of the payer, including private payers. Some state laws require pharmaceutical companies to report expenses relating to the marketing and promotion of pharmaceutical products and to report gifts and payments to individual health care providers in those states. Some of these states also prohibit certain marketing related activities including the provision of gifts, meals, or other items to certain health care providers. Some states restrict the ability of manufacturers to offer co-pay support to patients for certain prescription drugs. Some states require the posting of information relating to clinical studies and their outcomes. Other states and cities require identification or licensing of sales representatives. In addition, several states require pharmaceutical companies to implement compliance programs or marketing codes of conduct.
The FDCA and comparable foreign laws, in addition to prohibiting the promotionTable of the safety or effectiveness of product candidates not yet approved for commercialization, an act known as pre-approval promotion, also generally restrict companies from promoting approved products for indications other than those indications for which a product is approved, which is also referred to as off-label use. This means, for example, that we may not make claims about the use of our products, should they be approved for sale, outside of their approved indications, and we may not proactively discuss or provide information regarding any of their off-label uses subject to very specific and limited exceptions. If we or our business partners fail to comply with applicable laws and regulations governing off-label uses of our product candidates, if approved, then we could be subject to administrative or judicially imposed sanctions, including, but not limited to: (i) enforcement proceedings by regulatory agencies; (ii) reduced demand for our products; and (iii) civil or criminal sanctions. Furthermore, actions under the FCA have recently been brought against companies for allegedly promoting off-label uses of drugs, because such promotion induces the use and subsequent claims for reimbursement under Medicare and other federal programs. Similar actions for off-label promotion have been initiated by several states for Medicaid fraud.Contents
| ● | Analogous state laws and regulations, including anti-kickback and false claims laws, which apply to items and services reimbursed under Medicaid or, in several states, regardless of the payer, including private payers. Some state laws require pharmaceutical companies to report expenses relating to the marketing and promotion of pharmaceutical products and to report gifts and payments to individual health care providers in those states. Some of these states also prohibit certain marketing related activities including the provision of gifts, meals, or other items to certain health care providers. Some states restrict the ability of manufacturers to offer co-pay support to patients for certain prescription drugs. Some states require the posting of information relating to clinical studies and their outcomes. Other states and cities require identification or licensing of sales representatives. In addition, several states require pharmaceutical companies to implement compliance programs or marketing codes of conduct. |
| ● | The FD&C Act and comparable foreign laws, in addition to prohibiting the promotion of the safety or effectiveness of product candidates not yet approved for commercialization, an act known as pre-approval promotion, also generally restrict companies from promoting approved products for indications other than those indications for which a product is approved, which is also referred to as off-label use. This means, for example, that we may not make claims about the use of our products, should they be approved for sale, outside of their approved indications, and we may not proactively discuss or provide information regarding any of their off-label uses subject to very specific and limited exceptions. If we or our business partners fail to comply with applicable laws and regulations governing off-label uses of our product candidates, if approved, then we could be subject to administrative or judicially imposed sanctions, including, but not limited to: (i) enforcement proceedings by regulatory agencies; (ii) reduced demand for our products; and (iii) civil or criminal sanctions. Furthermore, actions under the FCA have recently been brought against companies for allegedly promoting off-label uses of drugs, because such promotion induces the use and subsequent claims for reimbursement under Medicare and other federal programs. Similar actions for off-label promotion have been initiated by several states for Medicaid fraud. |
The bringing of any FCA or other enforcement investigation or action, even if unsuccessful, could require us to devote resources to investigate and defend the action, as well as result in reputational harm. Failure to comply with the fraud and abuse laws could result in significant civil and criminal penalties and costs, including the loss of licenses and the ability to participate in federal and state health care programs, and could have a material adverse effect on our business. In addition, many of these laws are vague, subject to modification, and subject to evolving interpretation by prosecutorial and regulatory authorities, increasing the risk of noncompliance. We cannot predict whether changes in applicable law, or interpretation of laws, or changes in our services or marketing practices in response to changes in applicable law or interpretation of laws could have a material adverse effect on our business. If our product candidates are commercialized, then we would also be required to report detailed and complex pricing information, net of included discounts, rebates and other concessions, to CMS for the purpose of calculating national reimbursement levels, certain federal prices and certain federal and state rebate obligations, and we would need to develop the expertise, as well as the systems for collecting and reporting this data accurately to CMS and have instituted a compliance program to assure that the information collected is complete in all respects. Companies that fail to accurately report this kind of pricing information to the U.S. government could be subject to fines and other sanctions (including potential FCA liability) that could adversely affect their business. We must comply with data privacy and security laws and regulations, and failure to comply with these laws and regulations could expose us to significant liabilities. We must operate in compliance with various data privacy and security regulations in the United States by both the federal government and the states in which we conduct our business, as well as in other jurisdictions outside of the United States, such as the United Kingdom, the EU and Asia, where we conduct clinical trials.
In the U.S., we may be subject to data privacy and security laws and regulations by both the federal government and the states in which we conduct our business, including, for example, state data breach notification laws, state health information and/or genetic privacy laws and federal and state consumer protection laws (e.g., Section 5 of the FTC Act and the CCPA), govern the collection, use, disclosure, and protection of health-related and other personal information. Many of these laws differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts. Compliance with these laws is difficult, constantly evolving, and time consuming. Federal regulators, state attorneys general, and plaintiffs’ attorneys, including class action attorneys, have been and will likely continue to be active in this space. HIPAA, for example, imposes specific requirements relating to the privacy, security and transmission of individually identifiable health information. Failure to comply with these laws and regulations can result in substantial penalties and other liabilities. We may obtain health information from third parties, such as research institutions, that are subject to privacy and security requirements under HIPAA. Although we are not directly subject to HIPAA other than with respect to providing certain employee benefits, we could potentially be subject to criminal penalties if we, our affiliates, or our agents knowingly obtain, use, or disclose individually identifiable health information maintained by a HIPAA-covered entity in a manner that is not authorized or permitted by HIPAA. The CCPA became effective on January 1, 2020 and establishes certain requirements for data use and sharing transparency, and provides California residents certain rights concerning the use, disclosure, and retention of their personal data. The CCPA provides for civil penalties for violations, as well as a private right of action for data breaches that is expected to increase data breach litigation. The CCPA and its implementing regulations have already been amended multiple times since their enactment. The CCPA could mark the beginning of a trend toward more stringent state privacy legislation in the U.S., which could increase our potential liability and adversely affect our business. The obligations to comply with the CCPA and other evolving legislation may require us, among other things, to update our notices and develop new processes internally and with our partners. In addition, the European Parliament and the Council of the European Union have adopted a new pan-European General Data Protection Regulation (“GDPR”), effective May 25, 2018, which increases privacy rights for individuals in Europe, extends the scope of responsibilities for data controllers and data processors and imposes increased requirements and potential penalties on companies, offering goods or services to individuals who are located in the EU or monitoring the behavior of such individuals (including by companies based outside of the EU). The GDPR governs the collection, use, storage, disclosure, transfer, or other processing of personal data regarding individuals in the EU, including personal health data. It is wide-ranging in scope and imposes numerous requirements on companies that process personal data, including requirements relating to processing health and other sensitive data, providing information to individuals regarding data processing activities, implementing safeguards to protect the security and confidentiality of personal data, providing notification of data breaches, and taking certain measures when engaging third-party processors. The GDPR also imposes strict rules on the transfer of personal data to countries outside the EU, including the United States, and permits data protection authorities to impose large penalties for violations of the GDPR. Noncompliance can result in penalties of up to the greater of EUR 20 million, or 4% of annual global company revenues. The GDPR also confers a private right of action on data subjects and consumer associations to lodge complaints with supervisory authorities, seek judicial remedies, and obtain compensation for damages resulting from violations of the GDPR. In addition, the GDPR includes restrictions on cross-border data transfers. While we expect to have substantially compliant programs and controls in place to comply with the GDPR requirements, our compliance with the new regulation is likely to impose additional costs on us and we cannot predict whether the interpretations of the requirements, or changes in our practices in response to new requirements or interpretations of the requirements could have a material adverse effect on our business. There is a risk that we may be subject to fines and penalties, litigation, and reputational harm in connection with our European activities. Further, the United Kingdom’s decision to leave the EU, often referred to as Brexit, has created uncertainty with regard to data protection regulation in the United Kingdom. The EU-UK Trade and Cooperation Agreement of 24 December 2020 allows forOn June 28, 2021, the EU Commission adopted decisions on the U.K.’s adequacy under the EU’s GDPR. This means that most personal data can continue to be transferredflow from the EU and the EEA to the United KingdomU.K. without the need to use a transfer mechanism as set out in Chapter V GDPR until 30 June 2021. This means that personal data may flow freelyfor additional safeguards. However, restricted transfers from the EUU.K. to other countries, including to the United Kingdom until 30 June 2021. It is unclear howEEA, are subject to transfer rules under the U.K. regime. On February 2, 2022, the U.K. announced the implementation of a new series of U.K. Standard Contractual Clauses, which must be adopted by U.K. companies no later than March 21, 2024. These U.K. transfer rules broadly mirror the EU GDPR rules, but the U.K. has the independence to keep the framework under review, lending uncertainty to future data transfers in to and from the United Kingdom will be regulated after that date. However, the intentionout of the 6 month grace period is to give the European Commission sufficient time to adopt an adequacy decision in respect of the United Kingdom so that personal data may flow freely from the EU to the United Kingdom after 30 June 2021 as well.U.K. We must also ensure that we maintain adequate safeguards to enable the transfer of personal data outside of the EEA, in particular to the United States in compliance with European data protection laws. We expect that we will continue to face uncertainty as to whether our efforts to comply with our obligations under European privacy laws will be sufficient. If we are investigated by a European data protection authority, we may face fines and other penalties. Any such investigation or charges by European data protection authorities could have a negative effect on our existing business and on our ability to attract and retain new clients or pharmaceutical collaboration partners. We may also experience hesitancy, reluctance, or refusal by European or multi-national clients or pharmaceutical partners to continue to use our products due to the potential risk exposure as a result of the current (and, in particular, future) data protection obligations imposed on them by certain data protection authorities in interpretation of current law,
including the GDPR. Such clients or pharmaceutical collaboration partners may also view any alternative approaches to compliance as being too costly, too burdensome, too legally uncertain, or otherwise objectionable and therefore decide not to do business with us. Any of the foregoing could materially harm our business, prospects, financial condition and results of operations. We are subject to extensive government regulatory compliance and ethics oversight, and we will need to develop more extensive compliance and ethics policies in the future. Our business is subject to extensive government regulation and ethics oversight, which will become more complex and extensive if we succeed in commercializing products. We have enacted various compliance policies and procedures that govern our business practices as appropriate for a company in our stage of development. These policies and procedures are implemented through education, training and monitoring of our employees, distributors and suppliers. However, our adoption and enforcement of these various policies and procedures does not ensure that we will avoid investigation or the imposition of penalties by applicable government agencies. In addition, to enhance compliance with applicable health care laws and mitigate potential liability in the event of non-compliance, regulatory authorities, such as OIG, of the HHS have recommended the adoption and implementation of a comprehensive health care compliance program that generally contains the elements of an effective compliance and ethics program described in Section 8B2.1 of the U.S. Sentencing Commission Guidelines Manual. Increasing numbers of U.S.-based pharmaceutical companies have such programs. Although we believe our existing compliance policies and procedures are adequate for our current operations, these policies and procedures would not be considered a comprehensive health care compliance program consistent with the HHS OIG’s recommendations. Depending upon the nature of our future operations, we anticipate developing a more extensive compliance program in the future. Our employees, independent contractors, principal investigators, consultants, commercial partners and vendors may engage in misconduct or other improper activities, including non-compliance with regulatory standards and requirements. We are exposed to the risk of fraudulent or other illegal activity by our employees, independent contractors, principal investigators, consultants, commercial partners and vendors. Misconduct by these parties could include intentional, reckless and/or negligent conduct that fails to comply with the laws of the FDA and similar foreign regulatory bodies; fails to comply with manufacturing standards we have established, or with federal, state and foreign health care fraud and abuse laws and regulations; fails to report financial information or data accurately, including to our regulators, such as the FDA and similar foreign regulatory bodies; or fails to disclose unauthorized activities to the Company. In particular, the promotion, sale and marketing of health care items and services, as well as certain business arrangements in the health care industry are subject to extensive laws and regulations intended to prevent misconduct, including fraud, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and, structuring and commissions, certain customer incentive programs and other business arrangements. Activities subject to these laws also involve the improper use of information obtained in the course of patient recruitment for clinical trials. We have adopted a Code of Business Conduct and Ethics Policy and other policies and practices that are designed to help ensure that the Company, our employees, officers, agents, intermediaries and other third parties comply with applicable laws, but it is not always possible to identify and deter such misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against the Company, and in some cases regardless of the merits of those actions, those actions could have a significant impact on our business, including the costs of investigation, settlement arrangements, imposition of civil, criminal and administrative penalties (such as additional reporting requirements and oversight if we become subject to Corporate Integrity Agreements and other arrangements, damages, monetary fines, disgorgement, imprisonment, and possible exclusion from participation in Medicare, Medicaid and other federal health care programs), contractual damages, reputational harm, diminished profits and future earnings, and curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations. In the United States, legislation limiting or restricting liability for medical products used to fight bioterrorism is new, and it cannot be certain that any such protection will apply to our product candidates or if applied what the scope80
Table of any such coverage will be.Contents The Public Readiness Act creates general immunity for manufacturers of drug products used to address bioterrorism attacks, when the Secretary of HHS issues a declaration for their manufacture, administration or use. The declaration is meant to provide general immunity from all claims under state or federal law for loss arising out of the administration or use of a covered drug product, generally referred to as a “countermeasure.” Manufacturers are excluded from this protection in cases of willful misconduct. Although we believe that our anthrax vaccine product candidate is covered under the general immunity provisions of the Public Readiness Act, there can be no assurance that this coverage will continue, or that the Secretary of HHS will make other declarations in the future that would cover any of our other product candidates, or that the U.S. Congress will not act in the future to reduce coverage under the Public Readiness Act or to repeal it altogether.
In addition, under the Public Readiness Act, upon a declaration by the Secretary of HHS, a compensation fund would be created to provide “timely, uniform, and adequate compensation to eligible individuals for covered injuries directly caused by the administration or use of a covered countermeasure.” The “covered injuries” to which the program applies are defined as serious physical injuries or death. Individuals are permitted to bring a willful misconduct action against a manufacturer after they have exhausted their remedies under the compensation program. However, there is no assurance that the Secretary of HHS would issue under this act a declaration to establish a compensation fund.
Additionally, we are considering applying for liability protection under the Support Anti-terrorism by the SAFETY Act, which provides certain protections that would limit the damages potentially faced by companies who provide certain “qualified” anti-terrorism products. However, we cannot be certain that we will be able to obtain or maintain coverage under the SAFETY Act. If the U.S. Department of Homeland Security limits the scope of any coverage awarded to the Company, denies it coverage or continued coverage for a particular product or product candidate, or delays in making decisions about whether to grant it coverage, we may become exposed to legal claims.
We are required to comply with certain export control laws which may limit our ability to sell our products to non-U.S. persons and may subject us to regulatory requirements that may delay or limit our ability to develop and commercialize our products.
Our product candidates are subject to the Export Administration Regulations (“EAR”), administered by the U.S. Department of Commerce and are, in certain instances subject to the International Traffic in Arms Regulations (“ITAR”), administered by the U.S. Department of State. EAR restricts the export of dual-use products and technical data to certain countries, while ITAR restricts the export of defense products, technical data and defense services. In addition, EAR and ITAR may also regulate the disclosure to certain foreign nationals in the United States, such as research staff, of technical data about controlled commodities. The U.S. government agencies responsible for administering EAR and ITAR have significant discretion in the interpretation and enforcement of these regulations. Failure to comply with these regulations can result in criminal and civil penalties and may harm our ability to enter into contracts with the U.S. government. It is also possible that these regulations could adversely affect our ability to sell our products to non-U.S. customers.
Our product candidates may also be subject to export control laws within the United Kingdom and European Union resulting in the need for authorization from customs authorities before they can leave the United Kingdom or European Union customs territories and restrictions on export from these territories to certain countries. Again, such laws could adversely affect our ability to sell to customers in certain countries and non-compliance can result in civil and criminal penalties. Such restrictions exist across the European Union and within its member states individually and may vary between member states.
We must comply with environmental laws and regulations, and failure to comply with these laws and regulations could expose us to significant liabilities. We use hazardous chemicals and biological materials in certain aspects of our business and are subject to a variety of federal, state and local laws and regulations governing the use, generation, manufacture, distribution, storage, handling, treatment and disposal of these materials. We cannot eliminate the risk of accidental injury or contamination from the use, manufacture, distribution, storage, handling, treatment or disposal of hazardous materials. In the event of contamination or injury, or failure to comply with environmental, occupational health and safety and export control laws and regulations, we could be held liable for any resulting damages and any such liability could exceed our assets and resources. In addition, we may be required to pay damages or civil judgments related to third-party claims, for which we are uninsured, including those relating to personal injury (including exposure to hazardous chemicals and biological materials), product quality issues, property damage or contribution to remedial obligations. If we use biological and hazardous materials in a manner that causes contamination or injury or violates laws, we may be liable for damages.
Our research and development activities and clinical trials involve the use of potentially harmful biological materials, including anthrax, as well as hazardous materials and chemicals. We cannot completely eliminate the risk of accidental contamination or injury from the distribution, use, storage, handling or disposal of these materials. In the event of contamination or injury, we could be held liable for damages that result, and any liability could exceed our available financial resources. The Company, our collaborative partners, the third parties that conduct clinical trials on our behalf, and our third-party manufacturers are subject to federal, state, local or foreign laws and regulations governing the use, storage, handling and disposal of these materials and waste products. The cost of compliance with these laws and regulations could be significant. The failure to comply with any of these laws and regulations could result in significant fines and work stoppages.
Risks Related to our Securities The trading price of our common stock has been volatile with substantial price fluctuations on heavy volume, which could result in substantial losses for purchasers of our common stock and existing stockholders. Our stock price has been and, in the future, may be subject to substantial volatility. OnFor example, on September 13, 2018 we amended our Amended and Restated Certificate of Incorporation to effect a reverse stock split at a ratio 1-for-30 (the “Reverse Stock Split”). The Reverse Stock Split was effective on September 13, 2018, and our shares of common stock commenced trading on NASDAQ on a post-Reverse Stock Split basis on September 14, 2018. The volatility of our stock price has increased since we effected the Reverse Stock Split. Since our common stock began trading on a post-Reverse Stock Split basis on September 14, 2018 and through December 31, 2020,2022, our stock has traded in a range with a low of $1.51 and a high of $36.25. Furthermore, the stock market in general and the market for biopharmaceutical companies in particular have experienced extreme volatility that has often been unrelated to the operating performance of particular companies. As a result of this volatility, investors may not be able to sell their common stock at or above the price paid for the shares. The market price for our common stock may be influenced by many factors, including: | ● | announcements relating to development, regulatory approvals or commercialization of our product candidates or those of competitors; |
| ● | results of clinical trials of our product candidates or those of our competitors; |
| ● | announcements by us or our competitors of significant strategic partnerships or collaborations or terminations of such arrangements; |
| ● | actual or anticipated variations in our operating results and whether we have achieved key business targets; |
| ● | sales of our common stock, including sales by our directors and officers or specific stockholders; |
| ● | changes in, or our failure to meet, financial estimates by us or by any securities analysts who might cover our stock; |
| ● | changes in securities analysts’ buy and/or sell recommendations; |
| ● | general economic, political, or stock market conditions; |
| ● | conditions or trends in our industry; |
| ● | changes in laws or other regulatory actions affecting us or our industry; |
| ● | stock market price and volume fluctuations of comparable companies and, in particular, those that operate in the biopharmaceutical industry; |
results of clinical trials of our product candidates or those of our competitors;
| ● | announcements of investigations or regulatory scrutiny of our operations or lawsuits filed against us; |
| ● | investors’ general perception of our company, our business, and our prospects; |
| ● | disputes concerning our intellectual property or other proprietary rights; and |
| ● | recruitment or departure of key personnel. |
actual or anticipated variations in our operating results and whether we have achieved key business targets;
sales of our common stock, including sales by our directors and officers or specific stockholders;
changes in, or our failure to meet, financial estimates by us or by any securities analysts who might cover our stock;
changes in securities analysists’ buy and / or sell recommendations;
general economic, political, or stock market conditions;
conditions or trends in our industry;
changes in laws or other regulatory actions affecting us or our industry;
stock market price and volume fluctuations of comparable companies and, in particular, those that operate in the biopharmaceutical industry;
announcements of investigations or regulatory scrutiny of our operations or lawsuits filed against us;
investors’ general perception of our company, our business, and our prospects;
disputes concerning our intellectual property or other proprietary rights; and
recruitment or departure of key personnel.
In the past, stockholders have initiated class action lawsuits against pharmaceutical and biotechnology companies following periods of volatility in the market prices of these companies’ stock. Such litigation, if instituted against us, could cause us to incur substantial costs and divert management’s attention and resources from our business. Future sales and issuances of our common stock or rights to purchase common stock could result in substantial dilution to the percentage ownership of our stockholders. We expect that significant additional capital will be needed in the future to continue our planned operations. To raise capital, we may sell common stock or other securities convertible into or exchanged for our common stock in one or more transactions, and in a manner we determine from time to time and at prices that may not be the same as the price per share paid by other investors, and dilution to our stockholders could result. The price per share at which we sell additional shares of our common stock, or securities convertible or exchangeable into common stock, in future transactions may be higher or lower than the price per share paid by other investors. New investors could also receive rights, preferences and privileges senior to those of existing holders of our common stock. In addition, in the event of stock dividends, stock splits, reorganizations or similar events affecting our common stock, we may be
required to proportionally adjust the conversion price, exercise price or number of shares issuable upon exercise of our outstanding warrants. If we do not meet the continued listing standards of The NASDAQ Global Market our common stock could be delisted from trading, which could limit investors’ ability to make transactions in our common stock and subject us to additional trading restrictions. Our common stock is listed on Nasdaq Global Market, a national securities exchange, which imposes continued listing requirements with respect to listed shares. If we fail to satisfy the continued listing standards, including with respect to the maintenance of a minimum share price, or if NASDAQ in its discretion, determines that a condition exists that makes further dealings of our Company on the exchange unwarranted, NASDAQ may issue a non-compliance letter or initiate delisting proceedings. If our securities are delisted from trading on the NASDAQ exchange, our securities could be quoted on the OTCQB or on the Pink Open Market. As a result, we could face significant adverse consequences including: | ● | a limited availability of market quotations for our securities; |
| ● | a determination that our common stock is a “penny stock” which will require brokers trading in our common stock to adhere to more stringent rules and possibly result in a reduced level of trading activity in the secondary trading market for our securities; |
| ● | a limited amount of news and analyst coverage for us; and |
| ● | a decreased ability to issue additional securities (including pursuant to short-form registration statements on Form S-3) or obtain additional financing in the future. |
a determination that our common stock is a “penny stock” which will require brokers trading in our common stock to adhere to more stringent rules and possibly result in a reduced level of trading activity in the secondary trading market for our securities;
a decreased ability to issue additional securities (including pursuant to short-form registration statements on Form S-3) or obtain additional financing in the future.
Shares that we may issue in the future in connection with certain capital-raising transactions and shares available for future issuance upon exercise of warrants and options could dilute our stockholders and depress the market price of our common stock. The issuance or even the expected issuance of a large number of shares of our common stock upon purchase, conversion or exercise of the securities described above could depress the market price of our stock and the issuance of such shares will dilute the stock ownership of our existing stockholders. Shares that we may issue in the future in connection with certain capital-raising transactions and shares available for future issuance upon exercise of warrants and options could dilute our stockholders and depress the market price of our common stock and result in the adjustment of the conversion terms of our existing securities. We can give no assurances that we will ever again pay dividends. Other than for the PharmAthene board of directors’ declaration of a special one-time cash dividend paid in 2017, neither Private Altimmune nor PharmAthene has evernever paid any dividends on our common stock. While subject to periodic review, our current policy is to retain all earnings, if any, primarily to finance our future growth or ability to consummate strategic transactions, such as a merger or other business combination. We make no assurances that we will ever pay future dividends, cash or otherwise. Whether we pay any dividends in the future will depend on our financial condition, results of operations, and other factors that we will consider.
Item 1B. Unresolved Staff Comments None. Item 2. Properties Our principal executive offices are located in Gaithersburg, Maryland, where we occupy approximately 18,59819,699 square feet of laboratory and office space. For additional information, see Note 6 to our consolidated financial statements. Management believes that these facilities are suitable and adequate to meet our anticipated needs. Item 3. Legal Proceedings In December 2019, a complaint was filed by Dr. De-ChuDe-chu Christopher Tang (“Plaintiff”) against us, inwhich we removed to the United States District Court for the Eastern District of Texas. The Plaintiff amended the complaint in February 2020 to include Vipin K. Garg and David J. Drutz as defendants, in addition to the Company (Dr. Garg, Dr. Drutz, and the Company are collectively referred to as “Defendants”). See Note 17In March 2020 the Defendants filed a motion to dismiss the consolidated financial statements appearing in Item 8complaint. On March 25, 2021, the court granted the motion and dismissed the action for lack of this report for further details.
A prior lawsuit filed bypersonal jurisdiction. In December 2021, the Plaintiff against usrefiled the case in the United States District Court for the Northern District of Alabama, resultedMaryland, captioned Tang v. Altimmune, Inc., et al., Case No. 8:21-cv-03283 (D. Md.). Plaintiff, who is representing himself, asserts two causes of action: (1) breach of a prior settlement agreement by “robbing Plaintiff’s properties”; and (2) use by the Company of the “AdHigh system,” which Plaintiff claims is “proprietary.” On April 4, 2022, Defendants filed a motion to dismiss all claims in Plaintiff’s operative complaint, which motion is pending. We believe the allegations in the entry of a Final Consent Judgmentoperative complaint are without merit and Permanent Injunction on August 25, 2016 (the “Alabama Judgment”). Inintend to vigorously defend the Alabama Judgment, the court declared, among other things, that we owned the DVD technology that Plaintiff had developed during his employment with us, and enjoined Plaintiff from “using or disclosing any Proprietary Information or Innovations relating to the DVD technology and any associated intellectual property rights” without our written consent. litigation.
Item 4. Mine Safety Disclosures Not applicable. PART II PART IIItem 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities Market Information Our common stock trades on the NASDAQ Global Market under the symbol “ALT”. Prior to the completion of the Mergers, PharmAthene’s common stock traded on the NYSE American (formerly the NYSE MKT) under the symbol “PIP.” Holders As of February 23, 2021,24, 2023, we had 172178 record holders of our common stock. The number of record holders is based on the actual number of holders registered on the books of our transfer agent and does not reflect holders of shares in “street name” or persons, partnerships, associations, corporations or other entities identified in security position listings maintained by depository trust companies. Dividends Other than the special dividend immediately prior to the Mergers, we have never declared or paid any cash dividends on our capital stock. We intend to retain future earnings, if any, to finance the operation and expansion of our business and do not expect to pay any cash dividends in the foreseeable future. Any future determination related to our dividend policy will be made at the discretion of our board of directors after considering our financial condition, results of operations, capital requirements, business prospects and other factors the board of directors deems relevant, and subject to the restrictions contained in any future financing instruments. Securities Authorized for Issuance under Equity Compensation Plans Information about our equity compensation plans is contained in Part III, Item 12 of this Annual Report under the heading Equity Compensation Plans and is incorporated herein by reference. Recent Sales of Unregistered Securities Not applicable.
Use of Proceeds
Not applicable.None.
Purchases of Equity Securities by the Issuer and Affiliated Purchases Not applicable.None.
Item 6. Selected Financial Data[Reserved] We are a smaller reporting company and not required to provide this information.
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of OperationsThe following discussion and analysis of our financial condition and results of operations should be read together with our consolidated financial statements and related notes appearing elsewhere in this Annual Report. Some of the information contained in this discussion and analysis contains forward-looking statements that involve substantial risks and uncertainties. See “Forward-looking statements” in Part I of this Annual Report and the section entitled “Risk Factors” in Part I, Item 1A of this Annual Report for a discussion of certain factors that could cause actual results or events to differ materially from the forward-looking statements that we make. Overview Altimmune, Inc. is a clinical stage biopharmaceutical company focused on developing intranasal vaccines, immune modulating therapies and treatments for obesity and liver disease.diseases. Our diverse pipeline includes proprietary intranasal vaccineslead product candidate, pemvidutide (formerly known as ALT-801), is a GLP-1/glucagon dual receptor agonist that is being developed for COVID-19 (AdCOVID), anthrax (NasoShield)the treatment of obesity and influenza (NasoVAX); an intranasal immune modulating therapeutic for COVID-19 (T-COVID); and next generation peptide therapeutics for non-alcoholic steatohepatitis (“NASH”) (ALT-801) and. In addition, we are developing HepTcell, an immunotherapeutic agent designed to achieve a functional cure for chronic hepatitis B (HepTcell). Our business is the result of a merger between PharmAthene, Inc. (“PharmAthene”) and the business previously known as Altimmune, Inc. (“Private Altimmune”). On May 4, 2017, Private Altimmune merged with PharmAthene in a series of mergers and reorganizations (collectively, the “Mergers”) pursuant to an Agreement and Plan of Merger and Reorganization (the “PharmAthene Merger Agreement”) dated January 18, 2017, among Private Altimmune, PharmAthene, its wholly owned acquisition subsidiaries Mustang Merger Sub Corp I Inc. and Mustang Merger Sub II LLC. Upon closing of the Mergers, all equity instruments of Private Altimmune were exchanged for corresponding equity instruments of PharmAthene. Prior to the Mergers, PharmAthene was a publicly traded biodefense company engaged in Phase 2 clinical trials.B. Except where the context indicates otherwise, references to “we,” “us,” “our,” “Altimmune” or the “Company” refer for periods prior to the completion of the Mergers, to Private Altimmune and its subsidiaries, and for periods following the completion of the Mergers to the combined company and its subsidiaries.
AdCOVIDFiscal Year 2022 Business Update
AdCOVID isIn December 2021, an intranasal COVID-19 vaccine candidate designed to guard the respiratory tract from viral invasion and to provide downstream protection against viral spread through stimulation of both mucosal and systemic antibodies (IgA and IgG) as well as cell-mediated immunity. By stimulating mucosal immunity in the nasal cavity, a key point of entry and replication for the SARS-CoV-2 virus, AdCOVID has the potential to defend against both the infection and spread of the virus to others. AdCOVID’s intranasal delivery method provides an easier route of administration than an injection and may eliminate the need for administration by trained medical personnel. We believe nasal mucosal immunity has the potential to provide an effective protection at the site of viral entry and early replication, and may block transmission by shed virus. We believe AdCOVID has the potential to meet most of the World Health Organization’s published preferred attributes for a COVID-19 vaccine, including single dose, rapid onset of protection, immunity lasting one year, non-injected and temperature stability. In particular, data from our NasoVAX clinical trials demonstrated a strong serological response at two weeks that remained unchanged at 400 days, after a single dose. In addition, since it is expected to have extended stability at room temperature, AdCOVID may avoid the need for costly cold chain logistics. During 2020, we completed initial preclinical mouse studies in collaboration with the University of Alabama at Birmingham (“UAB”) and began manufacturing AdCOVID during the third quarter of 2020. We submitted an InvestigationInvestigational New Drug application (“IND”) withfor obesity was submitted to the U.S. Food and Drug Administration (“FDA”) and on January 31, 2022 we received FDA clearance for a Phase 2 trial in this indication.
In April 2022, we announced that we had enrolled the first subject in the fourth quarter of 2020 and received clearance from the FDA in February 2021 to initiate a48-week Phase 1 clinical2 MOMENTUM trial of AdCOVID. The Phase 1 clinical trial is currently enrolling and will evaluateevaluating the safety and immunogenicityefficacy of AdCOVIDpemvidutide in up to 180 healthy adult volunteers between the ages of 18 and 55,subjects with data expected in the second quarter of 2021. T-COVID
T-COVID is an intranasal immune modulating therapeutic candidate based on the same replication-deficient adenovirus 5 (“RD-Ad5”) vector technology behind our other intranasal vaccine candidates, but it acts through a different mechanism. In preclinical studies sponsored by the National Institute of Allergy and Infectious Diseases, intranasal administration of RD-Ad5 vectors modulated the innate immune response to lethal challenge with a respiratory virus in mice and protected them from death. The immunomodulatory effects resulted in significantly decreased cellular inflammation and lower concentrations of IL-6 and other inflammatory cytokines in the lungs of treated animals compared to controls. Excessive production of inflammatory cytokines like IL-6 has been associated with the lung pathology and death in COVID-19. The protective effects were independent of any specific immunityobesity or vaccine effects against the challenge virus. These protective effects were only observed with intranasal administration of RD-Ad5, and intramuscular administration provided no survival benefit.
With the support of the U.S. Army Medical Research & Development Command in collaboration with the Medical Technology Enterprise Consortium, we initiated a placebo-controlled Phase 1/2 double-blind clinical trial to evaluate the potential of T-COVID to
prevent clinical worsening in patients with early COVID-19 during 2020.overweight. The trial is expected to enroll 96 community-based patients who are 18 years and older that present with fever, cough, or shortness of breath, with onset of symptoms within 48 hours, and a diagnosis of COVID-19 within 24 hours.being conducted at approximately 30 sites in the U.S. The randomized, placebo-controlled trial consists of three cohorts of increasing age and risk for complications of COVID-19, and patients areenrolled approximately 320 non-diabetic subjects randomized 1:1:1:1 to NasoVAXreceive either 1.2 mg, 1.8 mg, 2.4 mg pemvidutide or placebo administered asweekly for 48 weeks. No dose titration is being used with the 1.2 mg or 1.8 mg dose, while a single 0.5ml nasal spray within 24 hours of diagnosis.short 4-week dose titration is being employed at the 2.4 mg dose. The primary endpoint of the trial is the proportionrelative (percent) change in body weight at 48 weeks compared to baseline, with additional readouts including metabolic and lipid profiles, cardiovascular measures and glucose homeostasis. The trial is being conducted with adjunctive diet and exercise interventions that is typical in weight loss studies.
In August 2022, we announced that 167 subjects had been randomized and we further announced on September 28, 2022 the first dosing of patients with clinical worsening, defined as a 4% decrease in pulse oxygen saturation, or the need for hospitalization. Secondary endpoints will measure the average decrease in resting pulse oxygen saturation, average increase in resting pulse rate and proportion of patients requiring oxygen supplementation and mechanical ventilation. In order to expedite the study, we have been permitted by the FDA to use existing lots of NasoVAX, which is an identical vector to T-COVID, in lieu of newly manufactured T-COVID. The protocol was recently amended such that 40% of patientsall subjects in the final cohort, in which efficacy will primarily be assessed, to be either 65 years and older or have risk for complications of COVID-19. Based on these protocol changes,MOMENTUM trial. In September 2022, we expect to receiveannounced the topline results from the Phase 1/2 trial in the second quarter of 2021. NasoShield
NasoShield is an anthrax vaccine product candidate designed to provide rapid and stable protection after a single intranasal administration. It is being developed with the support of the U.S. Biomedical Advanced Research and Development Authority (“BARDA”) for post-exposure prophylaxis against anthrax following exposure to aerosolized B. anthracis spores. After an individual has been exposed to the spores that cause anthrax, B. anthracis bacteria multiply and release toxins within the host. Although antibiotic therapy is effective at eliminating the actively growing bacteria, vaccination is necessary to protect against the germination of dormant spores after the cessation of antibiotic therapy. Because NasoShield is intended to protect against anthrax after a single intranasal dose, we believe it may be a convenient and simple alternative to the only approved vaccine, which must be given as a series of three injections over 1 month. We believe the simplified immunization route and schedule, together with the reliable stability at ambient temperature may allow NasoShield to be deployed in an anthrax event more easily and faster than the currently approved vaccine. We commenced aour 12-week Phase 1b clinical trial of NasoShieldpemvidutide in adults in 2020 which builds on the Phase 1a trial completed in 2018 and evaluates the effect of modified methods of intranasal dosing on NasoShield safety and immunogenicity. Results are expected in the first quarter of 2021.
NasoVAX
NasoVAX is a recombinant intranasal vaccine product candidate that is being developed for both seasonal and pandemic use. NasoVAX is believed to simultaneously activate the humoral, mucosal and cellular immune arms which may enable a more comprehensive immune response. The data from our Phase 2a trialsubjects with a monovalent NasoVAX vaccine indicated that NasoVAX was generally well-tolerated and achieved 100% seroprotection with serum antibody responses, which was comparable to published results of a licensed injected influenza vaccine. Statistically significant increases in mucosal antibody were noted as well as a robust T cell response directed against influenza. Approximately half of the subjects from the highest dose were evaluated between 12 and 14 months after initial dosing for additional immunogenicity assessment. The durability data show that the immune response elicited by NasoVAX was stable with no overall change in the antibody titer or level of seroprotection over an average of 13 months. The combination of serum antibody, mucosal antibody and T-cell response in combination with the durability data provides the potential for improved protection against influenza and suggests that NasoVAX could have a great impact on flu symptoms and shedding of the influenza virus. We are currently evaluating the development path of NasoVAX.
ALT-801
We completed an acquisition in July 2019 of all of the equity interests of Spitfire Pharma, Inc. (“Spitfire”). Spitfire was a privately held, preclinical pharmaceutical company with the primary asset being a novel peptide-based dual GLP-1/glucagon receptor agonist for the treatment of NASH. We refer to this product candidate as ALT-801, and it is designed to treat the obesity and metabolic dysfunction that causes NASH. NASH, the most severe form of non-alcoholic fatty liver disease (“NAFLD”), involves multiple metabolic pathways leading to the abnormal accumulation of liver fat, toxic lipid metabolites, and inflammation, leading to fibrosis or eventually liver cancer. NAFLD is present in up to 90% of obese patients, and approximately 20% of NAFLD patients progress to NASH. In addition, up to 40% of NASH patients develop NAFLD recurrence one year after liver transplant, which we believe indicates that the underlying metabolic disease is still present after transplant. We believe the treatment of obesity is the cornerstone of treating NASH and the principal morbidities of NASH.
ALT-801’s dual agonist mechanism of action is designed to combine the activity of GLP-1 for the reduction of appetite and inflammation, with the direct activity of glucagon on the liver, including increased energy expenditure, adipose browning, lipolysis and mobilization of the liver fat. ALT-801 incorporates the EuPort domain, which is designed to enhance pharmacokinetics for tolerability in the gastrointestinal tract and permit weekly dosing. As observed in a well-established preclinical model of the disease, ALT-801 is capable of inducing significant weight loss with concomitant decreases in liver fat, inflammation and fibrosis, with superior results compared to elafibranor and semaglutide. In addition, ALT-801 demonstrated improved metabolic function and exhibited pleiotropic effects in our preclinical testing across multiple metabolic pathways that are involved in NASH. We also
observed in preclinical studies that ALT-801 resulted in more profound suppression of genes associated with steatosis, inflammation and stellate cell fibrosis by RNA sequencing compared to elafibranor and semaglutide. We commenced a Phase 1 clinical trial in Australia in overweight and obese adult volunteers in the fourth quarter of 2020.. The trial is expected to have both single-ascending and multiple-ascending dose arms over a 6 week treatment duration, with data readouts planned for each arm in the second quarter of 2021. The endpoints of the Phase 1a trial are expected to be safety, tolerability and pharmacokinetics, with a preliminary readout on weight loss, resting energy expenditure, liver fat by MRI-PDFF and glucose homeostasis. Pending interim results of the single-ascending dose arms, we intend to extend the multiple-ascending dose arms by 6 weeks for a twelve-week, parallel-dosing Phase 1b clinical trial. The endpoints of the Phase 1b trial are expected to be safety, tolerability, pharmacokinetics, weight loss, decrease in liver fat (as measured by the MRI-PDFF standard) and lean body mass, as well as other metabolic biomarkers. If successful, we expect data from the Phase 1b study Q3 2021.
HepTcell
HepTcell is an immunotherapeutic product candidate for patients chronically infected with the hepatitis B virus (“HBV”). Approximately 300 million people worldwide live with chronic HBV infection, including approximately 2.2 million in the United States. Chronic HBV infection can lead to serious complications, including cirrhosis and liver cancer. Approximately 780,000 people die per year worldwide due to cirrhosis and liver cancer. HepTcell is designed to drive CD4+ and CD8+ T-cell responses against all HBV genotypes in patients of all ethnic backgrounds. Stimulating T-cell responses in chronically infected HBV patients has been challenging because chronic infection with HBV and elevated hepatitis B surface antigen (HBsAg) levels strongly diminishes T-cell immunity directed against the virus. HepTcell focuses the immune system on discrete highly conserved regions of the HBV proteome. We believe our approach allows HepTcell to break immune tolerance by activating T-cells against critical viral sequences with decreased probability of immune escape due to viral mutation. HepTcell is based on our synthetic peptide technology platform and is given by intramuscular injection. In 2018, we completed a Phase 1 trial in the United Kingdom and South Korea in adult patients with chronic HBV. The HepTcell Phase 1 trial was a double-blinded,double-blind, placebo-controlled study. Subjects were randomized dose-escalation study that enrolled 61 subjects1:1:1:1 to 1.2 mg, 1.8 mg, 2.4 mg pemvidutide or placebo administered weekly for 12 weeks. No dose titration was used with chronic HBV who were HBeAg-negative and well-controlled on licensed antivirals. A total of 41 patients received one of two1.2 mg or 1.8 mg dose, levels of HepTcell, with and without IC31TM,while a depot-forming TLR9 adjuvant developed by Valneva SE, while 20 control patients received either placebo or IC31 alone. Patients received three injections each 28 days apart and were followed for six months aftershort 4-week dose titration was employed at the final2.4 mg dose. All dose combinations showed excellent tolerability and met the primary endpoint of safety. In the two adjuvanted HepTcell arms, T-cell responses against HBV markedly increased over baseline compared to placebo.
We initiated a Phase 2 study during Q4 2020 in the United States, Canada and Europe that is a double-blind, randomized, placebo-controlled study of 80 adult patients with HBeAg-negative inactive CHB and HBsAg ≤ 100 IU/mL. HepTcell will be administered in 6 doses at 4-week intervals for 24 weeks, and patients will be followed for one year to evaluate safety and durability of response. The primary efficacy endpoint is virological response, defined as a 1-logwas the percent (%) reduction in HBsAg levelsliver fat content (“LFC”) from baseline, and the key secondary efficacy endpoint was the % weight loss from baseline, both at 24 weeks. Secondary12 weeks of treatment. The trial was conducted without adjunctive diet and exercise interventions that are the standard for obesity trials.
In December 2022 we announced the topline results from our 24-week (12-week extension) Phase 1b clinical trial of pemvidutide in subjects with NAFLD. The trial was conducted without adjunctive diet and exercise interventions and the double-blinding of the trial was maintained during the extension study. The same endpoints as the 12-week parent NAFLD trial were employed, with a primary efficacy endpoint of percent (%) reduction in liver fat content; key secondary endpoints include reactivation of anti-HBV T cell responses, HBsAg clearance,were reduction in liver inflammation, as measured by serum alanine aminotransferase (“ALT”) levels and other assessments of virologic response. We expect data from this study in the first quarter of 2022.corrected T1 (“cT1”), and percent weight loss. Impact of COVID-19 We are closely monitoring how the spread of COVID-19, including any resurgences or the coronavirus disease (“COVID-19”)emergence of new variants, is affecting our employees, business, preclinical studies and clinical trials. In responseWe have reopened our executive office to allow employees to return to the COVID-19 pandemic, we have closed our executive officesoffice based on an approach that is intended to comply with certain employees continuing theirfederal and state guidelines, with a focus on employee safety and optimal work outside of our offices and travel for all employees has been restricted. Essential laboratory staff continue to work onsite with enhanced safety measures.environment. We are continuing our regular interactions with the FDA and other regulatory agencies and, based on current information, we do not anticipate COVID-19 to materially affect our regulatory timelines for NasoShield, T-COVID, AdCOVID, ALT-801our ongoing clinical trials. Furthermore, as a government contractor, we are subject to the federal government vaccination mandate, which requires federal contractor employees, except in certain limited circumstances, to be vaccinated against COVID-19 by December 8, 2021. While the vaccination mandate remains subject to the interpretation of various government agencies and HepTcell.other entities, and questions remain regarding the specific application of the vaccination mandate, we are continuing to develop and implement health, safety, employment and operational protocols in order to timely comply with the vaccination mandate. As of and for the year ended December 31, 2022, the vaccination mandate has not had a material impact on our employees or operations. Although operations have not been materially affected by the COVID-19 pandemic as of and for the year ended December 31, 2022, at this time, however, there is significant uncertainty relating to the trajectory of the pandemic and the impact of related responses, and disruptions caused by the COVID-19 pandemic may result in difficulties or delays in initiating, enrolling, conducting or completing our planned and ongoing trials and the incurrence of unforeseen costs as a result of disruptions in clinical supply or preclinical study or clinical trial delays. The impact of COVID-19 on our future results will largely depend on future developments, which are highly uncertain and cannot be predicted with confidence, such as the ultimate geographic spread of the disease, the duration of the pandemic, travel restrictions and social distancing in the United States and other countries, business closures or business disruptions, the ultimate impact on financial markets and the global economy, and the effectiveness of actions taken in the United States and other countries to contain and treat the disease. In addition, a recurrence of COVID-19 cases, or variants thereof, could cause other widespread or more severe impacts depending on where infection rates are highest. We continue to monitor developments as we deal with the disruptions and uncertainties relating to the COVID-19 pandemic. See “Risk Factors— Our business, results of operations and financial condition may be adversely affected by the widespreadA pandemic, epidemic or outbreak of an illness or any other communicableinfectious disease or any other public health crisis, includingin the ongoingUnited States such as the COVID-19 pandemic.pandemic may adversely affect our business.” in Part I, Item 1A of this Annual Report on Form 10-K.
U.S. Government ContractsRecent Global Events
Russia and GrantsUkraine Conflict In June 2020,The military conflict in Russia and Ukraine that began in February 2022 continues as of the date of this annual report. As the conflict continues to evolve, we were awarded $4.7 millionare closely monitoring the impact on our business. The conflict, and the sanctions and counter-sanctions imposed in response to it, have created increased economic uncertainty and operational complexity globally. While we have no direct exposure to Russia and Ukraine, and do not at the moment believe the situation will have a material impact on our operating results, we are monitoring any broader economic impact from the U.S. Army Medicalsituation. Should the conflict continue or escalate, it could have a significant negative effect on the global economy or on our operations, including continued inflationary pressures on raw materials, supply chain and logistics disruptions, volatility in foreign exchange rates and interest rates and heightened cybersecurity threats.
Financial Operations Overview The consolidated financial information presented below includes the accounts of Altimmune, Inc., Altimmune UK, Ltd, Spitfire Pharma, LLC. and Altimmune AU Pty, Ltd. All intercompany accounts and transactions have been eliminated in consolidation. Revenue We have not generated any revenues from the sale of any products to date. Our revenue in previous years consisted primarily of government and foundation grants and contracts that support our efforts on specific research projects. These grants and contracts generally provided for reimbursement of approved costs as those costs were incurred by us. Research & Development Command (“USAMRDC”grants and contracts and the related accounts receivable were recognized as earned when reimbursable expenses are incurred and the performance obligation was complete. Payments received in advance of services being provided were recorded as deferred revenue. We are closing out one of the remaining such contracts and any revenue reported during the year ended December 31, 2022 was for indirect rate adjustments. Research and development expenses Research and development expenses consist primarily of costs incurred for the development of our product candidates, which include: | ● | expenses incurred under agreements with contract research organizations (“CROs”) and investigative sites that conduct our clinical trials; |
| ● | employee-related expenses, including salaries, benefits, travel and stock-based compensation expense; |
| ● | costs associated with preclinical and clinical activities and regulatory operations, including the cost of acquiring, developing and manufacturing clinical trial materials; and |
| ● | depreciation and other expenses, which include direct and allocated expenses for insurance, consultants, legal fees and other supplies. |
Research and development costs are expensed as incurred. Costs for certain development activities are recognized based on an evaluation of the progress to fundcompletion of specific tasks using information and data provided to us by our Phase 1/2vendors, CROs and clinical trialsites. We cannot determine with certainty the duration and completion costs of T-COVID.the current or future clinical trials of our product candidates or if, when or to what extent we will generate sales from the commercialization of any of our product candidates if they receive regulatory approval. The competitive award was granted by USAMRDCsuccessful development of our product candidates is highly uncertain and may never result in collaborationapproved products. The duration, costs and timing of clinical trials and development of our product candidates will depend on a variety of factors, including: | ● | scope, rate of enrollment and expense of our ongoing, as well as any additional, clinical trials, and other research and development activities; |
| ● | significant and potentially changing government regulation; and |
| ● | the timing and receipt of regulatory approvals, if any. |
A change in the outcome of any of these variables with respect to the development of a product candidate could mean a significant change in the costs and timing associated with the development of that product candidate. For example, if the FDA, or another regulatory authority were to require us to conduct clinical trials beyond those that we currently anticipate will be required for the completion of clinical development of a product candidate, we could be required to expend significant additional financial resources and time on the completion of clinical development. We plan to increase our research and development expenses for the foreseeable future as we continue the development of clinical and preclinical candidates. Our current active and planned research and development activities include the following: | ● | completion of a Phase 2 clinical trial for pemvidutide in obesity; |
| ● | initiation of a Phase 2 clinical trial for pemvidutide in NASH; |
| ● | conduct of clinical trials and nonclinical safety studies for pemvidutide; |
| ● | completion a Phase 2 clinical trial for HepTcell; and |
| ● | manufacture of clinical trial materials in support of our clinical trials. |
To date, a significant portion of our research and development efforts have been related to the development of pemvidutide and HepTcell, as well as for NasoShield, NasoVAX, AdCOVID and T-COVID programs which were discontinued in 2021. We do not allocate personnel-related costs, costs associated with our general research platform improvements, depreciation or other indirect costs to specific programs. General and Administrative Expenses General and administrative expenses consist primarily of salaries and related costs for personnel, including stock-based compensation and travel expenses for our employees in executive, operational, finance and human resource functions. Other general and administrative expenses include insurance expenses, facility-related costs and professional fees for directors, accounting and legal services, and expenses associated with obtaining and maintaining our intellectual property. We anticipate that our general and administrative expenses will increase in the future as we increase our headcount to support our continued research and development activities. We also anticipate increased expenses related to audit, legal, regulatory and tax-related services associated with maintaining compliance with exchange listing and the SEC requirements, director and officer insurance, investor relations costs and other costs associated with being a public company. Additionally, if and when we believe a regulatory approval of the first product candidate appears likely, we anticipate an increase in staffing and related expenses as a result of our preparation for commercial operations, especially as it relates to the sales and marketing of our product candidates. Results of Operations Comparison of years ended December 31, 2022 and December 31, 2021: | | | | | | | | | | | | | | | Year Ended December 31, | | (in thousands) | | 2022 | | 2021 | | Increase (Decrease) | | Revenue | | $ | (68) | | $ | 4,410 | | $ | (4,478) | | (102) | % | Operating expenses: | | | | | | | | | | | | | Research and development | | | 70,538 | | | 74,541 | | | (4,003) | | (5) | % | General and administrative | | | 17,134 | | | 15,413 | | | 1,721 | | 11 | % | Impairment loss on construction-in-progress | | | — | | | 11,370 | | | (11,370) | | 100 | % | Total operating expenses | | | 87,672 | | | 101,324 | | | (13,652) | | (13) | % | Loss from operations | | | (87,740) | | | (96,914) | | | 9,174 | | 9 | % | Other income (expense): | | | | | | | | | | | | | Interest expense | | | (8) | | | (5) | | | (3) | | (60) | % | Interest income | | | 2,870 | | | 203 | | | 2,667 | | 1,314 | % | Other income (expense), net | | | (32) | | | (374) | | | 342 | | 91 | % | Total other income (expense), net | | | 2,830 | | | (176) | | | 3,006 | | 1,708 | % | Net loss before income taxes | | | (84,910) | | | (97,090) | | | 12,180 | | 13 | % | Income tax expense (benefit) | | | (197) | | | — | | | (197) | | | | Net loss | | $ | (84,713) | | $ | (97,090) | | $ | 12,377 | | 13 | % |
Revenue For the year ended December 31, 2021, revenue consisted primarily of research grants in the United States from Medical Technology Enterprise Consortium (“MTEC”), a 501(c)(3) biomedical technology consortium working in partnership with the Department of Defense (“DoD”). Under the contract, MTEC pays us a firm fixed fee based upon the achievement of certain milestones for conductour T-COVID product candidate and completion of a Phase 1/2 study and research and development work on the replication-deficient adenovirus 5 (“RD-Ad5”) vector vaccine platform. For the year ended December 31, 2020, we have recognized approximately $4.2 million of grant revenue under this contract. In July 2016, we signed a five-year contract with Biomedical Advanced Research and Development Authority (“BARDA”). The contract, as amended, has a total value for our NasoShield vaccine product candidate. These grants consisted of up to $133.7 millionfirm fixed fee contracts based on milestones and is used to fund clinical development of NasoShield. Under the contract, BARDA pays uscost reimbursement contracts, with a fixed fee based on either costs incurred or milestones met. Our T-COVID and reimburses certain costs forNasoShield programs were discontinued as of the research and developmentend of an Ad5-vectored, protective antigen-based intranasal anthrax vaccine through cGMP manufacture and conduct of a Phase 1 clinical trial dose ranging assessment of safety and immunogenicity. The contract consists of an initial base performance period providing approximately $27.8 million in funding for the period July 2016 through June 2021. BARDA has seven options to extend the contract to fund certain continued development and manufacturing activities for the anthrax vaccine, including Phase 2 clinical trials. Each option, if exercised by BARDA, would provide additional funding ranging from approximately $1.1 million to $34.4 million for a three-year period beginning January 2021. For the
year ended December 31, 2022, as a result of adjustments to prior year cost reimbursements under BARDA, we reported negligible negative revenue. Revenue decreased by $4.5 million, or 102% for the year ended December 31, 2020,2022 as compared to the year ended December 31, 2021. The decrease was primarily the result of the discontinuation of development work on both T-COVID and NasoShield programs as described above. Research and development expenses Research and development expenses for the years ended December 31, 2022 and 2021 consisted primarily of expenses related to product candidate development. Research and development expenses for the years ended December 31, 2022 and 2021 are summarized as follows: Research and development expenses decreased by $4.0 million, or 5%, during the year ended December 31, 2022 as compared to the year ended December 31, 2021. The decrease in expense was primarily due to: | ● | a decrease of $35.1 million due primarily to development activities for our COVID-19 programs, which included AdCOVID and T-COVID (which were discontinued in 2021); |
| ● | a decrease of $0.6 due to change in the fair value of contingent consideration liability with respect to the acquisition of pemvidutide, which was fully paid on June 10, 2022; |
| ● | an increase of $26.2 due to the development activities for pemvidutide primarily due to the ongoing NAFLD trials and the MOMENTUM Phase 2 trial in obesity; |
| ● | an increase of $2.4 million due to development activities for HepTcell; and; |
| ● | a net increase of $3.1 million due primarily to costs associated with non-project specific research and development costs including employee compensation and facility costs. |
General and administrative expenses General and administrative expenses increased by $1.7 million, or 11%, during the year ended December 31, 2022 as compared to the year ended December 31, 2021. The increased expense is primarily due to a $1.7 million increase in stock compensation and other labor-related expenses. Impairment loss on construction-in-progress Impairment loss on construction-in-progress of $11.4 million reported during the year ended December 31, 2021 represents non-cash impairment charges recorded for assets that were previously capitalized in connection with the construction of the Lonza facility and subsequent discontinuation of AdCOVID. There were no impairment charges reported during the year ended December 31, 2022. Total other income (expense), net Total other income (expense), net increased by $3.0 million during the year ended December 31, 2022 as compared to the year ended December 31, 2021, primarily due to $2.7 million increase in interest income earned on our cash equivalents and short-term investments and $0.3 million decrease in loss from foreign currency exchange. Income tax benefit Income tax benefit of $0.2 million related to interest received and receivable on income tax refunds was recorded during the year ended December 31, 2022. Other than the income tax benefit related to interest, we did not record an income tax benefit in the year ended December 31, 2022 and 2021 due to a full valuation allowance. Liquidity and Capital Resources Overview Our primary sources of cash for the year ended December 31, 2022 were from equity transactions. Our cash, cash equivalents, restricted cash and short-term investments were $184.9 million as of December 31, 2022. We believe, based on the operating cash requirements and capital expenditures expected for 2023 and 2024, our cash on hand at December 31, 2022, together with expected cash receipts from our income tax refunds and R&D incentives, are sufficient to fund operations for at least a twelve-month period from issuance date of our December 31, 2022 consolidated financial statements. We have not generated any revenues from the sale of any products to date, and there is no assurance of any future revenues from product sales. In the past, our sources of revenue have consisted of grant revenues under our arrangements with BARDA for the development of NasoShield, MTEC for a clinical trial and development work on T-COVID, and to a lesser degree from other licensing arrangements. The MTEC contract was closed out in June 2021 and we are currently closing out the BARDA contract which was not renewed after December 31, 2021. We have incurred significant losses since we commenced operations. As of December 31, 2022, we had an accumulated deficit of $377.9 million. In addition, we have recognized approximately $3.1 millionnot generated positive cash flows from operations. We have had to rely on a variety of grant revenue underfinancing sources, including the current BARDA contract.issuance of debt and equity securities. As capital resources are consumed to fund our research and development activities, we may require additional capital beyond our currently anticipated amounts. In order to address our capital needs, including our planned clinical trials, we must continue to actively pursue additional equity or debt financing, government funding, and monetization of our existing programs through partnership arrangements or sales to third parties. FinancingSources of Liquidity
Public Offering On July 16, 2020, we offered and sold (i) 3,369,564 shares of our common stock, at a price to the public of $23.00 per share, and (ii) pre-funded warrants to purchase 1,630,436 shares of our common stock at an exercise price equal to $0.0001 per share (the “Pre-Funded Warrants”), at a price to the public of $22.9999 per share of common stock underlying the Pre-Funded Warrants (equal to the public offering price per share of Common Stock, minus the exercise price of each Pre-Funded Warrant). The Pre-Funded Warrants are exercisable at any time, provided that each Pre-Funded Warrant holder will be prohibited from exercising such Pre-Funded Warrants into shares of our common stock if, as a result of such exercise, the holder, together with its affiliates, would own more than 4.99% of the total number of shares of our common stock then issued and outstanding, which percentage may change at the holders’ election to any other number less than or equal to 19.99% upon 61 days’ notice to us. The gross proceeds of this offering were approximately $132.2 million, which includes the exercise in full of the underwriters’ option to purchase an additional 750,000 shares of common stock, before deducting underwriting discounts and commissions and offering expenses during the third quarter of 2020. The net proceeds of this offering were approximately $124.0 million, after deducting underwriting discounts and commissions and offering expenses payable by us. As of December 31, 2022, 760,870 of the Pre-Funded Warrants were exercised, leaving 869,566 remaining Pre-Funded Warrants unexercised. Shelf Registration On December 31, 2020, we filed a shelf registration statement on Form S-3, which was declared effective by the SEC on January 11, 2021. This shelf registration statement covered the offering, issuance and sale by us of up to an aggregate of $250.0 million of our common stock, preferred stock, debt securities, warrants, rights and units (the “2021 Shelf”). As of December 31, 2022, approximately $125.0 million remained available to be sold under the 2021 Shelf. At-the-Market Offering On March 27, 2020,February 25, 2021, we entered into an Equity Distribution Agreement (the “Agreement”“2021 Agreement”) with JMPPiper Sandler & Co., Evercore Group L.L.C. and B. Riley Securities, LLC,Inc., serving as placement agentsales agents (the “Placement Agent”“Sales Agents”) with respect to an at-the-market offeringofferings program under which we may offeroffered and sell, from time to time at our sole discretion,sold shares of our common stock, par value $0.0001 per share (the “Common Stock”), having an aggregate offering price of up to $50.0$125.0 million (the “Shares”) through the Placement Agent (the “Offering”). We offered Shares having an aggregate offering priceSale Agents from the 2021 Shelf. During the prospectus supplement filed with the SEC on March 27, 2020. On June 1, 2020, we filed an amendment to the Agreement which amended the prospectus supplement dated March 27, 2020 to increase the aggregate offering price to $50.0 million. As ofyear ended December 31, 2020,2022, we sold 5,594,4555,204,415 shares of Common Stock under the 2021 Agreement resulting in $48.2approximately $56.2 million in net proceeds. As of December 31, 2022, we sold 10,004,869 shares of Common Stock under the 2021 Agreement resulting in approximately $121.0 million in net proceeds. As of December 31, 2022, there were no remaining shares available for issuance under the 2021 Agreement. Cash Flows The following table provides information regarding our cash flows for the years ended December 31, 2022 and 2021: | | | | | | | | | | | | Year Ended December 31, | (in thousands) | | 2022 | | 2021 | | Increase (Decrease) | Net cash (used in) provided by: | | | | | | | | | | Operating activities | | $ | (62,586) | | $ | (78,238) | | $ | 15,652 | Investing activities | | | (73,399) | | | 87,523 | | | (160,922) | Financing activities | | | 56,781 | | | 65,098 | | | (8,317) | Net (decrease) increase in cash and cash equivalents and restricted cash | | $ | (79,204) | | $ | 74,383 | | $ | (153,587) |
Operating Activities Net cash used in operating activities was $62.6 million for the year ended December 31, 2022 compared to $78.2 million during the year ended December 31, 2021. Our sources of cash provided by operations during the year ended December 31, 2022 was primarily cash receipts of R&D incentive credits and income tax refunds. The primary uses of cash from our operating activities include payments for labor and labor-related costs, professional fees, research and development costs associated with our clinical trials and other general corporate expenditures. The decrease in cash used in operating activities of $15.7 million is due to a decrease in net loss as adjusted for noncash items of $1.3 million and changes in working capital accounts of $14.4 million. Investing Activities Net cash used in investing activities was $73.4 million for the year ended December 31, 2022 compared to $87.5 million during the year ended December 31, 2021. The net cash used in investing activities during 2022 was primarily due to purchase of short-term investments, net of maturities. The net cash provided by investing activities during the year ended December 31, 2021 was primarily due to net proceeds from short-term investments activities of $99.8 million, partially offset by purchases of property and equipment of $12.1 million related to Lonza. Financing Activities Net cash provided by financing activities was $56.8 million for the year ended December 31, 2022 compared to $65.1 million during the year ended December 31, 2021. The net cash provided by financing activities during 2022 was primarily the result of the receipt of $56.2 million in net proceeds which completedfrom the At-the-Market Offering. Registered Direct Offering
On March 12, 2019, we issued a combined totalissuance of 4,361,370 common units and pre-funded units to certain institutional investors in a registered direct offering (the “Registered Direct Offering”). Each common unit in the Registered Direct Offering was sold at a price of $3.21 and consisted of one share of our common stock and 0.70from our at-the-market offerings program. The net cash provided by financing activities during 2021 was primarily the result of a warrant to purchase one sharethe receipt of our$64.7 million in net proceeds from the issuance of common stock at an exercise price of $3.21. Each warrant sold in the Registered Direct Offering was exercisable immediately and expires five years from the date of issuance. Each pre-funded unit in the Registered Direct Offering was sold at a public offering price of $3.20 and consisted of a pre-funded warrant to purchase one share of our common stock at an exercise price of $0.01 per share and 0.70 of a warrant to purchase one share of our common stock at an exercise price of $3.21. The pre-funded warrants were immediately exercisable and were able to be exercised at any time. All of the pre-funded warrants were exercised prior to March 31, 2019. The net proceeds of the Registered Direct Offering were approximately $12.7 million, after deducting the underwriting discount and estimated offering expenses payable by us. The Registered Direct Offering triggered an adjustment to the exercise price of the warrants issued in the 2018 Unit Offering from $4.1798 to $2.7568.at-the-market offerings program.
CurrentCapital Resources
We have financed our operations to date principally through our equity offerings and proceeds from issuances of our preferred stock, common stock and warrants. We secured net proceeds of $213.2 million through our Public Offering, At-the-Market Offering, and exercise of warrants during the year endedAt December 31, 20202022, we had $184.9 million of cash, cash equivalents, restricted cash and $12.7 million through equity sales during the year ended December 31, 2019.short-term investments. Accordingly, management believes that we have sufficient capital to fund our plan of operations for at least a twelve-month period from the issuance date of our December 31, 20202022 consolidated financial statements. However, in order to address our capital needs in the long-term, including our planned clinical trials, we must continue to actively pursue additional equity or debt financing, government funding and monetization of our existing programs through partnership arrangements or sales to third parties. Financial Operations Overview
The consolidated financial information presented below includes the accounts of Altimmune, Inc., Altimmune UK, Ltd, Spitfire Pharma, LLC., and Altimmune AU Pty, Ltd. All intercompany accounts and transactions have been eliminated in consolidation.
Revenue
To date, we have not generated any product sales. Our revenue consists primarily of government and foundation grants and contracts that support our efforts on specific research projects. These grants and contracts generally provide for reimbursement of approved costs as those costs are incurred by us. Research grants and contracts and the related accounts receivable are recognized as earned when reimbursable expenses are incurred and the performance obligation is complete. Payments received in advance of services being provided are recorded as deferred revenue.
Research and development expenses
Research and development expenses consist primarily of costs incurred for the development of our product candidates, which include:
expenses incurred under agreements with CROs and investigative sites that conduct our clinical trials;
employee-related expenses, including salaries, benefits, travel and stock-based compensation expense;
costs associated with preclinical and clinical activities and regulatory operations, including the cost of acquiring, developing and manufacturing clinical trial materials; and
facilities, depreciation and other expenses, which include direct and allocated expenses for insurance and other supplies.
Research and development costs are expensed as incurred. Costs for certain development activities are recognized based on an evaluation of the progress to completion of specific tasks using information and data provided to us by our vendors, CROs and clinical sites.
We cannot determine with certainty the duration and completion costs of the current or future clinical trials of our product candidates or if, when or to what extent we will generate sales from the commercialization of any of our product candidates if they receive regulatory approval. The successful development of our product candidates is highly uncertain and may never result in approved products. The duration, costs and timing of clinical trials and development of our product candidates will depend on a variety of factors, including:
scope, rate of enrollment and expense of our ongoing, as well as any additional, clinical trials, and other research and development activities;
significant and potentially changing government regulation; and
the timing and receipt of regulatory approvals, if any.
A change in the outcome of any of these variables with respect to the development of a product candidate could mean a significant change in the costs and timing associated with the development of that product candidate. For example, if the FDA, or another regulatory authority were to require us to conduct clinical trials beyond those that we currently anticipate will be required for the completion of clinical development of a product candidate, we could be required to expend significant additional financial resources and time on the completion of clinical development.
We plan to increase our research and development expenses for the foreseeable future as we continue the development of clinical and preclinical candidates. Our current planned research and development activities include the following:
complete a Phase 2 clinical trial for HepTcell;
| •
| complete a Phase 1/2 clinical trial for T-COVID;
|
complete a Phase 1 clinical trial for ALT-801;
complete a Phase 1 clinical trial of AdCOVID;
commence Phase 2 development of NasoShield, contingent on BARDA exercising one or more options under our existing contract;
additional development of NasoVAX, contingent on non-dilutive funding from BARDA or other entities; and
manufacture clinical trial materials in support of our clinical trials.
To date, a significant portion of our research and development efforts have been related to the development of AdCOVID, ALT-801, HepTcell, NasoShield, NasoVAX and T-COVID product candidates. We do not allocate personnel-related costs, costs associated with our general research platform improvements, depreciation or other indirect costs to specific programs.
General and Administrative Expenses
General and administrative expenses consist primarily of salaries and related costs for personnel, including stock-based compensation and travel expenses for our employees in executive, operational, finance and human resource functions. Other general and administrative expenses include facility-related costs and professional fees for directors, accounting and legal services, and expenses associated with obtaining and maintaining our intellectual property.
We anticipate that our general and administrative expenses will increase in the future as we increase our headcount to support our continued research and development activities. We also anticipate increased expenses related to audit, legal, regulatory and tax-related services associated with maintaining compliance with exchange listing and the SEC requirements, director and officer insurance, investor relations costs and other costs associated with being a public company. Additionally, if and when we believe a regulatory approval of the first product candidate appears likely, we anticipate an increase in staffing and related expenses as a result of our preparation for commercial operations, especially as it relates to the sales and marketing of our product candidates.
Critical Accounting Policies and Significant Judgment and Estimates Our management’s discussion and analysis of our financial condition and results of operations is based on our consolidated financial statements, which have been prepared in accordance with U.S. generally accepted accounting principles. The preparation of our consolidated financial statements requires us to make judgments and estimates that affect the reported amounts of assets, liabilities, revenues and expenses, and the disclosure of contingent liabilities in our consolidated financial statements. We base our estimates on historical experience, known trends and events, and various other factors that we believe to be reasonable under the circumstances. Actual results may differ from these estimates under different assumptions or conditions. On an ongoing basis, we evaluate our judgments and estimates in light of changes in circumstances, facts and experience. The effects of material revisions in estimates, if any, will be reflected in the consolidated financial statements prospectively from the date of change in estimates. While our significant accounting policies are described in more detail in the notes to our consolidated financial statements appearing elsewhere in this Annual Report, we believe the following accounting policies used in the preparation of our consolidated financial statements require the most significant judgments and estimates. Impairment of long-lived assets We evaluate our long-lived tangible and intangible assets for impairment whenever events or changes in circumstances indicate that the carrying amount of such assets may not be recoverable. Impairment of long-lived assets other than indefinite lived intangibles is assessed by comparing the undiscounted cash flows expected to be generated by the asset group to its carrying value. We have one in-process research and development (“IPR&D”) asset, HepTcell, that was acquired in 2015. This candidate is a viral pathogen immunotherapy product for the treatment of chronic HBV. Our IPR&D assets areasset is currently non-amortizing. Until such time as the projects areproject is either completed or abandoned, we test those assetsthis asset for impairment at least annually at year end, or more frequently at interim periods, by evaluating qualitative factors which could be indicative of impairment. Qualitative factors being considered include, but are not limited to, the current project status, forecasted changes in the timing or amounts required to complete the project, forecasted changes in timing or changes in the future cash flows to be generated by the completed products, a probability of success of the ultimate project and changes to other market-based assumptions, such as discount rates. If impairment indicators are present as a result of our qualitative assessment, we test those assets for impairment by comparing the fair value of the assets to their carrying value. Upon completion or abandonment, the value of the IPR&D assets will be amortized to expense over the anticipated useful life of the developed products,product, if completed, or charged to expense when abandoned if no alternative future use exists. Key assumptions used in our impairment analysis tests include projected cash flows, a probability of success of the ultimate project and the discount rate.
For the year ended December 31, 2020, we have one IPR&D asset, HepTcell, that was acquired in 2015. This candidate is a viral pathogen immunotherapy product for the treatment of chronic HBV. We performed a qualitative assessment for the IPR&D impairment testing for 2020the years ended December 31, 2022 and 2021 and determined that no impairment indicators were present.
During 2019, we also carried a $1.0 million IPR&D asset for SparVax-L, a two-dose anthrax vaccine funded by the National Institute of Allergy and Infectious Diseases (“NIAID”). As a result of the completion of the NIAID contract and the U.S. government’s funding prioritization of only single dose anthrax vaccine candidates, we abandoned the project and concluded that the full remaining net book value of the SparVax-L IPR&D asset was impaired, resulting in an impairment charge of $1.0 million in Q3 2019.
Fair Value Measurements We follow the guidance in Financial Accounting Standards Board (“FASB”) Accounting Standard Codification Topic 820, Fair Value Measurements and Disclosures(“ASC 820”), which defines fair value and establishes a fair value hierarchy that prioritizes the inputs to valuation techniques used to measure fair value. The hierarchy gives the highest priority to unadjusted quoted prices in active markets for identical assets or liabilities (Level 1 measurements) and the lowest priority to unobservable inputs (Level 3 measurements). The three levels of the fair value hierarchy are described below: Level 1 — Quoted prices (unadjusted) in active markets for identical assets or liabilities that we can access at the measurement date. Level 2 — Inputs other than quoted prices included within Level 1 that are either directly or indirectly observable. If the asset or liability has a specified (contractual) term, a Level 2 input must be observable for substantially the full term. Level 3 — Unobservable inputs developed using estimates of assumptions developed by us, which reflect those that a market participant would use. To the extent that valuation is based on models or inputs that are less observable or unobservable in the market, the determination of fair value requires more judgment. Accordingly, the degree of judgment exercised by us in determining fair value is greatest for instruments categorized in Level 3. A financial instrument’s level within the fair value hierarchy is based on the lowest level of any input that is significant to the fair value measurement. Fair value is a market-based measure considered from the perspective of a market participant rather than an entity-specific measure. Therefore, even when market assumptions are not readily available, our own assumptions are set to reflect those that market participants would use in pricing the asset or liability at the measurement date. We use prices and inputs that are current as of the measurement date, including during periods of market dislocation. In periods of market dislocation, the observability of prices and inputs may change for many instruments. This condition could cause an instrument to be reclassified within levels in the fair value hierarchy. There were no transfers withininto or out of Level 3 of the fair value hierarchy during the years ended December 31, 20202022 and 2019.2021.
Contingent Consideration We record contingent consideration associated with development and regulatory milestones that meets the definition of a liability under FASB Accounting Standards Codification Topic 480, Distinguishing Liabilities From Equity (“ASC 480480”) at fair value. The fair value model used to calculate this obligation is based on the Monte Carlo simulation that has been risk adjusted based on the probability of achievement of the milestones. The inputs we use for determining the fair value of the contingent consideration associated with development and regulatory milestones are Level 3 fair value measurements. We re-evaluate the fair value on a quarterly basis. Changes in the fair value can result from adjustments to the probability of achievement of the milestones, stock price, volatility and the risk-free interest rate. The change in our estimates associated with payments which will become due and payable for development and regulatory milestones willcould change the fair value of contingent consideration, resulting in a charge or contra expense to research and development expense in the period in which the increase or decrease is determined. Research and Development Research and development costs are expensed as incurred. Research and development costs consist of payroll and personnel expense, consulting costs, external contract research and development expenses, which includes fees paid to other entities that conduct certain research and development activities on our behalf, such as clinical research organizations (“CROs”) and contract manufacturing organizations (“CMOs”), raw materials, drug product manufacturing costs, laboratory supplies and allocated overhead, including depreciation and amortization, rent and utilities. Material research and development costs that are paid in advance of performance are capitalized as a prepaid expense and amortized over the service period as the services are provided.
Clinical trial costs are a significant component of research and development expenses, and we outsource a significant portion of these costs to third parties. Third partyThird-party clinical trial expenses include investigator fees, site and patient costs, CRO costs, costs for central laboratory testing, data management and CMO costs. The accrual for site and patient costs includes inputs such as estimates of patient enrollment, patient cycles incurred, clinical site activations, and other pass-through costs. These inputs are required to be estimated due to a lag in receiving the actual clinical information from third parties. Payments for these activities are based on the terms of the individual arrangements, which may differ from the pattern of costs incurred, and are reflected on the consolidated balance sheets as a prepaid asset or accrued expenses. These third-party agreements are generally cancelable, and related costs are recorded as research and development expenses as incurred. Material advance payments for goods or services that will be used or rendered for future research and development activities are recorded as a prepaid asset and recognized as expense as the related goods are delivered or the related services are performed. When evaluating the adequacy of the accrued expenses, we analyze progress of the studies, including the phase or completion of events, invoices received and contracted costs. Significant judgments and estimates may be made in determining the accrued balances at the end of any reporting period. Results of Operations
Comparison of years ended December 31, 2020 and December 31, 2019:
| | Year Ended December 31, | (in thousands except percentages) | | 2020 | | | 2019 | | | Increase (Decrease) | Revenue | | $ | 8,185 | | | $ | 5,801 | | | $ | 2,384 | | | 41 | | % | Operating expenses: | | | | | | | | | | | | | | | | | Research and development | | | 49,774 | | | | 17,765 | | | | 32,009 | | | 180 | | % | General and administrative | | | 13,209 | | | | 8,501 | | | | 4,708 | | | 55 | | % | Impairment charge | | | — | | | | 1,000 | | | | (1,000 | ) | | (100 | ) | % | Total operating expenses | | | 62,983 | | | | 27,266 | | | | 35,717 | | | 131 | | % | Loss from operations | | | (54,798 | ) | | | (21,465 | ) | | | (33,333 | ) | | (155 | ) | % | Other income (expense): | | | | | | | | | | | | | | | | | Changes in fair value of warrant liability | | | — | | | | 30 | | | | (30 | ) | | (100 | ) | % | Interest expense | | | (9 | ) | | | (2 | ) | | | (7 | ) | | (350 | ) | % | Interest income | | | 322 | | | | 843 | | | | (521 | ) | | (62 | ) | % | Other income, net | | | 24 | | | | 15 | | | | 9 | | | 60 | | % | Total other income, net | | | 337 | | | | 886 | | | | (549 | ) | | (62 | ) | % | Net loss before income tax benefit | | | (54,461 | ) | | | (20,579 | ) | | | (33,882 | ) | | (165 | ) | % | Income tax benefit | | | 5,417 | | | | 59 | | | | 5,358 | | | 9,081 | | % | Net loss | | $ | (49,044 | ) | | $ | (20,520 | ) | | $ | (28,524 | ) | | (139 | ) | % |
Revenue
Revenue from contracts and grants for the years ended December 31, 2020 and 2019 consisted primarily of research grants in the United States from MTEC for our T-COVID product candidate and BARDA for our NasoShield vaccine candidate. These grants consist of firm fixed fee contracts based on milestones and cost reimbursement contracts, with a fixed fee based on either costs incurred or milestones met.
| | Year Ended December 31, | (in thousands except percentages) | | 2020 | | | 2019 | | | Increase (Decrease) | Revenue | | $ | 8,185 | | | $ | 5,801 | | | $ | 2,384 | | | 41 | | % |
Revenue increased by $2.4 million, or 41% for the year ended December 31, 2020 as compared to 2019. The increase was primarily the result of:
an increase of $4.2 million in MTEC revenue attributable to a clinical trial and development work on the T-COVID program; and
a decrease of $1.6 million in BARDA revenue due to timing of clinical trials and development activities on the NasoShield program.
Research and development expenses
Research and development expenses for the years ended December 31, 2020 and 2019 consisted primarily of expenses related to product candidate development. Research and development expenses for the years ended December 31, 2020 and 2019 are summarized as follows:
| | Year Ended December 31, | (in thousands except percentages) | | 2020 | | | 2019 | | | Increase (Decrease) | Research and development | | $ | 49,774 | | | $ | 17,765 | | | $ | 32,009 | | | 180 | | % |
Research and development expenses increased by $32.0 million, or 180%, during the year ended December 31, 2020 as compared to 2019. The increased expense was primarily due to:
an increase of $12.9 million due to development activities for the COVID-19 programs, which include AdCOVID and T-COVID;
an increase of $8.5 million due to an increase in the fair value of ALT-801 contingent consideration during 2020, that is partially offset by costs incurred in 2019 with respect to the acquisition of ALT-801;
an increase of $8.6 million due to development activities for ALT-801 which was acquired in July 2019; and
a net increase of $2.0 million due to the timing of clinical trial and development activities related to our other programs, along with costs associated with our pre-clinical projects and non-project specific research and development costs including employee compensation and facility costs.
General and administrative expenses
The following is a summary of general and administrative expenses for the years ended December 31, 2020 and 2019:
| | Year Ended December 31, | (in thousands except percentages) | | 2020 | | | 2019 | | | Increase (Decrease) | General and administrative | | $ | 13,209 | | | $ | 8,501 | | | $ | 4,708 | | | 55 | | % |
General and administrative expenses increased by $4.7 million, or 55%, during the year ended December 31, 2020 as compared to 2019. The increased expense is primarily due to an increase of $1.8 million in labor-related and other costs, $1.6 million in professional fees, and $1.3 million in stock compensation expense.
Impairment charges
There were no impairment charges reported during the year ended December 31, 2020. Impairment charges of $1.0 million reported during the year ended December 31, 2019 resulted from the completion of the SparVax-L NIAID contract with no future funding identified. As a result of the contract completion and the U.S. government’s funding prioritization of only single dose anthrax vaccine candidates, we abandoned the project and impaired the remaining net book value of the SparVax-L IPR&D asset.
Total other income, net
Total other income, net decreased by $0.5 million during the year ended December 31, 2020 as compared to the year ended December 31, 2019. The net decrease was primarily due to changes in interest income related to our short-term investments.
Income tax benefit
| | Year Ended December 31, | (in thousands except percentages) | | 2020 | | | 2019 | | | Increase (Decrease) | Income tax benefit | | $ | 5,417 | | | $ | 59 | | | $ | 5,358 | | | 9,081 | | % |
Income tax benefit increased by $5.4 million during the year ended December 31, 2020 as compared to the year ended December 31, 2019. The increase is due to the Coronavirus Aid, Relief, and Economic Security Act (the “CARES Act”) signed into law on March 27, 2020 which made temporary changes regarding the utilization and carry back of net operating losses. As of December 31, 2020, we filed a refund claim of $2.7 million with the U.S. Federal and Maryland tax authorities, reflecting a partial refund of our 2016 tax liability by carrying back a portion of our 2018 and 2019 tax losses. We plan to file additional refund claims during 2021 by carrying back our 2020 tax losses to claim the remaining refund available for the 2016 tax liability.
Liquidity and Capital Resources
Overview
Our primary sources of cash for the year ended December 31, 2020 were from equity transactions, maturities of short-term investments and cash receipts of revenue from our BARDA and MTEC contracts. Our cash, cash equivalents, restricted cash, and short-term investments were $216.0 million at December 31, 2020. We believe, based on the operating cash requirements and capital expenditures expected for 2021 and 2022, our cash on hand and short-term investments at December 31, 2020, together with expected revenue from our government sponsored contracts and tax refunds, are sufficient to fund operations for at least a twelve-month period from issuance date of our December 31, 2020 consolidated financial statements.
We have not generated any revenues from the sale of any products to date, and there is no assurance of any future revenues from product sales. Our sources of revenue have consisted of grant revenues under our arrangements with BARDA for the development of NasoShield, MTEC for a clinical trial and development work on T-COVID, and to a lesser degree from other licensing arrangements. We have incurred significant losses since we commenced operations. As of December 31, 2020, we had accumulated losses of $186.4 million since our inception. In addition, we have not generated positive cash flows from operations. We have had to rely on a variety of financing sources, including the issuance of debt and equity securities. As capital resources are consumed to fund our research and development activities, we may not have sufficient capital to fund our plan of operations. In order to address our capital needs, including our planned clinical trials, we must continue to actively pursue additional equity or debt financing, government funding, and monetization of our existing programs through partnership arrangements or sales to third parties.
In June 2020, we were awarded $4.7 million from the U.S. Army Medical Research & Development Command (“USAMRDC”) to fund our Phase 1/2 clinical trial of T-COVID. The competitive award was granted by USAMRDC in collaboration with the Medical Technology Enterprise Consortium (“MTEC”), a 501(c)(3) biomedical technology consortium working in partnership with the Department of Defense (“DoD”). Under the contract, MTEC pays us a firm fixed fee based upon the achievement of certain milestones for conduct and completion of a Phase 1/2 study and research and development work on the replication-deficient adenovirus 5 (“RD-Ad5”) vector vaccine platform. Through December 31, 2020, we have collected approximately $1.1 million in cash under the contract.
In July 2016, we signed a five-year contract with BARDA. The contract, as amended, has a total value of up to $133.7 million and is used to fund clinical development of NasoShield. Under the contract, BARDA pays us a fixed fee and reimburses certain costs for the research and development of an Ad5-vectored, protective antigen-based intranasal anthrax vaccine through cGMP manufacture and conduct of a Phase 1 clinical trial dose ranging assessment of safety and immunogenicity. The contract consists of an initial base performance period providing approximately $27.8 million in funding for the period July 2016 through June 2021. BARDA has seven options to extend the contract to fund certain continued development and manufacturing activities for the anthrax vaccine, including Phase 2 clinical studies. Each option, if exercised by BARDA, would provide additional funding ranging from approximately $1.1 million to $34.4 million for a three-year period beginning Q1 2021. Through December 31, 2020, we have collected approximately $24.7 million in cash under the current BARDA contract.
Indebtedness
Paycheck Protection Program
On April 7, 2020, we applied for a loan from ServisFirst Bank, as lender, pursuant to the Paycheck Protection Program of the Coronavirus Aid, Relief, and Economic Security Act (the “CARES Act”) as administered by the U.S. Small Business Administration (the "SBA"). On April 13, 2020, the Loan was approved and we received the proceeds from a loan in the amount of $632,000 (the “PPP Loan”).
The PPP Loan, which took the form of a promissory note (the “Promissory Note”), was set to mature on April 7, 2022 and bore interest at a rate of 1% per annum. Monthly principal and interest payments, less the amount of any potential forgiveness (discussed below), was to commence on November 7, 2020. The Promissory Note provided for customary events of default, including, among others, those relating to failure to make payment, bankruptcy, breaches of representations and material adverse effects. We could prepay the principal of the PPP Loan at any time without incurring any prepayment charges.
All or a portion of the Loan may be forgiven by the SBA and lender upon application by us upon documentation of expenditures in accordance with the SBA requirements. In July 2020, we voluntarily extinguished the Promissory Note by paying the outstanding principal and accrued interest in cash.
BPI France Notes
We had two non-interest-bearing research and development funding arrangements with BPI France that were entered into in December 2013 to provide up to €750,000 in research funding in the first arrangement and up to €250,000 in the second arrangement. We were permitted to draw 50% of the funds upon the signing of the arrangements, an additional 30% contingent upon a financial audit and technical progress report, and the remaining amounts at the completion of the research and development project being funded by the arrangements. In October 2016, we agreed to extend the term on the arrangement by two years. The total amount advanced under the arrangements was €500,000. In April 2019, we were notified that €102,951 exceeded the allowable funding in accordance with the arrangement and made a payment of this amount on June 5, 2019. In September 2019, we were notified that €238,229 ($265,540) was converted into a grant and we recognized this amount as grant revenue for the three and nine months ended September 30, 2019. In addition to the €102,951 amount paid in excess of the allowable funding, we paid €62,500 (total repayments of $186,940) during the nine months ended September 30, 2019. In October 2019, we paid the remaining balance on the BPI France notes.
Cash Flows
The following table provides information regarding our cash flows for the years ended December 31, 2020 and 2019:
| | Year Ended December 31, | (in thousands, except percentages) | | 2020 | | | 2019 | | | Increase (Decrease) | Net cash provided by (used in): | | | | | | | | | | | | | | | | | Operating activities | | $ | (34,437 | ) | | $ | (9,602 | ) | | $ | (24,835 | ) | | (259 | ) | % | Investing activities | | | (72,095 | ) | | | (28,286 | ) | | | (43,809 | ) | | (155 | ) | % | Financing activities | | | 213,487 | | | | 12,532 | | | | 200,955 | | | 1,604 | | % | Net increase (decrease) in cash and cash equivalents and restricted cash | | $ | 106,955 | | | $ | (25,356 | ) | | $ | 132,311 | | | 522 | | % |
Operating Activities
Net cash used in operating activities was $34.4 million for the year ended December 31, 2020 compared to $9.6 million during the year ended December 31, 2019. Our sources of cash provided by operations during the year ended December 31, 2020 were primarily cash receipts of revenue generated by our BARDA and MTEC contracts. The primary uses of cash from our operating activities include payments for labor and labor-related costs, professional fees, research and development costs associated with our clinical trials, and other general corporate expenditures. The increase in cash used in operations of $24.8 million year over year is due to an increase in net loss as adjusted for noncash items of $18.6 million and changes in working capital accounts of $6.3 million.
Investing Activities
Net cash used in investing activities was $72.1 million for the year ended December 31, 2020 compared to $28.3 million during the year ended December 31, 2019. The net cash used in investing activities during 2020 was primarily due to net purchases and maturities of short-term investments. The net cash used in investing activities in 2019 was primarily due to purchases of short-term investments.
Financing Activities
Net cash provided by financing activities was $213.5 million for the year ended December 31, 2020 compared to $12.5 million during the year ended December 31, 2019. The net cash provided by financing activities during 2020 was primarily the result of the receipt of $124.0 million in net proceeds from the Public Offering, $48.2 million in proceeds from the issuance of common stock from the At-the-Market Offering program and $41.0 million in proceeds from the exercise of warrants. The net cash provided by financing activities in 2019 was primarily the result of the receipt of $12.7 million in proceeds from the Registered Direct Offering of units that consisted of common stock and warrants.
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements.
Item 7A. Quantitative and Qualitative Disclosures about Market Risk WeAs a “smaller reporting company” as defined by Item 10 of Regulation S-K, we are a smaller reporting company and not required to provide the information required by this information.Item.
Item 8. Financial Statement sStatements and Supplementary Data ALTIMMUNE, INC. INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
REPORT OF INDEPENDENT REGIST EREDREGISTERED PUBLIC ACCOUNTING FIRMTheTo the Shareholders and the Board of Directors of Altimmune, Inc.
Opinion on the Financial Statements We have audited the accompanying consolidated balance sheets of Altimmune, Inc. (the Company) as of December 31, 20202022 and 2019,2021, the related consolidated statements of operations and comprehensive loss, changes in stockholders’stockholders' equity and cash flows for each of the two years in the period ended December 31, 2020,2022, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 20202022 and 2019,2021, and the results of its operations and its cash flows for each of the two years in the period ended December 31, 2020,2022, in conformity with U.S. generally accepted accounting principles. Basis for opinion These financial statements are the responsibility of the Company’sCompany's management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB. We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’sCompany's internal control over financial reporting. Accordingly, we express no such opinion. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion. Critical Audit MattersMatter The critical audit matter communicated below is a matter arising from the current period audit of the financial statements that was communicated or required to be communicated to the audit committee and that: (1) relates to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective or complex judgments. The communication of the critical audit matter does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the account or disclosure to which it relates. | | | | | Impairment assessment of Indefinite-lived Intangible AssetsAccrued Research and Development Expenses
|
| | | Description of the Matter | | At December 31, 2020, the Company's indefinite-lived intangible assets consisted of one acquired in-processThe Company’s accrued research and development asset (IPR&D) with a carrying value of approximately $12.4 million.expenses were $7.3 million at December 31, 2022. As explaineddiscussed in Note 2 toof the consolidated financial statements, the Company tests IPR&D for impairment at least annually at year end by evaluating qualitative factors which could be indicative of impairment. In evaluating indefinite-lived intangible assets for impairment, the Company considers the current project status, forecasted changes in the timing or amounts required to complete the project, forecasted timing or changes in the future cash flows to be generated by the completed products, probability of successanalyzes progress of the ultimate project, and changes to other market-based assumptions, such as discount rates.
Auditing the Company's qualitative impairment assessment was complex and judgmental due to the significant estimationstudies, including the evaluationphase or completion of assumptions that could indicate potential impairment. Management’s assumptions that affectevents, invoices received and contracted costs to estimate the evaluation included the forecasted changes in timing or amounts required to complete the project or in the future expected cash flows to be generated by the completed project
accrual for research and the probability of success of the ultimate project. These assumptions are forward looking and are sensitive to and affected by economic, industry, and company-specific qualitative factors.development expense at period end. | How We Addressed the Matter in Our Audit | | To assessAuditing the Company’s evaluationaccrued research and development expenses was especially challenging due to the application of impairment indicatorsmanagement judgment about the estimate of services provided but not yet invoiced. Specifically, the amount of accrued research and development expenses recognized is sensitive to the availability of information to make the estimate, including the estimate of the IPR&D asset, weperiod over which services will be performed, the associated cost of such services, and the level of services performed and progress in the period for which the Company has not yet received an invoice from the supplier. Additionally, due to the duration of clinical trials and the timing of invoicing received from third parties, the actual amounts incurred are not always known by the report date.
To evaluate the Company’s estimate of services incurred as of period end pursuant to its research and development activities, our audit procedures that included, among others, evaluating the Company's significant assumptions described above. We also, testedtesting the completeness and accuracy of the underlying data. For example, we compareddata used in the estimates and evaluating the significant assumptions stated above that are used by management to current industry, marketestimate the recorded amounts. To assess the reasonableness of the significant assumptions, we obtained information regarding the nature and economic trends,extent of progress of clinical trials and to other relevant data. In addition, toactivities from the Company’s research and development personnel that oversee the clinical trials. To evaluate the probability of success, we considered the phase of developmentcompleteness of the IPR&D project,accrued research and development expenses, we also compared payments made by the Company's historyCompany subsequent to December 31, 2022 to the amounts accrued by the Company as of obtaining regulatory approval, and third-party data regarding clinical trial success rates. We assessed the historical accuracy of management’s estimates and performed sensitivity analyses on significant assumptions to evaluate the effect on the impairment assessment that would result from changes in the assumptions. date. |
/s/ Ernst & Young LLP We have served as the Company’s auditor since 2017. Baltimore, MarylandTysons, Virginia
February 25, 202128, 2023 ALTIMMUNE, INC.
CONSOLIDATED BALANCE SHEETS (In thousands, except share and per-share) | | December 31, | | | | 2020 | | | 2019 | | ASSETS | | | | | | | | | Current assets: | | | | | | | | | Cash and cash equivalents | | $ | 115,917,807 | | | $ | 8,962,686 | | Restricted cash | | | 34,174 | | | | 34,174 | | Total cash, cash equivalents and restricted cash | | | 115,951,981 | | | | 8,996,860 | | Short-term investments | | | 100,005,558 | | | | 28,277,386 | | Accounts receivable | | | 4,610,202 | | | | 1,021,179 | | Tax refund receivable | | | 7,762,793 | | | | 629,096 | | Prepaid expenses and other current assets | | | 1,926,675 | | | | 470,228 | | Total current assets | | | 230,257,209 | | | | 39,394,749 | | Property and equipment, net | | | 1,056,920 | | | | 1,104,208 | | Right of use asset | | | 903,825 | | | | 698,321 | | Intangible assets, net | | | 12,823,846 | | | | 12,732,195 | | Other assets | | | 73,413 | | | | 128,547 | | Total assets | | $ | 245,115,213 | | | $ | 54,058,020 | | LIABILITIES AND STOCKHOLDERS’ EQUITY | | | | | | | | | Current liabilities: | | | | | | | | | Accounts payable | | $ | 612,293 | | | $ | 18,232 | | Accrued expenses and other current liabilities | | | 11,408,154 | | | | 3,904,767 | | Total current liabilities | | | 12,020,447 | | | | 3,922,999 | | Contingent consideration | | | 5,390,000 | | | | 2,750,000 | | Other long-term liabilities | | | 1,828,443 | | | | 1,864,875 | | Total liabilities | | | 19,238,890 | | | | 8,537,874 | | Commitments and contingencies (Note 17) | | | | | | | | | Stockholders’ equity: | | | | | | | | | Common stock, $0.0001 par value; 200,000,000 shares authorized; 37,142,946 and 15,312,381 shares issued; 37,142,946 and 15,312,167 shares outstanding at December 31, 2020 and 2019, respectively | | | 3,697 | | | | 1,508 | | Additional paid-in capital | | | 417,337,742 | | | | 187,914,916 | | Accumulated deficit | | | (186,420,599 | ) | | | (137,376,122 | ) | Accumulated other comprehensive loss, net | | | (5,044,517 | ) | | | (5,020,156 | ) | Total stockholders’ equity | | | 225,876,323 | | | | 45,520,146 | | Total liabilities and stockholders’ equity | | $ | 245,115,213 | | | $ | 54,058,020 | |
| | | | | | | | | December 31, | | | 2022 | | 2021 | ASSETS | | | | | | | Current assets: | | | | | | | Cash and cash equivalents | | $ | 111,097 | | $ | 190,301 | Restricted cash | | | 34 | | | 34 | Total cash, cash equivalents and restricted cash | | | 111,131 | | | 190,335 | Short-term investments | | | 73,783 | | | — | Accounts receivable | | | 173 | | | 429 | Income tax and R&D incentive receivables | | | 2,368 | | | 5,410 | Prepaid expenses and other current assets | | | 5,358 | | | 7,952 | Total current assets | | | 192,813 | | | 204,126 | Property and equipment, net | | | 1,081 | | | 1,448 | Intangible assets, net | | | 12,419 | | | 12,419 | Other assets | | | 615 | | | 872 | Total assets | | $ | 206,928 | | $ | 218,865 | LIABILITIES AND STOCKHOLDERS’ EQUITY | | | | | | | Current liabilities: | | | | | | | Accounts payable | | $ | 4,804 | | $ | 2,034 | Contingent consideration | | | — | | | 6,090 | Accrued expenses and other current liabilities | | | 12,250 | | | 10,152 | Total current liabilities | | | 17,054 | | | 18,276 | Other long-term liabilities | | | 4,581 | | | 1,454 | Total liabilities | | | 21,635 | | | 19,730 | Commitments and contingencies (Note 17) | | | | | | | Stockholders’ equity: | | | | | | | Common stock, $0.0001 par value; 200,000,000 shares authorized; 49,199,845 and 40,993,768 shares issued and outstanding as of December 31, 2022 and December 31, 2021, respectively | | | 5 | | | 4 | Additional paid-in capital | | | 568,399 | | | 497,342 | Accumulated deficit | | | (377,884) | | | (293,171) | Accumulated other comprehensive loss, net | | | (5,227) | | | (5,040) | Total stockholders’ equity | | | 185,293 | | | 199,135 | Total liabilities and stockholders’ equity | | $ | 206,928 | | $ | 218,865 |
The accompanying notes are an integral part of the consolidated financial statements. ALTIMMUNE, INC. CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS (In thousands, except share and per-share amounts) | | | | | | | | | Year Ended December 31, | | | 2022 | | 2021 | Revenues | | $ | (68) | | $ | 4,410 | Operating expenses: | | | | | | | Research and development | | | 70,538 | | | 74,541 | General and administrative | | | 17,134 | | | 15,413 | Impairment loss on construction-in-progress | | | — | | | 11,370 | Total operating expenses | | | 87,672 | | | 101,324 | Loss from operations | | | (87,740) | | | (96,914) | Other income (expense): | | | | | | | Interest expense | | | (8) | | | (5) | Interest income | | | 2,870 | | | 203 | Other income (expense), net | | | (32) | | | (374) | Total other income (expense), net | | | 2,830 | | | (176) | Net loss before income taxes | | | (84,910) | | | (97,090) | Income tax expense (benefit) | | | (197) | | | — | Net loss | | | (84,713) | | | (97,090) | Other comprehensive income — unrealized (loss) gain on short-term investments | | | (187) | | | 4 | Comprehensive loss | | $ | (84,900) | | $ | (97,086) | Net loss per share, basic and diluted | | $ | (1.81) | | $ | (2.35) | Weighted-average common shares outstanding, basic and diluted | | | 46,926,349 | | | 41,283,498 |
The accompanying notes are an integral part of the consolidated financial statements. ALTIMMUNE, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSSCHANGES IN STOCKHOLDERS’ EQUITY | | Year Ended December 31, | | | | 2020 | | | 2019 | | Revenues | | $ | 8,185,027 | | | $ | 5,801,401 | | Operating expenses: | | | | | | | | | Research and development | | | 49,774,328 | | | | 17,765,553 | | General and administrative | | | 13,209,440 | | | | 8,500,783 | | Impairment charge | | | — | | | | 1,000,000 | | Total operating expenses | | | 62,983,768 | | | | 27,266,336 | | Loss from operations | | | (54,798,741 | ) | | | (21,464,935 | ) | Other income (expense): | | | | | | | | | Changes in fair value of warrant liability | | | — | | | | 30,000 | | Interest expense | | | (9,421 | ) | | | (2,244 | ) | Interest income | | | 322,514 | | | | 843,409 | | Other income, net | | | 24,147 | | | | 15,139 | | Total other income, net | | | 337,240 | | | | 886,304 | | Net loss before income tax benefit | | | (54,461,501 | ) | | | (20,578,631 | ) | Income tax benefit | | | 5,417,024 | | | | 58,500 | | Net loss | | | (49,044,477 | ) | | | (20,520,131 | ) | Other comprehensive (loss) income — unrealized (loss) gain on investments | | | (24,361 | ) | | | 20,007 | | Comprehensive loss | | $ | (49,068,838 | ) | | $ | (20,500,124 | ) | Net loss | | $ | (49,044,477 | ) | | $ | (20,520,131 | ) | Deemed dividends | | | — | | | | (452,925 | ) | Net loss attributable to common stockholders | | $ | (49,044,477 | ) | | $ | (20,973,056 | ) | Net loss per share attributable to common stockholders, basic and diluted | | $ | (1.91 | ) | | $ | (1.60 | ) | Weighted-average common shares outstanding, basic and diluted | | | 25,637,023 | | | | 13,124,951 | |
(In thousands, except share amounts) | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | Accumulated | | | | | | | | | | | Additional | | | | | Other | | Total | | | Common Stock | | Paid-In | | Accumulated | | Comprehensive | | Stockholders’ | | | Shares | | Amount | | Capital | | Deficit | | Loss | | Equity | Balance at December 31, 2020 | | 37,142,946 | | $ | 4 | | $ | 417,337 | | $ | (186,421) | | $ | (5,044) | | $ | 225,876 | Stock-based compensation | | — | | | — | | | 5,519 | | | — | | | — | | | 5,519 | Exercise of stock options | | 54,068 | | | — | | | 176 | | | — | | | — | | | 176 | Vesting of restricted stock awards including withholding, net | | (28,850) | | | — | | | (400) | | | — | | | — | | | (400) | Issuance of common stock from Employee Stock Purchase Plan | | 24,100 | | | — | | | 225 | | | — | | | — | | | 225 | Retirement of common stock in exchange for common stock warrant | | (1,000,000) | | | — | | | (7,540) | | | (9,660) | | | — | | | (17,200) | Issuance of common stock warrant in exchange for retirement of common stock | | — | | | — | | | 17,200 | | | — | | | — | | | 17,200 | Issuance of common stock in at-the-market offerings, net | | 4,800,454 | | | — | | | 64,815 | | | — | | | — | | | 64,815 | Issuance of common stock upon cashless exercise of warrants | | 1,050 | | | — | | | 10 | | | — | | | — | | | 10 | Unrealized gain (loss) on short-term investments | | — | | | — | | | — | | | — | | | 4 | | | 4 | Net loss | | — | | | — | | | — | | | (97,090) | | | — | | | (97,090) | Balance at December 31, 2021 | | 40,993,768 | | | 4 | | | 497,342 | | | (293,171) | | | (5,040) | | | 199,135 | Stock-based compensation | | — | | | — | | — | 8,101 | | — | — | | — | — | | | 8,101 | Exercise of stock options | | 358,317 | | | — | | — | 950 | | — | — | | — | — | | | 950 | Vesting of restricted stock awards including withholding, net | | 8,695 | | | — | | — | (516) | | — | — | | — | — | | | (516) | Issuance of common stock from Employee Stock Purchase Plan | | 26,395 | | | — | | — | 181 | | — | — | | — | — | | | 181 | Issuance of common stock in at-the-market offerings, net | | 5,204,215 | | | 1 | | — | 56,165 | | — | — | | — | — | | | 56,166 | Issuance of common stock upon cashless exercise of warrants | | 1,760,854 | | | — | | — | — | | — | — | | — | — | | | — | Issuance of common stock related to contingent consideration liability | | 847,444 | | | — | | — | 6,176 | | — | — | | — | — | | | 6,176 | Other increase | | 157 | | | — | | — | — | | — | — | | — | — | | | — | Unrealized gain (loss) on short-term investments | | — | | | — | | — | — | | — | — | | — | (187) | | | (187) | Net loss | | — | | | — | | — | — | | — | (84,713) | | — | — | | | (84,713) | Balance at December 31, 2022 | | 49,199,845 | | $ | 5 | | $ | 568,399 | | $ | (377,884) | | $ | (5,227) | | $ | 185,293 |
The accompanying notes are an integral part of the consolidated financial statement ALTIMMUNE, INC. CONSOLIDATED STATEMENTS OF CASH FLOWS (In thousands) | | | | | | | | | Year Ended December 31, | | | 2022 | | 2021 | CASH FLOWS FROM OPERATING ACTIVITIES: | | | | | | | Net loss | | $ | (84,713) | | $ | (97,090) | Adjustments to reconcile net loss to net cash used in operating activities: | | | | | | | Change in fair value of contingent consideration liability | | | 86 | | | 700 | Impairment loss on construction-in-progress | | | — | | | 11,370 | Impairment loss on intangible assets | | | — | | | 579 | Stock-based compensation expense | | | 8,101 | | | 5,519 | Depreciation and amortization | | | (205) | | | 551 | Loss on foreign currency exchange | | | 34 | | | 384 | Changes in operating assets and liabilities: | | | | | | | Accounts receivable | | | 256 | | | 4,181 | Prepaid expenses and other assets | | | 2,650 | | | (5,908) | Accounts payable | | | 2,770 | | | 1,422 | Accrued expenses and other liabilities | | | 5,393 | | | (2,299) | Income tax and R&D incentive receivables | | | 3,042 | | | 2,353 | Net cash used in operating activities | | | (62,586) | | | (78,238) | CASH FLOWS FROM INVESTING ACTIVITIES: | | | | | | | Proceeds from sales and maturities of short-term investments | | | — | | | 107,427 | Purchases of short-term investments | | | (73,273) | | | (7,592) | Purchases of property and equipment, net | | | (126) | | | (12,117) | Cash paid for internally developed patents | | | — | | | (195) | Net cash (used in) provided by investing activities | | | (73,399) | | | 87,523 | CASH FLOWS FROM FINANCING ACTIVITIES: | | | | | | | Payments of deferred offering costs | | | — | | | (118) | Proceeds from issuance of common stock in at-the-market offerings, net | | | 56,166 | | | 64,815 | Proceeds from issuance of common stock from Employee Stock Purchase Plan | | | 181 | | | 225 | Proceeds from exercises of stock options, net | | | 434 | | | 176 | Net cash provided by financing activities | | | 56,781 | | | 65,098 | Net (decrease) increase in cash and cash equivalents and restricted cash | | | (79,204) | | | 74,383 | Cash, cash equivalents and restricted cash at beginning of period | | | 190,335 | | | 115,952 | Cash, cash equivalents and restricted cash at end of period | | $ | 111,131 | | $ | 190,335 | | | | | | | | SUPPLEMENTAL NON-CASH ACTIVITIES: | | | | | | | Fair value of common stock retired in exchange for issuance of common stock warrant | | $ | — | | $ | 17,200 | Common stock issued related to contingent consideration liability | | $ | 6,176 | | $ | — | Operating lease liability and right of use asset addition | | $ | — | | $ | 72 |
The accompanying notes are an integral part of the consolidated financial statements. ALTIMMUNE, INC. CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERS’ EQUITY
| | Common Stock | | | Additional Paid-In | | | Accumulated | | | Accumulated Other Comprehensive | | | Total Stockholders’ | | | | Shares | | | Amount | | | Capital | | | Deficit | | | Loss | | | Equity | | Balance at December 31, 2018 | | | 9,078,238 | | | $ | 876 | | | $ | 170,207,844 | | | $ | (116,855,991 | ) | | $ | (5,040,163 | ) | | $ | 48,312,566 | | Stock based compensation | | | — | | | | — | | | | 1,264,231 | | | | — | | | | — | | | | 1,264,231 | | Vesting of restricted stock awards including withholding, net | | | (25,691 | ) | | | 6 | | | | (47,126 | ) | | | — | | | | — | | | | (47,120 | ) | Issuance of common stock in registered direct offering, net | | | 4,361,370 | | | | 436 | | | | 12,668,348 | | | | — | | | | — | | | | 12,668,784 | | Issuance of common stock for acquired in-process research and development | | | 1,887,250 | | | | 189 | | | | 3,791,296 | | | | — | | | | — | | | | 3,791,485 | | Issuance of common stock upon exercise of warrants | | | 11,000 | | | | 1 | | | | 30,323 | | | | — | | | | — | | | | 30,324 | | Unrealized gain on short term investments | | | — | | | | — | | | | — | | | | — | | | | 20,007 | | | | 20,007 | | Net loss | | | — | | | | — | | | | — | | | | (20,520,131 | ) | | | — | | | | (20,520,131 | ) | Balance at December 31, 2019 | | | 15,312,167 | | | | 1,508 | | | | 187,914,916 | | | | (137,376,122 | ) | | | (5,020,156 | ) | | | 45,520,146 | | Stock based compensation | | | — | | | | — | | | | 2,576,006 | | | | — | | | | — | | | | 2,576,006 | | Issuance of common stock from exercise of stock options | | | 46,966 | | | | 4 | | | | 128,892 | | | | — | | | | — | | | | 128,896 | | Vesting of restricted stock awards including withholding, net | | | 84,320 | | | | 16 | | | | (262,729 | ) | | | — | | | | — | | | | (262,713 | ) | Issuance of common stock from Employee Stock Purchase Plan | | | 92,661 | | | | 8 | | | | 135,463 | | | | — | | | | — | | | | 135,471 | | Issuance of common stock and pre-funded warrants in public offering, net | | | 4,119,564 | | | | 412 | | | | 124,027,403 | | | | — | | | | — | | | | 124,027,815 | | Issuance of common stock in at-the-market offering, net | | | 5,594,455 | | | | 560 | | | | 48,155,512 | | | | — | | | | — | | | | 48,156,072 | | Issuance of common stock upon exercise of warrants | | | 10,197,907 | | | | 1,020 | | | | 41,037,438 | | | | — | | | | — | | | | 41,038,458 | | Issuance of common stock related to contingent consideration liability | | | 1,694,906 | | | | 169 | | | | 13,624,841 | | | | — | | | | — | | | | 13,625,010 | | Unrealized loss on short term investments | | | — | | | | — | | | | — | | | | — | | | | (24,361 | ) | | | (24,361 | ) | Net loss | | | — | | | | — | | | | — | | | | (49,044,477 | ) | | | — | | | | (49,044,477 | ) | Balance at December 31, 2020 | | | 37,142,946 | | | $ | 3,697 | | | $ | 417,337,742 | | | $ | (186,420,599 | ) | | $ | (5,044,517 | ) | | $ | 225,876,323 | |
The accompanying notes are an integral part of the consolidated financial statements.
ALTIMMUNE, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
| | Year Ended December 31, | | | | 2020 | | | 2019 | | CASH FLOWS FROM OPERATING ACTIVITIES: | | | | | | | | | Net loss | | $ | (49,044,477 | ) | | $ | (20,520,131 | ) | Adjustments to reconcile net loss to net cash used in operating activities: | | | | | | | | | Non-cash consideration for acquired in-process research and development | | | — | | | | 6,541,485 | | Changes in fair value of contingent consideration liability | | | 16,265,010 | | | | — | | Impairment charges | | | — | | | | 1,000,000 | | Stock-based compensation expense | | | 2,576,006 | | | | 1,264,231 | | Depreciation and amortization | | | 298,057 | | | | 387,321 | | Unrealized gain on foreign currency exchange | | | (19,073 | ) | | | (5,761 | ) | Changes in fair value of warrant liability | | | — | | | | (30,000 | ) | Changes in operating assets and liabilities: | | | | | | | | | Accounts receivable | | | (3,589,024 | ) | | | 2,440,759 | | Prepaid expenses and other current assets | | | (1,371,312 | ) | | | 133,001 | | Accounts payable | | | 594,060 | | | | (354,628 | ) | Accrued expenses and other liabilities | | | 6,987,815 | | | | (779,651 | ) | Tax refund receivable | | | (7,133,698 | ) | | | 379,877 | | Deferred taxes | | | — | | | | (58,500 | ) | Net cash used in operating activities | | | (34,436,636 | ) | | | (9,601,997 | ) | CASH FLOWS FROM INVESTING ACTIVITIES: | | | | | | | | | Proceeds from sales and maturities of short-term investments | | | 56,406,564 | | | | — | | Purchases of short-term investments | | | (128,159,098 | ) | | | (28,257,379 | ) | Purchases of property and equipment | | | (203,957 | ) | | | (1,227 | ) | Cash paid for internally developed patents | | | (138,464 | ) | | | (27,772 | ) | Net cash used in investing activities | | | (72,094,955 | ) | | | (28,286,378 | ) | CASH FLOWS FROM FINANCING ACTIVITIES: | | | | | | | | | Cash paid in conjunction with warrant exchange | | | — | | | | (25,000 | ) | Proceeds from exercises of warrants | | | 41,038,458 | | | | 30,324 | | Proceeds from issuance of common stock in at-the-market offering, net | | | 48,156,072 | | | | — | | Proceeds from issuance of common stock and pre-funded warrants in public offering, net | | | 124,027,815 | | | | — | | Proceeds from issuance of common stock in registered direct offering, net | | | — | | | | 12,668,784 | | Proceeds from issuance of notes payable | | | 632,000 | | | | — | | Payments of notes payable | | | (632,000 | ) | | | (292,002 | ) | Proceeds from conditional economic incentive | | | — | | | | 150,000 | | Proceeds from issuance of common stock from Employee Stock Purchase Plan | | | 135,471 | | | | — | | Proceeds from exercises of stock options | | | 128,896 | | | | — | | Net cash provided by financing activities | | | 213,486,712 | | | | 12,532,106 | | Net increase (decrease) in cash, cash equivalents and restricted cash | | | 106,955,121 | | | | (25,356,269 | ) | Cash, cash equivalents and restricted cash — beginning of year | | | 8,996,860 | | | | 34,353,129 | | Cash, cash equivalents and restricted cash — end of year | | $ | 115,951,981 | | | $ | 8,996,860 | | | | | | | | | | | SUPPLEMENTAL NON-CASH ACTIVITIES: | | | | | | | | | Common stock issued related to contingent consideration liability | | $ | 13,625,010 | | | $ | — | | Common stock issued for acquired in-process research and development | | $ | — | | | $ | 3,791,485 | | Operating lease liability and right of use asset addition | | $ | 338,212 | | | $ | 1,744,128 | |
The accompanying notes are an integral part of the consolidated financial statements.
ALTIMMUNE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS 1. Nature of Business and Organization Altimmune, Inc., headquartered in Gaithersburg, Maryland, United States, together with its subsidiaries (collectively, the “Company” or “Altimmune”) is a clinical stage biopharmaceutical company incorporated under the laws of the State of Delaware. The Company is focused on developing intranasal vaccines, immune modulating therapies and treatments for obesity and liver disease.diseases. The Company’s diverse pipeline includes proprietary intranasal vaccines for COVID-19 (AdCOVID), anthrax (NasoShield) and influenza (NasoVAX); an intranasal immune modulating therapeutic for COVID-19 (T-COVID); and next generation peptide therapeutics for NASH (ALT-801)obesity and non-alcoholic steatohepatitis (“NASH”) (for both, pemvidutide, formerly known as ALT-801), and for chronic hepatitis B (HepTcell). Since its inception, the Company has devoted substantially all of its efforts to business planning, research and development, recruiting management and technical staff and raising capital, and has financed its operations through the issuance of common and preferred stock, long-term debt and proceeds from research grants and government contracts. The Company has not generated any revenues from the sale of any products to date, and there is no assurance of any future revenues from product sales. 2. Summary of Significant Accounting Policies Basis of Presentation and Principles of Consolidation The accompanying consolidated financial statements are prepared in conformity with generally accepted accounting principles in the United States (“U.S. GAAP”) and in accordance with the rules and regulations of the United States Securities and Exchange Commission (“SEC”). The accompanying consolidated financial statements include the accounts of the Company and its wholly owned subsidiaries. All significant intercompany accounts and transactions have been eliminated in consolidation. Use of Estimates The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenue and expenses, and the disclosure of contingent assets and liabilities as of and during the reporting period. The Company bases estimates and assumptions on historical experience when available and on various factors that it believes to be reasonable under the circumstances. Significant estimates relied upon in preparing the accompanying consolidated financial statements were related to revenue recognition, the fair value of common stock and other equity instruments, accounting for stock-based compensation, income taxes, useful lives of long-lived assets, fair value of contingent consideration, impairment of long-lived assets, and accounting for project development and certain accruals. The Company assesses the above estimates on an ongoing basis; however, actual results could differ materially from those estimates. Comprehensive Loss
For the years presented, the total comprehensive loss includes net loss and other comprehensive income (loss) which represents unrealized gains or losses on investments.
Segment Information The Company is managed and operates as a single business focused on the research and development of immunotherapiestreatments for various diseases and disorders, and vaccines. The Company is managed by a single management team, and consistent with its organizational structure, the Chief Executive Officer manages and allocates resources at a consolidated level. Accordingly, the Company views its business as one operating segment. Cash Equivalents The Company considers all highly liquid investments purchased with remaining maturities of 90 days or less on the purchase date to be cash equivalents, and includes amounts held in money market funds which are actively traded (a Level 1 input). Restricted Cash The Company had restricted cash of $34,000 as of both December 31, 2022 and 2021, held in money market savings accounts as collateral. The restricted cash as of December 31, 2022 and 2021 is for the Company’s facility lease obligation. Restricted cash is classified as a component of cash, cash equivalents, and restricted cash in the accompanying consolidated balance sheets and consolidated statements of cash flows. Short-term Investments The Company’s short-term investments are comprised of U.S. Treasury, corporate debt securities and certificate of deposit that have original maturities less than or equal to one year and are classified as available-for-sale securities. Such securities are carried at estimated fair value, with any unrealized holding gains or losses reported as accumulated other comprehensive income or loss, which is a separate component of stockholders’ equity. Realized gains and losses and declines in value judged to be other-than-temporary, if any, are included in other income (expenses), net in the consolidated results of operations. The Company reviews its investment portfolio for impairment quarterly or more frequently if circumstances warrant. In determining whether a decline in the value of an investment is other-than-temporary, the Company evaluates currently available factors that may include, among others: (1) general market conditions; (2) the duration and extent to which fair value has been less than the carrying value; (3) the investment issuer'sissuer’s financial condition and business outlook; and (4) its assessment as to whether it is more likely than not that the Company will be required to sell a security prior to recovery of its amortized cost basis. A decline in the market value of any available-for-sale security below cost that is deemed to be other-than-temporary results in a reduction in fair value charged to earnings in that period, and a new cost basis for the security is established. Dividend and interest income are recognized in other income when earned. The cost of
securities sold is calculated using the specific identification method. The Company places all investments with government agencies, or corporate institutions whose debt is rated as investment grade. Intangible Assets
Intangible assets acquired in a business combination consist primarily of in-process research and development (“IPR&D”) assets. The value attributable to IPR&D projects at the time of acquisition is capitalized as an indefinite-lived intangible asset and tested for impairment until the project is completed or abandoned. Upon completion of the project, the indefinite-lived intangible asset will be accounted for as a finite-lived intangible asset and amortized on a straight-line basis over its estimated useful life. If the project is abandoned, the indefinite-lived intangible asset will be charged to expense. Intangible assets, including patents and licenses, acquired in other transactions are recorded at cost. Intangible assets with finite useful lives consist of legal costs incurred in the course of obtaining patents and license issuance fees for the use of proprietary technologies. Costs incurred for obtaining patents are amortized on a straight-line basis over the estimated useful lives of the assets from the time of approval of the patent. Prior to approval, these costs are carried on the balance sheets and not amortized. In the event approval is denied, the cost of the denied application is expensed. License issuance fees are amortized on a straight-line basis over the estimated useful lives of the underlying licensed technology. Intangible assets with finite useful lives are being amortized over 6 to 20 years. These amortization costs are classified as research and development expenses in the accompanying statements of operations and comprehensive loss.
Impairment of Long-lived Assets
The Company evaluates its long-lived tangible and intangible assets for impairment whenever events or changes in circumstances indicate that the carrying amount of such assets may not be recoverable. Impairment of long-lived assets other than goodwill and indefinite lived intangibles is assessed by comparing the undiscounted cash flows expected to be generated by the asset group to its carrying value.
As mentioned above, the Company’s IPR&D assets are currently non-amortizing. Until such time as the projects are either completed or abandoned, the Company test those assets for impairment at least annually at year end, or more frequently at interim periods, by evaluating qualitative factors which could be indicative of impairment. Qualitative factors being considered include, but are not limited to, the current project status, forecasted changes in the timing or amounts required to complete the project, forecasted changes in timing or changes in the future cash flows to be generated by the completed products, a probability of success of the ultimate project and changes to other market-based assumptions, such as discount rates. If impairment indicators are present as a result of the Company’s qualitative assessment, the Company will test those assets for impairment by comparing the fair value of the assets to their carrying value. Upon completion or abandonment, the value of the IPR&D assets will be amortized to expense over the anticipated useful life of the developed products, if completed, or charged to expense when abandoned if no alternative future use exists.
The Company has one IPR&D asset, HepTcell, that was acquired in 2015. The Company performed a qualitative assessment for the IPR&D impairment testing for 2020 and determined that no impairment indicators were present. During the third quarter of 2019, the Company abandoned its SparVax-L asset and recorded $1.0 million of impairment to fully write-off the balance of this IPR&D asset. See Note 5 for further details. The Company performed a quantitative assessment for the HepTcell IPR&D impairment test during 2019 and determined that there was no impairment.
Fair Value Measurements The Company records certain financial assets and liabilities at fair value in accordance with the guidance in Financial Accounting Standards Board (“FASB”) Accounting Standard Codification (“ASC”)Topic 820, Fair Value Measurements and Disclosures(“ASC 820”), which defines fair value and establishes a fair value hierarchy that prioritizes the inputs to valuation techniques used to measure fair value. The hierarchy gives the highest priority to unadjusted quoted prices in active markets for identical assets or liabilities (Level 1 measurements) and the lowest priority to unobservable inputs (Level 3 measurements). The three levels of the fair value hierarchy are described below: Level 1 — Quoted prices (unadjusted) in active markets for identical assets or liabilities that the Company can access at the measurement date. Level 2 — Inputs other than quoted prices included within Level 1 that are either directly or indirectly observable. If the asset or liability has a specified (contractual) term, a Level 2 input must be observable for substantially the full term. Level 3 — Unobservable inputs developed using estimates of assumptions developed by the Company, which reflect those that a market participant would use.
To the extent that valuation is based on models or inputs that are less observable or unobservable in the market, the determination of fair value requires more judgment. Accordingly, the degree of judgment exercised by the Company in determining fair value is greatest for instruments categorized in Level 3. A financial instrument’s level within the fair value hierarchy is based on the lowest level of any input that is significant to the fair value measurement. Fair value is a market-based measure considered from the perspective of a market participant rather than an entity-specific measure. Therefore, even when market assumptions are not readily available, the Company’s own assumptions are set to reflect those that market participants would use in pricing the asset or liability at the measurement date. The Company uses prices and inputs that are current as of the measurement date, including during periods of market dislocation. In periods of market dislocation, the observability of prices and inputs may change for many instruments. This condition could cause an instrument to be reclassified within levels in the fair value hierarchy. There were no transfers withininto or out of Level 3 of the fair value hierarchy during the years ended December 31, 20202022 and 2019.2021.
Financial Instruments The Company’s financial instruments consist of cash, cash equivalents, restricted cash, accounts receivable, short-term investments, notes payable, accounts payable, accrued expenses, contingent consideration, common stock warrants classified as a liability, and common stock warrants classified as equity. The carrying amounts of cash, cash equivalents, restricted cash, accounts receivable, accounts payable, and accrued expenses approximate their fair value due to the short-term nature of those financial instruments. Short-term investments are recorded at fair value, with any unrealized holding gains or losses reported as accumulated other comprehensive income or loss. Notes payable, prior to their redemption or cancellation, are recorded at their repayment value which approximates fair value. Contingent payments classified as a liability are recorded at fair value estimated using the Monte Carlo simulation valuation model. Common stock warrants classified as equity is initially recorded at their grant date fair value. For those warrants with a down round feature, if the down round feature is triggered, the Company would remeasure those instruments at that time with changes recorded as a deemed dividend all within equity. Accounts Receivable Accounts receivable includes both billed and unbilled amounts. The Company makes judgments as to its ability to collect outstanding receivables and provides an allowance for receivables when collection becomes doubtful. Provisions are made based upon a specific review of all significant outstanding invoices and the overall quality and age of those invoices not specifically reviewed. The Company’s receivables represent amounts reimbursed under its government grants and contracts. The Company believes that credit risks associated with these government grants and contracts is not significant. To date, the Company has not experienced any losses associated with accounts receivable and does not maintain an allowance for doubtful accounts. Concentrations of Credit Risk Financial instruments that potentially subject the Company to significant concentration of credit risk consist primarily of cash, cash equivalents, restricted cash, short-term investments and accounts receivable. Periodically, the Company maintains deposits in financial institutions in excess of government insured limits. Management believes that the Company is not exposed to significant credit risk as the Company’s deposits are held at financial institutions that management believes to be of high credit quality. The Company has not experienced any losses in these deposits. Property and Equipment, Net The Company records property and equipment at cost less accumulated depreciation and amortization. Expenditures for maintenance and repairs are charged to operations as incurred, whereas major improvements are capitalized as additions to property and equipment. Costs of assets under construction are capitalized but are not depreciated until the construction is substantially complete and the assets being constructed are ready for their intended use. Depreciation and amortization are recorded using the straight-line method over the estimated useful lives of the assets, as follows: | | | Asset Category |
| Estimated Useful Life | Computer and telecommunications | | 3 – 5 years | Software | | 3 years | Furniture, fixtures and equipment | | 5 years | Laboratory equipment | | 7 years | Leasehold improvements | | Lesser of lease term or estimated useful lives |
Intangible Assets The Company records intangible assets acquired in a business combination based on fair value on the date of acquisition. Acquired in-process research and development (“IPR&D”) assets that have alternative future use at the time of acquisition are capitalized as an indefinite-lived intangible asset and tested for impairment until the project is completed or abandoned. Upon completion of the project, the indefinite-lived intangible asset will be accounted for as a finite-lived intangible asset and amortized on a straight-line basis over its estimated useful life. If the project is abandoned, the indefinite-lived intangible asset will be charged to expense. Intangible assets acquired in other transactions are recorded at cost. The Company capitalizes costs incurred in the course of obtaining patents and license issuance fees for the use of proprietary technologies. Costs incurred for obtaining patents are amortized on a straight-line basis over the estimated useful lives of the assets from the time of approval of the patent. Prior to approval, these costs are carried on the balance sheets and not amortized. In the event approval is denied, the cost of the denied application is expensed. License issuance fees are amortized on a straight-line basis over the estimated useful lives of the underlying licensed technology. Amortization costs are classified as research and development expenses. Impairment of Long-lived Assets The Company evaluates its long-lived tangible and intangible assets for impairment whenever events or changes in circumstances indicate that the carrying amount of such assets may not be recoverable. Impairment of long-lived assets other than goodwill and indefinite lived intangibles is assessed by comparing the undiscounted cash flows expected to be generated by the asset group to its carrying value. The Company has one IPR&D asset, HepTcell, that was acquired in 2015. This candidate is a viral pathogen immunotherapy product for the treatment of chronic HBV. The IPR&D asset is currently non-amortizing. Until such time as the project is either completed or abandoned, the Company tests this asset for impairment at least annually at year end, or more frequently at interim periods, by evaluating qualitative factors which could be indicative of impairment. Qualitative factors being considered include, but are not limited to, the current project status, forecasted changes in the timing or amounts required to complete the project, forecasted changes in timing or changes in the future cash flows to be generated by the completed product, a probability of success of the ultimate project and changes to other market-based assumptions, such as discount rates. If impairment indicators are present as a result of the Company’s qualitative assessment, the Company will test the asset for impairment by comparing the fair value of the asset to its carrying value. Upon completion or abandonment, the value of the IPR&D asset will be amortized to expense over the anticipated useful life of the developed product, if completed, or charged to expense when abandoned if no alternative future use exists. Key assumptions used in the Company’s impairment analysis tests include projected cash flows, a probability of success of the ultimate project, and the discount rate. The Company performed a qualitative assessment for the IPR&D impairment testing for the year ended December 31, 2022 and 2021 and determined that no impairment indicators were present. Leases The Company determines if an arrangement is a lease at inception. Operating leases are recorded as a current and long-term lease obligation, with a corresponding right of use lease assets. Lease liabilities represent the Company’s obligation to make lease payments arising from leases. Right of use (“ROU”) assets represent the Company’s right to use an underlying asset for the lease term. Lease liabilities and ROU assets are recognized at the lease commencement date based on the present value of lease payments over the lease term. As most of the Company’s leases do not provide an implicit rate, the Company uses its incremental borrowing rate based on the information available at the lease commencement date in determining the present value of lease payments. The Company’s lease terms may include options to extend or terminate the lease when it is reasonably certain that the Company will exercise those options. Lease expense for lease payments is recognized on a straight-line basis over the lease term. Short-term leases are leases having a term of twelve months or less. The Company recognizes short-term leases on a straight-line basis and does not record a related lease asset or liability for such leases. Lease incentives and allowance provided by our landlord for the construction of leasehold improvements are recorded as lease incentive obligations as the related construction costs are incurred, up to the maximum allowance. Contingent Consideration The Company records contingent consideration associated with development and regulatory milestones that meets the definition of a liability under FASB Accounting Standards Codification Topic 480, Distinguishing Liabilities From Equity (“ASC 480”) at fair value. The fair value model used to calculate this obligation is based on the Monte Carlo simulation that has been risk adjusted based on the probability of achievement of the milestones. The inputs the Company uses for determining the fair value of the contingent consideration associated with development and regulatory milestones are Level 3 fair value measurements. The Company re-evaluates the fair value on a quarterly basis. Changes in the fair value can result from adjustments to the probability of achievement of the milestones, stock price, volatility and the risk-free interest rate. The change in the Company’s estimates associated with payments for development and regulatory milestones could change the fair value of contingent consideration, resulting in a charge or contra expense to research and development expense in the period in which the increase or decrease is determined. Warrants Common stock warrants classified as a liability are recorded at fair value and are remeasured every reporting periodissued in connection with the changes2018 Unit Offering, the 2018 and 2019 Registered Direct Offerings, and the 2020 Public Offering (all terms defined in fair value recordedNote 10 and Note 11), were classified as a component of permanent equity because they are freestanding financial instruments that were legally detachable and separately exercisable from other debt and equity instruments, are contingently exercisable, do not embody an obligation for the Company to repurchase its shares, and permits the holders to receive a fixed number of common shares upon exercise. In addition, such warrants did not provide any guarantee of value or return. The 2018 Registered Direct Offering and 2019 Registered Direct Offering triggered down round adjustments to the exercise price of warrants issued in connection with the 2018 Unit Offering. In the event that down round adjustment is triggered, the Company treats the value of the effect of the reduction in exercise price as a deemed dividend, resulting in a reduction to income (expenses),available to common shareholders. Stock-based Compensation The Company accounts for all stock-based compensation granted to employees and non-employees using a fair value method. The Company estimates the fair value of stock options granted using the Black-Scholes option pricing model on the dates of grant. For restricted stock and restricted stock units granted, fair value is determined based on the grant date closing price of the Company’s common stock. Stock-based compensation awarded to employees is measured at the grant date fair value of stock option grants and is recognized over the requisite service period of the awards, usually the vesting period, on a straight-line basis, net until their settlement or exercise.of estimated forfeitures. If awards are modified, the Company compares the fair value of the affected award measured immediately prior to modification to its value after modification. To the extent that the fair value of the modified award exceeds the original award, the incremental fair value of the modified award is recognized as compensation expense on the date of modification for vested awards, and over the remaining vesting period for unvested awards. Revenue OurThe Company’s revenue consists primarily of government and foundation grants and contracts that support the Company’s efforts on specific research projects. The Company has determined that the government agencies and foundations providing grants and contracts to the Company are not customers. These grants and contracts generally provide for reimbursement of approved costs as those costs are incurred by the Company. Research grants and contracts and the related accounts receivable are recognized as earned in proportion to when reimbursable expenses are incurred in performance of the contract. Payments received in advance of services being provided are recorded as deferred revenue. The Company anticipates that these government and foundation grants will decline in future periods.
Research and Development Research and development costs are expensed as incurred. Research and development costs consist of payroll and personnel expense, consulting costs, external contract research and development expenses, which includes fees paid to other entities that conduct certain research and development activities on the Company’s behalf, such as clinical research organizations (“CROs”) and contract manufacturing organizations (“CMOs”), raw materials, drug product manufacturing costs, laboratory supplies and allocated overhead, including depreciation and amortization, rent and utilities. Material research and development costs that are paid in advance of performance are capitalized as a prepaid expense and amortized over the service period as the services are provided. Clinical trial costs are a significant component of research and development expenses, and the Company outsources a significant portion of these costs to third parties. Third party clinical trial expenses include investigator fees, site and patient costs, CRO costs, costs for central laboratory testing, data management and CMO costs. The accrual for site and patient costs includes inputs such as estimates of patient enrollment, patient cycles incurred, clinical site activations and other pass-through costs. These inputs are required to be estimated due to a lag in receiving the actual clinical information from third parties. Payments for these activities are based on the terms of the individual arrangements, which may differ from the pattern of costs incurred, and are reflected on the consolidated balance sheets as a prepaid asset or accrued expenses. These third-party agreements are generally cancelable, and related costs are recorded as research and development expenses as incurred. Material advance payments for goods or services that will be used or rendered for future research and development activities are recorded as a prepaid asset and recognized as expense as the related goods are delivered or the related services are performed. When evaluating the adequacy of the accrued expenses, the Company analyzeanalyzes progress of the studies, including the phase or completion of events, invoices received and contracted costs. Significant judgments and estimates may be made in determining the accrued balances at the end of any reporting period.
Cash EquivalentsResearch and Development Incentive Credits
The Company considers all highly liquid investments purchased with remaining maturitiesis eligible to obtain certain research and development (“R&D”) incentive credits, through the participation in the U.K. R&D Small and Medium Enterprise tax relief program (“U.K. R&D credit”) and the Australian research and development incentive credit (the “Australia R&D credit”) program administered through the Australian Tax Office (the “ATO”). The U.K. R&D credits are calculated as a percentage of 90 days or lessqualifying R&D expenses and are payable in cash by the U.K. government to the Company. Qualifying R&D expenses consist of employment costs for research staff, consumables, a proportion of relevant, permitted sub-contract costs and certain internal overhead costs incurred as part of research projects for which the Company does not receive income. The Australia R&D credits provide for a cash refund based on a percentage of certain research and development activities undertaken in Australia by the Company’s wholly owned subsidiary, Altimmune AU Pty, Limited. Qualifying R&D expenses must be incurred within the country. The U.K. and Australian incentive credits are available on the purchase datebasis of specific criteria with which the Company must comply. The incentive credits are subject to future audits by the government authorities and a statute of limitations. Although the incentive credits may be cash equivalents, and include amounts held in money market funds which are actively traded (a Level 1 input). Restricted Cash
The Company had restricted cash of $34,174 at both December 31, 2020 and 2019, held in money market savings accounts as collateral. The restricted cash as of December 31, 2020 and 2019 is foradministered through the Company’s facility lease obligation. Restricted cash is classified as a component of cash, cash equivalents, and restricted cash in the accompanying consolidated balance sheets and consolidated statements of cash flows.
Accounts Receivable
Accounts receivable includes both billed and unbilled amounts. The Company makes judgments as to its ability to collect outstanding receivables and provides an allowance for receivables when collection becomes doubtful. Provisions are made based upon a specific review of all significant outstanding invoices and the overall quality and age of those invoices not specifically reviewed. The Company’s receivables represent amounts reimbursed under its government grants and contracts. The Company believes that credit risks associated with these government grants and contracts is not significant. To date,local tax authority, the Company has accounted for the incentives outside of the scope of FASB Accounting Standards Codification Topic 740, Income Taxes (“ASC 740”), since the incentives are not experienced any losses associatedlinked to the Company’s taxable income and can be realized regardless of whether the Company has generated taxable income in the respective jurisdictions. The Company accounts for these incentive credits as a government grant which analogizes with accountsInternational Accounting Standards 20 (“IAS 20”), Accounting for Government Grants and Disclosure of Government Assistance.
The Company records qualifying U.K. R&D expenses as receivable and does not maintain an allowance for doubtful accounts. Concentrations of Credit Risk
Financial instruments that potentially subject the Companya corresponding reduction to significant concentration of credit risk consist primarily of cash, cash equivalents, restricted cash, and accounts receivable. Periodically, the Company maintains deposits in financial institutions in excess of government insured limits. Management believes that the Company is not exposed to significant credit risk as the Company’s deposits are held at financial institutions that management believes to be of high credit quality. The Company has not experienced any losses in these deposits. The Company recognizes research grants and contracts earned in connection with the services provided on research and development projects. The Company provides creditR&D expense in the normal courseconsolidated statement of providing such services based on evaluations of the grantors’ financial conditionoperations and generally does not require collateral. To manage accounts receivable credit risk, the Company monitors the creditworthiness of its grantors. The U.S. government accounts for 97% and 95% of revenue forcomprehensive loss. During the years ended December 31, 20202022 and 2019, respectively. The U.S. government accounts for 100%2021, the Company recognized $1.8 million and $1.9 million, respectively, of accounts receivable for bothR&D credits as a reduction to R&D expense in the years endedconsolidated statement of operations and comprehensive loss. As of December 31, 20202022 and 2019.
Property2021, the Company had $1.6 million and Equipment, Net$1.8 million, respectively, of R&D credits included in “Income tax and R&D incentive receivables” on the accompanying consolidated balance sheets.
The Company records property and equipment at cost less accumulated depreciation and amortization. Expendituresqualifying Australian R&D as receivable with a full valuation reserve. Cash receipts for maintenance and repairs are charged to operations as incurred, whereas major improvements are capitalized as additions to property and equipment. Costs of assets under construction are capitalized but are not depreciated until the construction is substantially complete and the assets being constructed are ready for their intended use. Depreciation and amortizationAustralia R&D credits are recorded usingas long-term liability until it either passes an audit performed by the straight-line method overAustralian Tax Office, or the estimated useful livesstatute of limitations ends, whichever occurs first. Upon successfully passing an audit or the expiration of the assets, as follows:
Asset Category
| | Estimated Useful Life
| Computer and telecommunications
| | 3 – 5 years
| Software
| | 3 years
| Furniture, fixtures and equipment
| | 5 years
| Laboratory equipment
| | 7 years
| Leasehold improvements
| | Lesser of lease term or estimated useful lives
|
Warrants
Common stock warrants issued in connection with the 2018 Unit Offering, the 2018 and 2019 Registered Direct Offerings, and the 2020 Public Offering (all terms defined in Note 10 and Note 11), were classified as a componentstatute of permanent equity because they are freestanding financial instruments that were legally detachable and separately exercisable from other debt and equity instruments, are contingently exercisable, do not embody an obligation forlimitations, the Company will clear the liability and a corresponding reduction to repurchase its shares,R&D expense unless recognition criteria is met in a later year, in which case the R&D credit will be recorded as other income in the consolidated statement of operations and permitscomprehensive loss. During the holders to receive a fixed number of common shares upon exercise. In addition, such warrants did not provide any guarantee of value or return. The 2018 Registered Direct Offering and 2019 Registered Direct Offering triggered down round adjustments to the exercise price of warrants issued in connection with the 2018 Unit Offering. In 2019,year ended December 31, 2022, the Company treatedreceived a total of $3.6 million in cash for R&D incentive credit related to R&D costs that the value ofCompany incurred during the effect offiscal years 2021 and 2020 through the reductionparticipation in exercise price as a deemed dividend, resulting in a reduction to income available to common shareholders (see Note 11).
There was a reduction in exercise price in 2020 due to the At-the-Market Offering which triggered an anti-dilution provision under the warrant agreement with the Company’s holders of redeemable preferred stock (see Note 11).
Stock-based Compensation
The Company accounts for all stock-based compensation granted to employees and non-employees using a fair value method. Stock-based compensation awarded to employees is measured at the grant date fair value of stock option grantsAustralian R&D credit program, and is recognized over the requisite service period of the awards, usually the vesting period, on a straight-line basis, net of estimated forfeitures.
If awards are modified, the Company compares the fair value of the affected award measured immediately prior to modification to its value after modification. To the extent that the fair value of the modified award exceeds the original award, the incremental fair value of the modified award is recognized as compensationincluded in “Other long-term liabilities” on the date of modification for vested awards, and over the remaining vesting period for unvested awards.accompanying consolidated balance sheets.
Income Taxes The Company accounts for income taxes usingin accordance with ASC 740. ASC 740 uses the asset and liability approach, which requires the recognition of future tax benefits or liabilities on the temporary differences between the financial reporting and tax bases of our assets and liabilities. Deferred tax assets and liabilities represent future tax consequences of temporary differences between the financial statement carrying amounts and the tax basis of assets and liabilities and for loss carryforwards using enacted tax rates expected to be in effect in the years in which the differences reverse. A valuation allowance is established when necessary to reduce deferred tax assets to the amounts expected to be realized. The Company also recognizes a tax benefit from uncertain tax positions only if it is “more likely than not” that the position is sustainable based on its technical merits. The Company accounts for interest and penalties related to uncertain tax positions as part of its provision for income taxes. To date, the Company has not incurred interest and penalties related to uncertain tax positions. Should such costs be incurred, they would be classified as a component of provision for income taxes. The Company conducts R&D activities potentially qualified to claim research tax credits for U.S. federal and state purposes under Internal Revenue Code Section 41. The Company has not performed a formal study claiming these credits in the tax returns because the Company does not yet have taxable profits. Once the Company becomes profitable, it will likely have a study prepared, and the amount of R&D tax credits available could generate income tax benefit, subject to an annual Section 383 limitation and valuation allowance for realizability of the deferred tax asset. Comprehensive Loss For the years presented, the total comprehensive loss includes net loss and other comprehensive income (loss) which represents unrealized gains or losses on short-term investments. Net Loss per Share Basic net loss per share is computed by dividing the net loss attributable to common stockholders by the weighted-average number of common shares outstanding for the period without consideration for potentially dilutive securities. The Company computes diluted net loss per common share after giving consideration to all potentially dilutive common equivalents, including all unvested restricted stock, common stock warrants, and common stock options outstanding during the period except where the effect of such non-participating securities would be anti-dilutive. Diluted net loss per share is computed by dividing the net loss attributable to common stockholders by the weighted-average number of common shares and dilutive common stock equivalents outstanding for the period determined using the treasury-stock and if-converted methods. Leases
The Company’s headquarters lease is the primary lease, accounted for as an operating lease under the new lease accounting guidance, which the Company adopted on January 1, 2019 under the prospective optional transition method. The Company elected the package of practical expedients permitted. Accordingly, the Company accounted for its existing operating leases as operating leases under the new guidance, without reassessing (a) whether the contracts contain a lease, (b) whether classification of the operating leases would be different in accordance, or (c) whether the unamortized initial direct costs before transition adjustments would have met the definition of initial direct costs at lease commencement. In addition, the Company does not allocate the consideration between lease and non-lease components.
The Company determines if an arrangement is a lease at inception. Operating leases are recorded as a current and long-term lease obligation, with a corresponding right of use lease assets.
The lease obligations represent the Company’s obligation to make lease payments arising from the lease. The right of use lease assets represent the Company’s right to use an underlying asset for the lease term. The lease obligations and the operating right of use lease assets are recognized at the commencement date based on the present value of lease payments over the lease term. As most of the Company’s leases do not provide an implicit rate, the Company uses its incremental borrowing rate based on the information available at the commencement date in determining the present value of lease payments. The Company’s lease terms may include options to extend or terminate the lease when it is reasonably certain that the Company will exercise that option. Lease expense for lease payments is recognized on a straight-line basis over the lease term.
Short-term leases are leases having a term of twelve months or less. The Company recognizes short-term leases on a straight-line basis and does not record a related lease asset or liability for such leases.
Lease incentives and allowance provided by our landlord for the construction of leasehold improvements are recorded as lease incentive obligations as the related construction costs are incurred, up to the maximum allowance.
Contingent Consideration
The Company records contingent consideration associated with development and regulatory milestones that meets the definition of a liability under ASC 480 at fair value. The fair value model used to calculate this obligation is based on the Monte Carlo simulation that has been risk adjusted based on the probability of achievement of the milestones. The inputs the Company uses for determining the fair value of the contingent consideration associated with development and regulatory milestones are Level 3 fair value measurements. The Company re-evaluates the fair value on a quarterly basis. Changes in the fair value can result from adjustments to the probability of achievement of the milestones, stock price, volatility and the risk-free interest rate.
The change in Company’s estimates associated with payments which will become due and payable for development and regulatory milestones will change the fair value of contingent consideration, resulting in a charge or contra expense to research and development expense in the period in which the increase or decrease is determined.
Recently Issued Accounting Pronouncements Recently Adopted:
In August 2018, the FASB issued ASU No. 2018-13, Fair Value Measurement (Topic 820) – Disclosure Framework – Changes to the Disclosure Requirements for Fair Value Measurement (“ASU No. 2018-13”). ASU No. 2018-13 was issued to modify and enhance the disclosure requirements for fair value measurements and eliminates certain disclosure requirements, such as the amount of, and reasons for, transfers between Level 1 and Level 2 of the fair value hierarchy. This ASU adds new disclosure requirements for Level 3 measurements and is effective for fiscal years beginning after December 15, 2019 and interim periods within those fiscal years. The Company not yet adopted this guidance in the first quarter of 2020, which resulted in expanded disclosures in Note 3 regarding the Company’s recurring Level 3 fair value measurements.
Not Yet Adopted:
In June 2016, the FASB issued ASU No. 2016-13, Financial Instruments – Credit Losses (Topic 326): Measurement of Credit Losses on Financial Instruments (“ASU No. 2016-13”). ASU No. 2016-13 requires entitiesfinancial assets measured at amortized cost to measure allbe presented at the net amount expected to be collected and any unrealized loss relating to available-for-sale debt securities that is a result of credit losses be recorded through an allowance for financial assets held at the reporting date basedcredit losses. The Company will adopt this new accounting standard on historical experience, current conditions and reasonable and supportable forecasts. This update also required enhanced disclosures to help financial statement users better understand significant estimates and judgments used in estimating credit losses, as well as the credit quality and underwriting standards of an entity’s portfolio. This standard is effective for the Company as a smaller reporting company beginning January 1, 2023.2023 using a modified retrospective method. Adoption of this update is not expected to have a material impact on the Company’s consolidated financial statement disclosure requirements. In December 2019, the FASB issued ASU 2019-12, Income Taxes (Topic 740), Simplifying the Accounting for Income Taxes (“ASU No. 2019-12”). ASU 2019-12 amends the approachesstatements and methodologies in accounting for income taxes during interim periods and makes changes to certain income tax classifications. The new standard allows exceptions to the use of the incremental approach for intra-period tax allocation, when there is a loss from continuing operations and income or a gain from other items, and to the general methodology for calculating income taxes in an interim period, when a year-to-date loss exceeds the anticipated loss for the year. The standard also requires franchise or similar taxes partially based on income to be reported as income tax and the effects of enacted changes in tax laws or rates to be included in the annual effective tax rate computation from the date of enactment. Lastly, in any future acquisition, the Company would be required to evaluate when the step-up in the tax basis of goodwill is part of the business combination and when it should be considered a separate transaction. The standard will be effective for the Company beginning January 1, 2021, with early adoption of the amendments permitted. Adoption is not expected to have a material impact on the Company’s consolidated financial statement disclosure requirements.related disclosures.
3. Fair Value Measurement The Company records cash equivalents, short-term investments and contingent consideration and warrant liability at fair value on a recurring basis. Fair value is an exit price, representing the amount that would be received from the sale of an asset or paid to transfer a liability in an orderly transaction between market participants based on assumptions that market participants would use in pricing an asset or liability. The Company’s assets and liabilities measured at fair value on a recurring basis atas of December 31, 20202022 consisted of the following:following (in thousands): | | Fair Value Measurement at December 31, 2020 | | | | | Total | | | Level 1 | | | Level 2 | | | Level 3 | | | | | | | | | | | | | | | | | | | | Fair Value Measurement at December 31, 2022 | | | | Total | | Level 1 | | Level 2 | | Level 3 | Assets: | | | | | | | | | | | | | | Cash equivalents - money market funds | | $ | 90,389,473 | | | $ | 90,389,473 | | | $ | — | | | $ | — | | | $ | 105,794 | | $ | 105,794 | | $ | — | | $ | — | Short-term investments | | | 100,005,558 | | | | — | | | | 100,005,558 | | | | — | | | | 73,783 | | | — | | | 73,783 | | | — | Contingent consideration liability (see Note 8) | | | 5,390,000 | | | | — | | | | — | | | | 5,390,000 | | | Warrant liability | | | 10,000 | | | | — | | | | — | | | | 10,000 | | | Total | | | $ | 179,577 | | $ | 105,794 | | $ | 73,783 | | $ | — |
The warranty liability is included in Other long-term liabilities in the consolidated balance sheet at December 31, 2020.
The Company’s assets and liabilities measured at fair value on a recurring basis atas of December 31, 20192021 consisted of the following:following (in thousands): | | Fair Value Measurement at December 31, 2019 | | | | Total | | | Level 1 | | | Level 2 | | | Level 3 | | Cash equivalents — money market fund | | $ | 8,034,640 | | | $ | 8,034,640 | | | $ | — | | | $ | — | | Short-term investments | | | 28,277,386 | | | | — | | | | 28,277,386 | | | | — | | Contingent consideration liability (see Note 8) | | | 2,750,000 | | | | — | | | | — | | | | 2,750,000 | | Warrant liability | | | 10,000 | | | | — | | | | — | | | | 10,000 | |
| | | | | | | | | | | | | | | Fair Value Measurement at December 31,��2021 | | | Total | | Level 1 | | Level 2 | | Level 3 | Assets: | | | | | | | | | | | | | Cash equivalents - money market funds | | $ | 65,634 | | $ | 65,634 | | $ | — | | $ | — | Total | | $ | 65,634 | | $ | 65,634 | | $ | — | | $ | — | Liabilities: | | | | | | | | | | | | | Contingent consideration liability (see Note 8) | | $ | 6,090 | | $ | — | | $ | — | | $ | 6,090 | Total | | $ | 6,090 | | $ | — | | $ | — | | $ | 6,090 |
The warranty
As described in Note 8 the remaining milestone payment underlying the contingent consideration liability is includedwas fully settled during 2022 in Other long-term liabilities inshares of the consolidated balance sheet atCompany’s common stock. As of December 31, 2019.2022, the Company had no contingent consideration liability. Short-term investments have been initially valued at the transaction price and subsequently valued at the end of each reporting period utilizing third party pricing services or other market observable data (Level 2). The pricing services utilize industry standard valuation models, including both income and market-based approaches and observable market inputs to determine value. Short-term investments with quoted prices at December 31, 20202022 as shown below:below (in thousands): | | As of December 31, 2020 | | | | | Amortized Cost | | | Unrealized Gain | | | Market Value | | | | | | | | | | | | | | | | | December 31, 2022 | | | | Amortized Cost | | Unrealized (Loss) Gain | | Market Value | United States treasury securities | | $ | 20,052,757 | | | $ | 1,843 | | | $ | 20,054,600 | | | $ | 15,868 | | $ | (86) | | $ | 15,782 | Asset-backed and corporate debt securities | | | 54,935,963 | | | | (6,197 | ) | | | 54,929,766 | | | Certificates of deposit | | | 25,021,192 | | | | — | | | | 25,021,192 | | | Commercial paper and corporate debt securities | | | | 50,747 | | | (71) | | | 50,676 | Asset backed securities | | | | 5,427 | | | (35) | | | 5,392 | Agency debt securities | | | | 1,928 | | | 5 | | | 1,933 | Total | | $ | 100,009,912 | | | $ | (4,354 | ) | | $ | 100,005,558 | | | $ | 73,970 | | $ | (187) | | $ | 73,783 |
Short-term investments with quoted prices at
As of December 31, 2019 as shown below: | | As of December 31, 2019 | | | | Amortized Cost | | | Unrealized Gain | | | Market Value | | United States treasury securities | | $ | 3,394,579 | | | $ | 3,228 | | | $ | 3,397,807 | | Asset-backed and corporate debt securities | | | 24,862,800 | | | | 16,779 | | | | 24,879,579 | | Total | | $ | 28,257,379 | | | $ | 20,007 | | | $ | 28,277,386 | |
The2021, the fair value of contingent payments classified as a liability is was based on the regulatory milestones described in Note 8 and was estimated using the Monte Carlo simulation valuation model with Level 3 inputs.
The assumptions used to estimate the fair value of contingent payments that were classified as a liability at December 31, 2020 2021 include the following significant unobservable inputs:inputs: | | | | | Unobservable input | | Value or Range | | Weighted-Average | Expected volatility | | 80.1% | | 80.1% | Risk-free interest rate | | 0.26% | | 0.26% | Cost of capital | | 30% | | 30% | Discount for lack of marketability | | 8%‑13% | | 11% | Probability of payment | | 88% | | 88% | Projected year of payment | | 2022 | | 2022 |
Unobservable input | | Value or Range | | | Weighted Average | | Expected volatility | | 114.9% | | | 114.9% | | Risk-free interest rate | | 0.11% | | | 0.11% | | Cost of capital | | 30% | | | 30% | | Discount for lack of marketability | | 9%-15% | | | 12% | | Probability of payment | | 63% | | | 63% | | Projected year of payment | | 2022 | | | 2022 | |
108 Unobservable input | | Value or Range | | | Weighted Average | | Expected volatility | | 95.3% | | | 95.3% | | Risk-free interest rate | | 1.60% | | | 1.60% | | Cost of capital | | 30% | | | 30% | | Discount for lack of marketability | | 8%-12% | | | 10% | | Probability of payment | | 31%-50% | | | 46% | | Projected year of payment | | 2020-2022 | | | 2020 | |
The Company’s warrant liability is valued using the Monte Carlo simulation valuation model with Level 3 inputs.
If applicable, the Company will recognize transfers into and out of levels within the fair value hierarchy at the end of the reporting period in which the actual event or change in circumstance occurs. There were no transfers into and out of any of the levels of the fair value hierarchy during 2020 or 2019.
Separate disclosure is required for assets and liabilities measured at fair value on a recurring basis from those measured at fair value on a non-recurring basis. Assets recorded at fair value on a non-recurring basis, such as property and equipment and intangible assets are recognized at fair value when they are impaired. During the year ended December 31, 2020,2022, the Company had no significant assets or liabilities that were measured at fair value on a non-recurring basis. During the year ended December 31, 2019,2021, the Company recognized an intangible assetrecorded non-cash impairment (see Note 5) measured at fair valuecharges to property and equipment, net on a non-recurring basis.basis (see below). Lonza Manufacturing Agreement In March 2021, the Company expanded its manufacturing collaboration with Lonza Houston, Inc. (“Lonza”) for the manufacture of AdCOVID or other adenovirus-based vaccines. Under the expanded agreement, the Company had committed approximately $23.0 million to Lonza to procure long-lead equipment and construct a dedicated manufacturing suite for clinical and commercial production of adenovirus-based vaccines. In June 2021, the Company announced the discontinuation of further development of AdCOVID following the Company’s review of findings from its Phase 1 clinical trial. Construction continued at Lonza, and the Company assessed its strategic options with respect to the suite. This work was completed during the fourth quarter of 2021. The Company capitalized a total of $11.4 million as construction-in-progress (“CIP”) during the nine months ended September 30, 2021 under this expanded agreement. In connection with the discontinuation of further development of AdCOVID, the Company recorded a non-cash impairment charge of $8.1 million in the unaudited consolidated statements of operations and comprehensive loss for the nine months ended September 30, 2021 to write-down the CIP associated with the construction of the Lonza facility to its fair value of $3.3 million as of September 30, 2021. As of September 30, 2021, the fair value of the CIP related assets was primarily determined utilizing the cost approach, which reflected the replacement cost of the asset being appraised, adjusted for contractual restrictions on the assets, the probability of satisfying the contractual restrictions, physical deterioration, functional obsolescence and economic obsolescence. The fair value measurement was considered a Level 3 measurement within the valuation hierarchy. Furthermore, the remaining $3.3 million CIP was fully charged to impairment during the three months ended December 31, 2021 upon termination of the agreement. 4. Property and Equipment, Net Property and equipment, net consistedconsists of the following:following (in thousands): | | | | | | | | | December 31, | | | 2022 | | 2021 | Furniture, fixtures and equipment | | $ | 163 | | | 222 | Laboratory equipment | | | 295 | | | 1,040 | Computers and telecommunications | | | 194 | | | 291 | Software | | | 178 | | | 148 | Leasehold improvements | | | 1,749 | | | 1,794 | Property and equipment, at cost | | | 2,579 | | | 3,495 | Less: accumulated depreciation and amortization | | | (1,498) | | | (2,047) | Property and equipment, net | | $ | 1,081 | | $ | 1,448 |
| | December 31, | | | | 2020 | | | 2019 | | Furniture, fixtures and equipment | | $ | 125,538 | | | $ | 121,491 | | Laboratory equipment | | | 959,585 | | | | 926,590 | | Computers and telecommunications | | | 220,316 | | | | 150,517 | | Software | | | 64,409 | | | | 25,069 | | Leasehold improvements | | | 1,285,883 | | | | 1,228,108 | | Property and equipment, at cost | | | 2,655,731 | | | | 2,451,775 | | Less accumulated depreciation and amortization | | | (1,598,811 | ) | | | (1,347,567 | ) | Property and equipment, net | | $ | 1,056,920 | | | $ | 1,104,208 | |
Depreciation expense related to property and equipment for the years ended December 31, 20202022 and 20192021 was $251,244$0.5 million and $239,821,$0.4 million, respectively. 5. Intangible Assets The Company’s intangible assets consistedconsist of the following:following (in thousands): | | | | | | | | | | | | | | | | | | | Gross | | | | | | | | | | | | Estimated | | Carrying | | Accumulated | | | | | Net Book | | | Useful Lives | | Value | | Amortization | | Impairment | | Value | December 31, 2022: | | | | | | | | | | | | | | | IPR&D assets | | Indefinite | | $ | 12,419 | | $ | — | | $ | — | | $ | 12,419 | Total | | | | $ | 12,419 | | $ | — | | $ | — | | $ | 12,419 | December 31, 2021: | | | | | | | | | | | | | | | Internally developed patents | | 6–20 years | | $ | 1,079 | | $ | (500) | | $ | (579) | | $ | — | Acquired licenses | | 16–20 years | | | 285 | | | (285) | | | — | | | — | Total intangible assets subject to amortization | | | | | 1,364 | | | (785) | | | (579) | | | — | IPR&D assets | | Indefinite | | | 12,419 | | | — | | | — | | | 12,419 | Total | | | | $ | 13,783 | | $ | (785) | | $ | (579) | | $ | 12,419 |
| | December 31, 2020 | | | | Estimated Useful Lives | | Gross Carrying Value | | | Accumulated Amortization | | | Impairment | | | Net Book Value | | Internally developed patents | | 6 – 10 years | | $ | 884,787 | | | $ | (479,908 | ) | | $ | — | | | $ | 404,879 | | Acquired licenses | | 16 – 20 years | | | 285,000 | | | | (285,000 | ) | | | — | | | | — | | Total intangible assets subject to amortization | | | | | 1,169,787 | | | | (764,908 | ) | | | — | | | | 404,879 | | IPR&D assets | | Indefinite | | | 12,418,967 | | | | — | | | | — | | | | 12,418,967 | | Total | | | | $ | 13,588,754 | | | $ | (764,908 | ) | | $ | — | | | $ | 12,823,846 | |
| | December 31, 2019 | | | | Estimated Useful Lives | | Gross Carrying Value | | | Accumulated Amortization | | | Impairment | | | Net Book Value | | Internally developed patents | | 6 – 10 years | | $ | 746,323 | | | $ | (448,874 | ) | | $ | — | | | $ | 297,449 | | Acquired licenses | | 16 – 20 years | | | 285,000 | | | | (269,221 | ) | | | — | | | | 15,779 | | Total intangible assets subject to amortization | | | | | 1,031,323 | | | | (718,095 | ) | | | — | | | | 313,228 | | IPR&D assets | | Indefinite | | | 13,418,967 | | | | — | | | | (1,000,000 | ) | | | 12,418,967 | | Total | | | | $ | 14,450,290 | | | $ | (718,095 | ) | | $ | (1,000,000 | ) | | $ | 12,732,195 | |
Amortization expenseAs of intangible assets subject to amortization totaled $46,813 and $147,500December 31, 2021, the Company recorded an impairment loss of $0.6 million for the years ended December 31, 2020 and 2019, respectively, and was classified asremaining net book value of the internally developed patents that are associated with the Company's discontinued development programs. The impairment loss has been recorded to research and development expenses in the accompanying audited consolidated statements of operations and comprehensive loss.
As of December 31, 2020, future estimated amortization expense is as follows:
For the Year Ended December 31, | | | | | 2021 | | $ | 26,561 | | 2022 | | | 26,561 | | 2023 | | | 26,561 | | 2024 | | | 22,655 | | 2025 | | | 17,187 | | 2026 and thereafter | | | 285,354 | | Total | | $ | 404,879 | |
The above future estimated amortization expense does not include potential amortization charges related to the remaining carrying value of IPR&D assets as of December 31, 2020. Those assets, which represent incomplete technologies, will be amortized to expense once the underlying technologies are substantially complete over their estimated useful lives, expected to be 15 to 18 years. In the event that in the future the Company ceases the development of these assets, the remaining carrying value would be written off at that time. IPR&D assets are periodically assessed for impairment by considering the state of completion of the projects, the remaining activities required to complete development, the anticipated market for the completed products, and anticipated future cash required to complete development.
There was no IPR&D impairment loss during the yearyears ended December 31, 2020. During 2019, as a result of the SparVax-L NIAID contract completion2022 and the U.S. government’s funding prioritization of only single dose anthrax vaccine candidates, the Company abandoned the project and concluded that the full remaining net book value of the SparVax-L IPR&D asset was impaired. As a result, $1.0 million was written off as an impairment charge which was classified as a component of operating expenses during the year ended December 31, 2019.2021 6. Leases The Company rents office and laboratory space in the United States. The Company alsoStates, which are operating leases office equipment under a non-cancellable equipment lease through December 2022.and expire in April 2025. Rent expense under these leases during each of the years ended December 31, 20202022 and 2019 under all of
the Company’s operating leases2021 was $407,200 and $346,603, respectively,$0.5 million, which includes short-term leases and variable lease costs that are not included in the lease obligation.
The office space lease provides for increases in future minimum annual rental payments as defined in the lease agreements. The office space lease also includes an option to renew the lease as of the end of the term. The Company has determined that the lease renewal option is not reasonably certain of being exercised. The cash paid for operating lease liabilities for each of the yearyears ended December 31, 20202022 and 20192021 was $391,090 and $380,805, respectively. $0.5 million. Supplemental balance sheet information related to the operating leases is as follows:follows (in thousands): | | | | | | | | | | December 31, | | | | 2022 | | 2021 | | Operating lease obligations (see Note 7 and 9) | | $ | 1,124 | | $ | 1,535 | | Operating lease right-of-use assets (included in "Other assets" in Balance Sheet) | | $ | 596 | | $ | 798 | | Weighted-average remaining lease term (years) | | | 2.3 | | | 3.3 | | Weighted-average discount rate | | | 7.2 | % | | 7.2 | % |
| | December 31, | | | | 2020 | | | 2019 | | Operating lease obligations (see Note 7 and 9) | | $ | 1,824,840 | | | $ | 1,744,128 | | Right of use asset | | $ | 903,825 | | | $ | 698,321 | | Weighted-average remaining lease term | | | 4.33 | | | | 5.33 | | Weighted-average discount rate | | | 7.3 | % | | | 8.0 | % |
Maturities of operating lease liabilities are as follows:follows (in thousands): | | | | Year ending December 31, | | | | 2023 | | $ | 515 | 2024 | | | 526 | 2025 | | | 176 | Total operating lease payments | | | 1,217 | Less: imputed interest | | | (93) | Total operating lease liabilities (see Note 7 and 9) | | $ | 1,124 |
Year ending December 31, | | | | | 2021 | | $ | 475,372 | | 2022 | | | 484,484 | | 2023 | | | 493,868 | | 2024 | | | 503,535 | | 2025 | | | 168,929 | | Total operating lease payments | | | 2,126,188 | | Less: imputed interest | | | (301,348 | ) | Total operating lease liabilities (see Note 7 and 9) | | $ | 1,824,840 | |
110 7. Accrued Expenses and Other Current Liabilities Accrued expense and other current liabilities consist of the following:following (in thousands): | | | | | | | | | December 31, | | | 2022 | | 2021 | Accrued professional services | | $ | 276 | | $ | 396 | Accrued payroll and employee benefits | | | 2,955 | | | 2,313 | Accrued research and development | | | 7,295 | | | 6,988 | Lease obligation, current portion (see Note 6) | | | 452 | | | 411 | Excess tax refund payable | | | 1,169 | | | — | Accrued interest and other | | | 103 | | | 44 | Total accrued expenses and other current liabilities | | $ | 12,250 | | $ | 10,152 |
| | December 31, | | | | 2020 | | | 2019 | | Accrued professional services | | $ | 1,350,194 | | | $ | 429,467 | | Accrued payroll and employee benefits | | | 2,351,599 | | | | 1,183,130 | | Accrued interest | | | 13,016 | | | | 5,047 | | Accrued research and development | | | 7,316,876 | | | | 1,966,111 | | Lease obligation, current portion (see Note 6) | | | 356,716 | | | | 259,449 | | Deferred revenue | | | 19,753 | | | | 61,563 | | Total accrued expenses | | $ | 11,408,154 | | | $ | 3,904,767 | |
8. Contingent Consideration The Company entered into an Agreement and Plan of Merger and Reorganization, dated July 8, 2019, by and among the Company, Springfield Merger Sub, Inc., Springfield Merger Sub, LLC, Spitfire Pharma, Inc. and David Collier, as the Stockholder Representative (the “Spitfire Merger Agreement”) to acquire all of the equity interests of Spitfire Pharma, Inc. (“Spitfire”). Spitfire was a privately held, preclinical pharmaceutical company developing a novel dual GLP-1/glucagon receptor dual agonist for the treatment of non-alcoholic steatohepatitis. NASH. The transaction closed on July 12, 2019. The Company issued 1,887,250 unregistered shares of its common stock (the “shares”) as upfront consideration to certain former securityholders of Spitfire, (collectively, the “Spitfire Equityholders”), representing an amount equal to $5.0 million less working capital and transaction expense adjustment amounts as defined in the agreement.
The acquisition of Spitfire was accounted for as an asset acquisition instead of a business combination because substantially all of the fair value of the gross assets acquired was concentrated in a single identifiable asset or group of similar identifiable assets, and therefore, the asset was not considered a business. The Company expensed the acquired intellectual property as of the acquisition date as in-process research and development with no alternative future uses. During the year ended December 31, 2019, the Company recorded an in-process research and development expense of $4.3 million for the upfront consideration, which included the fair value of the common stock transferred and net liabilities assumed. Transaction costs of $0.7 million was recorded within research and development expense on the Company’s consolidated statements of operations and comprehensive loss during the year ended December 31, 2019. The Spitfire Merger Agreement also includes future contingent payments up to $88.0 million payable in either cash andor shares of the Company’s common stock as follows (each, a “Milestone Event”):follows: | ● | a one-time payment of $5.0 million (the “IND Milestone Consideration Amount”) within sixty days of the submission of an Investigational New Drug Application (“IND”) to the United States Food and Drug Administration (the “FDA”) or other applicable governmental authority in a foreign jurisdiction, which IND has not been rejected or placed on clinical hold by the FDA or such applicable foreign governmental authority within time specified in the Spitfire Merger Agreement; |
| ● | a one-time payment of $3.0 million (the “Phase 2 Milestone Consideration Amount” and together with the IND Milestone Consideration Amount, the “Regulatory Milestones”) within sixty days of the initiation (first patient, first dosing) of the first Phase 2 clinical trial of a product candidate anywhere in the world; and |
| ● | payments of up to $80.0 million upon the achievement of specified worldwide net sales (the “Sales Milestones”) of all products developed using the technology acquired in the License Agreement within ten years following the approval of a new drug application filed with the FDA. |
a one-time payment of $5.0 million (the “IND Milestone Consideration Amount”) within sixty days of the submission of an Investigational New Drug Application (“IND”) to the United States Food and Drug Administration (the “FDA”) or other applicable governmental authority in a foreign jurisdiction, which IND has not been rejected or placed on clinical hold by the FDA or such applicable foreign governmental authority within time specified in the Spitfire Merger Agreement;
a one-time payment of $3.0 million (the “Phase 2 Milestone Consideration Amount” and together with the IND Milestone Consideration Amount, the “Regulatory Milestones”) within sixty days of the initiation of a human clinical trial of a product candidate anywhere in the world; and
payments of up to $80.0 million upon the achievement of specified worldwide net sales (the “Sales Milestones”) of all products developed using the technology acquired in the License Agreement within ten years following the approval of a new drug application filed with the FDA.
The Regulatory Milestones will bewere payable in shares of the Company’s Common Stock, with the number of shares of the Company’s Common Stock to becommon stock issued in connection with each milestone amount, if any, are dependent on the share price at the time of achievement. The number of any shares issued in consideration for the IND Milestone Consideration Amount will bewas determined based on the lower of (A) the average of the closing prices of our Common Stockthe Company’s common stock as reported on the Nasdaq Global Market for the twenty (20) consecutive trading days prior to the IND Reference Date or (B) $2.95. The valuenumber of any shares issued in consideration for the Phase 2 Milestone Consideration Amount shall bewas determined based on the lower of (A) on the average of the closing trading prices of our Common Stockthe Company’s common stock as reported on the Nasdaq Global Market for the twenty (20) consecutive trading days immediately preceding the date of the occurrence of the Phase 2 Milestone Event or (B) $3.54. The future contingent payments related to the Regulatory Milestones are stock-based payments accounted for under FASB Accounting Standards Codification Topic 480, Distinguishing Liabilities From Equity (“ASC 480”). Such stock-based payments are subject to a lock-up whereby 50% of the shares are released at 3 months and 50% are released at 6 months. The future contingent payments related to the Sales Milestones are predominately cash-based payments accounted for under FASB Accounting Standards Codification Topic 450, Contingencies. Accordingly, the Company will recognize the Sales Milestones when the contingency is resolvedprobable and the amount is paid or payable.can be reasonably estimated. OnIn November 3, 2020, the Company received acknowledgement from the Australian Government Department of Health on the Company’s submitted clinical trial notification (“CTN”) which triggered the obligation to settle the IND Milestone payment to the former owners. As a result, on November 19, 2020, the Company issued 1,694,906 shares of its Common Stockcommon stock valued at $9.57 per share for the amount value of $13.6 million to the former Spitfire stockholders. Pursuantstockholders to satisfy the obligations under IND Milestone.
On April 26, 2022, the Company dosed the first patient in the Phase 2 MOMENTUM trial of pemvidutide in obesity, which triggered the obligation to pay the Phase 2 Milestone Consideration Amount to the Spitfire Merger Agreement,former owners. As a result, on June 10, 2022, the Company issued 847,444 shares of its common stock valued at $8.55 per share for the shares within sixty daysamount value of $7.2 million to the submission offormer Spitfire stockholders. From the CTN, which was October 29, 2020. From September 30, 2020last valuation date on March 31, 2022 through June 10, 2022, the date of issuance, the Company recognized a decreasean increase in the fair value of the INDPhase 2 Milestone paymentConsideration Amount of $5.4$1.9 million to research and development expense and reclassified the balance in the contingent consideration liability associated with the fair value of the IND Milestone payment to equity in the Company’s consolidated balance sheet. At the acquisition date, As of December 31, 2022, the Company estimated futurehad no contingent consideration of $2.8 million for the Regulatory Milestones which was based upon a Monte Carlo simulation that was risk adjusted based on the probability of achieving the milestones and a discount for lack of marketability, which was expensed to in-process research and development expenses during the third quarter of 2019. The Company remeasures the fair value of contingent consideration at each reporting date, and belowliability.
Below is a summary of the contingent consideration activity through December 31, 2020:(in thousands):
| | Year Ended December 31, | | | | 2020 | | | 2019 | | Beginning balance | | $ | 2,750,000 | | | $ | — | | Acquisition date fair value | | | — | | | | 2,750,000 | | Cumulative change in fair value | | | 16,265,010 | | | | — | | Fair value of payments settled in common stock (IND Milestone) | | | (13,625,010 | ) | | | — | | Ending balance | | $ | 5,390,000 | | | $ | 2,750,000 | |
| | | | | | | | | Year Ended December 31, | | | 2022 | | 2021 | Beginning balance | | $ | 6,090 | | $ | 5,390 | Change in fair value | | | 86 | | | 700 | Fair value of payments settled in common stock (Phase II Milestone) | | | (6,176) | | | — | Ending balance | | $ | — | | $ | 6,090 |
The net increase in fair value throughout 2020 was primarily attributable to an increase in the closing share price of the Company’s common stock and in the probability of milestone achievement, partially offset by the fair value of the IND Milestone payment settled in common stock. Any changes in fair value including the initial determination of acquisition date fair value have been recorded within research and development expense during the respective periods presented.
9. Notes Payable and Other Long-Term Liabilities Paycheck Protection Program
On April 7, 2020, the Company applied for a loan from ServisFirst Bank, as lender, pursuant to the Paycheck Protection Program of the Coronavirus Aid, Relief, and Economic Security Act (the “CARES Act”) as administered by the U.S. Small Business Administration (the "SBA"). On April 13, 2020, the Loan was approved and the Company received the proceeds from a loan in the amount of $632,000 (the “PPP Loan”).
The PPP Loan, which took the form of a promissory note (the “Promissory Note”), was set to mature on April 7, 2022 and bore interest at a rate of 1% per annum. Monthly principal and interest payments, less the amount of any potential forgiveness (discussed below), was to commence on November 7, 2020. The Promissory Note provided for customary events of default, including, among others, those relating to failure to make payment, bankruptcy, breaches of representations and material adverse effects. The Company could prepay the principal of the PPP Loan at any time without incurring any prepayment charges.
All or a portion of the Loan may be forgiven by the SBA and lender upon application by the Company upon documentation of expenditures in accordance with the SBA requirements. On July 21, 2020, the Company voluntarily extinguished the Promissory Note by paying the outstanding principal and accrued interest in cash.
The Company’s other long-term liabilities are summarized as follows:follows (in thousands): | | December 31, | | | | | 2020 | | | 2019 | | | | | | | | | | | | | | December 31, | | | | 2022 | | 2021 | Research and development incentive credit | | | $ | 3,599 | | $ | — | Lease obligation, long-term portion (see Note 6) | | $ | 1,468,124 | | | $ | 1,484,679 | | | | 672 | | | 1,124 | Conditional economic incentive grants | | | 250,000 | | | | 250,000 | | | | 250 | | | 250 | Other | | | 110,319 | | | | 130,196 | | | | 60 | | | 80 | Total other long-term liabilities | | $ | 1,828,443 | | | $ | 1,864,875 | | | $ | 4,581 | | $ | 1,454 |
BPI France Notes
The Company had two non-interest-bearing research and development funding arrangements with BPI France that were entered into in December 2013 to provide up to €750,000 in research funding in the first arrangement and up to €250,000 in the second arrangement. The Company was permitted to draw 50% of the funds upon the signing of the arrangements, an additional 30% contingent upon a financial audit and technical progress report, and the remaining amounts at the completion of the research and development project being funded by the arrangements. In October 2016, the Company and BPI France agreed to extend the term on the arrangement by two years. The total amount advanced under the arrangements was €500,000. In April 2019, the Company was notified that €102,951 exceeded the allowable funding in accordance with the arrangement and made payment of this amount on June 5, 2019. In September 2019, the Company was notified that €238,229 ($265,540) was converted into a grant and recognized as grant revenue forR&D incentive credit Program
During the year ended December 31, 2019. In October 2019,2022, the Company paidreceived a total of $3.6 million in cash for research and development (“R&D”) incentive credit related to R&D costs that the remaining balance on the BPI France notes. The total repaymentsCompany incurred during the year ended December 31, 2019 was $292,002.fiscal years 2021 and 2020 through the participation in the Australian research and development incentive credit program administered through the Australian Tax Office. The Company recorded the receipt as long-term liability until there is reasonable assurance that the Company will comply with the conditions attached to the incentive credit. Economic Incentive Grants The Company has two conditional economic incentive grants for a total of $250,000 from Montgomery County, Maryland and the State of Maryland. The Montgomery County grant was received in May of 2018, with a term expiring on February 28, 2028. The State of Maryland grant was received in October 2019, with a 10-year term expiring on December 31, 2029. These grants are conditional primarily based on the Company maintaining its current headquarter locations in addition to employing a required number
of employees at different reporting dates through the term of the grant. grants. The Company is accruing 3% interest on both grants and has recorded $12,625 and $5,042approximately $8,000 in interest expense for each of the years ended December 31, 20202022 and 2019, respectively.2021. 10. Common Stock The Amended and Restated Certificate of Incorporation, as amended (“Charter”), authorized the Company to issue 200,000,000 shares of common stock, par value $0.0001 per share. As of December 31, 2022, the Company had 49,199,845 shares of common stock issued and outstanding. The Charter also authorized the Company to issue 1,000,000 shares of preferred stock, par value $0.0001 per share. As of December 31, 2022, the Company had no shares of preferred stock issued and outstanding. At-the-Market Offerings On February 25, 2021, the Company entered into an Equity Distribution Agreement (the “2021 Agreement”) with Piper Sandler & Co., Evercore Group L.L.C., and B. Riley Securities, Inc., serving as sales agents (the “Sales Agents”) with respect to an at-the-market offerings program under which the Company offered and sold shares of its common stock, par value $0.0001 per share (the “Common Stock”), having an aggregate offering price of up to $125.0 million (the “Shares”) through the Sale Agents (the “2021 Offering”). All Shares offered and sold in the 2021 Offering were issued pursuant to the Company’s Registration Statement on Form S-3 filed with the SEC on December 31, 2020, which was declared effective on January 11, 2021 (“2021 Shelf”), the prospectus supplement relating to the 2021 Offering filed with the SEC on February 25, 2021 and any applicable additional prospectus supplements related to the 2021 Offering that form a part of the Registration Statement. During the year ended December 31, 2022, the Company sold 5,204,415 shares of Common Stock under the 2021 Agreement resulting in approximately $56.2 million in proceeds, net of $1.9 million commission and other offering cost. As of December 31, 2022, the Company has sold 10,004,869 shares of Common Stock under the 2021 Agreement resulting in approximately $121.0 million in proceeds, net of $4.0 million commission and other offering costs. As of December 31, 2022, there were no remaining shares available under the 2021 Agreement. The Company recorded approximately $0.3 million of other offering costs which offset the proceeds received from the shares sold through December 31, 2022. Exchange Agreement On February 25, 2021, the Company entered into an exchange agreement (the “Exchange Agreement”) with an Investor and its affiliates (the “Exchanging Stockholders”), pursuant to which the Company exchanged an aggregate of 1,000,000 shares of the Company’s common stock, par value $0.0001 per share, owned by the Exchanging Stockholders for pre-funded warrants (the “Exchange Warrants”) to purchase an aggregate of 1,000,000 shares of common stock (subject to adjustment in the event of any stock dividends and splits, reverse stock split, recapitalization, reorganization or similar transaction, as described in the Exchange Warrants), with an exercise price of $0.0001 per share. On January 24, 2022, the Exchange Warrants to purchase 1,000,000 shares were net exercised, resulting in the issuance of 999,984 shares of common stock, and no Exchange Warrants remain outstanding (see Note 11). Public Offering On July 16, 2020, the Company offered and sold (i) 3,369,564 shares of common stock, at a price to the public of $23.00 per share, and (ii) pre-funded warrants of the Company to purchase 1,630,436 shares of common stock at an exercise price equal to $0.0001 per share (the “Pre-Funded Warrants”), at a price to the public of $22.9999 per share of common stock underlying the Pre-Funded Warrants (equal to the public offering price per share of Common Stock, minus the exercise price of each Pre-Funded Warrant). The Pre-Funded Warrants are exercisable at any time, provided that each Pre-Funded Warrant holder will be prohibited from exercising such Pre-Funded Warrants into shares of the Company’s common stock if, as a result of such exercise, the holder, together with its affiliates, would own more than 4.99% of the total number of shares of the Company’s common stock then issued and outstanding, which percentage may change at the holders’ election to any other number less than or equal to 19.99% upon 61 days’ notice to the Company. The gross proceeds The Company has assessed the Pre-Funded Warrants for appropriate equity or liability classification and determined that the Pre-Funded Warrants are freestanding instruments that do not meet the definition of a liability pursuant to ASC 480 and do not meet the definition of a derivative pursuant to FASB Accounting Standards Codification Topic 815, Derivatives and Hedging (“ASC 815”). The Pre-Funded Warrants are indexed to the Company’s common stock and meet all other conditions for equity classification under ASC 480 and ASC 815. Accordingly, the Pre-Funded Warrants are classified as equity and arewere accounted for as a component of additional paid-in capital at the time of issuance. During the year ended December 31, 2020, no Pre-Funded Warrants were exercised.
At-the-Market Offering
On March 27, 2020, the Company entered into an Equity Distribution Agreement (the “Agreement”) with JMP Securities LLC, serving as placement agent (the “Placement Agent”) with respect to an at-the-market offering program under which the Company may offer and sell, from time to time at its sole discretion, shares of its common stock, par value $0.0001 per share (the “Common Stock”), having an aggregate offering price of up to $50.0 million (the “Shares”) through the Placement Agent (the “Offering”).
Any Shares offered and sold in the Offering were issued pursuant to the Company’s Registration Statement on Form S-3 filed with the Securities and Exchange Commission (the “SEC”) on April 4, 2019, which was declared effective on April 12, 2019, the prospectus supplement relating to the Offering filed with the SEC on March 27, 2020 and any applicable additional prospectus supplements related to the Offering that form a part of the Registration Statement. The aggregate market value of Shares eligible for sale in the Offering and under the Equity Distribution Agreement were subject to the limitations of General Instruction I.B.6 of Form S-3, to the extent required under such instruction. The Company offered Shares having an aggregate offering price of $18.9 million pursuant to the prospectus supplement filed with the SEC on March 27, 2020. On June 1, 2020, the Company filed an amendment to the Agreement which amended the prospectus supplement dated March 27, 2020 to increase the aggregate offering price to $50.0 million.
As of December 31, 2020,2022, 760,870 of the Company has sold 5,594,455 sharesPre-Funded Warrants were exercised, leaving 869,566 remaining Pre-Funded Warrants unexercised (see Note 11). 11. Warrants The following common stock warrants were outstanding at December 31, 2022: | | | | | | | | | | | | Number of | | | | | | | | | | Common | | Per Share | | | | | | | Stock | | Exercise | | | | | | | Warrants | | Price | | Issuance Date | | Expiration Date | Issued with common units in the 2018 Unit Offering | | 3,300 | | | 2.7568 | | October 2, 2018 | | October 2, 2023 | Issued with common units in the 2018 Registered Direct Offering | | 92,300 | | | 5.40 | | October 10, 2018 | | October 10, 2023 | Issued with common units in the 2019 Registered Direct Offering | | 50,000 | | | 3.21 | | March 12, 2019 | | March 12, 2024 | Issued with common units in the 2020 Public Offering (see Note 10) | | 869,566 | | | 0.0001 | | July 16, 2020 | | — | Total | | 1,015,166 | | | | | | | | | | | | | | | | | |
The following common stock warrants were outstanding at December 31, 2021: | | | | | | | | | | | | Number of | | | | | | | | | | Common | | Per Share | | | | | | | Stock | | Exercise | | | | | | | Warrants | | Price | | Issuance Date | | Expiration Date | Replacement warrants | | 155 | | $ | 483.00 | | March 3, 2012 | | March 3, 2022 | Issued with common units in the 2018 Unit Offering | | 3,300 | | | 2.7568 | | October 2, 2018 | | October 2, 2023 | Issued with common units in the 2018 Registered Direct Offering | | 92,300 | | | 5.40 | | October 10, 2018 | | October 10, 2023 | Issued with common units in the 2019 Registered Direct Offering | | 50,000 | | | 3.21 | | March 12, 2019 | | March 12, 2024 | Issued with common units in the 2020 Public Offering (see Note 10) | | 1,630,436 | | | 0.0001 | | July 16, 2020 | | — | Issued in exchange for retirement of common stock per the Exchange Agreement (see Note 10) | | 1,000,000 | | | 0.0001 | | February 25, 2021 | | — | Total | | 2,776,191 | | | | | | | |
The following is a description of Common Stock under the Agreement resulting in $48.2 million in net proceeds, which concluded this Offering. The Company recorded and recognized approximately $0.3 million of offering costs which offset the proceeds received from the shares sold associated with this program.common stock warrants issued prior to January 1, 2020: 2019 Registered Direct Offering On March 12, 2019, the Company issued a combined total of 4,361,370 common units and pre-funded units to certain institutional investors in a registered direct offering (the “Registered Direct Offering”). Each common unit in the Registered Direct Offering was sold at a price of $3.21 and consisted of one share of common stock and 0.70 of a warrant to purchase one share of common stock at an exercise price of $3.21. Each warrant sold in the Registered Direct Offering was exercisable immediately and expired five years from the date of issuance. Each pre-funded unit in the Registered Direct Offering was sold at a public offering price of $3.20 and consisted of a pre-funded warrant to purchase one share of common stock at an exercise price of $0.01 per share and 0.70 of a warrant to purchase one share of common stock at an exercise price of $3.21. All of the pre-funded warrants were exercised
during 2019. The net proceeds of the Registered Direct Offering were $12.7 million, after deducting the underwriting discount and estimated offering expenses payable by the Company. The warrants issued in the Registered Direct Offering were recognized as equity classified freestanding financial instruments. The Registered Direct Offering triggered a down round adjustment to the exercise price of the warrants issued in the 2018 Unit Offering (refer to Note 11) from $4.1798 to $2.7568. During the year ended December 31, 2019, the Company treated the value of the effect of the reduction in exercise price as a deemed dividend of $452,925 which reduced income available to common shareholders. The value of a down round feature is measured as the difference between the financial instrument’s fair value (without the down round feature) using the pre-trigger exercise price and the financial instrument’s fair value (without the down round feature) using the reduced exercise price. 11. Warrants
The following common stock warrants were outstanding at December 31, 2020:
| | Number of Common Stock Warrants | | | Per Share Exercise Price | | | Issuance Date | | Expiration Date | | Replacement warrants | | | 155 | | | $ | 483.00 | | | March 3, 2012 | | March 3, 2022 | | Issued with redeemable preferred stock* | | | 1,420 | | | | 3.50 | | | August 16, 2017 | | August 16, 2022 | | Issued with common units in the 2018 Unit Offering | | | 3,300 | | | | 2.7568 | | | October 2, 2018 | | October 2, 2023 | | Issued with common units in the 2018 Registered Direct Offering | | | 92,300 | | | | 5.40 | | | October 10, 2018 | | October 10, 2023 | | Issued with common units in the 2019 Registered Direct Offering (see Note 10) | | | 50,000 | | | | 3.21 | | | March 12, 2019 | | March 12, 2024 | | Issued with common units in the 2020 Public Offering (see Note 10) | | | 1,630,436 | | | | 0.0001 | | | July 16, 2020 | | | — | | Total | | | 1,777,611 | | | | | | | | | | | | *Liability classified warrants | | | | | | | | | | | | | | |
The following common stock warrants were outstanding at December 31, 2019:
| | Number of Common Stock Warrants | | | Per Share Exercise Price | | | Issuance Date | | Expiration Date | Replacement warrants | | | 155 | | | $ | 483.00 | | | March 3, 2012 | | March 3, 2022 | Issued with redeemable preferred stock* | | | 62 | | | | 80.10 | | | August 16, 2017 | | August 16, 2022 | Issued with common units in 2018 Unit Offering | | | 2,505,250 | | | | 2.7568 | | | October 2, 2018 | | October 2, 2023 | Underwriter warrant issued in 2018 Unit Offering | | | 196,650 | | | | 6.25 | | | October 2, 2018 | | September 28, 2021 | Issued with common units in the 2018 Registered Direct Offering | | | 4,629,630 | | | | 5.40 | | | October 10, 2018 | | October 10, 2023 | Issued with common units in the 2019 Registered Direct Offering (see Note 10) | | | 3,052,959 | | | | 3.21 | | | March 12, 2019 | | March 12, 2024 | Total | | | 10,384,706 | | | | | | | | | | *Liability classified warrants | | | | | | | | | | | | |
The following is a description of the common stock warrants issued prior to January 1, 2019:
Replacement Warrants
In May 2017, the Company issued 155 common stock warrants to replace outstanding common stock warrants in connection with the Company’s merger with PharmAthene, Inc.
Redeemable Preferred Stock Warrants
In August 2017, in connection with a redeemable preferred stock issuance, the Company granted warrants to holders of redeemable preferred stock to purchase up to 78,181 shares of the Company’s common stock. Warrants issued with the redeemable
preferred stock are classified as a liability and were initially recorded at their grant date fair value, and remeasured on each subsequent balance sheet date. The warrant liability is classified as a component of other long-term liabilities. During the year ended December 31, 2019, the Company exchanged 1,550 of these warrants for a combination of common stock and cash, leaving 62 of these warrants outstanding asAs of December 31, 2019. During the second quarter of 2020, the 62 common stock2022, there were 50,000 unexercised warrants were repriced to 1,420 common stock warrants due to the At-the-Market Offering which triggered an anti-dilution provisionissued under the warrant agreement with the Company’s holders of redeemable preferred stock. 2019 Registered Direct Offering.
2018 Unit Offering On October 2, 2018, the Company issued a combined total of 2,400,000 common units and pre-funded units in a public offering (the “2018 Unit Offering”). Each common unit in the 2018 Unit Offering was sold at a public offering price of $5.00 and consisted of one share of common stock and a warrant to purchase one share of common stock at an exercise price of $6.00. Each warrant sold in the 2018 Unit Offering was exercisable immediately and expired five years from the date of issuance. Each pre-funded unit in the 2018 Unit Offering was sold at a public offering price of $4.99 and consisted of a pre-funded warrant to purchase one share of common stock at an exercise price of $0.01 per share and a warrant to purchase one share of common stock at an exercise price of $6.00. The pre-funded warrants were immediately exercisable and were able to be exercised at any time until all of the pre-funded warrants were exercised in full. All of the pre-funded warrants were exercised prior to December 31, 2018. The warrants issued in the 2018 Unit Offering are each subject to anti-dilution protection. Accordingly, to the extent the Company was to issue additional common stock or securities convertible into common stock at an issuance price lower than exercise price of the warrants, the exercise price of the warrants would be adjusted to the lower of (i) the issuance price or (ii) the lowest volume weighted averageweighted-average price of the Company’s common stock on the five trading days following the announcement of the new offering. In conjunction withThe 2018 Registered Direct Offering triggered a down round adjustment to the exercise price of the warrants issued in the 2018 Unit Offering from $6.00 to $4.1798. In addition, the Company issued 196,650 warrants2019 Registered Direct Offering triggered a down round adjustment to the underwriter. The underwriter warrants had an exercise price per share equal to 125% of the public offering price per common unitwarrants issued in this offering and could be exercised on a cashless basis.the 2018 Unit Offering from $4.1798 to $2.7568. As of December 30, 2020, all of31, 2022, there were 3,300 unexercised warrants issued under the underwriter warrants were exercised in full.2018 Unit Offering.
2018 Registered Direct Offering On October 10, 2018, the Company issued a combined total of 4,629,630 common units and pre-funded units to certain institutional investors in a registered direct offering (the “2018 Registered Direct Offering”). Each common unit in the 2018 Registered Direct Offering was sold at a price of $5.40 and consisted of one share of common stock and a warrant to purchase one share of common stock at an exercise price of $5.40. Each warrant sold in the 2018 Registered Direct Offering was exercisable immediately and expired five years from the date of issuance. Each pre-funded unit in the 2018 Registered Direct Offering was sold at a public offering price of $5.39 and consisted of a pre-funded warrant to purchase one share of common stock at an exercise price of $0.01 per share and a warrant to purchase one share of common stock at an exercise price of $5.40. The pre-funded warrants were immediately exercisable and were able to be exercised at any time until all of the pre-funded warrants are exercised in full. All of the pre-funded warrants were exercised prior to December 31, 2018. The As of December 31, 2022, there were 92,300 unexercised warrants issued under the 2018 Registered Direct Offering triggered a down round adjustment to the exercise price of the warrants issued in the 2018 Unit Offering from $6.00 to $4.1798.Offering.
A summary of warrant activity is as follows: | | | | | | | | | | | | | | | Weighted-Average | | | | | | Weighted | | Remaining | | | Number of | | | Average | | Contractual Term | | | Warrants | | | Exercise Price | | (Years) | Warrants outstanding, December 31, 2021 | | 2,776,191 | | | | | | Expired | | (155) | | | | | | Exercises (see Note 10) | | (1,760,870) | | | | | | Warrants outstanding, December 31, 2022 | | 1,015,166 | | $ | 0.66 | | 0.9 |
| | Year Ended December 31, | | | | 2020 | | | 2019 | | Warrants outstanding, beginning | | | 10,384,706 | | | | 7,344,297 | | Issuances | | | 1,631,794 | | | | 3,052,959 | | Exercises | | | (10,238,889 | ) | | | (12,550 | ) | Warrants outstanding, ending | | | 1,777,611 | | | | 10,384,706 | |
12. Stock-Based Compensation Stock Options The Company established the 2001 Employee Stock Option Plan to provide incentive stock options and non-qualified stock options to employees, and the 2001 Non-employee Stock Option Plan to provide non-qualified stock options to the members of the board of directors and advisory board, and non-employees. The 2001 Employee Stock Option Plan and the 2001 Non-employee Stock Option Plan are collectively referred to as the “2001 Plans.” In connection with the Company’s merger with PharmAthene, Merger AgreementInc. in 2017, the
Company issued options from its 2001 Plans to replace options previously granted. The Company de-designated common stock available for issuance under the 2001 Plans. No additional options or restricted stock will be granted under these plans. Options outstanding and unvested restricted stock granted or replaced under these plans will continue to vest over the remaining vesting period through the earlier of exercise, expiration or forfeiture no additional options, restricted stock or other awards will be granted under these plans.forfeiture. The replacement options issued after the 2017 mergers will continue to vest over the remaining vesting period through the earlier of exercise, expiration or forfeiture. Also, in connection with the 2017 mergers, the 2001 Plans were assumed by the Company. In addition, the Company assumed the PharmAthene, Inc. Amended and Restated 2007 Long-Term Incentive Compensation Plan (the “2007 Plan”). Awards outstanding under the 2007 Plan remained outstanding in accordance with their applicable terms and conditions. No additional awards will be made under the 2007 Plan. The Company established the 2017 Omnibus Incentive Plan (the “Omnibus Plan”) to provide incentive stock options, non-qualified stock options, restricted stock, and other stock-based awards denominated in shares of the Company’s common stock, and performance-based cash awards to eligible employees, consultants and directors. In 2018, the Company’s shareholders approved an amendment to the Omnibus Plan to increase the number of shares reserved for issuance from 1,500,000 to 5,000,000. The aggregate share reserve will be increased on January 1 of each year commencing in 2018 and ending on and including January 1, 2027 up to an amount equal to the lowest of (i) 4% of the total number of shares of common stock outstanding on a fully diluted basis as of December 31 of the immediately preceding calendar year, and (ii) such number of shares of common stock, if any, determined by the Company’s board of directors. Accordingly, on January 1, 2023, the number of shares of Common Stock reserved and available for issuance under the Omnibus Plan increased by 2,160,537. The maximum shares of common stock that may be granted to each employee or consultant in any fiscal year under the Omnibus Plan is the lesser of 800,000 shares per type of award or a maximum compensation amount of $5,000,000 under a Black-Scholes valuation model. The maximum common stock that may be granted to directors under the Omnibus Plan during any fiscal year is 500,000 shares. On November 29, 2018, the Board approved and adopted the Altimmune Inc. 2018 Inducement Grant Plan (the “Inducement Plan”). The Inducement Plan provides for the grant of equity or equity-based awards in the form of non-qualified stock options, restricted stock awards and other stock-based awards. The Inducement Plan was adopted by the Board without stockholder approval pursuant to Rule 5635(c)(4) of the NASDAQ Listing Rules. The Board has reserved 2,000,000 shares of the Company’s common stock for issuance pursuant to awards granted under the Inducement Plan (subject to customary adjustments in the event of a change in capital structure of the Company), and the Inducement Plan will be administered by the Compensation Committee. In accordance with Rule 5635(c)(4) of the NASDAQ Listing Rules, awards under the Inducement Plan may be only made to an employee who has not previously been an employee or member of the Board or any parent or subsidiary, or following a bona fide period of non-employment by the Company or a parent or subsidiary, if he or she is granted such award in connection with his or her commencement of employment with the Company or a subsidiary and such grant is an inducement material to his or her entering into employment with the Company or such subsidiary. The 2001 Plans, the 2007 Plan, the Omnibus Plan and the Inducement Plan are collectively referred to as the “Plans.” During the year ended December 31, 20202022 under the Plans, a total of 708,0001,473,427 options to purchase shares of common stock were granted. As of December 31, 2020,2022, there were 324,7101,368,689 and 1,517,0011,309,275 shares of common stock available for future grants under the Omnibus Plan and the Inducement Plan, respectively. The fair value of stock option issued to employees was estimated at the date of grant using Black-Scholes with the following weighted-average assumptions: | | | | | | | | Year Ended December 31, | | | | 2022 | | 2021 | | Expected volatility | | 110.1 | % | 109.6 | % | Expected term (years) | | 6.0 | | 6.0 | | Risk-free interest rate | | 2.4 | % | 0.8 | % | Expected dividend yield | | 0.0 | % | 0.0 | % |
| | For the Year Ended December 31, | | | | 2020 | | | 2019 | | Expected volatility | | | 102.35 | % | | | 92.68 | % | Expected term (years) | | | 5.88 | | | | 5.89 | | Risk-free interest rate | | | 1.24 | % | | | 2.17 | % | Expected dividend yield | | | 0.00 | % | | | 0.00 | % |
Expected volatility: As there is not sufficient historical volatility for the expected term of the stock options, the Company uses an average historical share price volatility, inclusive of its own volatility, based on an analysis of reported data for a peer group of comparable companies which were selected based upon industry similarities. Expected term (years): Expected term represents the number of years that the Company’s option grants are expected to be outstanding. There is not sufficient historical share exercise data to calculate the expected term of the stock options, options; therefore, the
Company elected to utilize the simplified method to value option grants. Under this approach, the weighted averageweighted-average expected life is presumed to be the average of the vesting term and the contractual term of the option. Risk-free interest rate: The Company determined the risk-free interest rate by using a weighted-average equivalent to the expected term based on the daily U.S. Treasury yield curve rate in effect as of the date of grant. Expected dividend yield: The Company does not anticipate paying any dividends in the foreseeable future. The fair value of each non-employee stock option is estimated at the date of grant using Black-Scholes with assumptions generally consistent with those used for employee stock options, with the exception of expected term, which is over the contractual life. A summary of stock option activity under the Plans is presented below:below (in thousands, except share and per share data): | | | | | | | | | | | | | | | | | | Weighted-Average | | | | | | | | Weighted- | | Remaining | | | | | | Number of | | Average | | Contractual Term | | Aggregate Intrinsic | | | Stock Options | | Exercise Price | | (Years) | | Value | Outstanding, December 31, 2021 | | 2,583,443 | | $ | 8.63 | | 5.9 | | $ | 8,460 | Granted | | 1,473,427 | | $ | 8.48 | | | | | | Exercised | | (373,545) | | $ | 2.65 | | | | | | Forfeited or expired | | (299,388) | | $ | 8.92 | | | | | | Outstanding, December 31, 2022 | | 3,383,937 | | $ | 9.20 | | 5.9 | | $ | 25,724 | Exercisable, December 31, 2022 | | 1,523,504 | | $ | 9.00 | | 5.8 | | $ | 12,292 | Vested and expected to vest, December 31, 2022 | | 3,197,894 | | $ | 9.19 | | 5.9 | | $ | 25,724 |
| | Number of Stock Options | | | Weighted- Average Exercise Price | | | Weighted-Average Remaining Contractual Term (Years) | | | Aggregate Intrinsic Value | | Outstanding, December 31, 2019 | | | 973,172 | | | $ | 4.36 | | | | 5.91 | | | $ | — | | Granted | | | 708,000 | | | $ | 4.75 | | | | | | | | | | Exercised | | | (46,966 | ) | | $ | 2.74 | | | | | | | | | | Forfeited or expired | | | (7,454 | ) | | $ | 6.48 | | | | | | | | | | Outstanding, December 31, 2020 | | | 1,626,752 | | | $ | 4.58 | | | | 5.90 | | | $ | 12,234,740 | | Exercisable, December 31, 2020 | | | 550,518 | | | $ | 5.93 | | | | 5.74 | | | $ | 4,104,806 | | Vested and expected to vest, December 31, 2020 | | | 968,611 | | | $ | 3.88 | | | | 5.98 | | | $ | 8,129,935 | |
The per share weighted-average grant date fair value of stock options granted during the years ended December 31, 20202022 and 20192021 were $4.75$7.07 and $2.67$11.79 per share, respectively. The total intrinsic value of stock options exercised during the yearyears ended December 31, 20202022 and 2021 was $0.9 million. There were no shares exercised during the year ended December 31, 2019.$4.4 million and $0.7 million, respectively. The total fair value of awardsoptions vested during 2020the years ended December 31, 2022 and 20192021 was $1.1$6.8 million and $1.0$4.7 million, respectively. AtAs of December 31, 2020,2022, there was $2.7$11.7 million of unrecognized compensation cost related to stock options, which is expected to be recognized over a weighted-average period of 2.42.6 years. Restricted Stock In October 2016, the Company authorized and granted a restricted stock award of 2,651 shares at an aggregate purchase price of $1,067. The weighted average grant date fair value of the restricted stock award was $310.80 per share. The restricted stock vests ratably at the end of each quarter over four years starting on December 31, 2016 with 50% of the original issued shared subject to accelerated vesting upon a deemed liquidation event. During the year ended December 31, 2020, 213 of the restricted shares fully vested, which completed the vesting of this share award.
In November 2018, the Company authorized and granted the Chief Executive Officer a restricted stock award of 322,907 shares on his date of hire. The weighted averageweighted-average grant date fair value of the restricted stock award was $3.59 per share. The restricted stock vests over a four-year period, 25% of the shares vesting on the one-year anniversary, and the remaining 75% vesting in 36 substantially equal monthly installments and will be fully vested on December 1, 2022; provided, however, that the executive officer has not experienced a termination prior to the applicable vesting date. The fair value of the 80,727 restricted shares that vested during the year ended December 31, 2020 totaled $0.8 million. In June 2020, the Company authorized and granted 109,525 shares of restricted stock awards which vested over three months. The weighted average grant date fair value of the restricted stock award was $9.19 per share. The fair value of the 109,525 restricted shares that vested during the year ended December 31, 2020 totaled $1.3 million.
In September 2020, the Company authorized and granted 15,000 shares of restricted stock units which vested over a four-year period, 25% of the shares vesting on the one-year anniversary, and the remaining 75% vesting in 36 substantially equal monthly installments and will beinstallments. The restricted stock was fully vested on September 22, 2024. The weighted average grant date fair value of the restricted stock award was $14.35 per share. No restricted shares vested duringNovember 30, 2022. During the year ended December 31, 2020.2022, the Company issued 41,572 of unrestricted common stock as a result of the vesting of 74,000 restricted stock net of 32,428 shares of common stock withheld to satisfy tax withholding obligations. The fair value of restricted stock awards vested during the years ended December 31, 2022 and 2021 was $0.7 million and $1.0 million, respectively. As of December 31, 2022, there was no unrecognized compensation expense related to restricted stock.
A summary of restricted stock activities is presented below: | | | | | | | | | | Weighted- | | | | | average | | | | | Grant Date | | | Shares | | Fair Value | Unvested, December 31, 2021 | | 74,000 | | $ | 3.59 | Vested | | (74,000) | | | 3.59 | Unvested, December 31, 2022 | | — | | $ | |
| | Shares | | | Weighted- average Grant Date Fair Value | | Unvested, December 31, 2019 | | | 235,666 | | | $ | 3.87 | | Granted | | | 124,525 | | | | 9.81 | | Vested | | | (190,465 | ) | | | 7.15 | | Unvested, December 31, 2020 | | | 169,726 | | | $ | 4.54 | |
117 Restricted Stock Units (RSUs) During the year ended December 2022, the Company granted 285,000 shares of RSUs with a weighted-average grant date fair value of $7.11 per share which vest over four years. During the year ended December 31, 2022, the Company issued 41,123 shares of unrestricted common stock as a result of the vesting of 61,028 RSUs net of 19,905 shares of common stock withheld to satisfy tax withholding obligations. The fair value of RSUs vested during the years ended December 31, 2022 and 2021 was $0.6 million and $0.1 million, respectively. A summary of RSUs activities is presented below: | | | | | | | | | | Weighted- | | | | | average | | | | | Grant Date | | | Shares | | Fair Value | Unvested, December 31, 2021 | | 231,928 | | $ | 14.53 | Granted | | 285,000 | | | 7.11 | Vested | | (61,028) | | | 14.39 | Forfeited or expired | | (41,415) | | | 10.95 | Unvested, December 31, 2022 | | 414,485 | | $ | 9.81 |
As of December 31, 2020,2022, total unrecognized compensation expense related to restricted stock awardsRSUs was $0.7$2.8 million, which the Company expects to recognize over a weighted averageweighted-average period of approximately 2.12.8 years. 2019 Employee Stock Purchase Plan On March 29, 2019, the board of directorsBoard adopted the 2019 Employee Stock Purchase Plan (the “2019 ESPP”). A total of 403,500 shares of the Company’s common stock have been reserved for issuance under the 2019 ESPP. Subject to any plan limitations, the 2019 ESPP allows eligible employees to contribute through payroll deductions up to 10% of their earnings for the purchase of the Company’s common stock at a discounted price per share. The offering periods begin in February and August of each year, with the initial offering period started on August 1, 2019. The common shares issuable under the 2019 ESPP were registered pursuant to a registration statement on Form S-8 on April 4, 2019. Unless otherwise determined by the administrator, the Company’s common stock will be purchased for the accounts of employees participating in the 2019 ESPP at a price per share that is the lesser of 85% of the fair market value of the Company’s common stock on the first trading day of the offering period or 85% of the fair market value of the Company’s common stock on the last trading day of the offering period. The 2019 ESPP estimated shares to be purchased fair value is included in the stock-based compensation expense. Employees have the ability to purchase shares of the Company’s common stock at a price equal to the lower of the first or last trading day of the offering period, which represents an option and, therefore, the 2019 ESPP is a compensatory plan under ASC 718-50, Employee Stock Purchase Plans. Accordingly, stock-based compensation expense is determined based on the option’s grant-date fair value, employee contributions, and the Company’s stock price and is recognized over the requisite service period of the option. The Company used the Black-Scholes valuation model. model to determine the fair value of ESPP. During the year ended December 31, 2020,2022, employees purchased 92,66126,395 shares for $135,466$0.2 million under the 2019 ESPP. As of December 31, 2022, there were 260,344 shares of common stock available for future issuance under the 2019 ESPP Plan. The Company recognized stock-based compensation expense related to this plan of $158,177 and $21,608$0.3 million for each of the years ended December 31, 20202022 and 2019,2021, respectively. Stock-based Compensation Expense Stock-based compensation expense is classified in the accompanying consolidated statements of operations and comprehensive loss for the years ended December 31, 20202022 and 20192021 as follows:follows (in thousands): | | | | | | | | | | | | | | | | Year Ended December 31, | | | 2022 | | 2021 | Research and development | | $ | 2,835 | | $ | 1,656 | General and administrative | | | 5,266 | | | 3,863 | Total | | $ | 8,101 | | $ | 5,519 |
| | Year Ended December 31, | | | | 2020 | | | 2019 | | Research and development | | $ | 357,762 | | | $ | 356,718 | | General and administrative | | | 2,218,244 | | | | 907,513 | | Total | | $ | 2,576,006 | | | $ | 1,264,231 | |
13. U.S. Government Contracts and Grants In June 2020, the Company was awarded $4.7 million from the U.S. Army Medical Research & Development Command (“USAMRDC”) to fund its Phase 1/2 clinical trial of T-COVID. The competitive award was granted by USAMRDC in collaboration with the Medical Technology Enterprise Consortium (“MTEC”), a 501(c)(3) biomedical technology consortium working in partnership with the Department of Defense (“DoD”). Under the contract, MTEC will paypaid the Company a firm fixed fee based upon the achievement of certain milestones for conduct and completion of a Phase 1/2 study and research and development work on the replication-deficient adenovirus 5 (“RD-Ad5”) vector vaccine platform. For the year ended December 31, 2020,2021, the Company has recognized $4.2$0.5 million of grant revenue under this contract.contract, which completed the full recognition of this award. No revenue was recognized for this contract for the year ended December 31, 2022.
In July 2016, the Company signed a five-year contract with BARDA.Biomedical Advanced Research and Development Authority (“BARDA”). The contract, as amended, hashad a total value of up to $133.7$136.8 million and is used to fund clinical development of NasoShield. Under the contract, BARDA payspaid the Company a fixed fee and reimburses certain costs for the research and development of an Ad5-vectored, protective antigen-based intranasal anthrax vaccine through cGMP manufacture and conduct of a Phase 1 clinical trial dose ranging assessment of safety and immunogenicity. The contract consistsconsisted of an initial base performance period providing approximately $27.8$30.9 million in funding for the period July 2016 through JuneDecember 2021. BARDA hashad seven options to extend the contract to fund certain continued development and manufacturing activities for the anthrax vaccine, including Phase 2 clinical studies. Each option, if exercised by BARDA, would providehave provided additional funding ranging from approximately $1.1 million to $34.4 million for a three-year period beginning in 2021. For the yearsyear ended December 31, 2020 and 2019,2021, the Company has recognized $3.1 million and $5.3$3.7 million of grant revenue under the current BARDA contract. For the year ended December 31, 2022, the Company has recognized de minimis grant revenue related to the close-out of the BARDA contract. BARDA did not extend the contract respectively.beyond the end of December 2021. The Company accounts for these contracts as a government grant which analogizes with International Accounting Standards 20 (“IAS 20”), Accounting for Government Grants and Disclosure of Government Assistance. 14. Employee Benefit Plans The Company has a 401(k)-retirement plan in which substantially all of our employees in the United States are eligible to participate in. Eligible employees may elect to contribute up to the maximum limits, as set by the Internal Revenue Service, of their eligible compensation. During 2020each of the years ended December 31, 2022 and 2019,2021, the Company made discretionary plan contributions of $170,727 and $131,412, respectively.$0.3 million. 15. Income Taxes The components of net loss before income tax benefit are as follows:follows (in thousands): | | | | | | | | | Year Ended December 31, | | | 2022 | | 2021 | U.S. operations | | $ | (72,750) | | $ | (78,583) | Non-U.S. operations | | | (12,160) | | | (18,507) | Net loss before income tax benefit | | $ | (84,910) | | $ | (97,090) |
| | Year Ended December 31, | | | | 2020 | | | 2019 | | U.S. operations | | $ | (50,735,735 | ) | | $ | (18,562,738 | ) | Non-U.S. operations | | | (3,725,766 | ) | | | (2,015,893 | ) | Net loss before income tax benefit | | $ | (54,461,501 | ) | | $ | (20,578,631 | ) |
The components of the income tax benefitexpense (benefit) are as follows:follows (in thousands): | | | | | | | | | Year Ended December 31, | | | 2022 | | 2021 | U.S. federal | | | | | | | Current | | $ | (144) | | $ | — | U.S. state and local | | | | | | | Current | | | (53) | | | — | Income tax expense (benefit) | | $ | (197) | | $ | — |
| | Year Ended December 31, | | | | 2020 | | | 2019 | | U.S. federal | | | | | | | | | Current | | $ | 4,706,092 | | | $ | — | | Deferred | | | — | | | | 45,465 | | U.S. state and local | | | | | | | | | Current | | | 710,932 | | | | — | | Deferred | | | — | | | | 13,035 | | Income tax benefit | | $ | 5,417,024 | | | $ | 58,500 | |
Reconciliation between the effect of applying the federal statutory rate and the effective income tax rate used to calculate the Company’s income tax benefit is as follows: | | | | | | | | Year Ended December 31, | | | | 2022 | | 2021 | | Federal statutory rate | | 21.00 | % | 21.00 | % | State income taxes, net of federal benefit | | (0.12) | | 4.71 | | Research and development tax credit | | (2.22) | | (2.30) | | Acquired in process research and development | | (0.02) | | (0.15) | | Rate change | | 1.29 | | — | | Other | | 0.21 | | (0.47) | | Change in valuation allowance | | (19.91) | | (22.79) | | Effective tax rate | | 0.23 | % | — | % |
| | Year Ended December 31, | | | | 2020 | | | 2019 | | Federal statutory rate | | | 21.00 | % | | | 21.00 | % | State income taxes, net of federal benefit | | | 4.10 | | | | 3.12 | | Research and development tax credit | | | (0.84 | ) | | | (1.51 | ) | Acquired in process research and development | | | (6.27 | ) | | | (7.03 | ) | CARES Act U.S. federal and state carryback claim | | | 1.81 | | | | — | | Other | | | 0.29 | | | | (2.81 | ) | Change in valuation allowance | | | (10.14 | ) | | | (12.49 | ) | Effective tax rate | | | 9.95 | % | | | 0.28 | % |
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income and for tax carryforwards. Significant components of the Company’s deferred tax assets and liabilities are as follows:follows (in thousands): | | | | | | | | | December 31, | | | 2022 | | 2021 | Deferred tax assets: | | | | | | | Net operating losses | | $ | 44,606 | | $ | 37,229 | Accrued expenses | | | 523 | | | 352 | Amortization | | | 540 | | | 746 | Stock compensation | | | 2,193 | | | 1,276 | Lease liability | | | 309 | | | 422 | Asset impairment | | | — | | | 3,129 | Capitalized research and development costs | | | 11,996 | | | — | Other | | | 107 | | | 109 | Total deferred tax assets | | | 60,274 | | | 43,263 | Valuation allowance | | | (57,245) | | | (40,584) | Deferred tax assets, net | | | 3,029 | | | 2,679 | Deferred tax liabilities: | | | | | | | IPR&D assets | | | (2,847) | | | (2,360) | Right of use asset | | | (164) | | | (220) | Depreciation | | | (18) | | | (99) | Total deferred tax liabilities | | | (3,029) | | | (2,679) | Total deferred tax assets (liabilities), net | | $ | — | | $ | — |
| | December 31, | | | | 2020 | | | 2019 | | Deferred tax assets: | | | | | | | | | Net operating losses | | $ | 18,978,410 | | | $ | 13,493,760 | | Accrued expenses | | | 495,301 | | | | 208,604 | | Amortization | | | 785,523 | | | | 947,115 | | Stock compensation | | | 584,507 | | | | 622,789 | | Lease liability | | | 502,151 | | | | 479,940 | | Other | | | 106,197 | | | | 117,713 | | Valuation allowance | | | (18,671,086 | ) | | | (13,149,779 | ) | Total deferred tax assets | | | 2,781,003 | | | | 2,720,142 | | Deferred tax liabilities: | | | | | | | | | IPR&D assets | | | (2,386,667 | ) | | | (2,373,304 | ) | Right of use asset | | | (248,710 | ) | | | (192,160 | ) | Depreciation | | | (145,626 | ) | | | (154,678 | ) | Total deferred tax liabilities | | | (2,781,003 | ) | | | (2,720,142 | ) | Total deferred tax liabilities net | | $ | — | | | $ | — | |
The Company assesses the need for a valuation allowance against our deferred tax assets and considers both positive and negative evidence related to the likelihood of realization of the deferred tax assets to determine, based on the weight of available evidence, whether it is more-likely-than-not that some or all of the deferred tax assets will not be realized. This determination requires significant judgment, including assumptions about future taxable income that are based on historical and projected information. The increase in the valuation allowance during the year ended December 31, 20202022 primarily relates to increases for current year losses in both the U.S. and foreign locations which the Company concluded needed a full valuation allowance, but was partially offset by the utilization of a portion of the U.S. federal and state losses to offset the Company’s 2016 tax liability, due to provisions of the CARES Act enacted in 2020.locations. The Company has recorded a valuation allowance against its grossnet U.S. and net non-U.S. deferred tax assets as it believes are not more likely than not realizable and the net non-U.S. deferred tax assets, although, a portion of the current year loss was benefited for an additional U.S. Federal carryback from the CARES Act.realizable. Deferred tax liabilities, consist primarily of indefinite life IPR&D assets located in a foreign subsidiary, which will be applied in the future to offset against net operating losses (“NOLs”) that have an unlimitedindefinite life. The Company has U.S. federal and state net operating loss carryforwards of approximately $45.7$134.3 million and $103.7 million, respectively, as of December 31, 2020. Of this2022, of which a portion of the federal and state amount $6.6of $7.1 million and $103.7 million, respectively, has a 20-year carry forward period that will expire at various dates beginning in 2021.2024. Under current law, the remaining federal amount of $39.1$127.2 million has an unlimited life.indefinite life and amounts utilized in the future may not exceed 80% of taxable income. The Company also has foreign net operating loss carryforward of approximately $30.8$38.6 million which carryforward indefinitely. Under Section 382 of the Internal Revenue Code of 1986 (“IRC 382”), as amended, substantial changes in the Company’s ownership may limit the amount of NOLs that can be utilized annually in the future to offset its U.S. federal and state taxable income. Specifically, this limitation may arise in the event of a cumulative change in ownership of the Company of more than 50% within any three-year period. The amount of the annual limitation is determined based on the value of the Company immediately before the ownership change. The Company has reduced the NOL and related valuation allowance in historical periodsperiods. The Company’s existing NOLs are subject to limitations arising from previous ownership changes from 2020 and whileprior that impact the timing and amount. In addition, future changes in the Company’s stock ownership, many of which are outside of the Company’s control, could result in an ownership change. We have not determined if we have experienced Section 382 ownership changes in the past and if a portion of our NOL and tax credit carryforwards are subject to an annual limitation under Section 382. Accordingly, the Company is in the processmay not be able to utilize a material portion of completing its evaluation of whether an equity shift occurred in 2020, it is not expected to require an adjustment to the net NOLs and related valuation allowance. Subsequent ownership changes may further affectthis could harm the limitation inCompany’s future years. The Company continues to evaluate its losses for these provisions.operating results by effectively increasing the Company’s future tax obligations. Significant judgment is required in evaluating tax positions and determining the provision for income taxes. The Company establishes liabilities for tax-related uncertainties based on estimates of whether, and the extent to which, additional taxes may be due. These liabilities are established when the Company believes that certain positions might be challenged despite its belief that its tax return positions are fully supportable. The Company adjusts these liabilities in light of changing facts and circumstances, such as the outcome of a tax audit. The provision for income taxes includes the impact of changes to these liabilities. The amount of unrecognized tax benefits was $0.7 million and $0.0$0.2 million as of December 31, 20202022 and 2019,2021, respectively. Any changes in the next twelve months are not anticipated to have a significant impact on the results of operations, financial position or cash flows of the Company. All of the Company’s uncertain tax positions, if recognized, would affect its income tax expense.expense, although the net impact would be zero due to the Company’s valuation allowance position.
The Company has elected an accounting policy to classify interest and penalties related to unrecognized tax benefits as a component of income tax expense. During the year ended December 31, 2022, the company recorded income tax benefit of $0.2 million related to interest received and receivable on income tax refunds. As of December 31, 2020 and 2019,2021, potential interest and penalties on unrecognized tax benefits were not significant. The following is a tabular reconciliation of the total amounts of unrecognized tax benefits excluding related interest and penalties:penalties (in thousands): | | Year Ended December 31, | | | | 2020 | | | 2019 | | Beginning of year | | $ | — | | | $ | — | | Positions taken in the prior year | | | 710,783 | | | | — | | End of year | | $ | 710,783 | | | $ | — | |
| | | | | | | | | Year Ended December 31, | | | 2022 | | 2021 | Beginning balance | | $ | 237 | | $ | 711 | Increases for prior year tax positions | | | 474 | | | — | Decreases for prior year tax positions | | | — | | | (474) | Ending balance | | $ | 711 | | $ | 237 |
The Company files income tax returns in the United States, various U.S. states, U.K., and Australia. The Company is still open to examination by the applicable taxing authorities from 20092010 forward, although tax attributes that were generated prior to 20092010 may still be adjusted upon examination by federal, state, foreign or local tax authorities if they either have been or will be used in a future period. 16. Net Loss Per Share Because the Company has reported net loss attributable to common stockholders for the years ended December 31, 20202022 and 2019,2021, basic and diluted net loss per share attributable to common stockholders in each year are the same for both years.same. Basic net loss per share attributable to common stockholders is computed by dividing the net loss attributable to common stockholders by the weighted averageweighted-average numbers of shares of common stock outstanding for the period. Basic shares outstanding includes the weighted averageweighted-average effect of the Company’s outstanding pre-funded warrants, the exercise of which requires little or no consideration for the delivery of shares of common stock. Diluted net loss per share is calculated by adjusting weighted averageweighted-average shares outstanding for the dilutive effect of common stock equivalents outstanding for the period. As such, all unvested restricted stock, common stock warrants, and stock options have been excluded from the computation of diluted weighted averageweighted-average shares outstanding because such securities would have an anti-dilutive impact for all periods presented. Potential common shares issuable upon conversion, vesting or exercise of unvested restricted stock, common stock warrants, and stock options that are excluded from the computation of diluted weighted-average shares outstanding, as they are anti-dilutive, are as follows: | | | | | | | Year Ended December 31, | | | 2022 | | 2021 | Common stock warrants | | 145,600 | | 145,755 | Common stock options | | 3,397,998 | | 2,594,177 | Restricted stock units | | 414,485 | | 231,928 | Restricted stock | | — | | 74,000 |
| | December 31, | | | | 2020 | | | 2019 | | Common stock warrants | | | 147,175 | | | | 10,384,706 | | Common stock options | | | 1,631,898 | | | | 1,001,242 | | Restricted stock | | | 169,726 | | | | 235,666 | |
17. Commitments and Contingencies License Obligations University of Alabama at Birmingham Research Foundation
The Company had an agreement with the University of Alabama at Birmingham Research Foundation (“UABRF”) for the exclusive worldwide license to develop, manufacture, and commercialize certain proprietary technology developed at UABRF. That agreement expired in accordance with its terms in January 2020. Under the terms of the amended and restated agreement, the Company was obligated to pay an annual license fee of $20,000 and low single digit royalty fees upon the commencement of product sales. Fees incurred under the UABRF agreement totaled $20,000 for the year ended December 31, 2019 and are classified as a component of research and development expenses in the accompanying consolidated statements of operations and comprehensive loss.
PER.C6 Cell Line - Janssen Vaccines & Prevention B.V. The Company has a royalty-bearing, worldwide non-exclusive license agreement with Janssen Vaccines & Prevention B.V. (formerly(formerly known as Crucell Holland B.V.) (“Janssen”) for use of its vaccine technology. The Company may terminate the license agreement without cause, and the agreement contains customary provisions for either party to terminate prior to the expiration of the
agreement. The amended license agreement expires on a product-by-product and country-by-country basis on the later of the date upon which the last of the licensed patents applicable to the relevant product expires or 15 years from the date of first commercial sale of the relevant product. The Janssen patent rights include patents issued in the United States with an expected expiration date no earlier than April 2020, in each case not giving effect to any potential extensions and assuming payment of all associated fees. Upon expiration of the amended license agreement, or if the Company terminates the amended license agreement for Janssen’s material breach, the Company retains the right to exploit the rights granted. Under the agreement, the Company is required to pay an annual license fee and annual royalty fees upon reaching certain milestones in an amount that equals the greater of a low single digit percentage of net sales or $150,000. On April 2, 2020, the Company entered into Amendment No. 3 to the Second Restated License Agreement and additionally entered into Amendment No. 4, 5 and 6 throughout 2020 (collectively, the “Amendments”), by and between the Company and Janssen (as amended by Amendment No. 1 to Second Restated License Agreement and Amendment No. 2 to Second Restated License Agreement, together with the Amendments, the “License Agreement”). Pursuant to the Amendment, the field of licenses granted to the Company for the use of the PER.C6 cell line under the License Agreement is expanded to cover COVID-19 caused by SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2), in addition to the existing licenses related to Bacillus anthracis and influenza virus. All capitalized terms not defined herein shall have the meanings assigned to them in the Amendment or the License Agreement, as applicable. Pursuant to the Amendment, the Company agreed to pay certain additional development-based milestone payments through approval of licensed products by the FDA for the treatment or prevention of COVID-19, up to an aggregate amount of $1.2 million. The Company also agreed to pay royalty payments as a percentage of net sales of products to a royalty stacking reduction and minimum annual royalty payments, until the expiration of the term of the License Agreement, as amended. Fees incurred under the Janssen agreement totaled $310,502$0.1 million and $111,499$0.2 million for the years ended December 31, 20202022 and 2019,2021, respectively, and are included in research and development expenses in the accompanying consolidated statements of operations and comprehensive loss. The agreement’s current maintenance fee term is set to expire on April 2, 2023, and the Company has provided notice to Janssen that it does not intend to renew the agreement. Spitfire Acquisition As disclosed in Note 8, the Company is obligated to make payments of up to $80.0 million upon the achievement of specified worldwide net sales of all products developed using the technology acquired from Spitfire Pharma Inc. within ten years following the approval of a new drug application filed with the FDA. Litigation In December 2019, a complaint was filed by Dr. De-ChuDe-chu Christopher Tang (“Plaintiff”) against the Company, which the Company removed to the United States District Court for the Eastern District of Texas. The Plaintiff amended the complaint in February 2020 to include Vipin K. Garg and David J. Drutz as defendants, in addition to the Company (Dr. Garg, Dr. Drutz, and the Company are collectively referred to as “Defendants”). In March 2020 the Defendants filed a motion to dismiss the complaint. The Court deniedOn March 25, 2021, the court granted the motion without prejudice and alloweddismissed the action for lack of personal jurisdiction. In December 2021, the Plaintiff an opportunity to file an amended complaint.refiled the case in the United States District Court for the District of Maryland, captioned Tang v. Altimmune, Inc., et al., Case No. 8:21-cv-03283 (D. Md.). Plaintiff, who is representing himself, asserts two causes of action: (1) breach of a prior settlement agreement by “robbing Plaintiff’s second amended complaint was filed onproperties”; and (2) use by the Company of the “AdHigh system,” which Plaintiff claims is “proprietary.” On April 17, 2020, and4, 2022, Defendants filed a motion to dismiss thatall claims in Plaintiff’s operative complaint, on May 1, 2020. A hearing on Defendants’which motion to dismiss was held on May 20, 2020, and the motion is currently pending. Plaintiff, who is representing himself, alleges five causes of action as follows: (1) Defendants’ alleged retention of Plaintiff’s lab notebooks after the termination of his employment in 2012; (2) alleged plagiarism based on publishing an article without naming Plaintiff as an author; (3) use of the Adhigh System, which Plaintiff alleges he developed; (4) allegations that Defendants manipulated the Company’s stock and caused a decrease in value; and (5) allegations that the Defendants “wast[ed] government grant money and poison[ed] science by leaving data to rot.” On September 30, 2020, Plaintiff filed a motion titled “Motion to Proscribe Defendants’ Allegedly Illegal Use of Plaintiff’s AdHigh System in Altimmune’s Human Clinical Trials,” to which Defendants filed an opposition on October 13, 2020. The court has not yet ruled on that motion, which also remains pending. On November 6, 2020, Defendants filed a motion for summary judgment on the basis of lack of personal jurisdiction, insufficient service of process, and failure to state a claim. The court has not yet ruled on that motion, which also remains pending. On December 1, 2020, the magistrate judge assigned to the case issued a report and recommendation recommending that Defendants’ motion to dismiss of May 1, 2020 be granted and that this action be dismissed for lack of personal jurisdiction. Plaintiff filed objections to the report and recommendation on December 14, 2020, and the resolution of those objections by the district court remains pending. The Company believes the allegations in the operative complaint are without merit and intendsintend to vigorously defend the litigation. However, the outcome of this legal proceeding is uncertain at this time and the Company cannot reasonably estimate a range of loss, if any. Accordingly, the Company has not accrued any liability associated with this action. The Company is a party in various other contractualcontracts and subject to disputes, litigation, and potential claims arising in the ordinary course of business none of which are currently reasonably possible or probable of material loss. . Item 9. Changes in and Disagreements with Accou ntantsAccountants on Accounting and Financial DisclosureNone. Item 9A. Controls and Procedures Evaluation of Disclosure Controls and Procedures Our management, with the participation of our principal executive officer and principal financial officer, evaluated the effectiveness of our disclosure controls and procedures pursuant to Rule 13a-15 under the Securities Exchange Act of 1934, as of December 31, 2020.2022. In designing and evaluating our disclosure controls and procedures, our management recognized that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives, and our management necessarily applied its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on this evaluation, our principal executive officer and principal financial officer concluded that, as of December 31, 2020,2022, our disclosure controls and procedures were effective to provide reasonable assurance that information we are required to disclose in reports that we file or submit under the Exchange Act is recorded, processed, summarized, and reported within the time periods specified in the SEC’s rules and forms, and that such information is accumulated and communicated to our management, including our principal executive officer and principal financial officer, as appropriate, to allow timely decisions regarding required disclosure. Changes in Internal Control Over Financial Reporting Our management, including our principal executive and principal financial officer, has evaluated any changes in our internal control over financial reporting that occurred during the year ended December 31, 2020,2022, and has concluded that there was no change that occurred during the year ended December 31, 20202022 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting. Management’s Report on Internal Control over Financial Reporting Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as defined in Rule 13a-15(f) under the Exchange Act. Our management conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework in Internal Control — Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on this evaluation, our management has concluded that our internal control over financial reporting was effective as of December 31, 2020.2022. Limitations on Effectiveness of Controls and Procedures In designing and evaluating the disclosure controls and procedures, management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives. In addition, the design of disclosure controls and procedures must reflect the fact that there are resource constraints and that management is required to apply its judgment in evaluating the benefits of possible controls and procedures relative to their costs. We are a smaller reporting company, and therefore our independent registered public accounting firm has not issued a report on the effectiveness of internal control over financial reporting. Item 9B. Other Information None. Item 9C. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections Not Applicable. PART III PART IIIItem 10. Directors, Executive Officers and Corporate Governance Directors Our directors are elected at each annual meeting of stockholders and hold office until the next annual meeting of stockholders and until their successors have been elected and qualified. Our Bylaws provide that the number of Directors constituting the entire Board shall be not less than one nor more than nine as determined by resolution of the Board. Our Board currently has eight Directors, each of whom was elected at the Company’s 20202022 annual meeting of stockholders. The names and ages of our directors as of February 25, 202128, 2023 are set forth below: | | | | | Name |
| Age |
| Position | Vipin K. Garg, Ph.D. | | 6365
| | President, Chief Executive Officer, and Director | Mitchel Sayare, Ph.D. | | 7375
| | Chairman of the Board | David J. Drutz, M.D. | | 8284
| | Director | John M. Gill | | 6971
| | Director | Philip L. Hodges | | 5254
| | Director | Wayne Pisano | | 6668
| | Director | Diane Jorkasky, M.D. | | 6971
| | Director | Klaus O. Schafer, M.D., MPH | | 7173
| | Director |
Vipin K. Garg, Ph.D.currently serves as our President and Chief Executive Officer and is a member of the Board of Directors. Dr. Garg joined Altimmune in November 2018 with over three decades of experience in the biotechnology and pharmaceutical industries. He has a proven track record of building and managing both private and publicly traded companies. Before joining Altimmune, from October 2013 to June 2018, he served as President and Chief Executive Officer of Neos Therapeutics, Inc. (since acquired by Aytu BioPharma, Inc. (Nasdaq: NEOS)AYTU)), where he built a NASDAQ-listed commercial-stage biopharmaceutical company, launching three branded therapeutic products including Adzenys XR-ODTTMXR-ODTTM and Cotempla XR-ODTTM,XR-ODTTM, the first ever XR-ODTTMextended release, orally dissolving tablet medications for the treatment of ADHD. Prior to Neos, he served as president and Chief Executive Officer of Tranzyme, PharmaInc. until its acquisition by Ocera Therapeutics, Inc. (subsequently becoming a subsidiary of Mallinckrodt Pharmaceuticals (NYSEAmerican: MNK)) where he progressed a discovery-stage, emerging biotech company to a Nasdaq-listed clinical-stage, drug development company. Prior to joining Tranzyme, Dr. Garg served as Chief Operating Officer of Apex Bioscience, Inc. (acquired by Curacyte AG of Munich, Germany), and held senior management positions at DNX Bio-Therapeutics, Inc. (until its acquisition by Baxter Healthcare Corporation), Sunovion Pharmaceuticals, Inc. (formerly known as Sepracor Inc., now a subsidiary of Sumitomo Dainippon Pharma), and Bio-Response Inc. (acquired by Baxter Healthcare Corporation). Dr. Garg received his Ph.D. in Biochemistry in 1982 from the University of Adelaide, Australia, and his M.S. from IARI Nuclear Research Laboratory, New Delhi, India in 1978. We believe that Dr. Garg’s extensive experience in the biotechnology and pharmaceutical industries makes him well qualified to serve as a member of our Board of Directors.Directors. Mitchel Sayare, Ph.D.has been a member of the Board of Directors since April 2010. Dr. Sayare became Chairman of the Board in January 2018 and served as Executive Chairman from June 2018 to November 2018. Until 2010, Dr. Sayare served as the Chairman of the Board of public company ImmunoGen, Inc. (Nasdaq:IMGN) (a position he had held since 1989). In addition, he served as ImmunoGen’s Chief Executive Officer from 1986 to December 31, 2009, and as its President from 1986 to 1992, and from 1994 to July 2008. Prior to joining ImmunoGen, he served as Vice President of Development of Xenogen from 1982 to 1985. Prior to that he was Assistant Professor of Biophysics and Biochemistry at the University of Connecticut. Dr. Sayare earned a Ph.D. in biochemistry from Temple University School of Medicine. Dr. Sayare is a director of Boston IVF, Inc. and Advanced Aesthetic Technologies, Inc., both privately-held companies. We believe that Dr. Sayare’s substantial experience as a board member and executive officer of biotechnology companies makes him well qualified to serve as a member of our Board of Directors. David J. Drutz, M.D.has served as a member of our Board of Directors since May 2017, when he was appointed to the Board in connection with the completion of the Merger.merger with PharmAthene, Inc. Dr. Drutz was first elected to Privatepre- merger Altimmune’s Board of Directors in January 2010 and was elected Board Chairman in October 2011. Dr. Drutz is the President of Pacific Biopharma Associates, LLC, a biopharmaceutical consulting company that he founded in 1999. From 2008 to 2015, he held various positions at DARA BioSciences (Nasdaq: DARA), an oncology supportive care company which was acquired by Midatech Pharma plc, including Director, Chief Executive Officer, Executive Chairman and Chief Medical Officer. He also previously served as Chairman of Tranzyme, Inc. (Nasdaq:TZYM) from 2000 to 2010; and Director of MethylGene (TSX:MYG) from 2000 to 2010 and Gentris Corporation from 2007 to 2014. From 1999 to 2008 he was a general partner with Pacific Rim Ventures, a Tokyo-based venture capital firm. Dr. Drutz’s management experience includes tenures as VP Biological Sciences and VP Clinical Research at Smith Kline & French Laboratories; VP Clinical Development at Daiichi Pharmaceutical Corporation; and CEO of Inspire Pharmaceuticals (1995-1998)(1995 – 1998) and Sennes Drug Innovations (1994-1995)(1994 – 1995). Earlier, Dr. Drutz was Professor of Medicine, Chief of the Division of Infectious Diseases, and the founder of the NSF Center for Cell Regulation at the UT Health Science Center, San Antonio. Dr. Drutz received his M.D. from the University of Louisville School of Medicine and postgraduate training in internal medicine and infectious diseases at Vanderbilt University School of Medicine, serving
subsequently as a research medical officer in the U.S. Navy (LCDR, USNR). He is certified by the American Board of Internal Medicine; a fellow of the American College of Physicians and the Infectious Diseases Society of America; a member of Alpha Omega Alpha, the American Society of Clinical Oncology and the American Society for Clinical Investigation; and the author of more than 200 peer-reviewed publications in the area of infectious diseases. We believe Dr. Drutz’s significant experience in biotechnology investment and as a physician make him well qualified to serve as a member of our Board of Directors. John M. Gillhas served as a member of our Board of Directors since August 2004. Mr. Gill served as PharmAthene’s President and Chief Executive Officer of PharmAthene, Inc. from March 2015 until the completion of the Mergermerger with Altimmune in May 2017. From 2003 to 2013, Mr. Gill served as the President, Chief Executive Officer, co-founder and a Director of TetraLogic Pharmaceuticals Corporation, a public biopharmaceutical company. Mr. Gill has previously held positions at 3-Dimensional Pharmaceuticals and SmithKline Beecham. After serving in the United States Marine Corps, Mr. Gill earned a B.A. in Accounting and Economics from Rutgers University. We believe Mr. Gill’s executive and board experience in the pharmaceutical industry and his substantial financial knowledge and expertise make him well qualified to serve as a member of our Board of Directors. Philip L. Hodgeshas served as a member of our Board of Directors since May 2017, when he was appointed to the Board in connection with the completion of the Merger,merger with PharmAthene, Inc., and was first elected to Privatepre-merger Altimmune’s board of directors in September 2003. Mr. Hodges is Managing Partner of Redmont Capital, a private equity firm located in Birmingham, Alabama, which he joined at its inception in 1997. Redmont Capital is a co-founder of pre-merger Altimmune. Mr. Hodges’ investment strategy is focused on high-growth small businesses within the health care, life science and technology sectors. He currently serves as a director for several of the firm’s portfolio companies. Mr. Hodges holds a Bachelor of Science in Business Administration from the Brock School of Business at Samford University. We believe Mr. Hodges experience as a life science investor makes him well qualified to serve as a member of our Board of Directors. Wayne Pisanohas served as a member of our Board of Directors since August 2018. Mr. Pisano also serves on the board of directors of Provention Bio, Inc. (Nasdaq: PRVB), a biopharmaceutical company, since April 2018; and Oncolytics Biotech Inc. (Nasdaq: ONCY), a biotechnology company, since May 2013. Mr. Pisano served on the board of directors of IMV INC. (Nasdaq: IMV) a bio pharmaceutical company from October 2011 until March 2021. Mr. Pisano served as president and Chief Executive Officer of VaxInnate Corporation, a biotechnology company, from January 2012 until November 2016. Mr. Pisano joined Sanofi Pasteur in 1997 and was promoted to President and Chief Executive Officer in 2007, the position he successfully held until his retirement in 2011. He has a Bachelor of Science in biology from St. John Fisher College, New York and an MBA from the University of Dayton, Ohio. We believe Mr. Pisano’s depth of experience across the spectrum of commercial operations, public immunization policies and pipeline development makes him well qualified to serve as a member of our Board of Directors. Diane Jorkasky, M.D. has served as a member of our Board of Directors since May 2020. Dr. Jorkasky also has servedbeen serving on the board of directorsAPIE Therapeutics, a private biopharmaceutical company, since January 2021. Dr. Jorkasky has served as a Science and Advisory Board Member and a Board Member of Alzheon, Inc., a private biopharmaceutical company, since December 20192016 and also served on the board of directors of Q Therapeutics, Inc. from September 2013 until August 2016. From June 2014 to August 2019, she served as Executive Vice President, Chief Medical Officer and Head of Development at Complexa Inc., a clinical stated biopharmaceutical company. Dr. Jorkasky received her MDM.D. in 1977 from the University of Pennsylvania School of Medicine and is board certified in internal medicine, nephrology and clinical pharmacology. She is a member of the Connecticut Academy of Science and Technology. Dr. Jorkasky is on the faculties of University of California, San Francisco, and Uniformed Service of Health Sciences Medical Schools, with previous faculty appointments at Yale University and the University of Pennsylvania Schools of Medicine. We believe Dr. Jorkasky’s executive and board experience in the pharmaceutical industry and as a physician make her well qualified to serve as a member of our Board of Directors. Brigadier General (ret.), Klaus O. Schafer, M.D., MPH has served as a member of our Board of Directors since JulyMay 2017, upon completion of the merger with PharmAthene, Inc. Dr. Schafer was first elected to pre-merger Altimmune’s Board of Directors in 2012. Dr. Schafer has over 35 years of healthcare leadership experience, having held senior positions in government and industry. He previously held the position ofAs former, Acting Deputy Assistant to the Secretary of Defense for chemical and biological defense, overseeinghe oversaw the Department’s $1.0 billion program for vaccine, therapeutics, medical device and sensor development against biothreats.biological threats and was instrumental in advancing research in human immune response. He retired from the U.S. Air Force as a Brigadier General in the role of Assistant Surgeon General for medical readiness, science and technology. He has managed all aspects of large integrated health care delivery systems, from clinical care, to runningmanaging clinics and hospitals, managing budgets, professional staffs and oversaw large science and technology portfolios.S&T portfolios, including clinical trials. He has private sector business experience in imaging technology, aswas CEO and co-founder of TessArae LLC, a biotech medical sequencing device company. Most recently he held the position of Chief Medical Officer and client executive for health at CACI International, an information technology company.International. He has been an independent consultant since 2002 and has served as advisory board member toserving a number of biotech and health related companies.company advisory boards and Tadpole Ventures, a private venture capital firm. Dr. Schafer earned his Doctor of Medicine and SurgeryM.D. degree at the University of Iowa, medical boards in family practice and aerospace medicine in the Air Force, a Master of Public Health at the University of Texas, and a Master of Science at the Dwight D. Eisenhower School of National Security and Resource Strategy. Dr. Schafer recently became CERT certified in Cybersecurity Oversight from the Carnegie Mellon University Software Engineering Institute. We believe Dr. Schafer’s broad experience base relevant to Altimmune’s core technology and experience in cybersecurity makes him well qualified to serve as a member of our Board of Directors.
Executive Officers The names and ages of our executive officers as of February 25, 202128, 2023 are set forth below: | | | | | Name |
| Age |
| Position | Vipin K. Garg, Ph.D. | | 6365
| | President, Chief Executive Officer, and Director | William M. Brown, CPARichard Eisenstadt, M.B.A.
| | 3964
| | Chief Financial Officer | M. Scot Roberts, Ph.D. | | 6264
| | Chief Scientific Officer | M. Scott Harris, M.D. | | 6769
| | Chief Medical Officer |
Raymond M. Jordt | | 50 | | Chief Business Officer (effective 1/1/2023) | |

