Washington, D.C. 20549
FORM 10-K
(Mark One)
☒ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2023 | |
OR | |
☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Commission file number 00-50626 | |
CYCLACEL PHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
Delaware | 91-1707622 | ||||
(State or Other Jurisdiction | (I.R.S. Employer | ||||
| | ||||
200 Connell Drive | 07922 | ||||
(Address of principal executive offices) | (Zip Code) |
Registrant’s telephone number, including area code: (908) (908) 517-7330
Securities registered under Sectionpursuant to section 12(b) of the Exchange Act:
Title of each class | Trading Symbol(s) | Name of each exchange on which registered | |||
Common Stock, | CYCC | The | |||
Preferred Stock, $0.001 par value | CYCCP | The |
Securities registered pursuant to Sectionsection 12(g) of the Act: None.
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act. Yes ☐ No ☒
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically, and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer”filer,” “smaller reporting company,” and “smaller reporting“emerging growth company” in Rule 12b-2 of the Exchange Act:
Large accelerated filer | ☐ | Accelerated filer | ☐ | ||
Non-accelerated filer | ☒ | Smaller reporting company | ☒ | ||
Emerging growth company | ☐ | | |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☐
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). ☐Yes☒No
The aggregate market value of the registrant’s voting and non-voting common stock held by non-affiliates of the registrant (without admitting that any person whose shares are not included in such calculation is an affiliate), as of June 30, 20172023 (based upon the closing sale price of $3.77$8.84 of such shares on The NASDAQ Capital Market on June 30, 2017)2023), the last business day of the registrant’s most recently completed second fiscal quarter, was $12,719,813.
As of March 27, 2018,15, 2024, there were 11,997,4471,318,257 shares of the registrant’s common stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
The following documentsdocument (or parts thereof) are incorporated by reference into the following parts of thethis Form 10-K: Certain information required in Part III of this Annual Report on Form 10-K is incorporated from the Registrant’s Proxy Statementregistrant’s definitive proxy statement for the Annual Meeting2024 annual meeting of Stockholdersstockholders to be held on or about Mayfiled pursuant to Regulation 14A with the Securities and Exchange Commission within 120 days of the registrant’s fiscal year ended December 31, 2018.
| | |||||||
| | Page | ||||||
| | |||||||
| | |||||||
4 | ||||||||
27 | ||||||||
64 | ||||||||
64 | ||||||||
66 | ||||||||
66 | ||||||||
66 | ||||||||
| | | ||||||
| | |||||||
| | | ||||||
66 | ||||||||
66 | ||||||||
Management’s Discussion and Analysis of Financial Condition and Results of Operations | 67 | |||||||
76 | ||||||||
77 | ||||||||
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure | 106 | |||||||
106 | ||||||||
108 | ||||||||
Disclosure Regarding Foreign Jurisdictions that Prevent Inspections | 108 | |||||||
| | | ||||||
| | |||||||
| | | ||||||
108 | ||||||||
108 | ||||||||
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters | 108 | |||||||
Certain Relationships and Related Transactions, and Director Independence | 108 | |||||||
109 | ||||||||
| | | ||||||
| | |||||||
| | | ||||||
110 |
2
Summary of Principal Risk Factors
This summary briefly lists the principal risks and uncertainties facing our business, which are only a select portion of those risks. A more complete discussion of those risks and uncertainties is set forth in Part I, Item 1A of this Annual Report, entitled “Risk Factors”. Additional risks not presently known to us or that we currently deem immaterial may also affect us. If any of these risks occur, our business, financial condition or results of operations could be materially and adversely affected.
Our business is subject to the following principal risks and uncertainties:
Risks Associated with Development and Commercialization of Our Drug Candidates
| ● |
| ● |
| ● |
| ● |
| ● |
| ● |
| ● | Our drug candidates are subject to extensive regulation, which can be costly and time-consuming, and we may not obtain approvals for the commercialization of any of our drug candidates. |
| ● | Even if we successfully complete the clinical trials for one or more of our product candidates, the product candidates may fail for other reasons. |
| ● | We face intense competition and our competitors may develop drugs that are less expensive, safer, or more effective than our drug candidates. |
| ● | If we fail to enter into and maintain successful strategic alliances for our drug candidates, we may have to reduce or delay our drug candidate development or increase our expenditures. |
| ● | If our drug candidates or distribution partners’ products fail to achieve market acceptance, we may not be able to generate significant revenue and our business may suffer. Our business may be affected by the efforts of government and third-party payors to contain or reduce the cost of healthcare through various means. |
| ● | We face potential product liability exposure, and if successful claims are brought against us, we may incur substantial liability for a product candidate and may have to limit its commercialization. |
| ● | We may be subject to numerous and varying privacy and security laws, and our failure to comply could result in penalties and reputational damage. |
Risks Related to Our Business and Financial Condition
| ● | We have a history of operating losses and we may never become profitable. Our stock is a highly speculative investment. |
| ● | There is substantial doubt regarding our ability to continue as a going concern. We will need to raise additional capital in upcoming periods which may not be available to us on reasonable terms, if at all. |
| ● | Unstable market and economic conditions may have serious adverse consequences on our business, financial condition and stock price. |
| ● | If we fail to comply with the continued listing requirements of the Nasdaq Capital Market, our common stock may be delisted and the price of our common stock and our ability to access the capital markets could be negatively impacted. |
3
| ● | Funding constraints may negatively impact our research and development activities, forcing us to delay our efforts to develop certain product candidates in favor of developing others, which may prevent us from commercializing our product candidates as quickly as possible. |
| ● | Our business has been and may continue to be adversely affected by the ongoing coronavirus pandemic. |
| ● | We are experiencing an increasingly tight and competitive labor market with an increase in employee turnover rates and higher compensation and hiring costs. This may have an adverse effect on our ability to attract and retain skilled personnel and may harm our business. |
Risks Related to our Intellectual Property
| ● | If we fail to enforce adequately or defend our intellectual property rights, our business may be harmed. |
| ● | We may incur substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights. |
Risks Related to Securities Regulations and Investment in Our Securities
| ● | Failure to achieve and maintain internal controls in accordance with Sections 302 and 404 of the Sarbanes-Oxley Act of 2002 could have a material adverse effect on our business and stock price. |
| ● | We incur increased costs and management resources as a result of being a public company, and we may fail to comply with public company obligations. |
| ● | We may have limited ability to pay cash dividends on our preferred stock, and there is no assurance that future quarterly dividends will be declared. |
| ● | The future sale of our common and convertible preferred stock and future issuances of our common stock upon conversion of our preferred stock could negatively affect our stock price and cause dilution to existing holders of our common stock. |
| ● | The number of shares of common stock which are registered, including the shares to be issued upon exercise of our outstanding warrants, is significant in relation to our currently outstanding common stock and could cause downward pressure on the market price for our common stock. |
| ● | Our management team will have broad discretion over the use of the net proceeds from any sale of our securities. |
| ● | We have restated our previously issued consolidated financial statements and, as part of that process, have identified a material weakness in our internal control over financial reporting as of December 31, 2022. If we are unable to develop and maintain effective internal control over financial reporting, we may not be able to accurately report our financial results in a timely manner. We may also face litigation and other risks as a result of the restatement. |
PART I
The following Business Section contains forward-looking statements. Our actual results could differ materially from those anticipated in these forward-looking statements as a result of certain risks, uncertainties and other factors including the risk factors set forth in Part I, Item 1A of this Annual Report on Form 10-K. In this report, “Cyclacel,” the “Company,” “we,” “us,” and “our” refer to Cyclacel Pharmaceuticals, Inc.
General
We are a clinical-stage biopharmaceutical company using cell cycle control, transcriptional regulation and DNA damage response biologyworking to develop innovative targetedcancer medicines for cancerbased on cell cycle, transcriptional regulation, epigenetics and other proliferative diseases. Cyclacel ismitosis biology. We are a pioneer company in the field of cancer cell cycle biology with a vision to improve patient healthcare by translating insights in cancer biology into medicines.medicines that can overcome resistance and ultimately increase a patient’s overall survival.
The transcriptional regulation program is evaluating fadraciclib, a CDK2/9 inhibitor, in solid tumors and hematological malignancies. The epigenetic/anti-mitotic program is evaluating plogosertib, a PLK1 inhibitor, in solid
4
tumors and hematological malignancies. Our strategy is to build a diversified biopharmaceutical business focused in hematology and oncology based on a development pipeline of novel drug candidates. candidates addressing oncology and hematology indications.
We have retained rights to commercialize our clinical development candidates and our business objective is to enter into selective partnership arrangements with these programs. Substantially all our efforts of the Company to date have been devoted to performing research and development, conducting clinical trials, developing and acquiring intellectual property, raising capital and recruiting and training personnel.
Cell Cycle Control Biology
Loss of control of the cell cycle, the process by which cells grow and divide, lies at the heart of cancer. In normal cells, a complex set of interacting proteins tightly regulates progression through the phases of the cell cycle by which a cell grows, replicates its DNA and divides. This process also includes mechanisms known as cell cycle checkpoints, to ensure all necessary events of each cell cycle phase are completed before beginning the next phase. Specific isoforms of cyclin dependent kinases, or CDKs, and Polo-like Kinases, or PLKs, are some of the key regulators among the numerous genes and proteins involved in cell cycle checkpoints. If thecheckpoint control events are not completed correctly, the cancer cells may commit suicide by a process of programmed cell death called apoptosis. Cyclin dependent kinases, or We seek to enhance and facilitate apoptotic outcomes in cancer cells with the objective of containing the disease and benefiting patients with various cancers.
CDKs are key regulators among the numerous proteins involved in cell cycle control processes. CDKs connectinteract with proteins called cyclins to regulate cell cycle checkpoints and control transcription, DNA repair and metastatic spread. The discovery of CDKs and cyclins and their regulation of cell cycle checkpoint control were cited in the 2001 Nobel Prize in Physiology or Medicine.
The lead drug in our transcriptional regulation program is fadraciclib (formerly known as CYC065).
Polo Kinases and seliciclib.other mitotic kinases were first discovered in fruit flies by our former Chief Scientist, Professor David Glover, PhD. PLK1 is a serine/threonine kinase playing a central role in cell division, or mitosis. In particular, PLK1 regulates mitotic entry, spindle formation, mitotic exit, cytokinesis and is an important regulator of the DNA damage checkpoint. Cancer cells are much more sensitive to PLK1 depletion than normal cells with intact cell cycle checkpoints. Inhibiting PLK1 blocks proliferation by prolonged mitotic arrest followed by onset of cancer cell death.
The lead drug in our anti-mitotic program is plogosertib (formerly known as CYC140).
Clinical Development Pipeline
Our pipeline of innovative medicines aims to provide safe and effective anticancer treatment options to patients combined with the convenience of oral administration. Our preclinical and clinical studies suggests that daily dosing by the oral route is a preferred strategy for both our drugs. We believe that thesealso conducted certain early clinical studies using i.v. administration. The aim of the current streamlined studies is to assess safety and identify signals of clinical activity which may lead to registration-enabling outcomes.
5
The following table summarizes our current development programs:
PROGRAM | INDICATION | PHASE | ||
Transcriptional Regulation | ||||
Fadraciclib CDK inhibitor (oral) | Solid tumors – multiple cohorts defined by tumor histology and a basket cohort | Phase 1/2 to achieve proof of concept | ||
Mitosis Regulation | ||||
Plogosertib PLK inhibitor (oral) | Solid tumors – multiple cohorts defined by tumor histology and a basket cohort | Phase 1/2 to achieve proof of concept # | ||
CDK: cyclin-dependent kinase; PLK: polo-like kinase. # Study to resume recruitment following introduction of new oral formulation.
We currently retain all global marketing rights to the compounds associated with our clinical-stage drug candidates are differentiated from others in that they are orally-available and interact with unique target profiles and mechanisms and have the potential to treat multiple cancer indications.
Transcriptional Regulation:
Regulation Program
Fadraciclib — Cyclin Dependent Kinase (CDK) Inhibitors
CDKs are a family of enzymes first discovered as regulators of the cell cycle, but now understood to also provide pivotal functions in the regulation of transcription, DNA repair and metastatic spread. Different CDK inhibitor drugs selectively target different sets of CDKs. The precise selectivity of an individual CDK inhibitor molecule for certain specific CDKs is key to targeting particular tumor types and minimizing undesirable side effects through non-specific antiproliferativeor off-target activity.
The best characterized CDK enzymes include CDK2, -4, -6 and when dysregulated, can be drivers of particular cancer sub-sets. Modulating CDK activity with targeted therapies is an attractive strategy to reinforce cell cycle control and decrease the
Following Professor Sir David Lane, is an internationally recognized authority in cell cycle biology, who discovered p53, a key tumor suppressor that malfunctions in about two-thirds of human cancers. Under his guidance, Cyclacel’sLane’s insights, our drug discovery and development programs concentrated on the CDK2/9 isoforms, which operate as key components of the p53 pathway. These efforts resulted
Pharmacological inhibition of the CDK2/9 isoforms, by medicines like fadraciclib, has been shown to have potent anticancer effects in bringing two molecules intopreclinical and clinical trials: seliciclib, a first-generation CDK inhibitor, and CYC065, a second-generation CDK inhibitor, which has benefited from the Company’s clinical experiencestudies against certain cancer types, including some that are resistant to approved treatments. It is hoped that treatment with seliciclib.
The FDA approved CDK4/6 inhibitors, palbociclib, ribociclib and abemaciclib, represent an important therapeutic advance and are associated with very promising progression-freeclinically meaningful survival advantages with good tolerability when combined with standard of carehormone therapy versus standard of carehormone therapy alone in patients with certain gynecological tumors, including forms ofhormone receptor positive, HER2-negative breast cancer. Clinical data show that treatment failure after CDK4/6 inhibitors is associated with amplification of cyclin E (Turner NC et al, JCO, 2019). Treatment of patients failing CDK4/6 inhibitors with CDK2/9 inhibitors, such as palbociclib, induce senescence or dormancyfadraciclib, may provide extended benefit to these patients. Preclinical data suggest that treatment of HER2-positive breast cancer cells which may be associated with the emergence of resistance. If clinical manifestation of resistance becomes common, the therapeutic utility of CDK4/6 inhibitors could be hampered.
Different CDKs are responsible for controlling different aspects of proliferation which, when dysregulated, can be drivers of particular cancer sub-sets. Our CDK clinical candidates target:
6
Fadraciclib mechanism and leukemogenic pathways,potential biomarkers:
| ● | CDK2/cyclin E drives cell cycle transition: |
| o | The cyclin dependent kinase inhibitor 2A (CDKN2A) gene encodes two tumor suppressor proteins p16INK4A and p14ARF. p16INK4A inhbits the activity of CDK4/6 and p14ARF inhibits MDM2-mediated degradation of p53. CDKN2A gene mutations or deletions can lead to decreased function of p16INK4A and p14ARF and are associated with cancer development. Loss of CDK4/6 inhibition by p16INK4A can dysregulate G1 phase cell cycle control which could be restored via CDK2 inhibition. For example, in CDKN2A mutant cancer cells activity of the approved CDK4/6 inhibitor drug abemaciclib is limited by CDK2 bypass of CDK4/6i (Gong, Cancer Cell 2017). Loss of p53 activation by p14ARF also contributes to cancer development, and may be countered by suppressing MDM2 expression via CDK9 inhibition. |
| o | The cyclin dependent kinase inhibitor 2B (CDKN2B) gene encodes p15 INK4B and inhibits the activity of CDK4/6. Loss of function of CDKN2B can lead to potentially uncontrolled cell cycle progression which can lead to tumor growth. |
| o | Hypothesis generating data from fadraciclib Phase 1 studies suggest that CDKN2A and/or CDKN2B abnormalities are potential cancer vulnerabilities which can be exploited by a CDK2/9 inhibitor, like fadraciclib. We intend to test this hypothesis in patient cohorts during the proof of concept stage of our Phase 1/2 CYC065-101 study. |
| ● | CDK9/cyclin T regulates transcription of certain genes through phosphorylation of RNA polymerase II C-terminal domain Ser2. MCL1 mRNA and protein are labile (turn over rapidly). Transient blocking of CDK9-dependent transcription leads quickly to loss of MCL1 protein, resulting in apoptosis in MCL1-dependent cancer cells. Labile proteins rapidly depleted by short exposure to a CDK2/9 inhibitor, such as fadraciclib, include MCL1, MYCN, MYC, MYB, BCL2A1 and MDM2. |
| o | MCL1 is overexpressed in many types of cancer acting as a survival and drug resistance mechanism. |
| o | MYC proto-oncogenes encode MYC family proteins which are overexpressed in over 50% of human cancers often via gene amplification. MYC proteins are transcriptional regulators which promote cancer cell growth and survival by increasing the expression of target genes involved in cell metabolism and growth. |
| o | Multiple studies show that knockdown of MCL1 and/or MYC lead to cancer cell death and resensitization to drug treatment. |
Clinical development
Solid tumors
Phase 1/2 Study in advanced solid tumors and lymphomas (CYC065-101, dosed orally, NCT04983810)
The ongoing study is an open-label, multicenter, Phase 1/2 registration-directed trial using a streamlined design. Phase 1 explores both schedule and escalating doses of oral fadraciclib as a single-agent in a 28-day cycle with a primary objective of identifying maximum tolerated dose or MTD and/or the recommended Phase 2 dose or RP2D. Once RP2D has been established, the trial will immediately enter proof-of-concept, cohort stage, using a Simon 2-stage design, where single agent fadraciclib will be administered to patients in up to eight cohorts defined by histology or molecular subtype thought to be sensitive to the drug’s mechanism of action and informed by the clinical activity of fadraciclib in previous studies. The cohorts are expected to include patients with breast cancer (selected for metastatic, hormone receptor positive, HER-2 negative, post-CDK4/6 inhibitor; HER-2 refractory; or triple negative), colorectal (including KRAS mutant), endometrial, hepatobiliary, ovarian cancers, and certain lymphomas. An additional basket cohort will enroll patients with mechanistically relevant biomarkers, including MYCNCDKN2A and/or CDKN2B mutation or deletion, MCL1, MYC and/or cyclin E overexpression or amplification, regardless of histology. The protocol allows for
7
expansion of a cohort based on exceeding certain futility criteria. The primary objective of Phase 2 is to achieve proof of concept and mixed lineage leukemia rearrangements, or MLL-r. CYC065determine preliminary efficacy by overall response rate. Safety, pharmacokinetics and seliciclib both suppressefficacy will be investigated for all subjects. Exploratory objectives are to investigate clinical pharmacodynamics and pharmacogenomics of fadraciclib.
Encouraging preliminary data from the Mcl-1-mediated survival pathwaystudy were presented during a poster presentation at the 34th EORTC-NCI-AACR (ENA) Symposium on Molecular Targets and Cancer Therapeutics. At our R&D Day on October 31, 2022, a principal investigator from Seoul National University Hospital showed preclinical data demonstrating sensitivity to fadra in biliary tract and pancreatic cancer cells leadingobtained from patient specimens.
In the 065-101 study of oral fadraciclib, CDK2/9 inhibitor, a total of 47 patients have been dosed to rapid inductiondate as monotherapy through six dose levels, of apoptosiswhich 33 are evaluable for efficacy. Dose limiting toxicities of nausea and hyperglycemia were observed, which were controlled after dose interruption, with blood glucose levels returning to normal range after treatment.
Dose level 5 (100mg twice daily for 5 days per week, 4 out of 4 weeks) was the maximally tolerated dose (MTD) per protocol for the twice daily schedule and is safe for continued development.
Evaluation of once daily dosing at dose level 6B (150mg once daily for 7 days per week, 4 out of 4 weeks) continues accrual with two patients treated.
To date, single agent activity, including complete response, partial response and stable disease, has been observed in Mcl-1 dependentcertain patients with advanced endometrial, squamous non-small cell lung cancer cells and reversalT-cell lymphoma. Encouraging signals of drug resistance associatedactivity were also observed in certain patients with advanced cervical, hepatocellular, ovarian and pancreatic cancers. Fadraciclib tablets can be given orally with repeat dosing which has led to transient suppression of anti-apoptosis proteins with generally good tolerability and no Grade 3 or higher hematological toxicity in the addictionfirst cycle. We believe that fadraciclib’s inhibition of cancer cellsCDK2 and CDK9 may be superior to cyclin E,inhibiting either CDK2 or CDK9 alone.
Advanced cancers (CYC065-01, i.v., NCT02552953)
Fadraciclib, using i.v. administration, has been evaluated in a partner protein of CDK2.
Leukemias
Phase 1/2 study in hematological malignancies (CYC065-102, dosed orally, NCT05168904)
This is an open-label, multicenter, Phase 1/2 registration-directed trial using a streamlined design. Phase 1 explores both schedule and escalating doses of oral fadraciclib as a single-agent in a 28-day cycle with a primary objective of identifying MTD and/or the RP2D. Once RP2D has been established, the trial will immediately enter proof-of-concept, cohort stage, using a Simon 2-stage design. Oral fadraciclib, both as a single agent and in combinations, will be administered to patients in up to seven cohorts relevant to the drug’s mechanism of action and informed by the clinical activity of fadraciclib in previous studies. 14 patients have been enrolled in this study through five dose levels. The study is not currently recruiting.
8
Chronic lymphocytic leukemia (CYC065-02, i.v., NCT03739554)
CLL cell survival depends on the expression of anti-apoptotic proteins, including MCL1 and BCL2. In this context, targeting MCL1 or BCL2 releases pro-death signals and commits CLL cells to apoptosis. In preclinical studies, rapid cell death was induced in CLL and multiple myeloma patient-derived cell lines after short exposure to fadraciclib, even in the presence of stromal cells which confer protection from standard treatments. MCL1 down-regulation was observed, consistent with the pro-apoptotic mechanism of fadraciclib. Fadraciclib synergizes with venetoclax in 11 outpreclinical models at clinically achievable concentrations, supporting the clinical investigation of 13combination regimens of fadraciclib and venetoclax.
In a Phase 1 study, i.v. fadraciclib was evaluated in combination with venetoclax in patients treatedwith relapsed or refractory CLL. The study design and preliminary data were presented at a poster during the 2019 Annual Meeting of the American Society of Hematology. Fadraciclib was administered intravenously via four-hour infusion on days 1 and 15 in combination with daily oral venetoclax. Initial dose escalation is 33% and upon occurrence of the first dose limiting toxicity, or DLT, 25%. The primary objective is determination of a recommended Phase 2 dose, or RP2D, following adefined as the highest dose level at which less than one-third of at least six patients experience DLT during the first treatment cycle. Treatment continued until progression of disease, unacceptable toxicity or changes in patient condition that renders patients ineligible for further treatment. Laboratory tests and CT scans were performed regularly to assess response according to standard criteria.
Of the five R/R CLL patients enrolled in CYC065-02 all had failed ibrutinib and one had also failed CAR-T cell treatment. Patients remained minimal residual disease, or MRD, positive after treatment ramp with single doseagent venetoclax for up to 5 weeks. Continuing shrinkage of CYC065, which was generally well tolerated. Preliminary anticancer activityenlarged lymph nodes was observed by CT scan on the combination of venetoclax and fadraciclib dosed once every two weeks. The patient who failed CAR-T cell therapy and two additional patients achieved MRD negative status on the combination.
Acute myeloid leukemia, or AML (CYC065-03, i.v., NCT04017546)
Drug resistance in 6 patients,AML has been attributed among others to high levels of whichMCL1. AML cell lines are highly sensitive to fadraciclib and 5 were treated atto 8 hours of treatment is sufficient to achieve induction of cell death. Fadraciclib has single agent efficacy in AML xenografts and the RP2Dpotential to be combined with approved AML therapies. In leukemia cells harboring the rearranged MLL-r, fadraciclib reduced both MCL1 expression and 3CDK9 dependent transcription of which were reported by investigators to have molecular features of their cancers associated with CYC065’s mechanism of action, including overexpression or amplification of Mcl-1, MYCN and/or cyclin E. The trial is being conducted at the Dana Farber Cancer InstituteMLL-regulated leukemogenic genes.
We completed enrollment in Boston. Part 2 of thea Phase 1 translational study will evaluate additional dosing schedulesevaluating i.v. fadraciclib in combination with venetoclax in patients with advanced solid tumors, in particular those tumors with amplification of Mcl-1, MYCN and/relapsed or cyclin E, including subsets of high grade serous ovarianrefractory AML or MDS. The study design and uterine cancers. Biospecimens will be collected for assessment of biomarkers related to CYC065’s mechanism of action.
Four of twelve patients in CYC065-03 achieved decreases in vitro models of B-cell lymphoma, including double-hit lymphomas. Combinations of CYC065 with the Bcl-2 inhibitor, venetoclax (ABT-199), or BET inhibitors were both synergistic. Short exposure to CYC065 was sufficient to downregulate MYCN, an oncogene product aberrantly expressedleukemia blast cells in many cancers, and
Published preclinical data
Preclinical data suggest therethat fadraciclib may be a therapeutic rationale to combine CYC065benefit adults and venetoclax for the treatmentchildren with hematological malignancies, including AML, acute lymphocytic leukemias, or ALL, and in particular leukemias with MLL-r, CLL, B-cell lymphomas, multiple myelomas, and patients with certain solid tumors, including breast and uterine cancers, and neuroblastomas.
9
| ● | Prolonged survival and reduced tumor burden in MYCN-addicted neuroblastoma |
The MYCN oncogene is over-expressed in a number of differentseveral types of cancer, most notably neuroblastoma, butand also rhabdomyosarcoma, medulloblastoma, astrocytoma, Wilms’ tumor and small cell lung cancer. Amplification of the MYCN oncogene is the most common genomic alteration in aggressive neuroblastomas,neuroblastoma and is associated with poor clinical outcome. Preclinical data presented at the 2016 Childhood Cancer Meeting demonstrated that CYC065fadraciclib prolonged survival in MYCN-addicted neuroblastoma models and neuroblastomamodels. Neuroblastoma cells with MYCN amplification and overexpression were found to be particularly sensitive. The mechanism of action of CYC065 includedTreatment with fadraciclib was associated with inhibition of MYCN transcription, downregulation of N-MYCMYCN protein, blocking neuroblastoma cell proliferation and induction of apoptosis. There are no approved drugs that directly target MYCN, prompting the investigation of indirect approaches such as suppression of MYCN gene expression via CDK9 inhibition, or exploitation of a synthetic lethal relationship between MYCN amplification/overexpression and inhibition of CDK2.
| ● | May reverse drug resistance associated with addiction of cancer cells to cyclin E, the partner protein of CDK2 |
Fadraciclib as a single-agentsingle agent can induce tumor growth delay in HER2-positive breast cancer cells addicted to cyclin E and resistant to trastuzumab, while administration of CYC065fadraciclib in combination with trastuzumab resulted in regression or sustained tumor growth inhibition.
| ● | May have activity in KRAS-mutated cancers |
Researchers led by Frank McCormick, PhD of University of California San Francisco and NCI’s Frederick National Lab for Cancer Research reported that overactive KRAS mutants are impeded by CDK9 inhibition (Lai LP, et al, SLAS Discovery 2021). These data expand on previous publications which report that dual CDK2/9 inhibition is an optimal strategy to treat colorectal cancer (Somarelli JA, et al, Mol Cancer Ther, 2020), that KRAS mutant pancreatic cancer is sensitive to CDK9 inhibition (Blake DR, et al, Science Signalling, 2019), and that fadraciclib showed efficacy against KRAS mutant lung cancer in triple-negative breast cancerpreclinical PDX models (Kawakami M, et al J Natl Cancer Inst, 2017). Collectively these publications suggest the potential for the therapeutic use of fadraciclib in KRAS-mutated cancers, including colorectal, lung and pancreatic.
| ● | Induces leukemia cell death and can combine beneficially with other anti-cancer drugs |
Fadraciclib targets key CDK9-dependent oncogenic and leukemogenic survival pathways. Data presented at the 2015 San Antonio Breast Cancer Symposium demonstrated in particular the mechanistic rationale for clinical development of CYC065 in basal-like triple negative breast cancer, or TNBC, a cancer with poor prognosis frequently associated with BRCA mutations. Molecular characteristics of TNBC include amplification or overexpression of Cyclin E, the partner protein of CDK2, and MYCN. CYC065 directs a pro-apoptotic mechanism in breast cancer cell lines, which includes transcriptional down regulation of key pro-survival and oncogenic regulators, including Mcl-1 and MYCN. CYC065’s potent anticancer activity has been confirmed in breast cancer xenograft animal models. Like seliciclib, CYC065 combined effectively with sapacitabine in breast cancer cell lines.
Data from Part 1 and 2 of the study were presented at the 2016 Annual Meeting of the American SocietyAssociation of Clinical Oncology Annual Meeting. Antitumor activityCancer Research demonstrated that fadraciclib can induce cell death and combine beneficially with anti-cancer drugs from the BCL2 and BET (Bromodomain and Extra-Terminal domain) inhibitor classes, in in vitro models of B-cell lymphoma, including double-hit lymphomas. Combinations of fadraciclib with the BCL2 inhibitor venetoclax, or BET inhibitors were both synergistic. Short exposure to fadraciclib was observedsufficient to downregulate MYC and MCL1 and induce cell death. Fadraciclib treatment had no impact on BCL2 levels.
In April 2022, a publication in the subgroup of 45 patients with breast, ovarian and pancreatic cancers who tested positive for BRCA mutations (44 germline and 1 sporadic) with an overall disease control rate of over 30%. Best responses were as follows:
| | | | PART 1 | | | PART 2 | | ||||||||||||||||||
| | | | BRCA carriers (n=16) | | | Others (n=22) | | | BRCA carriers (n=28) | | | Others (n=1) | | ||||||||||||
| CR | | | | | 1 | | | | | | — | | | | | | — | | | | | | — | | |
| PR | | | | | 3 | | | | | | — | | | | | | 2 | | | | | | — | | |
| SD | | | | | 2 | | | | | | 6 | | | | | | 7 | | | | | | 1* | | |
| ORR (CR/PR) | | | | | 25% | | | | | | 0% | | | | | | 7% | | | | | | 0% | | |
| Disease Control (CR/PR/SD) | | | | | 6 (37.5)% | | | | | | 6 (27.3)% | | | | | | 9 (32.1)% | | | | | | 1 (100.0)% | | |
| | |||||||||||||||||||||||||
These findings support the study protocol until the prespecified 424 events had been observed.
10
Mitosis Regulation Program
Polo-Like-Kinase inhibitor: CYC140
In our polo-like kinase,Polo-like Kinase, or PLK, inhibitor program, we have discovered potent and selective small molecule inhibitors of PLK1, a kinase active during cell division, which target the mitotic phase of the cell cycle. PLKPLK1. Polo Kinase was discovered by Professor David Glover, our former Chief Scientist. We received
PLK1 is a grant award of approximately $3.5 million fromserine/threonine kinase with a central role in cell division, or the Biomedical Catalystmitotic phase of the United Kingdom government to complete IND-directed preclinicalcell cycle, and is an important regulator of the DNA damage checkpoint. PLK1 over-expressing tumors include colorectal, esophageal, gastric, leukemia, lung, lymphoma, ovarian and squamous cell cancers, as well as MYC amplified cancers including breast. Recent data with another PLK1 inhibitor in clinical development, suggest that PLK1 inhibition may be effective in KRAS-mutated metastatic colorectal cancer.
Plogosertib is a novel, small molecule, selective, PLK1 inhibitor which has demonstrated an epigenetic mechanism, potent and selective target inhibition (PLK1 IC50 approximately 3 nM) and impressive efficacy in human tumor xenografts at non-toxic doses. Plogosertib has improved pharmaceutical properties over earlier, clinical stage, PLK inhibitors. Our translational biology program supports the development of CYC140,plogosertib in solid tumor and hematological malignancy indications.
Clinical development
Phase 1/2 Study in advanced solid tumors and lymphomas (CYC140-101, orally dosed)
Similar to fadraciclib, this ongoing open-label Phase 1/2 registration-directed trial uses a streamlined design and seeks to first determine in a dose escalation stage the RP2D for single-agent plogosertib. Once RP2D has been established, the trial will immediately enter into proof-of-concept, cohort stage, using a Simon 2-stage design. In this stage plogosertib will be administered to patients in up to seven mechanistically relevant cohorts including patients with bladder, breast, colorectal (including KRAS mutant), hepatocellular and biliary tract, and lung cancers (both small cell and non-small cell), as well as lymphomas. An additional basket cohort will enroll patients with biomarkers relevant to the drug’s mechanism, including MYC amplified tumors. The protocol allows for expansion of individual cohorts based on response which was completed in November 2016. Without additional funding, wemay allow acceleration of the clinical development and registration plan for plogosertib. Fifteen patients have been treated at the first five dose escalation levels with no dose limiting toxicities observed. A new oral formulation of plogosertib is under development and further patients will not be ablerecruited to progress this program into clinical development. We have retained worldwide rights to commercialize CYC140.
Published preclinical data
Preclinical data presented at the 2016 28th28th EORTC-NCI-AACR Molecular Targets and Cancer Therapeutics Symposium and at the 2017 Annual Meeting of the American Association of Cancer Research demonstrated the therapeutic potential of CYC140plogosertib as a targeted anti-cancer agent. The data demonstrated that CYC140plogosertib is a selective PLK1 inhibitor which is highly active against both solid and liquid cancer models, preferentially induces growth inhibition and cell death in malignant versus non-malignant cells.
Treatment of proliferating cells with CYC140plogosertib resulted in reduced phosphorylation of the PLK1 substrate phospho-nucleophosmin, accumulation of cells in mitosis and an increase in the proportion of mitotic cells with monopolar spindles, which are all features consistent with PLK1 inhibition. In a cell line panel derived from esophageal cancer and various non-malignant solid tissues, CYC140plogosertib was preferentially cytotoxic to malignant cells. Its differential cytotoxicity is further increased through pulse treatment. Malignant cells which are sensitive to CYC140plogosertib undergo complete growth inhibition and induction of cell death in response to treatment. In contrast, non-malignant cells are only temporarily arrested and normal cell cycle transit is restored.
Business Strategy
We plan to continue to build a diversified biopharmaceutical business focused inon hematology and oncology based on a pipeline of novel drug candidates and utilizing our area of historical expertise in cancer cell cycle and mitosis biological mechanisms. Our clinical development strategy is focused on two ongoing programs in transcriptional
11
regulation and DNA damage response.
Focus on the cell cycle and cancer
Our core area of expertise is in cell cycle biology and our scientists include recognized leaders in this field. In addition, our senior management team has extensive experience in research, preclinical and clinical development and sales and marketing. The novel, mechanism-targeted cell cycle drugs we are developing are designed to be highly selective in comparison to conventional chemotherapies, potentially inducing death in cancer cells while sparing most normal cells which may give rise to fewer side-effects.
Thus, we believe that we are well placed to exploit the significant opportunities that this area offers for new drug discovery and development.
Develop anticancer drug candidates in all phases of the cell cycle and multiple compounds for particular cell cycle targets
Targeting a broad development program focused on multiple phases of the cell cycle allows us to minimize risk while maximizing the potential for success, and also to develop products that are complementary to one another.
Enter into partnering arrangements selectively, while developing our own sales and marketing capability
We currently retain virtually all marketing rights to the compounds associated with our clinical-stage drug programs. To optimize our commercial return, we intend to enter into selected partnering arrangements and to retain co-promotion rights as appropriate. Historically,Generally we have plannedplan to develop compounds through the Phase 2 proof-of-efficacy stage before seeking a partner. We may enter into partnering arrangements earlier than Phase 2 proof-of-concept trials where appropriate, or in connection with drug programs outside our core competency in oncology.
Licenses
Some of our programs are based on technology licensed from others. Our breach of an existing license or failure to obtain a license to technology required to develop, test and commercialize our products may seriously harm our business.
Patents and Proprietary Technology
Patents and Proprietary Rights
We own 2514 patents granted in the United States, 145 granted by the European Patent Office, or EPO, and 5930 granted in other countries worldwide. In addition, we have a license to 4811 patents granted in the US, by the EPO or worldwide.
We have 13 patent applicationapplications pending in the United States, 64 before the EPO 12and 25 pending patent applications in other countries and one pending PCT application still in the international application phase.countries. No assurances can be given that any patents will be issued with respect to the pending applications, nor that the claims will provide equivalent coverage in all jurisdictions. In addition to the pending patent applications referred to above that we own, there are 3 pending patent applications worldwide to which we have a license.
Intellectual Property Strategy
We consider intellectual property rights to be vital and use a variety of methods to secure, protect and evaluate these rights. These methods include ownership and enforcement of patent rights, patent applications, license agreements with third parties, invention assignment, confidentiality and non-compete agreements with key employees and consultants, material transfer agreements, and trademark protection.
We give priority to obtaining substance of matter claims in the United States, the EPO, Japan and other important markets if such protection is available. We prefer substancecomposition of matter claims because they provide us with rights to the compounds themselves, and not merely a particular use. In addition to
12
coverage for solid state forms, polymorphic and crystalline forms, medical uses, combination therapies, specific regimens, pharmaceutical forms of our compounds and synthetic routes where available and appropriate. Claims covering combination therapies, specific regimens and pharmaceutical forms can be valuable because the therapeutic effect of pharmaceuticals used in the anticancer field is often enhanced when individual therapeutics are used in particular combinations or dosed in a certain way. The availability of protection in these areas can, however, vary from jurisdiction to jurisdiction and combination claims are particularly difficult to obtain for many inventions.
Since publications in the scientific or patent literature often lag behind actual discoveries, we are not certain of being first to make the inventions covered by each of our pending patent applications or the first to file those patent applications. Generally, patent applications in the United States are maintained in secrecy for a period of 18 months or more, which increases the uncertainty we face. Moreover, the patent positions of biotechnology and pharmaceutical companies are highly uncertain and involve complex legal and factual questions. As a result, we cannot predict the breadth of claims allowed in biotechnology and pharmaceutical patents, or their enforceability. To date, there has been no consistent policy regarding the breadth of claims allowed in biotechnology patents. Third parties or competitors may challenge or circumvent our patents or patent applications, if issued. Because of the extensive time required for development, testing and regulatory review of a potential product, it is possible that before we commercialize any of our products, any related patent may expire, or remain in existence for only a short period following commercialization, thus reducing any advantage of the patent and the commercial opportunity of the product.
If patents are issued to others containing valid claims that cover our compounds or their manufacture or use or screening assays related thereto, we may be required to obtain licenses to these patents or to develop or obtain alternative technology. We are aware of several published patent applications, and understand that others may exist, that could support claims that, if granted and held valid, would cover various aspects of our developmental programs, including in some cases particular uses of our drug candidates sapacitabine, seliciclib, CYC065fadraciclib and plogosertib, or other therapeutic candidates, or substances, processes and techniques that we use in the course of our research and development and manufacturing operations.
In addition, we understand that other applications and patents exist relating to potential uses of sapacitabine, seliciclibfadraciclib and CYC065 thatplogosertib which are not part of our current clinical programs for those compounds. Although we intend to continue to monitor the pending applications, it is not possible to predict whether these claims will ultimately be allowed or if they were allowed what their breadth would be. In addition, we may need to commence litigation to enforce any patents issued to us or to determine the scope and validity of third-party proprietary rights. For example, in one case we opposed a European patent relating to human aurora kinase and the patent has beenwas finally revoked (no appeal was filed). Litigation would create substantial costs. We are aware that other patents exist that claim substances, processes, techniques and techniques,methods of use, which, if held valid, could potentially restrict the scope of our research, development or manufacturing operations. If competitors prepare and file patent applications in the United States that claim technology that we also claim, we may have to participate in interference proceedings in the United States Patent and Trademark Office to determine which invention has priority. These proceedings could result in substantial costs, even if the eventual outcome is favorable to us. An adverse outcome in litigation could subject us to significant liabilities to third parties and require us to seek licenses of the disputed rights from third parties or to cease using the technology, even a therapeutic product, if such licenses are unavailable or too expensive.
Issued patents for the CYC065 compoundfadraciclib cover the United States, EPO and nineeleven other countries.
Manufacturing
We have no in-house manufacturing capabilities and have no current plans to establish manufacturing facilities for significant clinical or commercial production. We have no direct experience in manufacturing commercial quantities of any of our products, and we currently lack the resources or capability to manufacture any of our products on a clinical or commercial scale. As a result, we are dependent on corporate partners, licensees or other third parties for the manufacturing of clinical and commercial(and eventually commercial) scale quantities of all of our products. We believe that this strategy will enable us to direct operational and financial resources to the development of our product candidates rather than diverting resources to establishing a manufacturing infrastructure.
13
Government Regulation
The FDA EMA and comparable regulatory agencies in state and local jurisdictions, as well as in foreign countries, impose substantial regulatory requirements upon the clinical development, manufacture, marketing and distribution of drugs. These agencies and other federal, state and local entitiesauthorities regulate research and development activities and the testing, manufacture, quality control, import, export, safety, effectiveness,efficacy, labeling, packaging, storage, distribution, record keeping, approval, advertising, promotion, marketing, post-approval monitoring, and promotionpost-approval reporting of drug products, such as those we are developing. Along with our drug candidatesthird-party contractors, we will be required to navigate the various preclinical, clinical and commercialized drugs.
For example, in the United States, the FDA regulates drugs under the Federal Food, Drug and Cosmetic Act and its implementing regulations. The process required by the FDA before our drug candidates may be marketed in the United States generally involves the following:
| ● | completion of extensive nonclinical laboratory tests, which may include animal studies and formulation studies, all performed in accordance with the FDA’s good laboratory practice, or GLP, regulations; |
| ● | submission to the FDA of an investigational new drug application, or IND, which must become effective before clinical trials may begin and must be updated annually or when significant changes are made; |
| ● | approval by an IRB or ethics committee at each clinical site before the trial is initiated at such sites; |
| ● | performance of adequate and well-controlled clinical trials in accordance with good clinical practice, or GCP, and other clinical-trial related regulations to establish the safety and efficacy of the drug candidate for each proposed indication; |
| ● | preparation and submission of a new drug application, or NDA, to the FDA; |
| ● | a determination by the FDA within 60 days of its receipt of an NDA to file the application for review; |
| ● | satisfactory completion of an FDA Advisory Committee review, if applicable; |
| ● | satisfactory completion of an FDA pre-approval inspection of the manufacturing facilities at which the product is produced to assess compliance with current good manufacturing practice requirements, or cGMP, regulations; |
| ● | potential audit of selected clinical trial sites to assess compliance with GCP and the integrity of the clinical data submitted in support of the NDA; and |
| ● | FDA review and approval of the NDA to permit commercial marketing of the drug product for particular approved indications for use in the United States. |
This testing and approval process requires substantial time, effort and financial resources, and we cannot be certain that any approvals for our drug candidates will be granted on a timely basis, if at all.
Preclinical development
Before testing any drug product candidate, including our product candidates, in humans, the product candidate must undergo rigorous preclinical testing. Preclinical and other nonclinical tests generally include laboratory evaluation of product chemistry, formulation and stability, as well as studies to evaluate toxicity in animals. The Consolidated Appropriations Act for 2023, signed into law on December 29, 2022, (P.L. 117-328) amended the FDCA and the Public Health Service Act to specify that nonclinical testing for drugs may, but is not required to, include in vivo animal testing. According to the amended language, a sponsor may fulfill nonclinical testing requirements by completing various in
14
vitro assays (e.g., cell-based assays, organ chips, or microphysiological systems), in silico studies (i.e., computer modeling), other human or nonhuman biology-based tests (e.g., bioprinting), or in vivo animal tests. The conduct of preclinical studies is subject to federal regulations and requirements, including GLP regulations for safety/toxicology studies. Some long-term preclinical testing, such as animal tests of reproductive adverse events and carcinogenicity, may continue after an IND for an investigational drug candidate is submitted to the FDA and human clinical trials have been initiated.
The results of preclinical tests, together with manufacturing information and analytical data, are submitted as part of an IND to the FDA. An IND must become effective before human clinical trials may begin. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, issues a notice expressly authorizing the proposed trial to proceed or raises concerns or questions about the adequacy or safety of the preclinical testing or the proposed conduct of the clinical trial, including concerns that human research subjects will be exposed to unreasonable health risks. In such a case,If FDA raises concerns or places the trial on clinical hold, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. Our submission of an IND, or those of our collaborators, may therefore not result in FDA authorization to commence a clinical trial. A separate submission to an existing IND must also be made for each successive clinical trial conducted during product development. Further, an independent institutional review board, or IRB, for each medical center proposing to conduct the clinical trial (or a central IRB) must review and
Clinical Trials
For purposes of an NDA submission, clinical trials are typically conducted in the following three sequential phases, which may overlap:
| ● | Phase 1: The clinical trials are initially conducted in a limited population to test the drug candidate for safety, dose tolerance, absorption, metabolism, distribution and excretion in healthy humans. In the case of some products for severe or life-threatening diseases, such as cancer, especially when the product may be too inherently toxic to ethically administer to healthy volunteers, the initial human testing is often conducted in patients. Phase 1 clinical trials can be designed to evaluate the impact of the drug candidate in combination with currently approved drugs. |
| ● | Phase 2: These clinical trials are generally conducted in a limited patient population to identify possible adverse effects and safety risks, to evaluate preliminary efficacy of the drug candidate for specific targeted indications and to determine dose tolerance and optimal dosage. Multiple Phase 2 clinical trials may be conducted by the sponsor to obtain information prior to beginning larger and more expensive Phase 3 clinical trial. |
| ● | Phase 3: These clinical trials are commonly referred to as pivotal clinical trials. If the Phase 2 clinical trials demonstrate that a dose range of the drug candidate is effective and has an acceptable safety profile, Phase 3 clinical trials are then undertaken in large patient populations to further evaluate dosage, to provide substantial evidence of clinical efficacy and to further test for safety in an expanded and diverse patient population at multiple, geographically dispersed clinical trial sites. These trials are intended to establish the overall risk-benefit ratio of the product candidate and provide, if appropriate, an adequate basis for product labeling. Phase 3 trials typically include comparisons with placebo and/or other comparator treatments. The duration of treatment is often extended to mimic the actual use of a product during marketing. |
In some cases, the FDA may condition approval of an NDA for a drug candidate on the sponsor’s agreement to conduct a Phase 4, which includes additional clinical trials to further assess the drug’s safety and effectiveness after NDA approval.
15
In the Consolidated Appropriations Act for 2023, Congress amended the FDCA to require sponsors of a Phase 3 clinical trial, or other “pivotal study” of a new drug to support marketing authorization, to submit a diversity action plan for such clinical trial. The action plan must include the sponsor’s diversity goals for enrollment, as well as a rationale for the goals and a description of how the sponsor will meet them. A sponsor must submit a diversity action plan to FDA by the time the sponsor submits the trial protocol to the agency for review. The FDA may grant a waiver for some or all of the requirements for a diversity action plan. It is unknown at this time how the diversity action plan may affect Phase 3 trial planning and timing or what specific information FDA will expect in such plans, but if FDA objects to a sponsor’s diversity action plan and requires the sponsor to amend the plan or take other actions, it may delay trial initiation.
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and more frequently if unexpected serious adverse events, or SAEs, occur. The FDA or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the clinical protocol, GCP, or other IRB requirements or if the drug candidatehas been associated with unexpected serious harm to patients.
NDA Submission and Review by the FDA
Assuming successful completion of all required testing in accordance with all applicable regulatory requirements, the results of product development, nonclinical testingpreclinical studies and clinical trials are submitted to the FDA as part of an NDA.NDA requesting approval to market the product for one or more indications. The NDA also must contain extensiveproof of the product candidate’s safety and substantial evidence of effectiveness for its proposed indication or indications in the form of relevant data available from pertinent preclinical and clinical studies, including negative or ambiguous results as well as positive findings, together with detailed information relating to the product’s chemistry, manufacturing, information. Oncecontrols, and proposed labeling, among other things. In particular, a marketing application must demonstrate that the manufacturing methods and quality controls used to produce the drug product are adequate to preserve the drug’s identity, strength, quality, and purity. Data can come from company-sponsored clinical studies intended to test the safety and effectiveness of a use of the product, or from a number of alternative sources, including studies initiated by investigators. FDA approval of an NDA must be obtained before the corresponding drug may be marketed in the United States.
The FDA reviews all NDAs submitted to determine if they are substantially complete before it accepts them for filing and may request additional information rather than accepting a submission for filing. The FDA must make a decision on accepting an NDA for filing within 60 days of receipt and must inform the sponsor by the 74th day after the FDA’s receipt of the submission has been accepted for filing,whether the application is sufficiently complete to permit substantive review. The FDA may refuse to file any submission that it deems incomplete or not properly reviewable at the time of submission and may request additional information. In this event, the marketing application must be resubmitted with the additional information requested by lawthe agency. The resubmitted application is also subject to review before the FDA has 180 days to reviewaccepts it for filing.
During the application and respond to the applicant. The review process, the FDA reviews the NDA to determine, among other things, whether the product is often significantly extended by FDA requests for additional informationsafe and effective and whether the facility in which it is manufactured, processed, packed, or clarification.held meets standards designed to assure the product’s continued strength, quality, and purity. The FDA may refer theany NDA, including applications for novel drug candidates which present difficult questions of safety or efficacy to an advisory committee forto provide clinical insight on application review evaluationquestions. Typically, an advisory committee is a panel of independent experts, including clinicians and other scientific experts that reviews, evaluates and provides a recommendation as to whether the application should be approved.approved and under what conditions. The FDA is not bound by the recommendation of an advisory committee, but it generally followsconsiders such recommendations.recommendations carefully when making final decisions on approval.
Under the Pediatric Research Equity Act, or PREA, amendments to the FDCA, an NDA or supplement to an NDA must contain data that are adequate to assess the safety and efficacy of the product candidate for the claimed indications in all relevant pediatric populations and to support dosing and administration for each pediatric population for which the product is safe and effective. The FDA may deny approvalgrant deferrals for submission of pediatric data or full or partial waivers. The PREA requires a sponsor that is planning to submit a marketing application for a product that includes a new active ingredient, new indication, new dosage form, new dosing regimen or new route of administration to submit an initial Pediatric Study Plan, or PSP, within sixty days of an NDAend-of-Phase 2 meeting or, if there is no such meeting, as early as
16
practicable before the applicable regulatory criteria are not satisfied,initiation of the Phase 3 or it may require additional clinical data or an additional pivotal Phase 2/3 clinical trial. EvenThe initial PSP must include an outline of the pediatric study or studies that the sponsor plans to conduct, including trial objectives and design, age groups, relevant endpoints and statistical approach, or a justification for not including such detailed information, and any request for a deferral of pediatric assessments or a full or partial waiver of the requirement to provide data from pediatric studies along with supporting information. The FDA and the sponsor must reach an agreement on the PSP. A sponsor can submit amendments to an agreed upon initial PSP at any time if changes to the pediatric plan need to be considered based on data collected from pre-clinical studies, early-phase clinical trials or other clinical development programs.
If regulatory approval of a product is granted, such data are submitted,approval is limited to the conditions of use (e.g., patient population, indication) described in the application and may entail further limitations on the indicated uses for which such product may be marketed. For example, the FDA may ultimately decide thatapprove the NDA does not satisfywith a Risk Evaluation and Mitigation Strategy, or REMS, plan to mitigate risks, which could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. The FDA determines the criteriarequirement for approval. Data from clinical trials are not always conclusive anda REMS, as well as the specific REMS provisions, on a case-by-case basis. If the FDA concludes a REMS plan is needed, the sponsor of the NDA must submit a proposed REMS. The FDA will not approve an NDA without a REMS, if one is required. The FDA also may interpret data differently than wecondition approval on, among other things, changes to proposed labeling (e.g., adding contraindications, warnings or our collaborators do.precautions) or the development of adequate controls and specifications. Once issued,approved, the FDA may withdraw a drugthe product approval if ongoingcompliance with pre- and post-marketing regulatory requirements arestandards is not metmaintained or if safety problems occur after the drugproduct reaches the market. In addition, themarketplace. The FDA may require further testing, includingone or more Phase 4 clinical trials,post-market studies and surveillance programs to further assess and monitor the effect of approved drugs which have been commercialized. The FDA has the power to prevent orproduct’s safety and effectiveness after commercialization and may limit further marketing of a drugthe product based on the results of these post-marketing programs. Drugsstudies. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further testing requirements and FDA review and approval. In addition, new government requirements, including those resulting from new legislation, may be marketed only forestablished, or the approved indicationsFDA’s policies may change, which could delay or indications and in accordance with the provisions of the approved label. Further, if there are any modifications to a drug, including changes in indications, labeling or manufacturing processes or facilities, we may be required to submit and obtain FDAprevent regulatory approval of a new NDA or NDA supplement, which may require us to develop additional data or conduct additional nonclinical studies and clinical trials.
Fast Track, Designation
A sponsor may seek approval of its product candidate under programs designed to accelerate FDA’s review and approval of new drugs and biological products that meet certain criteria. Specifically, new drugs and biological products are eligible for fast track program isdesignation if they are intended to facilitate the development and to expedite the review of drugs that are intended for the treatment oftreat a serious or life-threatening condition for which there is no effective treatment and which demonstrate the potential to address unmet medical needs for the condition. Under the fastFast track program, thedesignation provides increased opportunities for sponsor of a new drug candidate may requestinteractions with the FDA during preclinical and clinical development, in addition to designate the drug candidatepotential for rolling review once a specific indication as a fast track drug concurrent with or after the submission of the IND for the drug candidate. The FDA must determine if the drug candidate qualifies for fast track designation within 60 days of receipt of the sponsor’s request.
Priority review within a six-month time frame fromis granted when there is evidence that the time a complete NDA is accepted for filing, if the drug candidate providesproposed product would be a significant improvement compared to marketed drugs in the safety or effectiveness of the treatment, diagnosis, or prevention of a disease. We cannot guaranteeserious condition. Significant improvement may be illustrated by evidence of increased effectiveness in the treatment of a condition, elimination or substantial reduction of a treatment-limiting drug reaction, documented enhancement of patient compliance that anymay lead to improvement in serious outcomes, or evidence of our drug candidates will receivesafety and effectiveness in a new subpopulation. If criteria are not met for priority review, the application is subject to the standard FDA review period of 10 months after FDA accepts the application for filing.
In addition, a sponsor may seek FDA designation of its product candidate as a breakthrough therapy if the product candidate is intended, alone or in combination with one or more other drugs or biologics, to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the therapy may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. Breakthrough therapy designation provides all the features of fast track
17
designation in addition to intensive guidance on an efficient development program beginning as early as Phase 1, and FDA organizational commitment to expedited development, including involvement of senior managers and experienced review and regulatory staff in a proactive, collaborative, cross-disciplinary review, where appropriate. A drug designated as breakthrough therapy is also eligible for accelerated approval if the relevant criteria are met.
Even if a priority designation is received,product qualifies for one or more of these programs, the FDA may later decide that the product no longer meets the conditions for qualification or decide that the time period for FDA review or approval will not be faster than conventional FDA procedures,shortened. fast track, priority review and breakthrough therapy designations do not change the scientific or thatmedical standards for approval or the FDA will ultimately grant drug approval.
Accelerated Approval.
In addition, products studied for their safety and effectiveness in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefit to patients over existing treatments based upon eithermay receive accelerated approval from the FDA and may be approved on the basis of adequate and well-controlled clinical trials establishing that the drug product has an effect on a surrogate endpoint that is expectedreasonably likely to predict clinical benefit. The FDA may also grant accelerated approval for such a clinical benefitdrug or on the basis ofbiologic when it has an effect on aan intermediate clinical endpoint that can be measured earlier than an effect on irreversible morbidity or mortality, or IMM, and that is reasonably likely to predict an effect on IMM or other than patient survival. Inclinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. As a condition of approval, the FDA may require that a sponsor of a drug or biologic receiving accelerated approval perform post-marketing clinical trials surrogate endpoints are alternative measurements ofto verify and describe the symptoms ofpredicted effect on IMM or other clinical endpoint, and the product may be subject to expedited withdrawal procedures. Drugs and biologics granted accelerated approval must meet the same statutory standards for safety and effectiveness as those granted traditional approval.
The accelerated approval pathway is usually contingent on a disease or condition that are substituted for measurements of observablesponsor’s agreement to conduct, in a diligent manner, additional post-approval confirmatory studies to verify and describe the product candidate’s clinical symptoms. A drugbenefit. As a result, a product candidate approved on this basis is subject to rigorous post-marketing compliance requirements, including the completion of Phase 4 or post-approval clinical trials to validate the surrogate endpoint or confirm the effect on the clinical endpoint. Failure to conduct required post-approval studies, or to validate a surrogate endpoint or confirm athe predicted clinical benefit of the product during post-marketing studies, willwould allow the FDA to withdraw approval of the drugproduct. As part of the Consolidated Appropriations Act for 2023, Congress provided FDA additional statutory authority to mitigate potential risks to patients from continued marketing of ineffective drugs or biologics previously granted accelerated approval. Under the marketact’s amendments to the FDCA, FDA may require the sponsor of a product granted accelerated approval to have a confirmatory trial underway prior to approval. The sponsor must also submit progress reports on ana confirmatory trial every six months until the trial is complete, and such reports are published on FDA’s website. The amendments also give FDA the option of using expedited basis. procedures to withdraw product approval if the sponsor’s confirmatory trial fails to verify the claimed clinical benefits of the product.
All promotional materials for drugproduct candidates being considered and approved under the accelerated regulationsapproval program are subject to prior review by the FDA. In rare instances the FDA may grant accelerated approval of an NDA based on Phase 2 data and require confirmatory Phase 3 studies to be conducted after approval and/or as a condition of maintaining approval. We can give no assurance that any of our drugs will be reviewed under such procedures.
Special Protocol Assessment
If a Phase 2 clinical trial is the subject of discussion at an end-of-Phase 2 meeting with the FDA, a sponsor may be able to request a Special Protocol Assessment, or SPA, the purpose of which is to reach agreement with the FDA on the design of the Phase 3 clinical trial protocol design and analysis that will form the primary basis of an efficacy claim. If such an agreement is reached, it will be documented and made part of the administrative record, and it will be binding on the FDA and may not be changed unless the sponsor fails to follow the agreed-upon protocol, data supporting the request are found to be false or incomplete, or the FDA determines that a substantial scientific issue essential to determining the safety or effectiveness of the drug was identified after the testing began. Even if an SPA is agreed to, approval of the NDA is not guaranteed because a final determination that an agreed-upon protocol satisfies a specific objective, such as the demonstration of efficacy, or supports an approval decision, will be based on a complete review of all the data in the NDA.
18
Orphan Drugs
Under the Orphan Drug Act, the FDA may grant orphan drug designation to a drug intended to treat a rare disease or condition, defined as a disease or condition with a patient population of fewer than 200,000 individuals in the United States, or a patient population greater than 200,000 individuals in the United States and when there is no reasonable expectation that the cost of developing and making available the drug in the United States will be recovered from sales in the United States for that drug. Orphan drug designation must be requested before submitting a marketing application. After the FDA grants orphan drug designation, the generic identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA.
If a drug product that has orphan drug designation subsequently receives the first FDA approval for a particular active ingredient for the disease for which it has such designation, the product is entitled to orphan product exclusivity, which means that the FDA may not approve any other applications, including a full NDA, to market the same drug for the same indication for seven years, except in limited circumstances, such as a showing of clinical superiority to the product with orphan product exclusivity or if FDA finds that the holder of the orphan product exclusivity has not shown that it can assure the availability of sufficient quantities of the orphan product to meet the needs of patients with the disease or condition for which the drug was designated. Orphan product exclusivity does not prevent the FDA from approving a different drug for the same disease or condition, or the same drug for a different disease or condition. Among the other benefits of orphan drug designation are tax credits for certain research and a waiver of the NDA application user fee.
Patent term restoration
Depending upon the timing, duration and specifics of FDA approval of the use of our product candidates, some of our United States patents may be eligible for limited patent term extension under the Hatch-Waxman Act. The Hatch-Waxman Act permits a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory requirements
Post-approval requirements
Following approval of a new product, the manufacturer and the approved product are subject to pervasive and continuing regulation by the FDA, or EMA, including, record-keeping requirementsamong other things, monitoring and recordkeeping activities, reporting of adverse experiences associated with the drug. Drugproduct, product sampling and distribution restrictions, complying with promotion and advertising requirements, which include restrictions on promoting drugs for unapproved uses or patient populations (i.e., “off-label use”) and limitations on industry-sponsored scientific and educational activities. The manufacturer and its products are also subject to similar post-approval requirements by regulatory authorities comparable to FDA in jurisdictions outside of the United States where the products are approved. Although physicians may prescribe legally available products for off-label uses, manufacturers may not market or promote such uses.
FDA regulations require that products be manufactured in specific approved facilities and their subcontractorsin accordance with cGMPs. The cGMP regulations include requirements relating to organization of personnel, buildings and facilities, equipment, control of components and drug product containers and closures, production and process controls, packaging and labeling controls, holding and distribution, laboratory controls, records and reports and returned or salvaged products. The manufacturing facilities for our product candidates must meet applicable cGMP requirements to the FDA's or comparable foreign regulatory authorities' satisfaction before any product is approved and our commercial products can be manufactured. We rely, and expect to continue to rely, on third parties for the production of clinical (and
19
ultimately commercial) quantities of our products in accordance with cGMP regulations. These manufacturers must comply with cGMP regulations that require, among other things, quality control and quality assurance, the maintenance of records and documentation and the obligation to investigate and correct any deviations from cGMP. Manufacturers and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA or EMA and certain state agencies and are subject to periodic prescheduled or unannounced inspections by the FDA or EMA and certain state agencies for compliance with ongoingcGMP and other laws. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain cGMP compliance. Future inspections by the FDA and other regulatory agencies may identify compliance issues at the facilities of our CMOs that may disrupt production or distribution or require substantial resources to correct. In addition, the discovery of conditions that violate these rules, including failure to conform to cGMPs, could result in enforcement actions, and the discovery of problems with a product after approval may result in restrictions on a product, manufacturer or holder of an approved NDA, including voluntary recall and regulatory sanctions as described below.
Once an approval or clearance of a drug is granted, the FDA may withdraw the approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including cGMP,adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in mandatory revisions to the approved labeling to add new safety information; imposition of post-market or clinical trials to assess new safety risks; or imposition of distribution or other restrictions under a REMS program. Other potential consequences include, among other things:
| ● | restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls; |
| ● | fines, warning letters or other enforcement-related letters or clinical holds on post-approval clinical trials; |
| ● | refusal of the FDA to approve pending marketing applications or supplements to approved marketing authorizations, or suspension or revocation of product approvals; |
| ● | product seizure or detention, or refusal to permit the import or export of products; |
| ● | injunctions or the imposition of civil or criminal penalties; and |
| ● | consent decrees, corporate integrity agreements, debarment, or exclusion from federal health care programs; or mandated modification of promotional materials and labeling and the issuance of corrective information. |
In addition, the distribution of prescription pharmaceutical products is subject to the Prescription Drug Marketing Act, or PDMA, which regulates the distribution of drugs and drug samples at the federal level and sets minimum standards for the registration and regulation of drug distributors by the states. Both the PDMA and state laws limit the distribution of prescription pharmaceutical product samples and impose requirements to ensure accountability in distribution. Most recently, the Drug Supply Chain Security Act, or DSCSA, was enacted with the aim of building an electronic system to identify and trace certain proceduralprescription drugs distributed in the United States. The DSCSA mandates phased-in and documentation requirements upon usresource-intensive obligations for pharmaceutical manufacturers, wholesale distributors, and our third-party manufacturers. Failuredispensers over a 10-year period that is expected to culminate in November 2023. From time to time, new legislation and regulations may be implemented that could significantly change the statutory provisions governing the approval, manufacturing and marketing of products regulated by the FDA. It is impossible to predict whether further legislative or regulatory changes will be enacted, whether FDA regulations, guidance or interpretations will be changed or what the impact of such changes, if any, may be.
Other U.S. health care laws and regulations
The majority of states also have statutes or regulations similar to the aforementioned federal laws, some of which are broader in scope and apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor. Some state laws require pharmaceutical companies to comply with the statutorypharmaceutical industry’s voluntary compliance guidelines, or the relevant compliance guidance promulgated by the
20
federal government, in addition to requiring drug manufacturers to report information related to payments to physicians and other health care providers or marketing expenditures to the extent that those laws impose requirements that are more stringent than the Physician Payments Sunshine Act. State and foreign laws also govern the privacy and security of health information in some circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts.
Because of the breadth of these laws and the narrowness of their exceptions and safe harbors, it is possible that business activities can be subject to challenge under one or more of such laws. The scope and enforcement of each of these laws is uncertain and subject to rapid change in the current environment of healthcare reform, especially in light of the lack of applicable precedent and regulations. Federal and state enforcement bodies have recently increased their scrutiny of interactions between healthcare companies and healthcare providers, which has led to a number of investigations, prosecutions, convictions and settlements in the healthcare industry.
Ensuring that business arrangements with third parties comply with applicable healthcare laws and regulations is costly and time consuming. If business operations are found to be in violation of any of the laws described above or any other applicable governmental regulations a pharmaceutical manufacturer may be subject to penalties, including civil, criminal and administrative penalties, damages, fines, disgorgement, individual imprisonment, exclusion from governmental funded healthcare programs, such as Medicare and Medicaid, contractual damages, reputational harm, diminished profits and future earnings, additional reporting obligations and oversight if subject to a corporate integrity agreement or other agreement to resolve allegations of non-compliance with these laws, and curtailment or restructuring of operations, any of which could adversely affect a pharmaceutical manufacturer’s ability to operate its business and the results of its operations.
Regulation outside of the United States
In addition to regulations in the United States, we will be subject to a variety of foreign regulations governing clinical trials and commercial sales and distribution of our products outside of the United States. Whether or not we obtain FDA approval for a product candidate, we must obtain approval by the comparable regulatory authorities of foreign countries or economic areas, such as the 28-member European Union, before we may commence clinical trials or market products in those countries or areas. The approval process and requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly between countries and jurisdictions and can involve additional testing and additional administrative review periods. The time required to obtain approval in other countries and jurisdictions might differ from and be longer than that required to obtain FDA approval. Regulatory approval in one country or jurisdiction does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country or jurisdiction may negatively impact the regulatory process in others.
European Union drug development, review and approval
In the European Union, our product candidates also may be subject to extensive regulatory requirements. As in the United States, medicinal products can be marketed only if a marketing authorization from the competent regulatory agencies has been obtained. Similar to the United States, the various phases of pre-clinical and clinical research in the European Union are subject to significant regulatory controls.
The Clinical Trials Directive 2001/20/EC, the Directive 2005/28/EC on GCP, and the related national implementing provisions of the individual EU Member States govern the system for the approval of clinical trials in the European Union. Under this system, an applicant must obtain prior approval from the competent national authority of the EU Member States in which the clinical trial is to be conducted. Furthermore, the applicant may only start a clinical trial at a specific study site after the competent ethics committee has issued a favorable opinion. The clinical trial application must be accompanied by, among other documents, an IMPD (the Common Technical Document) with supporting information prescribed by Directive 2001/20/EC, Directive 2005/28/EC, and where relevant the implementing national provisions of the individual EU Member States and further detailed in applicable guidance documents. All suspected unexpected serious adverse reactions to the investigated drug that occur during the clinical trial have to be reported to the competent national authority and the Ethics Committee of the Member State where they occurred.
21
Under the EU’s new Clinical Trials Regulation, which took effect in January 2022, there will be a centralized application procedure where one EU Member State’s competent authority takes the lead in reviewing part I of the application, which contains scientific and medicinal product documentation, and the other national authorities only have limited involvement. Part II, which contains the national and patient-level documentation, will be assessed individually by each EU Member State. Strict deadlines have been established for the assessment of clinical trial applications. The role of the relevant ethics committees in the assessment procedure will continue to be governed by the national law of the concerned EU Member State. Any substantial changes to the trial protocol or other information submitted with the CTA must be notified to or approved by the relevant competent authorities and ethics committees. Medicines used in clinical trials must be manufactured in accordance with good manufacturing practices. Other national and EU-wide regulatory requirements can subject a manufacturermay also apply. Currently, the extent to possible legal or regulatory action, such as warning letters, suspension of manufacturing, seizure of product, injunctive action or possible civil penalties. We cannot be certain that we or our present or future third-party manufacturers or supplierswhich clinical trials will be able togoverned by the Clinical Trials Regulation will depend on when the clinical trial is initiated or on the duration of an ongoing trial. As of January 2023, all new clinical trials must comply with the cGMP regulationsClinical Trials Regulation. In addition, any clinical trial that was already under way as of January 1, 2023 and continues for more than three years from the day on which the Clinical Trials Regulation becomes applicable (i.e., January 31, 2025), the Clinical Trials Regulation will at that time begin to apply to the clinical trial.
To obtain a marketing authorization of a drug in the European Union, we may submit marketing authorization applications, or MAA, either under the so-called centralized or national authorization procedures.
Centralized procedure
The centralized procedure provides for the grant of a single marketing authorization following a favorable opinion by the European Medicines Agency, or EMA, that is valid in all EU member states, as well as Iceland, Liechtenstein and Norway. The centralized procedure is compulsory for medicines produced by specified biotechnological processes, products designated as orphan medicinal products, advanced-therapy medicines (such as gene-therapy, somatic cell-therapy or tissue-engineered medicines) and products with a new active substance indicated for the treatment of specified diseases, such as HIV/AIDS, cancer, diabetes, neurodegenerative disorders or autoimmune diseases and other immune dysfunctions and viral diseases. The centralized procedure is optional for products that represent a significant therapeutic, scientific or technical innovation, or whose authorization would be in the interest of public health. Under the centralized procedure the maximum timeframe for the evaluation of an MAA by the EMA is 210 days, excluding clock stops, when additional written or oral information is to be provided by the applicant in response to questions asked by the Committee for Medicinal Products for Human Use, or the CHMP. Accelerated assessment might be granted by the CHMP in exceptional cases, when a medicinal product is expected to be of a major public health interest, particularly from the point of view of therapeutic innovation. The timeframe for the evaluation of an MAA under the accelerated assessment procedure is of 150 days, excluding stop-clocks.
National authorization procedures
There are also two other possible routes to authorize medicinal products in several EU countries, which are available for investigational medicinal products that fall outside the scope of the centralized procedure:
| ● | Decentralized procedure. Using the decentralized procedure, an applicant may apply for simultaneous authorization in more than one EU country of medicinal products that have not yet been authorized in any EU country and that do not fall within the mandatory scope of the centralized procedure. |
| ● | Mutual recognition procedure. In the mutual recognition procedure, a medicine is first authorized in one EU Member State, in accordance with the national procedures of that country. Following this, further marketing authorizations can be sought from other EU countries in a procedure whereby the countries concerned agree to recognize the validity of the original, national marketing authorization. |
Under the above described procedures, before granting the marketing authorization, the EMA or the competent authorities of the Member States of the EEA make an assessment of the risk-benefit balance of the product on the basis of scientific criteria concerning its quality, safety and efficacy.
22
Conditional approval
In specific circumstances, E.U. legislation (Article 14(7) Regulation (EC) No 726/2004 and Regulation (EC) No 507/2006 on Conditional Marketing Authorizations for Medicinal Products for Human Use) enables applicants to obtain a conditional marketing authorization prior to obtaining the comprehensive clinical data required for an application for a full marketing authorization. Such conditional approvals may be granted for product candidates (including medicines designated as orphan medicinal products) if (1) the risk-benefit balance of the product candidate is positive, (2) it is likely that the applicant will be in a position to provide the required comprehensive clinical trial data, (3) the product fulfills unmet medical needs and (4) the benefit to public health of the immediate availability on the market of the medicinal product concerned outweighs the risk inherent in the fact that additional data are still required. A conditional marketing authorization may contain specific obligations to be fulfilled by the marketing authorization holder, including obligations with respect to the completion of ongoing or new studies, and with respect to the collection of pharmacovigilance data. Conditional marketing authorizations are valid for one year, and may be renewed annually, if the risk-benefit balance remains positive, and after an assessment of the need for additional or modified conditions or specific obligations. The timelines for the centralized procedure described above also apply with respect to the review by the CHMP of applications for a conditional marketing authorization.
European Union regulatory exclusivity
In the European Union, new products authorized for marketing (i.e., reference products) qualify for eight years of data exclusivity and an additional two years of market exclusivity upon marketing authorization. The data exclusivity period prevents generic or biosimilar applicants from relying on the pre-clinical and clinical trial data contained in the dossier of the reference product when applying for a generic or biosimilar marketing authorization in the European Union during a period of eight years from the date on which the reference product was first authorized in the European Union. The market exclusivity period prevents a successful generic or biosimilar applicant from commercializing its product in the EU until ten years have elapsed from the initial authorization of the reference product in the EU. The ten-year market exclusivity period can be extended to a maximum of eleven years if, during the first eight years of those ten years, the marketing authorization holder obtains an authorization for one or more new therapeutic indications which, during the scientific evaluation prior to their authorization, are held to bring a significant clinical benefit in comparison with existing therapies.
European Union orphan designation and exclusivity
The criteria for designating an orphan medicinal product in the European Union, are similar in principle to those in the United States. Under Article 3 of Regulation (EC) 141/2000, a medicinal product may be designated as orphan if (1) it is intended for the diagnosis, prevention or treatment of a life-threatening or chronically debilitating condition; (2) either (a) such condition affects no more than five in 10,000 persons in the European Union when the application is made, or (b) the product, without the benefits derived from orphan status, would not generate sufficient return in the European Union to justify investment; and (3) there exists no satisfactory method of diagnosis, prevention or treatment of such condition authorized for marketing in the European Union, or if such a method exists, the product will be of significant benefit to those affected by the condition, as defined in Regulation (EC) 847/2000. Orphan medicinal products are eligible for financial incentives such as reduction of fees or fee waivers and are, upon grant of a marketing authorization, entitled to ten years of market exclusivity for the approved therapeutic indication. The application for orphan designation must be submitted before the application for marketing authorization. The applicant will receive a fee reduction for the marketing authorization application if the orphan designation has been granted, but not if the designation is still pending at the time the marketing authorization is submitted. Orphan designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process.
The ten-year market exclusivity in the European Union may be reduced to six years if, at the end of the fifth year, it is established that the product no longer meets the criteria for orphan designation, for example, if the product is sufficiently profitable not to justify maintenance of market exclusivity. Additionally, marketing authorization may be granted to a similar product for the same indication at any time if:
| ● | the second applicant can establish that its product, although similar, is safer, more effective or otherwise clinically superior; |
23
| ● | the applicant consents to a second orphan medicinal product application; or |
| ● | the applicant cannot supply enough orphan medicinal product. |
PRIME designation
The EMA grants access to the Priority Medicines, or PRIME, program to investigational medicines for which it determines there to be preliminary data available showing the potential to address an unmet medical need and bring a major therapeutic advantage to patients. As part of the program, the EMA provides early and enhanced dialogue and support to optimize the development of eligible medicines and speed up their evaluation, aiming to bring promising treatments to patients sooner.
Periods of authorization and renewals
A marketing authorization is valid for five years in principle and the marketing authorization may be renewed after five years on the basis of a re-evaluation of the risk-benefit balance by the EMA or by the competent authority of the authorizing member state. To this end, the marketing authorization holder must provide the EMA or the competent authority with a consolidated version of the file in respect of quality, safety and efficacy, including all variations introduced since the marketing authorization was granted, at least six months before the marketing authorization ceases to be valid. Once renewed, the marketing authorization is valid for an unlimited period, unless the European Commission or the competent authority decides, on justified grounds relating to pharmacovigilance, to proceed with one additional five-year renewal. Any authorization which is not followed by the actual placing of the drug on the E.U. market (in case of centralized procedure) or on the market of the authorizing member state within three years after authorization ceases to be valid (the so-called sunset clause).
Health Care Reform
The FDA’s and other regulatory authorities’ policies may change, and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates. For example, Congress must reauthorize the FDA’s user fee programs every five years and often makes changes to those programs in addition to policy or procedural changes that may be negotiated between the FDA and industry stakeholders as part of this periodic reauthorization process. Congress most recently reauthorized the user fee programs in September 2022 but without any substantive policy changes. If we are slow or EMA regulatory requirements. If our presentunable to adapt to changes in existing requirements or future third-party manufacturersthe adoption of new requirements or supplierspolicies, or if we are not able to complymaintain regulatory compliance, we may lose any marketing approval that we otherwise may have obtained and we may not achieve or sustain profitability, which would adversely affect our business, prospects, financial condition and results of operations.
As previously mentioned, the primary trend in the U.S. health care industry and elsewhere is cost containment. Government authorities and other third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medical products and services, implementing reductions in Medicare and other health care funding and applying new payment methodologies. For example, the ACA, among other things, increased the minimum Medicaid rebates owed by most manufacturers under the Medicaid Drug Rebate Program; extended the Medicaid Drug Rebate Program to utilization of prescriptions of individuals enrolled in Medicaid managed care plans; imposed mandatory discounts for certain Medicare Part D beneficiaries as a condition for manufacturers’ outpatient drugs coverage under Medicare Part D; and established a Center for Medicare Innovation at the U.S. Centers for Medicare and Medicaid Services, or CMS, to test innovative payment and service delivery models to lower Medicare and Medicaid spending.
We expect that future changes or additions to the ACA, the Medicare and Medicaid programs and changes stemming from other healthcare reform measures, especially with these requirements,regard to healthcare access, financing or other legislation in individual states, could have a material adverse effect on the health care industry in the United States.
In addition, other legislative changes have been proposed and adopted in the United States since the ACA that affect health care expenditures. These changes include aggregate reductions to Medicare payments to providers of up to 2% per fiscal year pursuant to the Budget Control Act of 2011, which began in 2013 and was extended by the Consolidated Appropriations Act for 2023, and will remain in effect through 2032 unless additional Congressional action is taken.
24
Moreover, there has been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products, which has resulted in several Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drug products. Notably, on December 20, 2019, the Further Consolidated Appropriations Act for 2020 was signed into law (P.L. 116-94) that includes a piece of bipartisan legislation called the Creating and Restoring Equal Access to Equivalent Samples Act of 2019 (the CREATES Act). The CREATES Act aims to address the concern articulated by both the FDA or EMA may halt our clinical trials, require usand others in the industry that some brand manufacturers have improperly restricted the distribution of their products, including by invoking the existence of a REMS for certain products, to recall adeny generic and biosimilar product from distribution, or withdraw approvaldevelopers access to samples of that product.
In August 2022, the Inflation Reduction Act of 2022, or the IRA, was signed into law. Among other things, the IRA has multiple provisions that may impact the prices of drug products that are both sold into the Medicare program and throughout the United States. Starting in 2023, a manufacturer of a drug or biological product covered by Medicare Parts B or D must pay a rebate to the federal government if the drug product’s price increases faster than the rate of inflation. This calculation is made on a drug product by drug product basis and the amount of the rebate owed to the federal government is directly dependent on the volume of a drug product that is paid for by Medicare Parts B or D. Additionally, starting in payment year 2026, CMS will negotiate drug prices annually for a select number of single source Part D drugs without generic or biosimilar competition. CMS will also negotiate drug prices for a select number of Part B drugs starting for payment year 2028. If a drug product is selected by CMS for negotiation, it is expected that the revenue generated from such drug will decrease. In addition to the IRA’s drug price negotiation provisions, Executive Order 14087, issued in October 2022, called for the CMS innovation center to prepare and submit a report to the White House on potential payment and delivery modes that would complement to IRA, lower drug costs, and promote access to innovative drugs. As of February 3, 2023 the report had not been released but it is expected to further inform the current Administration’s priorities and activities in this area.
At the state level, individual states are increasingly aggressive in passing legislation and implementing regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. In December 2020, the U.S. Supreme Court held unanimously that federal law does not preempt the states’ ability to regulate pharmacy benefit managers (PBMs) and other members of the healthcare and pharmaceutical supply chain, an important decision that appears to be leading to further and more aggressive efforts by states in this area. The Federal Trade Commission in mid-2022 also launched sweeping investigations into the practices of the PBM industry that could lead to additional federal and state legislative or regulatory proposals targeting such entities’ operations, pharmacy networks, or financial arrangements. Significant efforts to change the PBM industry as it currently exists in the United States may affect the entire pharmaceutical supply chain and the business of other stakeholders, including biopharmaceutical developers like us. In addition, regional healthcare authorities and individual hospitals are increasingly using bidding procedures to determine what pharmaceutical products and which suppliers will be included in their prescription drug and other healthcare programs. These measures could reduce the ultimate demand for our products, once approved, or put pressure on our product pricing.
We expect that these requirements canand other healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on the price that we receive for any approved drug, which could have an adverse publicity, warning letters, corrective advertisingeffect on customers for our product candidates. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors.
25
There have been, and potential civillikely will continue to be, legislative and criminal penalties. Physiciansregulatory proposals at the foreign, federal and state levels directed at broadening the availability of healthcare and containing or lowering the cost of healthcare. The implementation of cost containment measures or other healthcare reforms may prescribe approved drugsprevent us from being able to generate revenue, attain profitability, or commercialize our products. Such reforms could have an adverse effect on anticipated revenue from product candidates that we may successfully develop and for uses that are not described in the drug’s labelingwhich we may obtain regulatory approval and that differ from those tested by usmay affect our overall financial condition and approved by the FDA or EMA. Such off-label uses are common across certain medical specialties. Physicians may believe that such off-label uses are the best treatment for many patients in varied circumstances. The FDA or EMA generally does not regulate the behavior of physicians in their choice of treatments. The FDA or EMA does, however, impose stringent restrictions on manufacturers’ communications regarding off-label use.
Competition
The biotechnology and biopharmaceutical industries are rapidly changing and highly competitive. We are seeking to develop and market drug candidates that will compete with other products and therapies that currently exist or are being developed. Other companies are actively seeking to develop products that have disease targets similar to those we are pursuing. We face competition from many different sources, including commercial, pharmaceutical and biotechnology companies, academic institutions, government agencies and private and public research institutions. Many of our competitors have significantly greater financial,
A large number of drug candidates are in development for the treatment of leukemia and lymphomas, MDS, breast, lung,gastrointestinal, genitourinary, gynecological and nasopharyngeal cancer.thoracic cancers and other advanced solid tumors. Several pharmaceutical and biotechnology companies have nucleoside analogs or other products on the market or in clinical trials which may be competitive to sapacitabine in both hematology and oncology indications. These include AbbVie, AstraZeneca, Onconova, Boehringer Ingelheim, BMS/Innate, Celgene, CTI Biopharma, Daiichi Sankyo, Eisai, Jazz, GlaxoSmithKline,Johnson & Johnson, Lilly, MEI Pharma, Otsuka, Pfizer, Sanofi, Sunesis and Teva. Several pharmaceutical and biotechnologybiopharmaceutical companies have CDK inhibitors in clinical trials including Bayer,Allorion Therapeutics, Amgen, AstraZeneca, Blueprint, Carrick, Dainippon Sumitomo, Eli Lilly, G1 Therapeutics, Lilly, Merck, MetaMax, Incyclix Bio, Incyte, Kronos Bio, MEI Pharma, Nerviano Medical Sciences/Merck, Novartis, Otsuka, Pfizer, Prelude, Servier, Syros, Tiziana Life Sciences, Novartis, Otsuka/Astex, Pfizer, Piramal, and Tragara.Vincerx. Cardiff Oncology has a PLK1 inhibitor in clinical trials and we believe that Arbutus, Boehringer Ingelheim, GlaxoSmithKline, Merck, Onconova, and Takeda have been and may continue to be evaluating PLK inhibitors for hemato-oncology indications. Several companies are pursuing discovery and research activities in each of the other areas that are the subject of our research and drug development programs.
Environmental Social and Government (“ESG”) Matters
We recognize the importance of ESG matters, with a specific focus on Human Capital Management, as integral to creating a sustainable foundation for our long-term business strategy. We support professional development at all levels. We also take reports of suspected violations of our codes of conduct and take seriously appropriate action.
As we do not operate laboratories or manufacture products, we believe that Amgen, AstraZeneca, CASI Pharmaceuticals, Nerviano Medical Sciences, Nemucore, Otsuka/Taiho Oncologyour environmental impact is relatively small. We are involved in office waste reduction practices. Our mostly remote workforce has further reduced our carbon footprint. We strive to offer excellent benefits and Takedalong-term incentives to help retain our workforce.
Our human capital resources and objectives include identifying, recruiting, retaining and incentivizing our existing and additional employees. The principal purposes of our equity incentive plans are conducting clinical developmentto attract, retain and reward personnel through the granting of Aurora kinase inhibitorsequity-based compensation awards in order to increase shareholder value and our success by motivating such individuals to perform to the best of their abilities to achieve our objectives.
We recognize that our industry is specialized and dynamic and a significant aspect of our success is our continued ability to execute our human capital strategy of attracting, engaging, developing and retaining highly skilled talent. There is fierce competition both within our industry and in the geographic locations in which we have offices for hemato-oncology indications.highly skilled talent, and we offer a robust set of benefits, career-enhancing learning experiences and initiatives aligned with our mission, vision, and values in order to attract qualified prospective employees and to retain and motivate our employees. We believe that Arbutus Biopharma, Boehringer Ingelheim, GlaxoSmithKline, Merck, Nerviano Medical Sciences/Trovageneoffer competitive compensation for our employees and Takedastrongly embrace a pay for performance philosophy in setting and adjusting compensation.
26
Our codes of conduct clearly outline our commitment to diversity and inclusion, where all employees are welcomed in an environment designed to make them feel comfortable, respected, and accepted regardless of their age, race, national origin, gender, religion, disability or sexual orientation. We have commenced clinical trials with PLK1 inhibitor candidatesa set of policies explicitly setting forth our expectations for hemato-oncology indications.
Legal Proceedings
From time to time, we may be involved in routine litigation incidental to the conduct of our business. As of December 31, 20172023, we were not party to any material legal proceedings.
Corporate information
We were incorporated in Delaware in August 1997. Our corporate headquarters are located at 200 Connell Drive, Suite 1500, Berkeley Heights, New Jersey 07922, and our telephone number is 908-517-7330. This is also where our medical and regulatory functionsOur employees are located. Our research facility is located in Dundee, Scotland, which is also the center of our translational workUnited States and development programs.
Available information
We file reports, proxy statements and other information with the Securities and Exchange Commission, or the SEC. Copies of our reports, proxy statements and other information may be inspected and copied at the public reference facilities maintained by the SEC at SEC Headquarters, Public Reference Room, 100 F Street, N.E., Washington D.C. 20549. The public may obtain information on the operation of the SEC’s Public Reference Room by calling the SEC at 1-800-SEC-0330. The SEC maintains a website that contains reports, proxy statements and other information regarding Cyclacel. The address of the SEC website is http://www.sec.gov.
We will also provide copies of our current reports on Form 8-K, annual reports on Form 10-K, quarterly reports on Form 10-Q and proxy statements, and all amendments to those reports at no charge through our website at www.cyclacel.com as soon as reasonably practicable after such material is electronically filed with, or furnished to, the SEC. We have not incorporated by reference in this Annual Report on Form 10-K the information on, or accessible through, our website. Copies are also available, without charge, from Cyclacel Pharmaceuticals, Inc., 200 Connell Drive, Suite 1500, Berkeley Heights, NJ 07922.
Item 1A. Risk Factors
In analyzing our company, you should carefully consider carefully the following risk factors. Factors that could cause or contribute to differences in our actual results include those discussed in the following subsection, as well as those discussed below in “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and elsewhere throughout this Annual Report on Form 10-K. Each of the following risk factors, either alone or taken together, could adversely affect our business, operating results and financial condition, as well as adversely affect the value of an investment in our Company. The risks and uncertainties described below are not the only ones we face. Additional risks not currently known to us or other factors not perceived by us to present significant risks to our business at this time also may impair our business operations.
Risks Associated with Development and Commercialization of Our Drug Candidates
Clinical trials are expensive, time consuming, subject to delay and may be required to continue beyond our available funding and we cannot be certain that we will be able to raise sufficient funds to complete the development and commercialize any of our product candidates currently in clinical development, should they succeed.
Clinical trials are expensive, complex, can take many years to conduct and may also have uncertain outcomes. We estimate that clinical trials of our most advanced drug candidates may be required to continue beyond our available funding and may take several more years to complete. The designs used in some of our trials have not been used widely by other pharmaceutical companies. We cannot guarantee that any clinical trials we undertake to conduct will be conducted as planned or completed on schedule or at all. Failure can occur at any stage of the testing and we may experience numerous unforeseen events during, or as a result of, the
27
clinical trial process that could delay or prevent commercialization of our current or future drug candidates, including, but not limited to:
| ● | delays in securing clinical investigators or trial sites for our clinical trials; |
| ● | delays in obtaining institutional review board, or IRB, and regulatory approvals to commence a clinical trial; |
| ● | failure to obtain regulatory authority permission to conduct a clinical trial, after review of an investigational new drug or equivalent foreign application or amendment; |
| ● | slower than anticipated rates of subject recruitment and enrollment, or not reaching the targeted number of subjects because of competition for patients from other trials; |
| ● | negative or inconclusive results from clinical trials, as demonstrated by our announcement on February 24, 2017 that our SEAMLESS Phase 3 study failed to reach its primary endpoint; |
| ● | inability to generate satisfactory preclinical or other nonclinical data, including, toxicology, or other in vivo or in vitro data or diagnostics to support the initiation or continuation of clinical trials; |
| ● | unforeseen safety issues; |
| ● | failure by clinical sites or contract research organizations, or CROs, or other third parties to adhere to clinical trial requirements, GCP, or other applicable regulatory requirements; |
| ● | subjects discontinuing participating in our clinical trials at a greater than expected rate; |
| ● | imposition by the FDA of a clinical hold or the requirement by other similar regulatory agencies that one or more clinical trials be delayed or halted; |
| ● | uncertain dosing issues that may or may not be related to incompletely explored pharmacokinetic and pharmacodynamics behaviors; |
| ● | approval and introduction of new therapies or changes in standards of practice or regulatory guidance that render our clinical trial endpoints or the targeting of our proposed indications less attractive; |
| ● | inability to monitor patients adequately during or after treatment or problems with investigator or patient compliance with the trial protocols; |
| ● | inability to replicate in large, controlled studies safety and efficacy data obtained from a limited number of patients in uncontrolled trials; |
| ● | the ultimate affordability of the cost of clinical trials of our product candidates; |
| ● | changes in regulatory requirements and guidance that require amending or submitting new clinical protocols or performing additional nonclinical studies; and |
| ● | unavailability of clinical trial supplies. |
Any inability to successfully complete clinical development and obtain regulatory approval for our product candidates could result in additional costs to us or impair our ability to generate revenue. In addition, if we make manufacturing or formulation changes to our product candidates, we may need to conduct additional nonclinical studies and/or clinical trials, or the results obtained from such new formulation may not be related to suboptimal pharmacokinetic and pharmacodynamics behaviors;
28
If we experience delays or prevent regulatory approval. The FDA, EMA or we may suspend or terminatedifficulties in the enrollment of research subjects in clinical trials, at any time. Any failurethose clinical trials could take longer than expected to complete and our receipt of necessary regulatory approvals could be delayed or significant delay in completingprevented.
We may not be able to initiate or continue clinical trials for our drug candidates if we are unable to locate and enroll a sufficient number of research subjects to participate in these trials. In particular, for some diseases and conditions we are or in receiving regulatory approvalwill be focusing on, our pool of suitable patients may be smaller and more selective and our ability to enroll a sufficient number of suitable patients may be limited or take longer than anticipated. In addition, some of our competitors have ongoing clinical trials for drug candidates that treat the commercialization ofsame indications as our drug candidates, and volunteers or patients who would otherwise be eligible for our clinical trials may severely harminstead enroll in clinical trials of our businesscompetitors’ drug candidates.
Patient enrollment for any of our clinical trials may also be affected by other factors, including without limitation:
| ● | the size and nature of the target patient population; |
| ● | the severity of the disease under investigation; |
| ● | the subject eligibility criteria for the clinical trial in question; |
| ● | our ability to recruit clinical trial investigators with the appropriate competencies and experience; |
| ● | the perceived risks and benefits of the drug candidate under study in the clinical trial; |
| ● | the approval and availability of other therapies to treat the disease or disorder that is being investigated in the clinical trial; |
| ● | the extent of the efforts to facilitate timely enrollment in clinical trials; |
| ● | the patient referral practices of physicians; |
| ● | the ability to monitor volunteers or subjects adequately during and after treatment; |
| ● | the presence of other drug candidates in clinical development for the same indication or against the same target; and |
| ● | the proximity and availability of clinical trial sites for prospective subjects. |
In addition, our clinical trials will compete with other clinical trials for product candidates that are in the same therapeutic areas as our product candidates, and reputation.this competition will reduce the number and types of patients available to us, because some patients who might have opted to enroll in our trials may instead opt to enroll in a trial being conducted by one of our competitors. Because the number of qualified clinical investigators is limited, we may conduct some of our clinical trials at the same clinical trial sites that some of our competitors use, which will reduce the number of patients who are available for our clinical trials at such clinical trial sites. Moreover, because our product candidates represent a departure from more commonly used methods for cancer treatment, potential patients and their doctors may be inclined to use conventional therapies rather than enroll patients in any future clinical trial.
Our inability to enroll a sufficient number of subjects for our clinical trials would result in significant delays and could require us to abandon one or more clinical trials altogether. Enrollment delays in our clinical trials may result in increased development costs for our drug candidates, and we may not have or be able to obtain sufficient cash to fund such increased costs when needed, which could result in the further delay or termination of clinical trials.
The outcome of preclinical testing and early clinical trials may not be predictive of the success of later clinical trials, and the results of our clinical trials may not satisfy the requirements of the FDA or comparable foreign regulatory authorities.
We currently have no products approved for sale and we cannot guarantee that we will ever have marketable products. Clinical failure can occur at any stage of clinical development. Clinical trials may produce negative or
29
inconclusive results, and we or any future collaborators may decide, or regulatory authorities may require us, to conduct additional clinical trials or nonclinical studies. We will be required to demonstrate with substantial evidence through well-controlled, adequate clinical trials that our product candidates are safe and effective for use in a diverse population before we can seek marketing approvals for their commercial sale. Success in preclinical studies and early-stage clinical trials does not mean that future larger registration clinical trials will be successful. This is because product candidates in later-stage clinical trials may fail to demonstrate sufficient safety and efficacy to the satisfaction of the FDA and comparable foreign regulatory authorities despite having progressed through nonclinical studies and early-stage clinical trials.
From time to time, we may publish or report interim or preliminary data from our clinical trials. Interim or preliminary data from clinical trials that we may conduct may not be indicative of the final results of such trials and are subject to the risk that one or more of the clinical outcomes may materially change as subject enrollment continues and more data from the trials become available. Interim or preliminary data also remain subject to audit and verification procedures that may result in the final data being materially different from the interim or preliminary data. As a result, interim or preliminary data should be viewed with caution until the final data are available.
We are making use of biomarkers, which are not scientifically validated, and our reliance on biomarker data may thus cause us to direct our resources inefficiently.
We are making some use of biomarkers in an effort to facilitate our drug development and to optimize our clinical trials. Biomarkers are proteins or other substances whose presence in the blood can serve as an indicator of specific cell processes. We believe that these biological markers serve a useful purpose in helping us to evaluate whether our drug candidates are having their intended effects through their assumed mechanisms, and that they may thus enable us to identify more promising drug candidates at an early stage and to direct our resources efficiently. We also believe that biomarkers may eventually allow us to improve patient selection in connection with clinical trials and monitor patient compliance with trial protocols.
For most purposes, however, the biomarkers we are currently evaluating have not been scientifically validated. If our understanding and use of biomarkers is inaccurate or flawed, or if our reliance on them is otherwise misplaced, then we will not only fail to realize any benefits from using biomarkers but may also be led to invest time and financial resources inefficiently in attempting to develop inappropriate drug candidates. For example, there is no assurance that exploiting CDKN2A and/or CDKN2B abnormalities with fadraciclib will result in clinical benefit for patients or lead to regulatory approval.
Moreover, although the FDA has issued for comment a draft guidance document on the potential use of biomarker data in clinical development, such data are not currently accepted by the FDA or other regulatory agencies in the United States, the European Union or elsewhere in applications for regulatory approval of drug candidates, and there is no guarantee that such data will ever be accepted by the relevant authorities in this connection. Our biomarker data should not be interpreted as evidence of efficacy.
The review processes of regulatory authorities are lengthy, time consuming, expensive and other third parties to conduct clinical trials,inherently unpredictable. If we may beare unable to directly control the timing, conduct and expense of our clinical trials.
The research, testing, manufacturing, labeling, approval, sale, marketing and distribution of drug products are, and will remain, subject to extensive regulation by the FDA in the United States and by the respective regulatory authorities in other countries where regulations differ. We are not permitted to market our product candidates in the United States until we receive the respective approval of an NDA from the FDA, or in any foreign countries until we receive the requisite approval from the respective regulatory authorities in such countries. The time required to obtain approval, if any, by the FDA, EMA and comparable foreign authorities is unpredictable, but typically takes many years following the commencement of clinical trials, if approval is obtained at all, and depends upon numerous factors, including the substantial discretion of the regulatory authorities and the type, complexity and novelty of the product candidates involved. Regulatory authorities have substantial discretion in the approval process and may refuse to accept any
30
application or may decide that our data are insufficient for approval and require additional nonclinical studies or clinical trials. We have not submitted a marketing application such as an NDA to the FDA, an MAA to the EMA or any similar application to any other jurisdiction. We have limited experience in planning and conducting the clinical trials required for marketing approvals, and we have relied, and expect to continue to rely on third-party CROs to assist us in this process. Obtaining marketing approval requires the submission of extensive nonclinical and clinical data and supporting information to regulatory authorities for each therapeutic indication to establish the product candidate’s safety and effiveness. Securing marketing approval also requires the submission of information about the product manufacturing process, and in many cases the inspection of manufacturing, processing and packaging facilities by the regulatory authorities. Our product candidates may not be effective, may be only moderately effective or may prove to have undesirable or unintended side effects, toxicities or other characteristics that may preclude our obtaining marketing approval or prevent or limit commercial use, or there may be deficiencies in cGMP compliance by us or by our contract manufacturers that could result in the candidate not being approved. Moreover, we have not obtained regulatory approval for any drug candidate in any jurisdiction and it is possible that none of our existing drug candidates or successfully commercialize our drug candidates.
Our drug candidates is entering into strategic alliances with pharmaceutical companies or other industry participants to advance our programs and enable us to maintain our financial and operational capacity.
| ● | the FDA, EMA or comparable foreign regulatory authorities may disagree with the design or implementation of our clinical trials; |
| ● | we may be unable to demonstrate to the satisfaction of the FDA, EMA or comparable foreign regulatory authorities that a drug candidate is safe and effective for its proposed indication; |
| ● | the results of clinical trials may not meet the level of statistical significance required by the FDA, EMA or comparable foreign regulatory authorities for approval; |
| ● | we may be unable to demonstrate that a drug candidate’s clinical and other benefits outweigh its safety risks; |
| ● | the FDA, EMA or comparable foreign regulatory authorities may disagree with our interpretation of data from preclinical studies or clinical trials; |
| ● | the data collected from clinical trials of our drug candidates may not be sufficient to support the submission of an NDA or other submission or to obtain regulatory approval in the United States or elsewhere; |
| ● | upon review of our clinical trial sites and data, the FDA or comparable foreign regulatory authorities may find our record keeping or the record keeping of our clinical trial sites to be inadequate; |
| ● | the manufacturing processes or facilities of third-party manufacturers with which we contract for clinical and commercial supplies may fail to meet the requirements of the FDA, EMA or comparable foreign regulatory authorities; |
| ● | the FDA, EMA or comparable foreign regulatory authorities may fail to approve the companion diagnostics we contemplate developing internally or with partners; and |
| ● | the change of the medical standard of care or the approval policies or regulations of the FDA, EMA or comparable foreign regulatory authorities may significantly change in a manner that renders our clinical data insufficient for approval. |
The time and expense of the approval process, as well as the unpredictability of future clinical trial results and other contributing factors, may result in our drug development or research programs. If we elect to fund drug development or research programs on our own, we will have to increase our expenditures and will needfailure to obtain additional funding, which may be unavailable or available only on unfavorable terms.
In addition, even if we are able,were to obtain regulatory approval in one or decide to, expand our operations to include manufacturing capacities. If the FDA or EMAmore jurisdictions, regulatory authorities may approve any of our drug candidates for fewer or more limited indications than we request, may not approve prices we may propose to charge for our products, may grant approval contingent on the performance of costly post-marketing
31
clinical trials (referred to as “conditional” or “accelerated” approval depending on the jurisdiction), or may approve a drug candidate with a label that does not include the labeling claims necessary or desirable for the successful commercialization of that drug candidate. Any of the foregoing circumstances could materially harm the commercial sale,prospects for our drug candidates.
Our product candidates may cause undesirable side effects that could delay or prevent their marketing approval, limit their commercial potential, or result in significant negative consequences following marketing approval, if we significantly expandmarketing approval is obtained.
Undesirable side effects caused by our product candidates could cause us or the FDA or other regulatory authorities to interrupt, delay or halt our clinical trials we will need to manufacture themand could result in larger quantities and will be required to secure additionala more restrictive label or alternative third-party suppliers tothe delay or denial of marketing approval by the FDA or other regulatory authorities of our current suppliers. To date,product candidates. In the event that our drug candidates have
Clinical trials by their nature utilize a sample of the potential patient population. With a limited number of patients, rare and severe side effects of our drugsproduct candidates may only be uncovered with a significantly larger number of patients exposed to the product candidate. If our product candidates receive marketing approval and we or others identify undesirable side effects caused by such product candidates (or any other similar products) after such approval, a number of potentially significant negative consequences could result, including:
| ● | regulatory authorities may withdraw or limit their approval of such product candidates; |
| ● | regulatory authorities may require the addition of labeling statements, specific warnings or a contraindication; |
| ● | we may be required to create a medication guide outlining the risks of such side effects for distribution to patients, or we may be required to implement a REMS to ensure that the benefits of the product outweigh the risks; |
| ● | we may be required to change the way such product candidates are distributed or administered, or change the labeling of the product candidates; |
| ● | we may be subject to regulatory investigations and government enforcement actions; |
| ● | the FDA or a comparable foreign regulatory authority may require us to conduct additional clinical trials or costly post-marketing testing and surveillance to monitor the safety and efficacy of the product; |
| ● | we may decide to recall such product candidates from the marketplace after they are approved; |
| ● | we could be sued and held liable for injury caused to individuals exposed to or taking our product candidates; and |
| ● | our reputation may suffer. |
Any of these events could prevent us from achieving or maintaining market acceptance of the affected product candidates and could substantially increase the costs of commercializing our product candidates, if approved, and significantly impact our ability to sellsuccessfully commercialize our commercial products, producing additional lossesproduct candidates and depriving us of potential productgenerate revenues.
As we evolve from a company primarily involved in discovery and development to one also involved in the commercialization of drugs and devices, we may encounter difficulties in managing our growth and expanding our operations successfully.
In order to execute our business strategy, we will need to expand our development, control and regulatory capabilities and develop financial, manufacturing, marketing and sales capabilities or contract with third parties to provide these capabilities for us. If our operations expand, we expect that we will need to manage additional
32
relationships with various collaborative partners, suppliers and other third parties. Our ability to manage our operations and any growth will require us to make appropriate changes and upgrades, as necessary, to our operational, financial and management controls, reporting systems and procedures wherever we may operate. Any inability to manage growth could delay the execution of our business plan or disrupt our operations.
Our applications for regulatory approval could be delayed or denied due to problems with studies conducted before we in-licensed the rights to some of our product candidates.
We currentlymay now or in the future license some of the compounds and drug candidates used in our research programs from third parties. These include sapacitabine, which was licensed from Daiichi Sankyo. Our present research involving these compounds relies upon previous research conducted by third parties over whom we had no control and before we in-licensed the drug candidates. In order to receive regulatory approval of a drug candidate, we must present all relevant data and information obtained during our research and development, including research conducted prior to our licensure of the drug candidate. Although we are not currently aware of any such problems, any problems that emerge with preclinical research and testing conducted prior to our in-licensing may affect future results or our ability to document prior research and to conduct clinical trials, which could delay, limit or prevent regulatory approval for our drug candidates.
Even if we obtain regulatory approval for a product candidate, we will remain subject to ongoing regulatory requirements.
If any of our product candidates receive regulatory approval,are approved for marketing, we may still face future development and regulatory difficulties.
Manufacturers and manufacturers’ facilities are required to continuously comply with extensive FDA and EMAcomparable foreign regulatory authority requirements, and requirements of other similar agencies, including ensuring that quality control and manufacturing procedures conform to the FDA’s or EMA’s cGMP.cGMP regulations and corresponding foreign regulatory manufacturing requirements. As such, we and our contract manufacturers arewill be subject to continual review and periodic inspections to assess compliance with cGMP.cGMP and adherence to commitments made in any marketing application. Accordingly, we and others with whom we work must continue to expend time, money, and effort in all areas of regulatory compliance, including manufacturing, production, and quality control.
Any regulatory approvals that we receive for our product candidates may be subject to limitations on the approved indicated uses for which the product may be marketed or to the conditions of approval, or contain requirements for potentially costly post-marketing testing, including Phase 4 clinical trials, and surveillance to monitor the safety and efficacy of the product candidate. The FDA may also require a REMS program as a condition of approval of our product candidates, which could entail requirements for long-term patient follow-up, a medication guide, physician communication plans or additional elements to ensure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. In addition, if the FDA or a comparable foreign regulatory authority approves our product candidates, we will have to comply with requirements including submissions of safety and other post-marketing information and reports, registration, as well as continued compliance with cGMP and GCP for any clinical trials that we conduct post-approval.
The FDA strictly regulates marketing, labeling, advertising, and promotion of products that are placed on the market. Drugs may be promoted only for the approved indications and in accordance with the provisions of the approved label. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability.
Failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical studies to assess new safety risks; or imposition of distribution restrictions or other restrictions under a REMS program. Other potential consequences include, among other things:
33
| ● | restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls; |
| ● | fines, warning letters or other enforcement-related letters or clinical holds on post-approval clinical trials; |
| ● | refusal of the FDA to approve pending NDAs or supplements to approved NDAs, or suspension or revocation of product approvals; |
| ● | product seizure or detention, or refusal to permit the import or export of products; |
| ● | injunctions or the imposition of civil or criminal penalties; and |
| ● | consent decrees, corporate integrity agreements, debarment, or exclusion from federal health care programs; or mandated modification of promotional materials and labeling and the issuance of corrective information. |
The policies of the FDA and of other regulatory authorities may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained and we may not achieve or sustain profitability.
Obtaining and maintaining marketing approval of our current and future product candidates in one jurisdiction does not mean that we will be successful in obtaining marketing approval of our current and future therapeutic product candidates in other jurisdictions.
Obtaining and maintaining marketing approval of our current and future product candidates in one jurisdiction does not guarantee that we will be able to obtain or maintain marketing approval in any other jurisdiction, while a failure or delay in obtaining marketing approval in one jurisdiction may have a negative effect on the marketing approval process in others. For example, even if the FDA grants marketing approval of a product candidate, comparable foreign regulatory authorities in foreign jurisdictions must also approve the manufacturing, marketing and promotion of the product candidate in those countries. Approval procedures vary among jurisdictions and can involve requirements and administrative review periods different from, and greater than, those in the United States, including additional nonclinical studies or clinical trials in the event that certain nonclinical studies or clinical trials conducted in one jurisdiction are not accepted by regulatory authorities in other jurisdictions. In many jurisdictions outside the United States, a product candidate must be approved for reimbursement before it can be approved for sale in that jurisdiction. In some cases, the price that we intend to charge for our future products will also be requiredsubject to report certain adverse reactions and production problems, if any,approval.
We may submit marketing applications in other countries in addition to the FDA and EMA and to comply with certain requirements concerning advertising and promotion for our products. Promotional communications with respect to prescription drugs are subject to a variety of legal and regulatory restrictions and must be consistent with the informationUnited States. Regulatory authorities in the product’s approved label. Accordingly, we may not promote our approved products, if any, for indications or uses for which they are not approved.
Even if we successfully complete the clinical trials for one or more of our product candidates, the product candidates may fail for other reasons.
Even if we successfully complete the clinical trials for one or more of our product candidates, the product candidates may fail for other reasons, including, without limitation, the possibilities that the product candidates will:
| ● | fail to receive the regulatory approvals required to market them as drugs; |
| ● | be subject to proprietary rights held by others requiring the negotiation of a license agreement prior to marketing; |
34
| ● | be difficult or expensive to manufacture on a commercial scale; |
| ● | have adverse side effects that make their use less desirable; or |
| ● | fail to compete effectively with product candidates or other treatments commercialized by our competitors. |
If we are unable to receive the required regulatory approvals, secure our intellectual property rights, minimize the incidence of any adverse side effects or if we fail to compete with our competitors’ products, our business, financial condition, and results of operations couldmay be materially and adversely affected.
We face intense competition and our competitors may develop drugs that are less expensive, safer, or more effective than our drug candidates.
A large number of drug candidates are in development for the treatment of leukemia, lung cancer, lymphomassolid tumors including breast, endometrial/uterine and nasopharyngeal cancer.ovarian cancers and lymphomas. Several pharmaceutical and biotechnology companies have nucleoside analogsCDK inhibitors, PLK1 inhibitors or other products on the market or in clinical trials which may be competitive to sapacitabineour drugs in both hematological and oncology indications. Our competitors, either alone or together with collaborators, may have substantially greater financial resources and research and development staff. Our competitors may also have more experience:
| ● | developing drug candidates; |
| ● | conducting preclinical and clinical trials; |
| ● | obtaining regulatory approvals; and |
| ● | commercializing product candidates. |
Our competitors may succeed in obtaining patent protection and regulatory approval and may market drugs before we do. If our competitors market drugs that are less expensive, safer, more effective or more convenient to administer than our potential drugs, or that reach the market sooner than our potential drugs, we may not achieve commercial success. Scientific, clinical or technical developments by our
Our future product candidates for which we obtain approval may face competition sooner than anticipated.
Even if we are successful in achieving regulatory approval to commercialize a product candidate ahead of our competitors, our future pharmaceutical products may face direct competition from generic and other follow-on drug products. Any of our product candidates that may achieve regulatory approval in the future may face competition from follow-on products earlier or more aggressively than anticipated, depending upon how well such approved products perform in the U.S. prescription drug market. Our ability to compete may also be affected in many cases by insurers or other third-party payors seeking to encourage the use of generic products.
The Hatch-Waxman Amendments to the FDCA authorized the FDA to approve generic drugs that are the same as drugs previously approved for marketing under the NDA provisions of the statute pursuant to ANDAs and, in addition, created the Section 505(b)(2) NDA pathway. An ANDA relies on the preclinical and clinical testing conducted for a previously approved reference listed drug and must demonstrate to the FDA that the generic drug product is identical to the RLD with respect to the active ingredients, the route of administration, the dosage form, and the strength of the drug and also that it is “bioequivalent” to the reference listed drug. In contrast, Section 505(b)(2) enables the applicant to rely, in part, on the FDA’s prior findings of safety and efficacy data for an existing product, or published literature, in support of its application. Section 505(b)(2) provides an alternate path to FDA approval for new or improved formulations or new uses of previously approved products; for example, a follow-on applicant may be seeking approval to market a previously approved drug for new indications or for a new patient population that would require new clinical data to
35
demonstrate safety or effectiveness. Such products, if approved and depending upon the scope of the changes made to the reference drug, may also compete with any product candidates for which we receive approval.
The FDA is prohibited by statute from approving an ANDA or 505(b)(2) NDA when certain marketing or data exclusivity protections apply to the reference listed drug. However, if any competitor or third party is able to demonstrate bioequivalence without infringing our patents, then such competitor or third party may then be able to gain approval of an ANDA and introduce a competing generic product onto the market.
Furthermore, the CREATES Act established a private cause of action that permits a generic product developer to sue the brand manufacturer to compel it to furnish necessary samples of an RLD on “commercially reasonable, market-based terms.” If generic developers request samples of any product candidates for which we receive marketing approval in order to conduct comparative testing to support one or more ANDAs for a generic version of our products, and we refuse any such request, we may be subject to litigation under the CREATES Act. Although lawsuits have been filed under the CREATES Act since its enactment, those lawsuits have settled privately; therefore, to date, no federal court has reviewed or opined on the statutory language and there continues to be uncertainty regarding the scope and application of the law.
We cannot predict the interest of potential follow-on competitors or how quickly others may seek to come to market with competing products, whether approved as a direct ANDA competitor or as a Section 505(b)(2) NDA referencing one of our future product candidates. If the FDA approves generic versions of any of our products in the future, should they be approved for commercial marketing, such competitive products may be able to immediately compete with us in each indication for which our product has received approval, which could negatively impact our future revenue, profitability and cash flows and substantially limit our ability to obtain a return on our investments.
The commercial success of our drug candidates depends upon their market acceptance among physicians, patients, healthcare providers and payors and the medical community.
If our drug candidates are approved, or are approved by the FDA or EMA, together with another agent such as decitabine, the resulting drugs, if any, must still gain market acceptance among physicians, healthcare providers and payors, patients and the medical community. The degree of market acceptance of any of our approved drugs will depend on a variety of factors, including:
| ● | timing of market introduction, number and clinical profile of competitive drugs; |
| ● | our ability to provide acceptable evidence of safety and efficacy; |
| ● | relative convenience and ease of administration; |
| ● | pricing and cost-effectiveness, which may be subject to regulatory control; |
| ● | availability of coverage, reimbursement and adequate payment from health maintenance organizations and other third-party payors; and |
| ● | prevalence and severity of adverse side effects; and other potential advantages over alternative treatment methods. |
If any product candidate that we develop does not provide a treatment regimen that is at least as beneficial as the current standard of care or otherwise does not provide some additional patient benefit over the current standard of care, that product will not achieve market acceptance and we will not generate sufficient revenues to achieve profitability.
If our drug candidates or distribution partners’ products fail to achieve market acceptance, we may not be able to generate significant revenue and our business would suffer.
Reimbursement decisions by third-party payors may have an adverse effect on pricing and market acceptance. If there is not sufficient reimbursement for our products, it is less likely that they will be widely used. Market acceptance
36
and sales of our product candidates that we develop, if approved, will depend on reimbursement policies, and may be affected by future healthcare reform measures. Government authorities and third-party payors, such as private health insurers and health maintenance organizations, decide which drugs they will cover and establish payment levels. We cannot be certain that reimbursement will be available for our product candidates that we develop. Also, we cannot be certain that reimbursement policies will not reduce the demand for, or the price paid for, our products. If reimbursement is not available or is available on a limited basis, we may not be able to successfully commercialize any of our product candidates.
Even if we succeed in bringing one or more products to the market, these products may not be considered medically necessary and/or cost-effective, and the amount reimbursed for any products may be insufficient to allow us to sell our products on a competitive basis. At this time, we are unable to determine their cost effectiveness or the likely level or method of reimbursement for our product candidates. Increasingly, third-party payors, such as government and private insurance plans, are requiring that biopharmaceutical companies provide them with predetermined discounts from list prices, and are seeking to reduce the prices charged or the amounts paid for biopharmaceutical products. If the price we are able to charge for any products we develop, or the payments provided for such products, is inadequate in light of our development and other costs, our return on investment could be adversely affected.
Discussions continue at the federal level regarding policies that would require manufacturers to pay higher rebates in Medicare Part D, give states more flexibility on drugs that are covered under the Medicaid program, and other policy proposals that could impact reimbursement for our products. The efforts of governments and third-party payors to contain or reduce the cost of health care and legislative and regulatory proposals to broaden the availability of health care will continue to affect the business and financial condition of pharmaceutical and biopharmaceutical companies. A number of legislative and regulatory changes in the health care system in the United States and other major health care markets have been proposed and/or adopted in the recent past, and such efforts have expanded substantially in the past several years.
Our business may be affected by the efforts of government and third-party payorspairs to contain or reduce the cost of healthcare through various means.
The failure to attract and retain skilled personnel and key relationships could impair our drug development and commercialization efforts.
We are highly dependent on our senior management and key clinical development, scientific and technical personnel. The loss of the services of any member of our senior management, clinical development, scientific or technical staff may significantly delay or prevent the achievement of drug development and other business objectives and could have a material adverse effect on our business, operating results and financial condition. We also rely on consultants and advisors to assist us in formulating our strategy. All of our consultants and advisors are either self-employed or employed by other organizations, and they may have conflicts of interest or other commitments, such as consulting or advisory contracts with other organizations, that may affect their ability to contribute to us. We intend to expand and develop new drug candidates. We will need to hire additional employees in order to continue our clinical trials and market our drug candidates. This strategy will require us to recruit additional executive management and clinical development, scientific, technical and sales and marketing personnel. There is currently intense competition for skilled executives and employees with relevant clinical development, scientific, technical and sales and marketing expertise, and this competition is likely to continue. The inability to attract and retain sufficient clinical development, scientific, technical and managerial personnel could limit or delay our product development efforts, which would adversely affect the development of our drug candidates and commercialization of our potential drugs and growth of our business.
We are experiencing an increasingly tight and competitive labor market and could face unforeseen challenges in the availability of labor, such as we have experienced since the outbreak of COVID-19. A sustained labor shortage or increased turnover rates within our employee base as a result of general macroeconomic factors have led and in the future could lead to increased costs, such as increased overtime to meet demand and increased wages to attract and retain employees. We have also been negatively affected and could continue to be negatively affected by labor shortages or constraints experienced by our partners. Failure to achieve and maintain a diverse workforce and leadership team,
37
compensate our employees competitively and fairly, maintain a safe and inclusive environment or promote the well-being of our employees could affect our reputation and also result in lower performance and an inability to retain valuable employees.
We may be exposed to product liability claims that may damage our reputation and we may not be able to obtain adequate insurance.
Because we conduct clinical trials in humans, we face the risk that the use of our drug candidates will result in adverse effects. We believe that we have obtained reasonably adequate product liability insurance coverage for our trials. We cannot predict, however, the possible harm or side effects that may result from our clinical trials. Such claims may damage our reputation and we may not have sufficient resources to pay for any liabilities resulting from a claim excluded from, or beyond the limit of, our insurance coverage or if the amount of the insurance coverage is insufficient to meet any liabilities resulting from any claims.
We may also be exposed to additional risks of product liability claims. These risks exist even with respect to drugs that are approved for commercial sale by the FDA or other regulatory authorities in the United States, the European Union or elsewhere and manufactured in facilities licensed and regulated by the FDA, EMA or other such regulatory authorities. We have secured limited product liability insurance coverage but may not be able to maintain such insurance on acceptable terms with adequate coverage, or at a reasonable cost. There is also a risk that third parties that we have agreed to indemnify could incur liability. Even if we were ultimately successful in product liability litigation, the litigation would consume substantial amounts of our financial and managerial resources and may exceed insurance coverage creating adverse publicity, all of which would impair our ability to generate sales of the litigated product as well as our other potential drugs.
If a supplier upon whom we rely fails to produce on a timely basis the finished goods in the volumes that we require or fails to meet quality standards and maintain necessary licensure from regulatory authorities, we may be unable to meet demand for our products, potentially resulting in lost revenues.
If any third-party manufacturer service providers do not meet our or our licensor’s requirements for quality, quantity or timeliness, or do not achieve and maintain compliance with all applicable regulations, demand for our products or our ability to continue supplying such products could substantially decline. As the third-party manufacturers are the sole supplier of the products, any delays may impact our sales.
In all the countries where we may sell our products, governmental regulations exist to define standards for manufacturing, packaging, labeling and storing. All of our suppliers of raw materials and contract manufacturers must comply with these regulations. Failure to do so could result in supply interruptions. In the United States, the FDA requires that all suppliers of pharmaceutical bulk material and all manufacturers of pharmaceuticals for sale in or from the United States achieve and maintain compliance with the FDA’s cGMP. Similar requirements exist in the European Union through the EMA. Failure of our third-party manufacturers to comply with applicable regulations could result in sanctions being imposed on them or us, including fines, injunctions, civil penalties, disgorgement, suspension or withdrawal of approvals, license revocation, seizures or recalls of products, operating restrictions and criminal prosecutions, any of which could significantly and adversely affect supplies of our products. In addition, before any product batch produced by our manufacturers can be shipped, it must conform to release specifications for the content of the pharmaceutical product. If the operations of one or more of our manufacturers were to become unavailable for any reason, any required FDA or EMA review and approval of the operations of an alternative supplier could cause a delay in the manufacture of our products.
The commercialization of our products will be substantially dependent on our ability to develop effective sales and marketing capabilities.
One of our primary strategies for product candidates under development is to develop compounds through the Phase 2 stage of clinical testing and market or co-promote certain of our drugs. We currently have no sales, marketing or distribution capabilities. We will depend primarily on strategic alliances with third parties, which have established distribution systems and sales forces, to commercialize our drugs. To the extent that we are unsuccessful in
38
commercializing any drugs ourselves or through a strategic alliance, product revenues may suffer, we may incur significant additional losses, and our share price would be negatively affected.
We face potential product liability exposure, and if successful claims are brought against us, we may incur substantial liability for a product candidate and may have to limit its commercialization.
The use of our product candidates in clinical trials and the sale of any products for which we may obtain marketing approval expose us to the risk of product liability claims. Product liability claims may be brought against us or our collaborators by participants enrolled in our clinical trials, patients, health care providers or others using, administering or selling our products. If we cannot successfully defend ourselves against any such claims, we would incur substantial liabilities. Regardless of merit or eventual outcome, product liability claims may result in:
| ● | withdrawal of clinical trial participants; |
| ● | termination of clinical trial sites or entire trial programs; |
| ● | costs of related litigation; |
| ● | substantial monetary awards to patients or other claimants; |
| ● | decreased demand for our product candidates and loss of revenues; |
| ● | impairment of our business reputation; |
| ● | diversion of management and scientific resources from our business operations; and |
| ● | the inability to commercialize our product candidates. |
We have obtained limited product liability insurance coverage for our clinical trials in the United States and in selected other jurisdictions where we are conducting clinical trials. Our primary product liability insurance coverage for clinical trials in the United States is at least $10.0 million and outside of the United States, we have coverage for lesser amounts that vary by country. As such, our insurance coverage may not reimburse us or may not be sufficient to reimburse us for any expenses or losses we may suffer. Moreover, insurance coverage is becoming increasingly expensive, and, in the future, we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses due to product liability. We intend to expand our insurance coverage for products to include the sale of commercial products if we obtain marketing approval for our product candidates in development, but we may be unable to obtain commercially reasonable product liability insurance for any products approved for marketing. Large judgments have been awarded in class action lawsuits based on drugs that had unanticipated side effects. A successful product liability claim or series of claims brought against us, particularly if judgments exceed our insurance coverage, could decrease our cash resources and adversely affect our business.
Healthcare legislative reform measures may have a material adverse effect on our business, financial condition or results of operations.
In the United States, there have been and continue to be a number of legislative initiatives to contain healthcare costs. For example, the Patient Protection and Affordable Care Act and the Health Care and Education Affordability Reconciliation Act of 2010, referred to jointly as ACA enacted in March 2010, substantially changed the way healthcare is financed by both governmental and private insurers, and significantly impacted the U.S. pharmaceutical industry. With regardThe ACA, among other things, increased the minimum Medicaid rebates owed by manufacturers under the Medicaid Drug Rebate Program and extended the rebate program to pharmaceutical products,individuals enrolled in Medicaid managed care organizations, established annual fees and taxes on manufacturers of certain branded prescription drugs and biologics, and created a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 70% (increased from 50% pursuant to the Bipartisan Budget Act of 2018, effective as of 2019) point-of-sale discounts off negotiated prices of applicable brand drugs and biologics to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs or biologics to be covered under Medicare Part D.
39
We expect that future changes or additions to the ACA, may have the effect of expandingMedicare and increasing industry rebates for drugs covered under Medicaid programs and make changes stemming from other healthcare reform measures, especially with regard to healthcare access, financing or other legislation in individual states, could have a material adverse effect on the health care industry in the United States.
Over the past several years there has been heightened governmental scrutiny over the manner in which biopharmaceutical manufacturers set prices for their marketed products, which has resulted in several U.S. Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drug products. The probability of success of these newly announced policies, many of which have been subjected to legal challenge in the federal court system, and their potential impact on the U.S. prescription drug marketplace is unknown. There are likely to be continued political and legal challenges associated with implementing these reforms as they are currently envisioned. For example, in July 2021, President Biden issued a sweeping executive order on promoting competition in the American economy that included several mandates pertaining to the coverage requirements underpharmaceutical and health care insurance industries and called on HHS to release a comprehensive plan to combat high prescription drug prices. The drug pricing plan released by HHS in September 2021 in response to the executive order makes clear that the Biden Administration supports aggressive action to address rising drug prices, including allowing HHS to negotiate the cost of Medicare Part B and D drugs, but such significant changes will require either new legislation to be passed by Congress or time-consuming administrative actions.
Most recently, in August 2022, President Biden signed into law the Inflation Reduction Act of 2022, or the IRA. Among other things, the IRA has multiple provisions that may impact the prices of drug products that are both sold into the Medicare program and throughout the United States. Starting in 2023, a manufacturer of a drug or biological product covered by Medicare Parts B or D must pay a rebate to the federal government if the product’s price increases faster than the rate of inflation. This calculation is made on a drug product by drug product basis and the amount of the rebate owed to the federal government is directly dependent on the volume of a drug product that is paid for by Medicare Parts B or D. Additionally, starting in payment year 2026, CMS will negotiate drug prices annually for a select number of single source Part D drugs without generic or biosimilar competition. CMS will also negotiate drug prices for a select number of Part B drugs starting for payment year 2028. If a drug product is selected by CMS for negotiation, it is expected that the revenue generated from such drug will decrease. Additional state and federal health care reform measures are expected to be adopted in the future, any of which could limit the amounts that federal and state governments will pay for health care products and services, which could result in reduced demand for certain biopharmaceutical products or additional pricing pressures.
In addition to the IRA’s drug price negotiation provisions, President Biden’s Executive Order 14087, issued in October 2022, called for the CMS Innovation Center to prepare and submit a report to the White House on potential payment and delivery modes that would complement to IRA, lower drug costs, and promote access to innovative drugs. In February 2023, CMS published its report which described three potential models focusing on affordability, accessibility and feasibility of implementation for further testing by the CMS Innovation Center. As of February 2024, the CMS Innovation Center continues to test the proposed models and has started to roll out plans for access model testing of certain product types (e.g., cell and gene therapies) by states and manufacturers.
At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. For example, California requires pharmaceutical manufacturers to notify certain purchasers, including health insurers and government health plans at least 60 days before any scheduled increase in the wholesale acquisition cost (WAC), of their product if the increase exceeds 16%, and further requires pharmaceutical manufacturers to explain whether a change or improvement in the product necessitates such an increase. Similarly, Vermont requires pharmaceutical manufacturers to disclose price information on certain prescription drugs, and to provide notification to the state if introducing a new drug with a WAC in excess of the Medicare Part D program. Additionally, sincespecialty drug threshold. In addition, in recent years, several states have formed prescription drug affordability boards (PDABs). Much like the 2016IRA’s drug price negotiation program, these PDABs have attempted to implement upper payment limits (UPLs) on drugs sold in their respective states in both public and commercial health plans. For example, in August 2023, Colorado’s PDAB announced a list of five prescription drugs that would undergo an
40
affordability review. The effects of these efforts remain uncertain pending the outcomes of several federal elections, which resultedlawsuits challenging state authority to regulate prescription drug payment limits.
In December 2020, the U.S. Supreme Court also held unanimously that federal law does not preempt the states’ ability to regulate pharmaceutical benefit managers, or PBMs, and other members of the healthcare and pharmaceutical supply chain, an important decision that appears to be leading to further and more aggressive efforts by states in this area. The Federal Trade Commission in mid-2022 also launched sweeping investigations into the practices of the PBM industry that could lead to additional federal and state legislative or regulatory proposals targeting such entities’ operations, pharmacy networks, or financial arrangements. Significant efforts to change the PBM industry as it currently exists in the election of President Trump,United States may affect the Republican presidential nominee, and Republican majorities in both houses of Congress, there have been judicial and Congressional challenges to certain aspects of the ACA. Both Congress and President Trump have expressed their intention to repeal or repeal and replace the ACA, and as a result, certain sections of the ACA have not been fully implemented or were effectively repealed. The uncertainty around the future of the ACA, and in particular the impact on reimbursement levelsentire pharmaceutical supply chain and the numberbusiness of insured individuals, may lead to uncertaintyother stakeholders, including biopharmaceutical developers like us. Legally mandated price controls on payment amounts by third-party payors or delay in the purchasing decisions ofother restrictions could harm our customers, which may in
In the appetite of investors to make investments in companies like ours. Regardless of whether or not ACA is overturned or repealed, we expect both government and private health plans to continue to require healthcare providers, including healthcare providers that may one day purchase our products, to contain costs and demonstrate the value of the therapies they provide.
We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative or executive action. We expect that additional federal and state health care reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for health care products and services, which could result in limited coverage and reimbursement and reduced demand for our products, profitably. Among policy makersonce approved, or additional pricing pressures.
We may be subject, directly or indirectly, to federal and state healthcare fraud and abuse laws, false claims laws, and health information privacy and security laws. If we are unable to comply, or have not fully complied, with such laws, we could face substantial penalties.
If we obtain FDA approval for any of our product candidates and begin commercializing those products in the United States, our operations may be subject to various federal and state fraud and abuse laws, including, without limitation, the federal Anti-Kickback Statute, the federal False Claims Act, and physician payments sunshine laws and regulations. These laws may impact, among other things, our proposed sales, marketing, and education programs. In addition, we may be subject to patient privacy regulation by both the federal government and the states in which we conduct our business. The laws that may affect our ability to operate include:
| ● | The federal Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, to induce, or in return for, the purchase or recommendation of an item or service reimbursable under a federal healthcare program, such as the Medicare and Medicaid programs; |
| ● | Federal civil and criminal false claims laws and civil monetary penalty laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other government payors that are false or fraudulent or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government; |
| ● | The federal Health Insurance Portability and Accountability Act of 1996 (HIPAA), which created new federal criminal statutes that prohibit executing a scheme to defraud any healthcare benefit program and making false statements relating to healthcare matters; |
41
| ● | HIPAA, as amended by the Health Information Technology and Clinical Health Act, and its implementing regulations, which imposes specified requirements relating to the privacy, security, and transmission of individually identifiable health information; |
| ● | The federal physician payments sunshine requirements under the ACA require manufacturers of drugs, devices, biologics, and medical supplies to report annually to the CMS information related to payments and other transfers of value to physicians, certain advanced non-physician healthcare providers, and teaching hospitals, and ownership and investment interests held by physicians and other healthcare providers and their immediate family members; and |
| ● | State law equivalents of each of the above federal laws, such as anti-kickback and false claims laws that may apply to items or services reimbursed by any third-party payor, including governmental and private payors, to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government, or otherwise restrict payments that may be made to healthcare providers and other potential referral sources; state laws that require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures, and state laws governing the privacy and security of health information in specified circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts. |
Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available, it is possible that some of our business activities could be subject to challenge under one or more of such laws. In addition, recent health care reform legislation has strengthened these laws. For example, the ACA, among other things, amended the intent requirement of the federal anti-kickback and criminal healthcare fraud statutes. A person or entity no longer needs to have actual knowledge of this statute or specific intent to violate it. Moreover, the ACA provides that the U.S. government may assert that a claim including items or services resulting from a violation of the federal anti-kickback statute constitutes a false or fraudulent claim for purposes of the False Claims Act.
If our operations are found to be in violation of any of the laws described above or any other governmental regulations that apply to us, we may be subject to penalties, including civil and criminal penalties, damages, fines, exclusion from participation in government health care programs, such as Medicare and Medicaid, imprisonment, and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations.
We may be subject to, or may in the future become subject to, U.S. federal and state, and international laws and regulations imposing obligations on how we collect, use, disclose, store and process personal information. Our actual or perceived failure to comply with such obligations could result in liability or reputational harm and could harm our business. Ensuring compliance with such laws could also impair our efforts to maintain and expand our customer base, and thereby decrease our revenue.
In many activities, including the conduct of clinical trials, we are subject to laws and regulations governing data privacy and the protection of health-related and other personal information. The regulatory framework for collecting, using, safeguarding, sharing, transfering and other processing of information worldwide is rapidly evolving and is likely to remain uncertain for the foreseeable future. The withdrawal of the United Kingdom from the European Union and the subsequent separation of the data protection regimes of these territories means we are required to comply with separate data protection laws in the European Union and the United Kingdom, which may lead to additional compliance costs and could increase our overall risk. Similar laws and regulations govern our processing of personal data, including the collection, access, use, analysis, modification, storage, transfer, security breach notification, destruction and disposal of personal data. For example, the collection, use, disclosure, transfer, or other processing of personal data regarding individuals in the European Union, including personal health data, is subject to the General Data Protection Regulation, or GDPR, which took effect across all Member States of the European Economic Area, or EEA, on May 25, 2018, and as still in effect in the United Kingdom as the UK GDPR. On June 28, 2021, the EU Commission adopted decisions on the UK’s adequacy under the EU GDRP, and the UK continues to operate under this adequacy decision. The GDPR imposed a broad data protection framework that expanded the scope of EU and UK data protection law, including to non-EU and non-UK entities meeting the jurisdictional requirements that process, or control the processing of, personal
42
data relating to individuals located in the EU or UK, including clinical trial data. The GDPR sets out a number of requirements for controllers and/or processors, as applicable, that must be complied with when handling the personal data of EU or UK based data subjects, including: providing expanded disclosures about how their personal data will be used; higher standards for organizations to demonstrate that they have obtained valid consent or have another legal basis in place to justify their data processing activities; the obligation to appoint data protection officers in certain circumstances; new rights for individuals to be “forgotten” and rights to data portability, as well as enhanced current rights (e.g., access requests); the principal of accountability and demonstrating compliance through policies, procedures, training and audit; and a new mandatory data breach regime. In particular, medical or health data, genetic data and biometric data are all classified as “special category” data under the GDPR and afford greater protection and require additional compliance obligations. Further, the UK and EU member states have a broad right to impose additional conditions—including restrictions—on these data categories. This is because the GDPR allows EU member states to derogate from the requirements of the GDPR mainly in regard to specific processing situations (including special category data and processing for scientific or statistical purposes). We must comply with laws and regulations associated with the international transfer of personal data based on the location in which the personal data originates and the location in which it is processed and/or controlled. Although there are legal mechanisms to facilitate the transfer of personal data from the UK, EEA, and Switzerland to the United States, the decision of the Court of Justice of the EU (CJEU) that invalidated the safe harbor framework has increased uncertainty around compliance with EU privacy law requirements. As a result of the decision, it was no longer possible to rely on safe harbor certification as a legal basis for the transfer of personal data from the European Union to entities in the United States. However, on July 10, 2023, the European Commission adopted an adequacy decision for a new mechanism for transferring data from the EU to the United States – the EU-U.S. Data Privacy Framework, which provides EU individuals with several new rights, including the right to obtain access to their data, or obtain correction or deletion of incorrect or unlawfully handled data. That being said, we have not yet self-certified under the Data Privacy Framework. The GDPR only permits exports of personal data outside of the EU to “non-adequate” countries where there is a suitable data transfer mechanism in place to safeguard personal data (e.g., the EU Commission approved Standard Contractual Clauses or certification under the newly-adopted Data Privacy Framework). On July 16, 2020, the Court of Justice of the EU, or the CJEU, issued a landmark opinion in the case Maximilian Schrems vs. Facebook (Case C-311/18) (Schrems II). This decision calls into question certain data transfer mechanisms as between the EU member states and the U.S. The CJEU is the highest court in Europe and the Schrems II decision heightened the burden to assess U.S. national security laws on their business, and future actions of EU data protection authorities are difficult to predict at this time. While the newly-adopted Data Privacy Framework was meant to address the concerns raised by the CJEU in Schrems II, it will likely be subject to future legal challenges. Consequently, there is some risk of any data transfers from the EU being halted. If we have to rely on third parties to carry out services for us, including processing personal data on our behalf, we are required under GDPR to enter into contractual arrangements to flow down or help ensure that these third parties only process such data according to our instructions and have sufficient security measures in place. Any security breach or non-compliance with our contractual terms or breach of applicable law by such third parties could result in enforcement actions, litigation, fines and penalties or adverse publicity and could cause customers to lose trust in us, which would have an adverse impact on our reputation and business. Any contractual arrangements requiring the processing of personal data from the EU to us in the U.S. will require greater scrutiny and assessments as required under Schrems II and may have an adverse impact on cross-border transfers of personal data or increase costs of compliance. The GDPR provides an enforcement authority to impose large penalties for noncompliance, including the potential for fines of up to €20 million or 4% of the annual global revenues of the noncompliant company, whichever is greater. Some customers or other service providers may respond to these evolving laws and regulations by asking us to make certain privacy or data-related contractual commitments that we are unable or unwilling to make. This could lead to the loss of current or prospective customers or other business relationships.
The privacy and security of personally identifiable information stored, maintained, received or transmitted, including electronically, is subject to significant regulation in the United States and elsewhere, there is significant interestabroad. While we strive to comply with all applicable privacy and security laws and regulations, legal standards for privacy continue to evolve and any failure or perceived failure to comply may result in promoting changesproceedings or actions against us by government entities or others, or could cause reputational harm, which could have a material adverse effect on our business.
Numerous foreign, federal and state laws and regulations govern collection, dissemination, use and confidentiality of personally identifiable health information, including state privacy and confidentiality laws (including state laws
43
requiring disclosure of breaches); federal and state consumer protection and employment laws; HIPAA; and European and other international data protection laws. These laws and regulations are increasing in complexity and number, may change frequently and sometimes conflict.
HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act (HITECH), establishes a set of U.S. national privacy and security standards for the protection of individually identifiable health information, including protected health information, or PHI, by health plans, certain healthcare systems with the stated goalsclearinghouses and healthcare providers that submit certain covered transactions electronically, or covered entities, and their “business associates,” which are persons or entities that perform certain services for, or on behalf of, containing healthcare costs, improving qualitya covered entity that involve creating, receiving, maintaining or transmitting PHI. While we are not currently a covered entity or business associate under HIPAA, we may receive identifiable information from these entities. Failure to receive this information properly could subject us to HIPAA’s criminal penalties, which may include fines up to $50,000 per violation and/or expanding accessimprisonment. In addition, responding to healthcare. government investigations regarding alleged violations of these and other laws and regulations, even if ultimately concluded with no findings of violations or no penalties imposed, can consume company resources and impact our business and, if public, harm our reputation.
In the United States, various federal and state regulators, including governmental agencies like the pharmaceutical industryFederal Trade Commission, have promulgated, or are considering promulgating, regulations concerning personal information and data securityIn addition to fines and penalties imposed upon violators, some of these state laws also afford private rights of action to individuals who believe their personal information has been misused. California’s patient privacy laws, for example, provide for penalties of up to $250,000 and permit injured parties to sue for damages. In addition, The California Consumer Privacy Act (“CCPA”) went into effect January 1, 2020, and is one of the most restrictive state privacy laws, protecting a particularwide variety of personal information and granting significant rights to California residents with respect to their personal information. Regulations under CCPA have been modified several times, and continue to be modified. Additionally, a new privacy law, the California Privacy Rights Act, (“CPRA”) was approved by California voters in the election of November 3, 2020 and went into effect in January of 2023. The CPRA modified the CCPA significantly, and may result in further uncertainty, additional costs and expenses stemming from efforts to comply with this law, and increases the potential for harm and liability for failure to comply. Among other things, the CPRA established a new regulatory authority, the California Privacy Protection Agency, which is enacting new regulations and has expanded enforcement authority. Other states have implemented similar laws protecting identifiable health and personal information, and most such laws differ from each other in significant ways and may not be preempted by HIPAA, thus complicating compliance efforts. In addition, various states, such as California, Colorado, Connecticut, New Jersey, Delaware, Utah, Virginia, Oregon, Indiana, Iowa, Tennessee, Montana, Florida and Texas , have implemented similar privacy laws and regulations.
The interplay of federal and state laws may be subject to varying interpretations by courts and government agencies, creating complex compliance issues for us and our clients and potentially exposing us to additional expense, adverse publicity and liability. Further, as regulatory focus on privacy issues continues to increase and laws and regulations concerning the protection of personal information expand and become more complex, these potential risks to our business could intensify.
The legislative and regulatory landscape for privacy and data security continues to evolve, and there has been an increasing focus on privacy and data security issues which may affect our business. Failure to comply with current and future laws and regulations could result in government enforcement actions (including the imposition of significant penalties), criminal and civil liability for us and our officers and directors, private litigation and/or adverse publicity that negatively affects our business.
Defending against claims relating to improper handling, storage or disposal of hazardous chemical, radioactive or biological materials could be time consuming and expensive.
Our research and development involves the controlled use of hazardous materials, including chemicals, radioactive and biological materials such as chemical solvents, phosphorus and bacteria. Our operations produce hazardous waste products. We cannot eliminate the risk of accidental contamination or discharge and any resultant injury from those materials. Various laws and regulations govern the use, manufacture, storage, handling and disposal of hazardous
44
materials. We may be sued for any injury or contamination that results from our use or the use by third parties of these materials. Compliance with environmental laws and regulations may be expensive, and current or future environmental regulations may impair our research, development and production efforts.
Our business and operations would suffer in the event of system failures.
Despite the implementation of security measures, our internal computer systems, and those of our CROs and other third parties on which we rely, are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. If such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our drug development programs. For example, the loss of clinical trial data from completed or ongoing or planned clinical trials could result in delays in our regulatory approval efforts and has been significantly affected by major legislative initiatives.increase our costs to recover or reproduce the data. To the extent that any disruption or security breach were to result in a loss of or damage to our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and the further development of our product candidates could be delayed.
Risks Related to Our Business and Financial Condition
We have a history of operating losses and we may never become profitable. Our stock is a highly speculative investment.
We have incurred operating losses in each year since beginning operations in 1996 due to costs incurred in connection with our research and development activities and selling, general and administrative costs associated with our operations, and we may never achieve profitability. As of December 31, 2022 and December 31, 2023, our accumulated deficit was $405.7 million and $428.3 million, respectively. Our net loss was $21.2 million and $22.5 million for the years ended December 31, 2022 and 2023, respectively. Our drug candidates are in the early- to mid-stages of clinical testing and we must conduct significant additional clinical trials before we can seek the regulatory approvals necessary to begin commercial sales of our drugs. We expect to incur continued losses for several years as we continue our research and development of our drug candidates, seek regulatory approvals and commercialize any approved drugs. If our drug candidates are unsuccessful in clinical trials or we are unable to obtain regulatory approvals, or if our drugs are unsuccessful in the market, we will not be profitable. If we fail to become and remain profitable, or if we are unable to fund our continuing losses, particularly in light of the current economic conditions, you could lose all or part of your investment.
There is substantial doubt regarding our ability to continue as a going concern. Our ability to raise additional capital in the future may not be available to us on reasonable terms, if at all, when or as we require additional funding. If we issue additional shares of our common stock or other securities that may be convertible into, or exercisable or exchangeable for, our common stock, our existing stockholders would experience pricing pressures in connectionfurther dilution. If we fail to obtain additional funding, we may be unable to complete the development and commercialization of our lead drug candidates, fadraciclib and plogosertib, or continue to fund our research and development programs.
We have funded all of our operations and capital expenditures with proceeds from the saleissuance of productspublic equity securities, private placements of our securities, interest on investments, licensing revenue, government grants, research and development tax credits and product revenue. In order to conduct the lengthy and expensive research, preclinical testing and clinical trials necessary to complete the development and marketing of our drug candidates, we will require substantial additional funds. We may have insufficient public equity available for issue to raise the required additional substantial funds to implement our operating plan and we may not be able to obtain the appropriate stockholder approvals necessary to increase our available public equity for issuance within a time that we develop,may require additional funding. As of December 31, 2023, our cash and cash equivalents were $3.4 million. Based on our current operating plan, there is substantial doubt regarding our ability to continue as a going concern for a period of one year after the date that our financial statements for the year ended December 31, 2023 are issued. To meet our long-term financing requirements, we may raise funds through public or private equity offerings, debt financings or strategic alliances. Raising additional funds by issuing equity or convertible debt securities may cause our stockholders to experience substantial dilution in their ownership interests and new investors may have rights superior to the rights of our other stockholders. To the extent equity valuations, including the trading price of our common stock, are depressed as a result
45
of economic disruptions or other uncertainties, for example due to rising inflationary pressures, ongoing military conflicts or other factors, the trend toward cost containmentpotential magnitude of this dilution will increase. Raising additional funds through debt financing, if available, may involve covenants that restrict our business activities and options. To the extent that we raise additional legislative proposals.funds through collaborations and licensing arrangements, we may have to relinquish valuable rights to our drug discovery and other technologies, research programs or drug candidates, or grant licenses on terms that may not be favorable to us. Additional funding may not be available to us on favorable terms, or at all, particularly in light of the current economic conditions. Changes to United Kingdom tax legislation related to research and development tax credits may reduce or eliminate the cash flow benefit we receive from these tax credits. If we are unable to obtain additional funds, we may be forced to delay or terminate our current clinical trials and the development and marketing of our drug candidates including fadraciclib and plogosertib.
Unstable market and economic conditions may have serious adverse consequences on our business, financial condition and stock price.
As widely reported, global credit and financial markets have experienced extreme disruptions in the past several years, including severely diminished liquidity and credit availability, declines in consumer confidence, declines in economic growth, increases in unemployment rates, and uncertainty about economic stability. There can be no assurance that further deterioration in credit and financial markets and confidence in economic conditions will not continue to occur. Our general business strategy may be adversely affected by any such economic downturn, volatile business environment or continued unpredictable and unstable market conditions. If the current financial markets deteriorate, or do not improve, it may make any necessary financing more difficult, more costly, and more dilutive. Failure to secure any necessary financing in a timely manner and on favorable terms could have a material adverse effect on our growth strategy, financial performance and stock price and could require us to delay or abandon clinical development or other operating or strategic plans for our business.
Inadequate funding for the FDA, the SEC and other government agencies could hinder their ability to hire and retain key leadership and other personnel, prevent new products and services from being developed or commercialized in a timely manner or otherwise prevent those agencies from performing normal business functions on which the operation of our business may rely, which could negatively impact our business.
The ability of the FDA to review and approve new products can be affected by a variety of factors, including government budget and funding levels, ability to hire and retain key personnel and accept the payment of user fees, and statutory, regulatory, and policy changes. Average review times at the agency have fluctuated in recent years as a result. In addition, government funding of the SEC and other government agencies on which our operations may rely, including those that fund research and development activities is subject to the political process, which is inherently fluid and unpredictable.
Disruptions at the FDA and other agencies may also slow the time necessary for new drugs to be reviewed and/or approved by necessary government agencies, which would adversely affect our business. If a prolonged government shutdown occurs, it could significantly impact the ability of the FDA to timely review and process our regulatory submissions, which could have a material adverse effect on our business. Further, upon completion of this offering and in our operations as a public company, future government shutdowns could impact our ability to access the public markets and obtain necessary capital in order to properly capitalize and continue our operations.
Our employees, independent contractors, principal investigators, contract research organizations, consultants or vendors may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements.
We are exposed to the risk that our employees, independent contractors, principal investigators, contract research organizations, consultants or vendors may engage in fraudulent or other illegal activity. Misconduct by these parties could include intentional, reckless and/or negligent conduct or disclosure of unauthorized activities to us that violates: FDA regulations, including those laws requiring the reporting of true, complete and accurate information to the FDA; manufacturing standards; federal and state health care fraud and abuse laws and regulations; or laws that require the true, complete and accurate reporting of financial information or data. In addition, sales, marketing and business arrangements
46
in the health care industry are subject to extensive laws and regulations intended to prevent fraud, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Activities subject to these laws also involve the improper use or misrepresentation of information obtained in the course of clinical trials or creating fraudulent data in our nonclinical studies or clinical trials, which could result in regulatory sanctions and serious harm to our reputation.
It is not always possible to identify and deter misconduct by our employees and other third parties, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. Additionally, we are subject to the risk that a person could allege such fraud or other misconduct, even if none occurred. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of civil, criminal and administrative penalties, damages, monetary fines, possible exclusion from participation in Medicare, Medicaid and other federal health care programs, contractual damages, reputational harm, diminished potential profits and future earnings, and curtailment of our operations, any of which could adversely affect our business, and financial condition.
If we are unable to compete successfully in our market place,marketplace, it will harm our business.
There are existing products in the marketplace that compete with our products. Companies may develop new products that compete with our products. Certain of these competitors and potential competitors have longer operating histories, substantially greater product development capabilities and financial, scientific, marketing and sales resources. Competitors and potential competitors may also develop products that are safer, more effective or have other potential advantages compared to our products. In addition, research, development and commercialization efforts by others could render our products obsolete or non-competitive. Certain of our competitors and potential competitors have broader product offerings and extensive customer bases, allowing them to adopt aggressive pricing policies that would enable them to gain market share. Competitive pressures could result in price reductions, reduced margins and loss of market share. We could encounter potential customers that, due to existing relationships with our competitors, are committed to products offered by those competitors. As a result, those potential customers may not consider purchasing our products.
We are at an early stage of development as a company and we do not have, and may never have, any products that generate significant revenues.
We are at an early stage of development as a company and have a limited operating history on which to evaluate our business and prospects. While we earned modest product revenues from the ALIGN business prior to terminating such operations effective September 30, 2012, we have not generated any product revenues from our product candidates currently in development. We cannot guarantee that any of our product candidates currently in development will ever become marketable products. We must demonstrate that our drug candidates satisfy rigorous standards of safety and efficacy for their intended uses before the FDA, EMA and other regulatory authorities in the United States, the European Union and elsewhere. Significant additional research, preclinical testing and clinical testing is required before we can file applications with the FDA or EMA for approval of our drug candidates. In addition, to compete effectively, our drugs must be easy to administer, cost-effective and economical to manufacture on a commercial scale. We may not achieve any of these objectives. As our Phase 3 study for AML did not meet its primary endpoint of demonstrating statistically significant improvement in overall survival for the experimental arm versus an active control our clinical development programs are now all at an early stage of testing in Phase 1/2. CYC065 is in a first-in-human Phase 1 study and a combination of sapacitabine and seliciclib, is currently in a Phase 1/2 clinical trial. We cannot be certain that the clinical development of these or any otherour drug candidates in preclinical testing or clinical development will be successful, that we will receive the regulatory approvals required to commercialize them or that any of our other research and drug discovery programs will yield a drug candidate suitable for investigation through clinical trials. Our commercial revenues from our product candidates currently in development, if any, will be derived from sales of drugs that will not become marketable for several years, if at all.
If we fail to comply with the continued listing requirements of the NASDAQNasdaq Capital Market, our common stock may be delisted and the price of our common stock and our ability to access the capital markets could be negatively impacted.
Our common stock is currently listed for trading on the NASDAQNasdaq Capital Market.Market (“Nasdaq”). We must satisfy NASDAQ’sNasdaq’s continued listing requirements, including, among other things, a minimum stockholders’ equity of $2.5 million and a minimum bid price for our common stock of $1.00 per share, or risk delisting, which would have a material adverse effect on our business. A delisting of our common stock from the NASDAQNasdaq Capital Market could materially
47
reduce the liquidity of our common stock and result in a corresponding material reduction in the price of our common stock. In addition, delisting could harm our ability to raise capital through alternative financing sources on terms acceptable to us, or at all, and may result in the potential loss of confidence by investors, suppliers, customers and employees and fewer business development opportunities.
We effected a letter from the Listing Qualifications Staff15:1 reverse stock split of our common stock on December 18, 2023 (the “Staff”) of The NASDAQ“Reverse Stock Market LLC indicating that the Company had not regained compliance with the
To the extent we elect to fund the development of a drug candidate or the commercialization of a drug at our expense, we will need substantial additional funding.
We plan to market drugs on our own, with or without a partner, that can be effectively commercialized and sold in concentrated markets that do not require a large sales force to be competitive. To achieve this goal, we will need to establish our own specialized sales force, marketing organization and supporting distribution capabilities. The development and commercialization of our drug candidates is very expensive. To the extent we elect to fund the full development of a drug candidate or the commercialization of a drug at our expense, we will need to raise substantial additional funding to:
| ● | fund research and development and clinical trials connected with our research; |
| ● | fund clinical trials and seek regulatory approvals; |
| ● | build or access manufacturing and commercialization capabilities; |
| ● | implement additional internal control systems and infrastructure; |
| ● | commercialize and secure coverage, payment and reimbursement of our drug candidates, if any such candidates receive regulatory approval; |
| ● | maintain, defend and expand the scope of our intellectual property; and |
| ● | hire additional management, sales and scientific personnel. |
Our future funding requirements will depend on many factors, including:
| ● | the scope, rate of progress and cost of our clinical trials and other research and development activities; |
| ● | the costs and timing of seeking and obtaining regulatory approvals; |
| ● | the costs of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights; |
| ● | the costs associated with establishing sales and marketing capabilities; |
| ● | the costs of acquiring or investing in businesses, products and technologies; |
| ● | the effect of competing technological and market developments; and |
48
| ● | the payment, other terms and timing of any strategic alliance, licensing or other arrangements that we may establish. |
If we are not able to secure additional funding when needed, especially in light of the current economic conditions and financial market turmoil, we may have to delay, reduce the scope of or eliminate one or more of our clinical trials or research and development programs or future commercialization efforts.
Our insurance policies are expensive and only protect us from some business risks, which will leave us exposed to significant uninsured liabilities.
We do not carry insurance for all categories of risk that our business may encounter. Some of the policies we currently maintain include property, general liability, employment benefits liability, workers’ compensation, products liability and clinical trials (U.S(U.S. and foreign), and directors’ and officers’, employment practices and fiduciary liability insurance. We do not know, however, if we will be able to maintain insurance with adequate levels of coverage. Any significant uninsured liability may require us to pay substantial amounts, which would adversely affect our financial position and results of operations.
Funding constraints may negatively impact our research and development, forcing us to delay our efforts to develop certain product candidates in favor of developing others, which may prevent us from commercializing our product candidates as quickly as possible.
Research and development is an expensive process. As part of our operating plan, since announcing that our SEAMLESS trial failed to meet its primary endpoint, we have decided to focusconcentrate our clinical development strategy in oncology on our two ongoing, hemato-oncology clinical programs in transcriptional regulation and
We are exposed to risks related to foreign currency exchange rates.
Some of our costs and expenses are denominated in foreign currencies. Most of our foreign expenses are associated with our research and development expenditures, including the operating costs of our United Kingdom-based wholly-ownedwholly owned subsidiary. When the United States dollar weakens against the British pound or the Euro, the United States dollar value of the foreign currency denominated expense increases, and when the United States dollar strengthens against the British pound or the Euro, the United States dollar value of the foreign currency denominated expense decreases. Consequently, changes in exchange rates, and in particular a weakening of the United States dollar, may adversely affect our results of operations.
Security breaches,incidents, loss of data and other disruptions could compromise sensitive information related to our business or prevent us from accessing critical information and expose us to liability, which could adversely affect our business and our reputation. Our business and operations would suffer in the event of system failures.
In the ordinary course of our business, we collect and store sensitive data, intellectual property and proprietary business information owned or controlled by ourselves or our customers. This data encompasses a wide variety of business-critical information including research and development information, commercial information, and business and financial information. We face four primary risks relative to protecting this critical information: loss of access; inappropriateunauthorized disclosure; inappropriateunauthorized modification; and inadequate monitoring of our controls over the first three risks.
We utilize information technology, or IT, systems and networks to process, transmit and store electronic information in connection with our business activities. The secure processing, storage, maintenance, and transmission of this critical information is vital to our operations and business strategy, and we devote significant resources to protecting such information. As use of digital technologies has increased, cyber incidents, including deliberate attacks and attempts to gain unauthorized access to computer systems and networks, have increased in frequency and sophistication. These threats pose a risk to the security of our systems and networks and the confidentiality, availability and integrity of our
49
data. There can be no assurance that we will be successful in preventing cyber-attackscybersecurity incidents or successfully mitigating their effects.
Despite the implementation of security measures, our internal and cloud-based computer systems and those of our contractors and consultants are vulnerable to damage from such cyber-attacks,cybersecurity incidents, including computer viruses, social engineering, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. Such an event could cause interruption of our operations. For example, the loss of data from ongoing or completed clinical trials for our product candidates could result in delays in our regulatory approval efforts and significantly increase our costs. In addition, there can be no assurance that we will promptly detect any such disruption or security breach, if at all. To the extent that any disruption or security breach were to result in a loss of or damage to our data, or inappropriate disclosure of confidential or proprietary information, we could suffer material legal claims and liability, damage to our reputation, suffer loss or harm to our intellectual property rights and the further research, development and commercial efforts of our products and product candidates could be delayed. The loss of drug development or clinical trial data could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data and could adversely impact our business and operations, and could result in financial, legal, operational or reputational harm to us, loss of competitive advantage or face litigationloss of consumer confidence.
Risks Related to our Reliance on Third Parties
We rely, and expect to continue to rely, on third parties to conduct some aspects of our product formulation, research, preclinical, and clinical studies, and those third parties may not perform satisfactorily, including by failing to meet deadlines for the completion of such formulation, research or adversetesting.
We have relied upon and plan to continue to rely upon third parties, including independent clinical investigators, contracted laboratories and third-party CROs, to conduct our preclinical studies and clinical trials, as well as certain product candidate discovery and development activities, in accordance with applicable regulatory actionrequirements and to monitor and manage data for our ongoing preclinical and clinical programs. We rely on these parties for execution of our preclinical studies and clinical trials, and control only certain aspects of their activities. Nevertheless, we are responsible for ensuring that each of our studies and trials is conducted in accordance with the applicable protocol, legal and regulatory requirements and scientific standards, and our reliance on these third parties does not relieve us of our regulatory responsibilities. We and our third-party contractors and CROs are required to comply with GLP and GCP requirements, as applicable, which are regulations and guidelines enforced by the FDA, the EMA and other comparable foreign regulatory authorities for all of our products in clinical development. Regulatory authorities enforce these GLP and GCP regulations through periodic inspections of laboratories conducting GLP studies, and clinical trial sponsors, principal investigators, CROs, and trial sites when auditing for GCP compliance. If we, our investigators or any of our CROs or contracted laboratories fail to comply with applicable GLP and GCP regulations, as applicable, the data generated in our preclinical studies and clinical trials may be deemed unreliable and the FDA, the EMA or other comparable foreign regulatory authorities may require us to perform additional preclinical studies or clinical trials before approving our marketing applications for our therapeutic product candidates. We cannot assure you that upon inspection by a given regulatory authority, such regulatory authority will determine that any of our preclinical studies or clinical trials comply with applicable GLP or GCP regulations. In addition, our clinical trials must be conducted with product manufactured in compliance with applicable cGMP regulations. Our failure to comply with these regulations may require us to repeat preclinical studies or clinical trials, which would delay the regulatory approval process.
Further, these laboratories, investigators and CROs are not our employees, and we will not be able to control, other than by contract, the amount of resources, including time, which they devote to our product candidates and clinical trials. If independent laboratories, investigators or CROs fail to devote sufficient resources to the development of our product candidates, or if their performance is substandard, it may delay or compromise the prospects for approval and commercialization of any product candidates that we develop. In addition, the use of third-party service providers requires us to disclose our proprietary information to these parties, which could increase the risk that this information will be misappropriated.
There is a limited number of third-party service providers that specialize or have the expertise required to achieve our business objectives. If any of our relationships with these third-party laboratories, CROs or clinical investigators
50
terminate, we may not be able to enter into arrangements with alternative laboratories, CROs or investigators or to do so in a timely manner or on commercially reasonable terms. If laboratories, CROs or clinical investigators do not successfully carry out their contractual duties or obligations or meet expected deadlines, if they need to be replaced or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our preclinical or clinical protocols, regulatory requirements or for other reasons, our preclinical studies or clinical trials may be extended, delayed or terminated and we may not be able to obtain regulatory approval for or successfully commercialize our product candidates. As a result, our results of operations and the commercial prospects for our product candidates would be harmed, our costs could increase and our ability to generate revenues could be delayed.
Switching or adding additional laboratories or CROs (or investigators) involves additional cost and requires management time and focus. In addition, there is a natural transition period when a new laboratory or CRO commences work. As a result, delays occur, which can materially impact our ability to meet our desired clinical development timelines. Though we carefully manage our relationships with our contracted laboratories and CROs, there can be no assurance that we will not encounter similar challenges or delays in the future or that these delays or challenges will not have a material adverse impact on our business, financial condition and results of operations.
In addition, clinical investigators may serve as scientific advisors or consultants to us from time to time and may receive cash compensation in connection with such services. If these relationships and any related compensation result in perceived or actual conflicts of interest, or the FDA concludes that the financial relationship may have affected the interpretation of the preclinical study or clinical trial, the integrity of the data generated at the applicable preclinical study or clinical trial site may be questioned and the utility of the preclinical study or clinical trial itself may be jeopardized, which could result in the delay or rejection by the FDA. Any such delay or rejection could prevent us from commercializing our clinical-stage product candidate or any future therapeutic product candidates it may develop.
We rely on third-party supply and manufacturing partners for drug supplies for our late-stage clinical activities and may do the same for any commercial supplies of our product candidates.
We rely on third-party contract manufacturing organizations, or CMOs, for our preclinical and future clinical trial product materials and commercial supplies. We do not intend to produce any meaningful quantity of our future product candidates for preclinical and clinical development through our internal resources, and we do not currently own manufacturing facilities for producing such supplies. While we intend to try to avoid sole-source arrangements with any of our manufacturing, supply and testing vendors, it may not always be possible to do so. We cannot assure you that our preclinical or future clinical development product supplies and commercial supplies will not be limited or interrupted, especially with respect to any sole source third-party manufacturing and supply partners or will be of satisfactory quality or continue to be available at acceptable prices. In particular, any replacement of our manufacturers could require significant effort and expertise because there may be a limited number of qualified replacements.
In complying with the manufacturing regulations of the FDA and other comparable foreign regulatory authorities, we and our third-party suppliers must spend significant time, money and effort in the areas of design and development, testing, production, record-keeping and quality control to assure that the products meet applicable specifications and other regulatory requirements. Suppliers and manufacturers must meet applicable manufacturing requirements and undergo rigorous facility and process validation tests required by regulatory authorities in order to comply with regulatory standards, such as cGMP. Although our agreements with our CMOs require them to perform according to certain cGMP requirements such as those relating to quality control, quality assurance and qualified personnel, we cannot control the conduct of our CMOs to implement and maintain these standards. In the event that any of our current or future manufacturers fails to comply with such requirements or to perform its obligations to us in relation to quality, timing or otherwise, or if our supply of components or other materials becomes limited or interrupted for other reasons, we may be forced to manufacture the materials ourselves, for which we currently do not have the capabilities or resources, or enter into an agreement with another third party, which we may not be able to do on commercially reasonable terms, or at all. In some cases, the technical skills or technology required to manufacture our future product candidates may be unique or proprietary to the original manufacturer and we may have difficulty transferring such skills or technology to another third party and a feasible alternative may not exist. These factors would increase our reliance on such manufacturer or require us to obtain a license from such manufacturer in order to have another third party manufacture our product candidates. If we are required to change manufacturers for any reason, we will be required to
51
verify that the new manufacturer maintains facilities and procedures that comply with quality standards and with all applicable regulations and guidelines. The delays associated with the verification of a new manufacturer could negatively affect our ability to develop product candidates in a timely manner or within budget.
In addition, our CMOs are subject to inspection and approval by regulatory authorities before we can commence the manufacture and sale of any of our product candidates, and thereafter are subject to ongoing inspection from time to time. Our CMOs may not be able to comply with applicable cGMP regulations or similar regulatory requirements outside of the United States. Our failure, or the failure of our third-party manufacturers, to comply with applicable regulations could result in regulatory actions, such as the issuance of FDA Form 483 notices of observations, warning letters or sanctions being imposed on us, including clinical holds, fines, injunctions, civil penalties, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of product candidates or drugs, operating restrictions and criminal prosecutions, any of which could significantly and adversely affect supplies of our products. Any such failure by us or any of our CMOs would significantly impact our ability to develop, obtain regulatory approval for or, if approved, market our product candidates.
We may rely on third-party manufacturers if we receive regulatory approval for any product candidate. To the extent that we have existing, or enter into future, manufacturing arrangements with third parties, we will depend on these third parties to perform their obligations in a timely manner consistent with contractual and regulatory requirements, including those related to quality control and assurance. If we are unable to obtain or maintain third-party manufacturing for product candidates, or to do so on commercially reasonable terms, we may not be able to develop and commercialize our product candidates successfully. Our or a third-party’s failure to execute on our manufacturing requirements could adversely affect our business in a number of ways, including:
| ● | an inability to initiate or continue clinical trials of product candidates under development, which may impact our potential economic benefits; |
| ● | delay in submitting regulatory applications, or receiving regulatory approvals, for product candidates; |
| ● | loss of the cooperation of a collaborator; |
| ● | subjecting our product candidates to additional inspections by regulatory authorities; |
| ● | requirements to cease distribution or to recall batches of our product candidates; and |
| ● | in the event of approval to market and commercialize a product candidate, an inability to meet commercial demands for our products. |
If we fail to enter into and maintain successful strategic alliances for our drug candidates, we may have to reduce or delay our drug candidate development or increase our expenditures.
An important element of our strategy for developing, manufacturing and commercializing our drug candidates is entering into strategic alliances with pharmaceutical companies, research institutions or other industry participants to advance our programs and enable us to maintain our financial and operational capacity.
We face significant competition in seeking appropriate alliances. We may not be able to negotiate alliances on acceptable terms, if at all. In addition, these alliances may be unsuccessful. If we fail to create and maintain suitable alliances, we may have to limit the size or scope of, or delay, one or more of our drug development or research programs. If we elect to fund drug development or research programs on our own, we will have to increase our expenditures and will need to obtain additional funding, which may be unavailable or available only on unfavorable terms.
To the extent we are able to enter into strategic transactions, we will be exposed to risks related to those collaborations and alliances.
We expect to enter into strategic transactions to complete the development and commercialization of some of our drug candidates, including but not limited to after the Phase 2 stage of clinical testing. These arrangements may place the
52
development of our drug candidates outside our control, may require us to relinquish important rights, or may otherwise be on terms unfavorable to us.
Dependence on collaborative arrangements or strategic alliances will subject us to a number of risks, including the risks that:
| ● | we may not be able to control the amount and timing of resources that our collaborators may devote to the drug candidates; |
| ● | our collaborators may experience financial difficulties; |
| ● | we may be required to relinquish important rights such as marketing and distribution rights; |
| ● | business combinations or significant changes in a collaborator’s business strategy may also adversely affect a collaborator’s willingness or ability to complete its obligations under any arrangement; |
| ● | a collaborator could independently move forward with a competing drug candidate developed either independently or in collaboration with others, including our competitors; and |
| ● | collaborative arrangements are often terminated or allowed to expire, which would delay development and may increase the cost of developing our drug candidates. |
Risks Related to our Intellectual Property
If we fail to enforce adequately or defend our intellectual property rights, our business may be harmed.
Our commercial success depends in large part on obtaining and maintaining patent and trade secret protection for our drug candidates, the methods used to manufacture those drug candidates and the methods for treating patients using those drug candidates.
Our ability to obtain patents is uncertain because legal means afford only limited protections and may not adequately protect our rights or permit us to gain or keep any competitive advantage. Some legal principles remain unresolved and the breadth or interpretation of claims allowed in patents in the United States, the European Union or elsewhere can still be difficult to ascertain or predict. In addition, the specific content of patents and patent applications that are necessary to support and interpret patent claims is highly uncertain due to the complex nature of the relevant legal, scientific and factual issues. Changes in either patent laws or in interpretations of patent laws in the United States, the European Union or elsewhere may diminish the value of our intellectual property or narrow the scope of our patent protection. Our existing patents and any future patents we obtain may not be sufficiently broad to prevent others from practicing our technologies or from developing competing products and technologies. In addition, we generally do not control the patent prosecution of subject matter that we license from others and have not controlled the earlier stages of the patent prosecution. Accordingly, we are unable to exercise the same degree of control over this intellectual property as we would over our own.
Even if patents are issued regarding our drug candidates or methods of using them, those patents can be challenged by our competitors who may argue such patents are invalid and/or unenforceable. Patents also will not protect our drug candidates if competitors devise ways of making or using these product candidates without legally infringing our patents. The FDA and FDA regulations and policies and equivalents in other jurisdictions provide incentives to manufacturers to challenge patent validity or create modified, non-infringing versions of a drug in order to facilitate the approval of abbreviated new drug applications for generic substitutes. These same types of incentives encourage manufacturers to submit NDAs that rely on literature and clinical data not prepared for or by the drug sponsor.
Proprietary trade secrets and unpatented know-how are also very important to our business. We rely on trade secrets to protect our technology, especially where we do not believe that patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. Our employees, consultants, contractors, outside scientific collaborators and other advisors may unintentionally or willfully disclose our confidential information to competitors, and confidentiality agreements may not provide an adequate remedy in the event of unauthorized disclosure of confidential
53
information. Enforcing a claim that a third-party obtained illegally and is using trade secrets is expensive and time consuming, and the outcome is unpredictable. Moreover, our competitors may independently develop equivalent knowledge, methods and know-how. Failure to obtain or maintain trade secret protection could adversely affect our competitive business position.
If we do not obtain protection under the Hatch-Waxman Act and similar legislation outside of the United States by extending the patent terms and obtaining data exclusivity for our product candidates, our business may be materially harmed.
Depending upon the timing, duration and specifics of FDA marketing approval of sapacitabine and our other product candidates, if any, one or more of our United States patents may be eligible for limited patent term restorationextension under the Drug Price Competition and Patent Term Restoration Act of 1984, referred to as the Hatch-Waxman Act. The Hatch-Waxman Act permits a patent restoration term extension of up to five years as compensation for patent term lost during product development and the FDA regulatory review
We may be subject to damages resulting from claims that our employees or we have wrongfully used or disclosed alleged trade secrets of their former employers.
Many of our employees were previously employed at universities or other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although no claims against us are currently pending, we may be subject to claims that these employees or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. If we fail in defending such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. A loss of key research personnel or their work product could hamper or prevent our ability to commercialize certain potential drugs, which could severely harm our business. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
Confidentiality agreements with employees and others may not adequately prevent disclosure of our trade secrets and other proprietary information and may not adequately protect our intellectual property, which could limit our ability to compete.
Because we operate in the highly technical field of drug discovery and development of small molecule drugs, we rely in part on trade secret protection in order to protect our proprietary technology and processes. However, trade secrets are difficult to protect. We enter into confidentiality and intellectual property assignment agreements with our corporate partners, employees, consultants, outside scientific collaborators, sponsored researchers, and other advisors. These agreements generally require that the other party keep confidential and not disclose to third parties all confidential information developed by the party or made known to the party by us during the course of the party’s relationship with us. These agreements also generally provide that inventions conceived by the party in the course of rendering services to us will be our exclusive property. However, these agreements may not be honored and may not effectively assign intellectual property rights to us. Enforcing a claim that a party illegally obtained and is using our trade secrets is difficult, expensive and time consuming, and the outcome is unpredictable. In addition, courts outside the United States may be less willing to protect trade secrets. The failure to obtain or maintain trade secret protection could adversely affect our competitive position.
54
Intellectual property rights of third parties may increase our costs or delay or prevent us from being able to commercialize our drug candidates.
There is a risk that we are infringing or will infringe on the proprietary rights of third parties because patents and pending applications belonging to third parties exist in the United States, the European Union and elsewhere in the world in the areas of our research. Others might have been the first to make the inventions covered by each of our or our licensors’ pending patent applications and issued patents and might have been the first to file patent applications for these inventions. We are aware of several published patent applications, and understand that others may exist, that could support claims that, if granted and held valid, could cover various aspects of our developmental programs, including in some cases particular uses of our drug candidates sapacitabine, seliciclib, CYC065 or other therapeutic candidates,fadraciclib, plogosertib, or substances, processes and techniques that we use in the course of our research and development and manufacturing processes. We are aware that other patents exist that claim substances, processes, techniques and techniques,methods of use, which, if held valid, could potentially restrict the scope of our research, development or manufacturing operations. In addition, we understand that other applications and patents exist relating to potential uses of sapacitabine, seliciclibfadraciclib and CYC065plogosertib that are not part of our current clinical programs for these compounds. Numerous third-party United States and foreign issued patents and pending applications exist in the area of kinases, including CDK and PLK for which we have research programs. For example, some pending patent
There has been substantial litigation and other proceedings regarding patent and other intellectual property rights in the pharmaceutical and biotechnology industries. Defending against third party claims, including litigation in particular, would be costly and time consumingtime-consuming and would divert management’s attention from our business, which could lead to delays in our development or commercialization efforts. If third parties are successful in their claims, we might have to pay substantial damages or take other actions that are adverse to our business. As a result of intellectual property infringement claims, or to avoid potential claims, we might:
| ● | be prohibited from selling or licensing any product that we may develop unless the patent holder licenses the patent to us, which it is not required to do; |
| ● | be required to pay substantial royalties or grant a cross license to our patents to another patent holder; decide to locate some of our research, development or manufacturing operations outside of Europe or the United States; |
| ● | be required to pay substantial damages for past infringement, which we may have to pay if a court determines that our product candidates or technologies infringe a competitor’s patent or other proprietary rights; or |
| ● | be required to redesign the manufacturing process or formulation of a drug candidate so it does not infringe, which may not be possible or could require substantial funds and time. |
We may incur substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights.
If we choose to go to court to stop another party from using the inventions claimed in any patents we obtain, that individual or company has the right to ask the court to rule that such patents are invalid or should not be enforced against that third party. These lawsuits are expensive and would consume time and resources and divert the attention of managerial and scientific personnel even if we were successful in stopping the infringement of such patents. In addition, there is a risk that the court will decide that such patents are not valid and that we do not have the right to stop the other party from using the inventions.
55
There is also a risk that, even if the validity of such patents is upheld, the court will refuse to stop the other party on the ground that such other party’s activities do not infringe our rights to such patents. In addition, the United States Supreme Court has recently modified some tests used by the United States Patent and Trademark Office, or USPTO, in granting patents over the past 20 years, which may decrease the likelihood that we will be able to obtain patents and increase the likelihood of challenge of any patents we obtain or license.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees, renewal fees, annuity fees and various other governmental fees on patents and/or applications will be due to be paid to the USPTO and various governmental patent agencies outside of the United States in several stages over the lifetime of the patents and/or applications. We have systems in place to remind us to pay these fees, and we employ an outside firm and rely on our outside counsel to
The patent applications of pharmaceutical and biotechnology companies involve highly complex legal and factual questions, which, if determined adversely to us, could negatively impact our patent position.
The patent positions of pharmaceutical and biotechnology companies can be highly uncertain and involve complex legal and factual questions. The U.S. Patent and Trademark Office’s, or USPTO’s, standards are uncertain and could change in the future. Consequently, the issuance and scope of patents cannot be predicted with certainty. Patents, if issued, may be challenged, invalidated or circumvented.
U.S. patents and patent applications may also be subject to interference proceedings, and U.S. patents may be subject to Inter PartesReview (IPR), Post Grant Review (PGR) or reexamination proceedings in the USPTO (and foreign patents may be subject to opposition or comparable proceedings in the corresponding foreign patent office), which proceedings could result in either loss of the patent or denial of the patent application or loss or reduction in the scope of one or more of the claims of the patent or patent application. Similarly, opposition or invalidity proceedings could result in loss of rights or reduction in the scope of one or more claims of a patent in foreign jurisdictions. In addition, such interference, reexamination and opposition proceedings may be costly. Accordingly, rights under any issued patents may not provide us with sufficient protection against competitive products or processes.
If we fail to obtain and maintain patent protection and trade secret protection of our product candidates, proprietary technologies and their uses, we could lose our competitive advantage and competition we face would increase, reducing our potential revenues and adversely affecting our ability to attain or maintain profitability.
Risks Related to Securities Regulations and Investment in Our Securities
Failure to achieve and maintain internal controls in accordance with Sections 302 and 404 of the Sarbanes-Oxley Act of 2002 could have a material adverse effect on our business and stock price.
Section 404 of the Sarbanes-Oxley Act of 2002 requires that we maintain internal control over financial reporting that meets applicable standards. As with many smaller companies with small staff, material weaknesses in our financial controls and procedures may be discovered. If we fail to maintain our internal controls or fail to implement required new or improved controls, as such control standards are modified, supplemented or amended from time to time, we may not be able to conclude on an ongoing basis that we have effective internal controls over financial reporting. Effective internal controls are necessary for us to produce reliable financial reports and are important in the prevention of financial
56
fraud. If we cannot produce reliable financial reports or prevent fraud, our business and operating results could be harmed.
We incur increased costs and management resources as a result of being a public company, and we may fail to comply with public company obligations.
As a public company, we face and will continue to face increased legal, accounting, administrative and other costs and expenses as a public company that we would not incur as a private company. Compliance with the Sarbanes Oxley Act of 2002, as well as other rules of the SEC, the Public Company Accounting Oversight Board and NASDAQNasdaq resulted in a significant initial cost to us as well as an ongoing compliance cost. As a public company, we are subject to Section 404 of the Sarbanes Oxley Act relating to internal control over financial reporting. We have completed a formal process to evaluate our internal controls for purposes of Section 404, and we concluded that as of December 31, 2017,2023, our internal control over financial reporting was effective. As our business grows and changes, there can be no assurances that we can maintain the effectiveness of our internal controls over financial reporting. In addition, our independent certified public accounting firm has not provided an opinion on the effectiveness of our internal controls over financial reporting for the year ended December 31, 20172023 because we are a smaller reporting company. In the event our independent auditor is required to provide an opinion on such controls in the future, there is a risk that the auditor would conclude that such controls are ineffective.
Effective internal controls over financial reporting are necessary for us to provide reliable financial reports and, together with adequate disclosure controls and procedures, are designed to prevent fraud. If we cannot provide reliable financial reports or prevent fraud, our operating results could be harmed. We have completed a formal process to evaluate our internal control over financial reporting. However, guidance from regulatory authorities in the area of internal controls continues to evolve and substantial uncertainty exists regarding our on-going ability to comply by applicable deadlines. Any failure to implement required new or improved controls, or difficulties encountered in their implementation, could harm our operating results or cause us to fail to meet our reporting obligations. Ineffective internal controls could also cause investors to lose confidence in our reported financial information, which could have a negative effect on the trading price of our common stock.
Our common stock may have a volatile public trading price.
An active public market for our common stock has not developed. Our stock can trade in small volumes which may make the price of our stock highly volatile. The last reported price of our stock may not represent the price at which you would be able to buy or sell the stock. The market prices for securities of companies comparable to us have been highly volatile. Often, these stocks have experienced significant price and volume fluctuations for reasons that are both related and unrelated to the operating performance of the individual companies. In addition, the stock market as a whole and biotechnology and other life science stocks in particular have experienced significant recent volatility. Like our common stock, these stocks have experienced significant price and volume fluctuations for reasons unrelated to the operating performance of the individual companies. Factors giving rise to this volatility may include:
| ● | disclosure of actual or potential clinical results with respect to product candidates we are developing; |
| ● | regulatory developments in both the United States and abroad; |
| ● | developments concerning proprietary rights, including patents and litigation matters; |
| ● | public concern about the safety or efficacy of our product candidates or technology, or related technology, or new technologies generally; |
| ● | concern about the safety or efficacy of our product candidates or technology, or related technology, or new technologies generally; |
| ● | public announcements by our competitors or others; and |
| ● | general market conditions and comments by securities analysts and investors. |
57
Fluctuations in our operating losses could adversely affect the price of our common stock.
Our operating losses may fluctuate significantly on a quarterly basis. Some of the factors that may cause our operating losses to fluctuate on a period-to-period basis include the status of our preclinical and clinical development programs, level of expenses incurred in connection with our preclinical and clinical development programs, implementation or termination of collaboration, licensing, manufacturing or other material agreements with third parties, non-recurring revenue or expenses under any such agreement, and compliance with regulatory requirements. Period-to-period comparisons of our historical and future financial results may not be meaningful, and investors should not rely on them as an indication of future performance. Our fluctuating losses may fail to meet the expectations of securities analysts or investors. Our failure to meet these expectations may cause the price of our common stock to decline.
If securities or industry analysts do not publish research or reports about us, if they change their recommendations regarding our stock adversely or if our operating results do not meet their expectations, our stock price and trading volume could decline.
The trading market for our common stock is influenced by the research and reports that industry or securities analysts publish about us. If analysts do not publish research reports or one or more of these analysts who were publishing research cease coverage of us or fail to regularly publish reports on us, we could lose visibility in the financial markets, which in turn could cause our stock price or trading volume to decline. Moreover, if one or more of the analysts who cover us downgrade our stock or if our operating results do not meet their expectations, our stock price could decline.
Anti-takeover provisions in our charter documents and provisions of Delaware law may make an acquisition more difficult and could result in the entrenchment of management.
We are incorporated in Delaware. Anti-takeover provisions of Delaware law and our amended and restated certificate of incorporation and amended and restated bylaws may make a change in control or efforts to remove management more difficult. Also, under Delaware law, our Board of Directors may adopt additional anti-takeover measures.
We have the authority to issue up to 5 million shares of preferred stock and to determine the terms of those shares of stock without any further action by our stockholders. If the Board of Directors exercises this power to issue preferred stock, it could be more difficult for a third party to acquire a majority of our outstanding voting stock and vote the stock they acquire to remove management or directors. Our amended and restated certificate of incorporation and amended and restated bylaws also provides staggered terms for the members of our Board of Directors. Under Section 141 of the Delaware General Corporation Law, our directors may be removed by stockholders only for cause and only by vote of the holders of a majority of voting shares then outstanding. These provisions may prevent stockholders from replacing the entire board in a single proxy contest, making it more difficult for a third-partythird party to acquire control of us without the consent of our Board of Directors. These provisions could also delay the removal of management by the Board of Directors with or without cause. In addition, our directors may only be removed for cause and amended and restated bylaws limit the ability of our stockholders to call special meetings of stockholders.
As at December 31, 2023, we had 335,273 shares of 6% Convertible Exchangeable Preferred Stock, 119,000 shares of Series B Preferred Stock and 264 shares of Series A Preferred Stock issued and outstanding.
Under Section 203 of the Delaware General Corporation Law, a corporation may not engage in a business combination with any holder of 15% or more of its capital stock until the holder has held the stock for three years unless, among other possibilities, the Board of Directors approves the transaction. Our Board of Directors could use this provision to prevent changes in management. The existence of the foregoing provisions could limit the price that investors might be willing to pay in the future for shares of our common stock.
58
Certain severance-related agreements in our executive employment agreements may make an acquisition more difficult and could result in the entrenchment of management.
In March 2008 (as subsequently amended, and most recently renewed as of January 1, 2017)2023), we entered into employment agreements with our President and Chief Executive Officer and our Executive Vice President, Finance, Chief Financial Officer and Chief Operating Officer, which contain severance arrangements in the event that such executive’s employment is terminated without “cause” or as a result of a “change of control” (as each such term is defined in each agreement). The financial obligations triggered by these provisions may prevent a business combination or acquisition that would be attractive to stockholders and could limit the price that investors would be willing to pay in the future for our stock.
In the event of an acquisition of our common stock, we cannot assure our common stockholders that we will be able to negotiate terms that would provide for a price equivalent to, or more favorable than, the price at which our shares of common stock may be trading at such time.
We may not effect a consolidation or merger with another entity without the vote or consent of the holders of at least a majority of the shares of our preferred stock (in addition to the approval of our common stockholders), unless the preferred stock that remains outstanding and its rights, privileges and preferences are unaffected or are converted into or exchanged for preferred stock of the surviving entity having rights, preferences and limitations substantially similar, but no less favorable, to our convertible preferred stock.
In addition, in the event a third party seeks to acquire our company or acquire control of our company by way of a merger, but the terms of such offer do not provide for our preferred stock to remain outstanding or be converted into or exchanged for preferred stock of the surviving entity having rights, preferences and limitations substantially similar, but no less favorable, to our preferred stock, the terms of the Certificate of Designations of our preferred stock provide for an adjustment to the conversion ratio of our preferred stock such that, depending on the terms of any such transaction, preferred stockholders may be entitled, by their terms, to receive up to $10.00 per share in common stock, causing our common stockholders not to receive as favorable a price as the price at which such shares may be trading at the time of any such transaction. As of December 31, 2017,2023, there were 335,273 shares of our preferred stock6% Convertible Exchangeable Preferred Stock issued and outstanding. If the transaction were one in which proceeds were received by the Companyus for distribution to stockholders, and the terms of the Certificate of Designations governing the preferred stock were strictly complied with, approximately $4.0 million would be paid to the preferred holders before any distribution to the common stockholders, although the form of transaction could affect how the holders of preferred stock are treated. In such an event, although such a transaction would be subject to the approval of our holders of common stock, we cannot assure our common stockholders that we will be able to negotiate terms that would provide for a price equivalent to, or more favorable than, the price at which our shares of common stock may be trading at such time. Thus, the terms of our preferred stock might hamper a third party’s acquisition of our company.
Our certificate of incorporation and bylaws and certain provisions of Delaware law may delay or prevent a change in our management and make it more difficult for a third-party to acquire us.
Our amended and restated certificate of incorporation and bylaws contain provisions that could delay or prevent a change in our Board of Directors and management teams. Some of these provisions:
| ● | authorize the issuance of preferred stock that can be created and issued by the Board of Directors without prior stockholder approval, commonly referred to as “blank check” preferred stock, with rights senior to those of our common stock; |
| ● | provide for the Board of Directors to be divided into three classes; and |
| ● | require that stockholder actions must be effected at a duly called stockholder meeting and prohibit stockholder action by written consent. |
In addition, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which limits the ability of large stockholders to complete a business combination
59
with, or acquisition of, us. These provisions may prevent a business combination or acquisition that would be attractive to stockholders and could limit the price that investors would be willing to pay in the future for our stock.
These provisions also make it more difficult for our stockholders to replace members of our Board of Directors. Because our Board of Directors is responsible for appointing the members of our management team, these provisions could in turn affect any attempt to replace our current management team. Additionally, these provisions may prevent an acquisition that would be attractive to stockholders and could limit the price that investors would be willing to pay in the future for our common stock.
We may have limited ability to pay cash dividends on our preferred stock, and there is no assurance that future quarterly dividends will be declared.
Delaware law may limit our ability to pay cash dividends on our preferred stock. Under Delaware law, cash dividends on our preferred stock may only be paid from surplus or, if there is no surplus, from the corporation’s net profits for the current or preceding fiscal year. Delaware law defines “surplus” as the amount by which the total assets of a corporation, after subtracting its total liabilities, exceed the corporation’s capital, as determined by its board of directors.
Since we are not profitable, our ability to pay cash dividends will require the availability of an adequate surplus. Even if adequate surplus is available to pay cash dividends on our preferred stock, we may not have sufficient cash to pay dividends on the preferred stock or we may choose not to declare the dividends.
Our common and preferred stock may experience extreme price and volume fluctuations, which could lead to costly securities-related litigation, including securities class action litigation or securities-related investigations, which could make an investment in us less appealing.
The market price of our common and preferred stock may fluctuate substantially due to a variety of factors, including:
| ● | announcements of technological innovations or new products or services by us or our competitors; announcements concerning our competitors or the biotechnology industry in general; |
| ● | new regulatory pronouncements and changes in regulatory guidelines; |
| ● | general and industry-specific economic conditions; |
| ● | additions to or departures of our key personnel; |
| ● | changes in financial estimates or recommendations by securities analysts; |
| ● | variations in our quarterly results; and |
| ● | announcements about our collaborators or licensors; and |
| ● | changes in accounting principles |
The stock markets have from time to timetime-to-time experienced significant price and volume fluctuations that have affected the market prices for publicly traded securities. The market prices of the securities of biotechnology companies, particularly companies like us without product revenues and earnings, have been highly volatile and are likely to remain highly volatile in the future. This volatility has often been unrelated to the performance of particular companies. In the past, companies that experience volatility in the market price of their securities have often faced securities class action and derivative litigation, and as a public company, we could be subject to sanctions or investigations by NASDAQ,Nasdaq, the SEC or other regulatory authorities. Moreover, market prices for stocks of biotechnology-related and technology companies frequently reach levels that bear no relationship to the performance of these companies. These market prices generally are not sustainable and are highly volatile.
60
Whether or not meritorious, litigation brought against us could result in substantial costs, divert our management’s attention and resources and harm our financial condition and results of operations.
The future sale of our common and convertible preferred stock and future issuances of our common stock upon conversion of our preferred stock could negatively affect our stock price and cause dilution to existing holders of our common stock.
If our common or preferred stockholders sell substantial amounts of our stock in the public market, or the market perceives that such sales may occur, the market price of our common and preferred stock could fall. If additional holders of convertible preferred stock elect to convert their shares to shares of common stock at renegotiated prices, such conversion as well as the sale of substantial amounts of our common stock, could cause dilution to existing holders of our common stock, thereby also negatively affecting the price of our common stock. For example, in 2013, we issued an aggregate of 140,3739,358 shares of our common stock in exchange for an aggregate of 877,869 shares of our preferred stock in arms-length negotiations between us and the other parties who had approached us to propose the exchanges.
If we exchange the convertible preferred stock for debentures, the exchange will be taxable, but we will not provide any cash to pay any tax liability that any convertible preferred stockholder may incur.
An exchange of convertible preferred stock for debentures, as well as any dividend make-whole or interest make-whole payments paid in our common stock, will be taxable events for United States federal income tax purposes, which may result in tax liability for the holder of convertible preferred stock without any corresponding receipt of cash by the holder. In addition, the debentures may be treated as having original issue discount, a portion of which would generally be required to be included in the holder’s gross income even though the cash to which such income is attributable would not be received until maturity or redemption of the debenture. We will not distribute any cash to the holders of the securities to pay these potential tax liabilities.
If we automatically convert the convertible preferred stock, there is a substantial risk of fluctuation in the price of our common stock from the date we elect to automatically convert to the conversion date.
We may automatically convert the convertible preferred stock into common stock if the closing price of our common stock exceeds $2,961$888,300 per share. There is a risk of fluctuation in the price of our common stock between the time when we may first elect to automatically convert the preferred and the automatic conversion date.
We do not intend to pay cash dividends on our common stock in the foreseeable future.
We do not anticipate paying cash dividends on our common stock in the foreseeable future. Any payment of cash dividends will depend on our financial condition, results of operations, capital requirements, the outcome of the review of our strategic alternatives and other factors and will be at the discretion of our Board of Directors. Accordingly, investors will have to rely on capital appreciation, if any, to earn a return on their investment in our common stock. Furthermore, we may in the future become subject to contractual restrictions on, or prohibitions against, the payment of dividends.
The number of shares of common stock which are registered, including the shares to be issued upon exercise of our outstanding warrants, is significant in relation to our currently outstanding common stock and could cause downward pressure on the market price for our common stock.
The number of shares of common stock registered for resale, including those shares which are to be issued upon exercise of our outstanding warrants, is significant in relation to the number of shares of common stock currently outstanding. If the security holder determines to sell a substantial number of shares into the market at any given time, there may not be sufficient demand in the market to purchase the shares without a decline in the market price for our common stock. Moreover, continuous sales into the market of a number of shares in excess of the typical trading volume for our common stock, or even the availability of such a large number of shares, could depress the trading market for our common stock over an extended period of time.
61
If persons engage in short sales of our common stock, including sales of shares to be issued upon exercise of our outstanding warrants, the price of our common stock may decline.
Selling short is a technique used by a stockholder to take advantage of an anticipated decline in the price of a security. In addition, holders of options and warrants will sometimes sell short knowing they can, in effect, cover through the exercise of an option or warrant, thus locking in a profit. A significant number of short sales or a large volume of other sales within a relatively short period of time can create downward pressure on the market price of a security. Further sales of common stock issued upon exercise of our outstanding warrants could cause even greater declines in the price of our common stock due to the number of additional shares available in the market upon such exercise, which could encourage short sales that could further undermine the value of our common stock. You could, therefore, experience a decline in the value of your investment as a result of short sales of our common stock.
We are exposed to riskrisks related to the marketable securities we may purchase.
We may invest cash not required to meet short termshort-term obligations in short term marketable securities. We may purchase securities in United States government, government-sponsored agencies and highly rated corporate and asset-backed securities subject to an approved investment policy. Historically, investment in these securities has been highly liquid and has experienced only very limited defaults. However, recent volatility in the financial markets has created additional uncertainty regarding the liquidity and safety of these investments. Although we believe our marketable securities investments are safe and highly liquid, we cannot guarantee that our investment portfolio will not be negatively impacted by recent or future market volatility or credit restrictions.
Claims for indemnification by our directors and officers may reduce our available funds to satisfy successful stockholder claims against us and may reduce the amount of money available to us.
As permitted by Section 102(b)(7) of the Delaware General Corporation Law, our restated certificate of incorporation limits the liability of our directors to the fullest extent permitted by law. In addition, as permitted by Section 145 of the Delaware General Corporation Law, our restated certificate of incorporation and restated bylaws provide that we shall indemnify, to the fullest extent authorized by the Delaware General Corporation Law, each person who is involved in any litigation or other proceeding because such person is or was a director or officer of our company or is or was serving as an officer or director of another entity at our request, against all expense, loss or liability reasonably incurred or suffered in connection therewith. Our restated certificate of incorporation provides that the right to indemnification includes the right to be paid expenses incurred in defending any proceeding in advance of its final disposition, provided, however, that such advance payment will only be made upon delivery to us of an undertaking, by or on behalf of the director or officer, to repay all amounts so advanced if it is ultimately determined that such director is not entitled to indemnification.
If we do not pay a proper claim for indemnification in full within 60 days after we receive a written claim for such indemnification, except in the case of a claim for an advancement of expenses, in which case such period is 20 days, our restated certificate of incorporation and our restated bylaws authorize the claimant to bring an action against us and prescribe what constitutes a defense to such action.
Section 145 of the Delaware General Corporation Law permits a corporation to indemnify any director or officer of the corporation against expenses (including attorney’s fees), judgments, fines and amounts paid in settlement actually and reasonably incurred in connection with any action, suit or proceeding brought by reason of the fact that such person is or was a director or officer of the corporation, if such person acted in good faith and in a manner that he reasonably believed to be in, or not opposed to, the best interests of the corporation, and, with respect to any criminal action or proceeding, if he or she had no reason to believe his or her conduct was unlawful. In a derivative action, (i.e., one brought by or on behalf of the corporation), indemnification may be provided only for expenses actually and reasonably incurred by any director or officer in connection with the defense or settlement of such an action or suit if such person acted in good faith and in a manner that he or she reasonably believed to be in, or not opposed to, the best interests of the corporation, except that no indemnification shall be provided if such person shall have been adjudged to be liable to the corporation, unless and only to the extent that the court in which the action or suit was brought shall determine that the defendant is fairly and reasonably entitled to indemnity for such expenses despite such adjudication of liability.
62
The rights conferred in the restated certificate of incorporation and the restated bylaws are not exclusive, and we are authorized to enter into indemnification agreements with our directors, officers, employees and agents and to obtain insurance to indemnify such persons. We have entered into indemnification agreements with each of our officers and directors.
The above limitations on liability and our indemnification obligations limit the personal liability of our directors and officers for monetary damages for breach of their fiduciary duty as directors by shifting the burden of such losses and expenses to us. Although we obtained coverage under our directors’ and officers’ liability insurance, certain liabilities or expenses covered by our indemnification obligations may not be covered by such insurance or the coverage limitation amounts may be exceeded. As a result, we may need to use a significant amount of our funds to satisfy our indemnification obligations, which could severely harm our business and financial condition and limit the funds available to stockholders who may choose to bring a claim against our company.
Risks Relating to Restatement of our Consolidated Financial Statements
We have had to restate our previously issued consolidated financial statements and, as part of that process, have identified a material weakness in our internal control over financial reporting as of December 31, 2022. If we are unable to develop and maintain effective internal control over financial reporting, we may not be able to accurately report our financial results in a timely manner, which may adversely affect investor confidence in us and may adversely affect our business, financial condition and results of operations.
A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of our annual or interim financial statements will not be prevented or detected on a timely basis. Effective internal control over financial reporting is necessary for us to provide reliable financial reporting and prevent fraud. We continue to evaluate steps to remediate the material weakness. These remediation measures may be time consuming and costly, and there is no assurance that these initiatives will ultimately have the intended effects. Any failure to maintain effective internal control over financial reporting could adversely impact our ability to report our financial position and results from operations on a timely and accurate basis. If our financial statements are not accurate, investors may not have a complete understanding of our operations. Likewise, if our financial statements are not filed on a timely basis, we could be subject to sanctions or investigations by the stock exchange on which our common stock is listed, the SEC or other regulatory authorities. In either case, there could be an adverse effect on our business, financial condition and results of operations. Ineffective internal control over financial reporting could also cause investors to lose confidence in our reported financial information, which could have a negative effect on the trading price of our stock.
We can provide no assurance that the measures we are taking and plan to take in the future will remediate the material weakness identified or that any additional material weaknesses or restatements of financial results will not arise in the future due to a failure to implement and maintain adequate internal control over financial reporting or circumvention of these controls. In addition, even if we are successful in strengthening our controls and procedures, in the future those controls and procedures may not be adequate to prevent or identify irregularities or errors or to facilitate the fair presentation of our consolidated financial statements.
We may face litigation and other risks as a result of the restatement of our previously issued consolidated financial statements contained in Amendment No. 1 to the Form 10-K for the fiscal year ended December 31, 2022 and material weakness in our internal control over financial reporting.
As part of the restatement of our previously issued consolidated financial statements contained in Amendment No. 1 to the Form 10-K for the fiscal year ended December 31, 2022, we identified a material weakness in our internal control over financial reporting. As a result of such material weakness, the restatement and other matters raised or that may in the future be raised by the SEC, we face potential for litigation or other disputes which may include, among others, claims invoking the federal and state securities laws, contractual claims or other claims arising from the restatement and the material weakness in our internal control over financial reporting and the preparation of our financial statements. As of the date of this report, we have no knowledge of any such litigation or dispute. However, we can provide no assurance
63
that such litigation or dispute will not arise in the future. Any such litigation or dispute, whether successful or not, could adversely affect our business, financial condition and results of operations.
Item 1B. Unresolved Staff Comments
None.
Item 1C. Cybersecurity
We recognize the critical importance of maintaining the trust and confidence of business partners, employees and patients, toward our business and are committed to protecting the confidentiality, integrity and availability of our business operations and systems. Our board of directors is actively involved in oversight of our risk management activities, and cybersecurity represents an important element of our overall approach to risk management. Our cybersecurity policies, standards, processes and practices are based on recognized frameworks established by the UK governments’ National Cyber Security Centre and other applicable industry standards. In general, we seek to address cybersecurity risks through a comprehensive, cross-functional approach that is focused on preserving the confidentiality, security and availability of the information that we collect and store by identifying, preventing and mitigating cybersecurity threats and effectively responding to cybersecurity incidents when they occur.
Cybersecurity Risk Management and Strategy; Effect of Risk
We face risks related to cybersecurity such as unauthorized access, cybersecurity attacks and other security incidents, including as perpetrated by hackers and unintentional damage or disruption to hardware and software systems, loss of data, and misappropriation of confidential information. To identify and assess material risks from cybersecurity threats, we maintain a comprehensive cybersecurity program to ensure our systems are effective and prepared for information security risks, including regular oversight of our programs for security monitoring for internal and external threats to ensure the confidentiality and integrity of our information assets. We consider risks from cybersecurity threats alongside other company risks as part of our overall risk assessment process. We employ a range of tools and services, including regular network and endpoint monitoring, audits, vulnerability assessments, and penetration testing to inform our risk identification and assessment. As discussed in more detail under “Cybersecurity Governance” below, our audit committee provides oversight of our cybersecurity risk management and strategy processes, which are led by our Chief Financial Officer.
We also identify our cybersecurity threat risks by comparing our processes to standards set by the UK governments’ National Cyber Security Centre. To provide for the availability of critical data and systems, maintain regulatory compliance, manage our material risks from cybersecurity threats, and protect against and respond to cybersecurity incidents, we undertake the following activities:
| ● | monitor emerging data protection laws and implement changes to our processes that are designed to comply with such laws; |
| ● | through our policies, practices and contracts (as applicable), require employees, as well as third parties that provide services on our behalf, to treat confidential information and data with care; |
| ● | employ technical safeguards that are designed to protect our information systems from cybersecurity threats, including firewalls, intrusion prevention and detection systems, anti-malware functionality and access controls, which are evaluated and improved through vulnerability assessments and cybersecurity threat intelligence; |
| ● | provide regular training for our employees regarding cybersecurity threats as a means to equip them with effective tools to address cybersecurity threats, and to communicate our evolving information security policies, standards, processes and practices; |
| ● | leverage the National Cyber Security Centre incident handling framework to help us identify, protect, detect, respond and recover when there is an actual or potential cybersecurity incident; and |
64
| ● | carry information security risk insurance that provides protection against the potential losses arising from a cybersecurity incident. |
Our response to an incident involves the coordination of activities to detect, respond to and recover from cybersecurity incidents, which include processes to triage, assess severity for, escalate, contain, investigate and remediate the incident, as well as to comply with potentially applicable legal obligations and mitigate damage to our business and reputation.
As part of the above processes, we regularly engage with consultants, auditors and other third parties, including having a third-party independent qualified and accredited advisor review our cybersecurity program to help identify areas for continued focus, improvement and compliance.
Our processes also address cybersecurity threat risks associated with our use of third-party service providers, including our suppliers and manufacturers or who have access to patient and employee data or our systems. In addition, cybersecurity considerations affect the selection and oversight of our third-party service providers. We perform diligence on third parties that have access to our systems, data or facilities that house such systems or data, and continually monitor cybersecurity threat risks identified through such diligence. Additionally, we would require those third parties, although there are currently none, that could introduce significant cybersecurity risk to us to agree by contract to manage their cybersecurity risks in specified ways, and to agree to be subject to cybersecurity audits, which we conduct as appropriate.
We describe whether and how risks from identified cybersecurity threats, including as a result of any previous cybersecurity incidents, have materially affected or are reasonably likely to materially affect us, including our business strategy, results of operations, or financial condition, under the risk factor heading “Security incidents, loss of data and other disruptions could compromise sensitive information related to our business or prevent us from accessing critical information and expose us to liability” which disclosures are incorporated by reference herein.
We have not experienced any material cybersecurity incidents and the expenses we have incurred from cybersecurity incidents were immaterial.
Cybersecurity Governance; Management
Cybersecurity is an important part of our risk management processes and an area of focus for our board of directors and management. The audit committee of our board of directors is responsible for the oversight of risks from cybersecurity threats.
At least annually, our audit committee receives an update from management of our cybersecurity threat risk management and strategy processes. In such sessions, our audit committee generally receives materials that include a cybersecurity dashboard and other materials discussing current and emerging material cybersecurity threat risks, and describing our ability to mitigate those risks, as well as recent developments, evolving standards, technological developments and information security considerations arising with respect to our peers and third parties. Our audit committee also receive prompt and timely information regarding any cybersecurity incident that meets establishing reporting thresholds, as well as ongoing updates regarding any such incident until it has been addressed. To date, we have not experience any cyber security incident.
Members of our audit committee are also encouraged to regularly engage in conversations with management on cybersecurity-related news events and discuss any updates to our cybersecurity risk management and strategy programs. Material cybersecurity threat risks are also considered during separate board meeting discussions of important matters like enterprise risk management, operational budgeting, business continuity planning, mergers and acquisitions, brand management, and other relevant matters.
Our cybersecurity risk management and strategy processes, which are discussed in greater detail above, are led by our Chief Financial Officer. Our Chief Financial Officer has been responsible for our IT functions for 20 years, and has managed the IT function of companies generally for the last 35 years. Our IT manager has managed our IT functions for over 20 years. The management team members are informed about and monitor the prevention, mitigation, detection, and remediation of cybersecurity incidents through their management of, and participation in, the cybersecurity risk
65
management and strategy processes described above, including the operation of our incident response plan. As discussed above, the management team members report to the audit committee of our board of directors about cybersecurity threat risks, among other cybersecurity related matters, periodically.
Item 2. Properties
We lease our corporate headquarters in Berkeley Heights, New Jersey and a research and development facility in Dundee, Scotland.Jersey. We believe that our existing facilities are adequate to accommodate our business needs.
Item 3. Legal Proceedings
From time to time, we may be involved in routine litigation incidental to the conduct of our business. As of December 31, 2017,2023, we were not a party to any material legal proceedings.
Item 4. Mine Safety Disclosures
Not applicable.
PART II
Market Information
Our common stock is traded on The NASDAQNasdaq Capital Market, or NASDAQ,Nasdaq, under the symbol “CYCC”. Our preferred stock currently trades on NASDAQNasdaq under the symbol “CYCCP”. The following table summarizes, for the periods indicated, the high and low sales prices for the common stock as reported by NASDAQ:
| | | | High | | | Low | | ||||||
| 2017 | | | | | | | | | | | | | |
| Quarter ended March 31, 2017 | | | | $ | 6.14 | | | | | $ | 3.13 | | |
| Quarter ended June 30, 2017 | | | | $ | 10.90 | | | | | $ | 3.77 | | |
| Quarter ended September 30, 2017 | | | | $ | 3.87 | | | | | $ | 1.56 | | |
| Quarter ended December 31, 2017 | | | | $ | 2.25 | | | | | $ | 1.50 | | |
| 2016 | | | | | | | | | | | | | |
| Quarter ended March 31, 2016 | | | | $ | 6.45 | | | | | $ | 3.60 | | |
| Quarter ended June 30, 2016 | | | | $ | 8.27 | | | | | $ | 3.84 | | |
| Quarter ended September 30, 2016 | | | | $ | 9.72 | | | | | $ | 4.21 | | |
| Quarter ended December 31, 2016 | | | | $ | 6.18 | | | | | $ | 3.05 | | |
Holders of Common Stock
We effected a 15:1 reverse stock split of the our common stock on December 18, 2023 (the “Reverse Stock Split”). All share and per share information has been retroactively adjusted to give effect to the Reverse Stock Split for all periods presented, unless otherwise indicated.
On March 27, 2018,15, 2024, we had approximately 2711 registered holders of record of our 11,997,4471,318,257 shares of common stock outstanding. On March 27, 2018,15, 2024, the closing sale price of our common stock as reported by NASDAQNasdaq was $1.34$2.43 per share.
Dividends
We have never declared nor paid any cash dividends on our common stock and do not currently anticipate declaring or paying any cash dividends on our outstanding shares of common stock in the foreseeable future. We are, however, required to make or accrue quarterly dividend payments on our Preferred Stock. Except for dividends that may be paid on the Preferred Stock, we currently intend to retain all of our future earnings, if any, to finance operations. Any future determination relating to our dividend policy will be made at the discretion of our Board of Directors and will depend on a number of factors, including future earnings, capital requirements, financial conditions, future prospects, contractual restrictions and other factors that our Board of Directors may deem relevant.
Unregistered Sales of Securities
None.
Issuer Purchases of Equity Securities
None.
Item 6. Selected Financial Data[Reserved]
66
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
Cautionary Statement Regarding Forward-Looking Statements
This report contains certain statements that may be deemed ‘forward-looking statements’ within the meaning of United States securities laws. All statements, other than statements of historical fact, that address activities, events or developments that we intend, expect, project, believe or anticipate will or may occur in the future are forward-looking statements. Such statements are based upon certain assumptions and assessments made by our management in light of their experience and their perception of historical trends, current
We encourage you to read those descriptions carefully. We caution you not to place undue reliance on the forward-looking statements contained in this report. These statements, like all statements in this report, speak only as of the date of this report (unless an earlier date is indicated) and we undertake no obligation to update or revise the statements except as required by law. Such forward-looking statements are not guarantees of future performance and actual results will likely differ, perhaps materially, from those suggested by such forward-looking statements.
We effected a 15:1 reverse stock split of our common stock on December 18, 2023 (the “Reverse Stock Split”). All share and per share information has been retroactively adjusted to give effect to the Reverse Stock Split for all periods presented, unless otherwise indicated.
Overview
We are a clinical-stage biopharmaceutical company developing innovative cancer medicines based on cell cycle, transcriptional regulation, epigenetics and mitosis control biology. We reported revenue of $0.4 million for the year ended December 31, 2023 and no revenues for the year ended December 31, 2022. We do not expect to report revenue for the foreseeable future.
During 2017,2023, our primary focus has been on our transcriptional regulation program, pursuantwhich is evaluating fadraciclib, a CDK2/9 inhibitor, in solid tumors and hematological malignancies. The epigenetic/anti-mitotic program is evaluating plogosertib, a PLK1 inhibitor, in advanced cancers.
We currently retain all marketing rights worldwide to which we are evaluating our CYC065, cyclin dependent kinase, or CDK, inhibitor and our DNA damage response, or DDR, program, in which we are evaluating sapacitabine in combinationthe compounds associated with our CDK inhibitor seliciclib in solid tumors in a Phase 1 study. Additionally, we have completed the analysis of data from SEAMLESS, the Phase 3 study in acute myeloid leukemia, or AML. Data from the SEAMLESS study was presented at the 59th American Society of Hematology Annual Meeting. Based on the outcome of prespecified and exploratory subgroup analyses, we plan to discuss the data with European and US regulatory authorities.
Revenue from Contracts with Customers
| | | | Year ended December 31, | | | Difference | | ||||||||||||||||||
| | | | 2016 | | | 2017 | | | $ | | | % | | ||||||||||||
| Grant revenue | | | | $ | 843 | | | | | | — | | | | | $ | (843) | | | | | | (100) | | |
| Collaboration and research and development revenue | | | | | — | | | | | | — | | | | | | — | | | | | | — | | |
| Total Revenue | | | | $ | 843 | | | | | | — | | | | | $ | (843) | | | | | | (100) | | |
| | |||||||||||||||||||||||||
We have entered into a collaboration, licensing and supply agreement with ManRos Therapeutics SA or ManRos. Receipt of further revenue under is dependent on the clinical progress of the program, which we do not control. We have received a $150,000 paymentgenerated any revenues from ManRos as of December 31, 2017 in anticipation of ManRos achieving a clinical milestone in 2018. This payment has been recorded as deferred revenues on the balance sheet (part of other current liabilities).
| | | | Year ended December 31, | | | Difference | | ||||||||||||||||||
| | | | 2016 | | | 2017 | | | $ | | | % | | ||||||||||||
| Sapacitabine | | | | $ | 6,239 | | | | | $ | 2,661 | | | | | $ | (3,578) | | | | | | (57) | | |
| Other costs related to research and development programs and management | | | | | 3,238 | | | | | | 1,576 | | | | | | (1,662) | | | | | | (51) | | |
| Total research and development | | | | $ | 9,477 | | | | | $ | 4,237 | | | | | $ | (5,240) | | | | | | (55) | | |
| | |||||||||||||||||||||||||
| | | | Year ended December 31, | | | Difference | | ||||||||||||||||||
| | | | 2016 | | | 2017 | | | $ | | | % | | ||||||||||||
| General and administrative | | | | $ | 5,516 | | | | | $ | 5,254 | | | | | $ | (262) | | | | | | (5) | | |
| Total General and administrative | | | | $ | 5,516 | | | | | $ | 5,254 | | | | | $ | (262) | | | | | | (5) | | |
| | |||||||||||||||||||||||||
| | | | Year ended December 31, | | | Difference | | ||||||||||||||||||
| | | | 2016 | | | 2017 | | | $ | | | % | | ||||||||||||
| Foreign exchange gains (losses) | | | | | 273 | | | | | | (39) | | | | | $ | (312) | | | | | | (114) | | |
| Interest income | | | | | 37 | | | | | | 118 | | | | | $ | 81 | | | | | | 219 | | |
| Other income, net | | | | | 66 | | | | | | 949 | | | | | $ | 883 | | | | | | 1,338 | | |
| Total other income, net | | | | $ | 376 | | | | | $ | 1,028 | | | | | $ | 652 | | | | | | 173 | | |
| | |||||||||||||||||||||||||
| | | | Year ended December 31, | | | Difference | | ||||||||||||||||||
| | | | 2016 | | | 2017 | | | $ | | | % | | ||||||||||||
| Income tax benefit | | | | $ | 1,983 | | | | | $ | 993 | | | | | $ | (990) | | | | | | (50) | | |
| Total Income tax benefit | | | | $ | 1,983 | | | | | $ | 993 | | | | | $ | (990) | | | | | | (50) | | |
| | |||||||||||||||||||||||||
Funding Requirements and will continue to elect to receive paymentGoing Concern
As of the tax credit. The amount of tax credits we will receive is entirely dependent on the amount of eligible expenses we incur. As we expect our eligible expenses to be higher in the fiscal year ended December 31, 2018, the level2023, we had cash and cash equivalents of tax credits recoverable is anticipated to be higher in 2018 compared to the fiscal year ended December 31, 2017. The new US tax reform Bill enacted in December 2017 is not expected to$3.4 million We have a material effect on the company’s results of operations or financial position.
| | | | Year ended December 31, | | |||||||||
| | | | 2016 | | | 2017 | | ||||||
| Cash and cash equivalents | | | | $ | 16,520 | | | | | $ | 23,910 | | |
| Working capital: | | | | | | | | | | | | | |
| Current assets | | | | | 19,617 | | | | | | 25,974 | | |
| Current liabilities | | | | | (5,259) | | | | | | (4,113) | | |
| Total working capital | | | | $ | 14,358 | | | | | $ | 21,861 | | |
| | |||||||||||||
| | | | Year Ended December 31, | | |||||||||
| | | | 2016 | | | 2017 | | ||||||
| Net cash used in operating activities | | | | $ | (10,079) | | | | | $ | (7,480) | | |
| Net cash provided by (used in) investing activities | | | | | 9 | | | | | | (13) | | |
| Net cash provided by financing activities | | | | | 6,594 | | | | | | 14,748 | | |
We believe that existing funds together with cash generated from operations, such as the R&D tax credit, and recent financing activities, are sufficient to satisfy our planned working capital, capital expenditures and other financial commitments through 2019. However, we do not currently have sufficient funds to complete development and commercialization of any of our drug candidates. Current business and capital market risks could have a detrimental effect on the availability of sources of
67
funding and our ability to access them in the future, which may delay or impede our progress of advancing our drugs currently in the clinical pipeline to approval by the FDA or EMA for commercialization. Additionally, we plan to continue to evaluate in-licensing and acquisition opportunities to gain access to new drugs or drug targets that would fit with our strategy. Any such transaction would likely increase our funding needs in the future.
Our future funding requirements will depend on many factors, including but not limited to:
| ● | the rate of progress and cost of our clinical trials, preclinical studies and other discovery and research and development activities; |
| ● | the costs associated with establishing manufacturing and commercialization capabilities; |
| ● | the costs of acquiring or investing in businesses, product candidates and technologies; |
| ● | the costs of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights; |
| ● | the costs and timing of seeking and obtaining FDA and EMA approvals; |
| ● | the effect of competing technological and market developments; and |
| ● | the economic and other terms and timing of any collaboration, licensing or other arrangements into which we may enter. |
Until we can generate a sufficient amount of product revenue to finance our cash requirements, which we may never do, we expect to finance future cash needs primarily through public or private equity offerings, debt financings or strategic collaborations. Although we are not reliant on institutional credit finance and therefore not subject to debt covenant compliance requirements or potential withdrawal of credit by banks, we are reliant on the availability of funds and activity in equity markets. We do not know whether additional funding will be available on acceptable terms, or at all. If we are not able to secure additional funding when needed, we may have to delay, reduce the scope of or eliminate one or more of our clinical trials or research and development programs or make changes to our operating plan. In addition, we may have to partner one or more of our product candidate programs at an earlier stage of development, which would lower the economic value of those programs to us.
Since our inception, we have relied primarily on the proceeds from sales of common and preferred equity securities to finance our operations and internal growth. Additional funding has come through research and development tax credits, government grants, the sale of product rights, interest on investments, licensing revenue, royalty income, and a limited amount of product revenue from operations discontinued in September 2012.
As discussed in Note 1 of the Notes to the Consolidated Financial Statements accompanying this Annual Report on Form 10-K, under ASC Topic 205-40, Presentation of Financial Statements - Going Concern, management is required at each reporting period to evaluate whether there are conditions and events, considered in the aggregate, that raise substantial doubt about an entity’s ability to continue as a going concern within one year after the date that the financial statements are issued. This evaluation initially does not take into consideration the potential mitigating effect of management’s plans that have not been fully implemented as of the date the financial statements are issued.
Based on our current operating plan, we anticipate that our cash and cash equivalents of $3.4 million as of December 31, 2023 will allow us to meet our liquidity requirements into April of 2024. As of March 21, 2024 our cash balance on hand was approximately $3.4 million. We continue to work to raise additional capital however as of the date of the Consolidated Financial Statements accompanying this Annual Report on Form 10-K, there is no guarantee that we will be able to raise additional funds to extend operations past April 2024. Our history of losses, our negative cash flows from operations, our liquidity resources currently on hand, and our dependence on the ability to obtain additional financing to fund our operations after the current resources are exhausted, about which there can be no certainty, have resulted in our assessment that there is substantial doubt about our ability to continue as a going concern for a period of at least twelve months from the issuance date of this Annual Report on Form 10-K. While we have plans in place to
68
mitigate this risk, which primarily consist of raising additional capital through a combination of public or private equity or debt financings or by entering into partnership agreements for further development of our drug candidates, there is no guarantee that we will be successful in these mitigation efforts.
Agreements to Sell Securities
On December 21, 2023, we entered into a securities purchase agreement (the “Securities Purchase Agreement”) with certain institutional investors (the “Purchasers”). Pursuant to the Securities Purchase Agreement, we agreed to sell in a registered direct offering (“Registered Direct Offering”) 168,500 shares (“Shares”) of our common stock, $0.001 par value per share ( “Common Stock”), and pre-funded warrants (“Pre-Funded Warrants”) to purchase up to 219,700 shares of Common Stock. The Pre-Funded Warrants have an exercise price of $0.001 per share and are immediately exercisable and can be exercised at any time after their original issuance until such Pre-Funded Warrants are exercised in full. Each Share was sold at a price of $3.315 and each Pre-Funded Warrant was sold at a price of $3.314 (equal to the purchase price per Share minus the exercise price of the Pre-Funded Warrant).
Pursuant to the Securities Purchase Agreement, in a concurrent private placement (together with the Registered Direct Offering, the “Offerings”), we also agreed to issue to the Purchasers unregistered warrants (“Common Warrants”) to purchase up to 388,200 shares of Common Stock. Each Common Warrant has an exercise price of $3.19 per share, is exercisable immediately following their original issuance and will expire seven years from the original issuance date. The closing of the offering occurred on December 26, 2023, and the net proceeds to us were approximately $1.0 million, after deducting placement agent fees and other offering expenses payable by us. Ladenburg Thalmann & Co. Inc. (the “Placement Agent”) acted as the exclusive placement agent for the Offerings, pursuant to a placement agency agreement dated December 21, 2023, by and between us and the Placement Agent.
On December 21, 2023, in a separate concurrent insider private placement (the “Insider Private Placement”), we also entered into a Securities Purchase Agreement with certain of our executive officers (the “Insider Securities Purchase Agreement”) pursuant to which we agreed to sell in a private placement (i) 6,070 shares of Common Stock and warrants to purchase 6,070 shares of Common Stock on the same terms as the Common Warrants issued to the Purchasers in the Offerings to Spiro Rombotis, our Chief Executive Officer, and (ii) 1,886 shares of Common Stock and warrants to purchase 1,886 shares of Common Stock on the same terms as the Common Warrants issued to the Purchasers in the Offerings to Paul McBarron, our Executive Vice President-Finance, Chief Financial Officer and Chief Operating Officer. Each such share of Common Stock and accompanying warrant was sold at a purchase price of $3.315, which was the same purchase price for the Shares sold in the Registered Direct Offering.
On August 12, 2021, we entered into a Controlled Equity Offering Sales Agreement (the “Sales Agreement”) with Cantor Fitzgerald & Co. ("Cantor"), pursuant to which we could issue and sell, from time to time, shares of our common stock having an aggregate offering price of up to $50.0 million through Cantor as the sales agent. Cantor could sell our common stock by any method permitted by law deemed to be an “at the market offering” as defined in Rule 415(a)(4) of the Securities Act.
On August 12, 2022, we became aware that the shelf registration statement on Form S-3 (file number 333-231923) (the “Registration Statement”) associated with this Sales Agreement had expired on June 21, 2022. Prior to becoming aware of the expiration, but following the expiration, we sold an aggregate of 132,473 shares of our common stock at market prices for aggregate proceeds of approximately $2,721,187. The sale of these shares were subject to potential rescission rights by certain stockholders. As a result of these rescission rights, we classified 207,807 shares (including 75,333 previously issued and outstanding shares sold for which the Company did not receive proceeds and which were reclassified to temporary equity as of September 30, 2022), with an aggregate redemption value of $4,494,496 of our common stock as stock outside stockholders equity. We also restated our loss per share as a result of $135,000 of associated fees not initially accounted for as accretion to the maximum redemption amount of the shares subject to potential rescission. During the third quarter of 2023, upon expiration of the rescission rights and with no claims or demands to exercise such rights, we reclassified all 207,807 shares back to permanent equity. In all periods presented, the shares subject to the rescission rights were treated as issued and outstanding for purposes of earnings per share and general financial reporting.
69
On August 15, 2022, due to expiry of the Registration Statement, the Sales Agreement was mutually terminated. A total of 218,738 shares, for gross proceeds of approximately $7.6 million, had been sold pursuant to the Sales Agreement, including 168,576 shares during the year ended December 31, 2022 for gross proceeds of approximately $3.4 million.
Dividend on Preferred Stock
On January 12, 2024, the Board of Directors declared a quarterly cash dividend in the amount of $0.15 per share on our 6% Convertible Exchangeable Preferred Stock. The cash dividend of approximately $50,000 was paid on February 1, 2024 to the holders of record of the 6% Convertible Exchangeable Preferred Stock as of the close of business on January 22, 2024.
Results of Operations
Years Ended December 31, 2023 and 2022
Results of Continuing Operations
Revenues
The following table summarizes the revenues for years ended December 31, 2023 and 2022 (in thousands except percentages):
| | | | | | | | | | | |
| | Year ended December 31, | | Difference | |||||||
|
| 2023 |
| 2022 | | $ |
| % | |||
Clinical trial supply | |
| 420 | |
| — | |
| 420 | | 100 |
Total Revenue | | $ | 420 | | $ | — | | $ | 420 | | 100 |
We recognize recognized $420,000 of revenue for the year ended December 31, 2023. This revenue relates to recovery of clinical manufacturing costs associated with an investigator sponsored study managed by Cedars-Sinai Medical Center. There were no revenues recognized for the comparative periods in 2022.
We do not expect to report revenue for the foreseeable future.
Research and development
We expense all research and development costs as they are incurred. Research and development expenses primarily include:
| ● | Clinical trial and regulatory-related costs; |
| ● | Payroll and personnel-related expenses, including consultants and contract research organizations; |
| ● | Preclinical studies and materials; |
| ● | Technology license costs; |
| ● | Stock-based compensation; and |
| ● | Rent and facility expenses for our office. |
70
The following table provides information with respect to our research and development expenditures for the years ended December 31, 2023 and 2022 (in thousands except percentages):
| | | | | | | | | | | |
| | Year Ended | |||||||||
| | December 31, | | Difference | |||||||
| | 2023 |
| 2022 |
| $ |
| % | |||
Transcriptional Regulation (fadraciclib) | | $ | 13,358 | | $ | 14,043 | | $ | (685) | | (5) |
Epigenetic/anti-mitotic (plogosertib) | | | 4,987 | | | 5,546 | | | (559) | | (10) |
Other research and development expenses | | | 810 | | | 685 | | | 125 | | 18 |
Total research and development expenses | | $ | 19,155 | | $ | 20,274 | | $ | (1,119) | | (6) |
Research and development expenses represented 74% and 73% of our operating expenses for the years ended December 31, 2023 and 2022, respectively.
Research and development expenses decreased by $1.1 million from $20.3 million for the year ended December 31, 2022 to $19.2 million for the year ended December 31, 2023. Expenditure for the transcriptional regulation program decreased by $0.7 million for the year ending December 31, 2023 relative to the respective comparative period. This decrease was primarily due to a decrease in clinical trial costs of $2.2 million associated with the temporary halt in the Phase 1/2 study in hematological malignancies and the completion of a bioequivalence study during the prior year, offset by an increase in manufacturing and other non-clinical expenditure of $1.6 million. Research and development expenses relating to plogosertib decreased by $0.6 million for the year ending December 31, 2023 relative to the respective comparative period. This decrease was primarily due to a decrease in clinical trial costs of $0.2 million associated with the progression of clinical trials for the evaluation of plogosertib in Phase 1/2 studies, and a decrease in manufacturing and other non-clinical expenditure of $0.4 million.
The future
We anticipate that overall research and development expenses for the year ended December 31, 2024 will decrease compared to the year ended December 31, 2023 as we focus on our Phase 1/2 programs in advanced solid tumors and lymphomas.
General and administrative
General and administrative expenses include costs for administrative personnel, legal and other professional expenses and general corporate expenses. The following table summarizes the total general and administrative expenses for the years ended December 31, 2023 and 2022 (in thousands except percentages):
| | | | | | | | | | | |
| | Year Ended | |||||||||
| | December 31, | | Difference | |||||||
|
| 2023 |
| 2022 |
| $ |
| % | |||
Total general and administrative expenses | | $ | 6,718 | | $ | 7,382 | | $ | (664) | | (9) |
Total general and administrative expenses represented 26% and 27% of our operating expenses for the years ended December 31, 2023 and 2022, respectively.
Our general and administrative expenditures decreased by $0.7 million from $7.4 million for the year ended December 31, 2022 to $6.7 million for the year ended December 31, 2023. This decrease was primarily due to a non-recurring $0.4 million cost associated with the Controlled Equity Offering Sales Agreement with Cantor Fitzgerald & Co. (the “Sales Agreement”) in the comparative prior period.
The future
We expect general and administrative expenditures for the year ended December 31, 2024 to remain relatively flat compared to the year ended December 31, 2023.
71
Other income (expense), net
The following table summarizes the other income (expense) for years ended December 31, 2023 and 2022 (in thousands except percentages):
| | | | | | | | | | | |
| | Year Ended | |||||||||
| | December 31, | | Difference | |||||||
| | 2023 |
| 2022 |
| $ |
| % | |||
Foreign exchange gains (losses) | | $ | (414) | | $ | 233 | | $ | (647) | | (278) |
Interest income | |
| 266 | |
| 210 | |
| 56 | | 27 |
Other income (expense), net | |
| 50 | |
| 1,298 | |
| (1,248) | | (96) |
Total other income (expense), net | | $ | (98) | | | 1,741 | | $ | (1,839) | | (106) |
Total other income, net, decreased by approximately $1.8 million from an income of approximately $1.7 million for the year ended December 31, 2022 to an expense of approximately $0.1 million for the year ended December 31, 2023. The decrease in other income primarily relates to royalties receivable under a December 2005 Asset Purchase Agreement, or APA, whereby Xcyte Therapies, Inc., or Xcyte (a business acquired by us in March 2006) sold through the APA and other related agreements certain assets and intellectual property which are not related to our product development plans to ThermoFisher Scientific Company, or TSC. Accordingly, we presented $50,000 and $1.3 million as other income received from TSC during the years ended December 31, 2023 and 2022 respectively. We have no knowledge of TSC’s activities and cannot predict when we may receive income under the APA, if any.
Foreign exchange gains (losses)
Foreign exchange gains decreased by $0.6 million to a loss of $0.4 million for the year ended December 31, 2023 compared to a gain of approximately $0.2 million for the year ended December 31, 2022.
We have intercompany loans in place between our parent company based in New Jersey and our subsidiary based in Scotland. The intercompany loans outstanding are not expected to be repaid in the foreseeable future and the nature of the funding advanced is of a long-term investment nature. Therefore, all unrealized foreign exchange gains or losses arising on the intercompany loans are recognized in other comprehensive income until repayment of the intercompany loan becomes foreseeable. Favorable unrealized foreign exchange movements related to intercompany loans resulted in a gain of $12.9 million for the year ended December 31, 2023 compared to a loss of $21.2 million for the year ended December 31, 2022.
The future
Other income (expense), net will continue to be impacted by changes in foreign exchange rates and the receipt of income under the APA. As we are not in control of sales made by TSC, we are unable to estimate the level and timing of income under the APA, if any.
As the funding advanced through intercompany loans is that of a long-term investment in nature, unrealized foreign exchange gains and losses on such funding will be recognized in other comprehensive income (loss) until repayment of the intercompany loan becomes foreseeable.
Income tax benefit
We record research and development tax credits within income taxes. Credit is taken for research and development tax credits, which are claimed from the United Kingdom’s taxation and customs authority (HMRC), in respect of qualifying research and development costs incurred.
72
The following table summarizes total income tax benefit from such credits for the years ended December 31, 2023 and 2022 (in thousands except percentages):
| | | | | | | | | | | |
| | Year Ended | |||||||||
| | December 31, | | Difference | |||||||
| | 2023 |
| 2022 |
| $ |
| % | |||
Total income tax benefit | | $ | 2,996 | | $ | 4,717 | | $ | (1,721) | | (36) |
The income tax benefit decreased significantly by approximately $1.7 million, from $4.7 million for the year ended December 31, 2022 to $3.0 million for the year ended December 31, 2023, due to legislative changes that took effect in April 2023. The level of tax credits recoverable is linked directly to qualifying research and development expenditure incurred in any one year and the availability of trading losses.
The future
We expect to continue to be eligible to receive United Kingdom research and development tax credits for the year ending December 31, 2024 and will continue to elect to receive payment of the tax credit. The legislative changes which took effect in April 2023 have been partially reversed in early 2024 resulting in additional income tax benefit of $0.8 million in relation to expenditure incurred during the year ended December 31, 2023 and receivable during the second quarter of 2024. Beyond 2024, there is no guarantee that we will be eligible to receive this tax credit or if eligible, any amount that may be receivable due to proposed changes by HMRC to the eligibility criteria would be correctly estimated. We expect research and development tax credits for the year ending December 31, 2024 to be lower compared to the year ended December 31, 2023.
Liquidity and Capital Resources
The following is a summary of our key liquidity measures as of December 31, 2023 and 2022 (in thousands):
| | | | | | |
| | | ||||
| | December 31, | ||||
|
| 2023 |
| 2022 | ||
Cash and cash equivalents | | $ | 3,378 | | $ | 18,345 |
Working capital: | | | | | | |
Current assets | | $ | 7,444 | | $ | 24,411 |
Current liabilities | |
| (8,161) | |
| (7,511) |
Total working capital | | $ | (717) | | $ | 16,900 |
Cash Flows
Cash provided by (used in) operating, investing and financing activities for the years ended December 31, 2023 and 2022 is summarized as follows (in thousands):
| | | | | | |
| | Year Ended December 31, | ||||
|
| 2023 |
| 2022 | ||
Net cash used in operating activities | | $ | (16,112) | | $ | (20,827) |
Net cash used in investing activities | |
| (6) | |
| (7) |
Net cash provided by financing activities | |
| 848 | |
| 2,998 |
73
Operating activities
Net cash used in operating activities decreased by $4.7 million, from $20.8 million for the year ended December 31, 2022 to $16.1 million for the year ended December 31, 2023. The decrease in cash used by operating activities was primarily the result of a change in working capital of $6.3 million, offset by an increase in net loss of $1.3 million. The $6.3 million change in working capital was primarily due to decreased balances in clinical trial deposits and receivables for research and development tax credits. A cash receipt of approximately $4.8 million in research and development tax credit was received during the year ended December 31, 2023.
Investing activities
Net cash used in investing activities decreased by $1,000 for the year ended December 31, 2023 due to a slight decrease in capital expenditures on information technology (“IT”) during the respective comparative period.
Financing activities
Net cash provided by financing activities was $0.8 million for the year ended December 31, 2023 as a direct result of receiving approximately:
| - | $1.0 million in net proceeds from the issuance of common stock and pre-funded warrants pursuant to the Registered Direct Offering, |
| - | $3.2 million in net proceeds from the issuance of common stock under a Controlled Equity Offering Sales Agreement with Cantor Fitzgerald & Co., |
The following table summarizes our long-term contractual obligations as of December 31, 20172023 (in thousands):
| | | | Payments Due by Period | | |||||||||||||||||||||||||||
| | | | Total | | | Less than 1 year | | | 1 – 3 years | | | 3 – 5 years | | | More than 5 years | | |||||||||||||||
Operating Lease Obligation(1) | | | | $ | 2,679 | | | | | $ | 348 | | | | | $ | 688 | | | | | $ | 687 | | | | | $ | 956 | | |
| | |||||||||||||||||||||||||||||||
| | | | | | | | | | | | | | | |
| | | | | Payments Due by Period | ||||||||||
|
| | |
| Less than |
| | |
| | |
| More than | ||
| | Total | | 1 year | | 1 – 3 years | | 3 – 5 years | | 5 years | |||||
Operating Lease Obligations (1) | | $ | 105 | | $ | 67 | | $ | 38 | | $ | — | | $ | — |
| (1) | Operating lease obligations relate primarily to leasing office space at our Berkeley Heights, New Jersey location. The lease for our Berkeley Heights location, which was entered into in April 2022, expires in July 2025. The table does not include any amouns relating to a short term lease for offices in Dundee, Scotland. |
Off-Balance Sheet Arrangements
Since our inception, we have not had any off-balance sheet arrangements or relationships with unconsolidated entities or financial partnerships, such as entities often referred to as structured finance or variable interest entities, which are typically established for the purpose of facilitating off-balance sheet arrangements or other contractually narrow or limited purposes.
Recently Issued Accounting Pronouncements
Please see Note 2 to the consolidated financial statements for a discussion of the potential effects that recently issued, but not yet effective, accounting standards will have on our financial statements when adopted in a future period.
74
Critical Accounting Policies
Our discussion and analysis of our financial condition and results of operations is based on our financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities and expenses and related disclosure of contingent assets and liabilities. We review our estimates on an ongoing basis. We base our estimates on historical experience and on various other factors that we believe to be reasonable under the circumstances. Actual results may differ from these estimates. We believe the judgments and estimates required by the following accounting policies to be critical in the preparation of our consolidated financial statements.
Our significant accounting policies are more fully described in Note 2 to our consolidated financial statements included elsewhere in this report. We believe the following critical accounting policies reflect our more significant estimates and assumptions used in the preparation of our consolidated financial statements.
Accrued Research and Development Costs
Accrued research and development costs comprise our best estimates related to the cost of clinical trials, laboratory, and manufacturing activities that were incurred, but not paid or invoiced, as of the end of a reporting period.
Data management and monitoring of our clinical trials are performed with the assistance of contract research organizations, or CROs, or clinical research associates, or CRAs, in accordance with our standard operating procedures. Typically, CROs and CRAs bill monthly for services performed, and others billor based upon milestones achieved. For outstanding amounts, weWe accrue unbilled clinical trial expenses based on estimates of the level of services performed each period. Costs of setting upMoreover, clinical trial sites for
We also perform outsourced laboratory and manufacturing activities. We accrue for unbilled laboratory and manufacturing activities performed by third parties based on estimates of their progress towards completing the clinical trial site is recognized upon executionrequested tasks.
As of the clinical trial agreements and expensed asDecember 31, 2023, we accrued $3.7 million research and development expenses.
When recording these accruals, we must make judgments about the progress of our various clinical activities. We (as well as our CROs and CRAs) are reliant on information being provided timely and accurately by the multitude of clinics and hospitals where the studies are being conducted, some of which are located internationally. We must also make estimates about the progress our third-party vendors are making towards completing laboratory and manufacturing activities.
Stock-based Compensation
We grant stock options, restricted stock units and restricted stock to officers, employees, directors and consultants under the Company’s 2015our 2018 Equity Incentive Plan or(the 2018 Plan) and the 2015 Plan. The 2015 Plan replaced the Company’s Amended and Restated2020 Inducement Equity Incentive Plan which expired in 2015.Plan. We measure compensation cost for all stock-based awards at fair value on date of grant and recognize compensation over the requisite service period. The fair value of restricted stock and restricted stock units is determined based on the number of shares granted and the quoted price of our common stock on the date of grant. The determination of grant-date fair value for stock option awards is estimated using an option-pricing model, which includes variables such as the expected volatility of our share price, the anticipated exercise behavior of our employees, interest rates, and dividend yields. These variables are projected based on our historical data, experience, and other factors. Changes in any of these variables could result in materialsignificant adjustments to the expensecosts recognized for share-based payments.
75
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
As a smaller reporting company, we are not required to provide information response to this item.
76
Item 8. Financial Statements and Supplementary Data
INDEX TO CYCLACEL PHARMACEUTICALS, INC. FINANCIAL STATEMENTS
| |||||||
| Page | ||||||
| | ||||||
Report of Independent Registered Public Accounting Firm (PCAOB ID: 49) | 78 | ||||||
80 | |||||||
81 | |||||||
82 | |||||||
83 | |||||||
84 | |||||||
85 | |||||||
77
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Stockholders and the Board of Directors of Cyclacel Pharmaceuticals, Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Cyclacel Pharmaceuticals, Inc. and its subsidiaries (the Company) as of December 31, 20162023 and 2017,2022, the related consolidated statements of operations (loss), other comprehensive loss, changes in stockholders’ equity and cash flows for the years then ended, and the related notes to the consolidated financial statements (collectively, the financial statements). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 20162023 and 2017,2022, and the results of its operations and its cash flows for the years then ended, in conformity with accounting principles generally accepted in the United States of America.
Substantial Doubt About the Company’s Ability to Continue as a Going Concern
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the financial statements, the Company does not currently have sufficient funds to complete development and commercialization and has a limited cash balance as of December 31, 2023. This raises substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matters
The critical audit matter communicated below is a matter arising from the current period audit of the financial statements that were communicated or required to be communicated to the audit committee and that: (1) relate to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the financial statements, taken as a whole, and we are not, by communicating the critical audit matters below, providing separate opinions on the critical audit matters or on the accounts or disclosures to which they relate.
Clinical Trial Accrual and Expenses
As discussed in Notes 2 and 9 of the consolidated financial statements, the Company’s total accrued expenses for research and development were $3.7 million at December 31, 2023, which included the estimated obligation for pre-clinical and clinical trial expenses incurred as of December 31, 2023, but not paid as of that date. The Company’s clinical trial expenses are based on the Company’s estimates of the level of services performed each period pursuant
78
agreements with third parties that conduct research and development on the Company’s behalf, which results in an accrual or prepaid at period end.
We identified the Company’s accrued clinical trial expenses as a critical audit matter because auditing the application of significant management judgment over the estimate of services provided but not yet invoiced required significant audit effort and a high degree of auditor judgment and subjectivity to evaluate the audit evidence obtained. Specifically, the amount of accrued clinical trial expenses recognized is dependent on the availability of information to make the estimate, including information from multiple sources, the level of effort expended as of the balance sheet date and the associated cost of such services. Additionally, due to the timing of invoicing received from third parties, the actual amounts incurred are not typically known on the date the Company issues its financial statements.
Our audit procedures to evaluate the Company’s estimate of services incurred as of period end pursuant to its clinical trials included, among others:
| ● | We tested the accuracy and completeness of the underlying data used in the estimates and evaluated the significant assumptions stated above that are used by management to estimate the recorded amounts. |
| ● | To assess the reasonableness of the significant assumptions, we obtained information regarding the nature and extent of progress of clinical trials from the Company’s research and development personnel that oversee the clinical trials and obtained information directly from third parties which indicated the third parties’ estimate of costs incurred to date. |
| ● | To evaluate the completeness and valuation of the accrual clinical trial expenses, we compared invoices received by the Company subsequent to December 31, 2023, to the amounts recognized by the Company as of that date. |
| ● | We inspected the Company’s contracts with third parties and any pending change orders to assess the impact to the amounts recorded. |
/s/ RSM US LLP
We have served as the Company’s auditor since 2013.
New York, New York
March 30, 201821, 2024
79
(In thousands, except share and per share amounts)
| | | | December 31, | | |||||||||
| | | | 2016 | | | 2017 | | ||||||
| ASSETS | | | | | | | | | | | | | |
| Current assets: | | | | | | | | | | | | | |
| Cash and cash equivalents | | | | $ | 16,520 | | | | | $ | 23,910 | | |
| Prepaid expenses and other current assets | | | | | 3,097 | | | | | $ | 2,064 | | |
| Total current assets | | | | | 19,617 | | | | | $ | 25,974 | | |
| Property and equipment, net | | | | | 45 | | | | | $ | 29 | | |
| Total assets | | | | $ | 19,662 | | | | | $ | 26,003 | | |
| LIABILITIES AND STOCKHOLDERS’ EQUITY | | | | | | | | | | | | | |
| Current liabilities: | | | | | | | | | | | | | |
| Accounts payable | | | | $ | 2,497 | | | | | $ | 1,558 | | |
| Accrued and other current liabilities | | | | | 2,762 | | | | | $ | 2,555 | | |
| Total current liabilities | | | | | 5,259 | | | | | $ | 4,113 | | |
| Other liabilities | | | | | 130 | | | | | $ | 124 | | |
| Total liabilities | | | | | 5,389 | | | | | $ | 4,237 | | |
| Commitments and contingencies (Note 9) | | | | | | | | | | | | | |
| Stockholders’ equity: | | | | | | | | | | | | | |
| Preferred stock, $0.001 par value; 5,000,000 shares authorized at December 31, 2016 and December 31, 2017; | | | | | | | | | | | | | |
| 6% Convertible Exchangeable preferred stock; 335,273 shares issued and outstanding at December 31, 2016 and December 31, 2017. Aggregate preference in liquidation of $4,006,512 at December 31, 2016 and December 31, 2017 | | | | | — | | | | | | — | | |
| Series A convertible preferred stock; 0 shares and 264 shares issued and outstanding at December 31, 2016 and December 31, 2017, respectively | | | | | — | | | | | | — | | |
| Common stock, $0.001 par value; 100,000,000 shares authorized at December 31, 2016 and December 31, 2017; 4,256,829 and 11,997,447 shares issued and outstanding at December 31, 2016 and December 31, 2017, respectively. | | | | | 4 | | | | | | 12 | | |
| Additional paid-in capital | | | | | 350,051 | | | | | $ | 365,057 | | |
| Accumulated other comprehensive loss | | | | | (743) | | | | | $ | (794) | | |
| Accumulated deficit | | | | | (335,039) | | | | | $ | (342,509) | | |
| Total stockholders’ equity | | | | | 14,273 | | | | | $ | 21,766 | | |
| Total liabilities and stockholders’ equity | | | | $ | 19,662 | | | | | $ | 26,003 | | |
| | |||||||||||||
| | | | | | |
|
| December 31, | | December 31, | ||
|
| 2023 |
| 2022 | ||
ASSETS | | | | | | |
Current assets: |
| |
|
| |
|
Cash and cash equivalents | | $ | 3,378 | | $ | 18,345 |
Prepaid expenses and other current assets | |
| 4,066 | |
| 6,066 |
Total current assets | |
| 7,444 | |
| 24,411 |
Property and equipment, net | |
| 9 | |
| 32 |
Right-of-use lease asset | | | 93 | | | 142 |
Non-current deposits | | | 1,259 | | | 3,465 |
Total assets | | $ | 8,805 | | $ | 28,050 |
LIABILITIES AND STOCKHOLDERS’ EQUITY | |
|
| |
|
|
Current liabilities: | |
|
| |
|
|
Accounts payable | | $ | 3,543 | | $ | 2,561 |
Accrued and other current liabilities | |
| 4,618 | |
| 4,950 |
Total current liabilities | |
| 8,161 | |
| 7,511 |
Lease liability | | | 37 | | | 106 |
Total liabilities | |
| 8,198 | |
| 7,617 |
Redeemable common stock, $0.001 par value; | | | | | | |
0 shares issued and outstanding at December 31, 2023 and 207,807 shares issued and outstanding at December 31, 2022 (Note 11) | | | — | |
| 4,494 |
| | | | | | |
Stockholders’ equity: | | | | | | |
Preferred stock, $0.001 par value; 5,000,000 shares authorized at December 31, 2023 and December 31, 2022; | |
| — | |
| — |
6% Convertible Exchangeable preferred stock; 335,273 shares issued and outstanding at December 31, 2023 and December 31, 2022. Aggregate preference in liquidation of $4,006,512 as of December 31, 2023 and December 31, 2022 | |
| — | |
| — |
Series A convertible preferred stock, $0.001 par value; 264 shares issued and outstanding at December 31, 2023 and December 31, 2022 | |
| — | |
| — |
Series B convertible preferred stock, $0.001 par value; 119,000 shares issued and outstanding at December 31, 2023 and 237,745 shares issued and outstanding at December 31, 2022 | |
| — | |
| — |
Common stock, $0.001 par value; 100,000,000 shares authorized at December 31, 2023 and December 31, 2022; 1,058,892 shares issued and outstanding at December 31, 2023 and 628,139 shares issued and outstanding at December 31, 2022 | |
| 1 | |
| 1 |
Additional paid-in capital | |
| 429,796 | |
| 422,981 |
Accumulated other comprehensive loss | |
| (908) | |
| (1,316) |
Accumulated deficit | |
| (428,282) | |
| (405,727) |
Total stockholders’ equity | |
| 607 | |
| 15,939 |
Total liabilities and stockholders’ equity | | $ | 8,805 | | $ | 28,050 |
The accompanying notes are an integral part of these consolidated financial statements.
80
(In thousands, except share and per share amounts)
| | | | Year Ended December 31, | | |||||||||
| | | | 2016 | | | 2017 | | ||||||
| Revenues: | | | | | | | | | | | | | |
| Grant revenue | | | | $ | 843 | | | | | $ | — | | |
| Operating expenses: | | | | | | | | | | | | | |
| Research and development | | | | | 9,477 | | | | | | 4,237 | | |
| General and administrative | | | | | 5,516 | | | | | | 5,254 | | |
| Total operating expenses | | | | | 14,993 | | | | | | 9,491 | | |
| Operating loss | | | | | (14,150) | | | | | | (9,491) | | |
| Other income (expense): | | | | | | | | | | | | | |
| Foreign exchange gains (losses) | | | | | 273 | | | | | | (39) | | |
| Interest income | | | | | 37 | | | | | | 118 | | |
| Other income, net | | | | | 66 | | | | | | 949 | | |
| Total other income (expense), net | | | | | 376 | | | | | | 1,028 | | |
| Loss from continuing operations before taxes | | | | | (13,774) | | | | | | (8,463) | | |
| Income tax benefit | | | | | 1,983 | | | | | | 993 | | |
| Net loss | | | | | (11,791) | | | | | | (7,470) | | |
| Dividend on convertible exchangeable preferred shares | | | | | (200) | | | | | | (201) | | |
| Beneficial conversion feature of Series A convertible stock | | | | | — | | | | | | (3,638) | | |
| Conversion of Series A convertible preferred stock | | | | | — | | | | | | (3,537) | | |
| Net loss applicable to common shareholders | | | | $ | (11,991) | | | | | $ | (14,846) | | |
| Basic and diluted earnings per common share: | | | | | | | | | | | | | |
| Net loss per share – basic and diluted | | | | $ | (3.50) | | | | | $ | (1.95) | | |
| Weighted average common shares outstanding | | | | | 3,424,976 | | | | | | 7,631,152 | | |
| | |||||||||||||
| | | | | | |
|
| Year Ended | ||||
| | December 31, | ||||
| | 2023 |
| 2022 | ||
Revenues: | | | | | | |
Clinical trial supply | | $ | 420 | | $ | — |
Revenues | | | 420 | | | — |
| | | | | | |
Operating expenses: | |
|
| |
|
|
Research and development | |
| 19,155 | |
| 20,274 |
General and administrative | |
| 6,718 | |
| 7,382 |
Total operating expenses | |
| 25,873 | |
| 27,656 |
Operating loss | |
| (25,453) | |
| (27,656) |
Other income (expense): | |
|
| |
|
|
Foreign exchange gains (losses) | |
| (414) | |
| 233 |
Interest income | |
| 266 | |
| 210 |
Other income (expense), net | |
| 50 | |
| 1,298 |
Total other income (expense), net | |
| (98) | |
| 1,741 |
Loss before taxes | |
| (25,551) | |
| (25,915) |
Income tax benefit | |
| 2,996 | |
| 4,717 |
Net loss | |
| (22,555) | |
| (21,198) |
Dividend on convertible exchangeable preferred shares | |
| (201) | |
| (201) |
Net loss applicable to common shareholders | | $ | (22,756) | | $ | (21,399) |
Basic and diluted earnings per common share: | |
|
| |
|
|
Net loss per share – basic and diluted (common shareholders) | | $ | (26.75) | | $ | (28.70) |
Net loss per share – basic and diluted (redeemable common shareholders) | | $ | — | | $ | (27.24) |
The accompanying notes are an integral part of these consolidated financial statements.
81
(In thousands)
| | | | Year Ended December 31, | | |||||||||
| | | | 2016 | | | 2017 | | ||||||
| Net loss | | | | $ | (11,791) | | | | | $ | (7,470) | | |
| Translation adjustment | | | | | 27,204 | | | | | | (14,687) | | |
| Unrealized foreign exchange gain (loss) on intercompany loans | | | | | (27,351) | | | | | | 14,636 | | |
| Comprehensive loss | | | | $ | (11,938) | | | | | $ | (7,521) | | |
| | |||||||||||||
| | | | | | |
|
| Year Ended | ||||
| | December 31, | ||||
|
| 2023 |
| 2022 | ||
Net loss | | $ | (22,555) | | $ | (21,198) |
Translation adjustment | |
| (12,142) | |
| 20,598 |
Unrealized foreign exchange gain (loss) on intercompany loans | |
| 12,550 | |
| (21,166) |
Comprehensive loss | | $ | (22,147) | | $ | (21,766) |
The accompanying notes are an integral part of these consolidated financial statements.
82
(In thousands, except share amounts)
| | | | Preferred Stock | | | Common Stock | | | Additional Paid-in Capital | | | Accumulated Other Comprehensive Loss | | | Accumulated Deficit | | | Total Stockholders’ Equity | | ||||||||||||||||||||||||||||||
| | | | Shares | | | Amount | | | Shares | | | Amount | | ||||||||||||||||||||||||||||||||||||
| Balances at December 31, 2015 | | | | | 335,273 | | | | | $ | — | | | | | | 2,965,208 | | | | | $ | 3 | | | | | $ | 342,587 | | | | | $ | (596) | | | | | $ | (323,159) | | | | | $ | 18,835 | | |
| Cumulative effect of change in Accounting | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Principle for ASU 2016-09 | | | | | — | | | | | | — | | | | | | — | | | | | | — | | | | | | 89 | | | | | | — | | | | | | (89) | | | | | | — | | |
| Issue of common stock on At Market Issuance sales agreement | | | | | — | | | | | | — | | | | | | 1,291,621 | | | | | | 1 | | | | | | 6,793 | | | | | | — | | | | | | — | | | | | | 6,794 | | |
| Stock-based compensation | | | | | — | | | | | | — | | | | | | — | | | | | | — | | | | | | 782 | | | | | | — | | | | | | — | | | | | | 782 | | |
| Preferred stock dividends | | | | | — | | | | | | — | | | | | | — | | | | | | — | | | | | | (200) | | | | | | — | | | | | | — | | | | | | (200) | | |
| Unrealized foreign exchange on intercompany loans | | | | | — | | | | | | — | | | | | | — | | | | | | — | | | | | | — | | | | | | (27,351) | | | | | | — | | | | | | (27,351) | | |
| Translation adjustment | | | | | — | | | | | | — | | | | | | — | | | | | | — | | | | | | — | | | | | | 27,204 | | | | | | — | | | | | | 27,204 | | |
| Net loss | | | | | — | | | | | | — | | | | | | — | | | | | | — | | | | | | — | | | | | | — | | | | | | (11,791) | | | | | | (11,791) | | |
| Balances at December 31, 2016 | | | | | 335,273 | | | | | $ | — | | | | | | 4,256,829 | | | | | $ | 4 | | | | | $ | 350,051 | | | | | $ | (743) | | | | | $ | (335,039) | | | | | $ | 14,273 | | |
| Issue of common stock, preferred stock and associated warrants on underwritten offering, net of expenses | | | | | 8,872 | | | | | | — | | | | | | 3,154,000 | | | | | | 3 | | | | | | 13,681 | | | | | | — | | | | | | — | | | | | | 13,684 | | |
| Series A Preferred stock conversions | | | | | (8,608) | | | | | | — | | | | | | 4,304,000 | | | | | | 4 | | | | | | (4) | | | | | | — | | | | | | — | | | | | | — | | |
| Warrant exercise | | | | | — | | | | | | — | | | | | | 99,500 | | | | | | — | | | | | | 199 | | | | | | — | | | | | | — | | | | | | 199 | | |
| Issue of common stock on At Market Issuance sales agreement | | | | | — | | | | | | — | | | | | | 183,118 | | | | | | 1 | | | | | | 1,065 | | | | | | — | | | | | | — | | | | | | 1,066 | | |
| Stock-based compensation | | | | | — | | | | | | — | | | | | | — | | | | | | — | | | | | | 266 | | | | | | — | | | | | | — | | | | | | 266 | | |
| Preferred stock dividends | | | | | — | | | | | | — | | | | | | — | | | | | | — | | | | | | (201) | | | | | | — | | | | | | — | | | | | | (201) | | |
| Unrealized foreign exchange on intercompany loans | | | | | — | | | | | | — | | | | | | — | | | | | | — | | | | | | — | | | | | | 14,636 | | | | | | — | | | | | | 14,636 | | |
| Translation adjustment | | | | | — | | | | | | — | | | | | | — | | | | | | — | | | | | | — | | | | | | (14,687) | | | | | | — | | | | | | (14,687) | | |
| Loss for the period | | | | | — | | | | | | — | | | | | | — | | | | | | — | | | | | | — | | | | | | — | | | | | | (7,470) | | | | | | (7,470) | | |
| Balances at December 31, 2017 | | | | | 335,537 | | | | | $ | — | | | | | | 11,997,447 | | | | | $ | 12 | | | | | $ | 365,057 | | | | | $ | (794) | | | | | $ | (342,509) | | | | | $ | 21,766 | | |
| | |||||||||||||||||||||||||||||||||||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Accumulated | | | | | | | |
| | | | | | | | | | |
| Additional |
| Other | | | |
| Total | |||
|
| Preferred Stock |
| Common Stock |
| Paid-in |
| Comprehensive |
| Accumulated |
| Stockholders’ | ||||||||||
|
| Shares |
| Amount |
| Shares |
| Amount |
| Capital |
| Loss |
| Deficit |
| Equity | ||||||
Balances at December 31, 2021 | | 573,282 | | $ | — |
| 666,209 | | $ | 1 | | $ | 422,969 | | $ | (748) | | $ | (384,529) | | $ | 37,693 |
| | | | | | | | | | | | | | | | | | | | | | |
Issue of common stock in At Market Issuance sales agreement, net of expenses | | — | |
| — |
| 36,103 | |
| — | |
| 478 | |
| — | |
| — | |
| 478 |
Reclassification of redeemable common stock |
| — | |
| — |
| (75,333) | |
| — | |
| (1,636) | |
| — | |
| — | |
| (1,636) |
Accretion on redeemable common stock |
| — | |
| — |
| — | |
| — | |
| (135) | |
| — | |
| — | |
| (135) |
Stock-based compensation |
| — | |
| — |
| 1,161 | |
| — | |
| 1,506 | |
| — | |
| — | |
| 1,506 |
Preferred stock dividends |
| — | |
| — |
| | |
| | |
| (201) | |
| — | |
| — | |
| (201) |
Unrealized foreign exchange on intercompany loans | | — | |
| — |
| — | |
| — | |
| — | |
| (21,166) | |
| — | |
| (21,166) |
Translation adjustment | | — | |
| — |
| — | |
| — | |
| — | |
| 20,598 | |
| — | |
| 20,598 |
Loss for the period | | — | |
| — |
| — | |
| — | |
| — | |
| — | |
| (21,198) | |
| (21,198) |
Balances at December 31, 2022 |
| 573,282 | | $ | — |
| 628,139 | | $ | 1 | | $ | 422,981 | | $ | (1,316) | | $ | (405,727) | | $ | 15,939 |
Issue of common stock and pre-funded warrants in Securities Purchase Agreement In Registered Direct Offering, net of expenses | | — | | | — | | 176,456 | | | — | | | 1,049 | | | — | | | — | | | 1,049 |
Conversion of series B Preferred stock | | (118,745) | | | — | | 39,582 | | | — | | | — | | | — | | | — | |
| — |
Reclassification of redeemable common stock |
| — | | | — | | 207,806 | | | — | | | 4,494 | | | — | | | — | |
| 4,494 |
Stock-based compensation | | — | | | — | | 6,909 | | | — | | | 1,473 | | | — | | | — | |
| 1,473 |
Preferred stock dividends |
| — | | | — | | — | | | — | | | (201) | | | — | | | — | |
| (201) |
Unrealized foreign exchange on intercompany loans |
| — | | | — | | — | | | — | | | — | | | 12,550 | | | — | |
| 12,550 |
Translation adjustment | | — | | | — | | — | | | — | | | — | | | (12,142) | | | — | | | (12,142) |
Loss for the period |
| — | | | — | | — | | | — | | | — | | | — | | | (22,555) | |
| (22,555) |
Balances at December 31, 2023 |
| 454,537 | | $ | — |
| 1,058,892 | | $ | 1 | | $ | 429,796 | | $ | (908) | | $ | (428,282) | | $ | 607 |
| | | | | | | | | | | | | | | | | | | | | | |
The accompanying notes are an integral part of these consolidated financial statements.
83
(In thousands)
| | | | Year Ended December 31, | | |||||||||
| | | | 2016 | | | 2017 | | ||||||
| Cash flows from operating activities: | | | | | | | | | | | | | |
| Net loss | | | | $ | (11,791) | | | | | $ | (7,470) | | |
| Adjustments to reconcile net loss to net cash used in operating activities: | | | | | | | | | | | | | |
| Depreciation expense | | | | | 133 | | | | | | 32 | | |
| Gain on disposal of property and equipment | | | | | (12) | | | | | | — | | |
| Stock-based compensation expense | | | | | 782 | | | | | | 266 | | |
| Changes in operating assets and liabilities: | | | | | | | | | | | | | |
| Prepaid expenses and other assets | | | | | 439 | | | | | | 1,232 | | |
| Accounts payable and other current liabilities | | | | | 370 | | | | | | (1,540) | | |
| Net cash used in operating activities | | | | | (10,079) | | | | | | (7,480) | | |
| Cash flows from investing activities: | | | | | | | | | | | | | |
| Purchases of property and equipment | | | | | (3) | | | | | | (13) | | |
| Proceeds from sale of property and equipment | | | | | 12 | | | | | | — | | |
| Net cash provided by (used in) investing activities | | | | | 9 | | | | | | (13) | | |
| Cash flows from financing activities: | | | | | | | | | | | | | |
| Proceeds from issuance of equity securities, net of issuance costs | | | | | 6,794 | | | | | | 14,749 | | |
| Proceeds from the exercise of stock options and warrants, net of issuance costs | | | | | — | | | | | | 200 | | |
| Payment of preferred stock dividend | | | | | (200) | | | | | | (201) | | |
| Net cash provided by financing activities | | | | | 6,594 | | | | | | 14,748 | | |
| Effect of exchange rate changes on cash and cash equivalents | | | | | (444) | | | | | | 135 | | |
| Net decrease in cash and cash equivalents | | | | | (3,920) | | | | | | 7,390 | | |
| Cash and cash equivalents at beginning of period | | | | | 20,440 | | | | | | 16,520 | | |
| Cash and cash equivalents at end of period | | | | $ | 16,520 | | | | | $ | 23,910 | | |
| Supplemental cash flow information: | | | | | | | | | | | | | |
| Cash received during the period for: | | | | | | | | | | | | | |
| Interest | | | | | 38 | | | | | | 118 | | |
| Taxes | | | | | 1,965 | | | | | | 1,815 | | |
| Non cash financing activities: | | | | | | | | | | | | | |
| Accrual of preferred stock dividends | | | | | 50 | | | | | | 50 | | |
| | | | | | |
| | Year Ended | ||||
| | December 31, | ||||
|
| 2023 |
| 2022 | ||
Operating activities: | | |
| | |
|
Net loss | | $ | (22,555) | | $ | (21,198) |
Adjustments to reconcile net loss to net cash used in operating activities: | | |
| | |
|
Depreciation | | | 31 | | | 32 |
Stock-based compensation | | | 1,473 | | | 1,506 |
Changes in lease liability | | | (69) | | | 40 |
Changes in operating assets and liabilities: | | | | | | |
Prepaid expenses and other assets | | | 4,712 | | | (3,784) |
Accounts payable, accrued and other current liabilities | | | 296 | | | 2,577 |
Net cash used in operating activities | | | (16,112) | | | (20,827) |
Investing activities: | | |
| | |
|
Purchase of property, plant and equipment | | | (6) | | | (7) |
Net cash used in investing activities | | | (6) | | | (7) |
Financing activities: | | |
| | |
|
Proceeds, net of issuance costs, from issuing common stock and pre-funded warrants | | | 1,049 | | | 3,199 |
Payment of preferred stock dividend | | | (201) | | | (201) |
Net cash (used in) provided by financing activities | | | 848 | | | 2,998 |
| | | | | | |
Effect of exchange rate changes on cash and cash equivalents | | | 303 | | | (378) |
Net (decrease) in cash and cash equivalents | | | (14,967) | | | (18,214) |
Cash and cash equivalents, beginning of period | | | 18,345 | | | 36,559 |
Cash and cash equivalents, end of period | | $ | 3,378 | | $ | 18,345 |
Supplemental cash flow information: | | |
| | |
|
Cash received during the period for: | | |
| | |
|
Interest | | $ | 266 | | $ | 211 |
Research & Development Tax Credits | | $ | 4,846 | | $ | 3,312 |
| | | | | | |
Cash paid during the period for: | | | | | | |
Taxes | | $ | 2 | | $ | 2 |
| | | | | | |
Non cash financing activities: | | |
| | |
|
Accrual of preferred stock dividends | | $ | 50 | | $ | 50 |
Accretion on redeemable common stock | | $ | — | | $ | 135 |
The accompanying notes are an integral part of these consolidated financial statements.
84
Cyclacel Pharmaceuticals, Inc., (“Cyclacel” or “the Company”) is a clinical-stage biopharmaceutical company usingdeveloping innovative cancer medicines based on cell cycle, transcriptional regulation and DNA damage response biology to develop innovative, targeted medicines for cancer and other proliferative diseases. Cyclacel’s transcriptional regulation program is evaluating CYC065, a CDK inhibitor, in patients with advanced cancers. The DNA damage response program is evaluating a sequential regimen of sapacitabine and seliciclib, a CDK inhibitor, in patients with BRCA positive, advanced solid cancers. mitosis control biology. Cyclacel is analyzing stratifieda pioneer company in the field of cancer cell cycle biology with a vision to improve patient healthcare by translating insights in cancer biology into medicines that can overcome resistance and exploratory subgroups fromultimately increase a Phase 3 study of sapacitabine in elderly patients with AML. Cyclacel’s strategy is to build a diversified biopharmaceutical business focused in hematology and oncology based on a pipeline of novel drug candidates.
As of December 31, 2017,2023, substantially all efforts of the Company to date have been devoted to performing research and development, conducting clinical trials, developing and acquiring intellectual property, raising capital and recruiting and training personnel.
The Company is subject to risks and uncertainties common to early-stage companies in the biopharmaceutical industry, including, but not limited to, the fact that drug candidates developed by the Company typically will require approvals or clearances from the FDA, EMAU.S. Food and Drug Administration, the European Medicines Agency or other similar regulatory agencies in other countries prior to commercial sales. There can be no assurance that the Company’s drug candidates will receive any of the required approvals or clearances. If any of the Company wasCompany’s drug candidates are denied approval or clearance or such approval wasis delayed, or if the Company is unable to obtain the necessary financing to complete development and approval, there will be a material adverse impact on the Company’s financial condition and results of operations. The Company has relied upon government grants to fund its earlier stage programs and does not expect to be able to continue to be successful in obtaining government grants to fund the Company’s research and development expenditure.
Through December 31, 2017,2023, the Company has funded all of its operations and capital expenditures with proceeds from the issuance of public equity securities, private placements of securities, government grants, research and development tax credits, interest on investments, royalty income, product revenue and licensing revenue. The Company has incurred recurring losses since its inception, including net losses of $11.8$22.5 million and $7.5$21.2 million for the years ended December 31, 20162023 and 2017,2022, respectively. As of December 31, 2017,2023, the Company had an accumulated deficit of $342.5$428.3 million. The Company expects to continue to generate operating losses for the foreseeable future due to, among other things, costs related to the clinical development of its drug candidates, its preclinical programs and its administrative organization.
Going Concern
Pursuant to the requirements of Accounting Standard Codification (ASC) 205-40, Presentation of Financial Statements-Going Concern, management is required at each reporting period to evaluate whether there are no conditions or events, considered in the aggregate, that raise substantial doubt about an entity’s ability to continue as a going concern within one year after the date that the financial statements are issued. This evaluation initially does not take into consideration the potential mitigating effect of management’s plans that have not been fully implemented as of the date the financial statements are issued. When substantial doubt exists under this methodology, management evaluates whether the mitigating effects of its plans sufficiently alleviate the substantial doubt about the Company’s ability to continue as a going concern. The mitigating effect of management’s plans, however, is only considered if both (1) it is probable that the plans will be effectively implemented within one year after the date that the financial statements are issued, and (2) it is probable that the plans, when implemented, will mitigate the relevant conditions or events that raise substantial doubt about the entity’s ability to continue as a going concern for one year after the date that these financial statements are issued. In performing its analysis, management excluded certain elements of its operating plan that cannot be considered probable. Under ASC 205-40, the future receipts of potential funding from future equity or debt issuances or by entering into partnership agreements cannot be considered probable at this time because these plans are not entirely within the Company’s control nor have they been approved by the Board of Directors as of the date of these consolidated financial statements.
85
Based on the Company’s current operating plan, it is anticipated that cash and cash equivalents of $3.4 million as of December 31, 2023 will allow it to meet liquidity requirements into April 2024. As of March 21, 2024 the Company’s cash balance on hand is approximately $3.4 million. The Company continues to work to raise additional capital however as of the date of these financial statements there is no guarantee that the Company will be able to raise additional funds to extend operations past April 2024. The Company’s history of losses, negative cash flows from operations, liquid resources currently on hand, and dependence on the ability to obtain additional financing to fund its operations, about which there can be no certainty, have resulted in the assessment that there is substantial doubt about the Company’s ability to continue as a going concern for a period of at least one yeartwelve months from the issuance date of these financial statements. While the Company has plans in place to mitigate this risk, which primarily consist of raising additional capital through a combination of public or private equity or debt financings or by entering into partnership agreements for further development of our drug candidates, there is no guarantee that it will be successful in these mitigation efforts.The accompanying consolidated financial statements are issued. The Company expects that its cashhave been prepared on a going concern basis, which contemplates realization of $23.9 million as of December 31, 2017 will be sufficient to fund its operating expenses and capital expenditure requirements through to the end of 2019.
Basis of Presentation
The accompanying consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America, (“GAAP”)or GAAP and include the financial statements of Cyclacel Pharmaceuticals, Inc. and all of the Company’s wholly owned subsidiaries. All intercompany accounts and transactions have been eliminated. Certain amounts in
Reverse Stock Split
The Company effected a 15:1 reverse stock split of the prior year’s financial statements haveCompany’s common stock on December 18, 2023 (the “Reverse Stock Split”). All share and per share information has been reclassifiedretroactively adjusted to conformgive effect to the current year’s presentation.
2.
Use of Estimates
The preparation of financial statements in accordance with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets, liabilities and related disclosures of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Critical estimates include inputs used to determine clinical trial accruals and stock-based compensation expense and the recognition of revenue.expense. Cyclacel reviews its estimates on an ongoing basis. The estimates are based on historical experience and on various other assumptions that the Company believes to be reasonable under the circumstances. Actual results may differ from these estimates. Cyclacel believes the judgments and estimates required by the following accounting policies to be significant in the preparation of the Company’s consolidated financial statements.
Foreign Currency and Currency Translation
Transactions that are denominated in a foreign currency are remeasured into the functional currency at the current exchange rate on the date of the transaction. Any foreign currency-denominated monetary assets and liabilities are subsequently remeasured at current exchange rates, with gains or losses recognized as foreign exchange (losses) gains in the statement of operations.
The assets and liabilities of the Company’s international subsidiary are translated from its functional currency into United States dollars at exchange rates prevailing at the balance sheet date. Average rates of exchange during the period are used to translate the statement of operations, while historical rates of exchange are used to translate any equity transactions. Translation adjustments arising on consolidation due to differences between average rates and balance sheet rates, as well as unrealized foreign exchange gains or losses arising from translation of intercompany loans for which
86
settlement is not not planned or anticipated in the foreseeable future and that are of a long-term-investment nature, are recorded in other comprehensive loss.
Cash and Cash Equivalents
Financial instruments that potentially expose the Company to concentrations of credit risk consist primarily of cash and cash equivalents. The Company considers all highly liquid investments with an original maturity of three months or less at the time of initial purchase to be cash equivalents. The objectives of the Company’s cash management policy are to safeguard and preserve funds, to maintain liquidity sufficient to meet Cyclacel’s cash flow requirements and to attain a market rate of return. The Company deposits its cash in financial institutions that it believes have high credit quality and has not experienced any losses on such accounts and does not believe it is exposed to any significant credit risk on cash and cash equivalents.
The Company’s cash and cash equivalents balance at December 31, 20172023 was $23.9$3.4 million and it maintains its cash accounts in several entities both within the United States and the United Kingdom. The total cash balances for amounts held in the United States are insured by the Federal Deposit Insurance Corporation, (“FDIC”)or FDIC up to $250,000 per account. The Company has cash balances exceeding the balance insured by the FDIC that totaled approximately $23.0$2.4 million at December 31, 2017.2023. The total cash balances for amounts held in the United Kingdom are insured by the UK Government Financial Services Compensation Scheme, (“FSCS”)or FSCS up to £75,000£85,000 per account. The Company has cash balances exceeding the balance insured by the FSCS that totaled approximately $0.5$0.6 million at December 31, 2017.
Property and Equipment
The components of property and equipment are stated at cost and depreciated on a straight-line basis over the estimated useful lives of the related assets, which are generally three to five years. Amortization of leasehold improvements is performed using the straight-line method over the shorter of the remaining lease term or the estimated useful life of the related assets, currently between five and fifteen years.assets. Upon sale or retirement of assets, the costs and related accumulated depreciation and amortization are removed from the balance sheet and the resulting gain or loss on sale is reflected as a component of operating income or loss. Expenditures for maintenance and repairs are charged to operating expenses as incurred.
Impairment of Long-lived Assets
The Company reviews property and equipment for impairment whenever events or changes in business circumstances indicate that the carrying amount of the assets may not be fully recoverable. The Company assesses the recoverability of the potentially affected long-lived assets by determining whether the carrying value of such assets can be recovered through undiscounted future operating cash flows.
Impairment, if any, is measured as the amount by which the carrying amount of a long-lived asset (oror asset group)group exceeds its fair value.
Fair Value of Financial Instruments
Fair value is defined as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. Valuation techniques used to measure fair value must maximize the use of observable inputs and minimize the use of unobservable inputs. Assets and liabilities measured at fair value are classified and disclosed in one of the following three levels of the fair value hierarchy, of which the first two are considered observable and the last is considered unobservable:
| ● | Level 1 — Quoted prices in active markets for identical assets or liabilities. |
| ● | Level 2 — Observable inputs (other than Level 1 quoted prices), such as quoted prices in active markets for similar assets or liabilities, quoted prices in markets that are not active for identical or similar assets or liabilities, or other inputs that are observable or can be corroborated by observable market data. |
87
| ● | Level 3 — Unobservable inputs that are supported by little or no market activity that are significant to determining the fair value of the assets or liabilities, including pricing models, discounted cash flow methodologies and similar techniques. |
The carrying values of cash and cash equivalents, other receivables, accounts payable and accrued expenses approximate their fair values due to the short-term nature of these assets and liabilities.
Segments
The Company is managed and operated as one business which is focused on using cell cycle, transcriptional regulation and DNA damage responsemitosis control biology to develop innovative, targeted medicines for cancer and other proliferative diseases. The entire business is managed by a single management team that reports to the Chief Executive Officer. The Company does not operate separate lines of business with respect to any of its products or product candidates and the Company does not prepare discrete financial information with respect to separate products or product candidates or by location. Accordingly, the Company views its business as one reportable operating segment with development operations in two geographic areas, namely the United States and the United Kingdom.
Revenue Recognition
The Company records as deferredrecognizes revenue any amounts received prior to satisfying the revenue recognition criteria. Deferred revenue not expected to be recognized within the next twelve months is reported as non-current deferred revenue.in accordance with Accounting Standards Codification (ASC) 606, Revenue from Contracts with Customers. The Company recognized $0 and $0.2 million deferred$420,000 of revenue as offor the year ended December 31, 2016 and 2017 respectively. The deferred2023. This revenue reported as of December 31, 2017 relates to a prepayment received from ManRos Therapeutics SArecovery of clinical manufacturing costs associated with an investigator sponsored study managed by Cedars-Sinai Medical Center. There were no revenues recognized for a development milestone that is not expected to be achieved until mid-2018 (see Note 3 for additional details).
Other Income
Other income is primarily related to royalty income received under a historical Asset Purchase Agreement for activities which are not part of the Company’s ongoing operations and activities.
Research and Development Expenditures
Research and development expenses consist primarily of costs associated with the development of the Company’s product candidates, including upfront fees, milestones, compensation and other expenses for research and development personnel, supplies and development materials, costs for consultants and related contract research, facility costs and depreciation. Expenditures relating to research and development are expensed as incurred.
Clinical Trial Accounting
Data management and monitoring of the Company’s clinical trials are performed with the assistance of contract research organizations, (“CROs”)or CROs or clinical research associates, (“CRAs”)or CRAs in accordance with the Company’s standard operating procedures. Typically, CROs and CRAs bill monthly for services performed, and others bill based upon milestones achieved. For outstanding amounts, theThe Company accrues unbilled clinical trial expenses based on estimates of the level of services performed each period. Costs of setting up clinical trial sites for participation in the trials are recognized upon execution of the clinical trial agreement and expensed immediately as research and development expenses. Clinical trial costs related to patient enrollment are accrued as patients are entered into and progress through the trial.
Patent Costs
Patent prosecution costs are charged to general and administrative expenses as incurred as recoverability of such expenditure is uncertain.
88
Leases
The costsCompany accounts for lease contracts in accordance with ASC 842. As of December 31, 2023 and 2022, all of the Company’s leases are classified as operating leases.
The Company recognizes an asset for the right to use an underlying leased asset for the lease term and records lease liabilities based on the present value of the Company’s obligation to make lease payments under the lease. As the Company’s leases do not indicate an implicit rate, the Company uses a best estimate of its incremental borrowing rate to discount the future lease payments. The Company estimates its incremental borrowing rate based on observable information about risk-free interest rates that are the same tenure as the lease term, adjusted for various factors, including the effects of assumed collateral, the nature of how a loan would be repaid (e.g., amortizing versus bullet), and the Company’s credit risk.
The Company evaluates options included in its lease agreements to extend or terminate the lease. The Company will reflect the effects of exercising those options in the lease term when it is reasonably certain that the Company will exercise that option. In assessing whether it is reasonably certain that the Company will exercise an option, the Company considers factors such as:
| ● | The lease payments due in any optional period; |
| ● | Penalties for failure to exercise (or not exercise) the option; |
| ● | Market factors, such as the availability of similar assets and current rental rates for such assets; |
| ● | The nature of the underlying leased asset and its importance to the Company’s operations; and |
| ● | The remaining useful lives of any related leasehold improvements. |
Lease expense for the Company’s operating leases are charged to operationsis recognized on a straight-line basis over the lease term. Operating leases relate primarilyterm and is reported as a component of general and administrative expense. Variable lease payments, if any, are recognized in the period when the obligation to make those payments is incurred. Lease incentives received prior to lease commencement are recorded as a reduction in the Company’s researchright-of-use asset. Fixed lease incentives received after lease commencement reduce both the lease liability and development facilitiesthe right-of-use asset.
The Company has elected an accounting policy to account for the lease and corporate headquarters.
Stock-based Compensation
The Company measures all stock options and other stock-based awards granted to employees and directors based on the fair value on the date of the grant and recognizes compensation expense of those awards over the requisite service period, which for the Company is the period between the grant date and the date the award vests or becomes exercisable. Most of theMany awards granted by the Company (and still outstanding) vest ratably over three or four years. However, certain awards granted to members of the Company’s Board of Directors vest in their entirety on the one-year anniversary following the date of grant. Generally, the Company issues stock options and restricted stock awards to employees with only service-based vesting conditions and records the expense for these awards using the straight-line method. However, in certain years, the Company granted restricted stock unitswill grant share-based payment awards to employees that wereare dependent upon the fulfillment of certain clinical and financial conditions. In such instances where the performance condition must be met for the award to vest, the company only recognizes compensation expense in the period thatwhen the award becomesis probable of vesting (See Note 1112 — Stock-Based Compensation).
The Company classifies stock-based compensation expenseexpenses in its statement of operations in the same manner in which the award recipient’s payroll costs are classified. The Company accounts for forfeitures as they occur.
89
The fair value of restricted stock and restricted stock units is determined based on the number of shares granted and the quoted price of the Company’s common stock on the date of grant. The determination of grant-date fair value for stock option awards is estimated using the Black-Scholes model, which includes variables such as the expected volatility of the Company’s share price, expected term of the award, interest rates, and dividend yields.
The Company relies exclusively on its historical volatility as an input to the option pricing model as management believes that this rate will be representative of future volatility over the expected term of the options.
The expected term assumption is estimated using past history of early exercise behavior and expectations about future behaviors.
The weighted average risk-free interest rate represents the interest rate for treasury constant maturities published by the Federal Reserve Board. If the term of available treasury constant maturity instruments is not equal to the expected term of an employee option, Cyclacel uses the weighted average ofinterpolates a discount rate based on the two Federal Reserve securities closest to the expected term of the employee option.
The expected dividend yield is based on the fact thatzero, as the Company has never paid cash dividends on common stock and does not expect to pay any cash dividends on common stock in the foreseeable future.
Income Taxes
The Company accounts for income taxes using the liability method. Under this method, deferred tax assets and liabilities are determined based on the difference between the financial statement and tax bases of assets and liabilities using enacted tax rates in effect for the year in which the differences are expected to affect taxable income. Valuation allowances are established when necessary to reduce deferred tax assets to the amounts expected to be realized.
The Company accounts for uncertainty in income taxes recognized in the financial statements by applying a two-step process to determine the amount of tax benefit to be recognized. First, the tax position must be evaluated to determine the likelihood that it will be sustained upon external examination by the taxing authorities. If the tax position is deemed more-likely-than-not tomore-likely-than-not-to be sustained, the tax position is then assessed to determine the amount of benefit to recognize in the financial statements. The amount of the benefit that may be recognized is the largest amount that has a greater than 50% likelihood of being realized upon ultimate settlement. The provision for income taxes includes the effects of any resulting tax reserves orfor unrecognized tax benefits that are considered appropriate as well as the related net interest and penalties.
The Company records research and development tax credits within income taxes. Credit is taken in the accounting period for research and development tax credits, which will be claimed from H.M. Revenue & Customs, (“HMRC”),or HMRC, the United Kingdom’s taxation and customs authority, in respect of qualifying research and development costs incurred in the same accounting period.
Net Loss Per Common Share
The Company calculates net loss per common share in accordance with ASC 260 “Earnings Per Share” (“ASC 260”). Basic and diluted net loss per common share was determined by dividing the net loss applicable to common stockholders by the weighted average number of common shares outstanding during the period.
In periods in which the Company reports a net loss attributable to common stockholders, diluted net loss per share attributable to common stockholders is the same as basic net loss per share attributable to common stockholders since potentially dilutive common shares are not assumed to have been issued if their effect is anti-dilutive. The Company reported a net loss attributable to common stockholders for the years ended December 31, 20162023 and 2017.2022.
For the year ended December 31, 2022, the Company calculated loss per share using the two-class method. The two-class method is an allocation formula that determines loss per share for each share of common stock and redeemable common stock (see Note 11), according to dividends declared and participation rights in undistributed earnings. The
90
reconciliation showing the loss per share attributable to common and redeemable common stockholders is presented in Note 15.
Comprehensive Income (Loss)
All components of comprehensive income (loss), including net income (loss), are reported in the financial statements in the period in which they are recognized. Comprehensive income (loss) is defined as the change in equity during a period from transactions and other events and circumstances from non-owner
Recently Issued Accounting Pronouncements
The Financial Accounting Standards Board (“FASB”) has issued Accounting Standards Update (“ASU”) No. 2017-11, Accounting2020-04, “Reference Rate Reform (Topic 848)”. This standard provides optional expedients and exceptions for Certain Financial Instruments with Down Round Features (“ASU 2017-11”), which simplifiesapplying GAAP to contracts, hedging relationships, and other transactions affected by reference rate reform initiatives that would replace interbank offered rates, including the accountingLondon Interbank Offered Rate (LIBOR). For example, modifications of lease contracts within the scope of ASC 842 solely for certain financial instruments with down-round features. A down round feature ischanges in reference rates would be accounted for as a provision in a financial instrument that reduces the strike price of an issued financial instrument if the issuer sells shares of its stock for an amount less than the currently stated strike pricecontinuation of the issued financial instrument or issues an equity-linked financial instrumentexisting contracts with a strike price below the currently stated strike priceno reassessments of the issued financial instrument.lease classification and the discount rate. Following the issuance of ASU 2017-11 is2022-06, “Reference Rate Reform (Topic 848): Deferral of the Sunset Date of Topic 848”, the relief remains effective for fiscal years,all entities as of March 12, 2020 through December 31, 2024. The Company does not currently have any contracts affected by this guidance.
The FASB has issued ASU 2023-07, “Segment Reporting (Topic 280)”. This standard will require all public entities – even those like the Company that have a single reportable segment – to disclose additional information about the title and interim periods within thoseposition of the Chief Operaing Decision Maker (“CODM”), the measure or measures of segment profit and loss used by the CODM in assessing segment performance and deciding how to allocate resources, an explanation of how the CODM uses the reported measure(s) in assessing segment performance, significant segment expenses that are regularly provided to the CODM, and a reconciliation of segment profit and loss to the closest consolidated totals prepared under United States GAAP. The amendments in ASU 2023-07 are effective for fiscal years beginning after December 15, 2018. Early adoption is permitted, including adoption in an2023, and interim period.periods within fiscal years beginning after December 15, 2024. ASU 2017-11 should be adopted retrospectively for each prior reporting period presented or retrospectively as of the beginning of the year of adoption. The Company anticipates this standard2023-07 will not have a material impactchange the way in which reportable segments are determined. However, the Company is currently evaluating the effects of ASU 2023-07 on its consolidated financial statements.
The FASB has issued ASU No. 2016-16, Income2023-09, “Income Taxes (Topic 740): Intra-Entity TransferImprovements to Income Tax Disclosures”. This standard will require all entities shall disclose the amount of Assets Other than Inventory (“income taxes paid (net of refunds received) disaggregated by federal (national), state, and foreign for each annual reporting period. The guidance in ASU 2016-16”), which requires the recognition of the income tax consequences of an intra-entity transfer of an asset, other than inventory, when the transfer occurs. The standard is2023-09 becomes effective for annual periods beginning after December 15, 2017, including interim periods within those fiscal years. This standard will not have a material impact on2024. The Company already discloses the company’s consolidated financial statements.
3.
Distribution, Licensing and Research Agreements
The Company has entered into licensing and similar agreements with academic and research organizations. Under the terms of these agreements, the Company has received licenses to technology and patent applications. The Company ismay be required to pay royalties on future sales of products employing the technology or falling under claims of patent applications.
91
Under a 2003 license agreement, is with Daiichi Sankyo. Pursuant to the Daiichi Sankyo license under which the Company licenseslicensed certain patent rights for sapacitabine. The Company will not be pursuing further development of sapacitabine and the license agreement was terminated effective as of March 23, 2023 for commercial reasons.
The three-year term of the Clinical Collaboration Agreement, or CCA with The University of Texas MD Anderson Cancer Center, or MD Anderson ended in September 2021, in accordance with its terms. The main objective of the CCA was to clinically evaluate the safety and efficacy of three Cyclacel medicines in patients with hematological malignancies, including chronic lymphocytic leukemias, acute myeloid leukemias, myelodysplastic syndromes and other advanced leukemias. Under the terms of the CCA, MD Anderson conducted four clinical studies and under the risk-sharing agreement MD Anderson assumed the patient costs for all studies and Cyclacel, who is the sponsor, provided investigational drugs and other limited support. The agreement provided that the Company is under an obligationwould make certain payments to use reasonable endeavors to develop a product and obtain regulatory approval to sell a product and has agreed to pay Daiichi Sankyo an up-front fee, reimbursement for Daiichi Sankyo’s enumerated expenses, milestone payments and royalties on a country-by-country basis. The up-front fee, Phase 3 entry milestone, and certain past reimbursements have been paid. A further $10.0 million in aggregate milestone payments could be payable subject to achievement of all the specific contractual milestones which are primarily related to regulatory approval in various territories, and the Company’s decision to continue with these projects. Royalties are payable in each country for the term of patent protection in the country or for ten years following theMD Anderson upon first commercial sale of licensed productsin specific indications studied in the country, whichever is later. Royalties are payable on net sales. Net sales are defined as the gross amount invoiced by the Company or its affiliates or licensees, less discounts, credits, taxes, shipping and bad debt losses. The agreement extends from its commencement date to the date on which no further amounts are owed under it. If the Company wishes to appoint a third party to develop or commercialize a sapacitabine-based product in Japan, within certain limitations, Daiichi Sankyo must be notified and given a right of first refusal, with the right of first refusal ending sixty days after notification, to develop and/or commercialize in Japan. In general, the license may be terminated by the Company for technical, scientific, efficacy, safety, or commercial reasons on six months’ notice, or twelve months’ notice, if after a launch of a sapacitabine-based product, or by either party for material default.alliance. There were no milestonessuch payments earned in 20162023 or 2017.
The following is a summary of cash and cash equivalents at December 31, 20162023 and 20172022 (in thousands):
| | | | December 31, | | |||||||||
| | | | 2016 | | | 2017 | | ||||||
| Cash | | | | $ | 5,332 | | | | | $ | 1,103 | | |
| Investments with original maturity of less than three months at the time of purchase | | | | | 11,188 | | | | | | 22,807 | | |
| Total cash and cash equivalents | | | | $ | 16,520 | | | | | $ | 23,910 | | |
| | |||||||||||||
| | | | | | |
|
| December 31, | ||||
|
| 2023 |
| 2022 | ||
Cash | | $ | 3,303 | | $ | 13,798 |
Cash equivalents | |
| 75 | |
| 4,547 |
Total cash and cash equivalents | | $ | 3,378 | | $ | 18,345 |
Cash equivalents are made up entirely of money market funds and commercial paper.
The following tables present information about the Company’s financial assets and liabilities measured at fair value on a recurring basis and indicate the level of the fair value hierarchy utilized to determine such fair values (in thousands):
| | | | | | | | | | | | |
|
| Fair Value Measurements | | | | |||||||
|
| as of December 31, 2023 Using: | | | | |||||||
|
| Level 1 |
| Level 2 |
| Level 3 |
| Total | ||||
Assets: |
| |
|
| |
|
| |
|
| |
|
Cash equivalents | | $ | 75 | | $ | — | | $ | — | | $ | 75 |
Total Assets | | $ | 75 | | $ | — | | $ | — | | $ | 75 |
| | | | | | | | | | | | |
|
| Fair Value Measurements | | | | |||||||
|
| as of December 31, 2022 Using: | | | | |||||||
|
| Level 1 |
| Level 2 |
| Level 3 |
| Total | ||||
Assets: |
| |
|
| |
|
| |
|
| |
|
Cash equivalents | | $ | 4,547 | | $ | — | | $ | — | | $ | 4,547 |
Total Assets | | $ | 4,547 | | $ | — | | $ | — | | $ | 4,547 |
| | | | Fair Value Measurements as of December 31, 2016 Using: | | |||||||||||||||||||||
| | | | Level 1 | | | Level 2 | | | Level 3 | | | Total | | ||||||||||||
| Assets: | | | | | | | | | | | | | | | | | | | | | | | | | |
| Cash equivalents | | | | $ | 11,188 | | | | | $ | — | | | | | $ | — | | | | | $ | 11,188 | | |
| Total Assets | | | | $ | 11,188 | | | | | $ | — | | | | | $ | — | | | | | $ | 11,188 | | |
| | |||||||||||||||||||||||||
| | | | Fair Value Measurements as of December 31, 2017 Using: | | |||||||||||||||||||||
| | | | Level 1 | | | Level 2 | | | Level 3 | | | Total | | ||||||||||||
| Assets: | | | | | | | | | | | | | | | | | | | | | | | | | |
| Cash equivalents | | | | $ | 22,807 | | | | | $ | — | | | | | $ | — | | | | | $ | 22,807 | | |
| Total Assets | | | | $ | 22,807 | | | | | $ | — | | | | | $ | — | | | | | $ | 22,807 | | |
| | |||||||||||||||||||||||||
92
The following is a summary of prepaid expenses and other current assets at December 31, 20162023 and 20172022 (in thousands):
| | | | December 31, | | |||||||||
| | | | 2016 | | | 2017 | | ||||||
| Research and development tax credit receivable | | | | $ | 1,730 | | | | | $ | 1,054 | | |
| Prepayments | | | | | 867 | | | | | | 363 | | |
| Delaware tax receivable | | | | | — | | | | | | 25 | | |
| VAT receivable | | | | | 327 | | | | | | 409 | | |
| Deposits | | | | | 132 | | | | | | 132 | | |
| Other current assets | | | | | 41 | | | | | | 81 | | |
| Prepaid expenses and other current assets | | | | $ | 3,097 | | | | | $ | 2,064 | | |
| | |||||||||||||
| | | | | | |
|
| December 31, | | December 31, | ||
|
| 2023 |
| 2022 | ||
Research and development tax credit receivable | | $ | 2,933 | | $ | 4,664 |
Prepayments and VAT receivable | | | 792 | |
| 976 |
Other current assets | |
| 341 | | | 426 |
| | $ | 4,066 | | $ | 6,066 |
7.
As at December 31, 2023, the Company had non-current assets of $1.3 million, which comprised clinical trial deposits held by a contract research organization in relation to the Company’s Phase 1/2 clinical trials.
8. Property and Equipment
Property and equipment consisted of the following at December 31, 20162023 and 20172022 (in thousands):
| | | | Lives in years | | | December 31, | | |||||||||
| | | | 2016 | | | 2017 | | |||||||||
| Leasehold improvements | | | 5 to 15 | | | | $ | 792 | | | | | $ | 835 | | |
| Research and laboratory equipment | | | 3 to 5 | | | | | 4,438 | | | | | | 4,854 | | |
| Office equipment and furniture | | | 3 to 5 | | | | | 1,157 | | | | | | 1,236 | | |
| | | | | | | | | 6,387 | | | | | | 6,925 | | |
| Less: accumulated depreciation and amortization | | | | | | | | (6,342) | | | | | | (6,896) | | |
| | | | | | | | $ | 45 | | | | | $ | 29 | | |
| | ||||||||||||||||
| | | | | | | | |
| | |
| December 31, | ||||
|
| Lives in years |
| 2023 |
| 2022 | ||
Leasehold improvements | | 2 to 5 | | $ | 6 | | $ | 6 |
Office equipment and furniture |
| 3 to 5 | |
| 423 | |
| 409 |
| | | |
| 429 | |
| 415 |
Less: accumulated depreciation and amortization |
|
| |
| (420) | |
| (383) |
| | | | $ | 9 | | $ | 32 |
9. Accrued and Other Current Liabilities
Accrued and other current liabilities consisted of the following at December 31, 20162023 and 20172022 (in thousands):
| | | | December 31, | | |||||||||
| | | | 2016 | | | 2017 | | ||||||
| Accrued research and development | | | | $ | 2,138 | | | | | $ | 1,645 | | |
| Accrued legal and professional fees | | | | | 194 | | | | | | 248 | | |
| Other current liabilities | | | | | 430 | | | | | | 662 | | |
| | | | | $ | 2,762 | | | | | $ | 2,555 | | |
| | |||||||||||||
| | | | | | |
|
| December 31, | | December 31, | ||
|
| 2023 |
| 2022 | ||
Accrued research and development | | $ | 3,668 | | $ | 3,611 |
Accrued legal and professional fees | |
| 570 | |
| 333 |
Other current liabilities | |
| 380 | |
| 1,006 |
| | $ | 4,618 | | $ | 4,950 |
Other current liabilities in 2017 include $150,000 deferred income in respectaccrued payroll costs of payment received in advance of achieving a milestone underapproximately $0 and $680,000 for the ManRos agreement (see Note 3 — Significant Contracts).
10. Commitments and Contingencies
General
Please refer to Note 3 — Significant Contracts for further discussion of certain of the Company’s commitments and contingencies.
93
Leases
In October 2000,April 2022, the Company entered into a twenty-five yearextended the lease for its research and developmentcorporate headquarters facility in Dundee, Scotland. Rent expense, which includes lease payments related to the Company’s research and development facilities and corporate headquarters and other rent related expenses was $0.5 and $0.5 millionBerkeley Heights, New Jersey for each ofa further three years, expiring in July 2025.
For the years ended December 31, 20162023 and 2017, respectively.
The following is a summary of the Company’s future contractual obligations and commitments relating to its facilities leaseslease as at December 31, 20172023 (in thousands):
| | | | Operating Lease Obligation | | |||
| 2018 | | | | $ | 348 | | |
| 2019 | | | | | 344 | | |
| 2020 | | | | | 344 | | |
| 2021 | | | | | 344 | | |
| 2022 | | | | | 343 | | |
| thereafter | | | | | 956 | | |
| Total future minimum lease obligations | | | | $ | 2,679 | | |
| | |||||||
| | | |
|
| Operating Lease | |
|
| Obligation | |
2024 | | $ | 66 |
2025 | |
| 38 |
Thereafter | |
| — |
Total future minimum lease obligation | | $ | 104 |
Less imputed interest | | | (11) |
Total | | $ | 93 |
11. Stockholders’ Equity
The Company has completed the following equity issuances during the periods presented in the consolidated financial statements.
December 2023 Registered Direct Offering
On JulyDecember 21, 2017,2023, the Company issued (i) 3,154,000 Class A Units for $2 per unit, each consisting of one shareentered into a securities purchase agreement (the “Securities Purchase Agreement”) with certain institutional investors (“Purchasers”). Pursuant to the Securities Purchase Agreement, the Company agreed to sell in a registered direct offering (“Registered Direct Offering”) 168,500 shares (“Shares”) of the Company’s common stock, $0.001 par value per share (“Common Stock”), and a warrantpre-funded warrants (“Pre-Funded Warrants”) to purchase one shareup to 219,700 shares of common stock (the “Class A Warrants”), and (ii) 8,872 Class B Units, each consistingCommon Stock. The Pre-Funded Warrants have an exercise price of one share of the Company’s Series A Convertible Preferred Stock, par value $0.001 per share (the “Series A Preferred Stock”and are immediately exercisable and can be exercised at any time after their original issuance until such Pre-Funded Warrants are exercised in full. Each Share is being sold at an offering price of $3.315 and each Pre-Funded Warrant is being sold at an offering price of $3.314 (equal to the purchase price per Share minus the exercise price of the Pre-Funded Warrant).
Pursuant to the Securities Purchase Agreement, in a concurrent private placement (together with the Registered Direct Offering, the “Offerings”), convertiblethe Company also agreed to issue to the Purchasers unregistered warrants (“Common Warrants”) to purchase up to 388,200 shares of Common Stock. Each Common Warrant has an exercise price of $3.19 per share, is exercisable immediately following their original issuance and will expire seven years from the original issuance date. The closing of the offering occurred on December 26, 2023, and the net proceeds to the Company were approximately $1.0 million, after deducting placement agent fees and other offering expenses payable by the Company.
On December 21, 2023, in a separate concurrent insider private placement (the “Insider Private Placement”), the Company also entered into 500a Securities Purchase Agreement with certain of its executive officers (the “Insider Securities Purchase Agreement”) pursuant to which the Company agreed to sell in a private placement (i) 6,070 shares of Common Stock and warrants to purchase 6,070 shares of Common Stock on the same terms as the Common Warrants issued to the Purchasers in the Offerings to Spiro Rombotis, the Company’s Chief Executive Officer, and (ii) 1,886 shares of Common Stock and warrants to purchase 1,886 shares of Common Stock on the same terms as the Common Warrants issued to the Purchasers in the Offerings to Paul McBarron, the Company’s Executive Vice President-Finance, Chief Financial Officer and Chief Operating Officer. Each such share of Common Stock and accompanying warrant was sold at a purchase price of $3.315, which was the same purchase price for the Shares sold in the Registered Direct Offering.
94
Ladenburg Thalmann & Co. Inc. (the “Placement Agent”) acted as the exclusive placement agent for the Offerings, pursuant to a placement agency agreement (the “Placement Agency Agreement”), dated December 21, 2023, by and between the Company and the Placement Agent.
Pursuant to the Placement Agency Agreement, the Company paid the Placement Agent a cash placement fee equal to 8.0% of the aggregate gross proceeds raised in the Offerings from sales arranged for by the Placement Agent. Subject to certain conditions, the Company also agreed to reimburse all reasonable travel and other out-of-pocket expenses of the Placement Agent in connection with the Offerings, including but not limited to legal fees, up to a maximum of $85,000. In addition, the Placement Agent also received warrants that have substantially the same terms as the Warrants issued in the concurrent private placement to the Purchasers in the Offerings to purchase that number of shares of Common Stock equal to 6.0% of the aggregate number of shares of Common Stock and Prefunded Warrants sold in the Offerings, or an aggregate of 23,769 shares of Common Stock, at an exercise price of $4.14375 per share (the “Placement Agent Warrants”). The Placement Agent Warrants will be exercisable immediately following the initial conversion price,date of issuance and a warrant to purchase a number of shares of common stock equal to $1,000.00 dividedwill expire five years from issuance. The Placement Agency Agreement contains customary representations, warranties and agreements by the conversion price (the “Class B Warrants”) for $1,000 per unit.Company and customary conditions to closing. The net proceedsCompany has agreed to indemnify the Company afterPlacement Agent against certain liabilities, including liabilities under the underwriters’ exerciseSecurities Act, and liabilities arising from breaches of representations and warranties contained in fullthe Placement Agency Agreement, or to contribute to payments that the Placement Agent may be required to make in respect of those liabilities.
Each of the over-allotment option were approximately $13,700,000, after deducting underwriting discounts, commissions and other estimated offering expenses. The Class A Units and Class B Units have no stand-alone rightsinstruments issued in the Offerings and the sharesInsider Private Placement have been classified and recorded as part of common stock, Series A Preferred Stock and the Class A and Class B Warrants comprising those units were immediately separable.
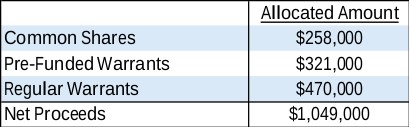
The aggregate fair value of the Placement Agent Warrants was $47,000. These have been accounted for as a direct cost of the Offerings and Inside Private Placement, resulting in no net effect to overall shareholders’ equity.
In determining the fair values of the Pre-Funded Warrants, Regular Warrants, and Placement Agent Warrants, the Company used a Black-Scholes Option Pricing model with the Class B units,following assumptions:
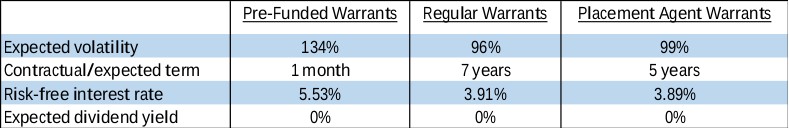
The fair value of the Companycommon shares was determined using the closing price of the Company’s common stock as of December 26, 2023, which is the date that the Series A Preferred Stock had a beneficial conversion feature with an aggregate intrinsic value of approximately $3,638,000.
August 2021 Controlled Equity Offering SM
On July 10, 2015,August 12, 2021, the Company entered into a Controlled Equity OfferingSM Sales Agreement (the “Sales Agreement”) with Cantor Fitzgerald (the “Cantor Sales Agreement”& Co. ("Cantor") under, pursuant to which the Company may,could issue and sell, from time to time, sell shares of its common stock having an aggregate offering price of up to $8.35$50.0 million through Cantor Fitzgerald. Underas the sales agent. Cantor Sales Agreement, Cantor Fitzgerald maycould sell shares of Cyclacelthe Company’s common stock by methodsany method permitted by law deemed to be an “at-the-market” offering“at the market offering” as defined in Rule 415 promulgated under415(a)(4) of the Securities ActAct.
On August 12, 2022, the Company became aware that the shelf registration statement on Form S-3 (file number 333-231923) (the “Registration Statement”) associated with this Sales Agreement had expired on June 21, 2022. Prior to
95
becoming aware of the gross sales price per share sold. Cyclacel is not obligated to make any sales under the Cantor Sales Agreement. Effective as of June 17, 2016, and prior to entering into the FBR Sales Agreement,expiration, the Company and Cantor agreed to terminate the Cantor Sales Agreement. The Company had issuedsold an aggregate of 114,078132,473 shares of its common stock at the market price, following the expiration of the Registration Statement and through August 12, 2022, for aggregate proceeds of approximately $2,721,187. There was no sale of shares post August 12, 2022. The sale of these shares were subject to potential rescission rights by certain shareholders. As a result of these potential rescission rights, the Company reclassified 207,807 shares (including 75,333 shares sold for which the Company did not receive any proceeds), with an aggregate purchase price of $4,494,496 of its common as stock outside stockholders’ equity as of December 31, 2022. These shares have been treated as issued and outstanding for financial reporting purposes. The rescission rights for these shares has now lapsed and the shares were reclassified back to permanent equity. As of December 31, 2023, there have been no claims or demands to exercise such rights.
On August 15, 2022, due to expiry of the Registration Statement, the Sales Agreement was mutually terminated. A total of 218,738 shares, for gross proceeds of approximately $7.6 million, had been sold pursuant to the Cantor Sales AgreementAgreement.
Warrants
December 2020 Warrants
As of which 41,996December 31, 2023, warrants to purchase 44,657 shares of common stock issued pursuant to a securities purchase agreement in a December 2020 financing transaction remained outstanding. Each warrant shall be exercisable beginning on the 12-month anniversary of the date of issuance for a period of five years after the date of issuance, at an exercise price of $61.95 per warrant share. The exercise price of the warrants will be subject to adjustment in the event of any stock dividends and splits, reverse stock split, recapitalization, reorganization or similar transaction, as described in the warrants. The warrants may be exercised on a “cashless” basis.
During the year ended December 31, 2023, 15,833 warrants lapsed upon conversion of the associated Series B Preferred Stock. There were issuedno exercises of these warrants during the year ended December 31, 2016 for net proceeds of approximately $0.2 million.
April 2020 Warrants
As of December 31, 2017, there were 7,490,5002023, 146,000 warrants issued in connection with an April 2020 equity financing remained outstanding, each with an exercise price of $2.00.$75.00. The common warrants are immediately exercisable and will expire on the fifth anniversary of the original issuance date. The exercise price and number of shares of common stock issuable upon exercise is subject to appropriate adjustment in the event of stock dividends, stock splits, reorganizations or similar events affecting the Company’s common stock. The common warrants were issued separately from the common stock and were eligible for transfer immediately after issuance. A common warrant to purchase one share of common stock was issued for every share of common stock purchased in this offering.
The common warrants are exercisable, at the option of each holder, in whole or in part, by delivering to the Company a duly executed exercise notice accompanied by payment in full for the number of shares of the Company’s common stock purchased upon such exercise (except in the case of a cashless exercise). A holder (together with its affiliates) may not exercise any portion of the common warrant to the extent that the holder would own more than 4.99% of the outstanding common stock immediately after exercise, except that upon at least 61 days prior notice from the holder to the Company, the holder may increase the amount of ownership of outstanding stock after exercising the holder’s common warrants up to 9.99% of the number of shares of the Company’s common stock outstanding immediately after giving effect to the exercise, as such percentage ownership is determined in accordance with the terms of the common warrants. No fractional shares of common stock will be issued in connection with the exercise of a common warrant. In lieu of fractional shares, the Company will round down to the next whole share.
There were no warrants exercised during the years ended December 31, 2023 or December 31, 2022.
July 2017 Warrants
As of December 31, 2023, 24,968 warrants issued in connection with a July 2017 underwritten public offering remained outstanding, each with an exercise price of $600. All such warrants were issued in connection with the
96
July 2017 Underwritten Public Offeringunderwritten public offering and are immediately exercisable. The Warrantswarrants expire in 2024. Subject to limited exceptions, a holder of warrants will not have the right to exercise any portion of its warrants if the holder (together with such holder’s affiliates, and any persons acting as a group together with such holder or any of such holder’s affiliates) would beneficially own a number of shares of common stock in excess of 4.99% (or, at the election of the purchaser, 9.99%) of the shares of our Common Stock then outstanding after giving effect to such exercise.
The exercise price and the number of shares issuable upon exercise of the warrants is subject to appropriate adjustment in the event of recapitalization events, stock dividends, stock splits, stock combinations, reclassifications, reorganizations or similar events affecting the Company’s common stock. The warrant holders must pay the exercise price in cash upon exercise of the warrants unless such warrant holders are utilizing the cashless exercise provision of the warrants. On the expiration date, unexercised warrants will automatically be exercised via the “cashless” exercise provision.
Prior to the exercise of any warrants to purchase common stock, holders of the warrants will not have any of the rights of holders of the common stock purchasable upon exercise, including the right to vote, except as set forth therein.
There were no portion of these warrants exercised during each of the years ended December 31, 2023 and 2022.
Series B Preferred Stock
237,745 shares of the Company’s Series B Preferred Stock were issued in a December 2020 Securities Purchase Agreement. Each share of Series B Preferred Stock shall initially be convertible into one third (1/3) share of Common Stock (the “Conversion Shares”), subject to adjustment in accordance with the Certificate of Designation.
Holders of Series B Preferred Stock are entitled to receive dividends on shares of Series B Preferred Stock equal, on an as-if-converted-to-Common-Stock basis, and in the same form as dividends actually paid on shares of the Common Stock. Except as otherwise required by law, the Series B Preferred Stock does not have voting rights. However, as long as any shares of Series B Preferred Stock are outstanding, the Company will not, without the affirmative vote of the holders of a majority of the then outstanding shares of the Series B Preferred Stock, (a) alter or change adversely the powers, preferences or rights given to the Series B Preferred Stock, (b) alter or amend the Certificate of Designation, (c) amend its certificate of incorporation or other charter documents in any manner that adversely affects any rights of the holders of Series B Preferred Stock, (d) increase the number of authorized shares of Series B Preferred Stock, (e) pay certain dividends or (f) enter into any agreement with respect to any of the foregoing. The Series B Preferred Stock does not have a preference upon any liquidation, dissolution or winding-up of the Company. The Purchaser may convert shares of Series B Preferred Stock through a conversion into shares of Common Stock if and solely to the extent that such conversion would not result in the Purchaser beneficially owning in excess of 9.99% of then-outstanding Common Stock or aggregate voting power of the Company (such limitation, the “Ownership Limitation”) and any portion in excess of such limitation will remain outstanding as Series B Preferred Stock.
During the year ended December 31, 20162023, 118,745 shares of Series B Preferred Stock was converted, at the option of the holder, into 39,582 shares of Common Stock. As of December 31, 2023, 119,000 shares of the Series B Preferred Stock remained issued and 2017 were 0outstanding and 99,500 respectively. Gross proceeds during the year for such exercises amounted to $199,000 with a weighted average exercise priceare convertible into 39,667 shares of $2.00 per share.
Series A Preferred Stock
8,872 shares of the Company’s Series A Preferred Stock were issued in thea July 2017 Underwritten Public Offering. Each share of Series A Preferred Stock is convertible at any time at the option of the holder thereof, into a number of shares of common stock determined by dividing $1,000 by the initial conversion price of $2.00$600.00 per share, subject to a 4.99% blocker provision, or, upon election by a holder prior to the issuance of shares of Series A Preferred Stock, 9.99%, and is subject to adjustment for stock splits, stock dividends, distributions, subdivisions and combinations.
As of December 31, 2017,2023 and 2022, 264 shares of the Series A Preferred Stock remain issued and outstanding. The 264 shares of Series A Preferred Stock issued and outstanding at December 31, 2017,2023, are convertible into 132,000440 shares of common stock.
97
In the event of a liquidation, the holders of shares of the Series A Preferred Stock may participate on an as-converted-to-common-stock basis in any distribution of assets of the Company. The Company shall not pay any dividends on shares of common stock (other than dividends in the form of common stock) unless and until such time as dividends on each share of Series A Preferred Stock are paid on an as-converted basis. There is no restriction on the Company’s ability to repurchase shares of Series A Preferred Stock while there is any arrearage in the payment of dividends on such shares, and there are no sinking fund provisions applicable to the Series A Preferred Stock.
Subject to certain conditions, at any time following the issuance of the Series A Preferred Stock, the Company has the right to cause each holder of the Series A Preferred Stock to convert all or part of such holder’s Series A Preferred Stock in the event that (i) the volume weighted average price of our common stock for 30 consecutive trading days, (the “Measurement Period”)or Measurement Period exceeds 300% of the initial conversion price of the Series A Preferred Stock (subject to adjustment for forward and reverse stock splits, recapitalizations, stock dividends and similar transactions), (ii) the daily trading volume on each Trading Day during such Measurement Period exceeds $500,000 per trading day and (iii) the holder is not in possession of any information that constitutes or might constitute, material non-public information which was provided by the Company. The right to cause each holder of the Series A Preferred Stock to convert all or part of such holder’s Series A Preferred Stock shall be exercised ratably among the holders of the then outstanding preferred stock.
The Series A Preferred Stock has no maturity date, will carry the same dividend rights as the common stock, and with certain exceptions contains no voting rights. In the event of any liquidation or dissolution of the Company, the Series A Preferred Stock ranks senior to the common stock in the distribution of assets, to the extent legally available for distribution.
6% Convertible Exchangeable Preferred Stock
As of December 31, 2017,2023, there were 335,273 shares of the Company’s 6% Convertible Exchangeable, (“or Preferred Stock”)Stock issued and outstanding at an issue price of $10.00 per share. Dividends on the Preferred Stock are cumulative from the date of original issuance at the annual rate of 6% of the liquidation preference of the Preferred Stock, and if declared, payable quarterly on the first day of February, May, August and November, commencing February 1, 2005. Any dividends must be declared by the Company’s Board of Directors and must come from funds that are legally available for dividend payments. The Preferred Stock has a liquidation preference of $10.00 per share, plus accrued and unpaid dividends.
The Company’s Board of Directors considers numerous factors in determining whether to declare the quarterly dividend pursuant to the Certificate of Designations governing the terms of the Company’s Preferred Stock, including the requisite financial analysis and determination of a surplus. Accrued andAccumulated but unpaid dividends in arrears on preferred stock were $0.7 million, or $1.95 per share, of preferred stock, as of December 31, 2016 and 2017.
The Preferred Stock is convertible at the option of the holder at any time into the Company’s shares of common stock at a conversion rate of approximately 0.005070.0000167 shares of common stock for each share of Preferred Stock based on a price of $1,974.$592,200. The Company has reserved 1,6986 shares of common stock for
The Company may automatically convert the Preferred Stock into common stock if the closing price of the Company’s common stock has exceeded $2,961,$888,300, which is 150% of the conversion price of the Preferred Stock, for at least 20 trading days during any 30-day trading period, ending within five trading days prior to notice of automatic conversion.
The Certificate of Designations governing the Preferred Stock provides that if the Company fails to pay dividends on its Preferred Stock for six quarterly periods, holders of Preferred Stock are entitled to nominate and elect two directors to the Company’s Board of Directors. This right accrued to the holders of Preferred Stock as of August 2, 2010 and two directors were nominated and elected at the annual meeting held on May 24, 2011.
98
The Preferred Stock has no maturity date and no voting rights prior to conversion into common stock, except under limited circumstances.
The Company may, at its option, redeem the Preferred Stock in whole or in part, out of funds legally available at the redemption price of $10.00 per share.
The Preferred Stock is exchangeable, in whole but not in part, at the option of the Company on any dividend payment date beginning on November 1, 2005, (the “Exchange Date”)or Exchange Date for the Company’s 6% Convertible Subordinated Debentures, (“Debentures”)or Debentures at the rate of $10.00 principal amount of Debentures for each share of Preferred Stock. The Debentures, if issued, will mature 25 years after the Exchange Date and have terms substantially similar to those of the Preferred Stock. No such exchanges have taken place as of December 31, 2017.
For the year ended December 31, 2017,2023, the company declared dividends of $0.15 per share quarterly on its 6% Convertible Exchangeable Preferred Stock. These dividends were paid on May 1, August 1 and November 1, 2017,2023, and February 1, 2018,2024, respectively.
Stock based compensation has been reported within the following expense line items on the consolidated statement of operations for the years ended 20162023 and 2017,2022 as shown in the following table (in thousands):
| | | | Year Ended December 31, 2016 | | | Year Ended December 31, 2017 | | ||||||
| Research and development | | | | $ | 305 | | | | | $ | 72 | | |
| General and administrative | | | | | 477 | | | | | | 194 | | |
| Stock-based compensation costs before income taxes | | | | $ | 782 | | | | | $ | 266 | | |
| | |||||||||||||
| | | | | | |
|
| Year Ended | ||||
|
| December 31, | ||||
| | 2023 |
| 2022 | ||
General and administrative | | $ | 1,039 | | $ | 1,051 |
Research and development | | | 434 | | | 455 |
Stock-based compensation costs before income taxes | | $ | 1,473 | | $ | 1,506 |
2018 Plan
In May 22, 2015,2018, the Company’s stockholders approved the 20152018 Equity Incentive Plan (the “2015“2018 Plan”), under which Cyclacel may make equity incentive grants to its officers, employees, directors and consultants. The company2018 Plan replaced the 2015 Equity Incentive Plan (the “2015 Plan”).
The 2018 Plan allows for various types of award grants, including stock options and restricted stock units.
On June 13, 2023, the Company’s stockholders approved an additional 60,000 shares of common stock that may be issued under the 2018 Plan. On June 14, 2022, the Company’s stockholders approved an additional 33,333 shares of common stock that may be issued under the 2018 Plan. As of December 31, 2023, the Company has reserved 458,75325,415 shares of the Company’s common stock under the 2015
2020 Inducement Equity Incentive Plan
In October 2020, the Inducement Equity Incentive Plan (the “Inducement Plan”), became effective. Under the Inducement Plan, Cyclacel may make equity incentive grants to new senior level Employees (persons to whom the Company granted 197,841 optionsmay issue securities without stockholder approval). The Inducement Plan allows for the issuance of up to employees and directors with a grant date fair value13,333 shares of approximately $0.7 million. Of these options, 189,091 are performance based, which will vest upon the fulfilmentCompany’s common stock (or the equivalent of certain clinical conditions and will terminate if they have not vested bysuch number). As of December 31, 2020. The Company determined that2023, 8,000 shares under the satisfactionInducement Plan have been issued, leaving a remaining reserve of the vesting criteria was not probable throughout 20165,333 shares.
99
Option Grants and 2017 and, as a result, did not record any expense related to these awards for the years ended December 31, 2016 and 2017.
There were 170,85343,342 options granted during the year ended December 31, 2017. Of these options, 158,853 are performance based, which will vest upon the fulfillment of certain clinical conditions. The Company determined that the satisfaction of one of the conditions was probable as of December 31, 2017, but that the other vesting criteria related to these awards were not probable as of December 31, 2017. As such, the Company recognized compensation cost for these grants2023, all issued under the expectation that 25% of these awards will vest.
Of the options granted during the year ended December 31, 2023, 25,633 shall vest on the third anniversary of their date of grant, or earlier if either of the certain performance conditions are met relating to enrollment goals for various clinical studies. The Company has assumed that these awards will vest after three years as satisfaction of the performance conditions is not probable at this time.
As of December 31, 2017,2023, the total remaining unrecognized compensation cost related to the non-vested stock options with service conditions amounted to approximately $0.2$1.0 million, which will be amortized over the weighted-average remaining requisite service period of 0.91.53 years.
Outstanding Options
A summary of the share option activity and related information is as follows:
| | | | Number of Options Outstanding | | | Weighted Average Exercise Price Per Share | | | Weighted Average Remaining Contractual Term (Years) | | | Aggregate Intrinsic Value ($000s) | | ||||||||||||
| Options outstanding at December 31, 2015 | | | | | 206,298 | | | | | $ | 72.60 | | | | | | 8.09 | | | | | $ | — | | |
| Granted | | | | | 197,841 | | | | | $ | 4.68 | | | | | | | | | | | | | | |
| Exercised | | | | | — | | | | | | | | | | | | | | | | | | | | |
| Cancelled/forfeited | | | | | (14,760) | | | | | $ | 396.83 | | | | | | | | | | | | | | |
| Options outstanding at December 31, 2016 | | | | | 389,379 | | | | | $ | 25.80 | | | | | | 5.83 | | | | | $ | 121 | | |
| Granted | | | | | 170,853 | | | | | $ | 1.93 | | | | | | | | | | | | | | |
| Exercised | | | | | — | | | | | | | | | | | | | | | | | | | | |
| Cancelled/forfeited | | | | | (24,615) | | | | | $ | 179.92 | | | | | | | | | | | | | | |
| Options outstanding at December 31, 2017 | | | | | 535,617 | | | | | $ | 11.10 | | | | | | 8.23 | | | | | $ | — | | |
| Unvested at December 31, 2017 | | | | | 386,682 | | | | | $ | 3.64 | | | | | | 8.84 | | | | | $ | — | | |
| Vested and exercisable at December 31, 2017 | | | | | 148,755 | | | | | $ | 30.49 | | | | | | 6.65 | | | | | $ | — | | |
| | |||||||||||||||||||||||||
| | | | | | | | | | |
|
| |
| | |
| Weighted |
| | |
|
| |
| Weighted |
| Average |
| | | |
|
| Number of |
| Average |
| Remaining |
| Aggregate | ||
| | Options |
| Exercise |
| Contractual | | Intrinsic | ||
| | Outstanding | | Price Per Share |
| Term (Years) | | Value ($000) | ||
Options outstanding at December 31, 2021 |
| 73,291 | | $ | 112.90 |
| 8.99 | | $ | 189 |
Granted | | 34,822 | | $ | 36.16 |
| — | | $ | — |
Cancelled/forfeited | | (740) | | $ | 157.33 |
| — | | $ | — |
Options outstanding at December 31, 2022 |
| 107,373 | | $ | 87.70 |
| 8.34 | | $ | — |
| | | | | | | | | | |
Granted |
| 43,342 | | $ | 8.82 |
| — | |
| — |
Cancelled/forfeited |
| (7,919) | | $ | 332.24 |
| — | |
| — |
Options outstanding at December 31, 2023 |
| 142,796 | | $ | 50.20 |
| 7.96 | | $ | — |
Unvested at December 31, 2023 |
| 59,126 | | $ | 19.81 |
| 9.10 | | $ | — |
Vested and exercisable at December 31, 2023 |
| 83,670 | | $ | 71.68 |
| 7.16 | | $ | — |
The fair value of the stock options granted is calculated using the Black-Scholes option-pricing model as prescribed by ASC 718 using the following assumptions:
| | | | Year ended December 31, 2016 | | | Year ended December 31, 2017 | |
| Expected term (years) | | | 5 – 6 | | | 6 | |
| Risk free interest rate | | | 1.370% – 1.500% | | | 1.890% – 2.265% | |
| Volatility | | | 98% – 104% | | | 108% | |
| Expected dividend yield over expected term | | | 0.00% | | | 0.00% | |
| Resulting weighted average grant date fair value | | | $3.66 | | | $1.59 | |
| | | | |
| | | | |
| | Year ended | | Year ended |
|
| December 31, 2023 |
| December 31, 2022 |
Expected term (years) |
| 5-6 |
| 5-6 |
Risk free interest rate |
| 3.660% – 4.160% | | 1.370% – 3.605% |
Volatility |
| 89% – 92% | | 86% – 93% |
Expected dividend yield over expected term |
| 0.00% | | 0.00% |
Resulting weighted average grant date fair value |
| $6.63 | | $26.44 |
There were no stock options exercised during each of the years ended December 31, 2022 and 2023, respectively. The Company does not expect to be able to benefit from the deduction for stock option exercises that may occur because the company has tax loss carryforwards from prior periods that would be expected to offset any potential taxable income.
Restricted Stock Units
The Company issued 25,621 restricted stock units during the year ended December 31, 2023.
8,488 restricted stock units issued in June 2023 vest on the first anniversary of the date of grant. Each of these restricted stock units were valued at $8.84 at the date of grant, which was equivalent to the market price of a share of the Company’s common stock on that date.
100
17,133 restricted stock units issued in January 2023 vest on the third anniversary of their date of grant, or earlier if certain defined clinical trial related performance targets are met. A three-year vesting assumption was applied to these restricted stock units as satisfaction of the performance conditions is not probable at this time. Each restricted stock unit was valued at $13.50 at the date of grant, which was equivalent to the market price of a share of the Company’s common stock on that date. As of December 31, 2023, 300 of these restricted stock units have been forfeited as the recipient’s employment was voluntarily terminated by the Company.
The Company issued 7,911 restricted stock units during the year ended December 31, 2022. These restricted stock units vest over a period of one year for awards granted to directors and three years for grants to employees. Each restricted stock unit was valued at $16.65 based on their fair value at the date of grant, which is equivalent to the market price of a share of the Company’s common stock.
Summarized information for restricted stock units as of December 31, 2023 is as follows:
| | | | | | | |
|
| |
| Weighted | | Weighted | |
|
| |
| Average | | Average | |
| | Restricted |
| Grant Date | | Remaining | |
| | Stock Units | | Value Per Share | | Term | |
Restricted Stock Units outstanding at December 31, 2021 |
| 1,266 | | $ | 100.35 | | |
Granted |
| 7,911 | | | 16.65 | | |
Restricted Stock Units outstanding at December 31, 2022 |
| 9,177 | | $ | 28.20 | | |
| | | | | | | |
Granted | | 25,621 | | | 11.96 | | |
Cancelled/forfeited | | (300) | | | 13.50 | | |
| | | | | | | |
Restricted Stock Units outstanding at December 31, 2023 | | 34,498 | | $ | 16.26 | | 8.96 years |
Unvested at December 31, 2023 |
| 26,428 | | $ | 12.30 | | 9.20 years |
Vested and exercisable at December 31, 2023 |
| 8,070 | | $ | 29.24 | | 8.17 years |
13. Employee Benefit Plans
Pension Plan
The Company operates a defined contribution group personal pension plan for all of its UK based employees. Company contributions to the plan totaled approximately $57,000$52,000 and $54,000$44,000 for the years ended December 31, 20162023 and 2017,2022, respectively.
401(k) Plan
The 401(k) Plan provides for matching contributions by the Company in an amount equal to the lesser of 100% of the employee’s deferral or 6% of the U.S. employee’s qualifying compensation. The 401(k) Plan is intended to qualify under Section 401(k) of the Internal Revenue Code, so that contributions to the 401(k) Plan by employees or by the Company, and the investment earnings thereon, are not taxable to the employees until withdrawn. Company matching contributions are tax deductible by the Company when made. In 2023, Company employees maycould elect to reduce their current compensation by up to the statutorily prescribed annual limit of $18,000$22,500 if under 50 years old and $24,000$30,000 if over 50 years old and to have those funds contributed to the 401(k) Plan. The Company made contributions of approximately $32,000$103,000 and $28,000$95,000 to the 401(k) Plan for the years ended December 31, 20162023 and 2017,2022, respectively.
101
14. Taxes
(Loss) income from continuing operations before taxes is comprised of the following components for the years ended December 31, 20162023 and 20172022 (in thousands):
| | | | Year Ended December 31, | | |||||||||
| | | | 2016 | | | 2017 | | ||||||
| Domestic | | | | $ | (2,011) | | | | | $ | (127) | | |
| Foreign | | | | | (11,763) | | | | | | (8,336) | | |
| Loss from continuing operations before taxes | | | | $ | (13,774) | | | | | $ | (8,463) | | |
| | |||||||||||||
| | | | | | |
|
| Year Ended December 31, | ||||
|
| 2023 |
| 2022 | ||
Domestic | | $ | (806) | | $ | (1,981) |
Foreign | |
| (24,745) | |
| (23,934) |
Loss from continuing operations before taxes | | $ | (25,551) | | $ | (25,915) |
The benefit (provision) for income taxes from continuing operations consists of the following (in thousands):
| | | | Year Ended December 31, | | |||||||||
| | | | 2016 | | | 2017 | | ||||||
| Current – domestic | | | | $ | — | | | | | $ | — | | |
| Current – foreign | | | | | 1,983 | | | | | | 993 | | |
| Current – total | | | | | 1,983 | | | | | | 993 | | |
| Deferred – domestic | | | | | — | | | | | | — | | |
| Income tax benefit | | | | $ | 1,983 | | | | | $ | 993 | | |
| | |||||||||||||
| | | | | | |
|
| Year Ended December 31, | ||||
|
| 2023 |
| 2022 | ||
Current – domestic | | $ | (2) | | $ | (2) |
Current – foreign | |
| 2,998 | |
| 4,719 |
Current – total | |
| 2,996 | |
| 4,717 |
Deferred – domestic | |
| — | |
| — |
Income tax benefit | | $ | 2,996 | | $ | 4,717 |
The Company has incurred a taxable loss in each of the operating periods since incorporation. The income tax credits of $2.0$3.0 million and $1.0$4.7 million for the years ended December 31, 20162023 and 2017,2022, respectively, represent UK research and development (“R&D”) tax credits for expenditures in the United Kingdom that are refundable.
A reconciliation of the (benefit) provision for income taxes from continuing operations with the amount computed by applying the statutory federal tax rate to loss from continuing operations before income taxes is as follows (in thousands):
| | | | Year Ended December 31, | | |||||||||
| | | | 2016 | | | 2017 | | ||||||
| Loss from continuing operations before taxes | | | | $ | (13,774) | | | | | $ | (8,463) | | |
| Income tax expense computed at statutory federal tax rate | | | | | (4,683) | | | | | | (2,877) | | |
| Disallowed expenses and non-taxable income | | | | | 12 | | | | | | 3 | | |
| Loss surrendered to generate R&D credit | | | | | 1,945 | | | | | | 856 | | |
| Additional research and development tax relief | | | | | (2,827) | | | | | | (1,262) | | |
| Change in valuation allowance | | | | | 2,029 | | | | | | (4,487) | | |
| Foreign items, including change in tax rates, and other | | | | | 1,238 | | | | | | 1,901 | | |
| Change in US Tax Rate | | | | | — | | | | | | 4,112 | | |
| Other foreign items | | | | | 303 | | | | | | 761 | | |
| | | | | $ | (1,983) | | | | | $ | (993) | | |
| | |||||||||||||
| | | | | | |
|
| Year Ended December 31, | ||||
|
| 2023 |
| 2022 | ||
Loss from continuing operations before taxes | | $ | (25,551) | | $ | (25,915) |
Income tax expense computed at statutory federal tax rate | |
| (5,366) | |
| (5,442) |
Additional research and development tax relief | |
| (5,409) | |
| (4,719) |
Loss surrendered to generate R&D credit | |
| 4,865 | |
| 6,169 |
Change in valuation allowance | |
| 1,261 | |
| 2,310 |
Other foreign items | |
| 809 | |
| (3,487) |
Disallowed expenses and non-taxable income | |
| 782 | |
| 369 |
Stock Compensation | | | 62 | | | 105 |
Foreign items, including change in tax rates, and other | |
| — | |
| (20) |
Change in UK Tax Rate | |
| — | |
| (2) |
| | $ | (2,996) | | $ | (4,717) |
Significant components of the Company’s deferred tax assets are shown below (in thousands):
| | | | | | |
|
| Year Ended December 31, | ||||
|
| 2023 |
| 2022 | ||
Net operating loss and tax credit carryforwards | | $ | 57,074 | | $ | 53,092 |
Depreciation, amortization and impairment of property and equipment | |
| 39 | |
| 61 |
Stock options | |
| 210 | |
| 153 |
Right of use asset | | | (26) | | | (40) |
Lease liability | | | 26 | | | 40 |
Total deferred tax assets | |
| 57,323 | |
| 53,306 |
Valuation allowance for deferred tax assets | |
| (57,323) | |
| (53,306) |
Net deferred tax assets | | $ | — | | $ | — |
| | | | Year Ended December 31, | | |||||||||
| | | | 2016 | | | 2017 | | ||||||
| Net operating loss and tax credit carryforwards | | | | $ | 42,851 | | | | | $ | 38,948 | | |
| Depreciation, amortization and impairment of property and equipment | | | | | 137 | | | | | | 111 | | |
| Stock options | | | | | 2,365 | | | | | | 1,807 | | |
| Research and development credits | | | | | 4,021 | | | | | | 4,021 | | |
| Other | | | | | — | | | | | | — | | |
| Deferred tax assets | | | | | 49,374 | | | | | | 44,887 | | |
| Valuation allowance for deferred tax assets | | | | | (49,374) | | | | | | (44,887) | | |
| Net deferred tax assets | | | | $ | — | | | | | $ | — | | |
| | |||||||||||||
102
A valuation allowance has been established, as realization of such assets is uncertain. The Company’s management evaluated the positive and negative evidence bearing upon the realizability of its deferred assets, and has determined that, at present, the Company may not be able to recognize the benefits of the deferred tax assets under the more likely than not criteria. Accordingly, a valuation allowance of approximately $44.9$57.3 million has been established at December 31, 2017.2023. The valuation allowance has decreasedincreased by approximately $4.5$4.0 million in 2017.
As specified in the Tax Reform Act of 1986, due to ownership changes, the Company’s ability to utilize its net operating loss (“NOL”) carryforwards may be limited. The benefit of deductions from the exercise of stock options is included in the NOL carryforwards. As of December 31, 2016 and 2017, the Company had federal NOLs of $28.4 million and $28.4 million and foreign NOLs of $178.2 million and $186.2 million, respectively. The Company’s federal NOLs will start to expire in 2026, and the state NOLs totaling $18.7 million will start expiring in 2023. The Company’s foreign NOL’s do not expire under UK tax law. However, for losses created after April 1, 2017 the use of these NOLs is restricted to an annual £5 million allowance in each standalone company or group and above this allowance, there will be a 50% restriction in the profits that can be covered by losses brought forward. The amount of profits restricted for the year ended December 31, 2017 is $0. As of December 31, 2017, the Company had a US R&D credit carryforward of $4.0 million which will start expiring in 2019.
As of December 31, 2017. 2023 and 2022, the Company has federal NOLs of $3.9 million and $3.6 million, respectively. The federal NOLs have an indefinite life. As of December 31, 2023 and 2022, the Company has state NOLs of $17.3 million and $22.1 million, respectively, which will begin to expire in 2028. As of December 31, 2023 and 2022, the Company had foreign NOLs of $220.1 million and $203.0 million, respectively. The Company’s foreign NOL’s do not expire under UK tax law however the use of these NOLs is restricted to an annual £5.0 million allowance in each standalone company or group and above this allowance, there will be a 50% restriction in the profits that can be covered by losses brought forward.
Management has evaluated all significant tax positions at December 31, 20162023 and 20172022 and concluded that there are no material uncertain tax positions. The Company would recognize both interest and penalties related to unrecognized benefits in income tax expense. The Company has not recorded any interest and penalties on any unrecognized tax benefits since its inception.
Tax year 2016 remainsyears 2020 - 2022 remain open to examination by major taxing jurisdictions to which the Company is subject, which are primarily in the United Kingdom and the United States, as carryforward attributes generated in years past may still be adjusted upon examination by the United Kingdom’s H.M. Revenue & Customs, the Internal Revenue Service (“IRS”) or state tax authorities. The Company is currently not under examination by the IRS or any other jurisdictions for any tax years.
We have not provided a deferred tax liability on the cumulative amount of unremitted foreign earnings of international subsidiaries because it is our intent to permanently reinvest such earnings outside of the United States.
The Company has an aggregate deficit in foreign earnings and therefore has not provided any deferred tax liability on its outside book-tax basis difference in its foreign subsidiaries and because it is also our intent to permanently reinvest any earnings outside of the United States. We would recognize this deferred tax liability if we were to experience a change in circumstances producing a change in that intention. As a result of the repeal of the Section 902 foreign tax credit under the Tax Act, future distributions would not be offset by a foreign tax credit.
Effective for tax years beginning after December 31, 2021, taxpayers are required to capitalize any expenses incurred that are considered incidental to research and experimentation (R&E) activities under IRC Section 174. While taxpayers historically had the option of deducting these expenses under IRC Section 174, the December 2017 Tax Cuts and Jobs Act mandates capitalization and amortization of R&E expenses for tax years tax years beginning after December 31, 2021. Expenses incurred in connection with R&E activities in the US must be amortized over a 5-year period if incurred, and R&E expenses incurred outside the US must be amortized over a 15-year period. R&E activities are broader in scope than qualified research activities that are considered under IRC Section 41 (relating to the research tax credit). For the year ended December 31, 2023, the Company performed an analysis based on available guidance and determined that the company does not have any R&E expenses in the US. The company will continue to monitor this issue for future developments, but it does not expect R&E capitalization and amortization to require it to pay cash taxes now or in the near future.
103
Basic and diluted net loss per share attributable to common stockholders was calculated as follows:
| | | | Years ended December 31, | | |||||||||
| | | | 2016 | | | 2017 | | ||||||
| Numerator: | | | | | | | | | | | | | |
| Net loss | | | | $ | (11,791) | | | | | $ | (7,470) | | |
| Dividend on convertible exchangeable preferred shares | | | | $ | (200) | | | | | $ | (201) | | |
| Beneficial conversion feature of Series A convertible stock | | | | $ | — | | | | | $ | (3,638) | | |
| Conversion of Series A convertible preferred stock | | | | $ | — | | | | | $ | (3,537) | | |
| Net loss attributable to common shareholders | | | | $ | (11,991) | | | | | $ | (14,846) | | |
| Denominator: | | | | | | | | | | | | | |
| Weighted-average number of common shares used in loss per share – basic and diluted | | | | | 3,424,976 | | | | | | 7,631,152 | | |
| Loss per share – basic and diluted | | | | $ | (3.50) | | | | | $ | (1.95) | | |
| | |||||||||||||
| | | | | | | | | | | | |
| | Years ended December 31, | | | | | | | ||||
| | 2023 | | 2022 |
| |
| | ||||
Numerator: | | | | | | | | | | | | |
Net loss | | $ | (22,555) | | $ | (21,198) | | | | | | |
Dividend on convertible exchangeable preferred shares | | | (201) | | | (201) | | | | | | |
Net loss attributable to common shareholders | | $ | (22,756) | | $ | (21,399) | | | | | | |
Deemed dividend on accretion of redeemable common stock | | | | | | (135) | | | | | | |
Remaining undistributed loss | | | (22,756) | | | (21,534) | | | | | | |
| | | | | | | | | | | | |
| | Years ended December 31, 2023 | | Years ended December 31, 2022 | ||||||||
| | Common Shareholders | | Redeemable Common Shareholders | | Common Shareholders |
| Redeemable Common Shareholders | ||||
Allocation of undistributed loss | | $ | (22,756) | | $ | — | | $ | (18,872) | | $ | (2,662) |
Deemed dividend on accretion of redeemable common stock | | | — | | | — | | | | | | 135 |
Net loss attributable to common shareholders | | | (22,756) | | | — | | | (18,872) | | | (2,527) |
| | | | | | | | | | | | |
Denominator: | | | | | | | | | | | | |
Weighted-average number of common shares used in loss per share – basic and diluted | | | 850,815 | | | — | | | 657,620 | | | 92,759 |
Loss per share - basic and diluted | | $ | (26.75) | | $ | — | | $ | (28.70) | | $ | (27.24) |
Distributed earnings | | | — | | | — | | | — | | | 1.46 |
Undistributed loss | | | (26.75) | | | — | | | (28.70) | | | (28.70) |
Net loss per share | | $ | (26.75) | | $ | — | | $ | (28.70) | | $ | (27.24) |
| | | | | | | | | | | | |
| | | | | | | | | | | | |
| | | | | | | | | | | | |
Potential dilutive securities have been excluded from the computation of diluted net loss per share as the effect would be to reduce the net loss per share. Therefore, the weighted average number of common shares outstanding used to calculate both basic and diluted net loss per share attributable to common stockholders is the same. The Company excluded the following potential common shares, presented based on amounts outstanding at each period end, from the computation of diluted net loss per share attributable to common stockholders for the periods indicated because including them would have had an anti-dilutive effect:
| | | | |
|
| December 31, | | December 31, |
|
| 2023 |
| 2022 |
Stock options |
| 142,796 |
| 107,373 |
Restricted Stock Units |
| 34,498 |
| 9,177 |
6% convertible exchangeable preferred stock |
| 6 |
| 6 |
Series A preferred stock |
| 440 |
| 440 |
Series B preferred stock |
| 39,667 |
| 79,248 |
Common stock warrants |
| 635,550 |
| 215,625 |
Total shares excluded from calculation |
| 852,957 |
| 411,869 |
104
| | | | Year ended December 31, | | |||||||||
| | | | 2016 | | | 2017 | | ||||||
| Stock options | | | | | 389,378 | | | | | | 535,616 | | |
| Convertible preferred stock | | | | | 1,698 | | | | | | 1,698 | | |
| Series A preferred stock | | | | | — | | | | | | 132,000 | | |
| Common stock warrants | | | | | — | | | | | | 7,490,500 | | |
| Total shares excluded from calculation | | | | | 391,076 | | | | | | 8,159,814 | | |
| | |||||||||||||
Geographic information for the years ended December 31, 20162023 and 20172022 is as follows (in thousands):
| | | | Year Ended December 31, | | |||||||||
| | | | 2016 | | | 2017 | | ||||||
| Revenue | | | | | | | | | | | | | |
| United Kingdom | | | | $ | 843 | | | | | $ | — | | |
| Total Revenue | | | | $ | 843 | | | | | $ | — | | |
| Net loss | | | | | | | | | | | | | |
| United States | | | | $ | (2,211) | | | | | $ | (127) | | |
| United Kingdom | | | | | (9,580) | | | | | | (7,343) | | |
| Total Net Loss | | | | $ | (11,791) | | | | | $ | (7,470) | | |
| | |||||||||||||
| | | | December 31, | | |||||||||
| | | | 2016 | | | 2017 | | ||||||
| Total Assets | | | | | | | | | | | | | |
| United States | | | | $ | 15,197 | | | | | $ | 23,522 | | |
| United Kingdom | | | | | 4,465 | | | | | | 2,481 | | |
| Total Assets | | | | $ | 19,662 | | | | | $ | 26,003 | | |
| Long Lived Assets, net | | | | | | | | | | | | | |
| United States | | | | $ | 2 | | | | | $ | 0 | | |
| United Kingdom | | | | | 43 | | | | | | 29 | | |
| Total Long Lived Assets, net | | | | $ | 45 | | | | | $ | 29 | | |
| | |||||||||||||
| | | | | | |
|
| Year Ended December 31, | ||||
|
| 2023 |
| 2022 | ||
Revenue |
| |
|
| |
|
United Kingdom | | $ | 420 | | $ | — |
Total Revenue | | $ | 420 | | $ | — |
Net loss | |
|
| |
|
|
United States | | $ | (808) | | $ | (1,983) |
United Kingdom | |
| (21,747) | |
| (19,215) |
Total Net Loss | | $ | (22,555) | | $ | (21,198) |
| | | | | | |
|
| December 31, | ||||
|
| 2023 |
| 2022 | ||
Total Assets |
| |
|
| |
|
United States | | $ | 2,938 | | $ | 18,220 |
United Kingdom | |
| 5,867 | |
| 9,830 |
Total Assets | | $ | 8,805 | | $ | 28,050 |
Long Lived Assets, net | |
|
| |
|
|
United States | | $ | — | | $ | — |
United Kingdom | |
| 9 | |
| 32 |
Total Long Lived Assets, net | | $ | 9 | | $ | 32 |
17. Subsequent Events
On March 7, 2018,January 12, 2024, the Board of Directors declared a quarterly cash dividend in the amount of $0.15 per share on the Company’s Preferred Stock. The cash dividend is due to be was paid on MayFebruary 1, 20182024 to the holders of record of the Preferred Stock as of the close of business on April 11, 2018.January 22, 2024.
105
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
(a) Disclosure Controls:
We maintain disclosure controls and procedures that are designed to ensure that information required to be disclosed in our Exchange Act reports is recorded, processed, summarized and reported within the time periods specified in the Securities and Exchange Commission’s rules and forms and that such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow for timely decisions regarding required disclosure. An evaluation was performed under the supervision and with the participation of the Company’s management, including the Chief Executive Officer and Chief Financial Officer, on the effectiveness of the Company’s disclosure controls and procedures as of December 31, 2017.
Pursuant to this evaluation, our Chief Executive Officer and Chief Financial Officer concluded that, as of December 31, 2017,2023, the end of the period covered by this report, our disclosure controls and procedures were effective.
(b) Management’s Annual Report on Internal Control Over Financial Reporting:
Internal control over financial reporting refers to the process designed by, or under the supervision of, our Chief Executive Officer and Chief Financial Officer, and effected by our Board of Directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles, and includes those policies and procedures that:
| (1) | Pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of the assets of the Company; |
| (2) | Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the Company are being made only in accordance with authorizations of management and directors of the Company; and |
| (3) | Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the Company’s assets that could have a material effect on the financial statements. |
Internal control over financial reporting cannot provide absolute assurance of achieving financial reporting objectives because of its inherent limitations. Internal control over financial reporting is a process that involves human diligence and compliance and is subject to lapses in judgment and breakdowns resulting from human failures. Internal control over financial reporting also can be circumvented by collusion or improper override. Because of such limitations, there is a risk that material misstatements may not be prevented or detected on a timely basis by internal control over financial reporting. However, these inherent limitations are known features of the financial reporting process, and it is possible to design into the process safeguards to reduce, though not eliminate, this risk.
106
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rules 13a-15(f) and 15d-15(f). Under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework in Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in 2013.
Management assessed the effectiveness of the Company’sour internal control over financial reporting as of December 31, 2017. Management based this assessment on criteria for effective internal control over financial reporting described in “Internal Control-Integrated Framework” issued by the Committee of Sponsoring Organizations of the Treadway Commission.2023. Management’s assessment included an evaluation of the design of the Company’sour internal control over financial reporting and testing of the operational effectiveness of itsour internal control over financial reporting. Management reviewed the results of its assessmentSubsequent to that evaluation, management, with the Audit Committee.
Material Weakness in Internal Control over Financial Reporting
A material weakness is a deficiency, or combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of a company’s annual and interim financial statements will not be detected or prevented on a timely basis. Management has identified material weaknesses in our internal control over financial reporting associated with the design of our controls over the accounting treatment of contractually required deposits and the preparation ofaccounting treatment for complex non-routine equity transactions. Specifically, we did not effectively design controls to properly account for contractually required deposits and complex non-routine equity transactions.
Remediation Plan
Our remediation process is ongoing and is focused on designing and implementing additional monitoring controls to ensure all complex non routine transactions are identified and properly accounted for in a timely manner. This remediation process cannot be considered complete at this time. There can be no assurance that we will be successful in remediating the material weaknesses. We plan to continue to assess internal controls and procedures and intend to take further action as necessary or appropriate to address any other matters as they are identified. Notwithstanding the identified material weaknesses, we have concluded that the consolidated financial statements in this Annual Report on Form 10 K present fairly, in all material respects, our financial position, results of operations and cash flows as of the dates, and for external purposesthe periods, presented, in accordanceconformity with accounting principles generally accepted in the United States of America.
This annual report does not include an attestation report of the Company’sour registered independent public accounting firm regarding internal control over financial reporting. Management’s report was not subject to attestation by the Company’sour registered public accounting firm pursuant to rules of the Securities and Exchange Commission that permit the Companyus to provide only management’s report in this annual report.
(c) Changes in Internal Control Over Financial Reporting
There have not been any changes in the Company’sour internal control over financial reporting (as defined in Rule 13a-15(f)) during the fiscal year ended December 31, 20172023 that have materially affected or are reasonably likely to materially affect, the Company’sour internal control over financial reporting.
107
Item 9B. Other information
Amendment to Amended and Restated Certificate of Incorporation for Reverse Stock Split
At the Special Meeting of Stockholders (the “Special Meeting”) of the Company held on December 8, 2023, the Company’s stockholders approved a proposal authorizing an amendment (the “Certificate of Amendment”) to the Company’s Amended and Restated Certificate of Incorporation, as amended, to effect a one-time reverse stock split of the Company’s outstanding shares of common stock at a ratio of not less than 1-for-3 and not greater than 1-for-15.
On December 15, 2023, the Company filed with the Secretary of State of the State of Delaware the Certificate of Amendment (and also a Certificate of Correction to Certificate of Amendment to the Amended and Restated Certificate of Incorporation) to effect a one-time reverse stock split of the Company’s common stock, at a ratio of 1-for-15 (the “Reverse Stock Split”). The Reverse Stock Split was effective at 5:00 p.m. Eastern Time, after the close of trading on The Nasdaq Capital Market, on December 15, 2023 (the “Effective Time”). At the Effective Time, every 15 shares of the Company’s issued and outstanding common stock were automatically converted into one share of common stock, without any change in the par value per share. In addition, proportionate adjustments were made to the per share exercise price and the number of shares issuable upon the exercise of all outstanding stock options, warrants and other convertible securities, and to the number of shares issued and issuable under the Company’s stock incentive plans.
Rule 10b5-1 Trading Arrangements
During the three months ended December 31, 2023, no director or officer of the Company adopted, modified or terminated a “Rule 10b5-1 trading arrangement” or “non-Rule 10b5-1 trading arrangement,” as each term is defined in Item 408(a) of Regulation S-K.
Item 9C. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections
Not applicable.
PART III
The information required by item 10 is incorporated herein by reference from the Company’sour Proxy Statement, which will be filed with the SEC within 120 days after the end of the Company’s 2017our 2023 fiscal year pursuant to Regulation 14A for its 2018our 2024 Annual Meeting of Stockholders.
Item 11.11. Executive Compensation
The information required by item 11 is incorporated herein by reference from the Company’sour Proxy Statement, which will be filed with the SEC within 120 days after the end of the Company’s 2017our 2023 fiscal year pursuant to Regulation 14A for its 2018our 2024 Annual Meeting of Stockholders.
The information required by item 12 is incorporated herein by reference from the Company’sour Proxy Statement, which will be filed with the SEC within 120 days after the end of the Company’s 2017our 2023 fiscal year pursuant to Regulation 14A for its 2018our 2024 Annual Meeting of Stockholders.
The information required by item 13 is incorporated herein by reference from the Company’sour Proxy Statement, which will be filed with the SEC within 120 days after the end of the Company’s 2017our 2023 fiscal year pursuant to Regulation 14A for its 2017our 2024 Annual Meeting of Stockholders.
108
The information required by item 14 is incorporated herein by reference from the Company’sour Proxy Statement, which will be filed with the SEC within 120 days after the end of the Company’s 2017our 2023 fiscal year pursuant to Regulation 14A for its 2018our 2024 Annual Meeting of Stockholders.
109
PART IV
Item 15. Exhibits and Financial Statement Schedules
| (a) | Documents filed as part of this report are as follows: |