UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K10-K/A
Amendment No. 1
| | | | | |
| ☒ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 20212023
OR
| | | | | |
| ☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 FOR THE TRANSITION PERIOD FROM TO |
Commission File Number 001-37449
ALPINE IMMUNE SCIENCES, INC.
(Exact name of Registrant as specified in its Charter)
| | | | | | | | |
| Delaware | 20-8969493 |
(State or other jurisdiction of
incorporation or organization) | (I.R.S. Employer
Identification No.) |
| | | |
| 188 East Blaine Street | Suite 200 | 98102 |
| Seattle, | WA |
| (Address of principal executive offices) | (Zip Code) |
Registrant’s telephone number, including area code: (206) 788-4545
Securities registered pursuant to Section 12(b) of the Act:
| | | | | | | | |
| Title of each class | Trading Symbol | Name of each exchange on which registered |
| Common Stock, par value $0.001 per share | ALPN | The Nasdaq Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐☒ No ☒☐
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the Registrantregistrant has submitted electronically every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| | | | | | | | | | | | | | | | | | | | |
Large accelerated filerAccelerated Filer | | ☐ | | Accelerated filerFiler | | ☐ |
| | | | | | | |
Non-accelerated filerFiler | | ☒ | | Smaller reporting companyReporting Company | | ☒ |
| | | | | | | |
| | | | Emerging growth companyGrowth Company | | ☐ | | | | |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report.☐
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
The aggregate market value of the voting and non-voting common equity held by non-affiliates of the registrant, based on the closing price of the shares of common stock on the Nasdaq Stock Market on June 30, 2021,2023, was approximately $104.8$349.1 million. Shares of common stock held by each executive officer and director and by each other person who may be deemed to be an affiliate of the registrant, have been excluded from this computation. The determination of affiliate status for this purpose is not necessarily a conclusive determination for other purposes.
The number of shares of the registrant’s common stock outstanding as of March 7, 2022April 9, 2024 was 30,294,434.65,560,484.
DOCUMENTS INCORPORATED BY REFERENCE
None.
| | | | | | | | | | | | | | | | | | | | | | | |
| Auditor Name: | Ernst & Young LLP | | Auditor Location: | Seattle, Washington | | Auditor Firm PCAOB ID: | 42 |
Portions ofEXPLANATORY NOTE
This Amendment No. 1 on Form 10-K/A (this “Amendment”), amends Alpine Immune Sciences, Inc.’s (the “Company,” “Alpine,” “we,” “us,” and “our”) Annual Report on Form 10-K for the Registrant’s Definitive Proxy Statement relating to the 2022 Annual Meeting of Stockholders, which will beyear ended December 31, 2023, originally filed with the Securities and Exchange Commission (the “SEC”) on March 20, 2024 (the “Original Filing”). We are amending and refiling Part III to include information required by Items 10, 11, 12, 13 and 14 because our definitive proxy statement will not be filed within 120 days after December 31, 2023, the end of the fiscal year covered by our Annual Report on Form 10-K.
Pursuant to the rules of the SEC, we have also included as exhibits currently dated certifications required under Section 302 of The Sarbanes-Oxley Act of 2002. Because no financial statements are contained within this Amendment, we are not including certifications pursuant to Section 906 of The Sarbanes-Oxley Act of 2002. We are amending and refiling Part IV to reflect the inclusion of those certifications, along with any changes to Part IV that occurred after the date of the Original Filing.
In addition, we made certain revisions to the cover page, including the deletion of the reference to our proxy statement and the amendment to our Annual Report on Form 10-K and inclusion of updated outstanding share information. Except as described above, no other changes have been made to the Original Filing.
Except as otherwise indicated herein, this Amendment continues to speak as of the date of the Original Filing, and we have not updated the disclosures contained therein to reflect any events that occurred subsequent to the date hereof, are incorporated by reference intoof the Original Filing. The filing of this Amendment is not a representation that any statements contained in items of our annual report on Form 10-K other than Part III, of this Report. Such Proxy Statement will be filed withItems 10 through 14, Part IV and the Securities and Exchange Commission not later than 120 days following the endaforementioned portions of the Registrant’s fiscal year ended December 31, 2021.cover page are true or complete as of any date subsequent to the Original Filing.
Table of Contents
In this report, unless otherwise stated or as the context otherwise requires, references to “Alpine,” “the Company,” “we,” “us,” “our” and similar references refer to Alpine Immune Sciences, Inc. All rights reserved.and its subsidiaries. “VIGD,” “NEON-1," "NEON-2," "Synergy,"SIP” and the Alpine logo“TIP” are registered trademarks orand “DENALI,” “NEON-1,” “NEON-2,” “RAINIER,” “RUBY,” “SYNERGY” and the Company logo are trademarks of Alpine Immune Sciences, Inc. in various jurisdictions.All rights reserved. This report also contains registered marks, trademarks, and trade names of other companies. All other trademarks, registered marks, and trade names appearing in this report are the property of their respective holders.
Summary Risk FactorsPART III
Our business is subject to numerous risks
Item 10. Directors, Executive Officers and uncertainties, including those highlighted in the section of this report captioned “Risk Factors.” Corporate Governance.
BOARD OF DIRECTORS
The following is a summarytable sets forth the names and certain other information for each of the principal risks we face:members of our board of directors as of April 9, 2024.
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Age | | Position | | Director Since(7) | | Current
Term
Expires |
| Robert Conway(2)(5) | | 70 | | Director | | 2015 | | 2024 |
| James N. Topper, M.D., Ph.D.(6) | | 62 | | Director | | 2017 | | 2024 |
| Christopher Peetz(4) | | 45 | | Director | | 2018 | | 2024 |
| Peter Thompson, M.D.(1)(3) | | 64 | | Director | | 2017 | | 2025 |
| Natasha Hernday(4) | | 52 | | Director | | 2020 | | 2025 |
| Mitchell H. Gold, M.D. | | 57 | | Executive Chairman and Chief Executive Officer | | 2017 | | 2026 |
| Xiangmin Cui, Ph.D.(1)(2) | | 55 | | Director | | 2019 | | 2026 |
| Jörn Drappa, M.D., Ph.D. | | 59 | | Director | | 2022 | | 2026 |
•(1)Our approach toMember of the discovery and development of innovative therapeutic treatments based on our technology is unproven and may not result in marketable products.compensation committee.
•(2)Our therapeutic candidates are in early stagesMember of developmentthe nominating and may fail in development or suffer delays that materially and adversely affect their commercial viability.corporate governance committee.
•(3)Product development involves a lengthyChairman of the nominating and expensive process with an uncertain outcome, and results of earlier preclinical and clinical trials may not be predictive of future clinical trial results.corporate governance committee.
•(4)We face competition from entities that have developed or may develop therapeutic candidates for our target disease indications, including companies developing novel treatments and technology platforms based on modalities and technology similar to us. If these companies develop technologies or therapeutic candidates more rapidly than we do, or their technologies, including delivery technologies, are more effective, our ability to develop and successfully commercialize therapeutic candidates may be adversely affected.Member of the audit committee.
•(5)To date, our revenue has been primarily derived from our collaboration agreements, and our success will be dependent, in part, on our collaborators’ efforts to develop our therapeutic candidates.Chairman of the audit committee.
•(6)If third parties on which we depend to conduct our clinical or preclinical studies, or any future clinical trials, do not perform as expected, fail to satisfy regulatory or legal requirements, or miss expected deadlines, our development program could be delayed, which may result in materially adverse effects on our business, financial condition, resultsChairman of operations, and prospects.the compensation committee.
•(7)We may not successfully engageRepresents first year of service on the board of directors of Nivalis Therapeutics, Inc. (which we refer to as Nivalis), renamed as Alpine Immune Sciences, Inc. (which we refer to as Alpine) in strategic transactions, including any additional collaborations we seek, which could adversely affect our ability to develop and commercialize therapeutic candidates, impact our cash position, increase our expenses, and present significant distractions to our management.
•If any of our therapeutic candidates are approved for marketing and commercialization and we are unable to develop sales, marketing and distribution capabilities on our own or enter into agreements with third parties to perform these functions on acceptable terms, we may be unable to successfully commercialize any such future products.
•The COVID-19 coronavirus could adversely impact our business, including our clinical trials.
•We depend on our information technology systems and any failure of these systems could harm our business.
•We will need to raise substantial additional funds to advance development of our therapeutic candidates, and we cannot guarantee we will have sufficient funds available in the future to develop and commercialize our current or future therapeutic candidates.
•We are an early stage biopharmaceutical company with a history of losses, we expect to continue to incur significant loses for the foreseeable future, we may never achieve or maintain profitability, and we have a limited operating history that may make it difficult for investors to evaluate the potential success of our business.
•If we are not able to obtain and enforce patent protection for our technology, including therapeutic candidates, therapeutic products, and platform technology, development of our therapeutic candidates and platform, and commercialization of our therapeutic products may be materially and adversely affected.
•We may license patent rights from third-party owners or licensees. If such owners or licensees do not properly or successfully obtain, maintain or enforce the patents underlying such licenses, or if they retain or license to others any competing rights, our competitive position and business prospects may be materially and adversely affected.
•We or our licensors, collaborators, or any future strategic partners may become subject to third-party claims or litigation alleging infringement of patents or other proprietary rights or seeking to invalidate patents or other proprietary rights, and we may need to resort to litigation to protect or enforce our patents or other proprietary rights, all of which could be costly, time consuming, delay or prevent the development of our therapeutic candidates and commercialization of our therapeutic products, or put our patents and other proprietary rights at risk.
•If we fail to comply with our obligations under any license, collaboration, or other agreements, we may be required to pay damages and could lose intellectual property rights necessary for developing and protecting our technology, including our platform technology, therapeutic candidates, and therapeutic products, or we could loseJuly 2017.
certain rightsNatasha Hernday has served as a member of our board of directors since December 2020. From November 2022 to grant sublicenses, eitherDecember 2023, Ms. Hernday served as Chief Business Officer and Executive Committee member of which could haveSeagen Inc., an oncology company that was acquired by Pfizer in December 2023, where she previously served as Executive Vice President, Corporate Development from October 2020 to November 2022, Senior Vice President of Corporate Development from September 2017 to October 2020 and as Vice President, Corporate Development from January 2011 to September 2017. She also serves on the board of directors of Xoma Corp. (Nasdaq: XOMA) and on the Knight Campus External Advisory Board for the University of Oregon. Ms. Hernday received her B.A. in Microbiology from the University of California at Santa Barbara and M.B.A. from Pepperdine University.
We believe that Ms. Hernday’s experience and expertise in the biotechnology and the pharmaceuticals industry including exploratory research, corporate development, and corporate strategy, as well as her record of successful asset licensing, collaborations, and M&A provide her with the qualifications necessary to serve as a material adverse effectmember of our board of directors.
Peter Thompson, M.D. has served as a member of our board of directors since the completion of the merger of Nivalis and Alpine in July 2017 and previously served as a member of the board of directors of Alpine since June 2016. Dr. Thompson is a Member at OrbiMed Advisors LLC, an investment firm focused on our resultsthe healthcare sector, where he has also served as venture partner since joining in September 2010. Dr. Thompson is a co-founder of operations and business prospects.
•We may be unable to obtain U.S. or foreign regulatory approvalhas served as a member of the board of directors of Corvus Pharmaceuticals, Inc. (Nasdaq: CRVS) since December 2014. Dr. Thompson served as a director of Silverback Therapeutics, Inc. (Nasdaq: SBTX) from April 2016 until Silverback's acquisition by ARS Pharmaceuticals Inc. (Nasdaq: SPRY) in November 2022, and has since continued as a director of ARS; Dr. Thompson has also served as a director of Decibel Therapeutics, Inc. (Nasdaq: DBTX) since November 2020 and as a result, may be unabledirector of PMV Pharmaceuticals, Inc. (Nasdaq; PMVP) since November 2014. Dr. Thompson also served as a member of the board of directors of Prevail Therapeutics Inc. (Nasdaq: PRVL) from October 2017 until January 2021, and as a member of the board of directors of Synthorx, Inc. (Nasdaq: THOR) from April 2018 to commercialize our therapeutic candidates.January 2020. He also currently serves on the boards of directors of several private companies. Dr. Thompson is a board-certified internist and oncologist and has served as Affiliate Professor of Neurosurgery at the University of Washington since 2010. Dr. Thompson previously held executive positions at Chiron Corporation and Becton Dickinson and served on the faculty of the National Cancer Institute following his medical staff fellowship there. Dr. Thompson holds a Sc. B. in Molecular Biology and Mathematics from Brown University and an M.D. from Brown University Medical School.
•The healthcare industry is heavily regulatedWe believe that Dr. Thompson’s venture capital and management experience in the U.S. atpharmaceuticals industry provides him with the federal, state,qualifications and local levels,skills necessary to serve as a member of our board of directors.
Mitchell H. Gold, M.D. has served as our executive chairman, chief executive officer and a member of our failure to comply with applicable requirements may subject us to penalties and negatively affect our financial condition.
•Our stock price may be volatile, and an active, liquid, and orderly trading market may not develop for our common stock. As a result, stockholders may not be able to resell shares at or above their purchase price.
•Our officers andboard of directors and their respective affiliates, have a controlling influence over our business affairs and may make business decisions with which stockholders disagree and which may adversely affectsince the value of their investment.
Our Risk Factors are not guarantees that no such conditions exist ascompletion of the datemerger of this reportNivalis and should not be interpretedAlpine in July 2017 and, prior to the merger, served as Alpine’s chief executive officer since June 2016 and as Alpine’s executive chairman and member of Alpine’s board of directors since January 2015. Prior to co-founding Alpine, Dr. Gold was Chairman and Founder of Alpine Biosciences, a privately-held biotech company, from 2012 to 2014. Dr. Gold is currently a Managing Partner at Alpine BioVentures, a venture capital firm. Dr. Gold holds an affirmative statementM.D. from Rush Medical College of Rush University Medical Center and a B.S. in Biology from the University of Wisconsin.
We believe that such risks or conditions have not materialized,Dr. Gold possesses specific attributes that qualify him to serve as a member of our board of directors, including more than 20 years of experience in whole orsenior executive management roles with both early stage and public biopharmaceutical companies.
Xiangmin Cui, Ph.D. has served as a member of our board of directors since January 2019. Dr. Cui has served as managing director of Decheng Capital, an investment firm focused on life sciences companies, since he founded the firm in part.2011. Prior to founding Decheng, Dr. Cui was an investment partner at Bay City Capital, an international life science venture capital firm in San Francisco. Dr. Cui was previously director of strategic investment for the Southern Research Institute, a not-for-profit research organization. Prior to that, Dr. Cui co-founded Pan Pacific Pharmaceuticals and Hucon Biopharmaceuticals, where he led efforts in discovery and development of several key technologies in the fields of oncology, cardiology, and infectious and inflammatory diseases. Dr. Cui served as a member of the board of directors of Cue Health (Nasdaq: HLTH) from June 2020 to July 2022 and also currently serves on the boards of directors of several private companies. Dr. Cui holds a Ph.D. in Cancer Biology from Stanford University and a B.S. and M.S. in Molecular Biology from Peking University.
We believe that Dr. Cui’s venture capital and management experience in the pharmaceuticals industry provides him with the qualifications and skills necessary to serve as a member of our board of directors. Dr. Cui was appointed to our board of directors pursuant to the terms of the securities purchase agreement relating to our January 2019 private placement.
Forward-Looking StatementsJörn Drappa, M.D., Ph.D. has served as a member of our board of directors since July 2022. Dr. Drappa has served as the Chief Medical Officer of Alumis Inc. since August 2022. Previously, Dr. Drappa served as Chief Medical Officer of Ventyx Biosciences from September 2021 to May 2022. Prior to Ventyx, Dr. Drappa co-founded Viela Bio, a biotechnology company focused on the discovery, development and commercialization of treatments for autoimmune and severe inflammatory diseases in March 2018 and served as its Head of R&D and Chief Medical Officer through its acquisition by Horizon Therapeutics in March 2021. At Viela Bio, Dr. Drappa led the development of UPLIZNA™ (inebilizumab-cdon) in multiple indications, culminating in its approval for the treatment of neuromyelitis optica spectrum disorder. Dr. Drappa received his M.D. and Ph.D. degrees from the University of Cologne in Germany. He completed a residency in internal medicine at New York Presbyterian Hospital and a fellowship in rheumatology at the Hospital for Special Surgery, NY.
This Annual Report on Form 10-K contains forward-looking statementsWe believe that Dr. Drappa's depth of experience leading the successful development of immunology drugs across both small biotech and information withinlarge pharmaceutical companies, and from early stage through commercialization, proven track record as a biotech executive and career as a board-certified internist and rheumatologist provide him with the meaningqualifications and skills necessary to serve as a member of Section 27Aour board of directors.
Robert Conway has served as a member of our board of directors since the completion of the Securities Actmerger of 1933,Nivalis and Alpine in July 2017 and previously served as amended, and Section 21Ea member of the Securities Exchange Actboard of 1934,directors of Nivalis since April 2015. Since 2013, Mr. Conway has served on the board of directors of ARCA BioPharma (Nasdaq: ABIO), a publicly traded biopharmaceutical company, and was elected Chairman in June 2014. Mr. Conway is also a member of the board of directors of Signant Health, a private clinical technology company, and was a member of the board of another private clinical technology company, Advarra, Inc. until August 2022, when Advarra was sold to Blackstone. In addition, Mr. Conway is a member of the strategic advisory committee of Genstar Capital. Mr. Conway received a B.S. in Accounting from Marquette University in 1976.
We believe that Mr. Conway’s experience and expertise in the pharmaceutical industry, pharmaceutical development and clinical trials, and corporate finance, governance, accounting and public company compliance give him the qualifications and skills to serve on our board of directors.
James N. Topper, M.D., Ph.D. has served as amended, which are subjecta member of our board of directors since the completion of the merger of Nivalis and Alpine in July 2017 and, prior to the “safe harbor” created by those sections. In some cases you can identify these statements by forward-looking words suchmerger, served as “believe,” “may,” “will,” “estimate,” “continue,” “anticipate,” “intend,” “could,” “would,” “project,” “plan,” “expect,” or similar expressions, ora member of the negative or pluralboard of these words or expressions. You should read these statements carefully because they discuss future expectations, contain projectionsdirectors of future resultsAlpine since June 2016. Dr. Topper has been a partner with Frazier Life Sciences since August 2003, serving as general partner since 2005. Dr. Topper currently serves as a member of operations or financial condition, or statethe boards of directors of Phantom Pharmaceuticals, Inc. (Nasdaq: PHAT), and NewAmsterdam Pharma Company NV (Nasdaq: NAMS), and has served on numerous other “forward-looking” information. These statements relateboards of directors, including Allena Pharmaceuticals (Nasdaq: ALNA), Aptinyx, Inc. (Nasdaq: APTX), Entasis Therapeutics Holdings Inc. (Nasdaq: ETTX), Sierra Oncology, Inc. (formerly ProNai) (Nasdaq: SRRA), Amicus Therapeutics, Inc. (Nasdaq: FOLD), Portola Pharmaceuticals, Inc. (Nasdaq: PTLA), and La Jolla Pharmaceutical Company (Nasdaq: LJPC). Dr. Topper received his M.D. and Ph.D. in Biophysics from Stanford University and his B.S. in Biology from the University of Michigan.
We believe that Dr. Topper’s experience overseeing Frazier Life Sciences’ investments in biotechnology, his experience in senior management positions, and his significant knowledge of industry, medical and scientific matters, provide Dr. Topper with the qualifications and skills to serve on our future plans, objectives, expectations, intentions and financial performance and the assumptions that underlie these statements. These forward-looking statements include, but are not limited to:board of directors.
•Christopher Peetz has served as a member of our abilityboard of directors since April 2018. Mr. Peetz has been the president and chief executive officer of Mirum Pharmaceuticals, Inc. since March 2019 and president since November 2018. He has served as an entrepreneur-in-residence at Frazier Healthcare Partners since May 2017. He served as the chief executive officer of Flashlight Therapeutics, Inc. from May 2017 until December 2019. Mr. Peetz received an M.B.A. from Stanford Graduate School of Business and a B.S.B.A. in Finance, International Business and French from Washington University in St. Louis.
We believe Mr. Peetz’ experience in senior management positions in both business and finance and his experience supporting various corporate and financing transactions provide him with the qualifications and skills to identify, develop and commercialize additional products or product candidates;serve on our board of directors.
•our estimates regarding our expenses, revenues, anticipated capital requirements and our needs for additional financing;
•our ability to obtain funding for our operations;
•
the implementation of our business model and strategic plans for our business and technology;
•the timing of the commencement, progress and receipt of data from any of our preclinical and clinical trials;
•the expected results of any preclinical or clinical trial and the impact on the likelihood or timing of any regulatory approval;
•the scope of protection we are able to establish and maintain for intellectual property rights covering our technology and product candidates;
•the anticipated impact of the COVID-19 pandemic on our business, research and clinical development plans and timelines and results of operations;
•the timing or likelihood of regulatory filings and approvals;
•the therapeutic benefits, effectiveness and safety of our product candidates;
•the rate and degree of market acceptance and clinical utility of any future products;
•our ability to maintain and establish collaborations;
•our ability to achieve milestones in our current and any future collaborations;
•our expectations regarding market risk, including interest rate changes;
•our expectations regarding the sufficiency of our cash and cash equivalents to fund operations for at least the next 12 months;
•developments relating to our competitors and our industry; and
•our expectations regarding licensing, acquisitions and strategic operations.
These forward-looking statements are subject to certain risks and uncertainties that could cause actual results to differ materially from those anticipated in the forward-looking statements. Factors that might cause such a difference include, but are not limited to, those discussed in this report in Part I, Item 1A. Risk Factors, and elsewhere in this report. Forward-looking statements are based on our management’s beliefs and assumptions and on information currently available to our management. These statements, like all statements in this report, speak only as of their date, and we undertake no obligation to update or revise these statements in light of future developments, except as required by law. In addition, statements that “we believe” and similar statements reflect our beliefs and opinions on the relevant subject. These statements are based upon information available to us as of the date of this report, and while we believe such information forms a reasonable basis for such statements, such information may be limited or incomplete, and our statements should not be read to indicate that we have conducted an exhaustive inquiry into, or review of, all potentially available relevant information. These statements are inherently uncertain and investors are cautioned not to unduly rely upon these statements.
PART IEXECUTIVE OFFICERS
The names of our executive officers, their ages, their positions, and other biographical information as of April 9, 2024, are set forth below. Executive officers are appointed by our board of directors to hold office until their successors are elected and qualified. There are no family relationships among our directors or executive officers.
| | | | | | | | | | | | | | |
| Name | | Age | | Position |
| Mitchell H. Gold, M.D. | | 57 | | | Executive Chairman and Chief Executive Officer |
| Stanford Peng, M.D., Ph.D. | | 53 | | | President and Head of Research and Development |
| Paul Rickey | | 45 | | | Senior Vice President, Chief Financial Officer and Corporate Secretary |
| Remy Durand, Ph.D. | | 39 | | | Chief Business Officer |
Item 1. Business
OverviewStanford Peng, M.D., Ph.D. has served as our president and head of research and development since April 2019, prior to which he served as our executive vice president of research and development and chief medical officer since the completion of the merger of Nivalis and Alpine in July 2017, as Alpine’s chief medical officer from September 2016 to February 2017 and as Alpine’s executive vice president of research and development and chief medical officer from February 2017 until completion of the merger. Dr. Peng received an M.D. and Ph.D. in Biology from the Yale University School of Medicine and a B.A. in Music and B.S. in Biological Sciences from Stanford University.
Paul Rickey has served as our senior vice president and chief financial officer since the completion of the merger of Nivalis and Alpine in July 2017, and prior to the merger, served as Alpine’s senior vice president and chief financial officer since April 2017. Mr. Rickey has served as our treasurer since December 2018, and our secretary since March 2019. Mr. Rickey graduated from the University of Washington with a B.A. and Masters in Professional Accounting and is a certified public accountant, inactive.
Remy Durand, Ph.D. has served as our chief business officer since January 2021, prior to which he served as our senior vice president of business development and corporate strategy from January 2020 to December 2020 and as vice president of business development from October 2018 to December 2019. Dr. Durand has also served as principal on the investment team at Alpine BioVentures since October 2018. Prior to joining Alpine, Dr. Durand served as vice president on the investment team at Frazier Healthcare Partners from February 2015 to August 2018. Previously, Dr. Durand served as senior associate at GE Ventures Healthcare, a corporate venture capital practice within General Electric, from 2013 to 2015, and as a senior life sciences specialist at L.E.K. Consulting, a global strategy consulting firm, from 2011 to 2013. Dr. Durand received a Ph.D. and M.S. in Bioengineering from Stanford University and a B.S. in Biomedical Engineering from Case Western Reserve University.
GOVERNANCE
Corporate Governance Guidelines
Our board of directors has adopted a set of guidelines that establish the corporate governance policies pursuant to which our board of directors intends to conduct its oversight of our business and affairs in accordance with its fiduciary responsibilities. These guidelines address, among other items, the responsibilities of our directors, the structure and composition of our board of directors and corporate governance policies and standards applicable to us in general. Our corporate governance guidelines are posted on the Corporate Governance portion of our website at http://ir.alpineimmunesciences.com/governance.
Code of Business Conduct and Ethics
We are a clinical-stage biopharmaceutical company dedicated to discovering and developing innovative, protein-based immunotherapies to treat cancer and autoimmune and inflammatory diseases. Our approach includes a proprietary scientific platform that converts native immune system proteins into differentiated, multi-targeted therapeutics. We believe our strategies are capable of meaningfully modulating the human immune system and significantly improving outcomes in patients with serious diseases.
Autoimmune/Inflammatory Diseases
ALPN-101, or acazicolcept, is a dual Inducible T cell Costimulator, or ICOS, and CD28 antagonist intended for the treatment of autoimmune and inflammatory diseases. Preclinical studies with acazicolcept have demonstrated efficacy in models of systemic lupus erythematosus, or SLE, Sjögren’s syndrome, or SjS, arthritis, inflammatory bowel disease, multiple sclerosis, type 1 diabetes, uveitis, and graft versus host disease. We have evaluated acazicolcept in a Phase 1 healthy volunteer study and initiated patient dosing in Synergy, a global, randomized, double-blind, placebo-controlled Phase 2 study of acazicolcept in adults with moderate-to-severe SLE. In June 2020, we entered into an Option and License Agreement with AbbVie Ireland Unlimited Company, or AbbVie, which grants AbbVie an exclusive option to take an exclusive license to acazicolcept. Through December 31, 2021, we have received $105.0 million in upfront and pre-option exercise development milestones as part of the Option and License Agreement with AbbVie, or the AbbVie Agreement.
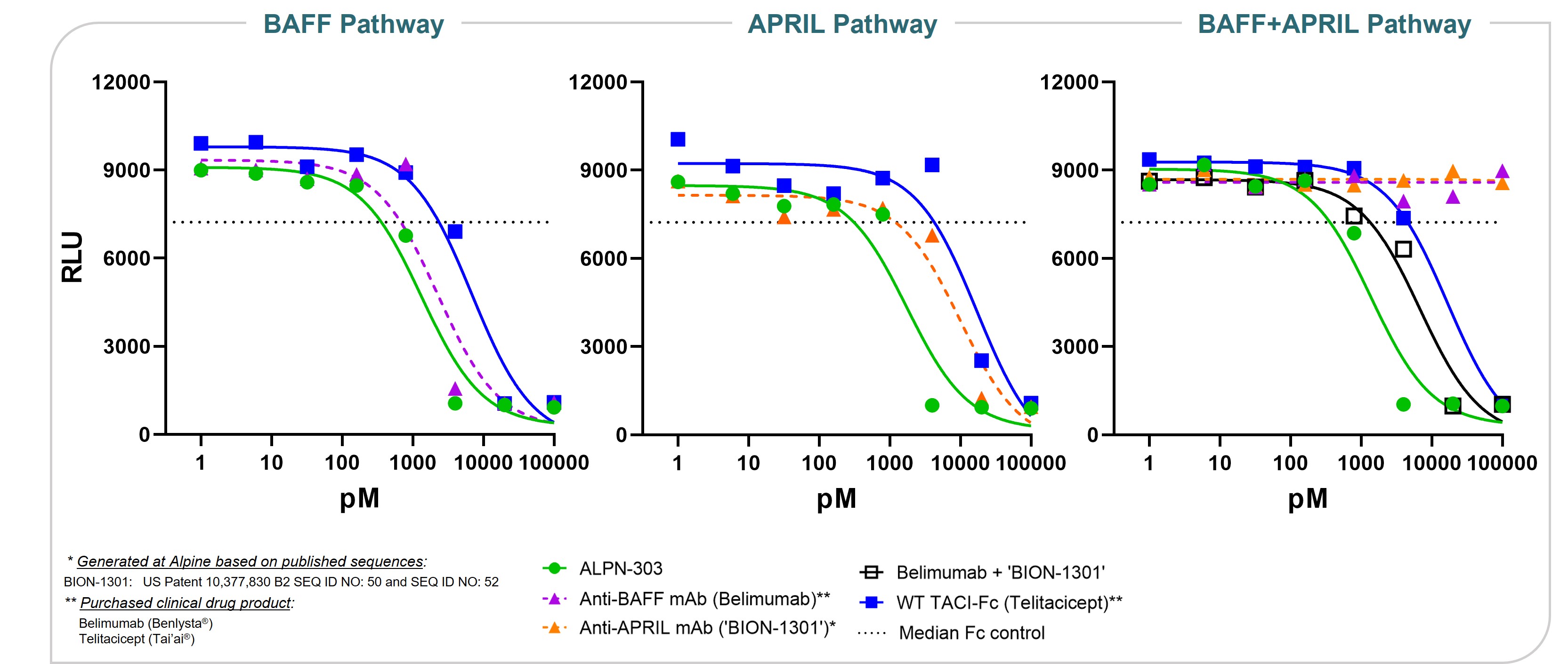
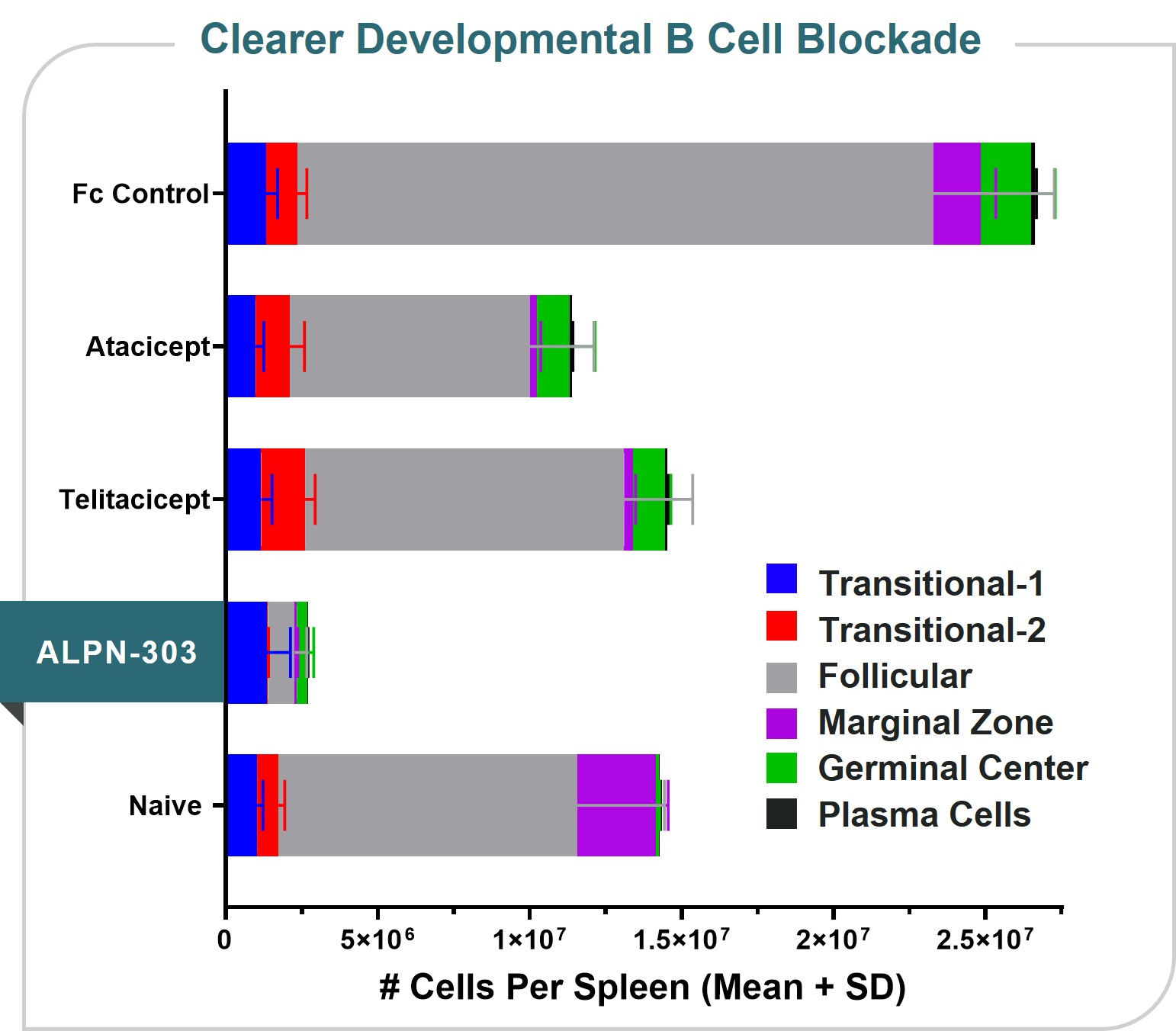
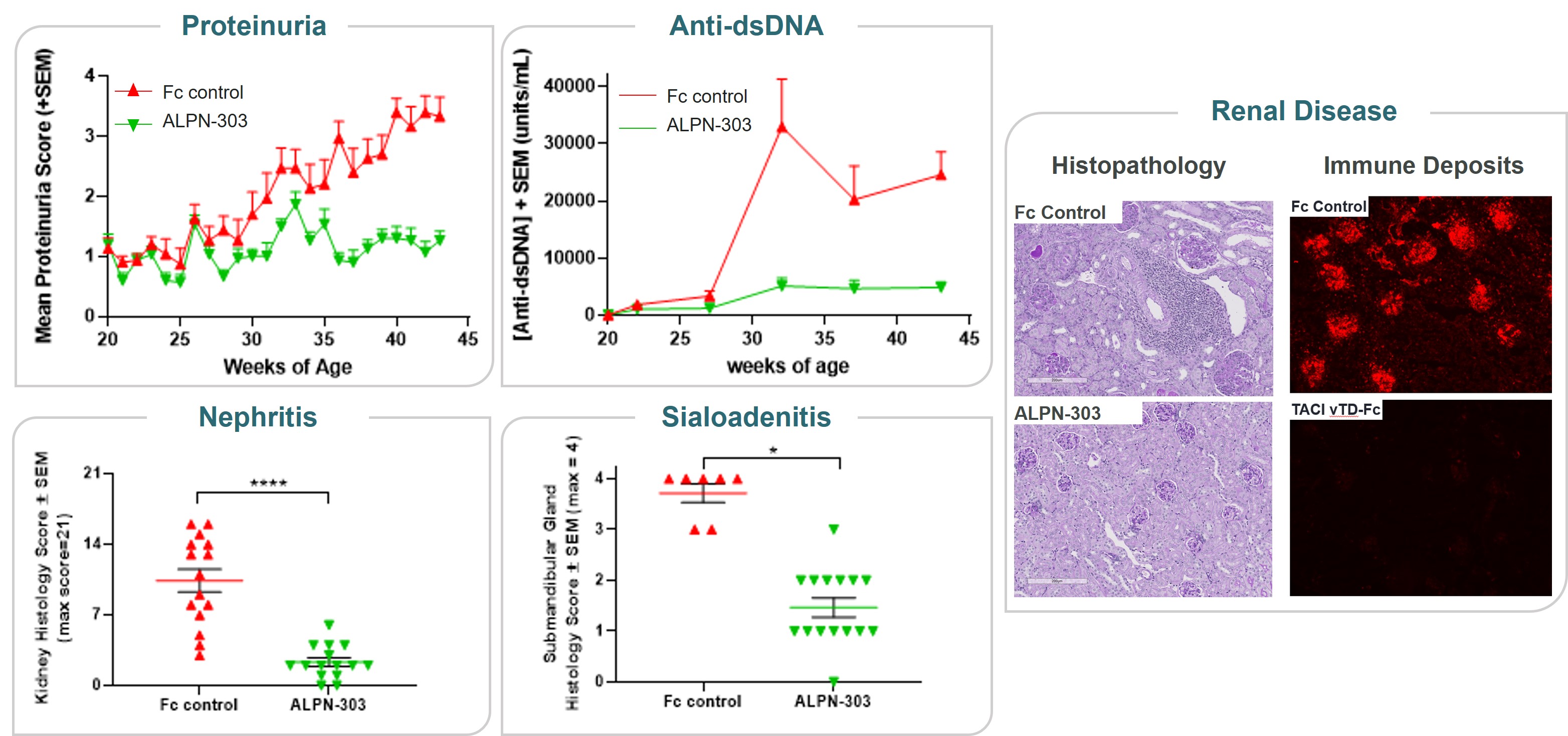
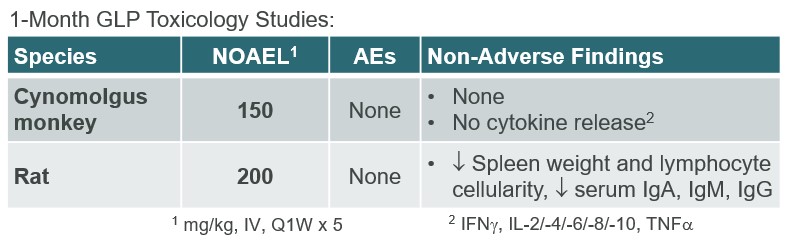
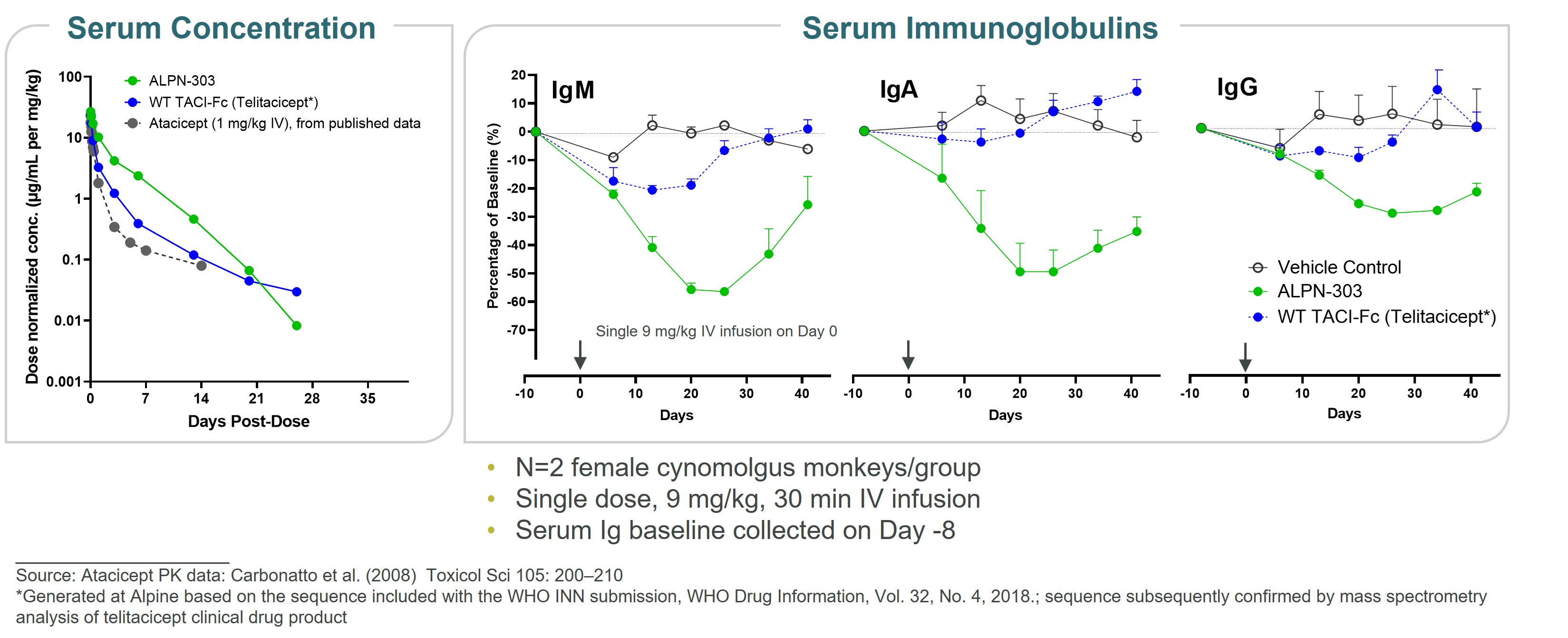
ALPN-303 is a dual B cell cytokine antagonist, being developed for the treatment of B cell mediated inflammatory and autoimmune diseases. Engineered using our proprietary directed evolution platform, ALPN-303 potently inhibits the pleiotropic B cell cytokines B cell activating factor (BAFF, BLyS) and a proliferation inducing ligand (APRIL), which play key roles in B cell development, differentiation, and survival, and together contributecommitted to the pathogenesishighest standards of multiple autoimmune diseases such as SLEintegrity and manyethics in the way we conduct our business. Our board of directors has adopted a Code of Business Conduct and Ethics, which applies to all of our employees, officers and directors, including our chief executive officer, chief financial officer, and other autoantibody-related inflammatory diseases. Data presented atexecutive and senior financial officers. Our Code of Business Conduct and Ethics establishes our policies and expectations with respect to a wide range of business conduct, including preparation and maintenance of financial and accounting information, compliance with laws and conflicts of interest. In accordance with our Code of Business Conduct and Ethics, each of our employees, officers and directors is required to report suspected or actual violations to the American College of Rheumatology (ACR) Convergence 2021 Annual Meeting demonstrated that ALPN-303 inhibits the activity of the B cell cytokines APRIL and BAFF more potently than wild-type TACI-Fc counterparts, as well as anti-BAFF and anti-APRIL monoclonal antibodies.extent permitted by law. In addition, ALPN-303our board of directors has been well-tolerated in preclinical modelsadopted separate policies and exhibited superior pharmacokineticsprocedures concerning the receipt and pharmacodynamics over wild-type TACI-Fc counterparts, including superior serum exposure, suppressioninvestigation of T-dependent antibody production, and/or serum immunoglobulins in mice and/or cynomolgus monkeys. A first-in-human, Phase 1 study of ALPN-303 in healthy volunteers was initiated in the fourth quarter of 2021, with top-line results targeted by mid-2022. This randomized, placebo-controlled study is designedcomplaints relating to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of ALPN-303 administered intravenously and subcutaneously. We are planning to initiate patient-based studies in the second half of 2022 including SLE, and possibly other autoantibody-related diseases in other therapeutic areas.
In December 2021, we entered into an exclusive license and collaboration agreement,accounting, internal accounting controls or the Horizon Agreement, with Horizon Therapeutics Ireland DAC, or Horizon, for the development and commercialization of up to four preclinical candidates generated from our unique discovery platform for autoimmune and inflammatory diseases. Under the terms of the agreement, Horizon made an upfront payment to us of $25.0 million as well as an equity investment for which they paid $15.0 million, a 25 percent premium to the 30-day volume-weighted average share price as of December 9, 2021. In addition, we are eligible to receive up to $381.0 million per program, or approximately $1.5 billion in total, in future success-based payments related to development, regulatory and commercial milestones as well as tiered royalties on global net sales.
Immuno-oncology
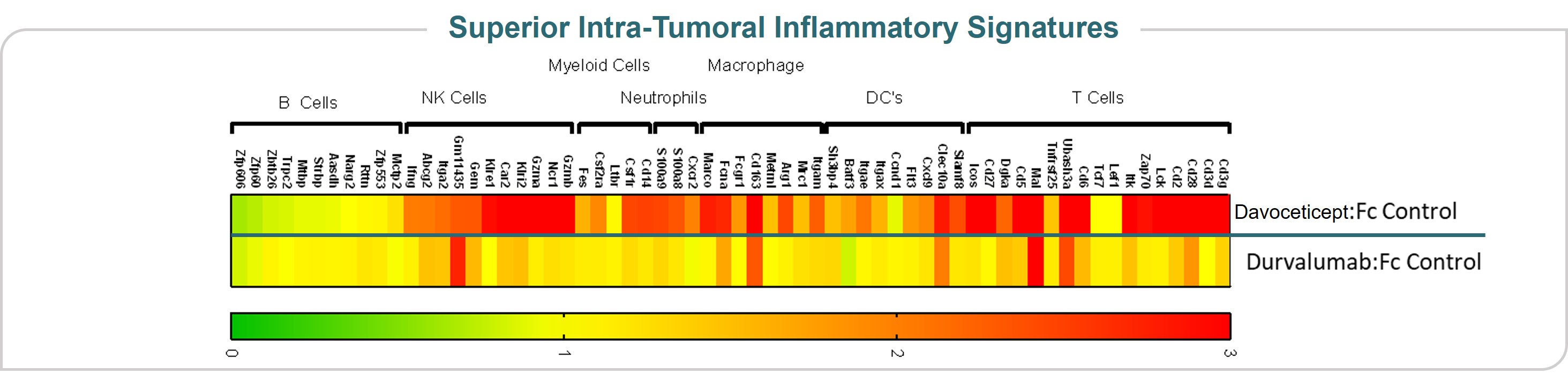
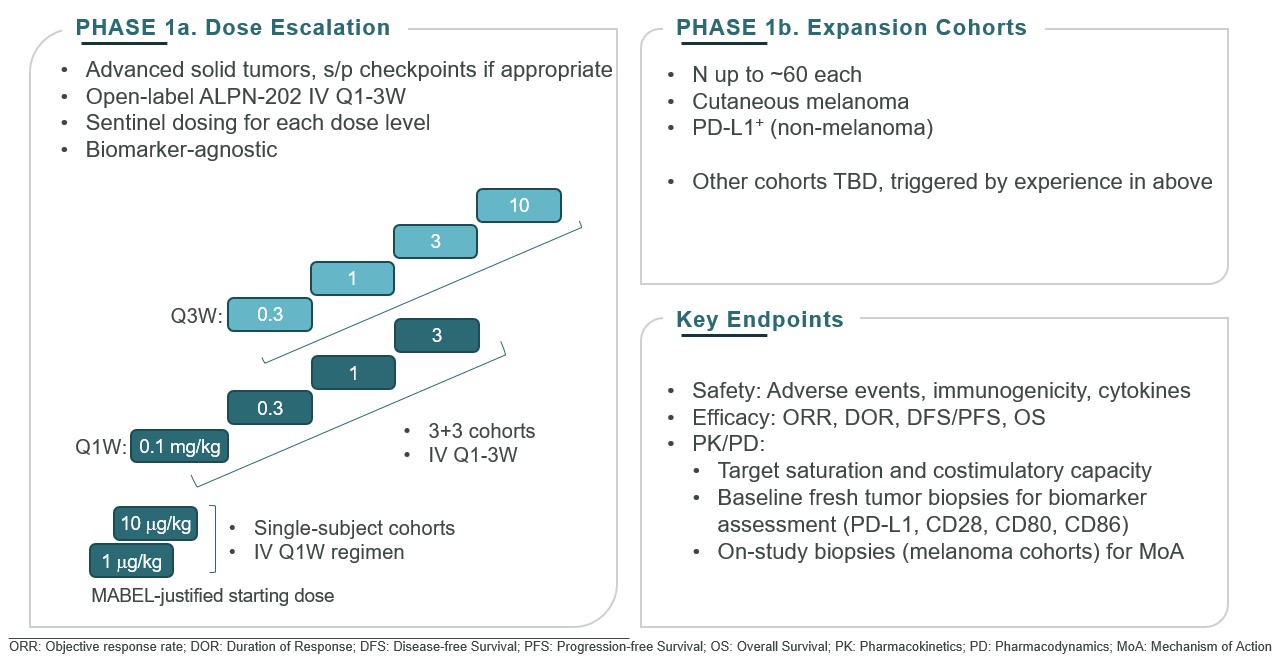
Our lead oncology program is ALPN-202, or davoceticept, a conditional CD28 costimulator and dual checkpoint inhibitor intended for the treatment of cancer. Preclinical in vivo data have demonstrated monotherapy efficacy in tumor models superior to approved therapies. In June 2020, we initiated NEON-1, a Phase 1 monotherapy dose escalation and expansion study in patients with advanced malignancies. Initial data from NEON-1 were presented at the 2021 American Society of Clinical Oncology (ASCO) Virtual Meeting demonstrating that davoceticept was well-tolerated as of the cutoff date (April 22, 2021) with evidence of peripheral T cell modulation consistent with CD28 agonism. In addition, although most enrolled participants had tumors considered classically non-responsive to immunotherapies, the majority derived clinical benefit as defined as a best outcome of stable disease or better. Completion of dose escalation for NEON-1 and initiation of expansion cohorts is anticipated in the first half of 2022.
In June 2021, we announced a collaborationauditing matters, which are administered by our audit committee. Our Code of Business Conduct and supply agreement with Merck to evaluate the safety and efficacy of davoceticept in combination with Merck’s anti-PD-1 therapy KEYTRUDA® (pembrolizumab) in a Phase 1 dose escalation and expansion study. The clinical trial, NEON-2, was initiated in June 2021andEthics is currently subject to a partial clinical hold. As discussed further below, on March 7, 2022, we announced that the FDA placed a partial clinical holdposted on the NEON-2 trial in responseCorporate Governance portion of our website at http://ir.alpineimmunesciences.com/governance. We will post amendments to our Code of Business Conduct and Ethics or waivers of our Code of Business Conduct and Ethics for directors and executive officers on the same website.
Audit Committee
The responsibilities of the audit committee include, but are not limited to, the death of a study participant in the NEON-2 trial. The participant’s death was attributedfollowing:
•meeting with our independent auditors, our management team and such other personnel as it deems appropriate to cardiogenic shock, considered by the treating physicians as likely related to immune-mediated myocarditis, or possibly infection. Participants who are currently enrolled in the NEON-2 trial may continue to receive davoceticeptconduct and pembrolizumab, although no new participants may be enrolled until the partial clinical hold is resolved. This partial clinical hold does not affect the ongoing NEON-1 clinical trial of davoceticept as monotherapy (NCT04186637).
Our scientific platform has also generated immune modulatory proteinsassist with the potential of improving engineered cellular therapies such as chimeric antigen receptor T cells, T cell receptor-engineered T cells, and tumor infiltrating lymphocytes. In May 2019, we signed a collaboration and license agreement with Adaptimmune Therapeutics plc, or Adaptimmune, to develop next-generation SPEAR™ T cell products which incorporate our secreted and transmembrane immunomodulatory protein (termed SIP™ and TIP™) technology. We intend to continue to leverage our existing pipeline and platform to actively explore and evaluate potential value-creating partnering opportunities.
Our Strategy
Our goal is to discover and develop modern therapies to treat patients with serious conditions such as cancer and autoimmune/inflammatory diseases. To achieve our goals, we intend to:certain audit committee functions;
•aggressively moveoverseeing our lead autoimmune/inflammatory program acazicolcept through clinical development as partaccounting and financial reporting processes and audits of the AbbVie Agreement, including conducting Synergy, our Phase 2 study for the treatment of SLE;its financial statements;
•aggressively movedeciding whether to appoint, retain or terminate our second autoimmune/inflammatory program ALPN-303 through a Phase 1 study in healthy volunteersindependent auditors, including the sole authority to approve all audit engagement fees and into patient-based studies forterms and to pre-approve all audit and permitted non-audit and tax services to be provided by the treatment of B cell mediated autoimmune/inflammatory diseases;independent auditors;
•aggressively movereviewing and discussing with management and our lead oncology program davoceticept through clinical development forindependent auditors the treatmentfinancial statements, including certain disclosures, addressing any issues encountered in the course of cancer, and;the audit work, and evaluating the performance of our independent auditors;
•maximizediscussing with management our earnings press releases, financial information and any earnings guidance provided to analysts and ratings agencies;
•discussing with management and the valueinternal auditors (if any) our disclosure controls, internal accounting and financial controls and accounting policies and practices;
•discussing with management any outsourcing of the internal audit function (if any), including selection of vendor, fees paid and areas to be audited;
•establishing procedures for the receipt, retention and treatment of complaints received by us regarding certain accounting or audit matters;
•establishing policies governing the hiring by us of any current or former employee of our pipelineindependent auditors;
•reviewing our compliance with applicable laws and platform viaregulations and to review and oversee our policies and procedures designed to promote and monitor regulatory compliance;
•obtaining assurance from the independent auditors that the audit of the financial statements was conducted in a manner consistent with Section 10A of the Exchange Act;
•reviewing, approving and overseeing transactions between us and any related person and any other potential partnering activities.conflict of interest situation;
•administering our Whistleblower and Non-Retaliation Policy and responding to and resolving related complaints or concerns;
•overseeing portions of our Code of Business Conduct and Ethics as designated by our board of directors;
•providing our board of directors with the results of its monitoring and recommendations derived from its responsibilities;
•reviewing and approving our investment policy;
•providing the independent and internal auditors with access to the board of directors; and
•producing the report required to be prepared for inclusion in our annual proxy statement.
Since December 2020, the audit committee has consisted of three directors: Messrs. Conway (chairman) and Peetz and Ms. Hernday. Our board of directors has determined that Mr. Conway is an “audit committee financial expert” as defined in the SEC rules and made a qualitative assessment of Mr. Conway’s level of knowledge and experience based on several factors, including his prior experience, business acumen and independence. Our board of directors has concluded that the composition of the audit committee meets the requirements for independence under the rules and regulations of Nasdaq and the SEC.
The audit committee met four times during 2023. The audit committee also meets periodically with our outside auditors without management present, at such times as it deems appropriate. Our board of directors has adopted a written charter for the
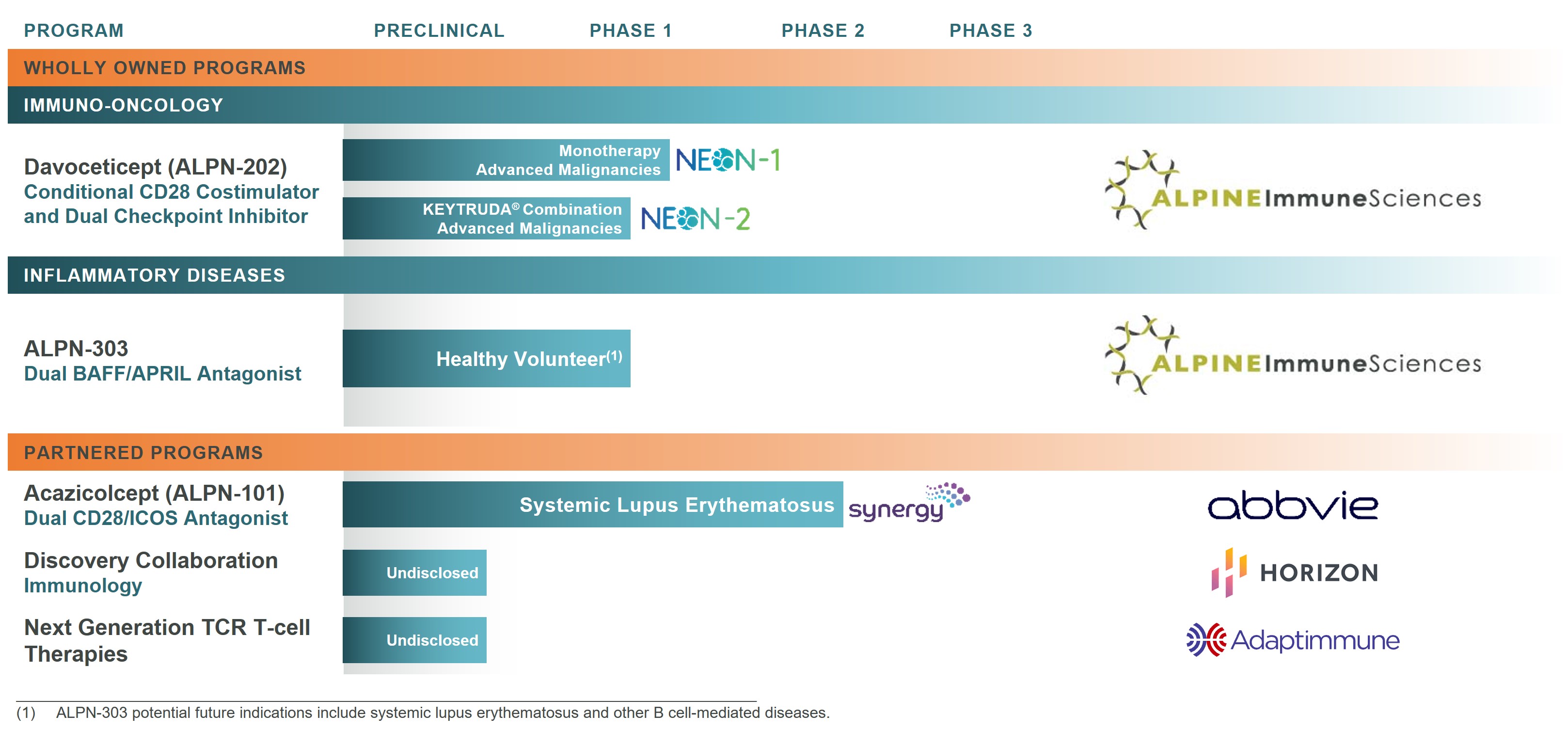
Product Pipelineaudit committee in compliance with the applicable rules of the SEC and the Nasdaq listing standards and which is available on our website at https://ir.alpineimmunesciences.com/governance.
Item 11. Executive Compensation.
Overview
Our compensation committee is responsible for the executive compensation programs for our executive officers and reports to our board of directors on its discussions, decisions and other actions. The compensation committee is authorized to retain the services of one or more executive compensation advisors, as it sees fit, in connection with the establishment of our compensation programs and related policies. Beginning in 2017, the compensation committee retained AON Radford, or AON, a compensation consultant, to provide it with information, recommendations and other advice relating to executive compensation on an ongoing basis. Accordingly, AON now serves at the discretion of the compensation committee. The compensation committee engaged AON to assist in developing a group of peer companies to help us determine the appropriate level of overall compensation for our executive officers, as well as assess each separate element of compensation, with a goal of ensuring that the compensation we offer to our executive officers is competitive and fair.
Summary Compensation Table
The following table provides information regarding the compensation of our named executive officers:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Name and Principal Position | | Year | | Salary
($) | | Option
Awards
($)(1) | | Non-Equity
Incentive Plan
Compensation
($)(2) | | All Other
Compensation
($)(3) | | Total
($) |
| Mitchell H. Gold, M.D. | | 2023 | | 599,000 | | | 1,897,924 | | | 419,300 | | | 5,000 | | | 2,921,224 | |
| Executive Chairman and Chief Executive Officer | | 2022 | | 544,000 | | | 1,970,079 | | | 231,200 | | | 3,000 | | | 2,748,279 | |
| Stanford Peng, M.D., Ph.D. | | 2023 | | 546,000 | | | 813,396 | | | 389,100 | | | 5,000 | | | 1,753,496 | |
| President and Head of Research and Development | | 2022 | | 525,000 | | | 1,970,079 | | | 223,200 | | | 3,000 | | | 2,721,279 | |
| Paul Rickey | | 2023 | | 437,000 | | | 591,068 | | | 249,100 | | | 5,000 | | | 1,282,168 | |
| Senior Vice President and Chief Financial Officer | | 2022 | | 416,000 | | | 1,030,856 | | | 158,100 | | | 3,000 | | | 1,607,956 | |
| Wolfgang Dummer, M.D., Ph.D.(4) | | 2023 | | 64,375 | | | 1,698,822 | | | — | | | — | | | 1,763,197 | |
| Former Chief Medical Officer | | | | | | | | | | | | |
We (1)have a diverse pipeline of novel therapies, asAmounts shown in this column do not reflect dollar amounts actually received by our named executive officers. Instead, these amounts reflect the aggregate grant date fair value of the stock options granted, computed in accordance with the provisions of FASB ASC Topic 718. For additional details regarding the assumptions and methodologies used to calculate the amounts reported, please see the discussion of equity awards contained in Note 11 to our consolidated financial statements contained in our annual report on Figure 1Form 10-K below.Figure 1
Our Scientific Platformfiled with the SEC on March 20, 2024.
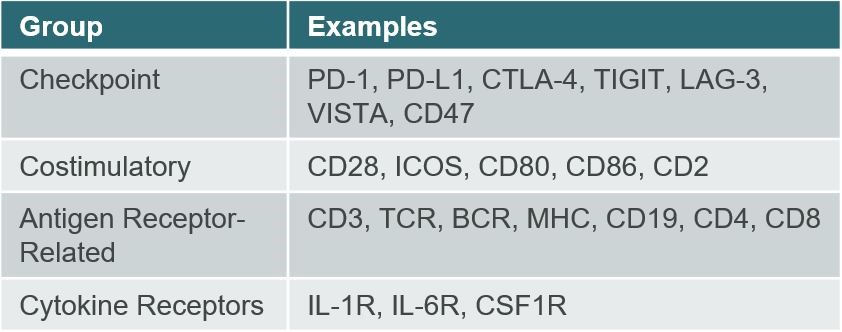
The(2) human immune system is a complex network of biological processesRepresents bonuses earned under our performance bonus plans. Bonuses earned with respect to our 2023 and structures evolved to protect humans from external infections2022 performance bonus plan were paid in 2024 and harmful changes of internal cells. Within2023, respectively. See further discussion described under the immune system, proteins play a key role in a variety of essential functions, including recognition of foreign and self-antigens, cell adhesion and trafficking, and modulation of cellular activity through costimulatory or inhibitory signaling. Our scientific platform seeks to develop novel therapeutics by engineering native, or so-called “wild-type,caption “Executive Compensation — Executive Incentive Compensation Plan.” proteins with unique properties that may benefit patients with cancer or inflammatory diseases. We have focused our efforts to-date on two major protein superfamilies that play critical roles in the regulation of immune cell signaling and activity: the immunoglobulin superfamily, or IgSF, and the tumor necrosis factor (receptor) superfamily, or TNFSF/TNFRSF.
The IgSF(3)All other compensation solely relates to 401(k) matching contributions provided by us during the applicable fiscal year. Perquisites and personal benefits are excluded as the total value of all perquisites and personal benefits for each named executive officer is the largest family of adhesion, costimulatory (activating),less than $10,000.
(4)Dr. Dummer commenced employment with us in October 2023 and inhibitory (blocking) proteins foundseparated from our employ on the surface of immunological, neurological, and other human cell types. These cell surface and soluble molecules are broadly involved with recognition of antigens, assisting in the formation of the immune synapse, and performing costimulatory, coinhibitory, and cytokine receptor signaling functions. This family includes many well-known targets, such as those seen in Figure 2. We believe the IgSF protein family members may be particularly valuable because many IgSF proteins naturally bind multiple binding partners, also referred to as “counterstructures.” Acazicolcept and davoceticept are both derived from members of the IgSF.November 13, 2023.
Figure 2
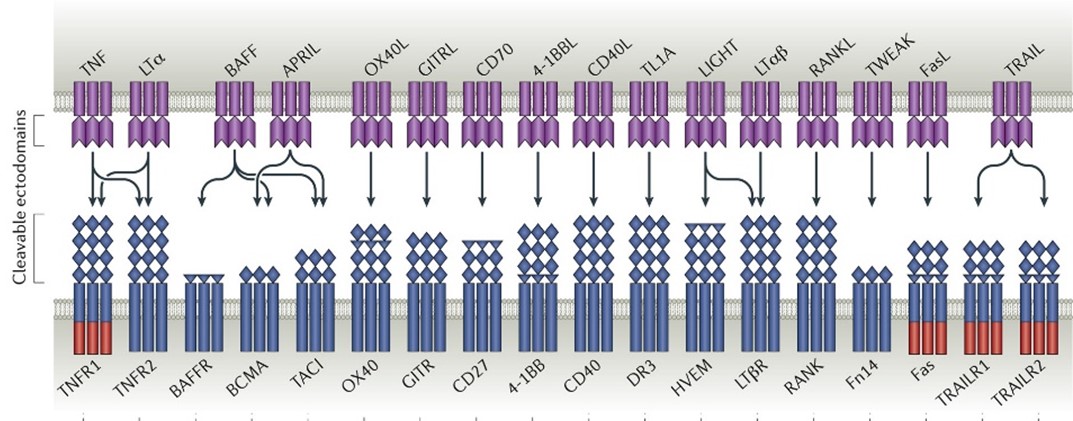
TNFSF/TNFRSF proteins are expressed broadly in the immune system and play a critical role in immune cell signaling and proliferation. TNFSF/TNFRSF members are composed of 48 unique proteins that are structurally similar and are
characterized byExecutive Employment Arrangements with Our Named Executive Officers
We entered into amended and restated executive employment agreements with each of Drs. Gold and Peng, and Mr. Rickey effective January 1, 2018. In January 2023, the compensation committee approved adjusted salaries for Drs. Gold and Peng, and Mr. Rickey of $599,000, $546,000 and $437,000, respectively. In January 2024, the compensation committee approved adjusted salaries for Drs. Gold and Peng, and Mr. Rickey of $635,000, $579,000 and $464,000, respectively. Additionally, Drs. Gold and Peng, and Mr. Rickey are eligible to earn cash bonuses of up to 50%, 50% and 40%, respectively, of their ability to bind to trimeric tumor necrosis factors (Figure 3). Membersbase salary under our Executive Incentive Compensation Plan described below. These agreements also provide for certain severance benefits upon the termination of employment or a change in control of the TNFSF/TNFRSF include many clinically relevant targets with applicationscompany pursuant to our Change in both autoimmune diseaseControl and immuno-oncology (e.g.Severance Policy, or the Severance Policy.
Pursuant to the Severance Policy, if we terminate the employment of any of Dr. Gold, Dr. Peng or Mr. Rickey, each an Eligible Employee, other than for cause, death or disability, or the Eligible Employee resigns for good reason on or within 12 months following a change of control, then, subject to the Eligible Employee signing and not revoking a separation agreement and release of claims and continuing to adhere to the Eligible Employee’s non-competition, non-disclosure and invention assignment agreement, the Severance Conditions (as defined in the Severance Policy), CD40, TACI, BCMA, 4-1BB, TNFαsuch Eligible Employee will be eligible to receive the following severance benefits, less applicable tax withholdings:
•A lump-sum payment totaling 100% (or, in the case of Dr. Gold, 150%) of the Eligible Employee’s applicable annual base salary.
•A lump-sum payment equal to (1) 100% of the Eligible Employee’s applicable target annual bonus (or, in the case of Dr. Gold, 150%) plus (2) a payment equal to the Eligible Employee’s pro-rated applicable target annual bonus.
•100% of the Eligible Employee’s then-outstanding and unvested time-based equity awards will become vested and exercisable.
•Payment or reimbursement of continued health coverage for the Eligible Employee and the Eligible Employee’s dependents under COBRA for a period of up to 12 months (or, in Dr. Gold’s case, 18 months).
The TNF Superfamily
Figure 3Further, under the Severance Policy, if we terminate an Eligible Employee’s employment other than for cause, death or disability or such Eligible Employee resigns for good reason at any time other than during the period lasting from the date of a change of control or within 12 months thereafter, then, subject to the Severance Conditions, such Eligible Employee will be eligible to receive the following severance benefits, less applicable tax withholdings:
Our scientists create engineered proteins from IgSF members (variant immunoglobulin domains,•Continued payments totaling 75% (or, in Dr. Gold’s case, 100%) of the Eligible Employee’s applicable annual base salary over a period of 9 months (or, in Dr. Gold’s case, 12 months).
•Payment or vIgDs)reimbursement of continued health coverage for the Eligible Employee and TNFSF/TNFRSF members (variant TNF domains, or vTDs). We use directed evolution, which is an iterative scientific engineering process purposefully conducted to modify an IgSF and TNFSF/TNFRSF proteinthe Eligible Employee’s dependents under COBRA for a desired therapeutic function. The potentialperiod of up to create therapies capable9 months (or, in Dr. Gold’s case, 12 months).
In addition, we entered into an employment agreement with Dr. Dummer on September 20, 2023. This employment agreement provided for an annual base salary of working within$515,000 as well as the grant of a formed immune synapse, forcing a synapsestock option to occur, or preventing a synapse from occurring are important, novel attributespurchase 200,000 shares of our scientific platform.common stock, and eligibility to earn a cash bonus of up to 45% of Dr. Dummer’s base pay under our Executive Incentive Compensation Plan. As previously disclosed, Dr. Dummer tendered his resignation from our service on November 13, 2023, effective immediately. Dr. Dummer was not eligible for any severance benefits in connection with his resignation.
Executive Incentive Compensation Plan
Each of our executive officers is eligible for bonuses under our Executive Incentive Compensation Plan, which plan was approved by our board of directors in March 2019, and has an established target bonus amount as set forth in the section “— Executive Employment Arrangements with our Named Executive Officers.” The actual amount of such bonuses is tied to the achievement of various objectives for each year.
Figure 4 illustratesFor 2023, the processcompensation committee of directed evolutionour board of directors determined that Dr. Gold's bonus would be based solely on achievement of corporate objectives (including advancement of our povetacicept, davoceticept and acazicolcept development programs, enhancement of our discovery portfolio and achievement of other corporate objectives, such as securing additional capital to support execution, and establishing infrastructure to support growth), and that the bonuses for Dr. Peng and Mr. Rickey would be based on 75% achievement of the previously noted corporate objectives and 25% individual objectives developed in our scientific platform. Our scientists utilize yeast display protein library strategies to identify variants of wild-type proteinsconsultation with desired binding characteristics. We start with a wild-type IgSF or TNFSF/TNFRSF protein and then enter a cycle of library generation and yeast display. Flow cytometry or other methods are used to sort for yeast clones displaying variants with desired binding characteristics. Biologic and biophysical assays of purified proteins assess biological function and manufacturing characteristics. The end product is an optimized vIgD or vTD. Additional cycles can be carried out by building next generation libraries from the output of prior libraries to result in further optimization.
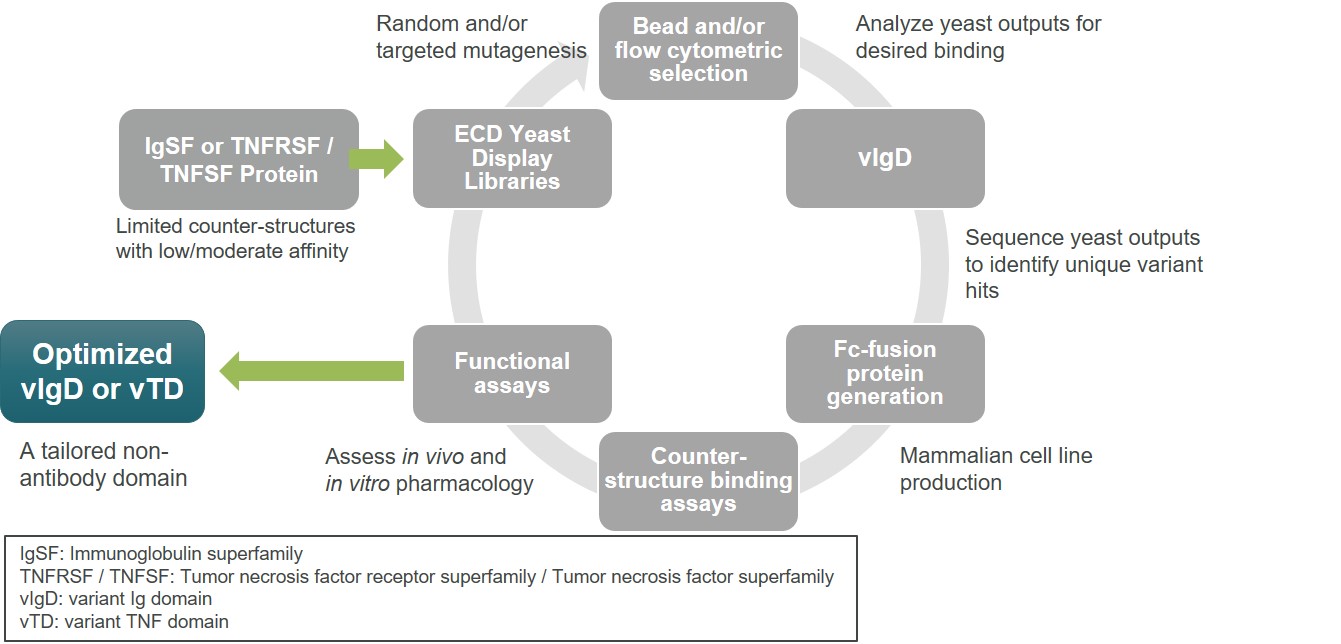
Figure 4Dr. Gold.
Our scientific platform is generally ableIn January 2024, our compensation committee determined that the 2023 corporate objectives had been achieved at the 140% level, and that Dr. Peng's and Mr. Rickey's individual goals had both been achieved at the 150% levels, resulting in payments of $389,100 and $249,100, respectively. The compensation committee recognized achievement of Dr. Peng’s and Mr. Rickey's individual goals above the 100% level in consideration of significant individual efforts to improveachieve certain milestones, including enrollment, clinical development and regulatory milestones for povetacicept for Dr. Peng, and securing additional capital and meeting certain budget targets for Mr. Rickey. Dr. Gold received a payment of $419,300. The amounts earned in fiscal 2023 were paid in 2024.
Outstanding Equity Awards at December 31, 2023
The following table provides information regarding the equity awards outstanding held by each of our named executive officers at December 31, 2023:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | Option Awards |
| Name | | Vesting
Commencement
Date | | Grant
Date | | Number of
Securities
Underlying
Unexercised
Options (#)
Exercisable | | Number of
Securities
Underlying
Unexercised
Options (#)
Unexercisable | | Equity Incentive Plan Awards: Number of Securities Underlying Unexercised Unearned Options (#) | | | | Option
Exercise
Price
($) | | Option
Expiration
Date |
| Mitchell H. Gold | | 01/20/2017 | | 03/14/2017 | | 247,951 | | | — | | | — | | | | | 0.65 | | | 03/13/2027 |
| | 01/20/2017 | | 04/12/2017 | | 208,916 | | | — | | | — | | | | | 5.02 | | | 04/11/2027 |
| | 01/02/2018 | | 01/02/2018 | | 70,000 | | | — | | | — | | | | | 11.31 | | | 01/01/2028 |
| | 02/06/2019 | | 02/06/2019 | | 200,000 | | | — | | | — | | | | | 6.51 | | | 02/05/2029 |
| | 01/23/2020 | | 01/23/2020 | | 264,375 | | | 5,625 | | | — | | | (1) | | 3.23 | | | 01/22/2030 |
| | 01/05/2021 | | 01/05/2021 | | 185,937 | | | 69,063 | | | — | | | (1) | | 13.20 | | | 01/04/2031 |
| | 01/04/2022 | | 01/04/2022 | | 103,020 | | | 111,980 | | | — | | | (1) | | 13.30 | | | 01/03/2032 |
| | 01/04/2023 | | 01/04/2023 | | — | | | 350,000 | | | — | | | (1) | | 7.55 | | | 01/03/2033 |
| Paul Rickey | | 04/01/2017 | | 04/12/2017 | | 535 | | | — | | | — | | | | | 5.02 | | | 04/11/2027 |
| | 01/02/2018 | | 01/02/2018 | | 45,000 | | | — | | | — | | | | | 11.31 | | | 01/01/2028 |
| | 02/06/2019 | | 02/06/2019 | | 75,000 | | | — | | | — | | | | | 6.51 | | | 02/05/2029 |
| | 01/23/2020 | | 01/23/2020 | | 17,375 | | | 2,125 | | | — | | | (1) | | 3.23 | | | 01/22/2030 |
| | 01/05/2021 | | 01/05/2021 | | 87,500 | | | 32,500 | | | — | | | (1) | | 13.20 | | | 01/04/2031 |
| | 01/04/2022 | | 01/04/2022 | | 53,906 | | | 58,594 | | | — | | | (1) | | 13.30 | | | 01/03/2032 |
| | 01/04/2023 | | 01/04/2023 | | — | | | 109,000 | | | — | | | (1) | | 7.55 | | | 01/03/2033 |
| Stanford Peng | | 09/06/2016 | | 09/22/2016 | | 161,492 | | | — | | | — | | | | | 0.65 | | | 09/21/2026 |
| | 09/06/2016 | | 03/14/2017 | | 37,267 | | | — | | | — | | | | | 0.65 | | | 03/13/2027 |
| | 01/02/2018 | | 01/02/2018 | | 65,000 | | | — | | | — | | | | | 11.31 | | | 01/01/2028 |
| | 09/28/2018 | | 09/28/2018 | | 250,000 | | | — | | | — | | | | | 6.33 | | | 09/27/2028 |
| | 02/06/2019 | | 02/06/2019 | | 75,000 | | | — | | | — | | | | | 6.51 | | | 02/05/2029 |
| | 04/22/2019 | | 04/22/2019 | | 50,000 | | | — | | | — | | | | | 7.20 | | | 04/21/2029 |
| | 01/23/2020 | | 01/23/2020 | | 105,750 | | | 2,250 | | | — | | | (1) | | 3.23 | | | 01/22/2030 |
| | 09/15/2020 | | 09/15/2020 | | — | | | — | | | 80,000 | | | (2) | | 8.28 | | | 09/14/2030 |
| | 01/05/2021 | | 01/05/2021 | | 109,375 | | | 40,625 | | | — | | | (1) | | 13.20 | | | 01/04/2031 |
| | 01/04/2022 | | 01/04/2022 | | 103,020 | | | 111,980 | | | — | | | (1) | | 13.30 | | | 01/03/2032 |
| | 01/04/2023 | | 01/04/2023 | | — | | | 150,000 | | | — | | | (1) | | 7.55 | | | 01/03/2033 |
(1)1/4th of the shares will vest on the one-year anniversary of the vesting commencement date, and 1/36th of the remaining shares shall vest on each monthly anniversary thereafter, such that 100% of the shares shall be vested and exercisable as of the four-year anniversary of the vesting commencement date.
(2)The shares underlying the option were to vest in two equal tranches upon native IgSFthe achievement of specified performance goals to be achieved on or TNFSF/TNFRSF activity regardless of whether natural binding affinity is weak or strong. When starting affinity is very weak, techniques employed by our scientists have accomplished several thousand-fold increases in binding affinity with sometimes as few as two library generation cycles. Even when starting affinity is very high, our scientific platform can still improve binding affinities. The same general strategies can be used when the desired therapeutic profile requires reduced affinity comparedprior to the wild-type protein. We have applied our scientific platform to several IgSF and TNFSF/TNFRSF protein targets.
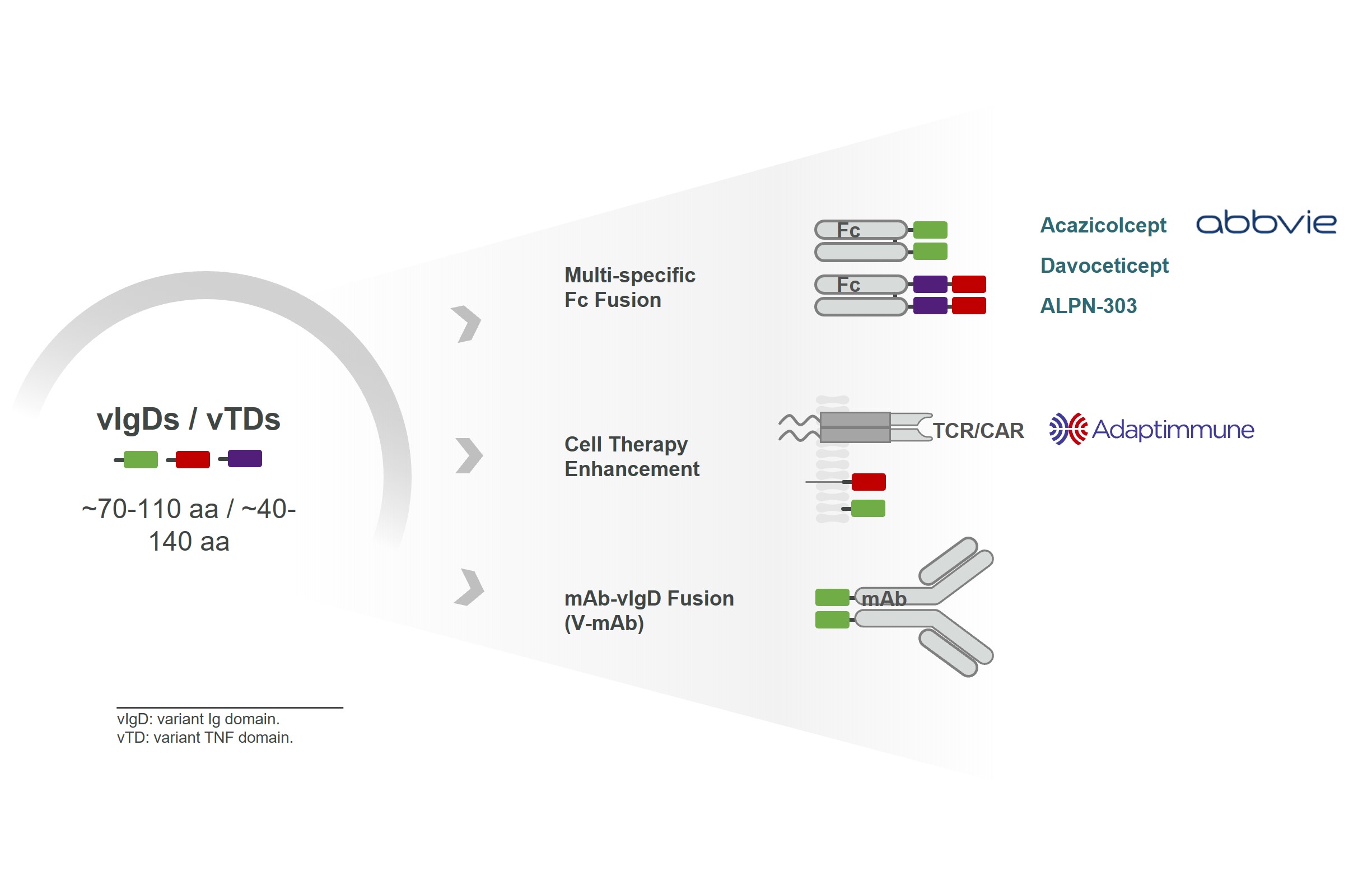
We believe our vIgDs and vTDs are highly flexible.expiration of the options, as determined by the board of directors or the compensation committee of the board of directors. In many cases, a single affinity-maturation campaign can result in multiple domains suitable for use inJanuary 2024, the formats such as those illustrated in Figure 5 and further described below.
Figure 5
vIgD-Fcor vTD-Fc
A vIgD- or vTD-Fc fusion protein iscompensation committee of the simplest format. Our lead autoimmune/inflammatory program, acazicolcept, and lead oncology program, davoceticept, are both examplesboard of vIgD-Fc formats. ALPN-303 is an example of a vTD-Fc format. The engineered vIgD or vTD protein is fused to an Fc backbone. Combining vIgDs or vTDs with antibody Fc domains to make Fc fusion proteins potentially allows for better expression, facilitates purification, and improves pharmacokinetic (dosing) properties. Fc fusion proteins are a standard format indirectors determined that the industry, with examples such as etanercept, abatacept, and belatacept. A vIgD- or vTD-Fc could potentially be administered intravenously, subcutaneously, topically, or via other methods of delivery.
Cell Therapy Enhancement
Our scientific platform has also generated immune modulatory proteins with the potential of improving engineered cellular therapies, such as CAR-Ts, TCR-Ts, or TILs.
Acazicolcept, a Dual ICOS/CD28 Antagonist for Autoimmune/Inflammatory Diseases
Our lead autoimmune/inflammatory disease program, acazicolcept, is an Fc fusion protein of a human inducible T cell costimulator ligand, or ICOSL, vIgD designed to inhibit simultaneously the CD28 and ICOS T cell costimulatory pathways (Figure 6). This vIgD is fused to an “effectorless” Fc backbone and is intended for the potential treatment of autoimmune/inflammatory diseases. Notably, acazicolcept is not a bispecific antibody construct. A traditional bispecific might be constructed of one domain binding ICOS and one domain binding CD28. Instead, acazicolcept makes use of a novel single vIgD domainspecified performance goals were no longer capable of binding both ICOSbeing achieved and CD28 engineered by our scientists using our proprietary scientific platform.the option was deemed terminated in full.
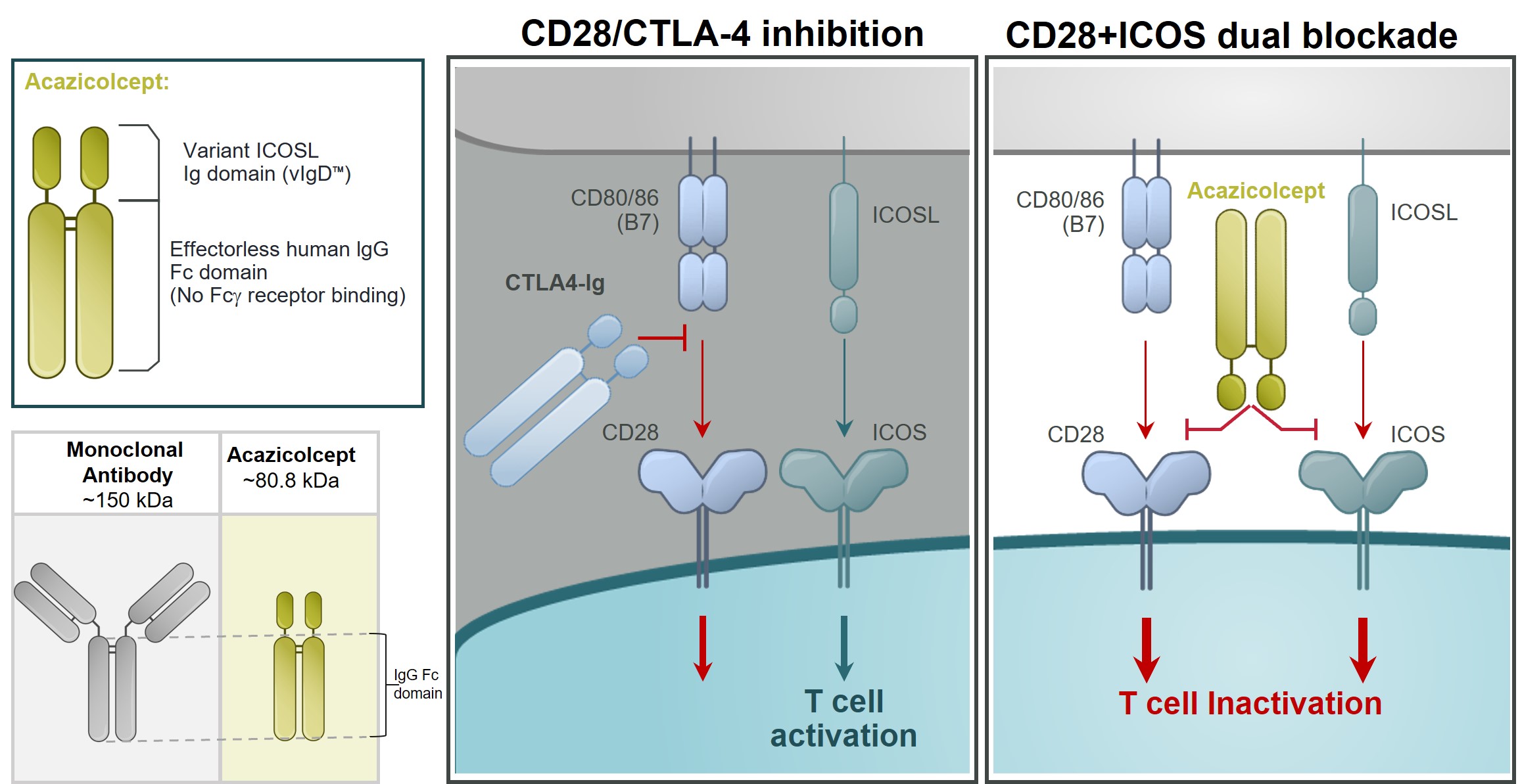
Figure 6
CD28 has been long recognizedWe have adopted the Alpine Immune Sciences 401(k) Plan, which is a defined contribution retirement plan, in which all non-temporary employees are eligible to participate immediately. This plan provides our eligible employees with an opportunity to save for retirement on a tax advantaged basis, and participants can defer a portion of their eligible compensation. All participants’ interests in their deferrals are 100% vested when contributed. The 401(k) plan permits us to make matching contributions and profit-sharing contributions to eligible participants. Pre-tax contributions are allocated to each participant’s individual account and are then invested in selected investment alternatives according to the participants’ directions. The 401(k) plan is intended to qualify under Sections 401(a) and 501(a) of the Internal Revenue Code. As a tax qualified retirement plan, contributions to the 401(k) plan and earnings on those contributions are not taxable to the employees until distributed from the 401k) plan and all employer contributions are deductible by AIS Operating Co., Inc. when made. The 401(k) plan also permits contributions to be requiredmade on a post-tax basis for naïve T cell activation. The therapeutic inhibitorsemployees participating in the Roth 401(k) plan component.
Beginning in 2022, we initiated matching contributions equal to 50% of each employee’s salary deferral contribution of up to 6% of the CD28 pathway (e.g., abatacept, CTLA4-Ig;employee’s compensation, subject to a maximum annual match of $3,000 per participant. For 2023, we increased the maximum annual match to $5,000 per participant.
Compensation Committee Interlocks and belatacept, a second generation CTLA4-Ig) have proven valuable forInsider Participation
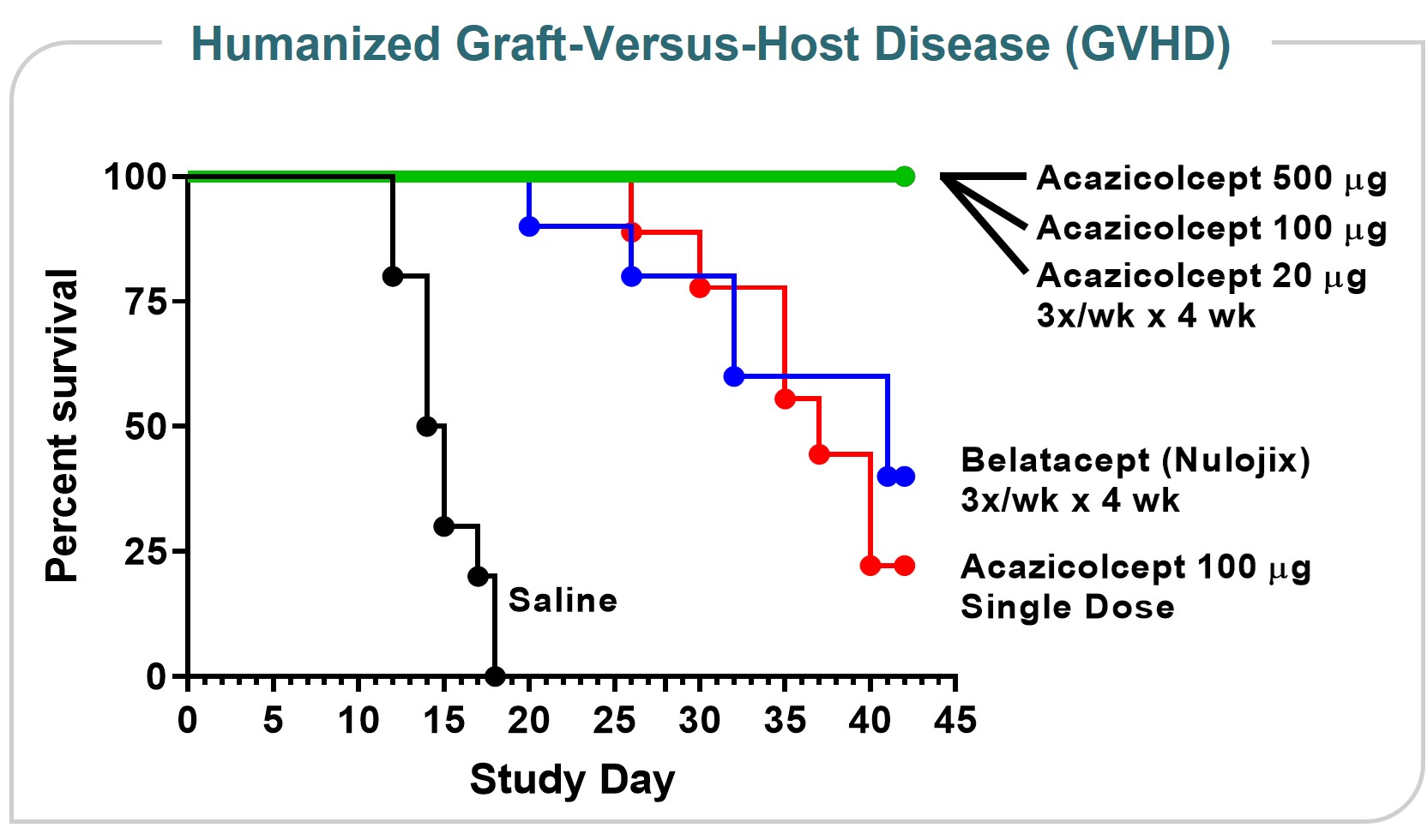
Currently, the treatmentmembers of some inflammatory arthritis conditions (rheumatoid arthritis, juvenile idiopathic arthritis, psoriatic arthritis)our compensation committee are Drs. Topper, Thompson and for the prevention of renal allograft rejection or graft versus host disease. However, therapeutic blockadeCui. None of the CD28 pathway, primarilyforegoing members of our compensation committee currently serves, or in the past year has served, as studied with abatacept,an officer or employee of Alpine Immune Sciences. None of our executive officers currently serves, or in the past year has not been successful in several other inflammatory diseases (e.g., Crohn’s disease, lupus nephritis, multiple sclerosis) despite extensive evidence implicating T cells in disease pathogenesis and evidence of clinical biological activity. This suggests an additional pathogenic costimulatory pathway(s) remains unaddressed.
ICOS is partserved, as a member of the CD28 costimulatory familyboard of molecules, including PD-1, CD28, and CTLA-4. ICOS is related to CD28, but, in contrast, is poorly expressed in naïve T cells. ICOS is, however, rapidly induced upon T cell activation. It appears to be a dominant costimulatory pathway in at least some effectordirectors or pathogenic T cells, such as potentiallycompensation committee (or other board committee performing equivalent functions or, in the absence of CD28. Elevated levelsany such committee, the entire board of ICOS‑expressing T cellsdirectors) of any entity that has one or more executive officers serving on our board of directors or compensation committee.
Policy on Hedging and Pledging of Alpine Securities
We have beenan insider trading policy, which, among other things, prohibits our directors, officers and employees from engaging in “short sales” of our securities, and from purchasing or selling derivative securities, or entering into derivatives contracts, with respect to our securities. In addition, our directors and certain of our senior officers are prohibited from pledging our securities as collateral for a loan or entering into any margin arrangement with respect to our securities.
Compensation Recovery Policy
We have a compensation recovery policy—also known as a clawback policy—that provides for the recovery of incentive-based compensation received by our current and former executive officers in the event of an accounting restatement, as further described in an increasing numberthe policy, in accordance with SEC and Nasdaq requirements.
Non-Employee Director Compensation
Directors who are also our employees receive no additional compensation for their service as a director. Compensation for Dr. Gold, who serves as our chief executive officer, is discussed under the caption “Executive Compensation.” We reimburse our directors for expenses associated with attending meetings of autoimmune/inflammatory diseases, correlating with disease activity. At the same time, inhibitionour board of ICOS is effective in several preclinical inflammatory disease models. The ICOS pathway may therefore represent a major costimulatory pathway, nonredundant with CD28,directors and highly relevant to autoimmune/inflammatory diseases.
We have performed a numbermeetings of preclinical experiments demonstrating acazicolcept is active in both in vitro and in vivo models, severalcommittees of which are described below.our board.
A potent immunomodulator of diseased cells
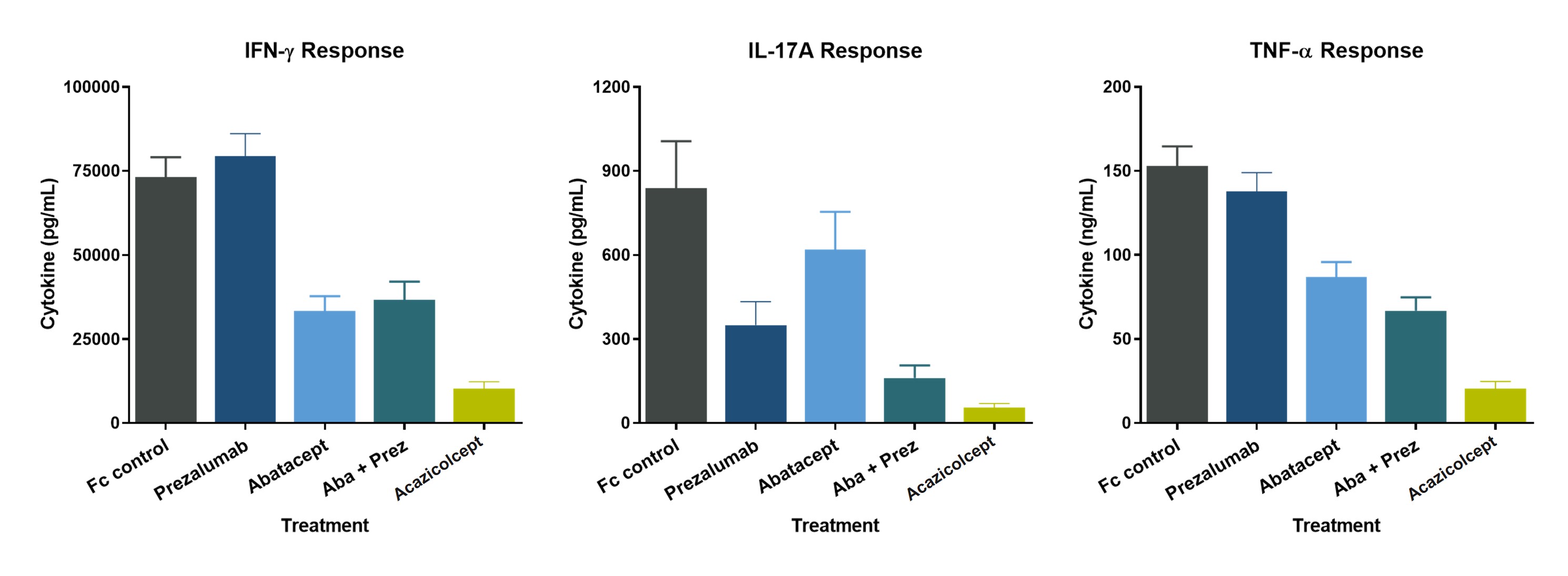
Acazicolcept inhibits cytokine production from human peripheral blood mononuclear cells in vitro more potently than single CD28 (abatacept, or CTLA4-Ig) or ICOS (prezalumab, or anti-ICOSL mAb) pathway inhibitors alone or in combination (Figure 7).
Figure 7
Sjögren’s Syndrome Model2023 Director Compensation Table
The following table shows for the fiscal year ended December 31, 2023 certain information with respect to the compensation of our non-employee directors who served on our board of directors during any part of 2023.
| | | | | | | | | | | | | | | | | | | | |
| Name | | Fees Earned
or paid in
Cash ($) | | Option Awards ($)(1) | | Total ($) |
| Robert Conway(2) | | 58,750 | | | 51,460 | | | 110,210 | |
| Xiangmin Cui, Ph.D.(3) | | 48,750 | | | 51,460 | | | 100,210 | |
| Jörn Drappa, M.D., Ph.D.(4) | | 40,000 | | | 51,460 | | | 91,460 | |
| Natasha Hernday(5) | | 47,500 | | | 51,460 | | | 98,960 | |
| Christopher Peetz(6) | | 47,500 | | | 51,460 | | | 98,960 | |
| Peter Thompson, M.D.(7) | | 52,500 | | | 51,460 | | | 103,960 | |
| James N. Topper, M.D., Ph.D.(8) | | 50,000 | | | 51,460 | | | 101,460 | |
Sjögren(1)’s syndromeAmounts shown in this column do not reflect dollar amounts actually received by our non-employee directors. Instead, these amounts reflect the aggregate grant date fair value of the stock options granted, computed in accordance with the provisions of Financial Accounting Standards Board, or FASB, Accounting Standards Codification, or ASC, Topic 718. For additional details regarding the assumptions and methodologies used to calculate the amounts reported, please see the discussion of equity awards as disclosed in Note 11 to our consolidated financial statements contained in our annual report on Form 10-K filed with the SEC on March 20, 2024. The assumptions and methodologies used to determine fair value for stock options granted to our non-employee directors are identical to those used for stock options granted to our employees and other service providers.(2)As of December 31, 2023, Mr. Conway held outstanding options to purchase 58,455 shares of common stock.
(3)As of December 31, 2023, Dr. Cui held outstanding options to purchase 42,950 shares of common stock.
(4)As of December 31, 2023, Dr. Drappa held outstanding options to purchase 30,000 shares of common stock.
(5)As of December 31, 2023, Ms. Hernday held outstanding options to purchase 42,650 shares of common stock.
(6)As of December 31, 2023, Mr. Peetz held outstanding options to purchase 50,600 shares of common stock.
(7)As of December 31, 2023, Dr. Thompson held outstanding options to purchase 50,600 shares of common stock
(8)As of December 31, 2023, Dr. Topper held outstanding options to purchase 50,600 shares of common stock.
Director Compensation Policy
Our board of directors has approved a director compensation policy for our non-employee directors, which was most recently amended in December 2023. For purposes of the policy, the board of directors classified each director into one of the two following categories: (1) an “employee director,” is a director who is employed by us; and (2) a “non-employee director,” is a director who is not an autoimmune diseaseemployee director. Only non-employee directors will receive compensation under the director compensation policy. Non-employee directors will receive compensation in which immune cells attack the glandsform of equity and cash under the director compensation policy, as described below. We believe our non-employee director compensation program provides reasonable compensation to our non-employee directors that produce salivais appropriately aligned with our peers and tears,is commensurate with the services and contributions of our non-employee directors.
Non-employee directors receive an initial stock option grant to purchase shares of our common stock upon appointment or election to the board of directors. During 2023, our policy provided for an initial stock option grant of 20,000 shares, as well as other internal organs. In an animal modeladditional, annual stock option grant of salivary gland inflammation (sialoadenitis),10,000 shares. The amendment of our policy in 2023 increased the size of these grants to 35,000 and 17,500 shares, respectively. The annual grants occur on the first trading day in January of each year. All options have an exercise price equal to the closing price of our common stock as reported by Nasdaq on the date of grant, are subject to vesting in 36 equal monthly installments over a key organ manifestation of Sjögren’s syndrome, acazicolcept appeared more efficacious in reducingthree-year period from the incidence and severity of sialadenitis as compared to abatacept or wild-type ICOSL-Fc alonegrant date for initial option grants, or in combination. These data were presented at12 equal monthly installments over a 12-month period from the 2019grant date for annual meeting ofstock option grants, subject to further evaluation by the ACR. (Figure 8)
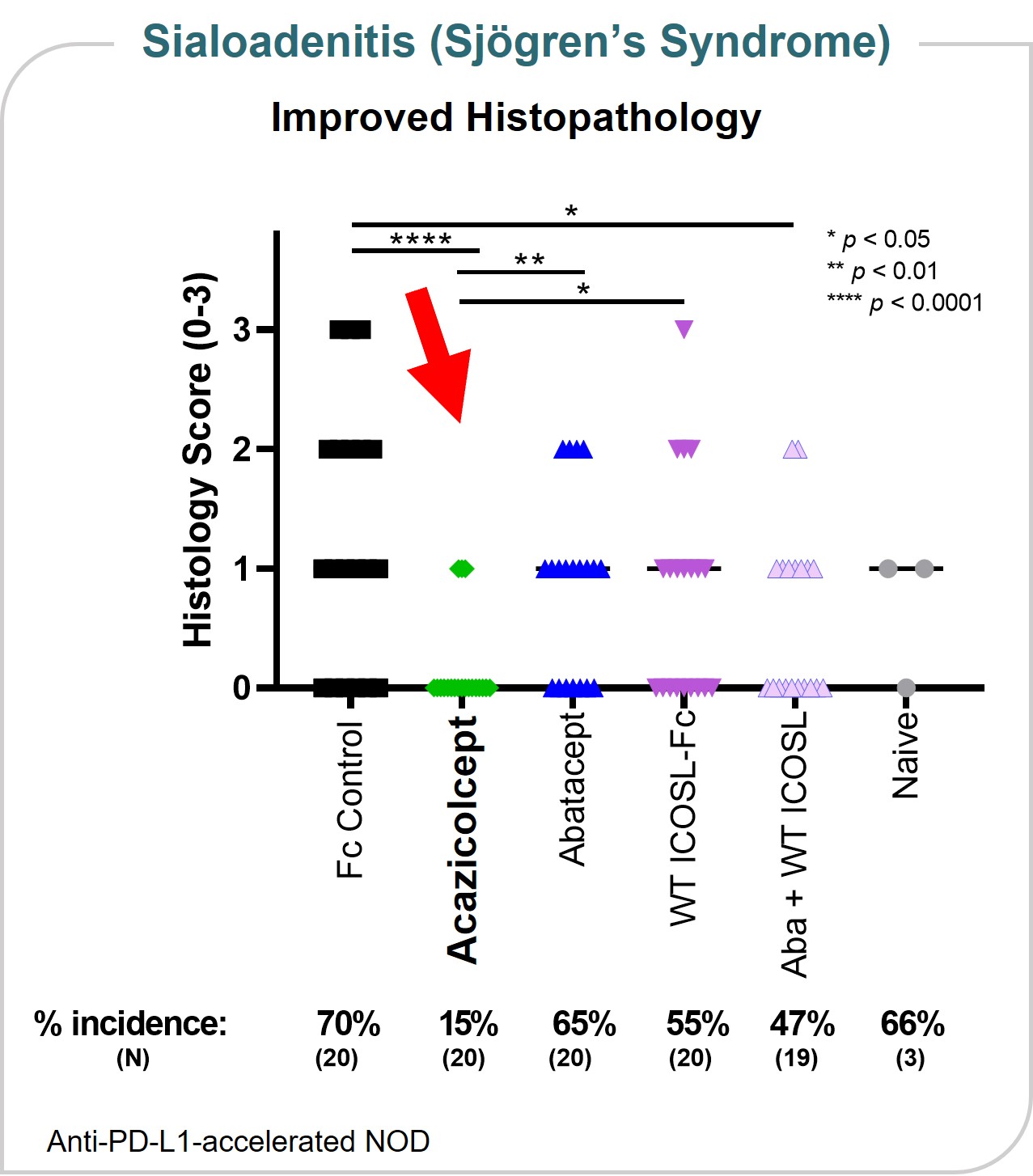
Figure 8
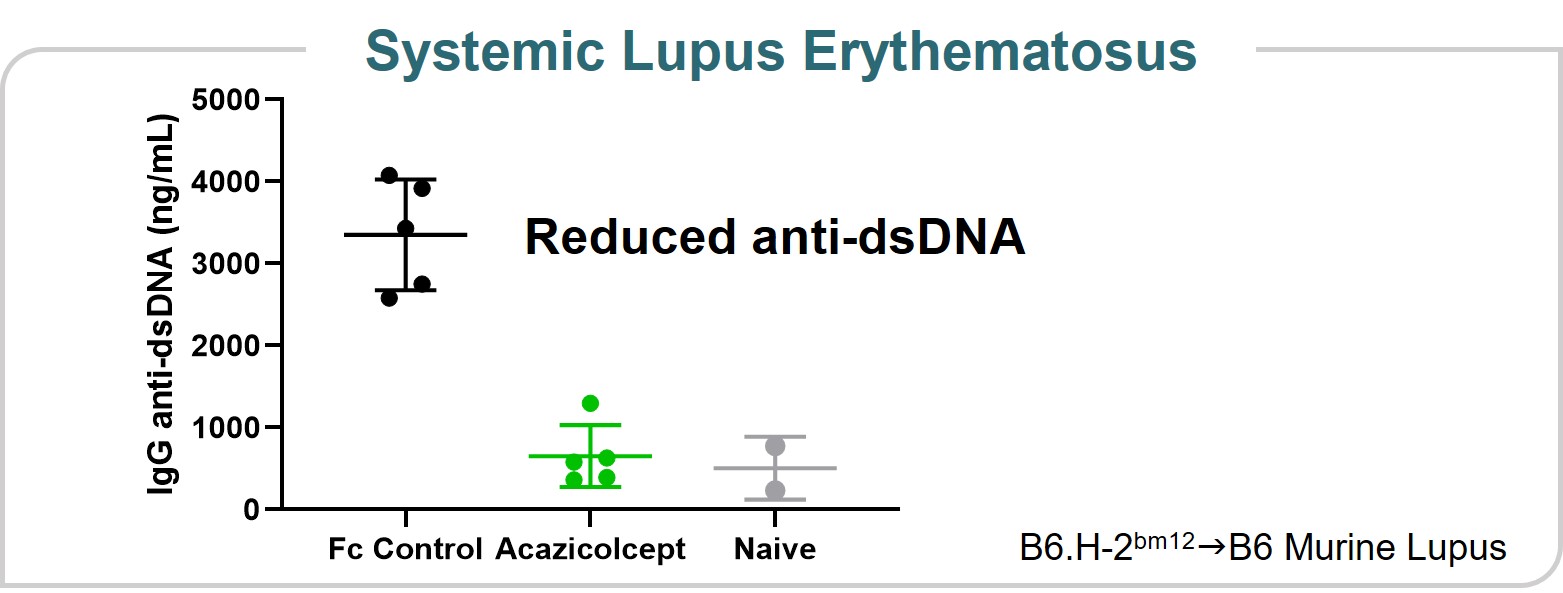
Systemic Lupus Erythematosus Model
Acazicolcept has demonstratedefficacycompensation committee. On a change in a preclinical model of SLE, a multiorgan autoimmune disease that that can leadcontrol, all outstanding, unvested options held by non-employee directors are expected to serious organ complications and death. In Figure 9, which evaluated acazicolceptvest in a bm12 inducible model of SLE, treatment with acazicolcept reduced serum titers of anti-dsDNA autoantibodies throughout the study compared to Fc control treatment. These data were presented at the 2019 annual meeting of the ACR. We evaluated acazicolcept in a Phase 1 healthy volunteer study and in the second quarter of 2021 initiated patient dosing in Synergy, a global, randomized, double-blind, placebo-controlled Phase 2 study of acazicolcept in adults with moderate-to-severe SLE.
Figure 9full.
Arthritis ModelEach non-employee director is eligible to receive the following cash annual retainer, which is paid quarterly in arrears on a prorated basis.
| | | | | |
| Annual retainer for board membership | $ | 40,000 | |
| Annual retainer for board chairperson | 25,000 | |
| Annual retainer for audit committee chairperson | 15,000 | |
| Annual retainer for audit committee member | 7,500 | |
| Annual retainer for compensation committee chairperson | 10,000 | |
| Annual retainer for compensation committee member | 5,000 | |
| Annual retainer for nominating and corporate governance committee chairperson | 7,500 | |
| Annual retainer for nominating and corporate governance committee member | 3,750 | |
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
Securities Authorized for Issuance Under Equity Compensation Plans
The following table provides information as of December 31, 2023, with respect to the shares of our common stock that may be issued under existing equity compensation plans:
| | | | | | | | | | | | | | | | | | | | |
| | A | | B | | C |
| Plan Category | | Number of
Securities to be
Issued Upon
Exercise of
Outstanding
Options and
Rights | | Weighted-
Average
Exercise
Price of
Outstanding
Options and
Rights(1) | | Number of
Securities
Remaining
Available for
Future
Issuance Under
Equity
Compensation
Plans
(Excluding
Securities
Reflected in
Column A)(2) |
| Equity compensation plans approved by security holders: | | | | | | |
| Amended and Restated 2015 Stock Plan, as amended, or the 2015 Stock Plan | | 810,702 | | | $ | 2.90 | | | — | |
| 2015 Equity Incentive Plan | | 278,955 | | | $ | 12.38 | | | — | |
| 2018 Equity Incentive Plan | | 6,579,167 | | | $ | 9.22 | | | 281,984 | |
| Employee Stock Purchase Plan | | — | | | N/A | | 45,211 | |
| Total equity compensation plans approved by security holders | | 7,668,824 | | | $ | 8.67 | | | 327,195 | |
| Equity compensation plans not approved by security holders (3) | | 345,000 | | | $ | 11.09 | | | — | |
| Total | | 8,013,824 | | | $ | 8.77 | | | 327,195 | |
Figure 10(1) shows data fromRepresents the outstanding options’ weighted-average exercise price and does not take into account the shares issuable upon vesting of outstanding performance stock units or restricted stock units, which do not have an exercise price.
(2)in vivo collagen-induced arthritis model. This modelRepresents the number of securities remaining available for future issuance under the 2015 Equity Incentive Plan, the 2015 Stock Plan, the 2018 Equity Incentive Plan and the Employee Stock Purchase Plan. The number of shares available for issuance under the 2018 Equity Incentive Plan is designedsubject to test a drug’s abilityan annual increase on the first day of each year equal to reduce inflammatory signals thought to play a role in rheumatoid arthritis, psoriatic arthritis, and other typesthe lesser of inflammatory arthritis conditions. In the data presented at the 2019 annual meeting(a) 1,500,000 shares or (b) 5% of the American Collegetotal number of Rheumatology, acazicolcept was superior to abatacept,shares of common stock outstanding on December 31 of the preceding calendar year or (c) a druglesser number of shares of common stock approved by the FDAboard of directors prior to treat rheumatoid, psoriatic, and juvenile idiopathic arthritis.
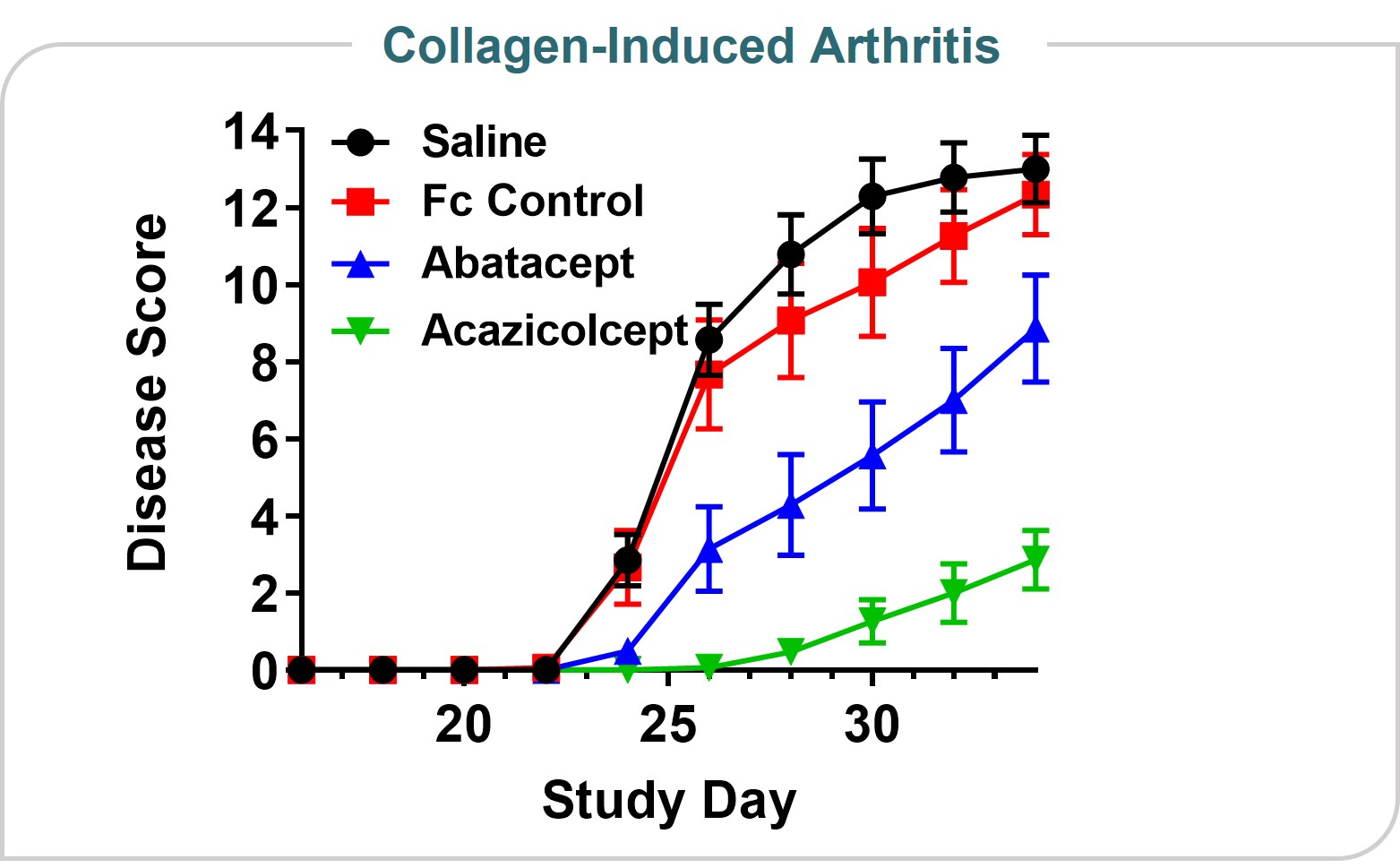
Figure 10
Human Xenograft GVHD Model
Acazicolcept has been studied in an in vivo mouse modelJanuary 1 of GVHD, a damaging and potentially fatal inflammatory disease frequently observed following allogeneic stem cell and/or bone marrow transplant treatments for cancer or other serious diseases. The results represented in Figure 11 show acazicolcept had superior survival when dosed three times per week for four weeks compared to belatacept (an FDA-approved drug for prevention of renal allograft rejection - a type of inflammation-related rejection process analogous to GVHD. Belatacept is a more potent variant of abatacept, which is FDA-approved for the prevention of GVHD). In fact, 100% of acazicolcept multi-dose treated animals across three different dose levels survived. Animals given only a single dose of acazicolcept performed comparably to animals treated with belatacept dosed 3x/week for four weeks, demonstrating the potency and efficacy of acazicolcept in this disease model.
Figure 11
Summary of acazicolcept Program Preclinical Data
Our scientists and collaborators have demonstrated in in vivo preclinical studies that acazicolcept:year.
•(3)demonstrates a lower incidenceRepresents 185,000 and severity160,000 shares of sialadenitis, a modelour common stock subject to the Stand-Alone Inducement Stock Option Grant approved by our compensation committee upon the hiring of Sjögren’s syndrome, as compared to abatacept or wild-type ICOSL-Fc alone orour Chief Technology Officer, M. Christina Yi, in combination;
•reduces levels of pathogenic anti-dsDNA autoantibodies comparedAugust 2023, and our Chief Medical Officer, Dr.Andrew Sandler, in August 2022, respectively. This stock option was not approved by security holders pursuant to an Fc control in an animal modelexemption permitted under the rules of SLE;Nasdaq.
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT
•reduces disease severity and delays disease onset time relativeThe following table sets forth certain information with respect to control in an in vivothe beneficial ownership of our common stock as of April 9, 2024 for: arthritis model with activity superior to abatacept, an FDA-approved drug for rheumatoid, psoriatic, and juvenile idiopathic arthritis;
•improves survival compared to belatacept in a humanized in vivo mouse GVHD model;each person who we know beneficially owns more than 5% of our common stock;
•demonstrates controleach of colitis in an animal modelour directors;
•each of inflammatory bowel disease, or IBD;our named executive officers; and
•shows improved disease scoresall of our directors and executive officers as a group.
The percentage of beneficial ownership shown in an animal modelthe table is based upon 65,560,484 shares outstanding as of multiple sclerosis, or MS, compared to abatacept.April 9, 2024.
Acazicolcept Clinical Development
We have completeddetermined beneficial ownership in accordance with the rules of the SEC. Except as indicated by the footnotes below, we believe, based on the information furnished to us, that the persons and entities named in the table below have sole voting and investment power with respect to all shares of common stock that they beneficially own, subject to applicable community property laws.
In computing the number of shares beneficially owned by a Phase 1 studyperson and the percentage ownership of acazicolcept in healthy volunteers (NCT03748836). This study was designedthat person, we take into account shares of common stock issuable pursuant to evaluatestock options and warrants that may be exercised or that are scheduled to vest on or before the safety60th day after April 9, 2024. These shares are deemed to be outstanding and tolerability of single and multiple ascending intravenous and/beneficially owned by the person holding those options, warrants or subcutaneous doses of acazicolcept. In addition, pharmacokinetics, pharmacodynamics and exploratory biomarkers were evaluated to help determine acazicolcept’s potentialrestricted stock units for the treatmentpurpose of autoimmune/inflammatory diseases. Resultscomputing the percentage ownership of that person, but they are not treated as outstanding for the study were presented atpurpose of computing the 2020 European Alliancepercentage ownership of Associationsany other person.
Except as otherwise noted below, the address for Rheumatology (EULAR) E-Congress and publishedeach person or entity listed in the peer-reviewed journal Clinical Translational Science (doi:10.1111/cts.12983). The first-in-human study randomized 96 healthy adults to receive single or multiple, intravenous or subcutaneous, placebo or acazicolcept at doses ranging from 1 μg/kg to 10 mg/kg. At all dose levels, acazicolcept was well-tolerated, with no severe adverse events, clinically-significant immunogenicity events, or evidence of cytokine release. Pharmacokinetics and pharmacodynamics (Figure 12) exhibited desirable dose dependence, with increasing doses corresponding to increasing duration of complete, or near-complete target saturation, as well as inhibition of antibody responses to keyhole limpet hemocyanin, or KLH, immunization.table is c/o Alpine Immune Sciences, Inc., 188 East Blaine Street, Suite 200, Seattle, Washington 98102.
| | | | | | | | | | | | | | |
| | | Common Stock Beneficially Owned |
| Name of Beneficial Owner | | Shares | | Percentage |
| 5% Stockholders: | | | | |
| Entities affiliated with Decheng Capital(1) | | 8,317,924 | | | 12.7% |
| RA Capital Management, L.P.(2) | | 5,903,234 | | | 8.9% |
| Lynx1 Capital Management LP(3) | | 3,813,926 | | | 5.8% |
| Great Point Partners, LLC(4) | | 3,703,433 | | | 5.6% |
| Directors and Named Executive Officers: | | | |
| Mitchell H. Gold(5) | | 4,095,689 | | | 6.1% |
| Stanford Peng(6) | | 1,032,275 | | | 1.6% |
| Paul Rickey(7) | | 231,107 | | | * |
| Peter Thompson(8) | | 2,738,387 | | | 4.2% |
| James N. Topper(9) | | 6,091,026 | | | 8.9% |
| Robert Conway(10) | | 115,746 | | | * |
| Christopher Peetz(11) | | 59,791 | | | * |
| Xiangmin Cui(12) | | 8,368,165 | | | 12.8% |
| Natasha Hernday(13) | | 49,941 | | | * |
| Jörn Drappa(14) | | 29,513 | | | * |
| Wolfgang Dummer | | — | | | * |
| All current directors and executive officers as a group (12 persons)(15) | | 23,108,449 | | | 32.2% |
Figure 12 | | | | | |
| (*) | Less than one percent. |
S(1)upportedAccording to a Schedule 13D/A filed on November 14, 2023 with the SEC, as of November 9, 2023, Decheng Capital Management III (Cayman), LLC (“Decheng Capital Management”) and Xiangmin Cui may be deemed to beneficially own 6,582,380 shares which are held by these results,Decheng Capital China Life Sciences USD Fund III, L.P. (“Decheng”). Decheng Capital Management is the general partner of Decheng. Dr. Cui is the sole manager of Decheng Capital Management and as part of the AbbVie Agreement, we initiated Synergy, an international, double-blind placebo-controlled Phase 2 study of acazicolcept in patients with SLE in mid-2021.
Davoceticept, a Conditional CD28 Costimulator and Dual PD-L1/CTLA-4 Inhibitor for Oncology
Immune checkpoint blockade using inhibitors of the cytotoxic T-lymphocyte antigen 4, or CTLA-4, or programmed death 1, or PD-1, pathways have been therapeutically successful for a wide variety of malignancies, dramatically altering the
treatment paradigmmay be deemed to have voting and investment power with respect to the shares held by Decheng and as a result may be deemed to have beneficial ownership of such shares. Additionally, Decheng Capital Global Healthcare Fund (Master), LP (“Decheng Healthcare”) directly holds an additional 1,735,544 shares of our common stock. Decheng Capital Global Healthcare GP, LLC (“Decheng Healthcare GP”) is the general partner of Decheng Healthcare and Dr. Cui is the indirect managing member and ultimate beneficial owner of Decheng Healthcare GP, and may be deemed to have beneficial ownership over the shares held by Decheng Healthcare. The address for each of the persons and entities referred to in oncology. However,this footnote is 3000 Sand Hill Road, Building 2, Suite 110, Menlo Park, California 94025.
(2)According to a Schedule 13G/A filed on February 14, 2024 with the majoritySEC, as of patients treated withDecember 31, 2023, RA Capital Management, L.P. ("RA Capital") may be deemed to beneficially own (i) 4,909,763 shares of our common stock and (ii) 993,471 shares of our common stock issuable upon the exercise of outstanding warrants. All of such shares are held by RA Capital Healthcare Fund, L.P. ("RA Healthcare Fund") and RA Capital serves as its investment advisor. RA Capital Healthcare Fund GP, LLC is the general partner of RA Healthcare Fund. The general partner of RA Capital is RA Capital Management GP, LLC, of which Peter Kolchinsky and Rajeev Shah are the controlling persons. RA Healthcare Fund has delegated to RA Capital the sole power to vote and the sole power to dispose of all securities held in RA Healthcare Fund's portfolios. Because RA Healthcare Fund has divested itself of voting and investment power over the reported securities it holds and may not revoke that delegation on less than 61 days’ notice, RA Healthcare Fund disclaims beneficial ownership of the shares it holds for purposes of Section 13(d) of the Act and, therefore, disclaims any obligation to report ownership of the reported securities under Section 13(d) of the Act. As managers of RA Capital, Dr. Kolchinsky and Mr. Shah may be deemed beneficial owners of the shares beneficially owned by RA Capital. RA Capital, Dr. Kolchinsky, and Mr. Shah disclaim beneficial ownership of the shares held by RA Healthcare Fund, except to the extent of its or his pecuniary interest therein, if any. The address for RA Capital is 200 Berkeley Street, 18th Floor, Boston MA 02116. After December 31, 2023, we issued to RA Capital 3,199,879 shares of our common stock upon the exercise of outstanding warrants to purchase an inhibitoraggregate of CTLA-4, PD-1, or programmed death-ligand 1, or PD-L1, fail3,200,000 shares of our common stock, pursuant to respond or develop resistance, indicatinga net exercise mechanism under the warrants. Such warrants were subject to a beneficial ownership limitation, which provided that additional strategiessuch warrants may not be exercised if, after such exercise, the reporting person would beneficially hold more than 9.99% of our company's common stock. Neither RA Capital nor RA Healthcare Fund have publicly disclosed their beneficial ownership of our securities as of any date subsequent to improve anti-tumor immunity and responses remain needed to improve outcomes. Because immune checkpoints like PD-1 and CTLA-4 appear to suppress anti-tumor immune responses in part by inhibiting the activating signals mediated by cluster of differentiation 28, or CD28 (Figure 13), next generation immunotherapeutic strategies may substantially improve anti-tumor responses by activating a T cell (co-)stimulatory pathway(s) while also inhibiting a checkpoint pathway(s). Preferably, such activity might be focused primarily within the tumor microenvironment to limit potential systemic immune activation and toxicity.
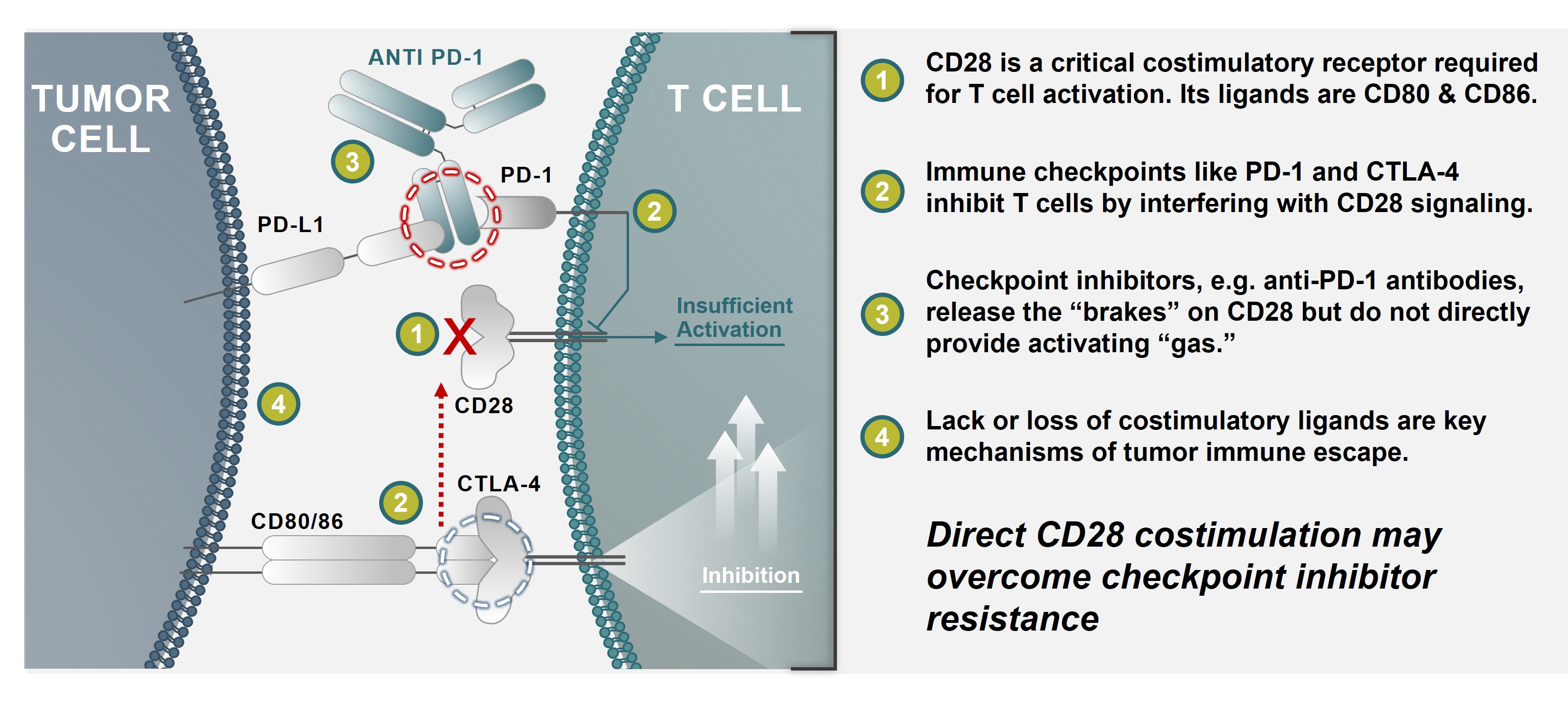
Figure 13December 31, 2023.Davoceticept(3)According to a Schedule 13G/A filed on February 14, 2024 with the SEC, as of December 31, 2023, Lynx1 Master Fund LP ("Lynx1 Fund") beneficially owns 3,813,926 shares of our common stock. Lynx1 Capital Management LP ("Lynx1 Management") is an Fc fusion proteinthe investment manager to Lynx1 Fund, and Weston Nichols is the sole member of Lynx1 Capital Management GP LLC, the general partner of Lynx1 Management. Mr. Nichols shares voting and investment power over the shares held by Lynx1 Fund and as a modified human clusterresult may be deemed to have beneficial ownership of differentiation 80, or CD80, vIgD designedsuch shares. The address for the foregoing persons is c/o Lynx1 Capital Management LP, 151 Calle de San Francisco, Suite 200, PMB 1237, San Juan, PR 00901.
(4)According to blocka Schedule 13G/A filed on February 14, 2024 with the inhibitory immune checkpoints PD-L1SEC, as of December 31, 2023, Great Point Partners, LLC ("Great Point") may be deemed to beneficially own (i) 2,103,550 shares which are held by Biomedical Value Fund, L.P, ("BVF") (ii) 1,348,048 shares held by Biomedical Offshore Value Fund, Ltd. ("BOVF"), and CTLA-4,(iii) 251,835 which are held by Cheyne Global Equity Fund (an Open-Ended Fund of Cheyne Select Master Fund ICAV) ("CGEF"). Each of Dr. Jeffrey R. Jay, M.D., as Senior Managing Member of Great Point, and to provide PD-L1-dependent T cell activation via CD28 costimulatory receptor. As illustrated in Figure 14, davoceticept binds PD-L1 expressed on the tumor, blocking PD-L1/PD-1 interactions. LocalizedMr. Ortav Yehudai, as Managing Director of Great Point, has voting and investment power with respect to the tumor, davoceticeptshares held by BVF, BOVF, and CGEF. Each of Dr. Jay and Dr. Yehudai disclaim beneficial ownership of the shares held by BVF, BOVF, and CGEF, except to the extent of their respective pecuniary interests. The address for Great Point is able165 Mason Street, 3rd Floor, Greenwich, CT 06830.
(5)Consists of (i) 1,468,532 shares of our common stock issuable upon the exercise of options within 60 days of April 9, 2024, and (ii) 2,627,157 shares of our common stock held directly by Alpine Immunosciences, L.P.. Dr. Gold and Dr. Venkatesan are the Managing Partners of Alpine BioVentures GP, LLC. Dr. Gold and Dr. Venkatesan are also limited partners of Alpine Immunosciences, L.P. By virtue of such relationships, Dr. Gold and Dr. Venkatesan may be deemed to providehave voting and investment power with respect to the shares held by Alpine Immunosciences, L.P. and as a CD28 signalresult may be deemed to T cells (PD-L1-dependent CD28 costimulation). Additionally, davoceticept binds CTLA-4 expressed on T cellshave beneficial ownership of such shares. Each of Dr. Gold and blocks CTLA4-CD80/CD86 interactions.Dr. Venkatesan disclaims beneficial ownership of the shares held by Alpine Immunosciences, L.P., except to the extent of his pecuniary interest therein, if any. The address for Alpine Immunosciences, L.P. is 117 E Louisa Street #256, Seattle, Washington 98101.
(6)Consists of (i) 24,371 shares of our common stock held directly by Dr. Peng and (ii) 1,007,904 shares of our common stock issuable upon the exercise of options within 60 days of April 9, 2024.
(7)Consists of 231,107 shares of our common stock issuable upon the exercise of options within 60 days of April 9, 2024.
(8)Consists of (i) 57,891 shares of our common stock issuable upon the exercise of options within 60 days of April 9, 2024, and (ii) 2,416,181 shares of our common stock held directly by OrbiMed Private Investments VI, LP (“OPI VI”), and (iii)

Figure 14(9)Consists of (i) 57,891 shares of our common stock issuable upon the exercise of options within 60 days of April 9, 2024, (ii) according to a Form 4 filed with the SEC on December 13, 2023, 1,629,366 shares of our common stock held directly by Frazier Life Sciences VIII, L.P. and (iii) according to a Schedule 13D/A filed with the SEC on November 22, 2023, 1,200,000 shares of our common stock issuable upon the exercise of warrants held by Frazier Life Sciences VIII, L.P. exercisable within 60 days of April 9, 2024, 1,501,642 shares of our common stock held directly by Frazier Life Sciences Public Fund, L.P. and 1,702,127 shares of our common stock issuable upon the exercise of warrants held by Frazier Life Sciences Public Fund, L.P. exercisable within 60 days of April 9, 2024. FHMLSP, L.P. is the general partner of Frazier Life Sciences Public Fund, L.P. and FHMLSP, L.L.C. is the general partner of FHMLSP, L.P., Dr. Topper, Patrick J. Heron, Albert Cha and James Brush are the members of FHMLSP, L.L.C. and, therefore, share voting and investment power over the shares held by Frazier Life Sciences Public Fund, L.P. Dr. Topper and Messrs. Heron, Cha and Brush disclaim beneficial ownership of the shares held by Frazier Life Sciences Public Fund, L.P., except to the extent of their pecuniary interests in such shares, if any. The address for Frazier Life Sciences Public Fund, L.P. is 601 Union Street, Suite 3200, Seattle, Washington 98101. FHM Life Sciences VIII, LP is the general partner of Frazier Life Sciences VIII, L.P. and FHM Life Sciences VIII, L.L.C. is the general partner of FHM Life Sciences VIII, LP. Dr. Topper and Patrick J. Heron are the sole members of FHM Life Sciences VIII, L.L.C. and, therefore, share voting and investment power over the shares held by Frazier Life Sciences VIII, L.P. Dr. Topper and Mr. Heron disclaim beneficial ownership of the shares held by Frazier Life Sciences VIII, L.P., except to the extent of their pecuniary interests in such shares, if any. The address for Frazier Life Sciences VIII, L.P. is 601 Union Street, Suite 3200, Seattle, Washington 98101.
Using (10)in vitro assays, we have characterizedConsists of (i) 50,000 shares of our common stock held indirectly through the Robert E. Conway Revocable Trust and validated the three primary mechanismsCarolyn J. Conway Revocable Trust, and (ii) 65,746 shares of actionour common stock issuable upon exercise of davoceticept: conditional CD28 costimulation and dual PD-L1/CTLA-4 inhibition (Figure 15). Importantly, CD28 costimulation with davoceticept requires both T cell receptor, or TCR, signaling and expressionoptions within 60 days of PD-L1 on the antigen presenting cell, or APC. PD-L1 dependent T cell costimulation is a unique and key attribute of davoceticept which we believe may limit the risk for systemic toxicity while providing potent anti-tumor activity.April 9, 2024.
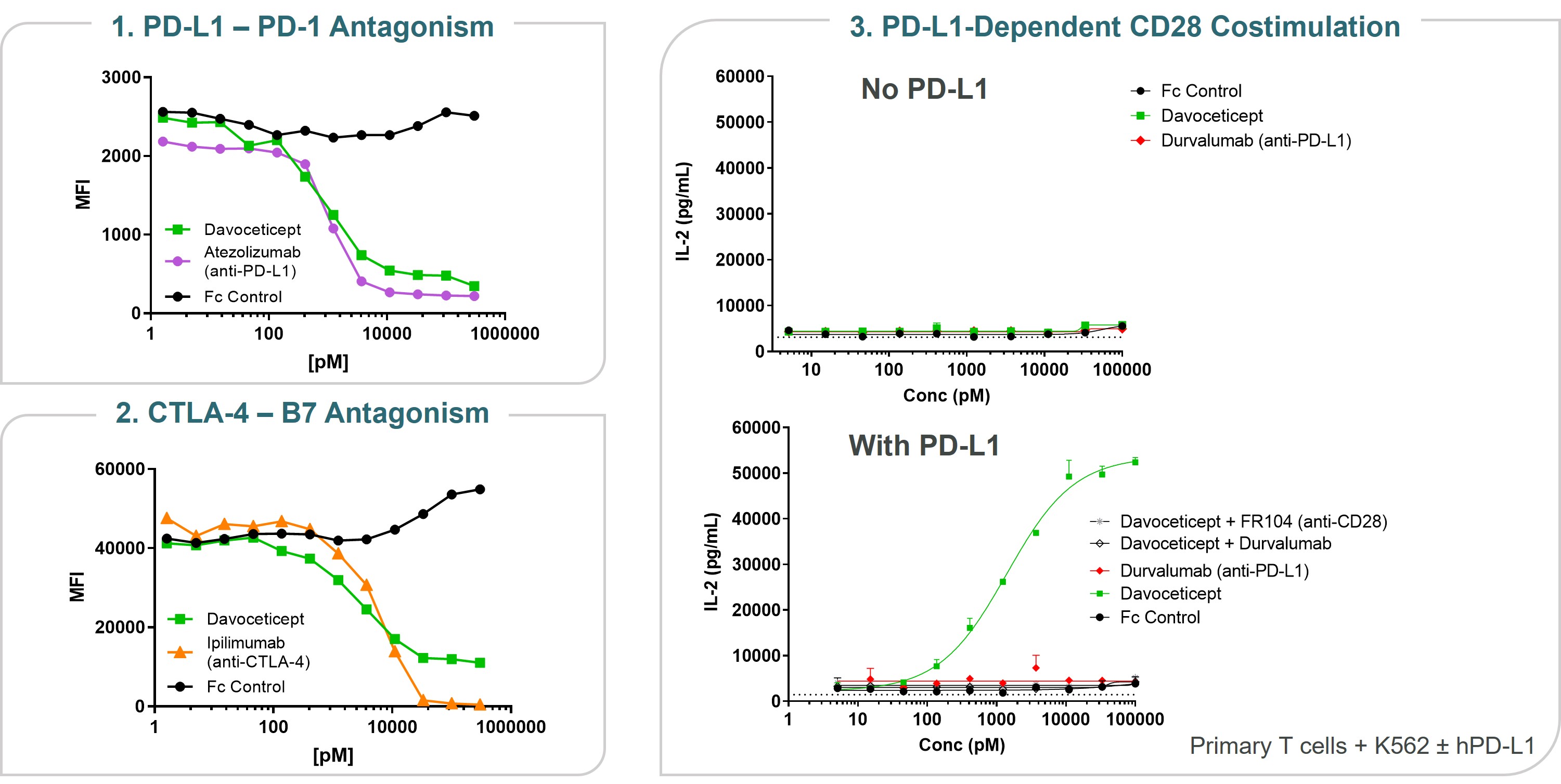
Figure 15(12)Consists of (i) 50,241 shares of our common stock issuable upon the exercise of options within 60 days of April 9, 2024, (ii) 6,582,380 shares of our common stock held directly by Decheng Capital China Life Sciences USD Fund III, L.P., and (iii) 1,735,544 shares of the Issuer’s Common Stock held directly by Decheng Healthcare. Decheng Healthcare GP is the general partner of Decheng Healthcare and Xiangmin Cui is the indirect managing member and ultimate beneficial owner of Decheng Healthcare GP. Decheng Healthcare, Decheng Healthcare GP and Dr. Cui may be deemed to share voting and dispositive power with respect to the shares held directly by Decheng Healthcare. Please see footnote 1 regarding Dr. Cui’s voting and investment power over the shares held by Decheng.
(13)Consists of 49,941 shares of our common stock issuable upon the exercise of options within 60 days of April 9, 2024.
(14)Consists of 29,513 shares of our common stock issuable upon the exercise of options within 60 days of April 9, 2024.
(15)Includes only current directors and executive officers serving in such capacity as of April 9, 2024. Includes 3,366,562 shares of our common stock issuable upon the exercise of options within 60 days of April 9, 2024 and 2,902,127 shares of our common stock issuable upon the exercise of warrants within 60 days of April 9, 2024.
Davoceticept has also been validatedItem 13. Certain Relationships and Related Transactions, and Director Independence.
In addition to the compensation arrangements, including employment, termination of employment and change-in-control arrangements discussed, when required, elsewhere in this report, the following is a description of each transaction since January 1, 2022 and each currently proposed transaction in vivo tumor models. In a mouse model where mice were implanted with MC38 mouse colon cancer tumors transfected with human PD-L1, davoceticept as a monotherapy demonstrated superior tumor control compared to durvalumab, an FDA-approved anti-PD-L1 monoclonal antibody (Figure 16).
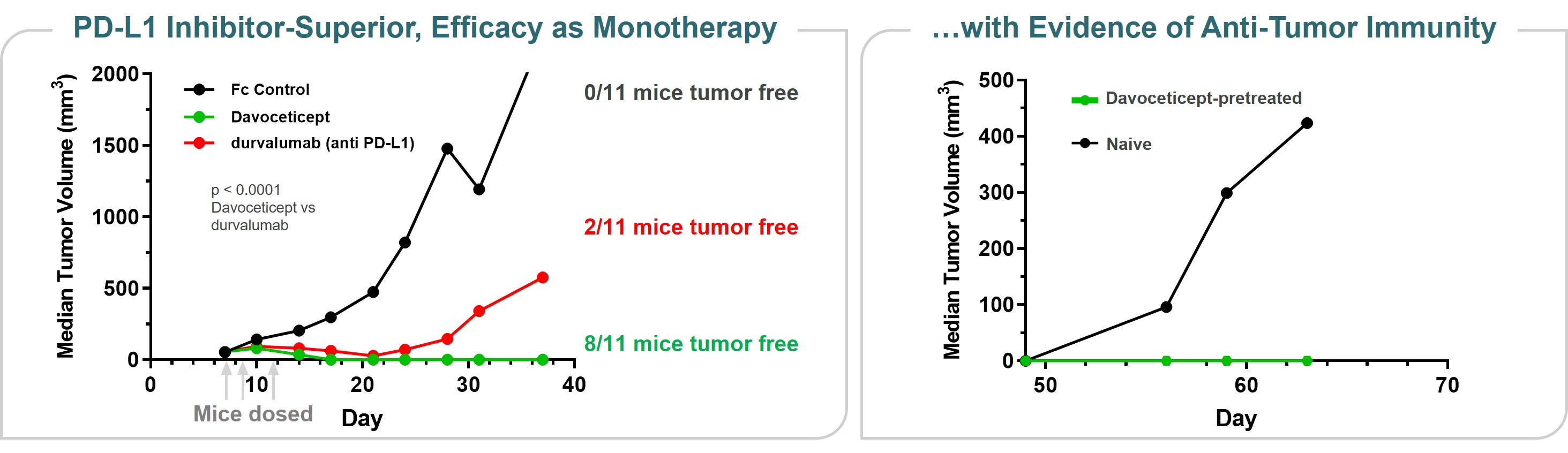
Figure 16 Figure 17which:When •tumor-free mice in the davoceticept arm were re-challenged with additional tumors, they continuedwe have been or are to be tumor free despite no additional doses of davoceticept. (Figure 17)
The effects of davoceticept or durvalumab on various components of the immune system were compared using a technique called RNA sequencing in the tumor model. This technique generates a display of inflammatory gene signatures where green represents lower or no upregulation of inflammatory genes and red represents higher upregulation of inflammatory genes. For the treatment of cancer, it is thought the more upregulation of the immune system - indicated by higher upregulation of inflammatory genes - potentially results in better outcomes for patients. As seen in Figure 18 below, davoceticept upregulates a broader variety of different inflammatory genes connected with several different types of immune cells compared to durvalumab.
Figure 18
Summary of davoceticept Preclinical Data
We have demonstrated in preclinical studies that davoceticept:participant;
•exhibits three primary mechanisms of action: conditional CD28 costimulation and dual PD-L1/CTLA-4 inhibition;
•improves tumor control in a human PD-L1 transduced mouse model of colon cancer compared to the FDA approved anti PD-L1 therapeutic durvalumab;
•demonstrates a more robust intra-tumor inflammatory signature in the mouse colon cancer model than durvalumab, potentially indicating superior immune system upregulation to fight cancer;amount involved exceeds $120,000; and
•hasany of our directors, executive officers or holders of more than 5% of our capital stock, or any immediate family member of or person sharing the potential to be used ashousehold with any of these individuals, had or will have a monotherapydirect or in combination with standard of care chemotherapy or checkpoint only inhibition.indirect material interest.
Davoceticept Clinical Development
In June 2020,addition to the arrangements described below, we initiated enrollmenthave also entered into the arrangements which are described under the caption “Executive Compensation — Executive Employment Arrangements with Our Named Executive Officers.”
Policies and Procedures for Related Party Transactions
Our board of directors has adopted a written policy governing the review and approval of related party transactions. The audit committee of our board of directors has the primary responsibility for reviewing and approving or disapproving related party transactions, as designated in NEON-1,the audit committee charter. In addition, our Phase 1 studyCode of Business Conduct and Ethics requires that each of our employees and directors inform his or her superior or the chairman of the audit committee, respectively, of any material transaction or relationship that comes to his or her attention that could reasonably be expected to create a conflict of interest. Further, at least annually, each director and executive officer will complete a detailed questionnaire that asks questions about any business relationship that may give rise to a conflict of interest and all transactions in patientswhich we are involved and in which an executive officer, a director or a related person has a direct or indirect material interest.
Affiliations with advanced malignancies (Figure 19). NEON-1 enrolls adultsPrincipal Stockholders
Dr. Gold is an executive officer, a member of our board of directors and, in his individual capacity, a limited partner of Alpine Immunosciences, L.P., Delaware limited partnership. In addition, Dr. Gold, in his individual capacities, is a Managing Partner of Alpine BioVentures, GP, LLC, a Delaware limited liability company, which is the general partner of Alpine Immunosciences, L.P.
Dr. James N. Topper is a member of our board of directors and, in his individual capacity, is a managing member of FHM Life Sciences VIII, LLC, a Delaware limited liability company. FHM Life Sciences VIII, LLC is the general partner of FHM Life Sciences VIII, LP, a Delaware limited partnership. FHM Life Sciences VIII, LP is the general partner of Frazier Life Sciences VIII, L.P., a Delaware limited partnership. Dr. Topper is also, in his individual capacity, a member of FHMLSP, L.L.C., a Delaware limited liability company, the general partner of FHMLSP, L.P., a Delaware limited partnership, which is the general partner of Frazier Life Sciences Public Fund L.P., a Delaware limited partnership. Frazier Life Sciences Public Fund L.P. is a holder of our common stock.
Mr. Peetz is a member of our board of directors and, in his individual capacity, is an Entrepreneur-in-Residence at Frazier Healthcare Partners, which is an affiliate of the Frazier entities described above.
Dr. Peter Thompson is a member of our board of directors and, in his individual capacity, is a member of OrbiMed Advisors LLC. OrbiMed Advisors LLC is the managing member of OrbiMed Capital GP VI LLC. OrbiMed Capital GP VI LLC is the general partner of OrbiMed Private Investments VI, LP.
Dr. Xiangmin Cui is a member of our board of directors and, in his individual capacity, is the manager of Decheng Capital Management III (Cayman), LLC, which in turn is the general partner of Decheng Capital China Life Sciences USD Fund III, L.P. Decheng Capital China Life Sciences USD Fund III, L.P.
Indemnification Agreements
We have entered into indemnification agreements with advanced solid tumorseach of our directors and with each of our executive officers. Pursuant to the indemnification agreements, we have agreed to indemnify and hold harmless these directors and officers to the fullest extent permitted by applicable law. The agreements generally cover expenses that a director or lymphoma refractoryofficer incurs or resistant to standard therapy, including checkpoint inhibitors when indicated. Measurable disease is required for most participants, as are an ECOG status of 0 to 2 and adequate hematological, renal, and hepatic function. Safety endpoints include dose-limiting toxicities, adverse events, andamounts
circulating cytokines. Objective responses willthat a director or officer becomes obligated to pay because of any proceeding to which he or she is made or threatened to be assessedmade a party or participant by RECIST v1.1 for solid tumors and Lugano criteria for lymphoma. Pharmacokinetics and pharmacodynamics will also be evaluated. More information is available at www.clinicaltrials.gov (NCT04186637).
Figure 19
Initial data from NEON-1 were presented at the 2021 ASCO Virtual Meeting demonstrating that davoceticept was well-tolerated asreason of the cutoff date (April 22, 2021) with evidence of peripheral T cell modulation consistent with CD28 agonism. In addition, although most enrolled participants had tumors considered classically non-responsive to immunotherapies, the majority derived clinical benefit as definedhis or her service as a best outcomecurrent or former director, officer, employee or agent of stable disease or better (Figure 20). Completion of dose escalation for NEON-1 and initiation of monotherapy expansion cohorts, initially cutaneous melanoma and PD-L1+ (non-melanoma) tumors, is anticipated in the first half of 2022.
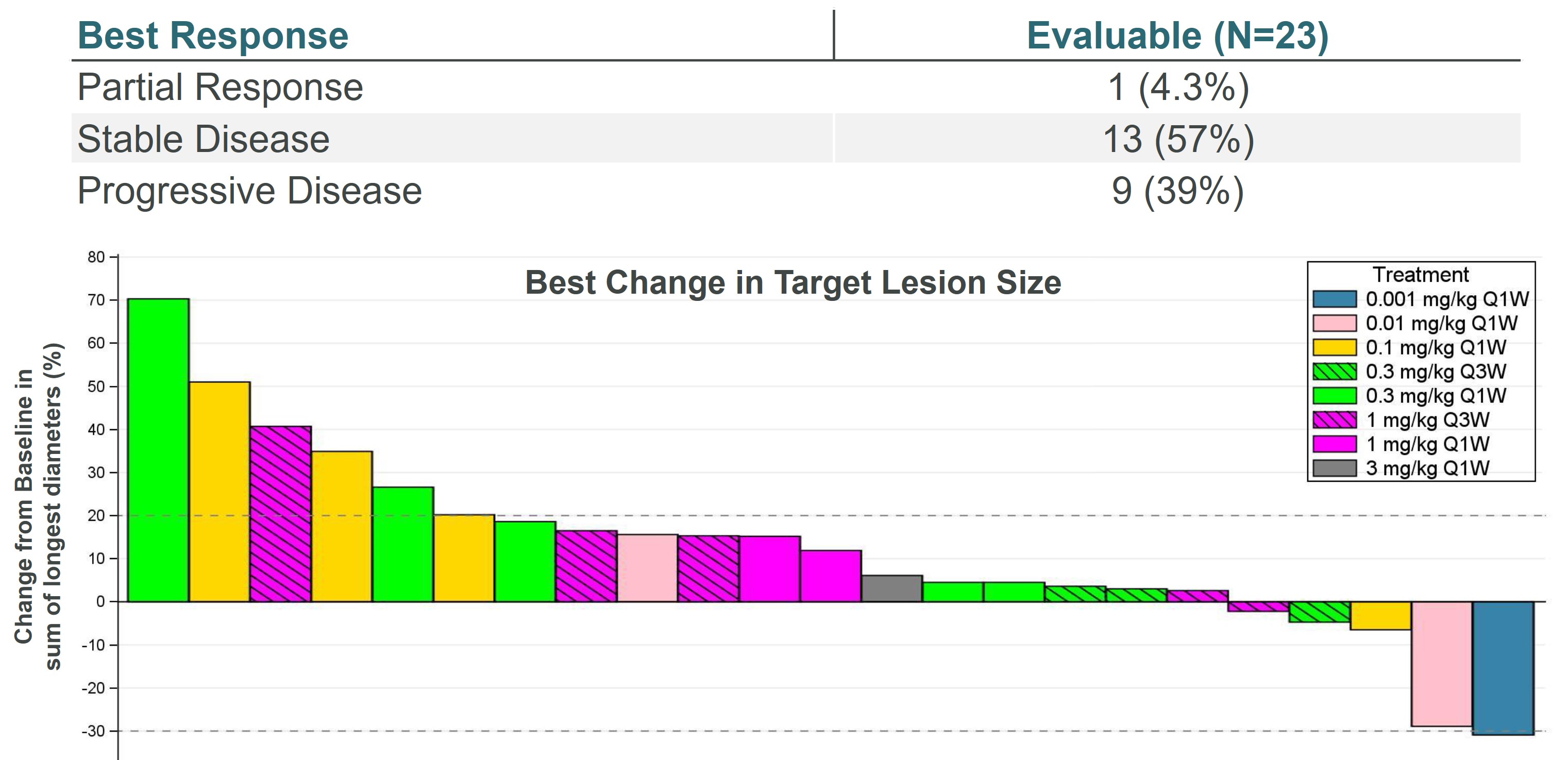
Figure 20In June 2021, we announced a collaboration and supply agreement with Merck to evaluate the safety and efficacy of davoceticept in combination with Merck’s anti-PD-1 therapy KEYTRUDA® (pembrolizumab) in a Phase 1 dose escalation and
expansion study.Alpine Immune Sciences. The clinical trial, NEON-2, was initiated in June 2021. More information for both NEON-1 (NCT04186637) and NEON-2 (NCT04920383) is available at www.clinicaltrials.gov.
On March 7, 2022, we announced that the FDA placed a partial clinical hold on the NEON-2 trial evaluating davoceticept in combination with pembrolizumab in adults with advanced malignancies. This partial clinical hold was prompted by report of a death of a study participant in the NEON-2 trial. The participant had choroidal melanoma previously treated with nivolumab and ipilimumab, and had received a single dose each of davoceticept and pembrolizumab. The participant’s death was attributed to cardiogenic shock, considered by the treating physicians as likely related to immune-mediated myocarditis, or possibly infection. We are working closely with the FDA, Merck, the study Safety Monitoring Committee, and the study investigators to further understand this event and to evaluate the appropriate safety precautions before we resume enrollment of additional participants. Participants who are currently enrolled in the NEON-2 trial may continue to receive davoceticept and pembrolizumab, although no new participants may be enrolled until the partial clinical hold is resolved. This partial clinical hold does not affect the ongoing NEON-1 clinical trial of davoceticept as monotherapy (NCT04186637).
ALPN-303, a Dual B Cell Cytokine Antagonist for B Cell-Mediated Autoimmune/Inflammatory Diseases
ALPN-303 is a dual B cell cytokine antagonist, being developedagreements also provide for the treatmentadvancement of B cell mediated inflammatoryexpenses to the directors and autoimmune diseases. Engineered using our proprietary directed evolution platform, ALPN-303 is an Fc fusion protein of a human transmembrane activator and CAML interactor, or TACI, variant TNFR domain, or vTD, that potently inhibits the pleiotropic B cell cytokines BAFF and APRIL. It mediates significantly improved combined BAFF and APRIL inhibition in vitro and enhanced pharmacokinetic and immunomodulatory properties in vivo, as comparedexecutive officers subject to wild-type, or WT, TACI-Fc molecules. BAFF and APRILspecified conditions. There are TNF superfamily members that bind TACI, BCMA, and/or BAFF-R on B cells and together support B cell development, differentiation, and survival. Their co-neutralization dramatically reduces B cell survival and function, including antibody production, whereas inhibition of either BAFF or APRIL alone mediates only modest effects (Figure 21). B cell targeting therapies like the WT TACI-Fc fusions atacicept and telitacicept have demonstrated promising clinical potential in B cell-related diseases like SLE but have not yet clearly exhibited long-term and/or complete disease remissions. ALPN-303, with enhanced inhibitory activity against BAFF & APRIL, could further improve clinical outcomes.
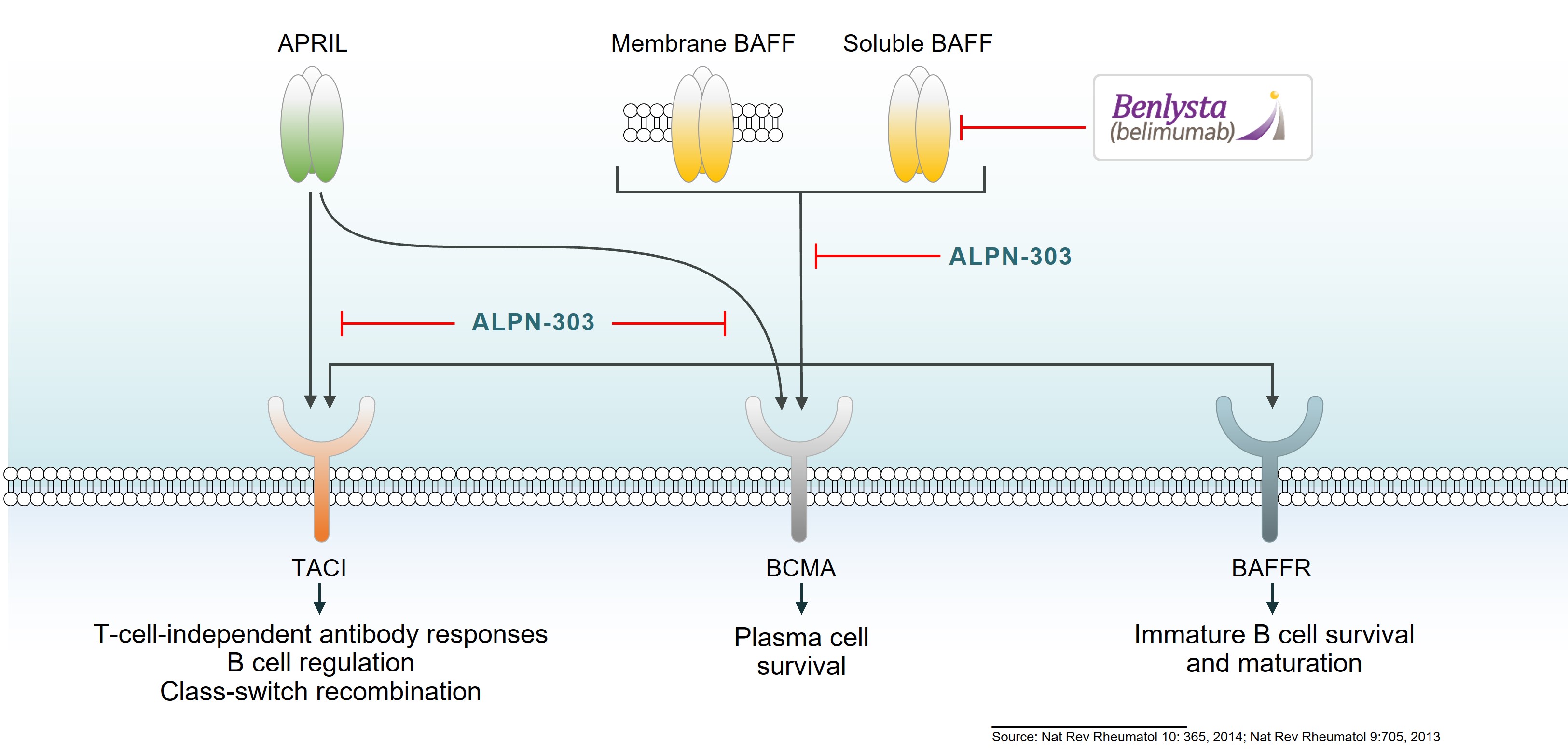
Figure 21
Data on ALPN-303 presented at the ACR Convergence 2021 Annual Meeting demonstrated that ALPN-303 inhibited the activity of the B cell cytokines APRIL and BAFF more potently than wild-type TACI-Fc counterparts, as well as anti-BAFF and anti-APRIL monoclonal antibodies. In Figure 22, ALPN-303 inhibited BAFF- and APRIL-mediated signaling in vitro with significantly lower IC50 values than WT TACI-Fc, anti-BAFF mAb, and anti-APRIL mAb comparators.
Figure 22
Administration of ALPN-303 rapidly and significantly reduced key B lymphocyte subsets including plasma cells, germinal center, and follicular B cells in a KLH immunization mouse model (Figure 23).
Figure 23
Furthermore, treatment with ALPN-303 potently suppressed anti-dsDNA auto antibodies, proteinuria, sialoadenitis, kidney lesions, and renal immune complex deposition in the (NZBxNZW)F1 lupus model (Figure 24).
Figure 24
ALPN-303 has been well-tolerated in nonclinical studies (Figure 25) and exhibited superior pharmacokinetics and pharmacodynamics than wild-type TACI-Fc counterparts, including superior serum exposure and pharmacodynamic reduction of serum immunoglobulins in mice and/or cynomolgus monkeys (Figure 26). Based on these data, we believe that ALPN-303 represents an attractive development candidate for the treatment of multiple autoimmune and inflammatory diseases, including SLE and other autoantibody-mediated diseases.
Figure 25
Figure 26
ALPN-303 Clinical Development
Enrollment in a first-in-human, Phase 1 study of ALPN-303 in healthy volunteers began in the fourth quarter of 2021, with top-line results targeted by mid-2022. This randomized, placebo-controlled study is designed to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of ALPN-303 administered intravenously and subcutaneously. We are planning to initiate patient-based studies in the second half of 2022 including SLE, and possibly other autoantibody-related diseases in other therapeutic areas.
Other Research Programs
In additioncertain exceptions to our acazicolcept, davoceticept,obligation to indemnify the directors and ALPN-303 programs, we have a number of other research efforts underway to address cancer and autoimmune/inflammatory diseases that we intend to continue to develop either internallyofficers, including any intentional misconduct or together with a partner.
Partnerships
In addition to advancing programs internally, we continue to seek partners who can bring therapeutic area experience, development expertise, commercialization capabilities, and funding to maximizeact where the potential ofdirector or officer did not in good faith believe he or she was acting in our existing programs and scientific platform.
Collaboration with Horizon (December 2021)
In December 2021, we entered into the Horizon Agreement, pursuant to which we granted to Horizon rights to one of our existing preclinical biologic therapeutic programs, or the Existing Program, and agreed to collaborate with Horizon in the discovery, research and preclinical development of up to three additional autoimmune and inflammatory disease programs for other designated biological targets, or the Research Programs, and products arising from the Existing Program and the Research Programs, or the Agreement Products.
Existing Program
We granted to Horizon an exclusive, worldwide, royalty-bearing, sublicensable license under our intellectual property rights for the development, manufacture and commercialization of Agreement Products associated with the Existing Program, or the Existing Program Products, for all applications; however, Horizon has agreed not to pursue the Existing Program Products for oncology. In addition, we granted to Horizon a non-exclusive license to access our libraries of proteins and molecules for research, discovery and identification of additional compounds meeting the agreed criteria (subject to certain exceptions, including compounds reserved by us from the libraries, as set forth in the Horizon Agreement) which would be included in Horizon’s license.
We will conduct our activities under the Existing Program and deliver compounds meeting the agreed criteria pursuant to one or more deliverables plans as mutually agreed by the parties. In addition, Horizon will pay us for the costs and expenses of conducting such activities under the deliverables plans.
Research Programs
Under the Horizon Agreement, during a limited research term, we will conduct, together with Horizon, up to three Research Programs for specified biological targets selected by Horizon (subject to certain exceptions, including targets reserved by us) to discover proteins and molecules with specified attributes and biological functions, pursuant to a mutually agreed research plan for each such Research Program. Each party has the right to reserve certain targets and to replace such reserved targets or reserve more targets (up to a fixed number) through a gatekeeper mechanism. We will deliver certain compounds meeting the agreed criteria for each Research Program as mutually agreed upon by the parties. Horizon will pay us for the costs and expenses for conducting its activities under the Research Programs. As agreed between the parties, Horizon will own the inventions and intellectual property developed from the Research Programs or otherwise associated with the Agreement Products arising therefrom.
Governance
The parties will establish a joint research committee composed of an equal number of representatives from each of Alpine and Horizon, which will, among other responsibilities, oversee and govern the Existing Program and the Research Programs and review and approve research plans. The parties will also form a joint patent committee comprised of one or two representatives of each party to discuss strategy and facilitate communication and coordination for prosecution and maintenance of the relevant patents under the Horizon Agreement.
Development, Manufacture, Regulatory and Commercialization.
Horizon will have the sole right and responsibility for the development, manufacturing, regulatory and commercialization of all the Agreement Products. We will provide Horizon reasonable assistance and cooperation. In addition, we have agreed during the term of the Horizon Agreement not to conduct or enable any third party to conduct the development, manufacture or commercialization of certain competing products as set forth therein.
Financial Terms
In connection with the transaction, Horizon made an upfront payment to us of $25.0 million as well as an equity investment for which they paid $15.0 million, a 25 percent premium to the 30-day volume-weighted average share price as of December 9, 2021.
In addition to the upfront payment and equity consideration, Horizon has agreed to make milestone payments to us upon our achievement of certain preclinical, clinical and regulatory and commercialization milestones, up to an aggregate amount of $381.0 million per program, or approximately $1.5 billion in total, if all milestones are met.
Horizon has further agreed to pay the Company royalties based on future net sales of the Agreement Products. For the Existing Program, such royalty percentages range from a mid-single digit percentage to a low double-digit percentage of net sales, with the specific royalty rate depending on the aggregate net sales. For each Research Program, such royalty percentages are in a range of mid-single digit percentages of net sales, with the specific royalty rate depending on the aggregate net sales. Horizon’s obligations to pay royaltiesbest interests, with respect to an Agreement Product“short-swing” profit claims under Section 16(b) of the 1934 Act and, country will expire after specific criteria for such Agreement Product in such country including it no longer being covered by valid claims of applicable patent rights in such country, or the Royalty Term. Royalty payments are subject to reduction in specified circumstances, including expiration of patent rights, biosimilar competition, or Horizon is required to make payments to a third partywith certain exceptions, with respect to an Agreement Product.proceedings that he or she initiates.
TermIndebtedness of Directors and TerminationOfficers
Unless earlier terminated, the Horizon Agreement remains in effect until the expiration of the Royalty Term for all Agreement Products. The Horizon Agreement is subject to customary termination provisions including termination by a party for the other party’s uncured, material breach. Additionally, Horizon may terminate the Horizon Agreement with specified prior notice in its entirety or on a Program-by-Program basis, for any or no reason.
In the event of certain terminations of the Horizon Agreement, at our request, the parties will negotiate one or more licenses for us to develop, manufacture and commercialize terminated Agreement Products.
The Horizon Agreement includes certain other customary terms and conditions, including mutual representations and warranties, indemnification and confidentiality provisions.
Collaboration with AbbVie (June 2020)
In June 2020, we entered into the AbbVie Agreement, which grants to AbbVie an exclusive option to take an exclusive license to acazicolcept, or the License Option.
Under the terms of the AbbVie Agreement we granted to AbbVie an exclusive option to obtain an exclusive, royalty-bearing, sublicensable license to certain intellectual property rights for the research, development and commercialization of acazicolcept and any other molecule owned or controlled by us that binds to or directly modulates or targets ICOS at certain agreed-upon levels, or the Compounds, on a worldwide basis for all human and non-human diagnostic, prophylactic and therapeutic uses, subject to certain exceptions set forth in the AbbVie Agreement. The License Option is immediately exercisable and will expire 90 days following our delivery of the data package described below to AbbVie, subject to certain exceptions, including clearance under the Hart-Scott-Rodino Antitrust Improvements Act of 1976, or the HSR Act, if required.
Financial Terms
In connection with the execution of the AbbVie Agreement, AbbVie paid us a nonrefundable upfront payment of $60.0 million in cash. If AbbVie exercises its License Option, they will pay a one-time cash payment of $75.0 million.
In addition to the upfront payment and License Option payment, AbbVie has agreed to make cash payments upon our achievement of certain development milestones, the Alpine Development Milestones, prior to the exercise of the License Option as set forth in a written development plan, up to an aggregate amount of $75.0 million. In the second quarter of 2021, we achieved $45.0 million of the Alpine Development Milestones. Following the exercise of the License Option, AbbVie has agreed to make cash payments of up to $205.0 million upon AbbVie’s achievement of certain development and commercial milestones and additional cash payments of up to $450.0 million upon AbbVie’s achievement of certain sales-based cash milestones. AbbVie has further agreed to pay royalties from a high-single digit percentage to a low double-digit percentage of net sales of any pharmaceutical product that contains a Compound, or a Licensed Product, with the specific royalty rate depending on the aggregate net sales. AbbVie’s obligations to pay royalties with respect to a Licensed Product and country will expire upon the latest of the expiration of the last to expire valid patent claim applicable to such Licensed Product in such country, 10 years from the first commercial sale of the Licensed Product in such country, and the expiration of regulatory exclusivity for such Licensed Product in such country. Royalty payments are subject to reduction in specified circumstances, including expiration of patent rights, if average net sales decrease by a certain percentage after the introduction of a generic product, or if AbbVie is required to pay amounts to a third party in order commercialize a Licensed Product in a particular country.
Development Activities
Prior to the exercise of the License Option, we will conduct acazicolcept development efforts as per a development plan providing for, among other things, the generation of a data package in order for AbbVie to evaluate exercising the License Option and an itemized budget for such activities, including all activities reasonably necessary to conduct a Phase 2 clinical study of acazicolcept for the treatment of systematic lupus erythematosus; all non-clinical activities; and all CMC activities agreed to under the development plan. We will be fully responsible for all costs incurred to conduct our activities, provided that, AbbVie may be responsible for increased costs under the development plan in connection with certain material amendments agreed upon with AbbVie.
Prior to the exercise of the License Option, we will be solely responsible, at our sole cost and expense, for preparing, filing and maintaining regulatory documentation, which AbbVie will be entitled to access and review. We will also be responsible for any and all correspondence with the applicable regulatory authorities and for maintaining all data related to acazicolcept. We will be solely responsible, at our sole cost and expense, for manufacturing the compounds necessary to complete the development activities consistent with the development plan.
Governance
The parties will establish a joint governance committee, or JGC, composed of an equal number of representatives from each of Alpine and AbbVie, which will, among other responsibilities, coordinate and oversee the development activities,
approve amendments to the development plan and discuss interactions with regulatory authorities. The chairperson of the JGC will be appointed by AbbVie. AbbVie may disband the JGC, at its sole discretion, following the exercise of the License Option.
Commercialization
Upon AbbVie’s exercise of the License Option, AbbVie and its affiliates will be solely responsible, at AbbVie’s sole cost and expense, for the development, manufacture, commercialization, and regulatory compliance of any Licensed Product. Following exercise of the License Option, AbbVie shall use commercially reasonable efforts to develop and seek regulatory approval for one of the compounds in one indication in each of the United States and one of the United Kingdom, Germany, France, Spain, or Italy, or the Major Markets, and, following receipt of any such regulatory approval, commercialize the compound in such country.
Changes in Control
We will notify AbbVie immediately upon the closing of any change in control (as defined in the AbbVie Agreement) during the term of the AbbVie Agreement. Following the delivery of such notice, AbbVie may, in its sole and absolute discretion, elect to continue the AbbVie Agreement subject to certain modifications as set forth in the AbbVie Agreement, including the assumption by AbbVie of responsibility to perform certain activities previously assigned to us.
Term and Termination
Unless earlier terminated, the AbbVie Agreement shall terminate either: (i) in the event that the License Option is not exercised by AbbVie, the first day following the last day of the License Option exercise period; or (ii) in the event that the License Option is exercised by AbbVie, the date of the expiration of the last Royalty Term for the last Licensed Product.
Both us and AbbVie may terminate the AbbVie Agreement upon written notice in the event of a material breach by the other party that has not been cured within a 90-day cure period. However, if the uncured material breach is with respect to AbbVie’s obligation to use commercially reasonable efforts to obtain regulatory approval for and commercialize a Licensed Product with respect to any Major Market (but not all Major Markets), then we will only be entitled to terminate the AbbVie Agreement with respect to such Major Market(s). Both AbbVie and us may also terminate the AbbVie Agreement upon written notice if the other party voluntarily or involuntarily files for bankruptcy or insolvency, makes an assignment for the benefit of creditors, has a receiver or trustee appointed over substantially all of such other party’s property, proposes or is party to any dissolution or liquidation, or admits in writing its inability generally to meet such other party’s obligations as they fall due in the general course.
AbbVie may terminate the AbbVie Agreement in its entirety or on a country-by-country basis, for any or no reason, by providing at least 90 days’ prior written notice to us. AbbVie may also terminate the AbbVie Agreement upon notice to us if (i) either we or AbbVie receives a second request for additional information under the HSR Act, provided AbbVie’s notice of termination is delivered within ten business days after AbbVie becomes aware of such request or receives notice from us regarding such request or (ii) the License Option has not been exercised or clearance under the HSR Act, if required, has not occurred within 180 days of submission of the parties’ request for such clearance, provided AbbVie’s notice of termination is delivered within ten business days after the end of such 180-day period.
Upon the termination of the AbbVie Agreement in its entirety for any reason, all licenses and other rights granted (i) to AbbVie by us and (ii) to us by AbbVie shall terminate. Upon termination in certain circumstances, AbbVie has agreed to grant to us licenses to certain intellectual property that is reasonably necessary, and that was actually used by AbbVie for the development, manufacturing or commercialization of the terminated products, to research, develop and commercialize the terminated products in the terminated countries.
In lieu of terminating the AbbVie Agreement in connection with an uncured material breach or the bankruptcy or insolvency of the Company, AbbVie may alternatively elect to continue the AbbVie Agreement subject to certain modifications, including that AbbVie will be entitled to conduct activities allocated to us under the Development Plan, subject to reimbursement by us for AbbVie’s out-of-pocket expenses in connection with such activities. If AbbVie’s right to terminate in connection with an uncured material breach or the bankruptcy or insolvency of the Company arises before exercise of the License Option, then the License Option exercise payment amount will be reduced by half and the amount of any then-unearned milestone payments will be reduced by half. If AbbVie’s right to terminate arises after exercise of the License Option, then the amount of any then-unearned milestone payments will be reduced by 25%.
The AbbVie Agreement includes certain other customary terms and conditions, including mutual representations and warranties, indemnification and confidentiality provisions.
Collaboration with Adaptimmune Therapeutics (May 2019)
In May 2019, we entered into a collaboration and license agreement with Adaptimmune, or the Adaptimmune Agreement, to develop next-generation SPEAR™ T cell products which incorporate our secreted and transmembrane immunomodulatory protein (termed SIP™ and TIP™) technology. We and Adaptimmune will collaborate on a specified number of programs to develop SIP and TIP candidates with tailored affinities and modulatory activities that may enhance the anti-tumor responses seen with Adaptimmune’s SPEAR™ T cells. For each program, Adaptimmune has an option to take a worldwide exclusive license for development and commercialization of SPEAR™ T cell products incorporating the developed SIP or TIP candidate for the treatment of cancer. Under the terms of the collaboration agreement, Adaptimmune provided an upfront payment and will provide research funding for ongoing programs. In addition, we may be eligible for downstream development and commercialization milestones up to $288.0 million, if all pre-specified milestones for each program are achieved. In addition, we are eligible to receive low-single digit royalties on worldwide net sales of the applicable products. In February 2022, Adaptimmune selected an additional research program, triggering a $1.0 million upfront payment, which will be recorded as deferred revenue upon receipt and recognized to revenue based on employee hours contributed.
Manufacturing
We have established in-house non-current good manufacturing practices, recombinant protein generation capabilities enabling our scientific platform, including validation of new scientific discoveries in vitro and in vivo. Having protein production capabilities in-house allows more rapid progression for vIgDs and vTDs generated by our scientific platform.
We have chosen U.S.-based contract drug substance and U.S. and Australian drug product manufacturers for our initial current good manufacturing practices, or cGMP, clinical trial supplies of acazicolcept, davoceticept and ALPN-303. We believe these contract manufacturers’ particular expertise in protein production, analytical development and fill/finish provide us with the capability to meet rapid timelines encompassing the development of production cell-lines to manufacturing of clinical trial quantities of the biopharmaceutical product.
We have successfully completed two cGMP manufacturing campaigns for acazicolcept and one cGMP campaign each for davoceticept and ALPN-303 and believe we have produced sufficient quantities of drug product necessary to execute our stated Synergy, NEON-1, NEON-2, and ALPN-303 Healthy volunteer clinical trials. We have not yet manufactured any of our proteins at commercial scale.
Competition
We participate in the highly competitive sector of biotechnology and pharmaceuticals and in the subsector of immune modulation. This subsector has undergone tremendous technological advancement over the last decade due to advancements in understanding the role of the immune system across multiple therapeutic areas, including oncology and autoimmune/inflammatory disease. While we believe our novel technology platform, discovery programs, knowledge, experience, and scientific resources offer competitive advantages, we face competition from major pharmaceutical and biotechnology companies, academic institutions, governmental agencies, public and private research institutions, and others.
Any products we successfully develop and commercialize will face competition from currently approved therapies and new therapies potentially available in the future.
The availability of reimbursement from government and other third-party payors will also significantly affect the pricing and competitiveness of our products. Our competitors also may obtain FDA or other regulatory approval for their products more rapidly than we may obtain approval for our products, which could result in our competitors establishing a strong market position before we are able to enter the market.
Many of the companies we compete against may have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals, and marketing approved products. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
Specifically, our competitors include companies developing therapies with the same target(s) as acazicolcept, davoceticept and ALPN-303 as well as companies building novel platforms to generate immunomodulatory multi-specific antibody or non-antibody-based targeting proteins, particularly in the area of oncology or autoimmunity.
See the risk factor “We face competition from entities that have developed or may develop therapeutic candidates for our target disease indications, including companies developing novel treatments and technology platforms based on modalities and technology similar to us. If these companies develop technologies or therapeutic candidates more rapidly than we do, or their technologies, including delivery technologies, are more effective, our ability to develop and successfully commercialize therapeutic candidates may be adversely affected.” in Part I, Item 1A of this report for more discussion of the effects of competition and competitors on our business.
Acazicolcept Program Competitors (ICOSL/CD28)
The competitors listed below have programs targeting either ICOS or CD28 (or one of their ligands) for autoimmune and inflammatory diseases. To our knowledge, there are currently no competitors with a single molecule targeting ICOS and CD28 simultaneously.
•an anti-BAFF, anti-ICOSL bispecific antibody being developed by Amgen, Inc. (rozibafusp alfa (AMG570/MEDI0700));
•an anti-CD28 monoclonal antibody fragment being developed by OSE ImmunoTherapeutics SA and Veloxis Pharmaceuticals Inc., a subsidiary of Asahi Kasei (FR104);
•an anti-CD28 peptide being developed by AtoxBio, Inc. (reltecimod (AB-103));
•an anti-CD28 monoclonal antibody being development by TheraMAB (TAB08); and
•CTLA-4-Fc fusion proteins targeting CD80 and CD86 being marketed Bristol Myers Squibb (abatacept and belatacept).
Davoceticept Program Competitors
There are numerous clinical trials for immuno-oncology products used as a single agent or in combination. One of the potentially novel attributes of the davoceticept program is that it has exhibited conditional CD28 costimulation and dual checkpoint inhibition in a single molecule interacting with multiple immune targets.
Examples of additional multi-target compounds for immuno-oncology are highlighted below. To our knowledge, there are currently no competitors with a single molecule capable of dual PD-L1/CTLA-4 antagonism and PD-L1-dependent CD28 agonism.
•wild-type CD80-Fc being developed by Five Prime Therapeutics, Inc., which was purchased by Amgen Inc. (FPT155);
•bispecific antibodies being developed by Regeneron targeting tumor specific antigens and CD28 (REGN5678 anti-PSMAxCD28, REGN5668 anti-MUC16xCD28, and REGN7075 anti-EGFRxCD28);
•trispecific antibodies being developed by Sanofi (CD3xCD38xCD28) (SAR442257) and SAR443216 (CD3xCD28xHER2);
•bifunctional fusion protein composed of monoclonal antibody against PD-L1 fused to the extracellular domain of human transforming growth factor-ß, or TGF-ß, receptor II being developed by EMD Serono, Inc. (bintrafusp alfa, or M7824);
•bifunctional fusion protein composed of PD-1 and OX40L developed by Shattuck Labs, Inc. (SL-279252);
•bispecific fusion protein targeting 4-1BBL and PD-1 being developed by Shattuck Labs, Inc. (SL-279137);
•bispecific fusion protein targeting 4-1BB and PD-L1 being developed by Pieris Pharmaceuticals, Inc. and Servier Laboratories (PRS-344/S095012);
•bispecific monoclonal antibody targeting 4-1BB and PD-L1 being developed by Genmab A/S and BioNTech SE (GEN1046);
•trispecific monoclonal antibody/fusion targeting 4-1BB and PD-L1 being developed by Numab Therapeutics AG and CStone Pharmaceuticals Co., Ltd. (NM021/NM21-1480);
•bispecific monoclonal antibody targeting 4-1BB and PD-L1 being developed by Merus NV and Incyte Corporation (MCLA-145);
•bispecific antibody 4-1BB and PD-L1 being developed by Inhibrx, Inc. and Elpiscience Biopharma Ltd. (INBRX-105);
•bispecific monoclonal antibody targeting 4-1BB and PD-L1 being developed by F-star Biotechnology Ltd. (FS-222);
•bispecific fusion protein targeting 4-1BB and PD-L1 being developed by Kahr Medical Ltd., (DSP105);
•bispecific monoclonal antibody/fusion protein targeting 4-1BB and PD-L1 being developed by ABL, Inc., and I-Mab Biopharma Co., Ltd. (ABL503/TJ-L14B);
•bispecific monoclonal antibody targeting PD-L1 and LAG-3 being developed by F-star Biotechnology Ltd. (FS118);
•bispecific monoclonal antibodies being developed by Xencor, Inc. including XmAb808 targeting CD28 and B7-H3, Vudalimab/XmAb20717 targeting CTLA-4 and PD-1, XmAb841/XmAb22841 targeting CTLA-4 and LAG-3, XmAb104/XmAb23104 targeting PD-1 and ICOS, and additional preclinical CD28 bispecific antibody programs;
•bispecific constructs called “DARTs” being developed by Macrogenics Inc., including Tebotelimab (MGD013) targeting PD-1 and LAG-3 and Lorigerlimab MGD019) targeting PD-1 and CTLA-4;
•DARPin® therapeutic candidate targeting 4-1BB and FAP being developed by Molecular Partners AG and Amgen, Inc (MP0310/AMG 506);
•bispecific monoclonal antibody targeting 4-1BB and FAP being developed by Tesaro, Inc., which was purchased by GlaxoSmithKline plc, targeting PD-1 and LAG-3;F. Hoffmann-La Roche AG (RO7122290);
•bispecific monoclonal antibody targeting 4-1BB and HER-2 being developed by Pieris Pharmaceuticals, Inc. (cinrebafusp alfa or PRS-343);
•small molecule antagonists being developed by Aurigene Ltd. and Curis, Inc., including CA-170 targeting PD-L1 and VISTA and CA-327 targeting PD-L1 and TIM-3;
•various combinations of separate anti PD-1/L1 and anti-CTLA-4 monoclonal antibodies; and
•various combinations of separate anti PD-1/L1 and costimulatory monoclonal antibodies such as OX-40, 4-1BB, and others.
ALPN-303 Program Competitors
The competitors listed below have programs targeting either the TACI, BCMA, or BAFF pathway for autoimmune disease.
•Anti-BAFF antibody marketed by GSK plc (belimumab);
•TACI-Fc being developed by Vera Therapeutics (atacicept);
•TACI-Fc being developed by RemeGen Ltd. (telitacicept (RC18));
•Anti-BAFF-R IgG1 being developed by Novartis AG (Ianalumab (VAY736));
•Anti-APRIL antibody being developed by Visterra, Inc., a subsidiary of Otsuka Pharmaceutical Co., Ltd. (sibeprenlimab (VIS649));
•Anti-BAFF, anti-ICOSL bispecific antibody being developed by Amgen, Inc. (rozibafusp alfa (AMG570/MEDI0700));
•Anti-APRIL antibody being developed by Chinook Therapeutics, Inc. (BION-1301); and
•a recombinant Fc fusion protein designed to block BAFF and APRIL cytokines being developed by Aurinia Pharmaceuticals Inc. (AUR200).
Novel Platform Competitors
Multifunctional therapeutic protein platforms potentially competitive with our platform include:
•Amgen Inc. (BiTE®): fusion proteins consisting of two single-chain variable fragments to link T cells to tumors;
•Macrogenics, Inc. (DART®): Dual-Affinity Re-Targeting and Trident technology platforms bind multiple targets with a single molecule;
•Xencor, Inc. (XmAb Bispecific): Optimized Fc domains for improved potency, half-life and stability;
•Zymeworks, Inc. (Azymetric™): Proprietary amino acid modifications to facilitate interaction of distinct heavy chains;
•Pieris Pharmaceuticals, Inc. (Anticalin®): Engineered proteins derived from natural lipocalins found in blood plasma;
•Compass Therapeutics, LLC (Targeted Immunomodulation™, StitchMabs™): Antibody discovery targeting the tumor-immune synapse;
•Harpoon Therapeutics, Inc.: TriTAC™ (Tri-specific T cell Activating Construct) contain CD3 binding domain, half-life extension domain, and antigen-binding domain;
•Shattuck Labs, Inc.: Agonist Redirected Antibody platform claimed to bind tumor-necrosis factor (“TNF”) and checkpoint targets;
•Ablynx NV (Nanobody®), purchased by Sanofi Pharma, Inc.: Platform technology of single-domain, heavy-chain antibody fragments derived from camelidae (e.g., camels and llamas);
•Regeneron, Inc.: VEGF Trap and VelociSuite® antibody technology platforms;
•Five Prime Therapeutics, Inc., purchased by Amgen Inc.: Proprietary protein library and rapid protein production and testing platform; and
•Merus N.V. (Merus Multiclonics® including Biclonics® and Triclonics®): Discovery, screening, and identification platform for bispecific and trispecific antibodies.
Intellectual Property
Our scientific platform and substantially all our intellectual property have been developed internally. As of December 31, 2021, our patent portfolioconsistsof 47 granted patents and over 155 pendingpatentapplications. Several of our initial patent applications are directed to our scientific platform itself. Other patent applications in our portfolio are directed to various target domains and research programs under development. Each of these patent applications is solely owned by us. As we continue the development of our scientific platform and target vIgDs, we intend to continue pursuing intellectual property protection for these technologies.
We have in-licensed some intellectual property and trade secret materials on a non-exclusive basis. To date, such non-exclusive in-licenses are solely related to commercially-available cell lines involved in the manufacture of our vIgD programs. To date, no other intellectual property related to our scientific platform has been in-licensed. We have out-licensed two programs under our TIP/SIP technology to Adaptimmune on an exclusive basis. Additionally, pursuant to the AbbVie Agreement, we have granted AbbVie an exclusive option to purchase an exclusive worldwide license to acazicolcept. If AbbVie exercises the License Option, AbbVie will take over the future development and commercialization. Finally, pursuant to the Horizon Agreement, we have granted Horizon an exclusive license of certain intellectual property and have entered into a collaboration for the development and commercialization of up to four preclinical candidates generated from Alpine’s unique discovery platform. These candidates include previously undisclosed multi-specific fusion protein-based therapeutic candidates for autoimmune and inflammatory diseases. No other out-licenses have been made.
Although we do not believe our technology infringes any intellectual property rights owned by third parties, we are aware of one or more patents and patent applications that may relate to our technology. Third parties may assert claims against us alleging infringement of their intellectual property rights regardless of whether their allegations have merit. Allegations of infringement could harm our reputation, may result in the expenditure of significant resources to defend and resolve such allegations, and could require us to pay monetary damages if we are found to have infringed any third-party intellectual property rights.
Government Regulation
The FDA and comparable regulatory authorities in state and local jurisdictions and in other countries impose substantial and burdensome requirements on the clinical development, manufacture, marketing, and distribution of therapeutic candidates. These agencies and other federal, state, and local entities regulate research and development activities and the testing, manufacture, quality control, safety, effectiveness, labeling, storage, record keeping, approval, advertising and promotion, and export and import of therapeutic candidates and products.
In the U.S., the FDA regulates drugs, medical devices, and biologic products under the Federal Food, Drug, and Cosmetic Act, or FFDCA, its implementing regulations and other laws, including, in the case of biologics, the Public Health
Service Act. Our potential therapeutic candidates and products will be subject to regulation by the FDA as biologics. Biologics require the submission of a Biologics License Application, or BLA, and approval by the FDA before being marketed in the U.S. None of our therapeutic candidates have been approved by the FDA for marketing in the U.S., and we currently have no BLAs pending. If we fail to comply with applicable FDA or other requirements at any time during the product development process, clinical testing, the approval process, or after approval, we may become subject to administrative or judicial sanctions. These sanctions could include the FDA’s refusal to approve pending applications, license suspension or revocation, withdrawal of an approval, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties, or criminal prosecution. Any FDA enforcement action could have a material adverse effect on us. The process required by the FDA before biologic therapeutic candidates may be marketed in the U.S. generally involves the following:
•completion of extensive preclinical laboratory tests, preclinical animal studies, and formulation studies all performed in accordance with the FDA’s current good laboratory practice, or cGLP, regulations;
•submission to the FDA of an Investigational New Drug, or IND, application which must become effective before human clinical trials in the U.S. may begin;
•performance of adequate and well-controlled human clinical trials to establish the safety and efficacy of the drug candidate for each proposed indication;
•submission to the FDA of a BLA;
•satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product is produced to assess compliance with cGMP regulations; and
•FDA review and approval of the BLA prior to any commercial marketing, sale, or shipment of the therapeutic product.
The testing and approval process requires substantial time, effort, and financial resources, and we cannot be certain any approvals for our therapeutic candidates will be granted on a timely basis, if at all.
Once a therapeutic candidate is identified for development, it enters the preclinical testing stage. Preclinical studies include laboratory evaluations of protein chemistry, formulation, and stability, as well as studies to evaluate toxicity in animals. The results of the preclinical studies, together with manufacturing information and analytical data, are submitted to the FDA as part of an IND application. Currently, the IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day period, raises concerns or questions about the conduct of the clinical trial, including concerns that human research subjects will be exposed to unreasonable health risks. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. Submission of an IND may result in the FDA not allowing the clinical trials to commence or not allowing the clinical trials to commence on the terms originally specified in the IND. A separate submission to an existing IND must also be made for each successive clinical trial conducted during drug development, and the FDA must grant permission, either explicitly or implicitly by not objecting, before each clinical trial can begin.
Clinical trials involve the administration of the therapeutic candidate to human subjects under the supervision of qualified investigators. Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical trial, the parameters to be used in monitoring safety, and the effectiveness criteria to be used. Each protocol must be submitted to the FDA as part of the IND. For each medical center proposing to conduct a clinical trial, an institutional review board, or IRB, must also review and approve a plan for any clinical trial before it can begin at that center and the IRB must monitor the clinical trial until it is completed. The FDA, an IRB, or the sponsor may suspend or discontinue a clinical trial at any time on various grounds, including a finding that the subjects are being exposed to an unacceptable health risk. Clinical testing also must satisfy extensive Good Clinical Practice requirements, including the requirements for informed consent.
All clinical research performed in the U.S. in support of a BLA must be authorized in advance by the FDA under the IND regulations and procedures described above. However, a sponsor who wishes to conduct a clinical trial outside the U.S. may, but need not, obtain FDA authorization to conduct the clinical trial under an IND. If a foreign clinical trial is not conducted under an IND, the sponsor may submit data from the clinical trial to the FDA in support of a BLA so long as the clinical trial is conducted in compliance with an international guideline for the ethical conduct of clinical research known as the Declaration of Helsinki and/or the laws and regulations of the country or countries in which the clinical trial is performed, whichever provides the greater protection to the participants in the clinical trial.
Clinical Trials
For purposes of BLA submission and approval, clinical trials are typically conducted in three sequential phases, which may overlap or be combined.
•Phase 1 clinical trials are initially conducted in a limited population of subjects to test the therapeutic candidate for safety, dose tolerance, absorption, metabolism, distribution, and excretion in healthy humans or, on occasion, in patients with severe problems or life-threatening diseases to gain an early indication of its effectiveness.
•Phase 2 clinical trials are generally conducted in a limited patient population to evaluate preliminary efficacy of the therapeutic candidate for specific targeted indications in patients with the disease or condition under study, evaluate dosage tolerance and appropriate dosage, determine a dosage schedule, and identify possible adverse effects and safety risks.
•Phase 3 clinical trials are commonly definitive efficacy studies of the experimental medication. Phase 3 trials are typically conducted when Phase 2 clinical trials demonstrate a dose range of the therapeutic candidate is effective and has an acceptable safety profile. Phase 3 clinical trials are generally undertaken with large numbers of patients, such as groups of several hundred to several thousand, to provide substantial evidence of clinical efficacy and to further test for safety in an expanded patient population at multiple, geographically-dispersed clinical trial sites.
In some cases, the FDA may condition approval of a BLA on the sponsor’s agreement to conduct additional post-approval clinical trials to further assess the biologic’s safety and effectiveness after BLA approval. Such post-approval clinical trials are typically referred to as Phase 4 clinical trials.
Concurrent with clinical trials, companies usually complete additional animal trials and must also develop additional information about the chemistry and physical characteristics of the biologic and finalize a process for manufacturing the biologic in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the therapeutic candidate and, among other things, the manufacturer must develop methods for testing the identity, strength, quality, and purity of the final biologic product. Additionally, appropriate packaging must be selected and tested, and stability studies must be conducted to demonstrate the therapeutic candidate does not undergo unacceptable deterioration over its shelf life.
Further, as a result of the COVID-19 pandemic, we may be required to develop and implement additional clinical trial policies and procedures designed to help protect subjects from the COVID-19 virus. For example, the FDA has issued guidance on conducting clinical trials during the pandemic, which describes a number of considerations for sponsors of clinical trials impacted by the pandemic, including certain reporting requirements, and additional guidance on the good manufacturing practice considerations for responding to COVID-19 infection and other topics. We may be required to make further adjustments to our clinical trials or business operations based on current or future guidance and regulatory requirements as a result of the COVID-19 pandemic.
Biologics License Applications
The results of preclinical studies and of the clinical trials, together with other detailed information, including extensive manufacturing information and information on the chemistry, pharmacology, clinical pharmacology, and the clinical effects of the biologic,former directors or executive officers is indebted to us, nor are submitted to the FDA in the form of a BLA requesting approval to market the biologic for one or more specified indications. The FDA reviews a BLA to determine, among other things, whether a biologic is safe, pure, and potent and whether the facility in which the biological product is manufactured, processed, packed, or held meets standards designed to assure the biological product continues to be safe, pure, and potent.
Once a BLA has been accepted for filing, by law the FDA will review the application and respond to the applicant, but the review process may be significantly delayed by FDA’s requests for additional information or clarification. Under the Prescription Drug User Fee Act, the FDA evaluates a standard original BLA submission within the first 60 days of its receipt to determine if it is sufficiently complete to conduct a full review, and the FDA has a goal of responding to the submission within ten months of the 60-day filing date, but this timeframe is often extended. The FDA may refer the application to an advisory committee for review, evaluation, and/or recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendations. The FDA may deny approval of a BLA if the applicable statutory and regulatory criteria are not satisfied, or for any reason, or it may require additional clinical data. Even if such data are submitted, the FDA may ultimately decide the BLA does not satisfy the criteria for approval. Data from clinical trials are not always conclusive and the FDA may interpret data differently than we interpret data. Once the FDA approves a BLA, or supplement thereto, the FDA may withdraw the approval if ongoing regulatory requirements are not met or if safety problems are identified after the biologic reaches the market. Where a withdrawal may not
be appropriate, the FDA still may seize existing inventory of such biologic or require a recall of any biologic already on the market. In addition, the FDA may require testing, including Phase 4 clinical trials and surveillance programs to monitor the effect of approved biologics which have been commercialized. The FDA has the authority to prevent or limit further marketing of a biologic based on the results of these post-marketing programs.
A sponsor may also seek approval of its therapeutic candidates under programs designedindividuals indebted to accelerate FDA review and approval of BLAs. For instance, a sponsor may seek FDA designation of a therapeutic candidate as a “fast track product.” Fast track products are those products intended for the treatment of a serious or life-threatening disease or condition andanother entity which demonstrate the potential to address unmet medical needs for such diseases or conditions. If fast track designationindebtedness is obtained, the FDA may initiate review of sections of a BLA before the application is complete. This “rolling review” is available if the applicant provides, and the FDA approves, a schedule for the remaining information. In some cases, a fast track product may be approved on the basis of either a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments, under the FDA’s accelerated approval program. Approvals of this kind typically include requirements for appropriate post-approval confirmatory clinical trials to validate the surrogate endpoint or otherwise confirm the effect of the clinical endpoint.
In addition, the Food and Drug Administration Safety and Innovation Act, or FDASIA, which was enacted and signed into law in 2012, established a new category of drugs referred to as “breakthrough therapies” that may be subject to accelerated approval. A sponsor may seek FDA designation of a drug candidate as a “breakthrough therapy” if the drug is intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development.
Therapeutic candidates may also be eligible for “priority review,” or review within a six-month timeframe from the 60-day filing date, if a sponsor provides sufficient clinical data demonstrating its therapeutic candidate provides a significant improvement compared to marketed products. Even if a therapeutic candidate qualifies for one or more of these programs, the FDA may later decide the therapeutic candidate no longer meets the conditions for qualification or that the period for FDA review or approval will be lengthened. When appropriate, we intend to seek fast track designation and/or accelerated approval for our biologics. We cannot predict whether any of our therapeutic candidates will obtain a fast track and/or accelerated approval designation and, if so, whether such designation will be maintained or rescinded by FDA, or the ultimate impact, if any, of the fast track or the accelerated approval process on the timing or likelihood of FDA approval of any of our proposed biologics.
Biologics may be marketed only for the FDA approved indications and in accordance with the provisions of the approved labeling. Further, if there are any modifications to the biologic, including changes in indications, labeling, or manufacturing processes, equipment, or facilities, the applicant may be required to submit and obtain FDA approval of a new BLA or BLA supplement, which may require us to develop additional data or conduct additional preclinical studies and clinical trials.
Before approving an application, the FDA will inspect the facility or the facilities at which the biologic product is manufactured and will not approve the product unless cGMP compliance is satisfactory. The FDA may also inspect the sites at which the clinical trials were conducted to assess their compliance and will not approve the biologic unless compliance with Good Clinical Practice requirements is satisfactory.
The testing and approval processes require substantial time, effort, and financial resources, and each may take several years to complete. The FDA may not grant approval on a timely basis, or at all. Even if we believe a clinical trial has demonstrated safety and efficacy of one of our therapeutic candidates for the treatment of a disease, the results may not be satisfactory to the FDA. Preclinical and clinical data may be interpreted by the FDA in different ways, which could delay, limit, or prevent regulatory approval. We may encounter difficulties or unanticipated costs in our efforts to secure necessary governmental approvals which could delay or preclude us from marketing our therapeutic candidates. The FDA may limit the indications for use or place other conditions on any approvals restricting the commercial application of the products. After approval, certain changes to the approved biologic, such as adding new indications, change in personnel, manufacturing changes, or additional labeling claims, are subject to further FDA review and approval. Depending on the nature of the change proposed, a BLA supplement which may require additional studies to evaluate the effect of such change on the identity, strength, quality, purity, or potency of the product as they may relate to the safety or effectiveness of the product—must be filed and approved before the change may be implemented. As with new BLAs, the review process for BLA supplements may be delayed by the FDA through requests for additional information or clarification.
We believe any of our therapeutic products approved as a biological product under a BLA might qualify for a 12-year period of exclusivity currently permitted by the Biologics Price Competition and Innovation Act, or BPCIA. Specifically, the BPCIA established an abbreviated pathway for the approval of biosimilar and interchangeable biological products. The new abbreviated regulatory pathway establishes legal authority for the FDA to review and approve biosimilar biologics, including the possible designation of a biosimilar as “interchangeable” based on its similarity to an existing brand product. Under the BPCIA, an application for a biosimilar product cannot be submitted by an applicant until four years after the date the reference product was first licensed and cannot be approved by the FDA until 12 years after the original branded product was first licensed under a BLA. There is a risk the U.S. Congress could amend the BPCIA to significantly shorten this exclusivity period or the FDA will not consider our therapeutic candidates to be reference products for competing products, potentially creating the opportunity for competition sooner than anticipated. Moreover, the extent to which a biosimilar, once approved, will be substituted for any one of our reference products in a way similar to traditional generic substitution for non-biological products is not yet clear, and will depend on a number of marketplace and regulatory factors that are still developing. The BPCIA is complex and is only beginning to be interpreted and implemented by the FDA and the courts. As a result, its ultimate impact, implementation, and meaning are subject to uncertainty. While it is uncertain when any such processes may be fully adopted by the FDA, any such processes operating to limit the scope or length of exclusivity afforded by the BPCIA could have a material adverse effect on the future commercial prospects for our biological products. In addition, foreign regulatory authorities may also provide for exclusivity periods for approved biological products or for abbreviated pathways for follow on biological products. For example, biological products in Europe may be eligible for a 10-year period of exclusivity.
Under the Orphan Drug Act, the FDA may grant orphan drug designation to therapeutic candidates intended to treat a rare disease or condition, which is generally a disease or condition affecting fewer than 200,000 individuals in the U.S. or more than 200,000 individuals in the U.S. and for which there is no reasonable expectation the cost of developing and making available in the U.S. a therapeutic candidate for this type of disease or condition will be recovered from sales in the U.S. for that therapeutic candidate. Orphan drug designation must be requested before submitting a marketing application for the therapeutic for that particular rare disease or condition. After the FDA grants orphan drug designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process. The FDA may revoke orphan drug designation, and if it does, it will publicize the drug is no longer designated as an orphan drug. If a therapeutic candidate with orphan drug designation subsequently receives the first FDA approval for the disease for which it has such designation, the therapeutic candidate is entitled to orphan product exclusivity, which means the FDA may not approve any other applications to market the same therapeutic candidate for the same indication, except in very limited circumstances, for seven years. Orphan drug exclusivity, however, could also block the approval of one of our therapeutic candidates for seven years if a competitor obtains approval of the same therapeutic candidate as defined by the FDA or if our therapeutic candidate is determined to be contained within the competitor’s therapeutic candidate for the same indication or disease.
Under the Best Pharmaceuticals for Children Act, certain therapeutic candidates may obtain an additional six months of exclusivity if the sponsor submits information requested in writing by the FDA, referred to as a “Written Request,” relating to the use of the active moiety of the therapeutic candidate in children. The FDA may not issue a Written Request for studies on unapproved or approved indications where it determines information relating to the use of a therapeutic candidate in a pediatric population, or part of the pediatric population, may not produce health benefits in that population. In addition, the Pediatric Research Equity Act, or PREA, requires a sponsor to conduct pediatric studies for most therapeutic candidates and biologics, for a new active ingredient, new indication, new dosage form, new dosing regimen, or new route of administration. Under PREA, original or New Drug Applications, or NDAs, BLAs and supplements thereto must contain a pediatric assessment unless the sponsor has received a deferral or waiver. The required assessment must assess the safety and effectiveness of the therapeutic candidate for the claimed indications in all relevant pediatric subpopulations and support dosing and administration for each pediatric subpopulation for which the therapeutic candidate is safe and effective. The sponsor or the FDA may request a deferral of pediatric studies for some or all of the pediatric subpopulations. A deferral may be granted for several reasons, including a finding that the drug or biologic is ready for approval for use in adults before pediatric studies are complete or additional safety or effectiveness data needs to be collected before the pediatric studies begin. The FDA must send a noncompliance letter to any sponsor that fails to submit the required assessment, keep a deferral current, or fails to submit a request for approval of a pediatric formulation.
Other Regulatory Requirements
Any biologics manufactured or distributed by us or our collaborators pursuant to FDA approvals would be subject to continuing regulation by the FDA, including recordkeeping requirements and reporting of adverse experiences associated with the product. Manufacturers and their subcontractors are required to register their establishments with the FDA and certain state agencies and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with ongoing regulatory requirements, including cGMPs, which impose certain procedural and documentation requirements upon us and third-party manufacturers. Failure to comply with the statutory and regulatory requirements can subject a manufacturer to
possible legal or regulatory action, such as warning letters, suspension of manufacturing, seizure of product, injunctive action or possible civil penalties. Our company cannot be certain it or its present or future third-party manufacturers or suppliers will be able to comply with the cGMP regulations and other ongoing FDA regulatory requirements. If our company or its present or future third-party manufacturers or suppliers are not able to comply with these requirements, the FDA may halt our clinical trials, require us to recall a drug from distribution, or withdraw approval of the BLA for the therapeutic product.
The FDA closely regulates the post-approval marketing and promotion of biologics, including standards and regulations for direct-to-consumer advertising, off-label promotion, industry-sponsored scientific and educational activities, and promotional activities involving the Internet. A company can make only those claims relating to safety and efficacy approved by the FDA. Failure to comply with these requirements can result in adverse publicity, warning letters, corrective advertising, and potential civil and criminal penalties. Physicians may prescribe legally available biologics for uses not described in the product’s labeling and different from those tested by us and approved by the FDA. Such off-label uses are common across medical specialties. Physicians may believe such off-label uses are the best treatment for many patients in varied circumstances. The FDA does not regulate the behavior of physicians in their choice of treatments. The FDA does, however, impose stringent restrictions on manufacturers’ communications regarding off-label use.
Healthcare Reform
In 2010, Congress passed the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, which we refer to as the ACA, a sweeping law intended to broaden access to health insurance, reduce or constrain the growth of health spending, enhance remedies against fraud and abuse, add new transparency requirements for the healthcare and health insurance industries, impose new taxes and fees on the health industry, and impose additional policy reforms. The ACA contains a number of provisions, including those governing enrollment in federal healthcare programs, reimbursement changes, and fraud and abuse, impacting existing government healthcare programs and resulting in the development of new programs, including Medicare payment for performance initiatives, and improvements to the physician quality reporting system and feedback program. The Affordable Care Act also does, among other things, the following:
•Increases pharmaceutical manufacturer rebate liability under the Medicaid Drug Rebate Program due to an increase in the minimum basic Medicaid rebate on most branded prescription drugs, and the application of Medicaid rebate liability to drugs used in risk-based Medicaid managed care plans.
•Expands the 340B Drug Pricing Program to require discounts for “covered outpatient drugs” sold to certain children’s hospitals, critical access hospitals, freestanding cancer hospitals, rural referral centers, and sole community hospital.
•Requires pharmaceutical companies to offer discounts on brand-name drugs to patients who fall within the Medicare Part D coverage gap, commonly referred to as the “Donut Hole.”
•Requires pharmaceutical companies to pay an annual non-tax-deductible fee to the federal government based on each company’s market share of prior year total sales of branded drugs to certain federal healthcare programs, such as Medicare, Medicaid, Department of Veterans Affairs, and Department of Defense.
•Establishes the Patient-Centered Outcomes Research Institute to identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research. The research conducted by the Patient-Centered Outcomes Research Institute may affect the market for certain pharmaceutical products.
•Establishes the Center for Medicare and Medicaid Innovation within the Centers for Medicare and Medicaid Services, or CMS to test innovative payment and service delivery models to lower Medicare and Medicaid spending, potentially including prescription drug spending.
From time to time, legislation is drafted, introduced, and passed in Congress that could significantly change the statutory provisions governing the sale, marketing, coverage, and reimbursement of products regulated by the CMS or other government agencies. In addition to new legislation, CMS regulations and policies are often revised or interpreted by the agency in ways significantly affecting our business and our products.
Since its enactment, there have been judicial and Congressional challenges to certain aspects of the ACA, and we expect there will be additional challenges and amendments to the ACA in the future. For example, in June 2021, the United States Supreme Court held that Texas and other challengers had no legal standing to challenge the ACA, dismissing the case without specifically ruling on the constitutionality of the ACA. Accordingly, the ACA remains in effect in its current form. It is unclear how this Supreme Court decision, future litigation, or healthcare measures promulgated by the Biden administration will impact our business, financial condition and results of operations. Complying with any new legislation or changes in healthcare regulation could be time-intensive and expensive, resulting in material adverse effect on our business.
Additionally, at the federal level, in 2020, the HHS and CMS issued various rules that are expected to impact, among others, price reductions from pharmaceutical manufacturers to plan sponsors under Part D, fee arrangements between pharmacy benefit managers and manufacturers, importation of prescription drugs from Canada and other countries, manufacturer price reporting requirements under the Medicaid Drug Rebate Program, including regulations that affect manufacturer-sponsored patient assistance programs subject to pharmacy benefit manager accumulator programs and Best Price reporting related to certain value-based purchasing arrangements. Multiple lawsuits have been brought against the HHS challenging various aspects of the rules implemented during the Trump administration. As a result, the Biden administration and HHS have delayed the implementation or published rules rescinding some of these Trump-era policies. In January 2021, President Biden also issued an executive order to initiate a special enrollment period for people to obtain health insurance coverage through the ACA marketplace, and instructs certain governmental agencies to review and reconsider their existing policies and rules that limit access to healthcare, among others. Under the American Rescue Plan Act of 2021, effective January 1, 2024, the statutory cap on Medicaid Drug Rebate Program rebates that manufacturers pay to state Medicaid programs will be eliminated. Elimination of this cap may require pharmaceutical manufacturers to pay more in rebates than it receives on the sale of products, which could have a material impact on our business. Additionally, in July 2021, the Biden administration released an executive order, “Promoting Competition in the American Economy,” with multiple provisions aimed at prescription drugs. In response to Biden’s executive order, on September 9, 2021, HHS released a Comprehensive Plan for Addressing High Drug Prices that outlines principles for drug pricing reform and sets out a variety of potential legislative policies that Congress could pursue as well as potential administrative actions HHS can take to advance these principles. Although no legislation or administrative actions have been finalized to implement these principles, Congress is considering legislation that, if passed, could have significant impact on prices of prescription drugs covered by Medicare, including limitations on drug price increases and allowing Medicare to negotiate pricing for certain drugs. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability, or commercialize our product candidates if approved. Complying with any new legislation and regulatory changes could be time-intensive and expensive, resulting in a material adverse effect on our business.
Further, many states have proposed or enacted legislation that seeks to indirectly or directly regulate pharmaceutical drug pricing, such as by requiring biopharmaceutical manufacturers to publicly report proprietary pricing information or to place a maximum price ceiling on pharmaceutical products purchased by state agencies. For example, a number of states are considering or have recently enacted state drug price transparency and reporting laws that could substantially increase our compliance burdens and expose us to greater liability under such state laws once we begin commercialization after obtaining regulatory approval for any of our products candidates. We cannot be sure to what extent these and future legislative and regulatory efforts, whether FDA regulations, guidance or interpretations will be changed, or what the impact of such changes on the marketing approvals of our product candidates, if any, may be. In addition, increased scrutiny by Congress of the FDA’s approval process may significantly delay or prevent marketing approval, as well as subject us to more stringent product labeling and post-marketing testing and other requirements. These measures could reduce the ultimate demand for our products, once approved, or put pressure on our product pricing. We expect that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could affect the prices we may obtain for any of our product candidates for which we may obtain regulatory approval or the frequency with which any such product candidate, if approved, is prescribed or used.
Furthermore, political, economic, and regulatory influences are subjecting the health care industry in the U.S. to fundamental change. Initiatives to reduce the federal budget and debt and to reform health care coverage are increasing cost-containment efforts. We anticipate federal agencies, Congress, state legislatures, and the private sector will continue to review and assess alternative health care benefits, controls on health care spending, and other fundamental changes to the healthcare delivery system. Any proposed or actual changes could limit coverage for, or the amounts federal and state governments will pay for, health care products and services, which could also result in reduced demand for our products or additional pricing pressures, and limit or eliminate our spending on development projects and affect our ultimate profitability.
Third-Party Payor Coverage and Reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of any products for which we may obtain regulatory approval. In the U.S., sales of any products for which we may receive regulatory approval for commercial sale will depend in part on the availability of coverage and reimbursement from third-party payors. Third-party payors include government authorities such as Medicare, Medicaid, TRICARE, and the Veterans Administration, managed care providers, private health insurers and other organizations.
The Medicaid Drug Rebate Program, which is part of the federal Medicaid program, a program for financially needy patients, among others, requires pharmaceutical manufacturers to enter into and have in effect a national rebate agreement with the Secretary of the Department of Health and Human Services as a condition for states to receive federal matching funds for the manufacturer’s outpatient drugs furnished to Medicaid patients.
In order for a pharmaceutical product to receive federal reimbursement under Medicare Part B, part of the federal Medicare program covering outpatient items and services for the aged and disabled, and Medicaid programs or to be sold directly to U.S. government agencies, the manufacturer must extend discounts to entities eligible to participate in the 340B drug pricing program, a federal program requiring manufacturers to provide discounts to certain safety-net providers. The required 340B discount on a given product is calculated based upon certain Medicaid Drug Rebate Program metrics reported by the manufacturer.
The process for determining whether a payor will provide coverage for a product is typically separate from the process for setting the reimbursement rate a payor will pay for the product. Third-party payors may limit coverage to specific products on an approved list or formulary which might not include all of the FDA-approved products for a particular indication. Also, third-party payors may refuse to include a particular branded product on their formularies or otherwise restrict patient access to a branded drug when a less costly generic equivalent or other alternative is available. However, under Medicare Part D - Medicare’s outpatient prescription drug benefit - there are protections in place to ensure coverage and reimbursement for oncology products and all Part D prescription drug plans are required to cover substantially all anti-cancer agents. Furthermore, a payor’s decision to provide coverage for a product does not imply an adequate reimbursement rate will be available. Adequate third-party reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development.
Third-party payors are increasingly challenging the price and examining the medical necessity and cost-effectiveness of medical products and services, in addition to their safety and efficacy. In order to obtain coverage and reimbursement for any product approved for sale, we may need to pursue compendia listings or conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and cost-effectiveness of any products, in addition to the costs required to obtain regulatory approvals. Our drug candidates may not be considered medically necessary or cost-effective. If third-party payors do not consider a product to be cost-effective compared to other available therapies, they may not cover an approved product as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow a company to sell its products at a profit.
Other Healthcare Laws and Regulations
In the United States, the research, manufacturing, distribution, sale and promotion of drug products are subject to regulation by various federal, state and local authorities in addition to the FDA, including the Centers for Medicare & Medicaid Services, other divisions of the U.S. Department of Health and Human Services (e.g., the Office of Inspector General), the U.S. Department of Justice, state attorneys general, and other state and local government agencies.
If we obtain regulatory approval of our products, we may be subject to various federal and state laws targeting fraud and abuse in the healthcare industry. These laws may impact, among other things, our proposed sales and marketing strategies. In addition, we may be subject to patient privacy regulation by both the federal government and the states in which we conduct our business. The laws affecting our ability to operate include, but are not limited to:
•the federal Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, receiving, offering, or paying remuneration (a term interpreted broadly to include anything of value, including, for example, gifts, discounts, and credits), directly or indirectly, in cash or in kind, to induce or reward, or in return for, either the referral of an individual for, or the purchase, order, or recommendation of, an item or service reimbursable under a federal health care program, such as the Medicare and Medicaid programs;
•federal civil and criminal false claims laws and civil monetary penalty laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment to Medicare, Medicaid, or other third-party payors that are false or fraudulent, or making a false statement or record material to payment of a false claim or avoiding, decreasing, or concealing an obligation to pay money owed to the federal government;
•provisions of HIPAA, prohibiting knowingly and willfully executing a scheme to defraud any health care benefit program and making false statements relating to health care matters;
•provisions of HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act and its implementing regulations, which imposes certain requirements relating to the privacy, security, and transmission of individually identifiable health information;
•the federal transparency laws, including the federal Physician Payment Sunshine Act, which was part of the Affordable Care Act, requiring applicable manufacturers of certain drugs and biologics, among other covered medical products for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program, with certain exceptions, to track and report annually certain payments and other transfers of value they
make to covered recipients, including U.S. physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors, other health care professionals (such as nurse practitioners and physician assistants, among others) and teaching hospitals, as defined by law, as well as physicians’ and physicians’ immediate family members’ ownership and investment interests in the applicable manufacturer, which are subsequently made publicly available in a searchable format on the CMS Open Payments website; and
•state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payor, including commercial insurers, state transparency reporting and compliance laws, and state and foreign laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts.
The ACA broadened the reach of the fraud and abuse laws by, among other things, amending the intent requirement of the federal Anti-Kickback Statute and the applicable criminal healthcare fraud statutes contained within 42 U.S.C. § 1320a-7b. Pursuant to the statutory amendment, a person or entity no longer needs to have actual knowledge of this statute or specific intent to violate it in order to have committed a violation. In addition, the ACA provides the government may assert a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the civil False Claims Act or the civil monetary penalties statute. Many states have adopted laws similar to the federal Anti-Kickback Statute, some of which apply to the referral of patients for healthcare items or services reimbursed by any source, not only the Medicare and Medicaid programs.
If our operations are found to be in violation of any of the U.S. federal and state laws described above or any other governmental regulations that apply to us, we may be subject to penalties, including criminal and significant civil monetary penalties, damages, fines, disgorgement, imprisonment, exclusion from participation in government healthcare programs, injunctions, recall or seizure of products, total or partial suspension of production, denial or withdrawal of pre-marketing product approvals, private qui tam actions brought by individual whistleblowers in the name of the government or refusal to allow us to enter into supply contracts, including government contracts, and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations. We may also be subject to additional reporting requirements and oversight if we become subject to a corporate integrity agreement or similar agreement with a governmental entity to resolve allegations that we have violated these laws. To the extent that any of our product candidates, once approved, are sold in a foreign country, we may be subject to similar foreign laws and regulations, which may include, for instance, applicable post-approval requirements, including safety surveillance, anti-fraud and abuse laws, and implementation of corporate compliance programs and reporting of payments or transfers of value to healthcare professionals.
Human Capital
Our employees share a passion for meaningful work and are committed to solving the most complex problems in creating immunotherapies to treat cancer and autoimmune and inflammatory diseases. Our culture is guided by our core values of innovative thinking, collaboration, flexibility, bias for action, and healthy debate. As of December 31, 2021, we had 85 employees, of which 67 are engaged in research and development activities and 18 in general and administrative. None of our employees are represented by labor unions or covered by collective bargaining agreements. We consider our relationship with our employees to be good.
We believe that we provide competitive compensation and benefits for our personnel and that our compensation and benefit packages are designed to attract and retain highly qualified personnel essential to our business. In addition to salary compensation, our compensation includes new equity grants, a 401(k) retirement plan, healthcare and insurance benefits and a flexible paid time off policy.
We are committed to diversity, equity and inclusion. We recruit the best qualified employees regardless of gender, ethnicity or other protected traits and it is our policy to comply with all applicable laws related to discrimination in the workplace.
Finally, in response to the COVID-19 pandemic, we continue to provide many employees with the ability to work from home and have implemented additional safety and infection prevention measures including enhanced cleaning, additional personal protective equipment, testing, and contact tracing protocols for employees who have transitioned back to critical work on site.
Corporate Information
On July 24, 2017, Alpine Immune Sciences, Inc., or Private Alpine, completed its business combination with Nivalis Therapeutics, Inc., a publicly held company. In connection with the merger, Nivalis Therapeutics, Inc. changed its name to Alpine Immune Sciences, Inc. Nivalis Therapeutics, Inc. was incorporated in Delaware in March 2007. Alpine Immune Sciences, Inc. (prior to its business combination with Nivalis Therapeutics, Inc.) was incorporated in Delaware on December 30, 2014.
Our principal executive office is located at 188 East Blaine Street, Suite 200, Seattle WA, 98102. Our telephone number is (206) 788-4545. Our website is www.alpineimmunesciences.com. Information contained in, or that can be accessed through, our website is not a part of, and is not incorporated into, this report.
Item 1A. Risk Factors
You should carefully consider the following risk factors, in addition to the other information contained in this Annual Report on Form 10-K, including Management’s Discussion and Analysis of Financial Condition and Results of Operations included in Part II, Item 7, and our consolidated financial statements and related notes. If any of the events described in the following risk factors and the risks described elsewhere in this report occurs, our business, operating results and financial condition could be seriously harmed. Our Risk Factors are not guarantees that no such conditions exist as of the date of this report and should not be interpreted as an affirmative statement that such risks or conditions have not materialized, in whole or in part. This report on Form 10-K also contains forward-looking statements that involve risks and uncertainties. Our actual results could differ materially from those anticipated in the forward-looking statements as a result of factors that are described below and elsewhere in this report.Risks Related to Our Pipeline and Product Development
Our approach to the discovery and development of innovative therapeutic treatments based on our technology is unproven and may not result in marketable products.
We plan to develop novel protein-based immunotherapies in part via our proprietary directed evolution platform for the treatment of cancer and autoimmune/inflammatory diseases. The potential to create therapies capable of working within and/or modulating an immune synapse, forcing a synapse to occur, or preventing a synapse from occurring is an important, novel attribute of the majority of our approaches. However, the scientific research forming the basis of our efforts to develop therapeutic candidates based on our platform is relatively new. Further, the scientific evidence to support the feasibility of developing therapeutic treatments based on our platform is both preliminary and limited.
Relatively few therapeutic candidates based on immunoglobulin superfamily, or IgSF, domains, or tumor necrosis factor receptor super family, or TNFRSF, domains, have been tested in humans. We may discover the therapeutic candidates developed using our scientific platform do not possess certain properties required for the therapeutic candidate to be effective. We currently have only limited data to suggest we can introduce these necessary therapeutic properties into variant Ig domain, or vIgD or variant TNF(R) domain, or vTD, based therapeutic candidates. In addition, vIgDs or vTDs may demonstrate different chemical and pharmacological properties in human subjects or patients than they do in laboratory studies. Even if our programs have successful results in animal studies, they may not demonstrate the same chemical and pharmacological properties in humans and may interact with human biological systems in unforeseen, ineffective, or harmful ways. While we continue to evaluate our vIgDs and vTDs preclinically and clinically, the risk profile in humans is still being fully assessed. Undesirable side effects that may be caused by our therapeutic candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA or other comparable foreign authorities. Such side effects could also affect patient recruitment or the ability of enrolled patients to complete clinical trials or result in potential product liability claims. Any of these occurrences may harm our business, financial condition and prospects significantly. As a result, we may never succeed in developing a marketable therapeutic, we may not become profitable, and the value of our common stock may decline.
Further, we believe that the FDA has little prior experience with vIgDs or vTDs, which may increase the complexity, uncertainty, and length of the regulatory approval process for our therapeutic candidates. Our company and our current collaborators, or any future collaborators, may never receive approval to market and commercialize any therapeutic candidate. Even if our company or a collaborator obtains regulatory approval, the approval may be for disease indications or patient populations not as broad as we intended or desired or may require labeling, including significant use or distribution restrictions or safety warnings. Our company or a collaborator may be required to perform additional or unanticipated clinical trials to obtain approval or be subject to post-marketing testing requirements to maintain regulatory approval. If therapeutic candidates we develop using our scientific platform prove to be ineffective, unsafe, or commercially unviable, our entire platform and pipeline would have little, if any, value, which could have a material adverse effect on our business, financial condition, results of operations, and prospects.
The market may not be receptive to our therapeutic products based on a novel therapeutic modality, and we may not generate any future revenue from the sale or licensing of therapeutic products.
Even if approval is obtained for a therapeutic candidate, we may not generate or sustain revenue from sales of the therapeutic product due to factors such as whether the therapeutic product can be sold at a competitive price and otherwise accepted in the market. Therefore, any revenue from sales of the therapeutic product may not offset the costs of development. The therapeutic candidates we are developing are based on new technologies and therapeutic approaches. Market participants with significant influence over acceptance of new treatments, such as physicians and third-party payors, may not adopt a
treatment based on our therapeutic products, and we may not be able to convince the medical community and third-party payors to accept and use, or to provide favorable coverage or reimbursement for, any therapeutic products developed by our company, our existing collaborator, or any future collaborators. Market acceptance of our therapeutic products will depend on, among other factors:
•the timing of our receipt of any marketing and commercialization approvals;
•the terms and scope of any approvals and the countries in which approvals are obtained;
•the safety and efficacy of our therapeutic products;
•the prevalence and severity of any adverse side effects associated with our therapeutic products;
•the prevalence and severity of any adverse side effects associated with therapeutics of the same type or class as our therapeutic products;
•limitations or warnings contained in any labeling approved by the FDA or other regulatory authority;
•relative convenience and ease of administration of our therapeutic products;
•the willingness of patients to accept, and the willingness of physicians to prescribe, any new methods of administration;
•the success of our physician education programs;
•the availability of adequate government and third-party payor coverage and reimbursement;
•the willingness of patients to pay out-of-pocket in the absence of coverage by government and third-party payors;
•the pricing of our products, particularly as compared to alternative treatments;
•our ability to compliantly and effectively market and sell our products;
•the timing of market introduction of our therapeutic products as well as alternative treatments; and
•availability of alternative effective treatments for the disease indications our therapeutic products are intended to treat and the relative risks, benefits, and costs of those treatments.
With our development focus, these risks may increase to the extent this field becomes more competitive or less favored in the commercial marketplace. Additional risks apply in relation to any disease indications we pursue which are classified as rare diseases and allow for orphan drug designation by regulatory agencies in major commercial markets, such as the United States, European Union, and Japan. Because of the small patient population for a rare disease, if pricing is not approved or accepted in the market at an appropriate level for an approved therapeutic product with orphan drug designation, such drug may not generate enough revenue to offset costs of development, manufacturing, marketing, and commercialization despite any benefits received from the orphan drug designation, such as market exclusivity, assistance in clinical trial design, or a reduction in user fees or tax credits related to development expense. Market size is also a variable in disease indications classified as rare. Our estimates regarding potential market size for any rare indication may be materially different from what we discover to exist at the time we commence commercialization, if any, for a therapeutic product, which could result in significant changes in our business plan and have a material adverse effect on our business, financial condition, results of operations, and prospects.
If a therapeutic product with orphan drug designation subsequently receives the first FDA approval for the indication for which it has such designation, the therapeutic product is entitled to orphan product exclusivity, which means the FDA may not approve any other applications to market the same therapeutic product for the same indication, except in very limited circumstances, for seven years. Orphan drug exclusivity, however, could also block the approval of one of our therapeutic products for seven years if a competitor obtains approval of the same therapeutic product as defined by the FDA or if our therapeutic product is determined to be within the same class as the competitor’s therapeutic product for the same indication or disease.
As in the United States, we may apply for designation of a therapeutic product as an orphan drug for the treatment of a specific indication in the European Union before the application for marketing authorization is made. Sponsors of orphan drugs in the European Union can enjoy economic and marketing benefits, including up to ten years of market exclusivity for the approved indication unless another applicant can show its therapeutic product is safer, more effective, or otherwise clinically superior to the orphan-designated therapeutic product. The respective orphan designation and exclusivity frameworks in the United States and in the European Union are subject to change, and any such changes may affect our ability to obtain EU or U.S. orphan designations in the future.
Our therapeutic candidates are in early stages of development and may fail in development or suffer delays that materially and adversely affect their commercial viability.
We have no products on the market and all of our therapeutic candidates are in early stages of development. Our ability to achieve and sustain profitability depends on obtaining regulatory approval and Institutional Review Board, or IRB, approval to conduct clinical trials at particular sites, obtaining regulatory approvals to market our therapeutic candidates and successfully commercializing our therapeutic candidates, either alone or with third parties, such as our collaborators. Before obtaining regulatory approval for the commercial distribution of our therapeutic candidates, we or a collaborator must conduct extensive preclinical tests and clinical trials to demonstrate the safety and efficacy in humans of our therapeutic candidates. Preclinical testing and clinical trials are expensive, difficult to design and implement, can take many years to complete, and are uncertain as to outcome. For example, we are currently advancing the development of acazicolcept, davoceticept and ALPN-303; however, even with the significant investment of time and funding to advance these product candidates, we cannot guarantee that our clinical and preclinical development efforts will be successful. The start or end of a clinical study is often delayed or halted due to delays in or failure to obtain regulatory approval to commence the study, delays in or failure to reach agreement on acceptable terms with prospective contract research organizations, or CROs, or clinical trial sites, delays in or failure to obtain IRB approval at each site, changing regulatory requirements, manufacturing challenges, clinical sites or CROs deviating from the trial protocol or failing to comply with regulatory requirements or meet contractual obligations, slower than anticipated patient enrollment, changing standards of care, availability or prevalence of use of a comparative therapeutic or required prior therapy, clinical outcomes, failure of patients to complete the trial or return for post-treatment follow-up, or financial constraints. For instance, delays or difficulties in patient enrollment or difficulties in retaining trial participants can result in increased costs, longer development times, or termination of a clinical trial. Clinical trials of a new therapeutic candidate require the enrollment of a sufficient number of patients, which may include patients who are suffering from the disease the therapeutic candidate is intended to treat and who meet other eligibility criteria. Rates of patient enrollment are affected by many factors, including the size of the patient population, the eligibility criteria for the clinical trial, the age and condition of the patients, the stage and severity of disease, the nature of the protocol, the proximity of patients to clinical sites, and the availability of effective treatments or competing academic and other clinical trials for the relevant disease.
A therapeutic candidate can unexpectedly fail at any stage of preclinical and clinical development. The historical failure rate for therapeutic candidates is high due to scientific feasibility, safety, efficacy, changing standards of medical care, and other variables. The novelty of our platform may mean our failure rates are higher than historical norms. The results from preclinical testing or early clinical trials of a therapeutic candidate may not predict the outcome of later phase clinical trials of the therapeutic candidate, particularly in immuno-oncology and autoimmune/inflammatory disorders. We will have to conduct additional trials in our proposed indications to verify the results obtained to date in our preclinical and clinical studies and to support any future regulatory submissions. A number of companies in the biopharmaceutical industry have suffered significant setbacks in advanced clinical trials due to lack of efficacy or adverse safety profiles despite promising results in earlier, smaller clinical trials. Moreover, clinical data are often susceptible to varying interpretations and analyses. We do not know whether Phase 1, Phase 2, Phase 3, or other clinical trials we may conduct will demonstrate consistent or adequate efficacy and safety with respect to the proposed indication for use sufficient to receive regulatory approval or market our therapeutic candidates.
We, the FDA, an IRB, an independent ethics committee, or other applicable regulatory authorities may suspend clinical trials of a therapeutic candidate at any time for various reasons, including a belief that subjects participating in such trials are being exposed to unacceptable health risks or adverse side effects. Similarly, an IRB or ethics committee may suspend a clinical trial at a particular trial site. We may not have the financial resources to continue development of, or to enter into collaborations for, a therapeutic candidate if we experience any problems or other unforeseen events delaying or preventing clinical development or regulatory approval of, or our ability to commercialize, therapeutic candidates, including:
•negative or inconclusive results from our clinical trials, or the clinical trials of others for therapeutic candidates similar to ours, leading to a decision or requirement to conduct additional preclinical testing or clinical trials or abandon a program;
•serious and unexpected drug-related side effects experienced by participants in our clinical trials or by individuals using therapeutics similar to our therapeutic candidates;
•serious drug-related side effects experienced in the past by individuals using therapeutics similar to our therapeutic candidates;
•delays in submitting IND applications or clinical trial applications, or comparable foreign applications, or delays or failure in obtaining the necessary approvals from regulators or IRBs to commence a clinical trial, or a suspension or termination of a clinical trial once commenced;
•conditions imposed by the FDA or comparable foreign authorities, such as the European Medicines Agency, or EMA, regarding the scope or design of our clinical trials;
•delays in enrolling research subjects in clinical trials;
•high drop-out rates of research subjects;
•inadequate supply or quality of therapeutic candidate or therapeutic candidate components, or materials or other supplies necessary for the conduct of our clinical trials, including those owned, manufactured, or provided by companies other than ours;
•greater than anticipated clinical trial costs, including the cost of any approved drugs used in combination with our therapeutic candidates;
•poor effectiveness of our therapeutic candidates during clinical trials;
•unfavorable FDA or other regulatory agency inspection and review of a clinical trial site;
•failure of our third-party contractors or investigators to comply with regulatory requirements or otherwise meet their contractual obligations in a timely manner, or at all;
•delays and changes in regulatory requirements, policies, and guidelines, including the imposition of additional regulatory oversight around clinical testing generally or with respect to our technology in particular; or
•varying interpretationsof databy theFDAand similarforeignregulatoryagencies.
Because we have limited financial and operational resources, we must prioritize our research programs and will need to focus our discovery and development on select product candidates and indications. Correctly prioritizing our research and development activities is particularly important for us due to the breadth of potential product candidates and indications that we intend to utilize with our clinical development strategy. As a result, we may forego or delay pursuit of opportunities with other product candidates or for other indications that later prove to have greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities. Our spending on current and future research and development programs and product candidates for specific indications may not yield any commercially viable products. If we do not accurately evaluate the commercial potential or target market for a particular product candidate, we may also relinquish valuable rights to that product candidate through collaboration, licensing or other royalty arrangements in cases in which it would have been more advantageous for us to retain sole development and commercialization rights to such product candidate.
Product development involves a lengthy and expensive process with an uncertain outcome, and results of earlier preclinical and clinical trials may not be predictive of future clinical trial results.
Clinical testing is expensive and generally takes many years to complete, and the outcome is inherently uncertain. Failure can occur at any time during the clinical trial process. The results of preclinical trials and early clinical trials of our product candidates may not be predictive of the results of larger, later-stage controlled clinical trials. Product candidates showing promising results in early-stage clinical trials may still suffer significant setbacks in subsequent clinical trials. We have evaluated acazicolcept in a Phase 1 healthy volunteer trial and previously initiated a Phase 1b/2 study of acazicolcept in patients with steroid-resistant or steroid-refractory active acute graft-versus-host disease, or SR-aGVHD. We terminated this Phase 1b/2 SR-aGVHD study in June 2020. Our Phase 2 study in SLE will materially increase our anticipated research and development spending. SLE is a challenging indication and a number of trials conducted by other companies have failed after significant investment of time and funding. We cannot predict whether our efforts in this indication will be successful. If we are unsuccessful, it is unlikely that AbbVie would exercise its option for acazicolcept pursuant to our option and license agreement and, as a result, we would not receive the option payment pursuant to this agreement and we would not be eligible for future milestones and royalties. In addition, we have initiated our Phase 1 studies of davoceticept as well as our Phase 1 study of ALPN-303 in healthy volunteers. We will have to conduct additional preclinical studies and human trials in our proposed indications to verify the results obtained to date and to support any regulatory submissions for further clinical development. A number of companies in the biopharmaceutical industry have suffered significant setbacks in advanced clinical trials due to lack of efficacy or adverse safety profiles despite promising results in earlier, smaller clinical trials. Moreover, clinical data are often susceptible to varying interpretations and analyses. We do not know whether Phase 1, Phase 2, Phase 3, or other clinical trials we may conduct will demonstrate consistent or adequate efficacy and safety with respect to the proposed indication for use sufficient to receive regulatory approval or market our therapeutic candidates.
Additionally, disruptions at the FDA and other agencies may also slow the time necessary for new drugs to be reviewed by necessary government agencies, which would adversely affect our business. For example, over the last several years, the U.S. government has shut down multiple times and certain regulatory agencies, such as the FDA, have had to furlough critical FDA and other government employees. In response to the COVID-19 public health emergency, the FDA has postponed some inspections and continues to conduct “mission-critical” inspections on a case-by-case basis, or, where possible to do so safely,
has resumed prioritized domestic inspections, such as pre-approval and surveillance inspections. In 2020 and 2021, a number of companies announced receipt of complete response letters due to the FDA’s inability to complete required inspections for their applications. While the FDA continues to ensure timely reviews of applications for medical products during the ongoing COVID-19 pandemic in line with its user fee performance goals and conducting mission critical domestic and foreign inspections to ensure compliance of manufacturing facilities with FDA Good Manufacturing Practices, the FDA may not be able to continue its current inspection pace or be unable to complete required inspections during the review period, or the review timelines could be extended. Regulatory authorities outside the U.S. may adopt similar restrictions or other policy measures in response to the COVID-19 pandemic and may experience delays in their regulatory activities. If a prolonged government shutdown or other disruption occurs, or if global health or other concerns continue to prevent the FDA or other regulatory authorities from conducting their regular inspections, reviews, or other regulatory activities in a timely manner, it could significantly impact the ability of the FDA to timely review and process our regulatory submissions, which could have a material adverse effect on our business.
If we encounter delays or difficulties enrolling patients in our clinical trials and/or retention of patients in clinical trials, our clinical development activities could be delayed or otherwise adversely affected.
We may experience difficulties in patient enrollment in our clinical trials for a variety of reasons, including supply chain disruptions, staffing shortages and other business and economic disruptions resulting from geopolitical actions, including war and terrorism, natural disasters, including earthquakes, typhoons, floods and fires, as well as other disruptions resulting from the impact of public health factors, including the COVID-19 pandemic, business disruptions of our strategic partners, third-party manufacturers, suppliers and other third parties upon which we rely. The timely completion of clinical trials in accordance with their protocols depends, among other things, on our ability to enroll a sufficient number of patients who remain in the trial until completion of treatment and adequate follow-up. The enrollment of patients depends on many factors, including:
•Inability to enroll, or delay in enrollment of, patients due to outbreaks and public health crises, such as the COVID-19 global pandemic, as further described under “Risk Factors — Risks Related to COVID-19 and Other Health Epidemics”;
•The patient eligibility criteria defined in the protocol;
•The perceived risks and benefits of the product candidate being studied;
•The size of the patient population required for analysis of the trial’s primary endpoints;
•The proximity of patients to trial sites;
•The design of the trial;
•Our ability to recruit clinical trial investigators with the appropriate competencies and experience;
•Our ability to obtain and maintain patient consent;
•Geopolitical events in countries where we have or seek to have clinical trial sites;
•Reporting of the preliminary results of any of our clinical trials; and
•The risk that patients enrolled in clinical trials will drop out of the trials before completion of treatment and adequate follow-up.
In addition, our clinical trials will compete with other clinical trials for product candidates that are in the same therapeutic areas as our product candidates, and this competition will reduce the number and types of patients available to us because some patients who might have opted to enroll in our trials may instead opt to enroll in a trial being conducted by one of our competitors. Since the number of qualified clinical investigation sites is limited, we expect to conduct some of our clinical trials at the same clinical trial sites that some of our competitors use, which will reduce the number of patients who are available for our clinical trials at such clinical trial sites. Geopolitical events in countries where we have or seek to have clinical trial sites can also negatively impact our ability to enroll patients in our trials.For example, we had intended to open trial sites in Russia for both our ongoing Synergy trial and our planned Phase 2 trial in SLE with ALPN-303. Following the start of the Russia-Ukraine conflict, we have decided to abandon our plans to open these sites.Although no sites in Russia had been opened, we must revise our plans and locate alternative trial sites in order to achieve targeted enrollment numbers for these trials.Any resulting delays in patient enrollment may increase our costs or may affect the timing or outcome of our ongoing and planned clinical trials, which could prevent completion or commencement of these trials and adversely affect our ability to advance the development of our product candidates.
Our product candidates may cause undesirable side effects or have other properties that could halt their clinical development, prevent their regulatory approval, require expansion of the trial size, limit their commercial potential, or result in significant negative consequences.
Clinical trials that involve cancer patients with significant co-morbidities are associated with increased risks as such participants may be particularly susceptible to safety and toxicity risks. In addition, these side effects may not be appropriately recognized or managed by the treating medical staff, as safety and toxicity monitoring may be complicated and difficult to manage, which could result in patient death or other significant issues. Additionally, it can be difficult to determine if the serious adverse or unexpected side effects were caused by the product candidate or another factors, especially in oncology subjects who may suffer from other medical conditions and take other medications. Such risks are increased in clinical trials that evaluate combination therapies.
If serious adverse events or undesirable side effects arise, we could be required to suspend, delay, or halt our clinical trials and regulatoryauthorities could deny approval or require us to limit development of that product candidate to certain uses or subpopulations in which the undesirable side effects or other characteristics are less prevalent, less severe or more acceptable from a risk-benefit perspective. Undesirable side effects could also result in an expansion in the size of our clinical trials, increasing the expected costs and timeline of our clinical trials. Side effects that are observed during the trial, whether treatment related or not, could also affect patient recruitment for future trials or the ability of enrolled patients to complete the trial or result in potential product liability claims.
Further, if serious adverse events or undesirable side effects are identified during development or after approval and are determined to be attributed to any of our product candidates, we may be required to develop Risk Evaluation and Mitigation Strategies, or REMS, to ensure that the benefits of treatment with such product candidate outweigh the risks for each potential patient, which may include, among other things, a communication plan to health care practitioners, patient education, extensive patient monitoring or distribution systems and processes that are highly controlled, restrictive and more costly than what is typical for the industry.
Any of these occurrences may harm our business, financial condition and prospects significantly. As discussed further in the risk factor below, FDA placed a partial clinical hold on the NEON-2 trial evaluating davoceticept in combination with Merck’s pembrolizumab in adults with advanced malignancies. This partial clinical hold was prompted by the death of a study participant in the NEON-2 trial. We are working closely with the FDA, Merck, the Safety Monitoring Committee, and the study investigators to further understand this event. FDA must lift the partial clinical hold before we can continue to enroll additional patients in this study. We can provide no assurances that FDA will lift the partial clinical hold for this study, or will do so in a timely manner. At this time, the full extent of this event on the NEON-2 trial is unclear.
Development of product candidates in combination with other therapies could expose us to additional risks.
Development of any of our product candidates in combination with one or more other therapies that have either been approved or not yet been approved for marketing by the FDA, EMA or comparable foreign regulatory authorities could expose us to additional risks, as combination therapies may increase the rate of serious or unexpected adverse events, which could result in a clinical hold as well as pre-approval and post-approval restrictions by the FDA or other regulatory authorities on the proposed combination therapy, including narrowing of the indication, warnings, additional safety data collection and monitoring procedures, and REMS, even if the cause of such serious or unexpected adverse events are not directly attributed to our product candidate. Any of these events or restrictions could have a material adverse effect on our business, development of our product candidates, delay our regulatory approval, and decrease the market acceptance and profitability of our product candidate if approved for a combination therapy.
We will not be able to market and sell any product candidate in combination with any unapproved therapies that do not ultimately obtain marketing approval. If the FDA, EMA or other comparable foreign regulatory authorities do not approve or revoke their approval of other therapies used in combination therapies, or if safety, efficacy, commercial adoption, manufacturing or supply issues arise with the therapies we choose to evaluate in combination with any of our product candidate, we may be unable to obtain approval of or successfully market any one or all of the product candidates we develop.
Even if any of our product candidates were to receive marketing approval or be commercialized for use in combination with other existing approved therapies, we would continue to be subject to the risks that the FDA, EMA or other comparable foreign regulatory authorities could revoke approval of the other therapy used in combination with any of our product candidates, or safety, efficacy, manufacturing or supply issues could arise with these existing therapies. In addition, it is possible that existing therapies with which our product candidates are approved for use could themselves fall out of favor or be relegated to later lines of treatment. This could result in the need to identify other combination therapies for our product candidates or our own products being removed from the market or being less successful commercially.
Additionally, if the third-party providers of therapies or therapies in development used in combination with our product candidates are unable to produce sufficient quantities for clinical trials or for commercialization of our product candidates, or if the cost of combination therapies is prohibitive, our development and commercialization efforts would be impaired, which would have an adverse effect on our business, financial condition, results of operations and growth prospects.
On March 7, 2022, we announced that the FDA placed a partial clinical hold on the NEON-2 trial evaluating davoceticept in combination with Merck’s pembrolizumab in adults with advanced malignancies. This partial clinical hold was prompted by the death of a study participant in the NEON-2 trial. The participant had choroidal melanoma previously treated with nivolumab and ipilimumab, and had received a single dose each of davoceticept and pembrolizumab. The participant’s death was attributed to cardiogenic shock, considered by the treating physicians as likely related to immune-mediated myocarditis, or possibly infection. We are working closely with the FDA, Merck, the Safety Monitoring Committee, and the study investigators to further understand this event and to evaluate the appropriate safety precautions before we resume enrollment of additional participants. Participants who are currently enrolled in the NEON-2 trial may continue to receive davoceticept and pembrolizumab, although no new participants may be enrolled until the partial clinical hold is resolved. We can provide no assurances that FDA will lift the partial clinical hold for this study, or will do so in a timely manner. This partial clinical hold does not affect the ongoing NEON-1 clinical trial of davoceticept as monotherapy (NCT04186637).
At this time, it is unclear what FDA will require before the partial hold can be removed or the full impact of this event. To the extent the FDA requires additional safety data to be collected in the NEON-2 trial, impose further or more stringent exclusion criteria, expand the size of the trial, or impose other restrictions or changes to our study protocol, or prolong the partial clinical hold for any reason, our clinical plans for the combination therapy would be delayed substantially and incur additional costs, which could have an adverse effect on our business, financial condition, results of operations and growth prospects. We may encounter further challenges with enrollment. If we fail to meet the additional requirements set by the FDA for resolving the partial clinical hold, we may be required to abandon the development of davoceticept in combination with pembrolizumab. Continued safety concerns could cause the FDA and other regulatory authorities or the IRB to terminate the NEON-2 trial, narrow the indication or impose other restrictions in the labeling even if we obtain regulatory approval for the combination therapy, such as implementation of REMS and addition of a black box warning, among other measures, any of which could decrease the demand, market acceptance and profitability of the combination therapy.
We face competition from entities that have developed or may develop therapeutic candidates for our target disease indications, including companies developing novel treatments and technology platforms based on modalities and technology similar to us. If these companies develop technologies or therapeutic candidates more rapidly than we do, or their technologies, including delivery technologies, are more effective, our ability to develop and successfully commercialize therapeutic candidates may be adversely affected.
We participate in the highly competitive sector of biotechnology and pharmaceuticals and in the subsector of immune modulation. This subsector has undergone tremendous technological advancement over the last decade due to advancements in understanding the role of the immune system across multiple therapeutic areas, including oncology and autoimmune/inflammatory disease. While we believe our novel technology platform, discovery programs, knowledge, experience, and scientific resources offer competitive advantages, we face competition from major pharmaceutical and biotechnology companies, academic institutions, governmental agencies, public and private research institutions, and others.
Any products we successfully develop and commercialize will face competition from currently approved therapies and new therapies potentially available in the future.
The availability of reimbursement from government and other third-party payors will also significantly affect the pricing and competitiveness of our products. Our competitors also may obtain FDA or other regulatory approval for their products more rapidly than we may obtain approval for our products, which could result in our competitors establishing a strong market position before we are able to enter the market.
Many of the companies we compete against may have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals, and marketing approved products. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs. For additional information regarding our competitors and the competitive landscape, please refer to the section of this report titled “Business — Competition.”
Many of our competitors have significantly greater financial, technical, manufacturing, marketing, sales, and supply resources or experience than we have. If we successfully obtain approval for any therapeutic candidate, we will face competition based on many different factors, including safety and effectiveness, ease with which our products can be administered and the extent to which patients accept relatively new routes of administration, timing and scope of regulatory approvals, availability and cost of manufacturing, marketing and sales capabilities, price, reimbursement coverage, and patent position of our products. Competing products could present superior treatment alternatives, including by being more effective, safer, less expensive, or marketed and sold more effectively than any products we may develop. Competitive products may make any products we develop obsolete or noncompetitive before we recover the expense of developing and commercializing our therapeutic candidates. Competitors could also recruit our employees, which could negatively impact our ability to execute our business plan.
We believe our development programs and platform have a particular mechanism of action, but this mechanism of action has not been proven conclusively.
Our scientific platform is novel, and the underlying science is not exhaustively understood nor conclusively proven. In particular, the interaction of vIgDs with the immune synapse, the ability of vIgDs to slow, stop, restart, or accelerate immune responses, and the ability of vIgD domains to interact with multiple counter structures is still largely theoretical. Graphical representations of proposed mechanisms of action of our therapies, the size, actual or relative, of our therapeutics, and how our therapeutics might interface with other cells within the human body, inside the immune synapse, or inside the disease and/or the tumor microenvironment are similarly theoretical and not yet conclusively proven. The lack of a proven mechanism of action may adversely affect our ability to raise sufficient capital, complete preclinical studies, adequately manufacture drug product, obtain regulatory clearance for clinical trials, gain marketing approval, or conclude collaborations, or interfere with our ability to market our product to patients and physicians or achieve reimbursement from payors.
Any inability to present our data in scientific journals or at scientific conferences could adversely impact our business and stock price.
We may from time to time submit data related to our research and development activities in peer-reviewed scientific publications or apply to present data related to our research and development activities at scientific or other conferences. We have no control over whether these submissions or applications are accepted. Even if accepted for a conference, we have no control over whether presentations at scientific conferences will be accepted for oral presentation, poster presentation, or abstract publication only. Even when accepted for publication, we have no control over the timing of the release of the publication. Rejection by publications, delays in publication, rejection for presentation, or a less-preferred format for a presentation may adversely impact our stock price, ability to raise capital, and business.
Our business may be affected by adverse scientific publications or editorial or discussant opinions.
We may from time to time publish data related to our research and development activities in peer-reviewed scientific publications or present data related to our research and development activities at scientific or other conferences. Editorials or discussants unrelated to us may provide opinions on our presented data unfavorable to us. In addition, scientific publications or presentations may be made which are critical of our science or research or the field of immunotherapy in general. This may adversely affect our ability to raise necessary capital, complete clinical and preclinical studies, adequately manufacture drug product, obtain regulatory clearance for clinical trials, or approval for marketing, or interfere with our ability to market our product to patients and physicians or achieve reimbursement from payors.
Risks Related to Our Relationships with Third Parties
To date, our revenue has been primarily derived from our collaboration agreements, and our success will be dependent, in part, on our collaborators’ efforts to develop our therapeutic candidates.
Our success is dependent, in part, on our collaborators’ efforts to develop our therapeutic candidates and, historically, our revenue has been primarily derived from our agreements with collaborators. For example, in June 2020, we entered into the AbbVie Agreement for the development of acazicolcept and in December 2021, we entered into the Horizon Agreement pursuant to which we granted to Horizon rights to one of our existing preclinical biologic therapeutic programs and we and Horizon agreed to collaborate in the discovery, research and preclinical development of up to three additional autoimmune and inflammatory disease programs for other designated biological targets.
Pursuant to the terms of the AbbVie Agreement, we received an upfront payment of $60.0 million in cash and are eligible to receive up to $75.0 million in development milestones (of which $45.0 million was achieved in the second quarter of 2021), an additional $75.0 million if AbbVie exercises its option with respect to acazicolcept following our completion of certain development activities, additional development, commercial and sales-based milestones up to an aggregate of $655.0 million and royalties on any future net sales. Pursuant to the AbbVie Agreement, we will conduct certain development activities under a development plan that provides for, among other things, the generation of a data package in order for AbbVie to evaluate exercising its exclusive option, including all activities reasonably necessary to complete our Phase 2 study of acazicolcept in SLE. If we successfully complete these activities, AbbVie may not exercise its option, which would make achievement of future milestones and receipt of future royalties unattainable. If AbbVie exercises its option, our realization of additional milestones and royalty payments will depend upon the efforts of AbbVie. If AbbVie fails to develop, obtain regulatory approval for, or ultimately commercialize acazicolcept or if AbbVie terminates the collaboration, our business, financial condition, results of operations, and prospects could be materially and adversely affected. For additional information regarding the AbbVie Agreement, please refer to the Partnerships section within “Item 1. Business” of this report.Pursuant to the terms of the Horizon Agreement, we received an upfront payment of $25.0 million as well as an equity investmentfor which they paid $15.0 million. We are also eligible to receive milestone payments upon our achievement of certain preclinical, clinical and regulatory and commercialization milestones, up to an aggregate amount of $381.0 million per program, or approximately $1.5 billion in total, if all milestones are met, as well as royalties on future product sales. Pursuant to the Horizon Agreement, we will conduct certain research activities under a research program; however, even if we successfully perform our obligations under the Horizon Agreement, there is no certainty that Horizon will continue the development of any of the programs, or, if development is continued, if such programs will ultimately succeed and receive regulatory approval. Horizon will have discretion in determining and directing its efforts and resources for future development activities and, if approval is obtained, commercialization and marketing of the approved drug. As a result, there can be no assurances that we will achieve additional milestones pursuant to the Horizon Agreement. For additional information regarding the Horizon Agreement, please refer to the Partnerships section within “Item 1. Business” of this report.Our collaborations may also result in reduced royalty revenues if we are unable to obtain and maintain patent protection, as well as if we are unable to obtain patent term extension, for therapeutic candidates or products developed under our agreements with collaborators. In the event of expiration or invalidation of patents covering a therapeutic candidate or product, for example, our collaborators may be entitled to a significant decrease in royalty revenues owed to us under the agreements. Invalidation of patents and failure to obtain patent term extension for one or more patents in our portfolio may occur as a result of factors beyond our control due to the complex legal and factual questions surrounding pharmaceutical and biotechnology patents. If we are unable to obtain and maintain patent protection, or if we unable to obtain patent term extension for therapeutic candidates or products developed under our agreements with collaborators, our revenue derived from our collaborators may be less than the full amount anticipated, and our business, financial condition, results of operations, and growth prospects could be materially harmed.
Continued advancement of our other product candidates and other development efforts depends, in part, upon the efforts of AbbVie, Horizon, and our other current or future collaborators. If our collaborators do not dedicate sufficient resources to the development of product candidates that are the subject of our agreements, such product candidates may never be successful and we may be ineligible to receive additional milestone payments or royalties pursuant to the termsa guarantee, support agreement, letter of our arrangements, which could have a material adverse impact on our financial results and operations. Even if we and our collaborators dedicate sufficient resources to our collaboration agreements, neither we nor our collaborators may be effective in obtaining approvals for any therapeutic candidates or, if approved, the successful commercialization of any approved products. Collaborators may change their strategic focus or pursue alternative technologies after entering into a collaboration agreement with us, which could result in reduced, delayed or no revenue to us. Disputes regarding collaboration agreements, including disputes pertaining to ownership of intellectual property, may also arise and if we and our collaborators are unable to resolve such disputes, litigation proceedings may occur, which could further delay development programs, create uncertainty as to ownership of intellectual property rights, distract management from other business activities and generate substantial expenses, any of which could materially and negatively impact our business.
If third parties on which we depend to conduct our clinical or preclinical studies, or any future clinical trials, do not perform as expected, fail to satisfy regulatory or legal requirements, or miss expected deadlines, our development program could be delayed, which may result in materially adverse effects on our business, financial condition, results of operations, and prospects.
We rely, in part, on third-party clinical investigators, CROs, clinical data management organizations, and consultants to design, conduct, supervise, and monitor clinical trials and preclinical studies of our therapeutic candidates and may do the same for future clinical trials. Because we rely on third parties to conduct preclinical studies or clinical trials, we have less control
over the timing, quality, compliance, and other aspects of preclinical studies and clinical trials than we would if we conducted all preclinical studies and clinical trials on our own. These investigators, CROs, and consultants are not our employees and we have limited control over the amount of time and resources they dedicate to our programs. These third parties may have contractual relationships with other entities, some of which may be our competitors, which may draw their time and resources away from our programs. The third parties with which we contract might not be diligent, careful, compliant, or timely in conducting our preclinical studies or clinical trials, resulting in the preclinical studies or clinical trials being delayed or unsuccessful. Further, if any of our relationships with third-party CROs terminate, we may not be able to enter into arrangements with alternative CROs or to do so on commercially reasonable terms.
If we cannot contract with acceptable third parties on commercially reasonable terms, or at all, or if these third parties do not carry out their expected duties, satisfy legal and regulatory requirements for the conduct of preclinical studies or clinical trials, or meet expected deadlines, our clinical development programs could be delayed and otherwise adversely affected. In all events, we are responsible for ensuring each of our preclinical studies and clinical trials is conducted in accordance with the general investigational plan and protocols for the trial and with legal, regulatory and scientific standards. The FDA and certain foreign regulatory authorities, such as the EMA, require preclinical studies to be conducted in accordance with applicable Good Laboratory Practices, or GLPs, and clinical trials to be conducted in accordance with applicable FDA regulations and Good Clinical Practices, or GCPs, including requirements for conducting, recording, and reporting the results of preclinical studies and clinical trials to assure data and reported results are credible and accurate and the rights, integrity, and confidentiality of clinical trial participants are protected. Our reliance on third parties we do not control does not relieve us of these responsibilities and requirements. If we or any of our CROs fail to comply with applicable GCPs, the clinical data generated in our clinical trials may be deemed unreliable and the FDA, the EMA or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. We cannot assure you that upon inspection by a given regulatory authority, such regulatory authority will determine that any of our clinical trials comply with GCP regulations. In addition, our clinical trials must be conducted with product produced under cGMP regulations. Our failure to comply with these regulations may require us to repeat clinical trials, which would delay the regulatory approval process. Any such event could have a material adverse effect on our business, financial condition, results of operations, and prospects.
In addition, switching or adding additional CROs involves additional cost and requires management time and focus. There is also a natural transition period when a new CRO commences work. As a result, delays may occur, which could materially impact our ability to meet our desired clinical development timelines. There can be no assurance that we will not encounter such challenges or delays in the future or that these delays or challenges will not have a material adverse impact on our business, financial condition and prospects.
Because we rely on third-party manufacturing and supply partners, our supply of clinical trial materials may become limited or interrupted or may not be of satisfactory quantity or quality, and our dependence on these third parties may impair the advancement of our research and development programs.
We have established in-house recombinant protein generation capabilities for producing sufficient protein materials to enable a portion of our current preclinical studies. We rely on third-party supply and manufacturing partners to supply the materials, components, and manufacturing services for a portion of preclinical studies and also rely on such third parties for all our clinical trial drug supplies. We do not own manufacturing facilities or supply sources for such components and materials for clinical trial supplies and our current manufacturing facilities are insufficient to supply such components and materials for all of our preclinical studies. Certain raw materials necessary for the manufacture of our therapeutic products, such as cell lines, are available from a single or limited number of source suppliers on a purchase order basis. There can be no assurance our supply of research and development, preclinical study, and clinical trial drugs and other materials will not be limited, interrupted, restricted in certain geographic regions, of satisfactory quality or quantity, or continue to be available at acceptable prices. In particular, any replacement of our therapeutic substance manufacturer could require significant effort and expertise and could result in significant delay of our preclinical or clinical activities because there may be a limited number of qualified replacements. In addition, disruptions to ports and other shipping infrastructure, due in part to the impact of the ongoing COVID-19 pandemic, may result in shortages or delays impacting the availability of materials and other supplies, which could negatively impact our manufacturers, suppliers and other third parties on whom we rely. While we have not yet suffered any direct, material negative impacts from these ongoing supply chain disruptions, we cannot be certain that we will not be impacted, which could increase our costs or negatively impact our development timelines.
The manufacturing process for a therapeutic candidate is subject to FDA and foreign regulatory authority review, and the facilities used by our contract manufacturers to manufacture our therapeutic candidates must be approved by the FDA pursuant to inspections that will be conducted after we submit our marketing application(s) to the FDA. Suppliers and manufacturers must meet applicable manufacturing requirements and undergo rigorous facility and process validation tests required by regulatory authorities in order to comply with cGMP regulationscredit or other regulatory standards. In the event anysimilar arrangement or understanding provided by us.
Sales of Securities
our suppliers or manufacturers fails to comply with such requirements or to perform its obligations to us in relation to quality, timing, or otherwise, or if our supply of components or other materials becomes limited or interrupted for other reasons, we may experience shortages resulting in delayed shipments, supply constraints, and/or stock-outs of our products, be forced to manufacture the materials alone, for which we currently do not have the capabilities or resources, or enter into an agreement with another third party, which we may not be able to do on reasonable terms, if at all. In some cases, the technical skills or technology required to manufacture our therapeutic candidates may be unique or proprietary to the original manufacturer and we may have difficulty, or there may be contractual and intellectual property restrictions prohibiting us from, transferring such skills or technology to another third party and a feasible alternative may not exist. These factors may increase our reliance on such manufacturer or require us to obtain a license from such manufacturer in order to have another third party manufacture our therapeutic candidates. If we are required to change manufacturers for any reason, we will be required to verify the new manufacturer maintains facilities and procedures complying with quality standards and with all applicable regulations. The delays associated with the verification of a new manufacturer could negatively affect our ability to develop therapeutic candidates in a timely manner, within budget, or at all.
We expect to continue to rely on third-party manufacturers if we receive regulatory approval for any therapeutic candidate. To the extent we have existing, or enter into future, manufacturing arrangements with third parties, we will depend on these third parties to perform their obligations in a timely manner consistent with contractual and regulatory requirements, including those related to quality control and assurance. If we are unable to obtain or maintain third-party manufacturing for therapeutic candidates, or to do so on commercially reasonable terms, we may not be able to develop and commercialize our therapeutic candidates successfully. Our, or a third party’s, failure to execute on our manufacturing requirements could adversely affect our business in a number of ways, including as a result of:
•an inability to initiate or continue preclinical studies or clinical trials of therapeutic candidates under development;
•delay in submitting regulatory applications, or receiving regulatory approvals, for therapeutic candidates;
•the loss of the cooperation of a collaborator;
•subjecting manufacturing facilities of our therapeutic candidates to additional inspections by regulatory authorities;
•requirements to cease distribution or to recall batches of our therapeutic candidates; and
•in the event of approval to market and commercialize a therapeutic candidate, an inability to meet commercial demandsforour products.
As product candidates progress from preclinical studies to late-stage clinical trials to marketing approval and commercialization, it is common that various aspects of the development program, such as manufacturing methods, materials and processes, are altered along the way in an effort to optimize yield, manufacturing batch size, minimize costs and achieve consistent purity, identity, potency, quality and results. Such changes carry the risk that they will not achieve these intended objectives. Any of these changes could cause our product candidates to perform differently and could affect planned or other clinical trials conducted with product candidates produced using the modified manufacturing methods, materials, and processes. This could delay completion of clinical trials and could require non-clinical or clinical bridging and comparability studies, which could increase costs, delay approval of our product candidates and jeopardize our ability to commercialize our product candidates, if approved.
We may not successfully engage in strategic transactions, including any additional collaborations we seek, which could adversely affect our ability to develop and commercialize therapeutic candidates, impact our cash position, increase our expenses, and present significant distractions to our management.
From time to time, we consider strategic transactions, such as collaborations, acquisitions of companies, asset purchases or divestitures, and out- or in-licensing of therapeutic candidates or technologies. In particular, we intend to evaluate and, if strategically attractive, seek to enter into additional collaborations, including with major biotechnology or pharmaceutical companies. The competition for collaborative partners is intense, and the negotiation process is time-consuming and complex. Any new collaboration may be on suboptimal terms for us, and ultimately may not maximize value for our stockholders. In addition, we may be unable to maintain any new or existing collaboration if, for example, development or approval of a therapeutic candidate is delayed, sales of an approved therapeutic product do not meet expectations, or the collaborator terminates the collaboration. Any such collaboration, or other strategic transaction, may require us to incur non-recurring or other charges, increase our near- and long-term expenditures and pose significant integration or implementation challenges or disrupt our management or business.
Thesetransactions would entail numerous operational and financial risks, including:
•exposure to unknown liabilities;
•disruption of our business and diversion of our management’s time and attention in order to manage a collaboration or develop acquired therapeutic candidates, or technologies;
•incurrence of substantial debt or dilutive issuances of equity securities to pay transaction consideration or costs;
•higher than expected collaboration, acquisition, or integration costs;
•write-downs of assets, or incurring impairment charges or increased amortization expenses; and
•difficulty and cost in facilitating the collaboration or combining the operations and personnel of any acquired business or impairment of relationships with key suppliers, manufacturers, or customers of any acquired business due to changesin managementand ownershipand theinabilityto retainkey employeesof any acquiredbusiness.
Accordingly, although there can be no assurance we will undertake or successfully complete any transactions of the nature described above, any transactions we do complete may be subject to the foregoing or other risks and have a material adverse effect on our business, results of operations, financial condition, and prospects. Conversely, any failure to enter any collaboration or other strategic transaction beneficial to us could delay the development and potential commercialization of our therapeutic candidates and have a negative impact on the competitiveness of any therapeutic candidate reaching market.
Risks Related to Our Ability to Commercialize Product Candidates
If any of our therapeutic candidates are approved for marketing and commercialization and we are unable to develop sales, marketing and distribution capabilities on our own or enter into agreements with third parties to perform these functions on acceptable terms, we may be unable to successfully commercialize any such future products.
We currently have no sales, marketing, or distribution capabilities or experience. If any of our therapeutic candidates are approved, we will need to develop internal sales, marketing, and distribution capabilities to commercialize such products, which may be expensive and time-consuming, or enter into collaborations with third parties to perform these services. If we decide to market our products directly, we will need to commit significant financial, legal, and managerial resources to develop a marketing and sales force with technical expertise and supporting distribution, administration, and compliance capabilities. If we rely on third parties with such capabilities to market our approved products, or decide to co-promote products with collaborators, we will need to establish and maintain marketing and distribution arrangements with third parties, and there can be no assurance we will be able to enter into such arrangements on acceptable, compliant terms or at all. In entering into third-party marketing or distribution arrangements, any revenue we receive will depend upon the efforts of the third parties and there can be no assurance such third parties will establish adequate sales and distribution capabilities or be successful in gaining market acceptance of any approved therapeutic. If we are not successful in commercializing any therapeutic approved in the future, either on our own or through third parties, our business, financial condition, results of operations, and prospects could be materially and adversely affected.
If we fail to comply with U.S. and foreign regulatory requirements, regulatory authorities could limit or withdraw any marketing or commercialization approvals we may receive and subject us to other penalties that could materially harm our business.
Our company, our therapeutic candidates, our suppliers, and our contract manufacturers, distributors, and contract testing laboratories are subject to extensive regulation by governmental authorities in the European Union, the United States, and other countries, with regulations differing from country to country.
Even if we receive marketing and commercialization approval of a therapeutic candidate, we and our third-party service providers will be subject to continuing regulatory requirements, including a broad array of regulations related to establishment registration and product listing, manufacturing processes, risk management measures, quality and pharmacovigilance systems, post-approval clinical studies, labeling and packaging, advertising and promotional activities, record keeping, distribution, adverse event reporting, import and export of pharmaceutical products, pricing, sales, and marketing, and fraud and abuse requirements.
Furthermore, the FDA strictly regulates manufacturers’ promotional claims of drug products. In particular, a drug product may not be promoted by manufacturers for uses that are not approved by the FDA, as reflected in the FDA-approved labeling, although healthcare professionals are permitted to use drug products for off-label uses. The FDA, the Department of
Justice, the Inspector General of the Department of Health and Human Services, among other government agencies, actively enforce the laws and regulations prohibiting manufacturers’ promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability, including large civil and criminal fines, penalties, and enforcement actions. The FDA has also imposed consent decrees or permanent injunctions under which specified promotional conduct is changed or curtailed for companies that engaged in such prohibited activities. If we cannot successfully manage the promotion of our approved product candidates, we could become subject to significant liability, which would materially adversely affect our business and financial condition.
We are required to submit safety and other post market information and reports, and are subject to continuing regulatory review, including in relation to adverse patient experiences with the product and clinical results reported after a product is made commercially available, both in the United States and in any foreign jurisdiction in which we seek regulatory approval. The FDA and certain foreign regulatory authorities, such as the EMA, have significant post-market authority, including the authority to require labeling changes based on new safety information and to require post-market studies or clinical trials to evaluate safety risks related to the use of a product or to require withdrawal of the product from the market.
The FDA also has the authority to require a REMS plan either before or after approval, which may impose further requirements or restrictions on the distribution or use of an approved therapeutic. The EMA now routinely requires risk management plans, or RMPs, as part of the marketing authorization application process, and such plans must be continually modified and updated throughout the lifetime of the product as new information becomes available. In addition, the relevant governmental authority of any EU member state can request an RMP whenever there is a concern about the risk/benefit balance of the product.
The manufacturers and manufacturing facilities we use to make a future product, if any, will also be subject to periodic review and inspection by the FDA and other regulatory agencies, including for continued compliance with cGMP requirements. The discovery of any new or previously unknown problems with our third-party manufacturers, manufacturing processes or facilities may result in restrictions on the product, manufacturers or facilities, including withdrawal of the product from the market. If we rely on third-party manufacturers, we will have limited control over compliance with applicable rules and regulations by such manufacturers.
If we or our collaborators, manufacturers, or service providers fail to comply with applicable continuing regulatory requirements in the U.S. or foreign jurisdictions in which we seek to market our products, we may be subject to, among other things, fines, warning and untitled letters, clinical holds, a requirement to conduct additional clinical trials, delay or refusal by the FDA or foreign regulatory authorities to approve pending applications or supplements to approved applications, suspension, refusal to renew or withdrawal of regulatory approval, product recalls, seizures, or administrative detention of products, refusal to permit the import or export of products, operating restrictions, inability to participate in government programs including Medicare and Medicaid, and total or partial suspension of production or distribution, injunction, restitution, disgorgement, debarment, civil penalties, and criminal prosecution.
Imposed price controls may adversely affect our future profitability.
In most countries, the pricing of prescription drugs is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take considerable time after receipt of marketing approval for a product. In addition, there can be considerable pressure by governments and other stakeholders on prices and reimbursement levels, including as part of cost containment measures. Political, economic, and regulatory developments may further complicate pricing and reimbursement negotiations, and pricing negotiations may continue after reimbursement has been obtained.
Reference pricing used by various EU member states and parallel distribution, or arbitrage between low-priced and high-priced member states, can further reduce prices. In some countries, we or our collaborators may be required to conduct a clinical trial or other studies comparing the cost-effectiveness of our therapeutic candidates to other available therapies in order to obtain or maintain reimbursement or pricing approval. Publication of discounts by third-party payors or authorities may lead to further pressure on the prices or reimbursement levels within the country of publication and other countries. If reimbursement of any product candidate approved for marketing is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, our business, financial condition, results of operations, or prospects could be adversely affected.
Our business may become subject to economic, political, regulatory and other risks associated with international operations.
Our business is subject to risks associated with conducting business internationally, including our use of foreign clinical trial sites. Some of our suppliers, collaborators and clinical trial relationships are located outside the United States. Accordingly, our future results could be harmed by a variety of factors, including:
•economic instability or weakness, including inflation, reduced growth, diminished credit availability, weakened consumer confidence or increased unemployment;
•sociopolitical instability in particular foreign economies and markets;
•difficulties in compliance with non-U.S. laws and regulations;
•changes in non-U.S. regulations and customs, tariffs and trade barriers, including any changes that China may impose as a result of political tensions between the United States and China;
•changes in non-U.S. currency exchange rates and currency controls;
•trade protection measures, import or export licensing requirements or other restrictive actions by U.S. or non-U.S. governments;
•workforce uncertainty in countries where labor unrest is more common than in the United States;
•production shortages resulting from any events affecting raw material supply or manufacturing capabilities outside the United States; and
•business interruptions resulting from geopolitical actions, including war and terrorism, or natural disasters including earthquakes, typhoons, floods and fires.
Risks Related to Our Personnel and Operations
We will need to raise substantial additional funds to advance development of our therapeutic candidates, and we cannot guarantee we will have sufficient funds available in the future to develop and commercialize our current or future therapeutic candidates.
We will need to raise substantial additional funds to expand our development, regulatory, manufacturing, marketing, and sales capabilities or contract with other organizations to provide these capabilities to us. We have used substantial funds to develop our therapeutic candidates and will require significant funds to conduct further research and development, preclinical testing, and clinical trials of our therapeutic candidates, to seek regulatory approvals for our therapeutic candidates, and to manufacture and market products, if any are approved for commercial sale. As of December 31, 2021, we had $215.4 million in cash and cash equivalents, restricted cash, and investments. Based on our current operating plan, we believe our available cash and cash equivalents, and investments will be sufficient to fund our planned level of operations, including anticipated capital expenditures, for at least the next 12 months. Our future capital requirements and the period for which we expect our existing resources to support our operations may vary significantly from what we expect. Our monthly spending levels vary based on new and ongoing development and corporate activities. Because the length of time and activities associated with successful development of our therapeutic candidates are highly uncertain, we are unable to estimate the actual funds we will require for development and any approved marketing and commercialization activities. To execute our business plan, we will need, among other things:
•to obtainthehumanand financialresourcesnecessaryto develop,test,obtainregulatoryapprovalfor, manufacture, and market our therapeutic candidates;
•to build and maintain a strong intellectual property portfolio and avoid infringing intellectual property of third parties;
•to establish and maintain successful licenses, collaborations, and alliances;
•to satisfy the requirements of clinical trial protocols, including patient enrollment;
•to establish and demonstrate the clinical efficacy and safety of our therapeutic candidates;
•to obtain regulatory approvals;
•to manage our spending as costs and expenses increase due to preclinical studies, clinical trials, regulatory approvals, manufacturing scale-up, and commercialization;
•to obtain additional capital to support and expand our operations; and
•to market our productsto achieveacceptanceand use by themedicalcommunityin general.
If we are unable to obtain necessary funding on a timely basis or on acceptable terms, we may have to delay, reduce, or terminate our research and development programs, preclinical studies, or clinical trials, if any, limit strategic opportunities, or undergo reductions in our workforce or other corporate restructuring activities. We also could be required to seek funds through
arrangements with collaborators or others requiring us to relinquish rights to some of our technologies or therapeutic candidates we would otherwise pursue on our own. We do not expect to realize revenue from product sales, or royalties in the foreseeable future, if at all. Our revenue sources are, and will remain, extremely limited unless and until our therapeutic candidates are clinically tested, approved for commercialization, and successfully marketed.
To date, we have financed our operations primarily through the sale of equity securities, debt, and payments received under our collaboration agreements. We will be required to seek additional funding in the future and intend to do so through a combination of public or private equity offerings, debt financings, credit and loan facilities, research collaborations, and license agreements. Our ability to raise additional funds from these or other sources will depend on financial, economic, and other factors, many of which are beyond our control. Additional funds may not be available to us on acceptable terms or at all.
If we raise additional funds by issuing equity securities, our stockholders will suffer dilution, and the terms of any financing may adversely affect the rights of our stockholders. For example, in January 2019, we issued in a private placement 4,706,700 shares of common stock and warrants to purchase an additional 1,835,610 shares of common stock for gross proceeds of approximately $25.3 million. In July 2020, we issued in a private placement 5,139,610 shares of common stock, prefunded warrants to purchase 790,710 shares of common stock and warrants to purchase an additional 1,779,096 shares of common stock for gross proceeds of approximately $60.0 million. In September 2021 we issued in a private placement 6,489,357 shares of common stock and prefunded warrants to purchase an additional 3,191,487 shares of common stock for gross proceeds of approximately $91.0 million. In December 2021, in connection with the Horizon Agreement, we sold 951,980 shares of our common stock to Horizon for which they paid $15.0 million. We also have a sales agreement in place with Cowen and Company, LLC, or Cowen, to sell up to $75.0 million of our common stock from time to time through an “at the market” equity offering under which Cowen will act as sales agent.
In addition, as a condition to providing additional funds to us, future investors may demand, and may be granted, rights superior to those of existing stockholders. Debt financing, if available, may involve restrictive covenants limiting our flexibility in conducting future business activities, and, in the event of a liquidation or insolvency, debt holders would be repaid before holders of equity securities receive any distribution of corporate assets. Our failure to raise capital or enter into such other arrangements within a reasonable timeframe would have a negative impact on our financial condition, and we may have to delay, reduce, or terminate our research and development programs, preclinical or clinical trials, or undergo reductions in our workforce or other corporate restructuring activities.
We are an early stage biopharmaceutical company with a history of losses, we expect to continue to incur significant losses for the foreseeable future, we may never achieve or maintain profitability, and we have a limited operating history that may make it difficult for investors to evaluate the potential success of our business.
We are a clinical-stage immunotherapy company, with a limited operating history, focused on developing treatments for autoimmune/inflammatory diseases and cancer. Since inception, we have devoted our resources to developing novel protein-based immunotherapies primarily using our proprietary directed evolution platform, which converts native immune system proteins into potential differentiated, multi-targeted therapeutics designed to modulate the immune system. We have had significant operating losses since inception. For the year ended December 31, 2021, our net loss was $50.3 million. Substantially all of our losses have resulted from expenses incurred in connection with our research programs and from general and administrative costs associated with our operations. In addition, inflationary pressure could adversely impact our financial results. Our operating costs have increased, and may continue to increase, due to the recent growth in inflation. Our technologies and therapeutic candidates are in early stages of development, and we are subject to the risks of failure inherent in the development of therapeutic candidates based on novel technologies.
We have historically generated revenue primarily from the receipt of research funding and upfront and other payments under our collaboration agreements. We have not generated, and do not expect to generate, any revenue from product sales for the foreseeable future, and we expect to continue to incur significant operating losses for the foreseeable future due to the cost of research and development, preclinical studies, clinical trials, and the regulatory approval process for therapeutic candidates. The amount of future losses is uncertain. Our ability to achieve profitability, if ever, will depend on, among other things, our or our existing collaborators, or any future collaborators, successfully developing therapeutic candidates, obtaining regulatory approvals to market and commercialize therapeutic candidates, manufacturing any approved products on commercially reasonable terms, establishing a sales and marketing organization or suitable third-party alternatives for any approved product, and raising sufficient funds to finance business activities. If we or our existing collaborators, or any future collaborators, are unable to develop and commercialize one or more of our therapeutic candidates or if sales revenue from any therapeutic candidate receiving approval is insufficient, we will not achieve profitability, which could have a material adverse effect on our business, financial condition, results of operations, and prospects.
Interim, preliminary or topline data from our clinical trials that we announce or publish from time to time may change as more patient data become available and are subject to audit and verification procedures that could result in material changes in the final data.
From time to time, we may publish interim, preliminary or topline data from clinical trials. Interim data from clinical trials that we may complete are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment continues and more patient data become available. Interim or preliminary data from clinical trials that we may conduct may not be indicative of the final results of the trial and are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment continues and more patient data becomes available. Interim or preliminary data also remains subject to audit and verification procedures that may result in the final data being materially different from the interim or preliminary data. As a result, interim or preliminary data should be viewed with caution until the final data are available. Adverse differences between interim, preliminary or topline data and final data could significantly harm our reputation and business prospects. We do not know whether any clinical trials we may conduct will demonstrate consistent or adequate efficacy and safety sufficient to obtain marketing approval to market our product candidates.
Moreover, preliminary, interim and topline data are subject to the risk that one or more of the clinical outcomes may materially change as more patient data become available when patients mature on study, patient enrollment continues or as other ongoing or future clinical trials with a product candidate further develop. Past results of clinical trials may not be predictive of future results. In addition, the information we choose to publicly disclose regarding a particular study or clinical trial is based on what is typically more extensive information, and you or others may not agree with what we determine is the material or otherwise appropriate information to include in our disclosure. Any information we determine not to disclose may ultimately be deemed significant with respect to future decisions, conclusions, views, activities or otherwise regarding a particular product candidate or our business. Similarly, even if we are able to complete our planned and ongoing preclinical studies and clinical trials of our product candidates according to our current development timeline, the positive results from such preclinical studies and clinical trials of our product candidates may not be replicated in subsequent preclinical studies or clinical trial results. Moreover, preclinical, nonclinical and clinical data are often susceptible to varying interpretations and analyses, and many companies that believed their product candidates performed satisfactorily in preclinical studies and clinical trials nonetheless failed to obtain FDA or other regulatory approval.
Any inability to attract and retain qualified key management and technical personnel would impair our ability to implement our business plan.
Our success largely depends on the continued service of key management and other specialized personnel, including Mitchell H. Gold, M.D., our Executive Chairman and Chief Executive Officer, Stanford Peng, M.D., Ph.D., our President and Head of Research and Development, and Paul Rickey, our Senior Vice President and Chief Financial Officer.
The loss of one or more members of our management team or other key employees or advisors could delay our research and development programs and materially harm our business, financial condition, results of operations, and prospects. The relationships our key managers have cultivated within our industry make us particularly dependent upon their continued employment with us. We are dependent on the continued service of our technical personnel because of the highly technical nature of our therapeutic candidates and technologies, and the specialized nature of the regulatory approval process. Because our management team and key employees are not obligated to provide us with continued service, they could terminate their employment with us at any time without penalty. We do not maintain key person life insurance policies on any of our management team members or key employees. Our future success will depend in large part on our continued ability to attract and retain other highly qualified scientific, technical, and management personnel, as well as personnel with expertise in clinical testing, manufacturing, governmental regulation, and commercialization. We face competition for personnel from other companies, universities, public and private research institutions, government entities, and other organizations, including significant competition in the Seattle employment market.
As our therapeutic candidates advance into clinical trials, we may experience difficulties in managing our growth and expanding our operations.
We have limited experience in therapeutic development and very limited experience with clinical trials of therapeutic candidates. As our therapeutic candidates enter and advance through preclinical studies and clinical trials, we will need to expand our development, regulatory, and manufacturing capabilities or contract with other organizations to provide these capabilities for us. For example, as we continue enrollment in our Phase 2 study in SLE and continue with the development of our other product candidates, we will need to hire additional personnel in clinical operations. We also must manage relationships with collaborators or partners, suppliers, and other organizations. Our ability to manage our operations and future growth will require us to continue to improve our operational, financial, and management controls, reporting systems, and
procedures. We may not be able to implement improvements to our management information and control systems in an efficient or timely manner and may discover deficiencies in existing systems and controls.
Our business entails a significant risk of product liability and our inability to obtain sufficient insurance coverage could harm our business, financial condition, results of operations, or prospects.
Our business exposes us to significant product liability risks inherent in the development, testing, manufacturing, and marketing of therapeutic treatments. Product liability claims could delay or prevent completion of our development programs. If we succeed in marketing products, such claims could result in an investigation by certain regulatory authorities, such as FDA or foreign regulatory authorities, of the safety and effectiveness of our products, our manufacturing processes and facilities, or our marketing programs and potentially a recall of our products or more serious enforcement action, limitations on the approved indications for which they may be used, or suspension or withdrawal of approvals. Regardless of the merits or eventual outcome, liability claims may also result in decreased demand for our products, injury to our reputation, costs to defend the related litigation, a diversion of management’s time and our resources, substantial monetary awards to trial participants or patients, and a decline in our valuation. We currently have product liability insurance we believe is appropriate for our stage of development and may need to obtain higher levels of product liability insurance prior to marketing any therapeutic candidates. Any insurance we have or may obtain may not provide sufficient coverage against potential liabilities. Furthermore, clinical trial and product liability insurance is becoming increasingly expensive. As a result, we may be unable to obtain sufficient insurance at a reasonable cost to protect us against losses caused by product liability claims with a potentially material adverse effect on our business.
Our employees may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements, which could have a material adverse effect on our business.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include, but is not limited to:
•intentional failures to comply with FDA or U.S. health care laws and regulations, or applicable laws, regulations, guidance, or codes of conduct set by foreign governmental authorities or self-regulatory industry organizations;
•a provision of inaccurate information to any governmental authorities such as FDA;
•noncompliance with manufacturing standards we may establish;
•noncompliance with federal and state healthcare fraud and abuse laws and regulations;
•noncompliance with the U.S. Foreign Corrupt Practices Act (the FCPA) and similar anti-bribery or anti-corruption laws, regulations or rules of other countries in which we operate; and
•a failureto reportfinancialinformationor dataaccuratelyor a failureto discloseunauthorized activitiesto us.
In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws, regulations, guidance and codes of conduct intended to prevent fraud, kickbacks, self-dealing and other abusive practices. These laws, regulations, guidance statements, and codes of conduct may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive program, health care professional, and other business arrangements. As our business is heavily regulated, it involves significant interaction with government officials, including potentially officials of non-U.S. governments. Additionally, in many countries, healthcare providers are employed by the government, and the purchasers of biopharmaceuticals are government entities. As a result, our dealings with are subject to regulation and such healthcare providers and employees of such purchasers may be considered “foreign officials” as defined in the FCPA. Recently, the Securities and Exchange Commission, or SEC, and Department of Justice have increased their FCPA enforcement activities with respect to biotechnology companies. We also may be subject to U.S. and foreign export controls, trade sanctions and import laws and regulations and if we fail to comply with these laws and regulations, we may face significant penalties, finds and/or denial of certain export privileges.
Employee misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions, including debarment or disqualification of those employees from participation in FDA regulated activities and serious harm to our reputation. This could include violations of provisions of the U.S. federal Health Insurance Portability and Accountability Act, or HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH, other U.S. federal and state law, and requirements of non-U.S. jurisdictions, including the European Union General Data Protection Regulation.
It is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws, regulations, guidance or codes of conduct. Furthermore, we may be held liable under the FCPA and similar laws in other jurisdictions for the corrupt or other illegal activities of our employees, our third-party business partners, representatives and agents, even if we do not explicitly authorize such activities. If any such governmental investigations or other actions or lawsuits are instituted against us, and we are not successful in defending such actions or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines, exclusion from government programs, or other sanctions.
Our business involves the use of hazardous materials and we and our third-party manufacturers must comply with environmental laws and regulations, which may be expensive and restrict how we conduct business.
Our third-party manufacturers’ activities and our own activities involve the controlled storage, use and disposal of hazardous and flammable materials, including the components of our pharmaceutical product candidates, test samples and reagents, biological materials and other hazardous compounds. We and our manufacturers are subject to federal, state, local, and foreign laws and regulations governing the use, generation, manufacture, storage, handling, and disposal of these hazardous materials. Although we believe our safety procedures for handling and disposing of these materials and waste products comply with the standards prescribed by these laws and regulations, we cannot eliminate the risk of accidental injury or contamination from the use, storage, handling, or disposal of hazardous materials. In the event of an accident, state, or federal or other applicable authorities may curtail our use of these materials and/or interrupt our business operations. In addition, if an accident or environmental discharge occurs, or if we discover contamination caused by prior operations, including by prior owners and operators of properties we acquire, we could be liable for cleanup obligations, damages, and fines. If such unexpected costs are substantial, this could significantly harm our financial condition and results of operations.
Compliance with governmental regulations regarding the treatment of animals used in research could increase our operating costs, which would adversely affect the commercialization of our technology.
The Animal Welfare Act, or AWA, is the federal law covering the treatment of certain animals used in research. Currently, the AWA imposes a wide variety of specific regulations governing the humane handling, care, treatment, and transportation of certain animals by producers and users of research animals, most notably relating to personnel, facilities, sanitation, cage size and feeding, watering, and shipping conditions. Third parties with whom we contract are subject to registration, inspections, and reporting requirements under the AWA. Furthermore, some states have their own regulations, including general anti-cruelty legislation, which establish certain standards in handling animals. Comparable rules, regulations, and or obligations exist in many foreign jurisdictions. If we or our contractors fail to comply with regulations concerning the treatment of animals used in research, we may be subject to fines and penalties and adverse publicity, and our operations could be adversely affected.
Our current operations are concentrated in one location and any events affecting this location may have material adverse consequences.
Our current operations are located in facilities situated in Seattle, Washington. Any unplanned event, such as flood, fire, explosion, earthquake, extreme weather condition, medical epidemics, power shortage, power outage, telecommunication failure, or other natural or man-made accidents or incidents resulting in our company being unable to fully utilize the facilities, may have a material adverse effect on our ability to operate our business, particularly on a daily basis, and have significant negative consequences on our financial and operating conditions. Loss of access to these facilities may result in increased costs, delays in the development of our therapeutic candidates, or interruption of our business operations. As part of our risk management policy, we maintain insurance coverage at levels we believe are appropriate for our business. However, in the event of an accident or incident at these facilities, we cannot assure you the amounts of insurance will be sufficient to satisfy any damages and losses or that the insurance covers all risks. If our facilities are unable to operate because of an accident or incident or for any other reason, even for a short period of time, any or all of our research and development programs may be harmed. Any business interruption may have a material adverse effect on our business, financial position, results of operations, and prospects.
Our business may be affected by litigation and government investigations.
We may from time to time receive inquiries and subpoenas and other types of information requests from government authorities and others and we may become subject to claims and other actions related to our business activities. While the ultimate outcome of investigations, inquiries, information requests, and legal proceedings is difficult to predict, defense of
litigation claims can be expensive, time-consuming and distracting, and adverse resolutions or settlements of those matters may result in, among other things, modification of our business practices, costs, and significant payments, any of which could have a material adverse effect on our business, financial condition, results of operations, and prospects.
Risks Related to Our Financial Position and Capital Needs
The investment of our cash and cash equivalents, in fixed income and other marketable securities is subject to risks which may cause losses and affect the liquidity of these investments.
As of December 31, 2021, we had $215.4 million in cash and cash equivalents, restricted cash, and investments. We expect to invest our excess cash in fixed income and other marketable securities. These investments are subject to general credit, liquidity, market and interest rate risks. We may realize losses in the fair value of these investments, an inability to access cash in these investments for a potentially meaningful period, or a complete loss of these investments, which would have a negative effect on our financial statements.
Our business may be materially affected by changes to fiscal and tax policies. Negative or unexpected tax consequences could adversely affect our results of operations.
New tax laws, statutes, rules, regulations or ordinances could be enacted at any time, which could adversely affect our business operations, and our business and financial performance. Further, existing tax laws, statutes, rules, regulations or ordinances could be interpreted, changed, modified or applied adversely to us. For example, the Tax Cuts and Jobs Act of 2017, or TCJA, enacted in December 2017, as modified by the Coronavirus Aid, Relief, and Economic Security Act, or the CARES Act, enacted in April 2020, significantly changed the U.S. Internal Revenue Code. Such changes include a reduction in the corporate tax rate and limitations on certain corporate deductions and credits, among other changes. We have generally accounted for changes related to the TCJA in accordance with our understanding of the legislation and guidance available as of the date of this filing as described in more detail in our financial statements and will continue to monitor and assess the impact of the federal legislation on our business and the extent to which various states conform to the newly enacted federal tax law. In addition, adverse changes in the financial outlook of our operations or further changes in tax laws or regulations could lead to changes in our valuation allowances against deferred tax assets on our accompanying Consolidated Balance Sheets, which could materially affect our results of operations.Nivalis’ pre-merger net operating loss carryforwards and certain other tax attributes are likely subject to limitations. The pre-merger net operating loss carryforwards and certain other tax attributes of Alpine and of the combined organization may also be subject to limitations as a result of ownership changes resulting from the merger.
In general, a corporation that undergoes an “ownership change” is subject to limitations on its ability to utilize its pre-change net operating loss carryforwards, or NOL carryforwards, to offset future taxable income. In general, an ownership change occurs if the aggregate stock ownership of certain stockholders, generally stockholders beneficially owning five percent or more of a corporation’s common stock, applying certain look-through and aggregation rules, increases by more than 50 percentage points over such stockholders’ lowest percentage ownership during the testing period, generally three years. Nivalis may have experienced ownership changes in the past and may experience ownership changes in the future. In addition, the closing of the merger in 2017 likely resulted in an ownership change for Nivalis. It is likely that, due to the method by which limitations on the utilization of NOL carryforwards are calculated, we will not be able to utilize any of Nivalis’ NOL carryforwards and certain other tax attributes. It is also possible that Alpine’s NOL carryforwards and certain other tax attributes may be subject to limitation as a result of ownership changes in the past and/or the closing of the merger. Consequently, even if we achieve profitability, we may not be able to utilize a material portion of Alpine’s, or any of Nivalis’ NOL carryforwards and certain other tax attributes, which could have a material adverse effect on cash flow and results of operations.
Provisions of our debt instruments may restrict our ability to pursue our business strategies.
Our term loan agreement requires us, and any debt financing we may obtain in the future may require us, to comply with various covenants that limit our ability to, among other things:
•dispose of assets;
•complete mergers or acquisitions;
•incur indebtedness;
•encumber assets;
•pay dividends or make other distributions to holders of our capital stock;
•make specified investments;
•engage in any new line or business; and
•engagement in certain transactions with our affiliates.
These restrictions could inhibit our ability to pursue our business strategies. If we default under our term loan agreement, including a material adverse change in our business, operations or condition (financial or otherwise), and such event of default is not cured or waived, the lenders could terminate commitments to lend and cause all amounts outstanding with respect to the debt to be due and payable immediately, which in turn could result in cross defaults under other debt instruments. Our assets and cash flow may not be sufficient to fully repay borrowings under our outstanding debt instruments if some or all of these instruments are accelerated upon a default. We may incur additional indebtedness in the future. The debt instruments governing such indebtedness could contain provisions that are as, or more, restrictive than our existing debt instruments. If we are unable to repay, refinance or restructure our indebtedness when payment is due, the lenders could proceed against the collateral granted to them to secure such indebtedness or force us into bankruptcy or liquidation.
Risks Related to COVID-19 and Other Health Epidemics
The COVID-19 coronavirus could adversely impact our business, including our clinical trials.
In December 2019, a novel strain of coronavirus, SARS-CoV-2, the causative agent of coronavirus disease 2019, or COVID-19, was first reported. Since then, SARS-CoV-2 has spread globally, including countries in which we have planned or active clinical trial sites. We have experienced and will likely continue to experience disruptions that could severely impact our business and clinical trials, including:
•delays or difficulties in enrolling patients in our clinical trials;
•delays or difficulties in clinical site initiation, including difficulties in recruiting clinical site investigators and clinical site staff;
•diversion of healthcare resources away from the conduct of clinical trials, including the diversion of hospitals serving as our clinical trial sites and hospital staff supporting the conduct of our clinical trials;
•interruption of key clinical trial activities, such as clinical trial site monitoring, due to limitations on travel imposed or recommended by federal or state governments, employers and others;
•limitations in employee resources that would otherwise be focused on the conduct of our clinical trials, including because of sickness of employees or their families or the desire of employees to avoid contact with large groups of people;
•delays in receiving approval from local regulatory authorities and ethics committees to initiate our planned clinical trials;
•delays in clinical sites receiving the supplies and materials needed to conduct our clinical trials;
•interruption in global shipping that may affect the transport of clinical trial materials, such as investigational drug product used in our clinical trials;
•changes in local regulations as part of a response to the COVID-19 coronavirus outbreak which may require us to change the ways in which our clinical trials are conducted, which may result in unexpected costs, or to discontinue the clinical trials altogether;
•delays in necessary interactions with local regulators, ethics committees and other important agencies and contractors due to limitations in employee resources or forced furlough of government employees; and
•refusal of the FDA to accept data from clinical trials whose conduct has been affected by the COVID-19 outbreak, such as due to missing data.
Further, we may be required to develop and implement additional clinical trial policies and procedures designed to help protect subjects from the COVID-19 virus. For example, in March 2020, the FDA issued a guidance, which the FDA subsequently updated, on conducting clinical trials during the pandemic, which describes a number of considerations for sponsors of clinical trials impacted by the pandemic. The FDA has also published other COVID-19-related industry guidance,
including updates to previous guidance documents, regarding Good Manufacturing Practices, remote interactive evaluations of drug manufacturing and bioresearch monitoring facilities, and drug product manufacturing and supply chain inspections, among others. It is possible that additional governmental action will be taken to address the COVID-19 pandemic. The ultimate impact of the COVID-19 pandemic on our business operations is highly uncertain and subject to change and will depend on future developments, including new regulatory requirements and changes to existing regulations.
The global outbreak of the COVID-19 coronavirus continues to rapidly evolve. The extent to which the COVID-19 coronavirus may impact our business and clinical trials will depend on future developments, which are highly uncertain and cannot be predicted with confidence, such as the duration of the outbreak, travel restrictions and social distancing in the United States and other countries, business closures or business disruptions and the effectiveness of actions taken in the United States and other countries to contain and treat the disease. For example, with the increased availability of vaccines in North America and certain countries around the world, the rate of additional COVID-19 infections and hospitalizations declined in certain locations for periods of time, resulting in relaxed restrictions and a general reopening of the economy and travel across many jurisdictions, however some jurisdictions continue to maintain or reimpose restrictions in view of increased hospitalization rates and cases due to COVID-19 variants. While certain of these developments are positive, reduced vaccine availability, resistance to vaccination by certain persons or ineffectiveness of vaccines against certain variants may result in increasing infection and hospitalization rates, which could be further complicated by the emergence of more virulent or infectious variants of the virus.
We face risks related to health epidemics and other outbreaks, which could significantly disrupt our operations and/or business.
Our business could be adversely impacted by the effects of the COVID-19 outbreak originating in China, or by other epidemics. Our supply chain for raw materials, drug substance or drug product is worldwide, including China, and accordingly could be subject to disruption. There may be restrictions on the export or shipment of raw materials, drug substance or drug product that could materially delay our business or clinical trials.
Certain of our research and development efforts are also conducted globally, for example the NEON-1 clinical trial and ALPN-303 healthy volunteer study include investigative sites in Australia and our Synergy trial includes investigative sites in Korea and Poland. A health epidemic or other outbreak, including the current COVID-19 outbreak, may materially and adversely affect our business, financial condition and results of operations. The extent to which the outbreak impacts our results will depend on future developments, which are highly uncertain and cannot be predicted, including new information which may emerge concerning the severity of the outbreak and the actions to contain the outbreak or treat its impact, among others.
Risks Related to Cybersecurity
Our computer systems, or those of any of our CROs, manufacturers, other contractors or consultants or potential future collaborators, may fail or suffer security or data privacy breaches or incidents or other unauthorized or improper access to, use of, or destruction of our proprietary or confidential data, employee data, or personal data, which could result in additional costs, loss of revenue, significant liabilities, harm to our brand and material disruption of our operations.
Despite the implementation of security measures in an effort to protect systems that store our information, given their size and complexity and the increasing amounts of information maintained on our internal information technology systems, and those of our third-party CROs, other contractors (including sites performing clinical trials) and consultants, these systems are potentially vulnerable to breakdown or other damage or interruption from service interruptions, system malfunction, natural disasters, terrorism, war and telecommunication and electrical failures, as well as security breaches and incidents from inadvertent or intentional actions by our employees, contractors, consultants, business partners, and/or other third parties, or from cyber-attacks by malicious third parties (including supply chain cyber-attacks or the deployment of harmful malware, ransomware, denial-of-service attacks, social engineering and other means to affect service reliability and threaten the confidentiality, integrity and availability of information), which may compromise our system infrastructure or lead to the loss, destruction, alteration, prevention of access to, disclosure, or dissemination of, or damage or unauthorized access to, our data (including trade secrets or other confidential information, intellectual property, proprietary business information, and personal information) or data that is processed or maintained on our behalf, or other assets, which could result in financial, legal, business and reputational harm to us. Companies have experienced an increase in phishing and social engineering attacks from third parties in connection with the COVID-19 pandemic, and the increase in remote working further increases security threats. To the extent that any disruption or security incident were to result in any loss, destruction, unavailability, alteration, disclosure, or dissemination of, or damage or unauthorized access to, our applications, any other data processed or maintained on our behalf or other assets, or for it to be believed or reported that any of these occurred, we could incur liability, financial harm and
reputational damage and the development and commercialization of our product candidates could be delayed. We cannot assure you that our data protection efforts and our investment in information technology, or the efforts or investments of CROs, consultants or other third parties, will prevent significant breakdowns or breaches in systems or other cyber incidents that cause loss, destruction, unavailability, alteration or dissemination of, or damage or unauthorized access to, our data and other data processed or maintained on our behalf or other assets that could have a material adverse effect upon our reputation, business, operations or financial condition. For example, if such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our programs and the development of our product candidates could be delayed. In addition, the loss of clinical trial data for our product candidates could result in delays in our marketing approval efforts and significantly increase our costs to recover or reproduce the data. Further, any such event that leads to loss, damage, or unauthorized access to, or use, alteration, or disclosure or dissemination of, personal information, including personal information regarding our clinical trial subjects or employees, could harm our reputation directly, compel us to comply with federal and/or state breach notification laws and foreign law equivalents, subject us to mandatory corrective action, and otherwise subject us to liability under laws and regulations that protect the privacy and security of personal information, which could result in significant legal and financial exposure and reputational damages that could potentially have an adverse effect on our business.
Notifications and follow-up actions related to a security breach or incident could impact our reputation and cause us to incur significant costs, including legal expenses and remediation costs. For example, the loss of clinical trial data from completed or future clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the lost data. We expect to incur significant costs in an effort to detect and prevent security breaches and incidents, and we may face increased costs and requirements to expend substantial resources in the event of an actual or perceived security breach or incident. We also rely on third parties to manufacture our product candidates, and similar events relating to their computer systems could also have a material adverse effect on our business. To the extent that any disruption or security incident were to result in any loss, destruction, or alteration of, unavailability of, or damage or unauthorized access to, our data or other information that is processed or maintained on our behalf, or inappropriate disclosure of or dissemination of any such information, or if any of these were perceived or reported to have occurred, we could be exposed to litigation and governmental investigations, the further development and commercialization of our product candidates could be delayed, and we could be subject to significant fines or penalties for any noncompliance with certain state, federal and/or international privacy and security laws.
Our insurance policies may not be adequate to compensate us for the potential losses arising from any such disruption in or, failure or security breach or incident of or impacting our systems or third-party systems where information important to our business operations or commercial development is stored. In addition, such insurance may not be available to us in the future on economically reasonable terms, or at all. Further, our insurance may not cover all claims made against us and could have high deductibles in any event, and defending a suit, regardless of its merit, could be costly and divert management attention.
Our information technology systems could face serious disruptions adversely affecting our business.
Our information technology and other internal infrastructure systems, including corporate firewalls, servers, leased lines, and connection to the Internet, face the risk of systemic failure potentially disruptive to our operations. A significant disruption in the availability of our information technology and other internal infrastructure systems, or those of third parties that perform services or supply materials to us, could cause interruptions in our collaborations with our partners and delays in our research and development work.
Our facility is located in Seattle, Washington. We have not undertaken a systematic analysis of the potential consequences to our business and financial results from a major flood, blizzard, fire, earthquake, power loss, terrorist activity, pandemics or other disasters and do not have a recovery plan for such disasters. Also, our contract development and manufacturing organizations’ and suppliers’ facilities are located in multiple locations where other natural disasters or similar events which could severely disrupt our operations, could expose us to liability and could have a material adverse effect on our business. The occurrence of any of these business disruptions could seriously harm our operations and financial condition and increase our costs and expenses.
Risks Related to Our Intellectual Property
If we are not able to obtain and enforce patent protection for our technology, including therapeutic candidates, therapeutic products, and platform technology, development of our therapeutic candidates and platform, and commercialization of our therapeutic products may be materially and adversely affected.
Our success depends in part on our ability to obtain and maintain patents and other forms of intellectual property rights, including in-licenses of intellectual property rights of others, for our technology, including platform technology and therapeutic candidates and products, methods used to manufacture our therapeutic candidates and products, and methods for treating patients using our therapeutic candidates and products, as well as our ability to preserve our trade secrets, to prevent third parties from infringing upon our proprietary rights, and to operate without infringing upon the proprietary rights of others. Our scientific platform and substantially all of our intellectual property have been developed internally. As of December 31, 2021, our patent portfolio consists of 47 granted patents and over 155 pending patent applications. We may not be able to apply for patents on certain aspects of our technology, including therapeutic candidates and products, in a timely fashion or at all. Any future patents we obtain may not be sufficiently broad to prevent others from using our technology or from developing competing therapeutics and technology. There is no guarantee that any of our pending patent applications will result in issued or granted patents, any of our issued or granted patents will not later be found to be invalid or unenforceable, or any issued or granted patents will include claims sufficiently broad to cover our technology, including platform technology and therapeutic candidates and products, or to provide meaningful protection from our competitors. Moreover, the patent position of pharmaceutical and biotechnology companies can be highly uncertain because it involves complex legal and factual questions. We will be able to protect our proprietary rights from unauthorized use by third parties only to the extent our current and future technology, including platform technology and therapeutic candidates and products, are covered by valid and enforceable patents or are effectively maintained as trade secrets. If third parties disclose or misappropriate our proprietary rights, it may materially and adversely impact our competitive position in the market.
The U.S. Patent and Trademark Office, or USPTO, and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment, and other provisions during the patent process. There are situations in which noncompliance can result in abandonment or lapse of a patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Patent offices may be affected by COVID-19 or other health epidemic shut-downs, resulting in, for example, non-essential administrative tasks being delayed or eliminated. This could affect patent rights, including the partial or complete loss of patent rights in jurisdictions such as the USPTO and international patent offices. In such an event, competitors might be able to enter the market earlier than would otherwise have been the case. Further, the standards applied by the USPTO and foreign patent offices in granting patents are not always applied uniformly or predictably. For example, there is no uniform worldwide policy regarding patentable subject matter or the scope of claims allowable in biotechnology and pharmaceutical patents. As such, we do not know the degree of future protection we will have on our technology, including platform technology and therapeutic candidates and products. While we will endeavor to try to protect our technology, including platform technology and therapeutic candidates and products, with intellectual property rights such as patents, as appropriate, the process of obtaining patents is time-consuming, expensive, and sometimes unpredictable, and we can provide no assurances our technology, including our platform technology, therapeutic candidates and products, will be adequately protected in the future against unauthorized uses or competing claims by third parties.
In addition, recent and future changes to the patent laws and to the rules of the USPTO or other foreign patent offices may have a significant impact on our ability to protect our technology, including therapeutic candidates and products, and enforce our intellectual property rights. For example, the Leahy-Smith America Invents Act enacted in 2011 involves significant changes in patent legislation. In addition, we cannot assure that court rulings or interpretations of any court decision will not adversely impact our patents or patent applications. In addition to increasing uncertainty with regard to our ability to obtain patents in the future, there also may be uncertainty with respect to the value of patents, once obtained. Depending on decisions by the U.S. Congress, the federal courts, the USPTO, or made in foreign jurisdictions, the laws and regulations governing patents could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce our existing patents and patents we might obtain in the future.
The issuance of a patent is not conclusive as to its inventorship, scope, validity, or enforceability. Once granted, patents may remain open to opposition, interference, re-examination, post-grant review, inter partes review, revocation, nullification, or derivation action in court or before patent offices or similar proceedings before or after allowance or grant, during which time third parties can raise objections against such initial grant. In the course of such proceedings, which may continue for a protracted period of time, the patent owner may be compelled to limit the scope of the pending, allowed or granted claims thus attacked or may lose the allowed or granted claims altogether. Our patent risks include that:
•others may, or may be able to, make, use, offer to sell, or sell compounds that are the same as or similar to our therapeutic candidates and products but that are not covered by the claims of the patents we own or license;
•we or our licensors, collaborators, or any future collaborators may not be the first to file patent applications covering certain aspects of our technology, including our platform technology, therapeutic candidates and products;
•others may independently develop similar or alternative technology or duplicate any of our technology without infringing our intellectual property rights;
•a third party may challenge our patents and, if challenged, a court may not hold that our patents are valid, enforceable, or that a third party is infringing;
•a third party may challenge our patents in various patent offices and, if challenged, we may be compelled to limit the scope of our pending, allowed or granted claims or lose the allowed or granted claims altogether;
•any issued patents we own or have licensed may not provide us with any competitive advantages, or may be challenged by third parties;
•we may not develop additional proprietary technologies that are patentable;
•the patents of others could harm our business; and
•our competitors could conduct research and development activities in countries where we do not or will not have enforceable patent rights and then use the information learned from such activities to develop competitive products for sale in major commercial markets where we do not or will not have enforceable patent rights.
We may license patent rights from third-party owners or licensors. If such owners or licensors do not properly or successfully obtain, maintain or enforce the patents underlying such licenses, or if they retain or license to others any competing rights, our competitive position and business prospects may be materially and adversely affected.
We may rely upon intellectual property rights licensed from third parties to protect our technology, including platform technology and therapeutic candidates and products. To date, we have in-licensed some intellectual property on a non-exclusive basis relating to commercially-available cell lines involved in the manufacture of our vIgD programs; however, we may also license additional third-party intellectual property in the future, to protect our technology, including intellectual property relating to our platform technology and therapeutic candidates and products. Our success will depend in part on the ability of our licensors to obtain, maintain, and enforce patent protection for our licensed intellectual property, in particular those patents to which we have secured exclusive rights. Our licensors may elect not to prosecute, or may be unsuccessful in prosecuting, any patent applications licensed to us. Even if patents issue or are granted, our licensors may fail to maintain these patents, may determine not to pursue litigation against other companies infringing these patents, or may pursue litigation less aggressively than we would. Further, any additional licenses we enter into may be non-exclusive and we may not be able to obtain exclusive rights, which would potentially allow third parties to develop competing products or technology. Without protection for, or exclusive right to, any intellectual property we may license, other companies might be able to offer substantially identical or similar product(s) for sale, which could adversely affect our competitive business position and harm our business prospects. In addition, we may need to sublicense any rights we have under third-party licenses to current or future collaborators or any future strategic partners. Any impairment of these sublicensed rights could result in reduced revenue under or result in termination of an agreement by one or more of our collaborators or any future strategic partners.
Patent terms may be inadequate to protect our competitive position on our platform technology and therapeutic candidates and products for an adequate amount of time.
Patents have a limited lifespan. In the United States and abroad, if all maintenance fees/annuity fees are timely paid, the natural expiration of a patent is generally 20 years from its earliest non-provisional filing date. The protection a patent affords is limited. Even if patents covering our products are obtained, once the patent life has expired, we may be open to competition from competitive products. Given the amount of time required for the development, testing and regulatory review of new products, patents protecting such products might expire before or shortly after such products are commercialized. As a result, our owned and licensed patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours.
We may be unable to protect our patent intellectual property rights throughout the world.
Obtaining a valid and enforceable issued or granted patent covering our technology, including therapeutic candidates and products, in the United States and worldwide can be extremely costly. In jurisdictions where we have not obtained patent protection, competitors may use our technology, including our platform technology and therapeutic candidates and products, to develop their own products, and further, may commercialize such products in those jurisdictions and export otherwise infringing products to territories where we have not obtained patent protection. In certain instances, a competitor may be able to export otherwise infringing products in territories where we will obtain patent protection. In jurisdictions outside the United States where we will obtain patent protection, it may be more difficult to enforce a patent as compared to the United States. Competitor products may compete with our future products in jurisdictions where we do not or will not have issued or granted patents or where our issued or granted patent claims or other intellectual property rights are not sufficient to prevent competitor activities in these jurisdictions. The legal systems of certain countries, particularly certain developing countries, make it difficult to enforce patents and such countries may not recognize other types of intellectual property protection, particularly relating to biopharmaceuticals. This could make it difficult for us to prevent the infringement of our patents or marketing of competing products in violation of our proprietary rights generally in certain jurisdictions. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial cost and divert our efforts and attention from other aspects of our business.
We generally file a provisional patent application first (a priority filing) at the USPTO. An international application under the Patent Cooperation Treaty, or PCT, is usually filed within twelve months after the priority filing, at times with a United States filing. Based on the PCT filing, national and regional patent applications may be filed in various international jurisdictions, such as in Europe, Japan, Australia, Canada, and the United States. We have so far not filed for patent protection in all national and regional jurisdictions where such protection may be available. In addition, we may decide to abandon national and regional patent applications before they are granted. Finally, the grant proceeding of each national or regional patent is an independent proceeding which may lead to situations in which applications might in some jurisdictions be refused by the relevant registration authorities, while granted by others. It is also quite common that, depending on the country, various scopes of patent protection may be granted on the same therapeutic candidate, product, or technology. The laws of some jurisdictions do not protect intellectual property rights to the same extent as the laws in the United States, and many companies have encountered significant difficulties in protecting and defending such rights in such jurisdictions. If we or our licensors encounter difficulties in protecting, or are otherwise precluded from effectively protecting, the intellectual property rights important for our business in such jurisdictions, the value of these rights may be diminished, and we may face additional competition from others in those jurisdictions. Many countries have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties. In addition, many countries limit the enforceability of patents against government agencies or government contractors. In these countries, the patent owner may have limited remedies, which could materially diminish the value of such patent. If we or any of our licensors are forced to grant a license to third parties with respect to any patents relevant to our business, our competitive position in the relevant jurisdiction may be impaired and our business and results of operations may be adversely affected.
We or our licensors, collaborators, or any future strategic partners may become subject to third-party claims or litigation alleging infringement of patents or other proprietary rights or seeking to invalidate patents or other proprietary rights, and we may need to resort to litigation to protect or enforce our patents or other proprietary rights, all of which could be costly, time consuming, delay or prevent the development of our therapeutic candidates and commercialization of our therapeutic products, or put our patents and other proprietary rights at risk.
We or our licensors, licensees, collaborators, or any future strategic partners may be subject to third-party claims for infringement or misappropriation of patent or other proprietary rights. We are generally obligated under our license or collaboration agreements to indemnify and hold harmless our licensors, licensees, or collaborators for damages arising from intellectual property infringement by us. If we or our licensors, licensees, collaborators, or any future strategic partners are found to infringe a third-party patent or other intellectual property rights, we could be required to pay damages, potentially including treble damages, if we are found to have willfully infringed. In addition, we or our licensors, licensees, collaborators, or any future strategic partners may choose to seek, or be required to seek, a license from a third party, which may not be available on acceptable terms, if at all. Even if a license can be obtained on acceptable terms, the rights may be non-exclusive, which could give our competitors access to the same technology or intellectual property rights licensed to or from us. If we fail to obtain a required license, we or our licensee or collaborator, or any future licensee or collaborator, may be unable to effectively market therapeutic products based on our technology, which could limit our ability to generate revenue or achieve profitability and possibly prevent us from generating revenue sufficient to sustain our operations. In addition, we may find it necessary to pursue claims or initiate lawsuits to protect or enforce our patent or other intellectual property rights. The cost to us in defending or initiating any litigation or other proceeding relating to patent or other proprietary rights, even if resolved in our favor, could be substantial, and litigation would divert our management’s attention. Some of our competitors may be able to
sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could delay our research and development efforts and limit our ability to continue our operations.
Although we do not believe our technology infringes the intellectual property rights of others, we are aware of one or more patents or patent applications that may relate to our technology, and third parties may assert against our products alleging infringement of their intellectual property rights regardless of whether their claims have merit. Infringement claims could harm our reputation, may result in the expenditure of significant resources to defend and resolve such claims, and could require us to pay monetary damages if we are found to have infringed the intellectual property rights of others.
If we were to initiate legal proceedings against a third party to enforce a patent covering our technology, including therapeutic candidates and products, the defendant could counterclaim that our patent is invalid or unenforceable. In patent litigation in the United States, defendant counterclaims alleging invalidity or unenforceability are commonplace. Grounds for a validity challenge could be an alleged failure to meet any of several statutory requirements, for example, patent ineligibility, lack of novelty, lack of written description, obviousness, or non-enablement. Grounds for an unenforceability assertion could be an allegation someone connected with prosecution of the patent withheld relevant information from the USPTO, or made a misleading statement, during prosecution. The outcome following legal assertions of invalidity and unenforceability during patent litigation is unpredictable. With respect to the validity question, for example, we cannot be certain there is no invalidating prior art, of which we and the patent examiner were unaware during prosecution. If a defendant were to prevail on a legal assertion of invalidity or unenforceability, we would lose at least part, and perhaps all, of the patent protection on our technology, including our platform technology and therapeutic candidates and products. Such a loss of patent protection could have a material adverse impact on our business. Patents and other intellectual property rights also will not protect our technology, including our platform technology and therapeutic candidates and products, if competitors design around our protected technology, including our platform technology and therapeutic candidates and products, without legally infringing our patents or other intellectual property rights.
It is also possible we have failed to identify relevant third-party patents or applications. For example, patent applications in the United States and elsewhere are published approximately 18 months after the earliest filing for which priority is claimed, with such earliest filing date being commonly referred to as the priority date. Therefore, patent applications covering our technology, including our platform technology and therapeutic candidates and products, could have been filed by others without our knowledge. Additionally, pending patent applications which have been published can, subject to certain limitations, be later amended in a manner that could cover our technology, including our platform technology and therapeutic candidates and products. Third-party intellectual property rights holders may also actively bring infringement claims against us. We cannot guarantee we will be able to successfully settle or otherwise resolve such infringement claims. If we are unable to successfully settle future claims on terms acceptable to us, we may be required to engage in or continue costly, unpredictable, and time-consuming litigation and may be prevented from, or experience substantial delays in, marketing our technology, including therapeutic candidates and products. If we fail in any such dispute, in addition to being forced to pay damages, we may be temporarily or permanently prohibited from commercializing our technology, including a therapeutic product, held to be infringing. We might, if possible, also be forced to redesign therapeutic candidates or products so we no longer infringe the third-party intellectual property rights. Any of these events, even if we were ultimately to prevail, could require us to divert substantial financial and management resources we would otherwise be able to devote to our business.
If we fail to comply with our obligations under any license, collaboration, or other agreements, we may be required to pay damages and could lose intellectual property rights necessary for developing and protecting our technology, including our platform technology, therapeutic candidates, and therapeutic products, or we could lose certain rights to grant sublicenses, either of which could have a material adverse effect on our results of operations and business prospects.
Our current licenses impose, and any future licenses we enter into are likely to impose, various development, commercialization, funding, milestone, royalty, diligence, sublicensing, insurance, patent prosecution and enforcement, and other obligations on us. If we breach any of these obligations, or use the intellectual property licensed to us in an unauthorized manner, we may be required to pay damages and the licensor may have the right to terminate the license, which could result in us being unable to develop, manufacture, and sell or offer to sell products covered by the licensed technology or enable a competitor to gain access to the licensed technology. Moreover, our licensors may own or control intellectual property that has not been licensed to us and, as a result, we may be subject to claims, regardless of their merit, that we are infringing or otherwise violating the licensor’s rights. In addition, while we cannot currently determine the amount of the royalty obligations we would be required to pay on future sales of licensed products, if any, the amounts may be significant. The amount of our future royalty obligations will depend on the technology and intellectual property we use in therapeutic products we successfully develop and commercialize, if any. Therefore, even if we successfully develop and commercialize therapeutic products, we may be unable to achieve or maintain profitability.
If we do not obtain patent term extension and data exclusivity for any therapeutic candidate or product we may develop, our business may be materially harmed.
Depending upon the timing, duration, and specifics of any FDA marketing approval of any therapeutic candidate or product we may develop, one or more of our or in-licensed U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984 (Hatch-Waxman Act). The Hatch-Waxman Act permits a patent term extension of up to five years as compensation for patent term lost during the FDA regulatory review process. A patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval, only one patent may be extended and only those claims covering the approved drug, a method for using it, or a method for manufacturing it may be extended. Similar extensions as compensation for patent term lost during regulatory review processes are also available in certain foreign countries and territories, such as in Europe under a Supplementary Patent Certificate. However, we may not be granted an extension in the United States and/or foreign countries and territories because of, for example, failing to exercise due diligence during the testing phase or regulatory review process, failing to apply within applicable deadlines, failing to apply prior to expiration of relevant patents, or otherwise failing to satisfy applicable requirements. Moreover, the applicable time period or the scope of patent protection afforded could be less than we request. If we are unable to obtain patent term extension or the term of any such extension is shorter than what we request, our competitors may obtain approval of competing products following our patent expiration, and our business, financial condition, results of operations, and growth prospects could be materially harmed.
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed.
In addition to seeking patent protection for certain aspects of our technology, including platform technology and therapeutic candidates and products, we also consider trade secrets, including confidential and unpatented know-how, important to the maintenance of our competitive position. We protect trade secrets and confidential and unpatented know-how, in part, by entering into non-disclosure and confidentiality agreements with parties who have access to such knowledge, such as our employees, corporate collaborators, outside scientific collaborators, CROs, contract manufacturers, consultants, advisors, and other third parties. We also enter into confidentiality and invention or patent assignment agreements with our employees and consultants obligating them to maintain confidentiality and assign their inventions to us. Despite these efforts, any of these parties may breach the agreements and disclose our proprietary information, including our trade secrets, and we may not be able to obtain adequate remedies for such breaches. We also cannot guarantee that we have entered into such agreements with each party that may have or has had access to our trade secrets or proprietary technology and processes. Enforcing a claim in the event of a party illegally disclosing or misappropriating a trade secret is difficult, expensive, and time-consuming, and the outcome is unpredictable. In addition, some courts in the United States and certain foreign jurisdictions are less willing or unwilling to protect trade secrets. If any of our trade secrets were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent them from using that technology or information to compete with us. If any of our trade secrets were to be disclosed to or independently developed by a competitor, our competitive position would be harmed.
We are also subject both in the United States and outside the United States to various regulatory schemes regarding requests for the information we provide to regulatory authorities, which may include, in whole or in part, trade secrets or confidential commercial information. While we are likely to be notified in advance of any disclosure of such information and would likely object to such disclosure, there can be no assurance our challenge to the request would be successful.
We may be in the future subject to claims we or our employees or consultants have wrongfully used or disclosed alleged trade secrets of our employees’ or consultants’ former employers or their clients. These claims may be costly to defend and if we do not successfully do so, we may be required to pay monetary damages, may be prohibited from using some of our research and development and may lose valuable intellectual property rights or personnel.
Many of our employees were previously employed at universities or biotechnology or pharmaceutical companies, including our current and potential competitors. We may receive correspondence from other companies alleging the improper use or disclosure, and have received, and may in the future receive, correspondence from other companies regarding the use or disclosure, by certain of our employees who have previously been employed elsewhere in our industry, including with our competitors, of their former employer’s trade secrets or other proprietary information. Responding to these allegations can be costly and disruptive to our business, even when the allegations are without merit, and can be a distraction to management. We may be subject to claims in the future that our employees have, or we have, inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. If we fail in defending current or future claims, in addition to paying monetary damages, we may lose valuable intellectual property rights, personnel, or the ability to use some of our research and development. A loss of intellectual property, key research personnel, or their work product could hamper our ability to commercialize, or prevent us from commercializing, our
therapeutic candidates, which could severely harm our business. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
If our trademarks and trade names are not adequately protected, then we may not be able to build name recognition in our markets of interest and our business may be materially and adversely affected.
Our trademarks or trade names may be challenged, infringed, circumvented, or declared generic or determined to be infringing on other marks. Any trademark litigation could be expensive. We may not be able to protect our rights to these trademarks and trade names or may be forced to stop using these names, which we need for name recognition by potential partners or customers in our markets of interest. If we are unable to establish name recognition based on our trademarks and trade names, we may not be able to compete effectively, and our business may be materially and adversely affected.
Third parties may independently develop similar or superior technology.
There can be no assurance others will not independently develop, or have not already developed, similar or more advanced technologies than our technology or that others will not design around, or have not already designed around, aspects of our technology or our trade secrets developed therefrom. If third parties develop technology similar or superior to our technology, or they successfully design around our current or future technology, our competitive position, business prospects, and results of operations could be materially and adversely affected.
Breaches of our internal computer systems, or those of our contractors, vendors, or consultants, may place our patents or proprietary rights at risk.
The loss of clinical or preclinical data or data from any future clinical trial involving our technology, including therapeutic candidates and products, could result in delays in our development and regulatory filing efforts and significantly increase our costs. In addition, theft or other exposure of data may interfere with our ability to protect our intellectual property, including trade secrets, and other information critical to our operations. We have experienced in the past, and may experience in the future, unauthorized intrusions into our internal computer systems, including portions of our internal computer systems storing information related to our platform technology, therapeutic candidates and products, and we can provide no assurances that certain sensitive and proprietary information relating to one or more of our therapeutic candidates or products has not been, or will not in the future be, compromised. Although we have invested significant resources to enhance the security of our computer systems, there can be no assurances we will not experience additional unauthorized intrusions into our computer systems, or those of our CROs, vendors, contractors, and consultants, that we will successfully detect future unauthorized intrusions in a timely manner, or that future unauthorized intrusions will not result in material adverse effects on our financial condition, reputation, or business prospects. Payments related to the elimination of ransomware may materially affect our financial condition and results of operations.
Certain data breaches must also be reported to affected individuals and the government, and in some cases to the media, under provisions of HIPAA, as amended by HITECH, other U.S. federal and state law, and requirements of non-U.S. jurisdictions, including the European Union General Data Protection Regulation, and financial penalties may also apply.
Risks Related to Government Regulation
We may be unable to obtain U.S. or foreign regulatory approval and, as a result, may be unable to commercialize our therapeutic candidates.
Our therapeutic candidates are subject to extensive governmental regulations relating to, among other things, research, development, testing, manufacture, quality control, approval, labeling, packaging, promotion, storage, record-keeping, advertising, distribution, sampling, pricing, sales and marketing, safety, post-approval monitoring and reporting, and export and import of drugs. Rigorous preclinical testing and clinical trials and an extensive regulatory approval process are required to be completed successfully in the United States and in many foreign jurisdictions before a new therapeutic can be marketed. Satisfaction of these and other regulatory requirements is costly, time consuming, uncertain, and subject to unanticipated delays. We have not obtained regulatory approval for any therapeutic candidates, and it is possible none of the therapeutic candidates we may develop will obtain the regulatory approvals necessary for us or our collaborators to begin selling them.
We have very limited experience in conducting and managing the clinical trials necessary to obtain regulatory approvals, including approval by the FDA as well as foreign regulatory authorities, such as the EMA. The time required to obtain FDA and foreign regulatory approvals is unpredictable but typically takes many years following the commencement of clinical trials,
depending upon the type, complexity, and novelty of the therapeutic candidate, and at the substantial discretion of the regulatory authorities. The standards the FDA and its foreign counterparts use when regulating us are not always applied predictably or uniformly and can change. Any analysis we perform of data from preclinical and clinical activities is subject to confirmation and interpretation by regulatory authorities, who could delay, limit, or prevent regulatory approval. We may also encounter unexpected delays or increased costs due to new government regulations, future legislation or administrative action, or from changes in the policy of FDA or foreign regulatory authorities during the period of product development, clinical trials, and regulatory review by the FDA or foreign regulatory authorities. It is impossible to predict whether legislative changes will be enacted, or whether FDA or foreign, regulations, guidance, or interpretations will be changed, or what the impact of such changes, if any, may be.
Because the therapeutic candidates we are developing may represent a new class of therapeutics, we are not aware of any definitive policies, practices, or guidelines that the FDA or its foreign counterparts have established in relation to these drugs. While we believe the therapeutic candidates we are currently developing are regulated as new biological products under the Public Health Service Act, or PHSA, the FDA could decide to regulate them or other products we may develop as drugs under the Federal Food, Drug, and Cosmetic Act, or FDCA. The lack of policies, practices, or guidelines may hinder or slow review by the FDA or foreign regulatory authorities of any regulatory filings we may submit. Moreover, the FDA or foreign regulatory authorities may respond to these submissions by defining requirements we may not have anticipated. Such responses could lead to significant delays in the clinical development of our therapeutic candidates.
Our therapeutic candidates could fail to receive regulatory approval for many reasons, including the following:
•the FDA or comparable foreign regulatory authorities may disagree with the design or implementation of our clinical trials;
•we may be unable to demonstrate to the satisfaction of the FDA or comparable foreign regulatory authorities that a therapeutic candidate is safe and effective for its proposed indication;
•the results of clinical trials may not meet the level of statistical significance required by the FDA or comparable foreign regulatory authorities for approval;
•we may be unable to demonstrate that a therapeutic candidate’s clinical and other benefits outweigh its safety risks;
•the FDA or comparable foreign regulatory authorities may disagree with our interpretation of data from preclinical studies or clinical trials;
•the data collected from clinical trials of our therapeutic candidates may not be sufficient to support the submission of a BLA or other submission or to obtain regulatory approval in the United States, the European Union or elsewhere;
•the FDA or comparable foreign regulatory authorities may fail to approve the manufacturing processes or facilities of third-party manufacturers with which we contract for clinical and commercial supplies; and
•the approval policies or regulations of the FDA or comparable foreign regulatory authorities may significantly change in a manner rendering our clinical data insufficient for approval.
Any delay or failure in obtaining required approvals could have a material adverse effect on our ability to generate revenues from the particular therapeutic candidate for which we are seeking approval. The FDA, the EMA and other regulatory authorities have substantial discretion in the approval process, and in determining when or whether regulatory approval will be obtained for any of our therapeutic candidates. Even if we believe the data collected from preclinical and clinical trials of our therapeutic candidates are promising, such data may not be sufficient to support approval by the FDA, the EMA or any other regulatory authority. Furthermore, any regulatory approval to market a product may be subject to limitations on the approved uses for which we may market the product or in the product labeling or be subject to other restrictions. Regulatory authorities also may impose requirements for costly post-marketing studies or clinical trials and surveillance to monitor the safety or efficacy of the therapeutic. In addition, the FDA has the authority to require a REMS plan as part of the approval of a BLA or NDA, or after approval, which may impose further requirements or restrictions on the distribution or use of an approved drug or biologic, such as limiting prescribing to certain physicians or medical centers that have undergone specialized training, limiting treatment to patients who meet certain safe-use criteria, and requiring treated patients to enroll in a registry. These limitations and restrictions may limit the size of the market for the therapeutic and affect coverage and reimbursement by third-party payors.
We are also subject to numerous foreign regulatory requirements governing, among other things, the conduct of clinical trials, manufacturing, marketing authorization, pricing, and third-party reimbursement. The foreign regulatory approval process varies among countries and may include all of the risks associated with FDA approval described above as well as risks
attributable to the satisfaction of local regulations in foreign jurisdictions. Moreover, the time required to obtain approval may differ from that required to obtain FDA approval. Approval by the FDA does not ensure approval by regulatory authorities outside the U.S. and vice versa.
If we fail to obtain orphan drug designation or obtain or maintain orphan drug exclusivity for certain of our products, our competitors may sell products to treat the same conditions and our revenue may be reduced.
Under the Orphan Drug Act, the FDA may designate a product as an orphan drug if it is intended to treat a rare disease or condition, defined as a patient population of fewer than 200,000 in the United States, or a patient population greater than 200,000 in the United States where there is no reasonable expectation that the cost of developing the drug will be recovered from sales in the United States. In the United States, orphan drug designation entitles a party to financial incentives such as opportunities for grant funding towards clinical trial costs, tax advantages and user-fee waivers. In addition, if a therapeutic product with orphan drug designation subsequently receives the first FDA approval for the indication for which it has such designation, the therapeutic product is entitled to orphan product exclusivity, which means the FDA may not approve any other applications to market the same therapeutic product for the same indication for seven years, except in limited circumstances such as a showing of clinical superiority over the product with orphan exclusivity or where the manufacturer is unable to assure sufficient product quantity.
As in the United States, we may apply for designation of a therapeutic candidate as an orphan drug for the treatment of a specific indication in the European Union before the application for marketing authorization is made. In the European Union, the EMA’s Committee for Orphan Medicinal Products, grants orphan drug designation to promote the development of products that are intended for the diagnosis, prevention or treatment of a life-threatening or chronically debilitating condition affecting not more than five in 10,000 persons in the European Union. Additionally, designation is granted for products intended for the diagnosis, prevention or treatment of a life-threatening, seriously debilitating or serious and chronic condition when, without incentives, it is unlikely that sales of the drug in the European Union would be sufficient to justify the necessary investment in developing the drug or biological product or where there is no satisfactory method of diagnosis, prevention or treatment, or, if such a method exists, the medicine must be of significant benefit to those affected by the condition. Sponsors of orphan drugs in the European Union can enjoy economic and marketing benefits, including reduction of fees or fee waivers and up to ten years of market exclusivity for the approved indication unless another applicant can show its therapeutic product is safer, more effective, or otherwise clinically superior to the orphan-designated therapeutic product. This period may be reduced to six years if the orphan drug designation criteria are no longer met, including where it is shown that the product is sufficiently profitable not to justify maintenance of market exclusivity.
We may seek orphan drug designation from the FDA and the EMA for certain of our product candidates. However, we may never receive such designation. Even if we are able to obtain orphan designation, we may not be the first to obtain marketing approval for any particular orphan indication due to the uncertainties associated with developing pharmaceutical products. In addition, exclusive marketing rights in the United States may be limited if we seek approval for an indication broader than the orphan-designated indication or may be lost if the FDA later determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantities of the product to meet the needs of patients with the rare disease or condition. Further, even if we obtain orphan drug exclusivity for a product, that exclusivity may not effectively protect the product from competition because different drugs with different active moieties can be approved for the same condition. Even after an orphan drug is approved, regulatory authorities may subsequently approve the same drug with the same active moiety for the same condition if they conclude that the later drug is safer, more effective, or makes a major contribution to patient care. Orphan drug designation neither shortens the development time or regulatory review time of a product candidate nor gives the product candidate any advantage in the regulatory review or approval process. In addition, orphan drug exclusivity could block the approval of one of our therapeutic candidates if a competitor obtains approval of the same therapeutic product as defined by the FDA before we do, or if our therapeutic candidate is determined to be within the same class as the competitor’s therapeutic product for the same indication or disease.
The respective orphan designation and exclusivity frameworks in the United States and in the European Union are subject to change, and any such changes may affect our ability to obtain EU or U.S. orphan designations in the future.
If we or our existing or future collaborators, manufacturers, or service providers fail to comply with healthcare laws and regulations, we or such other parties could be subject to enforcement actions, which could adversely affect our ability to develop, market, and sell our therapeutics and may harm our reputation.
Although we do not currently have any products on the market, once we begin commercializing our therapeutic candidates, if approved, we will be subject to additional healthcare statutory and regulatory requirements and enforcement by the federal, state, and foreign governments of the jurisdictions in which we conduct our business. Healthcare providers, physicians, and third-party payors play a primary role in the recommendation and prescription of any therapeutic candidates for which we obtain marketing approval. Our future arrangements with third-party payors and customers may expose us to broadly applicable fraud, abuse, and other healthcare laws and regulations constraining the business or financial arrangements and relationships through which we market, sell, and distribute the therapeutic candidates for which we obtain marketing approval. Restrictions under applicable federal and state healthcare laws and regulations include the following:
•the U.S. federal Anti-Kickback Statute, which prohibits, among other things, persons from soliciting, receiving, offering, or providing remuneration, directly or indirectly, to induce either the referral of an individual for a healthcare item or service, or the purchasing or ordering of an item or service, for which payment may be made, in whole or in part, under a federal healthcare program such as Medicare or Medicaid;
•the U.S. federal False Claims Act, which imposes criminal and civil penalties, including through civil whistleblower or qui tam actions, against individuals or entities for knowingly presenting, or causing to be presented, to the federal government, false or fraudulent claims for payment or making a false statement to avoid, decrease, or conceal an obligation to pay money to the federal government. In addition, the government may assert a claim including items and services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act;
•state all-payor fraud laws, which impose criminal and civil liability for executing a scheme to defraud any healthcare benefit program, or knowingly and willfully falsifying, concealing, or covering up a material fact or making any materially false statement in connection with the delivery of or payment for healthcare benefits, items or services; similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation;
•HIPAA, HITECH, and their implementing regulations, which impose obligations on certain covered entity healthcare providers, health plans, and healthcare clearinghouses as well as their business associates performing certain services involving the use or disclosure of individually identifiable health information, including mandatory contractual terms, with respect to safeguarding the privacy, security, and transmission of individually identifiable health information, and require notification to affected individuals and regulatory authorities of certain breaches of security of individually identifiable health information;
•the federal Physician Payment Sunshine Act and its implementing regulations, also referred to as “Open Payments,” require applicable manufacturers of pharmaceutical and biological drugs, among other covered medical products, reimbursable under Medicare, Medicaid, or Children’s Health Insurance Programs to track and report to the CMS certain payments and transfers of value made in the previous year, including but not limited to, consulting fees, travel reimbursements, and research grants made to cover recipients, including physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors), certain non-physician healthcare professionals (such as physician assistants and nurse practitioners, among others), and teaching hospitals, as well as information regarding physicians’ and their immediate family members’ ownership and investment interests in the applicable manufacturer, with limited exceptions; and
•analogous and similar state and foreign laws and regulations, such as, state anti-kickback and false claims laws potentially applicable to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payors, including private insurers; and some state laws require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government in addition to requiring drug manufacturers to report information related to payments to physicians and other healthcare providers or marketing expenditures, and state and foreign laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts.
Ensuring our future business arrangements with third parties comply with applicable healthcare laws and regulations could involve substantial costs. If our operations are found to be in violation of any such requirements, we may be subject to penalties, including civil or criminal penalties, monetary damages, the curtailment or restructuring of our operations, or exclusion from participation in government contracting, healthcare reimbursement, or other government programs, including
Medicare and Medicaid, any of which could adversely affect our financial results. Although effective compliance programs can mitigate the risk of investigation and prosecution for violations of these laws, these risks cannot be entirely eliminated. Any action against us for an alleged or suspected violation could cause our company to incur significant legal expenses and could divert our management’s attention from the operation of our business, even if our defense is successful. In addition, achieving and sustaining compliance with applicable laws and regulations may be costly to us in terms of money, time, and resources.
If we or our current or future collaborators, manufacturers, or service providers fail to comply with applicable federal, state, or foreign laws or regulations, we could be subject to enforcement actions, which could affect our ability to develop, market, and sell our therapeutics successfully and could harm our reputation and lead to reduced acceptance of our therapeutics by the market. These enforcement actions include, among others:
•adverse regulatory inspection findings;
•warning or untitled letters;
•voluntary product recalls with public notification or medical product safety alerts to healthcare professionals;
•restrictions on, or prohibitions against, marketing our therapeutics;
•restrictions on, or prohibitions against, importation or exportation of our therapeutics;
•suspension of review or refusal to approve pending applications or supplements to approved applications;
•exclusion from participation in government-funded healthcare programs;
•exclusion from eligibility for the award of government contracts for our therapeutics;
•FDA debarment;
•suspension or withdrawal of therapeutic approvals;
•seizures or administrative detention of therapeutics;
•injunctions; and
•restitution, disgorgement of profits, or civil and criminal penalties and fines.
Enacted and future legislation may increase the difficulty and cost for us to obtain marketing approval of our therapeutic candidates.
The policies of the FDA or similar regulatory authorities may change, and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates. For example, in 2016, the 21st Century Cures Act, or Cures Act, was signed into law. The Cures Act, among other things, is intended to modernize the regulation of drugs and biologics and spur innovation, but it is still being implemented and its ultimate implementation is unclear. If we or our collaborators are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we or our collaborators are not able to maintain regulatory compliance, our therapeutic candidates may not obtain or maintain regulatory approval, and we may not achieve or sustain profitability, which would adversely affect our business.
We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or executive or administrative action, either in the United States or abroad. The FDA’s and other regulatory authorities’ policies may change, and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained, and we may not achieve or sustain profitability. It is difficult to predict how current and future legislation, executive actions, and litigation, including the executive orders, will be implemented, and the extent to which they will impact our business, our clinical development, and the FDA’s and other agencies’ ability to exercise their regulatory authority, including FDA’s pre-approval inspections and timely review of any regulatory filings or applications we submit to the FDA. To the extent any legislative or executive actions impose constraints on FDA’s ability to engage in oversight and implementation activities in the normal course, our business may be negatively impacted.
Any therapeutics we develop may become subject to unfavorable pricing regulations, third-party coverage and reimbursement practices, or healthcare reform initiatives, thereby harming our business.
The regulations governing marketing approvals, pricing, coverage, and reimbursement for new drugs and biologics vary widely from country to country. Many countries require approval of the sale price of a drug before it can be marketed. In many
countries, the pricing review period begins after marketing or product licensing approval is granted. In some foreign markets, prescription pharmaceutical pricing remains subject to continuing governmental control even after initial approval is granted. Although we intend to monitor these regulations, our programs are currently in the early stages of development and we will not be able to assess the impact of price regulations for a number of years. As a result, we might obtain regulatory approval for a product in a particular country but then be subject to price regulations delaying our commercial launch of the product and negatively impacting the revenues we are able to generate from the sale of the product in that country.
Our ability to commercialize any therapeutics successfully also will depend in part on the extent to which coverage and reimbursement for these products and related treatments will be available from government health administration authorities, private health insurers, and other organizations. However, there may be significant delays in obtaining coverage for newly-approved therapeutics. Moreover, eligibility for coverage does not necessarily signify a therapeutic will be reimbursed in all cases or at a rate covering our costs, including research, development, manufacture, sale, and distribution costs. Also, interim payments for new therapeutics, if applicable, may be insufficient to cover our costs and may not be made permanent. Thus, even if we succeed in bringing one or more therapeutics to the market, these products may not be considered cost-effective, and the amount reimbursed for any of them may be insufficient to allow us to sell them on a competitive basis. Because our programs are in the early stages of development, we are unable at this time to determine their cost effectiveness, coverage prospects, potential compendia listings, or the likely level or method of reimbursement, if covered. It is equally difficult for us to predict how Medicare coverage and reimbursement policies will be applied to our products in the future, and coverage and reimbursement under different federal healthcare programs are not always consistent. Medicare reimbursement rates may also reflect budgetary constraints placed on the Medicare program.
Third-party payors often rely upon Medicare coverage policies and payment limitations in setting their own reimbursement rates. These coverage policies and limitations may rely, in part, on compendia listings for approved therapeutics. Our inability to promptly obtain relevant compendia listings, coverage, and adequate reimbursement from both government-funded and private payors for new therapeutics we develop and for which we obtain regulatory approval could have a material adverse effect on our operating results, our ability to raise capital needed to commercialize products, and our financial condition.
We believe the efforts of governments and third-party payors to contain or reduce the cost of healthcare, and legislative and regulatory proposals to broaden the availability of healthcare, will continue to affect the business and financial condition of pharmaceutical and biopharmaceutical companies. A number of legislative and regulatory changes in the healthcare system in the United States and other major healthcare markets have been proposed, and such efforts have expanded substantially in recent years. These developments could, directly or indirectly, affect our ability to sell our products, if approved, at a favorable price. In addition, third-party payors who reimburse patients or healthcare providers, such as government and private insurance plans, are seeking greater upfront discounts, additional rebates, and other concessions to reduce the prices for therapeutics. If the price we are able to charge for any therapeutics we develop, or the reimbursement provided for such products, is inadequate, our return on investment could be adversely affected.
Pursuant to health reform legislation and related initiatives, the Centers for Medicare and Medicaid Services, or CMS, are working with various healthcare providers to develop, refine, and implement Accountable Care Organizations, or ACOs, and other innovative models of care for Medicare and Medicaid beneficiaries, including the Bundled Payments for Care Improvement Initiative, the Comprehensive Primary Care Initiative, the Duals Demonstration, and other models. The continued development and expansion of ACOs and other innovative models of care will have an uncertain impact on any future reimbursement we may receive for approved therapeutics administered by such organizations.
In addition, in recent years, the U.S. Congress has enacted various laws seeking to reduce the federal debt level and contain healthcare expenditures. For example, as a result of the Budget Control Act of 2011 and the Bipartisan Budget Act of 2015, an annual 2% reduction to Medicare payments that took effect in 2013 and will remain in effect through 2031, with the exception of a temporary suspension implemented under various COVID-19 relief legislation from May 1, 2020 through March 31, 2022, unless additional Congressional action is taken. Under current legislation, the actual reduction in Medicare payments will vary from 1% in 2022 to up to 4% in the final fiscal year of this sequester. These across-the-board spending cuts could adversely affect our future revenues, earnings, and cash flows.
There has been increasing legislative and enforcement interest in the United States with respect to specialty drug pricing practices. Specifically, there have been several recent U.S. Congressional inquiries and proposed federal and state legislation designed to, among other things, bring more transparency to drug pricing, reduce the cost of prescription drugs under Medicare, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drugs. At the federal level, the Trump administration used several means to propose or implement drug pricing reform, including through federal budget proposals, executive orders and policy initiatives. For example, in 2020, HHS and CMS issued various rules that are expected to impact, among others, price reductions from pharmaceutical manufacturers to
plan sponsors under Part D, fee arrangements between pharmacy benefit managers and manufacturers, manufacturer price reporting requirements under the Medicaid Drug Rebate Program, including regulations that affect manufacturer-sponsored patient assistance programs subject to pharmacy benefit manager accumulator programs and Best Price reporting related to certain value-based purchasing arrangements. Multiple lawsuits have been brought against the HHS challenging various aspects of the rules implemented during the Trump administration. As a result, the Biden administration and HHS have delayed the implementation or published rules rescinding some of these Trump-era policies.
Under the American Rescue Plan Act of 2021, effective January 1, 2024, the statutory cap on Medicaid Drug Rebate Program rebates that manufacturers pay to state Medicaid programs will be eliminated. Elimination of this cap may require pharmaceutical manufacturers to pay more in rebates than it receives on the sale of products, which could have a material impact on our business. In July 2021, the Biden administration released an executive order, “Promoting Competition in the American Economy,” with multiple provisions aimed at increasing competition for prescription drugs. In response to this executive order, the HHS released a Comprehensive Plan for Addressing High Drug Prices that outlines principles for drug pricing reform and potential legislative policies that Congress could pursue to advance these principles. In addition, Congress is considering legislation that, if passed, could have significant impact on prices of prescription drugs covered by Medicare, including limitations on drug price increases. The impact of these legislative, executive, and administrative actions and any future healthcare measures and agency rules implemented by the Biden administration on us and the pharmaceutical industry as a whole is unclear. Any reduction in reimbursement from Medicare or other government programs may result in a reduction in payments from private payors. Additionally, a number of states are considering or have recently enacted state drug price transparency and reporting laws that could substantially increase our compliance burdens and expose us to greater liability under such state laws once we begin commercialization after obtaining regulatory approval for any of our products.
The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability or commercialize our product candidates. The impact of legislative, executive, and administrative actions of the Biden administration on us and the biopharmaceutical industry as a whole is unclear. We expect that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for our product candidates or additional pricing pressures.
The healthcare industry is heavily regulated in the U.S. at the federal, state, and local levelsand in other jurisdictions in which we may conduct trials or other activities, and our failure to comply with applicable requirements may subject us to penalties and negatively affect our financial condition.
As a healthcare company, our operations, clinical trial activities, and interactions with healthcare providers will be subject to extensive regulation in the United States, particularly if we receive FDA approval for any of our products in the future, and we may be subject to laws and regulations in other jurisdictions as we conduct clinical trials or engage in other activities in foreign jurisdictions. For example, if we receive FDA approval for a therapeutic for which reimbursement is available under a federal healthcare program, it would be subject to a variety of federal laws and regulations, including those prohibiting the filing of false or improper claims for payment by federal healthcare programs, prohibiting unlawful inducements for the referral of business reimbursable by federal healthcare programs, and requiring disclosure of certain payments and other transfers of value made to covered recipients in the previous year, including U.S.-licensed physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors), certain non-physician healthcare professionals (such as physician assistants and nurse practitioners, among others), and teaching hospitals, as well as information regarding certain physicians’ and their immediate family members’ ownership and investment interests in the applicable manufacturer, with limited exceptions. If our past or present operations, or those of our contractors or agents who conduct business on our behalf, are found to be in violation of any of these laws, we could be subject to enforcement action, government investigation, civil and criminal penalties, which could hurt our business, operations, and financial condition. It is not always possible to identify and deter misconduct by parties we may contract with, including employees, contractors, collaborators, CROs, and suppliers, and the precautions we take to detect and prevent misconduct may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant penalties, including civil, criminal and administrative penalties, damages, fines, disgorgement, imprisonment, exclusion from participation in government funded healthcare programs, such as Medicare and Medicaid, integrity oversight and reporting obligations, contractual damages, reputational harm, diminished profits and future earnings and the curtailment or restructuring of our operations
Similarly, some state laws prohibit, among other offenses, knowingly and willfully executing a scheme to defraud any health care benefit program, including private payors, or falsifying, concealing, or covering up a material fact or making any
materially false, fictitious, or fraudulent statement in connection with the delivery of or payment for items or services under a health care benefit program. We may also be subject to the privacy and security provisions of HIPAA, as amended by HITECH, which restricts the use and disclosure of patient-identifiable health information, mandates the adoption of standards relating to the privacy and security of patient-identifiable health information, and requires the reporting of certain security breaches to healthcare provider customers with respect to such information. Additionally, many states have enacted similar laws imposing more stringent requirements on entities like us. Failure to comply with applicable laws and regulations could result in substantial penalties and adversely affect our financial condition and results of operations. Complying with new regulatory requirements and changes in the laws and regulations will increase our compliance cost and exposure to potential liability.
Additionally, the collection and use of health data in the EU is governed by the General Data Protection Regulation, or GDPR, which extends the geographical scope of EU data protection law to non-EU entities under certain conditions and imposes substantial obligations upon companies and new rights for individuals.Further, state and foreign laws may apply generally to the privacy and security of information we maintain, and may differ from each other in significant ways, thus complicating compliance efforts, as discussed in the risk factor below titled, “Data collection is governed by restrictive regulations governing the use, processing and cross-border transfer of personal information.”
Data collection is governed by restrictive regulations governing the use, processing and cross-border transfer of personal information.
In conducting and/or enrolling patients in our current or future clinical trials, we are subject to restrictions relating to privacy, data protection and data security and may be subject to additional restrictions as our clinical operations expand. For example, the collection, use, storage, disclosure, transfer, or other processing of personal data regarding individuals in the European Union, or EU, including personal health data, is subject to the GDPR. The GDPR is wide-ranging in scope and imposes numerous requirements on companies that process personal data, including requirements relating to processing health and other sensitive data, obtaining consent of the individuals to whom the personal data relates, providing information to individuals regarding data processing activities, implementing safeguards to protect the security and confidentiality of personal data, providing notification of data breaches (initially to supervisory authorities and, if the breach is serious enough, to individuals), and taking certain measures when engaging third-party processors. The GDPR also imposes strict rules on the transfer of personal data to countries outside the EU, including the United States, and permits data protection authorities to impose large penalties for violations of the GDPR, including potential fines of up to €20 million or 4% of annual global revenues, whichever is greater, for the most serious of violations. The GDPR also confers a private right of action on data subjects and consumer associations to lodge complaints with supervisory authorities, seek judicial remedies, and obtain compensation for damages resulting from violations of the GDPR. In addition, the GDPR includes restrictions on cross-border data transfers. Certain aspects of cross-border data transfers under the GDPR are uncertain as the result of legal proceedings in the EU, including a July 2020 decision by the Court of Justice for the European Union, or CJEU, that invalidated the EU-U.S. Privacy Shield and, to some extent, called into question the efficacy and legality of using standard contractual clauses (SCCs). To address certain concerns of the CJEU, the European Commission issued revised SCCs in June 2021. Regulatory guidance and other developments relating to cross-border personal data transfers, including the necessity of putting in place those revised SCCs and the UK SCCs, as discussed below, may increase the complexity of transferring personal data across borders and may require us to engage in additional contractual negotiations and to modify our policies and practices relating to the transfer and other processing of personal data. The GDPR increased our responsibility and liability in relation to personal data that we process where such processing is subject to the GDPR, and we may be required to put in place additional mechanisms to ensure compliance with the GDPR, including as implemented by individual countries.
Further, the vote in the United Kingdom, or UK, in favor of exiting the EU, referred to as Brexit, has created uncertainty with regard to data protection regulation in the UK. The Data Protection Act of 2018, which “implements” and complements the GDPR, achieved Royal Assent on May 23, 2018 and is now effective in the UK along with a version of the GDPR referred to as the UK GDPR. Collectively, the Data Protection Act of 2018 and the UK GDPR authorize significant fines, up to the greater of £17.5 million or 4% of global turnover, and expose us to two parallel regimes and other potentially divergent enforcement actions for certain violations.Further, aspects of data protection in the UK remain uncertain. On June 28, 2021, the European Commission issued an adequacy decision under the GDPR and the Law Enforcement Directive, pursuant to which personal data generally may be transferred from the EU to the UK without restriction; however, this adequacy decision is subject to a four-year “sunset” period, after which the European Commission’s adequacy decision may be renewed, and this decision may be revoked or modified in the interim.Additionally, on February 2, 2022, the UK’s Information Commissioner’s Office issued new standard contractual clauses to support personal data transfers out of the UK. If approved by the UK Parliament, these standard contractual clauses become effective March 21, 2022, and we may, in addition to other impacts, experience additional costs associated with increased compliance burdens and be required to engage in new contract negotiations with third parties that aid in processing personal data on our behalf or localize certain personal data.
Other jurisdictions also increasingly maintain laws and regulations addressing privacy, data protection, and information security.We may incur liabilities, expenses, costs, and other operational losses under GDPR and local laws of applicable EU member states, the UK, and other regions in connection with any measures we take to comply with them. Working to comply with the GDPR and other laws and regulations to which we are subject in Europe and other regions outside the United States relating to privacy, data protection, and information security will be a rigorous and time-intensive process that may increase our cost of doing business or require us to change our business practices, and despite those efforts, there is a risk that we may be subject to fines and penalties, litigation, and reputational harm in connection with our activities in those regions.
In addition, in California, the CCPA creates new individual privacy rights for California consumers (as defined in the law) and places increased privacy and security obligations on entities handling personal data of consumers or households. The CCPA requires covered companies to provide new disclosure to consumers about such companies’ data collection, use and sharing practices, provide such consumers new ways to opt-out of certain sales or transfers of personal information, and provide consumers with additional causes of action in data breach situations. While it exempts some data regulated by HIPAA and certain clinical trials data, the CCPA, to the extent applicable to our business and operations, may increase our compliance costs and potential liability with respect to other personal information we collect about California residents. The CCPA went into effect on January 1, 2020, and the California Attorney General commenced enforcement actions for violations on July 1, 2020. Moreover, the CPRA was approved by California voters in the November 3, 2020 election. The CPRA will significantly modify the CCPA, creating obligations beginning on January 1, 2022, with implementing regulations expected on or before July 1, 2022, and enforcement commencing July 1, 2023. The CPRA creates further uncertainty and may require us to incur additional costs and expenses. The CCPA and CPRA could mark the beginning of a trend toward more stringent privacy legislation in the United States. The CCPA has prompted a number of proposals for federal and state privacy legislation. For example, in March 2021, Virginia enacted the Virginia Consumer Data Protection Act (CDPA), a comprehensive privacy statute that becomes effective on January 1, 2023 and shares similarities with the CCPA. In addition, on July 7, 2021, Colorado enacted the Colorado Privacy Act (CPA), becoming the third state to pass comprehensive consumer privacy legislation in the United States. These and other new laws that may be proposed or enacted could increase our potential liability and adversely affect our business.
Compliance with U.S. and international data protection laws and regulations could require us to take on more onerous obligations in our contracts, restrict our ability to collect, use and disclose data, or in some cases, impact our ability to operate in certain jurisdictions. Any actual or alleged failure to comply with U.S. or international laws and regulations relating to privacy, data protection, and data security could result in governmental investigations, proceedings and enforcement actions (which could include civil or criminal penalties), private litigation or adverse publicity, harm to our reputation, and could negatively affect our operating results and business. Moreover, clinical trial subjects about whom we or our potential collaborators obtain information, as well as the providers who share this information with us, may contractually limit our ability to use and disclose the information or impose other obligations or restrictions in connection with our use, retention and other processing of information, and we may otherwise face contractual restrictions applicable to our use, retention, and other processing of information. Claims that we have violated individuals’ privacy rights, failed to comply with data protection laws, or breached our contractual obligations, even if we are not found liable, could be expensive and time-consuming to defend and could result in adverse publicity that could harm our business.
Our ability to obtain services, reimbursement, or funding from the federal government may be impacted by possible reductions in federal spending.
The U.S. federal budget remains in flux and could, among other things, cut Medicare payments to providers. The Medicare program is frequently mentioned as a target for spending cuts. The full impact on our business of any future cuts in Medicare or other programs is uncertain. We cannot predict the extent of legislative, executive, and administrative actions of the Biden administration will have on us and the biopharmaceutical industry as a whole. If federal spending is reduced, anticipated budgetary shortfalls may also impact the ability of relevant agencies such as the FDA or the National Institutes of Health to continue to function at current levels. Amounts allocated to federal grants and contracts may be reduced or eliminated. These reductions may also impact the ability of relevant agencies to timely review and approve drug research and development, manufacturing, and marketing activities, which may delay our ability to develop, market, and sell any therapeutics we may develop.
If any of our therapeutic candidates receives marketing approval and we or others later identify undesirable side effects caused by the therapeutic product, our ability to market and derive revenue from the therapeutic products could be compromised.
In the event any of our therapeutic candidates receive regulatory approval and we or others identify undesirable side effects, adverse events, or other problems caused by one of our therapeutics, any of the following adverse events could occur, which could result in the loss of significant revenue to us and materially and adversely affect our results of operations and business:
•regulatory authorities may withdraw their approval of the product and require us to take the product off the market or seize the product;
•we may need to recall the therapeutic or change the way the therapeutic is administered to patients;
•additional restrictions may be imposed on the marketing and promotion of the particular therapeutic or the manufacturing processes for the therapeutic or any component thereof;
•we may not be able to secure or maintain adequate coverage and reimbursement for our proprietary therapeutic products from government (including U.S. federal health care programs) and private payors;
•we may lose or see adverse alterations to compendia listings or treatment protocols specified by accountable care organizations;
•we may be subject to fines, restitution, or disgorgement of profits or revenues, injunctions, or the imposition of civil penalties or criminal prosecution;
•regulatory authorities may require the addition of labeling statements, such as a “black box” warning, or equivalent, or a contraindication;
•regulatory authorities may require us to implement a REMS plan, or to conduct post-marketing studies or clinical trials and surveillance to monitor the safety or efficacy of the product;
•we may be required to create a Medication Guide outlining the risks of such side effects for distribution to patients;
•we could be sued and held liable for harm caused to patients;
•the therapeutic may become less competitive; and
•our reputation may suffer.
Our therapeutic candidates for which we intend to seek approval as biologic products may face competition sooner than anticipated.
The Patient Protection and Affordable Care Act, signed into law in 2010, includes a subtitle called the Biologics Price Competition and Innovation Act of 2009, or BPCIA, which created an abbreviated approval pathway for biological products that are biosimilar to or interchangeable with an FDA-licensed reference biological product. Under the BPCIA, an application for a biosimilar product may not be submitted to the FDA until four years following the date that the reference product was first licensed by the FDA. In addition, the approval of a biosimilar product may not be made effective by the FDA until 12 years from the date on which the reference product was first licensed. During this 12-year period of exclusivity, another company may still market a competing version of the reference product if the FDA approves a full BLA for the competing product containing the sponsor’s own preclinical data and data from adequate and well-controlled clinical trials to demonstrate the safety, purity and potency of their product. The law is complex and is still being interpreted and implemented by the FDA. As a result, its ultimate impact, implementation, and meaning are subject to uncertainty. While it is uncertain when such processes intended to implement BPCIA may be fully adopted by the FDA, any such processes could have a material adverse effect on the future commercial prospects for our biological products.
We believe that any of our therapeutic candidates approved as a biological product under a BLA should qualify for the 12-year period of exclusivity. However, there is a risk that this exclusivity could be shortened due to congressional action or otherwise, or that the FDA will not consider our therapeutic candidates to be reference products for competing products, potentially creating the opportunity for generic competition sooner than anticipated. Other aspects of the BPCIA, some of which may impact the BPCIA exclusivity provisions, have also been the subject of recent litigation. Moreover, the extent to which a biosimilar, once approved, will be substituted for any one of our reference products in a way that is similar to traditional generic substitution for non-biological products is not yet clear, and will depend on a number of marketplace and regulatory factors that are still developing.
Significant developments stemming from the United Kingdom’s recent withdrawal from the European Union could have a material adverse effect on us.
The United Kingdom’s recent withdrawal from the European Union and ongoing negotiations related to the United Kingdom’s trade and other relationships with the European Union continue to create political and economic uncertainty, particularly in the United Kingdom and the European Union. Any business we conduct, now and in the future, in the United Kingdom, the European Union, and worldwide could be affected as a result of this transition and there are multiple ways in which our business could be negatively affected.
Brexit is expected to disrupt the operation of pre- and post-authorization clinical trial infrastructure. As of January 1, 2021, European Union law applies only to the territory of Northern Ireland to the extent foreseen in the Protocol on Ireland / Northern Ireland. The rules around GMP and pharmacovigilance in the UK currently remain similar to the EU requirements. However, the Falsified Medicines Directive will not apply in Great Britain though it is likely that the UK will implement a procedure to minimize the risk of falsified medicines. Uncertainty in the regulatory framework and future legislation can lead to disruption in the execution of international multi-center clinical trials, the monitoring of adverse events in through pharmacovigilance programs, and determination of marketing authorization across different jurisdictions. There could also be disruption to the supply and distribution as well as the import/export both of active pharmaceutical ingredients and finished product. Such a disruption could create supply difficulties for ongoing clinical trials and may damage the integrity of the pharmacovigilance database for the safety of new products. The cumulative effects of the disruption to the regulatory framework, uncertainty in future regulation, and changes to existing regulations may add considerably to the development lead time to marketing authorization and commercialization of products in the European Union and/or the United Kingdom and increase our costs. We cannot predict the impact of such changes and future regulation on our business or the results of our operations. Exposure to different and changing regulations in multiple foreign jurisdictions may increase our liabilities, expenses, costs, and other operational losses.
Risks Related to Ownership of Our Common Stock
Our stock price may be volatile, and an active, liquid, and orderly trading market may not develop for our common stock. As a result, stockholders may not be able to resell shares at or above their purchase price.
Although our common stock is listed on Nasdaq, an active trading market for our common stock may not be sustained. The lack of an active market may impair the ability of our stockholders to sell their shares at the time they wish to sell them or at a price that they consider reasonable, which may reduce the fair market value of their shares. Further, an inactive market may also impair our ability to raise capital by selling our common stock should we determine additional funding is required.
The market price of our common stock could be subject to significant fluctuations. Market prices for securities of early-stage pharmaceutical, biotechnology, and other life sciences companies have historically been particularly volatile. Some of the factors that may cause the market price of our common stock to fluctuate include:
•our and our collaborators’ ability to obtain regulatory approvals for product candidates, and delays or failures to obtain such approvals;
•the failure of any of our product candidates, if approved, to achieve commercial success;
•issues in manufacturing our approved products, if any, or product candidates;
•the results of current, and any future, preclinical or clinical trials of our product candidates;
•our ability to achieve development milestones and receive associated milestone payments pursuant to the terms of our collaboration agreements;
•the entry into, or termination of, key agreements, including key licensing, collaboration or acquisition agreements;
•the initiation or material developments in, or conclusion of, litigation to enforce or defend any of our intellectual property rights or defend against the intellectual property rights of others;
•announcements by commercial partners or competitors of new commercial products, clinical progress (or the lack thereof), significant contracts, commercial relationships, or capital commitments;
•adverse publicity relating to our markets, including with respect to other products and potential products in such markets;
•adverse publicity about our company, employees, therapeutic candidates, and/or therapeutic products in the media or on social media;
•the impact of COVID-19 on our company or the economy generally;
•the introduction of technological innovations or new therapies competing with our potential products;
•the loss of key employees;
•changes in estimates or recommendations by securities analysts, if any, who cover our common stock;
•general and industry-specific economic conditions potentially affecting our research and development expenditures;
•changes in the structure of health care payment systems;
•unanticipated serious safety concerns related to the use of any of our product candidates;
•failure to meet or exceed financial and development projections we may provide to the public;
•failure to meet or exceed the financial and development projections of the investment community;
•the perception of the pharmaceutical industry by the public, legislators, regulators, and the investment community;
•adverse regulatory decisions;
•disputes or other developments relating to proprietary rights, including patents, litigation matters, and our ability to obtain patent protection for our technologies;
•commencement of, or our involvement in, litigation;
•sales of our common stock by us or our stockholders in the future;
•trading volume of our common stock;
•period-to-period fluctuations in our financial results; and
•the other factors described in this “Risk Factors” section.Moreover, the stock markets in general have experienced substantial volatility that has often been unrelated to the operating performance of individual companies or the biotechnology sector. These broad market fluctuations may also adversely affect the trading price of our common stock.
In the past, following periods of volatility in the market price of a company’s securities, stockholders have often instituted class action securities litigation against those companies. Such litigation, if instituted, could result in substantial costs and diversion of management attention and resources, which could significantly harm our business and reputation.
Our officers and directors, and their respective affiliates, have a controlling influence over our business affairs and may make business decisions with which stockholders disagree and which may adversely affect the value of their investment.
Our executive officers and directors together with their respective affiliates, beneficially own approximately 51% of our common stock as of December 31, 2021. As a result, if some of these persons or entities act together, they will have the ability to exercise significant influence over matters submitted to the stockholders for approval, including the election of directors, amendments to the certificate of incorporation and bylaws and the approval of any strategic transaction requiring the approval of the stockholders. These actions may be taken even if they are opposed by other stockholders. This concentration of ownership may also have the effect of delaying or preventing a change of control of our company or discouraging others from making tender offers for our shares, which could prevent our stockholders from receiving a premium for their shares. Some of these persons or entities who make up our principal stockholders may have interests different from other stockholders. The significant concentration of stock ownership may adversely affect the trading price of our common stock due to investors’ perception that conflicts of interest may exist or arise.
Future sales, or the perception of future sales, of a substantial amount of our common stock could depress the trading price of our common stock.
Our stock price could decline as a result of sales of a large number of shares of our common stock or the perception that these sales could occur. These sales, or the possibility that these sales may occur, also might make it more difficult for us to sell equity securities in the future at a time and at a price that we deem appropriate.
For example, in connection with our July 2020 private placement, we entered into a registration rights agreement with the private placement investors that required us to prepare and file a resale registration statement, which was declared effective by the SEC on August 18, 2020 and permits the resale by the private placement investors of approximately 5.1 million shares of our common stock as well as approximately 2.6 million shares of common stock issuable upon the exercise of prefunded warrants and warrants issued in the July 2020 private placement. Additionally, in connection with our September 2021 private placement, we entered into a registration rights agreement with the private placement investors that required us to prepare and file a resale registration statement, which was declared effective by the SEC on November 19, 2021 and permits the resale by the private placement investors of approximately 6.5 million shares of our common stock as well as approximately 3.2 million shares of common stock issuable upon the exercise of prefunded warrants issued in the September 2021 private placement. We have in the past and may again in the future sell shares of our common stock to strategic partners in connection with collaboration agreements, as we did in December 2021 in connection with our agreement with Horizon. The shares subject to outstanding options and warrants, of which options and warrants (including prefunded warrants) to purchase 3.0 million shares and 8.8 million shares, respectively, were exercisable as of December 31, 2021, and the shares reserved for future issuance under our equity incentive plans will become available for sale immediately upon the exercise of such options.
We also register the offer and sale of all shares of common stock that we may issue under our equity incentive plans. Once we register the offer and sale of shares for the holders of registration rights and option holders, they can be freely sold in the public market upon issuance, subject to any related lock-up agreements or applicable securities laws.
In addition, in the future, we may issue additional shares of common stock or other equity or debt securities convertible into common stock in connection with a financing, acquisition, litigation settlement, employee arrangements or otherwise. Any such future issuance, including any issuances pursuant to our “at the market” equity offering program with Cowen, could result in substantial dilution to our existing stockholders and could cause our stock price to decline.
We have broad discretion over the use of the proceeds to us from our financing activities and may apply the proceeds to uses that do not improve our operating results or the value of your securities.
We have broad discretion over the use of proceeds to us from our financing activities and our stockholders rely solely on the judgment of our board of directors and management regarding the application of these proceeds. Our use of proceeds may not improve our operating results or increase the value of our common stock. Any failure to apply these proceeds effectively could have a material adverse effect on our business, delay the development of our product candidates and cause the market price of our common stock to decline.
Anti-takeover provisions in our charter documents and under Delaware or Washington law could discourage, delay or prevent a change in control of our company, limit attempts by our stockholders to replace or remove our current management and may affect the trading price of our common stock.
Our corporate documents contain provisions that may delay or discourage transactions involving an actual or potential change in our control or change in our management, including transactions in which stockholders might otherwise receive a premium for their shares, or transactions that our stockholders might otherwise deem to be in their best interests. Therefore, these provisions could adversely affect the price of our stock. Among other things, our certificate of incorporation and bylaws:
•stagger the terms of our board of directors and require 66 and 2/3% stockholder voting to remove directors, who may only be removed for cause;
•provide that the authorized number of directors may be changed only by resolution of the board of directors;
•provide that all vacancies, including newly-created directorships, may, except as otherwise required by law, be filled by the affirmative vote of a majority of directors then in office, even if less than a quorum;
•authorize our board of directors to issue “blank check” preferred stock and to determine the rights and preferences of those shares, which may be senior to our common stock, without prior stockholder approval;
•establish advance notice requirements for nominating directors and proposing matters to be voted on by stockholders at stockholders’ meetings;
•prohibit our stockholders from calling a special meeting and prohibit stockholders from acting by written consent;
•require 66 and 2/3% stockholder voting to effect certain amendments to our certificate of incorporation and bylaws; and
•prohibit cumulative voting in the election of directors, which limits the ability of minority stockholders to elect director candidates.
In addition, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, or DGCL, which generally prohibits a Delaware corporation from engaging in any of a broad range of business combinations with any “interested” stockholder for a period of three years following the date on which the stockholder became an “interested” stockholder. Likewise, because our principal executive offices are located in Washington, the anti-takeover provisions of the Washington Business Corporation Act may apply to us under certain circumstances now or in the future. These provisions prohibit a “target corporation” from engaging in any of a broad range of business combinations with any stockholder constituting an “acquiring person” for a period of five years following the date on which the stockholder became an “acquiring person.” These provisions could discourage, delay or prevent a transaction involving a change in control of our company. These provisions could also discourage proxy contests and make it more difficult for stockholders to elect directors of their choosing and cause us to take other corporate actions our stockholders desire.
Claims for indemnification by our directors and officers may reduce our available funds to satisfy successful third-party claims against us and may reduce the amount of available cash.
Our amended and restated certificate of incorporation provides that we will indemnify our directors to the fullest extent permitted by Delaware law.
In addition, as permitted by Section 145 of the DGCL, our amended and restated bylaws and our indemnification agreements that we have entered into with our directors and officers provide that:
•We will indemnify our directors and officers for serving us in those capacities or for serving other business enterprises at our request, to the fullest extent permitted by Delaware law. Delaware law provides that a corporation may indemnify such person if such person acted in good faith and in a manner such person reasonably believed to be in or not opposed to the best interests of the corporation and, with respect to any criminal proceeding, had no reasonable cause to believe such person’s conduct was unlawful.
•We may, in our discretion, indemnify other employees and agents in those circumstances where indemnification is permitted by applicable law.
•We are required to advance expenses, as incurred, to our directors and officers in connection with defending a proceeding, except that such directors or officers shall undertake to repay such advances if it is ultimately determined that such person is not entitled to indemnification.
•We will not be obligated pursuant to our amended and restated bylaws to indemnify any director or officer in connection with any proceeding (or part thereof) initiated by such person unless the proceeding was authorized in the specific case by our board of directors or such indemnification is required to be made pursuant to our amended and restated bylaws.
•The rights conferred in our amended and restated bylaws are not exclusive, and we are authorized to enter into indemnification agreements with our directors, officers, employees and agents and to obtain insurance to indemnify such persons.
•We may not retroactively amend our amended and restated bylaw provisions to reduce our indemnification obligations to our directors or officers.
As a result, if we are required to indemnify one or more of our directors or officers, it may reduce our available funds to satisfy successful third-party claims against us, may reduce the amount of available cash and may have a material adverse effect on our business and financial condition.
Our amended and restated certificate of incorporation designates the Court of Chancery of the State of Delaware as the sole and exclusive forum for certain types of actions and proceedings that may be initiated by our stockholders, which could limit
our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers, employees or agents.
Our amended and restated certificate of incorporation provides that, unless we consent in writing to an alternative forum, the Court of Chancery of the State of Delaware will be the sole and exclusive forum for any derivative action or proceeding brought on our behalf, any action asserting a claim of breach of a fiduciary duty owed by any of our directors, officers, employees or agents to us or our stockholders, any action asserting a claim arising pursuant to any provision of the DGCL, our amended and restated certificate of incorporation or our amended and restated bylaws or any action asserting a claim that is governed by the internal affairs doctrine, in each case subject to the Court of Chancery having personal jurisdiction over the indispensable parties named as defendants therein and the claim not being one which is vested in the exclusive jurisdiction of a court or forum other than the Court of Chancery or for which the Court of Chancery does not have subject matter jurisdiction. Any person purchasing or otherwise acquiring any interest in any shares of our common stock shall be deemed to have notice of and to have consented to this provision of our amended and restated certificate of incorporation. In addition, our amended and restated bylaws provide that the U.S. federal district courts will be the exclusive forum for resolving any complaint asserting a cause of action arising under the Securities Act.
These choice of forum provision may limit our stockholders’ ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, officers, employees or agents, which may discourage such lawsuits against us and our directors, officers, employees and agents even though an action, if successful, might benefit our stockholders. Stockholders who do bring a claim in the Court of Chancery could face additional litigation costs in pursuing any such claim, particularly if they do not reside in or near Delaware. The Court of Chancery may also reach different judgments or results than would other courts, including courts where a stockholder considering an action may be located or would otherwise choose to bring the action, and such judgments or results may be more favorable to us than to our stockholders. Alternatively, if a court were to find these choice of forum provisions in our amended and restated certificate of incorporation and amended and restated bylaws inapplicable to, or unenforceable in respect of, one or more of the specified types of actions or proceedings, we may incur additional costs associated with resolving such matters in other jurisdictions, which could have a material adverse effect on our business, financial condition or results of operations.
We do not expect to pay any dividends on our common stock for the foreseeable future.
We currently expect to retain all future earnings, if any, for future operations and expansion, and have no current plans to pay any cash dividends to holders of our common stock for the foreseeable future. Any decision to declare and pay dividends in the future will be made at the discretion of our board of directors and will depend on, among other things, our results of operations, financial condition, cash requirements, contractual restrictions and other factors that our board of directors may deem relevant. As a result, stockholders may not receive any return on an investment in our common stock unless stockholders sell our common stock for a price greater than that which they paid for it.
Nasdaq may delist our common stock from its exchange, which could limit investors’ ability to make transactions in our securities and subject us to additional trading restrictions.
Our common shares are listed on Nasdaq under the trading symbol “ALPN.” Our securities may fail to meet the continued listing requirements to be listed on Nasdaq. If Nasdaq delists our common shares from trading on its exchange, we could face significant material adverse consequences, including:
•significant impairment of the liquidity for our common stock, which may substantially decrease the market price of our common stock;
•a limited availability of market quotations for our securities;
•a determination that our common stock qualifies as a “penny stock” which will require brokers trading in our common stock to adhere to more stringent rules and possibly resulting in a reduced level of trading activity in the secondary trading market for our common stock;
•a limited amount of news and analyst coverage for our company; and
•a decreased ability to issue additional securities or obtain additional financing in the future.
Risks Related to Our Financial Reporting and Disclosure
We are a smaller reporting company, and any decision on our part to comply only with reduced reporting and disclosure requirements applicable to such companies could make our common stock less attractive to investors.
We are a “smaller reporting company,” as defined in the Securities Exchange Act of 1934, as amended, or the Exchange Act. For as long as we continue to be a smaller reporting company, we may choose to take advantage of exemptions from various reporting requirements applicable to other public companies that are not smaller reporting companies, including, but not limited to, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements.
We will remain a smaller reporting company so long as, as of June 30 of the preceding year, (i) the market value of our common shares held by non-affiliates, or our public float, is less than $250 million; or (ii) we have annual revenues less than $100 million and either we have no public float or our public float is less than $700 million.
If we take advantage of some or all of the reduced disclosure requirements available to smaller reporting companies, investors may find our common stock less attractive, which may result in a less active trading market for our common stock and greater stock price volatility. For so long as we are a smaller reporting company and not classified as an “accelerated filer” or “large accelerated filer” pursuant to SEC rules, we will continue to be exempt from the auditor attestation requirements of Section 404(b) of Sarbanes-Oxley.
If we fail to maintain proper and effective internal controls, our ability to produce accurate financial statements on a timely basis could be impaired.
We are subject to the reporting requirements of the Exchange Act, the Sarbanes-Oxley Act and the rules and regulations of The Nasdaq Stock Market LLC. The Sarbanes-Oxley Act requires, among other things, that we maintain effective disclosure controls and procedures and internal control over financial reporting. We must perform system and process evaluation and testing of our internal control over financial reporting to allow management to report on the effectiveness of our internal controls over financial reporting in our Annual Report on Form 10-K filing for that year, as required by Section 404 of the Sarbanes-Oxley Act.
We may discover weaknesses in our system of internal financial and accounting controls and procedures that could result in a material misstatement of our financial statements. Our internal control over financial reporting will not prevent or detect all errors and all fraud. An internal control system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the internal control system’s objectives will be met. Because of the inherent limitations in all internal control systems, no evaluation of internal controls can provide absolute assurance that misstatements due to error or fraud will not occur or that all internal control issues and instances of fraud will be detected.
If we are not able to comply with the requirements of Section 404 of the Sarbanes-Oxley Act, or if we are unable to maintain proper and effective internal controls, we may not be able to produce timely and accurate financial statements. If that were to happen, the market price of our common stock could decline and we could be subject to sanctions or investigations by The Nasdaq Stock Market LLC, the SEC, or other regulatory authorities.
Our disclosure controls and procedures may not prevent or detect all errors or acts of fraud.
Our disclosure controls and procedures are designed to reasonably ensure that information required to be disclosed by us in reports we file or submits under the Exchange Act is accumulated and communicated to management and recorded, processed, summarized and reported within the time periods specified in the rules and forms of the SEC. We believe that any disclosure controls and procedures as well as internal controls and procedures, no matter how well-conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are and will be met. These inherent limitations include the realities that judgments in decision-making can be faulty and that breakdowns can occur because of simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people or by an unauthorized override of the controls. Accordingly, because of the inherent limitations in our control system, misstatements due to error or fraud may occur and not be detected.
We will continue to incur costs and demands upon management as a result of complying with the laws and regulations affecting public companies.
We will incur significant legal, accounting, and other expenses associated with public company reporting requirements. We will also incur costs associated with corporate governance requirements, including requirements under the Sarbanes-Oxley Act, as well as new rules implemented by the SEC and The Nasdaq Stock Market LLC. Although our status as a smaller reporting company may for a limited period of time somewhat lessen the cost of complying with these additional regulatory and other requirements, we nonetheless expect that these rules and regulations will increase our legal and financial compliance costs and to make some activities more time-consuming and costlier. Our executive officers and other personnel will need to devote substantial time to oversee our operations as a public company and compliance with applicable laws and regulations. These rules and regulations may also make it difficult and expensive for us to obtain directors and officer’s liability insurance. As a result, it may be more difficult for us to attract and retain qualified individuals to serve on our board of directors or as executive officers of our company, which may adversely affect investor confidence in us and could cause our business or stock price to suffer.
General Risk Factors
Changes in accounting rules and regulations, or interpretations thereof, could result in unfavorable accounting charges or require us to change our compensation policies.
Accounting methods and policies for biopharmaceutical companies, including policies governing revenue recognition, research and development and related expenses, and accounting for stock-based compensation, are subject to review, interpretation, and guidance from our auditors and relevant accounting authorities, including the SEC. Changes to accounting methods or policies, or interpretations thereof, may require us to reclassify, restate, or otherwise change or revise our financial statements.
If equity research analysts do not publish research or reports, or publish unfavorable research or reports, about us, our business or our market, our stock price and trading volume could decline.
The trading market for our common stock will be influenced by the research and reports that equity research analysts publish about us and our business. Equity research analysts may elect not to provide research coverage of our common stock or discontinue existing research coverage, and such lack of research coverage may adversely affect the market price of our common stock. We do not have any control over the analysts, or the content and opinions included in their reports. The price of our common stock could decline if one or more equity research analysts downgrade our stock or issue other unfavorable commentary or research. If one or more equity research analysts ceases coverage of us or fails to publish reports regularly, demand for our common stock could decrease, which in turn could cause our stock price or trading volume to decline.
Item 1B. Unresolved Staff Comments.
None.
Item 2. Properties.
In March 2019, we entered into a lease for 27,164 square feet of office and laboratory space located at 188 East Blaine Street, Seattle, Washington. The term of the lease is 10.8 years with one option to extend the term by 5.0 years. The lease term commenced in June 2019. We believe that our existing facility is adequate for our current needs as the facility has sufficient space to house additional personnel as we expand.
Item 3. Legal Proceedings.
We are not engaged in any material legal proceedings. From time to time, we may become involved in litigation relating to claims arising from the ordinary course of business. We believe that there are no claims or actions pending against us currently, the ultimate disposition of which would have a material adverse effect on our consolidated results of operation, financial condition or cash flows.
Item 4. Mine Safety Disclosures.
Not applicable.
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
Market Information
From June 17, 2015 through July 24, 2017, our common stock was traded under the symbol “NVLS.” On July 24, 2017, in connection with the business combination of Nivalis Therapeutics, Inc. and Alpine Immune Sciences, Inc., we completed a 1-for-4 reverse stock split. Commencing on July 25, 2017, our common stock began trading on The Nasdaq Global Market under the symbol “ALPN.” As of March 7, 2022, we have approximately 26 stockholders of record for our common stock, which excludes stockholders whose shares were held in nominee or street name accounts through brokers.
Dividends
We have never declared or paid cash dividends on our capital stock. We currently intend to retain any future earnings for use in the operation of our business and do not intend to declare or pay any cash dividends in the foreseeable future. Any further determination to pay dividends on our capital stock will be at the discretion of our board of directors, subject to applicable laws, and will depend on our financial condition, results of operations, capital requirements, general business conditions and other factors that our board of directors considers relevant.
Stock Performance Graph
As a “smaller reporting company,” as defined by Rule 12b-2 of the Exchange Act, and pursuant to Instruction 6 to Item 201(e) of Regulation S-K we are not required to provide the stock performance graph.
Item 6. [Reserved]
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
You should read the following discussion and analysis of our financial condition and results of operations together with our consolidated financial statements and the related notes appearing elsewhere in this Annual Report on Form 10-K. In addition to historical information, some of the information contained in this discussion and analysis or set forth elsewhere in this report, including information with respect to our plans and strategy for our business, future financial performance, expense levels and liquidity sources, includes forward-looking statements that involve risks and uncertainties. You should read Risk Factors in Part I, Item 1A of this report for a discussion of important factors that could cause actual results to differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis.Overview
We are a clinical-stage biopharmaceutical company dedicated to discovering and developing innovative, protein-based immunotherapies to treat cancer and autoimmune and inflammatory diseases. Our approach includes a proprietary scientific platform that converts native immune system proteins into differentiated, multi-targeted therapeutics. We believe our strategies are capable of meaningfully modulating the human immune system and significantly improving outcomes in patients with serious diseases.
Autoimmune/Inflammatory Diseases
Acazicolcept, is a dual ICOS and CD28 antagonist intended for the treatment of autoimmune and inflammatory diseases. Preclinical studies with acazicolcept have demonstrated efficacy in models of SLE, SjS, arthritis, inflammatory bowel disease, multiple sclerosis, type 1 diabetes, uveitis, and graft versus host disease. We have evaluated acazicolcept in a Phase 1 healthy volunteer study and initiated patient dosing in Synergy, a global, randomized, double-blind, placebo-controlled Phase 2 study of acazicolcept in adults with moderate-to-severe SLE. In June 2020, we entered into an Option and License Agreement with AbbVie Ireland Unlimited Company, or AbbVie, which grants AbbVie an exclusive option to take an exclusive license to acazicolcept. Through December 31, 2021, we have received $105.0 million in upfront and pre-option exercise development milestones as part of the AbbVie Agreement.
ALPN-303 is a dual B cell cytokine antagonist, being developed for the treatment of B cell mediated inflammatory and autoimmune diseases. Engineered using our proprietary directed evolution platform, ALPN-303 potently inhibits BAFF, BLyS and APRIL, which play key roles in B cell development, differentiation, and survival, and together contribute to the pathogenesis of multiple autoimmune diseases such as SLE and many other autoantibody-related inflammatory diseases. Data presented at the ACR Convergence 2021 Annual Meeting demonstrated that ALPN-303 inhibits the activity of the B cell cytokines APRIL and BAFF more potently than wild-type TACI-Fc counterparts, as well as anti-BAFF and anti-APRIL monoclonal antibodies. In addition, ALPN-303 has been well-tolerated in preclinical models and exhibited superior pharmacokinetics and pharmacodynamics over wild-type TACI-Fc counterparts, including superior serum exposure, suppression of T-dependent antibody production, and/or serum immunoglobulins in mice and/or cynomolgus monkeys. A first-in-human, Phase 1 study of ALPN-303 in healthy volunteers began in the fourth quarter of 2021, with top-line results targeted by mid-2022. This randomized, placebo-controlled study is designed to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of ALPN-303 administered intravenously and subcutaneously. We are planning to initiate patient-based studies in the second half of 2022 including SLE, and possibly other autoantibody-related diseases in other therapeutic areas.
In December 2021, we entered into the Horizon Agreement which grants Horizon an exclusive license for the development, manufacture and commercialization of one Existing Program and up to three additional Research Programs generated from our libraries of proteins and molecules for research, discovery and identification of additional compounds. Under the terms of the agreement, Horizon made an upfront payment to us of $25.0 million as well as an equity investment for which they paid $15.0 million, a 25 percent premium to the 30-day volume-weighted average share price as of December 9, 2021. In addition, we are eligible to receive up to $381.0 million per program, or approximately $1.5 billion in total, in future success-based payments related to development, regulatory and commercial milestones as well as tiered royalties on global net sales.
Immuno-oncology
Our lead oncology program is davoceticept, a conditional CD28 costimulator and dual checkpoint inhibitor intended for the treatment of cancer. Preclinical in vivo data have demonstrated monotherapy efficacy in tumor models superior to approved therapies. In June 2020, we initiated NEON-1, a Phase 1 monotherapy dose escalation and expansion study in patients with advanced malignancies. Initial data from NEON-1 were presented at the 2021 ASCO Virtual Meeting demonstrating that davoceticept was well-tolerated as of the cutoff date (April 22, 2021) with evidence of peripheral T cell modulation consistent with CD28 agonism. In addition, although most enrolled participants had tumors considered classically non-responsive to
immunotherapies, 61% (14 of 23 evaluable) derived clinical benefit as defined as a best outcome of stable disease or better. Completion of dose escalation for NEON-1 and initiation of expansion cohorts is anticipated in the first half of 2022.
In June 2021, we announced a collaboration and supply agreement with Merck to evaluate the safety and efficacy of davoceticept in combination with Merck’s anti-PD-1 therapy KEYTRUDA (pembrolizumab) in a Phase 1 dose escalation and expansion study. The clinical trial, NEON-2, was initiated in June 2021. As discussed above, on March 7, 2022, we announced that the FDA placed a partial clinical hold on the NEON-2 trial in response to the death of a study participant in the NEON-2 trial. The participant’s death was attributed to cardiogenic shock, considered by the treating physicians as likely related to immune-mediated myocarditis, or possibly infection. Participants who are currently enrolled in the NEON-2 trial may continue to receive davoceticept and pembrolizumab, although no new participants may be enrolled until the partial clinical hold is resolved. This partial clinical hold does not affect the ongoing NEON-1 clinical trial of davoceticept as monotherapy (NCT04186637).
Our scientific platform has also generated immune modulatory proteins with the potential of improving engineered cell therapies, such as CAR-T, TCR-T, and TILs. In May 2019, we signed a collaboration and license agreement with Adaptimmune to develop next-generation SPEARTM T cell products which incorporate our secreted and transmembrane immunomodulatory protein (termed SIP™ and TIP™) technology. We intend to continue to leverage our existing pipeline and platform to actively explore and evaluate potential value-creating partnering opportunities.
Our goal is to discover and develop modern therapies to treat patients with serious conditions such as cancer and autoimmune/inflammatory diseases. To achieve our goals, we intend to:
•aggressively move our lead autoimmune/inflammatory program acazicolcept through clinical development as part of the AbbVie Agreement, including conducting Synergy, our Phase 2 study for the treatment of SLE;
•aggressively move our second autoimmune/inflammatory program ALPN-303 through a Phase 1 study in healthy volunteers and into clinical studies for the treatment of B cell mediated autoimmune/inflammatory diseases;
•aggressively move our lead oncology program davoceticept through clinical development for the treatment of cancer; and
•maximize the value of our pipeline and platform via potential partnering activities.
Our operations to date have been limited to business planning, raising capital, developing our platform technology, identifying potential immunotherapy candidates, clinical studies, and other research and development activities. To date, we have financed operations primarily through private placements of common stock and convertible preferred stock, funds received from license and research agreements, debt financing and assets acquired upon the close of our merger with Nivalis Therapeutics Inc., or Nivalis. We do not have any products approved for sale and have not generated any product sales. Since inception and through December 31, 2021, excluding amounts borrowed through debt financing, we have raised an aggregate of $393.1 million to fund operations, of which $170.6 million was from the sale of common stock and warrants, $49.2 million was from the sale of convertible preferred stock, $129.2 million was through our license and collaboration agreements, and $44.1 million in cash, cash equivalents, and marketable securities acquired through the merger with Nivalis. As of December 31, 2021, we had cash, cash equivalents, restricted cash, and investments totaling $215.4 million.
Our net loss was $50.3 million, $27.9 million, and $41.9 million for the years ended December 31, 2021, 2020, and 2019, respectively. We expect to continue incurring significant expenses and operating losses for at least the next several years as we:
•initiate and complete nonclinical studies and clinical trials for our product candidates, including acazicolcept, a dual ICOS/CD28 antagonist program targeting autoimmune/inflammatory disorders, davoceticept, a conditional CD28 costimulator and dual checkpoint inhibitor intended for the treatment of cancer, and ALPN-303, a dual B cell cytokine antagonist for B cell-mediated autoimmune/inflammatory diseases;
•contract to manufacture and perform additional process development for our product candidates;
•continue research and development efforts to build our pipeline beyond the current product candidates;
•maintain, expand, and protect our intellectual property portfolio;
•hire additional clinical, quality control, scientific, and management personnel; and
•add operational and financial personnel to support our product development efforts and operational capabilities applicable to operating as a public company.
We do not expect to generate product revenue unless and until we successfully complete development of, obtain marketing approval for and commercialize our product candidates, either alone or in collaboration with third parties. We expect these activities will take a number of years and our success in these efforts is subject to significant uncertainty. Accordingly, we will need to raise additional capital prior to the regulatory approval and commercialization of any of our product candidates. Until such time, if ever, as we can generate substantial product revenues, we expect to finance our operating activities through equity or debt financings, collaborations or licenses, capital lease transactions, or other available financing transactions. However, additional capital may not be available on reasonable terms, if at all, and if we raise additional funds through the issuance of additional equity or debt securities, it could result in dilution to our existing stockholders and increased fixed payment obligations.
Financial Overview
Collaboration Revenue
We derive our collaboration revenue primarily from our collaboration and licensing agreements. We may generate revenue in the future from milestone payments received pursuant to our collaboration and licensing agreements with AbbVie, Horizon, Adaptimmune, or from payments from future license or collaboration agreements, product sales, or government contracts and grants. We expect revenue we generate, if any, will fluctuate from quarter to quarter.
Horizon
In December 2021, we entered into the Horizon Agreement which grants Horizon an exclusive license for the development, manufacture and commercialization of one Existing Program and up to three additional Research Programs generated from our libraries of proteins and molecules for research, discovery and identification of additional compounds.
Under the terms of the agreement, Horizon made an upfront payment to us of $25.0 million as well as an equity investment for which they paid $15.0 million, a 25 percent premium to the 30-day volume-weighted average share price as of December 9, 2021. In addition, we are eligible to receive up to $381.0 million per program, or approximately $1.5 billion in total, in future success-based payments related to development, regulatory and commercial milestones as well as tiered royalties on global net sales. We will conduct our activities under the Existing Program and up to three Research Programs to deliver compounds meeting agreed criteria. In addition, Horizon will pay us for the costs and expenses of conducting such activities under the deliverables plans. Horizon will then assume responsibility for development and commercialization activities and costs.
For revenue recognition purposes, we determined that the Existing Program and each Research Program are distinct performance obligations. We allocated revenue to each performance obligation based on its relative stand-alone selling price. The future success-based payments related to development and regulatory milestones are probable of significant revenue reversal as the achievement is highly dependent on factors outside our control. Therefore, these milestone payments are fully constrained and are not initially included in the transaction price. We will continue to re-evaluate the transaction price each reporting period and update as uncertain events are resolved or other changes in circumstances occur. Any consideration related to commercial milestones and royalties will be recognized when the related sales occur.
AbbVie
In June 2020, we entered into the AbbVie Agreement for the development of acazicolcept. The AbbVie Agreement grants AbbVie the exclusive option to purchase an exclusive worldwide license to acazicolcept, or the License Option. The License Option is exercisable by AbbVie at any time and will expire 90 days from the achievement of certain development milestones. If AbbVie exercises the License Option, AbbVie will take over the future development and commercialization. Prior to the exercise of the License Option, we will perform research and development services, including conducting our Phase 2 study in SLE, based on an agreed-upon development plan, or the Development Plan. We will be fully responsible for all costs incurred to conduct the activities under the Development Plan, provided that, AbbVie may be responsible for increased costs under the Development Plan in connection with certain material amendments proposed by AbbVie. We will also be solely responsible, at our sole cost and expense, for manufacturing and regulatory filings for acazicolcept necessary to complete activities under the Development Plan.
In June 2020, in connection with the execution of the AbbVie Agreement, AbbVie paid us a nonrefundable upfront payment of $60.0 million. Prior to the exercise of the License Option, AbbVie has agreed to make cash payments upon our achievement of certain predefined pre-option development milestones, or the Alpine Development Milestones, up to an aggregate amount of $75.0 million. In the second quarter of 2021, we achieved $45.0 million of the Alpine Development Milestones. If AbbVie exercises the License Option, they will pay a one-time cash payment of $75.0 million. Following the exercise of the License Option, AbbVie has also agreed to make aggregate cash payments of up to $205.0 million upon
AbbVie’s achievement of certain development and commercial milestones and additional aggregate cash payments of up to $450.0 million upon AbbVie’s achievement of certain sales-based cash milestones, collectively referred to as the AbbVie Milestones. Subsequent to commercialization, we are also eligible to receive high single-digit to low double-digit percentage royalties on worldwide net sales of licensed products.
For revenue recognition purposes, we determined that our contractual promises in the AbbVie Agreement are not distinct and are interdependent with our performance obligation to provide research and development services under the Development Plan. Thus, all contractual promises related to the upfront payment and Alpine’s Development Milestones were combined into a single performance obligation. We determined the Alpine Development Milestone payments are probable of significant revenue reversal as the achievement is highly dependent on factors outside our control. Therefore, these milestone payments were fully constrained and were not initially included in the transaction price. In June 2021, we re-evaluated and updated the transaction price to include the achieved portion of the Alpine Development Milestones. We will continue to re-evaluate the transaction price each reporting period and update as uncertain events are resolved or other changes in circumstances occur.
The License Option and the AbbVie Milestones were not determined to be performance obligations at the inception of the contract as they did not represent material rights. If exercised, the License Option and AbbVie Milestones will be accounted for as a separate contract and will be recognized as revenue if and when triggered. Any consideration related to sales-based royalties and profit-sharing payments will be recognized when the related sales occur.
We use a cost-based input method to measure progress toward completion of the performance obligation and to calculate the corresponding revenue to recognize each period. In applying the cost-based input, we use actual costs incurred relative to budgeted costs for the combined performance obligation. These costs consist primarily of internal personnel efforts and third-party contract costs relative to the level of patient enrollment in the study. Revenue will be recognized based on the level of costs incurred relative to the total budgeted costs for the performance obligation. A cost-based input method of revenue recognition requires management to make estimates of costs to complete our performance obligation. In making such estimates, significant judgment is required to evaluate assumptions related to cost estimates. The cumulative effect of revisions to estimated costs to complete our performance obligation will be recorded in the period in which changes are identified and amounts can be reasonably estimated. A significant change in these assumptions and estimates could have a material impact on the timing and amount of revenue recognized in future periods.
We recognized revenue from the AbbVie Agreement of $23.4 million and $7.0 million for the years ended December 31, 2021 and 2020, respectively. As of December 31, 2021 the remaining balance of the transaction price is $74.5 million and is recorded as current and noncurrent deferred revenue on our accompanying Consolidated Balance Sheets. We expect to recognize the remaining deferred revenue over the remainder of our Development Plan, which began in June 2020 and will end upon the later of the exercise or expiration of the option.Adaptimmune
In May 2019, we entered into the Adaptimmune Agreement with Adaptimmune, a clinical-stage biopharmaceutical company primarily focused on providing novel cell therapies to patients, particularly for the treatment of solid tumors, to develop next-generation SPEAR T cell products which incorporate our SIP and TIP technologies. Under the Adaptimmune Agreement, we are to perform certain research services and grant Adaptimmune an exclusive license to programs from our SIP and TIP technologies. In June 2019, under the terms of the Adaptimmune Agreement, we received an upfront license payment of $2.0 million and through December 31, 2021 we have received an additional $1.6 million in research support payments to fund ongoing programs. These payments were recorded to deferred revenue upon receipt and were recognized as revenue based on employee hours contributed to each performance obligation. In the fourth quarter of 2020, based on the completion of our initial research and development efforts in connection with our performance obligations, we recognized the remaining balance in deferred revenue associated with Adaptimmune on our accompanying Consolidated Balance Sheets. Under the Adaptimmune Agreement, we recognized no revenue for the year ended December 31, 2021, and $2.3 million and $1.3 million for the years ended December 31, 2020 and 2019, respectively. In addition, we are eligible for additional research support payments, one-time payments and downstream development and commercialization milestones of up to $288.0 million, if all pre-specified milestones for each program are achieved. We are also eligible to receive low-single digit percentage royalties on worldwide net sales of the applicable products. In February 2022, Adaptimmune selected an additional research program, triggering a $1.0 million upfront payment, which will be recorded as deferred revenue upon receipt and recognized to revenue based on employee hours contributed.
Research and Development Expenses
We focus our resources on research and development activities, including the conduct of preclinical studies, product development, regulatory support, and clinical trials for our product candidates. We recognize research and development expenses as they are incurred. Our research and development expenses consist of:
•employee-related expenses, including salaries, benefits, taxes, travel, and stock-based compensation expense for personnel in research and development functions;
•expenses related to process development and production of product candidates paid to contract manufacturing organizations;
•costs associated with preclinical activities and regulatory operations, including the cost of acquiring, developing, and manufacturing research material;
•clinical trials and activities related to regulatory filings for our product candidates; and
•allocation of facilities, overhead, depreciation, and amortization of laboratory equipment and other expenses.
The table below summarizes our research and development expenses for the periods indicated. Our direct research and development expenses consist primarily of expenses incurred pursuant to agreements with third-party manufacturing organizations for our product candidates, CROs, clinical trial sites, collaborators, and consultants. Other direct costs included direct research and development costs incurred before a selected product candidate begins clinical trials.
We use our employee and infrastructure resources across multiple research and development programs that we are advancing in parallel, and therefore do not allocate salaries, stock-based compensation, employee benefit expenses or other indirect costs related to our research and development to specific product candidates. These expenses are included in indirect research and development expense by type in the table below.
Our research and development expenses are summarized as follows (in thousands): | | | | | | | | | | | | | | | | | | | | |
| | | Years Ended December 31, |
| | | 2021 | | 2020 | | 2019 |
| Direct research and development expense by program: | | | | | | |
| Acazicolcept (ALPN-101) | | $ | 10,923 | | | $ | 4,814 | | | $ | 10,583 | |
| Davoceticept (ALPN-202) | | 10,640 | | | 4,697 | | | 8,393 | |
| ALPN-303 | | 13,311 | | | 1,047 | | | 31 | |
| Other | | 515 | | | 62 | | | 236 | |
| Total direct research and development expense by program | | 35,389 | | | 10,620 | | | 19,243 | |
| Indirect research and development expense by type: | | | | | | |
| Personnel-related costs | | 17,456 | | | 11,697 | | | 11,169 | |
| Research and development supplies and services | | 2,833 | | | 2,042 | | | 2,589 | |
| Allocated facility, equipment and other expenses | | 3,064 | | | 2,826 | | | 2,846 | |
| Total indirect research and development expense | | 23,353 | | | 16,565 | | | 16,604 | |
| Total research and development expense | | $ | 58,742 | | | $ | 27,185 | | | $ | 35,847 | |
We expect our research and development expenses to increase for the foreseeable future as we continue to develop our platform and product candidates.
The successful development of our platform and product candidates is highly uncertain. At this time, we cannot reasonably estimate the nature, timing, or costs of the efforts necessary to finish developing any of our product candidates or the period in which material net cash, if any, from these product candidates may commence. This is due to the numerous risks and uncertainties associated with developing therapeutics, including the uncertainty of:
•the scope, rate of progress, expense, and results of clinical trials;
•the scope, rate of progress, and expense of process development and manufacturing;
•preclinical and other research activities; and
•the timing of regulatory approvals.
General and Administrative Expenses
General and administrative expenses consist primarily of salaries and related costs for employees in executive, business development, finance, and administrative functions. Other significant general and administrative expenses include professional fees for accounting and legal services, expenses associated with obtaining and maintaining patents and other intellectual property, and allocation of facility and overhead costs.
We expect general and administrative expenses to increase as we expand infrastructure, headcount, and continue to prosecute our patents and other intellectual property. Other increases could potentially include increased costs for insurance, costs related to the hiring of additional personnel, and increased fees for directors, outside consultants, lawyers, and accountants. We expect to incur significant costs to comply with corporate governance, internal controls, and similar requirements applicable to public companies.
Interest Expense
Interest expense consists primarily of interest associated with our term loan with Silicon Valley Bank, or SVB, and the amortization of the related debt discount.
Interest Income
Interest income consists of interest earned on our cash, cash equivalents, and investments.
Other (Expense) Income
For the years ended December 31, 2020 and 2019, other (expense) income consists primarily of research and development tax credits received by our wholly-owned Australian subsidiary and an employee retention tax credit related to COVID-19 relief legislation. Other (expense) income was immaterial for the year ended December 31, 2021.
Results of Operations
Comparison of Years Ended December 31, 2021 and 2020
The following table summarizes our results of operations for the years ended December 31, 2021 and 2020 (in thousands):
| | | | | | | | | | | | | | | | | | | | |
| | | Years Ended December 31, | | |
| | | 2021 | | 2020 | | Change |
| Collaboration revenue | | $ | 23,443 | | | $ | 9,335 | | | $ | 14,108 | |
| Operating expenses: | | | | | | |
| Research and development | | 58,742 | | | 27,185 | | | 31,557 | |
| General and administrative | | 14,560 | | | 10,899 | | | 3,661 | |
| Total operating expenses | | 73,302 | | | 38,084 | | | 35,218 | |
| Loss from operations | | (49,859) | | | (28,749) | | | (21,110) | |
| Other income (expense): | | | | | | |
| Interest expense | | (816) | | | (775) | | | (41) | |
| Interest income | | 259 | | | 245 | | | 14 | |
| Other (expense) income | | (4) | | | 1,333 | | | (1,337) | |
| Loss before taxes | | (50,420) | | | (27,946) | | | (22,474) | |
| Income tax (expense) benefit | | 87 | | | 6 | | | 81 | |
| Net loss | | $ | (50,333) | | | $ | (27,940) | | | $ | (22,393) | |
Collaboration Revenue
Revenue for the year ended December 31, 2021 consists of $23.4 million recognized under the AbbVie Agreement, an increase from prior year as we execute on our AbbVie Development Plan. Revenue for the year ended December 31, 2020 consists of $7.0 million recognized under the AbbVie Agreement and $2.3 million recognized under the Adaptimmune Agreement.
Research and Development Expenses
The $31.6 million increase in research and development expenses was primarily attributable to increases of $10.8 million in clinical trial activity primarily related to our Synergy and NEON studies, $10.0 million in contract manufacturing and process development of our product candidates primarily for acazicolcept and ALPN-303, and $4.8 million in preclinical and other research activities. Additionally, we had an increase of $5.7 million related to personnel expenses, which includes $1.2 million in stock-based compensation expense, and an increase of $0.3 million related to overhead and facilities.
General and Administrative Expenses
The $3.7 million increase in general and administrative expenses was primarily attributable to increases of $2.5 million in personnel related expenses, which includes $0.9 million in stock-based compensation expense, $0.8 million related to professional and legal services, and $0.4 million in insurance, facility and other costs to support the growth and expansion of our business.
Interest Expense
Interest expense relates primarily to interest paid on our term loan with SVB and the related non-cash interest expense associated with the amortization of the debt discount. Interest expense during the 2021 period is slightly higher due to additional interest related to the drawdown of the second tranche of our SVB loan in March 2020.
Interest Income
Higher average investment balances during 2021, combined with lower interest rates produced relatively consistent interest income as compared to the 2020 period.
Other (Expense) Income
Other expense during 2021 consisted of a loss on an asset disposal. In 2020, other income consisted of a $1.0 million research and development tax credit received by our wholly-owned Australian subsidiary and a $0.3 million employee retention tax credit related to COVID-19 relief legislation.
Comparison of Years Ended December 31, 2020 and 2019
The following table summarizes our results of operations for the years ended December 31, 2020 and 2019 (in thousands):
| | | | | | | | | | | | | | | | | | | | |
| | | Years Ended December 31, | | |
| | | 2020 | | 2019 | | Change |
| Collaboration revenue | | $ | 9,335 | | | $ | 1,740 | | | $ | 7,595 | |
| Operating expenses: | | | | | | |
| Research and development | | 27,185 | | | 35,847 | | | (8,662) | |
| General and administrative | | 10,899 | | | 9,467 | | | 1,432 | |
| Total operating expenses | | 38,084 | | | 45,314 | | | (7,230) | |
| Loss from operations | | (28,749) | | | (43,574) | | | 14,825 | |
| Other income (expense): | | | | | | |
| Interest expense | | (775) | | | (338) | | | (437) | |
| Interest income | | 245 | | | 1,248 | | | (1,003) | |
| Other (expense) income | | 1,333 | | | 812 | | | 521 | |
| Loss before taxes | | (27,946) | | | (41,852) | | | 13,906 | |
| Income tax (expense) benefit | | 6 | | | — | | | 6 | |
| Net loss | | $ | (27,940) | | | $ | (41,852) | | | $ | 13,912 | |
Collaboration Revenue
Revenue for the year ended December 31, 2020 consists of $7.0 million recognized under the AbbVie Agreement and $2.3 million recognized under the Adaptimmune Agreement. Revenue for the year ended December 31, 2019 consists of $1.3 million related to the Adaptimmune Agreement and $0.4 million related to the milestone payment from Laurel from the sale of our GSNOR assets.
Research and Development Expenses
The $8.7 million decrease in research and development expenses was primarily attributable to decreases of $5.7 million in contract manufacturing and process development of our product candidates and $4.1 million in other research activities. The decreases were partially offset by increases of $0.6 million in clinical trial activity and $0.5 million in stock-based compensation.
General and Administrative Expenses
The $1.4 million increase in general and administrative expenses was primarily attributable increases of $0.8 million related to professional and legal services, $0.6 million in stock-based compensation and $0.4 million in insurance and facility costs to support the growth and expansion of our business. These increases were partially offset by a decrease of $0.4 million related primarily to personnel expenses.
Interest Expense
Interest expense relates primarily to interest paid on our term loan with SVB and the related non-cash interest expense associated with the amortization of the debt discount. The $0.4 million increase in interest expense is due to additional interest related to the drawdown of the second tranche of our SVB loan in March 2020.
Interest Income
The $1.0 million decrease in interest income is due primarily to less interest earned on our investments as we maintained a lower average investment balance during the 2020 period.
Other (Expense) Income
The $0.5 million increase in other (expense) income is attributable a $0.2 million increase in research and development tax credits received by our wholly-owned Australian subsidiary. Additionally, for the 2020 period, we recognized a $0.3 million employee retention tax credit related to COVID-19 relief legislation.
Liquidity and Capital Resources
Sources of Liquidity
To date, we have financed our operations primarily through the sale of equity securities, debt, and payments received under our collaboration agreements. As of December 31, 2021, we had cash, cash equivalents, restricted cash, and investments totaling $215.4 million. Except for any obligations of our collaborators to make milestone payments under our agreements with them, we do not have any committed external sources of capital. Until such time as we can generate substantial product revenue, if ever, we expect to finance our cash needs through a combination of collaboration agreements and equity or debt financings.
Equity Financing Agreements
In December 2021, in connection with the execution of the Horizon Agreement, we entered into a Stock Purchase Agreement with Horizon, or the Purchase Agreement, in which Horizon made an equity investment in Alpine of 951,980 shares of our common stock for approximately $15.76 per share, for an aggregate purchase price of $15.0 million. The purchase price represents a 25% premium to the volume-weighted average share price of our common stock for the 30-day period ended December 9, 2021. The shares are subject to lock-up restrictions, which, subject to certain exceptions, prohibit Horizon from selling the shares for a period of six months after the date of the Purchase Agreement. The shares were recorded at the fair value of our common stock on the effective date of the Horizon Agreement, with the excess of the proceeds recorded to deferred revenue.
In September 2021, we entered into a securities purchase agreement or the 2021 Securities Purchase Agreement, for a private placement with a select group of institutional investors, pursuant to which we sold 6,489,357 shares of our common stock or the Shares, and prefunded warrants to purchase 3,191,487 Shares, or the Prefunded Warrants.shares of common stock. The purchase price for each Shareshare of common stock and for each Prefunded Warrantprefunded warrant was $9.40 per share, for an aggregate purchase price of approximately $91.0 million.
The Prefunded Warrants became fully exercisablefollowing table summarizes the purchases in September 2021 of our securities by our 5% stockholders at the time of the September 2021 private placement:
| | | | | | | | | | | | | | | | | | | | |
| Name of Purchaser | | Number of Shares of Common Stock Purchased | | Number of Shares of Common Stock Subject to Warrants | | Aggregate Purchase Price |
| Decheng Capital China Life Sciences USD Fund III, L.P. | | 1,542,553 | | — | | $ | 14,500,000 | |
| Omega Fund VI, L.P. | | 744,680 | | — | | $ | 7,000,000 | |
| Entities affiliated with Avidity Partners Management LP | | 425,532 | | 638,297 | | $ | 9,999,993 | |
| OrbiMed Private Investments VI, LP | | 1,010,637 | | — | | $ | 9,500,000 | |
Additionally, as part of the September 2021 private placement, Frazier Life Sciences Public Fund L.P., an entity for which Dr. Topper shares voting control with others, acquired prefunded warrants to purchase 1,702,127 shares of common stock. The aggregate purchase price for such prefunded warrants was $16,000,000.
In September 2021, we also entered into an exchange agreement with Frazier Life Sciences VIII, L.P. (the “Exchanging Stockholder”), pursuant to which we exchanged an aggregate of 1,200,000 shares of common stock held by the Exchanging Stockholder for prefunded warrants (the “Exchange Warrants”) to purchase an aggregate of 1,200,000 shares of common stock.
We also entered into a registration rights agreement with the investors in the September 2021 private placement. The registration rights agreement required us to register the resale of the shares of common stock issued and issuable upon the closing dateexercise of prefunded warrants issued in the September 2021 private placement. We filed a registration statement on Form S-3 on November 10, 2021 covering the resale of such shares, which registration statement was declared effective by the Securities and haveExchange Commission on November 19, 2021.
September 2022 Follow-on Public Offering
In September 2022, we issued and sold 15,509,282 shares (including 1,903,282 shares sold pursuant to the underwriters’ partial exercise of their option to purchase additional shares) of our common stock at a public offering price of $7.35 per share for aggregate gross proceeds of approximately $114.0 million. Decheng Capital China Life Sciences USD Fund III, L.P. invested in the follow-on public offering, purchasing 639,456 shares of our common stock.
November 2023 Follow-on Public Offering
In November 2023, we issued and sold 8,800,000 shares of our common stock (plus an additional 991,832 shares sold pursuant to the underwriters’ partial exercise of their option to purchase additional shares) and 3,200,000 pre-funded warrants to purchase shares of common stock at a public offering price of $12.50 per share and $12.499 per pre-funded warrant, respectively, for an aggregate purchase price of $162.4 million. Decheng Capital Global Healthcare Fund (Master), LP invested in the follow-on public offering, purchasing 375,000 shares of our common stock.
Warrant Exercises
On December 11, 2023, the Company issued 145,251 shares of its common stock to Frazier Life Sciences VIII, L.P. upon their exercise of outstanding warrants to purchase an aggregate of 145,251 shares of the Company’s common stock for an aggregate total of $1,850,497.74 paid in cash. Each warrant had an exercise price of $12.74 per share. The issuances of the shares were exempt from registration under the Securities Act of 1933, as amended, pursuant to Section 4(a)(2) thereof as a transaction by an issuer not involving a public offering.
On December 29, 2023, the Company issued 26,177 shares of its common stock to Alpine Immunosciences, L.P. upon their exercise of outstanding warrants to purchase an aggregate of 74,441 shares of the Company’s common stock, pursuant to a net exercise mechanism under the warrants. Each warrant had an exercise price of $12.74 per share. The issuances of the shares were exempt from registration under the Securities Act of 1933, as amended, pursuant to Section 3(a)(9) thereof as an exchange with an existing security holder where no commission or other remuneration is paid or given for soliciting such exchange.
On January 11, 2024, the Company issued 145,251 shares of its common stock toOrbiMed Private Investments VI, LP upon their exercise of outstanding warrants to purchase an aggregate of 145,251 shares of the Company’s common stock for an aggregate total of $1,850,497.74 paid in cash. Each warrant had an exercise price of $12.74 per share. The issuances of the shares were exempt from registration under the Securities Act of 1933, as amended, pursuant to Section 4(a)(2) thereof as a transaction by an issuer not involving a public offering.
On January 19, 2024, the Company issued 1,234,636 shares of its common stock to Decheng Capital China Life Sciences USD Fund III, L.P. upon their exercise of outstanding warrants to purchase an aggregate of 1,234,636 shares of the Company’s common stock for an aggregate total of $15,729,262.64 paid in cash. Each warrant had an exercise price of $12.74 per share. The issuances of the shares were exempt from registration under the Securities Act of 1933, as amended, pursuant to Section 4(a)(2) thereof as a transaction by an issuer not involving a public offering.
On February 2, 2024, the Company issued 3,199,879 shares of its common stock to RA Capital Healthcare Fund, L.P. upon their exercise of outstanding warrants to purchase an aggregate of 3,200,000 shares of the Company’s common stock, pursuant to a net exercise mechanism under the warrants. Each warrant had an exercise price of $0.001 per share. In connection with the 2021 Securities Purchase Agreement approximately 3.7 million of the Shares issued and approximately 2.3 million of the Prefunded Warrants issued, for gross proceeds of approximately $57.0 million, were issued to certain stockholders whose beneficial ownership exceeded 5% prior to completion of the 2021 Securities Purchases Agreement.
In July 2021, we entered into a sales agreement, or the Sales Agreement, with Cowen and Company, LLC, or Cowen, pursuant to which we may sell shares of our common stock from time to time through an “at the market” equity offering for up to $75.0 million in gross cash proceeds. Cowen will act as the sales agent and will be entitled to compensation for services of up to 3.0% of the gross sales price per share of all shares sold through Cowen under the Sales Agreement. The shares would be issued pursuant to our effective shelf registration statement on Form S-3 (File No. 333-256107). We filed a prospectus supplement, dated July 2, 2021, with the SEC in connection with the offer and saleissuances of the shares were exempt from registration under the Securities Act of 1933, as amended, pursuant to Section 3(a)(9) thereof as an exchange with an existing security holder where no commission or other remuneration is paid or given for soliciting such exchange.
Other Transactions
We have granted stock options and/or restricted stock units to our named executive officers, other executive officers and our directors.
Director Independence
Our board of directors has undertaken a review of its composition, the Sales Agreement. Ascomposition of its committees and the independence of current directors and considered whether any such director has a material relationship with us that could compromise his or her ability to exercise independent judgment in carrying out his or her responsibilities. Based upon information requested from and provided by each current director concerning his or her background, employment and affiliations, including family relationships, the board of directors has determined that none of our current directors, except for Dr. Gold, have a relationship which would interfere with the exercise of independent judgment in carrying out the responsibilities of a director and that each of these directors is an “independent director” as defined under the rules of the dateNasdaq. The board of this report, we have made no such sales under the Sales Agreement.
In July 2020, we entered into a securities purchase agreement, or the 2020 Securities Purchase Agreement, for a private placement with a select group of institutional investors, pursuant to which we sold 5,139,610 units, or the Common Units,directors also determined that Messrs. Conway (chairman) and 790,710 units, or the Prefunded Warrant Units, for an aggregate purchase price of $60.0 million. Each Common Unit consists of one share ofPeetz and Ms. Hernday, who comprise our common stock plus a warrant to purchase 0.3 shares of common stock, or the Common Stock Warrants,audit committee, Drs. Topper (chairman), Thompson and each Prefunded Warrant Unit consists of one prefunded warrant to purchase one share of common stock, or the Prefunded Warrants, plus one Common Stock Warrant to purchase 0.3 shares of common stock. The Prefunded Warrant Units and the Common Units are collectively referred to as the Units and each Unit has a purchase price of $10.1175. The Common Stock Warrants have an exercise price of $12.74 and a term of 3.5 years. The Prefunded Warrants became fully exercisable upon the closing date and have an exercise price of $0.001 per share.
In January 2019, we entered into a securities purchase agreement, or the 2019 Securities Purchase Agreement, with a limited number of accredited investors, pursuant to which we sold approximately 4.7 million units, or the 2019 Units, for an aggregate purchase price of $25.3 million in a private placement, which we refer to as the Private Placement. Each 2019 Unit has a purchase price of $5.37 and consists of one share ofCui, who comprise our common stock and a warrant to purchase 0.39 shares of common stock. Pursuant to the terms of the 2019 Securities Purchase Agreement, we issued approximately 4.7 million shares of common stock and warrants to purchase an aggregate of approximately 1.8 million shares of common stock. The warrants have an exercise price of $12.74 and have a term of five years.
Debt Financing Agreements
In August 2019, we entered into an Amended and Restated Loan and Security Agreement, or the Loan Agreement, with SVB, pursuant to which SVB agreed to extend term loans to us with an aggregate principal amount of up to $15.0 million, or the Term Loans. Borrowings under the Loan Agreement consisted of up to three separate tranches. The initial tranche of $5.0 million was funded in August 2019, $3.0 million of which was used to repay amounts owing under our prior loan and security agreement with SVB. In March 2020, the second tranche of $5.0 million was funded to us. We did not draw down the final tranche of $5.0 million, which expired on July 31, 2020.
The Term Loans accrue interest at a floating per annum rate of 0.25% above the prime rate, subject to a floor of 5.75%, which interest is payable monthly commencing in September 2019. Upon the occurrence and during the continuance of an event of default, a default interest rate will apply that is 4.0% above the otherwise applicable interest rate. The Term Loans were interest only until September 30, 2020, however, under the Loan Agreement our interest only period automatically extended to June 30, 2021 if we received aggregate new capital of at least $40.0 million no later than June 30, 2020. We met this milestone in June 2020 in conjunction with the execution of the AbbVie Agreement, discussed in detail in Note 11. As a result of the interest only extension, the Term Loans will be payable in 25 equal monthly installments of principal plus interest, with the final installment due and payable on July 1, 2023.We may prepay all, but not less than all, of the Term Loans subject to a prepayment fee equal to $75,000, which represents the deferred portion of the final payment due under the Original Agreement, plus the outstanding principal balance under the Term Loans at the time of such prepayment multiplied by a prepayment fee of 2.0% in the first year, 1.0% in the second year, and 0.0% in the third year and thereafter. Additionally a final payment in the amount of 5.5% of the funded Term Loans is payable to SVB on the date on which the Term Loans are prepaid, paid or become due and payable in full. The final payment fees are recorded in long-term debt with an offsetting reduction to debt discount on our accompanying Consolidated Balance Sheets.compensation committee, Drs. Thompson
The Loan Agreement contains customary representations(chairman) and warranties, events of defaultCui and affirmative and negative covenants, including, among others, covenants that limit or restrictMr. Conway, who comprise our ability to, among other things, incur additional indebtedness, grant liens, merge or consolidate, make acquisitions, pay dividends or other distributions or repurchase equity, make investments, dispose of assets, engage in any new lines of business, and enter into certain transactions with affiliates, in each case subject to certain exceptions. Among other events, a failure to make a required loan payment, an uncured covenant breach or a material adverse change in our business, operations or condition (financial or otherwise) could lead to an event of default, and in such case, all amounts then outstanding may become due and payable immediately. We were in compliance with our covenants as of December 31, 2021. As security for its obligations under the Loan Agreement, we granted SVB a first priority security interest on substantially all of our assets, except intellectual property, and subject to certain other exceptions. As of December 31, 2021, we had $8.2 million in outstanding principal and final fees due under our term loan agreement. See Note 9 for further discussion of our Term Loans.Cash Flows
The following is a summary of our cash flows (in thousands):
| | | | | | | | | | | | | | | | | | | | |
| | | Years Ended December 31, |
| | | 2021 | | 2020 | | 2019 |
| Net cash (used in) provided by operating activities | | $ | (15,248) | | | $ | 30,084 | | | $ | (35,346) | |
| Net cash (used in) provided by investing activities | | (52,480) | | | (72,819) | | | 16,763 | |
| Net cash provided by (used in) financing activities | | 100,764 | | | 61,381 | | | 24,255 | |
Net cash (used in) provided by operating activities:
Net cash used in operating activities was $15.2 million for the year ended December 31, 2021 and consisted primarily of our net loss of $50.3 million. This was offset by an increase of $27.3 million in our net operating assets and liabilities. Additionally, we had a $7.8 million increase in our net non-cash adjustments, which primarily relates to stock-based compensation, depreciation and amortization.
Net cash provided by operating activities was $30.1 million for the year ended December 31, 2020 and consisted primarily of our net loss of $27.9 million. This was offset by an increase of $52.9 million in our net operating assets and liabilities. This increase was primarily driven by the receipt of a $60.0 million upfront payment from AbbVie, which was recorded as current and noncurrent deferred revenue on our accompanying Consolidated Balance Sheets. Additionally, we had an increase of $5.1 million in our net non-cash adjustments, which primarily relates to stock-based compensation, depreciation and amortization.Net cash used in operating activities was $35.3 million for the year ended December 31, 2019 and consisted primarily of our net loss of $41.9 million. This was offset by an increase of $3.2 million in our net operating assets and liabilities and an increase of $3.3 million in our net non-cash adjustments, which primarily relate to stock-based compensation, depreciation and amortization.
Net cash (used in) provided by investing activities:
Cash flows from investing activities primarily reflect cash used to purchase investments and proceeds from the maturities and sales of investments, thus causing a shift between our cash and cash equivalents and investment balances. We manage our cash usage with respect to our total cash, cash equivalents and investments.
Net cash used in investing activities was $52.5 million for the year ended December 31, 2021 and consisted primarily of our purchases and maturities of investments in U.S. Treasury securities, commercial paper,nominating and corporate debt securities, partially offsetgovernance committee, satisfy the independence standards for those committees established by purchases of property and equipment, primarily lab equipment, to support our research and development efforts.
Net cash used in investing activities was $72.8 million for the year ended December 31, 2020 and consisted primarily of our purchases and maturities of investments in U.S. Treasury securities, commercial paper, and corporate debt securities, partially offset by purchases of property and equipment, primarily lab equipment, to support our research and development efforts.
Net cash provided by investing activities was $16.8 million during the year ended December 31, 2019 and consisted primarily of the maturities, sales and purchases of short-term investments in U.S. Treasury securities, commercial paper, and corporate debt securities, partially offset by purchases of property and equipment, primarily lab equipment, to support our research and development efforts.
Net cash provided by financing activities:
Net cash provided by financing activities was $100.8 million for the year ended December 31, 2021 and consisted primarily of the net proceeds of $90.7 million related to the sale of approximately 6.5 million shares of common stock and 3.2 million prefunded warrants under our 2021 Securities Purchase Agreement, $11.9 million in proceeds related to the fair value of our common stock purchased by Horizon in December 2021, and $0.5 million related to the exercise of stock options. These were partially offset by $2.4 million in principal payments on our debt.
Net cash provided by financing activities was $61.4 million for the year ended December 31, 2020 and consisted primarily of the net proceeds of $56.3 million related to the sale of approximately 5.9 million Units under our 2020 Securities Purchase Agreement, $5.0 million in proceeds received from the draw down of the second tranche of our Loan Agreement in March 2020, and $0.1 million related to the exercise of stock options.
Net cash provided by financing activities was $24.3 million for the year ended December 31, 2019 and consisted primarily of the net proceeds of $23.6 million related to the sale of approximately 4.7 million Units under our 2019 Securities Purchase Agreement and $2.0 million in net proceeds from our debt refinancing, partially offset by $1.3 million in principal payments on our debt.
Funding Requirements
We have incurred operating losses since inception. We expect to continue to incur significant expenses and operating losses for the foreseeable future as we continue our research and preclinical and clinical development of our product candidates; expand the scope of our current studies for our product candidates; initiate additional preclinical, clinical or other studies for our product candidates, including under any collaboration agreements; change or add additional manufacturers or suppliers; seek regulatory and marketing approvals for any of our product candidates that successfully complete clinical studies; seek to identify, evaluate and validate additional product candidates; acquire or in-license other product candidates and technologies; maintain, protect and expand our intellectual property portfolio; attract and retain skilled personnel; and experience any delays or encounter issues with any of the above. Additionally, we have ongoing obligations with respect to our Loan Agreement, as described above, and our Operating Lease and certain contingencies, as described below.
Operating Lease
In March 2019, we entered into a lease with ARE-Seattle No. 28, LLC, or the Landlord, for 27,164 square feet of office and laboratory space located at 188 East Blaine Street, Seattle, Washington. The term of the lease is 10.8 years with one option to extend the term by 5 years. The lease term commenced in June 2019. The “Rent Commencement Date” began in March 2020, nine months after the commencement date. We were not required to pay base rent from the Rent Commencement Date through November 2020, the last day of the ninth month following the Rent Commencement Date. The annual base rent under the lease is $1.7 million for the first year and will increase by 3.0% each year thereafter. We received a tenant improvement allowance of $5.4 million, which is included in our base rent, and a maximum additional tenant improvement allowance of $1.8 million, which will result in additional rent amortized over the term of the lease at an annual rate of 8.0%. The lease also requires us to pay additional amounts for operating and maintenance expenses. In March 2019, in connection with the lease, we provided a $254,000 letter of credit as a security deposit, which is recorded as restricted cash in our accompanying Consolidated Balance Sheets. See further discussion of our operating lease in Note 10.Contingencies
Certain credits received related to our research and development expenditures and recorded within other income in our accompanying Consolidated Statements of Operations and Comprehensive Income (Loss) are subject to review by foreign taxing authorities. It is reasonably possible we may incur losses upon the completion of these reviews of up to approximately $1.8 million, which we would be required to repay to certain tax authorities.Until such time as we can generate substantial product revenue, if ever, we expect to finance our cash needs through a combination of equity or debt financings and collaboration agreements. To the extent that we raise additional capital through the future sale of equity or debt, the ownership interest of our stockholders will be diluted,applicable SEC rules and the termsrules of these securities may include liquidation or other preferences that adversely affect the rights of our existing common stockholders. If we raise additional funds through collaboration agreements in the future, we may have to relinquish valuable rights to our technologies, future revenue streams or product candidates or grant licenses on terms that may not be favorable to us. If we are unable to raise additional funds through equity or debt financings when needed, we may be required to delay, limit, reduce or terminate our product development or future commercialization efforts or grant rights to develop and market product candidates that we would otherwise prefer to develop and market ourselves.
Our future capital requirements are difficult to forecast and will depend on many factors, including:
•the number and characteristics of the future product candidates we pursue either from our internal research efforts or through acquiring or in-licensing other product candidates or technologies;
•the scope, progress, results and costs of independently researching and developing any of our future product candidates, including conducting preclinical research and clinical trials;
•whether our existing collaborations generate substantial milestone payments and, ultimately, royalties on future approved products for us;
•the timing of, and the costs involved in, obtaining regulatory approvals for any future product candidates we develop independently;
•the cost of future commercialization activities, if any;
•the cost of manufacturing our future product candidates and products, if any;
•our ability to maintain our existing collaborations and to establish new collaborations, licensing or other arrangements and the financial terms of such arrangements;
•the costs of preparing, filing, prosecuting, maintaining, defending and enforcing patents, including litigation costs and the outcome of such litigation; and
•the timing, receipt and amount of sales of, or royalties on, our current or future collaborators’ product candidates, and our future products, if any.
We have considered that our long-term operations anticipate continuing net losses and the need for potential equity or debt financing. We have also considered that new collaborations or selectively partnering our technology or programs may provide other sources of capital. However, there can be no assurances that additional funding or other sources of capital will be available on terms acceptable to us, or at all. Based on our current operating plan, we believe our available cash and cash equivalents and investments, will be sufficient to fund our planned level of operations for at least the next 12 months. We have based this estimate on assumptions that may prove to be wrong, and we could use our capital resources sooner than we expect. Additionally, the process of testing drug candidates in preclinical and clinical studies is costly, and the timing of progress in these studies remains uncertain. Further, inflation may affect our use of capital resources by increasing our cost of labor and clinical trial expenses. Our long-term funding requirements will consist of operational, capital, and manufacturing expenditures, including those contractual commitments described above. Because of the inherent risks and uncertainties associated with the development and commercialization of our product candidates, we are unable to estimate the amounts of capital outflows and operating expenditures associated with our long-term anticipated preclinical studies and clinical trials.
Critical Accounting Policies and Estimates
Our management’s discussion and analysis of our financial condition and results of operations is based on our consolidated financial statements, which we have prepared in accordance with generally accepted accounting principles in the United States, or GAAP. The preparation of these consolidated financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the consolidated financial statements, as well as the reported revenues and expenses during the reporting periods. We evaluate these estimates and judgments on an ongoing basis. We base our estimates on historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Our actual results may differ from these estimates under different assumptions or conditions.
While our significant accounting policies are more fully described in Note 2 to our consolidated financial statements, we believe that the following accounting policies are the most critical to fully understanding and evaluating our financial condition and results of operations.Accrued Liabilities
As part of the process of preparing our consolidated financial statements, we are required to estimate accruals for professional services and research and development expenses. This process involves reviewing contracts and vendor agreements and communicating with applicable personnel to identify services that have been performed on our behalf. We estimate the level of service performed and the associated cost incurred for the service when we have not yet been invoiced or otherwise notified of the actual cost. We estimate accrued liabilities as of each balance sheet date based on known facts and circumstances.
Although we do not expect our estimates to be materially different from amounts actually incurred, if our estimates of the status and timing of services performed differ from the actual status and timing of services performed, we may report amounts that are too high or too low in any particular period. To date, we have not experienced any significant adjustments to our estimates.
Revenue Recognition
Revenue recognition is a critical accounting estimate due to the magnitude and nature of the upfront cash and milestone payments we receive. Our primary sources of revenue is derived from our collaboration and licensing agreements.
Revenue is recognized when control of the promised goods or services is transferred to our customers, in an amount that reflects the consideration we expect to be entitled to in exchange for those goods or services. Our steps for recognizing revenue consist of; (1) identifying the contract, (2) identifying the performance obligations as either distinct or bundled goods and services, (3) determining the transaction price associated with each performance obligation for which we expect to be entitled in exchange for transferring such goods and services, (4) allocating the transaction price to the performance obligations in the contract and (5) recognizing revenue upon satisfaction of performance obligations.
Our collaboration agreements principally contain multiple performance obligations, which may include (1) grants of, or options to obtain, intellectual property licenses; (2) research and development services; and/or (3) manufacturing or supply services. Payments typically received under these arrangements include one or more of the following: non-refundable upfront license fees, option exercise fees, payment for research and/or development efforts, amounts due upon the achievement of specified objectives, and/or royalties on future product sales. Our revenue is primarily derived from our collaboration agreements with Adaptimmune, AbbVie, and Horizon. See further discussion of our collaboration agreements in Note 11.We allocate revenue to each performance obligation based on its relative stand-alone selling price. We generally determine stand-alone selling prices at the inception of the contract based on our best estimate of what the selling price would be if the deliverable was regularly sold by us on a stand-alone basis. Payments received prior to satisfying the relevant revenue recognition criteria are recorded as deferred revenue in the accompanying Consolidated Balance Sheets and recognized as revenue when the related revenue recognition criteria are met. We recognize revenue under our collaboration agreements by using a cost-based input method to measure progress toward completion of the performance obligation, including employee hours contributed to each performance obligation, and to calculate the corresponding revenue to recognize each period.Our collaboration agreements provide for non-refundable milestone payments. We recognize revenue that is contingent upon the achievement of a substantive milestone in its entirety in the period in which the milestone is achieved. A milestone is considered substantive when the consideration payable to us for such milestone (1) is consistent with our performance necessary to achieve the milestone or the increase in value to the collaboration resulting from our performance; (2) relates solely to our past performance; and (3) is reasonable relative to all of the other deliverables and payments within the arrangement. In making this assessment, we consider all facts and circumstances relevant to the arrangement, including factors such as the scientific, regulatory, commercial, and other risks that must be overcome to achieve the milestone, the level of effort and investment required to achieve the milestone and whether any portion of the milestone consideration is related to future performance or deliverables.
We review the contributed employee hours and progress towards completion for each performance obligation under our collaboration agreements and adjust the revenue recognized to reflect changes in assumptions relating to the estimated satisfaction of the performance obligation. Revenue recognition may be accelerated in the event of early termination of programs or if our expectations change. Alternatively, revenue recognition may be decelerated if programs are extended or delayed. While such changes to our estimates have no impact on our reported cash flows, the timing of revenue recorded in future periods could be materially impacted.
Stock-based Compensation
Stock-based compensation is recognized for all share-based payments based on the estimated fair value as of the date of grant. The fair value of our stock options is calculated using the Black-Scholes option pricing model, which requires us to apply our judgment regarding certain key assumptions including risk-free interest rate, expected term, volatility and dividend yield. For risk-free interest rate, we use the zero-coupon U.S. Treasury instruments security rate with a term equal to the expected life of the option. We use the “simplified method” for options to determine the expected term of stock options. Under this approach, the weighted-average expected life is presumed to be the average of the vesting term and the contractual term of the option. For volatility, we analyzed the stock price volatility of companies at a similar stage of development to estimate expected volatility of our stock price. Our assumed dividend yield is zero as we have never paid cash dividends and have no present intention to pay cash dividends. The fair value of restricted stock units, or RSUs, is based on the closing price of our common stock on the award date. Stock-based compensation expense is recognized over the requisite service period of the awards, usually the vesting
period, on a straight-line basis. For performance-based awards where the vesting of the options may be accelerated upon the achievement of certain milestones, the related stock-based compensation is recognized as expense when it is probable the milestone will be met. We recognize forfeiture of awards as they occur rather than estimating the expected forfeiture rate.
If factors change and we employ different assumptions for estimating stock-based compensation expense in future periods, or if we decide to use a different valuation model, the stock-based compensation expense we recognize in future periods may differ significantly from what we have recorded in the current period and could materially affect our financial statements.
Income Taxes
Income taxes are accounted for using an asset and liability approach that requires the recognition of deferred tax assets and liabilities for the expected future tax consequences of temporary differences between the consolidated financial statement and tax bases of assets and liabilities at the applicable enacted tax rates. We will establish a valuation allowance for deferred tax assets if it is more likely than not that these items will expire before we are able to realize their benefits or that future deductibility is uncertain.
We recognize the tax benefit from uncertain tax positions only if it is more likely than not that the tax position will be sustained on examination by the tax authorities, based on the technical merits of the position. The tax position is measured based on the largest benefit that has a greater than 50% likelihood of being realized upon ultimate settlement. We recognize interest and penalties related to income tax matters in income tax expense if incurred.
Recently Adopted Accounting Pronouncements
In December 2019, the Financial Accounting Standards Board, or FASB, issued Accounting Standards Update, or ASU, No. 2019-12, Income Taxes, or Topic 740: Simplifying the Accounting for Income Taxes. This ASU simplifies the accounting for income taxes by removing certain exceptions to the general principles in Topic 740 and also improves consistent application by clarifying and amending existing guidance. The standard is effective for fiscal years and interim periods within those fiscal years, beginning after December 15, 2020. We adopted this standard on January 1, 2021, and concluded that the adoption of the standard did not have a material impact on our consolidated financial statements and related disclosures.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk.
As a “smaller reporting company,” as defined by Rule 12b-2 of the Exchange Act, and pursuant to Item 305 of Regulation S-K we are not required to provide quantitative and qualitative disclosures about market risk.
Item 8. Financial Statements and Supplementary Data.
As a “smaller reporting company,” as defined by Rule 12b-2 of the Exchange Act and pursuant to Article 8, Regulation X and Item 302 of Regulation S-K, we are permitted to provide scaled Item 8 disclosure.
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure.
None.
Item 9A. Controls and Procedures.
Evaluation of Disclosure Controls and Procedures
Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, evaluated the effectiveness of our disclosure controls and procedures as of December 31, 2021. The term “disclosure controls and procedures,” as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, or the Exchange Act, means controls and other procedures of a company that are designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the Securities and Exchange Commission’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company
in the reports that it files or submits under the Exchange Act is accumulated and communicated to the company’s management, including its principal executive and principal financial officers, as appropriate to allow timely decisions regarding required disclosure. Our management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and our management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on the evaluation of our disclosure controls and procedures as of December 31, 2021, our Chief Executive Officer and Chief Financial Officer concluded that, as of such date, our disclosure controls and procedures were effective at the reasonable assurance level.
Management’s Annual Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rule 13a-15(f) and Rule 15d-15(f) of the Exchange Act. Our internal control over financial reporting is a process to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. Our internal control over financial reporting includes those policies and procedures that:
(i)pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of our assets;
(ii)provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorizations of our management and directors; and
(iii)provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements.
The effectiveness of any system of internal control over financial reporting, including ours, is subject to inherent limitations, including the exercise of judgment in designing, implementing, operating, and evaluating the controls and procedures, and the inability to eliminate misconduct completely. Accordingly, any system of internal control over financial reporting, including ours, no matter how well designed and operated, can only provide reasonable, not absolute, assurances. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions or that the degree of compliance with the policies or procedures may deteriorate. Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2021. In making this assessment, our management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission, or COSO, Internal Control-Integrated Framework (2013). Based on our assessment using those criteria, our management has concluded that, as of December 31, 2021, our internal control over financial reporting was effective.
Changes in Internal Control Over Financial Reporting
No significant changes in our internal control over financial reporting occurred during the fiscal quarter ended December 31, 2021, that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Auditor Attestation
We ceased to be an “emerging growth company” under the JOBS Act effective December 31, 2020. However, for so long as we are not classified as an “accelerated filer” or “large accelerated filer” pursuant to SEC rules, we will continue to be exempt from the auditor attestation requirements of Section 404(b) of Sarbanes-Oxley.
Item 9B. Other Information.
None.
Item 9C. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections.
Not applicable.
PART III
Item 10. Directors, Executive Officers and Corporate Governance.
The information required by Item 10 of Form 10-K is incorporated by reference to our Proxy Statement for the 2022 Annual Meeting of Stockholders to be filed with the SEC within 120 days after the end of the fiscal year ended December 31, 2021.
Item 11. Executive Compensation.
The information required by Item 11 of Form 10-K is incorporated by reference to our Proxy Statement for the 2022 Annual Meeting of Stockholders to be filed with the SEC within 120 days after the end of the fiscal year ended December 31, 2021.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
The information required by Item 12 of Form 10-K is incorporated by reference to our Proxy Statement for the 2022 Annual Meeting of Stockholders to be filed with the SEC within 120 days after the end of the fiscal year ended December 31, 2021.
Item 13. Certain Relationships and Related Transactions, and Director Independence.
The information required by Item 13 of Form 10-K is incorporated by reference to our Proxy Statement for the 2022 Annual Meeting of Stockholders to be filed with the SEC within 120 days after the end of the fiscal year ended December 31, 2021.Nasdaq.
Item 14. Principal Accounting Fees and Services.
Fees Paid to the Independent Registered Public Accounting Firm
The information required by Item 14 of Form 10-K is incorporated by referencefollowing table presents fees for professional audit services and other services rendered to our Proxy Statementcompany by Ernst & Young LLP, or EY, for the 2022 Annual Meetingyears ended December 31, 2023 and 2022.
| | | | | | | | | | | | | | |
| | | Year Ended December 31, |
| Fee Category | | 2023 | | 2022 |
| Audit fees(1) | | $ | 948,390 | | | $ | 849,510 | |
| Audit-related fees(2) | | — | | | — | |
| Tax fees(3) | | 44,617 | | | 31,918 | |
| All other fees(4) | | — | | | — | |
| Total fees | | $ | 993,007 | | | $ | 881,428 | |
(1)Audit fees consist of Stockholders to befees for professional services provided in connection with the audit of our annual consolidated financial statements, review of our quarterly consolidated financial statements, procedures for comfort letters, consents and assistance with and review of documents filed with the SEC within 120 days afterSEC.
(2)Audit-related fees consist of assurance and related services that are reasonably related to the endperformance of the fiscal year ended December 31, 2021.audit or review of our financial statements and are not reported under “Audit Fees”.
(3)Tax fees consist of fees associated with tax compliance, tax advice and tax planning fees.
(4)All other fees include any fees billed that are not audit fees, audit-related fees or tax fees.
Audit Committee Policy on Pre-Approval of Audit and Permissible Non-Audit Services of Independent Registered Public Accounting Firm
Pursuant to its charter, the audit committee must review and approve, in advance, the scope and plans for the audits and the audit fees and approve in advance (or, where permitted under the rules and regulations of the SEC, subsequently) all non-audit and tax services to be performed by the independent auditor that are not otherwise prohibited by law and any associated fees. All fees paid to EY for 2023 and 2022 were pre-approved by our audit committee. The audit committee may delegate to one or more members of the committee the authority to pre-approve audit and permissible non-audit services, as long as this pre-approval is presented to the full committee at scheduled meetings.
PART IV
Item 15. Exhibits, Financial Statement Schedules.
(a)The financial statements, schedules and exhibits filed as a part of this Annual Report on Form 10-K are as follows:
(a)(1)Financial statements – The financial statements included in Item 8 are filed as part of this Annual Report on Form 10-K.
(b)(2)Financial Statement Schedules – All schedules have been omitted because they are not applicable or required, or the information required to be set forth therein is included in the consolidated financial statements or notes thereto included in Item 8 of this Annual Report on Form 10-K.(c)(3)Exhibits – The exhibits required to be filed as part of this report are listed in the Exhibit List attached hereto and are incorporated herein by reference.
INDEX TO EXHIBITS
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | Incorporated by Reference |
| Exhibit Number | | Description | | Form | | File No. | | Exhibit | | Filing Date |
| 3.1 | | | | 10-K | | 001-37449 | | 3.1 | | March 28, 2018 |
| 3.2 | | | | 10-K | | 001-37449 | | 3.2 | | March 18, 2021 |
| 4.1 | | | | 10-K | | 001-37449 | | 4.1 | | March 28, 2018 |
| 4.2 | | | | 10-K | | 001-37449 | | 4.5 | | March 28, 2018 |
| 4.3 | | | | 10-K | | 001-37449 | | 4.6 | | March 28, 2018 |
| 4.4 | | | | 8-K | | 001-37449 | | 10.3 | | January 16, 2019 |
| 4.5 | | | | 8-K | | 001-37449 | | 4.1 | | August 28, 2019 |
| 4.6 | | | | 8-K | | 001-37449 | | 10.3 | | July 24, 2020 |
| 4.7 | | | | 8-K | | 001-37449 | | 10.4 | | July 24, 2020 |
| 4.8 | | | | 10-K | | 001-37449 | | 4.1 | | March 18, 2021 |
| 4.9 | | | | 8-K | | 001-37449 | | 10.3 | | September 14, 2021 |
| 10.1* | | | | S-8 | | 333-205220 | | 4.4 | | June 25, 2015 |
| 10.2* | | | | S-8 | | 333-205220 | | 4.5 | | June 25, 2015 |
| 10.3* | | | | S-8 | | 333-205220 | | 4.6 | | June 25, 2015 |
| 10.4* | | | | S-8 | | 333-205220 | | 4.7 | | June 25, 2015 |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | Incorporated by Reference |
| Exhibit Number | | Description | | Form | | File No. | | Exhibit | | Filing Date |
| 2.1 | | | | 8-K | | 001-37449 | | 2.1 | | April 10, 2024 |
| 3.1 | | | | 8-K | | 001-37449 | | 3.1 | | June 14, 2023 |
| 3.2 | | | | 8-K | | 001-37449 | | 3.1 | | January 26, 2023 |
| 4.1 | | | | 10-K | | 001-37449 | | 4.1 | | March 28, 2018 |
| 4.2 | | | | 10-K | | 001-37449 | | 4.5 | | March 28, 2018 |
| 4.3 | | | | 8-K | | 001-37449 | | 4.1 | | August 28, 2019 |
| 4.4 | | | | 10-K | | 001-37449 | | 4.4 | | March 20, 2024 |
| 4.5 | | | | 8-K | | 001-37449 | | 10.3 | | September 15, 2021 |
| 10.1* | | | | S-8 | | 333-205220 | | 4.4 | | June 25, 2015 |
| 10.2* | | | | S-8 | | 333-205220 | | 4.5 | | June 25, 2015 |
| 10.3* | | | | S-8 | | 333-205220 | | 4.6 | | June 25, 2015 |
| 10.4* | | | | S-8 | | 333-205220 | | 4.7 | | June 25, 2015 |
| 10.5* | | | | S-1 | | 333-204127 | | 10.18 | | May 13, 2015 |
| 10.6* | | | | 10-K | | 001-37449 | | 10.6 | | March 20, 2024 |
| 10.7* | | | | 8-K | | 001-37449 | | 10.1 | | January 5, 2024 |
| 10.8* | | | | 10-K | | 001-37449 | | 10.33 | | March 28, 2018 |
| 10.9* | | | | 10-K | | 001-37449 | | 10.35 | | March 28, 2018 |
| 10.10* | | | | 10-K | | 001-37449 | | 10.37 | | March 28, 2018 |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | Incorporated by Reference |
| Exhibit Number | | Description | | Form | | File No. | | Exhibit | | Filing Date |
| 10.5* | | | | S-1 | | 333-204127 | | 10.18 | | May 13, 2015 |
| 10.6* | | | | 10-K | | 001-37449 | | 10.6 | | March 18, 2021 |
| 10.7* | | | | 8-K | | 001-37449 | | 10.1 | | December 11, 2017 |
| 10.8* | | | | 10-K | | 001-37449 | | 10.33 | | March 28, 2018 |
| 10.9* | | | | 10-K | | 001-37449 | | 10.35 | | March 28, 2018 |
| 10.10* | | | | 10-K | | 001-37449 | | 10.37 | | March 28, 2018 |
| 10.11* | | | | 10-Q | | 001-37449 | | 10.2 | | August 10, 2021 |
| 10.12* | | | | 10-Q | | 001-37449 | | 10.1 | | August 10, 2021 |
| 10.13*+ | | | | | | | | | | |
| 10.14* | | | | S-8 POS | | 333-218134 | | 4.1 | | September 11, 2017 |
| 10.15* | | | | S-8 POS | | 333-218134 | | 4.2 | | September 11, 2017 |
| 10.16* | | | | 8-K | | 001-37449 | | 10.1 | | June 17, 2020 |
| 10.17* | | | | 8-K | | 001-37449 | | 10.2 | | June 14, 2018 |
| 10.18* | | | | 8-K | | 001-37449 | | 10.1 | | January 27, 2020 |
| 10.19 | | | | 8-K | | 001-37449 | | 10.1 | | January 16, 2019 |
| 10.20* | | | | 8-K | | 001-37449 | | 10.1 | | April 1, 2019 |
| 10.21 | | | | 10-Q | | 001-37449 | | 10.6 | | May 9, 2019 |
| 10.22 | | | | 8-K | | 001-37449 | | 10.1 | | August 28, 2019 |
| 10.23** | | | | 10-Q | | 001-37449 | | 10.2 | | August 11, 2020 |
| 10.24 | | | | 8-K | | 001-37449 | | 10.1 | | July 24, 2020 |
| 10.25 | | | | 8-K | | 001-37449 | | 10.2 | | July 24, 2020 |
| 10.26 | | | | 8-K | | 001-37449 | | 1.1 | | July 2, 2021 |
| 10.27 | | | | 8-K | | 001-37449 | | 10.1 | | September 14, 2021 |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | Incorporated by Reference |
| Exhibit Number | | Description | | Form | | File No. | | Exhibit | | Filing Date |
| 10.11* | | | | 10-Q | | 001-37449 | | 10.1 | | November 14, 2022 |
| 10.12* | | | | 10-Q | | 001-37449 | | 10.2 | | May 11, 2023 |
| 10.13* | | | | 10-Q | | 001-37449 | | 10.1 | | November 14, 2023 |
| 10.14* | | | | 10-Q | | 001-37449 | | 10.2 | | November 14, 2023 |
| 10.15* | | | | 10-Q | | 001-37449 | | 10.3 | | November 14, 2023 |
| 10.16* | | | | S-8 POS | | 333-218134 | | 4.1 | | September 11, 2017 |
| 10.17* | | | | S-8 POS | | 333-218134 | | 4.2 | | September 11, 2017 |
| 10.18* | | | | 8-K | | 001-37449 | | 10.1 | | June 17, 2020 |
| 10.19* | | | | 8-K | | 001-37449 | | 10.2 | | June 14, 2018 |
| 10.20* | | | | 8-K | | 001-37449 | | 10.1 | | January 27, 2020 |
| 10.21 | | | | 8-K | | 001-37449 | | 10.1 | | January 16, 2019 |
| 10.22* | | | | 8-K | | 001-37449 | | 10.1 | | April 1, 2019 |
| 10.23 | | | | 10-Q | | 001-37449 | | 10.6 | | May 9, 2019 |
| 10.24** | | | | 10-Q | | 001-37449 | | 10.2 | | August 11, 2020 |
| 10.25** | | | | 10-Q | | 001-37449 | | 10.2 | | August 11, 2022 |
| 10.26** | | | | 10-K | | 001-37449 | | 10.26 | | March 20, 2024 |
| 10.27 | | | | 8-K | | 001-37449 | | 10.1 | | September 15, 2021 |
| 10.28 | | | | 8-K | | 001-37449 | | 10.2 | | September 15, 2021 |
| 10.29 | | | | 8-K | | 001-37449 | | 10.4 | | September 15, 2021 |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | Incorporated by Reference |
| Exhibit Number | | Description | | Form | | File No. | | Exhibit | | Filing Date |
| 10.28 | | | | 8-K | | 001-37449 | | 10.2 | | September 14, 2021 |
| 10.29 | | | | 8-K | | 001-37449 | | 10.4 | | September 14, 2021 |
| 10.30+** | | | | | | | | | | |
| 10.31+ | | | | | | | | | | |
| 21.1 | | | | 10-K | | 001-37449 | | 21.1 | | March 28, 2018 |
| 23.1+ | | | | | | | | | | |
| 24.1+ | | | | | | | | | | |
| 31.1+ | | | | | | | | | | |
| 31.2+ | | | | | | | | | | |
| 32.1+ | | | | | | | | | | |
| 32.2+ | | | | | | | | | | |
| 101.INS+ | | Inline XBRL Instance Document | | | | | | | | |
| 101.SCH+ | | Inline XBRL Taxonomy Extension Schema Document | | | | | | | | |
| 101.CAL+ | | Inline XBRL Taxonomy Extension Calculation Linkbase Document | | | | | | | | |
| 101.LAB+ | | Inline XBRL Taxonomy Extension Label Linkbase Document | | | | | | | | |
| 101.PRE+ | | Inline XBRL Taxonomy Extension Presentation Linkbase Document | | | | | | | | |
| 101.DEF+ | | Inline XBRL Taxonomy Extension Definition Linkbase Document | | | | | | | | |
| 104 | | Cover page formatted as Inline XBRL and contained in Exhibit 101 | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | Incorporated by Reference |
| Exhibit Number | | Description | | Form | | File No. | | Exhibit | | Filing Date |
| 10.30** | | | | 10-K | | 001-37449 | | 10.30 | | March 17, 2022 |
| 10.31 | | | | 10-K | | 001-37449 | | 10.31 | | March 17, 2022 |
| 10.32 | | | | S-3 | | 333-271517 | | 1.2 | | April 28, 2023 |
| 10.33 | | | | 8-K | | 001-37449 | | 10.1 | | April 10, 2024 |
| 10.34 | | | | 8-K | | 001-37449 | | 10.2 | | April 10, 2024 |
| 21.1 | | | | 10-K | | 001-37449 | | 21.1 | | March 23, 2023 |
| 23.1 | | | | 10-K | | 001-37449 | | 23.1 | | March 20, 2024 |
| 24.1 | | | | 10-K | | 001-37449 | | 24.1 | | March 20, 2024 |
| 24.2+ | | | | | | | | | | |
| 31.1 | | | | 10-K | | 001-37449 | | 31.1 | | March 20, 2024 |
| 31.2 | | | | 10-K | | 001-37449 | | 31.2 | | March 20, 2024 |
| 31.3+ | | | | | | | | | | |
| 31.4+ | | | | | | | | | | |
| 32.1 | | | | 10-K | | 001-37449 | | 32.1 | | March 20, 2024 |
| 32.2 | | | | 10-K | | 001-37449 | | 32.2 | | March 20, 2024 |
| 97 | | | | 10-K | | 001-37449 | | 97 | | March 20, 2024 |
| 101.INS+ | | Inline XBRL Instance Document | | | | | | | | |
| 101.SCH+ | | Inline XBRL Taxonomy Extension Schema Document | | | | | | | | |
| 101.CAL+ | | Inline XBRL Taxonomy Extension Calculation Linkbase Document | | | | | | | | |
| 101.LAB+ | | Inline XBRL Taxonomy Extension Label Linkbase Document | | | | | | | | |
| 101.PRE+ | | Inline XBRL Taxonomy Extension Presentation Linkbase Document | | | | | | | | |
| 101.DEF+ | | Inline XBRL Taxonomy Extension Definition Linkbase Document | | | | | | | | |
| 104 | | Cover page formatted as Inline XBRL and contained in Exhibit 101 | | | | | | | | |
| | | | | |
| * | Indicates a management contract or a compensatory plan, contract or arrangement. |
| | | | | |
| ** | Portions of this exhibit have been omitted in accordance with Item 601(b)(10) of Regulation S-K because they are private, confidential and not material. |
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, as amended, the Registrant has duly caused this Report to be signed on its behalf by the undersigned, thereunto duly authorized.
| | | | | | | | | | | | | | |
| | | ALPINE IMMUNE SCIENCES, INC. |
| | | | |
Date: March 17, 2022 | April 25, 2024 | | By: | /s/ Mitchell H. Gold, M.D. |
| | | Name: | Mitchell H. Gold, M.D. |
| | | TitleTitle: | Executive Chairman and Chief Executive Officer |
| | | |
| | | ALPINE IMMUNE SCIENCES, INC. |
| | | | |
Date: March 17, 2022 | April 25, 2024 | | By: | /s/ Paul Rickey |
| | | Name: | Paul Rickey |
| | | Title: | Senior Vice President and Chief Financial Officer |
| | | | |
| | | ALPINE IMMUNE SCIENCES, INC. |
| | | | |
| Date: | April 25, 2024 | | By: | /s/ U. Martin Fuhs |
| | | Name: | U. Martin Fuhs |
| | | Title: | Vice President, Finance and Chief Accounting Officer |
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Mitchell H. Gold, M.D. and Paul Rickey, and each of them, with full power of substitution and resubstitution and full power to act without the other, as his or her true and lawful attorney-in-fact and agent to act in his or her name, place and stead and to execute in the name and on behalf of each person, individually and in each capacity stated below, and to file, any and all documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing, ratifying and confirming all that said attorneys-in-fact and agents or any of them or their and his or her substitute or substitutes, may lawfully do or cause to be done by virtue thereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, as amended, this Report has been signed below by the following persons on behalf of the Registrant in the capacities and on the dates indicated.
| | | | | | | | | | | | | | |
| Name | | Title | | Date |
| | | | |
/s/ Mitchell H. Gold, M.D. | | Chief Executive Officer and Executive Chairman of the Board of Directors (Principal Executive Officer) | | March 17, 2022April 25, 2024 |
| Mitchell H. Gold, M.D. | | | | |
| | | | |
/s/ Paul Rickey | | Senior Vice President and Chief Financial Officer (Principal Accounting and Financial Officer) | | March 17, 2022April 25, 2024 |
| Paul Rickey | | | | |
| | | | |
/s/ Peter Thomson, M.D.U. Martin Fuhs | | Vice President, Finance and Chief Accounting Officer (Principal Accounting Officer) | | April 25, 2024 |
| U. Martin Fuhs | | | | |
| | | | | | | | | | | | | | |
| | | | |
/s/ * | | Director | | March 17, 2022April 25, 2024 |
| Peter Thompson, M.D. | | | | |
| | | | |
/s/ James N. Topper, M.D., Ph.D.* | | Director | | March 17, 2022April 25, 2024 |
| James N. Topper, M.D., Ph.D. | | | | |
| | | | |
/s/ Jay Venkatesan, M.D.* | | Director | | March 17, 2022 |
Jay Venkatesan, M.D. | | | | |
| | | | |
/s/ Robert Conway
| | Director | | March 17, 2022April 25, 2024 |
| Robert Conway | | | | |
| | | | |
/s/ Natasha Hernday* | | Director | | March 17, 2022April 25, 2024 |
| Natasha Hernday | | | | |
| | | | |
/s/ Christopher Peetz* | | Director | | March 17, 2022April 25, 2024 |
| Christopher Peetz | | | | |
| | | | |
/s/ Xiangmin Cui, Ph.D.* | | Director | | March 17, 2022April 25, 2024 |
| Xiangmin Cui, Ph.D. | | | | |
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
| | | | | |
| | F-2Director | | April 25, 2024 |
| Jörn Drappa, M.D., Ph.D. | |
| |
| |
| |
| |
| |
| |
| |
| |
| |
Report of Independent Registered Public Accounting Firm
To the Stockholders and the Board of Directors of Alpine Immune Sciences, Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Alpine Immune Sciences, Inc. (the Company) as of December 31, 2021 and 2020, the related consolidated statements of operations and comprehensive income (loss), stockholder’s equity and cash flows for each of the three years in the period ended December 31, 2021, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2021 and 2020, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2021, in conformity with U.S. generally accepted accounting principles.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matter
The critical audit matter communicated below is a matter arising from the current and prior period audits of the financial statements that was communicated or required to be communicated to the audit committee and that: (1) relates to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective or complex judgments. The communication of the critical audit matter does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the account or disclosure to which it relates.
| | | | | |
Description of the Matter
| | | | | | | | | | | | | *By: | | Accounting for Revenue under the AbbVie Collaboration Agreement
As discussed in Note 11 to the consolidated financial statements, the Company entered into an option and license agreement with AbbVie Ireland Unlimited Company for the development of ALPN-101 in June 2020. Collaboration revenue recognized from the AbbVie Agreement was $23.4 million and $7.0 million for the year ended December 31, 2021 and 2020, respectively. The Company determined that all contractual promises related to the upfront payment and Alpine’s Development Milestone represent a single performance obligation and used a cost-based input method to measure progress toward completion of the performance obligation and to calculate the corresponding revenue to recognize each period.
Auditing the Company’s estimated measure of progress toward completion of the performance obligation is complex. A cost-based input method of revenue recognition requires management to make estimates of total expected costs to complete the performance obligation. In making such estimates, significant judgment is required to evaluate key assumptions related to cost estimates, including internal personnel efforts and third-party contract costs.
/s/ Mitchell H. Gold | | Description of the MatterAs discussed in Note 11 to the consolidated financial statements, the Company entered into an option and license agreement with AbbVie Ireland Unlimited Company for the development of ALPN-101 in June 2020. Collaboration revenue recognized from the AbbVie Agreement was $23.4 million and $7.0 million for the year ended December 31, 2021 and 2020, respectively. The Company determined that all contractual promises related to the upfront payment and Alpine’s Development Milestone represent a single performance obligation and used a cost-based input method to measure progress toward completion of the performance obligation and to calculate the corresponding revenue to recognize each period.Mitchell H. Gold, M.D.
Auditing the Company’s estimated measure of progress toward completion of the performance obligation is complex. A cost-based input method of revenue recognition requires management to make estimates of total expected costs to complete the performance obligation. In making such estimates, significant judgment is required to evaluate key assumptions related to cost estimates, including internal personnel efforts and third-party contract costs.Attorney-in-fact
| How We Addressed the Matter in Our Audit
| To test revenue recognized, we performed audit procedures that included, among others, gaining an understanding and testing the Company’s estimates of total expected costs including testing the completeness and accuracy of the underlying data, inspecting evidence of actual costs incurred and comparing with previous estimates. We evaluated any changes in the total expected costs, inspected communications between the Company and AbbVie regarding updates to estimated budgeted costs as well as any significant changes in its development plan, performed sensitivity analyses of key assumptions and compared the estimates to actual incurred costs for the same or similar activities. We discussed the basis for key assumptions with the Company's research and development personnel to assess management's key assumptions used in the Company's estimates of total expected costs. |
/s/ Ernst & Young LLP
We have served as the Company’s auditor since 2015.
Seattle, WA
March 17, 2022
ALPINE IMMUNE SCIENCES, INC.
CONSOLIDATED BALANCE SHEETS
(in thousands, except share and per share amounts)
| | | | | | | | | | | | | | | | | | December 31, | | | | 2021 | | 2020 | | Assets | | | | | | Current assets: | | | | | | Cash and cash equivalents | | $ | 67,907 | | | $ | 34,959 | | | Short-term investments | | 94,396 | | | 70,622 | | | Accounts receivable | | 25,000 | | | — | | | | | | | | Prepaid expenses and other current assets | | 4,710 | | | 1,520 | | | Total current assets | | 192,013 | | | 107,101 | | | Restricted cash, noncurrent | | 254 | | | 254 | | | Property and equipment, net | | 1,716 | | | 1,785 | | | Operating lease, right-of-use asset | | 8,837 | | | 9,401 | | | Long-term investments | | 52,866 | | | 25,549 | | | Deferred tax asset | | 214 | | | — | | | Total assets | | $ | 255,900 | | | $ | 144,090 | | | Liabilities and stockholders’ equity | | | | | | Current liabilities: | | | | | | Accounts payable | | $ | 3,349 | | | $ | 582 | | | Accrued liabilities | | 9,417 | | | 5,777 | | | Deferred revenue, current | | 51,773 | | | 31,627 | | | Operating lease liability, current | | 617 | | | 655 | | | Current portion of long-term debt | | 4,622 | | | 2,526 | | | Total current liabilities | | 69,778 | | | 41,167 | | | Deferred revenue, noncurrent | | 50,830 | | | 21,348 | | | Operating lease liability, noncurrent | | 11,009 | | | 11,815 | | | Long-term debt | | 3,380 | | | 7,602 | | | Total liabilities | | 134,997 | | | 81,932 | | | Commitments and contingencies | | 0 | | 0 | | Preferred stock, $0.001 par value per share; 10,000,000 shares authorized at December 31, 2021 and 2020; zero shares issued and outstanding at December 31, 2021 and 2020 | | — | | | — | | | Stockholders’ equity: | | | | | | Common stock, $0.001 par value per share; 200,000,000 shares authorized at December 31, 2021 and 2020; 31,444,746 shares issued and 30,194,279 shares outstanding at December 31, 2021; 23,853,650 shares issued and 23,803,183 shares outstanding at December 31, 2020 | | 30 | | | 24 | | | Treasury stock, at cost; 1,250,467 shares and 50,467 shares at December 31, 2021 and 2020, respectively | | — | | | — | | | Additional paid-in capital | | 287,345 | | | 177,947 | | | Accumulated other comprehensive (loss) gain | | (273) | | | 53 | | | Accumulated deficit | | (166,199) | | | (115,866) | | | Total stockholders’ equity | | 120,903 | | | 62,158 | | | Total liabilities and stockholders’ equity | | $ | 255,900 | | | $ | 144,090 | |
The accompanying notes are an integral part of these consolidated financial statements.
ALPINE IMMUNE SCIENCES, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE INCOME (LOSS)
(in thousands, except share and per share amounts)
| | | | | | | | | | | | | | | | | | | | Years Ended December 31, | | | 2021 | | 2020 | | 2019 | | Collaboration revenue | $ | 23,443 | | | $ | 9,335 | | | $ | 1,740 | | | Operating expenses: | | | | | | | Research and development | 58,742 | | | 27,185 | | | 35,847 | | | General and administrative | 14,560 | | | 10,899 | | | 9,467 | | | | | | | | | Total operating expenses | 73,302 | | | 38,084 | | | 45,314 | | | Loss from operations | (49,859) | | | (28,749) | | | (43,574) | | | Other income (expense): | | | | | | | Interest expense | (816) | | | (775) | | | (338) | | | Interest income | 259 | | | 245 | | | 1,248 | | | Other (expense) income | (4) | | | 1,333 | | | 812 | | | Loss before taxes | (50,420) | | | (27,946) | | | (41,852) | | | Income tax (expense) benefit | 87 | | | 6 | | | — | | | Net loss | $ | (50,333) | | | $ | (27,940) | | | $ | (41,852) | | | Comprehensive income (loss): | | | | | | | Unrealized loss on investments | (238) | | | (15) | | | 29 | | | Unrealized (loss) gain on foreign currency translation | (88) | | | 58 | | | (6) | | | Comprehensive loss | $ | (50,659) | | | $ | (27,897) | | | $ | (41,829) | | | Weighted-average shares used to compute basic and diluted net loss per share | 25,476,889 | | 20,826,466 | | 18,358,864 | | Basic and diluted net loss per share | $ | (1.98) | | | $ | (1.34) | | | $ | (2.28) | |
The accompanying notes are an integral part of these consolidated financial statements.
ALPINE IMMUNE SCIENCES, INC.
CONSOLIDATED STATEMENT OF STOCKHOLDERS’ EQUITY
(in thousands, except share and per share amounts)
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | Common Stock | | Treasury | | Additional
Paid-in Capital | | Accumulated
Other Comprehensive (Loss) Gain | | Accumulated Deficit | | Total
Stockholders’ Equity | | | | | | | | Shares | | Amount | | Shares | | Amount | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | Balance, December 31, 2018 | | | | | | 13,854,205 | | | $ | 14 | | | 50,467 | | | $ | — | | | $ | 90,664 | | | $ | (13) | | | $ | (46,074) | | | $ | 44,591 | | | Issuance of Units in Private Placement, net of offering costs | | | | | | 4,706,700 | | | 5 | | | — | | | — | | | 23,593 | | | — | | | — | | | 23,598 | | | Exercise of stock options | | | | | | 26,987 | | | — | | | — | | | — | | | 13 | | | — | | | — | | | 13 | | | Stock-based compensation | | | | | | — | | | — | | | — | | | — | | | 3,041 | | | — | | | — | | | 3,041 | | | Issuance of warrants | | | | | | | | | | | | | | 60 | | | | | | | 60 | | | Unrealized gain on investments | | | | | | — | | | — | | | — | | | — | | | — | | | 29 | | | — | | | 29 | | | Unrealized loss on foreign currency translation | | | | | | — | | | — | | | — | | | — | | | — | | | (6) | | | — | | | (6) | | | Net loss | | | | | | — | | | — | | | — | | | — | | | — | | | — | | | (41,852) | | | (41,852) | | | Balance, December 31, 2019 | | | | | | 18,587,892 | | | 19 | | | 50,467 | | | — | | | 117,371 | | | 10 | | | (87,926) | | | 29,474 | | | Issuance of Units in private offering, net of offering costs | | | | | | 5,139,610 | | | 5 | | | — | | | — | | | 56,253 | | | — | | | — | | | 56,258 | | | Issuance of warrants | | | | | | — | | | — | | | — | | | — | | | 60 | | | — | | | — | | | 60 | | | Issuance of common stock under equity incentive plans | | | | | | 75,681 | | | — | | | — | | | — | | | 123 | | | — | | | — | | | 123 | | | Stock-based compensation | | | | | | | | | | | | | | 4,140 | | | | | | | 4,140 | | | Unrealized loss on investments | | | | | | — | | | — | | | — | | | — | | | — | | | (15) | | | — | | | (15) | | | Unrealized gain on foreign currency translation | | | | | | — | | | — | | | — | | | — | | | — | | | 58 | | | — | | | 58 | | | Net loss | | | | | | — | | | — | | | — | | | — | | | — | | | — | | | (27,940) | | | (27,940) | | | Balance, December 31, 2020 | | | | | | 23,803,183 | | | 24 | | | 50,467 | | | — | | | 177,947 | | | 53 | | | (115,866) | | | 62,158 | | | Issuance of common stock in Private Placement, net of offering costs | | | | | | 6,489,357 | | | 6 | | | — | | | — | | | 90,727 | | — | | | — | | | 90,733 | | | Issuance of common stock to Horizon | | | | | | 951,980 | | | 1 | | | — | | | — | | | 11,928 | | | — | | | — | | | 11,929 | | | Exchange of common stock for prefunded warrants | | | | | | (1,200,000) | | | (1) | | | 1,200,000 | | | — | | | 1 | | | — | | | — | | | — | | | Stock-based compensation | | | | | | — | | | — | | | — | | | — | | | 6,240 | | | — | | | — | | | 6,240 | | | Issuance of common stock under equity incentive plans | | | | | | 149,759 | | | — | | | — | | | — | | | 502 | | | — | | | — | | | 502 | | | Unrealized loss on investments | | | | | | — | | | — | | | — | | | — | | | — | | | (238) | | | — | | | (238) | | | Unrealized loss on foreign currency translation | | | | | | — | | | — | | | — | | | — | | | — | | | (88) | | | — | | | (88) | | | Net loss | | | | | | — | | | — | | | — | | | — | | | — | | | — | | | (50,333) | | | (50,333) | | | Balance, December 31, 2021 | | | | | | 30,194,279 | | | $ | 30 | | | 1,250,467 | | | $ | — | | | $ | 287,345 | | | $ | (273) | | | $ | (166,199) | | | $ | 120,903 | |
The accompanying notes are an integral part of these consolidated financial statements.
ALPINE IMMUNE SCIENCES, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(in thousands)
| | | | | | | | | | | | | | | | | | | | Years Ended December 31, | | | 2021 | | 2020 | | 2019 | | Operating activities | | | | | | | Net loss | $ | (50,333) | | | $ | (27,940) | | | $ | (41,852) | | | Adjustments to reconcile net loss to net cash used in operating activities: | | | | | | | Loss on sale of property and equipment | 4 | | | 5 | | | 16 | | | Depreciation expense | 620 | | | 578 | | | 468 | | | Amortization of premium/discount on investments | 906 | | | 103 | | | (360) | | | Non-cash interest expense | 274 | | | 261 | | | 140 | | | Deferred income tax | (214) | | | — | | | — | | | Stock-based compensation expense | 6,240 | | | 4,140 | | | 3,041 | | | Changes in operating assets and liabilities: | | | | | | | Prepaid expenses and other current assets | (3,063) | | | 255 | | | (408) | | | Right-of-use asset | 564 | | | 592 | | | 1,553 | | | Accounts payable and accrued liabilities | 5,970 | | | (491) | | | 816 | | | Deferred revenue | 24,628 | | | 51,540 | | | 1,435 | | | Lease liabilities | (844) | | | 1,041 | | | (195) | | | Net cash (used in) provided by operating activities | (15,248) | | | 30,084 | | | (35,346) | | | Investing activities | | | | | | | Purchases of property and equipment | (118) | | | (802) | | | (821) | | | | | | | | | Purchase of investments | (133,518) | | | (101,328) | | | (59,382) | | | Maturities of investments | 81,156 | | | 29,311 | | | 75,575 | | | Proceeds from the sale of investments | — | | | — | | | 1,391 | | | Net cash (used in) provided by investing activities | (52,480) | | | (72,819) | | | 16,763 | | | Financing activities | | | | | | | Proceeds from sale of common stock and warrants, net of offering costs | 90,733 | | | 56,258 | | | 23,598 | | | Proceeds from issuance of common stock to Horizon | 11,929 | | | — | | | — | | | Proceeds from borrowings, net of issuance costs | — | | | 5,000 | | | 1,977 | | | Repayment of debt | (2,400) | | | — | | | (1,333) | | | Proceeds from exercise of stock options | 502 | | | 123 | | | 13 | | | Net cash provided by financing activities | 100,764 | | | 61,381 | | | 24,255 | | | Effect of exchange rate on cash, cash equivalents and restricted cash | (88) | | | 58 | | | (6) | | | Net increase in cash and cash equivalents and restricted cash | 32,948 | | | 18,704 | | | 5,666 | | | Cash and cash equivalents and restricted cash, beginning of period | 35,213 | | | 16,509 | | | 10,843 | | | Cash and cash equivalents and restricted cash, end of period | $ | 68,161 | | | $ | 35,213 | | | $ | 16,509 | | | Supplemental Information | | | | | | | Recognition of right-of-use asset | $ | — | | | $ | — | | | $ | 11,173 | | | Cash paid for interest | $ | 554 | | | $ | 490 | | | $ | 170 | | | Discount in connection with issuance of debt | $ | — | | | $ | 334 | | | $ | — | | | | | | | |
The accompanying notes are an integral part of these consolidated financial statements.
ALPINE IMMUNE SCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
1. Description of Business
Alpine Immune Sciences, Inc. (the “Company”, “Alpine”, “we”, “us”, or “our”), together with its consolidated subsidiaries, is a clinical-stage biopharmaceutical company dedicated to discovering and developing innovative, protein-based immunotherapies to treat cancer and autoimmune and inflammatory diseases. Our approach includes a proprietary scientific platform that converts native immune system proteins into differentiated, multi-targeted therapeutics. We believe our strategies are capable of meaningfully modulating the human immune system and significantly improving outcomes in patients with serious diseases. We were incorporated under the laws of the State of Delaware and are headquartered in Seattle, Washington.
A novel strain of coronavirus, SARS-CoV-2 (“COVID-19”), was first reported in December 2019, and subsequently declared a global pandemic by the World Health Organization in March 2020. As a result of the COVID-19 outbreak, many companies have experienced disruptions in their operations and in markets served. We have implemented some and may take additional temporary precautionary measures intended to help ensure the well-being of our employees and minimize business disruption. We considered the impact of COVID-19 on the assumptions and estimates used and determined that there were no material adverse impacts to our results of operations and financial position at December 31, 2021. The full extent of the future impacts of the continuing COVID-19 outbreak on our operations is uncertain and may adversely impact our business, including our clinical trials.
2. Summary of Significant Accounting Policies
Basis of Presentation and Use of Estimates
The accompanying consolidated financial statements have been prepared in accordance with the rules and regulations of the Securities and Exchange Commission (“SEC”) and generally accepted accounting principles in the United States of America (“GAAP”). The preparation of financial statements in conformity with GAAP requires management to make judgments, estimates and assumptions that affect the amounts reported in the consolidated financial statements and accompanying notes. Significant estimates inherent in the preparation of the accompanying consolidated financial statements include those used for revenue recognition, accruals for clinical trial activities and other accruals, and the estimated fair value of equity-based awards. We base our estimates and assumptions on historical experience when available and on various factors we believe to be reasonable under the circumstances. Actual results could differ materially from those estimates.
Principles of Consolidation
Our consolidated financial statements include the financial position and results of operations of Alpine Immune Sciences, Inc. and our wholly owned operating company and subsidiary, AIS Operating Co., Inc., and our wholly-owned subsidiary, Alpine Immune Sciences Australia PTY LTD. All inter-company balances and transactions have been eliminated in consolidation.
Segments
We operate as 1 operating segment and use cash flow as the primary financial measure to manage our business.
Cash and Cash Equivalents and Restricted Cash
We consider all highly liquid investments with an original maturity of 90 days or less at the time of purchase to be cash equivalents. Cash and cash equivalents consist of deposits with commercial banks in checking and interest-bearing accounts, and highly liquid money market funds.
Restricted cash represents cash drawn on our line of credit used to establish collateral to support the security deposit on our operating lease to rent office and laboratory space in Seattle, Washington.
Periodically, we maintain deposits in financial institutions in excess of government insured limits. We believe we are not exposed to significant credit risk as our deposits, which are held at financial institutions, are high credit quality securities such as money market funds, U.S. Treasury securities, and commercial paper. To date, we have not realized any losses on these deposits.
ALPINE IMMUNE SCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
Investments
Our investments include funds invested in highly liquid money market funds, U.S. Treasury securities, commercial paper, and corporate debt securities with a final maturity of each security of less than two years. These investments are classified as available-for-sale debt securities, which are recorded at fair value based on quoted prices in active markets. We classify our investments maturing within one year of the reporting date as short-term investments.
If the estimated fair value of a debt security is below its amortized cost basis, we evaluate whether it is more likely than not that we will sell the security before its anticipated recovery in market value and whether credit losses exist for the related securities. A credit loss exists if the present value of expected cash flows is less than the amortized cost basis of the security. Credit-related losses are recognized as an allowance for credit losses on the balance sheet with a corresponding adjustment to earnings. Unrealized gains and losses that are unrelated to credit deterioration are reported in other comprehensive income (loss). Purchase premiums and discounts are recognized as interest income using the interest method over the terms of the securities. Realized gains and losses and declines in fair value deemed to be other than temporary are reflected in the Consolidated Statements of Operations and Comprehensive Income (Loss) using the specific-identification method.Property and Equipment
Property and equipment are stated at cost, net of accumulated depreciation. Depreciation is recorded using the straight-line method over the estimated useful lives of the assets, generally three to five years, while leasehold improvements are amortized over the shorter of their estimated useful lives or the related lease term. Upon retirement or sale, the cost of assets disposed of and the related accumulated depreciation are removed from the accounts and any resulting gain or loss is credited or charged to operations. Maintenance and repairs are expensed as incurred. Major improvements are capitalized as additions to property and equipment.
Impairment of Long-lived Assets
We evaluate our long-lived tangible assets for impairment whenever events or changes in circumstances indicate that the carrying amount of such assets may not be recoverable. If the carrying value exceeds the undiscounted future cash flows estimated to result from the use and eventual disposition of the asset, we write down the asset to its estimated fair value. Impairment is assessed by comparing the undiscounted cash flows expected to be generated by the asset to its carrying value. We did not record any impairments in the years ended December 31, 2021, 2020 or 2019.
Accrued Liabilities
As part of the process of preparing our consolidated financial statements, we are required to estimate accruals for professional services and research and development expenses. This process involves reviewing contracts and vendor agreements and communicating with applicable personnel to identify services that have been performed on our behalf. We estimate the level of service performed and the associated cost incurred for the service when we have not yet been invoiced or otherwise notified of the actual cost. We estimate accrued liabilities as of each balance sheet date based on known facts and circumstances.
Although we do not expect our estimates to be materially different from amounts actually incurred, if our estimates of the status and timing of services performed differ from the actual status and timing of services performed, we may report amounts that are too high or too low in any particular period. To date, we have not experienced any significant adjustments to our estimates.
Leases
We account for our leases under Accounting Standards Codification (“ASC”) 842, Leases. Under this guidance, we applied the practical expedients regarding the identification of leases, lease classification, indirect costs, and the combination of lease and non-lease components. Arrangements meeting the definition of a lease are classified as operating or financing leases, and are recorded on the consolidated balance sheet as both a right-of-use asset and lease liability, calculated by discounting fixed lease payments over the lease term at the rate implicit in the lease or our incremental borrowing rate. As we do not know the lessor’s implicit rate, we use our incremental borrowing rate at the commencement date of the lease in determining the present value of lease payments. Lease liabilities are increased by interest and reduced by payments each period, and the right-of-use asset is amortized over the lease term. For operating leases, interest on the lease liability and the amortization of the right-of-use asset result in straight-line rent expense over the lease term. For finance leases, interest on the lease liability and the amortization of the right-of-use asset results is front-loaded expense over the lease term. Variable lease expenses are recorded when incurred.
ALPINE IMMUNE SCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
In calculating the right-of-use asset and lease liability, we elected to combine lease and non-lease components. We exclude short-term leases having initial terms of 12 months or less from the new guidance as an accounting policy election, and recognize rent expense on a straight-line basis over the lease term.
Derivative Financial Instruments
We evaluate all of our financial instruments, including prefunded warrants and warrants to purchase common stock, to determine if such instruments are derivatives or contain features qualifying as embedded derivatives. For derivative financial instruments accounted for as liabilities, the derivative instrument is initially recorded at its fair value and is then re-valued at each reporting date, with changes in the fair value reported in the Consolidated Statements of Operations and Comprehensive Income (Loss). We use the Black-Scholes option-pricing model to value the derivative instruments at inception and subsequent valuation dates. The classification of derivative instruments, including whether such instruments should be recorded as liabilities or as equity, is re-assessed at the end of each reporting period. We do not use derivative instruments to hedge exposures to cash flow, market, or foreign currency risks.Revenue Recognition
Revenue is recognized when control of the promised goods or services is transferred to our customers, in an amount that reflects the consideration we expect to be entitled to in exchange for those goods or services. Our steps for recognizing revenue consist of; (1) identifying the contract, (2) identifying the performance obligations as either distinct or bundled goods and services, (3) determining the transaction price associated with each performance obligation for which we expect to be entitled in exchange for transferring such goods and services, (4) allocating the transaction price to the performance obligations in the contract and (5) recognizing revenue upon satisfaction of performance obligations.
Our collaboration agreements principally contain multiple performance obligations, which may include (1) grants of, or options to obtain, intellectual property licenses; (2) research and development services; and/or (3) manufacturing or supply services. Payments typically received under these arrangements include one or more of the following: non-refundable upfront license fees, option exercise fees, payment for research and/or development efforts, amounts due upon the achievement of specified objectives, and/or royalties on future product sales. Our revenue is primarily derived from our collaboration agreements with Adaptimmune Therapeutics plc (“Adaptimmune”), AbbVie Ireland Unlimited Company (“AbbVie”), and Horizon Therapeutics Ireland DAC (“Horizon”). See further discussion of our collaboration agreements in Note 11.We allocate revenue to each performance obligation based on its relative stand-alone selling price. We generally determine stand-alone selling prices at the inception of the contract based on our best estimate of what the selling price would be if the deliverable was regularly sold by us on a stand-alone basis. Payments received prior to satisfying the relevant revenue recognition criteria are recorded as deferred revenue in the accompanying Consolidated Balance Sheets and recognized as revenue when the related revenue recognition criteria are met. We recognize revenue under our collaboration agreements by using a cost-based input method to measure progress toward completion of the performance obligation, including employee hours contributed to each performance obligation, and to calculate the corresponding revenue to recognize each period.Our collaboration agreements provide for non-refundable milestone payments. We recognize revenue that is contingent upon the achievement of a substantive milestone in its entirety in the period in which the milestone is achieved. A milestone is considered substantive when the consideration payable to us for such milestone (1) is consistent with our performance necessary to achieve the milestone or the increase in value to the collaboration resulting from our performance; (2) relates solely to our past performance; and (3) is reasonable relative to all of the other deliverables and payments within the arrangement. In making this assessment, we consider all facts and circumstances relevant to the arrangement, including factors such as the scientific, regulatory, commercial, and other risks that must be overcome to achieve the milestone, the level of effort and investment required to achieve the milestone and whether any portion of the milestone consideration is related to future performance or deliverables.
We review the contributed employee hours and progress towards completion for each performance obligation under our collaboration agreements, and adjust the revenue recognized to reflect changes in assumptions relating to the estimated satisfaction of the performance obligation. Revenue recognition may be accelerated in the event of early termination of programs or if our expectations change. Alternatively, revenue recognition may be decelerated if programs are extended or delayed. While such changes to our estimates have no impact on our reported cash flows, the timing of revenue recorded in future periods could be materially impacted.
Research and Development
Research and development costs are expensed as incurred. Research and development costs include personnel costs, clinical trials, external contract research and development expenses, raw materials, drug product manufacturing costs and
ALPINE IMMUNE SCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
allocated overhead – including depreciation, rent and utilities. Research and development costs that are paid in advance of performance are capitalized as a prepaid expense and amortized over the service period as the services are provided.
Stock-based Compensation
Stock-based compensation is recognized for all share-based payments based on the estimated fair value as of the date of grant. The fair value of our stock options is calculated using the Black-Scholes option pricing model, which requires judgmental assumptions including volatility, risk-free interest rate, expected term and dividend yield. The fair value of restricted stock units (“RSUs”) is based on the closing price of our common stock on the award date. Stock-based compensation is recognized over the requisite service period of the awards, usually the vesting period, on a straight-line basis. For performance-based awards where the vesting of the options may be accelerated upon the achievement of certain milestones, the related stock-based compensation is recognized as expense when it is probable the milestone will be met. We recognize forfeiture of awards as they occur rather than estimating the expected forfeiture rate.
Income Taxes
Income taxes are accounted for using an asset and liability approach that requires the recognition of deferred tax assets and liabilities for the expected future tax consequences of temporary differences between the consolidated financial statement and tax bases of assets and liabilities at the applicable enacted tax rates. We will establish a valuation allowance for deferred tax assets if it is more likely than not that these items will expire before we are able to realize their benefits or that future deductibility is uncertain.
We recognize the tax benefit from uncertain tax positions only if it is more likely than not that the tax position will be sustained on examination by the tax authorities, based on the technical merits of the position. The tax position is measured based on the largest benefit that has a greater than 50% likelihood of being realized upon ultimate settlement. We recognize interest and penalties related to income tax matters in income tax expense if incurred.
Comprehensive Income (Loss)
Comprehensive income (loss) is comprised of net loss and certain changes in equity excluded from net loss. For the years ended December 31, 2021, 2020, and 2019, other comprehensive loss consisted of unrealized gains and losses on our investments and unrealized gains and losses on foreign currency translation.
Foreign Currency Translation
Our wholly-owned Australian subsidiary uses the Australian dollar as its functional currency. All assets and liabilities related to this subsidiary are translated using period-end exchange rates and revenues and expenses are translated at average exchange rates for the year. Translation adjustments are included as components of comprehensive income (loss) in the Consolidated Statements of Operations and Comprehensive Income (Loss).Recently Adopted Accounting Pronouncements
In December 2019, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) No. 2019-12, Income Taxes (“Topic 740”): Simplifying the Accounting for Income Taxes. This ASU simplifies the accounting for income taxes by removing certain exceptions to the general principles in Topic 740 and also improves consistent application by clarifying and amending existing guidance. The standard is effective for fiscal years and interim periods within those fiscal years, beginning after December 15, 2020. We adopted this standard on January 1, 2021, and concluded that the adoption of the standard did not have a material impact on our consolidated financial statements and related disclosures.
3. Net Loss Per Share
Basic net loss per share is computed by dividing net loss by the weighted-average number of common shares outstanding during the period.
The net loss per share for the year ended December 31, 2021 reflects 6,489,357 shares of our common stock issued pursuant to a private placement financing completed in September 2021 and 951,980 shares of our common stock issued in December 2021 pursuant to a private placement financing with Horizon. The net loss per share for the year ended December 31, 2020 reflects 5,139,610 shares of our common stock issued pursuant to the securities offering completed in July 2020. The net loss per share for the year ended December 31, 2019 reflects 4,706,700 shares of our common stock issued pursuant to a private placement financing completed in January 2019. The increased number of shares issued in these periods has affected the year-over-year comparability of our net loss per share calculations.
ALPINE IMMUNE SCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
The common stock issuable upon the conversion or exercise of the following dilutive securities has been excluded from the diluted net loss per share calculation because their effect would have been anti-dilutive. Diluted net loss per share, therefore, does not differ from basic net loss per share for the periods presented.
| | | | | | | | | | | | | | | | | | | | December 31, | | | 2021 | | 2020 | | 2019 | | | | | | | | Common stock warrants | 3,666,435 | | | 3,673,551 | | | 1,877,094 | | | Prefunded warrants to purchase common stock | 5,182,197 | | | 790,710 | | | — | | | Stock options and RSUs outstanding | 5,877,309 | | | 4,175,345 | | | 3,252,144 | | | Total | 14,725,941 | | | 8,639,606 | | | 5,129,238 | |
4. Cash Equivalents and Investments
The amortized cost and fair value of our cash equivalents and investments are as follows (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | December 31, 2021 | | Amortized
Cost | | Gross
unrealized
gains | | Gross
unrealized
losses | | Fair market
value | | Money market funds | $ | 50,277 | | | $ | — | | | $ | — | | | $ | 50,277 | | | U.S. treasury bills | 30,006 | | | 1 | | | (100) | | | 29,907 | | | Corporate debt securities and commercial paper | 117,492 | | | 2 | | | (139) | | | 117,355 | | | Total | $ | 197,775 | | | $ | 3 | | | $ | (239) | | | $ | 197,539 | | | | | | | | | | | Classified as: | | | | | | | | | Cash equivalents | | | | | | | $ | 50,277 | | | Short-term investments | | | | | | | 94,396 | | | Long-term investments | | | | | | | 52,866 | | | Total | | | | | | | $ | 197,539 | |
| | | | | | | | | | | | | | | | | | | | | | | | | December 31, 2020 | | Amortized
Cost | | Gross
unrealized
gains | | Gross
unrealized
losses | | Fair market
value | | Money market funds | $ | 28,424 | | | $ | — | | | $ | — | | | $ | 28,424 | | | U.S. treasury bills | 18,122 | | | 8 | | | — | | | 18,130 | | | Corporate debt securities and commercial paper | 82,047 | | | 2 | | | (9) | | | 82,040 | | | Total | $ | 128,593 | | | $ | 10 | | | $ | (9) | | | $ | 128,594 | | | | | | | | | | | Classified as: | | | | | | | | | Cash equivalents | | | | | | | $ | 32,423 | | | Short-term investments | | | | | | | 70,622 | | | Long-term investments | | | | | | | 25,549 | | | Total | | | | | | | $ | 128,594 | |
All investments held as of December 31, 2021 and 2020 were classified as available-for-sale debt securities and had contractual maturities of less than two years. There were no realized gains and losses on these securities for the periods presented. The aggregate fair value of available-for-sale debt securities in an unrealized loss position as of December 31, 2021 was $83.0 million. We did not have any investments in a continuous unrealized loss position for more than twelve months as of December 31, 2021. Our available-for-sale debt securities are invested in highly liquid funds with high credit ratings that have a final maturity of two years or less on the date of purchase. Unrealized losses on these investments were primarily due to changes in interest rates. We evaluated our investments that are in an unrealized loss position and believe it is more likely than not that we will hold these investments until maturity and will recover the amortized cost basis of these investments.
ALPINE IMMUNE SCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
5. Fair Value Measurements
Cash and cash equivalents, restricted cash, receivables, accounts payable and accrued liabilities, which are recorded at invoiced amount or cost, approximate fair value based on the short-term nature of these financial instruments. Fair value is defined as the exchange price received for an asset or paid to transfer a liability, or an exit price, in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. Valuation techniques used to measure fair value must maximize the use of observable inputs and minimize the use of unobservable inputs. The three-tier fair value hierarchy, which prioritizes the inputs used in measuring fair value, is as follows:
Level 1: Quoted prices in active markets for identical assets or liabilities.
Level 2: Observable inputs other than Level 1 prices, such as quoted prices for similar assets or liabilities, quoted prices in inactive markets, or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities.
Level 3: Unobservable inputs supported by little or no market activity and significant to the fair value of the assets or liabilities.
As of December 31, 2021 and 2020, cash of $17.6 million and $2.5 million, respectively, is excluded from the fair value table below. The following tables summarize our financial assets and liabilities measured at fair value on a recurring basis (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | Assets: | December 31, 2021 | | | Level 1 | | Level 2 | | Level 3 | | Total | | Money market funds | $ | 50,277 | | | $ | — | | | $ | — | | | $ | 50,277 | | | U.S. treasury bills | 29,907 | | | — | | | — | | | 29,907 | | | Corporate debt securities and commercial paper | — | | | 117,355 | | | — | | | 117,355 | | | Total | $ | 80,184 | | | $ | 117,355 | | | $ | — | | | $ | 197,539 | |
| | | | | | | | | | | | | | | | | | | | | | | | | Assets: | December 31, 2020 | | | Level 1 | | Level 2 | | Level 3 | | Total | | Money market funds | $ | 28,424 | | | $ | — | | | $ | — | | | $ | 28,424 | | | U.S. treasury bills | 18,130 | | | — | | | — | | | 18,130 | | | Corporate debt securities and commercial paper | — | | | 82,040 | | | — | | | 82,040 | | | Total | $ | 46,554 | | | $ | 82,040 | | | $ | — | | | $ | 128,594 | |
Our Level 2 assets consist of commercial paper and corporate debt securities. We review trading activity and pricing for our available-for-sale securities as of the measurement date. When sufficient quoted pricing for identical securities is not available, we use market pricing and other observable market inputs for similar securities obtained from various third-party data providers. These inputs either represent quoted prices for similar assets in active markets or have been derived from observable market data.
6. Property and Equipment
Property and equipment, net, consists of the following (in thousands):
| | | | | | | | | | | | | | December 31, | | | 2021 | | 2020 | | Laboratory equipment | $ | 3,132 | | | $ | 2,604 | | | General equipment and furniture | 479 | | | 479 | | | Computer equipment and software | 211 | | | 211 | | | Leasehold improvements | 85 | | | 85 | | | Property and equipment, at cost | 3,907 | | | 3,379 | | | Less accumulated depreciation and amortization | (2,191) | | | (1,594) | | | Property and equipment, net | $ | 1,716 | | | $ | 1,785 | |
ALPINE IMMUNE SCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
Depreciation expense was $620,000, $578,000 and $468,000 for the years ended December 31, 2021, 2020 and 2019, respectively.
7. Sale of Intangible Asset
In February 2018, we entered into an Option License Agreement (“Option Agreement”) with Laurel Venture Capital Ltd. (“Laurel”), which granted Laurel a limited license to evaluate the indefinite-life GSNOR inhibitor IPR&D asset acquired as part of the merger with Nivalis in 2017. The IPR&D represents the processes, expertise, and technology employed in the development of GSNOR inhibitors and Nivalis’ lead product candidate, cavosonstat. Under the Option Agreement, we received an upfront non-refundable payment of $75,000, which was recognized as revenue in our accompanying Consolidated Statements of Operations and Comprehensive Income (Loss).In June 2018, we entered into an Asset Purchase Agreement (“Purchase Agreement”) with Laurel and completed the sale of global rights to the GSNOR asset. Upon the sale of the GSNOR assets, we derecognized the full carrying value of the intangible asset. As consideration under the Purchase Agreement, we received a non-refundable closing payment of $250,000, which was accounted for as a purchase of our intangible asset. In June 2019, we recognized as revenue an additional payment of $425,000, related to the asset purchase. In addition, we are eligible to receive milestone payments of up to $20.0 million, in the aggregate upon satisfaction by Laurel of certain regulatory approval milestones. We will also be eligible to receive royalty payments equal to a low single-digit percentage rate of worldwide net sales of any approved products.
8. Additional Balance Sheet Information
Prepaid expenses and other current assets consist of the following (in thousands): | | | | | | | | | | | | | | | | December 31, | | | | | 2021 | | 2020 | | | | Prepaid research and development | $ | 3,315 | | | $ | 517 | | | | | Prepaid insurance | 454 | | | 447 | | | | | Deferred financing | 352 | | | — | | | | | Tenant improvement allowance receivable | — | | | 84 | | | | | Prepaid other | 266 | | | 168 | | | | | Other receivables | 323 | | | 304 | | | | | Prepaid expenses and other current assets | $ | 4,710 | | | $ | 1,520 | | | |
Accrued liabilities consist of the following (in thousands): | | | | | | | | | | | | | | December 31, | | | 2021 | | 2020 | | Research and development services | $ | 5,536 | | | $ | 2,571 | | | Employee compensation | 3,084 | | | 2,619 | | | Legal and professional fees | 394 | | | 482 | | | | | | | | | | | | | | | Accrued taxes | 127 | | | — | | | Accrued other | 276 | | | 105 | | | Accrued Liabilities | $ | 9,417 | | | $ | 5,777 | |
9. Long-term Debt
In December 2016, we entered into a Loan and Security Agreement (the “Original Agreement”), with Silicon Valley Bank (“SVB”), under which we borrowed $5.0 million. The Original Agreement accrued interest at a floating per annum rate equal to the lender’s prime rate minus 1.75%. The Original Agreement had an interest-only period through July 2018.
In August 2019 (the “Effective Date”), we entered into an Amended and Restated Loan and Security Agreement (the “Loan Agreement”) with SVB, pursuant to which SVB agreed to extend term loans to us with an aggregate principal amount of up to $15.0 million (the “Term Loans”). Borrowings under the Loan Agreement consisted of up to 3 separate tranches. The initial tranche of $5.0 million was funded in August 2019, $3.0 million of which was used to repay amounts owing under our Original Agreement. In March 2020, the second tranche of $5.0 million was funded to us. We did not draw down the final tranche of $5.0 million, which expired on July 31, 2020. We intend to use the debt proceeds for working capital and other general corporate purposes, including the advancement of our development programs.
ALPINE IMMUNE SCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
The Term Loans accrue interest at a floating per annum rate of 0.25% above the prime rate, subject to a floor of 5.75%, which interest is payable monthly commencing in September 2019. Upon the occurrence and during the continuance of an event of default, a default interest rate will apply that is 4.0% above the otherwise applicable interest rate. The Term Loans were interest only until September 30, 2020, however, under the Loan Agreement our interest only period automatically extended to June 30, 2021 if we received aggregate new capital of at least $40.0 million no later than June 30, 2020. We met this milestone in June 2020 in conjunction with the execution of the AbbVie agreement, discussed in detail in Note 11. As a result of the interest only extension, the Term Loans will be payable in 25 equal monthly installments of principal plus interest, with the final installment due and payable on July 1, 2023.We may prepay all of the Term Loans subject to a prepayment fee equal to $75,000, which represents the deferred portion of the final payment due under the Original Agreement, plus the outstanding principal balance under the Term Loans at the time of such prepayment multiplied by a prepayment fee of 2.0% in the first year, 1.0% in the second year, and 0% in the third year and thereafter. Additionally, a final payment in the amount of 5.5% of the funded Term Loans is payable to SVB on the date on which the Term Loans are prepaid, paid or become due and payable in full. The final payment fees are recorded in long-term debt with an offsetting reduction to debt discount on our accompanying Consolidated Balance Sheets.The Loan Agreement contains customary representations and warranties, events of default and affirmative and negative covenants, including, among others, covenants that limit or restrict our ability to, among other things, incur additional indebtedness, grant liens, merge or consolidate, make acquisitions, pay dividends or other distributions or repurchase equity, make investments, dispose of assets, engage in any new lines of business, and enter into certain transactions with affiliates, in each case subject to certain exceptions. We assessed the likelihood of the lender accelerating payment of the loan due to a material adverse change in our business, operations, financial, or other condition as remote. We were in compliance with our covenants as of December 31, 2021. As such, as of December 31, 2021, the classification of the loan is split between current and noncurrent based on the timing of payment obligations. As security for its obligations under the Loan Agreement, we granted SVB a first priority security interest on substantially all of our assets, except intellectual property, and subject to certain other exceptions.
In connection with the Loan Agreement, we issued a warrant to SVB to purchase up to 52,083 shares of our common stock at a price of $4.32 per share, 17,361 shares of which became exercisable in August 2019 after we drew down the initial tranche. In March 2020, after we drew down the second tranche of our Term Loan, an additional 17,361 shares became exercisable. The remaining warrants did not vest and expired on July 31, 2020, upon the expiration of the third tranche of our Term Loan. The fair value of the warrants on the date of issuance for the initial tranche and second tranche was $60,000 and $60,000, respectively, determined using the Black-Scholes option-pricing model, and was recorded as a component of equity and as a debt discount on our accompanying Consolidated Balance Sheets. In connection with Original Agreement, SVB also holds 7,069 fully vested common stock warrants at an exercise price of $12.38 per share. The Term Loan was accounted for as a debt modification in a non-troubled debt restructuring, rather than a debt extinguishment, based on a comparison of the present value of the cash flows under the terms of the debt immediately before and after the Effective Date of the Term Loan, which resulted in a change of less than 10%. As a result, the remaining unamortized debt discount recorded in connection with the Original Agreement will be amortized to interest expense over the repayment term of Loan Agreement. In connection with the initial and second tranches of the Loan Agreement, we recorded a total debt discount of $812,000, which is being amortized to interest expense using the effective interest method over the repayment term of the loan. Non-cash interest expense associated with the amortization of the discount was $274,000, $261,000, and $140,000, for years ended December 31, 2021, 2020, and 2019, respectively. The unamortized discount was $223,000 as of December 31, 2021.
Scheduled principal payments on our outstanding debt as of December 31, 2021 under our Loan Agreement, excluding final fee amounts, are as follows (in thousands):
| | | | | | | | | | Year Ending December 31, | | Total | | 2022 | | $ | 4,800 | | | 2023 | | 2,800 | | | Total future principal payments | | $ | 7,600 | |
ALPINE IMMUNE SCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
10. Commitments and Contingencies
Operating Leases
We leased office and laboratory space located at 201 Elliott Avenue West, in Seattle, Washington, under an agreement classified as an operating lease that expired on December 31, 2019. In May 2017, as required by the terms of the lease, we entered into a line of credit to establish collateral to support the security deposit in an amount of $132,000, which was recorded as current restricted cash in our Consolidated Balance Sheets for the year ended December 31, 2019.
In March 2019, we entered into a lease for office and laboratory space located at 188 East Blaine Street, Seattle, Washington. The term of the lease is 10.8 years with 1 option to extend the term by 5.0 years. Our option to extend the rental term of our lease was not considered reasonably certain as of December 31, 2021. The lease term commenced in June 2019. The “Rent Commencement Date” began in March 2020, nine months after the commencement date. The annual base rent under the lease is $1.7 million for the first year and will increase by 3.0% each year thereafter. We were not required to pay base rent from the Rent Commencement Date through November 2020, the last day of the ninth month following the Rent Commencement Date. We received a tenant improvement allowance of $5.4 million, which is included in our base rent, and a maximum additional tenant improvement allowance of $1.8 million, which will result in additional rent amortized over the term of the lease at an annual rate of 8.0%. The lease also requires us to pay additional amounts for operating and maintenance expenses. In March 2019, in connection with the lease, we provided a $254,000 letter of credit as a security deposit, which is recorded as noncurrent restricted cash in our accompanying Consolidated Balance Sheets.As of December 31, 2021, our operating lease right-of-use assets and operating lease liability associated with our leases were $8.8 million and $11.6 million, respectively. Supplemental operating lease information for the year ended December 31, 2021 was as follows (in thousands):
| | | | | | | | | | | | | For the Year Ended December 31, | | 2021 | | 2020 | | Operating lease cost | $ | 1,837 | | $ | 1,872 | | Variable lease cost | 603 | | 475 | | Total lease cost | $ | 2,440 | | $ | 2,347 | | Other information: | | | | | Cash paid for amounts included in the measurement of lease liabilities | $ | 2,117 | | | $ | 213 | | | | | | Weighted-average remaining lease term (years) | 8.2 | | 9.2 | | Weighted-average discount rate | 10.7% | | 10.7% |
Variable lease costs represent our share of the landlord’s operating expenses. We do not act as a lessor or have any leases classified as financing leases. Maturities of our operating lease liabilities as of December 31, 2021 are as follows (in thousands): | | | | | | | | Minimum Lease Payments | | 2022 | $ | 1,841 | | | 2023 | 2,057 | | | 2024 | 2,111 | | | 2025 | 2,167 | | | 2026 | 2,224 | | | Thereafter | 7,438 | | | Total future minimum lease payments | 17,838 | | | Less: imputed interest | (6,212) | | | Operating lease liabilities | $ | 11,626 | |
Contingencies
Certain credits received related to our research and development expenditures and recorded within other income in our accompanying Consolidated Statements of Operations and Comprehensive Income (Loss) are subject to review by foreign taxing authorities. It is reasonably possible we may incur losses upon the completion of these reviews ranging from $0 to $1.8 million, which we could be required to repay to certain tax authorities.
ALPINE IMMUNE SCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
11. License and Collaboration Agreements
Horizon
In December 2021, we entered into an exclusive license and collaboration agreement with Horizon (the “Horizon Agreement”) for the development and commercialization of up to 4 preclinical candidates generated from our unique discovery platform. The agreement includes licensing of 1 of our existing preclinical biologic therapeutic programs (the “Existing Program”), as well as a research partnership to jointly develop candidates for up to 3 additional autoimmune and inflammatory disease programs for other designated biological targets, (the “Research Programs”). These candidates include previously undisclosed multi-specific fusion protein-based therapeutic candidates for autoimmune and inflammatory diseases. We will advance candidate molecules to predefined preclinical milestones while Horizon will be responsible for the respective costs and, ultimately, Horizon will assume responsibility for development and commercialization activities and costs.
In connection with the execution of the Horizon Agreement in December 2021, we entered into a stock purchase agreement under which Horizon purchased 951,980 shares of our common stock in a private placement for approximately $15.76 per share and aggregate proceeds of $15.0 million. The shares were sold at a 25% premium to the volume-weighted average share price of our common stock for a specified 30-day period prior to entering into the agreement. The fair value of the common stock issued to Horizon of $11.9 million was recorded to equity, based on the closing price of common stock on the effective date of the Horizon Agreement. For accounting purposes, the $3.1 million difference between the cash proceeds and the fair value of the common stock was treated as additional consideration attributable to the Horizon Agreement.
Under the terms of the agreements, Horizon also paid us a non-refundable upfront payment of $25.0 million in the first quarter of 2022. In addition, we are eligible to receive up to $381.0 million per program, or up to approximately $1.5 billion in total, in future success-based payments related to development, regulatory and commercial milestones. Furthermore, we are eligible to receive tiered royalties from a mid-single digit percentage to a low double-digit percentage on global net sales.
As of December 31, 2021, we recorded on our accompanying Consolidated Balance Sheets $28.1 million in current and noncurrent deferred revenue, of which $25.0 million relates to accounts receivable for the up-front payment and $3.1 million relates to the premium paid by Horizon on the stock issuance. As work under the Horizon license and collaboration agreement did not begin until 2022, we did not recognize any related revenue for the year ended December 31, 2021.For revenue recognition purposes, we determined that the Existing Program and each Research Program are distinct performance obligations. We allocated revenue to each performance obligation based on its relative stand-alone selling price. The future success-based payments related to development and regulatory milestones are probable of significant revenue reversal as the achievement is highly dependent on factors outside our control. Therefore, these milestone payments are fully constrained and are not initially included in the transaction price. We will continue to re-evaluate the transaction price each reporting period and update as uncertain events are resolved or other changes in circumstances occur. Any consideration related to commercial milestones and royalties will be recognized when the related sales occur.
AbbVie
In June 2020, we entered into an option and license agreement with AbbVie (the “AbbVie Agreement”) for the development of ALPN-101 (“acazicolcept”). The AbbVie Agreement grants AbbVie the exclusive option to purchase an exclusive worldwide license to acazicolcept (the “License Option”). The License Option is exercisable by AbbVie at any time and will expire 90 days from the achievement of certain development milestones. If AbbVie exercises the License Option, AbbVie will take over the future development and commercialization. Prior to the exercise of the License Option, we will perform research and development services, including conducting a Phase 2 study in systemic lupus erythematosus, based on an agreed-upon development plan (the “Development Plan”). We will be fully responsible for all costs incurred to conduct the activities under the Development Plan, provided that, AbbVie may be responsible for increased costs under the Development Plan in connection with certain material amendments proposed by AbbVie. We will also be solely responsible, at our sole cost and expense, for manufacturing and regulatory filings for acazicolcept necessary to complete activities under the Development Plan.
In June 2020, in connection with the execution of the AbbVie Agreement, AbbVie paid us a nonrefundable upfront payment of $60.0 million. Prior to the exercise of the License Option, AbbVie has agreed to make cash payments upon our achievement of certain predefined pre-option development milestones (the “Alpine Development Milestones”) up to an aggregate amount of $75.0 million. In the second quarter of 2021, we achieved $45.0 million of the Alpine Development Milestones. If AbbVie exercises the License Option, they will pay a 1-time cash payment of $75.0 million. Following the exercise of the License Option, AbbVie has also agreed to make aggregate cash payments of up to $205.0 million upon AbbVie’s achievement of certain development and commercial milestones and additional aggregate cash payments of up to $450.0 million upon AbbVie’s achievement of certain sales-based cash milestones, collectively referred to as (the “AbbVie
ALPINE IMMUNE SCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
Milestones”). Subsequent to commercialization, we are also eligible to receive high single-digit to low double-digit percentage royalties on worldwide net sales of licensed products.
For revenue recognition purposes, we determined that our contractual promises in the AbbVie Agreement are not distinct and are interdependent with our performance obligation to provide research and development services under the Development Plan. Thus, all contractual promises related to the upfront payment and Alpine’s Development Milestones were combined into a single performance obligation. We determined the Alpine Development Milestone payments are probable of significant revenue reversal as the achievement is highly dependent on factors outside our control. Therefore, these milestone payments were fully constrained and were not initially included in the transaction price. In June 2021, we re-evaluated and updated the transaction price to include the achieved portion of the Alpine Development Milestones. We will continue to re-evaluate the transaction price each reporting period and update as uncertain events are resolved or other changes in circumstances occur.
The License Option and the AbbVie Milestones were not determined to be performance obligations at the inception of the contract as they did not represent material rights. If exercised, the License Option and AbbVie Milestones will be accounted for as a separate contract and will be recognized as revenue if and when triggered. Any consideration related to sales-based royalties and profit-sharing payments will be recognized when the related sales occur.
We use a cost-based input method to measure progress toward completion of the performance obligation and to calculate the corresponding revenue to recognize each period. In applying the cost-based input, we use actual costs incurred relative to budgeted costs for the combined performance obligation. These costs consist primarily of internal personnel efforts and third-party contract costs relative to the level of patient enrollment in the study. Revenue will be recognized based on the level of costs incurred relative to the total budgeted costs for the performance obligation. A cost-based input method of revenue recognition requires management to make estimates of costs to complete our performance obligation. In making such estimates, significant judgment is required to evaluate assumptions related to cost estimates. The cumulative effect of revisions to estimated costs to complete our performance obligation will be recorded in the period in which changes are identified and amounts can be reasonably estimated. A significant change in these assumptions and estimates could have a material impact on the timing and amount of revenue recognized in future periods.
We recognized revenue from the AbbVie Agreement of $23.4 million and $7.0 million for the years ended December 31, 2021 and 2020, respectively. As of December 31, 2021 the remaining balance of the transaction price is $74.5 million and is recorded as current and noncurrent deferred revenue on our accompanying Consolidated Balance Sheets. We expect to recognize the remaining deferred revenue over the remainder of our Development Plan, which began in June 2020 and ends upon the later of the exercise or expiration of the option.Adaptimmune
In May 2019, we entered into a collaboration and licensing agreement with Adaptimmune (the “Adaptimmune Agreement”) to develop next-generation SPEAR T cell products. Under the Adaptimmune Agreement, we are to perform certain research services and grant Adaptimmune an exclusive license to programs from our secreted immunomodulatory protein (“SIP”) and transmembrane immunomodulatory protein (“TIP”) technologies. In June 2019, under the terms of the Adaptimmune Agreement, we received an upfront license payment of $2.0 million, and through December 31, 2021 we have received an additional $1.6 million in research support payments to fund ongoing programs. These payments were recorded as deferred revenue upon receipt and were recognized to revenue based on employee hours contributed to each performance obligation. In the fourth quarter of 2020, based on the completion of our initial research and development efforts in connection with our performance obligations, we recognized the remaining balance in deferred revenue associated with Adaptimmune on our accompanying Consolidated Balance Sheets. Under the Adaptimmune Agreement we recognized no revenue for the year ended December 31, 2021, and $2.3 million and $1.3 million for the years ended December 31, 2020, and 2019, respectively. In addition, we are eligible for additional research support payments, one-time payments and downstream development and commercialization milestones of up to $288.0 million, if all pre-specified milestones for each program are achieved. We are also eligible to receive low-single digit royalties on worldwide net sales of the applicable products. In February 2022, Adaptimmune selected an additional research program, triggering a $1.0 million upfront payment, which will be recorded as deferred revenue upon receipt and recognized to revenue based on employee hours contributed.
ALPINE IMMUNE SCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
12. Stockholders’ Equity
Common Stock
Shares of common stock reserved for future issuance were as follows: | | | | | | | | | | | | | | December 31, | | | 2021 | | 2020 | | Shares to be issued upon exercise of outstanding stock options | 5,611,743 | | | 4,175,345 | | | Shares to be issued upon release of RSUs | 265,566 | | | — | | | Shares to be issued upon conversion of common stock warrants | 3,666,435 | | | 3,673,551 | | | Shares to be issued upon conversion of prefunded warrants | 5,182,197 | | | 790,710 | | | Shares available for future stock grants | 284,906 | | | 887,901 | | | Shares to be issued under employee stock purchase plan | 45,211 | | | 45,211 | | | Shares of common stock reserved for future issuance | 15,056,058 | | | 9,572,718 | |
Securities Offerings
In September 2021, we entered into a securities purchase agreement (the “2021 Securities Purchase Agreement”) for a private placement with a select group of institutional investors, pursuant to which we sold 6,489,357 shares of our common stock (the “Shares”) and prefunded warrants to purchase 3,191,487 Shares (the “Prefunded Warrants”). The purchase price for each Share and for each Prefunded Warrant was $9.40 per share, for an aggregate purchase price of approximately $91.0 million. The Prefunded Warrants became fully exercisable upon the closing date and have an exercise price of $0.001 per share. We incurred $266,000 in financing costs associated with the 2021 Securities Purchase Agreement, which was netted against the proceeds within additional-paid-in-capital on our accompanying Consolidated Balance Sheets.In September 2021, we entered into an exchange agreement (the “Exchange Agreement”) with Frazier Life Sciences VIII, L.P. (the “Exchanging Stockholder”), which Exchanging Stockholder is affiliated with a member of our board of directors, pursuant to which we exchanged an aggregate of 1,200,000 shares of common stock held by the Exchanging Stockholder for Prefunded Warrants (the “Exchange Warrants”) to purchase an aggregate of 1,200,000 shares of common stock. Upon the closing of the exchange, we reclassified 1,200,000 shares of common stock into treasury stock on our accompanying Consolidated Balance Sheets.In July 2020, we entered into a securities purchase agreement (the “2020 Securities Purchase Agreement”) for a private placement with a select group of institutional investors, pursuant to which we sold 5,139,610 units (the “Common Units”) and 790,710 units (the “Prefunded Warrant Units”), for an aggregate purchase price of $60.0 million. Each Common Unit consists of 1 share of our common stock plus a warrant to purchase 0.3 shares of common stock (the “Common Stock Warrants”), and each Prefunded Warrant Unit consists of 1 prefunded warrant to purchase 1 share of common stock plus 0.3 Common Stock Warrants. The Prefunded Warrant Units and the Common Units are collectively referred to as the “Units” and each Unit has a purchase price of $10.1175. Pursuant to the terms of the 2020 Securities Purchase Agreement, we issued warrants to purchase 1,779,096 shares of common stock with an exercise price of $12.74 and a term of 3.5 years. Additionally, we issued 790,710 prefunded warrants, which became fully exercisable upon the closing date and have an exercise price of $0.001 per share. We incurred $3.7 million in financing costs associated with the 2020 Securities Purchase Agreement, which was netted against the proceeds within additional-paid-in-capital on our accompanying Consolidated Balance Sheets.In January 2019, we entered into a securities purchase agreement (the “2019 Securities Purchase Agreement”) with a limited number of accredited investors, pursuant to which we sold 4,706,700 units (the “2019 Units”) for an aggregate purchase price of $25.3 million in a private placement (the “Private Placement”). Each 2019 Unit has a purchase price of $5.37 and consists of 1 share of our common stock and a warrant to purchase 0.39 shares of common stock. Pursuant to the terms of the 2019 Securities Purchase Agreement, we issued 4,706,700 shares of common stock and warrants to purchase an aggregate of 1,835,610 shares of common stock. The warrants have an exercise price of $12.74 and have a term of five years. We incurred $1.7 million in financing costs associated with the 2019 Securities Purchase Agreement, which was netted against the proceeds within additional-paid-in-capital on our accompanying Consolidated Balance Sheets.The issuance of the securities sold under the 2021, 2020, and 2019 Securities Purchase Agreements have not been registered under the Securities Act of 1933, as amended, or state securities laws and may not be offered or sold in the United States absent registration with the SEC or an applicable exemption from such registration requirements. We filed registration statements for the 2021, 2020 and 2019 Securities Purchase Agreements with the SEC, which were declared effective by the SEC in November 2021, August 2020 and April 2019, respectively, which cover the resale of the shares of common stock issuable in connection with the private placements and upon exercise of the warrants. In May 2021, the registration statement
ALPINE IMMUNE SCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
for the 2019 Securities Purchase Agreement was deactivated following the expiration of our obligation to maintain its effectiveness under the related registration rights agreement.
Financing Agreements
In July 2021, we entered into a sales agreement (the “Sales Agreement”) with Cowen and Company, LLC (“Cowen”) pursuant to which we may sell shares of our common stock from time to time through an “at the market” equity offering for up to $75.0 million in gross cash proceeds. Cowen will act as the sales agent and will be entitled to compensation for services of up to 3.0% of the gross sales price per share of all shares sold through Cowen under the Sales Agreement. The shares would be issued pursuant to our effective shelf registration statement on Form S-3 (File No. 333-256107). We filed a prospectus supplement, dated July 2, 2021, with the SEC in connection with the offer and sale of the shares pursuant to the Sales Agreement. As of December 31, 2021, no sales have been made under the Sales Agreement.
Common Stock Warrants
We have issued warrants in connection with our securities offerings, SVB loans, and to certain non-employee professional advisers. Excluding the prefunded warrants we issued in connection with our securities offerings discussed above, the table below summarizes our warrant activity: | | | | | | | | | | | | | | | | | | | | Warrants
Outstanding | | Weighted-
average
Exercise
Price | | Weighted-
average
Remaining
Contract
Term
(in years) | | Outstanding at December 31, 2020 | 3,673,551 | | | $ | 12.74 | | | 3.12 | | Exercised | (2,484) | | | 5.02 | | | | | Expired | (4,632) | | | 97.12 | | | | | Outstanding at December 31, 2021 | 3,666,435 | | | $ | 12.64 | | | 2.12 | | Exercisable at December 31, 2021 | 3,666,435 | | | $ | 12.64 | | | 2.12 |
Equity Incentive Plans
In June 2018 our stockholders approved, the 2018 Equity Incentive Plan (“2018 Plan”). Upon adoption, we ceased granting stock awards under the Nivalis Therapeutics, Inc. 2015 Equity Incentive Plan (the “2015 EIP”) and the Amended and Restated 2015 Stock Plan (the “2015 Plan”), collectively, the “Legacy Plans”. All shares of common stock subject to awards under the Legacy Plans that expire or terminate without having been exercised in full, or are forfeited to or repurchased by the company, will be added to the 2018 Plan, up to a maximum of 1,972,784 shares. In June 2020, in conjunction with our annual meeting of stockholders, our stockholders approved an additional increase of 743,515 shares authorized under our 2018 Plan.
Under our 2018 Plan we may issue stock options, stock appreciation rights, restricted stock, RSUs or performance shares. As of December 31, 2021 we have only issued stock options and RSUs. Our 2018 Plan provides for an annual increase in the number of shares reserved for insurance equal to the lesser of (1) 5% of the number of shares of common stock outstanding as of the last day of the preceding calendar year or (2) 1,500,000. However, our board of directors may act prior to January 1 of a given year to provide that there will be no January 1 increase for such year or that the increase for such year will be a lesser number of shares. On January 1, 2022, a total of 1,500,000 additional shares were automatically added to the shares authorized under the 2018 Plan.
In July 2017, in connection with the merger, we assumed Nivalis’ Employee Stock Purchase Plan (the “ESPP”) and the 2015 EIP. Upon assumption of the ESPP, there were 45,211 shares available for issuance under the ESPP. As of December 31, 2021, we have not activated the ESPP.
Stock options granted under our equity plans generally vest within four years and vested options are exercisable from the grant date until ten years after the date of grant. Vesting of certain employee options may be accelerated in the event of a change in control of the Company. We grant stock options to employees with exercise prices equal to the fair value of our common stock on the date of grant. The term of incentive stock options may not exceed ten years from the date of grant.
ALPINE IMMUNE SCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
As of December 31, 2021, a total of 6,616,862 shares of common stock were authorized for issuance under our 2018 Plan, 2015 Plan and 2015 EIP. A summary of stock option activity under our plans is presented below:
| | | | | | | | | | | | | | | | | | | | | | | | | | Options
Outstanding | | Weighted-
average
Exercise
Price | | Weighted-
average
Remaining
Contract
Term
(in years) | | Aggregate
Intrinsic
Value
(in thousands) | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | Outstanding at December 31, 2020 | 4,175,345 | | | $ | 5.30 | | | | | | | Granted | 1,940,518 | | | $ | 12.38 | | | | | | | Exercised | (91,190) | | | $ | 5.37 | | | | | | | Forfeited | (412,930) | | | $ | 9.88 | | | | | | | Outstanding at December 31, 2021 | 5,611,743 | | | $ | 7.41 | | | 7.41 | | $ | 36,465 | | | Vested and expected to vest after December 31, 2021 | 5,471,743 | | | $ | 7.38 | | | 7.37 | | $ | 35,685 | | | Exercisable at December 31, 2021 | 2,944,273 | | | $ | 5.25 | | | 6.31 | | $ | 26,053 | |
The aggregate intrinsic value of stock options exercised during the years ended December 31, 2021, 2020 and 2019 was $546,000, $104,000 and $155,000, respectively. The fair value of stock options vested during the years ended December 31, 2021, 2020 and 2019 was $3.4 million, $4.2 million and $3.0 million, respectively.
A summary of our RSU activity under our plans is presented below: | | | | | | | | | | | | | | | | | | | | | | | | | | Number of Shares | | Weighted-
Average
Grant Date Fair Value
(per share) | | Weighted-
average
Remaining
Contract
Life
(in years) | | Aggregate
Intrinsic
Value
(in thousands) | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | Non-vested at December 31, 2020 | — | | | $ | — | | | | | | | Granted | 265,566 | | | $ | 12.00 | | | | | | | Non-vested at December 31, 2021 | 265,566 | | | $ | 12.00 | | | 1.95 | | $ | 491 | |
The aggregate intrinsic value of RSUs released during the years ended December 31, 2021 and 2020 was $0 and $1.6 million, respectively. The fair value of RSUs vested during the years ended December 31, 2021 and 2020 was $0 and $461,000, respectively. We had no RSU activity for the year ended December 31, 2019.
We utilize newly issued shares to satisfy option exercises and RSU releases. As of December 31, 2021, there was $16.4 million of unrecognized stock-based compensation expense related to approximately 2.9 million nonvested stock options and RSU awards that are expected to be recognized over a weighted-average period of 2.7 years.
Stock-Based Compensation Expense
The fair value of RSUs is equal to the closing stock price on the date of grant. We use the Black-Scholes option pricing model to estimate the fair value of stock options at the grant date. The Black-Scholes option pricing model requires us to make certain estimates and assumptions, including assumptions related to the expected price volatility of our stock, the period during which the options will be outstanding, the rate of return on risk-free investments, and the expected dividend yield of our stock. The fair values of stock options granted to employees were calculated using the following assumptions: | | | | | | | | | | | | | | | | | | | | Years Ended December 31, | | | 2021 | | 2020 | | 2019 | | Weighted-average estimated fair value at grant | $8.44 | | $3.03 | | $4.14 | | Risk-free interest rate (1) | 0.25% - 1.3% | | 0.38% - 1.68% | | 1.42% - 2.63% | | Expected term of options (in years) (2) | 3.49 - 6.08 | | 5.27 – 6.90 | | 5.27 – 6.08 | | Expected stock price volatility (3) | 79% - 83% | | 73% - 82% | | 70% - 77% | | Expected dividend yield (4) | —% | | —% | | —% |
(1)The risk-free interest rate assumption was based on zero-coupon U.S. Treasury instruments that had terms consistent with the expected term of our stock option grants.
(2)We used the “simplified method” for options to determine the expected term of stock options granted, since we do not have sufficient historical exercise data to provide a reasonable basis upon which to estimate expected term due to the
ALPINE IMMUNE SCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
limited time our shares have been publicly traded. Under this approach, the weighted-average expected life is presumed to be the average of the vesting term and the contractual term of the option.
(3)Volatility is a measure of the amount by which a financial variable, such as share price, has fluctuated or is expected to fluctuate during a period. We analyzed the stock price volatility of companies at a similar stage of development to estimate expected volatility of our stock price.
(4)We have never declared or paid any cash dividends and do not presently plan to pay cash dividends in the foreseeable future.
| | | | | | | | | | | | | | | | | | | | Years Ended December 31, | | | 2021 | | 2020 | | 2019 | | Employee: | | | | | | | Research and development | $ | 3,323 | | | $ | 2,145 | | | $ | 1,608 | | | General and administrative | 2,891 | | | 1,955 | | | 1,359 | | | Non-Employee: | | | | | | | Research and development | 24 | | | 34 | | | 68 | | | General and administrative | 2 | | | 6 | | | 6 | | | Total stock-based compensation expense | $ | 6,240 | | | $ | 4,140 | | | $ | 3,041 | |
13. Income Taxes
Our loss before taxes is derived from domestic (United States) and foreign (Australian) sources as follows (in thousands): | | | | | | | | | | | | | | | | | | | Years Ended December 31, | | 2021 | | 2020 | | 2019 | | Domestic | $ | (48,932) | | | $ | (28,356) | | | $ | (38,234) | | | Foreign | (1,488) | | | 410 | | | (3,618) | | | Total | $ | (50,420) | | | $ | (27,946) | | | $ | (41,852) | |
The provision for income taxes is composed of the following (in thousands): | | | | | | | | | | | | | | | | | | | | Years Ended December 31, | | | 2021 | | 2020 | | 2019 | | Current: | | | | | | | U.S. - Federal | $ | — | | | $ | — | | | $ | — | | | U.S. - State | — | | | (6) | | | — | | | Foreign | 129 | | | — | | | — | | | Total current | 129 | | | (6) | | | — | | | Deferred: | | | | | | | U.S. - Federal | — | | | — | | | — | | | U.S. - State | — | | | — | | | — | | | Foreign | (216) | | | — | | | — | | | Total deferred | (216) | | | — | | | — | | | Total income tax benefit | $ | (87) | | | $ | (6) | | | $ | — | |
ALPINE IMMUNE SCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
The effective tax rate of the provision for income taxes differs from the federal statutory rate as follows: | | | | | | | | | | | | | | | | | | | | Years Ended December 31, | | | 2021 | | 2020 | | 2019 | | U.S. Statutory rate | 21.0 | % | | 21.0 | % | | 21.0 | % | | Effect of: | | | | | | | Permanent differences | (0.4) | % | | 0.1 | % | | (0.1) | % | | Federal research and development credit | 4.5 | % | | 2.6 | % | | 1.1 | % | | Change in valuation allowance | (21.4) | % | | (20.7) | % | | (19.8) | % | | Global intangible low-taxed income recapture | (1.4) | % | | — | % | | — | % | | Stock-based compensation | (1.8) | % | | (2.8) | % | | (0.5) | % | | Foreign rate differential | 0.2 | % | | (0.1) | % | | 0.6 | % | | Other | (0.5) | % | | (0.1) | % | | (2.3) | % | | Effective income tax rate | 0.2 | % | | — | % | | — | % |
We recorded tax benefits of $87,000, $6,000, and $0, representing effective tax rates of 0.2% for the year ended December 31, 2021, and 0.0%, for the years ended December 31, 2020 and 2019, respectively. The $87,000 tax benefit for the year ended December 31, 2021 represents deferred tax benefits from true-ups and the removal of the valuation allowance in place against our foreign deferred tax assets, partially offset by income tax expense for current year activity.
The difference between the U.S. federal statutory tax rates of 21% and our effective tax rate in all periods is primarily due to changes in our valuation allowance related to our deferred tax assets and the generation and consumption of federal research and development tax credits. We have elected to treat taxes due on future U.S. inclusions in taxable income under the global intangible low-taxed income (“GILTI”) provision as a current-period expense when incurred. As such, expected future GILTI inclusions have not been factored into the measurement of our deferred taxes.
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. The following table represents the significant components of our deferred tax assets and liabilities for the periods presented (in thousands): | | | | | | | | | | | | | | December 31, | | | 2021 | | 2020 | | Deferred tax assets: | | | | | Net operating loss | $ | 22,054 | | | $ | 21,992 | | | | | | | Research and development credits | 6,297 | | | 4,044 | | | Intangible asset basis | 13 | | | 24 | | | Lease liability | 2,476 | | | 2,619 | | | Deferred revenue | 8,333 | | | — | | | Stock based compensation | 1,672 | | | 1,337 | | | Other | 217 | | | 72 | | | Gross deferred tax assets | 41,062 | | | 30,088 | | | Valuation allowance | (38,752) | | | (27,851) | | | Total deferred tax assets, net of valuation allowance | 2,310 | | | 2,237 | | | Deferred tax liabilities: | | | | | Prepaid expenses | (147) | | | (125) | | | Fixed asset basis | (93) | | | (137) | | | Right-of-use asset basis | (1,856) | | | (1,975) | | | | | | | Total deferred tax liability | (2,096) | | | (2,237) | | | Net deferred tax assets and liabilities | $ | 214 | | | $ | — | |
A valuation allowance is provided for deferred tax assets where the recoverability of the assets is uncertain. The determination to provide a valuation allowance is dependent upon the assessment of whether it is more likely than not that sufficient taxable income will be generated to utilize the deferred tax assets. For the year ended December 31, 2021, we determined that based on an evaluation of the four sources of income and all available evidence, both positive and negative, including our latest forecasts and cumulative losses in recent years, it was more likely than not that none of our domestic deferred tax assets would be realized and therefore we continued to record a full valuation allowance. For the year ended
ALPINE IMMUNE SCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
December 31, 2021, we determined that it was more likely than not that our foreign deferred tax assets would be realized. As such, we removed the valuation allowance previously in place against our foreign deferred tax assets as we have generated sufficient foreign taxable income to utilize all historical operating losses and are expecting future foreign taxable income. The valuation allowance increased by $10.9 million and $5.8 million during the year ended December 31, 2021 and 2020, respectively.
We have net operating loss (“NOL”) carryforwards as follows (in thousands): | | | | | | | | | | | | | | | | | | | | December 31, | | | 2021 | | 2020 | | 2019 | | Federal (before January 1, 2018) | $ | 11,094 | | | $ | 11,094 | | | $ | 11,094 | | | Federal (after January 1, 2018) | $ | 91,592 | | | $ | 91,868 | | | $ | 67,500 | | | State | $ | 6,433 | | | $ | 6,433 | | | $ | 6,433 | | | Foreign | $ | — | | | $ | — | | | $ | 787 | |
Federal NOL carryforwards created before January 1, 2018 begin to expire in 2037. Federal NOL carryforwards created after January 1, 2018 carryforward indefinitely. State NOL carryforwards begin to expire in 2038. Foreign NOLs carryforward indefinitely.
We have net research and development tax credit carryforwards as follows (in thousands): | | | | | | | | | | | | | | | | | | | | December 31, | | | 2021 | | 2020 | | 2019 | | Federal | $ | 7,880 | | | $ | 5,063 | | | $ | 3,986 | |
Federal research and development tax credit carryforwards begin to expire in 2035.
Current tax laws impose substantial restrictions on the utilization of research and development credit and NOL carryforwards in the event of an ownership change, as defined by the Internal Revenue Code Section 382 and 383. Such an event may limit our ability to utilize NOLs and research and development tax credit carryforwards. Under Internal Revenue Code Section 382 and 383, the 2017 merger with Nivalis is likely considered an ownership change with respect to the potential limitation of the Nivalis federal tax credits and NOLs. As such, it is likely that any future utilization of Nivalis federal tax credits and NOLs is substantially limited. Therefore, as of December 31, 2018, all Nivalis tax credit and NOL carryforwards have been reduced to zero.
We account for uncertainty in income taxes in accordance with ASC 740. Tax positions are evaluated in a two-step process, whereby we first determine whether it is more likely than not that a tax position will be sustained upon examination by the tax authority, including resolutions of any related appeals or litigation processes, based on technical merit. If a tax position meets the more-likely-than-not recognition threshold it is then measured to determine the amount of benefit to recognize in the financial statements. The tax position is measured as the largest amount of benefit that is greater than 50% likely of being realized upon ultimate settlement.
The following table summarizes the activity related to unrecognized tax benefits (in thousands): | | | | | | | | | | | | | | | | | | | | December 31, | | | 2021 | | 2020 | | 2019 | | Unrecognized benefits – beginning of year | $ | 990 | | | $ | 775 | | | $ | 469 | | | Gross increases (decreases) – prior year tax positions | — | | | (7) | | | — | | | Gross increases – current year tax positions | 563 | | | 222 | | | 306 | | | Unrecognized benefit – end of year | $ | 1,553 | | | $ | 990 | | | $ | 775 | |
All of the unrecognized tax benefits as of December 31, 2021 are accounted for as a reduction in our deferred tax assets. Due to our valuation allowance, none of the $1.6 million of unrecognized tax benefits would affect our effective tax rate, if recognized. We do not believe it is reasonably possible that our unrecognized tax benefits will significantly change in the next twelve months.
We recognize interest and penalties related to unrecognized tax benefits as income tax expense. There were no accrued interest or penalties related to unrecognized tax benefits for 2021, 2020 and 2019.
We do not expect any significant change in our unrecognized tax benefits during the next twelve months.
ALPINE IMMUNE SCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
Our material income tax jurisdictions are the United States (federal), California (state), and Australia (foreign). We are subject to audit for tax years 2012 and forward for federal purposes, 2017 and forward for California purposes, and 2019 and forward for foreign purposes.
14. Related Party Transactions
In September 2021, in connection with our 2021 Securities Purchase Agreement we issued 3,723,402 shares of common stock and 2,340,424 prefunded warrants to purchase shares of common stock for gross proceeds of approximately $57.0 million to certain of our stockholders whose beneficial ownership exceeded 5% prior to the completion of the 2021 Securities Purchases Agreement. Also in September 2021, we exchanged an aggregate of 1,200,000 shares of common stock held Frazier Life Sciences VIII, L.P. for Prefunded Warrants to purchase an aggregate of 1,200,000 shares of common stock.
None of the purchasers in the July 2020 private placement was a greater than 5% holder of our outstanding capital stock prior to the July 2020 private placement.
In January 2019, in connection with our 2019 Securities Purchase Agreement we sold an aggregate of 935,753 shares of common stock and issued warrants to purchase an aggregate of 364,943 shares of common stock for gross proceeds of approximately $5.0 million to certain of our 5% stockholders.
15. 401(k) Retirement Plan
We have adopted a 401(k) plan. All employees are eligible to participate, provided they meet the requirements of the plan. Through December 31, 2021, we have not matched employee contributions to the plan. Beginning in 2022, we will match the employees’ 401(k) contributions, at our discretion and not to exceed a prescribed annual limit.
|