UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
☒ | |
ANNUAL REPORT PURSUANT TO SECTION 13 OR | |
For the fiscal year ended
OR
TRANSITION REPORT PURSUANT TO SECTION 13 OR |
For the Transition Periodtransition period from ___ to ___
Commission file number
333-191083RASNA THERAPEUTICS, INC.
(Exact name of registrant as specified in its charter)
Nevada | 39-2080103 |
(State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification Number) |
420 Lexington Ave, Suite 2525, New York, NY 10170
(Address of principal executive offices) (Zip Code)
Telephone: (646) 396-4087
(Registrant’s telephone number)
Securities registered pursuant to Section 12(b)12(b) of the Act: None.
Securities registered pursuant to Section 12(g)12(g) of the Act: None.
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act Yes
Indicate by check if the registrant is not required to file reports pursuant to Section 13 or Section 15(d)15(d) of the Act. Yes
Indicate by check mark whether the registrant (1)(1) has filed all reports required to be filed by Section 13 or 15(d)15(d) of the Securities Exchange Act of 1934 during the preceding 12 months and (2) (2)has been subject to such filing requirements for the past 90 days. Yes
Indicate by check mark whetherwhether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or informationinformation statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K.
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definition of “large accelerated filer”, “accelerated filer” and “smaller reporting company” in Rule 12b-212b-2 of the Exchange Act. (Check one)one):
Large accelerated filer | Accelerated filer |
Non-accelerated filer | Smaller reporting company |
(Do not check if a smaller reporting company) | Emerging growth company |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financialfinancial accounting standards provided pursuant to Section 13(a)13(a) of the Exchange Act.
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-212b-2 of the Act). Yes
The aggregate market value of the voting and non-voting common equity held by non-affiliates of the registrant was $10,336,200 as of September 30, 2019, based upon the closing price on the OTCQB market reported for such date.
Indicate the number of shares outstanding of each of the issuer’s classes of common stock, as of the latest practicable date: 68,046,465 68,908,003 shares of common stock were issued and outstanding as of June 28, 2017.
DOCUMENTS INCORPORATED BY REFERENCE: None
| 1 |
Forward Looking Statements.
This annual report contains forward-looking statements within the meaning of Section 27A27A of the Securities Act of 1933, as amended, and Section 21E21E of the Securities Exchange Act of 1934, as amended. These statements relate to future events or our future financial performance. In some cases, you can identify forward-looking statements by terminology such as “may”, “should”, “expects”, “plans”, “anticipates”, “believes”, “estimates”, “predicts”, “potential” or “continue” or the negative of these terms or other comparable terminology. These statements are only predictions and involve known and unknown risks, uncertainties and other factors, including the risks in the section entitled “Risk Factors”, any of which may cause our or our industry’s actual results, levels of activity, performance or achievements to be materially different from any future results, levels of activity, performance or achievements expressed or implied by these forward-looking statements. These risks include, by way of example and not in limitation:.
This list is not an exhaustive list of the factors that may affect any of our forward-looking statements. These and other factors should be considered carefully and readers should not place undue reliance on our forward-looking statements.
Forward looking statements are made based on management’s beliefs, estimates and opinions on the date the statements are made and we undertake no obligation to update forward-looking statements if these beliefs, estimates and opinions or other circumstances should change. Although we believe that the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee future results, levels of activity, performance or achievements. Except as required by applicable law, including the securities laws of the United States, we do not intend to update any of the forward-looking statements to conform these statements to actual results.
Our financial statements are stated in United States dollars (US$) and are prepared in accordance with United States Generally Accepted Accounting Principles.
In this annual report, unless otherwise specified, all dollar amounts are expressed in United States dollars and all references to “common stock” refer to the common shares in our capital stock.
As used in this annual report, the terms “we”, “us”, “our”, “Rasna” and the “Company” mean Rasna Therapeutics, Inc. unless the context clearly requires otherwise.
General.
We are a highly experienced industry team together with field-leading scientists, to focus onleukemia-focused biotechnology company that has been developing therapeutics to address the unmet need that exists for Acute Myloid Leukemia ("AML")acute myeloid leukemia, or AML, and other forms of leukemia and lymphoma.lymphoma. AML is generally a disease of older adults, with onset typically after the age of 45 (average patient age approximately 68 years old). Our strategy is to develop small molecule drug candidates targeting two genes: NPM1 and LSD1, which are considered to control major pathways underlying etiology of majority of AML sub-types.
RASP-101 is our lead compound and is a controlled release, nanoparticulate, intravenous formulation of dactinomycin, which is an established anticancer therapeutic.Our consultants, Prof. Falini and Prof. Martelli conducted a Phase 2 clinical trial usingunformulated free dactinomycin, or ACT D, in refractory/relapse (R/R) AML patients carrying NPM1 gene mutation.Treatment with ACT D at 15 µg/kg/day for 5 days every 28 days induced CR in 4 out of the 9 evaluable patients (44.4%) with only 1 or 2 cycles of therapy. We believe these results demonstrated proof-of-concept for therapy with ACT D for NPM1 gene mutated AML patients. However, intravenous treatment with ACT D treatment also produced severe dose-limiting oral mucositis.Pursuant to a license agreement with Prof. Falini and Prof. Martelli, we have developed RASP-101 which is a nanoparticle controlled release formulation of ACT D which we believe will maximize therapeutic effectiveness while minimizing oral mucositis. After completion of animal safety toxicity and pharmacokinetic, or PK,studies in animals, we are planning to conduct a Phase 1 safety and PK study, followed by a single, bridging Phase 3 trial in AML patients to support a 505(b)(2) submission for registration (subject to FDAapproval), offering a shortened approval pathway by the end of 2023.
RASP-201 is a novel, orally dosed, selective reversible inhibitor of lysine specific demethylase, or LSD1, a pathway that blocks differentiation and confers a poor prognosis to AML. We expect RASP-201 to display a safer metabolic profile than competitor irreversible inhibitors such as GSK2879552 (GlaxoSmithKline) and ORY-1001 (Oryzon/Roche).RASP-201 when dosed orally shows in vivo therapeutic utility in murine (mouse) models of AML.
| 3 |
RASP-301 is our innovative, first- in-class, oral, small molecule inhibitor that in preclinical studies has been shown to disrupt NPM1 oligomerization (aggregation of individual subunits) and has the potential to treat refractory AML with reduced toxicity at low dose levels.RASP-301 exhibits cytotoxic effects at nanomolar concentrations against AML cells in culture and was not cytotoxic to normal cells at the same concentrations.In vivo usefulness of these compounds in AML murine models has been evaluated confirming the druggability of the target and its potential to treat refractory AML.
Our primary indication is AML which may be fatal within weeks to months, has a 5-year5-year survival rate of only about 25%25% and very poor prospects for long-term survival of patients.
Leukemia Overview and user-friendly information on activity-based travel.
Leukemia is a cancer of the blood or bone marrow involving abnormal proliferation of white blood cells, called WBCs or leukocytes. Leukemia is caused by a mutation of the DNA in bone marrow stem cells resulting in the abnormal multiplication of leukocytes. If untreated, surplus leukocytes will overwhelm the bone marrow, enter the bloodstream and eventually invade other parts of the body, such as the lymph nodes, spleen, liver, and central nervous system. In this way, the behavior of leukemia is different from that of other cancers, which usually begin in major organs and ultimately spread to the bone marrow. Leukemia is an umbrella term covering a large group of blood cancers.
All leukemia arises from mutations or damage to the DNA within the blood cells. These mutations may occur spontaneously or as a result of exposure to radiation or carcinogenic substances. Ionizing radiation, as well as exposure to chemicals such as benzene, increases the risk of AML, while agricultural chemicals have been linked to an increased incidence of chronic lymphoctic Leukemia (“CLL”).lymphocytic leukemia, or CLL. A weak immune system, some virus forms such as human T-cell Leukemia virus I (“HTLV-1”), or HTLV-1, genetic predisposition, cigarette smoking, and reactions to some therapeutic drugs are also implicated in the etiology of leukemia.
Diagnosis, Treatment, and Management
The first symptoms of leukemia are often vague and are correlated with other disorders. Common symptoms include fatigue and malaise, excessive bruising, and abnormal bleeding due to low platelet count. Further symptoms can include weight loss, bone and joint pain, infection and fever, and an enlarged spleen, lymph nodes and liver. After a blood test, several blood abnormalities such as anemia, or leucopenia may be observed, and in most cases a bone marrow test is required to confirm the diagnosis.
The preliminary diagnostic test for leukemia is a blood cell count, which is followed by immune-phenotyping to assess whether the abnormal lymphocyte levels are caused by inflammation or cancer. The physician may also require additional confirmatory tests such as cytogenetic analysis or bone marrow sampling.
The specific variety and combination of anticancer drugs prescribed depends on the form and stage of the disease. For example, treatment for AML, the most common form of leukemia, usually involves chemotherapy with cytotoxic cytarabine in conjunction with an anthracycline such as daunorubicin or idarubicin. Because of the severity of the cytotoxic treatment, bone marrow transplants, (“BMTs”)or BMTs are sometimes necessary. By transplanting healthy bone marrow into the body, BMTs help rebuild tissue damaged by the treatment. Interferon, (“INF”)or INF, therapy, particularly with INF-alpha, is an alternative or additional treatment offered to almost all newly diagnosed patients in these markets. However, it is very difficult to cure, even though early treatment indicates it will help people to live longer.
The standard first-line treatment strategy for CLL: PatientsCLL for patients who might not be able to tolerate the side effects of strong chemotherapy (chemo) are oftenis to be treated with chlorambucil alone or with a monoclonal antibody like rituximab (Rituxan) or obinutuzumab (Gazyva). Other options include ibrutinib (Imbruvica), rituximab alone, or a corticosteroid like prednisone.
Other drugs or combinations of drugs may also be also used. If the only problem is an enlarged spleen or swollen lymph nodes in one region of the body, localized treatment with low-dose radiation therapy may be used. Splenectomy (surgery to remove the spleen) is another option if the enlarged spleen is causing symptoms.
In addition, very high numbers of leukemia cells in the blood causes problems with normal circulation. This is called leukostasis. Leukapheresis is also sometimes used before chemochemotherapy if there are very high numbers of leukemia cells (even when they aren’t causing problems) to prevent tumor lysis syndrome.
| 4 |
Upon the failure of first line therapy, the standard therapy is usually to administer many of the drugs and combinations listed above as potential second-line treatments. For many people who have already had fludarabine, alemtuzumab seems to be helpful as second-line treatment, but it carries an increased risk of infections. Other purine analog drugs, such as pentostatin or cladribine (2-CdA)(2-CdA), may also be tried. Newer drugs such as ofatumumab, ibrutinib, idelalisib (Zydelig), and venetoclax (Venclexta) may be other options. If these types of chemotherapy fail, the next option is usually a bone marrow transplant. A stem cell transplant is a third treatment option depending on leukemia response.
Market Potential
According to the American Cancer Society, it is estimated that approximately 21,000 new AML cases will be diagnosed in the US each year and the projected cost of drug therapy for each patient over the course of one year of the disease is approximately $280,000 for chemotherapy alone (Preussler et. al. Biol. Blood Marrow Transplant 23 (2017) 1021-1028) amounting to a total cost of approximately $5.9 billion. Of this it is estimated that 50% of the costs are attributable to standard of care chemotherapy. Thus, the assessment is that the U.S. market potential for AML is approximate $3 billion annually. The rest of the world is estimated to constitute 50% of the AML market; thus, the total annual world market is approximately $6 billion. If our NPM1 targeted therapy proves beneficial to only the 20% of AML patients with mutated NPM1 genes, the addressable market will be diminished but we believe that our NPM1 targeted therapy would then be the only therapy to address that sub-population.
Table 1Total Estimated Number of New Leukemia Cases in
the United States for2017*
| |||||||
|
|
|
|
|
|
|
|
Type | Total | Male | Female |
| |||
Acute Lymphocytic Leukemia |
| 5,970 |
| 3,350 |
| 2,620 |
|
Chronic Lymphocytic Leukemia |
| 20,110 |
| 12,310 |
| 7,800 |
|
Acute Myeloid Leukemia |
| 21,380 |
| 11,960 |
| 9,420 |
|
Chronic Myeloid Leukemia |
| 8,950 |
| 5,230 |
| 3,720 |
|
Other Leukemia |
| 5,720 |
| 3,440 |
| 2,280 |
|
Total Estimated New Cases |
| 62,130 |
| 36,290 |
| 25,840 |
|
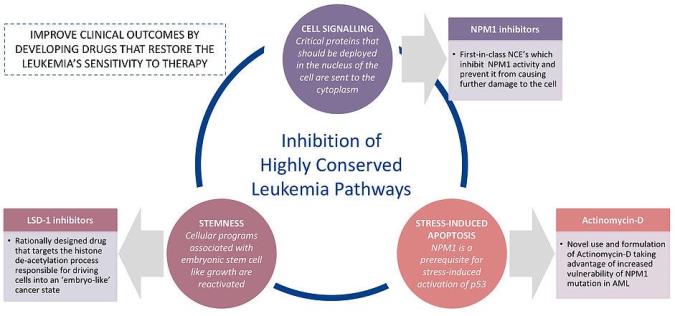
Figure 1Three Druggable Intervention Points for Developing AML Therapies
| 5 |
Our business strategy focuses on differentiated targeted therapies for genetic pathways which are known to be involved in etiologies of number of leukemia and other cancers. Our clinical program is based around three druggable intervention points with potential to improve safety and efficacy of current AML therapies, namely “stemness” (activation of stem cell-like growth patterns), cell signaling (between nucleus and cytoplasm) and stress-induced apoptosis (programmed cell death) (Figure 1). Our focus is to target two specific genetic pathways NPM1 and LSD1, which are known to be associated with etiologies of a number of cancers, including AML (Figure 2).
Figure 2Key Regulators for Developing Targeted AML Therapies
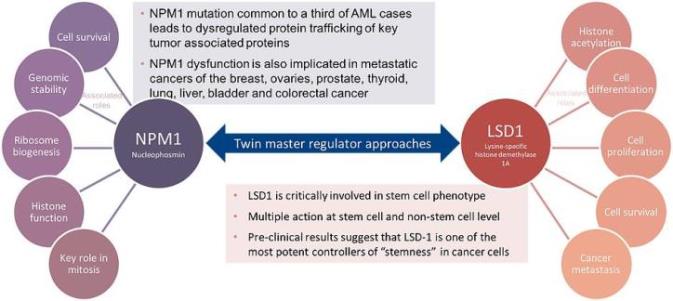
Roles of NPM1 and LSD1 in Tumorigenesis
Leukemia arises due to DNA damage or mutations. Chromosomal aberrations involving NPM1 wereNPM1 and LSD1 have been found in patients with non-Hodgkin lymphoma, acute promyelocytic leukemia, myelodysplastic syndrome, and AML. NPM1 has been foundThus, targeting NPM1 and LSD1 is an appropriate approach for treatment of AML. In addition, combination of therapies targeting these two genes could also be effective in the cytoplasm in patients with primarytreatment of relapse AML.
The NPM1 gene is up-regulated, mutated and chromosomally translocated in many tumor types. NPM1NPM1 is transferred from nucleolus to nucleoplasm and cytoplasm by anticancer drugs. When expressed at high level, NPM1NPM1 could promote tumor growth by inactivation of tumor suppressor p53/p53/ARF pathway; when expressed at low level, NPM1NPM1 could suppress tumor growth by inhibition of centrosome duplication. NPM1NPM1 is haplo insufficient in hemizygous mice that are vulnerable to tumor development. NPM1c+NPM1c+ (cytoplasm form) translocation into cytoplasm could serve as an AML remission signal. NPM1NPM1 forms a pentamer that could serve as a potential anticancer target.
The histone H3K4/K9 demethylase LSD1 can regulate gene activation and repression in epigenetic regulation and is a key effector of the differentiation block in MLL-rearranged leukemia. High LSD1 expression blocks differentiation and is associated with a poor prognosis in AML. LSD1 can be targeted by tranylcypromine analogs or downregulated by RNA interference which induces differentiation of MLL-rearranged leukemic cells. A number of different small molecular inhibitors of LSD1 have been in clinical evaluation. While this class of inhibitors has shown clinical activity in AML patients, toxicities associated with irreversible inhibitors prohibited their further development. Reversible inhibitors of LSD1 have shown good potential for further development.
Other Leukemias
Differences between the NPM1 mutation and possiblydifferent leukemias are dependent on the types of cells involved.Myeloid leukemias originate in the broader NPM1 population.bone marrow and involve granulocytes, white and red blood cells.Lymphoid leukemias, including lymphomas, originate in the lymph nodes and lymphoid tissue but involve immune cells including lymphocytes, T cells and B cells.MDS are group of bone marrow failure disorders that have various subtypes with variable onsets, prognoses and risks of developing leukemia.
| 6 |
RASP-101
Prof. Brunangelo Falini, Dr. Lorenzo Brunetti, and Prof. Maria Paola Martelli (N Engl J Med, 2015 373(12), 373(12):1180-2)1180-2) hypothesized that NPM1-mutatedNPM1-mutated AML cells might be vulnerable to a drug like dactinomycinthat triggers a nucleolar stress response. In an initial evaluation, a single patient was treated for six cycles of five consecutive daily intravenous doses of 12.5 µg/kg at intervals of 3 to 4 weeks. Morphologic and immunohistochemical complete remissionCR, was evident after the fourth cycle, and an assay for mutant copies of NPMA showed molecular complete remission after the fourth cycle. In a subsequent Phase II2 clinical trial (EudraCT number 2014-000693-18), completed in two and a half years,refractory/relapse (R/R) AML patients carrying NPM1 gene mutation, treatment with refractory or relapsed NPM1-mutated AML were treated with dactinomycinAct D at 15 µg/kg/day for 5 days every 28 days induced CR in4 out of the 9 evaluable patients (44.4%) with only 1 or 2 cycles of 5 consecutive days.therapy. One patient underwent haploidentical allogeneic peripheral blood stem cell, or PBSC, transplantation at 3 months after CR achievement and is alive in molecular CR [minimal residual disease (MRD)-negative] after 24 months..
RASP-101 is a nanoparticle-based formulation program that focuses on control release of ACT D to achieve therapeutic benefit.ACT D was previously approved by the FDA for treatment of Wilm’s Tumor, Ewing’s Sarcoma, Metastatic Nonseminomatous Testicular Cancer, Gestational Trophoblastic Neoplastic, Childhood Rhabdomyosarcoma and Regional Perfusion in Locally Recurrent and Locoregional Solid Malignancies under the trade name, Cosmegen® (Ovation Pharmaceuticals). Our approach is to reformulate ACT D with a nanoparticle-based formulation to reduce toxicity and maximize efficacy. We believe RASP-101 is a candidate for submission via the 505(b)(2) regulatory pathway for approval, which has shortened approval timelines for regulatory pathway for repurpose of already approved drugs for other disease indications.
RASP-101 is based on encapsulated polymeric nanoparticles with desirable safety and PK profile.Preclinical animal studies with all four prototypes were conducted to evaluate drug release profile, PK and tolerability side by side with unformulated ACT D.
The maximum plasma concentration of ACT D was higher for the encapsulated polymeric nanoparticle formulations compared to free ACT D dosing.Although the observed plasma concentration for the free ACT D was lower than the formulated versions, higher mortality was observed in the free drug group of animals.After 24 hours, substantial plasma levels were observed in nanoparticle formulations A complete responseand C.Formulation A had highest systemic levels of ACT D and minimal adverse effects and was achievedselected as the lead formulation.
A decrease in 40%body weights of animals dosed with free ACT D was recorded subsequent to dosing and animal did not recover from body weight loss even at 7 days post-dosing.Dosing with nanoparticle-formulated ACT D at the same dose showed no drug-related body weight loss.
The rationale for developing RASP-101 was to permit weekly or biweekly dosing to limit drug toxicity associated with daily dosing in human subjects. Animal studies performed to date suggest that at the dose at which 80% of rats die within 13 days of administering unformulated RASP-101, no mortality was observed in groups of animals administered the same dose of RASP-101 formulated with polymeric nanoparticles (Figure 3). Only 1 animal in the unformulated group of animals survived at day 13.
Figure 3Survival of Rats Administered free ACT D or nanoparticle formulated ACT
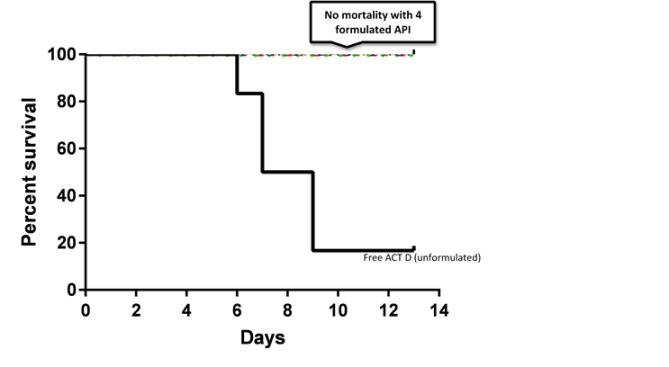
The total accumulation of ACT D in tissues was less than 2% of the patients.
RASP-201 (LSD1)
LSD1 is an enzyme that demethylates (remove methyl groups) lysine side chain of histones. The levels of LSD1 are elevated (overexpressed) in several human cancers as compared to healthy normal adults. In 2012, a paper publishedAML, elevated levels of LSD1 are observed in Nature Medicine established thatless differentiated blood cells (immature cells). Inhibition of the enzyme activity was found to promote differentiation. Hence, inhibitors of an enzyme involved in the epigenetic regulation of gene expression known as LSD1 or KDM1ALSD1 are promising therapeutic candidates that can make drug-insensitive forms of AML, due to reduced levels of methylated histones, responsive to treatment with all-trans-retinoic acid, (ATRA).or ATRA isan agent that induces differentiation (maturation)and used to treat a subtype of AML called acute promyelocytic leukemia, (APL), but it is normally not effective in non-APL AML because the drug does not cause proper transcriptional activation of retinoic acid receptor target genes. This is a result of reduced methylation (specifically histone 3lysine 4 (H3K4) demethylation) on the promoter regions of these target genes. Therefore, the authors of the article hypothesized that inhibiting LSD1 might facilitate ATRA-induced differentiation of AML cells, which is known to halt the division of these cells. The research highlights a crosstalk between the ATRA-induced myeloid differentiation pathway and H3K4 methylation, and suggests that ATRA combined with LSD1 inhibitors might be therapeutically beneficial in AML.
Working from this premise, Dr. Pelicci’s groupwe executed a research agreement with TES Pharma for screening of compounds and to conduct animal studies to identify lead compounds with suitable druggability properties. TES Pharma has developedscreened ~34000 compounds across 4 chemical series for their ability to inhibit LSD1in vitro. The hit rate was ~1%. The hits from this assay were expanded and IC50 (half maximal inhibitory concentration) values were determined for 115 compounds. One chemical class namely, Thienopyrrole series was prioritized for further expansion and in vitro characterization.
We have filed patent applications covering this novel irreversible and reversible LSD1 regulators which have shown appropriatepotent inhibitor class. The current lead compound DDP_43242 inhibitory effect on the LSD1 gene
These studies have facilitated the selection of aDDP_43242 as the current lead compound. Additional absorption, distribution, metabolism and then aexcretion, or ADME, and selectivity characterization is underway for drug candidate selection for clinical trials by Rasna.trials. We anticipate filing an investigation new drug application, or IND, in 2019. We believe that this breakthrough program may have significant benefits across all forms of leukemia.
RASP-301 (NPM1)
The NPM1 gene provides instructions for making a protein called nucleophosmin (NPM) which is found in a small region inside the nucleus of the cell called the nucleolus.Nucleophosmin shuttles back and forth between the nucleus and the fluid surrounding it (the cytoplasm).Nucleophosmin helps to transport other proteins from ribosomes (sites of protein synthesis) through the membrane surrounding the nucleus.The NPM-1 gene is mutated in approximately 35% of new cases of AML. However, mutations in the NPM1 gene, result in dislocation of NPM1 protein in the cytoplasm. NPM1 mutation may be an early event in development of leukemia.Given that NPM1 is thought to have a tumor-suppressor function, alterations in its localization in the cell (from the nucleus to the cytoplasm) may be crucial for the cell to change from normal to cancerous state. Our approach is to identify small molecule drugs that physically interfere with the aggregation of NPM1 which is important for normal function. RASP-301 is our innovative, first- in-class, oral, small molecule, potent inhibitor that disrupts NPM1-1 oligomerization (aggregation of individual subunits) and has the potential to treat refractory (resistant to treatment) AML with reduced toxicity at low dose levels The current lead candidate, TES-2169, exhibits cytotoxic (toxic to cancer cells) effects at nanomolar concentrations against AML cells in culture and was not cytotoxic to normal cells at the same concentrations.In vivo usefulness of these compounds in AML murine (mouse) models has been evaluated confirming the druggability of the target and its potential to treat refractory AML.
| 8 |
Strategy
Our strategy is to target master regulators of cancer through deep knowledge of highly conserved pathways that are estimatedcommon to leukemia sub-types. Employing a multi-pronged approach, our programs are focused on three druggable intervention points with a potential to improve safety and efficacy of current AML mono and/or combination therapies. For the RASP-101 program, We plan to seek approval from FDA for submission via the 505 (b)(2) regulatory pathway employing a single Phase 1 study and a bridging Phase 3 pivotal study.For the RASP-201 program, the lead reversible LSD-1 candidate has been identified and manufacturing will be scaled for production of cGMP supplies for animal safety/toxicology studies, Phase 1, Phase 2 and Phase 3 clinical studies to support submission via the traditional 505(b)(1) regulatory pathway used for new chemical entities, or NCEs.We expect to use a similar pathway for the RASP-301 program, following identification of the lead drug candidate.
| ● | Rapidly develop and seek approval for our lead drug candidate, RASP-101 taking advantage of shortened timelines associated with the 505(b)(2) regulatory pathway. Schedule meeting with FDA to discuss suitability for this pathway. The 505(b)(2) pathway is applicable to reformulation of an approved drug used for a different disease indication. RASP-101 is a nanoparticulate reformulation of Cosmegen ® (Ovation Pharamaceutical) used for treatment of Wilm’s tumor and other cancers. Development and approval via the 505(b)(2) pathway can result in shortened timelines (3-4 years from development to approval in some instances) compared to the traditional 505(b)(1) pathway used for regulatory approval of NCEs (10-15 years). | |
| ● | Determine feasibility of Orphan Drug Designation for RASP-101 and other pipeline products used for treating AML. Schedule meeting with FDA to discuss suitability for this pathway.Orphan Drug Designation provides special status for drug products used to treat rare disease (“orphan indications”) defined as affecting fewer than 200,000 people in US and less than 5 per 10,000 people in EU. Incentives for developing orphan drugs include periods of marketing exclusivity (7 years exclusivity in US, 10 years in EU), tax credits up to 50% of development costs, waived FDA fees and other clinical trial tax incentives. | |
| ● | Determine feasibility of Breakthrough Therapy and Accelerated Approval for RASP-101 and other pipeline products. Schedule meeting with FDA to discuss suitability for this pathway. Breakthrough therapy designation is available from the FDA to drugs or drug combinations used to treat serious or life-threatening disease conditions based on preliminary clinical evidence that the drug may offer substantial improvement over existing therapies.FDA may grant priority approval to breakthrough drug indications.FDA may also grant accelerated approval and priority review for drugs that fill an unmet medical need. An advantage to this designation is that clinical trials may use surrogate endpoints to predict clinical benefit, requiring less time than other objective endpoints such as overall survival, or OS. | |
| ● | Complete development of orally-available, small molecule, highly-selective, reversible inhibitor of LSD1 (RASP-201) and file IND by 2019. Manufacture of the lead RASP -201 candidate will be scaled to supply animal safety/toxicology studies and Phase 1, 2 and 3 clinical studies following the traditional 505(b)(1) pathway used for marketing approval of NCEs. | |
| ● | Selection of a lead candidate for first in class NPM1 inhibitor (RASP-301) by the first quarter of 2019. | |
| ● | Evaluate strategic opportunities to accelerate development timelines and maximize the commercial potential of the drug candidates. Such opportunities may include partnering to achieve business objectives and achieve maximum return on investment. |
License Agreement
On November 10, 2016, Prof. Brunangelo Falini and Prof. Maria Paola Martelli and us entered into an exclusive license agreement pursuant to which we licensed certain patent rights owned by Prof. Falini and Prof. Martelli related to the use or reformulation of ACT D for the treatment of cancer.The agreement provides for up to €500,000 of milestone payment to be approximately 19,000paid upon achievement of clinical and regulatory milestones.In addition, the agreement provides for a low single digit royalty to be paid on net sales.The agreement may be terminated by us at any time upon 90 days prior written notice.In addition, each party to the agreement may terminate the agreement at any time upon a material breach of the agreement by the other party; provided that the other party shall have a 90 day period to cure the material breach.
Research Agreement
On December 17, 2013 TES Pharma S.R.L. (“TES”) and our predecessor, Arna Therapeutics Limited, entered into a research agreement pursuant to which TES agreed to perform research related to the development of products and services associated with NPM1 and AML.The initial term of the agreement was for two years and was extended by amendment on May 3, 2016 for an additional year.On September 8, 2016, the agreement was assigned to our subsidiary, Rasna Research, Inc.The agreement was further extended on March 24, 2017 until August 30, 2017. We are currently in discussions with TES Pharma S.R.L, profiling the next stage of research activity to be included in a new AML casesresearch agreement.
Manufacturing
Wedonothaveanymanufacturingfacilitiesorpersonnel.Wecurrentlyrely,andexpecttocontinuetorely,onthird partiesforthemanufacturingofourproductcandidatesforpreclinical as wellasforcommercial manufacturing if our product candidatesreceive marketingapproval.
Commercialization
We plan to retain our worldwide commercialization rights for some of our key product candidates while for other product candidates we might consider partnership opportunities to maximize returns.
While we currently have no sales, marketing or commercial product distribution capabilities and have no experience as a company in commercializing products, we intend to build our own commercialization organization and capabilities over time. When appropriate, we will decide whether to build a specialty sales force to manage commercialization for these product candidates on our own or in combination with a larger pharmaceutical partner, to maximize patient coverage in the United States each year and to support global expansion especially as our programs have substantial opportunity for additional follow-up indications alone or in combinations.
As product candidates advance through our pipeline, our commercial plans may change. Clinical data, the projected cost of treatment attributable to drug administrations for each patient over the coursesize of the disease can be estimated at US $100,000 per patient or US $1,900,000,000development programs, the size of the target market, the size of a commercial infrastructure and manufacturing needs may all influence our United States, European Union and rest-of-world strategies
Government Regulation
The FDA and comparable regulatory authorities in state and local jurisdictions and in other countries impose substantial and burdensome requirements upon companies involved in the aggregate from the different treatment cycles. Of this it is estimated that 50%clinical development, manufacture, marketing and distribution of the costs will continue to be attributable to standard of care chemotherapy. Thus, it is estimated that the US market potential for AML is $1 billion annually. The rest of the world is estimated to constitute 50% of the AML market; thus, the total annual world market is estimated to be $2 billion. If Rasna’s NPM1 targeted therapy proves beneficial to only that 20% of AML patients with mutated NPM1 genes, the addressable market will be diminished. However, in that event, Rasna’s NPM1 targeted therapy would then be the only therapy which address that sub-population and may command higher pricing and reimbursement.
| Type | Total | Male | Female | |||||||||
| Acute Lymphoblasti Leukemia | 6,020 | 3,140 | 2,880 | |||||||||
| Chronic Lymphocytic Leukemia | 15,720 | 9,100 | 6,620 | |||||||||
| Acute Myeloid Leukemia | 18,860 | 11,530 | 7,330 | |||||||||
| Chronic Myeloid Leukemia | 5,980 | 3,130 | 2,850 | |||||||||
| Other Leukemia | 5,800 | 3,200 | 2,600 | |||||||||
| Total Estimated New Cases | 52,380 | 30,100 | 22,280 | |||||||||
U.S. Government Regulation of Drug Products
In the United States, the FDA regulates drugs under the Federal Food, Drug, and exportCosmetic Act, or FDCA, and its implementing regulations. The process of pharmaceutical products.obtaining regulatory approvals and the subsequent compliance with applicable federal, state, local and foreign statutes and regulations requires the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval, may subject a companyan applicant to a variety of administrative or judicial sanctions, such as FDAthe FDA’s refusal to approve pending new drug applications, or NDAs, or biologic license applications, or BLAs,withdrawal of an approval, imposition of a clinical hold, issuance of warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement or civil penalties, andor criminal prosecution.penalties.
| 10 |
The process required by the FDA before product developmentcandidates may be marketed in the United States typicallygenerally involves preclinical laboratory and animal tests, the following:
- submission to the
FDA/EMAFDA of anInvestigational New Drug (IND)IND application which must become effective before human clinical trials may begin and must be updated annually; - completion of extensive preclinical laboratory tests and preclinical animal studies, all performed in accordance with the FDA’s GLP regulations. Preclinical testing generally includes evaluation of our products in the laboratory or in animals to characterize the product and determine safety and efficacy;
- approval by an independent institutional review board, or IRB, at each clinical site before each trial may
commence, andbe initiated; - performance of adequate and well-controlled human clinical trials in accordance with good clinical practice, or GCP,requirements to establish the safety and
effectiveness of the drug or biologic for each indication for which FDA/EMA approval is sought. Satisfaction of FDA/EMA pre-market approval requirements typically takes many years (typically between 5-7 years post an IND submission) and the actual time required may vary substantially based upon the type, complexity and noveltyefficacy of the product candidate for each proposed indication; - submission to the FDA of a NDA after completion of all pivotal clinical trials;
- a determination by the FDA within 60 days of its receipt of an NDA to accept the filing for review;
- satisfactory completion of an FDA advisory committee review, if applicable;
- satisfactory completion of an FDA pre-approval inspection of the manufacturing facilities at which the active pharmaceutical ingredient, or
disease.
Preclinical testsstudies include laboratory evaluation of product chemistry, toxicity and formulation, as well as animal trialsstudies to assess potential safety and efficacy. An IND is a request for authorization from the characteristicsFDA to administer an investigational drug product to humans. The central focus of an IND submission is on the general investigational plan and potential pharmacology and toxicity of the product.protocol(s) for human studies. The conduct of the preclinical tests must comply with federal regulations and requirements including good laboratory practices. TheIND also includes results of preclinical testing are submittedstudies or other human studies, as appropriate, as well as manufacturing information, analytical data and any available clinical data or literature to support the use of the investigational new drug. An IND must become effective before human clinical trials may begin. An IND will automatically become effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions related to the proposed clinical trials. In such a case, the IND may be placed on clinical hold and the IND sponsor and the FDA as partmust resolve any outstanding concerns or questions before clinical trials can begin. Accordingly, submission of an IND along with other information, including information about product chemistry, manufacturing and controls, andmay or may not result in the FDA allowing clinical trials to commence. As a proposed clinical trial protocol. Long term preclinical tests, such as animal tests of reproductive toxicity and carcinogenicity, may continue after the IND is submitted.
Clinical trials involve the administration of the new investigational drug to healthy volunteers or patientshuman subjects under the supervision of a qualified investigator.investigators in accordance with GCPs, which include the requirement that all research subjects provide their informed consent for their participation in any clinical trial. Clinical trials must beare conducted in compliance with federal regulations and good clinical practices, or GCP, as well as under protocols detailing, among other things, the objectives of the trial,study, the parameters to be used in monitoring safety, and the effectivenessefficacy criteria to be evaluated. EachA protocol involving testing on U.S. patientsfor each clinical trial and any subsequent protocol amendments must be submitted to the FDA as part of the IND.
Additionally, approval must also be obtained from each clinical trial site’s Institutional Review Board, or IRB, before the trials may be initiated, and the IRB must monitor the study until completed. There are also requirements governing the reporting of ongoing clinical trials and clinical trial results to public registries.
The clinical investigation of a drug is generally divided into three phases. Although the phases are usually conducted sequentially, they may overlap or be combined. The three phases of an investigation are as follows:
| 1. | Phase 1. Phase 1 includes the initial introduction of an investigational new drug into humans. Phase 1 clinical trials are typically closely monitored and may be conducted in patients with the target disease or condition or in healthy volunteers. These studies are designed to evaluate the safety, dosage tolerance, metabolism and pharmacologic actions of the investigational drug in humans, the side effects associated with increasing doses, and if possible, to gain early evidence on effectiveness. During Phase 1 clinical trials, sufficient information about the investigational drug’s pharmacokinetics and pharmacological effects may be obtained to permit the design of well-controlled and scientifically valid Phase 2 clinical trials. | |
| 2. | Phase 2. Phase 2 includes controlled clinical trials conducted to preliminarily or further evaluate the effectiveness of the investigational drug for a particular indication(s) in patients with the disease or condition under study, to determine dosage tolerance and optimal dosage, and to identify possible adverse side effects and safety risks associated with the drug. Phase 2 clinical trials are typically well-controlled, closely monitored, and conducted in a limited patient population. |
| 11 |
| 3. | Phase 3. Phase 3 clinical trials are generally controlled clinical trials conducted in an expanded patient population generally at geographically dispersed clinical trial sites. They are performed after preliminary evidence suggesting effectiveness of the drug has been obtained, and are intended to further evaluate dosage, clinical effectiveness and safety, to establish the overall benefit-risk relationship of the investigational drug product, and to provide an adequate basis for product approval. |
A pivotal study is any clinical study, which adequately meets regulatory agency requirements for the evaluation of a drug candidate’s efficacy and safety such that it can be used to justify the approval of the product. Generally, pivotal studies are Phase 3 studies but may also be Phase 2 studies if the trial design provides a well-controlled and reliable assessment of clinical benefit, particularly in situations where there is an unmet medical need.
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and more frequently if serious adverse events occur. Phase 1, Phase 2 and Phase 3 clinical trials may not be completed successfully within any specified period, or at all.The FDA, the IRB, or the clinical trial sponsor may order the temporarysuspend or permanent discontinuation ofterminate a clinical trial at any time or impose other sanctions if it believeson various grounds, including a finding that the research subjects are being exposed to an unacceptable health risk. Additionally, some clinical trials are overseen by an independent group of qualified experts organized by the clinical trial issponsor, known as a data safety monitoring board or committee. This group provides authorization for whether or not being conducteda trial may move forward at designated check points based on access to certain data from the study. We may also suspend or terminate a clinical trial based on evolving business objectives and/or competitive climate.
Assuming successful completion of all required testing in accordance with FDAall applicable regulatory requirements, or presents an unacceptable riskdetailed investigational drug product information is submitted to the clinical trial patients. FDA in the form of an NDA requesting approval to market the product for one or more indications.
The clinical trial protocolapplication includes all relevant data available from pertinent preclinical and informed consent information for patients in clinical trials, must also be submittedincluding negative or ambiguous results as well as positive findings, together with detailed information relating to an institutional review board,the product’s chemistry, manufacturing, controls and proposed labeling, among other things. Data can come from company-sponsored clinical trials intended to test the safety and effectiveness of a use of a product, or IRB, for approval. An IRB may also require the clinical trial at the site to be halted, either temporarily or permanently, for failure to comply with the IRB’s requirements, or may impose other conditions.
In most cases, the submission of an NDA is subject to a substantial application user fee. Under the Prescription Drug User Fee Act, or PDUFA, guidelines that are currently in effect, the FDA has a goal of ten months from the date of “filing” of a standard NDA for a particular indication or indications, dosage tolerancenew molecular entity to review and optimum dosage, and identify common adverse effects and safety risks. Inact on the case of product candidates for severe or life-threatening diseases such as pneumonia,submission. This review typically takes twelve months from the initial human testingdate the NDA is often conducted in patients rather than in healthy volunteers.
In addition, under the overall benefit-risk relationshipPediatric Research Equity Act of 2003, or PREA, as amended and reauthorized, certain NDAs or supplements to an NDA must contain data that are adequate to assess the safety and effectiveness of the investigational drug for the claimed indications in all relevant pediatric subpopulations, and to provide adequate informationsupport dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may, on its labeling.
The FDA also may require submission of a risk evaluation and mitigation strategy, or REMS, plan to ensure that the benefits of the product may begin in the United States.drug outweigh its risks. The marketing application mustREMS plan could include the resultsmedication guides, physician communication plans, assessment plans, and/or elements to assure safe use, such as restricted distribution methods, patient registries, or other risk minimization tools.
The FDA conducts a preliminary review of all preclinical, clinical and other testing and a compilation of data relating toNDAs within the product’s pharmacology, chemistry, manufacture, and controls.
The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving a marketing application,an NDA, the FDA will typicallymay inspect one or more clinical trial sites to assure compliance with GCP.
| 12 |
After the FDA evaluates the NDA or BLA and theconducts inspections of manufacturing facilities where the drug product and/or its active pharmaceutical ingredient, or API, will be produced, it issuesmay issue an approval letter or a complete response letter. A complete response letter outlines the deficiencies in the submission and may require substantial additional testing or information in order for the FDA to reconsider the application. If and when those deficiencies have been addressed in a resubmission of the marketing application, the FDA will re-initiate review. If the FDA is satisfied that the deficiencies have been addressed, the agency will issue an approval letter. The FDA has committed to reviewing such resubmissions in two or six months depending on the type of information included. It is not unusual for the FDA to issue a complete response letter because it believes that the drug product is not safe enough or effective enough or because it does not believe that the data submitted are reliable or conclusive.
After regulatory approval of a drug product is obtained, a company is required to comply with a number of post-approval requirements. As a holder of an approved NDA, we would be required to report, among other things, certain adverse reactions and production problems to the FDA, to provide updated safety and efficacy information, and may imposeto comply with requirements concerning advertising and promotional labeling for any of our products. Also, quality control and manufacturing procedures must continue to conform to cGMP after approval to ensure and preserve the long-term stability of the drug product. In addition, drug manufacturers and other conditions, including labeling restrictions, which can materially affectentities involved in the product’s potential marketmanufacture and profitability. Once granted, product approvals may be withdrawn ifdistribution of approved drugs are required to register their establishments with the FDA and state agencies, and are subject to periodic unannounced inspections by the FDA and these state agencies for compliance with cGMP requirements. In addition, changes to the manufacturing process are strictly regulated, and, depending on the significance of the change, may require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP and impose reporting and documentation requirements upon us and any third-party manufacturers that we may decide to use. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with cGMP and other aspects of regulatory standards is not maintainedcompliance.
We rely, and expect to continue to rely, on third parties for the production of clinical and commercial quantities of our product candidates. Future FDA and state inspections may identify compliance issues at our facilities or at the facilities of our contract manufacturers that may disrupt production or distribution, or require substantial resources to correct. In addition, discovery of previously unknown problems are identified following initial marketing.
| • | restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls; | |
| • | fines, warning letters or holds on post-approval clinical trials; | |
| • | refusal of the FDA to approve pending NDAs or supplements to approved NDAs, or suspension or revocation of product approvals; | |
| • | product seizure or detention, or refusal to permit the import or export of products; or | |
| • | injunctions or the imposition of civil or criminal penalties. |
Newly discovered or developed safety or effectiveness data may require changes to a product’s approved labeling, including the addition of new warnings and contraindications, and also may require the implementation of other risk management measures. The FDA closelystrictly regulates the post-approval marketing, labeling, advertising and promotion of therapeutic products including standards and regulations for direct-to-consumer advertising, off-label promotion, industry-sponsored scientific and educational activities and promotional activities involvingthat are placed on the internet.
Marketing Exclusivity
Market exclusivity provisions under the FDCA also can delay the submission or the approval of certain applications. The FDCA provides a five-year periodofnon-patentmarketingexclusivitywithintheUnitedStatestothefirstapplicanttoobtainapprovalofanNDAforanewchemicalentity.Adrugis a new chemical entity if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsiblefortheactionofthedrugsubstance.Duringtheexclusivityperiod,theFDAmaynotacceptforreviewanabbreviatednewdrugapplication,or ANDA, or a 505(b)(2) NDA submitted by another company for another version of such drug where the applicant does not own or have a legal right of referencetoallthedatarequiredforapproval.However,anapplicationmaybesubmittedafterfouryearsifitcontainsacertificationofpatentinvalidityor non-infringement. The FDCA also provides three years of marketing exclusivity for an NDA, 505(b)(2) NDA or supplement to an approved NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example, for new indications, dosages or strengths of an existing drug. This three-year exclusivity covers only the conditions associated with the new clinical investigations and does not prohibit the FDA from approving ANDAs for drugs containing the original active pharmaceuticalingredient.Five-yearandthree-yearexclusivitywillnotdelaythesubmissionorapprovalofafullNDA;however,anapplicantsubmitting a full NDA would be required to conduct or obtain a right of reference to all of the preclinical studies and adequate and well-controlled clinical trials necessarytodemonstratesafetyandeffectiveness.
The Hatch-Waxman Amendments: 505(b)(2) approval process
Section 505(b)(2) of the FDCA provides an alternate regulatory pathway to FDA approval for new or improved formulations or new uses of previously approved drug products. Specifically, Section 505(b)(2) permits the filing of an NDA where at least some of the conditions established ininformation required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference. The applicant may rely upon the FDA’s findings of safety and effectiveness for an approved product that acts as the Reference Listed Drug, or RLD. If the 505(b)(2) applicant can establish that reliance on FDA’s previous findings of safety and effectiveness is scientifically appropriate, it may eliminate the need to conduct certain preclinical or clinical studies of the new product. The FDA may also require 505(b)(2) applicants to perform additional studies or measurements to support the change from the RLD. The FDA may then approve the new product candidate for all or some of the labeled indications for which the referenced product has been approved, as well as for any new indication sought by the Section 505(b)(2) applicant.
In seeking approval for a drug through an NDA, including a 505(b)(2) NDA, applicants are required to list with the FDA certain patents whose claims cover the applicant’s product. Upon approval of an NDA, each of the patents listed in the application including changesfor the drug is then published in indications, labeling, or manufacturing processes or facilities, require submission and FDAthe Orange Book. Any applicant who files an ANDA seeking approval of a generic equivalent version of a drug listed in the Orange Book or a 505(b)(2) NDA referencing a drug listed in the Orange Book must certify to the FDA that (1) no patent information on the drug product that is the subject of the application has been submitted to the FDA; (2) such patent has expired; (3) the date on which such patent expires; or (4) such patent is invalid or will not be infringed upon by the manufacture, use or sale of the drug product for which the application is submitted. This last certification is known as a paragraph IV certification. A notice of the paragraph IV certification must be provided to each owner of the patent that is the subject of the certification and to the holder of the approved NDA to which the ANDA or 505(b)(2) application refers. The applicant may also elect to submit a ‘‘section viii’’ statement certifying that its proposed label does not contain (or carves out) any language regarding the patented method-of-use rather than certify to a listed method-of-use patent. If the reference NDA holder and patent owners assert a patent challenge directed to one of the Orange Book listed patents within 45 days of the receipt of the paragraph IV certification notice, the FDA is prohibited from approving the application until the earlier of 30 months from the receipt of the paragraph IV certification expiration of the patent, settlement of the lawsuit or a decision in the infringement case that is favorable to the applicant.The ANDA or 505(b)(2) application also will not be approved until any applicable non-patent exclusivity listed in the Orange Book for the branded reference drug has expired.
Our current and anticipated product candidates will be based on already approved active pharmaceutical ingredients, orAPIs, rather than new BLAchemical entities, and a formulation that has been through Phase 1 studies. Accordingly, we expect to be able to rely on information from previously conducted formulation studies involving our clinical development plans and our NDA submissions. For product candidates that involve novel fixed-dose combinations of existing drugs or BLA supplement, before the change can be implemented. A BLA supplement for studies of an existing product or product candidate in a new indication, typically requires clinical data similarwe believe we generally will be able to that in the original application, andinitiate Phase 2/3 studies without conducting any new non-clinical or Phase 1 studies, though the FDA uses the same proceduresmay not agree with our conclusions and actions in reviewing BLA supplements as it does in reviewing BLAs. We cannot be certain that the FDAmay require us to conduct additional clinical or anypreclinical studies prior to initiating Phase 3 or other regulatory agency will grant approval forpivotal clinical trials. In those instances where our product candidates for any other indications or any other product candidate for any indication onis a timely basis, if at all.
U.S. Foreign Corrupt Practices Act
The U.S. Foreign Corrupt Practices Act, to cGMPs after approval. Manufacturers and certain of their subcontractors are required to register their establishments with the FDA and certain state agencies, andwhich we are subject, prohibits corporations and individuals from engaging in certain activities to periodic unannounced inspections byobtain or retain business or to influence a person working in an official capacity. It is illegal to pay, offer to pay or authorize the FDA during which the agency inspects manufacturing facilitiespayment of anything of value to assess compliance with cGMPs. Accordingly, manufacturers must continueany foreign government official, government staff member, political party or political candidate to expend time, money and effortobtain or retain business or to otherwise influence a person working in the areas of production and quality control to maintain compliance with cGMPs. Regulatory authorities may withdraw product approvals or request product recalls if a company fails to comply with regulatory standards, if it encounters problems following initial marketing, or if previously unrecognized problems are subsequently discovered.
Federal and State Fraud and Abuse Laws
Healthcare providers, physicians and third-party payors play a primary role in the recommendation and prescription of drug and biologic product candidates which obtain marketing approval. In addition to FDA restrictions on marketing of pharmaceutical products, severalpharmaceutical manufacturers are exposed, directly, or indirectly, through customers, to broadly applicable fraud and abuse and other types of statehealthcare laws and federal laws have been applied to restrict certain marketing practices inregulations that may affect the biopharmaceuticalbusiness or financial arrangements and medical device industries in recent years.relationships through which a pharmaceutical manufacturer can market, sell and distribute drug and biologic products. These laws include, anti-kickback statutes and false claims statutes.
The federal health care program anti-kickback statuteAnti-Kickback Statute which prohibits, any person or entity from, among other things, knowingly and willfully offering, paying, soliciting, or receiving any remuneration, directly or indirectly, overtly or covertly, in cash or in-kind, to induce or in returnreward either the referring of an individual for, or the purchasing, leasing, ordering, or arranging for the purchase, lease, or order of any health carehealthcare item or service reimbursable, in whole or in part, under Medicare, Medicaid, or any other federally financed healthcare programs.program. The term “remuneration” has been broadly interpreted to include anything of value. This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on one hand and prescribers, purchasers, and formulary managers on the other.other hand. Although there are a number of statutory exemptions and regulatory safe harbors protecting certain common activities from prosecution, the exemptions and safe harbors are drawn narrowly, and practices that involve remuneration intended to induce prescribing, purchases, or recommendations may be subject to scrutiny if they do not qualify for an exemption or safe harbor. Our practices may not in all cases meet all of the criteria for safe harbor protection from anti-kickback liability.
The federal false claims and civil monetary penalty laws, prohibitincluding the Federal False Claims Act, which imposes significant penalties and can be enforced by private citizens through civil qui tam actions, prohibits any person or entity from, among other things, knowingly presenting, or causing to be presented, a false, fictitious or fraudulent claim for payment to the federal government, or knowingly making, using or causing to be made, a false statement or record material to get a false or fraudulent claim paid.to avoid, decrease or conceal an obligation to pay money to the federal government. In addition, a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act. As a result of a modification made by the Fraud Enforcement and Recovery Act of 2009, a claim includes "any request or demand" for money or property presented to the U.S. government. In addition, manufacturers can be held liable under the False Claims Act even when they do not submit claims directly to government payors if they are deemed to "cause" the submission of false or fraudulent claims. Criminal prosecution is also possible for making or presenting a false, fictitious or fraudulent claim to the federal government. Recently, several pharmaceutical and other health carehealthcare companies have been prosecuted under these laws for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. Other companies have been prosecuted for causing false claims to be submitted because of the Company’scompany’s marketing of the product for unapproved, and thus non-reimbursable, uses.
The majorityfederal Health Insurance Portability and Accountability Act of states also have statutes1996, or HIPAA, which, among other things, imposes criminal liability for executing or attempting to execute a scheme to defraud any healthcare benefit program, including private third-party payors, knowingly and willfully embezzling or stealing from a healthcare benefit program, willfully obstructing a criminal investigation of a healthcare offense, and creates federal criminal laws that prohibit knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statements or representations, or making or using any false writing or document knowing the same to contain any materially false, fictitious or fraudulent statement or entry in connection with the delivery of, or payment for, benefits, items or services.
HIPAA, as amended by the Health Information Technology and Clinical Health Act of 2009, or HITECH, and its implementing regulations, similarwhich impose certain requirements relating to the privacy, security, transmission and breach reporting of individually identifiable health information upon entities subject to the law, such as health plans, healthcare clearinghouses and healthcare providers and their respective business associates that perform services for them that involve individually identifiable health information. HITECH also created new tiers of civil monetary penalties, amended HIPAA to make civil and criminal penalties directly applicable to business associates, and gave state attorneys general new authority to file civil actions for damages or injunctions in U.S. federal courts to enforce the federal HIPAA laws and seek attorneys' fees and costs associated with pursuing federal civil actions.
The federal physician payment transparency requirements, sometimes referred to as the “Physician Payments Sunshine Act,” and its implementing regulations, which require certain manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) to report annually to the United States Department of Health and Human Services, or HHS, information related to payments or other transfers of value made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members.
State and foreign law equivalents of each of the above federal laws, such as anti-kickback law and false claims laws, whichthat may impose similar or more prohibitive restrictions, and may apply to items andor services reimbursed under Medicaidby non-governmental third-party payors, including private insurers.
State and foreign laws that require pharmaceutical companies to implement compliance programs, comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government, or to track and report gifts, compensation and other remuneration provided to physicians and other healthcare providers, and other federal, state programs,and foreign laws that govern the privacy and security of health information or personally identifiable information in several states, apply regardlesscertain circumstances, including state health information privacy and data breach notification laws which govern the collection, use, disclosure, and protection of the payor. Sanctions under these federalhealth-related and state laws may include civil monetary penalties, exclusionother personal information, many of a manufacturer’s productswhich differ from reimbursement under government programs, criminal fines,each other in significant ways and imprisonment.
Because of the breadth of these laws and the narrowness of the safe harbors, it is possible that some of our business activities couldcan be subject to challenge under one or more of such laws. SuchThe scope and enforcement of each of these laws is uncertain and subject to rapid change in the current environment of healthcare reform, especially in light of the lack of applicable precedent and regulations.Federal and state enforcement bodies have recently increased their scrutiny of interactions between healthcare companies and healthcare providers, which has led to a challengenumber of investigations, prosecutions, convictions and settlements in the healthcare industry.
Ensuring that business arrangements with third parties comply with applicable healthcare laws and regulations is costly and time consuming. If business operations are found to be in violation of any of the laws described above or any other applicable governmental regulations a pharmaceutical manufacturer may be subject to penalties, including civil, criminal and administrative penalties, damages, fines, disgorgement, individual imprisonment, exclusion from governmental funded healthcare programs, such as Medicare and Medicaid, contractual damages, reputational harm, diminished profits and future earnings, additional reporting obligations and oversight if subject to a corporate integrity agreement or other agreement to resolve allegations of non-compliance with these laws, and curtailment or restructuring of operations, any of which could adversely affect a pharmaceutical manufacturer’s ability to operate its business and the results of its operations.
Healthcare Reform in the United States
In the United States, there have been, and continue to be, a material adversenumber of legislative and regulatory changes and proposed changes to the healthcare system that could affect the future results of pharmaceutical manufactures’ operations. In particular, there have been and continue to be a number of initiatives at the federal and state levels that seek to reduce healthcare costs. Most recently, the Patient Protection and Affordable Care Act, or PPACA, was enacted in March 2010, which includes measures to significantly change the way healthcare is financed by both governmental and private insurers. Among the provisions of the PPACA of greatest importance to the pharmaceutical and biotechnology industry are the following:
•an annual, nondeductible fee on any entity that manufactures or imports certain branded prescription drugs and biologic agents, apportioned among these entities according to their market share in certain government healthcare programs;
•implementation of the federal physician payment transparency requirements, sometimes referred to as the “Physician Payments Sunshine Act”;
•a licensure framework for follow-on biologic products;
•a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research;
•establishment of a Center for Medicare Innovation at the Centers for Medicare & Medicaid Services to test innovative payment and service delivery models to lower Medicare and Medicaid spending, potentially including prescription drug spending;
•an increase in the statutory minimum rebates a manufacturer must pay under the Medicaid Drug Rebate Program, to 23.1% and 13% of the average manufacturer price for most branded and generic drugs, respectively and capped the total rebate amount for innovator drugs at 100% of the Average Manufacturer Price, or AMP;
•a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for certain drugs and biologics, including our product candidates, that are inhaled, infused, instilled, implanted or injected;
•extension of manufacturers’ Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations;
•expansion of eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to additional individuals and by adding new mandatory eligibility categories for individuals with income at or below 133% of the federal poverty level, thereby potentially increasing manufacturers’ Medicaid rebate liability;
•a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50% point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D; and
•expansion of the entities eligible for discounts under the Public Health program.
Some of the provisions of the PPACA have yet to be implemented, and there have been legal and political challenges to certain aspects of the PPACA. Since January 2017, President Trump has signed two executive orders and other directives designed to delay, circumvent, or loosen certain requirements mandated by the PPACA. Concurrently, Congress has considered legislation that would repeal or repeal and replace all or part of the PPACA. While Congress has not passed repeal legislation, the Tax Cuts and Jobs Act of 2017 includes a provision repealing, effective January 1, 2019, the tax-based shared responsibility payment imposed by the PPACA on certain individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate”. Congress may consider other legislation to repeal or replace elements of the PPACA.
Many of the details regarding the implementation of the PPACA are yet to be determined, and at this time, the full effect that the PPACA would have on oura pharmaceutical manufacturer remains unclear. In particular, there is uncertainty surrounding the applicability of the biosimilars provisions under the PPACA. The FDA has issued several guidance documents, but no implementing regulations, on biosimilars. A number of biosimilar applications have been approved over the past few years. The regulations that are ultimately promulgated and their implementation are likely to have considerable impact on the way pharmaceutical manufacturers conduct their business and may require changes to current strategies. A biosimilar is a biological product that is highly similar to an approved drug notwithstanding minor differences in clinically inactive components, and for which there are no clinically meaningful differences between the biological product and the approved drug in terms of the safety, purity, and potency of the product.
Individual states have become increasingly aggressive in passing legislation and implementing regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access, and marketing cost disclosure and transparency measures, and to encourage importation from other countries and bulk purchasing. Legally mandated price controls on payment amounts by third-party payors or other restrictions could harm a pharmaceutical manufacturer’s business, results of operations, financial condition and prospects. In addition, regional healthcare authorities and individual hospitals are increasingly using bidding procedures to determine what pharmaceutical products and which suppliers will be included in their prescription drug and other healthcare programs. This could reduce ultimate demand for certain products or put pressure product pricing, which could negatively affect a pharmaceutical manufacturer’s business, results of operations.
In addition, given recent federal and state government initiatives directed at lowering the total cost of healthcare, Congress and state legislatures will likely continue to focus on healthcare reform, the cost of prescription drugs and biologics and the reform of the Medicare and Medicaid programs. While no one cannot predict the full outcome of any such legislation, it may result in decreased reimbursement for drugs and biologics, which may further exacerbate industry-wide pressure to reduce prescription drug prices. This could harm a pharmaceutical manufacturer’s ability to generate revenue. Increases in importation or re-importation of pharmaceutical products from foreign countries into the United States could put competitive pressure on a pharmaceutical manufacturer’s ability to profitably price products, which, in turn, could adversely affect business, results of operations, financial condition and prospects. A pharmaceutical manufacturer might elect not to seek approval for or market products in foreign jurisdictions in order to minimize the risk of re-importation, which could also reduce the revenue generated from product sales. It is also possible that other legislative proposals having similar effects will be adopted.
Furthermore, regulatory authorities’ assessment of the data and results required to demonstrate safety and efficacy can change over time and can be affected by many factors, such as the emergence of new information, including on other products, changing policies and agency funding, staffing and leadership. No one can be sure whether future changes to the regulatory environment will be favorable or unfavorable to business prospects. For example, average review times at the FDA for marketing approval applications can be affected by a variety of factors, including budget and funding levels and statutory, regulatory and policy changes.
International Regulations
In addition to regulations in the European Union
To obtain regulatory approval of an investigational drug under European Union for example, thereregulatory systems, we must submit a marketing authorization application. The application used to file the NDA in the United States is a centralized approval proceduresimilar to that authorizes marketingrequired in Europe, with the exception of, a product in all among other things, country-specific document requirements. For other countries outside of the European Union, which includes most majorsuch as countries in Europe. Eastern Europe, Latin America or Asia, the requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. In all cases, again, the clinical trials are conducted in accordance with GCP and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki.
If thiswe fail to comply with applicable foreign regulatory requirements, we may be subject to, among other things, fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
Centralized Procedure
The European Medicines Agency, or EMA, implemented the centralized procedure for the approval of human medicines to facilitate marketing authorizations that are valid throughout the EU. This procedure results in a single marketing authorization issued by the European Commission following a favorable opinion by the EMA that is not used, approval in one country ofvalid across the European Union, canas well as Iceland, Liechtenstein, and Norway. The centralized procedure is compulsory for human medicines that are: derived from biotechnology processes, such as genetic engineering, contain a new active substance indicated for the treatment of certain diseases, such as HIV/AIDS, cancer, diabetes, neurodegenerative disorders or autoimmune diseases and other immune dysfunctions, and officially designated orphan medicines. For medicines that do not fall within these categories, an applicant has the option of submitting an application for a centralized marketing authorization to the EMA, as long as the medicine concerned is a significant therapeutic, scientific or technical innovation, or if its authorization would be usedin the interest of public health.
There are also two other possible routes to obtain approvalauthorize medicinal products in another countryseveral European Union countries, which are available for investigational medicinal products that fall outside the scope of the European Union under two simplified application processes,centralized procedure: the decentralized procedure and the mutual recognition procedure. Under the decentralized procedure, an applicant may apply for simultaneous authorization in more than one EU country for medicinal products that have not yet been authorized in any EU country and that do not fall within the mandatory scope of the centralized procedure. Under the mutual recognition procedure, ora medicine is first authorized in one EU Member State, in accordance with the decentralizednational procedures of that country. Following a national authorization, the applicant may seek further marketing authorizations from other EU countries under a procedure both of which rely onwhereby the principle of mutual recognition. After receiving regulatory approval through anycountries concerned agree to recognize the validity of the European registration procedures, pricingoriginal, national marketing authorization.
In the EU, medicinal products designated as orphan drug products benefit from financial incentives such as reductions in marketing authorization application fees or fee waivers and reimbursement approvals are also required10 years of marketing exclusivity following medicinal product approval. For a medicinal product to qualify as orphan drugs: (i) it must be intended for the treatment, prevention or diagnosis of a disease that is life-threatening or chronically debilitating; (ii) the prevalence of the condition in most countries.
Other Regulations
We are also subject to numerous federal, state and local laws relating to such matters as safe working conditions, manufacturing practices, environmental protection, fire hazard control, and disposal of hazardous or potentially hazardous substances and biological materials. We may incur significant costs to comply with such laws and regulations now or in the future.
Reimbursement
Obtaining coverage and reimbursement approval for a product from a government or other third-party payor is a time-consuming and costly process that can require the provision of supporting scientific, clinical and cost effectiveness data for the use of drug or biologic products to the payor. There may be significant delays in obtaining such coverage and reimbursement for newly approved products, and coverage may be more limited than the purposes for which the product is approved by the FDA or similar regulatory authorities outside of the United States. Moreover, eligibility for coverage and reimbursement does not imply that a product will be paid for in all cases or at a rate that covers operating costs, including research, development, intellectual property, manufacture, sale and distribution expenses. Reimbursement rates may vary according to the use of the product and the clinical setting in which it is used, may be based on reimbursement levels already set for lower cost products and may be incorporated into existing payments for other services. Net prices for products may be reduced by mandatory discounts or rebates required by government healthcare programs or private payors and by any future relaxation of laws that presently restrict imports of product from countries where they may be sold at lower prices than in the United States.
| 18 |
There is significant uncertainty related to the insurance coverage and reimbursement of newly approved products. Third-party payors often rely upon Medicare coverage policy and payment limitations in setting their own reimbursement policies, but also have their own methods and approval process apart from Medicare coverage and reimbursement determinations. It is difficult to predict what third party payors will decide with respect to coverage and reimbursement for new drug and biologic product candidates. An inability to promptly obtain coverage and adequate reimbursement rates from both government-funded and private payors for any approved products could have a material adverse effect on a pharmaceutical manufacturer’s operating results, ability to raise capital needed to commercialize products and overall financial condition.
Reimbursement may impact the demand for, and/or the price of, any product which obtains marketing approval. Even if coverage is obtained for a given product by a third-party payor, the resulting reimbursement payment rates may not be adequate or may require co-payments that patients find unacceptably high. Patients who are prescribed medications for the treatment of their conditions, and their prescribing physicians, generally rely on third-party payors to reimburse all or part of the costs associated with those medications. Patients are unlikely to use products unless coverage is provided and reimbursement is adequate to cover all or a significant portion of the cost of the products. Therefore, coverage and adequate reimbursement is critical to new product acceptance. Coverage decisions may depend upon clinical and economic standards that disfavor new drug products when more established or lower cost therapeutic alternatives are already available or subsequently become available.
The U.S. government, state legislatures and foreign governmental entities have shown significant interest in implementing cost containment programs to limit the growth of government-paid healthcare costs, including price controls, restrictions on reimbursement and coverage and requirements for substitution of generic products for branded prescription drugs. Adoption of government controls and measures, and tightening of restrictive policies in jurisdictions with existing controls and measures, could exclude or limit our products from coverage and limit payments for pharmaceuticals.
In addition, it is expected that the increased emphasis on managed care and cost containment measures in the United States by third-party payors and government authorities will continue and place further pressure on pharmaceutical pricing and coverage. Coverage policies and third-party reimbursement rates may change at any time. Even if favorable coverage and reimbursement status is attained for one or more drug products that gain regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
Intellectual Property
Our success depends, in part, on our ability to remain competitive, we must developobtain, maintain, and enforce patents and other proprietary protections of our commercially important technologies and product candidates, to operate without infringing the proprietary rights of others, and to maintain protectiontrade secrets or other proprietary know-how, both in the United States and other countries. Our ability to stop third parties from making, using, selling, offering to sell or importing our products will depend on the proprietary aspects of our technologies.extent to which we have rights under valid and enforceable patents or trade secrets that protect these activities. We rely on a combination of patents, copyrights, trademarks, trade secret laws and confidentiality, material data transfer agreements, licenses and invention assignment agreementsseek to protect our intellectual property rights. We also rely upon unpatented trade secretsproprietary technology, inventions, and improvements unpatented know-howthat are commercially important to our business by seeking, maintaining, and continuing technological innovationdefending patent rights, whether developed internally or licensed from third parties.
We have two active patent families relating to develop and maintain our competitive position. We generally protect this information with reasonable security measures.
We also have an exclusive license to three pending USpatents and patent applications and one issued USacross seven different patent families relating to our LSD1 program.
The patent applications for LSD-1positions of biotechnology companies like ours are generally uncertain and Dactinomycin as of March 31, 2017involve complex legal, scientific and factual questions. In addition, the coverage claimed in a patent application can be significantly reduced before the patent is issued. Consequently, we do not know whether the product candidates we are set forth below:
The term of individual patents depends upon the legal term of the patents in the countries in which they are obtained. In most countries in which we file, the patent term is 20 years from the earliest date of filing a non-provisional patent application. In the United States, a patent term may be lengthened by patent term adjustment, which compensates a patentee for administrative delays by the U.S. Patent and Trademark Office in granting a patent, or may be shortened if a patent is terminally disclaimed over another patent. Certain of our patents currently benefit from patent term adjustment and some of our patents issuing in the future may benefit from patent term adjustment.
The patent term of a patent that covers an FDA-approved product may also be eligible for patent term extension, which permits patent term restoration as compensation for the patent term lost during the FDA regulatory review process. The Drug Price Competition and Patent Term Restoration Act of 1984, or the Hatch-Waxman Act, permits a patent term extension of up to five years beyond the expiration of the patent. The length of the patent term extension is related to the length of time the product is under regulatory review. Patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval and only one patent applicable to an approved product may be extended. Similar provisions are available in Europe and other non-U.S. jurisdictions to extend the term of a patent that covers an approved product. In the future, if and when our product candidates receive FDA approval, we expect to apply for patent-term extensions on patents covering those products.
To protect our rights to any of our issued patents and proprietary information, we may need to litigate against infringing third parties. Furthermore, if weparties, or avail ourselves of the courts or participate in hearings to determine the scope and validity of those patents or other proprietary rights. These types of proceedings are foundoften costly and could be very time-consuming to willfully infringe these patents, weus, and there can be no assurance that the deciding authorities will rule in our favor. An unfavorable decision could in additionallow third parties to other penalties, beuse our technology without being required to pay treble damages. Althoughus licensing fees or may compel us to license needed technologies to avoid infringing third-party patent and intellectual property disputesproprietary rights. Such a decision could even result in this area have often been settled through licensingthe invalidation or similar arrangements, costsa limitation in the scope of our patents or forfeiture of the rights associated with our patents or pending patent applications.
We intend to seek orphan drug status whenever it is available. If a product which has an orphan drug designation subsequently receives the first regulatory approval for the indication for which it has such arrangements may be substantial and could include ongoing royalties. We may be unabledesignation, the product is entitled to obtain necessary licenses on satisfactory or commercially feasible terms, if at all. If we do not obtain necessary licenses, weorphan exclusivity, meaning that the applicable regulatory authority may not approve any other applications to market the same drug for the same indication, except in certain very limited circumstances, for a period of seven years in the United States and 10 years in the European Union. Orphan drug designation does not prevent competitors from developing or marketing different drugs for an indication.
We also rely on trade secret protection for our confidential and proprietary information. No assurance can be ablegiven that others will not independently develop substantially equivalent proprietary information and techniques or otherwise gain access to complete our researchtrade secrets or disclose such technology, or that we can meaningfully protect our trade secrets. However, we believe that the substantial costs and development, or such research and development may take considerable time, and forceresources required to develop technological innovations will help us to reassessprotect the competitive advantage of our business plans. Adverse determinationsproducts.
It is our policy to require our employees, consultants, outside scientific collaborators, sponsored researchers and other advisors to execute confidentiality agreements upon the commencement of employment or consulting or collaborative relationships with us. These agreements provide that all confidential information developed or made known to the individual during the course of the individual’s relationship with us is to be kept confidential and not disclosed to third parties except in a judicialspecific circumstances. In the case of employees, the agreements provide that all inventions conceived by the individual shall be and are our exclusive property. There can be no assurance, however, that these agreements will provide meaningful protection or administrative proceedingadequate remedies for our trade secrets in the event of unauthorized use or failure to obtain necessary licenses could prevent us from developing our targets, which would have a significant adverse impact on our business.
Competition
The biotechnology and pharmaceutical industries are characterized by rapidly advancing technologies, intense competition and a strong emphasis on proprietary products. While we believe that our technology, development experience and scientific knowledge provide us with competitive advantages, we face potential competition from many different sources, including major pharmaceutical, specialty pharmaceutical and biotechnology companies, academic institutions and governmental agencies and public and private research institutions. Any product candidates that we successfully develop and commercialize will compete with existing therapies and new therapies that may become available in the future.
Several types of existing treatments may be used for people with AML. The main treatments include chemotherapy, bone marrow transplants, stem cell transplants and/or interferon therapy. In most cases AML can progress rapidly, so it is important to start treatment as soon as possible after the diagnosis is made. In addition to currently marketed therapies, there are also a number of products in late stage clinical development to treat AML. These products in development may provide efficacy, safety, convenience and other benefits that are not provided by currently marketed therapies. As a result, they may provide significant competition for any of our product candidates for which we obtain market approval.
| 20 |
There are other companies and research institutions working to develop therapies that target AML. Many of our competitors may have significantly greater financial resources and expertise in research and development, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. Mergers and acquisitions in the pharmaceutical, biotechnology and diagnostic industries may result in even more resources being concentrated among a smaller number of our competitors. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs. Smaller or early stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies.
A listing of approved products currently used for treatment of AML include the following: topoisomerase II inhibitors, e.g., Cerubidine® (daunorubicin HCL, Bedford Labs), Novantrone®(mitoxantrone HCL, Pfizer) and Idamycin®(idarubicin HCl, Pfizer); alkylating agents, e.g., Cytoxan®(cyclophosphamide, Baxter); DNA polymerase inhibitors, e.g,. Cytarabine®(cytarabine, Pfizer); DNA binding agents, e.g., Doxil®(doxorubicin HCl liposome, Alza); purine analogs, e.g., Tabloid®(thioguanine, GlaxoSmithKline); mitotic spindle inhibitors, e.g., Vincasar PFS®(vincristine sulfate, Teva); DNA fragmentation agents, e.g,. Trisenox®(arsenic trioxide, Cephalon); FLT3 inhibitors, e.g., Rydapt® (midostaurin, Novartis); IDH2 inhibitors, e.g., IDHIFA®(enasidenib mesylate, Celgene).
A listing of AML drugs in advanced clinical developmentby our competitors include the following: (irreversible) LSD-1 inhibitors, e.g. ORY-1001 (Oryzon); FLT3 inhibitors, e.g., quizartinib (Daiichi Sankyo), crenolanib (Arog Pharma) and gilteritinib (Astellas); topoisomerase inhibitors, e.g., Vosaroxin®(qinprezo, Sunesis Pharma), CPX-351 (vyxeos, Celator Pharma); DNA hypomethylating agents, e.g., guadecditabine (ASTX Pharma); PLK1 inhibitors, e.g., volasertib (Boehringer Ingelheim); IDH2 inhibitors, e.g., enasidenib (Agios Pharma);IDH1 inhibitors, e.g., ivosidenib (Agios Pharma); DOT1L ihibitors, e.g., pinometostat (Epizyme); BCL2 inhibitors, e.g., venetoclax (Roche); BET inhibitors, e.g., OTX-015 (OncoEthix) and HDAC inhibitors, e.g., pracinostat (MEI Pharma).
A large number of irreversible inhibitors of LSD1 have been developed by major pharmaceutical companies and research institutes.Among them, ORY-1001 and GSK2879552, two TCP derivatives, developed by Oryzon Genomics and GSK, respectively, have been in Phase 1 clinical trials for treatment of AMLWe have focused on development of reversible inhibitors instead of irreversible inhibitors with the expectation of better therapeutic effect,safety profiles and lower toxicity compared to the irreversible inhibitors
The key competitive factors affecting the success of all of our targets, if approved, are likely to be their efficacy, safety, convenience, price, the level of generic competition and the availability of reimbursement from government and other third-party payors.
Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient or are less expensive than any products that we may develop. Our competitors also may obtain FDA or other regulatory approval for their products more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market. In addition, our ability to compete may be affected in many cases by insurers or other third-party payors seeking to encourage the use of generic products. There are many generic products currently on the market for the indications that we are
Shared Services Agreement
On January 1, 2017, Tiziana Life Sciences plc, a company incorporated in England and Wales (“Tiziana”), entered into a Shared Services Agreement with us pursuant to which Tiziana agreed to provide certain services to us including various administrative, financial, legal, tax, insurance, facility and information technology services.The term of the agreement is until April 1, 2018 and is renewed automatically thereafter for successive three month terms with respect to any services still in effect unless either party elects not to renew the agreement or any specific service by notice in writing to the other party no later than 45 days prior to the end of any term.We may terminate any of the services provided under the agreement at any time upon 30 days prior written notice.Tiziana may terminate any of the services provided under the agreement if we shall have failed to perform any of our material obligations under the agreement and failed to cure any such failure within 30 days after receiving notice thereof.
Employees
As of March 31, 2017September 30, 2019 and September 30, 2018, we had 4 full-time employees.
Risks Relating to Our Business
We are a development stage leukemia-focused biotechnology company with minimal operating history.
We are a development stageleukemia-focused biotechnology company with minimal operating historylimited operations to date and no revenue. We currentlyhave not yet commenced clinical trials, have no product candidates ready for commercialization, have not generated any revenue from operations and expect to incur substantial net losses for the foreseeable future to further develop and commercialize our molecular targets.product candidates. We are unable to predict the extent of these future net losses, or when we may attain profitability, if at all. We may never be able to generate any revenues or royalties from the sales of our therapeutics or become profitable even if we do generate revenues or royalties.
We expect to continue to incur increasing net losses for the foreseeable future, and we may never achieve or maintain profitability.
Investment in biopharmaceutical product development is highly speculative because it entails substantial upfront capital expenditures and significant risk that any potential product candidate will fail to demonstrate adequate effect or an acceptable safety profile, gain regulatory approval and become commercially viable. We have no products approved for commercial sale and have not generated any revenue from product sales to date, and we continue to incur significant research and development and other expenses related to our ongoing operations. As a result, we are not profitable and have incurred losses in each period since our inception. For the year ended September 30, 2019 and 2018, we reported a net loss of $936,380 and $4,076,361, respectively. As of September 30, 2019, we had an accumulated deficit of approximately $17,311,809 .
To become and remain profitable, we or our partners must succeed in developing our product candidates, obtaining regulatory approval for them, and manufacturing, marketing and selling those products for which we or our partners may obtain regulatory approval. We or they may not succeed in these activities, and we may never generate revenue from product sales that is significant enough to achieve profitability. Because of the numerous risks and uncertainties associated with biopharmaceutical product development and commercialization, we are unable to accurately predict the timing or amount of future expenses or when, or if, we will be able to achieve or maintain profitability. Currently, we have no products approved for commercial sale, and to date we have not generated any product revenue. We have financed our operations primarily through the sale of equity securities. The size of our future net losses will depend, in part, on the rate of growth or contraction of our expenses and the level and rate of growth, if any, of our revenues. Our ability to achieve profitability is dependent on our ability, alone or with others, to complete the development of our products successfully, obtain the required regulatory approvals, manufacture and market our proposed products successfully or have such products manufactured and marketed by others, and gain market acceptance for such products. There can be no assurance as to whether or when we will achieve profitability.
We will require substantial additional funding which may not be available to us on acceptable terms, or at all. If we fail to raise the necessary additional capital, we may be unable to complete the development and commercialization of our product candidates, or continue our development programs.
We expect to significantly increaseescalate moderately our spending to advance the preclinicalpre-clinical and clinical development of our product candidates and launch and commercialize any product candidate for which we receive regulatory approval, including building our own commercial organizations to address certain markets. We will require additional capital for the further development and commercialization of our product candidates, as well as to fund our other operating expenses and capital expenditures.
We cannot be certain that additional funding will be available on acceptable terms, or at all. If we are unable to raise additional capital in sufficient amounts or on terms acceptable to us we may have to significantly delay, scale back or discontinue the development or commercialization of our product candidate. We may also seek collaborators for one or more of our current or future product candidates at an earlier stage than otherwise would be desirable or on terms that are less favorable than might otherwise be available. Any of these events could significantly harm our business, financial condition and prospects.
| 22 |
Our future capital requirements will depend on many factors, including:
• | the progress of the development of our product candidates; |
• | the number of product candidates we pursue; |
• | the time and costs involved in obtaining regulatory approvals; |
• | the costs involved in filing and prosecuting patent applications and enforcing or defending patent claims; |
• | our plans to establish sales, marketing and/or manufacturing capabilities; |
• | the effect of competing technological and market developments; |
• | the terms and timing of any collaborative, licensing and other arrangements that we may establish; |
• | general market conditions for offerings from biopharmaceutical companies; |
• | our ability to establish, enforce and maintain selected strategic alliances and activities required for product commercialization; and |
• | our revenues, if any, from successful development and commercialization of our product candidates. |
In order to carry out our business plan and implement our strategy, we anticipate that we will need to obtain additional financing from time to time and may choose to raise additional funds through strategic collaborations, licensing arrangements, public or private equity or debt financing, bank lines of credit, asset sales, government grants, or other arrangements. We cannot be sure that any additional funding, if needed, will be available on terms favorable to us or at all. Furthermore, any additional equity or equity-related financing may be dilutive to our stockholders, and debt or equity financing, if available, may subject us to restrictive covenants and significant interest costs. If we obtain funding through a strategic collaboration or licensing arrangement, we may be required to relinquish our rights to certain of our product candidate or marketing territories. Our inability to raise capital when needed would harm our business, financial condition and results of operations, and could cause our stock price to decline or require that we wind down our operations altogether.
Our independent registered public accounting firm has expressed substantial doubt about our ability to continue as a going concern, which may hinder our ability to obtain future financing.
Our consolidated financial statements as of September 30, 2019 were prepared under the assumption that we will continue as a going concern for the next twelve months. Due to our recurring losses from operations, we concluded that there is substantial doubt in our ability to continue as a going concern within one year after the financial statements are unableissued without additional capital becoming available. Our independent registered public accounting firm has issued an audit opinion that included an explanatory paragraph referring to manage our expected growth, we may not be ableprojected future losses along with recurring losses from operations and expressing substantial doubt in our ability to develop our business.
If we fail to select product candidates, fail to successfully complete clinical trials and commercialize product candidates or fail to obtain regulatory approval, our business would be harmed and the value of our securities would decline.
We must be evaluated in light of the uncertainties and complexities affecting a pre-commercial biopharmaceutical company. We have not completed preclinical or clinical research and have not yet selected product candidates or completed the development of suchour product candidates. Our failure to select product candidates and subsequently to develop and commercialize such product candidates successfully may cause us to cease operations. We are performing preclinical research on NPM1NPM1 and LSD1.LSD1. This research will require significant additional development efforts by us prior to selection of product candidates and significant additional development efforts by us and regulatory approvals prior to commercialization. We cannot be certain that our efforts in this regardwill lead to commercially viable therapeutics. We do not know what the final cost to select and commercialize product candidates will be.
We do not know whether any of our molecular targets under development ultimately will be selected as product candidates or whether our product candidates if any, will be shown to be effective. Moreover, governmental authorities may enact new legislation or regulations that could limit or restrict our development efforts. We may receive unfavorable results from pre-clinical studies or clinical studies on the molecular targets, which may cause us to abandon the product selection process and further development efforts. If we are unable to select product candidates and then to successfully develop such product candidates, we will not have a source of revenue and will not achieve profitability.
Regulatory agencies including the U.S. Food and Drug Administration, or FDA, must approve our product candidates, if any, before they can be marketed or sold. The approval process is lengthy, requires significant capital expenditures, and uncertain as to outcome. Our ability to obtain regulatory approval of any product candidate depends on, among other things, completion of additional clinical trials, whether our clinical trials demonstrate statistically significant efficacy with safety issues that do not potentially outweigh the therapeutic benefit of the product candidates, and whether the regulatory agencies agree that the data from our future clinical trials are sufficient to support approval for any of our product candidates. The final results of our current and future preclinical or clinical trials may not meet FDA or other regulatory agencies’ requirements to approve a product candidate for marketing, and the regulatory agencies may otherwise determine that our manufacturing processes or facilities are insufficient to support approval. We or our collaborators may need to conduct more preclinical or clinical trials than we currently anticipate. Even if we do receive FDA or other regulatory agency approval, we or our collaborators may not be successful in commercializing approved product candidates. If any of these events occur, our business could be materially harmed and the value of our securities would decline.
While ACT D is used to treat multiple cancers, the formulation of ACT D that we intend to use has not been proven to be safe or efficacious.
ACT D is an established anticancer therapeutic that has been used to treat a number of types of cancer. However, the formulation of ACT D that we intend to use for the treatment of AML in our lead compound, RASP-101, has not yet to tested in clinical trials and has not been proven to be safe or efficacious. Our consultants have only tested ACT D in clinical trials and we intend to use a nanoparticle controlled release formulation of ACT D in our clinical trials.
All of our current data for our lead product candidate are the result of Phase 2 clinical trials conducted by third parties and do not necessarily provide sufficient evidence that our product candidates will be viable as potential pharmaceutical products.
Through our proprietary access to relevant laboratory and clinical trial results of Phase 2 trials conducted by Professors Falini and Martelli, we possess toxicology, pharmacokinetic, and other preclinical data and clinical data on ACT D. Previous clinical trials using ACT D have had different trial designs, doses, parameters and endpoints than our planned clinical trial. There is no guarantee that Phase 2 results can or will be replicated by us with use of our formulation of RASP-101.
To date, long-term safety and efficacy have not yet been demonstrated in clinical trials for our product candidates. Favorable results in early studies or trials may not be repeated in later studies or trials. Even if our clinical trials are initiated and completed as planned, we cannot be certain that the results will support our product candidate claims. Success in preclinical testing and early clinical trials does not ensure that later clinical trials will be successful. We cannot be sure that the results of later clinical trials would replicate the results of prior clinical trials and preclinical testing, nor that they would satisfy the requirements of the FDA or other regulatory agencies. Clinical trials may fail to demonstrate that our product candidate is safe for humans and effective for indicated uses. Preclinical and clinical results are frequently susceptible to varying interpretations that may delay, limit or prevent regulatory approvals or commercialization. Any delay in, or termination of, our clinical trials would delay our obtaining FDA or EMA approval for the affected product candidate and, ultimately, our ability to commercialize that product candidate.
If we cannot demonstrate an acceptable toxicity profile for our product candidates, if any, in non-clinical studies, we will not be able to initiate or continue clinical trials or obtain approval for our product candidates.
To move a product candidate into human clinical trials, we must first demonstrate an acceptable toxicity profile in preclinical testing. Furthermore, to obtain approval, we must also demonstrate safety in various non-clinical tests. We may not have conducted or may not conduct the types of non-clinical testing required by regulatory authorities, or future non-clinical tests may indicate that our product candidates are not safe for use. Preclinical and non-clinical testing is expensive, time-consuming and has an uncertain outcome. In addition, success in initial non-clinical testing does not ensure that later non-clinical testing will be successful. We may experience numerous unforeseen events during the non-clinical testing process, which could delay or prevent our ability to develop or commercialize our product candidates, including:
• | our preclinical and non-clinical testing may produce inconclusive or negative safety results, which may require us to conduct additional non-clinical testing or to abandon product candidates; |
• | our product candidates may have unfavorable pharmacology or toxicity characteristics; |
• | RASP-101 has not been tested in humans; |
• | our product candidates may cause undesirable side effects such as negative immune responses that lead to complications; |
• | our enrolled patients may have allergies that lead to complications after treatment; and |
• | the FDA or other regulatory authorities may determine that additional safety testing is required. |
Any such events would increase our costs and could delay or prevent our ability to commercialize our product candidates, which could adversely impact our business, financial condition and results of operations.
We, or our collaborators, may face delays in completing our pre-clinical or clinical trials, and may not be able to complete them at all.
We have not completed the pre-clinical and clinical trials necessary to support an application for approval to market of our product candidates, if any. Our or our collaborators’ current and future clinical trials may be delayed, unsuccessful, or terminated as a result of many factors, including:
• | delays in designing an appropriate clinical trial protocol and reaching agreement on trial design with investigators and regulatory authorities; |
• | governmental or regulatory delays, failure to obtain regulatory approval or changes in regulatory requirements, policy or guidelines; |
• | adding new clinical trial sites |
• | reaching agreement on acceptable terms with prospective contract research organizations, or CROs, and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; |
• | the actual performance of CROs and clinical trial sites in ensuring the proper and timely conduct of our clinical trials; |
• | adverse effects experienced by subjects in clinical trials; |
• | manufacturing sufficient quantities of product candidates for use in clinical trials; and |
• | delays in achieving study endpoints and completing data analysis for a trial. |
In addition to these factors, our trials may be delayed, unsuccessful or terminated because:
• | regulators or institutional review boards, or IRBs, may not authorize us to commence a clinical trial; |
• | regulators or IRBs may suspend or terminate clinical research for various reasons, including noncompliance with regulatory requirements or concerns about patient safety; |
• | we may suspend or terminate our clinical trials if we believe that they expose the participating patients to unacceptable health risks; |
• | patients may not complete clinical trials due to safety issues, side effects, such as injection site discomfort, a belief that they are receiving placebo instead of our product candidates, or other reasons; |
• | patients with serious diseases included in our clinical trials may die or suffer other adverse medical events for reasons that may not be related to our product candidates; |
• | in those trials where our product candidate is being tested in combination with one or more other therapies, deaths may occur that may be attributable to the other therapies; |
• | we may have difficulty in maintaining contact with patients after treatment, preventing us from collecting the data required by our study protocol; |
• | product candidates may demonstrate a lack of efficacy during clinical trials; |
• | personnel conducting clinical trials may fail to properly administer our product candidates; and |
• | our collaborators may decide not to pursue further clinical trials. |
We could encounter delays if our clinical trials are suspended or terminated by us, by IRBs of the institutions in which such trials are being conducted, by the Data Safety Monitoring Boards for such trials or by the FDA or other regulatory authorities. Such authorities may impose such a suspension or termination due to a number of factors, including potential for unacceptable safety risks to patients, inspection of the clinical trial operation or trial site, changes in government regulations or administrative actions.
In addition, we rely on academic institutions, physician practices and CROs to conduct, supervise or monitor some or all aspects of clinical trials involving our product candidates. We have less control over the timing and other aspects of these clinical trials than if we conducted the monitoring and supervision entirely on our own. Third parties may not perform their responsibilities for our clinical trials on our anticipated schedule or consistent with a clinical trial protocol or applicable regulations. We also may rely on CROs to perform our data management and analysis. They may not provide these services as required or in a timely or compliant manner, and we may be held legally responsible for any or all of their performance failures or inadequacies.
Moreover, our development costs will increase because we will be required to complete additional or larger clinical trials for our product candidates prior to FDA or other regulatory approval. If we or our collaborators experience delays in the completion of, or termination of, any clinical trial of our product candidates, the commercial prospects of our product candidates will be harmed, and our ability to generate product revenues from any of these product candidates will be delayed or
We have limited experience in the development of therapeutic product candidates and therefore may encounter difficulties developing our product candidate or managing our operations in the future.
We have limited experience in the discovery, development and manufacturing of therapeutic compounds. In order to successfully develop our product candidate, we must continuously supplement our research, clinical development, regulatory, medicinal chemistry, and manufacturing capabilities through the addition of key employees, consultants or third-party contractors to provide certain capabilities and skill sets that we do not possess.
Furthermore, we have adopted an operating model that largely relies on the outsourcing of a number of responsibilities and key activities to third-party consultants, and contract research and manufacturing organizations in order to advance the development of our product candidates. Therefore, our success depends in part on our ability to retain highly qualified key management, personnel, and directors to develop, implement and execute our business strategy, operate the company and oversee the activities of our consultants and contractors, as well as academic and corporate advisors or consultants to assist us in this regard. We are currently highly dependent upon the efforts of our management team. In order to develop our product candidates, we need to retain or attract certain personnel, consultants or advisors with experience in drug development activities that include a number of disciplines, including research and development, clinical trials, medical matters, government regulation of pharmaceuticals, manufacturing, formulation and chemistry, business development, accounting, finance, regulatory affairs, human resources and information systems. We are highly dependent upon our senior management and scientific staff, particularly Kunwar Shailubhai, our Chief Executive Officer. The loss of services of Dr. Shailubhai or one or more of our other members of senior management could delay or prevent the successful completion of our planned clinical trials or the commercialization of our product candidate.
Our success depends in part on our continued ability to attract, retain and motivate highly qualified management, clinical and scientific personnel and on our ability to develop and maintain important relationships with leading academic institutions, clinicians and scientists. The competition for qualified personnel in the biotechnology and pharmaceuticals field is intense. We will need to hire additional personnel as we expand our clinical development and commercial activities. While we have not had difficulties recruiting qualified individuals, to date, we may not be able to attract and retain quality personnel on acceptable terms given the competition for such personnel among biotechnology, pharmaceutical and other companies. Although we have not experienced material difficulties in retaining key personnel in the past, we may not be able to continue to do so in the future on acceptable terms, if at all. If we lose any key managers or employees, or are unable to attract and retain qualified key personnel, directors, advisors or consultants, the development of our product candidate could be delayed or terminated and our business may be harmed.
We have limited capacity for recruiting and managing clinical trials, which could impair our timing to initiate or complete clinical trials of our product candidates and materially harm our business.
We have limited capacity to recruit and manage the clinical trials necessary to obtain FDA approval or approval by other regulatory authorities. By contrast, larger pharmaceutical and bio-pharmaceutical companies often have substantial staff with extensive experience in conducting clinical trials with multiple product candidates across multiple indications. In addition, they may have greater financial resources to compete for the same clinical investigators and patients that we are attempting to recruit for our clinical trials. If potential competitors are successful in completing drug development for their product candidates and obtain approval from the FDA, they could limit the demand for our product candidates.
As a result, we may be at a competitive disadvantage that could delay the initiation, recruitment, timing, completion of our clinical trials and obtaining regulatory approvals, if at all, for our product candidate.
If we encounter difficulties enrolling patients in our clinical trials, our clinical trials could be delayed or otherwise adversely affected.
Clinical trials for our product candidates, if any, will require us to identify and enroll a large number of patients with the disease under investigation. We may not be able to enroll a sufficient number of patients, or those with required or desired characteristics, in a timely manner. Patient enrollment is affected by factors including:
• | severity of the disease under investigation; |
• | design of the trial protocol; |
• | the size and nature of the patient population; |
• | eligibility criteria for the study in question; |
• | lack of a sufficient number of patients who meet the enrollment criteria for our clinical trials; |
• | delays required to characterize the infection to allow us to select a product candidate, which may lead patients to seek to enroll in other clinical trials or seek alternative treatments; |
• | perceived risks and benefits of the product candidate under study; |
• | availability of competing therapies and clinical trials; |
• | efforts to facilitate timely enrollment in clinical trials; |
• | scheduling conflicts with participating clinicians; |
• | patient referral practices of physicians; |
• | the ability to monitor patients adequately during and after treatment; and |
• | proximity and availability of clinical trial sites for prospective patients. |
If we have difficulty enrolling a sufficient number or diversity of patients to conduct our clinical trials as planned, we may need to delay or terminate ongoing or planned clinical trials, either of which would have an adverse effect on our business.
Results of earlier studies and clinical trials may not be predictive of future trial results.
Clinical testing is expensive and can take many years to complete, and its outcome is inherently uncertain. Failure can occur at any time during the clinical trial process. The results of preclinical studies and early clinical trials of our product candidates, if any, may not be predictive of the design or results of later-stage clinical trials. Any positive results generated to date do not ensure that later trials will demonstrate similar results. While we have observed statistically significant improvements in the outcomes of some of our clinical trials, many of the improvements we have seen have not reached statistical significance. Statistical significance is a statistical term that means that an effect is unlikely to have occurred by chance. In order to be approved, product candidates must demonstrate that their effect on patients’ diseases in the trial is statistically significant. Product candidates in later stages of clinical trials may fail to show the desired safety and efficacy traits despite having progressed through preclinical studies and initial clinical trials. Early clinical trials frequently enroll patient populations that are different from the patient populations in later trials, resulting in different outcomes in later clinical trials from those in earlier stage clinical trials. In addition, adverse events may not occur in early clinical trials and only emerge in larger, late-stage clinical trials or after commercialization. A number of companies in the biopharmaceutical industry have suffered significant setbacks in advanced clinical trials due to lack of efficacy or adverse safety profiles, notwithstanding promising results in earlier clinical trials. If later stage clinical trials do not demonstrate efficacy and safety of our product candidates we will not be able to market them and our business will be materially harmed.
Regulatory authorities may not approve our product candidates, if any, even if they meet safety and efficacy endpoints in clinical trials.
Under certain circumstances, regulatory authorities may revise or retract previous guidance during the course of our clinical activities or after the completion of our clinical trials. A regulatory authority may also disqualify a clinical trial in whole or in part from consideration in support of approval of a potential product for commercial sale or otherwise deny approval of that product. Prior to regulatory approval, a regulatory authority may elect to obtain advice from outside experts regarding scientific issues and/or marketing applications under a regulatory authority review. In the United States, these outside experts are convened through the FDA’s Advisory Committee process, which would report to the FDA and make recommendations that
| 27 |
The FDA and foreign regulatory agencies may delay, limit or deny marketing approval for many reasons, including:
• | a product candidate may not be considered safe or effective; |
• | our manufacturing processes or facilities may not meet the applicable requirements; |
• | changes in the agencies’ approval policies or adoption of new regulations may require additional work on our part, for example, the FDA may require us to change or expand the endpoints in our clinical trials; |
• | different divisions of the FDA are reviewing different product candidates and those divisions may have different requirements for approval; and |
• | changes in regulatory law, FDA or foreign regulatory agency organization, or personnel may result in different requirements for approval than anticipated. |
Our product candidates may not be approved even if they achieve their endpoints in clinical trials. Regulatory agencies, including the FDA, or their advisors may disagree with our trial design and our interpretations of data from preclinical studies and clinical trials. Regulatory agencies also may approve a product candidate for fewer or more limited indications than requested or may grant approval subject to the performance of post-marketing studies. In addition, regulatory agencies may not approve the labeling claims that are necessary or desirable for the successful commercialization of our product candidates.
Any delay in or failure to receive or maintain approval for any of our product candidates could prevent us from ever generating revenues or achieving profitability.
We may be required to suspend, repeat or terminate our clinical trials if they are not conducted in accordance with regulatory requirements, the results are negative or inconclusive, or the trials are not well designed.
Clinical trials must be conducted in accordance with FDA regulations governing clinical studies, or other applicable foreign government guidelines, and are subject to oversight by the FDA, other foreign governmental agencies and IRBs at the medical institutions where the clinical trials are conducted. In addition, clinical trials must be conducted with product candidates produced under current Good Manufacturing Practices, or cGMP, and may require large numbers of test subjects. Clinical trials may be suspended by the FDA, other foreign governmental agencies or us for various reasons, including:
• | deficiencies in the conduct of the clinical trials, including failure to conduct the clinical trial in accordance with regulatory requirements or clinical protocols; |
• | deficiencies in the clinical trial operations or trial sites; |
• | the product candidate may have unforeseen adverse side effects; |
• | the time required to determine whether the product candidate is effective may be longer than expected; |
• | deaths or other adverse events arising during a clinical trial due to medical problems that may not be related to clinical trial treatments; |
• | the product candidate may not appear to be more effective than current therapies; |
• | the quality or stability of the product candidate may fall below acceptable standards; and |
• | insufficient quantities of the product candidate might be available to complete the trials. |
In addition, changes in regulatory requirements and guidance may occur and we may need to amend clinical trial protocols to reflect these changes. Amendments may require us to resubmit our clinical trial protocols to IRBs for reexamination, which may impact the costs, timing or successful completion of a clinical trial. Due to these and other factors, our product candidates could take longer to gain regulatory approval than we expect or we may never gain approval for any product candidates, which could reduce or eliminate our revenue by delaying or terminating the commercialization of our product candidates.
Any product candidate for which we, or our collaborators, obtain marketing approval could be subject to restrictions or withdrawal from the market, and we may be subject to penalties if we fail to comply with regulatory requirements or if we experience unanticipated problems with our products, when and if any of them are approved.
Any product candidate that we or our collaborators obtain marketing approval for, along with the manufacturing processes, post-approval clinical data, labeling, advertising and promotional activities for such product, will be subject to continual requirements of the FDA and other regulatory authorities. These requirements include submissions of safety and other post-marketing information, reports, registration and listing requirements, cGMP requirements relating to quality control, quality assurance and corresponding maintenance of records and documents, requirements regarding the distribution of samples to physicians and recordkeeping. Even if marketing approval of a product candidate is granted, the approval may be subject to limitations on the indicated uses for which the product may be marketed or to conditions of approval, or contain requirements for costly post-marketing testing and surveillance to monitor the safety or efficacy of the product. The FDA closely regulates
| 28 |
In addition, later discovery of previously unknown problems with these products, manufacturers or manufacturing processes, or failure to comply with regulatory requirements, may yield various results, including:
• | restrictions on such products, manufacturers or manufacturing processes; |
• | restrictions on the labeling or marketing of a product; |
• | restrictions on product distribution or use; |
• | requirements to conduct post-marketing clinical trials; |
• | warning or untitled letters; |
• | withdrawal of the products from the market; |
• | refusal to approve pending applications or supplements to approved applications that we submit; |
• | recall of products, fines, restitution or disgorgement of profits or revenue; |
• | suspension or withdrawal of marketing approvals; |
• | refusal to permit the import or export of our products; |
• | product seizure; and |
• | injunctions or the imposition of civil or criminal penalties. |
The FDA’s policies may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates. If we, or our collaborators, are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we, or our collaborators, are not able to maintain regulatory compliance, any marketing approval that was obtained could be lost, which would adversely affect our business, prospects and ability to achieve or sustain profitability.
If we, or our collaborators, are unable to comply with foreign regulatory requirements or obtain foreign regulatory approvals, our ability to develop foreign markets for our products could be impaired.
Sales of our product candidates, if any, outside the United States will be subject to foreign regulatory requirements governing clinical trials, marketing approval, manufacturing, product licensing, pricing and reimbursement. These regulatory requirements vary greatly from country to country. As a result, the time required to obtain approvals outside the United States may differ from that required to obtain FDA approval and we may not be able to obtain foreign regulatory approvals on a timely basis or at all. Approval by the FDA does not ensure approval by regulatory authorities in other countries, and approval by one foreign regulatory authority does not ensure approval by regulatory authorities in other countries or by the FDA and foreign regulatory authorities could require additional testing. Failure to comply with these regulatory requirements or obtain required approvals could impair our ability to develop foreign markets for our products.
Competitive products for treatment of AML may reduce or eliminate the commercial opportunity for our product candidates, if any.
The clinical and commercial landscape for AML is rapidly changing. New data from commercial and clinical-stage products continue to emerge. It is possible that these data may alter current standards of care, completely precluding us from further developing our product candidates, if any, or getting them approved by regulatory agencies. Further, it is possible that we may initiate a clinical trial or trials for these product candidates, only to find that data from competing products make it impossible for us to complete enrollment in these trials, resulting in our inability to file for marketing approval with regulatory agencies. Even if these products are approved for marketing in a particular indication or indications, they may have limited sales due to particularly intense competition in these markets.
We will need to develop or acquire additional manufacturing and distribution capabilities in order to commercialize our product candidates, if any, that obtain marketing approval, and we may encounter unexpected costs or difficulties in doing so.
If we independently develop and commercialize one or more of our product candidates, if any, we will need to invest in acquiring or building additional capabilities and effectively manage our operations and facilities to successfully pursue and complete future research, development and commercialization efforts. We will require additional investment and validation process development in order to qualify our commercial-scale manufacturing process to manufacture clinical trial materials and commercial material if any of our products are approved for marketing. This investment and validation process development may be expensive and time-consuming. We will require additional personnel with experience in commercial-scale
• | recruit, hire, train, manage and motivate a growing employee base; |
• | accurately forecast demand for our products; |
• | assemble and manage the supply chain to ensure our ability to meet demand; and |
• | expand existing operational, manufacturing, financial and management information systems. |
| 29 |
We may seek FDA approval for our production process and facilities simultaneously with seeking approval for sale of our product candidates. Should we not complete the development of adequate capabilities, including manufacturing capacity, or fail to receive timely approval of our manufacturing process and facilities, our ability to supply clinical trial materials for planned clinical trials or supply products following regulatory approval for sale could be delayed, which would further delay our clinical trials or the period of time when we would be able to generate revenues from the sale of such products, if we are even able to obtain approval or generate revenues at all.
Additionally, we may decide to outsource some or all of our manufacturing activities to a third party commercial manufacturing organization, or CMO. Under any agreement with a CMO, we would have less control over the timing and quality of manufacturing than if we were to perform such manufacturing ourselves. A CMO would be manufacturing other pharmaceutical products in the same facilities as our product candidates, increasing the risk of cross product contamination. Further, there is no guarantee that any CMO will continue ongoing operations, causing potential delays in product supply, reduced revenues and other liabilities for us.
Any such events would increase our costs and could delay or prevent our ability to commercialize our product candidates, which could adversely impact our business, financial condition and results of operations.
Our product candidates may cause undesirable side effects or have other properties that could delay or prevent their regulatory approval, limit the commercial profile of an approved label, or result in significant negative consequences following marketing approval, if any.
Undesirable side effects caused by our product candidates, if any, could cause us, our collaborators, or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA or other comparable foreign regulatory authorities. As a result of any side effects, our clinical trials could be suspended or terminated and the FDA or comparable foreign regulatory authorities could order us to cease further development, or deny approval, of our product candidates for any or all targeted indications. The drug-related side effects could affect patient recruitment or the ability of enrolled patients to complete the trial or result in potential product liability claims. Any of these occurrences may harm our business, financial condition and prospects significantly.
Additionally if one or more of our product candidates receives marketing approval, and we, our collaborators, or others later identify undesirable side effects caused by such products, a number of potentially significant negative consequences could result, including:
• | regulatory authorities may withdraw approvals of such product; |
• | regulatory authorities may require additional warnings on the label; |
• | we may be required to create a medication guide outlining the risks of such side effects for distribution to patients; |
• | we may be sued and held liable for harm caused to patients; and |
• | our reputation may suffer. |
Any of these events could prevent us from achieving or maintaining market acceptance of the particular product candidate, if approved, and could significantly harm our business, results of operations and prospects.
If we are unable to establish sales and marketing capabilities or enter into agreements with third parties to sell and market our product candidates, we may not be successful in commercializing our product candidates if and when they are approved.
We do not have a sales and marketing infrastructure or any experience in the sales, marketing or distribution of pharmaceutical products. We may seek additional third-party collaborators for the commercialization of our other product candidates. In the future, we may choose to build a focused sales and marketing infrastructure to market or co-promote some of our product candidates if and when they are approved, which would be expensive and time-consuming. Alternatively, we may elect to outsource these functions to third parties. Either approach carries significant risks. For example, recruiting and training a sales force is expensive and time-consuming and, if done improperly, could delay a product launch and result in limited sales. If the commercial launch of a product candidate for which we recruit a sales force and establish marketing capabilities is delayed or does not occur for any reason, we would have prematurely or unnecessarily incurred these commercialization expenses. This may be costly, and our investment would be lost if we cannot retain or reposition our sales and marketing personnel.
Factors that may inhibit our efforts to commercialize our products on our own include:
• | our inability to recruit, manage and retain adequate numbers of effective sales and marketing personnel; |
• | the inability of marketing personnel to develop effective marketing materials; |
• | the inability of sales personnel to obtain access to or persuade adequate numbers of physicians to prescribe any future products; |
• | the lack of complementary products to be offered by sales personnel, which may put us at a competitive disadvantage relative to companies with more extensive product lines; and |
• | unforeseen costs and expenses associated with creating an independent sales and marketing organization. |
| 30 |
The availability and amount of reimbursement, if approved, for our product candidates, if any, and the manner in which government and private payors may reimburse for any potential products, are uncertain.
In both U.S. and foreign markets, sales of any products will depend in part on the availability of reimbursement from third-party payors such as government health administration authorities, private health insurers and other organizations. The future magnitude of our revenues and profitability may be affected by the continuing efforts of governmental and third-party payors to contain or reduce the costs of health care. We cannot predict the effect that private sector or governmental health care reforms may have on our business, and there can be no assurance that any such reforms will not have a material adverse effect on our business, financial condition and results of operations.
Third-party payors are increasingly challenging the price and cost-effectiveness of medical products and services. Significant uncertainty exists as to the reimbursement status of newly-approved health care products. There can be no assurance that our products will be considered cost-effective or that adequate third-party reimbursement will be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development. Legislation and regulations affecting the pricing of pharmaceuticals may change before any of our products are approved for marketing. Adoption of such legislation could further limit reimbursement for medical products and services. We, or our collaborators, may elect not to market future products in certain markets.
We may expend our limited resources to pursue a particular research program, product candidate or indication and fail to capitalize on product candidates or indications that may be more profitable or for which there is a greater likelihood of success.
Because we have limited financial and managerial resources, we focus on research programs and eventually product candidates for the indications that we believe are the most scientifically and commercially promising. Our resource allocation decisions may cause us to fail to capitalize on viable scientific or commercial products or profitable market opportunities. In addition, we may spend valuable time and managerial and financial resources on research programs and product candidates for specific indications that ultimately do not yield any scientifically or commercially viable products. If we do not accurately evaluate the scientific and commercial potential or target market for a particular product candidate, we may relinquish valuable rights to that product candidate through collaboration, licensing or other royalty arrangements in situations where it would have been more advantageous for us to retain sole rights to development and commercialization.
Failure to Our Stock
Because of the Exchange Act. The penny stock rules generally applyspecialized scientific nature of our business, our success is highly dependent upon our ability to companies whose common stock is not listedattract and retain qualified scientific and technical personnel, consultants and advisors. We depend greatly on the NASDAQ Stock Market or other national securities exchange and trades at less than $4.00 per share, other than companies that have had average revenue of at least $6,000,000 for the last three years or that have tangible net worth of at least $5,000,000 ($2,000,000 if the company has been operating for three or more years). These rules require, among other things, that brokers who trade penny stockour Chief Executive Officer, Dr. Kunwar Shailubhai. We will also need to persons other than “established customers” complete certain documentation, make suitability inquiries of investors and provide investors with certain information concerning trading in the security, includingrecruit a risk disclosure document and quote information under certain circumstances. Many brokers have decided not to trade penny stocks because of the requirements of the penny stock rules and, as a result, theconsiderable number of broker-dealers willingadditional personnel to actachieve our operating goals and financial reporting obligations. To pursue our product development and marketing and sales plans, we will need to hire additional qualified scientific personnel to perform research and development, as market makerswell as personnel with expertise in clinical testing, government regulation, manufacturing, marketing and sales, which may strain our existing managerial, operational, regulatory compliance, financial and other resources. We also rely on consultants and advisors to assist in formulating our research and development strategy and adhering to complex regulatory requirements. We face competition for qualified individuals from numerous pharmaceutical and biotechnology companies, universities and other research institutions. There can be no assurance that we will be able to attract and retain such securities is limited. If we remain subjectindividuals on acceptable terms, if at all. The failure to the penny stock rules for any significant period, itattract and retain qualified personnel, consultants and advisors could have ana material adverse effect on the market, if any, for our securities. If our securities are subject to the penny stock rules, investors will find it more difficult to dispose of our securities.
| 31 |
Risks Relating to Our Industry
The biopharmaceutical industry is subject to significant regulation and oversight in the United States, in addition to approval of products for sale and marketing.We may be subject, directly or indirectly, to federal and state healthcare fraud and abuse laws, andtransparency, health information privacy and security laws. laws and other healthcare laws and regulations. If we are unable to comply, or have not fully complied, with such laws, we could face substantial penalties.
Healthcare providers, physicians and third-party payors will play a primary role in the recommendation and prescription of any future product candidates we may develop and any product candidates for which we obtain regulatory approval for anymarketing approval. In addition to FDA restrictions on marketing of biopharmaceutical products, we are exposed, directly, or indirectly, through our product candidates and begin commercializing such product candidates in the United States, our operations may be subjectcustomers, to various federal and statebroadly applicable fraud and abuse and other healthcare laws including, without limitation,and regulations that may affect the federal anti-kickback statute. These laws may impact, among other things, our proposed sales, marketingbusiness or financial arrangements and education programs. In addition, we may be subject to patient privacy regulation by both the federal government and the states inrelationships through which we conductwould market, sell and distribute our business.products. The laws that may affect our ability to operate include:
The federalAnti-Kickback Statute which prohibits any person or entity from, among other things, knowingly and willfully offering, paying, soliciting or receiving any remuneration, directly or indirectly, overtly or covertly, in cash or in-kind, to induce or reward either the referring of an individual for, or thepurchasing, leasing, ordering or arranging for the purchase, lease or order of any health care item or service reimbursable, in whole or in part, under Medicare, Medicaid or any other federally financed healthcare program.The term “remuneration” has been broadly interpreted to include anything of value.This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on one hand and prescribers, purchasers and formulary managers on the other hand. Although there are many statutory exemptions and regulatory safe harbors protecting certain common activities from prosecution, the exemptions and safe harbors are drawn narrowly, and practices that involve remuneration intended to induce prescribing, purchases or recommendations may be subject to scrutiny if they do not qualify for an exemption or safe harbor. Our practices may not in all cases meet all of the criteria for safe harbor protection from anti-kickback liability.
The federal false claims and civil monetary penalty laws, including the Federal False Claims Act, which imposes significant penalties and can be enforced by private citizens through civil qui tam actions, prohibits any person or entity from, among other things, knowingly presenting, or causing to be presented, a false, fictitious or fraudulent claim for payment to the federal government, or knowingly making, using or causing to be made, a false statement or record material to a false or fraudulent claim to avoid, decrease or conceal an obligation to pay money to the federal government. In addition, a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act. As a result of a modification made by the Fraud Enforcement and Recovery Act of 2009, a claim includes "any request or demand" for money or property presented to the U.S. government. Further, manufacturers can be held liable under the False Claims Act even when they do not submit claims directly to government payors if they are deemed to "cause" the submission of false or fraudulent claims. Criminal prosecution is also possible for making or presenting a false, fictitious or fraudulent claim to the federal government. Several pharmaceutical and other health care companies have been prosecuted under these laws for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. Other companies have been prosecuted for causing false claims to be submitted because of marketing of the product for unapproved, and thus non-reimbursable, uses.
The federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which, created new federalamong other things, imposes criminal statutes that prohibitliability for executing or attempting to execute a scheme to defraud any healthcare benefit program, including private third-party payors, knowingly and willfully embezzling or stealing from a healthcare benefit program, willfully obstructing a criminal investigation of a healthcare offense, and creates federal criminal laws that prohibit knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statements relatingor representations, or making or using any false writing or document knowing the same to healthcare matters;
HIPAA, as amended by the Health Information Technology and Clinical Health Act of 2009, or HITECH, and its implementing regulations, which impose certain requirements relating to the privacy, security, transmission and transmissionbreach reporting of individually identifiable health information;information upon entities subject to the law, such as health plans, healthcare clearinghouses and healthcare providers and their respective business associates that perform services for them that involve individually identifiable health information. HITECH also created new tiers of civil monetary penalties, amended HIPAA to make civil and criminal penalties directly applicable to business associates, and gave state attorneys general new authority to file civil actions for damages or injunctions in U.S. federal courts to enforce the federal HIPAA laws and seek attorneys’ fees and costs associated with pursuing federal civil actions.
| 32 |
The federal physician payment transparency requirements, sometimes referred to as the “Physician Payments Sunshine Act,” and its implementing regulations, which require certain manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) to report annually to the United States Department of Health and Human Services, or HHS, information related to payments or other transfers of value made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members.
State and foreign law equivalents of each of the above federal laws, such as anti-kickback and false claims laws, whichthat may impose similar or more prohibitive restrictions, and may apply to items or services reimbursed by anynon-governmental third-party payor,payors, including commercial insurers,private insurers.
State and foreign laws that require pharmaceutical companies to implement compliance programs, comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government, or to track and report gifts, compensation and other remuneration provided to physicians and other healthcare providers, and other federal, state and foreign laws governingthat govern the privacy and security of health information or personally identifiable information in certain circumstances, including state health information privacy and data breach notification laws which govern the collection, use, disclosure, and protection of health-related and other personal information, many of which differ from each other in significant ways and mayoften are not have the same effect,pre-empted by HIPAA, thus complicatingrequiring additional compliance efforts.
Because of the breadth of these laws and the narrowness of the safe harbors, it is possible that some of our business activities could be subject to challenge under one or more of these laws.The scope and enforcement of each of these laws is uncertain and subject to rapid change in the current environment of healthcare reform, especially considering the lack of applicable precedent and regulations.Federal and state enforcement bodies have recently increased their scrutiny of interactions between healthcare companies and healthcare providers, which has led to several investigations, prosecutions, convictions and settlements in the healthcare industry.
Ensuring that our business arrangements with third parties comply with applicable healthcare laws and regulations will likely be costly and time consuming. If our operations are found to be in violation of any of the laws described above or any other governmental regulations that apply to us, we may be subject to penalties, including civil, criminal and criminaladministrative penalties, damages, fines, disgorgement, individual imprisonment, exclusion from governmental funded healthcare programs, such as Medicare and Medicaid, contractual damages, reputational harm, diminished profits and future earnings,additional reporting obligations and oversight if we become subject to a corporate integrity agreement or other agreement to resolve allegations of non-compliance with these laws, and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations.
Our employees may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements, which could have a material adverse effect on our business.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include intentional failures to comply with governmental regulations, comply with federal and state health-care fraud and abuse laws and regulations, report financial information or data accurately or disclose unauthorized activities to us. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Employee misconduct could also involve the improper use of information obtained in the course of clinical studies, which could result in regulatory sanctions and serious harm to our reputation. It is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent misconduct may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business and results of operations, including the imposition of significant fines or other sanctions.
In the United States and foreign jurisdictions, there have been, a number ofand continue to be, many legislative and regulatory changes and proposed changes to the healthcare system that could affect our future results of operations. In particular, thereThere have been and continue to be a number of initiatives at the U.S. federal and state levels that seek to reduce healthcare costs. In March 2010 the Patient Protection and Affordable Health Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, or collectively, the PPACA was enacted, which includes measures to significantly change the way health care is financed by both governmental and private insurers. Among the provisions of the PPACA of greatest importance to the pharmaceutical and biotechnology industry are the following:
· | an annual, nondeductible fee on any entity that manufactures or imports certain branded prescription drugs and biologic agents, apportioned among these entities according to their market share in certain government healthcare programs; |
· | implementation of the federal physician payment transparency requirements, sometimes referred to as the “Physician Payments Sunshine Act”; |
· | a licensure framework for follow-on biologic products; |
· | a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research; |
· | establishment of a Center for Medicare Innovation at the Centers for Medicare & Medicaid Services to test innovative payment and service delivery models to lower Medicare and Medicaid spending, potentially including prescription drug spending; |
· | an increase in the statutory minimum rebates a manufacturer must pay under the Medicaid Drug Rebate Program, to 23.1% and 13% of the average manufacturer price for most branded and generic drugs, respectively and capped the total rebate amount for innovator drugs at 100% of the Average Manufacturer Price, or AMP; |
· | a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for certain drugs and biologics, including our product candidates, that are inhaled, infused, instilled, implanted or injected; |
· | extension of manufacturers’ Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations; |
· | expansion of eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to additional individuals and by adding new mandatory eligibility categories for individuals with income at or below 133% of the federal poverty level, thereby potentially increasing manufacturers’ Medicaid rebate liability; |
· | a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50% point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D; and |
· | expansion of the entities eligible for discounts under the Public Health program. |
Some of the provisions of the PPACA have yet to be implemented, and there have been legal and political challenges to certain aspects of the PPACA. Since January 2017, President Trump has signed two executive orders and other directives designed to delay, circumvent, or loosen certain requirements mandated by the PPACA. Concurrently, Congress has considered legislation that would repeal or repeal and replace all or part of the PPACA. While Congress has not passed repeal legislation, the Tax Cuts and Jobs Act of 2017 includes a provision repealing, effective January 1, 2019, the tax-based shared responsibility payment imposed by the PPACA on certain individuals who fail to reportmaintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate”. Congress may consider other legislation to repeal or replace elements of the PPACA.
Many of the details regarding the implementation of the PPACA are yet to be determined, and at this time, the full effect that the PPACA would have on our business remains unclear. There is uncertainty surrounding the applicability of the biosimilars provisions under the PPACA to our product candidates. The FDA has issued several guidance documents, but no implementing regulations, on biosimilars. Many biosimilar applications have been approved over the past few years. It is not certain that we will receive 12 years of biologics marketing exclusivity for any of our products. The regulations that are ultimately promulgated and their implementation are likely to have considerable impact on the way we conduct our business and may require us to change current strategies. A biosimilar is a biological product that is highly similar to an approved drug notwithstanding minor differences in clinically inactive components, and for which there are no clinically meaningful differences between the biological product and the approved drug in terms of the safety, purity, and potency of the product.
Individual states have become increasingly aggressive in passing legislation and implementing regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access, and marketing cost disclosure and transparency measures, and to encourage importation from other countries and bulk purchasing. Legally mandated price controls on payment amounts by third-party payors or other restrictions could harm our business, results of operations, financial arrangements with physicianscondition and others, including reporting any “transfer of value” made or distributedprospects. In addition, regional healthcare authorities and individual hospitals are increasingly using bidding procedures to prescribersdetermine what pharmaceutical products and which suppliers will be included in their prescription drug and other healthcare providersprograms. This could reduce ultimate demand for our products or put pressure on our product pricing, which could negatively affect our business, results of operations, financial condition and reporting any investment interests held by physicians and their immediate family members;
In addition, given recent federal and state government initiatives directed at lowering the total cost of healthcare, Congress and state legislatures will likely continue to focus on healthcare reform, the cost of prescription drugs biologics, and diagnostic testsbiologics and the reform of the Medicare and Medicaid programs. While we cannot predict the full outcome of any such legislation, it may result in decreased reimbursement.reimbursement for drugs and biologics, which may further exacerbate industry-wide pressure to reduce prescription drug prices. This could harm our ability to generate revenues. Increases in importation or re-importation of pharmaceutical products from foreign countries into the United States could put competitive pressure on our ability to profitably price our products, which, in turn, could adversely affect our business, results of operations, financial condition and prospects. We might elect not to seek approval for or market our products in foreign jurisdictions to minimize the risk of re-importation, which could also reduce the revenue we generate from our product sales. It is also possible that other legislative proposals having similar effects will be adopted.
Furthermore, regulatory authorities’ assessment of the data and results required to demonstrate safety and efficacy can change over time and can be affected by many factors, such as the emergence of newadditional information, including on other products, changing policies and agency funding, staffing and leadership. We cannot be sure whether future changes to the regulatory environment will be favorable or unfavorable to our business prospects.
We are subject to U.S. and certain foreign export and import controls, sanctions, embargoes, anti-corruption laws, and anti-money laundering laws and regulations. Compliance with these legal standards could impair our ability to compete in domestic and international markets. We can face criminal liability and other serious consequences for violations, which can harm our business.
We are subject to export control and import laws and regulations, including the U.S. Export Administration Regulations, U.S. Customs regulations, various economic and trade sanctions regulations administered by the U.S. Treasury Department’s Office of Foreign Assets Controls, the U.S. Foreign Corrupt Practices Act of 1977, as amended, or FCPA, the U.S. domestic bribery statute contained in 18 U.S.C. § 201, the U.S. Travel Act, the USA PATRIOT Act, and other state and national anti-bribery and anti-money laundering laws in the countries in which we operate is highly regulatedconduct activities. Anti-corruption laws are interpreted broadly and prohibit companies and their employees, agents, contractors, and other collaborators from authorizing, promising, offering, or providing, directly or indirectly, improper payments or anything else of value to recipients in the failurepublic or private sector. We may engage third parties for clinical trials outside of the United States, to sell our products abroad once we enter a commercialization phase, and/or to obtain necessary permits, licenses, patent registrations, and other regulatory approvals. We have direct or indirect interactions with officials and employees of government agencies or government-affiliated hospitals, universities, and other organizations. We can be held liable for the corrupt or other illegal activities of our employees, agents, contractors, and other collaborators, even if we do not explicitly authorize or have actual knowledge of such activities. Any violations of the laws and regulations described above may result in substantial civil and criminal fines and penalties, imprisonment, the loss of export or import privileges, debarment, tax reassessments, breach of contract and fraud litigation, reputational harm, and other consequences.
An NDA submitted under Section 505(b)(2) subjects us to the risk that we may be subject to a patent infringement lawsuit that would delay or prevent the review or approval of our product candidate.
We plan to submit our RASP-101 product candidate to the FDA for approval under Section 505(b)(2) of the FDCA. Section 505(b)(2) permits the submission of an NDA where at least some of the information required for approval comes from studies that were not conducted by, or for, the applicant and on which the applicant has not obtained a right of reference. The 505(b)(2) application would enable us to reference published literature and/or the FDA’s previous findings of safety and effectiveness for the branded reference drug. For NDAs submitted under Section 505(b)(2) of the FDCA, the patent certification and related provisions of the Hatch-Waxman Act apply. In accordance with the Hatch- Waxman Act, such NDAs may be required to include certifications, known as paragraph IV certifications, that certify that any delayspatents listed in obtaining, regulatory approvals could adversely affect ourthe Patent and Exclusivity Information Addendum of the FDA’s publication, Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the Orange Book, with respect to any product referenced in the 505(b)(2) application, are invalid, unenforceable or will not be infringed by the manufacture, use or sale of the product that is the subject of the 505(b)(2) NDA.
Under the Hatch-Waxman Act, the holder of patents that the 505(b)(2) application references may file a patent infringement lawsuit after receiving notice of the paragraph IV certification. Filing of a patent infringement lawsuit against the filer of the 505(b)(2) applicant within 45 days of the patent owner’s receipt of notice triggers a one-time, automatic, 30-month stay of the FDA’s ability to commercialize ourapprove the 505(b)(2) NDA, unless patent litigation is resolved in the favor of the paragraph IV filer or the patent expires before that time. Accordingly, we may invest a significant amount of time and expense in the development of one or more product candidates if any,only to be subject to significant delay and generate revenue.
We may seek Fast Track designation for one or more of our product candidates, but we might not receive such designation, and even if we do, such designation may not actually lead to a faster development or regulatory review or approval process.
If a product is intended for the treatment of a serious condition and nonclinical or clinical data demonstrate the potential to address unmet medical need for this condition, a product sponsor may apply for FDA Fast Track designation. If we seek Fast Track designation for a product candidate, we may not receive it from the FDA. However, even if we receive Fast Track designation, Fast Track designation does not ensure that we will receive marketing approval or that approval will be granted within any particular timeframe. We may not experience a faster development or regulatory review or approval process with Fast Track designation compared to conventional FDA procedures. In addition, the FDA may withdraw Fast Track designation if it believes that the designation is no longer supported by data from our clinical development program. Fast Track designation alone does not guarantee qualification for the FDA’s priority review procedures.
If the FDA does not conclude that our product candidates satisfy the requirements for the 505(b)(2) regulatory approval pathway, or if the requirements for approval of any of our product candidates under Section 505(b)(2) are not as we expect, the approval pathway for our product candidates will likely take significantly longer, cost significantly more and encounter significantly greater complications and risks than anticipated, and in any case may not be successful.
We intend to seek FDA approval through the 505(b)(2) regulatory pathway for certain of our product candidates, including RASP-101. The Hatch-Waxman Act, added Section 505(b)(2) to the FDCA. Section 505(b)(2) permits the filing of an NDA where at least some of the information required for approval comes from studies that were not conducted by or for the applicant. If the FDA does not allow us to pursue the 505(b)(2) regulatory pathway for our product candidates as anticipated, we may need to conduct additional clinical trials, provide additional data and information and meet additional standards for regulatory approval. If this were to occur, the time and financial resources required to demonstrate throughobtain FDA approval for our product candidates would likely substantially increase. Moreover, the inability to pursue the 505(b)(2) regulatory pathway could result in new competitive products reaching the market faster than our product candidates, which could materially adversely impact our competitive position and prospects. Even if we are allowed to pursue the 505(b)(2) regulatory pathway for a product candidate, we cannot assure you that we will receive the requisite or timely approvals for commercialization of such product candidate. In addition, we expect that our competitors will file citizens’ petitions with the FDA in an attempt to persuade the FDA that our product candidates, or the clinical studies that support their approval, contain deficiencies. Such actions by our tests are effectivecompetitors could delay or even prevent the FDA from approving any NDA that we submit under Section 505(b)(2).
We expect to rely heavily on orphan drug status to commercialize some of our product candidates, if approved, but we might not receive such designation and any orphan drug designations we receive may not confer marketing exclusivity or other expected commercial benefits.
We expect to rely heavily on orphan drug exclusivity for their intended purpose. Thereour product candidates. Designation of orphan drug status is within the discretion of the FDA and the FDA may determine that we do not meet the criteria for orphan drug status. In the United States, orphan drug designation entitles a party to financial incentives such as opportunities for grant funding towards clinical trial costs, tax advantages and user-fee waivers. In addition, if a product that has orphan drug designation subsequently receives the first FDA approval for the disease for which it has such designation, the product is entitled to orphan drug exclusivity. Orphan drug exclusivity in the United States provides that the FDA may not approve any other applications, including a full NDA, to market the same drug for the same indication for seven years, except in limited circumstances the applicable exclusivity period is ten years in Europe. The European exclusivity period can be reduced to six years if a drug no longer meets the criteria for orphan drug designation or if the drug is sufficiently profitable so that market exclusivity is no guaranteelonger justified. Even if we, or any future collaborators, obtain orphan drug designation for a product candidate, we, or they, may not be able to obtain or maintain orphan drug exclusivity for that its testsproduct candidate. We may not be the first to obtain marketing approval of any product candidate for which we have obtained orphan drug designation for the orphan-designated indication due to the uncertainties associated with developing pharmaceutical products, and it is possible that another company also holding orphan drug designation for the same product candidate will receive marketing approval for the same indication before we do. If that were to happen, our applications for that indication may not be approved until the competing company’s period of exclusivity expires. In addition, exclusive marketing rights in the United States may be limited if we seek approval for an indication broader than the orphan-designated indication or processes willmay be lost if the FDA later determines that the request for designation was materially defective or if we are unable to assure sufficient quantities of the product to meet the applicableneeds of patients with the rare disease or condition. Further, even if we, or any future collaborators, obtain orphan drug exclusivity for a product, that exclusivity may not effectively protect the product from competition because different drugs with different active moieties may be approved for the same condition. Even after an orphan drug is approved, the FDA can subsequently approve the same drug with the same active moiety for the same condition if the FDA concludes that the later drug is clinically superior in that it is shown to be safer, more effective or makes a major contribution to patient care or the manufacturer of the product with orphan exclusivity is unable to maintain sufficient product quantity. Orphan drug designation neither shortens the development time or regulatory standards. Thereview time of a drug nor gives the drug any advantage in the regulatory clearancereview or approval process, nor does it prevent competitors from obtaining approval of the same product candidate as ours for indications other than those in which we have been granted orphan drug designation.
We may seek a breakthrough therapy designation for RASP-101 or one or more of our other product candidates, we might not receive such designation, and even if we do, such designation may not lead to a faster development or regulatory review or approval process.
We may seek a breakthrough therapy designation for RASP-101 or one or more of our other product candidates. A breakthrough therapy is defined as a drug that is intended, alone or in combination with one or more other drugs, to treat a serious condition, and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. For drugs and biologics that have been designated as breakthrough therapies, interaction and communication between the FDA and the sponsor of the trial can help to identify the most efficient path for clinical development while minimizing the number of patients placed in ineffective control regimens. Drugs designated as breakthrough therapies by the FDA may also requirebe eligible for priority review if supported by clinical data at the expendituretime the NDA is submitted to the FDA. Designation as a breakthrough therapy is within the discretion of substantial resources, is uncertainthe FDA. Accordingly, even if we believe that one of our product candidates meets the criteria for designation as a breakthrough therapy, the FDA may disagree and subjectinstead determine not to delays.make such designation. Even if we receive breakthrough therapy designation, the receipt of such designation for a product candidate may not result in a faster development or regulatory review or approval process compared to drugs considered for approval under conventional FDA procedures and does not assure ultimate approval by the FDA. In addition, even if one or more of our product candidates qualify as breakthrough therapies, the FDA may later decide that the product candidates no longer meet the conditions for qualification or decide that the time period for FDA review or approval bywill not be shortened.
We may seek priority review designation for RASP-101 or one or more of our other product candidates, but we might not receive such designation, and even if we do, such designation may not lead to a faster development or regulatory authorityreview or approval process.
If the FDA determines that a product candidate offers a treatment for a serious condition and, if approved, the product would provide a significant improvement in one countrysafety or effectiveness, the FDA may designate the product candidate for priority review. A priority review designation means that the goal for the FDA to review an application is six months, rather than the standard review period of ten months. We may request priority review for our product candidates. The FDA has broad discretion with respect to whether or not to grant priority review status to a product candidate, so even if we believe a particular product candidate is eligible for such designation or status, the FDA may decide not to grant it. Moreover, a priority review designation does not ensurenecessarily mean a faster development or regulatory review or approval process or necessarily confer any advantage with respect to approval compared to conventional FDA procedures. Receiving priority review from the FDA does not guarantee approval by regulatory authoritieswithin the six-month review cycle or at all.
| 37 |
Risks Related to Third Parties
We may rely on third parties to conduct our non-clinical studies and our clinical studies. If these third parties do not perform as contractually required or expected, we may not be able to select product candidates or obtain market acceptance for our product candidates, if any, or we may be delayed in doing so.
We often rely on third parties, such as CROs, medical institutions, academic institutions, clinical investigators and contract laboratories, to conduct our clinical studies. We are responsible for confirming that our clinical studies are conducted in accordance with applicable regulations and in accordance with its general investigational plan and protocol. Our reliance on third parties does not relieve us of these responsibilities. If the third parties conducting our clinical studies do not perform their contractual duties or obligations, do not meet expected deadlines, fail to comply with Good clinical practice (GCP), do not adhere to our clinical study protocols or otherwise fail to generate reliable clinical data, we may need to enter into new arrangements with alternative third parties and our clinical study may be more costly than expected or budgeted, extended, delayed or terminated or may need to be repeated, and we may not be able to obtain regulatory approval for or commercialize the target molecules or product candidates, if any, tested in such studies.
We may explore strategic collaborations that may never materialize or may fail.
We may, in the future, periodically explore a variety of possible strategic collaborations including international distributors and partners, in an effort to gain access to new product candidates or resources. At the current time, we cannot predict what form such a strategic collaboration might take. We are likely to face significant competition in seeking appropriate strategic collaborators, and these strategic collaborations can be complicated and time-consuming to negotiate and document. We may not be able to negotiate strategic collaborations on acceptable terms, or at all. We are unable to predict when, if ever, we will enter into any additional strategic collaborations because of the numerous risks and uncertainties associated with establishing strategic collaborations.
Risks Related to Our Intellectual Property
If we are unable to protect our proprietary rights or to defend against infringement claims, we may not be able to compete effectively or operate profitably.
Our success will depend, in part, on our ability to obtain patents, operate without infringing the proprietary rights of others and maintain trade secrets, both in the United States and other countries. Patent matters in the biotechnology and pharmaceutical industries can be highly uncertain and involve complex legal and factual questions. Accordingly, the validity, breadth and
There can be no assurance that we will discover or develop patentable products or processes or that patents will issue from any of the currently pending patent applications or that claims granted on issued patents will be sufficient to protect our technology or adequately cover the actual products we may actually sell. Potential competitors or other researchers in the field may have filed patent applications, been issued patents, published articles or otherwise created prior art that could restrict or block our efforts to obtain additional patents. There also can be no assurance that our issued patents or pending patent applications, if issued, will not be challenged, invalidated, rendered unenforceable or circumvented or that the rights granted hereunder will provide us with proprietary protection or competitive advantages. Our patent rights also depend on our compliance with technology and patent licenses upon which our patent rights are based and upon the validity of assignments of patent rights from consultants and other inventors that were, or are, not employed by us.
In addition, competitors may manufacture and sell our potential products in those foreign countries where we have not filed for patent protection or where patent protection may be unavailable, not obtainable or ultimately not enforceable. In addition, even where patent protection is obtained, third-party competitors may challenge our patent claims in the various patent offices, for example via opposition in the European Patent Office or reexamination or interference proceedings in the United States Patent and Trademark Office, or USPTO. The ability of such competitors to sell such products in the United States or in foreign countries where we have obtained patents is usually governed by the patent laws of the countries in which the product is sold.
We will incur significant ongoing expenses in maintaining our patent portfolio. Should we lack the funds to maintain our patent portfolio or to enforce our rights against infringers, we could be adversely impacted. Even if claims of infringement are without merit, any such action could divert the time and attention of management and impair our ability to access additional capital and/or cost us significant funds to defend.
| 38 |
Intellectual property rights do not necessarily address all potential threats to our competitive advantage.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations, and may not adequately protect our business, or permit us to maintain our competitive advantage. The following examples are illustrative:
• | Others may be able to make compounds that are similar to our product candidates but that are not covered by the claims of the patents that we own or have exclusively licensed. |
• | We or our licensors or strategic collaborators might not have been the first to make the inventions covered by the issued patent or pending patent application that we own or have exclusively licensed. |
• | We or our licensors or strategic collaborators might not have been the first to file patent applications covering certain of our inventions. |
• | Others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our intellectual property rights. |
• | It is possible that our pending patent applications will not lead to issued patents. |
• | Issued patents that we own or have exclusively licensed may not provide us with any competitive advantages, or may be held invalid or unenforceable, as a result of legal challenges by our competitors. |
• | Our competitors might conduct research and development activities in countries where we do not have patent rights and then use the information learned from such activities to develop competitive products for sale in our major commercial markets. |
• | We may not develop additional proprietary technologies that are patentable. |
• | The patents of others may have an adverse effect on our business. |
Should any of these events occur, they could significantly harm our business, results of operations and prospects.
Patent reform legislation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents.
On September 16, 2011, the Leahy-Smith America Invents Act, or the Leahy-Smith Act, was signed into law. The Leahy-Smith Act includes a number of significant changes to U.S. patent law. These include provisions that affect the way patent applications will be prosecuted and may also affect patent litigation. The USPTO is currently developing regulations and procedures to govern administration of the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Leahy-Smith Act will not become effective until one year or 18 months after its enactment. Accordingly, it is not clear what, if any, impact the Leahy-Smith Act will have on the operation of our business, our current and pending patent portfolio and future intellectual property strategy. However, the Leahy-Smith Act and its implementation could increase the uncertainties and
We may be subject to litigation with respect to the ownership and use of intellectual property that will be costly to defend or pursue and uncertain in its outcome.
Our success also will depend, in part, on our refraining from infringing patents or otherwise violating intellectual property owned or controlled by others. Pharmaceutical companies, biotechnology companies, universities, research institutions and others may have filed patent applications or have received, or may obtain, issued patents in the United States or elsewhere relating to aspects of our technology. It is uncertain whether the issuance of any third-party patents will require us to alter our products or processes, obtain licenses, or cease certain activities. Some third-party applications or patents may conflict with our issued patents or pending applications. Any such conflict could result in a significant reduction of the scope or value of our issued or licensed patents.
In addition, if patents issued to other companies contain blocking, dominating or conflicting claims and such claims are ultimately determined to be valid, we may be required to obtain licenses to these patents or to develop or obtain alternative non-infringing technology and cease practicing those activities, including potentially manufacturing or selling any products deemed to infringe those patents. If any licenses are required, there can be no assurance that we will be able to obtain any such licenses on commercially favorable terms, if at all, and if these licenses are not obtained, we might be prevented from pursuing the development and commercialization of certain of our potential products. Our failure to obtain a license to any technology that we may require to commercialize our products on favorable terms may have a material adverse impact on our business, financial condition and results of operations.
| 39 |
Litigation, which could result in substantial costs to us (even if determined in our favor), may also be necessary to enforce any patents issued or licensed to us or to determine the scope and validity of the proprietary rights of others. The FDA has only recently published draft guidance documents for implementation of the Biologics Price Competition and Innovation Act (BPCIA) under the PPACA, related to the development of follow-on biologics (biosimilars), and detailed guidance for patent litigation procedures under this act has not yet been provided. If another company files for approval to market a competing follow-on biologic, and/or if such approval is given to such a company, we may be required to promptly initiate patent litigation to prevent the marketing of such biosimilar version of our product prior to the normal expiration of the patent. There can be no assurance that our issued or licensed patents would be held valid by a court of competent jurisdiction or that any follow-on biologic would be found to infringe our patents.
In addition, if our competitors file or have filed patent applications in the United States that claim technology also claimed by us, we may have to participate in interference proceedings to determine priority of invention. These proceedings, if initiated by the USPTO, could result in substantial costs to us, even if the eventual outcome is favorable to us. Such proceedings can be lengthy, are costly to defend and involve complex questions of law and fact, the outcomes of which are difficult to predict. Moreover, we may have to participate in post-grant proceedings or third-party ex parte or inter partes reexamination proceedings under the USPTO. An adverse outcome with respect to a third-party claim or in an interference proceeding could subject us to significant liabilities, require us to license disputed rights from third parties, or require us to cease using such technology, any of which could have a material adverse effect on our business, financial condition and results of operations.
The patent protection and patent prosecution for some of our target molecules and product candidates, if any, is dependent or may be dependent in the future on third parties.
While we normally seek and gain the right to fully prosecute the patents relating to our target molecules and product candidates, if any, there may be times when platform technology patents or product-specific patents that relate to the target molecules or product candidates are controlled by our licensors. In addition, our licensors and/or licensees may have back-up rights to prosecute patent applications in the event that we do not do so or choose not to do so, and our licensees may have the right to assume patent prosecution rights after certain milestones are reached. If any of our licensing collaborators fails to appropriately
We have licensed, or will license, from third parties certain technology necessary to develop and commercialize its therapeutics. If these licenses terminate, or if these third parties do not comply with the terms of the license, or if the underlying licensed patents are found to be invalid, our business could be negatively impacted.
We have licensed, or will license, from third parties, technology necessary to research, develop and commercialize LSD1.our product candidiates. In return for the use of their technology, we have paid or agreed, or will agree, to pay the licensor certain fees. We may need to license additional technology to in the future. If these licenses terminate, if the licensors fail to abide by the terms of the license or fail to prevent infringement by third parties, if the licensed patents or other rights are found to be invalid or if we are unable to enter into necessary licenses on acceptable terms, our business could be negatively impacted.
We may incur significant expensesbecome involved in lawsuits to protect or enforce our patents or other intellectual property, which could be preventedexpensive, time consuming and unsuccessful.
Competitors may infringe our patents, trademarks, copyrights or other intellectual property. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time consuming and divert the time and attention of our management and scientific personnel. Any claims we assert against perceived infringers could provoke these parties to assert counterclaims against us alleging that we infringe their patents, in addition to counterclaims asserting that our patents are invalid or unenforceable, or both. In any patent infringement proceeding, there is a risk that a court will decide that a patent of ours is invalid or unenforceable, in whole or in part, and that we do not have the right to stop the other party from developingusing the invention at issue. There is also a risk that, even if the validity of such patents is upheld, the court will construe the patent’s claims narrowly or commercializingdecide that we do not have the right to stop the other party from using the invention at issue on the grounds that our product candidates,patent claims do not cover the invention. An adverse outcome in a litigation or proceeding involving our patents could limit our ability to assert our patents against those parties or other competitors, and may curtail or preclude our ability to exclude third parties from making and selling similar or competitive products. Any of these occurrences could adversely affect our competitive business position, business prospects and financial condition. Similarly, if we assert trademark infringement claims, a court may determine that the marks we have asserted are invalid or unenforceable, or that the party against whom we have asserted trademark infringement has superior rights to the marks in question. In this case, we could ultimately be forced to cease use of such trademarks.
Even if we establish infringement, the court may decide not to grant an injunction against further infringing activity and instead award only monetary damages, which may or may not be an adequate remedy. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during litigation. There could also be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a material adverse effect on the price of shares of our common stock. Moreover, there can be no assurance that we will have sufficient financial or other resources to file and pursue such infringement claims, which typically last for years before they are concluded. Even if we ultimately prevail in such claims, the monetary cost of such litigation and the diversion of the attention of our management and scientific personnel could outweigh any benefit we receive as a result of anthe proceedings.
We may rely on trade secret and proprietary know-how which can be difficult to trace and enforce and, if we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed.
We also rely on trade secrets to protect technology, especially where patent protection is not believed to be appropriate or obtainable or where patents have not issued. For example, our manufacturing process involves many trade secret steps, processes, and conditions. Trade secrets and know-how can be difficult to protect. We attempt to protect our proprietary technology and processes, in part, with confidentiality agreements and assignment of invention agreements with our employees and confidentiality agreements with our consultants and certain contractors. Despite these efforts, any of these parties may breach the agreements and disclose our proprietary information, including our trade secrets, and we may not be able to obtain adequate remedies for such breaches. Any disclosure, either intentional or unintentional, by our employees, the employees of third parties with whom we share our facilities or third party consultants and vendors that we engage to perform research, clinical studies or manufacturing activities, or misappropriation by third parties (such as through a cybersecurity breach) of our trade secrets or proprietary information could enable competitors to duplicate or surpass our technological achievements, thus eroding our competitive position in our market.
Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive and time-consuming, and the outcome is unpredictable. In addition, some courts inside and outside the United States are less willing or unwilling to protect trade secrets. If any of our trade secrets were to be lawfully obtained or independently developed by a competitor or other third party, we would have no right to prevent them from using that technology or information to compete with us. If any of our trade secrets were to be disclosed to or independently developed by a competitor or other third party, our competitive position would be harmed.
There can be no assurance that these agreements are valid and enforceable, will not be breached, that we would have adequate remedies for any breach, or that our trade secrets will not otherwise become known or be independently discovered by competitors. We may fail in certain circumstances to obtain the necessary confidentiality agreements or assignment of invention agreements, or their scope or term may not be sufficiently broad to protect our interests or transfer adequate rights to us.
If our trade secrets or other intellectual property infringement claim.
We may not be able to protect our intellectual property rights throughout the world.
Patents are of national or regional effect, and filing, prosecuting and defending patents on all of our product candidates throughout the world would be prohibitively expensive. As such, we may not be able to prevent third parties from practicing our inventions in all countries outside the United States, or from selling or importing products made using our inventions in and into the United States or other jurisdictions. Further, the legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection, particularly those relating to pharmaceuticals or biologics, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights ofgenerally. In addition, certain developing countries, including China and India, have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties. Infringement proceedingsIn those countries, we and our licensors may have limited remedies if patents are infringed or if we or our licensors are compelled to grant a license to a third party, which could materially diminish the value of those patents. This could limit our potential revenue opportunities. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
Patent terms may be inadequate to protect our competitive position on our product candidates for an adequate amount of time.
Patent rights are of limited duration. Given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such product candidates are commercialized. Even if patents covering our product candidates are obtained, once the patent life has expired for a product, we may be open to competition from biosimilar or generic products. A patent term extension based on regulatory delay may be available in the biopharmaceutical industryUnited States. However, only a single patent can be extended for each marketing approval, and any patent can be extended only once, for a single product. Moreover, the scope of protection during the period of the patent term extension does not extend to the full scope of the claim, but instead only to the scope of the product as approved. Laws governing analogous patent term extensions in foreign jurisdictions vary widely, as do laws governing the ability to obtain multiple patents from a single patent family. Additionally, we may not receive an extension if we fail to apply within applicable deadlines, fail to apply prior to expiration of relevant patents or otherwise fail to satisfy applicable requirements. If we are unable to obtain patent term extension or restoration, or the term of any such extension is less than we request, the period during which we will have the right to exclusively market our product will be shortened and our competitors may obtain approval of competing products following our patent expiration, and our revenue could be reduced, possibly materially.
We may be lengthy, costlysubject to claims by third parties asserting that our employees or we have misappropriated their intellectual property or claiming ownership of what we regard as our own intellectual property.
Some of our employees and time-consumingour licensors’ employees, including our senior management, were previously employed at universities or at other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Some of these employees may have executed proprietary rights, non-disclosure and non-competition agreements, or similar agreements, in connection with such previous employment. Although we try to ensure that our employees do not use the proprietary information or know-how of others in their outcome is uncertain.work for us, we may be subject to claims that we or these employees have used or disclosed intellectual property, including trade secrets or other proprietary information, of any such third party. Litigation may be necessary to defend against such claims. If we become involvedfail in defending any patent litigation, interferencesuch claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or other administrative proceedings, it will incur substantial expensepersonnel or sustain damages. Such intellectual property rights could be awarded to a third party, and the efforts of its technical and management personnel will be significantly diverted. As a result of such litigation or proceedings, we could lose our proprietary position and be restricted or prevented from developing, manufacturing and selling its product candidates, if any, incur significant damage awards, including punitive damages, or be required to seek third-party licenses thatobtain a license from such third party to commercialize our technology or products. Such a license may not be available on commercially acceptablereasonable terms ifor at all. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management.
In addition, while we typically require our employees, consultants and contractors who may be involved in the development of intellectual property to execute agreements assigning such intellectual property to us, we may be unsuccessful in executing such an agreement with each party who in fact develops intellectual property that we regard as our own, which may result in claims by or against us related to the ownership of such intellectual property. If we fail in prosecuting or defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights. Even if we are successful in prosecuting or defending against such claims, litigation could result in substantial costs and be a distraction to our senior management and scientific personnel.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submissions, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees on any issued patent are due to be paid to the USPTO and foreign patent agencies in several stages over the lifetime of the patent. The USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. While an inadvertent lapse can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Non-compliance events that could result in abandonment or lapse of a patent or patent application include, but are not limited to, failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. If we fail to maintain the patents and patent applications covering our product candidates, our competitive position would be adversely affected.
We are exposed to potential product liability or similar claims, and insurance against these claims may not be available to us at a reasonable rate in the future.
Our business exposes us to potential liability risks that are inherent in the testing, manufacturing and marketing of human therapeutic products. Clinical trials involve the testing of product candidates on human subjects or volunteers under a research plan, and carry a risk of liability for personal injury or death to patients due to unforeseen adverse side effects, improper administration of the product candidate, or other factors. Many of these patients are already seriously ill and are therefore particularly vulnerable to further illness or death.
We currently do not carry clinical trial liability insurance but we will need to once we begin clinical trials. There can be no assurance that we will be able to obtain such insurance or that the amount of such insurance will be adequate to cover claims. We could be materially and adversely affected if we were required to pay damages or incur defense costs in connection with a claim outside the scope of indemnity or insurance coverage, if the indemnity is not performed or enforced in accordance with its terms, or if our liability exceeds the amount of applicable insurance. In addition, there can be no assurance that insurance will continue to be available on terms acceptable to us, if at all, or that if obtained, the insurance coverage will be sufficient to cover any potential claims or liabilities. Similar risks would exist upon the commercialization or marketing of any products by us or our collaborators.
Regardless of their merit or eventual outcome, product liability claims may result in:
• | decreased demand for our product; |
• | injury to our reputation and significant negative media attention; |
• | withdrawal of clinical trial volunteers; |
• | costs of litigation; |
• | distraction of management; and |
• | substantial monetary awards to plaintiffs. |
Should any of these events occur, it could have a material adverse effect on our business and financial condition.
Risks Relating to Our Stock
Our common stock is deemed a “penny stock,” which would make it more difficult for our investors to sell their shares.
Our common stock is subject to the Company“penny stock” rules adopted under Section 15(g) of the Exchange Act. The penny stock rules generally apply to companies whose common stock is not listed on the NASDAQ Stock Market or other national securities exchange and trades at less than $4.00 per share, other than companies that have had average revenue of at least $6,000,000 for the last three years or that have tangible net worth of at least $5,000,000 ($2,000,000 if the company has been engagedoperating for three or more years). These rules require, among other things, that brokers who trade penny stock to persons other than “established customers” complete certain documentation, make suitability inquiries of investors and provide investors with certain information concerning trading in the security, including a risk disclosure document and quote information under certain circumstances. Many brokers have decided not to trade penny stocks because of the requirements of the penny stock rules and, as a result, the number of broker-dealers willing to act as market makers in such securities is limited. If we remain subject to the penny stock rules for any significant period, it could have an adverse effect on the market, if any, for our securities. If our securities are subject to the penny stock rules, investors will find it more difficult to dispose of our securities.
We have not paid cash dividends in the past and do not expect to pay dividends in the future. Any return on investment may be limited to the value of our common stock.
We have never paid cash dividends on our common stock and do not anticipate doing so in the foreseeable future. The payment of dividends on our common stock will depend on earnings, financial condition and other business and economic factors affecting us at such time as our board of directors may consider relevant. If we do not pay dividends, our common stock may be less valuable because a return on your investment will only occur if our stock price appreciates.
The price of our common stock may fluctuate substantially.
You should consider an investment in our common stock to be risky, and you should invest in our common stock only if you can withstand a significant loss and wide fluctuations in the market value of your investment. Some factors that may cause the market price of our common stock to fluctuate, in addition to the other risks mentioned in this “Risk Factors” section and elsewhere in this prospectus, are:
· | sale of our common stock by our stockholders, executives, and directors; |
· | volatility and limitations in trading volumes of our shares of common stock; |
· | our ability to obtain financings to conduct and complete research and development activities including, but not limited to, our human clinical trials, and other business activities; |
· | our announcements or our competitors’ announcements regarding new products or services, enhancements, significant contracts, acquisitions or strategic investments; |
· | failures to meet external expectations or management guidance; |
· | changes in our capital structure or dividend policy; |
· | our cash position; |
· | announcements and events surrounding financing efforts, including debt and equity securities; |
· | our inability to enter into new markets or develop new products; |
· | reputational issues; |
· | announcements of acquisitions, partnerships, collaborations, joint ventures, new products, capital commitments, or other events by us or our competitors; |
· | changes in general economic, political and market conditions in any of the regions in which we conduct our business; |
· | changes in industry conditions or perceptions; |
· | changes in valuations of similar companies or groups of companies; |
· | analyst research reports, recommendation and changes in recommendations, price targets, and withdrawals of coverage; |
· | departures and additions of key personnel; |
· | disputes and litigations related to contractual obligations; |
· | changes in applicable laws, rules, regulations, or accounting practices and other dynamics; or |
· | other events or factors, many of which may be out of our control. |
In addition, if the market for stocks in our industry or industries related to our industry, or the stock market in general, experiences a loss of investor confidence, the trading price of our common stock could decline for reasons unrelated to our business, financial condition and results of operations. If any of the foregoing occurs, it could cause our stock price to fall and may expose us to lawsuits that, even if unsuccessful, could be costly to defend and a distraction to management.
Our ability to use our net operating loss carry-forwards and certain other tax attributes is limited by Sections 382 and 383 of the Internal Revenue Code.
Net operating loss carryforwards allow companies to use past year net operating losses to offset against future years’ profits, if any, to reduce future tax liabilities. Sections 382 and 383 of the Internal Revenue Code of 1986 limit a corporation’s ability to utilize its net operating loss carryforwards and certain other tax attributes (including research credits) to offset any future taxable income or tax if the corporation experiences a cumulative ownership change of more than 50% over any rolling three-year period. State net operating loss carryforwards (and certain other tax attributes) may be similarly limited. An ownership change can therefore result in significantly greater tax liabilities than a corporation would incur in the absence of such a change and any increased liabilities could adversely affect the corporation’s business, results of operations, financial condition and cash flow.
Even if another ownership change has not occurred and does not occur as a result of this offering, additional ownership changes may occur in the future as a result of additional equity offerings or events over which we will have little or no control, including purchases and sales of our equity by our five percent security holders, the emergence of new five percent security holders, redemptions of our securities or certain changes in the ownership of any of our five percent security holders.
Our principal stockholders and management own a significant percentage of our stock and will be able to exercise significant influence over matters subject to stockholder approval.
As of September 30, 2019, our executive officers, directors and principal security holders, together with their respective affiliates, owned approximately 50.3% of our outstanding securities. Accordingly, this group of security holders will be able to exert a significant degree of influence over our management and affairs and over matters requiring security holder approval, including the election of our Board of Directors, future issuances of our securities, declaration of dividends and approval of other significant corporate transactions. This concentration of ownership could have the effect of delaying or preventing a change-of-control of our company or otherwise discouraging a potential acquirer from attempting to obtain control of us, which in turn could have a material and adverse effect on the fair market value of our securities. In addition, this significant concentration of share ownership may adversely affect the trading price for our common stock if investors perceive disadvantages in owning stock in a comment letter processcompany with such concentrated ownership.
U.S. federal income tax reform could adversely affect us.
On December 22, 2017, President Trump signed into law the “Tax Cuts and Jobs Act” (TCJA) that significantly reforms the Internal Revenue Code of 1986, as amended. The TCJA, among other things, includes changes to U.S. federal tax rates, imposes significant additional limitations on the deductibility of interest, allows for the expensing of capital expenditures, and puts into effect the migration from a “worldwide” system of taxation to a territorial system. The tax reform did not have a material impact to our projection of minimal cash taxes or to our net operating losses. Our net deferred tax assets and liabilities are revalued at the newly enacted U.S. corporate rate, and the impact has been recognized in our tax expense in the year of enactment. Further, any eligibility we may have or may someday have for tax credits associated with the Staffqualified clinical testing expenses arising out of the SEC relatingdevelopment of orphan drugs was reduced to questions raised by25% as a result of the SECTCJA; thus, our net taxable income is affected. We continue to examine the impact this tax reform legislation may have on our business. The impact of this tax reform on holders of our common stock is uncertain and could be adverse. This prospectus does not discuss any such tax legislation or the way it might affect purchasers of our common stock. We urge our stockholders, including purchasers of common stock in this offering, to consult with their legal and tax advisors with respect to such legislation and the potential tax consequences of investing in our common stock.
In preparing our consolidated financial statements, our management determined that our disclosure controls and procedures and internal controls were ineffective as of September 30, 2019 and if they continue to be ineffective could result in material misstatements in our financial statements.
Our management is responsible for establishing and maintaining adequate internal control over our financial reporting, as defined in Rule 13a-15(f) under the Securities Exchange Act of 1934, as amended, or the Exchange Act. As of September 30, 2019, our management has determined that our disclosure controls and procedures and internal controls were ineffective due to weaknesses in our financial closing process.
We intend to implement remedial measures designed to address the ineffectiveness of our disclosure controls and procedures and internal controls. If these remedial measures are insufficient to address the ineffectiveness of our disclosure controls and procedures and internal controls, or if material weaknesses or significant deficiencies in our internal control are discovered or occur in the future and the ineffectiveness of our disclosure controls and procedures and internal controls continues, we may fail to meet our future reporting obligations on a timely basis, our consolidated financial statements may contain material misstatements, we could be required to restate our prior period financial results, our operating results may be harmed, we may be subject to class action litigation, and our common stock could be delisted. Any failure to address the ineffectiveness of our disclosure controls and procedures could also adversely affect the results of the periodic management evaluations regarding the Company’s accounting for its acquired In- processeffectiveness of our internal control over financial reporting and our disclosure controls and procedures that are required to be included in our annual report on Form 10-K. Internal control deficiencies and ineffective disclosure controls and procedures could also cause investors to lose confidence in our reported financial information. We can give no assurance that the measures we plan to take in the future will remediate the ineffectiveness of our disclosure controls and procedures or that any material weaknesses or restatements of financial results will not arise in the future due to a failure to implement and maintain adequate internal control over financial reporting or adequate disclosure controls and procedures or circumvention of these controls. In addition, even if we are successful in strengthening our controls and procedures, in the future those controls, and procedures may not be adequate to prevent or identify irregularities or errors or to facilitate the fair presentation of our consolidated financial statements.
Future sales and issuances of our common stock or rights to purchase common stock pursuant to our equity incentive plan could result in additional dilution of the percentage ownership of our stockholders and could cause our share price to fall.
We expect that significant additional capital will be needed in the future to continue our planned operations, including expanding research and development, intangible asset.funding clinical trials, purchasing of capital equipment, hiring new personnel, commercializing our products, and continuing activities as an operating public company. To the extent we raise additional capital by issuing equity securities, our stockholders may experience substantial dilution. We may sell common stock, convertible securities or other equity securities in one or more transactions at prices and in a manner, we determine from time to time. If we sell common stock, convertible securities or other equity securities in more than one transaction, investors may be materially diluted by subsequent sales. Such sales may also result in material dilution to our existing stockholders, and new investors could gain rights superior to our existing stockholders.
We are an ‘‘emerging growth company’’ and will be able to avail ourselves of reduced disclosure requirements applicable to emerging growth companies, which could make our common stock less attractive to investors.
We are an ‘‘emerging growth company,’’ as defined in the JOBS Act and we intend to take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not ‘‘emerging growth companies’’ including not being required to comply with the auditor attestation requirements of Section 404(b) of the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. In addition, Section 107 of the JOBS Act also provides that an ‘‘emerging growth company’’ can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act, for complying with new or revised accounting standards. In other words, an ‘‘emerging growth company’’ can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We are electing to delay such adoption of new or revised accounting standards, and as a result, we may not comply with new or revised accounting standards on the relevant dates on which adoption of such standards is required for non-emerging growth companies. As a result of such election, our financial statements may not be comparable to the financial statements of other public companies. We cannot predict if investors will find our common stock less attractive because we may rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile. We may take advantage of these reporting exemptions until we are no longer an ‘‘emerging growth company.’’ We will remain an ‘‘emerging growth company’’ until the earliest of (i) the last day of the fiscal year in which we have total annual gross revenues of $1.07 billion or more; (ii) the last day of our fiscal year following the fifth anniversary of the date of the completion of this filing,offering; (iii) the Company hasdate on which we have issued more than $1 billion in nonconvertible debt during the previous three years; or (iv) the date on which we are deemed to be a large accelerated filer under the rules of the Securities and Exchange Commission.
We may be at risk of securities class action litigation.
We may be at risk of securities class action litigation. This risk is especially relevant for us due to our dependence on positive clinical trial outcomes and regulatory approvals of each of our product candidates. In the past, biotechnology and pharmaceutical companies have experienced significant stock price volatility, particularly when associated with binary events such as clinical trials and product approvals. If we face such litigation, it could result in substantial costs and a diversion of management’s attention and resources, which could harm our business and results in a decline in the market price of our common stock.
Our articles of incorporation and bylaws and Nevada law may have anti-takeover effects that could discourage, delay or prevent a change in control, which may cause our stock price to decline.
Our amended and restated articles of incorporation, our amended and restated bylaws and Nevada law could make it more difficult for a third party to acquire us, even if closing such a transaction would be beneficial to our stockholders. Although we currently do not yet resolved those questionshave shares of preferred stock outstanding, our board of directors could authorize the issuance of a series of preferred stock that would grant holders preferred rights to our assets upon liquidation, special voting rights, the right to receive dividends before dividends would be declared to common stockholders, and the right to the redemption of such shares, possibly together with a premium, prior to the redemption of the common stock. To the extent that we do issue preferred stock, the rights of holders of common stock could be impaired thereby, including without limitation, with respect to liquidation.
Provisions of our bylaws may also prevent or frustrate attempts by our stockholders to replace or remove our management. In particular, our bylaws, among other things:
- provide the board of directors with the
Staffability to alter the bylaws without stockholder approval; and - provide that vacancies on the board of directors may be filled by a majority of directors in office, although less than a quorum.
Reports published by securities or industry analysts, including projections in those reports that exceed our actual results, could adversely affect our common stock price and trading volume.
Securities research analysts, including those affiliated with our underwriters from this offering, establish and publish their own periodic projections for our business. These projections may vary widely from one another and may not accurately predict the results we actually achieve. Our stock price may decline if our actual results do not match securities research analysts’ projections. Similarly, if one or more of the SEC. Ifanalysts who writes reports on us downgrades our stock or publishes inaccurate or unfavorable research about our business or if one or more of these analysts ceases coverage of our company or fails to publish reports on us regularly, our stock price or trading volume could decline. While we expect securities research analyst coverage to continue going forward, if no securities or industry analysts begin to cover us, the Company is unable to satisfytrading price for our stock and the Staff’s questions, then the intangible assettrading volume could be written off in a yet to be determined period.
| 46 |
Item 1B. Unresolved Staff Comments
None.
Our executive offices are located at 420 Lexington Avenue, 25th25th Floor, New York, NY 10170.10170. We do not own any real property.
We are currently sharing approximately 3,011 square feet of laboratory and office space for our headquarters in Lexington Avenue, New York with Tiziana Therapeutics Inc. The lease is under Tiziana Therapeutic's Inc. name and we are being charged for a portion of the space via a Shared Services agreement.
We are also currently sharing approximately 652 square feet of office space in our office in London, United Kingdom. The lease is under Tiziana Pharma Limited's name and we are being charged for a portion of the space via a Shared Services agreement.
We also leasedlease approximately 451 square feet of lab and office space in Doylestown, Pennsylvania under a lease agreement that expiresexpired January 31, 2018.2019. The lease was renewed under the same terms and expires on 31 January 2020. The lease has a renewable option. We believe that our facilities are adequate for our needs for the immediate future and that, should it be needed, suitable additional space will be available to accommodate expansion of our operations on commercially reasonable terms.
We are not involved in any pending legal proceeding or litigationslitigation and, to the best of our knowledge, no governmental authority is contemplating any proceeding to which we are a party or to which any of our properties is subject, which would reasonably be likely to have a material adverse effect on us.
Market for Securities
Our common shares are currently quoted on the OTCQXOTCQB under the trading symbol “RASP”. Our shares were previously quoted on the OTCQB Market from September 27, 2016 to April 21, 2017.2017 and the OTCQX market from April 22, 2017 until October 5, 2018. Prior to September 27, 2016, our shares were quoted on the OTC Market under the trading symbol “ATVM.”“ATVM”. Prior to September 27, 2016, there were no trades in our common stock. The following table sets forth the range of high and low per share sales prices of our common stock during the periods indicated, as reported on the OTC Markets.
| 2017 | ||||
| High | Low | |||
| First Quarter | $1.31 | $1.31 | ||
| Second Quarter | $1.31 | $1.31 | ||
| Third Quarter | $2.80 | $1.05 | ||
| Fourth Quarter | $2.18 | $1.85 | ||
Our transfer agent is Philadelphia Stock Transfer, Inc., 2320 Haverford Road, Ardmore, PA 19003.19003.
As of June 15, 2017,September 30, 2019, there were 6460 registered stockholders of our issued and outstanding common stock.
Dividend Policy
We have never declared or paid any cash dividends on our common stock and we do not anticipate paying any dividends or making any other distributions in the foreseeable future. The payment by us of dividends, if any, in the future, rests within the discretion of our board of directors and will depend, among other things, upon our earnings, capital requirements and financial condition.
Securities Authorized for Issuance Under Equity Compensation Plans
The following table summarizes information about our equity compensation plans as of March 31, 2017.September 30, 2019.
|
|
|
|
| Number of Shares of Common Stock to be Issued upon Exercise of Outstanding Options, Warrants and Rights | Weighted- Average Exercise Price of Outstanding Options, Warrants and Rights | Number of Options Remaining Available for Future Issuance Under Equity Compensation Plans (excluding securities reflected in column (a)) |
|
|
|
|
Equity Compensation Plans Approved by Stockholders | 4,073,675 | $0.59 | 4,920,125 |
Equity Compensation Plans Not Approved by Stockholders | 1,926,501 | $0.43 | — |
Total | 6,000,176 |
| 4,920,125 |
Number of Shares of Common Stock to be Issued upon Exercise of Outstanding Options, Warrants and Rights | Weighted- Average Exercise Price of Outstanding Options, Warrants and Rights | Number of Options Remaining Available for Future Issuance Under Equity Compensation Plans (excluding securities reflected in column (a)) | |
| Equity Compensation Plans Approved by Stockholders | 3,162,375 | $0.29 | 6,587,625 |
| Equity Compensation Plans Not Approved by Stockholders | - | - | - |
| Total | 3,162,375 | $0.29 | 6,587,625 |
Not applicable.
| 49 |
Forward-Looking Statements
The following discussion and analysis of our financial condition and results of operations should be read in conjunction with the financial statements and the related notes and other information that are included elsewhere in this Form 10-K. This discussion contains forward-looking statements based upon current expectations that involve risks and uncertainties, such as our plans, objectives, expectations and intentions. Actual results and the timing of events could differ materially from those anticipated in these forward-looking statements as a result of a number of factors, including those set forth under the cautionary note regarding “Forward-Looking Statements” contained elsewhere in this Form 10-K. Additionally, you should read the “Risk Factors” section of this Form 10-K for a discussion of important factors that could cause actual results to differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis.
| 50 |
Our audited financial statements are stated in United States dollars and are prepared in accordance with United States generally accepted accounting principles.
We assume no obligation to revise or publicly release the results of any revision to these forward-looking statements, except as required by law. Given these risks and uncertainties, readers are cautioned not to place undue reliance on such forward-looking statements.
Unless expressly indicated or the context requires otherwise, the terms "Rasna,",” the “Company,” “we,” “us,” and “our” refer to Rasna Therapeutics, Inc., a Nevada corporation, and, where appropriate, its wholly owned subsidiaries.
Company Background
Overview
To date, we have devoted substantially all of our resources to research and development efforts relating to our therapeutic candidates, including conducting clinical trials and developing manufacturing capabilities, in-licensing related intellectual property, protecting our intellectual property and providing general and administrative support for these operations. Since our inception, we have funded our operations primarily through the issuance of equity securities.
We anticipate that our expenses will increase substantially if and as we:
| continue enrollment in our ongoing clinical trials; | ||
| initiate new clinical trials; | ||
| seek to identify, assess, acquire and develop other products, therapeutic candidates and technologies; | ||
| seek regulatory and marketing approvals in multiple jurisdictions for our therapeutic candidates that successfully complete clinical studies; | ||
| establish collaborations with third parties for the development and commercialization of our products and therapeutic candidates; | ||
| make milestone or other payments under our agreements pursuant to which we have licensed or acquired rights to intellectual property and technology | ||
| seek to maintain, protect, and expand our intellectual property portfolio; | ||
| seek to attract and retain skilled personnel; | ||
| incur the administrative costs associated with being a public company and related costs of compliance; | ||
| create additional infrastructure to support our operations as a commercial stage public company and our planned future commercialization efforts; and | ||
| experience any delays or encounter issues with any of the above. |
We expect to continue to incur significant expenses and increasing losses for at least the next several years. Accordingly, we anticipate that we will need to raise additional capital in addition to the net proceeds from this offering in order to obtain regulatory approval for, and the commercialization of our therapeutic candidates. Until such time that we can generate meaningful revenue from product sales, if ever, we expect to finance our operating activities through public or private equity or debt financings, government or other third-party funding, marketing and distribution arrangements and other collaborations, strategic alliances and licensing arrangements or a combination of these approaches. If we are unable to obtain funding on a timely basis, we may be required to significantly curtail, delay or discontinue one or more of our research or development programs or the commercialization of any approved therapies or products or be unable to expand our operations or otherwise capitalize on our business opportunities, as desired, which could materially adversely affect our business, financial condition and results of operations.
On October 11, 2017, we changed our fiscal year end from March 31 to September 30 and we filed a Form 10-KT on November 30, 2017.
On April 27, 2016, Rasna Therapeutics Limited, a private limited company incorporated in England and Wales under the StateU.K. Companies Act (“Rasna UK”) sold its stake in Falconridge Holdings Limited, or Falconridge,to Rasna DE for $1. This entity had no operations, assets or liabilities as of Nevada on December 6, 2012.
On May 17, 2016, Rasna DE and its subsidiary Falconridge entered into an Agreement of Merger and Plan of Reorganization (the “Merger Agreement”) with Arna Therapeutics Limited, (“Arna”) is a company incorporated in the British Virgin Islands under applicable lawcompany, or Arna, which was a clinical stage biotechnology company focused on drugs to treat diseases in oncology and regulationimmunology, mainly focusing on December 31, 2013.the treatment of leukemia. Pursuant to the Merger Agreement, Arna was merged into Falconridge and the shareholders of Arna were issued shares of Rasna DE in exchange for shares of Arna.
On August 15, 2016, Active With Me, Inc., or AWM, entered into an Agreement of Merger and Plan of Reorganization with Rasna DE, and Rasna Acquisition, providing for the merger of Rasna Acquisition with and into Rasna DE, (the “Merger”), with Rasna DE, surviving the Merger as a wholly-owned subsidiary of AWM. As a result of the Merger, the resulting company, Rasna Therapeutics, Inc., is a biotechnology company that is engaged in modulating the molecular targets NPM1 and LSD1, which are implicated in the disease progression of leukemia and lymphoma.
The Merger has been treated as a reverse recapitalization effected by a share exchange for financial accounting and reporting purposes since substantially all of AWM’s operations were disposed of prior to the consummation of the transaction. Rasna DE is treated as the accounting acquirer as its stockholders control us after the Merger Agreement, even though AWM was treated as the legal acquirer. As a result of the Merger, the assets and liabilities and the historical operations that are reflected in our financial statements are those of Rasna DE as if Rasna DE had always been the reporting company. Since the transaction was treated as a reverse recapitalization for accounting purposes, no goodwill or other intangible assets were recorded by us as a result of the Merger Agreement.
Thereafter, pursuant to a Stock Purchase Agreement, the Company transferred all of the outstanding capital stock of Rasna DE to a former officer and director of AWM in exchange for cancellation of an aggregate of 1,500,000 shares of Rasna DE’s common stock held by such person.
In connection with the share exchange, each share of Rasna DE was exchanged for the right to receive .33 shares in AWM. Once issued, the new shares were combined with the 3,305,000 common shares held by legacy AWM shareholders. Immediately following the Merger, 1,500,000 shares were canceled, which related to one legacy AWM shareholder that effectively spun off the remaining assets of AWM in connection with the transaction. Finally, subsequent to the transaction, the legal acquirer executed a 3.25 for 1 stock split on its common shares. Historical common stock amounts and additional paid-in capital have been retroactively adjusted for the effect of the share splits executive in connection with the Merger transaction.
On September 20, 2016, we filed a Certificate of Change in Nevada which effected a 3.25 for 1 forward stock split of our common stock for shareholders of record as of August 16, 2016 and increased the authorized number of shares of common stock to 200,000,000 shares.
We only has have one segment of activity which is that of a clinical stage biotechnology company focused on targeted drugs to treat diseases in oncology and immunology, mainly focusing on the treatment of leukemia.
Acquisitions
The following transactions were accounted for using the purchase accounting method which requires, among other things, that the assets acquired and liabilities assumed are recognized at their acquisition date fair value.
On May 5, 2016, Rasna UK sold its intellectual property to Falconridge, a subsidiary of Rasna DE, for a note payable in the amount of $236,269.$236,269. The fair value of the intellectual property was deemeddetermined to be the $236,269$236,269 based on the consideration received.
On May 17, 2016, Arna was merged into Falconridge and the shareholders of Arna were issued shares of Rasna DE, in exchange for shares of Arna. On this day, Rasna DE, and its subsidiary Falconridge entered into an Agreement of Merger and Plan of Reorganization with Arna. Pursuant to the agreement, Arna was merged into Falconridge and the shareholders of Arna were issued shares of Rasna DE, in exchange for shares of Arna. Arna was deemed to be the accounting acquirer because Rasna DE and Falconridge Holdings Limited were non-trading holding companies and Arna’s operations will comprise the ongoing operations of the combined entity and its senior management will serve as the senior management of the combined entity. Further, 65%65% of
The consideration transferred was measured based upon the share price recently received during a non-public equity raise in Rasna DE, during which non-related investors paid $0.40$0.40 per share of common stock. During the acquisition transaction, 19,187,500 of 54,837,790 shares were issued to legacy Rasna DE shareholders, which results in consideration transferred to the acquiree’s shareholders of $7,675,000.
In addition, $607,159$607,159 of a related party receivable due to Arna from Rasna UK, was forgiven as part of the consideration transferred.
Critical Accounting Policies and Estimates
This discussion and analysis of our financial condition and results of operations is based on our financial statements, which have been prepared in accordance with generally accepted accounting principles in the United States of America, or GAAP. The preparation of these financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities, disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenue and expenses during the reported period. In accordance with US GAAP, we base our estimates on historical experience and on various other assumptions that we believe are reasonable under the circumstances. Actual results may differ from these estimates under different assumptions or conditions.
While our significant accounting policies are more fully described in Note 2 to our audited financial statements appearing elsewhere in this Annual Report, we believe the following accounting policies are critical to the process of making significant judgments and estimates in the preparation of our financial statements.
Basis of preparation
The accompanying consolidated financial statements have been prepared in conformity with accounting principles generally accepted in the United States of America (“US GAAP”). Any reference in these notes to applicable guidance is meant to refer to GAAP as found in the Accounting Standards Codification (“ASC”) and Accounting Standards Updates (“ASU”) of the Financial Accounting Standards Board (“FASB”).
Principles of Consolidation
In accordance with ASC 810
,Consolidation, the Company consolidates any entity in which it has a controlling financial interest. Further, the Company consolidates any variable interest entity that it is deemed to be the primary beneficiary of, and have the power to direct its significant activities. Upon review of the relationship between Rasna Therapeutics (“Rasna UK”) and theThe consolidated financial statements include the financial statements of the Company and its wholly owned subsidiary, Rasna DE, and Rasna DE’s subsidiary, Arna Therapeutics Limited. All significant intercompany accounts and transactions have been eliminated in the preparation of the accompanying consolidated financial statements.
Business Combinations
Management accounts for business combinations under the provisions of Accounting Standards Codification ("ASC")ASC Topic 805-10,805-10, Business Combinations, ("ASC 805-10"), which requires that the purchase method of accounting be used for all business combinations. Assets acquired and liabilities assumed, including non-controlling interests, are recorded at the date of acquisition at their respective fair values. ASC 805-10805-10 also specifies criteria that intangible assets acquired in a business combination must meet to be recognized and reported apart from goodwill. Goodwill represents the excess purchase price over the fair value of the tangible net assets and intangible assets acquired in a business combination. Acquisition-related expenses are recognized separately from the business combinations and are expensed as incurred.
Management is required to complete the purchase price allocation within 12 months of the acquisition date. If such completion of the allocation results in a change in the preliminary values, the measurement period adjustment will be recognized in the period in which the adjustment amount is determined in accordance with ASU 2015-16.2015-16. The amounts reflected within the
Going Concern
We are subject to a number of risks similar to those of other pre-commercial stage companies, including its dependence on key individuals, uncertainty of product development and generation of revenues, dependence on outside sources of capital, risks associated with research, development, testing, and obtaining related regulatory approvals of its pipeline products, suppliers and collaborators, successful protection of intellectual property, competition with larger, better-capitalized companies, successful completion of the Company'sour development programs and, ultimately, the attainment of profitable operations are dependent on future events, including obtaining adequate financing to fulfill its development activities and generating a level of revenues adequate to support the Company's cost structure.
We have experienced net losses and significant cash outflows from cash used in operating activities over the past two years, and as at March 31, 2017,ofSeptember 30, 2019, had an accumulated deficit of $9,257,780,$17,311,809, a net lossfor the year ended March 31, 2017September 30, 2019 of $4,433,732$936,380 and net cash used in operating activities of $2,963,858.
We expect to continue to incur net losses and have significant cash outflows for at least the next twelve months. We have sufficient funds to continue operating until the end of the second fiscal quarter of 2019, December 2020, but will require significant additional cash resources to launch new development phases of existing products in its pipeline. In the event that the Company iswe are unable to secure the necessary additional cash resources needed, the Companywe may need to slow current development phases or halt new development phases in order to mitigate the effects of the costs of development.
| 53 |
Use of Estimates
The preparation of financial statements in conformity with US GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent liabilities at the date of the financial statements and the reported amounts of revenue and expenses during the reporting period. We evaluate our estimates on an ongoing basis, including those related to the fair values of stock based compensation awards, income taxes and contingent liabilities, among others. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable, the results of which form the basis for making judgments about the carrying valuesamounts of assets and liabilities. Actual results could differ from those estimates and such differences could be material to our consolidated financial position and results of operations.
Fair Value of Financial Instruments
The carrying value of the Company’sour financial instruments, including cash and cash equivalents, accounts payable and accrued liabilities, approximate fair value because of the short-term nature of such financial instruments. Management measures certain other assets, at fair value on a nonrecurring basis when they are deemed to be other-than-temporarily impaired.
Concentration of Credit Risk
Financial instruments that potentially subject the Companyus to concentrations of credit risk consist principally of related party receivables.
Deposits held with banks, including those held in foreign branches of global banks, may exceed the amount of insurance provided on such deposits. These deposits may be redeemed upon demand and bear minimal risk. Management believes that the institutions that hold our instruments are financially sound and are subject to minimal credit risk.
Cash and cash equivalents
Cash and cash equivalents consists of cash on deposit with banks with an original maturity of three months or less.
From time to time, the Company’sour balances in its bank accounts exceed Federal Deposit Insurance Corporation limits. The CompanyWe will periodically evaluate the risk of exceeding insured levels and might transfer funds if it deems appropriate. The Company hasWe have not experienced any losses with regards to balances in excess of insured limits or as a result of other concentrations of credit risk
Goodwill and Intangible assets
Intangible assets are made up of in-process research and development, (“IPR&D”) and certain intellectual property (“IP”). The balance of IPR&D represents IPR&D acquired in 2013, which, at the time, was determined to have alternative future uses. IPR&D assets also represent the fair value assigned to acquired technologies in a business combination, which at the time of the business combination have not reached technological feasibility and have no alternative future use. IP assets represent the fair value assigned to technologies, which at the time of acquisition have reached technological feasibility, however, have not yet been put into service. Intangible assets are considered to have an indefinite useful life until the completion or abandonment of the associated research and development projects.
Goodwill represents the premium paid over the fair value of the net tangible and intangible assets acquired in business combinations. Goodwill is not amortized; rather, it is subject to a periodic assessment for impairment by applying a fair value based test. Goodwill is assessed for impairment on an annual basis or more frequently if events or changes in circumstances indicate that the asset might be impaired. An impairment charge is recognized only when the implied fair value of the Company’sour reporting unit’s goodwill is less than its carrying amount.
Management evaluates indefinite life intangible assets for impairment on an annual basis and on an interim basis if events or changes in circumstances between annual impairment tests indicate that the asset might be impaired. The ongoing evaluation for impairment of its indefinite life intangible assets requires significant management estimates and judgment. Management reviews definite life intangible assets for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. There were no impairment charges as of the year ended March 31, 2017September 30, 2019 and March 31, 2016.
Risks and Uncertainties
We intend to operate in an industry that is subject to rapid change. The Company’sOur operations will be subject to significant risk and uncertainties including financial, operational, technological, regulatory, and other risks associated with an early stage company, including the potential risk of business failure.
| 54 |
Research and development
Expenditure on research and development is charged to the statementsstatement of operations in the year in which it is incurred with the exception of expenditures incurred in respect of the development of major new products where the outcome of those projects is assessed as being reasonably certain in regards to viability and technical feasibility. Such expenditure is capitalized and amortized straight line over the estimated period of sale for each product, commencing in the year that sales of the product are first made. To date, the Company haswe have not capitalized any such expenditures other than certain IPR&D & IP recorded in connection with certain acquisition or equity transactions.
Income Taxes
We account for income taxes under the asset and liability method. Deferred tax assets and liabilities are recognized for future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which the temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date. Management considers many factors when assessing the likelihood of future realization of deferred tax assets, including recent earnings experience by jurisdiction, expectations of future taxable income, and the carryforward periods available for tax reporting purposes, as well as other relevant factors. A valuation allowance may be established to reduce deferred tax assets to the amount that management believes is more likely than not to be realized. Due to inherent complexities arising from the nature of the business, future changes in income tax law and variances between actual and anticipated operating results, management makes certain judgments and estimates. Therefore, actual income taxes could materially vary from these estimates.
We recognize in the financial statements the impact of a tax position, if that position is more likely than not to be sustained upon an examination, based on the technical merits of the position. The Company recordsWe record a liability for the difference between the benefit recognized and measured and the tax position taken or expected to be taken on the Company’sour tax return. To the extent that the assessment of such tax positions changes, the change in estimate is recorded in the period in which the determination is made. To the extent interest and penalties are not assessed with respect to uncertain tax positions, amounts accrued will be reduced and reflected as a reduction of the overall income tax provision. The Companyprovision.We have incurred no liability and, therefore, did not need to record interest and penalties during the year ended March 31, 2017September 30, 2019 and 2016.
Foreign Currency
Items included in the financial statements are measured using their functional currency, beingwhich is the currency of the primary economic environment in which the company operates. The Company's consolidated financial statements are presented in United States Dollar (“USD”), which is the company’s functional and presentational currency.
Foreign currency transactions are translated using the rate of exchange applicable at the date of the transaction. Foreign exchange gains and losses resulting from the settlement of such transactions and from the re-translation at the year-end of monetary assets and liabilities denominated in foreign currencies are recognized in the statements of operations.
Net Loss per Share
Basic net loss per share is computed by dividing net loss available to common shareholders by the weighted average number of common shares outstanding during the period. Diluted net loss per share includes potentially dilutive securities such as outstanding options and warrants, using various methods such as the treasury stock or modified treasury stock method in the determination of dilutive shares outstanding during each reporting period.
| 55 |
The following table sets forth potential common shares issuable upon the exercise of outstanding options, and the exercise of warrants and conversion of loan notes, all of which have been excluded from the computation of diluted weighted average shares outstanding as they would be anti-dilutive, including the impact on dilutive net loss per share of in-the-money warrants as per ASC 260-10-45-35260-10-45-35 through ASC 260-10-45-37:260-10-45-37:
|
|
|
|
|
|
| September 30, 2019 |
| September 30, 2018 | ||
Stock options | 4,073,675 |
|
| 4,819,875 |
|
Warrants | 1,926,501 |
|
| 1,926,501 |
|
| Convertible Notes | 1,723,333 | 900,000 | |||
Total shares issuable upon exercise or conversion | 7,723,509 |
|
| 7,646,376 |
|
| March 31, 2017 | March 31, 2016 | ||||
| Stock options | 3,162,375 | 1,662,375 | |||
| Warrants | 1,440,501 | — | |||
| Total shares issuable upon exercise or conversion | 4,602,876 | 1,662,375 | |||
Warrants
In April 2016,August 2017, in connection with the issuance of equity, the Company committed to issue warrants as compensation to the placement agents. On February 28, 2017, the Companywe issued a ten year warrant to purchase 1,440,501112,000 shares of common stock at an exercise price of $0.37$0.65 per share.
In September 2017, in connection with a consultancy services agreement, we issued ten year warrants was marked to market each period until the grant date,purchase 374,000 shares of common stock at which point the Company an exercise price of $0.60 per share, in lieu of cash payments.
We determined that in accordance with ASC 815-40-25-7,815-40-25-7, all the warrants wereare to be classified as equity in stockholder’s equity.
Convertible Notes
In August 2018, we entered into a 12% Convertible Promissory Note. The Note has an original purchase amount of $135,000, bears interest at a rate of 12% per annum calculated on a 360-day year basis. In July 2019, the maturity of the note was extended from August 8, 2019 to August 8, 2020, unless earlier paid, redeemed or converted in accordance with the terms of the Agreement.
In October 2018, we entered into an additional12% Convertible Promissory Note. The Note has an original purchase amount of $100,000, bears interest at a rate of 12% per annum calculated on a 360-day year basis. In July 2019, the maturity of the note was extended from October 20, 2019 to October 19, 2020, unless earlier paid, redeemed or converted in accordance with the terms of the Agreement.
Equity-Based Payments
ASC Topic 718 “Compensation-Stock Compensation” requires companies to measure the cost of employee services received in exchange for the award of equity instruments based on the estimated fair value of the award at the date of grant. The expense is to be recognized over the period during which an employee is required to provide services in exchange for the award. The Company accountsWe account for shares of common stock, stock options and warrants issued to employees based on the fair value of the stock, stock option or warrant, if that value is more reliably measurable than the fair value of the consideration or services received.
We account for stock options issued and vesting to non-employees in accordance with ASC Topic 505-50505-50 “Equity -Based Payment to Non-Employees” and accordingly the value of the stock compensation to non-employees is based upon the measurement date as determined at either a) the date at which a performance commitment is reached, or b) at the date at which the necessary performance to earn the equity instruments is complete. Accordingly the fair value of these options is being “marked to market” quarterly until the measurement date is determined.
Accounting Pronouncements Not Yet Adopted
Recent Accounting Pronouncements
In July 2017, FASB, issued ASU, 2017-11, Earnings Per Share (Topic 260); Distinguishing Liabilities from Equity (Topic 480); Derivatives and Hedging (Topic 815): (Part I) Accounting for Certain Financial Instruments with Down Round Features, (Part II) Replacement of the Indefinite Deferral. The ASU applies to issuers of financial instruments with down-round features. It amends (1) the classification of such instruments as liabilities or equity by revising the guidance in ASC 815 on the evaluation of whether instruments or embedded features with down-round provisions must be accounted for as derivative instruments and (2) the guidance on recognition and measurement of the value transferred upon the trigger of a down-round feature for equity-classified instruments by revising ASC 260. The ASU is effective for public business entities for fiscal years, and interim periods within those fiscal years, beginning after December 15, 2018. Early adoption is permitted. The Company is currently evaluating the impact off adopting this guidance.
In January 2017, the FASB issued ASU 2017-04,2017-04, Intangibles -Goodwill and Other (Topic 350)350): Simplifying the Test for Goodwill Impairment, which addresses the concerns over the cost and complexity of the two-steptwo-step impairment test, and removes the second step of the test.An entity will apply a one-stepone-step quantitative test and record the amount of goodwill impairment as the excess of a reporting unit’s carrying amount over its fair value, not to exceed the total amount of good willgoodwill allocated to the reporting unit.The guidance is effective for annual and interim goodwill impairment tests performed for periods beginning after December 15, 2019 with early adoption permitted in January 2017.The2019. The Company is currently evaluating the impactimpact of adopting this guidance.guidance on our consolidated financial statements.
In June 2018, the FASB issued ASU 2018-07, Compensation - Stock Compensation (Topic 718): Improvements to Non employee Share Based Payment Accounting, which simplifies several aspects of the accounting for nonemployee share-based payment transactions resulting from expanding the scope of Topic 718, to include share-based payment transactions for acquiring goods and services from nonemployees. Some of the areas for simplification apply only to nonpublic entities. The amendments specify that Topic 718 applies to all share-based payment transactions in which a grantor acquires goods or services to be used or consumed in a grantor’s own operations by issuing share-based payment awards. The amendments in ASU 2018-07 also clarify that Topic 718does not apply to share-based payments used to effectively provide (1) financing to the issuer or (2) awards granted in conjunction with selling goods or services to customers as part of a contract accounted for under Topic 606, Revenue from Contracts with Customers. The amendments in this Update are effective for public business entities for fiscal years beginning after December 15, 2018, including interim periods within that fiscal year. Early adoption is permitted. We do not plan to early adopt this ASU. We are currently evaluating the potential impacts of this updated guidance on the Comapny's consolidated financial statements.
In August 2018, the FASB issued ASU 2018-13, Fair Value Measurement (Topic 820): Disclosure Framework-Changes to the Disclosure Requirements for Fair Value Measurement. ASU 2018-13 removes certain disclosures, modifies certain disclosures and adds additional disclosures. The ASU is effective for annual periods, including interim periods within those annual periods, beginning after December 15, 2019. Early adoption is permitted. We are currently evaluating the effect that this update will have on its financial statements and related disclosures.
| 57 |
Results of Operations
The following paragraphs set forth our results of operations for the periods presented. The period-to-period comparison of financial results is not necessarily indicative of future results.
Results of Operations for the Years Ended March 31, 2017years ended September 30, 2019 and 2016
| For the Year Ended March 31, | ||||||||
| 2017 | 2016 | |||||||
| Revenue | $ | — | $ | — | ||||
| Cost of revenue | — | — | ||||||
| Gross profit | — | — | ||||||
| Operating expenses: | ||||||||
| General and administrative | 1,009,240 | — | ||||||
| Research and development | 1,564,353 | — | ||||||
| Consultancy fees third parties | 1,071,777 | 128,176 | * | |||||
| Consultancy fees related parties | 150,000 | 350,000 | ||||||
| Legal and professional fees | 739,158 | 99,930 | ||||||
| Total operating expenses | 4,534,528 | 578,106 | ||||||
| Loss from operations | (4,534,528 | ) | (578,106 | ) | ||||
| Other income/(expense): | ||||||||
| Foreign currency transaction gain | 100,796 | — | ||||||
| Other income | 100,796 | — | ||||||
| Net loss | $ | (4,433,732 | ) | $ | (578,106 | ) | ||
Revenues
There were no revenues for the year ended March 31, 2017September 30, 2019 and 20162018 because Rasna Therapeutics, Inc. doeswe do not have any commercial biopharmaceutical products.
Operating Expenses
Operating expenses consisting of research and development costs, consultancy fees, legal and professional fees and general and administrative expenses for the year ended March 31, 2017 increasedSeptember 30, 2019decrease to $4,534,528 $915,170from $578,106$4,070,313 for the year ended March 31, 2016, an increaseSeptember 30, 2018, a decrease of $3,956,422.$3,155,143. The increasedecrease is primarily attributable to the pace of development of the LSD1, NPM1LSD1, NPM1 and ACT D projects which have led to an increasedecreased while the direction of the programs were being evaluated based on results achieved so far, along with a decrease in researchlegal and development, consultancyprofessional fees and general administrative costs.
Net Loss
Net loss for the year ended March 31, 2017 increased to $4,433,732September 30, 2019decrease to $936,380 from $578,106$4,076,361 for the year ended March 31, 2016, an increaseSeptember 30, 2018, a decrease of $3,855,626. $3,139,981. The increasedecrease is primarily attributable to the pace of development of the LSD1, NPM1LSD1, NPM1 and ACT D projects which have led to an increasedecreased while the direction of the programs are being evaluated. along with a decrease in researchlegal and development, consultancyprofessional fees and general administrative costs.
Liquidity and Capital Resources
On December 20, 2016, the Company issued an aggregate of 3,366,667 shares of common stock at $0.60 per share for aggregate gross proceeds of $2,020,000, in connectionAugust 8, 2018, we entered into a 12% Convertible Promissory Note with a securities purchase agreement with certain accredited investors, as defined in Regulation D promulgated under Securities Act of 1933. The net proceedsHolder pursuant to which we issued a Convertible Promissory Note to the Company were $2,007,500.
On October 19, 2018, we entered into a 12% Convertible Promissory Note with a Holder pursuant to carry out its activities untilwhich we issued a Convertible Promissory Note to the second fiscal quarter of 2019.Holder. The CompanyHolder provided us with $100,000 in cash, which we received in October 2018.
We will be required to raise additional capital to continue the development and commercialization of current product candidates and to fund operations. We cannot be certain that additional funding will be available on acceptable terms, or at all. To the extent that we raise additional funds by issuing equity securities, our shareholders may experience significant dilution. Any debt financing, if available, may (i) involve restrictive covenants that impact our ability to conduct, delay, scale back or discontinue the development and/or commercialization of one or more product candidates; (ii) seek collaborators for product candidates at an earlier stage than otherwise would be desirable and on terms that are less favorable than might otherwise be available; or (iii) relinquish or otherwise dispose of rights to technologies, product candidates or products that we would otherwise seek to develop or commercialize its self on unfavorable terms.
Capital Resources
The following table summarizes total current assets, liabilities and working capital as of the periods indicated:
|
|
|
|
|
|
|
|
|
|
|
|
| September 30, 2019 |
| September 30, 2018 |
| Change | ||||||
Current assets | $ | 71,579 |
|
| $ | 347,495 |
|
| $ | (275,916 | ) |
Current liabilities | $ | 2,405,496 |
|
| $ | 2,108,995 |
|
| $ | 296,501 | |
Working capital deficiency | $ | (2,333,917) |
| $ | (1,761,500 | ) |
| $ | (572,417) | ||
| March 31, 2017 | March 31, 2016 | Change | |||||||||
| Current assets | $ | 4,199,874 | $ | 607,159 | $ | 3,592,715 | |||||
| Current liabilities | $ | 1,528,002 | $ | 628,227 | $ | 899,775 | |||||
| Working capital | $ | 2,671,872 | $ | (21,068 | ) | $ | 2,692,940 | ||||
We had a cash balance of $4,048,962$50,068 and $0,$42,693 as of March 31, 2017September 30, 2019 and March 31, 2016,September 30, 2018, respectively.
The following table sets forth a summary of our cash flows for the periods indicated:
| For the year ended September 30, |
| For the year ended September 30, |
| |||||||
| 2019 |
| 2018 |
| Increase/(Decrease) | ||||||
Net cash used in operating activities | $ | (92,625) |
| $ | (2,629,800 | ) |
| $ | 2,537,175 | ||
Net cash used in investing activities | $ | — |
| $ | (4,073 | ) |
| $ | 4,073 | ||
Net cash provided by financing activities | $ | 100,000 |
|
| $ | 135,000 |
|
| $ | (35,000 | ) |
| For the Year Ended March 31, | |||||||||||
| 2017 | 2016 | Increase/(Decrease) | |||||||||
| Net cash used in operating activities | $ | (2,963,858 | ) | $ | — | $ | (2,963,858 | ) | |||
| Net cash provided by investing activities | $ | 5,106,116 | $ | — | $ | 5,106,116 | |||||
| Net cash provided by financing activities | $ | 2,007,500 | $ | — | $ | 2,007,500 | |||||
Net Cash Used in Operating Activities
Net cash used in operating activities was $2,963,858$92,625 for the year ended March 31, 2017September 30, 2019 compared to $0$2,629,800 for the year ended March 31, 2016. September 30, 2018.The change is principally attributable to net loss of $4,433,732$936,380 excluding non-cash items such as share based compensation of $1,023,555$368,076 and changes in operating assets and liabilities of $442,856$452,225 and for the year ended March 31, 2017September 30, 2019 as compared toa net loss of $578,106,$4,076,361, adjusted for non-cash share based compensation of $60,676$621,931 and changes in operating assets and liabilities of $517,430 $264,083for the year ended March 31, 2016.
Net Cash Provided by Investing Activities
Net cash provided byused in investing activities consists primarily of cashproperty and cash equivalents acquired in the business combination on May 17, 2016equipment purchases of $5,116,609$0 and $4,073 for the yearyears ended March 31, 2017 compared to $0 for the year ended March 31, 2016.
Net Cash Provided by Financing Activities
Net cash provided by financing activities consists of shares proceeds from the issuance of common stock issued in a private placement to accredited investorsconvertible note of $2,007,500 for $100,000during the year ended March 31, 2017September 30, 2019 compared to $0 for $135,000of proceeds from the issuance of a convertible note during the year ended March 31, 2016.September 30, 2018.
| 59 |
Off-Balance Sheet Arrangements
We consolidate variable interest entities (“VIE”) in which we hold a controlling financial interest as evidenced by the power to direct the activities of a VIE that most significantly impact its economic performance and the obligation to absorb losses of, or the right to receive benefits from, the VIE that could potentially be significant to the VIE and therefore are deemed to be the primary beneficiary. We take into account our entire involvement in a VIE (explicit or implicit) in identifying variable interests that individually or in the aggregate could be significant enough to warrant our designation as the primary beneficiary and hence require us to consolidate the VIE or otherwise require us to make appropriate disclosures.
Capital Market Risk.
We currently do not have any revenues because we do not have any commercial products and therefore depend on funds raised through other sources. One source of funding is through future debt or equity offerings. Our ability to raise funds in this manner depends upon, among other things, capital market forces affecting our stock price.
The full text of our audited consolidated financial statements as of March 31, 2017September 30, 2019 and 2016,2018, and for the years ended March 31, 2017September 30, 2019 and 20162018 begins on page F-1F-1 of this Annual Reportreport on Form 10-K.
None.
Evaluation of Disclosure Controls and Procedures
Disclosure Controls and Procedures
The Company’s management, with the participation of management’s assessment regardingthe Company’s Chief Executive Officer and Chief Financial Officer, have evaluated the effectiveness of the Company’s disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended (the“Exchange Act”)) as of September 30, 2019. Based on that evaluation, the Company’s Chief Executive Officer and the Company’s Chief Financial Officer have concluded that as of September 30, 2019, due to the existence of the material weaknesses in the Company’s internal control over financial reporting described below, the Company’s disclosure controls and procedures were not effective.
Management’s Annual Report on Internal Control over Financial Reporting
The Company’s management is responsible for establishing and maintaining adequate internal controls over financial reporting as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act. The Company’s internal control over financial reporting are a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles (“US GAAP”).
Because of their inherent limitations, internal controls over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Under the supervision and with the participation of management, the Company’s Chief Executive Officer and the Company’s
Chief Financial Officer, the Company conducted an attestation reportevaluation of the company’s registered public accounting firmeffectiveness of its internal control over financial over financial reporting based on the framework described in Internal Control-Integrated Framework issued by the Commission of Sponsoring Organizations of the Treadway Commission, as revised in 2013. Based on that evaluation, management has concluded that the Company did not maintain effective internal control over financial reporting as of the period ended September 30, 2019 due to a transition period established by rulesthe existence of the Securitiesmaterial weaknesses in internal control over financial reporting described below.
Material Weaknesses
A material weakness is a deficiency, or a combination of deficiencies, in internal controls over financial reporting, such that there is a reasonable possibility that a material misstatement of the Company’s annual or interim financial statements will not be prevented or detected on a timely basis.
Management has determined that the Company did not maintain effective internal control over financial reporting as of the period ended September 30, 2019 due to the existence of the following material weaknesses identified by management:
Lack of Accounting Resources
The Company had a lack of accounting resources resulting in inadequate monitoring controls and other oversight procedures, which resulted in corrected and uncorrected misstatements.
Control Environment
The Company did not maintain an effective control environment. The control environment, which is the responsibility of senior management, sets the tone of the organization, influences the control consciousness of its people, and is the foundation for all other components of internal control over financial reporting. Our control environment was ineffective because:
• | We did not implement a Whistleblower hotline in time for it to be effective; |
• | We developed an Insider Trading policy, which is within our code of ethics policy, but it should be a separate and more robust policy to include a definition of “insiders” versus “employees”, “black-out periods”, and how to contact the Compliance Officer if there are questions or concerns prior to executing a trade. In addition, it should be communicated and acknowledged by all employees, officers, and directors; |
• | We did not timely develop and communicate an employee handbook for employees to consult in the event an issue arises; and |
• | We did not complete formal performance evaluations for employees responsible for governance and internal controls. |
Remediation efforts to address the material weakness relating to the Control Environment
Management intends to remediate this item in the following manner:
i. | Develop and implement the policies as listed herein. These policies will be approved by the Audit Committee and posted on the Company’s website. |
ii. | Perform a recertification, at least annually, of all employees that they have read and understand the Company’s Code of Ethics. |
iii. | Develop and maintain an Employee Handbook, even if electronically, for employees to reference. |
iv. | Perform evaluations of all employees who impact Internal Controls over Financial Reporting. |
Accordingly, management has determined that this control deficiency constitutes a material weakness. Management will begin implementing the Remediation Plan described herein and intends to continue working on it through the year ended September 30, 2019.
Changes in internal control over financial reporting
Other than as noted above, there have been no changes in our internal control over financial reporting (as such term is defined in Rules 13a-15(f) and 15(d)-15(f) under the Exchange Commission for newly public companies.
None.
D
irectors and Executive OfficersThe following persons are our executive officers and directors as of June 29, 2017September 30, 2019 and hold the positions set forth opposite their respective names.
Name | Age | Position | ||
Kunwar Shailubhai | Chief Executive Officer, Director | |||
John Brancaccio | 71 | Director | ||
Alessandro Padova | 51 | Chairman | ||
Tiziano Lazzaretti | 60 | Chief Financial Officer |
Chief Executive Officer, Director
Dr Shailubhai has been a director of our company since August 2016, and CEO since April 2017. Dr. Shailubhai is a Co-Founder of Synergy Pharmaceuticals Inc. and has served as Chief Scientific Officer since 2008.from July 2008 until May 2017. From March 2004 until July 2008 he served as Senior Vice President, Drug Discovery, of Synergy which at that time was a subsidiary of Callisto Pharmaceuticals, Inc. (“Synergy DE”). From May 2003 until March 2004, Dr. Shailubhai served as Executive Vice President, Research and Development of Synergy DE. From 2001 to April 2003, Dr. Shailubhai held the position of Vice President, Drug Discovery at Synergy DE where he was chiefly responsible for the preclinical development of our GC-C agonist program for drugs to treat colon cancerDE. Between 1993 and GI inflammation. Between 1993 and 2000, he was with Monsanto Company, serving as Group Leader of the cancer chemoprevention group. Dr. Shailubhai previously served as a Senior Staff Fellow at the National Institutes of Health, and as an Assistant Professor at the University of Maryland. Dr. Shailubhai received his Ph.D. in microbiology in 1984 from the University of Baroda, India, and his M.B.A. in 2001 from the University of Missouri, St. Louis. We believe that Dr Shailubhai's scientific background and clinical experience make him well qualified to serve as a director on our board.
John Brancaccio
Director
Mr. Brancaccio has made fundamental contributions to the field of cancer, especially in the study of the molecular genetics of B cell malignancies. As a researcher, he has contributed much of the current knowledge on the genetic lesions responsible for human B cell lymphoma, which have led to the development of diagnostic tests and are being tested as targets in clinical trials with lymphoma patients. A Columbia faculty member for more than 15 years, Dr. Dalla-Favera helped found and has led the Institute for Cancer Genetics at Columbia University since 1999. He is the Percy and Joanne Uris Professor of Pathology and Professor of Genetics & Development at the Columbia University College of Physicians and Surgeons. He is alsobeen a director of the Specialised Center for research on Lymphoma at Columbia University funded by the Leukemia Lymphoma Society. Dr. Dalla-Favera joined Columbia’s College of Physicians and Surgeons in the Department of Pathology in 1989. He completed a fellowship at the National Cancer Institute and was previously a faculty member at New York University School of Medicine. Dr. Dalla-Favera currently serves as a director of Tiziana Life Sciences plc. Dr. Dalla-Favera co-founded Therasis, Inc. in 2007 and has since served as a director of that company. He has been a member of the Scientific Advisory Board of Trovagene, Inc. since April 2010. He serves as a member of the Scientific Advisory Board of Xigen SA. Dr. Dalla-Favera was a director of the Herbert Irving Comprehensive Cancer Centre (HICCC) from 2005 to 2011. He has served as the Chair of the Scientific Advisory Board of the Yale Cancer Centre. He served as the Co-Chair of the National Centre Institute Program Review Group for Leukemia, Lymphoma and Myeloma and as a member of the boards of the Scientific Counsellors of the National Institute of Environmental Health and the National Cancer Institute. We believe that Dr Dalla-Favera's medical experience and scientific background make him well qualified to serve as a director on our board.
Alessandro Padova
Chairman
Dr Padova has 25 years of R&D in pharma & biotech companies and public private initiatives, including appointments at Parke Davis Neuroscience (Cambridge, UK), Peptide Therapeutics (Cambridge, UK), Medivir UK (Cambridge, UK) and Astex Technology (Cambridge, UK).
Dr Padova has been appointed a director of our company in August 2016. Since October 2015, Dr. Padova has served as Chief Executive Officer of Fondazione Ri.MED, a US-Italy public-private research foundation based in Sicily, that promotes, supports and leads biomedicaltranslational medicine programs in oncology, aging and biotechnological research projects.organ insufficiency diseases. From July 2014June 2013 to September 2015, Dr. Padova wascovered senior roles at IRBM Group in Rome, Italy, including Senior Director, Strategy and Business Development and from June 2013 to June 2014, Director of Strategic Alliances for IRBM Group in Rome, Italy, a diversified industrial group operating within the biotech and pharmaceutical sector. From May 2004 to March 2013,Alliances. Dr. Padova was held various positions with Siena Biotech S.p.A., a drug discovery and development company, most recently as General Manager. During his career, he contributed to the progression of several programs from discovery to the clinic. Dr. Padova graduated from University of Kent with a degree in Chemistry and received his Ph.D. in Chemistry from University of Exeter. Dr Padova served as President of Scientific Advisory Board and member of Board of Director for Exosomics Siena and Externautics. At present Dr Padova also serves as a Board member of Tes Pharma. We believe that Dr Padova's business experience in the medial industry makes him well qualified to serve as a director on our board.
TizianoLazzaretti
Chief Financial Officer
Mr. MartinLazzaretti has been Chief Executive Officerthe CFO of Puricore Inc., a biopharmaceuticalour company focused on dermatology, ophthalmology, and rare diseases since 2015. From 2011 to 2015,November 2016. Mr. Martin was President of Moksha8 Pharmaceuticals, Inc., an emerging markets pharmaceutical company. From 2009 to 2011, Mr. Martin was Managing Director of The Sophocles Group, a private consulting company. From 2007 to 2009, Mr. Martin was Chief Operating Officer of Intercept Pharmaceuticals, Inc. Mr. Martin graduated from Cornell University with a BA degree in Government and received his MBA from Harvard Graduate School of Business Administration. We believe that Mr Martin's experience as a biotechnology company executive makes him well qualified to serve as a director on our board.
Before, he served as Corporate Finance Director and Deputy Chairman at Fiat GroupFIAT UK based in London for 15 years, spanning activities through all different sectors from automotive to financial services. MrMr. Lazzaretti has a Bachelor of Science (BSc Hons) in Economics and Accounting from the University of Turin, Italy, and he was awarded a Master in Business Administration (MBA) from Bocconi University (Milan ) on FIAT Group’s scholarship, and studied Corporate Finance at the London Business School.
Our directors are appointed for a one-year term to hold office until the next annual general meeting of our shareholders or until removed from office in accordance with our bylaws. Our officers are appointed by our board of directors and hold office until removed by the board.
| 63 |
Board Independence
We currently have seventhree directors serving on our board of directors. Our Board of Directors has adopted the definition of “independence” as described in NASDAQ Rules 4200 and 4350.4350. Independent directors would not include anyone who, within the past three years, be employed by our Company or any parent or subsidiary of our Companycompany or any of their family members; or any director who is, or who has a family member who is, a controlling shareholder. Our Board of Directors has determined that a majority of our directors do meet the independence requirements.
Family Relationships and Other Arrangements
There are no family relationships among our directors and executive officers. There are no arrangements or understandings between or among our executive officers and directors pursuant to which any director or executive officer was or is to be selected as a director or executive officer.
Audit Committee
We have a separately-designatedseparately designated standing Audit Committee established in accordance with Section 3(a)(58)3(a)(58)(A) of the Securities Exchange Act of 1934, as amended, or the Exchange Act. The Audit Committee’s responsibilities include, among other things: (i) selecting and retaining an independent registered public accounting firm to act as our independent auditors, setting the compensation for our independent auditors, overseeing the work done by our independent auditors and terminating our independent auditors, if necessary, (ii) periodically evaluating the qualifications, performance and independence of our independent auditors, (iii) pre-approving all auditing and permitted non-audit services to be provided by our independent auditors, (iv) reviewing with management and our independent auditors our annual audited financial statements and our quarterly reports prior to filing such reports with the Securities and Exchange Commission, or the SEC, including the results of our independent auditors’ review of our quarterly financial statements, and (v) reviewing with management and our independent auditors significant financial reporting issues and judgments made in connection with the preparation of our financial statements. The Audit Committee also prepares the Audit Committee report that is required to be included in our annual proxy statement pursuant to the rules of the SEC.
The Audit Committee currently consists of John P. Brancaccio, chairman of the Audit Committee, Jim Mervis and John Alex Martin.Committee. Under the applicable rules and regulations of NASDAQ, each member of a company’s audit committee must be considered independent in accordance with NASDAQ Listing Rule 5605(c)(2)5605(c)(2)(A)(i) and (ii) and Rule 10A-3(b)(1)10A-3(b)(1) under the Exchange Act. The Board has determined that each of Messrs.Mr. Brancaccio Mervis and Martin is “independent” as that term is defined under applicable NASDAQ and SEC rules. Mr. Brancaccio is our audit committee financial expert. The Board plans to adopthas adopted a written charter setting forth the authority and responsibilities of the Audit Committee.
Compensation Committee
The purpose of the Compensation Committee is to discharge the Board’s responsibilities relating to compensation of our directors and executive officers. The Compensation Committee has responsibility for, among other things, (i) recommending to the Board for approval the overall compensation philosophy for our company and periodically reviewing the overall compensation philosophy for all employees to ensure it is appropriate and does not incentivize unnecessary and excessive risk taking, (ii) reviewing annually and making recommendations to the Board for approval, as necessary or appropriate, with respect to our compensation plans, (iii) based on an annual review, determining and approving, or at the discretion of the Compensation Committee, recommending to the Board for determination and approval, the compensation and other terms of employment of each of our officers, (iv) reviewing and making recommendations to the Board with respect to the compensation of directors, (v) overseeing our regulatory compliance with respect to compensation matters, (vi) reviewing and discussing with management, prior to the filing of our annual proxy statement or annual report on Form 10-K, our disclosure relating to executive compensation, including our Compensation Discussion and Analysis and executive and director compensation tables as required by SEC rules, and (vii) preparing an annual report regarding executive compensation for inclusion in our annual proxy statement or our annual report on Form 10-K. The Compensation Committee has the power to form one or more subcommittees, each of which may take such actions as may be delegated by the Compensation Committee.
| 64 |
The Compensation Committee currently consists of John Bracaccio,Brancaccio, Chairman of the Compensation Committee, John Alex Martin and Riccardo Dalla-Favera.Committee. The Board has determined that all of the members are “independent” under NASDAQ Listing Rule 5602(a)(2), except for Dr Shailubhai.. The Board plans to adopthas adopted a written charter setting forth the authority and responsibilities of the Compensation Committee.
Corporate Governance/Nominating Committee
The Corporate Governance/Nominating Committee has responsibility for assisting the Board in, among other things, (i) effecting Board organization, membership and function, including identifying qualified board nominees, (ii) effecting the organization, membership and function of the committees of the Board, including the composition of the committees of the Board and recommending qualified candidates for the committees of the Board, (iii) evaluating and providing successor planning for the chief executive officer and our other executive officers, (iv) identifying and evaluating candidates for director in accordance with certain general and specific criteria, (v) developing and recommending to the Board Corporate Governance Guidelines and any changes thereto, setting forth the corporate governance principles applicable to us, and overseeing compliance with the our Corporate Governance Guidelines, and (vi) reviewing potential conflicts of interest involving directors and determining whether such directors may vote on issues as to which there may be a conflict. The Corporate Governance/Nominating Committee is responsible for identifying and evaluating candidates for director. Potential nominees are identified by the Board based on the criteria, skills and qualifications that are deemed appropriate by the Corporate Governance/Nominating Committee.
The Corporate Governance/Nominating Committee currently consists of John P. Brancaccio, chairman of the Corporate Governance/Nominating Committee and Dr. Kunwar Shailubhai.Alessandro Padova. The Board has determined that all of the members are “independent” under NASDAQ Listing Rule 5605(a)(2)5605(a)(2). The Board plans to adopthas adopted a written charter setting forth the authority and responsibilities of the Corporate Governance/Nominating Committee.
Code of Business Conduct and Ethics
We have adopted a formal Code of Business Conduct and Ethics applicable to all Board members, officers and employees. That code is available on our coporatecorporate website www.rasna.com. A copy will also be provided free of charge upon request to:
Rasna Therapeutics Inc, Inc., 420 Lexington Ave, Suite 2525, New York, NY 10170.
Summary Compensation Table
The following table sets forth summary information concerning the total compensation paid to our executive officers have received any compensation for the last two fiscal years.year ended September 30, 2019 for services to our company.
|
|
|
|
|
|
|
Executive Compensation |
|
|
|
|
| |
Name | Fees Earned or Paid in Cash ($) |
| Option Awards ($) (1) |
| Total ($) | |
Kunwar Shailubhai | 87,500 |
| — |
|
| 87,500 |
Tiziano Lazzaretti | 80,000 |
| — |
|
| 80,000 |
(1) Represents the fair value of incentive stock options granted during the year ended September 30, 2019 using the Black-Scholes model for computing stock-based compensation expense as of the date of grant.
Grants of Plan-Based Awards During Fiscal Year
No grants of plan-based awards to our executive officers:
| 65 |
All Other Option Awards: Number of Shares of Stock or Units (#) (#) | Exercise or Base Price Per Share of Option Awards($) (($/sh)($/Sh) | Grant Date Fair Value of Stock and Option Awards ($)(1) | |||
| Name Officer | Grant Date | ||||
| Tiziano Lazzaretti | 9/1/16 | 100,000 | 0.40 | 128,952 | |
| Tiziano Lazzaretti | 11/25/16 | 200,000 | 0.40 | 267,230 | |
| James Tripp | 9/1/16 | 125,000 | 0.40 | 161,190 | |
Outstanding Equity Awards at Fiscal Year-End
The following table sets forth information for the named executive officers regarding the number of shares subject to both exercisable and unexercisable stock options, as well as the exercise prices and expiration dates thereof, as of March 31, 2017. September 30, 2019.Except for the options set forth in the table below, no other equity awards were held by any our named executive officers as of March 31, 2017.September 30, 2019.
|
|
|
|
|
|
|
| Number of |
| Option Exercise Price ($) |
| Option Expiration Date | |
Name | Exercisable | Unexercisable |
|
| ||
Kunwar Shailubhai | 100,000 | — |
| 0.40 |
| 9/1/2026 |
Kunwar Shailubhai | 850,000 | 850,000 |
| 0.85 |
| 4/14/2027 |
Tiziano Lazzaretti | 100,000 | — |
| 0.40 |
| 9/1/2026 |
Tiziano Lazzaretti | 200,000 | — |
| 0.40 |
| 11/28/2026 |
Number of Securities Underlying Unexercised Options (#) | Option Exercise Price ($) | Option Expiration Date | ||||
| Name | Exercisable | Unexercisable | ||||
| Tiziano Lazaretti | — | 100,000 | 0.40 | 9/1/2026 | ||
| Tiziano Lazaretti | — | 200,000 | 0.40 | 11/28/2026 | ||
| James Tripp | — | 125,000 | 0.40 | 9/1/2026 | ||
Director Compensation
The following table sets forth summary information concerning the total compensation paid to our non-employee directors for the year ended March 31, 2017September 30, 2019 for services to our company.
|
|
|
|
|
|
|
Director Compensation |
|
|
|
|
| |
Name | Fees Earned or Paid in Cash ($) |
| Option Awards ($) (1) |
| Total ($) | |
John Brancaccio | 25,000 |
| — |
|
| 25,000 |
Alessandro Padova | 25,000 |
| — |
|
| 25,000 |
John Alex Martin (2) | 18,750 |
| — |
|
| 18,750 |
| Director Compensation for 2017 | ||||||
| Name | Fees Earned or Paid in Cash ($) | Option Awards ($) (1) | Total ($) | |||
| Kunwar Shalubhai | 12,500 | 128,952 | 141,452 | |||
| Riccardo Dalla-Favara | 12,500 | 256,499 | 268,999 | |||
| Jim Mervis | 12,500 | 256,499 | 268,999 | |||
| John Brancaccio | 12,500 | 128,952 | 141,452 | |||
| Alessandro Padova | 12,500 | 161,190 | 173,690 | |||
| John Alex Martin | 12,500 | 128,952 | 141,452 | |||
(1) Represents the fair value of incentive stock options granted during the year ended March 31, 2017September 30, 2019 using the Black-Scholes model for computing stock-based compensation expense as of the date of grant.
(2) Mr. Martin resigned as director on July 10, 2019.
| 66 |
Employment Agreements
Kunwar Shailubhai
On May 24, 2017, we entered into an employment agreement with Dr. Kunwar Shailubhai (the “Employment Agreement”) pursuant to which Dr. Shailubhai shall act as Chief Executive Officer and Chief Scientific Officer. Pursuant to the Employment Agreement, Dr. Shailubhai’s current base compensation is $300,000$300,000 per year.Dr. Shailubhai is eligible to receive a cash bonus of up to 35%35% of his base salary per year based on meeting certain performance objectives and bonus criteria.Pursuant to the Employment Agreement, Dr. Shailubhai received a grant of stock options to purchase 1,200,0001,700,000 shares of common stock which vest over 4 years.
If Dr. Shailubhai’s employment is terminated by us for cause or as a result of Dr. Shailubhai’s death or permanent disability or if Dr. Shailubhai terminates his employment agreement voluntarily, Dr. Shailubhai will be entitled to receive (i) any portion of unpaid base compensation then due for periods prior to termination, (ii) all business expenses reasonably and necessarily incurred by Dr. Shailubhai prior to the date of termination and (iii) benefits owed to Dr. Shailubhai under any qualified retirement plan or health and welfare benefit plan in which Dr. Shailubhai was a participant. If Dr. Shailubhai’s employment is terminated by us without cause or by Dr. Shailubhai for good reason, Dr. Shailubhai will be entitled to receive the amounts due upon termination of his employment by us for cause, or as a result of his death or permanent disability or upon termination by Dr. Shailubhai of his employment voluntarily, in addition to (provided that Dr. Shailubhai executes a written release with respect to certain matters) a severance payment equal to his base compensation for 12months from the date of termination and the benefits that Dr. Shailubhai would be eligible for during such 12-month12-month period.
Tiziano Lazzaretti
In October 2016, we entered into a consultancy agreement with Tiziano Lazzaretti in which he agreed to serve as Chief Financial Officer for a fee of $50,000 per year. This was increased to $80,000 a year in April 2017 by our compensation committee.
The following tables set forth certain information regarding the ownership of our common stock as of March 31,November 30, 2017, by:
• | each director; |
• | each person known by us to own beneficially 5% or more of our Common Stock; |
• | each executive officer; and |
• | all directors and executive officers as a group. |
The amounts and percentages of common stock beneficially owned are reported on the basis of regulations of the Securities and Exchange Commission governing the determination of beneficial ownership of securities. Under the rules of the SEC, a person is deemed to be a “beneficial owner” of a security if that person has or shares “voting power,” which includes the power to vote or to direct the voting of such security, or “investment power,” which includes the power to dispose of or to direct the disposition of such security. A person is also deemed to be a beneficial owner of any securities of which that person has the right to acquire beneficial ownership within 60 days. Under these rules more than one person may be deemed a beneficial owner of the same securities and a person may be deemed to be a beneficial owner of securities as to which such person has no economic interest.
Unless otherwise indicated below, to the best of our knowledge each beneficial owner named in the table has sole voting and sole investment power with respect to all shares beneficially owned, subject to community property laws where applicable. Unless otherwise indicated, the address of each beneficial owner is c/o 420 Lexington Avenue, New York, NY 10170.
| 67 |
| Name of Beneficial Owner | Number of Shares Beneficially Owned | Percentage Beneficially Owned (1) | |||||
| Executive Officers and Directors : | |||||||
| James Tripp | — | — | |||||
| Tiziano Lazzaretti | — | — | |||||
| Kunwar Shailubhai | 3,101 | * | |||||
| Riccardo Dalla-Favara | 328,700 | (2) | * | ||||
| Jim Mervis | 328,700 | (2) | * | ||||
| John Alex Martin | — | ||||||
| John Brancaccio | — | ||||||
| Alessandro Padova | — | ||||||
| All executive officers and directors as a group (8 persons) | 374,701 | * | |||||
| 5% or Greater Stockholders: | |||||||
| Panetta Partners Ltd.(3) | 8,909,054 | (4) | 13.0 | ||||
| TES Pharma Srl (5) | 5,684,250 | (6) | 8.3 | ||||
| Eurema Consulting Srl (7) | 5,684,250 | (6) | 8.3 | ||||
| MS Investment Holding, Inc. (8) | 7,773,167 | 11.4 | |||||
| Howard I. Freedberg Revocable Trust (9) | 6,758,188 | 9.9 | |||||
|
|
|
|
|
|
|
|
Name of Beneficial Owner |
| Number of Shares Beneficially Owned |
|
| Percentage |
| |
Executive Officers and Directors : |
|
|
|
|
|
| |
Tiziano Lazzaretti |
| 200,000 |
| (2) |
| * |
|
Kunwar Shailubhai |
| 494,434 |
| (3) |
| * |
|
John Brancaccio |
| 66,666 |
| (2) |
| * |
|
Alessandro Padova |
| 83,334 |
| (2) |
| * |
|
All executive officers and directors as a group (4 persons) |
| 711,100 |
| (4) |
| 1.0 |
|
5% or Greater Stockholders: |
|
|
|
|
|
| |
Panetta Partners Ltd.(5) |
| 9,016,307 |
| (6) |
| 13.0 |
|
TES Pharma Srl (7) |
| 5,684,250 |
| (8) |
| 8.4 |
|
Eurema Consulting Srl (9) |
| 5,684,250 |
| (8) |
| 8.4 |
|
MS Investment Holding, Inc. (10) |
| 7,773,167 |
|
|
| 11.4 |
|
Howard I. Freedberg Revocable Trust (11) |
| 6,758,188 |
|
|
| 9.9 |
|
*less than 1%
(1) | ||
Based on | ||
(2) | Consists of shares of common stock issuable upon exercise of vested stock options. | |
(3) | Includes 491,666 shares of common stock issuable upon exercise of stock options. | |
(4) | Includes 708,332 shares of common stock issuable upon exercise of vested stock options. | |
(5) | Gabriele Cerrone is a director of Panetta Partners Ltd. and in such capacity holds voting and dispositive power over our securities held by such entity. Panetta Partners Ltd. address is, c/o Cooley Services Limited, Dashwood, 60 Old Broad Street, London | |
(6) | Includes | |
(7) | Dr. Roberto Pellicciari holds voting and dispositive power over securities of the company held by such entity. | |
(8) | Includes | |
(9) | Brunangelo Falini holds voting and dispositive power over securities of the company held by such entity. | |
(10) | Morris Silverman holds voting and dispositive power over securities of the company held by such entity. | |
(11) | Howard Freedberg is the trustee of the Howard I. Freedberg Revocable Trust and in such capacity holds voting and dispositive power over securities of the company held by such entity. |
| 68 |
The following is a description of transactions or series of transactions since AprilOctober 1, 2015,2016, or any currently proposed transaction, to which we were or are to be a participant and in which the amount involved in the transaction or series of transactions exceeds $120,000,$120,000, and in which any of our directors, executive officers or persons who we know hold more than five percent of our common stock, including their immediate family members, had or will have a direct or indirect material interest, other than compensation arrangements with our directors and executive officers.
Eurema Consulting
Eurema Consulting S.r.l. is a principal stockholder.significant shareholder of the Company. During the twelve monthsyears ended March 31, 2017September 30, 2019 and twelve months ended March 31, 2016,2018, Eurema Consulting did not supply the Company with consulting services. As of September 30, 2019, and September 30, 2018, the balance due to Eurema Consulting S.r.l. supplied us with consulting services amounting to $50,000 and $100,000, respectively. As of March 31, 2017, and March 31, 2016, Eurema Consulting S.r.l was owed $275,000 and $225,000, respectively, by us.
Gabriele Cerrone
Gabriele Cerrone is affiliated witha majority shareholder of Panetta Partners, one of our principal stockholders.. shareholders and was a director of Arna Therapeutics Ltd. During the twelve monthsyears ended March 31, 2017September 30, 2019 and 2016, Mr.2018, Gabriele Cerrone charged us $50,000, and $75,000, respectively, in respect of consultancy fees. did not supply the Company with consulting services. As of March 31, 2017,September 30, 2019, and March 31, 2016,September 30, 2018, the balance due to Mr.Gabriele Cerrone was $175,000 for past consultancy services.
Roberto Pellicciari and $125,000, respectively.
Roberto Pellicceri
Tiziana Life Sciences Plc
As at March 31, 2017,September 30, 2019 and September 30, 2018, the Companybalance owed $103,672 toby Tiziana Life Sciences PLC,a CompanyPlc was $14,335 and $237,867 respectively. As of which the date these consolidated financial statements are issued, the related party receivable had not been fully collected.Kunwar Shailubhai, CEO and Riccardo Dalla Favera werea director of our Company, is also common directors with. This was in respecta director of Tiziana. In addition, Tiziano Lazzaretti, our CFO, is also CFO of Tiziana. The Company is party to a Shared Services agreement with Tiziana whereby the Company is charged for shared services such as the payroll and rent, see Note 139 for more details
| 69 |
The aggregate fees billed to the Company by Marcum LLP, the Company’s current independent registered public accounting firm, BDO and Grant Thornton, the Company’s previous independent registered public accounting firm, for the indicated services for each of the last two fiscal years were as follows:
|
|
|
| Year ended September 30, | |
| 2019 | 2018 |
Audit fees(1) | $116,081 | $256,092 |
Audit - related fees(2) | — | — |
Tax fees(3) | — | — |
All other fees(4) | — | — |
Total fees | $116,081 | $256,092 |
_______________
| 2017 | 2016 | |
Audit fees(1) | $354,431 | $246,500 |
Tax fees(2) | — | — |
Other fees(3) | — | — |
| Total fees | $354,431 | $246,500 |
(1) | |
Audit fees consist of fees for professional services performed by Marcum LLP, |
(2) | During the fiscal year ended September 30, 2019, our independent registered public accounting firm did not render any audit-related services. |
(3) | During the fiscal year ended September 30, 2019, our independent registered public accounting firm did not render services to us for tax compliance, tax advice and |
(4) | |
All other fees consist of fees for professional services performed in connection with consultations, due diligence procedures and related matters. |
(a) (1) Financial Statements
Reference is made to the Index to Consolidated Financial Statements of Rasna Therapeutics Inc. appearing on page 57 of this report.
(a) (2) Financial Statement Schedules
The schedules required to be filed by this item have been omitted because of the absence of conditions under which they were required on, because the required information is included in the consolidated financial statements or the notes thereto.
(b) Exhibits
Exhibit Number | Description | |
101 | Financial statements from the annual report on Form 10-K of Rasna Therapeutics, Inc. for the year ended |
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report on Form 10-K to be signed on its behalf by the undersigned, thereunto duly authorized.
Date: January 13, 2020 | ||||||
RASNA THERAPEUTICS, INC. | ||||||
By: | /s/ Kunwar Shailubhai | |||||
Kunwar Shailubhai | ||||||
Director and Chief Executive Officer | ||||||
| 72 |
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below hereby constitutes and appoints, jointly and severally, Kunwar Shailubhai, his attorney-in-fact, each with full power of substitution and resubstitution, for him in any and all capacities, to sign any and all amendments to this Annual Report on Form 10-K, and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorney-in-fact full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming all that said attorney-in-fact, or their substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
P
ursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.Signature | Title(s) | Date | ||
/s/ Kunwar Shailubhai | ||||
Kunwar Shailubhai | Director, Chief Executive Officer | January 13, 2020 | ||
(Principal Executive Officer) | ||||
/s/ Tiziano Lazzaretti | ||||
Tiziano Lazzaretti | Chief Financial Officer | January 13, 2020 | ||
(Principal Accounting and Financial Officer) | ||||
/s/ John Brancaccio | ||||
John Brancaccio | Director | January 13, 2020 | ||
/s/ Alessandro Padova | ||||
Alessandro Padova | Director | January 13, 2020 | ||
| 73 | ||||
RASNA THERAPEUTICS, INC.
To the Audit Committee of the
Opinion on the Financial Statements
We have audited the accompanying consolidated balanceExplanatory Paragraph – Going Concern
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As more fully discussed in Note 1 to the financial statements, the Company has a significant working capital deficiency, has incurred significant losses and needs to raise additional funds to meet its obligations and sustain its operations. These conditions raise substantial doubt about the Company's ability to continue as a going concern. Management's plans in regard to these matters are described in Note 1. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on thesethe Company’s financial statements based on our audit.
We conducted our auditaudits in accordance with the standards of the Public Company Accounting Oversight Board (United States).PCAOB. Those standards require that we plan and perform the auditaudits to obtain reasonable assurance about whether the financial statements are free of material misstatement.The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks.Such procedures include, examining, on a test basis, evidence supportingregarding the amounts and disclosures in the financial statements, assessing thestatements.Our audits also included evaluating accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. presentation of the financial statements.We believe that our audit providesaudits provide a reasonable basis for our opinion.
/s/Marcum LLP
Marcum llp
We have served as the Company’s auditor since 2016.
New York, NY
January 13, 2020
|
|
|
|
|
|
|
|
| As of September 30, | ||||||
| 2019 |
|
| 2018 | |||
ASSETS |
|
|
|
|
|
|
|
Current assets: |
|
|
|
|
|
|
|
Cash and cash equivalents | $ | 50,068 |
|
| $ | 42,693 |
|
Prepayments and other receivables |
| 7,176 |
|
|
| 66,936 |
|
Related party receivable |
| 14,335 |
|
|
| 237,866 |
|
Total current assets |
| 71,579 |
|
|
| 347,495 |
|
|
|
|
|
|
|
|
|
Property and equipment, net |
| 1,949 |
|
|
| 5,420 |
|
Intellectual property |
| 236,269 |
|
|
| 236,269 |
|
In-process research and development |
| 613,100 |
|
|
| 613,100 |
|
Indefinite lived intangible asset - platform technology |
| 1,300,000 |
|
|
| 1,300,000 |
|
Goodwill |
| 2,722,985 |
|
|
| 2,722,985 |
|
Total non-current assets |
| 4,874,303 |
|
|
| 4,877,774 |
|
|
|
|
|
|
|
|
|
Total assets | $ | 4,945,882 |
|
|
| 5,225,269 |
|
|
|
|
|
|
|
|
|
LIABILITIES AND SHAREHOLDERS' EQUITY |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Liabilities: |
|
|
|
|
|
|
|
Current liabilities: |
|
|
|
|
|
|
|
Accounts payable and accrued expenses | $ | 1,590,589 |
|
| $ | 1,421,655 |
|
Related party payables |
| 550,000 |
|
|
| 550,000 |
|
Convertible note payable |
| 264,907 |
|
|
| 137,340 |
|
|
|
|
|
|
|
|
|
Total current liabilities |
| 2,405,496 |
|
|
| 2,108,995 |
|
|
|
|
|
|
|
|
|
Deferred income taxes |
| 3,034 |
|
|
| 10,618 |
|
|
|
|
|
|
|
| |
Total liabilities |
| 2,408,530 |
|
|
| 2,119,613 |
|
|
|
|
|
|
|
|
|
Commitments and contingencies (Note 12) |
| — |
|
|
| — |
|
|
|
|
|
|
|
|
|
Shareholders' equity |
|
|
|
|
|
|
|
Common stock, $0.001 par value, 200,000,000 shares authorized; 68,908,003 are issued and outstanding |
| 68,909 |
|
|
| 68,909 |
|
Additional paid-in capital |
| 19,780,252 |
|
|
| 19,412,176 |
|
Accumulated deficit |
| (17,311,809 | ) |
| (16,375,429 | ) | |
Total shareholders' equity |
| 2,537,352 |
|
|
| 3,105,656 |
|
Total liabilities and shareholders' equity | $ | 4,945,882 |
|
| $ | 5,225,269 |
|
| March 31, 2017 | March 31, 2016 | ||||||
| ASSETS | |||||||
| Current assets: | |||||||
| Cash and cash equivalents | $ | 4,048,962 | $ | — | |||
| Prepayments and other receivables | 65,846 | — | |||||
| Related party receivable | 85,066 | 607,159 | |||||
| Total current assets | 4,199,874 | 607,159 | |||||
| Property and Equipment, net | 7,030 | — | |||||
| Other intangible assets | 236,269 | — | |||||
| In-process research and development | 1,913,100 | 1,300,000 | |||||
| Goodwill | 2,722,985 | — | |||||
| Total non-current assets | 4,879,384 | 1,300,000 | |||||
| Total assets | $ | 9,079,258 | $ | 1,907,159 | |||
| LIABILITIES AND SHAREHOLDERS' EQUITY | |||||||
| Liabilities: | |||||||
| Current liabilities: | |||||||
| Accounts payable and accrued expenses | $ | 903,002 | $ | 78,227 | |||
| Related party payables | 625,000 | 550,000 | |||||
| Total current liabilities | 1,528,002 | 628,227 | |||||
| Total liabilities | 1,528,002 | 628,227 | |||||
| Commitments and Contingencies | |||||||
| Shareholders' equity | |||||||
| Common stock, $0.001 and $0.01 par value, respectively; 200,000,000 shares and 100,000,000 shares authorized respectively; of which 68,046,465 and 35,650,289 are issued. and outstanding respectively at March 31, 2017 and 2016 | 68,047 | 356,503 | |||||
| Additional paid-in capital | 16,740,989 | 5,746,477 | |||||
| Accumulated deficit | (9,257,780 | ) | (4,824,048 | ) | |||
| Total shareholders' equity | 7,551,256 | 1,278,932 | |||||
| Total liabilities and shareholders' equity | $ | 9,079,258 | $ | 1,907,159 | |||
The accompanying notes are an integral part of these consolidated financial statements.
|
|
|
|
|
|
|
|
Year ended September 30, | |||||||
| 2019 |
|
| 2018 | |||
Revenue | $ | — |
|
| $ | — |
|
Cost of revenue | — |
|
| — |
| ||
Gross profit | — |
|
| — |
| ||
|
|
|
|
| |||
Operating expenses: |
|
|
|
|
| ||
General and administrative | 598,763 |
|
| 2,527,345 |
| ||
Research and development | 14,373 |
|
| 324,608 |
| ||
Consultancy fees third parties and related parties | 75,228 |
|
| 403,290 |
| ||
Legal and professional fees | 226,806 |
|
| 812,730 |
| ||
Total operating expenses | 915,170 |
|
| 4,067,973 |
| ||
|
|
|
|
| |||
Loss from operations | (915,170 | ) |
| (4,067,973 | ) | ||
|
|
|
|
| |||
Other expense: |
|
|
|
|
| ||
| Interest expense | (27,567 | ) | (2,340 | ) | |||
Foreign currency transaction loss | (1,227 | ) |
| (3,955 | ) | ||
Other expense | (28,794 | ) |
| (6,295 | ) | ||
|
|
|
|
| |||
Loss before provision for income taxes | (943,964 | ) |
| (4,074,268 | ) | ||
|
|
|
|
| |||
Income tax (benefit)/expense | (7,584 | ) |
| 2,093 |
| ||
|
|
|
|
| |||
Net loss | $ | (936,380 | ) |
| $ | (4,076,361 | ) |
|
|
|
|
| |||
Basic and diluted loss per share attributable to common shareholders | $ | (0.01 | ) |
| $ | (0.06 | ) |
|
|
|
|
| |||
Basic and diluted weighted average common shares outstanding | 68,908,003 |
|
| 68,908,003 |
| ||
| Year ended March 31, | ||||||||
| 2017 | 2016 | |||||||
| Revenue | $ | — | $ | — | ||||
| Cost of revenue | — | — | ||||||
| Gross profit | — | — | ||||||
| Operating expenses: | ||||||||
| General and administrative | 1,009,240 | — | ||||||
| Research and development | 1,564,353 | — | ||||||
| Consultancy fees third parties | 1,071,777 | 128,176 | ||||||
| Consultancy fees related parties | 150,000 | 350,000 | ||||||
| Legal and professional fees | 739,158 | 99,930 | ||||||
| Total operating expenses | 4,534,528 | 578,106 | ||||||
| Loss from operations | (4,534,528 | ) | (578,106 | ) | ||||
| Other income/(expense): | ||||||||
| Foreign currency transaction gain | 100,796 | — | ||||||
| Other income | 100,796 | — | ||||||
| Loss from operations before income taxes | (4,433,732 | ) | (578,106 | ) | ||||
| Income tax provision | — | — | ||||||
| Net loss | $ | (4,433,732 | ) | $ | (578,106 | ) | ||
| Basic and diluted loss per share attributable to common shareholders | $ | (0.07 | ) | $ | (0.02 | ) | ||
| Basic and diluted weighted average common shares outstanding | 60,816,068 | 35,650,289 | ||||||
The accompanying notes are an integral part of these consolidated financial statements.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Common Stock |
| Additional Paid-In |
| Accumulated |
| Total Shareholders’ | |||||||||||
| Shares |
| Amount |
| Capital |
| Deficit |
| Equity | |||||||||
Balance at October 1, 2017 | 68,908,003 |
|
| $ | 68,909 |
|
| $ | 18,267,895 |
|
| $ | (12,299,068 | ) |
| $ | 6,037,736 |
|
|
|
|
|
|
|
|
|
|
| |||||||||
Share based compensation | — |
|
| — |
|
| 621,931 |
|
| — |
|
| 621,931 |
| ||||
Net loss | — |
|
| — |
|
| — |
|
| (4,076,361 | ) |
| (4,076,361 | ) | ||||
Warrants issued for consultancy services | — |
|
| — |
|
| 522,350 |
|
| — |
|
| 522,350 |
| ||||
Balance at September 30, 2018 | 68,908,003 |
|
| $ | 68,909 |
|
| $ | 19,412,176 |
|
| $ | (16,375,429 | ) |
| $ | 3,105,656 |
|
|
|
|
|
|
|
|
|
|
| |||||||||
Share based compensation | — |
|
| — |
|
| 368,076 |
|
| — |
|
| 368,076 |
| ||||
Net loss | — |
|
| — |
|
| — |
|
| (936,380 | ) |
| (936,380 | ) | ||||
Warrants issued for consultancy services | — |
|
| — |
|
| — |
|
| — |
|
| — |
| ||||
Balance at September 30, 2019 | 68,908,003 |
|
| $ | 68,909 |
|
| $ | 19,780,252 |
|
| $ | (17,311,809 | ) |
| $ | 2,537,352 |
|
| Common Stock | Additional Paid-In | Accumulated | Total Shareholders’ | |||||||||||||||
| Shares | Amount | Capital | Deficit | Equity | ||||||||||||||
| Balance at March 31, 2015 | 35,650,289 | $ | 356,503 | $ | 5,685,801 | $ | (4,245,942 | ) | $ | 1,796,362 | ||||||||
| Share based compensation | — | — | 60,676 | — | 60,676 | |||||||||||||
| Net loss | — | — | — | (578,106 | ) | (578,106 | ) | |||||||||||
| Balance at March 31, 2016 | 35,650,289 | $ | 356,503 | $ | 5,746,477 | $ | (4,824,048 | ) | $ | 1,278,932 | ||||||||
| Shares cancelled pursuant to reverse merger transaction | (35,650,289 | ) | (356,503 | ) | 356,503 | — | — | |||||||||||
| Shares issued pursuant to reverse merger transaction | 54,837,790 | 548,378 | 7,126,622 | — | 7,675,000 | |||||||||||||
| .33 share exchange | (36,741,319 | ) | (367,413 | ) | — | — | (367,413 | ) | ||||||||||
| Recapitalization | 3,305,000 | (159,563 | ) | 528,477 | — | 368,914 | ||||||||||||
| Cancellation of shares | (1,500,000 | ) | (1,500 | ) | — | — | (1,500 | ) | ||||||||||
| 3.25 for 1 Stock Split | 44,778,327 | 44,778 | (44,778 | ) | — | — | ||||||||||||
| Common stock issued in connection with offering | 3,366,667 | 3,367 | 2,004,133 | — | 2,007,500 | |||||||||||||
| Share based compensation | — | — | 1,023,555 | — | 1,023,555 | |||||||||||||
| Obligation for warrants to be issued | — | — | (484,009 | ) | — | (484,009 | ) | |||||||||||
| Increase in fair value of warrants to date of issuance | — | — | (2,430,875 | ) | — | (2,430,875 | ) | |||||||||||
| Warrant obligation reclassified to additional paid-in capital upon warrant issuance | — | — | 2,914,884 | — | 2,914,884 | |||||||||||||
| Net loss | — | — | — | (4,433,732 | ) | (4,433,732 | ) | |||||||||||
| Balance at March 31, 2017 | 68,046,465 | $ | 68,047 | $ | 16,740,989 | $ | (9,257,780 | ) | $ | 7,551,256 | ||||||||
The accompanying notes are an integral part of these consolidated financial statements.
| Year Ended March 31, | |||||||
| 2017 | 2016 | ||||||
| CASH FLOWS FROM OPERATING ACTIVITIES: | |||||||
| Net loss | $ | (4,433,732 | ) | $ | (578,106 | ) | |
| Adjustments to reconcile net loss to net cash used in operating activities: | |||||||
| Share based compensation | 1,023,555 | 60,676 | |||||
| Depreciation | 3,463 | — | |||||
| Changes in operating assets and liabilities: | |||||||
| Other receivables and prepayments | (65,843 | ) | — | ||||
| Related party receivable | (85,412 | ) | 113,651 | ||||
| Accounts and other payables | 519,111 | 53,779 | |||||
| Related party payables | 75,000 | 350,000 | |||||
| Net cash used in operating activities | (2,963,858 | ) | — | ||||
| CASH FLOWS FROM INVESTING ACTIVITIES: | |||||||
| Purchase of property, plant and equipment | (10,493 | ) | — | ||||
| Cash and cash equivalents acquired in reverse merger/business combination | 5,116,609 | — | |||||
| Net cash provided by investing activities | 5,106,116 | — | |||||
| CASH FLOWS FROM FINANCING ACTIVITIES: | |||||||
| Net proceeds from issuance of shares of common stock | 2,007,500 | — | |||||
| Net cash provided by financing activities | 2,007,500 | — | |||||
| Effect of foreign exchange rate | (100,796 | ) | — | ||||
| Net increase in cash and cash equivalents | 4,048,962 | — | |||||
| Cash and cash equivalent, beginning of period | — | — | |||||
| Cash and cash equivalent, end of period | $ | 4,048,962 | $ | — | |||
| Non-cash investing and financing activities: | |||||||
| Expenses paid by Rasna Therapeutics Limited on behalf of the Company | $ | — | $ | 113,651 | |||
| Common stock issued for acquisition | 7,675,000 | — | |||||
| Shares cancelled pursuant to reverse merger transaction | (356,503 | ) | — | ||||
| Shares issued pursuant to reverse merger transaction | 548,378 | — | |||||
| .33 share exchange | (367,413 | ) | — | ||||
| Recapitalization | (159,563 | ) | — | ||||
| Cancellation of shares | (1,500 | ) | — | ||||
| Shares issued in 3.25 for 1 stock split | 44,778 | — | |||||
| Related party receivable balance canceled in acquisition | $ | 607,159 | $ | — | |||
|
|
|
|
|
|
|
|
Year Ended September 30, | |||||||
| 2019 |
| 2018 | ||||
CASH FLOWS FROM OPERATING ACTIVITIES: |
|
|
|
|
| ||
Net loss | $ | (936,380 | ) |
| $ | (4,076,361 | ) |
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
| ||
Share based compensation | 368,076 |
|
| 621,931 |
| ||
Warrants issued for consultancy services | — |
|
| 522,350 |
| ||
Depreciation | 3,471 |
|
| 4,833 |
| ||
Deferred income tax expense | (7,584 | ) |
| 2,093 |
| ||
Non cash interest expense | 27,567 |
|
| 2,340 |
| ||
Write off related party receivable | — |
|
| 28,931 |
| ||
Changes in operating assets and liabilities: |
|
|
|
|
| ||
Prepayments and other receivables | 59,760 |
| 62,221 | ||||
Related party receivable | 223,532 |
|
| (237,866 | ) | ||
Accounts payable and accrued expenses | 168,933 |
|
| 439,728 |
| ||
Related party payables | — |
| — |
| |||
Net cash used in operating activities | (92,625 | ) |
| (2,629,800 | ) | ||
|
|
|
|
|
| ||
CASH FLOWS FROM INVESTING ACTIVITIES: |
|
|
|
|
| ||
Purchase of property and equipment | — |
| (4,073 | ) | |||
Net cash used by investing activities | — |
| (4,073 | ) | |||
|
|
|
|
| |||
CASH FLOWS FROM FINANCING ACTIVITIES: |
|
|
|
|
| ||
Proceeds from issuance of convertible note | 100,000 |
|
| 135,000 |
| ||
Net cash provided by financing activities | 100,000 |
|
| 135,000 |
| ||
|
|
|
|
| |||
Effect of foreign exchange rate on cash | — |
|
| 3,955 | |||
|
|
|
|
| |||
Net increase/(decrease) in cash and cash equivalents | 7,375 |
| (2,494,918 | ) | |||
|
|
|
|
| |||
Cash and cash equivalent, beginning of period | 42,693 |
|
| 2,537,611 |
| ||
|
|
|
|
| |||
Cash and cash equivalents, end of period | $ | 50,068 |
|
| $ | 42,693 |
|
|
|
|
|
| |||
The accompanying notes are an integral part of these consolidated financial statements.
1. GENERAL INFORMATION
Rasna Therapeutics, Inc. (“Rasna DE.DE", "Rasna Inc.” or the "Company"), is a biotechnology company incorporated in the State of Delaware on March 28, 2016 . Prior to May 17,2016. The Company is engaged in modulating the molecular targets NPM1 and LSD1, which are implicated in the disease progression of leukemia and lymphoma.
On April 27, 2016, Rasna Therapeutics Inc. wasLimited, a non-trading holdingprivate limited company with an investmentincorporated in one subsidiary company,England and also controlled an entity, Rasna Therapeutics LimitedWales under the U.K. Companies Act (“Rasna UK”), sold its stake in which it was deemed the primary beneficiary.
On May 17, 2016, Rasna UKDE and its subsidiary Falconridge entered into an Agreement of Merger and Plan of Reorganization (“Merger Agreement”) with Arna.Arna Therapeutics Limited, a British Virgin Islands company, or Arna, which was a clinical stage biotechnology company focused on drugs to treat diseases in oncology and immunology, mainly focusing on the treatment of leukemia. Pursuant to the agreement,Merger Agreement, Arna was merged into Falconridge and the shareholders of Arna were issued shares of Rasna UKDE in exchange for shares of Arna.
On August 15, 2016, Active With Me, Inc., or AWM, entered into an Agreement of Merger and Plan of Reorganization (the “Merger Agreement”) with Rasna Inc.,DE, and Rasna Acquisition, Corp.providing for the merger of Rasna Acquisition with and into Rasna DE, (the “Merger”), a Delaware corporation andwith Rasna DE, surviving the Merger as a wholly owned subsidiary of Active With Me, Inc. (“Merger Sub”), providing for the merger of Merger Sub with and into Rasna, Inc. (the “Merger”), with Rasna, Inc. surviving the Merger as a wholly-owned subsidiary of Active With Me, Inc. As a result of the Merger, the resulting company, Rasna Therapeutics, Inc., is a biotechnology company that is focused on targeted drugs to treat diseases in oncology and immunology, mainly focusing on the treatment of Leukemia.
The Merger is beingwas treated as a reverse recapitalization effected by a share exchange for financial accounting and reporting purposes since substantially all of Active With Me’sAWM’s operations were disposed of prior to the consummation of the transaction. Rasna Successor isDE was treated as the accounting acquirer as its stockholdersshareholders control the Company after the Exchange Agreement, even though Active With Me, Inc.Merger and AWM was treated as the legal acquirer. As a result of the Merger, the assets and liabilities and the historical operations that are reflected in thesethe financial statements are those of Rasna SuccessorDE as if Rasna SuccessorDE had always been the reporting company. Since Active With Me, Inc. had no operations upon the Merger Agreement taking place, the transaction was treated as a reverse recapitalization for financial accounting and reporting purposes, and no goodwill or other intangible assets were recorded by the Company as a result of the Merger Agreement.
These financial statements are presented in United States dollars (“USD”) which is also the functional currency of the primary economic environment in which the Company operates. See Note 2 Foreign, foreign currency policy.
2. BASIS OF PRESENTATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
The principal accounting policies applied in the preparation of these consolidated financial statements are set out below. These policies have been applied consistently to all the periods presented unless otherwise stated.
Basis of preparation
These consolidated financial statements have been prepared followingin accordance with accounting principles generally accepted in the requirementsUnited States (“US GAAP”) including all pronouncements of the U.S. Securities and Exchange Commission (“SEC”) and United States generally accepted accounting principles (“GAAP”) forapplicable to annual reporting. In the opinion of management,
Principles of Consolidation
In accordance with ASC Accounting Standards Codification ("ASC") 810
The consolidated financial statements include the financial statements of the Company and its wholly owned subsidiary, Rasna DE, and Rasna DE's subsidiary, Arna Therapeutics Limited as well as the operations of Rasna Inc. for the period from May 17, 2016 through December 31, 2016.Limited. All significant intercompany accounts and transactions have been eliminated in the preparation of the accompanying consolidated financial statements.
Business Combinations
Management accounts for business combinations under the provisions of Accounting Standards Codification ("ASC")ASC Topic 805-10,805-10, Business Combinations, ("ASC 805-10"), which requires that the purchase method of accounting be used for all business combinations. Assets acquired and liabilities assumed, including non-controlling interests, are recorded at the date of acquisition at their respective fair values. ASC 805-10805-10 also specifies criteria that intangible assets acquired in a business combination must meet to be recognized and reported apart from goodwill. Goodwill represents the excess purchase price over the fair value of the tangible net assets and intangible assets acquired in a business combination. Acquisition-related expenses are recognized separately from the business combinations and are expensed as incurred.
Management is required to complete the purchase price allocation within 12 months of the acquisition date. If such completion of the allocation results in a change in the preliminary values, the measurement period adjustment will be recognized in the period in which the adjustment amount is determined in accordance with ASU 2015-16. The amounts reflected within the
Use of Estimates
The preparation of financial statements in conformity with US GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent liabilities at the date of the financial statements and the reported amounts of revenue and expenses during the reporting period. The Company evaluates its estimates on an ongoing basis, including those related to the carrying amount of intangible assets, to the fair values of stock based awards, income taxes and contingent liabilities, among others. The Company bases its estimates on historical experience and on various other assumptions that the Company believes to be reasonable, the results of which form the basis for making judgments about the carrying values of assets and liabilities. Actual results could differ from those estimates and such differences could be material to the consolidated financial position and results of operations.
Fair Value
The carrying value of the Company’s financial instruments, including cash and cash equivalents and accounts payable and accrued liabilities, approximate fair value because of the short-term nature of such financial instruments. Management measures certain other assets at fair value on a nonrecurring basis when they are deemed to be other-than-temporarily impaired.
Cash and cash equivalents
The Company considers all short-term investments with an original maturity of three months or less.
Property and Equipment
Expenditures for additions, renewals and improvements are capitalized at cost. Depreciation is computed in a straight linestraight-line method based on the estimated useful lives of the related assets. The estimated useful lives of the major classes of depreciable assets are 2 to 53 years for equipmentproperty and furniture and fixtures.equipment. Expenditures for repairs and maintenance are charged to operations as incurred. The Company periodically evaluates whether current events or circumstances indicate that the carrying life of the depreciable assets may not be recoverable.
Goodwill and Intangible assets
Intangible assets are made up of indefinite lived intangible assets, in-process research and development, (“IPR&D”) and certain intellectual property (“IP”). The balance of IPR&Dthe indefinite lived intangible assets represents IPR&Dthe platform technology that was acquired in 2013, which, at the time, was determined to have alternative future uses. IPR&D assets also represent the fair value assigned to acquired technologies in a business combination, which at the time of the business combination havehad not reached technological feasibility and havehad no alternative future use. IP assets represent the fair value assigned to technologies, which at the time of acquisition have reached technological feasibility, however, havehad not yet been put into service. Intangible assets are considered to have an indefinite useful life until the completion or abandonment of the associated research and development projects.
Management evaluates indefinite life intangible assets for impairment on an annual basis and on an interim basis if events or changes in circumstances between annual impairment tests indicate that the asset might be impaired. The ongoing evaluation for impairment of its indefinite life intangible assets requires significant management estimates and judgment. Management reviews definite life intangible assets for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. There were no0 impairment charges during the year ended March 31, 2017September 30, 2019 and 2016.2018.
Goodwill represents the premium paid over the fair value of the net tangible and intangible assets acquired in business combinations. Goodwill is not amortized; rather, it is subject to a periodic assessment for impairment by applying a fair value based test. Goodwill is assessed for impairment on an annual basis or more frequently if events or changes in circumstances indicate that the asset might be impaired. An impairment charge is recognized only when the implied fair value of the Company’s reporting unit’s goodwill is less than its carrying amount.
The Company intends to operate in an industry that is subject to rapid change. The Company’s operations will be subject to significant risk and uncertainties including financial, operational, technological, regulatory, and other risks associated with an early stage company, including the potential risk of business failure.
Research and development
Expenditures for research and development isare charged to the statements of operations in the year in which it isthey are incurred with the exception of expenditures incurred in respect of the development of major new products where the outcome of those projects is assessed as being reasonably certain in regards to viability and technical feasibility. Such expenditure isexpenditures are capitalized and amortized straight line over the estimated period of sale for each product, commencing in the year that sales of the product are first made. To date, the Company has not capitalized any such expenditures other than certain IPR&D & IP recorded in connection with certain acquisition or equity transactions.
Income Taxes
The Company accounts for income taxes under the asset and liability method. Deferred tax assets and liabilities are recognized for future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which the temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in incomeoperations in the period that includes the enactment date. Management considers many factors when assessing the likelihood of future realization of deferred tax assets, including recent earnings experience by jurisdiction, expectations of future taxable income, and the carryforward periods available for tax reporting purposes, as well as other relevant factors. A valuation allowance may be established to reduce deferred tax assets to the amount that management believes is more likely than not to be realized. Due to inherent complexities arising from the nature of the business, future changes in income tax law and variances between actual and anticipated operating results, management makes certain judgments and estimates. Therefore, actual income taxes could materially vary from these estimates.
The Company recognizes in the financial statements the impact of a tax position, if that position is more likely than not to be sustained upon an examination, based on the technical merits of the position. The Company records a liability for the difference between the benefit recognized and measured and the tax position taken or expected to be taken on the Company’s tax return. To the extent that the assessment of such tax positions changes, the change in estimate is recorded in the period in which the determination is made. To the extent interest and penalties are not assessed with respect to uncertain tax positions, amounts accrued will be reduced and reflected as a reduction of the overall income tax provision. The Company incurred no liability and, therefore, did not need to record interest and penalties during the year ended March 31, 2017September 30, 2019 and 2016.
Foreign Currency
Items included in the financial statements are measured using their functional currency, beingwhich is the currency of the primary economic environment in which the company operates. The accompanying financial statements are presented in United States Dollar (“USD”), which is the company’sCompany’s functional and presentational currency.
Foreign currency transactions are translated using the rate of exchange applicable at the date of the transaction. Foreign exchange gains and losses resulting from the settlement of such transactions and from the re-translation at the year-end of monetary assets and liabilities denominated in foreign currencies are recognized in the statements of operations.
| F-9 |
Net Loss per Share
Basic net loss per share is computed by dividing net loss available to common shareholders by the weighted average number of common shares outstanding during the period. Diluted loss per share includes potentially dilutive securities such as outstanding options, warrants and warrants,convertible loan notes, using various methods such as the treasury stock or modified treasury stockif-converted method in the determination of dilutive shares outstanding during each reporting period.
The following table sets forth potential common shares issuable upon the exercise of outstanding options and the exercise of warrants, all of which have been excluded from the computation of diluted weighted average shares outstanding as they would be anti-dilutive:
| September 30, |
| ||||
| 2019 |
| 2018 |
| ||
Stock options | 4,073,675 |
|
| 4,819,875 |
|
|
Warrants | 1,926,501 |
|
| 1,926,501 |
|
|
| Convertible notes & associated fees | 1,723,333 | 900,000 | ||||
Total shares issuable upon exercise or conversion | 7,723,509 |
|
| 7,646,376 |
|
|
| March 31, 2017 | March 31, 2016 | ||||
| Stock options | 3,162,375 | 1,662,375 | |||
| Warrants | 1,440,501 | — | |||
| Total shares issuable upon exercise or conversion | 4,602,876 | 1,662,375 | |||
The following is the computation of net loss per share for the following periods:
| For the Year Ended March 31, | ||||||||
| 2017 | 2016 | |||||||
| Net loss for the period | $ | (4,433,732 | ) | $ | (578,106 | ) | ||
| Weighted average number of shares | 60,816,068 | 35,650,289 | ||||||
| Net loss per share (basic and diluted) | $ | (0.07 | ) | $ | (0.02 | ) | ||
| For the Year Ended |
| For the Year Ended September 30, |
| ||||
| 2019 |
| 2018 |
| ||||
Net loss | $ | (936,380 | ) | $ | (4,076,361 | ) |
| |
Weighted average number of shares | 68,908,003 | 68,908,003 |
|
| ||||
Net loss per share (basic and diluted) | $ | (0.01 | ) | $ | (0.06 | ) |
| |
Equity-Based Payments
ASC Topic 718 “Compensation-Stock Compensation” requires companies to measure the cost of employee services received in exchange for the award of equity instruments based on the estimated fair value of the award at the date of grant. The expense is to be recognized over the period during which an employee is required to provide services in exchange for the award. The Company accounts for shares of common stock, stock options and warrants issued to employees based on the fair value of the stock, stock option or warrant, if that value is more reliably measurable than the fair value of the consideration or services received.
The Company accounts for stock options issued and vesting to non-employees in accordance with ASC Topic 505-50505-50 “Equity -Based Payment to Non-Employees” and accordingly the value of the stock compensation to non-employees is based upon the measurement date as determined at either a) the date at which a performance commitment is reached, or b) at the date at which
Accounting Pronouncements Not Yet Adopted
In January 2017, the FASBFinancial Accounting Standards Board (the “FASB”) issued ASU 2017-1,Accounting Standards Update (“ASU”) 2017-01, Business Combinations (Topic 805)805): Clarifying the Definition of a Business, which clarifies the definition of a business with the objective of adding guidance to assist entities with evaluating whether transactions should be accounted for as acquisitions (or disposals) of assets or businesses. The definition of a business affects many areas of accounting including acquisitions, disposals, goodwill, and consolidation. The Company adopted ASU 2017-01 on October 1, 2018 and it has not had a material impact on the Company’s consolidated financial statements.
Recent Accounting Pronouncements
In July 2017, the FASB, issued ASU, 2017-11, Earnings Per Share (Topic 260); Distinguishing Liabilities from Equity (Topic 480); Derivatives and Hedging (Topic 815): (Part I) Accounting for Certain Financial Instruments with Down Round Features, (Part II) Replacement of the Indefinite Deferral. The ASU applies to issuers of financial instruments with down-round features. It amends (1) the classification of such instruments as liabilities or equity by revising the guidance in ASC 815 on the evaluation of whether instruments or embedded features with down-round provisions must be accounted for as derivative instruments and (2) the guidance on recognition and measurement of the value transferred upon the trigger of a down-round feature for equity-classified instruments by revising ASC 260. The ASU is effective for annual financial reportingpublic business entities for fiscal years, and interim periods within those fiscal years, beginning after December 15, 2017.2018. Early adoption is permitted. The Company is currently evaluating the impact of adopting this guidance.on the Company's consolidated financial statements.
In January 2017, the FASB issued ASU 2017-04,2017-04, Intangibles -Goodwill and Other (Topic 350)350): Simplifying the Test for Goodwill Impairment, which addresses the concerns over the cost and complexity of the two-steptwo-step impairment test, and removes the second step of the test.An entity will apply a one-stepone-step quantitative test and record the amount of goodwill impairment as the excess of a reporting unit’s carrying amount over its fair value, not to exceed the total amount of good willgoodwill allocated to the reporting unit.The guidance is effective for annual and interim goodwill impairment tests performed for periods beginning after December 15, 2019 with early adoption permitted in January 2017.The2019. The Company is currently evaluating the impact of adopting this guidance.guidance on the Company's consolidated financial statements.
In June 2018, the FASB issued ASU 2018-07, Compensation - Stock Compensation (Topic 718): Improvements to Non employee Share Based Payment Accounting, which simplifies several aspects of the accounting for nonemployee share-based payment transactions resulting from expanding the scope of Topic 718, to include share-based payment transactions for acquiring goods and services from nonemployees. Some of the areas for simplification apply only to nonpublic entities. The amendments specify that Topic 718 applies to all share-based payment transactions in which a grantor acquires goods or services to be used or consumed in a grantor’s own operations by issuing share-based payment awards. The amendments in ASU 2018-07 also clarify that Topic 718 does not apply to share-based payments used to effectively provide (1) financing to the issuer or (2) awards granted in conjunction with selling goods or services to customers as part of a contract accounted for under Topic 606, Revenue from Contracts with Customers. The amendments in this Update are effective for public business entities for fiscal years beginning after December 15, 2018, including interim periods within that fiscal year. Early adoption is permitted. The Company does not plan to early adopt this ASU. The Company is currently evaluating the potential impacts of this updated guidance, and do not expect the adoption of this guidance to have a material impact on the Company's consolidated financial statements.
In August 2018, the FASB issued ASU 2018-13, Fair Value Measurement (Topic 820): Disclosure Framework-Changes to the Disclosure Requirements for Fair Value Measurement. ASU 2018-13 removes certain disclosures, modifies certain disclosures and adds additional disclosures. The ASU is effective for annual periods, including interim periods within those annual periods, beginning after December 15, 2019. Early adoption is permitted. The Company is currently evaluating the effect that this update will have on its financial statements and related disclosures.
3. GOING CONCERN
The Company is subject to a number of risks similar to those of other pre-commercial stage companies, including its dependence on key individuals, uncertainty of product development and generation of revenues, dependence on outside sources of capital, risks associated with research, development, testing, and obtaining related regulatory approvals of its pipeline products, suppliers and collaborators, successful protection of intellectual property, competition with larger, better-capitalized companies, successful completion of the Company's development programs and, ultimately, the attainment of profitable operations are dependent on future events, including obtaining adequate financing to fulfill its development activities and generating a level of revenues adequate to support the Company's cost structure.
The Company has experienced net losses and significant cash outflows from cash used in operating activities since inception, and as of September 30, 2019, had an accumulated deficit of $17,311,809, a net loss for the year ended September 30, 2019 of$936,380 and net cash used in operating activities of$92,625.
The Company expects to continue to incur net losses and have significant cash outflows for at least the next 12 months and will require significant additional cash resources to launch new development phases of existing products in its pipeline. In the event that the Company is unable to secure the necessary additional cash resources needed, the Company may slow current development phases or halt new development phases in order to mitigate the effects of the costs of development. These conditions, among others, raise substantial doubt about the Company's ability to continue as a going concern within twelve months from the date these financial statements are issued. The accompanying condensed consolidated financial statements have been prepared assuming that the Company will continue as a going concern. This basis of accounting contemplates the recovery of the Company's assets and the satisfaction of liabilities in the normal course of business. A successful transition to attaining profitable operations is dependent upon achieving a level of positive cash flows adequate to support the Company's cost structure.
4. GOODWILL AND INTANGIBLE ASSETS
On May 17, 2016, the Company acquired an entity and, at initial purchase price, it was determined that there was $236,269 of intellectual property, $613,100 of In-process research and development, and $2,722,985 of goodwill.
Goodwill
The following transactions weretable summarizes the Company’s goodwill for the periods indicated resulting from the acquisitions by the Company:
|
| September 30, | ||||||
|
| 2019 |
| 2018 | ||||
Goodwill |
| $ | 2,722,985 |
| $ | 2,722,985 |
| |
The Company performed an impairment analysis and no impairment was determined. Therefore 0 impairment was recorded for the periods presented.
Intangible Assets
On December 17, 2013, one of the Company’s shareholders, Panetta Partners Limited, transferred 5,000,000 of its shares in Arna Therapeutics Limited to Eurema Consulting S.r.l. and 5,000,000 shares in Arna Therapeutics Limited to TES Pharma S.r.l. In exchange for the shares, Panetta Partners Limited obtained intellectual property ("Platform Technology") from TES Pharma S.r.l and Eurema Consulting S.r.l. Panetta Partners Limited then assigned the Platform Technology to Arna Therapeutics Limited, which was accounted for usingas a capital contribution. The fair value of the purchase accounting method which requires, among other things, thatshares exchanged for the assets acquiredIPR&D was $0.13 per share; in addition the issue price for shares in October 2013 was $0.13 per share (shares issued post acquisition of the IPR&D were issued at $0.28) and liabilities assumed are recognizedaccordingly the Company valued the Platform Technology at their acquisition date fair value.
| Balance as of | |||
| May 17, 2016 | |||
| Share consideration transferred | $ | 7,675,000 | |
| Forgiveness of receivable | 607,159 | ||
| Consideration transferred | $ | 8,282,159 | |
| Less: Fair value of assets acquired | |||
| Cash and cash equivalents | (5,116,609 | ) | |
| Other receivables | (14,187 | ) | |
| Prepayment | (66,856 | ) | |
| Related party receivables | (20,412 | ) | |
| Intellectual property | (236,269 | ) | |
| In-Process research and development | (613,100 | ) | |
| Plus: Liabilities assumed | |||
| Accounts payable and accrued expenses | 492,603 | ||
| Related party payables | 15,656 | ||
| Goodwill | $ | 2,722,985 | |
The Company retained a Clinical Research Organisation ("CRO") to perform all related research and development associated with LSD-1. As all research and development associated with LSD‐1 was performed by the CRO and no other contributions to LSD‐1 IPR&D were made beyond payments to the CRO, the Company considered the payments made to estimate the fair value of LSD‐1.
| Year Ended March 31, | ||||||||
| 2017 | 2016 | |||||||
| Total Revenues, net | $ | — | $ | — | ||||
| Net Loss | (4,693,223 | ) | (2,080,061 | ) | ||||
| Basic and diluted net loss per share | $ | (0.08 | ) | $ | (1.05 | ) | ||
| Goodwill | |||
| Balance at March 31, 2016 | $ | — | |
| Acquisition of Rasna and its subsidiaries | 2,722,985 | ||
| Balance at March 31, 2017 | $ | 2,722,985 | |
At the time of the acquisition, the Company had reasonably expected to use the Platform Technology, in the asset’s then current state, in two independent research projects that had not commenced as of the date of the acquisition. The Company’s research projects applied the conclusions reached in the Platform Technology to develop treatments for AML through reformulation of certain available pharmaceuticals and independent development of a new pharmaceutical treatment. Both research projects were initiated shortly after the Platform Technology was acquired and continue through the date of the financial statements.
At the time of acquisition, and at present, no legal, regulatory, contractual, competitive, economic, or other factors were present that would constrain the useful life of the asset to the Company. The agreement to purchase the asset has no provisions that would limit the timeframe of use, legally, contractually or economically, and the asset remains a competitive platform for results in the treatment of Acute Myeloid Leukemia and lymphoma. Specifically, the agreement irrevocably assigns all rights and title to the asset, without limitation or contingencies. No limitations or alternative technology has emerged that would suggest obsolescence or a change in the competitive landscape for the Platform Technology as of the most recent reporting period. In addition, the Company has concluded that the useful life of the Platform Technology at the time of acquisition was beyond a foreseeable horizon, and therefore the asset is classified as an indefinite lived intangible asset.
The IPR&D and intellectual property are considered to have an indefinite life and there were no0 impairment charges recognized during the periods ended March 31, 2017 and March 31, 2016.
The following table summarizes the Company’s intangible assets as of the following periods:
|
| For the Year Ended September 30, | |||||||||
|
| 2019 |
| 2018 |
| Estimated Useful Life |
| ||||
In-process research and development |
| $ | 613,100 |
|
| $ | 613,100 |
|
| Indefinite |
|
Intellectual property |
| 236,269 |
|
| 236,269 |
|
| Indefinite |
| ||
Indefinite lived intangible asset - platform technology |
| 1,300,000 |
|
| 1,300,000 |
|
| Indefinite |
| ||
Total Intangible Assets |
| $ | 2,149,369 |
|
| $ | 2,149,369 |
|
|
|
|
| March 31, 2017 | |||||||||||||||||
| Estimated Useful Life | Gross Carrying Amount | Additions | Accumulated Amortization | Net Book Value | |||||||||||||
| In-process research and development | Indefinite | $ | 1,300,000 | $ | 613,100 | $ | — | $ | 1,913,100 | ||||||||
| Intellectual Property | Indefinite | — | 236,269 | — | 236,269 | ||||||||||||
| $ | 1,300,000 | $ | 849,369 | $ | — | $ | 2,149,369 | ||||||||||
| F-13 |
| March 31, 2016 | |||||||||||||||||
| Estimated | Gross | ||||||||||||||||
| Useful | Carrying | Accumulated | Net Book | ||||||||||||||
| Life | Amount | Additions | Amortization | Value | |||||||||||||
| In-process research and development | Indefinite | $ | 1,300,000 | $ | — | $ | — | $ | 1,300,000 | ||||||||
| $ | 1,300,000 | $ | — | $ | — | $ | 1,300,000 | ||||||||||
5. PROPERTY PLANT AND EQUIPMENT,
Property plant and equipment consists of office equipment which areis recorded at cost with an estimated useful life of 3 years, depreciated on a straight linestraight-line basis. For
|
|
|
|
|
|
|
|
|
|
|
|
|
| September 30, |
|
|
| ||||||
|
| 2019 |
| 2018 |
| Estimated Useful Life (years) |
| ||||
Office equipment |
| $ | 15,447 |
|
| $ | 15,447 |
|
| 3 |
|
Less: accumulated depreciation |
| 13,498 |
|
| 10,027 |
|
|
|
| ||
Property and equipment, net |
| $ | 1,949 |
|
| $ | 5,420 |
|
|
|
|
Depreciation expense was $3,471 and $4,833 for the year ended March 31, 2017, there were additions to property, plantSeptember 30, 2019 and equipment of $10,493.2018, respectively.
| March 31, 2017 | ||||||||
Estimated Useful Life | Gross carrying amount | Accumulated depreciation | Net book value | |||||
| Office Equipment | 3 years | $10,493 | $3,463 | $7,030 | ||||
6. ACCOUNTS PAYABLE AND ACCRUED EXPENSES
The following table summarizes the Company’s accounts payable and accrued expenses as of the following periods:
|
|
|
|
|
|
|
|
|
|
| September 30, | ||||||
|
| 2019 |
| 2018 | ||||
Accounts payable |
| $ | 863,536 |
|
| $ | 719,830 |
|
Accrued expenses |
| 727,053 |
|
| 701,825 |
| ||
|
| $ | 1,590,589 |
|
| $ | 1,421,655 |
|
| March 31, 2017 | March 31, 2016 | |||||||
| Accounts payable | $ | 548,514 | $ | — | ||||
| Accrued expenses | 354,488 | 78,227 | ||||||
| $ | 903,002 | $ | 78,227 | |||||
Accounts payable is predominantly made up of unpaid invoices relating to research and development, accounting and professional fees. Included within the accrued expenses balance of $727,053 at September 30, 2019is $103,672 owed to Tiziana Life Sciences PLC (“Tiziana”) under a shared services agreement (see Note 13), $114,736 relating to vendors for research and development expenses and approximately $136,080 $145,000 of accrued legal, accounting and professional fees.fees, $208,000 of research and development fees, $22,000 for payroll related expenses, $234,000 for Directors fees, $60,000 for Consultancy fees, and $58,000 for credit card expenses. Included within the accrued expenses balance at September 30, 2018 was $72,000 of accrued legal, accounting and professional fees,$193,000 of research and development fees, $22,000 for payroll related expenses and $165,000 for Director fees, $155000 for Consultancy and legal fees, $68,000 for credit card expenses and $55,000 for patent related expenses.
| F-14 |
7. WARRANTS
The Company incurred the obligation to issuehad issued warrants to placement agents relating to fundraising. The Company accountedin lieu of fees for the obligation based on an estimate of the fair value of warrants issued using the Black-Scholes Model (“BSM”).
The following table summarizes warrant activity for the years ended September 30, 2019 and 2018:
|
|
|
|
|
|
|
|
|
|
|
| |
| Number of Warrants |
| Weighted Average Exercise Price Per Option |
| Weighted Average remaining Contractual Life (years) |
| Aggregate Intrinsic Value | |||||
Outstanding balance at October 1, 2017 | 1,926,501 |
|
| 0.43 |
|
| 8.86 |
|
| $ | 6,875,819 |
|
|
|
|
|
|
|
|
| |||||
Granted | — |
|
| — |
|
| — |
|
| — |
| |
|
|
|
|
|
|
|
| |||||
Forfeited | — |
|
| — |
|
| — |
|
| — |
| |
|
|
|
|
|
|
|
| |||||
Outstanding balance at September 30, 2018 | 1,926,501 |
|
| 0.43 |
|
| 7.86 |
|
| $ | 6,875,819 |
|
|
|
|
|
|
|
|
| |||||
Warrants exercisable at September 30, 2018 | 1,926,501 |
|
| 0.43 |
|
| 7.86 |
|
| $ | 6,875,819 |
|
|
|
|
|
|
|
|
| |||||
Granted | — |
|
| — |
|
| — |
|
| — |
| |
|
|
|
|
|
|
|
| |||||
Forfeited | — |
|
| — |
|
| — |
|
| — |
| |
|
|
|
|
|
|
|
| |||||
Outstanding balance at September 30, 2019 | 1,926,501 |
|
| 0.43 |
|
| 6.86 |
|
| $ | — |
|
|
|
|
|
|
|
|
| |||||
Warrants exercisable at September 30, 2019 | 1,926,501 |
|
| 0.43 |
|
| 6.86 |
|
| $ | — |
|
| F-15 |
8. CONVERTIBLE NOTES
On August 8, 2018, the Company entered into a 12% Convertible Promissory Note with High Octane Bioresearch Ltd. (the “Holder”) pursuant to which the Company issued a Convertible Promissory Note to the Holder. The Holder provided the Company with $135,000 in capital.
In relation to the Convertible Promissory Note, the Company has also entered into an agreement with GarcerBioventures , a broker who introduced the Holder to the Company. Under the terms of this agreement, should the Holder convert its principal amount into common stock of the Company, the Company will issue to GarcerBioventures the number of shares equal to 10% of the number of shares of common stock issued to High Octane upon such conversion.
On October 19, 2018, the Company entered into a second 12% Convertible Promissory Note with the Holder with a maturity date of October 19, 2019. The Holder provided the Company with $100,000 in cash, which was received by the Company during the three months ended December 31, 2018, under the same terms as the first Note.
The Company has also entered into another agreement with GarcerBioventures in lieu of fees, under the same terms as the earlier agreement.
In July 2019, the Company extended the maturity date of the Convertible Promissory Notes from August 8, 2019 and October 19, 2019 to August 8, 2020 and October 19, 2020, respectively. All other terms of the Convertible Promissory Notes remained the same. The Amended Convertible Promissory Notes were accounted for as a modification of the original Convertible Promissory Notes as the change in the fair value of the warrants at February 28, 2017embedded conversion option featured in the Convertible Promissory Notes immediately before and after the amendment did not exceed 10% of the carrying amount of the Convertible Promissory Notes.
The Company also evaluated the conversion option embedded in the Amended Convertible Promissory Notes and determined that the change in the fair value of the embedded conversion option of the Convertible Promissory Notes immediately before and after the extension was $2,914,884 based onimmaterial. The Company utilized a Monte Carlo simulation model to determine the following inputsfair value of the Amended Notes. The key assumptions used in the simulation model were:
| August 8 Note | October 19 Note | |||||||
| Stock Price at date of valuation | $ | 0.15 | $ | 0.15 | ||||
| Exercise price | $ | 0.65 | $ | 0.65 | ||||
| Risk-free interest rate | 1.98 | % | 1.98 | % | ||||
| Expected dividend yield | 0 | % | 0 | % | ||||
| Expected term (in years) | 1.04 | 1.24 | ||||||
| Expected Volatility | 82 | % | 82 | % | ||||
At September 30, 2019, there were 1,566,667 shares reserved for the conversion of the Notes and assumptions using156,667 shares were reserved in lieu of fees due to Garcer Bioventures.
Interest expense associated with the Black Scholes Model:
| April 10, 2016 | February 28, 2017 | |
| Warrants to be issued and Issued respectively | 1,440,501 | 1,440,501 |
| Exercise Price | $0.40 | $0.37 |
| Stock Price | $0.40 | $2.10 |
| Expected Term (Years) | 10 | 10 |
| Volatility % | 104% | 105% |
| Discount Rate - Bond Equivalent Yield | 1.93% | 2.55% |
| Dividend Yield | —% | —% |
| F-16 |
9.
2016 EQUITY INCENTIVE PLAN
On July 19, 2016, the Company adopted its 2016 Equity Incentive Plan (the "Equity Incentive Plan"). The planEquity Incentive Plan was established to attract, motivate, retain and reward selected employees and other eligible persons. For the Equity Incentive Plan, employees, officers, directors and consultants who provide services to the Company or one of the Company’s subsidiaries may be selected to receive awards. A total of9,750,000 shares of the Company’s common stock was authorized for issuance with respect to awards granted under the Equity Incentive Plan. During
Stock-based compensation expense is the year ended March 31, 2017,estimated fair value of options granted amortized on a straight-line basis over the requisite service period for the entire portion of the award less an aggregateestimate for anticipated forfeitures. The Company uses the “simplified” method to estimate the expected term of 1,500,000 sharesthe options because the Company’s historical share option exercise experience does not provide a reasonable basis upon which to estimate expected term. No options were granted under the Equity Incentive Plan.
The Black-Scholes option pricing model. Compensation expense is recognized overmodel was developed for use in estimating the periodfair value of service, generallytraded options, which have no vesting restrictions and are fully transferable. In addition, option valuation models require the vesting period. Duringinput of highly subjective assumptions including the year ended March 31, 2017, 1,500,000 options were granted byexpected stock price volatility. Because the Company. NoCompany's stock options were grantedhave characteristics significantly different from those of traded options, and because changes in the year ended March 31, 2016. subjective input assumptions can materially affect the fair value estimate, in management's opinion, the existing models do not necessarily provide a reliable single measure of the fair value of the Company's stock options.
The following assumptions were used in the Black-Scholes options pricing model to estimate the fair value of stock options forthe yearyears ended March 31, 2017:
| Directors and Employees – Vesting period | Non – Employees – Vesting Period | ||||||||||||||||
| Immediate | 1 Year | 2 Years | 3 Years | Immediate | 1 Year | 2 Years | 3 Years | ||||||||||
| Stock Price | $1.495-$1.55 | $1.495-$1.55 | $1.495-$1.55 | $1.495-$1.55 | $1.495 | $1.495-$1.85 | $1.495-$1.85 | $1.495-$1.85 | |||||||||
| Expected life (years) | 5 | 5.5 | 5.75 | 6 | 5 | 5.5 | 5.75 | 6 | |||||||||
| Expected volatility | 85-89% | 85-89% | 85-89% | 85-89% | 85-89% | 85-89% | 85-89% | 85-89% | |||||||||
| Expected dividend yield | —% | —% | —% | —% | —% | —% | —% | —% | |||||||||
| Risk-free interest rate | 0.91% | 0.91% | 0.91% | 0.91% - 1.57% | 1.57% | 1.57% | 1.57% | 1.57% | |||||||||
| F-17 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||
| Directors and Employees - Vesting period |
|
|
| Non - Employees - Vestion Period | ||||||||||||||
| Immediate |
| 1 Year |
| 2 Years |
| 3 Years |
| 4 Years |
| Immediate |
| 1 Year |
| 2 Years |
| 3 Years | ||
Stock Price | $1.495-$1.55 |
| $1.495-$4.00 |
| $1.495-$4.00 |
| $1.495-$4.00 |
| $0.85-$4.00 |
| $0.48 |
| $0.480-$1.85 |
| $0.480-$1.85 |
| $0.150-$0.48 | ||
Expected life (years) | 5 |
| 5.5 |
| 5.75 |
| 6 |
| 6.25 |
| 3.97 |
| 3.97 |
| 3.97 |
| 5.00 | ||
Expected volatility | 85-89% |
| 81-89% |
| 81-89% |
| 81-89% |
| 81% |
| 85-89% |
| 85-89% |
| 85-89% |
| 62-62% | ||
Expected dividend yield | —% |
| —% |
| —% |
| —% |
| —% |
| —% |
| —% |
| —% |
| —% | ||
Risk-free interest rate | 0.91% |
| 0.91% - 1.57% |
| 0.91% - 1.57% |
| 0.91% - 1.57% |
| 0.91% - 1.57% |
| 104.80% |
| 104.80% |
| 104.80% |
| 1.56% | ||
The input assumptions used are as follows:
Discount rate —Based on the daily yield curve rates for U.S. Treasury obligations with maturities which correspond to the expected term of the Company’s stock options.
Dividend yield —The Company has not paid any dividends on common stock since its inception and does not anticipate paying dividends on its common stock in the foreseeable future.
Expected volatility —Based on the historical volatility of seven7 different entities which are comparable Companys’to the Company's stock.
Expected term —The—The Company has had minimal stock options exercised since inception. The expected option term represents the period that stock-based awards are expected to be outstanding based on the simplified method provided in Staff Accounting Bulletin (“SAB”) No. 107, Share-Based Payment , (“SAB No. 107”107”), which averages an award’s weighted-average vesting period and expected term for “plain vanilla” share options. Under SAB No. 107, options are considered to be “plain vanilla” if they have the following basic characteristics: (i) granted “at-the-money”; (ii) exercisability is conditioned upon service through the vesting date; (iii) termination of service prior to vesting results in forfeiture; (iv) limited exercise period following termination of service; and (v) options are non-transferable and non-hedgeable.
The Company will continue to use the simplified method for the expected term until it has the historical data necessary to provide a reasonable estimate of expected life in accordance with SAB No. 107, as amended by SAB No. 110.110. For the expected term, the
Forfeitures —ASC—ASC Topic 718 Compensation - Stock Compensation requires forfeitures to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. The Company has estimated zero forfeiture.
The following table summarizes stock option activity for the yearyears ended March 31, 2017:September 30, 2019and September 30, 2018
| F-18 |
|
|
|
|
|
|
|
|
|
|
| ||||||||||||||
Number of Options |
| Weighted Average Exercise Price Per Option |
| Weighted Average remaining Contractual Life (years) |
| Aggregate Intrinsic Value | ||||||||||||||||||
| Number of Options | Weighted Average Exercise Price Per Option | Weighted Average remaining Contractual Life (years) | Aggregate Intrinsic Value | |||||||||||||||||||||
| Outstanding balance at March 31, 2016 | 1,662,375 | 0.20 | 7.32 | |||||||||||||||||||||
Outstanding balance at October 1, 2017 | 4,829,875 |
|
| 4,829,875 |
|
| 8.22 |
|
| 16,639,397 | ||||||||||||||
|
|
|
|
|
|
| ||||||||||||||||||
| Granted | 1,500,000 | 0.40 | — |
|
| — |
|
| — |
|
| — |
| |||||||||||
| Exercised | — | — | — | — | — | — | ||||||||||||||||||
|
|
|
|
|
|
| ||||||||||||||||||
| Forfeited and Expired | — | — | 10,000 |
|
| 4.00 |
|
| — |
|
| — |
| |||||||||||
|
|
|
|
|
|
| ||||||||||||||||||
| Outstanding balance at March 31, 2017 | 3,162,375 | 0.29 | 8.09 | $ | 5,975,874 | |||||||||||||||||||
Outstanding balance at September 30, 2018 | 4,819,875 |
|
| 0.56 |
|
| 8.22 |
|
| $ | 16,639,397 |
| ||||||||||||
| Options exercisable at March 31, 2017 | 1,629,825 | 0.24 | 7.29 | $ | 3,162,350 | |||||||||||||||||||
| Options exercisable at September 30, 2018 | 3,082,995 | 0.37 | 6.62 | 557,836 | ||||||||||||||||||||
|
|
|
|
|
|
|
| |||||||||||||||||
Granted | — |
|
| — |
|
| — |
|
| — |
| |||||||||||||
|
|
|
|
|
|
|
| |||||||||||||||||
Exercised | — |
|
| — |
|
| — |
|
| — |
| |||||||||||||
|
|
|
|
|
|
|
| |||||||||||||||||
Forfeited and Expired | (746,200 | ) |
| 0.32 |
|
| — |
|
| — |
| |||||||||||||
|
|
|
|
|
|
|
| |||||||||||||||||
Outstanding balance at September 30, 2019 | 4,073,675 |
|
| 0.59 |
|
| 6.37 |
|
| $ | — |
| ||||||||||||
|
|
|
|
|
|
|
| |||||||||||||||||
Options exercisable at September 30, 2019 | 3,183,925 |
|
| 0.48 |
|
| 6.04 |
|
| $ | — |
| ||||||||||||
There were no options exercised during the year years ended March 31, 2017 or 2016.September 30, 2019and September 30, 2018 As of March 31, 2017,September 30, 2019, there was approximately $1,076,368 $187,453of total unrecognized compensation cost related to stock options. The cost is expected to be recognized over a weighted averageweighted-average period of 2.51.50 years.
The chargecharges related to share basedshare-based compensation to directors, officers and non employees isare included within the Consultancygeneral and administrative expense category in the statement of operations. For the years ended September 30, 2019 and September 30, 2018, the charges were $372,846 and $621,931 respectively.
An additional $(4,770) and $($222,591) related to non-employees respectively, has been included within the consultancy fees third parties and related parties expense category in the consolidated financial statements.
| Fourth Quarter | Third Quarter | Second Quarter | First Quarter | |||||||||||||
| Fiscal year ended March 31, 2017 | ||||||||||||||||
| Total revenues | $ | — | $ | — | $ | — | $ | — | ||||||||
| Costs of revenues | — | — | — | — | ||||||||||||
| Gross profit | — | — | — | — | ||||||||||||
| Operating expenses | 990,202 | 1,377,545 | 1,836,946 | 329,834 | ||||||||||||
| Loss from operations | (990,202 | ) | (1,377,545 | ) | (1,836,946 | ) | (329,834 | ) | ||||||||
| Other income (expense) | 42,274 | 21,461 | 34,400 | 2,661 | ||||||||||||
| Income tax provision | — | — | — | — | ||||||||||||
| Net (loss) income | $ | (947,928 | ) | $ | (1,356,084 | ) | $ | (1,802,546 | ) | $ | (327,173 | ) | ||||
| Earnings per share - Basic and diluted | (0.01 | ) | (0.02 | ) | (0.03 | ) | (0.01 | ) | ||||||||
| F-19 |
| Fourth Quarter | Third Quarter | Second Quarter | First Quarter | |||||||||||||
| Fiscal year ended March 31, 2016 | ||||||||||||||||
| Total revenues | $ | — | $ | — | $ | — | $ | — | ||||||||
| Costs of revenues | — | — | — | — | ||||||||||||
| Gross profit | — | — | — | — | ||||||||||||
| Operating expenses | 213,041 | 140,746 | 117,343 | 106,976 | ||||||||||||
| Loss from operations | (213,041 | ) | (140,746 | ) | (117,343 | ) | (106,976 | ) | ||||||||
| Other income (expense) | — | — | — | — | ||||||||||||
| Income tax provision | — | — | — | — | ||||||||||||
| Net (loss) income | $ | (213,041 | ) | $ | (140,746 | ) | $ | (117,343 | ) | $ | (106,976 | ) | ||||
| Earnings per share - Basic and diluted | 0.00 | 0.00 | 0.00 | 0.00 | ||||||||||||
10 INCOME TAXES
The components of Income/(loss)loss before income taxes consisted of the following:
|
| Year Ended September 30, |
|
| Year Ended September 30, | |||
|
| 2019 | 2018 | |||||
US |
| $ | (943,964 | ) |
| $ | (4,063,896 | ) |
Foreign |
| — |
|
| (10,372 | ) | ||
Total |
| $ | (943,964 | ) |
| $ | (4,074,268 | ) |
| March 31, 2017 | ||||
| US | $ | (2,913,073 | ) | |
| Foreign | (1,520,659 | ) | ||
| Total | $ | (4,433,732 | ) | |
As of March 31, 2017,September 30, 2019, the Company is expected to have net operating loss carryforwards of $2approximately $7.34 million for federal tax purposes, which will expire in 2037.
Income tax expenses attributable to income for continuing operations consists of:
| Year Ended September 30, | Year Ended September 30, | |||||
| 2019 | 2018 | |||||
Federal: |
|
| |||||
Current | — |
| — | ||||
Deferred | $ | ( 306,125 | ) | $ | (75,319 | ) | |
|
|
| |||||
Foreign: |
|
| |||||
Current | — |
| — |
| |||
Deferred | — |
| — |
| |||
|
|
| |||||
State and local: |
|
| |||||
Current | — |
| — |
| |||
Deferred | (83,193 | ) | (345,541 | ) | |||
|
|
| |||||
Change in valuation allowance | 381,734 |
| 422,953 | ||||
|
|
| |||||
Income tax (benefit)/expense | $ | (7,584 | ) | $ | 2,093 | ||
| F-20 | ||
Deferred income taxes reflect the net tax effects of temporary differences between the carrying value of the asset and liabilities for financial reporting purposes and amounts used for income tax purposes. The temporary differences that gave rise to the deferred tax assets and liabilities are as follows:
| September 30, | ||||||
| 2019 |
| 2018 | ||||
Deferred tax assets: |
|
|
|
| |||
Accrued Compensation | $ | 73,717 |
|
| $ | 53,854 | |
Stock Compensation | 453,767 |
|
| 365,607 |
| ||
Net operating losses | 2,120,177 |
|
| 2,019,018 |
| ||
| R&D Credit carryforward | 148,715 | — | |||||
Fixed assets | 561 |
|
| — |
| ||
Total gross deferred tax asset | 2,796,937 |
|
| 2,438,479 |
| ||
|
|
|
|
| |||
Less: valuation allowance | (2,784,801 | ) | (2,438,300 | ) | |||
|
|
|
|
| |||
Net deferred tax asset | 12,136 |
|
| 179 |
| ||
|
|
|
|
| |||
Deferred tax liabilities: |
|
| |||||
Intangible assets | (15,170 | ) | (10,619 | ) | |||
Fixed assets | — | (178 | ) | ||||
Total Deferred tax liabilities | (15,170 | ) | (10,797 | ) | |||
|
|
|
|
| |||
Net Deferred Income Tax Liability | $ | (3,034 | ) | $ | (10,618 | ) | |
In assessing the realizability of the deferred tax assets, management considers whether it is more likely than not that some portion or all of the deferred tax assets will not be realized. Management considers the scheduled reversal of deferred tax liabilities, projected future taxable income, net operating loss carryback potential, and tax planning strategies in making these assessments.
Based upon the above criteria, the Company believes that it is more likely than not that the full amount of the remaining net deferred tax assets will not be realized. Accordingly, the Company has recorded a full valuation allowance of approximately $1.0 $2.79million against the deferred tax asset that is not realizable.
The Company recognizes interest accrued to unrecognized tax benefits and penalties as income tax expense. The Company accrued no penalties andor interest during the yearyears ended March 31, 2017,September 30, 2019 and asSeptember 30, 2018.
| F-21 |
A reconciliation of the statutory Federal Income tax rate and effective tax rate of the provision for income taxes is as follows:
| Year ended September 30, | Year ended September 30, | ||
| 2019 | 2018 | ||
Federal statutory rate | 21.00 | % | 25.08 | % |
Permanent items | 6.91 | % | (4.41 | )% |
Foreign rate differential | — | % | (0.06 | )% |
2017 Stock based compensation true up | — | % | — | % |
State taxes | 6.96 | % | 6.41 | % |
Increase in valuation allowance | (40.42 | )% | (10.37 | )% |
Impact of the change to Federal Statutory Tax | 6.35 | % | (16.70 | )% |
|
|
| ||
Effective income tax rate | 0.80 | % | (0.05 | )% |
The Company files tax returns as prescribed by the tax laws of the jurisdictions in which they operate. In the normal course of business, the Company is subject to examination by federal and foreign jurisdictions where applicable based on the statute of limitations that apply in each jurisdiction. As of March 31, 2017,September 30, 2019, open years related to the Federalfederal jurisdiction are fiscal years ending 2016, 20152018, 2017 , and 2014. Open years related to the foreign jurisdiction are 2015 and 2014.
The Company has no open tax audits for the returns that were filed, with any tax authority as of March 31, 2017.September 30, 2019. Accordingly, there were no material uncertain tax positions in any of the jurisdictions that the Company operated in.
During the normal course of its business, the Company enters into various transactions with entities that are both businesses and individuals. The following is a summary of the related party transactions during the years ended September 30, 2019 and 2018.
Eurema Consulting
Eurema Consulting S.r.l. is a significant shareholder of the Company. During the years ended September 30, 2019 and 2018, Eurema Consulting did not supply the Company with consulting services. As of September 30,2019, and September 30, 2018, the balance due to Eurema Consulting S.r.l. was $200,000 for past consultancy services.
Gabriele Cerrone
Gabriele Cerrone is the majority shareholder of Panetta Partners, one of the Company's principal shareholders and was a director of Arna Therapeutics Ltd. During the years ended September 30, 2019 and 2018, Gabriele Cerrone did not supply the Company with consulting services. As of September 30,2019, and September 30, 2018, the balance due to Gabriele Cerrone was $175,000 for past consultancy services.
Roberto Pellicciari and TES Pharma
Roberto Pellicciari is the majority shareholder of TES Pharma Srl, one of the Company's principal shareholders. During the years ended September 30, 2019 and 2018,Roberto Pellicciari did not supply the Company with consulting services. As of September 30,2019, and September 30, 2018,the balance due to Roberto Pellicciari was $175,000 for past consultancy services. Alessandro Padova is the chairman of Rasna Therapeutics Inc. and also serves on the Board of Directors of TES Pharma, one of the Company's suppliers. At both September 30, 2019 and September 30, 2018, TES Pharma was owed $75,000.
| F-22 |
Tiziana Life Sciences Plc ("Tiziana")
As at September 30, 2019 and 2018, the balance owed by Tiziana Life Sciences Plc was $14,335 and $252,746 respectively. As of the date these condensed consolidated financial statements are issued, the related party receivable had not been fully collected.Kunwar Shailubhai, CEO and a director of the Company, is also a director of Tiziana. In addition, Tiziano Lazzaretti, the Company's CFO, is also CFO of Tiziana. The Company is party to a Shared Services agreement with Tiziana whereby the Company is charged for shared services such as the payroll and rent, see Note 9 for more details.
Panetta Partners
Panetta Partners Limited, a shareholder of Arna, is a company in which Gabriele Cerrone has significant interest and also serves as a director. At September 30, 2019 and September 30, 2018, there was no balance owed to or from Panetta Partners Limited.
There is no interest charged on the balances with related parties. There are no defined repayment terms and such amounts can be called for payment at any time.
12. COMMITMENTS AND CONTINGENCIES
License Agreements
In November 2016, the Company entered into a license agreement with Profs. Falini and Martellii, wherein it obtained the exclusive rights related to the use or reformulation of Actinomycin D and intends to utilize these rights for the development of new product. In connection with this agreement, the Company is committed to paying milestone payments, the first being a EUR 50,000 payment to be paid6 months after the agreement was signed. The payment was made to Profs. Falini and Martelli in June 2017.
The specific timing of the remaining milestones cannot be predicted and depend upon research and clinical developments.
Lease Agreements
In January 2017, the Company entered into a lease agreement with Bucks County Biotechnology Centre IncInc. in Doylestown Pennsylvania, where certain employees of the Company are based. The lease provides for annual basic lease payments from February 1, 2017 to January 31, 2018, of $13,480,$13,480, plus and utility expense estimate of $237$237 per month.
Consultancy Agreements
In October 2016, the Company entered into a consultancy agreement with Tiziano Lazzaretti in which he agreed to serve as Chief Financial Officer for a fee of $50,000$50,000 per year. This was increased to $80,000 a year in April 2017 by the Company's compensation committee.
Employment Agreements
On May 24, 2017, the Company entered into an executive employment agreement with Kunwar Shailubhai to serve as Chief Executive Officer and Chief Scientific Officer for a remuneration of $300,000 per annum. Also included within the agreement is a performance related bonus of 35% of base salary. Based on Board discretion, it is not probable that this performance obligation will be met, therefore no bonus has been accrued as of September 30, 2019. On January 16 2019, this employment contract was terminated in recognition of the reduction in time spent on Rasna activities.
In September 2016,June 2017, the boardBoard of Directors awarded Mr Lazzaretti 100,000Dr Shailubhai 1,700,000 options to vest over a 34 year period. An additional 200,000 optionsperiod, with a 3 year vesting period, were also awarded in November 2016. The options under both awards have an exercise price of $0.40.
In January 2019, the decision was taken to terminate all employment agreements commencingcontracts in Rasna Therapeutics Inc. effective January 2017. These appointments relate16, 2019. This was to clinical and non clinicalreflect the fact that employees and are reviewablewere spending an increasing amount of time working on an annual basis. The Company's committed to paying $278,500 forTiziana Life Sciences Plc. Moving forward, any time spent on Rasna activities will be charged by Tiziana Life Sciences PLC under the period to December 2017.shared services agreement detailed below.
The Company has entered into a shared services agreement with Tiziana Life Sciences Plc. Under the terms of this agreement, the Company will be charged for shared administrative services including payroll and rent for the LexingtomLexington Avenue, NY premises, on a monthly basis based on allocated costs incurred. This agreement is effective from January 1, 2017. At March 31, 2017, $103,672September 30, 2019,$0 is due toby Tiziana Life Sciences PLC.
Other Commitments
The Company has entered into certain licensing agreements for products currently under development. The Company may be obligated in future periods to make additional payments, which would become due and payable only upon the achievement of certain research and development, regulatory, and approval milestones. The specific timing of such milestones cannot be predicted and depend upon future discretionary research and clinical developments, as well as, regulatory agency actions. Further, under the terms of certain agreements the Company may be obligated to pay commercial milestones contingent upon the realization of sales revenues and sublicense revenues. Due to the long range nature of such commercial milestones, they are neither probable at this time nor predictable, and consequently are not considered contingent milestone payment amounts.
13. SUBSEQUENT EVENTS
On April 14, 2017
The Note contains an anti-dilution provision which adjusts the conversion price in the event of an issuance by the Company of common stock below the then effective conversion price.
In connection with his appointment, Dr. Shailubhai was granted incentive stock optionsthe promissory note, the Company has also agreed to purchase 1,700,000issue to the introductory agent a number of shares equal to 10% of the number of shares of common stock at $0.85 per share. 425,000 ofissued to noteholder upon such stock options vest on each of the 1st, 2nd, 3rd and 4
| F-24 |