
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTONWashington, D.C. 20549
FORM 10-K
☒ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended:ended December 31 2017, 2021
orOR
☐TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the transition period fromto
COMMISSION FILE NUMBER:Commission File Number: 001-36445
NanoVibronix, Inc.
(Exact name of Registrantregistrant as specified in its charter)
| Delaware | 01-0801232 | |
(State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification Number) |
| 525 Executive Blvd.Elmsford, New York | 10523 | |
| ||
| (Address of principal executive office) | (Zip Code) |
Registrant’s telephone number, including area code:(914)233-3004
Securities registered pursuant to Section 12(b) of the Exchange Act:
| Title of each class | Trading Symbol | Name of each exchange on which registered | ||
| Common | NOAV | NASDAQ |
Securities registered pursuant to Section 12(g) of the Exchange Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐No☑ ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐No☑ ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the Registrantregistrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes☑ ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes☑ ☒ No ☐
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ☑
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” and “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ☐ | Accelerated filer | ☐ |
| Non-accelerated filer | Smaller reporting company | ||
| Emerging growth company |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☐
Indicate by check mark whether the registrant is a shell company (as defined byin Rule 12b-2 of the Exchange Act). Yes ☐No☑ ☒
As of June 30, 2017, the last day of the registrant’s most recently completed second fiscal quarter, theThe aggregate market value of the voting and non-votingour common equitystock held by non-affiliates as of the registrant based on the average bid and asked price of the common equity on such date,June 30, 2021, was approximately $10.8 million. For purposes of this computation only, all officers, directors and 10% or greater stockholders of the registrant are deemed to be affiliates.$9,447,579.
The number of shares outstanding of the registrant’s common stock, par value $0.001 per share,Common Stock as of April 1, 201814, 2022 was 3,935,865 shares.
DOCUMENTS INCORPORATED BY REFERENCE
PortionsThe information required by Part III of this Form 10-K, to the extent not set forth herein, is incorporated by reference from the registrant’s Definitive Proxy Statement on Schedule 14A to be furnished to stockholders in connection withdefinitive proxy statement for its 20182021 Annual Meeting of Stockholders, whichStockholders. Such proxy statement shall be filed with the Securities and Exchange Commission within 120 days after the end of the fiscal year to which this Annual Report on Form 10-K relates, are incorporated by reference in Part III, Items 10-14 of this Annual Report on Form 10-K.report relates.
NANOVIBRONIX, INC.
TABLE OF CONTENTS
Cautionary Note Regarding Forward-Looking Statements; Risk Factor Summary
This Annual Report on Form 10-K contains “forward-looking statements,” which include information relating to future events, future financial performance, financial projections, strategies, expectations, competitive environment and regulation. Words such as “may,” “should,” “could,” “would,” “predicts,” “potential,” “continue,” “expects,” “anticipates,” “future,” “intends,” “plans,” “believes,” “estimates,” and similar expressions, as well as statements in future tense, identify forward-looking statements. Forward-looking statements should not be read as a guarantee of future performance or results and may not be accurate indications of when such performance or results will be achieved. Forward-looking statements are based on information we have when those statements are made or management’s good faith belief as of that time with respect to future events, and are subject to a number of risks, and uncertainties and assumptions that could cause actual performance or results to differ materially from those expressed in or suggested by the forward-looking statements. These risks are more fully described in the “Risk Factors” section of this Annual Report on Form 10-K. The following is a summary of such risks:
Overview
| ● | Our history of losses and expectation of continued losses. | |
| ● | Global economic and political instability and conflicts, such as the conflict between Russia and Ukraine, could adversely affect our business, financial condition or results of operations | |
| ● | Increasing inflation could adversely affect our business, financial condition, results of operations or cash flows. | |
| ● | The geographic, social and economic impact of COVID-19 on the Company’s business operations. | |
| ● | Our ability to raise funding for, and the timing of, clinical studies and eventual U.S. Food and Drug Administration approval of our product candidates. | |
| ● | Regulatory actions that could adversely affect the price of or demand for our approved products. | |
| ● | Market acceptance of existing and new products. | |
| ● | Favorable or unfavorable decisions about our products from government regulators, insurance companies or other third-party payers. | |
| ● | Risks of product liability claims and the availability of insurance. | |
| ● | Our ability to successfully develop and commercialize our products. | |
| ● | Our ability to generate internal growth. | |
| ● | Risks related to computer system failures and cyber-attacks. | |
| ● | Our ability to obtain regulatory approval in foreign jurisdictions. | |
| ● | Uncertainty regarding the success of our clinical trials for our products in development. | |
| ● | Risks related to our operations in Israel, including political, economic and military instability. | |
| ● | The price of our securities is volatile with limited trading volume | |
| ● | Our ability to comply with the continued listing requirements of the NASDAQ capital market. | |
| ● | Our ability to maintain effective internal control over financial reporting and to remedy identified material weaknesses. | |
| ● | We are a “smaller reporting company” and have reduced disclosure obligations that may make our stock less attractive to investors. | |
| ● | Our intellectual property portfolio and our ability to protect our intellectual property rights. | |
| ● | Our ability to recruit and retain qualified regulatory and research and development personnel. | |
| ● | Unforeseen changes in healthcare reimbursement for any of our approved products. | |
| ● | The adoption of health policy changes and health care reform. | |
| ● | Lack of financial resources to adequately support our operations. | |
| ● | Difficulties in maintaining commercial scale manufacturing capacity and capability. | |
| ● | Changes in our relationship with key collaborators. | |
| ● | Changes in the market valuation or earnings of our competitors or companies viewed as similar to us. | |
| ● | Our failure to comply with regulatory guidelines. | |
| ● | Uncertainty in industry demand and patient wellness behavior. | |
| ● | General economic conditions and market conditions in the medical device industry. | |
| ● | Future sales of large blocks of our common stock, which may adversely impact our stock price. | |
| ● | Depth of the trading market in our common stock. |
The foregoing does not represent an exhaustive list of matters that may be covered by the forward-looking statements contained herein or risk factors that we are faced with that may cause our actual results to differ from those anticipated in our forward-looking statements. Please see “Item 1A. Risk Factors” for additional risks which could adversely impact our business and financial performance. Moreover, new risks regularly emerge, and it is not possible for us to predict or articulate all risks we face, nor can we assess the impact of all risks on our business or the extent to which any risk, or combination of risks, may cause actual results to differ from those contained in any forward-looking statements. All forward-looking statements included in this Form 10-K are based on information available to us on the date hereof. Except to the extent required by applicable laws or rules, we undertake no obligation to publicly update or revise any forward-looking statement, whether as a result of new information, future events or otherwise.
| 1 |
Unless the context otherwise indicates or requires, the terms “we,” “our,” “us,” “NanoVibronix,” and the “Company,” as used in this Annual Report on Form 10-K, refer to NanoVibronix, Inc. and its subsidiaries as a combined entity, except where otherwise stated or where it is clear that the terms mean only NanoVibronix, Inc. exclusive of its subsidiaries.
*
Overview
We were organized as a Delaware corporation in October 2003. Through our wholly-owned subsidiary, NanoVibronix Ltd., a private company incorporated under the laws of the State of Israel, we focus on noninvasive biological response-activating devices that target biofilm prevention, pain therapy, and wound healing and pain therapy and can be administered at home, without the assistance of medical professionals. Our primary products, which are in various stages of clinical and market development, currently consist of:
| ● | UroShield™, an ultrasound-based product that is designed to prevent bacterial colonization and biofilm in urinary catheters, increase antibiotic efficacy and decrease pain and discomfort associated with urinary catheter |
| ● | PainShield™, a patch-based therapeutic ultrasound technology to treat pain, muscle spasm and joint contractures by delivering a localized ultrasound effect to treat pain and induce soft tissue healing in a targeted |
| ● | PainShield™ MD, a single patch-based therapeutic ultrasound technology to treat pain, muscle spasm and joint contractures by delivering a localized ultrasound effect to treat pain and induce soft tissue healing in a targeted area. | |
| ● | PainShield™ Plus, a dual patch-based therapeutic ultrasound technology to treat pain, muscle spasm and joint contractures by delivering a localized ultrasound effect to treat pain and induce soft tissue healing in a targeted area. Similar to PainShield MD, with a dual ultrasound delivery. (we anticipate submitting our application for FDA Clearance in the second quarter of 2022); and, | |
| ● | PainShield™Relief, an over-the-counter patch-based therapeutic ultrasound technology to treat pain, muscle spasm and joint contractures by delivering a localized ultrasound effect to treat pain and induce soft tissue healing in a targeted area. (we anticipate submitting our application for FDA Clearance in the second quarter of 2022); and |
| ● | WoundShield™, a patch-based therapeutic ultrasound device intended to facilitate tissue regeneration and wound healing by using ultrasound to increase local capillary perfusion and tissue oxygenation. |
Each of our UroShield, PainShield, UroShield, and WoundShield products employs a small, disposable transducer that transmits low frequency, low intensity ultrasound acoustic waves that seek to repair and regenerate tissue, musculoskeletal and vascular structures, and decrease biofilm formation on urinary catheters and associated urinary tract infections. Through their size, effectiveness and ease of use, these products are intended to eliminate the need for technicians and medical personnel to manually administer ultrasound treatment through large transducers, thereby promoting patient independence and enabling more cost-effective home-based care.
PainShieldPainShield™, MD is currently cleared for marketing in the United States by the U.S. Food and Drug Administration. In September 2020, the U.S. Food and Drug Administration although there has not been a significant sales and marketing effortexercised its Enforcement Discretion to date.allow distribution of the UroShield device in the U.S. during the COVID-19 pandemic. . While the permitted use is currently temporary, it does permit the import of the UroShield to the U.S. during the ongoing COVID-19 pandemic. All three of our products have CE Mark approval in the European Union, a Canadian medical device license and a certificate allowing us to sell PainShield, UroShield and WoundShield in Israel. We are able to sell PainShield, UroShield and WoundShield in India and Ecuador based on our CE Mark. We have consummated sales of PainShield and UroShield in the relevant markets, although to dateand we saw sales have been minimal;increase in 2021; WoundShield has not generated significant revenue to date. Outside of the United States we generally apply, through our distributor, for approval in a particular country for a particular product only when we have a distributor in place with respect to such product.
In the United States, PainShield and UroShield requires a prescription from a licensed healthcare practitioner. If U.S. Food and Drug Administration clearance is obtained, we anticipate that WoundShield and UroShield will require a prescription from a licensed healthcare practitioner in the United States. We anticipate that UroShield willhas been approved through the U.S. Food and Drug Administration under Enforcement Discretion for the duration of the Covid-19 health emergency and is intended to be sold directly to health care facilities and therefore will not require a prescription for these venues. However, in other countries in which we sell PainShield, UroShield, and WoundShield, such products are eligible for sale without a prescription.
| 2 |
In addition to the need to obtain regulatory approvals, we anticipate that sales volumes and prices of our UroShield, PainShield, and WoundShield products will depend in large part on the availability of insurance coverage and reimbursement from third party payers. Third party payers include governmental programs such as Medicare and Medicaid in the United States, private insurance plans and workers’ compensation plans. We do not currently have reimbursement codes for use of WoundShield in any of the markets in which we have regulatory authority to sell WoundShield. Of the markets in which we have regulatory authority to sell PainShield, prior to January 2020, we haveonly had reimbursement codes in the United States (i.e., CPT codes) for clinical use only, but do not have suchonly. Effective as of January 2020, the U.S. Centers for Medicare and Medicaid Services (CMS) approved our PainShield™ for reimbursement codes for at-home use ofMedicare beneficiaries on a national basis. However, the product, althoughcompany continues to work toward a reimbursement value from CMS. We are working with qualified legal representation toward that goal. We were notified on March 30, 2020 that our Medicare Enrollment Application was approved, and we are now an approved Medicare Supplier for Durable Medical Equipment, or DME, through the productNational Supplier Clearinghouse, Palmetto-GBA as well as Noridian Administrative Services, LLC, the two Medicare Administrative Contractors that handle DME reimbursement nationwide. PainShield is marketed and soldcurrently available for such use.Medicare reimbursement on a national level under new HCPCS (Healthcare Common Procedure Coding System) code K1004. With respect to UroShield, which may be used in a clinical and home setting, we do not currently have reimbursement codes in any of the markets in which we have regulatory authority to sell UroShield. We anticipate that we will begin to seekare seeking reimbursement codes for use of our products in the markets in which we have regulatory authority, including the United States, to sell such products; however, additional clinical data will be requiredproducts. We have made application for a dedicated CMS reimbursement code in order to obtain such reimbursement codes.2021 and are still awaiting final approval. Our current ongoing research and planned research may facilitate our ability to obtain reimbursement codes and there is no guarantee that we will be successful in obtaining such codes quickly, or at all. We have engaged a reimbursement expert, Redemption Revenue Cycle Solutions, LLC, and plan on using outside regulatory counsel, to help facilitate reimbursement.
We have completed 6six separate clinical studies with UroShield that together evaluated approximately 194 patients with urinary catheters. In patients where the UroShield product was used there were no serious adverse events reported, while a variety of clinical beneficial observations were seen including: catheter biofilm reduction, reduction in catheter associated pain, reduction in urinary tract infections, and a significant decrease in bacteriuria rates. We recently completed a double blind clinical trial for UroShield in the United States in orderOctober 2018. The results of the study, entitled “The Effect of Surface Acoustic Waves on Bacterial Load and Preventing Catheter-Associated Urinary Tract Infections (CAUTI) in Long Term Indwelling Catheters,” were published in the December 2018 issue of Medical & Surgical Urology, a peer-reviewed journal in the field of urology. In the study, 55 patients in a skilled nursing facility chain treated with long term indwelling catheters were evaluated. There was a significant difference between the treated group and the placebo group in the number of colony forming units (“CFU”) present upon evaluation, as well as on the number of treated urinary tract infections (“UTI”), and the effect lasted beyond the time of active treatment. The study concluded that the UroShield™ device was shown to obtain 510(k) clearance frombe effective in significantly reducing the U.S. Foodnumber of CFUs in patients with indwelling catheters. The study also concluded that the UroShield™ device was shown to be effective in reducing the number of treated UTIs in this patient population, and Drug Administration.surface acoustic waves in the form of the UroShield™ device is an effective tool in the prevention of catheter-associated UTI and while further evaluation is encouraged, can be safely utilized with a high likelihood of success. In July 2017, we engaged Idonea Solutions, Inc., an FDA consultant, to assist in our efforts.efforts to obtain clearance under the FDA’s Enforcement Discretion, and obtain 510(k) clearance which is still ongoing. If we are able to successfully obtain 510(k) clearance,successful, we intend to pursue obtaining reimbursement codes and to target completion of partnerships with leading catheter product companies for sales and marketing efforts in the United States. The Company has entered into recent distribution partnerships for UroShield in the United States, U.K., Switzerland, Israel, India, and New Zealand.
We have one clinical study which is ongoing for our product UroShield. We announced positive interim results from an independent, real world patient study of its UroShield at Southampton University Health Sciences in December 2021. The independent study, which was launched in the first half of 2021, was devised to evaluate how UroShield helps to reduce infection by preventing bacteria colonisation and the buildup of biofilms on long-term indwelling urinary catheters in real world patients and to better understand the patient benefits and experiences of using UroShield. The study consists of both laboratory and patient studies and is nearing completion. At the conclusion of the study, Southern Health will be able to purchase the UroShield devices for all patients that want to continue using the device and for additional patients in their care who have indwelling catheters. The research team have reported strong interest from both the clinical teams and patients who live with long term catheters. Full results of the study are expected to be published in the second quarter of 2022.
In addition, we are currently ramping upcontinue to expand our clinical development and marketing efforts in North America with respect to PainShield. In February 2018, we have completed a clinical trial to evaluate the effect of PainShield in patients with trigeminal neuralgia. The double blinded, crossover trial was conducted across the United States with the intent to recruit 60and included 59 patients with a diagnosis of unilateral trigeminal neuralgia. Positive interim resultsAmong the 59 patients, 30 were reported in the 4th quarter of 2017. 45 patients had completed the study, with 23 in the active treatment group and 2229 were in the control group. BasedThe values which were assessed include Visual Analog Scale (“VAS”) pain score, both baseline prior to trial and VAS pain score at the end of the study. The study also assessed breakthrough medications per week at the start of the trial and breakthrough medications per week at the end of the trial, with a particular focus on the interim data, thereuse of opioids. Breakthrough medications are used for chronic pain directly related to the pre-existing trigeminal neuralgia condition. There was a significant difference in the outcomes of the two groups relative to pain, quality of life, and breakthrough medications taken. In additiontaken, which was directly correlated to measurable differences in all aforementioned measurement categories, there was a generalpain experienced during treatment. Specifically, the control group saw an improvement in uninterrupted sleep.baseline scores of 2.3% versus the treatment group, which saw a 55.2% improvement in baseline scores. Additionally, the control group saw a reduction in breakthrough pain medication of 1.5% versus the treatment group, which saw a 46.4% reduction in breakthrough pain medication.
| 3 |
In 2019, the Company has completed a study which was intended to assess the PainShield’s ability to effectively treat Lateral Epicondylitis (Tennis Elbow). This is a double blinded, randomized control trial. The study has been completed and we are contemplating submission to an appropriate journal. The interim results have been further validatedwere reported as follows:
91% of the patients in the crossoverPainShield treatment group from placebo device tohad complete or partial resolution of symptoms. Patients used PainShield in conjunction with over-the-counter medication, as needed, but without the benefit of opioid-based prescription medication.
Results of the Birmingham study further reinforce that PainShield group. We have submitted comprehensive data from this clinical trial for expected publicationis safe, easy-to-use and highly effective in treating soft tissue pain. Patients in the second quarterstudy who wore our device reported marked reduction in pain and when combined with over-the-counter, anti-inflammatory medications, those same patients reported a complete resolution of 2018, at which time we plan to reportsymptoms within 10 days.
Dr. David Lemak, MD, Lead Investigator of the final results. We believe that a positive outcome in this trial will assist in our expandingBirmingham Study, added, “Patient outcomes were markedly improved with the commercial use of this product through a direct sales effort thatPainShield and importantly, no patients returned with signs or symptoms of an exacerbation. Most encouraging are the results we intendwere able to manage.achieve for our patients without the use of prescription opioid medications, which can often lead to prolonged use and addiction.”
We have also identified a marketThe Company has entered into distribution partnerships for PainShield in the professional sports industry, where in some cases reimbursement may be available from sports alumni organizations or, more likely, self-pay. In orderUnited States, Israel, India, Malta, Australia, and New Zealand. We also sell direct to pursue this market, we are exhibiting at sports trainers meetings, pursuing alumni associations, advertising in their media, and, on January 18, 2018, signed an agreement with a leading medical device distributor, which has a sports trainer focused sales organization. The PainShield device is offered for sale to practitioners with a provider rental program which was implemented in January 2017. We are also contemplating establishment of a direct rental program with a rent-to-own option. The PainShield product was also modified and enhanced through various accessories for use within the equine community. We are contemplating pursuing this market through prominent equine clinicians and independent sales representatives and distributors. We believe there is an attractive opportunity in this segment due to the lack of an expectation for reimbursement and the opportunity to sell at a premium price point. We are pursuing appropriate distributors in the U.S. market with resources and qualifications to sell PainShield in the different segments of the pain treatment market.consumer worldwide.
WoundShield has been evaluated in two published clinical studies done to-date that suggest improved localized blood flow and oxygenation, and improved topical oxygen saturation (Morykwas M, “Oxygen Therapy with Surface Acoustic Waveform Sonication,” European Wound Management Association 2011; Covington S, “Ultrasound-Mediated Oxygen Delivery to Lower Extremity Wounds,” Wounds 2012; 24(8))). We supplied devices for these studies but had no further involvement with them. We are pursuing licensing opportunities to develop commercial markets for the WoundShield product.
Recent Developments
In April 2021, the U.S. Centers for Medicare and Medicaid Services (CMS) expanded its reimbursement approval of the company’s PainShield™ product by adding the device to its Durable Medical Equipment (DME) schedule.
In August 2021, the Therapeutic Goods Administration (TGA), the Australian Regulatory body for Therapeutic Devices, granted approval for the Company’s PainShield for use by patients in Australia. We since have sold product through our Australian distributor.
In addition, August 2021, we announced our intention to enter the Over-the-Counter (OTC) pain treatment market with the introduction of PainShield Relief™, a non-prescription ultrasound therapy device that delivers fast pain relief for nerve and soft tissue damage. Submission to the FDA had been delayed, but is now anticipated to be filed in the second quarter of 2022.
In September 2021, we announced that we were contemplating launching a new, transdermal gel containing Nano-Cannabidiol (CBD) for the treatment of joint pain and reducing inflammation. An ongoing impact analysis is being conducted internally. The adoption of the CBD technology is currently be re-evaluated due to regulatory constraints and regulations.
In September 2021, we announced that we entered into a Provider Participation Agreement with Orchid Medical, a national provider of integrated ancillary and surgical cost containment solutions specifically for the workers’ compensation industry. The agreement facilitates the reimbursement of the company’s PainShield™ product for eligible patients receiving benefits under a worker’s compensation plan making it easier for plan beneficiaries to obtain the product for the treatment of pain. We continue to focus on this market, and we hope to add additional agreements with other providers for the workers’ compensation market.
| 4 |
In October 2021, we received positive results from a randomized, double-blind study conducted at Birmingham Orthopedic and Sports Specialists in Birmingham, Alabama for our Painshield™ product.
In December 2021, we signed an agreement with Applied Medical Solutions, LLC (“AMS”) for the sale and distribution of its PainShield products to Veterans facilities located throughout the United States.
Additionally, in December 2021 we received positive interim results from an independent, real world patient study of its UroShield at Southampton University Health Sciences.
Additionally, in December 2021 we filed three U.S. patent applications with the U.S. Patent & Trademark Office related to its SAW technology and indwelling medical devices to protect targeted new product launches and improvements to existing medical devices.
On February 26, 2021, Protrade Systems, Inc. (“Protrade”) filed a Request for Arbitration (the “Request”) with the International Court of Arbitration (the “ICA”) of the International Chamber of Commerce alleging the Company is in breach of an Exclusive Distribution Agreement dated March 7, 2019 (the “Agreement”) between Protrade and the Company. Protrade alleges, in part, that the Company has breached the Agreement by discontinuing the manufacture of the DV0057 Painshield MD device in favor of an updated 10-100-001 Painshield MD device. Protrade claims damages estimated at $3 million.
On March 15, 2022, the arbitrator issued a final award, which, although finding that Protrade’s claims failed as a matter of law or were unsupported by the evidence, nevertheless awarded Protrade $1,500,250, which consists of $1,432,000 for “lost profits” and $68,250 as reimbursement of arbitration costs, on the grounds that the Company allegedly failed to supply Protrade with “requested patches.” We continue to dispute the claims asserted by Protrade and intend to pursue the available options to vacate or seek correction of the award to Protrade. As of December 31, 2021, the Company accrued the amount of the award to Protrade amounting to $1,500,250 as part of “General and administrative expenses” and “Other accounts payable and accrued expenses”.
In March 2022, we announced that PainGuard and UroGuard were approved by Health Canada / Santé Canada as licensed medical devices.
Business Model
All of our products consist of a reusable controller device and a disposable component, or transducer.which includes a transducer, and in the case of PainShield, a 30 day supply of adhering patches. The controllers have a life expectancy of up to three years, while the disposable transducer has a life expectancy of up to a month and must be replaced to provide the intended therapy. The components are purchased by either the distributor or end user for use in any of the intended applications. Once the controller is purchased by the end user, recurring revenue will be realized by purchases of replacement transducersdisposables to the extent that the end user continues treatment with our product.
In all product categories, ourOur products are intended to be distributed both by independent distributors as well as by potential licensees. Distributor cost is discounted to account for their intended margins, based upon purchase volumes and/or periodic purchase commitments, with the disposable transducer sold and distributed in the same fashion. We currently have an established distributor network and are implementing certain criteria within such network to ensure the appropriate assignment of a distributor or licensee. We also intend to add additional distributors to our network.
We also have a direct sales component, where we sell directly to consumers, in order to satisfy customer demand generated through on-line advertising and social media. We expect that demand to increase once we are able to start marketing our PainShield Relief assuming it receives FDA clearance
In August 2019, we established our first license agreement with Medisana, Inc. (“Medisana”) with a total of 1,500 devices that was shipped to Medisana directly from China, in April 2020. The devices were designed to carry the product labeling specific to the Medisana brand, with the product name of PT100. The product labeling includes the words, “PAINSHIELD Ultrasonic therapy, Medisana”. All instructions for use and packaging are specific to Medisana.
| 5 |
Our business plan continues to focus on these types of transactions/agreements. We continue to focus on the foundational aspects of each respective product, including the design and performance of each, the reimbursement, regulatory status, and quality control, in order to strengthen our position with prospective partners.
Ultrasound Technology and Our Products
As noted above, our primary products are based on the use of low frequency ultrasound, which delivers energy through mechanical vibrations in the form of sound waves. Ultrasound has long been used in physical therapy, physical medicine, rehabilitation and sports medicine. Moreover, there is a growing body of research that supports the positive biological effects of ultrasound. A 2002 study indicates that low frequency ultrasound increases nerve regeneration (Crisci AR, Ferreira AL, “Low-intensity pulsed ultrasound accelerates the regeneration of the sciatic nerve after neurotomy in rats”, Ultrasound Med. Biol. 2002 October; 28(10):1335-41). According to Atland, et. al., low frequency ultrasound also has important therapeutic metabolic effects (Altland OD, Dalecki D, Suchkova VN, Francis CW, “Low-intensity ultrasound increases endothelial cell nitric oxide synthase activity and nitric oxide synthesis”, J. Thromb. Haemost. 2004 April; 2(4):637-43). In addition, there is evidence that ultrasound increases the healing of fractures (Warden SJ, Favaloro JM, Bennell KL, McMeeken JM, Ng KW, Zajac JD, Wark JD, “Low-intensity pulsed ultrasound stimulates the bone-forming response in UMR-106 cells”, Biochem. Biophys. Res. Commun. 2001 August 24; 286(3):443-50 and Warden SJ, Bennell KL, McMeeken JM, Wark JD, “Acceleration of fresh fracture repair using the sonic accelerated fracture healing system (SAFHS)”, Calcif. Tissue Int. 2000 February; 66(2):157-63).
Research has further shown that ultrasound therapy has resulted in increased collagen repair (Da Cunha A, Parizotto NA, Vidal BC, “The effect of therapeutic ultrasound on repair of the achilles tendon (tendo calcaneus) of the rat”, Ultrasound Med. Biol. 2001 December; 27(12):1691-6), improved resolution of inflammation (Young SR, Dyson M, “Macrophage responsiveness to therapeutic ultrasound”, Ultrasound Med. Biol. 1990; 16(8):809-16) and increased tissue healing (Young SR, Dyson M, “Effect of therapeutic ultrasound on the healing of full-thickness excised skin lesions”, Ultrasonics. 1990 May; 28(3):175-80), which are all important factors in the wound healing process. Furthermore, research has shown that ultrasound therapy can contribute to increased membrane permeability (Sundaram J, Mellein BR, Mitragotri S, “An experimental and theoretical analysis of ultrasound-induced permeabilization of cell membranes,” Biophys. J. 2003 May; 84(5):3087-101) and accelerated fibrinolysis, a process that prevents blood clots from growing and becoming problematic (Harpaz D, “Ultrasound enhancement of thrombolytic therapy: observations and mechanisms”, Int. J. Cardiovasc Intervent. 2000 June; 3(2):81-89), which collectively improve the tissue regeneration process and healing of wounds. Sonophoresis, a process that increases the absorption of semisolid topical compounds, including medications, into the skin, is an additional significant effect of ultrasound therapy (Tezel A, Paliwal S, Shen Z, Mitragotri S, “Low-frequency ultrasound as a transcutaneous immunization adjuvant”, Vaccine 2005 May 31; 23(29):3800-7).
In general, ultrasound offers the benefits cited above by increasing local blood circulation, increasing vascular wall permeability, promoting protein secretion, promoting enzymatic reactions, accelerating nitric oxide production, promoting angiogenesis (the formation of new blood vessels from pre-existing vessels) and promoting fibroblast proliferation (fibroblasts are a type of cell that play a critical role in wound healing). We believe that the body of evidence, and the positive therapeutic effect that ultrasound has for various indications, potentially provides for future product development opportunities for us.
Our proprietary technology consists of a small, thin (1 millimeter) transducer that is capable of transmitting ultrasonic acoustic waves onto treatment surfaces with a radius of up to 10 centimeters beyond the transducer. This technology allows us to treat wounds by implanting our transducers into a small, portable self-adhering acoustic patch, thereby eliminating the need for technicians and medical personnel to manually administer ultrasound therapy, which should reduce the cost of therapy. Moreover, we believe that, based upon the body of evidence, the delivery of ultrasound through our portable devices is equal to or more effective than existing competitive products, as our technology is better positioned to target the affected areas of the body.
While there are currently a number of products on the market that treat pain through ultrasound therapy, we believe that our products differentiate themselves because they are portable, without the requirement to be plugged into an outlet and they operate withhave a frequency of 100kHz (in contrast to other devices, which have a frequency of 1MHz)closer to 1MHz and above), which means our products, dowhen functioning as intended and in accordance with applicable design specifications, should not produce heat that can damage tissue. Our products can therefore (i) be self-administered by the patient without the need to be moved about the treated area by the patient or a clinician, (ii) be applied for a significantly longer period without the risk of tissue damage and (iii) do not require the use of gel. We are also aware of one competitive product, with similar ultrasound technology, the SAM® Sport4, which has recently received U.S. Food and Drug Administration approval and has CE Mark approval, marketed by a company called Zetroz Systems LLC, aka ZetrOz,ZetrOZ, Inc., that we understand may eliminate certain of these requirements and limitations, namely the requirement to be plugged in, the need for movement around the treated area and the relatively short safe treatment period. However, it is our beliefwe understand that this product does not generate surface acoustic waves as our products do, which means that the treatment area is generally limited to that ofunder the transducer’s diameter,transducer, that the use of transmission gel is still required, and that the transducer thickness is significantly greater than ours (approximately 1.5cm). ToIt is also our knowledge,understanding that the device only provides a battery life of 4 hoursU.S. Food and is continuous therapy versus intermittent therapy. WeDrug Administration has issued contraindications which do not apply to the PainShield product.
There has been an article published in 2019 on SAM® Sport4 regarding clinical evidence demonstrating that ultrasound dose timing (i.e. daily treatment) and duration significantly impact benefits and treatment results, we are also aware of a small clinicalprospective randomized, double-blinded, placebo-controlled study for which results were reported in August 2013, in whichon the effects of the long-duration low-intensity ultrasound treatment using SAM® Sport4 showed positive results insuggesting that ultrasound may be used as a conservative non-pharmaceutical and non-invasive treatment option for patients with knee osteoarthritis.
In general, ultrasound offers the treatmentbenefits by increasing local blood circulation, increasing vascular wall permeability, promoting protein secretion, promoting enzymatic reactions, accelerating nitric oxide production, promoting angiogenesis (the formation of venous ulcers,new blood vessels from pre-existing vessels) and promoting fibroblast proliferation (fibroblasts are a type of chronic wound.cell that play a critical role in wound healing). We believe that the body of evidence, and the positive therapeutic effect that ultrasound has for various indications, potentially provides for future product development opportunities for us.
Our proprietary technology consists of a small, thin (1 millimeter) transducer that is capable of transmitting ultrasonic acoustic waves onto treatment surfaces with a radius of up to 10 centimeters beyond the transducer. This technology allows us to treat wounds by implanting our transducers into a small, portable self-adhering acoustic patch, thereby eliminating the need for technicians and medical personnel to manually administer ultrasound therapy, which should reduce the cost of therapy. Moreover, we believe that, based upon the body of evidence, the delivery of ultrasound through our portable devices is equal to or more effective than existing competitive products, as our technology is better positioned to target the affected areas of the body.
| 6 |

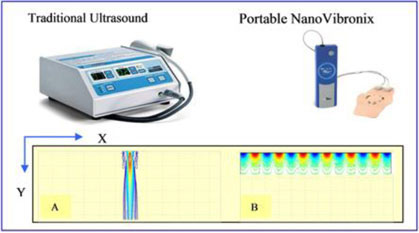
Traditional ultrasound device and our portable ultrasound patch-based device and a comparison of their energy distribution, where the X-axis represents treatment surface, and the Y-axis represents ultrasound energy penetration depth within tissue.
In a comparison of a traditional ultrasound device and our portable ultrasound patch-based device, the bulk wave conventional ultrasound machines with handheld transducers distribute the energy deeply into the body, as shown above in diagram (A) on the left. In comparison, our device distributes the energy on the surface, as shown in diagram (B), thereby meaningfully increasing the treatment area. Our transducers may also be incorporated into treatment patches, including patches that are designed to deliver medicine and other compounds through the skin. The generation and delivery of low frequency ultrasound over a period of time to a specific area has been termed “targeted slow-release ultrasound”. We believe that this delivery method of ultrasound may be comparable to that of slow release medication in the pharmaceutical industry. This “targeted slow-release” capability is intended to allow for more frequent targeting of the intended treatment area and thus may result in a more effective therapeutic response.
Micro Vibrations Technology and Our Products
It is well established that increasing blood flow to the wound and peri-wound area helps accelerate the healing of ischemic wounds. Micro-vibrations applied on the skin tissue increase local blood flow and oxygen delivery to the wound area and stimulate angiogenesis and growth factors that are helpful for the wound healing process. Vibration therapy has been found to stimulate blood flow due to mechanical stresses of endothelial cells resulting in increased production of nitric oxide and vasodilation, as well as increase soft tissue and skin circulationcirculation. (Maloney-Hinds et al., “The Role of Nitric Oxide in Skin Blood Flow Increases due to vibration in healthy adults and adults with type 2 diabetes,” School of Medicine, Loma Linda University. Ca. Diabetes Technology & Therapeutics, 2009 p. 39-43). In addition, micro vibrations induce skin surface nerve axon reflex and type IIa muscle fibers contraction rates, resulting in vasodilation (Nakagami et al., “Effect of vibration on skin blood flow in an in vivo microcirculatory model”, The University of Tokyo, Bio-Science Trends 2007; 1 (3): 161-166). Ten minutes of vibration therapy with laser doppler revealed a consistent increase in water content of the upper dermis (TJ Ryan et al., “The effect of mechanical forces (vibration or external compression) on the dermal water content of the upper dermis and epidermis, assessed by high frequency ultrasound”, Oxford Wound Healing Institute, Journal of Tissue Viability, 2001. In another study, mean blood flow increase was higher in the vibration group than the placebo group. Improvements in local blood flow may be beneficial in the therapeutic alleviation of pain or other symptoms resulting from acute or chronic injuries (C. Button et al., “The effect of multidirectional mechanical vibration on peripheral circulation of humans”, University of Otago New Zealand, Clinical Physiology and functional Imaging, 2007 27, p211-216). A study on the effect of whole body vibration on lower extremity skin blood flow suggests, that short duration vibration alone significantly increases lower extremity skin blood flow, doubling skin blood for a minimum of 10 minutes following treatment (Lohman et al., “The effect of whole body vibration on lower extremity skin blood flow in normal subjects”, Department of Physical Therapy, Loma Linda university, USA, Med Sci Monit, 2007; 13(2) 71-76). Vibration has also been shown to stimulate angiogenesis and growth factors such as vascular endothelial growth factor (Suhr F et al., “Effects of short-term vibration and hypoxia during high intensity cycling exercise on circulating level of angiogenic regulators in humans”, J Appl Physiol, 2007, 103:474-483,. Yue Z. et al., “On the cardiovascular effects of whole-body vibration I. Longitudinal effects: hydrodynamic analysis”, Studies Appl Math, 2007, 119:95-109). Of import with respect to diabetic wounds, in which a prolonged inflammatory phase occurs, vibration vasodilation has generated an indirect anti-inflammatory action, mainly by suppression of nuclear factor-kβ, the key gene for inflammatory mediators (Sackner, M.A., “Nitric Oxide is released into circulation with whole-body, periodic acceleration”, Chest 2005;127;30-39).
| 7 |
Urinary catheter usage is associated with pain and discomfort caused by the friction between the catheter surface and the urethral tissue. Generally, this friction is treated by applying lubricating gels and low friction catheter coatings. These methods are effective for a short term during the catheter insertion as the lubricating gel is quickly absorbed into the surrounding tissue and loses its effect and the catheter coatings lose their lubricity within a few days, as the coating is covered by a thin film of mucous.
Our UroShield product provides vibrations along the surface of the urinary catheter that is in contact with urethral tissue. We believe that these vibrations create a continuous acoustic lubrication effect along the surface of the indwelling catheter that is in contact with the surrounding tissue, thus reducing catheter-tissue contact time, which may lessen trauma from urethra abrasion and adhesion. We have also shown in animals and in humans that the micro-vibration technology can reduce the level of biofilm formation on urinary catheters.
Our Products
Product Design, Packaging, Identity
All products were redesigned in the fourth quarter 2019, with an updated look and improved performance. These new designs were coupled with new branding, packaging, instructional manuals, and marketing materials. Beginning in the fourth quarter of 2019, our manufacturers in China have commenced producing the redesigned products for distribution and delivered their first completed units in April 2020.
UroShield
UroShield is intended to prevent bacterial colonization and biofilm formation, increase antibiotic efficacy in the catheter lumen and decrease pain and discomfort associated with urinary catheter use. It is designed to be used with any type of indwelling urinary catheter regardless of the material or coating. We believe that if it is approved for marketing, UroShield couldmay be the first medical device on the market that attempts to simultaneously address all of the aforementioned catheter-related issues. UroShield is similar in design to WoundShield and PainShield, in that it uses a driver unit that produces low frequency, low intensity ultrasound. The driver unit connects to a disposable transducer that is clipped onto the external portion of the catheter to deliver ultrasound therapy to all catheter surfaces as well as the tissue surrounding the catheter.

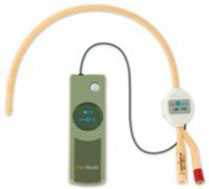
Picture of UroShield with actuator
| 8 |
The
We believe the UroShield system has the following advantageous effects:
| ● | Prevention or Reduction of Biofilm. The low frequency ultrasound generated by UroShield has been shown to decrease adherence of bacteria to catheter surfaces, thereby reducing biofilm. Biofilm is the complex matrix required for bacteria to grow and cause infection. See the discussion of our Heidelberg 1 trial below. |
| ● | Decreased Catheter Associated Pain and Discomfort. We believe that UroShield creates an acoustic envelope on the surfaces of the catheter, which decreases friction and tissue trauma, pain and discomfort caused by the catheter. In addition, in vivo (rabbit) studies have shown the tissue in contact with the catheter remains healthier and less traumatized as a result of the application of low frequency and low intensity ultrasound (Applebaum I, et.al., “The Effect of Acoustic Energy Induced By UroShield on Foley Catheter Related Trauma and Inflammation in a Rabbit Model” Department of Urology, Shaarey Zedek Medical Center and the Hadassah Hebrew University Medical School). |
| ● | Acoustically Augmented Antibiotic Therapy. Antibiotic resistance in biofilm bacteria is a well-known phenomenon. Although it has been known that ultrasound can increase antibiotic efficacy in in-vitro models, we do not believe that there has been a practical ultrasound-based medical device that was able to augment antibiotic efficacy in the clinical setting. In a clinical study, UroShield technology has been shown to eradicate biofilm-residing bacteria by greater than 85% when applied simultaneously with an antibiotic in three clinically relevant species, escherichia coli, staphylococcus epidermidis and pseudomonas aeruginosa (Banin E, et al., “Surface acoustic waves increase the susceptibility of Pseudomonas aeruginosa biofilms to antibiotic treatment,” Biofouling, August 2011; we supplied devices for this study, but had no further involvement with it). |
| ● | Preservation of the Patency of Catheters. We believe that low frequency ultrasound applied to catheters will add an anti-clogging effect and will preserve patency of catheters. This effect is achieved by ultrasound waves creating an acoustic layer on the inner lumen of the urinary catheter, thereby preventing adherence of biological material and biofilm formation. We believe that this anti-clogging benefit will help prevent local infection and sepsis secondary to catheter obstruction. |
UroShield has undergone a number of clinical trials. The Heidelberg 1 trial, conducted in 2005-2006, which we sponsored, was a 22 patient randomized, double blind, sham-controlled, independent trial that tested UroShield’s safety and ability to prevent biofilm in patients with an indwelling Foley catheter. The trial demonstrated that UroShield prevented biofilm in all patients with the active device as compared to biofilm being found in seven of eleven of the control patients. In addition, there was a marked decrease in pain, discomfort and spasm in the active UroShield patients, as evidenced by a statistically significant decrease in the requirement for the medications required to treat urinary catheter associated pain and discomfort (Ikinger U, “Biofilm Prevention by Surface Acoustic Nanowaves: A New Approach to Urinary Tract Infections?,” 25th World Congress of Endourology and SWL, Cancun, Mexico, October 2007).
In a subsequent physician-sponsored trial, known as Heidelberg 2, conducted in 2007, 40 patients who underwent radical prostatectomies were divided into two groups, with the active group receiving one intra-operative dose of antibiotics and UroShield and the control group receiving one intra-operative dose of antibiotics and then five subsequent doses over three days. At the end of the trial, the control group had four cases of bacteruria,bacteriuria, as compared to one in the active group. In a third trial, a physician-sponsored open label trial, ten10 patients who received emergency placement of a urinary catheter due to acute obstruction were given a UroShield device and followed with regard to their pain, discomfort, spasm and overall well-being. Within 24 hours, all patients showed improvement and increased toleration of the catheter (Zillich S., Ikinger U, “Biofilmprävention durch akustische Nanowellen: Ein neuer Aspekt bei katheterassoziierten Harnwegsinfektionen?,” Gesellschaft für Urologie, Heilbronn, Germany, May 2008). We supplied devices for this trial, but had no further involvement with it.
As recently announced, the Company submitted to The National Institute for Health and Care Excellence, for review, the findings from an independent evaluation of its UroShield® device on patients who had used the device for up to two years. Clinical data from the study conducted by Coventry University’s Assistant Professor, Ksenija Maravic da Silva, during 2020 reported statistically significant outcomes for the device including a reduced number of urinary tract infections (UTIs), reduced instances of prescribed antibiotics, reduced catheter blockages, reduced the need for unplanned catheter changes and reduced pain reported as a result of catheter associated complications. The study also provided important insights into the lives of those using the device including improvement of overall well-being, relating specifically to decreased levels of worry and increased ability to socialize. In addition, patient feedback on product improvements was addressed and has been incorporated in the present commercially available device.
| 9 |
Market for UroShield
According to the Centers for Disease Control and Prevention, urinary tract infection (UTI) is an infection involving any part of the urinary system, including urethra, bladder, ureters, and kidney. UTIs are the most common type of healthcare-associated infection reported to the National Healthcare Safety Network (NHSN). Among UTIs acquired in the hospital, approximately 75% are associated with a urinary catheter, which is a tube inserted into the bladder through the urethra to drain urine. Between 15-25% of hospitalized patients receive urinary catheters during their hospital stay. The most important risk factor for developing a catheter-acquired urinary tract infection (CAUTI) is prolonged use of the urinary catheter.
This study was written up in the December 2018 issue of “Medical & Surgical Urology”, a leading peer-reviewed journal in the field of urology.
Approximately 25%15-25% of patients who are admitted to a hospital will have an indwelling catheter at some point during their stay and 7% of nursing home residents are managed by long term catheterization.
Catheter acquired urinary tract infection (CAUTI)
CAUTI is the most common nosocomial infection in hospitals and nursing homes, representing over 40% of all hospital-acquired infections (HAIs) and 20% of intensive care unit HAIs (Maki, P and Tambyah, D. Engineering Out the Risk for Infection with Urinary Catheters., Emerging Infectious Diseases., Vol. 7, No. 2, March–April 2001). In addition, CAUTIs are the source for approximately 20% of healthcare acquired bacteremia in acute care and 50% in long-term care facilities (Nicolle, Lindsay E. “Catheter Associated Urinary Tract Infections.” Antimicrobial Resistance and Infection Control 3 (2014). The risk of acquiring CAUTI depends on the method and duration of catheterization and patient susceptibility. Patients requiring a urinary catheter have a daily risk of approximately five percent of developing bacteriuria and approximately 25% of patients develop nosocomial bacteriuria or candiduria over one week (Maki, P and Tambyah, D. Engineering Out the Risk for Infection with Urinary Catheters., Emerging Infectious Diseases., Vol. 7, No. 2, March–April 2001). Virtually all patients requiring indwelling urinary catheters for longer than a month become bacteriuric.
CAUTI occurs because urethral catheters inoculate organisms into the bladder and promote colonization by providing a surface for bacterial adhesion and causing mucosal irritation. The presence of a urinary catheter is the most important risk factor for bacteriuria. Once a catheter is placed, the daily incidence of bacteriuria is 3-10%. Between 10% and 30% of patients who undergo short-term catheterization (ie,(i.e., 2-4 days) develop bacteriuria and are asymptomatic. Between 90% and 100% of patients who undergo long-term catheterization develop bacteriuria. About 80% of nosocomial UTIs are related to urethral catheterization; only 5-10% are related to genitourinary manipulation. (John L. Brusch, Catheter-Related Urinary Tract Infection, Medscape, August 18, 2015).
According to a report by Zion Market Research, theThe global catheter market totaled approximately $26.6size was valued at USD 37.3 billion in 20152018 and is estimatedexpected to grow atwitness a CAGR of 9.7% through 2021. In2026. Rising prevalence of chronic disorders leading to hospitalization has fueled the United Statesgrowth of this market. Presence of multi-national manufacturers, improving medical facilities, supportive insurance policies are also some of the key factors propelling the market growth. North America is the largest regional market due to the presence of multi-national manufacturers and sophisticated healthcare infrastructure along with high product awareness levels. Asia Pacific is projected to expand at the maximum CAGR of 10.4%, over the study period. According to a Grandview research report published 2018, there are 25 million Foley catheters sold annually in the United States and there are 75 million catheters sold elsewhere yielding a total global Foley catheter market of 100 million units worldwide. The cost to treat a simple CAUTI has been estimated at $675$13,793 per case (AHRQ), and the cost of treating bacteremia has been estimated at $3,800$8,355 (NIH) per case, yielding a total healthcare burden of $830 million per year. While there are currently both antibiotic and silver coated catheters in the market, they often sell for approximately $10 above the non-antimicrobial equivalent.
In addition, as of October 1, 2008, Medicare stopped authorizing its payment to hospitals in which patients have developed a catheter-associated urinary tract infection that was not present on admission. This provides hospitals in the United States with a substantial financial incentive to reduce the occurrence of such infections through the use of products such as UroShield, which help prevent infections hospitals would otherwise have to treat without reimbursement. In addition, it has been noted that the Centers for Medicare & Medicaid Services may fine hospitals in the future when their patients develop CAUTI, which will likely increase the incentive of hospitals to invest in technologies that may prevent this complication (Brown J, et al. “Never Events: Not Every Hospital-Acquired Infection Is Preventable, Clinical Infectious Diseases, 2009, 49 (5)).
| 10 |
Competition for UroShield
Several types of products have been introduced to address the growing problem of catheter-acquired infection and biofilm formation on catheter surfaces. Manufacturers offer antibiotic-coated and antiseptic-impregnated catheters. In addition, manufacturers have produced silver-coated catheters, which have been shown in small studies to delay bacteruriabacteriuria for about two to four days. However, larger studies did not corroborate this result; on the contrary, silver hydrogel was associated with overgrowth of gram positive bacteria in the urine (Riley DK, Classen DC, “A large randomized clinical trial of a silver-impregnated urinary catheter: lack of efficacy and staphylococcal superinfection,” Am. J. Med. 1995 April; 98(4):349-56).
UroShield has been designed to be added to any type of catheter, including Foley catheters and silver-coated catheters, to improve a catheter’s infection prevention performance. UroShield is not intended to replace any existing products or technologies, but instead is intended to assist these existing products or technologies in preventing catheter-acquired urinary injury and catheter associated complications. While UroShield has been approved by the U.S. Food and Drug Administration (“FDA”) under Enforcement Discretion during the COVID-19 health emergency, if we do not obtain permanent clearance from the FDA, UroShield may be unable to successfully compete in this market due to an inability to obtain such permanent clearance from the U.S. Food and Drug AdministrationFDA and failure to be adopted by health care practitioners and facilities.
Regulatory Strategy
UroShield received CE Mark approval in September 2007 and was also approved for sale by the Israeli Ministry of Health in 2008. We are able to sell UroShield in India and Ecuador based on our CE Mark. UroShield was granted a Canadian medical device license in September 2016. 2016, although, due to a modification of regulatory standards in Canada, we have lost our Canadian license. We are working toward reinstatement of our Canadian license. To that extent, we passed an audit in November 2020 with a notified body and we are waiting on a certificate.
In the European Union, UroShield has been marketed for the prevention of biofilm, decreased pain and discomfort associated with urinary catheters and increased antibiotic efficacy.
In September 2020, the FDA exercised its Enforcement Discretion to allow distribution of the UroShield device in the United States,States. According to the FDA, “UroShield® device can use Intended Use Code (IUC) 081.006: Enforcement discretion per final guidance, and FDA product code QMK (extracorporeal acoustic wave generating accessory to urological indwelling catheter for use during the COVID-19 pandemic)”.
Accordingly, the FDA’s Enforcement Discretion clears the way for import of UroShield to the U.S. during the Covid-19 pandemic, immensely expanding the company’s addressable market for the device during this time period. The device is designed to aid in the prevention of CAUTI incidence in patients requiring long-term indwelling catheterization.
After reviewing the body of scientific evidence that we presented, the FDA took decisive action to clear the way for patient access to UroShield for the duration of the Covid-19 pandemic. The evidence presented to the FDA on UroShield demonstrated decreases in the risk of catheter-associated urinary tract infections and related complications in patients using UroShield who required long-term indwelling catheterization. Importantly, we are unaware of any other commercially available device that can prevent catheter-associated urinary tract infection incidence and achieve results comparable to UroShield.
We intend to seek 510(k) clearance from the U.S. Food and Drug Administration through the de novo classification process for UroShield. We submitted our application for 510(k) clearance on January 3, 2011. On March 11, 2011, we received a response from the U.S. Food and Drug Administration proposing that the approval go through the de novo route, which will require clinical trials with proposed study protocols to be pre-cleared by the U.S. Food and Drug Administration. We are currently seeking advice from the FDA prior to submission.
The FDA has made it clear that we will need to generate more clinical study data in order to achieve 510(k) clearance. Our intent is to conduct a strategic partner that is activecommunity based PRO study (Patient Reported Outcomes) measuring the impact UroShield will have on prevention of CAUTI, Prevention of Blockage, and prevention of Pain.
| 11 |
Studies completed to establish safety of UroShield for human use:
| ● | A large animal model (female sheep) study has been conducted to establish local tissue response from a urinary catheter with UroShield attached as compared to a control group of animals with a urinary catheter with no UroShield attached. The pre-clinical animal study was intended to demonstrate safety of UroShield device when used for 30-days with a urinary catheter. The study compared local tissue and organ response in two groups of 4 (female) sheep where one group was catheterized (urethral) using an uncoated silicone Foley catheter (only) and the other group was catheterized using an uncoated silicone Foley catheter with UroShield device attached to it. All catheters were identical in their size, material composition and manufacturer. After 30 days the animals were euthanized and local tissue and organs were examined. The results showed the group with UroShield device had fewer observations of swelling, redness or discharge at the vulva as compared to the group without UroShield. The animals did not exhibit signs of discomfort or pain during study period (of 30 days). The gross and histopathology findings were also very similar between the two groups. | |
| ● | A comparative study of leachables from a urinary catheter with and without UroShield attached has been performed to demonstrate that the leachables with UroShield attached do not exceed toxicological safe limits allowed for a medical device. The chemical characterization of leachables was intended to demonstrate safety for UroShield device for 30-day use with a urinary catheter. The study compared leachables from a group consisting of 3 uncoated silicone catheters with leachables from a group consisting of 3 uncoated silicone catheters with UroShield attached to it. All catheters were identical in their size, material composition and manufacturer. The exhaustive extractions were performed with non-polar, polar and aqueous solvents. An additional simulated use extraction using Saline and Ethanol was performed. Overall the extractables from both groups were comparable and toxicological evaluation showed that all compounds from extraction with UroShield were below the tolerable exposure limits. Most of compounds had a margin of safety greater than 10 and 4 compounds had margin of safety between 1.5 and 10. Overall, the toxicological risk for using UroShield with a urinary catheter is similar and at even lower as compared to a catheter without UroShield attached. |
Sales and Marketing
Since the FDA exercised its Enforcement Discretion to allow the distribution of the UroShield device in the urology market to coincide with the U.S. Food and Drug Administration clearance. WeUnited States, we have not made any further submissions to the U.S. Food and Drug Administration related to UroShield, but we recently completed a more robust study conducted at 5 different nursing facilitiesbeen actively seeking partnerships for marketing our product in the United States. This study was approved by the institutional review board, or IRB. In November 2017, we announced interim results of this study.
Sales and Marketing
We believe the business opportunity for UroShield is in the hundreds of millions in U.S. dollars to the extent that UroShield obtains permanent 510(k) clearance from the U.S. Food and Drug Administration,FDA, is recognized as effective and becomes widely adopted for use inon catheters. To that end, we are exploring sales distribution modelsseeking a strategic partnership with various companies which have an existing “footprint” in the United States through a distributor networkUrology market. Those discussions and direct sales. In order to have a distribution network in place if UroShield receives clearance from the U.S. Food and Drug Administration, wenegotiations are currently identifying distributors through several vehicles, including our sales staff, commissionable representation, and independent contractors.ongoing at this time. We have recently appointed distributors for UroShield in the United KingdomKingdom. Malta, and Australia. We recently appointed Med Tech Solutions Group (MTSG) to assist in our sales and marketing efforts in countries we are not represented. MTSG is based in San Antonio Texas and has sales representation around the globe.
We announced in May 2020, that we had expanded our license agreement with Ideal Medical International Limited to include exclusive rights to distribute the Company’s UroShield® and PainShield® technologies in Canada and an outside management organization, Morulaa Health, to assist with regulatory matters and distribution of UroShield in India. Each of these distributors is paid a small retainer and will be paid a commission between 10 to 20% of sales going forward.Turkey.
| 12 |
From time to time we have had interest from strategic companies in the catheter market to partner, license or acquire the UroShield technology. These strategic partners are active in the urology market and may be interested in integrating UroShield as an accessory, into its range of products. Discussions with these partners are ongoing. There has also been interest from other companies with various invasive line applications.
Clinical Trials
To date, we have conducted the clinical trials set forth below:
| Purpose | Doctor/Location | Time, subjects | Objectives | Results | ||||
To assess the safety of the UroShield Double Blind, Comparative, Randomized Study for the Safety Evaluation of the UroShield System (HD1) | Dr. U. Ikinger, Salem Academic Hospital, University of Heidelberg, Germany | 2005-2006 22 patients | To demonstrate that the use of the UroShield is safe and that the device is well tolerated by the patients and user friendly to the medical staff. Efficacy objectives were to demonstrate that the UroShield helps in prevention of biofilm formation in comparison with the urinary catheter alone, as well as bacteriuria. | UroShield was both safe and well tolerated. UroShield proved efficacious in prevention of biofilm. Subjects required significantly less medications than the control group for catheter related pain and discomfort. | ||||
Double Blind, Comparative, Randomized Study for the Safety Evaluation of the UroShield System (HD2 ) Physician initiated | Dr. U. Ikinger, Salem Academic Hospital, University of Heidelberg, Germany | 2007 40 patients | To demonstrate that the use of the UroShield is safe and helps in prevention of biofilm formation and UTI in comparison with the urinary catheter alone, as well as decrease antibiotic use. | In this trial, only 1/20 patients in UroShield device (no antibiotics) group developed urinary tract infection compared to 4/20 patients within control group treated with the antibiotic prophylaxis alone. |
The Effect of UroShield on Pain and Discomfort in Patients Released from the Emergency Room with Urinary Catheter Due to Urine Incontinence Physician initiated | Shaare Zedek Medical Center Jerusalem, Israel. | 2007 10 patients | The study aimed to assess the effectiveness of the UroShield in reducing pain and discomfort levels and improve the well-being of the subjects. Efficacy objectives included reduction of pain, spasm, burning and itching sensation levels of the subjects. | The results demonstrated a reduction in pain, itching, burning and spasm levels. Additionally, the well-being of the subjects showed a significant increase. | ||||
The Use of the UroShield Device in Patients with Indwelling Urinary Catheters Open labeled, comparative, randomized study | Dr. Shenfeld Shaare Zedek Medical Center Jerusalem, Israel. | 2007-2009 40 patients | Patient complaints related to catheter regarding pain according to VAS scale and discomfort according to 0-10 scale Presence of Clinically Significant UTI Presence of Bacteriuria Presence of Biofilm Use of medication | UroShield device was effective in reducing postoperative catheter related pain discomfort and bladder spasms. There was also a notable trend towards reduction of bacteriuria. |
| 13 |
| Purpose | Doctor/Location | Time, subjects | Objectives | Results | ||||||
Evaluation of the UroShield in urinary and nephrostomies to reduce | Prof. P.Tenke, Hungary |
|
| ● Pain, disability and QOL ● Catheter patency ● Bacteriuria / UTI ● Hospitalization period ● Analgesics and Antibiotics intake | Showed reduction in pain and significant decrease in bacteriuria rate. | |||||
| Double Blind, Randomized Control Study for Prevention of Bacterial Colonization and UTI associated with Indwelling Urinary Catheters | Dr. Shira Markowitz Buffalo, NY | 2017
| To demonstrate the use of the UroShield reduces bacterial colonization on the urinary catheter | Final results | ||||||
| Mean improvement advantage in treatment vs control was 87.2K CFU, (t (53) 18.1, p<0.001) at thirty days. At 60 days the mean improvement advantage in treatment vs control was 87.5K CFU, (t (53) 18.1, p<0.001). At 90 days the mean improvement advantage in treatment vs control was 79.3K CFU, (t (53) 12.4, p<0.001). |
| 14 |
| Purpose | Doctor/Location | Time, subjects | Objectives | Results | ||||
After cessation of treatment in the active group at 30 days, there was a minimal increase in CFU count at both 60 and 90 days. In the same group, there was no statistical difference in the decrease of CFU count from 30 to 60 days after treatment, t (28)=1. p= .326, however there was a marginally significant increase in CFU from 60 to 90 days for the active group (28)=1.7 p= 0.09.
At baseline, every enrolled patient had been treated for infection during the 90 days prior to enrollment. Compared to baseline, the treatment group showed significant statistical and clinical improvement (100%) at 30 days relative to the sham control (73%). There were no reported infections in the Treatment Group while in the control group there were seven reported infections. At 90 days after treatment, the treatment group showed a significantly stronger improvement (89.7%) compared to the sham control (46.2%). There were three reported infection in the Treatment group, while in the control group there were fourteen reported infections requiring antimicrobial therapy. (logistic regression B=2.3, Wald Chi-Square (df=1) =10.1, p=0.001.) |
| 15 |
| Purpose | Doctor/Location | Time, subjects | Objectives | Results | ||||
| UroShield Randomized Control trial | 5 different nursing facilities | 2017 - 2018 51 subjects | 51 subjects were evaluated with 26 in the active/treatment group and 25 in the control group. All patients had been treated for at least one incident of a catheter-acquired urinary tract infection (CAUTI) requiring antibiotics in the preceding 6 months prior to trial initiation. | At the 90-day At |
Recently Completed, Current, Ongoing and Planned Clinical Trial
If we are able to locate a strategic partner or otherwise obtain sufficient funding, we anticipate conducting the following clinical trial:
| Trial | Place | Start Date/Timing | Objectives | |||
| UroShield U.S. Food and Drug Administration trial 80 patient trial | To be determined | To be determined | Safety and efficacy of UroShield in urinary catheter related pain and infection and biofilm formation. The results of previous clinical trials may not be predictive of future results, and the results of our planned clinical trial, if we are able to locate a strategic partner or otherwise obtain sufficient funding, may not satisfy the requirements of the |
PainShield®
PainShield is an ultrasound device, consisting of a reusable driver unit and a disposable patch, which contains our proprietary therapeutic transducer. It delivers a localized ultrasound effect to treat pain and induce soft tissue healing in a targeted area, while keeping the level of ultrasound energy at a safe and consistent level of 0.4 watts. We believe that PainShield is the smallest and most portable therapeutic ultrasound device on the market and the only product in which the ultrasound transducer is integrated in a therapeutic disposable application patch.
The existing ultrasound therapy devices being used for pain reduction are primarily large devices used exclusively by clinicians in medical settings. PainShield is able to deliver ultrasound therapy without being located in a health care facility or clinic because it is portable, due to it being lightweight and battery operated. Because it is patch based and easy to apply, PainShield does not require medical personnel to apply ultrasound therapy to the patient. The patient benefits include ease of application and use, faster recovery time, high compliance, and increased safety and efficacy over existing devices that rely on higher-frequency ultrasound (Adahan M, et al, “A Sound Solution to Tendonitis: Healing Tendon Tears With a Novel Low-Intensity, Low-Frequency Surface Acoustic Ultrasound Patch,” American Academy of Physical Medicine and Rehabilitation Vol. 2, 685-687, July 2010). PainShield can be used by patients at home or work or in a clinical setting and can be used even while the patient is sleeping. Its range of applications includes acute and chronic pain reduction and anti-inflammatory treatment.
| 16 |
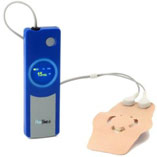
Picture of PainShield with Patch
In other countries outside the United States where the product is approved for such use, PainShield is used to treat tendon disease and trigeminal neuralgia (a chronic pain condition that affects the trigeminal or 5th cranial nerve, one of the most widely distributed nerves in the head); previously, the therapeutic options for these disorders have been very limited. In the United States, PainShield is only cleared to treat pain, muscle spasms, and joint contractures associated with or caused by various conditions or diseases. It has also been used to treat pelvic and abdominal pain. To date, to the best of our knowledge, the only treatment options for several of these conditions are pain medication and surgery. Several additional causes of pain, and the treatment of that pain with the PainShield product, can be explored through clinical trials.
Market for PainShield
Pain-related complaints are one of the most common reasons patients seek treatment from physicians (Prince V, “Pain Management in Patients with Substance-Use Disorders,” Pain Management, PSAP-VII, Chronic Illnesses). According to Landro L, “New Ways to Treat Pain: Tricking the Brain, Blocking the Nerves in Patients When all Else Has Failed,” Wall Street Journal, May 11, 2010, approximately 26% of adult Americans, or approximately 76.5 million people, suffer from chronic pain. The National Center for Health Statistics has estimated that approximately 54% of the adult population experiences musculoskeletal pain. Studies have shown that low-frequency ultrasound treatment has yielded positive results for a variety of indications, including tendon injuries and short-term pain relief (Warden SJ, “A new direction for ultrasound therapy in sports medicine,” Sports Med. 2003; 33 (2):95-107), chronic low back pain (Ansari NN, Ebadi S, Talebian S, Naghdi S, Mazaheri H, Olyaei G, Jalaie SA, “Randomized, single blind placebo controlled clinical trial on the effect of continuous ultrasound on low back pain,” Electromyogr Clin Neurophysiol. 2006 Nov; 46(6):329-36) and sinusitis (Ansari NN, Naghdi S, Farhadi M, Jalaie S, “A preliminary study into the effect of low-intensity pulsed ultrasound on chronic maxillary and frontal sinusitis,” Physiother Theory Pract. 2007 Jul-Aug; 23(4):211-8). We believe that PainShield’s technology, portability and ease of use may result in it becoming an attractive product in the pain management and therapy field.
Competition
There are numerous products and approaches currently utilized to treat chronic pain. The pharmacological approach, which may be the most common, focuses on drug-related treatments with the over-the-counter internal analgesic market estimated at $3.8$19 billion in 2013.2019. Alternatively, there are a large number of non-pharmacological pain treatment options available, such as ultrasound, transcutaneous electrical nerve stimulation, or TENS, laser therapy and pulsed electromagnetic treatment. In addition, there are some technologies and devices in the market that utilize low frequency ultrasound or patch technology. Many patients are initially prescribed anti-pain medication; however, ongoing use of drugs may cause substantial side effects and lead to addiction. Therefore, patients and clinicians have shown increased interest in alternative pain therapy using medical devices that do not carry these side effects.
| 17 |
The currently available ultrasound treatments for chronic pain have generally been accepted by the medical community as standard treatment for pain management. However, the traditional ultrasound treatments, such as those manufactured or distributed by Mettler Electronics Corp, Metron USA and Zimmer MedizinSysteme, are stationary devices found only in clinics and other health care facilities that need to be administered to patients by health care professionals. We are aware of three companies that market smaller ultrasound devices capable of certain self-administered use for the treatment of pain: Koalaty Products, Inc., Sun-Rain System Corp. and PhysioTEC. These devices generally function in the same manner, at the same frequency and with the same administration and safety requirements and limitations as traditional, larger ultrasound devices. We are also aware of one product, the SAM® Sport4, which has recently received U.S. Food and Drug Administration approval and also has CE Mark approval, marketed by ZetrOZ, Inc., that we understand may eliminate certain of these requirements and limitations, namely the requirement to be plugged in, the need for movement around the treated area and the relatively short safe treatment period. However, we understand that this product does not generate surface acoustic waves as our products do, which means that the treatment area is generally limited to that under the transducer, that the use of transmission gel is still required, and that the transducer thickness is significantly greater than ours (approximately 1.5cm). It is also our understanding that the U.S. Food and Drug Administration has prohibitedissued contraindications which do not apply to the manufacturer from labeling or promoting this product for use directly over bone that is near the skin surface. PainShield product.In addition, there are other patch-based methods of pain treatment, such as TENS therapy. TENS therapy may be painful and irritating for the patient due to the muscle contractions resulting from the electrical pulses. PainShield combines the efficacy of ultrasound treatment for pain with the ease of use and portability of a patch-based system. PainShield also may be self-administered by the patient, including while the patient is sleeping. However, if we are unable to obtain widespread insurance coverage and reimbursement for PainShield, its acceptance as a pain management treatment would likely be hindered, as patients may be reluctant to pay for the product out-of-pocket.
The CMS has approved PainShield for reimbursement for Medicare beneficiaries on a national basis effective January 2020.
Regulatory Strategy
PainShield received 510(k) clearance from the U.S. Food and Drug Administration in August 2008 for treatment of pain relief. PainShield received CE Mark approval in July 2008 and was also approved for sale by the Israeli Ministry of Health in 2010. We have a Canadian medical device license for PainShield and we are able to sell PainShield in India and Ecuador based on our CE Mark. We are in discussions with distributors in Southeast Asia, and, if a distributor is engaged, intend to seek regulatory approvals for PainShield in Southeast Asia through such distributor.
In the United States, a prescription from a licensed healthcare practitioner is required for the use of PainShield. We have engaged
Recently, we announced our intention to market a consultantPainShield Relief product. The PainShield Relief is intended to assist us in the process of reclassifying the PainShield device to remove thebe an Over-The Counter (OTC) product, not requiring a prescription requirement for the use of PainShield.from a medical professional. We believe that such reclassification will open up mass market opportunities which are currently not available to us due to the prescription requirement. However, there is no assurance that we will be able to remove the prescription requirement for the use of PainShield Relief or that, even if we accomplish such reclassification and the use of PainShiledPainShield Relief no longer requires a prescription, PainShield Relief will be successful commercially in the mass market or we will be able to generate significant revenues from the mass market opportunities, if any.
In order to eliminate the requirement for a physician prescription, proof of safety and consumer “usability” need to be established. With no adverse events reported on the PainShield MD device when we conmmenced the Usability study, we anticipated favorable results from such study. We engaged User-View, Inc to facilitate our Usability study and received the favorable results we expected. The product packaging and all instruction documents have been modified to meet OTC standards. We also engaged an outside laboratory to perform acoustic testing on all PainShield products. We previously anticipated submission of a 510(k) for PainShield Relief to the FDA, for OTC use as a class 1 device, in early April 2022, but we are reconsidering our target timeline for such submission and whether any additional data or action steps are needed. We currently expect to submit the 510(k) to the FDA in the second quarter of 2022.
The PainShield Plus, is a dual applicator device, which will also be submitted for specific clearance from the FDA. Submission for PainShield Plus was made in late February 2022.
In the United States, PainShield falls under the diathermy classification for the treatment of pain for initial reimbursement purposes. The permitted reimbursement codes can be used in the outpatient supervised medical setting. We intendcontinue to coordinatework with the Centers for Medicare and Medicaid Services and private insurers so that reimbursement can be extended to cover the administration of PainShield outside of health care facilities and clinics. We have engaged outside legal counsel to assist with all aspects of reimbursement. In addition, we intend to conduct clinical trials in order to effectively market PainShield for a larger range of indications. The targeted reimbursement would be based upon specific indications, where study data serves as justification for payment.
Sales and Marketing
PainShield was introduced in 2009 as a treatment for pain, such as tendonitis, sports injuries, pelvic pain, and neurologic pain, depending on the scope of the approval or clearance from each applicable jurisdiction, and we have sold approximately 1,700over 5,000 units and 15,000 treatment patches since its introduction. We have entered into distribution agreements in North America,United States, Europe, AsiaAustralia, and India for the distribution of PainShield. We intend to seek additional distribution opportunities in Europe, East Asia and Ecuador. In addition, we sell PainShield directly to patients through our website.website in jurisdictions where direct-to-consumer sale is permitted. We are currently ramping up our marketing efforts in North Americathe U.S. market and throughout the world. We anticipate that these efforts will include recruiting additional sales personnelworld to establish licensing and representatives, making in-office calls to physicians and attending trade shows and conferences. We intend to pursue the veterinary market with our equine PainShield device.private label partnerships as well.
| 18 |
We have identified a unique and effective application for PainShield in applicable foreign jurisdictions where such application is authorized, which is the treatment of a severe facial nerve pain called Trigeminal Neuralgia, otherwise known as tic douloureux. The FDA lists facial application as a contraindication and has not cleared or approved PainShield for such use in the United States. We are in the process of pursuing FDA approval of the PainShield for Trigeminal Neuralgia, which will likely require additional data and clinical investigation to support an application for premarket approval (“PMA”) for this indication, if such PMA is required by FDA. Two studies were performed in Israel, “a randomized control trial examining the efficacy of low intensity low frequency Surface Acoustic wave ultrasound in trigerminaltrigeminal neuralgia pain”, and “A sound solution for TrigerminalTrigeminal Neuralgia”. Two trials which enrolled a total of 16 and 15 patients respectively, both conducted at the Sheba Medical Center in Israel, concluded that this study supports the hypothesis that the application of Low Intensity Low Frequency Surface Acoustic Wave Ultrasound (LILF/SAW) may be associated with a clinically significant reduction of pain severity among patients suffering from trigeminal neuralgia disease. One of the studies showed a reduction in pain among 73% of the participants. We believe this to be an ideal market to address with the PainShield. With few existing treatment alternatives, we believe the PainShield’s effectiveness isPainShield could prove to be a practical and safe alternative. A broader RCT, targeting 60 patients suffering from unilateral trigeminal neuralgia, was recentlyalso completed. The article was published on January 22, 2019, in the Journal of Anesthesiology and Pain Research, under the title “The Effect of a Surface Acoustic Wave (SAW) Device on the Symptomatology of Trigeminal Neuralgia”. We cannot predict the success of any future trials, nor can we guarantee that FDA will be completed and readygrant approval for submission within 90 days.such use.
GlobalData’s epidemiological analysis forecasts that the total prevalent cases of trigeminal neuralgia in the seven major markets (United States, France, Germany, Italy, Spain, U.K and Japan) will grow at 15% between 2012 and 2022. According to an estimate by Ronald Brisman, M.D., in 2013 the prevalence of trigeminal neuralgia in the U.S. may have been as high as approximately 280,000 patients. With the favorable results from our current, ongoing study (explained in detail below), we continue to plan to aggressively pursue this market in the foreign jurisdictions where PainShield has been approved through direct marketing efforts and distributor relationships.
We have also identified a market for PainShield in the professional sports industry, where in some cases, reimbursement may be available from sports alumni organizations or, more likely, self-pay. In order to pursue this market, we are exhibiting at sports trainers meetings, pursuing alumni associations, advertising in their media, and have recently engaged a national distributor in the United States. Discussions and ongoing negotiations continue with other appropriate distributors in these various market segments.
Recently Completed Research
A double blind randomized control trial of a Painshield Surface Acoustic Wave Patch, the patch used in conjunction with the PainShield device, was completed in the first quarter of 2018.
After the enrollment and lead-in period, subjects were given a sham device to sleep with every night for a month. They were asked to fill out their pain and analgesic use logs, and undergo the bi weekly assessments. After a month they were crossed over to an active “Painshield SAW patch device” and continued to complete their pain and analgesic use logs as well as undergo biweekly assessments for months two and three of the study.
In the fourth quarter of 2017, the interim results were reported on 45 patients that had been evaluated as fully completing the study. Results from these patients showed a greater than 3 point difference in pain measured by the Visual Analog Scale between the active treatment group and the control group. This was further validated in the crossover group from sham to active groups. Patients also showed quality of life by greater than 35% in the treatment group versus the control group, which was validated in the crossover group. We have submitted comprehensive data from this clinical trial for expected publication in a peer review journal in the second quarter of 2018, at which time we plan to report the final results.
Clinical Trials
To date, we have conducted or are in the process of conducting the clinical trials set forth below:
| Purpose | Doctor/Location | Time, subjects | Objectives | Results | ||||
A sound solution for | Dr. Ch. Adahan Sheba Medical Center | 2009 15 patients | ●Reduction in pain ●Reduction in disability ●Improvement of function and quality of life ●Accelerating of healing | 73% of the subjects experienced complete or near complete relief. | ||||
| Randomized control trial examining the efficacy of low intensity low frequency Surface Acoustic wave ultrasound in | Dr. M. Zwecker Chaim Sheba Medical Center, Tel Hashomer, Israel | 2012-2012 16 patients | ●Reduction in pain ●Reduction in disability ●Improvement of function and quality of life ●Accelerating of healing | In conclusion this study supports the hypothesis that the application of Low Intensity Low Frequency Surface Acoustic Wave Ultrasound (LILF/SAW) may be associated with a clinically significant reduction of pain severity among patients suffering from trigeminal neuralgia disease. |
| 19 |
| Purpose | Doctor/Location | Time, subjects | Objectives | Results | ||||
| Treating Rutgers university athletic injuries with bandaid sized ultrasound unit PainShield | R. Monaco, G. Sherman, Rutgers University Athletic, Rutgers, New Jersey | 2011 35 patients | ●To assess the pain, functional capacity and discomfort of the subject ●To assess the subject’s quality of life ●To assess the injury status ●To assess the efficacy of the treatment ●To assess compliance factors | Active group: 74% had improvement, 26% no change Sham group: 56% no change, 44% had improvement This is an indication of the effectiveness of the device. Lack of funding for statistical analysis has stopped this trial prior to fulfillment. |
| Reduction of chronic abdominal and pelvic pain, urological and GI symptoms using wearable device delivering low frequency ultrasound | D. Wiseman, Synechion Institute for Pelvic Pain | 2011 19 patients | ●To assess the efficacy of PainShield for pelvic and related pain | Improvement in pain related symptoms noted for all symptoms. | ||||
| Dr. David Lemak, a leading orthopedic surgeon with Birmingham Orthopedic and Sports Specialists. | 2019, 24 patients | A randomized, double blinded study for 30 days that evaluated the effectiveness and safety of PainShield™ Surface Acoustic Wave (SAW) technology on patients suffering from pain and discomfort, as well as limited mobility caused by the effects of chronic or acute lateral epicondylitis (LE) (“tennis elbow”). | We plan to publish an article at the time and in conjunction with adding a marketing partner. | |||||
| The Effect of a Surface Acoustic Wave (SAW) Device on the Symptomatology of Trigeminal Neuralgia | Shira Markowitz, MD, New York, NY | Early 2018 |
| 20 |
If we are able to obtain sufficient funding, we anticipate conducting the following clinical trials:
| Trial | Place | Start Date/Timing | Objectives | |||
PainShield for Pelvic Pain 200 patient trial | To be determined | To be determined | Safety and Efficacy of PainShield in Chronic Pelvic Pain |
WoundShield®
Our WoundShield product was granted the European Wound Closure Customer Value Leadership Award, Ultrasound Therapy – Wound Closure in 2014. WoundShield is intended to treat acute and chronic wounds with a disposable treatment patch that delivers localized therapeutic low frequency ultrasound. The WoundShield patch has two configurations: one that is placed adjacent to the wound and another, called the instillation patch, that is placed on the wound to enable instillation through sonophoresis, a process that increases the absorption of semisolid topical compounds, including medications, into the skin. Based on studies conducted by BIO-EC Microbiology Laboratory and Rosenblum, we believe that our WoundShield product possesses significant potential for the treatment of, among other things, diabetic foot ulcers and burns (Gasser P, Study Report delivered by BIO-EC Microbiology Laboratory, Dec 2007, which we ordered, paid for, and provided devices for; Rosenblum J, “Surface Acoustic Wave Patch Diathermy Generates Healing In Hard To Heal Wounds,” European Wound Management Association 2011, for which we supplied devices but had no further involvement). In March 2020, we signed a license agreement with Sanuwave Health, Inc. (“Sanuwave”) for the manufacture and delivery of our WoundShield technology. Under the terms of the agreement, NanoVibronix received 127,000 warrants of Sanuwave stock upon signing, will receive a $250,000 milestone payment based on FDA approval, and 10% royalty on Sanuwave’s gross revenues from sales or rentals of WoundShield. In return, Sanuwave has received the worldwide, exclusive rights to the Company’s WoundShield product and technology. In addition, Sanuwave will bear the costs and clinical validation responsibilities associated with obtaining approval for WoundShield from the U.S. Food and Drug Administration and other regulatory agencies around the world.


Picture of WoundShield Driver and Instillation Patch
WoundShield delivers surface acoustic waves to the location of the wound. Surface acoustic waves move laterally across the surface of the wound, which enables the transfer of the acoustic energy of the waves along the entire wound surface in a continuous and consistent mode, providing access to the waves’ benefits for a longer treatment period than conventional ultrasound without the need for supervision or a treatment session by a clinician.
The technology has been found to have a positive effect on the epithelialization (healing by the growth of epithelial cells) of diabetic wounds, as well as on the stimulation of the precursors of dermal and epidermal (skin) growth. As such, it is a useful adjunct to wound care by increasing dermal and epidermal growth, including glycosaminoglycans, or GAGs (which bind to extracellular proteins like collagen, fibronectin, laminin, etc. and retain considerable amounts of water, thus preserving the skin structure) as well as the amount of collagen (a protein that helps skin heal) and decreasing the number of cells in mitosis (a type of cell division) (Rosenblum J, “Surface Acoustic Wave Patch Diathermy Generates Healing In Hard To Heal Wounds,” European Wound Management Association 2011, for which we supplied devices which were precursors to WoundShield, but had no further involvement). In addition, the WoundShield instillation patch allows for administration of therapeutic agents into the wound area through a sonophoresis effect.
| 21 |
Many key processes in wound healing are dependent upon an adequate supply of oxygen. Diabetic foot ulcers are particularly in need of an adequate oxygen supply because the disease often results from poor perfusion (blood flow) and decreased oxygen tension. Oxygen is also important for the immune system to combat bacteria, synthesize collagen, help with fibroblast proliferation (fibroblasts are a type of cell that play a critical role in wound healing), form oxidative (taking place in the presence of oxygen) pathways for adenosine triphosphate, or ATP, formation (ATP transports chemical energy within cells for metabolism), and the nitric oxide dependent signaling pathways. It is generally believed that a lack of available oxygen is a basic contributing factor in the perpetuation of these wounds. Recently, woundWound healing experts have developed a technique of perfusing ischemic wounds (which occur when blood flow is blocked) with hyper-oxygenated saline, while the wound is being treated with ultrasound, also known as sonication. This localized oxygenation therapy has many advantages over the use of hyperbaric chambers (large chambers in which the oxygen pressure is above normal), a common method for delivering oxygen to wounds, as it is more cost-effective, can be done at the patient’s bedside and can be administered more frequently. The WoundShield instillation patch was tested as a potential ultrasound technology for this localized oxygen therapy. In one study (Morykwas M, “Oxygen Therapy with Surface Acoustic Waveform Sonication,” European Wound Management Association 2011; we supplied devices for this study, but had no further involvement with it), oxygen sensors were placed in the wound bed to directly measure partial pressure of oxygen in an ischemic wound bed on a pig. The wound was perfused with hyperbaric oxygen and sonicated using the WoundShield instillation patch. With surface acoustic wave ultrasound technology, tissue oxygen levels (partial pressure of oxygen in the blood, or PaO2) were raised from a range of 20 mmHg (millimeters of mercury) to 60 mmHg in peripheral (periwound) areas, a 3 centimeter distance away from the transducer, and from 40 mmHg to greater than 100 mmHg in the central wound bed lying below the WoundShield instillation patch (see table below). The results of this study illustrated that the WoundShield instillation patch allowed oxygen to directly enter into the wound. The direct entry of the oxygen increased the amount of oxygen reaching the wound, which has been shown to advance the healing process. In addition, we believe that WoundShield’s small size, lower cost and ease of use makes localized oxygen treatment commercially viable.

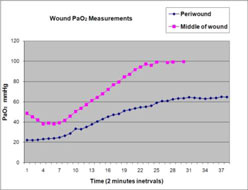
In 2012, results were published of a human feasibility trial for the WoundShield instillation patch that was performed at Duke University in North Carolina. Seven patients were treated with the WoundShield instillation patch for their wounds and average tissue oxygen levels (PaO2) increased by an average of 58% over baseline (Covington S, “Ultrasound-Mediated Oxygen Delivery to Lower Extremity Wounds,” Wounds 2012; 24(8)). We supplied devices for this trial, but had no further involvement with it. Based upon the results of this trial, we are planning a series of clinical trials, which we expect to begin in the fourth quarter of 2017, with an end point claim that our WoundShield product enhances perfusion in chronic wounds.
| 22 |
Market for Wound-Healing Devices
The global wound care device market totaled approximately $24 billion in 2015 and it is expected to grow at a CAGR of 6.7% during 2016-2022 (as reported by P&S Global Research in January 2017). According to the Global Report on Diabetes produced by the World Health Organization (“WHO”) in 2016, globally, an estimated 422 million adults were living with diabetes in 2014, compared to 108 million in 1980. According to a report entitled “Advances in Wound Closure Technology” by Frost and Sullivan (2005), foot complexities are the most frequent causes for patients with diabetes to get hospitalized, with complications usually starting with the formation of skin ulcers. In addition, according to the American Burn Association, approximately 486,000 patients received medical treatment annually for burn injuries in 2016 in the United States. There are also policy-based factors that may increase the size of the wound care market. We anticipate that reimbursement decisions with respect to hospital acquired wounds may create a large market opportunity for wound care products, including WoundShield. Furthermore, in 2009, the Centers for Medicare and Medicaid Services announced that they would stop reimbursements for treatment of certain complications that they believed were preventable with proper care. One such complication was surgical site infections after certain elective procedures, including some orthopedic surgeries and bariatric surgery. We believe that such developments incentivize medical care providers to invest in reducing the risk of infection through the use of wound care products, including WoundShield.
Competition for WoundShield
The market for advanced wound care includes a number of competitors, such as Kinetic Concepts, Inc. (a subsidiary of the 3M Company), or KCI, Smith and Nephew plc and Convatec Inc., all of whom market wound-healing medical devices. Due to their size, in general these companies may have significant advantages over us. These competitors have their own distribution networks for their products, which gives them an advantage over us in reaching potential customers. In addition, they are vertically-integrated, which may allow them to maximize efficiencies that we cannot achieve with our third-party suppliers and distributors. Finally, because of their significantly greater resources, they could potentially choose to focus on research and development of technology similar to ours, more than we are able to. In general, we believe that these competitors have, and will continue to have, substantially greater financial, technological, research and development, regulatory and clinical, manufacturing, marketing and sales, distribution and personnel resources than we do. However, we believe that our products differentiate us from these competitors, and we will be competitive on the basis of our technology. We believe that the strength of these competitors may create an opportunity through strategic partnerships.
At present, ultrasound treatment for wounds is limited only to wound debridement (removal of damaged tissue or foreign objects from a wound) and such products are marketed by Arobella Medical, LLC, which produces the Qoustic Wound Therapy System, Misonix Inc., which produces SonicOne products, and Alliqua Biomedical, Inc., which produces the MIST Therapy System. Due to their size, in general these companies may have the same advantages over us as discussed with respect to our competitors in the paragraph above. However, these ultrasound devices are indicated for use only in medical clinics and require an operator to deliver their treatment, thus limiting their use and application. The MIST Therapy System and Quostic Therapy System are a non-contact ultrasound device that delivers ultrasound through a mist that is applied directly on the wound.
We believe that these therapies are less advantageous than WoundShield because they require an operator to deliver the treatment and the removal of bandages to target the wound bed. In contrast, the WoundShield patch sits on normal skin bordering the open wound and no manipulation of the wound bandage is required. Moreover, WoundShield can be self-administered, without an operator, in both clinics and home settings. We also believe that WoundShield will prove to be an effective alternative to treating chronic wounds at a lower price than the existing products being used by medical practitioners. As such, we believe that facilities that are reimbursed based upon diagnosis-related groups will be more inclined to adopt WoundShield because it will provide the same therapeutic results at a significantly lower cost than traditional ultrasound therapies.
We are also aware of a small clinical study, for which results were reported in August 2013, in which a small ultrasound device showed positive results in the treatment of venous ulcers, a type of chronic wound. Based upon currently available information about this device, we believe it will be at least 2018 before this device is available on the market for treatment of venous ulcers. We understand that this product does not generate surface acoustic waves as our products do, which means that the treatment area is generally limited to that of the transducer’s diameter. We believe our products would have certain other advantages over this potential device, if developed, including that our products weigh less and are thinner. However, given the early stage of development of this potential device, we cannot say with certainty how our products would compare.
| 23 |
The most common method of oxygen administration for wound healing is hyperbaric oxygen therapy, especially to treat specific ulcerations in diabetic patients. Hyperbaric oxygen therapy has been shown to increase vascular endothelial growth factor expression, which measures the creation of new blood vessels (Fok TC, at el, “Hyperbaric oxygen results in increased vascular endothelial growth factor (VEGF) protein expression in rabbit calvarial critical-sized defects”, Schulich School of Medicine and Dentistry, University of Western Ontario, Canada). The activation of endothelial cells by VEGF sets in motion a series of steps toward the creation of new blood vessels (J Lewis et al, National Cancer Institute, Understanding Cancer and Related Topics, Understanding Angiogenesis). We believe that the WoundShield instillation patch, which can be used as an oxygen instillation system, will be complementary to, or in some cases an alternative to, the use of hyperbaric chamber therapy. This complementary treatment option will allow the treating physician greater therapeutic versatility in treating wounds. For a certain populace of patients, we believe that the WoundShield instillation patch could provide physicians with an alternative to hyperbaric oxygen therapy because it provides the same benefits as hyperbaric oxygen therapy at a lower cost to the patient. There are a number of competitors in the hyperbaric chamber therapy market, including approximately eight companies in the United States. Due to their size, in general these companies may have the same advantages over us discussed with respect to our competitors in the first paragraph of this section. However, we believe that the WoundShield instillation patch possesses certain advantages over the existing hyperbaric chamber therapy, including lower cost and greater ease of use. In addition, we do not believe that the WoundShield instillation patch will not necessarily compete with hyperbaric chamber therapy, but rather will often complement such therapy.
While we believe that WoundShield is well positioned to capture a share of the wound care market, WoundShield may be unable to achieve its anticipated place in the wound care market due to a number of factors, including, but not limited to, an inability to obtain the approval of the U.S. Food and Drug Administration, for which it is indicated and its failure to be adopted by health care practitioners and facilities or patients because of its status as a new product in a market that relies on patient-focused initiative to treat wounds.
Regulatory Strategy
For a general discussion of the U.S. Food and Drug Administration approval process with respect to our products, and regulation of our products in general, see “– Government Regulation” below.
Our general regulatory strategy for WoundShield ishas been focused on seeking U.S. Food and Drug Administration approval for a variety of indications. WoundShield obtained CE Mark approval in November 2012,2012. Sanuwave has received the worldwide, exclusive rights to the Company’s WoundShield product and obtained Canadian License approvaltechnology. Accordingly, the Company does not expect to continue to directly engage in November 2016, bothsales and marketing activities for use in wound healing.the WoundShield technology and expects Sanuwave to undertake such activities.
Sales and Marketing
WoundShield has generated minimal revenues to date. We intend to aggressively marketIn March 2020, we signed a license agreement with Sanuwave Health, Inc. for the manufacture and delivery of our WoundShield in Europe and Canada, and pursue the necessary approvals to commence marketing in the United States. Our strategy for selling WoundShield in the United States is to find a strategic partner in the wound care market. We are actively pursuing this strategy. WoundShield could be an effective adjunct to existing wound treatment devices or a stand alone wound treatment modality.technology.
Clinical Trials
With respect to WoundShield, to date, we have conducted the following evaluation studies:
| Purpose | Doctor/Location | Time, subjects | Objectives | Results | ||||
Clinical evaluation Physician initiated | Dr. J. Rosenblum, Shaare Zedek Medical Center | 2008 8 patients | To evaluate novel technology on wound healing in diabetic foot ulcers. | Therapy showed significant changes in wound, wound size was reduced, patients felt less pain, necrotic tissue was less adhesive, necrotic tissue decreased in size. The duration of the trial was one week. | ||||
Clinical evaluation Physician initiated | Dr. J. Rosenblum, Shaare Zedek Medical Center | 2010 8 patients | To evaluate novel technology on wound healing in diabetic foot ulcers. | The device, a precursor device to WoundShield using the same technology as | ||||
Clinical evaluation Physician initiated | Dr. S. Covington | 2010 7 patients | The study aimed to determine if hyper oxygenated saline delivered by surface acoustic waves improves tissue oxygenation in lower extremity wounds. | Surface acoustic wave technology in conjunction with oxygenated saline can increase interstitial oxygen in wound bed. This trial to validate proof of concept was put on hold due to financial constraints. The duration of the trial was two weeks. |
| 24 |
Third Party Reimbursement
NanoVibronix has entered into an agreement with Redemption Revenue Cycle Solutions LLC (“RRCS”), beginning on January 1, 2019. RRCS has an expertise in establishing reimbursement at a reasonable rate, and facilitating the billing for both NanoVibronix and its distributors. We have also retained McGuireWoods to assist in improving our PainShield reimbursement.
We anticipate that sales volumes and prices of the products we commercialize will depend in large part on the availability of coverage and reimbursement from third party payers. Third party payers include governmental programs such as Medicare and Medicaid, private insurance plans and workers’ compensation plans, among others. These third party-party payers may deny coverage and reimbursement for a product or therapy, in whole or in part, if they determine that the product or therapy was not medically appropriate or necessary. The third partythird-party payers also may place limitations on the types of physicians or clinicians that can perform specific types of procedures. In addition, third party payers are increasingly challenging the prices charged for medical products and services. Some third party-party payers must also pre-approve coverage for new or innovative devices or therapies before they will reimburse health care providers who use the products or therapies. Even though a new product may have been approved or cleared by the U.S. Food and Drug Administration for commercial distribution, we may find limited demand for the device until adequate reimbursement has been obtained from governmental and private third party-party payers.
Over-the-counter products, such as the anticipated PainShield Relief product that we are developing, are generally not reimbursed by any third-party payers.
In international markets, reimbursement and healthcare payment systems vary significantly by country, and many countries have instituted price ceilings on specific product lines and procedures. There can be no assurance that procedures using our products will be considered medically reasonable and necessary for a specific indication, that our products will be considered cost-effective by third party payers, that an adequate level of reimbursement will be available or that the third party-party payers’ reimbursement policies will not adversely affect our ability to sell our products profitably.
In the United States, some insured individuals are receiving their medical care through managed care programs, which monitor and often require pre-approval of the services that a member will receive. Some managed care programs are paying their providers on a per capita basis, which puts the providers at financial risk for the services provided to their patients by paying these providers a predetermined payment per member per month, and consequently, may limit the willingness of these providers to use certain products, including ours.
One of the components in the reimbursement decision by most private insurers and governmental payers, including the Centers for Medicare and Medicaid Services, which administers Medicare, is the assignment of a billing code. Billing codes are used to identify the procedures performed when providers submit claims to third party payers for reimbursement for medical services. They also generally form the basis for payment amounts.
Obtaining reimbursement approval for a product from any government or other third party-party payer is a time-consuming and costly process that could require us or our distributors to provide supporting scientific, clinical and cost-effectiveness data for the use of our product to each payer. Even if a code is obtained for a product, a third party-party payer must still make coverage and payment determinations. When a payer determines that a product that is eligible for reimbursement, the payer may impose coverage limitations that preclude payment for some uses that are approved by the FDA or other foreign regulatory authorities. We believe that the overall escalating costs of medical products and services has led to, and will continue to lead to, increased pressures on the health care industry to reduce the costs of products and services. In addition, recent health care reform measures, as well as legislative and regulatory initiatives at the federal and state levels, create significant additional uncertainties. There can be no assurance that third party coverage and reimbursement will be available or adequate, or that future legislation, regulation, or reimbursement policies of third party-party payers will not adversely affect the demand for our products or our ability to sell these products on a profitable basis. The unavailability or inadequacy of third party-party payer coverage or reimbursement would have a material adverse effect on our business, operating results and financial condition.
| 25 |
UroShield.We expect these products to be used in inpatient settings and therefore reimbursed under the DRGDiagnosis Related Group (DRG) or per diem reimbursement system. In addition, in an outpatient or home setting, we anticipate that these products will initially be purchased privately until a reimbursement code is obtained. However, we believe that if we can empirically demonstrate UroShield’s efficacy in preventing recurrent hospitals admission in chronic Foley catheter patients and reducing overall per-patient cost, third party payers may accelerate the reimbursement approval process since the device could reduce their overall per-patient cost. We believe the natural progression of the adoption of this technology will allow for use in the home setting. We intend to pursue reimbursement in the Medicare Part B code to support the use for long term catheter use and infection prevention in the home.
PainShield. Although it is a minimal amount, PainShield is presently reimbursed in the United States by many private insurers under the national umbrellaEffective as of January 2020, CMS approval for diathermy service, CPTMedicare reimbursement was added through code 97024, for useK1004. The value of the ultrasound device in a supervised medical setting and is reimbursed in 15-minute increments for up to an hour a day, 5 hours a week and 20 hours a month. The current reimbursement mechanism is inadequate to support the end user or distributor cost of the device. If the device is efficacious in the treatment of the patient’s condition, the treatment period can be extended in some cases for months. Presently, when used in an outpatient setting, such as by a clinic, PainShield is typically purchased by the clinic that then can bill the existing reimbursement codes. PainShield ishas not currently reimbursed for therapy in the home setting. When we have sufficient funding, we intendyet been confirmed. We continue to work to obtaintoward a favorable reimbursement in the home setting as well as codes that would allow forwith outside legal counsel and reimbursement for use of the non-disposable and disposable components of the PainShield device. Our anticipated clinical trials for PainShield would support this effort. In the United States, Painshield requires a prescription from a physician.consultants.
WoundShield.We believe that the initial usage of these products will be in the hospital setting. Reimbursement in the hospital setting is typically governed by the Diagnosis Related Group system, or DRG system, which is a prospective payment methodology that assigns a predetermined, fixed amount based on the patient’s diagnoses. In parallel to introducing these devices to hospitals, we intend to apply forSuch reimbursement codes for outpatient use. Although obtaining these codes can take years and may require extensive clinical data, we believe thatwill be sought by Sanuwave Health Inc. as the desirable characteristicslicensee of these products may serve as an incentive to insurance companies to grant these codes more quickly.this technology.
New ProductsProduct Under Development
Renooskin
In 2016, we started developing a device for the facial rejuvenation market called Renooskin. Previous in vitro studies on human skin were done showing that the SAW technology provided skin rejuvenation comparable to Retinol A which is a well-accepted anti-aging cream. We have developed a head band like applicator for the PainShield SAW treatment and are in the process of arranging for a pilot trial with a cosmetic dermatologist and/or plastic surgeon. We believe that, subject to proof of efficacy of the Renooskin and receiving regulatory approval, the device can be sold in a non-reimbursement market since cosmetic devices are private pay. We expectare still considering several paths towards commercialization.
Intellectual Property
Stemming from a combination of patent, copyright, trademark and trade secret laws, as well as non-disclosure agreements and other contracts, our intellectual property rights represent a vital resource to complete the developmentmanagement of our company. Therefore, we are continuing our practice of investing in obtaining appropriate legal protection for our innovations whenever possible and have adopted a more fully integrative approach to the prototypemanagement of the Renooskin product by June 2018,our intellectual property that mutually aligns with our ongoing R&D strategies, commercial opportunities based on market analyses, and Renooskin is currently under evaluation by a major cosmetics companies.
Lungshieldlonger-term business objectives.
A pilot study, adapting the UroShield technologyFrom our patented technologies to endotracheal tubes, is currently underway at Shaare Zedek Medical Center. The purpose of this study isour trademarked brands, we believe our intellectual property has substantial value and has significantly contributed to examine the effect of a device which generates low energy ultrasound waves like the UroShield product. The endpoint of the study isour success to show its effect on development of bacterial colonies on endotracheal tubes, in patient receiving mechanical ventilation, and to determine whether this effect lowers the rate of bacterial resistance to antibiotics. Results of this study are not known at this time. The targeted completion for the study is the second quarter of 2018.date.
Research and Development Expenses
During the years ended December 31, 2017 and 2016, we spent approximately $693,000 and $584,000, respectively, on research and development activities. None of the cost of such activities is borne directly by our customers.
Intellectual Property
Patents
We haveseek patent protection for our inventions not only to differentiate our products and technologies, but also to develop opportunities for licensing and secure our rights to sixprofits therefrom. With the aim of optimizing commercial and regulatory success, our proprietary technology and innovative applications thereof are protected by product, system, process, and method-of-use patent claims. We believe that our granted patents and pending applications collectively protect our technology, both in terms of our existing products, as well as our anticipated pipeline of new offerings.
Our patent portfolio includes at least the following issued patents, as well as a number of corresponding foreign patents in the United States. Grantedrelevant jurisdictions: (1) U.S. Patent No. 7,393,501 (having the following foreign counter-parts: China ZL03818327.7; Israel 165422; Japan 4504183; India 246351; Australia 2003231892; European Union 1511414 B), “Method, apparatusto “Method, Apparatus and systemSystem for treating biofilms associated with catheters” and grantedTreating Biofilms Associated With Catheters” (expiring on December 19, 2023); (2) U.S. Patent No. 7,829,029 (having the following foreign counter-parts: China ZL200780019732.3 and European Union 1998834), “Acoustic add-on deviceto “Acoustic Add-On Device for biofilm preventionBiofilm Prevention in urinary catheter,Urinary Catheter” both relate to the use of surface acoustic waves to prevent biofilm formation(expiring on indwelling catheters. These granted U.S. patents expire on December 19, 2023 and October 27, 2025, respectively. Granted U.S2025); (3) U.S. Patent No. 9,028,748 “Systemto “System and methodMethod for Surface Acoustic Wave Treatment of Medical Devices” (expiring on July 11, 2030); and (4) U.S. Patent No. 9,585,977 directed to “System and Method for Surface Acoustic Waves Treatment of Skin” (expiring on August 20, 2033). These patents cover a wide range of embodiments and applications of our proprietary surface acoustic wave treatment of medical devices,” relate to(SAW) technology, including our commercialized PAINSHIELD®, PAINSHIELD PLUSTM, WOUNDSHIELD® and UROSHIELD® devices. Specifically, the patents provide for methods of generating surface acoustic wavesSAW on medical device surfaces on bothof indwelling medical devices and implants to prevent biofilmtopical and urological applications therefor for alleviating pain, wound healing, and preventing formation This U.S. patent expiresof bacterial biofilms on July 11, 2030. Granted U.S. Patent No. 9,585,977 (havingcatheters.
In addition to the following foreign counter-parts: China ZL200780014875.5, European Union, and allowed Israel application), “System and method for surface acoustic waves treatment of skin,” relates to methods of using surface acoustic waves for treatment of skin for the purpose of wound-healing, reducing infection, pain reduction and cosmetic enhancements. This U.S. patent expires August 20, 2033.
We also license two in-forceabove patents, pursuant to a license agreement with Piezo-Top Ltd and PMG Medica Ltd., U.S. Patent No. 6,454,716 B1, “System and method for detection of fetal heartbeat,” and U.S. Patent No. 6,964,640 B2, “System and method for detection of motion,” which incorporate certain technology related to detecting in-vivo motion relating to biological parameters such as, for example, blood flow detection, heartbeat monitoring, fetal motion monitoring, fetal heartbeat monitoring, etc.. The configuration allows for an optimal scanning range at an unlimited number of angles. These patents expire on May 23, 2020 and January 22, 2023, respectively.
We believe the granted patents,our pending patent applications and license agreement (described below) collectively covernew filings are representative of our existing productsongoing efforts to broaden our portfolio as we continue developing new applications for our ultrasound technology. Although not yet granted, the extent necessary,aim of our growing number of patent applications is to secure our rights within additional industry sectors we foresee as most readily benefiting from our technology. Therefore, looking beyond just pain management and may be usefulurology, our patent applications relate to, inter alia: novel transdermal patches uniquely configured to work with our ultrasound technology to additionally provide for protectingimproved absorption and transdermal delivery of therapeutic agents during treatment; cosmetic applications of our futureultrasound technology developments. to provide anti-aging benefits; and certain new or improved stand-alone therapeutic medical devices or so-called “indwelling medical devices” (e.g., catheters, intravenous (IV) needle assemblies, and percutaneous endoscopic gastronomy (PEG) tubes) that include our SAW-generating technology to provide the accompanying antimicrobial effect for preventing infections typically associated with available indwelling devices.
We intend to continue patentingfurther grow our patent portfolio by continuing to patent new technology as it is developed, to defend intellectual property as we believe necessary by actively pursuing any infringements, to pursue the commercial opportunities our patents provide for our innovations, and to actively pursue any infringement of any ofcontinue to develop our patents.brands and trademarks.
To date, we are not aware of other companies that have patent rights to a comparable system and method for surface acoustic wave treatment for skin.
Trademarks
We believe thatIn addition to patent protection, we own numerous registered trademarks for our product brand names are an important factor in establishingcommercialized WOUNDSHIELD® (in the U.S. and maintaining brand recognition. We haveCanada), NanoVibronix® (in the following trademark registrations inU.S. and Canada), WOUNDSHIELD® (in the United States: NanoVibronix®U.S. and Canada), WoundShield®, PainShield®PAINSHIELD®. (in the U.S. and Canada), and UroShield®UROSHIELD® (in the U.S.). We intend to re-file and pursue our previously acquired trademark registration “Curing though prevention”®, which expired in July 2015. Generally, the protection afforded forby trademarks is perpetual, ifsubject to paying timely renewals and continuing proper use in commerce. In addition to the above, we expect to pursue additional trademark registrations to the extent we believe they are renewedwould be beneficial and cost-effective.
Other Rights
We regularly enter into, and rely on, a timely basis, if registered,confidentiality and continueproprietary rights agreements with our employees, consultants, contractors and business partners to be used properly as trademarks.
License Agreement
In October 2003, we entered into a license agreement with Piezo-Top Ltdprotect our trade secrets, proprietary technology and PMG Medica Ltd, pursuant to which we were granted an exclusive, worldwide license forother confidential information. We control the duration of the patent life of U.S. Patent No. 6,454,716 B1, U.S. Patent No. 6,964,640 B2 and U.S. Patent No. 7,431,892 B2 (see “—Patents” above). U.S. Patent No. 7,431,892 B2 has since expired. In exchange for the license, we paid Piezo-Top Ltd and PMG Medica Ltd payments of (i) $5,000 each after the first round of investment in us, (ii) $7,500 each after the second round of investment in us, and (iii) $25,000 each after either the third round of investment, the purchase of at least 40%use of our stock orproprietary technology through relevant provisions, notifications, and disclaimers provided on our initial public offering. We have made all threewebsite, our customer terms of the required payments under this agreement.use, and our vendor terms and conditions.
| 26 |
Government Regulation
U.S. Food and Drug Administration Regulation
Each of our products must be approved, cleared by, or registered with the U.S. Food and Drug Administration before it is marketed in the United States. Before and after approval or clearance in the United States, our products, approved or cleared products and product candidates, are subject to extensive regulation by the U.S. Food and Drug Administration under the Federal Food, Drug, and Cosmetic Act and/or the Public Health Service Act, as well as by other regulatory bodies. The U.S. Food and Drug Administration regulations govern, among other things, the development, testing, manufacturing, labeling, safety, storage, record-keeping, market clearance or approval, advertising and promotion, import and export, marketing and sales, distribution and distributionmarket withdrawal and recalls of medical devices and pharmaceutical products. PainShield has already obtained 510(k) marketing approval by the U.S. Food and Drug Administration.
In September 2020, the FDA exercised its Enforcement Discretion to allow distribution of the UroShield device in the United States. According to the FDA, “UroShield® device can use Intended Use Code (IUC) 081.006: Enforcement Discretion per final guidance, and FDA product code QMK (extracorporeal acoustic wave generating accessory to urological indwelling catheter for use during the COVID-19 pandemic)”. Accordingly, the FDA’s Enforcement Discretion clears the way for import of UroShield to the U.S. for limited use during the Covid-19 pandemic. The U.S. FDA may terminate or revoke this Enforcement Discretion at any time (after which the applicable products may no longer be used). The Enforcement Discretion does not ensure that UroShield will obtain 510(k) marketing approval.
U.S. Food and Drug Administration Approval or Clearance of Medical Devices
In the United States, medical devices are subject to varying degrees of regulatory control and are classified in one of three classes depending on the extent of controls the U.S. Food and Drug Administration determines are necessary to reasonably ensure their safety and efficacy:
| ● | Class I: general controls, such as labeling and adherence to quality system regulations, and a pre-market notification (510(k)) unless exempt; |
| ● | Class II: special controls, pre-market notification (510(k)) unless exempt, specific controls such as performance standards, patient registries and post-market surveillance and additional controls such as labeling and adherence to quality system regulations; and |
| ● | Class III: special controls and approval of a Pre-Market Approval, or PMA, application. |
WoundShield and PainShield are classified as Class II medical devices and require U.S. Food and Drug Administration authorization prior to marketing, by means of 510(k) clearance, except for our UroShield product, which we intend to seek clearance from the U.S. Food and Drug Administration through the de novo classification process, described below.
To request marketing authorization by means of a 510(k) clearance, we must submit a pre-market notification demonstrating that the proposed device is substantially equivalent to another legally marketed medical device, has the same intended use, and is as safe and effective as a legally marketed device and does not raise different questions of safety and effectiveness than a legally marketed device. 510(k) submissions generally include, among other things, a description of the device and its manufacturing, device labeling, medical devices to which the device is substantially equivalent, safety and biocompatibility information and the results of performance testing. In some cases, a 510(k) submission must include data from human clinical studies. Marketing may commence only when the U.S. Food and Drug Administration issues a clearance letter finding substantial equivalence. The typical duration to receive 510(k) approval is approximately nine months from the date of the initial 510(k) submission, although there is no guaranty that the timing will not be longer.
The U.S. Food and Drug Administration may require us to perform clinical studies to show a product candidate’s safety and efficacy in addition to technological equivalence in support of our filed 510(k). No matter which regulatory pathway we may take in the future towards marketing products in the United States, we believe we will be required to provide clinical proof of device effectiveness and safety.
After a device receives 510(k) clearance, any product modification that could significantly affect the safety or effectiveness of the product, or that would constitute a significant change in intended use, requires a new 510(k) clearance or, if the device would no longer be substantially equivalent, would require a PMA. If the U.S. Food and Drug Administration determines that the product does not qualify for 510(k) clearance, then a company must submit and the U.S. Food and Drug Administration must approve a PMA before marketing can begin.
| 27 |
A PMA application must provide a demonstration of safety and effectiveness, which generally requires extensive nonclinical and clinical trial data. Information about the device and its components, device design, manufacturing and labeling, among other information, must also be included in the PMA. As part of the PMA review, the U.S. Food and Drug Administration will inspect the manufacturer’s facilities for compliance with quality system regulation requirements, which govern testing, control, documentation and other aspects of quality assurance with respect to manufacturing. If the U.S. Food and Drug Administration determines the application or manufacturing facilities are not acceptable, the U.S. Food and Drug Administration may outline the deficiencies in the submission and often will request additional testing or information. Notwithstanding the submission of any requested additional information, the U.S. Food and Drug Administration ultimately may decide that the application does not satisfy the regulatory criteria for approval. During the review period, a U.S. Food and Drug Administration advisory committee, typically a panel of clinicians and statisticians, is likely to be convened to review the application and recommend to the U.S. Food and Drug Administration whether, or upon what conditions, the device should be approved. The U.S. Food and Drug Administration is not bound by the advisory panel decision. While the U.S. Food and Drug Administration often follows the panel’s recommendation, there have been instances where the U.S. Food and Drug Administration has not. If the U.S. Food and Drug Administration finds the information satisfactory, it will approve the PMA. The PMA approval can include post-approval conditions, including, among other things, restrictions on labeling, promotion, sale and distribution, or requirements to do additional clinical studies post-approval. Even after approval of a PMA, a new PMA or PMA supplement is required to authorize certain modifications to the device, its labeling or its manufacturing process. Supplements to a PMA often require the submission of the same type of information required for an original PMA, except that the supplement is generally limited to that information needed to support the proposed change from the product covered by the original PMA. The typical duration to receive PMA approval is approximately two years from the date of submission of the initial PMA application, although there is no guarantee that the timing will not be longer.
As describedstated above, we anticipate that our UroShield product will receive a de novo review from the U.S. Food and Drug Administration. De novo review is a two-step process that requires a company to submit a 510(k) and complete a standard review, including an analysis of the risk to the patient and operator associated with the use of the device and the substantial equivalence rationale. Once that has been accomplished, and the medical device in question has been determined to be not substantially equivalent to another approved device, the product is automatically classified as a Class III device. The manufacturer can then submit a request for an evaluation to have the product reclassified from Class III into Class I or Class II. The U.S. Food and Drug Administration will review the device classification proposal and either recommend special controls to create a new Class I or II device classification or determine that the product is a Class III device. If the U.S. Food and Drug Administration determines that the level of risk associated with the use of the device is appropriate for a Class II or Class I designation, then the product can be cleared as a 510(k) and the U.S. Food and Drug Administration will issue a new classification regulation and product code. If the device is not approved through de novo review, then it must go through the standard PMA process for Class III devices.
Clinical Trials of Medical Devices
One or more clinical trials are generally required to support a PMA application and more recently are becoming necessary to support a 510(k) submission. Clinical studies of unapproved or uncleared medical devices or devices being studied for uses for which they are not approved or cleared (investigational devices) must be conducted in compliance with U.S. Food and Drug Administration requirements. If an investigational device could pose a significant risk to patients, the sponsor company must submit an investigational device exemption application to the U.S. Food and Drug Administration prior to initiation of the clinical study. An investigational device exemption application must be supported by appropriate data, such as animal and laboratory test results, showing that it is safe to test the device on humans and that the testing protocol is scientifically sound. The investigational device exemption will automatically become effective 30 days after receipt by the U.S. Food and Drug Administration unless the U.S. Food and Drug Administration notifies the company that the investigation may not begin. Clinical studies of investigational devices may not begin until an institutional review board has approved the study.
| 28 |
During the study, the sponsor must comply with the U.S. Food and Drug Administration’s investigational device exemption requirements. These requirements include investigator selection, trial monitoring, adverse event reporting, and record keeping. The investigators must obtain patient informed consent, rigorously follow the investigational plan and study protocol, control the disposition of investigational devices, and comply with reporting and record keeping requirements. The sponsor, the U.S. Food and Drug Administration, or the institutional review board at each institution at which a clinical trial is being conducted may suspend a clinical trial at any time for various reasons, including a belief that the subjects are being exposed to an unacceptable risk. During the approval or clearance process, the U.S. Food and Drug Administration typically inspects the records relating to the conduct of one or more investigational sites participating in the study supporting the application.
Post-Approval Regulation of Medical Devices
After a device is cleared or approved for marketing, numerous and pervasive regulatory requirements continue to apply. These include:
| ● | the U.S. Food and Drug Administration quality systems regulation, which governs, among other things, how manufacturers design, test, manufacture, exercise quality control over, and document manufacturing of their products; |
| ● | labeling and claims regulations, which prohibit the promotion of products for unapproved or “off-label” uses and impose other restrictions on labeling; |
| ● | if applicable, the Electronic Product Regulations found in 21 CFR parts 1000-1050, which provide additional requirements applicable to electronic products, including records and reporting requirements; and | |
| ● | the Medical Device Reporting regulation, which requires reporting to the U.S. Food and Drug Administration of certain adverse experiences associated with use of the product. |
Good Manufacturing Practices Requirements
Manufacturers of medical devices are required to comply with the good manufacturing practices set forth in the quality system regulations promulgated under section 520 of the Food, Drug and Cosmetic Act as further set forth in the Code of Federal Regulations as 21 CFR Part 820. Current good manufacturing practices (“cGMP”CGMP”) regulations require, among other things, quality control and quality assurance as well as the corresponding maintenance of records and documentation. The manufacturing facility for an approved product must meet current good manufacturing practices requirements to the satisfaction of the U.S. Food and Drug Administration pursuant to a pre-PMA approval inspection before the facility can be used. Manufacturers, including third party contract manufacturers, are also subject to periodic inspections by the U.S. Food and Drug Administration and other authorities to assess compliance with applicable regulations. Failure to comply with or to promptly comply with statutory and regulatory requirements subjects a manufacturer, and possibly us, to possible legal or regulatory action, including the seizure or recall of products, injunctions, consent decrees placing significant restrictions on or suspending manufacturing operations, and civil and criminal penalties. Adverse experiences with the product must be reported to the U.S. Food and Drug Administration and could result in the imposition of marketing restrictions through labeling changes or in product withdrawal.recall. Product approvals may be withdrawn if compliance with regulatory requirements is not maintained or if problems concerning safety or efficacy of the product occur following the approval.
International Regulation
We are subject to regulations and product registration requirements in many foreign countries in which we may sell our products, including in the areas of product standards, packaging requirements, labeling requirements, import and export restrictions and tariff regulations, duties and tax requirements. The time required to obtain clearance required by foreign countries may be longer or shorter than that required for U.S. Food and Drug Administration clearance, and requirements for licensing a product in a foreign country may differ significantly from U.S. Food and Drug Administration requirements.
The primary regulatory environment in Europe is the European Union, which consists of 2527 member states and 4232 competent authorities encompassing most of the major countries in Europe. In the European Union, the European Medicines Agency and the European Union Commission determined that PainShield, UroShield, and WoundShield are to be regulated as medical device products. These products are classified as Class II devices. These devices are CE Marked and as such can be marketed and distributed within the European Economic Area. We are required to be recertified each year for CE by Intertek, which conducts an annual audit. The audit procedure, which includes on-site visits at our facility, requires us to provide Intertek with information and documentation concerning our management system and all applicable documents, policies, procedures, manuals, and other information.
| 29 |
The primary regulatory bodies and paths in Asia, Australia, Canada and Latin America are determined by the requisite country authority. In most cases, establishment registration and device licensing are applied for at the applicable Ministry of Health through a local intermediary. The requirements placed on the manufacturer are typically the same as those contained in ISO 9001 or ISO 13485, requirements for quality management systems published by the International Organization of Standardization. In some countries outside Europe, we are or will be able to sell on the basis of our CE Mark. We have the Health Canada medical device license for PainShield, WoundShield and UroShield, a certificate by the Israel Ministry of Health allowing us to sell PainShield, WoundShield and UroShield in Israel, a certificate allowing us to sell PainShield in Australia, and we are able to sell PainShield, WoundShield and UroShield in India and Ecuador based on our CE Mark. In addition, our distributor in Korea has applied for approval to sell PainShield and UroShield. We generally apply, through our distributor, for approval in a particular country for a particular product only when we have a distributor in place with respect to such product.
European Good Manufacturing Practices
In the European Union, the manufacture of medical devices is subject to good manufacturing practice, as set forth in the relevant laws and guidelines of the European Union and its member states. Compliance with good manufacturing practice is generally assessed by the competent regulatory authorities. Typically, quality system evaluation is performed by a notified body, which also recommends to the relevant competent authority for the European Community CE Marking of a device. The competent authority may conduct inspections of relevant facilities, and review manufacturing procedures, operating systems and personnel qualifications. In addition to obtaining approval for each product, in many cases each device manufacturing facility must be audited on a periodic basis by the notified body. Further inspections may occur over the life of the product.
U.S. Fraud and Abuse and Other Health Care Laws
In the United States, federal and state fraud and abuse laws prohibit the payment or receipt of kickbacks, bribes or other remuneration intended to induce the purchase or recommendation of health care products and services. Other provisions of federal and state laws prohibit presenting, or causing to be presented, to third party payers for reimbursement, claims that are false or fraudulent, or which are for items or services that were not provided as claimed. In addition, other health care laws and regulations may apply, such as transparency and reporting requirements, and privacy and security requirements. Violations of these laws can lead to civil and criminal penalties, including exclusion from participation in federal and state health care programs. These laws are potentially applicable to manufacturers of products regulated by the U.S. Food and Drug Administration as medical devices, such as us, and hospitals, physicians and other potential purchasers of such products. The health care laws that may be applicable to our business or operations include:
| ● | The federal Anti-Kickback Statute, which prohibits the offer, payment, solicitation or receipt of any form of remuneration in return for referring, ordering, leasing, purchasing or arranging for, or recommending the ordering, purchasing or leasing of, items or services payable by Medicare, Medicaid or any other federal health care program. | |
| ● | Federal false claims laws and civil monetary penalty laws, including the False Claims Act, that prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid or other government health care programs that are false or fraudulent, or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government. | |
| ● | The federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which prohibits knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program or obtain, by means of false or fraudulent pretenses, representations, or promises, any of the money or property owned by, or under the custody or control of, any health care benefit program, and for knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statements in connection with the delivery of or payment for health care benefits, items or services. |
| 30 |
● The federal Anti-Kickback Statute, which prohibits the offer, payment, solicitation or receipt of any form of remuneration in return for referring, ordering, leasing, purchasing or arranging for, or recommending the ordering, purchasing or leasing of, items or services payable by Medicare, Medicaid or any other federal health care program.
● Federal false claims laws and civil monetary penalty laws, including the False Claims Act, that prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid or other government health care programs that are false or fraudulent, or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government.
● The federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which prohibits knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program or obtain, by means of false or fraudulent pretenses, representations, or promises, any of the money or property owned by, or under the custody or control of, any health care benefit program, and for knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statements in connection with the delivery of or payment for health care benefits, items or services.
●
| ● | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, and its implementing regulations, which also impose obligations and requirements on health care providers, health plans, and healthcare clearinghouses as well as their respective business associates that perform certain services for them that involve the use or disclosure of individually identifiable health information, with respect to safeguarding the privacy and security of certain individually identifiable health information. | |
| ● | The federal transparency requirements under the Affordable Care Act, including the provision commonly referred to as the Physician Payments Sunshine Act, which requires certain manufacturers of drugs, devices, biologics and medical supplies that are reimbursable under Medicare, Medicaid or Children’s Health Insurance Program to report annually to Centers for Medicare and Medicaid Services, or CMS, information related to payments and other transfers of value to physicians and teaching hospitals, and ownership and investment interests held by physicians and their immediate family members. | |
| ● | Analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws, which may be broader in scope and apply to referrals and items or services reimbursed by both governmental and non-governmental third-party payers, including private insurers, many of which differ from each other in significant ways and often are not preempted by federal law, thus complicating compliance efforts. |
Manufacturing and Suppliers
In December 2018, we announced we appointed Quasar Engineering Ltd, as contract manufacturer for the PainShield®, UroShield® and WoundShield®, as well as their respective business associatesother devices. Following our agreement with Sanuwave, Quasar is no longer the manufacturer of the WoundShield®. Quasar is a medical device manufacturer, located in China, with over 30 years of experience, serving major brands worldwide, with complex catheters, disposables, and U.S. Food and Drug Administration regulated assemblies. Starting in the fourth quarter of 2019, we started using Quasar to manufacture all of our newly redesigned products. Quasar temporarily shut down for sixty days in early 2020, due to the COVID-19 outbreak which lead to a significant delay in the production of goods needed to fulfill our sales orders, and became fully operational in April 2020. Presently, we are no longer experiencing delays in the production of our products.
Quasar is anticipating the addition of a new manufacturing facility in Singapore late in the third quarter of 2022. Our product manufacturing will move to this plant when it is on-line. Several components will continue to be produced in China, but final production will be completed in Singapore.
We order certain component parts on an as-needed basis, generally from the manufacturer that perform certain servicesprovides us with the most competitive pricing. Our most significant suppliers for them that involve the use or disclosurethese components are B Star, Inc, Plastic One, Rotel Product Engineering Ltd., and Sinpro Electronics Co., Ltd. We do not have written agreements with any of individually identifiable health information, with respect to safeguarding the privacy and security of certain individually identifiable health information.these suppliers, but we believe anyone could be easily replaced if necessary.
● The federal transparency requirements under the Affordable Care Act, including the provision commonly referred to as the Physician Payments Sunshine Act, which requires certain manufacturers of drugs, devices, biologics and medical supplies that are reimbursable under Medicare, Medicaid or Children’s Health Insurance Program to report annually to Centers for Medicare and Medicaid Services, or CMS, information related to payments and other transfers of value to physicians and teaching hospitals, and ownership and investment interests held by physicians and their immediate family members.Customers
● Analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws, which may be broader in scope and apply to referrals and items or services reimbursed by both governmental and non-governmental third-party payers, including private insurers, many of which differ from each other in significant ways and often are not preempted by federal law, thus complicating compliance efforts.
Customers
We currently sell our products both directly, through our website, and indirectly via distribution agreements, with approximately 47%99% of our sales coming through distributors in 2017.2021. We expect that percentage to continue to grow significantly as we enter into additional distribution agreements.agreements, until we launch PainShield Relief. We have exclusive and non-exclusive distribution agreements for our products with medical product distributors based in the United States, in the United Kingdom and various countries throughout Europe, India, Canada and Asia. We have recently enlisted Diligence Wound Care Global LLC on a solely incentive based basis, to assist in identifying and obtaining distribution in various partsOur largest customer is Ultra Pain Products Inc, comprising approximately 77% of the world, in particular, Southeast Asia, China and Mexico.total sales.
We are currently in discussions with a number ofseveral distribution companies with access to various markets in the United States, Canada, Europe, and Asia, as well as a distributor which will allow access into Veterans Administration facilities. Our current agreements stipulate that distributors will be responsible for carrying out local marketing activities and sales. We are responsible for training, providing marketing guidance, marketing materials, and technical guidance. In addition, in most cases, all sales costs, including sales representatives, incentive programs, and marketing trials, will be borne by the distributor. We expect any future distribution agreements to contain substantially similar stipulations. Under our current agreements, distributors purchase our products from us at a fixed price. Our current agreements with distributors are generally for a term of approximately two to three years and automatically renew for an additional annual terms unless modified by either party.
| 31 |
Manufacturing
Employees
Our People and SuppliersHuman Capital Resources
We assemble our products in-house at our facilities in Nesher, Israel. AllEmployees
As of the component parts of our products are readily available from a number of manufacturersDecember 31, 2021, we had 12 full-time employees and suppliers. We order component parts onfour part-time employees, which is an as-needed basis, generallyincrease from the manufacturer11 full-time employees and one-part-time employee we had as of December 31, 2020, and as of March 15, 2022, we have added one additional full-time employee in 2022 We also regularly work with several independent consultants and other contract organizations to support our business and we regularly evaluate additional talent to help support our product manufacturing, development, financial, and other capabilities.
Diversity and Inclusion
We believe that providesan inclusive culture is required to understand and develop products that benefit all patients. By embracing differences, we aim to foster an environment of respect and trust in an effort to facilitate creativity, spark passion, and help us achieve better outcomes for all those who work at the Company. We are committed to creating and maintaining a workplace free from discrimination or harassment, including on the basis of any class protected by applicable law, and our recruitment, hiring, development, training, compensation, and advancement practices are based on qualifications, performance, skills, and experience without regard to gender, race, or ethnicity. Our management team and employees are expected to exhibit and promote honest, ethical, and respectful conduct in the workplace, including adhering to the standards for appropriate behavior set forth in our code of conduct.
Compensation and Benefits
We operate in a highly competitive environment for human capital, particularly as we seek to attract and retain talent with relevant experience in the mostmedical device sector. Therefore, we strive to provide a total rewards package to our employees that is competitive pricing. Our most significant suppliers are APC International, Ltd., R&D Medical Products, DI-EL Tack Ltd., Rotel Product Engineering Ltd.with our peer companies, including competitive healthcare benefits and Afinity. in certain cases, stock options. We also offer paid leave as mandated by government regulations, flexible work schedules, and other benefits as mandated by government regulations.
We also offer key employees the benefit of equity ownership in NanoVibronix through stock option grants. We believe these grants both help promote alignment between our employees and our stockholders and provide retention benefits, as the awards generally vest over a three-year period.
We do not have written agreementsany employees that are represented by a labor union or that have entered into a collective bargaining agreement with anythe Company.
Safety, Wellness, and Our Response to COVID-19
At NanoVibronix, we believe that health matters to everyone, and the safety health, and wellness of our employees is one of our top priorities. We are committed to developing and fostering a work environment that is safe, professional, and promotes teamwork, diversity, and trust in order to afford all of our employees the opportunity to contribute to the best of their abilities.
During 2020 and 2021, in response to the COVID-19 pandemic, we took certain measures and responded to changes in our operational needs, including actions designed to provide a safe work environment for our employees. These actions included investing in technology solutions to support increased work-from-home capabilities, shifting work schedules to reduce the number of people present in our offices, requiring mask wearing and social distancing, making hand sanitizer readily available, and other measures intended to comply with health and safety protocols as required by federal, state, and local governmental agencies, as well as guidance from the U.S. Centers for Disease Control and Prevention and similar public health authorities.
| 32 |
Available Information
The Company’s Annual Report on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, and amendments thereto, are filed with the SEC. The Company is subject to the informational requirements of the Securities Exchange Act of 1934, as amended (the “Exchange Act”) and files or furnishes reports, proxy statements and other information with the SEC. Such reports and other information filed by the Company with the SEC are available free of charge on the Company’s website at nanovibronix.com, as soon as reasonably practicable after we have electronically filed with, or furnished to, the SEC. The SEC maintains an internet site that contains reports, proxy and information statements and other information regarding issuers that file electronically with the SEC at www.sec.gov. The contents of these suppliers, but we believe anyone couldwebsites are not incorporated into this filing. Further, the Company’s references to website URLs are intended to be easily replaced if necessary.inactive textual references only.
An investment in our securities involves a high degree of risk. Before deciding whether to invest in our securities, you should consider carefully the risks described below, together with other information with the other information set forth in this Annual Report on Form 10-K. If any of these risks actually occur, our business, financial condition, results of operations or cash flow could be seriously harmed. This could cause the trading price of our common stock to decline, resulting in a loss of all or part of your investment. The risks and uncertainties described below are not the only ones facing us. Additional risks and uncertainties not presently known to us, or that we currently see as immaterial, may also harm our business. Please also read carefully the section below entitled “Cautionary Note Regarding Forward-Looking Statements.”
Risks Related to Our Business
We have a history of losses and we expect to continue to incur losses and may not achieve or maintain profitability.
For the fiscal year ended December 31, 2017,2021 we had a net loss of $4,965,approximately $14.3 million, with revenues of $239.approximately $1.7 million. As of December 31, 2017,2021, we had an accumulated deficit of $28,382.Weapproximately $57 million. We expect to incur losses for at least the next year, as we continue to incur expenses related to seeking U.S. Food and Drug Administration approval for UroShield, and WoundShield, and market acceptance and reclassification of PainShield, which will require costly additional clinical trials and research, further product development and professional fees associated with regulatory compliance. Even if we succeed in commercializing our new products, we may not be able to generate sufficient revenues to cover our expenses and achieve profitability or be able to maintain profitability.
Global economic and political instability and conflicts, such as the conflict between Russia and Ukraine, could adversely affect our business, financial condition or results of operations.
Our business could be adversely affected by unstable economic and political conditions within the United States and foreign jurisdictions and geopolitical conflicts, such as the conflict between Russia and Ukraine. While we do not have any customer or direct supplier relationships in either country at this time, the current military conflict, and related sanctions, as well as export controls or actions that may be initiated by nations including the United States, the European Union or Russia (e.g., potential cyberattacks, disruption of energy flows, etc.) and other potential uncertainties could adversely affect our business and/or our supply chain, business partners, employees or customers, and interrupt our ability to supply products, or otherwise adversely impact our business.
Increasing inflation could adversely affect our business, financial condition, results of operations or cash flows.
Inflation, as well as some of the measures taken by or that may be taken by the governments in countries where we operate in an attempt to curb inflation may have negative effects on the economies of those countries generally. If the United States or other countries where we operate experience substantial inflation in the future, our business may be adversely affected. This could have a material adverse effect on our business, financial condition, results of operations, or cash flows. Specifically, our existing distributor agreements limit the amount that we can increase the price that we sell our products to the distributors. Accordingly, an inflationary environment, including factors such as increasing freight and materials prices, could make it less profitable for us to do business.
The ongoing COVID-19 pandemic has and may continue to adversely impact our business.
The ongoing COVID-19 pandemic has and may continue to adversely impact our business, as our operations are based in and rely on third parties located in countries affected by the pandemic. Our third-party manufacturer, which is based in China, temporarily shut down for sixty days during 2020 due to the pandemic and became fully operational in April 2020 which led to a significant delay in the production of goods needed to fulfill our sales orders which were scheduled to be fulfilled in our first quarter of 2020. We were able to fulfill these orders in the second quarter of 2020. Additionally, the notified regulatory body we rely on to obtain European CE approval is located in Italy and was shut down for approximately six weeks from March to April 2020, which delayed our submission for CE mark approval for the year 2020. The CE Mark approval was subsequently approved in April 2020. The various precautionary measures taken by many governmental authorities around the world in order to limit the spread of COVID-19 have had and may continue to have an adverse effect on the global markets and global economy, including on the availability and pricing of employees, resources, materials, manufacturing and delivery efforts and other aspects of the global economy. The financial downturn had compelled us to furlough or reduce working hours for much of our operating staff in 2020, and continue to force remaining staff as well as third-party contractors, to work remotely from time to time. In addition, many staff members continue to operate remotely from their homes, which is continuing to result in delays in obtaining certain financial records. We also rely on third-party professionals to provide services such as the preparation of our financial statements and to conduct audits, and many of these parties have been affected by government-imposed precautionary measures, thereby delaying our receipt of these services. Such government-imposed precautionary measures may have been relaxed in certain countries or states, but there is no assurance that more strict measures will be put in place again due to a resurgence in COVID-19 cases. Therefore, the COVID-19 pandemic has and may again disrupt production and cause delays in the development, supply and delivery of our products, our operation, further divert the attention and efforts of the medical community coping with COVID-19 and disrupt the marketplace in which we operate. The extent to which COVID-19 impacts our results will depend on future developments, which are highly uncertain and cannot be predicted, including new information which may emerge concerning the severity of COVID-19, its variants and the actions to contain COVID-19 or treat its impact, among others. The COVID-19 pandemic could continue to materially disrupt our business and operations, hamper our ability to raise additional funds or sell or securities, continue to slow down the overall economy, curtail consumer spending, interrupt our sources of supply, and make it hard to adequately staff our operations.
| 33 |
If we are unable to raise additional capital, our clinical trials and product development will be limited and our long-term viability will be threatened; however, if we do raise additional capital, your percentage ownership as a stockholder could decrease and constraints could be placed on the operations of our business.
We have experienced negative operating cash flows since our inception and have funded our operations primarily from proceeds of the sale of our securities, with only limited revenue being generated from our product sales. In order to fully realize our business objectives, we may need to raise additional capital. We maywill seek to raise such additional funds through equity or debt financings, or strategic alliances with third parties, either alone or in combination with equity financings. These financings could result in substantial dilution to the holders of our common stock, or require contractual or other restrictions on our operations or on alternatives that may be available to us. If we raise additional funds by issuing debt securities, these debt securities could impose significant restrictions on our operations through the imposition of restrictive covenants and requiring us to pledge assets in order to secure repayment. In addition, if we raise funds through the sale of equity, we may issue equity securities with rights superior to our common stock, including voting rights, rights to proceeds upon our liquidation or sale, rights to dividends and rights to appoint board members. Any suchThere can be no assurance that we will be able to complete a required financing mayon acceptable terms or at all. If such financing is not beavailable on satisfactory terms, or is not available in sufficient amounts, we may be required to delay, limit or on terms acceptable to us, andeliminate the development of business opportunities. The failure to procure such required financing could have a material adverse effect on our business, financial condition and results of operations, or threaten our ability to continue as a going concern.
A variety of factors could impact the timing and amount of any required financings, including, without limitation:
| ● | unforeseen developments during our clinical trials; | |
| ● | delays in our receipt of required regulatory approvals; | |
| ● | delayed market acceptance of our products; | |
| ● | unanticipated expenditures in our acquisition and defense of intellectual property rights, and/or the loss of those rights; | |
| ● | the failure to develop strategic alliances for the marketing of some of our product candidates; | |
| ● | unforeseen changes in healthcare reimbursement for any of our approved products; | |
| ● | lack of financial resources to adequately support our operations; | |
| ● | difficulties in maintaining commercial scale manufacturing capacity and capability; | |
| ● | unanticipated difficulties in operating in international markets; | |
| ● | unanticipated financial resources needed to respond to technological changes and increased competition; | |
| ● | unforeseen problems in attracting and retaining qualified personnel; | |
| ● | enactment of new legislation or administrative regulations; | |
| ● | the application to our business of new regulatory interpretations; | |
| ● | claims that might be brought in excess of our insurance coverage; | |
| ● | the failure to comply with regulatory guidelines; and | |
| ● | the uncertainty in industry | |
| ● | the delisting of our common stock from the NASDAQ Capital Market; and | |
| ● | the geographic, social and economic impact of COVID-19 on the Company’s business operations. |
| 34 |
Any required financing efforts may divert our management from their day-to-day activities, which may adversely affect its ability to develop and commercialize our products Moreover, if we complete additional financing by issuing equity securities, the percentage ownership of its existing stockholders may be reduced, and accordingly these stockholders may experience substantial dilution. Given our need for cash and that equity issuances are the most common type of fundraising for similarly situated companies, the risk of dilution is particularly significant for our stockholders.
In addition, although we have no present commitments or understandings to do so, we may seek to expand our operations and product lines through acquisitions or joint ventures. Any acquisition or joint venture would likely increase our capital requirements.
If we fail to obtain an adequate level of reimbursement for our approved products by third party payers, there may be no commercially viable markets for our approved products or the markets may be much smaller than expected.
The availability and levels of reimbursement by governmental and other third party payers affect the market for our approved products. The efficacy, safety, performance and cost-effectiveness of our product and product candidates, and of any competing products, will determine the availability and level of reimbursement. Reimbursement and healthcare payment systems vary significantly by country, and include both government sponsored healthcare and private insurance. To obtain reimbursement or pricing approval in some countries, we may be required to produce clinical data, which may involve one or more clinical trials, that compares the cost-effectiveness of our approved products to other available therapies. We may not obtain reimbursement or pricing approvals in markets we seek to enter in a timely manner, if at all. Our failure to receive reimbursement or pricing approvals in target markets would negatively impact market acceptance of our products in these jurisdictions, placing us at a material cost disadvantage to our competitors.
Even if we obtain reimbursement approvals for our products, we believe that, in the future, reimbursement for any of our products or product candidates may be subject to increased restrictions both in the United States and in international markets. Future legislation, regulation or policies of third party payers that limit reimbursement may adversely affect the demand for our products currently under development and our ability to sell our products on a profitable basis. In addition, third party payers continually attempt to contain or reduce the costs of healthcare by challenging the prices charged for healthcare products and services.
In the United States, specifically, health care providers, such as hospitals and clinics, and individual patients, generally rely on third-party payers. Third-party reimbursement is dependent upon decisions by the Centers for Medicare and Medicaid Services, contracted Medicare carriers or intermediaries, individual managed care organizations, private insurers, other governmental health programs and other payers of health care costs. Failure to receive or maintain favorable coding, coverage and reimbursement determinations for our products by these organizations could discourage medical practitioners from using or prescribing our products due to their costs. In addition, with recent federal and state government initiatives directed at lowering the total cost of health care, the U.S. Congress and state legislatures will likely continue to focus on health care reform including the reform of the Medicare and Medicaid programs, and on the cost of medical products and services, which could limit reimbursement. Additionally, third-party payers are increasingly challenging the prices charged for medical products and services, and imposing conditions on payment. We may be unable to sell our products on a profitable basis if third-party payers deny coverage, provide low reimbursement rates or reduce their current levels of reimbursement.
The medical device and therapeutic product industries are highly competitive and subject to rapid technological change. If our competitors are better able to develop and market products that are safer and more effective than any products we may develop, our commercial opportunities will be reduced or eliminated.
Our success depends, in part, upon our ability to maintain a competitive position in the development of technologies and products. We face competition from established medical device companies, such as Neurometrix Inc., Zetrox, Kinetic Concepts, Inc., (a subsidiary of the 3M Company) and Smith & Nephew plc, manufacturers of certain portable ultrasound devices capable of self-administered use, as well as from academic institutions, government agencies, and private and public research institutions in the United States and abroad. Most, if not all, of our principal competitors have significantly greater financial resources and expertise than we do in research and development, manufacturing, pre-clinical testing, conducting clinical trials, obtaining regulatory approvals, marketing approved products, protecting and defending their intellectual property rights and designing around the intellectual property rights of others. Other small or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements, or mergers with, or acquisitions by, large and established companies, or through the development of novel products and technologies.
| 35 |
The industry in which we operate has undergone, and we expect it to continue to undergo, rapid and significant technological change, and we expect competition to intensify as technological advances are made. Our competitors may be able to respond to changes in technology or the marketplace faster than us. Our competitors may develop and commercialize medical devices that are safer or more effective or are less expensive than any products that we may develop. We also compete with our competitors in recruiting and retaining qualified scientific and management personnel, in establishing clinical trial sites and patient registration for clinical trials, and in acquiring technologies complementary to our programs or advantageous to our business. Given our small size and lack of resources, we are often at a disadvantage with our competitors in all of these areas, which could limit or eliminate our commercial opportunities.
We face the risk of product liability claims and may not be able to obtain insurance.
Our business exposes us to the risk of product liability claims that are inherent in the development of medical devices and products. If the use of one or more of our products harms people, we may be subject to costly and damaging product liability claims brought against us by clinical trial participants, consumers, health care providers, pharmaceutical companies or others selling our products. We currently carry clinical trial and product liability insurance for the products we sell. However, we cannot predict all of the possible harms or side effects that may result and, therefore, the amount of insurance coverage we hold may not be adequate to cover all liabilities we might incur. We intend to expand our insurance coverage to include the sale of additional commercial products as we obtain marketing approval for our product candidates in development and as our sales expand, but we may be unable to obtain commercially reasonable product liability insurance for such products. If we are unable to obtain insurance at an acceptable cost or otherwise protect against potential product liability claims and we continue to make sales, or if our coverages turns out to be insufficient, we may be exposed to significant liabilities, which may materially and adversely affect our business and financial position. If we are sued for any injury allegedly caused by our products and do not have sufficient insurance coverage, our liability could exceed our total assets and our ability to pay the liability. A product liability claim or series of claims brought against us would decrease our cash and could reduce our value or marketability.
Our product candidates may not be developed or commercialized successfully.
Our product candidates are based on a technology that has not been used previously in the manner we propose and must compete with more established treatments currently accepted as the standards of care. Market acceptance of our products will largely depend on our ability to demonstrate their relative safety, efficacy, cost-effectiveness and ease of use.
We are subject to the risks that:
| ● | the U.S. Food and Drug Administration or a foreign regulatory authority finds our product candidates ineffective or unsafe; | |
| ● | we do not receive necessary regulatory approvals; | |
| ● | the regulatory review and approval process may take much longer than anticipated, requiring additional time, effort and expense to respond to regulatory comments and/or directives; | |
| ● | we are unable to get our product candidates in commercial quantities at reasonable costs; and | |
| ● | the patient and physician community does not accept our product candidates. |
In addition, our product development program may be curtailed, redirected, eliminated or delayed at any time for many reasons, including:
| ● | adverse or ambiguous results; | |
| ● | undesirable side effects that delay or extend the trials; | |
| ● | the inability to locate, recruit, qualify and retain a sufficient number of clinical investigators or patients for our trials; and | |
| ● | regulatory delays or other regulatory actions. |
| 36 |
Additionally, we currently have limited experience in marketing or selling our products, and we have a limited marketing and sales staff and distribution capabilities. Developing a marketing and sales force is time-consuming and will involve the investment of significant amounts of financial and management resources, and could delay the launch of new products or expansion of existing product sales. In addition, we compete with many companies that currently have extensive and well-funded marketing and sales operations. If we fail to establish successful marketing and sales capabilities or fail to enter into successful marketing arrangements with third parties, our ability to generate revenues will suffer.
Furthermore, even if we enter into marketing and distributing arrangements with third parties, we may have limited or no control over the sales, marketing and distribution activities of these third parties, and these third parties may not be successful or effective in selling and marketing our products. If we fail to create successful and effective marketing and distribution channels, our ability to generate revenue and achieve our anticipated growth could be adversely affected. If these distributors experience financial or other difficulties, sales of our products could be reduced, and our business, financial condition and results of operations could be harmed.
We cannot predict whether we will successfully develop and commercialize our product candidates. If we fail to do so, we will not be able to generate substantial revenues, if any.
The loss ofIf we fail to retain our key management, would likely hinder our abilityor to attract and keep additional key personnel, we may be unable to successfully execute our business plan.
Our success depends on our ability to attract, retain and motivate highly qualified management and personnel. As a small company with eightten full-time employees and four contract employees, our success depends on the continuing contributions of our management team and qualified personnel and on our ability to attract and retain highly qualified personnel. We face intense competition in our hiring efforts from other medical device companies, as well as from universities and nonprofit research organizations, and we may have to pay higher salaries to attract and retain qualified personnel. We are also at a disadvantage in recruiting and retaining key personnel as our small size and limited resources may be viewed as providing a less stable environment, with fewer opportunities than would be the case at one of our larger competitors. The loss of one or more of these individuals, or our inability to attract additional qualified personnel, could substantially impair our ability to implement our business plan. In addition, the replacement of key personnel likely would involve significant time and costs, and may significantly delay or prevent the achievement of our business objectives.
Our need to increase the size of our organization and may not successfully manage our growth.
We are a clinical-stage company with a small number of planned employees, and our management systems currently in place are not likely to be adequate to support our future growth plans. Our ability to grow and to manage our growth effectively will require us to hire, train, retain, manage and motivate additional employees and to implement and improve its operational, financial and management systems. These demands also may require the hiring of additional senior management personnel or the development of additional expertise by our senior management personnel. Hiring a significant number of additional employees, particularly those at the management level, would increase our expenses significantly. Moreover, if we fail to expand and enhance its operational, financial and management systems in conjunction with its potential future growth, such failure could have a material adverse effect on our business, financial condition and results of operations.
Our failure to protect our intellectual property rights could diminish the value of our solutions, weaken our competitive position and reduce our revenue.
We regard the protection of our intellectual property, which includes patents and patent applications, trade secrets, trademarks and domain names, as critical to our success. We strive to protect our intellectual property rights by relying on federal, state and common law rights, as well as contractual restrictions. We enter into confidentiality and invention assignment agreements with our employees, consultants and contractors, and confidentiality agreements with parties with whom we conduct business in order to limit access to, and disclosure and use of, our proprietary information. However, these contractual arrangements and the other steps we have taken to protect our intellectual property may not prevent the misappropriation of our proprietary information or deter independent development of similar technologies by others.
| 37 |
We have obtained patents and we have patent applications pending in both the United States and foreign jurisdictions. There can be no assurance that our patent applications will be approved, that any patents issued will adequately protect our intellectual property, or that these patents will not be challenged by third parties or found to be invalid or unenforceable. We have also obtained trademark registration in the United States and in foreign jurisdictions. Effective trade secret, trademark and patent protection is expensive to develop and maintain, both in terms of initial and ongoing registration requirements and the costs of defending our rights. We may be required to protect our intellectual property in an increasing number of jurisdictions, a process that is expensive and may not be successful or which we may not pursue in every location. We may, over time, increase our investment in protecting our intellectual property through additional patent filings that could be expensive and time-consuming.
Monitoring unauthorized use of our intellectual property is difficult and costly. Our efforts to protect our proprietary rights may not be adequate to prevent misappropriation of our intellectual property. We may not be able to detect unauthorized use of, or take appropriate steps to enforce, our intellectual property rights. Further, our competitors may independently develop technologies that are similar to ours but which avoid the scope of our intellectual property rights. Further, the laws in the United States and elsewhere change rapidly, and any future changes could adversely affect us and our intellectual property. Our failure to meaningfully protect our intellectual property could result in competitors offering solutions that incorporate our most technologically advanced features, which could seriously reduce demand for our products. In addition, we may in the future need to initiate infringement claims or litigation. Litigation, whether we are a plaintiff or a defendant, can be expensive, time-consuming and may divert the efforts of our technical staff and managerial personnel, which could harm our business, whether or not the litigation results in a determination that is unfavorable to us. In addition, litigation is inherently uncertain, and thus we may not be able to stop our competitors from infringing our intellectual property rights.
We could incur substantial costs and disruption to our business as a result of any dispute related to, or claim of infringement of another party’s intellectual property rights, which could harm our business and operating results.
In recent years, there has been significant litigation in the United States over patents and other intellectual property rights. From time to time, we may face allegations that we or customers who use our products have infringed the trademarks, copyrights, patents and other intellectual property rights of third parties, including allegations made by our competitors or by non-practicing entities, or that we or our customers have misappropriated the intellectual property rights of such third parties. We cannot predict whether assertions of third party intellectual property rights or claims arising from these assertions will substantially harm our business and operating results. If we are forced to defend any infringement or misappropriation claims or attacks on the validity of our intellectual property rights, whether they are with or without merit or are ultimately determined in our favor, we may face costly litigation and diversion of technical and management personnel. Most of our competitors have substantially greater resources than we do and are able to sustain the cost of complex intellectual property litigation to a greater extent and for longer periods of time than we could. Furthermore, an adverse outcome of a dispute may require us, among other things: to pay damages, potentially including treble damages and attorneys’ fees, if we are found to have willfully infringed a party’s patent or other intellectual property rights; to cease making, licensing or using products that are alleged to incorporate or make use of the intellectual property of others; to expend additional development resources to redesign our products; and to enter into potentially unfavorable royalty or license agreements in order to obtain the rights to use necessary technologies. Royalty or licensing agreements, if required, may be unavailable on terms acceptable to us, or at all. In any event, we may need to license intellectual property which would require us to pay royalties or make one-time payments. Even if these matters do not result in litigation or are resolved in our favor or without significant cash settlements, the time and resources necessary to resolve them could harm our business, operating results, financial condition and reputation.
We face risks associated with litigation and claims.
We may, in the future, be involved in one or more lawsuits, claims or other proceedings. These suits could concern issues including contract disputes, employment actions, employee benefits, taxes, environmental, health and safety, fraud and abuse, personal injury and product liability matters.
| 38 |
Our business and operations would suffer in the event of computer system failures, cyber-attacks or deficiencies in our cyber-security.
In the ordinary course of our business, we collect and store sensitive data, including intellectual property, research data, our proprietary business information and that of our suppliers, technical information about our products, clinical trial plans and employee records. Similarly, our third-party providers possess certain of our sensitive data and confidential information. The secure maintenance of this information is critical to our operations and business strategy. Despite the implementation of security measures, our internal computer systems, and those of third parties on which we rely, are vulnerable to damage from computer viruses, malware, ransomware, cyber fraud, natural disasters, terrorism, war, telecommunication and electrical failures, cyber-attacks or cyber-intrusions over the Internet, attachments to emails, persons inside our organization, or persons with access to systems inside our organization. The risk of a security breach or disruption, particularly through cyber-attacks or cyber intrusion, including by computer hackers, foreign governments, and cyber terrorists, has generally increased as the number, intensity and sophistication of attempted attacks and intrusions from around the world have increased. Any such breach could compromise our networks and the information stored there could be accessed, publicly disclosed, encrypted, lost or stolen. Any such access, inappropriate disclosure of confidential or proprietary information or other loss of information, including our data being breached at third-party providers, could result in legal claims or proceedings, liability or financial loss under laws that protect the privacy of personal information, disruption of our operations or our product development programs and damage to our reputation, which could adversely affect our business.
Risks Related to the Regulation of Our Products
We are subject to extensive governmental regulation, including the requirement of U.S. Food and Drug Administration approval or clearance before our product candidates may be marketed.marketed and after approval or clearance and during the marketing of our products.
The process of obtaining U.S. Food and Drug Administration approval is lengthy, expensive and uncertain, and we cannot be sure that our additional product candidates will be approved in a timely fashion, or at all. If the U.S. Food and Drug Administration does not approve or clear our product candidates in a timely fashion, or at all, our business and financial condition would likely be adversely affected.
Both before and after approval or clearance of our product candidates, we, our product candidates, our suppliers and our contract manufacturers are subject to extensive regulation by governmental authorities in the United States and other countries. Failure to comply with applicable requirements could result in, among other things, any of the following actions:
| ● | FDA issuance of Form 483 or Warning Letters, which may be made public and may lead to further regulatory or enforcement actions, or similar letters by other regulatory authorities; | |
| ● | fines and other monetary penalties; | |
| ● | unanticipated expenditures; | |
| ● | delays in U.S. Food and Drug Administration approval and clearance, or U.S. Food and Drug Administration refusal to approve or clear a product candidate; | |
| ● | product recall or seizure; | |
| ● | interruption of manufacturing or clinical trials; | |
| ● | operating restrictions; | |
| ● | injunction or other restrictions imposed on our operations, including closing our facilities or our contract manufacturers’ facilities; or | |
| ● | criminal prosecutions. |
In addition to the approval and clearance requirements, numerous other regulatory requirements apply, both before and after approval or clearance, to us, our products and product candidates, and our suppliers and contract manufacturers. These include requirements related to the following:
| ● | testing and quality control; | |
| ● | manufacturing; | |
| ● | quality assurance |
| 39 |
| ● | ||
| ● | ||
| ● | ||
| ● | ||
| ● | ||
| ● | ||
| reporting to the U.S. Food and Drug Administration certain adverse experiences associated with the use of the | ||
| ● | obtaining additional approvals or clearances for certain modifications to the products or their labeling or claims. |
We are also subject to inspection by the U.S. Food and Drug Administration to determine our compliance with regulatory requirements, as are our suppliers and contract manufacturers, and we cannot be sure that the U.S. Food and Drug Administration will not identify compliance issues that may disrupt production or distribution, or require substantial resources to correct. We also cannot be sure that the U.S. Food and Drug Administration will agree with our analysis of, conclusions regarding, or handling of various situations that arise with our products. If it is determined that we failed to comply with any of our regulatory obligations, we could be subject to a wide range of enforcement actions that could limit our ability to continue to successfully commercialize impacted products or otherwise adversely impact us.
The U.S. Food and Drug Administration’s requirements may change and additional government regulations may be promulgated that could affect us, our product candidates, and our suppliers and contract manufacturers. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action. There can be no assurance that we will not be required to incur significant costs to comply with such laws and regulations in the future, or that such laws or regulations will not have a material adverse effect upon our business.
UroShield has not been cleared or approved by the FDA, nor has it undergone the same type of review as an FDA-approved or cleared device.
In September 2020, the FDA exercised its Enforcement Discretion to allow distribution of our UroShield device in the United States. This temporary authorization is limited to use as an extracorporeal acoustic wave generating accessory to urological indwelling catheter for use during the COVID-19 pandemic. The FDA may terminate or revoke this enforcement discretion policy at any time (after which the applicable products may no longer be used). There is no guarantee that our collaborators or customers will purchase or use the UroShield, that any sales of UroShield by us will generate any revenue or profits, or that we will ever be successful in obtaining FDA clearance or approval for the UroShield.
Failure to obtain regulatory approval in foreign jurisdictions will prevent us from marketing our products abroad.
International sales of our products and any of our product candidates that we commercialize are subject to the regulatory requirements of each country in which the products are sold. Accordingly, the introduction of our product candidates in markets outside the United States where we do not already possess regulatory approval will be subject to regulatory approvals in those jurisdictions. The regulatory review process varies from country to country. Many countries impose product standards, packaging and labeling requirements, and import restrictions on medical devices. In addition, each country has its own tariff regulations, duties and tax requirements, as well as reimbursement and healthcare payment systems. The approval by foreign government authorities is unpredictable and uncertain, and can be expensive. We may be required to perform additional pre-clinical, clinical or post-approval studies even if U.S. Food and Drug Administration approval has been obtained. Our ability to market our approved products could be substantially limited due to delays in receipt of, or failure to receive, the necessary approvals or clearances.
| 40 |
We are uncertain regarding the success of our clinical trials for our products in development.
We believe that all of our products in development, which consistcurrently consists of LungShield andonly RenooSkin, will require clinical trials to determine their safety and efficacy by regulatory bodies in their target markets, including the U.S. Food and Drug Administration and various foreign regulators. There can be no assurance that we will be able to successfully complete the U.S. and foreign regulatory approval processes for products in development. In addition, there can be no assurance that we will not encounter additional problems that will cause us to delay, suspend or terminate our clinical trials. In addition, we cannot make any assurance that clinical trials will be deemed sufficient in size and scope to satisfy regulatory approval requirements, or, if completed, will ultimately demonstrate our products to be safe and efficacious.
We depend on Sanuwave for developing and commercializing our WoundShield technology.
In March 2020, we entered into a license agreement with Sanuwave for the manufacture and delivery of our WoundShield technology. Under this agreement, Sanuwave has received the worldwide, exclusive rights to our WoundShield technology. Sanuwave will bear the cost and clinical validation responsibilities associated with obtaining approval for WoundShield from the FDA and other regulatory agencies around the world. Sanuwave is also responsible for manufacturing and commercializing the WoundShield product and technology. Our right to receive a milestone payment under the license agreement depends on the achievement of FDA approval by Sanuwave and our ability to receive royalties under the agreement depends on Sanuwave’s successful commercialization of the WoundShield product and technology.
The adoptiondevelopment and commercialization of health policy changesthe WoundShield product and health caretechnology and our ability to receive a potential milestone and royalty payments under the license agreement with Sanuwave, could be adversely affected if Sanuwave:
● lacks or does not devote sufficient time and resources to the development and commercialization of the WoundShield product and technology;
● lacks or does not devote sufficient capital to fund the development and commercialization of the WoundShield product and technology;
● develops, either alone or with others, products that compete with the WoundShield product and technology;
● fails to gain the requisite regulatory approvals for the WoundShield product and technology;
● does not successfully commercialize the WoundShield product and technology;
● does not conduct its activities in a timely manner;
● terminates its license with us; or
● does not effectively pursue and enforce intellectual property rights relating to the WoundShield product and technology.
We have limited or no control over the occurrence of any of the foregoing. Furthermore, disagreements with Sanuwave could lead to disputes, which could be time-consuming and expensive. If any of these issues arise, it may delay the development and commercialization milestone and royalties based on further development and sales of the WoundShield product and technology.
Healthcare reform inmeasures could adversely affect our business and financial results.
In the United States, there have been, and we expect there will continue to be, a number of legislative and regulatory changes to the healthcare system in ways that may adversely affect our business and financial results.
On March 23, 2010, President Obama signed into law major health care Federal and state lawmakers regularly propose and, at times, enact legislation that could result in significant changes to the healthcare system, some of which are intended to contain or reduce the costs of medical products and services. Current and future legislative proposals to further reform legislation underhealthcare or reduce healthcare costs may limit coverage of or lower reimbursement for our products. The cost containment measures that payers and providers are instituting and the effect of any healthcare reform initiative implemented in the future could impact our revenue from the sale of our products. For example, the Patient Protection and Affordable Care Act of 2010, commonly referred to as the Affordable Care Act, which was modified on March 30, 2010, by the enactmentcontains a number of the Health Care and Education Reconciliation Act of 2010. The Affordable Care Act contains numerous regulations regarding the payment for and provision of health care,provisions, including provisions aimed at improving quality, extending health care coverage to tens of millions of individuals, enhancing remedies for fraud and abuse, adding transparency requirements and conditions to reimbursement, and decreasing health care costs. The Affordable Care Act also includes significant provisions that encourage state and federal law enforcement agencies to increase activities related to preventing, detecting and prosecuting those who commit fraud, waste and abusegoverning enrollment in federal healthcare programs, including Medicare, Medicaidreimbursement changes and Tricare. This legislation is onefraud and abuse measures, all of which will impact existing government healthcare programs and will result in the development of new programs.
| 41 |
There have been executive, judicial and Congressional challenges to certain aspects of the mostAffordable Care Act. For example, President Trump signed several Executive Orders and other directives designed to delay the implementation of certain provisions of the Affordable Care Act. Concurrently, Congress considered legislation to repeal or repeal and replace all or part of the Affordable Care Act. While Congress has not passed comprehensive repeal legislation, it has enacted laws that modify certain provisions of the Affordable Care Act such as removing penalties, starting January 1, 2019, for not complying with the Affordable Care Act’s individual mandate to carry health insurance and significant reforms ever experienceddelaying the implementation of certain fees mandated by the Affordable Care Act. On December 14, 2018, a Texas U.S. District Court Judge ruled that the Affordable Care Act is unconstitutional in its entirety because the individual mandate was repealed by Congress as part of the Tax Cuts and Jobs Act of 2017. Additionally, on December 18, 2019, the U.S. Court of Appeals for the 5th Circuit upheld the District Court ruling that the individual mandate was unconstitutional and remanded the case back to the District Court to determine whether the remaining provisions of the Affordable Care Act are invalid as well. The United States Supreme Court is currently reviewing this case, but it is unknown when a decision will be reached. Although the Supreme Court has not yet ruled on the constitutionality of the Affordable Care Act, on January 28, 2021, President Biden issued an executive order to initiate a special enrollment period from February 15, 2021 through May 15, 2021 for purposes of obtaining health care industryinsurance coverage through the Affordable Care Act marketplace. The executive order also instructs certain governmental agencies to review and has significantly changedreconsider their existing policies and rules that limit access to healthcare, including among others, reexamining Medicaid demonstration projects and waiver programs that include work requirements, and policies that create unnecessary barriers to obtaining access to health insurance coverage through Medicaid or the way health careAffordable Care Act. It is financed by both governmentalunclear how the Supreme Court ruling, other such litigation and private insurers. Extending health care coverage to those who previously lacked coveragethe healthcare reform measures of the Biden administration will likely result in substantial cost toimpact the United States federal government, whichAffordable Care Act and negatively affect our business, financial condition and results of operations.
The current presidential administration and Congress may force additionalpursue significant changes to the health care system in the United States. Muchcurrent healthcare laws. We face uncertainties that might result from modifications or repeal of the funding for expanded health care coverage may be sought through cost savings. While some of these savings may come from realizing greater efficiencies in delivering care, improving the effectiveness of preventive care and enhancing the overall quality of care, much of the cost savings may come from reducing the cost of health care and increased enforcement activities. The cost of health care could be reduced by decreasing the level of reimbursement for medical services or products (including products we may sell or market), or by restricting coverage of medical services or products. A reduction in the use of or reimbursement for products we may sell in the United States could materially adversely affect our business and results of operations.
Someany of the provisions of the Affordable Care Act, have not yet been fully implementedincluding as a result of current and thefuture executive orders and legislative actions. The impact of those changes on us and potential effect of the legislationon our industry as a whole is difficultcurrently unknown. Any changes to predict. The Affordable Care Act continues to be implemented through regulation and government activity, and is subject to possible additional implementing regulations and interpretive guidelines. Further, the Affordable Care Act has been subjectare likely to judicialhave an impact on our results of operations, and Congressional challenges,may negatively affect our business, financial condition and legislative initiatives to modify, limit, or repeal the Affordable Care Act continue. It remains to be seen, however, precisely what new health care reform legislation will be enacted, if any, and what impact it will have on the availabilityresults of health care and containing or lowering the cost of health care. The manner in which the Affordable Care Act continues to evolve could materially affect the extent to which and the amount at which health care products and services are reimbursed by government programs such as Medicare, Medicaid and Tricare.operations. We cannot predict all impacts the Affordable Care Act orwhat other health care reform legislation may have on our products, but it may result in our products being chosen less frequently or the pricing being substantially lowered.
In addition, other health care reform proposals have emergedhealthcare programs and regulations will ultimately be implemented at the federal andor state levels, including those aimed at reducing health care costs and increasing transparency. We cannot predictlevel or the effect these newly enacted laws orof any future legislation or regulation will have on us. However,in the implementation of new legislation and regulationUnited States may lower reimbursements for our products, increase our compliance and other costs, and adverselynegatively affect our business.business, financial condition and results of operations.
We cannot predict whatexpect that additional state and federal healthcare reform initiatives maymeasures will be adopted in the future, or how federalparticularly in light of the new presidential administration. Changes in healthcare policy could increase our costs and state legislativesubject us to additional regulatory requirements that may interrupt commercialization of our current and regulatory developments are likely to evolve, but we expect ongoing initiativesfuture solutions. Changes in the United States tohealthcare policy could increase pressure on pricing for health care productsour costs, decrease our revenue and services. Such reforms could have an adverse effect on the pricingimpact sales of and marketreimbursement for our current and future products.
Further, it is possible that additional governmental action is taken in response to the COVID-19 pandemic.
If we fail to comply with the U.S. federal and state fraud and abuse and other health care laws and regulations, we could be subject to criminal and civil penalties and exclusion from the Medicare and Medicaid programs, which would have a material adverse effect on our business and results of operations.
All of our financial relationships with health care providers and others who provide products or services to federal health care program beneficiaries are potentially governed by the federal and state fraud and abuse laws, and other health care laws and regulations may be or become applicable to our business and operations and expose us to risk. For example:
| ● | The federal Anti-Kickback Statute, which prohibits the offer, payment, solicitation or receipt of any form of remuneration in return for referring, ordering, leasing, purchasing or arranging for, or recommending the ordering, purchasing or leasing of, items or services payable by Medicare, Medicaid or any other federal health care program. |
| 42 |
● The federal Anti-Kickback Statute, which prohibits the offer, payment, solicitation or receipt of any form of remuneration in return for referring, ordering, leasing, purchasing or arranging for, or recommending the ordering, purchasing or leasing of, items or services payable by Medicare, Medicaid or any other federal health care program.
●
| ● | Federal false claims laws and civil monetary penalty laws, including the False Claims Act, that prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid or other government health care programs that are false or fraudulent, or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government. | |
| ● | The federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which prohibits knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program or obtain, by means of false or fraudulent pretenses, representations, or promises, any of the money or property owned by, or under the custody or control of, any health care benefit program, and for knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statements in connection with the delivery of or payment for health care benefits, items or services. | |
| ● | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, and its implementing regulations, which also impose obligations and requirements on health care providers, health plans, and healthcare clearinghouses as well as their respective business associates that perform certain services for them that involve the use or disclosure of individually identifiable health information, with respect to safeguarding the privacy and security of certain individually identifiable health information. | |
| ● | The federal transparency requirements under the Affordable Care Act, including the provision commonly referred to as the Physician Payments Sunshine Act, which requires certain manufacturers of drugs, devices, biologics and medical supplies that are reimbursable under Medicare, Medicaid or Children’s Health Insurance Program to report annually to Centers for Medicare and Medicaid Services, or CMS, information related to payments and other transfers of value to physicians and teaching hospitals, and ownership and investment interests held by physicians and their immediate family members. | |
| ● | Analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws, which may be broader in scope and apply to referrals and items or services reimbursed by both governmental and non-governmental third-party payers, including private insurers, many of which differ from each other in significant ways and often are not preempted by federal law, thus complicating compliance efforts. |
● The federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which prohibits knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program or obtain, by means of false or fraudulent pretenses, representations, or promises, any of the money or property owned by, or under the custody or control of, any health care benefit program, and for knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statements in connection with the delivery of or payment for health care benefits, items or services.
● HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, and its implementing regulations, which also impose obligations and requirements on health care providers, health plans, and healthcare clearinghouses as well as their respective business associates that perform certain services for them that involve the use or disclosure of individually identifiable health information, with respect to safeguarding the privacy and security of certain individually identifiable health information.
● The federal transparency requirements under the Affordable Care Act, including the provision commonly referred to as the Physician Payments Sunshine Act, which requires certain manufacturers of drugs, devices, biologics and medical supplies that are reimbursable under Medicare, Medicaid or Children’s Health Insurance Program to report annually to Centers for Medicare and Medicaid Services, or CMS, information related to payments and other transfers of value to physicians and teaching hospitals, and ownership and investment interests held by physicians and their immediate family members.
● Analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws, which may be broader in scope and apply to referrals and items or services reimbursed by both governmental and non-governmental third-party payers, including private insurers, many of which differ from each other in significant ways and often are not preempted by federal law, thus complicating compliance efforts.
Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available, it is possible that some of our business activities could be subject to challenge under one or more of such laws. In addition, recent health care reform legislation has strengthened these laws. Efforts to ensure that our business arrangements with third parties and our operations are compliant with applicable health care laws and regulations will involve the expenditure of appropriate, and possibly significant, resources. If we are found to be in violation of any current or future statutes or regulations involving applicable fraud and abuse or other health care laws and regulations, we may be subject to significant civil, criminal and administrative penalties, damages, fines, disgorgement, imprisonment, exclusion from government funded health care programs, such as Medicare and Medicaid, contractual damages, reputational harm, diminished profits and future earnings, which could have a material adverse effect on our business, results of operations and financial condition. If any physicians or other health care providers or entities with whom we expect to do business are found to not be in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded health care programs, which could adversely affect our ability to operate our business and our results of operations.
| 43 |
Risks Related to our Operations in Israel
We conduct our operations in Israel and therefore our results may be adversely affected by political, economic and military instability in Israel and its region.
Our principal offices and manufacturing facilities are located in Israel and most of our officers and employees are residents of Israel. Accordingly, political, economic and military conditions in Israel and the surrounding region may directly affect our business. Since the establishment of the State of Israel in 1948, a number of armed conflicts have taken place between Israel and its Arab neighbors. Any hostilities involving Israel or the interruption or curtailment of trade within Israel or between Israel and its trading partners could adversely affect our operations and results of operations and could make it more difficult for us to raise capital. During the summer of 2014, Israel was engagedCivil unrest and political turbulence has occurred in an armed conflict with Hamas in Gaza, which involved missile strikes against civilian targets in various parts of Israel and negatively affected business conditions in Israel. In addition, recent political uprisings and conflicts in variousother countries in the Middle East,region, including EgyptSyria which shares a common border with Israel, and Syria, areis affecting the political stability of those countries. It is not clear howThe civil war that has been ongoing in Syria has escalated, and this instability will develop and how it will affect the political and security situationany intervention may lead to additional conflicts in the Middle East. This instability has raised concerns regarding security in the region and the potential for armed conflict.region. In addition, it is widely believed that Iran which has previously threatened to attack Israel has been stepping up its efforts to achieve nuclear capability. Iranand is alsowidely believed to havebe developing nuclear weapons. Iran also has a strong influence among extremist groups in the region, such as Hamas in Gaza and Hezbollah in Lebanon. Additionally, the Islamic State of Iraq and Levant (“ISIL”), a violent jihadist group, is involved in hostilities in Iraq and Syria. Although ISIL’s activities have not directly affected the political and economic conditions in Israel, ISIL’s stated purpose is to take control of the Middle East, including Israel. The tension between Israel and Iran and/or these groupsregion. These situations may potentially escalate in the future to more violent events which may affect Israel and turn violent, which could affect the Israeli economy in general and us in particular.our operations. Any armed conflicts, terrorist activities or political instability in the region could adversely affect business conditions and could harm our results of operations. For example, any major escalation in hostilities in the region could result in a portion of our employees being called up to perform military duty for an extended period of time. Our operations could be disrupted by the absence of a significant number of our employees. Parties with whom we do business have sometimes declined to travel to Israel during periods of heightened unrest or tension, forcing us to make alternative arrangements when necessary. In addition, the political and security situation in Israel may result in parties with whom we have agreements involving performance in Israel claiming that they are not obligated to perform their commitments under those agreements pursuant to force majeure provisions in such agreements.
Our commercial insurance does not cover losses that may occur as a result of events associated with the security situation in the Middle East. Although the Israeli government currently covers the reinstatement value of direct damages that are caused by terrorist attacks or acts of war, we cannot assure you that this government coverage will be maintained. Any losses or damages incurred by us could have a material adverse effect on our business. Any armed conflicts or political instability in the region would likely negatively affect business conditions and could harm our results of operations.
Further, in the past, the State of Israel and Israeli companies have been subjected to an economic boycott. Several countries still restrict business and trade activity with the State of Israel and with Israeli companies. These restrictive laws and policies may have an adverse impact on our operating results, financial condition or the expansion of our business.
Our operations may be disrupted as a result of the obligation of management or personnel to perform military service.
Many of our male employees in Israel, including members of our senior management, perform up to one month, and in some cases more, of annual military reserve duty until they reach the age of 45 or older and, in the event of a military conflict, may be called to active duty. There have also been periods of significant call-ups of military reservists, and it is possible that there will be military reserve duty call-ups in the future. Our operations could be disrupted by the absence of a significant number of our employees. Such disruption could materially adversely affect our business, financial condition and results of operations.
Because a certain portion of our expenses is incurred in currencies other than the U.S. dollar, our results of operations may be harmed by currency fluctuations and inflation.
We expect our revenues from future licensing agreements to be denominated mainly in U.S. dollars or in Euros. We pay a substantial portion of our expenses in U.S. dollars; however, a portion of our expenses, related to salaries of the employees in Israel and payment to part of the service providers in Israel and other territories, are paid in New Israeli Shekels, or NIS, and in other currencies. In addition, a portion of our financial assets is held in NIS and in other currencies. As a result, we are exposed to the currency fluctuation risks, and we do not attempt to hedge against such risks. For example, if the NIS strengthens against the U.S. dollar, our reported expenses in U.S. dollars may be higher than anticipated. In addition, if the NIS weakens against the U.S. dollar, the U.S. dollar value of our financial assets held in NIS will decline.
It may be difficult for investors in the United States to enforce any judgments obtained against us or any of our directors or officers.
Almost all of our assets are located outside the United States, although we do maintain a permanent place of business within the United States. In addition, some of our officers and directors are nationals and/or residents of countries other than the United States, and all or a substantial portion of such persons’ assets are located outside the United States. As a result, it may be difficult for investors to enforce within the United States any judgments obtained against us or any of our non-U.S. directors or officers, including judgments predicated upon the civil liability provisions of the securities laws of the United States or any state thereof. Additionally, it may be difficult to assert U.S. securities law claims in actions originally instituted outside of the United States. Israeli courts may refuse to hear a U.S. securities law claim because Israeli courts may not be the most appropriate forums in which to bring such a claim. Even if an Israeli court agrees to hear a claim, it may determine that the Israeli law, and not U.S. law, is applicable to the claim. Further, if U.S. law is found to be applicable, certain content of applicable U.S. law must be proved as a fact, which can be a time-consuming and costly process, and certain matters of procedure would still be governed by the Israeli law. Consequently, you may be effectively prevented from pursuing remedies under U.S. federal and state securities laws against us or any of our non-U.S. directors or officers.
| 44 |
Risks Related to Our Organization and Our Securities
The price of our securities may be volatile, and the market price of our securities may drop below the price you pay.
We expect that the price of our securities will fluctuate significantly. Market prices for securities of early-stage medical device companies have historically been particularly volatile. In addition to the factors discussed in this “Risk Factors” section and elsewhere in this report, these factors include:
| ● | progress, or lack of progress, in developing and commercializing our products; | |
| ● | favorable or unfavorable decisions about our products or intellectual property from government regulators, insurance companies or other third-party payers; | |
| ● | our ability to recruit and retain qualified regulatory and research and development personnel; | |
| ● | changes in investors’ and securities analysts’ perception of the business risks and conditions of our business; | |
| ● | changes in our relationship with key collaborators; | |
| ● | changes in the market valuation or earnings of our competitors or companies viewed as similar to us; | |
| ● | changes in key personnel; |
| ● | depth of the trading market in our common stock; | |
| ● | changes in our capital structure, such as future issuances of securities or the incurrence of additional debt; | |
| ● | the granting or exercise of employee stock options or other equity awards; | |
| ● | realization of any of the risks described under this section entitled “Risk Factors”; and | |
| ● | general market and economic conditions. |
| 45 |
In recent years, the stock markets, in general, have experienced extreme price and volume fluctuations especially in the biotechnology sector. Broad market and industry factors may materially harm the market price of shares of our common stock. In the past, following periods of volatility in the market price of a company’s securities, securities class action litigation has often been instituted against that company. If we were involved in any similar litigation, we could incur substantial costs and our management’s attention and resources could be diverted. On March 12, 2020, the WHO declared COVID-19 to be a pandemic, and the COVID-19 pandemic has resulted in significant financial market volatility and uncertainty in recent weeks. In addition, U.S. and global markets are experiencing volatility and disruption following the escalation of geopolitical tensions and the start of the military conflict between Russia and Ukraine. A continuation or worsening of the levels of market disruption and volatility could have an adverse effect on our ability to access capital, on our business, results of operations and financial condition, and on the market price of our common shares.
We have a significant number of warrants and options, and future sales of our common stock upon exercise of these options or warrants, or the perception that future sales may occur, may cause the market price of our common stock to decline, even if our business is doing well.
Sales of a significant number of shares of our common stock in the public market could harm the market price of our common stock and make it more difficult for us to raise funds through future offerings of common stock. Our stockholders and the holders of our outstanding warrants and options, upon exercise of these options or warrants, may sell substantial amounts of our common stock in the public market. The availability of these shares of our common stock for resale in the public market has the potential to cause the supply of our common stock to exceed investor demand, thereby decreasing the price of our common stock.
In addition, the fact that our stockholders and holders of our warrants and options can sell substantial amounts of our common stock in the public market, whether or not sales have occurred or are occurring, could make it more difficult for us to raise additional financing through the sale of equity or equity-related securities in the future at a time and price that we deem reasonable or appropriate.
Although our shares of common stock are now listed on the NASDAQ Capital Market, we currently have a limited trading volume, which results in higher price volatility for, and reduced liquidity of, our common stock.
Although our shares of common stock are now listed on the NASDAQ Capital Market under the symbol “NAOV,” trading volume in our common stock has been limited and an active trading market for our shares of common stock may never develop or be maintained. The absence of an active trading market increases price volatility and reduces the liquidity of our common stock. As long as this condition continues, the sale of a significant number of shares of common stock at any particular time could be difficult to achieve at the market prices prevailing immediately before such shares are offered.
If we cannot continuefail to satisfycomply with the continuingcontinued listing criteriarequirements of the NASDAQ Capital Market, the exchange may subsequently delist our common stock.
NASDAQ requires us to meet certain financial, public float, bid price and liquidity standards on an ongoing basis in order to continue the listing of our common stock. Generally, we must maintain a minimum amount of stockholders equity and a minimum number of holders of our securities. If we fail to meet any of the continuing listing requirements, our common stock may be subject to delisting. If our common stock is delisted and we are not able to list our common stock on another national securities exchange, we expect our securities would be quoted on an over-the-counter market. If this were to occur, our stockholders could face significant material adverse consequences, including limited availabilitythe price of market quotations for our common stock and reduced liquidity for the trading of our securities. In addition, we could experience a decreased ability to issue additional securities and obtain additional financing inaccess the future. There cancapital markets could be no assurance that an active trading market for ournegatively impacted.
Our common stock will develop or be sustained.
Complying with the laws and regulations affecting public companies has increased and will increase our costs and the demandsis currently listed for trading on management and could harm our operating results.
As a public company, we will continue to incur significant legal, accounting and other expenses that we did not incur as a private company, including costs associated with public company reporting requirements. We also anticipate that we will incur costs associated with relatively recently adopted corporate governance requirements, including requirements of the Securities Exchange Commission and the NASDAQ StockCapital Market. We expect these rules and regulations to increase our legal and financial compliance costs and to make some activities more time-consuming and costly. We also expect that these rules and regulations may make it more difficult and more expensive for us to obtain director and officer liability insurance and we may be required to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. As a result, it may be more difficult for us to attract and retain qualified individuals to serve on our board of directors or as executive officers. We are currently evaluating and monitoring developments with respect to these rules, and we cannot predict or estimate the amount of additional costs we may incur or the timing of such costs.
For example, the Sarbanes-Oxley Act requires,must satisfy NASDAQ’s continued listing requirements, including, among other things, that we assess the effectivenessa minimum stockholders’ equity of our internal control over financial reporting annually$2.5 million and the effectiveness of our disclosure controls and procedures quarterly. Section 404 of the Sarbanes-Oxley Act (“Section 404”) requires us to perform system and process evaluation and testing of our internal control over financial reporting to allow management to report on, and our independent registered public accounting firm potentially to attest to, the effectiveness of our internal control over financial reporting. Our compliance with applicable provisions of Section 404, including the requirement that our independent registered public accounting firm undertake an assessment of our internal control over financial reporting, will require that we incur substantial accounting expense and expend significant management time on compliance-related issues as we implement additional corporate governance practices and comply with reporting requirements. Moreover, if we are not able to comply with the requirements of Section 404 applicable to us in a timely manner, or if we or our independent registered public accounting firm identifies deficiencies in our internal control over financial reporting that are deemed to be material weaknesses, the marketminimum closing bid price of our stock could decline and we could be subject to sanctions$1.00 per share or investigations by the Securities Exchange Commission or other regulatory authorities,risk delisting, which would require additional financial and management resources. Furthermore, investor perceptions of our company may suffer if deficiencies are found, and this could cause a decline in the market price of our stock. Irrespective of compliance with Section 404, any failure of our internal control over financial reporting could have a material adverse effect on our stated operating resultsbusiness. A delisting of our common stock from the NASDAQ Capital Market could materially reduce the liquidity of our common stock and harmresult in a corresponding material reduction in the price of our reputation. If we are unable to implement these requirements effectively or efficiently, itcommon stock. In addition, delisting could harm our operations, financial reporting,ability to raise capital through alternative financing sources on terms acceptable to us, or financial resultsat all, and couldmay result in the potential loss of confidence by investors, suppliers, customers and employees and fewer business development opportunities.
| 46 |
On March 2 2022, the Company received notice from the Listing Qualifications Staff of Nasdaq indicating that, based upon the closing bid price of the Company’s common stock for the 30 consecutive business day period between January 14, 2022, through March 1, 2022, we did not meet the minimum bid price of $1.00 per share required for continued listing on the Nasdaq Capital Market pursuant to Nasdaq Listing Rule 555(a)(2). The letter also indicated that the Company will be provided with a compliance period of 180 calendar days, or until August 29, 2022 (the “Compliance Period”), in which to regain compliance pursuant to Nasdaq Listing Rule 5810(c)(3)(A).
There is no assurance that we can regain compliance with such minimum listing requirements. If our common stock were delisted from NASDAQ, trading of our common stock would most likely take place on an over-the-counter market established for unlisted securities, such as the OTCQB or the Pink Market maintained by OTC Markets Group Inc. An investor would likely find it less convenient to sell, or to obtain accurate quotations in seeking to buy, our common stock on an over-the-counter market, and many investors would likely not buy or sell our common stock due to difficulty in accessing over-the-counter markets, policies preventing them from trading in securities not listed on a national exchange or other reasons. In addition, as a delisted security, our common stock would be subject to SEC rules as a “penny stock,” which impose additional disclosure requirements on broker-dealers. The regulations relating to penny stocks, coupled with the typically higher cost per trade to the investor of penny stocks due to factors such as broker commissions generally representing a higher percentage of the price of a penny stock than of a higher-priced stock, would further limit the ability of investors to trade in our common stock. In addition, delisting could harm our ability to raise capital through alternative financing sources on terms acceptable to us, or at all, and may result in the potential loss of confidence by investors, suppliers, customers and employees and fewer business development opportunities. For these reasons and others, delisting would adversely affect the liquidity, trading volume and price of our common stock, causing the value of an investment in us to decrease and having an adverse opinioneffect on our internal control overbusiness, financial reporting fromcondition and results of operations, including our independent registered public accounting firm.ability to attract and retain qualified employees and to raise capital.
If we fail to maintain effective internal control over financial reporting, the market priceour business, financial condition or results of our securitiesoperations may be adversely affected.
As a public reporting company, we are required to establish and maintain effective internal control over financial reporting. Failure to establish such internal control, or any failure of such internal control once established, could adversely impact our public disclosures regarding our business, financial condition or results of operations. Any failure of our internal control over financial reporting could also prevent us from maintaining accurate accounting records and discovering accounting errors and financial frauds.
Rules adopted by the Securities and Exchange Commission pursuant to Section 404 of Sarbanes-Oxley Act of 2002 require annual assessment of our internal control over financial reporting. The standards that must be met for management to assess the internal control over financial reporting as effective are complex, and require significant documentation, testing and possible remediation to meet the detailed standards. We may encounter problems or delays in completing activities necessary to make an assessment of our internal control over financial reporting. If we cannot assess our internal control over financial reporting as effective, investor confidence and share value may be negatively impacted. In addition, management’s assessment of internal control over financial reporting may identify weaknesses and conditions that need to be addressed in our internal control over financial reporting or other matters that may raise concerns for investors. Any actual or perceived weaknesses and conditions that need to be addressed in our internal control over financial reporting (including those weaknesses identified in our periodic reports), or disclosure of management’s assessment of our internal control over financial reporting may have an adverse impact on the price of our securities.
| 47 |
While
As disclosed in Part II, Item 9A, “Controls and Procedures,” we currently qualifyhave identified material weaknesses in our internal control over financial reporting due to a lack of a full and complete testing of our disclosure controls and procedures. We concluded that our internal control over financial reporting and related disclosure controls and procedures were not effective as an “emerging growth company” under the Jumpstart of Business Startups Act of 2012, or the JOBS Act, we could lose that status, which may increase the costs and demands placed upon our management.
We are an “emerging growth company,” as definedDecember 31, 2021. Our management is in the Jumpstart Our Business Startups Act,process of implementing remediation measures with respect to the controls and would continuewritten policies and procedures as described in Part II, Item 9A, “Controls and Procedures,” and management expects that such measures, once fully implemented, will be sufficient to be an emerging growth company until theremediate such material weaknesses in our internal control over financial reporting that existed as of December 31, 2022, or until the earliest of (i) the last day of the fiscal year during which we had total annual gross revenues of $1.07 billion (as indexed for inflation); (ii) the date on which we have, during the previous 3-year period, issued more than $1 billion in non-convertible debt; or (iii) the date on which we2021.
We are deemed to be a ‘large accelerated filer,’ as defined by the Securities and Exchange Commission, which would generally occur upon our attaining a public float of at least $700 million. Once we lose emerging growth company status, we expect the costs and demands placed upon our management to increase, as we would have to comply with additional disclosure and accounting requirements, particularly if we would also no longer qualify as a smaller reporting company.
We are an “emerging growth company”company and we cannot be certain thatif the reduced disclosure requirements applicable to emerging growth companiesour filing status will make our common stock less attractive to investors.
The JOBS Act permits “emerging growth companies” like usWe are a “smaller reporting company” and, thus, have certain decreased disclosure obligations in our SEC filings, including, among other things, simplified executive compensation disclosures and only being required to rely on someprovide two years of the reduced disclosure requirements that are already available to smaller reporting companies. As long as we qualify as an emerging growth company or a smaller reporting company, we would be permitted to omit an auditor’s attestation on internal control overaudited financial reporting that would otherwise be required by the Sarbanes-Oxley Act, and are also exempt from the requirement to submit “say-on-pay”, “say-on-pay frequency” and “say-on-parachute” votesstatements in annual reports. Decreased disclosures in our SEC filings due to our stockholdersstatus as a “smaller reporting company” may make it harder for investors to analyze our results of operations and financial prospects and may avail ourselves of reduced executive compensation disclosure that is already available to smaller reporting companies.
In addition, Section 107 of the JOBS Act also provides that an emerging growth company can take advantage of the exemption from complying with new or revised accounting standards provided in Section 7(a)(2)(B) of the Securities Act of 1933, as amended, as long as we are an emerging growth company. An emerging growth company can therefore delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We have elected to take advantage of the benefits of this until we are no longer an emerging growth company or until we affirmatively and irrevocably opt out of this exemption. Our financial statements may therefore not be comparable to those of companies that comply with such new or revised accounting standards.
We will cease to be an emerging growth company at such time as described in the risk factor immediately above. Until such time, however, we cannot predict if investors will findmake our common stock a less attractive because we may rely on these exemptions.investment. If some investors find our common stock less attractive, as a result, there may be a less active trading market for our common stock and our stock price may be more volatile and could cause our stock price to decline.volatile.
Anti-takeover provisions of our certificate of incorporation, our bylaws and Delaware law could make an acquisition of us, which may be beneficial to our stockholders, more difficult and may prevent attempts by our stockholders to replace or remove the current members of our board and management.
Certain provisions of our amended and restated certificate of incorporation and bylaws could discourage, delay or prevent a merger, acquisition or other change of control that stockholders may consider favorable, including transactions in which you might otherwise receive a premium for your shares. Furthermore, these provisions could prevent or frustrate attempts by our stockholders to replace or remove members of our board of directors. These provisions also could limit the price that investors might be willing to pay in the future for our securities, thereby depressing the market price of our securities. Stockholders who wish to participate in these transactions may not have the opportunity to do so. These provisions, among other things:
| ● | allow the authorized number of directors to be changed only by resolution of our board of directors; |
| 48 |
| ● | authorize our board of directors to issue, without stockholder approval, preferred stock, the rights of which will be determined at the discretion of the board of directors and that, if issued, could operate as a “poison pill” to dilute the stock ownership of a potential hostile acquirer to prevent an acquisition that our board of directors does not approve; | |
| ● | establish advance notice requirements for stockholder nominations to our board of directors or for stockholder proposals that can be acted on at stockholder meetings; and | |
| ● | limit who may call a stockholder meeting. |
In addition, we are governed by the provisions of Section 203 of the Delaware General Corporation Law that may, unless certain criteria are met, prohibit large stockholders, in particular those owning 15% or more of the voting rights on our common stock, from merging or combining with us for a prescribed period of time.
If securities or industry analysts do not publish research or reports or publish unfavorable research about our business, the price of our securities and their trading volume could decline.
The trading market for our securities will depend in part on the research and reports that securities or industry analysts publish about us or our business. We do not currently have and may never obtainCurrently there is only one research coverage by a securities and industry analysts.analyst. If no securities or industry analysts commence coverage of us the trading price for our securities would be negatively affected. In the event we obtain securities or industry analyst coverage, if one or more of the analysts who covers us downgrades our securities, the price of our securities would likely decline. If one or more of these analysts ceases to cover us or fails to publish regular reports on us, interest in the purchase of our securities could decrease, which could cause the price of our securities and their trading volume to decline.
We may be subject to ongoing restrictions related to grants from the Israeli Office of the Chief Scientist.
Through our Israeli subsidiary, as of December 31, 2017, we received grants of $437,000 from the Office of the Chief Scientist of the Israeli Ministry of Industry, Trade and Labor, or the Office of the Chief Scientist, for research and development programs related to products that we are not currently commercializing or marketing. Because we are no longer developing the product to which the grants relate, we do not believe that we are subject to any material conditions with respect to the grants, except for the restrictions on our ability to make certain transfers of the technology or intellectual property related to these grants described below. We could in the future determine to apply for further grants. If we receive any such grants, we would have to comply with specified conditions, including paying royalties with respect to grants received. If we fail to comply with these conditions in the future, sanctions might be imposed on us, such as grants could be cancelled and we could be required to refund any payments previously received under these programs.
Pursuant to the Israeli Encouragement of Industrial Research and Development Law, any products developed with grants from the Office of the Chief Scientist are required to be manufactured in Israel and certain payments may be required in connection with the change of control of the grant recipient and the financing, mortgaging, production, exportation, licensing and transfer or sale of its technology and intellectual property to third parties, which will require the Office of the Chief Scientist’s prior consent and, in case such a third party is outside of Israel, extended royalties and/or other fees. This could have a material adverse effect on and significant cash flow consequences to us if, and when, any technologies, intellectual property or manufacturing rights are exported, transferred or licensed to third parties outside Israel. If the Office of the Chief Scientist does not wish to give its consent in any required situation or transaction, we would need to negotiate a resolution with the Office of the Chief Scientist. In any event, such a transaction, assuming it was approved by the Office of the Chief Scientist, would involve monetary payments, such as royalties or fees, of not less than the applicable funding received from the Office of the Chief Scientist plus interest, not to exceed, in aggregate, six times the applicable funding received from the Office of the Chief Scientist.
Because we do not expect to pay cash dividends for the foreseeable future, you must rely on appreciation of our common stock price for any return on your investment. Even if we change that policy, we may be restricted from paying dividends on our common stock.
We do not intend to pay cash dividends on shares of our common stock for the foreseeable future. Any determination to pay dividends in the future will be at the discretion of our board of directors and will depend upon results of operations, financial performance, contractual restrictions, restrictions imposed by applicable law and other factors our board of directors deems relevant. Accordingly, you will have to rely on capital appreciation, if any, to earn a return on your investment in our common stock. Investors seeking cash dividends in the foreseeable future should not purchase our common stock.
| 49 |
Our ability to use our net operating loss carry forwards and certain other tax attributes may be limited.
Our ability to utilize our federal net operating loss, carryforwards and federal tax credit may be limited under Sections 382 and 383 of the Internal Revenue Code of 1986, as amended. The limitations apply if an “ownership change,” as defined by Section 382, occurs. Generally, an ownership change occurs if the percentage of the value of the stock that is owned by one or more direct or indirect “five percent shareholders” increases by more than 50% over their lowest ownership percentage at any time during the applicable testing period (typically three years). If we have experienced an “ownership change” at any time since our formation, we may already be subject to limitations on our ability to utilize our existing net operating losses and other tax attributes to offset taxable income. In addition, future changes in our stock ownership, which may be outside of our control, may trigger an “ownership change” and, consequently, Section 382 and 383 limitations. As a result, if we earn net taxable income, our ability to use our pre-change net operating loss carryforwards and other tax attributes to offset U.S. federal taxable income may be subject to limitations, which could potentially result in increased future tax liability to us.
ITEM 1B. UNRESOLVED STAFF COMMENTS
None
We lease an office and manufacturing facility in Nesher, Israel and maintain an office in Elmsford, New York.Tyler, Texas. Our lease for the facility in Nesher expiredexpires on June 30, 2017 and we have continued to rent the facility on a month by month basis with the expectation to enter into a new lease in the second quarter of 2018.2022. The space is approximately 160 square meters. We pay approximately $2,500$3,600 per month under our lease. We are still in discussions with the landlord to extend the lease. We also use a small officefacility in Elmsford, New York. The use of this space is included in a services agreement pursuant toTyler, Texas from an unrelated party, for which we pay $7,000 perrent of $1,200 a month for, among other services, processing products for shipping, customer service, payment processing and maintenance of certain records.although we do not have a lease. This space is approximately 200 square meters. We believe that our facilities are adequate to meet our current and proposed needs.
From time to time, we may be involved in certain claims and litigation that arises througharising out of the normalordinary course and conduct of business. Management assesses such claims and, if it considers that it is probable that an asset had been impaired or a liability had been incurred and the amount of loss can be reasonably estimated, provisions for loss are made based on management’s assessment of the most likely outcome.
See “Item 8. Financial Statements and Supplementary Data – Note 12. Commitments and Contingencies,” which information is incorporated herein by reference, for a description of pending and recent litigation.
On February 26, 2021, Protrade Systems, Inc. (“Protrade”) filed a Request for Arbitration (the “Request”) with the International Court of Arbitration (the “ICA”) of the International Chamber of Commerce alleging the Company is in breach of an Exclusive Distribution Agreement dated March 7, 2019 (the “Agreement”) between Protrade and the Company. Protrade alleges, in part, that the Company has breached the Agreement by discontinuing the manufacture of the DV0057 Painshield MD device in favor of an updated 10-100-001 Painshield MD device. Protrade claims damages estimated at $3 million. On March 15, 2022, the arbitrator issued a final award, which, although finding that Protrade’s claims failed as a matter of law or were unsupported by the evidence, nevertheless awarded Protrade $1,500,250, which consists of $1,432,000 for “lost profits” and $68,250 as reimbursement of arbitration costs, on the grounds that the Company allegedly failed to supply Protrade with “requested patches.” The Company continues to dispute the claims asserted by Protrade and intends to pursue the available options to vacate or seek correction of the award to Protrade. As of December 31, 2021, the dateCompany accrued the amount of this filing, we are not a partythe award to any material litigation nor are we awareProtrade amounting to $1,500,250 as part of any such threatened or pending litigation.“General and administrative expenses” and “Other accounts payable and accrued expenses”.
ThereExcept as referenced above, there are no other material proceedings in which any of our directors, officers or affiliates or any registered or beneficial shareholder of more than 5% of our common stock, or any associate of any of the foregoing is an adverse party or has a material interest adverse to our interest.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
| 50 |
Not applicable.
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Our common stock has been quoted on the NASDAQ Capital Market under the symbol “NAOV” since November 8, 2017. Prior to that date, our common stock had been quoted on the OTCQB over-the-counter marketplace under the symbol “NAOV” since April 10, 2015. Prior to April 10, 2015, there was no established public trading market for our common stock.
The following table sets forth (i) the intra-day high and low sales price per share for our common stock, as reported on the NASDAQ Capital Market, for the period of November 8, 2017 to December 31, 2017, and (ii) the high and low bid prices of our common stock as reported on the OTCQB, for the period January 1, 2016, to November 7, 2018. The quotations reflect inter-dealer prices, without retail mark-up, mark-down or commission, and may not represent actual transactions.
| Common Stock | ||||||||
| High | Low | |||||||
| Fiscal Year Ending December 31, 2017 | ||||||||
| Fourth quarter | $ | 7.32 | $ | 3.68 | ||||
| Third quarter | $ | 6.75 | $ | 5.50 | ||||
| Second quarter | $ | 6.25 | $ | 5.80 | ||||
| First quarter | $ | 6.20 | $ | 5.95 | ||||
| Fiscal Year Ended December 31, 2016 | ||||||||
| Fourth quarter | $ | 7.00 | $ | 5.50 | ||||
| Third quarter | $ | 5.56 | $ | 5.20 | ||||
| Second quarter | $ | 5.50 | $ | 4.75 | ||||
| First quarter | $ | 4.95 | $ | 4.15 | ||||
The last reported sale price for our common stock on the NASDAQ as of March 29, 2018 was $4.76 per share. As of March 29, 2018,April 14, 2022, we had 3,935,86527,997,793 issued and outstanding shares of common stock. The common stock which werewas held by 129120 holders of record. The actual number of holders of our common stock is greater than the number of record holders, and includes stockholders who are beneficial owners, but whose shares are held in street names by brokers or other nominees.
On March 3, 2021, we filed a proxy statement in connection with a special meeting of stockholders that was held on March 31, 2021, and ultimately adjourned until May 6, 2021, to (i) ratify the increase in the number of authorized shares of common stock from 20,000,000 to 24,109,635 and the issuance of such 4,109,635 shares of common stock, and (ii) further increase the number of our authorized shares of common stock. On May 6, 2021, the Company’s stockholders voted to approve the ratification of the increase in the number of authorized shares of common stock from 20,000,000 to 24,109,635 and the issuance of such 4,109,635 shares of common stock to be effective as of December 4, 2020, but the stockholders did not approve a further increase in the number of its authorized shares of common stock.
On August 17, 2021, the Company’s stockholders voted to approve an amendment to our Amended and Restated Certificate of Incorporation to increase the number of shares of our common stock authorized for issuance from 24,109,635 shares to 40,000,000 shares.
As of March 29, 2018,April 14, 2022, we had a total of 2,483,142no shares of our Series C Preferred Stock issued and outstanding. Each share of our Series C Preferred Stock is convertible into one share of our common stock (subject to adjustment as provided in the related designation of preferences) at any time at the option of the holder, provided that the holder would be prohibited from converting Series C Preferred Stock into shares of our common stock if, as a result of such conversion, the holder, together with its affiliates, would beneficially own more than 9.99% of the total number of shares of our common stock then issued and outstanding. This limitation may be waived upon not less than 61 days’ prior written notice to us.
As of March 29, 2018,April 14, 2022, we had a total of 304no shares of our Series D Preferred Stock outstanding. Each share of our Series D Preferred Stock is convertible into one thousand shares of our common stock (subject to adjustment as provided in the related designation of preferences) at any time at the option of the holder, provided that the holder would be prohibited from converting Series D Preferred Stock into shares of our common stock if, as a result of such conversion, the holder, together with its affiliates, would own more than 9.99% of the total number of shares of our common stock then issued and outstanding. This limitation may be waived upon not less than 61 days’ prior written notice to us.
Dividend Policy
In the past,As of April 14, 2022, we have not declared or paid cash dividends onhad a total of no shares of our Series E Preferred Stock issued and outstanding. Each share of our Series E Preferred Stock is convertible into one share of our common stock (subject to adjustment as provided in the related designation of preferences) at any time at the option of the holder, provided that the holder would be prohibited from converting Series CE Preferred Stock or Series D Preferred Stock, and we do not intend to pay any cash dividends oninto shares of our common stock or preferred stock. Rather, we intend to retain future earnings (if any) to fundif, as a result of such conversion, the operation and expansionholder, together with its affiliates, would beneficially own more than 9.99% of the total number of shares of our businesscommon stock then issued and for general corporate purposes. Subjectoutstanding. This limitation may be waived upon not less than 61 days’ prior written notice to legal and contractual limits, our board of directors will make any decision as to whether to pay dividends in the future.
us.
Recent Sales of Unregistered Securities
NoneAll sales of unregistered securities during the year ended December 31, 2021 were previously disclosed in a Quarterly Report on Form 10-Q or a Current Report on Form 8-K.
Issuer Purchases of Equity Securities
We did not purchase any of our registered equity securities during the period covered by this Annual Report.
ITEM 6. SELECTED FINANCIAL DATARESERVED
| 51 |
Not applicable.
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
Management’s Discussion and Analysis of Financial Condition and Results of Operations is intended to provide a reader of our financial statements with a narrative from the perspective of our management on our financial condition, results of operations, liquidity, and certain other factors that may affect our future results. You should read the following discussion and analysis of financial condition and results of operations in conjunction with our consolidated financial statements and the related notes thereto included elsewhere in this Annual Report on Form 10-K. In addition to historical information, the following discussion and analysis includes forward-looking information that involves risks, uncertainties and assumptions. Our actual results and the timing of events could differ materially from those anticipated by these forward-looking statements as a result of many factors, including those discussed under “Item 1A. Risk Factors” and elsewhere in this Form 10 -K. See “Cautionary Note Regarding Forward-Looking Statements” included elsewhere in this Form 10 -K.
Overview
We are a medical device company focusing on noninvasive biological response-activating devices that target wound healing and pain therapy and can be administered at home, without the assistance of medical professionals. Our WoundShield, PainShield and UroShield products are backed by novel technology which relates to ultrasound delivery through surface acoustic waves.
Recent Events
Underwritten Public OfferingCOVID-19
On November 6, 2017,The ongoing COVID-19 pandemic has and may continue to adversely impact our business, as our operations are based in and rely on third parties located in countries affected by the pandemic. Our third-party manufacturer, which is based in China, temporarily shut down for sixty days during 2020 due to the pandemic and became fully operational in April 2020 which led to a significant delay in the production of goods needed to fulfill our sales orders which were scheduled to be fulfilled in our first quarter of 2020. We were able to fulfill these orders in the second quarter of 2020. Additionally, the notified regulatory body we closedrely on to obtain European CE approval is located in Italy and was shut down for approximately six weeks from March to April 2020, which delayed our submission for CE mark approval for the year 2020. The CE Mark approval was subsequently approved in April 2020. The various precautionary measures taken by many governmental authorities around the world in order to limit the spread of COVID-19 have had and may continue to have an underwritten public offeringadverse effect on the global markets and global economy, including on the availability and pricing of 1,224,488 sharesemployees, resources, materials, manufacturing and delivery efforts and other aspects of the global economy. During the first six months of 2020, the financial downturn compelled us to furlough or reduce working hours for much of our common stock (and Series D Convertible Preferred Stockoperating staff, and forced our remaining staff as well as third-party contractors, to work remotely. In addition, many staff members continue to operate remotely from their homes which is continuing to result in lieudelays in obtaining certain financial records. We also rely on third-party professionals to provide services such as the preparation of common stock ifour financial statements and to conduct audits, and many of these parties have been affected by government-imposed precautionary measures, thereby delaying our receipt of these services. Such government-imposed precautionary measures may have been relaxed in certain countries or states, but there is no assurance that more strict measures will be put in place again due to a resurgence in COVID-19 cases. Although there were no material disruptions during 2021, the purchase of common stockCOVID-19 pandemic again disrupt production and cause delays in the offering would otherwise result in a purchaser, together with its affiliatesdevelopment, supply and certain related parties, beneficially owning more than 4.99%delivery of our outstanding common stock immediately followingproducts, our operation, further divert the consummationattention and efforts of the offering), togethermedical community coping with COVID-19 and disrupt the marketplace in which we operate. The extent to which COVID-19 impacts our results will depend on future developments, which are highly uncertain and cannot be predicted, including new information which may emerge concerning the severity of COVID-19, its variants and the actions to contain COVID-19 or treat its impact, among others. The COVID-19 pandemic could continue to materially disrupt our business and operations, hamper our ability to raise additional funds or sell or securities, continue to slow down the overall economy, curtail consumer spending, interrupt our sources of supply, and make it hard to adequately staff our operations.
On February 26, 2021, Protrade Systems, Inc. (“Protrade”) filed a Request for Arbitration (the “Request”) with the International Court of Arbitration (the “ICA”) of the International Chamber of Commerce alleging the Company is in breach of an Exclusive Distribution Agreement dated March 7, 2019 (the “Agreement”) between Protrade and the Company. Protrade alleges, in part, that the Company has breached the Agreement by discontinuing the manufacture of the DV0057 Painshield MD device in favor of an updated 10-100-001 Painshield MD device. Protrade claims damages estimated at $3 million.
On March 15, 2022, the arbitrator issued a final award, which, although finding that Protrade’s claims failed as a matter of law or were unsupported by the evidence, nevertheless awarded Protrade $1,432,000 for “lost profits” on the grounds that the Company allegedly failed to supply Protrade with “requested patches.” We continue to dispute the claims asserted by Protrade and intend to pursue the available options to vacate or seek correction of the award to Protrade. As of December 31, 2021, the Company accrued the amount of the award to Protrade amounting to $1,500,250 as part of “General and administrative expenses” and “Other accounts payable and accrued expenses”.
Business Developments
Effective as of January 2020, the U.S. CMS approved our PainShield™ for reimbursement for Medicare beneficiaries on a national basis. We were notified on March 30, 2020 that our Medicare Enrollment Application was approved, and we are now an approved Medicare Supplier for DME through the National Supplier Clearinghouse, Palmetto-GBA as well as Noridian Administrative Services, LLC, the two Medicare Administrative Contractors that handle DME reimbursement nationwide. PainShield is currently available for Medicare reimbursement on a national level under new HCPCS (Healthcare Common Procedure Coding System) code K1004.
| 52 |
In March 2020, we signed a license agreement with Sanuwave Health, Inc. for the manufacture and delivery of our WoundShield technology. Under the terms of the agreement, we will receive warrants to purchase up to 918,366127,000 shares of Sanuwave stock, a $250,000 milestone payment based on receipt of U.S. Food and Drug Administration approval, and 10% royalty on Sanuwave’s gross revenues from sales or rentals of WoundShield. In return, Sanuwave has received the worldwide, exclusive rights to our WoundShield product and technology. In addition, Sanuwave will bear the costs and clinical validation responsibilities associated with obtaining approval for WoundShield from the U.S. Food and Drug Administration and other regulatory agencies around the world.
In September 2020, the FDA exercised its Enforcement Discretion to allow distribution of our UroShield device in the United States. This temporary authorization is limited to use as an extracorporeal acoustic wave generating accessory to urological indwelling catheter for use during the COVID-19 pandemic.
NASDAQ Delisting Procedure
On November 5, 2020, the Company received a subsequent letter from the Nasdaq Stock Market (“Nasdaq) indicating that, based upon the closing bid price of the Company’s common stock at an offeringfor the 30 consecutive business day period between September 24, 2020, through November 4, 2020, the Company did not meet the minimum bid price of $4.90$1.00 per share required for continued listing on the Nasdaq Capital Market pursuant to Nasdaq Listing Rule 5550(a)(2).
| 53 |
On January 4, 2021, the Company received formal notice that the Company has regained compliance with the equity requirement of Nasdaq Listing Rule 5550(b)(1), as required by the Panel decision dated October 6, 2020. Accordingly, the Panel determined to continue the listing of the Company’s securities on The Nasdaq Stock Market.
On February 2, 2021, the Company received formal notice that Nasdaq had determined that for the period from January 19 to February 1, 2021, the closing bid price of the Company’s common stock and accompanying warrant to purchase 0.75had been at $1.00 per share or greater. Accordingly, the Company regained compliance with Listing Rule 5550(a)(2).
On March 2, 2022, NanoVibronix, Inc. received a letter from Nasdaq indicating that, based upon the closing bid price of one share of common stock. We sold 327 shares of Series D Convertible Preferred Stock in lieu of 327,000 shares ofthe Company’s common stock for the 30 consecutive business day period between January 14, 2022, through March 1, 2022, the Company did not meet the minimum bid price of $1.00 per share required for continued listing on The Nasdaq Capital Market pursuant to Nasdaq Listing Rule 5550(a)(2). The letter also indicated that the Company will be provided with a compliance period of 180 calendar days, or until August 29, 2022 (the “Compliance Period”), in which to regain compliance pursuant to Nasdaq Listing Rule 5810(c)(3)(A).
In order to regain compliance with Nasdaq’s minimum bid price requirement, the offering. Additionally in connectionCompany’s common stock must maintain a minimum closing bid price of $1.00 for at least ten consecutive business days during the Compliance Period. In the event the Company does not regain compliance by the end of the Compliance Period, the Company may be eligible for additional time to regain compliance. To qualify, the Company will be required to meet the continued listing requirement for the market value of its publicly held shares and all other initial listing standards for The Nasdaq Capital Market, with this offering, we issuedthe exception of the bid price requirement, and will need to provide written notice of its intention to cure the deficiency during the second compliance period, by effecting a reverse stock split if necessary. If the Company meets these requirements, the Company may be granted an additional 180 calendar days to regain compliance. However, if it appears to Nasdaq that the Company will be unable to cure the deficiency, or if the Company is not otherwise eligible for the additional cure period, Nasdaq will provide notice that the Company’s common stock will be subject to delisting. The letter has no immediate impact on the listing of the Company’s common stock, which will continue to be listed and traded on The Nasdaq Capital Market, subject to the underwriters a unit purchase option to purchase units at an exercise price equal to $6.125 pursuant to which an aggregate of 61,224 shares and warrants to purchase 45,918 shares are issuable to the underwriters. Total gross proceeds from the offering totaled approximately $6,000,000, and net proceeds totaled approximately $5,056,000 after deducting underwriting and estimated offering expenses. Each warrant has an exercise price of $6.95 per full share of common stock with a life of five years. We intend to use the net proceeds from this offering: (i) to cover expenses related to listing our shares on The NASDAQ Capital Market; (ii) to expand our sales leadership and field level sales resources; (iii) for research and development; (iv) to implement our Surface Acoustic Wave platform to other applications; (v) to pursue complimentary acquisitions; and (vi) for general working capital. The securities were issued pursuant to our registration statement on Form S-1 originally filedCompany’s compliance with the Securities and Exchange Commission on June 21, 2017, and declared effective on November 1, 2017.other listing requirements of The Nasdaq Capital Market
| 54 |
Conversion of Convertible Promissory Notes
In September 2017, all of the holders of the convertible promissory notes issued in connection with a series of bridge financings between March 1, 2017 and September 30, 2017 (collectively, the “2017 Notes”) agreed to convert the full principal and accrued interest on the 2017 Notes into our equity securities in the event we consummated an equity financing pursuant to which we issue and sell shares of capital stock resulting in aggregate proceeds of at least $2,000,000 (a “Qualified Financing”) any time before December 31, 2017. The offering that closed on November 6, 2017, constituted a Qualified Financing, and based on the outstanding principal amount and all accrued but unpaid interest on the 2017 Notes at 80% of the offering price of $4.90 per share of common stock and accompanying warrant, we issued an aggregate of 361,462 shares of common stock (and common stock equivalents) and warrants to purchase an aggregate of 271,096 shares of common stock to the holders of the 2017 Notes, all of which are subject to lock-up agreements for 180 days from November 1, 2017.
Critical Accounting Policies and Significant Estimates
UseThis management’s discussion and analysis of estimates
financial condition and results of operations is based on our financial statements, which have been prepared in accordance with U.S. GAAP. The preparation of the consolidatedthese financial statements in conformity with U.S. GAAP requires managementus to make estimates judgments and assumptions. We believe that the estimates, judgments and assumptions used are reasonable based upon information available at the time they are made. These estimates, judgments and assumptions canthat affect the reported amounts of assets and liabilities, and disclosure of contingent assets and liabilities at the datesdate of the financial statements, and the reported amounts of revenue and expenses during the reportingreported period. In accordance with U.S. GAAP, we base our estimates on historical experience and on various other assumptions we believe to be reasonable under the circumstances. Actual results couldmay differ from those estimates.these estimates if conditions differ from our assumptions. While our significant accounting policies are more fully described in Note 3 in the “Notes to Financial Statements”, we believe the following accounting policies are critical to the process of making significant estimates in preparation of our financial statements.
Inventory
Inventories are stated at the lower of cost or net realizable value. Net realizable value is the estimated selling prices in the ordinary course of business, less reasonably predictable costs of completion, disposal, and transportation. Cost is determined using the “first-in, first-out” method.
Inventory write-offs are provided to cover risks arising from slow-moving items or technological obsolescence. The Company periodically evaluates the quantities on hand relative to current and historical selling prices and historical and projected sales volume. Based on this evaluation, provisions are made when required to write-down inventory to its net market value. As of December 31, 2021 and 2020, there was no allowance on inventory.
Impairment of Long-Lived Assets
Management reviews for impairment whenever events or changes in circumstances indicate that the carrying amount of property and equipment may not be recoverable under the provisions of accounting for the impairment of long-lived assets. If it is determined that an impairment loss has occurred based upon expected future cash flows, the loss is recognized in the Consolidated Statements of Operations.
Sequencing
The Company adopted a sequencing policy under ASC 815-40-35 whereby if reclassification of contracts from equity to liabilities is necessary pursuant to ASC 815 due to the Company’s inability to demonstrate it has sufficient authorized shares. This was due to the Company committing more shares than authorized. While temporary suspensions are in place to keep the potential exercises beneath the number authorized, certain instruments are classified as liabilities, after allocating available authorized shares on the basis of the most recent grant date of potentially dilutive instruments. Pursuant to ASC 815, issuances of securities granted as compensation in a share-based payment arrangement are not subject to the sequencing policy.
Derivative Liability
The Company’s derivative financial instruments are measured at fair value using the Black Scholes Model which takes into account, as of the valuation date, factors including the current exercise price, the expected life of the warrant, the current price of the underlying stock and its expected volatility, expected dividends on the stock and the risk-free interest rate for the term of the instrument. The Black Scholes Model is subject to uncertainty since the underlying calculation relies on historical data to project future stock prices, i.e. expected volatility, in order to estimate fair value. Because the expected volatility is based on historical stock prices, the volatility assumed for each period may vary. The liability is revalued at each reporting period and changes in fair value are recognized in the consolidated statements of operations and comprehensive loss under the caption “Change in fair value of derivative liabilities.” As of December 31, 2021 and 2020, there were $0 and $2,471 derivative liabilities on the consolidated balance sheet, respectively (see Note 8).
Revenue recognition
We generateIt is the Company’s policy that revenues from the sale of our products to distributors and patients. Revenues from those products areproduct sales is recognized in accordance with ASC 605,606 “Revenue Recognition,Recognition.” Five basic steps must be followed before revenue can be recognized; (1) Identifying the contract(s) with a customer that creates enforceable rights and obligations; (2) Identifying the performance obligations in the contract, such as promising to transfer goods or services to a customer; (3) Determining the transaction price, meaning the amount of consideration in a contract to which an entity expects to be entitled in exchange for transferring promised goods or services to a customer; (4) Allocating the transaction price to the performance obligations in the contract, which requires the company to allocate the transaction price to each performance obligation on the basis of the relative standalone selling prices of each distinct good or services promised in the contract; and (5) Recognizing revenue when delivery(or as) the entity satisfies a performance obligation by transferring a promised good or service to a customer. The amount of revenue recognized is the amount allocated to the satisfied performance obligation. Adoption of ASC 606 has occurred, persuasivenot changed the timing and nature of the Company’s revenue recognition and there has been no material effect on the Company’s financial statements.
Revenue from product sales is recorded at the net sales price, or “transaction price,” which includes estimates of variable consideration that result from coupons, discounts, chargebacks and distributor fees, processing fees, as well as allowances for returns and government rebates. The Company constrains revenue by giving consideration to factors that could otherwise lead to a probable reversal of revenue. Collectability of revenue is reasonably assured based on historical evidence of an agreement exists,collectability between the fee is fixed or determinable, no further obligation existsCompany and collectability is probable.its customers.
Revenues from sales to distributors are recognized at the time the products are delivered to the distributors (“sell-in”). The Company does not grant rights of return, credits, rebates, price protection, or other privileges on its products to distributors.
| 55 |
Revenues from sales to distributors are recognized at the time the products are delivered to the distributors (“sell-in”). The Company does not grant rights of return, credits, rebates, price protection, or other privileges on its products to distributors.
Stock-based compensation
We accountrely on the Black-Scholes option pricing model for stock-based compensation in accordance with ASC 718, “Compensation - Stock Compensation”, (“ASC 718”), which requires companies to estimateestimating the fair value of equity-based paymentstock-based awards on the date of grant using an option-pricing model. The value of the portion of the award that is ultimatelygranted, and expected to vest is recognized as an expense over the requisite service periods on a straight line method in our consolidated statement of comprehensive loss.
We have early adopted Accounting Standard Update (“ASU”) 2016-09, “Compensation - Stock Compensation”, in the 2016 consolidated financial statements and account for forfeitures as they occur.
We selected the Black-Scholes-Merton option pricing model as the most appropriate fair value method for our stock-options awards. The option-pricing model requires a number of assumptions, of which the most significant are the expected stock price volatility and the expected option term. Expected volatility was calculated based upon similar traded companies’ historical share price movements. The expected option term represents the period that our stock options are expected to be outstanding. We currently use the simplified method, in accordance with ASC No.718-10-S99-1 (SAB No. 110), and will continue to do so until sufficient historical exercise data supports using expected life assumptions. The risk-free interest rate is based on the yieldhistorical volatilities of peer company’s common stock. Stock options generally vest over one or two years from U.S. Treasury zero-coupon bonds with an equivalent term. The expected dividend yield assumption is based on our historical experiencethe grant date and expectation of no future dividend payouts. Wegenerally have historically not paid cash dividends and have no foreseeable plans to pay cash dividendsten-year contractual terms. Information about the assumptions used in the future.calculation of stock-based compensation expense is set forth in Notes 3 and 6 in the “Notes to Financial Statements”.
We apply ASC 505-50, “Equity-Based Payments to Non-Employees” (“ASC 505”) with respect to options and warrants issued to non-employees which requires the use of option valuation models to measure the fair value of the options and warrants at the measurement date.
Income taxes
On March 27, 2020, the Coronavirus Aid, Relief, and Economic Security Act (“CARES Act”) was enacted in response to the COVID-19 pandemic. The CARES Act, among other things, permits NOL carryovers and carrybacks to offset 100% of taxable income for taxable years beginning before 2021. In addition, the CARES Act allows NOLs incurred in 2018, 2019, and 2020 to be carried back to each of the five preceding taxable years to generate a refund of previously paid income taxes. The Company has been consistently in a loss position in the U.S. and at present does not expect that the NOL carryback provision of the CARES Act would result in a material cash benefit to the Company.
We account for income taxes in accordance with ASC 740, “Income Taxes”. This topic prescribes the use of the liability method whereby deferred tax assets and liability account balances are determined based on differences between financial reporting and tax bases of assets and liabilities and are measured using the enacted tax rates and laws that will be in effect when the differences are expected to reverse. We provide full valuation allowance, to reduce deferred tax assets to the amount that is more likely than not to be realized.
In November 2015, the FASB issued ASU 2015-17, Balance Sheet Classification of Deferred Taxes, related to balance sheet classification of deferred taxes. The ASU requires that deferred tax assets and liabilities be classified as noncurrent in the statement of financial position, thereby simplifying the current guidance that requires an entity to separate deferred assets and liabilities into current and noncurrent amounts. For public entities, this standard is effective for annual reporting periods beginning after December 15, 2016, including interim periods within that reporting period. For all other entities, this standard is effective for annual reporting periods beginning after December 15, 2017, and interim periods within annual periods beginning after December 15, 2018. We early adopted ASU 2015-17 as of December 31, 2017, which had no impact on our consolidated financial statements.
We implementsimplemented a two-step approach to recognize and measure uncertain tax positions. The first step is to evaluate the tax position taken or expected to be taken in a tax return by determining if the weight of available evidence indicates that it is more likely than not that, on an evaluation of the technical merits, the tax position will be sustained on audit, including resolution of any related appeals or litigation processes. The second step is to measure the tax benefit as the largest amount that is more than 50% (cumulative basis) likely to be realized upon ultimate settlement.
Warrants
We account for stock warrants held by investors as either equity instruments or liabilities in accordance with ASC 480, Distinguishing Liabilities from Equity (“ASC 480”), dependingrecognize interest and penalties related to uncertain tax positions on the specific terms of the warrant agreement.
Stock warrants are accounted for as a liability if they contain “down-round protection” or other terms that could potentially require “net cash settlement” in accordance with the provisions of ASC 815-40, Derivatives and Hedging - Contracts in Entity’s Own Equity (“ASC 815”), which provides a two-step model to be applied in determining whether a financial instrument or an embedded feature is indexed to an issuer’s own stock and thus able to qualify to be a derivative financial instrument. We measure such warrants at fair value by applying the Black-Scholes option pricing model in each reporting period until they are exercised or expired, with changesincome tax expense line in the fair value being recognizedaccompanying consolidated statement of operations. Accrued interest and penalties are included on the related tax liability line in the our statement of comprehensive loss as financial income or expense, as appropriate.consolidated balance sheet.
| 56 |
Debt Issued with Warrants
We consider guidance within ASC 470-20, Debt (ASC 470), ASC 480, and ASC 815 when accounting for the issuance of convertible debt with detachable warrants. As described above under the caption “Warrants”, we classify stock warrants as either equity instruments or liabilities depending on the specific terms of the warrant agreement. In circumstances in which debt is issued with liability-classified warrants, the proceeds from the issuance of convertible debt are first allocated to the warrants at their full estimated fair value and established as both a liability and a debt discount. The remaining proceeds, as further reduced by discounts created by the bifurcation of embedded derivatives and a beneficial conversion feature, is allocated to the debt. We account for debt as liabilities measured at amortized cost and amortize the resulting debt discount from the allocation of proceeds, to interest expense using the effective interest method over the expected term of the debt instrument pursuant to ASC 835, Interest (ASC 835).
Recently issued accounting standards
For a summary of recent accounting pronouncements applicable to our consolidated financial statements see Note 2, “Significant3, “Summary of Significant Accounting Policies” to the Consolidated Financial Statements included in Part IV, Item 15 of this Annual Report on Form 10-K.
Results of Operations
Year Ended December 31, 2021 Compared to Year Ended December 31, 2020
Revenues. For the years ended December 31, 2021 and 2020, our revenues were approximately $1,695,000 and $623,000, respectively, an increase of approximately 172%, or $1,072,000, between the periods. The increase was mainly attributable to increased sales from our Ultra Pain Products distributor as well as adding a new distributor who sells our products to Veteran Administration facilities. Our revenues may fluctuate as we add new customers or when existing distributors make large purchases of our products during one period and no purchases during another period. Our revenues by quarter may not be linear or consistent. We do not anticipate that our revenues will be impacted by inflation or changing prices in the foreseeable future.
For the years ended December 31, 2021, the percentage of revenues attributable to our products was: PainShield – 99% and UroShield – 1%. For the year ended December 31, 2020, the percentage of revenues attributable to our products was: PainShield – 92% WoundShield – 8%; and UroShield – 0. For both the years ended December 31, 2021 and 2020, the portion of our revenues that was derived from distributors was 93%.
Gross Profit. For the years ended December 31, 2021 and 2020, gross profit was approximately $770,000 and $214,000, respectively. The increase was due to the large increase in revenues in 2021, enhanced by decreased costs of manufacturing in 2021, as we incurred several one-time costs in the transition of our manufacturing of products to a third-party manufacturer in 2020.
| 57 |
Extended Transition Period
Gross profit as a percentage of revenues were approximately 45% and 34% for “Emerging Growth Companies”the years ended December 31, 2021 and 2020, respectively. The increase in gross profit as a percentage is mainly due to the reasons described above.
Research and Development Expenses. For the years ended December 31, 2021 and 2020, research and development expenses were approximately $293,000 and $171,000, respectively, an increase of approximately 71%, or $122,000 between the periods. This increase was mainly due to studies performed during 2021 and development of an over-the-counter PainShield product and a CBD application for our PainShield product.
Research and development expenses as a percentage of total revenues were approximately 17% and 27% for the years ended December 31, 2021 and 2020, respectively.
The significant difference during the year, as compared to last year’s percentage, was due to the variation of the number of clinical trials that the Company does per year. In particular, the Company only did one (1) clinical trial in 2021, which was considerably less than the previous years.
We have electedOur research and development expenses consist mainly of payroll expenses to useemployees involved in research and development activities, expenses related to subcontracting, patents, clinical trial and facilities expenses associated with and allocated to research and development activities.
Selling and Marketing Expenses. For the extended transition periodyears ended December 31, 2021 and 2020, selling and marketing expenses were approximately $1,101,000 and $993,000, respectively, an increase of approximately 11%, or $108,000, between the periods. The increase in selling and marketing expenses was mainly due to normalization of operations in 2021 after COVID-19 restrictions in 2020 which caused reduced payroll and curtailed marketing activities, such as less travel and trade show expenses.
Selling and marketing expenses as a percentage of total revenues were approximately 65% and 159% for complyingthe years ended December 31, 2021 and 2020, respectively. The decrease in our percentage was due to the increase in revenues.
Selling and marketing expenses consist mainly of payroll expenses to direct sales and marketing employees, stock-based compensation expenses, travel expenses, advertising and marketing expenses, rent and facilities expenses associated with newand allocated to selling and marketing activities.
General and Administrative Expenses. For the years ended December 31, 2021 and 2020, general and administrative expenses were approximately $5,059,000 and $3,769,000, respectively, an increase of approximately 34%, or revised accounting standards under Section 102(b)(1)$1,290,000, between the periods. The increase was mainly due to the recognition of a $1,500,000 arbitration settlement expense from final award of arbitration issued in favor of the Jumpstart Our Business ActCompany’s former distributor to cover for “lost profits” and reimbursement of 2012 (known asarbitration costs.
Interest expense. For the JOBS Act)years ended December 31, 2021 and 2020, were $0 and $147,000, respectively. All notes were paid off in the fourth quarter of 2020. There were no new notes in 2021.
Change in fair value of derivative liabilities. This election allows usFor the years ended December 31, 2021 and 2020, there was a change in fair value of derivative liabilities resulting in a loss of approximately $6,956,000 and gain of approximately $513,000, respectively. The change in fair value fluctuates based on the Company’s stock price.
Warrant modification expense. For the years ended December 31, 2021 and 2020, warrant modification expense was approximately $1,627,000 and $0, respectively. The warrant modification expense was due to delay the adoptionresolution of new or revised accounting standards that have different effective datesthe overissuance shares matter. The overissuance shares matter resulted in a reclassification of derivative liabilities to equity during 2021. There was no warrant modification in 2020.
| 58 |
Income tax expense. For the years ended December 31, 2021 and 2020, our income tax expense was approximately $35,000 and $15,000, respectively. The low tax expense for public and private companies until those standards apply to private companies. As2020 was a result of this election, our consolidated financial statements may not be comparablefavorable adjustments due to companies that comply with public company effective dates. Because our consolidated financial statements may not be comparablelapses of statutes of limitations on its Israel tax positions. In 2021, there was no such adjustment and large increase in revenues.
Net Loss. Our net loss increased by approximately $9,957,000, or 230%, to companies that comply with public company effective dates, investors mayapproximately $14,282,000 for the years ended December 31, 2021 from approximately $4,325,000 during the same period in 2020. The increase in net loss resulted primarily from the factors described above.
Liquidity and Capital Resources
We have difficulty evaluating or comparing our business, performance or prospects in comparison to other public companies, which may have a negative impact on the value and liquidity of our common stock.
Going Concern
The financial statements have been prepared assuming that we will continue as a going concern. Since our formation, we utilized funds generated from public offerings, private placement offerings and debt to fund our product development. We incurred losses in the amount of $4,965approximately $14,282,000 during the year ended December 31, 2017,2021, which primarily consisted of increased expenses from research studies and have an accumulated deficitproduct development, increase in payroll costs to normal levels since COVID furloughs in 2020, losses due to changes in fair value of $28,382derivative liabilities and warrant modifications. We also had negative cash flow from operating activities of $4,367,000 for the year ended December 31, 2021. Although we expect to continue to incur losses and negative cash flows from operating activities, we had a cash balance of just over $7,737,000 as of December 31, 2017.2021, and therefore, we expect to have sufficient resources to fund our operation for the next twelve months from the date of this filing. The Company may need to continue to raise additional capital to finance its losses and negative cash flows from operations beyond the next years and may continue to be dependent on additional capital raising as long as our products do not reach commercial profitability. If we are unable to obtain stockholder ratification of certain prior issuances of our common stock and approval of an increase in the number of authorized shares of our common stock, we will be unable to issue common stock or convertible instruments. As a result, the Company will be limited in its ability to raise additional capital.
During the year ended December 31, 2021, we met our short-term liquidity requirements from our existing cash reserves and proceeds from warrant exercises. Our future capital requirements and the adequacy of our available funds will depend on many factors, including our ability to successfully clear regulatory requirements and commercialize our products, our development of future products and competing technological and market developments.developments as well as our ability to overcome obstacles that may be presented due to developments caused by the coronavirus outbreak. We expect to continue to incur losses and negative flows from operations. We intend to use the proceeds generated from equity or debt financings, or strategic alliances with third parties, either alone or in combination with equity financing to meet our short-term liquidity requirements as well as to advance our long-term plans. While we believe we have sufficient capital to execute our business plan over the next twelve months, there are no assurances that we will not need to raise additional capital at a later time, or that we would be able to raise additional capital, if required, on terms favorable to us.
Results of Operations
Twelve Months Ended December 31, 2017 Compared to Twelve Months Ended December 31, 2016
Revenues. For the twelve months ended December 31, 2017 and 2016, our revenues were approximately $239,000 and $229,000, respectively, an increase of approximately 4.3%, or $10,000, between the periods. The increase was mainly attributable to increased sales from adding distributors. Our revenues may fluctuate as we add new customers or when existing distributors make large purchases of our products during one period and no purchases during another period. Our revenues may fluctuate from period to period and, as we continue to grow our business, growth in revenues by quarter may not be linear or consistent. We do not anticipate that our revenues will be impacted by inflation or changing prices in the forseeable future.
For the twelve months ended December 31, 2017, the percentage of revenues attributable to our products was: PainShield – 75.7% and UroShield – 24.3%. For the twelve months ended December 31, 2016, the percentage of revenues attributable to our products was: PainShield – 90.8% and UroShield – 9.2%. For the twelve months ended December 31, 2017 and 2016, the percentage of revenues attributable to our disposable products was 47.4% and 42.5%, respectively. For the twelve months ended December 31, 2017 and 2016, the portion of our revenues that was derived from distributors was 47% and 30.3%, respectively.
Gross Profit. For the twelve months ended December 31, 2017, gross profit increased by approximately 7.1%, or $10,000, to approximately $151,000 from approximately $141,000 during the same period in 2016.
Gross profit as a percentage of revenues were approximately 63.2% and 61.6% for the twelve months ended December 31, 2017 and 2016, respectively. The slight increase in gross profit as a percentage is mainly due to the increased percentage of our disposable product sales which typically have higher margins.
Our gross profit may be affected year-over-year by the mix of revenues between sales to distributers and sales directly to the end customers (where sales directly to the end customers generally have a higher margin). As a result, we are subject to year-over-year fluctuation in our gross profits.
Research and Development Expenses. For the twelve months ended December 31, 2017 and 2016, research and development expenses were $693,000 and $584,000, respectively, an increase of approximately 18.7%, or $109,000, between the periods. This increase was mainly due to an increased volume of clinical trial costs that took place in 2017 as well as increased development of new products.
Research and development expenses as a percentage of total revenues were approximately 290.0% and 255.0% for the twelve months ended December 31, 2017 and 2016, respectively.
Our research and development expenses consist mainly of payroll expenses to employees involved in research and development activities, stock based compensation expenses, expenses related to subcontracting, patents, clinical trial and facilities expenses associated with and allocated to research and development activities.
Selling and Marketing Expenses. For the twelve months ended December 31, 2017 and 2016, selling and marketing expenses were approximately $465,000 and $514,000, respectively, a decrease of approximately 9.5%, or $49,000, between the periods.
The decrease in selling and marketing expenses was mainly due to decreased selling and marketing activities, particularly decreased trade show expenses and marketing campaigns due to limited capital resources in 2017 and our focus on research and development.
Selling and marketing expenses as a percentage of total revenues were approximately 194.6% and 224.5% for the twelve months ended December 31, 2017 and 2016, respectively. The decrease in our percentage was due to the decreased spending mentioned above.
Selling and marketing expenses consist mainly of payroll expenses to direct sales and marketing employees, stock-based compensation expenses, travel expenses, advertising and marketing expenses, rent and facilities expenses associated with and allocated to selling and marketing activities.
General and Administrative Expenses. For the twelve months ended December 31, 2017 and 2016, general and administrative expenses were approximately $2,084,000 and $1,359,000, respectively, an increase of approximately 53.5%, or $725,000, between the periods.
The increase was mainly attributable to increased executive compensation for the management team that started in the fourth quarter of 2016 as well as the increased stock-based compensation of our new management as well as increased spending to position us to raise additional capital through the sale of our securities.
General and administrative expenses as a percentage of total revenues were approximately 872.0% and 593.4% for the twelve months ended December 31, 2017 and 2016, respectively. The increase was due to the increase in general and administrative expenses, described above.
Our general and administrative expenses consist mainly of payroll expenses for management and administrative employees, costs associated with being a publicly traded company, stock-based compensation expenses, accounting and facilities expenses associated with general and administrative activities.
Financial Expenses, net. For the twelve months ended December 31, 2017 and 2016, financial expenses, net were $1,836,000 and $398,000, respectively, an increase of approximately 361.3%, or $1,438,000, between the periods. The increase resulted primarily from an increased valuation adjustment of our warrants due to the increased stock price at the time of the exercise of the warrants and discount amortization of promissory notes issued in 2017.
Tax expenses. For the twelve months ended December 31, 2017 and 2016, tax expenses were $38,000 and $117,000 respectively. The tax expense is computed by multiplying income before taxes at our Israeli subsidiary by the appropriate tax rate and unrecognized tax benefits as a result of tax positions taken.
Net Loss. Our net loss increased by approximately $2,134,000, or 75.4%, to approximately $4,965,000 for the twelve months ended December 31, 2017 from approximately $2,831,000 during the same period in 2016. The increase in net loss resulted primarily from the factors described above.
52
Liquidity and Capital Resources
We have incurred losses in the amount of $4,965,000 during the year ended December 31, 2017, and have accumulated negative cash flow from operating activities of $2,182,000 for the year ended December 31, 2017.
During the year ended December 31, 2017, and through March 29, 2018, we met our short-term liquidity requirements from our existing cash reserves and from proceeds from the sales of convertible promissory notes in an aggregate amount of $1,380,000, as well as the net proceeds of $5,056,000 from our underwritten public offering of common stock and warrants which closed on November 6, 2017. Our future capital requirements and the adequacy of our available funds will depend on many factors, including our ability to successfully commercialize our products, our development of future products and competing technological and market developments. We intend to use these proceeds to meet our short-term liquidity requirements as well as to advance our long-term plans. It is our current belief that such proceeds will provide sufficient funding to meet our liquidity needs for the next twelve months. While we believe we have sufficient capital to execute our business plan over the next twelve months, there are no assurances that we will not need to raise additional capital at a later time, or that we would be able to raise additional capital, if required, on terms favorable to us.
We do not have any material commitments to capital expenditures as of December 31, 2017,2021, and we are not aware of any material trends in capital resources that would impact our business.
Twelve MonthsYears Ended December 31, 20172021 Compared to Twelve MonthsYears Ended December 31, 20162020
General. As of December 31, 2017,2021, we had cash and cash equivalents of approximately $4,360,000,$7,737,000, compared to approximately $106,000$7,533,000 as of December 31, 2016.2020. We have historically met our cash needs through a combination of issuance of equity, borrowing activities and sales. Our cash requirements are generally for product development, research and development cost, marketing and sales activities, general and administrative cost, capital expenditures and general working capital.
Cash used in our operating activities was approximately $2,182,000$4,367,000 for the twelve monthsyears ended December 31, 20172021 and approximately $1,533,000$3,391,000 for the same period in 2016.2020. The increase in our usage ofnet cash used in our operating activities in the amount of $649,000$975,000 is mainly attributable to the increasedecrease in costs associated the increasenoncash expense of change in researchfair value of derivative liability, legal settlement expense and development, increased compensation of the new management teams and the costs incurred in the raising ofwarrant modification expense, partially offset by other working capital described above.accounts.
No cash wasCash used in our investing activities was approximately $3,000 during the twelve monthsyears ended December 31, 20172021 compared to $8,000cash used in our investing activities was $2,000 during the twelve monthsyears ended December 31, 2016.2020.
| 59 |
Cash provided by financing activities during the years ended December 31, 2021 was approximately $6,436,000, from proceeds from the sales of the 2017 Notes in an aggregate amount of $1,380,000, as well as$4,580,000 which was mostly the net proceeds of $5,056,000 from our underwritten public offering which closed on November 6, 2017, compared to $33,000 for the twelve months ended December 31, 2016, which were derived from the proceedsreceived from the exercise of options.warrants completed in 2021 compared to $9,522,000 in 2020, which was the net proceeds received from the sale of common stock in private placements 2020. Our future capital requirements and the adequacy of available funds will depend on many factors, including our ability to successfully commercialize our products, our development of future products and competing technological and market developments.
Off Balance Sheet Arrangements
As of December 31, 2017, we have no off-balance sheet transactions, arrangements, obligations, or other relationships with unconsolidated entities or other persons that have, or may have, a material effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources.
Factors That May Affect Future Operations
We believe that our future operating results will continue to be subject to quarterly variations based upon a wide variety of factors, including the ordering patterns of our distributors, timing of regulatory approvals, the implementation of various phases of our clinical trials and manufacturing efficiencies due to the learning curve of utilizing new materials and equipment.equipment as well issues that may continue to occur due to the development of the coronavirus outbreak. While there were significant delays in the production of goods due to COVID-19 issues, presently, we are no longer experiencing such delays in the production of our products. That said, there are no assurances that if a second wave of the pandemic occurs that we will not experience significant delays in the future. Our operating results could also be impacted by a weakening of the Euro and strengthening of the New Israeli Shekel, or NIS, both against the U.S. dollar. Lastly, other economic conditions we cannot foresee may affect customer demand, such as individual country reimbursement policies pertaining to our products.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Not applicable.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLMENTARYSUPPLEMENTARY DATA
Our Consolidated Financial Statements and the relevant notes to those statements are attached to this report beginning on page F-1.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES.
Disclosure Controls and Procedures.
The Company maintains disclosure controls and procedures (as defined in Rule 13a-15(e) under the Securities Exchange Act) that are designed to ensure that information required to be disclosed in the Company’s Securities Exchange Act reports is recorded, processed, summarized and reported within the time periods specified in SEC rules and forms, and that such information is accumulated and communicated to the Company’s management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure.
| 60 |
Background and Remediation of Material Weakness
A material weakness is defined as a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of our annual or interim financial statements will not be prevented or detected on a timely basis. In previously filed 10-K’s, we disclosed material weaknesses related to the design and effectiveness of our internal control over financial reporting. We did not have adequate controls in place to ensure adequate review, including (1) effective controls over our information technology and information systems relevant to the preparation of our financial statements, (2) the controls over managements review procedures for processing, recording and reviewing transactions related to certain contracts, accounting memos and certain monthly closing procedures, (3) proper account for the number of shares of our common stock issued in connection with the conversion of shares of our preferred stock and the exercise of warrants, which resulted in the Company issuing more shares of common stock than are authorized under our governance documents, and (4) we lacked a formalized written set of policies and procedures including testing documentation to provide evidence that our system of internal controls over financial reporting meets the requirements of the COSO 2013 framework.
We have been able to remediate the material weakness identified above with respect to the issuance of shares in excess of the number of authorized shares in 2021 and implemented a plan in place to have adequate controls in place to avoid future issuances in excess of authorized shares.
During the period covered by this annual report on Form 10-K, with exception for issuance of excess shares, we have not been able to remediate the material weaknesses identified above. Although the Company has taken numerous steps, our remediation plan is not complete due to the lack of a written testing plan to conclude if our controls and procedures and management were operating effectively; and our remediation plan has not operated for a sufficient period of time for the Company to complete testing to conclude that our newly implemented controls and procedures were operating effectively as of December 31, 2021. Additionally, we did not maintain effective controls over the operating effectiveness of information technology (“IT”) general controls for information systems that are relevant to the preparation of their financial statements. Specifically, they did not establish or formalize appropriate IT policies, segregation of duties and monitoring procedures and without monitoring procedures over third-party service providers, did not evaluate whether the providers were appropriately managing its and the Company’s IT infrastructure, operations, and critical financial systems.
As of December 31, 2021, we did not have adequate controls in place to ensure adequate review, including (1) effective controls over our IT and information systems relevant to the issuance of securities, (2) the controls over managements review procedures for processing, recording and reviewing such issuances of securities, and (3) we implemented a new inventory system in the fourth quarter of 2021 which lacked adequate inventory control procedures, (4) we lacked a formalized written set of policies and procedures including testing documentation to provide evidence that our system of internal controls over our issuance of securities meets the requirements of the COSO 2013 framework, and (5) we did not maintain effective controls over the operating effectiveness of IT general controls for information systems that are relevant to the preparation of their financial statements.
Limitations on Effectiveness of Controls and Procedures
In designing and evaluating our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act), management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives. In addition, the design of disclosure controls and procedures must reflect the fact that there are resource constraints, and that management is required to apply judgment in evaluating the benefits of possible controls and procedures relative to their costs.
Evaluation of Disclosure Controls and Procedures
Under the PCAOB standards, a control deficiency exists when the design or operation of a control does not allow management or employees, in the normal course of performing their assigned functions, to prevent or detect misstatements on a timely basis. A significant deficiency is a deficiency, or a combination of deficiencies, in internal control over financial reporting that is less severe than a material weakness, yet important enough to merit the attention by those responsible for oversight of the company’s financial reporting. A material weakness is a deficiency, or combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of the company’s annual or interim financial statements will not be prevented or detected on a timely basis.
| 61 |
Under the supervision and with the participation of our management, including our principal executive officer and principal financial officer, we conducted an evaluation of the effectiveness of our “disclosuredisclosure controls and procedures” (“Disclosure Controls”),procedures, as such term is defined by Rulesunder Rule 13a-15(e) and Rule 15d-15(e) ofpromulgated under the Securities Exchange Act of 1934, as amended (the “Exchange Act”),(Exchange Act). Our management including the Chief Executive Officer and Chief Financial Officer has determined that, as of December 31, 2017,2021, the end of the period covered by this Annual Report on Form 10-K. The Disclosure Controls evaluation was done under the supervision and with the participation of management, including our chief executive officer and chief financial officer. There are inherent limitations to the effectiveness of any system of disclosure controls and procedures. Accordingly, even effectiveCompany’s disclosure controls and procedures can only provide reasonable assuranceare not effective due to a lack of achieving their control objectives. Based upon this evaluation, our chief executive officera full and chief financial officer have concluded that our Disclosure Controls were effective atcomplete testing plan of the reasonable assurance level as of December 31, 2017.Company’s disclosure controls and procedures.
Management’s Report on Internal Control overOver Financial Reporting.
ManagementOur management is responsible for establishing and maintaining adequate internal control over financial reporting, as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act. Our internalreporting. Internal control over financial reporting is defined in Rule 13a-15(f) and Rule 15d-15(f) under the Exchange Act as a process designed by, or under the supervision of, the Company’s principal executive and principal financial officers and effected by the company’s board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of the consolidated financial statements for external reporting purposes in accordance with generally accepted accounting principles.U.S. GAAP. Internal control over financial reporting includes policies and procedures that:
| 1) | Pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the Company; | |
| 2) | Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with U.S. generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the Company; and | |
| 3) | Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the Company’s assets that could have a material effect on the financial statements. |
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness of internal control over financial reporting to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies orand procedures may deteriorate over time.
deteriorate.
Management, including
With the participation of the Chief Executive Officer and Chief Financial Officer, our chief executive officer and chief financial officer, assessedmanagement conducted an evaluation of the effectiveness of our internal control over financial reporting as of December 31, 2017. In making this assessment, management used2021 based on the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission, (“COSO”)known as COSO, inInternal Control—Control — Integrated Framework (2013). Based on its assessmentthis evaluation, our management, including the Chief Executive Officer and those criteria, managementChief Financial Officer, has concluded that we maintained effectiveour internal control over financial reporting was not effective as of December 31, 2017.2021, as the result of the material weaknesses described above.
As a smaller reporting company, the Company is not required to include in this Annual Report on Form 10-K a report on the effectiveness of internal control over financial reporting by the Company’s independent registered public accounting firm.
Management’s Remediation Plans
Management developed a remediation plan, whereby we implemented changes to our internal controls for these material weaknesses. Our remediation activities included: (a) expanded consultations with third party specialists on complex accounting matters, financial reporting and regulatory filings, (b) enhanced documentation to support a more precise review process, (c) enhanced monitoring of the review process, and (d) review of inventory recording system. We will look to develop a full testing plan and document to determine that management designs, implements and maintains adequate controls over our financial processes and reporting in the future our controls and procedures and management are operating effectively. To address these internal control deficiencies, management will continue to perform additional analyses and other procedures to ensure that the financial statements included herein fairly present, in all material respects, our financial position, results of operations and cash flows for the periods presented.
| 62 |
Changes in Internal Control over Financial Reporting.
ThereOther than described above in this Item 9A, there have been no changes in our internal control over financial reporting during the year ended December 31, 2017,2021, that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
None.
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The information required in response to this Item 10 will be set forth in our definitive proxy statement on Schedule 14A for the 20182021 annual meeting of stockholders, which shall be filed with the Securities and Exchange Commission no later than 120 days after the end of the fiscal year covered by this Annual Report on Form 10-KMay 2, 2022 (the “Proxy Statement”), and is incorporated herein by reference..
We have adopted a code of ethics that applies to all of our directors, officers and employees, including the principal executive officer and the principal financial officer. The full text of our code of ethics was filed as Exhibit 14.1 to the annual report on Form 10-K for the year ended December 31, 2016, filed with the Securities and Exchange Commission on March 31, 2017.
| 63 |
ITEM 11. EXECUTIVE COMPENSATION
The information required in response to this Item 11 will be set forth in our Proxy Statement and is incorporated herein by reference.
ITEM 12.SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS.
The information required in response to this Item 12 will be set forth in our Proxy Statement and is incorporated herein by reference.
ITEM 13.CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE.
The information required in response to this Item 13 will be set forth in our Proxy Statement and is incorporated herein by reference.
ITEM 14.PRINCIPAL ACCOUNTING FEES AND SERVICES.
The information required in response to this Item 14 will be set forth in our Proxy Statement and is incorporated herein by reference.
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
The following documents are filed as part of this report:
| (1) | Financial Statements: |
| (2) | Financial Statement Schedules: |
None
| (3) | Exhibits: |
See “Index to Exhibits” for a description of our exhibits.
ITEM 16. FORM 10-K SUMMARY
None.
 |
|
|
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Shareholders and Board of Directors of
NanoVibronix, Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheetssheets of NanoVibronix, Inc. and its subsidiary (“the Company”Subsidiaries (the “Company”) as of December 31, 20172021 and 2016,2020, the related consolidated statements of operations, comprehensive loss, shareholders’stockholders’ equity (deficiency) and cash flows for each of the two years in the period ended December 31, 2017,2021, and the related notes (collectively referred to as the “consolidated financial“financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company atas of December 31, 20172021 and 2016,2020 and the results of its operations and its cash flows for each of the two years in the period ended December 31, 2017,2021, in conformity with U.S.accounting principles generally accepted accounting principles.in the United States of America.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB)(“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the auditaudits to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits audits provides a reasonable basis for our opinion.
/s/ KOST FORER GABBAY & KASIERERCritical Audit Matters
A Member
Critical audit matters are matters arising from the current period audit of Ernst & Young Globalthe financial statements that were communicated or required to be communicated to the audit committee and that: (1) relate to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective, or complex judgments. We determined that there are no critical audit matters.
We have served as the Company’s auditor since 2003.
Tel-Aviv, Israel
March 29, 2018
NANOVIBRONIX INC. AND ITS SUBSIDIARY
| Marcum LLP | |
| We have served as the Company’s auditor since 2018. | |
| New York, NY | |
| April 14, 2022 |
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| ASSETS | ||||||||
| CURRENT ASSETS: | ||||||||
| Cash and cash equivalents | $ | 4,360 | $ | 106 | ||||
| Trade receivables | 24 | 6 | ||||||
| Prepaid expenses and other accounts receivable (Note 3) | 56 | 42 | ||||||
| Inventories (Note 4) | 76 | 67 | ||||||
| Total current assets | 4,516 | 221 | ||||||
| NON-CURRENT ASSETS: | ||||||||
| Long-term prepaid expense | 5 | 5 | ||||||
| Severance pay fund | 338 | 257 | ||||||
| Property and equipment, net (Note 5) | 6 | 11 | ||||||
| Total non- current assets | 349 | 273 | ||||||
| Total assets | $ | 4,865 | $ | 494 | ||||
| F-1 |
NanoVibronix, Inc.
Consolidated Balance Sheets
(Amounts in thousands except share and per share data)
| December 31, 2021 | December 31, 2020 | |||||||
| ASSETS: | ||||||||
| Current assets: | ||||||||
| Cash | $ | 7,737 | $ | 7,142 | ||||
| Restricted cash | - | 391 | ||||||
| Trade receivables | 200 | 25 | ||||||
| Other accounts receivable and prepaid expenses | 230 | 267 | ||||||
| Inventory | 175 | 145 | ||||||
| Total current assets | 8,342 | 7,970 | ||||||
| Noncurrent assets: | ||||||||
| Fixed assets, net | 5 | 4 | ||||||
| Other assets | 19 | 25 | ||||||
| Severance pay fund | 207 | 199 | ||||||
| Operating lease right-of-use assets, net | 49 | 31 | ||||||
| Total non-current assets | 280 | 259 | ||||||
| Total assets | $ | 8,622 | $ | 8,229 | ||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY: | ||||||||
| Current liabilities: | ||||||||
| Trade payables | $ | 87 | $ | 144 | ||||
| Other accounts payable and accrued expenses | 1,723 | 488 | ||||||
| Shares issued in excess of authorized | - | 2,257 | ||||||
| Deferred revenue | 44 | - | ||||||
| Operating lease liabilities, current | 49 | 13 | ||||||
| Total current liabilities | 1,903 | 2,902 | ||||||
| Non-current liabilities: | ||||||||
| Accrued severance pay | 253 | 245 | ||||||
| Deferred licensing income | 153 | 199 | ||||||
| Operating lease liabilities, non-current | - | 18 | ||||||
| Derivative liabilities | - | 2,471 | ||||||
| Total liabilities | 2,309 | 5,835 | ||||||
| Commitments and contingencies | - | - | ||||||
| Stockholders’ equity: | ||||||||
| Series C Preferred stock of $ par value - Authorized: shares at both December 31, 2021 and 2020; Issued and outstanding: and at December 31, 2021 and 2020, respectively | - | 1 | ||||||
| Series D Preferred stock of $ par value - Authorized: shares at both December 31, 2021 and 2020; Issued and outstanding: and at December 31, 2021 and 2020, respectively | - | - | ||||||
| Series E Preferred stock of $ par value - Authorized: shares at both December 31, 2021 and 2020, respectively; Issued and outstanding: and at December 31, 2021 and 2020, respectively | - | 1 | ||||||
| Preferred stock, value | - | 1 | ||||||
| Common stock of $ par value - Authorized: and shares at December 31, 2021 and 2020, respectively; Issued and outstanding: and shares at December 31, 2021 and 2020, respectively | 28 | 22 | ||||||
| Additional paid in capital | 63,162 | 44,959 | ||||||
| Accumulated other comprehensive income | 60 | 66 | ||||||
| Accumulated deficit | (56,937 | ) | (42,655 | ) | ||||
| Total stockholders’ equity | 6,313 | 2,394 | ||||||
| Total liabilities and stockholders’ equity | $ | 8,622 | $ | 8,229 | ||||
The accompanying notes are an integral part of thethese consolidated financial statements.statements
F-3
| F-2 |
NANOVIBRONIX INC. AND ITS SUBSIDIARY
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY (DEFICIENCY) | ||||||||
| CURRENT LIABILITIES: | ||||||||
| Trade payables | $ | 168 | $ | 82 | ||||
| Other accounts payables (Note 6) | 629 | 483 | ||||||
| Total current liabilities | 797 | 565 | ||||||
| NON- CURRENT LIABILITIES: | ||||||||
| Warrants to purchase Common stock (Notes 8, 10i) | — | 2,079 | ||||||
| Accrued severance pay | 434 | 349 | ||||||
| Total long-term liabilities | 434 | 2,428 | ||||||
| COMMITMENTS AND CONTINGENT LIABILITIES (Note 9) | ||||||||
| STOCKHOLDERS’ EQUITY (DEFICIENCY) (Note 10): | ||||||||
| Stock capital - | ||||||||
| Common stock of $ 0.001 par value - Authorized: 20,000,000 shares at December 31, 2017 and 2016; Issued and outstanding: 3,935,865 and 2,632,710 shares at December 31, 2017 and 2016, respectively | 4 | 2 | ||||||
| Series C Preferred stock of $ 0.001 par value - Authorized: 5,000,000 shares at December 31, 2017 and 2016; Issued and outstanding: 2,483,142 and 1,951,261 at December 31, 2017 and 2016, respectively | 2 | 2 | ||||||
| Series D Preferred stock of $ 0.001 par value - Authorized: 5,000 and 0 shares at December 31, 2017 and 2016, respectively; Issued and outstanding: 304 and 0 at December 31, 2017 and 2016, respectively | *) | — | ||||||
| Additional paid-in capital | 32,010 | 20,073 | ||||||
| Accumulated deficit | (28,382 | ) | (22,576 | ) | ||||
| Total stockholders’ equity (deficiency) | 3,634 | (2,499 | ) | |||||
| Total liabilities and stockholders’ equity (deficiency) | $ | 4,865 | $ | 494 | ||||
NanoVibronix, Inc.
*) Represents an amount lower than $ 1 thousands.Consolidated Statements of Operations and Comprehensive Loss
(Amounts in thousands except share and per share data)
| Year Ended December 31, | ||||||||
| 2021 | 2020 | |||||||
| Revenues | $ | 1,695 | $ | 623 | ||||
| Cost of revenues | 925 | 409 | ||||||
| Gross profit | 770 | 214 | ||||||
| Operating expenses: | ||||||||
| Research and development | 293 | 171 | ||||||
| Selling and marketing | 1,101 | 993 | ||||||
| General and administrative | 5,059 | 3,769 | ||||||
| Total operating expenses | 6,453 | 4,933 | ||||||
| Loss from operations | (5,683 | ) | (4,719 | ) | ||||
| Interest expense | - | (147 | ) | |||||
| Gain on forgiveness of PPP loan | - | 42 | ||||||
| Financial income (expense), net | (48 | ) | - | |||||
| Change in fair value of derivative liabilities | (6,956 | ) | 513 | |||||
| Gain on purchase of warrants | 64 | - | ||||||
| Warrant modification expense | (1,627 | ) | - | |||||
| Loss before taxes | (14,250 | ) | (4,311 | ) | ||||
| Income tax expense | (32 | ) | (15 | ) | ||||
| Net loss | $ | (14,282 | ) | $ | (4,326 | ) | ||
| Basic and diluted net loss available for holders of common stock, Series C Preferred Stock and Series D Preferred Stock | $ | (0.57 | ) | $ | (0.42 | ) | ||
| Weighted average common shares outstanding: | ||||||||
| Basic and diluted | 25,162,823 | 10,298,117 | ||||||
| Comprehensive loss: | ||||||||
| Net loss available to common stockholders | (14,282 | ) | (4,326 | ) | ||||
| Change in foreign currency translation adjustments | (6 | ) | 66 | |||||
| Comprehensive loss available to common stockholders | (14,288 | ) | (4,260 | ) | ||||
The accompanying notes are an integral part of thethese consolidated financial statements.statements
| F-3 |
NANOVIBRONIX INC. AND ITS SUBSIDIARY
| Year ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| Revenues | $ | 239 | $ | 229 | ||||
| Cost of revenues | 88 | 88 | ||||||
| Gross profit | 151 | 141 | ||||||
| Operating expenses: | ||||||||
| Research and development | 693 | 584 | ||||||
| Selling and marketing | 465 | 514 | ||||||
| General and administrative | 2,084 | 1,359 | ||||||
| Total operating expenses | 3,242 | 2,457 | ||||||
| Operating loss | (3,091 | ) | (2,316 | ) | ||||
| Financial expense, net (Note 12) | 1,836 | 398 | ||||||
| Loss before taxes on income | (4,927 | ) | (2,714 | ) | ||||
| Taxes on income (Note 11) | 38 | 117 | ||||||
| Loss | $ | (4,965 | ) | $ | (2,831 | ) | ||
| Deemed dividend related to extension of February 2015 warrants to Common stock in January 2017 | 841 | — | ||||||
| Total comprehensive loss attributable to holders of Common stock, Preferred C stock and Preferred D stock | $ | (5,806 | ) | $ | (2,831 | ) | ||
| Common stock, Preferred C stock and Preferred D stock basic and diluted net loss per share (Note 2q) | $ | (1.17 | ) | $ | (0.62 | ) | ||
| Weighted average number of shares of Common stock, Preferred C stock and Preferred D stock used in computing basic and diluted net loss per share (Note 2q) | 4,964,077 | 4,578,470 | ||||||
NanoVibronix, Inc.
Consolidated Statement of Stockholders’ Equity
(Amounts in thousands except share and per share data)
| Series C Preferred Stock | Series D Preferred Stock | Series E Preferred Stock | Common Stock | Additional Paid - in | Accumulated Other Comprehensive | Accumulated | Total Stockholders’ | |||||||||||||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Shares | Amount | Shares | Amount | Capital | Income | Deficit | Equity | |||||||||||||||||||||||||||||||||||||
| Balance, December 31, 2019 | 2,993,142 | $ | 2 | 304 | $ | - | 1,825,000 | $ | 2 | 4,203,764 | $ | 5 | $ | 39,669 | $ | - | $ | (38,329 | ) | $ | 1,347 | |||||||||||||||||||||||||||
| Stock-based compensation | - | - | - | - | - | - | - | - | 376 | - | - | 376 | ||||||||||||||||||||||||||||||||||||
| Exercise of warrants | ||||||||||||||||||||||||||||||||||||||||||||||||
| Exercise of warrants, shares | ||||||||||||||||||||||||||||||||||||||||||||||||
| Reclass of derivative liabilities to APIC | ||||||||||||||||||||||||||||||||||||||||||||||||
| Conversion of Series C Preferred Stock into Common Stock | ||||||||||||||||||||||||||||||||||||||||||||||||
| Conversion of Series C Preferred Stock into Common Stock, shares | ||||||||||||||||||||||||||||||||||||||||||||||||
| Conversion of Series D Preferred Stock into Common Stock | ||||||||||||||||||||||||||||||||||||||||||||||||
| Conversion of Series D Preferred Stock into Common Stock, shares | ||||||||||||||||||||||||||||||||||||||||||||||||
| Conversion of Series E Preferred Stock into Common Stock | ||||||||||||||||||||||||||||||||||||||||||||||||
| Conversion of Series E Preferred Stock into Common Stock, shares | ||||||||||||||||||||||||||||||||||||||||||||||||
| Issuance of common stock for services | - | - | - | - | - | - | 375,000 | - | 566 | - | - | 566 | ||||||||||||||||||||||||||||||||||||
| Issuance of common stock for cash | - | - | - | - | - | - | 11,993,979 | 13 | 4,225 | - | - | 4,238 | ||||||||||||||||||||||||||||||||||||
| Reclass shares issued to liability due to lack of authorized shares | - | - | - | - | - | - | 1,246,523 | 1 | - | - | - | 1 | ||||||||||||||||||||||||||||||||||||
| Warrants issued with notes payable | - | - | - | - | - | - | - | - | 123 | - | - | 123 | ||||||||||||||||||||||||||||||||||||
| Exchange of Series C Preferred Stock into Common Stock | (2,326,475 | ) | (1 | ) | - | - | - | - | 2,326,475 | 1 | - | - | - | - | ||||||||||||||||||||||||||||||||||
| Exchange of Series D Preferred Stock into Common Stock | - | - | (151 | ) | - | - | - | 150,782 | - | - | - | - | - | |||||||||||||||||||||||||||||||||||
| Exchange of Series E Preferred Stock into Common Stock | - | - | - | - | (950,000 | ) | (1 | ) | 950,000 | 2 | - | - | - | 1 | ||||||||||||||||||||||||||||||||||
| Currency translation adjustment | - | - | - | - | - | - | - | - | - | 66 | - | 66 | ||||||||||||||||||||||||||||||||||||
| Net loss | - | - | - | - | - | - | - | - | - | - | (4,326 | ) | (4,326 | ) | ||||||||||||||||||||||||||||||||||
| Balance, December 31, 2020 | 666,667 | $ | 1 | 153 | $ | - | 875,000 | $ | 1 | 21,246,523 | $ | 22 | $ | 44,959 | $ | 66 | $ | (42,655 | ) | $ | 2,394 | |||||||||||||||||||||||||||
| Balance | 666,667 | $ | 1 | 153 | $ | - | 875,000 | $ | 1 | 21,246,523 | $ | 22 | $ | 44,959 | $ | 66 | $ | (42,655 | ) | $ | 2,394 | |||||||||||||||||||||||||||
| Stock-based compensation | - | - | - | - | - | - | - | - | 190 | - | - | 190 | ||||||||||||||||||||||||||||||||||||
| Exercise of warrants | - | - | - | - | - | - | 5,056,603 | 4 | 7,050 | - | - | 7,054 | ||||||||||||||||||||||||||||||||||||
| Reclass of derivative liabilities to APIC | - | - | - | - | - | - | - | - | 10,963 | - | - | 10,963 | ||||||||||||||||||||||||||||||||||||
| Conversion of Series C Preferred Stock into Common Stock | (666,667 | ) | (1 | ) | - | - | - | - | 666,667 | 1 | - | - | - | - | ||||||||||||||||||||||||||||||||||
| Conversion of Series D Preferred Stock into Common Stock | - | - | (153 | ) | - | - | - | 153,000 | - | - | - | - | - | |||||||||||||||||||||||||||||||||||
| Conversion of Series E Preferred Stock into Common Stock | - | - | - | - | (875,000 | ) | (1 | ) | 875,000 | 1 | - | - | - | - | ||||||||||||||||||||||||||||||||||
| Currency translation adjustment | - | - | - | - | - | - | - | - | - | (6 | ) | - | (6 | ) | ||||||||||||||||||||||||||||||||||
| Net loss | - | - | - | - | - | - | - | - | - | - | (14,282 | ) | (14,282 | ) | ||||||||||||||||||||||||||||||||||
| Balance, December 31, 2021 | - | $ | - | - | $ | - | - | $ | - | 27,997,793 | $ | 28 | $ | 63,162 | $ | 60 | $ | (56,937 | ) | $ | 6,313 | |||||||||||||||||||||||||||
| Balance | - | $ | - | - | $ | - | - | $ | - | 27,997,793 | $ | 28 | $ | 63,162 | $ | 60 | $ | (56,937 | ) | $ | 6,313 | |||||||||||||||||||||||||||
The accompanying notes are an integral part of thethese consolidated financial statements.statements
| F-4 |
NANOVIBRONIX INC. AND ITS SUBSIDIARY
NanoVibronix, Inc.
Consolidated Statements of Cash Flows
| Preferred C stocks | Preferred D stocks | Common stocks | Additional paid-in capital | Accumulated deficit | Total stockholders’ equity (deficiency) | |||||||||||||||||||||||||||||||
| Number | Amount | Number | Amount | Number | Amount | |||||||||||||||||||||||||||||||
| Balance as of January 1, 2016 | 1,951,261 | $ | — | — | — | 2,611,328 | $ | 2 | $ | 19,521 | $ | (19,734 | ) | $ | (209 | ) | ||||||||||||||||||||
| Issuance of Common stock upon exercise of options | — | — | — | — | 12,382 | *) | 33 | — | 33 | |||||||||||||||||||||||||||
| Issuance of Common stock to consultant | — | — | — | — | 9,000 | *) | — | — | — | |||||||||||||||||||||||||||
| Stock-based compensation related to options granted to employees | — | — | — | — | — | — | 459 | — | 459 | |||||||||||||||||||||||||||
| ASU 2016-09 adoption, Note 2o | — | — | — | — | — | — | 11 | (11 | ) | — | ||||||||||||||||||||||||||
| Stock-based compensation related to restricted stock granted to consultant | — | — | — | — | — | — | 49 | — | 49 | |||||||||||||||||||||||||||
| Total comprehensive loss | — | — | — | — | — | — | — | (2,831 | ) | (2,831 | ) | |||||||||||||||||||||||||
| Balance as of December 31, 2016 | 1,951,261 | $ | 2 | — | — | 2,632,710 | $ | 2 | $ | 20,073 | $ | (22,576 | ) | $ | (2,499 | ) | ||||||||||||||||||||
| Stock-based compensation related to options granted to employees | — | — | — | — | — | — | 800 | — | 800 | |||||||||||||||||||||||||||
| Issuance of warrants to Common stock | — | — | — | — | — | — | 852 | — | 852 | |||||||||||||||||||||||||||
| Deemed dividend related to extension of February 2015 warrants to Common stock in January 2017 | — | — | — | — | — | — | 841 | (841 | ) | — | ||||||||||||||||||||||||||
| Proceeds from issuance of Common stock, Preferred D stock and warrants, net of issuance costs | — | — | 327 | *) | 897,958 | 1 | 5,055 | — | 5056 | |||||||||||||||||||||||||||
| Conversion of convertible notes and accrued interest into Common and Preferred D stock | — | — | 131 | *) | 230,680 | 1 | 1,761 | — | 1,762 | |||||||||||||||||||||||||||
| Conversion of Preferred D into Common stock | — | — | (154 | ) | *) | 153,530 | *) | — | — | — | ||||||||||||||||||||||||||
| Issuance of Common and Preferred C stock upon cashless exercise of warrants and reclassification from liability to equity | 531,881 | *) | — | — | 20,987 | *) | 2,628 | — | 2,628 | |||||||||||||||||||||||||||
| Total comprehensive loss | — | — | — | — | — | — | — | (4,965 | ) | (4,965 | ) | |||||||||||||||||||||||||
| Balance as of December 31, 2017 | 2,483,142 | $ | 2 | 304 | $ | 0 | 3,935,865 | $ | 4 | $ | 32,010 | $ | (28,382 | ) | $ | 3,634 | ||||||||||||||||||||
(Amounts in thousands except share and per share data)
*) Represents an amount lower than $1.
| Year Ended December 31, | ||||||||
| 2021 | 2020 | |||||||
| Cash flows from operating activities: | ||||||||
| Net loss | $ | (14,282 | ) | $ | (4,326 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Depreciation and amortization | 2 | 2 | ||||||
| Stock-based compensation | 382 | 376 | ||||||
| Noncash interest expense | - | 123 | ||||||
| Gain on forgiveness of PPP Loan | - | (42 | ) | |||||
| Arbitration settlement expense | 1,500 | - | ||||||
| Warrant modification expense | 1,627 | - | ||||||
| Warrants received as licensing fee | - | (23 | ) | |||||
| Change in fair value of equity investment | 6 | - | ||||||
| Change in fair value of derivative liabilities | 6,956 | (513 | ) | |||||
| Other expense related to extension of warrants | - | - | ||||||
| Loss on extinguishment of derivative liability | - | - | ||||||
| Common stock payable to consultant | - | 566 | ||||||
| Gain on purchase of warrants | (64 | ) | - | |||||
| Changes in operating assets and liabilities: | ||||||||
| Trade receivable | (175 | ) | 86 | |||||
| Other accounts receivable and prepaid expenses | 37 | 1 | ||||||
| Inventory | (30 | ) | (26 | ) | ||||
| Trade payables | (59 | ) | 16 | |||||
| Other accounts payable and accrued expenses | (265 | ) | 209 | |||||
| Deferred revenue | (2 | ) | 199 | |||||
| Accrued severance pay, net | - | (39 | ) | |||||
| Net cash used in operating activities | (4,367 | ) | (3,391 | ) | ||||
| Cash flows from investing activities: | ||||||||
| Purchases of property plant and equipment | (3 | ) | (2 | ) | ||||
| Net cash used in investing activities | (3 | ) | (2 | ) | ||||
| Cash flows from financing activities: | ||||||||
| Proceeds from issuance of notes payable | - | 42 | ||||||
| Proceeds from note issued to related party | - | 200 | ||||||
| Payments of note payable to related party | - | (200 | ) | |||||
| Proceeds from sale of common stock, net | - | 9,479 | ||||||
| Proceeds from exercise of warrants | 4,968 | 1 | ||||||
| Buy back of warrants from investor | (388 | ) | - | |||||
| Net cash provided by financing activities | 4,580 | 9,522 | ||||||
| Effects of currency translation on cash | (6 | ) | 66 | |||||
| Net increase in cash, cash equivalents and restricted cash | 204 | 6,195 | ||||||
| Cash, cash equivalents and restricted cash at beginning of period | 7,533 | 1,338 | ||||||
| Cash, cash equivalents and restricted cash at end of period | $ | 7,737 | $ | 7,533 | ||||
| Supplemental disclosures of cash flow information: | ||||||||
| Cash paid for interest | $ | - | $ | - | ||||
| Cash paid for taxes | $ | - | $ | - | ||||
| Supplemental non-cash financing and investing activities: | ||||||||
| Reclass warrants to non-derivative instruments | $ | - | $ | - | ||||
| Discount on notes payable | $ | - | $ | 123 | ||||
| Exchange of common stock into Preferred Stock | $ | 1 | $ | - | ||||
| Shares issued from exercise of warrants previously classified as derivative liability | $ | 2,087 | $ | 2,984 | ||||
| Reclass derivative liability to equity due to increase in authorized shares | $ | 8,706 | $ | - | ||||
| Reclass equity to liability due to over issuance of shares | $ | - | $ | 2,257 | ||||
| Reclass liability to equity after increase in authorized shares | $ | 2,257 | $ | - | ||||
The accompanying notes are an integral part of the interimthese consolidated financial statements
| F-5 |
NANOVIBRONIX, INC. AND ITS SUBSIDIARY
Notes to Consolidated Financial Statements
(Amounts in thousands except share and per share data)
| Year ended | ||||||||
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Cash flows from operating activities: | ||||||||
| Loss | $ | (4,965 | ) | $ | (2,831 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Depreciation | 5 | 7 | ||||||
| Stock-based compensation | 800 | 508 | ||||||
| Discount amortization of promissory notes | 1,197 | — | ||||||
| Revaluation of warrants to purchase Common stock | 549 | 383 | ||||||
| Increase in trade receivables | (18 | ) | (1 | ) | ||||
| Increase (decrease) in prepaid expenses and other accounts receivable | (14 | ) | 44 | |||||
| Decrease (increase) in inventories | (9 | ) | 4 | |||||
| Increase in trade payables | 86 | 24 | ||||||
| Increase in other accounts payable | 183 | 244 | ||||||
| Increase in accrued severance pay, net | 4 | 90 | ||||||
| Increase in long-term prepaid expense | — | (5 | ) | |||||
| Net cash used in operating activities | (2,182 | ) | (1,533 | ) | ||||
| Cash flows from investment activities: | ||||||||
| Purchase of property and equipment | — | (8 | ) | |||||
| Net cash used in investment activities | — | (8 | ) | |||||
| Cash flows from financing activities: | ||||||||
| Proceeds from issuance of Convertible Promissory Notes and warrants | 1,380 | — | ||||||
| Proceeds from issuance of Common stock, Preferred D stock and warrants, net of issuance costs | 5,056 | — | ||||||
| Proceeds from exercise of options | — | 33 | ||||||
| Net cash provided by financing activities | 6,436 | 33 | ||||||
| Increase (decrease) in cash and cash equivalents | 4,254 | (1,508 | ) | |||||
| Cash and cash equivalents at the beginning of the year | 106 | 1,614 | ||||||
| Cash and cash equivalents at the end of the year | $ | 4,360 | $ | 106 | ||||
| Supplemental information and disclosure of non-cash financing transactions: | ||||||||
| Carve out of warrants’ fair value from Convertible Promissory Notes | $ | 852 | $ | — | ||||
| Conversion of convertible notes and accrued interest into Common and Preferred D stock | $ | 1,762 | $ | — | ||||
| Cashless exercise of warrants | $ | 2,628 | $ | — | ||||
| Stock-based compensation- ASU 2016-09 adoption | $ | — | $ | 11 | ||||
The accompanying notes are an integral partNOTE 1 - DESCRIPTION OF BUSINESS
NanoVibronix, Inc. (the “Company”), a Delaware corporation, commenced operations on October 20, 2003, and is a medical device company focusing on noninvasive biological response-activating devices that target wound healing and pain therapy and can be administered at home, without the assistance of the consolidated financial statements.medical professionals.
NANOVIBRONIX INC. AND ITS SUBSIDIARY
The Company’s principal research and development activities are conducted in Israel through its wholly-owned subsidiary, NanoVibronix (Israel 2003) Ltd., a company registered in Israel, which also commenced operations in October 2003.
NOTE 2 - LIQUIDITY AND PLAN OF OPERATIONS
The Company’s ability to continue to operate is dependent mainly on its ability to successfully market and sell its products and the receipt of additional financing until profitability is achieved. In 2021, the Company’s cash used in operations was $4,367 and received net proceeds of $4,968 from the sale of our equity securities, leaving a cash balance of $7,737 as of December 31, 2021. Because the Company has sufficient resources to fund our operation for the next twelve months from the date of this filing, there is no substantial doubt of the Company’s ability to continue as a going concern. The Company may need to continue to raise additional capital to finance its losses and negative cash flows from operations beyond the next years and may continue to be dependent on additional capital raising as long as our products do not reach commercial profitability.
NOTE 3 - SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Basis of presentation and principles of consolidation
The accompanying consolidated financial statements include the accounts of NanoVibronix, Inc. and its wholly owned subsidiary. Intercompany accounts and transactions have been preparedeliminated. The preparation of these consolidated financial statements and accompanying notes in accordanceconformity with U.S. generally accepted accounting principles generally accepted in(“US GAAP”) requires management to make estimates and assumptions that affect the United States (“U.S. GAAP”).amounts reported. Actual results could differ materially from those estimates.
Use of estimates
The preparation of the consolidated financial statements in conformity with U.S. GAAP requires management to make estimates, judgments and assumptions. The Company’s managementCompany believes that the estimates, judgments and assumptions used are reasonable based upon information available at the time they are made. These estimates, judgments and assumptions can affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the dates of the financial statements, and the reported amounts of revenue and expenses during the reporting period. Actual results could differ from those estimates.
The accompanying financial statements have been prepared in U.S. dollars.Foreign currency translation
The majority of the Company’s expenses, financing activities and revenues areNon-U.S. dollar denominated and determined in U.S. dollars. The Company’s management believes that the U.S. dollar is the currency of the primary economic environment in which the Company operates and expects to continue to operate in the foreseeable future. Thus, the functional and reporting currency of the Company is the U.S. dollar.
NANOVIBRONIX INC. AND ITS SUBSIDIARY
The Company’s transactions and balances denominated in U.S. dollars are presented at their original amounts. Non-dollar transactions and balances have been re-measured to U.S. dollars in accordance with the Accounting Standards Codification (ASC) 830, “Foreign Currency Matters”.dollars. All transaction gains and losses from re-measurement of monetary balance sheet items denominated in non-dollarnon-U.S. dollar currencies are reflected in the statements of operations as other comprehensive loss as financial income, or expenses, as appropriate.
The consolidated financial statements include the accountscumulative translation losses and gains as of the Companyyears ended December 31, 2021 and its wholly-owned subsidiary, NanoVibronix (Israel 2003) Ltd. All intercompany balances 2020 were $6 and transactions$66, respectively.
| F-6 |
Basic loss per share was computed using the weighted average number of common shares outstanding. Diluted loss per share includes the effect of diluted common stock equivalents. Potentially dilutive securities from the exercise of stock option, warrants and exercise of preferred stock as of December 31, 2021 and 2020, respectively, were excluded from the computation of diluted net loss per share because the effect of their inclusion would have been eliminated upon consolidation.antidilutive.
Cash equivalents are short-term highly liquid investments that are readily convertible to cash with original maturities of three months or less at acquisition. Inventory
Inventories are stated at the lower of cost or net realizable value. Net realizable value is the estimated selling prices in the ordinary course of business, less reasonably predictable costs of completion, disposal, and transportation. Cost is determined using the “first-in, first-out” method.
Inventory write-offs are provided to cover risks arising from slow-moving items or technological obsolescence. The Company periodically evaluates the quantities on hand relative to current and historical selling prices and historical and projected sales volume. Based on this evaluation, provisions are made when required to write-down inventory to its net market value. As of December 31, 20172021 and 2016, no provisions for inventory write-downs were recorded.2020, there was 0 allowance on inventory.
Non-current prepaid expenses consist of non-current lease deposits as security for the Company’s motor vehicles leases.Property and equipment
Property and equipment are stated at cost, net of accumulated depreciation. Depreciation is calculated using the straight-line method over the estimated useful lives of the assets, at the following annual rates:
SCHEDULE OF DEPRECIATION CALCULATED OVER ESTIMATED USEFUL LIVES OF ASSETS
| Computers and peripheral equipment | ||||
| Office furniture and equipment | 5-7 | |||
The Company’s long-lived assets are reviewedImpairment of Long-Lived Assets
Management reviews for impairment in accordance with Accounting Standard Codification (“ASC”) 360, “Property, Plant, and Equipment”, whenever events or changes in circumstances indicate that the carrying amount of an assetproperty and equipment may not be recoverable. Recoverabilityrecoverable under the provisions of assetsaccounting for the impairment of long-lived assets. If it is determined that an impairment loss has occurred based upon expected future cash flows, the loss is recognized in the Consolidated Statements of Operations.
Sequencing
The Company adopted a sequencing policy under ASC 815-40-35 whereby if reclassification of contracts from equity to be held and usedliabilities is measured by a comparisonnecessary pursuant to ASC 815 due to the Company’s inability to demonstrate it has sufficient authorized shares. This was due to the Company committing more shares than authorized. While temporary suspensions are in place to keep the potential exercises beneath the number authorized, certain instruments are classified as liabilities, after allocating available authorized shares on the basis of the carrying amountmost recent grant date of an assetpotentially dilutive instruments. Pursuant to ASC 815, issuances of securities granted as compensation in a share-based payment arrangement are not subject to the future undiscounted cash flows expected to be generated bysequencing policy.
Derivative Liability
The Company’s derivative financial instruments are measured at fair value using the assets. If such assets are considered to be impaired, the impairment to be recognized is measured by the amount byBlack Scholes Model which the carrying amounttakes into account, as of the assets exceedsvaluation date, factors including the current exercise price, the expected life of the warrant, the current price of the underlying stock and its expected volatility, expected dividends on the stock and the risk-free interest rate for the term of the instrument. The liability is revalued at each reporting period and changes in fair value are recognized in the consolidated statements of operations and comprehensive loss under the caption “Change in fair value of the assets. During the years endedderivative liabilities.” As of December 31, 20172021 and 2016, no impairment losses have been identified. 2020, there were $0 and $2,471 derivative liabilities on the consolidated balance sheet, respectively (see note 8).
| F-7 |
NANOVIBRONIX INC. AND ITS SUBSIDIARY
Severance pay
The Company’s liability for severance pay is for its Israeli employees and is calculated pursuant to Israeli Severance Pay Law based on the most recent salary of the employees multiplied by the number of years of employment as of the balance sheet date and is in large part covered by regular deposits with recognized pension funds, deposits with severance pay funds and purchases of insurance policies. The value of these deposits and policies is recorded as an asset in the Company’s balance sheet.
Severance expenses for the years ended Accrued severance pay liability at December 31, 20172021 and 2016 amounted to2020 was $ 85253 and $ 150,245, respectively.
The Company accounts for stock warrantsheld by investorsas either equity instruments or liabilities in accordance with ASC 480, Distinguishing Liabilities from EquityRevenue recognition (“ASC 480”), depending on the specific terms of the warrant agreement.
Stock warrants are accounted for as a liability if they contain “down-round protection” or other terms that could potentially require “net cash settlement” in accordance with the provisions of ASC 815-40,Derivatives and Hedging - Contracts in Entity’s Own Equity (“ASC 815”), which provides a two-step model to be applied in determining whether a financial instrument or an embedded featureIt is indexed to an issuer’s own stock and thus able to qualify to be a derivative financial instrument. The Company measures such warrants at fair value by applying the Black-Scholes option pricing model in each reporting period until they are exercised or expired, with changes in the fair value being recognized in the Company’s statement of comprehensive loss as financial income or expense, as appropriate.
The Company considers guidance within ASC 470-20, Debt (ASC 470), ASC 480, and ASC 815 when accounting for the issuance of convertible debt with detachable warrants. As described above under the caption “Warrants”, the Company classifies stock warrants as either equity instruments or liabilities depending on the specific terms of the warrant agreement. In circumstances in which debt is issued with liability-classified warrants, the proceeds from the issuance of convertible debt are first allocated to the warrants at their full estimated fair value and established as both a liability and a debt discount. The remaining proceeds, as further reduced by discounts created by the bifurcation of embedded derivatives and a beneficial conversion feature, is allocated to the debt. The Company accounts for debt as liabilities measured at amortized cost and amortizes the resulting debt discount from the allocation of proceeds, to interest expense using the effective interest method over the expected term of the debt instrument pursuant to ASC 835, Interest (ASC 835).
The Company applied ASC 470-20 and ASC 815 to the Convertible promissory notes (see Note 7).
|
The Company generatespolicy that revenues from the sale of its products to distributors and patients. Revenues from those products areproduct sales is recognized in accordance with ASC 605,606 “Revenue Recognition,Recognition.” Five basic steps must be followed before revenue can be recognized; (1) Identifying the contract(s) with a customer that creates enforceable rights and obligations; (2) Identifying the performance obligations in the contract, such as promising to transfer goods or services to a customer; (3) Determining the transaction price, meaning the amount of consideration in a contract to which an entity expects to be entitled in exchange for transferring promised goods or services to a customer; (4) Allocating the transaction price to the performance obligations in the contract, which requires the company to allocate the transaction price to each performance obligation on the basis of the relative standalone selling prices of each distinct good or services promised in the contract; and (5) Recognizing revenue when delivery(or as) the entity satisfies a performance obligation by transferring a promised good or service to a customer. The amount of revenue recognized is the amount allocated to the satisfied performance obligation. Adoption of ASC 606 has occurred, persuasivenot changed the timing and nature of the Company’s revenue recognition and there has been no material effect on the Company’s financial statements.
Revenue from product sales is recorded at the net sales price, or “transaction price,” which includes estimates of variable consideration that result from coupons, discounts, chargebacks and distributor fees, processing fees, as well as allowances for returns and government rebates. The Company constrains revenue by giving consideration to factors that could otherwise lead to a probable reversal of revenue. Collectability of revenue is reasonably assured based on historical evidence of an agreement exists,collectability between the fee is fixed or determinable, no further obligation existsCompany and collectability is probable.its customers.
Revenues from sales to distributors are recognized at the time the products are delivered to the distributors (“sell-in”). The Company does not grant rights of return, credits, rebates, price protection, or other privileges on its products to distributors.
Research and development costs are charged to the statement of comprehensive loss, as incurred.Income taxes
The Company accounts for income taxes in accordance with ASC 740, “Income Taxes”. This topic prescribes the use of the liability method whereby deferred tax assets and liability account balances are determined based on differences between financial reporting and tax bases of assets and liabilities and are measured using the enacted tax rates and laws that will be in effect when the differences are expected to reverse. The Company provides full valuation allowance, to reduce deferred tax assets to the amount that is more likely than not to be realized.
In November 2015, the FASB issued ASU 2015-17, Balance Sheet Classification of Deferred Taxes, related to balance sheet classification of deferred taxes. The ASU requires that deferred tax assets and liabilities be classified as noncurrent in the statement of financial position, thereby simplifying the current guidance that requires an entity to separate deferred assets and liabilities into current and noncurrent amounts. ASU 2015-17 is effective for financial statements issued for annual periods beginning after December 15, 2016 and interim periods within those annual periods. ASU 2015-17 was early adopted by the Company as of December 31, 2017, and had no impact on its consolidated financial statements.
The Company implements a two-step approach to recognize and measure uncertain tax positions. The first step is to evaluate the tax position taken or expected to be taken in a tax return by determining if the weight of available evidence indicates that it is more likely than not that, on an evaluation of the technical merits, the tax position will be sustained on audit, including resolution of any related appeals or litigation processes. The second step is to measure the tax benefit as the largest amount that is more than 50% (cumulative basis) likely to be realized upon ultimate settlement.
The Company recognizes interest and penalties related to uncertain tax positions on the income tax expense line in the accompanying consolidated statement of operations. Accrued interest and penalties are included on the related tax liability line in the consolidated balance sheet.
F-10
| F-8 |
NANOVIBRONIX INC. AND ITS SUBSIDIARYStock-based compensation
The Company accounts for stock-based compensation in accordance with ASC 718, “Compensation - Stock Compensation”, (“ASC 718”), which requires companies to estimate the fair value of equity-based payment awards on the date of grant using an option-pricing model. The value of the portion of the award that is ultimately expected to vest is recognized as an expense over the requisite service periods on a straight line method in the Company’s consolidated statement of comprehensive loss.
The Company has early adopted ASU 2016-09 in the 2016 consolidated financial statements using a modified retrospective transition method by means of a cumulative-effect adjustment to equity as of the beginning of the period in which the guidance is adopted. As a result of this adoption, the Company recorded an increase to accumulated deficit of $11 resulting from the election of accounting policy to account for forfeitures as they occur as of January 1, 2016.
The Company selected the Black-Scholes-Merton option pricing model as the most appropriate fair value method for its stock-options awards. The option-pricing model requires a number of assumptions, of which the most significant are the expected stock price volatility and the expected option term. Expected volatility was calculated based upon similar traded companies’ historical share price movements. The expected option term represents the period that the Company’s stock options are expected to be outstanding. The Company currently uses the simplified method and will continue to do so until sufficient historical exercise data supports using expected life assumptions. The risk-free interest rate is based on the yield from U.S. Treasury zero-coupon bonds with an equivalent term. The expected dividend yield assumption is based on the Company’s historical experience and expectation of no future dividend payouts. The Company has historically not paid cash dividends and has no foreseeable plans to pay cash dividends in the future.
There were no options granted in 2017. The fair value for options granted in 2016 is estimated at the date of grant using a Black-Scholes-Merton options pricing model with the following underlying assumptions:NOTE 4 - PREPAID EXPENSES AND OTHER RECEIVABLES
| Year ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| Risk free interest | — | 1.21%-1.88% | ||||||
| Dividend yields | — | 0% | ||||||
| Volatility | — | 61.3%-63.9% | ||||||
| Expected term (in years) | — | 5.5-6.25 | ||||||
The Company applies ASC 505-50, “Equity-Based Payments to Non-Employees” (“ASC 505”) with respect to optionsPrepaid expenses and warrants issued to non-employees which requires the use of option valuation models to measure the fair valueother receivables consist of the options and warrants at the measurement date. following:
SCHEDULE OF PREPAID EXPENSES AND OTHER RECEIVABLES
| December 31, | ||||||||
| 2021 | 2020 | |||||||
| Prepaid expenses | $ | 166 | $ | 199 | ||||
| Other receivables | 64 | 68 | ||||||
| Prepaid Expenses and Other Receivable | $ | 230 | $ | 267 | ||||
F-11
| F-9 |
NANOVIBRONIX INC. AND ITS SUBSIDIARY
ASC 820, “Fair Value Measurements and Disclosures,” defines fair value as the price that would be received to sell an asset or paid to transfer a liability (i.e., the “exit price”) in an orderly transaction between market participants at the measurement date.NOTE 5 – INVENTORY
In determining fair value, the Company uses various valuation approaches. ASC 820 establishes a hierarchy for inputs used in measuring fair value that maximizes the use of observable inputs and minimizes the use of unobservable inputs by requiring that the most observable inputs be used when available. Observable inputs are inputs that market participants would use in pricing the asset or liability developed based on market data obtained from sources independentInventory consists of the Company. Unobservable inputs are inputs that reflect the Company’s assumptions about the assumptions market participants would use in pricing the asset or liability developed based on the best information available in the circumstances.following components:
SCHEDULE OF INVENTORY
| December 31, | ||||||||
| 2021 | 2020 | |||||||
| Raw materials | $ | - | $ | 80 | ||||
| Finished goods | 175 | 65 | ||||||
| Inventory | $ | 175 | $ | 145 | ||||
NOTE 6 - STOCKHOLDERS’ EQUITY
Common Stock
The hierarchy is broken down into three levels based on the inputs as follows:
The carrying amounts of cash and cash equivalents, trade receivables, prepaid expenses and other accounts receivable, trade payables and other accounts payables approximate their fair value due to the short-term maturities of such instruments.
F-12
NANOVIBRONIX INC. AND ITS SUBSIDIARY
Basic net loss per share is computed based on the weighted average number of shares of Common stock, Preferred C and Preferred D stock outstanding during each year. Diluted net loss per share is computed based on the weighted average number of shares of Common stock, Preferred C and Preferred D stock outstanding during each year plus dilutive potential equivalent shares of Common stock, Preferred C and Preferred D stock considered outstanding during the year, in accordance with ASC 260, “Earnings per Share.”
For the years ended December 31, 2017 and 2016, all outstanding stock options and warrants have been excluded from the calculation of the diluted net loss per share as all such securities are anti-dilutive for all years presented.
Financial instruments that potentially subject the Company to concentrations of credit risk consist principally of cash and cash equivalents. Cash and cash equivalents are invested in major banks in U.S. and Israel. Management believes that the financial institutions that hold the Company’s investments are financially sound and, accordingly, minimal credit risk exists with respect to these investments.
The Company has no off-balance-sheet concentration of credit risk such as foreign exchange contracts, option contracts or other foreign hedging arrangements.
The Company accounts for its contingent liabilities in accordance with ASC 450 “Contingencies”. A provision is recorded when it is both probable that a liability has been incurred and the amount of the loss can be reasonably estimated. With respect to legal matters, provisions are reviewed and adjusted to reflect the impact of negotiations, estimated settlements, legal rulings, advice of legal counsel and other information and events pertaining to a particular matter. As of December 31, 2017 and 2016, the Company is not a party to any litigation that could have a material adverse effect on the Company’s business, financial position, results of operations or cash flows.
F-13
NANOVIBRONIX INC. AND ITS SUBSIDIARY
|
|
| ||
|
F-14
NANOVIBRONIX INC. AND ITS SUBSIDIARY
| ||
NANOVIBRONIX INC. AND ITS SUBSIDIARY
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Prepaid expenses | $ | 45 | $ | 34 | ||||
| Other accounts receivable | 11 | 8 | ||||||
| $ | 56 | $ | 42 | |||||
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Raw materials | $ | 68 | $ | 44 | ||||
| Work in process | — | 5 | ||||||
| Finished goods | 8 | 18 | ||||||
| $ | 76 | $ | 67 | |||||
F-16
NANOVIBRONIX INC. AND ITS SUBSIDIARY
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Cost: | ||||||||
| Computers and peripheral equipment | $ | 48 | $ | 48 | ||||
| Office furniture and equipment | 3 | 3 | ||||||
| 51 | 51 | |||||||
| Accumulated depreciation: | ||||||||
| Computers and peripheral equipment | 42 | 38 | ||||||
| Office furniture and equipment | 3 | 2 | ||||||
| 45 | 40 | |||||||
| Depreciated cost | $ | 6 | $ | 11 | ||||
During the years ended December 31, 2017 and 2016, fully depreciated assets that were no longer in use with a total cost and accumulated depreciation of $0 and $ 67 were disposed from the consolidated balance sheets.
Depreciation expenses for the years ended December 31, 2017 and 2016 were $5 and $7, respectively.
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Employees and payroll accruals | $ | 219 | $ | 170 | ||||
| Accrued expenses | 150 | 99 | ||||||
| Income tax accrual | 260 | 214 | ||||||
| 629 | 483 | |||||||
F-17
NANOVIBRONIX INC. AND ITS SUBSIDIARY
|
On November 6, 2017, the Company completed a public offering, which constituted a Qualified Financing, upon which the 2017 Notes were automatically converted. Based on the outstanding principal amount and all accrued but unpaid interest on the 2017 Notes, at 80% of the offering price of $4.90 per share of common stock and accompanying warrant, the Company issued an aggregate of 361,462 shares of common stock (and common stock equivalents) and warrants to purchase an aggregate of 271,096 shares of common stock to the holders of the 2017 Notes, all of which are subject to lock-up agreements for 180 days from November 1, 2017.
F-18
NANOVIBRONIX INC. AND ITS SUBSIDIARY
The Company accounted for these warrants as a liability according to the provisions of ASC 815-40 and measured the warrants at fair value by applying the Black-Scholes option pricing model in each reporting period until they were exercised, with changes in fair values being recognized in the Company’s consolidated statement of comprehensive loss as financial income or expenses.
In estimating the warrants’ fair value the Company used the following assumptions:
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Dividend yield (1) | 0% | 0% | ||||||
| Expected volatility (2) | 39.6%-52.2% | 54.07%-65.59% | ||||||
| Risk-free interest (3) | 1.13%-1.5% | 0.89%-1.47% | ||||||
| Expected term (years) (4) | 0.0-2.1 | 1.1-2.94 | ||||||
F-19
NANOVIBRONIX INC. AND ITS SUBSIDIARY
The level of inputs used to measure fair value was Level 2.
| Fair value of warrants to Common stock | ||||||||
| 2017 | 2016 | |||||||
| Balance at January 1 | $ | 2,079 | $ | 1,696 | ||||
| Change in fair value of warrants | 549 | 383 | ||||||
| Balance at October 4 | $ | 2,628 | $ | 2,079 | ||||
F-20
NANOVIBRONIX INC. AND ITS SUBSIDIARY
F-21
NANOVIBRONIX INC. AND ITS SUBSIDIARY
There are no future minimum lease commitments under non-cancelable operating lease agreements as of December 31, 2017.
The Company leases motor vehicles under cancelable lease agreements. The Company has an option to be released from this lease agreement, which may result in penalties in a maximum amount of approximately $5.
Rent and related expenses were $27 and $30 for the years ended December 31, 2017 and 2016, respectively.
Motor vehicle leases, and related expenses were $15 and $17 for the years ended December 31, 2017 and 2016, respectively.
Under the Company’s subsidiary research and development agreements with the IIA and pursuant to applicable laws, the Company is required to pay royalties at the rate of 3-3.5% of sales of products developed with funds provided by the IIA, up to an amount equal to 100% of the IIA research and development grants received, linked to the dollar including accrued interest at the LIBOR rate. The Company has received through the years grants in the amount of $ 437. The Company is obligated to repay the Israeli Government for the grants received only to the extent that there are sales of the funded products. As of December 31, 2017, there are no sales from the funded projects.
As of December 31, 2017, the Company has a contingent obligation to pay royalties in the principal amount of approximately $ 465. In addition, the IIA may impose certain conditions on any arrangement under which it permits the Company to transfer technology or development out of Israel.
F-22
NANOVIBRONIX INC. AND ITS SUBSIDIARY
The Common stock confers upon the holders the right to receive notice to participate and vote in general meetings of the Company, and the right to receive dividends, if declared, and to participate in the distribution of the surplus assets and funds of the Company in the event of liquidation, dissolution or winding up of the Company.
As of December 3, 2020, we had authorized shares of our common stock and shares of common stock outstanding resulting in shares of common stock being available for issuance. On December 4, 2020, certain holders of the Company’s Series C Preferred Stock converted shares of Series C Preferred Stock into shares of common stock, resulting in an overissue of shares of common stock. Beginning on December 17, 2020, through January 22, 2021, certain holders of warrants we had issued in December 2020 (the “December 2020 Warrants”) exercised a portion of the December 2020 Warrants for shares of Common Stock, resulting in an additional overissue of 2,657,144 shares of Common Stock. As of December 31, 2020, the aggregate number of shares of common stock that was overissued by the Company was . The shares issued in excess of the authorized amount are classified as liabilities. The common stock equivalents are subject to the Company’s sequencing policy and are classified as derivative liabilities (see Note 8). On March 3, 2021, the Company filed a proxy statement in connection with a special meeting of stockholders to was to be held on March 31, 2021, but postponed until May 6, 2021 to (i) ratify the increase in the number of authorized shares of common stock from to and the issuance of such shares of common stock, and (ii) further increase the number of our authorized shares of common stock. On May 6, 2021, the Company’s stockholders voted to approve the ratification of the increase in the number of authorized shares of common stock from to and the issuance of such shares of common stock to be effective as of December 4, 2020, but the stockholders did not approve a further increase in the number of its authorized shares of common stock.
On August 17, 2021, the Company’s stockholders voted to approve an amendment to the Company’s Amended and Restated Certificate of Incorporation to increase the number of shares of the Company’s Common Stock authorized for issuance from shares to shares. As a result of the vote to increase the number of shares authorized for issuance, the warrants that were previously accounted for as derivative liabilities were marked to market through the date of approval and then reclassified to additional paid in capital (equity), as the Company had sufficient authorized shares to settle the exercise of the warrants.
Issuance of common stock for cash
On August 24, 2020, the Company entered into an underwriting agreement with H.C. Wainwright & Co., LLC (“Wainwright”) (as amended and restated, the “August Underwriting Agreement”). Pursuant to the August Underwriting Agreement, the Company sold, in an upsized firm commitment offering, shares of the Company’s common stock, to Wainwright at an offering price to the public of $ per share, less underwriting discounts and commissions. The Company received net proceeds from the sale of such offering, after deducting underwriting discounts and commissions and other estimated offering expenses payable by the Company, of approximately $2.7 million. In addition, as partial compensation for Wainwright’s services as underwriter in the offering, the Company has issued to Wainwright’s designees warrants to purchase 339,858 shares of common stock. The warrants expire on August 24, 2025 and have an exercise price of $0.9375 per share.
| F-10 |
On September 22, 2020, the Company entered into an underwriting agreement with Wainwright (as amended and restated, the “September Underwriting Agreement”). Pursuant to the September Underwriting Agreement, the Company sold, in an upsized firm commitment offering, shares of common stock to Wainwright at an offering price to the public of $ per share, less underwriting discounts and commissions. The Company received net proceeds from the sale of such offering, after deducting underwriting discounts and commissions and other estimated offering expenses payable by the Company, of approximately $1.4 million. In addition, as partial compensation for Wainwright’s services as underwriter in the offering, the Company issued to Wainwright’s designees warrants to purchase 134,609 shares of common stock. The warrants expire on September 22, 2025 and have an exercise price of $1.25 per share.
On December 2, 2020, the Company entered into a Securities Purchase Agreement with certain institutional and accredited investors pursuant to which the Company issued and sold to such investors in a private placement (the “Private Placement”) an aggregate of (i) shares of the Company’s common stock at an offering price of $ per share and (ii) pre-funded warrants to purchase up to shares of common stock (the “Pre-funded Warrants”), at a purchase price of $ per Pre-funded Warrant, for gross proceeds of approximately $6.0 million.
The Pre-funded Warrants have an exercise price of $0.001 per share. The Pre-funded Warrants are immediately exercisable and may be exercised at any time after their original issuance until such Pre-funded Warrants are exercised in full. A holder of a Pre-funded Warrant may not exercise any portion of such holder’s Pre-funded Warrants to the extent that the holder, together with its affiliates, would beneficially own more than 4.99% (or, at the election of the holder, 9.99%) of the Company’s outstanding shares of common stock immediately after exercise, except that upon at least 61 days’ prior notice from the holder to the Company, the holder may increase the beneficial ownership limitation to up to 9.99% of the number of shares of common stock outstanding immediately after giving effect to the exercise.
The net proceeds to the Company from the Private Placement were approximately $5,400, after deducting placement agent fees and expenses and estimated offering expenses payable by the Company. The Company intends to use the net proceeds from the Private Placement for general corporate purposes. The Private Placement closed on December 7, 2020.
Series C, D and E Preferred Stock conversion to common stock
Each share of Series CE Preferred stockStock is convertible into one share of Common stock (subject to adjustment) at any time and from time to time at the option of a holder of Series E Preferred Stock into one share of the holders,Company’s common stock, provided that each holder would be prohibited from converting Series CE Preferred stockStock into shares of Commonthe Company’s common stock if, as a result of such conversion, any such holder, together with its affiliates, would own more than 9.99% of the total number of shares of Commonthe Company’s common stock then issued and outstanding. This limitation may be waived with respect to a holder upon such holder’s provision of not less than 61 days’ prior written notice to the Company.
InDuring the event of liquidation, dissolution, or winding up, each holder of Series C Preferred stock could elect to receive either (i) in preference to any payments made to the holders of Common stockyears ended December 31, 2021 and any other junior securities, a payment for each share of Series C Preferred stock then held equal $ 0.001, plus an additional amount equal to any dividends declared but unpaid on such shares,2020, shareholders converted and any other fees or liquidated damages then due and owing thereon or (ii) the amount of cash, securities or other property to which such holder would be entitled to receive with respect to each share of Series C Preferred stock if such share of Series C Preferred stock had been converted to Common stock immediately prior to such liquidation, dissolution, or winding up (without giving effect to any conversion limitations).
Shares of Series C Preferred stock are not entitled to receive any dividends, unless and until specifically declared by the board of directors. However, holders of Series C Preferred stock are entitled to receive dividends on shares of Series CE Preferred stock equal (on an as-if-converted-to-Common-stock basis) toStock into and in the same form as dividends actually paid on shares of the Commoncommon stock, when such dividends are specifically declared by the boardrespectively, at a conversion rate of directors. The Company is not obligated1 to redeem or repurchase any shares of Series C Preferred stock. Shares of Series C Preferred stock are not otherwise entitled1. No purchase was made in order to any redemption rights, or mandatory sinking fund or analogous fund provisions.convert these shares.
NANOVIBRONIX INC. AND ITS SUBSIDIARY
Each holder of Series C Preferred stock is entitled to the number of votes equal to the number of whole shares of Common stock into which the shares of Series C Preferred stock held by such holder are then convertible (subject to the beneficial ownership limitations) with respect to any and all matters presented to the stockholders for their action or consideration. Holders of Series C Preferred stock vote together with the holders of Common stock as a single class, except as provided by law and except that the consent of holders of a majority of the outstanding Series C Preferred stock is required to amend the terms of the Series C Preferred stock.
Each share of Series D Preferred Stock is convertible into 1,000 shares of common stock (subject to the beneficial ownership limitations and adjustment as provided in the certificate of designation) at any time at the option of the holders, provided that each holder would be prohibited from converting Series D Preferred Stock into shares of common stock if, as a result of such conversion, any such holder, together with its affiliates, would own more than 4.99% of the total number of shares of common stock then issued and outstanding. However, anyThis limitation may be waived with respect to a holder may increase or decreaseupon such percentage to any other percentageholder’s provision of not in excess of 9.99%, provided that any increase in such percentage shall not be effective until the 61st day after suchless than 61 days’ prior written notice to the Company.
| F-11 |
In
During the event of our liquidation, dissolution, or winding up, each holder of Series D Preferred Stock will be entitled to receive the amount of cash, securities or other property to which such holder would be entitled to receive with respect to suchyears ended December 31, 2021 and 2020, shareholders converted and shares of Series D Preferred Stock if such shares had been converted to common stock immediately prior to such event (without giving effect for such purposes to the 4.99% or 9.99% beneficial ownership limitation, as applicable) subject to the preferential rights of holders of any class or series of the Company’s capital stock specifically ranking by its terms senior to the Preferred D stock as to distributions of assets upon such event, whether voluntarily or involuntarily.
Shares of Series D Preferred Stock are not entitled to receive any dividends, unlessinto and until specifically declared by the board of directors. However, holders of Series D Preferred Stock are entitled to receive dividends on shares of Series D Preferred Stock equal (on an as-if-converted-to-common-stock basis) to and in the same form as dividends actually paid on shares of the common stock when such dividends are specifically declared by the board of directors, except for stock dividends or distributions payable in shares of common stock, onrespectively, at a conversion rate of 1 to 1,000. No purchase was made in order to convert these shares.
Each share of Series C Preferred Stock is convertible into one share of common stock at any time at the option of the holders, provided that each holder would be prohibited from converting Series C Preferred Stock into shares of common stock orif, as a result of such conversion, any other common stock equivalents for which the conversion price will be adjusted. The Company is not obligated to redeem or repurchase any shares of Series D Preferred Stock. Shares of Series D Preferred Stock are not otherwise entitled to any redemption rights, or mandatory sinking fund or analogous fund provision.
The holderssuch holder, together with its affiliates, would own more than 9.99% of the Series D Preferred Stock have no voting rights, except as required by law. The Company may not alter or change adversely the powers, preferences and rights of the Series D Preferred Stock or amend the certificate of designation or amend its certificate of incorporation or bylaws in any manner that adversely affects any right of the holders of the Series D Preferred Stock without the affirmative vote of the holders of a majority of the shares of Series D Preferred Stock then outstanding.
The Company is obligated to deliver shares of common stock upon conversion of the Series D Preferred Stock (the “Conversion Shares”), within the time period specified in the certificate of designation. Failure to comply with the timely delivery requirement triggers certain liquidated damages payable by the Company to each of the Series D Preferred Stock holders.
NANOVIBRONIX INC. AND ITS SUBSIDIARY
If, at any time while the Series D Preferred Stock is outstanding, the Company completed a Fundamental Transaction (as defined in the certificate of designation), then upon any subsequent conversion of the Series D Preferred Stock, the holder will receive, for each Conversion Share that would have been issuable upon such conversion immediately prior to the occurrence of such Fundamental Transaction, thetotal number of shares of common stock of the successor or acquiring corporation or of the Company, if it is the surviving corporation,then issued and any additional cash, securities and/or other property or consideration (the “Alternate Consideration”) receivable by holders of common stock as a result of such Fundamental Transaction for each share of common stock for which this Series D Preferred Stock is convertible immediately prior to such Fundamental Transaction. For purposes of any such conversion, the determination of the Conversion Price shall be appropriately adjusted to apply to such Alternate Consideration based on the amount of Alternate Consideration issuable in respect of one share of common stock in such Fundamental Transaction. If holders of common stock are given any choice as to the securities, cash or property to be received in a Fundamental Transaction, then the Holder shall be given the same choice as to the Alternate Consideration it receives upon any conversion of this Series D Preferred Stock following such Fundamental Transaction. If such Fundamental Transaction is also a Change of Control Transaction in which the Company is not the surviving entity, then all shares of Series D Preferred Stock shall, upon consummation of such Change of Control Transaction, automatically be converted into Conversion Shares.
Since the Company has sufficient authorized and unissued shares available to settle its commitments and since all holders of equally (both preferred stock and common stock) would receive the same form of consideration upon the consummation of a Fundamental Transaction, and the shares are not otherwise redeemable, the shares of Series D Preferred Stock are classified within permanent equity, consistent with the guidance of ASC 480.
F-25
NANOVIBRONIX INC. AND ITS SUBSIDIARY
| ||
| ||
NANOVIBRONIX INC. AND ITS SUBSIDIARY
| Warrants outstanding as of December 31, | ||||||||||||||
| 2017 | 2016 | Exercise price | Expiration date | |||||||||||
| $ | ||||||||||||||
| November 2011 Warrants (1) | 245,893 | 331,293 | 1.23 | November 15, 2018 | ||||||||||
| February 2013 Warrants (2) | — | 563,910 | 2.66 | February 2018 through December 2019 | ||||||||||
| February 2015 Warrants (3) | 686,667 | 840,000 | 3.00/6.00 | February 30, 2019 | ||||||||||
| March 2015 Warrants (4) | — | 61,000 | 2.57 | March 25, 2020 | ||||||||||
| March through September 2017 Warrants (5) | 552,000 | — | 5.90 | May through September 2022 | ||||||||||
| November 2017 Warrants (6) | 1,250,687 | — | 6.9 | November 1, 2022 | ||||||||||
| Total outstanding | 2,735,247 | 1,796,203 | ||||||||||||
NANOVIBRONIX INC. AND ITS SUBSIDIARY
| ||
| ||
| ||
|
In November 2004, the Board of Directors of the Company adopted a stock option plan (“the Plan”), according to which optionsoutstanding. This limitation may be granted to employees, directors and consultants.
Pursuant to the Plan, the Company reserved for issuance 400,000 shares of Common stock. Each option entitles the holder to purchase one share of Common stock of the Company and expires after 10 years from the date of grant. Any options that are terminated, cancelled, forfeited or not exercised, become available for future grants.
NANOVIBRONIX INC. AND ITS SUBSIDIARY
In November 2014, 10 years after it was adopted, the Plan expired.
In February 2014, the Board of Directors of the Company adopted a new stock option plan (“the New Plan”), according to which options may be granted to employees, directors and consultants.
Pursuant to the New Plan, the Company reserved for issuance 714,286 shares of Common stock. Each option entitles the holder to purchase one share of Common stock of the Company and expires after 10 years from the date of grant. Any options that are terminated, cancelled, forfeited or not exercised, become available for future grants.
As of December 31, 2017, under the New Plan, no options were available for future grants.
In addition, the Company issued options to purchase 275,038 shares of Common Stock outside of the New Plan.
A summary of the Company’s options activity and related informationwaived with respect to options granteda holder upon such holder’s provision of not less than 61 days’ prior written notice to employees and directors duringthe Company.
During the years ended December 31, 2017 are as follows:2021 and 2020, shareholders converted and shares of Series C Preferred Stock into and shares of common stock, respectively, at a conversion rate of 1 to 1. No purchase was made in order to convert these shares.
| Number of options | Weighted average exercise price | Weighted average contractual life | Aggregate intrinsic value | |||||||||||||
| Outstanding - beginning of the year | 1,202,006 | $ | 3.75 | 8.18 | 3,419 | |||||||||||
| Granted | — | $ | — | |||||||||||||
| Exercised | — | $ | — | |||||||||||||
| Expired or Forfeited | (7,160 | ) | $ | 10.08 | ||||||||||||
| Outstanding - end of the year | 1,194,846 | $ | 3.03 | 7.18 | 3,419 | |||||||||||
| Exercisable at end of year | 851,139 | $ | 2.23 | 7.09 | 2,896 | |||||||||||
Stock-based compensation and options
During the years ended December 31, 2021 and 2020, employee options were exercised, and and options were granted, respectively. The options granted during 2021 and 2020 vest at different schedules ranging from date granted to years and were recorded at fair values of $ and $, respectively. During the years ended December 31, 2021 and 2020, stock-based compensation expense of $ and $ was recorded for options that vested, respectively. During the year ended December 31, 2021, options expired.
On November 2, 2020, the Company entered into an option cancellation and release agreement with some of its option holders, pursuant to which the parties agreed to cancel options to purchase an aggregate of shares of common stock of the Company at exercise prices ranging from $ to $ (the “Options”) previously granted to each of the Option holders. In exchange for the cancellation of the Options, the Company paid $1.00 to each Option holder.
Shares Under Options | Weighted Average Exercise Price per Share | Weighted Average Remaining Life (Years) | ||||||||||
| Outstanding – December 31, 2020 | 1,748,544 | $ | 1.59 | |||||||||
| Granted | 877,500 | 1.02 | ||||||||||
| Forfeited | (72,200 | ) | 3.99 | |||||||||
| Expired | (13,845 | ) | 1.23 | - | ||||||||
| Exercised | - | - | - | |||||||||
| Outstanding – December 31, 2021 | 2,539,999 | $ | 1.33 | |||||||||
NANOVIBRONIX INC. AND ITS SUBSIDIARY
Weighted averageThe fair value offor options granted to employeesin 2021 and directors during the years 2017 and 2016 was $ 0 and $ 3.34 per option, respectively.
Aggregate intrinsic value of exercised options by employees and directors during the years 2017 and 2016 was $ 0 and $ 22, respectively. The Aggregate intrinsic value of the exercised options represents the total intrinsic value (the difference between the sale price of the Company’s share2020 is estimated at the date of exercise, andgrant using a Black-Scholes-Merton options pricing model with the exercise price) multiplied by the number of options exercised.following underlying assumptions:
The aggregate intrinsic value in the table above represents the total intrinsic value (the difference between the Company’s closing share price on the last trading day of calendar 2017 and the exercise price, multiplied by the number of in-the-money options) that would have been received by the option holders had all option holders exercised their options on December 31, 2017. This amount is impacted by the changes in the fair market value of the Company’s shares.
As of December 31, 2017, the total unrecognized estimated compensation cost related to non-vested options granted prior to that date was $ 729 which is expected to be recognized over a weighted average period of approximately 2.02 years.
SCHEDULE OF FAIR VALUE ASSUMPTIONS FOR OPTIONS GRANTED
| Price at valuation | – | |||
| Exercise price | $ | – | ||
| Risk free interest | – | % | ||
| Expected term (in years) | ||||
| Volatility | % - | % |
The Company’s outstanding options granted to consultants as of December 31, 2017 are as follows:
| F-12 |
| Issuance date | Options for Common stock | Weighted Average exercise price per share | Options exercisable | Expiration date | ||||||||||
| April 2009 | 1,071 | $ | 10.35 | 1,071 | April 2019 | |||||||||
| December 2010 | 786 | $ | 1.99 | 786 | December 2020 | |||||||||
| March 2013 | 30,000 | $ | 1.96 | 30,000 | March 2023 | |||||||||
| October 2013 | 1,000 | $ | 1.96 | 1,000 | October 2023 | |||||||||
| February 2015 | 714 | $ | 1.96 | 714 | February 2025 | |||||||||
| Total | 33,571 | $ | 2.23 | 33,571 | ||||||||||
NANOVIBRONIX INC. AND ITS SUBSIDIARY
The total stock basedstock-based expense recognized in the financial statements for services received from employees and non-employees is shown in the following table (refer alsotable.
SCHEDULE OF STOCK BASED EXPENSES RECOGNIZED FOR SERVICES FROM EMPLOYEES AND NON-EMPLOYEES
| Year Ended | ||||||||
| December 31, | ||||||||
| 2021 | 2020 | |||||||
| Research and development | $ | 17 | $ | 9 | ||||
| Selling and marketing | 51 | 43 | ||||||
| General and administrative | 382 | 324 | ||||||
| Total | $ | 450 | $ | 376 | ||||
As of December 31, 2021, the total unrecognized estimated compensation cost related to Note 10d):non-vested stock options granted prior to that date was $, which is expected to be recognized over a weighted average period of approximately years.
| F-13 |
Year ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| Research and development | $ | 30 | $ | 30 | ||||
| Selling and marketing | 13 | 12 | ||||||
| General and administrative | 757 | 466 | ||||||
| Total | $ | 800 | $ | 508 | ||||
In connectionWarrants
During the year ended December 31, 2020, the Company granted 3,774,468 warrants to purchase Company’s common in conjunction with the resignationprivate placements and a seven-year equity warrant to purchase 100,000 shares of the Company’s common stock in conjunction with notes payable (see note 7). On December 17, 2020, 1,000,000 pre-funded warrants were exercised at $0.001 per share.
On December 2, 2020, we entered into a director from the board of directors, on March 30, 2017,Securities Purchase Agreement with certain institutional and accredited investors pursuant to which the Company amendedissued and sold to such investors in a private placement an aggregate of (i) shares of the option agreement, dated March 25, 2015, for the grantCompany’s common stock at an offering price of an option$ per share and (ii) pre-funded warrants to purchase 30,000up to shares of common stock at a purchase price of $ per pre-funded warrant, for gross proceeds of approximately $6.0 million, and net proceeds of approximately $5.4 million. In January 2021, two investors exercised an aggregate of 1,657,144 warrants at $0.001 per share.
On January 21, 2021, Company entered into letter agreements (the “Letter Agreements”) with certain existing accredited investors to exercise certain outstanding warrants (the “Existing Warrants”) to purchase up to an aggregate of 1,205,968 shares of the Company’s common stock at an exercise price of $2.57 per share all of which have vested,$1.165 (the “Exercise”). Certain of the Existing Warrants (the “Registered Existing Warrants”) and the option agreement, dated July 18, 2016, for the grant of an option to purchase 40,000 shares of common stock underlying the Registered Existing Warrants have been registered pursuant to a registration statement on Form S-3 (File No. 333-251264) and a registration statement on Form S-1 (File No. 333-218871). In consideration for the exercise of the Existing Warrants for cash, the exercising holders received new unregistered warrants to purchase up to an aggregate of 1,205,967 shares of common stock (the “New Warrants”) at an exercise price of $5.35$1.04 per share alland with an exercise period of seven years from the initial closing date. The gross proceeds to the Company from the Exercise were approximately $1.4 million.
The New Warrants were accounted for in warrant modification expense, which were vesting on July 18, 2017,was measured at the amount equal to (i) accelerate the vestingincremental value reflecting the change in the fair value of the optionwarrants before and after the Warrant Amendment. Accordingly, warrant modification expense in the amount of $1,627 was recorded with a corresponding increase in additional paid in capital.
In August and September 2021, investors exercised warrants to purchase 2,193,492 shares of common stock between $0.88 and $2.50 per share for proceeds of approximately $3.6 million.
In estimating the warrants’ fair value, the Company used the following assumptions:
SCHEDULE OF FAIR VALUE ASSUMPTIONS FOR WARRANTS
| Risk free interest | 1.44 | % | ||
| Dividend yield | 0 | % | ||
| Volatility | 55.6% - 56.5 | % | ||
| Contractual term (in years) | 2 |
SCHEDULE OF WARRANTS ACTIVITY
| Warrants | ||||
| Outstanding – December 31, 2019 | 4,850,272 | |||
| Granted | 3,874,468 | |||
| Exercised | (1,000,000 | ) | ||
| Expired | - | |||
| Outstanding – December 31, 2020 | 7,724,740 | |||
| Granted | 1,205,967 | |||
| Exercised | (5,056,603 | ) | ||
| Exercised - cashless | ) | |||
| Expired | (620,001 | ) | ||
| Canceled | (663,332 | ) | ||
| Outstanding – December 31, 2021 | 2,309,347 | |||
| F-14 |
NOTE 7 - NOTES PAYABLE
PPP Loan
In May 2020, the Company was granted a loan (the “PPP Loan”) in the amount of $42, pursuant to the directorPaycheck Protection Program (the “PPP”) under Division A, Title I of the Coronavirus Aid, Relief, and Economic Securities (“CARES”) Act, which was enacted March 27, 2020. The application for these funds required the Company to, in 2016 sogood faith, certify that itthe current economic uncertainty made the loan request necessary to support the ongoing operations of the Company. This certification further required the Company to consider its current business activity and its ability to access other sources of liquidity sufficient to support ongoing operations in a manner that is not significantly detrimental to the business. The Company made this good faith assertion based upon the adverse impact the COVID-19 pandemic had on its business and the global economy. While the Company has made this assertion in good faith based upon all available guidance, management will continue to assess their continued qualification if and when updated guidance is released by the Treasury Department. The receipt of these funds, and the forgiveness of the loan attendant to these funds, is dependent on the Company having initially qualified for the loan and qualifying for the forgiveness of such loan based on its future adherence to the forgiveness criteria.
The PPP Loan, which was in the form of a note that was granted on May 14, 2020, matures in two years and accrues interest at a rate of 1.00% per annum, payable in monthly payments commencing six months after loan disbursement. The Company also has the option to negotiate with the lender to extend the maturity date to up to five years. The note may be fully vested as of March 30, 2017, and (ii) permitprepaid by the director to exercise the options granted in 2015 and 2016Company at any time prior to maturity with no prepayment penalties. Funds from the expirationPPP Loan may only be used for payroll costs and any payments of certain covered interest, lease and utility payments. The Company has used the entire PPP Loan amount for qualifying expenses in the covered period. Under the terms of the option periodPPP, certain amounts of the PPP Loan may be forgiven if they are used for qualifying expenses as set forthdescribed in the CARES Act. The ultimate forgiveness of the PPP Loan is also predicated upon regulatory authorities concurring with management’s good faith assessment that the current economic uncertainty made the loan request necessary to support ongoing operations. If, despite the Company’s good-faith belief that given the circumstances the Company satisfied all eligibility requirements for the PPP Loan, the Company is later determined to have violated any applicable option agreement. This modification resultedlaws or regulations or it is otherwise determined that the Company was ineligible to receive the PPP Loan, the Company may be required to repay the PPP Loan in its entirety and/or be subject to additional penalties. In the event the PPP Loan, or any portion thereof, is forgiven, the amount forgiven is applied to outstanding principal. The Company was granted full forgiveness for the loan in the 4th quarter of 2020 and recorded a gain on forgiveness of $42.
| F-15 |
Unsecured Note
On June 22, 2020, the Company issued and sold to a related party an unsecured promissory note in the principal amount of $200, which accrues interest at 10% per annum and matures in one year. On August 28, 2020, the Company paid the note in full including $4 of accrued interest.
SCHEDULE OF NOTES PAYABLE
| Notes payable: | ||||
| Total carrying value of notes payable at December 31, 2019 | $ | - | ||
| Principal value of unsecured note issued during year ended December 31, 2020 | $ | 242 | ||
| Forgiveness of notes payable | (42 | ) | ||
| Payoff of unsecured note | (200 | ) | ||
| Total carrying value of notes payable at December 31, 2020 | $ | - | ||
In addition to the promissory note, the Company granted a seven-year equity warrant to purchase 100,000 shares of the Company’s common stock. The exercise price for each warrant share is equal to $2.50, and the warrants may also be exercised, in whole or in part, by means of a cashless exercise. The warrants were recognized as a debt discount and is amortized over the life of the note. The warrants were valued at $123 using a Black Scholes Merton pricing model with the following underlying assumptions:
SUMMARY OF QUANTITATIVE INFORMATION TO VALUATION METHODOLOGY AND UNOBSERVABLE INPUTS
| Price at valuation | $ | 2.21 | ||
| Exercise price | $ | 2.50 | ||
| Risk free interest | 0.34 | % | ||
| Expected term (in years) | 7 | |||
| Volatility | 60.7 | % |
NOTE 8 - DERIVATIVE LIABILITIES
During 2020, the Company established a sequencing policy to which common stock equivalents are exercisable to shares of common stock more than the Company’s authorized limit. It was determined that all options and warrants by the end of the year were no longer permitted to be classified as equity and were valued at fair market value using Black Scholes and recorded as derivative liabilities.
On April 6, 2021, the Company agreed to buy back 663,332 warrants from investors for a total of $368. The warrants had exercise prices between $0.88 and $0.94 per share. The value of the derivative liabilities associated with these warrants was $451. The Company recorded a $64 gain in connection with the buyback of the warrants.
A summary of quantitative information with respect to valuation methodology and significant unobservable inputs used for the Company’s purchase warrants that were categorized within Level 3 of the fair value hierarchy during the year ended December 31, 2021 is as follows:
SCHEDULE OF QUANTITATIVE INFORMATION TO VALUATION METHODOLOGY AND UNOBSERVABLE INPUTS
| Stock price | $ | - $ | ||
| Conversion price | $ | - $ | ||
| Contractual term (in years) | 0.67 – 6.56 | |||
| Volatility (annual) | 82.7% - 211 | % | ||
| Risk-free rate | 0.09% - 1.21 | % |
| F-16 |
The foregoing assumptions were reviewed quarterly and were subject to change based compensationprimarily on management’s assessment of the probability of the events described occurring.
Financial Liabilities Measured at Fair Value on a Recurring Basis
The fair value accounting standards define fair value as the amount that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants. As such, fair value is determined based upon assumptions that market participants would use in pricing an asset or liability. Fair value measurements are rated on a three-tier hierarchy as follows:
| ● | Level 1 inputs: Quoted prices (unadjusted) for identical assets or liabilities in active markets; |
| ● | Level 2 inputs: Inputs, other than quoted prices included in Level 1, that are observable either directly or indirectly; and |
| ● | Level 3 inputs: Unobservable inputs for which there is little or no market data, which require the reporting entity to develop its own assumptions. |
There were no transfers between Level 3 during the years ended December 31, 2021 and 2020.
The following table presents changes in Level 3 asset and liability measured at fair value for the years ended December 31, 2021 and 2020:
SCHEDULE OF CHANGES IN LEVEL 3 AND LIABILITY MEASURED AT FAIR VALUE
| As of December 31, 2021 | ||||||||
| Asset | Liability | |||||||
| Balance – December 31, 2019 | $ | - | $ | - | ||||
| New Issuances | 25 | 2,983 | ||||||
| Change in fair value of warrant liability | - | (512 | ) | |||||
| Balance – December 31, 2020 | $ | 25 | $ | 2,471 | ||||
| New Issuances | - | 1,819 | ||||||
| Fair value adjustments – Sanuwave warrants | (6 | ) | - | |||||
| Fair value adjustments – Warrant liability | - | 6,956 | ||||||
| Reclassification liability to equity | - | (10,793 | ) | |||||
| Buy back of warrants | - | (453 | ) | |||||
| Balance – December 31, 2021 | $ | 19 | $ | - | ||||
The following table sets forth the Company’s assets and liabilities which are measured at fair value on a recurring basis by level within the fair value hierarchy:
SCHEDULE OF FAIR VALUE COMPANY’S ASSETS AND LIABILITIES
| Fair Value Measurements as of December 31, 2021 | ||||||||||||||||
| Level I | Level II | Level III | Total | |||||||||||||
| Asset: | ||||||||||||||||
| Other assets | $ | - | $ | - | $ | 19 | $ | 19 | ||||||||
| Liability: | ||||||||||||||||
| Derivative liabilities | $ | - | $ | - | $ | - | $ | - | ||||||||
Note 9 – LEASES
The Company has operating lease agreements with terms up to 3 years, including car leases.
The Company’s weighted-average remaining lease term relating to its operating leases is 2.13 years, with a weighted-average discount rate of 10%.
The Company incurred $38 of lease expense for its operating leases for the year ended December 31, 2021.
The following table presents information about the amount and timing of $98liabilities arising from the Company’s operating leases as of December 31, 2021:
SCHEDULE OF LIABILITIES ARISING FROM OPERATING LEASES
| 2022 | 26 | |||
| 2023 | 25 | |||
| 2024 | 4 | |||
| Total undiscounted operating lease payments | 55 | |||
| Less: Imputed interest | 6 | |||
| Present value of operating lease liabilities | $ | 49 |
| F-17 |
Basic net loss per common share (“Basic EPS”) is computed by dividing net loss available to common shareholders by the weighted average number of common shares outstanding during the period. All outstanding share options and warrants for the years ended December 31, 2021 and 2020 have been excluded from the calculation of the diluted net loss per share because all such securities are anti-dilutive for all periods presented.
SUMMARY OF COMMON SHARE EQUIVALENTS BEEN EXCLUDED FROM DILUTIVE LOSS PER SHARE AS ANTI- DILUTIVE
| December 31, 2021 | December 31, 2020 | |||||||
| Series D Preferred Stock Shares | - | 153,000 | ||||||
| Series E Preferred Stock Shares | - | 875,000 | ||||||
| Stock Options - employee and non-employee | 2,539,999 | 1,748,544 | ||||||
| Warrants | 2,309,347 | 7,724,740 | ||||||
| Total | 4,849,346 | 10,501,284 | ||||||
The diluted loss per share equals basic loss per share in the year ended December 31, 2017.2021 and 2020 because the Company had a net loss and the impact of the assumed exercise of stock options and the vesting of restricted stock would have been anti-dilutive.
F-31
| F-18 |
NANOVIBRONIX INC.
NOTE 11 - GEOGRAPHIC INFORMATION AND ITS SUBSIDIARYMAJOR CUSTOMER DATA
Summary information about geographic areas:
The Company manages its business on the basis of one reportable segment and derives revenues from selling its products directly to patients as well as through distributor agreements. The following is a summary of revenues within geographic areas:
SUMMARY OF REVENUE WITHIN GEOGRAPHIC AREAS
| Year Ended December 31, | ||||||||
| 2021 | 2020 | |||||||
| United States | $ | 1,627 | $ | 467 | ||||
| Europe | 18 | 147 | ||||||
| Israel | 5 | 3 | ||||||
| India | - | 1 | ||||||
| Other | 45 | 5 | ||||||
| Total | $ | 1,695 | $ | 623 | ||||
The Company’s long-lived assets are all located in Israel.
NOTE 12– OTHER ASSETS
On April 9, 2020, pursuant to a licensing agreement entered into in March 2020, the Company received 10-year warrants to purchase 127,000 shares of Sanuwave Health, Inc. at a price of $0.19 per share. The fair value for warrants received is estimated at the date of grant using a Black-Scholes-Merton pricing model with the following underlying assumptions:
SCHEDULE OF WARRANTS ASSUMPTIONS
| Price at valuation | $ | – | ||
| Exercise price | $ | |||
| Risk free interest | 0.66 - 0.73 | % | ||
| Expected term (in years) | 10 | |||
| Volatility | 140.6 - 143.9 | % |
The Company considers this to be level 3 inputs and is valued at each reporting period. The fair value of these warrants for the year ended December 31, 2021 was $18. There was a net $6 change in fair value during the year ended December 31, 2021.
NOTE 13 - COMMITMENTS AND CONTINGENCIES
Pending and settled litigation
On December 17, 2019, a lawsuit was filed by a former officer and director, Jona Zumeris, in the Haifa Israel District Financial Court, seeking damages of approximately $900 for breach of the Separation Agreement executed on July 4, 2018. The Israeli court issued a court order demanding that we restrict approximately $700 of the Company’s money until the matter is adjudicated. The Company appealed the court order and in February 2020, the Company agreed to restrict approximately 1,187 NIS (“New Israeli Shekel”) and agreed to try to settle the matter in mediation. On November 30, 2020, the Company funded the escrow account with $391. In January 2021, the parties reached a settlement in which the Company paid the plaintiff approximately $366 as settlement in full.
| F-19 |
On February 26, 2021, Protrade Systems, Inc. (“Protrade”) filed a Request for Arbitration (the “Request”) with the International Court of Arbitration (the “ICA”) of the International Chamber of Commerce alleging the Company is in breach of an Exclusive Distribution Agreement dated March 7, 2019 (the “Agreement”) between Protrade and the Company. Protrade alleges, in part, that the Company has breached the Agreement by discontinuing the manufacture of the DV0057 Painshield MD device in favor of an updated 10-100-001 Painshield MD device. Protrade claims damages estimated at $3 million.
On March 15, 2022, the arbitrator issued a final award, which, although finding that Protrade’s claims failed as a matter of law or were unsupported by the evidence, nevertheless awarded Protrade $1,500,250, which consists of $1,432,000 for “lost profits” and $68,250 as reimbursement of arbitration costs, on the grounds that the Company allegedly failed to supply Protrade with “requested patches.” The Company continues to dispute the claims asserted by Protrade and intend to pursue the available options to vacate or seek correction of the award to Protrade.
Other Risks
On March 12, 2020, the World Health Organization declared COVID-19 to be a pandemic, and the COVID-19 pandemic has resulted in significant financial market volatility and uncertainty. A continuation or worsening of the levels of market disruption and volatility seen in the recent past could have an adverse effect on our ability to access capital, on our business, results of operations and financial condition, and on the market price of our common shares.
NOTE 14 – INCOME TAXES
As of December 31, 2021, the U.S. Company had federal and pre-apportioned state net operating loss carry forward for tax purposes of approximately $30,010. $16,056of the federal net operating loss can be carried forward indefinitely but is limited to 80% utilization and $13,959 of the federal net operating loss can be offset against taxable income for 20 years. State net operating losses can also be carried forward for 20 years. Utilization of the U.S. net operating losses may be subject to substantial limitations in the event of a change of ownership under the provisions of the Internal Revenue Code of 1986. The Company has not performed an analysis but that the potential impact of any limitation would not be material to the financial statements since the respective DTAs are fully offset by a valuation allowance.
Income tax expense is comprised of the following:
SCHEDULE OF PROVISION FOR INCOME TAXES EXPENSES
| Year ended December 31, | ||||||||
| 2021 | 2020 | |||||||
| Current Tax | ||||||||
| Federal | ||||||||
| State | $ | - | $ | - | ||||
| Foreign | 32 | 15 | ||||||
| Total | $ | 32 | $ | 15 | ||||
| Deferred Tax | ||||||||
| Federal | $ | (1,263 | ) | $ | (584 | ) | ||
| State | (131 | ) | (5 | ) | ||||
| Foreign | $ | (4 | ) | 2 | ||||
| Total | $ | (1,398 | ) | $ | (587 | ) | ||
| Less: Valuation Allowance | 1,398 | 587 | ||||||
| Total Tax | $ | 32 | $ | 15 | ||||
The difference between the statutory tax rate of the Company and the effective tax rate is primarily the result of tax benefits generated by the Company and its subsidiary which have not been recognized due to the uncertainty that such tax benefits will ultimately be realized. A reconciliation of the statutory U.S Federal rate to the Company’s effective tax rate is as follows:
SCHEDULE OF RECONCILIATION OF STATUTORY U.S. FEDERAL RATE
| Year ended December 31, | ||||||||
| 2021 | 2020 | |||||||
| Federal income tax benefit at statutory rate | 21.00 | % | 21.00 | % | ||||
| State income taxes, net of federal benefit | 0.92 | % | 0.12 | % | ||||
| Foreign rate differential | 0.02 | % | -0.08 | % | ||||
| Permanent Items | -13.04 | % | 2.22 | % | ||||
| Change in valuation allowance | -9.81 | % | -13.63 | % | ||||
| Return to provision adjustments | -0.01 | % | -4.03 | % | ||||
| Forfeited options | -0.16 | % | -7.33 | % | ||||
| Other | 0.86 | % | -1.39 | % | ||||
| Effective tax rate | -0.22 | % | -0.34 | % | ||||
|
NANOVIBRONIX INC. AND ITS SUBSIDIARY
Foreign tax
| Tax rates applicable to the income of |
In December 2016, the Israeli Parliament approved the Economic Efficiency Law (Legislative Amendments for Applying the Economic Policy for the 2017 and 2018 Budget Years), 2016 which reduces thesubsidiary:
The Israeli corporate income tax rate to 24% (instead of 25%) effective from January 1, 2017in 2021 and to 23% effective from January 1, 2018.2020 is 23%.
| Year ended December 31, | |||||||||
| 2017 | 2016 | ||||||||
| Domestic | $ | 4,930 | $ | 2,738 | |||||
| Foreign | (3 | ) | (24 | ) | |||||
| $ | 4,927 | $ | 2,714 | ||||||
The subsidiary has final tax assessments through 2016.
Loss before taxes:
SCHEDULE OF INCOME BEFORE TAXES ON DOMESTIC AND FOREIGN
| Year ended December 31, | ||||||||
| 2021 | 2020 | |||||||
| Domestic | $ | 14,333 | $ | 4,380 | ||||
| Foreign | (82 | ) | (69 | ) | ||||
| Loss before taxes | $ | 14,250 | $ | 4,311 | ||||
Deferred income taxes
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial purposes and the amounts used for income tax purposes. Significant components of the Company’s deferred tax assets are as follows:
SCHEDULE OF DEFERRED TAX ASSETS
| December 31, | Year ended December 31, | |||||||||||||||
| 2017 | 2016 | 2021 | 2020 | |||||||||||||
| Deferred tax assets: | ||||||||||||||||
| Net operating loss carry forward | $ | 2,722 | $ | 3,894 | $ | 6,563 | $ | 5,712 | ||||||||
| Temporary differences | 35 | 34 | ||||||||||||||
| Arbitration accrual | 414 | - | ||||||||||||||
| Stock compensation and other | 327 | 175 | ||||||||||||||
| Deferred tax assets before valuation allowance | 2,757 | 3,928 | 7,304 | 5,887 | ||||||||||||
| Valuation allowance | (2,757 | ) | (3,928 | ) | (7,304 | ) | (5,887 | ) | ||||||||
| Net deferred tax asset | $ | — | $ | — | $ | - | $ | - | ||||||||
For the year ended December 31, 2021 and 2020, the net increases in valuation allowance of $1,417 and $596, respectively was primarily driven by the increase in net operating loss carryforwards.
In assessing the realization of deferred tax assets, management considers whether it is more likely than not that all or some portion of the deferred tax assets will not be realized.
The ultimate realization of the deferred tax assets is dependent upon the generation of future taxable income during the periods in which temporary differences are deductible and net operating losses are able to be utilized. Based on consideration of these factors, the Company concluded that all of its recorded deferred tax assets are not more likely than not realizable and recorded a full valuation allowance at December 31, 20172021 and 2016.2020.
The Company considers the earnings of its non-U.S. subsidiary to be indefinitely invested outside the United States on the basis of estimates that future domestic cash generation will be sufficient to meet future domestic cash needs and our specific plans for reinvestment of those subsidiary earnings. We have not recorded a deferred tax liability related to the U.S. federal and state income taxes as an estimate of undistributed earnings of foreign subsidiaries would not be practicable to estimate at this time. If the Company does decide to repatriate the foreign earnings, we would need to adjust our income tax provision in the period we determined that the earnings will no longer be indefinitely invested outside the United States.
| F-21 |
Reconciliation of the theoretical tax expense to the actual tax expense
The main reconciling items between the statutory tax rate of the Company and the effective tax rate are the non-recognition of tax benefits from accumulated net operating loss carryforward among the Company and its subsidiary due to the uncertainty of the realization of such tax benefits.
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Balance at beginning of the year | $ | 170 | $ | 97 | ||||
| Increases related to tax positions from prior years | 17 | 73 | ||||||
| Lapses of statutes of limitation | (19 | ) | — | |||||
| Balance at the end of the year | $ | 168 | $ | 170 | ||||
The Company recognizes interest and penalties related to unrecognized tax benefits in tax expense. During the year ended December 31, 2017,2021, the Company accrued $15$0 for interest and penalties expenses related to uncertain tax positions.
NANOVIBRONIX INC. AND ITS SUBSIDIARYU.S. federal and New York State income taxes are open for examination for years 2018-2021 and Israel tax returns are open for examination for years 2018-2021.
NOTE 15 - SUBSEQUENT EVENTS
| Year ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| Interest on promissory notes | $ | 37 | $ | — | ||||
| Discount amortization of promissory notes | 1,197 | — | ||||||
| Change in fair value of warrants | 549 | 383 | ||||||
| Other financial expense | 53 | 15 | ||||||
| $ | 1,836 | $ | 398 | |||||
Summary information about geographic areas:
ASC 280, “Segment Reporting,” establishes standards for reporting information about operating segments. Operating segments are defined as componentsOn March 2, 2022, the Company received a letter from the Listing Qualifications Department of an enterprise about which separate financial information is availablethe Nasdaq Stock Market (“Nasdaq”) indicating that, is evaluated regularly bybased upon the chief operating decision maker in deciding how to allocate resources and in assessing performance. The Company manages its business on the basis of one reportable segment, and derives revenues from selling its products mainly through distributor agreements. The following is a summary of revenues within geographic areas:
| Year ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| United States | $ | 90 | $ | 89 | ||||
| Israel | 8 | 13 | ||||||
| United Kingdom | 74 | 22 | ||||||
| European Union (excluding United Kingdom) | 14 | 30 | ||||||
| India | 14 | 24 | ||||||
| Other | 39 | 51 | ||||||
| $ | 239 | $ | 229 | |||||
During the year ended December 31, 2017, there were sales to two distributors each of which accounted for approximately 11% of total sales. During the year ended December 31, 2016, there were no sales to a single customer exceeding 10%closing bid price of the Company’s revenues.common stock for the 30 consecutive business day period between January 14, 2022, through March 1, 2022, the Company did not meet the minimum bid price of $ per share required for continued listing on The Nasdaq Capital Market pursuant to Nasdaq Listing Rule 5550(a)(2). The letter also indicated that the Company will be provided with a compliance period of 180 calendar days, or until August 29, 2022, in which to regain compliance pursuant to Nasdaq Listing Rule 5810(c)(3)(A).
On February 26, 2021, Protrade Systems, Inc. (“Protrade”) filed a Request for Arbitration (the “Request”) with the International Court of Arbitration (the “ICA”) of the International Chamber of Commerce alleging the Company is in breach of an Exclusive Distribution Agreement dated March 7, 2019 (the “Agreement”) between Protrade and the Company. Protrade alleges, in part, that the Company has breached the Agreement by discontinuing the manufacture of the DV0057 Painshield MD device in favor of an updated 10-100-001 Painshield MD device. Protrade claims damages estimated at $3 million.
On March 15, 2022, the arbitrator issued a final award, which, although finding that Protrade’s claims failed as a matter of law or were unsupported by the evidence, nevertheless awarded Protrade $1,500,250, which consists of $1,432,000 for “lost profits” and $68,250 as reimbursement of arbitration costs, on the grounds that the Company allegedly failed to supply Protrade with “requested patches.” The Company’s long-lived assets are all located in Israel.Company continues to dispute the claims asserted by Protrade and intends to pursue the available options to vacate or seek correction of the award to Protrade.As of December 31, 2021, the Company accrued the amount of the award to Protrade amounting to $1,500,250 as part of “General and administrative expenses” and “Other accounts payable and accrued expenses”.
F-34
| F-22 |
NANOVIBRONIX INC. AND ITS SUBSIDIARY
Balances with related parties:
| Year ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| Warrants to purchase Common stock (a) | $ | — | $ | 2,079 | ||||
Related parties’ expenses:
| Year ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| Financial expenses (a) | $ | 549 | $ | 383 | ||||
Index to Exhibits
| 65 |
| 66 |
| 67 |
| 68 |
| 69 |
| 101* | The following materials from the Company’s Annual Report on Form 10-K for the year ended December 31, | |
| 104 | Cover Page Interactive Data File (embedded within the Inline XBRL document) |
| * | Filed herewith. | |
| ** | Furnished herewith. |
| + | Management contract or compensatory plan or arrangement. | |
| # | Certain portions of this exhibit have been redacted pursuant to Item 601(b)(10) of Regulation S-K. The omitted information is (i) not material and (ii) would likely cause competitive harm to the Company if publicly disclosed. | |
| 70 |
SIGNATURES
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| NANOVIBRONIX, INC. | ||
| By: | /s/ Brian Murphy | |
| Brian Murphy | ||
Chief Executive Officer | ||
Date: March 29, 2018April 14, 2022
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Brian Murphy as his true and lawful attorneys-in-fact and agents, with full power of substitution and re-substitution, for him and in his name, place and stead, in any and all capacities, to sign any and all amendments to this Form 10-K, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the SEC, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming that all said attorneys-in-fact and agents, or any of them or their or his substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
| Signature | Title | Date | ||
| /s/ | Chief Executive Officer and Director | |||
| Brian Murphy | (principal executive officer) | |||
| /s/ STEPHEN BROWN | Chief Financial Officer, | |||
| Stephen Brown | (principal financial and accounting officer) | |||
| /s/ | Chairman of the Board of Directors | |||
| Christopher Fashek | ||||
| /s/ MARTIN GOLDSTEIN | Director | |||
| Martin Goldstein | ||||
| /s/ HAROLD JACOB M.D. | Director | |||
| Harold Jacob, M.D. | ||||
| /s/ MICHAEL FERGUSON | Director | |||
| Michael Ferguson | ||||
| /s/ THOMAS R. MIKA | Director | |||
| Thomas R. Mika |
| /s/ Aurora Cassirer | Director | April 14, 2022 | ||
| Aurora Cassirer | ||||
| /s/ Maria Schroeder | Director | April 14, 2022 | ||
| Maria Schroeder |
| 71 |
62